Настоящее изобретение относится к методу получения вакцины против ВИЧ, предотвращающей заражение ВИЧ и/или предотвращающей развитие ВИЧ-инфекции в организме человека. В частности, настоящее изобретение обеспечивает образование ВИЧ-специфических антител в ходе иммунного ответа в организме, которые связываются с существующими субтипами ВИЧ и его мутантами, отбирающимися после антиретровирусной терапии. Настоящее изобретение также относится к получению ВИЧ-специфических антител, способных узнавать и связываться практически со всеми изоформами ВИЧ.
ВВЕДЕНИЕ
Вирус иммунодефицита человека типа 1 (HIV-1) характеризуется огромной генетической вариабельностью, вызванной накоплением мутаций, возникающих во время репликации вируса, а также в ходе генетической рекомбинации [1, 18, 24]. Неудачи химиотерапевтических методов лечения ВИЧ-инфекции обусловлены высокой мутабельностью вирусных штаммов ВИЧ-1 [8]. Ранее было показано, что резистентные варианты вируса быстро появляются в организме пациентов после различных курсов антиретровирусной терапии и даже после комплексной высокоактивной антиретровирусной терапии (ВААРТ). Резистентные варианты вируса обладают специфическими изменениями в структуре и конформации вирусных белков. Обычно такие мутации, ответственные за ускользание ВИЧ-1 от используемой в данный момент лекарственной терапии, сохраняются и накапливаются в ходе отбора в условиях данной терапии.
Применение антиретровирусных препаратов против ВИЧ-1 не приводит к полному подавлению репликации вируса, что делает возможным отбор и накопление предсуществующих в его популяции мутаций резистентности, а также возникновение и накопление новых мутаций, и создает таким образом новые возможности для выживания вируса. Таким образом, все существующие классы антиретровирусных препаратов (NRTI, NNRTI, ингибиторы протеазы, ингибиторы слияния и даже комбинации различных препаратов, такие как ВААРТ) могут лишь замедлить репликацию вируса ВИЧ-1 на более или менее продолжительный период времени [7], до момента возникновения и размножения резистентных штаммов вируса. Широкое распространение резистентных вариантов ВИЧ-1, устойчивых к действию рутинно применяемых схем антиретровирусной терапии, стало серьезной проблемой, особенно для экономически развитых стран, в которых ВИЧ-инфицированные пациенты регулярно получают антиретровирусную терапию [8].
За 25-летнюю историю исследований ВИЧ было предложено несколько подходов к созданию иммунотерапевтической вакцины против ВИЧ и изучены практические результаты их использования. Эти подходы могут быть классифицированы на основе используемых активных компонентов вакцины, механизма их действия и способа получения вакцины:
Тип 1: ВИЧ/СПИД вакцины на основе ВИЧ-специфичных моноклональных
антител,
Тип 2: Вакцины на основе разрушенных вирусных частиц ВИЧ,
Тип 3: Вакцины на основе рекомбинантных белков/пептидов ВИЧ,
Тип 4: Вакцины на основе плазмидных или вирусных (адено-, адено-ассоциированных, вируса птичьей оспы, вируса оспы и других) ДНК векторов с клонированными в них генами белков ВИЧ.
Тип 1: Терапевтические ВИЧ/СПИД вакцины на основе ВИЧ-специфичных моноклональных антител, в их числе вакцины, основанные на использовании нейтрализующих антител в виде единичных моноклональных антител к ВИЧ или коктейля из 2-3 нейтрализующих антител к ВИЧ [5, 14, 28].
При изучении ВИЧ-инфекции сначала был открыт способ проникновения вируса в лимфоциты или другие клетки организма-хозяина посредством взаимодействия с CD4 рецептором и CCR5 и/или CXCR4 корецепторами. Затем была изучена структура белков оболочки ВИЧ (Рис.10 a-b), вариабельность третичной структуры петель gp120 и ключевая роль образования комплекса gp120-gp41 в распознавании и прикреплении вируса к CD4 рецептору и корецепторам. Моноклональные антитела, способные обнаруживать белки оболочки ВИЧ и связываться с их эпитопами, отвечающими за проникновение ВИЧ в клетки, либо антитела, способные связываться с доменами или эпитопами CD4 рецептора и корецепторов клеток, блокируя таким образом процесс ВИЧ-инфицирования на стадии прикрепления вируса к клетке, были названы нейтрализующими антителами.
Основной проблемой для создания вакцин на основе моноклональных антител, также обусловленной генетической вариабельностью ВИЧ, является неспособность рекомбинантных антител, выработанных на некоторый определенный антиген ВИЧ, нейтрализовать различные изоляты ВИЧ-1. Подавляющее большинство моноклональных антител против ВИЧ, вырабатывающихся в ходе иммунизации, обладают плохой способностью к перекрестной нейтрализации, либо не обладают ею вовсе, и обычно связываются с детерминантами, которые либо сильно различаются у разных вариантов вируса вследствие мутаций, либо плохо экспонированы на поверхности инфекционно активных вирионов. Были получены серии вариантов нейтрализующих моноклональных антител, но последующие клинические испытания показали, что вакцины, основанные на использовании нейтрализующих антител против белков оболочки gp120 и gp41, перестают работать в течение 1-2 месяцев (в тех редких случаях, когда они работали в лабораторных испытаниях вообще) по той же причине - вариабельность и изменения поверхностных эпитопов белков-мишеней у ВИЧ.
В основе описанного подхода к созданию вакцин лежит моноклональная селекция либо антител, либо антигенов вируса для иммунизации животных. Даже в случае создания панели нейтрализующих антител, специфичных к различным вариантам белков мишеней вируса, каждое антитело производится в виде рекомбинантного моноклона в бактериальной системе экспрессии. Более того, синтезированные в прокариотических клетках рекомбинантные антитела имеют по меньшей мере в 10 раз более низкую афинность к своим антигенам, по сравнению с нативными антителами из сыворотки крови животных или человека. Поликлональные ВИЧ-специфические иммуноглобулины, вырабатывающиеся у животных, обычно иммунотоксичны для других видов организмов, таких как человек. Их можно использовать для диагностических целей, но высокая вероятность возникновения анафилактических реакций является естественным барьером для их иммунотерапевтического применения. Гибридомная технология производства моноклональных антител (мАт) не решает проблему видовых биологических различий иммуноглобулинов. Технология создания гуманизированных или химерных мАт является очень трудоемкой, относительно долгой и дорогостоящей. Таким образом, с помощью этой технологии невозможно получить десятки или сотни вариантов мАт для использования в иммунотерапии против ВИЧ.
Тип 2: Вакцины на основе разрушенных вирусных частиц ВИЧ [9, 20].
Идея использовать природные вирионы и белки ВИЧ впервые возникла более 15 лет назад и с тех пор несколько раз возвращалась в различных формах. Среди них консервация вирусных частиц ВИЧ с помощью β-пропиолактона, псоралена или сходного агента, обладающего летальным действием на небольшие вирусы, но с относительно низким повреждающим действием на пептидные связи и конформацию белков. Вскоре стало очевидно, что концентрирование нативного вируса из крови пациентов с помощью ультрацентрифугирования не может обеспечить количества вирионов, пригодного для проведения иммунизации, оно с трудом может дать некоторое количество вируса для научно-исследовательских нужд. Таким образом, практические варианты создания вакцины такого типа предполагают либо in vitro инфицирование-культивирование лабораторных штаммов вируса, либо инфицирование первичными изолятами и их последующее культивирование с донорскими лимфоцитами. В обоих случаях необходимо крупномасштабное производство в рассчитанных на сотни литров ферментерах, чтобы обеспечить количество вирусных частиц, необходимое для формирования иммунного ответа на белки ВИЧ после иммунизации.
Сама по себе эта идея не совсем плоха, она даже имеет преимущества над другими тремя типами подходов к созданию вакцин против ВИЧ. Во-первых, безопасность использования инактивированных вирусных частиц для иммунизации становится более очевидной при проведении количественного определения числа копий РНК ВИЧ с помощью ПЦР в реальном времени после градиентного ультрацентрифугирования через сахарозную подушку. Вирусная РНК по большей части разрушается на небольшие фрагменты, и количество целых молекул падает до уровня в 104-105 раз ниже рассчитываемого на основе реального числа вирионов или их белков, полученных после концентрирования в градиенте сахарозы. Во-вторых, получение нативных вирусных белков, по-видимому, имеет больше шансов перекрыть существующее разнообразие эпитопов белков env ВИЧ. Однако это последнее утверждение показывает и реальную причину того, почему вакцины этого типа никогда не работали.
Создание вакцин против ВИЧ на основе разрушенных вирусных частиц является наилучшим примером того, как сильно условия отбора генетических мутаций in vitro отличаются от таковых в организмах животных или человека. Анализ вирусных белков выявил высокую вариабельность антигенных эпитопов, специфичных не только для различных субтипов вируса, но даже для разных вариантов вируса, выделенных из организма одного пациента. Вместе с тем все лабораторные штаммы, среди которых высокоинфекционные IIIB, U455, имеют константный и более гомогенный набор последовательностей белков env. Разнообразие пептидных библиотек env, проанализированных с помощью масс-спектрометрического или 3D-структурного методов для лабораторных штаммов ВИЧ, составляет до 5 процентов от такового для эквивалентных библиотек, полученных из вирусов из организма одного пациента. Та же тенденция наблюдается для первичных изолятов ВИЧ кокультивируемых in vitro с лимфоцитами донорской крови или с культурами человеческих клеточных линий, несущих CD4, CCR5 или CXCR4 рецепторы. Это означает, что условия отбора при инфекции in vitro очень сильно отличаются от естественных условий репликации вируса и образования вирионов в организме, и рамки для выживания вируса в организме человека на 95 процентов (или более) шире, чем при in vitro культивировании. Таким образом, все попытки использовать инактивированные вирусные частицы для получения вакцин после их крупномасштабной продукции в условиях in vitro не удались, так же как и попытки создания вакцин на основе вирусных пептидов, полученных из лабораторных штаммов ВИЧ.
Тип 3: вакцины на основе белков/пептидов ВИЧ [3, 6, 13, 15, 27, 33, 36].
Этот современный тип вакцин включает небольшие пептиды ВИЧ, множественные маленькие 15-20-аминокислотные фрагменты крупных белков ВИЧ, имитирующих эпитопы вирусных белков, ответственных за распознавание рецепторов и инфекционную активность, панели этих небольших пептидов. Как представитель небольшого семейства лентивирусов ВИЧ состоит из небольшого числа белков (всего 18), и большинство вакцин, основанных на использовании пептидов ВИЧ, содержат фрагменты gp120 (gp140, gp160) или обоих белков env gp120 и gp41, другие включают небольшие пептиды матрикса и фрагменты р24. Другая часть вакцин этого класса содержит полноразмерные белки env, pol, gag или их протяженные фрагменты, наработанные в дрожжах со способом гликозилирования, похожим на используемый в жизненном цикле ВИЧ, либо так называемые вакцины с использованием углеводных компонентов. Некоторые из вакцин на основе пептидов ВИЧ предназначаются для терапевтической иммунизации, в отношении других провозглашается, что они обладают профилактической активностью.
Однако ни коктейли из рекомбинантных пептидов ВИЧ, ни коктейли из синтетических 15-20-аминокислотных пептидов до настоящего времени не были способны обеспечить защиту от инфицирования вирусом и его репликации. Главную причину этого можно установить, анализируя принцип получения этих пептидов. Последовательности, кодирующие рекомбинантные пептиды, получают с помощью технологии автоматического секвенирования ДНК образцов, полученных с помощью РТ-ПЦР из вирусного материала от пациента, включая этап амплификации фрагментов генома ВИЧ полимеразой (обычно 1000-3000 пн), или секвенирования ПЦР фрагментов, полученных с использованием ВИЧ-специфических праймеров и ДНК из лимфоцитов пациента в качестве матрицы, с последующим клонированием в E.coli и отбором колоний. Существующая технология основана на моноклональной селекции генотипов ВИЧ, амплифицированных в случайном режиме с частотой единичного варианта последовательности из разнообразия вариантов 105-106, если не больше, при этом средний инфекционный титр вируса составляет 1%, то есть 103-104 копий инфекционно-активного вируса. Исследователям, секвенирующим и анализирующим геномные последовательности ВИЧ, хорошо известно, что две геномные последовательности вируса, полученные этим способом из одного и того же образца крови одного пациента, будут существенно различаться. Таким образом, иммунизация этими рекомбинантными пептидами либо коктейлями из 3-4 рекомбинантных пептидов, даже правильно гликозилированных в эукариотической экспрессионной системе, не может обеспечить формирование иммунного ответа, специфичного для инактивации вариантов вируса, с которыми приходится иметь дело. Следовательно, при разработке вакцин против ВИЧ следует отказаться от этих стандартов, информация о последовательностях рекомбинантных пептидов должна быть получена другим методом.
Маленькие синтетические пептиды ВИЧ [27] получают с использованием другого подхода - сотни их вариантов синтезируются в виде смеси в автоматическом синтезаторе пептидов, а на каждом цикле синтеза пептидов в реакционную смесь добавляется смесь возможных вариантов аминокислот, содержащихся в известных последовательностях ВИЧ в данном участке. Многие варианты вариабельных участков белков env могут быть получены с использованием синтезатора пептидов. Однако размер этих пептидов ограничен 15-20, максимум 30, аминокислотами, более протяженные варианты пептидов можно получить только в рекомбинантных системах. На практике иммунизация небольшими синтетическими пептидами и их коктейлями вызывает достаточно сильную, но низкоспецифичную или неспецифичную на ВИЧ иммунную реакцию. Соответственно, даже попытки иммунизации животных (макак-резус) синтетическими пептидами ВИЧ дали неудовлетворительные результаты, заключавшиеся в отсутствии ВИЧ-специфических антител в их крови при тестировании стандартными методами ELISPOT. Возможно, для терапевтических целей в комбинации с ВААРТ существующие вакцины, основанные на использовании пептидов ВИЧ, могли бы иметь некоторые шансы. Однако ни для одной из пептидных вакцин после иммунизации ими до сих пор не показан превентивный эффект предотвращения инфицирования ВИЧ.
Тип 4: вакцины на основе плазмидных или вирусных (адено-, адено-ассоциированные, птичьей оспы, вакциниа и другие) ДНК-векторов с клонированными в них генами белков ВИЧ [11, 12, 16, 21, 26, 29, 30].
Среди 55 вакцин против ВИЧ, получивших разрешение на 99 клинических испытаний в мире, большинство относятся к классу ДНК вакцин. Но только одна вакцина-кандидат прошла фазу IIb клинических испытаний и имеет некоторые шансы пройти фазу III [37, 42]. Идея использовать этот тип вакцин имеет здравую основу в том смысле, что ДНК при введении в организм не вызывает немедленных побочных эффектов, таких как аутоиммунные осложнения и анафилактические реакции и, таким образом, ее клиническое использование является простым и безопасным. Несмотря на это преимущество все вирусные и невирусные ДНК-вакцины имеют ряд недостатков, и надежда на их реальную эффективность против ВИЧ достаточно слабая.
Поскольку сама ДНК не вызывает никаких иммунологических реакций, эффективность вакцины складывается из трех условий, каждое из которых одинаково важно:
1) эффективность трансфекции/инфекции или количество клеток, приобретших необходимый генетический материал после однократного контакта с ДНК-вектором;
2) уровень экспрессии или количество синтеза данного белка в клетках, которые получили копию соответствующего гена/генов;
3) длительность иммунного ответа или как долго МНС будет продолжать стимулировать выработку мАт, узнающих патоген-мишень.
Эффективность трансфекции/инфекции in vitro представляет собой процент клеток, экспрессирующих определенный белок, подсчитанное через 24 часа после переноса соответствующего гена, до того момента, когда клетки могли бы пройти следующий цикл деления, процент считается для клеток, экспрессирующих флуоресцентный белок или LacZ, гены которых были перенесены одновременно в тех же условиях. Для невирусных плазмидных векторов эффективность in vitro может достигать 40-90%, но для тех же векторов при внутривенном введении in vivo она составляет в лучшем случае 1-5%. Из этих 40-90% (1-5% in vivo) 98-99% представляют собой краткосрочную преходящую либо эписомальную экспрессию, которая исчезает через 2 недели, и только 1-2% трансфицированного генетического материала встраивается в геном клетки и обеспечивает долговременную экспрессию. Количество плазмидной ДНК вакцины [16] ограничено максимальной допустимой дозой доставляющих ее агентов - катионных липидов и сделанных на их основе липосом, катионных полимеров (полиэтиленимин, полилизин), плюроников, а также их различных комбинаций. Практически все положительно заряженные агенты, способные связывать и переносить отрицательно заряженную ДНК, являются высокотоксичными в концентрациях 105-104 М и выше. Уровень экспрессии для невирусных векторов относительно высок по сравнению с уровнем экспрессии вирусных векторов.
Эффективность инфекции для вирусных ДНК векторов варьирует, но в норме не превышает 10-20% для in vitro экспериментов. Однако привлекательность вирусных векторов заключается в их способности обеспечивать доставку генетического материала непосредственно в геном. Таким образом, несмотря на то, что эффективность инфекции для вирусных векторов в in vivo экспериментах составляет в среднем 2-5%, экспрессия нужных белков оказывается преимущественно долговременной, а не преходящей. Таким образом считается, что вирусные ДНК-векторы обеспечивают достаточную длительность иммунного ответа и активности против ВИЧ для терапевтических или превентивных целей.
Рассмотрим вирусные ДНК-вакцины и как они работают. Первым классом ДНК-векторов, которые попали на клинические испытания, были аденовирусные конструкции. Хотя их современные варианты уже показывают эффективность инфекции, отличную от нуля, и титры вырабатывающихся после иммунизации антител детектируются иммунохимическим методом, они никогда не использовались в монорежиме. Дело заключается в том, что аденовирусные - ADV [11] или адено-ассоциированные вирусные векторы - AAV [29] обеспечивают лишь относительно низкую экспрессию доставляемых белков, обычно детектируемую с помощью ИФА, INF-γ ELISPOT или Вестерн-блоттинга через две недели после очередной иммунизации. Если сравнить эти данные для ADV и AAV с титрами антител через две недели после стандартной иммунизации любым рекомбинантным белком или смесью белков, становится очевидным, что абсолютные значения для ADV и AAV вакцинаций в 5-10 раз ниже. Глядя на эти значения, можно сделать некоторые выводы о возможной длительности иммунного ответа.
Единственная вакцина, дошедшая до фазы III клинических испытаний и введенная 16000 здоровых испытуемых в Таиланде с октября 2003, основана на многократной иммунизации плазмидной gag-pol-env-ДНК вакциной (AIDSVAX В/Е) с двумя последующими вакцинациями поксвирусом (вирусом коровьей оспы)-ВИЧ (ALVAC-HIV) [12]. Изучение данных патента показывает, что титры вырабатываемых антител в образцах крови вакцинированных макак-резус возрастают через одну-три недели после каждой иммунизации и в течение года продолжающихся вакцинаций умеренно отличаются в положительную сторону от контрольных вариантов [12]. Вопрос о том, как оценивать продолжительность иммунного ответа в данном случае, остается открытым. Следует также помнить, что аденовирусы и поксвирусы являются одними из самых крупных, они имеют сотни своих собственных белков на неверности и в матриксе вирионов. Это означает, что иммунный ответ, вырабатывающийся в короткий (одна-две недели) период после иммунизации, является сильным, но по большей части неспецифическим и помимо неспецифичности может вызывать иммунотоксические реакции в качестве побочных эффектов.
Единственным исключением в отношении эффективности вирусных вакцин является использование ретро- (ленти-) вирусных векторов [26]. ВИЧ сам по себе является хорошим представителем семейства лентивирусов. Ретровирусные векторы обеспечивают достаточно высокую (до 5%) эффективность инфекции in vivo, экспрессия доставляемых генов умеренно высока и долгосрочна или стабильна вследствие встраивания в геном клетки. Использование ретровирусных векторов дало существенно лучшие противоопухолевые ответы в клинических испытаниях при их использовании в онкологических терапевтических вакцинах, чем использование любых других генетических конструкций. Однако ретровирусы, включая ВИЧ, обладают свойством, которое делает проблематичным возможность их терапевтического применения и неприемлемым их использование для профилактической вакцинации - способностью встраиваться в геном человека в качестве мобильных генетических элементов и инициировать множественные генетические мутации, каскад которых через некоторый период времени становится неконтролируемым и вызывает множественные злокачественные трансформации в различных клетках и тканях.
Основное слабое место ДНК-вакцин против ВИЧ состоит в исходной нуклеотидной последовательности, полученной тем же методом, что был описан выше для рекомбинантных пептидов ВИЧ, и заключающимся в стандартном секвенировании ДНК после ПЦР и клонировании в монорежиме. Близко к истине считать среднее число генетических вариантов ВИЧ в крови одного пациента равным 105-106 вариантам. Генетические конструкции, созданные на основе данных по одной или нескольким последовательностям РНК вируса, полученных описанным выше способом в случайном режиме, в принципе не могут быть эффективны против большинства вариантов ВИЧ, даже для одного пациента. Все ДНК- вакцины против ВИЧ, основанные на плазмидной ДНК или вирусных векторах, содержат единичные последовательности env-, pol-, gag- участков генома ВИЧ и их комбинаций. До тех пор, пока эти конструкции состоят из моноклональных нуклеотидных последовательностей участков генома ВИЧ, это тупиковое направление для разработки вакцин против ВИЧ. Чтобы бороться с генетической вариабельностью и мутабельностью ВИЧ, необходимо проводить количественный анализ существующих форм вируса и получать вакцину против наиболее часто встречающихся вариантов.
Как было описано выше, другое основное ограничение эффективности ДНК-вакцин против ВИЧ заключается в слабости иммунного ответа, возникающей из-за несовершенства известных на сегодня методов доставки in vivo вирусных и невирусных генотерапевтических векторов. Чтобы понять, как низки шансы ДНК-вакцин обеспечить любой вид иммунизации против инфекции, можно провести следующее сравнение. Представьте гипотетические моноклональные антитела (мАт) против какого-либо белка или антигена и их рекомбинантную версию IgG из объединенных L-H цепей, продуцируемую в прокариотической системе экспрессии E.coli. Теперь мы попытаемся сравнить афинность к антигену для этих двух типов мАт с помощью всех возможных лабораторных иммунологических методов - ИФА, ELISPOT, иммуно-дот-блоттинга, Вестерн-блоттинга, проточной цитофлуориметрии, флуоресцентной микроскопии и т.д. Мы увидим, что афинность рекомбинантных мАт всегда по меньшей мере в 10 раз ниже афинности природных моноклональных антител из крови животного или полученных по гибридомной технологии. Более того, различия в минимальной связывающей активности во время титрования могут достигать 100-200 раз. Та же самая ситуация наблюдается при in vivo оценке иммуногенности вакцины, если анализируется активность композиций для иммунизации животных, основанных на ДНК и на белках. Эффективность специфического иммунного ответа, измеренная в виде титра мАт на определенный антиген в крови иммунизированных животных, будет во много раз ниже для антигена, доставляемого в составе генетического вектора, чем для исходного белка-антигена. Сила специфического иммунного ответа на антиген для ДНК варианта всегда в 5-20 раз ниже, чем для его "положительного контроля" - белкового варианта.
Существует еще одна небольшая категория потенциальных вакцин-кандидатов против ВИЧ - так называемые дендритные вакцины. Их разработка была основана на науке о стволовых клетках; дендритные вакцины используются для терапии некоторых опухолевых заболеваний в комбинации с химиотерапией или облучением с достаточно скромными терапевтическими результатами, несмотря на их достаточно высокую стоимость (45-60 тысяч долларов США на лечение одного пациента, в среднем). Однако поскольку дендритные клетки-предшественники макрофагов, «наученные» in situ отличать и убивать некоторый определенный патоген или микроорганизм, могут быть использованы в основном аутологически, в кровотоке того же самого пациента, их эффективность для терапии ВИЧ и, более того, для предотвращения инфекции, вызывает большие сомнения. Кроме того, остается открытым вопрос, где взять вирусные пептиды для «обучения» макрофагов, поскольку рекомбинантные пептиды представляют единичные последовательности вариантов вируса, а нативные не удается выделять в достаточно больших количествах. Следовательно, дендритные вакцины против ВИЧ не могут считаться серьезным кандидатом.
Единственно возможным путем контроля ВИЧ-пандемии является создание вакцины, способной предотвращать инфицирование вирусом ВИЧ-1 и/или блокировать его развитие посредством иммунизации неинфицированных людей, особенно из числа лиц, входящих в группы повышенного риска. Такая вакцина должна содержать смесь индивидуальных нативных пептидных эпитопов ВИЧ-1, а именно, основной белок оболочки ВИЧ-1 gp120, который является единственным экспонированным наружу белком на поверхности вируса, эпитопы его фрагментов, а также белок gp41, в качестве материала для env gp120-gp41 тетрамера с ЗАМЕНЯЮЩИЙ лист (ответ на запрос экспертизы от 5 августа 2010) соответствующими внешними частями и/или эпитопами, распознаваемыми иммунной системой вакцинированного индивидуума. Эти пептиды не могут быть нативными по причинам, рассмотренным выше (стр.3-4 строки 13-30, 1-20). Для рекомбинантных пептидов должна быть получена корректная информация о сиквенсе белков оболочки. Мы предложили альтернативный способ получения вакцины против ВИЧ, детально изложенный в данном патенте, включающий исследование последовательностей env, основанный на:
1) сборе и аффинной очистке нативных вирусных пептидов с помощью технологии фагового дисплея-обратного паннинга;
2) последующем количественном анализе и анализе последовательностей нативных вирусных пептидов, с использованием группы методов LC-MS, предоставляющих информацию о последовательностях gp120 и его фрагментов, представленных в большинстве вариантов в текущей когорте ВИЧ-инфицированных;
3) реконструкции нативных эпитопов пептидов env с использованием эукариотической системы экспрессии в L.tarentolae для продукции рекомбинантных пептидов env с идентичным для эукариот и ВИЧ-типом гликозилирования;
4) составлении профилактической вакцины против ВИЧ с использованием упаковки иммуногенных пептидов env либо в стерически стабилизированные липосомы, либо в виросомы, для обеспечения а) необходимой пролонгации периода иммунного ответа; b) контроля иммунотоксичности.
Протеомный анализ gp120 до настоящего времени производился очень редко и был неполным из-за отсутствия нативных вариантов пептидов, выделенных в чистом виде из смеси с другими вирусными пептидами и клеточными белками. Технология обратного паннинга с аффинной сорбцией вирусных пептидов env на колонках с достаточной сорбционной емкостью может решить эту проблему. Перед приготовлением состава для иммунизации против ВИЧ-инфекции необходимо отобрать изоформы пептидов env, преобладающих в текущей когорте ВИЧ-инфицированных.
Несмотря на огромную генетическую вариабельность вируса в 105 для одного пациента, в среднем, процесс отбора наиболее адаптированных и обладающих наибольшей инфекционной способностью вариантов происходит в организме каждого ВИЧ-инфицированного. Данные по эпидемиологической вариабельности доказывают, что распространение вариантов ВИЧ имеет территориальные границы, зависит от сексуальных контактов, либо от контактов внутривенных наркоманов через общие шприцы. Число доминирующих вариантов вирусных белков определенно значительно меньше, чем общее число генетических вариантов, хотя может меняться достаточно быстро. Нуклеотидная последовательность не может дать информации о том, какие именно варианты доминируют и инфекционно опасны; такую информацию может дать только протеомный количественный анализ и протеомный анализ последовательностей. Мы использовали метод квадрупольной ионной ловушки масс-спектрометрии, сопряженной с жидкостной хроматографической системой.
Нативные пептиды gp120 ВИЧ имеют высокую иммуногенность, но для сохранения такого же уровня иммуногенности у рекомбинантных вариантов без потери идентичности эпитопов требуется рекомбинантная экспрессионная система, обеспечивающая сходный способ гликозилирования. Для решения этой проблемы можно использовать культуры клеток, дрожжевые культуры или лейшмании. Продукция в культуре эукариотических клеток дает очень небольшое количество рекомбинантных белков из-за большого количества собственных клеточных белков - десятки миллионов по сравнению с одной тысячей у E.coli. Дрожжевые культуры обеспечивают достаточно высокий уровень продукции, но гликозилирование в дрожжах не так сильно похоже на гликозилирование высших эукариот, как это предполагалось ранее. Поэтому мы выбрали лейшманийную систему с возможностью индукции экспрессии, высокой экспрессией и с типичным для эукариот способом гликозилирования. Рекомбинантные варианты gp120, продуцируемые в лейшманиях, обеспечивают высокий и на 100% ВИЧ-специфичный иммунный ответ; следующий шаг состоял в том, чтобы сделать этот ответ достаточно пролонгированным для предотвращения развития инфекции.
Существует два возможных пути использования стерически стабилизированных липосом в качестве носителей для пептидных вакцин: либо пептиды включаются в водное содержимое липосомных везикул, либо связываются с активированными дистальными концами ПЭГ и презентируются на поверхности липосом. В обоих случаях пептиды env защищены от быстрого расщепления протеазами и деградации, поэтому продолжительность иммунного ответа увеличивается. Стерически стабилизированные липосомы (состоящие из нейтральных липидов) нетоксичны и безвредны сами по себе. Везикулы могут сохранять загруженные в них иммунногенные пептиды в течение нескольких недель или месяцев и способны высвобождать свое содержимое постепенно в течение достаточно долгого периода, а не одномоментно. Это дает возможность использовать большее количество белка для одной вакцинации. Более сильные и продолжительные иммунные ответы формируются в тех случаях, когда обеспечивается более длительный постоянный контакт НСС с чужеродными белками. Это может иметь решающее значение для профилактического блокирования ВИЧ-инфекции и успешной разработки вакцин.
И последнее, что следует учитывать при анализе вакцин-кандидатов против ВИЧ, состоит в том, что не существует адекватных моделей in vivo для предклинической оценки их эффективности. Все попытки использовать шимпанзе для моделирования ВИЧ-инфекции с последующим воздействием антиретровирусными химиотерапевтическими препаратами были убедительными и правомерными, но невозможно оценить иммунный ответ против ВИЧ у шимпанзе или макак-резус. Спектр иммуногенных реакций, которые могут быть вызваны у человекообразных обезьян и мартышек, сильно отличаются от спектра реакций у человека при иммунизации тем же антигеном. Более того, шимпанзе, например, могут быть инфицированы любым субтипом ВИЧ и жить до глубокой старости с летальными для человека уровнями вирусной нагрузки без малейших признаков развития симптомов болезни иммунодефицита, так же как при инфицировании их собственным обезьяньим вирусом. Таким образом, для тестирования любых иммунногенных композиций против ВИЧ обычные лабораторные мыши ничем не хуже обезьян, но, в отличие от последних, доступны в статистически значимых количествах и позволяют проводить более частый иммунологический анализ крови. Только клинические испытания могут подтвердить, может ли новая вакцина против ВИЧ обеспечить иммунопротективный эффект.
РИСУНКИ
Рис.1: Анализ наличия зрелых В-лимфоцитов ВИЧ-инфицированного пациента, проведенный с помощью моноклональных антител к CD-45, конфокальная микроскопия:
а,b) "хороший" источник для выделения РНК ВИЧ-специфических антител;
c,d) довольно "плохой" источник для выделения РНК - пациент, находящийся на поздней стадии развития заболевания (СПИД);
е) Т- и В- лимфоциты из крови инфицированного пациента, без флуоресцентного окрашивания.
Рис.2: Схема получения фагмидной ДНК-библиотеки в соответствии с предпочтительным способом исполнения настоящего изобретения.
Рис.3: Диаграмма, демонстрирующая отбор позитивных, продуцирующих антитела клонов с помощью ИФА в соответствии с предпочтительным вариантом исполнения настоящего изобретения.
Рис.4: Получение рекомбинантных фаговых библиотек и панинг.
Рис.5 a-b: Структура рекомбинантного фага М13 с презентированной на "головках" библиотекой антител, обогащенной специфическими антителами к env пептидам ВИЧ. Сканирующая атомная силовая микроскопия в контактном режиме выполнена с использованием NanoWizard (JPK Instruments, Германия) на основе флуоресцентного инвертированного микроскопа Nikon Eclipse 2000U длинным кантилевером CSC17/noAl(MicroMash, Эстония) с резонансной частотой 12 кГц. Длина фага в среднем 800 нм, толщина 40-50 нм, презентирование ВИЧ-специфической ScFv библиотеки составляет 1-5 молекул антител на одну фаговую частицу; измеренный размер "головки" составляет в среднем 200-250 нм:
а) рекомбинантный фаг М13 и его "головка" с презентироваными рекомбинантными ВИЧ-специфическими антителами;
b) контрольный фаг-помощник М13Ко7.
Рис.6 a-b: Структура афинной колонки с основой из крупнопористого эпокси-активированного криогеля, использовавшейся для проведения процедуры обратного панинга. Сканирующая атомная силовая микроскопия в контактном режиме выполнена с использованием NanoWizard (JPK Instruments, Германия) длинным кантилевером CSC17/noAl:
a) крупнопористый монолитный эпокси-активированный сорбент перед нанесением рекомбинантного фага;
b) крупнопористый монолитный эпокси-активированный сорбент после посадки М13 мАт и с презентированной на рекомбинантном фаге ВИЧ-специфической ScFv библиотекой.
Рис.7а,b: Технология обратного панинга для сбора пептидов env ВИЧ:
a) профиль фракции, элюированной с аффинной колонки для обратного панинга (пул изолятов субтипа А, для концентрирования использовали ПЭГ-преципитацию с последующим ультрацентрифугированием при 100000g в градиенте 20% сахарозы), пики А и В проверяли на наличие специфических пептидов env с помощью вестерн-блоттинга с использованием поликлональных антител против ВИЧ;
b) профиль фракции, элюированной с аффинной колонки для обратного панинга (пул изолятов субтипа А, для концентрирования супернатанта использовали ультрафильтрацию), пик проверяли с помощью Вестерн-блоттинга с использованием поликлональных антител против ВИЧ.
Рис.8 a-b: SDS-PAGE и Вестерн-блоттинг (ECL детекция) фракций пула пептидов env ВИЧ субтипа А, элюированных с колонки для обратного панинга:
a) 1 - белковые маркеры; 2 - фр.№4, 3 - фр.№5, 4 - фр.№6, 5 - фр.№7, 6 - фр.№8, 7 - фр.№11, 8- фр.№9, - все пробы были приготовлены с (3-меркаптоэтанолом ((3-МЕ);
b) 1 - фр.№1 с Р-МЕ; 2 - фр.№2 с β-МЕ; 3 - ВИЧ-ПЭГ с β-МЕ; 4 - ВИЧ-осадок (ультрацентрифугирование) с β-МЕ; 5 - ВИЧ-супернатант с β-МЕ; 6 - белковые маркеры; 7 - фр.№1 без Р-МЕ; 8 - фр.№2 без β-МЕ; 9 - фр.№6 без β-МЕ; 10 -ВИЧ-ПЭГ без β-МЕ; 11 -ВИЧ-осадок (ультрацентрифугирование) без β-МЕ; 12 - ВИЧ-супернатант без Р-МЕ.
Рис.9а-с: Реконструкция структуры gp 120 Env с помощью секвенирования и 2D анализа:
a) #А1.RU.03.03RU20_06_13_AY500393
MKAKGMQRNYQHLWRWGXMLFWXIIM;
b) B.RU.04.04RU128005_AY682547
MRARGIRKNYQGLLRWGTLLLGILMI;
c) #B.RU.04.04RU129005_AY751406
MRAKGTRKNYQRLWRWGIMLLGMLMI.
Рис.10 a-d: Схематическое изображение 3D структуры белков оболочки ВИЧ-1:
a) схематическое изображение 3D структуры коровой части gp120 [40,41];
b) схематическое изображение 3D структуры эпитопов gp120, связывающихся с CD4 - CCR5 [24];
c) схематическое изображение 3D структуры конформационного преобразования gp120 при образовании CD4-cвязывaющeй петли [22];
d) структура и вариабельность эктодомена gp41 [34].
Рис.11 a-b: ПЦР-амплификация фрагментов ДНК, кодирующих пептиды env ВИЧ:
a) полноразмерного gp120, внутреннего и внешнего доменов gp120 и петель V2, V3 и V4;
b) полноразмерного gp41 и эктодомена gp41.
Рис.12: Продукция пептидов env ВИЧ и их фрагментов в различных экспрессионных системах:
a) индуцируемая экспрессия внутреннего домена gp120, эктодомена gp41, SD-PAGE;
b) конститутивная экспрессия gp120, gp41, SDS-PAGE, Вестерн-блоттинг ECL-детекция.
Рис.13: Схема N-гликозилирования белков в клетках Leishmania tarentolae (экспрессионная система LEXSY) в сравнении с гликозилированием в других системах экспрессии белка. Типы гликозилирования, полученные в клетках млекопитающих и в Leishmania tarentolae, отличаются только присутствием N-сиаловой кислоты на концах углеводных цепей в клетках млекопитающих (Jena Bioscience GmbH).
Рис.14: Карта вектора семейства pLEXSY_I-2 с сайтами для клонирования генов, замещающих при клонировании фрагмент-вкладыш размером 1000 п.н. 5'odc и 3'odc участки нужны для интеграции в хромосому реципиента за счет гомологической рекомбинации после линеаризации экспрессионной плазмиды с помощью SwaI. Utrl, полученный из 0.4k-IR L. tarentolae aprt, utr2, полученный из 1.4k-IR camCB, и utr3, полученный из 1.7k-IR, представляют собой фланкирующие ген оптимизированные нетранслируемые участки, обеспечивающие сигналы сплайсинга для посттранскрипционного процессинга мРНК для обеспечения экспрессии целевых и маркерных генов в организме-реципиенте LEXSY T7-TR. SP обозначает сигнальный пептид секретируемой кислой фосфатазы L. mexicana LMSAP1 (7), а Н6 - последовательность из шести остатков гистидина. Использование альтернативных схем клонирования приводит либо к экспрессии нужного белка в цитозоль (с), либо к его секреторной экспрессии (s). Для обеспечения экспрессии в цитозоль при клонировании используют расположенные на 5' конце сайты BglII, NcoI, или SalI, а для секреторной экспрессии - SalI или XbaI. Использование для клонирования сайтов рестрикции NheI, MspCI или KpnI на 3'конце фрагмента-вкладыша приводит к синтезу белка с С-концевой His6 последовательностью, в то время как использование для клонирования сайта NotI дает белок без His6 последовательности. В качестве маркерных могут использоваться гены ble (устойчивости к блеомицину) и neo (аминоглюкозид фосфотрансферазы) (Jena Bioscience GmbH).
Рис 15 a-d: Этапы хроматографической очистки рекомбинантных пептидов env ВИЧ:
a) экспрессия 6Hisp120id1 в E.coli (SDS-PAGE 5-20%);
b) очистка 6Hisp120id1 на колонке Ni-NTA;
c) очистка 6Hisp120id1 на колонке Biosuite Q -PEEK 10um 4.6*50 мм (Waters, USA);
d) очистка 6Hisp120id1 с помощью гель-фильтрационной хроматографии на Superose 12 10/300 GL, до и после очистки.
Рис 16 a-b: Типы липосомного адъюванта для рекомбинантных пептидов env ВИЧ:
a) схематическое изображение стерически стабилизированных липосом (ССЛ, 150 нм, ПЭГ-400) с загруженными во внутреннюю водную фазу везикул рекомбинантными пептидами env ВИЧ;
b) схематическое изображение стерически стабилизированных липосом (ССЛ, 200 нм, ПЭГ-2000) с присоединенными к активированным дистальным концам ПЭГ рекомбинантными пептидами env ВИЧ.
Рис 17: Распределения по размеру по Гауссу и по Никомп для стерически стабилизированного липосомального (ССЛ) компонента вакцины: средний диаметр везикул составляет 155 нм.
Рис.18: Результаты ПЦР-I - вариабельные κ- и λ-цепи с частичными Сκ или CL фрагментами соответственно.
Вырезанные из геля нужные полосы отмечены стрелками.
Сокращения:
1VL - вариабельные λ цепи с частичным CL фрагментом;
2VL - вариабельные κ цепи с частичным CL фрагментом;
1VK - вариабельные κ цепи с частичным Сκ фрагментом;
1а - 10 - различные пары праймеров;
-VK - отрицательные контроли ПЦР;
100 bp - GeneRuler™ 100 bp DNA Ladder (Fermentas).
Рис.19: Результаты ПЦР-1 - вариабельные κ цепи с частичными Сκ фрагментами, вырезанные из геля нужные полосы отмечены стрелками.
1а-10 - различные пары праймеров;
1VK(2VK,3VK,4VK) - вариабельные κ цепи с частичным Сκ фрагментом;
100пн - GeneRuler™ 100пн ДНК-маркер (Fermentas).
Рис.20: Результаты ПЦР-I - вариабельные λ цепи с частичными CL фрагментами и вариабельные тяжелые (Н) IgM и IgG цепи с частичными СН1 фрагментами, вырезанные из геля нужные полосы отмечены стрелками.
1VL - библиотека 1, вариабельные λ цепи с частичным CL фрагментом;
1VHM (2VHM, 3VHM, 4VHM) - тяжелые вариабельные цепи с частичным СН1 фрагментом (IgM);
1VHG (2VHM, 3VHM, 4VHM) - библиотеки 1,2,3,4, тяжелые вариабельные цепи с частичным СН1 фрагментом (IgG);
1а-10 - различные пары праймеров;
-VL; -VHG; -VHM - отрицательные контроли ПЦР.
Рис.21: Результаты ПЦР-II - вариабельные тяжелые (Н) цепи с добавленными к ним линкерными фрагментами, кодирующими ((Сly)4Sеr)3. Вырезанные из геля нужные полосы отмечены стрелками.
1HG (2HG, 3HG, 4HG) - библиотеки 1,2,3,4, вариабельные тяжелые цепи из IgG кДНК пула;
1НМ (2НМ, 3НМ, 4НМ) - библиотеки 1,2,3,4, вариабельные тяжелые цепи из IgM кДНК пула;
1a-7a - пары различных содержащих линкеры праймеров;
-Н - отрицательные контроли ПЦР.
Рис.22: Результаты ПЦР-II - вариабельные κ- и λ- цепи с добавленными к ним линкерными фрагментами, кодирующими ((Gly)4Sеr)3. Вырезанные из геля нужные полосы отмечены стрелками.
1К (2К, 3К) - библиотеки 1,2,3, вариабельные κ цепи;
1L (2L, 3L, 4L) - библиотеки 1,2,3,4, вариабельные λ цепи;
1а-8 - пары различных содержащих линкеры праймеров;
-К и -L - отрицательные контроли ПЦР;
100 bp - GeneRuler™ 100 bp ДНК-лестница (Fermentas).
Рис.23: Результаты ПЦР-П - вариабельные κ-, λ- и тяжелые (Н) цепи с добавленными к ним линкерными фрагментами, кодирующими ((Gly)4Ser)3. Вырезанные из геля нужные полосы отмечены стрелками.
1К (2К, 3К, 4К) - вариабельные κ цепи;
1L - библиотека 1, вариабельные λ цепи;
2HG (3HG, 4HG) - библиотеки 2, 3 и 4, вариабельные тяжелые цепи из IgG к ДНК пула;
3НМ (4НМ) - библиотеки 3 и 4, вариабельные тяжелые цепи из IgM кДНК пула;
1a-8 - пары различных содержащих линкеры праймеров;
-К и -L - отрицательные контроли ПЦР;
100 bp - GeneRuler™ 100 bp ДНК-лестница (Fermentas).
Рис.24: Количественное определение в геле собранных ScFv фрагментов. Различные варианты Vн-линкер-Vкарра ScFv (библиотека 4);
H1a-VH1a-линкер-Vkappa mix вариант ScFv.
Рис.25: Реамплификация Vн-линкер-Vkарра смеси ScFv (из библиотеки 4). Вырезанные из геля нужные полосы отмечены стрелками.
Рис.26:
a) результаты ИФА для рекомбинантных mAb клонов с ВИЧ- позитивной сывороткой;
b) результаты ИФА для 38 фаговых моноклонов из фаговых библиотек, отобранных на вирусных пептидах А455 (статистический анализ в SigmaPlot 10.0);
c) результаты ИФА для 26 фаговых моноклонов из фаговых библиотек, отобранных на рекомбинантных gp110-, gp160 (статистический анализ в SigmaPlot 10.0).
Рис.27: Предварительные результаты иммунизации животных (статистический анализ в SigmaPlot 10.0).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает создание профилактической вакцины против ВИЧ, предпочтительно против ВИЧ-1 субтипов А и В, защитная активность которой обуславливается возникновением определенного иммунного ответа, который вырабатывается у людей после введения им настоящей вакцины. Следовательно, ее активной субстанцией являются смеси рекомбинантных полипептидов/пептидов, отобранных в соответствии с комплексной технологией, детально описанной ниже. Основные компоненты вакцины представлены белками оболочки вируса и их фрагментами, которые включают, в основном, белки ВИЧ gp120, gp140, gp160 (Рис.1) и gp41 на различных стадиях гликозилирования, консервативные домены в области V1-V3 петель gp120; антитела к ассоциирующимся с резистентностью вариабельным участкам gp120 V1-V5 петель, гликозилированным вариантам gp41; СD4-связывающие эпитопы вирусных белков оболочки gp120, gp140, gp160, проксимальные V1/V2 и V3 петли которых претерпевают конформационные изменения при контактировании CD4 рецептора с белками оболочки ВИЧ-1 и с внешней частью белка gp41; сайты связывания белков оболочки вируса с CXCR5 и CCR4 корецепторами; различные эпитопы р24.
Эти рекомбинантные полипептиды и их смеси собирают, идентифицируют и клонируют, используя библиотеку рекомбинантных фаг-презентированных антител, полученную из мРНК В-лимфоцитов различных доноров. Каждая созданная фаговая библиотека антител специфична в отношении связывания различных эпитопов рекомбинантных gp120-, gp41 и нативных полипептидов ВИЧ, и, предпочтительно, также эпитопов, присутствующих на рекомбинантных белках gp140-, gp160- и р24 ВИЧ-1 субтипа А.
Эти библиотеки рекомбинантных фаг-презентированных антител могут быть использованы в различных областях применения в качестве средств детектирования, анализа и/или очистки [23]. Области применения, использующие вышеупомянутые библиотеки антител, включают (но не ограничиваются ими) иммунологические анализы, иммуноблоты, хроматографию и др.
В соответствии с настоящим изобретением антитела также пригодны для разработки новых лекарственных средств для терапии и/или предотвращения ВИЧ-инфекции.
В предпочтительном варианте исполнения настоящего изобретения рекомбинантные антитела, презентированные на фаге М13, используются как стадия получения профилактической вакцины против ВИЧ.
Поскольку фрагменты антител фагмидной библиотеки связываются с достаточно консервативными конформационными эпитопами белков ВИЧ, мишени для вышеупомянутых антител квалифицируют как вакцину против ВИЧ-инфекции, поскольку после введения антигенов-мишеней иммунная система будет вырабатывать специфический иммунный ответ против этих эпитопов, то есть зрелые В-клетки и Т-клетки, способные в качестве клеток памяти передавать иммунитет следующим поколениям В-лимфоцитов.
Вакцина против ВИЧ, в соответствии с настоящим изобретением, включает рекомбинантные белки gp41 и р24 ВИЧ-1 субтипа А и фрагменты gp120, gp140 и gp160, фрагменты которых (Таблица 9 из Примера 3) связываются с антителом. В соответствии с методом настоящего изобретения вакцина может дополнительно включать обычные носители и наполнители и, по выбору, иммуностимуляторы.
Вакцина будет предотвращать инфицирование, а также дальнейшее прогрессирование ВИЧ-инфекции за счет обеспечения иммунной системы клетками памяти, специфичными по отношению к эпитопу, который может присутствовать на любом вирусе ВИЧ, а также на мутантных вирусах ВИЧ.
Вышеупомянутые рекомбинантные белки и/или их фрагменты получают на основе информации о последовательностях, полученной путем связывания и анализа нативных белков оболочки ВИЧ-1, которые отбираются с помощью ВИЧ-специфических фаг-презентированных антител.
Анализируемые белки оболочки выделяют при разрушении вирусных частиц соответствующими методами, такими как ультрацентрифугирование и лизис вирусных частиц.
Отбор подходящих белков может быть осуществлен любым подходящим методом скрининга, известным квалифицированному специалисту. В предпочтительном варианте исполнения изобретения отбор может быть произведен (i) панингом с использованием рекомбинантного фага с презентированными антителами для сбора белков оболочки вируса и/или (ii) аффинной сорбцией на ВИЧ-специфических антителах, прикрепленных к поверхности пластика для культивирования, и/или (iii) отбором белков оболочки вируса с помощью аффинной хроматографии с использованием закрепленных на колонке ВИЧ-специфических антител.
На следующем этапе могут быть идентифицированы последовательности полученных и отобранных нативных вирусных белков и/или 3D-конформации изоформ. Действуя таким образом, получают смесь многих вариантов высокоспецифичных вариабельных и/или константных фрагментов вирусных белков, таких как gp120, gp41 и р24, циркулирующих в крови ВИЧ-1-инфицированных, а также пациентов, которые получали различные режимы антиретровирусной терапии, включая различные варианты NRTI, NNRTI и ВААРТ. На основе вышеупомянутых последовательностей получают рекомбинантные полипептиды и/или фрагменты вирусных белков. Эти последовательности могут быть получены любым методом, пригодным для продукции полипептидов, которые могут быть распознаны иммунной системой, для индукции соответствующего иммунного ответа.
Рекомбинантные полипептиды могут быть получены в любой подходящей эукариотической системе экспрессии, такой как индуцируемая система экспрессии в лейшмании или любая дрожжевая система экспрессии, но обязательно с гликозилированием по принципу эукариотов.
Технология разработки различных вариантов профилактической вакцины против ВИЧ-1 субтипов А и В настоящего изобретения включает этапы 1-9, которые будут подробно рассмотрены ниже:
1. Создание фагмидной библиотеки рекомбинантных человеческих IgG антител, содержащей ВИЧ-специфические ScFv фрагменты антител (технология фагового дисплея).
2. Обогащение рекомбинантной фагмидной библиотеки фаг-презентированными ВИЧ-специфическими ScFv фрагментами антител (биопанинг).
3. Размножение не подвергшегося воздействию антиретровирусной терапии вирусного материала in situ методом РВМС-МТ инфекции.
4. Концентрирование частиц ВИЧ и пептидов ультрафильтрацией и/или ультрацентрифугированием; инактивация и разрушение вируса.
5. Сбор нативных пептидов env ВИЧ с помощью фагмидной библиотеки ВИЧ-специфических рекомбинантных ScFv фрагментов антител в ходе процедуры обратного панинга.
6. Количественный анализ и анализ вариабельности и частоты встречаемости последовательностей пептидов env методом жидкостной хроматографии - масс-спектрометрии (LC-MS).
7. Клонирование составляющих большинство пептидов env ВИЧ и продуцирование рекомбинантных пептидов в Leishmania tarentolae для создания вакцины.
8. Хроматографическая очистка рекомбинантных пептидов env ВИЧ и анализ их 3D-структуры.
9. Приготовление композиции профилактической вакцины против ВИЧ с использованием стерически стабилизированных липосом или виросом в качестве пролонгирующих имунный ответ средств доставки вакцины.
1. Создание фагмидной библиотеки человеческих рекомбинантных антител IgG, содержащей ВИЧ-специфические ScFv фрагменты антител (технология фагового дисплея)
В соответствии с методом настоящего изобретения фагмидная библиотека может дать получена на этапе 1) в соответствии со стадиями i) - iii), включающими:
i) амплификацию фрагментов ДНК, полученных из РНК, кодирующей вариабельные участки легких и тяжелых цепей иммуноглобулинов IgG, экспрессирующихся в В-лимфоцитах, полученных из крови группы ВИЧ-инфицированных;
ii) объединение фрагментов ДНК легкой и тяжелой цепей, полученных на стадии i), в единую конструкцию, состоящую из фрагмента ДНК, кодирующего вариабельный участок легкой цепи IgG, объединенного с фрагментом ДНК, кодирующим вариабельный участок тяжелой цепи IgG;
iii) клонирование в фагмидном векторе pCANTAB.
Из крови группы достаточно давно ВИЧ-инфицированных людей, у которых ожидается наличие ВИЧ-специфических антител, выделяют В- и Т-лимфоциты. Также могут быть использованы больные, инфицированные резистентными вариантами вируса ВИЧ. Выделение В-клеток может быть осуществлено любым известным методом, включая лейкофорез с последующим выделением В-/Т-клеток из популяции лимфоцитов [19]. Затем из В-/Т-лимфоцитов хорошо известными в литературе методами выделяют мРНК, например методом, описанным в [23]. Однако для успешного получения ScFv библиотек должна быть выделена мРНК, содержащая ВИЧ-специфические последовательности иммуноглобулинов. Поэтому перед выделением РНК мы оценивали число В-лимфоцитов в крови ВИЧ-инфицированных пациентов иммунологическим методом с использованием моноклональных антител к CD45 с помощью конфокальной или флуоресцентной микроскопии. Данные, представленные на Рис.1, показывают, что некоторые пациенты, находящиеся на поздних стадиях развития заболевания и имеющие симптомы СПИДа, имеют очень малое количество В-лимфоцитов по отношению к общему числу выделенных лимфоцитов (Рис.1 c,d). Обычно слабое окрашивание при использовании флуоресцентно-меченых антител к CD45 коррелирует с высокой вирусной нагрузкой и низким соотношением CD4: CD8. В отличие от остальных (Рис.1 а,b), такие пациенты являются довольно плохим источником биоматериала для получения фагмидных библиотек ВИЧ-специфических антител. Сама по себе высокая вирусная нагрузка или предшествовавшие курсы антиретровирусной терапии и частота последних не являются ограничением для возможности получения фагмидных библиотек ВИЧ-специфических ScFv фрагментов антител.
Из полученной таким образом мРНК в процессе обратной транскрипции может быть синтезирована кДНК с использованием олиго dT, или, как описано в этом патенте, с использованием олигонуклеотидных праймеров, специфичных для константных областей легких и тяжелых цепей иммуноглобулинов. Последовательности различных константных областей тяжелых и легких цепей иммуноглобулинов хорошо известны в литературе, так что соответствующие праймеры для синтеза кДНК могут быть легко подобраны. Метод позволяет осуществить первичный отбор иммуноглобулиновых транскриптов в пуле (пулах) РНК и упростить манипуляции с различными образцами РНК от различных доноров, поскольку не имеющий интереса материал может быть исключен на вышеупомянутом первом этапе. Также предусматривается и является предпочтительным объединение пулов РНК от различных доноров перед проведением обратной транскрипции мРНК в кДНК, поскольку, таким образом, может быть получено гораздо большее разнообразие вариантов (см. ниже). Вторые цепи в полученной кДНК синтезируются в соответствии со стандартными методиками.
Чтобы получить нужные фрагменты ДНК в достаточном количестве, интересующие участки могут быть амплифицированы с использованием в качестве матрицы непосредственно мРНК (в ходе РТ-ПЦР), полученной из В-/Т-лимфоцитов, кДНК, либо двухцепочечной ДНК, полученной из кДНК.
Для проведения ПЦР реакций могут быть использованы соответствующие праймеры, гибридизующиеся с концами амплифицируемых нуклеотидных последовательностей; праймеры обычно являются олигонуклеотидами длиной 10-40, предпочтительно 15-30, наиболее предпочтительно 20-30 нуклеотидов.
В качестве обратных праймеров могут быть использованы олигонуклеотиды, последовательности которых гибридизуются с константными участками иммуноглобулинов. В предпочтительном варианте вышеупомянутые обратные олигонуклеотидные праймеры гибридизуются с СН1 участком тяжелых цепей, либо Cλ или Сκ участками легких λ и κ цепей соответственно. Используемые прямые праймеры гибридизуются с противоположными концами вариабельных участков тяжелых и легких цепей.
Для ПЦР амплификации первого раунда используют прямые и обратные праймеры приведенные в таблицах 1-3, которые были взяты из базы данных V BASE (http://vbase.mrc-cpe.cam.ac.uk). В циклах ПЦР-амплификации в основном получаются фрагменты длиной около 750 п.н.
На следующем этапе происходит объединение фрагментов ДНК легкой и тяжелой цепей, полученных на этапе i), в единую конструкцию, состоящую из нуклеотидной последовательности, кодирующей вариабельную область легкой цепи IgG, объединенной с нуклеотидной последовательностью, кодирующей вариабельную область тяжелой цепи IgG, для обеспечения экспрессии полипептида, состоящего из вариабельных областей легкой и тяжелой цепей IgG соответственно.
Список олигонуклеотидных праймеров для ПЦР амплификации легких κ цепей человеческих иммуноглобулинов
Список олигонуклеотидных праймеров для ПЦР амплификации легких λ цепей человеческих иммуноглобулинов
Список олигонуклеотидных праймеров для ПЦР амплификации тяжелых цепей человеческих иммуноглобулинов (IgM, IgG, IgA)
Для объединения фрагментов ДНК, кодирующих вариабельные легкие и тяжелые цепи, количество образца, полученного на этапе i) может быть разделено на аликвоты, например, на две части, и при необходимости разбавлено. В одной из полученных частей образца вышеупомянутые фрагменты ДНК, полученные из РНК через этап получения кДНК, из кДНК или из двухцепочечной ДНК, полученной из кДНК, взаимодействуют с линкером, специфическим для легких цепей, а в другой части образца - с линкером, специфическим для тяжелых цепей, так что соответствующий линкер связывается с соответствующими фрагментами ДНК. Таким образом, в одной части будут фрагменты легких цепей с линкером для легких цепей, а в другой - фрагменты тяжелых цепей с линкером для тяжелых цепей. Комплементарные участки линкеров позволят им в определенных условиях гибридизоваться друг с другом, что приведет в присутствии полимеразы к синтезу фрагмента ДНК, состоящего из вариабельной области легкой цепи и вариабельной области тяжелой цепи. Объединение двух фрагментов ДНК происходит таким образом, что они оказываются в одной рамке считывания, что позволяет получить полипептид с аминокислотной последовательностью вариабельной области легкой цепи и вариабельной области тяжелой цепи. Таким же способом можно получить объединение двух тяжелых и двух легких цепей, если это требуется.
В таблицах 4 и 5 перечислены предпочтительные для использования праймеры.
Список обратных олигонуклеотидных праймеров для второго раунда ПЦР амплификации вариабельных фрагментов легких λ и κ цепей человеческих иммуноглобулинов
Список олигонуклеотидных праймеров для ПЦР амплификации и объединения легких и тяжелых цепей человеческих иммуноглобулинов
К нуклеотидным последовательностям, кодирующим вариабельные тяжелые и легкие цепи иммуноглобулинов, добавляются линкерные фрагменты, кодирующие полипептидный линкер ((Glу)4Sеr)3. Линкерные части тяжелых и легких цепей отжигаются друг на друга и служат затравками для реакции заполнения в присутствии Taq ДНК полимеразы, например TaqSE ДНК полимеразы. В конечном итоге тяжелые и легкие цепи объединяются в единый ген с помощью своих линкерных фрагментов.
Метод фагового дисплея позволяет получить огромное количество рекомбинантных антител, полученных путем случайного объединения последовательностей нуклеотидов, кодирующих вариабельные области легких и тяжелых цепей иммуноглобулинов друг с другом, а также позволяет получить комбинации легких и тяжелых цепей, образующих антигенсвязывающие центры, не присутствовавшие в полученном исходно РНК пуле. Как может быть показано, при случайном объединении предварительно сформированных природных частей антигенсвязывающих сайтов вариабельных участков иммуноглобулинов также можно получить антитела с усиленной и константной афинностью к белкам ВИЧ, по сравнению с нативными антителами, образующимися в организме ВИЧ-инфицированных индивидуумов.
Дополнительно в полученные таким образом ДНК-фрагменты могут быть введены сайты рестрикции, нужные для последующего использования, например для этапов клонирования. В принципе, может быть использован любой подходящий сайт рестрикции для последующей экспрессии в фаге или эукариотах. Добавление сайтов рестрикции может быть осуществлено любым подходящим описанным в литературе способом, например, с помощью олигонуклеотидных праймеров, содержащих нуклеотидную последовательность сайта рестрикции, либо с помощью адапторных молекул, содержащих нуклеотидную последовательность сайта рестрикции, объединенную с 5'- и/или 3'- концом, соответственно.
В концы объединенных фрагментов ДНК вводятся сайты рестрикции Sfi I и Not I, которые используются для последующих этапов клонирования в векторы. Sfi I и Not I сайты рестрикции добавляются к 5'- и 3'-концам объединенных фрагментов (ScFv генов), соответственно. Эти сайты рестрикции встречаются в последовательностях генов антител очень редко и позволяют клонировать большинство полученных объединенных фрагментов, состоящих из нуклеотидных последовательностей легкой и тяжелой цепей, в виде единого Sfi I/Not I фрагмента. Сайты рестрикции Sfi I и Not I вводятся с помощью олигонуклеотидных праймеров. Предпочтительные варианты олигонуклеотидных праймеров, содержащих Sfi I или Not I сайты рестрикции, подбираются на основе последовательностей праймеров из статьи [21]. Последовательности праймеров, используемые для введения Sfi I и Not I сайтов рестрикции в концы полученного слитого фрагмента, состоящего из нуклеотидных последовательностей легкой и тяжелой цепей, приведены в таблице 6.
Список олигонуклеотидных праймеров для введения Sfi I и Not I сайтов рестрикции в концы собранного ScFv гена
Для клонирования и экспрессии полученных объединенных фрагментов, состоящих из нуклеотидных последовательностей легких и тяжелых цепей, может быть использован любой подходящий клонирующий и/или экспрессионный вектор. В предпочтительном варианте используются фагмидные векторы, например фагмидный вектор pCANTAB 5 для Е.coli.
Фагмида pCANTAB 5E имеет ориджины репликации М13 и Co1E1 и может быть легко наработана в виде плазмиды, либо в виде рекомбинантного фага М13 с помощью фага-помощника М13К07. Объединенные вариабельные фрагменты генов иммуноглобулинов после рестрикции Sfi I и Not I клонируют между лидерной последовательностью и основной частью гена 3 фага М13 в фагмидном векторе pCANTAB 5E. Финальный слитый белок сохраняет свойства обоих родительских белков. Лидерная последовательность g3p направляет транспорт белка к внутренней мембране/периплазме Е.соli, где за счет основного домена g3p происходит прикрепление слитого белка к концу собирающегося фага. pCANTAB 5E также содержит амбер стоп кодон в области слияния клонированного ScFv и последовательности g3p. Конечный пул плазмидных производных pCANTAB, содержащих ScFv фрагменты, используется для трансформации supE штамма Е.coli, такого как TG1. В supE штаммах Е.coli при трансляция преодолевается амбер стоп кодон в pCANTAB 5E, что приводит к продукции слитого белка ScFv-g3p, расположенного на конце фага.
Олигонуклеотидные праймеры для реамплификации смесей ScFv фрагментов
Сайты узнавания для эндонуклеазы рестрикции Noti выделены синим цветом; сайты узнавания для эндонуклеазы рестрикции Sfil выделены зеленым цветом.
В несупрессорных штаммах, таких как НВ2151, амбер стоп кодон опознается, синтез белка прекращается в конце ScFv гена, и слитый с g3p белок не синтезируется. В этом случае, финальный ScFv белок транспортируется в периплазматическое пространство, но не соединяется с фаговой частицей, поскольку не имеет домена гена 3. Вместо этого растворимые фрагменты антител аккумулируются в периплазме и после продолжительной инкубации просачиваются в среду. Поэтому НВ2151 и сходные штаммы Е.coli используются для продукции растворимых антител после инфицирования отобранными антиген-позитивными фагами и не могут быть использованы при создании фаг-презентированных библиотек. Этапы создания ScFv библиотек представлены в Примере 1.
2. Обогащение рекомбинантной фагмидной библиотеки ВИЧ-специфическими scFv фрагментами антител (биопанинг)
Экспрессия антител может быть получена в подходящих организмах, чтобы получить высокоафинные антитела, способные связывать антигены - рекомбинантные gp120-, gp 41- и нативные полипептиды ВИЧ, полученные из изолятов различных доноров. Таким образом, полезно, но не необходимо, чтобы экспрессируемый полипептид был презентирован на поверхности клетки хозяина. Подходящие организмы-хозяева для экспрессии антител включают вирусные системы, прокариотические и эукариотические клетки и/или культуры клеток.
В варианте, когда фрагменты антител экспрессируются на поверхности бактериофагов М13, образуя фаговую библиотеку, что позволяет получить огромное число различных вариантов антител, каждый из которых представлен отдельной фаговой частицей - фаг дисплей. Технология фагового дисплея представляет собой мощное средство для клонирования генов иммуноглобулинов и для экспрессии и детекции функциональных антител. Она позволяет получить фрагменты вариабельных тяжелых и легких цепей антител в виде составных белков, расположенных на поверхности фагов; позволяет получить пул или библиотеку ВИЧ-специфических антител без этапа селекции моноклональных антител. Этот подход дает возможность быстро находить антитела к любому антигену и производить, в случае необходимости, их растворимые варианты с гликозилированием или без гликозилирования в других экспрессионных системах.
Панинг фагмидной библиотеки представляет собой in vitro технологию, которая позволяет очень быстро осуществить скрининг большого числа клонов, где фаги с презентированными на их поверхности антителами, имеющими афинность к выбранным полипептидам ВИЧ, могут быть идентифицированы и использованы для поддержания рекомбинантных фагмид и получения новых библиотек фагов для последующего этапа скрининга. Библиотека фаг-презентированных антител может быть проанализирована на афинность с помощью перекрестных циклов SDS-PAGE, вестерн-блоттинга и ИФА-скрининга для идентификации антиген-положительных клонов.
Поскольку фаг-презентированные ScFv фрагменты антител сохраняют свою антиген-связывающую способность, можно обогатить фракцию рекомбинантных фагов, экспрессирующих определенные специфические антитела, с помощью аффинной селекции. Используя этот подход, можно быстро отобрать из популяции антитела с определенной специфичностью и афинностью. Полученную фаговую библиотеку генов антител подвергают скринингу с целью усиления антиген-связывающей способности. Этот этап технологии называется панингом и заключается в том, что фаги с презентированными ВИЧ-специфическими ScFv фрагментами антител связываются и собираются:
i) рекомбинантными gp120-, gp140-, gp160-, gp41 пептидами env ВИЧ-1 субтипов А и В;
ii) нативными полипептидами env ВИЧ, выделенными из изолятов ВИЧ-инфицированных.
Поскольку отбирается фаговая библиотека, проявляющая афинность ко всем вышеперечисленным полипептидам, после многих циклов панинга число фаг-презентированных антител уменьшается с 107-1012 до 102-103. В результате независимых циклов взаимодействия вышеупомянутых полипептидов со специфическими рекомбинантными полипептидами ВИЧ, когда последовательности тех полипептидов (i) известны и (ii) неизменны, и с нативными полипептидами ВИЧ, выделенными из изолятов, когда в этих полипептидах могли произойти мутационные изменения, было возможно отобрать антитела, которые связывались практически со всеми известными мутантами ВИЧ, показывая тем самым, что антитела могут распознать константные конформации вышеупомянутых полипептидов ВИЧ.
Действуя таким образом, можно получить библиотеку ВИЧ-специфических рекомбинантных антител, презентированных на поверхности фагов, которые будучи отобранными из огромного пула антител у инфицированных индивидуумов, проявляют афинность к отобранным полипептидам ВИЧ, даже если в этих полипептидах произошли мутационные изменения, двумя различными методами:
i) стандартной процедурой биопанинга [4];
ii) посадкой на нитроцеллюлозную мембрану с иммобилизованными нативными пептидами ВИЧ.
В соответствии с первым методом i) для продукции 4×1010 PFU рекомбинантного фага фаг-помощник М13КO7 добавляют к приготовленной культуре Е.coli штамма TG1, находящейся в логарифмической фазе роста, для преинкубации в течение 1 часа и инкубации в течение 12 часов в присутствии 100 мкг/мл ампициллина и 50 мкг/мл канамицина при 37°С, с качанием при 250 об/мин (типичный выход фага составляет 1010-1011 ампициллин-трансдуцирующих единиц на 1 мл). Рекомендуется использовать полипропиленовые пробирки, поскольку фаг может неспецифически адсорбироваться на поверхности пластика другого вида.
Затем проводят ПЭГ-преципитацию. Бактериальную культуру центрифугируют при 1000g 10 минут, собирают супернатант и охлаждают. 1/5 v/v охлажденного раствора 20%ПЭГ/ 2,5 MNaCl добавляют к супернатанту и инкубируют при 0°С 60 минут, затем центрифугируют при 10000g на роторе Beckman JA-20 20 минут при 4°С. Супернатант отбрасывают. Осадок (может быть плохо различим) ресуспендируют в 16 мл среды 2х YT с 0.01% тимерозала. Мы рекомендуем профильтровать супернатант через 0.45 мкм фильтр, если он будет использоваться для хранения (при 4°С). Раствор, содержащий рекомбинантный фаг, используют для панинга.
ПЭГ-преципитация и циклы панинга должны быть проведены максимально быстро после процедуры сбора фага, поскольку некоторые препараты фаг-презентированных рекомбинантных антител могут быть нестабильными. Колонию клеток TG1 в логарифмической фазе роста переносят с чашки с минимальной средой в 5 мл среды 2х YT и инкубируют в течение ночи при 37°С с качанием при 250 об/мин. Затем 10 мл свежей среды 2х YT инокулируют 100 мкл ночной культуры и инкубируют при 37°С с качанием при 250 об/мин до тех пор, пока А600 культуры не достигнет значения 0.3.
В культуральный флакон 25 см2 сорбируют 5 мл антигена, разведенного до концентрации 10 мкг/мл в соответствующем буфере, например, PBS или 0.05М Nа2СО3 (рН 9.6). Сорбирование антигена может производиться в течение 1-2 часов при комнатной температуре или в течение ночи при 4°С. Условия для сорбирования антигеном чашки, а именно, буфер, температура и время инкубации, зависят от антигена и должны быть сходными с условиями иммунологического анализа, используемыми для исходного поликлонального или моноклонального антитела, из которого был получен новый рекомбинант. Концентрация антигена может варьировать в зависимости от требуемой афинности (антиген-связывающей способности) рекомбинантного фаг-презентированного антитела. Для высокоафинных антител требуется меньшее количество антигена, чем для антител с низкой афинностью. Однако жидкофазная селекция может быть предпочтительнее сорбционной селекции для изоляции антител со специфической афинностью, поскольку количество антигена, использующееся для селекции, можно контролировать более аккуратно.
Флакон три раза промывают PBS, полностью опорожняя его после каждой промывки. Затем флакон полностью заполняют блокирующим буфером, чтобы закрыть оставшуюся незасорбированной антигеном поверхность флакона, и инкубируют при комнатной температуре в течение 1 часа. Флакон снова промывают три раза PBS, полностью опорожняя его после каждой промывки.
Приготавливают свежий блокирующий буфер, содержащий 0.01% тимерозала, либо 0.01% азид натрия в качестве консерванта. 16 мл ПЭГ-преципитированного рекомбинантного фага разводят 14 мл блокирующего буфера (содержащего консервант) и инкубируют 10-15 минут при комнатной температуре. Неспецифические, гидрофобные белок-белковые взаимодействия могут возникать между нативными белками фага М13 и некоторыми антигенами на этапе панинга.
Эти взаимодействия могут быть уменьшены, если к разведенному фаговому супернатанту добавить Triton Х-100 до конечной концентрации 0.1%. Альтернативно, можно провести элюцию специфически связавшегося фага раствором глицина или трипсина. 20 мл разведенного рекомбинантного фага наливают в культуральный флакон и инкубируют 2 часа при 37°С. Затем флакон опорожняют и промывают 20 раз 30-50 мл PBS и 20 раз PBS, содержащим 0.1% Tween 20 (это удобнее делать при помощи пластиковой бутылки-промывателя). Каждый раз флакон опорожняют полностью.
Все 10 мл суспензии клеток TG1 в логарифмической фазе роста (см. шаг 1) добавляют в культуральный флакон или другой плоскодонный сосуд для панинга и инкубируют при 37°С в течение 1 часа. Через 1 час, 100 мкл из 10 мл клеточной суспензии удаляют. Из них готовят десятикратные разведения клеточной суспензии в среде 2х YT (1:10, 1:100, 1:1000). 100 мкл неразведенных клеток и 100 мкл из каждого разведения высевают на отдельные чашки со средой SOBAG, используя стерильную микробиологическую петлю для рассева колоний. Когда чашки подсохнут, их нужно перевернуть и инкубировать в течение ночи при 30°С. Если появляющиеся после инкубации колонии слишком малы, чашки могут быть дополнительно оставлены при 30°С на 4-8 часов. С чашками SOBAG можно обращаться следующим образом: а. Соскоблить клетки с чашки для приготовления стоковых культур. Налить в чашку 5 мл среды 2х YT и соскоблить клетки в среду стерильной микробиологической петлей. Добавить глицерин до конечной концентрации 15-30% и хранить при -70°С. b. Заклеить чашки парафилмом и хранить до 2 недель при 4°С для проведения отбора фага в более позднее время.
В соответствии со вторым методом и), модифицированным из [25], смесь нативных пептидов ВИЧ разгоняют в 10% SDS-PAGE геле, с последующим электропереносом на нитроцеллюлозную мембрану в Вестерн-трансфер буфере (25 мМ Трис, 193 мМ глицин и 20% метанол). Местоположение антигена определяют с помощью окрашивания мембраны либо обратимо Понсо красным, либо Кумасси бриллиантовым голубым. 7*30-мм2 участок мембраны, содержащий полосу белка, вырезают и блокируют инкубацией с 10% свиным желатином, 5×1011 PFU/мл фага- помощника в течение ночи при 4°С. После блокирования мембрану переносят в буфер для связывания (5% желатин, 3*1011 PFU/мл фага-помощника, 0.5 MNaCl) и добавляют 1012 PFU фагмидной библиотеки scFv антител. Фаговую библиотеку инкубируют с мембраной 4 часа при 4°С с легким покачиванием. Мембрану промывают 6 раз PBS, 0.1% Tween 20 (100-мл на каждую промывку) и 6 раз PBS (100- мл на каждую промывку). Альтернативно, пятна промывают три раза по 5 минут PBS, содержащим 0.1% Tween 20 (PBST), пять раз по 20 минут 10% MPBS, содержащим 25% глицерин, и, наконец, три раза по 5 минут PBS. Участок мембраны, содержащий полосу белка, вырезают лезвием и элюируют фаги с помощью 100 мМ TEA при комнатной температуре в течение 10 минут. После нейтрализации элюированные фаговые частицы инкубируют с блокированной желатином мембраной или покрытой желатином иммунотрубкой 30 минут при комнатной температуре. Супернатант затем используют для инфицирования TG1. Фаг для следующего раунда отбора получают из Е.coli, как описано ранее.
3. Размножение вирусного материала изолятов, полученных от пациентов, не подвергавшихся антиретровирусной терапии, РВМС-методом
Известно, что ВИЧ может успешно размножаться в культурах клеток с большим количеством CD4- или CCR5-CXCR4 рецепторов, однако на практике этот метод имеет много ограничений. Во-первых, инфекционный титр для нативного вирусного материала из пациентов или лабораторных штаммов для инфекции in vitro никогда не превышает 1-2 процента от общей концентрации вируса, определенной различными методами (РТ-ПЦР в реальном времени, р24 ИФА, и др.). Это означает, например, что если число копий вируса в инфекционном материале составляет 105, первоначальное число копий, которое исследователь смог бы проанализировать после in vitro размножения, составляет только 103, оставшиеся 102 первоначальных возможных вариантов ВИЧ будут утрачены для анализа. Во-вторых, число вариантов ВИЧ, прошедших через отбор инфекции in vitro, составляет в лучших случаях несколько вариантов последовательности из первоначальных 103 вариантов, поэтому лабораторные штаммы вируса никогда не могут воспроизвести реальную ситуацию с генетической и пептидной вариабельностью ВИЧ. И, в-третьих, ВИЧ из организма пациентов, получавших ВААРТ или другую антиретровирусную терапию, утрачивает способность размножаться in vitro, поэтому резистентные варианты ВИЧ нельзя культивировать in vitro. Наш опыт культивирования нативного вируса доказывает, что лучшие результаты получаются, когда:
i. Лимфоциты из ВИЧ инфицированных пациентов инкубируют с лимфоцитами здоровых доноров, выделенных из гепаринизированной свежей крови, с использованием раствора Фиколла, как описано в [19]. Стоит отметить, что для ВИЧ-1 субтипа А, широко распространенного на территории Российской Федерации, в большинстве случаев инфекция идет успешно, если ВИЧ инфицированные лимфоциты инкубируют с моноцитами, выделенными из свежей крови здорового донора, как описано в [19].
ii. MT-2, или МТ-4, или любую другую клеточную линию, использовавшуюся для инкубации с изолятами ВИЧ (CCR5F-CEM, РМ-1, HeLa, U937, etc.), приготовленную в концентрации 0,25х106 /мл, затем сокультивировали с равным числом зараженных ВИЧ моноцитов, отобранных дважды путем добавления RPMI-1640 среды к клеточному осадку до общего объема 50 мл и центрифугирования при 425g в течение 10 минут. Смесь клеток ресуспендируют в культуральной среде с добавлением IL-2 10 ul/ml и инкубируют при 37°С в вертикальном положении в 25 см2 культуральных флаконах. Содержащую вирус среду отбирают каждые 3-4 суток путем отбора половины культуральной среды и ее замещения равным объемом свежей среды (RPMI+10% FCS).
Эффективность инфекционной активности вируса контролируют анализом под микроскопом гибели клеток при окрашивании 1%-ным Трипановым синим и образования синцитиев, а также с помощью р24 ИФА-теста. Отобранную культуральную среду очищают от клеток центрифугированием при 3000 rpm (1000g) в течение 15 минут и хранят при -80°С.
4. Концентрирование осадков ВИЧ ультрафильтрацией, ультрацентрифугированием, инактивация и разрушение вируса
Стоковый раствор вирусных частиц получают из плазмы крови или культурального супернатанта. Сначала супернатант центрифугируют при 3000 об/мин (1000g) в течение 15 минут, затем полученный супернатант центрифугируют при 13200 об/мин (16000g) в течение следующих 15 минут. Около половины общего объема ультрацентрифужных пробирок заполняют 20%-ным р-ром сахарозы, который наливается на дно (плотность раствора сахарозы 1,16-1,18 г/см3), затем наслаивается супернатант, содержащий ретровирусные частицы. Пробирки центрифугируют при 38000 об/мин MLS-50 rotor Optima MAX, Beckmann (160000g) в течение 1 часа 35 минут [19]. Осадок растворяют в небольшом объеме культуральной среды (например, RPMI 1640).
Инактивация ВИЧ. Лизис вирусных осадков и получение белков ВИЧ. Первый метод осуществляется в соответствии с описанным в [1]. Состав буфера для лизиса ВИЧ (буфер для радиоиммунопреципитации) включает 20 мМ Трис-HCl, рН 8.0, 120 мМ NaCl, 2 мМ ЭДТА, 0.5% дезоксихолат, 0.5% NP-40, 2 мкг PMSF, 10 мкг/мл апопротеин, 10 мкг/мл пепстатин А. После добавления детергентов образцы осторожно перемешиваются на магнитной мешалке с небольшим нагреванием (50°С).
Второй метод является стандартным для приготовления пептидных смесей для масс-спектрометрического и кристаллографического анализов. рН полученной смеси белков ВИЧ-1 доводили до 2.5 с помощью 2 N НСl и инкубировали ее с 0.15% (wt/vol) свиного пепсина (Sigma Chemical Co., St Louis, МО) в течение 4 часов при 37°С. Гидролиз останавливали нагреванием при 80°С в течение 15 минут, затем рН доводили до 7.5-8 добавлением 2М NaOH. Затем смесь гидролизованных белков подвергали ультрафильтрации через 10 kDa гидролитическую мембрану, и пепсин с остальными негидролизованными белками удалялся. Фильтрованную смесь гидролизованных белков лиофилизировали и хранили при -80°С.
5. Сбор нативных пептидов env ВИЧ-1 с помощью фагмидной библиотеки ВИЧ-специфических рекомбинантных фаг-презентированных ScFv фрагментов антител в ходе процедуры обратного панинга
Подходы к использованию технологии фагового дисплея для создания вакцин с использованием библиотеки фаг-презентированных антигенов отчасти представлены в [35]. Перед началом процедуры иммобилизации на колонке рекомбинантных презентированных на М13 фаговых scFv библиотек их проверяют на специфичность методом модифицированного вестерн-блоттинга. Образцы разделяют в SDS-PAGE с последующим электропереносом на нитроцеллюлозу [25]. Мембраны сначала замачиваются в PBS, содержащем 1% Tween 20, в течение 1 часа для ренатурации перенесенных белков. Затем мембраны блокируют 4% раствором желатина в PBS при 37°С в течение 2 часов и инкубируют с 1012 PFU/мл фага (проинкубированного в течение 30 минут при комнатной температуре с 1.5% BSA в 4% растворе желатина) при комнатной температуре в течение 1 часа. Затем мембраны промывают три раза PBS, 0.1% Tween 20 и три раза PBS, связывание фага детектируют инкубацией с разведенными 1:8000 HRP-конъюгированными anti-M13 в 5% р-ре сухого молока/PBS при комнатной температуре в течение 1 часа. После трехкратной промывки PBS/0.1% Tween 20 и трехкратной промывки PBS, полосы визуализируют с помощью ECL детекции (Amersham Biosciences). После тщательной промывки в TPBS-блот буфере мембраны инкубируют 1 мин в ECL. Каждую мембрану затем инкубируют с рентгеновской пленкой Hyperfilm-ECL и проявляют.
Рекомбинантные моноклональные антитела в лучшем случае имеют афинность, составляющую 10-30% от афинности нативных антител, выделенных из организма. Однако полученная с помощью технологии фагового дисплея панель ВИЧ-специфических мАт (фагмидная библиотека), индивидуальных для каждой когорты вариантов вируса (пациентов), является достаточной для отбора большинства вариантов env и других пептидов и белков ВИЧ для создания превентивных вакцин против ВИЧ-1 (Рис. 7а, b, 8a, b).
Для получения фаг-презентированной библиотеки рекомбинантных антител фаг-помощник М13К07 добавляют к ночной культуре Е.coli TG1 для преинкубации в течение 1 часа и затем инкубируют 12 часов в присутствии 100 ug/мл ампициллина и 50 ug/мл канамицина при 37°С (типичный выход фага составляет 1010-1011 ампициллин-трансдуцирующих единиц в мл). Культуру центрифугируют при 1000 g 10 минут, супернатант собирают и охлаждают. Затем 1/5 v/v раствора ПЭГ8000/NаСl (20%ПЭГ/2,5MNaCl) добавляют к супернатанту и инкубируют 1 час на льду, затем проводят осаждение центрифугированием при 10000 g при температуре 4°С в течение 20 минут. Осадок растворяют в LB или 10 мМ Трис-НСl рН 8.0 и пропускают через 0.45 мкм фильтр. Рекомбинантный фаг можно хранить при 4°С, если добавить 0.01% тимерозала.
i) Иммобилизация М13-специфическими мАт на хроматографических колонках, содержащих крупнопористый монолитный эпокси-активированный криогель (Protista Biotechnology)
М13-специфические мАт закрепляют на крупнопористый монолитный эпокси-активированный криогель (Protista Biotechnology) следующим образом. Сухой сорбент ресуспендируют в 0.1 М NaHCO3 буфере, рН 8.3, содержащем 0.5 М NaCl. М13-специфические мАт растворяют в том же буфере до концентрации 10 мг/мл, добавляют к сорбенту и инкубируют 1 час при комнатной температуре с перемешиванием. После инкубации сорбент промывают 5 объемами того же буфера 0.1 М NaHCO3 рН 8.3 / 0.5 М NaCl. Для блокирования неспецифических реакционных групп сорбент инкубируют с 0.1М Трис-HCl буфером, рН 8.0, или с 1 М этаноламином, рН 8.0, в течение 2 часов при комнатной температуре, затем помещают его в 5-мл хроматографические колонки.
В обоих методах сначала частицы фага М13 с фрагментами антител, специфичными к gp120, gp140, gp160 и их фрагментам, gp41, p24, инкубировали при 37°С 40 минут со смесью гидролизованных пептидов ВИЧ-1, полученных, как описано выше (стадия 4). Затем фаговые частицы были сорбированы с помощью иммобилизованных М13 фаг-специфических антител (мАт) одним из двух способов.
Подготовленная колонка, содержащая крупнопористый монолитный эпокси-активированный криогель с иммобилизованными М13-специфическими мАт уравновешивается 0.05 М Трис-НСl буфером, рН 8.0, затем иммобилизуется рекомбинантный М13 в том же буфере в течение 5 часов со скоростью 0.5 мл/мин с использованием жидкостной хроматографической системы низкого давления ActaPrime Plus (GE Healthcare). Затем колонку промывают 5 объемами того же буфера 0.05 М Трис-HCl, рН 8.0.
Изображение сорбированных рекомбинантных фаговых частиц получили, используя метод АФМ (атомной силовой микроскопии). Криогель с успешно прикрепленной фаговой ВИЧ-специфичной библиотекой ScFv представлен на Рис.6b, структура контрольной колонки с крупнопористым монолитным эпокси-активированным криогелем (сухой сорбент) показана на Рис.6а.
Смесь гидролизованных в 0.05 М Трис-НСl буфере, рН 8.0, пептидов ВИЧ пропускают через хроматографическую колонку с иммобилизованными частицами рекомбинантного фага с презентированной ScFv библиотекой 5 часов со скоростью 0.5 мл/мин. Затем колонку промывают 5 объемами того же 0.05 М Трис-НСl буфера, рН 8.0.
Фаг со связанными пептидами ВИЧ элюируют 0.1М градиентом глицина, рН 2.2. Полученные фракции инкубируют в глициновом элюирующем буфере в присутствии 0.001М PMSF в течение 5 часов при комнатной температуре до тех пор, пока комплексы фаг-антиген не оказываются полностью разрушенными.
Пептиды ВИЧ анализируют [2] и чистят с помощью высокоэффективной жидкостной хроматографии (HPLC, Waters). Аналитическую обращенно-фазовую HPLC проводят на приборе Waters 1525 HPLC system с колонкой Symmetry С 18 (5 мкм, 4.6 мм × 150 мм, скорость тока 0.5 мл/мин). Препаративную обращенно-фазовую HPLC проводят на приборе Waters 1525 HPLC с UV-детектором на колонках Symmetry С-18 (10 мкм, 5.0 см × 25 см). Линейные градиенты ацетонитрила в воде/0.1% трифторуксусной кислоте (TFA) используют для элюирования связанных пептидов.
6. Количественный анализ и анализ вариабельности последовательностей и частоты встречаемости пептидов env методом жидкостной хроматографии-масс-спектрометрии (LC-MS);
Нативные пептиды ВИЧ для анализа собирали после концентрирования методом обратного панинга на фаг-презентированных ВИЧ-специфичных библиотеках ScFv. Количественный отбор, распределение по массе и определение характеристик пептидов env проводили с помощью одномерной жидкостной хроматографии-масс-спектрометрии (LC-MS-MS анализа). Для этого делали одномерный ПААГ-электрофорез (SDS-PAGE) смеси белков и пептидов ВИЧ, полученной на стадии обратного панинга.
Триптический гидролиз белков трипсином в полиакриламидном геле
После окраски Кумасси полосы в геле выглядели похожими на [10], белковые полосы интереса вырезали из геля, инкубировали их в деионизованной воде 15 мин при комнатной температуре, затем супернатант отбрасывали. К отмытым полосам геля добавляли 50-100 мкл ацетонитрила и инкубировали 20 мин при комнатной температуре, избыток ацетонитрила высушивали. К дегидратированным кусочкам геля добавляли раствор модифицированного трипсина (Promega) в 0.05 М NH4НСО3 с концентрацией 10 мкг/мл и инкубировали при 4°С во льду в течение часа. Реакцию расщепления проводили в течение ночи при 37°С. Экстракцию триптических пептидов из геля проводили при комнатной температуре добавлением 0.5% трифторуксусной кислоты в воде. Надгелевый раствор использовали для масс-спектрометрического анализа.
Одномерная жидкостная хроматография-масс-спектрометрия (ЖХ-МС-МС анализ)
Триптические пептиды из полос белков, полученные одномерным гель-электрофорезом, подвергались масс-спектрометрическому анализу на приборе Esquire 6000 plus (Bruker Daltonics, Бремен, Германия), оснащенном наноспрейным источником ионов с квадрупольной ионной ловушкой в качестве масс-анализатора. Данный прибор был сопряжен с нанохроматографом Ultimate LC Packings и системой отбора образцов Famos LC Packings. (Dionex, CA, USA) в режиме on-line. Хроматографическая часть установки представляла собой две последовательно соединенные колонки, между которыми располагался электромагнитный клапан.
Колонка №1 (100 мкм × 3см), заполненная полимерной фазой Poros R2 (гидрофобная полимерная фаза с большим диаметром пор, аналог C8), использовалась для предварительного концентрирования образца и его обессоливания. Вторая колонка (75 мкм × 25 см), заполненная сорбентом Phenomenex (C18) (размер зерна 5 мкм, диаметр пор ЗООА), использовалась непосредственно для разделения обессоленной смеси триптических пептидов. Хроматографическое разделение на описанной выше системе производилось со скоростью потока 200 мкл/мин (до сплиттера), при этом фактическая скорость потока при обессоливании составляла в среднем 900 нл/мин, а при разделении - 200 нл/мин. Для разделения пептидов использовался линейный градиент от 5% до 60% раствора В (75% - ацетонитрил, 25% - изопропанол с 0,1%-ной муравьиной кислотой) длительностью 48 минут. Измерения производились в диапазоне m/z 300-2500 с «массой оптимизации ловушки» 700. Тандемные эксперименты проводились только с ионами, зарядовое число которых превышало или было равно «2» с интенсивностью выше пороговой. Полученные масс-листы посылали в поисковую систему MASCOT. Поиск проводили по базе данных NCBI с выборкой по таксону HIV-1. Результаты поисков верифицировали с помощью программного комплекса Scaffold (версия 01-07-00) (http://www.proteomesoftware.com) для подтверждения правильности определения белков. В конечном списке оставлялись белки с вероятностью обнаружения более 95%. При этом количество определенных пептидов на белок составляло 2 и более.
Схемы представляют распределения гидрофобных (ось Y положительные индексы) и гидрофильных (отрицательные индексы) фрагментов полипептидной последовательности в белковой молекуле env.
onf: Confidence (0=low, 9=high)
Pred: Predicted secondary structure (H=helix, E=strand, C=coil) 30 AA: Target sequence
Относительные индексы гидрофобных аминокислот:
Ala: 1.800 Arg:-4.500 Asn:-3.500 Asp:-3.500 Cys: 2.500 Gin:-3.500 35
Glu: -3.500 Gly: -0.400 His: -3.200 He: 4.500 Leu: 3.800 Lys: -3.900

gp120 внутренний домен
GGXMRDNXRSELYKYXVVKIEPLGVXPTKAKRRVVQREKX
7. Клонирование мажорных пептидов оболочки ВИЧ и продукция рекомбинантных пептидов в Leishmania tarentolae для получения компонентов вакцины
Жизненный цикл ВИЧ может осуществляться в человеке, мартышках или грызунах и гликозилирование его белков близко к таковому у млекопитающих. Эукариотические экспрессионные системы включают дрожжевые системы, мицелиальные грибы, а также культуры клеток насекомых, млекопитающих и/или растений. Внешние домены gp120 и gp41 сильно гликозилированы. Если необходимо получить эукариотический белок или его фрагмент похожим на натуральный, его экспрессия должна осуществляться с гликозилированием в дрожжах, клеточных линиях млекопитающих, культурах клеток лейшмании, бакуловирусных экспрессионных системах. Экспрессия в клетках млекопитающих, таких как СНО-К1 (клетки китайского хомяка) или Cos-7 (клетки почечного эпителия африканской зеленой мартышки), возможна, но поскольку в метаболизме клеток млекопитающих участвуют миллионы белков, экспрессия рекомбинантных белков в них довольно низка, и трудно осуществить хроматографическую очистку достаточного количества продуцируемых рекомбинантных белков. Поэтому мы выбрали в качестве экспрессионной системы для продукции белков env Leishmania tarentolae.
После количественного масс-спектрометрического анализа последовательности, кодирующие варианты gp120, представляющие подавляющее большинство в пуле, амплифицируют, секвенируют и клонируют. Как было показано в ряде публикаций, вариации последовательности gp41 не являются критическими для ВИЧ-специфического иммунного ответа (Рис.10d, Пример 4). Поскольку уровень гликозилирования gp41 и связывание его с gp120 имеют гораздо большее значение для выработки ВИЧ специфических антител, чем вариабельность его последовательности [31], мы приняли решение взять один вариант этого гена из когорты пациентов в качестве стандартного компонента для клонирования. На основе полученных белковых последовательностей gp120, соответствующие фрагменты провирусной ДНК, кодирующие gp120 белки оболочки, амплифицируют в ходе двухраундовой гнездовой ПЦР с использованием в качестве матрицы кДНК из лимфоцитов пациентов и специфических пар праймеров (Таблица. 8). Последовательности праймеров и их наборы могут варьировать, в зависимости от результатов LC-MS анализа.
Возможно осуществить клонирование фрагментов ДНК, кодирующих полноразмерные gp120 и gp41, внутренний домен gp120, внешний домен gp120 и эктодомен gp41 (см. структуру gp120 на Рис.10 a,b,c). Схема ПЦР амплификации фрагментов ДНК ВИЧ-1, кодирующих gp120, gp41 и их основные домены, представлена на Рис.11 a, b. Набор праймеров для амплификации env gp120, gp41 и их основных доменов приведен в Таблице 8. Сайты рестрикции, выбранные в соответствии с вариантом клонирующего вектора, обозначены розовым для Ncol, синим - для XbaI, оранжевым - для NotI, зеленым - для Nhel.
Олигонуклеотидные праймеры, использовавшиеся для амплификации поледовательностей gp120, gp41 ВИЧ-1 и фрагментов ДНК, кодирующих их основные домены
120 For праймер использовался в качестве прямого праймера для амплификации внутреннего домена gp120; 41 For праймер использовался в качестве прямого праймера для амплификации эктодомена gp41.
Какие участки в наибольшей степени подходят для клонирования с целью получения лучших результатов иммунизации, в каждом случае является экспериментальным выбором исследователя.
Некоторые свойства являются критическими при выборе экспрессионной системы для продукции рекомбинантных белков для разработки вакцин. Их экспрессия должна быть: i) индуциемой; ii) белки должны быть гликозилированы сходным образом либо должны пройти посттрансляционную модификацию по типу млекопитающих.
i) Индуцируемая экспрессия необходима для получения надлежащего количества и концентрации рекомбинантных пептидов. Как показано на Рис. 12 в индуцируемой системе экспрессия рекомбинантного белка видна на сканах SDS-PA гель-электрофореза (Рис. 12а). В случае когда клетки трансфецируют вектором с неиндуцируемой экспрессией, наличие рекомбинантного белка обычно нужно детектировать с помощью Вестерн-блоттинга, поскольку она не видна на SDS-PAGE (Рис. 12b).
ii) Гликозилирование рекомбинантных белков, производимых для вакцинации, должно соответствовать естественному, типичному для хозяина вируса -эукариотических лимфоцитов - настолько, насколько это возможно. Получить достаточно высокую продукцию любых рекомбинантных белков в культурах эукариотических клеток среди миллионов их собственных белков трудно и очень дорого. Поэтому возможно осуществить продукцию белков оболочки ВИЧ-1 (gp120, gp41 и весь gp160) в штаммах дрожжей, клетках насекомых или в клетках эукариотических паразитов. Нашим выбором является трипаносома Leishmania tarentolae, которая сочетает наличие экспрессии/фолдинга/модификации белков по эукариотическому типу с легкостью манипулирования, и также не патогенна для млекопитающих. Основным преимуществом этой системы экспрессии является посттрансляционная модификация (гликозилирование, фосфорилирование, пренилирование) продуцируемых белков по типу клеток млекопитающих (Рис. 13).
Наиболее удобно клонировать гены белков оболочки ВИЧ-1 в семействе векторов pLEXSY из наборов LEXSYcon2 Expression и LEXSinduce2 Expression Kits, разработанных фирмой Jena Bioscience GmbH. У трипаносом мРНК транскрибируются в виде полицистронных предшественников, которые посттранскрипционно процессируются на индивидуальные мРНК посредством транссплайсинга и полиаденилирования внутри межгенных областей. Регуляция экспрессии белков у этих видов происходит, главным образом, на уровне РНК и на нее может влиять структура межгенных участков. В векторах pLEXSY используются межгенные участки, оптимизированные для экспрессии гетерологичных белков в L. tarentolae (Jena Bioscience GmbH).
Векторы pLEXSY-2 позволяют осуществлять конститутивную экспрессию белков либо с, либо без сигнального секреторного пептида (SP на рис. 14) после интеграции экспрессионной кассеты в локус, кодирующий хромосомную 18S рРНК (ssu). Таким образом, один и тот же вектор может быть использован для клонирования генов либо для цитозольной, либо для секреторной экспрессии. Последовательность, кодирующая сигнальный пептид LmSAP в этих векторах, происходит из гена, кодирующего секретируемую кислую фосфатазу (Imsapl) у Leishmania mexicana. Слияние в одной рамке считывания гена, кодирующего белок ВИЧ-1, с последовательностью этого сигнального пептида позволяет осуществить секреторную экспрессию в LEXSY реципиенте, в то время как клонирование в любой сайт рестрикции на 5' конце последовательности, кодирующей сигнальный пептид, приводит к экспрессии в цитозоль.
Клонирование целевого гена в экспрессионном векторе pLEXSY. Векторы pLEXSY-2 позволяют осуществить направленную инсерцию кассеты с целевым геном посредством замещения ею фрагмента-вкладыша размером 1 тпн. Полученная лигазная смесь используется для трансформации компетентных клеток Е.coli, устойчивых к последовательностям Leishmania (Stb12, Stb14, XL-1, XL-10, SURE и другие). Отбор рекомбинантных клонов Е.coli проводят с помощью ампициллина. После конструирования в Е.coli экспрессионную плазмиду линеаризуют расщеплением рестриктазой Swal, и после этого экспрессионная кассета с целевым геном интегрируется в хромосомный локус ssu 18S rRNA LEXSY реципиента PI 0 с помощью гомологической рекомбинации. Перед сайтом инсерции целевого гена в Е.coli нет сигналов для транскрипции и/или трансляции и, таким образом, отсутствие экспрессии гена в Е.coli является преимуществом для создания конструкций с генами белков, токсичных для Е.coli.
Для конститутивной экспрессии в цитозоль или конститутивной секреторной экспрессии за счет сигнального пептида env ВИЧ-1 гены белков оболочки ВИЧ-1 (gp120, gp41 полноразмерный ген env, кодирующий сигнальный пептид, gp120 и gp41) амплифицируют с помощью праймеров, содержащих сайты рестрикции Ncol (for) и Nhel (rev) (Таблица 8), разрезают рестриктазами Ncol I Nhel и клонируют в векторах pLEXSY-2. В такой конфигурации целевой белок ВИЧ-1 имеет на С-конце последовательность His6. В противном случае, гены белков оболочки ВИЧ-1 амплифицируют с помощью праймеров, содержащих сайты рестрикции Ncol (for) и NotI (rev), разрезают рестриктазами NcolI Nhel и клонируют в векторах pLEXSY-2. В этом случае полученный целевой белок ВИЧ-1 не имеет С-концевой последовательности His6.
Для конститутивной секреторной экспрессии за счет сигнального пептида LmSAP, кодирующегося последовательностью из pLEXSY-2 векторов, гены белков оболочки ВИЧ-1 (gp120, gp41 и весь ген env, но без последовательности, кодирующей сигнальный пептид) амплифицируют с помощью праймеров, содержащих сайты рестрикции XbaI (for) и NheI (rev) (Таблица 7), разрезают рестриктазами XbaI NheI и клонируют в pLEXSY-2 векторах. В такой конфигурации целевой белок ВИЧ-1 имеет на С-конце последовательность His6. В противном случае, гены белков оболочки ВИЧ-1 амплифицируют с помощью праймеров, содержащих сайты рестрикции XbaI (for) и NotI (rev), разрезают рестриктазами ХbаI NotI и клонируют в векторах pLEXSY-2. В этом случае полученный целевой белок ВИЧ-1 не имеет С-концевой последовательности His6.
Сайты рестрикции XbaI, NcoI, NheI и NotI относительно редко встречаются в env генах ВИЧ-1 субтипа А1 из стран бывшего СССР. Карта вектора pLEXSY-2 представлена на Рис. 14. LEXSinduce2 Expression Kit содержит pLEXSY_I-neo2 (кодирующий аминоглюкозид фосфотрансферазу) и пригоден для тетрациклин-индуцируемой экспрессии в LEXSY реципиенте T7-TR, осуществляемой полимеразой бактериофага Т7.
Экспрессия рекомбинантных белков. Векторы pLEXSY_I-2 позволяют осуществить индуцируемую экспрессию нужных белков либо с, либо без секреторного сигнального пептида. Таким образом, один и тот же вектор может быть использован для клонирования генов либо для индуцируемой цитозольной, либо для индуцируемой секреторной экспрессии. Последовательность, кодирующая сигнальный пептид LmSAP в этих векторах, происходит из гена, кодирующего секретируемую кислую фосфатазу (Imsapl) у Leishmania mexicana. Слияние в одной рамке считывания гена, кодирующего белок ВИЧ-1, с последовательностью этого сигнального пептида, позволяет осуществить индуцируемую секреторную экспрессию в LEXSY реципиенте, в то время как клонирование в любой сайт рестриции на 5' конце последовательности, кодирующей сигнальный пептид, приводит к индуцируемой экспрессии в цитозоль (Рис.5). Семейство векторов pLEXSYJ-2 обеспечивает индуцируемую экспрессию целевых белков после интеграции экспрессионной кассеты в хромосомный локус орнитиндекарбоксилазы (ode) T7-TR реципиентного штамма Leishmania tarentolae, который конститутивно экспрессирует РНК полимеразу бактериофага Т7 и ТЕТ репрессора под контролем РНК полимеразы I Leishmania. На первом этапе клонирования целевой ген снабжается линкерными последовательностями, содержащими сайты рестрикции, позволяющие осуществить инсерцию гена в pLEXSY_I-2 векторы за областью Т7 промотора/ТЕТ оператора. Эти векторы содержат оптимизированные не транслируемые области, фланкирующие сайты инсерции целевого гена, которые обеспечивают сигналы сплайсинга для посттранскрипционного процессинга мРНК. После конструирования в Е.coli экспрессионная плазмида линеаризуется и интегрируется в ode локус LEXSY реципиента T7-TR посредством гомологической рекомбинации.
Для тетрациклин-индуцируемой экспрессии в цитозоль, либо для тетрациклин-индуцируемой секреторной экспрессии, обеспечиваемой сигнальным пептидом белка env ВИЧ-1, гены белков оболочки ВИЧ-1 (gp120, gp41 и полноразмерный ген env, кодирующий сигнальный пептид, gp120 и gp41) амплифицируют с помощью праймеров, содержащих сайты рестрикции NcoI (for) и NheI (rev), разрезают эндонуклеазами рестрикции NcoI Nhel и клонируют в векторах pLEXSY-2. В такой конфигурации экспрессируемый белок ВИЧ-1 оказывается слитым с С-концевой последовательностью His6. Альтернативно, гены оболочки ВИЧ-1 амплифицируют с помощью праймеров, содержащих сайты рестрикции NcoI (for) и NotI (rev), разрезают эндонуклеазами рестрикции Ncol I NotI и клонируют в векторах pLEXSY-2. В этом случае экспрессируемый белок ВИЧ-1 не имеет С-концевой последовательности His6.
Для тетрациклин-индуцируемой секреторной экспрессии, обеспечиваемой кодируемым вектором сигнальным пептидом LmSAP, гены белков оболочки ВИЧ-1 (gp120, gp41 и весь ген env gene, но без сигнального пептида) амплифицируют с помощью праймеров, содержащих сайты рестрикции XbaI (for) и NheI (rev), разрезают эндонуклеазами рестрикции XbaI NheI и клонируют в векторах pLEXSY-2. В такой конфигурации экспрессируемый белок ВИЧ-1 оказывается слитым с С-концевой последовательностью His6. Альтернативно, гены оболочки ВИЧ-1 амплифицируют с помощью праймеров, содержащих сайты рестрикции XbaI (for) и NotI (rev), разрезают эндонуклеазами рестрикции XbaI NotI и клонируют в векторах pLEXSY-2. В этом случае экспрессируемый белок ВИЧ-1 не имеет С-концевой последовательности His6.
Полноразмерный ген env ВИЧ-1, клонированный в векторах pLEXSY семейства по NcoI/NheI или NcoI/NotI сайтам, либо ген env ВИЧ-1 без последовательности собственного сигнального пептида, вместо этого слитый с последовательностью, кодирующей сигнальный пептид LmSAP из pLEXSY векторов (клонирование в pLEXSY векторах по XbaI/NheI или XbaI/NotI сайтам), может быть использован для создания плазмидных конструкций, позволяющих осуществлять быструю I замену одного клонированного варианта последовательности gp120 на другие варианты последовательности gp120, полученные из других штаммов ВИЧ-1. Для этой цели с помощью сайт-специфического мутагенеза в последовательность гена env между концом gp120 и началом gp41 вводится дополнительный сайт XbaI. После этого плазмидная конструкция pLEXSY:: HI V-l env разрезается рестриктазами NcoI/XbaI (в том случае, когда ген env был клонирован по NcoI/NheI или NcoI/NotI сайтам), либо только рестриктазой XbaI (в том случае, когда ген env ВИЧ-1 без последовательности собственного сигнального пептида был клонирован по XbaI/NheI или XbaI/NotI сайтам) и последовательность gp120 удаляется. Оставшаяся часть плазмиды пригодна для клонирования последовательностей 1 gp120, полученных из других вариантов ВИЧ-1 с помощью ПЦР амплификации с праймерами, содержащими NcoI (for) или XbaI (for) и XbaI (rev) сайты.
Культивирование LEXSY-2 реципиента и экспрессионных штаммов. Leishmania растет в аэробных условиях в двух формах: промастиготной жгутиковой; форме (дикий тип в насекомых) и амастиготной форме в позвоночных. In vitro обе формы в T7-TR LEXSY-2 host можно культивировать в темноте при 26°С в комплексной среде (LEXSY BHI, или LEXSY YS) или синтетической среде (Synthetic LEXSY medium). Жидкую среду янтарного цвета готовят из порошка LEXSY BHI 37 г/л, автоклавируют и хранят до 6 месяцев. Непосредственно перед использованием в среду добавляют гемин до 5 мкг/мл, NTC (норзотрицин) 100 мкг/мл, а также пенициллин до 100 мкг/мл и стрептомицин до 50 мкг/мл для предотвращения бактериальных проростов. Среду можно хранить при 4°С в темноте и использовать в течение 2 недель после добавления вышеуказанных веществ. Нет необходимости добавлять сыворотку в комплексную среду, фетальная телячья сыворотка не вызывает усиления роста L.tarentolae. В случае ингибирования роста реципиента или LEXSY штаммов клетки следует отцентрифугировать 5 минут при 2000g, осторожно ресуспендировать в свежей среде и продолжить инкубирование. Штамм может поддерживаться в длительной суспензионной культуре с регулярными разведениями от 1:10 до 1:50 раз. Наилучшие результаты получаются при инокулировании во время среднепоздней фазы роста (OD 2-3; 8×107-1.4×108 клеток/мл). Для поддержания штамма удобно разводить 10 мл культуры 1:20 по понедельникам и пятницам и инкубировать культуральный флакон в вертикальном положении. Жизнеспособные клетки видны под микроскопом в виде подвижных промастигот с движущимися жгутиками; мертвые клетки имеют круглую или разорванную форму, они не движутся.
Для экспрессии рекомбинантных белков культивирование может быть осуществлено в вентилируемых культуральных флаконах для суспензионных культур, объем культуры от 10 до 200 мл или во флаконах Эрленмейера, в термостате с перемешиванием при 140 об/мин, объем культуры от 50 мл до 1 литра, в стандартных биореакторах, объем культуры до 100 литров. Отбор рекомбинантов при клонировании в векторе pLEXSY-neo2 проходит в присутствии 50 мкг/мл неомицина.
LEXSY реципиент и LEXSY экспрессионные штаммы можно хранить при -80°С в 20% глицерине в течение по меньшей мере года. 1/4 объема проавтоклавированного глицерина (80%) и % объема культуры, выращенной в LEXSY BHI* среде, на средней фазе роста 4-8×10 клеток/мл (OD 1.2-1.8) помещают в 15 мл пробирку Falcon, перемешивают и переносят в стерильные криопробирки. Пробирки держат 10 минут при комнатной температуре, затем 1 час на льду (wet ice), затем некоторое время при -20°С и помещают их на -80°С для длительного хранения. Для расконсервации глицериновые стоки в криопробирках размораживают на льду, их содержимое переливают в 10 мл готовой BHI среды и инкубируют в вертикальных вентилируемых флаконах при 26°С в неподвижном положении 2 суток до тех пор, пока культура не станет мутной.
Приготовление экспрессионной плазмиды для трансфекции LEXSY реципиента
1-5 мкг экспрессионной плазмиды, содержащей целевой ген, полученной из Е.соli, полностью разрезают рестриктазой Swal. Полученный 2.9 тпн фрагмент, часть которого представляет собой участок для клонирования в Е.coli, и больший фрагмент, представляющий собой линеаризованную экспрессионную кассету с целевым геном, который должен быть интегрирован в хромосомный локус ssu LEXSY реципиента, разделяют в агарозном геле. Больший фрагмент ДНК (экспрессионную кассету) выделяют из геля с помощью Agarose Gel Extraction Kit. Ферменты и соли из буфера могут быть удалены из образца с помощью PCR Purification Kit. Альтернативно, осаждают больший фрагмент этанолом, промывают 70% этанолом и растворяют максимум в 50 мкл стерильного бидистиллята или 10 мМ Трис, рН 8 на одну трансфекцию.
Трансфекция LEXSY реципиента с помощью электропорации
Для эффективной трансфекции L. tarentolae предкультуру инокулируют 1:20 в 10 мл среды LEXSY BHI и инкубируют в культуральном флаконе в вертикальном положении при 26°С, через 2 суток предкультуру разводят 1:10 в 10 мл среды и инкубируют в течение ночи в тех же условиях. Выращенная культура должна содержать 6×107 клеток/мл (OD 1.4 при длине волны 550-600 нм, 3% формалин); подтверждают микроскопированием, что клетки жизнеспособны и имеют каплеобразную форму. Клетки центрифугируют 5 минут при 2000g при комнатной температуре и удаляют 1/2 объема супернатанта. Осадок ресуспендируют в оставшейся среде (108 клеток/мл) и помещают на лед на 10 минут. 0.1-5 мкг трансформирующей ДНК в максимально 50 мкл воды или Трис-буфера держат на льду в параллельных пробирках. 350 мкл предварительно охлажденных клеток вносят в пробирку с ДНК и переносят в кювету для электропорации d=2 мм на лед, избегая образования везикул воздуха. Параметры электропорации 450 В, 450 мкФ, время импульса 5-6 мс. После электропорации кювету снова помещают в лед точно на 10 минут. После этого подвергшиеся электропорации клетки переносят в 10 мл LEXSY BHI и инкубируют ночь при 26°С (20 часов, OD 0.3-0.4).
Отбор трансгенных штаммов LEXSY
Для отбора трансформированных штаммов можно использовать один из двух методов, описанных ниже. Сходные уровни экспрессии обычно наблюдаются при сопоставлении культур, полученных в результате клонального или не клонального отбора, после трансфекции линеаризованными экспрессионными кассетами, предназначенными для интеграции в хромосому. Однако при трансфекции кольцевыми экспрессионными плазмидами необходим клональный отбор, поскольку эписомы имеют склонность амплифицироваться и, со временем, интегрироваться в геном гетерогенным способом. Неклональный отбор в суспензионных культурах после трансфекции кольцевой ДНК обычно приводит к пониженным уровням экспрессии.
Клональный отбор посредством посева на твердую среду
Клетки LEXSY host отбирают на свежеприготовленных чашках с агаризованной средой. 1-4 порции по 2 мл из трансфецированной 10 мл о/n культуры отбирают, оставшуюся культуру можно использовать параллельно для неклонального отбора. Клетки центрифугируют 5 минут при 2000g и 20°С, осадок ресуспендируют в 50-100 мкл оставшейся среды, ресуспендированные клетки распределяют по поверхности свежеприготовленного LEXSY BHI агара с 50 мкг/мл неомицина методом посева штрихом на нитроцеллюлозные фильтры, помещенные на поверхность агара. Посев на эти мембраны легче осуществить, чем посев непосредственно на 1% агар, и уменьшается кучность клеток. Кроме того, посев на мембраны позволяет использовать дополнительные фильтры для тестирования профилей экспрессии клональных популяций, например, с помощью флуоресцентного сканирования или специфических методов детекции для данного целевого белка. Чашки заклеивают парафильмом и инкубируют в перевернутом состоянии при 26°С.
Через 5-7 суток после посева начинают появляться маленькие колонии, на 7-9 сутки после посева, когда колонии вырастают до 1-2 мм в диаметре, их можно переносить в 0.2 мл селективной среды роста в 96-луночном планшете, с помощью наконечника для автоматической пипетки, через 1 сутки инкубации - в 1 мл селективной среды в 24-луночном планшете. После следующих 24-48 часов инкубации при 26°С культуры переносят в 10 мл селективной среды в культуральные флаконы, и их можно использовать для анализа и криоконсервации.
Отбор в суспензионной культуре
Как только 10 мл ночной культуры, полученной после трансфекции (см. 4.4.), начинают становиться немного мутными (OD 0.4, 10 клеток/мл; обычно приблизительно через 20 часов после электропорации), в них добавляют неомицин до 50 мкг/мл и продолжают инкубацию в течение 7 суток при 26°С. Рекомбинантные клетки выглядят подвижными под микроскопом, имеют каплеобразную форму и растут в виде «облакообразной» суспензионной культуры, в то время как клетки в отрицательном контроле начинают гибнуть в течение периода отбора и под микроскопом выглядят сферическими, либо имеют неправильную форму без жгутика. Обычно один последовательный перенос в свежую среду с неомицином при разведении 1:10 при инокуляции на 7 сутки отбора достаточен для получения мутной культуры устойчивой к антибиотику рекомбинантной клеточной линии.
Подтверждение геномной интеграции и экспрессии рекомбинантных белков env
Интеграция экспрессионной кассеты в локус SSU может быть подтверждена с помощью диагностической ПЦР, либо секвенирования, при использовании в качестве матрицы геномной ДНК трансгенных штаммов. Для векторов pLEXSY 1-2 диагностическую ПЦР проводят (температура отжига 55°С) с использованием прямого праймера из кассеты устойчивости к антибиотику и ode обратного праймера Р1510 (Таблица 9). Интеграция экспрессионной кассеты в ode локус приведет к появлению характерного фрагмента (1.9 или 2.0 тпн, соответственно), который отсутствует в контрольных реакциях. Кроме того, можно проводить диагностическую ПЦР (температура отжига 60°С) с ode прямым праймером А1304 и aprt обратным праймером А1715 (гибридизуется с областью внутри 5'utr целевого гена). Интеграция экспрессионной кассеты в ode локус приведет к появлению характерного фрагмента длиной 1.1 тпн, который отсутствует в контрольных реакциях, когда матрицей служит экспрессионная плазмида или геномная ДНК из LEXSY реципиента.
Экспрессию целевого белка в рекомбинантных LEXSY штаммах оценивают с
помощью SDS-PAGE и Вестерн-блоттинга клеточных экстрактов или, в случае секреторной экспрессии, аликвот из супернатантов. Для получения оптимальной экспрессии дополнительно можно калибровать среднюю индукцию экспрессии 1 мкг/мл тетрациклина при различных концентрациях тетрациклина, условиях культивирования и времени сбора для каждого индивидуального белка.
Последовательности праймеров из LEXSinduce2 kits (Jena Biosciences)
8. Продукция рекомбинантных белков оболочки ВИЧ и хроматографическая очистка
Для продукции белка рекомбинантный штамм выращивают в Complex LEXSY broth BHI (Jena Bioscience) до OD600 1-4 (108 клеток/мл). Продукцию белка индуцируют добавлением тетрациклина до 5 мг/л через 1 час после переноса клеток в свежую среду и инкубируют культуры при 26°С с перемешиванием при 140 об/мин в МultitronII термостате-шейкере (Infors AG, Switzerland) в течение 24-72 часов до тех пор, пока OD не достигнет значения 1.8. Присутствие рекомбинантного gp120 в культуральных супернатантах и в клетках определяли с помощью гель-электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия (SDS-PAGE) и с помощью Вестерн-блоттинга. Для подтверждения наличия N- гликозилирования культуральный супернатант или клетки обрабатываются N-гликозидазой и анализируют электрофоретическую подвижность обработанного гликозидазой белка.
Экспрессирующие белок клетки Leishmania центрифугируют 10 минут при 2500g, и ресуспендируют осадок в 20 мМ Трис, рН 8.0, 150 мМ NaCl, 5 мМ ЭДТА, 1 мМ PMSF. Лизис клеток осуществляют в ультразвуковом соникаторе при 20 кГц и длине импульса 19 мм, используя 10 импульсов продолжительностью в 1 минуту во льду, с 2 минутными интервалами между импульсами. Осветленный супернатант собирают, пропускают через 0.45 мкм фильтр и используют для аффинной очистки рекомбинантного gp120 на хроматографической колонке, содержащей иммобилизованные на агарозе положительно заряженные ионы никеля (GE Healthcare), используя нитрилтрехуксусную кислоту (Ni-NTA). Вкратце, Ni-NTA колонку промывают тремя объемами буфера 20 мМ Tris, рН 8.0, 150 мМ NaCl, 5 мМ ЭДТА, 1 мМ PMSF при скорости тока 1 мл/мин (LC Akta Prime Plus, GE Healthcare). Затем через колонку пропускают профильтрованный супернатант, содержащий рекомбинантный gp120 (r-gp120), используя скорость тока 0.25 мл/мин. После нанесения супернатанта колонку промывают тремя объемами промывочного буфера (20 мМ Tris-HCl, 500 мМ NaCl, 5 мМ имидазол, рН 7.4). r-gp120 расщепляют на колонке энтерокиназой, чтобы удалить полигистидиновый хвост. Для этого одну международную единицу (IU) Ek наносят на колонку в буфере, содержащем 10 мМ Трис-HCl, 10 мМ СаС1з, рН 8.0, и проводят реакцию расщепления в течение 18 часов при 25 С. Иначе, целевой белок с полигистидиновой последовательностью на конце элюируют в градиенте имидазола (0-0,5М имидазол в 100 мМ Tris-HCl, рН 8.0, содержащий 150 мМ NaCl). Белоксодержащие фракции объединяют и концентрируют ультрафильтрацией. Фракции анализируют с помощью SDS-PAGE/окрашивания серебром и Вестерн-блоттинга с антителами к человеческому (ВИЧ) gp120. Фракции, содержащие R-gp120, собирают, подвергают диализу против 0.1 М Трис-ЭДТА буфера (рН 8.0) и наносят на анион-обменную колонку (Q-PEEK 10um AXC Biosuite, Waters), уравновешенную тем же буфером стандартным способом [2]. Белок элюируют градиентом 0-1М NaCl. В конечном итоге фракцию, содержащую R-gp120, дочищают гель-фильтрацией с использованием Sephacryl S-200HR (GE-Healthcare).
N-концевое секвенирование очищенного rhEPO проводят на автоматическом секвенаторе по Эдману. Концентрацию очищенного белка определяют с помощью ВСА-анализа. Анализ фракций, полученных на различных этапах экспрессии и очистки белка, проводят с помощью SDS-PAGE. Белковые полосы визуализируют с помощью окрашивания кумасси Бриллиантовым голубым R-250 или серебром.
9. Приготовление композиции профилактической вакцины против ВИЧ с использованием стерически стабилизированных липосом (ССЛ) в качестве адъювантов для доставки вакцины
Любой фармацевтический агент для перорального, либо подкожного, внутримышечного, внутривенного введения с целью обеспечения специфического иммунного ответа против некоторой бактериальной или вирусной инфекции, при возможном контакте с этой инфекцией в будущем, обычно называемый "вакциной", должен удовлетворять ряду требований. Основными из этих требований являются:
iii) иммунный ответ должен быть высокоспецифичным по отношению к определенному патогенному микроорганизму или инфекции;
iv) иммунный ответ должен быть достаточно сильным для того, чтобы бороться с развитием данной частной инфекции, блокируя появление симптомов заболевания;
v) иммунный ответ должен наблюдаться в течение продолжительного периода времени, месяцев и лет;
vi) несмотря на все вышеперечисленные условия, вакцина не должна быть реактогенной (иммунотоксичной) для организма человека.
Эффективность профилактической вакцины против ВИЧ, в соответствии с настоящим изобретением, усиливается за счет ее связывания с иммуностимуляторным или иммуногенным носителем, адъювантом. Гликозилированные рекомбинантные варианты gp120 так же, как и смеси нативных белков env ВИЧ, высоко иммуногенны, довольно плохо переносятся при подкожном введении и сами по себе дают сильную иммунную реакцию (Пример 4). Однако, как и в случае всех чистых белков, происходит их быстрая биодеградация в организме, и иммунный ответ затухает в течение одной-двух недель в том случае, если пептиды не связаны с каким-либо предохраняющим или ингибирующим протеазы адъювантом. Нашей первой идеей было защитить пептиды env от деградации, поместив их внутрь стерически стабилизированных липосом (SSL, ССЛ), невидимых для ретикулоэндотелиальной системы. Но уже после самых первых экспериментов на мышах выяснилось, что SSL могут сохранять пептиды внутри себя или, связанными в течение достаточно долгого периода времени и уменьшать их иммунотоксичность. Для доставки пептидов стерически стабилизированные липосомы сочетают в себе преимущества низкой общей иммунотоксичности и лучшей фармакодинамики (своевременного высвобождения препарата), как это было показано для заключенных внутрь липосом лекарственных форм антрациклинов. Заключенные в липосомы пептиды могут медленно высвобождаться из ССЛ, в точности как цитостатики или другие низкомолекулярные агенты. Ингибирование немедленной иммунной реакции дает возможность увеличить дозу пептидов для однократного введения и пролонгировать контакт вирусных пептидов env с МНС для достаточно долговременного иммунного ответа. Таким образом, ССЛ используются одновременно и как иммуностимулирующий адъювант, и как система доставки вакцины.
Приготовление эффективной композиции вакцины может быть сделано подходящим образом с использованием адъюванта, способного к самобиодеградации, безопасного и эффективного при медицинском применении. Два подхода описываются ниже:
i) смесь пептидов оболочки ВИЧ помещается внутрь стерически стабилизированных липосом (ССЛ);
ii) пептиды оболочки ВИЧ ковалентно связываются с активированными
группами ПЭГ ССЛ;
iii) пептиды оболочки ВИЧ презентируются на виросомах возможных конструкций (pNL3-4, IRIV и другие).
i) Приготовление стерически стабилизированных липосом и загрузка в них пептидов
Стерически стабилизированные липосомы получают методом роторного вакуумного выпаривания хлороформа из смеси, состоящей из:
фосфолипид:холестерол в соотношении 7:3 и 0.2-0.5 Мо1/% полиэтиленгликоль-дистеароил (фосфатидил)этаноламин (ПЭГ-DSPE) при образовании везикул в потоке азота [40]. Используются смеси липидов: DОРС/Сhоl/DSРЕ-ПЭГ350, DОРС/Сhоl/DSРЕ-ПЭГ400 и другие (Avanti Polar Lipids, Birmingham, AL). Основным компонентом липосом является диолеоил-фосфатидилхолин (DOPC), который можно получить из природных источников, таких как яичный желток, ткани мозга или соевые бобы, либо можно получить синтетически. Холестерол необходим для стабилизации фосфолипидных бислоев в мембранах липосом, РЕ-ПЭГ обеспечивает стабилизацию и прочность мембран, он предотвращает слияние и деградацию липосом в суспензии и дает им возможность сохранять распределение по размерам и предохранять заключенный в них агент от просачивания наружу в течение месяцев. Идеальный молекулярный вес ПЭГ в ССЛ составляет 400-700, более длинные цепи ПЭГ 1000-2000 не стоит использовать для получения ССЛ, потому что прочность липосомных мембран становится больше требуемой для доставки их содержимого, и ССЛ с длинными цепями ПЭГ не удовлетворяют требованию самостоятельной биодеградации. Изменение процента DSPE-ПЭГ является основным способом тонкой регуляции для получения липосом с требуемыми характеристиками.
Навески лиофолизированных липидов смешивают в органическом растворителе - хлороформе или этанол-хлороформе, который затем выпаривают в роторном испарителе (Buchi R-200), в результате образуется тонкая липидная пленка. Суспензия липосом приготавливается во время дальнейшей гидратации в водном буфере с растворенным в нем агентом (например, 50 мМ NaH2PO4, 400 мМ NaCl, pH 8.0), перемешивания при 300-400 об/мин и температуре +45°С в течение 30 минут. Образуется смесь больших мультиламеллярных (MLV, 300nm -1µm) и маленьких моноламеллярных (SUV, 80-250nm) липосом. Для доставки любых растворимых в воде агентов, таких как пептиды, необходимы маленькие моноламеллярные липосомы, поэтому проводят обработку ультразвуком в водяной бане (600 т V, Avanti Polar Lipids), и несколько циклов фильтрования через поликарбонатную 0.4- 0.2-0.1 мкм мембрану (Avanti Polar Lipids). Проводимое дополнительно в стерильных условиях (в ламинаре через стерильные Гамильтоновские шприцы и поликарбонатные мембраны) фильтрование через 0.2-0.1 мкм мембраны дает препарат, пригодный для иммунизации. Распределение липосом по размеру и их стабильность в водной суспензии определяют методом динамического корреляционного лазерного светорассеяния на субмикронном анализаторе размеров частиц DLS Nicomp-380 (Рис.17).
Смесь рекомбинантных пептидов для иммунизации вводят в состав липосом на стадии гидратации липидной пленки, - смесь пептидов растворяется в фосфатном буфере и загружается в виде внутренней водной фазы в липосомные везикулы [40]. После экструзии через мембраны ССЛ очищают методом сортирующей по размерам гель-фильтрационной хроматографии с использованием Sephacryl S-200HR и Akta Prime LC системы (GE Healthcare). Белки и пептиды, не попавшие внутрь везикул, отделяются и остаются на колонке. Затем при необходимости суспензия ССЛ может быть сконцентрирована с помощью диализа, и после этого она представляет собой вакцинный состав для проведения иммунизации.
Подкожное введение вакцинного состава ССЛ может ингибировать эффект иммунизации из-за замедления процесса высвобождения рекомбинантных пептидов env из липосом, которые сами по себе нейтральны по отношению к МНС. Этот процесс можно регулировать с помощью термочувствительных липосом - tCCЛ. tCCЛ отличаются от других особым количественным соотношением мембранных компонентов или наличием некоторых дополнительных фосфолипидных компонентов, за счет которых липосомная мембрана расплавляется при достижении определенной температуры, обычно 40-45°С. В момент локального нагревания термочувствительные липосомы разрушаются и их содержимое - пептиды -высвобождается в ткань. Например, обычные стерически стабилизированные липосомы имеют температуру плавления около 54-58°С и сухой вес смеси для формирования липидной пленки состоит из
фосфатидилхолин:холестерол:дистеароил-фосфатидилэтаноламин-ПЭГ в соотношении: РС:Сhol:DSРЕ-ПЭГ-400 6.85: 2.75: 0.4 (до 0.5) Моl/% и для более длинных цепей ПЭГ - РС:Сhоl:DSРЕ-ПЭГ-2000 6.9: 2.95: 0.15 (до 0.25) Mol/%. Чтобы приготовить термочувствительные липосомы, можно изменить некоторые параметры, во-первых, соотношение липидов в смеси: увеличить количество холестерола с 27-29 до 30-35 Mol/%, уменьшить процент РЕ-ПЭГ с 2-5 Mol/% до 1.5-2 Mol/%, соответственно. Другой метод сделать мембраны липосом более пластичными и сдвинуть точку их плавления к более низким значениям температуры заключается в использовании более коротких хвостов жирных кислот в фосфолипидах: димиристоил-фосфатидилхолин (DMPC, С-14), дистеароил-фосфатидилхолин (DSPC, С-16), или, предпочтительнее, 30-40 Mol/% DMPC, либо DSPC вместо эквивалентной части DOPC.
ii) Связывание пептидов env с активированными группами ПЭГ ССЛ
Второй тип липосомного адъюванта для смеси рекомбинантных пептидов env представляет собой более длинные версии DSРЕ-ПЭГ2000, активированные для связывания пептидов: PDP-ПЭГ2000-DSPE/Chol/DOPC, малеимид(фенилбутират)-ПЭГ2000-DSРЕ/СhоlDOPC, р-нитрофенил(карбонил)-ПЭГ2000-DSРЕ/Сhol/DОРС. Концентрация ПЭГ-2000 в этих липидных смесях не должна превышать 1.5-2 Mol/%, потому что более длинные цепи полиэтиленгликоля эффективнее усиливают стабилизацию липосом, чем короткие версии ПЭГ, и те же самые концентрации ПЭГ могут сделать мембраны липосом слишком прочными для высвобождения активных пептидных компонентов и безвредной биодеградации липидов.
Первым способом соединения пептидов с активированным дистальным концом ПЭГ [38] является реакция р-нитрофенил(карбонил)-ПЭГ-2000-В8РЕ с аминогруппами пептидов в липосомной суспензии, в соотношении 1 мг пептидов на 25-40 мг липидов в 0.1М цитратном буфере с рН 4.0-5.0 (общий объем суспензии составляет 5.5-9 мл). Реакция прекращается при увеличении рН до 7.5-8.5 при добавлении NaOH, какой-либо специальной обработки пептидов не требуется.
Метод с малеимид-ПЭГ-2000-DSPE [39] требует предварительного тиолирования пептидов реагентом Траутт (2-иминотиоланом). 1 мг смеси пептидов растворяют в боратном буфере, содержащем Nа3ВО3 и ЭДТА, затем добавляют 50-70 мкг сухого реагента Траутт, смесь инкубируют 1 час при комнатной температуре, затем удаляют избыточный белок ультрафильтрацией с одновременной сменой буфера на PBS рН 8.0. Гомогенные по размеру фракции липосом, загруженные пептидной смесью, отделяют с помощью жидкостной хроматографии на Sepharose CL-6B (GE Healthcare, Sweden).
Соединение ODN-HIV env пептидов с PDP-PEG-РЕ-содержащими липосомами (метод рН градиента)
Для приготовления PDP-пептидных производных пептиды растворяют в 25 мМ HEPES, 140 мМ NaCl, рН 7.4 до концентрации 10мг/мл, затем к раствору пептидов медленно добавляют 25 мМ раствор сукцинимидил-4-МРВ (SMPB) в DMF до достижения молярного соотношения 20:1 (SMPB: пептиды) и инкубируют 30 минут при комнатной температуре. Несвязавшийся SMPB удаляют при понижении рН гель-фильтрацией, используя Sephacryl S-200HR колонку (GE-Healthcare), в 25 мМ HEPES, 25 мМ MES, 140 мМ NaCl рН 6.7 буфере. Пиридилдитио-группы на дистальных концах цепей PEG восстанавливают добавлением дитиотрейтола (DTT) до конечной концентрации 20 мМ и инкубируют 30 минут при комнатной температуре. DTT отделяется при возрастании рН пропусканием липосом через Sephadex G-25 колонку и их снятием буферной смесью 25 мМ HEPES, 25 мМ MES, 140 мМ NaCl, рН 6.7. Тиолированные липосомы инкубируют ночь при комнатной температуре с МРВ-пептидными производными при соотношении молекул пептид/липид приблизительно 1:1000. Несвязавшиеся пептиды удаляют гель-фильтрацией, пропуская липосомы через Sephacryl S-200HR колонку с 25 мМ HEPES, 140 мМ NaCl рН 7.4.
Соединение ODN-HIV env пептидов с COOH-PEG-PE-липосомами. К 300 µл суспензии HO2C-PEG-PE-содержащей липосомы в MES-буфере рН 4-5.5 (всего 3 µМоль липидов) добавляют 120 µл 0.25 М раствора 1-этил-3-(3-диметиламинопропил)карбодиимида и 120 мкл 0.25 М N-гидроксисульфосукцинимида в воде. Смесь инкубируют 10 минут при комнатной температуре и нейтрализуют до рН 7.5 с помощью 1М NaOH. 15 µM env пептидов ВИЧ добавляют к активированным липосомам, и инкубируют реакционную смесь 8 часов при 4°С, аккуратно перемешивая. Связавшие пептиды липосомы отделяют от несвязавшихся пептидов на колонке Sephacryl S-200HR (GE Healthcare), предварительно уравновешенной PBS. Основные фракции связанных с липосомами петидов/белков собирают, объединяют и при необходимости разводят буфером до требуемого объема.
Также возможно связать пептиды оболочки ВИЧ с никель-модифицированными фосфолипидами, их можно встраивать в мембрану между PC и хвостами PEG-DSPE в липосомной композиции DOPC/DOGS-NTA-Ni/Chol/DSPE-PEG-2000. Однако несмотря на то что DOGS-NTA стимулируют мукозный и остальные виды В-лимфоцитарного иммунного ответа, положение пептидов env, спрятанных под PEG, ослабляет их специфичность против ВИЧ. Этот метод описывается ниже.
В реакции связывания рекомбинантный (Нis)6-пептид (10-80 мкг) инкубируют с липосомами (1 μМ) в общем объеме фосфатного буфера 50 µл (50 мМ NаН2РO4, 400 мМ Nad, pH 8) при 37°С или при комнатной температуре 30 минут с круговым перемешиванием [17]. Связывание пептидов с липосомами определяют количественно косвенным путем, измеряя количество свободного белка в конце реакции связывания. Несвязавшийся белок отделяют с помощью центрифугирования на Microcon-100. Перед центрифугированием липосомно-пептидную смесь разводят до конечного объема 250 µл в фосфатном буфере. После центрифугирования при 12,000 g в течение 13 минут 20 µл фильтрата берут для оценки содержания свободного белка с помощью микро-ВСА анализа. Количество пептидов, связавшихся с липосомами, определяют вычитанием количества свободного белка из общего его количества. Этот непрямой метод количественного определения His-белкового связывания с липосомами дает примерно те же результаты, что и прямой метод, когда количество связанных с липосомами пептидов определяют напрямую с помощью микро-ВСА анализа после отделения свободного белка с помощью разделяющей по размерам гель-фильтрационной хроматографии на Sephacryl S-200HR гель-матриксе (GE Healthcare).
iii) Пептиды env, презентированные на виросомах возможных конструкций (pNL3-4, IRTV и другие)
Небольшие вирусы и сконструированные на их основе векторы можно использовать для экспрессии и презентирования пептидов env при создании вакцины против ВИЧ. Виросомы в меньшей степени подходят для создания превентивных вакцин, и эффект возникновения ВИЧ-специфического иммунного ответа при их использовании оказывается в любом случае ниже, чем при использовании технологии доставки вакцины с помощью ССЛ. В состав основного иммуногенного компонента вакцины входят env пептиды ВИЧ, экспрессированные на поверхности небольших вирусов - виросомы, - а не кодирующие пептиды env гены, доставляемые в вирусных векторах. Большие вирусы, такие как аденовирусы, аденоассоциированные вирусы, вирус коровьей оспы, являются плохими кандидатами для создания виросомы, потому что они имеют сотни белков на поверхности капсида, и иммунный ответ, который они вызывают после введения, в большей степени является неспецифическим, чем специфическим. Виросомы-векторы включают дефектное производное ВИЧ pNL3-4, вектор на основе вируса гриппа IRIV, доказавший свою эффективность при использовании в вакцинах против малярии и вируса гепатита А, производные вируса кори, альфавирусы различных энцефалитных патогенов, векторы на основе вируса желтой лихорадки и другие возможные варианты.
Животные, пригодные для тестирования адъюванта и адъювант-содержащей вакцины настоящего изобретения, включают приматов, а также грызунов или других млекопитающих. Мыши породы BalbC были использованы для первой проверки возникновения иммунного ответа. Два типа загруки ССЛ-адъюванта могут быть использованы отдельно или могут быть смешаны в различных соотношениях.
Трехнедельных мышей BalbC иммунизируют подкожно дозами в 20-50 мкг чистых пептидов на животное, концентрация адъюванта в суспензии в пересчете на массу сухих липидов MW составляет 5 мг/мл, в каждую группу берут 7-8 животных или более. Для иммунизации берут мышат, которые начали питаться твердой пищей и весят 11-14 г. Первый раз иммунизируют трехнедельных животных, второй раз проводят иммунизацию через две недели, когда они достигнут пятинедельного возраста, третий раз - через 1 месяц, когда мыши достигнут возраста 9 недель.
Варианты рекомбинантных gp120 и его доменов и рекомбинантный gp41 и его эктодомен используются для составления композиций по отдельности или вместе. Титр антител к env пептидам ВИЧ определяют с помощью ИФА на сорбированных r-gp120 (gp110, gp160) или r-gp41, которые использовались ранее для биопанинга фагмидных библиотек, а также на смеси нативных белков ВИЧ. ИФА проводят на 3, 14 и 28 сутки после последнего подкожного введения. Некоторые результаты представлены в Примере 4.
Из наших экспериментов с мышами можно сделать два основных заключения:
1. gp120 и его производные, как рекомбинантные, так и нативные пептиды, высокоиммуногенны и вызывают сильный, ВИЧ-специфический и продолжительный по времени иммунный ответ при подкожном введении мышам BalbC. Рекомбинантный gp41 и его эктодомен при введении тем же способом вызывают выработку в несколько раз меньшего титра специфических моноклональных и поликлональных антител. Та же ситуация с титром антител наблюдается в сыворотке крови ВИЧ-инфицированных пациентов и при презентировании библиотек антител на фаге М13. В крови пациентов эта ситуация возникает за счет внутреннего положения gp41 под gp120 в оболочке вируса. Однако тот же феномен при иммунизации рекомбинантными белками доказывает правоту нашей позиции, что для разработки профилактической вакцины против ВИЧ важна правильная идентификация последовательностей gp120 и их воспроизведение в виде рекомбинантных белков, а gp41 следует использовать как «композитный» пептидный "материал", но вариации его последовательностей не очень важны.
2. Липосомальные адъюванты могут обеспечить выработку иммунного ответа, сохраняющегося в течение более длительного периода, чем при иммунизации только одними пептидами, и одновременно могут уменьшить немедленную реакцию иммунотоксичности, что позволяет увеличить дозы пептидов для выработки защитного иммунного ответа против ВИЧ.
Величина дозы и подходящие формы дозировки для адъюванта и вакцинных композиций данного изобретения могут быть легко определены специалистом при использовании стандартных методов определения титра антител и стандартных протоколов определения биоэффективности/биосовместимости, и в зависимости от конкретного типа адъюванта, желаемого терапевтического эффекта и желаемого промежутка времени для биоактивности. Введение вакцины и ее компонентов может осуществляться парентеральными способами, такими как подкожная инъекция, чрескожное, трансдермальное, интраназальное и внутримышечное введение.
Разработанная профилактическая вакцина против ВИЧ является шагом на пути к индивидуализированной медицине в смысле ее активности против распространения инфекции, вызываемой вариантами ВИЧ в когортах инфицированных людей, библиотеки антител против ВИЧ которых были получены. Эта вакцина не может работать как универсальное оружие против распространения ВИЧ инфекции в форме единичной, однажды разработанной композиции. Однако все знания по эпидемиологии ВИЧ, накопленные за последние 25 лет исследований ВИЧ и борьбы со СПИДом, будут очень полезны при практическом получении вакцины.
ПРИМЕРЫ
Пример 1: Электрофоретические данные на Рис.18-25 иллюстрируют этапы создания фагмидной IgG библиотеки человеческих рекомбинантных антител, содержащих ВИЧ-специфические ScFv фрагменты антител (технология фагового дисплея).
Пример 2: Результаты количественного определения специфичности для рекомбинантных ВИЧ-специфических библиотек представлены на Рис.26.
Пример 3: вариабельность env пептидов ВИЧ
ВИЧ отличается от других патогенных вирусов огромной гетерогенностью пептидных последовательностей. 3D структура пептидов с измененной последовательностью также становится другой, и во многих случаях эти изменения вызываются теми же мутациями, которые обусловливают появления резистентных фенотипов вируса. Следовательно, возможно собрать часто встречающиеся варианты с помощью библиотеки моноклональных антител и получить рекомбинантные формы поверхностных вирусных белков. Некоторые распространенные варианты последовательностей пептидов env ВИЧ субтипов А и В представлены ниже. Вариабельные аминокислоты отмечены синим цветом, консервативные - красным. Некоторые из этих последовательностей были ранее получены в нашей лаборатории.
Пример 4: Предварительные результаты иммунизации животных (статистический анализ в SigmaPlot 10.0).
3-недельных мышей BalbC, весящих 11-14 г, иммунизируют подкожно дозами в 20-50 мкг чистых пептидов на одно животное, при концентрации липидов MW 5 мг/мл. Иммунизируют первый раз 3-недельных мышей, второй раз - 2 недели спустя, когда мыши достигнут 5-недельного возраста, третий раз - через 1 месяц, когда мыши достигнут 9-недельного возраста. Рекомбинантный gp120 вызывает в среднем в 5 раз более высокие уровни иммунного ответа, чем рекомбинантный эктодомен gp41. Те же различия в титре специфических антител наблюдаются, когда используются человеческие поликлональные антитела, выделенные из сыворотки крови пациентов, при ИФА-тестировании на одинаковых концентрациях сорбированных рекомбинантных gp120 и gp41. Результаты представлены на Рис.27.
ЛИТЕРАТУРА
1. Abram М.Е., Pamiak M.A. "Virion Instability of Human Immunodeficiency Virus Type 1 Reverse Transcriptase (RT) Mutated in the Protease Cleavage Site between RT p51 and the RT RNase H Domain" Journal of virology, September 2005, v. 79, Nol8,11952-11961;
2. Aguilar M-I. "HPLC ofPeptides and Proteins" "Methods in Molecular Biology Sen", v.251, Humana Press, 2004;
3. Amicosante M., Cappelli G. "Method ofAntigenic Peptide Identification and Relative Use for the Preparation of a Vaccine Anti HIV-1" Uni Degli Studi di Roma Tor Ve (IT), W02005075679,2005-08-18;
4. Barbados C.F. et al. "Phage Display: A Laboratory Manual", 2001, Cold Spring Harbor Laboratory Press;
5. Berman P.W., Fendly B.M. et al. "HIV Env Antibodies" Applicant Genetech INC, US patent US 7041293 Bl, 2006-05-09;
6. Berman P.W., Nakamura G.R. "Preparation of an HIV GP 120 Subunit Vaccine Involving Determining a Neutralising Epitope in the V2 and/or C4 Domains", Applicant Genetech INC, US patent NZ267838, 1997-12-19;
7. Bongiovanni M. et al., "Long Term Immunologic Outcome in HAART-Experienced Subjects Receiving Lopinavir/Ritonavir", AIDS Research and Human Retrovimses, 2006, 22(1 l),pp 1099-1105;
8. Butera S.T. "HIV Chemotherapy. A Critical Review", Caister Academic Press, 2005, U.K;
9. Capodici J. et al., "Large-Scale Beta-Propiolactone Inactivation of HIV for Vaccines" Bioprocess Int., 2006, Feb., pp 36-41;
10. Chertova E, Chertov 0. Coren LV, Roser JD, Trubev CM, et al. "Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages", J ViroL, 2006 Sep;80(18):9039-52;
11. Emini, E., Shiver J., Casimiro, D. R. et al. "Therapeutic Immunization ofHIV-infected individuals" Appl.: MERCK&CO., INC. A01N 43/04 (2006.01), WO/2005/027835, 31.03.2005;
12. Franchini G., Hel Z., Pavlakis G. et al. "Improved immunogenicity using a combination ofDNA and Vaccinia virus vaccines" Appl.: The Government of USA, A61K 39/21 (2006.01), WO/2001/082964, 08.11.2001;
13. Haynes В., Desaire F., Heather F. et al. "Carbohydrate-based vaccines for HLV" Appl.: Duke University, US Patent A61K 39/00 (2006.01), WO/2008/033500, 20.03.2008 ;
14. Herschhom A. et al., 2003, "Recombinant human antibodies against the reverse transcriptase of human immunodeficiency virus type-1" Biochim Biophys Acta., 1648:154-163;
15. Honda M., Matsuo K. "A Method of Prime-boost Vaccination", Appl.: Japan Science&Tech Agency (JP); JP Nat Inst of Infectious Disease (JP), Patent W02006057454, 2006.06.01;
16. Humphreys R., Macmillan D., ZinckgrafJ. „li-key enhanced vaccine potency" US Patent A61K 48/00, Applicant: Antigen Express, Inc., WO 2008/060385, 22.05.2008;
17. Ghania G. et al., "Attaching histidine-tagged peptides and proteins to lipid-based carriers through use of metal-ion-chelating lipids", Biochimica et Biophysica Acta 1567(2002)204-212;
18. Grundner,C. et al., "Factors limiting the immunogenicity of HIV-1 gp120 envelope glycoproteins" J. Virology, 2004.Dec.5.;330.(l.):233.-48;
19. Kam J. "HIV Volume 1: Virology and Immunology" Oxford University Press, 1995, reprinted 2001,63-75;
20. Karp N.M. "Immunogenic Composition and Method of Developing a Vaccine Based on Psoralen Inactivated HIV" Appl.: NMK RES LLC (US), US Patent W02005040353,2005-05-06;
21. Kirschner M., Monrose V., Paluch M., Techodamrongsin N., Rethwilm A., Moore J. „ The production of cleaved, trimeric human immunodeficiency virus type 1 (HIV-1) envelop glycoprotein vaccine antigens and infectious pseudovimses using linear polyethylenimine as a transfection reagent" Prot. Expres. Purification, Jul. 2006, 48(1), 61-68;-
22. Kwong P.D. et al. "Structure of an HIV-1 gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody". Nature, (1998) 393:649-59;
23. Maniatis et al., "Molecular Cloning, a Laboratory Manual", 1991,Cold Spring Harbor Laboratory Press;
24. Leitner T. "HIV Sequence Compendium 2006-2007", Los Alamos National Laboratory annual issue;
25. Liu В., Marks J.D. "Applying phage antibodies to proteomics: selecting single chain Fv antibodies blotted on nitrocellulose" Analitical Biochem. 2000, 286, 119-128;
26. Lu X., Dropulic B. "Lentivirus vector-based approaches for generating an immune response to HIV humans" US Patent, Appl.: VIRXSYS Corporation, WO/2005/023313, 17.03.2005;
27. Martin L., Stricher F., Descours A. et al. "CD4 mimic peptides and their usues" Appl.:
Commisariat A L'Energie Atomique, C07K 14/705, WO/2007/144685, 21.12.2007;
28. Marks J. D. et al. "By-passing Immunization Human Antibodies from V-gene Libraries Displayed on Phage" 1991, J. Mol. Biol., 222: 581-597;
29. Peluso R., Arnold S., Wustner J. Et al. "Recombinant AAV-based vaccine methods" US Patent C12N 15/864, Applicant: Targeted Genetics Corporation, WO 2006/073496, 13.07.2006;
30. Picker, L. J., Jarvis, M., Nelson, J, A. "SIV and HIV vaccination using RHCMV- and HCMV-based vaccine vectors" US Patent C12N 15/63 (2006.01), Appl.: Oregon Health and Science University, WO/2006/031264, 23.03.2006;
31. Poignard P. et al., "Heterogeneity of envelope molecules expressed on primary human immunodeficiency vims type 1 particles as probed by the binding of neutralizing and non-neutralizing antibodies" J. Virol.2003.Jan.; 77.(L): 353-65;
32. http://iavireport.org/specials/OngoingTrialsofPreventiveHIVVaccines.pdf NIAID' s report 2007 " Ongoing Trials of Preventive AIDS Vaccines";
33. Sala M. Greco, R. et al. "Polynucleotides allowing the expression and secretion of recombinant HbSag virus-like particles containing a foreign peptide, their production and use" Appl.: Institute Pasteur, WO Patent 2008/020331, C07K 14/43, 21.02.2008;
34. Sanders R.W. et al., "Mutational Analyses and Natural Variability of the gp41-Ectodomain", The 2002 HIV Sequence Compendium;
35. Sidhu S.S. "Phage Display in Biotechnology and Drug Discovery", 2005, Taylor & Francis group;
36. Soerensen B. "HP/ peptides, antigens, vaccine compositions, immunoassay kit and a method of detecting antibodies induced by HIV", Appl.: BIONOR AS (NO), Patent W00052040, 2000.09.08;
37. Steinbrook R. "One Step Forward, Two Steps Back - Will There Ever Be an AIDS Vaccine?" New England Journal of Medicine, Dec. 27, 2007(26), 357:2653-2655;
38. Torchilin VP, Levchenko TS, Lukyanov AN, Khaw BA, Klibanov AL, Rammohan R, Samokhin GP, Whiteman KR. "p-Nitrophenylcarbonyl-PEG-PE-liposomes: fast and simple attachment of specific ligands, including monoclonal antibodies, to distal ends of PEG chains via p-nitrophenylcarbonyl groups", Biochim Biophys Acta. 2001 Apr2; 1511(2):397-411;
39. Torchilin V.P. "Liposomes: A Practical Approach" Edition: 2, Publisher: Oxford Univ. Press, publ. 08/01/2003;
40. Wyatt R. et al., Sodroski J. The antigenic structure of the human immunodeficiency virus gp120 envelope glycoprotein. Nature, (1998) 393:705-11;
41. Wyatt R. et al., Sodroski J.G. "Structure of the Core of the HTV-1 gp120 Exterior Envelope Glycoprotein" The Human Retroviruses and AIDS Compendium, 1998;
42. http://www.hiv.lanl. gov/cgi-bin/vaccine/public/ Los Alamos HIV vaccine trials database.
| название | год | авторы | номер документа |
|---|---|---|---|
| Антитела против белка р17 ВИЧ-1 субтипа А | 2019 |
|
RU2727673C1 |
| Способ усиления направленного редактирования гена CXCR4 в CD4-лимфоцитах человека в целях создания резистентности клеток к ВИЧ-1 | 2021 |
|
RU2779300C1 |
| ИММУНОГЕННАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ХИМЕРНЫЙ ПОЛИПЕПТИД ВИЧ, ПОЛИПЕПТИДЫ И ТЕСТ-СИСТЕМА ДЛЯ ОБНАРУЖЕНИЯ АНТИТЕЛ К ВИЧ | 2001 |
|
RU2214274C2 |
| АНТИТЕЛО ПРОТИВ С5 И СПОСОБ ПРЕДУПРЕЖДЕНИЯ И ЛЕЧЕНИЯ ОБУСЛОВЛЕННЫХ КОМПЛЕМЕНТОМ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2663349C2 |
| Вакцина против герпеса | 2019 |
|
RU2731073C1 |
| СИНТЕТИЧЕСКИЙ ГЕН, КОДИРУЮЩИЙ ЗЕЛЕНЫЙ ФЛУОРЕСЦЕНТНЫЙ БЕЛОК, СПОСОБ ПОЛУЧЕНИЯ ГЕНА, ЭКСПРЕССИРУЮЩИЙ ВЕКТОР, СПОСОБ ПОЛУЧЕНИЯ КУЛЬТУРЫ КЛЕТОК | 1997 |
|
RU2233329C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ И/ИЛИ ПРЕДОТВРАЩЕНИЯ ЗЛОКАЧЕСТВЕННОЙ ОПУХОЛИ | 2016 |
|
RU2714205C2 |
| Искусственные гены, кодирующие белки-иммуногены EV.CTL и EV.Th, рекомбинантные плазмидные ДНК pEV.CTL и pEV.Th, обеспечивающие экспрессию искусственных генов, и искусственные Т-клеточные полиэпитопные белки-иммуногены EV.CTL и EV.Th, содержащие эпитопы антигенов вируса Эбола, используемые для создания вакцины против вируса Эбола | 2018 |
|
RU2713723C1 |
| ИММУНОИНДУЦИРУЮЩЕЕ СРЕДСТВО | 2016 |
|
RU2744843C2 |
| Генетическая конструкция, адаптированная для доставки гена SMN1 человека с помощью аденоассоциированного вируса серотипа 2 для обеспечения нейроспецифичной экспрессии | 2022 |
|
RU2801848C1 |






























































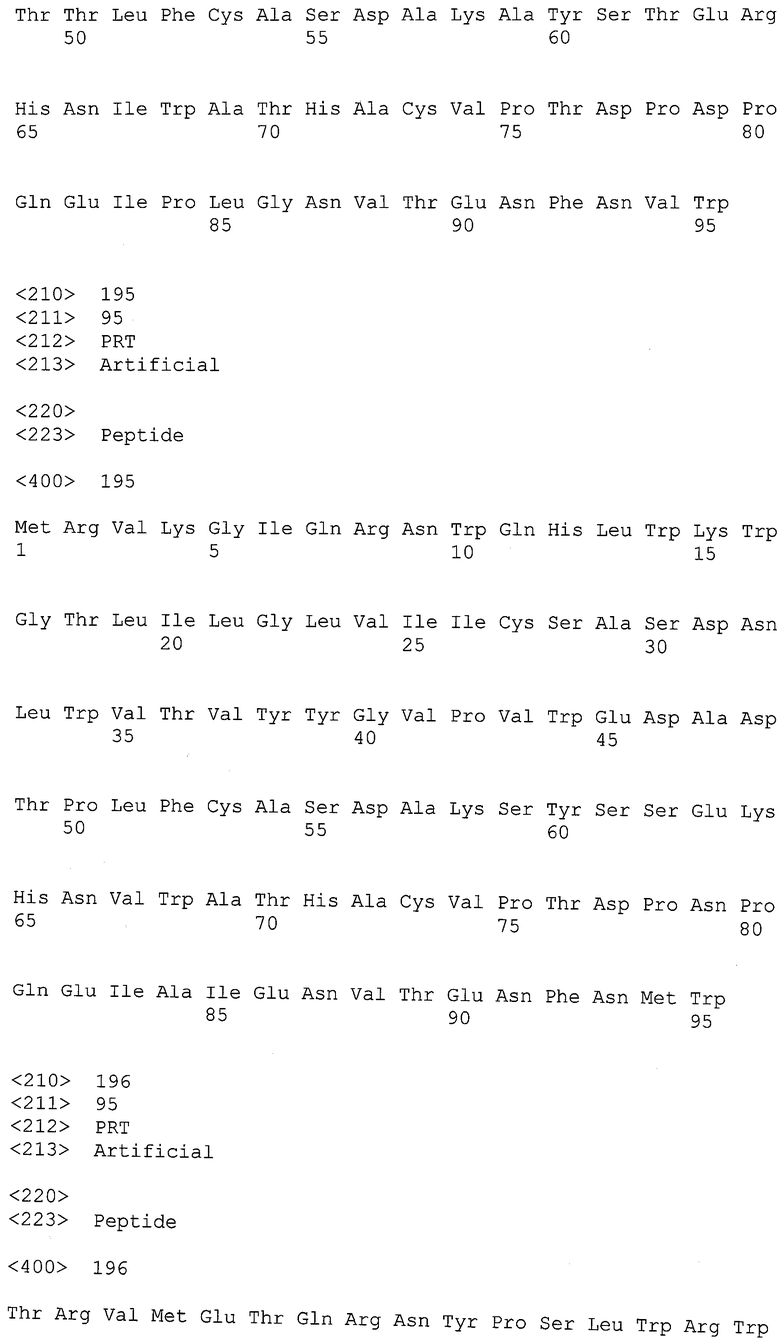




































































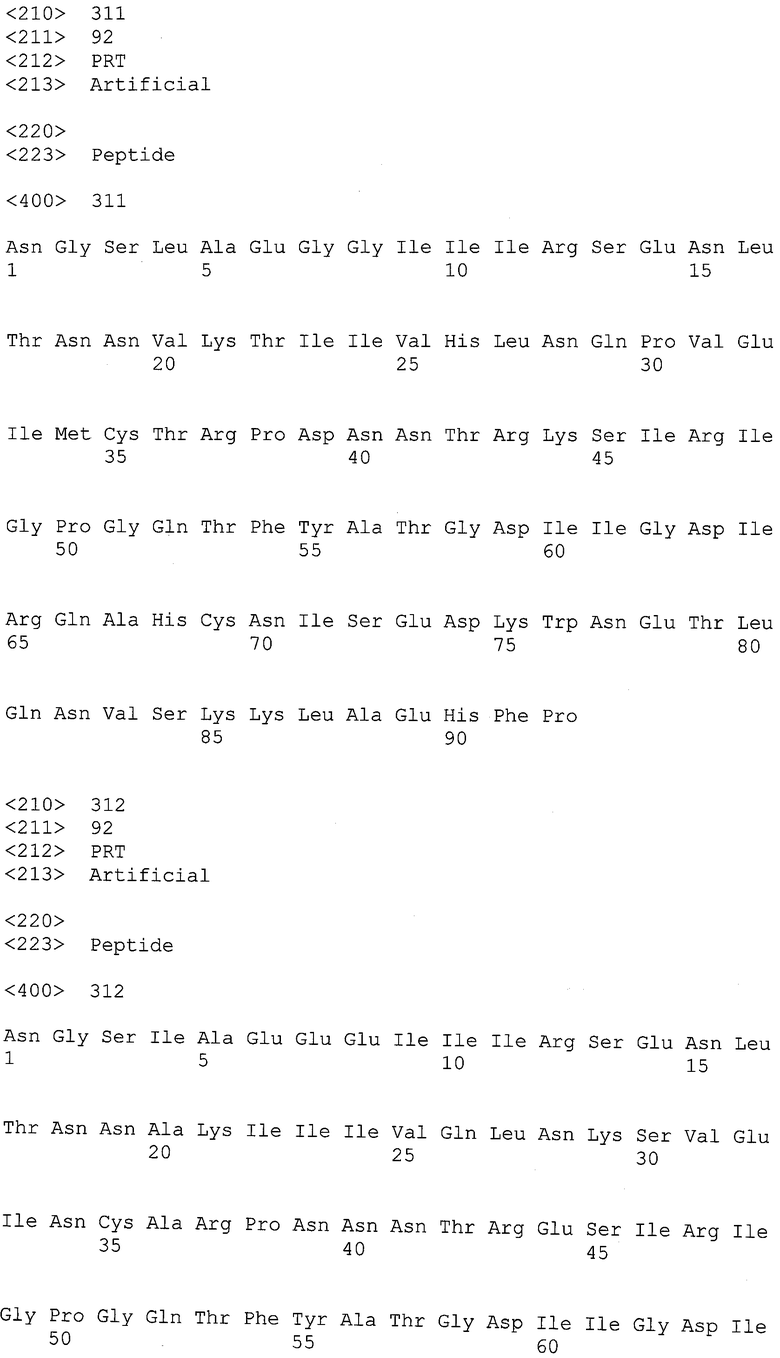








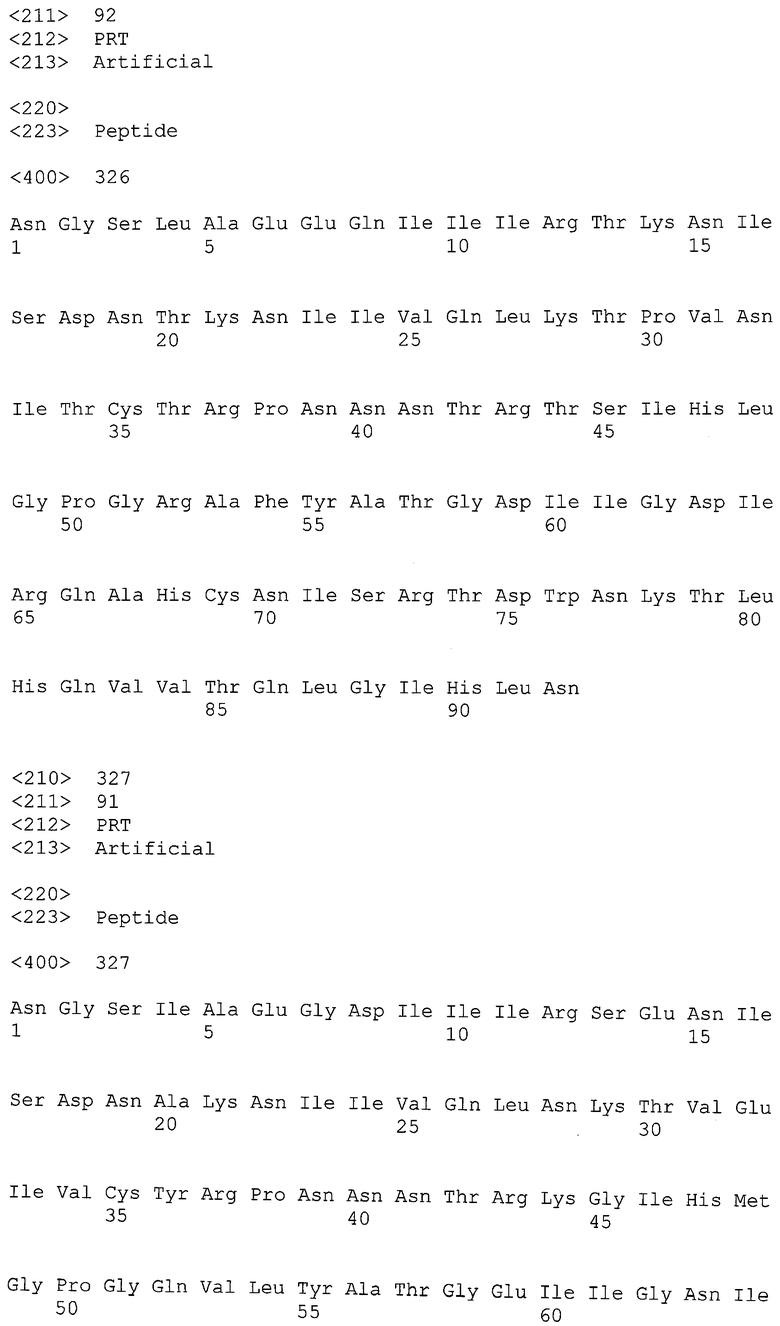













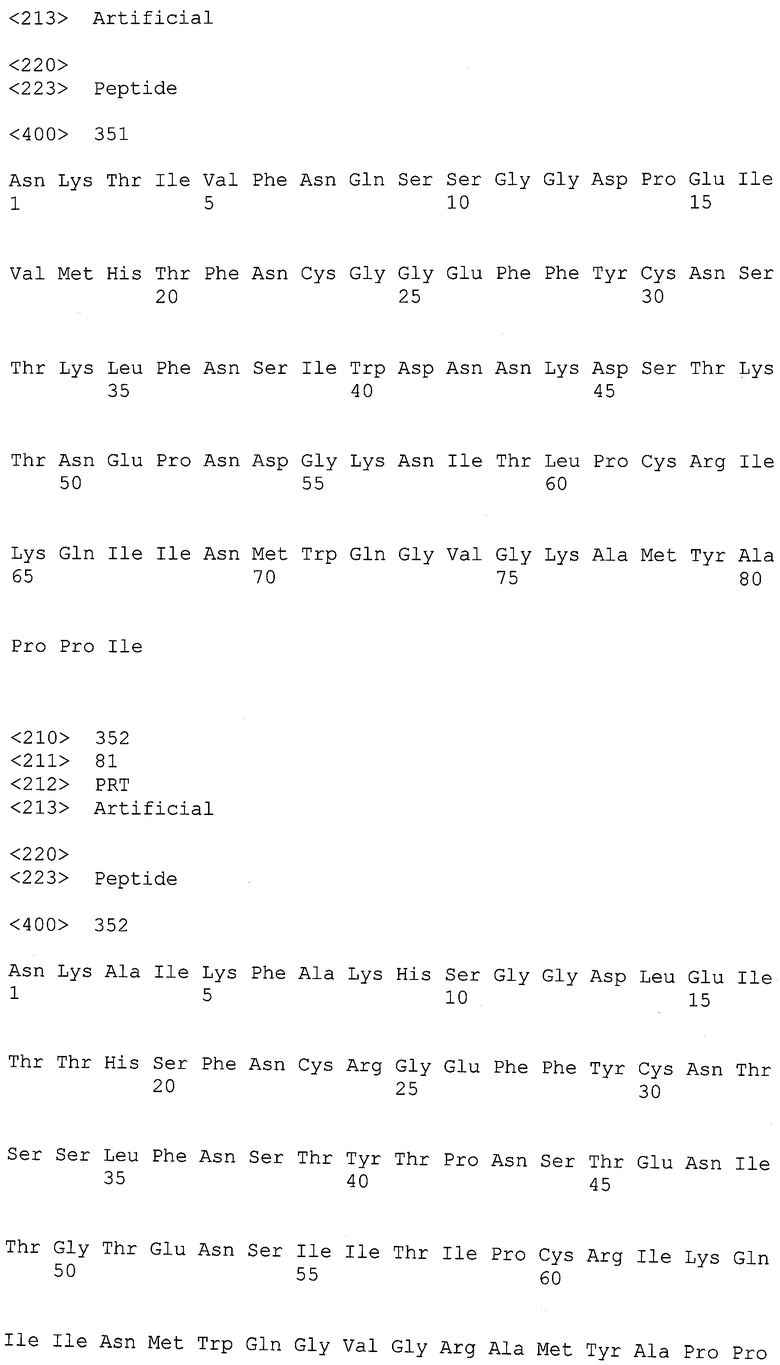








































































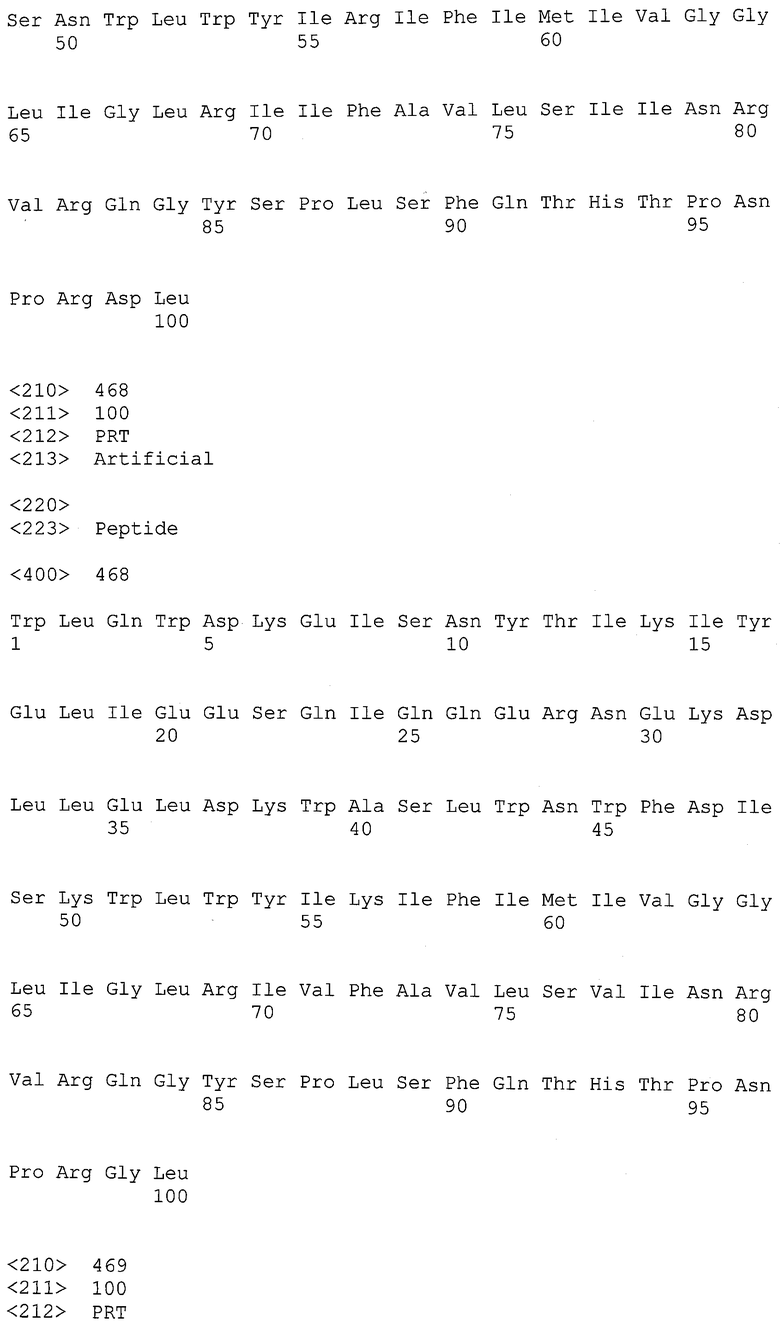















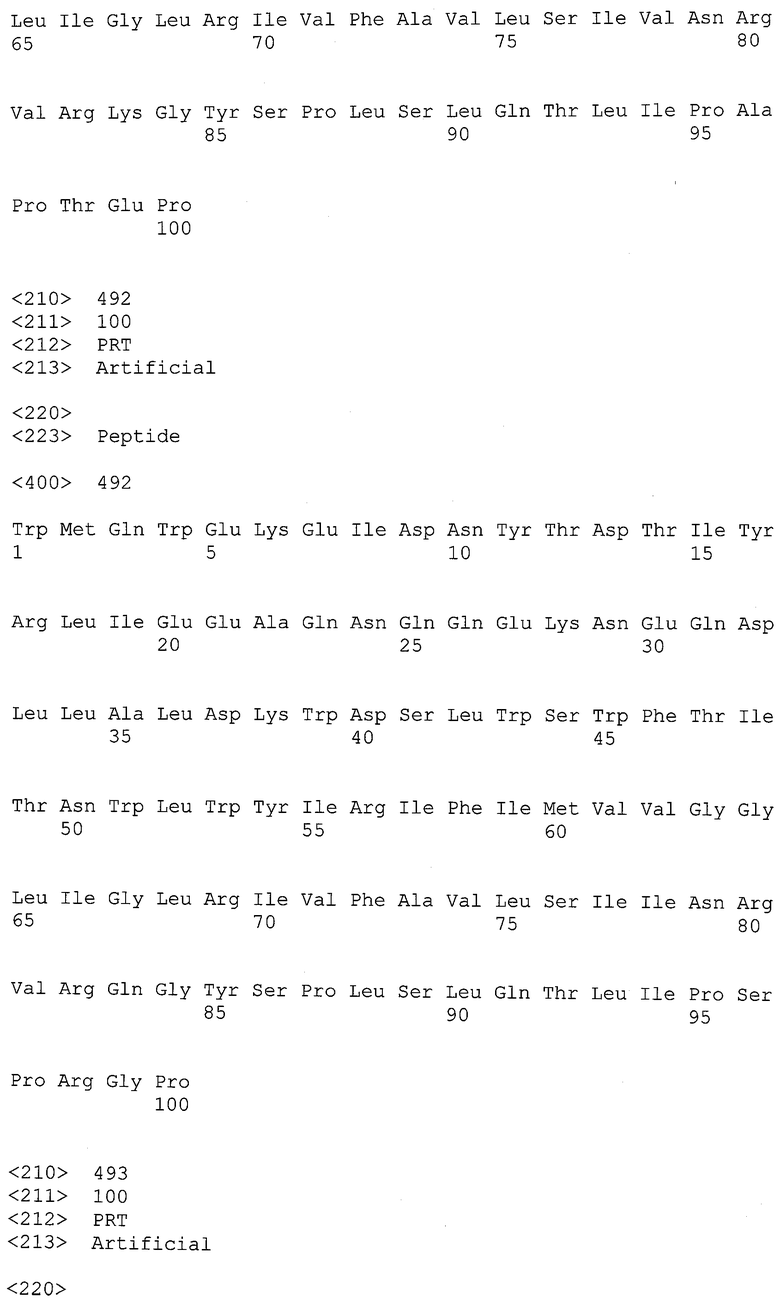



















































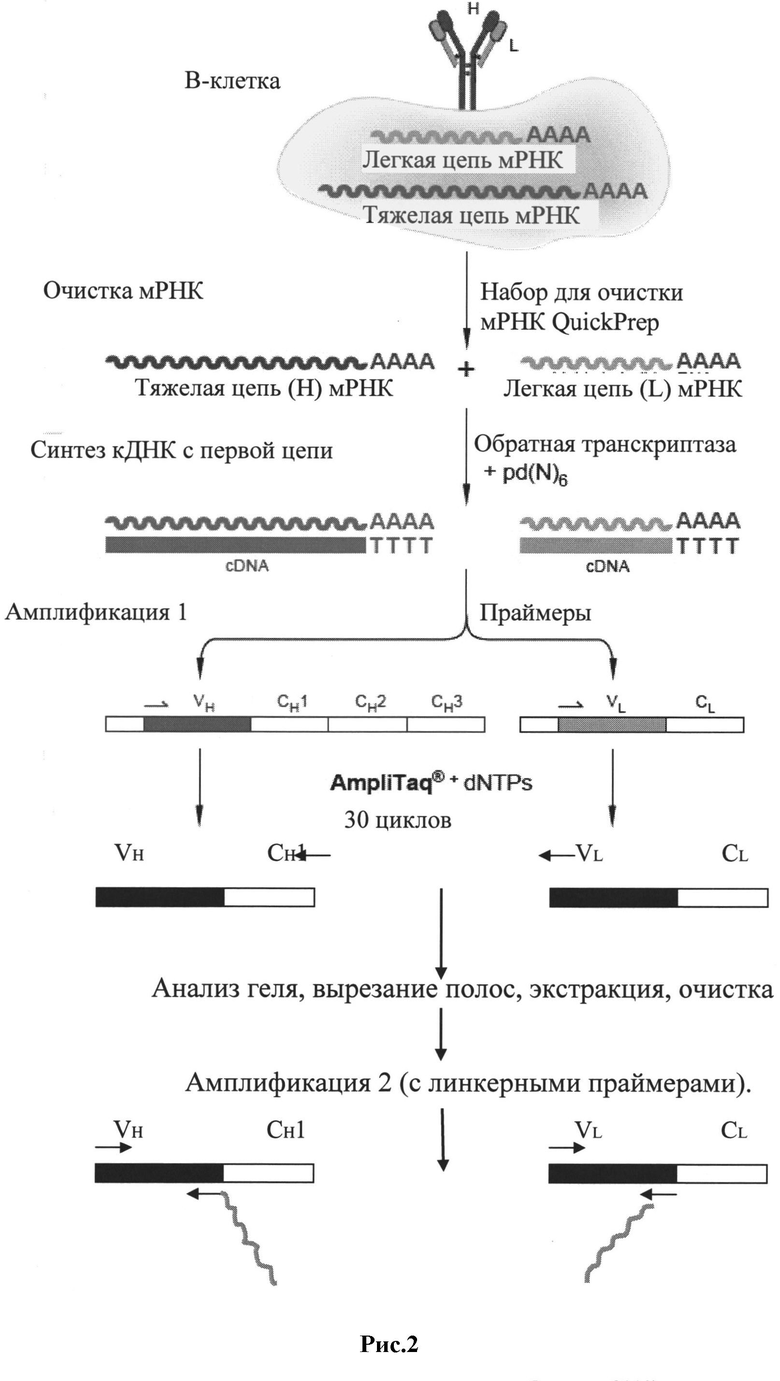
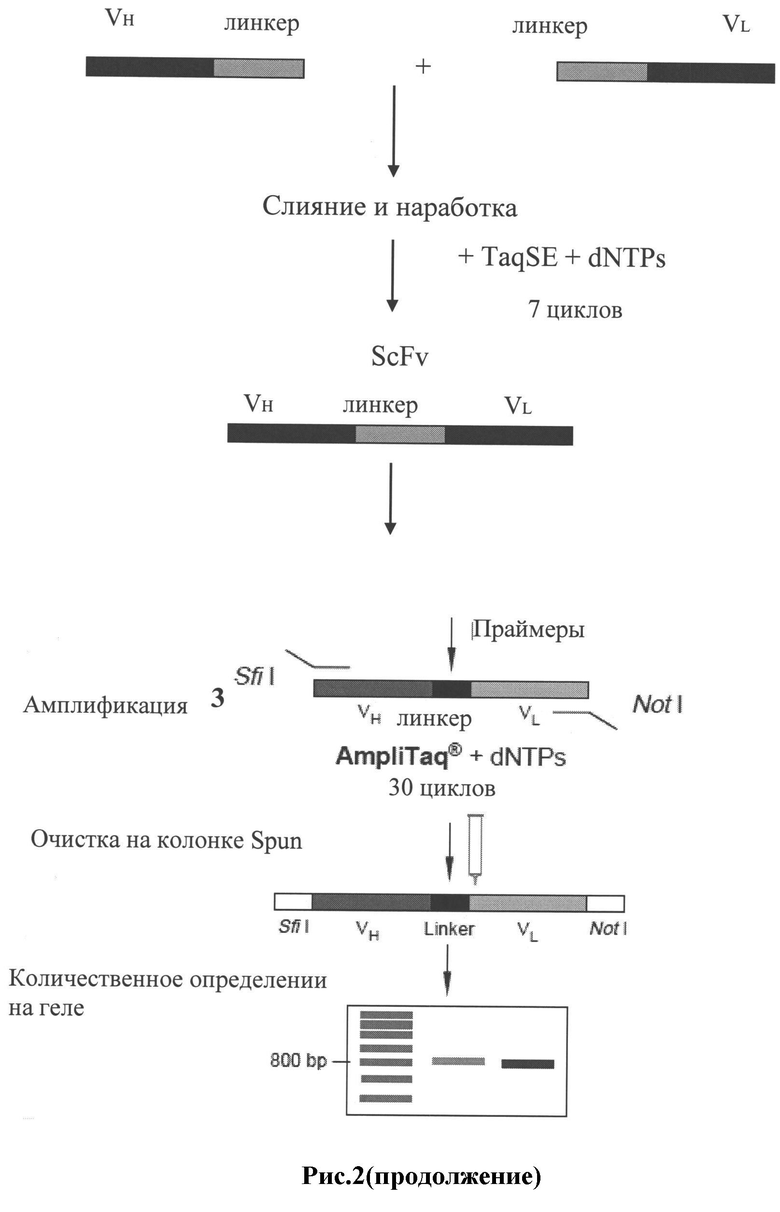
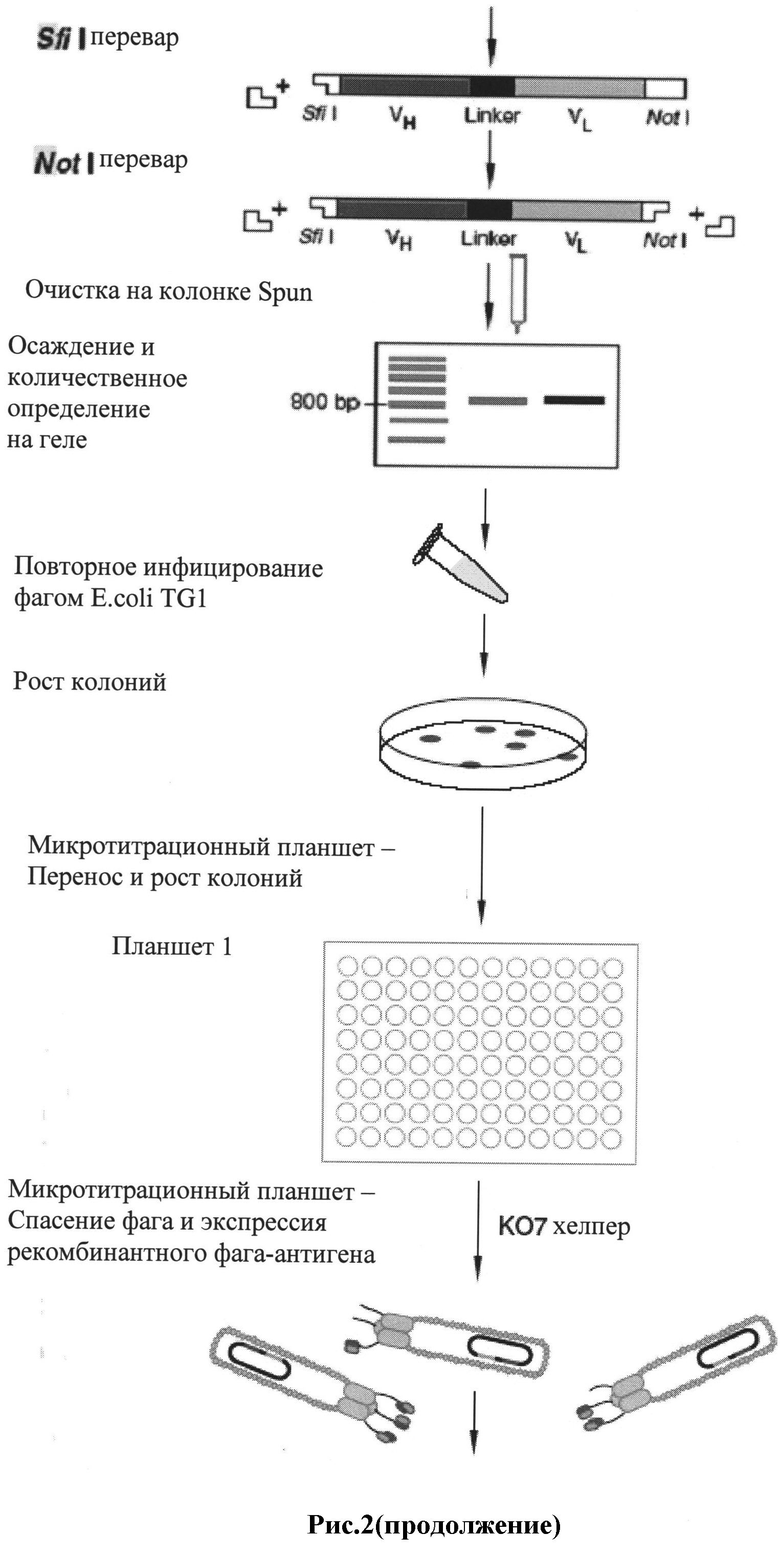
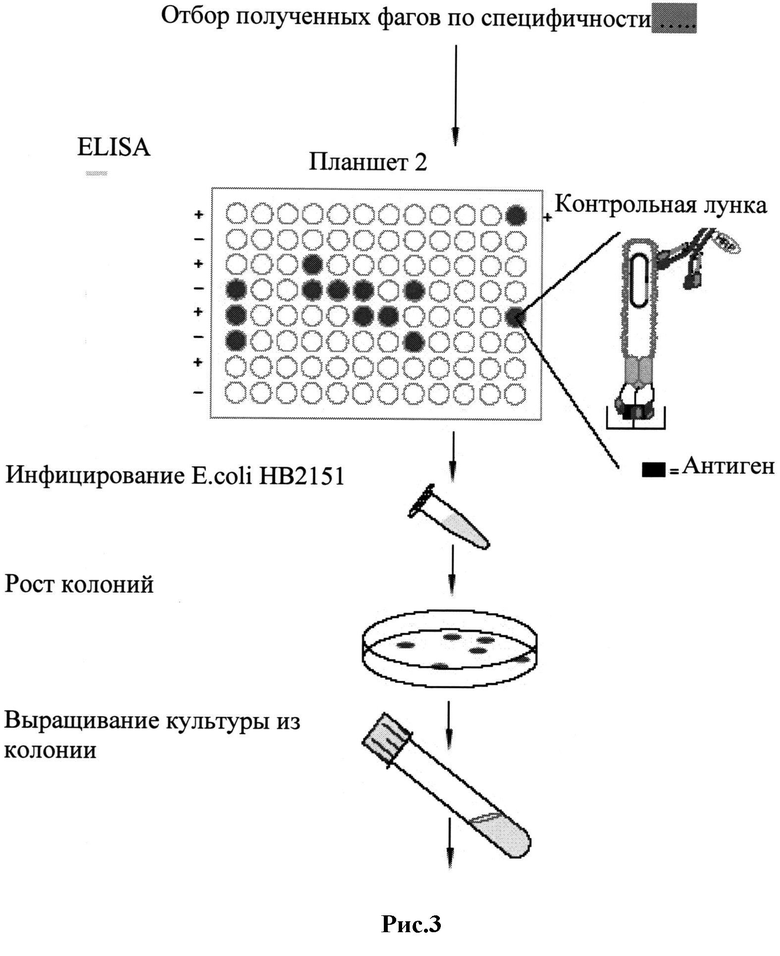
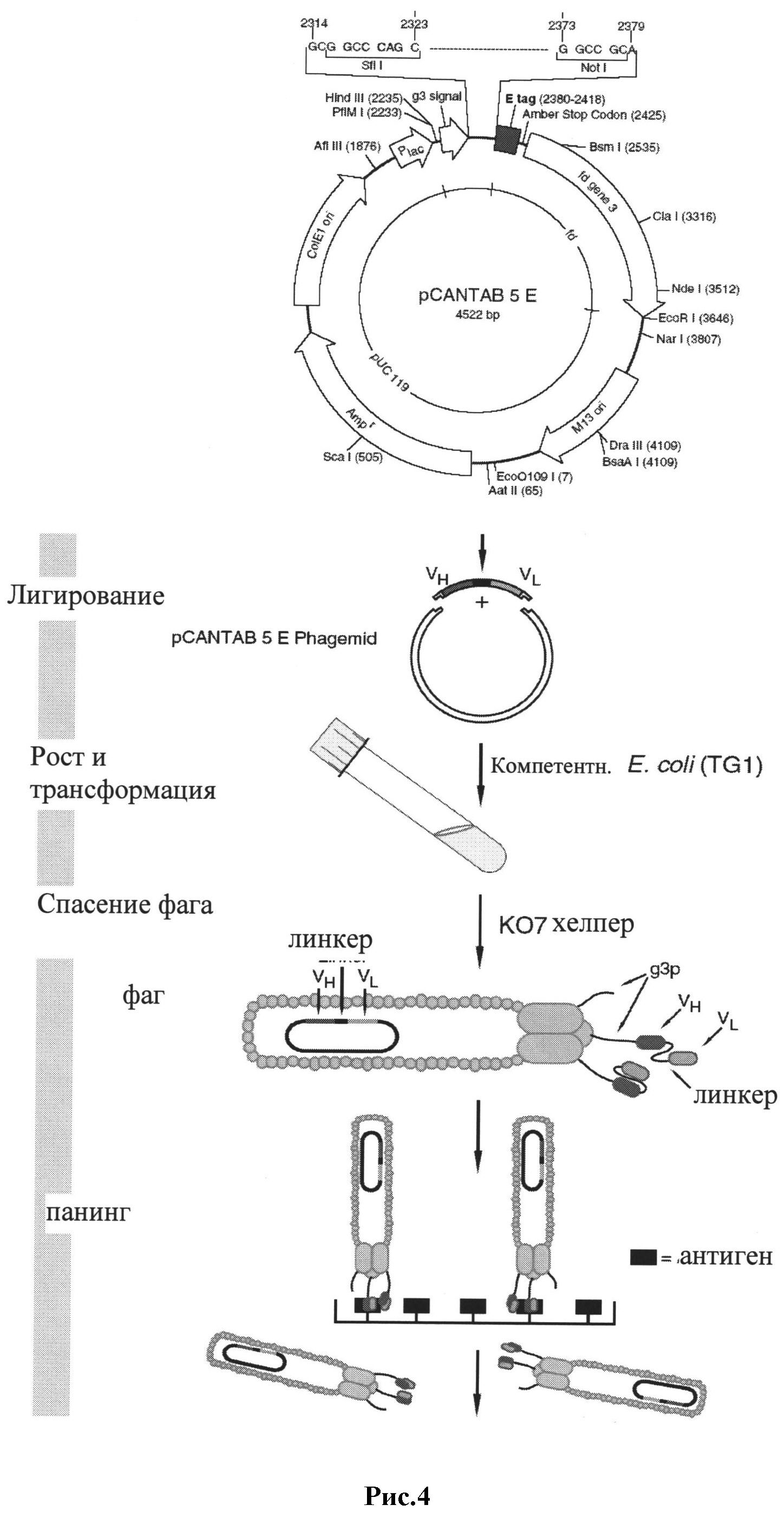
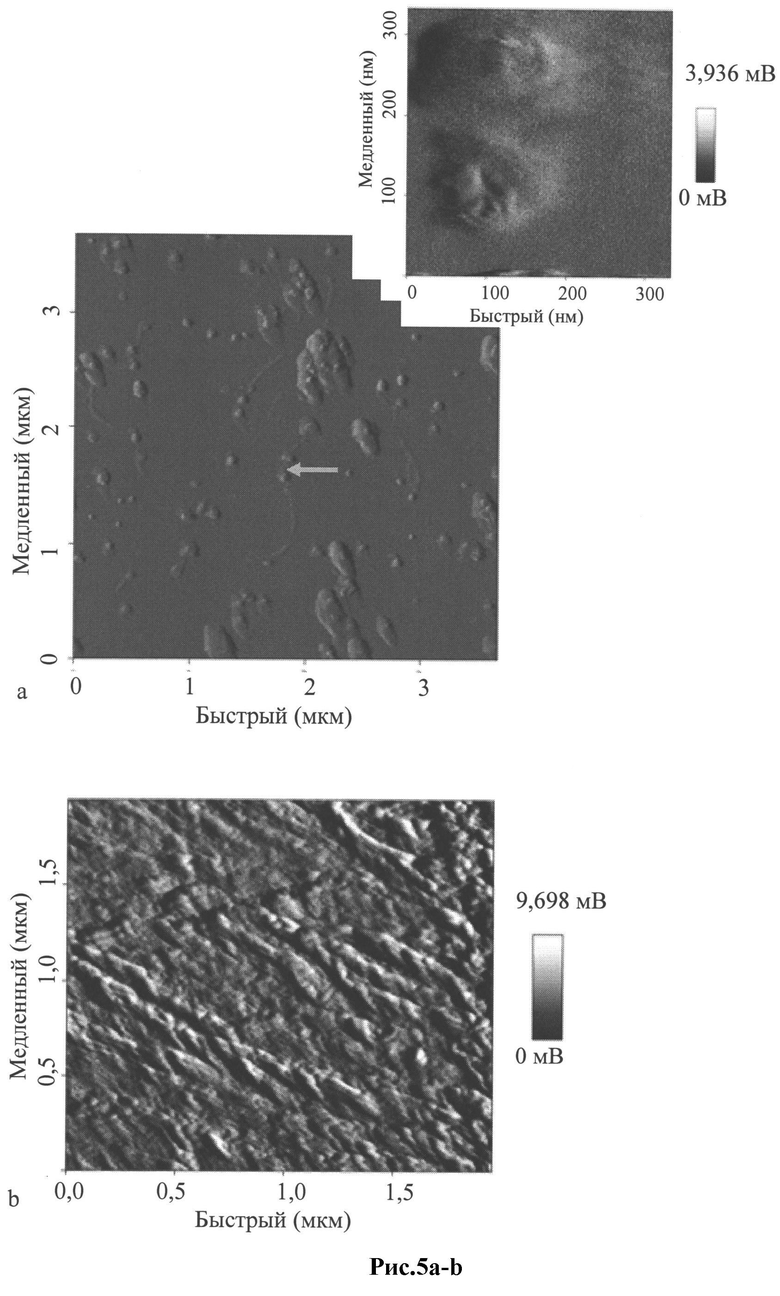

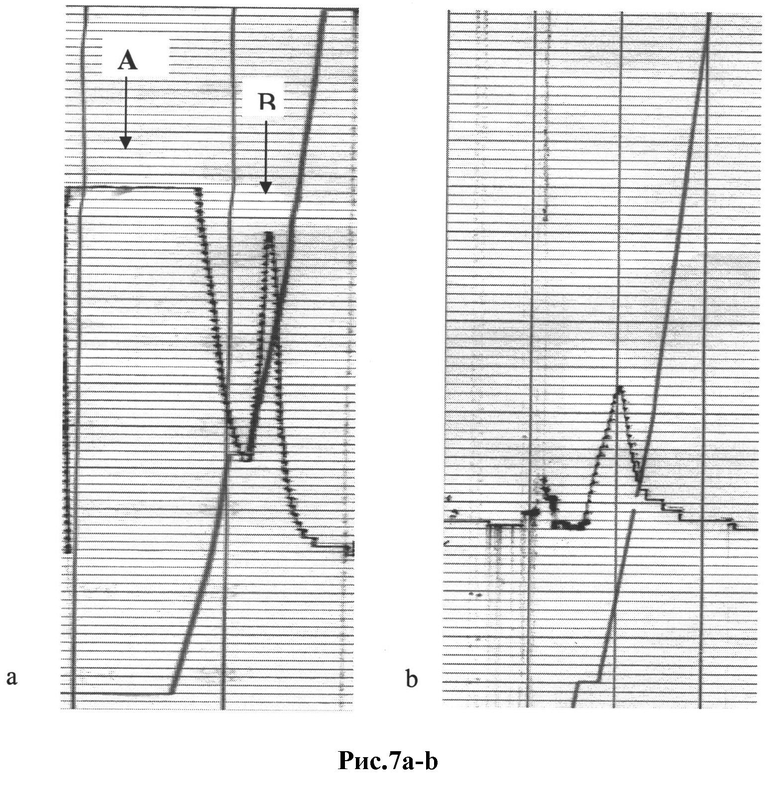
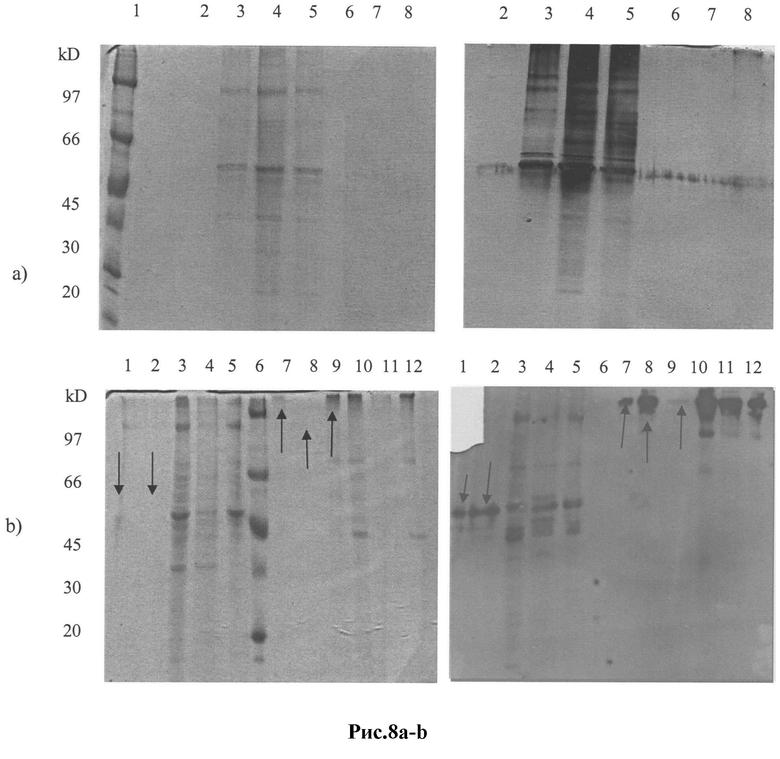
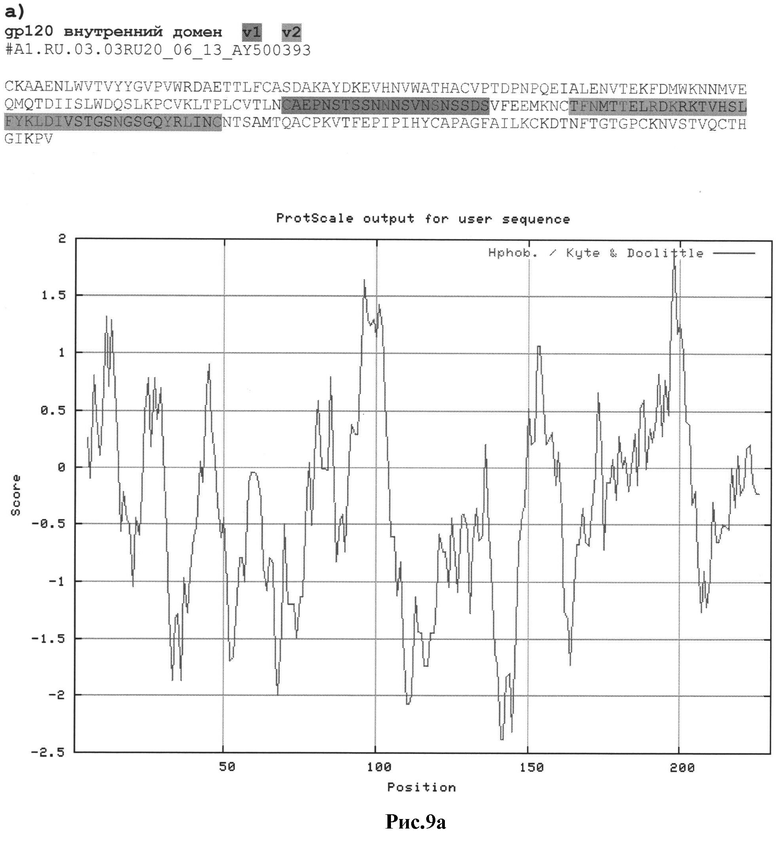

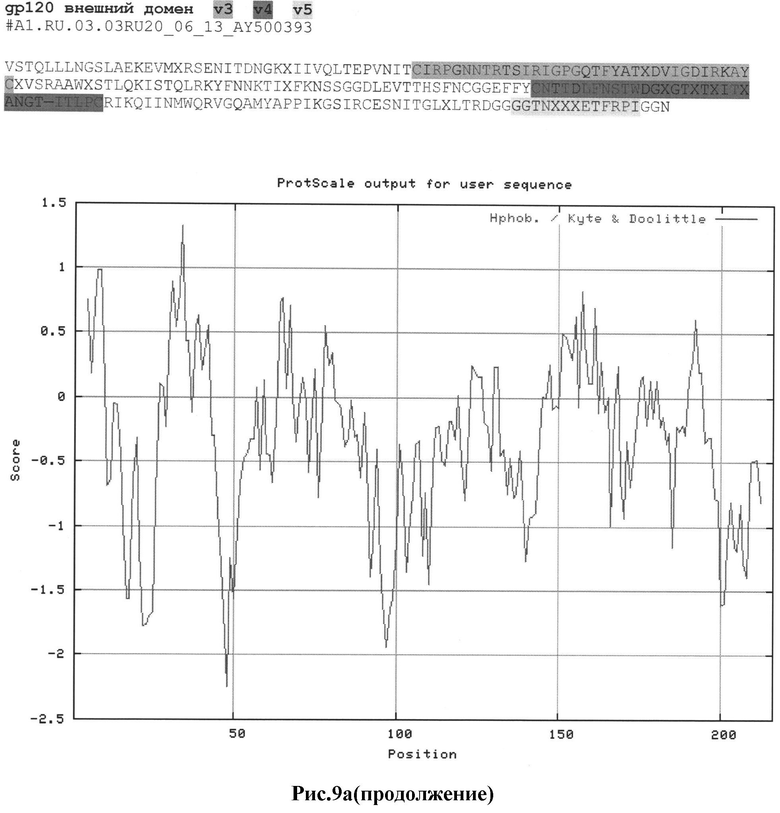
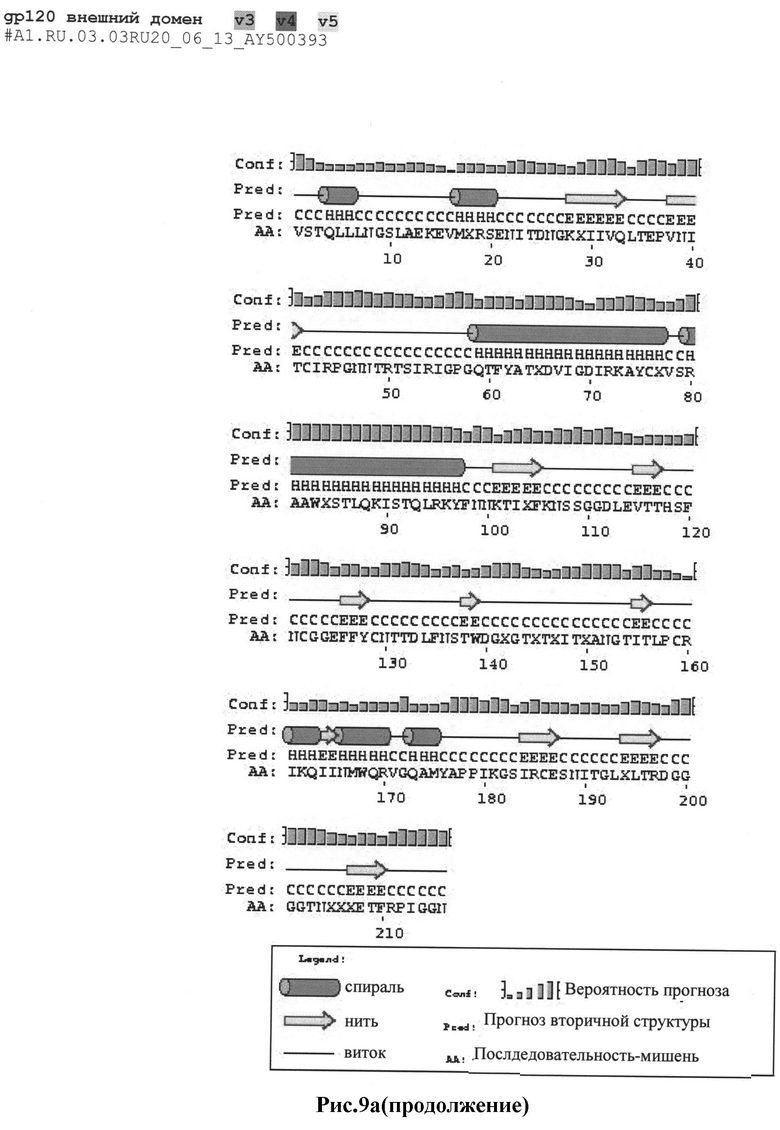
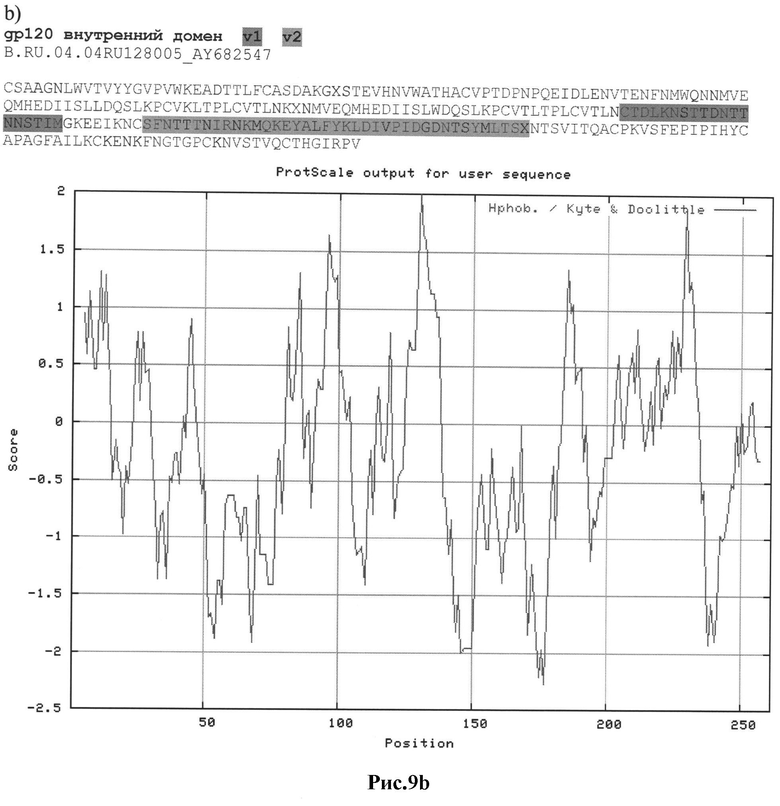

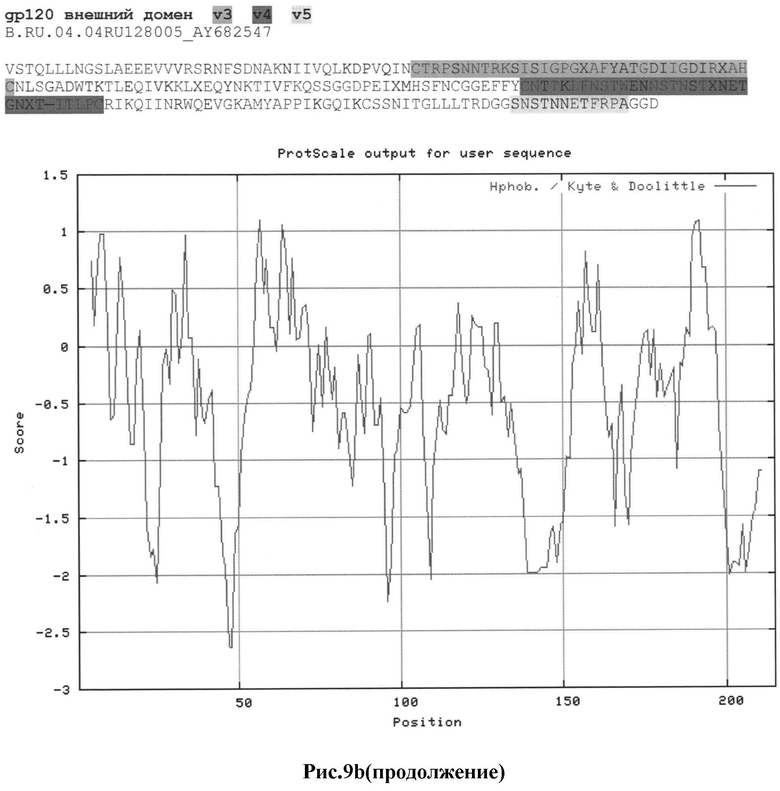
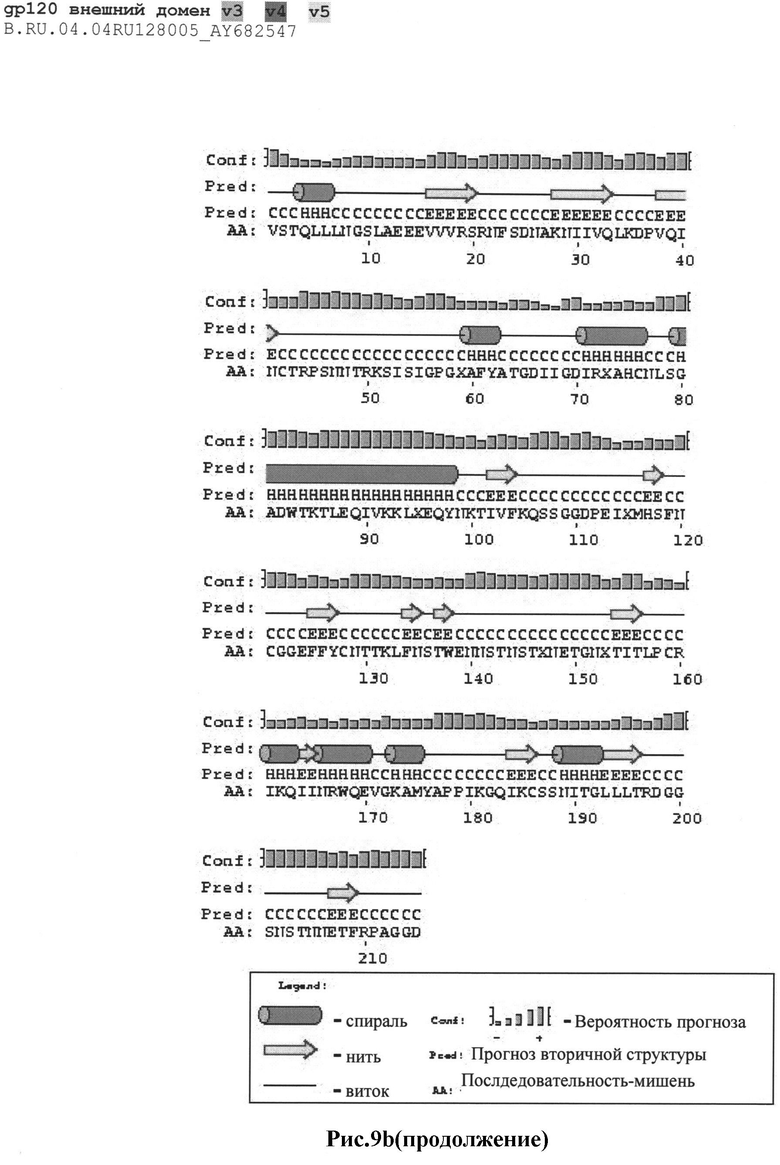
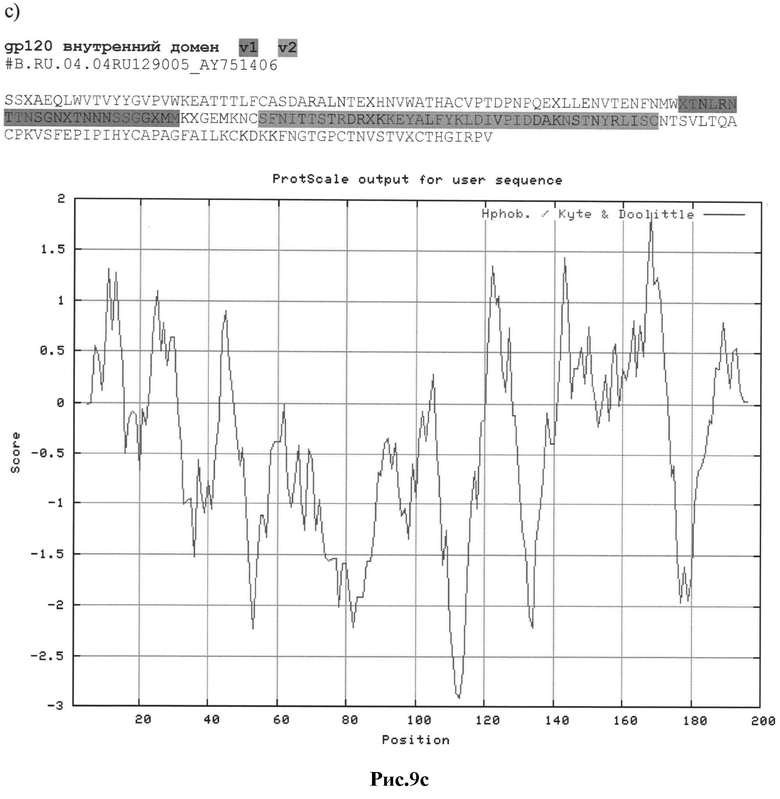
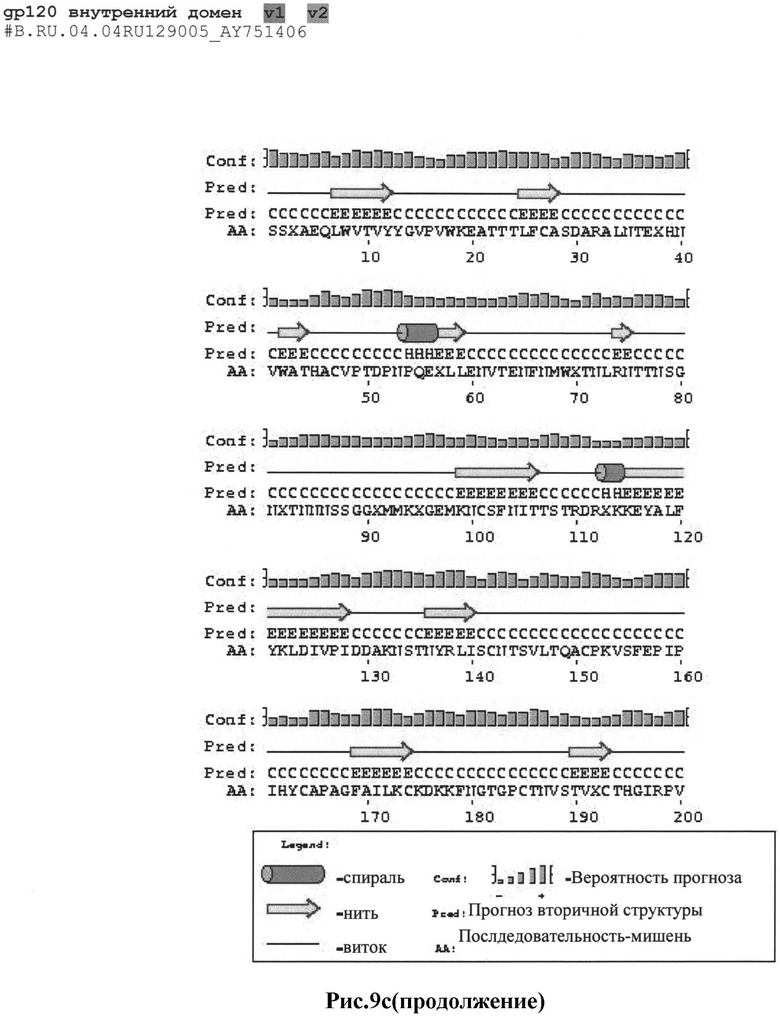
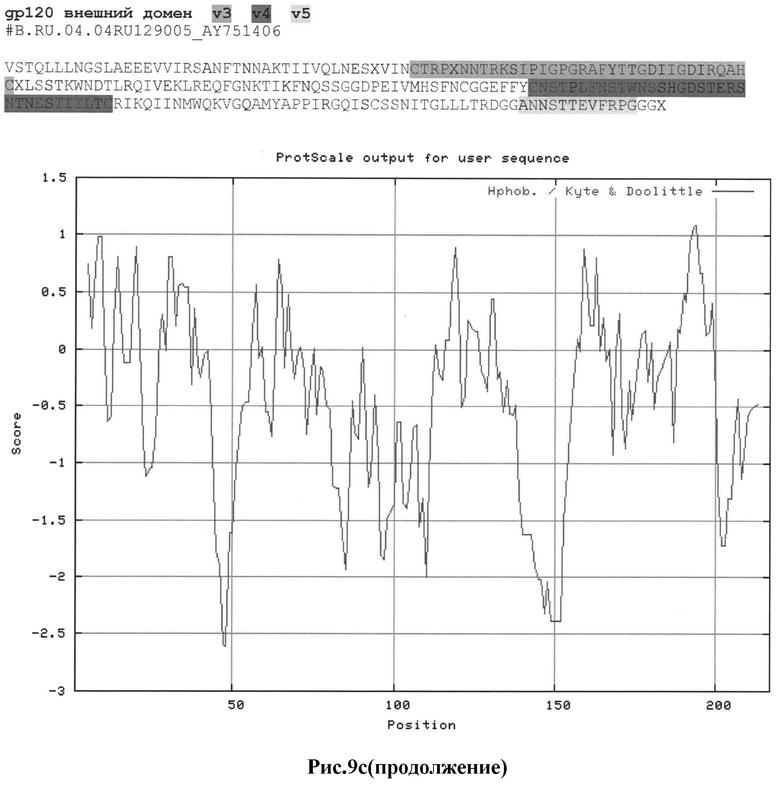
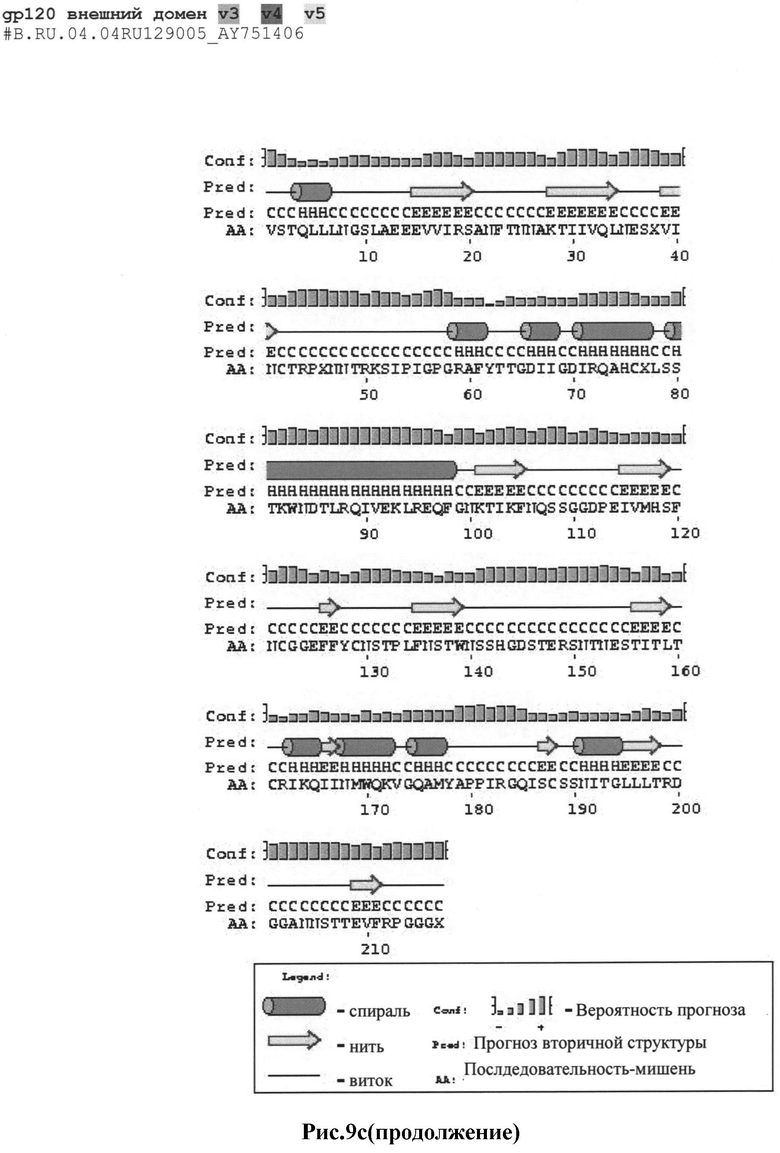
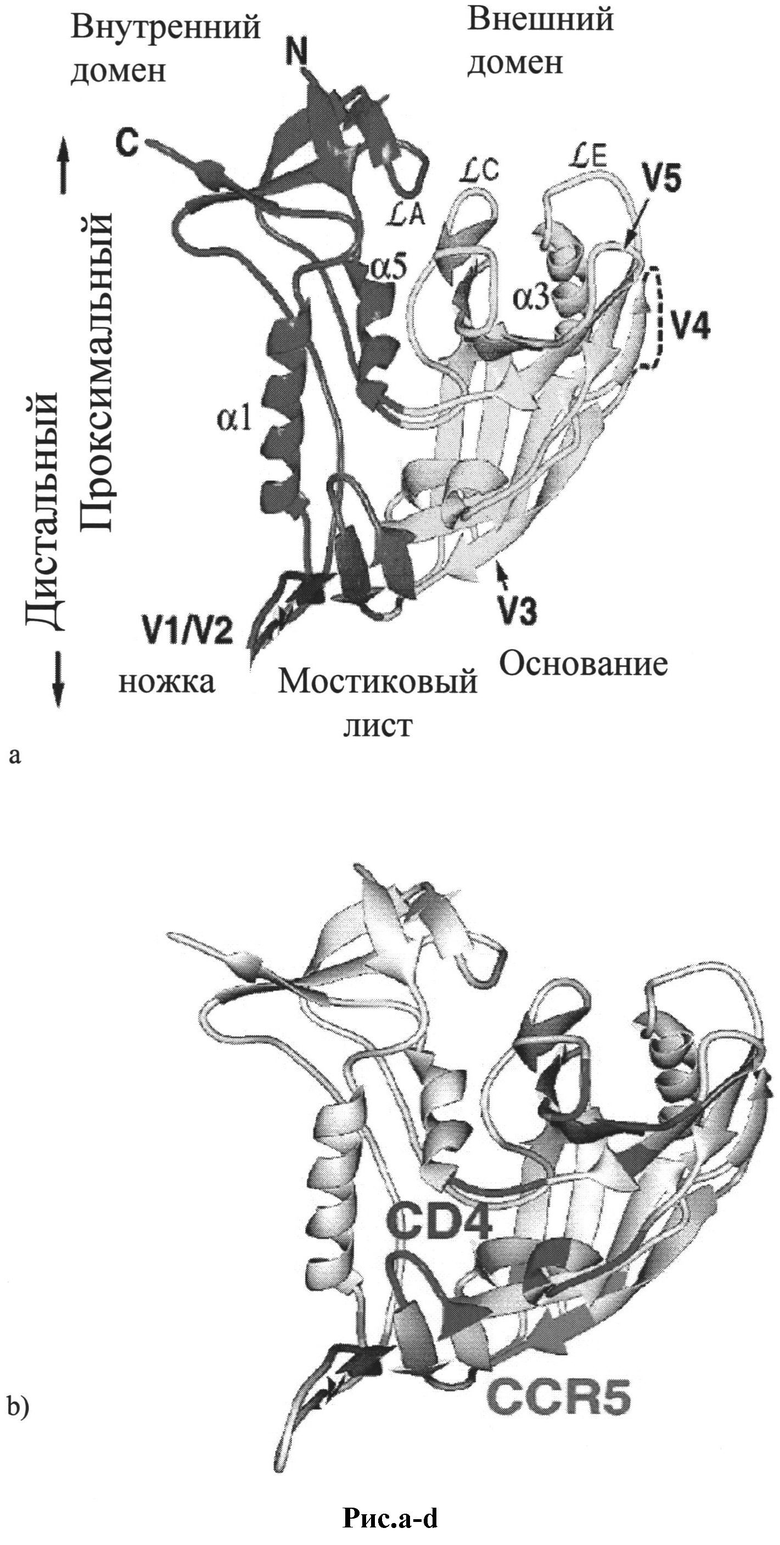
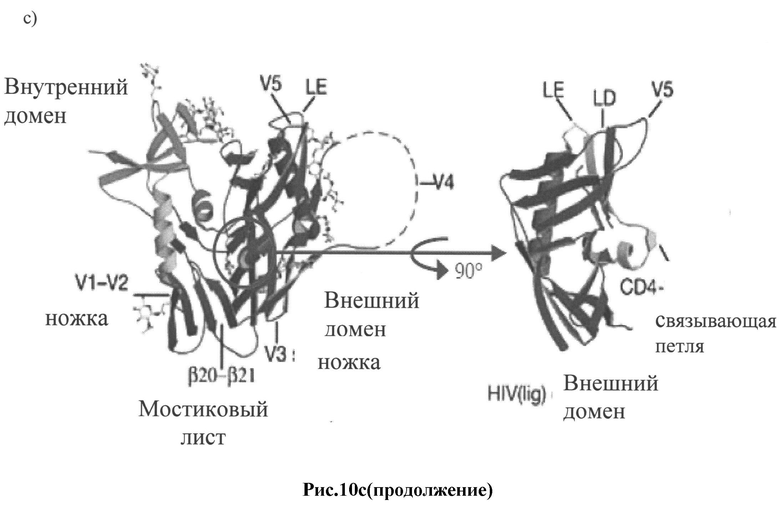
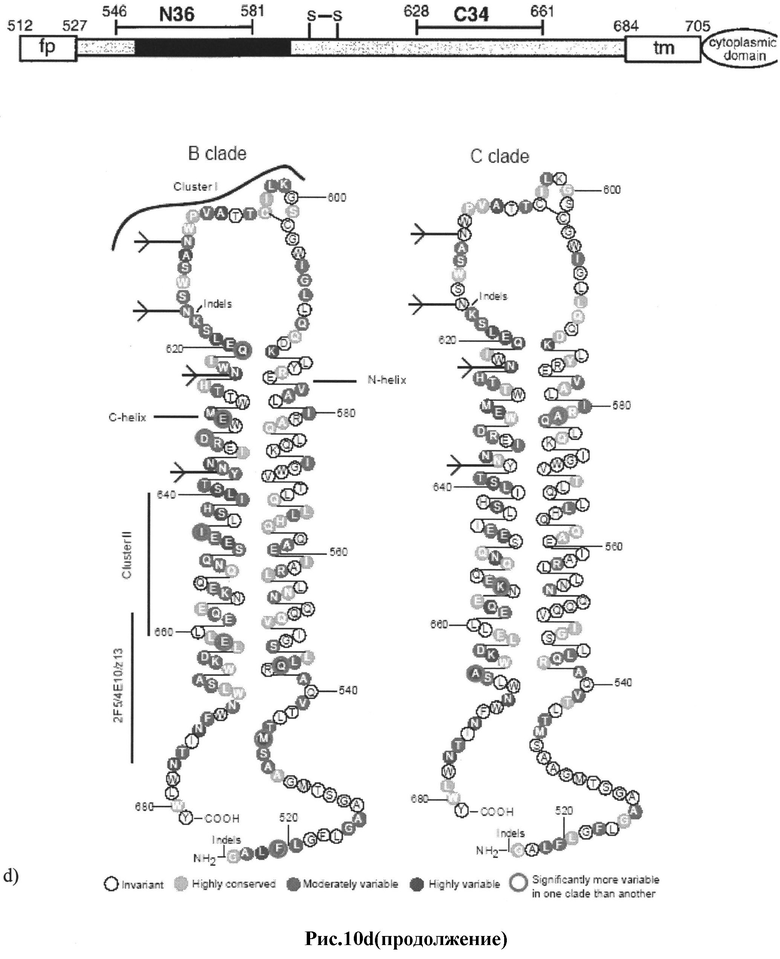
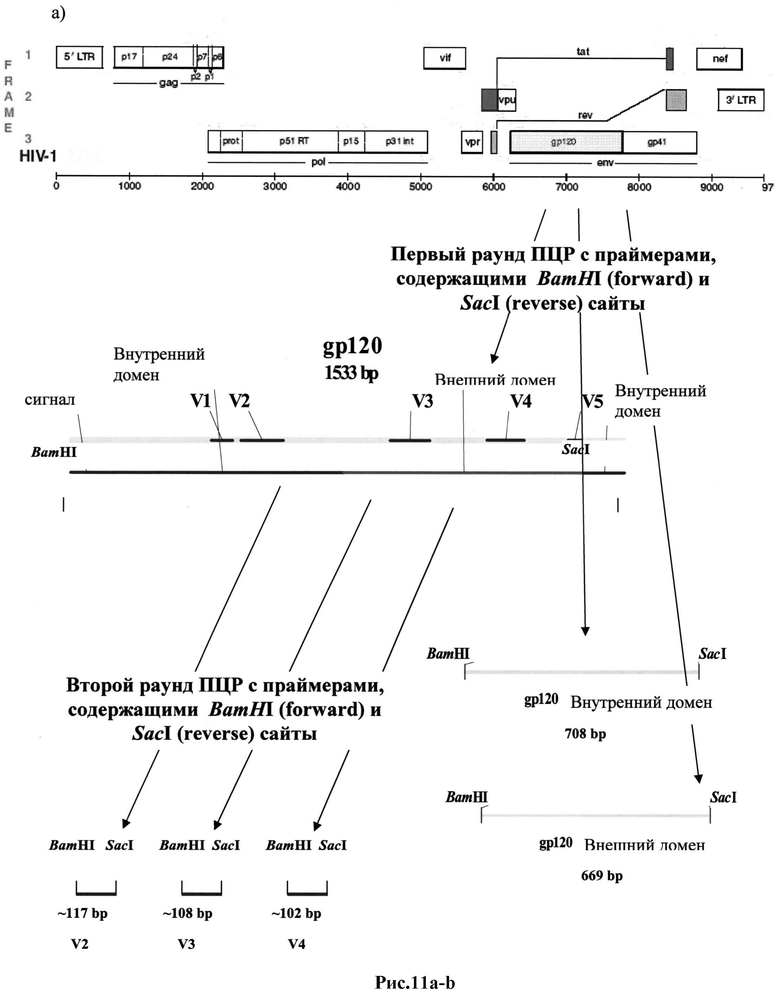
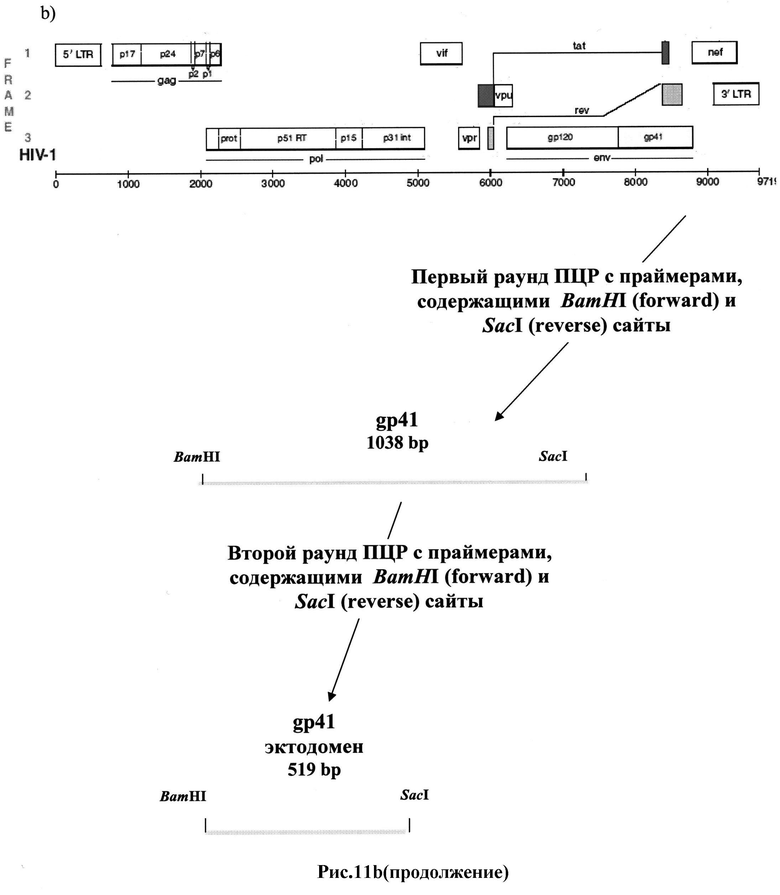
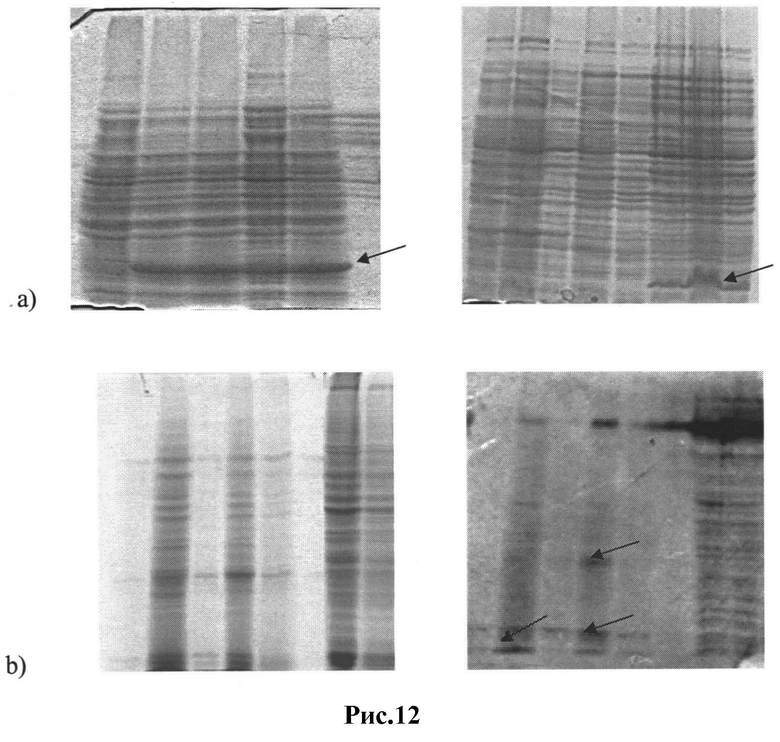
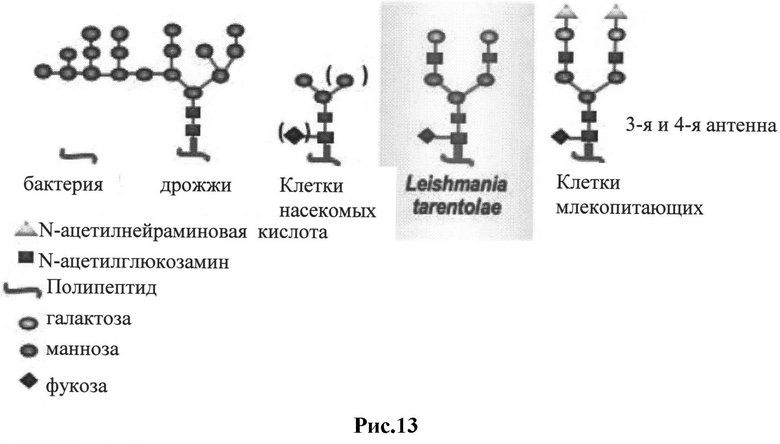
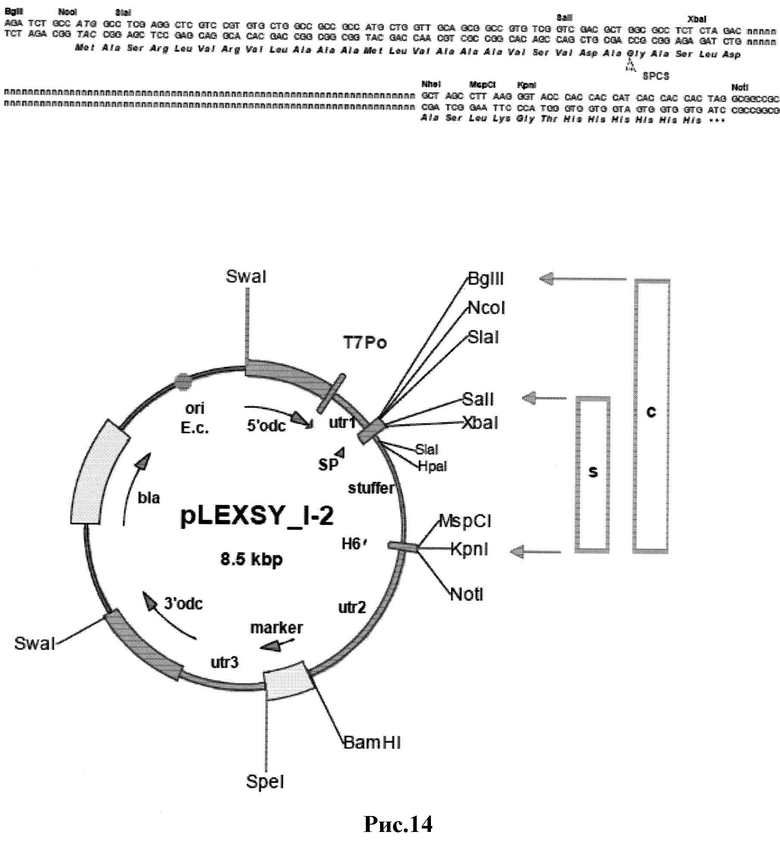
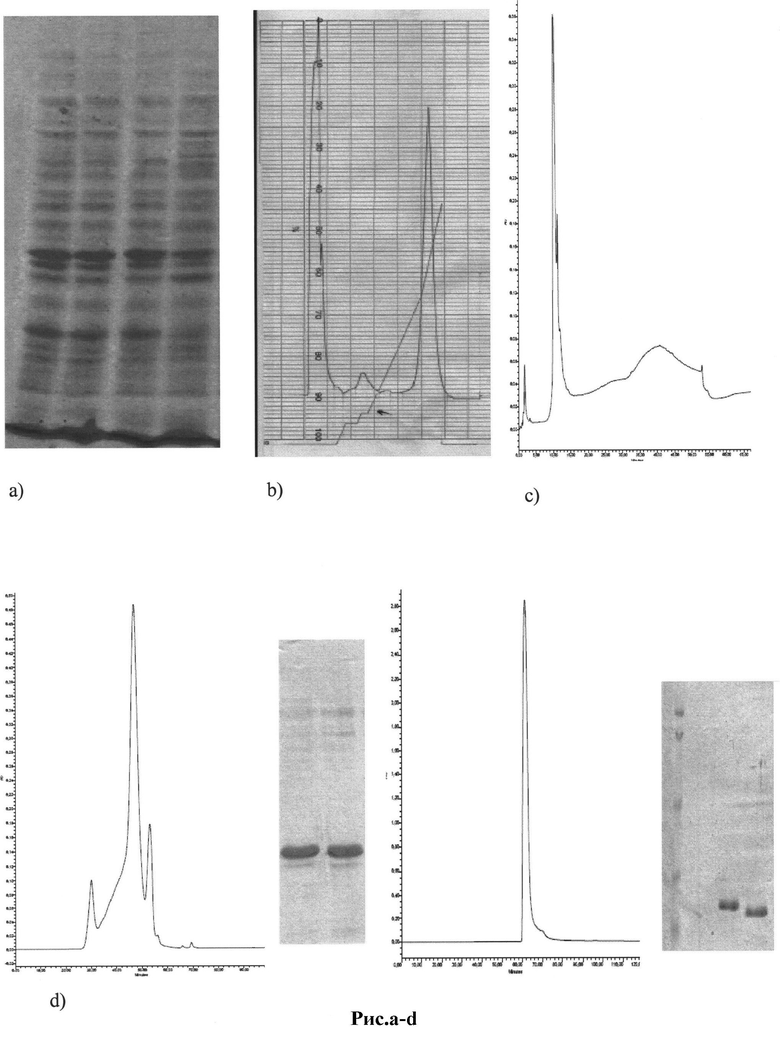
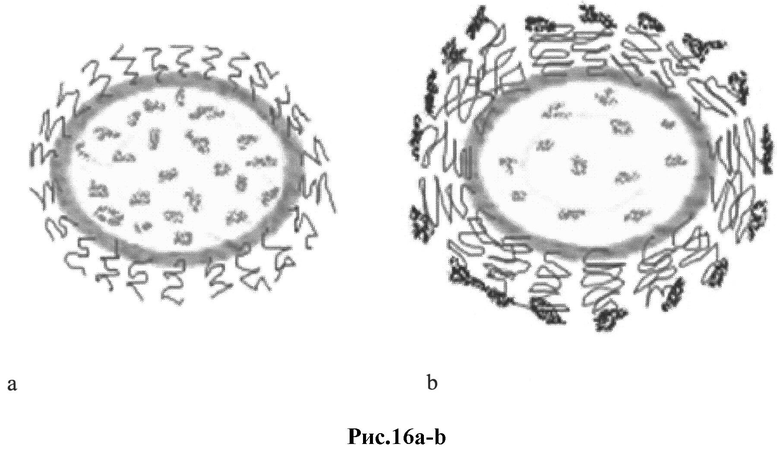
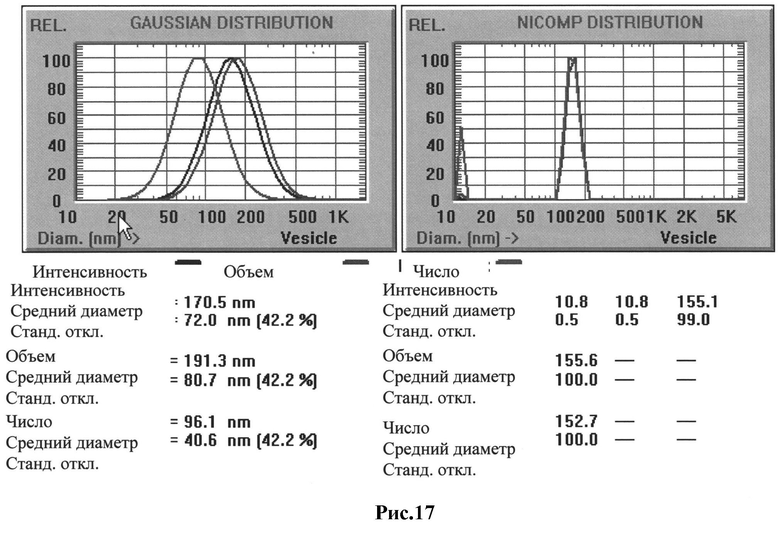
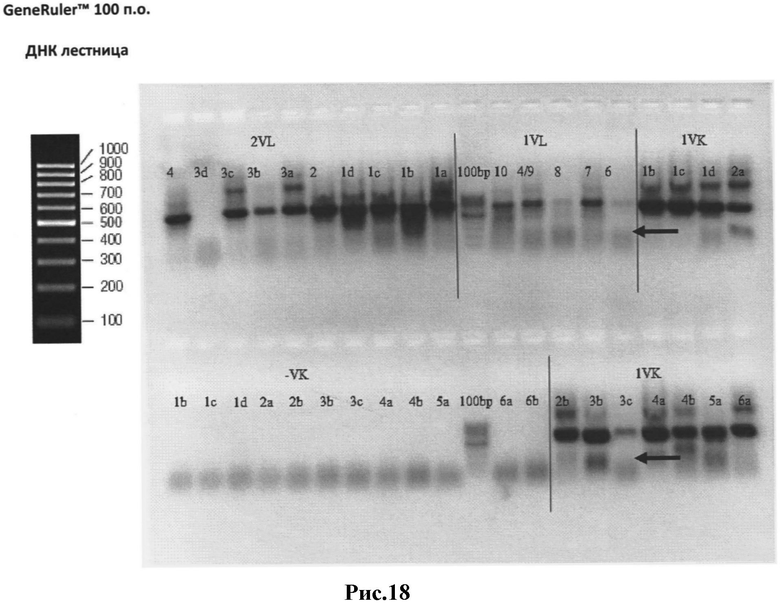
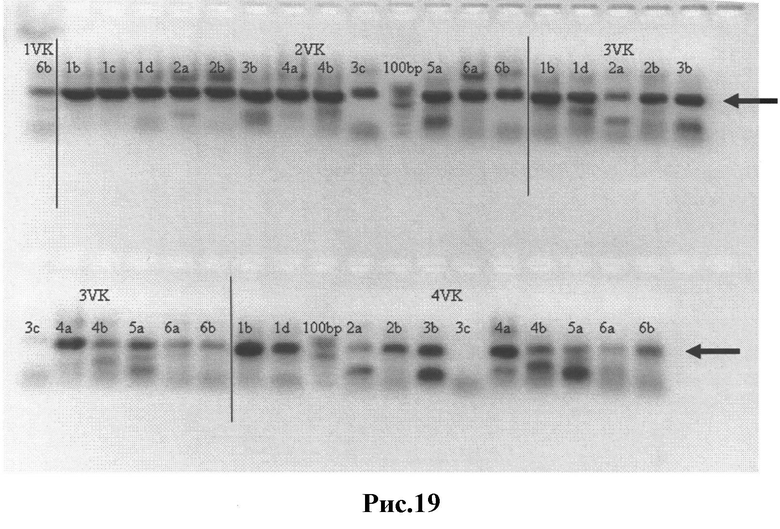
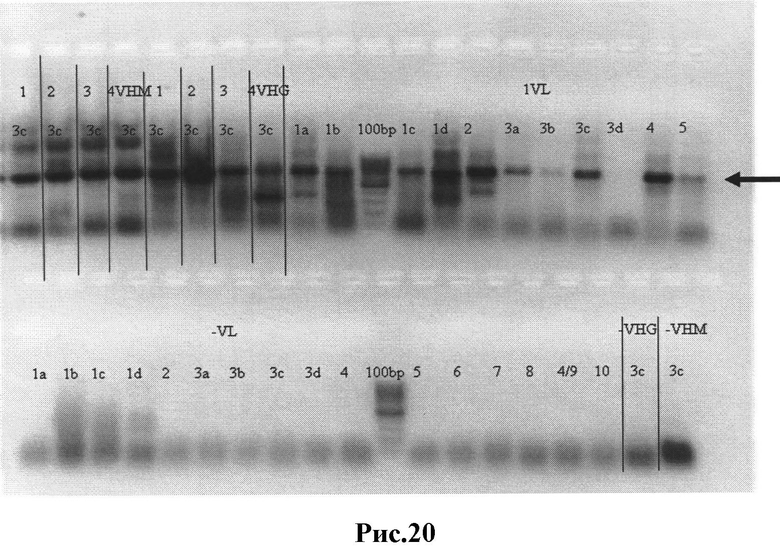
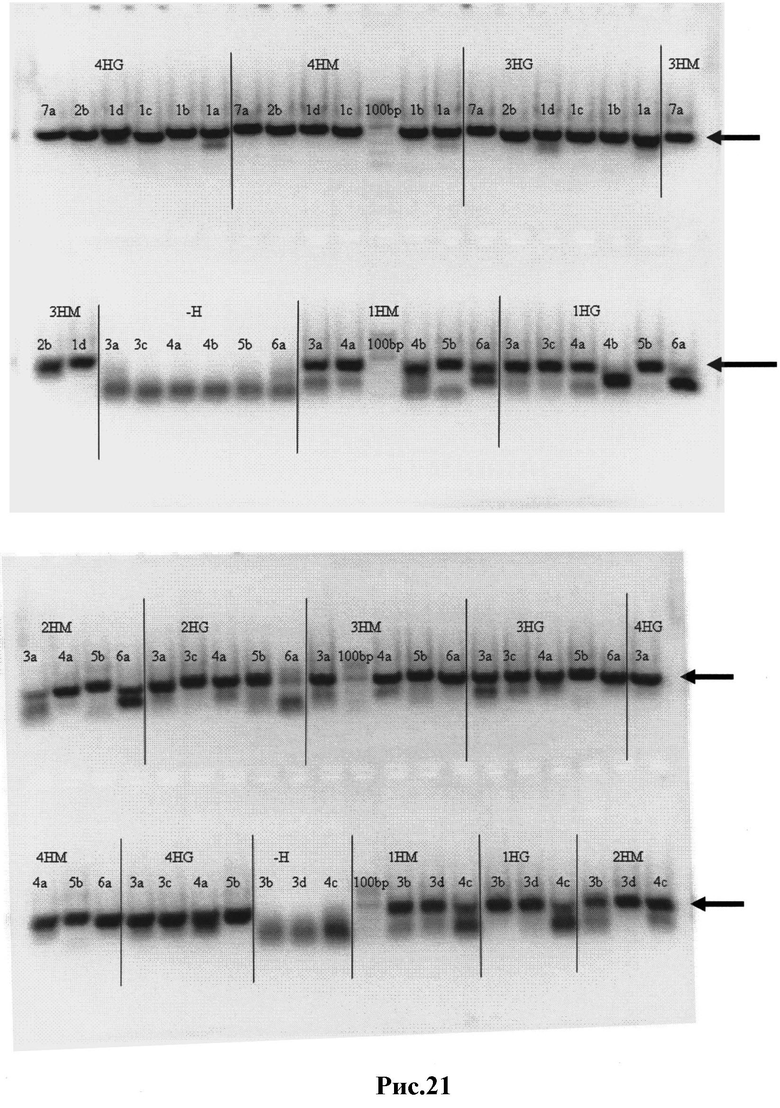
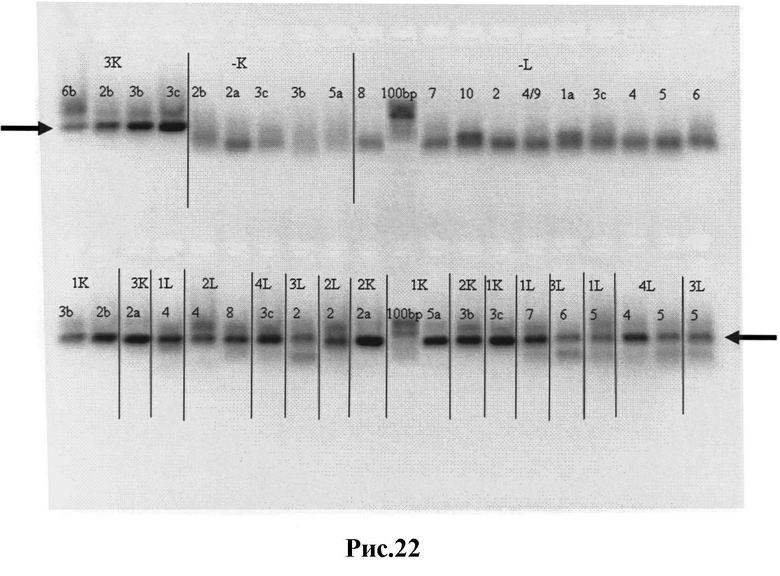
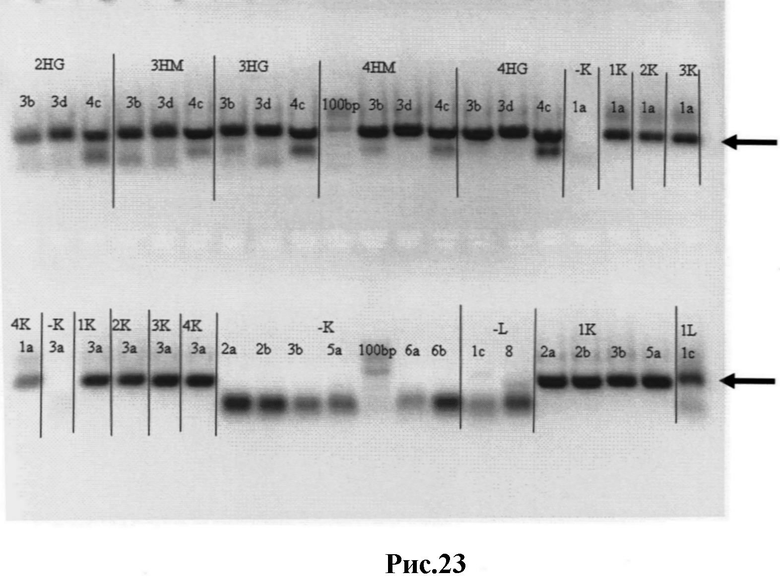
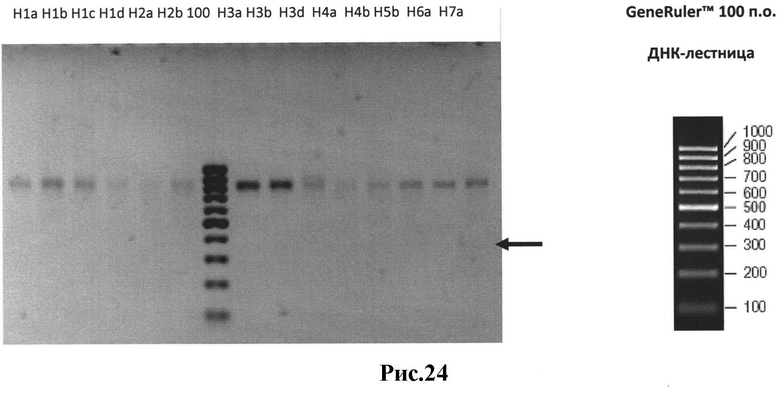
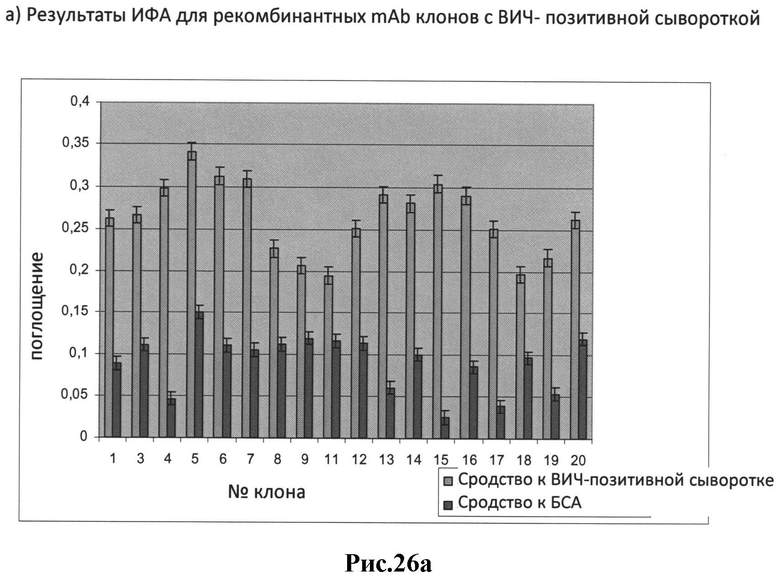
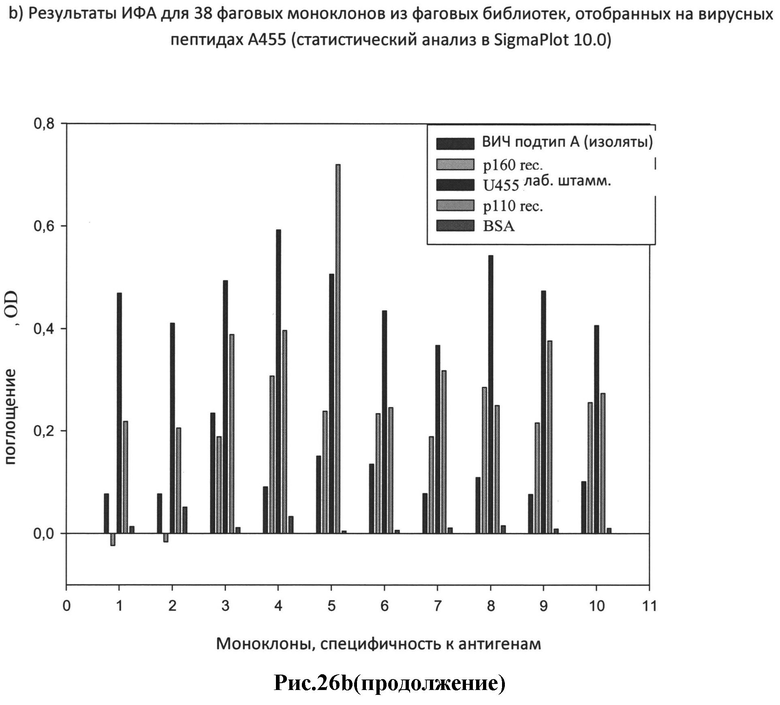
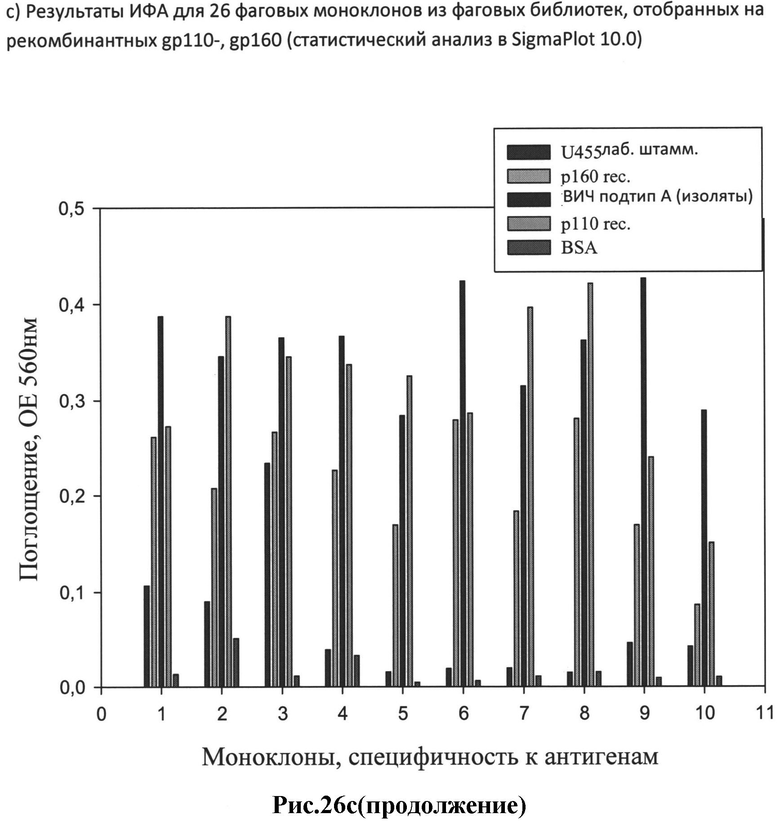
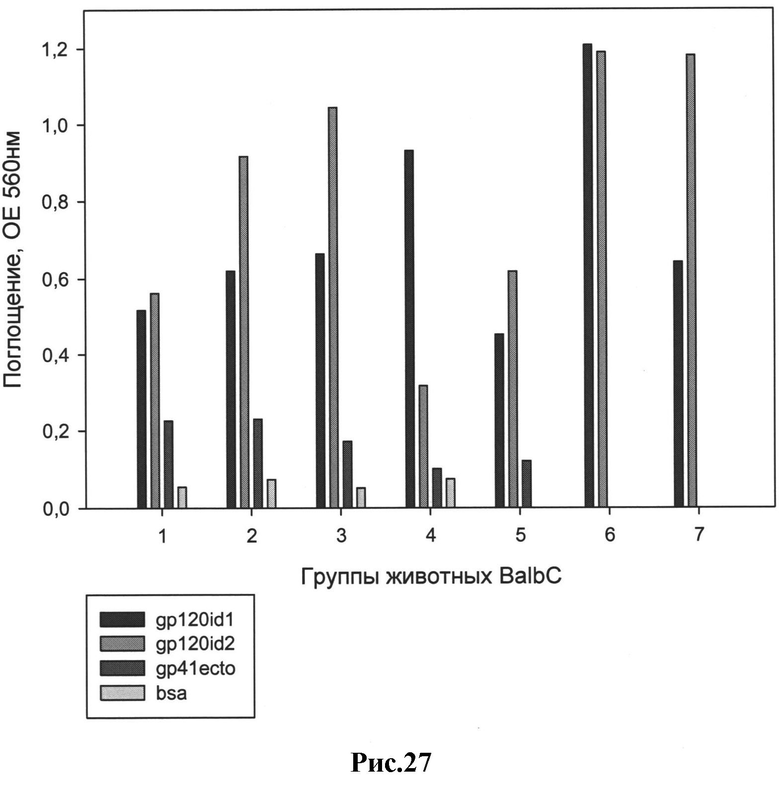
Настоящее изобретение относится к иммунологии. Предложен способ получения вакцины против ВИЧ-1 с использованием метода обратного панинга на фаг-презентированной библиотеке ВИЧ-1-специфических антител scFv, полученной на основе мРНК В-лимфоцитов больных, инфицированных ВИЧ-1, и обогащенной с помощью панинга на пептидах ВИЧ-1, и индуцируемых систем экспрессии рекомбинантных белков с эукариотическим гликозилированием. Также рассмотрена вакцина против ВИЧ-1 и ее применение для иммунизации неинфицированных людей против заражения и развития ВИЧ-инфекции. Вакцина ВИЧ-1, полученная в соответствии с данным изобретением, обеспечивает возникновение иммунного ответа в организме с образованием ВИЧ-1-специфических антител. 3 н. и 9 з.п. ф-лы, 27 ил., 9 табл., 4 пр.
1. Способ получения вакцины против ВИЧ-1, предотвращающей развитие инфекции посредством иммунизации неинфицированных людей, включающий этапы:
i) создание фагмидной библиотеки моноклональных антител, последовательности которых содержатся в мРНК В-лимфоцитов, полученных от группы из как минимум нескольких больных, инфицированных ВИЧ-1 субтипа А или В, по технологии фагового дисплея,
ii) обогащение фаг-презентированных scFv библиотек рекомбинантных антител, полученных на этапе i), на специфичность к белкам оболочки ВИЧ-1 с помощью панинга на нативных или рекомбинантных пептидах ВИЧ-1,
iii) увеличение числа копий биоматериала ВИЧ-1, содержащего пептиды, полипептиды и белки оболочки ВИЧ-1 (env ВИЧ-1), путем культивирования изолятов вируса in vitro на человеческих мононуклеарах периферической крови,
iv) сбор материала нативных пептидов/полипептидов/белков оболочки ВИЧ-1, полученных на этапе iii), с помощью метода обратного панинга с использованием ВИЧ-1 специфичной фаг-презентированной библиотеки, полученной на этапе ii), закрепленной на твердом носителе,
v) идентификация и количественная оценка пептидного материала, полученного на этапе iii), и его вариабельности с использованием масс-спектрометрического (PEAKS-LC-MS) анализа, секвенирование пептидов ВИЧ-1 и клонирование кодирующих их последовательностей, и
vi) использование результатов этапа v) для экспрессии гликозилированных рекомбинантных белков/пептидов оболочки ВИЧ-1 в индуцируемой системе экспрессии с эукариотическим гликозилированием (Leishmania tarentolae, дрожжи),
vii) очистка рекомбинантных пептидов env ВИЧ-1 и приготовление иммуноактивирующего состава для вакцинирования с использованием подходящего адъюванта в качестве системы доставки (стерически стабилизированных липосом, виросом).
2. Способ в соответствии с п.1 формулы изобретения, при котором носители ВИЧ, используемые для получения биоматериала, инфицированы ВИЧ одного или разных субтипов.
3. Способ в соответствии с любым из предшествующих пунктов, при котором ВИЧ-инфицированные для получения вирусного материала по п.1 i) могут быть как нелеченными, так и подвергавшимися лечению антиретровирусными препаратами.
4. Способ в соответствии с любым из предшествующих пунктов, при котором носитель презентирует множество различных ВИЧ-специфичных антител и/или фрагментов антител, отобранных как фагмидная библиотека.
5. Способ в соответствии с п.1, когда фагмидная библиотека создается на этапах:
a) получения фрагментов ДНК из нуклеиновых кислот, кодирующих вариабельные области легкой и тяжелой цепей иммуноглобулинов соответственно, экспрессирующихся в В-лимфоцитах, полученных от группы ВИЧ- инфицированных;
b) объединения фрагментов ДНК, кодирующих легкую и тяжелую цепь иммуноглобулинов, чтобы получить экспрессию полипептида, состоящего из вариабельных участков тяжелой и легкой цепей иммуноглобулинов соответственно, для того чтобы создать множество различных по специфичности вариантов;
c) клонирования объединенных фрагментов в фагмидном векторе и трансформации бактериального штамма для получения экспрессии на поверхности бактериофага, преимущественно когда амплификация выполняется любой из комбинаций праймеров, приведенных в таблицах 1-7, и когда фаг-презентированные scFv фрагменты специфичны на резистентные варианты ВИЧ-1 пациентов, подвергавшихся лечению ВААРТ или другими режимами антиретровирусной терапии.
6 Способ в соответствии с п.1, когда этап ii) дополнительно включает обогащение фагмидной библиотеки с презентированными scFv фрагментами антител в ходе процедуры панинга, связывания ВИЧ-специфических антител с рекомбинантными gp120-, gp41- и нативными полипептидами ВИЧ, выделенными из различных изолятов доноров.
7. Способ в соответствии с любым из предшествующих пунктов формулы изобретения, когда выделение и обогащение пептидов/полипептидов/белков env ВИЧ-1 осуществляется с использованием обратного панинга.
8. Способ в соответствии с любым из предшествующих пунктов формулы изобретения, когда жидкостная хроматография-масс-спектрометрия с подходящим программным обеспечением используется для количественного анализа, идентификации и/или секвенирования gp120 ВИЧ-1, его консервативных и вариабельных фрагментов.
9. Способ в соответствии с любым из предшествующих пунктов формулы изобретения, когда этап производства включает получение рекомбинантных пептидов/полипептидов/белков ВИЧ-1 в клетках подходящего организма-хозяина с типом гликозилирования, близким к эукариотическому.
10. Способ в соответствии с любым из предшествующих пунктов формулы изобретения, который включает приготовление состава профилактической вакцины против ВИЧ путем добавления/связывания с дополнительными иммуногенными стимуляторами, адъювантами или носителями, в частности стерически стабилизированными липосомами.
11. Вакцина против ВИЧ-1, которую получают в соответствии с любым из предшествующих пунктов формулы изобретения, тестированную in vivo на породе мышей с тяжелым комбинированным Т- и В- иммунодефицитом (SCID) на формирование иммунного ответа по человеческому типу, а также иммунизацию, способную предотвратить развитие ВИЧ-инфекции в человеческих лимфоцитах.
12. Применение вакцины против ВИЧ-1 в соответствии с п.1 формулы изобретения для иммунизации неинфицированных людей против заражения и развития ВИЧ-инфекции и СПИДа.
| PARREN P | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| BRETON M | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

