Область техники
Настоящее изобретение относится к новому амидному производному, обладающему гипогликемическим действием и/или защитным действием в отношении β-клеток или поджелудочной железы, или к его фармацевтически приемлемой соли и к фармацевтической композиции, содержащей их в качестве активных ингредиентов.
Уровень техники
Сахарный диабет представляет собой метаболическое заболевание, отличающееся главным образом хроническим гипергликемическим статусом вследствие недостаточного инсулинового действия. Лечение диабета обычно проводят с помощью лекарственной терапии вместе с лечебной диетой и лечебной физкультурой. Примерами находящихся в употреблении пероральных гипогликемических средств, которые составляют класс терапевтических лекарственных средств для лечения диабета, являются бигуанидиновые средства и тиазолидиндионовые средства, которые улучшают резистентность к инсулину; сульфонилмочевинные средства и глинидные лекарства, которые стимулируют выделение инсулина из панкреатических β-клеток; и ингибиторы α-глюкозидазы, которые ингибируют поглощение сахара.
Однако сообщается, что бигуанидиновые средства имеют вредные побочные действия, такие как пищеварительные симптомы и лактоцидоз; тиазолидиндионовые средства вызывают такие вредные побочные процессы, как увеличение веса и отек; сульфонилмочевинные средства и глинидные лекарства обладают такими вредными побочными действиями, как гипогликемия или вторичная недостаточность из-за длительного применения; и ингибиторы α-глюкозидазы обладают такими вредными побочными действиями, как диарея.
Таким образом, необходима разработка перорального гипогликемического средства, которое может решать такие проблемы.
Кроме того, в последние годы в качестве пероральных гипогликемических средств разработаны пиперидиновые соединения, имеющие новые структуры (см., например, патентные документы 1-4).
Список цитирования
Патентные документы
Патентный документ 1: WO 07/116229;
Патентный документ 2: WO 07/003960;
Патентный документ 3: WO 07/003962;
Патентный документ 4: WO 05/061489.
Суть изобретения
Техническая проблема
Однако у соединений, описанных в упомянутых выше патентных документах, есть проблема в том, что достаточное гипогликемическое действие и защитное действие в отношении β-клеток или поджелудочной железы, не может быть получено легко. Кроме того, патентные документы, описанные выше, раскрывают соединения, содержащие циклогексановое кольцо или пиперидиновое кольцо, но не описывают и не подразумевают какие-либо соединения, содержащие бензольное кольцо, пиридиновое кольцо или пиридазиновое кольцо в их структурах, вместо циклогексанового кольца или пиперидинового кольца. Таким образом, объектом настоящего изобретения является создание соединений, имеющих новую структуру, которая не описана и не предполагается в приведенных выше патентных документах, и обладающих прекрасным гипогликемическим действием и защитным действием в отношении β-клеток или поджелудочной железы, или их фармацевтически приемлемых солей; фармацевтической композиции, имеющей прекрасный терапевтический эффект и/или профилактический эффект при диабете 1 типа, диабете 2 типа и т.д., которые вызывают повышение уровня сахара в крови вследствие аномального метаболизма сахара; и фармацевтической композиции, обладающей защитным действием в отношении β-клеток или поджелудочной железы.
Решение проблемы
Настоящее изобретение предлагает:
(1) Соединение, представленное общей формулой (I):

где R1 представляет собой атом водорода или С1-С6-алкильную группу, замещенную одним или двумя заместителями, выбранными из подгруппы заместителя α;
подгруппа заместителя α представляет собой группу, включающую С1-С6-алкокси-группу, С1-С6-алкоксикарбонильную группу, гидроксильную группу, которая может быть замещена заместителем, выбранным из подгруппы заместителя β, и карбоксильную группу;
подгруппа заместителя β представляет собой группу, включающую С1-С6-алкилкарбонильную группу, замещенную одним или двумя заместителями, выбранными из подгруппы заместителя γ, и 4-6-членную гетероциклическую карбонильную группу, которая может быть замещена одной С1-С6-алкильной группой;
подгруппа заместителя γ представляет собой группу, включающую гидроксильную группу, амино-группу, (С1-С6-алкил)амино-группу, ди(С1-С6-алкил)амино-группу, карбамоильную группу, фенильную группу и 4-6-членную гетероциклическую группу;
R2 представляет собой атом водорода или С1-С6алкильную группу, которая может быть замещена одной гидроксильной группой;
или R1 и R2 вместе с атомом азота, с которым R1 и R2 связаны, могут быть объединены с образованием азетидино-группы, пирролидино-группы или морфолино-группы, где азетидино-группа, пирролидино-группа или морфолино-группа могут быть замещены одной гидроксильной группой или одной гидрокси-С1-С6-алкильной группой;
R3 и R4 каждый независимо друг от друга представляет собой С1-С6-алкильную группу;
R5 представляет собой атом галогена или С1-С6-алкильную группу;
R6 представляет собой атом галогена;
m и n каждый независимо друг от друга представляют собой целое число от 0 до 4; и
V, W, X, Y и Z каждый независимо друг от друга представляют собой СН или N,
или его фармацевтически приемлемую соль;
(2) соединение по пункту (1), где Y и Z оба представляют собой СН;
(3) соединение по пунктам (1) или (2), где V и W оба представляют собой СН;
(4) соединение по любому из пунктов (1)-(3), где Х представляет собой N;
(5) соединение по любому из пунктов (1)-(4), где R1 представляет собой С1-С4-алкильную группу, замещенную одной или двумя гидроксильными группами;
(6) соединение по любому из пунктов (1)-(4), где R1 представляет собой гидроксиэтильную группу, гидроксиизопропильную группу, гидрокси-1,1-диметилэтильную группу или 2-гидрокси-1-(гидроксиметил)этильную группу;
(7) соединение по любому из пунктов (1)-(4), где R1 представляет собой С1-С4-алкильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя α, где подгруппа заместителя α представляет собой гидроксильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя β, подгруппа заместителя β представляет собой С1-С4-алкилкарбонильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя γ, и подгруппа заместителя γ представляет собой амино-группу;
(8) соединение по любому из пунктов (1)-(4), где R1 представляет собой аминометилкарбонилоксиэтильную группу, аминометилкарбонилоксиизопропильную группу или аминометилкарбонилокси-1,1-диметилэтильную группу;
(9) соединение по любому из пунктов (1)-(8), где R2 представляет собой атом водорода;
(10) соединение по любому из пунктов (1)-(9), где R3 представляет собой С1-С3-алкильную группу;
(11) соединение по любому из пунктов (1)-(9), где R3 представляет собой этильную группу;
(12) соединение по любому из пунктов (1)-(11), где R4 представляет собой С1-С3-алкильную группу;
(13) соединение по любому из пунктов (1)-(11), где R4 представляет собой этильную группу или изопропильную группу;
(14) соединение по любому из пунктов (1)-(13), где R5 представляет собой атом галогена, и m принимает значение 1;
(15) соединение по любому из пунктов (1)-(13), где R5 представляет собой атом фтора, и m принимает значение 1;
(16) соединение по любому из пунктов (1)-(15), где n принимает значение 0;
(17) соединение, выбранное из группы, включающей следующие соединения:
2-фтор-N-(2-гидроксиэтил)-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид;
2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид;
2-фтор-N-(2-гидрокси-1,1-диметилэтил)-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид;
гидрохлорид (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропилглицината;
гидрохлорид 2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}-2-метилпропилглицината;
2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]-4-(2-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,3-оксадиазол-4-ил)бензамид;
гидрохлорид (2S)-2-{[2-фтор-4-(2-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,3-оксадиазол-4-ил)бензоил]амино}пропилглицината;
4-(5-{1-[4-(5-этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]бензамид;
гидрохлорид (2S)-2-{[4-(5-{1-[4-(5-этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензоил]амино}пропилглицината;
2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{(1R)-1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид; и
N-(циклопропилметил)-2-фтор-4-(5-{(1R)-1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид;
(18) фармацевтическая композиция, содержащая в качестве активного ингредиента соединение по любому из пунктов (1)-(17) или его фармацевтически приемлемую соль;
(19) фармацевтическая композиция по пункту (18) для лечения и/или предупреждения сахарного диабета 1 типа, сахарного диабета 2 типа, ассоциирующегося с диабетом заболевания или ожирения;
(20) фармацевтическая композиция по пункту (18) для защиты β-клеток или поджелудочной железы;
(21) применение соединения по любому из пунктов (1)-(17) или его фармацевтически приемлемой соли для получения фармацевтической композиции;
(22) способ лечения и/или предупреждения заболевания, включающий введение млекопитающему фармакологически эффективного количества соединения по любому из пунктов (1)-(17) или его фармацевтически приемлемой соли;
(23) способ по пункту (22), где заболеванием является сахарный диабет 1 типа, сахарный диабет 2 типа, ассоциирующееся с диабетом заболевание или ожирение;
(24) способ защиты β-клеток или поджелудочной железы, включающий введение млекопитающему фармакологически эффективного количества соединения по любому из пунктов (1)-(17) или его фармацевтически приемлемой соли; и
(25) способ по любому из пунктов (22)-(24), где млекопитающим является человек.
Положительные эффекты изобретения
В соответствии с настоящим изобретением могут быть получены оксадиазолиновое соединение, обладающее прекрасным гипогликемическим действием, или его фармацевтически приемлемая соль, фармацевтическая композиция, обладающая прекрасным терапевтическим действием и/или профилактическим действием на диабет 1 типа, диабет 2 типа и т.д., которые вызывают повышение уровня сахара в крови, и фармацевтическая композиция, обладающая защитным действием в отношении β-клеток или поджелудочной железы.
Описание вариантов осуществления изобретения
Определение «С1-С6-алкильная группа», используемое в данном описании, означает линейную, разветвленную или циклическую алкильную группу, содержащую от 1 до 6 атомов углерода. Конкретными примерами являются метильная группа, этильная группа, пропильная группа, изопропильная группа, циклопропильная группа, бутильная группа, изобутильная группа, втор-бутильная группа, трет.-бутильная группа, пентильная группа, 1,2-диметилпропильная группа, изопентильная группа, гексильная группа и изогексильная группа.
Определение «С1-С6-алкильная группа, замещенная одним или двумя заместителями», используемое в данном описании, означает группу, полученную замещением одного или двух атомов водорода «С1-С6-алкильной группы» одинаковыми или разными заместителями.
Определение «С1-С6-алкокси-группа», используемое в данном описании, означает группу, в которой «С1-С6-алкильная группа» связана с атомом кислорода. Конкретными примерами являются метокси-группа, этокси-группа, пропокси-группа, бутокси-группа, пентилокси-группа и гексилокси-группа.
Определение «С1-С6-алкоксикарбонильная группа», используемая в данном описании, означает группу, в которой «С1-С6-алкокси-группа» связана с карбонильной группой. Конкретными примерами являются метоксикарбонильная группа, этоксикарбонильная группа, пропоксикарбонильная группа, бутоксикарбонильная группа, пентилоксикарбонильная группа и гексилоксикарбонильня группа.
Определение «С1-С6-алкилкарбонильная группа», используемое в данном описании, означает группу, в которой «С1-С6-алкильная группа» связана с карбонильной группой. Конкретными примерами являются метилкарбонильная группа, этилкарбонильная группа, пропилкарбонильная группа, изопропилкарбонильная группа, бутилкарбонильная группа, изобутилкарбонильная группа, втор.-бутилкарбонильная группа и трет.-бутилкарбонильная группа.
Определение «гидроксильная группа, которая может быть замещена заместителем», используемое в данном описании, означает гидроксильную группу или группу, полученную замещением атома водорода гидроксильной группы заместителем.
Определение «С1-С6-алкилкарбонильная группа, замещенная одним или двумя заместителями», используемое в данном описании, означает группу, полученную замещением одного или двух атомов водорода «С1-С6-алкильной группой» в «С1-С6-алкилкарбонильной группе» одинаковыми или разными заместителями.
Определение «4-6-членная гетероциклическая группа», используемое в данном описании, означает 4-6-членную насыщенную моноциклическую или ненасыщенную моноциклическую, одновалентную группу, содержащую 1-3 гетероатома, выбранные из группы, включающей атом кислорода, атом азота и атом серы. Конкретные примеры включают фуранильную группу, тетрагидрофуранильную группу, пиранильную группу, тетрагидропиранильную группу, тиенильную группу, тиопиранильную группу, пирролильную группу, пирролидинильную группу, имидазолильную группу, имидазолидинильную группу, пиразолильную группу, пиразолидинильную группу, тиазолильную группу, тиазолидинильную группу, изотиазолильную группу, изотиазолидинильную группу, оксазолильную группу, оксазолидинильную группу, изоксазолильную группу, изоксазолидинильную группу, пиридинильную группу, пиперидинильную группу, пиразинильную группу, пиперазинильную группу, пиримидинильную группу, пиридазинильную группу, тиоморфолинильную группу и морфолинильную группу.
Определение «4-6-членная гетероциклическая карбонильная группа», используемое в данном описании, означает группу, в которой «4-6-членная гетероциклическая группа» связана с карбонильной группой. Конкретными примерами являются пирролидин-1-илкарбонильная группа, пирролидин-2-ил-карбонильная группа, имидазолидин-2-илкарбонильная группа и морфолин-4-илкарбонильная группа.
Определение «4-6-членная гетероциклическая карбонильная группа, которая замещена одной С1-С6-алкильной группой», используемое в данном описании, означает «4-6-членную гетероциклическую карбонильную группу» или группу, полученную замещением одного атома водорода «4-6-членной гетероциклической карбонильной группы» «С1-С6-алкильной группой».
Определение «(С1-С6-алкил)амино-группа», используемое в данном описании, означает группу, полученную замещением одного атома водорода амино-группы «С1-С6-алкильной группой». Конкретными примерами являются метиламино-группа, этиламино-группа, пропиламино-группа, изопропиламино-группа, бутиламино-группа, изобутиламино-группа, втор.-бутиламино-группа и трет.-бутиламино-группа.
Определение «ди(С1-С6-алкил)амино-группа», используемое в данном описании, означает группу, полученную замещением двух атомов водорода амино-группы «С1-С6-алкильными» группами, которые могут быть одинаковыми или могут отличаться друг от друга. Конкретными примерами являются N,N-диметиламино-группа, N,N-диэтиламино-группа, N,N-дипропиламино-группа, диизопропиламино-группа, N,N-дибутиламино-группа, N-метил-N-этиламино-группа, N-метил-N-пропиламино-группа и N-этил-N-пропиламино-группа.
Определение «атом галогена», используемое в данном описании, означает атом фтора, атом хлора, атом брома или атом йода.
Определение «С1-С6-алкиленовая группа», используемое в данном описании, означает двухвалентную группу, полученную удалением одного атома водорода из «С1-С6-алкильной группы». Конкретными примерами являются метиленовая группа, этиленовая группа, пропиленовая группа, бутиленовая группа, пентиленовая группа и гексиленовая группа.
Определение «фармацевтически приемлемая соль», используемое в данном описании, означает соль, образованную за счет введения в реакцию соединения настоящего изобретения с кислотой или основанием.
Примеры соли включают соли галогенводородных кислот, такие как гидрофториды, гидрохлориды, гидробромиды и гидройодиды; соли неорганических кислот, такие как гидрохлориды, нитраты, перхлораты, сульфаты и фосфаты; соли низших алкансульфоновых кислот, такие как метансульфонаты, трифторметансульфонаты и этансульфонаты; соли арилсульфоновых кислот, такие как бензолсульфонаты и п-толуолсульфонаты; соли органических кислот, такие как ацетаты, малаты, фумараты, сукцинаты, цитраты, аскорбаты, тартраты, оксалаты и малеаты; соли щелочных металлов, такие как натриевые соли, калиевые соли и литиевые соли; соли щелочноземельных металлов, такие как кальциевые соли и магниевые соли; соли металлов, такие как алюминиевые соли и железные соли; неорганические соли, такие как аммонийные соли; аминные соли, включая органические соли, такие как трет.-октиламинные соли, дибензиламинные соли, морфолиновые соли, глюкозаминные соли, соли фенилглициналкиловых эфиров, этилендиаминные соли, N-метилглюкаминовые соли, гуанидиновые соли, диэтиламинные соли, триэтиламинные соли, дициклогексиламинные соли, N,N'-дибензилэтилендиаминные соли, хлорпрокаиновые соли, прокаиновые соли, диэтаноламинные соли, N-бензилфенетиламинные соли, пиперазиновые соли, тетраметиламмонийные соли и трис(гидроксиметил)аминометановые соли; и соли аминокислот, такие как глициновые соли, лизиновые соли, аргининовые соли, орнитиновые соли, глутаматы и аспартаты.
Соединение настоящего изобретения абсорбирует воду, когда, например, его оставляют стоять в атмосфере или др. так, что абсорбированная вода может присоединяться к соединению и может образовываться гидрат. Следовательно, такие гидраты также включены в понятие соли настоящего изобретения.
Так как соединения настоящего изобретения могут иметь в молекуле асимметричные атомы углерода, соединение имеет оптические изомеры. Такие изомеры и смеси таких изомеров все представлены одной формулой, то есть, общей формулой (I). Следовательно, настоящее изобретение охватывает все оптические изомеры соединения, представленного общей формулой (I), и смеси оптических изомеров в любых соотношениях. Такой оптический изомер может быть получен, например, с использованием сырьевых материалов, имеющих оптическую активность, вместо сырьевых материалов, используемых в способах получения справочных примеров и примеров, которые будут описаны ниже; или могут быть получены воздействием на соединения, полученные со ссылкой на способы получения справочных примеров и примеров и др., которые будут описаны ниже, способа оптического разрешения, известного в соответствующей области техники, например, диастереомерного метода, метода ферментативной реакции или метода оптического разрешения, основанного на хроматографии.
Настоящее изобретение также может охватывать соединения, в которых один или несколько атомов, составляющих соединение, представленное общей формулой (I), замещены изотопами атомов. Изотопы включают два класса, такие как радиоактивные изотопы и стабильные изотопы, и примерами изотопов являются, например, изотопы водорода (2Н и 3Н), изотопы углерода (11С, 13С и 14С), изотопы азота (13N и 15N), изотопы кислорода (15О, 17С и 18О) и изотопы фтора (18F). Композиция, содержащая соединение, меченное изотопом, может быть полезна, например, в качестве терапевтического агента, профилактического агента, исследовательского реагента, реагента для оценки, диагностического агента или in vivo диагностического визуализирующего агента. Соединения, меченные изотопами, и смеси меченных изотопами соединений в любых соотношениях все включены в настоящее изобретение. Соединение, меченное изотопом, может быть получено способами, известными в соответствующей области техники, например, с использованием сырьевых материалов, меченных изотопами, вместо сырьевых материалов, используемых в способах получения настоящего изобретения, которые будут описаны ниже.
Настоящее изобретение также может охватывать пролекарства соединения, представленного общей формулой (I). Пролекарство представляет собой производное соединения, представленного общей формулой (I), и означает соединение, которое в живом организме ферментативным путем или химически превращается в соединение настоящего изобретения.
Примерами пролекарства являются соединения, в которых амино-группа в молекуле ацилирована, алкилирована или фосфорилирована; соединения, в которых карбоксильная группа в молекуле этерифицирована или амидирована; и соединения, в которых гидроксильная группа в молекуле ацилирована, алкилирована или фосфорилирована (см., например, публикацию Povl Krogsgaard-Larsen et al., «A Textbook of Drug Design and Development», Second Edition, Harwood Academic Publishers, 1996, pp. 351-385). Такое пролекарство может быть получено из соединения, представленного общей формулой (I), способами, известными в соответствующей области техники.
V предпочтительно представляет собой СН.
W предпочтительно представляет собой СН.
X предпочтительно представляет собой N.
Y предпочтительно представляет собой СН.
Z предпочтительно представляет собой СН.
Заместитель R1 предпочтительно представляет собой атом водорода или С1-С4-алкильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя α; и более предпочтительно представляет собой атом водорода, этильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя α, пропильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя α, изопропильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя α, изобутильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя α, втор.-бутильную группу, трет.-бутильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя α, или 1,1-диметилэтильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя α.
Подгруппа заместителя α предпочтительно представляет собой С1-С3-алкокси-группу и гидроксильную группу, которая может быть замещена заместителем, выбранным из подгруппы заместителя β; и более предпочтительно метокси-группу, гидроксильную группу и гидроксильную группу, замещенную заместителем, выбранным из подгруппы заместителя β.
Подгруппа заместителя β предпочтительно представляет собой С1-С3-алкилкарбонильную группу, замещенную одним или двумя заместителями, выбранными из подгруппы заместителя γ, и 5-членную гетероциклическую карбонильную группу, которая может быть замещена одной С1-С3-алкильной группой; и более предпочтительно метилкарбонильную группу, замещенную заместителем, выбранным из подгруппы заместителя γ, и пирролидинкарбонильную группу.
Подгруппа заместителя γ предпочтительно представляет собой гидроксильную группу, амино-группу и ди(С1-С3-алкил)амино-группу; и более предпочтительно гидроксильную группу, амино-группу и ди(метил)амино-группу.
Заместитель R1 даже более предпочтительно представляет собой С1-С4-алкильную группу, замещенную одной гидроксильной группой, или С1-С4-алкильную группу, замещенную одной гидроксильной группой, которая замещена одной С1-С4-алкилкарбонильной группой, которая замещена одной амино-группой; и особенно предпочтительно гидроксиэтильную группу, гидроксиизопропильную группу, гидрокси-1,1-диметилэтильную группу, аминометилкарбонилоксиэтильную группу, аминометилкарбонилоксиизопропильную группу или аминометилкарбонилокси-1,1-диметилэтильную группу.
Заместитель R2 предпочтительно представляет собой атом водорода или С1-С3-алкильную группу; и более предпочтительно атом водорода или метильную группу.
Заместитель R3 предпочтительно представляет собой С1-С3-алкильную группу; и более предпочтительно метильную группу или этильную группу.
Заместитель R4 предпочтительно представляет собой С1-С3-алкильную группу; и более предпочтительно этильную группу или изопропильную группу.
Заместитель R5 предпочтительно представляет собой атом водорода или С1-С3-алкильную группу; и более предпочтительно атом фтора или метильную группу.
Предпочтительно m принимает значения 0 или 1; и более предпочтительно 1.
Заместитель R6 предпочтительно представляет собой атом галогена; и более предпочтительно атом брома.
Предпочтительно n принимает значения 0 или 1; и более предпочтительно 0.
Предпочтительная комбинация V, W, X, Y, Z, R1, R2, R3, R4, R5, R6, m и n в общей формуле (I) представляет собой комбинацию, в которой: V представляет собой СН; W представляет собой СН; X представляет собой N; Y представляет собой СН; Z представляет собой СН; R1 представляет собой С1-С4-алкильную группу, замещенную одной гидроксильной группой, или С1-С4-алкильную группу, замещенную одной гидроксильной группой, которая замещена одной C1-C4-алкилкарбонильной группой, которая замещена одной амино-группой; R2 представляет собой атом водорода; R3 представляет собой С1-С3-алкильную группу; R4 представляет собой С1-С3-алкильную группу; R5 представляет собой атом галогена; m равно 1; и n равно 0.
Более предпочтительной комбинацией является комбинация, в которой: V представляет собой СН; W представляет собой СН; X представляет собой N; Y представляет собой СН; Z представляет собой СН; R1 представляет собой гидроксиэтильную группу, гидроксиизопропильную группу, гидрокси-1,1-диметилэтильную группу, аминометилкарбонилоксиэтильную группу, аминометилкарбонилоксиизопропильную группу или аминометилкарбонилокси-1,1-диметилэтильную группу; R2 представляет собой атом водорода; R3 представляет собой этильную группу; R4 представляет собой С1-С3-алкильную группу; R5 представляет собой атом галогена; m равно 1; и n равно 0.
Соединение настоящего изобретения может быть получено, например, следующими способами А-С. Кроме того, например, соединения на основе бензола, соединения на основе пиридина, соединения на основе пиридазина или соединения на основе амина, которые используют в качестве исходных сырьевых материалов в приведенных ниже способах получения, могут быть использованы продаваемые на рынке соединения.
Способ А представляет собой способ получения соединения (Ia) настоящего изобретения, представленного общей формулой (I), в которой Х представляет собой N; и R1 представляет собой атом водорода или С1-С6-алкильную группу, замещенную одним или двумя заместителями, выбранными из подгруппы заместителя α', где подгруппа заместителя α' представляет собой группу, включающую С1-С6-алкокси-группу, С1-С6-алкоксикарбонильную группу, гидроксильную группу и карбоксильную группу.
Способ В представляет собой способ получения соединения (Ib) настоящего изобретения, представленного общей формулой (I), в которой Х представляет собой СН; и R1 представляет собой атом водорода или С1-С6-алкильную группу, замещенную одним или двумя заместителями, выбранными из подгруппы заместителя α', где подгруппа заместителя α' имеет те же значения, как и определенные выше.
Способ С представляет собой способ получения соединения (Ic) настоящего изобретения, представленного общей формулой (I), в которой R1 представляет собой С1-С6-алкильную группу, замещенную одной гидроксильной группой, замещенной заместителем, выбранным из подгруппы заместителя β.
В реакциях различных стадий способов, описанных ниже, когда соединение, выступающее как реакционный субстрат, имеет группу, которая ингибирует предполагаемую реакцию (например, амино-группа, гидроксильная группа или карбоксильная группа), если необходимо, можно провести введение защитной группы для такой группы и удаление введенной защитной группы. Не существует особенных ограничений по таким защитным группам, пока они представляют собой обычно используемые защитные группы, а примеры включают защитные группы, описанные в публикации T.H. Greene, P.G. Wuts, Protective Groups in Organic Synthesis. Third Edition, 1999, John Wiley & Sons, Inc., или др. Реакция введения таких защитных групп и реакция снятия защитных групп могут быть проведены в соответствии с рутинными методами, таким как методы, описанные в упомянутой выше публикации.
Рассмотрение различных стадий в способах А-С представлено ниже.
Способ А
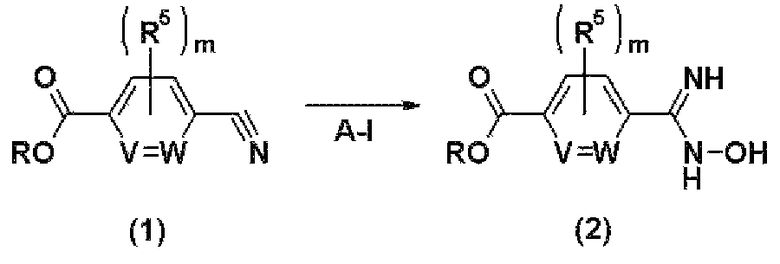
где R представляет собой защитную группу для карбоксильной группы; Halo представляет собой атом галогена; R1a представляет собой атом водорода или С1-С6-алкильную группу, замещенную одним или двумя заместителями, выбранными из подгруппы заместителя α'; V, W, Y, Z, R2, R3, R4, R5, R6, m, n и α' соответственно имеют те же самые значения, как и описанные выше.
Стадия А-I представляет собой стадию получения соединения (2) за счет введения в реакцию соединения (1) с гидроксиламином.
Примеры растворителя, используемого на данной стадии, включают метанол, этанол, смесь растворителей метанол/толуол, диметилформамид (ДМФА) и диметилсульфоксид, и предпочтительным примером является этанол.
Примерами гидроксиламина, используемого в данном случае, являются 50%-ный (масс./масс.) водный раствор гидроксиламина и гидрохлорид гидроксиламина, и предпочтительным примером является 50%-ный (масс./масс.) водный раствор гидроксиламина.
Примеры реагента, используемого в данном случае, включают карбонат натрия, карбонат калия, карбонат цезия, трет.-бутоксид калия, триэтиламин и диизопропилэтиламин.
Температура реакции составляет от 0 до 150ºС, и предпочтительно от 50 до 100ºС. Время реакции составляет от 10 минут до 24 часов, и предпочтительно от 30 минут до 5 часов.
Когда необходима обработка реакционной смеси, то такая обработка может быть проведена в соответствии со следующей методикой, например. Реакционную смесь охлаждают до комнатной температуры, затем растворитель отгоняют при пониженном давлении и полученный остаток промывают гексаном.
Стадия А-II представляет собой стадию получения соединения (5) путем введения в реакцию соединения (3) с соединением (4) в присутствии основания.
Примеры растворителя, используемого в данном случае, включают тетрагидрофуран (ТГФ), 1,4-диоксан, ацетонитрил и ацетон, и предпочтительным примером является ацетонитрил.
Примеры основания, используемого в данном случае, включают карбонат натрия, карбонат калия, карбонат цезия, трет.-бутоксид калия и гидроксид натрия, и предпочтительным примером является карбонат калия.
Температура реакции составляет от 0 до 150ºС, предпочтительно от 20 до 130ºС. Время реакции составляет от 30 минут до 24 часов, и предпочтительно от 30 минут до 6 часов.
Когда необходима обработка реакционной смеси, то такая обработка может быть проведена в соответствии со следующей методикой, например. Реакционную смесь охлаждают до комнатной температуры, и затем нерастворимое вещество удаляют с использованием Celite. Растворитель отгоняют при пониженном давлении из реакционной смеси, из которой удалено нерастворимое вещество. Полученный остаток очищают хроматографией на силикагеле или промывают органическим растворителем, водой или др.
Стадия А-III представляет собой стадию получения соединения (6) путем введения в реакцию соединения (5), полученного на стадии А-II, с гидроксиламином.
Примерами растворителя, используемого в данном случае, являются те же самые растворители, что и растворители, используемые на стадии А-I, и предпочтительным примером является этанол.
Примерами гидроксиламина, используемого в данном случае, являются те же самые гидроксиламины, что и гидроксиламины, используемые на стадии А-I, и предпочтительным примером является 50%-ный (масс./масс.) водный раствор гидроксиламина.
Примеры реагента, используемого в данном случае, включают те же самые реагенты, что и реагенты, используемые на стадии А-I.
Температура реакции составляет от 0 до 150ºС, предпочтительно от 50 до 100ºС. Время реакции составляет от 10 минут до 24 часов, и предпочтительно от 30 минут до 5 часов.
Когда необходима обработка реакционной смеси, то такая обработка может быть проведена в соответствии со следующей методикой, например. Реакционную смесь охлаждают до комнатной температуры, затем растворитель отгоняют при пониженном давлении и полученный остаток промывают гексаном.
Стадия А-IV представляет собой стадию получения оксадиазолинового соединения (8) путем введения в реакцию соединения (6), полученного на стадии А-III, с галогенангидридом кислоты (7).
Примеры растворителя, используемого в данном случае, включают ТГФ, ДМФА, толуол и пиридин, и предпочтительным примером является пиридин.
Примеры реагента, используемого в данном случае, включают пиридин, триэтиламин, диизопропилэтиламин и гидрид натрия.
Температура реакции составляет от 20 до 150ºС, предпочтительно от 40 до 100ºС. Время реакции составляет от 30 минут до 24 часов, и предпочтительно от 30 минут до 10 часов.
Когда необходима обработка реакционной смеси, то такая обработка может быть проведена в соответствии со следующей методикой, например. К реакционной смеси добавляют насыщенный раствор хлорида аммония, воду или насыщенный рассол, продукт экстрагируют с использованием органического растворителя, такого как этилацетат, и полученный в результате органический слой сушат над сульфатом натрия. После удаления нерастворимого вещества растворитель отгоняют при пониженном давлении.
Стадия А-V представляет собой стадию получения соединения (9) путем гидролиза соединения (8), полученного на стадии А-IV.
Примеры растворителя, используемого в данном случае, включают ТГФ, метанол, этанол и изопропиловый спирт, и предпочтительным примером является метанол.
Примеры реагента, используемого в данном случае, включают водный раствор гидроксида натрия, водный раствор гидроксида калия и водный раствор гидроксида лития, и предпочтительным примером является водный раствор гидроксида натрия.
Температура реакции составляет от 0 до 130ºС, предпочтительно от 20 до 70ºС. Время реакции составляет от 30 минут до 12 часов, и предпочтительно от 30 минут до 4 часов.
Когда необходима обработка реакционной смеси, то такая обработка может быть проведена в соответствии со следующей методикой, например. Кислоту, такую как соляная кислота, добавляют к реакционной смеси, чтобы сделать реакционную смесь кислой или нейтральной, и продукт экстрагируют с использованием органического растворителя, такого как этилацетат. Полученный в результате органический слой сушат над осушителем, таким как сульфат натрия. Нерастворимое вещество удаляют, и затем растворитель отгоняют при пониженном давлении.
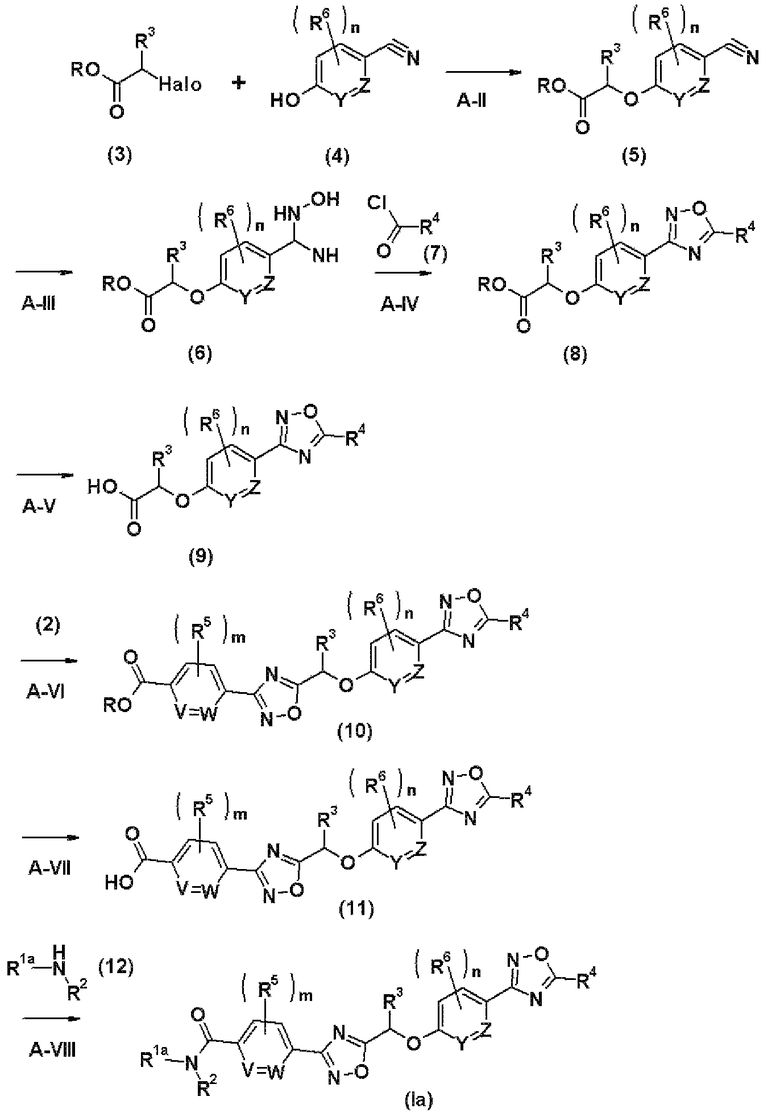
Стадия А-VI представляет собой стадию получения соединения (10) за счет введения в реакцию соединения (2), полученного на стадии А-I, с соединением (9), полученным на стадии А-V.
Примерами растворителя, используемого в данном случае, являются 3-диметил-2-имидазолидинон и ДМФА.
Примерами реагента, используемого в данном случае, являются 1-(3-диметиламинопропил)-3-этилкарбодиимид и 1-гидроксибензотриазол.
Температура реакции составляет от 30 до 130ºС, предпочтительно от 50 до 70ºС. Время реакции составляет от 30 минут до 12 часов, и предпочтительно от 30 минут до 6 часов.
Когда необходима обработка реакционной смеси, то такая обработка может быть проведена в соответствии со следующей методикой, например. К реакционной смеси добавляют воду, и затем продукт экстрагируют с использованием органического растворителя, такого как этилацетат. Полученный в результате органический слой промывают водой, насыщенным рассолом или др. и сушат над осушителем, таким как сульфат натрия. Растворитель отгоняют при пониженном давлении, и остаток очищают хроматографией на силикагеле.
Стадия A-VII представляет собой стадию получения соединения (11) путем гидролиза соединения (10), полученного на стадии А-VI.
Растворитель, реагент, реакционная температура, время реакции и обработка реакционной смеси, используемые в данном случае, являются такими же, как и на стадии А-V.
Стадия А-VIII представляет собой стадию получения соединения (Ia) настоящего изобретения за счет введения соединения (11), полученного на стадии А-VII в реакцию с аминным соединением (12) в присутствии конденсирующего агента.
Примерами растворителя, используемого в данном случае, являются метиленхлорид, тетрагидрофуран, 1,4-диоксан, ДМФА и диметилацетамид. Предпочтительными примерами являются метиленхлорид и ДМФА, и более предпочтительным примером является ДМФА.
Конденсирующий агент, используемый в данном случае, не имеет особенных ограничений, пока он представляет собой агент, используемый в реакциях амидирования, и можно использовать конденсирующие агенты, описанные в публикации R.C. Larock, Comprehensive Organic Transformations, Second Edition, 1999, John Wiley & Sons, Inc., и др. Конкретными примерами являются (i) эфиры фосфорной кислоты, такие как диэтилфосфорилцианид; (ii) карбодиимиды, такие как 1,3-дициклогексилкарбодиимид, 1,3-диизопропилкарбодиимид и 1-этил-3-(3-диметиламинопропил)-карбодиимид (WSC), и комбинации таких карбодиимидов и N-гидроксисоединений, таких как 4-гидроксибензотриазол; (iii) имидазолы, такие как 1,1'-карбонилдиимидазол (CDI); (iv) 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолинийхлорид (DMT-MM); и (v) фосфаты, такие как гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) и гексафторфосфат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HВTU). Предпочтительным примером является комбинация WSC и 4-гидроксибензотриазола.
Температура реакции составляет от 0 до 100ºС, и предпочтительно от 0 до 50ºС. Время реакции составляет от 30 минут до 96 часов, и предпочтительно от 1 до 12 часов.
Когда необходима обработка реакционной смеси, то такая обработка может быть проведена в соответствии со следующей методикой, например. К реакционной смеси добавляют воду, и затем продукт экстрагируют с использованием органического растворителя, такого как этилацетат. Полученный в результате органический слой промывают водой, насыщенным рассолом и т.д. и сушат над осушителем, таким как сульфат натрия. Растворитель отгоняют при пониженном давлении, и остаток очищают хроматографией на силикагеле.
Способ В
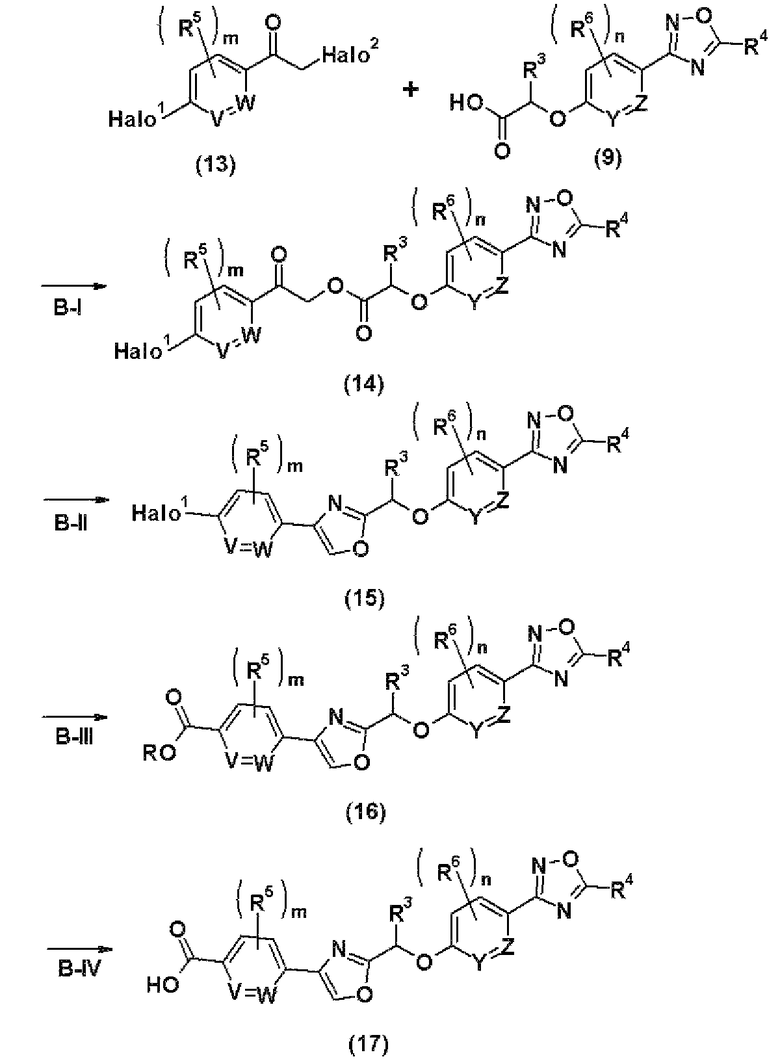
где Halo1 и Halo2 каждый независимо друг от друга представляют собой атом галогена; и R, R1a, V, W, Y, Z, R2, R3, R4, R5, R6, m и n соответственно имеют те же самые значения, как и описанные выше.
Стадия В-I представляет собой стадию получения соединения (14) путем конденсации соединения (9), полученного на стадии А-V, описанной выше, с соединением (13).
Примерами растворителя, используемого в данном случае, являются ТГФ, ДМФА, 1,4-диоксан, ацетонитрил и ацетон, и предпочтительным примером является ДМФА или ацетон.
Примерами реагента, используемого в данном случае, являются трет.-бутоксид калия, карбонат цезия, карбонат калия, карбонат натрия, гидрид натрия, триэтиламин и диизопропилэтиламин, и предпочтительным примером является триэтиламин.
Температура реакции составляет от 0 до 100ºС, предпочтительно от 20 до 80ºС. Время реакции составляет от 30 минут до 24 часов, и предпочтительно от 30 минут до 6 часов.
Когда необходима обработка реакционной смеси, то такая обработка может быть проведена в соответствии со следующей методикой, например. Реакционную смесь экстрагируют органическим растворителем, таким как этилацетат, и полученный в результате органический слой промывают последовательно водой и насыщенным рассолом. Затем органический слой сушат над осушителем, таким как сульфат натрия или безводный сульфат натрия, а затем полученный остаток очищают хроматографией на силикагеле.
Стадия В-II представляет собой стадию получения соединения (15) циклизацией соединения (14), полученного на стадии В-I.
Примерами растворителя, используемого в данном случае, являются толуол и уксусная кислота.
Примерами реагента, используемого в данном случае, являются трифторацетат аммония и ацетат аммония, и предпочтительным примером является трифторацетат аммония.
Температура реакции составляет от 80 до 200ºС, предпочтительно от 100 до 160ºС. Время реакции составляет от 30 минут до 24 часов, и предпочтительно от 30 минут до 12 часов.
Когда необходима обработка реакционной смеси, то такая обработка может быть проведена в соответствии со следующей методикой, например. К реакционной смеси добавляют воду, и проводят экстракцию органическим растворителем, таким как этилацетат. Полученный в результате органический слой промывают последовательно водой и насыщенным рассолом. Затем растворитель отгоняют при пониженном давлении, и полученный остаток очищают хроматографией на силикагеле.
Стадия В-III представляет собой стадию получения соединения (16) из соединения (15), полученного на стадии В-II, в атмосфере монооксида углерода в присутствии основания с использованием палладиевого катализатора.
Примерами растворителя, используемого в данном случае, являются метанол и смешанный растворитель метанол/ДМФА, и предпочтительным примером является смешанный растворитель метанол/ДМФА.
Примерами основания, используемого в данном случае, являются триэтиламин, диизопропиэтиламин и трибутиламин, и предпочтительным примером является триэтиламин.
Примерами палладиевого катализатора, используемого в данном случае, являются ацетат палладия(II), дибензилиденацетон палладия(0), тетракис(трифенилфосфин)палладий(0), хлорид палладия(II), хлорид бис(трифенилфосфин)палладия(II) и (дифенилфосфиноферроцен)палладийхлорид(II), и предпочтительным примером является ацетат палладия(II) или хлорид (дифенилфосфиноферроцен)палладия (II).
Примерами реагента, используемого в данном случае, являются трифенилфосфин, трициклогексилфосфин, 1,2-бис(дифенилфосфоно)этан, 1,3-бис(дифенилфосфоно)пропан, 2,2'-бис(дифенилфосфанил)-1,1'-бинафтил, 2-(дициклогексилфосфоно)бифенил и 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил, и предпочтительными примерами являются трифенилфосфин или 1,3-бис(дифенилфосфоно)пропан.
Температура реакции составляет от 0 до 130ºС, предпочтительно от 20 до 90ºС. Время реакции составляет от 30 минут до 12 часов, и более предпочтительно от 30 минут до 4 часов.
Когда необходима обработка реакционной смеси, то такая обработка может быть проведена в соответствии со следующей методикой, например. К реакционной смеси добавляют воду, и экстракцию проводят органическим растворителем, таким как этилацетат. Полученный в результате органический слой промывают последовательно водой и насыщенным рассолом. Затем растворитель отгоняют при пониженном давлении, и полученный остаток очищают хроматографией на силикагеле.
Стадия В-IV представляет собой стадию получения соединения (17) гидролизом соединения (16), полученного на стадии В-III.
Растворитель, реагент, реакционная температура, время реакции и обработка реакционной смеси, используемые в данном случае, являются теми же самыми, что и на стадии А-VII, описанной выше.
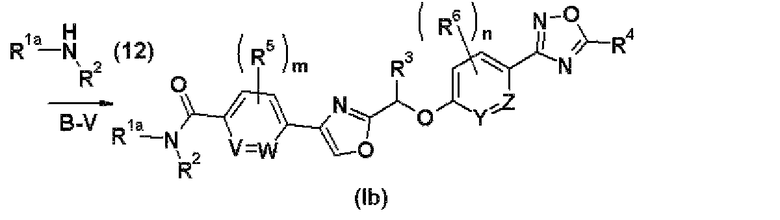
Стадия В-V представляет собой стадию получения соединения (Ib) настоящего изобретения путем введения соединения (17), полученного на стадии В-IV, в реакцию с аминным соединением (12) в присутствии конденсирующего агента.
Растворитель, конденсирующий агент, реакционная температура, время реакции и обработка реакционной смеси, используемые в данном случае, являются теми же самыми, что и на стадии А-VIII.
Способ С
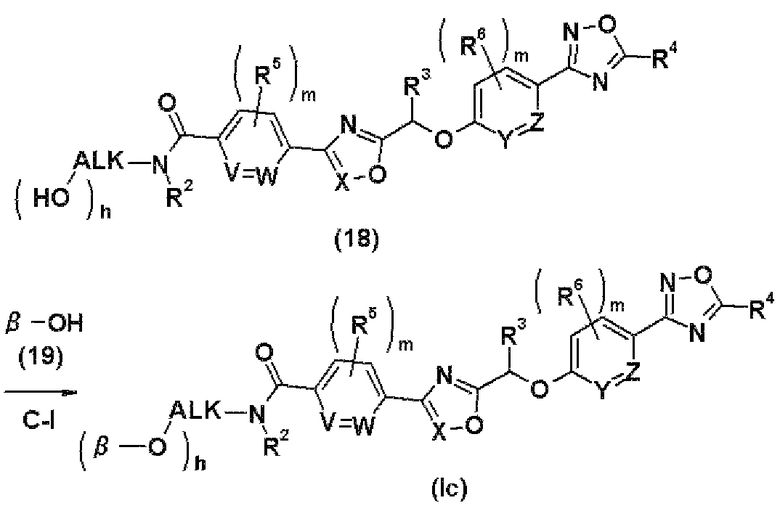
где ALK представляет собой С1-С6-алкиленовую группу; h имеет значение 1 или 2; и V, W, Y, Z, R2, R3, R4, R5, R6, m, n и β соответственно имеют те же самые значения, как и описанные выше.
Стадия С-I представляет собой стадию получения соединения (Ic) настоящего изобретения путем введения соединения (18) между соединением (Ia) и соединением (Ib) настоящего изобретения, в котором R1 представляет собой С1-С6-алкильную группу, замещенную одной или двумя гидроксильными группами, в реакцию с соединением (18) в присутствии конденсирующего агента.
Растворитель, конденсирующий агент, реакционная температура, время реакции и обработка реакционной смеси, используемые в данном случае, являются теми же самыми, что и на стадии А-VIII.
Соединение настоящего изобретения может быть получено путем использования способов, описанных выше, и также может быть легко получено из известных соединений в соответствии со справочными примерами и примерами, которые будут описаны ниже.
Соединение настоящего изобретения или его фармацевтически приемлемая соль, полученные способами, описанными выше, обладают прекрасным гипогликемическим действием, и могут быть использованы в качестве активного ингредиента фармацевтической композиции, которая может быть использована при лечении и/или предупреждении диабета 1 типа, диабета 2 типа, гестационного диабета, гипергликемии вследствие других факторов, приобретенной непереносимости глюкозы (IGT), ожирения, ассоциирующихся с диабетом заболеваний (например, гиперлипидемии, гиперхолестеринемии, аномального липидного метаболизма, гипертензии, ожирения печени, метаболического синдрома, водянки, сердечной недостаточности, стенокардии, инфаркта миокарда, атеросклероза, гиперурикемии и подагры) или диабетических осложнений (например, рестеноза, почечной недостаточности, невропатии, катаракты, гангрены ноги, инфекций и кетоза).
Кроме того, соединение настоящего изобретения или его фармацевтически приемлемая соль обладают прекрасным защитным действием в отношении β-клеток или поджелудочной железы, и, следовательно, могут быть использованы в качестве активного ингредиента фармацевтической композиции, которая может быть использована для защиты β-клеток или поджелудочной железы.
Соединение настоящего изобретения также может быть использовано в комбинации с терапевтическим лекарственным средством для диабета, терапевтическим лекарственным средством для диабетических осложнений, терапевтическим лекарственным средством для гиперлипидемии, терапевтическим лекарственным средством для гипертензии и подобными средствами, отличными от соединения настоящего изобретения.
При введении фармацевтической композиции, содержащей соединение настоящего изобретения или его фармацевтически приемлемую соль, млекопитающему (например, человеку, лошади, корове или свинье; предпочтительно человеку) фармацевтическая композиция может быть введена системно или местно и перорально или парентерально.
Фармацевтическая композиция настоящего изобретения может быть получена в соответствии со способами получения различных обычно используемых препаратов путем выбора соответствующих дозированных лекарственных форм в зависимости от способа введения.
Примерами дозированных лекарственных форм фармацевтической композиции для перорального применения являются таблетки, пилюли, порошки, гранулы, капсулы, жидкости, суспензии, эмульсии, сиропы и эликсиры. Фармацевтические композиции таких дозированных лекарственных форм могут быть получены обычными способами путем соответствующего выбора по необходимости наполнителей, связующих веществ, диспергирующих добавок, смазывающих агентов, набухающих агентов, способствующих набуханию добавок, покрывающих агентов, пластификаторов, стабилизаторов, антисептиков, антиоксидантов, красящих веществ, способствующих растворению добавок, суспендирующих агентов, эмульгаторов, подсластителей, консервантов, буферов, разбавителей, смачивающих агентов и т.д., которые обычно используют в качестве добавок.
Примерами дозированных лекарственных форм фармацевтических композиций для парентерального применения являются инъецируемые препараты, мази, гели, кремы, примочки, пластыри, аэрозоли, средства для ингаляции, спреи, глазные капли, капли для носа и свечи. Фармацевтические композиции таких дозированных лекарственных форм могут быть получены обычными способами путем соответствующего выбора по необходимости стабилизаторов, антисептиков, способствующих растворению добавок, увлажнителей, консервантов, антиоксидантов, ароматизаторов, желирующих агентов, нейтрализующих агентов, буферов, изотонических агентов, поверхностно-активных веществ, красящих веществ, буферных агентов, загустителей, смачивающих агентов, наполнителей, стимулирующих абсорбцию агентов, суспендирующих агентов, связующих веществ и т.д., которые обычно используют в качестве добавок.
Количество вводимого соединения настоящего изобретения или его фармацевтически приемлемой соли может меняться в зависимости от симптомов, возраста, массы тела или др. Однако в случае перорального введения соединение или соль вводят один раз или несколько раз в день в количестве от 1 до 2000 мг и предпочтительно от 1 до 400 мг из расчета на соединение на дозу для взрослых; и в случае парентерального введения соединение или соль вводят один раз или несколько раз в день в количестве от 0,01 до 500 мг и предпочтительно от 0,1 до 300 мг из расчета на соединение на дозу для взрослых.
Далее настоящее изобретение будет описано более подробно с помощью справочных примеров, примеров, рецептурного примера и примеров испытаний, но объем настоящего изобретения, как подразумевают, ими не ограничен.
ПРИМЕРЫ
Справочный пример 1
Метил-4-циано-2-фторбензоат
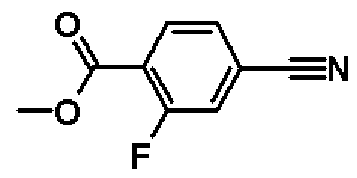
К раствору в метаноле (70,0 мл) 4-циано-2-фторбензойной кислоты (10,0 г, 60,6 ммоль) добавляют 4 М раствор соляной кислоты в диоксане (70,0 мл, 280 ммоль), и смесь перемешивают один час при 70ºС. Реакционную смесь охлаждают до комнатной температуры, и затем растворитель отгоняют при пониженном давлении. В результате получают сырой продукт названного соединения.
Справочный пример 2
Метил-4-амино(гидроксиимино)метил-2-фторбензоат
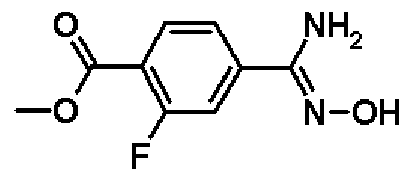
К раствору в этаноле (100 мл) соединения, полученного в справочном примере 1 (11,0 г, 66,6 ммоль), добавляют 50%-ный водный раствор гидроксиламина (3,20 мл, 100 ммоль), и смесь перемешивают 3 часа при 70ºС. Реакционную смесь охлаждают до комнатной температуры, затем растворитель отгоняют при пониженном давлении и остаток промывают гексаном. В результате получают названное соединение (9,10 г, выход 71%).
1H-ЯМР (500 МГц, CD3OD) δ м.д.: 7,93 (1Н, т, J=8 Гц), 7,56 (1Н, д, J=8 Гц), 7,50 (1Н, д, J=12 Гц), 3,91 (3Н, с).
Справочный пример 3
Этил-2-(4-цианофенокси)бутаноат

Карбонат калия (14,5 г, 105 ммоль) добавляют при комнатной температуре к раствору в ацетонитриле (80,0 мл) 4-цианофенола (5,00 г, 42,0 ммоль) и этил-2-бромбутирата (9,83 г, 50,4 ммоль), и смесь перемешивают 3 часа при 80ºС. После охлаждения смеси до комнатной температуры к реакционной смеси добавляют воду и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 9:1 → 2:1, об./об.). В результате получают названное соединение (9,79 г, выход 100%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 7,58 (2H, д, J=9 Гц), 6,92 (2H, д, J=9 Гц), 4,61 (1H, т, J=6 Гц), 4,25-4,20 (2H, м), 2,06-1,99 (2H, м), 1,25 (3H, т, J=7 Гц), 1,08 (3H, т, J=7 Гц).
Справочный пример 4
Этил-2-{4-[амино(гидроксиимино)метил]фенокси}бутаноат
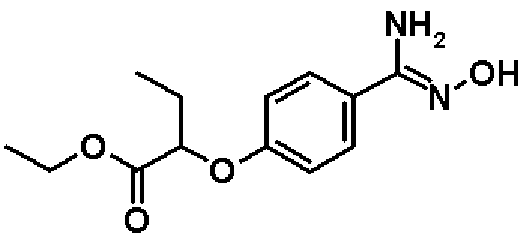
К раствору в этаноле (42,0 мл) соединения, полученного в справочном примере 3 (9,97 г, 42,0 ммоль), при комнатной температуре добавляют 50%-ный водный раствор гидроксиламина (8,32 мл, 126 ммоль), и смесь перемешивают 2,5 часа при 80ºС. После охлаждения реакционной смеси до комнатной температуры к реакционной смеси добавляют воду и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 1:1 → 0:1, об./об.). В результате получают названное соединение (9,82 г, выход 88%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 7,54 (2H, д, J=9 Гц), 6,89 (2H, д, J=9 Гц), 4,81 (2H, с), 4,58 (1H, т, J=6 Гц), 4,22 (2H, кв, J=7 Гц), 2,03-1,97 (2H, м), 1,24 (3H, т, J=7 Гц), 1,08 (3H, т, J=8 Гц).
Справочный пример 5
Этил-2-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]бутаноат
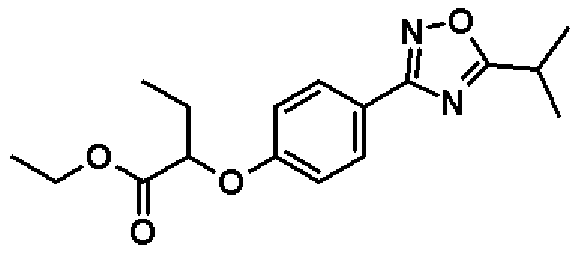
К раствору в пиридине (16,0 мл) соединения, полученного в справочном примере 4 (3,00 г, 11,3 ммоль), при комнатной температуре добавляют хлорангидрид изомасляной кислоты (1,29 мл, 12,4 ммоль), и смесь перемешивают 2 часа при 100ºС. После охлаждения смеси до комнатной температуры реакционную смесь концентрируют при пониженном давлении и добавляют воду. Смесь экстрагируют два раза этилацетатом, и полученный таким образом органический слой промывают 1 М водным раствором соляной кислоты и насыщенным рассолом и сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этиацетат = 3:1, об./об.). В результате получают названное соединение (3,25 г, выход 91%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,00 (2H, д, J=9 Гц), 6,95 (2H, д, J=9 Гц), 4,62 (1H, т, J=6 Гц), 4,22 (2H, кв, J=7 Гц), 3,31-3,22 (1H, м), 2,05-1,99 (2H, м), 1,45 (6H, д, J=7 Гц), 1,24 (3H, т, J=7 Гц), 1,10 (3H, т, J=7 Гц).
Справочный пример 6
2-[4-(5-Изопропил-1,2,4-оксадиазол-3-ил)фенокси]бутановая кислота

Соединение, полученное в справочном примере 5 (1,50 г, 4,71 ммоль), растворяют в смеси тетрагидрофурана (6,00 мл) и метанола (6,00 мл) и добавляют 1 М водный раствор гидроксида натрия (5,65 мл, 5,65 ммоль). Смесь перемешивают 30 минут при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении и добавляют воду и 1 М водный раствор соляной кислоты. Смесь экстрагируют два раза этилацетатом, и полученный таким образом органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении, и полученный остаток промывают гексаном. В результате получают названное соединение (1,30 г, выход 95%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,00 (2H, д, J=9 Гц), 6,98 (2H, д, J=9 Гц), 4,69 (1H, дд, J=6 Гц, 5 Гц), 3,31-3,25 (1H, м), 2,10-2,03 (2H, м), 1,45 (6H, д, J=7 Гц), 1,13 (3H, т, J=7 Гц).
Справочный пример 7
Метил-2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоат
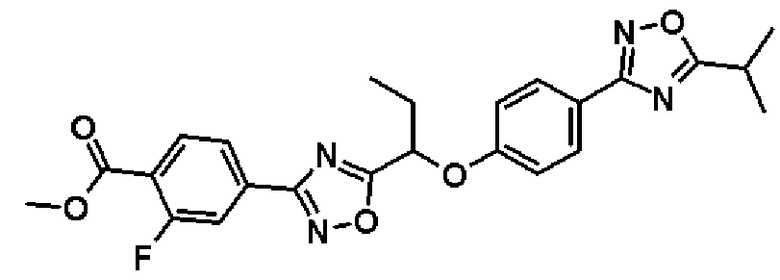
К раствору в диметилформамиде (24,0 мл) соединения, полученного в справочном примере 6 (1,37 г, 4,71 ммоль), при комнатной температуре добавляют моногидрат 1-гидроксибензотриазола (722 мг, 4,71 ммоль) и N-(3-диметиламинопропил)-N'-этилкарбодиимид (1,81 г, 9,43 ммоль). Смесь перемешивают 15 минут при той же температуре. Добавляют соединение, полученное в справочном примере 2 (1,00 г, 4,71 ммоль), полученную смесь перемешивают 15 минут и дополнительно перемешивают 3 часа при 100ºС. Реакционную смесь охлаждают до комнатной температуры, затем к реакционной смеси добавляют воду, и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным рассолом, и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 1:0 → 3:1, об./об.). В результате получают названное соединение (1,65 г, выход 75%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,05 (1H, дд, J=10 Гц, 8 Гц), 8,01 (2H, д, J=9 Гц), 7,92 (1H, дд, J=8 Гц, 2 Гц), 7,87 (1H, дд, J=10 Гц, 2 Гц), 7,06 (2H, д, J=9 Гц), 5,51 (1H, дд, J=7 Гц, 6 Гц), 3,96 (3H, с), 3,29-3,22 (1H, м), 2,34-2,20 (2H, м), 1,43 (6H, д, J=7 Гц), 1,14 (3H, т, J=7 Гц).
Справочный пример 8
2-Фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензойная кислота

К раствору в смеси тетрагидрофурана (4,00 мл) и метанола (4,00 мл) соединения, полученного в справочном примере 7 (1,64 г, 3,52 ммоль) добавляют 1 М водный раствор гидроксида натрия (4,22 мл, 4,22 ммоль), и смесь перемешивают один час при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении, добавляют воду и водный слой промывают диэтиловым эфиром. Затем добавляют 1 М водный раствор соляной кислоты, и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении, и полученный остаток промывают смесью гексана и диэтилового эфира (10:1, об./об.). В результате получают названное соединение (1,44 г, выход 91%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,14 (1H, дд, J=10 Гц, 8 Гц), 8,01 (2H, д, J=9 Гц), 7,97 (1H, дд, J=8 Гц, 1 Гц), 7,91 (1H, дд, J=10 Гц, 1 Гц), 7,07 (2H, д, J=9 Гц), 5,52 (1H, дд, J=7 Гц, 6 Гц), 3,29-3,23 (1H, м), 2,35-2,20 (2H, м), 1,44 (6H, д, J=7 Гц), 1,15 (3H, т, J=8 Гц).
Справочный пример 9
Метил-4-бром-2-метилбензоат

К раствору в метаноле (20,0 мл) 4-бром-2-метилбензойной кислоты (2,00 г, 9,30 ммоль) добавляют концентрированную серную кислоту (500 мкл) и смесь перемешивают 5 часов при 80ºС. Реакционную смесь охлаждают до комнатной температуры, затем к реакционной смеси добавляют насыщенный водный раствор гидрокарбоната натрия, и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают один раз насыщенным рассолом, и сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении, и в результате получают названное соединение (2,02 г, выход 95%).
1Н-ЯМР (400 МГц, CDCl3) δ м.д.: 7,78 (1H, д, J=9 Гц), 7,41 (1H, д, J=2 Гц), 7,38 (1H, дд, J=9 Гц, 2 Гц), 3,89 (3H, с), 2,58 (3H, с).
Справочный пример 10
Метил-4-циано-2-метилбензоат
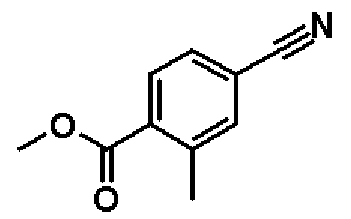
К раствору в диметилацетамиде (4,62 мл) соединения, полученного в справочном примере 9 (498 мг, 2,32 ммоль), добавляют цианид цинка (163 мг, 1,39 ммоль), трис(дибензилиденацетон)дипалладий(0) (42 мг, 46,3 мкмоль) и 1,1'-бис(дифенилфосфино)ферроцен (51 мг, 92,6 мкмоль), и смесь перемешивают 2 часа при 90ºС. Реакционную смесь охлаждают до комнатной температуры, затем к реакционной смеси добавляют насыщенный водный раствор хлорида аммония, и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают водой и насыщенным рассолом и сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении, и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этиацетат = 4:1 → 1:1, об./об.). В результате получают названное соединение (294 мг, выход 73%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 7,97 (1H, д, J=8 Гц), 7,55-7,53 (2H, м), 3,93 (3H, с), 2,62 (3H, с).
Справочный пример 11
Метил-4-[амино(гидроксиимино)метил]-2-метилбензоат

Синтез проводят таким же образом, как и в справочном примере 2, за исключением того, что соединение, полученное в справочном примере 10 (294 мг, 1,68 ммоль), используют вместо метил-4-циано-2-фторбензоата. В результате получают названное соединение (255 мг, выход 73%).
1H-ЯМР (400 МГц, CD3OD) δ м.д.: 7,89 (1H, д, J=8 Гц), 7,58 (1H, с), 7,55 (1H, д, J=8 Гц), 3,88 (3H, с), 2,59 (3H, с).
Справочный пример 12
Метил-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-метилбензоат

Синтез проводят таким же образом, как и в справочном примере 7, за исключением того, что соединение, полученное в справочном примере 11 (120 мг, 0,576 ммоль), используют вместо метил-4-амино(гидроксиимино)метил-2-фторбензоата. В результате получают названное соединение (200 мг, выход 75%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,02-7,93 (5H, м), 7,07 (2H, д, J=9 Гц), 5,51 (1H, т, J=6 Гц), 3,92 (3H, с), 3,25 (1H, септ, J=7 Гц), 2,66 (3H, с), 2,34-2,20 (2H, м), 1,43 (6H, д, J=7 Гц), 1,14 (3H, т, J=7 Гц).
Справочный пример 13
4-(5-{1-[4-(5-Изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-метилбензойная кислота
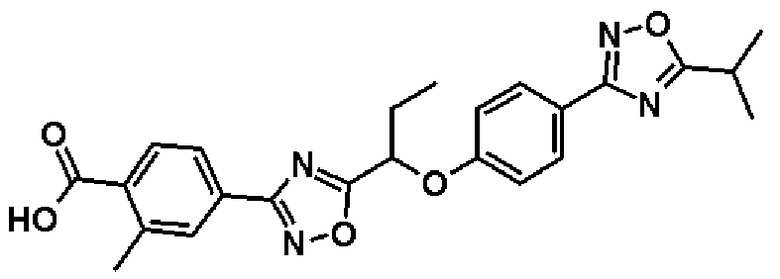
Синтез проводят таким же образом, как и в справочном примере 8, за исключением того, что соединение, полученное в справочном примере 12 (154 мг, 0,333 ммоль), используют вместо метил-2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоата. В результате получают названное соединение (113 мг, выход 76%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,15 (1H, д, J=8 Гц), 8,02-7,98 (4H, м), 7,08 (2H, д, J=9 Гц), 5,52 (1H, т, J=6 Гц), 3,26 (1H, септ, J=7 Гц), 2,72 (3H, с), 2,35-2,21 (2H, м), 1,44 (6H, д, J=7 Гц), 1,15 (3H, т, J=7 Гц).
Справочный пример 14
(2R)-2-{[2-Фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицинат
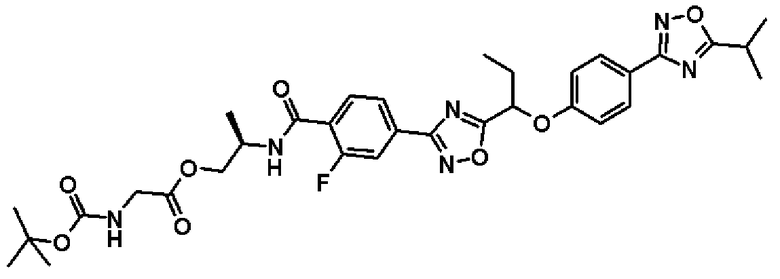
К раствору в диметилформамиде (1,00 мл) соединения, полученного в примере 7, который будет описан ниже, (69,2 мг, 0,136 ммоль) добавляют N-(трет.-бутоксикарбонил)глицин (47,6 мг, 0,272 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид (78,1 мг, 0,405 ммоль) и 4-диметиламинопиридин (1,70 мг, 0,0183 ммоль) и смесь перемешивают один час при комнатной температуре. К реакционной смеси добавляют воду, смесь экстрагируют два раза этилацетатом, полученный таким образом органический слой последовательно промывают водой и насыщенным рассолом и сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (метиленхлорид:метанол = 99:1 → 90:10, об./об.). В результате получают названное соединение (91 мг, выход 100%).
Справочный пример 15
(2S)-2-{[2-Фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицинат
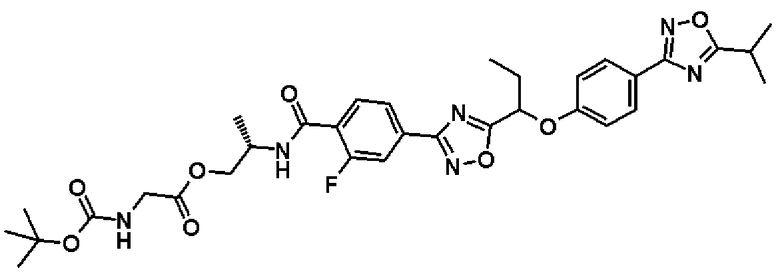
Синтез проводят таким же образом, как и в справочном примере 14, за исключением того, что соединение, полученное в примере 6, который будет описан ниже, (100 мг, 0,196 ммоль), используют вместо 2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамида. В результате получают сырой продукт названного соединения (125 мг).
Справочный пример 16
(2R)-2-{[2-Фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}бутил-N-(трет.-бутоксикарбонил)глицинат
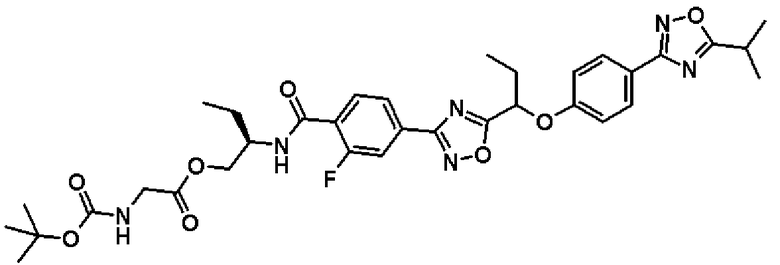
Синтез проводят таким же образом, как и в справочном примере 14, за исключением того, что соединение, полученное в примере 10, который будет описан ниже, (274 мг, 0,523 ммоль), используют вместо 2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамида. В результате получают сырой продукт названного соединения (358 мг).
Справочный пример 17
(2S)-2-{[2-Фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}бутил-N-(трет.-бутоксикарбонил)глицинат
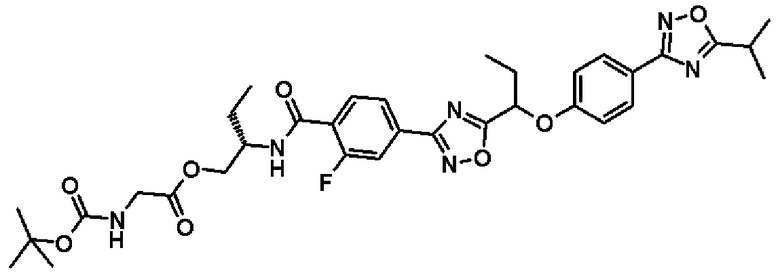
Синтез проводят таким же образом, как и в справочном примере 14, за исключением того, что соединение, полученное в примере 9, который будет описан ниже, (220 мг, 0,420 ммоль), используют вместо 2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамида. В результате получают сырой продукт названного соединения (274 мг).
Справочный пример 18
2-{[2-Фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}-2-метилпропил-N-(трет.-бутоксикарбонил)глицинат
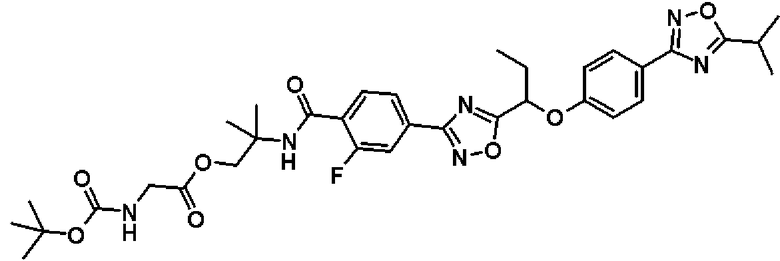
Синтез проводят таким же образом, как и в справочном примере 14, за исключением того, что соединение, полученное в примере 8, который будет описан ниже, (133 мг, 0,254 ммоль), используют вместо 2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамида. В результате получают названное соединение (173 мг, выход 100%).
Справочный пример 19
4-Метоксибензилгликолят

Гидрокарбонат натрия (2,20 г, 26,3 мг) добавляют к раствору гликолевой кислоты (2,00 г, 26,3 ммоль) в воде (2,00 мл), и смесь перемешивают один час при комнатной температуре. Растворитель отгоняют из реакционной смеси при пониженном давлении, и к раствору в диметилформамиде (10 мл) полученного остатка добавляют п-метоксибензилхлорид (4,10 г, 26,3 ммоль). Смесь перемешивают 2 часа при комнатной температуре. К реакционной смеси добавляют воду, и смесь экстрагируют этилацетатом. Полученный таким образом органический слой последовательно промывают водой и насыщенным рассолом и сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении. В результате получают сырой продукт названного соединения (4,24 г).
Справочный пример 20
4-Метоксибензил-{[(аллилокси)карбонил]окси}ацетат

К раствору в метиленхлориде (70,0 мл) соединения, полученного в справочном примере 19 (4,25 г, 25,7 ммоль), добавляют 4-диметиламинопиридин (3,80 г, 2,57 ммоль) и раствор в метиленхлориде (10,0 мл) аллилхлорформиата (3,30 мл, 30,8 ммоль), и смесь перемешивают 30 минут при комнатной температуре. К реакционной смеси добавляют воду и смесь экстрагируют метиленхлоридом. Полученный таким образом органический слой промывают водой и сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 19:1 → 1:1, об./об.). В результате получают сырой продукт названного соединения (4,69 г).
Справочный пример 21
[(Аллилокси)карбонил]оксиуксусная кислота
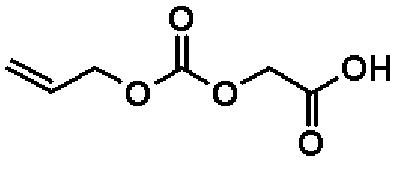
Трифторметансульфоновую кислоту (20,0 мл) и анизол (3,40 мл, 49,6 ммоль) добавляют к соединению, полученному в справочном примере 20 (4,69 г, 16,5 ммоль), и смесь перемешивают 2 часа при комнатной температуре. К реакционной смеси добавляют толуол (100 мл) и растворитель отгоняют при пониженном давлении. В результате получают сырой продукт названного соединения (2,90 г).
Справочный пример 22
(2R)-2-{[2-Фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-{[(аллилокси)карбонил]окси}ацетат
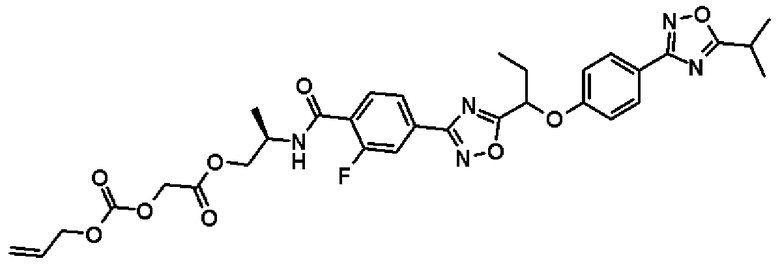
К раствору в диметилформамиде (7,00 мл) соединения, полученного в примере 7, который будет описан ниже, (344 мг, 0,676 ммоль), добавляют соединение, полученное в справочном примере 21 (163 мг, 1,01 ммоль), 4-диметиламинопиридин (8,26 мг, 0,0676 ммоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимид (389 мг, 2,03 ммоль), и смесь перемешивают 2 часа при комнатной температуре. К реакционной смеси добавляют воду и смесь экстрагируют этилацетатом. Полученный таким образом органический слой последовательно промывают водой и насыщенным рассолом и сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 9:1 → 1:1, об./об.). В результате получают названное соединение (300 мг, выход 75%).
Справочный пример 23
2-Диазо-1-(4-бром-3-фторфенил)этанон

К раствору в метиленхлориде (50,0 мл) 4-бром-3-фторбензойной кислоты (5,00 г, 22,8 ммоль) при комнатной температуре добавляют диметилформамид (5 капель) и оксалилхлорид (2,39 мл, 27,4 ммоль), и смесь перемешивают 1,5 часа при той же температуре. Затем отгоняют растворитель при пониженном давлении. Полученный остаток растворяют в смеси тетрагидрофурана (15,0 мл) и ацетонитрила (15,0 мл) и к раствору при комнатной температуре добавляют триэтиламин (6,36 мл, 45,7 ммоль) и 2 М раствор триметилсилилдиазометана в тетрагидрофуране (22,8 мл, 45,7 ммоль). Смесь перемешивают 30 минут. К реакционной смеси добавляют воду, и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении. Полученный остаток промывают смесью гексан:этилацетат (10:1 об./об.). В результате получают сырой продукт названного соединения (3,36 г).
Справочный пример 24
2-Бром-1-(4-бром-3-фторфенил)этанон

Добавляют 30% раствор (бромистый водород)-(уксусная кислота) (3,50 мл, избыток) к раствору в метиленхлориде (14,0 мл) соединения, полученного в справочном примере 23 (3,35 г, приблизительно 13,8 ммоль) при охлаждении водой со льдом, и смесь перемешивают 10 минут при той же температуре. К реакционной смеси добавляют воду, и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным рассолом и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении. Полученный остаток промывают смесью гексан:этилацетат (10:1 об./об.). В результате получают сырой продукт названного соединения (3,92 г).
Справочный пример 25
2-(4-Бром-3-фторфенил)-2-оксоэтил-2-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]бутаноат

Триэтиламин (353 мкл, 2,53 ммоль) при комнатной температуре добавляют к раствору в ацетоне (9,00 мл) соединения, полученного в справочном примере 24, (500 мг, 1,69 ммоль), и соединения, полученного в справочном примере 6 (540 мг, 1,86 ммоль), и смесь перемешивают 2 часа при той же температуре. К реакционной смеси добавляют воду, и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают насыщенным раствором гидрокарбоната натрия и насыщенным рассолом и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении. В результате получают сырой продукт названного соединения (854 мг).
Справочный пример 26
3-(4-{1-[4-(4-Бром-3-фторфенил)-1,3-оксазол-2-ил]пропокси}фенил)-5-изопропил-1,2,4-оксадиазол

Трифторацетат аммония (1,70 г, избыток) добавляют к соединению, полученному в справочном примере 25 (854 мг, 1,69 ммоль), и смесь перемешивают 6 часов при 150ºС. Реакционную смесь охлаждают до комнатной температуры, затем к реакционной смеси добавляют воду и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают водой и насыщенным рассолом, а затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 9:1 → 2:1, об./об.). В результате получают названное соединение (554 мг).
Справочный пример 27
2-Фтор-4-(2-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,3-оксазол-4-ил)бензойная кислота

К раствору в смеси диметилформамида (2,50 мл) и метанола (2,50 мл) соединения, полученного в справочном примере 26 (496 мг, 1,02 ммоль), при комнатной температуре добавляют триэтиламин (426 мкл, 3,06 ммоль) и комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) и метиленхлорида (167 мг, 0,204 ммоль), и смесь перемешивают 5 часов при 80ºС в токе монооксида углерода. Реакционную смесь охлаждают до комнатной температуры, затем к реакционной смеси добавляют воду и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток (300 мг) растворяют в смеси тетрагидрофурана (1,60 мл) и метанола (800 мкл). К раствору добавляют 1 М водный раствор гидроксида натрия (773 мкл, 0,773 ммоль) и смесь перемешивают 2,5 часа при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении и добавляют воду. Водный слой промывают диэтиловым эфиром и затем добавляют 1 М водный раствор соляной кислоты. Смесь экстрагируют два раза этилацетатом, полученный таким образом органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток промывают смесью гексан:(диэтиловый эфир) (10:1 об./об.). В результате получают сырой продукт названного соединения (262 мг).
Справочный пример 28
(2S)-2-{[2-Фтор-4-(2-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,3-оксазол-4-ил]бензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицинат
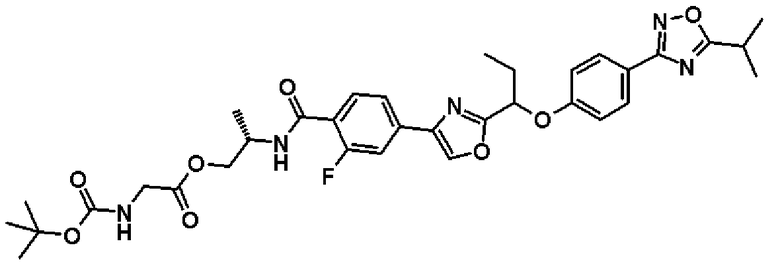
Синтез проводят таким же образом, как и в справочном примере 14, за исключением того, что соединение, полученное в примере 18, который будет описан ниже, (20,0 мг, 39,3 мкмоль), используют вместо 2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамида. В результате получают названное соединение (26 мг).
Справочный пример 29
Этил-2-[4-(5-этил-1,2,4-оксадиазол-3-ил)фенокси]бутаноат
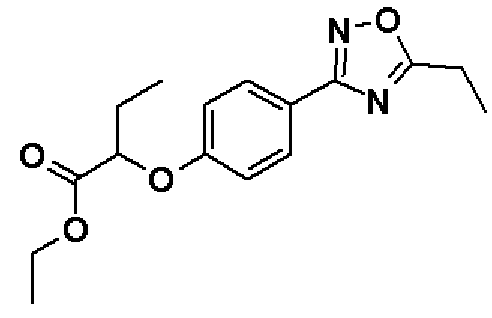
Синтез проводят таким же образом, как и в справочном примере 5, за исключением того, что используют соединение, полученное в справочном примере 4 (4,10 г, 15,4 ммоль), и хлорангидрид пропионовой кислоты (1,47 мл, 17,0 ммоль) используют вместо хлорангидрида изобутановой кислоты. В результате получают названное соединение (1,95 мг, выход 42%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,00 (2H, д, J=8 Гц), 6,96 (2H, д, J=8 Гц), 4,63 (1H, т, J=6 Гц), 4,23 (2H, кв, J=7 Гц), 2,96 (2H, кв, J=7 Гц), 2,02 (2H, кв, J=7 Гц), 1,44 (3H, т, J=7 Гц), 1,25 (3H, т, J=7 Гц), 1,10 (3H, т, J=7 Гц).
Справочный пример 30
2-[4-(5-Этил-1,2,4-оксадиазол-3-ил)фенокси]бутановая кислота

Синтез проводят таким же образом, как и в справочном примере 6, за исключением того, что соединение, полученное в справочном примере 29 (1,95 г, 6,41 ммоль), используют вместо этил-2-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]бутаноата. В результате получают названное соединение (1,77 г, выход 100%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,00 (2H, д, J=9 Гц), 6,99 (2H, д, J=9 Гц), 4,69 (1H, т, J=6 Гц), 2,97 (2H, кв, J=8 Гц), 2,11-2,05 (2H, м), 1,44 (3H, т, J=8 Гц), 1,13 (3H, т, J=8 Гц).
Справочный пример 31
Метил-4-(5-{1-[4-(5-этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензоат

Синтез проводят таким же образом, как и в справочном примере 7, за исключением того, что соединение, полученное в справочном примере 30 (1,97 г, 7,13 ммоль), используют вместо 2-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]бутановой кислоты. В результате получают названное соединение (1,94 г, выход 60%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,07-7,99 (3H, м), 7,93 (1H, дд, J=8 Гц, 2 Гц), 7,88 (1H, дд, J=11 Гц, 2 Гц), 7,07 (2H, д, J=9 Гц), 5,51 (1H, т, J=6 Гц), 3,96 (3H, с), 2,96 (2Н, кв, J=7 Гц), 2,34-2,18 (2H, м), 1,43 (3H, т, J=7 Гц), 1,15 (3H, т, J=7 Гц).
Справочный пример 32
4-(5-{1-[4-(5-Этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензойная кислота
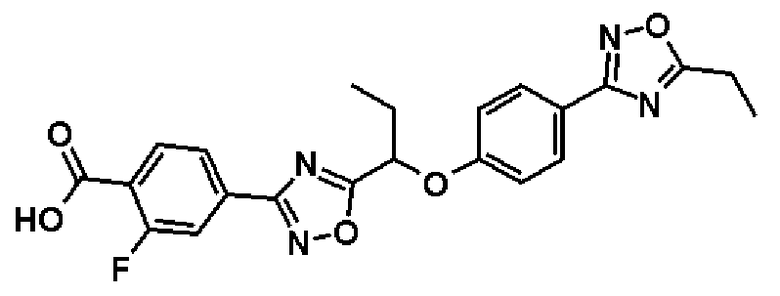
Синтез проводят таким же образом, как и в справочном примере 8, за исключением того, что соединение, полученное в справочном примере 31 (1,94 г, 4,29 ммоль), используют вместо метил-2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоата. В результате получают названное соединение (1,66 г, выход 88%).
1Н-ЯМР (400 МГц, CDCl3) δ м.д.: 8,16-8,14 (1H, м), 8,01 (2H, д, J=9 Гц), 7,97 (1H, дд, J=8 Гц, 2 Гц), 7,92 (1H, дд, J=11 Гц, 2 Гц), 7,07 (2H, д, J=9 Гц), 5,52 (1H, т, J=6 Гц), 2,96 (2H, кв, J=8 Гц), 2,31-2,24 (2H, м), 1,43 (3H, т, J=8 Гц), 1,15 (3Н, т, J=7 Гц).
Справочный пример 33
(2S)-2-{[4-(5-{1-[4-(5-Этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил]-2-фторбензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицинат
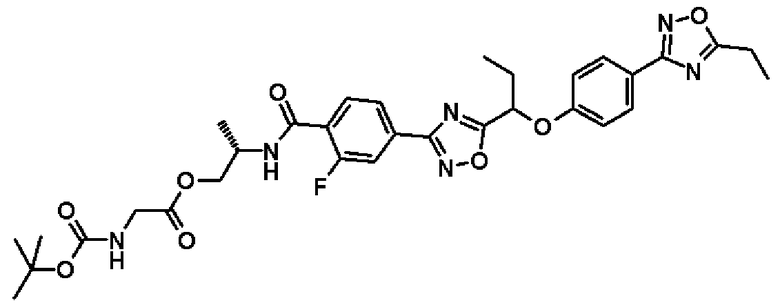
Синтез проводят таким же образом, как и в справочном примере 14, за исключением того, что соединение, полученное в примере 20, который будет описан ниже, (280 мг, 0,565 ммоль), используют вместо 2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамида. В результате получают названное соединение (210 мг, выход 98%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,19-8,15 (1H, м), 8,02-7,97 (3H, м), 7,86 (1H, дд, J=11 Гц, 2 Гц), 7,07 (2H, д, J=9 Гц), 6,83-6,78 (1H, м), 5,51 (1H, т, J=6 Гц), 5,06-5,01 (1H, м), 4,59-4,52 (1H, м), 4,28 (2H, шир.дд, J=12 Гц, 4 Гц), 3,95 (2H, шир.д, J=5 Гц), 2,95 (2H, кв, J=8 Гц), 2,32-2,25 (2H, м), 1,45-1,41 (12H, м), 1,32 (3H, д, J=7 Гц), 1,15 (3H, т, J=7 Гц).
Справочный пример 34
(2R)-2-{[4-(5-{1-[4-(5-Этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил]-2-фторбензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицинат

Синтез проводят таким же образом, как и в справочном примере 14, за исключением того, что соединение, полученное в примере 21, который будет описан ниже, (183 мг, 0,369 ммоль), используют вместо 2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамида. В результате получают названное соединение (210 мг, выход 87%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,20-8,18 (1H, м), 8,02-7,97 (3H, м), 7,86 (1H, дд, J=11 Гц, 2 Гц), 7,07 (2H, д, J=9 Гц), 6,82-6,77 (1H, м), 5,51 (1H, т, J=6 Гц), 5,04-5,00 (1H, м), 4,59-4,52 (1H, м), 4,28 (2H, шир.дд, J=12 Гц, 4 Гц), 3,95 (2H, шир.д, J=5 Гц), 2,95 (2H, кв, J=8 Гц), 2,34-2,20 (2H, м), 1,45-1,41 (12H, м), 1,32 (3H, д, J=7 Гц), 1,15 (3H, т, J=7 Гц).
Справочный пример 35
2-{[4-(5-{1-[4-(5-Этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил]-2-фторбензоил]амино}-2-метилпропил-N-(трет.-бутоксикарбонил)глицинат

Синтез проводят таким же образом, как и в справочном примере 14, за исключением того, что соединение, полученное в примере 22, который будет описан ниже, (355 мг, 0,697 ммоль), используют вместо 2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамида. В результате получают названное соединение (360 мг, выход 77%).
1Н-ЯМР (400 МГц, CDCl3) δ м.д.: 8,15-8,11 (1H, м), 8,02-7,96 (3H, м), 7,84 (1H, дд, J=11 Гц, 2 Гц), 7,0 7 (2H, д, J=9 Гц), 6,68-6,65 (1H, м), 5,51 (1H, т, J=6 Гц), 5,04-4,99 (1H, м), 4,42 (2H, с), 3,95 (2H, шир.д, J=6 Гц), 2,95 (2H, кв, J=8 Гц), 2,34-2,20 (2H, м), 1,49 (6H, с), 1,45-1,41 (12H, м), 1,15 (3H, т, J=7 Гц).
Справочный пример 36
Этил-2-[4-(5-Циклопропил-1,2,4-оксадиазол-3-ил)фенокси]бутаноат
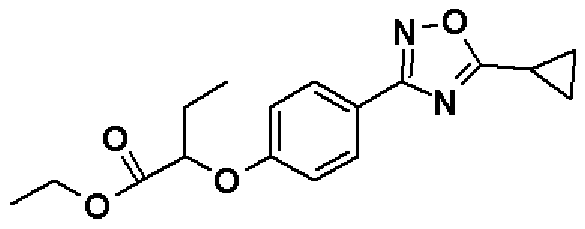
Синтез проводят таким же образом, как и в справочном примере 5, за исключением того, что используют соединение, полученное в справочном примере 4 (4,00 г, 15,0 ммоль), и циклопропанкарбонилхлорид (1,52 мл, 16,5 ммоль) используют вместо хлорангидрида изобутановой кислоты. В результате получают названное соединение (2,01 г, выход 42%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 7,96 (2H, д, J=9 Гц), 6,94 (2H, д, J=9 Гц), 4,62 (1H, т, J=6 Гц), 4,23 (2H, кв, J=7 Гц), 2,26-2,21 (1H, м), 2,04-1,99 (2H, м), 1,31-1,27 (4H, м), 1,24 (3H, т, J=7 Гц), 1,09 (3H, т, J=7 Гц).
Справочный пример 37
2-[4-(5-Циклопропил-1,2,4-оксадиазол-3-ил)фенокси]бутановая кислота
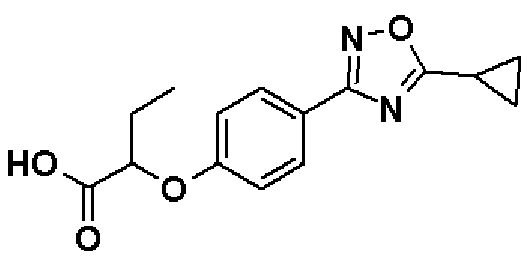
Синтез проводят таким же образом, как и в справочном примере 6, за исключением того, что соединение, полученное в справочном примере 36 (2,01 г, 6,35 ммоль), используют вместо этил-2-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]бутаноата. В результате получают названное соединение (1,83 г, выход 100%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 7,96 (2H, д, J=9 Гц), 6,97 (2H, д, J=9 Гц), 4,68 (1H, т, J=6 Гц), 2,28-2,24 (1H, м), 2,11-2,03 (2H, м), 1,33-1,22 (4H, м), 1,13 (3H, т, J=7 Гц).
Справочный пример 38
Метил-4-(5-{1-[4-(5-циклопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил]-2-фторбензоат
Синтез проводят таким же образом, как и в справочном примере 7, за исключением того, что соединение, полученное в справочном примере 37 (951 мг, 3,30 ммоль), используют вместо 2-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]бутановой кислоты. В результате получают названное соединение (930 мг, выход 61%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,07-8,03 (1H, м), 7,97 (2H, д, J=9 Гц), 7,94-7,86 (2H, м), 7,06 (2H, д, J=9 Гц), 5,51 (1H, т, J=6 Гц), 3,97 (3H, с), 2,34-2,20 (3H, м), 1,31-1,21 (4H, м), 1,15 (3H, т, J=7 Гц).
Справочный пример 39
4-(5-{1-[4-(5-Циклопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил]-2-фторбензойная кислота
Синтез проводят таким же образом, как и в справочном примере 8, за исключением того, что соединение, полученное в справочном примере 38 (930 мг, 2,00 ммоль), используют вместо метил-2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоата. В результате получают названное соединение (900 мг, выход 100%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,16-8,12 (1H, м), 7,98-7,95 (3H, м), 7,91 (1H, дд, J=11 Гц, 2 Гц), 7,06 (2H, д, J=9 Гц), 5,51 (1H, т, J=7 Гц), 2,34-2,20 (3H, 4H, м), 1,31-1,21 (4Н, м), 1,15 (3H, т, J=7 Гц)
Справочный пример 40
(2R)-2-{[4-(5-{1-[4-(5-Циклопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил]-2-фторбензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицинат
Синтез проводят таким же образом, как и в справочном примере 14, за исключением того, что соединение, полученное в примере 27, который будет описан ниже (243 мг, 0,479 ммоль), используют вместо 2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамида. В результате получают названное соединение (260 мг, выход 82%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,20-8,16 (1H, м), 7,99-7,96 (3H, м), 7,86 (1H, дд, J=12 Гц, 2 Гц), 7,06 (2H, д, J=9 Гц), 6,82-6,77 (1H, м), 5,50 (1H, т, J=6 Гц), 5,05-5,00 (1H, м), 4,60-4,51 (1H, м), 4,28 (2H, шир.дд, J=12 Гц, 4 Гц), 3,95 (2H, шир.д, J=6 Гц), 2,34-2,19 (3H, м), 1,43 (9H, с), 1,32 (3H, д, J=7 Гц), 1,29-1,29 (4H, м), 1,14 (3H, т, J=7 Гц).
Справочный пример 41
трет.-Бутил-4-циано-2-фторбензоат
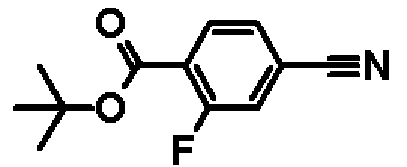
Добавляют ди-трет.-бутилдикарбонат (145,4 г, 666,2 ммоль) к раствору в смеси трет.-бутилового спирта (1000 мл) и тетрагидрофурана (500 мл) 4-циано-2-фторбензойной кислоты (100,0 г, 605,6 ммоль), и реакционную смесь перемешивают 3 часа при 60ºС. Реакционную смесь охлаждают до комнатной температуры, и нерастворимое вещество отфильтровывают через Celite. Растворитель отгоняют при пониженном давлении. В результате получают названное соединение.
Справочный пример 42
трет.-Бутил-4-амино(гидроксиимино)метил-2-фторбензоат
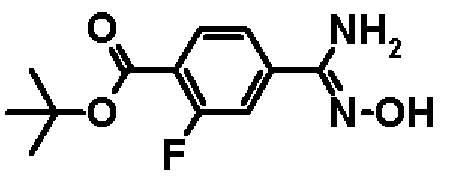
Добавляют 50%-ный водный раствор гидроксиламина (60 мл, 100 ммоль) к раствору в смеси этанола (100 мл) и тетрагидрофурана (50 мл) соединения, полученного в справочном примере 41 (11,0 г, 66,6 ммоль), и смесь перемешивают 2 часа при 80ºС. Реакционную смесь охлаждают до комнатной температуры, и затем растворитель отгоняют при пониженном давлении. Полученный остаток промывают водой и сушат при пониженном давлении при 40ºС в течение двух дней. В результате получают названное соединение (150,0 г, выход 98%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 7,89 (1H, т, J=8 Гц), 7,44 (2H, дд, J=8, 2 Гц), 7,39 (2H, дд, J=11, 2 Гц), 4,90 (2H, с), 1,60 (9H, с).
Справочный пример 43
N',4-Дигидроксибензолкарбоксиимидамид

При комнатной температуре добавляют 50%-ный водный раствор гидроксиламина (13,3 мл, 202 ммоль) к раствору в этаноле (300 мл) 4-цианофенола (20,0 г, 168 ммоль), и смесь перемешивают 2 часа при 80ºС. Реакционную смесь охлаждают до комнатной температуры, затем растворитель отгоняют при пониженном давлении и полученный остаток промывают водой и этилацетатом. В результате получают названное соединение (21,9 г, выход 86%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 9,59 (1H, с), 9,35 (1H, с), 7,47 (2H, д, J=9 Гц), 6,73 (2H, д, J=9 Гц) 5,63 (2H, с).
Справочный пример 44
4-(5-Изопропил-1,2,4-оксадиазол-3-ил)фенол
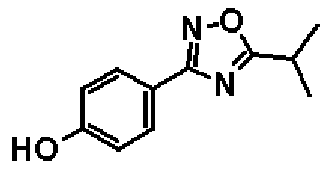
Изобутановый ангидрид (16,6 мл, 99,3 ммоль) добавляют при комнатной температуре к раствору в N,N-диметилформамиде (150 мл) соединения, полученного в справочном примере 43 (15,0 г, 99,3 ммоль), и смесь перемешивают 30 минут при 0ºС. Затем смесь нагревают до 100ºС и перемешивают 7 часов. Реакционную смесь охлаждают до комнатной температуры, затем добавляют воду и смесь экстрагируют 2 раза этилацетатом. Полученный таким образом органический слой промывают насыщенным водным раствором гидрокарбоната натрия и 10%-ным рассолом, а затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток промывают смесью гексана и этилацетата (10:1, об./об.). В результате получают названное соединение (18,9 г, выход 93%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 7,97 (2H, д, J=9 Гц), 6,91 (2H, д, J=9 Гц), 5,39 (1H, с), 3,33-3,22 (1H, м), 1,45 (6H, д, J=7 Гц).
Справочный пример 45
(2R)-2-[4-(5-Изопропил-1,2,4-оксадиазол-3-ил)фенокси]бутановая кислота

Раствор в 1,4-диоксане (100 мл) соединения, полученного в справочном примере 44, добавляют к 60%-ному раствору в диоксане (700 мл) гидрида натрия (18,8 г, 0,49 моль) при 0ºС, и смесь перемешивают 10 минут при комнатной температуре. Затем раствор в диоксане (100 мл) (S)-2-хлорбутановой кислоты (18,0 г, 147 ммоль) добавляют к реакционной смеси и смесь перемешивают 4 часа при 80ºС. Реакционную смесь охлаждают до 0ºС и затем добавляют насыщенный водный раствор хлорида аммония. К смеси добавляют 2 н. соляную кислоту до достижения значения рН 3. Смесь экстрагируют три раза этилацетатом и полученный таким образом органический слой промывают водой и насыщенным рассолом, а затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток промывают смесью гексана и этилацетата (1:1, об./об.). В результате получают названное соединение (30,5 г, выход 86%).
1Н-ЯМР (500 МГц, CDCl3) δ м.д.: 8,00 (2H, д, J=9 Гц), 6,98 (2H, д, J=9 Гц), 4,69 (1H, т, J=6 Гц), 3,32-3,24 (1H, м), 2,08-2,05 (2H, м), 1,45 (6H, д, J=7 Гц), 1,13 (3H, т, J=7 Гц).
Справочный пример 46
трет.-Бутил-2-фтор-4-(5-{(1R)-1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил]бензоат
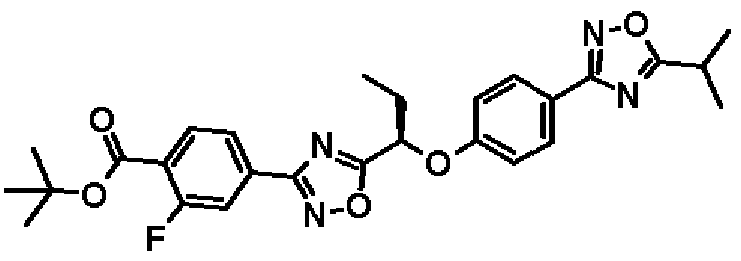
К раствору в N,N-диметилформамиде (500 мл) соединения, полученного в справочном примере 45 (30,6 г, 105 ммоль) добавляют при комнатной температуре моногидрат 1-гидроксибензотриазола (14,2 мг, 105 ммоль) и N-(3-диметиламинопропил)-N'-этилкарбодиимид (40,3 г, 210 ммоль), и смесь перемешивают 30 минут при той же температуре. Добавляют соединение, полученное в справочном примере 42 (26,8 г, 105 ммоль), и смесь перемешивают 30 минут, а затем дополнительно перемешивают 4 часа при 100ºС. Реакционную смесь охлаждают до комнатной температуры, затем к реакционной смеси добавляют воду и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают водой и 10%-ным рассолом, а затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 95:5 → 85:15, об./об.). В результате получают названное соединение (45,2 г, выход 84%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,01 (2H, д, J=9 Гц), 7,96 (1H, т, J=8 Гц), 7,89 (1H, дд, J=8, 2 Гц), 7,83 (1H, дд, J=11, 1 Гц), 7,07 (2H, д, J=9 Гц), 5,50 (1H, т, J=6 Гц), 3,31-3,20 (1H, м), 2,36-2,18 (2H, м), 1,61 (9H, с), 1,43 (6H, д, J=7 Гц), 1,15 (3H, т, J=7 Гц).
Справочный пример 47
2-Фтор-4-(5-{(1R)-1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил]бензойная кислота
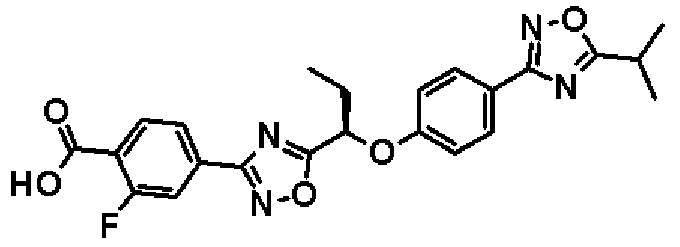
Трифторуксусную кислоту (300 мл) добавляют к раствору в дихлорметане (100 мл) соединения, полученного в справочном примере 46 (45,2 г, 89,4 ммоль), и смесь перемешивают 2 часа при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении и азеотропно кипятят с толуолом (100 мл) два раза. В результате получают названное соединение (33,1 г, выход 82%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,13 (1H, т, J=8 Гц), 8,01 (2H, д, J=9 Гц), 7,96 (1H, д, J=8 Гц), 7,90 (1H, д, J=11 Гц), 7,07 (2H, д, J=9 Гц), 5,51 (1H, т, J=7 Гц), 3,28-3,23 (1H, м), 2,34-2,20 (2H, м), 1,43 (6H, д, J=7 Гц), 1,15 (3H, т, J=7 Гц).
Пример 1
4-[2-Фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]морфолин
К раствору в метиленхлориде (1,60 мл) соединения, полученного в справочном примере 8 (70,0 мг, 0,115 ммоль), при комнатной температуре добавляют моногидрат 1-гидроксибензотриазола (28,4 мг, 0,186 ммоль) и N-(3-диметиламинопропил)-N'-этилкарбодиимид (35,6 мг, 0,186 ммоль), и смесь перемешивают 15 минут. Добавляют морфолин (16,2 мкл, 0,186 ммоль), и смесь перемешивают еще один час. К реакционной смеси добавляют насыщенный водный раствор гидрокарбоната натрия и смесь экстрагируют два раза метиленхлоридом. Экстракт сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 1:1 об./об.). В результате получают названное соединение (80 г, выход 100%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,04-7,85 (4H, м), 7,58-7,52 (1H, м), 7,11-7,07 (2H, м), 5,53 (1H, м), 3,90-3,63 (6H, м), 3,42-3,23 (3H, м), 2,38-2,22 (2H, м), 1,46 (6H, д, J=7 Гц), 1,17 (3H, т, J=7 Гц); МС (ББА+) [(бомбардировка быстрыми атомами) (FAB+)] m/z: 522 [M+H]+.
Пример 2
N-[(1S)-2-Гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-метилбензамид

Синтез проводят таким же образом, как и в примере 1, за исключением того, что соединение, полученное в справочном примере 13 (101 мг, 0,225 ммоль), используют вместо 2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензойной кислоты, а (S)-(+)-2-амино-1-пропанол (26 мкл, 0,338 ммоль) используют вместо морфолина. В результате получают названное соединение (17 мг, выход 15%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,04-7,92 (4H, м), 7,49 (1H, д, J=7 Гц), 7,10 (2H, д, J=8 Гц), 6,10-6,00 (1H, шир.с), 5,52 (1H, т, J=7 Гц), 4,37-4,25 (1H, м), 3,87-3,64 (2H, м), 3,31-3,25 (1H, м), 2,53 (3H, с), 2,38-2,20 (2H, м), 1,70-1,50 (1H, шир.с), 1,46 (6H, д, J=7 Гц), 1,32 (3H, д, J=7 Гц), 1,17 (3H, т, J=7 Гц); МС (ESI) (ионизация в электроспрее) m/z: 507 [M+H]+.
Пример 3
2-Фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-(2-метоксиэтил)бензамид
Синтез проводят таким же образом, как и в примере 1, за исключением того, что 2-метоксиэтиламин (16,0 мкл, 0,186 ммоль) используют вместо морфолина. В результате получают названное соединение (78 мг, выход 100%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,23 (1H, т, J=8 Гц), 8,05-7,86 (4H, м), 7,13-7,08 (3H, м), 5,53 (1H, д, J=7 Гц), 3,78-3,59 (4H, м), 3,44 (3H, с), 3,32-3,23 (1H, м), 2,38-2,22 (2H, м), 1,46 (6H, д, J=7 Гц), 1,18 (3H, т, J=7 Гц); МС (ББА+) m/z: 510 [M+H]+.
Пример 4
2-Фтор-N-(2-гидроксиэтил)-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид

Синтез проводят таким же образом, как и в примере 1, за исключением того, что этаноламин (11,1 мкл, 0,186 ммоль) используют вместо морфолина. В результате получают названное соединение (71 мг, выход 93%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,24 (1H, т, J=8 Гц), 8,05-7,88 (4H, м), 7,12-7,08 (3H, м), 5,53 (1H, д, J=7 Гц), 3,93-3,68 (4H, м), 3,31-3,25 (1H, м), 2,36-2,24 (3H, м), 1,46 (6H, д, J=7 Гц), 1,17 (3H, т, J=7 Гц); МС (ББА+) m/z: 496 [M+H]+.
Пример 5
2-Фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид
При комнатной температуре добавляют 1,1'-карбонилдиимидазол (20,2 мг, 0,124 ммоль) к раствору в тетрагидрофуране (0,50 мл) соединения, полученного в справочном примере 8 (37,5 мг, 0,0829 ммоль), и смесь перемешивают 30 минут при той же температуре. Затем добавляют 28%-ный (масс./масс.) водный раствор аммиака (0,50 мл, избыток) и смесь дополнительно перемешивают 10 минут при той же температуре. К реакционной смеси добавляют воду и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 3:1 → 1:2, об./об.). В результате получают названное соединение (37 мг, выход 100%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,27 (1H, т, J=8 Гц), 8,07-7,88 (4H, м), 7,11-7,06 (2H, м), 6,76-6,70 (1H, м), 5,97-5,90 (1H, м), 5,54 (1H, д, J=7 Гц), 3,31-3,25 (1H, м), 2,38-2,20 (2H, м), 1,46 (6H, д, J=7 Гц), 1,17 (3H, т, J=7 Гц); МС (ББА+) m/z: 452 [M+H]+.
Пример 6
2-Фтор-N-[(1S)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид
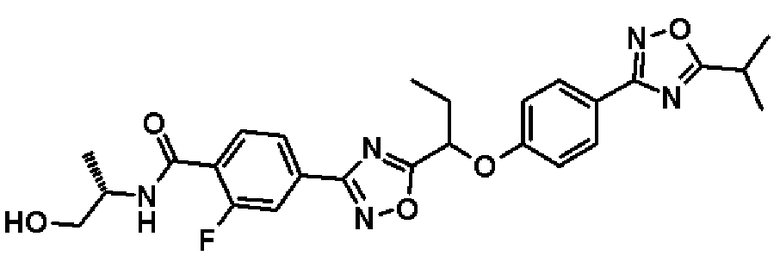
Синтез проводят таким же образом, как и в примере 1, за исключением того, что (S)-(-)-2-амино-1-пропанол (16,0 мкл, 0,273 ммоль) используют вместо морфолина. В результате получают названное соединение (82 мг, выход 88%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,20-7,72 (5H, м), 7,75-7,72 (2H, м), 5,97 (1H, т, J=7 Гц), 4,74 (1H, т, J=7 Гц), 3,98-3,92 (1H, м), 3,45-3,20 (4H, м), 2,20-2,15 (2H, м), 1,33 (6H, д, J=7 Гц), 1,10 (3H, д, J=7 Гц), 1,02 (3H, т, J=7 Гц); МС (ББА+) m/z: 510 [M+H]+.
Пример 7
2-Фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид
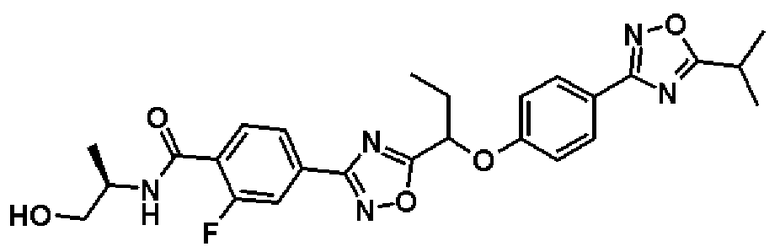
Синтез проводят таким же образом, как и в примере 1, за исключением того, что (R)-(-)-2-амино-1-пропанол (23,0 мкл, 0,290 ммоль) используют вместо морфолина. В результате получают названное соединение (85 мг, выход 86%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,18 (1H, д, J=8 Гц), 7,93 (2H, д, J=9 Гц), 7,87-7,73 (2H, м), 7,22 (2H, д, J=9 Гц), 5,99 (1H, т, J=7 Гц), 4,75 (1H, т, J=6 Гц), 4,03-3,90 (1H, м), 3,48-3,38 (1H, м), 3,37-3,20 (3H, м), 2,22-2,15 (2H, м), 1,34 (6H, д, J=7 Гц), 1,11 (3H, д, J=7 Гц), 1,02 (3H, т, J=7 Гц); МС (ББА+) m/z: 510 [M+H]+.
Пример 8
2-Фтор-N-(2-гидрокси-1,1-диметилэтил)-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид
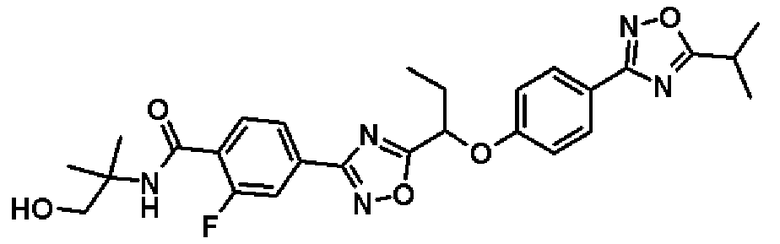
Синтез проводят таким же образом, как и в примере 1, за исключением того, что 2-амино-2-метил-1-пропанол (29,5 мкл, 0,332 ммоль) используют вместо морфолина. В результате получают названное соединение (82 мг, выход 72%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 7,95-7,70 (7H, м), 7,25-7,20 (2H, м), 5,97 (1H, т, J=7 Гц), 4,88 (1H, т, J=6 Гц), 3,60-3,42 (2H, м), 3,33-3,25 (1H, м), 2,20-2,13 (1H, м), 1,32 (3H, д, J=7 Гц), 1,28 (6H, с), 1,03 (6H, д, J=7 Гц); МС (ББА+) m/z: 524 [M+H]+.
Пример 9
2-Фтор-N-[(1S)-1-(гидроксиметил)пропил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид
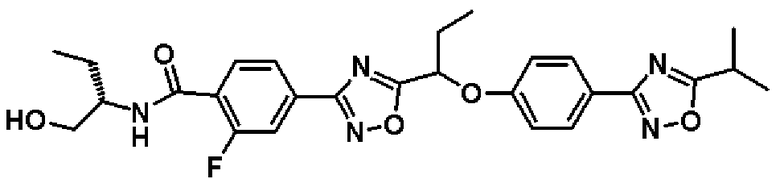
Синтез проводят таким же образом, как и в примере 1, за исключением того, что (S)-2-аминобутан-1-ол (42,0 мкл, 0,444 ммоль) используют вместо морфолина. В результате получают названное соединение (147 мг, выход 63%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,10 (1H, д, J=8 Гц), 7,95-7,70 (5H, м), 7,24-7,19 (2H, м), 5,97 (1H, т, J=6 Гц), 4,69 (1H, т, J=6 Гц), 3,85-3,75 (1H, м), 3,47-3,22 (3H, м), 2,20-2,13 (2H, м), 1,69-1,54 (1H, м), 1,41-1,32 (7H, м), 1,42 (3H, т, J=7 Гц), 1,09 (3H, т, J=7 Гц); МС (ББА+) m/z: 524 [M+H]+.
Пример 10
2-Фтор-N-[(1R)-1-(гидроксиметил)пропил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид

Синтез проводят таким же образом, как и в примере 1, за исключением того, что (R)-2-аминобутан-1-ол (16,0 мкл, 0,186 ммоль) используют вместо морфолина. В результате получают названное соединение (297 мг, выход 86%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,23 (1H, т, J=8 Гц), 8,05-7,87 (4H, м), 7,13-6,85 (3H, м), 5,53 (1H, д, J=7 Гц), 4,21-4,13 (1H, м), 3,86 (1H, дд, J=11 Гц, 4 Гц), 3,76 (1H, дд, J=11 Гц, 7 Гц), 3,31-3,25 (1H, м), 2,38-2,22 (2H, м), 1,80-1,48 (3H, м), 1,46 (6H, д, J=7 Гц), 1,17 (3H, т, J=7 Гц), 1,07 (3H, т, J=7 Гц); МС (ББА+) m/z: 524 [M+H]+.
Пример 11
2-Фтор-N-(3-гидроксипропил)-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид
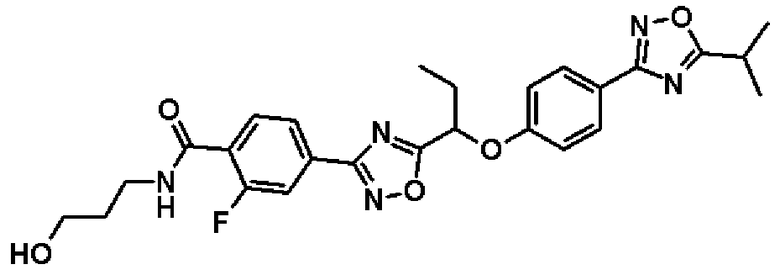
Синтез проводят таким же образом, как и в примере 1, за исключением того, что 3-амино-1-пропанол (50 мкл, 0,663 ммоль) используют вместо морфолина. В результате получают названное соединение (210 мг, выход 93%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,17 (1H, т, J=8 Гц), 8,01-7,78 (4H, м), 7,24-7,00 (3H, м), 5,47 (1H, т, J=7 Гц), 3,78-3,63 (4H, м), 3,25-3,18 (1H, м), 2,35-2,15 (2H, м), 1,84-1,75 (2H, м), 1,41 (6H, д, J=7 Гц), 1,12 (3H, т, J=7 Гц); МС (ESI) m/z: 511 [M+H]+.
Пример 12
Гидрохлорид (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропилглицината
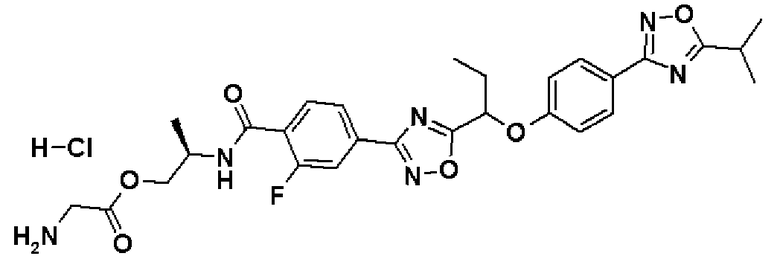
Добавляют 4 М водный раствор соляной кислоты (1,00 мл) к раствору в этилацетате (1,00 мл) соединения, полученного в справочном примере 14 (90,6 мг, 0,136 ммоль), и смесь перемешивают 4 часа при комнатной температуре. Растворитель отгоняют из реакционной смеси при пониженном давлении и остаток промывают диэтиловым эфиром. В результате получают названное соединение (52 мг, выход 64%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,49 (1H, д, J=8 Гц), 8,36-8,23 (2H, м), 7,95-7,71 (4H, м), 7,25-7,18 (2H, м), 5,98 (1H, т, J=7 Гц), 4,33-4,07 (3H, м), 3,79 (2H, с), 3,84-3,87 (3H, м), 2,20-2,14 (2H, м), 1,33 (6H, д, J=7 Гц), 1,17 (3H, д, J=7 Гц), 1,03 (3H, д, J=7 Гц); МС (ББА+) m/z: 567 [M+H]+.
Пример 13
Гидрохлорид (2S)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропилглицината
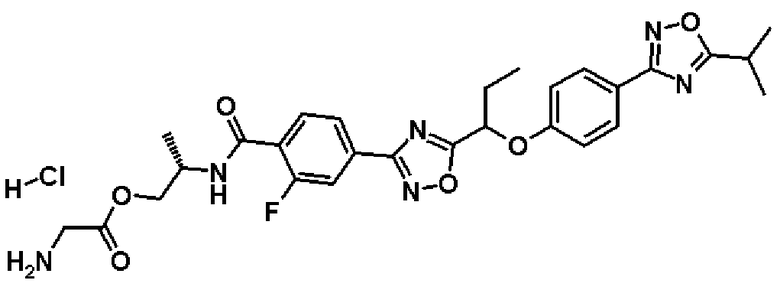
Синтез проводят таким же образом, как и в примере 12, за исключением того, что соединение, полученное в справочном примере 15 (123 мг, 0,184 ммоль), используют вместо (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицината. В результате получают названное соединение (63 мг, выход 57%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,49 (1H, д, J=8 Гц), 8,46-8,23 (2H, м), 7,99-7,71 (4H, м), 7,30-7,25 (2H, м), 6,04 (1H, т, J=7 Гц), 4,37-4,16 (3H, м), 3,89 (2H, с), 3,84-3,87 (3H, м), 2,20-2,14 (2H, с), 1,39 (6H, д, J=7 Гц), 1,24 (3H, д, J=7 Гц), 1,09 (3H, д, J=7 Гц); МС (ББА+) m/z: 567 [M+H]+.
Пример 14
Гидрохлорид (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}бутилглицината
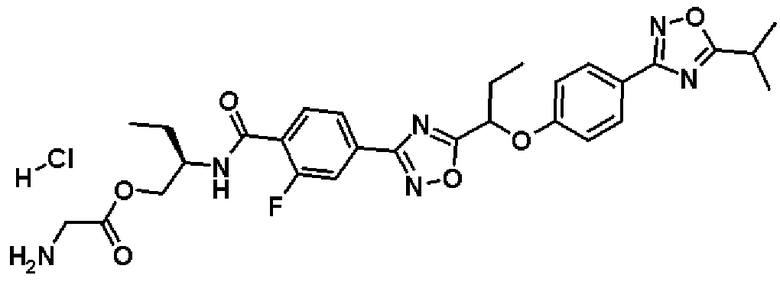
Синтез проводят таким же образом, как и в примере 12, за исключением того, что соединение, полученное в справочном примере 16 (358 мг, 0,523 ммоль), используют вместо (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицината. В результате получают названное соединение (308 мг, выход 95%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,63-8,33 (3H, м), 8,02-7,75 (5H, м), 7,32-7,24 (2H, м), 6,04 (1H, т, J=7 Гц), 4,37-4,09 (3H, м), 3,83 (2H, с), 2,26-2,21 (2H, м), 1,75-1,49 (2H, м), 1,34 (6H, д, J=7 Гц), 1,36-1,25 (1H, м), 1,13-0,83 (7H, м); МС (ББА+) m/z: 581 [M+H]+.
Пример 15
Гидрохлорид (2S)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}бутилглицината
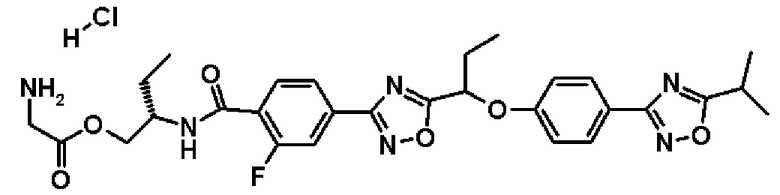
Синтез проводят таким же образом, как и в примере 12, за исключением того, что соединение, полученное в справочном примере 17 (274 мг, 0,403 ммоль), используют вместо (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицината. В результате получают названное соединение (49 мг, выход 20%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,39-8,22 (3H, м), 8,02-7,75 (4H, м), 7,29-7,27 (2H, м), 6,03 (1Н, т, J=7 Гц), 4,37-4,17 (3H, м), 3,85 (2H, с), 2,28-2,19 (2H, м), 1,75-1,55 (2H, м), 1,39 (6H, д, J=7 Гц), 1,09 (3H, т, J=7 Гц), 0,97 (3H, т, J=7 Гц); МС (ESI) m/z: 581 [M+H]+.
Пример 16
Гидрохлорид 2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}-2-метилпропилглицината
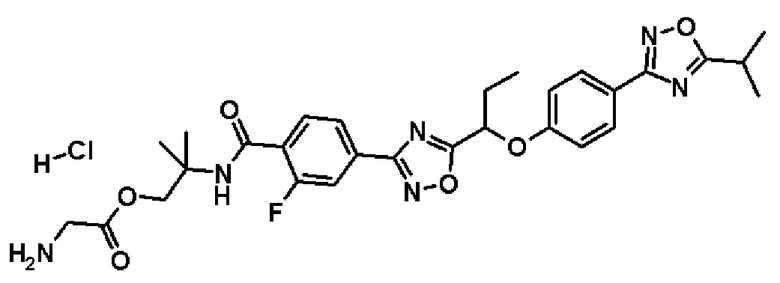
Синтез проводят таким же образом, как и в примере 12, за исключением того, что соединение, полученное в справочном примере 18 (169 мг, 0,523 ммоль), используют вместо (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицината. В результате получают названное соединение (129 мг, выход 84%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,52-8,34 (3H, м), 8,20 (1H, с), 8,01-7,72 (5H, м), 7,30-7,26 (2H, м), 6,04 (1H, т, J=7 Гц), 4,40 (2H, с), 3,87 (2H, с), 2,26-2,20 (2H, м), 1,41 (6H, с), 1,38 (6H, д, J=7 Гц), 1,38-1,36 (1H, шир.с), 1,09 (3H, т, J=7 Гц); МС (ББА+) m/z: 581 [M+H]+.
Пример 17
(2R)-2-{[2-Фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}-2-метилпропилглицинат
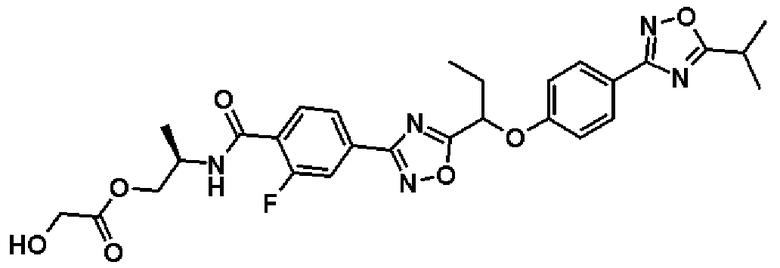
К раствору в тетрагидрофуране (5,00 мл) соединения, полученного в справочном примере 22 (300 мг, 0,505 ммоль), добавляют трифенилфосфин (58,3 мг, 0,224 ммоль), димедон (46,0 мг, 0,329 ммоль) и тетракистрифенилфосфинпалладий (58,4 мг, 0,0505 ммоль), и смесь перемешивают 4 часа при комнатной температуре. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 9:1 → 1:1, об./об.). В результате получают названное соединение (252 мг, выход 77%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,49 (1H, д, J=8 Гц), 8,00-7,75 (5H, м), 7,25-7,18 (2H, м), 6,04 (1H, т, J=7 Гц), 5,35 (1H, т, J=7 Гц), 4,32-4,04 (6H, м), 2,26-2,22 (2H, м), 1,40 (6H, д, J=7 Гц), 1,19 (3H, д, J=7 Гц), 1,09 (3H, т, J=7 Гц); МС (ББА+) m/z: 568 [M+H]+.
Пример 18
2-Фтор-N-[(1S)-2-гидрокси-1-метилэтил]-4-(2-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,3-оксазол-4-ил)бензамид
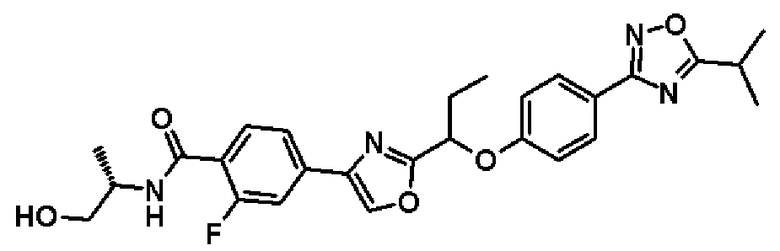
Синтез проводят таким же образом, как и в примере 1, за исключением того, что соединение, полученное в справочном примере 27 (70,0 мг, 0,155 ммоль), используют вместо 2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензойной кислоты, и (R)-2-амино-1-пропанол (14,4 мкл, 0,186 ммоль) используют вместо морфолина. В результате получают названное соединение (56 мг, выход 72%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,14 (1H, т, J=8 Гц), 8,03-7,99 (3H, м), 7,60-7,56 (2H, м), 7,15-7,10 (2H, м), 6,96-6,85 (1H, м), 5,37 (1H, т, J=7 Гц), 4,40-4,32 (1H, м), 3,83 (1H, дд, J=11 Гц, 4 Гц), 3,70 (1H, дд, J=11 Гц, 6 Гц), 3,30-3,25 (1H, м), 2,33-2,16 (3H, м), 1,46 (6H, д, J=7 Гц), 1,34 (3H, д, J=7 Гц), 1,12 (3H, т, J=7 Гц).
МС (ББА+) m/z: 509 [M+H]+.
Пример 19
Гидрохлорид (2S)-2-{[2-фтор-4-(2-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,3-оксазол-4-ил)бензоил]амино}пропилглицината
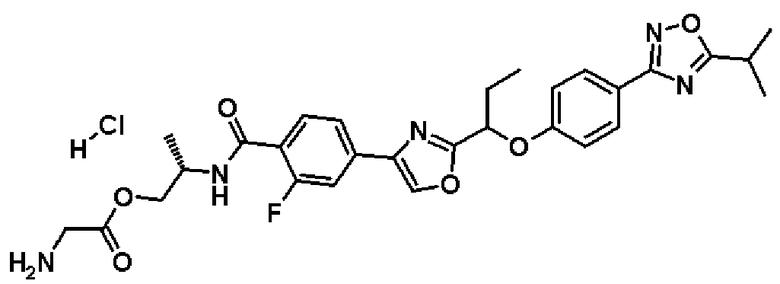
Синтез проводят таким же образом, как и в примере 12, за исключением того, что соединение, полученное в справочном примере 28 (123 мг, 0,184 ммоль), используют вместо (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицината. В результате получают названное соединение (19 мг, выход 85%).
1Н-ЯМР (400 МГц, CDCl3) δ м.д.: 8,73-8,57 (2H, шир.с), 8,00-7,88 (4H, м), 7,46-7,02 (5H, м), 5,32 (1H, т, J=7 Гц), 4,47-4,05 (4H, м), 3,30-3,19 (1H, м), 2,33-2,09 (5H, м), 1,43 (6H, д, J=7 Гц), 1,31-1,25 (2H, м), 1,07 (3H, т, J=7 Гц); МС (ББА+) m/z: 566 [M+H]+.
Пример 20
4-(5-{1-[4-(5-Этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]бензамид
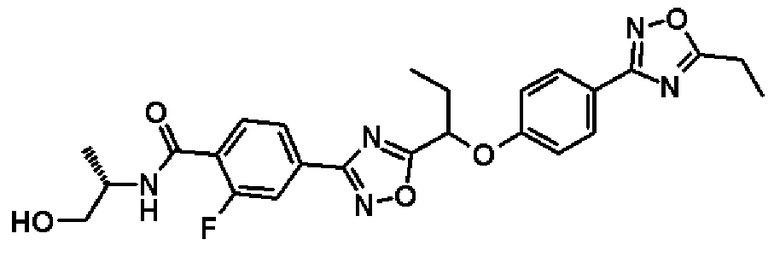
Синтез проводят таким же образом, как и в примере 6, за исключением того, что соединение, полученное в справочном примере 32 (300 мг, 0,684 ммоль), используют вместо 2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензойной кислоты. В результате получают названное соединение (280 мг, выход 83%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,18 (1H, т, J=8 Гц), 8,00-7,82 (4H, м), 7,08-6,86 (3H, м), 5,49 (1H, т, J=7 Гц), 4,38-4,27 (1H, м), 3,83-3,62 (2H, м), 2,93 (2H, кв, J=7 Гц), 2,48-2,40 (1H, шир.с), 2,33-2,15 (2H, м), 1,41 (3H, т, J=7 Гц), 1,30 (3H, д, J=7 Гц), 0,86 (3H, т, J=7 Гц); МС (ESI) m/z: 497 [M+H]+.
Пример 21
4-(5-{1-[4-(5-Этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]бензамид

Синтез проводят таким же образом, как и в примере 7, за исключением того, что соединение, полученное в справочном примере 32 (300 мг, 0,684 ммоль), используют вместо 2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензойной кислоты. В результате получают названное соединение (183 мг, выход 54%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,18 (1H, т, J=8 Гц), 8,02-7,81 (4H, м), 7,08-6,86 (3H, м), 5,49 (1H, т, J=7 Гц), 4,39-4,26 (1H, м), 3,79 (1H, дд, J=11 Гц, 4 Гц), 3,66 (1H, дд, J=11 Гц, 7 Гц), 2,93 (2H, кв, J=7 Гц), 2,34-2,20 (2H, м), 1,61 (1H, шир.с), 1,41 (3H, т, J=7 Гц), 1,30 (3H, д, J=7 Гц), 1,13 (3H, т, J=7 Гц); МС (ESI) m/z: 497 [M+H]+.
Пример 22
4-(5-{1-[4-(5-Этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-(2-гидрокси-1,1-диметилэтил)бензамид
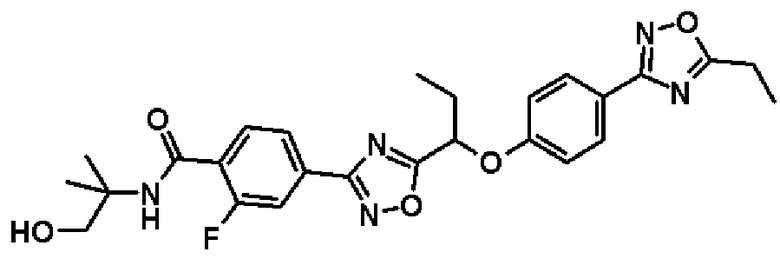
Синтез проводят таким же образом, как и в примере 8, за исключением того, что соединение, полученное в справочном примере 32 (400 мг, 0,912 ммоль), используют вместо 2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензойной кислоты. В результате получают названное соединение (355 мг, выход 76%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,13 (1H, т, J=7 Гц), 8,01-7,80 (4H, м), 7,08-6,83 (3H, м), 5,49 (1H, т, J=7 Гц), 4,39-4,26 (1H, шир.с), 3,70 (2H, с), 2,93 (2H, кв, J=7 Гц), 2,36-2,15 (2H, м), 1,43-1,37 (9H, м), 1,13 (3H, т, J=7 Гц); МС (ESI) m/z: 511 [M+H]+.
Пример 23
4-(5-{1-[4-(5-Этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-(2-гидроксиэтил)бензамид

Синтез проводят таким же образом, как и в примере 4, за исключением того, что соединение, полученное в справочном примере 32 (200 мг, 0,456 ммоль), используют вместо 2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензойной кислоты. В результате получают названное соединение (157 мг, выход 72%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,20 (1H, т, J=8 Гц), 8,00-7,83 (4H, м), 7,24-6,98 (3H, м), 5,49 (1H, д, J=7 Гц), 3,89-3,63 (4H, м), 2,93 (2H, кв, J=7 Гц), 2,35-2,15 (2H, м), 1,60-1,52 (1H, шир.с), 1,41 (3H, т, J=7 Гц), 1,12 (3H, т, J=7 Гц); МС (ESI) m/z: 482 [M+H]+.
Пример 24
Гидрохлорид (2S)-2-{[4-(5-{1-[4-(5-этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензоил]амино}пропилглицината

Синтез проводят таким же образом, как и в примере 12, за исключением того, что соединение, полученное в справочном примере 33 (360 мг, 0,552 ммоль), используют вместо (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицината. В результате получают названное соединение (244 мг, выход 75%).
1H-ЯМР (400 МГц, DMSО-d6) δ м.д.: 8,54 (1H, д, J=8 Гц), 8,46-8,23 (2H, м), 7,99-7,71 (5H, м), 7,30-7,25 (2H, м), 5,99 (1H, т, J=7 Гц), 4,37-4,08 (3H, м), 3,78 (2H, с), 3,43-3,34 (1H, м), 2,92 (2H, кв, J=7 Гц), 2,24-2,14 (2H, м), 1,30 (3H, т, J=7 Гц), 1,19 (3H, д, J=7 Гц), 1,04 (3H, т, J=7 Гц); МС (ESI) m/z: 554 [M+H]+.
Пример 25
Гидрохлорид (2R)-2-{[4-(5-{1-[4-(5-этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензоил]амино}пропилглицината
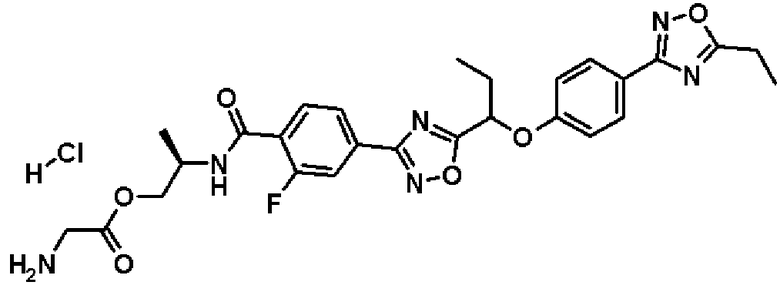
Синтез проводят таким же образом, как и в примере 12, за исключением того, что соединение, полученное в справочном примере 34 (210 мг, 0,322 ммоль), используют вместо (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицината. В результате получают названное соединение (170 мг, выход 90%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,54 (1H, д, J=8 Гц), 8,46-8,23 (3H, м), 7,95-7,74 (5H, м), 7,27-7,19 (2H, м), 6,00 (1H, т, J=7 Гц), 4,37-4,08 (3H, м), 3,78 (2H, с), 2,97 (2H, кв, J=7 Гц), 2,24-2,14 (2H, м), 1,30 (3H, т, J=7 Гц), 1,19 (3H, д, J=7 Гц), 1,04 (3H, т, J=7 Гц); МС (ESI) m/z: 554 [M+H]+.
Пример 26
Гидрохлорид 2-{[4-(5-{1-[4-(5-этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензоил]амино}-2-метилпропилглицината

Синтез проводят таким же образом, как и в примере 12, за исключением того, что соединение, полученное в справочном примере 35 (360 мг, 0,539 ммоль), используют вместо (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N-(трет.-бутоксикарбонил)глицината. В результате получают названное соединение (224 мг, выход 69%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,50-8,40 (2H, шир.с), 8,20 (1H, с), 8,00-7,24 (7H, м), 6,04 (1H, т, J=7 Гц), 4,40 (2H, с), 3,87 (2H, с), 3,66-3,60 (1H, м), 3,01 (2H, кв, J=7 Гц), 2,26-2,20 (2H, м), 1,50 (6H, с), 1,35 (3H, т, J=7 Гц), 1,09 (3H, т, J=7 Гц); МС (ESI) m/z: 568 [M+H]+.
Пример 27
4-(5-{1-[4-(5-Циклопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]бензамид
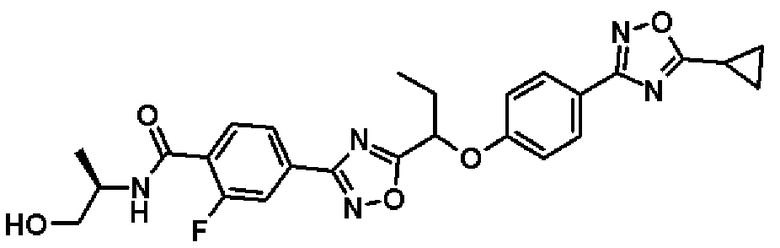
Синтез проводят таким же образом, как и в примере 7, за исключением того, что соединение, полученное в справочном примере 39 (357 мг, 0,797 ммоль), используют вместо 2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензойной кислоты. В результате получают названное соединение (280 мг, выход 69%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,18 (1H, т, J=8 Гц), 8,00-7,79 (4H, м), 7,05-6,84 (3H, м), 5,48 (1H, т, J=7 Гц), 4,37-4,25 (1H, м), 3,78 (1H, дд, J=11 Гц, 4 Гц), 3,66 (1H, дд, J=11 Гц, 7 Гц), 2,33-2,15 (3H, м), 1,73-1,65 (1H, шир.с), 1,32-1,02 (10H, м); МС (ESI) m/z: 509 [M+H]+.
Пример 28
Гидрохлорид (2R)-2-{[4-(5-{1-[4-(5-циклопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензоил]амино}пропилглицината
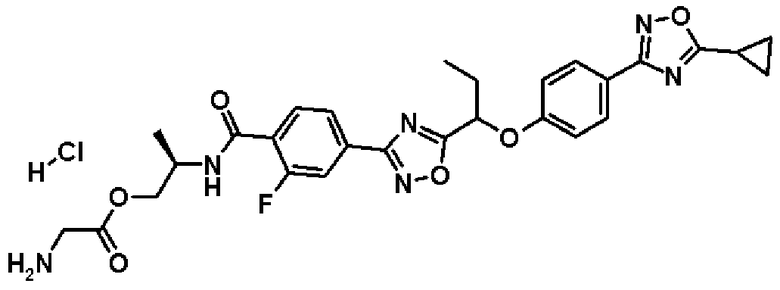
Трифторуксусную кислоту (1,00 мл) добавляют к раствору в метиленхлориде (1,00 мл) соединения, полученного в справочном примере 40 (260 мг, 0,391 ммоль), и смесь перемешивают один час при комнатной температуре. К реакционной смеси добавляют насыщенный водный раствор гидрокарбоната натрия и смесь экстрагируют два раза этилацетатом. Полученный таким образом органический слой промывают насыщенным рассолом и затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и полученный остаток очищают колоночной хроматографией на силикагеле (этилацетат:метанол = 9:1 → 4:1, об./об.). К раствору в этилацетате (500 мкл) полученного (2R)-2-{[4-(5-{1-[4-(5-циклопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензоил]амино}пропилглицината (187 мг, 0,331 ммоль) добавляют 4 н. раствор соляной кислоты в этилацетате (248 мкл, 0,994 ммоль) и растворитель отгоняют в вакууме при пониженном давлении. К полученному остатку добавляют диэтиловый эфир и смесь фильтруют. В результате получают названное соединение (170 мг, выход 72%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,52 (1H, д, J=8 Гц), 8,34-8,28 (3H, м), 7,93-7,71 (5H, м), 7,25-7,18 (2H, м), 5,99 (1H, т, J=7 Гц), 4,35-4,09 (3H, м), 3,80 (2H, с), 2,43-2,12 (3Н, м), 1,30-1,02 (10Н, м); МС (ESI) m/z: 566 [M+H]+.
Пример 29
Гидрохлорид (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N,N-диметилглицината

Синтез проводят таким же образом, как и в справочном примере 14, за исключением того, что N,N-диметилглицин (68,0 мг, 0,659 ммоль) используют вместо N-(трет.-бутоксикарбонил)глицина, и в результате получают (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропил-N,N-диметилглицинат. Добавляют 4 М водный раствор соляной кислоты (1,00 мл) и растворитель отгоняют из реакционной смеси при пониженном давлении. В результате получают названное соединение (280 мг, выход 52%).
1Н-ЯМР (400 МГц, DMSO-d6) δ м.д.: 10,3-10,2 (1H, шир.с), 8,55 (1H, д, J=8 Гц), 7,97-7,71 (5H, м), 7,23 (2H, д, J=8 Гц), 6,00 (1H, т, J=7 Гц), 4,38-3,27 (5H, м), 3,34-3,29 (1H, м), 2,82 (6H, с), 2,23-2,13 (2H, м), 1,18 (6H, д, J=7 Гц), 1,20 (3H, д, J=7 Гц), 1,10-1,00 (3H, м); МС (ESI) m/z: 596 [M+H]+.
Пример 30
Гидрохлорид 2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}этил-N,N-диметилглицината

Синтез проводят таким же образом, как и в примере 29, за исключением того, что соединение, полученное в примере 4 (390 мг, 0,787 ммоль), используют вместо 2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамида. В результате получают названное соединение (363 мг, выход 74%).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 10,3-10,2 (1H, шир.с), 8,72-8,65 (1H, м), 7,95-7,76 (5H, м), 7,21 (2H, д, J=8 Гц), 5,85 (1H, т, J=7 Гц), 4,29 (2H, т, J=7 Гц), 4,13 (2H, с), 3,58-3,56 (2H, м), 3,28-3,26 (1H, м), 2,80 (6H, с), 2,20-2,13 (2H, м), 1,33 (6H, д, J=7 Гц), 1,03 (3H, т, J=7 Гц); МС (ESI) m/z: 582 [M+H]+.
Соединения примеров 31-66 получают с учетом справочных примеров и примеров, описанных выше.

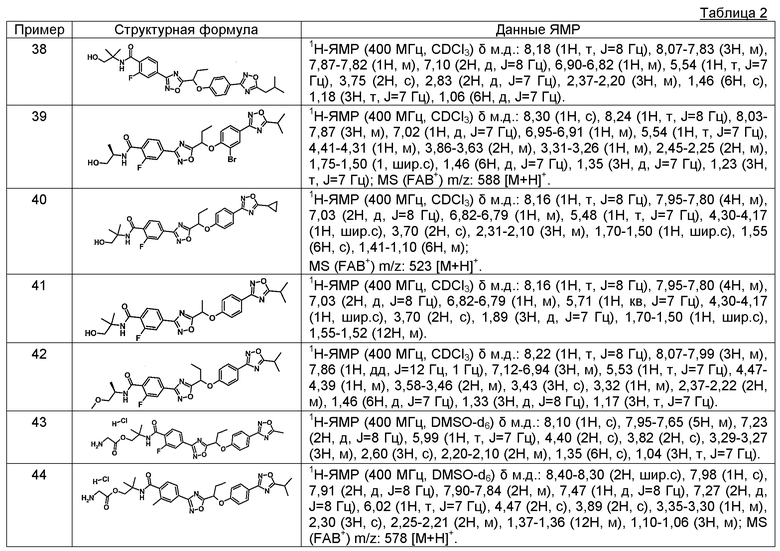
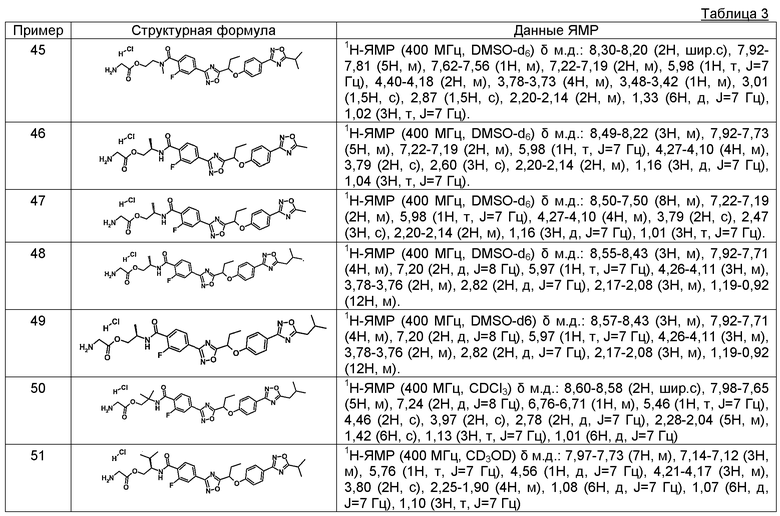
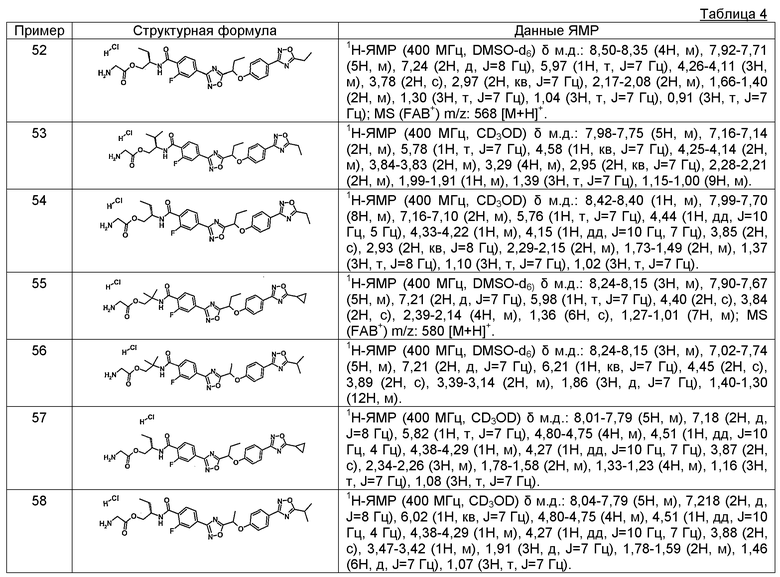


Пример 67
2-Фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{(1R)-1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид
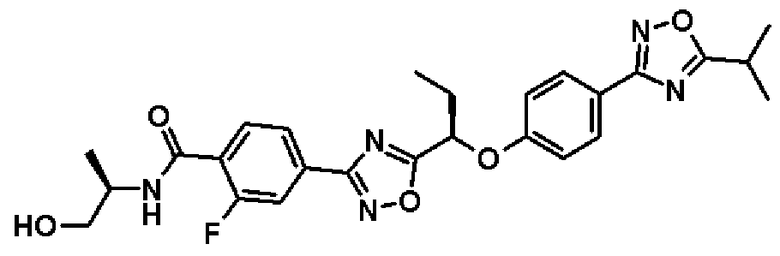
К раствору в N,N-диметилформамиде (2 мл) соединения, полученного в справочном примере 47 (100 мг, 0,222 ммоль), при комнатной температуре добавляют моногидрат 1-гидроксибензотриазола (29,9 мг, 0,222 моль), N-(3-диметиламинопропил)-N'-этилкарбодиимида (127,4 мг, 0,665 моль) и (R)-2-амино-1-пропанол (24,9 мг, 0,332 моль), и смесь перемешивают 3 часа при той же температуре. Затем к реакционной смеси добавляют воду и смесь экстрагируют один раз этилацетатом. Полученный таким образом органический слой промывают водой и 10%-ным рассолом, а затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении, и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 50:50 → 30:70, об./об.). В результате получают названное соединение (92,9 мг, выход 83%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.: 8,20 (1H, т, J=8 Гц), 8,01-7,98 (3H, м), 7,86 (1H, дд, J=12, 2 Гц), 7,06 (2H, дд, J=12, 3 Гц), 6,90 (1H, дд, J=12, 8 Гц), 5,51 (1H, т, J=7 Гц), 4,37-4,32 (1H, м), 3,81 (1H, дд, J=11, 3 Гц), 3,68 (1H, дд, J=11, 6 Гц), 3,30-3,21 (1H, м), 2,43 (1H, с), 2,31-2,23 (2H, м), 1,43 (6H, д, J=7 Гц), 1,32 (3H, д, J=7 Гц), 1,15 (3H, т, J=8 Гц); МС (ББА+) m/z: 510 [M+H]+.
Пример 68
N-Циклопропилметил-2-фтор-4-(5-{(1R)-1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид

К раствору в дихлорметане (2 мл) соединения, полученного в справочном примере 47 (103 мг, 0,228 ммоль), при комнатной температуре добавляют моногидрат 1-гидроксибензотриазола (34,9 мг, 0,228 ммоль), N-(3-диметиламинопропил)-N'-этилкарбодиимид (65,5 мг, 0,342 ммоль) и циклопропилметиламин (40 мкл, 0,46 ммоль), и смесь перемешивают 30 минут при той же температуре. Затем к реакционной смеси добавляют воду и смесь экстрагируют три раза дихлорметаном. Полученный таким образом органический слой промывают насыщенным водным раствором гидрокарбоната натрия, а затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении, и полученный остаток очищают колоночной хроматографией на силикагеле (гексан:этилацетат = 95:5 → 50:50, об./об.). В результате получают названное соединение (73,5 мг, выход 64%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 8,22 (1H, т, J=8 Гц), 8,01 (2H, д, J=9 Гц), 7,97-8,00 (1H, м), 7,86 (1H, д, J=13 Гц), 7,06 (2H, д, J=9 Гц), 6,82-6,90 (1H, м), 5,51 (1H, т, J=6 Гц), 3,36 (2H, дд, J=7, 7 Гц), 3,29-3,22 (1H, м), 2,34-2,20 (2H, м), 1,44 (6H, д, J=7 Гц), 1,15 (3H, т, J=7 Гц), 1,15-1,02 (1H, м), 0,61-0,56 (2H, м), 0,30 (2H, дд, J=11, 5 Гц); МС (ББА+) m/z: 506 [M+H]+.
Рецептурный пример
По 5 г каждого из соединений, полученных в примерах, 90 г лактозы, 34 г кукурузного крахмала, 20 г кристаллической целлюлозы и 1 г стеарата магния смешивают в смесителе, а затем смесь таблетируют с помощью таблетирующей машины. В результате получают таблетки.
Пример испытания 1
Тест oGTT на мышах (пероральный тест толерантности к глюкозе)
Взвешивают от 2,0 до 10,0 мг испытуемого соединения и затем добавляют 0,5%-ный (масс./об.) раствор метилцеллюлозы, чтобы получить жидкость для введения (1 мг/мл). Альтернативно, взвешивают от 1,0 до 10,0 мг испытуемого соединения и затем добавляют N,N-диметилформамид, чтобы получить раствор соединения (20 мг/мл). Раствор дополнительно разбавляют в 20 раз с использованием 0,5%-ного (масс./об.) раствора метилцеллюлозы, в результате получают жидкость для введения при конечной концентрации 1 мг/мл. От фирмы Charles River Laboratories Japan, Inc. получают мышей C57/BL6J (самцы возраста от 6 до 8 недель) и выращивают, пока они не достигнут возраста 9-13 недель. Мышей не кормят, начиная с временной точки от 17-ого до 18-ого часа за день до дня испытания, и до испытания не кормят непрерывно. На день испытания из каудальной вены отбирают кровь, а затем предварительно приготовленную жидкость для введения вводят перорально. Из каудальной вены снова отбирают кровь через тридцать минут после введения (уровень сахара в крови в это время обозначают как предварительное значение). Затем перорально вводят 30%-ный раствор глюкозы в количестве 10 мл/кг, в результате мыши подвергаются глюкозной нагрузке. После глюкозной нагрузки из каудальной вены отбирают кровь через временные точки 15, 30, 60 и 120 минут. Каждый из отобранных образцов крови центрифугируют, чтобы отделить плазму крови. Предварительное значение и значения уровня глюкозы в крови через 15, 30, 60 и 120 минут после глюкозной нагрузки измеряют с помощью Glucoloader GXT (A&T Corp.) с использованием образцов отделенной плазмы крови, и рассчитывают пониженную норму (%) уровня сахара в крови AUC относительно группы с введенным носителем. Тем временем группе с введенным носителем вводят 0,5%-ный (масс./об.) раствор метилцеллюлозы или смешанный раствор (5% (об./об.) N,N-диметилформамида)/(0,5 (масс./об.) метилцеллюлозы).
В результате установлено, что соединения примеров 5, 7, 14, 47-49, 52, 57, 58 и 64 снижают AUC на 5% или более и менее чем на 20%, а соединения примеров 1, 4, 6, 8, 10-13, 15-19, 23-26, 28, 31, 33, 43-45, 55-56, 61-63 и 66-68 снижают AUC на 20% или более.
Пример испытания 2
Тест oGTT на крысах (пероральный тест толерантности к глюкозе) и тест по измерению концентрации соединения в крови крыс
Взвешивают испытуемое соединение и затем готовят его суспензионную жидкость с использованием 0,5%-ного (масс./об.) раствора метилцеллюлозы. От фирмы Charles River Laboratories Japan, Inc. получают крыс Zucker Fatty и крыс Zucker Diabetic Fatty (самцы возраста от 8 до 20 недель) и перед проведением испытания крыс разбивают на группы на основе уровней сахара в крови и массы тела установленных групп. Крыс не кормят, начиная от временной точки от 15-ого до 18-ого часа за день до дня испытания, и до испытания не кормят непрерывно. На день испытания из каудальной вены отбирают кровь, а затем предварительно приготовленную суспензионную жидкость вводят перорально. Из каудальной вены снова отбирают кровь через тридцать минут после введения (уровень сахара в крови в это время обозначают как предварительное значение). Затем перорально вводят 50%-ный раствор глюкозы в количестве 4 мл/кг, в результате крысы подвергаются глюкозной нагрузке. После глюкозной нагрузки из каудальной вены отбирают кровь через временные точки 30 минут, 1, 2 и 3 часа. Каждый из отобранных образцов крови центрифугируют, чтобы отделить плазму крови. Предварительное значение и значения уровня глюкозы в крови через 30 минут, 1, 2 и 3 часа после глюкозной нагрузки измеряют с помощью Glucoloader GXT (A&T Corp.) с использованием образцов отделенной плазмы крови, и рассчитывают пониженную норму (%) уровня сахара в крови AUC относительно группы с введенным носителем. Тем временем группе с введенным носителем вводят 0,5%-ный (масс./об.) раствор метилцеллюлозы.
Образцы плазмы крови, полученные описанным выше способом, используют для измерения концентрации испытуемого соединения в плазме. Для измерения концентрации испытуемого соединения в плазме в течение дня кровь отбирают через 4-8 часов после введения и точно через 24 часа. Из плазмы крови удаляют белки и затем подают в анализатор (жидкостная хроматография)/масс-спектрометрия, чтобы рассчитать концентрацию соединения в плазме крови.
Пример испытания 3
Тест на защиту β-клеток (поджелудочной железы)
Защитное действие в отношении β-клеток (поджелудочной железы) испытуемого соединения подтверждают с учетом метода, описанного в публикации Junko Ogawa et al., Life Sciences, Vol. 65, № 12, pp. 1287-1296 (1999).
Применение в промышленности
Соединение настоящего изобретения или его фармацевтически приемлемая соль полезны в качестве активного ингредиента фармацевтической композиции для лечения и/или предупреждения диабета 1 типа, диабета 2 типа, гестационного диабета, гипергликемии вследствие других факторов, приобретенной толерантности к глюкозе, ассоциирующихся с диабетом заболеваний, диабетических осложнений и т.д., и в отношении защиты β-клеток или поджелудочной железы.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АЦИЛБЕНЗОЛА | 2011 |
|
RU2585765C2 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2010 |
|
RU2574410C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2006 |
|
RU2382781C2 |
| ПИПЕРАЗИНОВОЕ СОЕДИНЕНИЕ, ИНГИБИРУЮЩЕЕ ПРОСТАГЛАНДИН-D-СИНТАЗУ | 2010 |
|
RU2496778C2 |
| ПРОИЗВОДНЫЕ [1,8]НАФТИРИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕПЛИКАЦИИ ВИРУСА HCV | 2006 |
|
RU2467007C2 |
| ДИГИДРОПИРИДОФТАЛАЗИНОНОВЫЕ ИНГИБИТОРЫ ПОЛИ(АДФ-РИБОЗА)ПОЛИМЕРАЗЫ | 2009 |
|
RU2514937C2 |
| АГОНИСТЫ РЕЦЕПТОРА СФИНГОЗИН-1-ФОСФАТА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВНОГО СРЕДСТВА | 2014 |
|
RU2654483C2 |
| ИНГИБИТОРЫ ФЕРМЕНТА ДИАЦИЛГЛИЦЕРИН О-АЦИЛТРАНСФЕРАЗА ТИПА 1 | 2007 |
|
RU2474576C2 |
| Соединение, обладающее агонистической активностью в отношении GPR119, способ его получения и фармацевтическая композиция, содержащая его в качестве эффективного компонента | 2015 |
|
RU2670197C1 |
| ПРЕДОТВРАЩАЮЩИЙ ПРЕЖДЕВРЕМЕННУЮ ОВУЛЯЦИЮ АГЕНТ | 2006 |
|
RU2419435C2 |
Изобретение относится к соединениям формулы (I), где R1 представляет собой атом водорода или С1-С6-алкильную группу, замещенную одним или двумя заместителями, выбранными из С1-С6-алкокси-группы, гидроксильной группы, которые могут быть замещены С1-С6-алкилкарбонильной группой (замещенной одним или двумя заместителями γ), и 4-6-членной насыщенной моноциклической гетероциклической карбонильной группой, содержащей атом N; γ представляет собой гидроксильную группу, амино-группу, ди(С1-С6-алкил)амино-группу и карбамоильную группу; R2 представляет собой атом Н или С1-С6алкильную группу, которая может быть замещена гидроксильной группой; или R1 и R2 вместе с атомом азота, с которым они связаны, могут быть объединены с образованием азетидино-группы, пирролидино-группы или морфолино-группы, которые могут быть замещены одной гидроксильной группой или гидрокси-С1-С6-алкильной группой; R3 и R4 представляют собой С1-С6-алкильную группу; R5 представляет собой атом галогена или С1-С6-алкильную группу; R6 представляет собой атом галогена; m и n представляют собой целое число от 0 до 1; V и W представляют собой СН; X, Y и Z каждый независимо друг от друга представляет собой СН или N. Также изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), применению соединения формулы (I) и способу лечения сахарного диабета и заболевания, ассоциирующегося с диабетом. Технический результат - соединения формулы (I), обладающие гипогликемическим действием. 4 н. и 17 з.п. ф-лы, 6 табл., 72 пр.
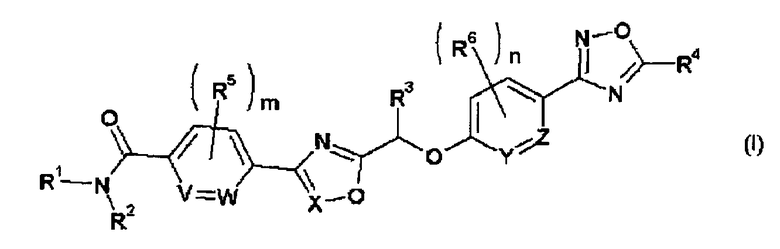
1. Соединение, представленное общей формулой (I):
где R1 представляет собой атом водорода или С1-С6-алкильную группу, замещенную одним или двумя заместителями, выбранными из подгруппы заместителя α;
подгруппа заместителя α представляет собой группу, включающую С1-С6-алкокси-группу и гидроксильную группу, которая может быть замещена заместителем, выбранным из подгруппы заместителя β;
подгруппа заместителя β представляет собой группу, включающую С1-С6-алкилкарбонильную группу, замещенную одним или двумя заместителями, выбранными из подгруппы заместителя γ, и 4-6-членную насыщенную моноциклическую гетероциклическую карбонильную группу, содержащую атом азота;
подгруппа заместителя γ представляет собой группу, включающую гидроксильную группу, амино-группу, ди(С1-С6-алкил)амино-группу и карбамоильную группу;
R2 представляет собой атом водорода или С1-С6алкильную группу, которая может быть замещена одной гидроксильной группой;
или R1 и R2 вместе с атомом азота, с которым R1 и R2 связаны, могут быть объединены с образованием азетидино-группы, пирролидино-группы или морфолино-группы, где азетидино-группа, пирролидино-группа или морфолино-группа могут быть замещены одной гидроксильной группой или одной гидрокси-С1-С6-алкильной группой;
R3 и R4 каждый независимо друг от друга представляет собой С1-С6-алкильную группу;
R5 представляет собой атом галогена или С1-С6-алкильную группу;
R6 представляет собой атом галогена;
m и n каждый независимо друг от друга представляет собой целое число от 0 до 1; и
V и W каждый представляет собой СН,
X, Y и Z каждый независимо друг от друга представляет собой СН или N,
или его фармацевтически приемлемая соль.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где Y и Z оба представляют собой СН.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где X представляет собой N.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 представляет собой С1-С4-алкильную группу, замещенную одной или двумя гидроксильными группами.
5. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 представляет собой гидроксиэтильную группу, гидроксиизопропильную группу, гидрокси-1,1-диметилэтильную группу или 2-гидрокси-1-(гидроксиметил)этильную группу.
6. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 представляет собой С1-С4-алкильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя α, где подгруппа заместителя α представляет собой гидроксильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя β, подгруппа заместителя β представляет собой C1-C4-алкилкарбонильную группу, замещенную одним заместителем, выбранным из подгруппы заместителя γ, и подгруппа заместителя γ представляет собой амино-группу.
7. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 представляет собой аминометилкарбонилоксиэтильную группу, аминометилкарбонилоксиизопропильную группу или аминометилкарбонилокси-1,1-диметилэтильную группу.
8. Соединение или его фармацевтически приемлемая соль по п. 1, где R2 представляет собой атом водорода.
9. Соединение или его фармацевтически приемлемая соль по п. 1, где R3 представляет собой C1-С3-алкильную группу.
10. Соединение или его фармацевтически приемлемая соль по п. 1, где R3 представляет собой этильную группу.
11. Соединение или его фармацевтически приемлемая соль по п. 1, где R4 представляет собой C1-С3-алкильную группу.
12. Соединение или его фармацевтически приемлемая соль по п. 1, где R4 представляет собой этильную группу или изопропильную группу.
13. Соединение или его фармацевтически приемлемая соль по п.1, где R5 представляет собой атом галогена и m принимает значение 1.
14. Соединение или его фармацевтически приемлемая соль по п. 1, где R5 представляет собой атом фтора и m принимает значение 1.
15. Соединение или его фармацевтически приемлемая соль по п. 1, где n принимает значение 0.
16. Соединение или его фармацевтически приемлемая соль по п. 1, выбранное из группы, включающей следующие соединения:
2-фтор-N-(2-гидроксиэтил)-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид;
2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид;
2-фтор-N-(2-гидрокси-1,1-диметилэтил)-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид;
гидрохлорид (2R)-2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}пропилглицината;
гидрохлорид 2-{[2-фтор-4-(5-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензоил]амино}-2-метилпропилглицината;
2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]-4-(2-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,3-оксазол-4-ил)бензамид;
гидрохлорид (2S)-2-{[2-фтор-4-(2-{1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,3-оксазол-4-ил)бензоил]амино}пропилглицината;
4-(5-{1-[4-(5-этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]бензамид;
гидрохлорид (2S)-2-{[4-(5-{1-[4-(5-этил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензоил]амино}пропилглицината;
2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-(5-{(1R)-1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)бензамид; и
N-(циклопропилметил)-2-фтор-4-(5-{(1R)-1-[4-(5-изопропил-1,2,4-оксадиазол-3-ил)фенокси]пропил}-1,2,4-оксадиазол-3-ил) бензамид.
17. Фармацевтическая композиция, обладающая гипогликемическим действием, содержащая в качестве активного ингредиента фармацевтически эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-16 и фармацевтически приемлемый наполнитель.
18. Фармацевтическая композиция по п. 17 для лечения и/или предупреждения сахарного диабета 1 типа, сахарного диабета 2 типа, ассоциирующегося с диабетом заболевания или ожирения.
19. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-16 для производства лекарственного средства для лечения и/или предупреждения сахарного диабета 1 типа, сахарного диабета 2 типа, ассоциирующегося с диабетом заболевания или ожирения.
20. Способ лечения и/или предупреждения сахарного диабета 1 типа, сахарного диабета 2 типа, ассоциирующегося с диабетом заболевания или ожирения, включающий введение млекопитающему фармакологически эффективного количества соединения по любому из пп. 1-16 или его фармацевтически приемлемой соли.
21. Способ по п. 20, где млекопитающим является человек.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Электрическое сопротивление для нагревательных приборов и нагревательный элемент для этих приборов | 1922 |
|
SU1997A1 |
| RU 2006138498 A, 10.05.2008 | |||

