Область техники
Настоящее изобретение относится к пиперазиновому соединению или его соли и фармацевтической композиции, содержащей пиперазиновое соединение или его соль в качестве активного компонента, и, в частности, к средству для предотвращения и/или лечения аллергического заболевания, воспалительного заболевания и миодегенеративного заболевания, обусловленному его ингибирующим действием в отношении гематопоэтической простагландин-D-синтазы.
Уровень техники
Простагландин D2 (PGD2) представляет собой воспалительный медиатор, продуцируемый и в больших количествах высвобождаемый тучными клетками, которые активируются при связывании антигенов с иммуноглобулином E (НПЛ 1), и, как предполагается, играет важную роль при объяснении механизма аллергических реакций. Высокие концентрации PGD2 обнаружены в бронхоальвеолярной жидкости астматиков (НПЛ 2), и сообщалось о том, что бронхоспазм был индуцирован ингаляцией простагландина D2 у пациентов с астмой, в отличие от здоровых субъектов (НПЛ 3).
С другой стороны, синтазы, которые генерируют PGD2, называются простагландин-D-синтазами. Известно два различных типа: гематопоэтическая простагландин-D-синтаза и простагландин-D-синтаза липокаинового типа. PGD2 участвует в начале и обострении различных заболеваний, включая аллергии, и в регуляторных механизмах организма; поэтому фармацевтические составы, которые могут повысить качество избыточного продуцирования, рассматриваются как очень эффективные в лечении различных заболеваний.
Человеческие гематопоэтические простагландин-D-синтазы (H-PGDS) в основном распределены в плаценте, легких, печени плода, лимфатических узлах, мозге, сердце, тимусе, костном мозге и селезенке. Кроме того, на клеточном уровне, они, как сообщают, экспрессируются в микроглии в мозге, мегакариоцитах и клетках Лангерганса в коже; клетках Купфера в печени; макрофагах; и многих других антиген-презентирующих клетках, таких как дендритные клетки, тучные клетки и Th2-клетки.
Кроме того, из-за того что H-PGDS в больших количествах экспрессируются в тучных клетках или воспалительных клетках в слизистой оболочке носа при аллергическом рините или в полипах носа при хроническом синусите, считается, что PGD2, продуцированный H-PGDS, играет важную роль в начале и обострении аллергических заболеваний, таких как астма, риносинусит, дерматит и хроническое обструктивное заболевание легких (НПЛ 4). Дополнительно была подтверждена экспрессия H-PGDS в некротизированной части скелетных мышц, в которых экспрессия H-PGDS обычно не встречается (НПЛ 5). На основании изложенного было предположено, что PGD2, продуцируемый гематопоэтической простагландин-D-синтазой, принимает участие в заболеваниях, сопровождаемых повреждением ткани, таких как мышечная дистрофия, амиотрофический боковой склероз, рассеянный склероз, неспецифический язвенный колит, ревматоидный артрит и хроническое обструктивное артериальное заболевание (НПЛ 6).
Поэтому ожидается, что ингибитор H-PGDS может быть использован в качестве фармацевтического препарата, который применяется в качестве средства предотвращения и/или лечения заболеваний, таких как аллергическое заболевание и воспалительное заболевание, в котором участвует PGD2, продуцированный гематопоэтической простагландин-D-синтазой, или его метаболит, а также некроз мышц и травматические повреждения головного мозга.
В некоторых источниках, касающихся ингибитора H-PGDS (например, ПТЛ 1 и 2) и источник Патентной Литературы 3, раскрывается ингибитор H-PGDS, имеющий структуру, подобную соединению по настоящему изобретению. Кроме того, широко изучались фармакологические свойства пиперазиновых соединений, помимо ингибиторов H-PGDS.
Источник Патентной Литературы 4 раскрывает в качестве hedgehog сигнального ингибитора пиперазиновое соединение, имеющее фурилкарбонильную пиперазиновую структуру. Источник Патентной Литературы 5 раскрывает широкий ряд пиперазиновых соединений в качестве соединений, взаимодействующих с калиевыми каналами.
Источник Патентной Литературы 6 раскрывает соединение мочевины с пиперазиновым кольцом в качестве соединения, полезного для лечения заболеваний, в которые вовлечена гидролаза амидов жирных кислот.
Список цитируемых источников
Патентная Литература
ПТЛ 1: WO2007-007778
ПТЛ 2: WO2007-041634
ПТЛ 3: WO2008-122787
ПТЛ 4: WO2007-054623
ПТЛ 5: WO99/007672
ПТЛ 6: WO2008-023720
Непатентная Литература
НПЛ 1: J. Immunol., 129, 1627-1631 (1982)
НПЛ 2: N. Eng. J. Med., 315, 800-804 (1986)
НПЛ 3: N. Eng. J. Med., 311, 209-213 (1984)
НПЛ 4: Arch. Otolaryngol Head Neck Surg., 133, 693-700 (2007)
НПЛ 5: Acta Neuropathol., 104, 377-384 (2002)
НПЛ 6: Am J Pathol., 174(5), 1735-1744 (2009)
Краткое описание изобретения
Техническая задача
Основная цель настоящего изобретения заключается в том, чтобы обеспечить новое соединение, которое показывает, в низкой дозе, высокую ингибирующую активность в отношении простагландин-D-синтазы, и, в частности, H-PGDS.
Другая дополнительная цель настоящего изобретения заключается в том, чтобы обеспечить лекарственное средство с незначительным числом побочных эффектов и высокой безопасностью, где эффективность лекарственного средства обусловлена его ингибирующим H-PGDS действием, при предотвращении и/или лечении заболеваний, опосредованных PGD2, который генерируется синтазой, или его метаболитом.
Решение задачи
Авторы настоящего изобретения провели обширное исследование соединений, проявляющих ингибирующее H-PGDS действие, и обнаружили, что новое пиперазиновое соединение, представленное формулой (I), обладает крайне высокой ингибирующей активностью в отношении H-PGDS. Авторы провели дальнейшие исследования и осуществили настоящее изобретение.
Настоящее изобретение обеспечивает пиперазиновое соединение, фармацевтическую композицию, ингибитор простагландин-D-синтазы и средство для предотвращения и/или лечения заболевания, в котором участвует простагландин D2 или его метаболит, как описано ниже.
1. Пиперазиновое соединение, представленное формулой (I),
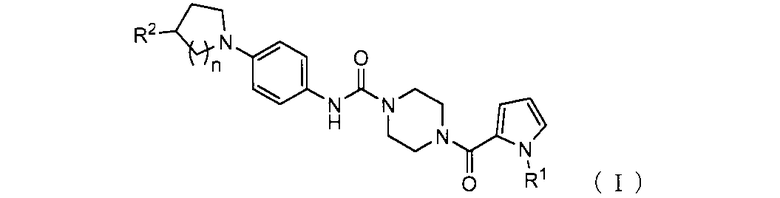
где
R1 представляет собой C1-6 алкил;
R2 представляет собой гидрокси, C1-6 алкил, который может содержать один или более заместителей, -(C=O)-N(R3)(R4) или -(C=O)-OR5;
R3 и R4 могут быть одинаковыми или отличаться, и каждый представляет собой водород или C1-6 алкил, который может содержать один или более заместителей, или
R3 и R4, связанные через атом азота, к которому присоединены R3 и R4, могут образовывать насыщенную гетероциклическую группу;
R5 представляет собой водород или C1-6 алкил, который может содержать один или более заместителей; и
n представляет собой 1 или 2;
или его соль.
2. Пиперазиновое соединение или его соль в соответствии с пунктом 1, где
R1 представляет собой метил или этил;
R2 представляет собой гидрокси, C1-6 алкил, который может иметь одну или более насыщенных или ненасыщенных гетероциклических групп в качестве заместителей, -(C=O)-N(R3)(R4) или -(C=O)-OR5;
R3 и R4 могут быть одинаковыми или отличаться, и каждый представляет собой водород или C1-6 алкил, который может иметь одну или более насыщенных или ненасыщенных гетероциклических групп в качестве заместителей, или
R3 и R4, связанные через атом азота, к которому присоединены R3 и R4, могут образовать пирролидинил, пиперидинил, пиперазинил и морфолино;
R5 представляет собой водород, метил, этил, трет-бутил или бензил; и
n представляет собой 1 или 2.
3. Пиперазиновое соединение или его соль в соответствии с пунктом 1 или 2, где
R1 представляет собой метил;
R2 представляет собой C1-3 алкил, который может содержать морфолино, пиразолил или триазолил в качестве заместителей, -(C=O)-N(R3)(R4) или -(C=O)-OR5; и триазолил может содержать один или два заместителя;
R3 и R4, связанные через атом азота, к которому присоединены R3 и R4, могут образовывать морфолино;
R5 представляет собой водород, метил или этил; и
n представляет собой 2.
4. Пиперазиновое соединение или его соль по любому из пунктов 1-3, где
R1 представляет собой метил;
R2 представляет собой линейный C1-3 алкил, который может содержать любой из 1,2,3-триазолила, 3,5-диметил-1,2,4-триазолила и морфолино в качестве заместителя, -(C=O)-N(R3)(R4) или -(C=O)-OR5;
R3 и R4, связанные через атом азота, к которому присоединены R3 и R4, могут образовать морфолино;
R5 представляет собой водород или этил; и
n представляет собой 2.
5. Пиперазиновое соединение или его соль в соответствии с пунктом 1, выбранное из группы, состоящей из следующего:
N-(4-(4-гидроксипиперидин-1-ил)фенил)-4-((1-метилпиррол-2-ил)карбонил)-1-пиперазинкарбоксамид,
4-((((1-метилпиррол-2-ил)карбонил)-1-пиперазинил)карбонил)амино-4-фенилпиперидин-4-карбоновая кислота,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолинометилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(3-(2-(1,2,4-триазол-1-ил)этил)пирролидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолиноэтилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-(1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(1,2,3-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(1,2,4-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(3,5-диметил-1,2,4-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(пиразол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(3-(1,2,4-триазол-1-ил)пропил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-этилпиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-пиперидин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(4-метилпиперазин-1-ил-карбонил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-морфолиноэтилкарбамоил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид и
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(пиридин-3-илметилкарбамоил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид.
6. Фармацевтическая композиция, включающая эффективное количество по меньшей мере одного из соединений по пунктам 1-5 или его фармацевтически приемлемые соли и фармацевтически приемлемый носитель.
7. Ингибитор простагландин-D-синтазы, включающий эффективное количество соединения по любому из пунктов 1-5 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
8. Средство для предотвращения и/или лечения заболевания, в котором участвует простагландин D2 или его метаболит, где средство включает эффективное количество соединения по любому из пунктов 1-5 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
9. Средство в соответствии с пунктом 8, где заболевание, в котором участвует простагландин D2 или его метаболит, является аллергическим заболеванием, воспалительным заболеванием или миодегенеративным заболеванием.
10. Способ предотвращения и/или лечения заболевания, в котором участвует простагландин D2 или его метаболит, где способ включает введение млекопитающему соединения по любому из пунктов 1-5 в количестве, эффективном для предотвращения или лечения заболевания.
11. Применение соединения по любому из пунктов 1-5 для получения средства для предотвращения и/или лечения заболевания, в котором участвуют простагландин D2 или его метаболит.
12. Соединение по любому из пунктов 1-5 для применения в способе предотвращения и/или лечения заболевания, в котором участвуют простагландин D2 или его метаболит.
Полезные эффекты изобретения
Настоящее изобретение обеспечивает новое пиперазиновое соединение, представленное вышеприведенной формулой (I), или его соль, которое является ингибитором простагландин-D-синтазы и, в частности, ингибитором H-PGDS.
Пиперазиновое соединение или его соль в соответствии с настоящим изобретением проявляет превосходную активность в отношении ингибирования H-PGDS in vitro. Кроме того, поскольку у морских свинок с антиген-индуцированным ринитом наблюдали ингибирующую H-PGDS активность на промывную жидкость из полости носа и ингибирующую эозинофильную инфильтрацию активность, было показано, что пиперазиновое соединение или его соль обладают превосходной активностью в отношении улучшения состояний заложенности носа и ингибирующей эозинофильное воспаление активностью. Кроме того, в исследованиях на силу захвата передней части конечностей мышей mdx было показано значительное улучшение при потере мышечной силы, что демонстрирует, что пиперазиновое соединение или его соль по настоящему изобретению могут применяться для лечения миодегенеративного заболевания, такого как мышечная дистрофия.
Таким образом, поскольку пиперазиновое соединение или его соль по настоящему изобретению проявляет превосходную ингибирующую H-PGDS активность, оно может применяться в качестве средства для предотвращения и/или лечения заболевания, в котором участвуют PGD2 или его метаболит, такого как аллергическое заболевание, воспалительное заболевание и миодегенеративное заболевание, и предполагаются также и другие области применения.
Описание вариантов осуществления
Пиперазиновое соединение, представленное формулой (I),
,
где
R1 представляет собой C1-6 алкил;
R2 представляет собой гидрокси, C1-6 алкил, который может содержать один или более заместителей, -(C=O)-N(R3)(R4) или -(C=O)-OR5;
R3 и R4 могут быть одинаковыми или отличаться, и каждый представляет собой водород или C1-6 алкил, который может содержать один или более заместителей, или
R3 и R4, связанные через атом азота, к которому присоединены R3 и R4, могут образовывать насыщенную гетероциклическую группу;
R5 представляет собой водород или C1-6 алкил, который может содержать один или более заместителей; и
n представляет собой 1 или 2;
или его соль.
Пиперазиновое соединение по настоящему изобретению, которое представлено формулой (I), представляет собой соединение с (N-алкилпиррол-2-ил)карбонилом и фениламинокарбонилом, которое является новым соединением и конкретно не раскрывается в вышеуказанных ссылках.
Например, в источнике Патентной Литературы 3 (WO2008/122787) раскрывается широкий ряд пиперазиновых соединений, ингибирующих H-PGDS; однако полностью умалчивается о пиперазиновом соединении с (N-алкилпиррол-2-ил)карбонилом, который содержится в соединении по настоящему изобретению. Дополнительно, как показано в контрольных примерах, описанных ниже, соединения, продемонстрированные в примерах (Справочные примеры 13-8) источника Патентной Литературы 3, не проявляют ингибирующего PGD2 действия на промывную жидкость из полости носа у морских свинок с антиген-индуцированным ринитом.
Источник Патентной Литературы 4 (WO2007/054623) раскрывает ингибирующее hedgehog сигнальный путь пиперазиновое соединение, имеющее фурилкарбонильную пиперазиновую структуру; однако, в отличие от источника Патентной Литературы 4, настоящее изобретение заключается в том, что (N-алкилпиррол-2-ил)карбонил, содержащийся в соединении по настоящему изобретению, ограничен фурилкарбонилом. Кроме того, источник Патентной Литературы 4 полностью умалчивает об ингибирующей H-PGDS активности.
Источник Патентной Литературы 5 (WO99/007672) раскрывает фурилкарбонильное пиперазиновое соединение, бензоилпиперазиновое соединение и т.д. в качестве соединения, которое взаимодействует с калиевыми каналами. Однако в источнике патентной литературе 5 не раскрывается соединение, содержащее (N-алкилпиррол-2-ил)карбонил, как в настоящем соединении, и полностью умалчивается об ингибирующей H-PGDS активности.
Источник Патентной Литературы 6 (WO2008/023720) раскрывает соединение мочевины, содержащее пиперидиновое кольцо, в качестве соединения для лечения заболевания, ассоциируемого с гидролазой амидов жирных кислот. Однако источник Патентной Литературы 6 не раскрывает соединение, содержащее (N-алкилпиррол-2-ил)карбонил, как в настоящем соединении, и полностью умалчивает об ингибирующей H-PGDS активности.
Как показано в контрольных примерах ниже, пиперазиновое соединение, не содержащее (N-алкилпиррол-2-ил)карбонил, практически не проявляет ингибирующего H-PGDS действия.
Примеры «заместителей» в настоящем описании включают галоген, гидрокси, циано, нитро, алкил, галогеналкил, циклоалкил, циклоалкилалкил, аралкил, алкенил, алкинил, алкокси, галогеналкокси, циклоалкокси, циклоалкилалкокси, аралкилокси, алкилтио, циклоалкилалкилтио, амино, моно- или диалкиламино, циклоалкилалкиламино, ацил, ацилокси, оксо, карбокси, алкоксикарбонил, аралкилоксикарбонил, карбамоил, насыщенные или ненасыщенные гетероциклические группы, ароматический углеводород, насыщенную гетероциклоксигруппу и т.д. Если такие заместители присутствуют, то их количество, как правило, составляет от 1 до 3.
В заместителях примеры галогена включают хлор, бром, фтор и йод.
В заместителях алкил или галогеналкил представляют собой предпочтительно линейную или разветвленную C1-6 алкильную группу или группу, в которой от одного до всех водородных атомов алкильной группы замещены галогеном, описанным выше. Примеры включают алкильные группы, такие как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил и гексил, и галогеналкильные группы, такие как трифторметил.
В заместителях циклоалкил представляет собой предпочтительно C3-7 циклоалкильную группу, и его примеры включают циклопропил, циклобутил, циклофенил, циклогексил и циклофенил.
В заместителях циклоалкилалкил представляет собой предпочтительно C1-6 алкильную группу, замещенную C3-7 циклоалкилом, и его примеры включают циклопропилметил, циклопропилэтил, циклобутилметил, циклопентилметил и циклогексилметил.
В заместителях аралкил представляет собой предпочтительно линейную или разветвленную C1-6 алкильную группу, замещенную C6-14 ароматической углеводородной группой, и его примеры включают бензил, фенилэтил, фенилпропил, нафтилметил и нафтилэтил.
В заместителях алкенил представляет собой предпочтительно C2-6 алкенильную группу, содержащую двойную углерод-углеродную связь, и его примеры включают винил, аллил, метилвинил, пропенил, бутенил, пентенил и гексенил.
В заместителях алкинил представляет собой предпочтительно C2-6 алкинильную группу, содержащую тройную углерод-углеродную связь, и его примеры включают этинил и пропаргил.
В заместителях алкокси или галогеналкокси представляет собой предпочтительно линейную или разветвленную C1-6 алкокси группу или алкокси группу, замещенную галогеном, описанным выше, и его примеры включают метокси, этокси, н-пропокси, изопропокси, 1-метилпропокси, н-бутокси, изобутокси, трет-бутокси, 2-метилбутокси, неопентилокси, пентан-2-илокси, фторметокси, дифторметокси, трифторметокси, 1,1-дифторэтокси, 2,2-дифторэтокси, 2,2,2-трифторэтокси, 1,1,2,2-тетрафторэтокси, перфторэтокси, 3-фтор-2-(фторметил)пропокси, 1,3-дифторпропан-2-илокси и 2,2,3,3,3-пентафтор-1-пропокси.
В заместителях циклоалкокси представляет собой предпочтительно C3-7 циклоалкокси группу, и его примеры включают циклопропокси, циклобутокси, циклопентилокси, циклогексилокси и циклогептилокси.
В заместителях циклоалкилалкокси представляет собой предпочтительно C1-6 алкокси группу, замещенную C3-7 циклоалкилом, и его примеры включают циклопропилметокси, циклопропилэтокси, циклобутилметокси, циклопентилметокси и циклогексилметокси.
В заместителях алкилтио представляет собой предпочтительно линейную или разветвленную C1-6 алкилтио группу, и его примеры включают метилтио, этилтио, н-пропилтио, изопропилтио, н-бутилтио, изобутилтио, втор-бутилтио, трет-бутилтио, пентилтио и гексилтио.
В заместителях циклоалкилалкилтио представляет собой предпочтительно C1-6 алкилтио группу, замещенную C3-7 циклоалкилом, и его примеры включают циклопропилметилтио, циклопропилэтилтио, циклобутилметилтио, циклопентилметилтио и циклогексилметилтио.
В заместителях аралкилокси представляет собой предпочтительно окси группу с вышеупомянутой аралкил группой, и их примеры включают бензилокси, фенэтилокси, фенилпропилокси, нафтилметилокси и нафтилэтилокси.
В заместителях моно- или диалкиламино представляют собой моно- или диаминогруппы, замещенные линейной или разветвленной C1-6 алкильной группой, и его примеры включают метиламино, диметиламино, этиламино, диэтиламино и метилэтиламино.
В заместителях циклоалкилалкиламино представляет собой алкиламино группу, замещенную вышеупомянутой циклоалкильной группой, и его примеры включают циклопропилметиламино, циклобутилметиламино и циклопентилметиламино.
В заместителях ацил представляет собой линейную или разветвленную C1-6 ацильную группу или бензоильную группу, и его примеры включают формил, ацетил, пропионил, н-бутирил, изобутирил, валерил, изовалерил и пивалоил.
В заместителях ацилокси представляет собой линейную или разветвленную C1-6 ацилокси группу или бензоилокси группу, и его примеры включают формилокси, ацетокси, пропионилокси, н-бутирилокси, изобутирилокси, валерилокси, изовалерилокси и пивалоилокси.
В заместителях алкоксикарбонил представляет собой карбонильную группу, замещенную вышеупомянутой алкокси группой, и его примеры включают метоксикарбонил, этоксикарбонил, н-пропоксикарбонил, изопропоксикарбонил, 1-метилпропоксикарбонил, н-бутоксикарбонил, изобутоксикарбонил, трет-бутоксикарбонил, 2-метилбутоксикарбонил, неопентилоксикарбонил и пентан-2-илоксикарбонил.
В заместителях аралкилоксикарбонил представляет собой предпочтительно карбонильную группу, замещенную вышеупомянутой аралкилокси группой, и его примеры включают бензилоксикарбонил, фенэтилоксикарбонил, фенилпропилоксикарбонил, нафтилметилоксикарбонил и нафтилэтилоксикарбонил.
В заместителях примеры карбамоила включают -CONH2, (моно- или диалкил)карбамоил, (моно- или диарил)карбамоил, (N-алкил-N-арил)карбамоил, пирролидинокарбамоил, пиперидинокарбамоил, пиперазинокарбамоил и морфолинокарбамоил.
В заместителях насыщенные или ненасыщенные гетероциклические группы представляют собой предпочтительно моноциклические или бициклические насыщенные или ненасыщенные гетероциклические группы, которые могут содержать любой из следующего: кислород, азот или сера, предпочтительно в количестве от 1 до 4. Их примеры включают пирролидинил, пиперидинил, пиперазинил, гексаметиленимино, морфолино, тиоморфолино, гомопиперазинил, тетрагидрофуранил, тетрагидропиранил, имидазолил, тиенил, фурил, пирролил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиразолил, триазолил, тетразолил, пиридил, пиразил, пиримидинил, пиридазинил, индолил, изоиндолил, индазолил, метилендиоксифенил, этилендиоксифенил, бензофуранил, дигидробензофуранил, бензоимидазолил, бензоксазолил, бензотиазолил, пуринил, хинолил, изохинолил, хиназолил и хиноксалил.
В заместителях ароматический углеводород представляет собой предпочтительно C6-14 ароматическую углеводородную группу, и его примеры включают фенил и нафтил.
В заместителях насыщенная гетероциклокси группа представляет собой моноциклическую насыщенную гетероциклическую группу, содержащую любое из следующего: кислород, азот и сера, в количестве одного или двух, и его примеры включают окси группы, содержащие пирролидинил, пиперидинил, пиперазинил, гексаметиленимино, морфолино, тиоморфолино или гомопиперазинил, такие как тетрагидрофуранилокси и тетрагидропиранилокси.
"C1-6 алкил", представленный как R1 в формуле (I), представляет собой линейную или разветвленную C1-6 алкильную группу, и его примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил и н-гексил. Из них метил и этил являются предпочтительными, а метил более предпочтителен.
Примеры «C1-6 алкила, которые могут иметь один или более заместителей» из «Ci-6 алкила», представленные как R2 в формуле (I), включают C1-6 алкил, представленный как R1 в формуле (I). Из них C1-3 алкил является предпочтительным, a C1-3 алкил, такой как метил, этил и н-пропил, более предпочтителен.
Примеры «заместителей» «C1-6 алкила, которые могут иметь один или более заместителей», представленных R2, включают вышеупомянутые заместители. Гидрокси и насыщенные или ненасыщенные гетероциклические группы являются предпочтительными; морфолино, пиразолил и триазолил более предпочтительны; и морфолино, 1,2,3-триазолил и 1,2,4-триазолил особенно предпочтительны. Ненасыщенные гетероциклические группы могут содержать заместители. Предпочтительным заместителем является метил, а количество заместителей составляет 1 или 2. Предпочтительные примеры «C1-6 алкила, которые могут содержать один или более заместителей», представленного R2, включают морфолино, 1,2,3-триазолил и 3,5-диметил-1,2,4-триазолил.
Примеры «С1-6 алкила» из «C1-6 алкила, который может содержать один или более заместителей», представленного как R3 и R4 в формуле (I), включают C1-6, алкил, представленный как R1 в формуле (I). Из них С1-3 алкил является предпочтительным.
Примеры «заместителей» «C1-6 алкила, который может содержать один или более заместителей», представленного как R3 и R4, включают вышеупомянутые заместители. Из них насыщенные или ненасыщенные гетероциклические группы являются предпочтительными.
Примеры «насыщенных гетероциклических групп», которые могут быть образованы из R3 и R4 в формуле (I) и атома азота, к которому присоединены R3 и R4, включают пирролидинил, пиперидинил, пиперазинил и морфолино, и морфолино является предпочтительным.
Предпочтительные примеры «C1-6 алкила, которые могут иметь один или более заместителей», представленного как R5 в формуле (I), включают метил, этил, трет-бутил и бензил.
В формуле (I) n представляет собой 1 или 2, и предпочтительно составляет 2.
Пиперазиновое соединение по настоящему изобретению может быть получено в соответствии с нижеприведенными схемами реакции от 1 до 5.
Схема реакции 1
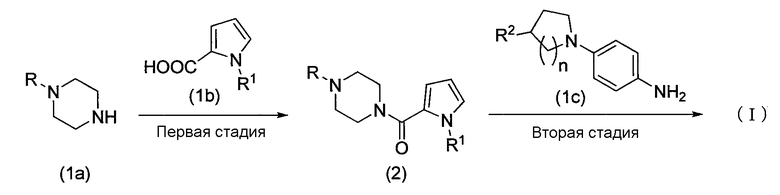
В приведенной выше схеме реакции 1 R1 и R2 являются такими, как указано выше, R представляет собой защитную группу аминогруппы или водорода, и n представляет собой 1 или 2.
Способ по настоящему изобретению включает первую стадию, в которой пиперазиновое соединение, представленное формулой (1a), или его соль конденсируется с соединением пирролкарбоновой кислоты, представленным формулой (1b), или его активным производным обычным методом с образованием амидного соединения, представленного формулой (2), и вторую стадию, в которой соединение амина, полученное путем удаления защитной группы аминогруппы, или его соль конденсируется с соединением амина, представленным формулой (1c), или его активным производным обычным методом с образованием соединения, представленного формулой (I).
Первая стадия
На первой стадии пиперазиновое соединение, представленное формулой (1a), или его соль конденсируется с соединением пирролкарбоновой кислоты, представленным формулой (1b), или его активным производным обычным методом, таким образом, получая соединение амида, представленное формулой (2).
Примеры активных производных соединения (1b) включают активные сложные эфиры, например обычные сложные эфиры, такие как метиловые сложные эфиры; галоидангидриды, такие как хлорангидриды; и Н-гидроксибензотриазол; и симметричные ангидриды кислот; и смеси ангидридов кислот с алкилкарбоновыми кислотами.
Если соединение (1b) вступает в реакцию со свободной кислотой или если активный сложный эфир или галогенангидрид вступают в реакцию без выделения, то могут быть использованы конденсирующие агенты, такие как 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид.
Если на 1 моль пиперазинового соединения, представленного формулой (1a), или его соли используется от 0,5 до 10 моль, и предпочтительно от 0,8 до 2 моль, соединения карбоновой кислоты, представленного формулой (1b), или его активного производного, то количество конденсирующего агента составляет от 0,5 до 20 моль, и предпочтительно от 0,8 до 3 моль на 1 моль пиперазинового соединения, представленного формулой (1a), или его соли.
Хотя в зависимости от используемых активных производных или конденсирующего агента реакцию обычно проводят в растворителе, который является неактивным в реакции при от -20 до 150°C, и предпочтительно от 0 до 100°C. Примеры такого растворителя включают галогенированый углеводород, такой как дихлорметан и хлороформ; ароматический углеводород, такой как толуол; простой эфир, такой как тетрагидрофуран; сложный эфир, такой как этилацетат; спирт, такой как метанол и этанол; воду; ацетонитрил; N,N-диметилформамид; N,N-диметилацетамид; диметилсульфоксид и пиридин.
Реакция может проходить спокойно, если ее проводить в присутствии приблизительно от 0,5 до 20 моль, и предпочтительно от 0,8 до 5 моль, основания, такого как триэтиламин, диизопропилэтиламин, N-метилморфолин, N,N-диэтиланилин, 4-(N,N-диметиламино)пиридин и пиридин, на 1 моль пиперазинового соединения, представленного формулой (1a), или его соли.
Вторая стадия
На второй стадии защитную группу R аминогруппы в соединении амида, представленного формулой (2), удаляют обычным известным методом, и полученное соединение и соединение амина, представленное формулой (1c), или его активное производное конденсируются обычным методом, образуя соединение, представленное формулой (I).
Удаление защитной группы может быть выполнено в кислой среде, если защитная группа R представляет собой формил или трет-бутоксикарбонил, и удаление защитной группы может быть выполнено каталитическим восстановительным методом, если защитная группа R представляет собой бензил или бензилоксикарбонил.
Для конденсации предпочтительно использовать активные производные c отщепляющимися группами, полученными в результате реакции соединения амина, представленного формулой (1c), с трифосгеном, 1,1'-карбонилдиимидазолом (CDI), фенилхлорформиатом, 4-нитрофенилхлорформиатом, этилхлорформиатом или т.п., в неактивном в реакции растворителе, таком как дихлорметан, тетрагидрофуран, ацетонитрил, этилацетат или N,N-диметилацетамид, при от -20 до 150°C, и предпочтительно от 0 до 100°C, в присутствии органического основания, такого как триэтиламин или пиридин, или без него.
Активные производные формулы (1c) могут содержать группу отщепления. Активные производные могут быть использованы в реакции после выделения или могут быть приготовлены в реакционной системе и использованы без выделения. Примеры групп отщепления включают хлор, имидазолил, фенокси, 4-нитрофенокси и этокси.
Примеры солей соединения амина, представленного формулой (2), включают кислотно-аддитивные соли с неорганическими кислотами, такими как соляная кислота, бромоводородная кислота и серная кислота, или с органическими кислотами, такими как угольная кислота и метансульфоновая кислота.
Если на 1 моль соединения амина, представленного формулой (1c), или его активного производного используют от 0,5 до 10 моль, и предпочтительно от 0,8 до 2 моль, соединения амина, представленного формулой (3), или его соли, то количество конденсирующего агента составляет от 0,5 до 20 моль, и предпочтительно от 0,8 до 3 моль, на 1 моль соединения амина, представленного формулой (1c), или его соли.
Хотя в зависимости от используемых активных производных или конденсирующего агента, реакцию обычно проводят в растворителе, который является неактивным в реакции при от -20 до 150°C, и предпочтительно от 0 до 100°C. Примеры такого растворителя включают галогенированый углеводород, такой как дихлорметан и хлороформ; ароматический углеводород, такой как толуол; простой эфир, такой как тетрагидрофуран; сложный эфир, такой как этилацетат; спирт, такой как метанол и этанол; воду; ацетонитрил; N,N-диметилформамид; N,N-диметилацетамид; диметилсульфоксид и пиридин.
Реакция может проходить спокойно, если ее проводить в присутствии приблизительно от 0,5 до 20 моль, и предпочтительно от 0,8 до 5 моль, основания, такого как триэтиламин, диизопропилэтиламин, N-метилморфолин, N,N-диэтиланилин, 4-(N,N-диметиламино)пиридин и пиридин, на 1 моль пиперазинового соединения, представленного формулой (1с), или его активного производного.
Соединение (I) по настоящему изобретению может быть получено при выполнении первой стадии и второй стадии.
Пиперазиновое соединение, представленное формулой (1a), или его соль, соединение пирролкарбоновой кислоты, представленное формулой (1b), или его активное производное и соединение амина, представленное формулой (1c), или его соль известны в данной области или могут быть получены известными в данной области методами.
Схема реакции 2
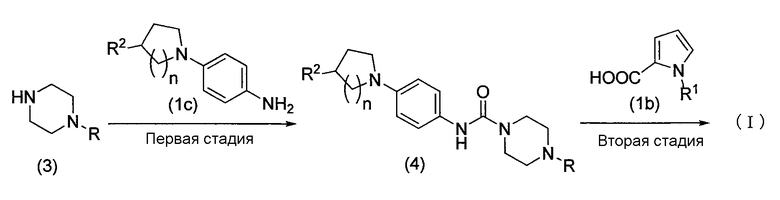
В приведенной схеме реакции 2 R1 и R2 определены выше, R представляет собой защитную группу аминогруппы или водорода, и n представляет собой 1 или 2.
Способ по настоящему изобретению включает первую стадию, на которой пиперазиновое соединение, представленное формулой (3), или его соль конденсируется обычным методом с соединением амина, представленным формулой (1c), или его активным производным, образуя соединение мочевины, представленное формулой (4), и вторую стадию, на которой соединение амина, полученное удалением защитной группы аминогруппы, или его соль конденсируется обычным методом с соединением пирролкарбоновой кислоты, представленным формулой (1b), или его активным производным, получая соединение, представленное формулой (I).
Первая стадия
На первой стадии пиперазиновое соединение, представленное формулой (3), и его соль конденсируется с соединением амина, представленным формулой (1c), в соответствии с обычным методом, как в реакции конденсации, выполненной на второй стадии схемы реакции 1, таким образом, получая соединение мочевины, представленное формулой (4).
Вторая стадия
На второй стадии защитная группа R аминогруппы в соединении мочевины, представленном формулой (4), удаляется, как в реакции удаления защитной группы, выполняемой на второй стадии схемы реакции 1, и затем соединение пирролкарбоновой кислоты, представленное формулой (1b), или его активное производное конденсируется обычным методом, как на первой стадии схемы реакции 1, таким образом, получая соединение, представленное формулой (I).
Схема реакции 3
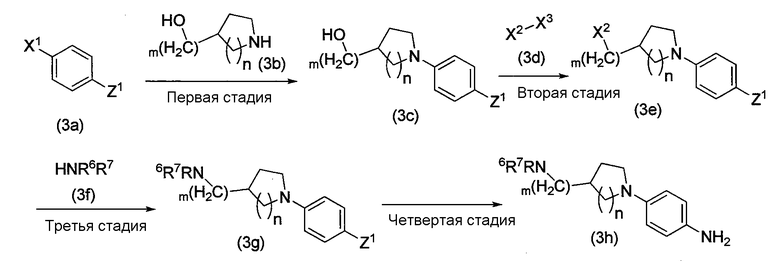
В приведенной схеме реакции 3 каждый из X1, X2 и X3 представляет собой функциональную группу отщепления, Z1 представляет собой нитрогруппу или защитную аминогруппу, а R6 и R7 является такими, как «заместители» в «C1-6 алкильной группе, которая может содержать один или более заместителей», представленных R2. R6 и R7, в частности, представляет собой замещенные или незамещенные гетероциклические группы, m представляет собой от 0 до 3, и n представляет собой 1 или 2.
Способ получения, показанный на схеме реакции 3, является таким же, как и для соединения амина (1c) в схемах реакции 1 и 2. Способ включает следующие четыре стадии:
первая стадия, на которой аминосодержащее соединение, представленное формулой (3b), вступает в реакцию с отщеплением функциональной группы с соединением, содержащим X1, представленным формулой (3a), с образованием гидроксисодержащего соединения, представленного формулой (3c);
вторая стадия, на которой соединение, представленное формулой (3d), вступает в реакцию с гидроксисодержащим соединением (3c) для введения функциональной группы отщепления X2, таким образом образуется соединение, представленное формулой (3е);
третья стадия, на которой соединение амина (3f) вступает в реакцию конденсации с функциональной группой отщепления X2, таким образом образуется соединение, представленное формулой (3g); и
четвертая стадия, в которой обычным методом осуществляют восстановление нитрогруппы или удаление защитной группы аминогруппы соединения (3g), полученного на третьей стадии, таким образом образуется соединение амина (3h).
Первая стадия
Любая функциональная группа отщепления может быть использована в качестве X1 соединения (3a) на первой стадии. Примеры включают галоген, такой как фтор и хлор, метансульфонилокси, п-толуолсульфонилокси и трифторметансульфонилокси.
Реакция проводится в подходящем растворителе, с использованием от 0,5 до 10 моль, и предпочтительно от 0,8 до 2 моль, соединения амина, представленного формулой (3b), или его соли на 1 моль соединения, представленного (3a), в присутствии от 0,5 до 10 моль, и предпочтительно от 0,8 до 3 моль, основания на 1 моль соединения, представленного формулой (3a), при от -20 до 180°C, и предпочтительно от 0 до 150°C, таким образом получая гидроксисодержащее соединение, представленное формулой (3c).
Могут быть использованы любые растворители, не оказывающие неблагоприятного воздействия на реакцию. Примеры подходящих растворителей включают галогенированный углеводород, такой как дихлорметан и хлороформ; ароматический углеводород, такой как толуол; простой эфир, такой как тетрагидрофуран; сложный эфир, такой как этилацетат; спирт, такой как метанол и этанол; воду; ацетонитрил; N,N-диметилформамид; N,N-диметилацетамид; N-метилпирролидон; диметилсульфоксид и пиридин. Может быть использован как один растворитель, так и их комбинация.
Примеры оснований включают неорганические основания, такие как гидроксид натрия, гидроксид кальция, карбонат натрия, карбонат калия и гидрид натрия, и органические основания, такие как пиридин, 4-(N,N-диметиламино)пиридин, триэтиламин, диизопропилэтиламин, 1,8-диазабицикло[5.4.0]ундец-7-ен и трет-бутилат калия.
Вторая стадия
Реакция проводится в подходящем растворителе, с использованием от 0,5 до 10 моль, и предпочтительно от 0,8 до 2 моль, соединения, содержащего функциональную группу отщепления, представленного формулой (3d), на 1 моль гидроксисодержащего соединения, представленного (3c), в присутствии от 0,5 до 10 моль, и предпочтительно от 0,8 до 3 моль, основания на 1 моль соединения, представленного формулой (3c), при от -20 до 180°C, и предпочтительно от 0 до 150°C, таким образом получая соединение, представленное формулой (3e).
Могут быть использованы любые растворители, не оказывающие неблагоприятного воздействия на реакцию. Примеры подходящих растворителей включают галогенированный углеводород, такой как дихлорметан и хлороформ; ароматический углеводород, такой как толуол; простой эфир, такой как тетрагидрофуран; сложный эфир, такой как этилацетат; ацетонитрил; N,N-диметилформамид; N,N-диметилацетамид; N-метилпирролидон; диметилсульфоксид и пиридин. Может быть использован как один растворитель, так и их комбинация.
Примеры основания включают неорганические основания, такие как гидроксид натрия, гидроксид кальция, карбонат натрия, карбонат калия и гидрид натрия, и органические основания, такие как пиридин, 4-(N,N-диметиламино)пиридин, триэтиламин, диизопропилэтиламин, 1,8-диазабицикло[5.4.0]ундец-7-ен и трет-бутилат калия.
Третья стадия
Реакция проводится в подходящем растворителе, с использованием от 0,5 до 10 моль, и предпочтительно от 0,8 до 3 моль, соединения амина, представленного формулой (3f), на 1 моль соединения, представленного формулой (3e), реакция проводится в присутствии от 0,5 до 10 моль, и предпочтительно от 0,8 до 3 моль, основания на 1 моль соединения, представленного формулой (3e), при от -20 до 180°C, и предпочтительно при от 0 до 150°C, таким образом получая соединение, представленное формулой (3g).
Могут быть использованы любые растворители, не оказывающие неблагоприятного воздействия на реакцию. Примеры подходящих растворителей включают галогенированный углеводород, такой как дихлорметан и хлороформ; ароматический углеводород, такой как толуол; простой эфир, такой как тетрагидрофуран; сложный эфир, такой как этилацетат; ацетонитрил; N,N-диметилформамид; N,N-диметилацетамид; N-метилпирролидон; диметилсульфоксид и пиридин. Может быть использован как один растворитель, так и их комбинация.
Примеры основания включают неорганические основания, такие как гидроксид натрия, гидроксид кальция, карбонат натрия, карбонат калия и гидрид натрия, и органические основания, такие как пиридин, 4-(N,N-диметиламино)пиридин, триэтиламин, диизопропилэтиламин, 1,8-диазабицикло[5.4.0]ундец-7-ен и трет-бутилат калия.
Четвертая стадия
Если Z1 представляет собой нитрогруппу, то восстановление может быть поведено при любых условиях, подходящих для восстановления нитрогруппы до аминогруппы. Предпочтительно, что условия реакции выбираются с учетом свойств других функциональных групп нитросоединения (3g).
Обычные методы восстановления включают следующее:
(A) От 0,01 до 10 моля, и предпочтительно 0,03 до 5 моля, катализатора восстановления, такого как восстановленное железо, хлорид олова или хлорид железа, в воде, спирте, таком как метанол или этанол, простом эфире, таком как тетрагидрофуран, или смеси его растворителей, вступает в реакцию в присутствии от 0,3 до 30 молей, и предпочтительно от 0,5 до 20 молей, соли аммония, такой как хлорид аммония, гидразина гидрат, соляная кислота и т.д., на 1 моль нитросоединения, представленного формулой (3g), при от 0 до 150°C, и предпочтительно от 20 до 120°C.
(B) Газообразный водород в спиртах, простых эфирах, сложных эфирах, таких как этилацетат, органические кислоты, такие как муравьиная кислота и уксусная кислота, или смеси его растворителей, вступает в реакцию при нормальном давлении или повышенном давлении в присутствии от 0,001 до 1 молей, и предпочтительно от 0,01 до 0,3 молей, катализатора восстановления, такого как палладиевый катализатор на углероде, оксид платины и ренеевский никелевый катализатор, на 1 моль нитросоединения, представленного формулой (3g), при от 0 до 120°C, и предпочтительно от 20 до 100°C; или от 0,5 до 20 моль, и предпочтительно от 1 до 10 моль, муравьиной кислоты, формиата аммония или циклогексена на 1 моль нитросоединения, представленного формулой (3g), использовали в качестве источника водорода вместо газообразного водорода.
Если Z1 представляет собой защитную аминогруппу, ее удаление может быть выполнено так же, как на второй стадии схемы реакции 1, для получения соединения амина (3h).
Соединения (3a), (3b), (3d) и (3f), используемые в схеме реакции 3, известны или могут быть получены известными методами.
Соединение амина (1c) в схеме реакции 1 или 2, т.е. соединение амина, представленное формулой (4f), может быть получено методом, показанным на схеме реакции 4 ниже.
Схема реакции 4
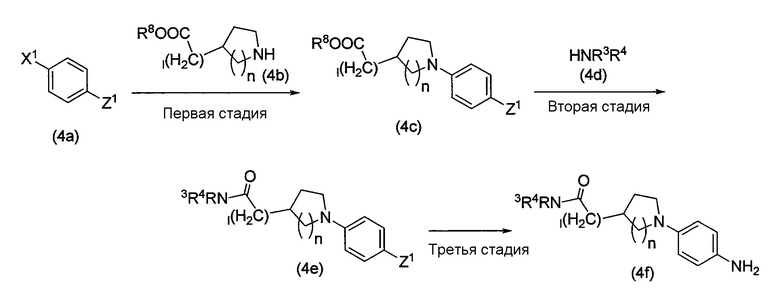
В схеме реакции 4 X1, Z1, R3 и R4 определены выше, R8 является таким, как R5, или силильной защитной группой, такой как трет-бутилдиметилсилил, l представляет собой от 0 до 3, и n представляет собой 1 или 2.
Метод получения, показанный на схеме реакции 4, включает первую стадию, на которой функциональная группа отщепления соединения (4a), содержащего X1, вступает в реакцию с аминосодержащим соединением сложного эфира, представленным формулой (4b), с образованием содержащего сложный эфир соединения, представленного формулой (4c); вторую стадию, на которой удаляется сложноэфирная группа соединения сложного эфира (4c), которое затем конденсируется с соединением амина, представленным формулой (4d), обычным методом с образованием соединения амида, представленного формулой (4e); и третью стадию, на которой нитрогруппа соединения (4e) восстанавливается или защитная группа аминогруппы удаляется обычным методом, таким образом образуется соединение амина (4f).
Первая стадия
На этой стадии содержащее сложный эфир соединение, представленное формулой (4c), может быть получено тем же методом, что и на первой стадии схемы реакции 3.
Вторая стадия
На этой стадии сложный эфир соединения, содержащего сложный эфир, представленного формулой (4c), удаляется обычным известным методом, а затем конденсируется с соединением амина, представленным формулой (4d), или его солью тем же методом, что и на первой стадии схемы реакции 1, с образованием соединения амида, представленного формулой (4e).
Третья стадия
На этой стадии Z1 может быть превращен в аминогруппу таким же методом, что и на четвертой стадии схемы реакции 3.
Соединения (4a), (4b) и (4d), используемые в схеме реакции 4, известны или могут быть получены известным методом.
Из соединений по настоящему изобретению, соединения, имеющие конкретные функциональные группы, могут быть превращены в другие соединения по изобретению химической модификацией этих групп, как показано ниже на схеме реакции 5.
Схема реакции 5
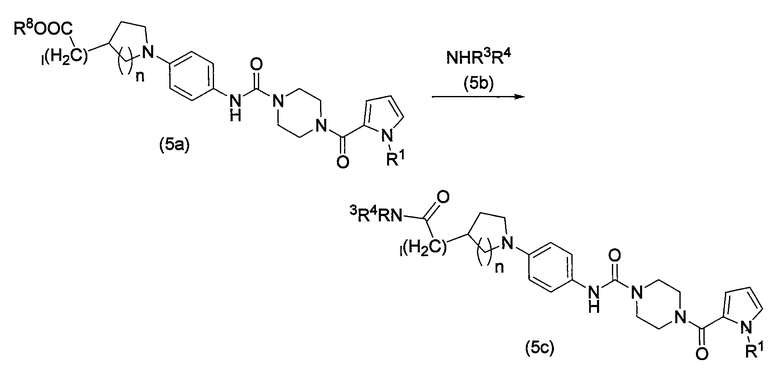
В схеме реакции 5 R1, R3, R4, R8, n и l определены выше.
В способе получения, показанном на схеме реакции 5, соединение карбоновой кислоты, полученное удалением защитной группы соединения сложного эфира, представленного формулой (5a), или его активного производного конденсируется тем же самым методом, как на первой стадии схемы реакции 1, с соединением амина, представленным формулой (5b), с образованием соединения амида (5c).
Если в соединении (I), которое может использоваться в качестве активного компонента лекарственного средства по настоящему изобретению, содержится один или более асимметричных углеродов, то из-за наличия таких симметричных атомов углерода могут образовываться оптические изомеры (энантиомеры и диастереомеры) и другие изомеры. Настоящее изобретение охватывает изомеры, которые могут быть выделены, и их смеси.
Соединение (I), которое может использоваться в качестве активного компонента лекарственного средства по настоящему изобретению, охватывает фармацевтически приемлемые пролекарства. Фармацевтически приемлемые пролекарства представляют собой соединения с функциональными группами, которые могут превращаться при химическом воздействии, таком как сольволиз, или в физиологических условиях, в амино, гидрокси, карбокси, карбонил или подобные функциональные группы соединения (I), которое является активным компонентом лекарственного средства по настоящему изобретению. Примеры функциональных групп пролекарств включают группы, упомянутые в «Iyakuhin no Kaihatsu Development of Pharmaceuticals)», Vol. 7, pp. 163-198, Hirokawa Publishing (1990).
Соединение (I), которое может использоваться в качестве активного компонента лекарственного средства по настоящему изобретению, может образовать кислотно-аддитивную соль или соль с основанием. Такие соли включены в настоящее изобретение, если они являются фармацевтически приемлемыми. Конкретные примеры включают кислотно-аддитивные соли с неорганическими кислотами, такими как соляная кислота, бромоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.д., или органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, угольная кислота, пикриновая кислота, метансульфоновая кислота, пара-толуолсульфоновая кислота, глутаминовая кислота и т.д.; соли с неорганическими основаниями, такими как натрий, калий, магний, кальций, алюминий и т.д., органическими основаниями, такими как метиламин, этиламин, меглумин, этаноламин и т.д., или основными аминокислотыами, такими как лизин, аргинин, орнитин и т.д.; и соли аммония.
Настоящее изобретение дополнительно охватывает гидраты, сольваты и кристаллические полиморфы соединения (I), которое может использоваться в качестве активного компонента лекарственного средства по настоящему изобретению, и его фармацевтически приемлемых солей.
В фармацевтическую композицию, содержащую пиперазиновое соединение или его соль по настоящему изобретению, при необходимости может быть добавлен фармацевтический носитель для создания подходящей дозированной формы в соответствии с целями лечения и предотвращения. Примеры дозированной формы включают пероральные составы, инъекции, свечи, мази, пластыри и т.д. Из перечисленного пероральные составы являются предпочтительными. Такие дозированные формы могут быть получены обычными способами, известными специалистам в данной области.
В качестве фармацевтического носителя, как составляющее препарата, могут быть использованы различные обычные органические или неорганические носители, которые добавляются как вспомогательное вещество, связующее, дезинтегрант, смазывающее вещество или краситель в твердых препаратах, или как растворитель, солюбилизирующий компонент, суспендирующий компонент, изотоничный компонент, буфер или смягчающее средство в жидких препаратах. Кроме того, если необходимо, могут быть использованы добавки фармацевтических препаратов, такие как антисептик, антиоксидант, краситель, подсластитель и стабилизатор.
Пероральные твердые составы готовят следующим образом. Вспомогательное вещество, необязательно вместе со связующим, дезинтегрантом, смазывающим веществом, красителем, подсластителем/вкусовой добавкой и т.д., добавляют к соединению по настоящему изобретению для получения обычными способами таблеток, таблеток, покрытых оболочкой, гранул, порошков, капсул или т.п.
Примеры вспомогательных веществ включают лактозу, сахарозу, D-маннит, глюкозу, крахмал, карбонат кальция, каолин, микрокристаллическую целлюлозу и кремниевую кислоту.
Примеры связующих включают воду, этанол, 1-пропанол, 2-пропанол, простой сироп, раствор глюкозы, жидкий α-крахмал, жидкий желатин, D-маннит, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилкрахмал, метилцеллюлозу, этилцеллюлозу, шеллак, фосфат кальция и поливинилпирролидон.
Примеры дезинтегрантов включают сухой крахмал, альгинат натрия, порошок агара, бикарбонат натрия, карбонат кальция, лаурилсульфат натрия, моноглицерид стеариновой кислоты и лактозу.
Примеры смазывающих веществ включают очищенный тальк, стеарат натрия, стеарат магния, буру и полиэтиленгликоль.
Примеры красителей включают оксид титана и оксид железа.
Примеры подсластителей/вкусовых добавок включают сахарозу, цедру дикого апельсина, лимонную кислоту и винную кислоту.
Пероральные жидкие препараты готовят следующим образом. Подсластитель, буфер, стабилизатор, подслащивающую вкусовую добавку и т.д. добавляют к соединению по настоящему изобретению для получения обычными методами жидкого лекарственного средства для употребления внутрь, сиропа, эликсира или т.п. В данном случае пригодные подсластители/вкусовые добавки такие, как описано выше. Примеры буферов включают цитрат натрия, и примеры стабилизаторов включают трагакант, гуммиарабик и желатин. Если требуется, кишечнорастворимое покрытие или покрытие для более длительного эффекта могут быть нанесены методами, известными в области пероральных препаратов. Примеры компонентов покрытий включают гидроксипропилметилцеллюлозу, этилцеллюлозу, гидроксиметилцеллюлозу, гидроксипропилцеллюлозу, полиоксиэтиленгликоль и Tween 80 (зарегистрированная торговая марка).
Инъекционные формы готовят следующим образом. Средство для доведения рН, буфер, стабилизатор, изотоничный компонент, местный анестетик и т.п. добавляют к соединению по настоящему изобретению, получая обычными методами инъекционный препарат для подкожного введения, инъекционный препарат для внутримышечного введения или инъекционный препарат для внутривенного введения. Примеры подходящих средств для доведения pH и буферов в данном случае включают цитрат натрия, ацетат натрия и фосфат натрия. Примеры пригодных стабилизаторов включают пиросульфит натрия, ЭДТА, тиогликолевую кислоту и тиомолочную кислоту. Примеры пригодных местных анестетиков включают новокаин и лидокаин. Примеры пригодных изотоничных компонентов включают хлорид натрия, глюкозу, D-маннит и глицерин.
Свечи готовят следующим образом. Фармацевтический носитель, известный в данной области, такой как полиэтиленгликоль, ланолин, масло какао и триглицериды жирных кислот, добавляют к соединению по настоящему изобретению, необязательно вместе с Tween 80 (зарегистрированная торговая марка) или подобным поверхностно-активным веществом, с последующим приготовлением обычным методом.
Мази готовят следующим образом. Обычную основу, стабилизатор, увлажняющий компонент, консервант и т.п. добавляют в необходимых количествах к соединению по настоящему изобретению, смешивают и готовят лекарственную форму обычным методом. Примеры основ включают жидкий парафин, медицинский вазелин, осветленный пчелиный воск, октилдодециловый спирт и парафин. Примеры консервантов включают метилпарагидроксибензоат, этилпарагидроксибензоат и пропилпарагидроксибензоат.
Пластыри могут быть приготовлены обычным методом путем покрытия основной подложки вышеуказанной мазью, кремом, гелем, пастой и т.д. Примеры подложек включают тканые или нетканые материалы, изготовленные из хлопка, штапельного волокна и химических волокон; как пленки, так и вспененные листы из мягкого винилхлорида, полиэтилена и полиуретана.
Количество соединения по настоящему изобретению, содержащееся в такой единичной дозированной форме, зависит от состояния пациента или вида дозированной формы. Желательное количество в одной единичной дозированной форме составляет от приблизительно 0,05 до приблизительно 1000 мг для перорального состава, от приблизительно 0,01 до приблизительно 500 мг для инъекционной формы и от приблизительно 1 до приблизительно 1000 мг для свечей.
Суточная доза лекарственного средства в такой дозированной форме зависит от состояния, веса тела, возраста, пола и т.д. пациента. Например, суточная доза для взрослого (вес тела: 50 кг) может в общем случае составлять от приблизительно 0,05 до приблизительно 5000 мг, и предпочтительно от 0,1 до 1000 мг, и предпочтительно вводится в одной или в двух-трех разделенных дозах в день.
Так как при введении лекарственного средства, содержащего соединение по настоящему изобретению, ингибирующая H-PGDS активность проявляется у млекопитающих и, в частности, у человека, то соединение по настоящему изобретению может использоваться для лечения, предотвращения или улучшения заболеваний, вызванных PGD2, продуцируемого синтазой, или его метаболитом. Соединение по настоящему изобретению может быть введено млекопитающим, включая людей, обезьян, мышей, крыс, кроликов, собак, кошек, коров, лошадей, свиней и овец. Соединение может быть введено млекопитающему перорально или неперорально обычными методами.
Примеры заболеваний для лечения, предотвращения или улучшения фармацевтическим средством, содержащим соединение по настоящему изобретению, включают аллергическое заболевание, воспалительное заболевание и миодегенеративное заболевание.
Примеры аллергических заболеваний включают бронхиальную астму, поллиноз, аллергический ринит, синусит, средний отит, аллергический конъюнктивит, весеннее катаральное воспаление, аллергический дерматит, контактный дерматит и аллергии на пищевые продукты. Из них предпочтительными являются бронхиальная астма, поллиноз, аллергический ринит и синусит. Примеры воспалительных заболеваний включают хроническое обструктивное заболевание легких, интерстициальную пневмонию, гиперчувствительный пневмонит, эозинофильную пневмонию, суставной ревматизм, дегенеративный артрит, рассеянный склероз, воспалительное заболевание кишечника, кожные заболевания (псориаз, экзема, эритема, синдром зуда, папула и т.д.), пост-PTCA рестеноз, хроническое обструктивное заболевание артерий, реперфузионное повреждение и реакцию на отторжение трансплантата.
Примеры миодегенеративных заболеваний включают мышечную дистрофию, такую как мышечная дистрофия Дюшенна, которая является миогенным заболеванием, мышечную дистрофию Беккера, мышечную дистрофию тазового пояса и врожденную мышечную дистрофию; различные миопатии, такие как врожденная миопатия; боковой амиотрофический склероз, который является неврогенной мышечной атрофией; сокращение мышц; кардиомиопатия (инфаркт миокарда) и диабетические периферические сосудистые нарушения (сосудистые нарушения гладких мышц). Предпочтительные примеры миодегенеративных заболеваний для лечения, предотвращения или улучшения лекарственным средством, содержащим соединение по настоящему изобретению, включают мышечную дистрофию Дюшена, мышечную дистрофию Беккера и боковой амиотрофический склероз.
Кроме того, предполагается, что лекарственное средство, содержащее соединение по настоящему изобретению, улучшит лечение и предотвращение проблем секреции слизи, репродуктивных проблем, нарушений коагуляции крови, нарушений сна, боли, проблем зрения, ожирения, иммунопатии и аутоиммунных заболеваний; предотвратит обострение заболевания Альцгеймера или повреждения головного мозга и/или улучшит прогноз после повреждения головного мозга. Кроме того, это может привести к ингибированию малигнизации клеток и метастатического роста опухоли, они также могут использоваться для терапии рака.
Кроме того, они могут применяться для лечения и/или предотвращения пролиферативных нарушений, вызванных PGD2 или его метаболитами, таких как пролиферация фибробласта, диабетической ретинопатии и ангиогенеза опухоли. Кроме того, поскольку они могут подавить PGD2-индуцированное сокращение гладких мышц, они также могут применяться для лечения и/или предотвращения бесплодия, дисменореи, преждевременных родов и лейкоцит-эозинофильных нарушений.
ПРИМЕРЫ
Настоящее изобретение описывается ниже детально с помощью Справочных примеров, Примеров и Примеров исследований, которые не ограничивают объема изобретения.
В дальнейшем описании спектры 1H-ЯМР измеряют с использованием TMS (тетраметилсилан) в качестве внутреннего стандарта, и химические сдвиги указаны δ (м.д.). Что касается химических сдвигов, спектров поглощения, постоянных связывания (J) и количества протонов, они указаны в скобках.
Следующие символы используют для спектров поглощения: с - синглет, д - дублет, т - триплет, кв. - квартет, дд - двойной дублет, м - мультиплет, уш. - широкий и уш.с - широкий синглет.
Кроме того, следующие символы используют в структурных формулах соединений: Me = метил и Et = этил.
Пример 1
N-(4-(4-гидроксипиперидиин-1-ил)фенил-4-((1-метилпиррол-2-ил)карбонил)-1-пиперазинкарбоксамид
Пример 1(1)
4-гидрокси-N-(4-нитрофенил)пиперидин
4-хлорнитробензол (31,5 г, 200 ммоль) растворяли в N,N-диметилацетамиде (80 мл) и добавляли карбонат калия (35,9 г, 260 ммоль) и 4-гидроксипиперидин (22,3 г, 220 ммоль), затем перемешивали при нагревании до 130°C в течение 3 часов. После охлаждения до комнатной температуры к смеси добавляли воду и собирали осадок фильтрацией. Полученное твердое вещество сушили при пониженном давлении, таким образом получая 4-гидрокси-N-(4-нитрофенил)пиперидин (41,3 г, 93%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,52-1,74 (м, 2H), 1,92-2,04 (м, 2H), 3,14-3,35 (м, 2H), 3,73-4,08 (м, 3H), 6,82 (д, J=9,6 Гц, 2H), 8,11 (д, J=9,6 Гц, 2H)
Пример 1(2)
4-гидрокси-N-(4-аминофенил)пиперидин
4-гидрокси-N-(4-нитрофенил)пиперидин (11,1 г, 50 ммоль), полученный в примере 1(1), растворяли в метаноле (100 мл) и тетрагидрофуране (50 мл) и добавляли 10% палладий на угле (8,0 г), затем перемешивали при комнатной температуре в среде газообразного водорода в течение 7 часов. После того как нерастворимые вещества отфильтровали через целит, фильтрат концентрировали при пониженном давлении, добавляли диэтиловый простой эфир к полученному остатку и осадок собирали фильтрацией, таким образом получая 4-гидрокси-N-(4-аминофенил)пиперидин (9,25 г, 94%) в виде красно-фиолетового твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,65-1,77 (м, 2H), 1,94-2,11 (м, 2H), 2,71-2,85 (м, 2H), 3,23-3,92 (м, 5H), 6,64 (д, J=8,9 Гц, 2H), 6,83 (д, J=8,9 Гц, 2H)
Пример 1(3)
N-(4-(4-гидроксипиперидин-1-ил)фенил)-4-((1-метилпиррол-2-ил)карбонил)-1-пиперазинкарбоксамид
4-нитрофенил хлорформиат (242 мг, 1,2 ммоль) растворяли в тетрагидрофуране (3 мл) и тетрагидрофурановый раствор (4 мл) 4-гидрокси-N-(4-аминофенил)пиперидина (211 мг, 1,0 ммоль), полученный в примере 1(2), добавляли по каплям при -30°C. После перемешивания в течение 30 минут при этой температуре к смеси добавляли 1-[(1-метил-1Н-пиррол-2-ил)карбонил]пиперазина гидрохлорид (252 мг, 1,1 ммоль) и триэтиламин (0,49 мл, 3,5 ммоль), затем перемешивали при комнатной температуре в течение 17 часов. В реакционную смесь добавляли насыщенный водный раствор бикарбоната натрия, затем проводили экстракцию этилацетатом. Органический слой промывали водой и насыщенным хлоридом натрия и сушили над безводным сульфатом натрия. Десикант отфильтровывали и после этого остаток, полученный выпариванием при пониженном давлении, очищали флэш-хроматографией среднего давления на колонке с силикагелем (силикагель NH, метанол:хлороформ = от 0:1 до 1:50), таким образом получая N-(4-(4-гидроксипиперидин-1-ил)фенил)-4-((1-метилпиррол-2-ил)карбонил)-1-пиперазинкарбоксамид (256 мг, 62%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,60-1,78 (м, 2H), 1,93-2,08 (м, 2H), 2,80-2,95 (м, 2H), 3,40-3,63 (м, 6H), 3,75-3,90 (м, 5H), 3,80 (с, 3H), 6,08-6,15 (м, 1H), 6,25 (уш.с, 1H), 6,32-6,40 (м, 1H), 6,70-6,77 (м, 1H), 6,90 (д, J=8,9 Гц, 2H), 7,21 (д, J=8,9 Гц, 2H)
Пример 2
4-((((1-метилпиррол-2-ил)карбонил)-1-пиперазинил)карбонил)амино-4-фенилпиперидин-4-карбоновая кислота
Пример 2 (1)
Этиловый эфир 1-(4-нитрофенил)пиперидин-4-карбоновой кислоты
В соответствии с методикой примера 1(1), используя этиловый эфир изонипекотиновой кислоты вместо 4-гидроксипиперидина, получали этиловый эфир 1-(4-нитрофенил)пиперидин-4-карбоновой кислоты (95%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,27 (т, J=7,2 Гц, 3H), 1,80-1,90 (м, 2H), 2,00-2,08 (м, 2H), 2,54-2,62 (м, 1H), 3,01-3,14 (м, 2H), 3,84-3,92 (м, 2H), 4,17 (кв., J=7,2 Гц, 2H), 6,83 (д, J=7,2 Гц, 2H), 8,11 (д, J=7,2 Гц, 2H)
Пример 2(2)
Этиловый эфир 1-(4-аминофенил)пиперидин-4-карбоновой кислоты
В соответствии с методикой примера 1(2), используя этиловый эфир 1-(4-нитрофенил)пиперидин-4-карбоновой кислоты, полученный в примере 2(1), вместо 4-гидрокси-N-(4-нитрофенил)пиперидина, получали этиловый эфир 1-(4-аминофенил)пиперидин-4-карбоновой кислоты (колич.) в виде красновато-коричневого масла.
1H-ЯМР (CDCl3): δ (м.д.) 1,27 (т, J=7,1 Гц, 3H), 1,78-2,13 (м, 4H), 2,30-2,47 (м, 1H), 2,55-2,75 (м, 2H), 3,20-3,64 (м, 4H), 4,15 (кв., J=7,1 Гц, 2H), 6,64 (д, J=8,9 Гц, 2H), 6,82 (д, J=8,9 Гц, 2H)
Пример 2(3)
Этиловый эфир 4-((((1-метилпиррол-2-ил)карбонил)-1-пиперазинил)карбонил)амино-4-фенилпиперидин-4-карбоновой кислоты
В соответствии с методикой примера 1(3), используя этиловый эфир 1-(4-аминофенил)пиперидин-4-карбоновой кислоты, полученный в примере 2(2), вместо 4-гидрокси-N-(4-аминофенил)пиперидина, получали этиловый эфир 4-((((1-метилпиррол-2-ил)карбонил)-1-пиперазинил)карбонил)амино-4-фенилпиперидин-4-карбоновой кислоты (80%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,27 (т, J=7,1 Гц, 3H), 1,75-2,12 (м, 4H), 2,33-2,48 (м, 1H), 2,67-2,84 (м, 2H), 3,42-3,65 (м, 6H), 3,70-3,93 (м, 4H), 3,79 (с, 3H), 4,15 (кв., J=7,1 Гц, 2H), 6,06-6,15 (м, 1H), 6,30-6,40 (м, 1H), 6,41 (уш.с, 1H), 6,69-6,76 (м, 1H), 6,88 (д, J=9,0 Гц, 2H), 7,22 (д, J=9,0 Гц, 2H)
Пример 2(4)
4-((((1-метилпиррол-2-ил)карбонил)-1-пиперазинил)карбонил)амино-4-фенилпиперидин-4-карбоновая кислота
Этиловый эфир 4-((((1-метилпиррол-2-ил)-карбонил)-1-пиперазинил)-карбонил)амино-4-фенилпиперидин-4-карбоновой кислоты (234 мг, 0,5 ммоль), полученный в примере 2(3), растворяли в тетрагидрофуране (1,5 мл) и этаноле (1,5 мл), и добавляли 2 н. водный раствор гидроксида натрия (1,4 мл, 2,8 ммоль), затем перемешивали при комнатной температуре в течение 5 часов. После охлаждения реакционной смеси до 0°C добавляли в реакционную смесь 2 н. соляную кислоту (1,4 мл, 2,8 ммоль), затем проводили экстракцию смешанным растворителем метанол:хлороформ (1:10). Органический слой промывали водой и насыщенным хлоридом натрия и сушили над безводным сульфатом натрия. Десикант отфильтровывали и после этого остаток, полученный выпариванием при пониженном давлении, промывали диэтиловым простым эфиром, таким образом получали 4-((((1-метилпиррол-2-ил)карбонил)-1-пиперазинил)карбонил)амино-4-фенилпиперидин-4-карбоновую кислоту (167 мг, 75%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,79-2,13 (м, 4H), 2,38-2,54 (м, 1H), 2,65-2,87 (м, 2H), 3,45-3,67 (м, 6H), 3,71-3,94 (м, 4H), 3,80 (с, 3H), 6,05-6,14 (м, 1H), 6,28-6,46 (м, 2H), 6,68-6,77 (м, 1H), 6,90 (д, J=8,9 Гц, 2H), 7,21 (д, J=8,9 Гц, 2H)
Пример 3
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Пример 3(1)
1-(4-нитрофенил)пиперидин-4-карбоновая кислота
Этиловый эфир 1-(4-нитрофенил)пиперидин-4-карбоновой кислоты (2,78 г, 10 ммоль), полученный в примере 2(1), растворяли в этаноле (10 мл) и добавляли 4 н. водный раствор гидроксида натрия (5 мл, 20 ммоль). Смесь нагревали с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры к смеси добавляли воду (30 мл) и 2 н. соляную кислоту (10 мл), и осадок собирали фильтрацией, таким образом получали 1-(4-нитрофенил)пиперидин-4-карбоновую кислоту (2,47 г, 97%) в виде желтого твердого вещества.
1H-ЯМР (ДМСО-d6): δ (м.д.) 1,49-1,68 (м, 2H), 1,84-2,00 (м, 2H), 2,50-2,66 (м, 1H), 3,01-3,19 (м, 2H), 3,90-4,05 (м, 2H), 7,02 (д, J=9,4 Гц, 2H), 8,03 (д, J=9,4 Гц, 2H), 12,28 (уш., 1H)
Пример 3(2)
1-(4-нитрофенил)пиперидин-4-морфолинкарбоксамид
1-(4-нитрофенил)пиперидин-4-карбоновую кислоту (10,1 г, 40 ммоль), полученную в примере 3(1), растворяли в N,N-диметилформамиде (25 мл) и добавляли морфолин (5,2 г, 60 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (9,2 г, 48 ммоль) и 1-гидроксибензотриазола моногидрат (6,7 г, 44 ммоль), затем перемешивали в течение ночи при нагревании до 70°C. После охлаждения до комнатной температуры добавляли воду, осадок собирали фильтрованием и сушили при нагревании при пониженном давлении, таким образом получая 1-(4-нитрофенил)пиперидин-4-морфолинкарбоксамид (12,1 г, 95%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,80-2,08 (м, 4H), 2,68-2,81 (м, 1H), 2,95-3,13 (м, 2H), 3,46-3,78 (м, 8H), 3,89-4,07 (м, 2H), 6,81 (д, J=8,4 Гц, 2H), 8,11 (д, J=8,4 Гц, 2H)
Пример 3(3)
1-(4-аминофенил)пиперидин-4-морфолинкарбоксамид
В соответствии с методикой примера 1(2), используя 1-(4-нитрофенил)пиперидин-4-морфолинкарбоксамид, полученный в примере 3(2), вместо 4-гидрокси-N-(4-нитрофенил)пиперидина, получали 1-(4-аминофенил)пиперидин-4-морфолинкарбоксамид (90%) в виде красновато-фиолетового твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,72-1,89 (м, 2H), 1,92-2,15 (м, 2H), 2,45-2,74 (м, 3H), 3,28-3,80 (м, 12Н), 6,65 (д, J=8,9 Гц, 2H), 6,82 (д, J=8,9 Гц, 2H)
Пример 3(4)
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 1(3), используя 1-(4-аминофенил)пиперидин-4-морфолинкарбоксамид, полученный в примере 3(3), вместо 4-гидрокси-N-(4-аминофенил)пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (82%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,72-2,12 (м, 4H), 2,47-2,78 (м, 3H), 3,43-3,90 (м, 18Н), 3,80 (с, 3H), 6,07-6,16 (м, 1H), 6,33-6,39 (м, 1H), 6,44 (уш.с, 1H), 6,69-6,78 (м, 1H), 6,88 (д, J=8,9 Гц, 2H), 7,22 (д, J=8,9 Гц, 2H)
Пример 4
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолинометилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Пример 4(1)
4-гидроксиметил-N-(4-нитрофенил)пиперидин
В соответствии с методикой примера 1(1), используя 4-гидроксиметилпиперидин вместо 4-гидроксипиперидина, получали 4-гидроксиметил-N-(4-нитрофенил)пиперидин (97%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,23-1,48 (м, 3H), 1,70-1,97 (м, 3H), 2,90-3,07 (м, 2H), 3,56 (т, J=5,7 Гц, 2H), 3,93-4,07 (м, 2H), 6,81 (д, J=9,4 Гц, 2H), 8,11 (д, J=9,4 Гц, 2H)
Пример 4(2)
4-тозилоксиметил-N-(4-нитрофенил)пиперидин
4-гидроксиметил-N-(4-нитрофенил)пиперидин (47,3 г, 200 ммоль), полученный в примере 4(1), растворяли в пиридине (300 мл), и добавляли п-толуолсульфонилхлорид (45,8 г, 240 ммоль) при охлаждении на льду, с последующим перемешиванием в течение 4 часов. В реакционную смесь добавляли воду и осадок собирали фильтрацией. Полученное твердое вещество сушили при пониженном давлении, таким образом получали 4-тозилоксиметил-N-(4-нитрофенил)пиперидин (72,7 г, 93%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,20-1,40 (м, 2H), 1,75-1,90 (м, 2H), 1,90-2,12 (м, 1H), 2,46 (с, 3H), 2,87-3,03 (м, 2H), 3,90 (д, J=6,4 Гц, 2H), 3,90-4,02 (м, 2H), 6,78 (д, J=9,4 Гц, 2H), 7,36 (д, J=8,4 Гц, 2H), 7,79 (д, J=8,4 Гц, 2H), 8,09 (д, J=9,4 Гц, 2H)
Пример 4(3)
4-морфолинометил-N-(4-нитрофенил)пиперидин
4-тозилоксиметил-N-(4-нитрофенил)пиперидин (39,0 г, 100 ммоль), полученный в примере 4(2), растворяли в метилэтилкетоне (150 мл) и добавляли йодид натрия (45,0 г, 300 ммоль), затем перемешивали при комнатной температуре в течение 4 дней. В реакционную смесь добавляли этилацетат, и нерастворимое вещество фильтровали через целит. Затем органический слой промывали водой и насыщенным хлоридом натрия и сушили над безводным сульфатом натрия. После этого десикант отфильтровывали и добавляли диизопропиловый простой эфир к остатку, полученному выпариванием при пониженном давлении, и осадок собирали фильтрацией, таким образом получая неочищенный йодид. Полученный неочищенный йодид растворяли в ацетонитриле (150 мл) и карбонате калия (19,7 г, 143 ммоль) и добавляли морфолин (12,4 мл, 143 ммоль), затем перемешивали при комнатной температуре в течение 2 дней. Добавляли воду в реакционную смесь, затем проводили экстракцию этилацетатом. Затем органический слой промывали водой и насыщенным хлоридом натрия и сушили над безводным сульфатом натрия. Десикант отфильтровывали и после этого остаток, полученный выпариванием при пониженном давлении, очищали флэш-хроматографией среднего давления на колонке с силикагелем (метанол:хлороформ = от 0:1 до 1:30), таким образом получали 4-морфолинометил-N-(4-нитрофенил)пиперидин (16,2 г, 53% на 2 стадии) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,17-1,38 (м, 2H), 1,70-2,00 (м, 3H), 2,21 (д, J=7,1 Гц, 2H), 2,32-2,53 (м, 4H), 2,86-3,05 (м, 2H), 3,62-3,78 (м, 4H), 3,86-4,05 (м, 2H), 6,80 (д, J=9,6 Гц, 2H), 8,10 (д, J=9,6 Гц, 2H)
Пример 4(4)
4-морфолинометил-N-(4-аминофенил)пиперидин
В соответствии с методикой примера 1(2), используя 4-морфолинометил-N-(4-нитрофенил)пиперидин, полученный в примере 4(3), вместо 4-гидрокси-N-(4-нитрофенил)пиперидина, получали 4-морфолинометил-N-(4-аминофенил)пиперидин (97%) в виде красновато-фиолетового твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,25-1,73 (м, 3H), 1,80-1,95 (м, 2H), 2,16-2,70 (м, 8H), 3,27-3,85 (м, 8H), 6,64 (д, J=8,6 Гц, 2H), 6,84 (д, J=8,6 Гц, 2H)
Пример 4(5)
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолинометилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 1(3), используя 4-морфолинометил-N-(4-аминофенил)пиперидин, полученный в примере 4(4), вместо 4-гидрокси-N-(4-аминофенил)пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолинометилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (72%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,22-1,46 (м, 2H), 1,53-1,75 (м, 1H), 1,81-1,98 (м, 2H), 2,22 (д, J=7,1 Гц, 2H), 2,33-2,51 (м, 4H), 2,57-2,74 (м, 2H), 3,45-3,93 (м, 14Н), 3,80 (с, 3H), 6,08-6,14 (м, 1H), 6,32 (уш.с, 1H), 6,30-6,41 (м, 1H), 6,70-6,75 (м, 1H), 6,89 (д, J=9,1 Гц, 2H), 7,20 (д, J=9,1 Гц, 2H)
Пример 5
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(3-(2-(1,2,4-триазол-1-ил)этил)пирролидин-1-ил)фенил)-1-пиперазинкарбоксамид
Пример 5 (1)
3-[2-(1,2,4-триазол-1-ил)этил]-N-(4-нитрофенил)пирролидин
4-тозилоксиметил-N-(4-нитрофенил)пиперидин (39,0 г, 100 ммоль), полученный в примере 4(2), растворяли в метилэтилкетоне (150 мл) и добавляли йодид натрия (45,0 г, 300 ммоль), затем перемешивали при комнатной температуре в течение 5 дней. В реакционную смесь добавляли этилацетат, и нерастворимые вещества отфильтровывали через целит. Затем органический слой промывали водой и насыщенным хлоридом натрия и сушили над безводным сульфатом натрия. Десикант отфильтровывали и после этого диизопропиловый простой эфир добавляли к остатку, полученному выпариванием при пониженном давлении, и осадок собирали фильтрацией, таким образом получали неочищенный йодид. Полученный неочищенный йодид растворяли в ацетонитриле (160 мл) и воде (40 мл) и добавляли карбонат калия (24,9 г, 180 ммоль) и 1,2,4-триазол (9,32 г, 134 ммоль), затем перемешивали при 80°C в течение 6 часов. После охлаждения реакционной смеси до комнатной температуры в нее добавляли воду и осадок собирали фильтрацией и сушили. Полученное твердое вещество очищали флэш-хроматографией среднего давления на колонке с силикагелем (метанол:хлороформ = от 0:1 до 1:30), таким образом получая 3-[2-(1,2,4-триазол-1-ил)этил]-N-(4-нитрофенил)пирролидин (16,4 г, 63% на 2 стадии) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,55-1,80 (м, 1H), 2,00-2,35 (м, 4H), 2,97-3,14 (м, 1H), 3,31-3,64 (м, 3H), 4,30 (т, J=7,0 Гц, 2H), 6,44 (д, J=9,4 Гц, 2H), 7,97 (с, 1H), 8,11 (д, J=9,4 Гц, 2H), 8,12 (с, 1H)
Пример 5(2)
3-[2-(1,2,4-триазол-1-ил)этил]-N-(4-аминофенил)пирролидин
В соответствии с методикой примера 1(2), используя 3-[2-(1,2,4-триазол-1-ил)этил]-N-(4-нитрофенил)пирролидин, полученный в примере 5(1), вместо 4-гидрокси-N-(4-нитрофенил)пиперидина, получали 3-[2-(1,2,4-триазол-1-ил)этил]-N-(4-аминофенил)пирролидин (96%) в виде красновато-фиолетового твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,65-1,87 (м, 1H), 2,00-2,43 (м, 4H), 2,75-3,70 (м, 6H), 4,25 (т, J=7,1 Гц, 2H), 6,44 (д, J=8,4 Гц, 2H), 6,68 (д, J=8,4 Гц, 2H), 8,00 (с, 1H), 8,08 (с, 1H)
Пример 5(3)
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(3-(2-(1,2,4-триазол-1-ил)этил)пирролидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 1(3), использовали 3-[2-(1,2,4-триазол-1-ил)этил]-N-(4-аминофенил)пирролидин, полученный в примере 5(2), вместо 4-гидрокси-N-(4-аминофенил)пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(3-(2-(1,2,4-триазол-1-ил)этил)пирролидин-1-ил)фенил)-1-пиперазинкарбоксамид (62%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,62-1,78 (м, 1H), 2,02-2,38 (м, 4H), 2,89-3,03 (м, 1H), 3,22-3,67 (м, 7H), 3,77-3,94 (м, 4H), 3,80 (с, 3H), 4,26 (т, J=7,1 Гц, 2H), 6,07-6,15 (м, 1H), 6,19 (уш.с, 1H), 6,33-6,41 (м, 1H), 6,48 (д, J=8,9 Гц, 2H), 6,70-6,78 (м, 1H), 7,16 (д, J=8,9 Гц, 2H), 7,96 (с, 1H), 8,09 (с, 1H)
Пример 6
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолиноэтилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Пример 6(1)
4-гидроксиэтил-N-(4-нитрофенил)пиперидин
В соответствии с методикой примера 1(1), используя 4-гидроксиэтилпиперидин вместо 4-гидроксипиперидина, получали 4-гидроксиэтил-N-(4-нитрофенил)пиперидин (100%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,18-1,40 (м, 3H), 1,47-1,92 (м, 5H), 2,85-3,03 (м, 2H), 3,63-3,78 (м, 2H), 3,85-4,02 (м, 2H), 6,77 (д, J=9,4 Гц, 2H), 8,07 (д, J=9,4 Гц, 2H)
Пример 6(2)
4-тозилоксиэтил-N-(4-нитрофенил)пиперидин
В соответствии с методикой примера 4(2) использовали 4-гидроксиэтил-N-(4-нитрофенил)пиперидин, полученный в примере 6(1), вместо 4-гидроксиметил-N-(4-нитрофенил)пиперидина, получали 4-тозилоксиэтил-N-(4-нитрофенил)пиперидин (93%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,13-1,35 (м, 2H), 1,55-1,84 (м, 5H), 2,46 (с, 3H), 2,82-3,01 (м, 2H), 3,84-4,00 (м, 2H), 4,10 (т, J=6,1 Гц, 2H), 6,78 (д, J=9,4 Гц, 2H), 7,36 (д, J=8,4 Гц, 2H), 7,80 (д, J=8,4 Гц, 2H), 8,10 (д, J=9,4 Гц, 2H)
Пример 6(3)
4-морфолиноэтил-N-(4-нитрофенил)пиперидин
4-тозилоксиэтил-N-(4-нитрофенил)пиперидин (2,02 г, 5,0 ммоль), полученный в примере 6(2), растворяли в ацетонитриле (20 мл) и добавляли карбонат калия (1,38 г, 10 ммоль) и морфолин (0,65 мл, 7,5 ммоль), затем перемешивали при 80°C в течение 15 часов. После охлаждения реакционной смеси до комнатной температуры в реакционную смесь добавляли этилацетат и органический слой промывали водой и насыщенным хлоридом натрия и сушили над безводным сульфатом натрия. Десикант отфильтровывали и после этого остаток, полученный выпариванием при пониженном давлении, очищали флэш-хроматографией среднего давления на колонке с силикагелем (метанол:хлороформ = от 1:50 до 1:20), таким образом получали 4-морфолиноэтил-N-(4-нитрофенил)пиперидин (1,23 г, 77%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,17-1,70 (м, 5H), 1,73-1,88 (м, 2H), 2,29-2,52 (м, 6H), 2,84-3,03 (м, 2H), 3,60-3,78 (м, 4H), 3,85-4,03 (м, 2H), 6,77 (д, J=9,4 Гц, 2H), 8,08 (д, J=9,4 Гц, 2H)
Пример 6(4)
4-морфолиноэтил-N-(4-аминофенил)пиперидин
В соответствии с методикой примера 1(2), используя 4-морфолиноэтил-N-(4-нитрофенил)пиперидин, полученный в примере 6(3), вместо 4-гидрокси-N-(4-нитрофенил)пиперидина, получали 4-морфолиноэтил-N-(4-аминофенил)пиперидин (83%) в виде красновато-фиолетового твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,28-1,98 (м, 7H), 2,32-2,73 (м, 8H), 3,20-3,90 (м, 8H), 6,62 (д, J=8,8 Гц, 2H), 6,80 (д, J=8,8 Гц, 2H)
Пример 6(5)
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолиноэтилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 1(3), используя 4-морфолиноэтил-N-(4-аминофенил)пиперидин, полученный в примере 6(4), вместо 4-гидрокси-N-(4-аминофенил)пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолиноэтилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (88%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,30-1,55 (м, 5H), 1,70-1,89 (м, 2H), 2,34-2,75 (м, 8H), 3,47-3,90 (м, 14Н), 3,80 (с, 3H), 6,05-6,13 (м, 1H), 6,27 (уш.с, 1H), 6,32-6,40 (м, 1H), 6,68-6,75 (м, 1H), 6,89 (д, J=8,9 Гц, 2H), 7,20 (д, J=8,9 Гц, 2H)
Пример 7
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(1,2,3-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Пример 7(1)
4-[2-(1,2,3-триазол-1-ил)этил]-N-(4-нитрофенил)пиперидин
В соответствии с методикой примера 6(3), используя 1,2,3-триазол вместо морфолина, получали 4-[2-(1,2,3-триазол-1-ил)этил]-N-(4-нитрофенил)пиперидин (39%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,25-1,72 (м, 3H), 1,77-2,05 (м, 4H), 2,84-3,05 (м, 2H), 3,87-4,04 (м, 2H), 4,49 (т, J=7,1 Гц, 2H), 6,80 (д, J=9,5 Гц, 2H), 7,56 (д, J=0,8 Гц, 1H), 7,73 (д, J=0,8 Гц, 1H), 8,10 (д, J=9,5 Гц, 2H)
Пример 7(2)
4-[2-(1,2,3-триазол-1-ил)этил]-N-(4-аминофенил)пиперидин
В соответствии с методикой примера 1(2), используя 4-[2-(1,2,3-триазол-1-ил)этил]-N-(4-нитрофенил)пиперидин, полученный в примере 7(1), вместо 4-гидрокси-N-(4-нитрофенил)пиперидина, получали 4-[2-(1,2,3-триазол-1-ил)этил]-N-(4-аминофенил)пиперидин (91%) в виде красновато-фиолетового твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,23-1,60 (м, 3H), 1,65-2,10 (м, 4H), 2,44-2,73 (м, 2H), 3,10-3,75 (м, 4H), 4,46 (т, J=7,4 Гц, 2H), 6,63 (д, J=8,7 Гц, 2H), 6,81 (д, J=8,7 Гц, 2H), 7,55 (с, 1H), 7,71 (с, 1H)
Пример 7(3)
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(1,2,3-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 1(3), используя 4-[2-(1,2,3-триазол-1-ил)этил]-N-(4-аминофенил)пиперидин, полученный в примере 7(2), вместо 4-гидрокси-N-(4-аминофенил)пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(1,2,3-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (50%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,31-1,53 (м, 3H), 1,72-2,00 (м, 4H), 2,55-2,78 (м, 2H), 3,46-3,66 (м, 6H), 3,73-3,92 (м, 4H), 3,80 (с, 3H), 4,47 (т, J=7,4 Гц, 2H), 6,05-6,14 (м, 1H), 6,32 (уш.с, 1H), 6,28-6,43 (м, 1H), 6,69-6,76 (м, 1H), 6,88 (д, J=9,0 Гц, 2H), 7,21 (д, J=9,0 Гц, 2H), 7,56 (д, J=0,7 Гц, 1H), 7,72 (д, J=0,7 Гц, 1H)
Пример 8
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(1,2,4-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Пример 8(1)
4-[2-(1,2,4-триазол-1-ил)этил]-N-(4-нитрофенил)пиперидин
В соответствии с методикой примера 6(3), используя 1,2,4-триазол вместо морфолина, получали 4-[2-(1,2,4-триазол-1-ил)этил]-N-(4-нитрофенил)пиперидин (78%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,23-1,65 (м, 3H), 1,74-2,03 (м, 4H), 2,82-3,09 (м, 2H), 3,85-4,08 (м, 2H), 4,26 (т, J=7,1 Гц, 2H), 6,80 (д, J=9,4 Гц, 2H), 7,95 (с, 1H), 8,08 (с, 1H), 8,10 (д, J=9,4 Гц, 2H)
Пример 8(2)
4-[2-(1,2,4-триазол-1-ил)этил]-N-(4-аминофенил)пиперидин
В соответствии с методикой примера 1(2), используя 4-[2-(1,2,4-триазол-1-ил)этил]-N-(4-нитрофенил)пиперидин, полученный в примере 8 (1), вместо 4-гидрокси-N-(4-нитрофенил)пиперидина, получали 4-[2-(1,2,4-триазол-1-ил)этил]-N-(4-аминофенил)пиперидин (99%) в виде красновато-фиолетового твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,20-1,55 (м, 3H), 1,66-1,90 (м, 4H), 2,43-2,64 (м, 2H), 3,27-3,44 (м, 2H), 3,45-4,05 (уш., 2H), 4,16 (т, J=7,3 Гц, 2H), 6,55 (д, J=8,6 Гц, 2H), 6,77 (д, J=8,6 Гц, 2H), 7,85 (с, 1H), 8,01 (с, 1H)
Пример 8(3)
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(1,2,4-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 1(3), используя 4-[2-(1,2,4-триазол-1-ил)этил]-N-(4-аминофенил)пиперидин, полученный в примере 8(2), вместо 4-гидрокси-N-(4-аминофенил)пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(1,2,4-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (45%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,30-1,98 (м, 7H), 2,53-2,75 (м, 2H), 3,43-3,67 (м, 6H), 3,71-3,92 (м, 4H), 3,80 (с, 3H), 4,25 (т, J=7,3 Гц, 2H), 6,07-6,15 (м, 1H), 6,27 (уш.с, 1H), 6,29-6,42 (м, 1H), 6,67-6,74 (м, 1H), 6,88 (д, J=9,0 Гц, 2H), 7,21 (д, J=9,0 Гц, 2H), 7,95 (с, 1H), 8,07 (с, 1H)
Пример 9
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(3,5-диметил-1,2,4-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Пример 9(1)
4-[2-(3,5-диметил-1,2,4-триазол-1-ил)этил]-N-(4-нитрофенил)пиперидин
В соответствии с методикой примера 6(3), используя 3,5-диметил-1,2,4-триазол вместо морфолина, получали 4-[2-(3,5-диметил-1,2,4-триазол-1-ил)этил]-N-(4-нитрофенил)пиперидин (84%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,25-1,90 (м, 7H), 2,33 (с, 3H), 2,41 (с, 3H), 2,86-3,05 (м, 2H), 3,88-4,10 (м, 4H), 6,80 (д, J=9,4 Гц, 2H), 8,10 (д, J=9,4 Гц, 2H)
Пример 9(2)
4-[2-(3,5-диметил-1,2,4-триазол-1-ил)этил]-N-(4-аминофенил)пиперидин
В соответствии с методикой примера 1(2), используя 4-[2-(3,5-диметил-1,2,4-триазол-1-ил)этил]-N-(4-нитрофенил)пиперидин, полученный в примере 9(1), вместо 4-гидрокси-N-(4-нитрофенил)пиперидина, получали 4-[2-(3,5-диметил-1,2,4-триазол-1-ил)этил]-N-(4-аминофенил)пиперидин (97%) в виде красновато-фиолетового твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,28-1,55 (м, 3H), 1,62-1,89 (м, 4H), 2,33 (с, 3H), 2,40 (с, 3H), 2,47-2,66 (м, 2H), 3,10-3,60 (м, 4H), 3,98-4,10 (м, 2H), 6,64 (д, J=8,7 Гц, 2H), 6,81 (д, J=8,7 Гц, 2H)
Пример 9(3)
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(3,5-диметил-1,2,4-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 1(3), используя 4-[2-(3,5-диметил-1,2,4-триазол-1-ил)-этил]-N-(4-аминофенил)пиперидин, полученный в примере 9(2), вместо 4-гидрокси-N-(4-аминофенил)пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(3,5-диметил-1,2,4-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (76%) в виде белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,30-1,55 (м, 3H), 1,72-1,95 (м, 4H), 2,33 (с, 3H), 2,40 (с, 3H), 2,57-2,75 (м, 2H), 3,48-3,65 (м, 6H), 3,73-3,90 (м, 4H), 3,80 (с, 3H), 4,03 (т, J=7,5 Гц, 2H), 6,06-6,13 (м, 1H), 6,31-6,38 (м, 1H), 6,40 (уш.с, 1H), 6,68-6,74 (м, 1H), 6,88 (д, J=9,1 Гц, 2H), 7,21 (д, J=9,1 Гц, 2H)
Пример 10
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(пиразол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Пример 10 (1)
4-[2-(пиразол-1-ил)этил]-N-(4-нитрофенил)пиперидин
В соответствии с методикой примера 6(3), используя пиразол вместо морфолина, получали 4-[2-(пиразол-1-ил)этил]-N-(4-нитрофенил)пиперидин (72%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,23-1,67 (м, 3H), 1,76-1,98 (м, 4H), 2,84-3,05 (м, 2H), 3,85-4,03 (м, 2H), 4,21 (т, J=7,1 Гц, 2H), 6,21-6,34 (м, 1H), 6,79 (д, J=9,4 Гц, 2H), 7,36-7,45 (м, 1H), 7,48-7,57 (м, 1H), 8,10 (д, J=9,4 Гц, 2H)
Пример 10(2)
4-[2-(пиразол-1-ил)этил]-N-(4-аминофенил)пиперидин
В соответствии с методикой примера 1(2), используя 4-[2-(пиразол-1-ил)этил]-N-(4-нитрофенил)пиперидин, полученный в примере 10(1), вместо 4-гидрокси-N-(4-нитрофенил)пиперидина, получали 4-[2-(пиразол-1-ил)этил]-N-(4-аминофенил)пиперидин (83%) в виде красновато-фиолетового твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,25-1,64 (м, 3H), 1,73-1,96 (м, 4H), 2,51-2,70 (м, 2H), 3,38-3,53 (м, 2H), 4,10-2,70 (уш., 2H), 4,20 (т, J=7,3 Гц, 2H), 6,25 (т, J=1,8 Гц, 1H), 6,64 (д, J=8,7 Гц, 2H), 6,86 (д, J=8,7 Гц, 2H), 7,39 (д, J=1,8 Гц, 1H), 7,50 (д, J=1,8 Гц, 1H)
Пример 10(3)
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(пиразол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 1(3), используя 4-[2-(пиразол-1-ил)этил]-N-(4-аминофенил)пиперидин, полученный в примере 10(2), вместо 4-гидрокси-N-(4-аминофенил)пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(пиразол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (56%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,30-1,52 (м, 3H), 1,70-1,97 (м, 4H), 2,53-2,74 (м, 2H), 3,45-3,67 (м, 6H), 3,71-3,93 (м, 4H), 3,80 (с, 3H), 4,20 (т, J=7,3 Гц, 2H), 6,06-6,18 (м, 1H), 6,22-6,40 (м, 3H), 6,67-6,78 (м, 1H), 6,88 (д, J=9,0 Гц, 2H), 7,20 (д, J=9,0 Гц, 2H), 7,39 (д, J=2,3 Гц, 1H), 7,51 (д, J=1,6 Гц, 1H)
Пример 11
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(3-(1,2,4-триазол-1-ил)пропил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Пример 11(1)
4-гидроксипропил-N-(4-нитрофенил)пиперидин
В соответствии с методикой примера 1(1), используя 4-гидроксипропилпиперидин вместо 4-гидроксипиперидина, получали 4-гидроксипропил-N-(4-нитрофенил)пиперидин (46%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,19-1,97 (м, 10Н), 2,85-3,08 (м, 2H), 3,57-3,76 (м, 2H), 3,88-4,05 (м, 2H), 6,79 (д, J=9,6 Гц, 2H), 8,10 (д, J=9,6 Гц, 2H)
Пример 11(2)
4-тозилоксипропил-N-(4-нитрофенил)пиперидин
В соответствии с методикой примера 4(2), используя 4-гидроксипропил-N-(4-нитрофенил)пиперидин, полученный в примере 11(1), вместо 4-гидроксиметил-N-(4-нитрофенил)пиперидина, получали 4-тозилоксипропил-N-(4-нитрофенил)пиперидин (97%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,15-1,84 (м, 9H), 2,45 (с, 3H), 2,82-3,03 (м, 2H), 3,84-4,00 (м, 2H), 4,04 (т, J=6,3 Гц, 2H), 6,78 (д, J=9,6 Гц, 2H), 7,35 (д, J=7,9 Гц, 2H), 7,79 (д, J=7,9 Гц, 2H), 8,10 (д, J=9,6 Гц, 2H)
Пример 11(3)
4-[3-(1,2,4-триазол-1-ил)пропил]-N-(4-нитрофенил)пиперидин
4-тозилоксипропил-N-(4-нитрофенил)пиперидин (4,19 г, 10,0 ммоль), полученный в примере 11(2), растворяли в ацетонитриле (40 мл) и добавляли карбонат калия (2,76 г, 20 ммоль) и 1,2,4-триазол (1,04 г, 15,0 ммоль), затем перемешивали при 80°C в течение 18 часов. После охлаждения реакционной смеси до комнатной температуры добавляли этилацетат в реакционную смесь, и органический слой промывали водой и насыщенным хлоридом натрия и сушили над безводным сульфатом натрия. Десикант отфильтровывали и после этого остаток, полученный выпариванием при пониженном давлении, очищали флэш-хроматографией среднего давления на колонке с силикагелем (метанол:хлороформ = от 1:100 до 1:30), получали 4-[3-(1,2,4-триазол-1-ил)пропил]-N-(4-нитрофенил)пиперидин (2,47 г, 78%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,17-1,39 (м, 4H), 1,45-1,65 (м, 1H), 1,70-2,05 (м, 4H), 2,83-3,05 (м, 2H), 3,84-4,02 (м, 2H), 4,18 (т, J=7,1 Гц, 2H), 6,79 (д, J=9,6 Гц, 2H), 7,95 (с, 1H), 8,05 (с, 1H), 8,10 (д, J=9,6 Гц, 2H)
Пример 11(4)
4-[3-(1,2,4-триазол-1-ил)пропил]-N-(4-аминофенил)пиперидин
В соответствии с методикой примера 1(2), используя 4-[3-(1,2,4-триазол-1-ил)пропил]-N-(4-нитрофенил)пиперидин, полученный в примере 11(3), вместо 4-гидрокси-N-(4-нитрофенил)пиперидина, получали 4-[3-(1,2,4-триазол-1-ил)пропил]-N-(4-аминофенил)пиперидин (95%) в виде красновато-фиолетового твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,20-1,48 (м, 5H), 1,65-2,07 (м, 4H), 2,43-2,68 (м, 2H), 3,06-3,77 (м, 4H), 4,17 (т, J=7,1 Гц, 2H), 6,63 (д, J=8,9 Гц, 2H), 6,82 (д, J=8,9 Гц, 2H), 7,94 (с, 1H), 8,05 (с, 1H)
Пример 11(5)
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(3-(1,2,4-триазол-1-ил)пропил)пиперидин-1-ил)фенил)пиперазинкарбоксамид
В соответствии с методикой примера 1(3), использовали 4-[3-(1,2,4-триазол-1-ил)пропил]-N-(4-аминофенил)пиперидин, полученный в примере 11(4), вместо 4-гидрокси-N-(4-аминофенил)пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(3-(1,2,4-триазол-1-ил)пропил)пиперидин-1-ил)фенил)пиперазинкарбоксамид (67%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,20-1,48 (м, 5H), 1,66-2,05 (м, 4H), 2,52-2,74 (м, 2H), 3,44-3,65 (м, 6H), 3,80 (с, 3H), 3,75-3,92 (м, 4H), 4,17 (т, J=7,1 Гц, 2H), 6,05-6,14 (м, 1H), 6,31 (уш.с, 1H), 6,30-6,44 (м, 1H), 6,67-6,79 (м, 1H), 6,88 (д, J=9,1 Гц, 2H), 7,20 (д, J=9,1 Гц, 2H), 7,95 (с, 1H), 8,05 (с, 1H)
Пример 12
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Пример 12 (1)
4-[3-(1,2,3-триазол-1-ил)пропил]-N-(4-нитрофенил)пиперидин
В соответствии с методикой примера 7(1), используя 4-тозилоксипропил-N-(4-нитрофенил)пиперидин, полученный в примере 11(2), вместо 4-тозилоксиэтил-N-(4-нитрофенил)пиперидина, получали 4-[2-(1,2,3-триазол-1-ил)пропил]-N-(4-аминофенил)пиперидин (69%) в виде желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,16-1,40 (м, 4H), 1,48-1,67 (м, 1H), 1,70-2,09 (м, 4H), 2,83-3,05 (м, 2H), 3,85-4,03 (м, 2H), 4,40 (т, J=7,1 Гц, 2H), 6,79 (д, J=9,6 Гц, 2H), 7,54 (д, J=0,9 Гц, 1H), 7,71 (д, J=0,9 Гц, 1H), 8,10 (д, J=9,6 Гц, 2H)
Пример 12(2)
4-[2-(1,2,3-триазол-1-ил)пропил]-N-(4-аминофенил)пиперидин
В соответствии с методикой примера 1(2), используя 4-[2-(1,2,3-триазол-1-ил)пропил]-N-(4-нитрофенил)пиперидин, полученный в примере 12(1), вместо 4-гидрокси-N-(4-нитрофенил)пиперидина, получали 4-[2-(1,2,3-триазол-1-ил)пропил]-N-(4-аминофенил)пиперидин (94%) в виде красновато-фиолетового твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,23-1,46 (м, 5H), 1,59-1,87 (м, 2H), 1,90-2,05 (м, 2H), 2,45-2,63 (м, 2H), 3,30-3,52 (м, 4H), 4,40 (т, J=7,2 Гц, 2H), 6,64 (д, J=8,8 Гц, 2H), 6,81 (д, J=8,8 Гц, 2H), 7,53 (д, J=0,7 Гц, 1H), 7,71 (д, J=0,7 Гц, 1H)
Пример 12(3)
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 1(3), используя 4-[2-(1,2,3-триазол-1-ил)пропил]-N-(4-аминофенил)пиперидин, полученный в примере 12(2), вместо 4-гидрокси-N-(4-аминофенил)пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (57%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,22-1,48 (м, 5H), 1,68-2,08 (м, 4H), 2,50-2,72 (м, 2H), 3,45-3,66 (м, 6H), 3,80 (с, 3H), 3,73-3,90 (м, 4H), 4,39 (т, J=7,1 Гц, 2H), 6,07-6,14 (м, 1H), 6,30-6,45 (м, 2H), 6,68-6,77 (м, 1H), 6,87 (д, J=8,9 Гц, 2H), 7,21 (д, J=8,9 Гц, 2H), 7,55 (д, J=0,8 Гц, 1H), 7,71 (д, J=0,8 Гц, 1H)
Пример 13
4-((1-этилпиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Пример 13(1)
4-(бензилоксикарбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
4-нитрофенилхлорформиат (7,26 г, 36,0 ммоль) растворяли в тетрагидрофуране (100 мл), и тетрагидрофурановый раствор (50 мл) 1-(4-аминофенил)пиперидин-4-морфолинкарбоксамида (8,68 г, 30,0 ммоль), полученный в примере 3(3), добавляли по каплям при -30°C. После перемешивания в течение 30 минут при этой температуре добавляли в смесь тетрагидрофурановый раствор (30 мл) N-бензилоксикарбонилпиперазина (7,27 г, 33,0 ммоль) и триэтиламин (14,0 мл, 100 ммоль), затем перемешивали при комнатной температуре в течение 13 часов и после этого при 60°C в течение 4 часов. После охлаждения реакционной смеси до комнатной температуры, добавляли разбавленный водный раствор гидроксида натрия, затем проводили экстракцию этилацетатом. Органический слой промывали водой и насыщенным хлоридом натрия и сушили над безводным сульфатом натрия. Десикант отфильтровывали и после этого остаток, полученный выпариванием при пониженном давлении, очищали флэш-хроматографией среднего давления на колонке с силикагелем (силикагель, метанол:хлороформ = от 0:1 до 1:30), таким образом получая 4-(бензилоксикарбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (9,02 г, 56%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,72-1,87 (м, 2H), 1,90-2,12 (м, 2H), 2,47-2,78 (м, 3H), 3,38-3,77 (м, 18Н), 5,16 (с, 2H), 6,24 (с, 1H), 6,88 (д, J=8,9 Гц, 2H), 7,20 (д, J=8,9 Гц, 2H), 7,29-7,44 (м, 5H)
Пример 13 (2)
N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
4-(бензилоксикарбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (5,89 г, 11,0 ммоль), полученный в примере 13(1), суспендировали в тетрагидрофуране (80 мл) и метаноле (80 мл) и добавляли 10% палладий на углероде (1,5 г), затем перемешивали при комнатной температуре в среде водородного газа в течение 18 часов. После фильтрации через целит нерастворимых веществ, фильтрат концентрировали при пониженном давлении, и полученный остаток очищали флэш-хроматографией среднего давления на колонке с силикагелем (силикагель NH, метанол:хлороформ = от 1:50 до 1:15), таким образом получая N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (3,62 г, 82%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,73-1,86 (м, 2H), 1,90-2,10 (м, 2H), 2,48-2,76 (м, 3H), 2,83-3,00 (м, 4H), 3,35-3,78 (м, 15H), 6,22 (с, 1H), 6,88 (д, J=9,1 Гц, 2H), 7,22 (д, J=9,1 Гц, 2H)
Пример 13(3)
4-((1-этилпиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
1-этилпиррол-2-карбоновую кислоту (153 мг, 1,1 ммоль) растворяли в N,N-диметилформамиде (3,0 мл) и добавляли 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (253 мг, 1,3 ммоль), 1-гидроксибензотриазола моногидрат (185 мг, 1,2 ммоль) и N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (201 мг, 0,5 ммоль), полученный в примере 13(2), затем перемешивали при нагревании при 60°C в течение 15 часов. После охлаждения реакционной смеси до комнатной температуры, насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь, затем проводили экстракцию хлороформом. Экстракт промывали водой и насыщенным хлоридом натрия и сушили над безводным сульфатом натрия. Десикант отфильтровывали и после этого остаток, полученный выпариванием при пониженном давлении, очищали флэш-хроматографией среднего давления на колонке с силикагелем (метанол:хлороформ = от 1:50 до 1:30), таким образом получая 4-((1-этилпиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (228 мг, 44%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,38 (т, J=7,3 Гц, 3H), 1,72-2,10 (м, 4H), 2,48-2,80 (м, 3H), 3,40-3,92 (м, 18Н), 4,18 (кв, J=7,3 Гц, 2H), 6,08-6,17 (м, 1H), 6,28-6,40 (м, 1H), 6,37 (уш.с, 1H), 6,75-6,83 (м, 1H), 6,89 (д, J=8,9 Гц, 2H), 7,22 (д, J=8,9 Гц, 2H)
Пример 14
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-пиперидин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
4-((((1-метилпиррол-2-ил)карбонил)-1-пиперазинил)карбонил)амино-4-фенилпиперидин-4-карбоновую кислоту (440 мг, 1,0 ммоль), полученную в примере 2(4), растворяли в N,N-диметилформамиде (3 мл) и добавляли 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (230 мг, 1,2 ммоль), 1-гидроксибензотриазола моногидрат (168 мг, 1,1 ммоль) и пиперидин (0,12 мл, 1,2 ммоль), затем перемешивали при нагревании при 60°C в течение 6 часов. После охлаждения реакционной смеси до комнатной температуры, добавляли насыщенный водный раствор бикарбоната натрия, затем проводили экстракцию этилацетатом. Органический слой промывали водой и насыщенным хлоридом натрия и сушили над безводным сульфатом натрия. Десикант отфильтровывали и после этого остаток, полученный выпариванием при пониженном давлении, очищали флэш-хроматографией среднего давления на колонке с силикагелем (метанол:хлороформ = 1:50 к 1:20), таким образом получая 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-пиперидин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (226 мг, 45%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,58-1,67 (м, 6H), 1,77-2,00 (м, 4H), 2,54-2,74 (м, 3H), 3,34-3,93 (м, 17Н), 6,05-6,15 (м, 1H), 6,32-6,50 (м, 2H), 6,68-6,77 (м, 1H), 6,89 (д, J=8,9 Гц, 2H), 7,21 (д, J=8,9 Гц, 2H)
Пример 15
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(4-метилпиперазин-1-ил-карбонил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 14, используя 1-метилпиперазин вместо пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(4-метилпиперазин-1-ил-карбонил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (14%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,63-2,15 (м, 4H), 2,30-2,77 (м, 10Н), 3,52-3,93 (м, 17Н), 6,08-6,15 (м, 1H), 6,30-6,45 (м, 2H), 6,68-6,75 (м, 1H), 6,88 (д, J=8,7 Гц, 2H), 7,22 (д, J=8,7 Гц, 2H)
Пример 16
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-морфолиноэтилкарбамоил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 14, используя 2-аминоэтилморфолин вместо пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-морфолиноэтилкарбамоил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (38%) в виде бледно-желтого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,75-2,03 (м, 4H), 2,14-2,29 (м, 1H), 2,34-2,55 (м, 6H), 2,62-2,78 (м, 2H), 3,28-3,41 (м, 2H), 3,49-3,87 (м, 17Н), 6,02-6,18 (м, 2H), 6,31-6,40 (м, 1H), 6,43 (с, 1H), 6,67-6,75 (м, 1H), 6,88 (д, J=8,8 Гц, 2H), 7,22 (д, J=8,8 Гц, 2H)
Пример 17
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(пиридин-3-илметилкарбамоил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
В соответствии с методикой примера 14, используя 3-аминометилпиридин вместо пиперидина, получали 4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(пиридин-3-илметилкарбамоил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид (33%) в виде молочно-белого твердого вещества.
1H-ЯМР (CDCl3): δ (м.д.) 1,76-1,99 (м, 4H), 2,13-2,34 (м, 1H), 2,55-2,74 (м, 2H), 3,44-3,88 (м, 13Н), 4,44 (д, J=5,9 Гц, 2H), 6,10 (дд, J=2,7, 3,8 Гц, 1H), 6,25-6,40 (м, 2H), 6,56 (с, 1H), 6,69-6,76 (м, 1H), 6,84 (д, J=8,9 Гц, 2H), 7,13-7,34 (м, 3H), 7,55-7,67 (м, 1H), 8,45-8,60 (м, 2H)
Справочные примеры
Способ A
В соответствии с методикой примера 13(3), используя соответствующую карбоновую кислоту вместо 1-этилпиррол-2-карбоновой кислоты, получали указанное соединение.
Способ B
N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид, полученный в примере 13(2), суспендировали в тетрагидрофуране и хлороформе и добавляли триэтиламин и соответствующий хлорангидрид, затем перемешивали при комнатной температуре. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь, затем выполняли экстракцию хлороформом. Экстракт промывали водой и насыщенным хлоридом натрия и сушили над безводным сульфатом натрия. Десикант отфильтровывали и после этого остаток, полученный выпариванием при пониженном давлении, очищали флэш-хроматографией среднего давления на колонке с силикагелем, получая указанное соединение.
Справочный пример 1
4-((пиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ A, выход: 53%
1H-ЯМР (CDCl3): δ (м.д.) 1,72-2,13 (м, 4H), 2,49-2,80 (м, 3H), 3,46-4,08 (м, 18Н), 6,23-6,32 (м, 1H), 6,33 (уш.с, 1H), 6,50-6,63 (м, 1H), 6,89 (д, J=8,9 Гц, 2H), 6,90-7,05 (м, 1H), 7,23 (д, J=8,9 Гц, 2H), 9,50 (уш.с, 1H)
Справочный пример 2
4-((3,5-диметилпиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ A, выход: 25%
1H-ЯМР (CDCl3): δ (м.д.) 1,60-2,15 (м, 4H), 2,11 (с, 3H), 2,23 (с, 3H), 2,47-2,79 (м, 3H), 3,40-3,82 (м, 18Н), 5,74 (д, J=2,6 Гц, 1H), 6,37-6,54 (м, 1H), 6,88 (д, J=8,9 Гц, 2H), 7,21 (д, J=8,9 Гц, 2H), 8,63 (уш.с, 1H)
Справочный пример 3
4-((1-метилпиррол-3-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ A, выход: 42%
1H-ЯМР (CDCl3): δ (м.д.) 1,65-2,12 (м, 4H), 2,47-2,80 (м, 3H), 3,45-3,87 (м, 18Н), 3,67 (с, 3H), 6,24-6,32 (м, 1H), 6,47 (уш.с, 1H), 6,53-6,62 (м, 1H), 6,88 (д, J=8,9 Гц, 2H), 6,98-7,05 (м, 1H), 7,22 (д, J=8,9 Гц, 2H)
Справочный пример 4
4-((тиофен-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ A, выход: 54%
1H-ЯМР (CDCl3): δ (м.д.) 1,70-2,10 (м, 4H), 2,45-2,80 (м, 3H), 3,43-3,91 (м, 18Н), 6,37 (уш.с, 1H), 6,89 (д, J=8,9 Гц, 2H), 7,02-7,13 (м, 1H), 7,22 (д, J=8,9 Гц, 2H), 7,25-7,37 (м, 1H), 7,40-7,54 (м, 1H)
Справочный пример 5
4-((тиофен-3-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ B, выход: 70%
1H-ЯМР (CDCl3): δ (м.д.) 1,72-2,10 (м, 4H), 2,48-2,80 (м, 3H), 3,38-3,89 (м, 18Н), 6,44 (уш.с, 1H), 6,88 (д, J=8,9 Гц, 2H), 7,15-7,27 (м, 3H), 7,37 (дд, J=4,9, 3,0 Гц, 1H), 7,55 (дд, J=3,0, 1,3 Гц, 1H)
Справочный пример 6
4-((фуран-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ B, выход: 76%
1H-ЯМР (CDCl3): δ (м.д.) 1,71-2,10 (м, 4H), 2,48-2,81 (м, 3H), 3,45-4,00 (м, 18Н), 6,34-6,46 (м, 1H), 6,51 (дд, J=3,5, 1,8 Гц, 1H), 6,89 (д, J=8,9 Гц, 2H), 7,06 (дд, J=3,5, 0,8 Гц, 1H), 7,22 (д, J=8,9 Гц, 2H), 7,51 (дд, J=1,8, 0,8 Гц, 1H)
Справочный пример 7
4-((фуран-3-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ A, выход: 58%
1H-ЯМР (CDCl3): δ (м.д.) 1,73-2,07 (м, 4H), 2,50-2,84 (м, 3H), 3,47-3,85 (м, 18Н), 6,53-6,62 (м, 1H), 6,87 (д, J=8,9 Гц, 2H), 7,29 (д, J=8,9 Гц, 2H), 7,45-7,53 (м, 1H), 7,70-7,78 (м, 1H), 7,85 (уш.с, 1H)
Справочный пример 8
4-((изоксазол-5-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ B, выход: 56%
1H-ЯМР (CDCl3): δ (м.д.) 1,72-2,09 (м, 4H), 2,49-2,78 (м, 3H), 3,48-3,90 (м, 18Н), 6,37 (уш.с, 1H), 6,84 (д, J=1,8 Гц, 1H), 6,89 (д, J=9,1 Гц, 2H), 7,21 (д, J=9,1 Гц, 2H), 8,34 (д, J=1,8 Гц, 1H)
Справочный пример 9
4-((1-метилимидазол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ A, выход: 27%
1H-ЯМР (CDCl3): δ (м.д.) 1,73-2,10 (м, 4H), 2,47-2,80 (м, 3H), 3,47-3,88 (м, 16Н), 3,91 (с, 3H), 4,14-4,34 (м, 2H), 6,27 (уш.с, 1H), 6,89 (д, J=8,9 Гц, 2H), 6,96 (д, J=1,2 Гц, 1H), 7,05 (д, J=1,2 Гц, 1H), 7,22 (д, J=8,9 Гц, 2H)
Справочный пример 10
4-(циклопентилкарбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ B, выход: 84%
1H-ЯМР (CDCl3): δ (м.д.) 1,50-2,11 (м, 12Н), 2,47-2,98 (м, 4H), 3,34-3,80 (м, 18Н), 6,44 (уш.с, 1H), 6,88 (д, J=8,9 Гц, 2H), 7,21 (д, J=8,9 Гц, 2H)
Справочный пример 11
4-(бензоил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ B, выход: 69%
1H-ЯМР (CDCl3): δ (м.д.) 1,70-2,10 (м, 4H), 2,47-2,77 (м, 3H), 3,30-3,95 (м, 18Н), 6,49 (уш.с, 1H), 6,87 (д, J=9,1 Гц, 2H), 7,20 (д, J=9,1 Гц, 2H), 7,32-7,50 (м, 5H)
Справочный пример 12
4-((1-метилиндол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид
Способ A, выход: 59%
1H-ЯМР (CDCl3): δ (м.д.) 1,72-2,10 (м, 4H), 2,47-2,79 (м, 3H), 3,47-3,95 (м, 21H), 6,30 (с, 1H), 6,63 (д, J=0,5 Гц, 1H), 6,89 (д, J=8,9 Гц, 2H), 7,11-7,43 (м, 5H), 7,64 (д, J=7,9 Гц, 1H)
Справочный пример 13
4-(3-фторбензоил)пиперазин-1-карбоновая кислота-(6-бромбензотиазол-2-ил)амид
Справочный пример 14
4-(3-фторбензоил)пиперазин-1-карбоновая кислота-(5,6-диметилбензотиазол-2-ил)амид
Справочный пример 15
4-(3-фторбензоил)пиперазин-1-карбоновая кислота-(6-метилбензотиазол-2-ил)амид
Справочный пример 16
4-(3-фторбензоил)пиперазин-1-карбоновая кислота-(6-метоксибензотиазол-2-ил)амид
Справочный пример 17
4-(3-фторбензоил)пиперазин-1-карбоновая кислота-(6-хлорбензотиазол-2-ил)амид
Справочный пример 18
4-(6-фторпиридин-2-карбонил)пиперазин-1-карбоновая кислота-(4-трифторметилфенил)амид
Справочные примеры 13-18 синтезировали по методикам способов, раскрытых в международной публикации WO2008-122787.
Справочный пример 19
N-метокси-N-метил-4-(5-бензоилбензимидазол-2-ил)-3,5-диметилпиррол-2-карбоксамид
Синтез выполняли способом, раскрытым в международной публикации WO2007-007778.
Примеры исследования
Пример исследования 1: Ингибирующая активность в отношении гематопоэтической простагландин-D-синтазы (H-PGDS)
Тесты выполняли в соответствии со способом Urade, Y. et al. (J. Biol. Chem., 262, 3820-3825, (1987)). Более конкретно, реакционную смесь (49 мкл), содержащую 100 мМ Трис-HCl (pH 8,0), 1 мМ восстановленный глутатион, 0,1 мг/мл γ-глобулина и человеческий H-PGDS (q.s), и соединение (конечная концентрация: 0,01-100 мкм), предварительно инкубировали при 25°C в течение 5 минут. Раствор ДМСО (конечная концентрация: 1%) добавляли в растворитель контрольной группы. Затем добавляли 1 мкл [14C] простагландин-H2 (конечная концентрация: 10 мкм) для начала реакции. Через одну минуту после начала реакции, добавляли 250 мкл останавливающего реакцию раствора (диэтиловый эфир/метанол/1М лимонной кислоты (30/4/1)) при температуре -20°C для прекращения реакции. После прекращения реакции 50 мкл верхней части слоя (фаза органического растворителя) наносили на пластину для тонкослойной хроматографии и разгоняли при -20°C в течение 45 минут (подвижная фаза: диэтиловый эфир/метанол/уксусная кислота (90/2/1)). После сушки ТСХ пластины, ТСХ-пластины проявляли в течение 1-24 часов и анализировали радиоактивность, соответствующую простагландину-D2 (PGD2), используя анализатор изображения (производитель Fujifilm Corporation). Площадь (%), соответствующую связыванию PGD2 на проход, рассчитывали для определения степени ингибирования (%) 0,1 мкМ соединения по каждому примеру по отношению к контрольной группе в каждом эксперименте, а также ингибирующую концентрацию при 50% (значение IC50, нМ) в отношении H-PGDS. Результаты показаны в таблице 1.
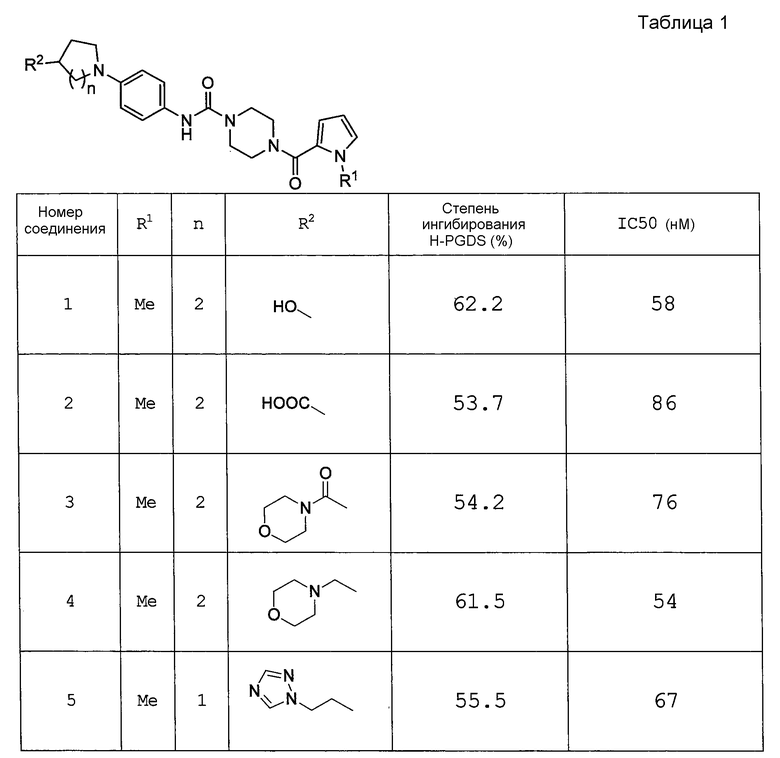
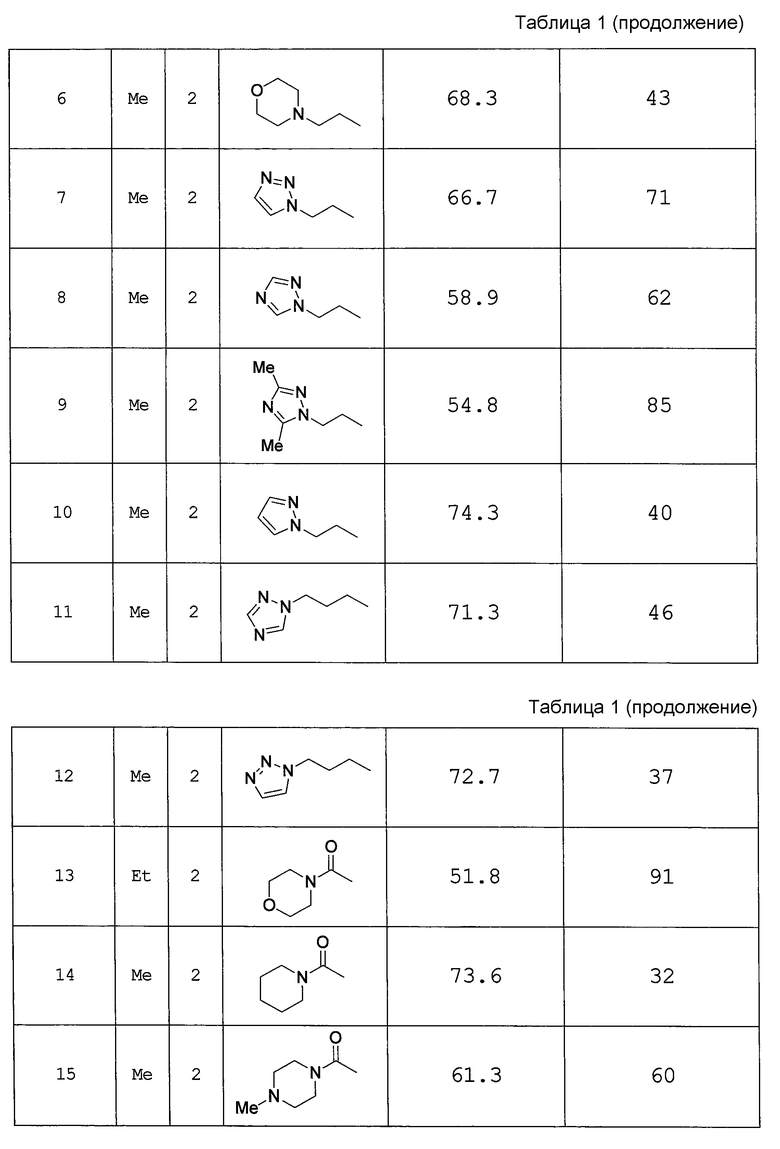
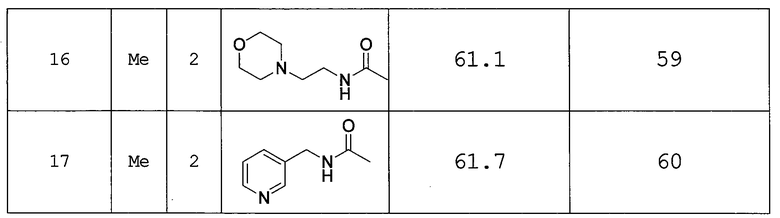
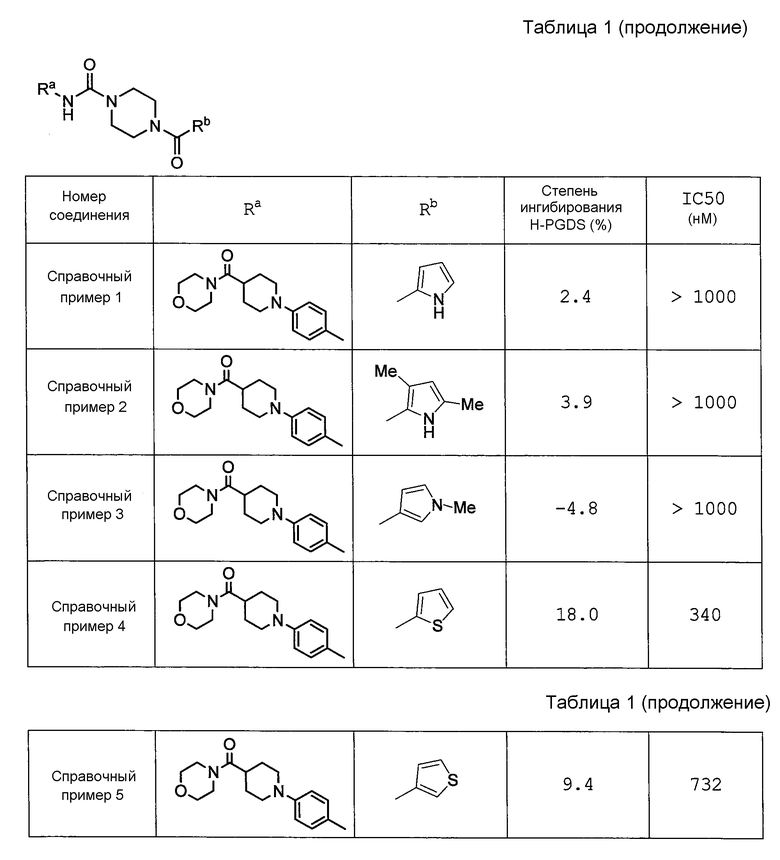
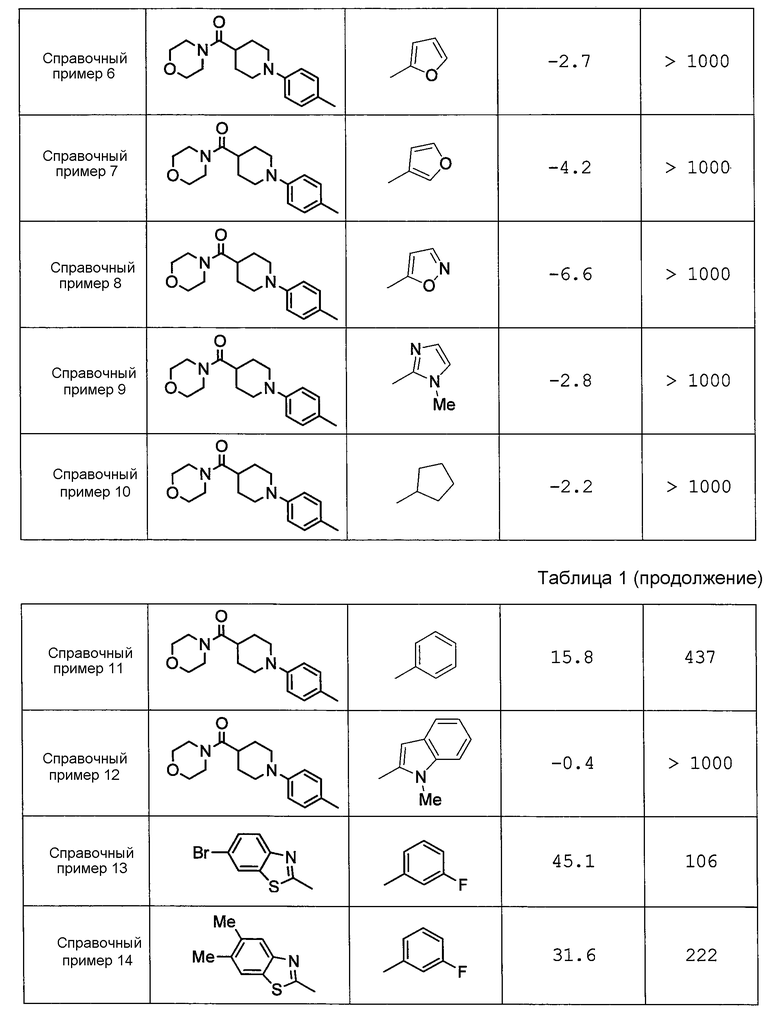
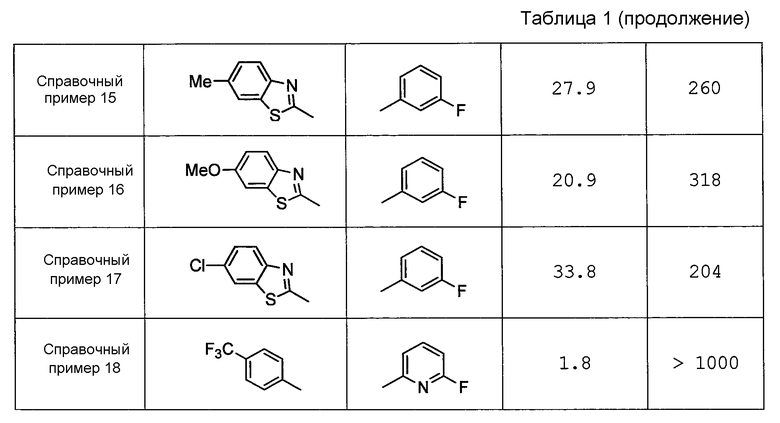
Соединения по справочным примерам 1-12 представляют собой соединения, в которых (N-алкилпиррол-2-ил)карбонильная группа, которая является отличительной для соединений по настоящему изобретению, замещена другим заместителем, таким как гетероциклическое кольцо. Как показано в таблице 1, пиперазиновое соединение с (N-алкилпиррол-2-ил)карбонильной группой, как в соединениях по настоящему изобретению, проявляло сильную ингибирующую H-PGDS активность, тогда как соединения по справочным примерам 1-12 показывали незначительную ингибирующую активность.
Далее соединения по справочным примерам 13-17 являются соединениями со структурой, подобной структуре соединений по настоящему изобретению, то есть имеют структуру, включающую фторбензоильную группу и аминокарбонильную группу, и показывают высокую ингибирующую GST2 (Диапазон A) активность. Соединение по справочному примеру 18 представляет собой соединение, включающее фторпиридинкарбонильную группу и аминокарбонильную группу, и эффективно против метаболического синдрома у мышей. Все эти соединения раскрыты в источнике Патентной Литературы 3.
Видно, что соединения по настоящему изобретению проявляют более сильную ингибирующую H-PGDS активность, чем по справочным примерам 13-18.
Пример исследования 2
Активность в отношении ингибирования продуцирования PGD2 в носовой полости морских свинок с антиген-индуцированным ринитом
Физиологический солевой раствор, содержащий 1 мг/мл овальбумина, подкожно вводили в спину 5-недельным самцам морских свинок Std:Hartley в количестве 1 мл/тело для активной сенсибилизации (первоначальная сенсибилизация). Через одну и две недели после первоначальной сенсибилизации 20 мкл физиологического солевого раствора, содержащего 10 мг/мл овальбумина, вводили в каждую носовую полость микропипеткой (сенсибилизация назальным введением). Через три недели после первоначальной сенсибилизации, 20 мкл физиологического солевого раствора, содержащего 10 мг/мл овальбумина, вводили в каждую носовую полость микропипеткой, чтобы вызвать реакцию ринита.
Исследуемое соединение (30 мг/кг) перорально вводили за 1 час до индукции реакции ринита. Животные в контрольной группе получали только среду перорально.
Через 30 минут после индукции реакции ринита носовые полости промывали под анестезией пентобарбиталом натрия. Промывную жидкость из полости носа (фосфатно-солевой буфер, содержащий 3 мМ ЭДТА и 10 мкм индометацина) подавали, используя перистальтический насос (Gilson, Inc), в направлении от трахеи до верхней части респираторного тракта со скоростью потока 1 мл/минута, и жидкость из носовых полостей собирали в течение 1 минуты. Собранную жидкость центрифугировали для отделения супернатанта как промывную жидкость из полости носа. Концентрацию PGD2 в промывной жидкости из полости носа определяли, используя набор EIA (набор простагландин-D2-MOX EIA, Cayman Chemical).
Показатель снижения PGD2 в промывной жидкости из полости носа рассчитывали по следующей формуле. Результаты показаны в таблице 2.
Показатель снижения в PGD2 в промывной жидкости из полости носа (%) =
{(Концентрация PGD2 в контрольной группе - концентрация PGD2 в группе, которой вводили соединение) ÷ (концентрация PGD2 в контрольной группе - концентрация PGD2 в нормальной группе)} × 100
8 или более образцов отбирали от каждой группы для определения наличия экспрессии активности в отношении ингибирования PGD2 активности, и сравнивали концентрации PGD2 в промывной жидкости из полости носа контрольной группы и каждым значением из группы, которой вводили соединение. Следует отметить, что случаи, в которых уровень значимости составлял ниже 0,05, рассматривали как наличие активности, и они отмечены в таблице символами (*). Справочный пример 19, известный как ингибитор H-PGDS, использовали в качестве положительного контроля.
В соответствии с результатами, представленными в таблице 2, соединение по настоящему изобретению проявляет показатель снижения концентрации PGD2, подобный таковому справочного примера 19 (эти соединения проявляют активность). Напротив, справочные примеры 13-18, раскрытые в источнике Патентной Литературы 3, не показывали значительного уменьшения концентрации PGD2.
Пример исследования 3
Активность в отношении ингибирования эозинофильной инфильтрации у морских свинок с антиген-индуцированным ринитом
Физиологический солевой раствор, содержащий 1 мг/мл овальбумина, вводили подкожно в спину самцов 5-недельных морских свинок Std:Hartley в количестве 1 мл/тело для активной сенсибилизации (первоначальная сенсибилизация). Через одну и две недели после первоначальной сенсибилизации, 20 мкл физиологического солевого раствора, содержащего 10 мг/мл овальбумина, вводили в каждую носовую полость микропипеткой (сенсибилизация назальным введением). Через три недели после начальной сенсибилизации, 20 мкл физиологического солевого раствора, содержащего 10 мг/мл овальбумина, вводили в каждую носовую полость микропипеткой, чтобы индуцировать реакцию ринита.
Исследуемое соединение (30 мг/кг) вводили в общей сложности перорально 3 раза (2 раза при сенсибилизации назальным введением и 1 раз при индукции реакции ринита) за 1 час до сенсибилизации или индукции. Животные в контрольной группе получали только среду перорально.
Через 6 часов после индукции реакции ринита, носовые полости промывали под анестезией пентобарбиталом натрия. Промывную жидкость из полости носа (фосфатно-солевой буфер, содержащий 3 мМ ЭДТА и 10 мкм индометацина) подавали перистальтическим насосом (Gilson, Inc) в направлении от трахеи до верхней части респираторного тракта при скорости потока 1 мл/минута и жидкость из полостей носа собирали в течение 3 минут. Собранную жидкость фильтровали через клеточный фильтр (40 мкм) и центрифугировали при 4°C, 300×g в течение 10 минут. Затем супернатант удаляли отсасыванием. 250 мкл промывной жидкости из полости носа добавляли к клеточным пеллетам, формируя суспензию. Общее количество клеток (×105 клеток) в клеточной суспензии определяли при помощи автоматизированного гематологического счетчика (F-820, Sysmex Corporation).
Клеточный мазок готовили на центрифужном устройстве для сбора клеток (Cytospin-3, Shandon). После окраски клеток окрашивающим раствором подсчитывали количество моноцитов, количество эозинофилов и количество нейтрофилов, так чтобы общее количество клеток составляло 300 клеток при наблюдении под микроскопом (с 200-кратным увеличением). Рассчитывали соотношение компонентов каждой клетки и число эозинофилов в промывной жидкости из полости носа определяли по следующей формуле.
Число эозинофилов (×105 клеток) =
(Количество эозинофилов ÷ общее расчетное количество (300)) × общее количество клеток
Показатель уменьшения количества эозинофилов в промывной жидкости полости носа рассчитывали как показатель ингибирования эозинофльной инфильтрации и определяли по следующей формуле. Результаты показаны в таблице 3.
Показатель ингибирования (%) =
{(Число эозинофилов в контрольной группе - число эозинофилов в группе, которой вводили соединения) ÷ (число эозинофилов в контрольной группе - число эозинофилов в нормальной группе)} × 100
16 образцов отбирали от каждой группы, и сравнивали количество эозинофилов в промывной жидкости из полости носа из контрольной группы с каждым значением из группы, в которой вводили соединение. Следует отметить, что случаи, в которых уровень значимости составлял ниже 0,05, рассматривали как наличие активности, и они отмечены в таблице символами (*).
Как видно из результатов, представленных в таблице 3, соединения по настоящему изобретению проявляют значительную активность в отношении ингибирования эозинофильной инфильтрации. Напротив, справочные примеры 13-17, раскрытые в источнике Патентной Литературы 3, как соединения, обладающие высокой ингибирующей GST2 (Диапазон A) активностью или повышенной эозинофильной инфильтрацией, не показали значительной активности в отношении ингибирования эозинофильной инфильтрации.
Пример исследования 4
Исследование на силу захвата передних конечностей мышей
В исследованиях использовали 4-недельных самцов мышей C57BL/10-mdx (mdx) в качестве группы с заболеванием, и 4-недельных самцов мышей C57BL/10Sn (дикого типа) в качестве нормальной группы. После транспортировки мышей выдерживали период акклиматизации. Исходные параметры каждого субъекта измеряли в возрасте 5 недель и отбирали 10 мышей из каждой группы. На следующий день исследуемое соединение (30 мг/кг) вводили мышам перорально один раз в день последовательно в течение 4 недель. Животные в контрольной группе получали только носитель перорально. Силу захвата передней конечности измеряли на 4 неделю после начала введения. Значение, полученное делением среднего из 5 измеренных значений (кг) силы захвата передней конечности, полученных с использованием малого устройства измерения силы захвата животного (GPM-100M, Melquest), на массу тела (кг), использовали как показатель для оценки (справочная литература: Muscle Nerve., 35, 43-48 (2007)).
9 или более образцов отбирали из каждой группы для определения наличия экспрессии активности в отношении увеличения силы захвата передней конечности. Во-первых, было подтверждено значительное уменьшение значений контрольной группы по сравнению с таковыми у нормальной группы, и затем при сравнении значений контрольной группы со значениями группы, которой вводили соединение. Случаи, в которых уровень значимости составлял ниже 0,05, рассматривали как наличие активности, и они отмечены в таблице символами (#) и (*). Результаты показаны в таблице 4.
В соответствии с результатами, представленными в таблице 4, наблюдали значительное снижение мышечной силы в контрольной группе по сравнению с нормальной группой. Наглядно показано, что соединение по настоящему изобретению проявляет активность в отношении усиления силы захвата передней конечности по сравнению с контрольной группой. Тогда как в справочных примерах 13-17 какая-либо значительная активность отсутствовала.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ 1,2,4-ТРИАЗОЛОНА | 2011 |
|
RU2566754C2 |
| ПРОИЗВОДНЫЕ БЕТА-КАРБОЛИНА, ОБЛАДАЮЩИЕ ДЕЙСТВИЕМ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), СПОСОБ ЕЕ ПОЛУЧЕНИЯ, СПОСОБ ИНГИБИРОВАНИЯ ДЕЙСТВИЯ ФОСФОДИЭСТЕРАЗЫ (ВАРИАНТЫ) И СПОСОБ УВЕЛИЧЕНИЯ КОНЦЕНТРАЦИИ ЦГМФ | 2001 |
|
RU2271358C2 |
| СОЕДИНЕНИЕ ДИКАРБОНОВОЙ КИСЛОТЫ | 2014 |
|
RU2662817C2 |
| ИНГИБИТОРЫ ГЛИКОЛАТОКСИДАЗЫ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2805308C2 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2019 |
|
RU2809763C2 |
| АЗОТСОДЕРЖАЩИЕ АРОМАТИЧЕСКИЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ, СПОСОБ ЛЕЧЕНИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2310651C2 |
| ПРОИЗВОДНЫЕ 2,3-ДИГИДРО-6-НИТРОИМИДАЗО [2, 1-b] ОКСАЗОЛА | 2003 |
|
RU2326121C2 |
| ИНГИБИТОРЫ ПРОЛИЛГИДРОКСИЛАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2429226C9 |
| КОНДЕНСИРОВАННОЕ 4-ОКСОПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ | 2005 |
|
RU2358969C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПИРИДИНА И ПРОИЗВОДНОЕ ПИРИМИДИНА (3) | 2006 |
|
RU2362771C1 |
Настоящее изобретение относится к пиперазиновому соединению, представленному формулой (I),
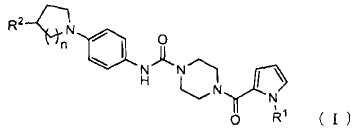
где R1 представляет собой С1-6алкил; R2 представляет собой гидрокси, C1-6алкил, который может содержать заместитель, выбранный из насыщенного или ненасыщенного 5-6 членного гетероцикла с 1-3 гетероатомами, выбранными из кислорода и азота, -(C=O)-N(R3)(R4) или -(C=O)-OR5; R3 и R4 могут быть одинаковыми или отличаться, и каждый представляет собой водород или С1-6алкил, который может содержать заместитель, выбранный из насыщенного или ненасыщенного 5-6 членного гетероцикла с 1-3 гетероатомами, выбранными из кислорода и азота, или R3 и R4, связанные через атом азота, к которому присоединены R3 и R4, могут образовывать насыщенную гетероциклическую группу, выбранную из 5-6 членного гетероцикла с 1-3 гетероатомами, выбранными из кислорода и азота; R5 представляет собой водород или C1-6алкил; и n представляет собой 1 или 2; или его соль. Также изобретение относится к фармацевтической композиции и средству, проявляющим активность в отношении простагландин-D-синтазы, на основе соединения формулы I, а также к способу предотвращения и лечения заболевания, в котором участвует простагландин D2. Технический результат: получены и описаны новые соединения, которые могут быть полезны в лечении заболеваний, в котором участвует простагландин D2. 6 н. и 5 з.п. ф-лы, 19 пр., 4 табл.
1. Пиперазиновое соединение, представленное формулой (I),
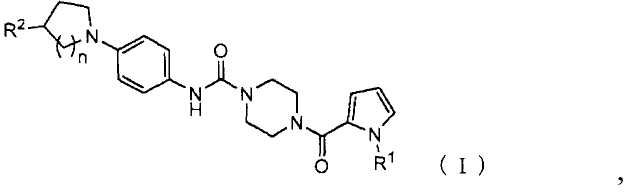
где R1 представляет собой C1-6алкил;
R2 представляет собой гидрокси, C1-6алкил, который может содержать заместитель выбранный из насыщенного или ненасыщенного 5-6 членного гетероцикла с 1-3 гетероатомами, выбранными из кислорода и азота,
-(C=O)-N(R3)(R4) или -(C=O)-OR5;
R3 и R4 могут быть одинаковыми или отличаться, и каждый представляет собой водород или С1-6алкил, который может содержать заместитель, выбранный из насыщенного или ненасыщенного 5-6 членного гетероцикла с 1-3 гетероатомами, выбранными из кислорода и азота, или
R3 и R4, связанные через атом азота, к которому присоединены R3 и R4,
могут образовывать насыщенную гетероциклическую группу, выбранную из 5-6 членного гетероцикла с 1-3 гетероатомами, выбранными из кислорода и азота;
R5 представляет собой водород или С1-6алкил; и
n представляет собой 1 или 2; или его соль.
2. Пиперазиновое соединение или его соль в соответствии с п.1, где
R1 представляет собой метил или этил;
R2 представляет собой гидрокси, C1-6алкил, который может иметь одну или более насыщенных или ненасыщенных 5-6 членных гетероциклических групп с 1-3 гетероатомами, выбранными из кислорода и азота в качестве заместителей, -(C=O)-N(R3)(R4) или -(C=O)-OR5; R3 и R4 могут быть одинаковыми или отличаться, и каждый представляет собой водород или С1-6алкил, который может иметь одну или более насыщенных или ненасыщенных 5-6 членных гетероциклических групп с 1-3 гетероатомами, выбранными из кислорода и азота, в качестве заместителей, или
R3 и R4, связанные через атом азота, к которому присоединены R3 и R4, могут образовать пирролидинил, пиперидинил, пиперазинил и морфолино;
R5 представляет собой водород, метил, этил, трет-бутил; и n представляет собой 1 или 2.
3. Пиперазиновое соединение или его соль в соответствии с п.1, где R1 представляет собой метил;
R2 представляет собой C1-3алкил, который может содержать морфолино,
пиразолил или триазолил в качестве заместителей, -(C=O)-N(R3)(R4) или -(С=O)-OR5; и триазолил может содержать один или два заместителя;
R3 и R4, связанные через атом азота, к которому присоединены R3 и R4,
могут образовывать морфолино;
R5 представляет собой водород, метил или этил; и
n представляет собой 2.
4. Пиперазиновое соединение или его соль по п.1, где
R1 представляет собой метил;
R2 представляет собой линейный С1-3алкил, который может содержать любой из 1,2,3-триазолила, 3,5-диметил-1,2,4-триазолила и морфолино в качестве заместителя, -(C=O)-N(R3)(R4) или -(C=O)-OR5;
R3 и R4, связанные через атом азота, к которому присоединены R3 и R4,
могут образовать морфолино;
R5 представляет собой водород или этил; и
n представляет собой 2.
5. Пиперазиновое соединение или его соль в соответствии с п.1, выбранное из группы, состоящей из следующего:
N-(4-(4-гидроксипиперидин-1-ил)фенил)-4-((1-метилпиррол-2-ил)карбонил)-1-пиперазинкарбоксамид,
4-((((1-метилпиррол-2-ил)карбонил)-1-пиперазинил)карбонил)амино-4-фенилпиперидин-4-карбоновая кислота,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолинометилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(3-(2-(1,2,4-триазол-1-ил)этил)пирролидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-морфолиноэтилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-(1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(1,2,3-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(1,2,4-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(3,5-диметил-1,2,4-триазол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-(пиразол-1-ил)этил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(3-(1,2,4-триазол-1-ил)пропил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-этилпиррол-2-ил)карбонил)-N-(4-(4-морфолин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-пиперидин-1-ил-карбонилпиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(4-метилпиперазин-1-ил-карбонил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид,
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(2-морфолиноэтилкарбамоил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид и
4-((1-метилпиррол-2-ил)карбонил)-N-(4-(4-(пиридин-3-илметилкарбамоил)пиперидин-1-ил)фенил)-1-пиперазинкарбоксамид.
6. Фармацевтическая композиция, проявляющая ингибирующую активность в отношении простагландин-D-синтазы, включающая эффективное количество по меньшей мере одного из соединений по пп.1-5 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
7. Средство для предотвращения и/или лечения заболевания, в котором участвует простагландин D2 или его метаболит, включающее эффективное количество соединения по любому из пп.1-5 или его фармацевтически приемлемой соли.
8. Средство в соответствии с п.7, где заболевание, в котором участвует простагландин D2 или его метаболит, является аллергическим заболеванием, воспалительным заболеванием или миодегенеративным заболеванием.
9. Способ предотвращения и/или лечения заболевания, в котором участвует простагландин D2 или его метаболит, включающий введение млекопитающему соединения по любому из пп.1-5 в количестве, эффективном для предотвращения или лечения заболевания.
10. Применение соединения по любому из пп.1-5 для получения средства для предотвращения и/или лечения заболевания, в котором участвуют простагландин D2 или его метаболит.
11. Применение соединения по любому из пп.1-5 для предотвращения и/или лечения заболевания, в котором участвуют простагландин D2 или его метаболит.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| ЗАМЕЩЕННЫЕ АЗЕТИДИНОНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2148056C1 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ АГЕНТОВ | 2000 |
|
RU2242474C2 |

