ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к фармацевтически приемлемым солям новых бициклозамещенных производных азопиразолона, к способам их получения, фармацевтическим композициям, содержащим их, и к их применению в качестве терапевтического агента, в частности в качестве миметиков тромбопоэтина (ТПО) и агонистов тромбопоэтинового рецептора.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
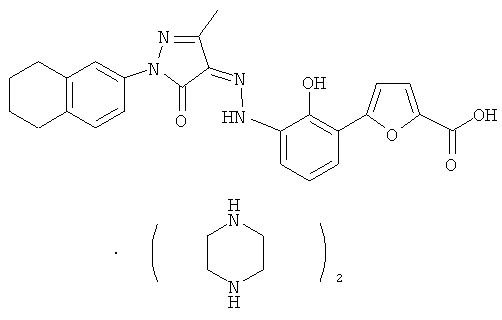
Тромбопоэтин (ТПО), также называемый фактором роста и развития мегакариоцитов (MGDF), стимулирующим фактором тромбоцитопоэза (TSF), лигандом с-миелопролиферативной лейкемии (с-Мр1), лигандом mpl или мегапоэтином, представляет собой гликопротеин, который описан как продуцирующий тромбоциты. См. Wendling, F., et. al., Biotherapy 10(4): 269-77 (1998); Kuter D.J. et al., The Oncologist, 1: 98-106 (1996); Metcalf, Nature 369: 519-520 (1994).
При определенных обстоятельствах активность ТПО является результатом связывания ТПО с рецептором ТПО (также называемым Mpl). Рецептор ТПО клонирован, и его аминокислотная последовательность описана. См. Vigon et al., Proc. Nat. Acad. Sci., 89: 5640-5644 (1992).
ТПО представляет собой 332-аминокислотный гликозилированный полипептид, который играет ключевую роль в регуляции мегакариоцитопоэза и в процессе, при котором тромбоциты продуцируются мегакариоцитами костного мозга. См. Kuter et al., Proc. Nat. Acad. Sci. USA 91: 11104-11108 (1994); Barley et al., Cell 77:1117-1124 (1994); Kaushansky et al., Nature 369:568-571 (1994); Wendling et al., Nature 369: 571-574 (1994); и Sauvage et al., Nature 369: 533-538 (1994). ТПО продуцируется в печени, но функционирует в основном в костном мозге, где он стимулирует дифференциацию стволовых клеток в предшественники мегакариоцитов и стимулирует пролиферацию мегакариоцитов, их полиплоидизацию и в конечном счете поступает в циркуляцию тромбоцитов в организме. ТПО также является основным регулятором в ситуациях, включающих тромбоцитопению, и в ряде исследований, которые включают повышенное количество тромбоцитов, размер тромбоцитов и включение изотопов в тромбоциты животных-реципиентов. См. Metcalf Nature 369: 519-520 (1994). Конкретно считают, что ТПО влияет на мегакариоцитопоэз следующими путями: (1) он вызывает увеличение размера и числа мегакариоцитов; (2) он увеличивает содержание ДНК в форме полиплоидии и числа мегакариоцитов; (3) он повышает эндомитоз мегакариоцитов; (4) он увеличивает число зрелых мегакариоцитов; (5) он повышает процент клеток-предшественников, число малых клеток, положительных по ацетилхолинэстеразе, число клеток костного мозга.
Тромбоциты необходимы для свертывания крови. Когда количество тромбоцитов является очень низким, пациент имеет риск гибели в результате катастрофического кровотечения. Таким образом, ТПО применяют как для диагностики, так и для лечения различных гематологических расстройств, например заболеваний, первично вызванных дефектами тромбоцитов. Подобным образом, ТПО может быть полезен для лечения тромбоцитопенических состояний, в частности состояний, возникающих в результате химиотерапии, лучевой терапии или трансплантации костного мозга для лечения рака или лимфомы.
Медленное восстановление уровней тромбоцитов у пациентов, страдающих тромбоцитопенией, является серьезной проблемой, поэтому желательно получить соединение для лечения тромбоцитопении посредством его действия в качестве миметика ТПО. Эти пептиды были разработаны для связывания и активации рецептора ТПО (ТПО-R), но они не обладают гомологией последовательности с природным ТПО. В последние годы описан ряд активных низкомолекулярных миметиков ТПО, включая производные циклических полиаминов (WO 00/28987), тиазол-2-илбензамиды (WO 01/07423, WO 01/53267), производные азоарила (WO 00/35446, WO 01/17349), 2-арилнафтимидазолы (WO 01/39773, WO 01/53267) и производные семикарбазонов (WO 01/34585). В системах на основе клеток все эти молекулы могут активировать биохимические пути преобразования сигнала, которые являются зависимыми от присутствия рецептора ТПО на клеточной мембране. Определенные типы соединений могут действовать непосредственно на сам рецептор ТПО. Было обнаружено, что некоторые из наиболее предпочтительных соединений этой серии стимулируют пролиферацию и дифференциацию ТПО-реагирующих линий клеток человека и ТПО в культурах костного мозга человека при их концентрации ниже 100 нМ.
В нескольких патентах на имя GlaxoSmithKline описан аналог тромбопоэтина-элтромбопаг (WO-2003098992/WO-01089457), с хорошей активностью.
В настоящем изобретении предложен ряд фармацевтически приемлемых солей бициклозамещенных производных азопиразолона, которые являются более эффективными миметиками ТПО и агонистами ТПО рецептора.
В международной заявке PCT/CN2009/000001, поданной автором настоящего изобретения 4 января 2009, описаны новые бициклозамещенные производные азопиразолона и их применение в качестве миметиков тромбопоэтина (ТПО) и агонистов рецептора тромбопоэтина. В шести примерах (Пример 1, Пример 9, Пример 15, Пример 28, Пример 43 и Пример 52), описанных в этой международной заявке, предложены соответственно нижеследующие соединения:
2'-гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-дифенил-3-карбоновая кислота,
5-{2-гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-фуран-2-карбоновая кислота,
5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновая кислота,
4-(2-гидрокси-3-[N'-(3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден)-гидразино]дифенилфуран-2-карбоновая кислота,
5-(3-{N'-[1-(3,3-диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-илиден]-гидразино}-2-гидроксифенил)-фуран-2-карбоновая кислота,
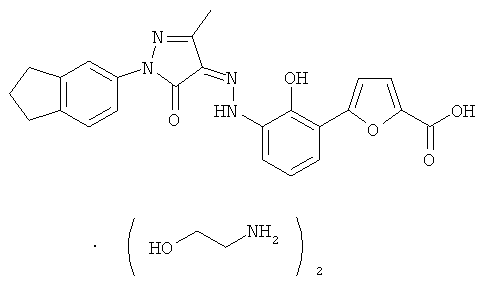
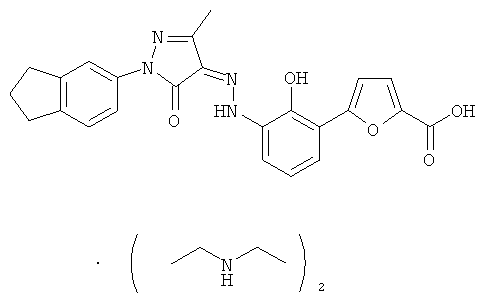
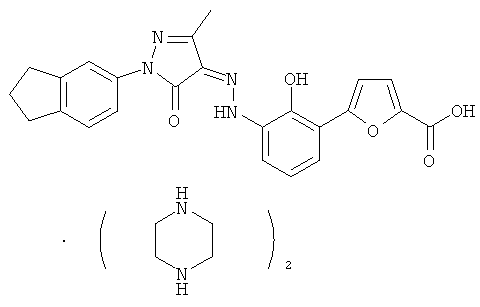
4-{2-гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-тиофен-2-карбоновая кислота,
и их сложные эфиры.
Эти соединения были протестированы и показали хорошую активность в качестве агонистов ТПО рецептора. Поэтому данная международная заявка полностью включена в данную заявку посредством ссылки. Однако в международной заявке №PCT/CN2009/000001 не описаны фармацевтически приемлемые соли этих соединений.
Автор изобретения открыл, что форма свободной кислоты бициклозамещенных производных азопиразолона слаборастворима в общепринятых растворителях и, следовательно, непригодна для получения лекарственной формы, поскольку ограничена ее биодоступность in vivo. Необходимо разрабатывать новые формы бициклозамещенных производных азопиразолона, которые можно использовать при общепринятом получении лекарственных форм, чтобы улучшить их растворимость и фармакокинетическую абсорбцию.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
С целью преодоления недостатков предшествующего уровня техники в настоящем изобретении предложены фармацевтически приемлемые соли новых бициклозамещенных производных азопиразолона, способы их получения, фармацевтические композиции, содержащие их, и их применение в качестве терапевтического агента, в частности в качестве миметиков тромбопоэтина (ТПО) и агонистов тромбопоэтинового рецептора. Эти соли обладают хорошими активностями для лечения тромбоцитопении, улучшенной растворимостью, хорошими активностями in vivo, лучшей биодоступностью, более низкой токсичностью, являясь хорошим кандидатом для получения лекарственного средства для лечения тромбоцитопении.
"Соединения по настоящему изобретению" и "соли по настоящему изобретению" взаимозаменяемы, и оба эти термина представляют собой фармацевтически приемлемые соли бициклозамещенных производных азопиразолона формулы (I)
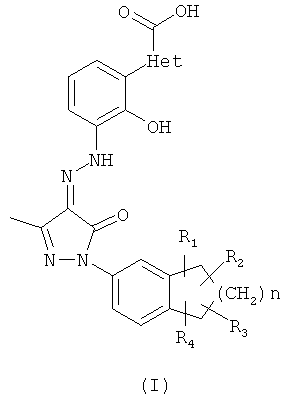
где:
Het выбран из группы, состоящей из фенила, фурила и тиенила;
R1, R2, R3 и R4 каждый независимо выбран из группы, состоящей из атома водорода и алкила;
n равно 0,1 или 2;
соли являются солями присоединения основания.
Кроме того, настоящее изобретение относится к солям соединений формулы (IA)

где:
Het выбран из группы, состоящей из фенила, фурила и тиенила;
R1, R2, R3 и R4 каждый независимо выбран из группы, состоящей из атома водорода и алкила;
M выбран из группы, состоящей из иона металла, иона аммония и основной аминокислоты;
m равно 1 или 2;
n равно 0,1 или 2;
соли являются солями присоединения основания.
Термин "свободная кислота" относится к бициклозамещенным производным азопиразолона формулы (I).
Понятие «эквивалент» относится к тем таутомерам соединений формулы (I), которые хорошо известны специалистам в данной области техники. Таутомеры соединений формулы (I) включают соединения формулы (II) и формулы (III), но не ограничены ими:
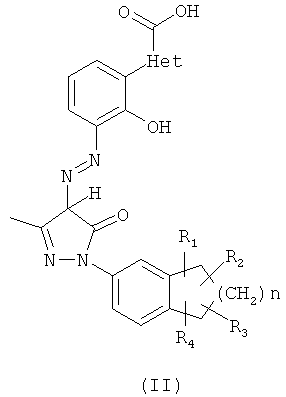

Все таутомеры соединений формулы (I) включены в объем настоящего изобретения, и все они включены в определение соединений формулы (I).
Термин "фармацевтически приемлемая соль" в настоящем изобретении относится к фармацевтически нетоксичным солям присоединения основания. Эти соли представляют собой соли, образованные между соединениями формулы (I) и подходящими основаниями, такими как гидроксид щелочного металла, основная аминокислота, амин или четвертичный аммоний, включая соль натрия, соль лития, соль калия, соль кальция, соль магния, соль аргинина, соль лизина, соль метанамина, соль диметиламина, соль триметиламина, соль этиламина, соль диэтиламина, соль триэтиламина, соль этаноламина, соль пиперазина, соль дибензилэтилендиамина, соль меглумина, соль трометамина, соль четвертичного тетраметиламмония, соль четвертичного тетраэтиламмония и соль холина, предпочтительно соль диэтиламина, соль этаноламина, соль холина, соль пиперазина, соль меглумина и соль трометамина, более предпочтительно соль этаноламина, соль холина, соль меглумина и соль трометамина, и наиболее предпочтительно соль этаноламина.
Фармацевтически приемлемые соли соединений формулы (I) настоящего изобретения предпочтительно включают следующие соединения, но не ограничены ими:














Настоящее изобретение относится к способу получения фармацевтически приемлемых солей соединений формулы (I), включающему стадии:
(a) растворение или суспендирование свободной кислоты по настоящему изобретению (соединения формулы (I)) в органическом растворителе, где органический растворитель выбран из группы, состоящей из метанола, этанола, ацетона, этилацетата и тетрагидрофурана, предпочтительно тетрагидрофурана;
(b) добавление основания к смеси при перемешивании, где основание может представлять собой органическое или неорганическое основание, такое как гидроксид щелочного металла или гидроксид щелочноземельного металла, основная аминокислота, амин или четвертичный аммоний;
(c) получение фармацевтически приемлемых солей соединения формулы (I),
где неорганические основания включают гидроксиды щелочных металлов, которые выбраны из группы, состоящей из гидроксида натрия, гидроксида лития, гидроксида калия, гидроксида кальция, гидроксида магния; основные аминокислоты выбраны из группы, состоящей из лизина и аргинина; амины выбраны из группы, состоящей из метанамина, диметиламина, триметиламина, этиламина, диэтиламина, триэтиламина, этаноламина, пиперазина, дибензилэтилендиамина, меглумина и трометамина; и четвертичные аммонии выбраны из группы, состоящей из четвертичного тетраметиламмония, четвертичного тетраэтиламмония, гидроксида холина, предпочтительно диэтиламин, этаноламин, гидроксид холина, пиперазин, меглумин и трометамин, более предпочтительно этаноламин, гидроксид холина, меглумин и трометамин, наиболее предпочтительно этаноламин.
На стадии (b) соотношение эквивалентов свободной кислоты и гидроксида щелочного металла, основной аминокислоты, амина и четвертичного аммония предпочтительно составляет 1:5~5:1, более предпочтительно 1:1~1:3 и наиболее предпочтительно 1:1~1:2.
На стадии (с) выделение солей предпочтительно включает прямое фильтрование из реакционной смеси, концентрирование из реакционной смеси и перекристаллизацию из органического растворителя. Соли можно высушивать в таких условиях, как вакуумная сушка или высокотемпературная воздушная сушка.
Реакции образования соли обычно проводят в условиях охлаждения, комнатной температуры или нагревания. Однако стоит отметить, что температура реакции связана с образованием соли, что хорошо известно специалистам в данной области техники.
Диапазон температур реакции по настоящему изобретению составляет диапазон от комнатной температуры до точки кипения растворителя реакционной смеси, предпочтительно 0~40°C. Специалист в данной области техники может легко определить наиболее предпочтительную температуру реакции для реакций образования соли общепринятыми методами.
Настоящее изобретение относится к применению фармацевтически приемлемых солей соединений формулы (I) для получения агониста тромбопоэтинового рецептора.
Настоящее изобретение относится к применению фармацевтически приемлемых солей соединений формулы (I) для получения лекарственного средства для лечения тромбоцитопении, где это лекарственное средство вводят совместно с лекарственным средством, выбранным из группы, состоящей из колониестимулирующего фактора, цитокина, хемокина, агониста или антагониста рецептора интерлейкина или цитокина, растворимого рецептора, агонистического или антагонистического антитела к рецептору или одного или более пептида или низкомолекулярного соединения, которое обладает одним и тем же механизмом с лекарственным средством.
Настоящее изобретение относится к фармацевтически приемлемым солям соединений формулы (I) для применения в качестве лекарственного средства для лечения тромбоцитопении, где это лекарственное средство вводят совместно с лекарственным средством, выбранным из группы, состоящей из колониестимулирующего фактора, цитокина, хемокина, агониста или антагониста рецептора интерлейкина или цитокина, растворимого рецептора, агонистического или антагонистического антитела к рецептору или одного или более пептида или низкомолекулярного соединения, которое обладает одним и тем же механизмом с лекарственным средством, где это лекарственное средство находится в форме пероральной лекарственной формы, либо лекарственное средство находится в форме парентеральной лекарственной формы.
Настоящее изобретение относится к способу лечения тромбоцитопении, включающему введение субъекту, нуждающемуся в этом, терапевтически эффективного количества фармацевтически приемлемой соли соединения формулы (I), где эту фармацевтически приемлемую соль вводят совместно с лекарственным средством, выбранным из группы, состоящей из колониестимулирующего фактора, цитокина, хемокина, агониста или антагониста рецептора интерлейкина или цитокина, растворимого рецептора, агонистического или антагонистического антитела к рецептору или одного или более пептида или низкомолекулярного соединения, которое обладает одинаковым механизмом действия с лекарственным средством, где фармацевтически приемлемая соль находится в форме пероральной лекарственной формы, либо фармацевтически приемлемая соль находится в форме парентеральной лекарственной формы.
Настоящее изобретение относится к фармацевтическим композициям, содержащим терапевтически эффективное количество фармацевтически приемлемой соли соединений формулы (I) и фармацевтически приемлемые носители или разбавители, где эту композицию вводят совместно с лекарственным средством, выбранным из группы, состоящей из колониестимулирующего фактора, цитокина, хемокина, интерлейкина и агониста рецептора цитокина. Настоящее изобретение также относится к применению этих композиций для получения лекарственного средства для лечения тромбоцитопении, где совместное введение включает применение лекарств по настоящему изобретению одновременно или последовательно.
Настоящее изобретение относится к способу получения фармацевтических композиций, содержащих терапевтически эффективное количество фармацевтически приемлемой соли соединений формулы (I) и фармацевтически приемлемые носители или разбавители, где этот способ включает стадию объединения соединений формулы (I) с фармацевтически приемлемыми носителями или разбавителями.
В способе получения фармацевтических композиций важно получить лекарственное средство в соответствующей форме, удобной в работе и в обращении, не только в плане коммерчески доступного препарата, но также в плане получения фармацевтических лекарственных форм, содержащих активные соединения.
В другом аспекте в способе получения фармацевтических композиций важно обеспечить надежную, воспроизводимую и постоянную кривую концентрации лекарства в плазме после введения субъекту.
Другие важные факторы включают химическую устойчивость, стабильность в твердом состоянии и срок хранения активного ингредиента. Лекарственные средства, содержащие эти композиции, можно предпочтительно хранить в течение относительно длительного времени без видимого изменения физических и химических свойств их активных ингредиентов, таких как химический состав, плотность, гигроскопичность и растворимость.
Важно также получить по возможности химически чистое лекарственное средство.
В типичном случае лекарственное средство может обеспечить следующие преимущества: удобную обработку, получение подходящих лекарственных форм и надежную растворимость, если это лекарственное средство может быть получено в стабильной форме, такой как стабильная кристаллическая форма, которая хорошо известна специалистам в данной области техники.
Эффективное количество активного ингредиента в фармацевтической стандартной лекарственной форме, как описано выше, должно быть нетоксичным, предпочтительно выбранным в интервале 0,001-100 мг/кг суммарной массы, более предпочтительно 0,001-50 мг/кг. При лечении субъекта, нуждающегося в миметиках ТПО, выбранную дозу предпочтительно вводят перорально или парентерально. Предпочтительные парентеральные формы включают формы местного, ректального, чрескожного введения, инъекции и непрерывной инфузии. Пероральные стандартные лекарственные формы для введения человеку предпочтительно содержат от 0,05 до 3500 мг активного ингредиента, наиболее предпочтительно от 0,5 до 1000 мг активного ингредиента. Предпочтительно пероральное введение, при котором используют более низкую дозировку. Парентеральное введение при высоких дозировках, однако, также можно применять, когда оно безопасно и приемлемо для пациента. Вышеописанные дозировки относятся к предпочтительному количеству активного ингредиента, в пересчете на свободную кислоту.
Специалистам в данной области техники понятно, что оптимальное количество и интервалы индивидуальных дозировок активного ингредиента зависят от природы и степени состояния, подлежащего лечению, формы, пути и сайта введения и от конкретного пациента, подлежащего лечению, и что такие оптимумы могут быть определены общепринятыми методами. Специалистам в данной области техники также понятно, что оптимальный курс лечения, то есть число доз активного ингредиента, даваемое за сутки в течение определенного числа суток, может быть определен специалистом в данной области техники при использовании общепринятых тестов определения курса лечения.
Соединения по настоящему изобретению можно вводить перорально или парентерально, причем эти соединения можно готовить в виде таблеток, пилюль, порошка и гранул, применяемых при различных путях введения. В вышеописанных твердых лекарственных формах активные ингредиенты смешаны по меньшей мере с одним видом инертного разбавителя. В соответствии с общепринятыми действиями, кроме инертного разбавителя, в пероральные лекарственные формы также включают другое вещество, такое как смазывающие вещества, глиданты и антиоксиданты. Приготовленные в виде капсул, таблеток и пилюль лекарственные формы содержат буферные агенты. Таблетки и пилюли можно готовить в лекарственных формах пролонгированного высвобождения.
Хотя можно использовать неводные эмульсии, парентеральные лекарственные формы по настоящему изобретению содержат стерильный водный раствор, и эти лекарственные формы также содержат адъюванты, например антисептики, увлажняющие агенты, агенты, способствующие проницаемости, буферные агенты, эмульгирующие агенты и диспергирующие агенты. В процессе стерилизации можно использовать фильтр, задерживающий бактерии, и в композиции, которые были подвергнуты облучению или нагреванию для стерилизации, можно добавлять стерилизующие агенты.
По сравнению со свободными кислотами соли по настоящему изобретению имеют описанные ниже преимущества:
(1) Соли по настоящему изобретению легко растворяются в общепринятых растворителях, таких как вода, метанол, 0,1% соляная кислота, и приспособлены для получения общепринятых лекарственных форм, где растворимость солей этаноламина заметно лучше в 0,1% соляной кислоте.
(2) Соли по настоящему изобретению обладают улучшенной стабильностью.
(3) Соли по настоящему изобретению обладают лучшей биологической активностью in vitro.
(4) Соли по настоящему изобретению обладают лучшими фармакокинетическими свойствами in vivo, лучшим всасыванием, более высокой биодоступностью и лучшей кривой фармакокинетики, где соль этаноламина, соль холина, соль пиперазина, соль меглумина и соль трометамина, предпочтительно соль этаноламина, обладают лучшими фармакокинетическими свойствами,.
(5) Способ получения солей по настоящему изобретению имеет преимущества высокого выхода, высокой чистоты, скорости, удобства и низких затрат, причем в способах получения более предпочтительны те соли этаноламина, соли холина, соли диэтиламина и соли пиперазина, которые можно кристаллизовать непосредственно.
По сравнению со свободными кислотами соли по настоящему изобретению, предпочтительно соль диэтиламина, соль этаноламина, соль холина, соль пиперазина, соль меглумина и соль трометамина, более предпочтительно соль этаноламина, соль холина, соль меглумина и соль трометамина, наиболее предпочтительно соль этаноламина, обладают лучшими свойствами растворимости, стабильности, биологической активности in vitro и фармакокинетики.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, приведенные ниже термины, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
Термин "этаноламин" относится к "2-аминоэтанолу".
Термин "холин" относится к "(2-гидроксиэтил)триметиламину".
Термин "меглумин" относится к "N-метил-D-меглумину".
Термин "фармацевтическая композиция" относится к смеси одной или более фармацевтически приемлемых солей соединения, описанного в данной заявке, или его пролекарств с другими химическими ингредиентами, такими как физиологически/фармацевтически приемлемые носители. Целью фармацевтической композиции является облегчение введения соединения в организм.
Термин "стабильность" относится к химической стабильности и стабильности твердого состояния.
Термин "химическая стабильность" относится к хранению соединений по настоящему изобретению, включая изолированные формы или лекарственные формы, смешанные с фармацевтически приемлемыми носителями или разбавителями (например, пероральные лекарственные формы, такие как таблетки, капсулы и т.д.), в стандартных условиях с незначительным химическим разложением или химическим распадом.
Термин "стабильность твердого состояния" относится к хранению соединений по настоящему изобретению, включая изолированные твердые формы или лекарственные формы, смешанные с фармацевтически приемлемыми носителями или разбавителями (например, пероральные лекарственные формы, такие как таблетки, капсулы и т.д.), в стандартных условиях с незначительным полиморфным превращением (например, кристаллизацией, перекристаллизацией, изменением твердофазного состояния, гидратацией, дегидратацией, сольватацией или удалением растворителя).
Примеры термина "хранимый в стандартных условиях" включают диапазон температуры от -80°C до +50°C (предпочтительно от 0°C до 40°C, более предпочтительно при комнатной температуре, такой как 15°C~30°C), диапазон давления от 0,1 Па до 2 Па (предпочтительно при атмосферном давлении), диапазон относительной влажности от 5% до 95% (предпочтительно 10%~60%) и/или воздействие 460 люкс УФ/видимого света в течение длительного времени (более длительного или равного шести месяцам).
Термин "парентеральное введение" включает внутривенное, внутримышечное, подкожное, интраназальное, интраректальное, интравагинальное или внутрибрюшинное введение, предпочтительно пероральное введение.
Термин "гигроскопичность" относится к свойству способности или степени, с которой вещество может поглощать воду при определенной температуре и влажности. Испытуемыми образцами являются твердые ингредиенты, удовлетворяющие стандарту контроля качества лекарственного средства. Упаковки лекарств и условия хранения могут относиться к результатам вышеописанного испытания.
СПОСОБ СИНТЕЗА СОЕДИНЕНИЯ ПО ИЗОБРЕТЕНИЮ
В целях достижения цели изобретения в изобретении используют описанные ниже технические решения:
Способ синтеза соединения формулы (I) раскрыт в примерах 1, 9,15, 28, 43 и 52 международной заявки PCT/CN2009/000001, поданной 4 января 2009. Эта заявка полностью включена в данную заявку посредством ссылки.
Способ получения фармацевтически приемлемых солей соединений формулы (I) включает следующие стадии:
(a) растворение или суспендирование свободной кислоты по настоящему изобретению (соединения формулы (I)) в органическом растворителе, где органический растворитель выбран из группы, состоящей из метанола, этанола, ацетона, этилацетата и тетрагидрофурана, предпочтительно тетрагидрофурана;
(b) добавление к смеси основания при перемешивании, где основание может представлять собой органическое или неорганическое основание, такое как гидроксид щелочного металла или гидроксид щелочноземельного металла, основная аминокислота, амин или четвертичный аммоний;
(c) получение фармацевтически приемлемой соли соединения формулы (I), где неорганические основания включают гидроксиды щелочных металлов, которые выбраны из группы, состоящей из гидроксида натрия, гидроксида лития, гидроксида калия, гидроксида кальция, гидроксида магния; амины и четвертичные аммонии выбраны из группы, состоящей из четвертичного тетраметиламмония, четвертичного тетраэтиламмония, этаноламина, холина, лизина, аргинина, метанамина, диметиламина, триметиламина, этиламина, диэтиламина, триэтиламина, дибензилэтилендиамина, меглумина, пиперазина и трометамина; предпочтительно диэтиламина, этаноламина, пиперазина, гидроксида холина, меглумина и трометамина, более предпочтительно этаноламина, гидроксида холина, меглумина и трометамина, наиболее предпочтительно этаноламина.
На стадии (b) соотношение эквивалентов свободной кислоты и основания предпочтительно составляет 1:5~5:1, более предпочтительно 1:1~1:3 и наиболее предпочтительно 1:1~1:2.
На стадии (с) выделение солей предпочтительно включает прямое фильтрование из реакционной смеси, концентрированно из реакционной смеси и перекристаллизацию из органического растворителя. Соли можно высушивать при условиях, таких как вакуумная сушка или высокотемпературная воздушная сушка.
Вышеописанные реакции образования соли обычно проводят в условиях охлаждения, комнатной температуры или нагревания. Однако следует отметить, что температура реакции обладает влиянием на реакцию образования соли, что хорошо известно специалистам в данной области техники. Диапазон температуры реакции по настоящему изобретению составляет от комнатной температуры до точки кипения растворителя реакционной смеси, предпочтительно 0~40°C. Специалист в данной области техники может легко определить наиболее предпочтительную температуру реакции из реакций образования соли общепринятыми методами.
Настоящее изобретение далее описано приведенными ниже Примерами, которые не предназначены для ограничения объема изобретения.
ПРИМЕРЫ
Структуры всех соединений были идентифицированы с помощью ядерного магнитного резонанса (1Н ЯМР) или масс-спектрометрии (МС).
ЯМР проводили на спектрометре Bruker AVANCE-400. Соответствующие растворители включали дейтерированный метанол (CD3OD), дейтерированный хлороформ (CDCl3) и дейтерированный диметилсульфоксид (ДМСО-d6) с тетраметилсиланом (ТМС) в качестве внутреннего стандарта, и химические сдвиги регистрировали в виде млн-1 (10-6).
МС определяли на масс-спектрометре FINNIGAN LCQ Ad (ESI) (Thermo, Model: Finnigan LCQ усовершенствованный MAX).
EC50 определяли на NovoStar ELIASA (BMG Co. German).
Понятие «тонкослойный гель» относится к пластине силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размер пластин, используемых при ТСХ, составляет от 0,15 мм~0,2 мм, и размер пластин, используемых при очистке препарата, составлял 0,4 мм~0,5 мм.
При колоночной хроматографии обычно использовали силикагель Yantai Huanghai 200-300 меш в качестве носителя.
ВЭЖХ на высокоэффективном жидкостном хроматографе Agilent 1200DAD (хроматографическая колонка Sunfire C18 150×4,6 мм) и высокоэффективном жидкостном хроматографе Waters 2695-2996 (хроматографическая колонка Gimini C18 150×4,6 мм).
Реакции гидрогенизации под давлением проводили с помощью гидрогенизатора Parr 3916EKX и генератора водорода QL. Микроволновые реакции проводили с помощью микроволнового реактора СЕМ Discover-S 908860.
При реакциях гидрогенизации реакционную систему обычно помещали в вакуум и заполняли водородом, повторяя операцию три раза.
Известный исходный материал по изобретению может быть получен способом, общепринятым в данной области техники, либо приобретен от фирмы ABCR GmbH & Со. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc или Dari chemical Company и т.д.
Если не указано иное, нижеописанные реакции проводили в атмосфере азота.
Термин "атмосфера азота" относится к тому, что реакционная колба оборудована 1 л баллоном азота.
Термин "атмосфера водорода" относится к тому, что реакционная колба оборудована 1 л баллоном водорода.
Если не указано иное, раствор, используемый в нижеописанной реакции, относится к водному раствору.
Термин "ТСХ" относится к тонкослойной хроматографии.
Термин "ВЭЖХ" относится к высокоэффективной жидкостной хроматографии.
Опытные условия ВЭЖХ: время прогона: 30 мин, температура колонки: 30°C ФДМ: 230 нм, подвижная фаза: ацетонитрил: вода (0,1% трифторуксусная кислота)=25:75, скорость тока: 1,0 мл/минута.
Хроматографическая колонка: C18,150*4,6 мм Gemini.
Пример 1
(Z)-2'-Гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-дифенил-3-карбоновой кислоты бис-(этаноламин)


Стадия 1
2-Бром-6-нитрофенол
Раствор 60 мл концентрированной серной кислоты, разведенной 186 мл воды, охлаждали до комнатной температуры. К этому раствору добавляли нитрат натрия (79,2 г, 0,93 моль). 2-Бромфенол 1а (60 мл, 0,52 моль) добавляли по каплям с такой скоростью, чтобы поддерживать температуру реакции ниже 25°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Осадок растворяли в 320 мл этилацетата. Смесь промывали водой и насыщенным рассолом, высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 2-бром-6-нитрофенола 1b (48,2 г, выход 42,8%) в виде желтого твердого вещества.
МС m/z (ИЭР):218[М+1]
1H ЯМР (400 МГц, CDCl3): δ 11.18 (s, 1Н), 8.12-8.15 (m, 1Н), 7.89-7.91 (m, 1Н), 6.88-7.02 (m, 1Н)
Стадия 2
1-Бром-2-метокси-3-нитробензол
2-Бром-6-нитрофенол 1b (46,55 г, 0,214 моль) растворяли в 500 мл ацетона с последующим добавлением карбоната калия (35,36 г, 0,26 моль) и йодометана (20,1 мл, 0,32 моль). Реакционную смесь кипятили с обратным холодильником при 70°C в течение 40 часов. Реакционную смесь концентрировали при пониженном давлении и разбавляли 1300 мл этилацетата и 500 мл воды. Водный слой экстрагировали этилацетатом (300 мл×2). Объединенные органические экстракты промывали 4 М соляной кислотой и насыщенным раствором бикарбоната натрия, а затем высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 1-бром-2-метокси-3-нитробензола 1 с (44,59 г, выход 90,0%) в виде коричневого твердого вещества.
МС m/z (ИЭР):234[М+1]
Стадия 3
2'-Метокси-3'-нитродифенил-3-карбоновая кислота
1-Бром-2-метокси-3-нитробензол 1с (23,25 г, 0,10 моль), 3-карбоксифенилбороновую кислоту (19,5 г, 117 ммоль) и тетракис(трифенилфосфин)палладий (8,86 г, 7,7 моль) растворяли в смеси растворителей 100 мл 2 М раствора карбоната натрия и 500 мл 1,4-диоксана. Реакционную смесь кипятили с обратным холодильником при 105°C в течение 43 часов. Смесь концентрировали при пониженном давлении, а затем добавляли 300 мл 6 н. соляной кислоты и 400 мл этилацетата. Водный слой экстрагировали этилацетатом (200 мл×2). Объединенные органические экстракты высушивали над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 2'-метокси-3'-нитродифенил-3-карбоновой кислоты 1d (53,93 г) в виде светло-желтого твердого вещества.
MC m/z (ИЭР): 272 [M-1]
1H ЯМР (400 МГц, CDCl3): δ 8.11 (s, 1H), 8.02 (d, J=8.0 Гц, 1H), 7.90-7.92 (m, 1H), 7.82-7.84 (m, 1H), 7.21-7.75 (m, 1H), 7.63-7.67 (m, 1H), 7.42-7.46 (m, 1H), 3.45 (s, 3H)
Стадия 4
2'-Метокси-3'-аминодифенил-3-карбоновая кислота
2'-Метокси-3'-нитродифенил-3-карбоновую кислоту 1d (0,48 г, 1,74 ммоль) растворяли в 60 мл этанола с последующим добавлением 0,5 г палладия на углероде (10%) и формиата аммония (1,1 г, 17,4 ммоль). Реакционную смесь кипятили с обратным холодильником при 80°C в течение 20 минут. Смесь фильтровали и фильтрат концентрировали при пониженном давлении и высушивали с получением соединения, указанного в заголовке, 2'-метокси-3'-аминодифенил-3-карбоновой кислоты 1е (0,42 г, выход 93,3%) в виде белого твердого вещества.
МС m/z (ИЭР): 242[М-1]
Стадия 5
3'-Амино-2'-гидроксидифенил-3-карбоновой кислоты гидробромид
Используя известный способ, описанный в заявке на патент WO 0189457: 2'-метокси-3'-аминодифенил-3-карбоновую кислоту 1е (2,5 г, 10,3 ммоль) растворяли в 100 мл бромисто-водородной кислоты (40%). Реакционную смесь кипятили с обратным холодильником при 120°C в течение ночи. Смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 3'-амино-2'-гидроксидифенил-3-карбоновой кислоты гидробромида 1f (2,4 г, 88,8%) в виде твердого вещества цвета хаки.
МС m/z (ИЭР): 230 [М+1]
Стадия 6
Индан-5-илгидразин
Индан-5-иламин 1g (3,59 г, 27,0 ммоль) растворяли в 20 мл концентрированной соляной кислоты при охлаждении в бане лед-вода и смесь перемешивали в течение 10 минут. Добавляли по каплям 10 мл раствора нитрита натрия (1,86 г, 27,0 ммоль) и смесь перемешивали еще в течение 15 минут и использовали в последующей реакции.
При охлаждении в бане лед-соль дигидрат хлорида олова (24,4 г, 108,0 ммоль) растворяли в 10 мл концентрированной соляной кислоты с последующим добавлением вышеупомянутой сохраненной смеси. Реакционную смесь нагревали до комнатной температуры и подвергали взаимодействию в течение 1,5 часов. Затем смесь доводили до pH 9 40% раствором гидроксида натрия при охлаждении в бане лед-вода. Смесь экстрагировали 400 мл этилацетата, объединенные органические экстракты концентрировали при пониженном давлении и высушивали с получением соединения, указанного в заголовке, индан-5-илгидразина 1h (2,05 г, выход 51,3%) в виде красновато-коричневого твердого вещества.
МС m/z (ИЭР): 149 [М+1]
Стадия 7
2-Индан-5-ил-5-метил-2,4-дигидропиразол-3-он
Индан-5-илгидразин 1h (2,05 г, 13,8 ммоль) растворяли в 50 мл уксусной кислоты с последующим добавлением этилацетоацетата (1,76 мл, 13,8 ммоль). Реакционную смесь нагревали при 100°C в течение ночи. Смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 2-индан-5-ил-5-метил-2,4-дигидропиразол-3-она 1i (1,84 г, выход 62,3%) в виде желтого твердого вещества.
МС m/z (ИЭР): 215 [М+1]
1H ЯМР (400 МГц, CDCl3): δ 7.69 (s, 1H), 7.60 (d, J=8.0 Гц, 1H), 7.24 (d, J=8 Гц, 1H), 3.44 (s, 2H), 2.90-2.97 (m, 4H), 3.21 (s, 3Н), 2.07-2.14 (m, 2H)
Стадия 8
(Z)-2'-Гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-дифенил-3-карбоновая кислота
При охлаждении в бане лед-вода 3'-амино-2'-гидроксидифенил-3-карбоновой кислоты гидробромид 1f (267 мг, 1,16 ммоль) растворяли в 10 мл 1 М соляной кислоты с последующим добавлением по каплям 10 мл раствора нитрита натрия (88 мг, 1,28 ммоль) и 2-индан-5-ил-5-метил-2,4-дигидропиразол-3-она 1i (249 мг, 1,16 ммоль). Смесь доводили до pH 8 насыщенным раствором бикарбоната натрия с последующим добавлением 10 мл этанола. Реакционную смесь нагревали до комнатной температуры в течение ночи. Смесь фильтровали, высушивали и перекристаллизовали из метанола с получением соединения, указанного в заголовке, (Z)-2'-гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-карбонил-1,5-дигидропиразол-4-илиден)-гидразино]-дифенил-3-карбоновой кислоты 1j (60 мг, выход 11,4%) в виде желтого твердого вещества.
МС m/z (ИЭР): 453 [М-1]
1H ЯМР (400 МГц, ДМСО-d6): δ 13.76 (br. s, 1Н), 13.03 (br.s, 1H), 9.66 (br.s, 1H), 8.13 (s, 1H), 7.96-7.98 (d, J=8.1 Гц, 1H), 7.60-7.82 (m, 5H), 7.28-7.30 (d, J=8.1 Гц, 1H), 7.13-7.17 (m, 2H), 2.86-2.93 (m, 4H), 2.34 (s, 3Н), 2.03-2.10 (m, 2H)
Стадия 9
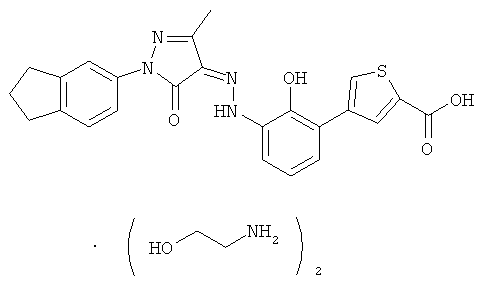
(Z)-2'-Гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-дифенил-3-карбоновой кислоты бис-(этаноламин)
(Z)-2'-гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-карбонил-1,5-дигидропиразол-4-ил иден)-гидразино]-дифенил-3-карбоновую кислоту 1j (454 мг, 1,0 ммоль) растворяли в 16 мл тетрагидрофурана. К реакционной смеси добавляли этаноламин (143 мг, 2,35 ммоль) и перемешивали в течение 3 часов. Смесь фильтровали, фильтрационный кек промывали тетрагидрофураном (2 мл×3) и твердое вещество высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-2'-гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-карбонил-1,5-дигидропиразол-4-илиден)-гидразино]-дифенил-3-карбоновой кислоты бис-(этаноламин) 1 (553 мг, выход:
96,0%) в виде темно-красного твердого вещества.
MC m/z (ИЭР): 453 [M-1]
1H ЯМР (400 МГц, CD3OD): δ 8.13 (s, 1H), 7.92 (d, J=7.6 Гц, 1H), 7.69 (m, 3H), 7.61 (d, J=8.0 Гц, 1H), 7.45 (t, J=7.6 Гц, 1H), 7.25 (d, J=8.0 Гц, 1H), 7.17 (d, J=8.0 Гц, 1H), 6.98 (t, J=8.0 Гц, 1H), 3.65 (t, J=5.2 Гц, 4H), 2.95 (m, 4H), 2.86 (t, J=5.2 Гц, 4H), 2.41 (s, 3H),2.12 (m, 2H).
Пример 2
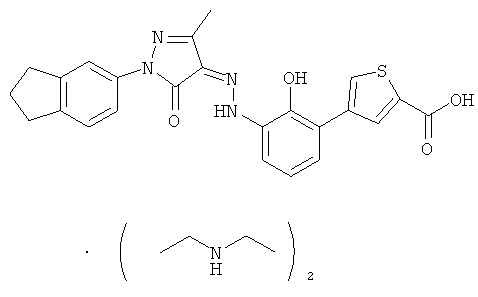
(Z)-2'-Гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино1-дифенил-3-карбоновой кислоты бис-(диэтиламин)

(Z)-2'-Гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-карбонил-1,5-дигидропиразол-4-ил иден)-гидразино]-дифенил-3-карбоновую кислоту 1j (150 мг, 0,33 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красного раствора. К этому раствору добавляли по каплям диэтиламин (48 мг, 0,66 ммоль) с образованием пурпурного раствора и перемешивали в течение 2 часов. Твердое вещество осаждали из раствора. Смесь фильтровали, фильтрационный кек промывали тетрагидрофураном (1 мл×3) и твердое вещество высушивали в вакууме с получением соединения, указанного в заголовке,
(Z)-2'-гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-карбонил-1,5-дигидропиразол-4-илиден)-гидразино]-дифенил-3-карбоновой кислоты бис-(диэтиламин) 2 (132 мг, выход: 66,7%) в виде красного твердого вещества.
ВЭЖХ: 99,2%
МС m/z (ИЭР): 452,9 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 8.08 (m, 1Н), 7.94 (d, J=7.6 Гц, 1Н), 7.72 (m, 2Н), 7.62 (m, 2Н), 7.55 (m, 1Н), 7.25 (d, J=8.4 Гц, 1Н), 7.14 (d, J=8.4 Гц, 1Н), 7.07 (m, 1Н), 2.89-2.98 (m, 12Н), 2.38 (s, 3H), 2.09-2.14 (m, 2Н), 1.34 (m, 12Н).
Пример 3
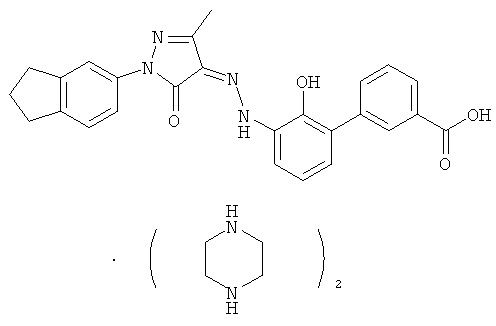
(Z)-2'-Гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино1-дифенил-3-карбоновой кислоты бис-(пиперазин)

(Z)-2'-Гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-карбонил-1,5-дигидропиразол-4-илиден)-гидразино]-дифенил-3-карбоновую кислоту 1] (150 мг, 0,33 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли пиперазин (57 мг, 0,66 ммоль) с образованием пурпурного раствора и перемешивали при комнатной температуре в течение 2 часов. Твердое вещество осаждали из раствора, фильтровали, затем фильтрационный кек промывали тетрагидрофураном (1 мл×3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-2'-гидрокси-3'-[N'-(1-индан-5-ил-3-метил-5-карбонил-1,5-дигидропиразол-4-илиден)-гидразино]-дифенил-3-карбоновой кислоты бис-(пиперазин) 3 (130 мг, выход:
62,8%) в виде темно-красного твердого вещества.
ВЭЖХ: 98,5%
МС m/z (ИЭР): 452,8 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 8.10 (s, 1Н), 7.92 (d, J=7.6 Гц, 1Н), 7.68 (m, 3H), 7.61 (m, 1Н), 7.43 (m, 1Н), 7.24 (d, J=8.0 Гц, 1Н), 7.15 (m, 1Н), 7.00 (m, 1Н), 2.89-2.95(m, 4Н), 2.84 (s, 16H), 2.39 (s, 3H), 2.09-2.12 (m, 2Н)
Пример 4
(Z)-5-(3-(N'-[1-(3,3-Диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-илиден]-гидразино}-2-гидроксифенил)-фуран-2-карбоновой кислоты бис-(этаноламин)


Стадия 1 Ди-трет-бутил-1-(3,3-диметилинден-5-ил)гидразин-1,2-дикарбоксилат
6-Бром-1,1-диметилиндан (полученный путем использования хорошо известного способа: заявка на патент WO 2005066115) 4а (4,32 г, 19,27 ммоль) растворяли в 40 мл тетрагидрофурана, а затем добавляли по каплям бутиллитий (15,67 мл, 1,6 М, 25,05 ммоль) при -78°C. После взаимодействия реакционной смеси в течение 40 минут затем добавляли раствор ди-трет-бутилазодикарбоксилата (5,32 г, 23,12 ммоль) в 30 мл тетрагидрофурана. Реакционную смесь подвергали взаимодействию еще в течение 3 часов при -78°C. К реакционной смеси добавляли 5 мл метанола, затем нагревали до комнатной температуры и фильтровали на силикагеле. Фильтрат концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, ди-трет-бутил-1-(3,3-диметил-1Н-инден-5-ил)гидразин-1,2-дикарбоксилата 4b (2,70 г, выход 37,2%) в виде желтого твердого вещества.
Стадия 2
2-(3,3-Диметилиндан-5-ил)-5-метил-2,4-дигидропиразол-3-он
Ди-трет-бутил-1-(3,3-диметил-1H-инден-5-ил)гидразин-1,2-дикарбоксилат 4b (2,70 г, 7,18 ммоль) растворяли в 100 мл уксусной кислоты с последующим добавлением 20 мл трифторуксусной кислоты. После взаимодействия смеси при комнатной температуре в течение 2 часов добавляли этилацетоацетат (0,98 г, 7,54 ммоль). Затем смесь нагревали до 100°C и подвергали взаимодействию в течение 2 часов. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении, чтобы удалить уксусную кислоту. Реакционную смесь нейтрализовали насыщенным раствором бикарбоната натрия, а затем экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным рассолом, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 2-(3,3-диметилиндан-5-ил)-5-метил-2,4-дигидропиразол-3-она 4 с (1,0 г, выход 47,7%) в виде светло-коричневого твердого вещества.
МС m/z (ИЭР):243[М+1]
Стадия 3
2-(2-Метокси-3-нитрофенил)-4,4,5,5-тетраметил-[1,3,2]диоксаборолан
1-Бром-2-метокси-3-нитробензол 1с (67 г, 289 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би-1,3,2-диоксаборолан (110 г, 433 ммоль), тетракис(трифенилфосфин)палладий (11,80 г, 14,44 ммоль) и ацетат калия (71 г, 724 ммоль) растворяли в 600 мл щавелевой кислоты диметилового эфира. Смесь кипятили с обратным холодильником в течение 17 часов. Смесь концентрировали при пониженном давлении, полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 2-(2-метокси-3-нитрофенил)-4,4,5,5-тетраметил-[1,3,2]диоксаборолана 4d (50,5 г, 61,9%) в виде желтых кристаллов.
Стадия 4
5-(2-Метокси-3-нитрофенил)фуран-2-карбоновая кислота
2-(2-Метокси-3-нитрофенил)-4,4,5,5-тетраметил-[1,3,2]диоксаборолан 4d (10 г, 35,85 ммоль), 5-бромфуран-2-карбоновую кислоту (5,47 г, 28,66 ммоль), тетракис(трифенилфосфин)палладий (2,07 г, 1,79 ммоль) и карбонат натрия (7,60 г, 71,66 ммоль) растворяли в смеси растворителей 200 мл 1,4-диоксана и 30 мл воды. Реакционную смесь кипятили с обратным холодильником в течение 2,5 часов. Смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток разбавляли 150 мл воды и доводили до pH 3 1 М соляной кислотой. Затем смесь фильтровали и фильтрационный кек промывали 50 мл смеси растворителей н-гексана/этилацетата (об/об=1:1). Остаток высушивали с получением соединения, указанного в заголовке, 5-(2-метокси-3-нитрофенил) фуран-2-карбоновой кислоты 4е (4,23 г, выход 56,1%) в виде серого твердого вещества.
МС m/z (ИЭР): 262[М-1]
Стадия 5
5-(3-Амино-2-метоксифенил)-фуран-2-карбоновая кислота
5-(2-Метокси-3-нитрофенил)фуран-2-карбоновую кислоту 4е (4,23 г, 16,09 ммоль) растворяли в 125 мл этилацетата с последующим добавлением 423 мг палладия на углероде (10%) и формиата аммония (4,054 г, 64,35 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 3,5 часов. Смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 5-(3-амино-2-метоксифенил)-фуран-2-карбоновой кислоты 4f (2,79 г, выход 74,4%) в виде светло-зеленого твердого вещества.
МС m/z(ИЭР):232[М-1]
Стадия 6
5-(3-Амино-2-гидроксифенил)-фуран-2-карбоновой кислоты гидробромид
5-(3-Амино-2-метоксифенил)-фуран-2-карбоновую кислоту 4f (2,79 г, 11,97 ммоль) растворяли в 25 мл дихлорметана с последующим добавлением по каплям трибромида бора (23,9 мл, 2,0 М). Реакционную смесь подвергали взаимодействию при комнатной температуре в течение 1 часа. Смесь концентрировали при пониженном давлении после добавления 5 мл метанола. Остаток разбавляли 100 мл этилацетата и перемешивали в течение 1 часа. Затем смесь фильтровали и фильтрационный кек высушивали с получением соединения, указанного в заголовке, 5-(3-амино-2-гидроксифенил)-фуран-2-карбоновой кислоты гидробромида 4д (1,24 г, выход 47,2%) в виде желтого твердого вещества.
MC m/z (ИЭР): 218[M-1]
Стадия 7
(Z)-5-(3-{N'-[1-(3,3-Диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-илиден]-гидразино}-2-гидроксифенил)-фуран-2-карбоновая кислота
(Z)-5-(3-Амино-2-гидроксифенил)-фуран-2-карбоновой кислоты гидробромид 4g (333 мг, 1,1 ммоль) растворяли в соляной кислоте (3,7 мл, 1 М) при охлаждении в бане лед-вода с последующим добавлением по каплям 1,5 мл раствора нитрита натрия (85 мг, 1,22 ммоль). После того как смесь подвергали взаимодействию в течение 20 минут, последовательно добавляли 2-(3,3-диметилиндан-5-ил)-5-метил-2,4-дигидропиразол-3-он 4 с (242 мг, 1,0 ммоль), бикарбонат натрия (1,4 г, 16,67 ммоль) и 3 мл этанола. Реакционную смесь подвергали взаимодействию в течение ночи при комнатной температуре. Смесь фильтровали и к фильтрационному кеку добавляли 20 мл воды. Смесь доводили до pH 3-4 концентрированной соляной кислотой. Смесь фильтровали и фильтрационный кек высушивали и очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (Z)-5-(3-{N'-[1-(3,3-диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-илиден]-гидразино}-2-гидроксифенил)-фуран-2-карбоновой кислоты 4h (190 мг, выход 40,3%) в виде красного твердого вещества.
МС m/z (ИЭР): 470,9 [М-1]
1H ЯМР (400 МГц, ДМСО-d6): δ 13.74 (br. s, 1H), 13.15 (br. s, 1H), 9.99 (br. s, 1H), 7.71 (т, 3Н), 7.55 (d, J=6.8 Гц, 1H), 7.37 (d, J=3.6 Гц, 1H), 7.20 (m, 2Н), 7.15 (m, 1H), 2.86 (t, J=7.2 Гц, 2Н), 2.33 (s, 3Н), 1.92 (t, J=7.2 Гц, 2Н), 1.26 (s, 6H)
Стадия 8
(Z)-5-(3-{N'-[1-(3,3-Диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-илиден]-гидразино}-2-гидроксифенил)-фуран-2-карбоновой кислоты бис-(этаноламин)
(Z)-5-(3-{N'-[1-(3,3-Диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-или ден]-гидразино}-2-гидроксифенил)-фуран-2-карбоновую кислоту 4h (2,3 г, 4,87 ммоль) растворяли в 20 мл тетрагидрофурана. К раствору добавляли этаноламин (594 мг, 9,75 ммоль) и перемешивали в течение 1 часа при комнатной температуре. Смесь фильтровали, фильтрационный кек промывали тетрагидрофураном (1 мл×3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-5-(3-{N'-[1-(3,3-диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-илиден]-гидразино}-2-гидроксифенил)-фуран-2-карбоновой кислоты бис-(этаноламин) 4 (2,5 г, выход: 86,4%) в виде черного твердого вещества.
MC m/z (ИЭР): 470,8 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.57 (m, 4Н), 7.19 (m, 1Н), 7.03 (d, J=3.6 Гц, 1Н), 6.95 (d, J=3.6 Гц, 1Н), 6.71 (t, J=8.0 Гц, 1Н), 3.73 (t, J=5.2 Гц, 4Н), 2.98 (m, 4Н), 2.88 (t, J=7.2 Гц, 2H), 2.36 (s, 3H), 1.96 (t, J=7.2 Гц, 2Н), 1.29 (s, 6H)
Пример 5
(Z)-5-(3-{N'-[1-(3,3-Диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-илиден1-гидразино}-2-гидроксифенил)-фуран-2-карбоновой кислоты бис-(диэтиламин)

(Z)-5-(3-{N'-[1-(3,3-Диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-или ден]-гидразино}-2-гидроксифенил)-фуран-2-карбоновую кислоту 4h (150 мг, 0,32 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли диэтиламин (46 мг, 0,63 ммоль) с образованием пурпурного раствора и перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, полученный в результате остаток очищали колоночной хроматографией на силикагеле (гексан: этилацетат=10:1) и твердое вещество высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-5-(3-{N'-[1-(3,3-диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-илиден]-гидразино}-2-гидроксифенил)-фуран-2-карбоновой кислоты бис-(диэтиламин) 5 (170 мг, выход: 86,7%) в виде темно-красного твердого вещества.
ВЭЖХ: 94,6%
МС m/z (ИЭР): 471,9 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.60 (m, 4Н), 7.19 (m, 1Н), 7.04 (m, 1Н), 6.87 (m, 2H), 2.98 (q, J=7.2 Гц, 8Н), 2.89 (t, J=7.2 Гц, 2H), 2.36 (s, 3H), 1.96 (t, J=7.2 Гц, 2H), 1.27 (t, J=7.2 Гц, 12Н), 1.25 (s, 6Н)
Пример 6
(Z)-5-(3-[N'-[1-(3,3-Диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-илиден1-гидразино}-2-гидроксифенил)-фуран-2-карбоновой кислоты бис-(пиперазин)

(Z)-5-(3-{N'-[1-(3,3-Диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-или ден]-гидразино}-2-гидроксифенил)-фуран-2-карбоновую кислоту 4h (150 мг, 0,32 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли пиперазин (55 мг, 0,64 ммоль) с образованием пурпурного раствора и перемешивали при комнатной температуре в течение ночи. Смесь фильтровали, фильтрационный кек промывали тетрагидрофураном (1 мл×3) и твердое вещество высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-5-(3-{N'-[1-(3,3-диметилиндан-5-ил)-3-метил-5-оксо-1,5-дигидропиразол-4-илиден]-гидразино}-2-гидроксифенил)-фуран-2-карбоновой кислоты бис-(пиперазин) 6 (158 мг, выход: 77,1%) в виде красного твердого вещества.
ВЭЖХ: 99,28%
MC m/z (ИЭР): 471,8 [M-1]
1H ЯМР (400 МГц, CD3OD): δ 7.64-7.66 (m, 3Н), 7.55 (d, J=8.0 Гц, 1Н), 7.21 (d, J=8.0 Гц, 1Н), 7.04 (m, 1Н), 6.87-6.88 (m, 2Н), 3.01 (s, 16H), 2.90 (t, J=7.2 Гц, 2Н), 2.38 (s, 3H), 1.97 (t, J=7.2 Гц, 2Н), 1.29 (s, 6H)
Пример 7
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(этаноламин)

Стадия 1
(5,6,7,8-Тетрагидронафталин-2-ил)-гидразин
5,6,7,8-Тетрагидронафталин-2-иламин 7а (3,68 г, 25,0 ммоль) растворяли в 20 мл концентрированной соляной кислоты и смесь перемешивали в течение 10 минут при охлаждении в бане лед-вода. Добавляли по каплям 10 мл раствора нитрита натрия (1,72 г, 25,0 ммоль), смесь перемешивали еще в течение 15 минут и использовали в следующей реакции.
При охлаждении в бане лед-соль дигидрат хлорида олова (22,6 г, 100 ммоль) растворяли в 10 мл концентрированной соляной кислоты с последующим добавлением вышеупомянутой сохраненной смеси. Реакционную смесь нагревали до комнатной температуры и подвергали взаимодействию в течение 1,5 часов. Затем смесь доводили до pH 9 40% раствором гидроксида натрия. Смесь экстрагировали 400 мл этилацетата, затем объединенные органические экстракты концентрировали при пониженном давлении и высушивали с получением соединения, указанного в заголовке, (5,6,7,8-тетрагидронафталин-2-ил)-гидразина 7b (2,19 г, выход 53,7%) в виде желтого масла.
МС m/z (ИЭР): 163[М+1]
Стадия 2
5-Метил-2-(5,6,7,8-тетрагидронафталин-2-ил)-2,4-дигидропиразол-3-он
(5,6,7,8-Тетрагидронафталин-2-ил)-гидразин 7b (2,0 г, 12,3 ммоль) растворяли в 50 мл уксусной кислоты с последующим добавлением этилацетоацетата (1,57 мл, 12,3 ммоль). Реакционную смесь нагревали до 100°C в течение ночи. Смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 5-метил-2-(5,6,7,8-тетрагидронафталин-2-ил)-2,4-дигидропиразол-3-она 7с (1,58 г, выход 56,2%) в виде бесцветного масла.
МС m/z (ИЭР): 457 [2М+1]
1H ЯМР (CDCl3): δ 7.54-7.58 (m, 2H), 7.09 (d, J=8 Гц, 1Н), 3.43 (s, 2H), 2.77-2.81 (m, 4H), 2.21 (s, 3H), 1.80-1.83 (m, 4H).
Стадия 3
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновая кислота
5-(3-Амино-2-гидроксифенил)-фуран-2-карбоновой кислоты гидробромид 4g (292 мг, 0,98 ммоль) растворяли в 3,3 мл 1 М соляной кислоты при охлаждении в бане лед-вода с последующим добавлением по каплям 1,3 мл раствора нитрита натрия (74 мг, 1,07 ммоль). После того как смесь перемешивали в течение 20 минут, добавляли 5-метил-2-(5,6,7,8-тетрагидронафталин-2-ил)-2,4-дигидропиразол-3-он 7с (200 мг, 0,88 ммоль). Смесь доводили до pH 8~9 путем добавления порциями раствора бикарбоната натрия (1,226 г, 14,6 ммоль). Образующиеся пузырьки гасили 2 мл этанола. Реакционную смесь нагревали до комнатной температуры и подвергали взаимодействию в течение ночи. Смесь фильтровали и фильтрационный кек растворяли в 20 мл воды. После хорошего перемешивания смесь доводили до pH 3-4 концентрированной соляной кислотой, фильтровали и высушивали. Сырой продукт очищали ВЭЖХ с получением соединения, указанного в заголовке, (Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты 7d (160 мг, выход 39,8%) в виде красного твердого вещества.
МС m/z (ИЭР): 457 [М-1]
1H ЯМР (400 МГц, ДМСО-d6): δ 7.71 (d, J=8.4 Гц, 1 Н), 7.63 (m, 2Н), 7.56 (d, J=7.6 Гц, 1H), 7.37 (d, J=3.2 Гц, 1H), 7.22 (t, J=8.0 Гц, 1H), 7.13 (m, 2Н), 2.75 (m, 4Н), 2.33 (s, 3Н), 1.76 (m, 4Н)
Стадия 4
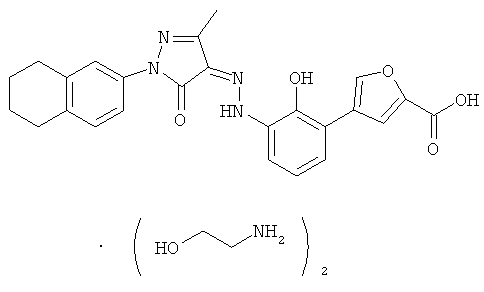
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(этаноламин)
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновую кислоту 7d (3,3 г, 7,2 ммоль) растворяли в 15 мл тетрагидрофурана. К реакционному раствору медленно добавляли по каплям этаноламин (0,88 г, 13 ммоль) и перемешивали в течение 1,5 часов при 15~20°C. Из раствора осаждалось большое количество твердого вещества, его фильтровали, затем фильтрационный кек промывали тетрагидрофураном (10 млх3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(этаноламин) 7 (3 г, выход: 74%) в виде темно-красного твердого вещества.
ВЭЖХ: 99,3%
МС m/z (ИЭР): 456,8 [М-1]
1H ЯМР (400 МГц, CH3OD): δ 7.51 (d, J=8.0 Гц, 1H), 7.44-7.46 (m, 3Н), 6.93-6.98 (m, 2Н), 6.88 (d, J=3.6 Гц, 1 Н), 6.67 (t, J=8.0 Гц, 1 Н), 3.61 (t, J=5.2 Гц, 4Н), 2.86 (t, J=5.2 Гц, 4Н), 2.65-2.70 (m, 4Н), 2.24 (s, 3Н), 1.70-1.72 (s, 3Н)
Пример 8
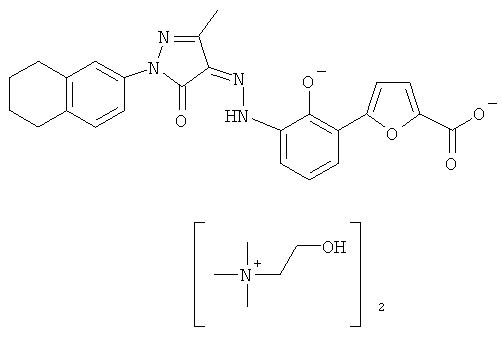
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5.6.7.8-тетрагидронафталин-2-ил)-1.5-дигидропиразол-4-илиден1-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(холин)

(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновую кислоту 7d (100 мг, 0,22 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли 45% раствор гидроксида холина в метаноле (45 мг, 0,44 ммоль) с образованием пурпурного раствора и перемешивали в течение 1 часа при комнатной температуре. Твердое вещество осаждали из раствора, фильтровали, затем фильтрационный кек промывали тетрагидрофураном (1 мл×3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(холин) 8 (140 мг, выход: 96,6%) в виде темно-красного твердого вещества.
ВЭЖХ: 98,82%
МС m/z (ИЭР): 457,8 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.74 (d, J=8.0 Гц, 1Н), 7.60 (m, 3Н), 7.08 (m, 3Н), 6.91 (t, J=8.0 Гц, 1Н), 3.96 (m, 4Н), 3.45 (t, J=4.8 Гц, 4H), 3.18 (s, 18H), 2.80 (m, 4Н), 2.38 (s, 3Н), 1.84 (m, 4Н)
Пример 9
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(диэтиламин)

(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновую кислоту 7d (100 мг, 0,22 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли по каплям диэтиламин (32 мг, 0,44 ммоль) с образованием пурпурного раствора и перемешивали при комнатной температуре в течение ночи. Твердое вещество осаждали из раствора, фильтровали и фильтрационный кек промывали тетрагидрофураном (1 млх3), твердое вещество высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(диэтиламин) 9 (77 мг, выход: 58,3%) в виде темно-красного твердого вещества. ВЭЖХ:99,1% МС m/z (ИЭР): 457,9 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.59 (т, 4Н), 7.08 (d, J=8.0 Гц, 1Н), 7.04 (d, J=3.6 Гц, 1 H), 6.94 (d, J=3.6 Гц, 1 H), 6.82 (t, J=8.0 Гц, 1 H), 2.99 (q, J=7.2 Гц, 8Н), 2.79 (т, 4Н), 2.36 (s, 3Н), 1.82 (t, J=3.2 Гц, 4Н), 1.27 (t, J=7.2 Гц, 12Н)
Пример 10
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(меглумин)

(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновую кислоту 7d (100 мг, 0,22 ммоль) суспендировали в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли меглумин (85 мг, 0,44 ммоль) и перемешивали при комнатной температуре в течение ночи. К полученному в результате раствору добавляли 4 мл метанола и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(меглумин) 10 (168 мг, выход: 90,8%) в виде темно-красного твердого вещества.
ВЭЖХ: 97,7%
MC m/z (ИЭР): 457,8 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.56 (m, 4Н), 7.06 (m, 2Н), 6.98 (d, J=3.2 Гц, 1Н), 6.75 (t, J=7.6 Гц, 1Н), 4.08 (m, 2Н), 3.81 (m, 2Н), 3.77 (m, 2Н), 3.63 (m, 6Н), 3.11 (m, 4Н), 2.76 (m, 4Н), 2.64 (s, 6Н), 2.33 (s, 3Н), 1.79 (m, 4Н)
Пример 11
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден1-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(пиперазин)

(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновую кислоту 7d (100 мг, 0,22 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли пиперазин (37 мг, 0,44 ммоль) с образованием пурпурного раствора и перемешивали в течение 2 часов при комнатной температуре. Твердое вещество осаждали из раствора, фильтровали, затем фильтрационный кек промывали тетрагидрофураном (1 мл×3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(пиперазин) 11 (120 мг, выход: 87,6%) в виде темно-красного твердого вещества.
ВЭЖХ: 98,8%
МС m/z (ИЭР): 457,8 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.59 (m, 4Н), 7.08 (d, J=8.0 Гц, 1Н), 7.04 (d, J=3.2 Гц, 1H), 6.87 (d, J=3.2 Гц, 1Н), 6.82 (t, J=8.0 Гц, 1Н), 3.00 (s, 16H), 2.78 (m, 4Н), 2.36 (s, 3H), 1.81 (m, 4H)
Пример 12
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(трометамол)

(Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновая кислота 7d (100 мг, 0.22 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли трометамол (53 мг, 0,44 ммоль) с образованием коричневого раствора и перемешивали при комнатной температуре в течение ночи. Твердое вещество осаждали из раствора, фильтровали, затем фильтрационный кек промывали тетрагидрофураном (1 мл×3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(трометамол) 12 (142 мг, выход: 92,8%) в виде темного твердого вещества. ВЭЖХ: 94,0% МС m/z (ИЭР): 457,8 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.58 (m, 4Н), 7.05 (m, 2Н), 6.96 (d, J=3.6 Гц, 1Н), 6.90 (t, J=8.0 Гц, 1Н), 3.65 (s, 12H), 2.76 (m, 4Н), 2.33 (s, 3Н), 1.80 (m, 4Н)
Пример 13
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(дибензилэтилендиамин)

(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновую кислоту 7d (100 мг, 0,22 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли дибензилэтилендиамин (104 мг, 0,44 ммоль) с образованием коричневого раствора и перемешивали в течение 2 часов при комнатной температуре. К полученному в результате раствору добавляли 4 мл метанола и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(дибензилэтилендиамин) 13 (167 мг, выход: 81,8%) в виде темного твердого вещества.
ВЭЖХ: 96,8%
МС m/z (ИЭР): 457,8 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.52-7.54 (m, 3Н), 7.39 (d, J=7.2 Гц, 1Н), 7.24-7.28 (m, 20Н), 7.01-7.04 (m, 2Н), 6.65-6.72 (m, 1Н), 3.89 (s, 8H), 3.01 (s, 8H), 2.73 (m, 4Н), 2.32 (s, 3H), 1.78 (m, 4H)
Пример 14
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты динатриевая соль
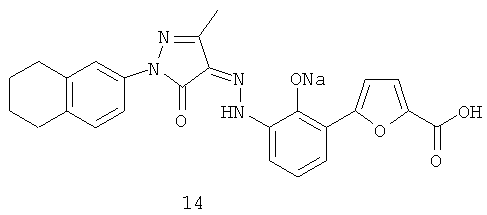
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновую кислоту 7d (110 мг, 0,24 ммоль) растворяли в 4 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли по каплям 1 М раствор гидроксида натрия (0,4 мл, 0,44 ммоль), перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь фильтровали, затем к фильтрату добавляли 4 мл метанола и концентрировали при пониженном давлении. Полученное в результате твердое вещество промывали гексаном с получением соединения, указанного в заголовке, (Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты динатриевой соли 14 (115 мг, выход: 81,8%) в виде темного твердого вещества. ВЭЖХ: 96,8% МС m/z (ИЭР): 457,8 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.79 (dd, J1=7.6 Гц, J2=1.2 Гц, 1Н), 7.52 (m, 3Н), 7.18 (d, J=3.6 Гц, 1Н), 7.05 (m, 2Н), 6.70 (m, 1Н), 2.78 (m, 4Н), 2.41 (s, 3Н), 1.82 (m, 4Н)
Пример 15
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден1-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(L-аргинин)

(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновую кислоту 7d (100 мг, 0,22 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли L-аргинин (76 мг, 0,44 ммоль) и 2 мл воды, перемешивали в течение 2 часов при комнатной температуре. Реакционный раствор концентрировали при пониженном давлении, добавляли 5 мл этилацетата. Твердое вещество осаждали из раствора, фильтровали, затем фильтрационный кек высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(L-аргинин) 15 (168 мг, выход: 95,5%) в виде темного твердого вещества.
ВЭЖХ: 97,5%
MC m/z (ИЭР): 457,9 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.59 (m, 4Н), 7.06 (m, 2Н), 6.98 (d, J=3.6 Гц, 1Н), 6.92 (t, J=8.0 Гц, 1Н), 3.57 (t, J=6.4 Гц, 2Н), 3.19 (m, 4Н), 2.78 (m, 4Н), 2.36 (s, 3Н), 1.83 (m, 8Н), 1.73 (m, 4Н)
Пример 16
(Z)-5-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-фуран-2-карбоновой кислоты бис-(этаноламин)
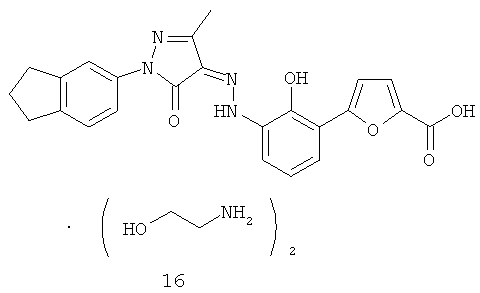

Стадия 1
(Z)-5-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-фуран-2-карбоновая кислота 5-(3-Амино-2-гидроксифенил)-фуран-2-карбоновой кислоты гидробромид 4g (300 мг, 1,0 ммоль) растворяли в соляной кислоте (3,4 мл, 1 М) с последующим добавлением по каплям 1,2 мл раствора нитрита натрия (73 мг, 1,05 ммоль) при охлаждении в бане лед-вода. После того как смесь подвергали взаимодействию в течение 10 минут, последовательно добавляли 2-индан-5-ил-5-метил-2,4-дигидропиразол-3-он 1i (193 мг, 0,9 ммоль), бикарбонат натрия (1,26 г, 15 ммоль) и 4,4 мл этанола. Смесь подвергали взаимодействию при комнатной температуре в течение 24 часов. Смесь фильтровали и фильтрационный кек промывали 20 мл воды, а затем растворяли в 20 мл воды. При охлаждении в бане лед-вода смесь доводили до pH<5 концентрированной соляной кислотой, фильтровали и высушивали с получением соединения, указанного в заголовке, (Z)-5-{2-гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-фуран-2-карбоновой кислоты 16а (287 мг, выход 71,8%) в виде желтого твердого вещества.
MC m/z (ИЭР): 443 [M-1]
1H ЯМР (400 МГц, ДМСО-d6): δ 13.73 (br.s, 1Н), 9.97 (br.s, 1H), 7.78 (s, 1H), 7.70 (m, 2H), 7.57 (m, 1H), 7.36 (d, J=3.6 Гц, 1H), 7.29 (d, J=8.0 Гц, 1H), 7.22 (t, J=8.0 Гц, 1H), 7.15 (m, 1H), 2.89 (m, 4H), 2.32 (s, 3H), 2.03 (m, 2H)
Стадия 2
(Z)-5-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-фуран-2-карбоновой кислоты бис-(этаноламин)
(Z)-5-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-фуран-2-карбоновую кислоту 16а (1,825 г, 4,11 ммоль) растворяли в 20 мл тетрагидрофурана. К реакционной смеси добавляли этаноламин (501 мг, 8,22 ммоль) и перемешивали в течение 2 часов при комнатной температуре. Твердое вещество осаждали из раствора, фильтровали, затем фильтрационный кек промывали тетрагидрофураном (1 млх3) и высушивали в вакууме с получением соединения, указанного в заголовке, (N)-5-{2-гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидр азино]-фенил}-фуран-2-карбоновой кислоты бис-(этаноламин) 16 (1,615 г, выход: 69,4%) в виде темно-красного твердого вещества.
MC m/z (ИЭР): 443 [M-1]
1H ЯМР (400 МГц, CD3OD): δ 7.67 (s, 1H), 7.53 (m, 3Н), 7.21 (d, J=8.0 Гц, 1H), 7.02 (m, 1 Н), 6.97 (d, J=3.2 Гц, 1 Н), 6.70 (m, 1 Н), 3.70 (m, 4H), 2.92 (m, 4H), 2.88 (m, 4H), 2.35 (s, 3H), 2.08 (m, 2H)
Пример 17
(Z)-5-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино1-фенил}-фуран-2-карбоновой кислоты бис-(диэтиламин)

(Z)-5-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-фуран-2-карбоновую кислоту 16а (150 мг, 0,38 ммоль) суспендировали в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли по каплям диэтиламин (49 мг, 0,67 ммоль) с образованием пурпурного раствора и перемешивали в течение 2 часов при комнатной температуре. Твердое вещество осаждали из раствора, фильтровали, затем фильтрационный кек промывали тетрагидрофураном (1 млх3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-5-{2-гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-фуран-2-карбоновой кислоты бис-(диэтиламин) 17 (163 мг, выход: 81,9%) в виде темно-красного твердого вещества.
ВЭЖХ:99,18%
МС m/z (ИЭР): 442,7 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.71 (s, 1Н), 7.60 (m, 3Н), 7.24 (d, J=8.0 Гц, 1Н), 7.04 (d, J=8.0 Гц, 1Н), 6.95 (d, J=8.0 Гц, 1Н), 6.82 (m, 1Н), 3.73 (m, 2Н), 2.95 (m, 8Н), 2.37 (s, 3Н), 2.13 (m, 2Н), 1.87 (m, 2Н), 1.28 (m, 12Н)
Пример 18
(Z)-5-{2-Гидрокси-3[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-фуран-2-карбоновой кислоты бис-(пиперазин)

(Z)-5-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-фуран-2-карбоновую кислоту 16а (150 мг, 0,38 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли пиперазин (58 мг, 0,68 ммоль) с образованием пурпурного раствора и перемешивали в течение 3 часов при комнатной температуре. Твердое вещество осаждали из раствора, фильтровали, затем фильтрационный кек промывали тетрагидрофураном (1 млх3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-5-{2-гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-фуран-2-карбоновой кислоты бис-(пиперазин) 18 (185 мг, выход: 88,9%) в виде темно-красного твердого вещества.
ВЭЖХ: 96,52%
МС m/z (ИЭР): 443,2 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.73 (s, 1Н), 7.61-7.64 (m, 2Н), 7.55 (d, J=8.4 Гц, 1Н), 7.23 (d, J=8.4 Гц, 1Н), 7.05 (d, J=8.8 Гц, 1Н), 6.78-6.90 (m, 2Н), 3.03 (s, 16H), 2.89-2.95 (т, 4Н), 2.35 (s, 3Н), 2.12 (t, J=7.2 Гц, 4Н)
Пример 19
(Z)-4-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразиноъ-фенил}-тиофен-2-карбоновой кислоты бис-(этаноламин)


Стадия 1 4-(3-Нитро-2-метоксифенил)-тиофен-2-карбоновая кислота
2-(2-Метокси-3-нитрофенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан 4d (0,81 г, 2,9 ммоль), 4-бромтиофен-2-карбоновую кислоту (0,3 г, 1,45 ммоль), тетракис(трифенилфосфин)палладий (80 мг, 0,073 ммоль) и карбонат натрия (0,31 г, 2,9 ммоль) растворяли в смеси растворителей 20 мл 1,4-диоксана и 10 мл воды. Реакционную смесь кипятили с обратным холодильником в течение 0,5 часа. Смесь доводили до pH 3 1 н. соляной кислотой и экстрагировали этилацетатом (20 мл×3). Объединенные органические экстракты концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 4-(3-нитро-2-метоксифенил)-тиофен-2-карбоновой кислоты 19а (0,54 г) в виде коричневого масла, которое непосредственно использовали на следующей стадии.
МС m/z (ИЭР): 277,6 [М-1]
Стадия 2 4-(3-Амино-2-метоксифенил)-тиофен-2-карбоновая кислота
4-(3-Нитро-2-метоксифенил)-тиофен-2-карбоновую кислоту 19а (400 мг, 1,45 ммоль) растворяли в 30 мл этилацетата с последующим добавлением 100 мг палладия на углероде (10%) и формиата аммония (360 мг, 5,8 ммоль). Смесь кипятили с обратным холодильником в течение 3 часов. Смесь фильтровали и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 4-(3-амино-2-метоксифенил)-тиофен-2-карбоновой кислоты 19b (410 мг) в виде коричневого масла, которое непосредственно использовали на следующей стадии.
МС m/z (ИЭР): 247,8 [М-1]
Стадия 3
4-(3-Амино-2-гидроксифенил)-тиофен-2-карбоновой кислоты гидробромид
4-(3-Амино-2-метоксифенил)-тиофен-2-карбоновой кислоты гидробромид 19b (360 мг, 1,45 ммоль) растворяли в 5 мл дихлорметана с последующим добавлением по каплям трибромида бора (2,8 мл, 5,6 ммоль). Реакционную смесь подвергали взаимодействию при комнатной температуре в течение 4,5 часов. К реакционной смеси добавляли 5 мл метанола и концентрировали при пониженном давлении. Остаток разбавляли 10 мл этилацетата и перемешивали в течение 0,5 часа. Смесь фильтровали и фильтрационный кек высушивали с получением соединения, указанного в заголовке, 4-(3-амино-2-гидроксифенил)-тиофен-2-карбоновой кислоты гидробромида 19с (80 мг, выход 17,5%) в виде серого твердого вещества.
МС m/z (ИЭР): 236,1 [М+1]
Стадия 4
(Z)-4-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-тиофен-2-карбоновая кислота
4-(3-Амино-2-гидроксифенил)-тиофен-2-карбоновой кислоты гидробромид 19 с (120 мг, 0,38 ммоль) растворяли в 2,7 мл 1 М соляной кислоты при охлаждении в бане лед-вода с последующим добавлением по каплям 0,45 мл раствора нитрита натрия (29 мг, 0,42 ммоль). После того как смесь подвергали взаимодействию в течение 20 минут, добавляли 2-индан-5-ил-5-метил-2,4-дигидропиразол-3-он 1i (73 мг, 0,34 ммоль). Смесь доводили до pH 8 насыщенным раствором бикарбоната натрия с последующим добавлением 2 мл этанола. Реакционную смесь подвергали взаимодействию в течение ночи при комнатной температуре. Смесь фильтровали и фильтрационный кек добавляли к 20 мл воды. Смесь доводили до pH 3-4 концентрированной соляной кислотой и фильтровали. Затем к фильтрационному кеку добавляли 5 мл этилацетата и смесь перемешивали в течение 1 часа. Смесь фильтровали и фильтрационный кек высушивали с получением соединения, указанного в заголовке, (Z)-4-{2-гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-тиофен-2-карбоновой кислоты 19d (45 мг, выход 28,7%) в виде желтого твердого вещества.
МС m/z (ИЭР): 458,8 [М-1]
1H ЯМР (400 МГц, ДМСО-d6): δ 13.79 (br.s, 1Н), 9.68 (br.s, 1H), 8.13 (d, J=1.2 Гц, 1H), 8.05 (d, J=1.6 Гц, 1H), 7.78 (s, 1H), 7.67 (m, 2H), 7.32 (m, 2Н), 7.13 (t, J=8.0 Гц, 1H), 2.87 (m, 4Н), 2.32 (s, 3H), 2.05 (m, 2H)
Стадия 5
(Z)-4-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-тиофен-2-карбоновой кислоты бис-(этаноламин)
(Z)-4-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-тиофен-2-карбоновую кислоту 19d (1,3 г, 2,83 ммоль) растворяли в 40 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли этаноламин (344 мг, 5,65 ммоль) с образованием пурпурного раствора и перемешивали в течение 2 часов при комнатной температуре. Из раствора осаждалось большое количество твердого вещества, его фильтровали, затем фильтрационный кек промывали тетрагидрофураном (1 мл×3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-4-{2-гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-тиофен-2-карбоновой кислоты бис-(этаноламин) 19 (1,513 г, выход: 92,0%) в виде темно-красного твердого вещества.
ВЭЖХ: 98,65%
МС m/z (ИЭР): 458,7 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.94 (s, 1H), 7.88 (s, 1H), 7.68 (s, 1H), 7.55-7.59 (m, 2Н), 7.28-7.30 (m, 1H), 7.22 (d, J=8.4 Гц, 1H), 6.83 (t, J=8.0 Гц, 3Н), 3.65-3.68 (m, 4Н), 2.88-2.92 (m, 8Н), 2.38 (s, 3Н), 2.06-2.14 (m, 2Н)
Пример 20
(Z)-4-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-тиофен-2-карбоновой кислоты бис-(диэтиламин)

(Z)-4-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-тиофен-2-карбоновую кислоту 19d (150 мг, 0,33 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли по каплям диэтиламин (49 мг, 0,66 ммоль) с образованием пурпурного раствора и перемешивали в течение 2 часов при комнатной температуре. Твердое вещество осаждали из раствора, фильтровали, затем фильтрационный кек промывали тетрагидрофураном (1 млх3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-4-{2-гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-тиофен-2-карбоновой кислоты бис-(диэтиламин) 20 (157 мг, в виде темно-красного твердого вещества). Выход: 79,3%.
ВЭЖХ: 98,98%
МС m/z(ИЭР): 458,8 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.81 (s, 1H), 7.73 (s, 1H), 7.68-7.70 (m, 2Н), 7.62 (d, J=8.8 Гц, 1H), 7.22-7.26 (m, 2Н), 7.06 (t, J=8.0 Гц, 1H), 3.03 (q, J=7.2 Гц, 8Н), 2.90-2.97 (т, 4Н), 2.37 (s, 3Н), 2.07-2.15 (m, 2Н), 1.29 (t, J=7.2 Гц, 12Н)
Пример 21
(Z)-4-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1.5-дигидропиразол-4-илиден)-гидразино1-фенил}-тиофен-2-карбоновой кислоты бис-(пиперазин)

(Z)-4-{2-Гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-тиофен-2-карбоновую кислоту 19d (150 мг, 0,33 ммоль) растворяли в 5 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли пиперазин (56 мг, 0,65 ммоль) с образованием пурпурного раствора и перемешивали в течение 2 часов при комнатной температуре. Твердое вещество осаждали из раствора, фильтровали, затем фильтрационный кек промывали тетрагидрофураном (1 млх3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-4-{2-гидрокси-3-[N'-(1-индан-5-ил-3-метил-5-оксо-1,5-дигидропиразол-4-илиден)-гидразино]-фенил}-тиофен-2-карбоновой кислоты бис-(пиперазин) 21 (195 мг, выход: 94,7%) в виде темно-красного твердого вещества.
ВЭЖХ:98,17%
МС m/z(ИЭР): 458,8 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.85 (s, 1Н), 7.75 (s, 1Н), 7.71 (s, 1H), 7.62 (m, 2Н), 7.26 (m, 2Н), 6.95 (t, 1Н), 2.96 (m, 16Н), 2.91 (m, 4Н), 2.37 (s, 3Н), 2.11 (m, 2Н)
Пример 22
(Z)-4-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафтал-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(этаноламин)


Стадия 1
4-Бромфуран-2-карбоновая кислота
Смесь 4,5-дибромфуран-2-карбоновой кислоты 22а (5,5 г, 20,3 ммоль) и 18 мл гидроксида аммония добавляли к 63 мл воды с последующим добавлением порошка цинка (1,46 г, 22,33 ммоль). После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Смесь доводили до pH 3 1 М соляной кислотой с образованием большого количества осадков. Смесь фильтровали и фильтрационный кек промывали н-гексаном (15 млх4) и высушивали с получением соединения, указанного в заголовке, 4-бромфуран-2-карбоновой кислоты 22b (3,2 г, выход 83,1%) в виде белого твердого вещества.
МС m/z (ИЭР): 188,7 [М-1]
Стадия 2
4-(3-Нитро-2-метоксифенил)-фуран-2-карбоновая кислота
2-(2-Метокси-3-нитрофенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан 4d (4 г, 14,34 ммоль), 4-бромфуран-2-карбоновую кислоту 22b (2,18 г, 11,47 ммоль), тетракис(трифенилфосфин)палладий (829 мг, 0,717 ммоль) и карбонат калия (3,96 г, 28,68 ммоль) растворяли в смеси растворителей 80 мл 1,4-диоксана и 30 мл воды. Реакционную смесь кипятили с обратным холодильником в течение 2,5 часов. Смесь доводили до pH 3 1 М соляной кислотой, а затем экстрагировали этилацетатом (80 мл×3). Объединенные органические экстракты концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, 4-(3-нитро-2-метоксифенил)-фуран-2-карбоновой кислоты 22с (3,42 г, выход 90,7%) в виде коричневого масла.
МС m/z (ИЭР): 261,8 [М-1]
Стадия 3 4-(3-Амино-2-метоксифенил)-фуран-2-карбоновая кислота
4-(3-Нитро-2-метоксифенил)-фуран-2-карбоновую кислоту 22с (500 мг, 1,9 ммоль) растворяли в 15 мл этилацетата с последующим добавлением 100 мг палладия на углероде и формиата аммония (429 мг, 7,6 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 3 часов. Смесь фильтровали для удаления палладия на углероде и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 4-(3-амино-2-метоксифенил)-фуран-2-карбоновой кислоты 22d (325 мг, выход 73,4%) в виде желтого масла.
МС m/z (ИЭР): 231,8 [М-1]
Стадия 4
4-(3-Амино-2-гидроксифенил)-фуран-2-карбоновой кислоты гидробромид
4-(3-Амино-2-метоксифенил)-фуран-2-карбоновую кислоту 22d (325 мг, 1,4 ммоль) растворяли в 5 мл дихлорметана с последующим добавлением по каплям трибромида бора (2,8 мл, 5,6 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 4,5 часов. Добавляли 5 мл метанола, а затем смесь концентрировали при пониженном давлении. Остаток разбавляли 10 мл этилацетата и перемешивали в течение 0,5 часа. Смесь фильтровали и фильтрационный кек высушивали с получением соединения, указанного в заголовке, 4-(3-амино-2-гидроксифенил)-фуран-2-карбоновой кислоты гидробромида 22е (174 мг, выход 57,1%) в виде серого твердого вещества.
МС m/z (ИЭР): 217,7 [М-1]
Стадия 5
(Z)-4-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафтал-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновая кислота
4-(3-Амино-2-гидроксифенил)-фуран-2-карбоновую кислоту 22е (170 мг, 0,57 ммоль) растворяли в соляной кислоте (1,9 мл, 1 М) при охлаждении в бане лед-вода с последующим добавлением по каплям 0,7 мл раствора нитрита натрия (43 мг, 0,63 ммоль). После того как смесь подвергали взаимодействию в течение 20 минут, добавляли 5-метил-2-(5,6,7,8-тетрагидронафталин-2-ил)-2,4-дигидропиразол-3-он 7с (116 мг, 0,51 ммоль). Смесь доводили до pH 8-9 насыщенным раствором бикарбоната натрия с последующим добавлением 2 мл этанола. Смесь подвергали взаимодействию при комнатной температуре в течение 24 часов. Смесь фильтровали, а затем к фильтрационному кеку добавляли 15 мл воды. При охлаждении в бане лед-вода смесь доводили до pH 2~3 концентрированной соляной кислотой и фильтровали. Фильтрационный кек промывали этилацетатом и высушивали с получением соединения, указанного в заголовке, (Z)-4-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафтал-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты 22f(13 мг, выход 5,5%) в виде красного твердого вещества.
MC m/z (ИЭР): 456,7 [М-1]
1H ЯМР (400 МГц, ДМСО-d6): δ 13.75 (br. s, 1Н), 13.20 (br. s, 1H), 9.68 (br. s, 1H), 8.37 (s, 1H), 7.62-7.68 (m, 4H), 7.41-7.43 (m, 1H), 7.11-7.15 (m, 2H), 2.67-2.76 (m, 2H), 2.31 (s,3H), 1.75 (m,4H)
Стадия 6
(Z)-4-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафтал-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(этаноламин)
(Z)-4-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафтал-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновую кислоту 22f (1,2 г, 2,6 ммоль) растворяли в 20 мл тетрагидрофурана с образованием темно-красной суспензии. К реакционной смеси добавляли этаноламин (399 мг, 6,5 ммоль) с образованием пурпурного раствора и перемешивали в течение 6 часов при комнатной температуре. Из раствора осаждалось большое количество твердого вещества, его фильтровали, затем фильтрационный кек промывали тетрагидрофураном (1 мл×3) и высушивали в вакууме с получением соединения, указанного в заголовке, (Z)-4-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафтал-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты бис-(этаноламин) 22 (1,51 г, в виде красного твердого вещества), выход 72,8%.
ВЭЖХ:97,16%
МС m/z (ИЭР): 456,7 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 8.20 (s, 1Н), 7.51-7.56 (m, 3Н), 7.31-7.35 (m, 2Н), 7.07 (d, J=9.2 Гц, 1Н), 6.89 (t, J=8.0 Гц, 1Н), 3.68-3.71 (m, 4Н), 2.90-2.95 (m, 4Н), 2.76-2.81 (m, 4Н), 2.39 (s, 3Н), 1.80-1.85 (m, 4Н)
Пример 23
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты холин
(Z)-5-(2-Гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновую кислоту (1,1 г, 2,4 ммоль) растворяли в 19 мл смеси растворителей этилацетата и этанола (об/об=12:7), затем реакционную смесь нагревали до 40°C и перемешивали в течение 15 минут с образованием суспензии каштанового цвета. К реакционной смеси медленно добавляли 1 М раствор холина в метаноле (2,4 мл, 2,4 ммоль) с образованием черного раствора до тех пор, пока твердое вещество не исчезло. К реакционному раствору добавляли 1 мл воды, затем охлаждали до 35°C и подвергали взаимодействию в течение 3 часов, затем перемешивали еще в течение 72 часов при комнатной температуре. Оранжевое твердое вещество осаждали, фильтровали, затем фильтрационный кек промывали этилацетатом (5 мл×3) и высушивали с получением соединения, указанного в заголовке, (Z)-5-(2-гидрокси-3-{N'-[3-метил-5-оксо-1-(5,6,7,8-тетрагидронафталин-2-ил)-1,5-дигидропиразол-4-илиден]-гидразино}-фенил)-фуран-2-карбоновой кислоты холина (620 мг, выход: 46,0%) в виде оранжевого твердого вещества.
МС m/z (ИЭР): 456,7 [М-1]
1H ЯМР (400 МГц, CD3OD): δ 7.66-7.57 (m, 4Н), 7.09-7.04 (m, 3Н), 6.92 (d, J=3.6 Hz, 1Н), 4.03-3.99 (m, 2H), 3.51-3.48 (m, 2H), 3.22 (s, 9H), 2.81-2.75 (m, 4H), 2.33 (s, 3H), 1.83-1.81 (m, 4H)
Пример 24
Композиция в виде таблетки
Лактозу, микрокристаллическую целлюлозу, натриевую соль гликолята крахмала, стеарат магния и соединение Примера 7 смешивают в соотношениях, показанных в приведенной ниже таблице 1. Затем смесь прессуют в таблетки.
Пример 25
Инъекционная парентеральная композиция
Инъекционную форму введения соединения Примера 7 получают путем перемешивания 5,0 мг соединения в 1,0 мл нормального физиологического раствора.
Тестовый пример
Анализ растворимости
В соответствии с общепринятым способом определения растворимости тестировали растворимость соединений Примеров и их солей в трех различных системах: вода, 0,1% соляная кислота и метанол.
Растворимость характеризовали, как описано ниже:
Термин "очень хорошо растворимый" означает, что 1 г (мл) растворенного вещества можно растворить менее чем в 1 мл растворителя;
Термин "свободно растворимый" означает, что 1 г (мл) растворенного вещества можно растворить в количестве растворителя от 1 мл до 10 мл, не включая 10 мл растворителя;
Термин "растворимый" означает, что 1 г (мл) растворенного вещества можно растворить в количестве растворителя от 10 мл до 30 мл, не включая 30 мл растворителя;
Термин "умеренно растворимый" означает, что 1 г (мл) растворенного вещества можно растворить в количестве растворителя от 30 мл до 100 мл, не включая 100 мл растворителя;
Термин "малорастворимый" означает, что 1 г (мл) растворенного вещества можно растворить в количестве растворителя от 100 мл до 1000 мл, не включая 1000 мл растворителя;
Термин "очень малорастворимый" означает, что 1 г (мл) растворенного вещества можно растворить в количестве растворителя от 1000 мл до 10000 мл, не включая 10000 мл растворителя;
Термин "практически нерастворимый или нерастворимый" означает, что 1 г (мл) растворенного вещества невозможно растворить полностью в 10000 мл растворителя.
Результаты растворимости представлены ниже:
Этот результат показал, что по сравнению с соединением Примера 1j, Примера 4h, Примера 7d, Примера 16a и Примера 19d растворимость их солей заметно повышена, особенно в воде и в метаноле. Конкретно, соли Примера 4 и Примера 7 обладали относительно лучшей растворимостью в 0,1% соляной кислоте, и две соли были очень малорастворимыми, тогда как другие были практически нерастворимыми или нерастворимыми.
Структура соединений Примера 1j, Примера 4h, Примера 7d, Примера 16a и Примера 19d показана ниже:




Анализ гигроскопичности
Тест на гигроскопичность соединений по настоящему изобретению после стояния в течение 48 часов Протокол:
1. Сухой стеклянный сосуд для взвешивания с пробкой (наружный диаметр 50 мм, высота 15 мм) соответствующим образом помещали при 25°C±1°C в термостатированную сушилку (насыщенный раствор сульфата аммония заливали в нижнюю часть, влажность составляла 79%) за сутки до анализа, точную массу измеряли как (m1);
2. Вышеописанный сосуд для взвешивания покрывали соединениями по изобретению (примерно 1 г), и толщина соединений обычно составляла 1 мм, точную массу измеряли как (m2);
3. Горлышко сосуда для взвешивания держали открытым и помещали с пробкой в вышеописанные условия при постоянной температуре и влажности на 48 часов;
4. Помещали пробку обратно на сосуд, точную массу измеряли как (m3)

Степень гигроскопичности определяли, как описано ниже:
Растворение: превращение в жидкость за счет поглощения достаточного количества влаги.
Высокая гигроскопичность: процент увеличения массы за счет гигроскопичности составляет не менее 15%.
Гигроскопичность: процент увеличения массы за счет гигроскопичности составляет менее 15%, но не менее 2%.
Умеренная гигроскопичность: процент увеличения массы за счет гигроскопичности составляет менее 2%, но не менее 0,2%.
Нет гигроскопичности или почти нет гигроскопичности: процент увеличения массы за счет гигроскопичности составляет менее 0,2%.
Результаты гигроскопичности соединений по настоящему изобретению представлены ниже:
Эти результаты показали, что по сравнению с другими солями соль Примера 7, то есть соль бис-(этаноламин) соединения 7d, менее гигроскопична и обладает лучшей стабильностью во влажной среде, что может позволить избежать потенциальных проблем изменения массы активных ингредиентов во время приготовления капсул или таблеток, устойчива к газу и пригодна для получения общепринятого препарата, и может храниться в течение длительного срока.
БИОЛОГИЧЕСКИЙ АНАЛИЗ
Тестовый Пример 1: Эффект пролиферации серии соединений ТПО на клетках BAF3-TPOR
1. Материал и реагенты
а) Среда RPM11640, порошок, 10*1 л, содержащая HEPES (Gibco, №по каталогу 23400021).
b) Фетальная бычья сыворотка (Gibco, №по каталогу 10099-141).
c) РАСТВОР ПЕНИЦИЛЛИНА И СТРЕПТОМИЦИНА (Gibco, № по каталогу 15140-122).
d) Генетицин (G418) (Gibco, № по каталогу 11811-098).
e) Рекомбинантный IL-3 мыши (Chemicon, № по каталогу IL015).
f) R Mab к тромбопоэтину (ТПО) человека (R&D, № по каталогу МАВ1016).
g) ДМСО, (AppliChem, № по каталогу А3672).
h) Набор для множественного сайт-направленного мутагенеза QuikChange®, 10 реакций (Stratagene ST200515).
i) Набор-8 для подсчета клеток (Dojindo, № по каталогу СК04-13)
j) Клетки BaF3 (Объединенный центр клеточных культур, № по каталогу 0095)
k) EX-EGFP-M02 (FulenGen, № по каталогу EX- EGFP-M02 Control)
l) EX-B0010-M02 (FulenGen, № по каталогу ЕХ-В0010-М02)
2. Процесс обработки:
(1) Плазмидные конструкции: на основе информации последовательности рецептора ТПО (TPOR) от Entrez (Gene ID: 4325, Refseq: NM_005373) двойную точечную мутацию осуществляли в плазмиде EX-B0010-M02 путем использования набора для множественного сайт-направленного мутагенеза QuikChange® (Stratagene). Были разработаны последовательности праймеров, содержащих множественную точечную мутацию:
g491a: 5'-gggaacttcagatcagctgggaggagccg-3'
g491a_antisense: 5'-cggctcctcccagctgatctgaagttccc-3';
c965t: 5'-caggaccatgctagctcccaaggcttcttct-3',
c965t_antisense: 5'-agaagaagccttgggagctagcatggtcctg-3'.
Компетентные клетки E.coli DH5α трансформировали мутированной плазмидой и положительные колонии собирали посредством селекции на ампициллине. Результат мутации был подтвержден анализом последовательности.
(2) Стабильная трансфицированная клеточная линия BAF3-TPOR:
нижеописанный способ использовали для конструирования клеток BaF3, которые стабильно гиперэкспрессировали TPOR человека. Успешно мутированную плазмиду EX-B0010-M02 (25 мкг), которая экспрессировала TPOR человека и ген неомицина для скрининга, трансфицировали в клетки BaF3 дикого типа (1×107) путем электропорации при 250 B в течение 18 мс, используя электроимпульсатор (Electro Square Porator ЕСМ830, ВТХ Division of Genetronic, Inc. US). Стабильно трансфицированные клетки BAF3-TPOR отбирали G418 (Gibco, US), затем инкубировали в среде RPMI1640 с добавлением 10% ФБС (Gibco, US), 800 нг/мл G418, 5 нг/мл rmlL-3 (Chemicon, US).
3. Скрининг соединений
(1) Промывание клеток центрифугированием: подходящее количество клеточной суспензии центрифугировали при 1000 об/мин в течение 5 минут и супернатант отбрасывали. Добавляли 10 мл среды для клеточной культуры без IL-3. Затем полученную в результате суспензию клеток центрифугировали при 1000 об/мин в течение 5 минут и супернатант отбрасывали.
(2) Добавляли 1 мл среды для клеточной культуры без IL-3, чтобы разбить клетки до однородности, и после разведения считали число клеток в подходящем количестве суспензии клеток.
(3) В соответствии с результатом подсчета клеток готовили суспензию клеток при концентрации 100000 клеток/мл.
(4) 100 мкл суспензии клеток переносили в каждую лунку 96-луночного культурального планшета и ставили 3 параллельные лунки, то есть пустую контрольную группу (В), отрицательную контрольную группу (N), положительную контрольную группу ТПО (Р) и группу тестируемого соединения (S).
(5) Тестируемое соединение растворяли в ДМСО с получением 10 мМ исходного раствора, а затем этот раствор разводили средой RPMI 1640 с получением серии тестируемых образцов при различной концентрации: 30 мкМ, 10 мкМ, 3 мкМ, 1 мкМ, 0,3 мкМ, 0,1 мкМ, 0,03 мкМ, 0,01 мкМ, 0,003 мкМ, 0,001 мкМ.
(6) 10 мкл раствора тестируемого соединения переносили в каждую лунку соответственно; 1 мкл гПТРО (10 мкг/мл) добавляли в положительную контрольную лунку.
(7) Планшеты инкубировали в инкубаторе при 5% CO2 и 37°C в течение 24 часов.
(8) После инкубации в каждую лунку добавляли 10 мкл раствора ССК-8 и планшеты инкубировали в инкубаторе еще в течение 24 часов.
(9) Определяли значение OD при 450 нм с помощью считывающего устройства для планшетов VICTOR3 (Perkin Elmer 1420-120).
4. Аналитические вычисления
(1) Скорость пролиферации вычисляли, как описано ниже:
Скорость = [(S-B)/ (Р-В)] * 100%
S: Значение OD лунок, которые содержат тестируемое соединение.
В: Значение OD пустых контрольных лунок
Р: Значение OD положительных контрольных лунок
(2) Значение ЕС50 вычисляли с помощью программного обеспечения Origin 7.0. 5. Результаты:
ЕС50 активности ТПО соединений по настоящему изобретению
Результаты этого исследования показали, что по сравнению со свободной кислотой соли по настоящему изобретению обладают более сильными пропролиферативными эффектами на клетки BAF3-TPOR. Порядок был следующим: соли по настоящему изобретению > свободная кислота > Элтромбопаг, и активность солей по настоящему изобретению была большей, чем активность Элтромбопага.
ФАРМАКОКИНЕТИЧЕСКИЙ АНАЛИЗ
Тестовый Пример 1: Фармакокинетический анализ соединений по настоящему изобретению у крыс
1.Цель
Соединения по настоящему изобретению вводили внутрижелудочно крысам, чтобы определить концентрацию лекарства в плазме в различные моменты времени с помощью ВЭЖХ-УФ. Фармакокинетические свойства соединений по настоящему изобретению исследовали и оценивали на крысах.
2. Протокол
2.1. Образцы
Соединения Примера 1j, Примера 1-3, Примера 4h, Примера 4, Примера 5, Примера 6, Примера 7d, Примера 7-15, Примера 16a, Примера 16-18, Примера 19d, Примера 19, Примера 20, Примера 21, Примера 22f, Примера 22 и Примера 23.
2.2. Подопытные животные
Здоровых взрослых крыс SD, половину самцов и половину самок, покупали у S1NO-BRITSH SIPPR/BK LAB.ANIMAL LTD., CO, номер лицензии: SCXK (Shanghai) 2003-0002.
2.3. Прибор
Высокоэффективный жидкостный хроматограф Waters 2695-2996, Waters Corp., USA.
2.4. Получение тестируемых соединений
Тестируемое соединение разводили 1% натриевой солью карбоксиметилцеллюлозы до 5 мг/мл (в перерасчете на свободную кислоту) суспензии перед использованием.
2.5. Введение
Здоровых взрослых крыс SD, половину самцов и половину самок, делили на 23 группы. После голодания в течение ночи крысам вводили соединение внутрижелудочно в дозе 50,0 мг/кг (в перерасчете на свободную кислоту) в объеме 10 мл/кг.
2.6. Отбор образцов
Образцы крови (0,2 мл) брали из глазницы перед введением и через 0,5,1,0, 2,0, 3,0, 4,0, 5,0, 6,0, 8,0, 11,0, 14,0, 24,0, 36,0 и 48,0 часов после введения, хранили их в гепаринизированных пробирках и центрифугировали в течение 10 минут при 3500 об/мин. Образцы плазмы хранили при -20°C до анализа. Крыс кормили через 2 часа после введения.
2.7. Аналитические методы
50 мкл плазмы крыс, полученной в различные моменты времени после введения, 50 мкл раствора внутреннего стандарта и 20 мкл смеси растворителей метанола и воды (80:20, об/об) хорошо смешивали, а затем добавляли 100 мкл метанола, что привело в результате к осаждению белка. Затем смесь перемешивали в течение 3 минут, используя вортекс, и центрифугировали в течение 10 минут при 13500 об/мин. 40 мкл супернатанта анализировали с помощью ВЭЖХ-УФ.
2.8. Вычисление фармакокинетических параметров
Камерную модель фармакокинетики приводили в соответствие с тестируемыми соединениями и вычисляли основные фармакокинетические параметры, в которых Cmax и tmax были действительно измеренными значениями.
3. Результаты фармакокинетических параметров
Фармакокинетические параметры соединений по настоящему изобретению представлены ниже:
Результаты исследования показали, что после введения крысам по сравнению со свободной кислотой фармакокинетика и биодоступность солей по настоящему изобретению заметно улучшена. Фармакокинетические данные соли Примера 7, то есть соли бис-(моноэтаноламин) Примера 7d, являются лучшими, и она обладает хорошими фармакокинетическими свойствами.
Тестовый Пример 2: Фармакокинетический анализ соединений по настоящему изобретению у собак породы бигль
1. Цель
Соединения Примера 7, Примера 8, Примера 10 и Примера 12 вводили внутрижелудочно собакам породы бигль для определения концентрации лекарства в плазме в различные моменты времени с помощью ВЭЖХ-УФ. Фармакокинетические свойства соединений по настоящему изобретению исследовали и оценивали на собаках породы бигль.
2. Протокол
2.1. Образцы
Соединения Примера 7, Примера 8, Примера 10 и Примера 12.
2.2. Подопытные животные
12 здоровых взрослых собак породы бигль, самцов, покупали у Suzhou Xishan Drug Research and Development CO., LTD. Номер лицензии: SCXK(Suzhou)2007-0005.
2.3. Прибор
Высокоэффективный жидкостный хроматограф Agilent 1100, Agilent Corp., USA.
2.4. Получение тестируемых соединений
Тестируемое соединение разводили 0,5% натриевой солью карбоксиметилцеллюлозы до 2,5 мг/мл (в перерасчете на свободную кислоту) суспензии перед использованием.
2.5. Введение
12 здоровых взрослых собак породы бигль, самцов, делили на 4 группы. После голодания в течение ночи собакам вводили соединения внутрижелудочно в дозе 5,0 мг/кг (в перерасчете на свободную кислоту) в объеме 2 мл/кг.
2.6. Отбор образцов
Образцы крови (1,2 мл) брали из вены передних конечностей перед введением и через 0,25, 0,5, 1,0, 1,5, 2,0, 3,0, 5,0, 6,0, 8,0, 12,0, 14,0, 24,0 и 48,0 часов после введения, хранили их в гепаринизированных пробирках и центрифугировали в течение 10 минут при 3500 об/мин. Образцы плазмы хранили при -20°C до анализа. Собак бигль кормили через 2 часа после введения.
2.7. Аналитические методы
50 мкл плазмы собак бигль, полученных в различные моменты времени после введения, 20 мкл раствора внутреннего стандарта и 150 мкл метанола добавляли, чтобы привести в результате к осаждению белков. Затем смесь перемешивали в течение 1 минуты, используя вортекс, и центрифугировали в течение 5 минут при 11000 об/мин. 50 мкл супернатанта анализировали с помощью ВЭЖХ-УФ.
2.8. Вычисление фармакокинетических параметров
Камерную модель фармакокинетики приводили в соответствие с тестируемыми соединениями и вычисляли основные фармакокинетические параметры, в которых Cmax и tmax были действительно измеренными значениями.

Данные четырех солей в настоящем исследовании показали, что соединение Примера 7 было заметно лучшим в отношении фармакокинетики.
В заключение получение соединений по настоящему изобретению было простым и давало хороший выход. Конкретно, соли этаноламина, соли холина, соли диэтиламина и соли пиперазина обладали преимуществом в способе синтеза, поскольку их можно кристаллизовать непосредственно. По сравнению со свободными кислотами растворимость солей по настоящему изобретению заметно повышена в общепринятых растворителях. Соли этаноламина были менее гигроскопичными и были пригодны для получения общепринятого препарата, а также легко хранились. Биологическая активность солей по настоящему изобретению заметно улучшена. Фармакокинетика также заметно улучшена у крыс и собак бигль, и эти соли обладали лучшими фармакокинетическими свойствами, особенно соли этаноламина.
Изобретение относится к фармацевтически приемлемым солям, выбранным из группы, состоящей из соли натрия, соли лития, соли калия, соли кальция, соли магния, соли аргинина, соли лизина, соли метанамина, соли диметиламина, соли триметиламина, соли этиламина, соли диэтиламина, соли триэтиламина, соли этаноламина, соли пиперазина, соли дибензилэтилендиамина, соли меглумина, соли трометамина, соли четвертичного тетраметиламмония, соли четвертичного тетраэтиламмония и соли холина, бициклозамещенных производных азопиразолона общей формулы (I). Изобретение также относится к способу их получения, фармацевтической композиции, содержащей их, и их применению в качестве терапевтического агента, в частности в качестве миметиков тромбопоэтина (ТПО), и их применению в качестве агонистов ТПО. В общей формуле (I) Het выбран из группы, состоящей из фенила, фурила и тиенила; R1, R2, R3 и R4 каждый независимо выбран из группы, состоящей из водорода и алкила, n равно 0, 1 или 2. Технический результат - улучшение фармокинетических свойств соединения формулы (I) благодаря повышению растворимости. 6 н. и 13 з.п. ф-лы, 1 табл., 25 пр.
1. Фармацевтически приемлемые соли соединения формулы (I):

где:
Het выбран из группы, состоящей из фенила, фурила и тиенила;
R1, R2, R3 и R4 каждый независимо выбран из группы, состоящей из водорода и алкила;
n равно 0, 1 или 2;
где соли выбраны из группы, состоящей из соли натрия, соли лития, соли калия, соли кальция, соли магния, соли аргинина, соли лизина, соли метанамина, соли диметиламина, соли триметиламина, соли этиламина, соли диэтиламина, соли триэтиламина, соли этаноламина, соли пиперазина, соли дибензилэтилендиамина, соли меглумина, соли трометамина, соли четвертичного тетраметиламмония, соли четвертичного тетраэтиламмония и соли холина.
2. Фармацевтически приемлемые соли по п.1, где соли выбраны из группы, состоящей из соли диэтиламина, соли этаноламина, соли холина, соли пиперазина, соли меглумина и соли трометамина.
3. Фармацевтически приемлемые соли по п. 1, где соли выбраны из группы, состоящей из соли этаноламина, соли холина, соли меглумина и соли трометамина.
4. Фармацевтически приемлемые соли по п.1, где соль представляет собой соль этаноламина.
5. Фармацевтически приемлемые соли по п.1, где соли выбраны из группы, состоящей из:


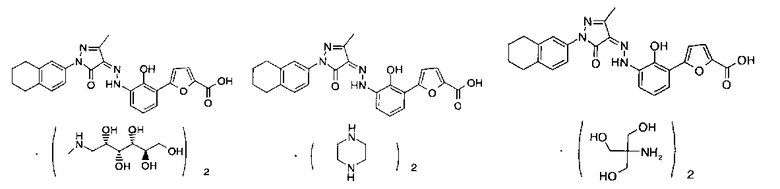
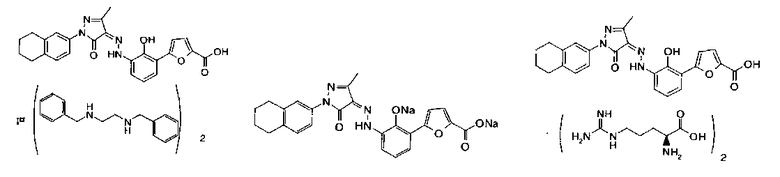


 .
.
6. Способ получения фармацевтически приемлемых солей по любому из пп.1-5, включающий следующие стадии:
(a) растворение или суспендирование соединения формулы (I) в органическом растворителе, где органический растворитель выбран из группы, состоящей из метанола, этанола, ацетона, этилацетата и тетрагидрофурана;
(b) добавление к полученной смеси основания при перемешивании;
(c) получение фармацевтически приемлемых солей соединения формулы (I),
где основание выбрано из группы, состоящей из гидроксида натрия, гидроксида лития, гидроксида калия, гидроксида кальция, гидроксида магния, лизина, аргинина, метанамина, диметиламина, триметиламина, этиламина, диэтиламина, триэтиламина, этаноламина, пиперазина, дибензилэтилендиамина, меглумина, трометамина, четвертичного тетраметиламмония, четвертичного тетраэтиламмония и гидроксида холина.
7. Способ по п.6, где указанный органический растворитель представляет собой тетрагидрофуран.
8. Способ по п.6, где основание выбрано из группы, состоящей из диэтиламина, этаноламина, пиперазина, меглумина, трометамина и гидроксида холина.
9. Способ по п.6, где основание выбрано из группы, состоящей из этаноламина, меглумина, трометамина и гидроксида холина.
10. Способ по п.6, где основание представляет собой этаноламин.
11. Способ по п.6, где соотношение эквивалентов соединения формулы (I) и основания составляет 1:5~5:1.
12. Способ по п.6, где соотношение эквивалентов соединения формулы (I) и основания составляет 1:1~1:3.
13. Способ по п.6, где соотношение эквивалентов соединения формулы (I) и основания составляет 1:1~1:2.
14. Фармацевтическая композиция для лечения тромбоцитопении, содержащая терапевтически эффективное количество фармацевтически приемлемых солей по п.1 и фармацевтически приемлемые носители или разбавители.
15. Способ получения композиции по п.14, включающий стадию объединения соединения по п.1 с фармацевтически приемлемыми носителями или разбавителями.
16. Применение фармацевтически приемлемых солей соединения формулы (I) по п.1 для получения агониста тромбопоэтинового рецептора.
17. Применение фармацевтически приемлемых солей соединения формулы (I) по п.1 для получения лекарственного средства для лечения тромбоцитопении.
18. Применение по п.17, где лекарственное средство находится в форме пероральной лекарственной формы.
19. Применение по п.17, где лекарственное средство находится в форме парентеральной лекарственной формы.
| БИЦИКЛОЗАМЕЩЕННЫЕ АЗОПРОИЗВОДНЫЕ ПИРАЗОЛОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2009 |
|
RU2488582C2 |
| БИС-(МОНОЭТАНОЛАМИН) 3`-[(2Z)-[1-(3,4-ДИМЕТИЛФЕНИЛ)-1,5-ДИГИДРО-3-МЕТИЛ-5-ОКСО-4H-ПИРАЗОЛ-4-ИЛИДЕН] ГИДРАЗИНО]-2`-ГИДРОКСИ-[1,1`-БИФЕНИЛ]-3-КАРБОНОВОЙ КИСЛОТЫ | 2003 |
|
RU2284994C2 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

