[Область техники]
Настоящее изобретение относится к кристаллическому 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамиду (далее обозначенному как "соединение А").
[Формула 1]
[Уровень техники]
Соединение А обладает очень сильным агонистическим воздействием на простациклин PGI2 и проявляет ингибирующее воздействие на агрегацию тромбоцитов, сосудорасширяющий эффект, бронхорасширяющий эффект, эффект ингибирования депонирования липидов, эффект ингибирования активации лейкоцитов и др. (см., например, патентную ссылку 1).
А именно, соединение А применимо в качестве профилактического или терапевтического средств в случае транзиторной ишемической атаки (TIA), диабетической нейропатии, диабетической гангрены, нарушения периферического кровообращения (например, хроническая закупорка артерии, перемежающаяся хромота, закупорка периферических кровеносных сосудов, вибрационный синдром, болезнь Рейно), заболеваний соединительных тканей (например, системная эритематозная волчанка, склеродермия, смешанное заболевание соединительной ткани, синдром воспаления кровеносных сосудов), реокклюзии/рестеноза после чрескожной транслюминальной коронарной ангиопластики (РТСА), артериосклероза, тромбообразования (например, острая фаза тромбоза сосудов головного мозга, эмболия легочной артерии), повышенного кровяного давления, легочной гипертензии, ищемического заболевания (например, церебральный инфаркт, инфаркт миокарда), стенокардии (например, стабильная стенокардия, нестабильная стенокардия), гломерулонефрита, диабетической нефропатии, хронической почечной недостаточности, аллергической реакции, бронхиальной астмы, язвы, пролежневой язвы (пролежня), рестеноза после коронарной ангиопластики, такой как атерэктомия и имплантация стента, тромбоцитопении вследствие диализа, в случае заболеваний, которые вызывают фиброз органов или тканей [например, почечная недостаточность (например, тубулоинтерстициальный нефрит), респираторные заболевания (например, интерстициальная пневмония (легочный фиброз), хроническое обструктивное заболевание легких), болезни пищеварительного тракта (например, цирроз печени, вирусный гепатит, хронический панкреатит и скиррозная злокачественная опухоль желудка), заболевания сердечно-сосудистой системы (например, миокардиальный фиброз), заболевания костей и суставов (например, фиброз костного мозга и ревматоидный артрит), заболевания кожи (например, послеоперационный шрам, ожоговый шрам, келоид и гипертрофический рубец), послеродовые болезни (например, гистеромиома), заболевания мочевых путей (например, гипертрофия предстательной железы), другие заболевания (например, болезнь Альцгеймера, склеродирующий перитонит, диабет I типа и послеоперационная спайка органов)], в случае эректильной дисфункции (например, диабетическая эректильная дисфункция, психогенная эректильная дисфункция, психотическая эректильная дисфункция, эректильная дисфункция, связанная с хронической почечной недостаточностью, эректильная дисфункция после внутритазовой операции удаления предстательной железы и васкулярная эректильная дисфункция, связанная со старением и артериосклерозом), воспалительных заболеваний кишечника (например, язвенный колит, болезнь Крона, интестинальный туберкулез, ишемический колит и интестинальная язва, связанная с болезнью Бехчета), в случае гастрита, язвенной болезни желудка, ишемической офтальмопатии (например, окклюзия артерии сетчатки, окклюзия вены сетчатки, ишемическая оптическая нейропатия), в случае внезапной потери слуха, аваскулярного некроза кости, желудочно-кишечных поражений, вызванных введением нестероидных противовоспалительных средств (например, таких как диклофенак, мелоксикам, оксапрозин, набуметон, индометацин, ибупрофен, кетопрофен, напроксен, целекоксиб) (не существует специфического ограничения желудочно-кишечного поражения, так как это поражение, появляющееся в двенадцатиперстной кишке, тонком кишечнике и толстом кишечнике, и примеры его включают поражение слизистой оболочки, такое как эрозия и язва, возникшие в двенадцатиперстной кишке, тонком кишечнике и толстом кишечнике), и в случае симптомов, связанных со стенозом спинномозгового канала поясничной области (например, паралич, слабая активность сенсорного восприятия, боль, онемение, снижение возможности движения и др., связанных со стенозом цервикального спинномозгового канала, со стенозом торакального спинномозгового канала, со стенозом поясничного спинномозгового канала, с недифференцированным стенозом спинномозгового канала или крестцовым стенозом) и др. (см., например, в патентных ссылках с 1 по 6). Кроме того, соединение А применимо в качестве ускоряющего средства при ангиогенной терапии, такой как генная терапия или аутогенная трансплантация костного мозга, в качестве ускоряющего средства для ангиогенеза при восстановлении периферической артерии или при ангиогенной терапии и др. (см., например, в патентной ссылке 1).
Как отмечено выше, несмотря на то что полезность соединения А в качестве терапевтических средств при вышеуказанных заболеваниях известна, не существует ссылок, где описано или высказано предположение про возможность существования кристаллов соединения А.
[Ссылки на предшествующий уровень техники]
[Патентные ссылки]
[Патентная ссылка 1] WO2002/088084
[Патентная ссылка 2] WO2009/157396
[Патентная ссылка 3] WO2009/107736
[Патентная ссылка 4] WO2009/154246
[Патентная ссылка 5] WO2009/157397
[Патентная ссылка 6] WO2009/157398
[Сущность изобретения]
[Проблема, решаемая изобретением]
Главная цель настоящего изобретения заключается в предоставлении новой кристаллической формы соединения А. Кроме того, целью настоящего изобретения является предоставление способа получения кристалла и фармацевтической композиции, содержащей кристалл в качестве активного ингредиента.
[Средства решения проблемы]
Существует надежда на то, что содержимое лекарственного средства представляет собой объект высокого качества, неизменное воздействие которого всегда может быть продемонстрировано, и является формой, с которой легко работать в промышленности. Настоящие изобретатели серьезно подошли к изучению этого аспекта. В результате настоящие изобретатели открыли новую кристаллическую форму соединения А и выполнили настоящее изобретение.
Настоящее изобретение включает, например, нижеследующие пункты (1)-(4).
(1) Форма-I кристалла соединения А, которая демонстрирует пики дифракции в спектре порошковой рентгеновской дифракции соединения А (далее в этом документе обозначаемая как "Форма-I кристалла изобретения") при следующих значениях углов дифракции 2θ: 9,4 градуса, 9,8 градуса, 17,2 градуса и 19,4 градуса, при этом спектр порошковой рентгеновской дифракции получали с использованием Cu Кα излучения (λ=1,54 Å),
(2) Форма-II кристалла соединения А, которая демонстрирует пики дифракции в спектре порошковой рентгеновской дифракции соединения А (далее в этом документе обозначаемая как "Форма-II кристалла изобретения") при следующих значениях углов дифракции 2θ: 9,0 градусов, 12,9 градуса, 20,7 градуса и 22,6 градуса, при этом спектр порошковой рентгеновской дифракции получали с использованием Cu Кα излучения (λ=1,54 Å),
(3) Форма-III кристалла соединения А, которая демонстрирует пики дифракции в спектре порошковой рентгеновской дифракции соединения А (далее в этом документе обозначаемая как "Форма-III кристалла изобретения") при следующих значениях углов дифракции 2θ: 9,3 градуса, 9,7 градуса, 16,8 градуса, 20,6 градуса и 23,5 градуса, при этом спектр порошковой рентгеновской дифракции получали с использованием Cu Кα излучения (λ=1,54 Å),
(4) Фармацевтическая композиция, содержащая кристалл, один из вышеуказанных с (1) по (3), в качестве активного ингредиента (далее в этом документе обозначаемая как "фармацевтическая композиция изобретения").
В случае, когда указывается угол дифракции 2 theta (2θ) для пика в примерах осуществления изобретения и в формуле изобретения, следует понимать, что данная величина истолковывается как интервал от значения, равного указанной величине минус 0,2°, до значения, равного указанной величине плюс 0,2°, и предпочтительно от значения, равного указанной величине минус 0,1°, до значения, равного указанной величине плюс 0,1°.
[Краткое описание чертежей]
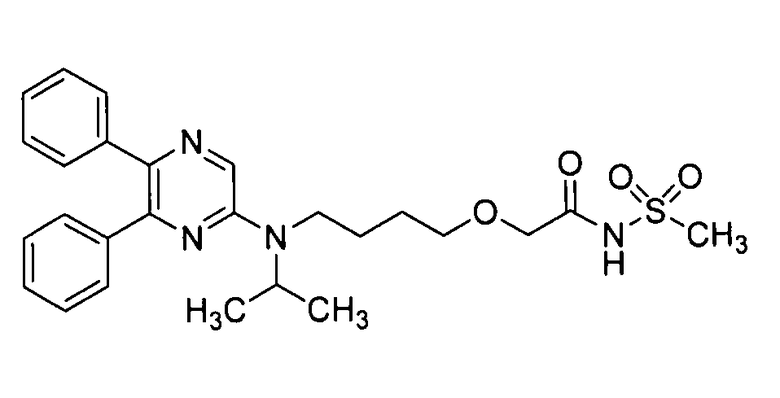
[Фиг.1] показывает спектр порошковой рентгеновской дифракции Формы-I кристалла изобретения. На вертикальной оси указана интенсивность пика (cps), и на горизонтальной оси указан дифракционный угол (2θ [°]).
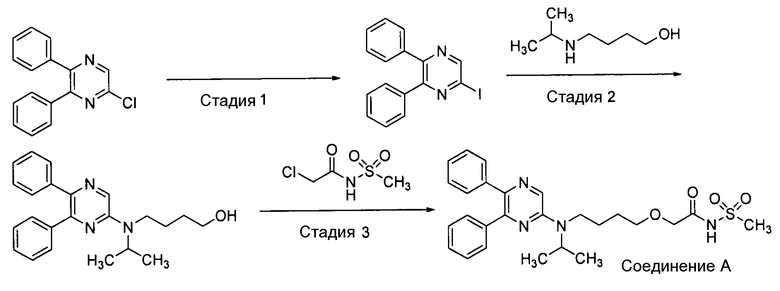
[Фиг.2] показывает спектр порошковой рентгеновской дифракции Формы-II кристалла изобретения. На вертикальной оси указана интенсивность пика (cps), и на горизонтальной оси указан дифракционный угол (2θ [°]).
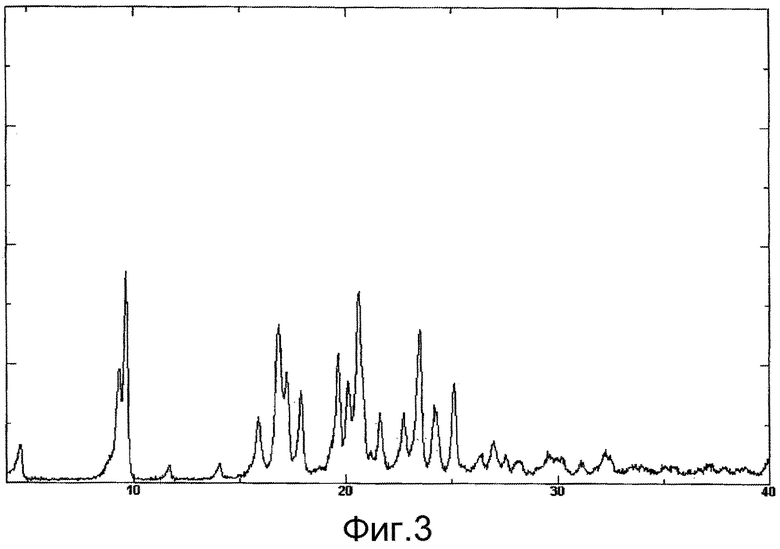
[Фиг.3] показывает спектр порошковой рентгеновской дифракции Формы-III кристалла изобретения. На вертикальной оси указана интенсивность пика (cps), и на горизонтальной оси указан дифракционный угол (2θ [°]).
[Фиг.4] показывает сканирующую электронную микрофотографию Формы-I кристалла изобретения.
[Фиг.5] показывает сканирующую электронную микрофотографию Формы-II кристалла изобретения.
[Фиг.6] показывает сканирующую электронную микрофотографию Формы-III кристалла изобретения.
[Способ осуществления изобретения]
Форма-I кристалла изобретения характеризуется тем, что демонстрирует пики дифракции при 9,4 градуса, 9,8 градуса, 17,2 градуса и 19,4 градуса в спектре порошковой рентгеновской дифракции соединения А.
Форма-II кристалла изобретения характеризуется тем, что демонстрирует пики дифракции при 9,0 градусах, 12,9 градуса, 20,7 градуса и 22,6 градуса в спектре порошковой рентгеновской дифракции соединения А.
Форма-III кристалла изобретения характеризуется тем, что демонстрирует пики дифракции при 9,3 градуса, 9,7 градуса, 16,8 градуса, 20,6 градуса и 23,5 градуса в спектре порошковой рентгеновской дифракции соединения А.
А. Получение соединения А
Соединение А может быть получено, например, способом, описанным в патентной ссылке 1, и оно также может быть получено согласно указанному ниже способу получения.
[Формула 2]
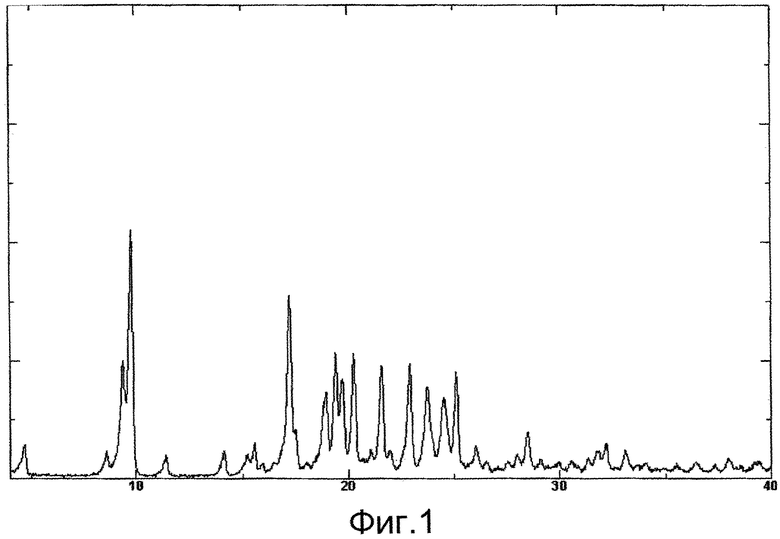
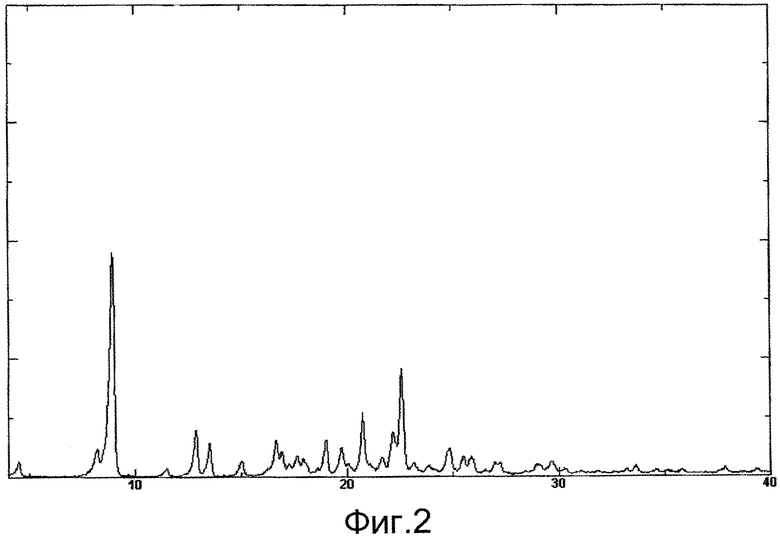
Стадия 1:
6-Йод-2,З-дифенилпиразин может быть получен из 6-хлор-2,3-дифенилпиразина посредством его реакции с иодидом натрия. Реакцию проводят в присутствии кислоты в органическом растворителе (например, этилацетате, ацетонитриле, ацетоне, метилэтилкетоне или в растворителе, полученном путем их смешивания). Используемая кислота представляет собой, например, уксусную кислоту, серную кислоту или кислоту, полученную путем их смешивания. Используемое количество иодида натрия составляет, как правило, величину в диапазоне от 1 до 10 молей, кратных по отношению к 6-хлор-2,3-дифенилпиразину, предпочтительно в диапазоне от 2 до 3 молярного кратного соотношения. Температура реакции изменяется в зависимости от природы используемых веществ и от используемой кислоты, но, как правило, может быть в диапазоне от 60°С до 90°С. Время реакции изменяется в зависимости от природы используемых веществ и от используемой кислоты, и от температуры реакции, но, как правило, может быть в диапазоне от 9 часов до 15 часов.
Стадия 2:
5,6-дифенил-2-[4-гидроксибутил(изопропил)амино]пиразин может быть получен из 6-йод-2,3-дифенилпиразина посредством его реакции с 4-гидроксибутил(изопропил)амином. Реакцию проводят в присутствии основания в органическом растворителе (например, сульфолане, N-метилпирролидоне, N,N-диметилимидазолидиноне, диметилсульфоксиде или в растворителе, полученном путем их смешивания). Используемое основание представляет собой, например, гидрокарбонат натрия, гидрокарбонат калия, карбонат калия, карбонат натрия или основание, полученное путем их смешивания. Количество используемого 4-гидроксибутил(изопропил)амина может составлять, как правило, величину в диапазоне от 1,5 до 5,0 молей, кратных по отношению к 6-йод-2,3-дифенилпиразину, предпочтительно в диапазоне от 2 до 3 молярного кратного соотношения. Температура реакции изменяется в зависимости от природы используемых веществ и от используемого основания, но, как правило, может быть в диапазоне от 170°С до 200°С. Время реакции изменяется в зависимости от природы используемых веществ и от используемого основания, и от температуры реакции, но, как правило, может быть в диапазоне от 5 часов до 9 часов.
Стадия 3:
Соединение А может быть получено из 5,6-дифенил-2-[4-гидроксибутил(изопропил)амино]пиразина посредством его реакции с N-(2-хлорацетил)метансульфонамидом. Реакцию проводят в присутствии основания в растворителе (N-метилпирролидоне, 2-метил-2-пропаноле или в растворителе, полученном путем их смешивания). Используемое основание представляет собой, например, трет-бутоксид калия, трет-бутоксид натрия или основание, полученное путем их смешивания. Количество используемого N-(2-хлорацетил)метансульфонамида может составлять, как правило, величину в диапазоне от 2 до 4 молей, кратных по отношению к 5,6-дифенил-2-[4-гидроксибутил(изопропил)амино]пиразину, предпочтительно в диапазоне от 2 до 3 молярного кратного соотношения. Температура реакции изменяется в зависимости от природы используемых веществ и от используемого основания, но, как правило, может быть в диапазоне от -20°С до 20°С. Время реакции изменяется в зависимости от природы используемых веществ и от используемого основания, и от температуры реакции, но, как правило, может быть в диапазоне от 0,5 часов до 2 часов.
Соединения, используемые в качестве исходных веществ в вышеуказанном способе получения соединения А, представляют собой известные соединения или могут быть получены путем известных способов.
В. Получения Формы-I кристалла изобретения, Формы-II кристалла изобретения и Формы-III кристалла изобретения (далее в этом документе все вместе обозначаемые как "кристаллы изобретения")
(I) Получение Формы-I кристалла изобретения
Форма-I кристалла изобретения может быть получена, например, в соответствии со способом, описанным ниже.
(1) Стадия растворения
Данная стадия представляет собой стадию растворения соединения А в растворителе при нагревании. Подходящий для использования растворитель на этой стадии представляет собой, например, спиртовой растворитель, смешанный растворитель из спиртового растворителя и кетонового растворителя. Подходящий для использования спиртовой растворитель на этой стадии представляет собой, например, метанол, этанол, 2-пропанол, предпочтительно этанол. Подходящий для использования кетоновый растворитель на этой стадии представляет собой, например, метилэтилкетон.
В особенности предпочтительный для использования растворитель на данной стадии представляет собой этанол или смешаный растворитель из этанола и метилэтилкетона. В случае смешаного растворителя из этанола и метилэтилкетона этанол составляет количество в диапазоне от 1,5-кратного (об./об.) до 100-кратного (об./об.) по отношению к количеству метилэтилкетона, предпочтительно в диапазоне от 3-кратного (об./об.) до 50-кратного (об./об.), более предпочтительно в диапазоне от 6-кратного (об./об.) до 20-кратного (об./об.).
Общий объем используемого растворителя на стадии составляет, предпочтительно, в диапазоне от 2-кратного (мл/г) до 30-кратного (мл/г) по отношению к количеству соединения А, более предпочтительно в диапазоне от 3-кратного (мл/г) до 20-кратного (мл/г), еще более предпочтительно в диапазоне от 4-кратного (мл/г) до 15-кратного (мл/г). Температура нагрева изменяется в зависимости от природы растворителя и от используемого объема растворителя, но, как правило, она ниже, чем точка кипения используемого растворителя, и составляет величину, предпочтительно, в диапазоне от 60°С до 100°С, более предпочтительно в диапазоне от 70°С до 90°С.
На этой стадии раствор можно отфильтровать для удаления нерастворимых компонентов, если это необходимо. Для предотвращения в процессе фильтрования выпадения в осадок кристаллов предпочтительно проводить фильтрование под давлением, используя воронку, оснащенную подогревательным устройством. В том случае, когда в фильтрате наблюдается выпадение в осадок кристаллов, предпочтительно осадок растворить снова путем повторного нагревания после фильтрования.
(2) Стадия охлаждения
Данная стадия представляет собой стадию осаждения Формы-I кристалла изобретения из раствора, полученного на вышеуказанной стадии (1), посредством охлаждения. Стадию предпочтительно проводить с использованием кристаллизатора, оснащенного функцией подогрева и функцией перемешивания.
Температура охлаждения (температура, при которой накапливаются выпавшие в осадок кристаллы) имеет подходящие значения в диапазоне от -10°С до 50°С, предпочтительно в диапазоне от 0°С до 20°С и более предпочтительно в диапазоне от 0°С до 10°С. Стадию предпочтительно проводить при охлаждении в течение времени в диапазоне от 3 часов до 95 часов, медленно достигая температуры охлаждения.
Кроме того, на этой стадии можно добавить кристалл-затравку Формы-I кристалла изобретения. В этом случае предпочтительно, чтобы кристалл-затравку Формы-I кристалла изобретения добавляли тогда, когда раствор охлажден до температуры в диапазоне от 60°С до 90°С. Количество кристалла-затравки Формы-I кристалла изобретения составляет, предпочтительно, в диапазоне от 1% до 10% масс. относительно количества соединения А.
(3) Стадия накопления кристаллов и сушки
Данная стадия представляет собой стадию накопления выпавших в осадок кристаллов на вышеописанной стадии (2) с использованием известных способов, таких как фильтрование и центрифугирование, и высушивания накопленных кристаллов.
Стадию сушки можно осуществить стандартным способом, таким как сушка при пониженном давлении и посредством осушающего реагента. Предпочтительно проведение сушки при пониженном давлении (например, 10 мм рт.ст. или менее), при температуре в диапазоне от 20°С до 70°С в течение времени от одного часа до 48 часов.
Кроме того, исходя из вышеуказанной стадии (1), после частичного осаждения кристаллов посредством удаления растворителя в процессе нагревания и перемешивания раствора, полученного на вышеуказанной стадии (1), Форма-I кристалла изобретения может быть получена посредством осуществления вышеуказанных стадий (2) и (3). На стадии удаления растворителя можно добавить кристалл-затравку Формы-I кристалла изобретения. Количество кристалла-затравки Формы-I кристалла изобретения составляет, предпочтительно, величину в диапазоне от 0,1% до 10% масс. относительно количества соединения А, использованного на вышеуказанной стадии (1).
(II) Получение Формы-II кристалла изобретения
Форма-II кристалла изобретения может быть получена, например, в соответствии со способом, описанным ниже.
(1) Стадия растворения
Данная стадия представляет собой стадию растворения соединения А в растворителе при нагревании. Подходящий для использования растворитель на этой стадии представляет собой, например, спиртовой растворитель, кетоновый растворитель, растворитель в виде насыщенного углеводорода, растворитель в виде простого эфира и воды или растворитель, полученный путем их смешивания. Предпочтительный смешанный растворитель представляет собой смесь растворителя в виде простого эфира и растворителя в виде насыщенного углеводорода или воды, или смесь спиртового растворителя и кетонового растворителя или воды.
Используемый на этой стадии спиртовой растворитель представляет собой, например, спирт с прямой или разветвленной цепью, имеющий от одного до 8 атомов углерода. А именно, спиртовой растворитель может включать метанол, этанол, н-пропанол, изопропанол, 1-бутанол, 2-бутанол, трет-бутанол, 1-пентанол, 1-гексанол, 1-гептанол, 1-октанол. Используемый на этой стадии растворитель в виде простого эфира может включать тетрагидрофуран, 1,4-диоксан. Используемый на этой стадии растворитель в виде насыщенного углеводорода представляет собой, например, алкан с прямой или разветвленной цепью, имеющий от 6 до 8 атомов углерода, или циклоалкан, имеющий от 6 до 8 атомов углерода. А именно, растворитель в виде насыщенного углеводорода может включать гептан, октан, циклогексан, циклогептан, циклооктан. Используемый на этой стадии кетоновый растворитель представляет собой, например, прямоцепный или с разветвленной цепью, имеющий от 3 до 8 атомов углерода. А именно, кетоновый растворитель может включать ацетон, метилэтилкетон.
Общий объем используемого на этой стадии растворителя составляет, целесообразно, величину в диапазоне от 2-кратного (мл/г) до 20-кратного (мл/г) соотношения относительно количества соединения А, предпочтительно в диапазоне от 3-кратного (мл/г) до 15-кратного (мл/г), более предпочтительно в диапазоне от 5-кратного (мл/г) до 10-кратного (мл/г). Температура нагрева изменяется в зависимости от природы растворителя и используемого объема растворителя, но, как правило, она ниже точки кипения используемого растворителя и представляет собой величину предпочтительно в диапазоне от 60°С до 90°С, более предпочтительно в диапазоне от 70°С до 80°С.
На этой стадии раствор можно отфильтровать для удаления нерастворимых компонентов, если это необходимо. Для предотвращения в процессе фильтрования выпадения в осадок кристаллов предпочтительно проводить фильтрование под давлением, используя воронку, оснащенную подогревательным устройством. В том случае, когда в фильтрате наблюдается выпадение в осадок кристаллов, предпочтительно осадок растворить снова путем повторного нагревания после фильтрования.
(2) Стадия охлаждения
Данная стадия представляет собой стадию осаждения Формы-II кристалла изобретения из раствора, полученного на вышеуказанной стадии (1), посредством охлаждения. Стадию предпочтительно проводить с использованием кристаллизатора, оснащенного функцией подогрева и функцией перемешивания.
Температура охлаждения (температура, при которой накапливаются выпавшие в осадок кристаллы) имеет подходящие значения в диапазоне от -10°С до 50°С, предпочтительно в диапазоне от 0°С до 20°С и более предпочтительно в диапазоне от 0°С до 10°С.
Кроме того, на этой стадии можно добавить кристалл-затравку Формы-II кристалла изобретения. Количество кристалла-затравки Формы-II кристалла изобретения составляет, предпочтительно, в диапазоне от 1% до 10% масс. относительно количества соединения А.
В том случае, когда Форму-II кристалла изобретения получают при использовании спиртового растворителя или при использовании в качестве растворителя смеси спиртового растворителя и кетонового растворителя, тогда необходимо добавить кристалл-затравку Формы-II кристалла изобретения в раствор, полученный на вышеуказанной стадии (1), и раствор охладить или необходимо раствор, полученный на вышеуказанной стадии (1), быстро охладить. Подходящей является скорость охлаждения в диапазоне от 60°C/час до 600°C/час.
(3) Стадия накопления кристаллов и сушки
Данная стадия представляет собой такую же стадию, как способ, описанный в "(3) Стадия накопления кристаллов и сушки" в вышеуказанном "(I) Получение Формы-I кристалла изобретения".
(III) Получение Формы-III кристалла изобретения
Форма-III кристалла изобретения может быть получена, например, в соответствии со способом, описанным ниже.
(1) Стадия растворения
Данная стадия представляет собой стадию растворения соединения А в растворителе при нагревании. Подходящий для использования растворитель на этой стадии представляет собой, например, растворитель в виде сложного эфира, ароматический углеводородный растворитель. Подходящий для использования растворитель в виде сложного эфира на этой стадии представляет собой, например, диэтилкарбонат, н-бутилацетат, изоамилацетат, н-амилацетат, предпочтительно н-бутилацетат. Подходящий для использования ароматический углеводородный растворитель на этой стадии представляет собой, например, этилбензол.
Общий объем используемого растворителя на стадии составляет, предпочтительно, в диапазоне от 5-кратного (мл/г) до 30-кратного (мл/г) по отношению к количеству соединения А, более предпочтительно в диапазоне от 7-кратного (мл/г) до 20-кратного (мл/г), еще более предпочтительно в диапазоне от 10-кратного (мл/г) до 15-кратного (мл/г). Температура нагрева изменяется в зависимости от природы растворителя и от используемого объема растворителя, но, как правило, она ниже, чем точка кипения используемого растворителя, и составляет величину предпочтительно в диапазоне от 40°С до 90°С, более предпочтительно в диапазоне от 50°С до 80°С.
На этой стадии раствор можно отфильтровать для удаления нерастворимых компонентов, если это необходимо. Для предотвращения в процессе фильтрования выпадения в осадок кристаллов предпочтительно проводить фильтрование под давлением, используя воронку, оснащенную подогревательным устройством. В том случае, когда в фильтрате наблюдается выпадение в осадок кристаллов, предпочтительно осадок растворить снова путем повторного нагревания после фильтрования.
(2) Стадия охлаждения
Данная стадия представляет собой стадию осаждения Формы-III кристалла изобретения из раствора, полученного на вышеуказанной стадии (1), посредством охлаждения. Стадию предпочтительно проводить с использованием кристаллизатора, оснащенного функцией подогрева и функцией перемешивания.
Подходящая скорость охлаждения находится в диапазоне от 0,5°C/час до 120°C/час. Температура охлаждения (температура, при которой накапливаются выпавшие в осадок кристаллы) имеет подходящие значения в диапазоне от -10°С до 30°С, предпочтительно в диапазоне от 0°С до 20°С и более предпочтительно в диапазоне от 0°С до 10°С.
(3) Стадия накопления кристаллов и сушки
Эта стадия представляет собой такую же стадию, как способ, описанный в "(3) Стадия накопления кристаллов и сушки" в вышеуказанном "(I) Получение Формы-I кристалла изобретения".
С. Медицинское применение и фармацевтическая композиция изобретения
Соединение А в соответствии с настоящим изобретением обладает очень сильным агонистическим воздействием на простациклин PGI2 и проявляет ингибирующее воздействие на агрегацию тромбоцитов, сосудорасширяющий эффект, бронхорасширяющий эффект, эффект ингибирования депонирования липидов, эффект ингибирования активации лейкоцитов и др.
В силу вышесказанного кристаллы изобретения или фармацевтическая композиция изобретения пригодны для применения в качестве профилактических или терапевтических средств в случае транзиторной ишемической атаки (TIA), диабетической нейропатии, диабетической гангрены, нарушения периферического кровообращения (например, хроническая закупорка артерии, перемежающаяся хромота, закупорка периферических кровеносных сосудов, вибрационный синдром, болезнь Рейно), заболеваний соединительных тканей (например, системная эритематозная волчанка, склеродермия, смешанное заболевание соединительной ткани, синдром воспаления кровеносных сосудов), реокклюзии/рестеноза после чрескожной транслюминальной коронарной ангиопластики (РТСА), артериосклероза, тромбообразования (например, острая фаза тромбоза сосудов головного мозга, эмболия легочной артерии), повышенного кровяного давления, легочной гипертензии, ишемического заболевания (например, церебральный инфаркт, инфаркт миокарда), стенокардии (например, стабильная стенокардия, нестабильная стенокардия), гломерулонефрита, диабетической нефропатии, хронической почечной недостаточности, аллергической реакции, бронхиальной астмы, язвы, пролежневой язвы (пролежня), рестеноза после коронарной ангиопластики, такой как атерэктомия и имплантация стента, тромбоцитопении вследствие диализа, в случае заболеваний, которые вызывают фиброз органов или тканей [например, почечная недостаточность (например, тубулоинтерстициальный нефрит), респираторные заболевания (например, интерстициальная пневмония (легочный фиброз), хроническое обструктивное заболевание легких), болезни пищеварительного тракта (например, цирроз печени, вирусный гепатит, хронический панкреатит и скиррозная злокачественная опухоль желудка), заболевания сердечно-сосудистой системы (например, миокардиальный фиброз), заболевания костей и суставов (например, фиброз костного мозга и ревматоидный артрит), заболевания кожи (например, послеоперационный шрам, ожоговый шрам, келоид и гипертрофический рубец), послеродовые болезни (например, гистеромиома), заболевания мочевых путей (например, гипертрофия предстательной железы), другие заболевания (например, болезнь Альцгеймера, склеродирующий перитонит, диабет I типа и послеоперационная спайка органов)], в случае эректильной дисфункции (например, диабетическая эректильная дисфункция, психогенная эректильная дисфункция, психотическая эректильная дисфункция, эректильная дисфункция, связанная с хронической почечной недостаточностью, эректильная дисфункция после внутритазовой операции удаления предстательной железы и васкулярная эректильная дисфункция, связанная со старением и артериосклерозом), воспалительных заболеваний кишечника (например, язвенный колит, болезнь Крона, интестинальный туберкулез, ишемический колит и интестинальная язва, связанная с болезнью Бехчета), в случае гастрита, язвенной болезни желудка, ишемической офтальмопатии (например, окклюзия артерии сетчатки, окклюзия вены сетчатки, ишемическая оптическая нейропатия), в случае внезапной потери слуха, аваскулярного некроза кости, желудочно-кишечных поражений, вызванных введением нестероидных противовоспалительных средств (например, таких как диклофенак, мелоксикам, оксапрозин, набуметон, индометацин, ибупрофен, кетопрофен, напроксен, целекоксиб) (не существует специфического ограничения желудочно-кишечного поражения, так как это поражение, появляющееся в двенадцатиперстной кишке, тонком кишечнике и толстом кишечнике, и примеры его включают поражение слизистой оболочки, такое как эрозия и язва, возникшие в двенадцатиперстной кишке, тонком кишечнике и толстом кишечнике), и в случае симптомов, связанных со стенозом спинномозгового канала поясничной области (например, паралич, слабая активность сенсорного восприятия, боль, онемение, снижение возможности движения и др., связанных со стенозом цервикального спинномозгового канала, со стенозом торакального спинномозгового канала, со стенозом поясничного спинномозгового канала, с недифференцированным стенозом спинномозгового канала или крестцовым стенозом) и др. Кроме того, кристаллы изобретения или фармацевтическая композиция изобретения также пригодны к применению в качестве ускоряющего средства при ангиогенной терапии, в случае генной терапии или аутогенной трансплантации костного мозга, в качестве ускоряющего средства для ангиогенеза при восстановлении периферической артерии или при ангиогенной терапии и др. Если кристаллы изобретения применяются как лекарственное средство, то фармацевтическая композиция изобретения представляет собой кристаллы изобретения сами по себе или содержит кристаллы изобретения в фармацевтически приемлемом, нетоксичном и инертном носителе в количестве, находящемся в диапазоне от 0,1% до 99,5%, предпочтительно в диапазоне от 0,5% до 90%.
Примеры носителя включают твердый, полутвердый или жидкий разбавитель, наполнитель и другие вспомогательные средства фармацевтического препарата. Они могут применяться по отдельности или в виде смеси двух или более их представителей.
Фармацевтическая композиция изобретения может быть в виде любой готовой лекарственной формы для перорального применения, такой как порошок, капсулы, таблетки, покрытые сахарной оболочкой таблетки, гранулы, разведенный порошок, суспензия, жидкость, сироп, эликсир или пастилка, и в виде лекарственного препарата для парентерального применения, такого как инъекция или суппозиторий в твердой или жидкой лекарственной форме с единичной дозой. Она также может быть в виде лекарственного препарата с замедленным высвобождением. Среди них пероральные лекарственные препараты, такие как таблетки, представляют собой особо предпочтительные.
Порошок может быть изготовлен посредством превращения кристаллов изобретения в соответствующий мелкий размер.
Разведенный порошок может быть изготовлен посредством такого же метода превращения кристаллов изобретения в соответствующий мелкий размер и с последующим смешиванием с фармацевтическим носителем, который аналогичным образом превращают в мелкий размер, таким как пищевой углевод (например, крахмал и маннитол). Вкусоароматизирующее вещество, консервирующее вещество, диспергирующий агент, окрашивающее вещество, душистое вещество и др. необязательно могут быть туда же добавлены.
Капсулы могут быть изготовлены посредством такого метода, когда порошок или разведенный порошок, при этом получение порошка осуществляют как указано выше, или гранулы, о которых будет сказано в связи с таблетками, используют для наполнения капсульной оболочки, такой как в случае желатиновых капсул. Также возможно изготовление таким методом, когда порошок или разведенный порошок в порошкообразной форме смешивают со скользящим веществом или с разжижающей добавкой, такими как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль, затем следует процесс наполнения. В том случае, когда добавляют вещество для улучшения распадаемости таблеток или солюбилизирующий агент, такие как кальциевая соль карбоксиметилцеллюлозы, малозамещенная гидроксипропилцеллюлоза, натриевая соль кроскармеллозы, натриевая соль карбоксиметилкрахмала, карбонат кальция или карбонат натрия, эффективность лекарственного препарата при приеме капсул внутрь может быть улучшена. Также возможно, чтобы тонкоизмельченный порошок кристаллов изобретения был суспендирован/диспергирован в растительном масле, полиэтиленгликоле, глицерине или в поверхностно-активном веществе и заключен в желатиновое покрытие с получением лекарственного препарата в виде твердой капсулы.
Таблетки можно получать таким методом, при котором порошкообразную смесь готовят посредством прибавления наполнителя к кристаллам изобретения, которые были превращены в порошок и сформированы в гранулы или крупинки, и затем вещество для улучшения распадаемости таблеток или скользящее вещество добавляют туда же и после этого формируют таблетки.
Порошкообразную смесь можно получить путем смешивания соответственно измельченных в порошок кристаллов изобретения с разбавителем или основой. В случае необходимости туда же можно добавить связующее вещество (такое как натриевая соль карбоксиметилцеллюлозы, метилцеллюлоза, гидроксипропилметилцеллюлоза, желатин, поливинилпирролидон или поливиниловый спирт), разжижающую добавку для замедления затвердевания (такая как парафин), реабсорбирующую добавку (такая как четвертичная соль), абсорбент (такой как бентонит или каолин) и др.
Порошкообразную смесь можно сформировать в гранулы таким методом, что сначала увлажняют посредством использования связующего вещества, например сиропа, крахмальной пасты, камеди, раствора целлюлозы или полимерного раствора, получают смесь при перемешивании и сушат, после чего перемалывают. Вместо превращения порошка в гранулы как таковые также возможно использование порошка в таблетирующей машине, и получающиеся в результате крупинки незавершенной формы представляют собой основу для получения гранул. В случае добавления скользящего вещества, такого как стеариновая кислота, стеарат, тальк или минеральное масло, в полученные таким образом гранулы можно предотвратить прилипание гранул друг к другу.
Таблетки также можно получить таким методом, при котором кристаллы изобретения смешивают с жидким инертным носителем и затем сразу же изготавливают из них таблетки, без осуществления вышеуказанных стадий получения из них гранул или крупинок.
Полученные таким образом таблетки могут быть покрыты пленкой или сахарной оболочкой. Также возможно применение прозрачного или полупрозрачного защитного покрытия, изготовленного из герметичной шеллаковой пленки, покрытия, изготовленного из сахара или полимерного материала, или гладкого покрытия, изготовленного из воска.
В случае других пероральных лекарственных препаратов, таких как жидкий раствор, сироп, пастилка или эликсир, также возможно их изготовление в виде готовой лекарственной формы с одноразовой дозой, в которой ее заранее определенное количество содержит заранее определенное количество кристалла настоящего изобретения.
Сироп можно получить посредством растворения кристаллов изобретения в подходящем водном растворе вкусоароматизирующего вещества. Эликсир можно получить посредством использования нетоксичного спиртового носителя.
Суспензию можно получить посредством диспергирования кристаллов изобретения в нетоксичном носителе. В случае необходимости туда же можно добавить солюбилизирующий агент или эмульгатор (такой как этоксилированный изостеариловый спирт и сложный эфир полиоксиэтиленсорбитола), консервирующее вещество или вещество, обеспечивающее вкусоароматические свойства (такое как масло мяты перечной или сахарин).
В случае необходимости лекарственная форма с одноразовой дозой для перорального введения может быть получена в виде микрокапсул. Лекарственная форма также может быть покрыта оболочкой или заключена в полимер или воск для получения пролонгированного действия или замедленного высвобождения активного ингредиента.
Парентеральный лекарственный препарат может быть в виде жидкой готовой лекарственной формы с одноразовой дозой для подкожной, внутримышечной или внутривенной инъекции в виде раствора или суспензии. Парентеральный лекарственный препарат может быть получен таким методом, при котором заранее определенное количество кристаллов изобретения суспендируют или растворяют в нетоксичном жидком носителе, который соответствует цели инъекцирования, таком как водная или масляная среда, и затем суспензию или раствор стерилизуют. Туда же могут быть добавлены нетоксичная соль или ее раствор для придания инъекционному раствору изотонических свойств. Также можно добавить стабилизатор, консервирующее вещество, эмульгатор и им подобные добавки.
Суппозитории можно получить посредством растворения или суспендирования кристаллов изобретения в легкоплавком и водорастворимом или нерастворимом твердом веществе, таком как полиэтиленгликоль, какао-масло, полусинтетический жир/масло (такой как Witepsol (зарегистрированный товарный знак)), высокомолекулярный сложный эфир (такой как сложный эфир миристилпальмитат) или их смесь.
Несмотря на то что доза может изменяться в зависимости от характеристики состояния пациента, такой как масса тела или возраст, способа введения или степени тяжести симптомов, величина дозы в диапазоне от 0,001 мг до 100 мг в день в пересчете на количество кристаллов изобретения, как правило, представляет собой приемлемую дозу для взрослых, и величина дозы в диапазоне от 0,01 мг до 10 мг представляет собой более предпочтительную дозу. В некоторых случаях меньшая доза, чем указано выше, может быть достаточной, или, с другой стороны, более значительная доза, чем указано выше, может быть необходима. Также возможно вводить ее за несколько раз в день или вводить ее с интервалом от одного до нескольких дней.
[Примеры]
Настоящее изобретение описано более подробно посредством ссылки на примеры и тестовые примеры, приведенные ниже; при этом настоящее изобретение никоим образом не должно ограничиваться этими примерами.
Для порошковой рентгеновской дифрактометрии использовали прибор Rigaku Corporation's RINT-Ultima III (зеркало анода рентгеновской трубки: Cu, напряжение: 40 кВ, ток: 40 мА, скорость сканирования: 4 градуса/мин).
Пример 1: Получение Формы-I кристалла изобретения
Этанол (440 мл) добавляли к соединению А (40 г) и смесь перемешивали и нагревали на масляной бане до температуры от 100°С до 110°С. После растворения соединения А этанол (280 мл) удаляли. Полученный концентрат перемешивали и нагревали до кипения на водяной бане с температурой 80°С в течение 1 часа. Раствор постепенно охлаждали до 10°С за 20 часов при перемешивании и выпавшее в осадок кристаллическое вещество отделяли посредством фильтрования. Полученное кристаллическое вещество промывали небольшим количеством этанола (48 мл) и сушили при пониженном давлении при 60°С, получая Форму-I кристалла изобретения (38,93 г, 97,3%). Спектр порошковой рентгеновской дифракции Формы-I кристалла изобретения представлен на фиг.1.
Т.пл.: 140,4°С (Фармакопея Японии, способ 1 Определения температуры плавления)
Пример 2: Получение Формы-I кристалла изобретения
Этанол (99 г) и метилэтилкетон (11 г) добавляли к соединению А (20 г) и нагревали до 77°С для растворения соединения А и затем раствор постепенно охлаждали до 10°С за 20 часов. В процессе охлаждения к раствору добавляли небольшое количество Формы-I кристалла изобретения. После охлаждения выпавшее в осадок кристаллическое вещество отделяли посредством фильтрования, промывали этанолом и сушили при пониженном давлении, получая Форму-I кристалла изобретения (18,72 г, 93,6%).
Пример 3: Получение Формы-II кристалла изобретения
Этанол (550 г) и метилэтилкетон (55 г) добавляли к соединению А (100 г), нагревали до 77°С и фильтровали под давлением, при этом поддерживая нагревание. При перемешивании полученный в результате фильтрат охлаждали от 70°С до 0°С, затратив на это 30 минут, и после достижения 0°С его перемешивали при 0°С в течение 2,5 часов. Выпавшее в осадок кристаллическое вещество отделяли посредством фильтрования, промывали этанолом (200 мл) и сушили при пониженном давлении. Этанол (99 г) и метилэтилкетон (11 г) добавляли к полученному кристаллическому веществу (20 г), нагревали до 70°С и выдерживали при 70°С в течение 1 часа, и постепенно охлаждали до 10°С, затратив на это 20 часов; и после достижения 10°С все перемешивали при 10°С в течение 1 часа. Выпавшее в осадок кристаллическое вещество отделяли посредством фильтрования, промывали этанолом (40 мл) и сушили при пониженном давлении, получая Форму-II кристалла изобретения (18,73 г, 93,7%). Спектр порошковой рентгеновской дифракции Формы-I кристалла изобретения представлен на фиг.2.
Т.пл.: 135,2°С (Фармакопея Японии, способ 1 Определения температуры плавления)
Пример 4: Получение Формы-II кристалла изобретения
Этанол (99 г) и метилэтилкетон (11 г) добавляли к соединению А (20 г) и нагревали до 77°С для растворения соединения А, и затем раствор постепенно охлаждали до 10°С за 20 часов. В процессе охлаждения к раствору добавляли небольшое количество Формы-II кристалла изобретения. После охлаждения выпавшее в осадок кристаллическое вещество отделяли посредством фильтрования, промывали этанолом и сушили при пониженном давлении, получая Форму-II кристалла изобретения (19,70 г, 98,5%).
Пример 5: Получение Формы-III кристалла изобретения
н-бутилацетат (500 мл) добавляли к соединению А (36,7 г) и нагревали до 75°С для растворения соединения А, и затем охлаждали до 5°С. Затем осуществляли процесс нагревания до 60°С и охлаждения до 5°С и этот процесс повторяли. Выпавшее в осадок кристаллическое вещество отделяли посредством фильтрования, промывали небольшим количеством изопропилацетата (50 мл) и сушили при пониженном давлении, получая Форму-III кристалла изобретения (29,0 г, 79,0%). Спектр порошковой рентгеновской дифракции Формы-III кристалла изобретения представлен на фиг.3.
Т.пл.: 138,0°С (Фармакопея Японии, способ 1 Определения температуры плавления)
Кристаллические вещества изобретения, использованные в нижеприведенных тестовых примерах 1-3, получали нижеприведенным способом.
Форму-I кристалла изобретения, Форму-II кристалла изобретения и Форму-III кристалла изобретения получали такими же способами, как в примере 2, примере 4 и примере 5 соответственно.
Тестовый пример 1: Измерение размера частиц
(1) Измерение распределения размера частиц для кристаллических веществ изобретения
После того как диспергирующее вещество (10 мл) добавили к кристаллам изобретения (20 мг) и затем встряхнули, кристаллы изобретения диспергировали с помощью ультразвуковых волн. Распределения размеров частиц кристаллов изобретения измеряли, используя HORIBA LA-910. Результат представлен в таблице 1.
Применяемое диспергирующее вещество представляет собой фильтрованный насыщенный раствор соединения А в 0,1 об./об.% водном растворе Полисорбата 80.
D50: Кумулятивный диаметр частиц, определенный ситовым анализом, при 50% объемном соотношении [мкм]
D90: Кумулятивный диаметр частиц, определенный ситовым анализом, при 90% объемном соотношении [мкм]
(2) Наблюдение кристаллических веществ изобретения с помощью сканирующего электронного микроскопа
Кристаллы наблюдали в сканирующем электронном микроскопе (HITACHI HIGH TECHNOLOGIES ТМ-1000 Miniscope).
Фиг.4 демонстрирует микроизображение сканирующего электронного микроскопа для Формы-I кристалла изобретения. Фиг.5 демонстрирует такое же микроизображение для Формы-II кристалла, и фиг.6 демонстрирует такое же микроизображение для Формы-III кристалла.
Исходя из результатов, вышеуказанных (1) и (2), пришли к заключению, что размер частиц Формы-I кристалла изобретения больше, чем в случае Формы-II и Формы-III кристаллов.
Тестовый пример 2: Определение остаточного содержания растворителя в кристаллах изобретения
Концентрацию остаточного растворителя, содержащегося в кристаллах изобретения, определяли с использованием нижеприведенных условий измерения. Результат представлен в таблице 2.
(Условия измерения)
Инструментальные средства для газовой хроматографии
Детектор: Пламенно-ионизационный детектор
Колонка: Капиллярная колонка
Температура колонки: 150-230°С
Температура инжектора: 200°С
Температура детектора: 300°С
Газ-носитель: Гелий
(м.д.)
Метилэтилкетон
82
Метилэтилкетон
246
н-Бутилацетат
2781
Хотя каждая кристаллическая форма и не содержит сколь-нибудь значительного количества остаточных растворителей, количество остаточных растворителей в Форме-I кристалла изобретения было меньше, чем в случае Формы-II и Формы-III.
Тестовый пример 3: Эффект удаления примесей при перекристаллизации
Эффективность удаления примесей в процессе перекристаллизации каждой кристаллической формы определяли, используя следующие условия измерений (обращенно-фазовая жидкостная хроматография)
(Условия измерений)
Инструментальные средства для ВЭЖХ
Детектор: Спектрофотометрический абсорбционный детектор в ультрафиолетовой области
Колонка: октадецилсиликагелевая (ODS) колонка
Температура колонки: 40°С
Подвижная фаза: Смесь воды, ацетонитрила и метансульфоновой кислоты.
Чистоту (%) каждого кристалла соединения А рассчитывали по нижеприведенному уравнению
Чистота (%)=(Площадь пика соединения А)/(Общая площадь)×100.
Относительную величину удаляемых примесей (%) для каждого кристалла рассчитывали по нижеприведенному уравнению
Относительная величина удаляемых примесей (%)={[(Чистота каждого кристалла соединения А)-(Чистота неочищенного соединения А)]/[100-(Чистота неочищенного соединения А)]}×100.
Результат представлен в таблице 3.
Из результатов, представленных в таблице 3, видно, что эффективность удаления примесей в случае Формы-I кристалла изобретения была выше в сравнении с этим показателем для Формы-II и III кристаллов.
Тестовый пример 4: Исследование по подбору растворителя для кристаллизации соединения А
Исследования кристаллизации соединения А проводили в соответствии с приведенными ниже способами (1) и (2).
(1) Растворитель для кристаллизации (см. таблицы 4 и 5) добавляли к соединению А и смесь перемешивали при 50°С в течение 60 минут. Полученную в результате смесь фильтровали. После фильтрования выделенный маточный раствор перемешивали при 60°С в течение 30 минут и охлаждали до 5°С на протяжении 11 часов. После перемешивания при 5°С в течение 72 часов выпавшее в осадок твердое вещество отделяли фильтрованием. Твердое вещество сушили при 20°С при пониженном давлении, в результате чего получали твердое вещество.
Регистрировали спектры порошковой рентгеновской дифракции полученных кристаллов и определяли форму каждого кристалла.
Результаты представлены в таблице 4 (исследования индивидуальных растворителей) и в таблице 5 (исследования смешанных растворителей).
При исследовании смешанных растворителей (таблица 5) каждый растворитель смешивался и использовался в одинаково равном количестве.
(2) Дальнейшие исследования проводили с использованием нижеприведенного способа с такими условиями, при которых кристаллические вещества не выпадали в осадок (см. таблицы 4 и 5), и с условиями, аналогичными им. Растворители, используемые в дальнейших экспериментах, выбирали с учетом токсичности, растворимости соединения А и возможности применения в промышленности.
Количество растворителя, меньшее по сравнению с вышеуказанным тестом (1), добавляли к соединению А и смесь нагревали до 75°С с перемешиванием. После растворения соединения А смесь перемешивали при 65°С в течение времени от 5 до 8 часов. Смесь охлаждали до 20°С на протяжении 9 часов. Выпавшее в осадок кристаллическое вещество отделяли фильтрованием и сушили при 70°С при пониженном давлении, в результате чего получали кристаллическое вещество. Результаты представлены в таблице 6.
При исследовании смешанных растворителей каждый растворитель смешивался и использовался в одинаково равном количестве.
По результатам вышеуказанных экспериментов (1) и (2) пришли к заключению, что Форма-II кристалла изобретения и Форма-III кристалла изобретения могут быть получены из различных растворителей.
С другой стороны, кристаллические вещества, которые содержат Форму-I кристалла изобретения, могут быть получены только из спиртовых растворителей, и Форма-I кристалла изобретения с высокой чистотой может быть получена из этанола.



Изобретение описывает кристаллы 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамида ("соединение А"), в виде формы-I кристалла соединения А, которая демонстрирует пики дифракции при 9,4 градуса, 9,8 градуса, 17,2 градуса и 19,4 градуса в ее спектре порошковой рентгеновской дифракции, в виде формы-II кристалла соединения А, которая демонстрирует пики дифракции при 9,0 градусах, 12,9 градуса, 20,7 градуса и 22,6 градуса в ее спектре порошковой рентгеновской дифракции, и в виде формы-III кристалла соединения А, которая демонстрирует пики дифракции при 9,3 градуса, 9,7 градуса, 16,8 градуса, 20,6 градуса и 23,5 градуса в ее спектре порошковой рентгеновской дифракции. Также описываются способы получения форм-I, II и III кристалла соединения А, фармацевтическая композиция и агонистический агент в отношении рецептора PGI2 на их основе, ускоряющий агент при ангиогенной терапии, генной инженерии или аутоиммунной трансплантации костного мозга и ускоряющий агент для ангиогенеза при восстановлении периферической артерии или при ангиогенной терапии на их основе, а также описывается профилактическое или терапевтическое средство против целого ряда заболеваний и состояний. 11 н.п. ф-лы, 6 ил., 6 табл., 5 пр.
1. Форма-I кристалла 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамида, демонстрирующая пики дифракции в спектре порошковой рентгеновской дифракции, по крайней мере, при следующих значениях углов дифракции 2θ: 9,4 градуса, 9,8 градуса, 17,2 градуса и 19,4 градуса, где спектр порошковой рентгеновской дифракции получали с использованием Cu Кα излучения.
2. Форма-II кристалла 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамида, демонстрирующая пики дифракции в спектре порошковой рентгеновской дифракции, по крайней мере, при следующих значениях углов дифракции 2θ: 9,0 градусов, 12,9 градуса, 20,7 градуса и 22,6 градуса, где спектр порошковой рентгеновской дифракции получали с использованием Cu Кα излучения.
3. Форма-III кристалла 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамида, демонстрирующая пики дифракции в спектре порошковой рентгеновской дифракции, по крайней мере, при следующих значениях углов дифракции 2θ: 9,3 градуса, 9,7 градуса, 16,8 градуса, 20,6 градуса и 23,5 градуса, где спектр порошковой рентгеновской дифракции получали с использованием Cu Кα излучения.
4. Фармацевтическая композиция, содержащая кристаллическое вещество по любому из пп.1-3 в качестве активного ингредиента.
5. Агонистический агент в отношении рецептора PGI2, содержащий кристаллическое вещество по любому из пп.1-3 в качестве активного ингредиента.
6. Профилактическое или терапевтическое средство против транзиторной ишемической атаки, диабетической нейропатии, диабетической гангрены, нарушения периферического кровообращения, заболеваний соединительных тканей, реокклюзии/рестеноза после чрескожной транслюминальной коронарной ангиопластики (РТСА), артериосклероза, тромбообразования, повышенного кровяного давления, легочной гипертензии, ишемического заболевания, стенокардии, гломерулонефрита, диабетической нефропатии, хронической почечной недостаточности, аллергической реакции, бронхиальной астмы, язвы, пролежневой язвы (пролежня), рестеноза после коронарной ангиопластики, такой как атерэктомия и имплантация стента, тромбоцитопении вследствие диализа, заболеваний, которые вызывают фиброз органов или тканей, эректильной дисфункции, воспалительных заболеваний кишечника, гастрита, язвенной болезни желудка, ишемической офтальмопатии, внезапной потери слуха, аваскулярного некроза кости, желудочно-кишечных поражений, вызванных введением нестероидного противовоспалительного средства, и в случае симптомов, связанных со стенозом спинномозгового канала поясничной области, включающее в себя кристаллическое вещество по любому из пп.1-3 в качестве активного ингредиента.
7. Ускоряющий агент при ангиогенной терапии, генной терапии или аутогенной трансплантации костного мозга, содержащий кристаллическое вещество по любому из пп.1-3 в качестве активного ингредиента.
8. Ускоряющий агент для ангиогенеза при восстановлении периферической артерии или при ангиогенной терапии, содержащий кристаллическое вещество по любому из пп.1-3 в качестве активного ингредиента.
9. Способ получения Формы-I кристалла 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамида, отличающийся тем, что 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамид растворяют в этаноле при нагревании, и затем кристаллизуют посредством постепенного охлаждения раствора от 80°С до 10°С за 20 часов.
10. Способ получения Формы-II кристалла 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамида, отличающийся тем, что 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамид кристаллизуют из спиртового растворителя, кетонового растворителя, растворителя, представляющего собой насыщенный углеводород, растворителя, представляющего собой простой эфир, и воды или из растворителя, полученного их смешиванием.
11. Способ получения Формы-III кристалла 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамида, отличающийся тем, что 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамид кристаллизуют из растворителя, представляющего собой сложный эфир, или из растворителя, представляющего собой ароматический углеводород.
| ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2002 |
|
RU2283835C2 |
| US 20040116530 A1, 17.06.2004 | |||

