Область техники, к которой относится изобретение
Настоящее изобретение относится к ингибитору фиброза, содержащему гетероциклическое производное (в настоящем описании далее обозначаемое как "настоящее гетероциклическое производное (1)"), представленное следующей общей формулой (1) или ее фармацевтически приемлемой солью в качестве активного компонента;
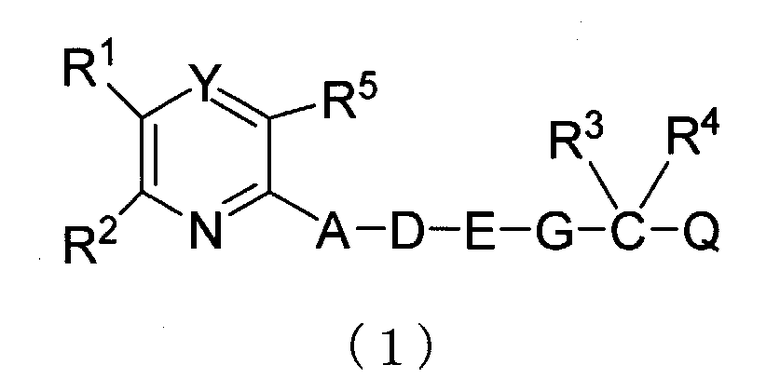
В формуле (1) R1 и R2 являются одинаковыми или различными и каждый представляет собой необязательно замещенный арил, и заместители являются одинаковыми или различными и от одного до трех заместителей выбирают из группы, состоящей из атома галогена, алкила, галогеналкила, арилалкила, алкокси, алкилтио, алкоксиалкила, алкилсульфонила, гидрокси, амино, моноалкиламино, диалкиламино, карбокси, циано и нитро;
R3 и R4 являются одинаковыми или различными и каждый представляет собой атом водорода или алкил;
R5 представляет собой атом водорода, алкил или атома галогена;
Y представляет собой N или N→O;
А представляет собой NR6, и R6 представляет собой атом водорода, алкил, алкенил или циклоалкил;
D представляет собой алкилен или алкенилен, который необязательно замещен гидрокси, или А и D объединяют вместе с образованием дивалентной группы, представленной следующей формулой (2)
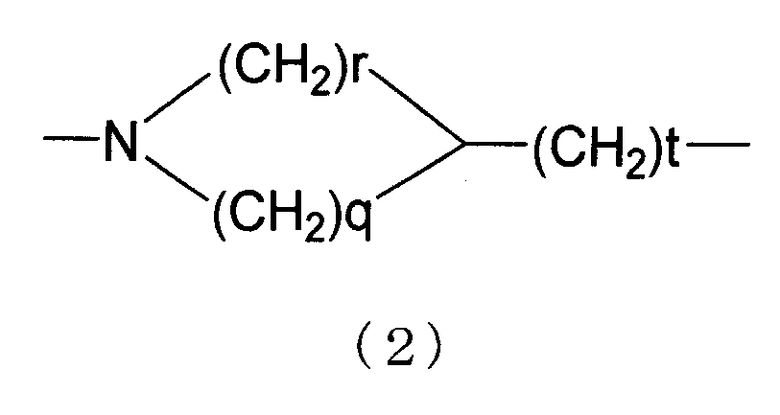
[в формуле (2) r представляет собой целое число от 0 до 2, q представляет собой 2 или 3 и t представляет собой целое число от 0 до 4];
E представляет собой фенилен или простую связь, или D и E объединяют вместе с образованием дивалентной группы, представленной следующей формулой (3)
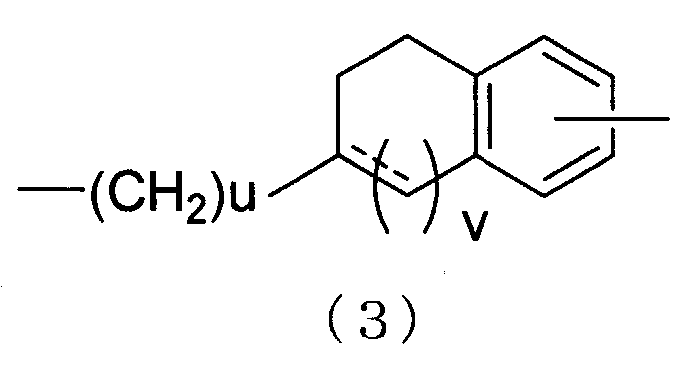
(--- представляет собой простую связь или двойную связь.)
[в формуле (3) u представляет собой целое число от 0 до 2 и v представляет собой 0 или 1];
G представляет собой O, S, SO или SO2; и
Q представляет собой карбокси, алкоксикарбонил, тетразолил, карбамоил, моноалкилкарбамоил, диалкилкарбамоил или группу, представленную следующей формулой (4).
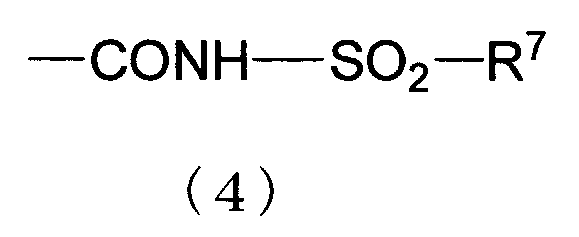
[В формуле (4), R7 представляет собой амино, моноалкиламино, диалкиламино, гидрокси, любую из следующих групп 1)-4), которые необязательно замещены от 1 до 3 заместителями, выбранными из группы, состоящей из атома галогена, алкила, галогеналкила, арилалкила, алкокси, алкилтио, алкоксиалкила, алкилсульфонила, гидрокси, амино, моноалкиламино, диалкиламино, карбокси, циано и нитро;
1) алкил,
2) арил,
3) арилокси и
4) гетероциклическая группа.]
Уровень техники
Фиброз органов возникает таким образом, что внеклеточный матрикс чрезмерно накапливается в органах посредством инвазии или повреждения органов из-за какой-либо причины. Когда степень повреждения органов из-за инвазии или повреждения является легкой, шрам не остается, и органы возвращаются к нормальному состоянию. Однако когда степень повреждения органов из-за инвазии или повреждения является тяжелой или устойчивой, фиброз шрама приводит к ухудшению присущей ему функции. Кроме того, он вызывает новый фиброз и образует замкнутый круг фиброза, и, в конце концов, возникает дисфункция органов.
В качестве заболевания, вызванного фиброзом органов, была известна интерстициальная пневмония (фиброз легких) и т.д. Интерстициальная пневмония является заболеванием, при котором воспаление происходит в альвеолярной стенке из-за некоторых причин; фибробласты пролиферируют в промежуточной ткани; легкое уплотняется благодаря избыточному осаждению коллагеновых волокон; газообмен нарушается; и, в конце концов, возникает дыхательная недостаточность. После возникновения заболевания, пациент умирает, в среднем, в течение трех-пяти лет. Подробный механизм патогенеза интерстициальной пневмонии еще не был разъяснен и так же еще не был доступен установленный способ лечения.
Недавно было сообщено, что в мышиной модели интерстициальной пневмонии, вызванной блеомицином, ONO-1301, который является агонистом рецептора простагландина I2 (здесь далее обозначаемый как "PGI2"), имеет ингибирующий эффект в отношении интерстициальной пневмонии (см., например, Непатентый документ 1).
О том, что настоящее гетероциклическое производное (1) или ее фармацевтически приемлемая соль пригодно для лечения легочной гипертензии и обструктивного атеросклероза в качестве агониста рецептора PGI2, уже сообщалось (см., например, Патентный документ 1).
Патентный документ 1: Международная публикация WO 02/088084
Непатентый документ 1: American J. Physiology, 2006, vol. 290, pages 59 to 65
Описание изобретения
Проблемы, решаемые изобретением
Основной целью настоящего изобретения является предоставление новых ингибиторов фиброза.
Средства для решения проблем
Авторами настоящего изобретения было обнаружено, что настоящее гетероциклическое производное (1) имеет ингибирующий эффект на рост фибробластов, и достигнуто настоящее изобретение.
Примером настоящего изобретения является ингибитор фиброза, содержащий от настоящее гетероциклическое производное (1) или ее фармацевтически приемлемую соль в качестве активного компонента.
Лучший вариант осуществления настоящего изобретения
В настоящем гетероциклическом производном (1), предпочтительным является то, что:
R1 и R2 являются одинаковыми или различными и каждый представляет собой необязательно замещенный фенил, и заместители являются одинаковыми или различными и от одного до трех заместителей выбраны из группы, состоящей из атома галогена, алкила и алкокси;
R3 и R4 являются одинаковыми или различными и каждый представляет собой атом водорода или алкил;
R5 представляет собой атом водорода;
Y представляет собой N;
А представляет собой NR6, и R6 представляет собой алкил;
D представляет собой алкилен;
E представляет собой простую связь;
G представляет собой O; и
Q представляет собой карбокси или группу, представленную следующей формулой (4), и R7 представляет собой амино, моноалкиламино, диалкиламино, гидрокси или любую из следующих групп 1) -4), которые необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из атома галогена, алкила, галогеналкила, арилалкила, алкокси, алкилтио, алкоксиалкила, алкилсульфонила, гидрокси, амино, моноалкиламино, диалкиламино, карбокси, циано и нитро;
1) алкил,
2) арил,
3) арилокси, и
4) гетероциклическая группа.
Более конкретно, 2-{4-[N-(5,6-дифенил-пиразин-2-ил)-N-изопропиламино]бутилокси}уксусная кислота (здесь далее обозначаемая как "соединение А") и 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамид (здесь далее обозначаемое как "соединение B") являются, например, предпочтительными.
Что касается "алкила" в настоящем изобретении, который имеет неразветвленную или разветвленную цепь, содержащую 1-6 атомов углерода, то, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, н-гексил или изогексил могут быть приведены в качестве примера. В частности, алкил, содержащий 1-4 атомов углерода, является предпочтительным.
Что касается алкильной группы в "галогеналкиле", "арилалкиле", "алкилтио", "алкоксиалкиле", "алкилсульфониле", "моноалкиламино", "диалкиламино", "моноалкилкарбазоиле" и "диалкилкарбамоиле" в настоящем изобретении, которая является такой же, как уже упомянутый алкил, который может быть приведен в качестве примера.
Что касается "алкокси" в настоящем изобретении, который имеет неразветвленную или разветвленную цепь, содержащую 1-6 атомов углерода, то, например, метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, изопентилокси, н-гексилокси или изогексилокси могут быть приведены в качестве примера. В частности, алкокси, содержащий 1-4 атомов углерода, является предпочтительным.
Что касается алкоксигруппы в "алкоксикарбониле" и "алкоксиалкиле" которая является такой же, как уже упомянутый алкокси, который может быть приведен в качестве примера.
Что касается "алкенила" в настоящем изобретении, который имеет неразветвленную или разветвленную цепь, содержащую 2-6 атомов углерода, например, винил, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил, 3-бутенил, 1-пентенил, 2-пентенил, 3-пентенил, 4-пентенил, 4-метил-3-пентенил, 1-гексенил, 2-гексенил, 3-гексенил, 4-гексенил или 5-гексенил могут быть приведены в качестве примера. В частности, алкенил, содержащий от 3 или 4 атомов углерода, является предпочтительным.
Что касается "циклоалкила" в настоящем изобретении, который имеет 3-8 атомов углерода, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил могут быть приведены в качестве примера. В частности, циклоалкил, содержащий от 5 до 7 атомов углерода, является предпочтительным.
Что касается "атома галогена" в настоящем изобретении, атом фтора, атом хлора, атом брома и атом иода могут быть приведены в качестве примера.
Что касается "арила" в настоящем изобретении, который имеет 6-10 атомов углерода, например, фенил, 1-нафтил или 2-нафтил могут быть приведены в качестве примера. В частности, фенил является предпочтительным.
Что касается арильной группы в "арилалкиле" и "арилокси" в настоящем изобретении, которая является такой же, как уже упомянутый арил, который может быть приведен в качестве примера.
Что касается "алкилена" в настоящем изобретении, который имеет неразветвленную или разветвленную цепь, содержащую 1-8 атомов углерода, например, метилен, этилен, 1-метилэтилен, 2-метилэтилен, триметилен, тетраметилен, пентаметилен, гексаметилен, гептаметилен или октаметилен могут быть приведены в качестве примера. В частности, алкилен, содержащий 3-6 атомов углерода, является предпочтительным, и алкилен, содержащий 4 атома углерода, является более предпочтительным.
Что касается "алкенилена" в настоящем изобретении, который имеет неразветвленную или разветвленную цепь, содержащую 2-8 атомов углерода, например, этенилен, 1-пропенилен, 2-пропенилен, 1-бутенилен, 2-бутенилен, 3-бутенилен, 1-пентенилен, 2-пентенилен, 3-пентенилен, 4-пентенилен, 4-метил-3-пентенилен, 1-гексенилен, 2-гексенилен, 3-гексенилен, 4-гексенилен, 5-гексенилен, 1-гептенилен, 2-гептенилен, 3-гептенилен, 4-гептенилен, 5-гептенилен, 6-гептенилен, 1-октенилен, 2-октенилен, 3-октенилен, 4-октенилен, 5-октенилен, 6-октенилен или 7-октенилен могут быть приведены в качестве примера. В частности, алкенилен, содержащий 3-6 атомов углерода, является предпочтительным, и алкенилен, содержащий 4 атома углерода, является более предпочтительным.
Что касается "гетероциклической группы" в настоящем изобретении, следующие (1) или (2) могут быть приведены в качестве примера.
(1) пяти-шестичленная ароматическая кольцевая группа, содержащая 1-4 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, или ее бензольное конденсированное кольцо и атом азота и атом серы могут образовывать оксид, когда атом, содержащийся в кольце, представляет собой атом азота или атом серы. Ее примеры включают 1-пирролил, 2-пирролил, 3-пирролил, 3-индолил, 2-фуранил, 3-фуранил, 3-бензофуранил, 2-тиенил, 3-тиенил, 3-бензотиенил, 1,3-оксазол-2-ил, 4-изооксазолил, 2-тиазолил, 5-тиазолил, 2-бензотиазолил, 1-имидазолил, 2-имидазолил, 4-имидазолил, 2-бензимидазолил, 1H-1,2,4-триазол-1-ил, 1H-тетразол-5-ил, 2H-тетразол-5-ил, 2-пиридил, 3-пиридил, 4-пиридил, 3-пиразолил, 2-пиримидинил 4-пиримидинил, 2-пиразинил, и 1,3,5-триазин-2-ил.
(2) четырех-восьмичленная насыщенная кольцевая группа, которая необязательно имеет от одного до четырех одинаковых или различных атомов азота, атомов кислорода или атомов серы, или ее бензольное конденсированное кольцо и атом азота и атом серы могут образовывать оксид, когда атом, содержащийся в кольце, представляет собой атом азота или атом серы. Ее примеры включают пиперидино, пиперазинил, 3-метилпиперазин-1-ил, гомопиперазинил, морфолино, тиоморфолино, 1-пирролидинил, 2-пирролидинил и 2-тетрагидрофуранил.
Настоящее гетероциклическое производное (1) может быть синтезировано способом, упомянутым в Патентном документе 1 (международная публикация WO 02/088084).
Несмотря на то, что настоящее гетероциклическое производное (1) может быть использовано в качестве лекарственного средства только в форме свободного основания или кислоты, его также возможно использовать путем преобразования в форму фармацевтически приемлемой соли с помощью известного способа.
Примеры "соли", когда настоящее гетероциклическое производное (1) показывает основность, включают соли с неорганической кислотой, такой как хлороводородная кислота, серная кислота, азотная кислота, фосфорная кислота, фтористоводородная кислота или бромистоводородная кислота, и с органической кислотой, такой как уксусная кислота, винная кислота, молочная кислота, лимонная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, метансульфокислота, этансульфокислота, бензолсульфокислота, п-толуолсульфокислота, нафталинсульфокислота или камфорсульфокислота.
Примеры "соли", когда настоящее гетероциклическое производное (1) показывает кислотность, включают соли щелочного металла, такие как соли натрия или соли калия и соли щелочноземельного металла, такие как соли кальция.
У настоящего гетероциклического производного (1) имеются геометрические изомеры (Z и E вещества), и каждый из геометрических изомеров и их смесь также включены в настоящее гетероциклическое производное (1). Некоторые из настоящего гетероциклического производного (1) имеют ассиметричный углерод(ы), и каждый из оптических изомеров и его рацемическое вещество также включены в настоящее гетероциклическое производное (1). Оптический изомер может быть получен путем подвергания рацемического вещества, полученного как указано выше, разделением оптических изомеров с помощью известного способа, используя оптически активные кислоты (такие как винная кислота, бензоилвинная кислота, миндальная кислота или 10-камфорсульфокислота), используя основность или используя ранее полученное оптически активное соединение в качестве материала.
Ингибитор фиброза по настоящему изобретению может быть использован для лечения следующих заболеваний, например, в которые вовлечены фиброз органов или тканей.
(1) Почечная недостаточность
Тубулоинтерстициальный нефрит
(2) Респираторные заболевания
Интерстициальная пневмония (фиброз легких)
(3) Желудочно-кишечные заболевания
Цирроз печени, хронический панкреатит и скиррозный рак желудка
(4) Сердечно-сосудистые заболевания
Миокардиальный фиброз
(5) Заболевания костей и суставов
Фиброз костного мозга и ревматоидный артрит
(6) Заболевания кожи
Послеоперационный рубец, ожоговый рубец, келоид, гипертрофический рубец и склеродермия
(7) Акушерские заболевания
Миома матки
(8) Урологические заболевания
Гипертрофия предстательной железы
(9) Другие заболевания
Болезнь Альцгеймера, склерозирующий перитонит, диабет I типа и послеоперационная спайка.
Ингибитор фиброза по настоящему изобретению представляет собой настоящее гетероциклическое производное (1) само по себе или содержит его в фармацевтически приемлемом, нетоксичном и инертном носителе в количестве в диапазоне от 0,01 до 99,5% или, предпочтительно, в диапазоне от 0,5 до 90%.
Примеры носителя включают твердый, полутвердый или жидкий разбавитель, наполнитель и другие дополнительные агенты для фармацевтической композиции. Они могут быть использованы одни или в виде смеси двух или более агентов.
Ингибитор фиброза по настоящему изобретению может быть в любых формах пероральных препаратов, таких как порошок, капсулы, таблетки, таблетки, покрытые сахаром, гранулы, разбавленный порошок, суспензия, жидкость, сироп, эликсир или пастилка и парентеральные препараты, такие как инъекционный раствор или суппозиторий в твердой или жидкой стандартной дозе. Он так же может быть в форме препарата с замедленным высвобождением. Среди них пероральные препараты, такие как таблетки, являются особенно предпочтительными.
Порошок может быть изготовлен путем измельчения настоящего гетероциклического производного (1) в соответствующий мелкий размер.
Разбавленный порошок может быть изготовлен таким образом, что настоящее гетероциклическое производное (1) делают соответствующего мелкого размера и затем смешивают с фармацевтическим носителем, который подобным образом делают мелкого размера, таким как пищевой карбогидрат (например, крахмал и маннит). Ароматизирующее вещество, консервант, диспергирующий агент, краситель, отдушка, и т.д. могут быть необязательно в него добавлены.
Капсулы могут быть изготовлены таким образом, что порошком или разбавленным порошком, который выполнен в виде порошка, как упомянуто выше, или гранулами, которые будут упомянуты под наименованием таблеток, заполняют оболочку капсулы, такой как желатиновая капсула. Также их можно изготовить таким образом, что порошкообразное вещество смешивают со смазкой или разжижающей добавкой, таким как коллоидная окись кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль, с последующим подверганием операции наполнения. Когда вещество для улучшения распадаемости таблеток или солюбилизирующий компонент, такой как карбоксиметилцеллюлоза, кальцийкарбоксиметилцеллюлоза, гидроксипропилцеллюлоза с низкой степенью замещения, кроскармеллоза натрия, натрия карбоксиметилкрахмал, карбонат кальция или карбонат натрия, добавляют, эффективность лекарственного средства, при проглатывании капсулы, может быть улучшена. Также возможно, чтобы мелкий порошок настоящего гетероциклического производного (1) был суспендирован/диспергирован в растительном масле, полиэтиленгликоле, глицерине или поверхностно-активном веществе и упакован в листовой желатин с получением препарата из мягкой капсулы.
Таблетки могут быть изготовлены таким образом, что порошкообразную смесь получают добавлением наполнителя и превращают в гранулы или заготовки, и затем вещество для улучшения распадаемости таблеток или смазку добавляют в него с последующим преобразованием в таблетки.
Порошкообразная смесь может быть получена путем смешения соответствующим образом превращенного в порошок вещества с вышеописанным разбавителем или основанием. При необходимости, в нее можно добавить связывающий компонент (такой, как карбоксиметилцеллюлоза натрия, метилцеллюлоза, гидроксипропилметилцеллюлоза, желатин, поливинилпирролидон или поливиниловый спирт), добавку, замедляющую процесс разжижения (такую, как парафин), реабсорбирующее вещество (такое, как четвертичные соли), адсорбент (такой, как бентонит или каолин), и т.д.
Порошкообразная смесь может быть превращена в гранулы таким образом, что сначала ее делают сырой, используя, например, сироп, крахмальную пасту, акацию, раствор целлюлозы или раствор полимера, смешивают при перемешивании и сушат с последующим растиранием. Вместо того чтобы превращать порошок в гранулы как таковые, также возможно поместить порошок в таблеточную машину, и заготовку незавершенной формы растирать с получением гранулы. Когда смазку, такую как стеариновая кислота, стеарат, тальк или минеральное масло, добавляют к гранулам, полученным таким образом, прилипание гранул друг к другу может быть предотвращено.
Таблетки могут также быть изготовлены таким образом, что настоящее гетероциклическое производное (1) смешивают с жидким инертным носителем и затем напрямую изготовляют таблетки без проведения вышеописанных стадий получения гранул или заготовок.
Таблетки, полученные таким образом, могут быть покрыты пленкой или сахаром. Так же можно применить прозрачное или полупрозрачное защитное покрытие, включающее плотно закрытую шеллакову пленку, покрытие, включающее сахар или полимерный материал или полированное покрытие, включающее воск.
Другие пероральные препараты, такие как жидкость, сироп, пастилка или эликсир, так же возможно превратить в стандартную лекарственную дозу, в которой ее заранее определенное количество содержит заранее определенное количество настоящего гетероциклического производного (1).
Сироп может быть изготовлен путем растворения настоящего гетероциклического производного (1) в соответствующем водном растворе ароматической добавки. Эликсир может быть изготовлен, используя нетоксичный спиртовой носитель.
Суспензия может быть изготовлена путем диспергирования настоящего гетероциклического производного (1) в нетоксичный носитель. При необходимости, в нее можно добавить солюбилизирующий компонент или эмульсификатор (такой как этоксилириванный изостеариловый спирт или полиоксиэтиленовый сложный эфир сорбита), консервант или компонент, обеспечивающий аромат (такой как масло мяты перечной или сахарин).
При необходимости, стандартная лекарственная форма для перорального введения может быть превращена в микрокапсулы. Описанная выше форма также может быть покрыта или залита полимером или воском с получением пролонгированного действия или замедленного высвобождения активного компонента.
Парентеральный препарат может быть в жидкой стандартной лекарственной форме для подкожного, внутримышечного или внутривенного введения, такой как в форме раствора или суспензии. Парентеральный препарат может быть изготовлен таким образом, что заранее определенное количество настоящего гетероциклического производного (1) суспендируют или растворяют в нетоксичном жидком носителе, учитывая цель введения, таком как водная или маслообразная среда, и затем суспензию или раствор стерилизуют. Нетоксичную соль или ее раствор могут добавить в него для того, чтобы сделать раствор для введения изотоническим. Так же возможно добавить стабилизатор, консервант, эмульсификатор и подобное.
Суппозиторий может быть изготовлен путем растворения или суспендирования настоящего гетероциклического производного (1) в легкоплавком и водорастворимом или нерастворимом твердом веществе, таком как полиэтиленгликоль, масло какао, полусинтетический жир/масло (такие, как Witepsol (зарегестрированный товарный знак)), высший сложный эфир (такой, как миристилпальмитат) или их смесь.
Несмотря на то, что доза ингибитора фиброза по настоящему изобретению может изменяться в зависимости от состояния пациента, такого как масса тела или возраст, путь введения или степень симптома, диапазон от 0,01 мг до 1000 мг/день в качестве количества настоящего гетероциклического производного (1) в основном пригоден для взрослого, и диапазон от 0,1 мг до 100 мг является более предпочтительным. В некоторых случаях, доза менее чем вышеуказанная, может быть достаточной или, с другой стороны, доза более чем вышеуказанная, может быть необходима. Так же возможно вводить один или несколько раз в день или вводить с интервалом в один или несколько дней.
Примеры
Настоящее изобретение далее будет проиллюстрировано более детально следующими тестовыми примерами, несмотря на то, что настоящее изобретение не ограничено объемом притязаний, упомянутым в примерах.
Тестовый пример 1
(1) Способы
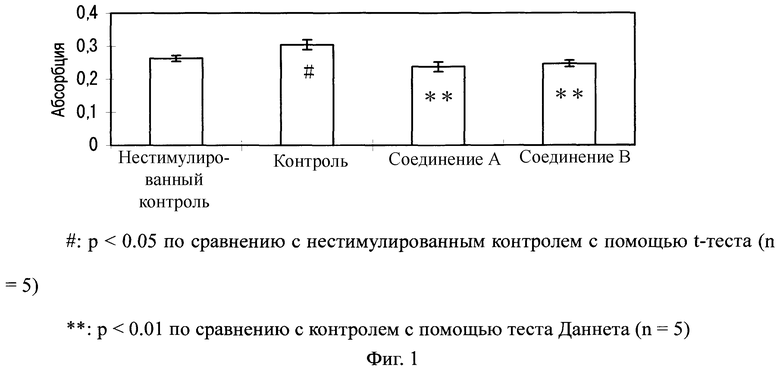
Фибробласты легкого человека (изготовленные Lonza Walkersville; далее будет использован такой же продукт) культивировали в питательной среде, которая состоит из базальной среды для фибробластов легкого человека (изготовленной Lonza Walkersville; далее будет использован такой же продукт и называется "базальная среда"), с набором дополнительных факторов FGM-2 (изготовленных Lonza Walkersville; далее будет использован такой же продукт) при условиях 37°C и 5% CO2. Фибробласты легкого человека высеивали в 96-луночный планшет при 1×103 клеток/лунка и инкубировали всю ночь в питательной среде.
Фибробласты промывали один раз с помощью 100 мкл физиологического раствора с фосфатным буфером (изготовлен Nissui Seiyaku; далее будет использован такой же продукт), и в него было добавлено 100 мкл базальной среды с последующей инкубацией в течение 24 часов. Их дополнительно промывали однократно 100 мкл физиологического раствора с фосфатным буфером и затем добавляли базальную среду в количестве 80 мкл/лунку. Десять мкл 100 мкМ раствора соединения А или соединения B добавляли к каждой лунке и инкубировали в течение 2 часов. Добавленный в них раствор получали таким образом, что соединение А или B сначала растворяли в диметилсульфоксиде (ДМСО), и полученные 10 ммоль раствора разбавляли 100-кратно базальной средой. Для группы нестимулированного контроля и контрольной группы, использовали ДМСО, который разбавляли 100-кратно базальной средой.
Затем 100 нг/мл раствора эпидермального фактора роста (EGF) (изготовлен Pepro Tech) добавляли в количестве 10 мкл к каждой лунке и инкубировали в течение 48 часов. Для группы нестимулированного контроля использовали базальную среду.
После инкубации в течение 48 часов измеряли абсорбцию при 490 нм, используя Cell Titer 96, AQueous Assay (изготовлен Promega) для анализа активности пролиферации клеток. Для измерения абсорбции использовали считывающее устройство для микропланшетов (Benchmark, изготовлено Bio-Rad; далее будет использован такой же прибор).
(2) Результаты
Как показано на Фиг.1, активность пролиферации клеток фибробластов легкого человека значительно возросла благодаря стимуляции с помощью EGF. В противоположность этому, в клетках, обработанных соединениями А или B, активность пролиферации клеток значительно понизилась по сравнению с контрольной группой.
Тестовый пример 2
(1) Способы
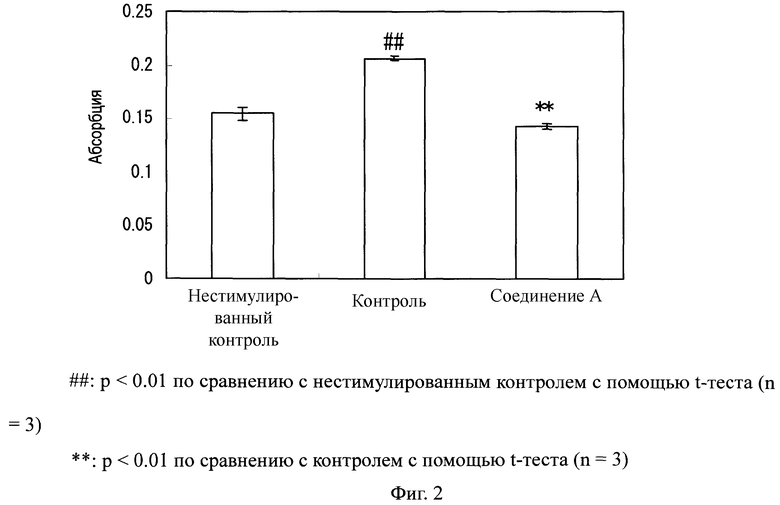
Фибробласты легкого человека инкубировали в питательной среде при условиях 37°C и 5% CO2. Фибробласты легкого человека высеивали в 96-луночный планшет при 3×103 клеток/лунка и инкубировали всю ночь в питательной среде.
Фибробласты промывали один раз с помощью 100 мкл физиологического раствора с фосфатным буфером. Затем в него добавляли базальную среду, и инкубацию проводили в течение 24 часов. Их дополнительно промывали 100 мкл физиологического раствора с фосфатным буфером один раз и затем добавляли базальную среду в количестве 80 мкл/лунку. Десять мкл 1 мкМ раствора соединения добавляли к каждой лунке и инкубировали в течение 2 часов. Использованный в них раствор получали таким образом, что соединение А сначала растворяли в ДМСО, и полученный 1 ммоль раствор разбавляли 100-кратно базальной средой. Для группы нестимулированного контроля и контрольной группы использовали ДМСО, который разбавляли 100-кратно базальной средой.
Затем 100 нг/мл раствора трансформирующего фактора-α(TGF-α) (изготовлен Chemicon) добавляли в количестве 10 мкл к каждой лунке и инкубировали в течение 48 часов. Для группы нестимулированного контроля использовали базальную среду.
После инкубации в течение 48 часов измеряли абсорбцию при 490 нм, таким же образом, как и в тестовом примере 1, используя реагент для анализа активности пролиферации клеток. Для измерения абсорбции использовали считывающее устройство для микропланшетов.
(2) Результаты
Как показано на Фиг.2, активность пролиферации клеток фибробластов легкого человека значительно возросла благодаря стимуляции с помощью TGF-α. В противоположность этому, в клетках, обработанных соединением А, активность пролиферации клеток значительно понизилась по сравнению с таковой у контрольной группы.
Тестовый пример 3
(1) Способы
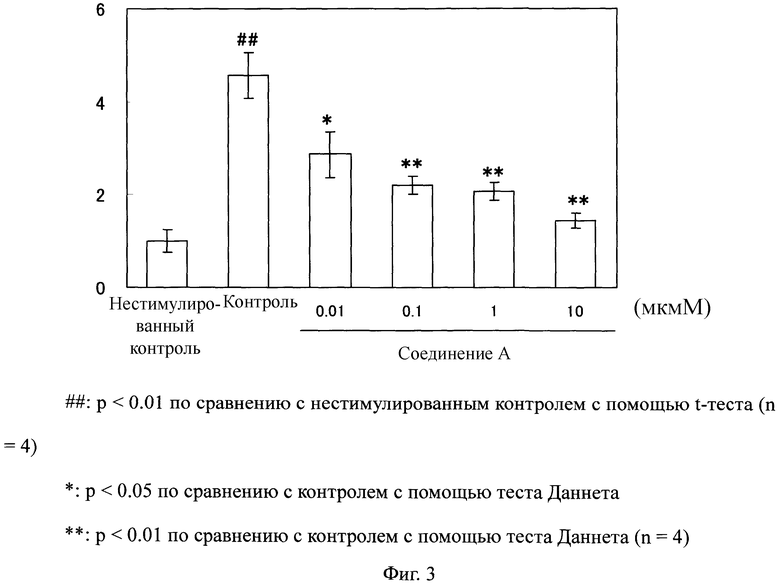
Фибробласты легкого человека высеивали в 96-луночный планшет при 5×103 клетках/лунку и инкубировали в питательной среде таким же образом, как и в тестовом примере 1 всю ночь. Фибробласты промывали один раз с помощью 100 мкл базальной среды, и 100 мкл базальной среды было в него добавлено с последующей инкубацией в течение 24 часов. Их дополнительно промывали один раз с помощью 100 мкл базальной среды, и затем 80 мкл/лунку добавляли базальную среду. 0,1, 1, 10 или 100 мкМ раствора соединения добавляли в количестве 10 мкл к каждой лунке. Добавленный в них раствор получали таким образом, что соединение А сначала растворяли в ДМСО и доводили до концентрации 10 мкМ, 100 мкМ, 1 ммоль или 10 ммоль, с последующим 100-кратным разбавлением базальной средой. Для группы нестимулированного контроля и контрольной группы, ДМСО, который разбавляли 100-кратно базальной средой, добавляли в количестве 10 мкл. После инкубации в течение 2 часов, 10 мкл 100 нг/мл трансформирующего фактора β1 (TGFβ1) (изготовлен Pepro Tech; далее будет использован такой же продукт) добавляли и, для группы нестимулированного контроля добавляли 10 мкл базальной среды. После инкубации в течение 48 часов, среду извлекали для измерения концентрации С концевого пептида проколлагена I типа (PIP) в среде, и после добавления 100 мкл базальной среды, измеряли активность пролиферации клеток, используя 3-(4,5-диметил-2-тиазолил)-2,5-дифенил-2H-тетразолия бромид (MTT) (изготовлен Nacalai Tesque). Измерение активности пролиферации клеток проводили таким образом, что 10 мкл 5 мг/мл MTT маточного раствора добавляли к каждой лунке, и после инкубации в течение 4 часов, 100 мкл 0,04M изопропанольного раствора хлороводородной кислоты добавляли к каждой лунке, с последующим измерением абсорбции при 595 нм (опорная длина волны: 655 нм), используя считывающее устройство для микропланшетов. Концентрацию PIP после инкубации в течение 48 часов измеряли, используя EIA набор С-пептида проколлагена I типа (PIP) (изготовлен Takara; далее будет использован такой же набор) в соответствии с приложенной к нему инструкцией. Оценку этого теста проводили, используя значение (индекс для продуцирования коллагена), вычисленное как относительное значение, где среднее значение группы нестимулированного контроля определяли как 1 после корректировки измеренного значения концентрации PIP для значения активности пролиферации клеток (абсорбция).
(2) Результаты
Как показано на Фиг.3, продуцирование коллагена значительно возросло в фибробластах легкого человека благодаря стимуляции с помощью TGFβ1. В противоположность этому, в клетках, обработанных соединением А, продуцирование коллагена значительно понизилось по сравнению с таковой у контрольной группы.
Тестовый пример 4
(1) Способы
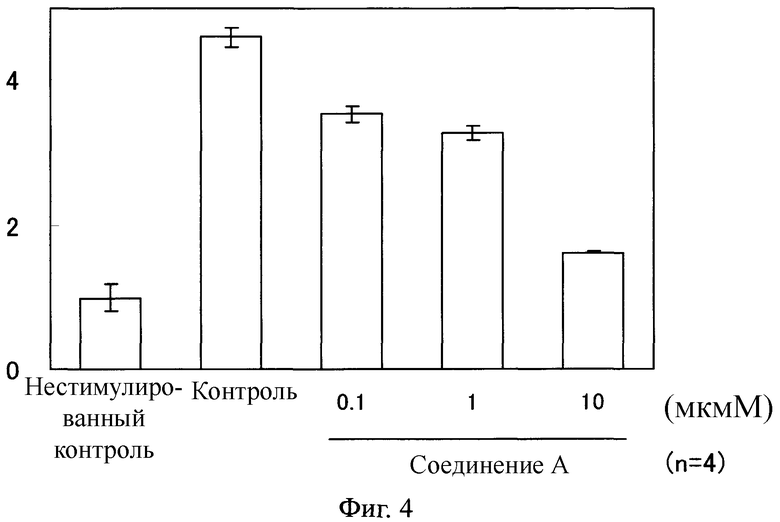
Фибробласты легкого человека высеивали в 24-луночный планшет при 1×105 клетках/лунку и инкубировали в питательной среде таким же образом, как и в тестовом примере 1 всю ночь. Фибробласты промывали один раз с помощью 500 мкл базальной среды, и добавляли500 мкл базальной среды с последующей инкубацией в течение 24 часов. Их дополнительно промывали один раз с помощью 500 мкл базальной среды и затем 400 мкл/лунку добавляли базальную среду. 1, 10 или 100 мкМ раствора соединения, полученного таким же образом, как в тестовом примере 3, добавляли в количестве 50 мкл к каждой лунке. Для группы нестимулированного контроля и контрольной группы, ДМСО, который разбавляли 100-кратно базальной средой, добавляли в количестве 50 мкл. После инкубации в течение 2 часов, 50 мкл 100 нг/мл TGFβ1 раствора добавляли к каждой лунке, и для группы нестимулированного контроля добавляли 50 мкл базальной среды. После инкубации в течение 24 часов экстрагировали РНК, используя систему выделения тотальной РНК SV (изготовлена Invitrogen), и первую цепь кДНК синтезировали из РНК, используя SuperScript III (изготовлен Invitrogen). Используя кДНК, полученную как описано выше, в качестве шаблона, экспрессированное количество мРНК для 1 цепи коллагена I типа (COL 1α1), 2 цепи коллагена I типа (COL 1α2), (актина гладкой мышцы (ACTA), TGF β1 и глицеральдегидов 3-фосфатдегидрогеназы (GAPDH) было измерено при помощи способа количественной ПЦР, выполняемого в реальном времени. Количественную ПЦР, выполняемую в реальном времени, проводили, используя Platinum SYBR Green qPCR Super-Mix-UDG с ROX (изготовлен Invitrogen) и праймер, специфичный к каждому гену в соответствии с инструкцией, приложенной к Platinum SYBR Green qPCR Super-Mix-UDG с ROX, используя ABI PRISM 7000 (Applied Biosystems). Оценку этого теста проводили, используя значение (уровень экспрессии мРНК), вычисленное как относительное значение, в котором среднее значение группы нестимулированного контроля определяли как 1 после корректировки экспрессированного значения мРНК каждого гена для экспрессированного количества мРНК GAPDH.
(2) Результаты
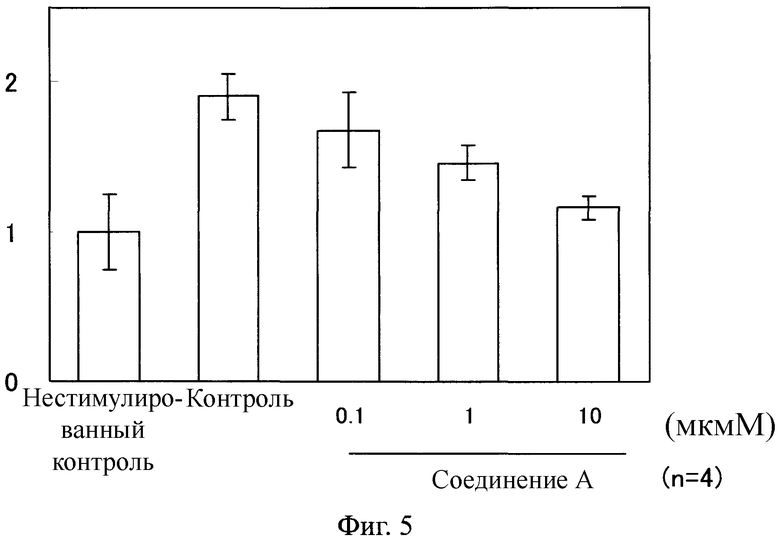
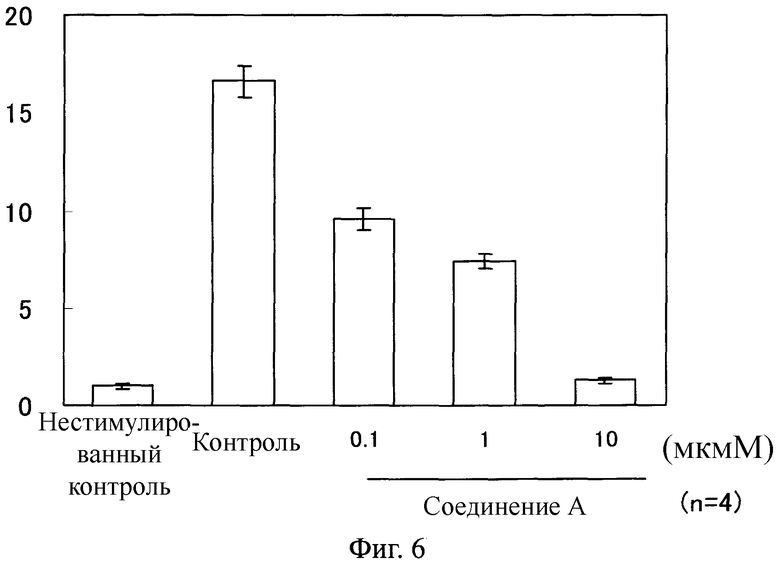
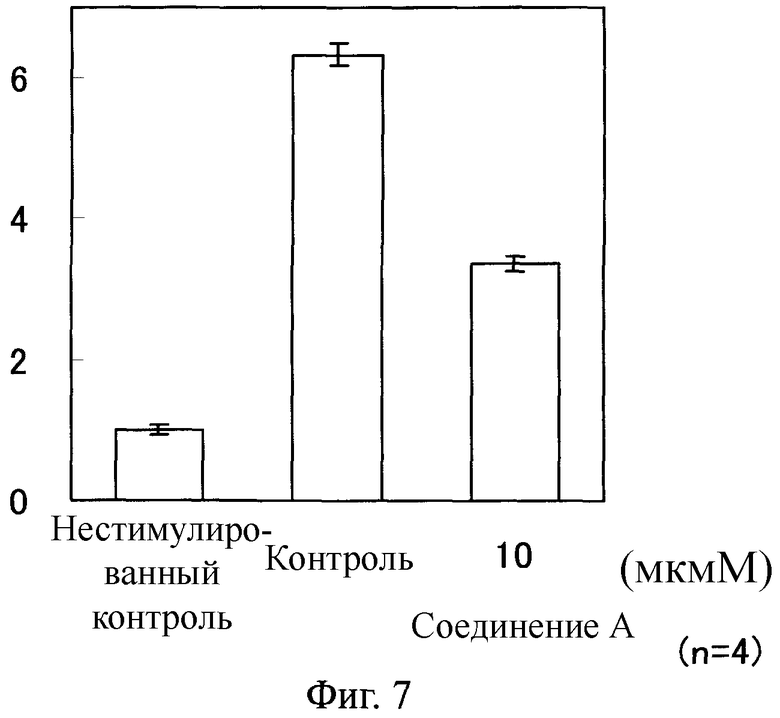
Как показано на Фигурах 4-7, в фибробластах легкого человека, каждый уровень экспрессии мРНК возрастал благодаря стимуляции с помощью TGF β1. В противоположность этому, в клетках, обработанных соединением А, каждый уровень мРНК понизился по сравнению с таковым у контрольной группы.
Тестовый пример 5
(1) Способы
Крысиные почечные интерстициоциты (клетки NRF 49F) высеивали в 96-луночный планшет при 1×104 клетках/лунку и инкубировали в минимальной эссенциальной среде (MEM среда; изготовлена Nippon Seiyaku; далее будет использован такой же продукт), содержащей 10% фетальную бычью сыворотку (изготовлена JRH Bioscience; далее будет использован такой же продукт) при условиях 37°C и 5% CO2 всю ночь. Клетки промывали один раз с помощью MEM среды, не содержащей фетальной бычьей сыворотки (здесь далее обозначаемой как "MEM среда без сыворотки"), и 100 мкл MEM среды без сыворотки было в них добавлено с последующей инкубацией в течение 24 часов. После промывки MEM средой без сыворотки один раз, MEM среду без сыворотки добавляли в количестве 80 мкл/лунку. 100 мкМ раствора соединения, полученного с помощью такого же способа, как в тестовом примере 1, добавляли в количестве 10 мкл к каждой лунке. Для группы нестимулированного контроля и контрольной группы, ДМСО, который разбавляли 100-кратно с помощью MEM среды без сыворотки, добавляли в количестве 10 мкл. После инкубации в течение 2 часов, 10 мкл 100 нг/мл раствора фактора роста тромбоцитов BB (PDGF BB; изготовлен Sigma) добавляли к каждой лунке, и, для группы нестимулированного контроля 10 мкл MEM среды без сыворотки добавляли с последующей инкубацией. После инкубации в течение 48 часов, активность пролиферации клеток (абсорбцию) измеряли с помощью MTT способа таким же образом, как и в тестовом примере 3. Оценку этого теста проводили, используя значение (уровень пролиферации клеток), выраженное как относительное значение, в котором среднее значение абсорбции нестимулированной контрольной группы определяли как 1.
(2) Результаты
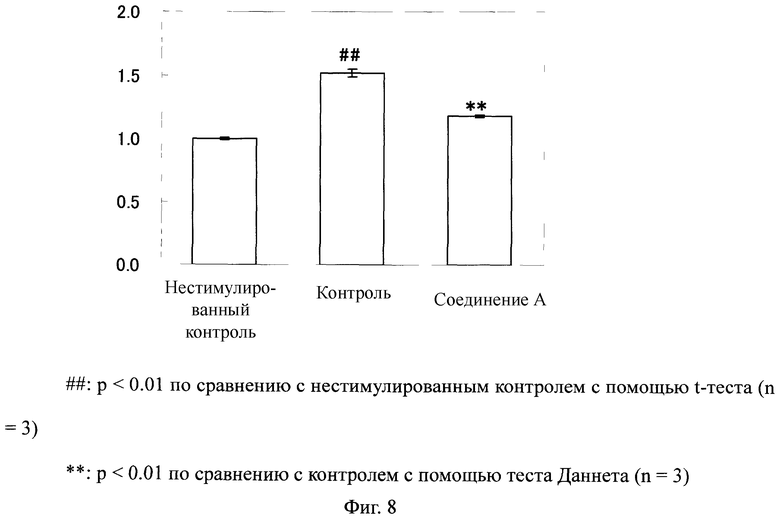
Как показано на Фиг.8, в крысиных почечных интерстициоцитах уровень пролиферации клеток значительно возрос благодаря стимуляции с помощью PDGF BB. В противоположность этому, в клетках, обработанных соединением А, уровень пролиферации клеток значительно понизился по сравнению с таковым у контрольной группы.
Тестовый пример 6
(1) Способы
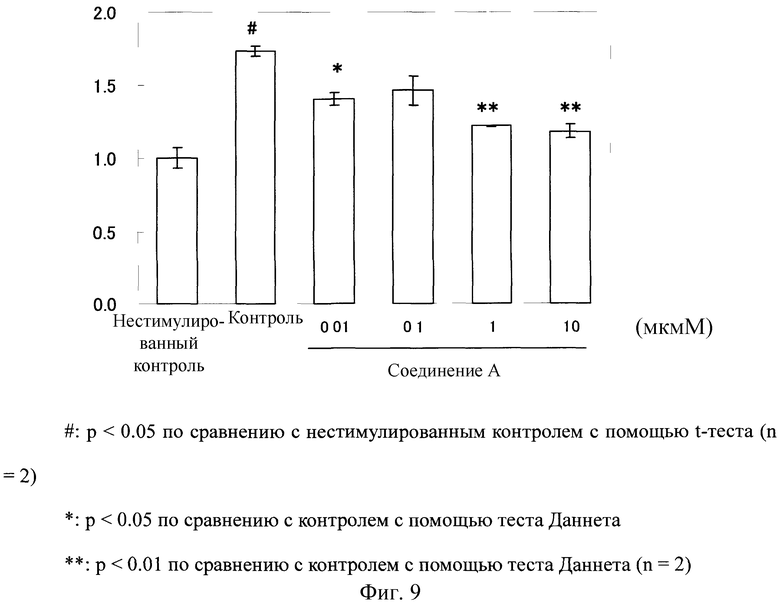
Фибробласты кожи человека (изготовлены Kurabo; далее будет использован такой же продукт) высеивали в 96-луночный планшет при 5×103 клетках/лунку и инкубировали в модифицированной способом Дульбекко среде Игла (DMEM среда; изготовлена Nippon Seiyaku; далее будет использован такой же продукт), содержащей 10% фетальную бычью сыворотку, при условиях 37°C и 5% CO2 всю ночь. Фибробласты кожи человека промывали один раз с помощью DMEM среды, не содержащей фетальной бычьей сыворотки (здесь далее обозначаемое как "DMEM среда без сыворотки"), и в них было добавлено 100 мкл DMEM среды без сыворотки с последующей инкубацией в течение 24 часов. После промывки с помощью DMEM среды без сыворотки еще раз, DMEM среду без сыворотки добавляли в количестве 80 мкл/лунку. Десять мкл 0,1, 1, 10 или 100 мкМ раствора соединения, полученного с помощью такого же способа, как в тестовом примере 3, добавляли к каждой лунке. Для группы нестимулированного контроля и контрольной группы, ДМСО, который разбавляли 100-кратно с помощью DMEM среды без сыворотки, добавляли в количестве 10 мкл в каждую с последующей инкубацией. После инкубации в течение 2 часов добавляли 10 мкл 100 нг/мл TGF β1 раствора к каждой лунке, и для группы нестимулированного контроля добавляли каждые 10 мкл MEM среды без сыворотки. После инкубации в течение 48 часов активность пролиферации клеток (абсорбцию) измеряли с помощью MTT способа, и концентрацию PIP в среде измеряли таким же образом, как и в тестовом примере 3. Оценку этого теста проводили, используя значение (индекс для продуцирования коллагена), вычисленное как относительное значение, в котором среднее значение в группе нестимулированного контроля определяли как 1 после корректировки измеренного значения концентрации PIP, используя значение активности пролиферации клеток (абсорбцию).
(2) Результаты
Как показано на Фиг.9, в фибробластах кожи человека продуцирование коллагена значительно возросло благодаря стимуляции с помощью TGF β1. В противоположность этому, в клетках, обработанных соединением А, продуцирование коллагена значительно понизилось по сравнению с таковой у контрольной группы.
Краткое описание чертежей
Фиг.1
На Фиг.1 показаны ингибирующие эффекты соединения А и соединения B на пролиферацию фибробластов легкого человека, стимулированных с помощью EGF. На ординате показана абсорбция.
Фиг.2
На Фиг.2 показаны ингибирующие эффекты соединения А и соединения B на пролиферацию фибробластов легкого человека, стимулированных с помощью TGFα. На ординате показана абсорбция.
Фиг.3
На Фиг.3 показан ингибирующий эффект соединения А на продуцирование коллагена в фибробластах легкого человека, стимулированных с помощью TGF β1. На ординате показано значение, вычисленное как относительное значение, в котором среднее значение в группе нестимулированного контроля определяли как 1 после корректировки измеренного значения для концентрации PIP с помощью значения активности пролиферации клеток (абсорбция).
Фиг.4
На Фиг.4 показан ингибирующий эффект соединения А на экспрессию COL 1α1 мРНК в фибробластах легкого человека, стимулированных с помощью TGF β1. На ординате показано относительное значение, в котором среднее значение в группе нестимулированного контроля определяли как 1 после корректировки экспрессированного количества мРНК с помощью COL 1α1, используя экспрессированное количество мРНК с помощью GAPDH.
Фиг.5
На Фиг.5 показан ингибирующий эффект соединения А на экспрессию COL 1α2 мРНК в фибробластах легкого человека, стимулированных с помощью TGF β1. На ординате показано относительное значение, в котором среднее значение в группе нестимулированного контроля определяли как 1 после корректировки экспрессированного количества мРНК с помощью COL 1α2, используя экспрессированное количество мРНК с помощью GAPDH.
Фиг.6
На Фиг.6 показан ингибирующий эффект соединения А на экспрессию ACTA мРНК в фибробластах легкого человека, стимулированных с помощью TGF β1. На ординате показано относительное значение, в котором среднее значение в группе нестимулированного контроля определяли как 1 после корректировки экспрессированного количества мРНК с помощью ACTA, используя экспрессированное количество мРНК с помощью GAPDH.
Фиг.7
На Фиг.7 показан ингибирующий эффект соединения А на экспрессию TGF β1 мРНК в фибробластах легкого человека, стимулированных с помощью TGF β1. На ординате показано относительное значение, в котором среднее значение в группе нестимулированного контроля определяли как 1 после корректировки экспрессированного количества мРНК с помощью TGF β1, используя экспрессированное количество мРНК с помощью GAPDH.
Фиг.8
На Фиг.8 показан ингибирующий эффект соединения А на пролиферацию крысиных почечных интерстициоцитов, стимулированных с помощью PDGF BB. На ординате показано относительное значение, в котором среднее значение абсорбции в группе нестимулированного контроля определяли как 1.
Фиг.9
На Фиг.9 показан ингибирующий эффект соединения А на продуцирование коллагена в фибробластах кожи человека, стимулированных с помощью TGF β1. На ординате показано значение, вычисленное как относительное значение, в котором среднее значение в группе нестимулированного контроля определяли как 1 после корректировки измеренного значения концентрации PIP с помощью значения активности пролиферации клеток (абсорбция).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИД, СПОСОБНЫЙ СВЯЗЫВАТЬСЯ С TGF-β 1 И ИНГИБИРОВАТЬ БИОЛОГИЧЕСКУЮ АКТИВНОСТЬ TGF-β 1 IN VITRO И/ИЛИ IN VIVO, ПРИМЕНЕНИЕ ПЕПТИДА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОСЛЕДОВАТЕЛЬНОСТЬ ДНК, КОНСТРУКЦИЯ ДНК, ВЕКТОР ЭКСПРЕССИИ, КЛЕТКА-ХОЗЯИН И СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА | 2004 |
|
RU2333917C2 |
| ПРИМЕНЕНИЕ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК, КОТОРАЯ КОДИРУЕТ ПЕПТИД, СПОСОБНЫЙ СВЯЗЫВАТЬСЯ С ТРАНСФОРМИРУЮЩИМ ФАКТОРОМ РОСТА β1 (ВАРИАНТЫ) | 2008 |
|
RU2455358C2 |
| ПРОФИЛАКТИЧЕСКИЙ ИЛИ ТЕРАПЕВТИЧЕСКИЙ АГЕНТ ДЛЯ ЛЕЧЕНИЯ ФИБРОЗА | 2011 |
|
RU2583290C2 |
| АНТИСМЫСЛОВЫЕ ОЛИГОНУКЛЕОТИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ СИГНАЛЬНОГО ПУТИ TGF-R | 2015 |
|
RU2717454C2 |
| ПРИМЕНЕНИЕ АНТАГОНИСТОВ РЕЦЕПТОРА СВ1 ДЛЯ ПОЛУЧЕНИЯ КОМПОЗИЦИИ, ПРИГОДНОЙ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ПЕЧЕНИ | 2005 |
|
RU2402328C2 |
| ИНГИБИТОРЫ ПЕРЕДАЧИ СИГНАЛА ТРАНСФОРМИРУЮЩИХ ФАКТОРОВ РОСТА (TGF-R) ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ ЦНС | 2005 |
|
RU2385933C2 |
| ПРИМЕНЕНИЕ ЧИГЛИТАЗАРА И РОДСТВЕННЫХ ЕМУ СОЕДИНЕНИЙ | 2019 |
|
RU2769446C1 |
| ПРИМЕНЕНИЕ ИНГИБИРУЮЩИХ TGF-бета1 ПЕПТИДОВ ДЛЯ ПРИГОТОВЛЕНИЯ МОДУЛИРУЮЩЕГО ИММУННЫЙ ОТВЕТ АГЕНТА | 2005 |
|
RU2420307C2 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ФИБРОЗА ТКАНЕЙ НА ОСНОВЕ КОМПОНЕНТОВ СЕКРЕТОМА МЕЗЕНХИМНЫХ СТРОМАЛЬНЫХ КЛЕТОК, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ СРЕДСТВА | 2020 |
|
RU2766707C1 |
| АНТИФИБРОГЕННЫЕ СОЕДИНЕНИЯ, СПОСОБЫ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2726416C2 |
Изобретение относится к области фармацевтики и медицины и касается ингибитора фиброза, содержащего 2-{4-[N-(5,6-дифенил-пиразин-2-ил)-N-изопропиламино]бутилокси} уксусную кислоту или ее фармацевтически приемлемую соль или 2-{4-[N-(5,6-дифенил-пиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил) ацетамид или его фармацевтически приемлемую соль в качестве активного компонента, для лечения интерстициальной пневмонии, фиброза легких, склеродермии и цирроза печени. Ингибитор обладает высокой эффективностью. 2 н. и 4 з.п. ф-лы, 9 ил., 6 пр.
1. Ингибитор фиброза, содержащий 2-{4-[N-(5,6-дифенил-пиразин-2-ил)-N-изопропиламино]бутилокси} уксусную кислоту или ее фармацевтически приемлемую соль в качестве активного компонента.
2. Ингибитор фиброза по п.1 для лечения заболевания, выбранного из группы, состоящей из интерстициальной пневмонии, фиброза легких, склеродермии и цирроза печени.
3. Ингибитор фиброза по п.2 для лечения интерстициальной пневмонии.
4. Ингибитор фиброза, содержащий 2-{4-[N-(5,6-дифенил-пиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамид или его фармацевтически приемлемую соль в качестве активного компонента.
5. Ингибитор фиброза по п.4 для лечения заболевания, выбранного из группы, состоящей из интерстициальной пневмонии, фиброза легких, склеродермии и цирроза печени.
6. Ингибитор фиброза по п.5 для лечения интерстициальной пневмонии.
| ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2002 |
|
RU2283835C2 |
| RU 2005134652 А, 10.05.2006 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |

