Настоящее изобретение касается применения некоторых производных 5-гидроксиметилоксазолидин-2-она для профилактики или лечения кишечных заболеваний, которые вызываются Clostridium difficile, Clostridium perfringens или Staphylococcus aureus.
Определенные штаммы Clostridium difficile, Clostridium perfringens или Staphylococcus aureus продуцируют токсины, которые вызывают диарею. Распространение штаммов, продуцирующих эти токсины, часто связывают с применением антибиотиков.
Среди этих штаммов, Clostridium difficile, анаэробная, формирующая споры, грамположительная бактерия, которая продуцирует два энтеротоксина (А и В), вызывает не только диарею, но также более серьезные кишечные расстройства такие, как угрожающий жизни фибринозно-пленчатый колит. В настоящее время общим источником инфекционной диареи в индустриализованном мире является длительное нахождение в больничных условиях. За последние годы скорость распространения заболевания прогрессивно возрастает и является важнейшей клинической проблемой в Северной Америке и Европе. Факторы риска включают предшествующее лечение антибиотиками, возраст и подверженную риску иммунную систему, являющуюся следствием, например, цитотоксичной химиотерапии или трансплантации органа.
Распространенным лечением Clostridium difficile ассоциированной диареи (CDAD) является лечение с помощью ванкомицина или метронидазола. В обоих случаях наблюдаются высокие скорости рецидива и невозможность предотвратить продуцирование токсинов и спор. Кроме того, лечение ванкомицином или метронидазолом служит дополнительным фактором, способствующим возникновению ванкомицин-резистентных Enterococcus spp.(Е. faecalis и Е. faecium) и Staphylococcus aureus штаммов (VRE и VRSA, соответственно) в кишечнике (см. W.N. Al-Nassir et al., Antimicrob. Agents Chemother., published ahead of print on 28 April 2008). Энтерококки являются грамположительными кокками, которые представляют собой нормальные микроорганизмы, населяющие желудочно-кишечный тракт. Однако они также могут становиться серьезными патогенами, вызывающими эндокардит и инфекции мочевого тракта и органов кровообращения. Установлено, что кишечная колонизация VRE может продолжаться в течение нескольких лет, исполняя роль резервуара потенциальной инфекции для инфицированного пациента и источника распространения VRE для других пациентов. VRE может являться сопутствующей инфекцией у пациентов, инфицированных С.difficile, или более обобщенно, инфицировать пациентов с высокой степенью риска таких, как гематологические и онкологические больные, пациенты интенсивной терапии, пожилые пациенты, находящиеся на долговременном больничном лечении, и пациенты, перенесшие трансплантацию твердого органа.
Поэтому существует необходимость в улучшении лечения CDAD и родственных инфекций, в частности, путем получения соединений с высокой активностью против С.Difficile, которые значительно снижают токсиновую нагрузку, продуцирование спор, и которые преодолевают VRE, оказывая при этом минимальное влияние на флору, вырабатываемую в кишечнике.
В публикациях WO 03/032962, WO 2004/096221 и WO 2005/058888 раскрываются антибактериальные соединения, содержащие оксазолидиновый фрагмент и 1-циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту или 1-циклопропил-6-фтор-4-оксо-1,4-дигидро-[1,8]нафтиридин-3-карбоновую кислоту.
Кроме того, соединения формулы (I) по определению, приведенному далее, и их антибактериальная активность были ранее описаны в заявке на изобретение PCT/IB 2007/054557.
Различные варианты осуществления настоящего изобретения представлены далее:
i) Согласно первому основному варианту настоящее изобретение относится к соединениям формулы (I)

А обозначает N или СН; и
n равен 0 или 1;
или их фармацевтически приемлемым солям, для профилактики или лечения кишечных заболеваний, вызываемых бактерией, выбранной из Clostridium difficile, Clostridium perfringens или Staphylococcus aureus.
В следующих параграфах представлены определения различных химических фрагментов для соединений согласно изобретению. Указанные определения предназначены для единообразного применения во всем описании и в формуле изобретения, если не указано иначе и если определения не подлежат более широкому или, наоборот, более узкому толкованию.
Термин "предупреждающий", "предупреждать" или "профилактика", используемый со ссылкой на заболевание, подразумевает, что указанное заболевание не диагностируется у пациента или животного, или что, хотя животное или человек заражены болезнью, часть или все симптомы заболевания либо ослаблены, либо отсутствуют.
Термин "лечащий", "обработка" или "лечение", используемый со ссылкой на заболевание, подразумевает, что указанное заболевание имеет место у пациента или животного, или что, хотя животное или человек остаются зараженными болезнью, часть или все симптомы заболевания либо ослаблены, либо устранены.
Термин "фармацевтически приемлемые соли" относится к нетоксичным, неорганическим или органическим кислотно - и/или основно-аддитивным солям. Ссылка может быть сделана на публикацию: "Salt selection for basic drugs", Int. J. Pharm., (1986), 33, 201-217.
Термин «галоген» подразумевает фтор, хлор, бром или йод, предпочтительно фтор или хлор.
Когда речь не идет о температурах, термин "приблизительно", расположенный перед численной величиной "X", относится в обычном применении к интервалу, составляющему от Х минус 10% Х к Х плюс 10% X, и предпочтительно, к интервалу, составляющему от Х минус 5% Х к Х плюс 5% X. В особом случае температур, термин "приблизительно", расположенный перед температурой "Y", относится в обычном применении к температурному интервалу, составляющему от Y минус 10°С до Y плюс 10°С, и предпочтительно, к интервалу, составляющему от Y минус 5°С до Y плюс 5°С; кроме того, термин «комнатная температура» в настоящей патентной заявке подразумевает температуру 25°С.
ii) Предпочтительно, соединение формулы (I), представленное в варианте i), или его фармацевтически приемлемая соль позволяет эффективно предупреждать или лечить диарейные заболевания, вызываемые энтеротоксиногенными штаммами Clostridium difficile, Clostridium perfringens или Staphylococcus aureus без увеличения концентрации ванкомицин-резистентных энтерококков (VRE) в кишечнике.
iii) Более предпочтительно, соединение формулы (I), представленное в варианте i), или его фармацевтически приемлемая соль, позволяет эффективно предупреждать или лечить диарейные заболевания, вызываемые энтеротоксиногенными штаммами Clostridium difficile, Clostridium perfringens или Staphylococcus aureus, и снижать концентрацию VRE в кишечнике.
iv) Предпочтительно, соединение формулы (I), представленное в одном из вариантов осуществления изобретения i)-iii), или их фармацевтически приемлемые соли должны быть такими, где А обозначает СН.
v) Предпочтительно также, что соединение формулы (I), представленное в одном из вариантов осуществления изобретения i)-iv), или их фармацевтически приемлемые соли должны быть такими, где n равен 1.
vi) Согласно предпочтительным подвариантам вариантов осуществления изобретения i)-iii) соединение формулы (I) или его фармацевтически приемлемая соль должны быть выбраны из группы, включающей:
1-циклопропил-6-фтор-7-{4-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-4-гидроксипиперидин-1-ил}-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту,
1-циклопропил-6-фтор-7-{4-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-4-гидроксипиперидин-1-ил}-4-оксо-1,4-дигидро-[1,8]нафтиридин-3-карбоновую кислоту,
1-циклопропил-6-фтор-7-{3-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-3-гидроксипирролидин-1-ил}-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту;
и их фармацевтически приемлемые соли.
vii) Согласно более предпочтительным подвариантам вариантов осуществления изобретения i)-iii) соединение формулы (I) или его фармацевтически приемлемая соль должны представлять собой 1-циклопропил-6-фтор-7-{4-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-4-гидроксипиперидин-1-ил}-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту или ее фармацевтически приемлемую соль.
viii) Согласно одному аспекту вариантов осуществления изобретения i)-vii) настоящее изобретение относится к соединениям формулы (I), представленным в одном из вариантов i)-vii), или их фармацевтически приемлемым солям, для лечения кишечных заболеваний, вызываемых бактерией, выбранной из Clostridium difficile, Clostridium perfringens и Staphylococcus aureus.
ix) Согласно одному предпочтительному аспекту варианта viii) настоящее изобретение относится к соединениям формулы (I), представленным в одном из вариантов осуществления изобретения i)-vii), или их фармацевтически приемлемым солям, для лечения кишечных заболеваний, вызываемых Clostridium difficile (предпочтительно, продуцирующим токсин штаммом Clostridium difficile).
х) Согласно другому главному аспекту вариантов осуществления изобретения i)-vii) настоящее изобретение относится к соединениям формулы (I), представленным в одном из вариантов осуществления изобретения i)-vii), или их фармацевтически приемлемым солям, для профилактики кишечных заболеваний, вызываемых бактерией, выбранной из Clostridium difficile, Clostridium perfringens и Staphylococcus aureus.
xi) Согласно одному предпочтительному аспекту варианта х) настоящее изобретение относится к соединениям формулы (I), представленным в одном из вариантов осуществления изобретения i)-vii), или их фармацевтически приемлемым солям, для профилактики кишечных заболеваний, вызываемых Clostridium difficile (предпочтительно, продуцирующим токсин штаммом Clostridium difficile).
xii) Пациенты с кишечными заболеваниями, отмеченными в вариантах i)-xi), предпочтительно, относятся к группе пациентов, включающей пациентов, прошедших лечение другими антибиотиками или антивирусную терапию, иммунологически «скомпрометированных» пациентов вследствие проведения цитотоксической химиотерапии или трансплантации органа, пациентов старшего возраста (65 лет или старше) или пациентов, подвергающихся интенсивной терапии, или долговременно пребывающих в больничных стационарах.
xiii) Еще один главный вариант осуществления настоящего изобретения относится к способу профилактики или лечения пациента с кишечным заболеванием, которое вызвано бактерией, выбранной из Clostridium difficile, Clostridium perfringens и Staphylococcus aureus, заключающемуся во введении указанному пациенту эффективного количества соединения формулы (I), представленного в одном из вариантов осуществления изобретения i)-vii), или фармацевтически приемлемой соли такого соединения, для продолжительной успешной профилактики или лечения кишечного заболевания.
xiv) Способ по варианту xiii) должен, предпочтительно, предотвращать или лечить диарейные заболевания, связанные с энтеротоксиногенными штаммами Clostridium difficile, Clostridium perfringens или Staphylococcus aureus, без увеличения концентрации ванкомицин-резистентных энтерококков (VRE) в кишечнике.
xv) Более предпочтительно, способ по варианту xiii) должен предотвращать или лечить диарейные заболевания, связанные с энтеротоксиногенными штаммами Clostridium difficile, Clostridium perfringens или Staphylococcus aureus, и снижать концентрацию VRE в кишечнике.
xvi) Согласно одному из аспектов вариантов осуществления изобретения xhi)-xv) настоящее изобретение относится к способу лечения пациента с кишечным заболеванием, вызываемым бактерией, выбранной из Clostridium difficile, Clostridium perfringens и Staphylococcus aureus, заключающемуся во введении указанному пациенту эффективного количества соединения формулы (I), представленного в одном из вариантов осуществления изобретения i)-vii), или фармацевтически приемлемой соли такого соединения, для длительного положительного эффекта при лечении кишечного заболевания.
xvii) Согласно одному предпочтительному аспекту варианта xvi) настоящее изобретение относится к способу лечения пациента с кишечным заболеванием, вызываемым бактерией, выбранной из Clostridium difficile (предпочтительно, продуцирующим токсин штаммом Clostridium difficile).
xviii) Согласно другому аспекту вариантов xih)-xv) настоящее изобретение относится к способу профилактики пациента с кишечным заболеванием, вызываемым бактерией, выбранной из Clostridium difficile, Clostridium perfringens и Staphylococcus aureus, заключающемуся во введении указанному пациенту эффективного количества соединения формулы (I), представленного в одном из вариантов осуществления изобретения i)-vii), или фармацевтически приемлемой соли такого соединения, для продолжительной успешной профилактики кишечного заболевания.
xix) Согласно одному предпочтительному аспекту варианта xviii) настоящее изобретение относится к способу профилактики пациента с кишечным заболеванием, вызываемым бактерией, выбранной из Clostridium difficile (предпочтительно, токсином, продуцируемым штаммом Clostridium difficile).
хх) Предпочтительно, пациенты, подвергаемые лечению способом по одному из вариантов xiii)-xix), относятся к группе пациентов, которые прошли лечение другими антибиотиками или антивирусную терапию, иммунологически «скомпрометированные» пациенты вследствие проведения цитотоксической химиотерапии или трансплантации органа, пациенты старшего возраста (65 лет или старше) или пациенты, подвергающиеся интенсивной терапии, или долговременно пребывающие в больничных стационарах.
xxi) Следующий аспект настоящего изобретения относится к применению соединения формулы (I), представленного в одном из вариантов осуществления изобретения i)-vii), или фармацевтически приемлемой соли такого соединения для получения лекарственного средства, предназначенного для профилактики или лечения кишечного заболевания, вызываемого бактерией, выбранной из Clostridium difficile, Clostridium perfringens и Staphylococcus aureus.
xxii) Согласно одному из аспектов варианта xxi) настоящее изобретение относится к применению соединения формулы (I), представленного в одном из вариантов осуществления изобретения i)-vii), или фармацевтически приемлемой соли такого соединения для получения лекарственного средства, предназначенного для лечения кишечного заболевания, вызываемого бактерией, выбранной из Clostridium difficile, Clostridium perfringens и Staphylococcus aureus.
xxiii) Согласно одному из предпочтительных аспектов варианта xxii) кишечные заболевании, предназначенные для лечения, должны быть вызваны Clostridium difficile (предпочтительно, токсином, продуцируемым штаммом Clostridium difficile).
xxiv) Согласно другому аспекту варианта xxi) настоящее изобретение должно относится к применению соединения формулы (I), представленного в одном из вариантов осуществления изобретения i)-vii), или фармацевтически приемлемой соли такого соединения, для получения лекарственного средства, предназначенного для профилактики кишечного заболевания, вызываемого бактерией, выбранной из Clostridium difficile, Clostridium perfringens и Staphylococcus aureus.
xxv) Согласно одному предпочтительному аспекту варианта xxiv) кишечное заболевание, предназначенное для профилактики, должны быть вызвано Clostridium difficile (предпочтительно, токсином, продуцируемым штаммом Clostridium difficile).
xxvi) Пациенты, для которых получают лекарственное средство согласно одному из вариантов осуществления изобретения xxi)-xxv), должны предпочтительно относиться к группе пациентов, которые прошли лечение другими антибиотиками или антивирусную терапию, иммунологически «скомпрометированным» пациентам вследствие проведения цитотоксической химиотерапии или трансплантации органа, пациентам старшего возраста (65 лет или старше) или пациентам, подвергающимся интенсивной терапии, или долговременно пребывающим в больничных стационарах.
xxvii) Другой аспект настоящего изобретения относится к способу профилактики или лечения животного (например, собаки, кошки, свиньи, коровы или лошади), страдающего кишечным заболеванием, вызываемым бактерией, выбранной из Clostridium difficile, Clostridium perfringens и Staphylococcus aureus, заключающемуся во введении указанному животному эффективного количества соединения формулы (I), представленного в одном из вариантов осуществления изобретения i)-vii), или фармацевтически приемлемой соли такого соединения, для длительного положительного эффекта при лечении кишечного заболевания.
xxviii) Согласно одному аспекту варианта xxvii) настоящее изобретение относится к способу лечения животного (например, собаки, кошки, свиньи, коровы или лошади), страдающего кишечным заболеванием, вызываемым бактерией, выбранной из Clostridium difficile, Clostridium perfringens и Staphylococcus aureus, заключающемуся во введении указанному животному эффективного количества соединения формулы (I), представленного в одном из вариантов осуществления изобретения i)-vii), или фармацевтически приемлемой соли такого соединения, для длительного положительного эффекта при лечении кишечного заболевания.
xxix) Согласно другому аспекту варианта xxvii) настоящее изобретение должно относится к способу профилактики животного (например, собаки, кошки, свиньи, коровы или лошади), страдающего кишечным заболеванием, вызываемым бактерией, выбранной из Clostridium difficile, Clostridium perfringens и Staphylococcus aureus, заключающемуся во введении указанному животному эффективного количества соединения формулы (I), представленного в одном из вариантов осуществления изобретения i)-vii), или фармацевтически приемлемой соли такого соединения, для продолжительной успешной профилактики кишечного заболевания.
Кишечные заболевания, подлежащие профилактике или лечению согласно вариантам осуществления настоящего изобретения i)-xxix) включают, предпочтительно диарею, колит и фибринозно-пленчатый колит.Предпочтительно, указанные кишечные заболевания вызываются Clostridium difficile (и, в частности, продуцирующим токсин штаммом Clostridium difficile).
Наиболее подходящим путем введения соединений формулы (I), применяемых согласно вариантам настоящего изобретения i)-xxix), является оральный путь. Введение может быть ежедневным (например, от одного до четырех раз в день) или осуществляться с меньшей частотой (например, однократно через день или однократно или дважды в неделю).
Точные дозы введения соединения формулы (I), представленного в одном из вариантов осуществления изобретения i)-vii), или его фармацевтически приемлемой соли должны определяться лечащим врачом или ветеринаром. Предполагается, что количество от 0,5 до 50 мг соединения формулы (I) или его фармацевтически приемлемой соли на кг массы тела пациента в день (например, количество от 1 до 5 мг соединения формулы (I) или его фармацевтически приемлемой соли на кг массы тела пациента в день), принимаемое один или два раза ежедневно в течение от 3 до 15 дней (например, в течение от 7 до 14 дней), должно быть приемлемым.
Получение фармацевтических композиций, содержащих соединения формулы (I), представленных в одном из вариантов осуществления изобретения i)-vii) или их фармацевтически приемлемых солей, может быть осуществлено методом, известным любому специалисту в области техники (см., например, Remington, The Science and Practice of Pharmacy, 21st Edition (2005), Part 5, "Pharmaceutical Manufacturing" [published by Lippincott Williams & Wilkins]), путем внесения описанного соединения формулы (I) или его фармацевтически приемлемых солей, необязательно в комбинации с другими терапевтически приемлемыми веществами, в лекарственную форму для введения вместе с приемлемым, нетоксичным, инертным, терапевтически совместимым твердым или жидким носителем и, если необходимо, стандартными фармацевтическими наполнителями.
Соединения формулы (I) могут быть получены в соответствии с настоящим изобретением при использовании методов, описанных далее.
Получение соединений формулы (I)
Аббревиатуры:
Следующие аббревиатуры используются в описании и примерах:
АсОН - уксусная кислота, AD-mix α - 1,4-бис-(дигидрохинин)фталазин, K3Fe(CN)6. К2СО3 и K2OsO4·2H2O, AD-mix β - 1,4-бис-(дигидрохинидин)фталазин, K3Fe(CN)6, К2СО3 и K2OsO4·2H2O, Alloc - аллилоксикарбонил. Бок - трет-бутоксикарбонил, t-ВиОК - трет-бутипат калия, Кбз - бензилоксикарбонил, CDAD - Clostridium difficile, вызывающая диарею, CFU - колониеобразующие единицы, CLSI - Клинический лабораторный институт стандартов, ДБУ - 1,8-диазабицикло[5.4.0]удец-7-ен, ДХМ - дихлорметан, ДИАД - диизопропилазадикарбоксилат, ДИПЭА - N,N-диизопропилэтиламин, ДМФ - 'М,Н-диметилформамид, ДМСО -диметилсульфоксид, ЭА - этилацетат, ЭДК - гидрохлорид 1-(диметиламинопропил)-3-этилкарбодиимида, ЭСИ - электрораспылительная ионизация, эфир или Et2O - диэтиловый эфир, УХ - ускоренная хроматография, ч - час(ы), гекс.- н-гексан, IC50 - концентрация, снижающая воздействие на 50%, LZD-R - линезолид-резистентный, MeCN - ацетонитрил, м-ХПБК - м-хлорпербензойная кислота, МИК - минимальная ингибирующая концентрация для ингибирования роста бактерий, МИК90 - минимальная ингибирующая концентрация для ингибирования роста ≥90% штамма, MRSA - метициллин резистентный Staphylococcus aureus, MeOH - метанол, МС - масс-спектроскопия, NaOMe - метилат натрия, N-МП - N-метилпирролидинон, OD595 - оптическая плотность, измеряемая при 595 нМ, орг.- органический, Pd/C или Pd(OH)2/C - палладий или дигидроксипалладий на угле, РРh3 - трифенилфосфин, КТ - комнатная температура, насыщ. - насыщенный, SiO2 - силикагель, ТБДМСС1 - тpeт-бутилдиметилсилилхлорид, ТЭА - триэтиламин, ТФК -трифторуксусная кислота, ТГФ - тетрагидрофуран, ТМСС1 -триметилсилилхлорид.
Общие препаративные методы:
Соединения формулы (I) могут быть получены в соответствии с настоящим изобретением посредством
а) реакции соединения формулы (II)

с соединением формулы (III)
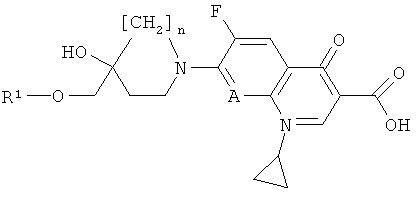
где n и А имеют значения, представленные в формуле (I), и R1 обозначает C1-3-залкилсульфонил (например, метилсульфонил), трифторметилсульфонил или арилсульфонил (например, фенил- или n-толилсульфонил), предпочтительно в температурном интервале приблизительно от 10°С до 100°С (более предпочтительно приблизительно от 40°С до 80°С), в присутствии неорганического основания такого, как К2СО3, или органического основания такого, как ТЭА, в органическом растворителе (например, ДМФ); или
б) реакции соединения формулы (II) с соединением формулы (IV)
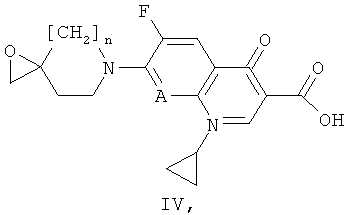
где n и А имеют значения, представленные в формуле (I) по описанию в пункте а). Указанные соединения формулы (IV) могут быть получены путем обработки соединения формулы (III) в присутствии органического основания (например, ТЭА) или неорганического основания (например, К2СО3 или метилата щелочного металла такого, как NaOMe, или гидрида щелочного металла такого, как NaH) в органическом растворителе (например, ДМФ); или
в) реакции соединения формулы (V)
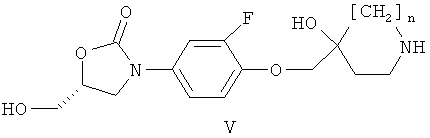
где n имеет значение, указанное в формуле (I), с соединением формулы (VI)
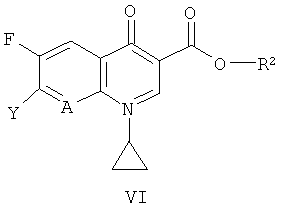
где А имеет значение, указанное в формуле (I), Y обозначает галоген и R2 обозначает водород, BF2 или В(ОС(=O)С1-4алкил)2, С1-5алкил (например, метил, этил, н-пропил, изопропил или трет-бутил), аллил, арил-С1-5алкил (например, бензил, n-нитробензил или n-метоксибензил), три-(С1-5алкил)силил (например, триметилсилил или трет-бутилдиметилсилил) или диарил-С1-5алкилсилил (например, трет-бутилдифенилсилил), предпочтительно, в температурном интервале приблизительно от 10°С до 100°С, более предпочтительно приблизительно от 40°С до 80°С, в присутствии органического основания такого, как ТЭА или ДИПЭА, в органическом растворителе, например, N-МП; или
г) превращения соединения формулы (VII)
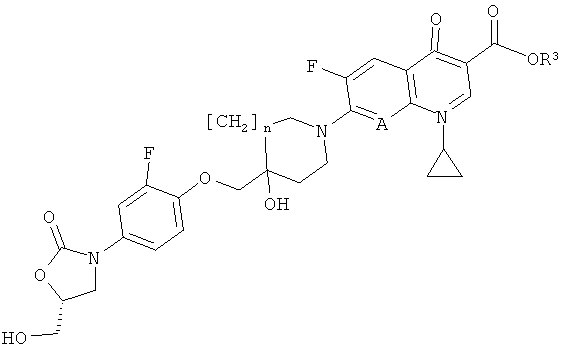
где R3 обозначает С1-5алкил (например, метил, этил, н-пропил, изопропил или трет-бутил), арил-С1-5алкил (например, бензил, п-нитробензил или п-метоксибензил), аллил, три-С1-5алкилсилил (например, триметилсилил или трет-бутилдиметилсилил) или диарил-С1-5алкилсилил (например, трет-бутилдифенилсилил) и n и А представлены в формулы (I), в соответствующее соединение формулы (I) посредством гидролиза, омыления или гидрирования (общие методы осуществления таких реакций можно найти в публикации:
Protecting groups, Kocienski, P.J., Thieme (1994)).
Рассматривая приведенные выше методы, следует отметить:
- относительно варианта а): соединение формулы (III) может быть также заменено его сложным эфиром, то есть соединением формулы (IIIE)
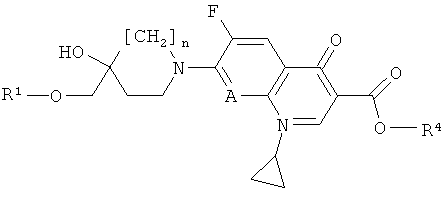 ,
,
где n, А и R1, представлены в формулы (III), и R4 представляет собой алкил, аллил или арилалкил, при этом в каждом случае стадия удаления сложноэфирной защитной группы должна следовать за реакцией соединения формулы (IIIE) с соединением формулы (II) (общие методы удаления сложноэфирной защитной группы можно найти в публикации: Protecting groups, Kocienski, P.J., Thieme (1994));
- относительно варианта а); соединение формулы (II) может быть также заменено его силильным сложным эфиром, то есть соединением формулы (IIPG)
 ,
,
где PG2 представляют собой силильную защитную группу для спиртовой функции такую, как три-C1-5алкилсилил (например, триметилсилил или трет-бутилдиметилсилил) или диарил-С1-5алкилсилил (например, трет-бутилдифенилсилил), при этом в каждом случае стадия удаления защитной группы должна следовать за реакцией соединения формулы (III) или (IIIE) с соединением формулы (IIPG) (общие методы осуществления таких реакций можно найти в публикации: Protecting groups, Kocienski, P.J., Thieme (1994));
- относительно варианта в): соединение формулы (V) может быть также заменено соединением формулы (Vp)

где n представлено в формуле (V) и PG2 представляет собой защитную группу для спиртовой функции (например, алкилсилил или диарилалкилсилильную группу такую, как триметилсилил, трет-бутилдиметилсилил или трет-бутилдифенилсилил), при этом в каждом случае стадия удаления защитной группы должна следовать за реакцией соединения формулы (Vp) с соединением формулы (VI) (общие методы осуществления таких реакций можно найти в публикации: Protecting groups, Kocienski, P.J., Thieme (1994));
- относительно варианта в): когда R2 не является водородом, требуется дополнительная стадия по удалению сложноэфирной защитной группы (общие методы осуществления таких реакций можно найти в публикации: Protecting groups, Kocienski, P.J., Thieme (1994)), за исключением случаев, где R2 обозначает BF2 или В(ОС(=O)(С1-4алкил)2, когда гидролиз происходит уже при кислотной обработке.
Соединения формулы (I), полученные таким образом, при необходимости могут быть превращены в их соли, и предпочтительно в их фармацевтически приемлемые соли.
Кроме того, когда соединения формулы (I) получают в виде смесей энантиомеров, энантиомеры могут быть разделены с использованием методов, известных любому специалисту в области техники (например, путем получения и разделения диастереомерных солей или с помощью хроматографии на хиральной стационарной фазе). Когда соединения формулы (I) получают в виде смесей диастереомеров, они могут быть разделены путем соответствующей комбинации хроматографии на силикагеле, ВЭЖХ и кристаллизационной техники.
Получение различных синтетических промежуточных соединений:
Получение соединений формулы (II)
Соединения формулы (II) могут быть получены гидрированием соединений формулы (VIII)
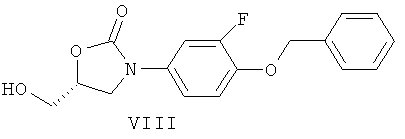
над катализатором из благородного металла такого, как палладий или платина, на угле, в растворителе таком, как ТГФ, МеОН или ЭА, в температурном интервале от 0°С до 40°С, или гидролизом в присутствии раствора НВr в воде или АсОН, в температурном интервале от 0°С до 80°С, в растворителе таком, как АсОН.
Получение соединения формулы (III)
Соединения формулы (III) могут быть получены в соответствии со схемой 1, представленной ниже:
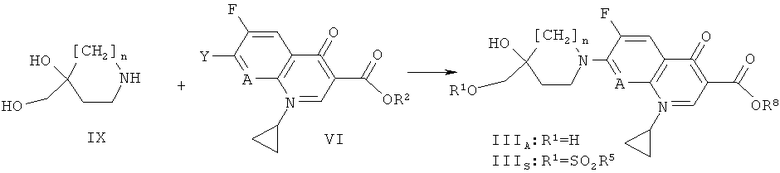
Схема 1
На схеме 1 R2 обозначает Н, алкил, аллил или арилалкил, а другие символы соответствуют символам, приведенным ранее.
Соединения формулы (III), где R1 обозначает SO2R5, R5 при этом обозначает алкил, трифторметил или арил, подобный фенилу или п-толилу, могут быть получены (схема 1) из соединений формулы (IIIA), где R обозначает водород, реакцией с соответствующими сульфонилхлоридами в присутствии органического основания такого, как ТЭА, в растворителе таком, как ДХМ или ТГФ, в температурном интервале от -10°С до 50°С. Соединения формулы (IIIA) могут быть получены реакцией соединений формулы (VI) с пиперидинами или пирролидинами формулы (IX) в присутствии органического основания такого, как ТЭА или ДИПЭА, в температурном интервале от 40°С до 100°С, в растворителе, таком, как ТГФ, ДМФ или N-МП. Если R обозначает бензил, карбоновая кислота формулы (IIIS) может быть освобождена от защитной группы согласно стандартным методам, описанным в публикации: Protecting groups, Kocienski, P.J., Thieme (1994) (например, гидрированием над Pd/C).
Получение соединений формулы (V)
Соединения формулы (V) могут быть получены, как представлено на приведенной ниже схеме 2.

Схема 2
Соединения формулы (V) могут быть получены удалением защитной группы с соединения формулы (XI), где PG представляет собой аминозащитную группу такую, как алкоксикарбонил (например, Бок), бензилоксикарбонил (например, Кбз), Аллок или бензил. Общие методы осуществления такой последовательности введения/удаления защитных групп вторичных аминов описаны в публикации: Protecting groups, Kocienski, P.J., Thieme (1994).
Соединения формулы (XI) могут быть получены реакцией соединений формулы (II) с соединениями формулы (X), где R1 обозначает SO2R5, R5 при этом обозначает алкил, трифторметил или арил, подобный фенилу или п-толилу, или реакцией соединения формулы (II) с соответствующими эпоксидами, полученными из соединений формулы (X). Указанные эпоксиды могут быть получены из соединений формулы (X), где R1 обозначает SO2R5, посредством обработки либо органическим основанием таким, как ТЭА, пиридин или ДБУ, либо неорганическим основанием таким, как К2СО3, в растворителе таком, как ТГФ, эфир или ДХМ, в температурном интервале от -10°С до 40°С. Соединения формулы (X), где R1 обозначает Н, являются либо коммерческими продуктами (например, соединениями (X), где PG3 обозначает Бок, n равен 1 и R1 обозначает Н или эпоксид, полученный из соединений формулы (X), где PG3=Бок, n равен 0), или могут быть получены, как описано далее. Альтернативно, соединения формулы (XI) могут быть получены реакцией соединения формулы (II) с соединениями формулы (X), где R1 обозначает Н, в условиях проведения реакции Митцунобу.
Получение соединения формулы (VI)
Соединения формулы (VI), где R2 обозначает водород, Me или Et, являются коммерческими продуктами (например, соединениями, где А обозначает СН, Y обозначает С1 и R2 обозначает Н, Me или Et, или Y обозначает F и R2 обозначает ВF2, или соединениями, где А обозначает N, Y обозначает С1 и R2 обозначает Н или Et). Соединения формулы (VI), где R2 обозначает В(ОС(=O)(С1-4алкил)2, могут быть получены из соединений формулы (VI), где R2 обозначает Н, согласно публикации WO 88/07998. Другие соединения формулы (VI) могут быть получены согласно стандартным методам из соединений формулы (VI), где R обозначает Н.
Получение соединений формулы (VII)
Соединения формулы (VII) могут быть получены реакцией конденсации соединений формулы (V) или, альтернативно, соединений формулы (Vp), описанных ранее, с соединениями формулы (VI), описанными ранее, за исключением того, что R2 представляет собой С1-5алкил (например, метил, этил, н-пропил, изопропил или трет-бугил), арил-С1-5алкил (например, бензил, п-нитробензил или п-метоксибензил), аллил, три-(С1-5алкил)силил (например, триметилсилил или wpew-бутилдиметилсилил) или диарил-С1-5алкилсилил (например, Wjpew-бутилдифенилсилил), в тех же условиях, которые описаны для реакции соединений формулы (V) с соединениями формулы (VI). Если используются соединения формулы (Vp), стадия удаления защитной группы может быть проведена после реакции конденсации.
Получение соединений формулы (VIII)
Соединения формулы (VIII) могут быть получены согласно методу, описанному в WO 2004/096221.
Получение соединений формулы (IX)
Соединения формулы (IX) могут быть получены путем удаления защитной группы у соединений формулы (X) (R1 обозначает Н), например, обработкой соответствующих Бок-защищенных соединений с помощью ТФК или гидрированием соответствующих Кбз-защищенных соединений над Pd/C.
Получение соединений формулы (X)
Соединения формулы (X) могут быть получены из метилиденпроизводных формулы (XII), как представлено на приведенной ниже схеме 3.

Схема 3
Соединения формулы (Хб), то есть соединения формулы (X), где R обозначает SO2R5, получают из соответствующих соединений формулы (Ха), где R1 обозначает Н, используя те же самые методы превращения соединений формулы (IIIA) в соединения формулы (IIIS). Соединения формулы (Ха) получают либо из известных метилиденовых производных формулы (XII) (например, таких, где n равен 0 и PG3 обозначает бензил, Бок или бензилоксикарбонил, см. ЕР 241206 и ЕР 550025; или таких, где n равен 1 и PG3 обозначает бензил, Бок, которые являются коммерческими продуктами), либо путем цис-дигидроксилирования, катализируемого тетроксидом осмия, или путем его асимметрической версии (дигидроксилирование по Чарлессу с использованием AD-mix α или β), как описано в J. Am. Chem. Soc., (1988), 110, 1968. Соединения формулы (Хв), то есть эпоксиды, получаемые из соединений формулы (Хб), получают либо внутримолекулярной циклизацией соединений формулы (Хб) с помощью неорганического основания такого, как К2СО3 или NaH, или органического основания такого, как ТЭА или ДБУ, либо путем эпоксидирования метиленовой двойной связи соединений формулы (XII) с помощью перкислоты такой, как м-ХПБК. Альтернативно, соединения формулы (Хв) могут быть также получены реакцией соответствующих оксопроизводных (являющихся коммерческими продуктами, когда n равен 0 или 1 и PG3 обозначает Кбз или Бок) с йодидом триметилсульфоксония или йодидом триметилсульфония в присутствии щелочи такой, как гидроксид калия, в полярном растворителе таком, как MeCN, в температурном интервале от 20 до 100°С (как описано в J.Am.Chem. Soc., (1965), 87, 1353-1364 и Tetrahedron Lett., (1987), 28, 1877-1878).
Некоторые варианты осуществления изобретения описаны в следующих примерах, которые служат для иллюстрации изобретения, ни в коей мере не лимитируя его объема.
Примеры
Все температуры представлены в °С. Все аналитические и препаративные исследования с помощью ВЭЖХ на нехиральных фазах осуществляются с использованием колонок на основе RP-C18. Аналитические ВЭЖХ исследования осуществляются на двух различных приборах с временным циклом приблизительно 2,5 мин и 3,5 мин, соответственно. Если не указано иначе, величины, приведенные для МС, соответствуют главному пику ((М+Н)+ с отклонением +/- 0,5 единицы). В ЯМР-спектрах константы сопряжения J представлены в Гц.
Стандартные методики обработки:
После растворения в подходящем органическом растворителе (см. соответствующие примеры) органическую фазу отделяют и последовательно промывают водой и рассолом. В случае, когда реакция протекает в водорастворимом растворителе (например, МеОН, ТГФ или ДМФ), объединенные водные слои снова промывают тем же самым растворителем, а затем объединенные органические фазы высушивают над MgSO4, фильтруют и выпаривают при пониженном давлении.
Стандартная хроматографическая методика:
Сырое вещество растворяют в минимальном количестве элюента (см. соответствующие примеры) и хроматографируют на SiO2. Соответствующие фракции объединяют и выпаривают при пониженном давлении.
Пример 1: 1-Циклопропил-6-фтор-7-{4-2-фтор-4-((R-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-4-гидроксипиперидин-1-ил}-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота:
1.i. (R)-3-(3-Фтор-4-гидроксифенил)-5-гидроксиметилоксазолидин-2-он:
Раствор (R)-3-(4-бензилокси-3-фторфенил)-5-гидроксиметилоксазолидин-2-она (6,34 г, получен согласно WO 2004/096221) в смеси ТГФ/МеОН (в соотношении 1:1; 200 мл) гидрируют над 10%-ным Pd/C (1 г) в течение ночи. Катализатор отфильтровывают, фильтрат выпаривают при пониженном давлении, а остаток переносят в этилацетат. Кристаллы отделяют фильтрованием, получая 3,16 г (70%-ный выход) продукта в виде бесцветного твердого вещества.
1Н ЯМР (ДМСОд6; δ м.д.): 3,5 (m, 1H), 3,64 (m, 1H), 3,74 (dd, J=8,8, 6,4, 1H), 3,99 (t, J=8,8, 1H), 4,64 (m, 1H), 5,16 (t, J=5,6, 1H), 6,93 (dd, J=9,7, 8,8, 1H), 7,08 (ddd, J=8,8, 2,6, 1,2. 1H), 7,45 (dd, J=13,5, 2,6, 1H), 9,66 (s, 1H).
МС(ЭСИ): 228,1.
l.ii. Бензиловый эфир 4-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-4-гидроксипиперидин-1-карбоновой кислоты
Раствор промежуточного соединения (l.i) (1,27 г) и бензиловый эфир 1-окса-6-аза-спиро[2.5]октан-6-карбоновой кислоты (1,60 г; получен согласно пат. США 4244961) растворяют в ДМФ (15 мл) и обрабатывают Nа2СО3 (1,16 г). Смесь нагревают при температуре 100°С в течение ночи. Остаток, полученный после обработки (ДХМ), перемешивают в этилацетате, и твердое вещество отделяют фильтрованием и последовательно промывают этилацетатом и гексаном, получая 2,52 г (94,5%-ный выход) продукта в виде бежевого твердого вещества.
1H ЯМР (ДМСОd6; δ м.д.): 1,57 (m, 4H), 3,14 (m, 2H), 3,54 (m, 1H), 3,64 (m, 1H). 3,79 (m, 5 H), 4,03 (t, J=9,1, 1 H), 4,66 (m, 1 H), 4,78 (s, 1 H), 5,05 (s, 2 H), 5,16 (t, J=5,6, 1 H), 7,18 (m, 2 H), 7,32 (m, 5 H), 7,55 (d, J=12, 1 H).
МС (ЭСИ): 475,0.
1.iii. (R)-3-[3-Фтор-4-(4-гидроксипиперидин-4-илметокси)фенил]-5-гидроксиметилоксазолидин-2-он
Суспензию промежуточного соединения (1.ii) (2,5 г) в смеси ЭА/МеОН (в соотношении 1:1; 100 мл) гидрируют над Pd/C в течение 48 ч, после чего нагревают при температуре 40°С и катализатор отфильтровывают. Фильтрат выпаривают при пониженном давлении, получая 1,61 г (89%-ный выход) продукта в виде желтого порошка.
1Н ЯМР (ДМСОd6; δ м.д.): 1,4-1,63 (m, 4H), 2,67 (m, 2H), 2,83 (m, 2H), 3,53 (dd, J=4,0, 12,0, 1H); 3,66 (dd, J=3,3, 12,0, 1H), 3,71 (s, 2H); 3,80 (m, 1H), 4,05 (t, J=9,0, 1H), 4,48 (s. 1H), 4,68 (m, 1H), 5,20 (s, 1H), 7,20 (m, 2H), 7,57 (d, 1H).
МС(ЭСИ): 341,5.
1.iv. 1-Циклопропил-6-фтор-7- {4-[2-фтор-4-((К)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-4-гидроксипиперидин-1-ил}-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
Раствор промежуточного соединения (1.iii) (200 мг), комплекса 7-хлор-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты с диацетатом бора (241 мг; получен по описанию в WO 88/07998) и ДИПЭА (100 м кл) в N-MH (2 мл) перемешивают при температуре 85°С в течение 5 ч. Реакционную смесь выпаривают при пониженном давлении, остаток переносят в 5-молярный раствор НСl в МеОН (3 мл) и перемешивают.Образовавшееся твердое вещество отделяют фильтрованием и промывают МеОН, получая 230 мг (67%-ный выход) продукта в виде желтого твердого вещества.
1Н ЯМР (ДМСОd6; δ 8 м.д.): 1.66-1.35 (m, 4H), 1,75 (d, J=12,8, 2H), 1,95 (m, 2H), 3,33 (t расширенный, J=11,0, 2H), 3,57 (m, 3H), 3,67 (dd, J=12,3, 3,3, 1H), 3,83 (m, 2H), 3,92 (s, 2H), 4,06 (t, J=9,0, 1H), 4,69 (m, 1H), 7,24 (m, 2H), 7,60 (m, 2H), 7,90 (d, J=13,3, 1H), 8,66 (s, 1H).
МС(ЭСИ): 585,9.
Пример 2: 1-Циклопропил-6-фтор-7-{4-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-4-гидроксипиперидин-1-ил}-4-оксо-1,4-дигидро-[1,8]нафтиридин-3-карбоновая кислота
Раствор 7-хлор-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты (166 мг; коммерческий продукт) и промежуточного соединения (1.iii) (200 мг) в N-МП (5 мл) обрабатывают ТЭА (0,32 мл) и ТМСС1 и нагревают при температуре 85°С в течение 5 ч. Реакционную смесь выпаривают при пониженном давлении, а остаток переносят в 5-молярный раствор НС1 в МеОН (3 мл) и перемешивают в течение 30 мин. Затем раствор выпаривают при пониженном давлении, а остаток переносят в этилацетат. Образовавшееся твердое вещество отделяют фильтрованием и промывают этилацетатом, получая 271 мг (78%-ный выход) продукта в виде желтого твердого вещества.
1Н ЯМР (ДМСОd6; δ м.д.): 0,89-1,27 (m, 4Н); 1,78 (d, J - 12,8, 2H); 1,90-2,04 (m, 2H); 3,53-3,88 (m, 6H); 3,88 (s, 2H), 4,06 (t, J=9,0, 1H), 4,42 (d расширенный, J=13,2, 2H), 4,44 (m, 1H); 7,11 (m, 2H); 7,55 (d, J=14,5, 1H); 8,05 (d, J=13,5, 1H); 8,60 (s, 1H).
МС(ЭСИ): 586,8.
Пример 3: 1-Циклопропил-6-фтор-7-{(RS)-3-Г2-фтор-4-(R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-3-гидроксипирролидин-1-ил}-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
3.i. Бензиловый эфир диаллилкарбаминовой кислоты
Бензоилхлорид (15,5 мл) добавляют по каплям в течение 30 мин к раствору диаллиламина (12,3 мл) и ТЭА (21 мл) в ДХМ (100 мл) при температуре 0°С. Реакционную смесь затем перемешивают при комнатной температуре в течение 16 ч. Остаток, полученный после обработки (ДХМ), очищают с помощью колоночной хроматографии (гексан/ЭА в соотношении 95:5), получая 20,71 г (88%-ный выход) продукта в виде бесцветной жидкости.
1Н ЯМР (ДМСОd6; δ м.д.): 3,83 (dt, J=1 и J=6, 4Н); 5,05-5,18 (m, 6H); 5.70-5,86 (m, 2H); 7,27-7,48 (m, 5H).
3.ii. Бензиловый эфир 2,5-дигидропиррол-1-карбоновой кислоты
Бензилиден-бис(трициклогексилфосфин)дихлоррутений (5 г) добавляют к раствору промежуточного соединения (3.i) (17,56 г) в ДХМ (1,5 л) при комнатной температуре в атмосфере азота. Реакционную смесь затем перемешивают при температуре 40°С в течение 2 ч, после чего концентрируют в вакууме. Остаток очищают с помощью ускоренной хроматографии (гексан/ЭА в соотношении 90:10), получая 14,08 г (91%-ный выход) продукта в виде желтой жидкости.
1Н ЯМР (ДМСОd6; δ м.д.): 4,05-4,16 (m, 4H); 5,08 (s, 2H); 5,81-5,92 (m, 2H); 7,27-7,41 (m. 5H).
3.iii. Бензиловый эфир (RS)-3-гидроксипирролидин-1-карбоновой кислоты
1-Молярный раствор борана в ТГФ (9 мл) прибавляют к раствору промежуточного соединения (3.ii) (1,81 г) в ТГФ (25 мл) при температуре 0°С в атмосфере азота. Реакционную смесь перемешивают при комнатной температуре в течение 16 ч, после чего охлаждают до температуры 0°С, осторожно по каплям прибавляют 20%-ный водный раствор NaOH (1,8 мл), а затем 35%-ный водный раствор перекиси водорода (1,2 мл). Смесь перемешивают при температуре 0°С в течение 30 мин и при комнатной температуре в течение 2 ч. После этого добавляют EtzO и 40%-ный водный раствор бисульфита натрия и реакционную смесь энергично перемешивают в течение 15 мин. Остаток, выделенный после обработки (EtzO), очищают с помощью ускоренной хроматографии (гексан/ЭА в соотношении от 5:5 до 3:7), получая 1,01 г (51%-ный выход) продукта в виде бесцветного масла.
1Н ЯМР (ДМСОd6; δ м.д.): 1,67-1,82 (m, 1H); 1,82-1,96 (m, 1H); 3,16-3,25 (m, 1Н); 3,28-3,44 (m, ЗН); 4,20-4,29 (расширенный, 1H); 4,92 (d, J=3, 1H); 5,06 (s, 2H); 7,27-7,41 (m, 5H).
3.iv. Бензиловый эфир 3-оксопирролидин-1-карбоновой кислоты
Раствор промежуточного соединения (3.iii) (1,10 г) в ДХМ (8 мл) охлаждают до температуры 0°С и прикалывают ДИПЭА (2,5 мл), а затем раствор комплекса триоксида серы с пиридином (1,79 г) в ДМСО (6,5 мл). Реакционную смесь перемешивают при температуре 0°С в течение 1 ч, после чего гасят, добавляя воду (6 мл). Водный слой (трижды по 5 мл) экстрагируют смесью Et2O/reKcaH (в соотношении 1:1) и объединенные органические слои концентрируют в вакууме. Остаток, выделенный после обработки (Et2О/гексан в соотношении 1:1), очищают с помощью ускоренной хроматографии (гексан/ЭА в соотношении 5:5), получая 1,05 г (96%-ный выход) продукта в виде желтоватого масла.
1Н ЯМР (ДМСОd6; δ м.д.): 2,48-2,61 (m, 2H); 3,61-3,80 (m, 4H); 5,09 (s, 2H); 7,27-7,41 (m, 5H).
3.v. Бензиловый эфир 3-метиленпирролидин-1-карбоновой кислоты
t-ВuОК (617 мг) одной порцией прибавляют к белой суспензии метилтрифенилфосфонийбромида (1,98 г) в ТГФ (10 мл) при комнатной температуре в атмосфере азота. Образовавшуюся желтую суспензию перемешивают при комнатной температуре в течение 1 ч, после чего охлаждают до температуры -10°С. Раствор промежуточного соединения (3.iv) (1,05 г) в ТГФ (2 мл) прикапывают в течение 10 мин, после чего реакционную смесь оставляют самопроизвольно нагреваться до комнатной температуры в течение 2 ч. Реакционную смесь гасят, добавляя насыщенный водный раствор NH4C1 (1 мл), и разбавляют этилацетатом. Остаток, выделенный после обработки (ЭА), очищают с помощью хроматография (гексан/ЭА в соотношении 90:10), получая 633 мг (64%-ный выход) продукта в виде желтоватой жидкости.
1Н ЯМР (ДМСОd6; δ м.д.): 2.48-2.61 (m, 2H); 3.36-3.53 (m, 2H); 3.84-4.01 (m, 2H); 4.97-5.03 (m, 2H); 5.08 (s, 2H); 7.27-7.41 (m, 5H).
3.vi. Бензиловый эфир 1-окса-5-аза-спиро[2.4]гептан-5-карбоновой кислоты
Раствор промежуточного соединения (3.v) (7,21 г) в ДХМ (400 мл) обрабатывают, добавляя м-ХПБК (20,1 г) и NaHCO3 (22,3 г) при комнатной температуре. Реакцию перемешивают при комнатной температуре в течение 2 ч, разбавляют ДХМ (200 мл) и переносят в раствор Na2SO3 (45 г) в воде (400 мл). Смесь перемешивают в течение 10 мин и органический слой отделяют. Остаток, выделенный после обработки (ДХМ), очищают с помощью ускоренной хроматографии (гексан/ЭА в соотношении 6:4), получая 4,37 г (56%-ный выход) продукта в виде желтого масла.
1Н ЯМР (ДМСОd6; δ м.д.): 1,70-1,83 (m, 1H); 2,22-2,37 (m, 1H); 2,90-2,94 (m, 1Н); 2,95-2.99 (m, 1H); 3,15 (t, J=11, 1H); 3,39-3,77 (m, 3H); 5,09 (s, 2H); 7,27-7,41 (m, 5H).
3.vii. Бензиловый эфир (RS)-3-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-3-гидроксипирролидин-1-карбоновой кислоты
К2СО3 (274 мг) прибавляют к суспензии промежуточного соединения (1.i) (300 мг) и промежуточного соединения (3.vi) (338 мг) в ДМФ (3 мл). Реакционную смесь перемешивают при температуре 80°С в течение 3 ч, после чего растворитель удаляют в вакууме. Остаток, выделенный после обработки (ДХМ), очищают с помощью ускоренной хроматографии (ДХМ/МеОН в соотношении 95:5), получая 531 мг (87%-ный выход) продукта в виде бежевой пены.
1Н ЯМР (ДМСОd6; δ м.д.): 1,80-1,92 (m, 1H); 1,96-2,08 (m, 1H); 3,32-3,59 (m, 5H); 3.66 (ddd, J=3, J=6 и J=13, 1H); 3,80 (dd, J=6 и J=9, 1H); 3,97-4,09 (m, 3Н); 4,64-4,72 (m, 1H); 5,07 (s, 2H); 5,19 (t, J=6, 1H); 5,23 (s. 1H); 7,18-7,23 (m, 2H); 7,27-7,38 (m, 5H); 7,57 (dd. J - 2 и J=14, 1H).
MC (ЭСИ): 460,9.
3.viii. (R)-3-[3-Фтор-4-((К8)-3-гидроксипирролидин-3-илметокси)фенил]-5-гидроксиметилоксазолидин-2-он
Раствор промежуточного соединения (3.vii) (259 мг) в смеси ТГФ/МеОН (в соотношении 1:1; 20 мл) гидрируют над 10%-ным Pd/C (60 мг) в течение 20 ч при комнатной температуре. Реакционную смесь концентрируют в вакууме, переносят в смесь ДХМ/МеОН (в соотношении 90:10; 20 мл) и перемешивают при комнатной температуре в течение 30 мин. Катализатор отфильтровывают и фильтрат концентрируют в вакууме, получая 184 мг (100%-ный выход) продукта в виде оранжевой пены.
MC (ЭСИ): 327,3.
3.ix. 1-Циклопропил-6-фтор-7-{(RS)-3-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-3-гидроксипирролидин-1-ил}-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
Раствор промежуточного соединения (3.viii) (226 мг) и комплекса 7-хлор-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты с диацетатом бора (270 мг; получен согласно WO 88/07998) в N-МП (5 мл) обрабатывают ДИПЭА (120 мкл) и перемешивают при температуре 60°С в течение 2 ч. Реакционную смесь концентрируют в вакууме и остаток переносят в 5-молярный раствор НСl в МеОН (2 мл). Раствор перемешивают при комнатной температуре в течение 1 ч, концентрируют в вакууме и остаток очищают с помощью ускоренной хроматографии (ДХМ/МеОН/АсОН в соотношении от 95:4:1 до 90:9:1). Остаток в виде пены переносят в МеОН (2 мл), перемешивают в течение 1 ч и фильтруют. Кристаллы отделяют и высушивают в вакууме, получая 23 мг (6%-ный выход) продукта в виде бежевого твердого вещества.
1H ЯМР (ДМСОd6; δ м.д.): 1,10-1,34 (m, 4H); 1,98-2,10 (m, 1H); 2,14-2,26 (m, 1H); 3,48-3,70 (m, 3H); 3,71-3,89 (m, 5H); 4,05 (t, J=9, 1H); 4,09-4,18 (m, 2H);
4,66-4,74 (m, 1H); 5,19 (t, J=6, 1H); 5,40 (s, 1H); 7,09 (d, J - 8, 1H); 7,18-7,31 (m, 2H); 7,59 (dd, J=2 и J=14, 1H); 7,82 (d, J=14, 1H); 8,59 (s, 1H); 15,52 (s, 1H).
МС(ЭСИ): 572,3.
Пример 4: Антибактериальная активность в условиях in vitro соединений по изобретению против штаммов Clostridium difficile или Clostridium perfrinsens
4.i. Экспериментальный метод
Минимальные ингибирующие концентрации (МИК) определялись посредством анализа микроразбавления бульонной среды. Была использована питательная среда Brucella, обогащенная дополнительно витамином K1 в концентрации 0,5 мг/л и 5 мг/л гемина (далее «обогащенная среда»), в качестве контрольной питательной среды. Кратко, исходные растворы для разведения соединений были приготовлены в ДМСО (5,12 мг/мл), а затем 5 мкл раствора в диапазоне двукратных разведении в смеси 50% ДМСО:50% Н2О были распределены в 96-ячеистый планшет для микротитрования, содержащий 45 мкл обогащенной среды Brucella, после чего его встряхивали в течение 5 мин. Для проведения инокуляции старые (24 ч) колонии С. Difficile, выращенные на агаре Brucella, обогащенным 5% гомолизированной овечьей крови, 5 мкг гемина/мг и 1 мкг витамина K1/мл, суспендировались в обогащенную среду Brucella и корректировались до плотности, соответствующей 0,5 по стандарту McFarland. Для инокуляции использовали 50 мкл этой суспензии с 50-кратным разбавлением, добавляя ее в ячейки 96-ячеистого планшета и получая в результате приблизительно 104 колониеобразующих единиц (CFU) на ячейку. Конечная концентрация ДМСО составляла 2,5%. Конечная концентрация всей зоны составляла 0,03-16 мг/мл. Планшеты инкубировали в анаэробных условиях в течение 48 ч при температуре 37°С.После инкубирования планшеты считывались в планшете-ридере при 00595 (Ultramark, фирма Biorad Laboratories). МИК сначала считывались при самой низкой концентрации, показавшей >90% роста ингибирования по сравнению с контрольными ячейками.
Планшеты также контролировались визуально с помощью считывающего зеркала, и полученные МИК были подтверждены отсутствием визуального роста.
4.ii. Результаты:
Соединения примеров 1-3 были протестированы в условиях in vitro для определения активности роста ингибирования трех стандартных штаммов С. difficile или Clostridium perfringens. Полученные результаты приведены ниже в таблице 1.
Соединения примеров 1-3 проявляли в условиях in vitro активность против штаммов С.difficile и С.perfringens. Значительная активность наблюдалась также против хинолон-резистентного гипервирулентного штамма NC13366. Соединения примеров 1 и 2 были немного более активны по сравнению с соединениями примера 3, и все соединения примеров были явно более активны, чем ципрофлоксацин и линезолид.
Пример 5: Антибактериальная активность в условиях in vitro соединений примера 1 против коллектированных клинических изолятов Clostridium difficile:
5.i. Экспериментальный метод:
Рекомендованный институтом клинических лабораторных стандартов (CLSI) метод разбавления в агаре для анаэробов (M11-A6) был использован для чувствительности тестирования. Агар Brucella, обогащенный 5% гомолизированной овечьей крови, 5 мкг гемина/мг и 1 мкг витамина К1/мл, применялся в качестве тестируемой среды. Тестируемые соединения были сериально разбавлены и добавлены к обогащенному расплавленному агару. Контроль роста осуществлялся на свободных от лекарственного препарата планшетах. Перед тестированием все изоляты были дважды пассированы в обогащенные Brucella агаром планшеты. Бактериальные колонии суспендировались в среду Brucella. Стандартизация с помощью Vitek колориметра была использована для получения каждого посевного материала, эквивалентного 0,5 по стандарту McFarland, соответствующему 104-105 CFU на пятно, после аппликации с помощью Steers репликатора. Затем планшеты инкубировались в анаэробных условиях в течение 48 ч при температуре 37°С. Минимальная ингибирующая концентрация (МИК) соответствовала концентрации, которая полностью ингибирует визуальный рост по сравнению со свободным от лекарственного средства контролем. Все антибиотики получены и протестированы наряду с ванкомицином и метронидазолом в качестве контрольных препаратов.
5.ii. Результаты:
Соединение примера 1 было протестировано на его активность относительно роста ингибирования коллекции 209 разновидностей клинических изолятов С.difficile в условиях in vitro. Соединение примера 1 показало МИК90 (минимальная ингибирующая концентрация для ингибирования роста 90% или более штаммов), равную 0,25 мкг/мл, полученные МИК лежали в диапазоне от 0,06 до 0,5 мкг/мл. В среднем эти величины указывают на большую активность по сравнению с ванкомицином, метронидазолом и линезолидом, МИК90 которых составляли 2, 1 и 8 мкг/мл, соответственно.
Пример 6: Антибактериальная активность в условиях in vitro соединений по изобретению против аэробных грамположительных бактерий:
6.i. Экспериментальный метод:
Минимальные ингибирующая концентрации (МИК) определялись с помощью метода разбавления в бульоне, рекомендованного институтом клинических лабораторных стандартов [CLSI, раньше называемым NCCLS, 1997]. Кратко, исходные растворы для разведения соединений были приготовлены в ДМСО (5,12 мг/мл), сериально разбавлены в катион-регулируемом Mueller-Hinton Broth II (СаМНВ), после чего перенесены в планшеты для титрования с помощью Biomek 2000 пипетирующего робота (фирма Beckman Coulter). Конечная концентрация ДМСО составляла 2,5% или ниже. Планшеты затем были проинкубированы для достижения концентрации, составляющей в общем 3-6×105 CFU/мл. После инкубирования при температуре 37°С в течение 18-24 ч планшеты считывались в планшете-ридере при OD595 (Ultramark, Biorad Laboratories). МИК сначала считывались при самой низкой концентрации, показавшей >90% роста ингибирования по сравнению с контрольными ячейками. Планшеты также контролировались визуально с помощью считывающего зеркала, и полученные МИК были подтверждены отсутствием визуального роста.
6.ii. Результаты:
Соединение примера 1 было протестировано против грамположительных аэробных бактерий. Полученные результаты приведены ниже в таблице 2.
В общем, соединение примера 1 имело в условиях in vitro потенциальную активность против штаммов Staphylococcus aureus, S. aureus MRSA, Enterococcus faecalis и Enterococcus faecium. Оно активно также против штаммов, резистентных к ванкомицину, ципрофлоксацину или линезолиду.
Пример 7: Антибактериальная активность в условиях in vitro соединения примера 1 против, в частности, анаэробных бактериальных штаммов:
Используя экспериментальный метод, описанный в примере 5 (см. 5.i), соединение примера 1 было протестировано в условиях in vitro на способность ингибировать рост некоторых анаэробных бактерий, о которых известно или предполагается, что они играют положительную роль в нормальной кишечной флоре, защищая против чрезмерного роста С. Difficile. Далее эти бактерии обобщенно называются "комменсальная кишечная бактерия". В качестве стандарта в то же самое время были протестированы соединение примера 5 публикации WO 2005/058888 (R1) и ципрофлоксацин (СР). Полученные МИК приведены ниже в таблицах 3 и 3а.
В общем, соединение примера 1 проявило меньшую активность по отношению к комменсальной кишечной бактерии по сравнению с С. difficile. В отношении Bacteroides spp., которые являются важными компонентами защитной кишечной флоры человека, активность соединения примера 1 оказалась в 30-200 раз ниже. Такая селективная активность предоставляет, таким образом, потенциальную возможность для селективного уничтожения С. difficile в кишечнике при ограниченном воздействии на важные бактерии кишечной флоры.
Кроме того, при сравнении с соединением примера 5 в публикации WO 2005/058888 соединение примера 1 проявляло в 2-4 раза меньшую активность против тестируемой Bacteroides spp., в 4 раза меньшую активность против Eubacterium limosum A-1259 или Finegoldia magna A-1254 штаммов, в 4-8 раз меньшую активность против тестируемой Bifidobacterium spp. и в 8 раз меньшую активность против Lactobacillus acidophilus A-1255 и Lactococcus lactis A-1256 штаммов.
Вследствие этого, при сравнении с соединением примера 5 в публикации WO 2005/058888 (R1), соединение примера 1 оказалось значительно менее активно против комменсальной кишечной бактерии, что указывает на преимущество при селективном уничтожении С. difficile в кишечнике.
Пример 8: Воздействие соединения примера 1 на продуцирование токсина С. difficile:
8.i. Экспериментальный метод:
Гипертоксигенный С. difficile штамм NC13366 выращивался в анаэробных условиях в обогащенной питательной среде Brucella в течение 24 ч при температуре 37°С для создания клеточной плотности, равной приблизительно 1х 108 CFU/мл. Бактерии далее отмывались посредством центрифугирования и ресуспендировались в том же объеме обогащенной питательной среды Brucella, содержащей антибиотики с концентрацией 1 и 8 мкг/мл, соответственно. Контрольный образец не содержал антибиотиков. Культуры далее инкубировались в анаэробных условиях при температуре 37°С. На пятый день супернатанты отделялись и анализировались на токсин А и токсин Б, как описано далее.
Токсин А детектировался с помощью Western Blotting с использованием NuPage Large системы для анализа белков (Invitrogen LP0001, согласно набору инструкций). Использовался Western Breeze антимышиный иммунодетекторный набор (Invitrogen WB7103) с анти-CdTA мышиными антиклональными антителами (PCG4.1, Biodesign C70517M).
С. difficile цитотоксический токсин (токсин В) полуколичественно детектировали путем тестирования супернатанта С. difficile культур с использованием анализа круговой клеточной активности в СНО клетках. Для этого СНО клетки высеивались в 96-ячеистый плоскодонный планшет (1х10 клеток/ячейку) и оставлялись для прикрепления на 3 ч. Затем к клеткам добавлялись стерилизованные супернатанты в соответствующих разбавлениях, после чего они инкубировались при температуре 37°С в 5% СО2. Через 20 ч инкубирования определялось клеточное вращение, с использованием при этом инверсионного микроскопа. Токсиновый титр определялся в виде наивысшего сериального 10-кратного разбавления супернатанта, встречающегося в >90% клеточного вращения.
Колониеобразующие единицы (CFU) определялись окрашиванием тестируемых образцов соответствующих разбавлений на обогащенном питательным бульоном Brucella агаре и отсчетом колоний после 48 ч инкубирования при температуре 37°С в анаэробных условиях.
8.ii. Результаты:
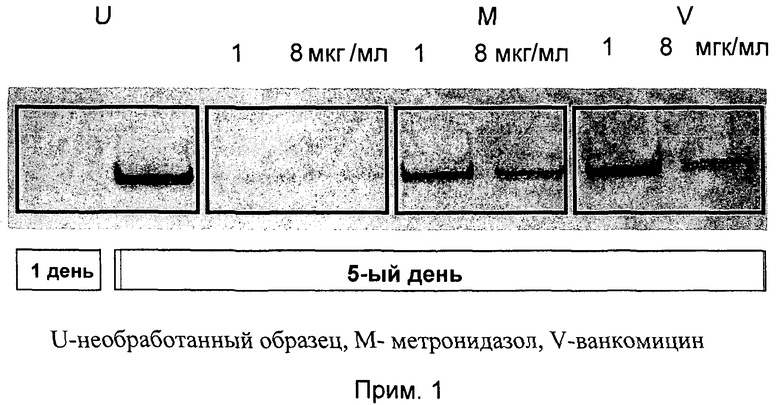
Данные, полученные на Western Blot plate при анализе продуцирования токсинов в супернатантах статистически высокой плотности клеточных культур С. difficile NC 13366 (гиперпродуцируемый токсин штамма NAPI/027-типа, вызывающий госпитальную вспышку с высокой смертностью), представлены на фиг.1.
Экспериментальный метод, отмеченный в параграфе 8.i, был использован для получения результатов, представленных на фиг.1. В первый день (D1) культуры обрабатывались либо соединением примера 1 (прим. 1), ванкомицином (V), метронидазолом (М) с концентрацией 1 и 8 мкг/мл, либо оставались необработанными (U); на пятый день (D5) проанализированное содержание токсина А в супернатантах сравнивалось с данными, полученными в D1 (см. фиг.1). В имеющей через 5 дней высокую плотность стационарной фазе культур токсиногенного С. difficile соединение примера 1 ингибировало новое продуцирование токсина А при концентрациях 1 и 8 мкг/мл, то есть при 2 и 16х МИК (см. фиг.1). По контрасту, метронидазол и ванкомицин совсем не ингибировали или только в малой степени ингибировали продуцирование токсина А в этих условиях.
Супернатанты культуры анализировались также на цитотоксичный токсин В с использованием анализа круговой клеточной активности, представленного в параграфе 8.i. Активности супернатантов культур, обработанных соединением примера 1, были снижены до >90% по сравнению с необработанными образцами. По контрасту, активности супернатантов ванкомицин-обработанных культур не понижались (при концентрации 1 мкм/мл) или понижались только до 30% (при концентрации 8 мкг/мл).
В общем, соединение примера 1 обладает способностью эффективно останавливать синтез С. difficile токсина А и токсина В даже в высокоплотных статичных культурах и в отсутствии цитолиза, которая не проявляется в случае ванкомицина и метронидазола, которые являются преимущественно используемыми в настоящее время лекарственными средствами для лечения инфекций, вызываемых С. difficile.
Пример 9: Воздействие соединения примера 1 на С. difficile спорообразование:
9.i Экспериментальный метод:
С. difficile A-1050 (АТСС 43596) культивировались при росте в логарифмической прогрессии в обогащенной среде Brucella, приводящем к помутнению, наблюдаемому при совместимости со стандартом McFarland 0,5 (6-7 ч при температуре 37°С в анаэробных условиях). Культуры разбавлялись затем в соотношении 1:50 в трубках, содержащих 1 мл обогащенной питательной среды Brucella и антибиотики в различных концентрациях при 2-кратном разбавлении. Трубки затем инкубировались в течение 4 дней при температуре 37°С в анаэробных условиях. МИК определялись в виде концентрации, которая полностью ингибирует видимый рост (помутнение), при этом 0,5х МИК является наивысшей концентрацией лекарственного средства, допускающей видимый рост.
Оценка колониеобразующих остатков (CFUs) общих клеток осуществлялась путем окрашивания соответствующих 10-кратно разбавленных культур на Brazier агаре, содержащем 4% суспензии яичного желтка и 1% лизированной лошадиной крови (Lab M). Количество колоний определялось после инкубирования при температуре 37°С в течение 48 ч в анаэробных условиях. Подсчет спор проводился путем селективного убийства вегетативных клеток посредством обработки 50%-ным этанолом в течение 1 ч при комнатной температуре, и последующий отсчет CFU осуществлялся на Brazier агаре, как описано выше.
9.ii. Результаты:
С. difficile является спорообразующим организмом, при этом споры могут долгое время существовать в госпитальных условиях. Споры чрезвычайно резистентны к общим дезинфикационным процедурам. Вследствие этого споры играют важную роль в трансмиссии и персистентности С. difficile инфекций. Выращиваемые в условиях in vitro культуры С. difficile обрабатывались различными суб-МИК концентрациями соединения примера 1, ванкомицином или метронидазолом и исследовались затем на предмет продуцирования спор после инкубирования в анаэробных условиях в течение 4 дней при температуре 37°С (см. параграф 9.i). При обработке соединением примера 1 с концентрацией, соответствующей 0,5х МИК, было обнаружено низкое количество спор (<1% спор от общего числа клеток). В противоположность этому, необработанные культуры и культуры, обработанные метронидазолом или ванкомицином при концентрациях 0,5х МИК, показали высокую степень спорообразования (>90% спор от общего числа клеток). Это свидетельствует о том, что соединение примера 1 имеет потенциальную возможность более эффективно снижать спорообразование при обработке С. difficile инфекции, чем ванкомицин или метронидазол.
Пример 10: Активность в условиях in vivo соединения по изобретению в С.difficile моделе хомяка:
10.i. Экспериментальный метод:
Золотистым сирийским хомячкам делают одноразовую инъекцию клиндамицинфосфата (10 мг/кг) и через день инфицируют, внося 10 CFU токсигеничного С difficile штамма 10465. У большинства животных (-95%) быстро развивается колит через 1-2 дня после введения С.difficile. У необработанных животных заболевание быстро прогрессировало и приводило к серьезному колиту, геморрагическому некрозу слепой кишки и смерти. Соединение примера 1 было орально введено в виде суспензии при трех уровнях дозирования (10 мг/кг, 30 мг/кг и 100 мг/кг; n=10 на группу). Ванкомицин (50 мг/кг) использовался в качестве контроля. Животным вводилась разовая доза ежедневно в течение 5 дней, после чего они находились под наблюдение дополнительно в течение 21 дня.
Животные обследовались трижды в день с целью выявления случаев заболеваемости и наличия или отсутствия диареи. Финальной точкой исследования была выживаемость. Животные, проверяемые на наличие предсмертного состояния, распространяющегося на следующие симптомы:
прогрессирующая потеря веса до состояния истощения, анорексия в течение 24-48 ч, длительная летаргия (более 3 дней), симптомы паралича, эрозия кожи или травма, образования горба, растяжение брюшной полости, были подвергнуты эвтаназии путем одноразовой инъекции пентобарбиталом натрия.
10. ii. Результаты:
Терапевтическая полезность соединения примера 1 тестировалась согласно протоколу параграфа 10.i. Все инфицированные необработанные животные умирали в течение 2-5 дней после инфицирования, тогда как все животные, обработанные 10, 30 или 100 мг/кг соединения примера 1, оставались в живых в течение 5 дней обработки. При самой низшей дозе обработки 40% животных оставались в живых с рецидивом через 21 день после обработки, тогда как в группах, обработанных дозами 30 и 100 мг/кг, 80 и 100% животных выживали без рецидива. Ванкомицин в дозе 50 мг был использован в качестве контроля. Эти результаты продемонстрировали, что соединение примера 1 обладает способностью лечения животных с С. difficile инфекциями, аналогичной ванкомицину.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГИБРИДНЫЕ АНТИБАКТЕРИАЛЬНЫЕ СРЕДСТВА ОКСАЗОЛИДИНОН-ХИНОЛОН, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ПАРЕНТЕРАЛЬНОГО ВВЕДЕНИЯ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ БАКТЕРИАЛЬНЫХ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2702364C2 |
| АНТИБАКТЕРИАЛЬНЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2525915C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С CLOSTRIDIUM DIFFICILE | 2011 |
|
RU2575477C2 |
| ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ ГИРАЗЫ И ТОПОИЗОМЕРАЗЫ IV | 2012 |
|
RU2609259C2 |
| ТВЕРДЫЕ ФОРМЫ ИНГИБИТОРА ГИРАЗЫ (R)-1-ЭТИЛ-3-[6-ФТОР-5[2-(1-ГИДРОКСИ-1-МЕТИЛ-ЭТИЛ) ПИРИМИДИН-5-ИЛ]-7-(ТЕТРАГИДРОФУРАН-2-ИЛ)-1Н-БЕНЗИМИДАЗОЛ-2-ИЛ] МОЧЕВИНЫ | 2012 |
|
RU2625305C2 |
| НОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ГРАМПОЛОЖИТЕЛЬНЫХ ИНФЕКЦИЙ | 2009 |
|
RU2512396C2 |
| ГИДРОКСИАЛКИЛТИАДИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2737892C1 |
| ЭКСТРАКТЫ KIBDELOS PORANGIUM В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2010 |
|
RU2572621C2 |
| ПРОИЗВОДНЫЕ 5-ГИДРОКСИМЕТИЛОКСАЗОЛИДИН-2-ОНА | 2007 |
|
RU2453546C2 |
| ОКСАЗОЛИДИНИЛОВЫЕ АНТИБИОТИКИ | 2009 |
|
RU2516701C2 |
Изобретение относится к соединениям формулы (I) 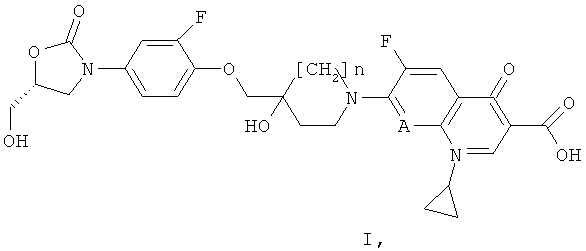 где А обозначает N или СН; и n равен 0 или 1; или их фармацевтически приемлемым солям для профилактики или лечения кишечных заболеваний, вызываемых бактерией, выбранной из Clostridium difficile, Clostridium perfringens или Staphylococcus aureus. Настоящее изобретение обладает высокой активностью против возбудителей кишечных инфекций, уменьшает токсиновую нагрузку, продуцирование спор, вызывая при этом минимальное влияние на вырабатываемую в кишечнике флору. 8 з.п. ф-лы, 1 ил., 3 табл., 10 пр.
где А обозначает N или СН; и n равен 0 или 1; или их фармацевтически приемлемым солям для профилактики или лечения кишечных заболеваний, вызываемых бактерией, выбранной из Clostridium difficile, Clostridium perfringens или Staphylococcus aureus. Настоящее изобретение обладает высокой активностью против возбудителей кишечных инфекций, уменьшает токсиновую нагрузку, продуцирование спор, вызывая при этом минимальное влияние на вырабатываемую в кишечнике флору. 8 з.п. ф-лы, 1 ил., 3 табл., 10 пр.
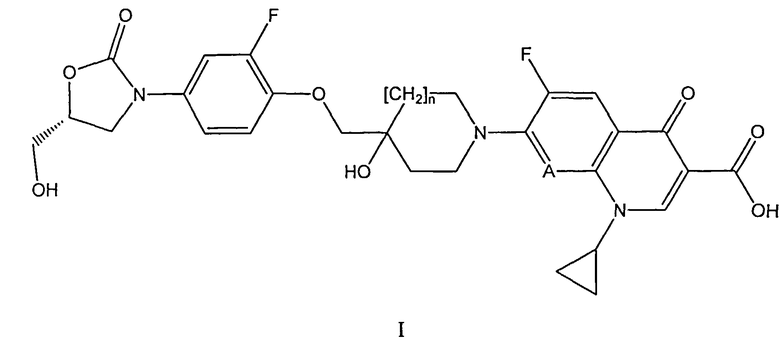
1. Применение соединения формулы (I)
 ,
,
где
А обозначает N или CH; и
n равен 0 или 1;
или его фармацевтически приемлемой соли, для получения лекарственного средства, предназначенного для профилактики или лечения кишечных заболеваний, вызываемых бактерией, выбранной из Clostridium difficile, Clostridium perfringens или Staphylococcus aureus.
2. Применение по п.1, где в соединении формулы (I) или его фармацевтически приемлемой соли A обозначает CH.
3. Применение по п.1, где в соединении формулы (I) или его фармацевтически приемлемой соли n равен 1.
4. Применение по п.1, где соединение формулы (I), выбрано из группы, включающей:
1-циклопропил-6-фтор-7-{4-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-4-гидроксипиперидин-1-ил}-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту;
1-циклопропил-6-фтор-7-{4-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-4-гидроксипиперидин-1-ил}-4-оксо-1,4-дигидро-[1,8]нафтиридин-3-карбоновую кислоту; и
1-циклопропил-6-фтор-7-{3-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-3-гидроксипирролидин-1-ил}-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту,
и фармацевтически приемлемые соли этих соединений.
5. Применение по п.1, где соединение формулы (I) представляет собой 1-циклопропил-6-фтор-7-{4-[2-фтор-4-((R)-5-гидроксиметил-2-оксооксазолидин-3-ил)феноксиметил]-4-гидроксипиперидин-1-ил}-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту или его фармацевтически приемлемую соль.
6. Применение по одному из пп.1-5 для получения лекарственного средства, предназначенного для профилактики или лечения кишечных заболеваний, вызываемых Clostridium difficile.
7. Применение по одному из пп.1-5 для получения лекарственного средства, предназначенного для профилактики или лечения кишечных заболеваний, вызываемых продуцирущим токсин штаммом Clostridium difficile.
8. Применение по одному из пп.1-5, где полученное лекарственное средство предназначено для лечения кишечного заболевания.
9. Применение по одному из пп.1-5, где полученное лекарственное средство предназначено для профилактики кишечного заболевания.
| WO2004096221 A1, 2004-11-11 | |||
| WO02059116 A2, 2002-08-01 | |||
| RU97108000 A, 10.05.1999 |

