Перекрестная ссылка на родственные заявки
По настоящей заявке испрашивается приоритет в соответствии с 35 U.S.C. § 119 на основании Временной патентной заявки США, серийный номер 61/432965, поданной 14 января 2011 года; Временной патентной заявки США, серийный номер 61/515174, поданной 4 августа 2011 года; Временной патентной заявки США, серийный номер 61/515249, поданной 4 августа 2011 года; и Временной патентной заявки США, серийный номер 61/499134, поданной 20 июня 2011 года; полное содержание каждой заявки включено в настоящую заявку посредством ссылки.
Предпосылки изобретения
Бактериальная резистентность к антибиотикам давно известна, и в настоящее время считается серьезной проблемой здравоохранения во всем мире. В результате резистентности некоторые бактериальные инфекции либо трудно поддаются лечению антибиотиками, либо вообще не поддаются лечению. Эта проблема стала особенно серьезной в связи с возникновением в последнее время множественной лекарственной резистентности в некоторых штаммах бактерий, таких как Streptococcus pneumoniae (SP), Mycobacterium tuberculosis и Enterococcus. Возникновение Ванкомицин-резистентного enterococcus вызвало особую тревогу, поскольку ванкомицин был ранее единственным эффективным антибиотиком для лечения этой инфекции, и для многих инфекций его считали "последним средством". В то время как другие лекарственно-резистентные бактерии не вызывают угрожающих жизни заболеваний, такие как энтерококки, существуют опасения, что гены, которые вызывают резистентность, могут распространиться на более смертельно опасные организмы, такие как Staphylococcus aureus, где резистентность к метициллину уже преобладает (De Clerq, et al., Current Opinion в Анти-infective Investigational Drugs, 1999, 1, 1; Levy, "The Challenge of Antibiotic Resistance", Scientific American, March, 1998).
Другое опасение связано с быстрым распространением резистентности к антибиотикам. Например, до 1960-х SP был чувствительным к пенициллину повсеместно, и в 1987 только 0,02% SP штаммов в США были резистентными. Однако к 1995 году, как сообщалось, SP резистентность к пенициллину составила около семи процентов, и даже 30% в некоторых районах США (Lewis, FDA Consumer magazine (September, 1995); Gershman в The Medical Reporter, 1997).
Больницы особенно являются центрами образования и переноса лекарственно-резистентных организмов. Инфекции, возникающие в больницах, известные как внутрибольничные инфекции, становятся все более серьезной проблемой. Два миллиона американцев заражаются в больницах каждый год, и более половины этих инфекций являются резистентными, по меньшей мере, к одному антибиотику. Согласно отчету Центра Контроля Заболеваний, в 1992 году более 13000 больничных пациентов умерли от бактериальных инфекций, которые были резистентными к лечению антибиотиками (Lewis, "The Rise of Antiviotic-Resistant Infections", FDA Consumer magazine, September 1995).
Как результат необходимости борьбы с лекарственно-резистентными бактериями и все возрастающей недостаточности доступных лекарственных средств, снова возрождается интерес к разработке новых антибиотиков. Одной привлекательной стратегией для разработки новых антибиотиков является ингибирование ДНК гиразы и/или топоизомеразы IV, бактериальных ферментов, необходимых для ДНК репликации, и поэтому необходимых для бактериального клеточного роста и деления. Активность гиразы и/или топоизомеразы IV также связывают с событиями в ДНК транскрипции, репарации и рекомбинации.
Гираза представляет собой одну из топоизомераз, группы ферментов, которые катализируют взаимное превращение топологических изомеров ДНК (смотрите, в общем смысле, Kornberg and Baker, DNA Replication, 2d Ed., Chapter 12, 1992, W. H. Freeman and Co.; Drlica, Molecular Microbiology, 1992, 6, 425; Drlica and Zhao, Microbiology and Molecular Biology Reviews, 1997, 61, pp. 377-392). Гираза сама по себе контролирует сверхскручивание ДНК и облегчает топологический стресс, который возникает, когда цепочки ДНК родительского дуплекса являются раскрученными в процессе репликации. Гираза также катализирует конверсию ослабленной замкнутой кольцевой дуплексной ДНК в отрицательно сверхспиральную форму, которая является более благоприятной для рекомбинации. Механизм реакции сверхскручивания включает закручивание гиразы вокруг одной области ДНК, с разрывом двойной цепи в этой области, проход во вторую область ДНК через разрыв и снова соединение разорванных цепей. Такой механизм расщепления является характерным для типа II топоизомеразы. Реакция сверхскручивания является управляемой через связывание АТФ с гиразой. АТФ затем гидролизуется в процессе этой реакции. Это связывание АТФ и последующий гидролиз вызывают конформационные изменения в ДНК-связанной гиразе, которые необходимы для ее активности. Также было обнаружено, что уровень сверхскручивания ДНК (или релаксации) зависит от отношения АТФ/АДФ. В отсутствие АТФ, гираза способна только к релаксации сверхскрученной ДНК.
Бактериальная ДНК гираза представляет собой 400 кДа тетрамерный белок, состоящий из двух A (GyrA) и двух B субъединиц (GyrB). Связывание и расщепление ДНК связано с GyrA, тогда как АТФ связывается и гидролизуется GyrB белком. GyrB состоит из амино-концевого домена, который обладает активностью АТФазы, и карбокси-концевого домена, который взаимодействует с GyrA и ДНК. В отличие от этого, эукариотные топоизомеразы типа II представляют собой гомодимеры, которые могут расслаблять отрицательные и положительные суперспирали, но не могут вводить отрицательные суперспирали. В идеальном случае, антибиотик, основанный на ингибировании бактериальной ДНК гиразы и/или топоизомеразы IV, мог бы быть селективным в отношении этих ферментов и относительно неактивным против эукариотных топоизомераз типа II.
Топоизомераза IV главным образом расщепляет связанные хромосомные димеры на этапе завершения ДНК репликации.
Широко используемые хинолоновые антибиотики ингибируют бактериальную ДНК гиразу (GyrA) и/или топоизомеразу IV (ParC). Примеры хинолонов включают ранние соединения, такие как налидиксиновая кислота и оксолиновая кислота, а также более поздние, обладающие более высокой активностью фторхинолоны, такие как норфлоксацин, ципрофлоксацин и тровафлоксацин. Эти соединения связываются с GyrA и/или ParC и стабилизируют расщепленный комплекс, таким образом, ингибируя функцию гиразы в целом, приводя к клеточной гибели. Фторхинолоны ингибируют каталитические субъединицы гиразы (GyrA) и/или топоизомеразы IV (Par C) (смотрите Drlica and Zhao, Microbiology and Molecular Biology Reviews, 1997, 61, 377-392). Однако было признано, что резистентность микроорганизмов к лекарственному средству также является проблемой для этого класса соединений (WHO Report, "Use of Quinolones in Food Animals and Potential Impact on Human Health", 1998). С хинолонами, так же как и с другими классами антибиотиков, бактерии, подверженные действию более ранних соединений, очень быстро развивают перекрестную резистентность к более сильным соединениям в этом же классе.
Ассоциированные субъединицы, ответственные за снабжение энергией, необходимой для каталитического обновления/восстановления ферментов через АТФ гидролиз, представляют собой GyrB (гираза) и ParE (топоизомераза IV), соответственно (смотрите Champoux, J. J., Annu. Rev. Biochem., 2001, 70, pp. 369-413). Соединения, которые прицельно действуют на эти самые сайты связывания АТФ в GyrB и ParE субъединицах, могли бы быть полезными для лечения различных бактериальных инфекций (смотрите Charifson et al., J. Med. Chem., 2008, 51, pp. 5243-5263).
Известно меньшее число ингибиторов, которые связываются с GyrB. Примеры включают кумарины, новобиоцин и кумермицин A1, циклотиалидин, цинодин и клероцидин. Было показано, что кумарины очень сильно связываются с GyrB. Например, новобиоцин образует сеть водородных связей с этим белком и несколько гидрофобных контактов. Тогда как новобиоцин и АТФ, как действительно оказалось, связываются в пределах сайта связывания АТФ, существует минимальное совпадение в ориентации связывания этих двух соединений. Перекрывающиеся части представляют собой сахарное звено новобиоцина и аденин из АТФ (Maxwell, Trends in Microbiology, 1997, 5, 102).
Что касается кумарин-резистентных бактерий, наиболее распространенная точечная мутация находится на поверхности аргининового остатка, который связывается с карбонилом кумаринового кольца (Arg136 в E. coli GyrB). При том, что ферменты с этой мутацией демонстрируют более низкое сверхскручивание и АТФазную активность, они также менее чувствительны к ингибированию кумариновыми лекарственными средствами (Maxwell, Mol. Microbiol., 1993, 9, 681).
Несмотря на то что они являются сильными ингибиторами сверхскручивания гиразы, кумарины не нашли широкого применения в качестве антибиотиков. В основном, они не являются подходящими из-за их низкой способности проникновения в бактерии, эукариотной токсичности и плохой водорастворимости (Maxwell, Trends in Microbiology, 1997, 5, 102). Было бы желательным получить новый эффективный ингибитор GyrB и ParE, который преодолевает эти недостатки, и, предпочтительно, его активность не зависит от связывания с Arg136. Такой ингибитор был бы привлекательным антибиотическим средством-кандидатом, без истории проблем резистентности, которая является вызывающей беспокойство проблемой других классов антибиотиков.
Поскольку бактериальная резистентность к антибиотикам стала важной проблемой здравоохранения, существует настоятельная необходимость в разработке более новых и более сильных антибиотиков. Более конкретно, существует необходимость в антибиотиках, которые представляют новый класс соединений, ранее не используемых для лечения бактериальной инфекции. Соединения, которые нацелены на сайты связывания АТФ в обеих субъединицах GyrB (гираза) и ParE (топоизомераза IV), будут полезными для лечения различных бактериальных инфекций. Такие соединения были бы особенно полезными для лечения внутрибольничных инфекций в госпиталях, где образование и перенос резистентных бактерий становится все более распространенным. Кроме того, существует потребность в новых антибиотиках, имеющих широкий спектр действия, с выгодными токсикологическими свойствами.
Краткое описание изобретения
Настоящее изобретение направлено на соединения и их фармацевтически приемлемые соли, полезные в качестве ингибиторов гиразы и/или топоизомеразы IV. Ингибиторы гиразы и/или топоизомеразы IV по настоящему изобретению могут быть представлены формулой (I) или солями таких соединений:
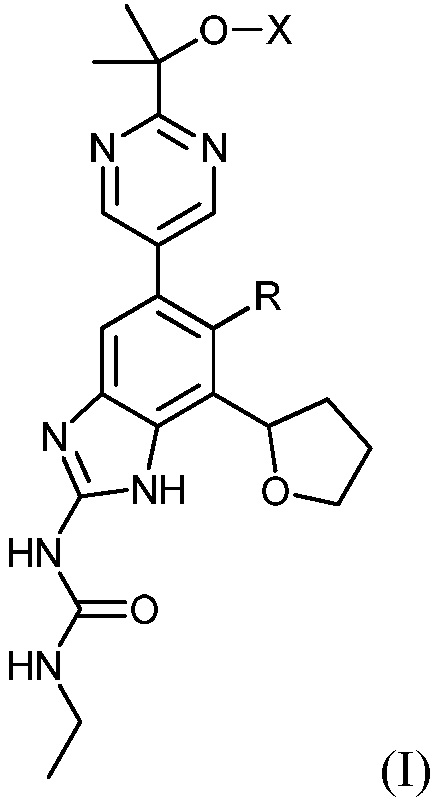
где R представляет собой водород или фтор; X представляет собой водород, -PO(OH)2, -PO(OH)O-M+, -PO(O-)2·2M+ или -PO(O-)2·D2+; M+ представляет собой фармацевтически приемлемый одновалентный катион; и D2+ представляет собой фармацевтически приемлемый двухвалентный катион. Соединения формулы (I) либо обладают широким спектром антибактериальной активности и выгодными токсикологическими свойствами, либо представляют собой пролекарства соединений, обладающих указанной активностью.
Настоящее изобретение также относится к соединениям формулы (IA) или их фармацевтически приемлемым солям, полезным в качестве ингибиторов гиразы и/или топоизомеразы IV. Соединения формулы (IA) охватываются формулой (I). Соединения формулы (IA) могут быть представлены как:
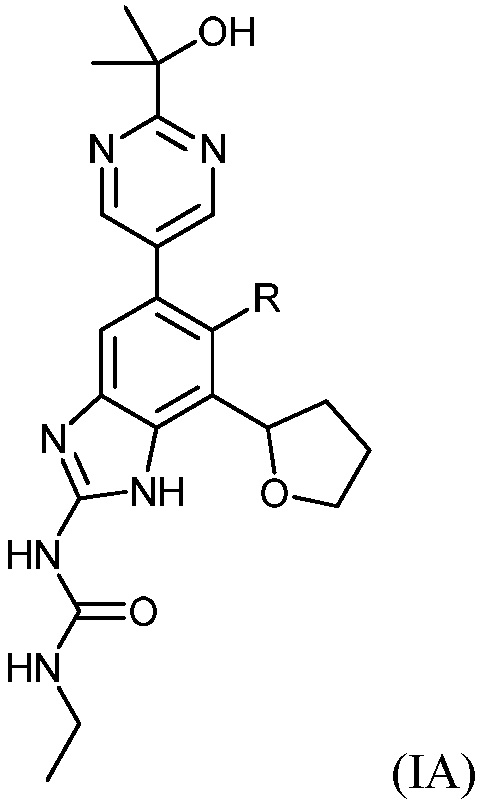
где R представляет собой водород или фтор. Соединения формулы (IA) обладают широким спектром антибактериальной активности и выгодными токсикологическими свойствами.
Настоящее изобретение также относится к соединениям формулы (IB) или их фармацевтически приемлем солям, полезным в качестве пролекарств для ингибиторов гиразы и/или топоизомеразы IV. Соединения формулы (IB) охватываются формулой (I). Соединения формулы (IB) могут быть представлены как:
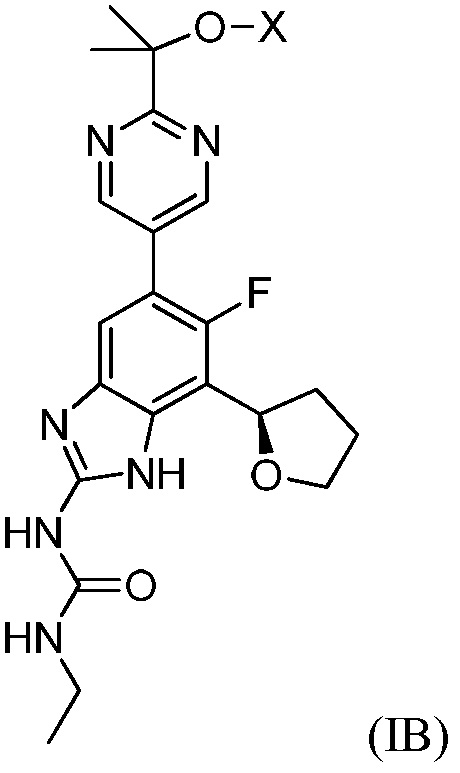
где X представляет собой -PO(OH)2, -PO(OH)O-M+, -PO(O-)2·2M+ или -PO(O-)2·D2+; M+ представляет собой фармацевтически приемлемый одновалентный катион; и D2+ представляет собой фармацевтически приемлемый двухвалентный катион. Соединения формулы (IB) представляют собой пролекарства в виде фосфатных сложных эфиров соединения (R)-1-этил-3-(6-фтор-5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевины, которое обладает широким спектром антибактериальной активности и выгодными токсикологическими свойствами. В дополнение к соединениям, представленным в настоящем изобретении, настоящее изобретение также обеспечивает фармацевтическую композицию, включающую соединение формулы (I) (которое включает другие формулы, охватываемые формулой (I), такие как формулы (IA), (IB), (IC) и (ID)) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
В другом варианте воплощения настоящее изобретение относится к фармацевтической композиции, включающей соединение формулы (I) или его фармацевтически приемлемую соль, фармацевтически приемлемый носитель и дополнительное терапевтическое средство, выбранное из антибиотика, противовоспалительного средства, ингибитора металлопротеиназы матрикса, ингибитора липоксигеназы, антагониста цитокина, иммуносупрессанта, противоракового средства, противовирусного средства, цитокина, фактора роста, иммуномодулятора, простагландина или соединения против гиперпролиферации сосудов.
В другом варианте воплощения настоящее изобретение относится к способу лечения бактериальной инфекции у млекопитающего, нуждающегося в этом, включающему введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В следующем варианте воплощения настоящее изобретение относится к способу лечения бактериальной инфекции у млекопитающего, нуждающегося в этом, включающему введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли и антибиотика, противовоспалительного средств, ингибитора металлопротеиназы матрикса, ингибитора липоксигеназы, антагониста цитокина, иммуносупрессанта, противоракового средства, противовирусного средства, цитокина, фактора роста, иммуномодулятора, простагландина или соединения против гиперпролиферации сосудов, либо в виде части составной лекарственной формы вместе с указанным соединением, либо в виде отдельной лекарственной формы.
Краткое описание чертежей
Фиг.1 графически представляет термальный эллипсоид из двух симметричных отдельных молекул соединения 12.
Фиг.2 графически представляет термальный эллипсоид из двух симметричных отдельных молекул соединения 23.
Подробное описание изобретения
Как используется в настоящей заявке, термин “галоген” означает F, Cl, Br или I.
Если не указано иное, структуры, описанные в настоящей заявке, также включают все стереохимические формы этих структур; т.е. R и S конфигурации для каждого асимметрического центра. Поэтому отдельные стереохимические изомеры, а также энантиомерные и диастереомерные смеси соединений по настоящему изобретению охватываются объемом настоящего изобретения.
Изотопно-меченные формы соединений формулы (I), где один или несколько атомов замещены атомом, имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, обычно присутствующих в природе, также включены в настоящую заявку. Примеры изотопов, которые могут быть включены в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода и фтора, такие как 2H, 3H, 13C, 14C, 15N, 18O и 17O. Радиоактивно-меченные и меченные стабильными изотопами соединения являются полезными, например, в качестве исследовательских или диагностических инструментов или ингибиторов гиразы и/или топоизомеразы IV с улучшенным терапевтическим профилем. Структуры также охватывают цвиттерионные формы соединений или солей, где это является подходящим.
В одном варианте воплощения соединения формулы (I) включают соединения формулы (IC)
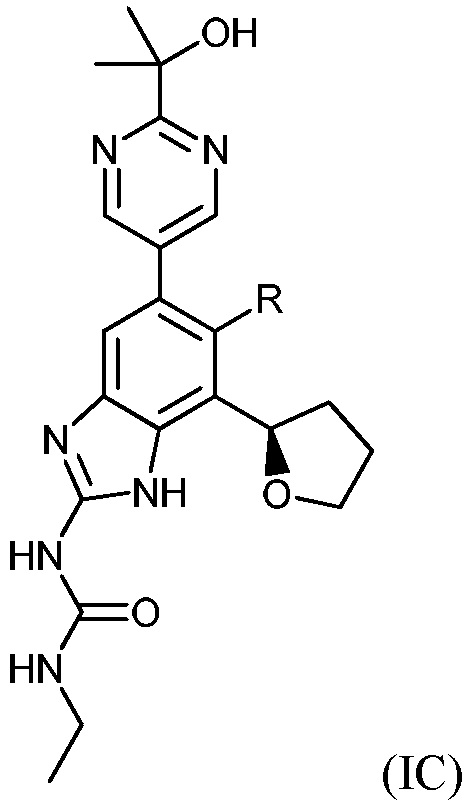
где R имеет значение, определенное выше.
В другом варианте воплощения соединения формулы (I) включают соединения формул (ID) и (IE), описанных ниже:
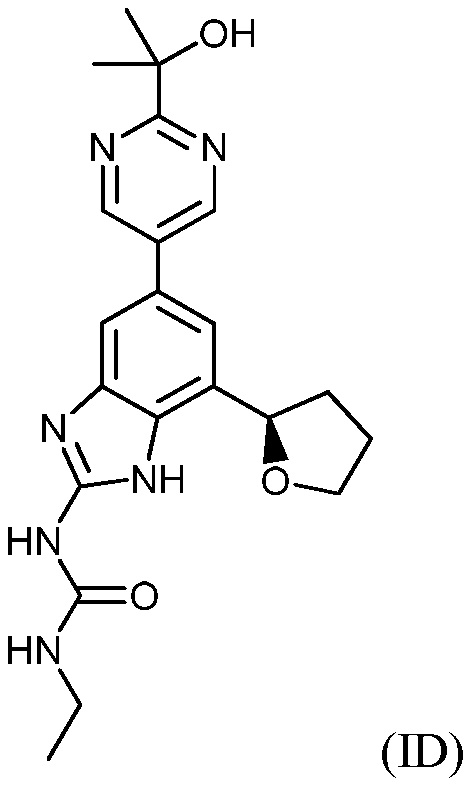
(R)-1-этил-3-(5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевина или ее фармацевтически приемлемая соль; и
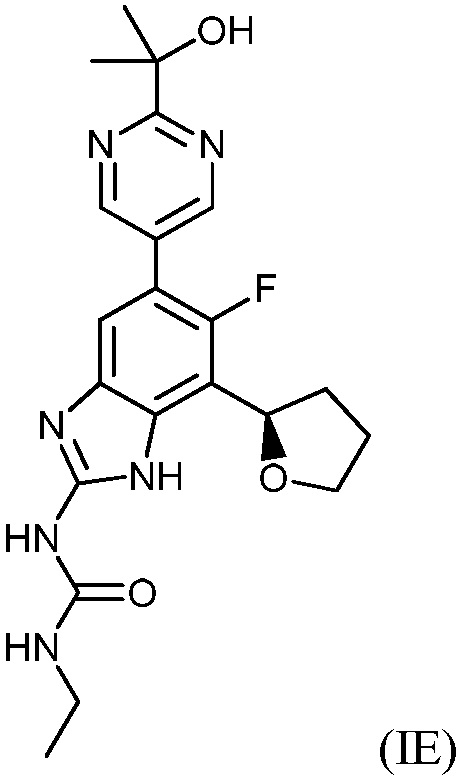
(R)-1-этил-3-(6-фтор-5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевина или ее фармацевтически приемлемая соль. Если не указано иное, фраза “соединения формулы (I)” включает другие формулы, описанные в настоящей заявке, которые охватываются формулой (I), включая формулы (IA), (IB), (IC), (ID) и (IE).
Соединения формулы (IB) представляют собой пролекарства их исходного соединения, 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины. Таким образом, активность, проявляемая после введения пролекарства, в принципе, проявляется благодаря присутствию исходного соединения, которое образуется при расщеплении пролекарства.
Термин “пролекарство” относится к соединениям, которые являются предшественниками лекарственного средства, которые после введения и абсорбции высвобождают лекарственное средство in vivo через некоторый метаболический процесс. Как правило, пролекарство обладает меньшей биологической активностью по сравнению с его исходным лекарственным средством. Пролекарство также может улучшать физические свойства исходного лекарственного средства, и/или оно также может улучшать общую эффективность лекарственного средства, например, через уменьшение токсичности и нежелательных эффектов лекарственного средства путем контроля его абсорбции, уровней в крови, метаболической дистрибуции и клеточного поглощения.
Термин “исходное соединение” или “исходное лекарственное средство” относится к биологически активному веществу, которое высвобождается через ферментативное действие метаболического или катаболического процесса или через химический процесс после введения пролекарства. Исходное соединение также может представлять собой исходное вещество для получения его соответствующего пролекарства.
Одновалентные катионы, обозначаемые как M+, включают ионы аммония, щелочных металлов, такие как ионы натрия, лития и калия, ион дициклогексиламина и ион N-метил-D-глюкамина. Двухвалентные катионы, обозначаемые как D2+, включают, ионы щелочно-земельных металлов, такие как ионы алюминия, кальция и магния. Также включены аминокислотные катионы, такие как ионы аргинина, лизина, орнитина и т. п. Когда M+ представляет собой одновалентный катион, должно быть понятно, что, если присутствует определение 2M+, каждый из M+ могут быть одинаковыми или отличными друг от друга. Кроме того, также должно быть понятно, что, если присутствует определение 2M+, может вместо этого присутствовать двухвалентный катион D2+. Также, щелочные азот-содержащие группы могут быть кватернизированы с использованием таких веществ, как: низшие алкилгалогениды, такие как метил, этил, пропил и бутил хлориды, бромиды и иодиды; диалкилсульфаты, такие как диметил, диэтил, дибутил, диамил сульфаты; длинноцепочечные галогениды, такие как децил, лаурил, миристил и стеарил хлориды, бромиды и иодиды; аралкилгалогениды, такие как бензилбромид и др.
Различные варианты воплощения настоящего изобретения включают соединения или соли формулы (IB), описанные ниже:
(1) соединения, где X представляет собой
(a) -PO(OH)O-M+;
(b) -PO(O-)2·2M+; или
(c) -PO(O-)2·D2+;
(2) соединения, где M+ представляет собой
(a) Li+, Na+, K+, N-метил-D-глюкамин или N(R9)4+; или
(b) Na+;
(c) каждый R9 независимо представляет собой водород или C1-C4 алкильную группу;
(3) соединения, где D2+ представляет собой
(a) Mg2+, Ca2+ и Ba2+; или
(b) Ca2+;
(4) соединение (R)-2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан-2-илфосфат; и
(5) соединение динатрий (R)-2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан-2-илфосфат.
Должно быть понятно, что различные альтернативные варианты воплощения соединений или солей формулы (IB) можно выбрать с использованием одного или нескольких альтернативных вариантов воплощения, описанных в (1)-(3) выше. Например, дополнительные варианты воплощения настоящего изобретения можно получить путем сочетания (1)(a) и (2)(a); (1)(a) и (2)(b); (1)(c) и (3)(a); (1)(c) и (3)(b); (1)(b) и (2)(a); (1)(b) и (2)(b) и т.п.
Пролекарства по настоящему изобретению характеризуются неожиданно высокой водорастворимостью. Эта растворимость способствует введению более высоких доз пролекарства, обеспечивая большую нагрузку лекарственного средства на стандартную дозу.
Один вариант воплощения настоящего изобретения относится к способу лечения бактериальной инфекции у млекопитающего, нуждающегося в этом, включающему введение указанному млекопитающему терапевтически эффективного количества соединения, имеющего формулу (I), или его фармацевтически приемлемой соли.
В соответствии с другим вариантом воплощения изобретение обеспечивает способ снижения или ингибирования количества бактерий в биологическом образце. Этот способ включает контактирование указанного биологического образца с соединением формулы (I) или его фармацевтически приемлемой солью.
Термин "биологический образец", как используется в настоящей заявке, включает клеточные культуры или их экстракты; материал биопсии, полученный от млекопитающего или его экстракты; и кровь, слюну, мочу, экскременты, сперму, слезы или другие жидкости организма или их экстракты. Термин "биологический образец" также включает живые организмы, и в этом случае термин "контактирование соединения по настоящему изобретению с биологическим образцом" синонимичен термину "введение указанного соединения или композиции, включающей указанное соединение) млекопитающему".
Один вариант воплощения включает контактирование указанного биологического образца с соединением, выбранным из группы, включающей (R)-1-этил-3-(5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевину или ее фармацевтически приемлемую соль и (R)-1-этил-3-(6-фтор-5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевину или ее фармацевтически приемлемую соль. Фармацевтические композиции, полезные для таких способов, описаны ниже.
Один вариант воплощения включает контактирование указанного биологического образца с пролекарством в форме фосфатного сложного эфира (R)-1-этил-3-(6-фтор-5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевины формулы (IB). Фармацевтические композиции, полезные для таких способов, описаны ниже.
Антимикробная активность соединений формулы (I) может быть продемонстрирована в анализе чувствительности к антимикробному средству. Условия, используемые для анализа чувствительности к антимикробному средству, подробно описаны в примерах ниже.
Ингибиторы гиразы и/или топоизомеразы IV по настоящему изобретению или их фармацевтические соли можно сформулировать в фармацевтические композиции для введения животным или человеку. Эти фармацевтические композиции, эффективные для лечения или профилактики бактериальной инфекции, которые включают ингибитор гиразы и/или топоизомеразы IV в количестве, достаточном для определяемого снижения количества бактерий, и фармацевтически приемлемый носитель, представляют собой еще один вариант воплощения настоящего изобретения. Термин "определяемое снижение количества бактерий", как он используется в настоящей заявке, означает поддающееся измерению изменение количества бактерий между образцом, содержащим указанный ингибитор, и образцом, содержащим только бактерии.
Средства, которые повышают чувствительность бактериальных организмов к антибиотикам, известны. Например, патент США № 5523288, патент США № 5783561 и патент США № 6140306 описывают способы с использованием бактерицидного/повышающего проникающую способность белка (BPI) для повышения чувствительности к антибиотикам грамположительных и грамотрицательных бактерий. Средства, которые повышают проникновение через внешнюю мембрану бактериальных организмов описаны Vaara, M. Microbiological Reviews (1992) pp. 395-411, и сенсибилизация грамотрицательных бактерии описана Tsubery, H., et al, J. Med. Chem. (2000) pp. 3085-3092.
Другой вариант воплощения настоящего изобретения относится к способу, описанному выше, профилактики, контроля, лечения или уменьшения прогрессирования, тяжести или эффектов бактериальной инфекции у млекопитающего, нуждающегося в этом, но, кроме того, включающему стадию введения указанному млекопитающему средства, которое усиливает чувствительность бактериальных организмов к антибиотикам.
В соответствии с другим вариантом воплощения способы по настоящему изобретению являются полезными для лечения пациентов в области ветеринарии, включая, но не ограничиваясь этим, животных зоопарков, лабораторных, домашних и сельскохозяйственных животных, включая приматов, грызунов, рептилий и птиц. Примерами указанных животных являются, но не ограничиваются этим, морские свинки, хомяки, песчанки, крысы, мыши, кролики, собаки, кошки, лошади, свиньи, овцы, коровы, козы, олени, макаки-резус, обезьяны, тамарины, человекообразные обезьяны, павианы, гориллы, шимпанзе, орангутаны, гиббоны, страусы, куры, индюки, утки и гуси.
Фармацевтические композиции и способы по настоящему изобретению, в основном, будут полезны для контроля бактериальных инфекций in vivo. Примеры бактериальных организмов, которые можно контролировать при помощи композиций и способов по настоящему изобретению, включают, но не ограничиваются этим, следующие организмы: Streptococcus pneumoniae, Streptococcus pyogenes, Enterococcus faecalis, Enterococcus faecium, Klebsiella pneumoniae, Enterobacter spp. Proteus spp. Pseudomonas aeruginosa, E. coli, Serratia marcescens, Staphylococcus aureus, Coag. Neg. Staphylococci, Haemophilus influenzae, Bacillus anthracis, Mycoplasma pneumoniae, Moraxella catarrhalis, Chlamydophila pneumoniae, Chlamydia trachomatis, Legionella pneumophila, Mycobacterium tuberculosis, Helicobacter pylori, Staphylococcus saprophyticus, Staphylococcus epidermidis, Francisella tularensis, Yersinia pestis, Clostridium difficile, Neisseria gonorrhoeae, Neisseria meningitidis, Mycobacterium avium комплекс, Mycobacterium abscessus, Mycobacterium kansasii и Mycobacterium ulcerans.
Композиции и способы поэтому будут полезны для контроля, лечения или уменьшения прогрессирования, тяжести или эффектов внутрибольничных или невнутрибольничных инфекций. Примеры внутрибольничных и невнутрибольничных инфекций, включают, но не ограничиваются этим, инфекции верхних дыхательных путей, инфекции нижних дыхательных путей, ушные инфекции, плевролегочные и бронхиальные инфекции, осложненные инфекции мочевых путей, неосложненные инфекции мочевых путей, интраабдоминальные инфекции, сердечно-сосудистые инфекции, заражение крови, сепсис, бактеремию, ЦНС инфекции, инфекции кожи и мягких тканей, желудочно-кишечные инфекции, инфекции, поражающие кости и суставы, половые инфекции, глазные инфекции или грануломатозные инфекции. Примеры конкретных бактериальных инфекций, включают но не ограничиваются этим, неосложненные инфекции кожи и кожных структур (uSSSI), осложненные инфекции кожи и кожных структур (cSSSI), катетеризационные инфекции, фарингит, синусит, отит наружного уха, отит среднего уха, бронхит, эмпиему, пневмонию, приобретенные в общественных местах бактериальные пневмонии (CABP), приобретенную в больнице пневмонию (HAP), приобретенную в больнице бактериальную пневмонию, связанную с искусственной вентиляцией легких пневмонию (VAP), связанные с диабетом инфекционные поражения стоп, ванкомицин-резистентные энтерококковые инфекции, цистит и пиелонефрит, почечные конкременты, простатит, перитонит, осложненные интраабдоминальные инфекции (cIAI) и другие интерабдоминальные инфекции, связанный с диализом перитонит, висцеральные абсцессы, эндокардит, миокардит, перикардит, связанный с переливанием крови сепсис, менингит, энцефалит, абсцесс головного мозга, остеомиелит, артрит, язвы половых органов, уретрит, вагинит, цервицит, гингивит, конъюнктивит, кератит, эндофтальмит, инфекцию у пациентов с кистозным фиброзом или инфекцию у пациентов с лихорадочным нейтропеническим состоянием.
Термин "невнутрибольничные инфекции" также относится к приобретенным в общественных местах инфекциям.
В одном варианте воплощения композиции и способы, поэтому, будут полезны для контроля, лечения или уменьшения прогрессирования, тяжести или эффектов приобретенной в общественных местах бактериальной пневмонии (CABP), приобретенной в больнице пневмонии (HAP), приобретенной в больнице бактериальной пневмонии, связанной с искусственной вентиляцией легких пневмонии (VAP), бактеремии, связанных с диабетом инфекционных поражений стоп, катетеризационных инфекций, неосложненных инфекций кожи и кожных структур (uSSSI), осложненных инфекций кожи и кожных структур (cSSSI), ванкомицин-резистентных энтерококковых инфекций или остеомиелита.
В другом варианте воплощения композиции и способы, поэтому, будут полезны для контроля, лечения или уменьшения прогрессирования, тяжести или эффектов инфекций верхних дыхательных путей, инфекций нижних дыхательных путей, ушных инфекций, плевролегочных и бронхиальных инфекций, осложненных инфекций мочевых путей, неосложненных инфекций мочевых путей, интраабдоминальных инфекций, сердечно-сосудистых инфекций, заражения крови, сепсиса, бактеремии, ЦНС инфекций, инфекций кожи и мягких тканей, желудочно-кишечных инфекций, инфекций, поражающих кости и суставы, половых инфекций, глазных инфекций или грануломатозных инфекций, неосложненных инфекций кожи и кожных структур (uSSSI), осложненных инфекций кожи и кожных структур (cSSSI), катетеризационных инфекций, фарингита, синусита, отита наружного уха, отита среднего уха, бронхита, эмпиемы, пневмонии, приобретенных в общественных местах бактериальных пневмоний (CABP), приобретенной в больнице пневмонии (HAP), приобретенной в больнице бактериальной пневмонии, связанной с искусственной вентиляцией легких пневмонии (VAP), связанных с диабетом инфекционных поражений стоп, ванкомицин-резистентных энтерококковых инфекций, цистита и пиелонефрита, почечных конкрементов, простатита, перитонита, осложненных интраабдоминальных инфекций (cIAI) и других интер-абдоминальных инфекций, связанного с диализом перитонита, висцеральных абсцессов, эндокардита, миокардита, перикардита, связанного с переливанием крови сепсиса, менингита, энцефалита, абсцесса головного мозга, остеомиелита, артрита, язв половых органов, уретрита, вагинита, цервицита, гингивита, конъюнктивита, кератита, эндофтальмита, инфекции у пациентов с кистозным фиброзом или инфекции у пациентов с лихорадочным нейтропеническим состоянием.
В другом варианте воплощения бактериальная инфекция характеризуется присутствием одного или нескольких из Streptococcus pneumoniae, Streptococcus pyogenes, Enterococcus faecalis, Enterococcus faecium, Staphylococcus aureus, Coag. Neg. Staphlococci, Bacillus anthracis, Staphylococcus epidermidis, Staphylococcus saprophyticus или Mycobacterium tuberculosis.
В другом варианте воплощения бактериальная инфекция характеризуется присутствием одного или нескольких из Streptococcus pneumoniae, Enterococcus faecalis или Staphylococcus aureus.
В другом варианте воплощения бактериальная инфекция характеризуется присутствием одного или нескольких из E. coli, Moraxella catarrhalis или Haemophilus influenzae.
В другом варианте воплощения бактериальная инфекция характеризуется присутствием одного или нескольких из Clostridium difficile, Neisseria gonorrhoeae, Neisseria meningitidis, Mycobacterium avium комплекса, Mycobacterium abscessus, Mycobacterium kansasii, Mycobacterium ulcerans, Chlamydophila pneumoniae и Chlamydia tracomatis.
В другом варианте воплощения бактериальная инфекция характеризуется присутствием одного или нескольких из Streptococcus pneumoniae, Staphylococcus epidermidis, Enterococcus faecalis, Staphylococcus aureus, Clostridium difficile, Moraxella catarrhalis, Neisseria gonorrhoeae, Neisseria meningitidis, Mycobacterium avium комплекса, Mycobacterium abscessus, Mycobacterium kansasii, Mycobacterium ulcerans, Chlamydophila pneumoniae, Chlamydia trachomatis, Haemophilus influenzae, Streptococcus pyogenes или β-гемолитических стрептококков.
В некоторых вариантах воплощения бактериальная инфекция характеризуется присутствием одного или нескольких из Метициллин-резистентного Staphylococcus aureus, Фторхинолон-резистентного Staphylococcus aureus, Ванкомицин-частично-резистентного Staphylococcus aureus, Линезолид-резистентного Staphylococcus aureus, Пенициллин-резистентного Streptococcus pneumoniae, Макролид-резистентного Streptococcus pneumoniae, Фторхинолон-резистентного Streptococcus pneumoniae, Ванкомицин-резистентного Enterococcus faecalis, Линезолид-резистентного Enterococcus faecalis, Фторхинолон-резистентного Enterococcus faecalis, Ванкомицин-резистентного Enterococcus faecium, Линезолид-резистентного Enterococcus faecium, Фторхинолон-резистентного Enterococcus faecium, Ампициллин-резистентного Enterococcus faecium, Макролид-резистентного Haemophilus influenzae, β-лактам-резистентного Haemophilus influenzae, Фторхинолон-резистентного Haemophilus influenzae, β-лактам-резистентного Moraxella catarrhalis, Метициллин-резистентного Staphylococcus epidermidis, Метициллин-резистентного Staphylococcus epidermidis, Ванкомицин-резистентного Staphylococcus epidermidis, Фторхинолон-резистентного Staphylococcus epidermidis, Макролид-резистентного Mycoplasma pneumoniae, Изониазид-резистентного Mycobacterium tuberculosis, Рифампин-резистентного Mycobacterium tuberculosis, Метициллин-резистентного Coagulase negative staphylococcus, Фторхинолон-резистентного Coagulase negative staphylococcus, Гликопептид-частично-резистентного Staphylococcus aureus, Ванкомицин-резистентного Staphylococcus aureus, Гетерованкомицин частично-резистентного Staphylococcus aureus, Гетерованкомицин-резистентного Staphylococcus aureus, Макролид-Линкосамид-Стрептограмин-резистентного Staphylococcus, β-лактам-резистентного Enterococcus faecalis, β-лактам-резистентного Enterococcus faecium, Кетолид-резистентного Streptococcus pneumoniae, Кетолид-резистентного Streptococcus pyogenes, Макролид-резистентного Streptococcus pyogenes, Ванкомицин-резистентного staphylococcus epidermidis, Фторхинолон-резистентного Neisseria gonorrhoeae, полирезистентного Pseudomonas aeruginosa или Цефалоспорин-резистентного Neisseria gonorrhoeae.
В соответствии с другим вариантом воплощения метициллин-резистентные стафилококки выбраны из Метициллин-резистентного Staphylococcus aureus, Метициллин-резистентного Staphylococcus epidermidis или Метициллин-резистентного Coagulase negative staphylococcus.
В некоторых вариантах воплощения форму соединения формулы (I) используют для лечения приобретенного в общественном месте MRSA (т.е. cMRSA).
В других вариантах воплощения форму соединения формулы (I) используют для лечения даптомицин-резистентного организма, включая, но не ограничиваясь этим, даптомицин-резистентного Enterococcus faecium и даптомицин-резистентного Staphylococcus aureus.
В соответствии с другим вариантом воплощения фторхинолон-резистентные стафилококки выбраны из Фторхинолон-резистентного Staphylococcus aureus, Фторхинолон-резистентного Staphylococcus epidermidis или Фторхинолон-резистентного Coagulase negative staphylococcus.
В соответствии с другим вариантом воплощения Гликопептид-резистентные стафилококки выбраны из Гликопептид-частично резистентного Staphylococcus aureus, Ванкомицин-резистентного Staphylococcus aureus, Ванкомицин-частично резистентного Staphylococcus aureus, Гетерованкомицин-частично резистентного Staphylococcus aureus или Гетерованкомицин-резистентного Staphylococcus aureus.
В соответствии с другим вариантом воплощения Макролид-Линкосамид-Стрептограмин-резистентные стафилококки представляют собой Макролид-Линкосамид-Стрептограмин-резистентный Staphylococcus aureus.
В соответствии с другим вариантом воплощения Линезолид-резистентные энтерококки выбраны из Линезолид-резистентного Enterococcus faecalis или Линезолид-резистентного Enterococcus faecium.
В соответствии с другим вариантом воплощения Гликопептид-резистентные энтерококки выбраны из Ванкомицин-резистентного Enterococcus faecium или Ванкомицин-резистентного Enterococcus faecalis.
В соответствии с другим вариантом воплощения β-лактам-резистентный фекальный стрептококк представляет собой β-лактам резистентный Enterococcus faecium.
В соответствии с другим вариантом воплощения пенициллин-резистентный стрептококк представляет собой Пенициллин-резистентный Streptococcus pneumoniae.
В соответствии с другим вариантом воплощения Макролид-резистентный стрептококк представляет собой Макролид-резистентный Streptococcus pneumoniae.
В соответствии с другим вариантом воплощения Кетолид-резистентые стрептококки выбраны из Макролид-резистентного Streptococcus pneumoniae и Кетолид-резистентого Streptococcus pyogenes.
В соответствии с другим вариантом воплощения фторхинолон-резистентный стрептококк представляет собой Фторхинолон-резистентный Streptococcus pneumoniae.
В соответствии с другим вариантом воплощения β-лактам-резистентный Haemophilus представляет собой β-лактам-резистентный Haemophilus influenzae.
В соответствии с другим вариантом воплощения фторхинолон-резистентный Haemophilus представляет собой Фторхинолон-резистентный Haemophilus influenzae.
В соответствии с другим вариантом воплощения Макролид-резистентный Haemophilus представляет собой Макролид-резистентный Haemophilus influenzae.
В соответствии с другим вариантом воплощения Макролид-резистентный Mycoplasma представляет собой Макролид-резистентный Mycoplasma pneumoniae.
В соответствии с другим вариантом воплощения Изониазид-резистентный Mycobacterium представляет собой Изониазид-резистентный Mycobacterium tuberculosis.
В соответствии с другим вариантом воплощения Рифампин-резистентный Mycobacterium представляет собой Рифампин-резистентный Mycobacterium tuberculosis.
В соответствии с другим вариантом воплощения β-лактам-резистентный Moraxella представляет собой β-лактам-резистентный Moraxella catarrhalis.
В соответствии с другим вариантом воплощения бактериальная инфекция характеризуется присутствием одного или нескольких из следующих: Метициллин-резистентный Staphylococcus aureus, Фторхинолон-резистентный Staphylococcus aureus, Ванкомицин-частично резистентный Staphylococcus aureus, Линезолид-резистентный Staphylococcus aureus, Пенициллин-резистентный Streptococcus pneumoniae, Макролид-резистентный Streptococcus pneumoniae, Фторхинолон-резистентный Streptococcus pneumoniae, Ванкомицин-резистентный Enterococcus faecalis, Линезолид-резистентный Enterococcus faecalis, Фторхинолон-резистентный Enterococcus faecalis, Ванкомицин-резистентный Enterococcus faecium, Линезолид-резистентный Enterococcus faecium, Фторхинолон-резистентный Enterococcus faecium, Ампициллин-резистентный Enterococcus faecium, Макролид-резистентный Haemophilus influenzae, β-лактам-резистентный Haemophilus influenzae, Фторхинолон-резистентный Haemophilus influenzae, β-лактам-резистентный Moraxella catarrhalis, Метициллин-резистентный Staphylococcus epidermidis, Метициллин-резистентный Staphylococcus epidermidis, Ванкомицин-резистентный Staphylococcus epidermidis, Фторхинолон-резистентный Staphylococcus epidermidis, Макролид-резистентный Mycoplasma pneumoniae, Изониазид-резистентный Mycobacterium tuberculosis, Рифампин-резистентный Mycobacterium tuberculosis, Фторхинолон-резистентный Neisseria gonorrhoeae или Цефалоспорин-резистентный Neisseria gonorrhoeae.
В соответствии с другим вариантом воплощения бактериальная инфекция характеризуется присутствием одного или нескольких из следующих: Метициллин-резистентный Staphylococcus aureus, Метициллин-резистентный Staphylococcus epidermidis, Метициллин-резистентный Coagulase negative staphylococcus, Фторхинолон-резистентный Staphylococcus aureus, Фторхинолон-резистентный Staphylococcus epidermidis, Фторхинолон-резистентный Coagulase negative staphylococcus, Ванкомицин-резистентный Staphylococcus aureus, Гликопептид-частично резистентный Staphylococcus aureus, Ванкомицин-резистентный Staphylococcus aureus, Ванкомицин-частично резистентный Staphylococcus aureus, Гетеро ванкомицин-частично резистентный Staphylococcus aureus, Гетеро ванкомицин-резистентный Staphylococcus aureus, Ванкомицин-резистентный Enterococcus faecium, Ванкомицин-резистентный Enterococcus faecalis, Пенициллин-резистентный Streptococcus pneumoniae, Макролид-резистентный Streptococcus pneumoniae, Фторхинолон-резистентный Streptococcus pneumoniae, Макролид-резистентный Streptococcus pyogenes или β-лактам-резистентный Haemophilus influenzae.
В соответствии с другим вариантом воплощения бактериальная инфекция характеризуется присутствием одного или нескольких из следующих: Метициллин-резистентный Staphylococcus aureus, Ванкомицин-резистентный Enterococcus faecium, Ванкомицин-резистентный Enterococcus faecalis, Ванкомицин-резистентный Staphylococcus aureus, Ванкомицин-частично резистентный Staphylococcus aureus, Гетеро ванкомицин-частично резистентный Staphylococcus aureus, Гетеро ванкомицин-резистентный Staphylococcus aureus, полирезистентный Pseudomonas aeruginosa, Изониазид-резистентный Mycobacterium tuberculosis и Рифампин-резистентный Mycobacterium tuberculosis.
В дополнение к соединениям по настоящему изобретению, фармацевтически приемлемые производные или пролекарства соединений по настоящему изобретению также можно использовать в композициях для лечения или профилактики указанных выше расстройств.
"Фармацевтически приемлемое производное или пролекарство" означает любую фармацевтически приемлемую соль, сложный эфир, соль сложного эфира или другое производное соединения по настоящему изобретению, которое при введении реципиенту способно обеспечивать, либо непосредственно, либо опосредованно, соединение по настоящему изобретению или его ингибиторно-активный метаболит или остаток. Особенно предпочтительные производные или пролекарства представляют собой такие, которые повышают биодоступность соединений по настоящему изобретению, когда такие соединения вводят млекопитающему (например, обеспечивая возможность более легкой абсорбции перорально вводимого соединения в кровотоке), или которые усиливают доставку исходного соединения в биологический компартмент (например, головной мозг или лимфатическую систему) по сравнению с исходными веществами.
Фармацевтически приемлемые пролекарства соединений по настоящему изобретению включают, без ограничения, сложные эфиры, аминокислотные сложные эфиры, фосфатные сложные эфиры, соли металлов и сульфонатные сложные эфиры.
Фармацевтически приемлемые соли соединений по настоящему изобретению включают соли, образованные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры подходящих кислотных солей включают ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, глицерофосфат, гликолят, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, пальмоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат, сульфат, тартрат, тиоцианат, тозилат и ундеканоат. Другие кислоты, такие как щавелевая кислота, хотя они и не являются фармацевтически приемлемыми как таковые, можно использовать в получении солей, полезных в качестве промежуточных соединений, для получения соединений по настоящему изобретению и их фармацевтически приемлемых кислотно-аддитивных солей.
Соли, образованные из подходящих оснований, включают соли щелочных металлов (например, натрия и калия), щелочно-земельных металлов (например, магния), соли аммония и N+(C1-4 алкил)4. Настоящее изобретение также предусматривает кватернизацию щелочных азот-содержащих групп соединений, раскрытых в настоящей заявке. Водо- или маслорастворимые или диспергируемые продукты можно получить путем такой кватернизации.
Фармацевтические композиции по настоящему изобретению включают соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Такие композиции, необязательно, могут включать дополнительное терапевтическое средство. Такие средства включают, но не ограничиваются этим, антибиотик, противовоспалительное средство, ингибитор металлопротеазы матрикса, ингибитор липоксигеназы, антагонист цитокина, иммуносупрессант, противораковое средство, противовирусное средство, цитокин, фактор роста, иммуномодулятор, простагландин или соединения против гиперпролиферации сосудов.
Термин "фармацевтически приемлемый носитель" относится к нетоксичному носителю, который можно вводить пациенту вместе с соединением по настоящему изобретению, и который не разрушает его фармакологическую активность.
Фармацевтически приемлемые носители, которые можно использовать в фармацевтических композициях по настоящему изобретению, включают, но не ограничиваются этим, ионообменники, оксид алюминия, стеарат алюминия, лецитин, белки сыворотки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протаминсульфат, динатрий гидрофосфат, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрий карбоксиметилцеллюлозу, полиакрилаты, воски, полиэтилен-полиоксипропиленовые блок-полимеры, ланолин и самоэмульгирующие системы доставки лекарственных средств (SEDDS), такие как альфа-токоферол, полиэтиленгликоль 1000 сукцинат или другие подобные используемые для доставки полимерные матрицы.
Термин "фармацевтически эффективное количество" относится к количеству, эффективному для лечения или облегчения бактериальной инфекции у пациента. Термин "профилактически эффективное количество" относится к количеству, эффективному для профилактики или существенного ослабления бактериальной инфекции у пациента.
В зависимости от конкретного состояния или заболевания, подлежащего лечению или профилактике, дополнительные терапевтические средства, которые обычно вводят для лечения или профилактики такого состояния, можно вводить вместе с ингибиторами по настоящему изобретению. Такие терапевтические средства включают, но не ограничиваются этим, антибиотик, противовоспалительное средство, ингибитор металлопротеазы матрикса, ингибитор липоксигеназы, антагонист цитокина, иммуносупрессант, противораковое средство, противовирусное средство, цитокин, фактор роста, иммуномодулятор, простагландин или соединения против гиперпролиферации сосудов.
Соединения по настоящему изобретению можно использовать обычным образом для контроля уровней бактериальной инфекциии in vivo и для лечения заболеваний или уменьшения прогрессирования или тяжести эффектов, которые опосредованы бактериями. Такие способы лечения, уровни дозирования и необходимые дозы сможет выбрать рядовой специалист в данной области из доступных способов и процедур.
Например, соединение по настоящему изобретению можно объединить с фармацевтически приемлемым адъювантом для введения пациенту, страдающему от бактериальной инфекции или заболевания, фармацевтически приемлемым способом и в количестве, эффективном для облегчения тяжести такой инфекции или заболевания.
Альтернативно, соединения по настоящему изобретению можно использовать в композициях и способах для лечения или защиты субъектов от бактериальных инфекций или заболеваний в течение продолжительных периодов времени. В одном варианте воплощения соединения по настоящему изобретению можно использовать в композициях и способах для лечения или защиты субъектов от бактериальных инфекций или заболеваний в течение 1-2-недельного периода. В другом варианте воплощения соединения по настоящему изобретению можно использовать в композициях и способах для лечения или защиты субъектов от бактериальных инфекций или заболеваний в течение 4-8-недельного периода (например, в лечении пациентов с эндокардитом или остеомиелитом или имеющих риск развития такого заболевания). В другом варианте воплощения соединения по настоящему изобретению можно использовать в композициях и способах для лечения или защиты субъектов от бактериальных инфекций или заболеваний в течение 8-12-недельного периода. Соединения можно использовать в таких композициях либо отдельно, либо вместе с другими соединениями по настоящему изобретению таким путем, который соответствует традиционному использованию ингибиторов ферментов в фармацевтических композициях. Например, соединение по настоящему изобретению можно объединить с фармацевтически приемлемыми адъювантами, традиционно используемыми в вакцинах, и вводить в профилактически эффективных количествах для защиты субъектов в течение продолжительного периода времени от бактериальных инфекций или заболеваний.
В некоторых вариантах воплощения соединения формулы (I) можно использовать профилактически для предотвращения бактериальной инфекции. В некоторых вариантах воплощения соединения формулы (I) можно использовать до, в процессе или после стоматологической или хирургической процедуры для профилактики инфекций, вызываемых условно-патогенными организмами, таких как в случае бактериального эндокардита. В других вариантах воплощения соединения формулы (I) можно использовать профилактически в стоматологических процедурах, включая, но не ограничиваясь этим, удаление зубов, периодонтальные процедуры, введение внутричелюстных зубных имплантатов и эндодонтическую операцию. В других вариантах воплощения соединения формулы (I) можно использовать профилактически в хирургических процедурах, включая, но не ограничиваясь этим, общую хирургию, хирургическую операцию в области дыхательных путей (тонзилэктомию/аденоидэктомию), хирургическую операцию в области желудочно-кишечного тракта (хирургическую операцию в верхней области желудочно-кишечного тракта и элективной области тонкого кишечника, эзофагеальную склеротерапию и дилатацию, резекции толстой кишки, удаление острого апендицита), связанную с травмой хирургическую операцию (проникающую хирургическую операцию брюшной полости), хирургическую операцию мочеполовой системы (удаление предстательной железы, расширение уретры, цистоскопию, вагинальную или абдоминальную гистерэктомию, кесарево сечение), хирургическую трансплантацию (трансплантацию почки, печени или поджелудочной железы), хирургическую операцию в области головы и шеи (иссечения кожи, диссекции шеи, ларингэктомию, хирургическое лечение рака головы и шеи, переломы нижней челюсти), ортопедическую хирургическую операцию (полную замену сустава, травматические открытые переломы), хирургическую операцию на сосудах (процедуры на периферических сосудах), хирургическую операцию на сердце, хирургическую процедуру коронарного шунтирования, резекцию легкого и нейрохирургическую операцию.
Термин "профилактика бактериальной инфекции", как он используется в настоящей заявке, если не указано иное, означает профилактическое применение антибиотика, такого как ингибитор гиразы и/или топоизомеразы IV по настоящему изобретению, для профилактики бактериальной инфекции. Лечение ингибитором гиразы и/или топоизомеразы IV можно осуществлять профилактически для профилактики инфекции, вызываемой организмом, который чувствителен к ингибитору гиразы и/или топоизомеразы IV. Одна общая группа состояний, где может рассматриваться профилактическое лечение, включает такие состояния, когда субъект является более чувствительным к инфекции из-за, например, ослабленного иммунитета, хирургической операции, травмы, присутствия искусственного устройства в организме (временного или постоянного), анатомического дефекта, воздействия высоких уровней бактерий или возможного воздействия болезнетворного патогена. Примеры факторов, которые могут привести к ослабленному иммунитету, включают химиотерапию, лучевую терапию, диабет, пожилой возраст, ВИЧ инфекцию и трансплантацию. Примером анатомического дефекта может быть дефект сердечного клапана, который повышает риск бактериального эндокардита. Примеры искусственных устройств включают искусственные суставы, хирургические штифты, катетеры и т.п. Другая группа включает ситуации, где профилактическое применение ингибитора гиразы и/или топоизомеразы IV может быть подходящим дря использования для профилактики распространения патогена между субъектами (непосредственно или опосредованно). Конкретным примером профилактического применения для предотвращения распространения патогена является использование ингибитора гиразы и/или топоизомеразы IV субъектами в учреждении здравоохранения (например, в больнице или частной лечебнице).
Соединения формулы (I) также можно вводить вместе с другими антибиотиками для усиления эффекта лечения или профилактики против различных бактериальных инфекций. Когда соединения по настоящему изобретению вводят в комбинированных терапиях с другими средствами, их можно вводить пациенту последовательно или одновременно. Альтернативно, фармацевтические или профилактические композиции в соответствии с настоящим изобретением включают комбинацию соединения формулы (I) и другого терапевтического или профилактического средства.
В некоторых вариантах воплощения дополнительное терапевтическое средство, или средства, представляет собой антибиотик, выбранный из природного пенициллина, пенициллиназа-резистентного пенициллина, антипсевдомонадного пенициллина, аминопенициллина, цефалоспорина первого поколения, цефалоспорина второго поколения, цефалоспорина третьего поколения, цефалоспорина четвертого поколения, карбапенема, цефамицина, хинолона, фторхинолона, аминогликозида, макролида, кетолида, полимиксина, тетрациклина, гликопептида, стрептограмина, оксазолидинона, рифамицина или сульфонамида.
В некоторых вариантах воплощения дополнительное терапевтическое средство, или средства, представляет собой антибиотик, выбранный из пенициллина, цефалоспорина, хинолона, аминогликозида или оксазолидинона.
В других вариантах воплощения дополнительные терапевтические средства выбраны из природного пенициллина, включая Бензатин пенициллин G, Пенициллин G и Пенициллин V, из пенициллиназа-резистентного пенициллина, включая Клоксациллин, Диклоксациллин, Нафциллин и Оксациллин, из антипсевдомонадного пенициллина, включая Карбенициллин, Мезлоциллин, Пиперциллин, Пиперциллин/тазобактам, Тикарициллин и Тикарициллин/Клавуланат, из аминопенициллина, включая Амоксициллин, Ампициллин и Ампициллин/Сулбактам, из цефалоспорина первого поколения, включая Цефазолин, Цефадроксил, Цефалексин и Цефадрин, из цефалоспорина второго поколения, включая Цефаклор, Цефаклор-CD, Цефамандол, Цефонацид, Цефпрозил, Лоракарбеф и Цефуроксим, из цефалоспорина третьего поколения, включая Цефдинир, Цефиксим, Цефоперазон, Цефотаксим, Цефподоксим, Цефтазидим, Цефтибутен, Цефтизоксим и Цефтриаксон, из цефалоспорина четвертого поколения, включая Цефепим, Цефтаролин и Цефтобипрол, из Цефамицина, включая Цефотетан и Цефокситин, из карбапенема, включая Дорипенем, Имипенем и Меропенем, из монобактама, включая Азтреонам, из хинолона, включая Циноксацин, Наликсидиновую кислоту, Оксолиновую кислоту и Пиппемидиновую кислоту, из фторхинолона, включая Бесифлоксацин, Ципрофлоксацин, Эноксацин, Гатифлоксацин, Грепафлоксацин, Левофлоксацин, Ломефлоксацин, Моксифлоксацин, Норфлоксацин, Офлоксацин и Спарфлоксацин, из аминогликозида, включая Амикацин, гентамицин, Канамицин, Неомицин, Нетилмицин, Спектиномицин, Стрептомицин и Торбамицин, из макролида, включая Азитромицин, Кларитромицин и Эритромицин, из кетолида, включая Телитромицин, из Тетрациклина, включая Хлортетрациклин, Домеклоциклин, Доксициклин, Миноциклин и Тетрациклин, из гликопептида, включая Оритаванцин, Далбаванцин, Телаванцин, Тейкопланин и Ванкомицин, из стрептограмина, включая Далфопристин/хинупристин, из оксазолидона, включая Линезолид, из Рифамицина, включая Рифабутин и Рифампин, и из других антибиотиков, включая бактитрацин, колистин, Тигацил, Даптомицин, хлорамфеникол, клиндамицин, изониазид, метронидазол, мупироцин, полимиксин B, пиразинамид, триметоприм/сульфаметоксазол и сульфисоксазол.
В других вариантах воплощения дополнительные терапевтические средства выбраны из природного пенициллина включая Пенициллин G, из пенициллиназа-резистентного пенициллина, включая Нафциллин и Оксациллин, из антипсевдомонадного пенициллина, включая Пиперциллин/тазобактам, из аминопенициллина, включая Амоксициллин, из цефалоспорина первого поколения, включая Цефалексин, из цефалоспорина второго поколения, включая Цефаклор, Цефаклор-CD и Цефуроксим, из цефалоспорина третьего поколения, включая Цефтазидим и Цефтриаксон, из цефалоспорина четвертого поколения, включая Цефепим, из карбапенема, включая Имепенем, Меропенем, Эртапенем, Дорипенем, Панипенем и Биапенем, фторхинолона, включая Ципрофлоксацин, Гатифлоксацин, Левофлоксацин и Моксифлоксацин, из аминогликозида, включая Торбамицин, из макролида, включая Азитромицин и Кларитромицин, из Тетрациклина, включая Доксициклин, из гликопептида, включая Ванкомицин, из Рифамицина, включая Рифампин, и из других антибиотиков, включая изониазид, пиразинамид, Тигацил, Даптомицин или триметоприм/сульфаметоксазол.
В некоторых вариантах воплощения твердую форму соединения формулы (I) можно вводить для лечения грамположительной инфекции. В некоторых вариантах воплощения композиция представляет собой твердую, жидкую (например, суспензию) или внутривенную (например, форму соединения формулы (I) растворяют в жидкости и вводят внутривенно) композицию. В некоторых вариантах воплощения композицию, включающую соединение формулы (I), вводят в комбинации с дополнительным антибиотиком, например, природным пенициллином, пенициллиназа-резистентным пенициллином, антипсевдомонадным пенициллином, аминопенициллином, цефалоспорином первого поколения, цефалоспорином второго поколения, цефалоспорином третьего поколения, цефалоспорином четвертого поколения, карбапенемом, цефамицином, хинолоном, фторхинолоном, аминогликозидом, макролидом, кетолидом, полимиксином, тетрациклином, гликопептидом, стрептограмином, оксазолидиноном, рифамицином или сульфонамидом. В некоторых вариантах воплощения композицию, включающую твердую форму соединения формулы (I), вводят перорально, дополнительный антибиотик, например, природный пенициллин, пенициллиназа-резистентный пенициллин, антипсевдомонадный пенициллин, аминопенициллин, цефалоспорин первого поколения, цефалоспорин второго поколения, цефалоспорин третьего поколения, цефалоспорин четвертого поколения, карбапенем, цефамицин, хинолон, фторхинолон, аминогликозид, макролид, кетолид, полимиксин, тетрациклин, гликопептид, стрептограмин, оксазолидинон, рифамицин или сульфонамид, вводят внутривенно.
В некоторых вариантах воплощения твердую форму соединения формулы (I) можно вводить для лечения грамотрицательной инфекции. В некоторых вариантах воплощения композиция представляет собой твердую, жидкую (например, суспензию) или внутривенную (например, форму соединения формулы (I) растворяют в жидкости и вводят внутривенно) композицию. В некоторых вариантах воплощения композицию, включающую соединение формулы (I), вводят в комбинации с дополнительным антибиотиком, выбранным из природного пенициллина, пенициллиназа-резистентного пенициллина, антипсевдомонадного пенициллина, аминопенициллина, цефалоспорина первого поколения, цефалоспорина второго поколения, цефалоспорина третьего поколения, цефалоспорина четвертого поколения, карбапенема, цефамицина, монобактама, хинолона, фторхинолона, аминогликозида, макролида, кетолида, полимиксина, тетрациклина или сульфонамида. В некоторых вариантах воплощения композицию, включающую твердую форму соединения формулы (I), вводят перорально, и дополнительный антибиотик, например, природный пенициллин, пенициллиназа-резистентный пенициллин, антипсевдомонадный пенициллин, аминопенициллин, цефалоспорин первого поколения, цефалоспорин второго поколения, цефалоспорин третьего поколения, цефалоспорин четвертого поколения, карбапенем, цефамицин, монобактам, хинолон, фторхинолон, аминогликозид, макролид, кетолид, полимиксин, тетрациклин или сульфонамид, вводят перорально. В некоторых вариантах воплощения дополнительное терапевтическое средство вводят внутривенно.
Дополнительные терапевтические средства, описанные выше, можно вводить отдельно, как часть схемы многократного введения, из ингибитор-содержащей композиции. Альтернативно, эти средства могут быть частью одной лекарственной формы, смешанные вместе с ингибитором в одной композиции.
Фармацевтические композиции по настоящему изобретению можно вводить перорально, парентерально, при помощи спрея для ингаляции, местным путем, ректально, назально, буккально, вагинально или через имплантируемый резервуар. Фармацевтические композиции по настоящему изобретению могут содержать любые традиционные нетоксичные фармацевтически приемлемые носители, адъюванты или наполнители. В некоторых случаях, pH композиции можно регулировать с использованием фармацевтически приемлемых кислот, оснований или буферов для повышения стабильности сформулированного в композицию соединения или формы его доставки. Термин “парентеральный”, как он используется в настоящей заявке, включает подкожную, внутрикожную, внутривенную, внутримышечную, внутрисуставную, интрасиновиальную, интрастернальную, интратекальную, внутриочаговую и интракраниальную инъекцию или инфузию.
Фармацевтические композиции могут быть в форме стерильного препарата для инъекций, например, в виде стерильной водной или масляной суспензии для инъекций. Такая суспензия может быть сформулирована в соответствии со способами, известными из уровня техники, с использованием подходящих диспергирующих или смачивающих веществ (таких как, например, Tween 80) и суспендирующих веществ. Стерильный препарат для инъекций также может представлять собой стерильный раствор или суспензию для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Из приемлемых носителей и растворителей, которые можно использовать, можно указать маннит, воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла традиционно используют в качестве растворителя или среды для суспендирования. Для этих целей можно использовать любое светлое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, являются полезными для получения препаратов для инъекций, так же как и природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных формах. Эти масляные растворы или суспензии также могут содержать длинноцепочечный спиртовой разбавитель или диспергирующий агент, такой как вещества, описанные в Pharmacopeia Helvetica, или подобный спирт.
Фармацевтические композиции по настоящему изобретению можно вводить перорально в любой перорально приемлемой лекарственной форме, включая, но не ограничиваясь этим, капсулы, таблетки и водные суспензии и растворы. В случае таблеток для перорального использования, носители, которые обычно используют, включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие вещества, такие как стеарат магния. Для перорального введения в форме капсулы, полезные разбавители включают лактозу и высушенный кукурузный крахмал. Когда водные суспензии и растворы и пропиленгликоль вводят перорально, активный ингредиент объединяют с эмульгаторами и суспендирующими веществами. Если желательно, можно добавить некоторые подсластители и/или отдушки и/или красители.
Фармацевтические композиции по настоящему изобретению также можно вводить в форме суппозиториев для ректального введения. Эти композиции можно получить путем смешивания соединения по настоящему изобретению с подходящим нераздражающим эксципиентом, который является твердым при комнатной температуре, но жидким при ректальной температуре, и поэтому будет плавиться в прямой кишке с высвобождением активных компонентов. Такие вещества включают, но не ограничиваются этим, масло какао, пчелиный воск и полиэтиленгликоли.
Местное введение фармацевтических композиций по настоящему изобретению является особенно полезным, когда желаемое лечение затрагивает области или органы, легко доступные путем местного нанесения. Для местного нанесения на кожу фармацевтическая композиция должна быть сформулирована в виде подходящей мази, содержащей активные компоненты, суспендированные или растворенные в носителе. Носители для местного введения соединений по настоящему изобретению включают, но не ограничиваются этим, минеральное масло, вазелиновое масло, медицинский вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропилен, эмульгирующий воск и воду. Альтернативно, фармацевтическая композиция может быть сформулирована в виде подходящего лосьона или крема, содержащего активное соединение, суспендированное или растворенное в носителе. Подходящие носители включают, но не ограничиваются этим, минеральное масло, сорбитанмоностеарат, полисорбат 60, содержащий сложные цетиловые эфиры воск, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду. Фармацевтические композиции по настоящему изобретению также можно наносить местно в нижней области кишечного тракта с использованием препарата в форме ректального суппозитория или подходящей композиции для введения через клизму. Чрескожные пластыри для местного введения также включены в настоящее изобретение.
Фармацевтические композиции по настоящему изобретению можно вводить при помощи назального аэрозоля или путем ингаляции. Такие композиции получают в соответствии со способами, хорошо известными в области фармацевтического формулирования, и их можно получить в виде растворов в физиологическом растворе, с использованием бензилового спирта или других подходящих консервантов, промоторов абсорбции для повышения биодоступности, фторуглеродов и/или других солюбилизирующих или диспергирующих веществ, известных из уровня техники.
В соответствии с другим вариантом воплощения соединения формулы (I) также могут быть доставлены путем имплантации (например, хирургическим путем), например, с использованием имплантируемого или находящегося внутри организма устройства. Имплантируемое или находящееся внутри организма устройство может быть предназначено либо для постоянного, либо для временного нахождения в организме субъекта. Примеры имплантируемых или находящихся внутри организма устройств включают, но не ограничиваются этим, контактные линзы, центральные венозные катетеры и безыгольные коннекторы, эндотрахеальные трубки, внутриматочные устройства, механические сердечные клапаны, сердечные ритмоводители, перитонеальные диализные катетеры, протезы суставов, такие как бедренные и коленные заместители, миринготомические трубки, мочевыводящие катетеры, голосовые протезы, стенты, питающие насосы, сосудистые фильтры и имплантируемые композиции контролируемого высвобождения. Биопленки могут быть вредными для здоровья пациентов с имплантируемым или находящимся внутри организма медицинским устройством, поскольку они вводят искусственный субстрат в организм и могут вызывать постоянные инфекции. Таким образом, обеспечение соединений формулы (I) в или на имплантируемом или находящемся внутри организма устройстве может предотвращать или уменьшать продукцию биопленки. Кроме того, имплантируемые или находящиеся внутри организма устройства можно использовать в качестве депо или резервуара соединений формулы (I). Любое имплантируемое или находящееся внутри организма устройство можно использовать для доставки соединения формулы (I), при условии, что a) устройство, соединения формулы (I) и любая фармацевтическая композиция, включая соединения формулы (I), являются биосовместимыми, и b), что устройство может доставлять или высвобождать эффективное количество соединений формулы (I) для обеспечения терапевтического эффекта принимающему лечение пациенту.
Доставка терапевтических средств через имплантируемые или находящиеся внутри организма устройства известна из уровня техники. Смотрите, например, “Recent Developments in Coated Stents” by Hofma et al., опубликованный в Current Interventional Cardiology Reports 2001, 3:28-36, полное содержание которого, включая документы, на которые в нем имеются ссылки, включено в настоящую заявку посредством ссылки. Другие описания имплантируемых устройств можно найти в патентах США №№ 6569195 и 6322847 и патентных заявках США №№ 2004/0044405, 2004/0018228, 2003/0229390, 2003/0225450, 2003/0216699 и 2003/0204168, каждый из этих документов включен в настоящую заявку посредством ссылки во всей его полноте.
В некоторых вариантах воплощения имплантируемое устройство представляет собой стент. В одном конкретном варианте воплощения стент может включать сцепленные между собой кабели. Каждый кабель может включать металлические проволоки для структурной поддержки и полимерные проволоки для доставки терапевтического средства. Включение доз терапевтического средства в полимерную проволоку можно осуществить путем погружения полимера в раствор терапевтического средства. Альтернативно, терапевтическое средство может быть включено в полимерную проволоку в процессе образования проволоки из растворов полимерных предшественников.
В других вариантах воплощения имплантируемые или находящиеся внутри организма устройства могут иметь полимерные покрытия, которые включают терапевтическое средство. Полимерное покрытие может быть предназначено для контроля скорости высвобождения терапевтического средства. Для контролируемого высвобождения терапевтических средств можно использовать различные технологии. Известны устройства, которые имеют монолитный слой или покрытие, включающее гетерогенный раствор и/или дисперсию активного агента в полимерном веществе, где имеется ограничение скорости диффузии агента, поскольку диффузия агента происходит через полимер к границе раздела полимер-жидкость с высвобождением в окружающую жидкость. В некоторых устройствах растворимое вещество также растворяют или диспергируют в полимерном материале, так чтобы оставались дополнительные поры или каналы после того, как материал растворяется. Матричное устройство обычно также является диффузионно-ограниченным, но при этом каналы или другая внутренняя геометрия такого устройства также играют роль в высвобождении агента в жидкую среду. Каналы могут представлять собой ранее существовавшие каналы или каналы, остающиеся после высвобождения агента или других растворимых веществ.
Эродируемые или разлагаемые устройства типично содержат активный агент, физически иммобилизованный в полимере. Активный агент может быть растворен и/или диспергирован по всему полимерному материалу. Полимерный материал часто гидролитически разлагается с течением времени через гидролиз лабильных связей, давая полимеру эродировать в жидкой среде, с высвобождением активного агента в жидкую среду. Гидрофильные полимеры, как правило, имеют более быструю скорость эрозии по сравнению с гидрофобными полимерами. Считается, что гидрофобные полимеры имеют почти чисто поверхностную диффузию активного агента, имея эрозию от поверхности внутрь. Считается, что гидрофильные полимеры позволяют воде проникать через поверхность полимера, обеспечивая возможность гидролиза лабильных связей под поверхностью, что может привести к гомогенной или объемной эрозии полимера.
Покрытие имплантируемого или находящегося внутри организма устройства может включать смесь полимеров, каждый из которых имеет разную скорость высвобождения терапевтического средства. Например, покрытие может включать сополимер полимолочной кислоты/полиэтиленоксида (PLA-PEO) и сополимер полимолочной кислоты/поликапролактона (PLA-PCL). Сополимер полимолочной кислоты/полиэтиленоксида (PLA-PEO) может демонстрировать более высокую скорость высвобождения терапевтического средства по сравнению с сополимером полимолочной кислоты/поликапролактона (PLA-PCL). Относительные количества и величину доз терапевтического средства, доставляемые в течение времени, можно контролировать путем контроля относительных количеств более быстро высвобождающих полимеров относительно более медленно высвобождающих полимеров. Для более высоких начальных скоростей высвобождения долю более быстро высвобождающего полимера можно увеличить по сравнению с более медленно высвобождающим полимером. Если желательно, чтобы большая часть дозы высвобождалась в течение длительного периода времени, полимер должен большей частью состоять из более медленно высвобождающего полимера. На устройство можно наносить покрытие путем разбрызгивания на устройство раствора или дисперсии полимера, активного вещества и растворителя. Растворитель может испаряться, оставляя покрытие из полимера и активного вещества. Активное вещество может быть растворено и/или диспергировано в полимере. В некоторых вариантах воплощения сополимеры можно наносить на устройство методом экструзии.
Уровни доз соединения в качестве активного ингредиента в пределах от около 0,01 до около 100 мг/кг массы тела в день, предпочтительно в пределах от 0, 5 до около 75 мг/кг массы тела в день, и наиболее предпочтительно в пределах от около 1 до 50 мг/кг массы тела в день являются полезными в монотерапии для профилактики и лечения бактериальных инфекций.
Обычно, фармацевтические композиции по настоящему изобретению можно вводить примерно 1-5 раз в день или, альтернативно, в виде непрерывной инфузии. Альтернативно, композиции по настоящему изобретению можно вводить в виде композиции импульсного высвобождения. Такое введение можно использовать в виде длительной или неотложной терапии. Количество активного ингредиента, которое можно объединить с веществами-носителями для получения одноразовой лекарственной формы, будет варьировать в зависимости от хозяина, подлежащего лечению, и конкретного способа введения. Типичный препарат будет содержать от около 5% до около 95% активного соединения (масс/масс). Предпочтительно, такие препараты содержат от около 20% до около 80% активного соединения.
Когда композиции по настоящему изобретению включают комбинацию соединения формулы (I) и одного или нескольких дополнительных терапевтических или профилактических средств, как соединение, так и дополнительное средство должны присутствовать в дозах на уровне приблизительно от 10% до 80% от дозы, обычно вводимой в режиме монотерапии.
После улучшения состояния пациента можно вводить поддерживающую дозу соединения, композиции или комбинации по настоящему изобретению, если необходимо. Впоследствии дозы или частоту введения, или и то и другое, можно уменьшить, в зависимости от симптомов, до уровня, при котором улучшенное состояние сохраняется, когда симптомы облегчаются до желаемого уровня, лечение следует прекратить. Пациентам, однако, может быть необходимо периодическое лечение на длительной основе после какого-либо рецидива или проявления симптомов заболевания.
Как должно быть понятно специалистам, могут потребоваться более низкие или более высокие дозы, чем те, которые указаны выше. Конкретные дозы и схемы лечения для любого конкретного пациента зависят от различных факторов, включая активность конкретного используемого соединения, возраст, массу тела, общее состояние здоровья, пол, режим питания, время введения, скорость выведения из организма, комбинацию лекарственных средств, тяжесть и протекание болезни и предрасположенность пациента к заболеванию, и от решения лечащего врача.
В соответствии с другим вариантом воплощения изобретение обеспечивает способы лечения или профилактики бактериальной инфекции или болезненного состояния, включающие стадию введения пациенту любого соединения, фармацевтической композиции или комбинации, описанных в настоящей заявке. Термин "пациент", как он используется в настоящей заявке, означает животное, предпочтительно млекопитающее, и наиболее предпочтительно человека.
Соединения по настоящему изобретению также являются полезными в качестве коммерческих реагентов, которые эффективно связываются с ферментами гиразой B и/или топоизомеразой IV. В качестве коммерческих реагентов, соединения по настоящему изобретению и их производные можно использовать для блокирования активности гиразы B и/или топоизомеразы IV в биохимических или клеточных анализах бактериальной гиразы B и/или топоизомеразы IV или их гомологов, или они могут быть дериватизированы для связывания со стабильной смолой в качестве связанного субстрата для применения в аффинной хроматографии. Эти и другие применения, которые характеризуют коммерческие ингибиторы гиразы B и/или топоизомеразы IV, будут очевидны для специалистов в данной области.
Соединения по настоящему изобретению можно получить в соответствии с общими способами, известными специалистам в данной области для аналогичных соединений, которые раскрыты в патенте США № RE40245 E; патенте США № 7495014 B2; патенте США № 7569591 B2; патенте США № 7582641 B2; патенте США № 7618974 B2 и патенте США № 7727992 B2. Все шесть указанных патентов включены посредством ссылки, как если бы они были полностью изложены в настоящей заявке. Условия, используемые для получения соединений по настоящему изобретению, подробно описаны далее в примерах.
В целях лучшего понимания настоящего изобретения далее представлены примеры. Эти примеры представлены только в целях иллюстрации, и не должны рассматриваться как ограничивающие каким-либо образом объем настоящего изобретения.
Далее описаны термины и аббревиатуры, используемые в настоящей заявке:
Ac - ацетил
Bu - бутил
Et - этил
Ph - фенил
Me - метил
ТГФ - тетрагидрофуран
DCM - дихлорметан
CH2Cl2 - дихлорметан
EtOAc - этилацетат
CH3CN - ацетонитрил
EtOH - этанол
Et2O - диэтиловый эфир
MeOH - метанол
MTBE - метил трет-бутиловый эфир
DMF - N,N-диметилформамид
DMA - N,N-диметилацетамид
ДМСО - диметилсульфоксид
HOAc - уксусная кислота
TEA - триэтиламин
TFA - трифторуксусная кислота
TFAA - трифторуксусный ангидрид
Et3N - триэтиламин
DIPEA - диизопропилэтиламин
DIEA - диизопропилэтиламин
K2CO3 - карбонат калия
Na2CO3 - карбонат натрия
Na2S2O3 - тиосульфат натрия
Cs2CO3 - карбонат цезия
NaHCO3 - бикарбонат натрия
NaOH - гидроксид натрия
Na2SO4 - сульфат натрия
MgSO4 - сульфат магния
K3PO4 - фосфат калия
NH4Cl - хлорид аммония
ЖХ/MS - жидкостная хроматография/масс-спектры
ГХ/MS - газовая хроматография/масс-спектры
ВЭЖХ - высокоэффективная жидкостная хроматография
ГХ - газовая хроматография
ЖХ - жидкостная хроматография
IC - ионная хроматография
IM - внутримышечный
КОЕ/кое колониеобразующие единицы
MIC - минимальная ингибиторная концентрация
Hr или h - часы
атм - атмосфер
rt или RT - комнатная температура
ТСХ - тонкослойная хроматография
HCl - хлористоводородная кислота
H2O - вода
EtNCO - этилизоцианат
Pd/C - палладий на углероде
NaOAc - ацетат натрия
H2SO4 - серная кислота
N2 - газообразный азот
H2 - газообразный водород
n-BuLi - н-бутиллитий
DI - деионизированный
Pd(OAc)2 - ацетат палладия(II)
PPh3 - трифенилфосфин
i-PrOH - изопропиловый спирт
NBS - N-бромсукцинимид
Pd[(Ph3)P]4 - тетракис(трифенилфосфин)палладий(0)
PTFE - политетрафторэтилен
об/мин - оборотов в минуту
SM - исходное вещество
Эквив. - эквиваленты
1H-ЯМР - протонный ядерный магнитный резонанс
HPMCAS - ацетат гидроксипропилметилцеллюлозы
PVP - поливинилпирролидон
EDTA - этилендиаминтетрауксусная кислота
K2EDTA - дикалий этилендиаминтетраацетат
mCPBA - мета-хлорпероксибензойная кислота
водн. - водный
Boc2O - ди-трет-бутилдикарбонат
DMAP - N,N-диметиламинопиридин
мл - миллилитры
л - литры
моль - моли
г - граммы
ЖХ/MS - жидкостная хроматография/масс-спектрометрия
МГц - мегагерцы
CDCl3 - дейтерохлороформ
NEt3 - триэтиламин
ммоль - миллимоли
ф/дюйм2 - фунтов на квадратный дюйм
iPrOH - изопропиловый спирт
м.д. - миллионные доли
NH4NO3 - нитрат аммония
Гц - герцы
Pd(dppf)Cl2 - [1,1′-
бис(дифенилфосфино)ферроцен]дихлорпалладий(II)
л - литры
MeOD - дейтеро-метанол
CD3OD - дейтеро-метанол
эи - энантиомерный избыток
мин - минуты
Bn - бензил
RBF - круглодонная колба
MeCN - ацетонитрил
PES - полиэфирсульфон
мм - миллиметры
мкм - микрометры
M - молярный
н - нормальный
Boc - трет-бутоксикарбонил
ESMS - масс-спектрометрия с электроспреем
CV - объем колонки
D2O - оксид дейтерия
NH3 - аммиак
OBD - оптимальная плотность слоя
мг - миллиграммы
CLSI - Clinical and Laboratory Standards Institute
ATCC - American Type Culture Collection
MHII - Mueller Hinton II
мкл - микролитры
WT - дикий тип
CGSC - Coli Genetic Stock Center
MS - масс-спектрометрия
IS - внутренний стандарт
APCI - химические ионизация атмосферного давления
MRM - многократный контроль реакции
m/z - отношение массы к заряду
LLOQ - нижний предел количественного определения
нг - нанограммы
УФ - ультрафиолет
SD - стандартное отклонение
% CV - коэффициент вариаций
PO - пероральный
MC - микрокристаллическая целлюлоза
EDTA - этилендиаминтетрауксусная кислота или
этилендиаминтетраацетат
PK - фармакокинетический
IV - внутривенный
D5W - 5% водный раствор декстрозы
HPMC-AS - ацетилсукцинат гидроксипропилметилцеллюлозы
PVP - поливинилпирролидон
CAPT - каптисол
АТФ - аденозинтрифосфат
АДФ - аденозиндифосфат
NADH - никотинамидаденин динуклеотид (восстановленная форма)
NAD+ - никотинамид аденин динуклеотид (окисленная форма)
TRIS - трис(гидроксиметил)аминометан
мМ - миллимолярный
MgCl2 - хлорид магния
KCl - хлорид калия
мкМ - микромолярный
DTT - дитиотреитол
нМ - наномолярный
Ki - константа диссоциации
ИК50 - полумаксимальная ингибиторная концентрация
мкг - микрограммы
BSA - бычий сывороточный альбумин
LDH - лактатдегидрогеназа
PVDF - поливинилиденфторид
AcN - ацетонитрил
VMAX - максимальная начальная скорость или интенсивность реакции
Пример 1
Получение 2-(2-нитрофенил)-2,5-дигидрофурана и 2-(2-нитроф енил)-2,3-дигидрофурана (3a & 3b)
Смешивали 1-бром-2-нитро-бензол (1) (600 г, 99%, 2,941 моль, Alfa Aesar A11686), 1,3-бис(дифенилфосфино)пропан (62,50 г, 97%, 147,0 ммоль, Alfa Aesar A12931), 1,4-диоксан (2, 970 л, Sigma-Aldrich 360481), карбонат калия (812,9 г, 5,882 моль, JT-Baker 301201) и 2,3-дигидрофуран (2) (1,041 кг, 99%, 1,124 л, 14,70 моль, Aldrich 200018). Поток азота барботировали через перемешиваемую смесь в течение 4 часов, с последующим добавлением ацетата палладия (II) (16,51 г, 73,52 ммоль, Strem 461780) и продолжением деоксигенирования еще в течение 10 минут. Реакционную смесь перемешивали при температуре кипения с обратным холодильником в атмосфере азота в течение ночи (ЯМР обработанной аликвоты показал полное поглощение арилбромида). Смеси давали охладиться, разбавляли гексаном (1 л), фильтровали через короткую пробку из Florisil® (500 г, -200 меш) и элюировали при помощи EtOAc. Фильтрат концентрировали при пониженном давлении (2-(2-нитрофенил)-2,3-дигидрофуран является летучим в условиях высокого вакуума и может быть несколько нестабильным при комнатной температуре) с получением смеси (3a) и (3b) в виде темно-коричневого масла (654,0 г). Неочищенное вещество хранили в холодильнике и использовали далее без дополнительной очистки.
Пример 2
Получение 2-т етрагидрофуран-2-ил-анилина (4)
5% Палладий на углероде (16,3 г, влажность 50%, 3,83 ммоль, Aldrich 330116) помещали в сосуд Парра в атмосфере азота, затем добавляли MeOH (100 мл, JT-Baker 909333). Добавляли неочищенную смесь 2-(2-нитрофенил)-2,5-дигидрофурана и 2-(2-нитрофенил)-2,3-дигидрофурана (3a & 3b)) (163 г), растворенную в MeOH (389 мл), с последующим добавлением NEt3 (237, 6 мл, 1,705 моль, Sigma-Aldrich 471283). Помещали сосуд на встряхивающее устройство Парра и насыщали H2. Добавляли 30 ф/дюйм2(2,109 кг/см2) H2 и продолжали встряхивание вплоть до полного поглощения (ЖХ/MS и ЯМР показали завершение реакции). Реакционную смесь продували азотом, фильтровали через Целит™ и промывали при помощи EtOAc. Фильтрат концентрировали на роторном испарителе с получением коричневого масла. Реакцию осуществляли еще три раза в таком же масштабе и партии объединяли для очистки. Неочищенный продукт подвергали вакуумной дистилляции (приблизительно 15 Торр), собирая дистиллят при 108-129°C, с получением соединения (4) в виде прозрачного бледно-желтого масла (427,9 г, выход составил в среднем 84%; ГХ/MS чистота 98%). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 163,95 (1,46 мин).
1H-ЯМР (300 МГц, CDCl3): δ 7,15-7,04 (м, 2H), 6,77-6,62 (м, 2H), 4,85-4,77 (м, 1H), 4,18 (с, 2H), 4,12-4,02 (м, 1H), 3,94-3,85 (м, 1H), 2,25-1,95 (м, 4H) м.д.
Пример 2a
Получение (R)-2-(те трагидрофуран-2-ил)анилина (4a)
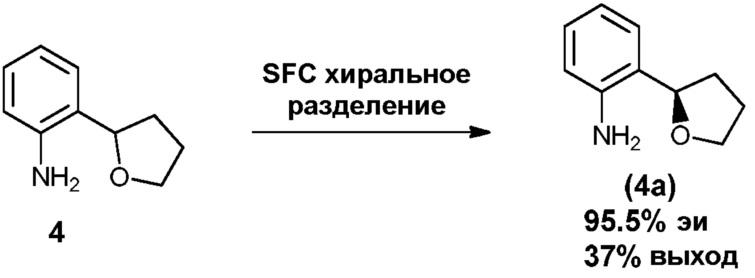
Растворяли 33 г соединения (4) в MeOH (265 мл) с получением концентрации приблизительно 125 мг/мл. Смесь фильтровали через мембранный фильтр 0,2 микрон, затем хроматографировали на колонке ChiralPak® IC (30×150 мм, температура колонки 35°C, Chiral Technologies) при 100 бар с использованием системы Berger для мультиграммовой сверхкритической жидкостной хроматографии. Подвижная фаза представляла собой (90:10) CO2:CH3OH с элюированием при 350 мл/мин, с УФ мониторингом при 220 нанометрах. Получали 15,64 г желаемого продукта (4a) в виде зеленого масла. Аналитическая SFC ([90:10] CO2:CH3OH, при 5 мл/мин на колонке ChiralPak IC (4,6×100 мм) при 35°C и при давлении 100 бар с УФ мониторингом при 220 нм) показала 95,5% эи с общей чистотой 95%.
Пример 3
Получение 4-бром-2-т етрагидрофуран-2-ил-анилина (5)
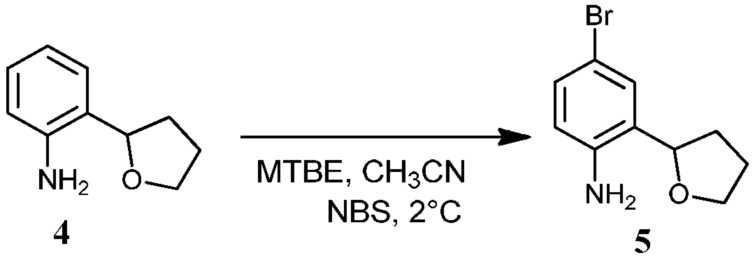
К перемешиваемому раствору 2-тетрагидрофуран-2-ил-анилина (4) (53,45 г, 327,5 ммоль) в метил трет-бутиловом эфире (MTBE, 641,4 мл) и ацетонитриле (213,8 мл), охлажденному до 2°C, добавляли N-бромсукцинимид (58,88 г, 99%,327,5 ммоль, Aldrich B81255) в 4 порциями, поддерживая внутреннюю температуру ниже чем около 8°C. Реакционную смесь перемешивали при охлаждении на ледяной бане в течение 30 минут (ЯМР обработанной аликвоты показал полное поглощение исходного вещества). Добавляли 1н водный раствор Na2S2O3 (330 мл), охлаждающую баню удаляли и перемешивали в течение 20 минут. Смесь разбавляли при помощи EtOAc и слои разделяли. Органическую фазу промывали насыщенным водным раствором NaHCO3 (2×), водой, насыщенным солевым раствором, сушили над MgSO4, фильтровали через короткую пробку из диоксида кремния, элюировали при помощи EtOAc и концентрировали при пониженном давлении с получением соединения (5) в виде масла очень темного янтарного цвета (82,25 г, ВЭЖХ чистота 77-94%). Продукт использовали далее без дополнительной очистки. ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 242,10 (2,89 мин).
1H-ЯМР (300 МГц, CDCl3) δ 7,22 (д, J=2,3 Гц, 1H), 7,16 (дд, J=8,4, 2,3 Гц, 1H), 6,54 (д, J=8,4 Гц, 1H), 4,79-4,73 (м, 1H), 4,15 (с, 2H), 4,10-4,01 (м, 1H), 3,93-3,85 (м, 1H), 2,26-2,13 (м, 1H), 2,12-1,97 (м, 3H) м.д.
Пример 4
Получение N-(4-бром-2-нитро-6-тетрагидрофуран-2-ил-фен ил)-2,2,2-трифтор-ацетамида (6)

К трифторуксусному ангидриду (455,3 мл, 3,275 моль, Sigma-Aldrich 106232) при перемешивании при 2°C медленно добавляли 4-бром-2-тетрагидрофуран-2-ил-анилин (5) (79,29 г, 327,5 ммоль) в виде вязкого масла через капельную воронку в течение 15 минут (температура реакции повышалась до 14°C). Оставшееся масло промывали в реакционную смесь безводным 2-метилтетрагидрофураном (39,6 мл, Sigma-Aldrich 414247). Охлаждающую баню удаляли и добавляли нитрат аммония (34,08 г, 425,8 ммоль, Aldrich 467758). Температура реакции повышалась до 40°C в течение примерно 30 минут, в это время использовали баню с холодной водой для контроля экзотермы и доведения реакции до комнатной температуры. Охлаждающую баню затем удаляли и перемешивание продолжали еще в течение 40 минут (ВЭЖХ показала очень небольшое количество оставшегося ненитрированного вещества). Реакционную смесь медленно выливали в перемешиваемую смесь измельченного льда (800 г). Осажденное твердое вещество собирали фильтрованием, промывали водой, насыщенным водным раствором NaHCO3 (до pH 8), снова водой и гексаном. Мокрое твердое вещество сушили сначала в конвекционной печи при 50°C в течение нескольких часов и затем при пониженном давлении в печи при 40°C в течение ночи с получением соединения (6) в виде светло-коричневого твердого вещества (77,86 г, выход 62%; ВЭЖХ чистота 98%). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 383,19 (3,27 мин).
1H-ЯМР (300 МГц, CDCl3) δ 9,81 (с, 1H), 8,08 (д, J=2,2 Гц, 1H), 7,73 (д, J=2,2 Гц, 1H), 4,88 (дд, J=9,0, 6,5 Гц, 1H), 4,17-4,08 (м, 1H), 4,03-3,95 (м, 1H), 2,45-2,34 (м, 1H), 2,17-2,06 (м, 2H), 1,96-1,83 (м, 1H) м.д.
Пример 5
Получение 4-бром-2-нитро-6-те трагидрофуран-2-ил-анилина (6a)
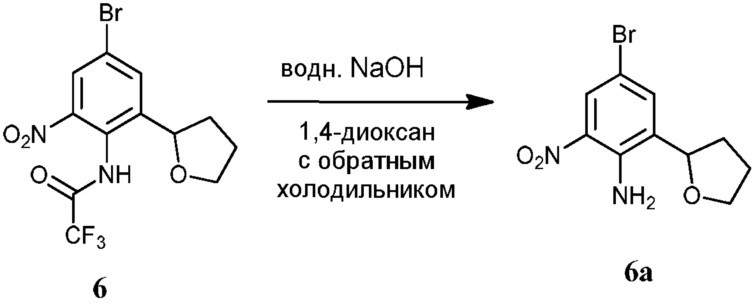
Растворяли N-(4-бром-2-нитро-6-тетрагидрофуран-2-ил-фенил)-2,2,2-трифтор-ацетамид (6) (54,00 г, 140,9 ммоль) в 1,4-диоксане (162 мл) и добавляли водный раствор 6 M NaOH (70,45 мл, 422,7 ммоль, JT-Baker 567202). Реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение 2 дней (ВЭЖХ показала полную конверсию), давали охладиться, разбавляли при помощи MTBE (800 мл), промывали водой (2×200 мл), насыщенным водным раствором NH4Cl, водой и насыщенным солевым раствором. Смесь сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением соединения (6a) в виде масла темного янтарного цвета (40,96 г, выход 93%; общая чистота 92% ВЭЖХ плюс ЯМР). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% MeOH/вода 3-5 минут с муравьиной кислотой в качестве модификатора) M+1: 287,28 (3,44 мин).
1H-ЯМР (300 МГц, CDCl3) δ 8,24 (д, J=2,4 Гц, 1H), 7,41 (д, J=2,3 Гц, 1H), 6,91 (с, 2H), 4,80 (т, J=7,2 Гц, 1H), 4,14-4,05 (м, 1H), 3,98-3,90 (м, 1H), 2,36-2,19 (м, 1H), 2,15-2,01 (м, 3H) м.д.
Пример 6
Получение 2-[5-(4-амино-3-нитро-5-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]про пан-2-ола (8)
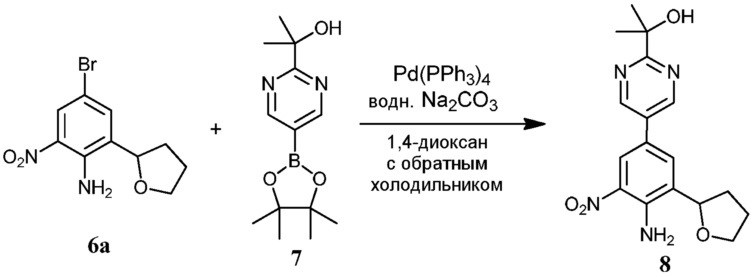
Смешивали 4-бром-2-нитро-6-тетрагидрофуран-2-ил-анилин (6a) (40,40 г, 92%, 129,5 ммоль), 1,4-диоксан (260 мл, Sigma-Aldrich 360481), 2-[5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-ил]пропан-2-ол (7) (41,05 г, 155,4 ммоль) и водный раствор 2,7 M Na2CO3 (143,9 мл, 388,5 ммоль). Через перемешиваемую смесь барботировали поток азота в течение 1 часа, с последующим добавлением тетракис(трифенилфосфин)палладия (0) (7,48 г, 6,47 ммоль, Strem 462150). Реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение 2 часов (ВЭЖХ показала завершение реакции), давали охладиться, разбавляли при помощи EtOAc, промывали водой, насыщенным водным раствором NH4Cl, насыщенным солевым раствором, сушили над MgSO4 и фильтровали через короткую пробку из Florisil® с элюированием при помощи EtOAc. Фильтрат концентрировали при пониженном давлении с получением масла темно-коричневого цвета. Масло растворяли в CH2Cl2 и элюировали через короткую пробку из силикагеля при помощи CH2Cl2 и затем EtOAc. Желаемую фракцию концентрировали на роторном испарителе до образования осадка с получением густой коричневой суспензии, которую растирали в порошок с MTBE. Твердое вещество собирали фильтрованием, промывали при помощи MTBE и сушили в условиях высокого вакуума с получением соединения (8) в виде желтого твердого вещества (35,14 г, чистота 99+% ВЭЖХ). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 345,00 (2,69 мин).
1H-ЯМР (300 МГц, CDCl3) δ 8,88 (с, 2H), 8,36 (д, J=2,2 Гц, 1H), 7,56 (д, J=2,1 Гц, 1H), 7,09 (с, 2H), 4,92 (т, J=7,2 Гц, 1H), 4,62 (с, 1H), 4,20-4,11 (м, 1H), 4,03-3,94 (м, 1H), 2,39-2,26 (м, 1H), 2,23-2,08 (м, 3H), 1,64 (с, 6H) м.д.
Фильтрат далее концентрировали и очищали при помощи хроматографии на силикагеле ISCO с элюированием смесью 0 до 80% EtOAc/гексан, с получением второй партии продукта (8) в виде твердого вещества янтарного цвета (4,46 г, общий выход 88%; ВЭЖХ чистота 88%).
Пример 7
Альтернативное получение 2-[5-(4-амино-3-нитро-5-тетрагидрофуран-2-ил-фенил) пиримидин-2-ил]пропан-2-ола (8)
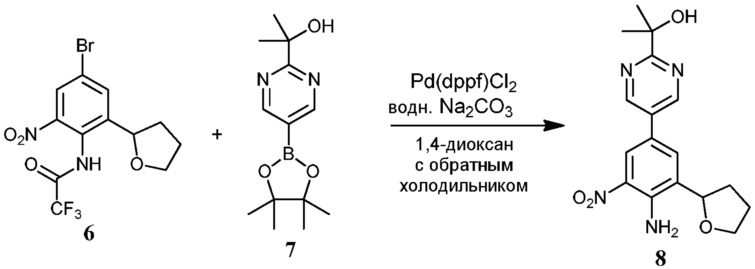
Смешивали N-(4-бром-2-нитро-6-тетрагидрофуран-2-ил-фенил)-2,2,2-трифтор-ацетамид (6) (19,00 г, 49,59 ммоль), 2-[5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-ил]пропан-2-ол (7) (14,41 г, 54,55 ммоль), водный раствор 2,7 M карбоната натрия (73,48 мл, 198,4 ммоль) и 1,4-диоксан (190 мл, Sigma-Aldrich 360481). Через перемешиваемую смесь барботировали поток азота в течение 40 минут, с последующим добавлением продукта присоединения 1,1’-бис(дифенилфосфино)ферроцендихлорпалладия и дихлорметана (2,025 г, 2,480 ммоль, Strem 460450). Реакционную смесь перемешивали при температуре кипения с обратным холодильником в атмосфере N2 в течение 7 часов, добавляли еще 50 мл насыщенного водного раствора карбоната натрия и нагревали при температуре кипения с обратным холодильником еще в течение 16 часов. Смеси давали охладиться, затем разбавляли EtOAc (500 мл) и водой (200 мл). Слои разделяли и водную фазу экстрагировали при помощи EtOAc (200 мл). Объединенную органическую фазу промывали водой (500 мл), насыщенным солевым раствором (500 мл), сушили над Na2SO4, фильтровали через Florisil® plug и концентрировали на роторном испарителе с получением неочищенного соединения (8) в виде масла оранжевого цвета. Неочищенный продукт очищали хроматографией на силикагеле ISCO с элюированием при помощи 20-90% EtOAc/гексан, с получением соединения (8) в виде оранжевого твердого вещества (15,00 г, выход 87%; чистота 81-88%). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 345,35 (2,68 мин).
1H-ЯМР (300 МГц, CDCl3) δ 8,88 (с, 2H), 8,36 (д, J=2,2 Гц, 1H), 7,56 (д, J=2,1 Гц, 1H), 7,09 (с, 2H), 4,92 (т, J=7,2 Гц, 1H), 4,62 (с, 1H), 4,20-4,11 (м, 1H), 4,03-3,94 (м, 1H), 2,39-2,26 (м, 1H), 2,23-2,08 (м, 3H), 1,64 (с, 6H) м.д.
Пример 8
Получение 2-[5-(3,4-диамино-5-тетрагидрофуран-2-ил-фенил) пиримидин-2-ил]пропан-2-ола (9)
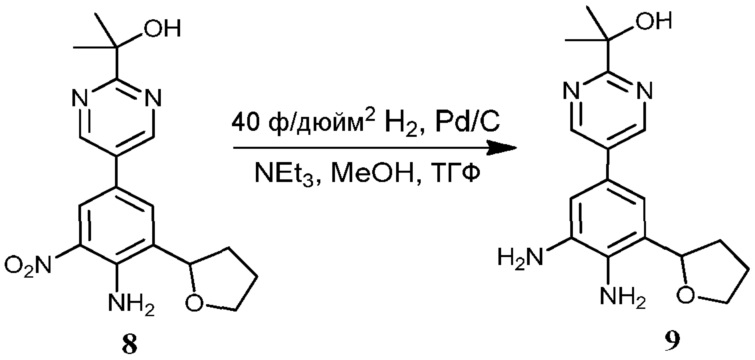
К суспензии 2-[5-(4-амино-3-нитро-5-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]пропан-2-ола (8) (30,10 г, 87,41 ммоль) и ТГФ (90 мл) в сосуде Парра в атмосфере азота добавляли суспензию 5% палладия на углероде (3,01 г, влажность 50%, 0,707 ммоль, Aldrich 330116) в MeOH (90 мл, JT-Baker 909333), затем добавляли NEt3 (24,37 мл, 174,8 ммоль, Sigma-Aldrich 471283). Помещали сосуд на встряхивающее устройство Парра и насыщали H2. Добавляли 45 ф/дюйм2 (3,164 кг/см2) H2 и продолжали встряхивание до полного поглощения (ВЭЖХ показала полную конверсию). Реакционную смесь продували азотом, фильтровали через Целит™ и промывали при помощи EtOAc. Фильтрат снова фильтровали через фильтровальную бумагу с 0,5-микронным стекловолокном, проложенную в виде сэндвичевой конструкции между двумя P5 бумагами, и концентрировали при пониженном давлении с получением соединения (9) в виде светло-коричневого пенистого вещества (28,96 г, выход 98%; ЯМР чистота 93%). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 315,32 (1,54 мин).
1H-ЯМР (300 МГц, CDCl3) δ 8,83 (с, 2H), 6,92 (д, J=1,8 Гц, 1H), 6,88 (д, J=1,8 Гц, 1H), 4,90 (дд, J=7,9, 6,2 Гц, 1H), 4,72 (с, 1H), 4,18 (с, 2H), 4,17-4,08 (м, 1H), 3,99-3,89 (м, 1H), 3,46 (с, 2H), 2,34-2,19 (м, 1H), 2,17-2,05 (м, 3H), 1,63 (с, 6H) м.д.
Пример 9
Получение 1-этил-3-[5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-тетрагидрофуран-2-ил-1H- бензимидазол-2-ил]мочевины (11)
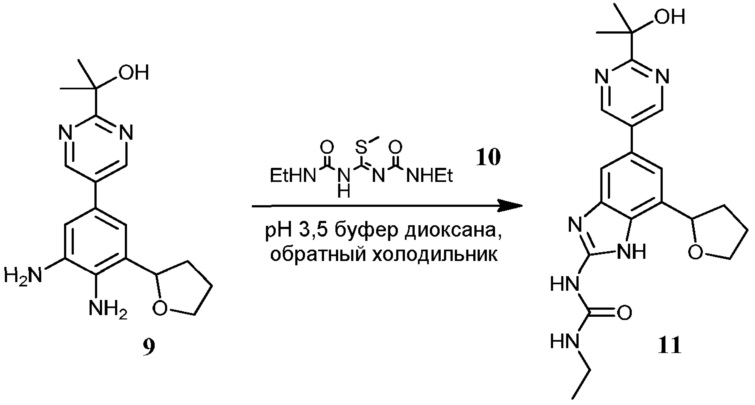
К перемешиваемому раствору 2-[5-(3,4-диамино-5-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]пропан-2-ола (9) (32,10 г, 102,1 ммоль) в 1,4-диоксане (160,5 мл, Sigma-Aldrich 360481) добавляли pH 3,5 буфер (240,8 мл), полученный путем растворения NaOAc тригидрата (34,5 г) в 1н водном растворе H2SO4 (240 мл). Добавляли 1-этил-3-(N-(этилкарбамоил)-C-метилсульфанил-карбонимидоил)мочевину (10) (28,46 г, 122,5 ммоль, CB Research and Development) и перемешивали при температуре кипения с обратным холодильником в течение ночи (ВЭЖХ показала 99% поглощение исходного диамина). Реакционную смесь охлаждали до комнатной температуры и выливали по порциям (вспенивание) в перемешиваемый насыщенный водный раствор NaHCO3 (480 мл) и воду (120 мл) с получением pH 8-9. Смесь перемешивали в течение 30 минут, твердое вещество собирали фильтрованием, промывали водой, используемой в обильном количестве, для получения нейтрального pH и затем небольшим количеством EtOH. Твердое вещество сушили при пониженном давлении с получением соединения (11) в виде не совсем белого твердого вещества (34,48 г, выход 82%; ВЭЖХ чистота 99,4%). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 411,41 (1,73 мин).
1H-ЯМР (300 МГц, MeOD) δ 9,02 (с, 2H), 7,62 (с, 1H), 7,37 (с, 1H), 5,31 (с, 1H), 4,23 (дд, J=14,5, 7,3 Гц, 1H), 4,01 (дд, J=15,0, 7,1 Гц, 1H), 3,38-3,28 (м, 2H), 2,58-2,46 (м, 1H), 2,16-2,05 (м, 2H), 2,02-1,88 (м, 1H), 1,63 (с, 6H), 1,22 (т, J=7,2 Гц, 3H) м.д.
Пример 10
Хиральное хроматографическое выделение 1-этил-3-[5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H- бензимидазол-2-ил]мочевины (12)
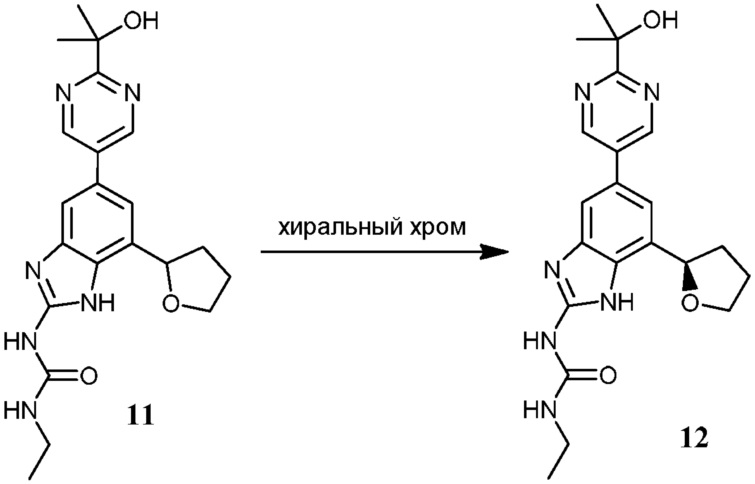
Рацемический образец 1-этил-3-[5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-тетрагидрофуран-2-ил-1H-бензимидазол-2-ил]мочевины (11) (24,60 г) разделяли на колонке CHIRALPAK® IC® (от Chiral Technologies) с элюированием при помощи DCM/MeOH/TEA (60/40/0,1) при 35°C с получением желаемого энантиомера (12) в виде белого твердого вещества (11,35 г, выход 45%; ВЭЖХ чистота 99+%, 99+% эи). Аналитическая хиральная ВЭЖХ, время удерживания 6,2 мин (колонка CHIRALPAK® IC® 4,6×250 мм, скорость потока 1 мл/мин, 30°C).
Структуру и абсолютную стереохимию соединения 12 подтверждали при помощи рентгено-структурного анализа монокристалла. Данные дифракции на монокристалле получали на Bruker Apex II дифрактометре, снабженном запаянной трубкой с Cu K-альфа источником (Cu Kα излучение, γ=1,54178 Å) и Apex II CCD детектором. Выбирали кристалл, имеющий размеры 0,5×0,05×0,05 мм, очищали с использованием минерального масла, закрепляли на MicroMount и центрировали на Bruker APEXII системе. Были получены три группы из 40 каркасов, разделенных в обратном пространстве, для получения матрицы ориентации и исходных параметров ячейки. Конечные параметры ячейки получали и уточняли после завершения сбора данных на основании полного набора данных. На основании статистических данных отсутствий и интенсивностей структура была определена и уточнена в не имеющей центра симметрии P21 пространственной группе.
Набор данных дифракции обратного пространства получали для разрешения 0,9 Å с использованием ступеней 0,5° с использованием 60 сек экспонирования для каждого каркаса. Данные собирали при 100 (2) K. Интеграцию интенсивностей и уточнение параметров ячейки осуществляли с использованием программы APEXII. Наблюдение кристалла после сбора данных не показало никаких признаков разложения. Как показано на фиг.1, присутствуют две симметричные независимые молекулы в структуре, и обе симметричные независимые молекулы представляют собой R изомеры.
Данные собирали, уточняли и обрабатывали с использованием программы Apex II. Структуру определяли с использованием SHELXS97 (Sheldrick,1990); программу(программы) и структуру уточняли с использованием программы SHELXL97 (Sheldrick, 1997). Кристалл показал моноклинную ячейку с P21 пространственной группой. Параметры решетки были следующими a=9,8423(4) Å, b=10,8426(3) Å, c=19,4441 (7) Å, β=102,966(3)°. Объем=2022,09(12) Å3.
Пример 11
Получение 1-этил-3-[5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины в форме сол и метансульфоновой кислоты (13)
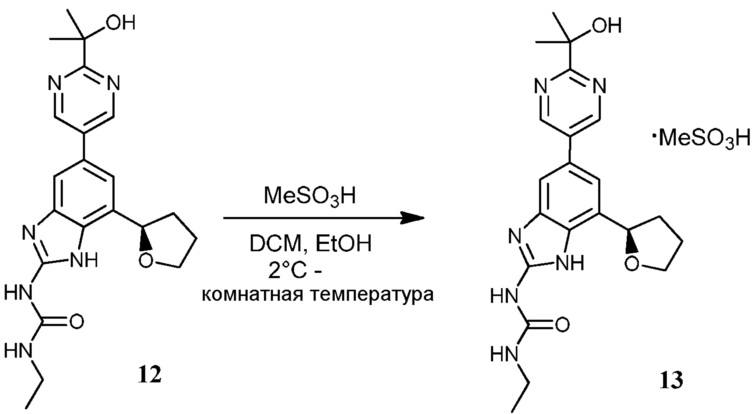
Перемешиваемую суспензию 1-этил-3-[5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины (12) (9,32 г, 22,71 ммоль) в абсолютном этаноле (93,2 мл) охлаждали на ледяной бане. Добавляли метансульфоновую кислоту (1,548 мл, 23,85 ммоль, Sigma-Aldrich 471356), охлаждающую баню и перемешивали при комнатной температуре в течение 20 минут. Смесь концентрировали на роторном испарителе при 35°C с получением густой суспензии, разбавляли при помощи EtOAc, твердое вещество собирали фильтрованием, промывали при помощи EtOAc и сушили при пониженном давлении с получением начальной партии соединения (13) в виде белого твердого вещества (8,10 г). Фильтрат концентрировали на роторном испарителе с получением желтоватого прозрачного пенистого вещества, которое растворяли в EtOH, концентрировали с получением твердой суспензии, растирали в порошок с EtOAc/Et2O и собирали фильтрованием. Твердое вещество промывали при помощи EtOAc/Et2O, объединяли с начальной партией и сушили при пониженном давлении с получением соединения (13) в виде белого твердого вещества (9,89 г, выход 86%; ВЭЖХ чистота 99+%, эи 99+%). Аналитическая хиральная ВЭЖХ показала один энантиомер с временем удерживания 6,3 мин, с элюированием смесью DCM/MeOH/TEA (60/40/0,1) на колонке CHIRALPAK® IC® 4,6×250 мм со скоростью потока 1 мл/мин при 30°C. ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 411,53 (1,74 мин).
1H-ЯМР (300 МГц, MeOD) δ 9,07 (с, 2H), 7,79 (с, 1H), 7,62 (с, 1H), 5,30 (т, J=7,3 Гц, 1H), 4,24 (дд, J=14,6, 7,3 Гц, 1H), 4,04 (дд, J=15,0, 7,6 Гц, 1H), 3,40-3,30 (м, 2H), 2,72 (с, 3H), 2,65-2,54 (м, 1H), 2,20-2,07 (м, 2H), 2,04-1,90 (м, 1H), 1,64 (с, 6H), 1,23 (т, J=7,2 Гц, 3H) м.д.
Пример 12
Получение 2-(2-фтор-6-нитро-фенил)-2,3-дигидрофурана (15A) и 2-(2-фтор-6-нитро -фенил)-2,5-дигидрофурана (15B)
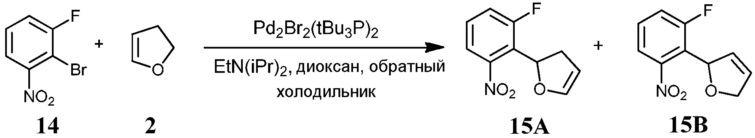
2-бром-1-фтор-3-нитро-бензол (14) (200,3 г, 98%, 892,3 ммоль, Bosche F6657), 1,4-диоксан (981,5 мл, Sigma-Aldrich 360481) и 2,3-дигидрофуран (2) (341,1 мл, 99%, 4,462 моль, Aldrich 200018) загружали в реакционную колбу, с последующим добавлением N,N-диизопропилэтиламина (155,4 мл, 892,3 ммоль, Sigma-Aldrich 550043) и димера бром(три-трет-бутилфосфин)палладия(I) (6,936 г, 8,923 ммоль, Johnson Matthey C4099). Смесь перемешивали при температуре кипения с обратным холодильником в течение 2 часов (ВЭЖХ показала 98% поглощение исходного арилбромида). Смеси давали охладиться, осадок удаляли фильтрованием, промывали при помощи EtOAc и фильтрат концентрировали в вакууме до темного красновато-коричневого полутвердого масла. Смесь растворяли в CH2Cl2, элюировали через пробку из диоксида кремния при помощи CH2Cl2 и концентрировали в вакууме с получением смеси 15A и 15B в виде масла темного янтарного цвета (291,3 г). Неочищенный продукт использовали далее без дополнительной очистки. Основной продукт представлял собой 2-(2-фтор-6-нитро-фенил)-2,3-дигидрофуран (15A) (96%): ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 210,23 (3,13 мин);
1H-ЯМР (300 МГц, CDCl3) δ 7,54 (дт, J=8,0, 1,2 Гц, 1H), 7,43 (тд, J=8,2, 5,2 Гц, 1H), 7,32 (ддд, J=9,7, 8,3, 1,3 Гц, 1H), 6,33 (дд, J=4,9, 2,4 Гц, 1H), 5,80 (т, J=10,9 Гц, 1H), 5,06 (кв., J=2,4 Гц, 1H), 3,18-3,07 (м, 1H), 2,94-2,82 (м, 1H) м.д.
Минорный продукт представлял собой 2-(2-фтор-6-нитро-фенил)-2,5-дигидрофуран (15B) (4%): ГХ/MS (Agilent HP-5MS 30 м × 250 мкм × 0,25 мкм колонка с нагреванием при 60°C в течение 2 мин до 300°C в течение 15 мин со скоростью потока 1 мл/мин) M+1: 210 (11,95 мин).
1H-ЯМР (300 МГц, CDCl3) δ 7,47 (д, J=8,0 Гц, 1H), 7,43-7,34 (м, 1H), 7,30-7,23 (м, 1H), 6,21-6,15 (м, 1H), 6,11-6,06 (м, 1H), 5,97-5,91 (м, 1H), 4,89-4,73 (м, 2H) м.д.
Пример 13
Получение 3-фтор-2-те трагидрофуран-2-ил-анилина (16)
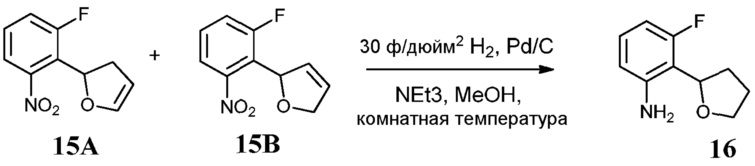
5% Палладий на углероде (37,3 г, влажность 50%, 8,76 ммоль, Aldrich 330116) помещали в сосуд Парра в атмосфере азота, с последующим добавлением MeOH (70 мл, JT-Baker 909333). Добавляли неочищенную смесь 2-(2-фтор-6-нитро-фенил)-2,3-дигидрофурана и 2-(2-фтор-6-нитро-фенил)-2,5-дигидрофурана (15A & 15B) (186,6 г, 892,1 ммоль), растворенную в MeOH (117 мл), с последующим добавлением NEt3 (124,3 мл, 892,1 ммоль, Sigma-Aldrich 471283). Сосуд помещали на встряхивающее устройство Парра и насыщали H2. После добавления 45 ф/дюйм2(3,164 кг/см2) H2, реакционную смесь встряхивали до полного поглощения исходного вещества (ВЭЖХ и ЖХ/MS показали завершение реакции). Реакционную смесь продували азотом, фильтровали через Целит™ и промывали при помощи EtOAc. Фильтрат концентрировали на роторном испарителе с получением коричневого масла, которое растворяли в Et2O и промывали водой (2×). Эфирную фазу экстрагировали при помощи 1н водного раствора HCl (5×250 мл), промывали при помощи Et2O (3×) и затем подщелачивали при помощи 6 н водного раствора NaOH до pH 12-14. Щелочную водную фазу экстрагировали при помощи CH2Cl2 (4×) и объединенный органический экстракт промывали насыщенным водным раствором NH4Cl, сушили над MgSO4 и фильтровали через слой диоксида кремния с элюированием при помощи CH2Cl2 → 25% EtOAc/гексан. Желаемый фильтрат концентрировали при пониженном давлении с получением соединения 16 в виде светло-коричневого масла (121,8 г, чистота 84% ГХ/MS плюс ЯМР). ГХ/MS (Agilent HP-5MS 30 м × 250 мкм × 0,25 мкм колонка с нагреванием при 60°C в течение 2 мин до 300°C в течение 15 мин со скоростью потока 1 мл/мин) M+1: 182,0 (11,44 мин). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 182,10 (2,61 мин).
1H-ЯМР (300 МГц, CDCl3) δ 6,97 (тд, J=8,1, 6,3 Гц, 1H), 6,43-6,35 (м, 2H), 5,21-5,13 (м, 1H), 4,54 (с, 2H), 4,16-4,07 (м, 1H), 3,90-3,81 (м, 1H), 2,23-2,00 (м, 4H) м.д.
Дополнительные партии получали следующим образом: объединенную эфирную фазу промывали насыщенным водным раствором NaHCO3, насыщенным солевым раствором, сушили над Na2SO4, декантировали и концентрировали при пониженном давлении. Масло подвергали вакуумной дистилляции (приблизительно 15 Торр), собирая дистиллят при 101-108°C. К перемешиваемому раствору дистиллированного масла в EtOH (1 объем) при 2°C медленно добавляли 5 M HCl (1 экв.) в iPrOH. Полученную суспензию доводили до комнатной температуры, разбавляли при помощи EtOAc (3 объема, об/об) и перемешивали в течение 2 часов. Белое твердое вещество собирали фильтрованием, промывали при помощи EtOAc и сушили при пониженном давлении с получением второй партии продукта в виде HCl соли. Маточный раствор концентрировали до получения суспензии, разбавляли при помощи EtOAc и твердое вещество собирали фильтрованием, промывали при помощи EtOAc и сушили в вакууме с получением HCl соли в качестве третьей партии продукта. ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 182,10 (2,58 мин).
1H-ЯМР (300 МГц, CDCl3) δ 10,73 (шир.с, 3H), 7,66 (д, J=8,1 Гц, 1H), 7,33 (тд, J=8,2, 5,9 Гц, 1H), 7,13-7,05 (м, 1H), 5,26 (дд, J=9,0, 6,5 Гц, 1H), 4,38-4,28 (м, 1H), 4,00-3,91 (м, 1H), 2,59-2,46 (м, 1H), 2,30-1,95 (м, 3H) м.д.
Общий выход от трех партий составил 76%.
Пример 14
Получение 4-бром-3-фтор-2-те трагидрофуран-2-ил-анилина (17)
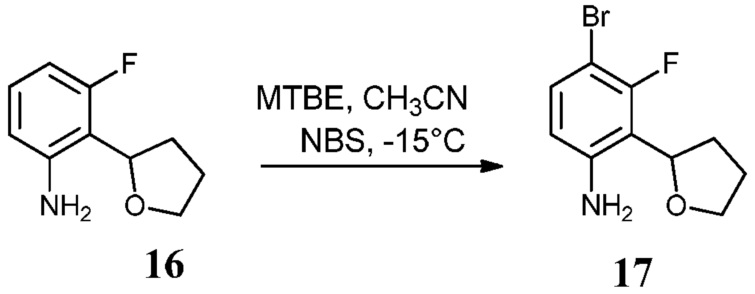
К перемешиваемому раствору 3-фтор-2-тетрагидрофуран-2-ил-анилина (16) (131,9 г, 92%, 669,7 ммоль) в метил трет-бутиловом эфире (1,456 л) и ацетонитриле (485 мл), охлажденному до -20°C, добавляли N-бромсукцинимид (120,4 г, 99%, 669,7 ммоль, Aldrich B81255) 3 порциями, поддерживая температуру реакции ниже около -15°C. После завершения добавления перемешивание продолжали при температуре в пределах от -15 до -10°C в течение 30 минут. 1H ЯМР обработанной аликвоты показал 96% поглощение исходного анилина, поэтому добавляли еще 4,82 г NBS и перемешивали при -10°C еще в течение 30 минут. К реакционной смеси добавляли 1н водный раствор Na2S2O3 (670 мл). Охлаждающую баню удаляли, смесь перемешивали в течение 20 минут, затем разбавляли при помощи EtOAc. Слои разделяли и органическую фазу промывали насыщенным водным раствором NaHCO3 (2×), водой, насыщенным солевым раствором, сушили над Na2SO4, декантировали и концентрировали при пониженном давлении с получением масла темно-янтарного цвета. Остаток разбавляли гексаном и элюировали через короткую пробку из диоксида кремния, элюируя смесью 25% EtOAc/гексан до 50% EtOAc/гексан. Желаемый фильтрат концентрировали в вакууме с получением соединения 17 в виде масла темного янтарного цвета (182,9 г, выход 90%; ЯМР чистота 86%). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 260,12 (3,20 мин).
1H-ЯМР (300 МГц, CDCl3) δ 7,15 (дд, J=8,6, 7,6 Гц, 1H), 6,30 (дд, J=8,7, 1,3 Гц, 1H), 5,19-5,12 (м, 1H), 4,58 (с, 2H), 4,16-4,07 (м, 1H), 3,90-3,81 (м, 1H), 2,23-1,99 (м, 4H) м.д.
Пример 15
Получение N-(4-бром-3-фтор-6-нитро-2-тетрагидрофуран-2-ил-фени л)-2,2,2-трифтор-ацетамида (18)
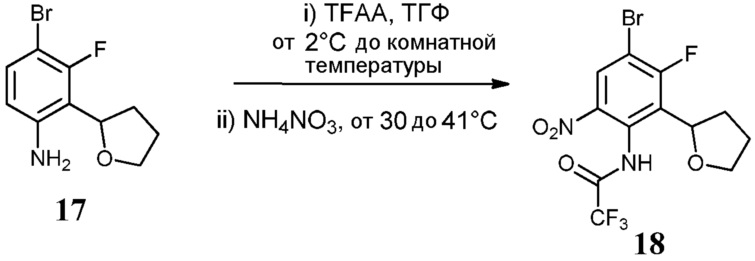
К трифторуксусному ангидриду (565,3 мл, 4,067 моль, Sigma-Aldrich 106232) при перемешивании при 2°C медленно добавляли чистый 4-бром-3-фтор-2-тетрагидрофуран-2-ил-анилин (17) (123,0 г, 86%, 406,7 ммоль) в виде вязкого масла через капельную воронку в течение примерно 20 минут (температура реакции повышалась до 13°C). Оставшееся масло промывали в реакционную смесь при помощи безводного ТГФ (35 мл). Охлаждающую баню удаляли и реакционную смесь нагревали до 35°C, с последующим добавлением по порциям NH4NO3 (4,88 г × 20 порций, 1,22 моль, Sigma-Aldrich A7455) в течение 2,5 часов, поддерживая температуру реакции между 30 и 41°C с использованием бани с ледяной водой только по мере необходимости для контроля экзотермы. После завершения добавления реакционную смесь перемешивали еще в течение 10 минут (ВЭЖХ показала 99% завершение реакции). Смесь медленно выливали в измельченный лед (1,23 кг) и перемешивали в течение 1 часа для образования фильтруемого твердого осадка, который собирали и промывали водой, небольшим количеством насыщенного водного раствора NaHCO3 и снова водой (до pH 7). Продукт сушили в конвекционной печи в течение ночи при 40°C и затем при пониженном давлении в печи при 50°C в течение ночи с получением соединения 18 в виде бежевого твердого вещества (152,5 г, выход 90%; ВЭЖХ чистота 96%). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 401,30 (3,41 мин).
1H-ЯМР (300 МГц, CDCl3) δ 10,56 (с, 1H), 8,19 (д, J=6,6 Гц, 1H), 5,22 (дд, J=10,3, 6,4 Гц, 1H), 4,22 (дд, J=15,8, 7,2 Гц, 1H), 3,99 (дд, J=16,1, 7,5 Гц, 1H), 2,50-2,38 (м, 1H), 2,22-2,11 (м, 2H), 1,86-1,71 (м, 1H) м.д.
Пример 16
Получение 4-бром-3-фтор-6-нитро-2-те трагидрофуран-2-ил-анилина (19)
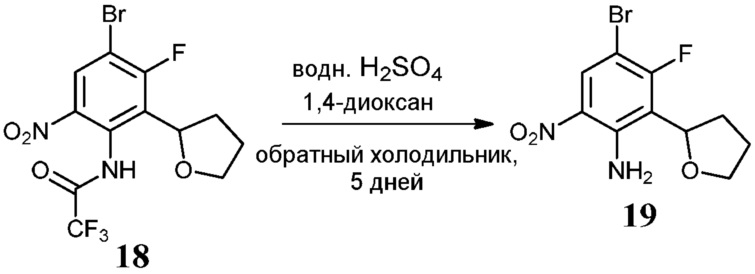
В реакционную колбу загружали N-(4-бром-3-фтор-6-нитро-2-тетрагидрофуран-2-ил-фенил)-2,2,2-трифтор-ацетамид (18) (242,3 г, 604,1 ммоль), 1,4-диоксан (1,212 л), водный раствор 2 M серной кислоты (362,4 мл, 724,9 ммоль) и перемешивали при температуре кипения с обратным холодильником в течение 5 дней (ВЭЖХ показала конверсию 98%). Смеси давали охладиться, разбавляли при помощи EtOAc, нейтрализовали насыщенным водным раствором NaHCO3, слои разделяли и снова экстрагировали водную фазу при помощи EtOAc (2×). Объединенную органическую фазу промывали насыщенным солевым раствором (2×), сушили над MgSO4, фильтровали и концентрировали в вакууме с получением соединения 19 в виде зеленовато-коричневого твердого вещества (181,7 г, выход 94%; ВЭЖХ чистота 95%). Продукт использовали на следующей стадии без дополнительной очистки. ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 305,20 (3,63 мин).
1H-ЯМР (300 МГц, CDCl3) δ 8,35 (д, J=7,3 Гц, 1H), 7,45 (с, 2H), 5,23-5,16 (м, 1H), 4,23-4,14 (м, 1H), 3,93-3,84 (м, 1H), 2,31-1,96 (м, 4H) м.д.
Пример 17
Получение 2-[5-(4-амино-2-фтор-5-нитро-3-тетрагидрофуран-2-ил-фенил)п иримидин-2-ил]пропан-2-ола (20)
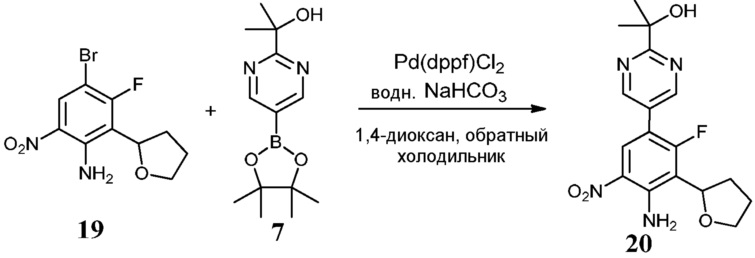
К перемешиваемому раствору 4-бром-3-фтор-6-нитро-2-тетрагидрофуран-2-ил-анилина (19) (525,0 г, 1,721 моль, Bridge Organics Co.) в 1,4-диоксане (4,20 л, Sigma-Aldrich 360481) добавляли 1,2 M водный раствор NaHCO3 (4,302 л, 5,163 моль). Через перемешиваемую смесь барботировали поток азота в течение 2 часов, с последующим добавлением 2-[5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-ил]пропан-2-ола (7) (545,4 г, 2,065 моль, Bridge Organics Co.) и продукта присоединения 1,1’-бис(дифенилфосфино)ферроцендихлорпалладия и дихлорметана (42,16 г, 51,63 ммоль, Strem 460450). Реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение ночи, давали охладиться, разбавляли при помощи EtOAc (8,4 л) и слои разделяли. Органическую фазу промывали насыщенным водным раствором NH4Cl и затем насыщенным солевым раствором. Водную фазу снова экстрагировали при помощи EtOAc (4 л) и органический экстракт промывали насыщенным солевым раствором. Объединенную органическую фазу сушили над MgSO4, фильтровали через короткую пробку из Florisil®, элюировали при помощи EtOAc и фильтрат концентрировали на роторном испарителе с получением темно-коричневого мокрого твердого вещества. Смесь растворяли в CH2Cl2, загружали на слой силикагеля, элюировали гексаном, затем 25% EtOAc/гексан и затем 50% EtOAc/гексан. Желаемый фильтрат концентрировали на роторном испарителе до получения густой суспензии и твердое вещество собирали фильтрованием, растирали в порошок с MTBE и сушили в вакууме с получением соединения 20 в виде ярко-желтого твердого вещества (выход 55,8%, ВЭЖХ чистота 90-97%). Фильтрат концентрировали и описанную выше очистку повторяли с получением второй партии соединения 20 в виде ярко-желтого твердого вещества (выход 19,7%). Фильтрат снова концентрировали с получением темно-коричневого масла и смесь загружали на колонку с диоксидом кремния с толуолом и минимальным количеством CH2Cl2. Смесь элюировали при помощи EtOAc/гексан (0% до 50%). Желаемые фракции концентрировали до получения суспензии и разбавляли при помощи MTBE/гексан. Твердое вещество собирали фильтрованием и промывали минимальным количеством MTBE с получением третьей партии соединения 20 в виде ярко-желтого твердого вещества (выход 4,9%) с общим выходом 80% от трех партий. ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 363,48 (2,95 мин).
1H-ЯМР (300 МГц, CDCl3) δ 8,84 (д, J=1,6 Гц, 2H), 8,27 (д, J=8,0 Гц, 1H), 7,62 (с, 2H), 5,31-5,24 (м, 1H), 4,63 (с, 1H), 4,27-4,18 (м, 1H), 3,97-3,87 (м, 1H), 2,33-2,05 (м, 4H), 1,64 (с, 6H) м.д.
Пример 18
Получение 2-[5-(4,5-диамино-2-фтор-3-тетрагидрофуран-2-ил-фенил)п иримидин-2-ил]пропан-2-ола (21)
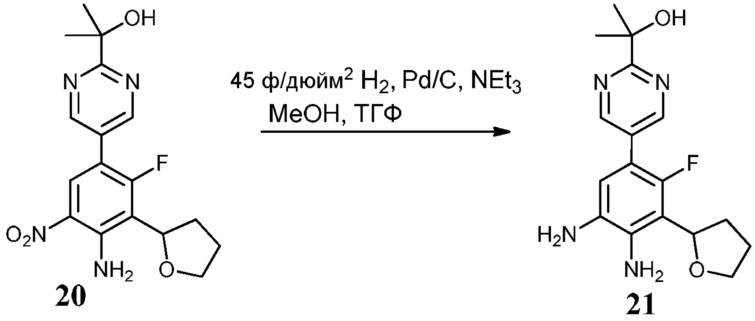
5% Палладий на углероде (14,21 г, влажность 50%, 3,339 ммоль, Aldrich 330116) помещали в сосуд Парра в атмосфере азота, с последующим добавлением MeOH (242 мл, JT-Baker 909333) и NEt3 (46,54 мл, 333,9 ммоль, Sigma-Aldrich 471283). 2-[5-(4-Амино-2-фтор-5-нитро-3-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]пропан-2-ол (20) (121,0 г, 333,9 ммоль) растворяли в горячем ТГФ (360 мл), давали охладиться, добавляли к реакционной смеси и промывали еще одной порцией ТГФ (124 мл). Помещали сосуд на встряхивающее устройство Парра и насыщали H2. Добавляли 45 ф/дюйм2(3,164 кг/см2) H2 и продолжали встряхивание до полного поглощения (ВЭЖХ и ЖХ/MS показали завершение реакции). Реакционную смесь продували азотом, фильтровали через Целит™ и промывали при помощи EtOAc. Смесь снова фильтровали через бумажный фильтр (стекломикроволокно) и фильтрат концентрировали в вакууме. Реакцию осуществляли еще три раза в таком же масштабе и партии объединяли с получением соединения 21 в виде коричневого твердого вещества (447 г, выход 99%; ВЭЖХ чистота 93%). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 333,46 (1,79 мин).
1H-ЯМР (300 МГц, CDCl3) δ 8,81 (д, J=1,4 Гц, 2H), 6,69 (д, J=7,3 Гц, 1H), 5,27-5,20 (м, 1H), 4,73 (с, 1H), 4,70 (с, 2H), 4,23-4,14 (м, 1H), 3,94-3,86 (м, 1H), 3,22 (с, 2H), 2,32-2,22 (м, 1H), 2,18-1,99 (м, 3H), 1,63 (с, 6H) м.д.
Пример 19
Получение 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-тетрагидрофуран-2-ил-1H- бензимидазол-2-ил]мочевины (22)
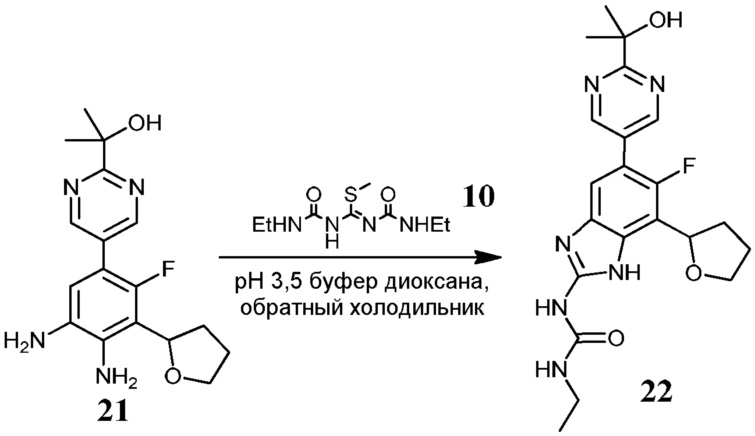
К перемешиваемой суспензии 2-[5-(4,5-диамино-2-фтор-3-тетрагидрофуран-2-ил-фенил)пиримидин-2-ил]пропан-2-ола (21) (111,3 г, 334,9 ммоль) и 1,4-диоксана (556,5 мл, Sigma-Aldrich 360481) добавляли 1-этил-3-(N-(этилкарбамоил)-C-метилсульфанил-карбонимидоил)мочевину (10) (93,36 г, 401,9 ммоль, CB Research and Development) с последующим добавлением pH 3,5 буфера (1,113 л), полученного путем растворения NaOAc тригидрата (158,1 г) в 1н водном растворе H2SO4 (1,100 л). Реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение ночи (ВЭЖХ показала полную конверсию), охлаждали до комнатной температуры и выливали по порциям (вспенивание) в перемешиваемый насыщенный водный раствор NaHCO3 (2,23 л) с получением pH 8-9. Смесь перемешивали в течение 30 минут, твердое вещество собирали фильтрованием, промывали обильно водой для получения нейтрального pH и затем небольшим количеством EtOH. Твердое вещество сушили при пониженном давлении с получением соединения 22 в виде беловато-желтоватого твердого вещества (135,2 г, выход 94%; ВЭЖХ чистота 99%). ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 429,58 (2,03 мин).
1H-ЯМР (300 МГц, MeOD) δ 8,95 (д, J=1,6 Гц, 2H), 7,45 (д, J=6,5 Гц, 1H), 5,38 (шир.с, 1H), 4,27 (дд, J=14,9, 7,1 Гц, 1H), 4,01 (дд, J=15,1, 7,0 Гц, 1H), 3,37-3,29 (м, 2H), 2,55 (шир.с, 1H), 2,19-2,07 (м, 2H), 2,02-1,82 (шир.с, 1H), 1,63 (с, 6H), 1,21 (т, J=7,2 Гц, 3H) м.д.
Пример 20
Хиральное хроматографическое выделение 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H -бензимидазол-2-ил]мочевины(23)
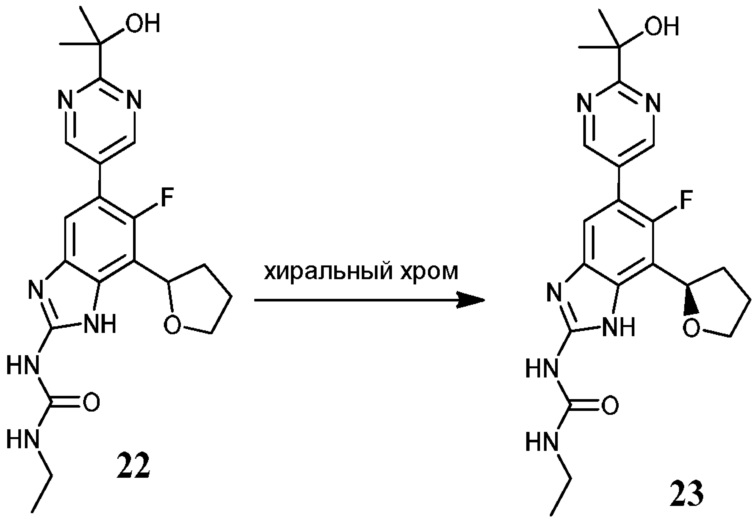
Рацемический образец 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-тетрагидрофуран-2-ил-1H-бензимидазол-2-ил]мочевины (22) (133,60 г) разделяли на колонке CHIRALPAK® IC® (от Chiral Technologies) с элюированием смесью DCM/MeOH/TEA (60/40/0,1) при 25°C с получением желаемого энантиомера 23 в виде не совсем белого твердого вещества (66,8 г, выход 45%; ВЭЖХ чистота 99,8 %, эи 99+%). Аналитическая хиральная ВЭЖХ, время удерживания 7,7 мин (колонка CHIRALPAK® IC® 4,6×250 мм, скорость потока 1 мл/мин, 30°C). Твердое вещество суспендировали в 2:1 EtOH/Et2O (5 объемов), перемешивали в течение 10 минут, собирали фильтрованием, промывали смесью 2:1 EtOH/Et2O и сушили при пониженном давлении с получением белого твердого вещества (60,6 г).
Структуру и абсолютную стереохимию соединения 23 подтверждали при помощи рентгеноструктурного анализа монокристалла. Данные дифракции на монокристалле получали на Bruker Apex II дифрактометре, снабженном запаянной трубкой с Cu K-альфа источником (Cu Kα излучение, γ=1,54178 Å) и Apex II CCD детектором. Выбирали кристалл, имеющий размеры 0,15×0,15×0,10 мм, очищали с использованием минерального масла, закрепляли на MicroMount и центрировали на Bruker APEXII системе. Были получены три группы из 40 каркасов, разделенных в обратном пространстве, для получения матрицы ориентации и исходных параметров ячейки. Конечные параметры ячейки получали и уточняли после завершения сбора данных на основании полного набора данных. На основании статистических данных отсутствий и интенсивностей структура была определена и уточнена в не имеющей центра симметрии P21 пространственной группе.
Набор данных дифракции обратного пространства получали для разрешения 0,85 Å с использованием ступеней 0,5° и 30 сек экспонирования для каждого каркаса. Данные собирали при 100 (2) K. Интеграцию интенсивностей уточнение параметров ячейки осуществляли с использованием программы APEXII. Наблюдение кристалла после сбора данных не показало никаких признаков разложения. Как показано на фиг.2, присутствуют две симметричные независимые молекулы в структуре, и обе симметричные независимые молекулы представляют собой R изомеры.
Данные собирали, уточняли и обрабатывали с использованием программы Apex II. Структуру определяли с использованием SHELXS97 (Sheldrick, 1990); программу(программы) и структуру уточняли с использованием программы SHELXL97 (Sheldrick, 1997). Кристалл показал моноклинную ячейку с P21 пространственной группой. Параметры решетки были следующими a=9,9016(2) Å, b=10,9184(2) Å, c=19,2975(4) Å, β=102,826(1)°. Объем=2034,19(7) Å3.
Пример 21
Получение 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины в виде соли метансульфоновой кислоты (23A)
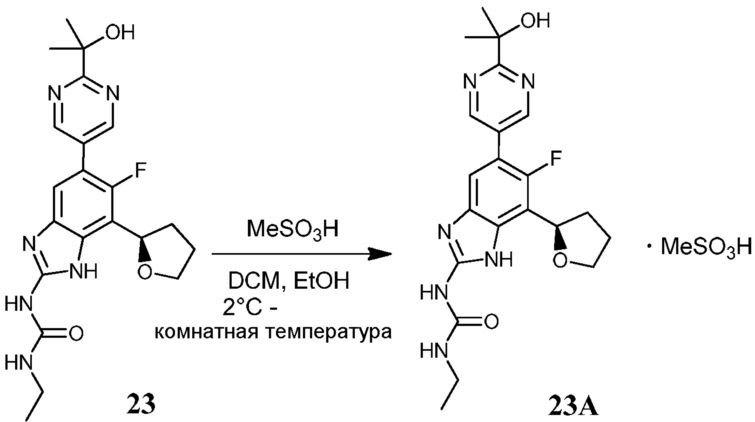
К перемешиваемой суспензии 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины (23) (15,05 г, 35,13 ммоль) в дихлорметане (60 мл, J. T. Baker 931533) и абсолютном этаноле (15 мл, Pharmco-AAPER 111000200) добавляли метансульфоновую кислоту (2,392 мл, 36,89 ммоль, Sigma-Aldrich 471356). Смесь перемешивали при комнатной температуре до получения прозрачного раствора. Медленно добавляли гептан (300 мл) в течение примерно 1 часа и осажденное твердое вещество собирали фильтрованием (с использованием Whatman qualitative # 3 бумаги поверх Whatman GF/F бумаги со стекломикроволокном). Сушили при пониженном давлении в вакуумной печи (осушали при помощи сульфата кальция и гидроксида калия) в течение ночи при 40°C с получением соединения 23A в виде белого твердого вещества (13,46 г, ВЭЖХ чистота 99+%, эи 99+%). Аналитическая хиральная ВЭЖХ показала один энантиомер с временем удерживания 8,6 мин с элюированием смесью CH2Cl2/MeOH/TEA (60/40/0,1) на колонке CHIRALPAK® IC® 4,6 × 250 мм со скоростью потока 1 мл/мин при 30°C. Вторую партию белого твердого продукта 23A (4,36 г, ВЭЖХ чистота 98%, эи 99+%) получали из фильтрата. ЖХ/MS (C18 колонка, элюирование с использованием градиента 10-90% CH3CN/вода в течение 5 минут с муравьиной кислотой в качестве модификатора) M+1: 429,58 (2,03 мин).
1H-ЯМР (300 МГц, MeOD) δ 9,00 (д, J=1,6 Гц, 2H), 7,67 (д, J=6,1 Гц, 1H), 5,39 (т, J=7,7 Гц, 1H), 4,30 (дд, J=14,9, 6,9 Гц, 1H), 4,03 (дд, J=14,8, 7,7 Гц, 1H), 3,40-3,31 (м, 2H), 2,72 (с, 3H), 2,70-2,60 (м, 1H), 2,21-2,08 (м, 2H), 1,98-1,84 (м, 1H), 1,65 (с, 6H), 1,22 (т, J=7,2 Гц, 3H) м.д.
(R)-1-этил-3-(6-фтор-5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-2-ил)мочевину 23 затем можно преобразовать в пролекарства в виде фосфата или фосфатной соли в соответствии со схемами, представленными ниже.
Схема 1
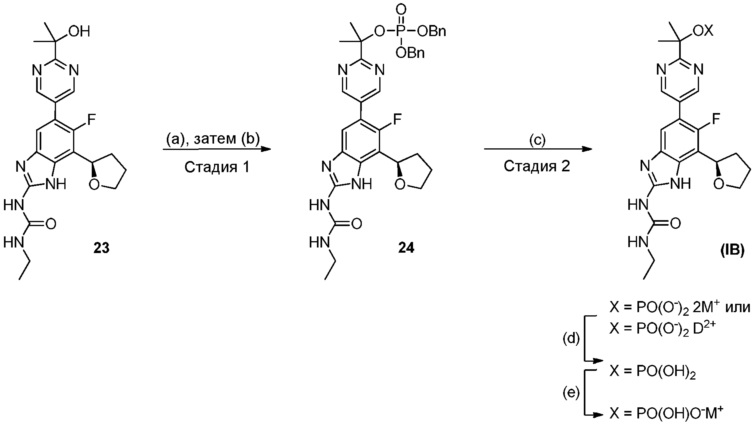
Реагенты и условия: (a) дибензил N,N-диизопропилфосфорамидит, тетразол, 23°C, DMF; (b) mCPBA, 0-23°C, DMF; (c) H2, Pd/C, M+OH- или D2+(OH-)2, EtOH, H2O; (d) водн. H+; (e) водн. M+OH-.
Соединения формулы (IB) можно получить из соединения 23, как показано на Схеме 1. На стадии 1, соединение 23 обрабатывают дибензил N,N-диизопропилфосфорамидитом и тетразолом, с последующей обработкой мета-хлорпероксибензойной кислотой (mCPBA), с получением дибензилфосфата 24. На стадии 2, гидрогенолиз соединения 24 в присутстви M+OH- или D2+(OH-)2 дает дианионную форму соединения формулы (IB) (X=-PO(O-)2·2M+ или -PO(O-)2·D2+). Свободнокислотную форму соединения формулы (IB) (X=PO(OH)2) можно получить путем обработки дианионной формы водным раствором кислоты. Моноанионную форму соединения формулы (IB) (X=PO(OH)O-M+) можно получить путем обработки свободнокислотной формы одним эквивалентом M+OH-.
Схема 2
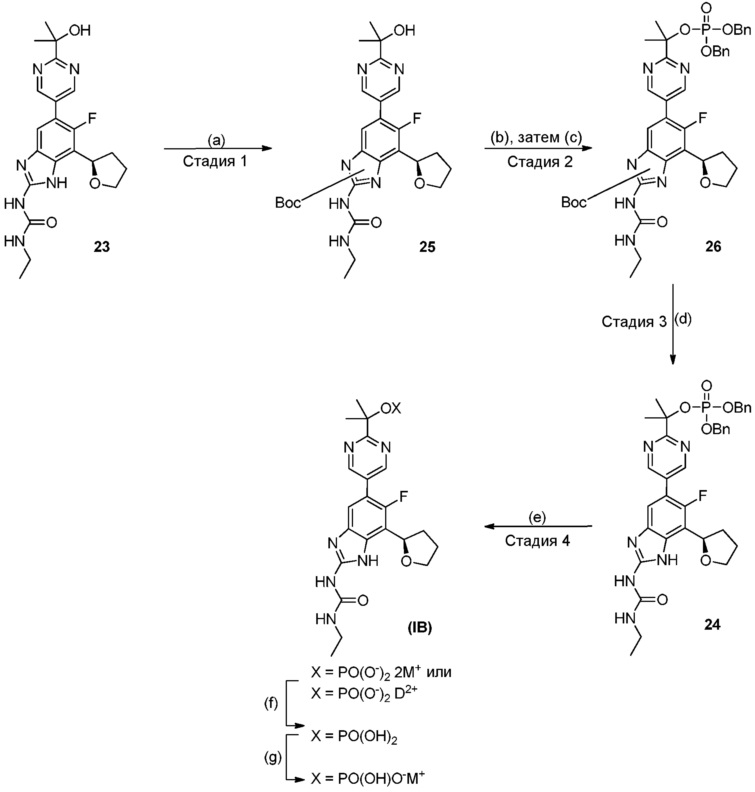
Реагенты и условия:(a) Boc2O, DMF, 23°C; (b) дибензил N,N-диизопропилфосфорамидит, тетразол, 23°C, DMF; (c) mCPBA, 0-23°C, DMF; (d) TFA, H2O, MeOH, DCM, 23°C; (e) H2, Pd/C, M+OH- или D2+(OH-)2, EtOH, H2O; (f) водн. H+; (g) водн. M+OH-.
Альтернативно, соединения формулы (IB) можно получить из соединения 23, как показано на Схеме 2. На стадии 1, соединение 23 обрабатывают ди-трет-бутилдикарбонатом (Boc2O) с получением защищенного бензимидазольного соединения 25. На стадии 2, соединение 25 обрабатывают дибензил N,N-диизопропилфосфорамидитом и тетразолом, с последующей обработкой при помощи mCPBA, с получением защищенного дибензилфосфата 26. На стадии 3, соединение 26 обрабатывают трифторуксусной кислотой (TFA) для удаления защитной группы и получения дибензилфосфата 24. На стадии 4, гидрогенолиз соединения 24 в присутстви M+OH- или D2+(OH-)2 дает дианионную форму соединения формулы (IB) (X=-PO(O-)2·2M+ или -PO(O-)2·D2+). Свободнокислотную форму соединения формулы (IB) (X=PO(OH)2) можно получить путем обработки дианионной формы водным раствором кислоты. Моноанионную форму соединения формулы (I) (X=PO(OH)O-M+) можно получить путем обработки свободнокислотной формы одним эквивалентом M+OH-.
Схема 3
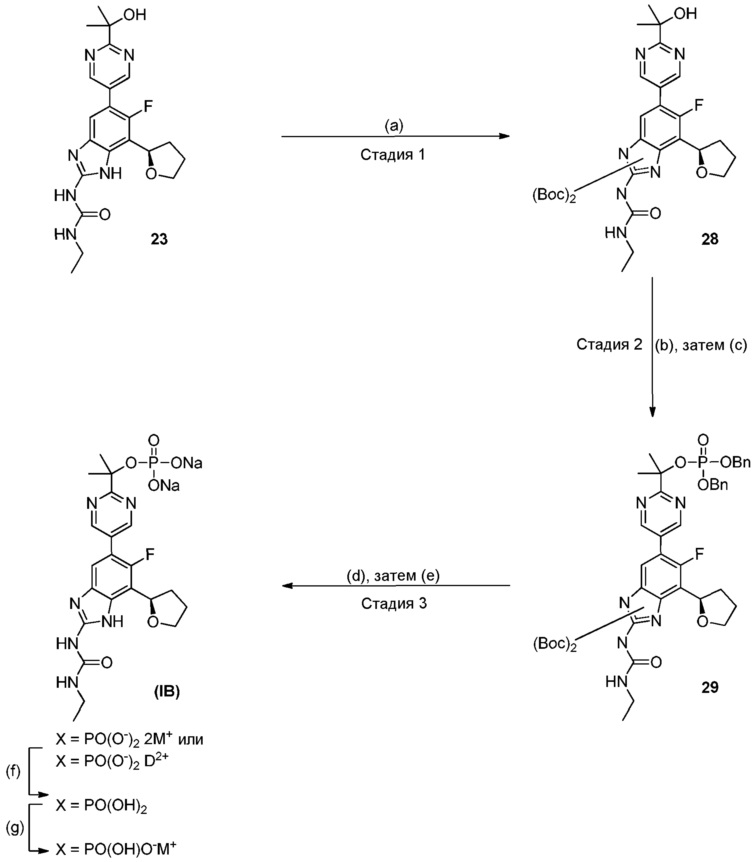
Реагенты и условия: (a)Boc2O, DMAP, DMF, 23°C; (b) дибензил N,N-диизопропилфосфорамидит, тетразол, 23°C, DMF; (c) mCPBA, 0-23°C, DMF; (d) TFA, DCM; (e) водн. M+OH- или D2+(OH-)2; (f) водн. H+; (g) водн. M+OH-.
Соединения формулы (IB) также можно получить из соединения 23, как показано на Схеме 3. На стадии 1, соединение 23 обрабатывают двумя эквивалентами Boc2O в присутствии N,N-диметиламинопиридина (DMAP) с получением бис-защищенного бензимидазольного соединения 28. На стадии 2, соединение 28 обрабатывают дибензил N,N-диизопропилфосфорамидитом и тетразолом, с последующей обработкой при помощи mCPBA, с получением бис-защищенного дибензилфосфата 29. На стадии 3, соединение 29 обрабатывают при помощи TFA для удаления защитных групп. Обработка полученного промежуточного соединения водным раствором M+OH- или D2+(OH-)2 дает дианионную форму соединения формулы (IB) (X=-PO(O-)2·2M+ или -PO(O-)2·D2+). Свободнокислотную форму соединения формулы (IB) (X=PO(OH)2) можно получить путем обработки дианионной формы водным раствором кислоты. Моноанионную форму соединения формулы (I) (X=PO(OH)O-M+) можно получить путем обработки свободнокислотной формы одним эквивалентом M+OH-.
Пример 22
Получение (R)-дибензил 2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримид ин-2-ил)пропан-2-илфосфата (24)
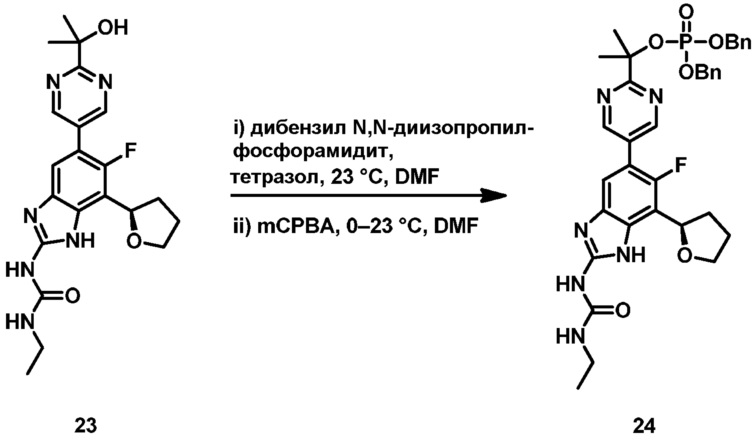
К 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевине (23) (10,24 г, 23,66 ммоль) в 1-л круглодонной колбе в атмосфере N2 при 23°C добавляли DMF (200 мл) с последующим добавлением раствора тетразола (105,2 мл раствора 0,45 M в MeCN, 47,32 ммоль) с последующим добавлением N-дибензилоксифосфанил-N-изопропил-пропан-2-амина (12,26 г, 11,93 мл, 35,49 ммоль). Через 4,5 часа снова добавляли N-дибензилоксифосфанил-N-изопропил-пропан-2-амин (4 мл). После перемешивания еще в течение 16 часов реакционную смесь охлаждали до 0°C (баня с ледяной водой), затем обрабатывали при помощи mCPBA (15,17 г, 61,52 ммоль). Смесь перемешивали при 0°C в течение 30 минут, затем при 23°C в течение 30 минут, после чего реакционную смесь распределяли между водой (400 мл) и EtOAc (700 мл). Органический слой отделяли, промывали насыщенным водным раствором бикарбоната натрия (500 мл), 10% водным раствором бисульфита натрия (500 мл), насыщенным водным раствором бикарбоната натрия (500 мл) и насыщенным солевым раствором (500 мл), затем сушили (сульфат магния), фильтровали и концентрировали. Остаток очищали при помощи MPLC с использованием системы флэш-хроматографической очистки ISCO COMBIFLASH (330 г колонка), с использованием для элюирования линейного градиента 0-10% EtOH в DCM более 16,5 колоночных объемов при скорости потока 200 мл/мин. После концентрирования в вакууме получали (R)-дибензил 2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан-2-илфосфат(24) (13,89 г, 20,17 ммоль, 85,27%) в виде белого твердого вещества. ESMS (M+1)=689,5;
1H-ЯМР (300 МГц, CD3OD) δ 8,88 (д, J=1,6 Гц, 2H), 7,37 (д, J=6 Гц, 1H), 7,30 (м, 10H), 5,38-5,33 (м, 1H), 5,12-5,01 (м, 4H), 4,24 (дд, J=6,8, 14,9 Гц, 1H), 3,98 (дд, J=6,9, 15,1 Гц, 1H), 3,35-3,27 (м, 3H), 2,52 (кв., J=5,9 Гц, 1H), 2,14-2,05 (м, 2H), 1,91 (с, 6H) и 1,22-1,14 (м, 3H) м.д.
Пример 23
Получение динатрий (R)-2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан- 2-илфосфата (W)
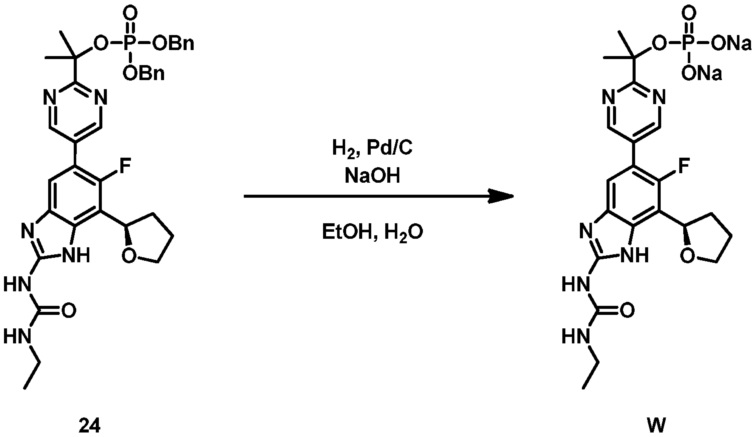
В 1-л сосуд Парра загружали воду (200 мл), Pd/C (4 г, 10% масс. в расчете на сухое вещество, влажный, тип Degussa), (R)-дибензил 2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан-2-илфосфат(24) (13,89 г, 20,17 ммоль), EtOH (400 мл) и водный раствор 1 M NaOH (40,34 мл, 40,34 ммоль). Полученную смесь гидрировали при давлении 50 ф/дюйм2(3,515 кг/см2) H2 на встряхивающем устройстве Парра в течение 40 мин. Реакционную смесь фильтровали через 0,22 мкм полиэфирсульфоновую (PES) мембрану с получением фильтрата темного цвета. Водная промывка давала еще более темное вещество, проходящее через фильтровальную мембрану. Полученный фильтрат пропускали через слой Целита и темное вещество не элюировало до тех пор, пока слой не промывали водой. Полученный темный раствор (приблизительно 700 мл) разбавляли тремя объемами EtOH (2100 мл), фильтровали через 0,22 мкм PES мембрану (с использованием 4 полистирольных фильтровальных систем Corning одноразового использования, #431098) и фильтрат концентрировали в вакууме. Полученный остаток растворяли в воде (100 мл) и EtOH (300 мл), фильтровали через 0,22 мкм PES мембрану с получением прозрачного желтого раствора, затем пропускали через пробку из Целита (26 мм в диаметре × 60 мм в высоту, предварительно смоченную EtOH), промывая при помощи EtOH (50 мл), и фильтрат затем концентрировали. Полученный остаток растворяли в воде (250 мл) в 1-л круглодонной колбе, затем медленно добавляли 1 M водный раствор HCl (40 мл) в течение 15 минут при перемешивании с получением суспензии белого твердого вещества. Через двадцать минут после завершения добавления HCl твердое вещество собирали фильтрованием через 0,22 мкм PES мембрану. Собранное твердое вещество промывали водой (100 мл), затем переносили (все еще мокрое) в 1-л круглодонную колбу и суспендировали в MeOH (150 мл) в течение 30 минут. Полученный мелкий белый осадок собирали фильтрованием, затем сушили в вакууме в течение ночи. Полученную свободную кислоту (9,17 г, 18,0 ммоль) обрабатывали водой (80 мл), затем 1,0 н водным раствором NaOH (36,0 мл, 2 экв.). Полученный раствор замораживали и лиофилизировали с получением динатрий [1-[5-[2-(этилкарбамоиламино)-6-фтор-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-5-ил]пиримидин-2-ил]-1-метил-этил]фосфата (W) (10,206 г, 18,08 ммоль, 89,66%) в виде бледного кремового твердого вещества с соответствующими аналитическими данными. ESMS (M+1)=509,4;
1H-ЯМР (300 МГц, D2O) δ 8,58 (с, 2H), 6,92 (д, J=6,3 Гц, 1H), 5,13 (т, J=7,5 Гц, 1H), 3,98-3,81 (м, 2H), 3,04 (кв., J=7,2 Гц, 2H), 2,26 (т, J=5,7 Гц, 1H), 1,97-1,92 (м, 2H), 1,67 (с, 6H) и 1,01 (т, J=7,2 Гц, 3H) м.д.
Пример 24
Получение Boc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H- бензимидазол-2-ил]мочевины (25)
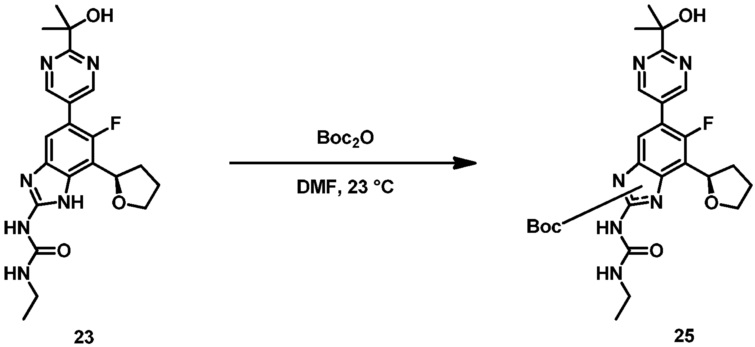
К раствору/суспензии 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины (23) (10,72 г, 25,02 ммоль) в DMF (250 мл) при 23°C добавляли Boc2O (6,11 г, 28,00 ммоль). Через 2 часа добавляли раствор 2 M аммиака в MeOH (2 мл) для гашения какого-либо избытка Boc2O. Гашеную реакционную смесь распределяли между EtOAc и водой (по 400 мл каждого), органический слой отделяли, промывали водой (2×400 мл) и насыщенным солевым раствором (400 мл), затем сушили над MgSO4, фильтровали и концентрировали с получением Boc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины (25) (12,69 г, 23,58 ммоль, 94,26%), которую использовали без дополнительной очистки. ESMS (M+1)=529,3;
1H-ЯМР (300,0 МГц, CDCl3) δ 9,50 (с, 1H), 9,02 (т, J=5,3 Гц, 1H), 8,91 (д, J=1,6 Гц, 2H), 7,74 (д, J=6,5 Гц, 1H), 5,58 (т, J=7,8 Гц, 1H), 4,64 (с, 1H), 4,22 (кв., J=7,4 Гц, 1H), 4,05 (тд, J=7,8, 4,3 Гц, 1H), 3,47 (тд, J=7,2, 4,3 Гц, 2H), 2,42-2,35 (м, 2H), 2,28-2,16 (м, 2H), 1,75 (с, 9H), 1,68 (с, 6H) и 1,31 (т, J=7,3 Гц, 3H) м.д.
Пример 25
Получение Boc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-и л]мочевина дибензилфосфата (26)
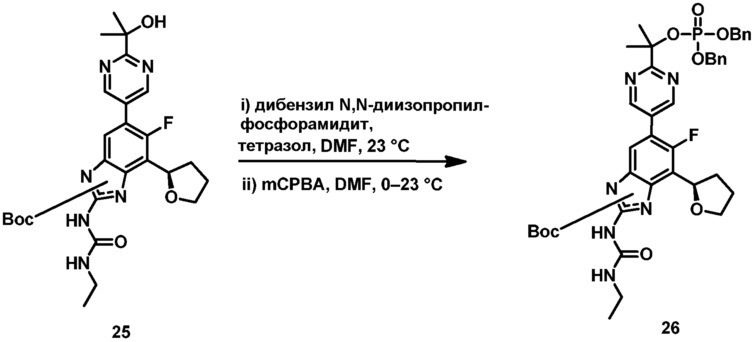
К Boc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевине (25) (12,69 г, 23,58 ммоль) и тетразолу (3,304 г, 47,16 ммоль) в атмосфере N2 при 23°C добавляли DCM (240 мл) с последующим добавлением N-дибензилоксифосфанил-N-изопропил-пропан-2-амина (9,775 г, 9,509 мл, 28,30 ммоль). После 3 часов при 23°C реакционную смесь охлаждали до 0°C, затем добавляли mCPBA (6,977 г, 28,30 ммоль). Полученный раствор перемешивали в течение 45 минут при 0°C, затем в течение 20 минут при 23°C. Реакционную смесь затем распределяли между DCM (50 мл) и насыщенным водным раствором бикарбонатанатрия (400 мл). Органические слой отделяли, затем промывали последовательно водным раствором бисульфита натрия (63 г в 350 мл воды) и насыщенным водным раствором бикарбоната натрия (400 мл), затем сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали при помощи MPLC с использованием системы флэш-хроматографической очистки ISCO COMBIFLASH (колонка с 330 г диоксида кремния), с использованием для элюирования линейного градиента 0-100% EtOAc в гексане более 16 колоночных объемов при 200 мл/мин. Фракции, содержащие продукт, упаривали в вакууме с получением Boc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевина дибензилфосфата (26) (11,92 г, 15,11 ммоль,64,09%). ESMS (M+1)=789,2;
1H-ЯМР (300,0 МГц, CDCl3) δ 9,51 (с, 1H), 9,03 (т, J=5,4 Гц, 1H), 8,91 (д, J=1,6 Гц, 2H), 7,73 (д, J=6,5 Гц, 1H), 7,37-7,28 (м, 10H), 5,58 (т, J=7,8 Гц, 1H), 5,17-5,05 (м, 4H), 4,23 (т, J=7,5 Гц, 1H), 4,05 (тд, J=7,8, 4,3 Гц, 1H), 3,53-3,44 (м, 2H), 2,39 (дд, J=7,9, 14,5 Гц, 2H), 2,28-2,15 (м, 2H), 1,98 (с, 6H), 1,72 (м, 9H) и 1,31 (т, J=7,2 Гц, 3H) м.д.
Пример 26
Получение (R)-дибензил 2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пирими дин-2-ил)пропан-2-илфосфат (24)
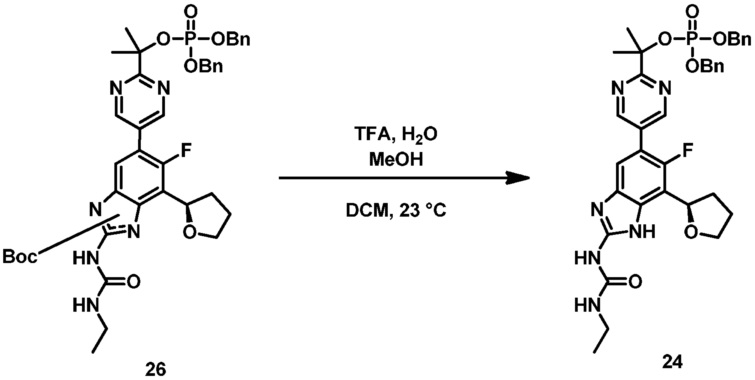
К раствору дибензилфосфата Boc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины (26) (11,9 г, 15,09 ммоль) в DCM (300 мл) при 23°C добавляли воду (2,325 мл, 129,1 ммоль), затем TFA (3,441 г, 2,325 мл, 30,18 ммоль). Через 1 час наблюдали только частичную конверсию, согласно данным ТСХ, поэтому снова добавляли TFA (3,441 г, 2,325 мл, 30,18 ммоль). Еще через 2,5 часа добавляли MeOH (2 мл) и смесь перемешивали еще в течение 18 часов. Реакционную смесь промывали 1:1 насыщенным солевым раствором:насыщенным водным раствором бикарбоната натрия (200 мл). Водный слой снова экстрагировали при помощи DCM (150 мл), органические слои объединяли, затем сушили (над сульфатом магния), фильтровали и концентрировали в вакууме. Полученный остаток снова растворяли в EtOAc (200 мл), промывали водой (150 мл) и насыщенным солевым раствором (100 мл), затем сушили (сульфат магния), фильтровали и концентрировали с получением (R)-дибензил 2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан-2-илфосфата (24) (10,21 г, 14,83 ммоль, 98,27%) в виде белого твердого вещества. ESMS (M+1)=689,4;
1H-ЯМР (300 МГц, CD3OD) δ 8,88 (д, J=1,6 Гц, 2H), 7,37 (д, J=6 Гц, 1H), 7,30 (м, 10H), 5,38-5,33 (м, 1H), 5,12-5,01 (м, 4H), 4,24 (дд, J=6,8, 14,9 Гц, 1H), 3,98 (дд, J=6,9, 15,1 Гц, 1H), 3,35-3,27 (м, 3H), 2,52 (кв., J=5,9 Гц, 1H), 2,14-2,05 (м, 2H), 1,91 (с, 6H) и 1,22-1,14 (м, 3H) м.д.
Пример 27
Получение динатрий (R)-2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин -2-ил)пропан-2-илфосфата (W)
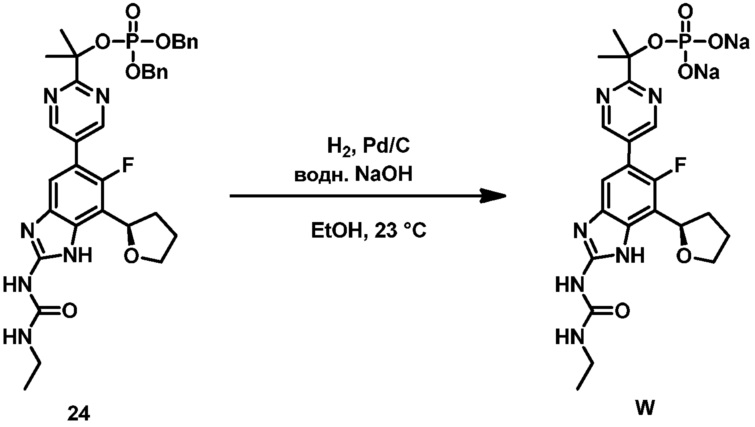
В 1-л круглодонную колбу загружали (R)-дибензил 2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан-2-илфосфат (24) (9,37 г, 13,61 ммоль), EtOH (300 мл), воду (150 мл), Pd/C (10% масс. в расчете на сухое вещество, влажный, тип Degussa, 3 г) и 1 M водный раствор NaOH (27,22 мл, 27,22 ммоль). Из суспензии откачивали газ в течение 3 минут (шприц для откачивания), затем помещали в атмосферу газообразного водорода (баллон). После перемешивания в течение 2,5 часов при 23°C реакционную смесь фильтровали через 0,22 мкм PES мембрану (фильтровальная система Corning одноразового использования, 1 л, полистирол, #431098) для удаления катализатора и промывали при помощи EtOH (50 мл). Полученный раствор концентрировали, остаток растворяли в воде (80 мл), обрабатывали при помощи MeCN (80 мл), затем замораживали и лиофилизировали с получением динатрий (R)-2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан-2-илфосфата (W) (7,10 г, 12,81 ммоль, 94,12%) в виде белого твердого вещества. ESMS (M+1)=509,3;
1H-ЯМР (300 МГц, D2O) δ 8,58 (с, 2H), 6,92 (д, J=6,3 Гц, 1H), 5,13 (т, J=7,5 Гц, 1H), 3,98-3,81 (м, 2H), 3,04 (кв., J=7,2 Гц, 2H), 2,26 (т, J=5,7 Гц, 1H), 1,97-1,92 (м, 2H), 1,67 (с, 6H) и 1,01 (т, J=7,2 Гц, 3H) м.д.
Пример 28
Получение диBoc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины (28)
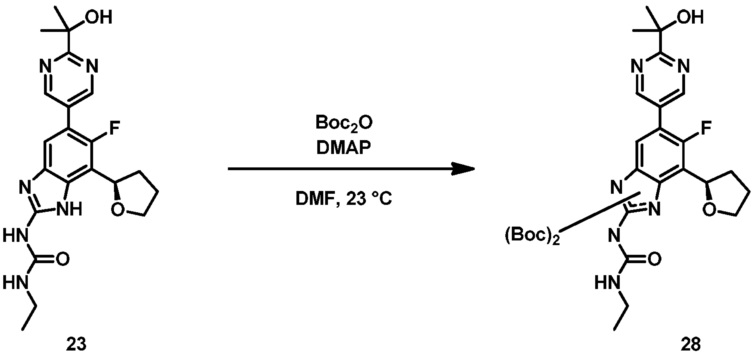
К раствору/суспензии 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины (23) (1,333 г, 3,111 ммоль) в DMF (30 мл) добавляли DMAP (38,01 мг, 0,3111 ммоль) с последующим добавлением Boc2O (1,426 г, 1,501 мл, 6,533 ммоль). Через 30 минут реакционную смесь разбавляли водой и EtOAc (по 300 мл каждого), органический слой отделяли, промывали водой и насыщенным солевым раствором (по 300 мл каждого), затем сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали при помощи MPLC с использованием системы флэш-хроматографической очистки ISCO COMBIFLASH (колонка с 80 г диоксида кремния), с использованием для элюирования линейного градиента 0-60% EtOAc в гексане более 20 колоночных объемов при скорости потока 60 мл/мин. Содержащие желаемый продукт фракции объединяли и упаривали с получением диBoc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевины (28) (1,43 г, 2,275 ммоль, 73,11%) в виде прозрачного пенистого вещества. ESMS (M+1)=629,3;
1H-ЯМР (300,0 МГц, CDCl3) δ 8,95 (д, J=1,6 Гц, 2H), 8,31-8,27 (м, 1H), 8,05 (д, J=6,5 Гц, 1H), 5,80-5,68 (м, 1H), 4,70 (с, 1H), 4,21-4,09 (м, 1H), 3,98 (д, J=6,4 Гц, 1H), 3,42-3,37 (м, 2H), 2,45-2,00 (м, 4H), 1,65 (с, 6H), 1,62 (с, 9H), 1,37 (с, 9H) и 1,28-1,21 (м, 3H) м.д.
Пример 29
Получение дибензилфосфата диBoc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-и л]мочевин ы (29)
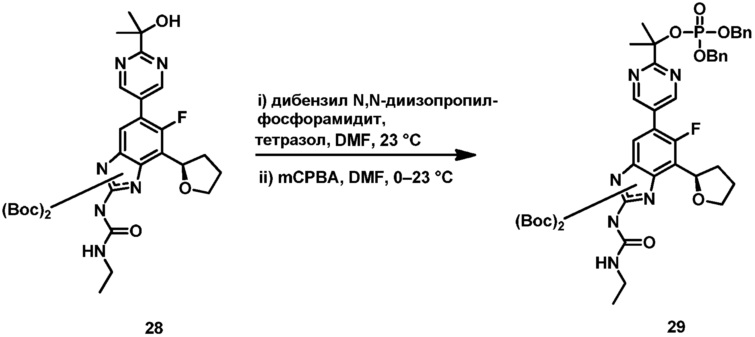
К диBoc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевине (28) (1,13 г, 1,797 ммоль) и тетразолу (251,8 мг, 3,594 ммоль) при 23°C в атмосфере N2 добавляли DCM (30 мл) с последующим добавлением N-дибензилоксифосфанил-N-изопропил-пропан-2-амина (744,7 мг, 724,4 мкл, 2,156 ммоль). После перемешивания в течение 18 часов реакционную смесь охлаждали до 0°C, затем обрабатывали при помощи mCPBA (531,5 мг, 2,156 ммоль). Реакционную смесь перемешивали в течение 15 минут при 0°C, затем в течение 30 минут при 23°C. Полученный раствор затем распределяли между EtOAc и насыщенным водным раствором бикарбоната натрия (по 300 мл каждого), органический слой отделяли, затем промывали 10% водным раствором бисульфита натрия, насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором (по 300 мл каждого), затем сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали при помощи MPLC с использованием системы флэш-хроматографической очистки ISCO COMBIFLASH (колонка с 80 г диоксида кремния), с использованием для элюирования линейного градиента 0-80% EtOAc в гексане более 20 колоночных объемов при скорости потока 60 мл/мин. Содержащие желаемый продукт фракции объединяли и упаривали с получением диBoc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевина дибензилфосфата (29) (1,03 г, 1,159 ммоль, 64,50%) в виде прозрачного блестящего масла. ESMS (M+1)=889,5;
1H-ЯМР (300,0 МГц, CDCl3) δ 8,93 (д, J=1,5 Гц, 2H), 8,31 (с, 1H), 8,04 (д, J=6,4 Гц, 1H), 7,36-7,26 (м, 10H), 5,83-5,70 (м, 1H), 5,16-5,05 (м, 4H), 4,24-4,18 (м, 1H), 4,03-3,97 (м, 1H), 3,42-3,36 (м, 2H), 2,43-2,05 (м, 4H), 1,98 (с, 6H), 1,64 (с, 9H), 1,40 (с, 9H) и 1,26 (т, J=7,2 Гц, 3H) м.д.
Пример 30
Получение натрий (R)-2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан-2-илфосфата (W)
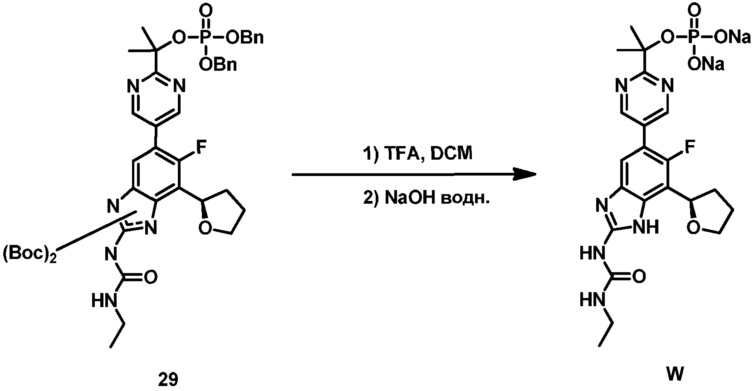
К раствору диBoc-1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевина дибензилфосфата (29) (121 мг, 0,1361 ммоль) в DCM (10 мл) при 23°C добавляли TFA (5 мл). Через 2 часа реакционную смесь концентрировали в вакууме. Остаток растворяли в MeOH (6 мл) и обрабатывали при помощи приблизительно 0,5 мл 2 M NH3 в MeOH (для полного растворения вещества). Полученный раствор очищали при помощи препаративной ВЭЖХ, 6 вводимых проб, обращено-фазовая, колонка Sunfire prep C18 OBD 5 мкм 19×100 мм; элюирование при помощи 10-90% водн. MeCN w/0,1% TFA буфер, линейный градиент в течение 15 минут при скорости потока 20 мл/мин. Фракции, содержащие продукт, объединяли и лиофилизировали. Полученное вещество суспендировали в MeOH (3 мл), перемешивали при 23°C в течение 30 минут, затем осадок собирали фильтрованием через пластиковую фритту. Полученное белое твердое вещество снова суспендировали в MeOH (3 мл), затем собирали фильтрованием с получением после сушки 68 мг белого твердого вещества. Белое твердое вещество обрабатывали при помощи 0,10 M водного раствора NaOH (2,68 мл, 2 экв. NaOH) с получением раствора, который затем пропускали через Acrodisc CR 13 мм фильтровальный шприц с 0,45 мкм PTFE мембраной, промывая водой (2 мл). Полученный раствор обрабатывали при помощи MeCN (3 мл), замораживали и лиофилизировали с получением натрий (R)-2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан-2-илфосфата (W) в виде белого порошка. ESMS (M+1)=509,2;
1H-ЯМР (300 МГц, D2O) δ 8,58 (с, 2H), 6,92 (д, J=6,3 Гц, 1H), 5,13 (т, J=7,5 Гц, 1H), 3,98-3,81 (м, 2H), 3,04 (кв., J=7,2 Гц, 2H), 2,26 (т, J=5,7 Гц, 1H), 1,97-1,92 (м, 2H), 1,67 (с, 6H) и 1,01 (т, J=7,2 Гц, 3H) м.д.
Пример 31
Испытание чувствительности в жидкой среде
Соединения по настоящему изобретению были испытаны на антимикробную активность в испытании чувствительности в жидкой среде. Такие анализы осуществляли в соответствии с указаниями последнего CLSI документа, являющегося руководством для практического осуществления таких анализов: "M07-A8 Methods for Dilution Antimicrobial Susceptibility Tests dor Bacteria that Grow Aerobically; Approved Standard--Eighth Edition (2009)". Другие публикации, такие как "Antibiotics in Laboratory Medicine" (Edited by V. Lorian, Publishers Williams and Wilkins, 1996) представляют основные практические методы лабораторных испытаний антибиотиков. Использовали следующие конкретные протоколы:
Протокол #1: Определение гираза MIC для соединений методом микроразведений с использованием бульонной питательной среды
Материалы:
Круглодонные 96-луночные микротитровальные планшеты (Costar 3788)
Агаровые пластинки Mueller Hinton II (MHII; BBL premix)
Жидкая бульонная питательная среда Mueller Hinton II (MHII; BBL premix)
Система быстрой инокуляции BBL (Fisher B26306)
Зеркальный агглютинометр (Fisher)
Агаровые пластинки с бактериями, посеянными штрихом для разделения колоний, свежеприготовленные
Стерильный ДМСО
Человеческая сыворотка (U.S. Biologicals S1010-51)
Лаковая лошадиная кровь (Quad Five 270-100)
Ресазурин 0,01%
Сыворотка крыс Sprague Dawley (U.S. Biologicals 1011-90B или Valley BioMedical AS3061SD)
Объединенная мышиная сыворотка (Valley BioMedical AS3054)
Штаммы (среда, бульон и агар):
1. Staphylococcus aureus ATCC #29213
а. MHII
b. MHII+50% человеческая сыворотка
c. MHII+50% крысиная сыворотка
d. MHII+50% мышиная сыворотка
2. Staphylococcus aureus ATCC #29213 GyrB T173I (MHII)
3. Staphylococcus aureus, JMI коллекция штаммов; смотрите таблицу 5 (MHII)
4. Staphylococcus epidermidis, JMI коллекция штаммов; смотрите таблицу 5 (MHII)
5. Enterococcus faecalis ATCC #29212 (MHII+3% лаковая лошадиная кровь)
6. Enterococcus faecium ATCC #49624 (MHII+3% лаковая лошадиная кровь)
7. Enterococus faecalis, JMI коллекция штаммов; смотрите таблицу 5 (MHII+3% лаковая лошадиная кровь)
8. Enterococus faecium, JMI коллекция штаммов; смотрите таблицу 5 (MHII+3% лаковая лошадиная кровь)
9. Streptococcus pneumoniae ATCC #10015 (MHII+3% лаковая лошадиная кровь)
10. Streptococcus pneumoniae, JMI коллекция штаммов; смотрите таблицу 5 (MHII+3% лаковая лошадиная кровь)
11. β-гемолитические стрептококки, Группы A, B, C, G) JMI коллекция штаммов; смотрите таблицу 5 (MHII+3% лаковая лошадиная кровь)
12. Bacillus cereus ATCC 10987 (MHII)
13. Bacillus cereus ATCC 14579(MHII)
14. Bacillus subtilis ATCC 6638 (MHII)
15. Bacillus subtilis (168) ATCC 6051 (MHII)
Получение инокулята (для всех штаммов, кроме S. aureus + 50% сыворотка)
1. Использовали набор BBL Prompt, отбирали 5 больших или 10 малых четко разделенных колоний из культуры, выращенной на подходящей агаровой среде, как указано выше, и инокулировали 1 мл стерильного физиологического раствора из набора.
2. Лунки встряхивали для перемешивания в течение приблизительно 30 сек с получением суспензии приблизительно 108 клеток/мл. Действительная плотность может быть подтверждена путем выделения разведений этой суспензии.
3. Разбавляли суспензию 1/100 путем переноса 0,15 мл клеток в 15 мл (приблизительно 106 клеток/мл) стерильного бульона (или смотрите ниже) для каждого планшета с испытываемыми соединениями, затем осуществляли вращение с завихрением для смешивания. Если испытывали более 1 планшета с соединениями (>8 соединений), объемы соответственно увеличивали.
а. Для E. faecalis, E. faecium и S. pneumoniae: использовали 14,1 мл MHII+0,9 мл лаковой лошадиной крови.
4. Использовали 50 мкл клеток (приблизительно 5⋅104 клеток) для инокуляции каждой лунки микротитровального планшета, содержащей 50 мкл лекарственного средства, разведенного в бульоне (смотрите ниже).
Разведения лекарственного средст ва, инокуляция, определение MIC:
1. Все исходные растворы лекарственного средства/соединения получали при концентрации 12,8 мг/мл, обычно в 100% ДМСО.
2. Разбавляли исходные растворы лекарственного средства/соединения до 200x желаемой конечной концентрации в 50 мкл ДМСО. Если исходная концентрация MIC представляла собой 8 мкг/мл конечную концентрацию, тогда требовалось 6,25 мкл исходного раствора + 43,75 мкл ДМСО. Каждый 200x исходный раствор помещали в отдельный ряд колонки 1 нового 96-луночного микротитровального планшета.
3. Добавляли 25 мкл ДМСО к колонкам 2-12 всех рядов микротитровального планшета, содержащего 200x исходные растворы соединений, и осуществляли серийные разведения 25 мкл из колонок с 1 по 11, заменяли насадки после каждой колонки. т. е. 25 мкл соединения + 25 мкл ДМСО=2x разведение. Оставляли ДМСО лунку “без соединения” в конце рядов для контроля.
4. Для каждого испытываемого штамма (за исключением S. aureus + 50% человеческая сыворотка), подготавливали два микротитровальных планшета с 50 мкл MHII бульона с использованием устройства для пипетирования Matrix.
5. Переносили 0,5 мкл каждого разведения (w/автоматическое устройство для пипетирования Matrix) в 50 мкл среды/лунку на микротитровальном планшете перед добавлением 50 мкл клеток. Обычная исходная концентрация соединения была 8 мкг/мл после 1/200 разведения в среде + клетки - концентрации соединения, сниженные в 2x этапа по всем рядам микротитровального планшета. Все MIC анализы осуществляли в двух повторах.
6. Все лунки инокулировали 50 мкл разбавленной клеточной суспензии (смотрите выше) до конечного объема 100 мкл.
7. После добавления инокулята осуществляли тщательное смешивание в каждой лунке с использованием ручного многоканального устройства для пипетирования; использовали одни и те же насадки, идя в направлении от низкой до высокой концентрации лекарственного средства в одном и том же микротитровальном планшете.
8. Планшеты инкубировали при 37°C, по меньшей мере, в течение 18 часов.
9. Планшеты оценивали с использованием зеркального агглютинометра через 18 часов, и регистрировали MIC как наименьшую концентрацию лекарственного средства, где не наблюдали никакого роста (оптическая чистота в лунке).
Получение S. aureus + 50% человеческая сыворотка, S. aureus + 50% крысиная сыворотка или S. aureus + 50% мышиная сыворотка
1. Получали 50% сывороточную среду путем объединения 15 мл MHII+15 мл человеческой сыворотки - всего 30 мл. Увеличивали объем с 30 мл приращениями, когда испытывали более 1 планшета с соединениями.
2. Использовали тот же самый BBL Prompt инокулят S. aureus ATCC #29213, описанный выше, разбавляли 1/200 путем переноса 0,15 мл клеток в 30 мл (приблизительно 5⋅105 клеток/мл) 50% сывороточной среды, полученной выше, и осуществляли вращение с завихрением для смешивания.
3. Заполняли все испытываемые лунки желаемого количества микротитровальных планшетов 100 мкл клеток в 50% сывороточной среде.
4. Переносили 0,5 мкл каждого разведения соединения (w/Matrix auto-устройство для пипетирования) в 100 мкл клеток/среда. Обычная исходная концентрация соединения была 8 мкг/мл после 1/200 разведения в среде + клетки - концентрации соединения, сниженные в 2x этапа во всех рядах микротитровального планшета. Все MIC анализы осуществляли в двух повторах.
5. Осуществляли тщательное смешивание в каждой лунке с использованием ручного многоканального устройства для пипетирования; использовали одни и те же насадки, идя в направлении от низкой до высокой концентрации лекарственного средства в одном и том же микротитровальном планшете.
6. Планшеты инкубировали при 37°C, по меньшей мере, в течение 18 часов. После инкубации добавляли 25 мкл 0,01% Ресазурина к каждой лунке и продолжали инкубирование при 37°C, по меньшей мере, в течение еще 1 часа или до изменения цвета Ресазурина.
7. Планшеты оценивали с использованием зеркального агглютинометра и регистрировали MIC. Когда использовали Ресазурин, цвет красителя менялся от темно-синего до ярко-розового в лунках с отсутствием какого-либо роста. Наименьшая концентрация лекарственного средства, которая изменяла цвет красителя на розовый, представляла собой MIC.
Протокол 2: Определение гираза MIC для соединений против Грамотр ицательных бактерий методом микроразведений с использованием бульонной питательной среды
Материалы:
Круглодонные 96-луночные микротитровальные планшеты (Costar 3788)
Агаровые пластинки Mueller Hinton II (MHII; BBL premix)
Жидкая бульонная питательная среда Mueller Hinton II (MHII; BBL premix)
Система инокуляции BBL Prompt (Fisher b26306)
Зеркальный агглютинометр (Fisher)
Агаровые пластинки с бактериями, посеянными штрихом для разделения колоний, свежеприготовленные
Стерильный ДМСО
Штаммы (MHII среда для всех; бульон и агар):
1. Escherichia coli ATCC # 25922
2. Escherichia coli, JMI коллекция штаммов, смотрите таблицу 5
3 Escherichia coliAG100 WT
4. Escherichia coli AG100 tolC
5. Acinetobacter baumannii ATCC # BAA-1710
6. Acinetobacter baumannii ATCC # 19606
7. Acinetobacter baumannii, JMI коллекция штаммов, смотрите таблицу 5
8. Klebsiella pneumoniae ATCC # BAA-1705
9. Klebsiella pneumoniae ATCC # 700603
10. Klebsiella pneumoniae, JMI коллекция штаммов, смотрите таблицу 5
11. Moraxella catarrhalis ATCC# 25238
12. Moraxella catarrhalis ATCC# 49143
13. Moraxella catarrhalis, JMI коллекция штаммов, смотрите таблицу 5
14. Haemophilus influenzae ATCC 49247
15. Haemophilus influenzae (Rd1 KW20) ATCC 51907
16. Haemophilus influenzae Rd0894 (AcrA-)
17. Haemophilus influenzae, JMI коллекция штаммов, смотрите таблицу 5
18. Pseudomonas aeruginosa PAO1
19. Pseudomonas aeruginosa, JMI коллекция штаммов, смотрите таблицу 5
20. Proteus mirabilis, JMI коллекция штаммов, смотрите таблицу 5
21. Enterobacter cloacae, JMI коллекция штаммов, смотрите таблицу 5
22. Stenotrophomonas maltophilia ATCC BAA-84
23. Stenotrophomonas maltophilia ATCC13637
Получение инокулята:
1. С использованием BBL Prompt набора отбирали 5 больших или 10 малых четко разделенных колоний из культур, выращенных на агаровой среде, и инокулировали 1 мл стерильного физиологического раствора, который был в наборе.
2. Лунки встряхивали для перемешивания в течение приблизительно 30 сек с получением суспензии приблизительно 108 клеток/мл. Действительная плотность может быть подтверждена путем выделения разведений этой суспензии.
3. Разбавляли суспензию 1/100 путем переноса 0,15 мл клеток в 15 мл (приблизительно 106 клеток/мл) стерильного бульона (смотрите ниже) для каждого планшета с испытываемыми соединениями, осуществляли вращение с завихрением для смешивания. Если испытывали более 1 планшета соединений (>8 соединений), объемы соответственно увеличивали.
4. Использовали 50 мкл клеток (приблизительно 5⋅104 клеток) для инокуляции каждой лунки микротитровального планшета, содержащей 50 мкл лекарственного средства, разведенного в бульоне (смотрите ниже).
Разведения лекарственного средства, инокуляция, определение MIC:
1. Все исходные растворы лекарственного средства/соединения получали при концентрации 12,8 мг/мл, обычно в 100% ДМСО.
2. Разбавляли исходные растворы лекарственного средства/соединения до 200x желаемой конечной концентрации в 50 мкл ДМСО. Если исходная концентрация MICs составляла 8 мкг/мл конечной концентрации, тогда требовалось 6,25 мкл исходного раствора + 43,75 мкл ДМСО. Каждый 200x исходный раствор помещали в отдельный ряд колонки 1 нового 96-луночного микротитровального планшета.
3. Добавляли 25 мкл ДМСО к колонкам 2-12 всех рядов микротитровального планшета, содержащего 200x исходные растворы соединений, и осуществляли серийные разведения 25 мкл из колонок с 1 по 11, заменяли насадки после каждой колонки. т. е. 25 мкл соединения + 25 мкл ДМСО = 2x разведение. Оставляли ДМСО лунку “без соединения” в конце рядов для контроля.
4. Для каждого испытываемого штамма подготавливали два микротитровальных планшета с 50 мкл MHII бульона с использованием устройства для пипетирования Matrix.
5. Переносили 0,5 мкл каждого разведения (w/автоматическое устройство для пипетирования Matrix) в 50 мкл среды/лунку на микротитровальном планшете перед добавлением 50 мкл клеток. Обычная исходная концентрация соединения была 8 мкг/мл после 1/200 разведения в среде + клетки - концентрации соединения, сниженные в 2x этапа во всех рядах микротитровального планшета. Все MIC анализы осуществляли в двух повторах.
6. Все лунки инокулировали 50 мкл разбавленной клеточной суспензии (смотрите выше) до конечного объема 100 мкл.
7. После добавления инокулята осуществляли тщательное смешивание в каждой лунке с использованием ручного многоканального устройства для пипетирования; использовали одни и те же насадки, идя в направлении от низкой до высокой концентрации лекарственного средства в одном и том же микротитровальном планшете.
8. Планшеты инкубировали при 37°C, по меньшей мере, в течение 18 часов.
9. Планшеты оценивали с использованием зеркального агглютинометра через 18 часов и регистрировали MIC как наименьшую концентрацию лекарственного средства, где не наблюдали никакого роста (оптическая чистота в лунке).
Протокол #3: Определение гираз ы MIC для соединений методом разведения с использованием агара
Материалы:
Чашки Петри 60×15 мм (Thermo Scientific Cat. # 12567100)
Пробирки для центрифугирования, 15 мл (Costar)
Система инокуляции BBL Prompt(Fisher b26306)
Агаровые пластинки с бактериями, посеянными штрихом для разделения колоний, свежеприготовленные
Стерильный ДМСО
GasPakTM инкубационные контейнеры (BD Cat. #260672)
GasPak TM EZ Анаэробная контейнерная система в виде пакетов-саше (BD Cat. #260678)
GasPak TM EZ C02 контейнерная система в виде пакетов-саше (BD Cat. #260679)
GasPak TM EZ Campy контейнерная система в виде пакетов-саше (BD Cat. #260680)
Штаммы:
1. Clostridium difficile ATCC BAA-1382;
2. Clostridium difficile, CMI коллекция штаммов, смотрите таблицу 4
3. Clostridium perfringens, CMI коллекция штаммов, смотрите таблицу 4
4. Bacteroides fragilis и Bacteroides spp., CMI коллекция штаммов, смотрите таблицу 4
5. Fusobacterium spp., CMI коллекция штаммов, смотрите таблицу 4
6. Peptostreptococcus, spp., CMI коллекция штаммов, смотрите таблицу 4
7. Prevotella spp., CMI коллекция штаммов, смотрите таблицу 4
8. N. gonorrhoeae ATCC 35541
9. N. gonorrhoeae ATCC 49226
10. Neisseria gonorrhoeae, JMI коллекция штаммов, смотрите таблицу 4
11. Neisseria meningitidis, JMI коллекция штаммов, смотрите таблицу 4
Получение среды и условия роста:
Питательную среду, рекомендованную для каждого вида бактерий получали в соответствии с CLSI публикацией ‘M11-A7 Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard - Seventh Edition (2007)’ за исключением N. gonorrhoeae и N. meningitidis, для которых среду получали в соответствии с "M07-A8 Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard--Eighth Edition (2009)".
Заполнение чашек Петри:
1. Получали 100x исходные растворы лекарственного средства с каждым испытываемым соединением, как описано в таблице 1. Использовали 15-мл пробирку для центрифугирования, добавляли 100 мкл каждого исходного раствора лекарственного средства к 10 мл расплавленного агара (охлажденного до приблизительно 55°C на водяной бане). Смешивали, переворачивая пробирку 2-3x, затем выливали в индивидуально меченную 60×15 мм чашку Петри.
2. Рутинные испытываемые концентрации были: 0,002 мкг/мл - 16 мкг/мл (14 чашек).
3. Заполняли 4 чашки, не содержащие лекарственного средства: 2 в качестве положительного контроля, 2 в качестве аэробного контроля.
4. Давали чашкам высохнуть. Использовали в тот же день или хранили в течение ночи при комнатной температуре или хранили до 3 дней при 4°C.
5. Чашки соответственно метили, указывая концентрацию лекарственного средства и помещенный в них штамм.
Рост клеток, требующий поддержание анаэробных условий:
1. Всю работу с анаэробными бактериями осуществляли настолько быстро, насколько это было возможно; работу, осуществляемую в биологически безопасных камерах (т.е. аэробные условия), завершали менее чем за 30 минут до того, как клетки возвращали в анаэробные камеры.
2. Инкубацию анаэробных бактерий осуществляли с использованием GasPakTM камер. Для камер типа большого бокса (VWR 90003-636) требовалось 2 анаэробных саше (VWR 90003-642), тогда как для высоких цилиндрических камер (VWR 90003-602) требовалось только 1 саше.
Инокуляция чашек (осуществляли в биологически безопасной камере):
1. Наносили полоски каждого штамма на индивидуальные агаровые пластинки, как описано выше. Инкубировали в течение необходимого для этого времени и в необходимых для этого окружающих условиях (т. е. анаэробные, микроаэрофильные и т.п.).
2. Использовали метод непосредственного суспендирования колоний для суспендирования полных петель для пересевов свеженанесенных полосками клеток в приблизительно 4 мл 0,9% NaCl2 и подвергали вихревому перемешиванию.
3. Доводили суспензию до O.D.600 0,05 (5⋅107 кое/мл). Подвергали вихревому перемешиванию для смешивания.
4. Переносили приблизительно 0,2 мл приспособленных к условиям смешанных культур в 96-луночный планшет. Когда испытывали ≤5 штаммов, все штаммы выстраивали вместе в один ряд. При испытании >5 штаммов, осуществляли перенос штаммов в планшет не более 5 штаммов в одном ряду. Это было необходимо для нанесения на небольшие пластины.
5. Использовали многоканальное устройство для пипетирования, наносили пятнами по 0,002 мл каждого штамма из подготовленных 96-луночных планшетов на каждую пластину для испытания MIC. Это дало приблизительно 1⋅105 кое/пятно. При испытании C. difficile, происходило скопление штаммов, когда они вырастали, однако расстояние между многоканальным устройством для пипетирования в виде пятен было достаточным, чтобы скопления клеток не ухудшало результаты анализа.
а. Сначала инокулировали 2 пластины без лекарственного средства, тогда как другие 2 пластины без лекарственного средства инокулировали позднее после пластин для испытания MIC. Первые и последние служили в качестве контролей роста и инокуляции. Инкубировали пластину из каждой группы не содержащих лекарственное средство контролей в необходимых атмосферных условиях с MIC пластинами и одну в аэробных условиях для испытания на загрязнение аэробными бактериями. Аэробная культура была негативной для роста при работе со строго анаэробным или микроаэрофильным штаммом. Некоторый рост наблюдали с N. gonorrhoeae.
6. Давали инокуляту высохнуть (по возможности быстро), затем помещали верхней стороной вниз в GasPak с подходящим количеством саше и инкубировали.
7. Neisseria spp. инкубировали при 37°C в условиях 5% CO2 в течение 24 часов.
Определение MIC:
Исследовали испытываемые пластины после необходимого времени инкубации и считывали конечную точку MIC при концентрации, при которой происходило заметное снижение появления роста на испытываемой пластине по сравнению с появлением роста на пластинах с положительным контролем.
Разведения соединений для определения MIC методом разведения с использованием агаровой среды
**соединение, растворенное и разведенное 100% ДМСО
Протокол 4. Процедура определения MIC для Mycobacterium species
Вещества
Круглодонные 96-луночные микротитровальные планшеты (Costar 3788) или подобные
Пленочные уплотнения для планшетов (PerkinElmer, TopSeal-A #6005250 или подобные)
Middlebrook 7H10 бульон с 0,2% глицерина
Middlebrook 7H10 агар с 0,2% глицерина
Middlebrook OADC Обогащение
Получение инокулята для M. tuberculosis:
1. Использовали полученный замороженный исходный раствор M. Tuberculosis, который хранился при -700C. M. Tuberculosis выращивали в 7H10 бульоне + 10% OADC, затем замораживали при концентрации 100 Klett или 5⋅107 кое/мл,
2. Получали 1:20 разведение, беря 1 мл замороженного исходного раствора и добавляя его к 19 мл 7H10 бульона + 10% OADC (конечная концентрация 2,5⋅106 кое/мл).
3. Из этого разведения получали второе 1:20 разведение, брали 1 мл и добавляли к 19 мл свежего бульона. Это был конечный инокулят для добавления в 96-луночные планшеты.
Получение инокулята для M. kansasii, M. avium, M. abscessus и Nocardia spc.:
1. Использовали полученный замороженный исходный раствор культуры или свежую культуру, выращенную в 7H10 бульоне при концентрации 10 Klett или 5⋅107/мл.
2. Получали 1:20 разведение, беря 1 мл замороженного исходного раствора и добавляя его к 19 мл 7H10 бульона (конечная концентрация 2,5⋅106 кое/мл).
3. Из этого разведения получали 1:20 разведение, брали 1 мл и добавляли к 19 мл свежего бульона (конечная суспензия).
Подготовка планшетов:
1. Меченные планшеты
2. Добавляли 50 мкл 7H10 бульона + 10% OADC во все лунки, используемые для определения MIC, с использованием многоканального электронного устройства для пипетирования.
3. Получали исходные растворы лекарственного средства (например, концентрация 1 мг/мл) для испытания.
4. Оттаивали и разбавляли замороженные исходные растворы с использованием 7H10 бульона + 10% OADC, с получением рабочего раствора 4x максимальной испытываемой концентрации (например, конечная концентрация 32 мкг/мл, самая высокая испытываемая концентрация была 8 мкг/мл). Разведения осуществляли из исходного раствора. Для начала испытания при концентрации 1 мкг/мл, лекарственные средства получали при 4 мкг/мл, таким образом, исходная концентрация была 1 мкг/мл. Брали 25 мкл 1 мг/мл исходного раствора и добавляли к 6,2 мл бульона. Все разведения лекарственных средств осуществляли в бульоне.
5. Добавляли 50 мкл 4x рабочего раствора в первую лунку обозначенного ряда. Продолжали для всех испытываемых соединений. С использованием многоканального электронного устройство для пипетирования осуществляли смешивание 4x и серийное разведение соединений во всех лунках вплоть до 11-й лунки. Оставшиеся 50 мкл сливали. Использовали 12-ю лунку в качестве положительного контроля.
6. Инкубировали планшеты при 37°C M. tuberculosis в течение приблизительно 18 дней; M. avium и M. kansasii в течение приблизительно 7 дней; Nocardia и M. abcessus в течение приблизительно 4 дней; с использованием пленочных уплотнений.
7. Считывали визуально и регистрировали результаты. Регистрировали MIC как наименьшую концентрацию лекарственного средства, где не наблюдали никакого роста (оптическая чистота в лунке).
Протокол 5. Протокол MIC анализа со смещением сыворотки для Mycobacterium tuberculosis
Материалы и реагенты:
Costar #3904 96-луночные микротитровальные планшеты с черными стенками и плоским дном
Middlebrook 7H9 бульон (BD271310) с 0,2% глицерина
Middlebrook OADC Обогащение
Фетальная бычья сыворотка
Каталаза (Sigma C1345)
Декстроза
NaCl2
Система инокуляции BBL Prompt(Fisher b26306)
Агаровые пластинки (Middlebrook 7H11 с 0,2% глицерина и OADC обогащением) с бактериями, посеянными штрихом для разделения колоний
Стерильный ДМСО
Получение среды:
1. Для MIC анализов со смещением сыворотки необходимо три разных среды, которые все имели основу 7H9+0,2% глицерина. Важно, чтобы все среды и дополнения были стерилизованы до осуществления MIC анализов.
2. Получали все среды, указанные ниже, и инокулировали, как описано в следующем разделе. Испытывали все соединения против Mtb с использованием каждой среды.
а. 7H9+0,2% глицерина + 10% OADC (“стандартная” MIC среда).
b. 7H9+0,2% глицерина + 2 г/л декстрозы + 0,85 г/л NaCl + 0,003 г/л каталазы (0% FBS).
с. 2x 7H9+0,2% глицерина + 2 г/л декстрозы + 0,85 г/л NaCl + 0,003 г/л каталазы объединяли с равным объемом фетальной бычьей сыворотки (50% FBS).
Получение инокулята:
1. С использованием BBL Prompt, отбирали 5-10 четко разделенных колоний и инокулировали 1 мл стерильного физиологического раствора, который был в наборе. Типично, планшеты были двух-трехнедельными, когда их использовали для этого анализа, из-за медленного роста этого организма в культуре.
2. Подвергали вихревому перемешиванию, затем обрабатывали ультразвуком на водяной бане в течение 30 сек с получением суспензии приблизительно 108 клеток/мл. Действительная плотность может быть подтверждена путем выделения разведений этой суспензии.
3. Получали инокулят в каждом из трех составов среды путем разведения BBL Prompt суспензии 1/200 (например: переносили 0,2 мл клеток в 40 мл среды) с получением начальной плотности клеток приблизительно 106 клеток/мл.
4. Использовали 100 мкл клеток (приблизительно 5⋅104 клеток) для инокуляции каждой лунки микротитровального планшета, содержащей 1 мкл лекарственного средства в ДМСО (смотрите ниже).
Разведения лекарственного средства, инокуляция, определение MIC:
1. Контрольные исходные растворы лекарственных средств Изониазид и Новобиоцин получали при 10 мМ в 100% ДМСО, тогда как Ципрофлоксацин и Рифампин получали при 1 мМ в 50% ДМСО и 100% ДМСО, соответственно. Получение разведений - распределяли 100 мкл исходного раствора в первой колонке 96-луночного планшета. Получали 11-ступенчатые 2-кратные серийные разведения по всему ряду для каждого соединения путем переноса 50 мкл из колонки 1 в 50 мкл ДМСО в колонке 2. Продолжали переносить 50 мкл из колонки 2 вплоть до колонки 11 при смешивании и замене насадок для каждой колонки. Оставляли колонку 12 только с ДМСО в качестве контроля.
2. Переносили 1 мкл каждого разведения в пустую лунку на микротитровальном планшете перед добавлением 100 мкл клеток. Исходная концентрация Изониазида и Новобиоцина была 100 мкМ после разведения в среде + клетки; исходная концентрация Ципрофлоксацина и Рифампина была 10 мкМ после разведения в среде + клетки. Концентрации соединения снижали в 2x этапа перемещения по рядам микротитровального планшета. Все MIC анализы осуществляли в двух повторах в условиях каждой из трех сред.
3. Испытываемые группы соединений типично использовали при 10 мМ и в объеме 50 мкл.
4. Использовали многоканальное устройство для пипетирования, брали весь объем из каждой колонки исходного планшета и переносили в первую колонку нового 96-луночного микротитровального планшета. Повторяли для каждой колонки соединений на исходном планшете, перенося в колонку 1 нового 96-луночного планшета.
5. Как описано выше для контрольных соединений, получали 2-кратные 11-точечные разведения каждого соединения с использованием ДМСО в качестве разбавителя. В любом случае, оставляли колонку 12 как содержащую только ДМСО для контроля. После завершения всех разведений снова переносили 1 мкл каждого разведения в пустую лунку на микротитровальном планшете перед добавлением 100 мкл клеток, как делали для контрольных соединений.
6. Все лунки инокулировали 100 мкл разбавленной клеточной суспензии (смотрите выше).
7. После добавления инокулята смешивали содержимое планшетов путем осторожного постукивания по стенкам планшета.
8. Планшеты инкубировали в увлажненной 37°C камере в течение 9 дней.
9. Через 9 дней добавляли 25 мкл 0,01% стерильного ресазурина к каждой лунке. Измеряли фоновое значение флуоресценции при длине волны возбуждения 492 нм и длине волны эмиссии 595 нм и возвращали планшет в инкубатор еще на 24 часа.
Через 24 часа флуоресценцию в каждой лунке измеряли при длине волны возбуждения 492 нм и длине волны эмиссии 595 нм.
Процент ингибирования определенным соединением рассчитывали следующим образом: Процент ингибирования=100-([флуоресценция лунки-среднее фоновое значение флуоресценции]/[ДМСО контроль-среднее фоновое значение флуоресценции]×100). MICs рассчитывали для всех трех условий среды как наименьшую концентрацию соединения, которая ингибировала сигнал восстановления ресазурина (‘%-ингибирования’) ≥70% при данных условиях среды.
Таблица 2 представляет результаты MIC анализа для выборочных соединений по настоящему изобретению.
В таблице 2 и в следующих далее таблицах и Примерах, “Соединение 12” соответствует 1-этил-3-[5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевине, а “Соединение 13” относится к мезилатной соли соединения 12. Подобным образом, “Соединение 23” соответствует 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевине, а “Соединение 23A” относится к мезилатной соли соединения 23. Это те же самые номера, которые использовали для идентификации указанных соединений и солей в примерах выше.
MIC значения для выборочных соединений
Таблица 3 представляет результаты MIC90 анализа для выборочных соединений по настоящему изобретению.
MIC90 значения для выборочных соединений с группами грамположительных, грамотрицательных и анаэробных патогенов
Таблица 3A также представляет результаты MIC анализов для выборочных соединений по настоящему изобретению. В таблице 3A “Соединение 23A” соответствует 1-этил-3-[6-фтор-5-[2-(1-гидрокси-1-метил-этил)пиримидин-5-ил]-7-[(2R)-тетрагидрофуран-2-ил]-1H-бензимидазол-2-ил]мочевине в форме соли метансульфоновой кислоты (23A). Подобным образом, “Соединение W” соответствует динатрий (R)-2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1H-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан-2-илфосфату (W). Это те же самые номера, которые использовали для идентификации указанных соединений в Примерах синтеза выше.
MIC значения для выборочных соединений
2Сконструирован Vertex
3Coli Genetic Stock Center
4все tolC конструкции представляют собой tolC::Tn10, образованные из CAG12184 (Coli Genetic Stock Center)
В таблице 4 ниже термин “CMI” означает The Clinical Microbiology Institute, расположенный в Вилсонвилле, Орегон.
Анаэробные организмы, используемые для получения MIC90 данных
В таблице 5 ниже, термин “JMI” означает The Jones Microbiology Institute, расположенный в Норт Либерти, Айова.
Грамположительные и грамотрицательные организмы, используемые для получения MIC90 данных
Пример 32
Мышиная модель почечной инфекции S. aureus
Животные: самок CD-1 мышей (возраст 8-10 недель; 6/группа) получали от Charles River Laboratories, и животных размещали и содержали в соответствии с Руководством по содержанию и использованию экспериментальных животных (the Guide to the Care and Use of Experimental Animals).
Бактериальный штамм и исходные растворы
Метициллин-чувствительный S. aureus (MSSA) штамм ATCC 29213 получали от American Type Culture Collection. Для получения исходных растворов для испытаний на животных S. aureus высевали на Mueller Hinton агаровые пластинки и инкубировали при 37°C в течение ночи. 3-4 колонии из пластинки использовали для инокуляции 5 мл Mueller Hinton Бульона (MHB), который инкубировали в течение 8 часов при 37°C при встряхивании при 300 об/мин. 5 мл 8-часовой культуры разводили 20-кратно до 100 мл и инкубировали в течение ночи (12-14 часов). Бактерии осаждали центрифугированием в течение 20 минут при 3000х и промывали два раза фосфатно-буферным солевым раствором (PBS), содержащим 0,5% Бычьего Сывороточного Альбумина (BSA). Аликвоты (1 мл), содержащие приблизительно 1⋅1010кое в PBS/20% глицерина, замораживали и хранили при -80°C до дня их использования. Титры исходного раствора подтверждали путем серийного разведения и посева на Mueller Hinton агаровые пластинки.
Мышиная модель почечной инфекции S. aureus
Перед инокуляцией бактериальные исходные растворы оттаивали и промывали один раз при помощи PBS/BSA. Исходные растворы затем разбавляли при помощи PBS/BSA до конечной концентрации 1⋅109 кое/мл с получением инокулята приблизительно 1-2⋅108кое на мышь в объеме 100 мкл. Этот инокулят, как было обнаружено, является оптимальным в предварительных экспериментах, в которых использовали контрольные вызывающие заражение дозы 106-109 S. aureus организмов/мышь для установления нагрузки приблизительно 106 кое/почки через 26 часов после инфицирования без смертности (данные не показаны). В этих предварительных испытаниях вызывающие заражение дозы S. aureus меньше чем 107 кое/мышь вызывали неустойчивую инфекцию или не вызывали никаких хронических инфекций, тогда как доза 109 кое/мышь приводила к быстрому заболеванию и смертности большого процента мышей. Инъекции вводили внутривенно через хвостовую вену с использованием стерильной иглы размера 30. После завершения инфицирования мышей бактериальные исходные растворы, используемые для заражения мышей, высевали и подсчитывали для контроля концентрации инокулята.
Для определения бактериальной нагрузки в указанные моменты времени после инфицирования (обычно 2-26 часов), мышей умерщвляли и почки асептически извлекали и помещали в стерильный PBS/BSA (5 мл/пара почек). Почки гомогенизировали в стерильных условиях с использованием ручного гомогенизатора (Powergen 125; Fisher Scientific). В процессе сбора и гомогенизации все образцы оставались на льду, и гомогенизатор тщательно промывали и стерилизовали перед каждым следующим образцом. Осуществляли серийные разведения гомогенатов в стерильном PBS/BSA и высевали на MH агар для подсчета бактерий на пару почек.
Соединение 23A снижает нагрузки S. aureus в мышиной модели почечной инфекции S. aureus
CD-1 мышей (6/группа) инфицировали внутривенно S. aureus (ATCC 29213) при 2⋅108 кое/мышь. Через два часа после инфицирования мышей обрабатывали вводили перорально через зонд носитель (10% VitE TPGS) при 10 мл/кг или Соединение 23A при 10, 30, 100 мг/кг. 30-минутную и 6-часовую группы обработки обрабатывали один раз, тогда как для 24-часовой группы осуществляли вторую обработку через 10 часов после первой дозы. После продления периода времени после обработки (30 минут, 6 часов или 24 часа) мышей умерщвляли и почки извлекали, гомогенизировали и высевали на чашки для количественного определения нагрузок S. aureus. Рассчитывали нагрузки из пары почек для каждой мыши и среднее значение для каждой группы мышей.
Результаты: Перорально вводимое Соединение 23A показало in vivo эффективность против экспериментально-индуцированной почечной MSSA (SA 29213) инфекции. Через 30 минут после начальной обработки не было никакой разницы в нагрузке на почки между обработанными соединением и носителем мышами. 30 мин группа обработки носителем служила в качестве раннего контроля для сравнения эффектов соединения в более поздние моменты времени. Через 6 часов после первой дозы все обработки соединением снижали бактериальные нагрузки в почках на 0,4-0,6 log против согласующихся во времени показателей в контрольной группе введения носителя. Кроме того, Соединение 23A, вводимое при 30 и 100 мг/кг, обеспечивало 0,3-0,4 log снижение против 30-мин раннего контроля.
После периода обработки 24 час (который включал вторую дозу обработки, вводимую на момент времени 10 час), снижение бактериальной плотности по сравнению с контрольной группой введения носителя с согласованием во времени наблюдали для всех групп обработки. Соединение 23A, вводимое при 30 и 100 мг/кг BID, обеспечивало 2,8-2,9 log уменьшение, тогда как доза 10 мг/кг Соединения 23A два раза в день была более вариабельной и обеспечивала 1,8 log уменьшение против 24-часового контроля обработки носителем. Кроме того, Соединение 23A, вводимое при 30 и 100 мг/кг, показало приблизительно 1,5 log уменьшения против 30-мин контроля обработки носителем, что является показателем бактерицидной активности.
Обобщая вышесказанное, и как показано выше в таблице 6, введение два раза в день 30 и 100 мг/кг Соединения 23A уменьшало бактериальный рост метициллин-чувствительного S. aureus (MSSA) штамма ATCC 29213 по сравнению с 30 мин контрольной группой при оценке на оба момента времени 6 часов и 24 часа, тогда как обработка 10 мг/кг Соединения 23A ограничивала бактериальный рост на момент времени 6 часов, но была менее эффективной по сравнению с другими группами обработки на момент времени 24 часа.
Одна доза Соединения 23A снижает нагрузки S. aureus в мышиной модели почечной инфекции S. aureus
CD-1 мышей (6/группа) инфицировали внутривенно S. aureus (ATCC 29213) при 2⋅108 кое/мышь. Через 2 часа одну группу мышей (ранний контроль (EC)) умерщвляли и почки извлекали, гомогенизировали и высевали на чашки для количественного определения нагрузок S. aureus. Дополнительные группы инфицированных мышей обрабатывали путем перорального введения через зонд носителя при 10 мл/кг (10% VitE TPGS; поздний контроль, ЖХ), Соединения 23A при 10, 30, 60, 100 мг/кг. Через 24 часа группы обработанных мышей умерщвляли и почки извлекали, гомогенизировали и высевали на чашки для количественного определения нагрузок S. aureus. Нагрузки из пар почек для каждой мыши и среднее значение для каждой группы мышей суммировали.
Результаты: Обобщая вышесказанное, и как показано выше в таблице 7, одна пероральная доза соединения 23A показала in vivo эффективность против экспериментально-индуцированной почечной MSSA (SA 29213) инфекции. Через 24 часа все обработки показали снижение бактериальной плотности по сравнению с согласующимися во времени показателями в контрольной группе введения носителя. Соединение 23A продемонстрировало уменьшения, которые были дозозависимыми, 1,9, 2,3, 3,1 и 3,3 log уменьшения против контрольной группы введения носителя при введении доз 10, 30, 60 или 100 мг/кг. Кроме того, дозы 60 и 100 мг/кг Соединения 23A снижали бактериальные нагрузки против раннего контроля на 1,2-1,4 log, и это говорит о том, что Соединение 23A обладает бактерицидной активностью.
Одна доза Соединения W снижает бактериальные нагрузки в мышиной модели почечной инфекции MSSA
CD-1 мышей (8/группа) инфицировали внутривенно S. aureus (ATCC 29213) при 2⋅108 кое/мышь. Через 2 часа одну группу мышей (ранний контроль (EC)) умерщвляли и почки извлекали, гомогенизировали и высевали на чашки для количественного определения нагрузок S. aureus. Дополнительные группы инфицированных мышей обрабатывали путем перорального введения через зонд носителя при 10 мл/кг (вода; поздний контроль, ЖХ), Соединение W вводили при номинальных уровнях доз 16, 49, 99 или 166 мг/кг, которые, как ожидалось, должны доставлять 10, 30, 60 или 100 мг/кг соединения 23A, дозовые эквиваленты активной группы при полной конверсии при 10, 30, 60, 100 мг/кг. Через 24 часа группы обработанных мышей умерщвляли и почки извлекали, гомогенизировали и высевали на чашки для количественного определения нагрузок S. aureus. Нагрузки из пар почек для каждой мыши и среднее значение для каждой группы мышей суммировали.
Результаты: Обобщая вышесказанное, и как показано выше в таблице 8, одна пероральная доза соединения W показала in vivo эффективность против экспериментально-индуцированной почечной MSSA (SA 29213) инфекции. Через 24 часа все обработки показали снижение бактериальной плотности по сравнению с согласующимися во времени показателями в контрольной группе введения носителя. Соединение W продемонстрировало уменьшения, которые были дозозависимыми, 1,9, 2,3, 2,7, 2,6 и 3,0 log уменьшения против контрольной группы введения носителя при введении при 16, 49, 99 и 166 мг/кг, что обеспечивало эквивалентное воздействие 10, 30, 60 или 100 мг/кг соединения 24. Кроме того, дозы 16, 49, 99 и 166 мг/кг Соединения W снижали бактериальные нагрузки против раннего контроля на 0,7-1,5 log, и это говорит о том, что Соединение 23A обладает бактерицидной активностью.
Пример 33
Нейтропеническая крысиная модель инфекции бедренной области
Животные: Свободных от специфического патогена самцов крыс Sprague Dawley с массой тела между 76-100 грамм получали от Charles River Laboratories, Inc. (Wilmington, MA) и использовали в этом эксперименте. Животным давали привыкнуть к окружающим условиям минимум в течение семи (7) дней до начала исследования.
Бактерии: Использовали метициллин-чувствительный Staphylococcus aureus(MSSA) ATCC штамм 29213 для in vivo экспериментов. Испытываемый изолят субкультивировали два раза на стандартной микробиологической агаровой среде (содержащий триптиказу соевый агар с 5% овечьей крови). Второй пересев осуществляли меньше чем за 24 часа до использования в получении инокулята для модели инфекции бедренной области.
Нейтропеническая крысиная модель инфекции бедренной области: Для индукции нейтропении крыс обрабатывали иммуносупрессантом циклофосфамидом при 150 мг/кг, вводили в объеме 1 мл путем интраперитонеальной (IP) инъекции за три дня до инфицирования. Крыс инфицировали путем внутримышечной (IM) 0,2 мл инъекции в обе задние бедренные области с использованием суспензии 107 кое/мл метициллин-чувствительного Staphylococcus aureus 29213 в изотоническом растворе. Через увеличивающиеся промежутки времени (2-26 часов) обе задние бедренные области каждого животного извлекали, промывали стерильным физиологическим раствором, взвешивали, затем помещали в 50 мл стерильного изотонического раствора и помещали на мокрый лед до гомогенизации. Приблизительно половину общего объема гомогенизированного образца пропускали через фильтр с крупными порами (для удаления хряща и больших агломерированных кусков ткани), разбавляли в физиологическом растворе и культивировали на пластинах с агаровой средой (содержащий трипсиказу соевый агар с 5% овечьей крови). Все пластины с культурой инкубировали при приблизительно 37°C в течение 18-24 часов. Колониеобразующие единицы подсчитывали (в кое/мл гомогената) и рассчитывали среднее значение для каждой группы обработки и контроля. Типично, каждая группа имела n=6; каждую бедренную область рассматривали как дискретное количество. Среднее значение кое/мл на группу сравнивали с начальной бактериальной плотностью по прошествии 2 часов (ранний контроль) или с согласующимися во времени показателями в контрольной группе введения носителя (поздний контроль; ЖХ), которые получали одновременно.
Соединение 23А демонстрирует дозозависимое и зависимое от времени снижение нагрузок Staphylococcus aureus в бедренной области нейтропенических крыс
Нейтропенических крыс (3/группа) инфицировали путем внутримышечного (IM) введения S. aureus (ATCC 29213) при приблизительно 2⋅106 кое/бедро. Через 2 часа одну группу крыс (ранний контроль (EC) умерщвляли и бедренные области отделяли, гомогенизировали и высевали на чашки для количественного определения нагрузки S. aureus. Дополнительных инфицированных крыс обрабатывали путем перорального введения через зонд носителя при 10 мл/кг (20% Кавитрон/1% HPMCAS-MG; поздний контроль, ЖХ) или Соединения 23A при 10, 30, 60 мг/кг. 8-час группы обработки получали одну обработку (QD), и их умерщвляли и бедренные области отделяли для определения КОЕ через 8 часов после обработки (QD), тогда как 24-час группы обработки получали 2 дозы с интервалом 12 часов (q12h), и животных умерщвляли и бедренные области отделяли через 24 часа после обработки. Определяли нагрузки в каждой индивидуальной бедренной области, и кое/мл и среднее значение от группы из 3 крыс суммировали.
Результаты: Как показано выше в таблице 8, перорально вводимое Соединение 23A показало in vivo эффективность против MSSA (SA 29213). Через 8 часов после введения первой дозы все группы имели уменьшение нагрузок по сравнению с согласующимся во времени контролем, приблизительно 1,3 log уменьшение для Соединени 23A при 10 мг/кг и приблизительно 2 log уменьшение для Соединени 23A при 30 и 60 мг/кг. По сравнению с ранним контролем, Соединение 23A при 10 мг/кг сдерживало бактериальный рост SA 29213 по меньшей мере до точки остановки роста, тогда как Соединение 23A при 60 и 100 мг/кг слегка снижало бактериальную нагрузку на приблизительно 0,5-0,6 log.
Через 24 часа и после второй дозы обработки, введенной с интервалом в 12 часов, уменьшение приблизительно 2,4-2,8 log бактериальной плотности по сравнению с поздним контролем наблюдали для всех групп обработки. По сравнению с ранним контролем приблизительно 0,8 log уменьшение наблюдали для Соединения 23A при 10 мг/кг, тогда как Соединение 23A при 30 и 60 мг/кг обеспечивало уменьшения приблизительно 1,1-1,2 log.
Соединение 13 демонстрирует дозозависимое и зависимое от времени снижение нагрузок Staphylococcus aureus в бедренной области нейтропенических крыс
Нейтропенических крыс (3/группа) инфицировали путем внутримышечного (IM) введения S. aureus (ATCC 29213) при приблизительно 2⋅106 кое/бедро. Через 2 часа одну группу крыс (ранний контроль (EC) умерщвляли и бедренные области отделяли, гомогенизировали и высевали на чашки для количественного определения нагрузки S. aureus. Дополнительных инфицированных крыс обрабатывали путем перорального введения через зонд носителя при 10 мл/кг (10% Витамин E/TPGS; поздний контроль, ЖХ) или Соединения 13 при 30, 60, 100 мг/кг. 8-час группы обработки получали одну обработку (QD), и их умерщвляли и бедренные области отделяли для определения КОЕ через 8 часов после обработки (QD), тогда как 24-час группы обработки получали 2 дозы с интервалом 12 часов (q12h), и животных умерщвляли и бедренные области отделяли через 24 часа после обработки. Определяли нагрузки в каждой индивидуальной бедренной области, и кое/индивидуальное бедренная область и средние значения от группы из 3 крыс суммировали для каждой группы.
Результаты: Как показано выше в таблице 9, перорально вводимое Соединение 13 показало in vivo эффективность против MSSA (SA 29213). Разницу уровней антибактериальной активности наблюдали между тремя группами обработки. Через 8 часов после первой дозы все группы имели уменьшение нагрузок по сравнению с согласующимся во времени контролем, приблизительно 1,5 log уменьшение для Соединения 13 при 10 мг/кг, и приблизительно 1,7 и 1,8 log уменьшение для Соединения 13 при 60 и 100 мг/кг. Через 8 часов после первой дозы Соединение 13 при 10 мг/кг сдерживало бактериальный рост SA 29213 по меньшей мере до точки остановки роста, тогда как Соединение 13 при 60 и 100 мг/кг слегка снижало бактериальную нагрузку против раннего контроля на приблизительно 0,4 и приблизительно 0,5 log, соответственно. Через 24 часа и после второй дозы обработки, введенной с интервалом в 12 часов, снижение приблизительно 1 log бактериальной плотности по сравнению с ранним контролем показало Соединение 13 при 60 и 100 мг/кг. В отличие от этого, Соединение 13, вводимое при 30 мг/кг, оказалось неэффективным с варьирующимися уровнями кое в среднем 0,3 log больше раннего контроля. Однако все уровни доз снижали бактериальную плотность в сравнении с поздним контролем. Уменьшение приблизительно 1,1 log наблюдали для группы обработки 10 мг/кг, тогда как уменьшение 2,85 и приблизительно 3 log наблюдали для уровней доз 60 и 100 мг/кг.
Обобщая вышесказанное, q12h дозы 60 и 100 мг/кг Соединения 13 уменьшали бактериальный рост SA 29213 по сравнению с исходной контрольной группой в точках оценки как 8 часов, так и 24 часа, тогда как обработка при дозе 30 мг/кг ограничивала бактериальный рост на момент времени 8 часов, но была менее эффективной по сравнению с другими группами обработки на момент времени 24 часов.
Пример 34
Семидневное исследование токсичности и токсикокинетики при пероральном введении (через зонд) крысам
Целью этого испытания было: 1) определение потенциальной токсичности Соединения 13 и Соединения 23A при пероральном введении через зонд самцам крысы в течение 7 последовательных дней, и 2) определение токсикокинетики Соединения 13 и Соединения 23A после первой и седьмой дозы.
Животные
Виды , источник, история и обоснование
Crl:CD(SD) крыс получали от Charles River Laboratories of Stone Ridge, NY. Животные были выращены в лаборатории и ранее не подвергались экспериментам. Крысы были выбраны, поскольку этот вид традиционно используют для неклинических оценок токсичности.
Количество, пол, возраст и диапазон массы тела
Были заказаны сорок крыс (20 неканюлированных самцов и 20 самцов с канюлями в яремной вене). Из этих животных использовали 15 неканюлированных самцов и 15 канюлированных самцов. Животные были настолько одинаковы по возрасту, насколько это было возможно. Использовали крыс от препубертального возраста до молодых взрослых особей, приблизительно 9-недельного возраста на момент начала обработки. Рассчитанная поставщиком дата их рождения была зафиксирована в материалах исследования. Диапазон массы тела для животных на момент распределения по группам составлял 218,5-306,3 г.
План исследования
Крыс распределяли, как показано в таблица 10 ниже. Животные получал испытываемое вещество или носитель путем перорального введения через зонд в течение 7 последовательных дней, и их умерщвляли в день после завершения введения средства. Первый день введения средства обозначали как День 1 исследования. Животных оценивали на изменения в клинических признаков, массы тела и других параметров, как описано ниже.
Распределение по группам и уровни доз
Путь/Доза
Носитель и испытываемое вещество вводили перорально через зонд один раз в день в течение 7 последовательных дней при дозе объемом 10 мл/кг массы тела для Группы A и Групп B-D, соответственно. Испытываемое вещество и носитель вводили перорально через зонд два раза в день, приблизительно с 8 часовым интервалом, в течение 7 последовательных дней при дозе объемом 10 мл/кг массы тела для Группы E и Группы F, соответственно. Действительный объем, вводимый каждому животному, рассчитывали и корректировали на основании последних показателей массы тела каждого животного.
Прижизненные наблюдения и измерения
Наблюдения
Животных наблюдали на жизнеспособность по меньшей мере один раз утром и один раз после полудня, по меньшей мере с 4-часовым интервалом, на протяжении всего испытания. В период обработки ежедневно наблюдали животных в клетке и регистрировали наблюдения перед введением дозы и после введения дозы (только после первой дозы). Наблюдения после введения дозы в процессе обработки осуществляли в следующие моменты времени на основании Cmax/Tmax для двух соединений из предыдущих исследований:
1 час после введения дозы для Групп A-F.
Одно наблюдение поведения животных в клетке осуществляли в день аутопсии.
Незапланированные наблюдения
Любые признаки, наблюдаемые в другое время, в отличие от запланированных для наблюдения моментов времени, должны были быть регистрированы как незапланированное наблюдение или в Provantis; однако никаких отклонений не наблюдали на протяжении всего испытания. Provantis представляет собой электронную систему сбора, обработки и представления данных, которую традиционно используют в данной области.
Показатели массы тела
До начала обработки массу тела животных измеряли для рандомизации в День 1. В процессе обработки массу тела измеряли в День 1 и День 7. Кроме того, массу тела натощак измеряли перед аутопсией для расчета отношений орган/масса тела.
Потребление пищи
На протяжении всего испытания потребление пищи измеряли ежедневно, начиная за 3 дня до начала введения средства.
Оценка клинической патологии
Образцы крови для определения гематологии, коагуляции и химического состава сыворотки собирали у всех животных из ретро-орбитального сплетения (под CO2/O2 анестезией, для основного исследования животных) или из яремной вены через канюлю (для токсикокинетических животных) перед аутопсией. Из-за присутствия остаточного гепарина, используемого чтобы поддерживать канюли доступными для токсикокинетических животных, образцы для определения коагуляции от этих крыс не были пригодны для анализа. Животных содержали без пищи в течение ночи перед сбором крови. В день сбора крови для анализов на клиническую патологию животных не подвергали аутопсии до тех пор, пока после взятия крови образцы не были признаны приемлемыми группой клинической патологии.
Гематологи я
Подходящее количество крови собирали в EDTA-содержащие пробирки. Образцы цельной крови анализировали для определения параметров указанных ниже в таблице 11.
Параметры цельной крови
Коагуляция
Подходящее количество крови собирали в пробирки, содержащие цитрат натрия, и затем центрифугировали для получения плазмы для определения протромбинового времени (PT) и активированного частичного тромбопластинового времени (APTT).
Химический состав сыворотки
Подходящее количество крови собирали в пробирки без антикоагулянта. Пробирки оставляли для свертывания образца и затем центрифугировали для получения сыворотки. Сыворотку анализировали для определения параметров, указанных ниже в таблице 12.
Параметры сыворотки
Токсикокинетика
В 1-й и 7-й День введения средства, образцы крови (приблизительно 0,5 мл/образец) собирали из яремной вены через канюлю у всех токсикокинетических животных в моменты времени, указанные ниже, в K3EDTA-содержащие пробирки. Токсикокинетические животные из контрольной группы (Группа F) кровь брали только по одному разу в день на момент времени 1 час (после введения первой дозы в этот день). Перед каждым сбором крови, небольшой образец крови (с гепариновым блокирующим раствором) удаляли из канюли и выбрасывали. Новый шприц вставляли в канюлю и брали определенный протоколом образец. Шприц с образцом крови удаляли и новый шприц с физиологическим раствором подсоединяли к канюле. Объем крови вытесняли объемом физиологического раствора и затем блокирующий раствор вводили в канюлю. Каждое животное возвращали в его клетку до следующего времени взятия крови.
Все образцы, собранные в процессе этого исследования, помещали в меченые контейнеры. Каждая метка содержала следующую информацию: 1) Номер испытания, 2) номер животного, 3) временной интервал сбора крови, 4) группа и пол животного, и 5) Дата взятия образца.
Образцы крови незамедлительно смешивали, переворачивая пробирки, затем помещали на мокрый лед и центрифугировали в охлажденном состоянии (приблизительно 1500g, приблизительно 10 минут, приблизительно 5°C) для получения плазмы. Плазму распределяли в 96-луночные 1,4-мл полипропиленовые пробирки с прокалываемым TPE колпачками с кластерной сертифицированной РНКазой, без ДНКазы, в виде 2 аликвот и хранили замораженной (≤-70°C).
Время взятия образцов
2Только после 1 дня введения.
3Получены от Групп E и F перед PM введением дозы.
4Только после 7 дня введения.
Окончание обработки
Никакое животное не считалось умирающим в процессе испытания. Всех испытываемых животных умерщвляли и подвергали аутопсии после соответствующих дней обработки, предписанных протоколом. Всех животных умерщвляли путем обескровливания (разрезание абдоминальной аорты под сильной CO2/O2 анестезией).
Аутопси я
Аутопсию со сбором ткани осуществляли на всех животных, умерщвленных в процессе испытания. Аутопсия включала исследование:
скелета и мышечной/скелетной системы; всех внешних поверхностей и проходов;
полости черепа и внешней поверхности головного мозга;
шеи с соответствующими органами и тканями; и
торакальной, абдоминальной и тазовой полостей с их соответствующими органами и тканями.
Все отклонения описывали и регистрировали.
Масса органов
Для всех животных, умерщвленных в предусмотренное протоколом время проведения аутопсии, почки, печень и предстательную железу взвешивали. После взвешивания, отвешивали приблизительно 1 грамм образца печени и почки, переносили в Precellys 7-мл CK28 пробирки для гомогенизации тканей (Cat. No. 0904-01), быстро замораживали и анализировали.
Отношения орган/масса тела рассчитывали с использованием конечного значения массы тела натощак, полученного перед аутопсией.
Консервация тканей и сбор костного мозга
Ткани и органы, указанные ниже в таблице 14, собирали у всех животных, и они были законсервированы в 10% нейтрально-забуференном формалине, за исключением яичек, эпидидимиса и глаз. Яички, эпидидимис и глаза вместе со зрительным нервом фиксировали в Модифицированном Растворе Дэвидсона в течение приблизительно 24-48 часов, промывали водой и затем переносили в 10% нейтрально-забуференный формалин для хранения.
Сбор тканей
Гистопатологи я
Для всех животных, запланированных для конечной аутопсии, почки, печень и предстательную железу заделывали в парафин, секционировали и окрашивали гематоксилином и эозином для дальнейшего исследования методом световой микроскопии. Только для Групп A, D, E и F, остальные ткани, перечисленные выше, заделывали в парафин, секционировали и окрашивали гематоксилином и эозином для дальнейшего исследования методом световой микроскопии.
Статистический анализ
Если это является подходящим, цифровые данные, полученные для животных, оценивали статистически.
Для сравнительной статистики, Группу A (контрольная группа) сравнивали с Группами B и C (группы обработки, введение доз QD) и Группу F (контрольная группа, введение доз BID) сравнивали с Группой E (группа обработки, введение доз BID). Данные оценивали с использованием критерия Levene для однородности дисперсии и критерия Shapiro-Wilks для нормальности распределения, со значимостью при p ≤0,05. Данные, определенные как гомогенные и с нормальным распределением, оценивали при помощи дисперсионного анализа (ANOVA). Если ANOVA удостоверял значимость при p ≤0,05, осуществляли попарные сравнения каждой группы обработки с соответствующей контрольной группой с использованием параметрического критерия (критерий Dunnett) для определения статистической разницы (p ≤0,05). Данные, определенные как негомогенные или с ненормальным распределением, оценивали с использованием критерия Kruskal-Wallis для определения значимости группового фактора. Если значимость (p ≤0,05) существовала между группами, использовали непараметрический критерий (Wilcoxon и Bonferroni-Holm) для сравнения групп обработки с контрольной группой. Данные потребления пищи животными, где произошел разброс данных, были исключены из релевантного периода времени. Сравнительная статистика данных потребления пищи ограничивалась критерием Dunnett (параметрический). Не было статистических данных о потреблении пищи в дни, предшествующие испытанию (День 4 по День 1).
Результаты
Воздействия различных уровней введения Соединения 23A и Соединения 13 были дозозависимыми. Не наблюдали никаких неблагоприятных признаков или эффектов на среднюю массу тела у животных, которых обрабатывали либо Соединением 13, либо Соединением 23A. Среднее потребление пищи уменьшалось в различные периоды времени в процессе исследования у животных, которых обрабатывали один раз в день Соединением 13 (100 или 200 мг/кг) и два раза в день Соединением 23A (300 мг/кг). Однако, поскольку уменьшение потребления пищи никак не было связано с изменением массы тела в группах введения Соединения 13 и Соединения 23A, эти эффекты не рассматривали как неблагоприятные или биологически значимые. Средняя концентрация ионов кальция (CA) была статистически ниже, тогда как средние ALT и AST для групп крыс, которым вводили 300 мг/кг Соединения 23A два раза в день, были статистически выше по сравнению с контролями, которых обрабатывали два раза в день. Никакие связанные с испытываемым веществом гистопатологические изменения не были обнаружены у животных, получавших либо Соединение 13, либо Соединение 23A при любой схеме введения.
В рамках этого исследования и на основании отсутствия изменения массы тела, клинической патологии и гистопатологии, NOEL (уровень, не вызывающий видимого эффекта) для Соединения 13, которое вводили самцам крыс один раз в день в течение 7 дней перорально через зонд, составил 200 мг/кг (844 мкг⋅час/мл День 7 AUC), тогда как NOEL для Соединения 23A, которое вводили один раз в день, составил 100 мг/кг (82 мкг⋅час/мл AUC). NOAEL (уровень, не вызывающий видимого неблагоприятного эффекта) для Соединения 23A, которое вводили самцам крыс два раза в день в течение 7 дней перорально через зонд, составил 300 мг/кг (291 мкг⋅час/мл AUC).
Поэтому Соединения 13 и 23A не продемонстрировали неблагоприятные токсические эффекты в рамках настоящего исследования на уровне доз до 200 мг/кг/день и 600 мг/кг/день, соответственно.
Пример 35
Исследование токсичности и токсикокинетики у самцов обезьян Cynomolgus в рамках перорального введения
Целью испытания было 1) определение потенциальной токсичности соединения 23 при введении перорально через зонд самцам обезьян Cynomolgus в течение 7 последовательных дней; и 2) определение токсикокинетики соединения 23 после первой и седьмой дозы.
Животные
Виды, источник, история и обоснование
Обезьян Cynomolgus (Macaca Fascicularis) получали от Primus Bio-Resources Inc. of PinCourt, Quebec, Canada. Обезьяны Cynomolguss были выбраны, потому что они относятся к отличному от грызунов виду, которых традиционно используют для неклинической оценки токсичности.
Количество, пол, возраст и диапазон массы тела
В испытания использовали восемь (2 не используемых ранее в экспериментах и 6 ранее используемых в экспериментах) самцов. Животные представляли собой молодые взрослые особи и имели массу тела от 2 до 4 кг в день начала обработки.
План исследования
Животных распределяли, как показано в таблице 15 ниже. Животные получали Соединение 23 или носитель путем перорального введения через зонд один раз в день в течение 7 последовательных дней, и их умерщвляли в день после завершения введения средства. Первый день введения средства обозначали как День 1 исследования. Действительный объем, вводимый каждому животному, рассчитывали и корректировали на основании последних показателей массы тела каждого животного.
Распределение по группам и уровни доз
Прижизненные наблюдения и измерения
Наблюдения
Клинические признаки у находящихся в клетке животных (ухудшение здоровья, поведенческие изменения и т.п.) регистрировали по меньшей мере один раз в день в процессе испытания.
Масса тела
Массу тела регистрировали для всех животных перед распределением по группам и в Дни 1 (до начала обработки), 3 и 7, а также последний раз перед аутопсией (натощак).
Электрокардиография (ЭКГ)
Электрокардиограммы (биполярные отведения I, II и III и увеличенные однополярные отведения aVR, aVL и aVF) получали для всех обезьян один раз в период предварительной обработки и еще раз в День 7 (после завершения обработки).
Показания считывали для определения значительных изменений, которые указывают на сердечную электрическую дисфункцию. Были определены потенциально присутствующие отклонения, включающие частоту сердечных сокращений (отведение II), синусовый и антривентрикулярный ритм или проводимость. Измеряли частоту сердечных сокращений, PR интервал, QRS длительность, QT и QTc интервалы.
Токсикокинетика
Собирали серию из 7 образцов крови (приблизительно по 0,5 мл каждый) от каждой обезьяны в Дни 1 и 7 в следующие моменты времени: 30 минут до введения дозы и 2, 3, 6, 12 и 24 часов после введения дозы. Для этой цели у каждой обезьяны брали кровь при помощи венопункции и образцы собирали в пробирки, содержащие антикоагулянт K2EDTA. Пробирки помещали на мокрый лед до момента, когда они были готовы для обработки.
Клиническая патология
Лабораторные исследования (гематология, коагуляция, клиническая химия и анализ мочи) осуществляли для всех животных перед началом обработки и перед завершением периода обработки в День 8.
Образцы крови собирали при помощи венопункции после периода голодания в течение ночи, представляющего собой по меньшей мере 12-часовой период, но не более чем 20 часов. Мочу собирали у животных, содержавшихся без пищи и воды в течение ночи (по меньшей мере 12 часов, но не более чем 20 часов).
Гематология
Следующие параметры измеряли в образцах крови, собранных в EDTA антикоагулянт: количество эритроцитов, средний эритроцитарный гемоглобин (рассчитанный), гематокритное число (рассчитанное), среднее гематокритное число, гемоглобин, морфология клеток, количество лейкоцитов, количество тромбоцитов, лейкоцитный дифференциал (абсолютный), ретикулоциты (абсолютное количество и проценты) и средняя концентрация эристроцитарного гемоглобина (рассчитанная).
Коагуляция
Активированное частичное тромбопластиновое время и протромбиновое время измеряли в образцах крови, собранных в цитратный антикоагулянт.
Клиническая химия
Следующие параметры измеряли в образцах крови, собранных в пробирки, содержащие активатор свертывания: a/g отношение (рассчитанное), креатинин, аланинаминотрансфераза, глобулин (рассчитанный), альбумин, глюкоза, щелочная фосфатаза, фосфор (неорганический), аспарагинаминотрансфераза, калий, билирубин (общий), натрий, кальций, общее содержание белка, хлорид, триглицериды, холестерин (общий), мочевина, гаммаглутамилтрансфераза и сорбитдегидрогеназа.
Анализ мочи
Следующие параметры измеряли в образцах мочи: билирубин, белок, кровь, седиментационная микроскопия, цвет и внешний вид, удельный вес, глюкоза, уробилиноген, кетоны, объем и pH.
Завершение периода обработки
Всех животных умерщвляли после завершения периода обработки в День 8 после периода голодания в течение ночи. Обезьян предварительно анестезировали кетамином и затем умерщвляли путем внутривенной передозировки пентобарбитала натрия с последующим обескровливанием путем рассечения главных кровеносных сосудов.
Аутопсия
Аутопсию со сбором ткани осуществляли на всех животных, умерщвленных в процессе испытания. Аутопсия включала исследование:
скелета и мышечной/скелетной системы;
всех внешних поверхностей и проходов;
полости черепа и внешней поверхности головного мозга;
шеи с соответствующими органами и тканями; и
торакальной, абдоминальной и тазовой полостей с их соответствующими органами и тканями.
Все отклонения описывали и регистрировали.
Консервация тканей
После завершения общего исследования и взвешивания выборочных органов, ткани и органы сохраняли, как указано ниже в таблице 16. Нейтрально забуференный 10% формалин использовали для фиксации и консервации, если не указано иное.
Сохранение тканей и органов
b легкие заливали 10% нейтрально забуференным формалином, используемым для фиксации и консервации
c легкие взвешивали с трахеей
d использовали жидкость Боуина для фиксации и консервации
є исследовали микроскопически
Гистопатология
Для всех животных, все указанные выше ткани заделывали в парафин, секционировали и окрашивали гематоксилином и эозином и исследовали методом световой микроскопии.
Результаты
Воздействия различных уровней введения соединения 23 были дозозависимыми.
Не было никаких клинических признаков или изменений массы тела, параметров электрокардиографии, параметров клинической патологии или массы органов, которые можно было бы отнести за счет введения соединения 23 при дозах до 200 мг/кг/день. Подобным образом, не было никаких макроскопических или микроскопических изменений, которые явным образом были бы связаны с введением соединения 23 в дозах до 200 мг/кг/день. Уровень, не вызывающий видимого эффекта (NOEL) для Соединения 23 у самцов обезьян Cynomolguss был определен как 200 мг/кг/день.
Пример 36
ФАРМАКОКИНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ
Фармакокинетические параметры 4 выборочных соединений по настоящему изобретению определяли в экспериментах, описанных ниже. Использовали следующие общие аналитические процедуры и специальные протоколы экспериментов:
Общ ие аналитические процедуры
В фармакокинетических экспериментах, описанных ниже, использовали следующие общие аналитические процедуры:
Анализ образцов. Концентрации соединения 23 и Соединения W в плазме определяли с использованием метода высоко-эффективной жидкостной хроматографии/тандемной масс-спектррометрии (ВЭЖХ/MS/MS). До экстракции образцы плазмы разбавляли с использованием 2-, 4-, 5- или 10-кратных разведений чистой плазмы, по мере необходимости, в зависимости от уровня дозы или композиции. Соединение 23 и Соединение W вместе с внутренним стандартом (IS) экстрагировали из (разбавленной) плазмы, 100 мкл каждого, путем прямого осаждения белка ацетонитрилом (1:4 отношение плазма/ацетонитрил). После центрифугирования, экстракт супернатанта (10 мкл) впрыскивали в ЖХ/MS/MS систему. ВЭЖХ система включала Waters Xterra MS C18 колонку, 5 микрон, 2,1 мм в диаметре × 50 мм в длину, с использованием для градиентного элюирования подвижной фазы, состоящей из 0,1% муравьиной кислоты в воде или в ацетонитриле.
Детекцию аналитов осуществляли методом MS/MS с Химической Ионизацией Атмосферного Давления (APCI) в режиме многократного контроля реакции (MRM). Нижний предел количественного определения (LLOQ) составлял 1, 2, 4, 5, 10 или 20 нг/мл, в зависимости от коэффициента разбавления образца. Линейный диапазон анализа находился в пределах от 1 до 5000 нг/мл. Суточная и междусуточная точность оценки была в пределах 2% от номинального значения. Суточная и междусуточная изменчивость оценки составляла <10%.
Образцы вводимой в форме суспензии композиции соединения W анализировали методом ВЭЖХ/УФ после 10-кратного до 500- или 1000-кратного разведения с использованием смеси ДМСО:ацетонитрил:вода (33:33:33), в зависимости от уровня дозы или композиции. Образцы вводимой в форме раствора композиции соединения W анализировали методом ВЭЖХ/УФ после 10-кратного до 500- или 1000-кратного разведения с использованием смеси ДМСО:вода (50:50), в зависимости от уровня дозы или композиции.
Фармакокинетический анализ данных. Профили концентрация в плазме-время Соединения 23 и Соединения W анализировали некомпартментными фармакокинетическими методами с использованием программы WinNonlin® Professional Edition, Version 5.1.1 (Pharsight Corporation, Mountain View, CA).
Определяли ключевые фармакокинетические параметры, включая AUCall, AUCэкстрап., Cmax, tmax, Cl_obs, Vss_obs и t1/2.
Статистический анализ данных. Были рассчитаны описательные статистические данные концентраций в плазме и оценки фармакокинетических параметров, включая средние значения, стандартные отклонения (SD) и коэффициент вариаций (%CV), с использованием программы WinNonlin, Version 5.1.,1 или Microsoft Excel 2000.
Испытание перорального(PO) введения обезьянам
Самцам cynomolgus обезьян (n=3 на группу дозирования) вводили разовые номинальные PO дозы 3, 30 и 300 мг/кг соединения W через зонд. Соединение W было сформулировано в 0,5% MC (микрокристаллическая целлюлоза). Животные имели свободный доступ к пище и воде до и после введения дозы.
Образцы крови (приблизительно 0,25 мл каждый) собирали через катетер из сонной артерии перед введением дозы и в момент времени 0 (перед введением), 0,25, 0,5, 1, 2, 3, 4, 6, 8, 12, 24, 48 часов после введения дозы. Каждый образец крови собирали в пробирку, которая хранилась на мокром льду и содержала калий EDTA в качестве антикоагулянта. Плазму отделяли и хранили при приблизительно -70°C до использования в анализе.
Образцы плазмы анализировали с использованием метода жидкостной хроматографии/тандемной масс-спектрометрии (ЖХ/MS/MS) для определения концентраций соединения 23 и Соединения W с нижним пределом количественного определения (LLOQ) от 1 до 20 нг/мл, в зависимости от коэффициента разбавления образца. Данные концентрация в плазме vs. время подвергали некомпартментному фармакокинетическому (PK) анализу. Результаты этого анализа представлены в таблице 17.
Фармакокинетические данные испытания перорального введения обезьянам
Самцам cynomolgus обезьян (n=3 на группу дозирования) вводили разовую номинальную внутривенную (IV) болюсную дозу 1 мг/кг соединения W в яремную вену через канюлю. Соединение W было сформулировано в D5W (5% раствор декстрозы в воде). Животные имели свободный доступ к пище и воде до и после введения дозы.
Образцы крови (приблизительно 0,25 мл каждый) собирали через катетер из сонной артерии перед введением дозы и в момент времени 0 (до введения дозы), 5 мин, 10 мин, 0,25, 0,5, 1, 2, 3, 4, 6, 8, 12, 24, 48 часов после введения дозы. Каждый образец крови собирали в пробирку, которая хранилась на мокром льду и содержала калия EDTA в качестве антикоагулянта. Плазму отделяли и хранили при приблизительно -70°C до использования в анализе.
Образцы плазмы анализировали с использованием метода жидкостной хроматографии/тандемной масс-спектррометрии (ЖХ/MS/MS) для определения концентраций соединения 23 и Соединения W, с нижним пределом количественного определения (LLOQ) от 1 до 20 нг/мл, в зависимости от коэффициента разбавления образца. Данные концентрация в плазме vs. время подвергали некомпартментному фармакокинетическому (PK) анализу. Результаты этого анализа представлены в таблице 18.
Фармакокинетические данные испытания внутривенного введения обезьянам
Испытание перорального введения крысам
Группам самцов крыс Sprague Dawley (n=3 на группу дозирования) вводили разовые номинальные пероральные дозы 3, 10, 30, 300 мг/кг соединения W через зонд. Соединение W было сформулировано либо в 0,5% MC (микрокристаллическая целлюлоза), либо в 20% Каптисола, 1% HPMC-AS (ацетилсукцинат гидроксипропилметилцеллюлозы), 1% PVP (поливинилпирролидон). Животные имели свободный доступ к пище и воде до и после введения дозы. Образцы крови (приблизительно 0,25 мл каждый) собирали через катетер из сонной артерии перед введением дозы и в момент времени 0 (до введения дозы), 0,25, 0,5, 1, 1,5, 2, 4, 6, 8, 12, 24 часа после введения дозы. Каждый образец крови собирали в пробирку, которая хранилась на мокром льду и содержала калий EDTA в качестве антикоагулянта. Плазму отделяли и хранили при приблизительно -70°C до использования в анализе.
Образцы плазмы анализировали с использованием метода жидкостной хроматографии/тандемной масс-спектррометрии (ЖХ/MS/MS) для определения концентраций соединения 23 и Соединения W с нижним пределом количественного определения (LLOQ) от 1 до 20 нг/мл, в зависимости от коэффициента разбавления образца. Данные концентрация в плазме vs. время подвергали некомпартментному фармакокинетическому (PK) анализу. Результаты этого анализа представлены в таблице 19.
Фармакокинетические данные испытания перорального введения крысам
Испытание IV введения крысам
Группам самцов крыс Sprague Dawley (n=3 на группу дозирования) вводили разовые номинальные IV болюсные дозы 1 и 5 мг/кг соединения W в яремную вену через канюля. Соединение W было сформулировано в D5W. Животные имели свободный доступ к пище и воде до и после введения дозы. Образцы крови (приблизительно 0,25 мл каждый) собирали через катетер из сонной артерии перед введением дозы и в момент времени 0 (до введения дозы), 5 мин, 10 мин, 0,25, 0,5, 1, 1,5, 2, 4, 6, 8, 12, 24 часа после введения дозы. Каждый образец крови собирали в пробирку, которая хранилась на мокром льду и содержала калий EDTA в качестве антикоагулянта. Плазму отделяли и хранили при приблизительно -70°C до использования в анализе.
Образцы плазмы анализировали с использованием метода жидкостной хроматографии/тандемной масс-спектррометрии (ЖХ/MS/MS) для определения концентраций соединения 23 и Соединения W с нижним пределом количественного определения (LLOQ) от 1 до 20 нг/мл, в зависимости от коэффициента разбавления образца. Данные концентрация в плазме vs. время подвергали некомпартментному фармакокинетическому (PK) анализу. Результаты этого анализа представлены в таблице 20.
Фармакокинетические данные испытания внутривенного введения крысам
Испытание перорального введения мышам
Группам самцов CD-1 мышей (n=3 на группу дозирования) вводили разовые номинальные пероральные дозы 10, 30, 100 мг/кг соединения W через зонд. Соединение W было сформулировано в 0,5% MC. Животные имели свободный доступ к пище и воде до и после введения дозы. Образцы крови (приблизительно 0,025 мл каждый) собирали из поднижнечелюстной вены перед введением дозы и в момент времени 0 (до введения дозы), 0,25, 0,5, 1, 1,5, 2, 4, 6, 8, 12, 24 часа после введения дозы. Каждый образец крови собирали в пробирку, которая хранилась на мокром льду и содержала калий EDTA в качестве антикоагулянта. Плазму отделяли и хранили при приблизительно -70°C до использования в анализе.
Образцы плазмы анализировали с использованием метода жидкостной хроматографии/тандемной масс-спектррометрии (ЖХ/MS/MS) с нижним пределом количественного определения (LLOQ) от 1 до 20 нг/мл, в зависимости от коэффициента разбавления образца. Данные концентрация в плазме vs. время подвергали некомпартментному фармакокинетическому (PK) анализу. Результаты этого анализа представлены в таблице 21.
Фармакокинетические данные испытания перорального введения мышам
Исследования, описанные выше, демонстрируют, что Соединение W преобразуется in vivo в Соединение 23, по меньшей мере, у крыс, собак и обезьян.
Пример 37
ЭНЗИМОЛОГИЧЕСКИЕ ИССЛЕДОВАНИЯ
Активности ингибирования фермента выборочных соединений по настоящему изобретению определяли в экспериментах, описанных ниже:
АТФазный анализ ДНК гиразы
АТФ гидролизную активность ДНК гиразы S. aureus измеряли путем объединения продукции АДФ через пируваткиназа/лактатдегидрогеназу с окислением NADH. Этот способ был описан ранее (Tamura and Gellert, 1990, J. Biol. Chem., 265, 21342).
АТФазные анализы осуществляли при 30°C в забуференных растворах, содержащих 100 мМ ТРИС pH 7,6, 1,5 мМ MgCl2, 150 мМ KCl. Объединенная система содержит конечные концентрации 2,5 мМ фосфоенолпирувата, 200 мкМ никотинамидадениндинуклеотида (NADH), 1 мМ DTT, 30 мкг/мл пируваткиназы и 10 мкг/мл лактатдегидрогеназы. Добавляли фермент (конечная концентрация 90 нМ) и ДМСО раствор (конечная концентрация 3%) выборочного соединения. Осуществляли инкубирование реакционной смеси в течение 10 минут при 30°C. Реакцию инициировали добавлением АТФ до конечной концентрации 0,9 мМ и скорость исчезновения NADH отслеживали при 340 нм в течение 10 минут. Значения Ki и ИК50 определяли на основании профилей скорости против концентрации.
Было определено, что выборочные соединения по настоящему изобретению ингибируют ДНК гиразу S. aureus. Таблица 22 показывает ингибиторную активность этих соединений в анализе ингибирования ДНК гиразы S. aureus.
Ингибирование ДНК гиразы S. aureus
АТФазный анализ ДНК топо IV
Конверсию АТФ в АДФ под действием фермента Топо IV S. aureus объединяли с конверсией NADH в NAD+ и развитие реакции измеряли по изменению поглощательной способности при 340 нм. Топо IV (64 нМ) инкубировали с выборочным соединением (конечная концентрация 3% ДМСО) в буфере в течение 10 минут при 30°C. Буфер состоял из 100 мМ Трис 7,5, 1,5 мМ MgCl2, 200 мМ K·Глутамата, 2,5 мМ фосфоенолпирувата, 0,2 мМ NADH, 1 мМ DTT, 5 мкг/мл линеаризованной ДНК, 50 мкг/мл BSA, 30 мкг/мл пируваткиназы и 10 мкг/мл лактатдегидрогеназы (LDH). Реакцию инициировали при помощи АТФ и скорости отслеживали непрерывно в течение 20 минут при 30°C на планшет-ридере Molecular Devices SpectraMAX. Константу ингибирования Ki и ИК50 определяли из графиков скорость против концентрации для выборочного соединения в соответствии с уравнением Моррисона для сильно связывающихся ингибиторов.
Было определено, что выборочные соединения по настоящему изобретению ингибируют ДНК Топо IV S. aureus. Таблица 23 показывает ингибиторную активность этих соединений в анализе ингибирования ДНК гиразы S. aureus.
Ингибирование ДНК Топо IV S. aureus
Пример 38
ИСПЫТАНИЕ ВОДОРАСТВОРИМОСТИ
Водорастворимость соединения 23 и соединения W определяли в соответствии со следующей процедурой.
Получение образцов. Водные образцы каждого соединения получали следующим образом. Соединения взвешивали (20-30 мг соединения) в 4-мл прозрачных сосудах перед добавлением воды (0,5 мл) и при перемешивании магнитной мешалкой. К суспензии добавляли 1,0н раствор HCl для доведения pH до желаемого уровня. После перемешивания в течение 96 часов при комнатной температуре суспензии фильтровали через 0,22-микронный фильтр (Millipore, Ultrafree центрифужные фильтры, Durapore PVDF 0,22 мкм, Cat# UFC30GVNB). Фильтрат собирали и pH измеряли при помощи pH-метра. Фильтрат, содержащий соединение W, разбавляли 10-кратно с получением подходящих концентраций для ВЭЖХ анализа. Фильтрат, содержащий соединение 23, не требовал разбавления.
Получение стандартных растворов. Стандартные растворы каждого соединения получали в соответствии со следующей процедурой. 1-2 мг каждого соединения точно отвешивали в 10-мл волюметрическую колбу и добавляли либо воду (для соединения W), либо 1:1 метанол:0,1н HCl (для соединения 23) для полного растворения соединений. Для Соединения 23 осуществляли обработку ультразвуком, что способствовало растворению в 1:1 метанол:0,1н HCl. Когда все твердые вещества растворялись, добавляли дополнительный растворитель для доведения объема каждого раствора до 10 мл. Полученные растворы тщательно смешивали с получением стандартных растворов каждого соединения. Каждый стандартный раствор затем разбавляли растворителем 2-кратно, 10-кратно и 100-кратно.
Анализ растворимости. Аликвоты каждого образца и каждого стандартного раствора анализировали методом ВЭЖХ (Agilent 1100, объем вводимой пробы 10 мкл, длина волны 271 нм, колонка XTerra® Phenyl 5 мкм, 4,6x50 мм, Part No. 186001144, подвижная фаза: A: 0,1% TFA в воде 0,1% TFA в AcN). Пробу каждого стандартного раствора вводили три раза, и каждый из образцов вводили два раза. Стандартные кривые получали путем построения графика средняя площадь пика по данным ВЭЖХ против концентраций стандартных растворов (с подходящими корректировками массы стандартов на основании общего содержания воды в твердом веществе, определенного при помощи элементного анализа). Концентрацию каждого образца рассчитывали из площади пика водного образца на основании результатов ВЭЖХ и угла наклона и отсекаемой дуги стандартных кривых. Показатели растворимости, представленные в таблице 24 ниже, были получены на основании концентрации продукта в образце и коэффициента разбавления образца.
Водорастворимость соединений 23 и W
Пример 39
ИСПЫТАНИЕ МЕТАБОЛИЗМА IN VIVO В ГЕПАТИЧЕСКИХ КЛЕТКАХ И КЛЕТКАХ ПЕЧЕНИ S9
Конверсию соединения W в Соединение 23 исследовали в печеночных и интестинальных S9 фракциях крысы, собак, обезьян и человека. Соединение W инкубировали при 0,1, 0,3, 1, 3, 10, 20, 40, 100, 200, 300 мкМ в печеночных S9 фракциях и при 1, 3, 10, 20, 100, 300, 500, 1000 мкМ в интестинальных S9 фракциях. Инкубации осуществляли в течение 0, 5, 10, 15, 30, 45 или 60 минут. Образование соединения 23 количественно определяли методом ЖХ/MS-MS и данные использовали в уравнении Michaelis Menten. Данные, приведенные в таблице 25 ниже, показывают, что Соединение W быстро преобразовывалось в Соединение 23 в этих гепатических и интестинальных S9 фракциях.
Скорость образования (VMAX) Соединения 23 из Соединения W в печеночных и интестинальных S9
| название | год | авторы | номер документа |
|---|---|---|---|
| ТВЕРДЫЕ ФОРМЫ ИНГИБИТОРА ГИРАЗЫ (R)-1-ЭТИЛ-3-[6-ФТОР-5[2-(1-ГИДРОКСИ-1-МЕТИЛ-ЭТИЛ) ПИРИМИДИН-5-ИЛ]-7-(ТЕТРАГИДРОФУРАН-2-ИЛ)-1Н-БЕНЗИМИДАЗОЛ-2-ИЛ] МОЧЕВИНЫ | 2012 |
|
RU2625305C2 |
| ЭКСТРАКТЫ KIBDELOS PORANGIUM В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2010 |
|
RU2572621C2 |
| ПРИМЕНЕНИЕ ТИГЕЦИКЛИНА, В ОТДЕЛЬНОСТИ ИЛИ В КОМБИНАЦИИ С РИФАМПИНОМ, ДЛЯ ЛЕЧЕНИЯ ОСТЕОМИЕЛИТА И/ИЛИ СЕПТИЧЕСКОГО АРТРИТА | 2004 |
|
RU2329047C2 |
| ГИДРОКСИАЛКИЛТИАДИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2737892C1 |
| Фторхинолоны на основе 4-дезоксипиридоксина | 2016 |
|
RU2634122C1 |
| Отологические композиции | 2020 |
|
RU2837853C2 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ТЯЖЕСТИ ХРОНИЧЕСКИХ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ НИЖНИХ ДЫХАТЕЛЬНЫХ ПУТЕЙ МИКРОБНОЙ ЭТИОЛОГИИ | 2004 |
|
RU2276361C2 |
| Способ профилактики инфекций, связанных с оказанием медицинской помощи, у пациентов, находящихся в хроническом критическом состоянии | 2023 |
|
RU2818910C1 |
| НОВЫЕ АНТИБАКТЕРИАЛЬНЫЕ СОЕДИНЕНИЯ | 2007 |
|
RU2444526C2 |
| СПОСОБ ЛЕЧЕНИЯ ИНФЕКЦИИ, ВЫЗЫВАЕМОЙ ШТАММАМИ РОДА STAPHYLOCOCCUS, STREPTOCOCCUS ИЛИ ENTEROCOCCUS С МНОЖЕСТВЕННОЙ ЛЕКАРСТВЕННОЙ УСТОЙЧИВОСТЬЮ | 1996 |
|
RU2202360C2 |
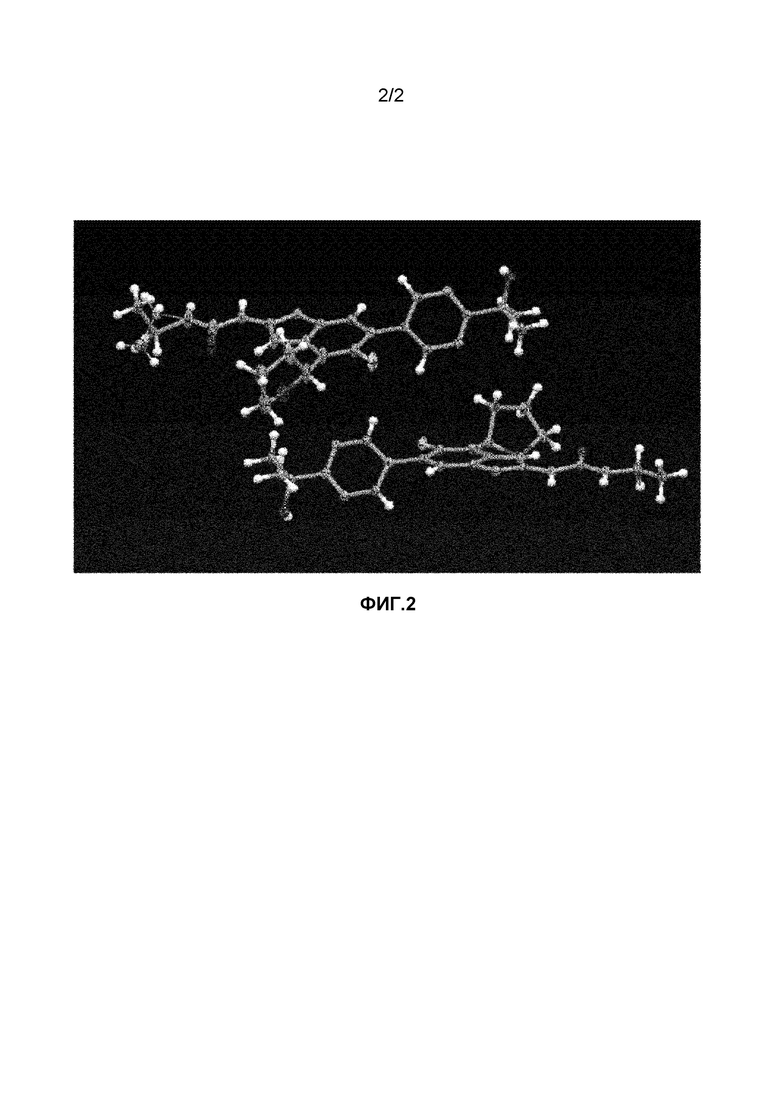
Изобретение относится к новому соединению формулы (I) или его фармацевтически приемлемой соли, которые обладают свойствами ингибиторов гиразы MIC и топоизомеразы IV и могут быть использованы для лечения бактериальных инфекций, опосредованных одной или несколькими бактериями из Streptococcus pneumoniae, Staphylococcus epidermidis, Enterococcus faecalis, Staphylococcus aureus, Clostridium difficile, Moraxella catarrhalis, Neisseria gonorrhoeae, Neisseria meningitidis, Mycobacterium avium комплекс, Mycobacterium abscessus, Mycobacterium kansasii, Mycobacterium ulcerans, Chlamydophila pneumoniae, Chlamydia trachomatis, Haemophilus influenzae, Streptococcus pyogenes или β-гемолитических стрептококков. Инфекции представляют собой инфекции нижних и верхних дыхательных путей, ушные инфекции, плевролегочные и бронхиальные инфекции, инфекции мочевых путей, интраабдоминальные инфекции, сердечно-сосудистые инфекции, заражение крови, сепсис, бактеремия, ЦНС инфекции, инфекции кожи и мягких тканей, желудочно-кишечные инфекции, инфекции, поражающие кости и суставы, половые инфекции, глазные инфекции или грануломатозные инфекции, инфекции кожи и кожных структур (uSSSI), катетеризационные инфекции, фарингит, синусит и др. В формуле (I)
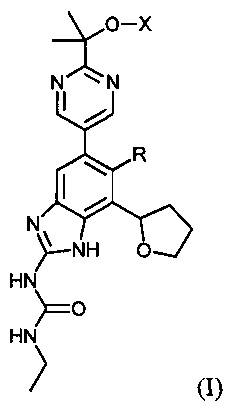
R представляет собой водород или фтор; X представляет собой водород, -РО(ОН)2, -РО(ОН)O-М+, -РО(O-)2⋅2М+ или -PO(O-)2⋅D2+; М+ представляет собой фармацевтически приемлемый одновалентный катион, такой как Li+, Na+, K+, N-метил-D-глюкамин и N(R9)4+, где каждый R9 независимо представляет собой водород или C1-C4 алкильную группу; D2+ представляет собой фармацевтически приемлемый двухвалентный катион, такой как Mg2+, Са2+ и Ва2+. 6 н. и 15 з.п. ф-лы, 2 ил., 27 табл., 39 пр.
1. Соединение формулы
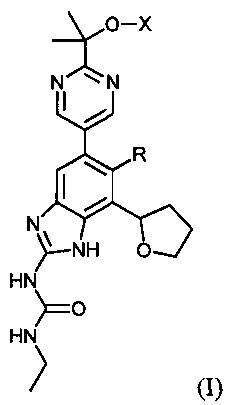
где R представляет собой водород или фтор; X представляет собой водород, -РО(ОН)2, -РО(ОН)O-М+, -РО(O-)2⋅2М+ или -PO(O-)2⋅D2+; М+ представляет собой фармацевтически приемлемый одновалентный катион и D2+ представляет собой фармацевтически приемлемый двухвалентный катион; или его фармацевтически приемлемая соль.
2. Соединение по п. 1, имеющее формулу
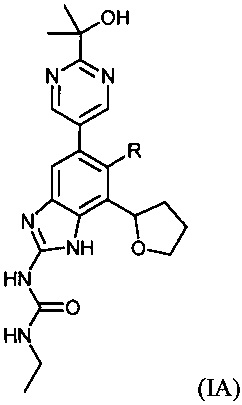
где R представляет собой водород или фтор; или его фармацевтически приемлемая соль.
3. Соединение по п. 1, имеющее формулу
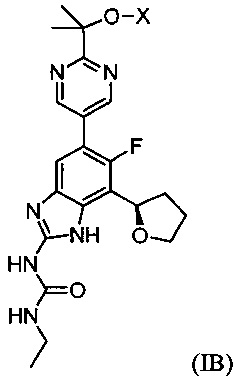
где X представляет собой -РО(ОН)2, -РО(ОН)O-М+, -РО(О-)2⋅2М+ или -РО(О-)2⋅D2+; М+ представляет собой фармацевтически приемлемый одновалентный катион и D2+ представляет собой фармацевтически приемлемый двухвалентный катион; или его фармацевтически приемлемая соль.
4. Соединение по п. 1, имеющее формулу
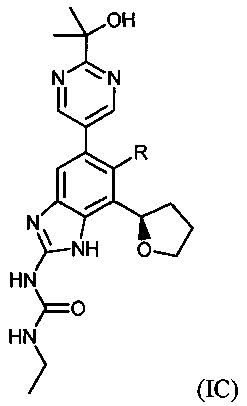
где R представляет собой водород или фтор; или его фармацевтически приемлемая соль.
5. Соединение по п. 4, где соединение представляет собой (R)-1-этил-3-(5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1Н-бензо[d]имидазол-2-ил)мочевину или ее фармацевтически приемлемую соль.
6. Соединение по п. 4, где соединение представляет собой (R)-1-этил-3-(6-фтор-5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1Н-бензо[d]имидазол-2-ил)мочевину или ее фармацевтически приемлемую соль.
7. Соединение по п. 4, где соль представляет собой соль метансульфоновой кислоты (R)-1-этил-3-(5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1Н-бензо[d]имидазол-2-ил)мочевины.
8. Соединение по п. 4, где соль представляет собой соль метансульфоновой кислоты (R)-1-этил-3-(6-фтор-5-(2-(2-гидроксипропан-2-ил)пиримидин-5-ил)-7-(тетрагидрофуран-2-ил)-1Н-бензо[d]имидазол-2-ил)мочевины.
9. Соединение по п. 3, где X представляет собой -РО(ОН)O-М+, -РО(O-)2⋅2М+ или -PO(O-)2⋅D2+; М+ выбран из группы, включающей Li+, Na+, K+, N-метил-D-глюкамин и N(R9)4+, где каждый R9 независимо представляет собой водород или C1-C4 алкильную группу; D2+ выбран из группы, включающей Mg2+, Са2+ и Ва2+.
10. Соединение по п. 9, где X представляет собой -РО(ОН)O-М+ или -РО(О-)2⋅2М+; М+ выбран из группы, включающей Li+, Na+, K+, N-метил-D-глюкамин и N(R9)4+, где каждый R9 независимо представляет собой водород или C1-C4 алкильную группу.
11. Соединение по п. 9, где X представляет собой -РО(О-)2⋅2М+; М+ выбран из группы, включающей Li+, Na+, K+, N-метил-D-глюкамин и N(R9)4+, где каждый R9 независимо представляет собой водород или C1-C4 алкильную группу.
12. Соединение по п. 9, где М+ представляет собой Na+.
13. Соединение по п.3, где соединение представляет собой динатрий (R)-2-(5-(2-(3-этилуреидо)-6-фтор-7-(тетрагидрофуран-2-ил)-1Н-бензо[d]имидазол-5-ил)пиримидин-2-ил)пропан-2-ил фосфат.
14. Фармацевтическая композиция, обладающая свойствами ингибитора гиразы MIC и топоизомеразы IV, включающая терапевтически эффективное количество соединения по п. 1 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, адъювант или наполнитель.
15. Фармацевтическая композиция, обладающая свойствами ингибитора гиразы MIC и топоизомеразы IV, включающая терапевтически эффективное количество соединения по п. 2 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, адъювант или наполнитель.
16. Фармацевтическая композиция, обладающая свойствами ингибитора гиразы MIC и топоизомеразы IV, включающая терапевтически эффективное количество соединения по п. 3 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, адъювант или наполнитель.
17. Способ снижения или ингибирования бактериального количества Streptococcus pneumoniae, Staphylococcus epidermidis, Enterococcus faecalis, Staphylococcus aureus, Clostridium difficile, Moraxella catarrhalis, Neisseria gonorrhoeae, Neisseria meningitidis, Mycobacterium avium комплекс, Mycobacterium abscessus, Mycobacterium kansasii, Mycobacterium ulcerans, Chlamydophila pneumoniae, Chlamydia trachomatis, Haemophilus influenzae, Streptococcus pyogenes или β-гемолитических стрептококков в биологическом образце, включающий контактирование указанного биологического образца с терапевтически эффективным количеством соединения по п. 1.
18. Способ контроля, лечения или уменьшения прогрессирования, тяжести или воздействий внутрибольничной или невнутрибольничной бактериальной инфекции, опосредованной активностью гиразы MIC и/или топоизомеразы IV, у пациента, включающий введение указанному пациенту терапевтически эффективного количества соединения по п. 1.
19. Способ по п. 18, где бактериальная инфекция характеризуется присутствием одного или нескольких из Streptococcus pneumoniae, Staphylococcus epidermidis, Enterococcus faecalis, Staphylococcus aureus, Clostridium difficile, Moraxella catarrhalis, Neisseria gonorrhoeae, Neisseria meningitidis, Mycobacterium avium комплекс, Mycobacterium abscessus, Mycobacterium kansasii, Mycobacterium ulcerans, Chlamydophila pneumoniae, Chlamydia trachomatis, Haemophilus influenzae, Streptococcus pyogenes или (β-гемолитических стрептококков.
20. Способ по п. 19, где бактериальная инфекция представляет собой одну или несколько инфекций, выбранных из следующих: инфекции верхних дыхательных путей, инфекции нижних дыхательных путей, ушные инфекции, плевролегочные и бронхиальные инфекции, осложненные инфекции мочевых путей, неосложненные инфекции мочевых путей, интраабдоминальные инфекции, сердечно-сосудистые инфекции, заражение крови, сепсис, бактеремия, ЦНС инфекции, инфекции кожи и мягких тканей, желудочно-кишечные инфекции, инфекции, поражающие кости и суставы, половые инфекции, глазные инфекции или грануломатозные инфекции, неосложненные инфекции кожи и кожных структур (uSSSI), осложненные инфекции кожи и кожных структур (cSSSI), катетеризационные инфекции, фарингит, синусит, отит наружного уха, отит среднего уха, бронхит, эмпиема, пневмония, приобретенные в общественных местах бактериальные пневмонии (САВР), приобретенная в больнице пневмония (НАР), приобретенная в больнице бактериальная пневмония, связанная с искусственной вентиляцией легких пневмония (VAP), связанные с диабетом инфекционные поражения стоп, ванкомицин-резистентные энтерококковые инфекции, цистит и пиелонефрит, почечные конкременты, простатит, перитонит, осложненные интраабдоминальные инфекции (cIAI) и другие интерабдоминальные инфекции, связанный с диализом перитонит, висцеральные абсцессы, эндокардит, миокардит, перикардит, связанный с переливанием крови сепсис, менингит, энцефалит, абсцесс головного мозга, остеомиелит, артрит, язвы половых органов, уретрит, вагинит, цервицит, гингивит, конъюнктивит, кератит, эндофтальмит, инфекция у пациентов с кистозным фиброзом или инфекция у пациентов с лихорадочным нейтропеническим состоянием.
21. Способ по п. 20, где бактериальная инфекция выбрана из одной или нескольких из следующих: приобретенные в общественных местах бактериальные пневмонии (САВР), приобретенная в больнице пневмония (НАР), приобретенная в больнице бактериальная пневмония, связанная с искусственной вентиляцией легких пневмония (VAP), бактеремия, связанные с диабетом инфекционные поражения стоп, катетеризационные инфекции, неосложненные инфекции кожи и кожных структур (uSSSI), осложненные инфекции кожи и кожных структур (cSSSI), ванкомицин-резистентные энтерококковые инфекции или остеомиелит.
| WO 2005012292 A1, 10.05.2005 | |||
| WO 2006022773 A1, 02.03.2006 | |||
| PAUL S CHARIFSON et al | |||
| Novel Dual-Targeting Benzimidazole Urea Inhibitors of DNA Gyrase and Topoisomerase IV Possessing Potent Antibacterial Activity: Intelligent Design and Evolution through the Judicious Use of Structure-Guided Design and Stucture-Activity Relationships.Journal of Medicinal Chemistry, 2008, Vol:51, Nr:17, Page(s):5243 - 5263 | |||
| US 5523288 A, 04.06.1996 | |||
| ИНГИБИТОРЫ ГИРАЗЫ БАКТЕРИЙ И ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2262932C2 |
| US 2003225450 A1, 04.12.2003 | |||
| RU 2013137751 А, 20.02.2015, приоритет 14.01.2011. | |||

