Область техники, к которой относится изобретение
Настоящее изобретение относится к новым кристаллическим формам 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона, т.е. к R-энантиомеру соединения:

и к способам его получения.
Предпосылки создания изобретения и известный уровень техники
Интерес к разработке ингибиторов дофамин-β-гидроксилазы (DβΗ) сосредоточен на гипотезе о том, что ингибирование этого фермента может способствовать значительному улучшению клинического состояния пациентов, страдающих сердечно-сосудистыми расстройствами, такими как гипертензия или хроническая сердечная недостаточность. Целесообразность применения ингибиторов DβΗ обосновывается их способностью ингибировать биосинтез норадреналина, который осуществляется посредством ферментативного гидроксилирования дофамина. Главным клиническим проявлением застойной сердечной недостаточности является активация нейрогуморальных систем, преимущественносимпатической нервной системы (Parmley, W.W., Clinical Cardiology, 18: 440-445, 1995). Пациенты с застойной сердечной недостаточностью имеют повышенные концентрации норадреналина в плазме (Levine, T.B. et al., Am. J. Cardiol., 49: 1659-1666, 1982), у них усилено влияние центральных симпатических факторов (Leimbach, W.N. et al., Circulation, 73: 913-919, 1986) и увеличен кардиоренальный выброс (spillover) норадреналина (Hasking, G.J. et al., Circulation, 73: 615-621, 1966). Продолжительное и избыточное воздействие норадреналина на миокард может приводить к понижающей регуляции сердечных β1-адренорецепторов, ремоделированию левого желудочка, аритмиям и некрозу, и все эти эффекты могут уменьшать функциональную целостность сердца. Пациенты с застойной сердечной недостаточностью, которые имеют высокие концентрации норадреналина в плазме, имеют и самый неблагоприятный отдаленный прогноз (Cohn, J.N. et al., N. Engl. J. Med., 311: 819-823, 1984). Более важным является наблюдение, что в плазме бессимптомных пациентов без явных признаков сердечной недостаточности концентрации норадреналина являются уже повышенными и это можно использовать для прогноза возможной заболеваемости и летальности (Benedict, C.R. et al., Circulation, 94: 690-697, 1996). Поэтому активированный симпатический эффект не является всего лишь клиническим маркером застойной сердечной недостаточности, но может вносить свой вклад и в прогрессирующее ухудшение заболевания.
Сильные ингибиторы дофамин-β-гидроксилазы, обладающие высокой активностью и значительно пониженным доступом в мозг, раскрыты в WO 2008/136695. WO 2008/136695 описывает соединения формулы I:
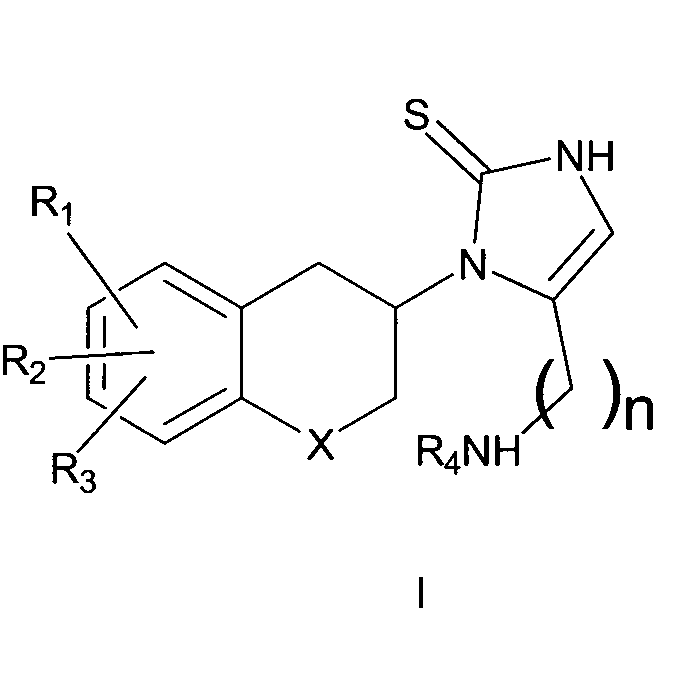
где R1, R2 и R3 являются одинаковыми или разными и обозначают водороды, галогены, алкил, нитрогруппу, аминогруппу, алкилкарбониламиногруппу, алкиламиногруппу или диалкиламиногруппу; R4 обозначает алкиларил или алкилгетероарил; X обозначает CH2, атом кислорода или атом серы; n равно 2 или 3; включая их индивидуальные (R)- и (S)-энантиомеры или смеси их энантиомеров и включая их фармацевтически приемлемые соли и сложные эфиры, где термин «алкил» означает углеводородные цепи, прямые или разветвленные, содержащие от одного до шести углеродных атомов, необязательно, замещенных арильными, алкоксильными, галогенными, алкоксикарбонильными или гидроксикарбонильными группами; термин «арил» означает фенильную или нафтильную группу, необязательно, замещенную алкильной, алкилоксильной, галогенной или нитрогруппой; термин «галоген» означает фтор, хлор, бром или йод; термин гетероарил означает гетероароматическую группу. В частности, WO 2008/136695 описывает 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тион.
Способы получения соединений формулы I и конкретно 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона описаны в WO 2008/136695, они включены в настоящий документ посредством ссылки.
Известно, что полиморфные формы одного и того же лекарственного средства могут иметь существенно разные фармацевтически важные свойства, такие как характеристики растворимости и биодоступность, а также стабильность лекарственного средства. Кроме того, разные формы могут иметь разный размер частиц, твердость и температуру стеклования. В частности, одна форма может предоставлять значительно большие преимущества по сравнению с другими формами того же лекарственного средства в способах изготовления твердых дозированных форм, такие как более точное измерение количества активных ингредиентов, более легкое фильтрование или повышенная стабильность при гранулировании или хранении. Кроме того, конкретный способ, подходящий для какой-либо одной формы, может также предоставить изготовителям такие преимущества, как экономически или экологически подходящие растворители и технологические процессы, или обеспечить более высокую чистоту или более высокий выход желаемого продукта.
Сущность изобретения
Настоящее изобретение предоставляет кристаллические полиморфы 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона и способы их получения. Новые полиморфные формы 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона демонстрируют высокую стабильность при интенсивном механическом воздействии и/или интенсивном воздействии водяным паром. Настоящее изобретение также предоставляет аморфную форму 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона и способы ее получения. Указанная аморфная форма также является частью настоящего изобретения.
Далее в настоящем документе 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тион будет указываться либо как таковойлибо как «соединение 2».
В нижеследующем описании настоящего изобретения полиморфные формы описаны как обладающие дифрактограммой XRPD с пиками в положениях, перечисленных в соответствующих таблицах. Следует также понимать, что в одном варианте осуществления настоящего изобретения полиморфная форма имеет дифрактограмму XRPD с пиками в положениях со значениями °2θ, приведенными с точностью до±0,2°2θ при любом значении интенсивности (% (I/I0)); или в другом варианте осуществления настоящего изобретения она имеет дифрактограмму XRPD с пиками в положениях со значениями °2θ, приведенными с точностью до ±0,1°2θ. Следует также заметить, что значения интенсивности приведены только для информации и определение каждого из пиков не подразумевает их ограничения конкретными значениями интенсивности.
Согласно одному аспекту настоящего изобретения предоставлена кристаллическая Форма А 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона.
В варианте осуществления настоящего изобретения кристаллическая Форма А соединения 2 не является сольватом, т.е. Форма А соединения 2 является несольватированной формой.
Согласно настоящему изобретению термин «несольватированная» означает, что термогравиметрическая (TGA) кривая кристаллической Формы A соединения 2 показывает потерю массы менее чем приблизительно на 1% по массе, предпочтительно, менее чем приблизительно на 0,6%, более предпочтительно, не показывает никакой потери массы при температуре, более низкой, чем приблизительно 200°C.
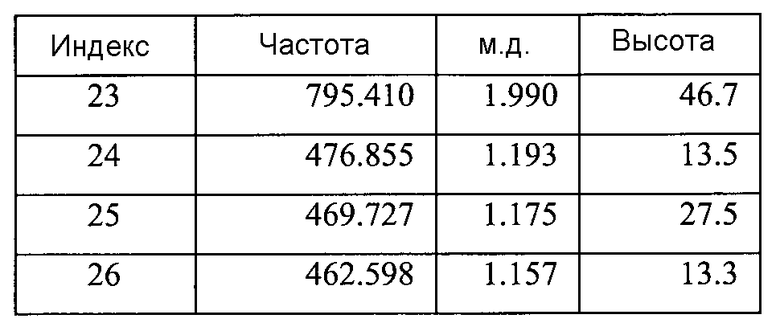
Согласно другому аспекту настоящего изобретения, предоставлена Форма A соединения 2, имеющая дифрактограмму XRPD с пиками при 14,0, 16,1, 16,6, 19,2 и 20,4°2θ±0,2°2θ. Дифрактограмма XRPD может иметь дополнительные пики при 15,6 и 18,4°2θ±0,2°2θ.
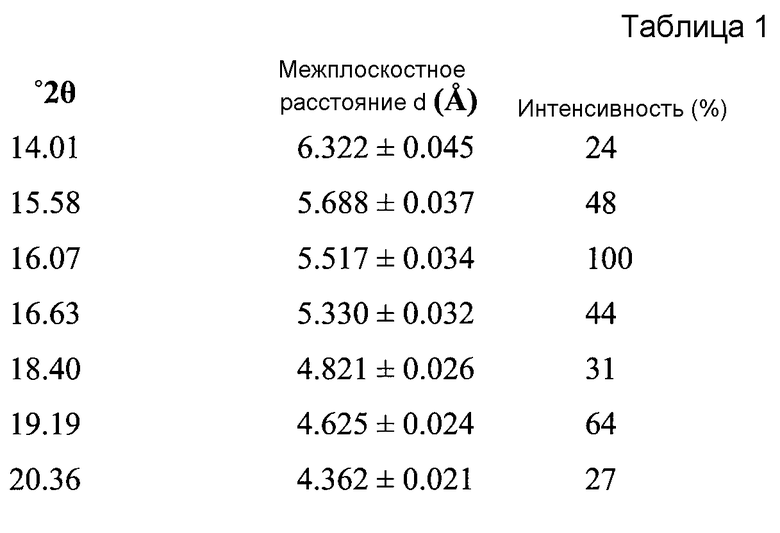
Форма A может быть охарактеризована как имеющая дифрактограмму рентгеновской порошковой дифракции (X-ray powder diffraction, XRPD) с пиками, представленными в Таблице 1.
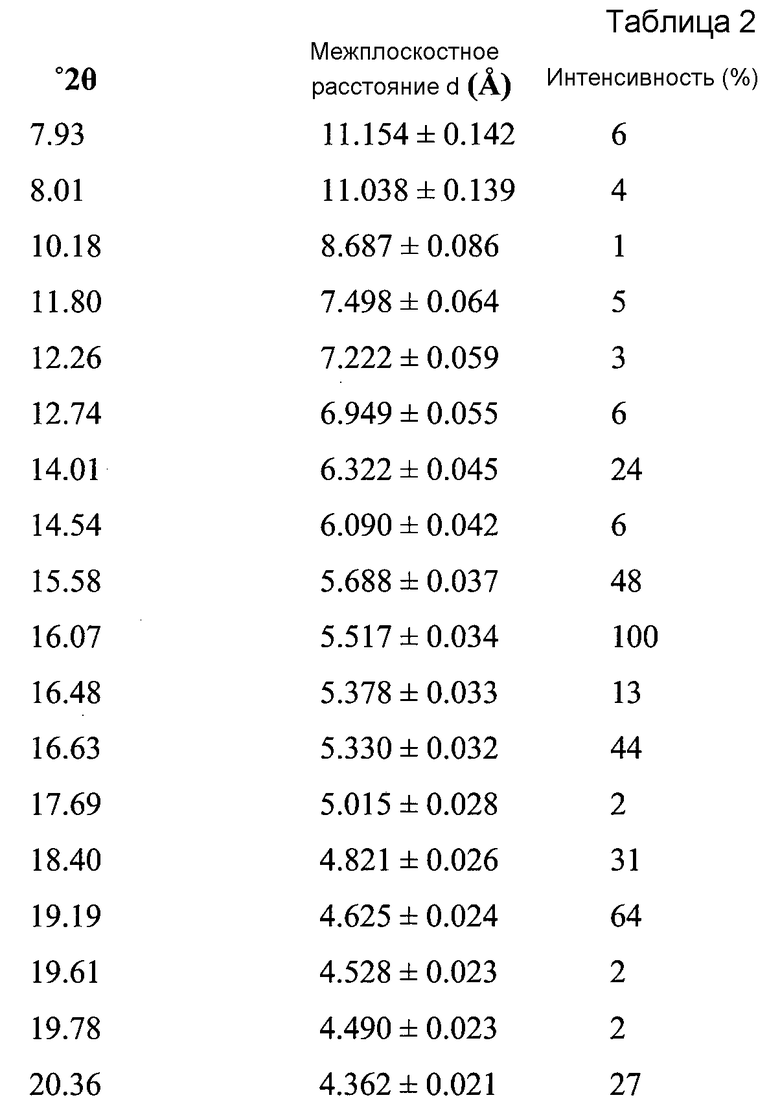
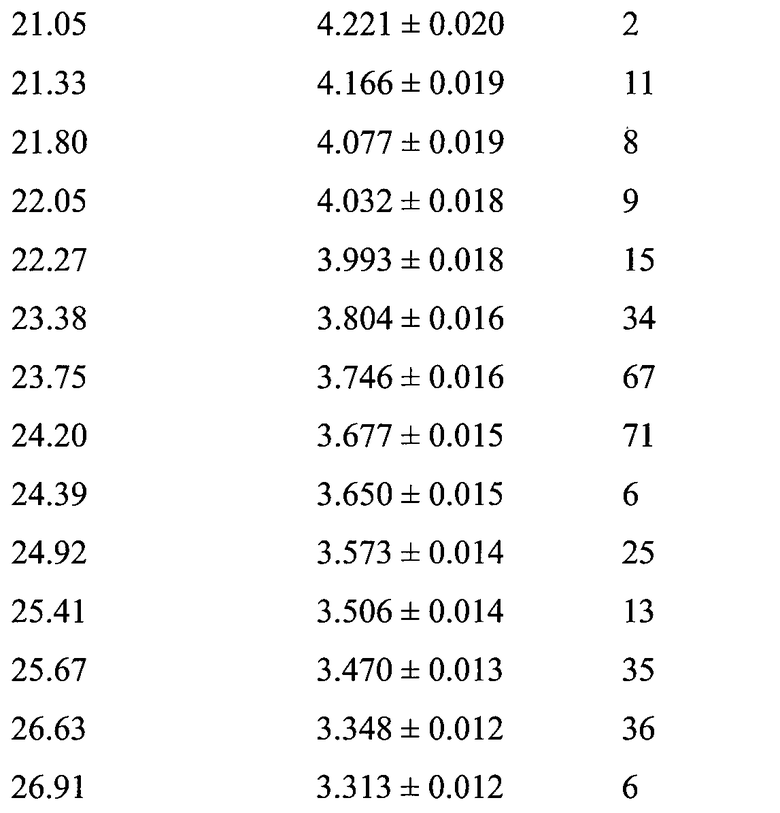
В варианте осуществления настоящего изобретения Форма А охарактеризована как имеющая дифрактограмму рентгеновской порошковой дифракции (XRPD) с одним или более из пиков, представленных в Таблице 2.

В другом варианте осуществления настоящего изобретения Форма A имеет дифрактограмму XRPD, как показано на Фигуре 2.
Согласно другому варианту осуществления настоящего изобретения предоставлена кристаллическая Форма A 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона, имеющая термограмму термогравиметрического анализа (thermogravimetric analysis, TGA), показывающую потерю массы, начинающуюся при температуре 259°C±5°C. В варианте осуществления настоящего изобретения термограмма TGA показывает потерю массы, начинающуюся при температуре в диапазоне от приблизительно 257°C до приблизительно 262°C. В варианте осуществления настоящего изобретения Форма A имеет термограмму TGA, показывающую потерю массы, начинающуюся при температуре приблизительно 259°C.
В варианте осуществления настоящего изобретения Форма A имеет термограмму TGA, как показано на Фигуре 3.
Согласно другому варианту осуществления настоящего изобретения предоставлена кристаллическая Форма A 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона, имеющая термограмму дифференциальной сканирующей калориметрии (differential scanning calorimetry, DSC), показывающую эндотермический пик, начинающийся при температуре 192°C±2°C и с максимумом при 193°C±2°C. В варианте осуществления настоящего изобретения термограмма DSC показывает эндотермический пик, начинающийся при температуре в диапазоне от приблизительно 190°C до приблизительно 192°C. В варианте осуществления настоящего изобретения DSC показывает эндотермический пик с максимумом в диапазоне от приблизительно 193°C до приблизительно 194°C. В варианте осуществления настоящего изобретения Форма A имеет термограмму DSC, имеющую эндотермический пик, начинающийся при температуре приблизительно 192°C и с максимумом приблизительно при 193°C. В варианте осуществления настоящего изобретения термограмма DSC показывает теплоту плавления 141 Дж/г±10 Дж/г. В варианте осуществления настоящего изобретения термограмма DSC показывает теплоту плавления в диапазоне от приблизительно 139 Дж/г до приблизительно 147 Дж/г. В варианте осуществления настоящего изобретения термограмма DSC показывает теплоту плавления приблизительно 147 Дж/г.
В варианте осуществления настоящего изобретения Форма A соединения 2 имеет термограмму DSC, как показано на Фигуре 4.
В следующем варианте осуществления настоящего изобретения Форма A соединения 2 представляет собой материал, демонстрирующий низкую гигроскопичность в диапазоне от 5% до 95% относительной влажности (relative humidity, RH). Материал с малой гигроскопичностью может быть определен как материал, который демонстрирует <0,5 масс. % поглощения воды в указанном диапазоне относительной влажности.
В следующем варианте осуществления настоящего изобретения Форма A демонстрирует незначительную потерю массы при уравновешивании при RH, составляющей ~5%. В контексте настоящей спецификации«незначительный» означает менее 0,5 масс.%.
В другом варианте осуществления настоящего изобретения Форма A демонстрирует приблизительно 0,02%-ное увеличение массы в диапазоне RH от приблизительно 5% до приблизительно 75%. В варианте осуществления настоящего изобретения Форма A демонстрирует приблизительно 0,19%-ное увеличение массы в диапазоне RH от приблизительно 75% до приблизительно 95%. В другом варианте осуществления настоящего изобретения Форма A демонстрирует приблизительно 0,20%-ное уменьшение массы в диапазоне RH от приблизительно 95% до приблизительно 5% с гистерезисом в диапазоне RH от приблизительно 85% до приблизительно 45% после десорбции.
Выгодным образом, Форма A имеет низкую гигроскопичность и остается стабильной кристаллической формой после интенсивного механического воздействия и интенсивного воздействия водяным паром.
В другом аспекте настоящее изобретение предоставляет кристаллическую Форму B 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона.
Кристаллическая Форма B соединения 2 представляет собой этилацетатный сольват. В варианте осуществления настоящего изобретения Форма B содержит 0,1-0,2 моля этилацетата. В варианте осуществления настоящего изобретения Форма B содержит приблизительно 0,1 моля этилацетата. В другом варианте осуществления настоящего изобретения форма B содержит приблизительно 0,2 моля этилацетата.
Согласно другому аспекту настоящего изобретения предоставлена Форма B 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона, которая представляет собой этилацетатный сольват, предпочтительно содержащий 0,1-0,2 моля этилацетата, и имеющая дифрактограмму XRPD с пиками при 7,9-8,0, 14,0, 16,0-16,1, 19,2 и 20,4°2θ±0,2°2θ. Дифрактограмма XRPD может иметь дополнительные пики при 15,6, 16,7 и 18,4°2θ±0,2°2θ.
В другом варианте осуществления настоящего изобретения Форма B соединения 2 охарактеризована как имеющая дифрактограмму рентгеновской порошковой дифракции (XRPD) с пиками, представленными в Таблице 3.
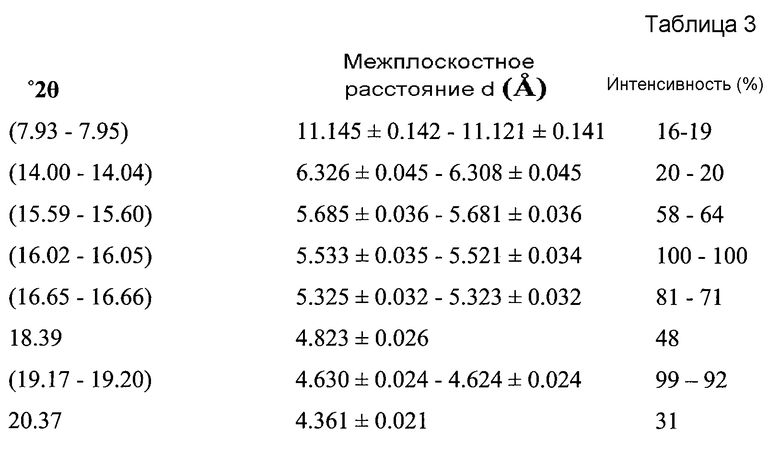
В варианте осуществления настоящего изобретения Форма B охарактеризована как имеющая дифрактограмму рентгеновской порошковой дифракции (XRPD) с одним или более из пиков, представленных в Таблице 4.
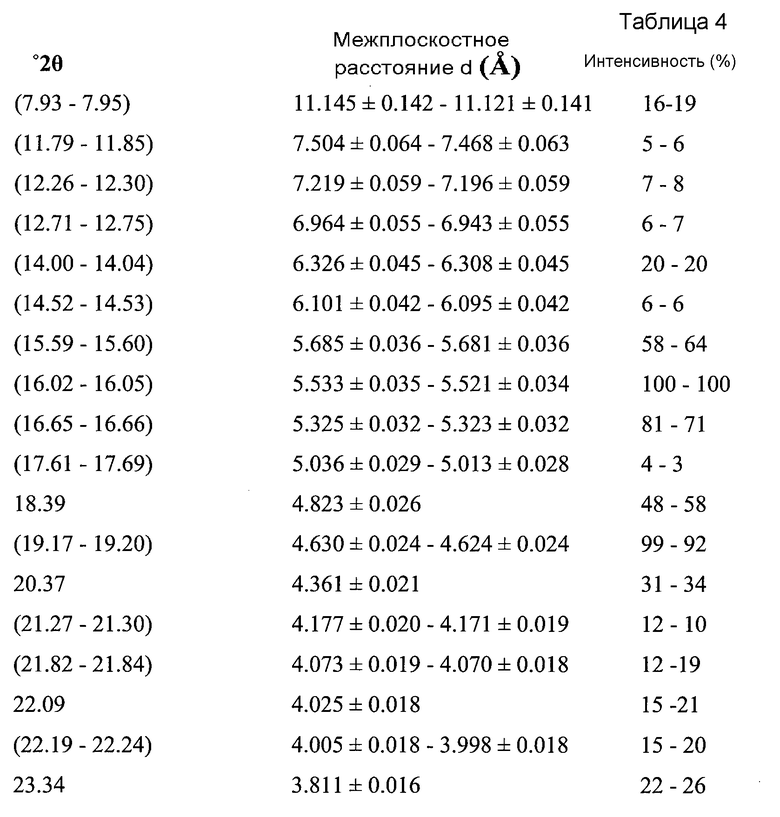

Как можно видеть из Таблиц 3 и 4, положения некоторых пиков указаны в виде некоторых диапазонов. Причиной этого является то, что материал представляет собой переменный сольват (в варианте осуществления настоящего изобретения содержащий 0,1-0,2 моль этилацетата).
В варианте осуществления настоящего изобретения Форма B имеет дифрактограмму XRPD, как показано на Фигуре 5.
Согласно другому варианту осуществления настоящего изобретения предоставлена кристаллическая Форма B соединения 2, имеющая термограмму TGA, показывающую уменьшение массы, начинающееся при температуре 257°C±5°C, и уменьшение массы в диапазоне от приблизительно 130°C до приблизительно 200°C. В варианте осуществления настоящего изобретения термограмма TGA, кроме того, имеет приблизительно 2,3%-ное уменьшение массы в диапазоне от приблизительно 162°C до приблизительно 200°C или приблизительно 4,7%-ное уменьшение массы в диапазоне от приблизительно 138°C до приблизительно 190°C.
В другом варианте осуществления настоящего изобретения Форма B соединения 2 имеет термограмму TGA, как показано на Фигуре 6.
Согласно другому аспекту настоящего изобретения предоставлена кристаллическая Форма B соединения 2, имеющая термограмму DSC, показывающую эндотермический пик, начинающийся при температуре приблизительно 190°C±2°C и с максимумом приблизительно при 192°C±2°C. В варианте осуществления настоящего изобретения термограмма DSC показывает теплоту плавления, составляющую приблизительно 141 Дж/г±10 Дж/г.
В варианте осуществления настоящего изобретения Форма B соединения 2 имеет термограмму DSC, как показано на Фигуре 7.
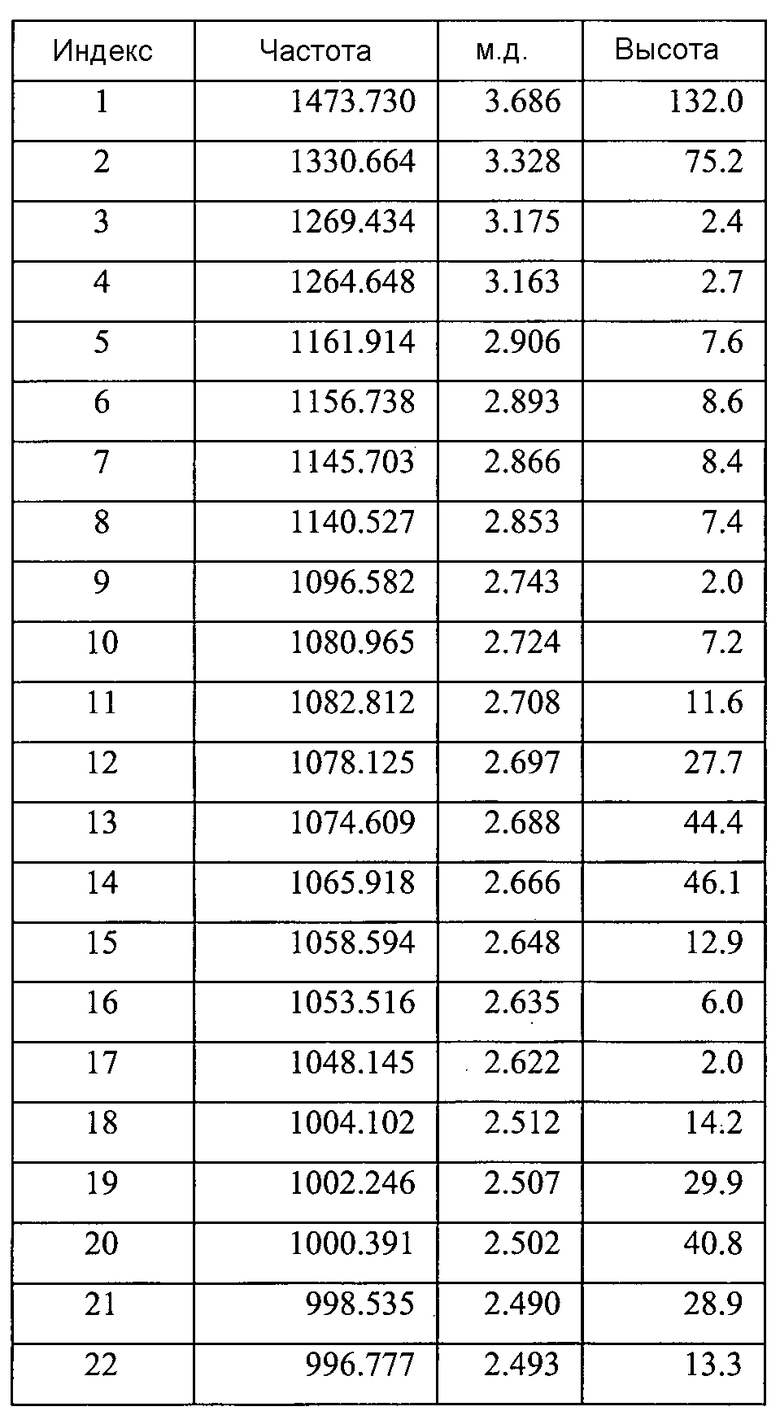
Согласно другому аспекту настоящего изобретения предоставлена кристаллическая Форма B соединения 2, имеющая спектр 1H ЯМР, содержащий пики, которые можно признать принадлежащими этилацетату. В варианте осуществления настоящего изобретения пики, которые можно приписать этилацетату, находятся приблизительно при 4,0 м.д., приблизительно при 2,0 м.д. и приблизительно при 1,2 м.д. Как будет понятно квалифицированному специалисту, спектр 1Н ЯМР будет также содержать пики, которые можно признать принадлежащими протонам 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона.
В варианте осуществления настоящего изобретения кристаллическая Форма B соединения 2 имеет спектр 1H ЯМР, как показано на Фигуре 8A. В варианте осуществления настоящего изобретения спектр 1H ЯМР является таким, как показано на Фигурах 8A-8E.
Согласно другому аспекту настоящего изобретения имеется 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тион в аморфной форме.
В контексте настоящей спецификации термин «аморфный» означает «рентгеновски аморфный», что означает, что в дифрактограмме XRPD материала отсутствуют пики рентгеновской дифракции. В варианте осуществления настоящего изобретения рентгеновски аморфные материалы являются:
- нанокристаллическими;
- кристаллическими с очень большой плотностью дефектов;
- кинетическими аморфными материалами; или
- термодинамическими аморфными материалами;
или комбинацией вышеуказанного.
В варианте осуществления настоящего изобретения аморфная форма имеет дифрактограмму XRPD, демонстрирующую гало.
В варианте осуществления настоящего изобретения аморфная форма имеет дифрактограмму XRPD, как показано на Фигуре 9.
Согласно другому аспекту настоящего изобретенияпредоставлен аморфный 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тион, имеющий термограмму термогравиметрического анализа (TGA), показывающую уменьшение массы, начинающееся при температуре 258°C±5°C, и приблизительно 1,2%-ное уменьшение массы в диапазоне от приблизительно 26°C до приблизительно 71°C.
В варианте осуществления настоящего изобретения аморфная форма имеет термограмму TGA, как показано на Фигуре 10.
В варианте осуществления настоящего изобретения аморфный 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тион имеет термограмму циклической дифференциальной сканирующей калориметрии (цикло-DSC), показывающую ступенчатое изменение, обусловленное стеклованием. В варианте осуществления настоящего изобретения указанное ступенчатое изменение происходит при температуре 50°C±2°C. В варианте осуществления настоящего изобретения циклическая процедура включает в себя 2 цикла, причем стеклование происходит в цикле 2.
В варианте осуществления настоящего изобретения аморфный 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тион имеет термограмму циклической дифференциальной сканирующей калориметрии (цикло-ДСК), показывающую экзотермический пик с максимумом в диапазоне от приблизительно 115°C и приблизительно 124°C. В варианте осуществления настоящего изобретения циклическая процедура включает в себя 2 цикла, причем экзотермический пик проявляется в цикле 2. В варианте осуществления настоящего изобретения процесс кристаллизации продуцирует Форму А.
В варианте осуществления настоящего изобретения аморфная форма имеет термограмму цикло-ДСК, как показано на Фигурах 11 и 12 (циклы 1 и 2 соответственно).
В следующем варианте осуществления настоящего изобретения аморфное соединение 2 представляет собой материал, демонстрирующий значительную гигроскопичность в диапазоне RH от приблизительно 5% до приблизительно 75%. Материал может быть определен как значительно гигроскопичный, если он демонстрирует поглощение воды, составляющее не менее 2,0 масс.% в указанном диапазоне RH.
В другом варианте осуществления настоящего изобретения аморфное соединение 2 демонстрирует приблизительно 0,08%-ное увеличение массы после уравновешивания при RH, составляющей приблизительно 5%.
В варианте осуществления настоящего изобретения аморфное соединение 2 демонстрирует приблизительно 1,2%-ное увеличение массы в диапазоне RH от приблизительно 5% до приблизительно 75%. В варианте осуществления настоящего изобретения аморфное соединение 2 демонстрирует приблизительно 8,7%-ное увеличение массы в диапазоне RH от приблизительно 75% до приблизительно 95%. В варианте осуществления настоящего изобретения аморфное соединение 2 демонстрирует приблизительно 8,6%-ное уменьшение массы в диапазоне RH от приблизительно 95% до приблизительно 5% и гистерезис при RH более 50%. Гистерезис может иметь место после десорбции в диапазоне RH от приблизительно 85% до приблизительно 15%.
Аморфная форма выгодна тем, что она представляет собой универсальную промежуточную форму, которую можно применять для изготовления других форм соединения 2. Например, аморфное соединение 2 можно применять для получения Формы B соединения 2, используя этилацетат в качестве растворителя при кристаллизации, и аморфное соединение 2 можно применять для получения Формы А соединения 2, используя при кристаллизации растворители, отличные от этилацетата. Если кристаллические формы соединения 2 обладают низкой растворимостью в воде, полезными материалами могут оказаться аморфные формы. При сольватировании аморфным формам не требуется энергия для разрушения кристаллической решетки (в отличие от соответствующих кристаллических форм), и поэтому они лучше подходят для приготовления фармацевтических композиций с более высокой растворимостью и с более высокой биодоступностью.
Кристаллические Формы A и B и аморфная форма описаны выше в связи с указанием данных XRPD, данных DSC, данных TGA и/или данных 1Н ЯМР (и мольных процентов сольвата в случае Формы В). Следует понимать, что эти формы можно охарактеризовать каждым набором данных индивидуально или комбинацией одного или более наборов данных.
Следует понимать, что положения пиков могут в небольшой степени меняться в зависимости от того, какой аппарат применяют для анализа образца. Поэтому все определения полиморфов, которые ссылаются на положения пиков с указанием значений °2θ, подразумевают их вариации в пределах ±0,2°2θ. Если не указано иначе (как например, в тех таблицах, где значения даны с указанием плюс-минус-вариаций), значения °2θ для положений пиков даны с вариациями, составляющими ±0,2°2θ.
Выгодным образом кристаллическая Форма A, как она описана в настоящем документе, кристаллическая Форма B, как она описана в настоящем документе, и/или аморфная форма, как она описана в настоящем документе, соединения 2 могут показывать дополнительные улучшенные свойства, такие как улучшенные биодоступность, растворимость, гигроскопичность, скорость растворения, профиль безопасности, стабильность (устойчивость к теплу, воздуху, давлению, свету), совместимость с эксципиентами в фармацевтическом препарате, (более высокую) точку плавления, плотность, твердость, более продолжительное ингибирование DβΗ, большую степень ингибирования DβΗ и/или более высокую периферическую селективность, когда их применяют в лекарственном средстве, по сравнению с другими формами соединения 2. Также выгодным образом кристаллическая Форма A, как она описана в настоящем документе, кристаллическая Форма B, как она описана в настоящем документе, и/или аморфная форма, как она описана в настоящем документе, соединения 2 могут показывать дополнительные улучшенные свойства, такие как стабильность при хранении, фильтруемость в технологическом процессе (размеры кристаллов, распределение частиц по размерам), технологичность (например, отсутствие налипания на оборудование), легкость высушивания, чистоту и величину выхода и/или смачиваемость (в терминах технологии изготовления) по сравнению с другими формами соединения 2.
Согласно другому аспекту настоящего изобретения предоставлен способ очистки кристаллической Формы A соединения 2, который включает в себя перекристаллизацию гидрохлорида (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона по меньшей мере в одном органическом растворителе. Предпочтительно, указанный органический растворитель представляет собой смесь толуола и метанола. В предпочтительном варианте осуществления настоящего изобретения толуол и метанол присутствуют в этой смеси в соотношении 62:38 по массе. В варианте осуществления настоящего изобретения органический растворитель отгоняют и заменяют толуолом.
Подходящим образом указанный способ очистки дополнительно включает в себя преобразование гидрохлорида (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона в (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тион. В варианте осуществления настоящего изобретения преобразование гидрохлорида (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона осуществляют, применяя гидроксид щелочного металла. Предпочтительно, указанный гидроксид щелочного металла представляет собой гидроксид натрия. В варианте осуществления настоящего изобретения указанное преобразование проводят в смеси метанола и воды.
Выгодным образом чистота (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона, полученного с применением способа очистки согласно настоящему изобретению, составляет, по меньшей мере, 95% (предпочтительно, по меньшей мере, 98%, наиболее предпочтительно ≥99,0%).
Согласно другому аспекту настоящего изобретения предоставлено применение Формы A, как она описана в настоящем документе, Формы B, как она описана в настоящем документе, или аморфной формы, как она описана в настоящем документе, 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона и одного или более фармацевтически приемлемых носителей или эксципиентов.
Фармацевтический препарат может также включать в себя по меньшей мере один другой активный фармацевтический ингредиент.
В другом аспекте настоящее изобретение также предоставляет способы лечения расстройств, при которых терапевтически полезно уменьшение гидроксилирования дофамина до норадреналина, которое включает в себя введение млекопитающему, нуждающемуся в этом, эффективного количества Формы А, как она описана в настоящем документе, Формы B, как она описана в настоящем документе, или аморфной формы, как она описана в настоящем документе, 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона.
Согласно другому аспекту настоящего изобретения предоставлено применение Формы А, как она описана в настоящем документе, Формы B, как она описана в настоящем документе, или аморфной формы, как она описана в настоящем документе, 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона при изготовлении лекарственного средства для лечения тревожных расстройств, мигреней, сердечно-сосудистых расстройств, гипертензии, хронической или застойной сердечной недостаточности, стенокардии, аритмий и расстройств кровообращения, таких как феномен Рейно (виброболезнь).
В вышеуказанных способах Форма А как она описана в настоящем документе, Форма B, как она описана в настоящем документе, или аморфная форма, как она описана в настоящем документе, 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона предоставлены для применения при лечении тревожных расстройств, мигреней, сердечно-сосудистых расстройств, гипертензии, хронической или застойной сердечной недостаточности, стенокардии, аритмий и расстройств кровообращения, таких как феномен Рейно (виброболезнь).
Согласно другому аспекту настоящего изобретения предоставлено применение Формы A, как она описана в настоящем документе, Формы B, как она описана в настоящем документе, или аморфной формы, как она описана в настоящем документе, 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона при изготовлении лекарственного средства для ингибирования DβΗ.
Согласно другому аспекту настоящего изобретения, предоставлена Форма A, как она описана в настоящем документе, Форма B, как она описана в настоящем документе, или аморфная форма, как она описана в настоящем документе 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона для применения при ингибировании DβΗ.
В вышеуказанных аспектах, Форму A, как она описана в настоящем документе, Форму B, как она описана в настоящем документе, или аморфную форму, как она описана в настоящем документе 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона, можно применять в комбинации, по меньшей мере, с одним другим активным фармацевтическим ингредиентом.
В другом аспекте настоящее изобретение также предоставляет способы лечения расстройств, при которых терапевтически полезно уменьшение гидроксилирования дофамина до норадреналина, которое включает в себя введение млекопитающему, нуждающемуся в этом, эффективного количества Формы А, как она описана в настоящем документе, Формы B, как она описана в настоящем документе, или аморфной формы, как она описана в настоящем документе, 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона.
В другом аспекте настоящее изобретение также предоставляет способы лечения по одному или более из следующих показаний: тревожных расстройств, мигреней, сердечнососудистых расстройств, гипертензии, хронической или застойной сердечной недостаточности, стенокардии, аритмий и расстройств кровообращения, таких как феномен Рейно (виброболезнь), которое включает в себя введение млекопитающему, нуждающемуся в этом, эффективного количества Формы А, как она описана в настоящем документе, Формы B, как она описана в настоящем документе, или аморфной формы, как она описана в настоящем документе, 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона.
В вышеуказанных способах Форму A, как она описана в настоящем документе, Форму B, как она описана в настоящем документе, или аморфную форму, как она описана в настоящем документе 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона, можно вводить в комбинации, по меньшей мере, с одним другим активным фармацевтическим ингредиентом.
Комбинированное лечение или применение, описанные в настоящем документе, можно проводить с одновременным или неодновременным введением.
К тревожным расстройствам относятся, но не ограничиваются ими, генерализованные тревожные расстройства, социальные тревожные расстройства, посттравматическое стрессовое расстройство, расстройство, обусловленное острой реакцией на стресс, обсессивно-компульсивные расстройства, панические расстройства, такие как панические атаки, и фобии, такие как агорафобия, социальные фобии, специфические фобии. Другие тревожные расстройства, которые можно лечить, применяя соединения согласно настоящему изобретению, можно найти на стр. 429-484 публикации American Psychiatric Association: Diagnostic and Statistic Manual of Mental Disorders, 4th edition, Text Revision, Washington, DC, American Psychiatric Association, 2000.
Термин «лечение» и его варианты, такие как «лечить» или «лечащий», используемые в настоящем документе, относятся к любому режиму, который может быть благоприятным для человека или животного, отличного от человека. Лечение может производиться в отношении существующего состояния или может быть профилактическим (превентивное лечение). Лечение может включать в себя лечебные, облегчающие или профилактические эффекты.
Краткое описание фигур
Ссылки даны на приложенные Фигуры, в которых:
Фигура 1 показывает ориентировочную индикацию для соединения 2 - линии указывают разрешенные рефлексы, соответствующие размерам элементарной ячейки и приписанной пространственной группе (P1, #1).
Фигура 2 показывает дифрактограмму XRPD кристаллической Формы A соединения 2
ПАРАМЕТРЫ:
Panalytical X-Pert Pro MPD PW3040 Pro
Рентгеновская трубка: Cu (1,54059 Å)
Напряжение: 45 кВ
Сила тока: 40 мА
Диапазон сканирования: 1,01-40,00°2θ
Размер шага: 0,017°2θ
Продолжительность сбора данных: 1940 с
Скорость сканирования: 1,2°/мин
Щель: Щель расходимости, DS перед зеркалом: 1/2°
Антирассеивающая щель падающего луча, SS: 1/4°
Время одного оборота: 0,5 с
Режим: пропускание
Обработка изображения File Monkey v3.2.3
Фигура 3 показывает термограмму TGA Формы A соединения 2
Методика: 00-350-10
Инструмент: AutoTGA 2950 V5.4A
Universal V4.4A TA Instruments
Фигура 4 показывает термограмму DSC кристаллической Формы A соединения 2
Методика: (-30)-250-10
Инструмент: DSC Q2000 V23.10 Build 79
Universal V4.4A TA Instruments
Фигура 5 показывает дифрактограмму XRPD Формы B соединения 2
ПАРАМЕТРЫ:
Panalytical X-Pert Pro MPD PW3040 Pro
Рентгеновская трубка: Cu (1,54059 Å)
Напряжение: 45 кВ
Сила тока: 40 мА
Диапазон сканирования: 1,01-39,99°2θ
Размер шага: 0,017°2θ
Продолжительность сбора данных: 1939 с
Скорость сканирования: 1,2°/мин
Щель: Щель расходимости (DS) перед зеркалом: 1/2°
Антирассеивающая щель падающего луча (SS): 1/4°
Время одного оборота: 0,5 с
Режим: пропускание
Обработка изображения - File Monkey v3.2.3
Фигура 6 показывает термограмму TGA Формы B соединения 2
Методика: 00-350-10
Инструмент: AutoTGA 2950 V5.4A
Universal V4.4A TA Instruments
Фигура 7 показывает термограмму DSC Формы B соединения 2
Методика: (-30)-250-10
Инструмент: 2920 MDSC V2.6A
Universal V4.4A TA Instruments
Фигура 8 A показывает спектр 1Н ЯМР для Формы B соединения 2
ПАРАМЕТРЫ:
в DMSO-d6 с TMS относительно TMS при 0,0 м.д.
Зонд: 5 мм VIDP
Растворитель: DMSO
Температура окружающей среды
Скорость вращения: 20 Гц
Последовательность импульсов: s2pul
Задержка релаксации: 5,000 с
Ширина импульса: 8,0 мкс (90,0°)
Время сбора данных: 2,500 с
Ширина спектра: 6400,0 Гц (16,008 м.д.)
40 сканирований
Число точек при сборе данных: 32000
Наблюдаемое ядро: H1 (399,7957232 МГц)
ОБРАБОТКА ДАННЫХ:
Уширение линий: 0,2 Гц
Размер преобразования Фурье (FT) 131072
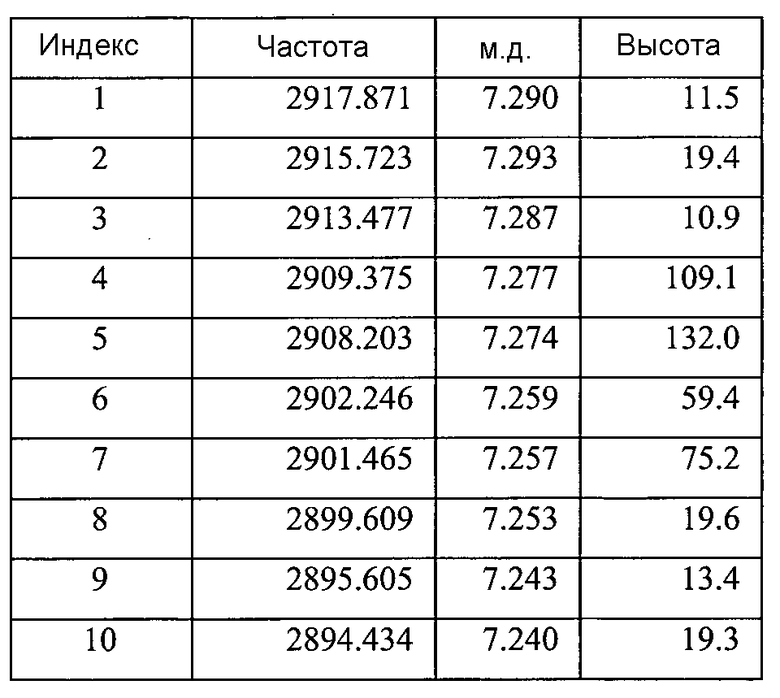
Список пиков на Фигуре 8A:
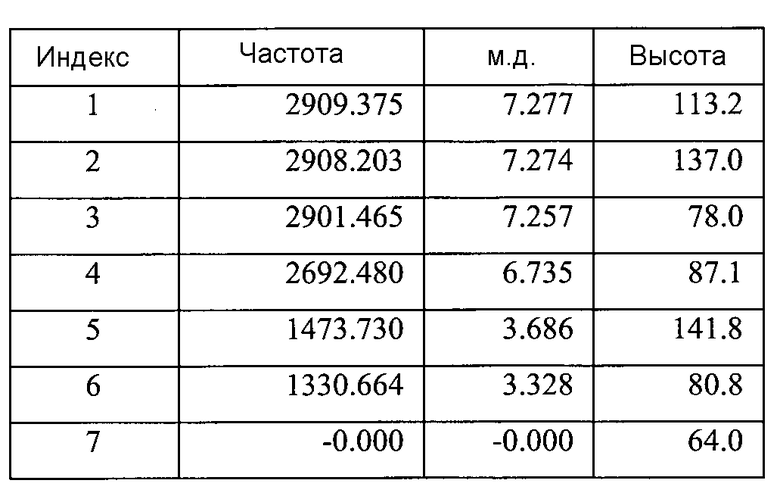
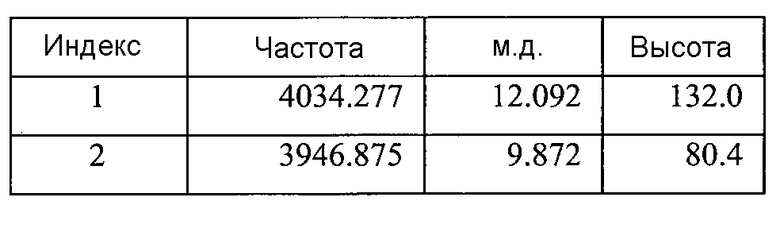
Фигура 8B показывает увеличенный фрагмент спектра 1Н ЯМР для Формы B соединения 2 (как показано на Фигуре 8A)
Параметры: как для Фигуры 8A
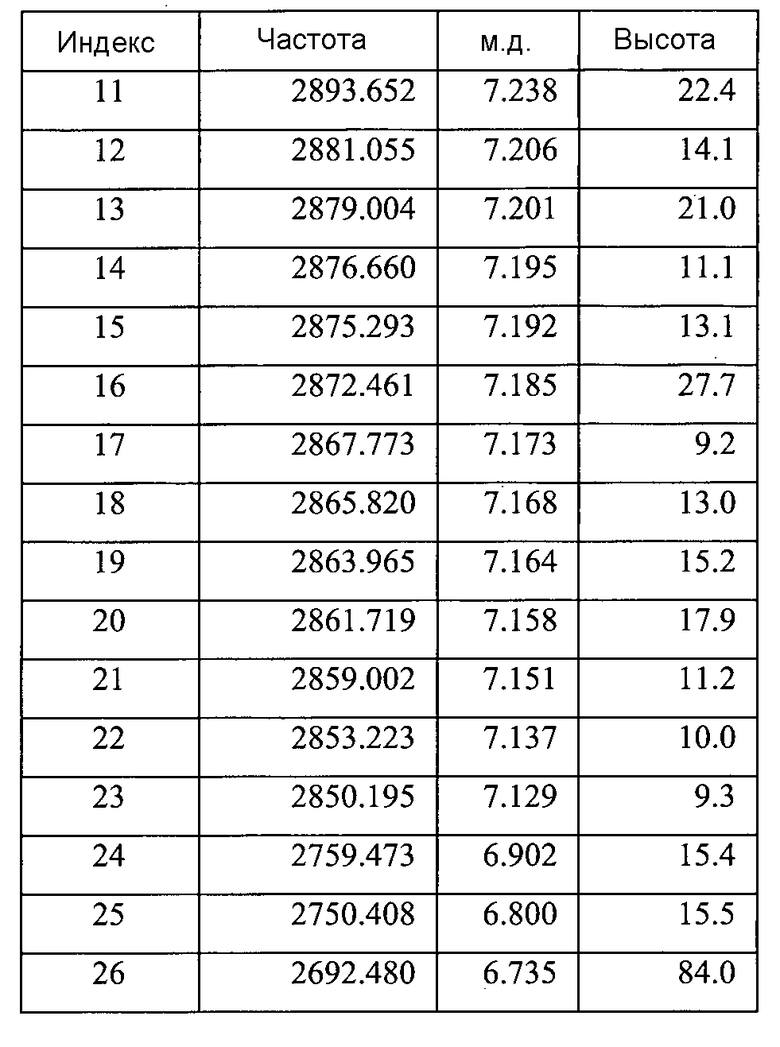
Список пиков на Фигуре 8B:
Фигура 8C показывает увеличенный фрагмент спектра 1Н ЯМР для Формы B соединения 2 (как показано на Фигуре 8A)
Параметры: как для Фигуры 8A
Список пиков на Фигуре 8C:
(продолжение)
Фигура 8D показывает увеличенный фрагмент спектра 1Н ЯМР для Формы B соединения 2 (как показано на Фигуре 8A)
Параметры: как для Фигуры 8A
Список пиков на Фигуре 8D:
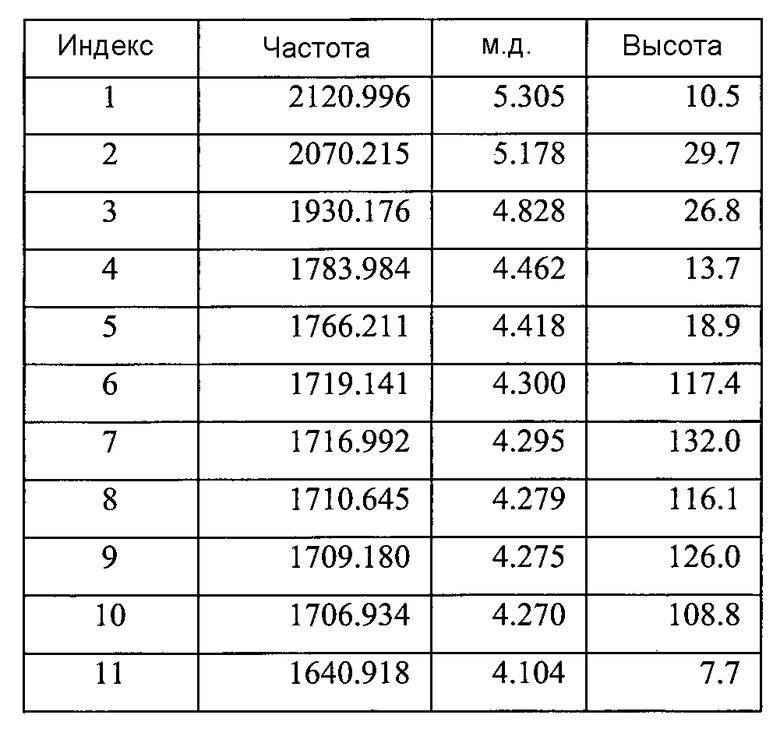
(продолжение)
Фигура 8E показывает увеличенный фрагмент спектра 1Н ЯМР для Формы B соединения 2 (как показано на Фигуре 8A)
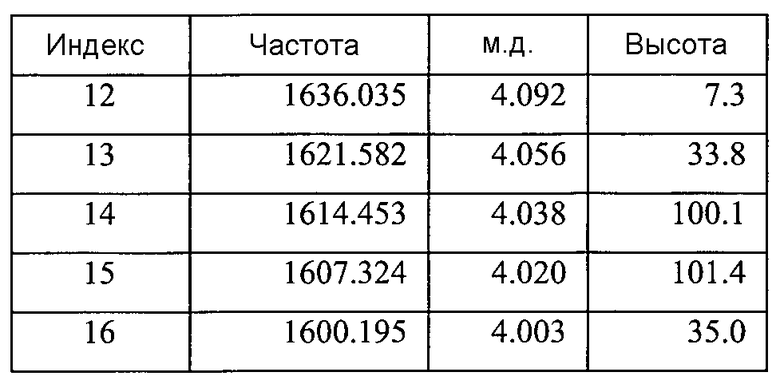
Параметры: как для Фигуры 8A
Список пиков на Фигуре 8E:
(продолжение)
Фигура 9 показывает XRPD соединения 2 в аморфной форме
Фигура 10 показывает термограмму TGA соединения 2 в аморфной форме
Методика: 00-350-10
Инструмент: TGA Q5000 V3.3 Build 250
Universal V4.4A TA Instruments
Фигура 11 показывает анализ циклической дифференциальной сканирующей калориметрии соединения 2 в аморфной форме - цикл 1
Методика: (-50)-(70-(-50)-250)-20
Инструмент: 2920 MDSC V2.6A
Universal V4.4A TA Instruments
Фигура 12 показывает анализ циклической дифференциальной сканирующей калориметрии соединения 2 в аморфной форме - цикл 2
Методика: (-50)-(70-(-50)-250)-20
Инструмент: 2920 MDSC V2.6A
Universal V4.4A TA Instruments
Фигура 13 показывает дифрактограмму XRPD материала C соединения 2
ПАРАМЕТРЫ:
Bruker Discovery D8
Рентгеновская трубка: Cu (1,54059 Å)
Диапазон сканирования: 2,12-37,00°2θ
Размер шага: 0,02°2θ
Продолжительность сбора данных: 900 с
Обработка изображений - File Monkey v3.2.3
Фигура 14 показывает дифрактограмму XRPD материала D соединения 2
ПАРАМЕТРЫ:
INEL XRG-3000
Рентгеновская трубка: 1,54187000 Å
Напряжение: 40 кВ
Сила тока: 30 мА
Размер шага: приблизительно 0,03°2θ
Продолжительность сбора данных: 300 с
Вращающийся капилляр
Обработка изображений - File Monkey v3.2.3
Подробное описание
Анализ, выполненный авторами настоящего изобретения, показал, что Форма А представляет собой несольватированный материал с низкой гигроскопичностью, плавящийся при ~187,9-192,2°C, а Форма B представляет собой нестехиометрический этилацетатный сольват. Данные для обеих форм соответствовали материалам, состоящим, главным образом, из единственной кристаллической фазы. Форма А была охарактеризована с использованием рентгеновской порошковой дифракции (XRPD), термогравиметрического анализа (TGA) и дифференциальной сканирующей калориметрии (DSC). Форма B была охарактеризована с использованием XRPD, TGA, DSC и спектроскопии 1H ЯМР.
Форма A оставалась стабильной кристаллической формой после интенсивного механического воздействия и интенсивного воздействия водяным паром.
Для облегчения лучшего понимания настоящего изобретения ниже приведены примеры определенных аспектов некоторых вариантов его осуществления. Однако приведенные примеры не следует понимать как ограничивающие или определяющие объем настоящего изобретения каким бы то ни было образом.
Экспериментальная часть
Экспериментальные методики
Получение соединения 2
Получено шесть партий соединения 2 (обозначенных как партии 1, 2, 3, 4, 5 и 6). Исходные материалы получали по приведенным ниже протоколам экспериментов.
Партия 1 (форма A)
К суспензии (R)-5-(2-аминоэтил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона (6,23 г, 20 ммоль) в смеси дихлорметана (DCM - 40 мл) и метанола (40,0 мл) добавляли бензальдегид (2,230 мл, 22,00 ммоль). К прозрачному раствору, полученному в результате этого, порциями добавляли цианоборгидрид натрия (1,9 г, 28,7 ммоль) при 20-25°C, чтобы избежать интенсивного вспенивания, и раствор перемешивали при 20-25°C в течение 40 ч. Раствор гасили при 20-25°C, добавляя 1 н. HCl (35 мл), нейтрализовывали, добавляя 3 н. NaOH (35 мл), и смесь экстрагировали DCM (200 мл). Органическую фазу промывали раствором соли, сушили (MgSO4) и выпаривали досуха. Маслянистый осадок кристаллизовали из 2-пропанола (40 мл) при 20-25°C в течение выходных дней. Кристаллы собирали, промывали 2-пропанолом и сушили, получая 5,2 г неочищенного продукта. Перекристаллизация из смеси 2-пропанола с DCM не смогла удалить все загрязняющие примеси. Собирали весь материал, выпаривали с диоксидом кремния, наносили на колонку, элюировали этилацетатом (EA) с метанолом (9:1→4:1), собирая фракции 8-25, получая 3,8 г. Перекристаллизовывали из 2-пропанола (45 мл) и DCM (120 мл, удаляли на роторном испарителе), получая 2,77 г начальной партии (a) (98,3% HPLC площади) и 0,3 г отфильтрованного нерастворенного вещества, которое по результатам ТСХ представляло собой истинный продукт. Начальную партию (a) перекристаллизовывали из 2-пропанола (35 мл) и DCM (95 мл удаляли на роторном испарителе), получая 2,51 г начальной партии (b) (98,3% площади HPLC). Вещество, объединенное с вышеуказанным нерастворенным материалом, перекристаллизовывали из ацетонитрила (200 мл, сливая на ледяную баню), получая 2,57 г начальной партии (c) (HPLC 98,8% площади). Перекристаллизация из ацетонитрила (180 мл, охлаждение до 15°C) давала 2,25 г партии 1 (99,2% площади HPLC), т.пл. 190-192°C.
Партия 2 (Форма A)
В тетрагидрофуране (300 мл), нагревая до кипения с обратным холодильником, растворяли (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тион (12 г, 29,9 ммоль); раствор охлаждали до 5-10°C и медленно (приблизительно 10 мин) с перемешиванием добавляли воду (510 мл). Смесь перемешивали в течение 1 ч, собирали твердое вещество, промывали водой и сушили, получая 11,73 г продукта, содержавшего, по данным HPLC, 1% гидрохлорида (R)-5-(2-аминоэтил)-1-(6,8-дифторхроман-3-ил)-1,3-дигидроимидазол-2-тиона и 1% менее полярных загрязняющих примесей. Продукт растворяли в тетрагидрофуране (300 мл) с нагреванием до кипения с обратным холодильником, добавляли 2-пропанол (150 мл), раствор концентрировали приблизительно до 100 мл (происходила кристаллизация) и перемешивали на льду в течение 1,5 ч. Собирали твердое вещество, промывали 2-пропанолом и сушили, получая 11,2 г продукта, содержавшего, по данным HPLC, 0,8% гидрохлорида (R)-5-(2-аминоэтил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона и 0,5% менее полярных загрязняющих примесей. Продукт растворяли в тетрагидрофуране (300 мл) с нагреванием до кипения с обратным холодильником, добавляли 2-пропанол (150 мл), раствор концентрировали приблизительно до 100 мл (происходила кристаллизация) и перемешивали при 20-25°C в течение 1 ч. Собирали твердое вещество, промывали 2-пропанолом и сушили, получая (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тион (10,22 г, 25,5 ммоль, выход 85%).
Партия 3 (форма B)
К (R)-5-(2-аминоэтил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиону (2,36 г, 7,58 ммоль) в смеси метанола (15,00 мл) с дихлорметаном (15 мл) добавляли бензальдегид (0,845 мл, 8,34 ммоль). К прозрачному раствору, полученному в результате этого, порциями добавляли цианоборгидрид натрия (0,702 г, 10,61 ммоль) при 20-25°C, стараясь избегать интенсивного вспенивания, и раствор перемешивали при 20-25°C в течение 40 ч. Раствор гасили при 20-25°C, добавляя 1 н HCl (12 мл), нейтрализовывали, добавляя 3 н NaOH (12 мл), и смесь экстрагировали DCM (100 мл). Органическую фазу промывали раствором соли, сушили (MgSO4) и выпаривали досуха. Остаток очищали на колонке со смесью EA-MeOH (9:1) в качестве элюэнта, собирали фракции, концентрировали приблизительно до 20 мл и охлаждали на льду. Осадок собирали, промывали смесью этилацетата с петролейным эфиром (1:1) и сушили на воздухе, получая (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тион (1,55 г, 3,86 ммоль, выход 50,9%).
Партия 4 (Форма A)
В 500-мл колбу, подготовленную для перегонки при атмосферном давлении, добавляли (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тион (20 г, 49,8 ммоль) и тетрагидрофуран (400 мл), образуя суспензию. Суспензию нагревали до полного растворения (61°C), после чего фильтровали. Затем раствор, полученный в результате этого, нагревали до 66°C до завершения перегонки. С той же скоростью, с какой собирали дистиллят, добавляли смесь воды (125 мл) с 2-пропанолом (125 мл). Перегонку продолжали до тех пор, пока не собирали 400 мл дистиллята. После сбора ~320 мл дистиллята начинали кристаллизацию. Суспензию охлаждали до 20°C и выдерживали в течение 45 мин для созревания осадка, после чего фильтровали, промывали добавочным 2-пропанолом (80 мл) и затем сушили в вакууме при 50°C в течение ночи, получая (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тион (18,79 г, 94%).
Партия 5 (Форма A)
К смеси метанола (66 л) с водой (10 л) при 20°C добавляли очищенный гидрохлорид (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона (4,37 кг, 9,98 моль), образуя суспензию. Затем реакционную смесь нагревали до 67°C для полного растворения, после чего одной порцией добавляли 1 н гидроксид натрия (10,48 л, 10,48 моль, 1,05 экв). Реакционную смесь опять доводили до 67°C и выдерживали при 67°C в течение 30 мин. Затем реакционную смесь охлаждали до 20°C и инкубировали для созревания осадка при 20°C, по меньшей мере, в течение 30 мин. После этого реакционную смесь фильтровали и слой на фильтре промывали водным метанолом (1:1 об/об, 20 л), отсасывали в течение 15 мин и затем сушили при 45°C в вакууме, получая (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тион (3,855 кг, 96%) в виде светло-коричневого кристаллического твердого вещества.
Партия 6 (Форма A)
В 250-л реактор загружали 10,22 кг очищенного гидрохлорида (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона, добавляли 113,0 кг метанола и реакционную смесь нагревали до 47,3°C с перемешиванием при 120 об/мин. Прозрачный раствор коричневого цвета, полученный в результате этого, в теплом состоянии фильтровали через фильтр GAF в 200-л бак, ополаскивая фильтр 8,0 кг метанола. Реактор очищали 45,0 кг метанола, профильтрованный метанольный раствор переносили из 200-л бака в 250-л реактор и раствор нагревали до 46,3°C, перемешивая со скоростью 120 об/мин. При этой температуре в течение 10 мин добавляли 23,5 кг воды, раствор нагревали до 64,3°C в течение 60 мин (с обратным холодильником) и при 64,5-65,3°C в течение 90 мин добавляли 26,1 кг раствора, содержавшего 1,2 кг гидроксида натрия в 30,6 кг воды (с обратным холодильником; экзотермический эффект). Суспензию бежевого цвета, полученную в результате этого, в течение 45 мин перемешивали при 65,2-66,9°C, охлаждали до 58-60°C и подвергали pH-контролю (pH 11). В течение 1 ч 55 мин суспензию охлаждали до 24,8°C и в течение 13 ч перемешивали при этой температуре. Суспензию переносили в центрифугу (Lanz Anliker PP20, осуществляющую фильтрование через матерчатый фильтр) и центрифугировали одной порцией. В 250-л реактор загружали 20,0 кг воды и 16,0 кг метанола и перемешивали при 23,0°C в течение 10 мин. Слой на фильтре промывали этой метанольной смесью, влажный продукт (8,84 кг) переносили в лотковую сушилку и сушили при 52,2°C и 290-1 мбар в течение 68 ч 44 мин, получая 8,45 кг (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона.
В контексте настоящей заявки на патент термин «очищенный гидрохлорид (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона» означает, что указанное соединение имеет степень чистоты не менее 95% (предпочтительно, не менее 98%, наиболее предпочтительно не менее 99,0%).
A. Получение очищенного гидрохлорида (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона:
Стадия 1: Неочищенный (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тион («неочищенное соединение 2»)
В 250-л реактор загружали 12,25 кг гидрохлорида (R)-5-(2-аминоэтил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона, добавляли 114,82 кг 2-пропанола и смесь перемешивали с максимальной скоростью (140 об/мин). Через капельную воронку добавляли 1,856 кг бензальдегида, а затем 3,945 кг триацетоксиборгидрида натрия в пяти порциях, добавленных при Ti=20-25°C в следующем порядке:
1/5 часть бензальдегида (приблизительно 0,36 л);
перемешивание в течение 5-10 мин;
добавление 1/5 части триацетоксиборгидрида натрия (приблизительно 0,79 кг);
перемешивание в течение 20-30 мин;
повторение этой процедуры 4 раза.
Отбирали образец для внутрипроизводственного контроля («in process control», IPC - только для информации) и через капельную воронку добавляли еще 1,856 кг бензальдегида, а затем 3,946 кг триацетоксиборгидрида натрия в пяти порциях, добавленных при Ti=20-25°C в следующем порядке:
1/5 часть бензальдегида (приблизительно 0,36 л);
перемешивание в течение 5-10 мин;
добавление 1/5 части триацетоксиборгидрида натрия (приблизительно 0,79 кг);
перемешивание в течение 20-30 мин;
повторение этой процедуры 4 раза.
Смесь оставляли на 60 мин при Ti=22,1°C.
В 250-л реактор загружали 79,9 кг воды и перемешивали со скоростью 140 об/мин, затем добавляли 4,48 кг гидроксида натрия и смесь перемешивали со скоростью 140 об/мин при Ti=24,8°C в течение 25 мин, получая прозрачный раствор (экзотермический эффект). В реакционную смесь добавляли раствор гидроксида натрия, перемешивали с максимальной скоростью (170 об/мин) в течение 90 мин при Ti=22,1-22,9°C (слабый экзотермический эффект и выделение H2 при начале добавления), получая суспензию светло-коричневого цвета. Суспензию перемешивали в течение 60 мин при Ti=22,9-22,1°C со скоростью 120 об/мин, охлаждали до Ti=3,2°C и перемешивали в течение 16,5 ч при этой температуре со скоростью 120 об/мин. Суспензию переносили в центрифугу и центрифугировали одной порцией.
В 250-л реактор загружали 19,3 кг 2-пропанола и 24,3 кг воды и охлаждали до Ti=3,5°C. Слой на фильтре промывали охлажденным раствором 2-пропанола в воде, влажный продукт (18,4 кг) переносили в лотковую сушилку и сушили в течение 2-3 дней при Te=55°C, p=400→1 мбар в течение 67 ч 45 мин). Сухой продукт (14,08 кг - неочищенное соединение 2) переносили в барабан с двойным полимерным вкладышем, гомогенизировали в течение 1 часа при вращении со скоростью 6 об/мин на колесном смесителе и хранили при температуре окружающей среды в атмосфере аргона до дальнейшей переработки.
Стадия 2: Неочищенный гидрохлорид (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона
В 400-л реактор загружали 280,0 кг воды и перемешивали при Ti=16,0→21,0°C со скоростью 120 об/мин, затем при Ti=21-21,1°C добавляли 14,02 кг неочищенного соединения 2, образуя суспензию. К этой суспензии тремя порциями добавляли 5,28 кг 37%-ной HCl при Ti=21,1-22,0°C в течение 23 мин (слабый экзотермический эффект), смесь нагревали до Ti=81,5°C в течение 120 мин и перемешивали при Ti=82,0°C в течение 60 мин, затем при перемешивании охлаждали до Ti=47,1°C в течение 150-180 мин со скоростью охлаждения 0,2-0,25°C/мин и перемешивали с умеренной скоростью при Ti=47,0°C в течение 60 мин. Суспензию центрифугировали и слой на фильтре промывали 64,5 кг воды. Влажный продукт (33,5 кг) переносили в лотковую сушилку и сушили при Te=48→53°C; p=300→1 мбар в течение 68 ч 20 мин. Сухой продукт (12,40 кг) переносили в барабан с двойным полимерным вкладышем, гомогенизировали в течение 3 часов при вращении со скоростью 13 об/мин на колесном смесителе, получая 12,4 кг (81%) неочищенного гидрохлорида (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона. Хранили при температуре окружающей среды в атмосфере аргона до дальнейшей переработки.
Стадия 3: Очищенный гидрохлорид (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона
В 400-л реактор загружали 12,3 кг неочищенного гидрохлорида (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона, добавляли 160,5 кг толуола и смесь перемешивали со скоростью 130 об/мин. Добавляли 98,0 кг метанола и смесь нагревали до Ti=62°C в течение 1 ч и затем медленно нагревали до кипения с обратным холодильником (Ti=65,9°C). Растворитель удаляли отгонкой (17,5-21 л/ч в течение 6-7 ч), одновременно заменяя его толуолом (17,5-21 л/час в течение 6-7 ч). Реакционную смесь перемешивали в течение 45 мин со скоростью 120 об/мин при Ti=63,9°C, в результате чего получали светло-серую суспензию. Суспензию охлаждали до Ti=23,0°C в течение 90 мин и перемешивали при этой температуре в течение 10 ч (в течение ночи).
Суспензию переносили в центрифугу (Lanz Anliker PP20, осуществляющую фильтрование через матерчатый фильтр) и центрифугировали одной порцией, слой на фильтре промывали смесью 48,0 кг толуола и 5,0 кг метанола (которые предварительно перемешивали в реакторе при Ti=20-25°C в течение 5-20 мин). Влажный продукт (17,8 кг) пропускали через барабан с двойным полимерным вкладышем в атмосфере аргона, переносили в лотковую сушилку (используя пластмассовые вкладыши, чтобы избежать контакта с металлом) и сушили при Ti=48-53°C, p=300→1 мбар в течение 67 ч, а затем дополнительно сушили при Te=50°C→53°C; p=300→1 мбар в течение 47 ч 20 мин. Продукт (10,286 кг) выгружали и помещали в 30-л барабан с двойным пластмассовым вкладышем. Сухой продукт (очищенный гидрохлорид (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1H-имидазол-2(3H)-тиона) гомогенизировали на колесном смесителе (7 об/мин) в течение 2 ч и хранили при температуре окружающей среды в атмосфере аргона до дальнейшей переработки. (HPLC-чистота ≥ 99,0%)
B. Получение соединения 2 в виде аморфного материала
Три образца соединения 2 в виде аморфного материала получали посредством лиофилизации, исходя из ~100 мг, ~500 мг и ~1 г соединения 2 партии 5. Растворы исходного материала готовили в 1,4-диоксане при повышенной температуре (~70-71°C) в концентрации приблизительно 7 мг/мл. После этого растворы фильтровали в горячем виде, используя 0,2-мкм нейлоновый фильтр и давая им возможность медленно охлаждаться до температуры окружающей среды после выключения нагревателя. Растворы, охлажденные до температуры окружающей среды, замораживали на бане с сухим льдом в ацетоне и переносили в лиофильную сушилку с вакуумным насосом, установленную на -50°С. Образцы, приготовленные из ~100 мг, ~500 мг исходного материала, сушили в течение приблизительно 2 дней. Образец, приготовленный из ~1 г исходного материала, сушили в течение ~5 дней. Твердые вещества, полученные после высушивания, хранили в морозильной камере над поглотителем влаги.
Подробности экспериментов
1. Скрининг полиморфов - среднемасштабные эксперименты
Эксперименты по скринингу полиморфов проводили, главным образом, используя партию 5 соединения 2 в качестве исходного материала. Эксперименты с дополнительной кристаллизацией проводили, используя три образца аморфного материала, образованного при скрининге (образцы № 1, 2 и 3). Для экспериментов обычно использовали ~10-80 мг. Как правило, произведенные твердые вещества выделяли вакуумным фильтрованием и обследовали в поляризованном свете.
a. Эксперименты с испарением
Растворы исходного материала получали при температуре окружающей среды посредством добавления данной системы растворителя для растворения твердых веществ. Как правило, растворы фильтровали, используя 0,2-мкм нейлоновый фильтр. Растворители удаляли в роторном испарителе при температуре окружающей среды или при повышенной температуре («роторное испарение» - rotary evaporation, RE), или давая возможность испаряться при температуре окружающей среды либо из открытого флакона («быстрое испарение» - fast evaporation, FE), либо из флакона, закрытого алюминиевой фольгой с отверстиями, проколотыми булавкой («медленное испарение» - slow evaporation, SE).
b. Эксперименты с быстрым, медленным и мгновенным охлаждением
Образцы исходного материала приводили в контакт с данной системой растворителя и подвергали воздействию повышенной температуры на масляной бане. Растворы, полученные в результате этого, обычно еще горячими фильтровали через 0,2-мкм нейлоновый фильтр. После этого растворы либо извлекали из нагревателя, давая возможность быстро охлаждаться до температуры окружающей среды («быстрое охлаждение» - fast cooling, FC), либо, отключив нагреватель, оставляли на масляной бане для медленного охлаждения до температуры окружающей среды («медленное охлаждение», slow cooling, SC), либо помещали на баню с сухим льдом в ацетоне для мгновенного охлаждения (crash cooling, CC). Если твердые вещества не образовывали растворов, их, как правило, обрабатывали ультразвуком и/или помещали в холодильник или в морозильную камеру.
c. Эксперименты с суспензией
Растворы готовили, добавляя растворитель или смесь растворителей к исходному материалу с избытком присутствующего твердого вещества. Затем смеси интенсивно перемешивали в закрытых флаконах - либо при температуре окружающей среды, либо при некоторой заданной температуре. Для перемешивания при температуре ниже температуры окружающей среды добавляли охлажденный растворитель и образец немедленно переносили в морозильную камеру. Через определенное время выделяли твердые вещества.
d. Эксперименты с обработкой водяным паром
Образцы исходного материала подвергали воздействию ~85% и ~97% относительной влажности при температуре окружающей среды и ~75% относительной влажности при ~40°C в течение указанного времени.
e. Эксперименты с обработкой парами органических растворителей
Образцы исходного материала подвергали воздействию паров конкретного органического растворителя в течение заданного времени, помещая открытые флаконы с испытуемыми твердыми веществами в 20-мл флаконы, содержавшие указанный растворитель. Эксперименты с обработкой парами органических растворителей проводили при температуре окружающей среды.
f. Эксперименты с осаждением антирастворителем
Растворы исходного материала готовили при температуре окружающей среды или при повышенной температуре, добавляя минимальное количество данного растворителя (solvent - S). После этого растворы либо фильтровали (возможно, в горячем состоянии) непосредственно в избыток антирастворителя (antisolvent - AS) либо антирастворитель быстро добавляли к профильтрованным растворам. Осажденные твердые вещества либо немедленно выделяли либо интенсивно перемешивали. Если твердые вещества не образовывали растворов, их, как правило, обрабатывали ультразвуком и/или помещали в холодильник или в морозильную камеру.
g. Эксперименты с диффузией паров
Растворы исходного материала готовили при температуре окружающей среды, добавляя минимальное количество соответствующего растворителя. Как правило, образцы фильтровали, используя 0,2-мкм нейлоновый фильтр. Открытые флаконы с профильтрованными растворами помещали в 20-мл флаконы, содержавшие соответствующий антирастворитель. Эти 20-мл флаконы закрывали крышками и оставляли в неподвижном состоянии.
h. Эксперименты с механическим стрессом
Образцы исходного материала помещали в шаровую мельницу Retsch и размалывали в двух пятиминутных циклах без растворителя (сухой размол) или с небольшим количеством добавленного растворителя (влажный размол), соскабливая твердые вещества между циклами. Десятиминутные циклы применяли для размола отобранного образца без добавления растворителя.
i. Эксперименты с тепловым стрессом
Образцы аморфного материала помещали в нагревательные печи при температурах, установленных ниже или выше температуры стеклования, или интенсивно перемешивали на шейкере при повышенной температуре в течение заданного времени.
j. Эксперименты с медленным охлаждением расплава
Образцы аморфного материала нагревали на горячей плитке при температуре выше температуры стеклования. Затем образцу давали возможность медленно охлаждаться до температуры окружающей среды с выключенным нагревателем.
2. Скрининг полиморфов - эксперименты с луночными планшетами (без подтверждения соответствия cGMP - клиническим Правилам организации производства и контроля качества лекарственных средств)
Микроаналитические эксперименты проводили, используя 96-луночный планшет. Эти эксперименты выполняли в условиях, не имевших подтверждения соответствия правилам cGMP. Полученные образцы обследовали в поляризованном свете.
Готовили запасные растворы партии 5 соединения 2 в гексафторизопропаноле (~22 мг/мл). В каждую лунку микропланшета добавляли по 100 мкл запасного раствора (~2,2 мг соединения 2 на лунку). Каждый второй и третий растворитель добавляли в количестве 25 мкл. Когда третий растворитель не применяли, тогда второй растворитель добавляли в количестве 50 мкл. Быстрое испарение производилось из лунок, оставленных открытыми. Для экспериментов с медленным испарением лунки накрывали алюминиевой фольгой, в которой были проколоты отверстия (по одному на лунку).
В контексте настоящей спецификации «комнатная температура» означает то же самое, что и «температура окружающей среды». Соответствующая комнатная температура является температурой в диапазоне от приблизительно 10°C до приблизительно 35°C, предпочтительно в диапазоне от приблизительно 15°C до приблизительно 30°C, более предпочтительно в диапазоне от приблизительно 20°C до приблизительно 25°C.
Инструментальная техника
3. Рентгеновская порошковая дифракция (XRPD)
a. Inel
Избранные дифрактограммы XRPD получали на дифрактометре Inel XRG-3000. Падающий луч излучения Cu Kα получали с помощью острофокусной трубки и параболического многослойного зеркала. Перед анализом проводили измерения с кремниевым стандартом (NIST SRM 640c) для уточнения положения пика Si(111). Фрагмент образца помещали в тонкостенный стеклянный капилляр и использовали отсечение луча для минимизирования фонового сигнала воздуха. Дифрактограммы регистрировали в геометрии пропускания, используя программу Windif v. 6.6 и криволинейный позиционно-чувствительный детектор Equinox со 120-градусным диапазоном 2θ. Параметры сбора данных для каждой дифрактограммы указаны выше в разделе «Краткое описание фигур».
b. Bruker
Избранные дифрактограммы XRPD получали, используя дифрактометр Bruker D8 DISCOVER и Bruker′c General Area-Detector Diffraction System (GADDS, v. 4.1.20). Падающий луч излучения Cu Kα получали с помощью длинной острофокусной трубки (40 кВ, 40 мА), параболического многослойного зеркала и двойного точечного (0,5 мм) коллиматора. Перед анализом проводили измерения с кремниевым стандартом (NIST SRM 640c) для уточнения положения пика Si(111). Фрагмент образца помещали между пленками толщиной 3 мкм, формируя портативный дискообразный образец. Подготовленный образец помещали в держатель, закрепленный для стадии трансляции. Видеокамеру и лазер размещали так, чтобы интересующая область пересекалась с падающим лучом в геометрии пропускания. Падающий луч сканировали и/или растрировали для оптимизации сбора данных и статистики ориентации. Для минимизирования фонового сигнала воздуха использовали отсечение луча. Дифракционную картину регистрировали двумерным детектором HISTAR™, расположенным в 15 см от образца, и обрабатывали с помощью GADDS. Интенсивность на GADDS-изображениях дифрактограмм интегрировали и представляли как функцию 2θ. Параметры сбора данных для каждой дифрактограммы указаны выше в разделе «Краткое описание фигур».
c. Bruker (с держателем луночных планшетов)
Дифрактограммы для микропланшентых образцов получали с использованием дифрактометра Bruker D8 DISCOVER и Bruker′c General Area-Detector Diffraction System (GADDS, v. 4.1.20). Падающий луч излучения Cu Kα получали с помощью длинной острофокусной трубки (40 кВ, 40 мА), параболического многослойного зеркала и двойного точечного (0,5 мм) коллиматора. Перед анализом проводили измерения с кремниевым стандартом (NIST SRM 640c) для уточнения положения пика Si(111). Образцы размещали для анализа, закрепляя луночный планшет для стадии трансляции и перемещая каждый образец для пересечения с падающим лучом в геометрии пропускания. Падающий луч сканировали и растрировали в процессе анализа для оптимизации статистики ориентации. Для минимизирования фонового сигнала воздуха использовали отсечение луча. Дифракционную картину регистрировали двумерным детектором HISTAR™, расположенным в 15 см от образца, и обрабатывали с помощью GADDS. Интенсивность на GADDS-изображениях дифрактограмм интегрировали и представляли как функцию 2θ. Прибор работал в условиях, не имевших подтверждения соответствия правилам GMP, и результаты также ни имеют такого подтверждения. Параметры сбора данных для каждой дифрактограммы указаны выше в разделе «Краткое описание фигур».
d. PANalytical
Избранные дифрактограммы XRPD получали, используя дифрактометр PANalytical X′Pert PRO MPD с падающим лучом излучения Cu, производимого длинным острофокусным источником Optix. Для фокусировки рентгеновских лучей Cu Kα через образец и на детектор применяли эллиптическое многослойное зеркало. Перед анализом проводили измерения с кремниевым стандартом (NIST SRM 640c) для уточнения положения пика Si(111). Фрагмент образца помещали между пленками толщиной 3 мкм и анализировали в геометрии пропускания. Для минимизирования фонового сигнала, создаваемого воздухом, использовали отсекающую луч короткую антирассеивающую насадку и, как правило, атмосферу гелия. Для минимизирования расширения, обусловленного аксиальной расходимостью, использовали щели Соллера для падающего и дифракционного луча. Дифрактограммы регистрировали с помощью сканирующего позиционно-чувствительного детектора (X′Celerator), расположенного на расстоянии 240 мм от образца, используя программу Data Collector v. 2.2b. Параметры сбора данных для каждой дифрактограммы указаны выше в разделе «Краткое описание фигур», включая щель расходимости (DS) перед зеркалом и антирассеивающую щель падающего луча (SS).
a. Индексация
Индексация и уточнение структуры являются вычислительными процедурами, которые не проводят по правилам cGMP.
Индексацию рентгенограммы XRPD соединения 2 проводили с использованием проприетарного программного обеспечения. Результаты индексации верифицированы и проиллюстрированы с использованием программы CheckCell версии 11/01/04. (Пакет программ LMGP-Suite для интерпретации рентгеноструктурных экспериментов, авторами которых являются Jean Laugier и Bernard Bochu, ENSP/Laboratoire des Materiaux et du Genie Physique, BP 46. 38042 Saint Martin d′Heres, France. WWW: http://www.inpg.fr/LMGP и http://www.ccp14.ac.uk/tutorial/lmgp/)
4. Термогравиметрический анализ (TGA)
Термогравиметрические анализы проводили с использованием термогравиметрического анализатора модели Q5000 и 2950 от TA Instruments. Для температурной калибровки применяли никель и Alumel™. Каждый образец помещали в алюминиевую чашку и вносили в TG-печь. Печь нагревали в потоке азота. Параметры сбора данных показаны над каждой термограммой.
Код методики для термограммы (указанный выше в перечне фигур в разделе «Краткое описание фигур») представляет собой аббревиатуру для начальной и конечной температуры, а также для скорости нагрева; например, обозначение 25-350-10 означает «от 25°С до 350°С со скоростью 10°С/мин».
5. Корреляционный термогравиметрический инфракрасный анализ (TG-IR)
Термогравиметрический инфракрасный (TG-IR) анализ проводили на термогравиметрическом (TG) анализаторе модели 2050 от TA Instruments, сопряженном с инфракрасным спектрометром с Фурье-преобразованием (FT-IR) модели Magna-IR 560® от Thermo Nicolet, оснащенным источником среднего и дальнего ИК-излучения Ever-Glo, светоделителем из бромида калия (KBr) и ртутно-кадмиевым теллуридным детектором (MCT-A). Калибровку FT-IR спектрометра по длинам волн проводили с полистиролом, стандартами для термогравиметрической калибровки служили никель и Alumel™. Образец помещали в платиновую чашку и вносили в TG-печь. Сначала включали TG-анализатор, затем FTIR-спектрометр. TG-анализатор работал в потоке гелия, пропускаемого со скоростью 90 и 10 см3/мин для продувки и для баланса соответственно. Печь нагревали в токе азота со скоростью 20°C/мин до конечной температуры 250°С. ИК-спектры регистрировали каждые 32 сек в течение приблизительно 13 мин. Каждый ИК-спектр представляет собой наложение 32 сканирований со спектральным разрешением 4 см-1. Летучие вещества идентифицировали по базе данных High Resolution Nicolet Vapor Phase spectral library.
6. Дифференциальная сканирующая калориметрия (DSC)
Для проведения DSC использовали дифференциальный сканирующий калориметр TA Instruments модели 2920 /или/ Q2000. Для температурной калибровки применяли металлический индий, контролируемый NIST. Образец помещали в алюминиевую чашку для DSC, закрывали крышкой и записывали точную массу. Подготовленную и взвешенную алюминиевую чашку с образцом помещали на референсную сторону ячейки. Параметры сбора данных и конфигурация чашки для каждой термограммы показаны на изображении каждой термограммы.
Код методики на термограмме представляет собой аббревиатуру для начальной и конечной температуры, а также для скорости нагрева; например обозначение 25-250-10 означает «от 25°С до 250°С со скоростью 10°С/мин».
7. Циклическая дифференциальная сканирующая калориметрия (цикло-DSC)
Для измерений температуры стеклования (Tg) аморфного материала ячейку образца уравновешивали при -50°C, затем нагревали в атмосфере азота со скоростью 20°C/мин до 70°C и уравновешивали при этой температуре. После этого ячейке образца давали возможность охлаждаться и уравновешиваться при -50°C. Затем ее опять нагревали со скоростью 20°С/мин до конечной температуры 250°С. Значение Tg соответствует полувысоте в точке перехода (в точке перегиба).
8. Высокотемпературная микроскопия (HSM)
Для высокотемпературной микроскопии использовали высокотемпературный прибор Linkam FTIR 600, смонтированный на микроскопе Leica DM LP, оснащенном цветной цифровой камерой SPOT Insight™. Температурную калибровку выполняли по стандартам температур плавления Фармакопеи США. Образцы помещали на покровное стекло и накрывали вторым покровным стеклом. Каждый нагретый образец обследовали визуально, используя 20-кратный объектив со скрещенными поляризаторами и красным компенсатором первого порядка. Изображения записывали с помощью программного обеспечения SPOT (v. 4.5.9).
9. Анализ сорбции влаги
Данные о сорбции/десорбции влаги получали с использованием прибора VTI SGA-100 Vapor Sorption Analyzer. В качестве стандартов применяли NaCl и PVP. Перед анализом образцы не сушили. Величины сорбции и десорбции измеряли в диапазоне RH от 5 до 95% с инкрементом 10% при продувке азотом. В качестве критерия равновесия принимали отсутствие изменения массы, превышающего 0,0100% в течение 5 мин с максимальным временем уравновешивания, составлявшим 3 ч. Корректировку данных с учетом начальной влажности образцов не производили.
10. Спектроскопия протонного ядерного магнитного резонанса (1H ЯМР) в растворе
Все образцы получали в дейтерированном DMSO. Конкретные параметры сбора данных указаны выше в разделе «Краткое описание фигур» для Фигуры 8А.
Характеристики партий 1, 2, 3, 4, 6 и 5 соединения 2 сведены в Таблице 5.
Физическая характеризация материалов соединения 2
(продолжение)
Резкое уменьшение массы при ~257°C (начало)
Резкое уменьшение массы при ~260°C (начало)
Содержит ~0,13 моль этилацетата (на основе пиков при ~4,03 м.д., ~1,99 м.д. и ~1,18 м.д.)
Небольшие неидентифицированные пики при ~9,87 м.д., ~5,31 м.д., ~4,09 м.д., и ~3,17 м.д.a
(высокое разрешение)
(высокое разрешение)
(высокое разрешение)
(продолжение)
143,0°C - без заметных изменений
187,9°C - начало перехода твердой фазы в жидкую
189,2°C - в процессе перехода твердой фазы в жидкую
192,2°C - завершен переход твердой фазы в жидкую. Охлаждение (неконтролируемое)
35,6°C - отсутствие видимой повторной кристаллизации
~0,02%-ное увеличение массы между ~5% и ~75% RH
~0,19%-ное увеличение массы между ~75% и ~95% RH
~0,20%-ное уменьшение массы между ~95% и ~5% RH. Небольшой гистерезис между ~85% и ~45% RH после десорбции
Материалы характеризовали с помощью рентгеновской порошковой дифрактографии высокого разрешения (XRPD), термогравиметрии (TGA) и дифференциальной сканирующей калориметрии (DSC). Высокотемпературную микроскопию и анализ сорбции влаги проводили на партии 5. Партию 3 дополнительно характеризовали с помощью спектроскопии протонного ядерного магнитного резонанса (1H ЯМР). Дифрактограмму XRPD партии 5 индексировали. При проведении индексирования не предпринимали попыток определения молекулярной упаковки для подтверждения ориентировочной индексации.
Ориентировочная индексация для партии 5 показана на Фигуре 1. Пространственные группы, согласующиеся с приписанным символом экстинкции, параметры элементарной ячейки и производные величины приведены в Таблице 6.
Ориентировочное индексирование и производные величины для материалов соединения 2
(продолжение)
В целом, данные для партий 1, 4, 6 и 5 указывают, что материалы являются несольватированными и состоят, главным образом, из одной и той же твердой формы, обозначенной как Форма А. Данные для партии 3 соответствуют нестехиометрическому этилацетатному сольвату соединения 2, обозначенному как Форма В. По данным XRPD, партия 2 соответствует Форме А, однако данные TGA свидетельствуют о некоторой степени сольватации. В целом, указанные шесть партий состоят преимущественно из единственной кристаллической фазы.
Дифрактограмма XRPD, продемонстрированная партией 4, была успешно проиндексирована, что указывает на то, что этот материал состоит преимущественно из единственной кристаллической фазы (Таблица 6). Согласуемость между разрешенными положениями пиков и наблюдаемыми пиками свидетельствует о корректном определении элементарной ячейки (Фигура 1). Для углов α и β предложены два значения. Если величина угла γ составляет несколько менее 90°, тогда для α и β следует использовать острые углы. Когда угол γ слегка превышает или равняется 90°, тогда для α и β следует использовать тупые углы. Приведены как острые, так и тупые ячейки, поскольку уточненная величина угла γ составляет 90,00°, однако в пределах погрешности она может быть на несколько сотых долей градуса ниже 90°.
Термогравиметрические кривые (TGA) для партий 1, 4, 6 и 5 были сходными и показывали отсутствие уменьшения массы при температурах ниже ~257-262°C, свидетельствуя о том, что эти материалы несольватированы. Партия 3, однако, демонстрировала ~2,3%-ное уменьшение массы между ~162°C и ~200°C, обусловленное высвобождением ~0,13 моль этилацетата (по данным ЯМР для этой партии и по данным TG-IR, полученным для материала, образованного при скрининге). Высокая температура этого высвобождения свидетельствуют о включении растворителя в кристаллическую решетку. Сходные, но менее выраженные различия наблюдали после нагревания партии 2. Этот материал демонстрировал меньшую степень уменьшения массы (~0,6 масс.%) между ~185°C и ~200°C. Резкое уменьшение массы, наблюдавшееся при ~257-262°C у всех шести партий, предположительно объясняется разложением этих материалов.
Термограммы DSC, полученные на партиях 1-5, демонстрировали интенсивные эндотермические эффекты в диапазоне ~191,9-193,8°C (максимумы пиков), соответствующие плавлению, что подтверждено данными высокотемпературной микроскопии, полученными на партии 5. Наблюдали небольшую асимметрию эндотермического пика, продемонстрированного партией 3, возможно, обусловленную десольватацией, о чем свидетельствуют данные TGA и ЯМР, показывающие присутствие этилацетата в этом материале.
Данные высокотемпературной микроскопии получали на партии 5, использованной в качестве первичного исходного материала для скрининга полиморфов. Этот материал первоначально показывал двойное лучепреломление с экстинкцией, свидетельствующее о его кристалличности. Никаких визуальных изменений не наблюдали при нагревании ниже ~143,0°C. Переход из твердого состояния в жидкое, свидетельствующий о плавлении материала, наблюдали в температурном диапазоне ~187,9-192,2°C. При охлаждении до ~35,6°C не было видно никакой кристаллизации.
Данные анализа сорбции влаги были получены на партии 5. Эти данные соответствуют материалу с низкой гигроскопичностью. Этот материал показывал незначительное уменьшение массы после уравновешивания при RH, составлявшей ~5%. Незначительный прирост массы (~0,02 масс.%) наблюдали при RH ниже ~75%, выше чего материал приобретал дополнительно ~0,19 масс.%, причем при увеличении RH от ~5% до ~95% общее поглощение воды составляло ~0,21 масс.%. Почти полная десорбция наблюдалась с небольшими гистерезисом при снижении относительной влажности с ~85% до ~45% (~0,20%-ное уменьшение массы при уменьшении RH от ~95% до ~5%).
Спектр протонного ЯМР был получен на партии 3 как вспомогательное средство для понимания различий, выявленных при сравнении этого материала с другими партиями. Химические сдвиги ЯМР и интегральные значения для этого материала согласуются с химической структурой соединения 2. Спектр демонстрировал дополнительные пики при ~4,03 м.д., ~1,99 м.д. и ~1,18 м.д., которые можно объяснить наличием ~0,13 моль этилацетата, присутствие которого можно было бы ожидать в соответствии с общими условиями. Небольшие неидентифицированные пики (вероятно, обусловленные присутствием загрязняющих примесей) наблюдали также при ~9,87 м.д., ~5,31 м.д., ~4,09 м.д. и ~3,17 м.д.
C. Скрининг полиморфов соединения 2
Выделенные твердые вещества анализировали посредством рентгеновской порошковой дифрактографии (XRPD), сравнивая дифрактограммы дуг с другом и с XRPD-дифрактограммами партии 5, обозначенной как Форма А. Для сравнения также использовали дифрактограммы, полученные на партии 3 и обозначенные как Форма В.
Условия и результаты микромасштабных и среднемасштабных экспериментов по кристаллизации, проведенных в органических растворителях с использованием партии 5 соединения 2, сведены в Таблице 7 и Таблице 8 соответственно. Таблица 9 представляет результаты интенсивного воздействия на материал органических паров и интенсивного механического воздействия.
Каждый из нижеследующих способов, результатом которых является Форма А соединения 2, является другим аспектом настоящего изобретения, и каждый из нижеследующих способов, результатом которых является Форма В соединения 2, является другим аспектом настоящего изобретения.
Кристаллизация соединения 2 из органических растворителей с использованием партии 5 (Форма A) (микромасштабное испарение)
(продолжение)
b Присутствует один пик, не видимый на других XRPD-дифрактограммах для формы А, но соответствующий Форме А (по результатам индексации для этого материала).
Кристаллизация соединения 2 из органических растворителей с использованием партии 5 (Форма A) (микромасштабное испарение)
(продолжение)
Кристаллизация соединения 2 из органических растворителей с использованием партии 5 (Форма A) (микромасштабное испарение)
(продолжение)
HFIPA - гексафторизопропанол; ACN - ацетонитрил; MTBE - метил-трет-бутиловый эфир; IPA - изопропиловый спирт; IPE - изопропиловый эфир; MCH - метилциклогексан; TFE - 2,2,2-трифторэтанол; EtOAc - этилацетат; MEK - метилэтилкетон; DCM - дихлорметан; UM - неизвестная морфология; BE - двойное лучепреломление и экстинкция; w/ - союз «с»; part. - частица/частицы.
Кристаллизация соединения 2 из органических растворителей с использованием партии 5 (Форма A) (средний масштаб)
(X/Y)
образца
кая техника
тые частицы, аглом., BE
(прозрачный), УЗ, (прозрачный). Выдержано при 2-8°C
тые, BE
(прозрачный), УЗ, (прозрачный). Выдержано при 2-8°C
(продолжение)
(X/Y)
образца
кая техника
AS в раствор (прозрачный), УЗ.
(прозрачный). PSE
b Температура и длительность экспериментов являются приблизительными
c Исходный материал (возможно, аморфный) без пиков на XRPD-дифрактограмме
Кристаллизация соединения 2 из органических растворителей с использованием партии 5 (Форма A) (средний масштаб)
кая техника
Твердые веще-ства через 1 неделю
от 70°C (небольшой осадок). Выдержан на сухом льду с ацетоном, 2 ч.
Выдержано при темп. от -25 до -10°C, 1 день. RE, ~½ об., 50°C. Выдержано при темп. от -25°C до -10°C, 1 нед. с
(продолжение)
кая техника
Выдержано при темп. от -25 до -10°C, 2 недели
(1/1)
(~2/1)
b Температура и длительность экспериментов являются приблизительными
c Проводили в масштабе ~500 мг в попытке увеличить количество Материала D при температуре ниже температуры окружающей среды
d Наблюдали, что твердые вещества, образованные при температуре ниже температуры окружающей среды, растворялись при температуре окружающей среды и снова осаждались при возвращении к температуре ниже температуры окружающей среды. Материал фильтровали с вакуумом в холодном состоянии непосредственно после извлечения из морозильной камеры.
Кристаллизация соединения 2 из органических растворителей с использованием партии 5 (Форма A) (средний масштаб)
тельa (X/Y)
ческая
техника
5 дней
Выдержано при темп. от -25 до -10°C, 4 дня
EtOAc
Добавл. EtOAc (прозрачный).
Частично RE (некоторое количество осадка). Оставлено при комн.т. в течение ночи
гептан
b Температура и длительность экспериментов являются приблизительными
DMSO - диметилсульфоксид; DEE - диэтиловый эфир; DMF - диметилформамид; RT - room temperature (комн.т.); diss. - dissolution/dissolved (растворение/растворенный); add. - addition/added (добавление/добавленный); agglom. - agglomerates (агломераты, аглом.); ppt - precipitation (осаждение, осадок); soln. - solution/solutions (раствор/растворы); S/AS - solvent/antisolvent precipitation (осаждение растворителем/антирастворителем); OM - optical microscopy (оптическая микроскопия); VD - vapor diffusion (диффузия пара); PSE - partial slow evaporation (частичное медленное испарение).
Интенсивное воздействие органическими парами и интенсивное механическое воздействие на соединение 2 с использованием партии 5 (Форма A)
Интенсивное воздействие органическими парами и интенсивное механическое воздействие на соединение 2 с использованием партии 5 (Форма A)
Условия и результаты кристаллизации в неводных средах, интенсивного воздействия органическими парами и термического стресса, полученных с использованием различных образцов исходно аморфного соединения 2, представлены в Таблице 10, Таблице 11 и Таблице 12 соответственно.
Кристаллизация соединения 2 из органических растворителей с использованием аморфного материала
тельa
(X/Y)
ческая техника
6 дней
b Исходный материал - образец № 2, если не указано иначе. Температура и длительность экспериментов приблизительные
c Исходный материал - образец № 3
d Исходный материал - образец № 14
e Небольшие сдвиги пиков можно объяснить особенностями использованного рентгеновского аппарата.
Кристаллизация соединения 2 из органических растворителей
с использованием аморфного материала
рительа (X/Y)
образца
ческая
техника
Промыто смесью MEK/толуол (4/1)
6 дней
b Исходный материал - образец № 2, если не указано иначе. Температура и длительность экспериментов приблизительные.
c Исходный материал - образец № 3
d Добавляли охлажденный растворитель, получая прозрачный раствор. Образец немедленно переносили в условия с температурой ниже температуры окружающей среды и интенсивно перемешивали.
e Небольшие сдвиги пиков можно объяснить особенностями использованного рентгеновского аппарата.
Интенсивное воздействие органическими парами на аморфный материал соединения 2
b Исходный материал - образец № 3
Термический стресс аморфного материала соединения 2 и охлаждение из расплава
b Нагрев с интенсивным перемешиванием
c Небольшие сдвиги пиков могут быть обусловленными особенностями использованного рентгеновского аппарата.
BR - двойное лучепреломление; E - экстинкция
Серия экспериментов, направленных на образование гидратов с использованием как партии 5, так и аморфного материала, образованного при скрининге, в качестве исходного материала, представлена в Таблицах 13-16. В частности, Таблица 13 и Таблица 15 представляют результаты различных экспериментов с суспензиями с разной активностью воды. Условия и результаты осаждения антирастворителем с водой с использованием партии 5 сведены в Таблице 14. Таблица 16 представляет результаты экспериментов с интенсивным воздействием водяным паром.
Кристаллизация соединения 2 в водных условиях с использованием партии 5 (Форма A) - суспензии в разной активностью воды
5 дней
5 дней
комн.т.,
5 дней. Промыто водой
комн.т.,
5 дней. Промыто водой
5 дней. Промыто водой
(продолжение)
комн.т., 5 дней. Промыто EtOH
b Значения активности воды рассчитывали при 25°C, используя калькулятор UNIFAC (v. 3.0). Эти оценки произведены без подтверждения соответствия правилам cGMP.
Кристаллизация соединения 2 в водных условиях с использованием партии 5 (Форма A) - Осаждение в системах растворитель/антирастворитель (S/AS)
Раств. при 50°C, FC до комн.т.,
Комн.т., раств. в холодном AS (осаждение)
Раств. при 50°C, FC до комн.т.
Комн.т., раств. в холодном AS (осаждение, липкое вещество). Суспензия, комн.т., 4 ч
AS в раствор (осаждение)
Раств. при 50°C, FC до комн.т.
Комн.т. раств. в холодный AS (осаждение)
Горячий раствор при 70°C до комн.т. AS (осаждение).
Центрифугировали, декантировали, сушили с N2
b Небольшие сдвиги пиков могут быть обусловлены особенностями использованного рентгеновского аппарата.
Кристаллизация соединения 2 в водных условиях с использованием аморфного материала - суспензии с разной активностью воды
b Значения активности воды рассчитывали при 25°C, используя калькулятор UNIFAC (v. 3.0). Эти оценки произведены без подтверждения соответствия правилам cGMP.
c Небольшие сдвиги пиков могут быть обусловлены особенностями использованного рентгеновского аппарата.
Кристаллизация соединения 2 в водных условиях с использованием аморфного материала - суспензии с разной активностью воды
комн.т., 5 дней. Промыто водой
b Значения активности воды рассчитывали при 25°C, используя калькулятор UNIFAC (v. 3.0). Эти оценки произведены без подтверждения соответствия правилам cGMP.
c Небольшие сдвиги пиков могут быть обусловлены особенностями использованного рентгеновского аппарата.
Кристаллизация соединения 2 в водных условиях с использованием аморфного материала - суспензии с разной активностью воды
3 дняb
Резкое уменьшение массы при -256°C (начало)
4 дня c
b Исходный материал - образец № 2
c В качестве смачивающего средства использовали поверхностно-активное вещество
d Значения активности воды рассчитывали при 25°C, используя калькулятор UNIFAC (v. 3.0). Эти оценки произведены без подтверждения соответствия правилам cGMP.
e TGA проводили на материале, который ранее исследовали посредством рентгеноструктурного анализа (образец № 120)
VO - vacuum oven (вакуумная печь)
Интенсивное воздействие водяного пара на соединение 2
ал a
85% RH, 3 дня
b Значения относительной влажности, температуры и длительности экспериментов являются приблизительными
c Образец, исследованный без подтверждения соответствия cGMP вследствие недостаточной документации для камеры RH.
d Из образца № 127 - часть образца № 14
В контексте настоящей спецификации, термин «неупорядоченно кристаллический» означает, что дифрактограмма XRPD для данного материала имеет широкие пики (по сравнению с шириной инструментальных пиков) и/или сильное диффузное рассеяние (по сравнению с пиками). В варианте осуществления настоящего изобретения неупорядоченные материалы являются:
- микрокристаллическими;
- кристаллическими с большими дефектами плотности; или
- смесями кристаллических и рентгеновски аморфных фаз; или
комбинацией вышеуказанного.
a. Материал C
Материал C продуцировала водная суспензия аморфного материала в исключительных условиях при повышенной температуре (~50°C). Эти экспериментальные условия и данные TGA для Материала С дают возможность предположить, что возможно образование гидратированного материала. Однако все дальнейшие эксперименты в водных средах, включая суспензии в смесях органических растворителей с водой при разной активности воды, а также суспензию в воде с использованием поверхностно-активного вещества, оказались неспособными произвести Материал С - их результатом была Форма А. Поэтому природа материала С неизвестна. Этот материал потенциально мог быть кристаллическим продуктом разложения соединения 2, произведенным водной суспензией при повышенной температуре. Альтернативно, Материал C мог быть нестабильным гидратом соединения 2, которые легко дегидратируется при температуре окружающей среды. Попытку XRPD-анализа аморфного материала в камере с переменной относительной влажностью не предпринимали, хотя она могла бы представлять потенциальный интерес.
b. Материал D
Материал D был произведен в единственном эксперименте посредством быстрого охлаждения раствора в смеси метилэтилкетона с толуолом (~4/1) до температуры ниже температуры окружающей среды. Наблюдали, что твердые вещества, первоначально образованные при температуре ниже температуры окружающей среды, растворялись после уравновешивания с температурой окружающей среды и снова осаждались, когда возвращались к температуре ниже температуры окружающей среды. Исключительность условий растворителя позволяет предположить, что возможно образование сольватированного материала. В частности, материал, выделенный холодным и первоначально демонстрировавший двойное лучепреломление с экстинкцией, свидетельствующих о кристалличности, будучи проанализированным при комнатной температуре демонстрировал неупорядоченную XRPD-дифрактограмму (попыток проведения XRPD при температуре ниже температуры окружающей среды не предпринимали). Это свидетельствует о частичной утрате кристалличности - возможно, вследствие быстрой потери растворителя при хранении в условиях окружающей среды. Сольватированная природа материала D не была подтверждена, поскольку все дополнительные эксперименты, проведенные с этим материалом, включая кристаллизацию аморфного материала и представленной партии соединения 2 из различных смесей метилэтилкетона с толуолом, а также из соответствующих индивидуальных растворителей, не смогли произвести Материал D - их результатом была Форма А.
D. Получение и характеризация аморфного материала соединения 2
Аморфный материал, первоначально образованный при скрининге полиморфов, был произведен в увеличенном масштабе как альтернативный исходный материал для скрининга. Эксперименты по увеличенному масштабированию проводили на уровне ~500 мг и ~1 г исходного материала, используя партию 5 соединения 2. Два образца были получены посредством лиофилизации раствора в 1,4-диоксане, первоначально приготовленного при температуре, повышенной для облегчения растворения, и охлажденного до температуры окружающей среды.
Образец № 2 был охарактеризован посредством XRPD, TGA, DSC (стандартной и циклической), высокотемпературной микроскопии, анализа влагопоглощения и 1H ЯМР. Циклическую DSC выполняли на образце № 3 для подтверждения аморфности материала. Условия производства в увеличенном масштабе приведены в виде сводки в Таблице 17. Результаты характеризации представлены в Таблице 18.
Производство аморфного соединения 2 в увеличенном масштабе
образца
техника
Раств. в 1,4-диоксане при 70°C, SC до комн.т. Лиофилизация при -50°C
Раств. в 1,4-диоксане при 70°C, SC до комн.т. Лиофилизация при 50°C
Цикл 2: ступенчатое изменение при ~50,6°C (между ~46,6°C и ~55,0°C). Экзотермический эффект при ~123,7°C с последующим интенсивным эндотермическим эффектом при ~195,0°C (максимум пика)
b Цикл 1: нагревание от -50°C до 70°C, цикл 2: охлаждение от 70°C назад до -50°C, затем нагревание до 250°C
Физическая характеризация аморфного соединения 2, произведенного в увеличенном масштабе
Цикл 2: ступенчатое изменение при ~48,8°C (между ~44,4°C и ~53,2°C).
31,2°C-нагревание, 10°c/мин.
41,4°C - без заметных изменений
48,9°C - без заметных изменений
60,2°C - без заметных изменений
70,1°C - без заметных изменений
79,6°C - без заметных изменений
90,5°C - 105,4°c - без заметных изменений
116,2°C - без заметных изменений
160,5°C - без заметных изменений
183,5°C - несколько частиц выглядят обладающими двойным лучепреломлением
186,0°C - начало перехода твердого вещества в жидкое
186,6°C - завершен переход твердого вещества в жидкое, охлаждение
27,5°C - без заметной кристаллизации
~1,18%-ное увеличение массы между ~5% и ~75% RH
~8,56%-ное уменьшение массы между ~95% и ~5% RH
Большой гистерезис между ~85% и ~15% RH после десорбции
Физическая характеризация аморфного соединения 2, произведенного в увеличенном масштабе
Вероятно, содержит остаточный диоксан (судя по пику при ~3,57 м.д.). Небольшие неидентифицированные пики при ~5,08 м.д., ~1,36 м.д., ~1,23 м.д. и ~0,85 м.д. a
Цикл 2: ступенчатое изменение при ~50,6°C (между ~46,6 °C и ~55,0 °C).
b Цикл 1: нагревание от -50°C до 70°C; цикл 2: охлаждение от 70°C назад до -50°C, затем нагревание до 250°C
В целом, данные для двух образцов согласуются с представлением об аморфном материале c температурой стеклования в диапазоне ~49-51°C. Дополнительные данные, полученные с образцом № 2, свидетельствуют о том, что этот материал содержит остаточный растворитель и обладает значительной гигроскопичностью, удерживая значительное количество влаги после десорбции в широком диапазоне относительной влажности (~85-15% RH).
a. Образец № 2
Данные XRPD для этого образца согласуются с представлением об аморфном материале, демонстрирующем характерные гало на своих дифрактограммах без признаков острых пиков.
Термические данные согласуются с представлением о материале, содержащем остаточный растворитель. Кривая TGA показала ~1,2%-ное уменьшение массы между ~26°C и ~71°C, что могло быть связанным с потерей удерживаемой влаги (если основываться на данных о гигроскопичности этого материала). Вносить свой вклад в это уменьшение массы может и высвобождение остаточного диоксана, поскольку присутствие небольшого количества диоксана подтверждают результаты ЯМР. Резкое уменьшение массы, наблюдаемое при ~258°C (начало эффекта), по-видимому, обусловлено разложением этого материала.
Термограмма DSC продемонстрировала небольшой широкий эндотермический эффект при ~54,3°C (максимум пика) в диапазоне ~34,0-68,0°C с одновременной потерей массы, выявленной TGA. Этот эффект может быть связанным в десольватацией материала, перекрывающейся с явлением стеклования (вероятно, с релаксацией), о чем свидетельствует DSC с температурным циклом. Наблюдавшийся слегка расширенный экзотермический эффект при ~111,7°C (максимум пика) с последующим интенсивным эндотермический эффектом при ~190,7°C (максимум пика), судя по данным высокотемпературной микроскопии, обусловлен кристаллизацией, за которой следует плавление. Результаты экспериментов с термическим стрессом, проведенных с аморфным материалом, а также данные DSC, полученные на Форме А, указывают, что этот материал повторно кристаллизуется в виде Формы А.
Кривая DSC с температурными циклами демонстрировала небольшой широкий эндотермический эффект при ~54,5°C (максимум пика) в цикле первого нагревания, который, судя по данным TGA, можно объяснить десольватацией. Температуру стеклования (Tg) этого материала наблюдали при ~48,8°C (средняя точка) в виде ступенчатого изменения базовой линии на втором цикле нагревания. Кроме того, термограмма продемонстрировала и расширенный экзотермический эффект при ~114,8°C (максимум пика) с последующим интенсивным эндотермическим эффектом при ~194,5°C (максимум пика), что обусловлено кристаллизацией Формы А, за которой следовало ее плавление, как предполагалось выше.
Высокотемпературная микроскопия не показала никаких видимых изменений при температуре ниже ~183,5°C, возможно, вследствие малого размера частиц, естественного для аморфного вещества. Некоторое двойное лучепреломление, видимое при ~183,5°C, согласуется с кристаллизацией Формы А (как можно заключить по данным DSC и результатам экспериментов с термическим стрессом, проведенных с аморфным материалом). Переход твердого вещества в жидкое, обусловленный плавлением перекристаллизованного материала, наблюдали между ~186,0°C и ~186,6°C.
Результаты анализа влагопоглощения, полученные с этим образцом, согласуются с представлением о материале, обладающем значительной гигроскопичностью. Приблизительно 0,1%-ное увеличение массы наблюдали после уравновешивания при ~5% RH. Материал набирал приблизительно 1,2 масс.% воды при RH ниже ~75% и дополнительно поглощал ~8,7 масс.% воды, после увеличения относительной влажности до ~95% (с общим увеличением массы приблизительно на 9,9 масс.%). После снижения относительной влажности до ~5% происходила частичная десорбция (~8,6%-ное уменьшение массы между ~95% и ~5% RH) [5]. После десорбции в интервале RH между ~85% и ~15% наблюдали большой гистерезис, указывающий на то, что этот материал удерживает значительное количество влаги (~3,6-5,8 масс.%) в широком диапазоне относительной влажности. Такое поведение может свидетельствовать о существовании гидрата. Однако эксперименты, направленные на обнаружение гидратированной формы (Материала C), были безуспешными, возможно, вследствие нестабильности гидрата. Ни в интервале RH между ~85% и ~95%, ни в интервале RH между ~15% и ~5% равновесие не достигалось, поэтому можно предположить, что возможно даже еще большее поглощение воды.
Химические сдвиги ЯМР и интегральные значения для этого материала согласуются с химической структурой соединения 2. Спектр демонстрирует небольшой дополнительный пик при ~3,57 м.д., приписываемый остаточному диоксану. Наблюдавшиеся небольшие неидентифицированные пики при ~5,08 м.д., ~1,36 м.д., ~1,23 м.д. и ~0,85 м.д., вероятно, обусловлены присутствием загрязняющих примесей.
b. Образец № 3
Данные DSC с температурными циклами, полученные для этого образца, были подобны данным, полученным для образца № 2. Термограмма цикло-DSC демонстрировала небольшой широкий эндотермический эффект при ~57,2°C (максимум пика) на первом цикле нагревания (возможно, обусловленный десольватацией). Температура стеклования (Tg) этого материала наблюдалась при ~50,6°C (средняя точка) в виде ступенчатого изменения базовой линии на втором цикле нагревания. Кроме того, термограмма продемонстрировала расширенный экзотермический эффект при ~123,7°C (максимум пика) с последующим интенсивным эндотермическим эффектом при ~195,0°C (максимум пика), что обусловлено кристаллизацией Формы А, за которой следовало ее плавление, как предполагалось выше.
E. Дополнительная характеризация Формы В соединения 2 (этилацетатного сольвата)
В дополнение к физической характеризации Формы В (партия 3) проводили и частичную характеризацию образцов Формы В, образованных при скрининге. Дополнительные данные были получены на материале, произведенном при интенсивном воздействии органических паров этилацетата на аморфный материал (образец № 79), и на материале, полученном посредством кристаллизации раствора Формы A в смеси тетрагидрофурана с этилацетатом после частичного выпаривания в роторном испарителе (образец № 48). Эти два материала были охарактеризованы посредством XRPD. Форма B, приготовленная из аморфного материала (образец № 79), была, кроме того, охарактеризована посредством TGA и коррелированным термогравиметрическим и ИК-спектроскопическим анализом (TG-IR). Эти данные представлены в Таблице 19.
Дополнительная физическая характеризация Формы В соединения 2 (этилацетатного сольвата)
Совокупность данных для этих двух материалов согласуется с ранее охарактеризованной Формой В - кристаллическим нестехиометрическим этилацетатным сольватом соединения 2.
Данные XRPD для этих образцов демонстрировали раздельные пики, характерные для кристаллического материала. Эти два материала демонстрировали дифрактограммы, согласующиеся с дифрактограммой ранее охарактеризованной партии 3, в которой наличие этилацетата подтверждено ЯМР спектроскопией.
Данные TGA согласуются с представлением о сольватированном материале. Кривая TGA для этого материала показывала ~4,7%-ное уменьшение массы между ~138°C и ~190°C, связанное, судя по данным TG-IR, с высвобождением этилацетата. Как указано выше для партии 3, высокая температура этого высвобождения согласуется с представлением о включении растворителя в кристаллическую решетку. Резкое уменьшение массы, наблюдавшееся при ~254°C (начало эффекта), можно объяснить разложением материала.
Данные коррелированного TG-IR согласуются с нестехиометрическим этилацетатным сольватом. И коррелированные данные TGA, и график Грама-Шмидта показывали утрату летучего вещества при нагревании. График Грама-Шмидта демонстрировал максимум интенсивности при ~9,1 мин, обусловленный высвобождением летучего вещества. Кривая TGA показывала ~4,8%-ное уменьшение массы между ~144°C и ~190°C, которое можно объяснить потерей приблизительно 0,23 моль этилацетата на моль соединения 2, что подтверждается ассоциированным ИК-спектром при ~9,1 мин.
F. Частичная характеризация материала C
Данные частичной характеризации были получены на материале, произведенном водной суспензией аморфного материала в исключительных условиях при ~50°C (образец № 119). Этот материал был охарактеризован посредством XRPD и TGA. Данные сведены в Таблице 15.
Данные XRPD для этого материала демонстрировали раздельные пики, характерные для кристаллического материала, обозначенного как Материал С. Дифрактограмма Материала С, сходная с дифрактограммой XRPD Формы А, демонстрировала дополнительные острые пики, свидетельствующие о возможной смеси с новым кристаллическим материалом. Данные TGA показывали небольшое уменьшение массы (~1,0% по массе) между ~28°C и ~192°C. Хотя природа этого уменьшения не установлена, оно, по-видимому, связано с высвобождением воды, что можно обосновать условиями образования этого материала, позволяющими предположить, что возможна некоторая степень гидратации. Резкое уменьшение массы, наблюдавшееся при ~256°C (начало эффекта), можно объяснить разложением материала.
Наблюдаемые и наиболее интенсивные пики XRPD для Материала С указаны ниже в Таблицах 20 и 21:
Наблюдаемые пики для Материала C
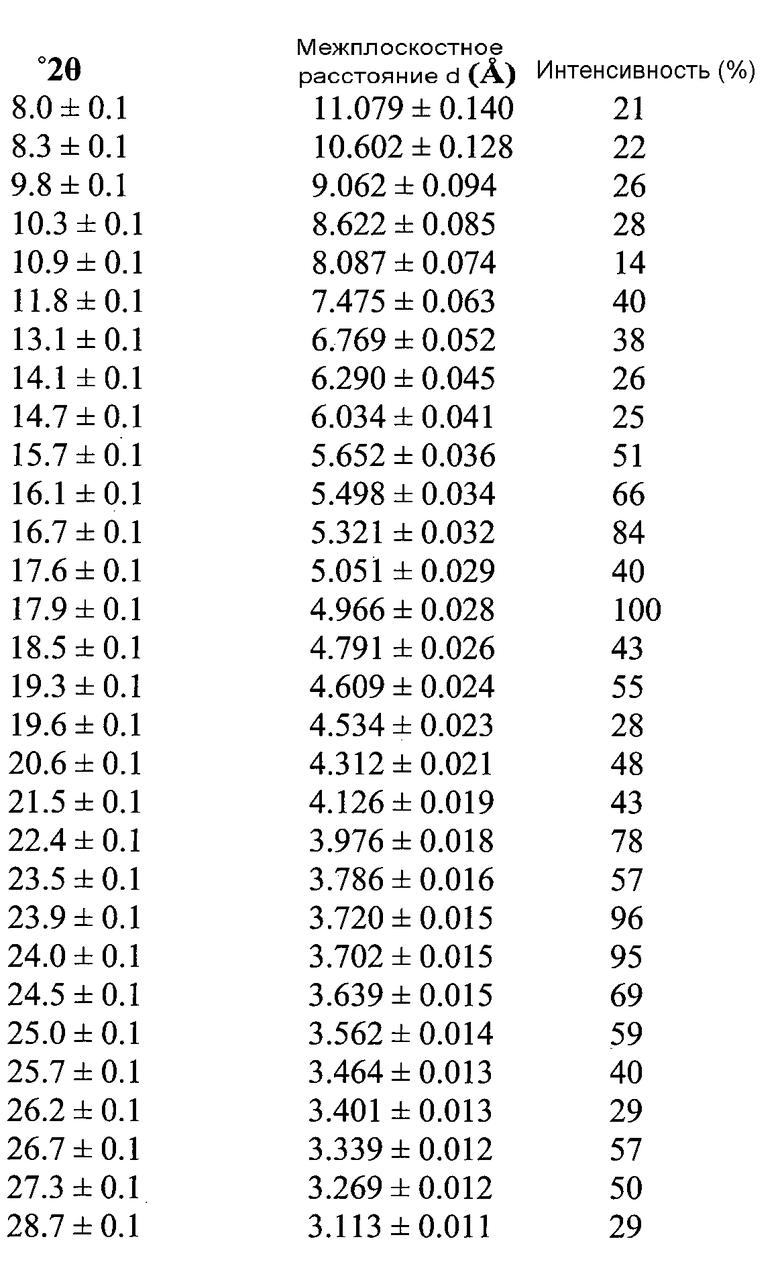
Наиболее интенсивные пики для Материала С

Материал C был произведен водной суспензией аморфного материала в исключительных условиях при повышенной температуре (~50°C). Дифрактограмма Материала С, сходная с дифрактограммой XRPD Формы А, демонстрировала дополнительные острые пики, свидетельствующие о возможной смеси с новым кристаллическим материалом. Небольшое уменьшение массы (~1,0 масс.%), наблюдавшееся после нагревания, наряду с условиями образования этого материала позволяет предположить, что Материал С представлял собой гидрат соединения 2. Однако такая природа этого уменьшения массы не была подтверждена, поскольку результатом всех дальнейших экспериментов, проведенных в водных средах и направленных на получение Материала С, включая суспензии в смесях органических растворителей с водой с разной активностью воды, а также суспензию в воде с использованием поверхностно-активного вещества, была Форма А.
Материал D был произведен в исключительных условиях, в частности посредством кристаллизации из раствора в смеси метилэтилкетона с толуолом (~4/1) при температуре ниже температуры окружающей среды. Исключительность этой системы растворителей позволяла предположить, что возможно образование сольватированного материала. Основываясь на данных XRPD для Материала D, можно было предположить, что частичная утрата кристалличности, происходящая при хранении, является следствием быстрой потери растворителя, но это предположение не было подтверждено, т.к. все дальнейшие эксперименты, направленные на получение этого материала, включая кристаллизацию из разных смесей метилэтилкетона с толуолом, а также из соответствующих индивидуальных растворителей, имели результатом Форму А.
Наблюдаемые и наиболее интенсивные пики XRPD для Материала D указаны ниже в Таблицах 22и 23:
Наблюдаемые пики для Материала D
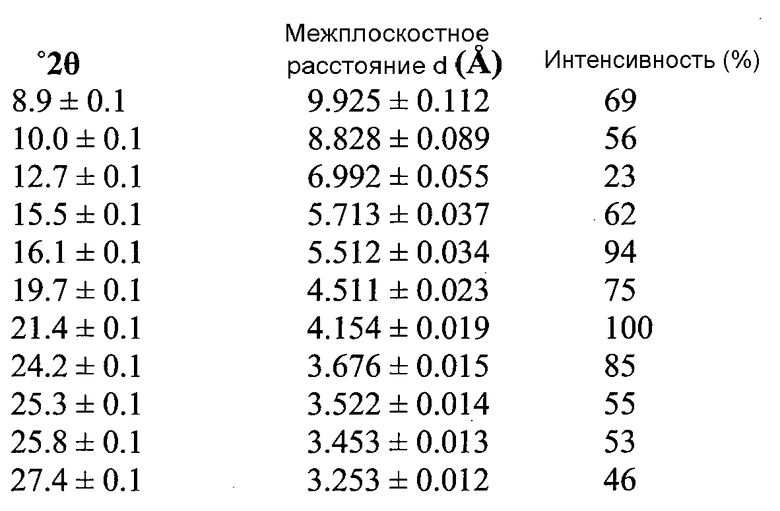
Наиболее интенсивные пики для Материала D

Следует понимать, что настоящее изобретение может быть модифицировано в пределах объема прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЕ СОЛИ И ПОЛИМОРФЫ АНТАГОНИСТА P2X3 | 2017 |
|
RU2782068C2 |
| КРИСТАЛЛИЧЕСКАЯ ИЛИ АМОРФНАЯ ФОРМА АГОНИСТОВ FXR, ПРЕДСТАВЛЯЮЩИХ СОБОЙ ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2800751C2 |
| АМИННЫЙ СОЛЬВАТ ИНГИБИТОРА НАТРИЙ-ГЛЮКОЗНОГО КОТРАНСПОРТЕРА И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2710230C1 |
| Твёрдые формы цефтолозана | 2014 |
|
RU2703457C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА TLR7/TLR8 | 2019 |
|
RU2792005C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО ТИОФЕНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2022 |
|
RU2830948C1 |
| ТВЕРДЫЕ ФОРМЫ 3-(5-АМИНО-2-МЕТИЛ-4-ОКСО-4Н-ХИНАЗОЛИН-3-ИЛ)ПИПЕРИДИН-2, 6-ДИОНА И ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ПРИМЕНЕНИЕ | 2012 |
|
RU2611007C2 |
| СОЛИ И ПОЛИМОРФЫ ЗАМЕЩЕННОГО ИМИДАЗОПИРИДИНИЛ-АМИНОПИРИДИНА | 2015 |
|
RU2732125C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА LTA4H | 2019 |
|
RU2808992C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА BTK | 2020 |
|
RU2828460C2 |
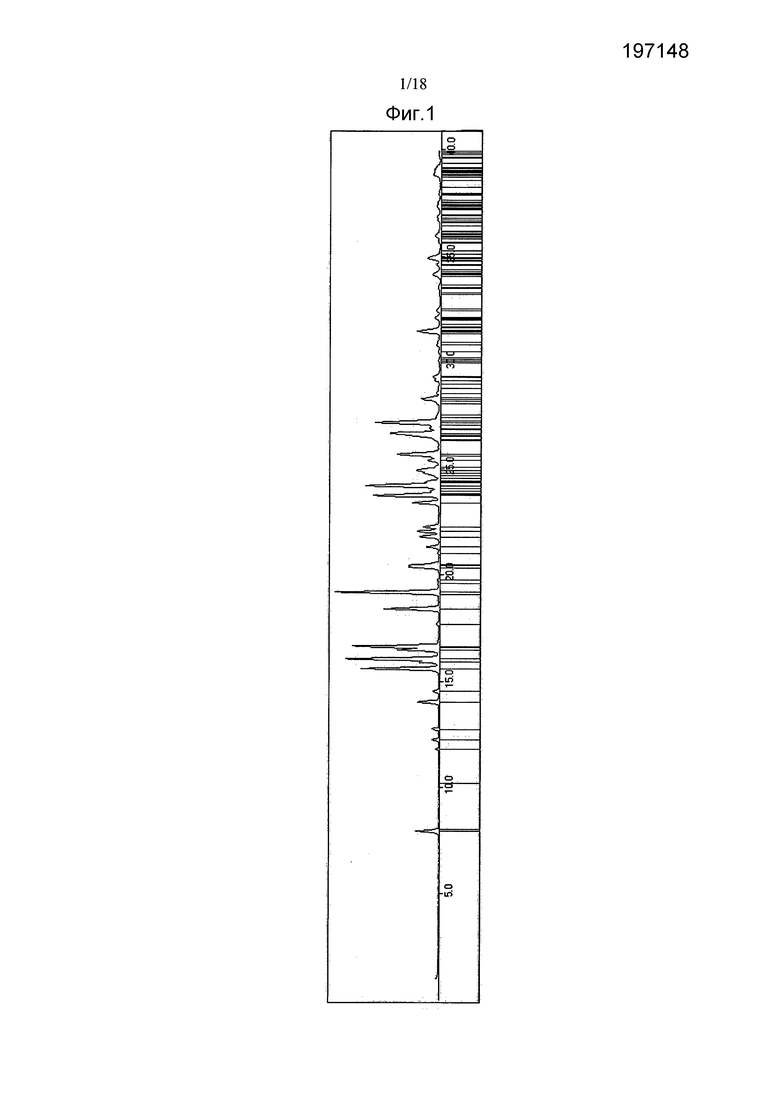
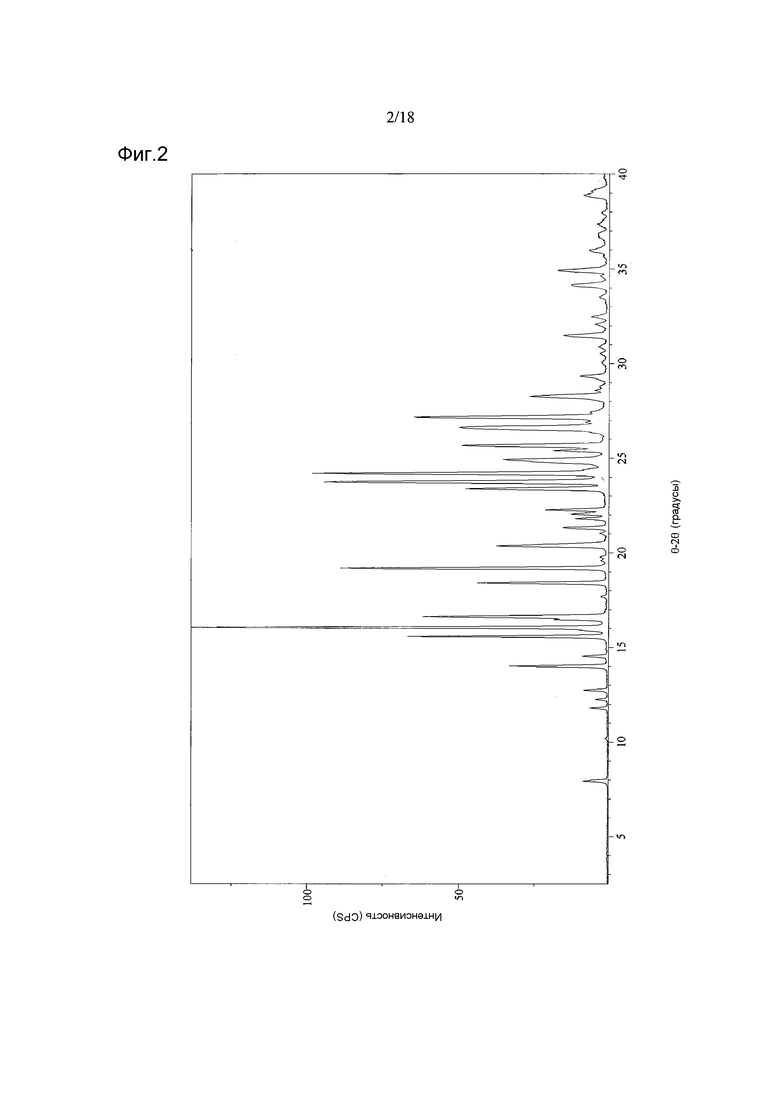
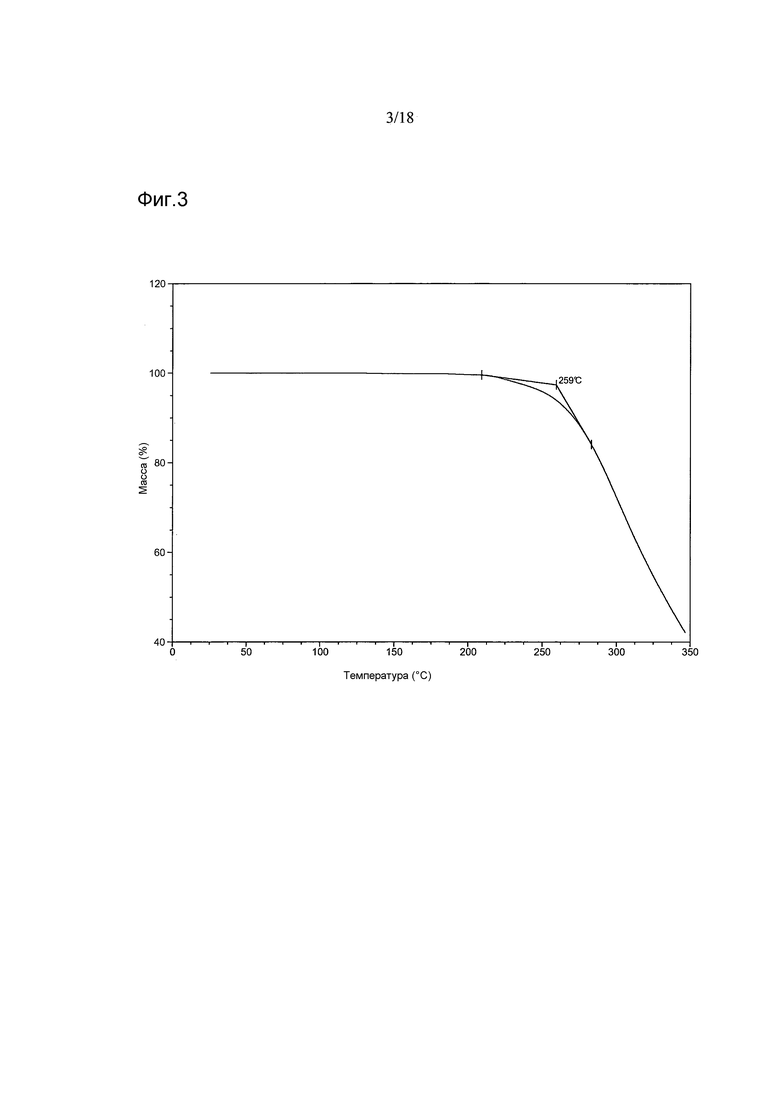
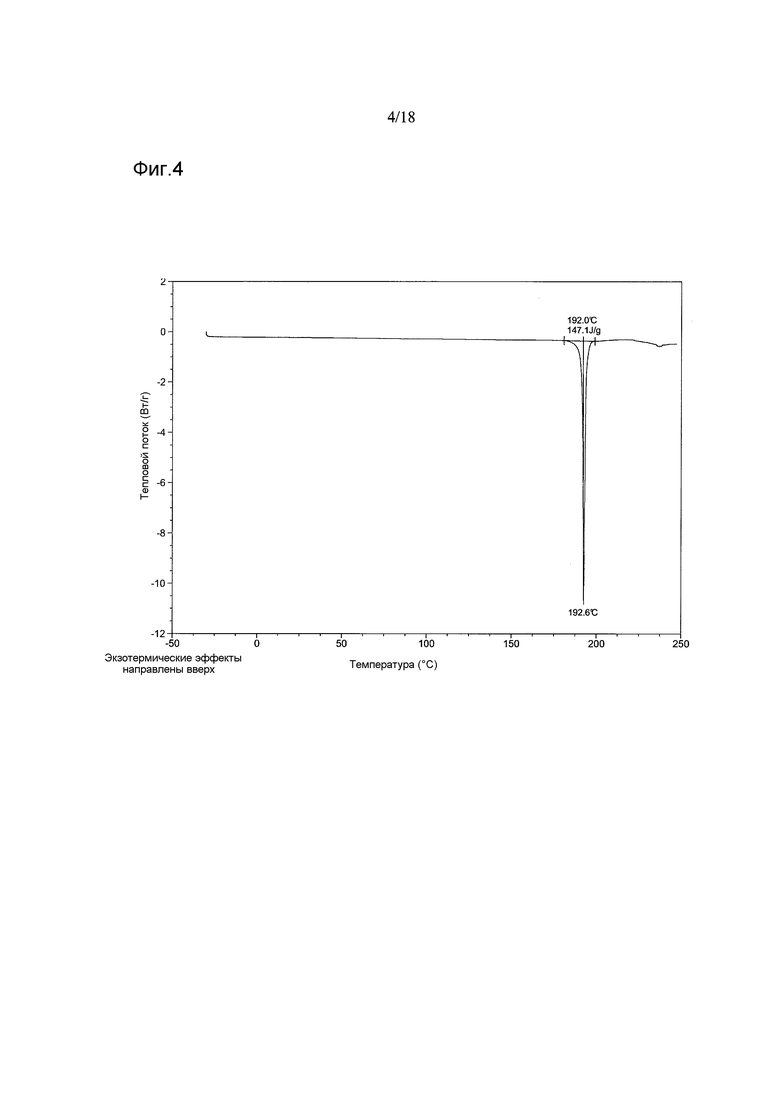
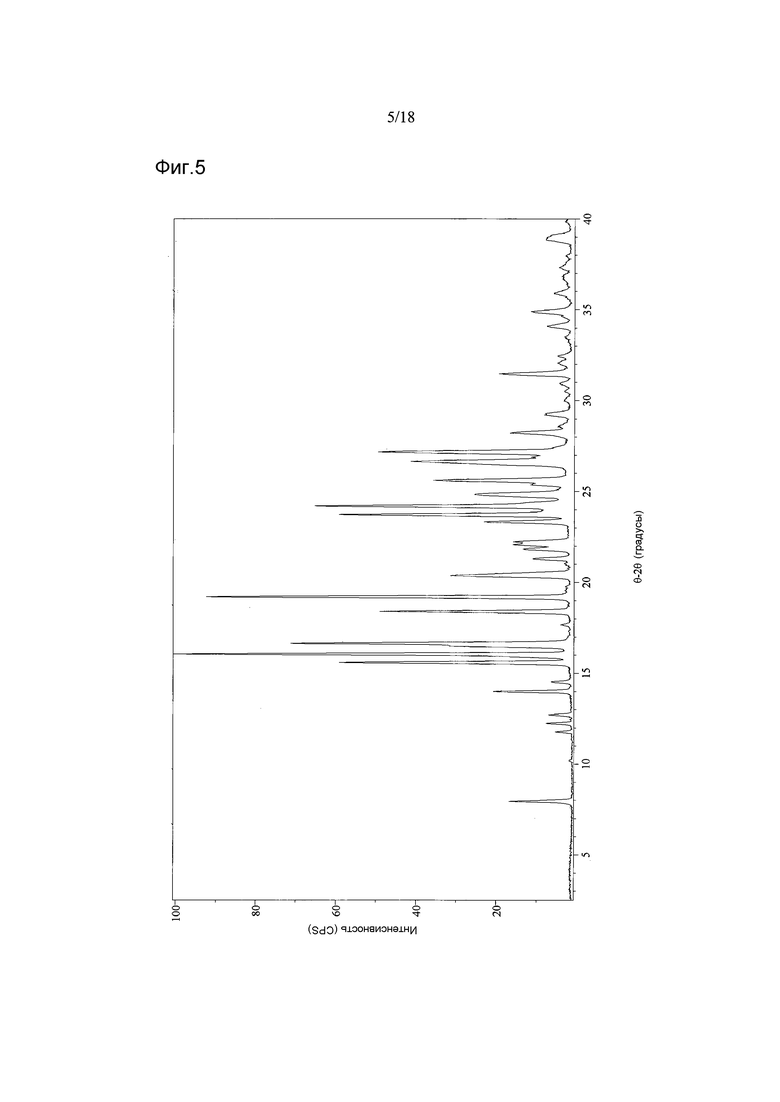
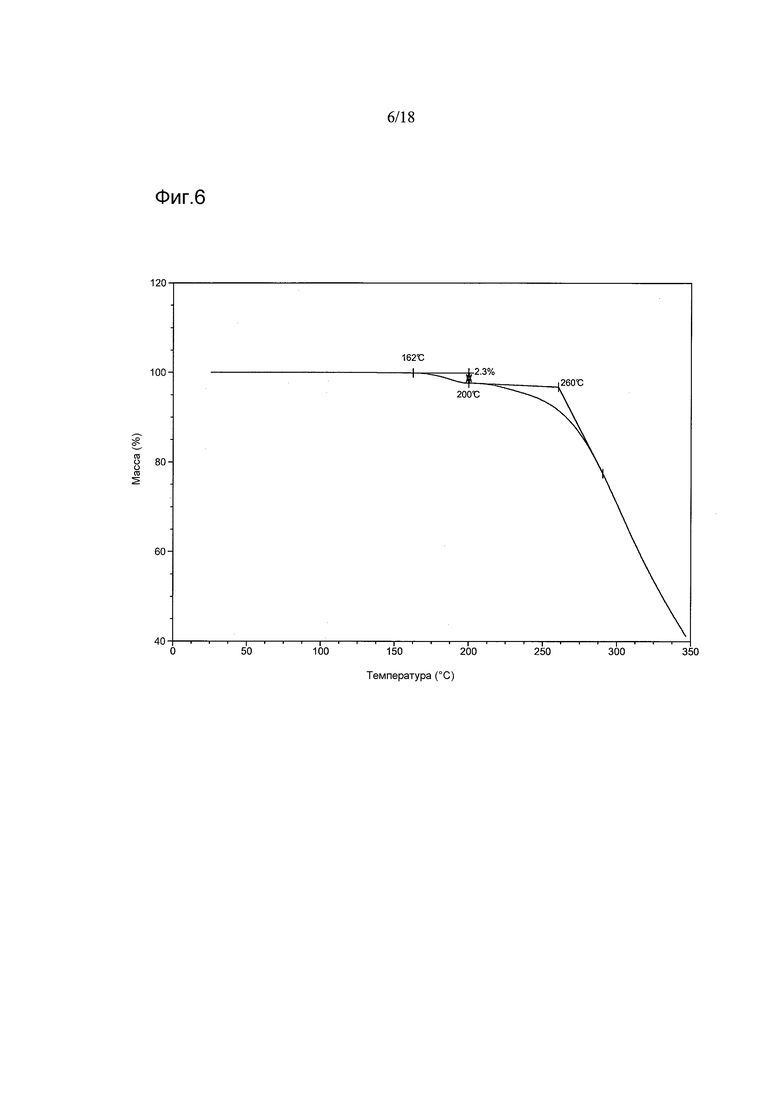
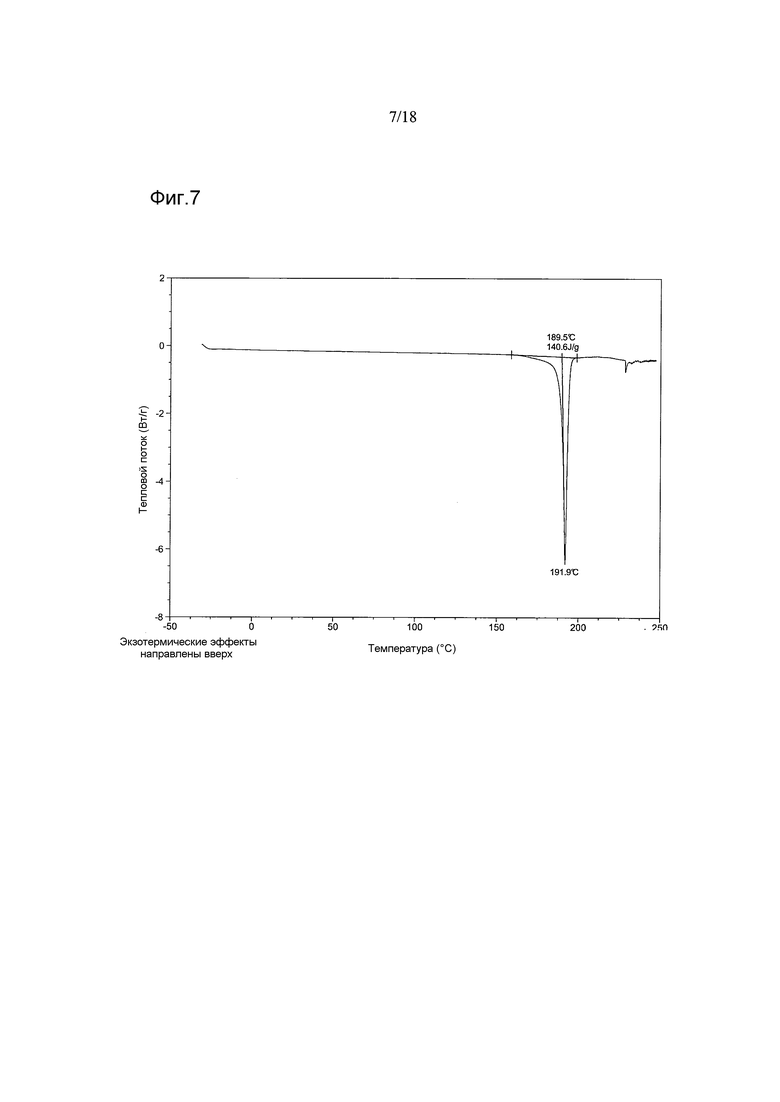


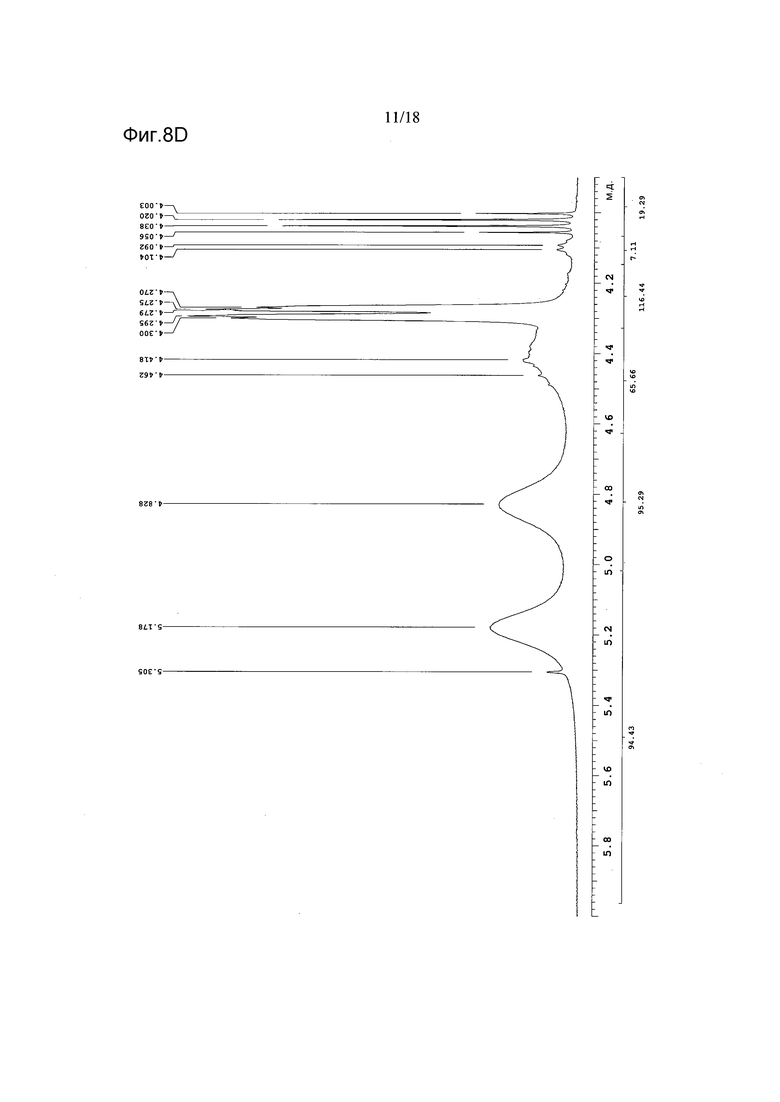

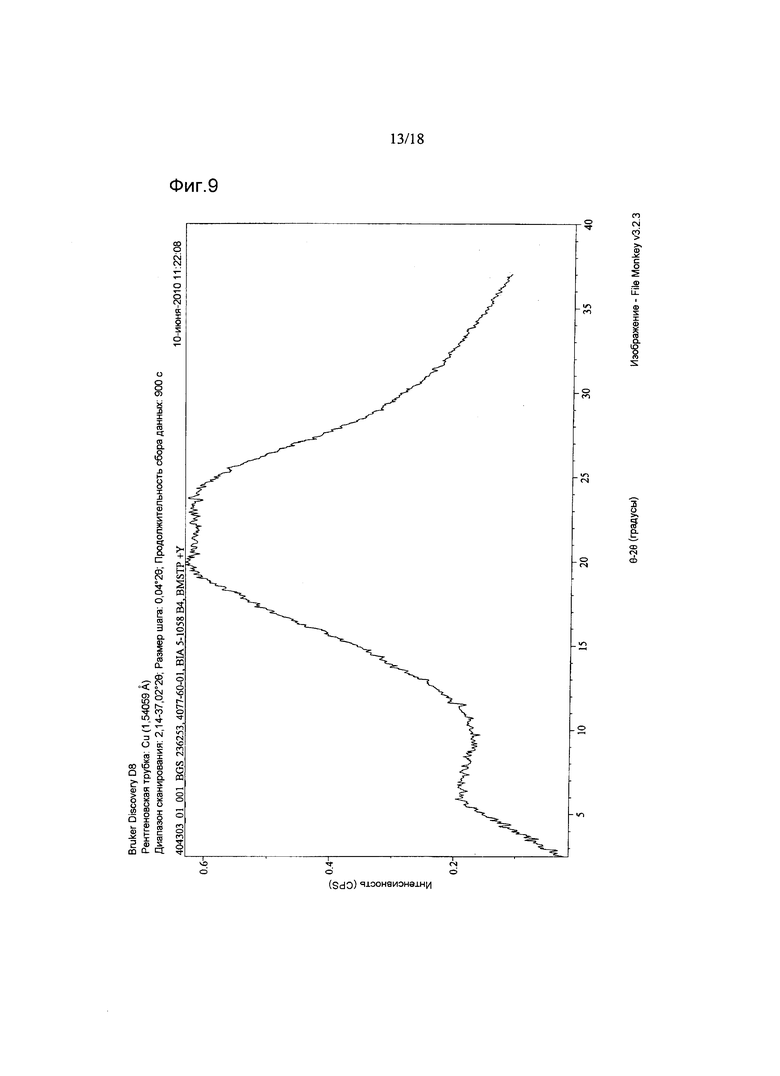
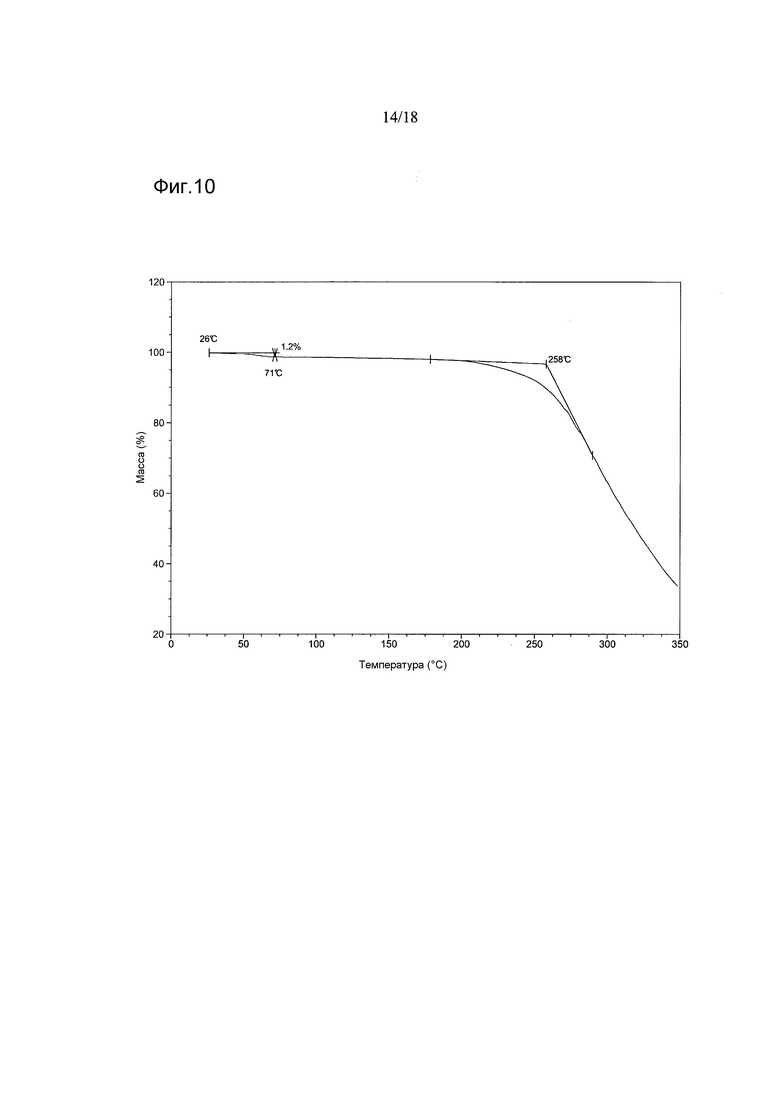
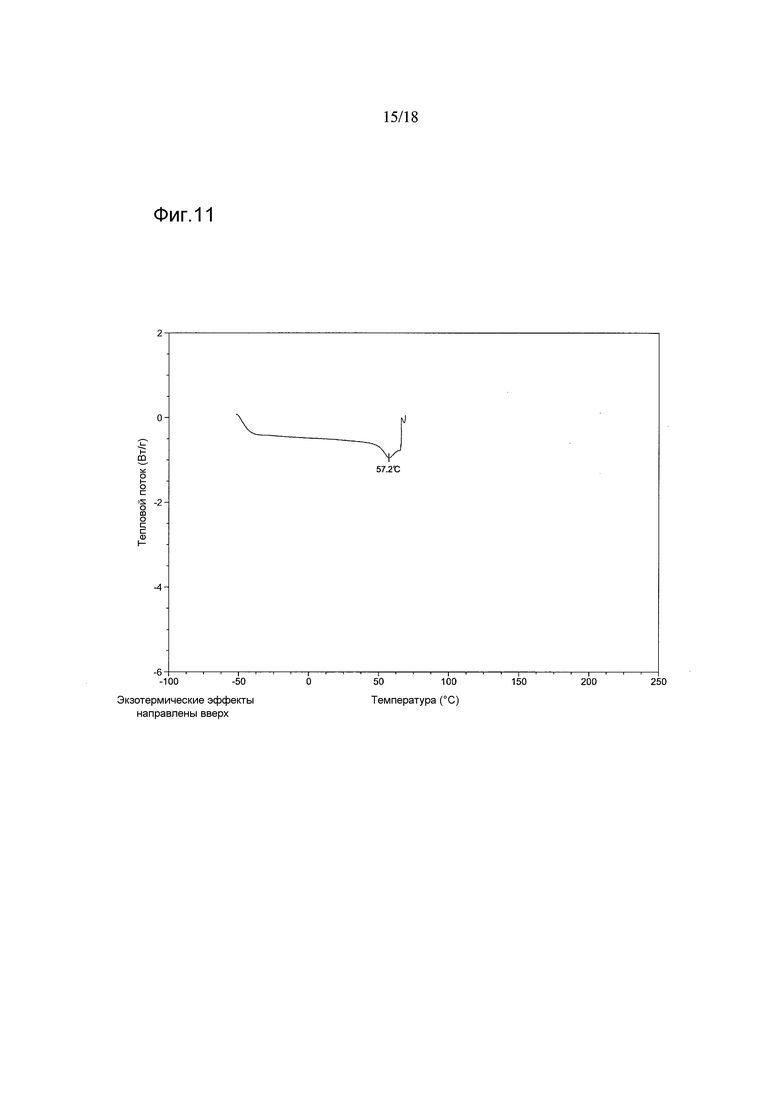
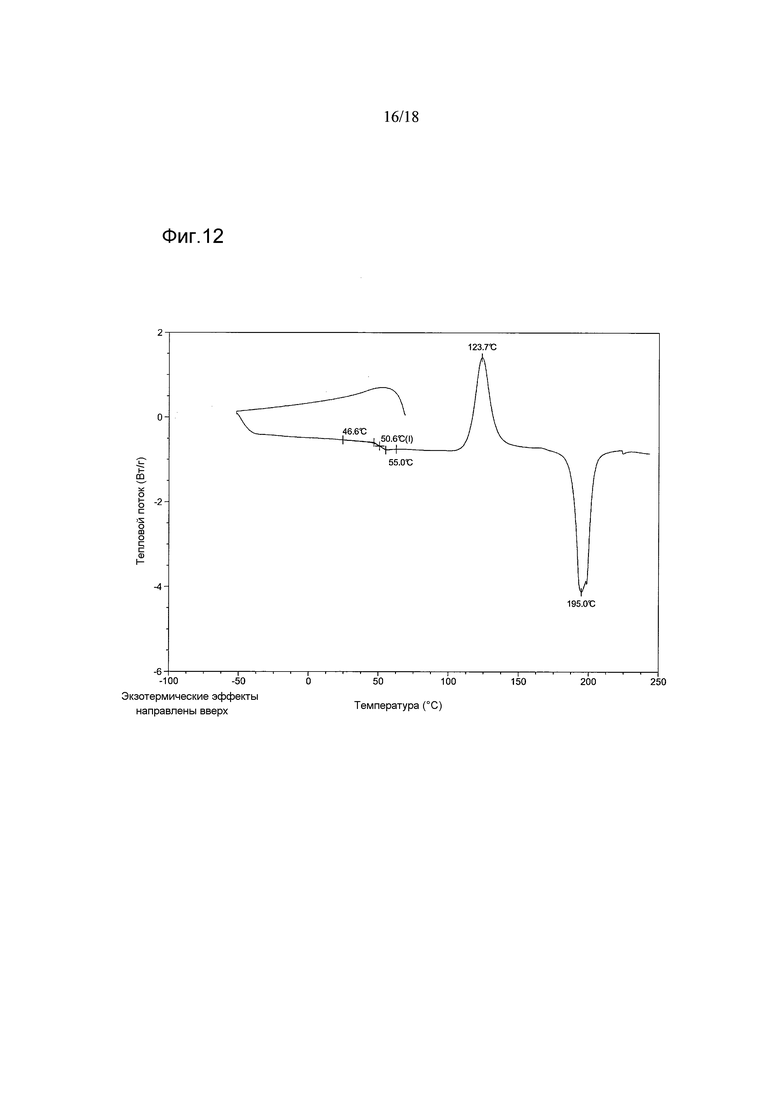
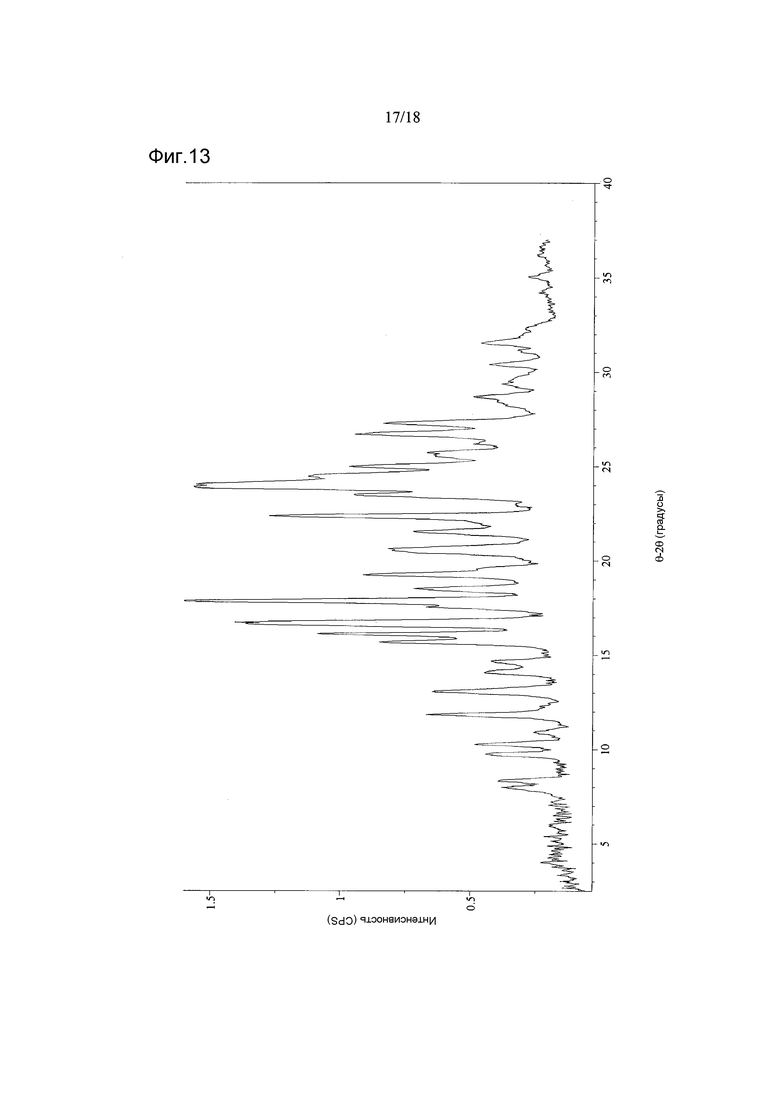
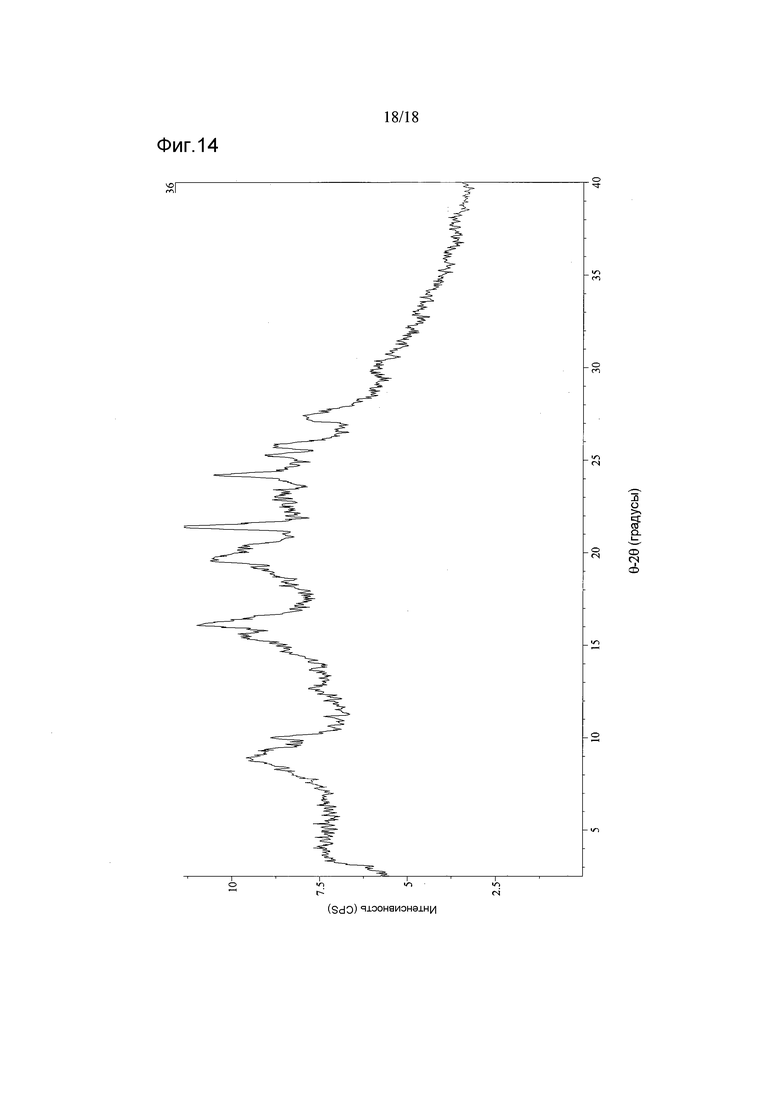
Настоящее изобретение относится к новой кристаллической Форме А 1-[(3R)-6,8-дифтор-3,4-дигидро-2H-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2H-имидазол-2-тиона, которая обладает действием ингибитора дофамин-β-гидроксилазы, и к способу ее очистки. Технический результат - высокий выход целевого продукта и его высокая стабильность. 2 н. и 14 з.п. ф-лы, 18 ил., 23 табл.
1. Кристаллическая Форма А 1-[(3R)-6,8-дифтор-3,4-дигидро-2Н-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2Н-имидазол-2-тиона, которая является несольватированной формой, имеющая дифрактограмму XRPD с пиками при 14,0, 16,1, 16,6, 19,2 и 20,4°2θ±0,2°2θ.
2. Форма А 1-[(3R)-6,8-дифтор-3,4-дигидро-2Н-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2Н-имидазол-2-тиона по п. 1, имеющая дифрактограмму XRPD с дополнительными пиками при 15,6 и 18,4°2θ±0,2°2θ.
3. Форма А 1-[(3R)-6,8-дифтор-3,4-дигидро-2Н-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2Н-имидазол-2-тиона по п. 1, имеющая дифрактограмму XRPD, как показано на Фигуре 2.
4. Форма А 1-[(3R)-6,8-дифтор-3,4-дигидро-2Н-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2Н-имидазол-2-тиона по п. 1, имеющая термограмму термогравиметрического анализа (TGA), показывающую уменьшение массы с начальной температурой 259°С±5°С.
5. Форма А 1-[(3R)-6,8-дифтор-3,4-дигидро-2Н-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2Н-имидазол-2-тиона по п. 4, имеющая термограмму TGA, как показано на Фигуре 3.
6. Форма А 1-[(3R)-6,8-дифтор-3,4-дигидро-2Н-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2Н-имидазол-2-тиона по п. 1, имеющая термограмму дифференциальной сканирующей калориметрии (DSC), показывающую эндотермический пик с начальной температурой 192°С±2°С и с максимумом пика при 193°С±2°С.
7. Форма А 1-[(3R)-6,8-дифтор-3,4-дигидро-2Н-1-бензопиран-3-ил]-1,3-дигидро-5-[2-[(фенилметил)-амино]-этил]-2Н-имидазол-2-тиона по п. 1, имеющая термограмму DSC, как показано на Фигуре 4.
8. Способ очистки кристаллической Формы A (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1Н-имидазол-2(3Н)-тиона, включающий перекристаллизацию гидрохлорида (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1Н-имидазол-2(3Н)-тиона по меньшей мере в одном органическом растворителе.
9. Способ по п. 8, в котором указанный органический растворитель представляет собой смесь толуола и метанола.
10. Способ по п. 9, в котором толуол и метанол присутствуют в смеси в пропорции 62:38 по массе.
11. Способ по п. 9 или 10, в котором указанный органический растворитель отгоняют и заменяют толуолом.
12. Способ по п. 8, в котором указанный способ очистки дополнительно включает преобразование гидрохлорида (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1Н-имидазол-2(3Н)-тиона в (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1Н-имидазол-2(3Н)-тион.
13. Способ по п. 12, в котором указанное преобразование гидрохлорида (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1Н-имидазол-2(3Н)-тиона осуществляют, применяя гидроксид щелочного металла.
14. Способ по п. 13, в котором указанный гидроксид щелочного металла представляет собой гидроксид натрия.
15. Способ по п. 12, в котором указанное преобразование проводят в смеси метанола и воды.
16. Способ по п. 8, в котором чистота (R)-5-(2-(бензиламино)этил)-1-(6,8-дифторхроман-3-ил)-1Н-имидазол-2(3Н)-тиона составляет, по меньшей мере, 95%, предпочтительно, по меньшей мере, 98%, наиболее предпочтительно ≥99,0%.
| WO 2008136695 A1 13.11.2008 | |||
| Устройство для монтажа колонн с консолями | 1986 |
|
SU1408038A1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПЕРИФЕРИЧЕСКИ-СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ДОФАМИН-БЕТА-ГИДРОЛАЗЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2003 |
|
RU2332416C2 |
| US 4631282 A1 23.12.1986. | |||

