УРОВЕНЬ ТЕХНИКИ, ПРЕДШЕСТВУЮЩИЙ ИЗОБРЕТЕНИЮ
Настоящее изобретение относится к кристаллическим формам 5-(2,4-диаминопиримидин-5-илокси)-4-изопропил-2-метокси-бензолсульфонамида ("соединения A") или его солей.
Респираторный тракт, или дыхательные пути, участвует в жизненно важном процессе газообмена для удовлетворения потребности в потреблении кислорода и удаления углекислого газа. Блуждающие вегетативные нервы контролируют гладкие мышцы трахеобронхиального дерева и, таким образом, диаметр дыхательных путей, а также высвобождение и перемещение выделений (слизи и жидкости). Контроль координируют ядра ствола головного мозга, которые регулируют произвольный и вегетативный выход, опираясь на интенсивный вход сенсорных сигналов блуждающего нерва от тканей дыхательных путей, которые, в свою очередь, передают соматические сенсорные сигналы и запускают вегетативные рефлексы. Чувствительные волокна блуждающего нерва отходят главным образом от клеток яремного и нодозного ганглиев, а их активность регулирует ряд химических веществ, таких как АТФ, который сенсибилизирует афферентные отростки блуждающего нерва и служит конвергентным сигналом механочувствительных рецепторов дыхательных путей.
АТФ активирует пуринергические рецепторы (например, P2X3 и P2X2/3), которые опосредуют множество физиологических и патологических ролей. АТФ стимулирует и сенсибилизирует окончания чувствительных нервов, что приводит к интенсивным ощущениям, таким как боль, дискомфорт, неотложный позыв к мочеиспусканию, зуд и позывы и выраженное возрастание разрядки чувствительных нервов, во многом посредством активации рецепторов P2X3 на афферентных нервных волокнах, иннервирующих ткани и органы грызунов и человека, особенно полые внутренние органы.
Соединение A описано в международной публикации WO2005/095359 (опубл. 13 октября 2005 года) и патенте США № 7858632 (опубл. 22 сентября 2005 года), которые, таким образом, полностью включены в настоящее описание посредством ссылки. Соединение A представляет собой ингибитор P2X3 и/или P2X2/3 и потенциально пригодно для лечения кашля, хронического кашля и позыва к кашлю при респираторных патологических состояниях и нарушениях, наряду с другими патологическими состояниями.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем документе описаны формы солей и полиморфы соединения A. В одном из вариантов осуществления полиморф представляет собой кристаллическую форму свободного основания A соединения A. В другом варианте осуществления полиморф представляет собой кристаллическую форму соли лимонной кислоты A соединения A. В другом варианте осуществления полиморф представляет собой кристаллическую форму соли лимонной кислоты B соединения A. В другом варианте осуществления полиморф представляет собой кристаллическую форму соли винной кислоты A соединения A. В еще одном варианте осуществления полиморф представляет собой кристаллическую форму соли винной кислоты F соединения A.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
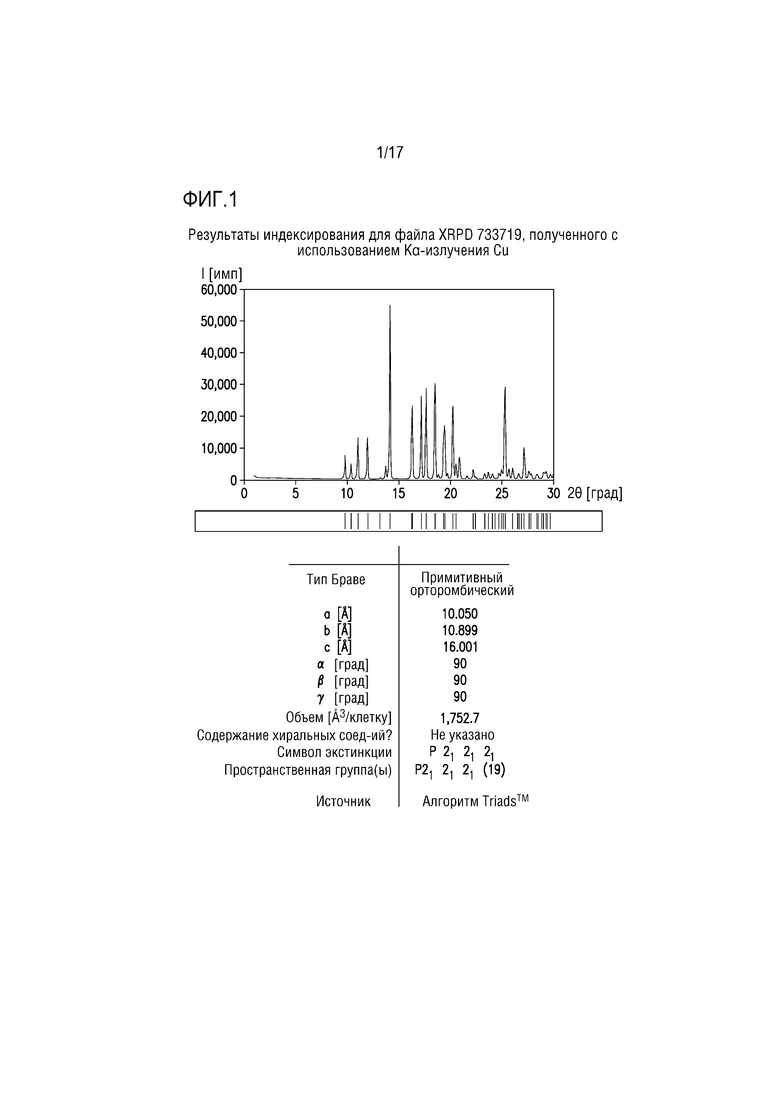
Фиг. 1 представляет собой характерный профиль XRPD для формы свободного основания A соединения A.
Фиг. 2 представляет собой характерный профиль XRPD для формы свободного основания B соединения A.
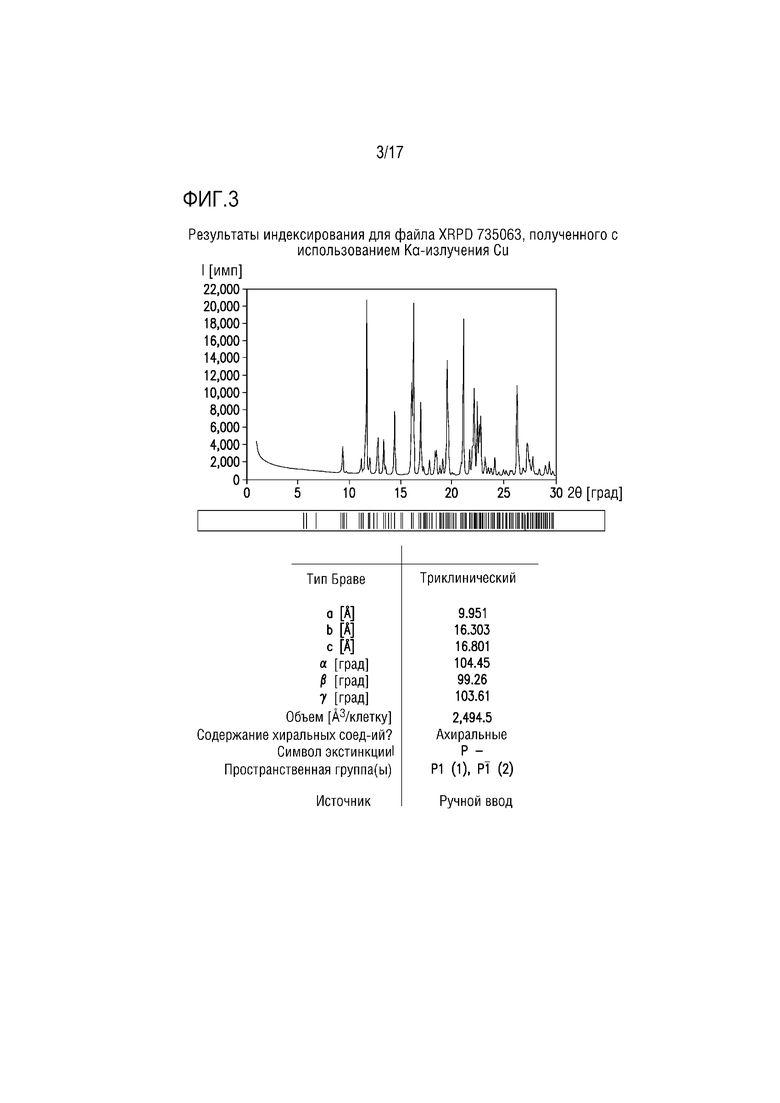
Фиг. 3 представляет собой характерный профиль XRPD для формы соли лимонной кислоты A соединения A.
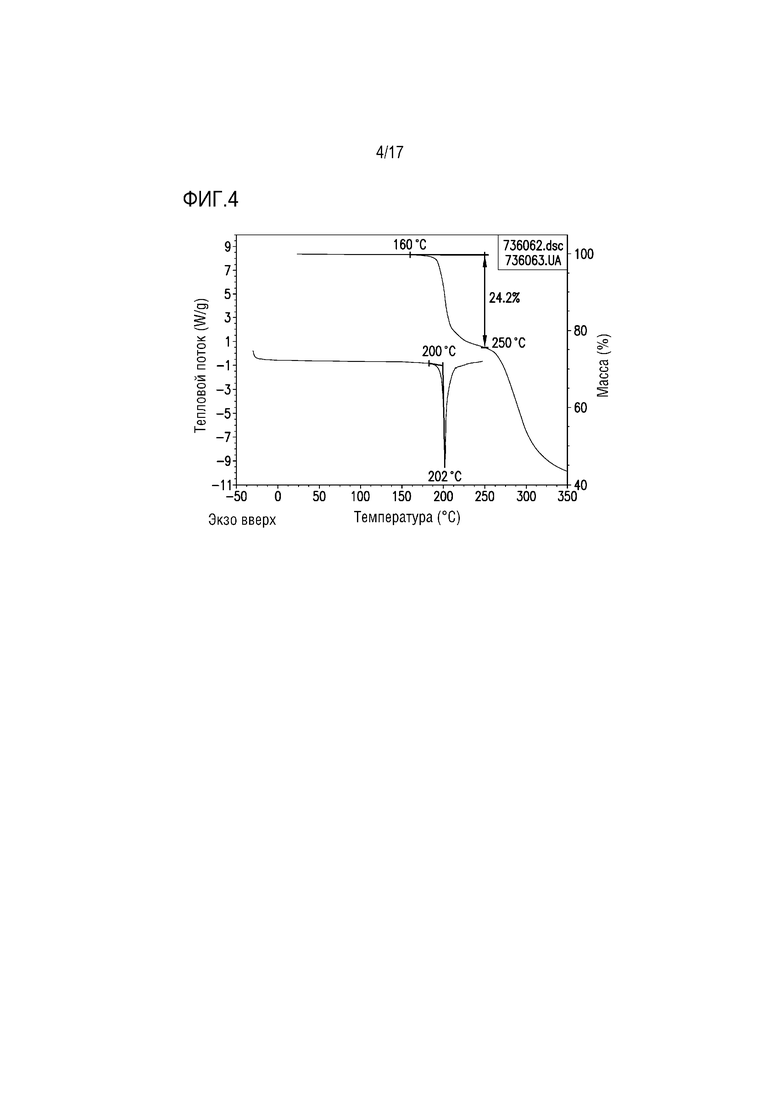
Фиг. 4 представляет собой наложение DSC и TGA для формы соли лимонной кислоты A соединения A.
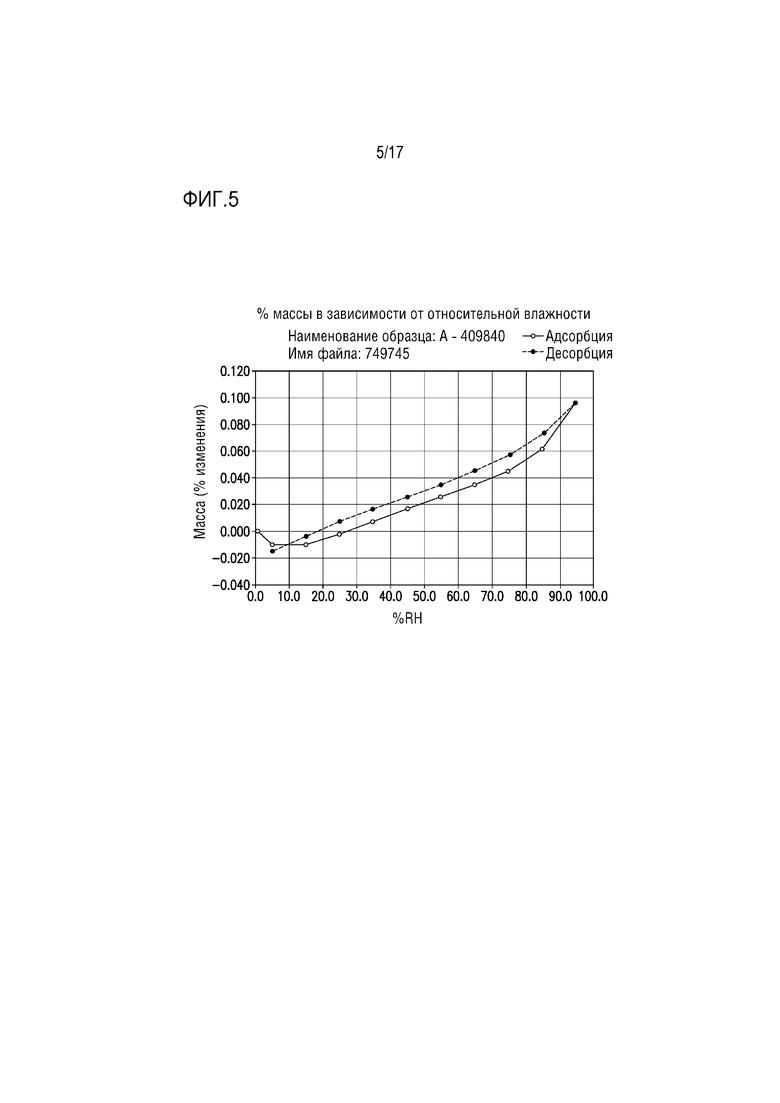
Фиг. 5 представляет собой изотерму DVS для формы соли лимонной кислоты A соединения A.
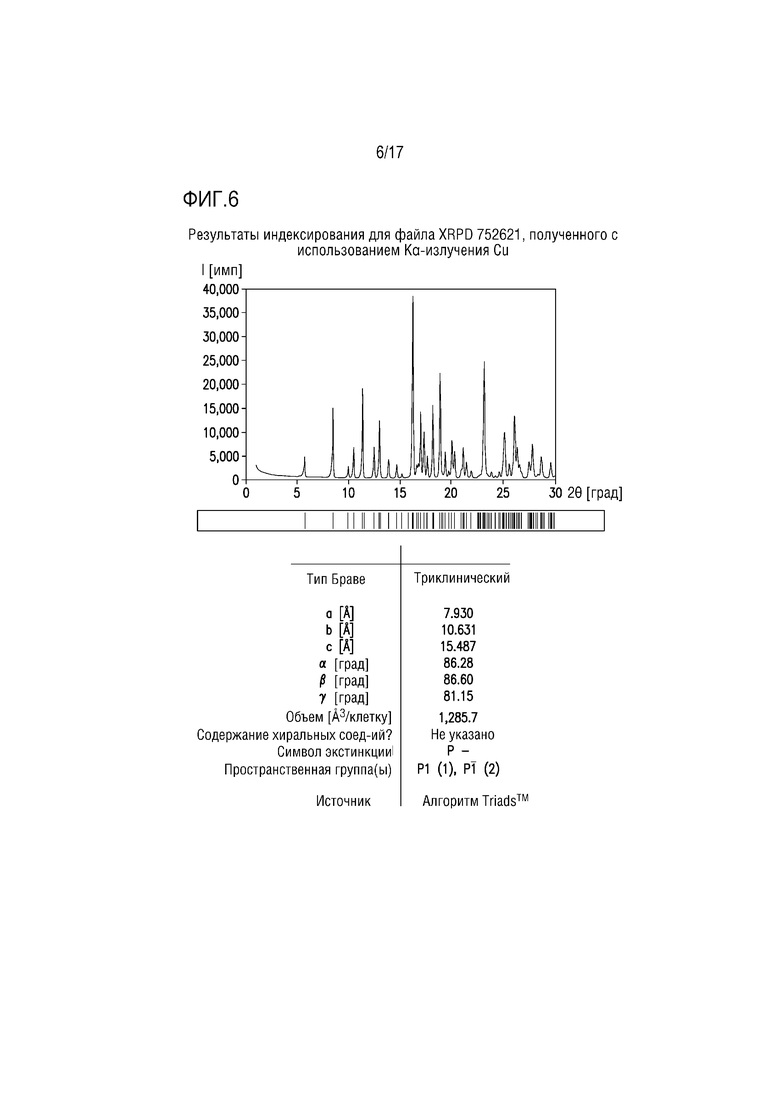
Фиг. 6 представляет собой характерный профиль XRPD для формы соли лимонной кислоты B соединения A.
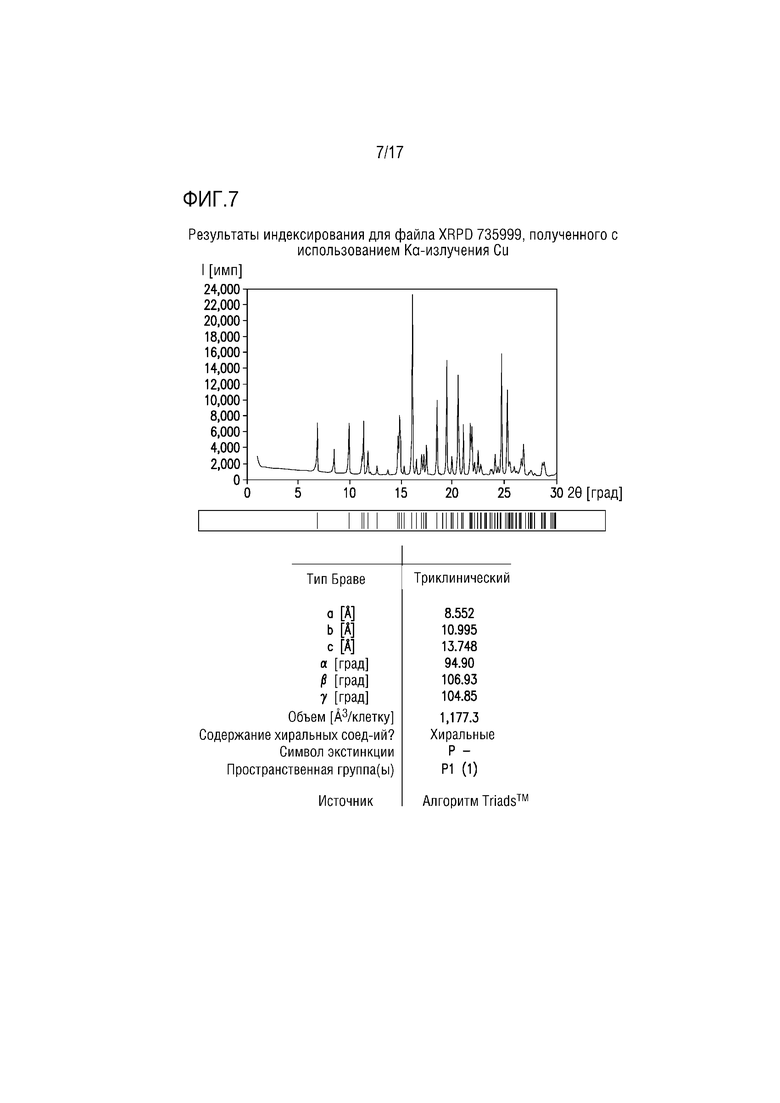
Фиг. 7 представляет собой характерный профиль XRPD для формы соли винной кислоты A соединения A.
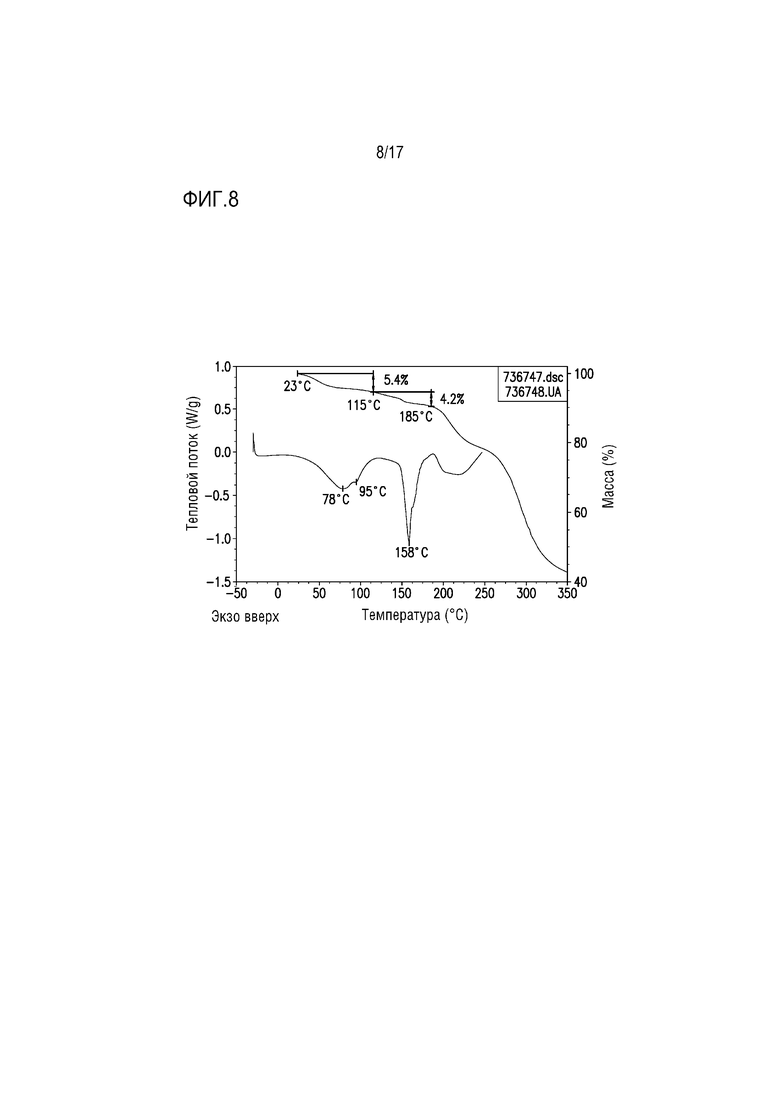
Фиг. 8 представляет собой наложение DSC и TGA для формы соли винной кислоты A соединения A.
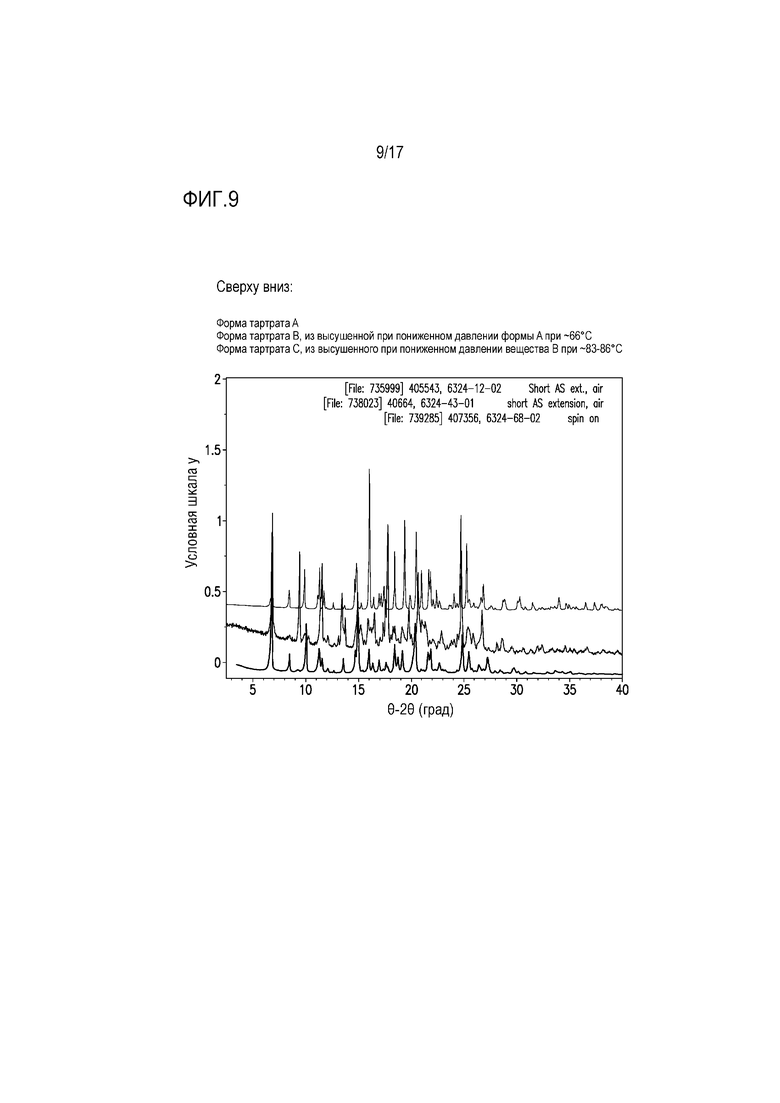
Фиг. 9 представляет собой наложение XRPD для соли винной кислоты формы A, формы B и формы C соединения A.
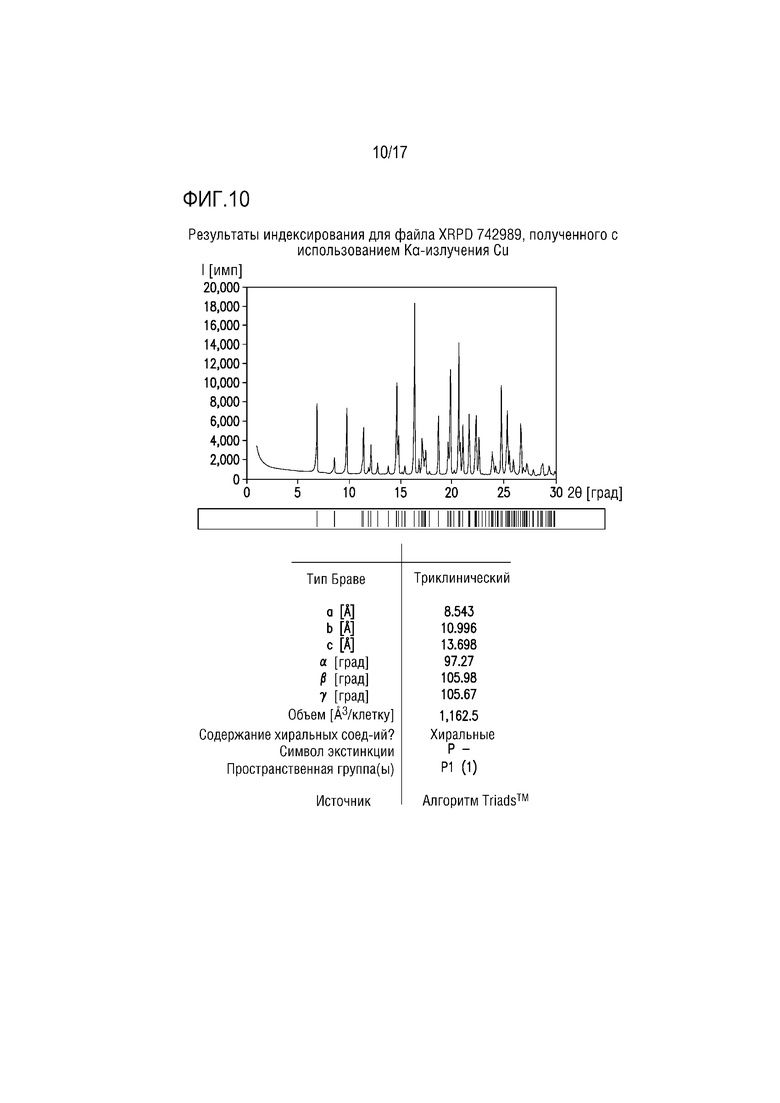
Фиг. 10 представляет собой характерный профиль XRPD для формы соли винной кислоты D соединения A.
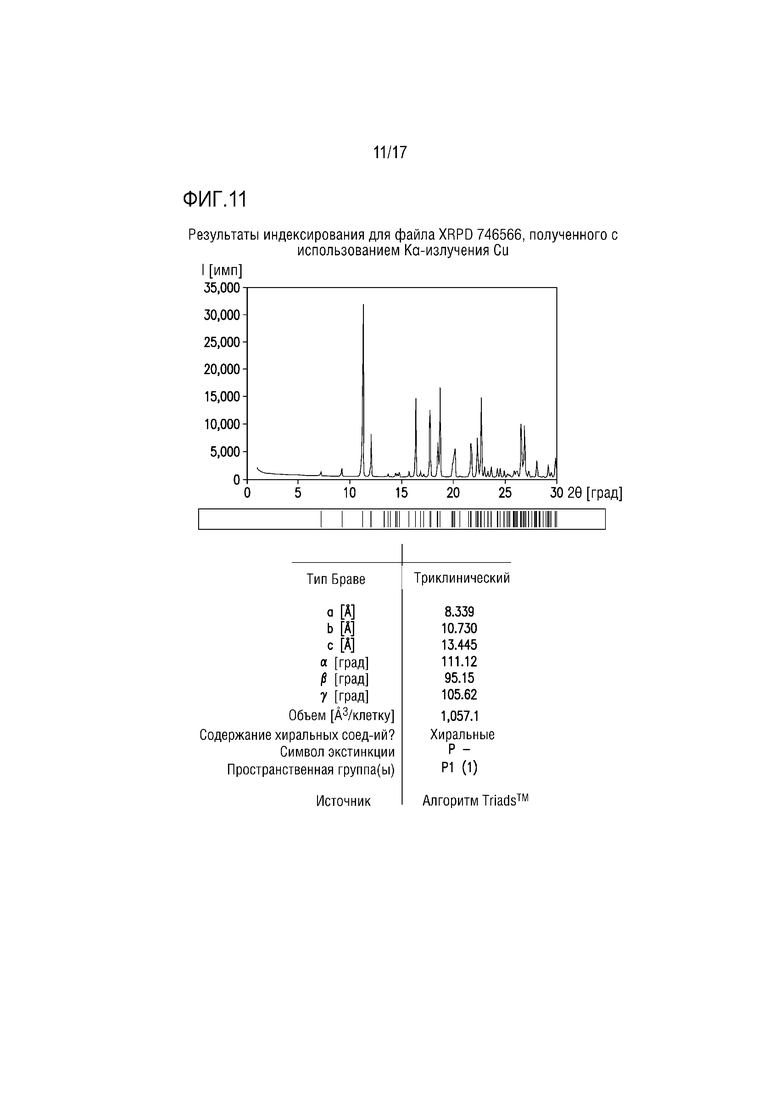
Фиг. 11 представляет собой характерный профиль XRPD для формы соли винной кислоты F соединения A.
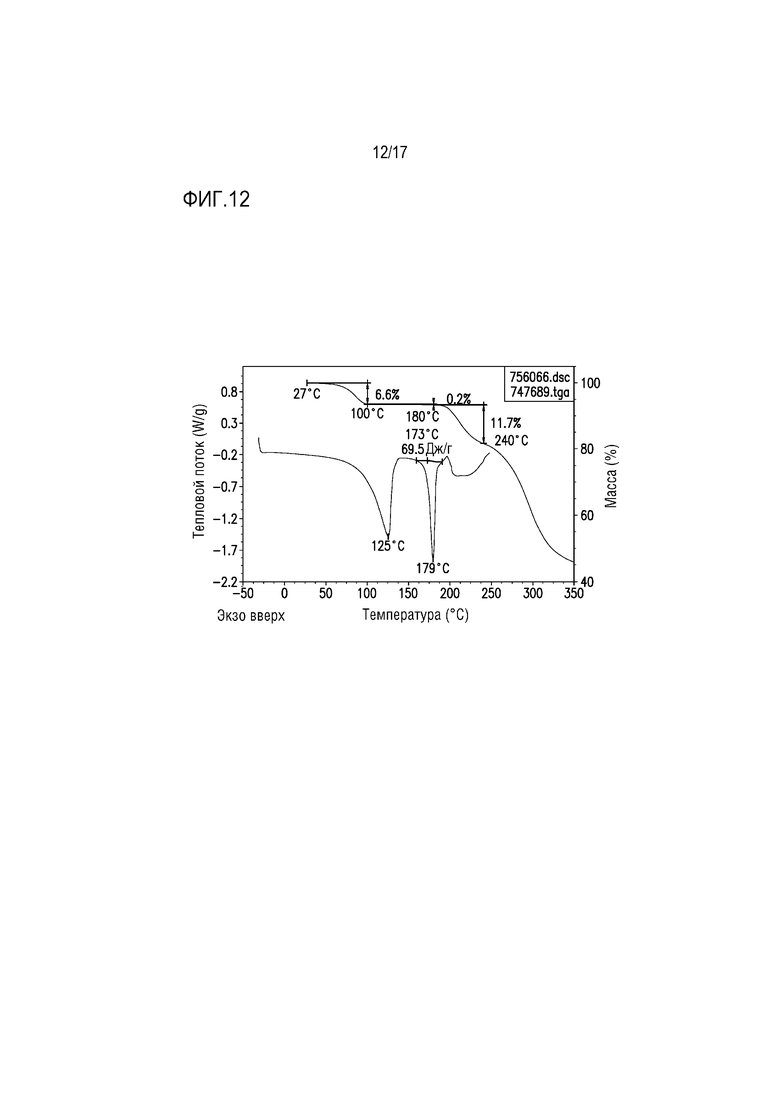
Фиг. 12 представляет собой наложение DSC и TGA для формы соли винной кислоты F соединения A.
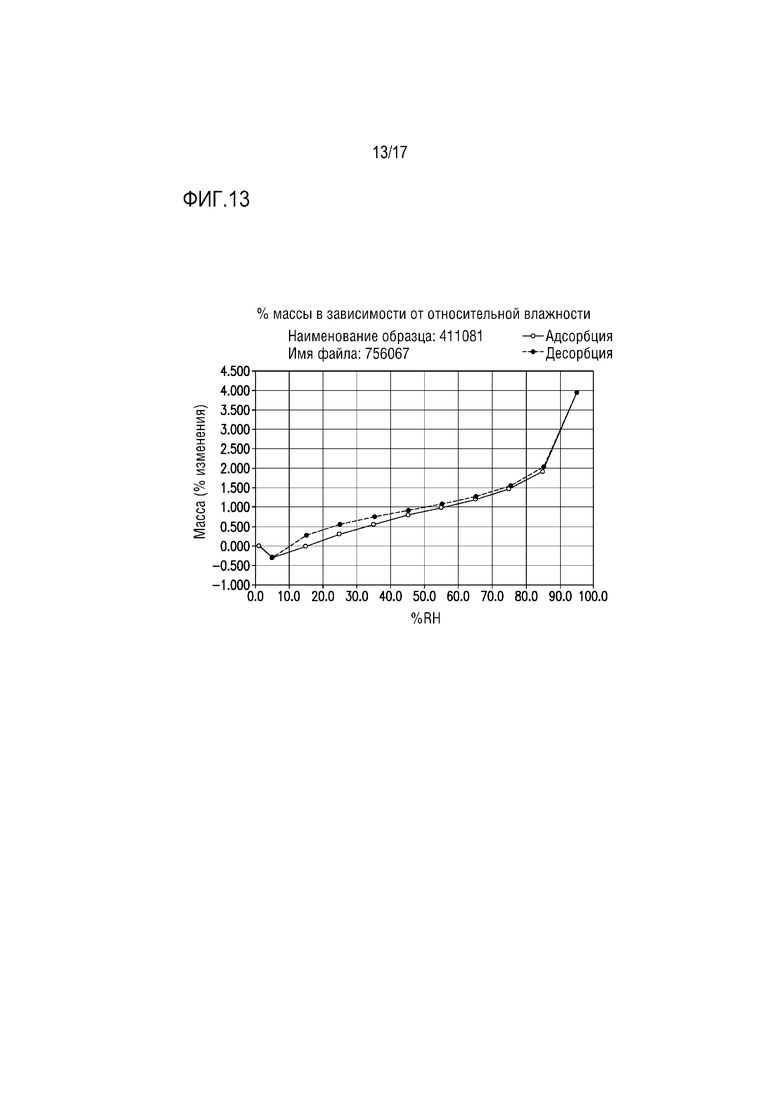
Фиг. 13 представляет собой изотерму DVS для формы соли винной кислоты F соединения A.
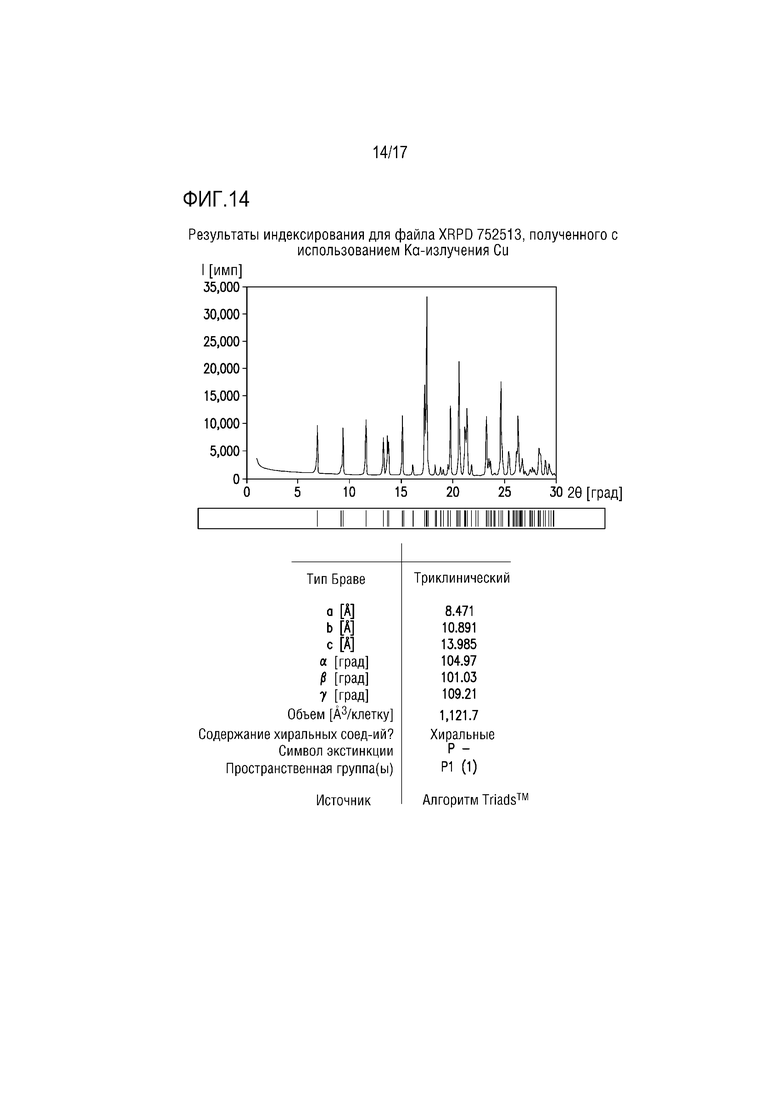
Фиг. 14 представляет собой характерный профиль XRPD для формы соли винной кислоты G соединения A.
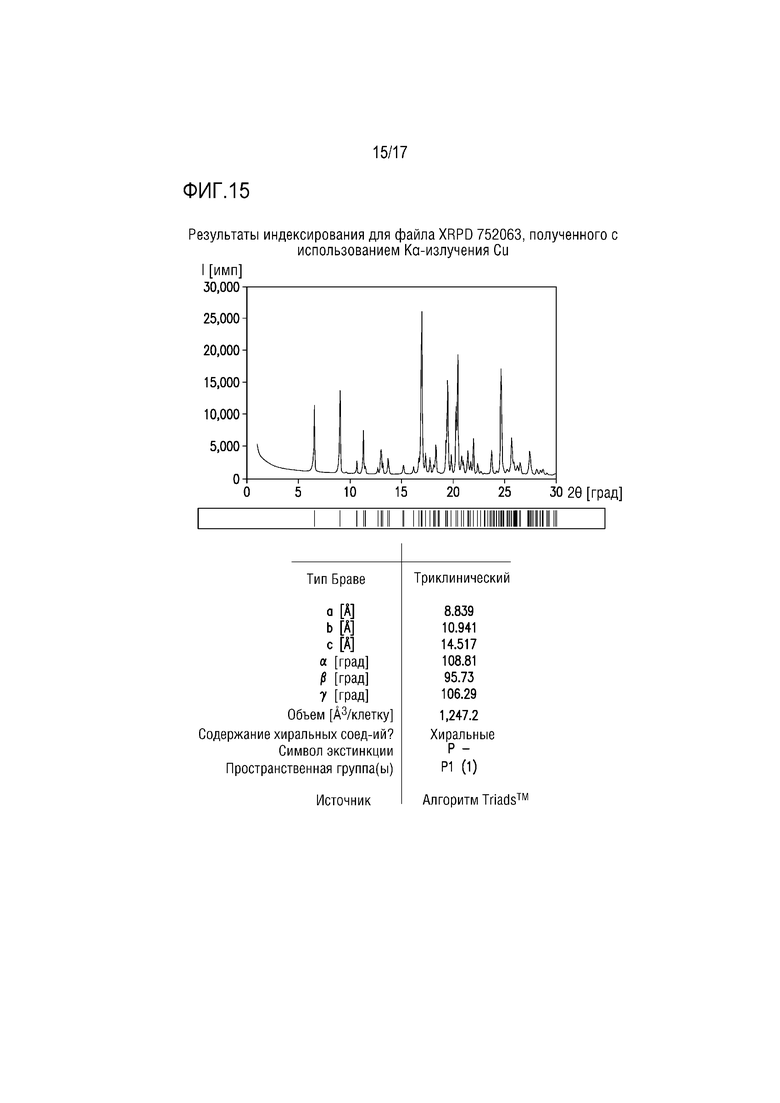
Фиг. 15. представляет собой характерный профиль XRPD для формы соли винной кислоты H соединения A.
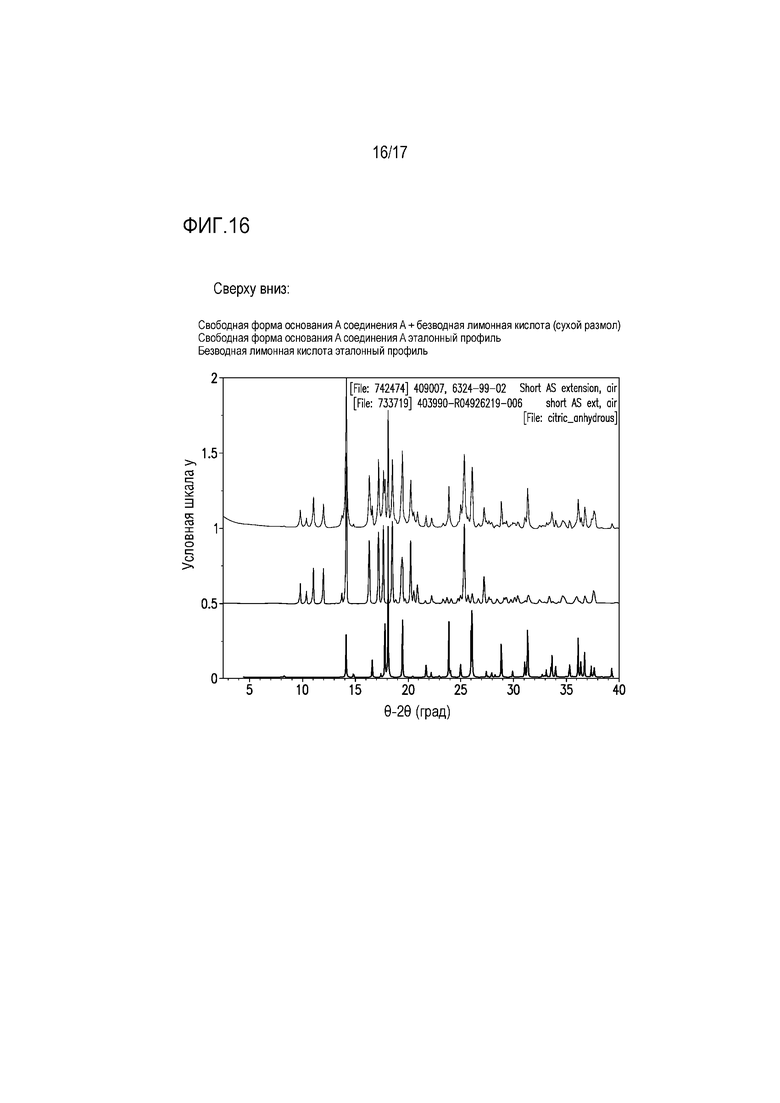
Фиг. 16 представляет собой наложение XRPD сухого размола соединения A с молярным эквивалентом лимонной кислоты.
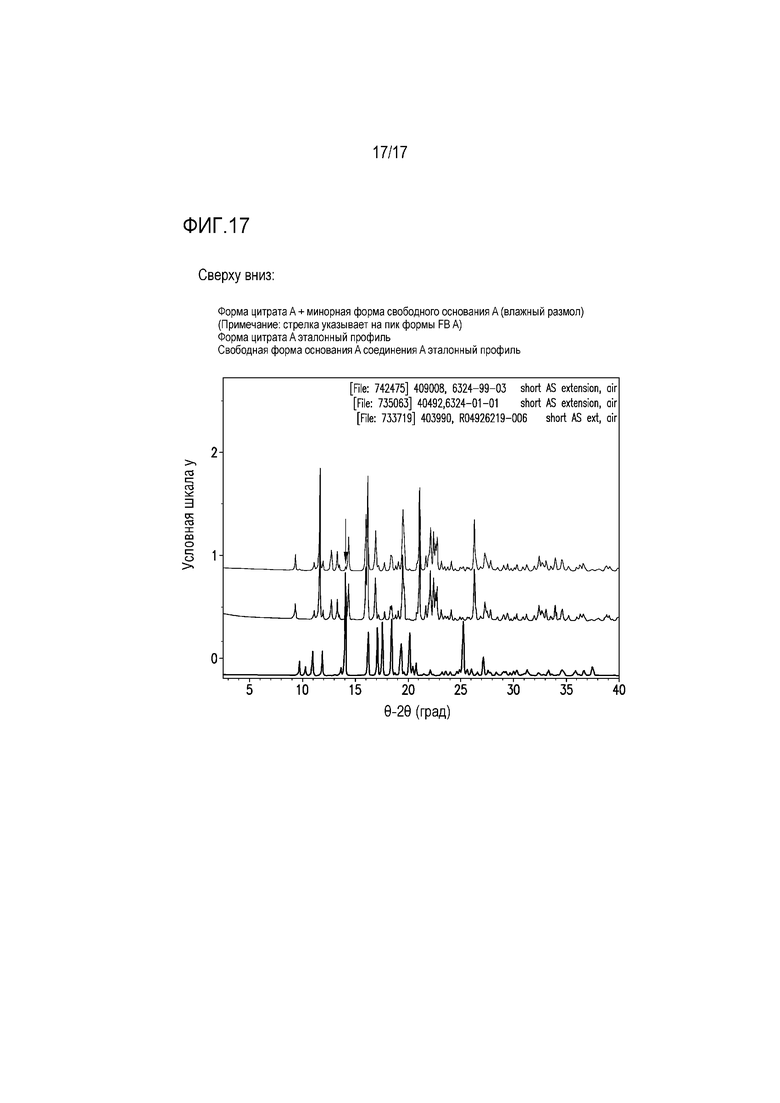
Фиг. 17 представляет собой наложение XRPD влажного размола соединения A с молярным эквивалентом лимонной кислоты.
Подробное описание изобретения
В настоящем документе описаны новые соли и кристаллические формы соединения A, ингибитор рецептора(ов) P2X3 и/или P2X2/3. Соединение A, 5-(2,4-диаминопиримидин-5-илокси)-4-изопропил-2-метокси-бензолсульфонамид, имеет следующую формулу:
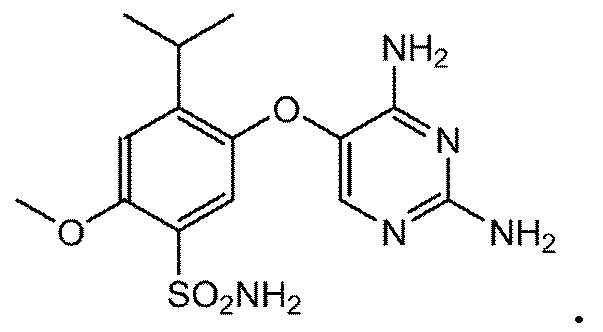
Соединение A можно получать способами, описанными в международной публикации WO2005/095359 (опубл. 13 октября 2005 года), патенте США №7858632 (опубл. 22 сентября 2005 года) и патенте США №7741484 (опубл. 1 марта 2007 года).
Новые кристаллические формы соединения A или его соли, особенно формы соли лимонной кислоты A, формы соли винной кислоты A и формы соли винной кислоты F, описываемые в настоящем документе, можно стабильно и постоянно получать с точки зрения производственного процесса, и они пригодны при потенциальном лечении опосредуемых P2X3 и/или P2X2/3 состояний. Эти кристаллические полиморфы обладают множеством неожиданных свойств по сравнению с формами свободного основания, как более подробно описано ниже.
В одном из вариантов осуществления кристаллические полиморфы, описываемые в настоящем документе, обладают улучшенными по сравнению с другими формами свойствами.
Новые кристаллические формы соединения A, описываемые в настоящем документе, обладают ингибирующим действием в отношении P2X3 и/или P2X2/3 и, таким образом, потенциально пригодны в качестве фармацевтических средств для лечения патологических состояний или нарушений, включая в качестве неограничивающих примеров нарушения мочевыводящих путей (также известные как уропатии), болезненные состояния, ассоциированные с мочевыводящими путями (также известные как заболевания мочевыводящих путей), гиперактивность мочевого пузыря (также известная как гиперактивность детрузора или недержание мочи), обструкцию выводящих путей (также известная как доброкачественная гипертрофия предстательной железы), недостаточность выводящих путей, тазовую гиперчувствительность, болевой синдром мочевого пузыря, эндометриоз, респираторные симптомы, кашель или позыв к кашлю, ассоциированные с заболеваниями органов дыхания, астму, гипертензию, сердечную недостаточность, одышку (также известную как укорочение дыхания), приступы апноэ во сне, признаки и симптомы гипертоничности и гиперрефлексии каротидных телец (такие как одышка и усталость) и гиперактивность симпатической нервной системы у индивидуума. Кроме того, соединением A, описываемым в настоящем документе, потенциально можно лечить признаки и симптомы инфекций верхних дыхательных путей, включая катарральные и респираторные симптомы фарингита, ринита, гиперемии слизистой оболочки носа, синдрома кашлевой гиперчувствительности, ринореи и затрагиваемых чиханием состояний.
В частности, новые кристаллические формы соединения A или его солей, а также само соединение A потенциально пригодны в качестве фармацевтических средств, например, для лечения респираторных симптомов, кашля или позывов к кашлю, ассоциированных с заболеваниями органов дыхания, и астмы.
Для характеристики молекулярных структур, степени кристаллизации и полиморфизма широко используют исследования рентгеновской порошковой дифракции (XRPD). Профили XRPD новых полиморфов, описываемых в настоящем документе, получали на дифрактометре PANalytical X'Pert PRO MPD с использованием падающий луч излучения Cu, получаемый с использованием острофокусного источника Optix long. Более подробно условия XRPD описаны в разделе примеров.
Профиль XRPD формы свободного основания A соединения A приведен на фиг. 1. Это вещество можно использовать в качестве исходного вещества для получения других форм, как более подробно описано в разделе примеров.
В одном из вариантов осуществления профиль XRPD формы соли лимонной кислоты A соединения A приведен на фиг. 3. В одном из вариантов осуществления форма цитрата A демонстрирует характеристические пики дифракции, соответствующие межплоскостным расстояниям d 11,69, 16,22 и 21,14 ангстрем. В другом варианте осуществления форма цитрата A дополнительно характеризуется межплоскостными расстояниями d 9,38 и 26,31 ангстрем. В другом варианте осуществления форма цитрата A дополнительно характеризуется межплоскостными расстояниями d 14,41 и 19,51 ангстрем. В другом варианте осуществления форма цитрата A дополнительно характеризуется межплоскостными расстояниями d 16,95 и 22,18 ангстрем.
В одном из вариантов осуществления профиль XRPD формы цитрата A демонстрирует характеристические пики дифракции, соответствующие межплоскостным расстояниям d 9,38, 11,69, 14,41, 16,22, 16,95, 19,54, 21,14, 22,18 и 26,31 ангстрем.
В дополнение к профилю XRPD, описанному выше, форма цитрата A также характеризуется спектрами протонного ядерного магнитного резонанса (ЯМР). В одном из вариантов осуществления данные протонного ЯМР позволили выявить, что отношение соединения A и цитрата составляет 1:1.
Форму цитрата A дополнительно охарактеризовывали с использованием DSC (дифференциальной сканирующей калориметрии) и TGA (термогравиметрического анализа). В одном из вариантов осуществления термограммы DSC/TGA для формы цитрата A приведены на фиг. 4. При TGA до 160°C наблюдали незначительную потерю массы, что соответствовало безводному/несольватированному веществу. Скачкообразная потеря массы приблизительно на 24 мас.% при температурах от 160°C до 250°C и эндотермический процесс при DSC, начиная с 200°C, вероятно, указывают на одновременное плавление и разрушение вещества.
В одном из вариантов осуществления изображения на высокотемпературном микроскопе для формы цитрата A подтвердили начало плавления приблизительно при 193°C, незначительно меньше, чем начало плавления отмеченное на термограмме DSC на фиг. 4 (200°C).
Форму цитрата A дополнительно охарактеризовывали посредством анализа DVS (динамической сорбции паров). В одном из вариантов осуществления изотерма DVS, приведенная на фиг. 5, демонстрировала низкую кинетическую гигроскопичность (0,11% общего увеличения/потери массы от 5% до 95% относительной влажности).
Форма цитрата A по сравнению с формой свободного основания A обладала неожиданными свойствами. В одном из вариантов осуществления форма цитрата A по сравнению с формой свободного основания A демонстрировала улучшенную водорастворимость и не демонстрировала признаков диспропорционирования при концентрации 6 мг/мл в течение периода продолжительностью до месяца. Кроме того, при подвергании соли нагрузке ≈97% относительной влажности в течение 14 суток не выявлено смачивания за счет поглощения влаги из воздуха.
В одном из вариантов осуществления профиль XRPD формы соли лимонной кислоты B соединения A приведен на фиг. 6. В одном из вариантов осуществления форма цитрата B демонстрирует характеристические пики дифракции, соответствующие межплоскостным расстояниям d 8,42, 16,15 и 23,04 ангстрем. В другом варианте осуществления форма цитрата A дополнительно характеризуется межплоскостными расстояниями d 5,71 и 18,77 ангстрем. В другом варианте осуществления форма цитрата A дополнительно характеризуется межплоскостными расстояниями d 11,30 и 26,01 ангстрем. В другом варианте осуществления форма цитрата A дополнительно характеризуется межплоскостными расстояниями d 12,93 и 25,02 ангстрем.
В одном из вариантов осуществления профиль XRPD формы цитрата B демонстрирует характеристические пики дифракции, соответствующие межплоскостным расстояниям d 5,71, 8,42, 11,30, 12,93, 16,15, 18,77, 23,04, 25,02 и 26,01 ангстрем.
В одном из вариантов осуществления данные протонного ЯМР формы цитрата B позволили выявить, что отношение соединения A и соли лимонной кислоты составляет 1:1 лишь с небольшим присутствующим количеством остаточного этанола. Никакое количество воды, которое может присутствовать, невозможно количественно определить посредством протонного ЯМР вследствие изначального присутствия воды в растворителе ЯМР (дейтерированный DMSO).
В одном из вариантов осуществления титрование по Карлу Фишеру продемонстрировало, что образец, содержащий форму цитрата B, содержал приблизительно 3 мас.% воды. Это количество воды коррелирует с моногидратом соли лимонной кислоты 1:1.
В одном из вариантов осуществления профиль XRPD формы соли винной кислоты A соединения A приведен на фиг. 7.
В одном из вариантов осуществления спектр протонного ЯМР для формы тартрата A позволил выявить соль 2:1 с 0,4 моль EtOH на моль соединения A, из чего можно заключить, что стехиометрия соединения A/кислоты/EtOH составляет ≈2:1:1.
В одном из вариантов осуществления наложение термограмм DSC и TGA для формы тартрата A приведено на фиг. 8. Широкая эндотерма с пиковым максимумом при 78°C и плечевым пиком при 95°C на термограмме DSC соответствует приблизительно 5% потере массы от 23 до 115°C при TGA в соответствии с потерей растворителя. Уровень потери массы соответствует ≈1 моль EtOH на моль соли винной кислоты соединения A 2:1 в соответствии с количеством EtOH, определяемого посредством протонного ЯМР. Этап дополнительной потери массы приблизительно 4 мас.% соответствует эндотермическому процессу с пиковым максимумом при 158°C, вероятно, соответствующему одновременным плавлению, диссоциации и разрушению соли.
В одном из вариантов осуществления профиль XRPD формы соли винной кислоты F соединения A приведен на фиг. 11. В одном из вариантов осуществления форма тартрата F демонстрирует характеристические пики дифракции, соответствующие межплоскостным расстояниям d 11,25, 18,73 и 22,67 ангстрем. В другом варианте осуществления форма тартрата F дополнительно характеризуется межплоскостными расстояниями d 12,06 и 17,74 ангстрем. В другом варианте осуществления форма тартрата F дополнительно характеризуется межплоскостными расстояниями d 9,22 и 26,52 ангстрем. В другом варианте осуществления форма тартрата F дополнительно характеризуется межплоскостными расстояниями d 16,37 и 21,69 ангстрем.
В одном из вариантов осуществления профиль XRPD формы тартрата F демонстрирует характеристические пики дифракции, соответствующие межплоскостным расстояниям d 9,22, 11,25, 12,06, 16,37, 17,74, 18,73, 21,69, 22,67 и 26,52 ангстрем.
В дополнение к профилю XRPD, описанному выше, форму тартрата F также охарактеризовывали посредством анализа протонного ядерного магнитного резонанса (ЯМР). В одном из вариантов осуществления спектр протонного ЯМР позволил выявить отношение соли винной кислоты соединения A 2:1.
Форму тартрата F дополнительно охарактеризовывали посредством DSC и TGA. В одном из вариантов осуществления наложение термограмм DSC и TGA для формы тартрата F представлено на фиг. 12. Этап начальной потери массы приблизительно 6,6 мас.% при температурах 27-100°C при TGA соответствует широкой эндотерме при DSC при 125°C, вероятно соответствующей потере растворителя. Потеря массы соответствует ≈3 моль воды на моль соли 2:1 в соответствии с количеством воды, допускаемым объемом элементарной ячейки. Фактически, потери массы при 100-180°C не выявлено. За относительно крутой эндотермой при 179°C сразу следует резкая потеря массы при температурах выше 180°C, вероятно свидетельствующая об одновременных плавлении и разрушении дегидратированного вещества.
В одном из вариантов осуществления анализ микрофотографий при высокотемпературной микроскопии формы тартрата F иллюстрирует изменения двойного лучепреломления, выявляемые в интервале от 70°C до 98°C, соответствующие постепенной потере массы и широкой эндотерме, выявляемой при анализе данных TGA и DSC, что, вероятно, свидетельствует о дегидратации образца при нагревании. При температурах ≈171°C-176°C наблюдали плавление, подтверждающее, что крутая эндотерма DSC с началом 173°C соответствует плавлению дегидратированного вещества.
В одном из вариантов осуществления титрование по Карлу Фишеру формы тартрата F выявило приблизительно 7,3% воды, что эквивалентно приблизительно 3,7 моль воды на моль соли 2:1. Это содержание воды несколько больше, чем ≈3 моль воды, выявляемое при анализе потери массы при TGA.
В одном из вариантов осуществления изотерма DVS для формы тартрата F приведена на фиг. 13. Вещество демонстрировало относительно небольшую потерю массы при уравновешивании до 5% относительной влажности (0,29 мас.%), что свидетельствует, что гидрат вероятно оставался интактным в начале адсорбции. При 5%-95% относительной влажности выявляли значительную гигроскопичность, где образец приобретал всего приблизительно 4,23% массы. Приблизительно половина увеличения массы происходила постепенно от 5% до 85% относительной влажности, тогда как другая половина происходила в течение одного этапа от 85% до 95% относительной влажности.
Хотя по данным эксперимента DVS наблюдали значительное увеличение массы, гидратированное вещество не продемонстрировало признаков смачивания за счет поглощения влаги из воздуха при ≈97% относительной влажности в течение скрининга. Профиль десорбции отражает профиль сорбции с очень небольшим наблюдаемым гистерезисом. Общая потеря массы приблизительно 4,25 мас.% происходила при относительной влажности от 95% до 5%, приблизительно с половиной потери массы, происходящей за один этап от 95% до 85% относительной влажности. XRPD образца после DVS не выявил изменения формы.
Форма тартрата F продемонстрировала улучшенную водорастворимость при добавлении растворителя по сравнению с формой свободного основания A соединения A, хотя через 1 сутки наблюдали начало диспропорционирования в чистой воде.
ПРИМЕРЫ
Пример 1: Получение формы свободного основания A соединения A
Этап 1. Получение сульфоновой кислоты соединения
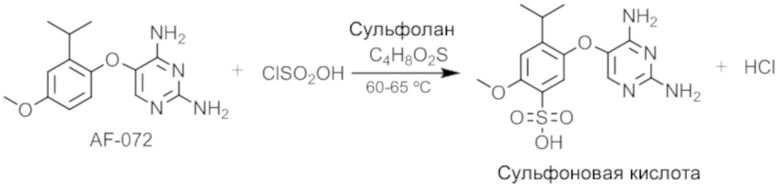
AF-072 и сульфолан раздельно помещали в реактор и температуру доводили до 50-55°C. Медленно добавляли хлорсульфоновую кислоту, поддерживая температуру на уровне 50-62°C, для начала реакции сульфонирования. Температуру доводили до 58-62°C и удерживали в течение 4 часов с последующим отбором образца для завершения реакции. Если необходимо, можно использовать импульсное введение хлорсульфоновой кислоты.
Этап 2. Получение сульфоналхлорида соединения
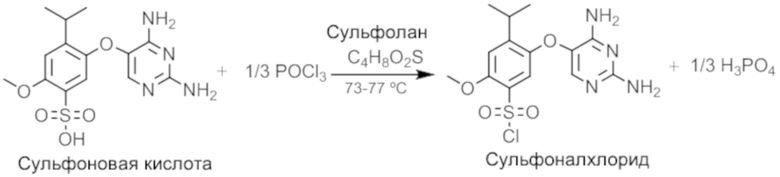
Партию после этапа 1 доводили до 65-70°C, а затем добавляли оксихлорид фосфора, поддерживая температуру при 60-70°C. Партию доводили до 73-77°C и удерживали в течение 17 часов с последующим отбором образца для завершения реакции. После дополнительных 3 часов выдерживания отбирали образец. Если необходимо, можно использовать импульсное введение оксихлорида фосфора.
Этап 3. Получение соединения A
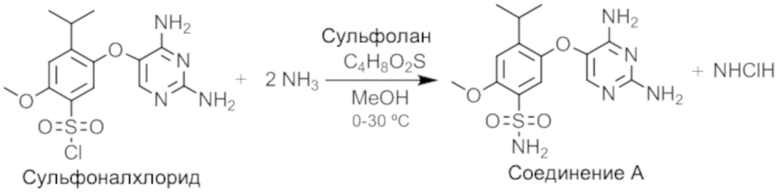
В другой сосуд помещали метанол и охлаждали до температуры от -10°C до 0°C. К метанолу медленно добавляли безводный аммиак, поддерживая температуру ниже 0°C. После завершения добавления раствор аммиака в метаноле доводили до 0-5°C.
Раствор после реакции хлорирования переносили в метанольный аммиак, поддерживая температуру при 0-30°C. Для прочистки канала переноса использовали сульфолан, а затем партию доводили до 18-22°C и удерживали в течение 2,5 часов. В дополнение к получению неочищенного соединения A, метанольный аммиак также действует, гася остаточные хлорсульфоновую кислоту, оксихлорид фосфора и фосфорную кислоту, с формированием водорастворимых солей, которые удаляли при выделении.
После завершения гашения партию подвергали перегонке при пониженном давлении с охлажденной водой на кожухе с удалением метанола и остаточного аммиака. В баню добавляли воду, а затем температуру доводили до 93-97°C и удерживали в течение 1 часа. Партию охлаждали в течение приблизительно 13 часов до 18-22°C и продукт выделяли на центрифуге с получением отфильтрованного осадка соединения A.
Этап 4. Перегонка и выделение соединения A
Содержимое после этапа 3 охлаждали до 0-10°C. Применяли пониженное давление с перегонкой и удалением метанола и аммиака. Перегонку начинали при 5,4°C и нагревание продолжали в течение приблизительно 8 часов до достижения температуры партии 67,7°C. В этот момент дистиллят переставал поступать, что свидетельствовало об удалении всего метанола и аммиака. После остановки перегонки партию доводили до 60-70°C.
В отдельный реактор помещали воду и нагревали до 60-70°C. Затем вещество переносили в другой реактор, поддерживая температуру 60-70°C. В этот момент добавляли воду, нагревали до 60-70°C и переносили в другой блок. Затем блок нагревали до 93-97°C и удерживали в течение 2 часов. После завершения этой инкубации начинали охлаждение для кристаллизации. Затем вещество охлаждали до 20°C в течение приблизительно 12 часов и выдерживали в течение 1 часа.
Затем вещество центрифугировали. После перенесения всего вещества в центрифугу, через проводящий канал для взвеси пропускали 53 литра прогоняемого раствора с выделением оставшегося продукта. Всего центрифугирование занимало приблизительно 6 часов, и из блока выделяли отфильтрованный осадок. Затем вещество высушивали в сушке, получая форму свободного основания соединения A.
Форму свободного основания A соединения A использовали в качестве исходного вещества для получения других солей и форм.
Пример 2: Характеристика формы свободного основания A соединения A
Форму свободного основания A охарактеризовывали посредством XRPD и спектроскопии протонного ЯМР. Профиль XRPD (фиг. 1) демонстрировал острые пики в соответствии с кристаллическим веществом, и его успешно проиндексировали. Объем элементарной ячейки, получаемый на основе индексирующего разрешения, соответствует безводному/несольватированному соединению A. Спектр протонного ЯМР для вещества соответствует химической структуре свободному основанию соединения A.
С использованием формы свободного основания А проведен скрининг солей, в результате чего получены соли и полиморфы, более подробно описанные ниже.
В таблице 1 предоставлены расчеты растворимости (при добавлении растворителя) формы свободного основания A соединения A в различных растворителях. Растворимости рассчитывали на основе всего растворителя с использованием данного раствора; фактические растворимости могут быть выше ввиду используемых объемов порций растворителя или низкой скорости растворения. Значения округлены до ближайшего целого числа. Если растворения не происходило, что определяли посредством визуальной оценки, значение представлено как "<".
Таблица 1. Приблизительные растворимости формы свободного основания A соединения A в различных растворителях
Пример 3: Форма свободного основания B соединения A
Форма свободного основания B может состоять из сольвата свободного основания соединения A с ацетоном, хотя химический состав не подтвержден. Профиль XRPD (фиг. 2) успешно проиндексирован, подтверждая, что образец преимущественно или исключительно состоит из одной кристаллической фазы. Объем элементарной ячейки, получаемый на основе индексирующего разрешения, обеспечивает до ≈1 моль ацетона на моль свободного основания соединения A.
Пример 4: Скрининг солей соединения A
Скрининг солей проводили с использованием формы свободного основания A и лимонной кислоты и L-винной кислоты, соответственно. Скрининг начинали, комбинируя свободное основание соединения A и выбранные кислоты в молярном соотношении 1:1. Вследствие низкой растворимости свободного основания, большинство экспериментов включали добавление к взвеси свободного основания кислоты (или в виде твердого вещества, или в виде раствора). В определенных экспериментах после контакта получали прозрачные или почти прозрачные растворы с последующим осаждением, означающим формирование соли. Подробные экспериментальные условия для получения солей лимонной и винной кислоты приведены в таблице 2.
Как использовано в таблице 2, молярное соотношение приведено в виде API/кислота, и API представляет собой форму свободного основания A соединения A. Температуры (°C), приведенные для DSC, представляют собой максимумы переходов, если не указано иначе, и округлены до ближайшего градуса. Потерю массы (%), приводимую для TGA при определенной температуре, округляют до 1 десятичного знака.
Таблица 2. Скрининг солей соединения A с использованием формы свободного основания A в качестве исходного вещества
2) взвесь, комнатная температура, 1 сутки
3) вакуумн. фильтр
2) непрозрачная белая суспензия
3) белые твердые вещества
2) добавить лимонной кислоты (1 M, водн.) с перемешиванием
3) взвесь, комнатная температура, 1 сутки
4) вакуумн. фильтр
3) непрозрачная белая суспензия
4) белые твердые вещества
2) добавить L-винной кислоты (1 M, водн.) с перемешиванием
3) взвесь, комнатная температура, 1 сутки
4) вакуумн. фильтр
3) непрозрачная белая суспензия
4) белые твердые вещества
2) добавить L-винной кислоты (1 M, водн.) с перемешиванием
3) взвесь, комнатная температура, 1 сутки
4) вакуумн. фильтр
3) непрозрачная белая суспензия
4) белые твердые вещества
2) добавить затравки виннокислого вещества C), взвесь при ≈70-71°C, 1 сутки
3) вакуумн. фильтр при теплом веществе
3) белые твердые вещества
2) добавить затравок виннокислого вещество C, взвесь при комнатной температуре, 12 суток
3) вакуумн. фильтр
3) белые твердые вещества
2) дополнительно добавить воды, перемешивание, ≈49-50°C, 2 суток
3) вакуумн. фильтр при теплом веществе
2) непрозрачная белая суспензия
3) белые твердые вещества
потеря 0,2 мас.% 100-180°C
потеря 11,7 мас.% 180-240°C
После первого цикла экспериментов выявлены несколько новых материалов, но для многих профилей XRPD наблюдали избыток непрореагировавшего свободного основания. В следующем цикле экспериментов добавляли избыток кислоты (соединение A/кислота от 1:1,2 до 1:1,5) с целью избежать осаждения свободного основания. Этот способ был более успешным для получения новых веществ в виде одной твердой фазы. Как правило, если индексирование было успешным, уникальные вещества подвергали индексированию XRPD для оценки чистоты фазы и возможных стехиометрических отношений, обеспечиваемых объемом элементарной ячейки.
Выбранные вещества дополнительно охарактеризовывали посредством протонного ЯМР для подтверждения химического состава. Определенные представляющие интерес вещества, для которых выявлено сольватирование, сушили в различных условиях с целью получения безводных/несольватированных формы солей. Кроме того, для выбранных солей приблизительно оценивали водорастворимость и физическую стабильность.
На основе исходного скрининга получали уникальные формы, включая форму цитрата A и формы тартрата A, D и F. В дополнение к найденным подтвержденным и потенциальным солям, проводили несколько экспериментов по скринингу солей в ацетоне с получением вещества, обозначенного как вероятная форма свободного основания B. Объем элементарной ячейки, получаемый на основе индексирующего разрешения XRPD, мог позволять обеспечивать свободному основанию соединения A до 1 моль ацетона на моль соединения A. Учитывая несколько получений, все содержащие ацетон системы растворителей и объем элементарной ячейки, вещество вероятно состоит из сольвата свободного основания соединения A с ацетоном.
Наблюдали несколько форм солей винной кислоты. Тартрат соединения A демонстрировал свойство формировать несколько сольватированных форм, а также гидрат.
Форму тартрата A получали из эксперимента по получению соли в EtOH с использованием 1 M водной L-винной кислоты. Данные характеристики для формы тартрата A свидетельствуют о сольватированной EtOH полусоли винной кислоты. На основе этого форму тартрата A сушили при пониженном давлении при ≈66°C в течение 1 суток, что приводило к преобразованию в новое вещество, обозначенное как форма тартрата B, и ≈9% гравиметрической потере массы. Наблюдали сходство в некоторых положениях пика XRPD с положением для формы A, что, возможно, указывает на частичное десольватацию в тестируемых условиях, поэтому образец формы B дополнительно сушили при пониженном давлении при ≈83-86°C в течение 1 суток, что приводило к преобразованию в другое новое вещество, обозначенный как форма тартрата С. Как и для формы B, наблюдали сходства профилей XRPD для формы C и форма A, возможно указывающие на неполную сушку. Форму C использовали в качестве затравок для нескольких экспериментов по получению солей винной кислоты с целью получения безводной/несольватированной формы.
Учитывая стремление соли винной кислоты к существованию в виде сольвата, проведено несколько дополнительных экспериментов с целью получения безводной/несольватированной соли винной кислоты. Эксперимент по получению соли проводили в EtOH при ≈70°C с использованием затравок формы тартрата C и молярного соотношения соединения A/кислоты 2:1 (вероятно, предпочтительная стехиометрия, основанная на характеристике ранее поученных образцов соли винной кислоты). Полученную взвесь перемешивали при ≈70-71°C в течение 1 суток, и получали форму тартрата D, подтвержденный сольват с EtOH. Проводили дополнительный эксперимент по получению солей в 2-BuOH, также с затравками из формы C, с целью предотвращения образования сольвата посредством использования химически более объемного растворителя. Этот эксперимент приводил к получению формы E, сольватированную 2-BuOH соль.
С целью получению гидрата соли винной кислоты проводили эксперимент по получению солей в воде при ≈50°C. Для увеличения растворимости свободного основания использовали немного повышенную температуру, что вероятно может увеличить кинетику реакции и облегчить получение соли. В эксперименте получали гидратированную полусоль винной кислоты, обозначенную как форму тартрата F, и выбирали для дальнейшего исследования.
Выбранные вещества цитраты и тартраты оценивали посредством нагрузки при высокой относительной влажности, и рассчитывали водорастворимость при температуре окружающей среды. Форма цитрата A и форма тартрата B (высушенные сольваты с EtOH) не продемонстрировали признаков смачивания за счет поглощения влаги из воздуха при ≈97% относительной влажности через 7-14 суток. Форма цитрата A и форма тартрата F продемонстрировали улучшенную водорастворимость по сравнению с формой свободного основания A соединения A при добавлении растворителя (6 мг/мл для цитрата, 2 мг/мл для тартрата, < 1 мг/мл для свободного основания). Форма цитрата A не продемонстрировала признаков диспропорционирования, поддерживая прозрачный раствор при концентрации 6 мг/мл до 1 месяц с определенным pH ≈4. Вероятно диспропорционирование наблюдали для растворяемого образца формы тартрата F, так как наблюдали небольшое количество белого осадка после пребывания в условиях окружающей среды в течение 1 суток.
Форму цитрата A (безводная/несольватированная соль лимонной кислоты 1:1) и форму тартрата F (гидратированная соль винной кислоты соединения A 2:1) дополнительно охарактеризовывали. Оба вещества успешно воспроизводили в масштабе ≈1-1,2 г. Форму цитрата A получали, комбинируя 1 M водную лимонную кислоту со взвесью формы свободного основания A соединения A в EtOH. Эксперимент повторяли в масштабе ≈5,6 г, успешно получая форму цитрата A. Полученное вещество использовали в сокращенном скрининге стабильной формы цитрата A. Масштабирование формы тартрата F для масштаба ≈1 г также было успешным, с комбинацией водного раствора L-винной кислоты с формой свободного основания A при ≈51°C, с затравкой в виде формы тартрата F и оставлением смеси для перемешивания при ≈51°C в течение 1 суток. Вещество масштабированной формы тартрата F использовали для сушки и исследований взвеси, более подробно описанных ниже. Подробные процедуры для масштабирования обоих веществ приведены ниже.
Пример 5: Форма цитрата A соединения A
Форма цитрата A состоит из безводной/несольватированной соли лимонной кислоты соединения A 1:1, и ее воспроизводимо получали посредством добавления водной лимонной кислоты ко взвеси свободного основания соединения A в EtOH и перемешивания в течение длительного периода. В одном из вариантов осуществления форму цитрата A получали с использованием условий, приведенных в таблице 2.
Профиль XRPD для формы цитрата A соединения A приведен на фиг. 3. Списки наблюдаемых и выраженных пиков приведены в таблицах 3 и 4, соответственно.
Таблица 3. Список наблюдаемых пиков XRPD для формы цитрата A
Таблица 4. Список выраженных пиков XRPD для формы цитрата A
Форму цитрата A анализировали посредством протонного ЯМР, и спектр соответствовал соли лимонной кислоты соединения A 1:1 с присутствием незначительного остаточного EtOH.
Наложение термограмм DSC и TGA для формы цитрата A приведено на фиг. 4. Наблюдали незначительную потерю массы при TGA до 160°C в соответствии с безводным/несольватированным веществом. Постепенная потеря массы приблизительно 24 мас.% от 160°C до 250°C соответствует эндотермическому процессу при DSC с началом при 200°C, вероятно свидетельствующим об одновременных плавлении и разрушении вещества.
Изображения на высокотемпературном микроскопе для вещества подтверждают начало плавления приблизительно при 193°C, немного менее, чем начало плавления, видимое на термограмме DSC на фиг. 4 (200°C).
Изотерма DVS (динамической сорбции паров), приведенная на фиг. 5, иллюстрирует низкую кинетическую гигроскопичность (приблизительно 0,11% общего увеличения/потери массы при относительной влажности от 5% до 95%).
Форма цитрата A обладает несколькими неожиданными свойствами. Она продемонстрировала улучшенную водорастворимость при добавлении растворителя (6 мг/мл) по сравнению с формой свободного основания A и не продемонстрировала признаков диспропорционирования при концентрации 6 мг/мл в течение периода ≈1 месяц. Она обладает улучшенной физической стабильностью при ряде условий, и не продемонстрировала смачивания за счет поглощения влаги из воздуха при нагрузке соли при ≈97% относительной влажности в течение 14 суток. Кроме того, объем элементарной ячейки, получаемый на основе индексирующего разрешения XRPD соответствует безводной/несольватированной соли лимонной кислоты соединения A 1:1.
Пример 6. Форма цитрата B соединения A
Форма цитрата B вероятно состоит из моногидрата соли лимонной кислоты соединения A 1:1, и ее получали из полученной в увеличенном масштабе соли лимонной кислоты. Образец анализировали посредством XRPD (с индексированием) и протонного ЯМР.
Профиль XRPD для формы цитрата B (фиг. 6) успешно проиндексирован, что свидетельствует, что образец преимущественно или исключительно состоит из одной кристаллической фазы. Объем элементарной ячейки соответствует соли лимонной кислоты соединения A 1:1 и может обеспечивать до 1 моля воды.
В профиле XRPD для формы цитрата B выбрали пики, и списки наблюдаемых и выраженных пиков приведены в таблицах 5 и 6, соответственно.
Таблица 5. Список наблюдаемых пиков XRPD для формы цитрата B
Таблица 6. Список выраженных пиков XRPD для формы цитрата B
Протонный ЯМР образца подтверждает соль лимонной кислоты соединения A 1:1 с присутствием только небольшого количества остаточного этанола.
Данные титрования по Карлу Фишеру продемонстрировали, что образец, содержащий форму цитрата B, содержал приблизительно 3 мас.% воды. Это количество воды коррелирует с моногидратом соли лимонной кислоты 1:1.
Пример 7. Форма тартрата A соединения A
Форма тартрата A состоит из соединения A/кислоты/сольватированной EtOH соли винной кислоты соединения A ≈2:1:1. В одном из вариантов осуществления вещество получали в эксперименте по получению солей с L-винной кислотой в EtOH, как приведено в таблице 2.
Профиль XRPD формы тартрата A (фиг. 7) успешно проиндексирован, что свидетельствует, что образец преимущественно или исключительно состоит из одной кристаллической фазы. Объем элементарной ячейки, полученный на основе индексирующего разрешения, может обеспечивать соль винной кислоты соединения A 2:1 с присутствием 2 моль EtOH на моль соли 2:1.
Спектр протонного ЯМР для формы тартрата A соответствует соли 2:1 с 0,4 моль EtOH на моль соединения A, что свидетельствует о стехиометрии соединения A/кислоты/EtOH ≈2:1:1.
Наложение термограмм DSC и TGA для формы тартрата A приведено на фиг. 8. Широкая эндотерма с пиковым максимумом при 78°C и плечевом пик при 95°C на термограмме DSC соответствует приблизительно 5% потере массы от 23 до 115°C при TGA в соответствии с потерей растворителя. Уровень потери массы соответствует ≈1 моль EtOH на моль соли винной кислоты соединения A 2:1 в соответствии с количеством EtOH, определяемым посредством протонного ЯМР. Этап дополнительной потери массы величиной приблизительно 4 мас.% соответствует эндотермическому процессу с пиковым максимумом при 158°C, вероятно соответствующим одновременным плавлению, диссоциации и разрушению соли.
Пример 8. Форма B и форма C тартрата соединения A
Выявлено, что форма тартрата A после сушки при низком давлении при ≈66°C в течение 1 суток преобразуется в новую форму, обозначенную как форма тартрата B. Выявлено сходство положений пиков XRPD между формой A (фиг. 9, верхний профиль) и формой B (фиг. 9, средний профиль), что возможно свидетельствует, о частичной десольватации формы A в этих условиях и о смеси веществ.
Выявлено, что форма тартрата B при дополнительной сушке при низком давлении при ≈83-86°C преобразуется в форму C (фиг. 9, нижний профиль). Форму тартрата B подвергали нагрузке при ≈97% относительной влажности в течение 14 суток, и она не продемонстрировала признаков смачивания за счет поглощения влаги из воздуха.
Пример 9. Форма тартрата D соединения A
Форма тартрата D состоит из соединения A/кислоты/сольватированной EtOH соли винной кислоты соединения A ≈2:1:1, и исходно ее получали в эксперименте по получению солей в EtOH при ≈70°C (таблица 2). Профиль XRPD успешно проиндексирован, что свидетельствует, что вещество преимущественно или исключительно состоит из одной кристаллической фазы (фиг. 10). Объем элементарной ячейки может обеспечивать соль винной кислоты соединения A 2:1 с содержанием ≈2 моль EtOH на моль соли 2:1.
Протонный ЯМР формы тартрата D соответствует сольватированной EtOH полусоли винной кислоты в молярном соотношении соединение A/кислота/EtOH 2:1:1.
Пример 10. Форма тартрата F соединения A
Форма тартрата F состоит из соединения A/кислоты/гидратированной водной соли винной кислоты соединения A ≈2:1:3, хотя содержание воды может варьировать. Вещество воспроизводимо получали, комбинируя форму свободного основания A соединения A с водной L-винной кислотой при ≈50°C (таблица 7).
Таблица 7. Форма тартрата F соединения A
2) непрозрачная белая суспензия
3) белые твердые вещества
Форма тартрата F обладала улучшенной водорастворимостью по сравнению со свободным основанием соединения A (2 мг/мл в отличие от <1 мг/мл, соответственно). Это вещество демонстрирует определенное диспропорционирование в чистой воде, гигроскопичность и предрасположенность к преобразованию в несколько соливатированных форм. Форму тартрата F охарактеризовывали посредством XRPD (фиг. 11), протонного ЯМР, DSC, TGA, высокотемпературной микроскопии, DVS и титрования по Карлу Фишеру.
Объем элементарной ячейки на основе индексирующего разрешения соответствует соли винной кислоты соединения A2:1 с присутствием до ≈3 моль воды (т.е. элементарная ячейка может обеспечивать соединение A/кислоту/воду 2:1:3). В нескольких случаях получения выявлены сдвиги минорных пиков для выбранных пиков, что может свидетельствовать о различном содержании воды.
В профиле XRPD для формы тартрата F выбрали пики, и списки наблюдаемых и выраженных пиков приведены в таблицах 8 и 9, соответственно.
Таблица 8. Список наблюдаемых пиков XRPD для формы тартрата F
Таблица 9. Список выраженных пиков XRPD для формы тартрата F
Спектр протонного ЯМР для формы тартрата F соответствует соли винной кислоты соединения A 2:1.
Наложение термограмм DSC и TGA для формы тартрата F приведено на фиг. 12. Этапу исходной потери массы величиной приблизительно 6,6 мас.% при 27-100°C при TGA соответствует широкая эндотерма при DSC при 125°C, вероятно соответствующая потере растворителя. Потеря массы соответствует ≈3 моль воды на моль соли 2:1 в соответствии с количеством воды, обеспечиваемой объемом элементарной ячейки. Фактически потери массы при 100-180°C не выявлено.
Анализ микрофотографий форма тартрата F при высокотемпературной микроскопии проиллюстрировал изменения двойного лучепреломления, выявляемые в интервале от 70°C до 98°C, соответствующие постепенной потере массы и широкой эндотерме, выявленной по данным TGA и DSC, что, вероятно, свидетельствует о дегидратации образца при нагревании. Наблюдали плавление при температурах от ≈171°C до 176°C, что подтверждает, что крутая эндотерма DSC с началом при 173°C соответствует плавлению дегидратированного вещества.
Титрование формы тартрата F по Карлу Фишеру выявило приблизительно 7,298% воды, эквивалентно приблизительно 3,7 моль воды на моль соли 2:1. Это содержание воды незначительно выше, чем ≈3 моль воды, выявленных при анализе потери массы TGA и допускаемых объемом элементарной ячейки.
Изотерма DVS для формы тартрата F приведена на фиг. 13. Вещество демонстрировало относительно небольшую потерю массы при уравновешивании до 5% относительной влажности (0,29 мас.%), что свидетельствует, что гидрат вероятно остается интактным вначале адсорбции. Отмечена значительная гигроскопичность от 5% до 95% относительной влажности, где образец приобретал всего приблизительно 4,23 мас.%. Приблизительно половина увеличения массы происходила постепенно от 5% до 85% относительной влажности, тогда как другая половина происходила в течение одного этапа от 85% до 95% относительной влажности.
Хотя в эксперименте DVS наблюдали значительное увеличение массы, гидратированное вещество в течение скрининга не продемонстрировало признаков смачивания за счет поглощения влаги из воздуха при ≈97% относительной влажности. Профиль десорбции зеркально отражает профиль сорбции с очень небольшим наблюдаемым гистерезисом. Общая потеря массы приблизительно 4,25 мас.% происходила в промежутке от 95% до 5% относительной влажности, с приблизительно половиной потери массы, происходившей за один этап от 95% до 85% относительной влажности. XRPD образца после DVS не выявила изменения формы.
Форма тартрата F демонстрировала улучшенную водорастворимость при добавлении растворителя по сравнению с формой свободного основания A соединения A, хотя через 1 сутки наблюдали начало диспропорционирования в чистой воде.
Выявлено, что форма тартрата F после дегидратации преобразуется в другую форму тартрата и обратно преобразуется в форму F (с минорными дополнительными пиками XRPD) при нагрузке при ≈97% относительной влажности.
Пример 11. Форма тартрата G соединения A
Форма тартрата G состоит из соединения A/кислоты/сольватированной ACN соли винной кислоты ≈2:1:2, и ее получают из взвеси формы тартрата F в ACN при ≈76°C. Профиль XRPD формы G приведен на фиг. 14, свидетельствуя, что образец преимущественно или исключительно состоит из одной кристаллической фазы. Объем элементарной ячейки, полученный на основе индексирующего разрешения, может обеспечивать соль винной кислоты соединения A 2:1 с 2 моль ACN на моль соли 2:1.
Спектр протонного ЯМР для формы тартрата G соответствует о соединении A/тартрате/сольватированной ACN соли ≈2:1:2.
Пример 12. Форма тартрата H соединения A
Форма тартрата H состоит из соединения A/кислоты/сольватированной THF соли винной кислоты ≈2:1:1,5 и вероятно изоморфно сольватировано IPA. Вещество получали в экспериментах со взвесями, начиная с формы тартрата F в THF (получая форму H) и в IPA (получая смесь формы H и формы D). Профиль XRPD формы H, полученный на основе THF успешно проиндексирован, что свидетельствует, что вещество преимущественно или исключительно состоит из одной кристаллической фазы (фиг. 15). Параметры элементарной ячейки для формы H являются сходными с параметрами формы тартрата F (гидрат), возможно свидетельствуя об изоструктурных веществах, хотя профили XRPD демонстрируют значимым отличия положений пиков и интенсивности пиков. Объем элементарной ячейки формы H является значительно большим, чем объем элементарной ячейки формы F и может обеспечивать до 2 моль THF на соединение A/соль винной кислоты 2:1.
Спектр протонного ЯМР образца формы H после THF соответствует соединению A/тартрату/сольватированной THF соли ≈2:1:1,5.
Учитывая множество растворителей, из которых она получена, и сравнение параметров элементарной ячейки, форма Н может состоять из семейства изоструктурных сольватов полусоли винной кислоты, включающей THF, IPA и воду.
Пример 13. Исследования измельчения и сокращенный скрининг стабильных форм
Для оценки возможного формирования соли лимонной кислоты при формулировании составов, проводили два эксперимента по измельчению с использованием формы свободного основания A и одного молярного эквивалента лимонной кислоты, как представлено в таблице 10.
Таблица 10. Эксперименты по измельчению с использованием формы свободного основания A соединения A и лимонной кислоты
В одном из экспериментов компоненты измельчали друг с другом в сухих условиях, тогда как другой эксперимент по измельчению включал добавление небольшого количества воды для стимуляции влажного гранулирования. Сухой размол приводил к получению физической смеси свободного основания и лимонной кислоты (фиг. 16), тогда как влажный размол приводил к получению формы цитрата A с незначительным количеством непрореагировавшего свободного основания (фиг. 17). Эти результаты свидетельствуют, что в составе, содержащем свободное основание соединения A и лимонную кислоту такая обработка состава, как влажное гранулирование, вероятно облегчает образование соли лимонной кислоты. Кроме того, формирование соли в составе подтверждали посредством анализа XRPD нескольких партий формулируемых таблеток, который в дополнение к другим кристаллическим компонентам состава демонстрировал пики, соответствующие формам солей лимонной кислоты.
Приблизительные значения растворимости для формы цитрата A приведены в таблице 11. Растворимости рассчитывали на основе всего растворителя, используемого для получения раствора; фактические растворимости могут быть большими вследствие объема используемых порций растворителя или низкой скорости растворения. Значения округлены до ближайшего целого числа. Если при определении посредством визуальной оценке растворения не происходило, значение приведено, как "<". Если при определении посредством визуальной оценке после добавления первой аликвоты растворение происходило, значение приведено, как ">".
Таблица 11. Приблизительные растворимости солей соединения A в различных растворителях при температуре окружающей среды
Форма цитрата A в большинстве систем органических растворителей продемонстрировала от низкой до ограниченной растворимости, с наибольшими значениями растворимости, выявленными в DMSO (≈33 мг/мл), HFIPA (≈10 мг/мл), MeOH (≈9 мг/мл) и воде (6 мг/мл). Эти значения облегчали выбор систем растворителей для применения при скрининге стабильных форм. Исследовали смеси растворителей для поиска оптимальных значений растворимости для длительных экспериментов со взвесями.
Проводили двенадцать экспериментов со взвесями с использованием формы цитрата A в различных системах органических растворителей и в чистой воде (таблица 12). Все системы растворителей исследовали при комнатной температуре (КТ), и исследовали дополнительные взвеси в MeOH и в воде при комнатной температуре.
Таблица 12. Сокращенный скрининг стабильных форм форма цитрата A соединения A
2) вакуумн. фильтр
2) белые твердые вещества
2) вакуумн. фильтр
2) белые твердые вещества
2) вакуумн. фильтр
2) белые твердые вещества
2) вакуумн. фильтр
2) белые твердые вещества
3) вакуумн. фильтр (0,2 мкм PTFE)
2) прозрачная жидкая фаза, небольшое количество белых твердых веществ на флаконе в проводнике на линии растворителя
3) твердых веществ не собрано, фильтрат становился очень вязким и выпаривался до очень небольшого количества (восстановление было невозможно)
2) вакуумн. фильтр
2) белые твердые вещества
2) вакуумн. фильтр
2) белые твердые вещества
2) вакуумн. фильтр в холодном состоянии
2) белые твердые вещества
2) вакуумн. фильтр
2) белые твердые вещества
2) вакуумн. фильтр
2) белые твердые вещества
2) вакуумн. фильтр
2) влажные белые твердые вещества
2) вакуумн. фильтр while cool
2) немного влажные белые твердые вещества
Все взвеси приводили к получению формы цитрата A. Соль демонстрировала значительную растворимость при добавлении растворителя во многих из используемых систем растворителей, что способствовало бы кинетике преобразования форм, если бы существовала более стабильная форма. Кроме того, все взвеси можно было перемешивать в течение 24-25 суток, длительный период, который, вероятно, мог бы обеспечить достаточное время для преобразования в более стабильную форму, если бы она существовала. Учитывая эти факторы, форма цитрата A, вероятно, является наиболее термодинамически устойчивой безводной формой соли лимонной кислоты в тестируемых условиях.
Пример 14. Получение выбранных солей соединения A
В дополнение к описанным выше способам, можно получать соли соединения A с использованием приводимых ниже способов.
Форма цитрата A соединения A - твердую форму свободного основания A соединения A (1,1957 г) комбинировали с EtOH (70 мл) в концентрации 17 мг/мл, получая взвесь. Во взвесь добавляли водный раствор 1 M лимонной кислоты (1,2 молярных эквивалентов, 4,06 мл) и видимых изменений не наблюдали. Смесь оставляли перемешиваться при температуре окружающей среды в течение 12 суток, получая непрозрачную белую суспензию. Твердые вещества собирали на бумажный фильтр посредством фильтрации при пониженном давлении и высушивали на воздухе на фильтре при пониженном давлении в течение приблизительно 4 минут. Твердые вещества переносили в чистый флакон, получая выход приблизительно 98%.
Альтернативно, твердые формы соединения A (5,5987 г) комбинировали с EtOH (330 мл) в концентрации 17 мг/мл, получая взвесь. К взвеси добавляли водный раствор 1 M лимонной кислоты (1,2 молярных эквивалентов, 19 мл) и видимых изменений не наблюдали. Добавляли затравку формы цитрата A с добавлением затравки на уровне 1% (55,6 мг), и видимых изменений не наблюдали. Смесь оставляли перемешиваться при температуре окружающей среды в течение 3 суток, получая непрозрачную белую суспензию. Твердые вещества собирали на бумажный фильтр посредством фильтрации при пониженном давлении и высушивали на воздухе на фильтре при пониженном давлении в течение приблизительно 10 мин и твердые вещества переносили в чистый флакон.
Форма тартрата F соединения A - твердую форму свободного основания A соединения A (1,0022 г) комбинировали с 1,5 молярных эквивалентов водной L-винной кислоты (0,6319 г кислоты, разбавленной в 12 мл вода) с перемешиванием при ≈51°C, получая непрозрачную белую суспензию. Добавляли небольшое количество затравки формы тартрата F и смесь перемешивали при ≈51°C в течение 1 суток, получая непрозрачную белую суспензию. Твердые вещества собирали на бумажный фильтр посредством фильтрации при пониженном давлении, поддерживая нагретыми, и высушивали на воздухе на фильтре при пониженном давлении в течение приблизительно 4 мин.
Пример 15. Способы кристаллизации
Ниже более подробно описаны способы кристаллизации, используемые при скрининге солей и/или способах получения.
Быстрое охлаждение (FC) - Насыщенные растворы данного вещества соли соединение A получали в данном растворителе при повышенной температуре. Сосуд закрывали и помещали на лабораторный стол для быстрого охлаждения до температуры окружающей среды. Твердые вещества выделяли и анализировали.
Измельчение - Взвешенные количества веществ данного соединения A (например, свободного основания соединения A с данными кислотами) переносили в контейнер для измельчения из агата. В контейнер добавляли агатовый шар и небольшое количество данного растворителя (если указан), после чего контейнер закрепляли на мельнице Retsch. Смесь измельчали в течение трех циклов длительностью по 10 минуту при 30 Гц, и твердые вещества между циклами соскребали со стенок резервуара. Полученные твердые вещества переносили в чистый флакон и анализировали.
Эксперименты со взвесями - Суспензии веществ данного соединения A (например, полученные соли или смеси свободного основания соединения A с различными кислотами) получали посредством добавления достаточного количества твердых веществ к данному растворителю или системе растворителей при указанной температуре так, что присутствуют нерастворенные твердые вещества. Когда указано, добавляли затравки из указанных веществ. Затем смесь перемешивали (как правило, посредством встряхивания или качания) в выбранном флаконе в указанных условиях в течение длительного периода времени. Твердые вещества выделяли и анализировали.
Нагрузка относительной влажностью - Твердые вещества данного соединения A переносили в сосуд, который затем открывали и помещали внутри контейнера, содержащего насыщенный водный раствор сульфата калий для создания ≈97% относительной влажности. Эксперименты по нагрузке относительной влажностью проводили при температуре окружающей среды.
Фильтрация при пониженном давлении - В кратком изложении, твердые вещества перед перенесением в сосуд собирали на бумажные или нейлоновые фильтры посредством фильтрации при пониженном давлении и высушивали на воздухе на фильтрах при пониженном давлении.
Определение пиков XRPD - на фиг. 1-3, 6-7, 9-11 и 14-17 в настоящем описании приведены профили рентгеновской дифракции, некоторые с отмеченными пиками и/или таблицами со списками пиков. Выбирали пики в диапазоне приблизительно до 30° 2θ. Использовали алгоритмы округления, округляя каждый пик до ближайшей 0,01° 2θ. Расположение пиков по оси x (° 2θ) на всех фиг. и в списках определяли с использованием проприетарного программного обеспечения (TRIADSTM v2.0) и округляли до двух значащих цифр после десятичной запятой. Вариабельности положения пиков приведены в пределах ±0,2° 2θ на основе рекомендаций изложенных в обсуждении USP вариабельности в рентгеновской порошковой дифракции.
Для регистрации межплоскостных расстояний d длина волны, используемая для расчета межплоскостных расстояний d, составляла 1,5405929Å, длину волны Cu-Kα1. Вариабельность, ассоциированную с расчетами межплоскостных расстояний d, рассчитывали по рекомендациям USP, для каждого межплоскостного расстояния d и предоставляли соответствующие таблицы данных.
По рекомендациям USP различные гидраты и сольваты могут демонстрировать колебания пиков более 0,2° 2θ и, таким образом, вариабельность пиков 0,2° 2θ неприменима для этих веществ.
Если доступно несколько профилей дифракции, тогда доступны оценки статистики частиц (PS) и/или предпочтительной ориентации (PO) частиц. Воспроизводимость профилей XRPD нескольких образцов, анализируемых на одном дифрактометре, означает, что статистика частиц является подходящей. Соответствие относительной интенсивности профилей XRPD, полученных на нескольких дифрактометрах, означает хорошую статистику ориентировки, наблюдаемый профиль XRPD можно сравнивать с рассчитанным профилем XRPD на основе одной кристаллической структуры, если доступно. Также для оценки PS/PO можно использовать двухмерные диаграммы рассеяния с использованием площадных детекторов. Если определено, что эффекты PS и PO незначительны, то профиль XRPD представляет среднюю интенсивность порошка для образца, и выраженные пики можно идентифицированы как "характерные пики".
"Характеристические пики", в тех случаях, когда они существуют, представляют собой подмножество характерных пиков, и их используют для различения одного кристаллического полиморфа от другого кристаллического полиморфа (полиморфы являются кристаллическими формами с одинаковым химическим составом). Характеристические пики определяют, оценивая, какие характерные пики, если они присутствуют, присутствуют в одном кристаллическом полиморфе соединения в отличие от всех других известных кристаллических полиморфов этого соединения в пределах ±0,2°2θ. Не у всех кристаллических полиморфов соединения обязательно существует по меньшей мере один характеристический пик.
Пример 16. Инструментальные способы
Ниже более подробно описаны инструментальные способы, используемые в скрининге солей и характеристика процессов.
Дифференциальная сканирующая калориметрия (DSC) - DSC проводили с использованием дифференциального сканирующего калориметра TA Instruments Q2000. Калибровку температуры проводили с использованием пригодного для анализа NIST металлического индия. Образец помещали в алюминиевый поддон для DSC, накрывали крышкой и точно регистрировали массу. На контрольную сторону ячейки помещали взвешенный алюминиевый поддон, сконфигурированный в качестве поддона для образца. Параметры сбора данных и конфигурация поддона для каждой термограммы приведены на изображении в разделе "Данные" настоящего описания. Способ кодирования на термограмме представляют собой сокращенное обозначение стартовой и конечной температуры, а также скорости нагревания; например, -30-250-10 означает "от -30°C до 250°C, при 10°C/мин". В приводимой ниже таблице перечислены сокращения, используемые в каждом изображении для конфигураций поддонов.
Динамическая сорбция паров (DVS) - Данные динамической сорбции паров (DVS) получали на анализаторе собранных паров VTI SGA-100. В качестве калибровочных стандартов использовали NaCl и PVP. Образцы перед анализом не сушили. Данные сорбции и десорбции собирали в диапазоне от 5% до 95% относительной влажности с приращениями относительной влажности 10% с промывкой азотом. Критерием равновесия, используемым для анализа, являлось изменение массы менее 0,0100% в течение 5 минут с максимальным временем уравновешивания в течение 3 часов. Данные на исходное содержание влаги в образцах не корректировали.
Высокотемпературная микроскопия - Высокотемпературную микроскопию проводили с использованием высокотемпературного устройства Linkam (FTIR 600), закрепленного на микроскопе Leica DM LP, оборудованного цветовой цифровой камерой SPOT Insight™. Калибровки температур проводили с использованием стандартов температур плавления USP. Образцы помещали на покровное стекло и сверху образца помещали второе покровное стекло. После ступенчатого нагревания, каждый образец визуально обследовали с использованием длиннофокусного объектива 20´ 0,40 N. A. со скрещенными поляризаторами и компенсатором красного цвета первого порядка. Изображения получали с использованием программного обеспечения SPOT (v. 4.5.9).
Оптическая микроскопия - Образцы наблюдали в оптический микроскоп Wolfe со скрещенными поляризаторами с объективами 2´ или 4´ или в стереомикроскоп Leica с компенсатором красного цвета первого порядка со скрещенными поляризаторами с объективами от 0,8x до 10x.
1H ЯМР спектроскопия в растворе - Спектр ЯМР раствора получали на спектрометре Agilent DD2-400. Образец получали, растворяя приблизительно 5-10 мг образца в содержащем DMSO-d6 TMS. Параметры получения данных приведены на первой диаграмме спектра в разделе данных настоящего описания. Остаточный пик от неполностью дейтерированного DMSO составляет приблизительно 2,50 м.д. Относительно широкий пик приблизительно при 3,3 м.д., если присутствует, появляется вследствие воды.
Альтернативно, параметры получения данных приведены на первой странице каждого спектра в разделе данных настоящего описания. Остаточный пик от неполностью дейтерированного DMSO составляет приблизительно 2,50 м.д. Относительно широкий пик приблизительно 3,3 м.д., если присутствует, появляется вследствие воды.
Термогравиметрия (TGA) - анализы TG проводили с использованием термогравиметрического анализатора TA Instruments Discovery. Калибровку температур проводили с использованием никеля и AlumelÔ. Каждый образец помещали в алюминиевый поддон и вставляли в калориметрическую камеру TG. Калориметрическую камеру нагревали в потоке азота. Параметры получения данных приведены выше каждой термограммы в разделе данных настоящего описания. Способ кодирования на термограмме представляет собой сокращения для начальной и конечной температур, а также скорости нагревания; например, 25-350-10 означает "от 25°C до 350°C, при 10°C/мин".
Рентгеновская порошковая дифракции (XRPD) - Профили XRPD получали на дифрактометре PANalytical X'Pert PRO MPD с использованием падающего пучка света излучения Cu, получаемого с использованием острофокусного источника Optix long. Для фокусировки рентгеновских лучей Cu Kα через образец и на детектор использовали эллиптически градуированное многослойное зеркало. Перед анализом анализировали образец кремния (NIST SRM 640d), чтобы проверить, что наблюдаемое положение пика Si 111 соответствует сертифицированному NIST положению. Образец образца помещали между пленками толщиной 3 мкм и анализировали в геометрии пропускания. Для минимизации фона, создаваемого воздухом, использовались ограничитель луча, расширение против рассеяния коротких волн и антирассеивающий ножевой коллиматор. Для минимизации расширения от осевой расходимости для падающего и дифрагированного пучков использовали щели Соллера. Профили дифракции получали с использованием сканирующего позиционно-чувствительного детектора (X'Celerator), расположенного на 240 мм от образца, и программного обеспечения Data Collector v. 2.2b. Параметры получения данных для каждого профиля приведены выше изображения в разделе данных настоящего описания, включая щель расходимости (DS) перед зеркалом и антирассеивающую щель для падающего пучка света (SS), если применимо.
Альтернативно, профили XRPD получали с использованием дифрактометра PANalytical X'Pert PRO MPD с использованием падающего пучка излучения Cu Kα, получаемого с использованием длиннофокусного, острофокусного источника и никелевого фильтра. Дифрактометр конфигурировали с использованием симметричной геометрии Брэгга-Брентано. Перед анализом анализировали образец кремния (NIST SRM 640d), чтобы проверить, что наблюдаемое положение пика Si 111 соответствует сертифицированному NIST положению. Образец образца получали в виде тонкого, циркулярного слоя, центрированного на кремниевом субстрате с нулевым фоном. Для минимизации расширения от осевой расходимости для падающего и дифрагированного пучков использовали щели Соллера. Профили дифракции получали с использованием сканирующего позиционно-чувствительного детектора (X'Celerator), расположенного на 240 мм от образца, и программного обеспечения Data Collector v. 2.2b. Параметры получения данных для каждого профиля приведены выше изображения в разделе данных настоящего описания, включая щель расходимости (DS) перед зеркалом и антирассеивающую щель для падающего пучка света (SS).
Пример 17: Получение таблеток с соединением A и винной кислотой
Теми же способами, с использованием влажного гранулирования получали две партии таблеток, содержащих соединение A и винную кислоту. Формула партии и способ получения предоставлены в таблице 13, в которой продемонстрированы количества ингредиентов, использованных в составах и последующих составах таблеток в партии 1 и партии 2.
Таблица 13
IG - внутригранулярный;
EG – экстрагранулярный.
Способ получения таблеток партии 1 и партии 2:
1. Все внутригранулярные ингредиенты (IG) навешивали и смешивали в ступке с использованием шпателя в течение по меньшей мере 1 минуты.
2. При смешивании медленно добавляли воду с шагом 5% до достижения подходящей влажности при гранулировании. Общее добавляемое количество воды составило приблизительно 20% мас./мас. смеси ингредиентов.
3. Влажную массу пропускали через сито 18 меш и гранулы получали и сушили в термостате в течение ночи при 35°C.
4. Полученные гранулы измеряли и, таким образом, рассчитывали количества каждого из экстрагранулярных ингредиентов (EG).
5. Навешивали экстрагранулярные Ac-di-sol и авицел и смешивали с гранулами в течение 2 минут.
6. Навешивали стеарат магния и смешивали со смесью после этапа 5 в течение 0,5 минут.
7. Полученную смесь прессовали с получением таблетки с использованием стандартного круглого вогнутого устройства 3/8" с массой таблетки 400 мг. Диапазон масс смеси для прессования составлял от 390 до 410 мг. Силу прессования доводили до достижения жесткости 10 до 15 кПа.
Пример 18 - Получение таблеток с соединением A и лимонной кислотой
Таблетки, содержащие соединение A и лимонную кислоту получали с использованием влажного гранулирования теми же способами, что и таблетки с винной кислотой в примере 16. Формула партии и способ получения приведены в таблице 14, в которой представлены количества ингредиентов, используемых в составе и последующем составе таблеток в партии 1.
Таблица 14
IG - внутригранулярный;
EG – экстрагранулярный.
Способ получения таблеток партии 1:
1. Все внутригранулярные ингредиенты (IG) навешивали и смешивали в грануляторе в течение по меньшей мере 1 минуты.
2. При смешивании медленно ступенчато добавляли воду до достижения подходящей влажности при гранулировании. Общее добавляемое количество воды составило приблизительно 20% мас./мас. смеси ингредиентов.
3. Влажную массу сушили в течение ночи при 25°C, затем в сушке псевдоожиженного слоя в течение приблизительно 40 минут при 50°C.
4. Гранулы собирали, измельчали и измеряли и, таким образом, рассчитывали количества экстрагранулярных ингредиентов (EG).
5. Навешивали экстрагранулярные Acdisol и авицел и смешивали с гранулами в течение 2 минут.
6. Навешивали стеарат магния и смешивали со смесью после этапа 5 в течение 0,5 минут.
7. Полученную смесь прессовали с получением таблеток с использованием 7 мм с использованием стандартного круглого вогнутого устройства с массой таблеток 150 мг. Силу прессования доводили до достижения жесткости 10 до 15 кПа.
8. Полученные сердцевины таблеток покрывали водным опадрай в баковом устройстве для нанесения покрытий до достижения приблизительно до 3 процентов увеличение массы.
Пример 19 - Растворение таблеток с соединением A и лимонной или винной кислотами при pH 6,8
Таблетки, используемые в этом примере, получали с использованием способов, описанных в примерах 17 и 18. Таблетки получали до общей массы ≈150 мг с 50 мг соединения A.
В таблице 15 приведены скорости растворения таблеток, содержащих соединение A и лактозу с лимонной кислотой или винной кислотой или без в 500 мл 50 мМ фосфатно-солевого буфера, pH 6,8. Каждую композицию (лимонная кислота, лактоза и винная кислота) тестировали в трех отдельных экспериментах.
Таблица 15. Скорости растворения
Растворение, приведенное для каждого образца в экспериментах, указано в % растворенного соединения A.
Как можно видеть из результатов в таблице 15, соединение A в таблетках с лимонной кислотой и в таблетках с винной кислотой растворялось при pH 6,8 более легко, чем в таблетках с лактозой.
Пример 20 - тестирование таблеток с соединением A и винной кислотой in vivo
Для проведения этого исследования использовали таблетки из партии 2 (пример 17, таблица 13). Таблетки вводили перорально одиннадцати собакам. Четырем собакам за 30 минут до таблетки внутримышечно вводили пентагастрин (6 мкг/кг). Четырем собакам за 2 часа до таблетки перорально вводили фамотидин (40 мг/собаке). Трем собакам не проводили предварительной обработки.
Фамотидин ингибирует продукцию кислоты в желудке, и его используют для повышения желудочного pH (т.е. для того, чтобы сделать его более нейтральным). Пентагастрин стимулирует продукцию кислоты в желудке, и его используют для снижения желудочного pH (т.е. для того, чтобы сделать его более кислотным).
Собаки голодали в течение ночи, и каждой собаке вводили одну таблетку, содержащую 150 мг соединения A. Собаки весили приблизительно 10 кг каждая, конечный уровень дозирования составлял 15 мг/кг.
Подобное предварительное исследование проводили с использованием суспензии соединения A (т.е. с винной или лимонной кислотой) и результаты настоящего исследования сравнивают с результатами этого предварительного исследования.
Собирали образцы крови перед дозированием и через 0,083, 0,25, 0,5, 1, 2, 4, 6, 8, 24, 32 и 48 часов после дозирования. Кровь центрифугировали с получением плазмы, которую анализировали на уровни соединения A посредством LC-MS и рассчитывали фармакокинетические параметры. Фармакокинетические параметры приводили по фактически измеренным концентрациям, а также приводили к массе каждой отдельной собаки. Концентрации ниже предела детекции (BLQ) для расчетов считали равными нулю.
В таблице 16 приведены средние фармакокинетические параметры соединения A в таблетках с винной кислотой партии 2, вводимым собакам перорально.
Таблица 16
У собак, предварительно обработанных пентагастрином, фамотидином, и собак, без предварительной обработки, наблюдали приблизительно равные групповые значения средних Cmax и AUC (таблица 16).
Результаты этого исследования позволяет предполагать, что вариабельность всасывания соединения A вследствие различий pH желудочно-кишечного тракта можно преодолевать, формулируя соединение A в таблетки, содержащие винную кислоту. Выдвинута гипотеза, что это происходит вследствие улучшенных свойств соли винной кислоты, образующейся в составе, содержащем винную кислоту.
Специалист в данной области легко поймет, что настоящее описание легко адаптировать для достижения указанных целей и преимуществ, а также свойственных ему. Способы, варианты и композиции, описываемые в настоящем документе, как приведены в настоящем документе как предпочтительные варианты осуществления, являются иллюстративными и не предназначены для ограничений объема. Специалисты в данной области могут проводить их изменения и применять их другими способами, которые включены в сущность настоящего изобретения, определены объемом формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛЫ АЛКИНИЛСОДЕРЖАЩЕГО СОЕДИНЕНИЯ, ЕГО СОЛИ И СОЛЬВАТА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПУТИ ПРИМЕНЕНИЯ | 2021 |
|
RU2829609C1 |
| КРИСТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2016 |
|
RU2789672C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И СПОСОБ ЕЕ ОЧИСТКИ | 2011 |
|
RU2604734C2 |
| ТВЕРДЫЕ ФОРМЫ 2-(5-(4-(2-МОРФОЛИНОЭТОКСИ)ФЕНИЛ)ПИРИДИН-2-ИЛ)-N-БЕНЗИЛАЦЕТАМИДА | 2018 |
|
RU2802964C2 |
| СОЛИ И ПОЛИМОРФЫ ЗАМЕЩЕННОГО ИМИДАЗОПИРИДИНИЛ-АМИНОПИРИДИНА | 2015 |
|
RU2732125C2 |
| КРИСТАЛЛИЧЕСКИЕ СОЛИ ИНГИБИТОРА B-RAF-КИНАЗЫ | 2018 |
|
RU2798091C2 |
| ТВЕРДЫЕ ФОРМЫ РОМИДЕПСИНА И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2607634C2 |
| ТАРТРАТ 3-((1R,3R)-1-(2,6-ДИФТОР-4-((1-(3-ФТОРПРОПИЛ)АЗЕТИДИН-3-ИЛ)АМИНО)ФЕНИЛ)-3-МЕТИЛ-1,3,4,9-ТЕТРАГИДРО-2H-ПИРИДО[3,4-b]ИНДОЛ-2-ИЛ)-2,2-ДИФТОРПРОПАН-1-ОЛА, ЕГО ТВЕРДЫЕ ФОРМЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2809220C2 |
| КРИСТАЛЛЫ СОЛИ | 2013 |
|
RU2652116C2 |
| ПОЛЕЗНЫЕ ФАРМАЦЕВТИЧЕСКИЕ СОЛИ 7-[(3R,4R) -3-ГИДРОКСИ-4- ГИДРОКСИМЕТИЛ- ПИРРОЛИДИН -1- ИЛМЕТИЛ ]- 3,5- ДИГИДРО-ПИРРОЛО [ 3,2-D] ПИРИМИДИН-4-ОНА | 2010 |
|
RU2489435C2 |
Изобретение относится к способу получения кристаллической формы цитрата A 5-(2,4-диаминопиримидин-5-илокси)-4-изопропил-2-метокси-бензолсульфонамида, которая характеризуется наличием профиля рентгеновской порошковой дифракции, полученного с использованием Kα-излучения меди, имеющего пики при значениях 2Θ 9,38 ± 0,20, 11,69 ± 0,20, 14,41 ± 0,20, 16,22 ± 0,20, 19,54 ± 0,20, 21,14 ± 0,20 и 26,31 ± 0,20 градусов. Способ осуществляют путем a) добавления этанола к форме свободного основания A 5-(2,4-диаминопиримидин-5-илокси)-4-изопропил-2-метокси-бензолсульфонамида с образованием раствора; b) добавления лимонной кислоты к раствору стадии a) с образованием взвеси; c) перемешивания взвеси стадии b); и d) высушивания перемешанной взвеси стадии c) посредством фильтрации при пониженном давлении. Технический результат - кристаллическая форма цитрата A 5-(2,4-диаминопиримидин-5-илокси)-4-изопропил-2-метокси-бензолсульфонамида, обладающая улучшенной водорастворимостью, без демонстрации признаков диспропорционирования и смачивания за счет поглощения влаги из воздуха. 17 ил., 16 табл., 20 пр.
Способ получения кристаллической формы цитрата A 5-(2,4-диаминопиримидин-5-илокси)-4-изопропил-2-метокси-бензолсульфонамида, которая характеризуется наличием профиля рентгеновской порошковой дифракции, полученного с использованием Kα-излучения меди, имеющего пики при значениях 2Θ 9,38 ± 0,20, 11,69 ± 0,20, 14,41 ± 0,20, 16,22 ± 0,20, 19,54 ± 0,20, 21,14 ± 0,20 и 26,31 ± 0,20 градусов, включающий:
a) добавление этанола к форме свободного основания A 5-(2,4-диаминопиримидин-5-илокси)-4-изопропил-2-метокси-бензолсульфонамида с образованием раствора;
b) добавление лимонной кислоты к раствору стадии a) с образованием взвеси;
c) перемешивание взвеси стадии b); и
d) высушивание перемешанной взвеси стадии c) посредством фильтрации при пониженном давлении.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ДИАМИНОПИРИМИДИНЫ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ Р2Х | 2005 |
|
RU2422441C2 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Abu T.M.Serajuddin: "Salt formation to improve drug solubility", Advanced Drug Reviews, 2007, vol.59, p.603-616 | |||
| Richard J.Bastin et al.: "Salt selection and Optimisation Procedures for | |||

