Область техники
Настоящее изобретение относится к новым соединениям миметиков обратного действия, к способу получения этих новых соединений и к их применению при лечении заболеваний, таких как острый миелолейкоз.
Уровень техники
Неупорядоченный поиск молекул, у которых возможна активность в качестве терапевтических агентов, проводился много лет и в результате привел к разработке ряда важных лекарственных препаратов. В последнее время были обнаружены непептидные соединения, которые наиболее близко имитируют вторичную структуру соединений обратного действия, найденных среди биологически активных белков или пептидов. Например, патент США № 5440013 и заявки PCT, опубликованные под № WO 94/03494, WO 01/00210 A1 и WO 01/16135 A2, все от Kahn, раскрывают конформационно ограниченные непептидные соединения, которые имитируют вторичную структуру соединений обратного действия. Кроме того, патенты США № 5929237 и 6013458, оба от автора Kahn, описывают конформационно ограниченные непептидные соединения, которые имитируют вторичную структуру участков соединений обратного действия из числа биологически активных пептидов и белков. Синтез и идентификация конформационно ограниченных миметиков обратного действия и применение их в случае заболеваний хорошо обзорно описаны автором Obrecht (Advances in Med. Chem., 4, 1-68, 1999).
Наряду со значительными достижениями в области синтеза и идентификации конформационно ограниченных миметиков обратного действия происходит развитие технологии для обеспечения синтетического получения и скрининга представителей библиотеки малых молекул, которые имитируют вторичную структуру пептидов с целью выявления биологически активных представителей из библиотеки. Таким образом, были предприняты попытки поиска конформационно ограниченных соединений и соединений с высокой биологической активностью, которые имитируют вторичную структуру участков соединений обратного действия из числа биологически активных пептидов и белков. Например, миметики обратного действия, способы получения этих соединений и их биологическая активность раскрыты в заявках PCT, опубликованных под № WO 04/093828 A2, WO 05/116032 A2 и WO 07/139346 A1.
Несмотря на то, что производится большое количество миметиков обратного действия, не так уж много соединений, как установлено, имеют высокую биоактивность. Таким образом, продолжаются исследования по получению соединений, пригодных для лечения заболеваний, таких как рак.
В частности, исследования сфокусированы на разработке соединений, которые сильно блокируют сигнальный путь Wnt, эффективно подавляя рост раковых клеток при остром миелолейкозе (AML), при котором, как известно, активируется сигнальный путь Wnt.
Также существует потребность в способах крупномасштабного производства соединений с высокой биологической активностью в том случае, когда они найдены.
Сущность изобретения
Вследствие этого задачей настоящего изобретения является предоставление новых биологически активных соединений, применения их в качестве терапевтических средств или пролекарств при раке, в частности при остром миелолейкозе, и способа крупномасштабного производства этих соединений.
Техническое решение
В соответствии с данным аспектом настоящее изобретение предоставляет новые соединения, представленные нижеприведенной химической формулой I:
Химическая формула I
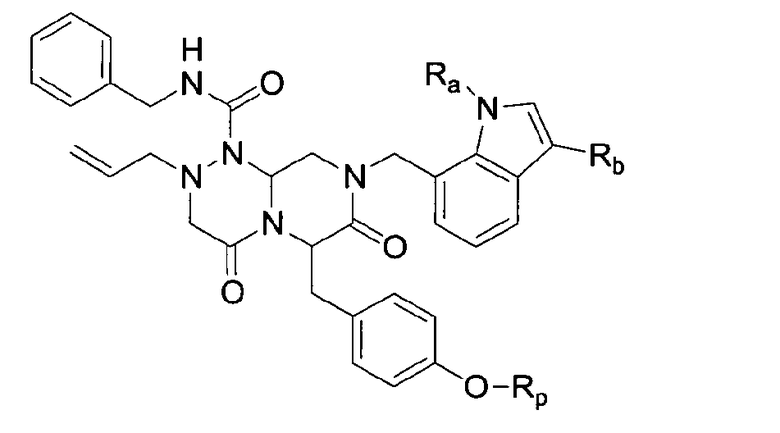
в которой
Ra представляет собой C1-C6 алкильную группу, C2-C6 алкенил или C2-C6 алкинильную группу;
Rb представляет собой арильную группу, замещенную арильную группу, или -С(=О)Re, в которой Re представляет собой С1-С6 алкильную группу, С2-С6 алкенильную группу или С2-С6 алкинильную группу;
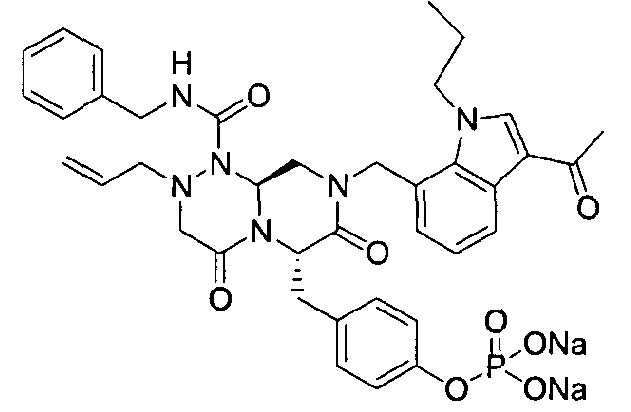
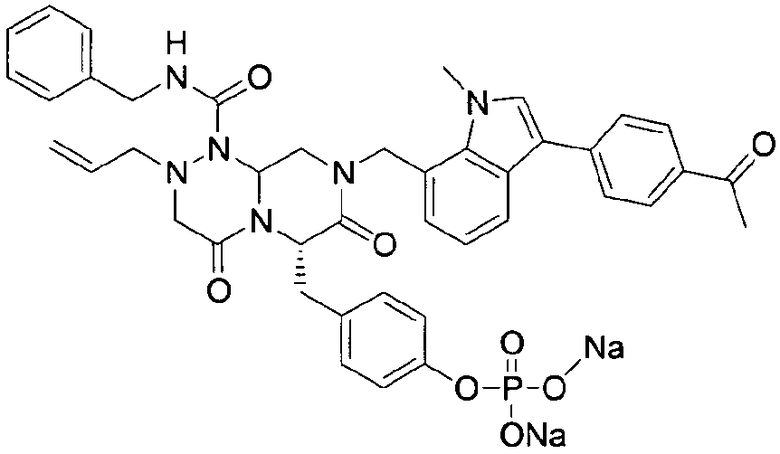
Rp представляет собой -Н, -РО3Н2, -НРО3 -Na+, -РО3 2-Na2 +, -РО3 2-К2 +, -РО3 2-Mg2+, -РО3 2-Са2+,

Замещенный арил может быть ацилзамещенным арилом (как определено в этом документе).
В одном примере осуществления изобретения в химической формуле I Ra представляет собой С1-С6 алкильную группу или C2-C6 алкенильную группу; Rb представляет собой -С(=О)Re, в которой Re представляет собой С1-С6 алкил; и Rp представляет собой -H, -PO3H2, -HPO3 -Na+ или -PO3 2-Na2 +.
В другом примере осуществления изобретения в химической формуле I Ra представляет собой метил; Rb представляет собой -С(=О)Re, в которой Re представляет собой С1-С6 алкил; и Rp представляет собой -H.
В еще одном примере осуществления изобретения в химической формуле I Ra представляет собой метил; Rb представляет собой -С(=О)Re, в которой Re представляет собой С1-С6 алкил; и Rp представляет собой -PO3H2, -HPO3 -Na+ или -PO3 2-Na2 +.
В одном аспекте настоящее раскрытие изобретения предоставляет фармацевтическую композицию, включающую соединение, предоставленное в настоящем описании, и фармацевтически приемлемый эксципиент.
В другом аспекте настоящее раскрытие изобретения предоставляет способ лечения острого миелолейкоза (AML), включающий введение пациенту, страдающему AML, эффективного количества соединения или композиции, предоставленных в настоящем описании. В некоторых примерах осуществления изобретения способ включает инъецирование эффективного количества соединения или композиции пациенту, страдающему AML.
В другом аспекте настоящее раскрытие изобретения предоставляет способ получения соединения, предоставленного в настоящем описании, включающий нижеприведенные последовательные стадии: (a) введение ацильной группы в индол-7-карбальдегид посредством ацилирования по Фриделю-Крафтсу с получением 3-ацилиндол-7-карбальдегида; (b) введение алкильной группы и аминоацетальной группы в 3-ацилиндол-7-карбальдегид с получением 1-алкил-3-ацилиндольного производного; (c) стереоселективное амидирование 1-алкил-3-ацилиндольного производного с помощью трет-бутилового эфира бензилоксикарбонилтирозина (Cbz-тирозин-OtBu) и 2-(1-аллил-4-бензилсемикарбазид)уксусной кислоты с получением реакционного промежуточного соединения; (d) циклизация реакционного промежуточного соединения в присутствии муравьиной кислоты с получением циклического промежуточного соединения; и (e) фосфорилирование циклического промежуточного соединения с получением соединения химической формулы (I). В некоторых примерах осуществления изобретения 2-(1-аллил-4-бензилсемикарбазид)уксусную кислоту синтезируют посредством нижеприведенных последовательных стадий: (1) добавление ТЭА (триэтиламина) к раствору этилгидразинацетата с получением реакционного раствора; (2) добавление аллилбромида к реакционному раствору и (3) добавление бензилизоцианата. В некоторых дополнительных примерах осуществления изобретения аллилбромид и бензилизоцианат добавляли путем прикапывания.
В связанном аспекте настоящее раскрытие изобретения предоставляет способ получения соединения химической формулы (I), включающий (a) превращение индол-7-карбальдегида в 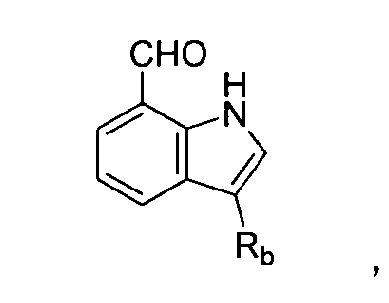 где Rb представляет собой арильную группу, замещенную арильную группу или -C(=O)Re, где Re представляет собой С1-С6 алкильную группу, C2-C6 алкенильную группу или С2-С6 алкинильную группу; (b) превращение
где Rb представляет собой арильную группу, замещенную арильную группу или -C(=O)Re, где Re представляет собой С1-С6 алкильную группу, C2-C6 алкенильную группу или С2-С6 алкинильную группу; (b) превращение 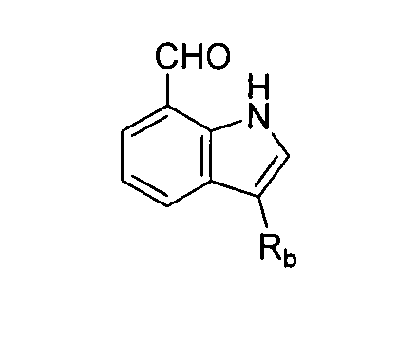 в
в  , где Ra представляет собой C1-C6 алкильную группу, C2-C6 алкенил или C2-C6 алкинильную группу; (c) стереоселективное амидирование
, где Ra представляет собой C1-C6 алкильную группу, C2-C6 алкенил или C2-C6 алкинильную группу; (c) стереоселективное амидирование  в присутствии Cbz-тирозин-OtBu и 2-(1-аллил-4-бензилсемикарбазид)уксусной кислоты с получением
в присутствии Cbz-тирозин-OtBu и 2-(1-аллил-4-бензилсемикарбазид)уксусной кислоты с получением 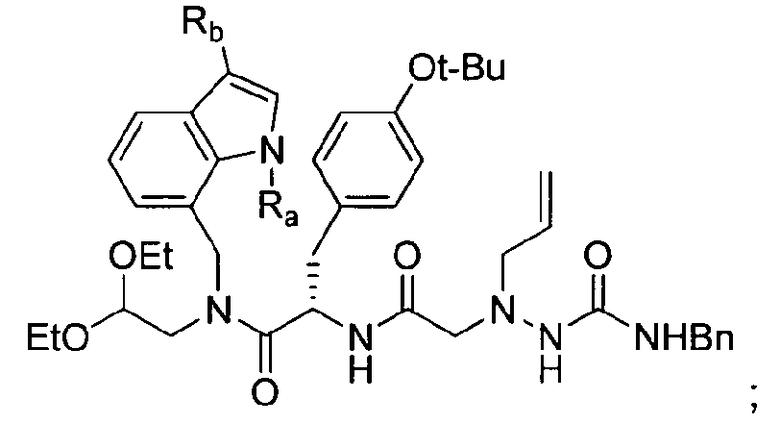 (d) циклизация
(d) циклизация  в присутствии муравьиной кислоты с получением
в присутствии муравьиной кислоты с получением  ; и (e) превращение
; и (e) превращение  , где Rp представляет собой -PO3H2, -HPO3 -Na+, -PO3 2-Na2 +, -PO3 2-K2 +, -PO3 2-Mg2+, -PO3 2-Са2+. В некоторых примерах осуществления изобретения Ra представляет собой метил, Rb представляет собой -C(=O)Re и Re представляет собой метил или циклопропил.
, где Rp представляет собой -PO3H2, -HPO3 -Na+, -PO3 2-Na2 +, -PO3 2-K2 +, -PO3 2-Mg2+, -PO3 2-Са2+. В некоторых примерах осуществления изобретения Ra представляет собой метил, Rb представляет собой -C(=O)Re и Re представляет собой метил или циклопропил.
Полезный эффект
Новые миметики обратного действия в соответствии с настоящим изобретением, как отмечено, эффективно ингибируют in vitro рост раковых клеток AML. Также для них отмечено при тестировании на модельных мышах с острым миелолейкозом эффективное ингибирование роста опухолей.
Не подводя теоретическую базу, считают, что в случае, когда уходящая группа (Rp), которую также обозначают как функциональную группу пролекарства, удаляется, тогда соединения химической формулы I превращаются в активные формы. Однако эти активные формы трудно приготовить в виде водного раствора из-за их плохой растворимости в воде. Пролекарственные формы соединений химической формулы I в соответствии с настоящим изобретением обладают хорошей растворимостью и высокой стабильностью, и из них легко приготовить лекарственный препарат для инъекции.
Тесты на животных показали, что соединения настоящего изобретения обладают исключительной фармацевтической эффективностью. Создается впечатление, что это обусловлено быстрым превращением соединений в их активные формы сразу же после внутривенной инъекции и, в этой связи, возрастанием на начальном этапе концентрации лекарственного препарата. Таким образом, скорость, с которой пролекарственные соединения изменяются в активные формы, оказывает влияние на их медицинскую эффективность, поэтому важно подбирать пролекарственные функциональные группы, обеспечивающие оптимальные эффекты.
В предпочтительном примере осуществления изобретения пролекарственные функциональные группы представлены в форме фосфата, потому что фосфатные пролекарства быстрее превращаются in vivo в активные формы в сравнении с другими пролекарствами, имеющими иные функциональные группы.
В том случае, когда пролекарственные функциональные группы представлены в форме натриевых солей, они легко получаются и имеют высокую растворимость в воде. Кроме того, они являются высокостабильными в процессе хранения при комнатной температуре.
Обычно подходящая для применения инъекционная композиция, как известно, имеет значение pH в диапазоне от 4 до 9 и, предпочтительно, имеет значение pH, близкое к этому показателю для человеческой крови, равному 7,4. Композиция, которая является сильно кислотной или сильно щелочной, не является предпочтительной в качестве композиции для инъекции. В случае фосфатной функциональной группы, итоговые пролекарства настоящего изобретения могут быть в форме мононатриевого или динатриевого фосфата, в зависимости от количества гидроксида натрия. Эти соединения предпочтительны для получения композиции, имеющей значения pH, подходящие для инъекции.
Кроме того, способ получения в соответствии с настоящим изобретением позволяет получать не только соединения химической формулы I, но также и их миметики обратного действия в промышленном масштабе.
Описание чертежей
Фиг.1 представляет собой графическое изображение корреляции между изменениями pH и потенциалом, сопровождающим проведение конечной стадии способа получения соединения, в которой 0,5н NaOH добавляют по каплям к 4-(((6S,9aS)-1-(бензилкарбамоил)-8-((3-ацетил-1-метил-1H-индол-7-ил)метил)-2-аллилоктагидро-4,7-диоксо-1H-пиразино[2,1-c][1,2,4]триазин-6-ил)метил)фенилдигидрофосфат (Соединение Р2). На этом графике на горизонтальной оси представлены добавленные количества NaOH. Первая и вторая точки перегиба кривой соответствуют началу получения мононатриевого и динатриевого производных, соответственно.
Лучший вариант осуществления изобретения
Таким образом, один из примеров осуществления изобретения предоставляет новые миметики обратного действия, представленные нижеприведенной химической формулой 1, которые применимы в качестве терапевтических средств при раке, а именно при остром миелолейкозе.
Химическая формула I

в которой
Rp может представлять собой какую-либо группу из обычных функциональных групп, приемлемых для пролекарств. Примеры функциональных групп включают фосфат, карбокси и C1-C6 алкиламино, и ациламино, такие как -PO3H2, -HPO3 -Na+, -PO3 2-Na2 +, -PO3 2-K2 +, -PO3 2-Mg2+, -PO3 2-Са2+,  , или
, или
Предпочтительно, когда Rp представляет собой фосфатную функциональную группу  , в которой Rc и Rd представляют собой, независимо друг от друга, H, Na, Mg, Ca или K. Предпочтительно, когда и Rc, и Rd представляют собой H или Na или когда один из них представляет собой Na, в то время как другой представляет собой H.
, в которой Rc и Rd представляют собой, независимо друг от друга, H, Na, Mg, Ca или K. Предпочтительно, когда и Rc, и Rd представляют собой H или Na или когда один из них представляет собой Na, в то время как другой представляет собой H.
Rp также может быть -H, химической структурой в активной форме получающейся из соответствующего пролекарства, учитывая, что пролекарственная функциональная группа удаляется.
Ra представляет собой алкильную группу, алкенильную группу или алкинильную группу, предпочтительно, C1-C6 алкильную группу, C2-C6 алкенильную или C2-C6 алкинильную группу, и более предпочтительно, C1-C6 алкильную группу.
Rb представляет собой арильную группу, замещенную арильную группу или С(=О)Re, в которой Re представляет собой С1-С6 алкильную группу, C2-C6 алкенильную группу или C2-C6 алкинил, и замещенная арильная группа представляет собой ацилзамещенную арильную группу и, предпочтительно, арилзамещенный фенил.
Пролекарства превращаются в активные формы в организме. В том случае когда пролекарства имеют фосфатную функциональную группу в качестве уходящей группы, тогда -PO3RcRd группа быстро расщепляется с помощью фосфатазы и пролекарства превращаются в их активные формы. В это же время Rp превращается в -H (химическая структура активной формы, с учетом того, что пролекарственная функциональная группа выбыла из структуры).
Использованный в этом документе термин "алкил" или "алкильная группа" предназначается, чтобы охватить линейный, разветвленный или циклический углеводородный радикал, включающий углеродные и водородные атомы, в котором углеродные атомы соединены вместе одинарными связями. В одном примере осуществления изобретения алкил содержит до 20 углеродных атомов. В предпочтительных примерах осуществления изобретения алкил может включать от одного до шести углеродных атомов и представляется как "C1-C6 алкил". Алкил присоединен к остатку молекулы посредством одинарной связи. Примеры алкилов включают, не ограничиваясь только приведенными, метил, этил, н-пропил, 1-метилэтил(изопропил), н-бутил, н-пентил, н-гексил, 1,1-диметилэтил(трет-бутил), 2,2-диметилпропил(неопентил), 3-метилгексил, 2-метилгексил и им подобные. Алкил также может быть моноциклическим или бициклическим углеводородным кольцевым радикалом, который может включать конденсированные или связанные кольцевые системы. Циклический алкил также обозначают как "циклоалкил". В некоторых примерах осуществления изобретения циклоалкил может включать от трех до шести углеродных атомов и обозначается как "C3-6циклоалкил". Примеры моноциклических циклоалкильных радикалов включают, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
"Алкенил" или "алкенильная группа" относится к линейному, разветвленному или циклическому углеводородному радикалу, включающему углеродные и водородные атомы, в котором по меньшей мере два углеродных атома связаны двойной связью. В одном из примеров осуществления изобретения алкенил содержит до 20 углеродных атомов. В предпочтительных примерах осуществления изобретения алкенил может включать от двух до шести углеродных атомов и обозначается как "C2-C6 алкенил". Алкенил присоединен к остатку молекулы одинарной или двойной связью. Примеры алкенилов включают, не ограничиваясь только приведенными, этенил, аллил, бутенил и им подобные.
"Алкинил" или "алкинильная группа" относится к линейному, разветвленному или циклическому углеводородному радикалу, включающему углеродные и водородные атомы, в котором по меньшей мере два углеродных атома связаны тройной связью. В одном из примеров осуществления изобретения алкинил содержит до 20 атомов углерода. В предпочтительных примерах осуществления изобретения алкинил может включать от двух до шести углеродных атомов и обозначается как "C2-С6 алкинил". Алкинил присоединен к остатку молекулы одинарной связью. Примеры алкинилов включают, не ограничиваясь только приведенными, этинил, 1-пропинил или 2-пропинил и им подобные.
Если специально не оговорено иное в описании изобретения, то подразумевается, что термин "алкил" включает алкил, имеющий только углеродные и водородные атомы, также как и "замещенный алкил", который обозначает алкильный радикал, в котором один или несколько водородных атомов замещены одним или несколькими заместителями, независимо выбираемыми из ацила, алкокси, арила, циано, циклоалкила, галогена, гидроксила, нитро, -OC(O)-R11, -N(R11)2, -C(O)OR11, -C(O)N(R11)2, -N(R11)С(O)OR11, -N(R11)С(O)R11, -N(R11)S(O)tR11 (где t представляет собой 1 или 2), -S(O)tOR11 (где t представляет собой 1 или 2), -S(O)pR11 (где p представляет собой 0, 1 или 2), и -S(O)tN(R11)2 (где t представляет 1 или 2), в которых каждый R11 независимо представляет собой водород, алкил, арил, как определено в настоящем описании. Термины "алкенил" и "алкинил" аналогичным образом определяются как включающие "замещенный алкенил" и "замещенный алкинил", соответственно.
"Алкокси" относится к радикалу, представленному формулой алкил-O-, в которой алкил представляет собой такой, как определено ранее. Алкильная часть может быть дополнительно замещена одним или несколькими галогенами. Алкокси также может быть обозначен с указанием числа углеродных атомов в алкильной группе, например, C1-6алкокси или C1-3алкокси.
"Ацил" относится к радикалу, представленному формулой R12C(=O)-, в которой R12 представляет собой алкил или арил, как определено в этом документе. Алкил или арил могут быть, необязательно, замещены такими заместителями, которые описаны для алкильной или арильной группы, соответственно. Примеры ацильных групп включают, не ограничиваясь только приведенными, метилацил (т.e. ацетил), фенилацил, циклопропилацил и т.п.
"Арил" относится к радикалу, полученному из ароматической моноциклической или бициклической кольцевой системы путем удаления водородного атома от кольцевого углеродного атома. Ароматическая моноциклическая или бициклическая углеводородная кольцевая система включает от шести до двенадцати углеродных атомов (т.e. C6-12арил), в которой по меньшей мере одно из колец в кольцевой системе представляет собой полностью ненасыщенное, т.e. оно содержит циклическую, делокализованную (4n+2) π-электронную систему в соответствии с теорией Хюккеля (Hϋckel). Примеры арильных радикалов включают, но не ограничиваются только приведенными, фенил и нафтил. Если специально не оговорено иное в описании изобретения, то подразумевается, что термин "арил" включает как арил, так и "замещенный арил", который относится к арильному радикалу, в котором один или несколько водородных атомов замещены одним или несколькими заместителями, независимо выбранными из ацила, алкокси, арила, циано, циклоалкила, галогена, гидроксила, нитро, -OC(O)-R11, -N(R11)2, -C(O)OR11, -C(O)N(R11)2, -N(R11)C(O)OR11, -N(R11)C(O)R11, -N(R11)S(O)tR11 (где t представляет собой 1 или 2), -S(O)tOR11 (где t представляет собой 1 или 2), -S(O)pR11 (где p представляет собой 0, 1 или 2) и -S(O)tN(R11)2 (где t представляет собой 1 или 2), в которых каждый R11 независимо представляет собой водород, алкил, арил, как определено в этом документе.
"Галоген" обозначает фтор, хлор, бром и йод.
Активная форма соединений не подходит для внутривенной инъекции из-за ее низкой растворимости в водной среде (например, солевом растворе или воде). Пролекарственные формы, описанные в настоящем описании, подходят для внутривенной инъекции благодаря их улучшенной растворимости в водной среде. В предпочтительном примере осуществления изобретения используют фосфатное пролекарство, и в том случае, когда один или два атома Na введены в фосфатный фрагмент, тогда растворимость дополнительно улучшена. Для введения атомов Na добавляют (например, прикапывают) гидроксид натрия к фосфатному соединению до определенного значения pH, чтобы осуществить замещение одного или двух протонов в фосфатном фрагменте ионами натрия.
Таким образом, следующий пример осуществления изобретения предоставляет фармацевтическую композицию, включающую соединение химической формулы (I) и фармацевтически приемлемый эксципиент. Соединения или композиции настоящего изобретения могут использоваться при лечении AML, что подробно описано ниже.
Фармацевтическую композицию настоящего изобретения составляют таким образом, чтобы была согласованность с ее предполагаемым способом введения. Примеры способов введения включают парентеральное, например внутривенное, внутрикожное, подкожное, пероральное (например, ингаляция), трансдермальное (топическое), трансмукозальное и ректальное введение. Растворы или суспензии, применяемые для парентерального, внутрикожного или подкожного нанесения, могут включать нижеперечисленные компоненты: стерильный разбавитель, такой как вода для инъекций, солевой раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные средства, такие как бензиловый спирт или метиловые эфиры пара-оксибензойной кислоты; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферные растворы, такие как ацетатные, цитратные или фосфатные и агенты для регулирования тоничности, такие как хлорид натрия или декстроза.
В предпочтительном примере осуществления изобретения фармацевтически приемлемый эксципиент подходит для применения при внутривенном введении, таком как внутривенная инъекция или инфузия. Удобные носители для внутривенного введения включают физиологический раствор, бактериостатическую воду, кремофор Cremophor EL™ (BASF, Parsippany, NJ) или солевой фосфатный буфер (PBS). Во всех случаях композиция должна быть стерильной и текучей в такой степени, чтобы существовала возможность простого введения через шприц. Она должна быть стабильной в условиях получения и хранения и должна быть защищена от инфицирующего воздействия со стороны микроорганизмов, таких как бактерии и микроскопические грибы.
В других примерах осуществления изобретения предоставляются пероральные композиции, которые обычно включают инертный разбавитель или пригодный к употреблению в пищу носитель. Такие композиции могут быть заключены в желатиновые капсулы или запрессованы в таблетки. Для целей перорального терапевтического введения соединение, описанное в этом документе, может быть объединено с эксципиентами и может применяться в форме таблеток, пастилок или капсул. Пероральные композиции также могут быть приготовлены с использованием жидкого носителя, чтобы использовать их в качестве жидкости для полоскания ротовой полости, в которых соединение в жидком носителе применяется перорально и путем полоскания рта, и как отхаркивающее средство или средство с возможностью проглатывания. Фармацевтически совместимые связывающие агенты и/или вещества-адъюванты могут быть включены в композицию в качестве ее составной части. Таблетки, пилюли, капсулы, пастилки и им подобные могут содержать любой из нижеприведенных ингредиентов или соединения аналогичной природы: связующее вещество, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; эксципиент, такой как крахмал или лактоза, вещество для улучшения распадаемости таблеток, такое как альгиновая кислота, Primogel или кукурузный крахмал; скользящее вещество, такое как стеарат магния или Sterotes; регулятор сыпучести, такой как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или ароматизирующее вещество, такое как мятное масло, метилсалицилат или органический ароматизатор.
В соответствии с другим аспектом настоящее раскрытие изобретения предоставляет способ лечения заболевания, в частности рака, а именно, острого миелолейкоза (AML), включающий введение пациенту, имеющему раковое заболевание (например, пациенту с AML), эффективного количества соединения химической формулы (I) или фармацевтической композиции, включающей это соединение. Пример 23, представленный ниже, демонстрирует, что типичные соединения настоящего раскрытия изобретения эффективны при лечении AML на животной модели.
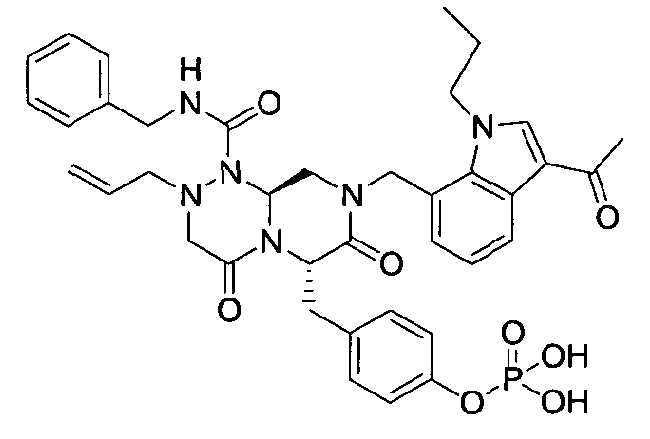
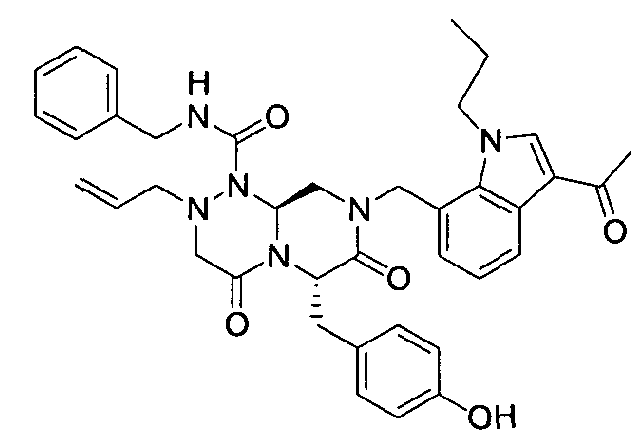
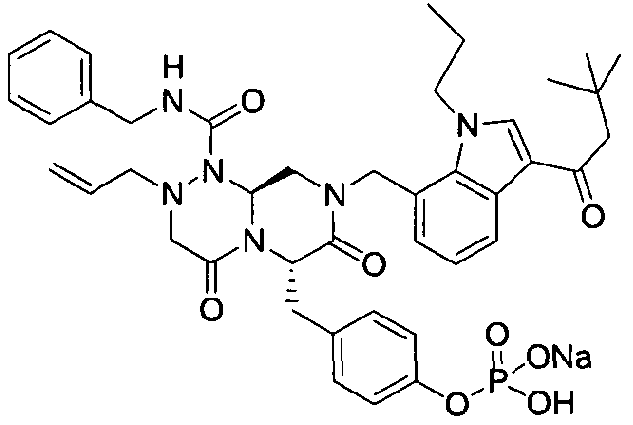
Примеры соединений химической формулы (I) приведены в нижерасположенной таблице 1. Из-за того, что четыре соединения в таблице 1 различаются только во фрагменте Rp, который представляет собой H или фосфатную функциональную группу, они имеют одинаковые данные ЯМР-спектра, что в общем виде и приведено таким образом в таблице 1 (Rp не наблюдали в 1H ЯМР-спектре из-за того, что он замещен дейтерием).
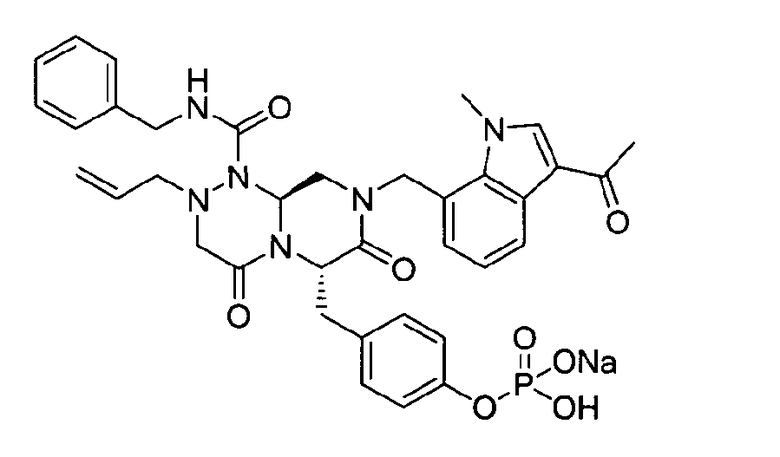
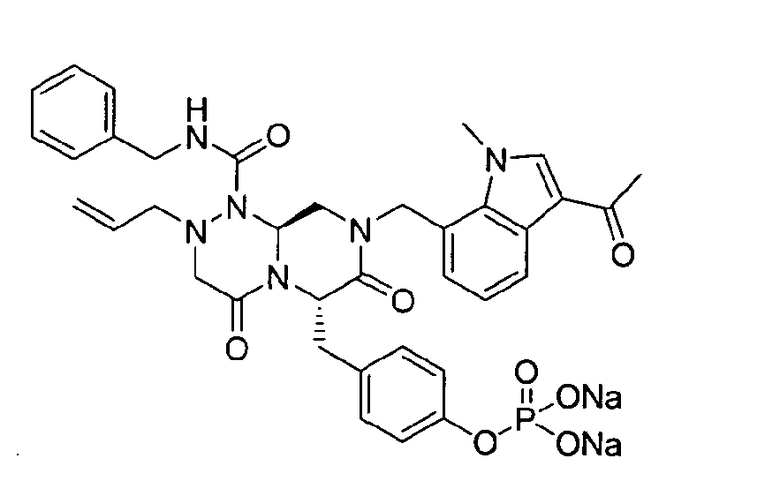





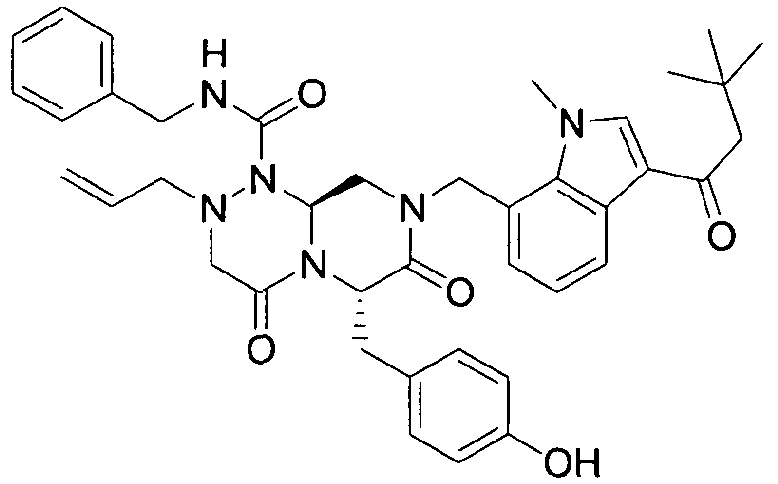
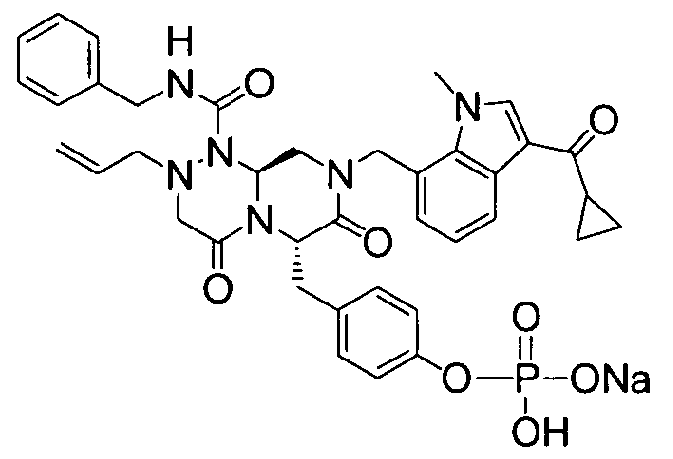
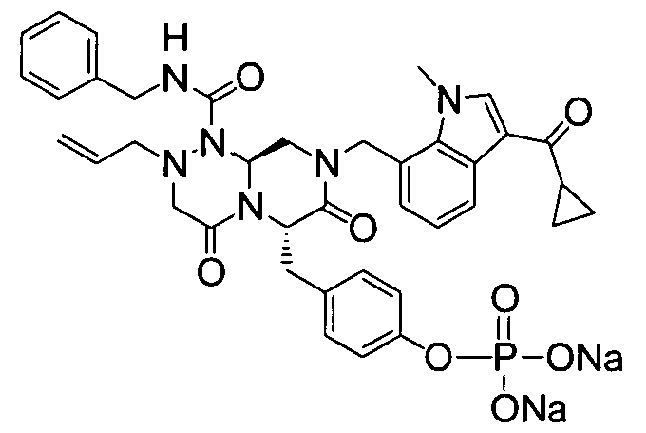


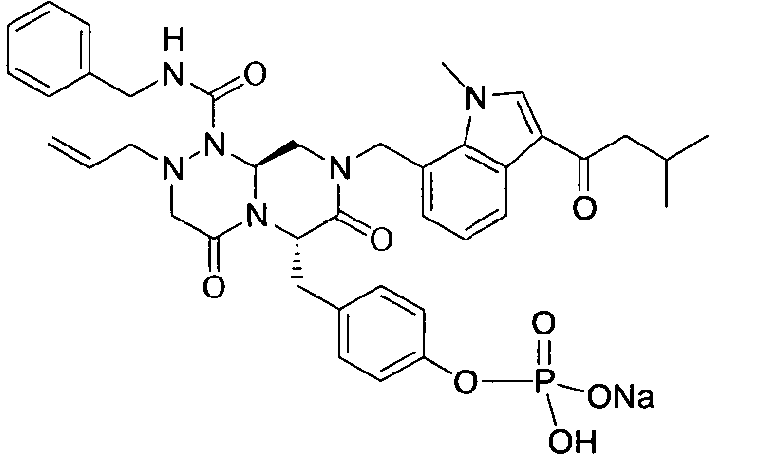
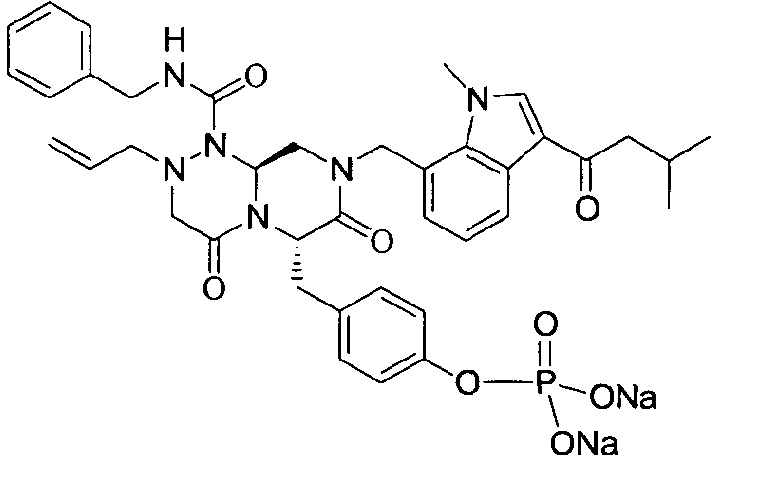

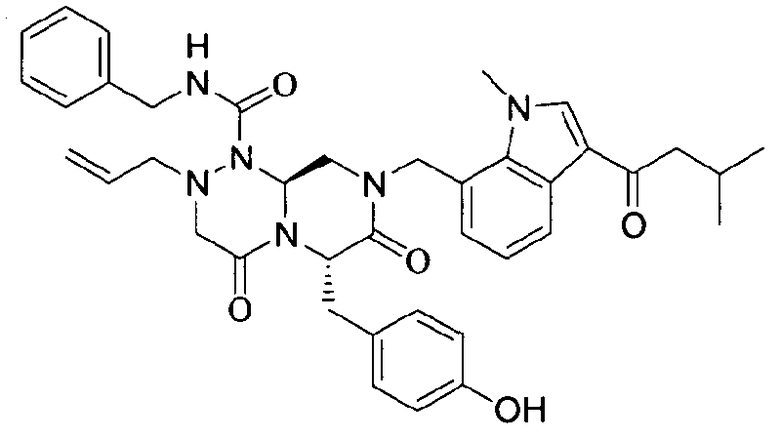
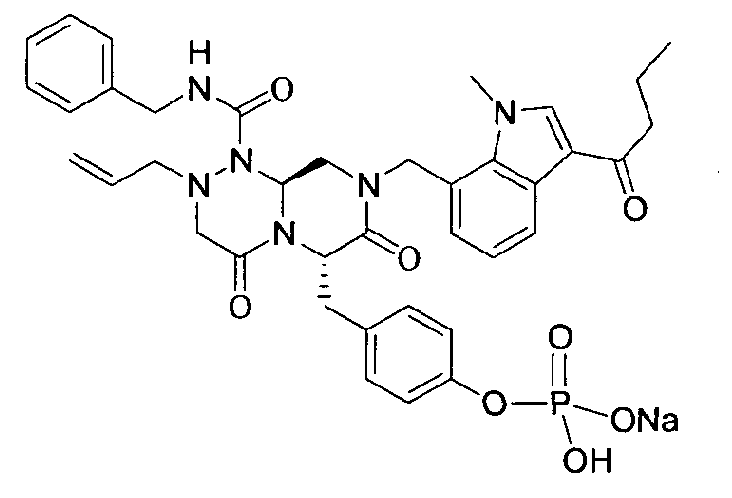

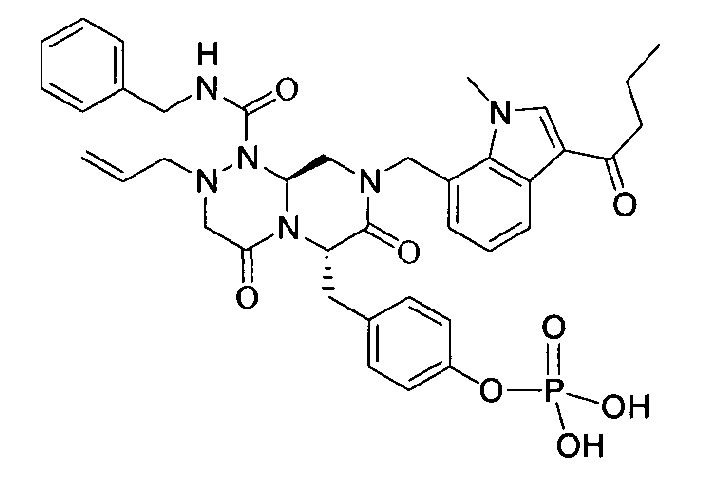





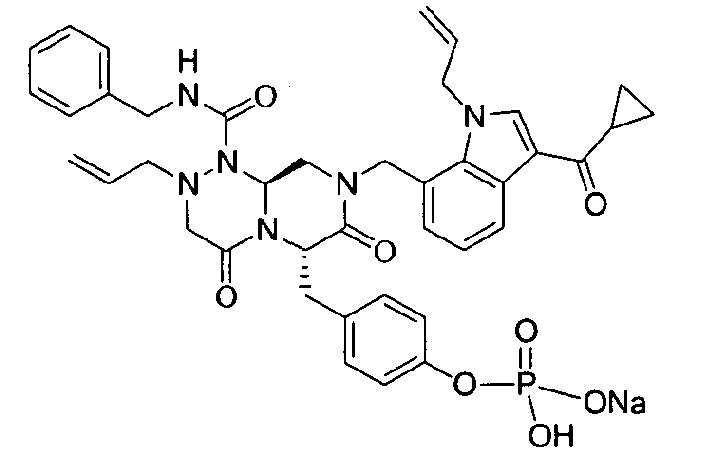
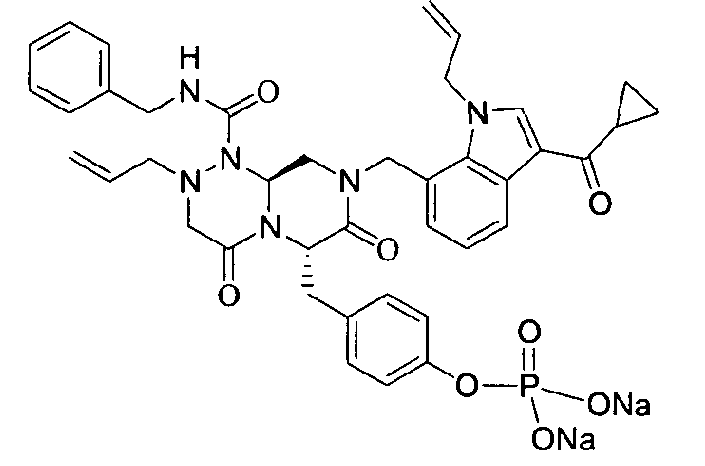

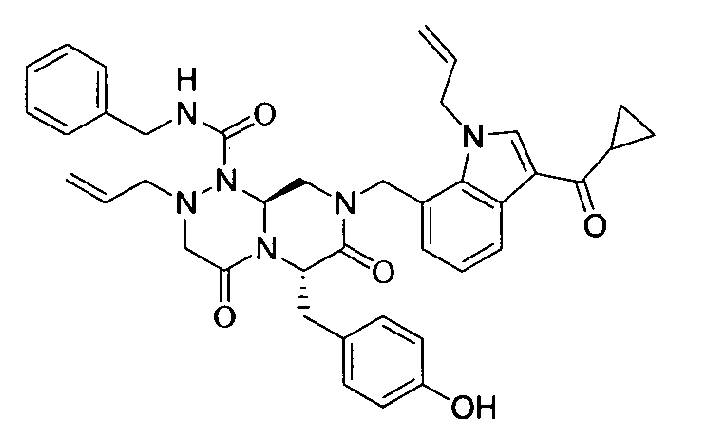
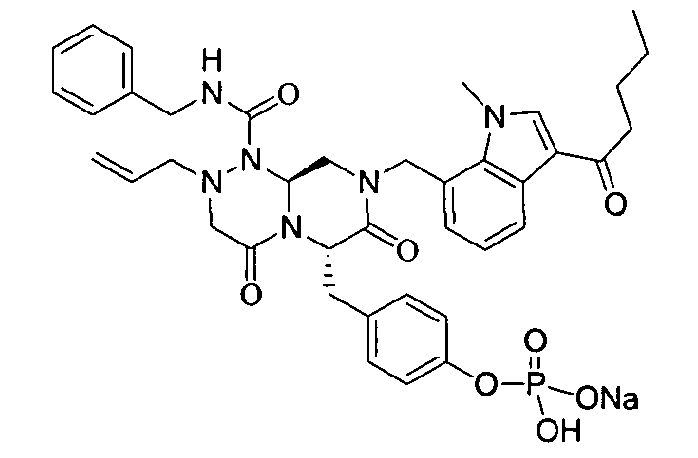

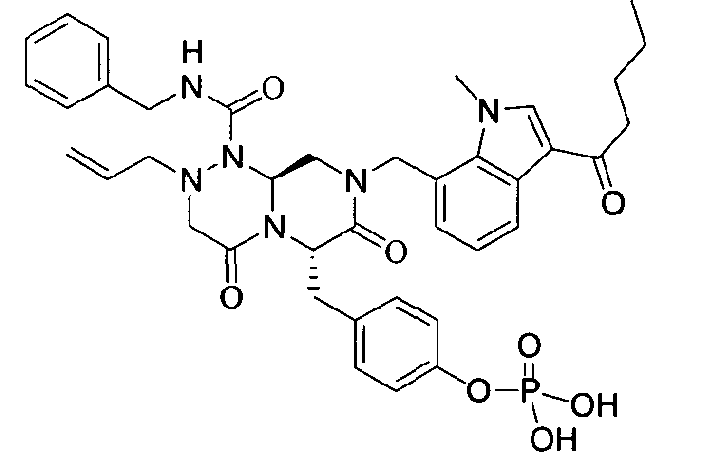
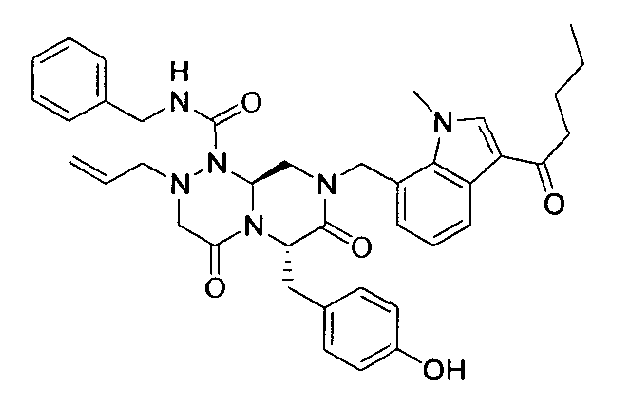
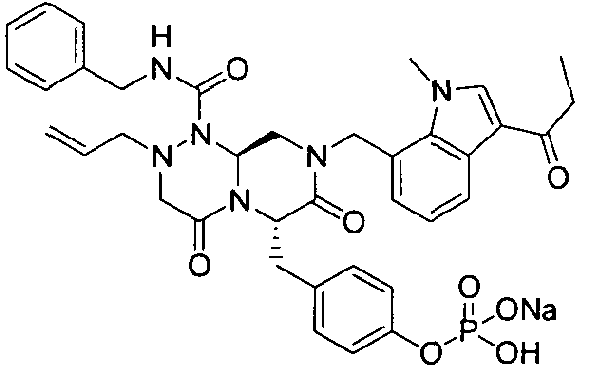
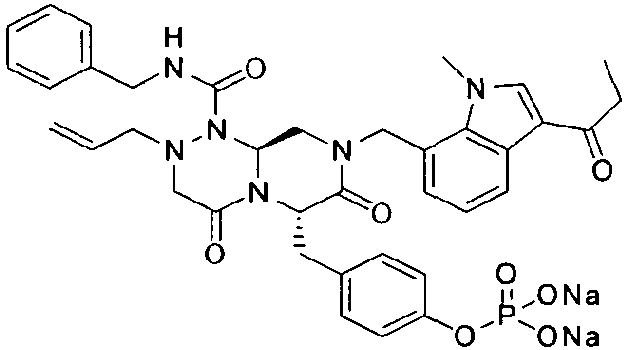
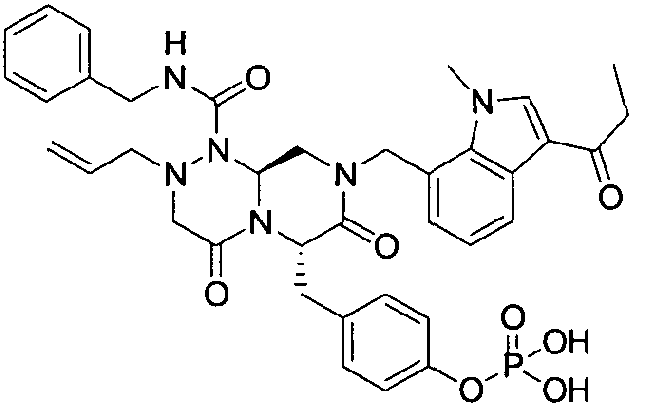

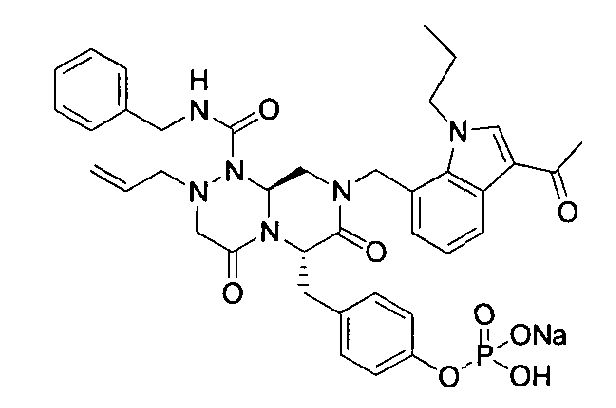
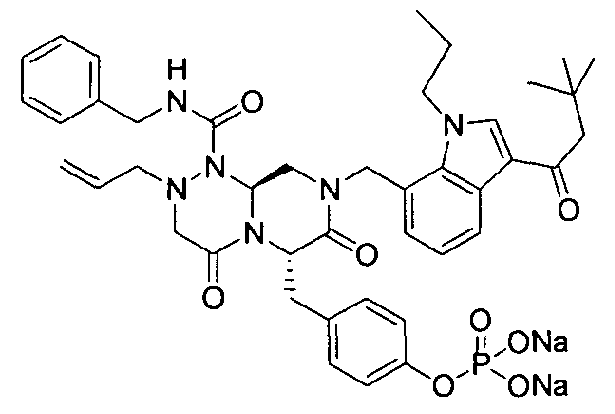
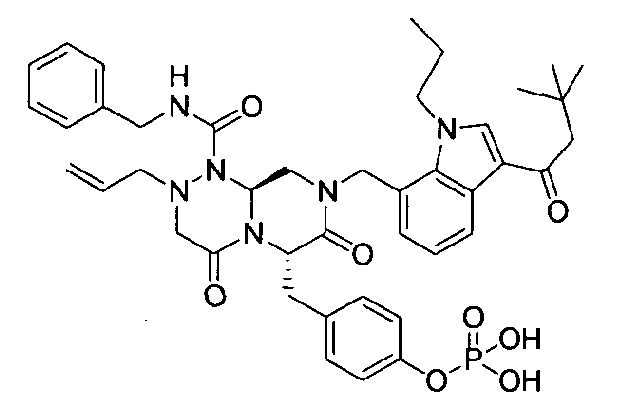
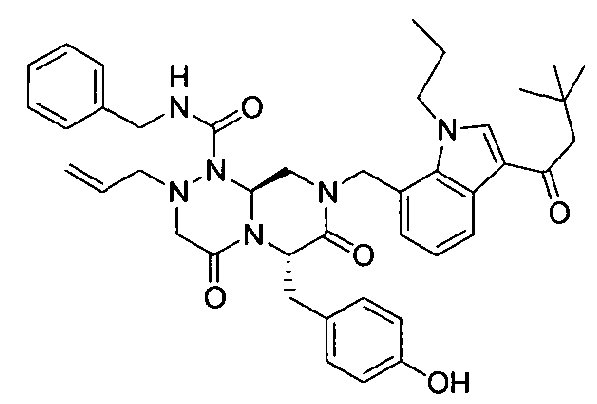
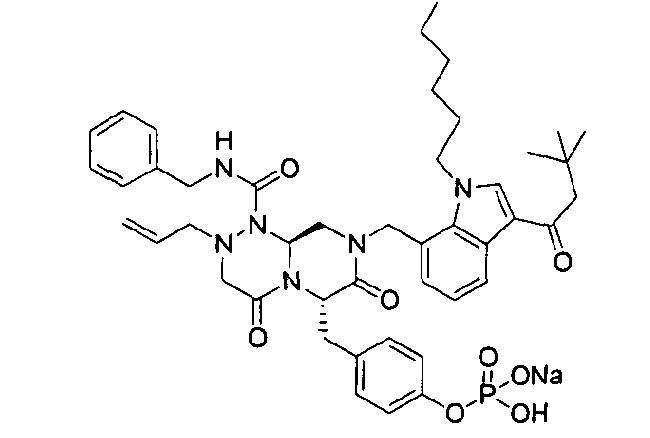
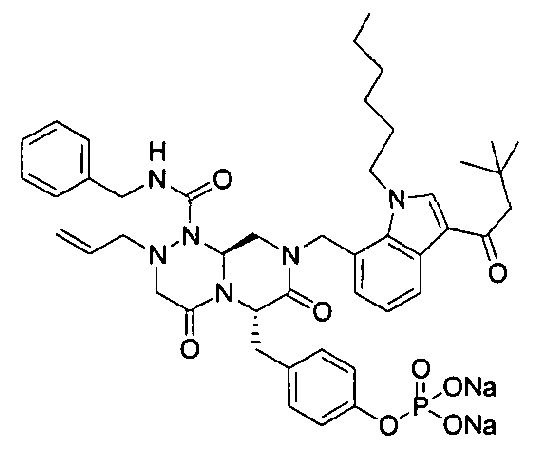
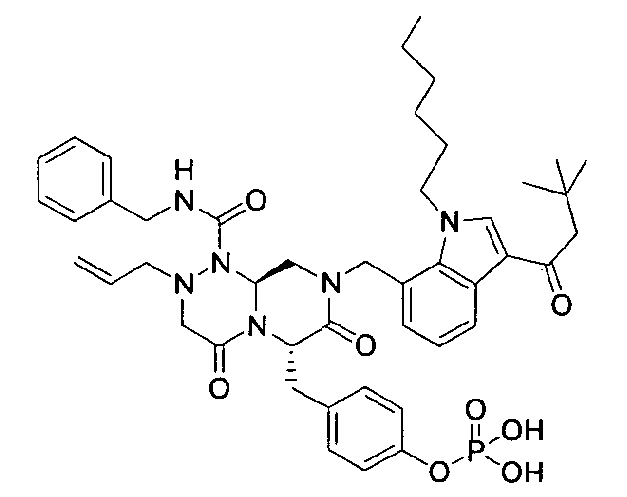
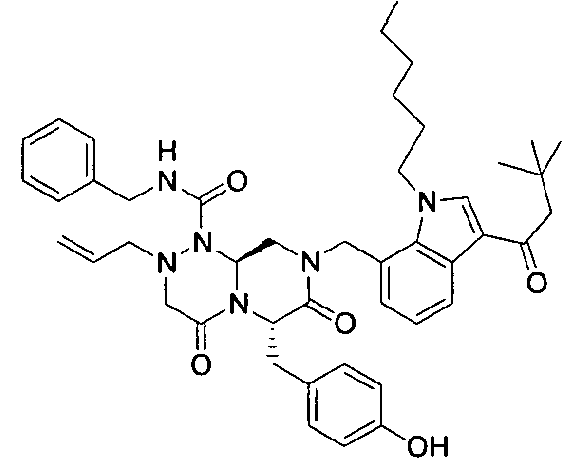

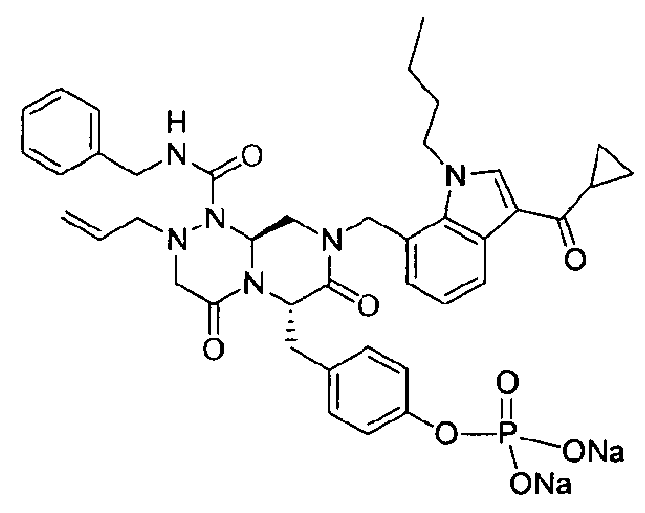



Способы, известные в данной области техники, могут применяться для определения эффективности соединения, предложенного в этом документе, при лечении ракового заболевания, такого как AML. Например, способ, описанный в примере 23, может применяться для оценки противораковой активности данного соединения. Дополнительные примеры способов оценки активности соединения при лечении AML включают описанные в статьях Bishop et al., Blood 87: 1710-7, 1996; Bishop, Semin Oncol 24:57-69, 1997; и Estey, Oncology 16: 343-52, 2002.
Соединения из настоящего раскрытия изобретения могут вводиться нуждающемуся в этом пациенту посредством различных способов, таких как пероральный, топический, трансдермальный или парентеральный. В одном примере осуществления изобретения соединения или их композиции вводятся патентерально. Термин "парентеральный", как он используется в этом документе, включает подкожные инъекции, внутривенные, внутримышечные, интрацистернальные инъекции и внутривенные инфузии. В предпочтительных примерах осуществления изобретения соединения или композиции вводятся посредством инъецирования, такого как внутривенные инъекции.
Токсичность и терапевтическая эффективность соединений, исходя из раскрытия настоящего изобретения, можно определить посредством стандартных фармацевтических испытаний на клеточных культурах или на подопытных животных, например, для определения LD50 (дозы, летальной для 50% популяции) и ED50 (дозы, терапевтически эффективной для 50% популяции). Соотношение доз между токсическим и терапевтическим воздействиями представляет собой терапевтический индекс, и его можно выразить в виде соотношения LD50/ED50. Соединения, которые демонстрируют большие значения величин терапевтических индексов, являются предпочтительными. Несмотря на то, что соединения проявляют побочные токсические эффекты, они могут использоваться при тщательной разработке системы доставки, нацеливающей такие соединения на участок пораженной ткани, с целью минимизации потенциальной угрозы для неинфицированных клеток и, посредством этого, уменьшения побочных эффектов.
Данные, полученные при исследованиях клеточной культуры и при изучении на животных, могут использоваться при установлении диапазона доз, применяемых для людей. Дозировка таких соединений находится, предпочтительно, в диапазоне вокруг концентраций, включающих ED50, с небольшой или вовсе отсутствующей токсичностью. In vitro кардиотоксичность соединений можно определить в соответствии со способом, описанным в Примере 24, ниже. Доза может изменяться в пределах этого диапазона в зависимости от применяемой дозированной формы и используемого способа введения. Для любого соединения, применяемого в способе изобретения, терапевтически эффективную дозу можно первоначально оценить из исследований на клеточной культуре. Доза может быть установлена на животных моделях для достижения диапазона циркулирующих в плазме концентраций, который включает IC50 (т.e. концентрация тестируемого соединения, при которой достигается половинная величина от максимального ингибирования симптомов), как это определено на клеточной культуре. Такую информацию можно использовать для более точного определения применяемых доз, подходящих для людей. Уровни концентраций в плазме могут быть определены, например, с помощью высокоэффективной жидкостной хроматографии.
Эффективная доза зависит от типа заболевания, от используемой композиции, от способа введения, от типа подвергаемого лечению субъекта, от физических характеристик конкретного субъекта, для которого рассматривается лечение, конкурирующие лекарственные препараты и другие факторы, которые принимаются во внимание при рассмотрении специалистом в этой медицинской области. Например, для лечения AML соединение настоящего раскрытия изобретения может быть введено посредством внутривенной инъекции или инфузии в количестве от 0,5 мг/кг до 500 мг/кг (например, от 0,5 до 10 мг/кг, от 10 до 100 мг/кг, приблизительно от 100 до 500 мг/кг массы тела), которое может быть введено в виде однократной дозы, в течение дня, недели, месяца или за любой приемлемый интервал времени. В некоторых примерах осуществления изобретения раскрытые соединения могут применяться при лечении AML способом, аналогичным тому, который применяется для Ara-C.
В соответствии со следующим аспектом настоящее изобретение предоставляет крупномасштабный способ производства миметиков обратного действия настоящего изобретения. Способ включает нижеприведенные последовательные стадии:
введение ацильной группы в индол-7-карбальдегид посредством ацилирования по Фриделю-Крафтсу с получением 3-ацилиндол-7-карбальдегида;
введение алкильной группы и аминоацетальной группы в 3-ацилиндол-7-карбальдегид с получением 1-алкил-3-ацилиндольного производного;
стереоселективное амидирование l-алкил-3-ацилиндольного производного с помощью Cbz-Tyr(OtBu) (т.е. (S)-2-(бензилоксикарбониламино)-3-(4-трет-бутоксифенил)пропановая кислота) и 2-(1-аллил-4-бензилсемикарбазид)уксусной кислоты с получением реакционного промежуточного соединения;
циклизация реакционного промежуточного соединения в присутствии муравьиной кислоты с получением циклического промежуточного соединения и
фосфорилирование циклического промежуточного соединения.
В вышеприведенном способе 2-(1-аллил-4-бензилсемикарбазид)уксусной кислоты может быть получен посредством нижеприведенных последовательных стадий:
добавление ТЭА (ТЕА) (триэтиламина) к раствору этилгидразинацетата с получением реакционного раствора;
добавление аллилбромида (например, прикапыванием) к реакционному раствору и
добавление бензилизоцианата (например, прикапыванием).
Типичные представители соединений изобретения могут быть получены, как показано на нижеприведенной реакционной схеме.

В некоторых примерах осуществления изобретения Ra представляет собой метил, Rb представляет собой -C(=O)Re, и Re представляет собой метил или циклопропил.
Как показано в настоящем описании, схема реакций направлена на новые миметики обратного действия, представленные химической формулой I.
Соединения в соответствии с настоящим изобретением основываются на структуре пиразинотриазинона с четырьмя различными функциональными группами, присоединенными к ней. Из-за их двух хиральных центров соединения должны быть синтезированы стереоселективно.
Ацильную группу вводят в индол-7-карбальдегид AA1 посредством ацилирования по Фриделю-Крафтсу, затем следует введение алкила и аминоацетальных групп.
После реакции AA2 с хиральным соединением (Cbz-тирозин-OtBu) получившееся в результате промежуточное соединение подвергают стереоселективному амидированию с помощью PivCl (пивалоилхлорида) и iBCF (изобутилхлорформиата), что позволяет получить AA3. После этого AA3 циклизуют с помощью муравьиной кислоты, получая AA4, далее следуют фосфорилирование, введение соли (присоединение Na к фосфату с использованием 0,5н NaOH) и лиофилизация, что и позволяет синтезировать с высокой чистотой пиразинотриазоновые соединения, AA5.
Вариант осуществления изобретения
Лучшее понимание настоящего изобретения может быть достигнуто посредством нижеприведенных примеров, которые приведены для иллюстрации, но не должны рассматриваться как ограничивающие настоящее изобретение.
Как продемонстрировано в настоящем описании, соединения химической формулы I проявляют противораковую активность.
Далее подробно проиллюстрирован способ получения согласно настоящему изобретению.

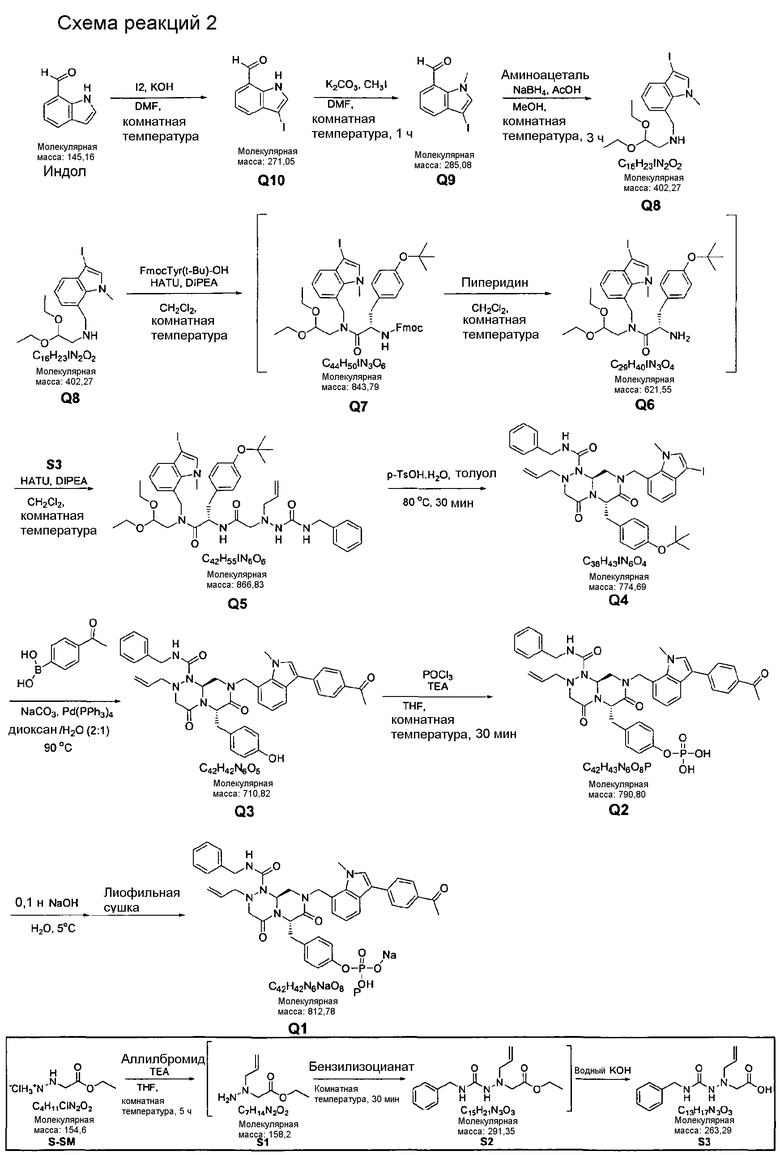
На Схеме реакций 1 S3 c боковой цепью может быть получен, как проиллюстрировано ниже.

Каждая стадия способа получения, показанная на Схеме реакций 1, ниже будет описана подробно в примерах 1-10.
Пример 1
Синтез S3
2-(1-Аллил-4-бензилсемикарбазид)уксусная кислота
67 г этилгидразинацетата растворяли в 673 мл ТГФ (тетрагидрофурана) и перемешивали со 121 мл ТЭА (триэтиламина). К этой реакционной смеси по каплям добавляли 41 мл аллилбромида в течение 20 мин. Этот раствор перемешивали в течение 5 ч и фильтровали. К фильтрату по каплям добавляли 53 мл бензилизоцианата в течение 15 мин, затем перемешивали в течение 30 мин при комнатной температуре. После этого перед перемешиванием в течение 30 минут по каплям добавляли раствор 48 г KOH (гидроксида калия) в 673 мл дистиллированной воды. Разделение слоев создавали посредством добавления 403 мл MC (дихлорметана) и 269 мл гексана с перемешиванием. Водный раствор один раз промывали 201 мл MC (дихлорметана). Для водного раствора доводили значение pH до 2~3 путем использования 100 мл концентрированной HCl. После перемешивания в течение 30 мин рН-регулируемого раствора проводили его экстракцию с использованием 1009 мл MC (дихлорметана). Слой MC (дихлорметана), полученный таким образом, сушили с помощью 269 г Na2SO4, фильтровали и затем концентрировали в вакууме. Концентрат кристаллизовали с использованием 134 мл EA (этилацетата) и 269 мл гексана, с последующим фильтрованием. Полученное таким образом твердое вещество суспендировали в 134 мл EA (этилацетата), фильтровали при 0°C и сушили в вакууме, получая 40 г S3 в виде белого твердого вещества (выход 35%).
1H ЯМР (500 МГц, CDCl3) δ 10,84 (ушир.с, 1H), δ 7,90 (с, 1H), δ 7,4-7,3 (м, 5H), δ 6,42 (т, J=5,0 Гц, 1H), δ 5,85-5,72 (м, 1H), δ 5,28 (дд, J=28,5, 2,0 Гц, 1H), δ 5,19 (д, J=17 Гц, 1H), δ 4,47-4,42 (м, 2H), δ 3,70 (дд, J=40,0, 2,5 Гц, 1H).
Пример 2
Синтез P9
3-Ацетил-1H-индол-7-карбальдегид
23,5 мл AcCl (ацетилхлорида) по каплям добавляли к раствору 55 г AlCl3 в 400 мл MC (дихлорметана) при перемешивании. К этому раствору по каплям добавляли раствор 40 г исходного вещества (индол-7-карбальдегида) в 400 мл MC (дихлорметана). Температуру раствора необходимо поддерживать равной 0~5°C при добавлении и затем оставить раствор до повышения температуры до комнатной. Протекание реакции контролировали с использованием тонкослойной хроматографии (ТСХ) и высокоэффективной жидкостной хроматографии. После завершения реакции раствор разделяли на слои при добавлении воды. Органический слой, образовавшийся таким образом, сушили над MgSO4 (сульфатом магния), фильтровали и затем концентрировали при 40°C, получая 41 г P9 в виде концентрированного остатка (выход 80%).
Пример 3
Синтез P8
3-Ацетил-1-метил-1H-индол-7-карбальдегид
41 г P9 растворяли в 412 мл ДМФА (диметилформамида) и перемешивали. После охлаждения раствора до 10°C туда же добавляли 91 г K2CO3 (карбоната калия), и 20 мл MeI (метилиодида) по каплям добавляли. Полученный в результате раствор оставляли до повышения температуры до комнатной и перемешивали в течение 4-5 ч. После подтверждения исчезновения исходного вещества отфильтровывали K2CO3, затем кристаллизовали из гексана, получая 35 г P8 в виде желтоватого твердого вещества (выход 80%).
Пример 4
Синтез P7
1-(7-((2,2-диэтоксиэтиламино)метил)-1-метил-1H-индол-3-ил)этанон
К раствору 35 г P8 в 354 мл MeOH (метанола) добавляли 3,5 мл AcOH (уксусной кислоты). Раствор смешивали с 33 мл аминоацетальдегида диэтилацеталя при комнатной температуре и перемешивали в течение 3-4 ч. После охлаждения раствора до 10°C туда же медленно добавляли 3,3 г восстанавливающего агента NaBH4 (натрийборгидрида). При этом следует соблюдать меры предосторожности, поскольку образуется газообразный водород и протекает экзотермическая реакция. Раствор перемешивали при комнатной температуре в течение 1 ч. После завершения реакции добавляли 354 мл EA (этилацетата) и 354 мл дистиллированной воды, таким образом разделяя слои. Образованный таким образом органический слой сушили над 141 г MgSO4 (сульфата магния) и кристаллизовали из гексана, что позволило получить 85 г P7 в виде желтоватого твердого вещества (выход 80%).
1H ЯМР (500 МГц, CDCl3), δ 8,36 (д, J=4,8 Гц, 1H), δ 7,61 (с, 1H), δ 7,17 (д, J=4,2 Гц, 1H), δ 7,10 (д, J=4,2 Гц, 1H), δ 4,58 (т, J=3,3, 1H), δ 4,21 (с, 3H), δ 4,07 (с, 3H), δ 3,68 (м, 2H), δ 3,51 (м, 2H), δ 2,82 (д, J=3,3 Гц, 2H), δ 2,48 (с, 3H), δ 1,19 (т, J=4,2 Гц, 6H).
Пример 5
Синтез P6
Бензил (S)-1-(N-((3-ацетил-1-метил-1H-индол-7-ил)метил)-N-(2,2-диэтоксиэтил)карбамоил)-2-(4-трет-бутоксифенил)этилкарбамат
85 г Cbz-Tyr(OtBu) растворяли в 449 мл EA (этилацетата) при перемешивании. После охлаждения раствора до 0~5°C туда же по каплям добавляли 31 мл NMM (N-метилморфолина) и 19 мл пивалоилхлорида. Раствор перемешивали в течение 1~2 ч и затем туда же добавляли 44,9 г P7 при 0~5°C. Раствор нагревали до комнатной температуры, затем перемешивали в течение 2~3 ч. После окончания реакции добавляли дистиллированную воду для обеспечения разделения слоев. Образовавшийся таким образом органический слой промывали 898 мл 5% водного раствора лимонной кислоты и 898 мл 5% водного раствора NaHCO3 и затем сушили над 179 г MgSO4 (сульфата магния), после концентрирования в остатке получали 85 г P6 (выход 90%).
Пример 6
Синтез P5
(S)-3-(4-трет-бутоксифенил)-N-(3-ацетил-1-метил-1H-индол-7-ил)метил)-2-амино-N-(2,2-диэтоксиэтил)пропанамид
К 85 г P6 в 853 мл MeOH добавляли 8,5 г 10% масс. Pd/C. Добавляли 16 г формиата аммония и затем кипятили с обратным холодильником в течение 2 ч. После завершения реакции раствор охлаждали до комнатной температуры и отфильтровывали Pd/C. Раствор концентрировали перед проведением разделения слоем при добавлении 853 мл EA (этилацетата) и 1706 мл дистиллированной воды. Образовавшийся таким образом органический слой промывали 850 мл 5% водного раствора лимонной кислоты и 850 мл 5% водного раствора NaHCO3, и концентрировали, получая 56 г P5 (выход 90%).
Пример 7
Синтез P4
Растворяли 40 г имеющего боковую цепь S3 в 426 мл EA (этилацетата) и охлаждали до -10°C. К раствору по каплям добавляли 41 мл NMM (N-метилморфолина) и 20 мл iBCF (изобутилхлорформиата) при той же температуре. Реакционную смесь перемешивали в течение 2~3 ч при -10°C, после этого туда же по каплям добавляли раствор 56 г P5 в 200 мл EA (этилацетата). Реакционную смесь нагревали до комнатной температуры и затем перемешивали в течение 1~2 ч. Затем реакцию заканчивали, добавляли EA (этилацетат) и 850 мл дистиллированной воды для разделения слоев. Образовавшейся таким образом органический слой промывали 850 мл 5% водного раствора лимонной кислоты и 850 мл 5% водного раствора NaHCO3, и сушили над 340 г MgSO4 (сульфата магния) с последующим концентрированием. Получали 81 г P4 в виде концентрированного остатка (выход 90%).
Пример 8
Синтез P3
(6S,9aS)-6-(4-гидроксибензил)-8-((3-ацетил-1-метил-1H-индол-7-ил)метил)-2-аллил-N-бензилгексагидро-4,7-диоксо-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамид
Растворяли 81 г P4 в 383 мл 85% муравьиной кислоты и нагревали до 50°C. После перемешивания в течение 1~2 ч при той же температуре раствор охлаждали до комнатной температуры и смешивали с ацетоном. Для этого раствора доводили значение pH до 4,0~4,2, добавляя по каплям 5 н NaOH с образованием неочищенного кристалла. После охлаждения до 10~15°C отфильтровывали твердое вещество и полностью растворяли его в 767 мл MeOH при нагревании. При медленном охлаждении выпадали кристаллы, которые отфильтровывали, что позволяло получить P3 в виде розовато-белого кристаллического вещества (40 г, выход 60%).
1H ЯМР (500 МГц, CDCl3) 8,43 (д, J=4,8 Гц, 1H), 7,63 (с, 1H), 7,38-7,35 (м, 2H), 7,31-7,30 (м, 1H), 7,29-7,21 (м, 2H), 7,00 (д, J=4,8 Гц, 2H), 6,97 (д, J=4,8 Гц, 1H), 6,69-6,65 (м, 3H), 5,87 (с, 1H), 5,55-5,44 (м, 3H), 5,34 (т, J=4,6 Гц, 1H), 5,03 (д, J=6,3 Гц, 1H), 4,87 (д, J=9,0 Гц, 1H), 4,79 (д, J=7,5 Гц, 1H), 4,42 (дд, J=9,0, 3,6 Гц, 1H), 4,29 (дд, J=9,0, 3,6 Гц, 1H), 4,02 (с, 3H), 3,43 (д, J=7,2 Гц, 1H), 3,38-3,33 (м, 3H), 3,27 (д, J=7,2 Гц, 1H), 3,29-3,24 (м, 1H), 3,18 (дд, J=7,2, 2,4 Гц, 1H), 2,51 (с, 3H).
Пример 9
Синтез P2
4-(((6S,9aS)-1-(бензилкарбамоил)-8-((3-ацетил-1-метил-1H-индол-7-ил)метил)-2-аллилоктагидро-4,7-диоксо-1H-пиразино[2,l-c][1,2,4]триазин-6-ил)метил)фенилдигидрофосфат
40 г P3 растворяли в 217 мл ТГФ (тетрагидрофурана), охлаждали до 0~5°C и смешивали с 25 мл POCl3. При той же температуре по каплям добавляли 28 мл ТЭА (триэтиламина). Перемешивали 1 ч, затем медленно добавляли 87 мл дистиллированной воды. К раствору добавляли 348 мл насыщенного водного раствора NaHCO3, затем перемешивали в течение 30 мин. После проведения разделения слоев раствора при добавлении 217 мл EA (этилацетата) к водному слою добавляли 217 мл MC (метиленхлорида) и затем доводили значение pH до 1~3, используя 14 мл концентрированную HCl, разделив слои. Образованный таким образом органический слой подвергали дегидратации, используя 174 г Na2SO4 (сульфат натрия), и концентрировали в вакууме. Концентрат кристаллизовали из 130 мл ТГФ (тетрагидрофурана) и 435 мл н-гексана, фильтровали, сушили в вакууме, что позволило получить 40 г P2 в виде белого твердого вещества (выход 90%).
1H ЯМР (500 МГц, ДМСО-d6) 8,27 (с, 1H), 8,16 (д, J=7,5 Гц, 1H), 7,85 (т, J=6,3 Гц, 1H), 7,34-7,29 (м, 3H), 7,22-7,01 (м, 9H), 6,79 (д, J=6,9 Гц, 1H), 5,84-5,75 (м, 1H), 5,52 (дд, J=8,1, 3,6 Гц, 1H), 5,38 (д, J=15,6 Гц, 1H), 5,17-5,13 (м, 1H), 5,09-5,03 (м, 2H), 4,90 (д, J=15,6 Гц, 1H), 4,22 (д, J=6,3 Гц, 2H), 4,06 (с, 3H), 3,76-3,68 (м, 1H), 3,61-3,55 (м, 2H), 3,33-3,27 (м, 4H), 3,07-3,02 (м, 2H), 2,41 (с, 3H).
Пример 10
Синтез P1
4-(((6S,9aS)-1-(бензилкарбамоил)-8-((3-ацетил-1-метил-1H-индол-7-ил)метил)-2-аллилоктагидро-4,7-диоксо-1H-пиразино[2,1-c][1,2,4]триазин-6-ил)метил)фенилгидрофосфат натрия
40 г высушенного P2 растворяли в 2000 мл дистиллированной воды при перемешивании. Раствор охлаждали до 0~5°C, затем доводили его pH до 4,6~4,8 (130~110 мВ) путем медленного добавления 0,1 н водного раствора NaOH и затем проводили лиофилизацию, что позволяло получить 40 г P1 в виде белого твердого вещества (выход 95%).
1H ЯМР (300 МГц, D2O) 7,86 (д, J=7,8 Гц, 1H), 7,60 (с, 1H), 7,07-6,93 (м, 10Н), 6,56 (д, J=7,2 Гц, 1Н), 5,39-5,32 (м, 2Н), 5,09 (т, J=5,4 Гц, 1Н), 4,95 (д, J=15,6 Гц, 1Н), 4,70-4,53 (м, 2Н), 4,14 (д, J=15,6 Гц, 1Н), 3,97 (д, J=15,6 Гц, 1H), 3,57 (с, 3H), 3,56-3,49 (м, 1H), 3,30-2,81 (м, 6H), 2,84-2,81 (м, 1H), 2,18 (с, 3H).
Другие примеры получения типичных соединений представлены ниже.
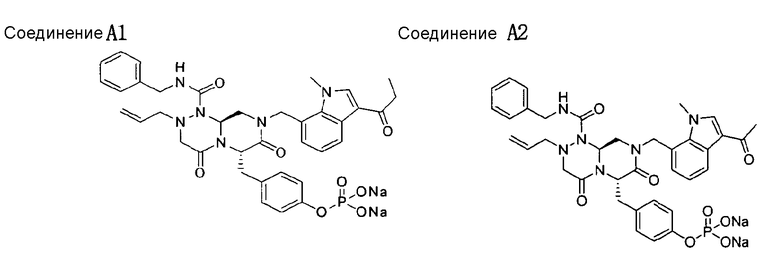
Способ, отображенный на Схеме реакций 2, ниже описан подробно в примерах 11-21.
Пример 11
Синтез S3 (Боковая цепь)
S3 получали таким же образом, как в примере 1.
Пример 12
Синтез Q10
3-Йод-1H-индол-7-карбальдегид
Раствор 24 г I2 в 125 мл ДМФА (диметилформамида) добавляли к исходному веществу (индол-7-карбальдегиду) и вводили в реакцию 5,3 г KOH при перемешивании. Протекание реакции контролировали с помощью ТСХ. После завершения реакции добавляли 354 мл EA (этилацетата) и 354 мл дистиллированной воды для обеспечения разделения слоев. Образованный таким образом органический слой промывали 10% водным раствором Na2S2O3, сушили над Na2SO4 (сульфатом натрия), фильтровали и концентрировали при 40°C, получая Q10 в виде концентрированного остатка.
1Н ЯМР (CDCl3, 300 МГц) δ 10,3 (ушир.с, 1Н), 10,2 (с, 1Н), 7,79 (д, 1Н, J=7,8 Гц), 7,75 (д, 1Н, J=7,2 Гц), 7,44 (д, 1Н, J=2,1 Гц), 7,37 (т, 1Н, J=7,2 Гц);
m/z 272,14 [M+1]+
Пример 13
Синтез Q9
Растворяли 17 г Q10 в 100 мл ДМФА (диметилформамида) при перемешивании. Полученный в результате раствор охлаждали до 10°C и смешивали с 18 г K2CO3 (карбоната калия). После того, как туда же по каплям добавили 6 мл MeI (метилиодида), раствор нагревали до комнатной температуры и перемешивали в течение 4~5 ч. После подтверждения исчезновения исходного вещества отфильтровывали K2CO3, затем проводили кристаллизацию из гексана, получая Q9.
1Н ЯМР (CDCl3, 300 МГц) δ 10,2 (с, 1Н), 7,76 (тд, 1Н, J=7,8, 1,2 Гц), 7,31 (т, 1Н, J=7,8 Гц), 7,12 (с, 1Н), 4,14 (с, 3Н).
Пример 14
Синтез Q8
К раствору 18 г Q9 в 600 мл MeOH (метанола) добавляли 0,4 мл AcOH (уксусной кислоты). При комнатной температуре добавляли к раствору 14 мл аминоацетальдегида диацеталя, затем перемешивали в течение 3~4 ч. Раствор охлаждали до 10°C перед проведением медленного добавления 3,3 г восстанавливающего агента NaCNBH3 (натрийцианоборгидрида). При этом следует соблюдать меры предосторожности, поскольку выделяются газообразный водород и тепло. После перемешивания реакционной смеси при комнатной температуре в течение 1 ч, контролировали протекание реакции. В случае завершения реакции 354 мл EA (этилацетата) и 354 мл дистиллированной воды использовали для разделения слоев. Образовавшийся таким образом органический слой подвергали дегидратации с использованием 141 г Na2SO4 (сульфата натрия) и кристаллизацией из гексана получали Q8.
Пример 15
Синтез Q7
27 г Fmoc-Tyr(OtBu) растворяли в 200 мл MC (дихлорметана) при перемешивании. К этому раствору добавляли 23 г HATU (О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфата) и 20 мл DIPEA (диизопропилэтиламина) при комнатной температуре. Раствор перемешивали в течение 1~2 ч, смешивали с 15,8 г Q9 и далее перемешивали в течение 2~3 ч. После завершения реакции добавляли дистиллированную воду для разделения слоев. Образовавшийся таким образом органический слой промывали 898 мл 5% водного раствора лимонной кислоты и 898 мл 5% водного раствора NaHCO3, дегидратацию проводили с помощью Na2SO4 (сульфата натрия) и концентрировали, что приводило к получению Q7 в виде концентрированного остатка.
Пример 16
Синтез Q6
К раствору 34 г Q7 в 400 мл MC (дихлорметана) добавляли 20 мл пиперидина. После завершения реакции раствор концентрировали, затем разделяли слои: 400 мл MC (дихлорметана) и 800 мл дистиллированной воды. Образовавшийся таким образом органический слой промывали 850 мл 5% водного раствора лимонной кислоты и 850 мл 5% водного раствора NaHCO3 и затем концентрировали, получая Q6.
Пример 17
Синтез Q5
К раствору 13 г S3 в 400 мл MC (дихлорметана) по каплям добавляли 19 г HATU (О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфата) и 16 мл DIPEA (диизопропилэтиламина) при комнатной температуре. После перемешивания раствора в течение 2~3 ч туда же по каплям добавляли раствор 28 г Q6 в 200 мл MC (дихлорметана). Его перемешивали при комнатной температуре в течение 1~2 ч. После завершения реакции использовали 200 мл MC (дихлорметана) и 200 мл дистиллированной воды для обеспечения разделения слоев. Образовавшийся таким образом органический слой промывали 200 мл 5% водного раствора лимонной кислоты и 200 мл водного раствора 5% NaHCO3, дегидратацию проводили с помощью 340 г Na2SO4 (сульфата натрия) и затем концентрировали, что позволяло получить Q5 в виде концентрированного остатка.
Пример 18
Синтез Q4
289 мг пара-TsOH·H2O добавляли к раствору 4 г Q5 в 100 мл толуола, затем нагревали до 80°C. Полученный в результате раствор перемешивали при той же температуре в течение 30 мин, охлаждали до комнатной температуры и концентрировали. Разделение слоев обеспечивали добавлением EA (этилацетата) и дистиллированной воды. Органический слой промывали 200 мл 5% водного раствора лимонной кислоты и 200 мл 5% водного раствора NaHCO3, дегидратацию проводили с использованием 340 г Na2SO4 (сульфата натрия) и затем концентрировали, получая Q4 в виде концентрированного остатка.
1Н ЯМР (CDCl3, 300 МГц) δ 7,43~7,27 (м, 2Н), 7,23~7,21 (м, 2Н), 7,12 (т, 1Н, J=7,2 Гц), 7,08 (с, 1Н), 7,05 (д, 2Н, J=7,8 Гц), 6,97 (д, 1Н, J=7,2 Гц), 6,90 (д, 2Н, J=8,4 Гц), 6,59 (т, 1Н, J=6,0 Гц), 5,62 (дд, 1Н, J=10,2, 4,8 Гц), 5,53~5,39 (м, 3Н), 5,37 (т, 1Н, J=6,0 Гц), 5,02 (д, 1Н, J=10,2 Гц), 4,93 (д, 1Н, J=16,5 Гц), 4,77 (д, 1Н, J=17,1 Гц), 4,44 (дд, 1Н, J=15,0, 6,3 Гц), 4,32 (дд, 1Н, J=15,0, 6,0 Гц), 3,97 (с, 3Н), 3,49~3,19 (м, 8Н), 1,33 (с, 9Н)
Пример 19
Синтез Q3
К раствору 100 мг Q4 в смеси 8 мл 1,4-диоксана и 4 мл воды добавляли 33 мг 4-ацетилфенилбороновой кислоты, 41 мг Na2CO3 (карбоната натрия) и 15 мг Pd(PPh3)4 (тетракистрифенилфосфинпалладий), затем температуру повышали до 90°C. После перемешивания в течение 2 ч при той же температуре раствор охлаждали до комнатной температуры и концентрировали. EA (этилацетат) и дистиллированную воду применяли для обеспечения разделения слоев. Образовавшийся таким образом органический слой подвергали дегидратации с помощью Na2SO4 (сульфата натрия) с последующим концентрированием. Концентрат растворяли в MC (дихлорметане), к которому затем по каплям добавляли 1 мл TFA (трифторуксусной кислоты), далее перемешивали при комнатной температуре. После завершения реакции реакционную смесь промывали 10 мл 5% водного раствора NaHCO3 и проводили дегидратацию с помощью Na2SO4 (сульфата натрия), получая Q3 в виде концентрированного остатка.
1Н ЯМР (CDCl3, 300 МГц) δ 8,05 (д, 2Н, J=8,4 Гц), 7,91 (д, 1Н, J=7,2 Гц), 7,71 (д, 2Н, J=8,4 Гц), 7,40~7,20 (м, 4Н), 7,16 (т, 1Н, J=7,2 Гц), 7,05 (д, 2Н, J=8,4 Гц), 6,96 (д, 1Н, J=6,9 Гц), 6,69 (д, 2Н, J=8,4 Гц), 6,68 (м, 1Н), 5,58~5,44 (м, 3Н), 5,37 (т, 1Н, J=5,7 Гц), 5,03 (д, 1Н, J=10,8 Гц), 4,97 (д, 1Н, J=14,7 Гц), 4,81 (д, 1Н, J=17,1 Гц), 4,47 (дд, 1Н, J=15,3, 6,3 Гц), 4,33 (дд, 1Н, J=15,3, 6,3 Гц), 4,33 (с, 3Н), 3,47~3,24 (м, 8Н), 2,64 (с, 3Н);
m/z 711,56 [M+1]+
Пример 20
Синтез Q2
Раствор 50 г Q3 в 217 мл ТГФ (тетрагидрофурана) охлаждали до 0~5°C и смешивали с 25 мл POCl3. При той же температуре по каплям добавляли 28 мл ТЭА (триэтиламина) к раствору, который затем перемешивали в течение 1 ч. Затем медленно добавляли 87 мл дистиллированной воды. Добавляли 348 мл насыщенного водного раствора NaHCO3 и раствор перемешивали в течение 30 мин. Добавление 217 мл EA (этилацетата) в результате привело к разделению слоев. К водному слою добавляли 217 мл MC (метиленхлорида), затем доводили значение pH раствора до 1~3 с помощью 14 мл концентрированной HCl. Образовавшийся таким образом органический слой подвергали дегидратации с помощью Na2SO4 (сульфата натрия) и концентрировали в вакууме. Концентрат кристаллизовали из 130 мл ТГФ (тетрагидрофурана) и 435 мл н-гексана, и твердое вещество отфильтровывали и сушили в вакууме.
Пример 21
Синтез Q1
44 г высушенного Q2 растворяли в 200 мл дистиллированной воды при перемешивании. После охлаждения до 0~5°C медленно добавляли 0,1н NaOH до доведения значения pH раствора до 4,6~4,8 (130~110 мВ), далее после лиофилизации получали Q1.
Подробное описание будет предоставлено ниже в связи с эффектом полученных соединений.
Пример 22
Соединения получали в форме пролекарств для улучшения их растворимости. Фосфат может быть введен в качестве возможного пролекарственного заместителя, который может быть либо в форме мононатрийфосфата, либо в форме динатрийфосфата.
Это пролекарство получали путем добавления гидроксида натрия к P2, которое синтезировали в соответствии с примером 9. Как мононатриевая, так и динатриевая формы пролекарства демонстрируют растворимость до 400 мг/мл. Обе формы имеют подходящие свойства для композиции для внутривенного введения, в которой мононатриевая форма имеет значение pH 4,45 и динатриевая форма имеет значение pH 7,62.
Фиг.1 графически представляет изменения pH и потенциала при прикапывании 0,5 н NaOH к соединению настоящего изобретения. На графике горизонтальная ось показывает добавленные количества гидроксида натрия. На графике первая и вторая точки перегиба соответствуют моменту образования мононатриевой и динатриевой форм, соответственно.
Пример 23
Противораковая активность в случае острого миелолейкоза (AML) (модель на животных)
Тестируемые вещества были получены в форме пролекарств для повышения растворимости представляющих интерес соединений. Фосфатная функциональная группа, которая может быть либо в мононатриевой форме, либо в динатриевой форме, вводилась как пролекарственный заместитель.
Вещество сравнения: Ara-C (коммерчески доступный лекарственный препарат для лечения острого миелолейкоза)
Использовали купленную клеточную линию человеческой AML, MV4-11 (ATCC, U.S.A.), и культивирование проводили при 37°C в условиях 5% CO2 окружающей атмосферы на питательной среде Дульбекко, модифицированной по способу Исков (GIBCO, № по каталогу 21056), дополненной 10% фетальной бычьей сывороткой (GIBCO, № по каталогу 25030-081). Самки Balb/C бестимусных мышей (OrientBio, Sungnam-city, Korea) в возрасте 5-6 недель подвергались акклиматизации в комнате для разведения. Посредством использования стерильного шприца смесь 1:1 MV4-11 клетки:матригель (об./об.) имплантировали в количестве 5×106/мышь под подмышечными впадинами для каждой мыши. Когда опухоль образовалась спустя 2 недели после имплантации, мышей разделяли на пять (5) групп таким образом, чтобы получить минимум отклонений между группами в размере опухоли и в массе тела. Тестируемые вещества растворяли в физиологическом растворе и внутривенно инъецировали в дозе 10 мл/кг один раз в день и пять раз в неделю в течение двух недель (дни введения тестируемых веществ, D1~D5, D8~D12). В качестве контроля использовали только физиологический раствор. Размер опухоли определяли как рассчитанный по нижеприведенному уравнению: длинная ось × короткая ось × короткая ось/2. Длинную и короткую оси опухоли измеряли в размерности отрезка, используя штангенциркуль с цифровой индикацией (Mitsutoyo, Japan). Противораковую активность тестируемых веществ подсчитывали по нижеприведенному уравнению.
Показатель ингибирования роста опухоли A (%) = 100 × [1-(b-a)/(Ref b-Ref a)],
где
a = средний размер опухоли в группе, которой вводили лекарственный препарат в день 1
b = средний размер опухоли в группе, которой вводили лекарственный препарат в день 12
Ref a = средний размер опухоли в контрольной группе в день 1
Ref b = средний размер опухоли в контрольной группе в день 12
В том случае, когда средний размер опухоли в группе, которой вводили лекарственный препарат, в день 12 был меньше этого показателя до введения тестируемых веществ, это обозначали как регрессия (>100%). Показатели ингибирования роста опухоли для растущей опухоли в присутствии тестируемых веществ суммированы в таблице 2, приведенной ниже.
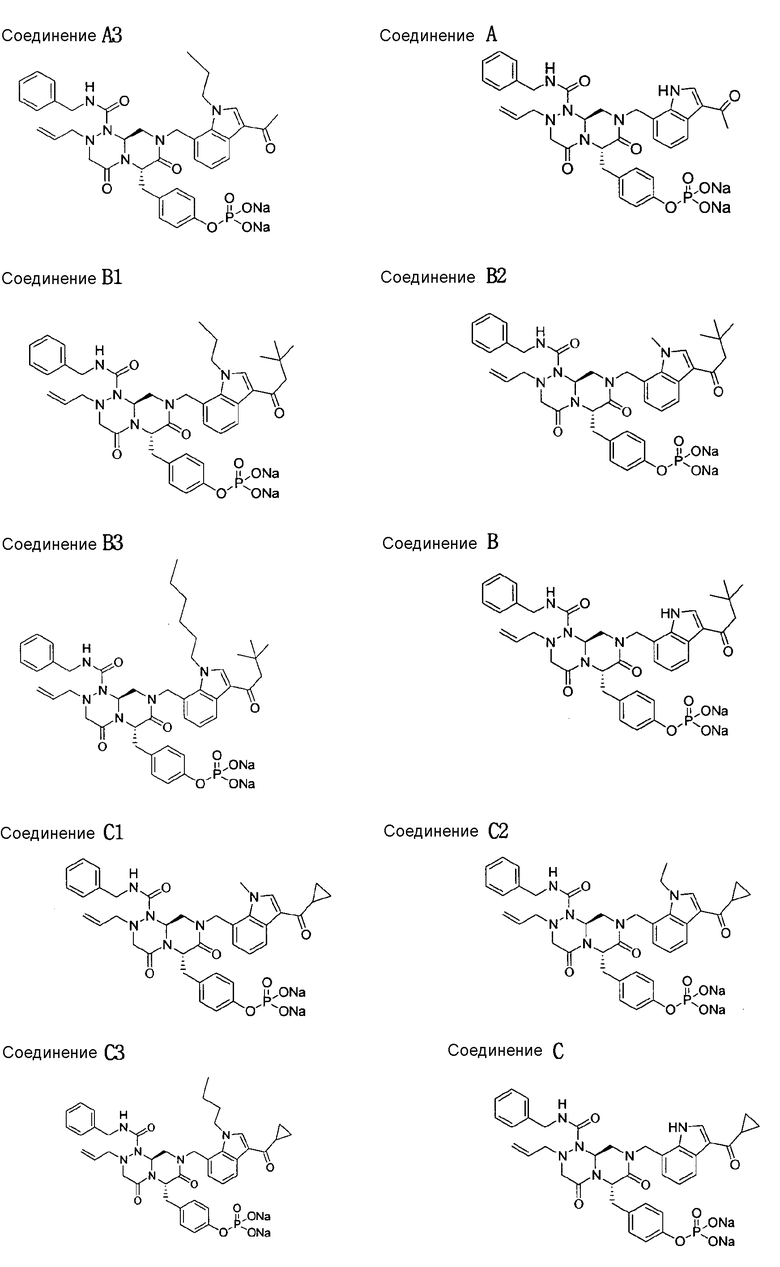
Результаты тестирования демонстрируют, что все тестируемые соединения обладают ингибирующей активностью в отношении роста опухоли. В случае соединений A1-A3, B1-B3 и C1-C3, в соответствии с настоящим изобретением, показатели ингибирования опухоли имеют значения в диапазоне от 70% до регрессии (>100%). В противоположность этому, Ara-C, широко применяемый лекарственный препарат при AML, как было найдено, имеет показатель ингибирования опухоли 66%. Взятые в совокупности, результаты показывают, что соединения настоящего изобретения обладают высокой ингибирующей способностью в отношении роста опухоли.
Пример 24
In vitro испытание кардиотоксичности: испытание ингибирующей активности в отношении человеческого гена, кодирующего белок калиевых каналов сердца (hERG)
Клетки линии HEK293 подвергали трансфекции с использованием hERG (human Ether-à-go-go Related Gene) кДНК в течение 48 ч, применяя при этом липофектамин 2000 (Invitrogen, USA). Трансфицированные клетки HEK293 культивировали на модифицированной среде Дульбекко (MEM, Gibco, 1 л), дополненной 10% фетальной бычьей сывороткой (FBS), пируватом натрия (10 мл), пенициллин/стрептомицином (10 мл) и зеоцином (100 мкг/мл, Invitrogen) при 37°C в атмосфере 5% CO2. После выделения из инкубационных сосудов с помощью обработки трипсином клетки HEK293 помещали в камеру для проведения процесса записи фиксирования потенциала. Применяли способ фиксации потенциала для всей совокупности клеток, чтобы зарегистрировать hERG K+ потоки в клетках HEK293, используя нижеприведенные внутри/внеклеточные растворы. После этого проводили наблюдение влияний на K+ потоки со стороны соединений, применяемых вне клеток.
• внутриклеточный раствор: K-аспартат 100 мМ, KCl 25 мМ, NaCl 5 мМ, MgCl2 1 мМ, Mg-АТФ 4 мМ, 1,2-бис(o-аминофенокси)этан-N,N,N',N'-тетрауксусная кислота (BAPTA) 10 мМ, 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота (HEPES) 10 мМ, нормализованную магнезию (NMG) применяли для доведения значения pH до 7,2;
• внеклеточный раствор: NaCl 145 мМ, KCl 5 мМ, глюкоза 10 мМ, MgCl2 1 мМ, CaCl2 2 мМ, HEPES 10 мМ, HCl применяли для доведения значения pH до 7,4.
Мембранный потенциал деполяризовался от -80 мВ до +20 мВ в течение 1000 мс в методе фиксации потенциала для всей совокупности клеток и затем реполяризовался до -40 мВ в течение 1000 мс, за это время следовый ток выходящих наружу hERG K+ потоков зафиксировали. В связи с этим концентрации соединений, которые требуются для 50% ингибирования тока, представляли как IC50.
(Испытание активности ингибирования hERG, IC50)
Риск кардиотоксичности возрастает в случае многих лекарственных препаратов. Некоторые из них были изъяты из рыночного оборота, потому что они вызывали внезапную смерть из-за их кардиотоксичности. Кардиотоксичность лекарственных препаратов сопровождается увеличением QT интервалов на электрокардиограммах. В частности, для большинства лекарственных препаратов, удлиняющих QT интервалы, известна способность ингибирования IKr (hERG K+ потоков) каналов (Bernard Fermini and Anthony A. Fossa, Nature Reviews Drug Discovery, 2003, 2, 439-447). hERG канал демонстрирует наиболее существенное влияние на кардиотоксичность среди IKr каналов. В этом примере риск кардиотоксичности оценивали, используя клетки млекопитающих, в которых экспрессировался человеческий канальный ген hERG, которые на международном уровне выявляются как система (ICH guideline, S7B, Step4, 12, May, 2005). Несмотря на то, что фармацевтическую активность лекарственных препаратов следует принимать во внимание, лекарственный препарат оценивают по наличию низкого риска кардиотоксичности, когда его IC50 составляет 10 мкМ или выше. В этих испытаниях большинство тестируемых соединений, как было установлено, превосходят этот критерий. Имеющее наивысшее значение IC50, соединение Al оценивалось как более безопасное, чем соединение A, и соединения B1, B2 и B3 оценивались как более безопасные, чем соединение B.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ МИМЕТИКИ ОБРАТНОГО ПОВОРОТА И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2457210C2 |
| ПЯТИЧЛЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ ОКСОКАРБОНОВОЙ КИСЛОТЫ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2832934C1 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА | 2002 |
|
RU2320658C2 |
| НОВОЕ ПРОИЗВОДНОЕ ОКСАЗОЛИДИНОНА И ВКЛЮЧАЮЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2012 |
|
RU2617408C2 |
| СЛОЖНОЭФИРНЫЕ ПРОИЗВОДНЫЕ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2005 |
|
RU2431480C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2726622C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛПИРИМИДИНОНА, СПОСОБ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2001 |
|
RU2263676C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ СИГМА РЕЦЕПТОРА | 2007 |
|
RU2451015C2 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ТЕРАПЕВТИЧЕСКОГО ЭФФЕКТА ИНГИБИТОРА LSD1 НА ОСНОВЕ ЭКСПРЕССИИ INSM1 | 2018 |
|
RU2789449C2 |
| ТЕТРАГИДРОИНДЕНОИНДОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИОКСИДАНТНОЙ АКТИВНОСТЬЮ, ПРОТИВООКИСЛИТЕЛЬНАЯ КОМПОЗИЦИЯ И СПОСОБ СТАБИЛИЗАЦИИ СОЕДИНЕНИЙ, ВОСПРИИМЧИВЫХ К ОКИСЛЕНИЮ | 1990 |
|
RU2104270C1 |

Описываются новые производные пиразинтриазинона общей формулы
 ,
,
в которой Ra - C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил; Rb - фенил, замещенный ацилом, алкокси, гидроксилом, нитро, NH2, тиоалкильной группой или -C(=O)Re, где Re - C1-C6 алкил, или группой -C(=O)Re, где Re представляет собой C1-C6 алкил или циклопропил и Rp представляет собой -H, -РО3Н2, -HPO3 -Na+, -PO3 2-Na2 +, фармацевтическая композиция, их содержащая, и способ лечения острого миелолейкоза. Новые соединения являются миметиками обратного действия и могут найти применение в медицине. 3 н. и 6 з.п. ф-лы, 1 ил., 3 табл., 24 пр.
1. Соединение химической формулы I:
Химическая формула I
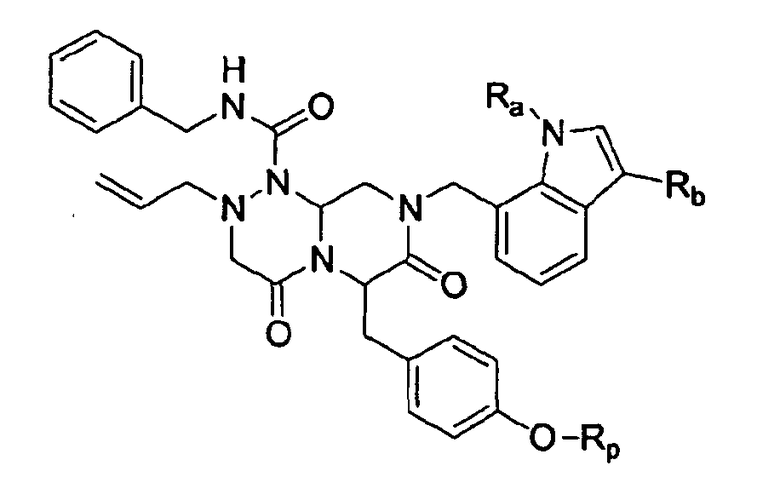
в которой
Ra представляет собой C1-C6 алкильную группу, C2-C6 алкенил или C2-C6 алкинильную группу;
Rb представляет фенил, замещенный ацилом, алкокси, гидроксилом, нитро, NH2, тиоалкильной группой или -C(=O)Re, в которой Re представляет собой C1-C6 алкильную группу, или
-C(=O)Re, в которой Re представляет собой C1-C6 алкильную группу или циклопропильную группу; и
Rp представляет собой -H, -РО3Н2, -HPO3 -Na+, -PO3 2-Na2 +.
2. Соединение по п.1, в котором
Ra представляет собой C1-C6 алкильную группу или С2-С6 алкенильную группу;
Rb представляет собой -C(=O)Re, в которой Re представляет собой C1-C6 алкил; и
Rp представляет собой -H, -РО3Н2, -HPO3 -Na+ или -PO3 2-Na2 +.
3. Соединение по п.1, в котором
Ra представляет собой метил;
Rb представляет собой -C(=O)Re, в которой Re представляет собой C1-C6 алкил; и
Rp представляет собой -H.
4. Соединение по п.1, в котором
Ra представляет собой метил;
Rb представляет собой -C(=O)Re, в которой Re представляет собой C1-C6 алкил; и
Rp представляет собой -РО3Н2, -HPO3 -Na+ или -PO3 2-Na2 +.
5. Соединение по п.1, в котором соединение, представленное химической формулой I, представляет собой
бензиламид 8-(3-ацетил-1-метил-1Н-индол-7-илметил)-2-аллил-6-(4-гидроксибензил)-4,7-диоксогексагидропиразино[2,1-с][1,2,4]триазин-1-карбоновой кислоты,
бензиламид 2-аллил-8-[3-(3,3-диметилбутирил)-1-метил-1Н-индол-7-илметил]-6-(4-гидроксибензил)-4,7-диоксогексагидропиразино[2,1-c][1,2,4]триазин-1-карбоновой кислоты,
бензиламид 2-аллил-8-(3-циклопропанкарбонил-1-метил-1Н-индол-7-илметил)-6-(4-гидроксибензил)-4,7-диоксогексагидропиразино[2,1-c][1,2,4]триазин-1-карбоновой кислоты,
бензиламид 2-аллил-6-(4-гидроксибензил)-8-[1-метил-3-(3-метилбутирил)-1Н-индол-7-илметил]-4,7-диоксогексагидропиразино[2,1-c][1,2,4]триазин-1-карбоновой кислоты,
бензиламид 2-аллил-8-(3-бутирил-1-метил-1Н-индол-7-илметил)-6-(4-гидроксибензил)-4,7-диоксогексагидропиразино[2,1-с][1,2,4]триазин-1-карбоновой кислоты,
бензиламид 2-аллил-8-(3-циклопропанкарбонил-1-этил-1Н-индол-7-илметил)-6-(4-гидроксибензил)-4,7-диоксогексагидропиразино[2,1-c][1,2,4]триазин-1-карбоновой кислоты,
бензиламид 2-аллил-8-(1-аллил-3-циклопропанкарбонил-1Н-индол-7-илметил)-6-(4-гидроксибензил)-4,7-диоксогексагидропиразино[2,1-c][1,2,4]триазин-1-карбоновой кислоты,
бензиламид 2-аллил-6-(4-гидроксибензил)-8-(1-метил-3-пентаноил-1H-индол-7-илметил)-4,7-диоксогексагидропиразино[2,1-с][1,2,4]триазин-1-карбоновой кислоты,
бензиламид 2-аллил-6-(4-гидроксибензил)-8-(1-метил-3-пропионил-1Н-индол-7-илметил)-4,7-диоксогексагидропиразино[2,1-с][1,2,4]триазин-1-карбоновой кислоты,
бензиламид 8-(3-ацетил-1-пропил-1Н-индол-7-илметил)-2-аллил-6-(4-гидроксибензил)-4,7-диоксогексагидропиразино[2,1-с][1,2,4]триазин-1-карбоновой кислоты,
бензиламид 2-аллил-8-[3-(3,3-диметилбутирил)-1-пропил-1Н-индол-7-илметил]-6-(4-гидроксибензил)-4,7-диоксогексагидропиразино[2,1-c][1,2,4]триазин-1-карбоновой кислоты,
бензиламид 2-аллил-8-[3-(3,3-диметилбутирил)-1-гексил-1Н-индол-7-илметил]-6-(4-гидроксибензил)-4,7-диоксогексагидропиразино[2,1-c][1,2,4]триазин-1-карбоновой кислоты или
бензиламид 2-аллил-8-(1-бутил-3-циклопропанкарбонил-1Н-индол-7-илметил)-6-(4-гидроксибензил)-4,7-диоксогексагидропиразино[2,1-c][1,2,4]триазин-1-карбоновой кислоты.
6. Фармацевтическая композиция для лечения рака, содержащая соединение по любому из пп.1-5 и фармацевтически приемлемый наполнитель.
7. Фармацевтическая композиция по п.6, где рак представляет собой острый миелолейкоз (AML).
8. Способ лечения острого миелолейкоза (AML), включающий введение пациенту с AML эффективного количества фармацевтической композиции по п.6.
9. Способ по п.8, в котором введение включает парентеральное введение фармацевтической композиции пациенту.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| МИМЕТИКИ С ОБРАТНОЙ КОНФИГУРАЦИЕЙ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2004 |
|
RU2342387C2 |
| RU 2010119447 A, 27.11.2011 | |||

