Изобретение относится к области синтетической органической химии, точнее к получению (3-гидроксипропил)-нафтолов, структурно относящихся к отдельному классу фенилпропаноидов - эффективных биологически активных веществ нейротропного действия [В.А Куркин, А.В. Дубрищев, И.Н. Титова, А.В. Волоцуева, Е.С. Петрова, Н.В. Жесткова, И.Ю. Климова. // Растительные ресурсы, 2003, вып. 3. C. 115-121]. 1-(3-Гидроксипропил)-2-нафтол(1) и 2-(3-гидроксипропил)-1-нафтол (2) предложены для получения ионных жидкостей [Dong Jin Hong, Dong Wook Kim // Tetrahedron Letters 2010, 51, 54-56]. На основе соединения (2) синтезирован ряд биологически активных продуктов, обладающих антиканцерогенным действием [Ngamhong Kongkathip, Suwaporn Luangkamin, Chak Sangma at all. // J. Med. Chem. 2004, 47, 4427-4438].
Все известные способы получения гидроксипропилнафтолов многостадийны. Схема получения 1-(3-гидроксипропил)-2-нафтола(1) из 2-нафтола с общим выходом 30% представлена ниже:

Эфир (3), образующийся при кипячении 2-нафтола с аллилбромидом в присутствии K2CO3, перегруппировывают в 1-(аллил)-2-нафтол (4). Gladfeld J.W.E., Rietz E.J.J. Am. Chem. Soc. 1940. В. 62. 974. На основе соединения (4) получают боран-тетрагидрофурановый комплекс, который окисляют перекисью водорода в щелочной среде, и из продуктов выделяют соединение 1 с выходом на этой стадии 75%. Tolbert L.M., Harvey L.C., Lum R.C. // J. Phys. Chem. 1993. V. 97. 13335-133340.
Подобный по исполнению трехстадийный подход использован для получения соединения 2 исходя из нафтола-1 [Kongkathip N., Luangkamin S., Kongkathip B. Songma Ch., Grigg R., Kongsaere P., Samran P.N., Piyaviriyagyl S., Siripong P. Bioorganic & Medicinal Chemistry 2003. V. 11. N 14. P. 3179-3191].

Известен способ получения соединения 1 путем конденсации эфира (3) с получением производного хромана (7), который далее восстанавливают с получением соединения 1 [Marcinkiewicz S., Green J., Mamalis P. // Tetrahedron 1961. T. 14. C. 208-222], представлен на схеме:

Abeywickrema A.N., Beckwith A.L.J., Gerba S / // J Org. Chem. 1987, 52, 4072-4078. Недостатком этого способа является многостадийность процесса.
Известен способ получения соединения 1 с выходом 46% путем восстановления сплитомицина (8) по схеме:

Недостатком метода Posakony J., Hirao M., Sam S., Simon J.A., Bedalov A. // J. Med. Chem. 2004, 47, 2635-2644 является сложность синтеза исходного сплитомицина.
Значительное число работ направлено на получение соединения 1 с использованием производного пропионовой кислоты (9) по схеме:

Ranade A.A., Joseph A.R., Kumbhar V.B., Paradkar M.V. // J. Chem. Researh, Miniprint; nb. 11; (2003); p. 1175-1184. Основным недостатком способа является многостадийность и использование гидридов металлов в качестве восстановителей.
Современный подход к получению гидроксипропилнафтолов может быть сокращен на одну стадию с использованием известного способа получения хроменов взаимодействием нафтола-2 с пропаргиловым спиртом в присутствии рутенийсодержащего катализатора. Kanao, Keiichiro; Miyake, Yoshihiro; Nishibayashi, Yoshiaki // Organometallics (2010), 29(9), 2126-2131. Хромены, как известно, могут быть подвергнуты восстановлению литийалюминийгидридом с получением соединения 1.
Этот подход затруднен сложностью синтеза и использования рутенийсодержащего катализатора.
Анализ всех известных способов получения гидроксипропилнафтолов указывает на их одну ограничивающую особенность: в процессе синтеза гидроксипропильная группа всегда образуется в орто-положение к гидроксигруппе нафтолов. Другим недостатком известных процессов с учетом доступности α и β-нафтола является их многостадийность и использование дорогих реагентов.
Предлагаемый для реализации одностадийный способ получения гидроксипропилнафтолов из нафтолов заключается во взаимодействии их солей щелочных металлов с аллиловым спиртом в присутствии избытка щелочи. Процесс может проходить в присутствии или отсутствии в качестве ароматических углеводородов толуола или ксилола. Он может быть осуществлен при атмосферном давлении или под давлением до 10 атм в автоклаве.
При проведении процесса при атмосферном давлении целесообразно присутствие ароматических углеводородов (наиболее удобными для использования являются толуол или ксилол), что обеспечивает удаление из реакционной массы образующейся в ходе реакции воды. В присутствии щелочи вода реагирует с аллиловым спиртом, что приводит к образованию побочных наиболее трудно отделяемых метилированных продуктов.
Из углеводородов наиболее удобными для использования является толуол или ксилол.
Ввиду высокой окисляемости на воздухе щелочной реакционной массы она защищается током инертного газа в процессе синтеза и ее нейтрализации на заключительной стадии реакции. Отклонение от указанных параметров взаимодействия нафтолов с аллиловым спиртом с использованием катализаторов изменяет природу образующихся продуктов и приводит к преимущественному образованию аллилнафтолов [Das, Biswanath; Veeranjaneyulu, Boyapati; Krishnaiah, Maddeboina; Balasubramanyam, P. // Synthetic Communications (2009), 39(11), 1929-1935], эфиров [Gladfeld J.W.E., Rietz E.J. J. Am. Chem. Soc. 1940. В. 62. 974] и продуктов циклизации [Kimura, Masanari; Fukasaka, Miki; Tamaru, Yoshinao // Synthesis (2006), (21), 3611-3616].
Синтез 1-(3-гидроксипропил)нафтола-2 (1) из нафтола-2 осуществляется одностадийно по указанной схеме:

Этот процесс может быть осуществлен под давлением 10 атм в автоклаве или же при атмосферном давлении в колбе. Структура соединения 1 доказана спектральными методами и превращением его в известное соединение 7.
Более экономичен автоклавный вариант, но он ограничен доступностью специальной аппаратуры, эксплуатируемой под давлением. В этом случае реагенты, указанные на схеме, выдерживают в инертной атмосфере в автоклаве при температуре 170°C в течение 15 часов. Полученную реакционную массу нейтрализуют и получают смесь продуктов, содержащую 85% соединения (1). Очистку продукта 1 проводят перегонкой в вакууме.
Показана возможность одностадийного получения соединения 1 при атмосферном давлении. Этот вариант исполнения осуществляется в кипящем аллиловом спирте действием его на β-нафтолят натрия, предварительно приготовленного из нафтола и щелочи с удалением из реактора реакционной воды с использованием инертных растворителей. Протекание реакции и нейтрализация реакционной массы проводится в токе инертного газа из-за высокой склонности ее к окислению.
Образующиеся побочно в процессе реакции нафтохиноны являются катализаторами окисления реакционной массы на воздухе, что требует удаления их из реакционной массы без промедления. Это достигается промывкой продуктов реакции хлороформом, в котором гидроксипропилнафтол слабо растворяется. Одновременно упрощается выделение продукта из реакционной массы. Взаимодействие α-нафтола с аллиловым спиртом в щелочной среде протекает по схеме:
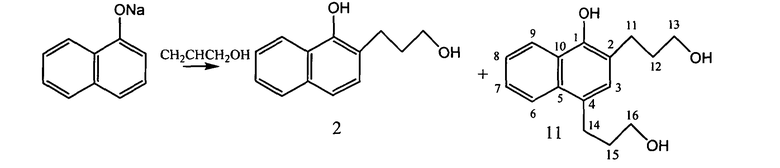
Процесс осуществляется при атмосферном давлении в токе инертного газа - аргона. Последовательность стадий такова: сначала в реакторе готовится α-нафталят натрия с удалением реакционной воды с помощью кипящего ксилола, а затем в реактор вводится аллиловый спирт и реакция осуществляется при кипении реакционной массы. Соединение 2 выделено из реакционной массы с выходом 40% в процессе ее вакуумной перегонки.
Избыток аллилового спирта способствует накоплению соединения 11 в продуктах реакции.
Примером введения гидроксипропильной группы в пара-положение к гидроксильной группе нафтола является взаимодействие 2-трет-бутилнафтола (12) с аллиловым спиртом с получением 2-трет-бутил-4-гидроксипропилнафтола-1(13):

Указанные стадии проводятся в одном сосуде: сначала готовится трет-бутилнафталят натрия кипячением соединения 12 и щелочи в ксилоле с отгонкой воды, затем полученная соль кипятится в аллиловом спирте с получением соединения 13 без заметного образования побочных продуктов.
Предлагаемый способ обладает следующими преимуществами:
1. Он одностадиен и реализован из доступных продуктов.
2. В отличие от известных методов позволяет ввести в одну стадию гидроксипропильную группу не только в орто-, но и в пара-положение к гидроксильной группе нафтола.
3. Способ позволяет получать нафтолы, содержащие в структуре две гидроксипропильные группы. Сказанные выше идеи подтверждаются следующими примерами.
Пример 1. Получение соединения (1) при давлении 10 атм (автоклавный метод)
В стальной вращающийся автоклав вместимостью 50 мл помещают 10 г (0.069 моль) 2-нафтола, 15 мл аллилового спирта и 2.9 г (0.073 моль) гранулированного едкого натра, растертого в порошок, вытесняют воздух инертным газом и содержимое нагревают при вращении автоклава в течение 15 часов при температуре 170°С. Затем автоклав охлаждают до комнатной температуры, вскрывают и в реакционную массу дополнительно помещают 7 мл соляной кислоты, 7 мл воды и 20 мл эфира. Автоклав герметизируют и нагревают до 100°C в течение часа и после охлаждения вскрывают, эфирный раствор промывают водой и упаривают. Получают 14.4 г кристаллической массы, содержащей по данным ГЖХ 86% 1-(3-гидроксипропил)-2-нафтола (1). После ее перегонки под вакуумом получают 6.7 г фракции с т. кип. 175-185°C / 3-5 мм рт.ст., содержащей по данным ГЖХ 97% основного вещества 1. Выход 48%. Т. пл. 127-129°C. После кристаллизации из смеси эфир - гексан, т. пл. 132-133°C (по данным [1] т. пл. 132.5-133.5°C). М+ 202 (масс-спектрометрически).
Сигналы ПМР раствора соединения 1 в CD3OD: 2.02 м, CH2; 3.22 т., CH2Ar; 3.60 т. CH2OH; 7.14 д. 1Н; 7.32 т. 1Н; 7.46 т. 1Н; 7.77 д., 1Н; 7.79 д., 1Н; 7.88-7.91 м. 1Н CH2 совпали с таковыми, приведенными для спектра ПМР соединения (1), снятого в растворе CD3CN. [J. Posakony, M. Hirao, S. Stevens, J.A. Simon, A. Bedalov // J. Med. Chem. 2004. V. 47. P. 2635-2644].
Дополнительно идентификацию соединения 1 провели превращением его в известное соединение 7. К раствору 0.5 г 98%-ного соединения 1 в 0.2 мл ДМФА прибавили при комнатной температуре при перемешивании 0.2 мл SOCl2 (бурное выделение SO2). Реакционную массу выдержали при температуре 80°C в течение 20 минут, после чего добавили 2 мл воды и кипятили еще 30 минут. Продукт экстрагировали эфиром и упарили растворитель. Получили 0.4 г вещества, содержащего по данным ГЖХ 90% 2,3,4-тригидронафтопирана(7). Оно очищено методом тонкослойной хроматографии, т. пл. 40-42°C. По литературным данным т. пл. 40-42°C [A.N. Abeywickrema, A. L.J. Beckwith, S. Gerba // J. Org. Chem. 1987. V. 52. P. 4072-4078].
ПМР-спектр соединения 7 в CDCI3, δ: 2.15-2.30 (m, CH2), 3.10 (t, J=6.1 Гц, CH2), 4.3 (t, J=5.1 Гц, CH2), 7.0-7.9 (m, 6H, Ar-H).
Пример 2. Получение соединения 1 при атмосферном давлении. В колбу с вводом инертного газа, мешалкой и дефлегматором помещают 2.84 г (0.02 моль) β-нафтола, 2 г (0.05 моль) растертого NaOH и 15 мл орто-ксилола. Из реактора при нагревании и перемешивании отгоняют в течение 30 минут 12 мл влажного орто-ксилола, после чего в реактор добавляют 2.5 мл аллилового спирта и кипятят с обратным холодильником в течение 1 часа 30 минут, добавляют порцию 1.5 мл аллилового спирта и кипятят еще 30 минут. Реакционную массу разбавляют подкисленной водой и экстрагируют метил-трет-бутиловым эфиром. Экстракт отделяют, промывают водой и упаривают. Получают 3.7 г твердого продукта, содержащего по данным ГЖХ 80% соединения 1. После его промывки теплым хлороформом получают 2.0 г соединения 1(выход 50%) с т. пл. 127-129°C.
Пример 3. Получение соединений 2 и 11 при атмосферном давлении
В стеклянную колбу помещают 2.4 г растертого порошка NaOH, 15 мл свежеперегнанного толуола и 1.4 г нафтола-1. При перемешивании в токе инертного газа в течение 1 часа из реактора отгоняют 8 мл влажного толуола. Затем в реактор добавляют 15 мл аллилового спирта и в токе инертного газа продолжают в течение 2 часов медленно отгонять из реактора смесь толуола и аллилового спирта. Реакционную массу охлаждают, растворяют в метил-трет-бутиловом эфире и раствор промывают разбавленной соляной кислотой до нейтральной реакции. После упаривания растворителя получают 2.1 г вязкого масла, содержащего по данным ГЖХ 31% исходного 1-нафтола, 62% 2-(3-гидроксипропил)-1-нафтола(2), и 17% 2, 4-ди-(3-гидроксипропил)-1-нафтола (11). Это масло перегоняют под вакуумом, собирая 1.0 г фракцию с т. кип 185°C/ 3 мм рт.ст. Продукт очищают кристаллизацией из гексана с добавкой метил-трет-бутилового эфира. Получают 0.8 г соединения 2 (выход 40%) с т. пл. 84-86°C. По литературным данным т. пл. 86-87°C [J. Am. Chem. Soc. 1940. V. 62. Р. 3067-3069] Найдено: M 202.1., m/z(%): 202(M+ 98), 184 (99), 156 (69), 128(100).
Соединение 11 выделено из реакционной массы методом тонкослойной хроматографии на силикагеле. Элюент спирт:хлороформ в соотношении 10:1. Т. пл. 161-164°C. Найдено: m/z=260.1405(M+ 44), 216 (39), 197 (100), 171 (75), 141 (45) 115 (37), 73 (62). C16H20O3. Вычислено: m/z=260.1407 (масс-спектрометрически). ПМР-спектр в CD3OD, δ: 1.84 m(-CH2-); 1.88 m. (-CH2-); 2.82 t. J=7.0 Гц (-CH2-); 3.10 t. J=7.0 Гц(-CH2-); 3.57 t. J=7.0 Гц (-CH2OH); 3.69 t. J=7.0 Гц (-CH2OH); 7.10 с. ArH; 7.3-7.4 m. 2H, 7.90-7.95 m. 1H, 8.15-8.22 m. 1H, ArH.
ЯМР-спектр 13C: C12 25.61; C15 28.13; C11 32.27; C14 33.42; C13 60.38; C16 61.00 C3 121.20; 121.9; 123.07; 123.81; 123.43; 124.49; C5 126.12; C4 129.33; C10 131.22; C1 147.65.
Пример 4. Получение 2-трет-бутил-4-(3-гидроксипропил)нафтола-1 (13)
В колбу с мешалкой, дефлегматором и вводом в реакционную массу инертного газа (аргон) помещают 1.2 г (0.006 моль) 2-трет-бутил-1-нафтола, 1.5 г растертого NaOH (0.038 моль) и 15 мл толуола. Содержимое нагревают до кипения и в течение 30 минут отгоняют 12 мл влажного толуола, затем добавляют 3.5 мл (0.06 моль) аллилового спирта и полученный раствор кипятят в течение одного часа.
В реактор одной порцией добавляют раствор 5 мл концентрированной соляной кислоты в 20 мл воды. Отделяется масло, из которого выпадает 0.65 г (выход 42%) бесцветных кристаллов соединения 13. Т. пл. 120-122.5°C (из CHCI3).
Найдено: М-1 257. 155. C17H22O2 (хроматомасс-спектрометрически). Вычислено: М-1 257. 154. УФ-спектр в метаноле: λмакс 242 и 302 нм; s 325 нм.
ПМР-спектр в CD3OD: δ 1.43 с. 9Н. C4H9-tert; 1.88 m. 2H. -CH2-; 2.98 t. J=7.5 Гц, 2H; 3.30 с. OH; 3.62 t. J=6.2 Гц, 2H. -CH2OH; 7.25 с. ArH; 7.34-7.36 m. 2H, 7.87 m. 1H, 8.02 m. 1H, ArH.
ЯМР-спектр 13С: C12 28.97; C15 29.98; C11 33.61; C14 34.27; C13 121.02; C3 121.02; 123.56; 124.81; 124.9; 125.33; C5 127; C4 129.12; C10 130.3; C2 130.92; C1 147.64.
Настоящее изобретение относится к способу получения в одну стадию (3-гидроксипропил)нафтолов указанной структуры
 , эффективных биологически активных веществ нейротропного действия. Способ заключается в реакции α-, β-нафтолов, их производных или их натриевых солей с аллиловым спиртом и щелочью при температуре 100-170°С. Выход очищенных продуктов 40-60%. 4 пр.
, эффективных биологически активных веществ нейротропного действия. Способ заключается в реакции α-, β-нафтолов, их производных или их натриевых солей с аллиловым спиртом и щелочью при температуре 100-170°С. Выход очищенных продуктов 40-60%. 4 пр.
Способ получения в одну стадию (3-гидроксипропил)нафтолов указанной структуры реакцией α-, β-нафтолов, их производных или их натриевых солей с аллиловым спиртом и щелочью при температуре 100-170°С.
| US 4833164A1, 23.05.1989 | |||
| Guss C.O | |||
| Intramolecular displacement of carboxylate ion | |||
| II | |||
| The effect of some variations in structure of phenol-alcohols | |||
| Journal of the American Chemical Society, 1951, 73, 608-611 | |||
| De Benneville P.L | |||
| et al, Hydrogenation of some substituted coumarins | |||
| Journal of the American Chemical Society, 1940, 62, 3067-3070 | |||
| Способ получения нафталиновых производных | 1986 |
|
SU1581217A3 |

