ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к селективным ингибиторам c-Met. Изобретение также обеспечивает фармацевтически приемлемые композиции, содержащие ингибитор c-Met, и способы применения композиций при лечении различных пролиферативных нарушений.
УРОВЕНЬ ТЕХНИКИ
Фактор роста гепатоцитов (HGF), также известный как рассеивающий фактор, представляет собой многофункциональный фактор роста, который увеличивает превращение и развитие опухоли, вызывая митогенез и подвижность клеток. Дополнительно HGF обеспечивает метастаз, стимулируя подвижность клетки и инвазию с помощью различных сигнальных путей. Для получения клеточных эффектов HGF должен связаться с его рецептором, рецептором тирозинкиназы c-Met. c-Met, широко экспрессированный гетеродимерный протеин, содержащий 50 килодальтон (кДа) α-субъединицы и 145 кДа альфа-субъединицы (Maggiora et al., J. Cell Physiol., 173:183-186, 1997), сверхэкспрессирован в значительном процентном соотношении раковых образований человека и амплифицируется в течение перехода между первичными опухолями и метастазами. Различные раковые образования, с которыми связана сверхэкспрессия c-Met, включают, но не ограничиваются ими, аденокарциному желудка, рак почек, мелкоклеточный рак легкого, колоректальный рак, рак простаты, рак мозга, рак печени, рак поджелудочной железы и рак груди. c-Met также связан с атеросклерозом и фиброзом легких.
Соответственно, существует большая потребность в развитии соединений, пригодных в качестве ингибиторов рецептора протеинкиназы c-Met. В частности, предпочтительные соединения должны иметь высокую аффинность к рецептору c-Met и проявлять функциональную активность в качестве антагонистов, при этом обладая небольшой аффинностью к другим рецепторам киназы или целям, которые, как известно, связаны с побочными эффектами.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Было обнаружено, что 3-(хинолин-6-ил)метил-N-(1H-пиррол-3-ил)-[1,2,4]триазоло[3,4-b][1,3,4]тиадиазол-6-амины эффективны при ингибировании c-Met.
Соответственно, изобретение обеспечивает соединение, имеющее формулу:
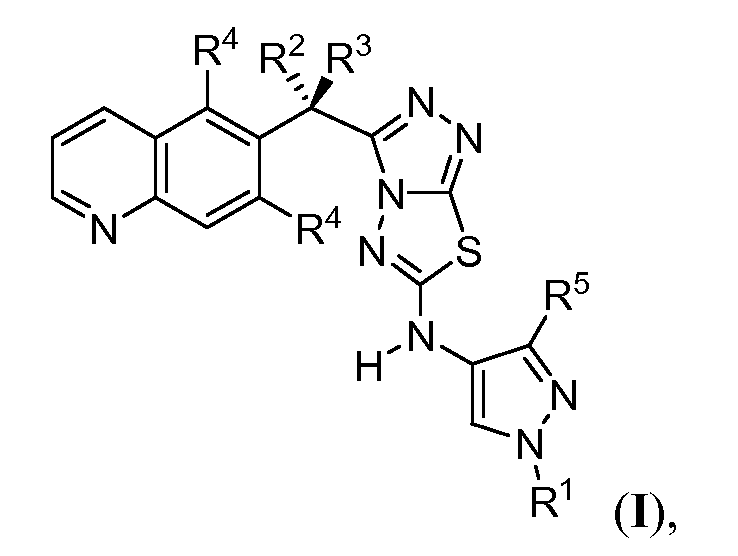
или его фармацевтически приемлемую соль, в которой каждый или R1, R2, R3, R4 и R5 представляет собой, как определено в другом месте в настоящем описании.
Изобретение также обеспечивает фармацевтические композиции, которые включают соединение формулы I и фармацевтически приемлемый носитель, адъювант или наполнитель. Кроме того, изобретение обеспечивает способы лечения или уменьшения степени тяжести пролиферативного заболевания, состояния или нарушения у пациента, которые включают стадию введения пациенту терапевтически эффективной дозы соединения формулы I или его фармацевтической композиции.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения и общая терминология
Используемые в настоящем описании следующие определения следует применять, если не указано иначе. В целях данного изобретения химические элементы идентифицированы в соответствии с Периодической Системой Химических Элементов, версией CAS и Handbook of Chemistry and Physics, 75th Ed. 1994. Дополнительно, общие принципы органической химии описаны в «Organic Chemistry», Thomas Sorrell, University Science Books, Sausalito: 1999 и «March's Advanced Organic Chemistry», 5th Ed., Smith, M.B. and March, J., eds. John Wiley & Sons, New York: 2001, все содержание которых включено в настоящее описание в полном объеме посредством ссылки.
Описание соединения изобретения
В первом аспекте изобретение обеспечивает следующие соединения формулы I:
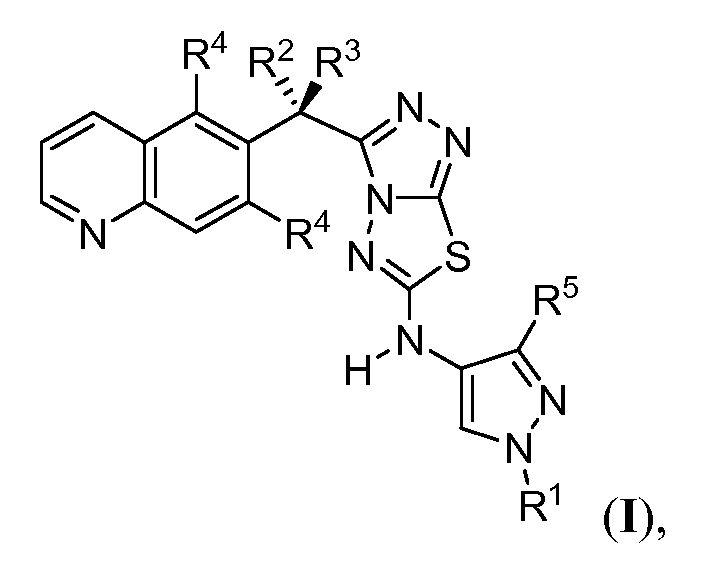
или их фармацевтически приемлемую соль, в которой
R1 представляет собой C1-3алифатическую группу;
R2 представляет собой водород, фтор или метил;
R3 представляет собой водород, фтор или метил;
каждый R4 представляет собой независимо водород или фтор; и
R5 представляет собой водород, хлор, циклопропил или C1-4алифатическую группу, необязательно замещенную 1-3 атомами фтора.
В одном варианте осуществления R2 представляет собой метил и R3 представляет собой водород. В другом варианте осуществления R2 представляет собой водород и R3 представляет собой метил.
В другом варианте осуществления каждый из R2 и R3 представляет собой фтор.
В другом варианте осуществления соединений изобретения R4 представляет собой водород.
В другом варианте осуществления соединений изобретения R1 представляет собой метил и R5 представляет собой водород.
В дополнительном варианте осуществления R1 представляет собой метил, каждый из R2 и R3 представляет собой фтор и каждый из R4 и R5 представляет собой водород.
В другом варианте осуществления соединение представляет собой соль гидрохлорида.
Соединения формулы I включают следующие:
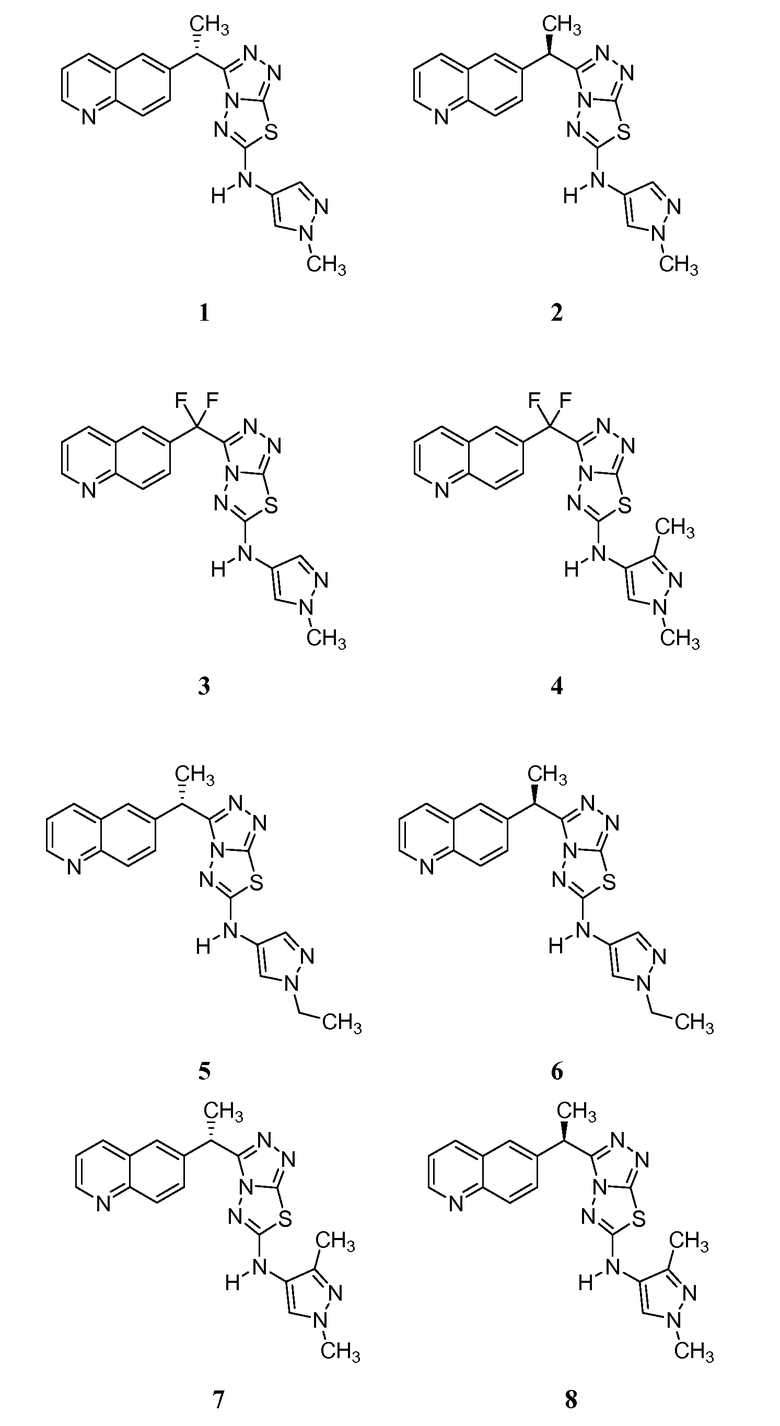
В другом аспекте изобретение обеспечивает фармацевтическую композицию, содержащую соединение формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, адъювант или наполнитель. В одном варианте осуществления композиция содержит дополнительный химиотерапевтический или антипролиферативный агент, противовоспалительный агент, агент для лечения атеросклероза или агент для лечения фиброза легких.
В другом аспекте изобретение обеспечивает способ лечения или уменьшения степени тяжести пролиферативного нарушения у пациента, включающий введение соединения формулы I в количестве, достаточном для лечения или уменьшения степени тяжести пролиферативного нарушения у указанного пациента. В одном варианте осуществления пролиферативное нарушение представляет собой метастатический рак. В другом варианте осуществления пролиферативное нарушение представляет собой глиобластому; гепатоцеллюлярную карциному, карциному желудка или рак, выбранный из рака толстой кишки, груди, простаты, мозга, печени, поджелудочной железы или легкого.
В другом варианте осуществления пролиферативное нарушение представляет собой метастатический рак.
Композиции, составы и введение соединений изобретения
В другом аспекте изобретение обеспечивает композицию, содержащую соединение формулы I или его фармацевтически приемлемое производное и фармацевтически приемлемый носитель, адъювант или наполнитель. В одном варианте осуществления количество соединения в композиции этого изобретения представляет собой такое, которое является эффективным в известной мере для ингибирования c-Met в биологическом образце или у пациента. Предпочтительно композицию этого изобретения получают для введения пациенту, нуждающемуся в такой композиции. Наиболее предпочтительно композицию этого изобретения получают для перорального введения пациенту.
Используемый в настоящем описании термин «пациент» означает животное, предпочтительно млекопитающее и наиболее предпочтительно человека.
Следует также отметить, что соединения формулы I могут существовать в свободной форме для лечения или при необходимости в качестве его фармацевтически приемлемого производного. Согласно настоящему изобретению, фармацевтически приемлемое производное включает, но не ограничивается ими, фармацевтически приемлемые пролекарства, соли, сложные эфиры, соли таких сложных эфиров или любой другой аддукт или производное, которое при введении пациенту, нуждающемуся в таком лечении, является способным к получению, прямо или косвенно, соединения формулы I, как иначе описано в настоящем изобретении, или его метаболита или остатка.
Используемый в настоящем описании термин «фармацевтически приемлемая соль» относится к таким солям, которые в рамках результатов тщательной медицинской оценки являются подходящими для применения при взаимодействии с тканями людей и низших животных без неспецифической токсичности, болезненной чувствительности, аллергического ответа и подобного.
Фармацевтически приемлемые соли хорошо известны в данной области техники. Например, S. М. Berge и др. описывают фармацевтически приемлемые соли подробно в J. Pharmaceutical Sciences, 66:1-19, 1977, который включен в настоящее описание посредством ссылки. Фармацевтически приемлемые соли соединений формулы I включают полученные из подходящих неорганических и органических кислот и оснований. Примеры фармацевтически приемлемых нетоксичных кислотно-аддитивных солей представляют собой соли аминогруппы, полученные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и хлорная кислота или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота или при использовании других способов, применяемых в данной области техники, таких как ионный обмен. Другие фармацевтически приемлемые соли включают адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептаноат, гексаноат, гидроиодид, 2-гидроксиэтансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканоат, валерат соли и подобные. Соли, полученные из соответствующих оснований, включают соли щелочного металла, щелочноземельного металла, аммония и N+(C1-4алкила)4.
Выше описано, что фармацевтически приемлемые композиции настоящего изобретения дополнительно включают фармацевтически приемлемый носитель, адъювант или наполнитель, который, как используется в настоящем описании, включает всевозможные растворители, разбавители или другой жидкий наполнитель, дисперсионные или суспензионные вспомогательные средства, поверхностно-активные вещества, изотонические агенты, загущающие или эмульгирующие агенты, консерванты, твердые связующие вещества, смазывающие вещества и подобные, как подходящие для определенной желательной формы дозировки. В Remington: The Science and Practice of Pharmacy, 21st edition, 2005, ed. D.B. Troy, Lippincott Williams & Wilkins, Philadelphia и Encyclopedia of Pharmaceutical Technology, eds. J. Swarbrick and J. C. Boylan, 1988-1999, Marcel Dekker, New York, содержание каждого из которых включено в настоящее описание посредством ссылки, раскрыты различные носители, используемые в составе фармацевтически приемлемых композиций и известные методики их получения. За исключением случаев, когда любая стандартная среда носителя является несовместимой с соединением формулы I так, как получение любого нежелательного биологического эффекта или иначе взаимодействия во вредной манере с любым другим компонентом(ами) фармацевтически приемлемой композиции, его использование рассматривают, чтобы находиться в объеме данного изобретения.
Некоторые примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают, но не ограничиваются ими, ионообменную смолу, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота или сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протаминсульфат, гидрофосфат натрия, гидрофосфат калия, хлорид натрия, соли цинка, диоксид кремния, трисиликат магния, поливинилпирролидон, полиакрилаты, воски, блок полимеры полиэтилен-полиоксипропилен, ланолин, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу и ее производные, такие как натрий карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошковый трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и воски для суппозиторий; масла, такие как арахисовое масло, хлопковое масло; сафлоровое масло; сезамовое масло; оливковое масло; кукурузное масло и соевое масло; гликоли; такие как пропиленгликоль или полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновую кислоту; апирогенную воду; изотонический солевой раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, так же как другие нетоксичные совместимые смазывающие вещества, такие как лаурилсульфат натрия и стеарат магния, так же как красители, высвобождающие агенты, покрывающие агенты, подслащивающие агенты, ароматизаторы и ароматизирующие добавки, консерванты и антиоксиданты могут также присутствовать в композиции согласно решению разработчика рецептур.
Композиции настоящего изобретения могут вводить перорально, парентерально, с помощью ингаляции, местно, ректально, назально, буккально, вагинально или с помощью имплантированного резервуара. Используемый в настоящем описании термин «парентеральный» включает подкожную, внутривенную, внутримышечную, внутрисуставную, интрасиновиальную, надчревную, интратекальную, внутриглазную, внутрипеченочную, вводимую внутрь пораженных тканей и внутричерепную инъекцию или методы инфузии. Предпочтительно, композиции вводят перорально, внутрибрюшинно или внутривенно. Стерильные инъекционные формы композиций данного изобретения могут быть водной или маслянистой суспензией. Эти суспензии могут получить согласно способам, известным в данной области техники, используя подходящие диспергирующие или смачивающие агенты и суспендирующие агенты. Стерильное инъекционное лекарственное средство может также быть стерильным инъекционным раствором или суспензией в нетоксичном парентерально приемлемом разбавителе или растворителе, например, как раствор в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей, которые могут применять, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла традиционно используют в качестве растворителя или суспендирующей среды.
С этой целью могут использовать любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее производные глицериды пригодны при получении инъекционных лекарственных средств, поскольку представляют собой природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных вариантах. Эти масляные растворы или суспензии могут также содержать длинноцепочечный спиртовой разбавитель или диспергирующее вещество, такое как карбоксиметилцеллюлоза или подобные диспергирующие агенты, которые обычно используют в составе фармацевтически приемлемых форм дозировки, включая эмульсии и суспензии. Другие обычно используемые поверхностно-активные вещества, такие как твины, спаны и другие эмульгирующие агенты или увеличивающие биодоступность агенты, которые обычно используют при получении фармацевтически приемлемого твердого вещества, жидкости или других форм дозировки, могут также применять для целей состава.
Фармацевтически приемлемые композиции этого изобретения могут вводить перорально в любой перорально приемлемой форме дозировки, включая но, не ограничиваясь ими, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения, стандартно используемые носители включают лактозу и кукурузный крахмал. Также обычно добавляют скользящие вещества, такие как стеарат магния. Для перорального введения в форме капсул пригодные разбавители включают лактозу и высушенный кукурузный крахмал. Когда требуются водные суспензии для перорального применения, активный ингредиент объединяют с эмульгирующими и суспендирующими агентами. При желании могут также добавить определенные подслащивающие агенты, ароматизаторы или красители.
Альтернативно, фармацевтически приемлемые композиции этого изобретения могут вводить в форме суппозиторий для ректального введения. Их могут получить с помощью смешивания агента с подходящим не раздражающим эксципиентом, который представляет собой твердое вещество при комнатной температуре, но является жидкостью при ректальной температуре и следовательно, будет таять в прямой кишке для высвобождения лекарственного средства. Такие материалы включают масло какао, воск и полиэтиленгликоли.
Фармацевтически приемлемые композиции этого изобретения могут также вводить местно, особенно когда цель лечения включает области или органы, легко доступные для местного применения, включая заболевания глаз, кожи или нижней части кишечника. Подходящие местные составы легко получают для каждого из этих областей или органов.
Местное применение для нижней части кишечника могут осуществить в составе ректальной суппозитории (см. выше) или в составе подходящей клизмы. Могут также применять местно-трансдермальные пластыри.
Для местного применения фармацевтически приемлемые композиции могут получить в форме подходящей мази, содержащей активный компонент, суспендированный или растворенный в одном или более носителях. Носители для местного введения соединений формулы I включают, но не ограничиваются ими, минеральное масло, жидкий петролатум, белый петролатум, пропиленгликоль, полиоксиэтилен, соединение полиоксипропилена, эмульгирующий воск и воду. Альтернативно, фармацевтически приемлемые композиции могут получить в форме подходящего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или более фармацевтически приемлемых носителях. Подходящие носители включают, но не ограничиваются ими, минеральное масло, сорбитан моностеарат, полисорбат 60, цетиловый сложноэфирный воск, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
Для офтальмологического применения фармацевтически приемлемые композиции могут получить, например, в виде тонкодисперсных суспензий в изотоническом с установленным pH стерильном солевом растворе или другом водном растворе или предпочтительно, в виде растворов в изотоническом с установленным pH стерильном солевом растворе или другом водном растворе; в присутствии или в отсутствии консерванта, такого как бензилалкония хлорид. Альтернативно, для офтальмологических применений фармацевтически приемлемые композиции могут получить в форме мази, такой как петролатум. Фармацевтически приемлемые композиции этого изобретения могут также вводить с помощью назального аэрозоля или ингаляции. Такие композиции получают согласно методам, хорошо известным в области фармацевтических составов, и могут получить в качестве растворов в солевом растворе, используя бензиловый спирт или другие подходящие консерванты, способствующие абсорбции вещества для увеличения биодоступности, фторуглероды и/или другие стандартные солюбилизирующие или диспергирующие агенты.
Наиболее предпочтительно фармацевтически приемлемые композиции этого изобретения получают для перорального введения.
Жидкие формы дозировки для перорального введения включают, но не ограничиваются ими, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям, жидкие формы дозировки могут содержать инертные разбавители, обычно используемые в данной области техники, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгирующие агенты, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и сезамовое масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоль и сложные эфиры сорбитана и жирных кислот и их смеси. Помимо инертных разбавителей пероральные композиции могут также включать адъюванты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подслащивающие агенты, ароматизаторы и ароматизирующие агенты.
Инъекционные лекарственные средства, например, стерильные инъекционные водные или маслянистые суспензии могут получить согласно способу, известному в данной области техники, используя подходящие диспергирующие или смачивающие агенты и суспендирующие агенты. Стерильное инъекционное лекарственное средство может также быть стерильным инъекционным раствором, суспензией или эмульсией в нетоксичном парентерально приемлемом разбавителе или растворителе, например, как раствор в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей, которые могут применять, находятся вода, раствор Рингера, U.S.P. и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла традиционно используют в качестве растворителя или суспендирующей среды. С этой целью могут использовать любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, используют при получении инъекционных лекарственных средств.
Инъекционные составы могут стерилизовать, например, с помощью фильтрования через сдерживающий бактерии фильтр или с помощью внедрения стерилизующих агентов в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или другой стерильной инъекционной среде до использования.
Для продления эффекта соединения формулы I часто желательно замедлить абсорбцию этого соединения из подкожной или внутримышечной инъекции. Это может быть достигнуто при помощи использования жидкой суспензии кристаллического или аморфного материала с плохой растворимостью в воде. Скорость абсорбции соединения формулы I тогда зависит от его скорости растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно, растворение или суспендирование соединения формулы I в маслянистом наполнителе обеспечивает замедленную абсорбцию парентерально вводимой формы соединения. Инъекционные формы депо получают с помощью образования микроинкапсулированных матриц соединения формулы I в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения соединения к полимеру и природы применяемого определенного полимера могут контролировать скорость высвобождения соединения. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъекционные составы депо также получают с помощью захватывания соединения формулы I в липосомах или микроэмульсиях, которые являются совместимыми с тканями тела.
Композиции для ректального или вагинального введения представляют собой предпочтительно суппозитории, которые могут получить с помощью смешивания соединение формулы I с подходящими нераздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиторий, которые представляют собой твердые вещества при температуре окружающей среды, но являются жидкостями при температуре тела и, следовательно, тают в прямой кишке или вагинальной полости и высвобождают активное соединение.
Твердые формы дозировки для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых формах дозировки активное соединение смешивают, по меньшей мере, с одним инертным фармацевтически приемлемым эксципиентом или носителем, таким как цитрат натрия или фосфат дикальция и/или a) наполнителями или утяжелителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремневая кислота, b) связующими веществами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и акация, c) увлажняющими веществами, такими как глицерин, d) дезинтегрирующими агентами, такими как агар-агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновая кислота, определенные силикаты и карбонат натрия, e) замедляющими растворение агентами, такими как парафин, f) ускоряющими абсорбцию агентами, такими как соединения четвертичного аммония, g) смачивающими агентами, такими как, например, цетиловый спирт и моностеарат глицерина, h) абсорбентами, такими как каолин и бентонитовая глина и i) скользящими агентами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль форма дозировки может также включать буферные агенты.
Твердые композиции подобного типа могут также использовать в качестве наполнителей в мягких и твердо-заполненных желатиновых капсулах, используя такие эксципиенты, как лактоза или молочный сахар, так же как полиэтиленгликоли с высокой молекулярной массой и подобные. Твердые формы дозировки таблеток, драже, капсул, пилюль и гранул могут получить с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в области фармацевтических составов. Они могут необязательно содержать рентгеноконтрастные агенты и могут также быть из композиции, из которой они высвобождают активный ингредиент(ы) только, или предпочтительно, в определенной части кишечного тракта, необязательно, замедленным способом. Примеры капсулирующих композиций, которые могут использовать, включают полимерные вещества и воски. Твердые композиции подобного типа могут также использовать в качестве наполнителей в мягких и твердо-заполненных желатиновых капсулах, используя такие эксципиенты, как лактоза или молочный сахар, так же как полиэтиленгликоли с высокой молекулярной массой и подобные.
Активные соединения могут также быть в микроинкапсулированной форме с одним или более эксципиентами, как отмечено выше. Твердые формы дозировки таблеток, драже, капсул, пилюль и гранул могут получить с покрытиями и оболочками, такими как энтеросолюбильные покрытия, контролирующие высвобождение покрытия и другие покрытия, хорошо известные в области фармацевтических составов. В таких твердых формах дозировки активное соединение могут смешать, по меньшей мере, с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие формы дозировки могут также содержать, что является обычной практикой, дополнительные вещества, кроме инертных разбавителей, например, таблетирующие скользящие вещества и другие таблетирующие вспомогательные вещества, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль формы дозировки могут также содержать буферные агенты. Они могут необязательно содержать рентгеноконтрастные агенты и могут также быть из композиции, из которой они высвобождают активный ингредиент(ы) только, или предпочтительно, в определенной части кишечного тракта, необязательно, замедленным способом. Примеры капсулирующих композиций, которые могут использовать, включают полимерные вещества и воски.
Формы дозировки для местного или трансдермального введения соединения формулы I включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, ингаляторы или пластыри. Активный компонент смешивают при стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферными агентами, как может потребоваться. Офтальмологический состав, ушные капли, и глазные капли также рассматривают в рамках этого изобретения. Дополнительно, настоящее изобретение рассматривает использование трансдермальных пластырей, которые имеют дополнительное преимущество обеспечения контролируемой доставки, соединения формулы I к телу. Такие формы дозировки могут получить с помощью растворения или распределения соединения формулы I в соответствующей среде. Усиливающие абсорбцию агенты могут также использовать для увеличения потока соединения формулы I через кожу. Скорость могут контролировать или с помощью обеспечения мембраны, контролирующей скорость, или с помощью диспергирования соединения формулы I в полимерной матрице или геле.
Соединения формулы I предпочтительно получают в форме единицы дозировки для простоты введения и однородности дозировки. Используемое в настоящем описании выражение «форма единицы дозировки» относится к физически дискретной единице агента, соответствующего для пациента, которого лечат. Следует понимать, однако, что полное ежедневное применение соединения формулы I и композиций, содержащих соединение формулы I, будет решено лечащим врачом по результатам тщательной медицинской оценки. Определенный эффективный уровень дозы для любого определенного пациента или организма будет зависеть от разнообразия факторов, включая нарушение, которое лечат, и степень тяжести нарушения; активность определенного применяемого соединения; определенное применяемое соединение; возраст, массу тела, общее состояние здоровья, пол и режим питания пациента; время введения, путь введения и скорость экскреции определенного применяемого соединения; продолжительность лечения; лекарственные средства, используемые в комбинации или совпадающие с определенным применяемым соединением, и подобные факторы, хорошо известные в области медицины.
Количество соединения формулы I, которое могут объединить с материалами носителя для получения композиции в единичной форме дозировки, будет изменяться в зависимости от хозяина, которого лечат, определенного режима введения. Предпочтительно, композиции должны быть составлены так, что дозировку от 0,01 до на 100 мг/кг массы тела/сутки ингибитора могут вводить пациенту, получающему эти композиции. В одном примере композиции получены так, что дозировка соединения формулы I может быть от 3 до 30 мг/кг массы тела/сутки. В другом примере композиции получены так, что дозировка соединения формулы I может быть от 5 до 60 мг/кг массы тела/сутки.
В зависимости от определенного состояния или заболевания, которое лечат или предотвращают, дополнительные терапевтические агенты, которые обычно вводят для лечения или предотвращения такого состояния, могут также присутствовать в композициях этого изобретения. Используемые в настоящем описании дополнительные терапевтические агенты, которые обычно вводят для лечения или предотвращения определенного заболевания или состояния, известны как «соответствующие для заболевания или состояния, которое лечат». Примеры дополнительных терапевтических агентов обеспечивают ниже.
Количество дополнительного терапевтического агента, присутствующего в композициях этого изобретения, будет не более, чем количество, которое обычно вводили бы в композицию, содержащую такой терапевтический агент в качестве единственного активного агента. Предпочтительно количество дополнительного терапевтического агента в настоящее время в раскрытых композициях располагается приблизительно от 50% до 100% количества, обычно присутствующего в композиции, содержащей такой агент в качестве единственного терапевтически активного агента.
Применения соединений Формулы I и композиций, содержащих соединения Формулы I
Согласно одному варианту осуществления изобретение относится к способу ингибирования активности протеинкиназы c-Met в биологическом образце, включающему стадию взаимодействия указанного биологического образца с соединением формулы I или композицией, содержащей указанное соединение. Используемый в настоящем описании термин «биологический образец» означает типовой внешний живой организм и включает, не ограничивается ими, клеточные культуры или их экстракты; биопсийный материал, полученный от млекопитающего или его экстракты; и кровь, слюну, мочу, экскременты, сперму, слезы или другие жидкости тела или их экстракты. Ингибирование активности киназы в биологическом образце полезно для разнообразия целей, известных специалисту в данной области техники. Примеры таких целей включают, но не ограничиваются ими, хранение биологического образца и биологические исследования. В одном варианте осуществления способ ингибирования активности киназы в биологическом образце ограничивают не терапевтическими способами.
Термин «c-Met» является синонимичным «c-MET», «cMet», «MET», «Met» или другим обозначениям, известным специалисту в данной области техники.
Согласно другому варианту осуществления изобретение относится к способу ингибирования активности киназы c-Met у пациента, включающему стадию введения указанному пациенту соединения формулы I или композиции, содержащей указанное соединение.
Используемый в настоящем описании термин «c-Met-опосредованное заболевание» или «c-Met-опосредованное состояние» означает любое состояние заболевания или другое болезнетворное состояние, при котором известно, что c-Met играет роль. Термины «c-Met-опосредованное заболевание» или «c-Met-опосредованное состояние» также означают такие заболевания или состояния, симптомы которых смягчаются при лечении с помощью ингибитора c-Met. Такие состояния включают, но не ограничиваются ими, рак почек, желудка, толстой кишки, мозга, груди, простаты, печени, поджелудочной железы или легких, глиобластому, атеросклероз или фиброз легких.
В одном аспекте настоящее изобретение обеспечивает способ лечения пролиферативного нарушения у пациента, включающий стадию введения пациенту терапевтически эффективной дозы соединения формулы I или композиции, содержащей соединение формулы I.
Согласно одному варианту осуществления пролиферативное нарушение представляет собой рак, такой как, например, рак почек, желудка, толстой кишки, мозга, груди, печени, простаты и легких или глиобластому.
В другом варианте осуществления настоящее изобретение относится к способу лечения или уменьшения степени тяжести гепатоцеллюлярной карциномы у пациента, нуждающегося в таком лечении, включающему введение указанному пациенту соединения формулы I или его композиции.
В другом варианте осуществления пролиферативное нарушение представляет собой истинную полицитемию, эссенциальную тромбоцитемию, хронический идиопатический миелофиброз, миелоидную метаплазию с миелофиброзом, хронический гранулоцитарный лейкоз (CML), хронический миеломоноцитарный лейкоз, хронический эозинофильный лейкоз, гипереозинофильный синдром, системный мастоцитоз, нетипичный CML или ювенильный миеломоноцитарный лейкоз.
В другом варианте осуществления пролиферативное нарушение представляет собой атеросклероз или фиброз легких.
Другой аспект настоящего изобретения относится к способу ингибирования метастаза опухоли у пациента, нуждающегося в таком лечении, включающему введение указанному пациенту соединения формулы I или его композиции.
В зависимости от определенного состояния или заболевания, которое лечат, дополнительные терапевтические агенты, которые обычно вводят для лечения такого состояния, могут также присутствовать в композициях этого изобретения. Используемые в настоящем описании дополнительные терапевтические агенты, которые обычно вводят для лечения определенного заболевания или состояния, известны как «соответствующие для заболевания или состояния, которое лечат».
В одном варианте осуществления химиотерапевтические агенты или другие антипролиферативные агенты могут объединить с соединением формулы I для лечения пролиферативных заболеваний и рака. Примеры известных химиотерапевтических агентов включают, но не ограничиваются ими, алкилирующие агенты, такие как, например, циклофосфамид, ломустин, бусульфан прокарбазин, ифосфамид, алтретамин, мелфалан, эстрамустина фосфат, гексаметилмеламин, хлорметин, тиотепа, стрептозоцин, хлорамбуцил, темозоломид, дакарбазин, семустин или кармустин; платиновые агенты, такие как, например, цисплатин, карбоплатин, оксалиплатин, ZD-0473 (AnorMED), спироплатин, лобаплатин (Aeterna), карбоксифталатоплатин, сатраплатин (Johnson Matthey), тетраплатин BBR-3464, (Hoffmann-La Roche), ормиплатин, SM-11355 (Sumitomo), ипроплатин или AP-5280 (Access); антиметаболиты, такие как, например, азацитидин, томудекс, гемцитабин, триметрексат, капецитабин, дезоксикоформицин, 5-фторурацил, флударабин, флоксуридин, пентостатин, 2-хлордеоксиаденозин, ралтитрексед, 6-меркаптопурин, гидроксимочевина, 6-тиогуанин, децитабин (SuperGen), цитарабин, клофарабин (Bioenvision), 2-фтордеоксицитидин, ирофулвен (MGI Pharma), метотрексат, DMDC (Hoffmann-La-Roche), идатрексат или этинилцитидин (Taiho); ингибиторы топоизомеразы, такие как, например, амсакрин, рубитекан (SuperGen), эпирубицин, эксатекана мезилат (Daiichi), этопозид, квинамед (ChemGenex), тенипозид, митоксантрон, гиматекан (Sigma-Tau), иринотекан (CPT-11), дифломотекан (Beaufour-Ipsen), 7-этил-10-гидрокси-камптотецин, TAS-103 (Taiho), топотекан, эльсамитруцин (Spectrum), дексразоксанет (TopoTarget), J-107088 (Merck and Co), пиксантрон (Novuspharma), BNP-1350 (BioNumerik), аналог ребеккамицина (Exelixis), CKD-602 (Chong Kun Dang), BBR-3576 (Novuspharma) или KW-2170 (Kyowa Hakko); противоопухолевые антибиотики, такие как, например, дактиномицин (актиномицин D), амонафид, доксорубицин (адриамицин), азонафид, деоксирубицин, антрапиразол, валрубицин, оксантразол, даунорубицин (дауномицин), лозоксантрон, эпирубицин, блеомицин, сульфат (бленоксан), терарубицин, блеомициновая кислота, идарубицин, блеомицин A, рубидазон, блеомицин B, пликамицин, митомицин C, порфиромицин, MEN-10755 (Menarini), цианоморфолинодоксорубицин, GPX-100 (Gem Pharmaceuticals) или митоксантрон (новантрон), антимитотические агенты, такие как, например, паклитаксел, SB 408075 (GlaxoSmithKline), доцетаксел, E7010 (Abbott), колхицины, PG-TXL (Cell Therapeutics), винбластин, IDN 5109 (Bayer), винкристин A, 105972 (Abbott), винорелбин, А 204197 (Abbott), виндезин, LU 223651 (BASF), доластатин 10 (NCI), D 24851 (ASTAMedica), ризоксин (Fujisawa), ER-86526 (Eisai), мивобулин (Warner-Lambert), комбретастатин A4 (BMS), цемадотин (BASF), изогомогалихондрин-B (PharmaMar), RPR 109881A (Aventis), ZD 6126 (AstraZeneca), TXD 258 (Aventis), PEG-паклитаксел (Enzon), эпотилон B (Novartis), AZ10992 (Asahi), T 900607 (Tularik), IDN-5109 (Indena), T 138067 (Tularik), AVLB (Prescient NeuroPharma), криптофицин 52 (Eli Lilly), азаэпотилон B (BMS), винфлунин (Fabre), BNP-7787 (BioNumerik), ауристатин PE (Teikoku Hormone), CA-4 пролекарство (OXiGENE), BMS 247550 (BMS), доластатин-10 (NIH), BMS 184476 (BMS), CA-4 (OXiGENE), BMS 188797 (BMS) или таксопрексин (Protarga); ингибиторы ароматазы, такие как, например, аминоглутетимид, эксеместан, летрозол, атаместан (BioMedicines), анастрозол YM-511 (Yamanouchi) или форместан; ингибиторы тимидилатсинтазы, такие как, например, пеметрексед (Eli Lilly), нолатрексед (Eximias), ZD-9331 (BTG) или CoFactor™ (BioKeys); антагонисты ДНК, такие как, например, трабектедин (PharmaMar), мафосфамид (Baxter International), глюфосфамид (Baxter International), апазиквон (Spectrum Pharmaceuticals), альбумин + 32P (Isotope Solutions), О6 бензилгуанин (Paligent), тимектацин (NewBiotics) или эдотреотид (Novartis); ингибиторы фарнезилтрансферазы, такие как, например, арглабин (NuOncology Labs), типифарниб (Johnson & Johnson), лонафарниб (Schering-Plough), периллиловый спирт (DOR BioPharma) или BAY-43-9006 (Bayer); ингибиторы насоса, такие как, например, CBT-1 (CBA Pharma), зосуквидар тригидрохлорид (Eli Lilly), тариквидар (Xenova), бирикодар дицитрат (Vertex) или MS-209 (Schering AG); ингибиторы гистон-ацетилтрансферазы, такие как, например, такединалин (Pfizer), пивалоилоксиметил бутират (Titan), SAHA (Aton Pharma), депсипептид (Fujisawa) или MS-275 (Schering AG); ингибиторы металлопротеиназы, такие как, например, Неовастат (Aeterna Laboratories), CMT-3 (CollaGenex), маримастат (British Biotech) или BMS-275291 (Celltech); ингибиторы рибонуклеозид-редуктазы, такие как, например, галлия мальтолат (Titan), тезацитабин (Aventis), триапин (Vion) или дидокс (Molecules for Health); агонисты/антагонисты TNF альфа, такие как, например, вирулизин (Lorus Therapeutics), ревимид (Celgene), CDC-394 (Celgene), энтанерцепт (Immunex Corp.), инфликсимаб (Centocor, Inc.) или адалимумаб (Abbott Laboratories); антагонисты рецептора эндотелина А, такие как, например, атрасентан (Abbott) YM-598 (Yamanouchi) или ZD-4054 (AstraZeneca); агонисты рецептора ретиноевой кислоты, такие как, например, фенретинид (Johnson & Johnson) алитретиноин (Ligand) или LGD-1550 (Ligand); иммуномодуляторы, такие как, например, терапия интерферон декзосома (Anosys), онкофаг (Antigenics), пентрикс (Australian Cancer Technology), GMK (Progenics), ISF-154 (Tragen), вакцина против аденокарциномы (Biomira), вакцина против рака (Intercell), CTP-37 (AVI BioPharma), норелин (Biostar), IRX-2 (Immuno-Rx), BLP-25 (Biomira), PEP-005 (Peplin Biotech), MGV (Progenics), вакцины синхровакс (CTL Immuno), бета-алетин (Dovetail), вакцина против меланомы (CTL Immuno), терапия CLL (Vasogen) или p21 RAS вакцина (GemVax); гормональные и антигормональные агенты, такие как, например, эстрогены, преднизон, конъюгированные эстрогены, метилпреднизолон, этинилэстрадиол, преднизолон, хлортрианизен, аминоглутетимид, иденестрол, лейпролид, гидроксипрогестерона капроат, гозерелин, медроксипрогестерон, лейпрорелин, тестостерон, бикалутамид, тестостерона пропионат, флуоксиместерон, флутамид, метилтестостерон, октреотид, диэтилстильбэстрол, нилутамид, мегестрол, митотан, тамоксифен, P-04 (Novogen), торемофин, 2-метоксиэстрадиол (EntreMed), дексаметазон или арзоксифен (Eli Lilly); фотодинамические агенты, такие как, например, талапорфин (Light Sciences), Pd-бактериофеофорбид (Yeda), Тералюкс (Theratechnologies), лютеция тексафирин (Pharmacyclics), мотексафин гадолиния (Pharmacyclics) или гиперицин; и ингибиторы тирозинкиназы, такие как, например, иматиниб (Novartis), кахалид F (PharmaMar), лефлуномид (Sugen/Pharmacia), CEP-701 (Cephalon), ZD1839 (AstraZeneca), CEP-751 (Cephalon), эрлотиниб (Oncogene Science), MLN518 (Millenium), канертиниб (Pfizer), PKC412 (Novartis), скваламин (Genaera), феноксодиол, SU5416 (Pharmacia), трастузумаб (Genentech), SU6668 (Pharmacia), C225 (ImClone), ZD4190 (AstraZeneca), ру-Маб (Genentech), ZD6474 (AstraZeneca), MDX-H210 (Medarex), ваталаниб (Novartis), 2C4 (Genentech), PKI166 (Novartis), MDX-447 (Medarex), GW2016 (GlaxoSmithKline), ABX-EGF (Abgenix), EKB-509 (Wyeth), IMC-1C11 (ImClone) или EKB-569 (Wyeth).
В дополнительном варианте осуществления дополнительный терапевтический агент не метаболизируется на более чем 90% с помощью Цитохрома P450 3A4 (CYP3A4).
Такие дополнительные агенты могут вводить отдельно от композиции, содержащей соединение формулы I, как часть режима многократной дозировки. Альтернативно, такие агенты могут быть частью единичной формы дозировки, смешанной вместе с соединением формулы I в единичной композиции. При введении как части режима многократной дозировки два активных агента могут быть представлены одновременно, последовательно или в пределах периода времени относительно друг друга, обычно в течение пяти часов относительно друг друга.
Количество обоих, соединения формулы I и дополнительного терапевтического агента (в таких композициях, которые содержат дополнительный терапевтический агент, как описано выше), которое могут объединить с материалами носителя для получения единичной формы дозировки, изменяется в зависимости от объекта, которого лечат, и определенного способа введения. Предпочтительно, композиции этого изобретения должны быть составлены так, что могут вводить дозировку соединения формулы I между 0,01-100 мг/кг массы тела/сутки. В одном примере композиции составлены так, что дозировка соединения формулы I может быть от 3 до 30 мг/кг массы тела/сутки. В другом примере композиции составлены так, что дозировка соединения формулы I может быть от 5 до 60 мг/кг массы тела/сутки.
В таких композициях, которые содержат дополнительный терапевтический агент, такой дополнительный терапевтический агент и соединение формулы I могут действовать синергетически. Следовательно, количество дополнительного терапевтического агента в таких композициях будет меньше, чем требуемое в монотерапии при использовании только такого терапевтического агента. В таких композициях могут вводить дозировку дополнительного терапевтического агента между 0,01-100 мг/кг массы тела/сутки.
Количество дополнительного терапевтического агента, присутствующего в композициях этого изобретения будет не более, чем количество, которое обычно вводили бы в композицию, содержащую такой терапевтический агент в качестве единственного активного агента. Предпочтительно количество дополнительного терапевтического агента в настоящее время в раскрытых композициях будет располагаться приблизительно от 50% до 100% количества, обычно присутствующего в композиции, содержащей такой агент в качестве единственного терапевтически активного агента.
Соединения формулы I или их фармацевтические композиции могут также быть включены в композиции для покрытия имплантируемых медицинских устройств, таких как протезы, искусственные клапаны, сосудистые трансплантаты, стенты и катетеры. Сосудистые стенты, например, использовали для предотвращения рестеноза (повторное сужение стенки сосуда после раны). Однако, пациенты, использующие стенты или другие имплантируемые устройства, рискуют образованием сгустков крови или активацией тромбоцитов. Эти нежелательные эффекты могут быть предотвращены или смягчены с помощью предварительного покрытия устройства фармацевтически приемлемой композиции, включающей ингибитор киназы. Подходящие покрытия и общий способ получения покрытых имплантируемых устройств описаны в Патентах США 6099562; 5886026; и 5304121. Покрытия представляют собой обычно биологически совместимые полимерные материалы, такие как гидрогелевый полимер, полиметилдисилоксан, поликапролактон, полиэтиленгликоль, полимолочная кислота, этиленвинилацетат и их смеси. Покрытия могут необязательно быть дополнительно покрыты с помощью подходящего верхнего слоя фторсиликона, полисахаридов, полиэтиленгликоля, фосфолипидов или их комбинаций для придания свойств контролируемого высвобождения композиции. Имплантируемые устройства, покрытые с помощью соединения формулы I, представляют собой другой вариант осуществления настоящего изобретения.
Получение соединений Формулы I
Чтобы изобретение, описанное в настоящей заявке, было более понятным, ниже представлены следующие примеры. Следует понимать, что эти примеры являются только иллюстративными и не должны быть рассмотрены как ограничение этого изобретения никаким способом.
Используемые в настоящем описании другие сокращения, символы и условные обозначения совместимы с используемыми в современной научной литературе. См., например, Janet S. Dodd, ed., The ACS Style Guide: A Manual for Authors and Editors, 2nd Ed., Washington, D. C: American Chemical Society, 1997, содержание которых включено в настоящее описание в полном объеме посредством ссылки. Следующие определения описывают термины и сокращения, используемые в настоящем описании:
Пример 1. Соединения формулы II
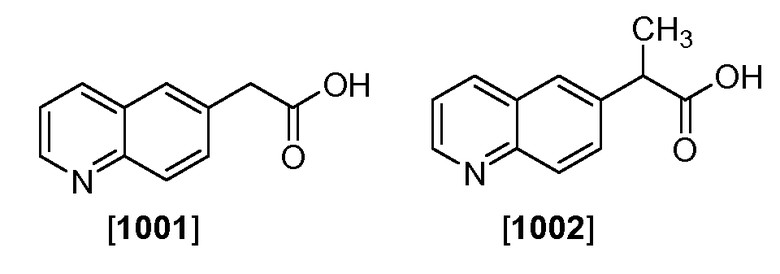
Соединения 1001 и 1002 получали от Okeanos Tech, Beijing, China (Каталожные номера OK-J-05024 и OK-J-05025, соответственно).
Соединение 1004 получали, как показано на Схеме 1. Соответственно, как показано в стадии 1-i, к суспензии NaH (60% в минеральном масле, 8,47 г, 212 ммоль) в ДМСО при 0°C (260 мл) медленно добавляли диэтил 2-метилмалонат (Соединение 1005, 29,5 г, 169,4 ммоль). Смесь перемешивали при 0°C в течение 2 часов, и добавляли 3,4,5-трифторнитробензол (25,0 г, 141,2 ммоль). Результирующую смесь нагревали до КТ, и перемешивали в течение 12 часов. Реакционную смесь выливали в насыщенный водный раствор NH4Cl, и осадок собирали с помощью фильтрации. После промывания водой 3 раза результирующий диэтил 2-(2,6-дифтор-4-нитрофенил)-2-метилмалонат (Соединение 1006 [R=CH3], 44,5 г, 95% выход) высушивали под пониженным давлением, и использовали в таком виде в следующей реакции.
Как показано в стадии 1-ii, к раствору диэтил 2-(2,6-дифтор-4-нитро-фенил)-2-метилмалоната (44,5 г, 135 ммоль) в MeOH добавляли Pd/C (10%, 4,0 г) под атмосферой азота. Атмосферу замещали H2 и смесь гидрировали при 50 psi в течение 3 дней. Атмосферу замещали азотом, смесь отфильтровывали через диатомовую землю, и летучие компоненты удаляли под пониженным давлением. Результирующий диэтил 2-(4-амино-2,6-дифторфенил)-2-метилмалонат (Соединение 1007 [R=CH3], 40,5 г, 99% выход) высушивали под пониженным давлением, и использовали в таком виде в следующей реакции.
Как показано в стадии 1-iii, к раствору диэтил 2-(4-амино-2,6-дифторфенил)-2-метилмалоната (40,0 г, 132,8 ммоль) в метаноле (200 мл) добавляли 6M NaOH (110,7 мл, 664,0 ммоль). Смесь нагревали при 100°C в течение 4 часов, охлаждали до 0°C, и окисляли конц. HCl до получения pH 3. Смесь нагревали до КТ, и перемешивали в течение 3 часов. Результирующий осадок собирали с помощью фильтрации, промывали водой и высушивали под высоким вакуумом при 50°C в течение 20 часов для получения 2-(4-амино-2,6-дифторфенил)пропионовой кислоты (Соединение 1008 [R=CH3], 22 г, 84% выход): 1H ЯМР (300,0 МГц, DMSO) δ 12,25 (уш.с, 1H), 6,16 (д, J=10,8 Гц, 2H), 5,58 (с, 2H), 3,74 (кв, J=7,2 Гц, 1H) и 1,28 (д, J=7,2 Гц, 3H) м.д.
Как показано в стадии 1-iv, смесь 2-(4-амино-2,6-дифторфенил)пропионовой кислоты (19,0 г, 94,45 ммоль), глицерина (35,83 г, 28,41 мл, 389,1 ммоль), нитробензола (7,209 г, 6,028 мл, 58,56 ммоль) и концентрированной серной кислоты (30,57 г, 16,61 мл, 311,7 ммоль) мягко нагревали. После прекращения исходной интенсивной реакции смесь нагревали до 170°C в течение 16 часов. После охлаждения летучие компоненты удаляли под пониженным давлением, осадок растворяли в MeOH (150 мл), добавляли 150 мл 6N NaOH и смесь нагревали при 110°C в течение 3 часов. После охлаждения до КТ смесь окисляли концентрированной HCl до pH 3. Результирующий темный осадок собирали с помощью фильтрации и промывали водой. Осадок поглощали в этаноле, и осторожно по каплям добавляли тионилхлорид (11,24 г, 6,891 мл, 94,45 ммоль). После окончания добавления смесь нагревали при 50°C в течение 20 часов. После охлаждения до КТ летучие компоненты удаляли под пониженным давлением, и осадок растворяли в смеси насыщ. NaHCO3 и ДХМ. Слои разделяли, и водный слой экстрагировали ДХМ. Объединенные органические слои высушивали над MgSO4, уменьшали в объеме под пониженным давлением и подвергали хроматографии на силикагеле среднего давления (0% EtOAc/гексан до 30% через 36 минут) для получения метил 2-(5,7-дифторхинолин-6-ил)пропионата (14,0 г, 56% выход за две стадии). Метиловый эфир (5,0 г) омыляли с помощью поглощения в метаноле (30 мл), обрабатывая результирующий раствор с помощью NaOH (16,58 мл 6 М, 99,50 ммоль) и перемешивая при КТ в течение 20 часов. После осторожного окисления с помощью конц. HCl до pH 2 результирующий осадок собирали с помощью фильтрации и высушивали под высоким вакуумом для получения 2-(5,7-дифторхинолин-6-ил)пропионовой кислоты, которую использовали в таком виде в последующих реакциях. Соединение 1003 могут получить в соответствии с такой же методикой, как используется при получении соединения 1004, заменяя диэтил 2-метилмалонат на диэтил малонат.
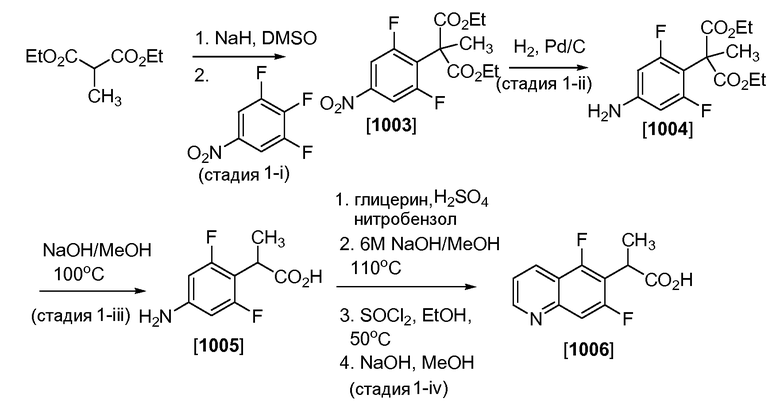
Схема 1
Пример 2. Получение соединений Формулы III
Соединения формулы III, в которой R2 и R3 представляют собой водород или метил, могут получить, как показано на Схеме 2. Соответственно, как показано в стадии 2-i Схемы 2, соответствующим образом замещенное хинолин уксусной кислотой соединение формулы II (248,5 ммоль, 1,0 эквивалент) и 1,3-диаминотиомочевину (273,4 ммоль, 1,1 эквивалента) суспендируют в смеси тетраметиленсульфона (сульфолан, 38 мл) и воды (57 мл). Метансульфоновую кислоту (546,7 ммоль, 2,2 эквивалента) добавляют к смеси, вследствие чего все твердые вещества растворяются. Реакционную смесь медленно нагревают до 90°C, и реакцию нагревают при 90°C в течение 40 часов. Реакционную смесь охлаждают на ледяной бане, и добавляют воду (75 мл) с последующим осторожным добавлением насыщенного раствора бикарбоната натрия (500 мл), пока не установится pH 8. Результирующий осадок собирают с помощью вакуумной фильтрации, промывают водой, насыщенным бикарбонатом натрия, водой и метил трет-бутиловым эфиром соответственно. Продукт высушивают в вакуумной печи при 55єC для получения соединения формулы III.
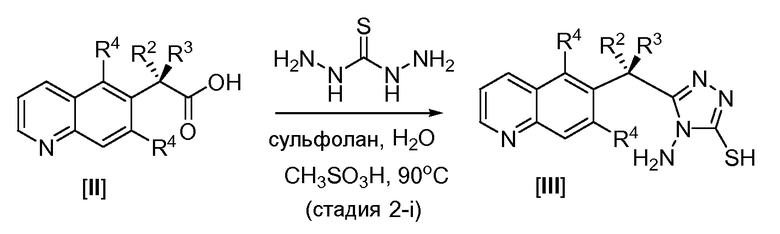
Схема 2
Пример 3. Получение 5-(дифтор(хинолин-6-ил)метил)-4-(иминотрифенилфосфорано)-4H-1,2,4-триазол-3-тиола (Соединение 1011)
Соединения Формулы III, в которой каждый из R2 и R3 представляет собой фтор, и R4 представляет собой водород, могут получить, как показано на Схеме 3. Соответственно, как показано в стадии 3-i, к смеси 6-иодхинолина (10,0 г, 39,21 ммоль, полученного от Ηangzhou Trylead Chemical Technology Co., Ltd., Китай) и меди (нанопорошок) (9,964 г, 156,8 ммоль) в ДМСО (150 мл) добавляли этил 2-бром-2,2-дифтор-ацетат (10,35 г, 50,97 ммоль). Смесь нагревали при 60°C в течение 6 часов, в течение этого времени смесь превратилась из красной медной суспензии в темно-красный почти гомогенный раствор. После охлаждения до комнатной температуры смесь разбавляли этилацетатом (300 мл) и водн. насыщенным раствором NH4Cl (450 мл). После перемешивания в течение 30 минут органический слой отделяли, промывали водой, промывали солевым раствором, и высушивали над сульфатом магния. Удаление летучих компонентов под пониженным давлением давало неочищенный продукт в виде красной жидкости. Очистка с помощью хроматографии на силикагеле среднего давления (ДХМ/этилацетат: 100% до 30% через 25 мин.) давала этил 2,2-дифтор-2-(хинолин-6-ил)ацетат (Соединение 1009, 51% выход): 1H ЯМР (300,0 МГц, CDCl3) δ 9,04-9,03 (м, 1H), 8,29-8,21 (м, 2H), 8,15 (с, 1H), 7,93 (дд, J=2,1, 8,9 Гц, 1H), 7,52 (кв, J=4,2 Гц, 1H), 4,35 (кв, J=7,1 Гц, 2H) и 1,34 (т, J=7,1 Гц, 3H) м.д.
Как показано в стадии 3-ii на Схеме 3, Соединение 1009 (10,0 г, 39,80 ммоль) растворяли в этаноле (100 мл), добавляли гидразин (7,65 г, 7,50 мл, 239 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 10 минут. После выливания смеси в раствор 2N HCl водную смесь промывали дважды ДХМ, и устанавливали pH 8, все время барботируя азот через раствор. Результирующий водный раствор тщательно экстрагировали ДХМ (10x) и объединенные органические слои высушивали над MgSO4, отфильтровывали и летучие вещества удаляли под пониженным давлением для получения 2,2-дифтор-2-(хинолин-6-ил)ацетогидразида в виде желтого твердого вещества (Соединение 1010, 91% выход). Это соединение использовали непосредственно без дополнительной очистки.
Как показано в стадии 3-iii Схемы 3, Соединение 1010 (3,55 г, 14,97 ммоль) в EtOH (71 мл) обрабатывали с помощью гидроксида калия (924 мг, 16,5 ммоль) и реакционную смесь мягко нагревали для достижения однородности. Добавляли дисульфид углерода (1,38 г, 1,09 мл, 18,2 ммоль) и смесь перемешивали при 90°C в течение 4 часов, в это время образовывался интермедиат соединения 5-(дифтор(хинолин-6-ил)метил)-1,3,4-оксадиазол-2-тиол натриевая соль. К раствору, нагревающемуся с обратным холодильником, добавляли гидразин (4,80 г, 4,70 мл, 150 ммоль) с последующим добавлением 3A молекулярных сит (3г). После нагревания с обратным холодильником в течение 2 часов сита удаляли с помощью фильтрации, и промывали EtOH. Объединенные органические слои охлаждали до 0°C на ледяной бане, и обрабатывали конц. HCl под атмосферой азота до установления pH 6,5. Осадок удаляли с помощью фильтрации, и фильтрат нагревали с обратным холодильником в течение 4 часов, используя ловушку Дина-Старка, для сбора любого избытка воды. Летучие компоненты удаляли под пониженным давлением, осадок поглощали водой, и устанавливали pH 6,5. Результирующее твердое вещество собирали с помощью фильтрации, промывали водой и высушивали для получения 5-(дифтор(хинолин-6-ил)метил)-4-амино-4H-1,2,4-триазол-3-тиола (Соединение 1011, 61% выход): 1H ЯМР (300,0 МГц, DMCO) δ 14,28 (с, 1H), 9,03-9,02 (м, 1H), 8,56 (д, J=8,0 Гц, 1H), 8,31 (с, 1H), 8,16 (д, J=8,8 Гц, 1H), 7,90 (дд, J=1,9, 8,8 Гц, 1H), 7,65 (кв, J=4,2 Гц, 1H) и 5,69 (с, 2H) м.д.
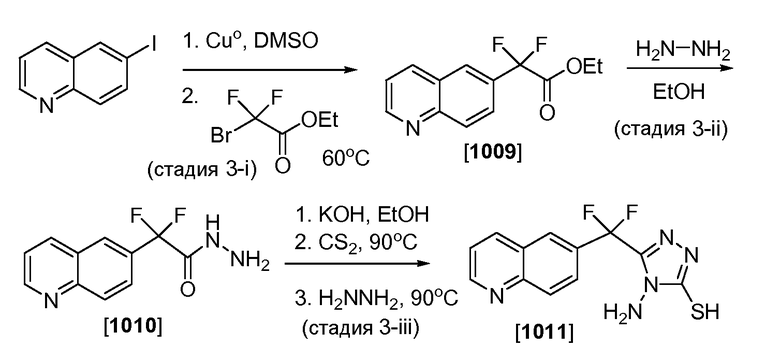
Схема 3
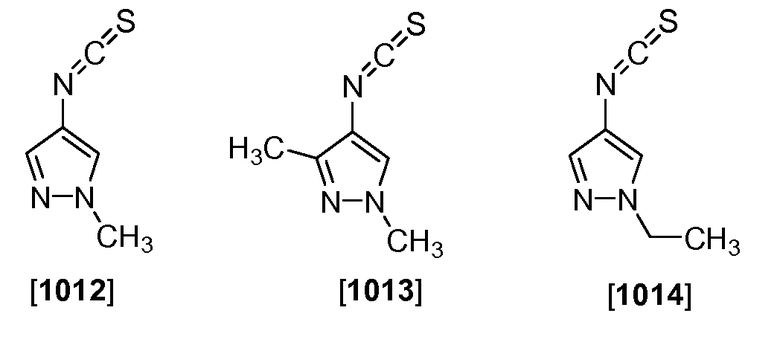
Пример 4. Соединения формулы IV
4-Изотиоцианато-1-метил-1Н-пиразол (Соединение 1012), 4-изотиоцианато-1,3-диметил-1Н-пиразол (Соединение 1013) и 1-этил-4-изотиоцианато-1Н-пиразол (Соединение 1014) получали из 1-метил-1Н-пиразол-4-амина, 1,3-диметил-1Н-пиразол-4-амина (от Matrix Chemical Co.) и 1-этил-1Н-пиразол-4-амина (от Oakwood Products), соответственно, с помощью взаимодействия пиразоламина с тиофосгеном при 0єC в присутствии пиридина.
Пример 5. Получение соединений Формулы I
Соединения формулы I могут получить, как показано на Схеме 5. Как показано в стадии 5-i Схемы 5, соединение формулы III (453,3 ммоль, 1,00 эквивалент) и соединение формулы IV нагревали вместе в пиридине при 110°C в течение 15 часов. После охлаждения до комнатной температуры реакционную смесь выливали в раствор 1 N HCl, осадок собирали с помощью фильтрации, промывали водой, и очищали с помощью хроматографии на силикагеле среднего давления. При желании рацемические смеси соединений могут разделить на их соответствующие энантиомеры с помощью сверхкритической флюидной хроматографии, используя колонку ChiralPak® AD-H (20 мм×250 мм, 5 микрон колонка) или колонку ChiralCel® OJ-H (20 мм×250 мм, 5 микрон колонка), элюируя соответствующим соотношением MeOH (0,1% DEA)/CO2 при соответствующей скорости потока.
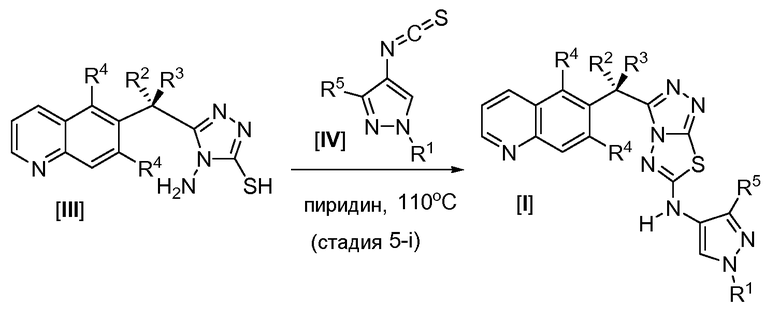
Схема 5
Пример 6. Альтернативное получение соединений Формулы I - получение 3-(дифтор(хинолин-6-ил)метил)-N-(1-метил-1H-пиразол-4-ил)-[1,2,4]триазоло[3,4-b][1,3,4]тиадиазол-6-амина (Соединение 3)
Соединения формулы I могут также получить с помощью взаимодействия 4-(иминотрифенилфосфорано)-4H-1,2,4-триазол-3-тиолов с изоцианатами. Соответственно, как показано в стадии 6-i Схемы 6, 6-иодхинолин (750 г, 2,94 моль) загружали в продутую сжатым азотом 22 л круглодонную колбу, оборудованную механической мешалкой, температурным датчиком, считывателем температуры, входным отверстием для азота и охлаждающей баней. Добавляли безводный ТГФ (5,25 л) и результирующий раствор охлаждали до -27°C, используя iPrOΗ/сухая ледяная баня. Добавляли i-PrMgCl∙LiCl (2,45 л, 1,3 М в ТГФ, 1,1 экв.) более 1 часа 17 минут с помощью капельной воронки, поддерживая температуру между -26°C и -29°C. Реакционную смесь затем перемешивали в течение 2,5 часов при температуре, поддерживаемой между -20°C и -29°C. Коричневую суспензию охлаждали до -53°C более 25 минут, используя i-PrOΗ/сухая ледяная баня, и добавляли диэтилоксалат (469 г, 0,44 л, 1,1 экв.) более 1 часа 15 минут с помощью капельной воронки, поддерживая температуру между -51°C и -53°C. Результирующий темный раствор нагревали до КТ в течение ночи (~18 часов) до получения суспензии горчичного цвета. Получали раствор хлорида аммония (500 г, 9,35 моль, 3,18 экв.) в воде (4,5 л) и охлаждали до 10°C, используя ледяную баню. Реакционную смесь переносили в раствор хлорида аммония более 37 минут с помощью линии перетока, поддерживая небольшой вакуум в 22 л колбе, содержащей перемешивающийся раствор хлорида аммония. После окончания переноса ледяную баню удаляли, добавляли EtOAc (3,75 л) и начинали перемешивание. После приблизительно 15 минут перемешивание останавливали и слои разделяли. Водную фазу (pH 8) экстрагировали EtOAc (3,75 л). Два органических слоя объединяли, и промывали раствором NaCl (112 г в 2,5 л воды). Органическую фазу концентрировали под вакуумом при 25°C для получения маслянистого вещества (763 г), которое очищали с помощью хроматографии на силикагеле (7:1 до 1:1 гексан/EtOAc). Фракции, содержащие чистый продукт, объединяли и концентрировали под вакуумом с получением этил 2-оксо-2-(хинолин-6-ил)ацетата в виде коричневого маслянистого вещества (Соединение 1015, 503 г, выход 74,5%): 1H ЯМР (500 МГц, DMCO-d6) δ 1,40 (т, 3H), 4,51 (кв, 2H), 7,71 (дд, 1H), 8,21 (д, 1H), 8,24 (дд, 1H), 8,68 (дд,lH), 8,77 (дд, 1H), 9,11 (дд, 1H).
Как показано в стадии 6-ii Схемы 6, соединение 1015 (282 г, 1,230 моль) и ДХМ (2,82 л) объединяли в продутой сжатым азотом 12 л круглодонной колбе, оборудованной механической мешалкой, входным отверстием для азота, температурным датчиком и водяной баней комнатной температуры. К результирующему раствору добавляли бис-(2-метоксиэтил)аминосеры трифторид (DeoxoFluor™, 615 г, 0,50 л, 2,26 экв.) более 45 минут с помощью капельной воронки. Добавляли абсолютный EtOH (12,8 г, 15 мл, 0,21 экв.) с помощью шприца частями более 3 минут и реакцию перемешивали в течение ночи при температуре окружающей среды. Обрабатываемые образцы забирали, обрабатывали, и анализировали с помощью 1H-ЯМР для контролирования протекания реакции. Типичное молярное отношение исходного материала к продукту после первого добавления этанола составляло 2:3. Соответственно, дополнительные порции EtOH (12,3 г, 0,2 экв.) последовательно добавляли с помощью шприца с периодами 10-20 часов между добавлениями, пока наблюдаемое содержание исходного материала не было ниже, чем 10%. Гашеный раствор получали с помощью смешивания бикарбоната натрия (827 г, 8 эквив.) в воде (8,3 л) и охлаждения до 13°C на ледяной бане. Реакционную смесь переносили в гашеный раствор бикарбоната натрия более 0,5 часа с помощью линии перетока, поддерживая вакуум в 22 л колбе, содержащей перемешивающийся раствор бикарбоната натрия. Наблюдали интенсивное выделение газа. Температуру поддерживали между 10°C-13°C в течение гашения, после этого ледяную баню удаляли, и смесь перемешивали в течение 2 часов при 12°C-15°C. Слой ДХМ отделяли, и водный слой экстрагировали ДХМ (2×1 л). Слои ДХМ объединяли и концентрировали при 26°C под вакуумом для получения 349 г неочищенного маслянистого вещества, которое очищали с помощью хроматографии на силикагеле (7:1 до 4:1 гексан/EtOAc). Фракции, содержащие чистый продукт, объединяли, и концентрировали с получением маслянистого вещества, которое поглощали в 2×180 мл абс. EtOH и концентрировали с помощью роторного испарения для получения этил 2,2-дифтор-2-(хинолин-6-ил)ацетата в виде маслянистого вещества (Соединение 1009, 164 г, 53% выход): 1H ЯМР (500 МГц, DMCO-d6) δ 1,24 (т, 3H), 4,35 (кв, 2H), 7,67 (дд, 1H), 7,91 (дд, 1H), 8,20 (д, 1H), 8,37 (с,lH), 8,60 (д,lH), 9,05 (дд, lH); 19F ЯМР (470 МГц, DMCO-d6) δ-101,2.
Как показано в стадии 6-iii Схемы 6, в 1 л круглодонную колбу, оборудованную магнитной мешалкой и термопарой, добавляли соединение 1009 (164 г, 633,9 ммоль) и EtOH (398 мл). Желтый раствор охлаждали до 0°C, используя ледяную/водяную баню. Гидроксид натрия (570,5 мл 2 М водного раствора, 1,141 моль) медленно добавляли более 1 часа к реакционной смеси, в то же время, поддерживая внутреннюю температуру ниже 20°C. Ледяную/водяную баню удаляли, и смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали под вакуумом и желтое твердое вещество высушивали в вакуумной печи (50°C, 20-25 мм Hg, очистка N2) с получением натрий 2,2-дифтор-2-(хинолин-6-ил)ацетата (Соединение 1016, 156,0 г, 99% выход): 1H ЯМР (500 МГц, DMCO-d6) δ 7,50-7,55 (дд, 1H), 7,90-7,85 (дд, 1H), 8,10-8,15 (д, 1H), 8,10 (с, 1H), 8,40-8,45 (д, 1H), 8,95-8,90 (дд, 1H); 19F ЯМР (470 МГц, DMCO-d6) δ-98,15.
Как показано в стадии 6-iv Схемы 6, в 3 л круглодонную колбу, оборудованную нагревающейся опорой, конденсатором обратного холодильника, термопарой, механической мешалкой и продутую N2, добавляли Соединение 1016 (98,6 г, 326,4 ммоль), 1,3-диметил-2-имидазолидинон (1,607 л) и пиридин (38,73 г, 39,60 мл, 489,6 ммоль). Добавляли 50% ангидрид пропанфосфоновой кислоты (T3P®) в 2-метилтетрагидрофуране (415,4 г, 652,8 ммоль) в единичной порции и наблюдали экзотермический эффект при 15-20°C. Реакционную смесь нагревали до 70°C в течение 1 часа, в это время добавляли тиокарбогидразин (53,03 г, 489,6 ммоль) в одной порции. Реакционную смесь перемешивали в течение дополнительных 3 часов и затем добавляли дополнительную порцию 50% T3P в 2-МеТГФ (207,7 г, 326,4 ммоль) с последующим перемешиванием при 70°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры. В отдельной колбе охлаждали раствор бикарбоната натрия (219,3 г, 2,611 моль) в воде (2,41 л), используя ледяную/водяную баню. Реакционную смесь медленно добавляли к гашеному раствору с помощью полой иглы более 45 минут, в течение которых наблюдали пенообразование и осаждение продукта. Раствор перемешивали при 5°C в течение дополнительного часа при pH=7. Результирующие твердые вещества собирали с помощью фильтрации с отсасыванием, и отфильтрованный осадок промывали водой (3,2 л) и МТБЭ (3,2 л). Белое твердое вещество высушивали в вакуумной печи (50°C, 20-25 мм Hg) с получением 4-амино-5-(дифтор(хинолин-6-ил)метил)-4H-1,2,4-триазол-3-тиола (Соединение 1011, 57 г, 58%): 1H ЯМР (500 МГц, DMCO-d6) δ 5,70-5,65 (с, 2H), 7,50-7,55 (дд, 1H), 7,90-7,85 (дд, 1H), 8,10-8,15 (д, 1H), 8,10 (с, 1H), 8,40-8,45 (д, 1H), 8,95-8,90 (дд, 1H), 14,3-14,25 (с, 1H); 19F ЯМР (470 МГц, DMCO-d6) δ-92,50.
Как показано в стадии 6-v Схемы 6, трифенилфосфин (17,66 г, 67,35 ммоль), 1,1,1,2,2,2-гексахлорэтан (15,94 г, 67,35 ммоль), Соединение 1011 (13,37 г, 44,90 ммоль) объединяли под атмосферой азота в 500 мл круглодонной колбе, оборудованной механической мешалкой, термопарой, в атмосфере азота. К перемешиваемой смеси добавляли безводный ацетонитрил (461,0 мл) с последующим добавлением Et3N (14,09 г, 19,41 мл, 139,2 ммоль), в то же время, поддерживая температуру между 21,4-25,1°C. Реакционная смесь становилась прозрачным раствором и затем становилась суспензией, как только образовывался продукт (в течение приблизительно 2 минут). Затем добавляли воду (808,9 мг, 808,9 мкл, 44,90 ммоль) с последующим добавлением MeOH (14,39 г, 18,19 мл, 449,0 ммоль) и реакцию затем перемешивали в течение дополнительных 45 мин. Твердое вещество собирали с помощью фильтрации, и отфильтрованный осадок промывали CH3CN (132 мл). Отфильтрованный осадок высушивали в вакуумной печи при 45°C с потоком азота для получения 5-(дифтор(хинолин-6-ил)метил)-4-(иминотрифенилфосфорано)-4H-1,2,4-триазол-3-тиола (Соединение 1017, 25,57 г, 98,8% выход) в виде бежевого твердого вещества: 1H ЯМР (400 МГц, DMCO-d6) δ 7,51-7,42 (м, 6H), 7,70-7,56 (м, 12H), 8,11 (д, 1H), 8,16 (м, 1H), 8,49 (дд, 1H), 9,03 (дд, 1H), 13,64 (уш. с, 1H); 19F ЯМР(376 МГц, DMCO-d6) δ-91,77; 31P ЯМР (162 МГц, DMCO-d6) δ 19,71.
Как показано в стадии 6-vi Схемы 6, в 2 л 4-горлую круглодонную колбу, оборудованную верхнеприводной мешалкой, термопарой, конденсатором обратного холодильника и барботажным устройством для азота, добавляли 1-метилпиразол-4-карбоновую кислоту (27,33 г, 216,7 ммоль). Толуол (600 мл) и триэтиламин (30,70 г, 42,29 мл, 303,4 ммоль) добавляли при 20,1°C без наблюдаемого увеличения температуры. Результирующая белая суспензия становилась бесцветным раствором после нагревания до 103°C. Азид дифенилфосфорила (DPPA, 61,48 г, 48,14 мл, 216,7 ммоль) добавляли в течение периода 30 минут, поддерживая температуру между 103,1 и 107°C. Нагревание прекращали и охлаждали до комнатной температуры. Результирующий 4-изоцианато-1-метил-1Н-пиразол не выделяли, и вместо этого к нему добавляли Соединение 1017 (120 г, 216,7 ммоль) за одну порцию при комнатной температуре. Аналитический анализ ВЭЖХ сразу после добавления показал 51,2% превращение исходного материала в Соединение 3. Исходя из 216,7 ммоль 1-метилпиразол-4-карбоновой кислоты, дополнительно 4-изоцианато-1-метил-1Н-пиразол получали в отдельной колбе, как показано выше. После охлаждения до комнатной температуры эту реакционную смесь переносили в первую реакционную смесь с помощью полой иглы. Анализ ВЭЖХ указал 100% превращение после добавления. К реакционной смеси добавляли EtOAc (240 мл) и образовывался белый осадок. Реакцию перемешивали в течение 30 минут, и твердое вещество собирали с помощью фильтрации с отсасыванием. Отфильтрованный осадок [(содержащий 1-(3-(дифтор(хинолин-6-ил)метил)-[1,2,4]триазоло[3,4-b][1,3,4]тиадиазол-6-ил)-1,3-бис(1-метил-1H-пиразол-4-ил)мочевину (Соединение 1018) в качестве побочного продукта] промывали EtOAc (600,0 мл). Фильтрат концентрировали под вакуумом с помощью роторного испарителя при 35°C с получением 272,4 г коричневого маслянистого вещества. Маслянистое вещество высушивали под высоким вакуумом и очищали с помощью колоночной хроматографии, используя соотношение 8:1 SiO2 к неочищенному маслянистому веществу и элюируя градиентом от 1% до 5% EtOH в ДХМ для получения 3-(дифтор(хинолин-6-ил)метил)-N-(1-метил-1H-пиразол-4-ил)-[1,2,4]триазоло[3,4-b][1,3,4]тиадиазол-6-амина (Соединение 3, 111 г), которое дополнительно очищали с помощью кристаллизации. Соответственно, порцию 33,5 г этого материала переносили в 250 мл 3-горлую круглодонную колбу, оборудованную механической мешалкой и барботажным устройством для азота. Твердое вещество было оранжево-коричневого цвета. В общей сложности 135 мл CΗ3CN добавляли для получения сгущенного остатка. После 2,5 часов твердое вещество собирали с помощью фильтрации с отсасыванием после 3 часов. Влажный отфильтрованный осадок промывали CH3CN (67 мл) с получением 13,9 г влажного твердого вещества. Сушку под вакуумом проводили (43°C, 20-25 мм Hg, очистка N2) более чем 15,5 часов с получением 10,25 г чистого Соединения 3 (>99,9% чистота с помощью анализа ВЭЖХ, <0,1% PPh3O). Фильтрат MeCN обрабатывали равным количеством воды. Осажденное твердое вещество и суспензию перемешивали в течение 2 часов. Твердое вещество собирали с помощью фильтрации с отсасыванием. Влажный отфильтрованный осадок промывали 35 мл воды. Отфильтрованный осадок высушивали (43°C, 20-25 мм Hg, очистка N2) с получением 9,5 г твердого материала, который обрабатывали CH3CN, как указано выше, для получения дополнительных 4,78 г чистого Соединения 3 (общее количество=15,03 г, 57,7% общий выход из Соединения 1017). Выход могут увеличить дополнительно с помощью аминолизирования побочного продукта мочевины (Соединение 1018) NH3/MeOH для восстановления дополнительного Соединения 3.
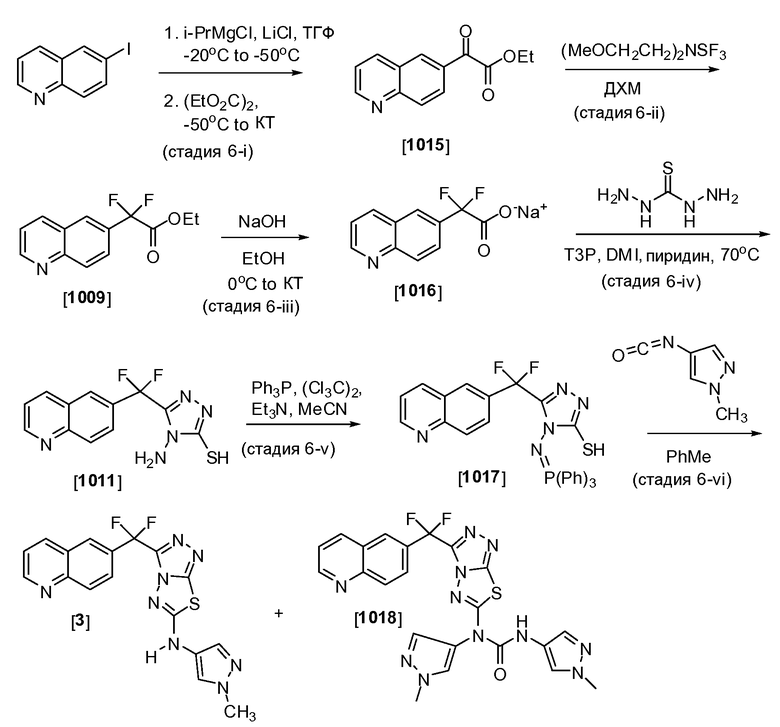
Схема 6
Аналитические данные для Соединений 1-8 показаны в Таблице 1.
Физические Характеристики Соединений Формулы I
Биологическое исследование Соединений Формулы I
Пример 3. Исследование ингибирования киназы c-Met
Соединения изобретения подвергали скринингу на их способность ингибировать киназу c-Met, используя стандартный радиометрический анализ. Кратко, в этом анализе киназы детально исследовали перенос конечного 33P-фосфата в 33P-АТФ к субстрату polyE4Y. Анализ проводили в 96-луночных планшетах до конечного объема 100 мкл в лунке, содержащей 0,5 нМ c-Met, 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 0,01% БСА, 1 мМ DTT, 0,5 мг/мл polyE4Y и 35 мкМ АТФ. Соответственно, соединения изобретения растворяли в ДМСО для создания 10 мМ исходных базовых растворов. Затем проводили последовательные разбавления в ДМСО для получения конечных растворов для исследования. В каждую лунку добавляли аликвоту 1,5 мкл ДМСО или ингибитора в ДМСО с последующим добавлением 33P-АТФ и окончательно добавлением c-Met и polyE4Y (полученный от Sigma). После 20 минут реакцию гасили 50 мкл 30% трихлоруксусной кислоты (ТХУК), содержащей 4 мМ АТФ. Реакционную смесь переносили на 0,66 мм GF пластины фильтра (Corning) и промывали три раза 5% ТХУК. После добавления 50 мкл Ultimate Gold™ высокоэффективного сцинтиллятора (Packard Bioscience) образцы подсчитывали на Packard TopCount NXT сцинтилляционных микропластинах и измерителе люминесценции (Packard BioScience). Величины Ki вычисляли, используя макрос Microsoft Excel Solver, для соответствия данным кинетической модели для конкурентного ингибирования прочного связывания. Каждое из Соединений 1-8 имело величину Ki для ингибирования c-Met менее чем 200 нм.
Пример 4. Ингибирование активности c-Met в клетках карциномы желудка Snu5
Соединения формулы I также подвергали скринингу на их способность ингибировать люцефераза-индуцированный сигнал в созданной линии клеток Snu5. Snu5 [полученный от American Type Culture Collection (Каталожный номер CRL-5973)] представляет собой карциному желудка человека, известную для сверхэкспрессии c-Met, которая является конститутивно активной. Клеточную линию трансфектировали ретровирусом, pCLPCX, который содержит генетическую конструкцию, состоящую из 6xAPl элементов отклика промотора и гена люциферазы, содержащего C-конечную PEST последовательность (протеолитический сигнал от мышиной орнитиндекарбоксилазы, которая сокращает половину жизни люциферазы). Конститутивно активная c-Met активирует клеточные пути (преимущественно MAP киназы), приводя к АР-1-индуцированной транскрипции люциферазы-PEST и трансляции в конечный продукт, активность которого является измеримой как хемилюминесцентное считывание после добавления люциферина (Steady-Glo от Promega). Остаточную люминесценцию строго коррелируют с ингибированием c-Met. Стабильную клеточную линию получали с помощью выбора новой клеточной линии (Snu5-AP1-Luc-Pest) с пуромицином. Клетки выращивали в полных средах [среда Исков (Invitrogen), содержащая 10% эмбриональную бычью сыворотку (FBS, Hyclone) и пенициллин/гентамицин (Invitrogen)]. Соединения изобретения растворяли в ДМСО для создания 10 мМ исходных базовых растворов. Затем проводили последовательные разбавления в ДМСО и переносили в полную среду для создания 10x раствора. Клетки Snu5-AP1-Luc-Pest подсчитывали, и разбавляли до 200000-клетки/мл раствора. Клетки (90 мкл) добавляли в каждую лунку 96-луночного черного планшета с прозрачным дном (Costar). Затем 10 мкл 10x раствора соединения добавляли к клеткам в три повтора. Планшеты инкубировали в инкубаторе 37°C/5% CO2. После 6 часов в каждую лунку добавляли 50 мкл реагента Steady-Glo (Promega) и помещали на шейкер для планшетов в течение 5 минут для гарантии того, что клетки были полностью лизированы. Планшет прочитывали на 1450 Microbeta жидкостно-сцинтилляционном и люминесцентном измерителе (Perkin-Elmer). Каждое из Соединений 1-8 имело величину IC50 для ингибирования активности c-Met в клетках карциномы желудка Snu5 менее чем 200 нм.
Пример 5. Ингибирование роста опухоли на модели мыши
Соединение 3 исследовали на его способность ингибировать рост опухоли подкожно имплантированных клеток рака желудка SNU-5 у мышей с тяжелым комбинированным иммунодефицитом (ТКИД). Клетки SNU-5 (CRL-5973, American Type Culture Collection, Manassas, VA) культурировали в среде Дульбекко, модифицированной по способу Исков (IMDM) (Invitrogen, Carlsbad, CA) дополненной 10% эмбриональной бычьей сывороткой (FBS) (Hyclone, Logan, UT), 100 единиц/мл пенициллина, 100 мг/мл стрептомицина (Invitrogen, Carlsbad, CA) и 2 мМ L-глутамина. Клетки культурировали в течение менее 4 циклов обработки до имплантации. Мышам-самкам с ТКИД (Fox Chase SCID, CB-17, мыши, весящие 17-19 г, полученные от Charles River Laboratories, Wilmington, MA) вводили подкожно (п.к.) 5×106 клеток SNU-5 в правую спинную подмышечную область на 0 День. Обработки начинали проводить на 25 День, когда средний объем опухоли достигал приблизительно 358 мм3.
Соединение 3, полученное в наполнителе, содержащем 30% (вес./об.) пропиленгликоля и 10% Солютола (Sigma-Aldrich, St Louis, MO) в виде суспендированной гомогенной формы, вводили перорально (п.o) один раз в день (QD) в полных дневных дозах 3, 10 и 30 мг/кг/сутки в течение 14 дней. Объемы опухоли (вычисленные, используя эллиптическую формулу (длина×ширина2)/2, где длина и ширина представляли собой наибольшие и наименьшие измерения опухоли, соответственно) регистрировали в течение двух недель после начала лечения. Исследование завершали спустя 38 дней после имплантации опухоли. Средние объемы опухоли представлены в Таблице 2. Веса опухоли при завершении исследования представлены в Таблице 3.
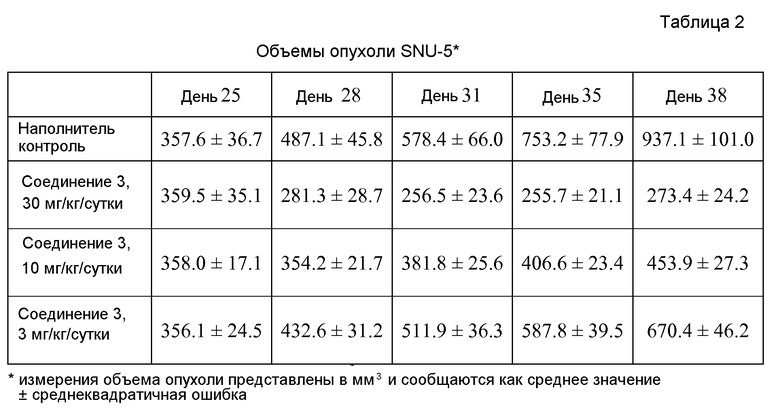
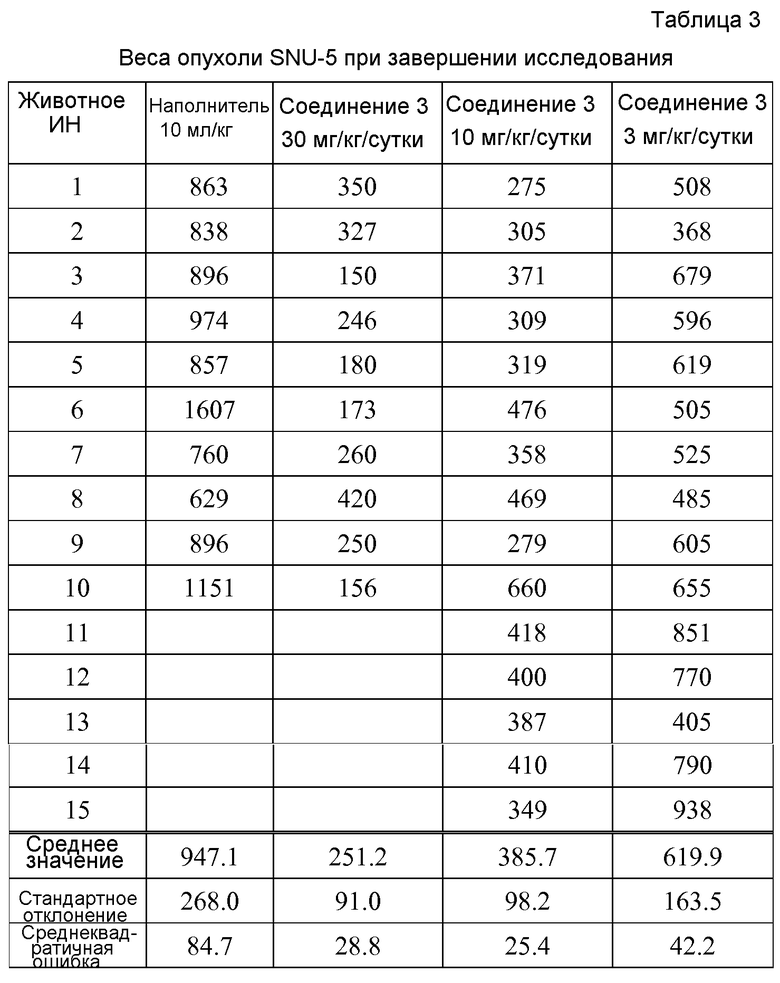
В Таблицах 2 и 3 показано, что Соединение 3 продемонстрировало значительную и дозозависимую противоопухолевую активность при всех трех исследуемых уровнях дозы. Доза 30 мг/кг/сутки приводила к регрессу опухоли -23,9% (P <0,001) с помощью анализа объема опухоли. Опухоли, полученные из групп лечения 3, 10 и 30 мг/кг/сутки VRT-846198, были значительно меньше, чем полученные от контрольной группы наполнителя, с процентными сокращениями веса 34,5%, 59,3% и 73,5% соответственно (все P<0,002).
Пример 6. Ингибирование метастаза опухоли на модели мыши
Соединение 3 исследовали на его способность ингибировать метастаз подкожно имплантированных опухолей легких у мышей с тяжелым комбинированным иммунодефицитом (ТКИД). Соответственно, клетки A549 (A549HGF-lm1115, трансфицированные фактором роста гепатоцитов, люциферазой и зеленым флуоресцентным белком) культурировали в среде RPMI 1640 (Invitrogen, Carlsbad, CA), дополненной 10% эмбриональной бычьей сывороткой (FBS) (Hyclone, Logan, UT), 100 единиц/мл пенициллина, 100 мг/мл стрептомицина (Invitrogen, Carlsbad, CA) и 2 мМ L-глутамина, в течение менее чем 4 циклов обработки до имплантации. 3-(Дифтор(хинолин-6-ил)метил)-N-(1-метил-1H-пиразол-4-ил)-[1,2,4]триазоло[3,4-b][1,3,4]тиадиазол-6-амин (Соединение 3) получали в наполнителе, содержащем 0,5% (вес./об.) метилцеллюлозы (Sigma-Aldrich, St Louis, MO) и 0,1% (об./об.) Tween 80™, в виде растворенной гомогенной формы, которую готовили свежей каждый день и вводили мышам с помощью перорального введения питания в объеме дозирования 10 мл/кг.
Мышам-самкам с ТКИД вводили подкожно (п.к.) 5×106 клеток A549HGF в правую спинную подмышечную область на 0 День. Лечение начинали в тот же самый день с помощью перорального введения (п.o.) Соединения 3 один раз в день (QD) в полных ежедневных дозах 30 и 60 мг/кг/сутки в течение 22 дней. Измерения эктопической опухоли регистрировали два раза в неделю в течение 3 недель после начала лечения. Было обнаружено, что Соединение 3 не приводило ни к какому, значительному изменению в первичном росте клеток опухоли A549 на участке имплантации для мышей, дозируемых 30 или 60 мг/кг/сутки, относительно роста клеток опухоли у мышей, дозируемых отдельно наполнителем.
Для оценки антиметастатического потенциала Соединения 3 при завершении исследования все ткани легкого животных собирали и лизировали с помощью гомогенизации для количественного определения ex vivo с помощью люминесценции люциферазы. Таблица 4 иллюстрирует содержание клеток опухоли в ткани легкого при завершении исследования, и данные там указывают, что Соединение 3 значительно ингибирует образование метастазов легкого у мышей, обработанных Соединением 3 при 60 мг/кг/сутки (средний флуоресцентный показатель 6672,3±1986,1 SEM) по сравнению с контролем наполнителем (средний флуоресцентный показатель 23531,5±8278,2 SEM, p<0,02).
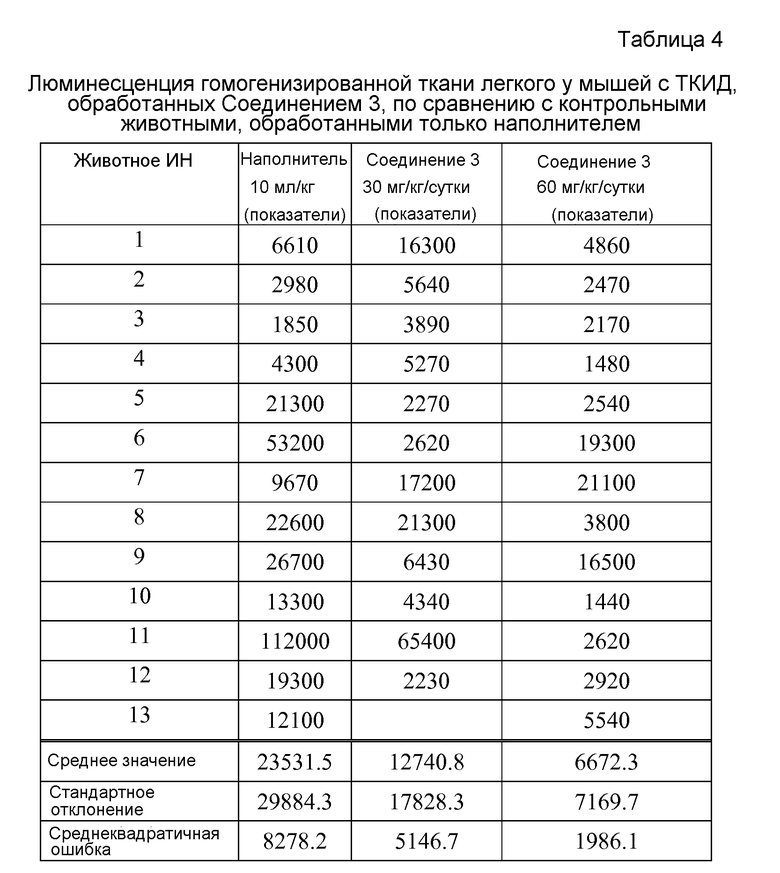
Все публикации и патенты, процитированные в этой спецификации, включены в настоящее описание посредством ссылки, как будто каждая индивидуальная публикация или патент были определенно и индивидуально показаны, чтобы быть включенными в настоящее описание посредством ссылки. Несмотря на то, что вышеупомянутое изобретение было описано в некоторых деталях посредством иллюстрации и примера в целях ясности или понимания, специалисту в данной области техники будет ясно очевидно в свете изучения этого изобретения, что определенные изменения и модификации могут проводить, не отступая от сущности или объема прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ СПИРО[ИЗОХИНОЛИН-4(1Н),3-ПИРРОЛИДИН]-1,2',3,5'[2Н]ТЕТРОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДОТВРАЩЕНИЯ ИЛИ ЛЕЧЕНИЯ ОСЛОЖНЕНИЙ, СВЯЗАННЫХ С САХАРНЫМ ДИАБЕТОМ | 1991 |
|
RU2110518C1 |
| СОЕДИНЕНИЯ АЗАЛАКТАМА В КАЧЕСТВЕ ИНГИБИТОРОВ HPK1 | 2021 |
|
RU2819642C1 |
| 1Н-ПИРАЗОЛО[3,4-B]ПИРИДИНЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2013 |
|
RU2689141C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЗОКСАЗОЛСУЛЬФОНАМИДА | 2020 |
|
RU2813356C2 |
| ИМИДАЗОПИРИМИДИНЫ КАК ИНГИБИТОРЫ EED И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2836176C2 |
| НОВОЕ ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ, ОБЛАДАЮЩЕЕ ЭФФЕКТОМ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2744168C1 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2838028C2 |
| ГЕТЕРОАРИЛ-БИФЕНИЛ АМИНЫ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2837843C1 |
| 2,5-ДИЗАМЕЩЕННЫЕ АРИЛСУЛЬФОНАМИДНЫЕ АНТАГОНИСТЫ CCR3 | 2010 |
|
RU2532515C2 |
| ПИРИДИЛОКСИ ПРОИЗВОДНЫЕ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АКТИВАТОРА/МОДУЛЯТОРА ГАММА-РЕЦЕПТОРА, АКТИВИРУЕМОГО ПРОЛИФЕРАТОРОМ ПЕРОКСИСОМ (PPAR) ГАММА | 2010 |
|
RU2480463C1 |
Изобретение относится к соединениям формулы (I), в которой R1 представляет собой метил или этил, R2 представляет собой водород, фтор или метил, R3 представляет собой водород, фтор или метил, каждый R4 представляет собой водород и R5 представляет собой водород, метил. Также изобретение относится к фармацевтической композиции для лечения или уменьшения степени тяжести пролиферативного нарушения на основе соединений формулы (I). Технический результат - соединения полезны при получении лекарственного средства для лечения или уменьшения степени тяжести метастатического рака, глиобластомы, карциномы желудка или рака, выбранного из рака толстой кишки, груди, простаты, мозга, печени, поджелудочной железы или легкого и гепатоцеллюлярной карциномы. 3 н. и 13 з.п. ф-лы, 4 табл., 6 пр.
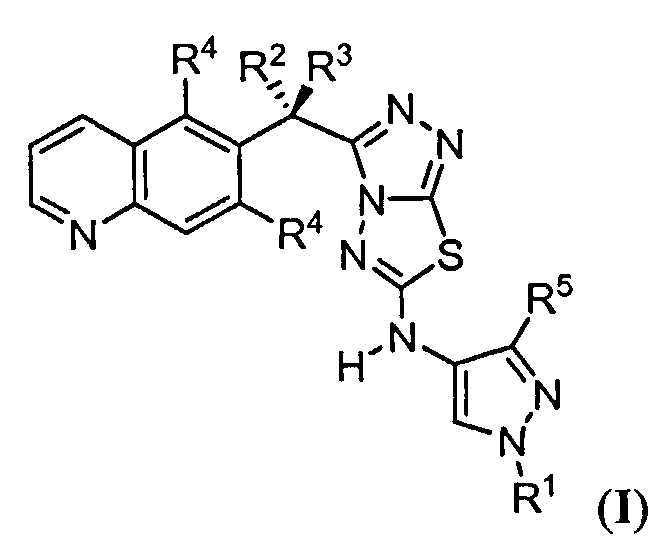
1. Соединение, имеющее формулу:
,
в которой
R1 представляет собой метил или этил;
R2 представляет собой водород, фтор или метил;
R3 представляет собой водород, фтор или метил;
каждый R4 представляет собой независимо водород; и
R5 представляет собой водород, метил.
2. Соединение по п.1, в котором R2 представляет собой метил и R3 представляет собой водород.
3. Соединение по п.2, в котором R1 представляет собой метил и R5 представляет собой водород.
4. Соединение по п.1, в котором R2 представляет собой водород и R3 представляет собой метил.
5. Соединение по п.4, в котором R1 представляет собой метил и R5 представляет собой водород.
6. Соединение по п.1, в котором каждый из R2 и R3 представляет собой фтор.
7. Соединение по п.6, в котором R1 представляет собой метил и R5 представляет собой водород.
8. Соединение по любому одному из пп.1-7, в котором R4 представляет собой водород.
9. Соединение по п.1, имеющее следующую структуру:
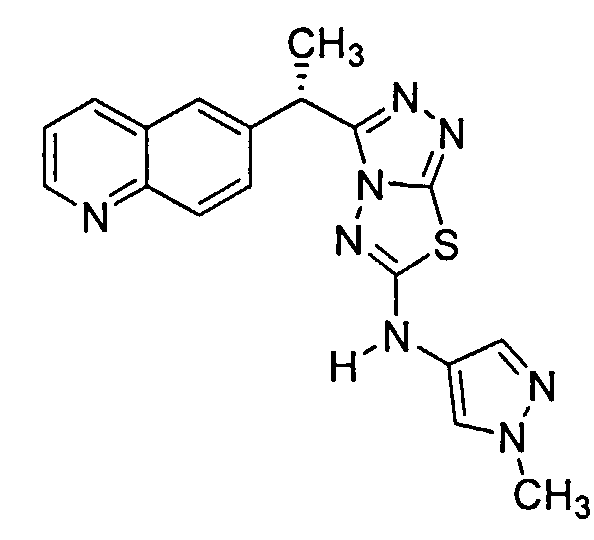 ,
,  ,
, 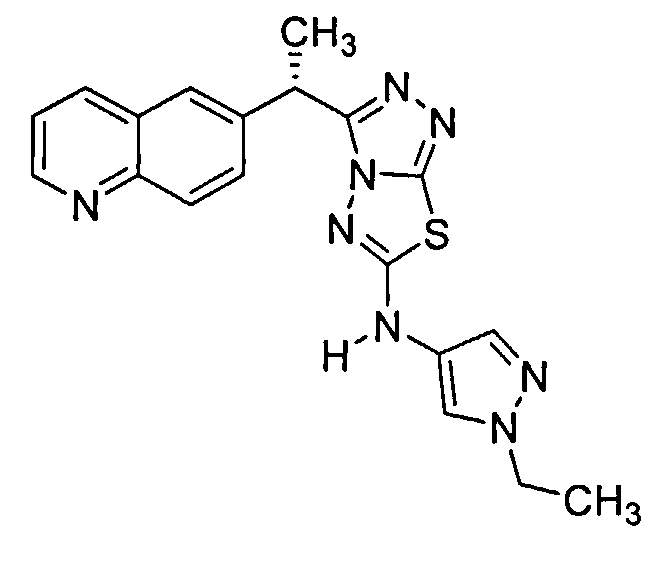 ,
,
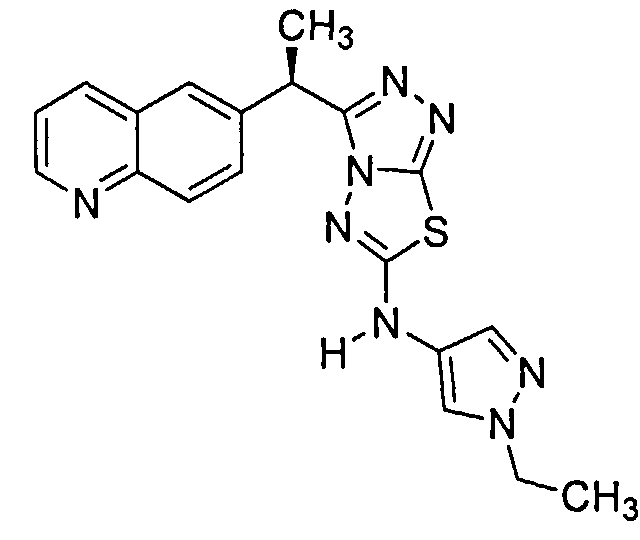 ,
, 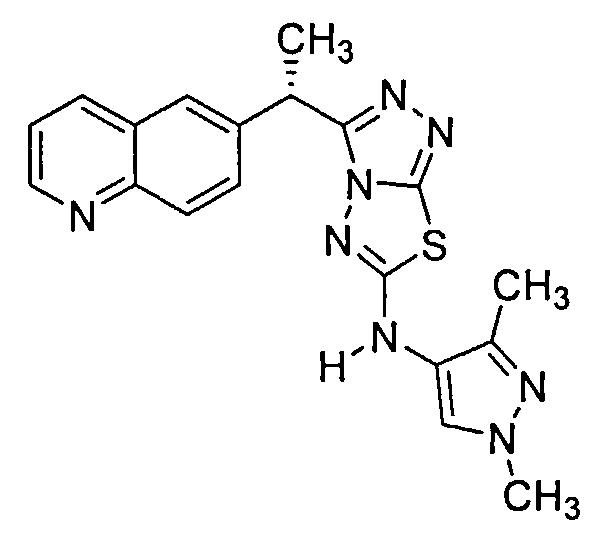 или
или
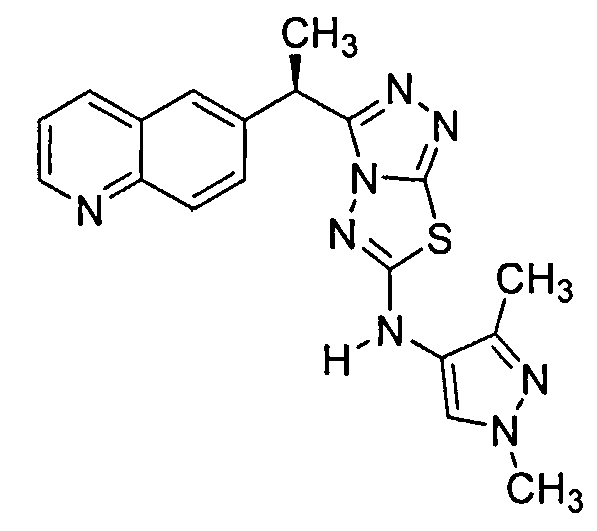 .
.
10. Соединение по п.1, имеющее следующую структуру:
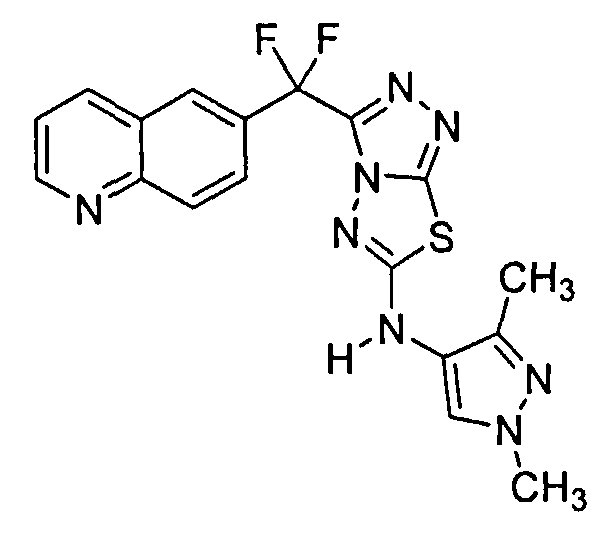 .
.
11. Соединение по п.1, имеющее следующую структуру:
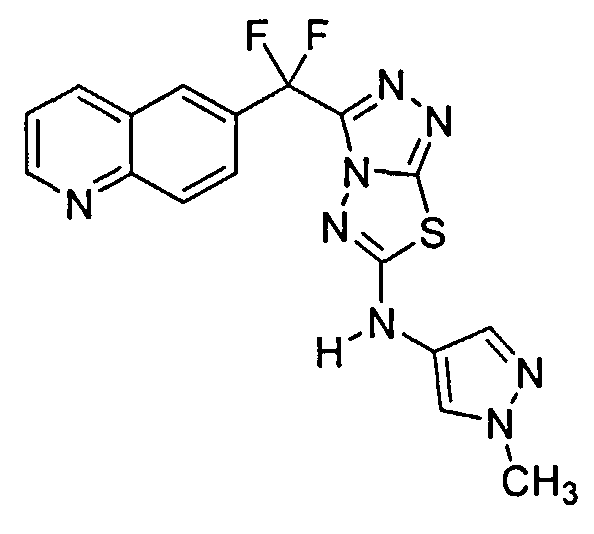 .
.
12. Фармацевтическая композиция для лечения или уменьшения степени тяжести пролиферативного нарушения, содержащая терапевтически эффективное количество соединения по пп.1-11 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, адъювант или наполнитель.
13. Применение соединения по любому одному из пп.1-11 при получении лекарственного средства для лечения или уменьшения степени тяжести пролиферативного нарушения у пациента.
14. Применение по п.13, в котором указанное нарушение представляет собой метастатический рак.
15. Применение по п.13, в котором указанное нарушение представляет собой глиобластому; карциному желудка; или рак, выбранный из рака толстой кишки, груди, простаты, мозга, печени, поджелудочной железы или легкого.
16. Применение по п.13, в котором указанное нарушение представляет собой гепатоцеллюлярную карциному.
| WO 2008144767 А1, 27.11.2008 | |||
| WO 2007064797 A2, 07.06.2007 | |||
| RU 2007109812 А, 27.09.2008 |

