Область техники, к которой относится изобретение
Настоящее изобретение относится к новому полипептиду, используемому для получения D-молочной кислоты, где указанный полипептид обладает активностью D-лактатдегидрогеназы; к полинуклеотиду, кодирующему данный полипептид; и к трансформанту, в который был встроен полинуклеотид. Настоящее изобретение также относится к способу получения D-молочной кислоты, который включает культивирование трансформанта.
Предпосылки создания изобретения
Исходя из положения нейтральности углерода, уделялось особое внимание полимолочным кислотам (ПМК), которые в качестве исходного материала используют возобновляемый источник, биомассу. ПМК характеризуются относительно низкой стоимостью и термоустойчивостью, поскольку имеют точку плавления примерно 170°C; и в этой связи ожидается, что они будут представлять собой биодеградируемый полимер, который можно формовать в расплавленном состоянии. Кроме того, известно, что стереокомплексные полимолочные кислоты могут быть получены при смешивании поли-L-молочной кислоты и поли-D-молочной кислоты (патентные документы 1-3). Известны стереокомплексные полимолочные кислоты, которые имеют более высокую точку плавления и повышенную способность к кристаллизации, в сравнении с единичными полимерами, и позволяют создавать формованные изделия, такие как изделия, изготовленные из волокна, пленок и смол. В случае обоих указанных исходных материалов, L-молочной кислоты и D-молочной кислоты, имеется потребность в способе их получения с высокой чистотой и высокой эффективностью.
В природе существуют бактерии, которые эффективно продуцируют молочные кислоты, такие как молочнокислые бактерии, и некоторые из соответствующих способов продукции молочной кислоты с использованием таких бактерий уже нашло практическое применение. Примеры бактерий, которые эффективно продуцируют L-молочную кислоту, включают Lactobacillus delbrueckii. Дополнительно, в качестве бактерий, которые эффективно продуцируют D-молочную кислоту, известны такие микроорганизмы, как Sporolactobacillus laevolacticus (патентный документ 4). Во всех этих случаях количество накопленной молочной кислоты достигало высокого уровня в условиях анаэробной культуры; однако, поскольку D-молочная кислота и L-молочная кислота образуются как побочный продукт при ферментации L-молочной кислоты и ферментации D-молочной кислоты, соответственно, оптическая чистота снижается. Кроме того, чрезвычайно трудно разделять эти молочные кислоты.
В этой связи в качестве способа получения молочной кислоты с высокой оптической плотностью, рассматривался вариант встраивания гена, кодирующего L-лактатдегидрогеназу или D-лактатдегидрогеназу в дрожжи, которые не обладали природной способностью к продукции молочной кислоты, и далее проводили ферментацию L-молочной кислоты и D-молочной кислоты (патентные документы 4-6 и непатентный документ 1). В том, что касается ферментации L-молочной генетически сконструированными дрожжами, то введение высоко активного гена из Xenopus laevis, который кодирует L-лактатдегидрогеназу, позволяет эффективно осуществлять ферментацию молочной кислоты с достижением высокой оптической чистоты (патентный документ 4). С другой стороны, в том, что касается ферментации D-молочной кислоты с использованием генетически сконструированных дрожжей, то, хотя D-молочная кислота с высокой оптической чистотой может быть получена таким же способом, как и в случае ферментации L-молочной кислоты, достижение хорошего выхода представляет собой проблему (патентные документы 5 и 6 и непатентный документ 1).
Документы, описывающие уровень техники
Патентные документы
Патентный документ 1: JP S61-36321A
Патентный документ 2: JP S63-241024A
Патентный документ 3: JP 2000-17163A
Патентный документ 4: WO2007/043253
Патентный документ 5: JP 2007-074939A
Патентный документ 6: WO2004/104202
Непатентные документы
Непатентный документ 1: Ishida N et al., Journal of Bioscience and Bioengineering (2006), 101, 172-7.
Краткое описание сущности изобретения
Проблемы, которые разрешает настоящее изобретение
Целью настоящего изобретения является достижение высокопродуктивной ферментации D-молочной кислоты с использованием полипептида, обладающего D-лактатдегидрогеназной активностью, которая превышает таковую для обычных полипептидов и полинуклеотида, кодирующего соответствующий полипептид.
Способы решения проблем
Авторы настоящего изобретения провели масштабное и интенсивное изучение большого числа D-лактатдегидрогеназ с целью выявления полипептида, который усиливает продукцию D-молочной кислоты и обладает D-лактатдегидрогеназной активностью с образованием при этом небольшого количества побочного продукта, а также полинуклеотида, кодирующего полипептид, что составило суть настоящего изобретения.
Соответственно, настоящее изобретение относится к следующим элементам.
(1) Полипептид, который представляет собой любой из указанных ниже полипептидов (A)-(C):
(A) полипептид с аминокислотной последовательностью, показанной в SEQ ID NO:1 или 2;
(B) полипептид с той же аминокислотной последовательностью, которая показана в SEQ ID NO:1 или 2; с тем исключением, что одна или несколько аминокислот замещены, делетированы, встроены и/или добавлены, где указанный полипептид обладает D-лактатдегидрогеназной активностью; и
(С) полипептид с аминокислотной последовательностью, которая характеризуется идентичностью по последовательности на уровне менее 80% с аминокислотной последовательностью, показанной в SEQ ID NO:1 или 2, где указанный полипептид обладает D-лактатдегидрогеназной активностью.
(2) Полинуклеотид, который представляет собой один из указанных ниже полинуклеотидов (a)-(e):
(a) полинуклеотид с нуклеотидной последовательностью, показанной в SEQ ID NO:3 или 4;
(b) полинуклеотид с той же нуклеотидной последовательностью, которая показана в SEQ ID NO:3 или 4, с тем исключением, что в ней один или несколько нуклеотидов замещены, делетированы, встроены и/или добавлены, где указанный полинуклеотид кодирует полипептид, обладающий D-лактатдегидрогеназной активностью;
(c) полинуклеотид, который гибридизуется в жестких условиях с полноразмерным полинуклеотидом или его частью, где указанный полинуклеотид имеет нуклеотидную последовательность, показанную в SEQ ID NO:3 или 4, или с его комплементарной цепью, где указанный полинуклеотид кодирует полипептид, обладающий D-ЛДГ активностью;
(d) полинуклеотид с нуклеотидной последовательностью, которая характеризуется идентичностью по последовательности на уровне не менее чем 80% с нуклеотидной последовательностью, показанной в SEQ ID NO:3 или 4, где указанный полинуклеотид кодирует полипептид, обладающий D-лактатдегидрогеназной активностью; и
(e) полинуклеотид, кодирующий полипептид, соответствующий п.(1).
(3) Конструкция ДНК, в которой соединены полинуклеотид по п.(2) и промотор, способный осуществлять экспрессию полинуклеотида.
(4) Конструкция ДНК по п.(3), отличающаяся тем, что указанный выше промотор представляет собой промотор гена пируватдекарбоксилазы 1 (ген PDC1), гена супрессии экспоненциального дефекта 1 (ген SED1) или гена глицеральдегид-3-фосфатдегидрогеназы 3 (ген TDH3).
(5) Конструкция ДНК по п.(3) или (4), где указанный выше промотор выбран из любого промотора (I)-(III):
(1) промотор с нуклеотидной последовательностью, показанной в любой из последовательностей SEQ ID NO: 5-7;
(II) промотор с нуклеотидной последовательностью, которая гибридизуется в жестких условиях с нуклеотидной последовательностью, показанной в любой из SEQ ID NO: 5-7, или с нуклеотидной последовательностью, включающей ее часть; и
(III) промотор с той же нуклеотидной последовательностью, которая была показана в любой из последовательностей SEQ ID NO: 5-7, с тем исключением, что в ней один или несколько нуклеотидов делетированы, замещены и/или добавлены.
(6) Трансформант, в который введен полинуклеотид по п.(2) или введена конструкция ДНК по любому из (3)-(5).
(7) Трансформированные дрожжи, в которые введен полинуклеотид по (2) или введена конструкция ДНК по любому из пп.(3)-(5).
(8) Трансформированные дрожжи по п.(7), в которых по меньшей мере один ген из группы, включающей ген пируватдегидрогеназы 1 (ген PDC1), ген супрессии экспоненциального дефекта 1 (ген SED1) и ген глицеральдегид-3-фосфатдегидрогеназы 3 (ген TDH3) из указанных выше трансформированных дрожжей, замещен полинуклеотидом по п.(2) или конструкцией ДНК по любому из пп.(3)-(5).
(9) Трансформированные дрожжи по п.(8), в которых по меньшей мере один из указанных выше генов представляет собой ген PDC1.
(10) Способ получения D-молочной кислоты, который включает стадию культивирования трансформанта по п.(6) или трансформированных дрожжей по любому из пп.(7)-(9).
(11) Трансформант, в котором полинуклеотид, кодирующий D-лактатдегидрогеназу, получен из семейства Limulidae или конструкции ДНК, в которую встроены слитый полинуклеотид и промотор, способный осуществлять экспрессию указанного полинуклеотида.
(12) Трансформированные дрожжи, в которых полинуклеотид, кодирующий D-лактатдегидрогеназу, получен из рода Limulus или конструкции ДНК, в которую встроены слитый полинуклеотид и промотор, способный осуществлять экспрессию указанного полинуклеотида.
(13) Трансформированные дрожжи по п.(12), в которых по меньшей мере один ген из группы, включающей ген пируватдегидрогеназы 1 (ген PDC1), ген супрессии экспоненциального дефекта 1 (ген SED1) и ген глицеральдегид-3-фосфатдегидрогеназы 3 (ген TDH3) из указанных выше трансформированных дрожжей, замещен указанным выше полинуклеотидом, кодирующим D-лактатдегидрогеназу из семейства Limulidae, или конструкцией ДНК, в которую встроены слитые полинуклеотид и промотор, способный осуществлять экспрессию полинуклеотида.
(14) Способ получения D-молочной кислоты, включающий стадию культивирования трансформанта по п.(11) или трансформированных дрожжей по п.(12) или (13).
Эффекты, достигаемые при осуществлении настоящего изобретения
Настоящее изобретение относится к полипептиду, обладающему D-лактатдегидрогеназной активностью, подходящей для энзиматической продукции D-молочной кислоты, и к полинуклеотиду, кодирующему указанный полипептид. Дополнительно, при использовании полинуклеотида по настоящему изобретению может быть легко получен трансформант с высокой продуктивной способностью по D-молочной кислоте и, в свою очередь, может быть эффективно получена D-молочная кислота с высокой чистотой при культивировании данного трансформанта.
Краткое описание фигур
На фиг.1 проиллюстрированы процедуры создания штаммов дрожжей SU042 по настоящему изобретению.
На фиг.2 показаны результаты, получаемые при непрерывном культивировании штамма дрожжей со слитыми мембранами SU042 по настоящему изобретению.
Способ осуществления настоящего изобретения
Ниже приводится подробное описание способов осуществления настоящего изобретения.
Термин "D-лактатдегидрогеназная (далее обозначаемая как "D-ЛДГ") активность" (или, альтернативно, “активность "D-лактатдегидрогеназы (D-ЛДГ”)) представляет собой активность, направленную на превращение восстановленного никотинамидадениндинуклеотида (НАДН) и пировиноградной кислоты в D-молочную кислоту и окисленный никотинамидадениндинуклеотид (НАД+).
Настоящее изобретение относится к полипептиду с аминокислотной последовательностью, которая соответствует SEQ ID NO:1 или 2 или их гомологу, где указанный полипептид обладает D-ЛДГ активностью.
Полипептид с аминокислотной последовательностью, показанной в SEQ ID NO:1 или 2, представляет собой полипептид. полученный из Limulus polyphemus, который относится к семейству Limulidae и роду Limulus.
Примеры гомолога полипептида с аминокислотной последовательностью, показанной в SEQ ID NO:1 или 2, включают полипептид с той же аминокислотной последовательностью, которая показана в SEQ ID NO:1 или 2, с тем исключением, что одна или несколько, предпочтительно 1-10, более предпочтительно 1-5, еще более предпочтительно 1 или 2 аминокислоты замещены, делетированы, встроены и/или добавлены, где указанный полипептид также обладает D-ЛДГ активностью.
Кроме того, гомолог полипептида, показанного в SEQ ID NO:3 или 4, может также представлять собой полипептид с аминокислотной последовательностью, которая характеризуется идентичностью по последовательности на уровне не менее чем 80%, предпочтительно 90%, более предпочтительно не менее чем 95%, к аминокислотной последовательности, показанной в SEQ ID NO:1 или 2, где указанный полипептид также обладает D-ЛДГ активностью. Согласно настоящему изобретению, идентичность на уровне последовательности любой рассматриваемой аминокислотной последовательности может быть легко определена с использованием хорошо известной в данной области программы BLAST. Любой специалист со средним уровнем знаний в данной области может с помощью программы BLAST, доступной через сайт Национального Центра Биотехнологической Информации (NCBI; National Center for Biotechnology Information), легко проверить идентичность, используя заданные параметры.
Кроме того, гомолог полипептида с аминокислотной последовательностью, показанной в SEQ ID NO:1 или 2, может также представлять собой полипептид, полученный из живого организма из семейства Limulidae, предпочтительно, живого организма из рода Limulus или Tachypleus, более предпочтительно, из Limulus polyphemus, относящегося к роду Limulus, или из Tachypleus tridentatus, Tachypleus gigas или Tachypleus rotundicauda, относящихся к роду Tachypleus, где указанный полипептид обладает D-ЛДГ активностью.
Полипептид с аминокислотной последовательностью, показанной в SEQ ID NO:1 или 2, может быть экстрагирован из Limulus polyphemus по известному способу или может быть получен с использованием известного метода пептидного синтеза. Кроме того, указанный полипептид может быть также получен в рамках методов рекомбинантной технологии с использованием полинуклеотида, кодирующего аминокислотную последовательность, показанную в SEQ ID NO:1 или 2. Указанный полипептид с аминокислотной последовательностью, показанной в SEQ ID NO:1 или 2, может быть также экстрагирован из живого организма, относящегося к роду Tachypleus, по известному способу или может быть получен по известному методу пептидного синтеза. Кроме того, указанный полипептид может быть также получен в рамках методов рекомбинантной генной технологии с использованием полинуклеотида, кодирующего аминокислотную последовательность, соответствующую данному полипептиду.
Гомолог полипептида с аминокислотной последовательностью, показанной в SEQ ID NO:1 или 2, может обладать улучшенной D-ЛДГ активностью и/или повышенной термостабильностью, в сравнении с полипептидом, имеющим аминокислотную последовательность, показанную в SEQ ID NO:1 или 2. В контексте настоящего описания термин "улучшенная D-ЛДГ активность" обозначает более высокие показатели по сродству к субстрату и молекулярной активности (Kcat) применительно к пировиноградной кислоте и НАДН, а также смещение оптимального для ферментативной активности полипептида значения pH ближе в тому значению pH, которое подходит для роста клеток, экспрессирующих данный полипептид. Такой полипептид может быть искусственно сконструирован на основе полипептида аминокислотной последовательности, показанной в SEQ ID NO:1 или 2, или может быть выделен из природного источника. Кроме того, в ген D-лактатдегидрогеназы может быть введена случайная мутация по методу эволюционной молекулярной инженерии для скрининга предпочтительного полипептида.
Настоящее изобретение относится к полинуклеотиду с нуклеотидной последовательностью, показанной в SEQ ID NO:3 или 4, или к ее гомологу, где указанный полинуклеотид кодирует полипептид, обладающий D-ЛДГ активностью. В контексте настоящего описания источник "полинуклеотида" не имеет значения, и такой полинуклеотид может представлять собой кДНК, геномную ДНК, синтетическую ДНК, мРНК, синтетическую РНК, репликон РНК; однако, предпочтительно, - это ДНК или РНК, более предпочтительно, ДНК. Кроме того, рассматриваемый "полинуклеотид" может быть одноцепочечным или двуцепочечным, содержащим комплементарную цепь. Дополнительно, указанный "полинуклеотид" может также содержать природное или искусственно полученное нуклеотидное производное.
Полинуклеотид с нуклеотидной последовательностью, показанной в SEQ ID NO:3 или 4, представляет собой такой полинуклеотид, который был получен из Limulus polyphemus и который характеризуется тем, что он кодирует полипептид, обладающий указанной выше D-ЛДГ активностью.
Примеры гомолога полинуклеотида, показанного в SEQ ID NO:3 или 4, включают полинуклеотид с той же нуклеотидной последовательностью, которая соответствует SEQ ID NO:3 или 4, с тем исключением, что один или несколько, предпочтительно 1-40, более предпочтительно, 1-30, еще более предпочтительно, 1-20, особенно предпочтительно, 1-10, и наиболее предпочтительно, от 1 до 5 нуклеотидов замещены, делетированы, встроены и/или добавлены в нее, где указанный полинуклеотид также кодирует полипептид, обладающий D-ЛДГ активностью.
Другие примеры гомолога полинуклеотида, показанного в SEQ ID NO:3 или 4, также включают полинуклеотид, который гибридизуется в жестких условиях с полноразмерным полинуклеотидом или его частью, где указанный полинуклеотид имеет нуклеотидную последовательность, показанную в SEQ ID NO:3 или 4, или соответствует ее комплементарной цепи, и где указанный полинуклеотид также кодирует полипептид, обладающий D-ЛДГ активностью. В контексте настоящего описания фраза "полинуклеотид, который гибридизуется в жестких условиях" относится, например, к полинуклеотиду, который гибридизуется в рамках известного метода гибридизации (Current Protocols I Molecular Biology edit. Ausubel et al., (1987) Publish. John Wily & Sons, Section 6.3-6.4) или аналогичной процедуры, при использовании в качестве одного или нескольких зондов одного или большего числа полинуклеотидов, которые включают по меньшей мере 20, предпочтительно 25, более предпочтительно, по меньшей мере 30 произвольно выбранных последовательных нуклеотидов из исходной нуклеотидной последовательности. В контексте настоящего описания гибридизация в жестких условиях может быть достигнута, например, при проведении гибридизации в 50% формамиде при температуре гибридизации 37°C, а также при 42°C для создания более жестких условий, и при 65°C для создания еще более жестких условий, с последующей промывкой полученной смеси с использованием 0,1x-2x раствора SSC (солевой раствор-цитрат натрия) (состав 1 × SSC раствора: 150 мМ хлорида натрия и 15 мМ цитрата натрия).
Кроме того, гомолог полинуклеотида, показанного в SEQ ID NO:3 или 4, может также представлять собой полинуклеотид с нуклеотидной последовательностью, которая характеризуется идентичностью по последовательности на уровне не менее чем 80%, более предпочтительно, не менее 90%, еще более предпочтительно, не менее 95%, к нуклеотидной последовательности, показанной в SEQ ID NO:3 или 4, где указанный полипептид, обладающий D-ЛДГ активностью. Идентичность используемой в настоящем изобретении нуклеотидной последовательности полинуклеотида на уровне последовательности может быть определена с использованием описанной выше программы для генного анализа BLAST или другой аналогичной программы.
Дополнительно, гомолог полинуклеотида, показанного в SEQ ID NO:3 или 4, может также представлять собой полинуклеотид, полученный из живого организма, относящегося к семейству Limulidae, предпочтительно живого организма, относящегося к роду Limulus или Tachypleus, более предпочтительно, Limulus polyphemus, Tachypleus tridentatus, Tachypleus gigas или Tachypleus rotundicauda, где указанный полипептид обладает D-ЛДГ активностью.
Хотя описанный выше полинуклеотид может быть получен при клонировании из живого организма, относящегося к семейству Limulidae, предпочтительно, из живого организма, относящегося к семейству Limulidae и роду Limulus (которые далее в целом обозначаются как "Limulus"), он также может быть синтезирован химически или по методу Fujimoto et al. (Hideya Fujimoto, Production Method of Synthetic Gene, Plant Cell Engineering Series 7, Plant PCR Experimental Protocol, 1997, Shujunsha Co., Ltd., рр.95-100), известному как метод синтеза длинноцепочечной ДНК. Обычно полинуклеотид, клонированный из Limulus, может быть выделен по известному методу. Примеры таких методов включают любой метод, в соответствии с которым с использованием праймера, синтезированного на основе последовательности кДНК, которая характеризуется высокой консервативностью по D-лактатдегидрогеназе, где указанная кДНК была получена из поли A(+)РНК, выделенной из Limulus hemocytes, амплифицируют и секвенируют частичный фрагмент и затем определяют полную последовательность ORF по процедурам 5'-RACE и 3'-RACE, с последующей амплификацией в рамках реакции ПЦР с получением полинуклеотида. Дополнительно, клонирование из Limulus может быть также осуществлено при комплементировании функции D-лактатдегидрогеназы. Соответствующий принцип метода подробно описан в работе DOMINIQUE, G., Appl Environ. Microbiol, United States (1995), 61, 266-272. Таким же образом, полинуклеотид по настоящему изобретению, нуклеотидная последовательность которого уже определена, может быть получен из Limulus hemocytes; однако он может быть также напрямую синтезирован химически с использованием синтезатора ДНК.
В настоящем описании указанный выше полинуклеотид может быть модифицирован в нужном направлении при введении соответствующих мутаций за счет одного или нескольких замещений, одной или нескольких делеций, одной или нескольких вставок и/или добавлений к его нуклеотидной последовательности или соответствующей аминокислотной последовательности по стратегии сайт-специфического мутагенеза (Current Protocols I Molecular Biology edit. Ausubel et al., (1987) Publish. John Wily & Sons Section 8.1-8.5) или аналогичной стратегии. При этом, такая модификация аминокислотной последовательности полипептида или нуклеотидной последовательности полинуклеотида не ограничивается процедурами искусственного введения или синтеза мутации, но охватывает не только модификации, достигаемые за счет искусственного введения мутации, но также природные модификации, возникающие в результате мутации аминокислот.
Описанный выше полинуклеотид может использоваться для получения клеток, продуцирующих D-молочную кислоту, путем генной рекомбинации. Для того, чтобы клетка-хозяин, в которую полинуклеотид по настоящему изобретению был введен путем генной рекомбинации (далее называемая просто "трансформант"), стала способной продуцировать D-молочную кислоту, необходимо, чтобы полипептид, обладающий D-ЛДГ активностью и который кодируется данным полинуклеотидом, экспрессировался в трансформанте. В этом случае в рамках способа, позволяющего достичь экспрессии полипептида, обладающего D-ЛДГ активностью, можно использовать конструкцию ДНК, в которой связаны полинуклеотид и промотор, способный осуществлять его экспрессию, то есть конструкцию ДНК, в которой полинуклеотид связан с 3'-концом промотора, и такая конструкция ДНК также включается в область настоящего изобретения. Указанная конструкция ДНК по настоящему изобретению может быть получена по способу, в соответствии с которым полинуклеотид (ДНК) по настоящему изобретению и промотор, способный экспрессировать указанный полинуклеотид, каждый расщепляют, одним или несколькими, ферментом(ами) рестрикции и затем встраивают в сайт фермента рестрикции или в сайт множественного клонирования описанного ниже вектора ДНК, подлежащего слиянию, или проводят связывание полинуклеотида (ДНК) по настоящему изобретению и промотора в рамках реакции ПЦР.
В предпочтительном варианте описанной выше конструкции ДНК указанная конструкция ДНК переносится вектором, таким как плазмида (ДНК), бактериофаг (ДНК), ретротранспозон (ДНК) или искусственная хромосома (например, YAC, PAC, BAC или MAC). В зависимости от способа введения конструкции ДНК (внутрь генома хозяина или за его пределами) и типа клетки-хозяина, выбирают прокариотический вектор, эукариотический вектор, вектор на основе животной клетки или вектор на основе растительной клетки из числа известных в данной области.
Согласно настоящему описанию, примеры плазмиды включают шаттл-векторы YCp-типа Escherichia coli-дрожжей, такие как pRS413, pRS415, pRS416, YCp50, pAUR112 и pAUR123; шаттл-векторы YEp-типа Escherichia coli-дрожжей, такие как pYES32 и Yep13; шаттл-векторы YIp-типа Escherichia coli-дрожжей, такие как pRS403, pRS404, pRS405, pRS406, pAUR101 и pAUR135; плазмиды, происходящие из Escherichia coli (например, плазмиды ColE-типа, такие как pBR322, pBR325, pUC18, pUC19, pUC119, pTV118N, pTV119N, pBluescript, pHSG298, pHSG396 и pTrc99A; плазмиды p1A-типа, такие как pACYC177 и pACYC184; и плазмиды pSC101-типа, такие как pMW118, pMW119, pMW218 и pMW219); а также плазмиды, происходящие из Bacillus subtilis (такие как pUB110 и pTP5). Примеры бактериофага включают λ-фаги (такие как Charon4A, Charon21A, EMBL3, EMBL4, λgt100, gt11 и zap), φX174, M13mp18 и M13mp19. Примером ретротранспозона является Ty фактор и пример YAC включает pYACC2.
Термин "промотор" в описанной выше конструкции ДНК означает нуклеотидную последовательность, вовлекаемую в инициацию транскрипции мРНК для данного рассматриваемого гена и обычно относится к последовательности против направления считывания информации от 5'-конца гена, имеющегося в хромосоме. Длина нуклеотидной последовательности промотора составляет предпочтительно 1-3000 п.н., более предпочтительно, 1-1000 п.н.; однако она особым образом не ограничивается, главное, чтобы такая нуклеотидная последовательность выполняла функцию инициации транскрипции мРНК гена, расположенного по направлению считывания информации. Кроме того, известны мутации и действия, направленные на улучшение транскрипционной активности промотора, и термин "промотор" также включает такие промоторы, которые были модифицированы по известному в данной области способу. Промотор в указанной выше конструкции ДНК особым образом не ограничивается, главное, чтобы он обладал промоторной активностью в трансформанте, при введении в составе конструкции ДНК; однако, как будет описано ниже, поскольку предпочтительно, чтобы конструкция ДНК по настоящему изобретению вводилась в дрожжи, промотор предпочтительно функционирует в дрожжах.
Предпочтительные примеры промотора, функционирующего в дрожжах, включают промоторы гена кислой фосфатазы (PH05), генов глицеральдегид-3-фосфатдегидрогеназы (TDH1, 2 и 3), генов алкогольдегидрогеназы (ADH1, 2, 3, 4, 5, 6 и 7), генов, связанных с метаболизмом галактозы (GAL1, 7 и 10), гена цитохрома c (CYC1), гена триозофосфатизомеразы (TP11), гена фосфоглицераткиназы (PGK1), гена фосфофруктокиназы (PFK1) и генов пируватдекарбоксилазы (PDC1, 5, 6), а также промоторы гена энолазы-1 (ENO1), гена белка клеточной стенки 2 (CWP2) и гена супрессии экспоненциального дефекта 1 (SED1), описание которых и варианты использования которых приведены в международной заявке на патент PCT/JP 2008/072129, и которые демонстрируют экспрессию гена на уровне, не менее чем в 5 раз превышающем средний относительный уровень экспрессии всех генов через 50 часов или позже после начала роста культуры дрожжей.
Более предпочтительные примеры промотора, функционирующего в дрожжах, включают промотор PDC1 и промотор TDH3, которые экспрессируются на высоком уровне в дрожжах на пути продуцирования этанола, и промотор SED1, который экспрессируется на высоком уровне в дрожжах, культивируемых в течение длительного периода времени, и более конкретные примеры включают промотор PDC1 с последовательностью SEQ ID NO:5, промотор SED1 с последовательностью SEQ ID NO:6 и промотор TDH3 с последовательностью SEQ ID NO:7.
При этом промотор PDC1, промотор SED1 или промотор TDH3 могут также иметь, при условии сохранения промоторной активности, нуклеотидную последовательность, соответствующую любой из последовательностей SEQ ID NO: 5-7, соответственно, с тем исключением, что в них один или несколько, предпочтительно 1-40, более предпочтительно 1-30, еще более предпочтительно 1 или 20, особенно предпочтительно 1-10 и наиболее предпочтительно, 1-5 нуклеотидов делетированы, замещены и/или добавлены.
Кроме того, промотор PDC1, промотор SED1 или промотор TDH3 могут также иметь, при условии сохранения промоторной активности, нуклеотидную последовательность, которая гибридизуется в жестких условиях с нуклеотидной последовательностью, показанной в SEQ ID NO:5-7, или с ее частью, соответственно. В контексте настоящего описания фраза нуклеотидная последовательность, которая «гибридизуется в жестких условиях», относится, например, к полинуклеотиду, который гибридизуется в рамках известной процедуры гибридизации (Current Protocols I Molecular Biology edit. Ausubel et al., (1987) Publish. John Wily & Sons, Section 6.3-6.4) или аналогичной методики, с использованием в качестве одного или нескольких зондов одного или большего числа выбранных полинуклеотидов, содержащих по меньшей мере 20, предпочтительно 25, более предпочтительно по меньшей мере 30 произвольно отобранных, непрерывно следующих друг за другом нуклеотидов из исходной нуклеотидной последовательности. Для целей настоящего изобретения жесткие условия могут быть созданы, например, при проведении гибридизации в присутствии 50% формамида при температуре гибридизации 37°C, 42°C - для достижения более жестких условий, 65°C - для еще более жестких условий, с последующей промывкой полученной смеси в растворе 0,1x-2xSSC (солевой раствор-цитрат натрия) (состав раствора 1 × SSC: 150 мМ хлорида натрия и 15 мМ цитрата натрия).
Настоящее изобретение также включает трансформант, полученный при встраивании описанного выше полинуклеотида или конструкции ДНК в клетку-хозяина. Клетка-хозяин особым образом не ограничивается, при условии, что она стабильно удерживает полинуклеотид или конструкцию ДНК, и соответствующие ее примеры включают бактерии, такие как Escherichia coli, Bacillus subtilis и молочнокислые бактерии; дрожжи; клетки насекомых; клетки животных и клетки растений, и предпочтительно используются дрожжи, поскольку они являются кислотоустойчивыми и способны к росту даже в том случае, когда D-молочная кислота продуцируется на высоком уровне при экспрессии полипептида, обладающего D-ЛДГ активностью.
Примеры дрожжей включают дрожжи, относящиеся к роду Saccharomyces, Schizosaccharomyces, Kluyveromyces, Candida, Pichia, Hansenura, Yarrowia, Zygosaccharomyces, Torulopsis, Debaryomyces, Issachenkia и Fellomyces; и предпочтительно, дрожжи, которые относятся к роду Saccharomyces, Candida или Kluyveromyces, более предпочтительно, Saccharomyces cerevisiae, Candida utilis, Candida glabrata, Candida albicans, Candida boidinii, Candida sonorensis, Kluyveromyces lactis или Kluyveromyces marxianus.
Дополнительно, согласно настоящему изобретению, предпочтительно используются полиплоидные дрожжи, поскольку они могут поддерживать высокий уровень продукции D-молочной кислоты в течение длительного периода времени в простых условиях культивирования, так что можно поддерживать стабильную продукцию D-молочной кислоты с низкими затратами. В контексте настоящего описания термин "полиплоидные дрожжи" относится к дрожжам, включающим 2 или больше наборов хромосом в клетке. Число наборов хромосом в полиплоидных дрожжах особо не ограничивается; однако предпочтительными являются диплоидные дрожжи, содержащие 2 набора хромосом. Примеры полиплоидных дрожжей включают пекарские дрожжи, дрожжи для сакэ, винные дрожжи и пивные дрожжи, которые часто используются в бродильном производстве. Подходящие для использования полиплоидные дрожжи могут быть выделены из природной среды или из такого источника, свойства которого были частично модифицированы путем мутации или генной рекомбинации.
Кроме того, в настоящем изобретении предпочтительно используются прототрофные дрожжи, поскольку они не только позволяют использовать культуральную среду, содержащую меньше, чем в случае традиционных сред, количество питательных компонентов, т.е. представляют собой дешевую среду, но также позволяют стабильно поддерживать высокий уровень продукции D-молочной кислоты в течение длительного периода времени в простых условиях культивирования, так что может поддерживаться стабильная продукция молочной кислоты с низкими затратами. В контексте настоящего описания термин "ауксотрофный" означает, что мутация по какой-либо причине присутствует в гене, ответственном за синтез питательного компонента, в дрожжах дикого типа, что сказывается на недостаточной способности синтезировать этот питательный компонент. Соответственно, термин "прототрофные дрожжи" означает такие дрожжи, которые не содержат ауксотрофный генотип или иной генотип, где такой генотип комплементирован. Примеры способа получения прототрофных дрожжей за счет реверсирования ауксотрофной способности у ауксотрофных дрожжей включают способ, согласно которому ауксотрофность реверсируется за счет встраивания гена, отвечающего за синтез питательного компонента, по методу генной рекомбинации; а также способ, согласно которому процессы спаривания дрожжей с разной ауксотрофностью, позволяют формировать сумку для спороношения, и процедуру реверсии ауксотрофности повторяют до тех пор, когда будет достигнута реверсия всех видов ауксотрофности с получением прототрофных дрожжей. Следует также отметить, что определить, являются ли дрожжи прототрофными, можно на основании данных о том, могут ли рассматриваемые дрожжи расти в SD среде, которая представляет собой минимальную ростовую среду для дрожжей.
Дополнительно, дрожжи представляют собой микроорганизм, который осуществляет энергичную ферментацию этанола, и на этом метаболическом пути пировиноградная кислота, являющаяся продуктом гликолиза, превращается пируватдекарбоксилазой в ацетальдегид, который затем превращается в этанол алкогольдегидрогеназой. В этой связи предпочтительно, чтобы в качестве организма-хозяина использовался штамм дрожжей, в котором разрушен ген пируватдекарбоксилазы, служащий исходной точкой на пути метаболизма этанола, поскольку это позволяет достичь естественной метаболизации пировиноградной кислоты на пути метаболизма этанола с последующим использованием на пути метаболизма D-молочной кислоты.
В частности, в случае Saccharomyces cerevisiae, имеются три типа генов, кодирующих пируватдекарбоксилазу: ген пируватдекарбоксилазы 1 (PDC1), ген пируватдекарбоксилазы 5 (PDC5) и ген пируватдекарбоксилазы 6 (PDC6). Известно, что среди этих генов только PDC1 и PDC5 обладают пируватдегидрогеназной активностью в клетках дрожжей; в этой связи предпочтительно использовать штамм с разрушенным геном PDC1 или геном PDC5 и более предпочтительно, использовать штамм, содержащий разрушенный ген PDC1. Кроме того, как описано в JP 2008-048726A, в случае штамма, содержащего разрушенный ген PDC1, предпочтителен штамм, содержащий мутантный ген PDC5, который включает мутацию, приводящую к снижению активности PDC5, и более предпочтителен штамм, содержащий чувствительный к температуре ген PDC5 мутантного типа. В контексте настоящего описания фраза "чувствительный к температуре ген PDC5 мутантного типа" относится к гену PDC5 мутантного типа, который обладает способностью проявлять пируватдекарбоксилазную активность, в отличие от штамма с PDC5 дикого типа, при определенной температуре культивирования, но демонстрирует потерю или снижение активности PDC5 в случае изменения температуры культивирования до уровня, который не ниже определенной температуры культивирования. Нормальная температура культивирования Saccharomyces cerevisiae составляет от 28 до 30°C, и предпочтительно, чтобы температура, при которой проявляется такая температурная чувствительность, была ближе к нормальной температуре культивирования, поскольку это снижает количество тепла, необходимое для изменения температуры культивирования, так что в результате снижается стоимость культивирования. В частности, предпочтительно, чтобы чувствительный к температуре штамм с мутантным типом PDC5 проявлял температурную чувствительность при температуре не ниже 34°C.
Примеры способа, позволяющего достичь экспрессии полипептида с D-ЛДГ активностью в трансформированных дрожжах путем введения описанного(ой) выше полинуклеотида или конструкции ДНК в дрожжи, включают, как уже указывалось, способ, в рамках которого в дрожжи вводится внехромосомно вектор, содержащий конструкцию ДНК, и способ, согласно которому указанный(ую) полинуклеотид или конструкцию ДНК вводят в хромосому дрожжей, и оба этих способа могут использоваться.
В способе введения описанного выше полинуклеотида в хромосому дрожжей предпочтительно используется конструкция, которая содержит полинуклеотид по настоящему изобретению и сегмент ДНК для гомологичной рекомбинации, последовательность которого гомологична последовательности в хромосоме дрожжей (далее, часто обозначаемой как "конструкция ДНК для гомологичной рекомбинации"). Сегмент ДНК для гомологичной рекомбинации содержит последовательность ДНК, которая гомологична последовательности вблизи целевого сайта в хромосоме дрожжей, в которую вводят полинуклеотид по настоящему изобретению. Указанная конструкция содержит по меньшей мере один, предпочтительно два таких сегмента ДНК для гомологичной рекомбинации. В тех случаях, когда указанная конструкция содержит два сегмента ДНК для гомологичной рекомбинации, предпочтительно, чтобы каждый из этих двух сегментов ДНК для гомологичной рекомбинации имел последовательность ДНК, гомологичную таковой ДНК против направления или по направлению считывания информации от целевого сайта, локализованного на хромосоме, и чтобы полинуклеотид по настоящему изобретению был связующим элементом между этими сегментами ДНК.
Способ проведения гомологичной рекомбинации с использованием конструкции ДНК для гомологичной рекомбинации конкретно не ограничивается, и может применяться способ, согласно которому конструкцию ДНК для гомологичной рекомбинации амплифицируют путем ПЦР и полученный фрагмент ДНК встраивают в плазмиду, которую затем вводят в дрожжи, или способ, в соответствии с которым полученный при ПЦР фрагмент вводят в дрожжи. Кроме того, предпочтительно, конструкция ДНК для гомологичной рекомбинации содержит терминирующий элемент для регуляции экспрессии полинуклеотида по настоящему изобретению. В контексте настоящего описания термин "терминирующий элемент" означает последовательность, которая терминирует транскрипцию мРНК из гена и обычно относится к последовательности, локализованной по направлению считывания информации от 3'-конца гена, имеющегося в данной хромосоме.
Дополнительно, также предпочтительно, чтобы конструкция ДНК для гомологичной рекомбинации содержала, кроме полинуклеотида или конструкции ДНК по настоящему изобретению, селективный маркер для облегчения отбора трансформированных дрожжей. Примеры используемого в этом случае селективного маркера включают ауксотрофные комплементарные гены, такие как URA3 и TRP1, и гены, определяющие лекарственную устойчивость, такие как ген резистентности к G418 и ген резистентности к неомицину.
Конструкция ДНК для гомологичной рекомбинации, содержащая указанные выше полинуклеотид, терминирующий элемент и селективный маркер, может быть получена путем ПЦР, например, в рамках стадий 1-3.
Стадия 1: С использованием плазмиды, в которой терминирующий элемент связан по направлению считывания информации с полинуклеотидом по настоящему изобретению, в качестве матрицы, и праймерами 1 и 2, в качестве праймерного набора, фрагмент, содержащий полинуклеотид по настоящему изобретению и терминирующий элемент, амплифицируют в реакции ПЦР. Праймер 1 сконструирован таким образом, чтобы добавлять последовательность размером не менее 40 комплементарных п.н. против направления считывания информации для целевого сайта введения, а праймер 2 создан на основе последовательности плазмиды, локализованной по направлению считывания информации для терминирующего элемента.
Стадия 2: С использованием плазмиды, содержащей селективный маркер, такой как pRS400, pRS424 или pRS426, в качестве матрицы, и праймеры 3 и 4, в качестве праймерного набора, фрагмент, содержащий селективный маркер, амплифицируют в реакции ПЦР. Праймер 3 сконструирован таким образом, чтобы добавлять последовательность размером не менее 30 п.н., которые гомологичны последовательности, расположенной по направлению считывания информации в терминирующем элементе фрагмента ПЦР, полученного на стадии 1, а праймер 4 сконструирован таким образом, чтобы добавлять последовательность размером не менее 40 п.н., которая соответствует направлению считывания информации для целевого сайта введения.
Стадия 3: С использованием смеси фрагментов ПЦР, полученных на стадиях 1 и 2, в качестве матрицы, и праймеров 1 и 4, в качестве праймерного набора, проводят реакцию ПЦР с получением конструкции ДНК для гомологичной рекомбинации, содержащей полинуклеотид по настоящему изобретению, терминирующий элемент и селективный маркер дрожжей, в которой последовательности, как соответствующие направлению считывания информации, так и против направления считывания информации для целевого сайта введения, добавлены к обоим соответствующим концам.
С целью введения описанной выше конструкции ДНК для гомологичной рекомбинации в дрожжи может применяться способ, такой как трансфекция, котрансфекция или электропорация. Конкретные примеры таких способов включают способ с использованием ацетата лития и способ с использованием протопластов.
В случае введения описанного выше полинуклеотида в дрожжевую хромосому путем гомологичной рекомбинации, указанное введение проводят таким образом, чтобы полинуклеотид мог регулироваться промотором эндогенного гена, локализованного на хромосоме дрожжей. В этом случае, при введении описанного выше полинуклеотида, эндогенный ген, регулируемый в естественном состоянии промотором, может быть разрушен, и в то же время вместо указанного эндогенного гена может экспрессироваться экзогенный ген по настоящему изобретению. Этот способ особенно полезен в тех случаях, когда промотором является промотор с высоким уровнем экспрессии в дрожжах, как описано выше, и когда разрушен эндогенный ген, функционирующий в направлении ингибирования метаболического пути D-молочной кислоты в дрожжах.
Предпочтительные примеры эндогенного гена, который служит в качестве мишени при введении вышеописанного полинуклеотида в дрожжевую хромосому, включают ген кислой фосфатазы (PHO5), гены глицеральдегид-3-фосфатдегидрогеназы (TDH1, 2 и 3), гены алкогольдегидрогеназы (ADH1, 2, 3, 4, 5, 6 и 7), гены, связанные с метаболизмом галактозы (GAL1, 7 и 10), ген цитохрома с (CYC1), ген триозофосфатизомеразы (TPI1), ген фосфоглицераткиназы (PGK1), ген фосфофруктокиназы (PFK1) и гены пируватдекарбоксилазы (PDC1, 5, 6), а также промоторы гена энолазы-1 (ENO1), гена, связанного с белком клеточной стенки 2 (CWP2), и гена супрессии экспоненциального дефекта 1 (SED1), которые описаны, включая их применение, в международной заявке на патент PCT/JP 2008/072129, и при этом достигается генная экспрессия на уровне, превосходящем не менее чем в 5 раз средний относительный уровень экспрессии всех генов через 50 часов или позже от начала культивирования дрожжей. В частности, более предпочтительные примеры включают ген PDC1 и ген TDH3, которые экспрессируются на высоком уровне на пути ферментации этанола в дрожжах; и ген SED1, который экспрессируется на высоком уровне в дрожжах, культивируемых в течение длительного периода времени. Как указывалось выше, еще более предпочтительно, описанный выше полинуклеотид вводят таким образом, чтобы разрушить по меньшей мере ген PDC1, служащий в качестве исходной точки на пути ферментации этанола, или ген PDC5, предпочтительно ген PDC1, поскольку это приводит к метаболизации пировиноградной кислоты на метаболическом пути этанола, используемого при метаболизме D-молочной кислоты.
Далее, в качестве способа введения указанной выше конструкции ДНК в дрожжевую хромосому таким же способом, как и в случае введения описанного выше полинуклеотида в дрожжевую хромосому, может быть получена конструкция ДНК для гомологичной рекомбинации и введена в нужный сайт, расположенный на хромосоме, путем гомологичной рекомбинации. В этой связи следует отметить, что описанная выше конструкция ДНК уже содержит промотор, способный экспрессировать описанный выше полинуклеотид, нет необходимости осуществлять встраивание конструкции ДНК таким образом, чтобы указанный выше полинуклеотид мог регулироваться промотором эндогенного гена, имеющегося в дрожжевой хромосоме.
Согласно настоящему изобретению, подтвердить встроился/встроилась ли указанный/ая выше полинуклеотид или конструкция ДНК в нужное положение на дрожжевой хромосоме, можно по методу ПЦР или по методу саузерн-гибридизации. Например, указанное подтверждение можно осуществить при получении ДНК из трансформированных дрожжей, с проведением впоследствии ПЦР со встроенным сайт-специфичным праймером и далее детекции ожидаемой полосы путем электрофореза полученного продукта ПЦР. Альтернативно, указанное подтверждение можно получить при проведении ПЦР с праймером, меченным флуоресцентным красителем или т.п. Такие способы известны специалистам в данной области.
Настоящее изобретение также относится к способу получения D-молочной кислоты, который включает стадию культивирования указанного выше трансформанта. При культивировании описанного выше трансформанта вновь достигается или усиливается D-ЛДГ активность в данном трансформанте и в этой связи заново осуществляется или усиливается метаболический путь D-молочной кислоты. В результате, D-молочная кислота может быть получена на стадии отделения D-молочной кислоты от культуры. В контексте настоящего описания термин "культура" включает культуральный супернатант, а также трансформант и его гомогенат.
Способ культивирования описанного выше трансформанта особо не ограничивается и может применяться уже известный в данной области способ. Например, в качестве способа культивирования указанных выше дрожжей может использоваться способ, описанный в работе «M.D. Rose et al., 'Methods In Yeast Genetics', Cold Spring Harbor Laboratory Press (1990)», или аналогичный способ. Согласно настоящему изобретению, в тех случаях, когда плазмида, содержащая указанную выше конструкцию ДНК, или конструкцию ДНК для гомологичной рекомбинации содержит селектируемый маркер, при культивировании описанного выше трансформанта в среде без добавленных питательных компонентов или в среде с лекарственным средством, в зависимости от селектируемого маркера, может быть отобран желательный трансформант.
В качестве среды, в которой культивируют описанный выше трансформант, может использоваться любая природная среда или синтетическая среда, которая содержит источник углерода, источник азота, неорганические соли и т.п., которые может утилизировать трансформант и которые позволяют осуществлять его эффективное культивирование. Например, в случае трансформированных дрожжей, в качестве источника углерода, могут использоваться углеводы, такие как глюкоза, фруктоза, сахароза, галактоза, мальтоза, раффиноза, трегалоза, сорбоза, целлобиоза, лактоза, мелибиоза, мелезитоза, инулин, ксилоза, арабиноза, рибоза, рамноза, глюкозамин, эритрит, рибитол, маннит, глюцитол, салицин и крахмал; органические кислоты, такие как уксусная кислота, пропионовая кислота и лимонная кислота; и спирты, такие как этанол и пропанол. В качестве источника азота, кроме аммониевых солей органических или неорганических кислот, таких как аммиак, хлорид аммония, сульфат аммония, ацетат аммония и фосфат аммония, могут использоваться и другие азот-содержащие соединения, пептон, мясной экстракт, кукурузный экстракт и т.п. В качестве неорганического соединения, могут использоваться монофосфат калия, фосфат магния, сульфат магния, хлорид натрия, сульфат железа, сульфат марганца, сульфат меди, карбонат кальция и т.п.
Обычно при культивировании описанного выше трансформанта условия подбирают таким образом, чтобы достигалась хорошая продукция молочной кислоты, где указанные условия могут варьировать от аэробного культивирования, когда выращиваемая культура подвергается встряхиванию или аэрации, перемешивается до анаэробных условий, когда аэрация не проводится, и культивирование предпочтительно проводят в микроанаэробных условиях или анаэробных условиях. Культивирование может проводиться при температуре культивирования от 25 до 37°C; однако предпочтительной является температура в диапазоне 28-35°C. Поскольку pH культуры снижается по мере накопления D-молочной кислоты в среде, среда может быть также нейтрализована добавлением щелочного вещества, такого как гидроксид кальция, карбонат кальция, гидроксид натрия, гидроксид калия, водный раствор аммиака или газообразный аммиак. Однако для облегчения последующей очистки молочной кислоты культивирование может также проводиться без такой нейтрализации. При этом способ культивирования особо не ограничивается, главное, чтобы он позволял осуществлять ферментативную продукцию D-молочной кислоты, представляющей интерес, может применяться периодическое культивирование, культивирование с подпиткой, хемостатное культивирование, непрерывное культивирование или т.п. Предпочтительно, культивирование проводится в режиме периодической культуры или непрерывной культуры с использованием мембраны, описанной в WO2007/097260, где ферментационная культура разделяется на фильтрат и не-фильтрат с использованием разделяющей мембраны, устойчивой к засорению, и желательный продукт ферментации собирают с фильтра, тогда как не-фильтрат в ней задерживается или дальше направляется в ферментационнную среду для рециркуляции.
Способ определения уровня D-молочной кислоты, полученной при выращивании описанной выше культуры, особо не ограничивается, и его примеры включают способ с использованием ВЭЖХ и способ с использованием F-набора (производство компании Roche).
D-молочная кислота, содержащаяся в описанной выше ферментационной среде, может быть выделена и очищена с использованием сочетания известных способов, основанных на концентрировании, перегонке и кристаллизации и т.п., примеры которых включают способ, в соответствии с которым рН фильтруемой и отделяемой жидкости корректируют до уровня не выше 1, с проведением впоследствии экстракции диэтиловым эфиром, этилацетатом или т.п.; способ, в соответствии с которым молочную кислоту адсорбируют на ионообменной смоле, с последующими промывкой и элюцией; способ, в соответствии с которым молочную кислоту подвергают реакции со спиртом в присутствии кислотного катализатора с получением сложного эфира, который затем подвергают перегонке; способ, в соответствии с которым молочную кислоту кристаллизуют в виде соли кальция или соли лития; и способ отделения и очистки, в рамках которого используют в сочетании нанофильтр, описанный в WO2009/004922, и дистилляцию.
ПРИМЕРЫ
Ниже описываются способы осуществления настоящего изобретения с помощью примеров; однако все эти способы приведены лишь как иллюстрация некоторых вариантов осуществления настоящего изобретения, и настоящее изобретение ими никоим образом не ограничивается.
Пример 1
Нуклеотидное секвенирование полинуклеотидов из Limulus polyphemus, которые кодируют полипептид с D-ЛДГ активностью (далее обозначаемые как "D-ЛДГ гены из Limulus polyphemus")
Для получения кДНК из Limulus polyphemus выделяют и очищают поли A(+)РНК. Поли A(+)РНК выделяют из гемоцитов Limulus polyphemus. Суммарную РНК выделяют из гемоцитов Limulus polyphemus (полученных от Marine Biological Laboratory (USA)) по протоколу AGPC (см. Experimental Medicine Vol.9 (1991), pp.1937-1940). Далее, выделяют поли А(+)РНК из полученной в результате суммарной РНК с использованием набора "Oligotex-dT30 SuperKit" (производство компании Takara Bio Inc.). После этого, с использованием обратной транскриптазы "SuperScript II Reverse Transcriptase" (производство компании Invitrogen) и олиго d(T) в качестве праймера, синтезируют кДНК. Далее, с использованием указанного реакционного раствора в качестве матрицы и олиго ДНК с последовательностями, показанными в SEQ ID NO:8 и SEQ ID NO:9 в качестве праймеров, где указанные олиго ДНК были разработаны с учетом последовательностей ДНК, характеризующихся высокой консервативностью в D-ЛДГ генах, проводили ПЦР для амплификации генов D-ЛДГ из Limulus polyphemus. Указанную ПЦР проводили в следующих условиях: 95°C (3 минуты); {95°C (30 секунд) - 45°C (30 секунд) - 72°C (30 секунд)}×35 циклов; 72°C (5 минут). Далее, в 1,0% геле агарозы проводили электрофорез амплифицированных таким образом ДНК для подтверждения их структуры, и после очистки амплифицированного фрагмента размером 600 п.н. с использованием набора для быстрой экстракции из геля "QIA quick Gel extraction kit" (производство компании Qiagen), указанный фрагмент клонировали в плазмидном векторе "pGEMt-easy" (производство компании Promega) и секвенировали ДНК. Нуклеотидное секвенирование проводили с использованием набора "Taq DyeDeoxy Terminator Cycle Sequencing Kit" (производство компании Applied Biosystems) по методу Сэнгера (Sanger). В результате, были получены два вида частичных последовательностей (D-ЛДГ1 и D-ЛДГ2), которые, предположительно, соответствуют D-ЛДГ генам из Limulus polyphemus.
Затем, по процедурам 5'-RACE и 3'-RACE методов, определяли полноразмерные последовательности ORF для D-LDH1 и D-LDH2 генов. Анализ в рамках 5'-RACE проводили по следующей ниже процедуре. Вначале, 50 мкл олиго ДНК, показанных в SEQ ID NO:10 (для D-LDH1) или SEQ ID NO:11 (для D-LDH2), откорректированных до количества 100 мкМ, 50 единиц полинуклеотидкиназы T4 (производство компании Takara Bio Inc.), 10 мкл 10х киназного буфера (500 мМ Трис-HCl pH7,6, 100 мМ MgCl2, 50 мМ ДТТ, 1 мМ спермидина, 1 мМ ЭДТА), 1 мкл 100 мМ АТФ и 34 мкл дистиллированной воды смешивали и оставляли на 1 час при температуре 37°C для фосфорилирования 5'-конца олиго ДНК, и указанную олиго ДНК очищали при проведении обработки фенолом-хлороформом-изоамиловым спиртом и осаждали этанолом. Далее, с использованием поли A(+)РНК из Limulus polyphemus в качестве матрицы, обратной транскриптазы "SuperScript II Reverse Transcriptase" (производство компании Invitrogen) и описанных выше фосфорилированных олиго ДНК, в качестве праймеров, синтезировали кДНК. После этого конкатемеризовали одноцепочечную кДНК в реакции лигирования с использованием набора "5'-Full RACE Core Set" (производство компании Takara Bio Inc.) и проводили первую ПЦР с использованием полученной таким образом конкатемеризованной ДНК в качестве матрицы и олиго ДНК, показанных в SEQ ID NO:12 и 13 (для D-LDH1) или в SEQ ID NO:14 и 15 (для D-LDH2) в качестве праймеров. Затем проводили вторую ПЦР с использованием полученного реакционного раствора в качестве матрицы и олиго ДНК, показанных в SEQ ID NO:16 и 17 (для D-LDH1) или в SEQ ID NO:18 и 19 (для D-LDH2) в качестве праймеров. Подтверждение идентичности полученных фрагментов и их очистку, а также их нуклеотидное секвенирование проводили по описанной выше процедуре.
Анализ 3'-RACE проводили по следующей процедуре. Вначале, с использованием поли A(+)РНК из Limulus polyphemus в качестве матрицы, обратной транскриптазы "SuperScript II Reverse Transcriptase" (производство компании Invitrogen) и олиго d(T) в качестве праймера, синтезировали кДНК. Далее, с использованием этого реакционного раствора в качестве матрицы и олиго d(T) и олиго ДНК с последовательностью, соответствующей SEQ ID NO:13 (для D-LDH1) или SEQ ID NO:15 (для D-LDH2) в качестве праймеров, проводили первую ПЦР. Затем проводили вторую реакцию ПЦР с использованием полученного реакционного раствора в качестве матрицы и олиго d(T) и олиго ДНК с последовательностью, соответствующей SEQ ID NO:17 (для D-LDH1) или SEQ ID NO:19 (для D-LDH2) в качестве праймеров. Подтверждение идентичности полученных фрагментов и их очистку, а также их нуклеотидное секвенирование проводили по описанной выше процедуре.
Аминокислотную последовательность (SEQ ID NO:1), кодируемую геном D-LDH1 из Limulus polyphemus, и последовательность ORF для кДНК (SEQ ID NO:3) или аминокислотную последовательность (SEQ ID NO:2), кодируемую геном D-LDH2, и последовательность ORF для кДНК (SEQ ID NO:4) определяли при лигировании последовательностей, полученных по указанному выше способу. Следует отметить, что идентичность на уровне последовательности аминокислотных последовательностей, соответствующих SEQ ID NO:1, а также нуклеотидных последовательностей, показанных в SEQ ID NO:3, определенная с помощью программы BLAST, составляла 93% и 82%, соответственно.
Пример 2
Конструирование плазмиды, экспрессирующей гены D-LDH из Limulus polyphemus
С использованием, в качестве праймерного набора, олиго ДНК с последовательностью SEQ ID NO:20 и 21 или с последовательностью SEQ ID NO:22 и 23, которые, соответственно, относятся к 5'-положению ORF и 3'-положению ORF гена D-LDH1 и гена D-LDH2, которые были секвенированы, в рамках примера 1 и которые, как считается, представляют собой гены D-LDH из Limulus polyphemus (далее обозначаемые как "ген Lp.D-LDH1" и "ген Lp.D-LDH2", соответственно), и, в качестве матрицы, кДНК, синтезированной на основе поли A(+)РНК из Limulus polyphemus, проводили ПЦР для клонирования этих генов. Полученные при этом фрагменты ДНК расщепляли рестриктазами XhoI и NotI и вводили в сайты расщепления вектора экспрессии для дрожжей pTRS11 (который содержит промотор TDH3 и селективный маркер гена URA3; см. JP 2006-280368A; следует отметить, что приведенный в указанной ссылке TDH3 промотор и терминатор указаны как GAPDH промотор и терминатор), который таким же образом расщепляли рестриктазами XhoI и NotI для получения плазмидных векторов, содержащих конструкцию ДНК, в которой связаны TDH3 промотор и ген Lp.D-LDH 1 или ген Lp.D-LDH2. Далее, такие плазмидные векторы обозначаются как "pTRS205" (которые содержат Lp.D-LDH1) и "pTRS206" (которые содержат Lp.D-LDH2). В настоящем изобретении вектор pTRS11 получали при расщеплении вектора pNV11 (см. Nature, vol.357, 25 JUNE 1992, p.700) рестриктазой XhoI и после удаления вставок оставляли для самолигирования.
Пример 3
Введение плазмиды, экспрессирующей гены D-LDH из Limulus polyphemus, в дрожжи
Векторы pTRS205 и pTRS206, полученные по процедуре примера 2, вводили в дрожжи Saccharomyces cerevisiae, штамм SW029-1B (генотип: MATa ura TRP(Δpdc1::TRP1) his ade lys leu) (далее обозначаемый как "штамм SW029-IB"). Введение плазмид проводили по методу с использованием ацетата лития с использованием системы для трансформации дрожжей "YEASTMAKER Yeast Transformation System" (производство компании Clonetech) (подробнее описано в прилагаемом протоколе). Используемый в качестве хозяина штамм SW029-1B представляет собой штамм, в котором утрачена способность к синтезу урацила, и за счет селективного маркера для гена URA3 из pTRS205 и 206 на среде без урацила могут быть отобраны трансформированные дрожжи, в которые были встроены pTRS205 и pTRS206. Введение векторов, экспрессирующих ген D-LDH, в полученные таким образом трансформанты подтверждали путем экстракции геномной ДНК, содержащей плазмидную ДНК из трансформантов, культивированных на среде без урацила, с использованием набора для экстракции геномной ДНК "Dr. GenTLE" (производство компании Takara Bio Inc.) и путем проведения далее ПЦР с использованием экстрагированной таким образом геномной ДНК в качестве матрицы. В качестве праймеров использовали такие праймеры, которые применяли при клонировании генов D-LDH из Limulus polyphemus, соответственно. В результате, было доказано, что каждый из генов D-LDH, полученных из Limulus polyphemus, соответственно, был встроен во все трансформированные клетки дрожжей.
Пример 4
Тест 1 на продукционную способность по D-молочной кислоте
С использованием штаммов Saccharomyces cerevisiae SW029-1B, полученных по процедуре примера 3, в которые были встроены векторы pTRS205 и 206 (далее обозначаемые как "штамм SW029-1B/pTRS205" и "штамм SW029-1B/pTRS206", соответственно), проводили тест на продукционную способность по D-молочной кислоте.
В опытные пробирки добавляли по 10 мл среды SC3, состав которой показан в таблице 1, из которой был удален урацил (среда SC3-Ura). В указанную среду инокулировали небольшое количество каждого из исследуемых штаммов и проводили культивирование в течение ночи при температуре 30°C (предварительная культура). Далее, в опытные пробирки добавляли по 10 мл среды SC3-Ura и по 100 мкл каждой из полученных сред для предварительной культуры и затем культивировали при температуре 30°C на качалке (основная культура). После 40-часового роста основной культуры культуральные среды центрифугировали и количество содержащейся в полученных супернатантах молочной кислоты определяли по методу ВЭЖХ в следующих условиях: колонка: "Shim-Pack SPR-H" (производство компании Shimadzu Corporation); подвижная фаза: 5 мМ п-толуолсульфоновая кислота (скорость течения = 0,8 мл/мин); реакционный раствор: 5 мМ п-толуолсульфоновая кислота, 20 мМ Bistris, 0,1 мМ ЭДТА-2Na (скорость течения = 0,8 мл/мин).
Способ детекции: основан на измерении электропроводности
Температура: 45°C
Далее, на основании результатов измерения концентраций D-молочной кислоты и L-молочной кислоты по методу ВЭЖХ в указанных ниже условиях рассчитывали оптическую чистоту D-молочной кислоты по приведенным ниже уравнениям.
Колонка: "TSK-gel Enantio LI" (зарегистрированная торговая марка: производство компании Tosoh Corporation)
Подвижная фаза: 1 мМ водный раствор сульфата меди
Скорость течения: 1,0 мл/мин
Метод детекции: в УФ области при длине волны 254 нм
Температура: 30°C.
Оптическая чистота (% e.e.)=100×(D-L)/(D+L)
Оптическая чистота (%)=100×D/(D+L)
В указанных выше уравнениях L обозначает концентрацию L-молочной кислоты, и D обозначает концентрацию D-молочной кислоты.
В результате, было показано, что концентрация накопленной D-молочной кислоты составляла 13 г/л для штамма SW029-1B/pTRS205 и 12 г/л для штамма SW029-1B/pTRS206. Кроме того, в культуральной среде была обнаружена только D-молочная кислота и уровень L-молочной кислоты не превышал предела определения. Было подтверждено, что трансформированные дрожжи были способны продуцировать D-молочную кислоту в случае встраивания в них D-LDH генов, клонированных из Limulus polyphemus.
Пример 5
Встраивание D-LDH генов из Limulus polyphemus в хромосому дрожжей
При проведении стадий 1-3 получали конструкцию ДНК для гомологичной рекомбинации, которая содержит D-LDH гены, полученные из Limulus polyphemus.
[Стадия 1]
С использованием pTRS205, содержащего ген Lp.D-LDH1, и pTRS206, содержащего Lp.D-LDH2, которые были получены по процедуре примера 2 в качестве матриц и олиго ДНК с последовательностями SEQ ID NO:24 и 25 или с последовательностями SEQ ID NO:26 и 25 в качестве праймерного набора проводили ПЦР для амплификации фрагментов ДНК размером примерно 1,1 тыс. пар оснований, содержащих каждый из D-LDH генов. В настоящем изобретении указанные олиго ДНК, показанные в SEQ ID NO:24 и 26, были сконструированы таким образом, чтобы последовательность была гомологична участку размером 65 пар оснований в гене PDC1 против направления считывания информации.
[Стадия 2]
Далее, с использованием плазмиды pRS424 (номер доступа в GenBank: U03453) в качестве матрицы и олиго ДНК, соответствующих SEQ ID NO:27 и 28, в качестве праймерного набора, проводили ПЦР для амплификации фрагмента ДНК размером 1,4 тыс. пар оснований, содержащих ген TRP1, который представляет собой маркер селекции для дрожжей. В настоящем изобретении олиго ДНК, показанная в SEQ ID NO:28, была сконструирована таким образом, что к ней добавляется последовательность, гомологичная участку размером 65 пар оснований в гене PDC1 против направления считывания информации.
[Стадия 3]
После разделения фрагментов ДНК путем электрофореза в 1% геле агарозы указанные фрагменты очищали с использованием набора для быстрой экстракции из геля "QIA quick Gel extraction kit" (производство компании Qiagen). С использованием смеси полученных таким образом фрагментов размером 1,1 тыс. пар оснований, содержащих соответствующие D-LDH гены, и фрагмент размером 1,4 тыс. пар оснований, содержащий ген TRP1 в качестве матрицы и олиго ДНК, показанные в SEQ ID NO:24 и 28, или в SEQ ID NO:26 и 28, в качестве праймерного набора, проводили ПЦР для амплификации конструкции ДНК размером примерно 2,5 тыс. пар оснований, используемой для гомологичной рекомбинации, где связаны все указанные гены: гены D-LDH, терминатор TDH3 и ген TRP1.
Конструкцию ДНК для гомологичной рекомбинации, полученную описанным выше способом, трансформировали штаммом Saccharomyces cerevisiae SW092-2D (далее обозначаемым как "штамм SW092-2D"), который представляет собой штамм Saccharomyces cerevisiae NBRC10505 (далее обозначаемый как "штамм NBRC10505"), в котором реверсирована ауксотрофность по лизину.
Штамм SW092-2D получали по следующей процедуре. С использованием геномной ДНК из штамма Saccharomyces cerevisiae BY4741, которую производит компания Funakoshi Corporation в качестве матриц и олигонуклеотидов (SEQ ID NO:29 и 30) в качестве праймерного набора, проводили ПЦР для амплификации ПЦР фрагмента размером примерно 2 тыс. пар оснований, соответствующего прежней половине LYS2 гена. После выделения ПЦР фрагмента электрофорезом в 1% геле агарозы и очистки по стандартной методике указанный ПЦР фрагмент трансформировали штаммом NBRC10505 для удаления amber мутации в LYS2 гене. При культивировании штамма дрожжей в не содержащей лизина среде, был отобран трансформант, в котором была достигнута реверсия способности к синтезу лизина (штамм NBRC10505(LYS2)). Для подтверждения того, что полученный таким образом трансформант представляет собой дрожжи, в которых удалена amber мутация в LYS2, проводили следующие стадии. Вначале, получали диплоидные клетки при спаривании полученного трансформанта со штаммом Saccharomyces cerevisiae L0GY7, содержащим ген LYS2 дикого типа, и указанные диплоидные клетки культивировали в среде для образования сумки, с тем, чтобы получить сформированную сумку. Полученные сумки анатомировали с использованием микроманипулятора с целью получения соответствующих гаплоидных клеток (тетрад), и ауксотрофность оценивали для каждой из гаплоидных клеток. Все полученные таким образом клетки способны синтезировать лизин. Полученные, таким образом, штамм NBRC10505(LYS2) и штамм NBRC10506 спаривали и образовавшиеся сумки для спороношения анатомировали с использованием микроманипулятора с получением штамма SW092-2D (генотип: MATa ura3 leu2 trp1 his3 ade2 LYS2).
Штамм SW092-2D трансформировали описанной выше конструкцией ДНК для гомологичной рекомбинации и культивировали в среде без триптофана для отбора трансформированных дрожжей. Полученные таким образом трансформированные дрожжи далее обозначали как "штамм SW092-2D (Δpdc1::Lp.D-LDH1-TRP1)" и "штамм SW092-2D (Δpdc1::Lp.D-DLH2-TRP1)".
Пример 6
Тест 2 на продукцию D-молочной кислоты
С использованием SW092-2D (штамм Δpdc1::Lp.D-LDH1-TRP1), SW092-2D (штамм Δpdc1::Lp.D-LDH2-TRP1) ферментер с минирезервуаром (производство компании B.E.Marubishi Co., Ltd., 5 л) оценивали их ферментацию.
Среду SC3, состав которой приведен в таблице 1, вносили в опытную пробирку в количестве 10 мл и инокулировали в нее небольшое количество штамма и культивировали в течение ночи при температуре 30°C (предварительная прекультура). Затем, к 45 мл среды SC3, внесенной в колбу Эрленмейера, добавляли 5 мл полученной таким образом среды с предварительной прекультурой и культивировали при температуре 30°C еще в течение 8 часов (прекультура). Общее количество полученной таким образом среды с прекультурой инокулировали в 1 л среды SC3, помещенной в ферментер с минирезервуаром, и культивировали в течение 30 часов. Ниже описаны условия культивирования::
pH: pH5
Аэрация: 100 мл/мин
Перемешивание: 120 об/мин
Нейтрализующее вещество: 1н гидроксид натрия.
Количество культуральной среды в конце периода культивирования и концентрации D-молочной кислоты и глюкозы в культуральной среде измеряли с помощью тестера на глюкозу "Glucose Test Wako C" (зарегистрированная торговая марка) (производство компании Wako Pure Chemical Industries, Ltd.) для определения выхода молочной кислоты по сахариду относительно количества внесенной глюкозы, который рассчитывается на основании определенных таким образом концентраций молочной кислоты и глюкозы. В результате, было показано, что SW092-2D (Δpdc1::Lp.D-LDH1-TRP1) и SW092-2D (Δpdcl::Lp.D-LDH2-TRP1) продуцируют D-молочную кислоту с выходом молочной кислоты по сахариду 45% и 42%, соответственно.
Сравнительный пример 1
Клонирование гена D-LDH из молочной кислой бактерии и оценка ферментативной способности
В качестве гена D-LDH, способного эффективно осуществлять ферментативную продукцию D-молочной кислоты в трансформированных дрожжах, известен ген D-LDH из штамма Leuconostoc mesenteroides ATCC 9135, и этот ген был использован в данном примере (см. WO2004/104202). При клонировании данного гена D-LDH (далее называемого как "ген Lm.D-LDH") и встраивании его в дрожжевые клетки для последующей ферментации оценивали D-ЛДГ активность гена Lm.D-LDH при сравнении ее с активностью гена Lp.D-LDH1 или гена Lp.D-LDH2 из Limulus polyphemus.
С использованием праймерного набора (SEQ ID NO:31 и 32), который был разработан применительно к нуклеотидным последовательностям, описанным в WO2004/104202, и с учетом использования для этого штамма ATCC 9135 в качестве матрицы ген Lm.D-LDH клонировали по методу молекулярных колоний ПЦР в геле (с помощью полимеразы "KOD-Plus-polymerase", производство компании Toyobo Co., Ltd.). После очистки полученного таким образом ПЦР- амплифицированного фрагмента и фосфорилирования его конца с использованием полинуклеотидкиназы из фага Т4 "T4 Polynucleotide Kinase" (производство компании Takara Bio Inc.), указанный фрагмент лигировали с вектором pUC118 (который был расщеплен рестриктазой HincII и полученную после разрыва поверхность дефосфорилировали). Лигирование проводили с использованием набора для лигирования ДНК "DNA Ligation Kit Ver.2" (производство компании Takara Bio Inc.). Штамм Escherichia coli DH5a трансформировали продуктом лигирования с плазмидой и собирали плазмидную ДНК с получением впоследствии плазмидной ДНК, в которой проводилось субклонирование гена Lm.D-LDH. Полученную таким образом плазмиду pUC118, в которую был встроен ген Lm.D-LDH, расщепляли рестриктазами XhoI и NotI, и образованные при этом фрагменты ДНК, каждый, встраивали в сайт расщепления XhoI/NotI в векторе pTRS11 для экспрессии в клетках дрожжей. Таким образом, была получена плазмида pTRS207, экспрессирующая ген Lm.D-LDH.
Далее, ген Lm.D-LDH встраивали в локус гена PDC1 на хромосоме штамма Saccharomyces cerevisiae SW092-2D, аналогично тому, как это было описано в примере 5, с тем исключением, что использовали pTRS207 в качестве матрицы и праймерный набор с последовательностью, соответствующей SEQ ID NO:33, вместо праймера, показанного в SEQ ID NO:24 или 26. Полученные таким образом дрожжи далее по тексту будут обозначаться как "SW092-2D (Δpdc1::Lm.D-LDH-TRP1)". Затем по процедуре, описанной в примере 6, оценивали продуктивность по D-лактату при проведении периодического культивирования. В результате, было показано, что выход по сахариду для штамма SW092-2D (Δpdc1::Lm.D-LDH-TRP1) составляет 38%. Полученные данные, как и данные, относящиеся к примеру 6, показаны в таблице 2.
(Δpdc1::Lp.D-LDH1-TRP1)
(Δpdc1::Lp.D-LDH2-TRP1)
Δpdc1::Lm.D-LDH-TRP1)
Стандартный пример 1
Получение дрожжей с введенным в них геном L-LDH, взятым из Xenopus laevis
В соответствии со способом, описанным в JP 2008-029329A, были получены дрожжи, которые содержали ген L-LDH из Xenopus laevis (ген X.L-LDH), введенный в локус гена PDC1. В настоящем изобретении в качестве дрожжей, используемых для введения указанного гена в их хромосому, был взят штамм NBRC10506, в котором была реверсирована ауксотрофность по аденину (штамм SU013-1D). Штамм SU013-1D получали по описанному ниже способу. С использованием плазмиды pRS422 в качестве матрицы и олигонуклеотидов (SEQ ID NO:34 и 35) в качестве праймерного набора, проводили реакцию ПЦР для амплификации с целью получения фрагмента ПЦР размером примерно 2 тыс. п.н., соответствующего гену ADE2. После отделения фрагмента ПЦР электрофорезом на 1% геле агарозы и его очистки по стандартной методике, указанный фрагмент ПЦР трансформировали для удаления мутации в гене ADE2. При культивировании трансформированного штамма в среде без аденина отбирали трансформант, в котором была реверсирована способность к синтезу аденина.
Для подтверждения того факта, что полученный таким образом трансформант представляет собой дрожжи с удаленной мутацией в гене AED2 проводились следующие стадии анализа. Вначале, получали диплоидные клетки при спаривании указанного трансформанта со штаммом Saccharomyces cerevisiae L0GY77, содержащего ген ADE2 дикого типа, и далее диплоидные клетки культивировали в среде, способствующей образованию сумки для спороношения. Полученные сумки для спороношения анатомировали с использованием микроманипулятора, получая гаплоидные клетки (тетрады), и для каждой из таких гаплоидных клеток оценивали ауксотрофную способность. Было показано, что все указанные гаплоидные клетки способны синтезировать аденин. Полученные таким образом штамм NBRC 10506(ADE2) и штамм NBRC 10505 спаривали, и образовавшиеся при этом сумки анатомировали с использованием микроманипулятора с получением штамм SU013-1D (генотип: MATα ura3 leu2 trp1 his3 ADE2 lys2). Далее по тексту такие трансформированные дрожжи будут обозначаться как "штамм SU013-1D (Δpdc1::X.L-LDH-TRP1)".
Пример 7
Сравнительный пример 2. Тест 3 на продукционную способность по D-молочной кислоте
Штамм SU013-1D (Δpdc1::X.L-LDH-TRP1), полученный по процедуре стандартного примера 1, спаривали с каждым из описанных ниже штаммов: штамм SW092-2D (Δpdc1::Lp.D-LDH1-TRP1), штамм SW092-2D (Δpdc1::Lp.D-LDH2-TRP1) и штамм SW092-2D (Δpdc1::Lm.D-LDH-TRP1), полученных согласно способу, описанному в примере 5 и в сравнительном примере 1, с образованием в результате диплоидных дрожжей, содержащих ген L-LDH и гетерозиготный ген D-LDH в локусе гена PDC1. Далее по тексту полученные таким образом диплоидные дрожжи будут называться как "штамм Lp1-X, штамм Lp2-X и штамм Lm-X, соответственно".
Далее, указанные штаммы Lp1-X, Lp2-X и Lm-X культивировали в периодическом режиме в тех же условиях, которые были описаны в примере 5, и далее оценивали оптическую чистоту полученной молочной кислоты (таблица 3).
Таким образом, было показано, что в результате получены дрожжи, которые содержат ген D-LDH из Limulus polyphemus и которые продуцируют D-молочную кислоту с оптической чистотой не менее 50%, тогда как оптическая чистота D-молочной кислоты, продуцируемой дрожжами, содержащими ген D-LDH из Leuconostoc mesenteroides, составляла 44,4%. Поскольку выход относительно сахарида, при котором каждый из оцениваемых вариантов дрожжей продуцирует D-молочную кислоту, и ее оптическая чистота пропорциональны D-ЛДГ активности в дрожжах, трансформированных геном D-LDH, то из данных таблиц 2 и 3 видно, что при встраивании в клетки дрожжей ген D-LDH, взятый из Limulus polyphemus, кодирует полипептид, обладающий более высокой активностью, чем полипептид, кодируемый D-LDH геном из Leuconostoc mesenteroides.
Пример 8
Введение множественных копий гена D-LDH в дрожжевую хромосому и получение температурочувствительного мутанта дрожжей PDC5
Далее, рассматривали вариант получения дрожжей с геном D-LDH из Limulus polyphemus, который вводили также в генный локус, отличный от локуса гена PDC1, с тем, чтобы еще больше повысить выход D-молочной кислоты, и ген D-LDH вводили в ген TDH3, где эффект такого введения был описан в JP 2008-029329A, и в локус гена SED1, где эффект такого введения был описан в Международной заявке на патент PCT/JP 2008/072129. Затем ввели также ген температурочувствительного мутанта дрожжей PDC5, описанный в JP 2008-048726A.
[Введение гена D-LDH в локус гена TDH3]
Для введения в локус гена TDH3 по способу, аналогичному способу получения pTRS150, описанному в JP 2008-029329A, была получена плазмида pTRS208, в которой терминатор TDH3 в pTRS205, содержащей ген Lp.D-LDH1, был замещен терминатором ADH1. Далее, с использованием последовательности SEQ ID NO:36 вместо последовательности SEQ ID NO:8, описанной в JP 2008-029329A, в качестве праймера, и pTRS208, в качестве матрицы, использовали конструкцию ДНК для гомологичной рекомбинации с целью введения в локус гена TDH3.
В качестве дрожжей, в которые была встроена указанная выше конструкция ДНК для гомологичной рекомбинации, использовали прототрофный по лейцину штамм SU013-1D (штамм SW087-2C). Ниже приводится описание способа получения штамма SW087-2C. С использованием плазмиды pRS425 в качестве матрицы и олигонуклеотидов (SEQ ID NO:37 и 38) в качестве праймерного набора проводили реакцию ПЦР для амплификации ПЦР-фрагмента с размером примером 2 тыс. п.н., соответствующего гену LEU2. После отделения фрагмента ПЦР путем электрофореза в 1% геле агарозы и очистки по стандартной методике, указанный очищенный фрагмент ПЦР трансформировали в штамм SU013-1D с целью удаления мутации в гене LEU2. При культивировании указанного штамма дрожжей в не содержащей лейцина среде отбирали трансформант, в котором была реверсирована способность к синтезу лейцина. Для подтверждения того факта, что полученный трансформант представляет собой дрожжи, в которых удалена мутация в гене LEU2, проводили следующие стадии анализа. Вначале, получали диплоидные клетки при спаривании полученного таким образом трансформанта со штаммом Saccharomyces cerevisiae L0GY77, содержащим ген LEU2 дикого типа, и диплоидные клетки культивировали в среде, подходящей для образования сумки. Образованные сумки для спороношения анатомировали с использованием микроманипулятора с получением соответствующих гаплоидных клеток (тетрад) и оценивали ауксотрофную способность каждой из таких полученных гаплоидных клеток. Было показано, что все исследованные гаплоидные клетки обладают способностью к синтезу лейцина. Далее, спаривали полученный штамм SU013-1D(LEU2) и штамм NBRC10505 и полученные сумки для спороношения анатомировали с использованием микроманипулятора с получением штамма SW087-2C (генотип: МАТα ura3 LEU2 trp1 his3 ADE2 lys2).
Штамм SW087-2C трансформировали указанной выше конструкцией ДНК для гомологичной рекомбинации и культивировали в не содержащей урацила селективной среде с получением трансформированных дрожжей, содержащих ген Lp.D-LDH1, введенный в локус гена TDH3. Далее, в тексте полученные таким образом трансформированные дрожжи будут обозначаться как "штамм SW087-2C (ΔTDH3::Lp.D-LDH-URA3)".
[Введение гена D-LDH в локус гена SED1]
Введение в локус гена SED1 осуществляли при модификации способа, описанного в примере 2 в Международной заявке на патент PCT/JP 2008/072129. А именно: с использованием pTRS205 вместо pTRS102, в качестве матрицы для ПЦР, и последовательности SEQ ID NO:39 вместо последовательности SEQ ID NO:14, как было описано в приведенной выше заявке на международный патент в качестве праймера, получали конструкцию ДНК для гомологичной рекомбинации, подходящую для введения в локус гена SED1.
В качестве дрожжей, в которые была встроена указанная выше конструкция ДНК для гомологичной рекомбинации, использовали штамм SW092-7D (генотип: MATa ura3 leu2 trp1 his3 ADE2 LYS2), который был получен при спаривании штамма NBRC10505(LYS2), полученного по процедуре примера 5 со штаммом NBRC10506(ADE2), полученным по процедуре стандартного примера 1, и проводили отделение нужного штамма при анатомировании полученных сумок для спороношения с помощью микроманипулятора.
Штамм SW092-7D трансформировали описанной выше конструкцией ДНК для гомологичной рекомбинации и культивировали в не содержащей гистидина селективной среде с получением трансформированных дрожжей, содержащих введенный в них ген Lp.D-LDH1 в локусе гена SED1. Далее, в тексте полученные таким образом трансформированные дрожжи будут называться как "штамм SW092-7D (ΔSED1::Lp.D-LDH-HIS3)".
[Введение гена температурочувствительного мутантного типа PDC5 (pdc5ts-9)]
В качестве дрожжей, содержащих температурочувствительный ген мутантного типа PDC5, использовали штамм дрожжей SW015, содержащий температурочувствительный ген мутантного типа PDC5 (pdc5ts-9), описанный в JP 2008-048726A. При спаривании штамма SW015 со штаммом SW087-2C и последующем отделении образованных сумок для спороношения путем анатомирования с использованием микроманипулятора, получали штамм SW095-4B (генотип: MATa ura3 LEU2 trp1 his3 ADE2 lys2 pdc5ts-9 Δpdcl::TRP1). Далее, при спаривании штамма SW095-4B со штаммом SW092-2D и при отделении образованных сумок для спороношения путем анатомирования с использованием микроманипулятора, получали штамм SW098-21B (генотип: MATα ura3 LEU2 trp1 his3 ADE2 LYS2 pdc5ts-9).
[Введение гена D-LDH в локусы генов PDC1, TDH3 и SED1 и введение температурочувствительного гена мутантного типа PDC5 (pdc5ts-9)]
При использовании полученного по описанной выше методике штамма SW092-2D (ΔPDC1::Lp.D-LDH1-TRP1), SW087-2C (ΔTDH3::Lp.D-LDH1-URA3), штаммов SW092-7D (ΔTDH3::Lp.D-LDH1-HIS3) и SW098-21B, повторяли спаривание диплоидных клеток и образование тетрад с получением штамма SU042, который содержит ген Lp.D-LDH1 в генных локусах PDC1, TDH3 и SED1, и штамма SU042 c температурочувствительным геном мутантного типа PDC5 (pdc5ts-9), где указанный штамм является диплоидным и не ауксотрофным по аденину, лейцину и лизину. Процедуры получения штамма SU042 проиллюстрированы на фиг.1.
Пример 9
Тест 4 на продукционную способность по D-молочной кислоте
С использованием штамма SU042, полученного по описанной в примере 8 методике, оценивали его продукционную способность по D-молочной кислоте в режиме периодического культивирования. В качестве среды была использована среда SC3, описанная в таблице 1, или среда с неочищенным сахаром (100 г/л "Yutosei" (производство компании Muso Co., Ltd.), 1,5 г/л сульфата аммония). Вначале, штамм SU042 выращивали в течение ночи на качалке в 5 мл среды SC3 или среды с неочищенным сахаром в опытной пробирке (предварительная прекультура). В колбу Сакагучи объемом 500 мл в 50 мл свежей среды SC3 или среды с неочищенным сахаром инокулировали среду с указанной предварительной прекультурой и культивировали в течение 24 часов на качалке (прекультура). В ферментер с минирезервуаром (производство компании Able Co., Ltd., 2 л) добавляли 1 л среды SC3 или среды с неочищенным сахаром, корректировали температуру до нужного уровня (32°C) при контроле рН (с использованием pH 5, 5н гидроксида кальция). Культивирование проводили при аэрации и перемешивании (с соответствующими показателями 200 мл/мин и 400 об/мин). В результате, среда SC3 была потреблена за 30 часов и выход относительно сахарида составил 63%. Тогда как культура в среде с неочищенным сахаром закончила рост через 50 часов и выход относительно сахарида составил 70%. Исходя из этих результатов, был сделан вывод, что течение ферментации D-молочной кислоты улучшается при введении гена Lp.D-LDH1 в генные локусы PDC1, TDH3 и SED1 и при введении мутации температурочувствительности в ген PDC5.
Пример 10
Тест на продукционную способность по D-молочной кислоте, осуществляемую в режиме непрерывного культивирования
Исследовали в режиме непрерывного культивирования штамм SU042 с использованием сепарирующей мембраны, описанной в WO2007/097260. В качестве среды использовали среду с неочищенным сахаром (75 г/л "Yutosei" (производство компании Muso Co., Ltd.), 1,5 г/л сульфата аммония).
Условия культивирования описаны ниже:
Емкость резервуара для ферментации: 2 л
Объем культуральной среды: 1,5 л
Использованная сепарирующая мембрана: мембранный фильтр из PVDF (описанный в стандартном примере 2 в WO2007/097260)
Эффективная площадь фильтрации сепарирующего мембранного элемента: 120 см2
Контролируемая температура: 32°C
Объем аэрации, поддерживаемый в реакционном сосуде ферментера: воздух 0,02 л/мин, газообразный азот 0,18 л/мин
Скорость перемешивания в реакционном сосуде при ферментации: 800 об/мин
Корректировка уровня pH: корректировка до pH 5 с использованием 5н гидроксида кальция
Стерилизация: сосуд для культивирования, включающий сепарирующий мембранный элемент и среду, автоклавировали при температуре 121°C в течение 20 минут.
Вначале, штамм SU042 культивировали при температуре 30°C в течение ночи на качалке в 10 мл среды с неочищенным сахаром в опытной пробирке (предварительная пре-прекультура). Все количество полученной при этом культуральной среды инокулировали в 100 мл свежей среды с неочищенным сахаром и культивировали в колбе Сакагучи объемом 500 мл в течение 24 часов при температуре 30°C на качалке (предварительная прекультура). Полученную среду с предварительной прекультурой инокулировали в 1,5 л среды с неочищенным сахаром, внесенной в резервуар ферментера для непрерывного культивирования со встроенной мембраной (указанный прибор показан на фиг.2 в WO2007/097260). К отбору культуральной среды с помощью перистальтического насоса (200 мл/час) приступали через 50 часов после начала роста культуры и продолжали культивирование до 400 часов, с тем, чтобы определить концентрацию молочной кислоты, как продуцируемого в ходе культивирования вещества, а также уровень продукции молочной кислоты. Полученные результаты проиллюстрированы на фиг.2. Следует в этой связи отметить, что уровень продукции молочной кислоты при непрерывном культивировании рассчитывали по уравнению 1, приведенному ниже.
Уровень продукции молочной кислоты (г/л·час) = концентрация продукта в отобранной жидкости (г/л) × скорость отбора ферментационной культуральной среды (л/час)÷ рабочий объем жидкости в приборе (л) (Уравнение 1)
В результате непрерывного культивирования с использованием сепарирующей мембраны было показано, что выход D-молочной кислоты относительно сахарида дополнительно улучшился примерно на 85% и уровень продукции D-молочной кислоты возрос до 7,5 г/л·час.
Пример 11
Введение D-LDH из Limulus polyphemus в Candida utilis
Далее, с использованием Candida utilis, представляющей собой дрожжи, отрицательные по эффекту Крэбтри, исследовали образование D-молочной кислоты.
(a) Получение вектора для введения гена D-LDH из Limulus polyphemus D-LDH
Вначале, конструировали вектор для введения гена D-LDH из Limulus polyphemus в локус PDC1 гена в хромосоме Candida utilis. Штамм Candida utilis NBRC0988 (далее обозначаемый как "NBRC0988") инокулировали в среду YPD (1% бакто-дрожжевой экстракт (Bacto Yeast Extract), 2% бакто-пептон, 2% глюкоза) и культивировали в течение ночи при температуре 30°C. Из полученных таким образом дрожжевых клеток экстрагировали геномную ДНК по стандартной методике и использовали ее в качестве матрицы в последующей реакции ПЦР. Вначале, с использованием олиго ДНК, показанных в SEQ ID NO:40 и 41 в качестве праймерного набора амплифицировали фрагмент, содержащий терминирующую область PDC1 гена. В этой связи следует отметить, что, если не указано иное, то в качестве ДНК-полимеразы использовали KOD-plus- (производство компании Toyobo Co., Ltd.) по прилагаемому протоколу. После расщепления полученного таким образом фрагмента размером примерно 1 тыс. п.н. рестриктазой BssHI и его очистки, полученный фрагмент лигировали с pBluescriptIISK(+), ранее также расщепленной рестриктазой BssHI по тому же методу. Полученная при этом плазмида далее будет обозначаться как "pKSO1".
Далее, по такому же способу, с использованием геномной ДНК из NBRC0988 в качестве матрицы и олиго ДНК, показанных в SEQ ID NO:42 и 43 в качестве праймерного набора проводили реакцию ПЦР для амплификации фрагмента ДНК, содержащего промотор PGK. Полученный при этом фрагмент размером примерно 1 тыс. п.н. очищали и использовали в последующих экспериментах. Затем, с использованием плазмиды pLC1-hph, содержащей ген hph, способный придавать резистентность к гигромицину В, представляющему собой вещество антибиотической природы в качестве матрицы и олиго ДНК, показанных в SEQ ID NO:44 и 45 в качестве праймерного набора, проводили ПЦР для амплификации фрагмента, содержащего ген hph, который затем очищали с получением фрагмента размером примерно 1,1 тыс. п.н. Далее, с использованием геномной ДНК из NBRC0988 в качестве матрицы и олиго ДНК, показанных в SEQ ID NO:46 и 47 в качестве праймерного набора, проводили ПЦР для амплификации фрагмента, содержащего терминирующую последовательность GAP, которую очищали с получением фрагмента размером примерно 500 тыс. п.н.
Фрагмент, содержащий PGK промотор, фрагмент, содержащий ген hph, и фрагмент, содержащий GAP терминатор, которые были получены описанным выше способом, объединяли и с использованием олиго ДНК, показанных в SEQ ID NO:48 и 49, в качестве праймерного набора, проводили ПЦР для амплификации фрагмента, в котором связаны PGK промотор, ген hph и GAP терминатор. После фосфорилирования конца полученного таким образом фрагмента размером примерно 2,6 тыс. п.н. указанный фрагмент лигировали с pUC118, которая была расщеплена HincII, и далее дефосфорилировали. Полученная, в итоге, плазмида далее по тексту будет обозначаться как "pKS02".
Далее, таким же образом с использованием геномной ДНК из NBRC0988 в качестве матрицы и олиго ДНК, показанных в SEQ ID NO:50 и 51, в качестве праймерного набора, проводили ПЦР для амплификации фрагмента, содержащего PGK терминатор, который затем очищали с получением фрагмента размером примерно 500 тыс. п.н. Затем, с использованием pKS02, полученной описанным выше способом, в качестве матрицы и олиго ДНК, показанных в SEQ ID NO:52 и 53, в качестве праймерного набора, проводили ПЦР для амплификации фрагмента, в котором связаны PGK промотор, ген hph и GAP терминатор, который затем очищали с получением фрагмента размером примерно 2,6 тыс. п.н.
Фрагмент, содержащий PGK терминатор, и фрагмент, в котором связаны PGK промотор, ген hph и GAP терминатор, где указанные фрагменты были получены описанным выше способом, объединяли и с использованием олиго ДНК, показанных в SEQ ID NO:50 и 53, в качестве праймерного набора, проводили ПЦР для амплификации фрагмента, в котором связаны PGK терминатор, PGK промотор, ген hph и GAP терминатор. После расщепления полученного таким образом фрагмента размером примерно 3 тыс. п.н. рестриктазами BamHI и ClaI и его очистки указанный фрагмент лигировали с pKS02, которая предварительно аналогичным образом была расщеплена рестриктазами BamHI и ClaI. Полученная при этом плазмида далее по тексту будет обозначаться как "pKS03".
Далее, таким же образом, с использованием геномной ДНК из NBRC0988 в качестве матрицы и олиго ДНК, показанных в SEQ ID NO:54 и 55, в качестве праймерного набора, проводили ПЦР для амплификации фрагмента, содержащего промоторный участок PDC1, который затем очищали с получением фрагмента размером примерно 2,1 тыс. п.н. После этого с использованием pTRS205, полученной по методу примера 2, в качестве матрицы, и олиго ДНК, показанных в SEQ ID NO: 56 и 57, в качестве праймерного набора, проводили ПЦР для амплификации фрагмента, содержащего ген D-LDH, полученный из Limulus polyphemus, который затем очищали с получением фрагмента размером примерно 1 тыс. п.н.
Фрагмент, содержащий промоторный участок PDC1, и фрагмент, содержащий ген D-LDH из Limulus polyphemus, которые были получены по описанной выше процедуре, объединяли и с использованием олиго ДНК, показанных в SEQ ID NO: 54 и 57, в качестве праймерного набора, проводили ПЦР для амплификации фрагмента, в котором связаны фрагмент, содержащий промоторный участок PDC1, и фрагмент, содержащий ген D-LDH из Limulus polyphemus. После расщепления полученного таким образом фрагмента размером примерно 3,1 тыс.п.н. рестриктазами NotI и BglII и его очистки указанный продукт лигировали с pKS03, которая была аналогичным способом предварительно расщеплена рестриктазами NotI и BamHI. Далее, по тексту полученная при этом плазмида будет обозначаться как "pKS04".
(b) Введение гена D-LDH из Limulus polyphemus в генный локус PDC1
Полученную описанным выше способом плазмиду pKS04 расщепляли рестриктазой BglII и встраивали в NBRC0988 по методу электропорации. Небольшое количество клеток инокулировали в среду YPD и культивировали в течение ночи при температуре 30°C. После центрифугирования культуру отделяли от супернатанта, клетки промывали стерилизованной водой и 1M сорбитом и, в итоге, суспендировали в 1M сорбите. Затем, после объединения клеток и pKS04 проводили расщепление рестриктазой BglII и оставляли полученную смесь на 5 минут, после чего указанную смесь переносили в кювету для электропорации и проводили электропорацию при емкости 25 мкФ, напряжении 0,75 кВ и сопротивлении 800 Ом. После этого смесь переносили в среду YPD, содержащую 1M сорбит, и культивировали при температуре 30°C примерно в течение 4 часов, после чего вносили в среду YPD с добавкой 600 мкг/л гигромицина B. Из полученного резистентного к гигромицину В штамма экстрагировали геном и после подтверждения размеров полученных фрагментов, равных примерно 3 тыс. п.н. и 2 тыс. п.н., указанные фрагменты амплифицировали в реакции ПЦР с использованием олиго ДНК, показанных в SEQ ID NO:54 и 57 и SEQ ID NO:56 и 51, в качестве праймерного набора, было показано, что получены трансформированные дрожжи, в которых в генный локус PDC1 был введен ген D-LDH из Limulus polyphemus. Далее, по тексту полученные таким образом дрожжи будут обозначаться как "штамм CuLpLDH".
Сравнительный пример 2
Введение D-LDH из молочнокислой бактерии в Candida utilis
Аналогично описанию, приведенному в примере 11, вводили ген D-LDH, взятый из молочнокислой бактерии Leuconostoc mesenteroides. При этом следует отметить, однако, что в данном случае использовали олиго ДНК с последовательностью SEQ ID NO:58, вместо последовательности, соответствующей SEQ ID NO:55, которая использовалась в примере 11. Кроме того, в качестве матрицы использовали pTRS207, полученную в рамках сравнительного примера 1, вместо pTRS205, полученной по процедуре примера 2, использовали олиго ДНК, показанные в SEQ ID NO:59 и 60, вместо SEQ ID NO:56 и 57. Далее, по тексту полученная, таким образом, плазмида будет обозначаться как "pKS05".
Плазмиду, которая была получена описанным выше способом, вводили в штамм NBRC0988 по процедуре примера 11. Далее, по тексту полученный таким образом штамм, в котором ген D-LDH, взятый из молочнокислой бактерии, Leuconostoc mesenteroides, был встроен в генный локус PDC1, будет обозначаться как "штамм CuLmLDH".
Пример 12
Сравнительный пример 3. Тест на продуктивность по D-молочной кислоте в Candida utilis
С использованием штамма CuLpLDH и штамма CuLmLDH, полученного по процедуре примера 11 и сравнительного примера 2, соответственно, оценивали их продукционную способность по D-молочной кислоте. В колбу Сакагучи объемом 500 мл вносили среду YPD в количестве 50 мл и в небольших количествах инокулировали в нее штаммы CuLpLDH и CuLmLDH и культивировали при температуре 30°C в течение ночи на качалке (прекультура). После сбора культуральной среды и промывки ее свежей средой YPD указанную культуральную среду помещали для культивирования в ферментер с мини-резервуаром, в который был добавлен 1 л среды YPD (содержащей 10% глюкозы). Ниже описаны условия культивирования:
Количество исходного инокулята: инокулировали при ОП600=10
pH: pH6
Аэрация: 100 мл/мин
Перемешивание: 120 об/мин
Нейтрализация: с использованием 1н гидроксида кальция
Температура культивирования: 35°C.
Культивирование проводили в течение 40 часов, и в культуральной среде определяли концентрацию молочной кислоты и концентрацию глюкозы в точке 40 часов. К этому времени глюкоза была полностью потреблена. Кроме того, как следует из таблицы 4, результаты анализа продуктивности по потребленной глюкозе (выход по сахариду) и оптической чистоты D-молочной кислоты показали, что дрожжи (Candida utilis) с введенным в них геном D-LDH способны к ферментативной продукции D-молочной кислоты, и штамм CuLpLDH с введенным геном D-LDH из Limulus polyphemus характеризовался более высоким выходом данного продукта.
Пример 13
Сравнительный пример 4. Тест 2 на продуктивность Candida utilis по D-молочной кислоте
Далее, с использованием штаммов CuLpLDH и CuLmLDH определяли их продукционную способность по D-молочной кислоте с использованием в качестве источника сахаров пентозный сахар ксилозу. В колбу Сакагучи объемом 500 мл добавляли среду YPD в количестве 50 мл и инокулировали в нее небольшие количества штаммов CuLpLDH и CuLmLDH с последующим культивированием в течение ночи при температуре 30°C на качалке (прекультура). После сбора культуральной среды и ее промывки свежей средой YPD указанную культуральную среду вносили в ферментер с мини-резервуаром, в который был добавлен 1 л среды YPX10 (1% дрожжевой экстракт, 2% бактопептон, 4% ксилоза). Ниже описаны условия культивирования:
Количество исходного инокулята: инокулировали при ОП600=10
pH: pH6
Аэрация: 100 мл/мин
Перемешивание: 120 об/мин
Нейтрализация: для нейтрализации использовали 1н гидроксид кальция
Температура культивирования: 30°C.
Культивирование проводили в течение 60 часов, и в указанной временной точке 60 часов в культуральной среде определяли концентрацию молочной кислоты и концентрацию ксилозы при проведении соответствующих анализов. К этому моменту ксилоза была полностью потреблена. Дополнительно, как видно из данных таблицы 4, результаты анализа продуктивности на основе потребленной ксилозы (выход по сахариду) и оценки оптической чистоты D-молочной кислоты показывают, что дрожжи (Candida utilis) с введенным в них геном D-LDH обладают способностью к ферментативной продукции D-молочной кислоты с использованием ксилозы в качестве исходного материала, и при этом штамм CuLpLDH с введенным в него геном D-LDH из Limulus polyphemus характеризовался более высоким выходом.
Промышленная применимость
При использовании настоящего изобретения становится возможной стабильная продукция D-молочной кислоты с низкой стоимостью, и, поскольку образуемая при этом D-молочная кислота обладает высокой оптической чистотой, она предпочтительно используется в качестве сырья при получении полимеров.


















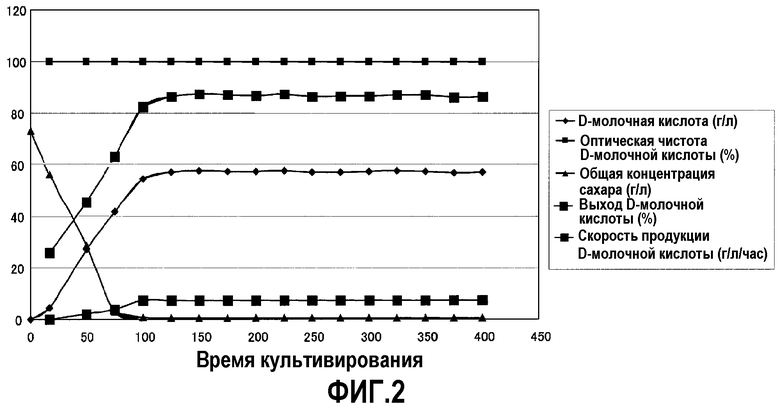
Изобретение относится к области биотехнологии. Представлен полипептид D-лактатдегидрогеназы, который содержит полипептид с аминокислотной последовательностью, которая характеризуется идентичностью по последовательности на уровне не менее 80% с аминокислотной последовательностью, показанной в SEQ ID NO:1 или 2, раскрытые в описании, где указанный полипептид обладает D-лактатдегидрогеназной активностью. Также представлены полинуклеотиды, кодирующие указанные полипептиды D-лактатдегидрогеназы; конструкция ДНК для экспрессии полинуклеотида, кодирующего D-лактатдегидрогеназу, в которой связаны указанный полинуклеотид и промотор, способный осуществлять экспрессию полинуклеотида. Описан трансформант, в частности трансформированные дрожжи, для получения D-молочной кислоты, в который введен полинуклеотид или конструкция ДНК по настоящему изобретению. Предложен способ получения D-молочной кислоты, который включает стадию культивирования указанного трансформанта. Изобретение позволяет получать D-молочную кислоту на повышенном уровне при помощи трансформанта по настоящему изобретению по сравнению с уровнем D-молочной кислоты, полученным с использованием нетрансформированной родительской клетки. 9 н. и 6 з.п. ф-лы, 2 ил., 4 табл., 13 пр.
1. Полипептид D-лактатдегидрогеназы, который содержит любой из указанных ниже полипептидов (а)-(b):
(a) полипептид с аминокислотной последовательностью, показанной в SEQ ID NO:1 или 2; и
(b) полипептид с аминокислотной последовательностью, которая характеризуется идентичностью по последовательности на уровне не менее 80% с аминокислотной последовательностью, показанной в SEQ ID NO:1 или 2, где указанный полипептид обладает D-лактатдегидрогеназной активностью.
2. Полинуклеотид, кодирующий полипептид D-лактатдегидрогеназы, где полинуклеотид содержит любой из указанных ниже полинуклеотидов (а)-(с):
(a) полинуклеотид с нуклеотидной последовательностью, показанной в SEQ ID NO:3 или 4;
(b) полинуклеотид с нуклеотидной последовательностью, которая характеризуется идентичностью по последовательности на уровне не менее 80% с нуклеотидной последовательностью, показанной в SEQ ID NO:3 или 4, где указанный полинуклеотид кодирует полипептид, обладающий D-лактатдегидрогеназной активностью; и
(c) полинуклеотид, кодирующий полипептид по п.1.
3. Конструкция ДНК для экспрессии полинуклеотида, кодирующего D-лактатдегидрогеназу, в которой связаны полинуклеотид по п. 2 и промотор, способный осуществлять экспрессию указанного полинуклеотида.
4. Конструкция ДНК по п.3, где промотор представляет собой промотор гена пируватдекарбоксилазы 1 (ген PDC1), гена супрессии экспоненциального дефекта 1 (ген SED1) или гена глицеральдегид-3-фосфатдегидрогеназы 3 (ген TDH3).
5. Конструкция ДНК по п.3 или 4, отличающаяся тем, что указанный промотор выбран из любого указанного ниже промотора (I)-(III):
(I) промотор с нуклеотидной последовательностью, показанной в любой из SEQ ID NO: 5-7;
(II) промотор с нуклеотидной последовательностью, которая гибридизуется в жестких условиях с нуклеотидной последовательностью, показанной в любой из SEQ ID NO: 5-7, или с нуклеотидной последовательностью, включающей ее часть; и
(III) промотор с нуклеотидной последовательностью, показанной в любой из SEQ ID NO: 5-7, с тем исключением, что в нем один или несколько нуклеотидов замещены, делетированы, встроены и/или добавлены.
6. Трансформант для получения D-молочной кислоты, в который введен полинуклеотид по п.2.
7. Трансформированные дрожжи для получения D-молочной кислоты, в которые введен полинуклеотид по п.2.
8. Трансформированные дрожжи по п.7, где по меньшей мере один ген из указанных ниже: ген пируватдекарбоксилазы 1 (ген PDC1), ген супрессии экспоненциального дефекта 1 (ген SED1) или ген глицеральдегид-3-фосфатдегидрогеназы 3 (ген TDH3) в указанных трансформированных дрожжах замещен геном D-лактатдегидрогеназы.
9. Трансформированные дрожжи по п.8, где по меньшей мере один из указанных генов представляет собой ген PDC1.
10. Способ получения D-молочной кислоты, который включает стадию культивирования трансформанта по п.6.
11. Трансформант для получения D-молочной кислоты, в который введена ДНК-конструкция по п.3.
12. Трансформированные дрожжи для получения D-молочной кислоты, в которые введена конструкция ДНК по п.3.
13. Трансформированные дрожжи по п.7, где по меньшей мере один ген из указанных ниже: ген пируватдекарбоксилазы 1 (ген PDC1), ген супрессии экспоненциального дефекта 1 (ген SED1) и ген глицеральдегид-3-фосфатдегидрогеназы 3 (ген TDH3) в указанных трансформированных дрожжах замещен введенной в них конструкцией ДНК, содержащей связанный ген D-лактатдегидрогеназы и промотор, способный осуществлять экспрессию гена D-лактатдегидрогеназы.
14. Трансформированные дрожжи по п.13, где по меньшей мере один из указанных генов представляет собой ген PDC1.
15. Способ получения D-молочной кислоты, который включает стадию культивирования трансформированных дрожжей по п.7.
| WO 2004104202 A1, 02.12.2004 | |||
| Устройство для наполнения жидкостью сосудов | 1948 |
|
SU80976A1 |
| JOSEPH F | |||
| SIEBENALLER, et al | |||
| "COMPARISON OF THE D-LACTATE STEREOSPECIFIC DEHYDROGENASE OF LIMUL US POL YPHEMUS WITH ACTIVE-SITE REGIONS OF L-LACTATE DEHYDROGENASES", Biochimica et Biophysica Acta, 749 (1983) pp | |||
| Паровозный золотник (байпас) | 1921 |
|
SU153A1 |
| ШТАММ БАКТЕРИЙ LACTOBACILLUS BULGARICUS, ИМЕЮЩИЙ АКТИВНОСТЬ ЛАКТАТДЕГИДРОГЕНАЗЫ И β-ГАЛАКТОЗИДАЗЫ | 1994 |
|
RU2141521C1 |

