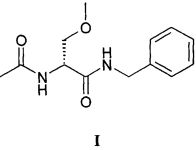
Настоящее изобретение относится к новому способу получения (2R)-2-(ацетиламино)-N-бензил-3-метоксипропанамида, активного ингредиента, используемого для лечения нейропатий, известного под названием лакосамид и представляемого приведенной ниже структурной формулой I
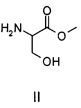
Способ согласно настоящему изобретению осуществляют, используя в качестве исходного материала метиловый эфир D,L-серина, т.е. молекулу, характеризующуюся приведенной ниже структурной формулой II,
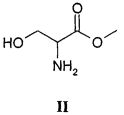
причем указанное соединение предпочтительно используют в форме гидрохлорида.
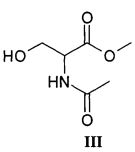
В альтернативном варианте способ может быть осуществлен с использованием в качестве исходного материала ацетамида метилового эфира D,L-серина, т.е. молекулы, соответствующей приведенной ниже структурной формуле III:
УРОВЕНЬ ТЕХНИКИ
Лакосамид представляет собой активный ингредиент, используемый в лечении боли и для лечения различных заболеваний нервной системы, включая эпилепсию. Хотя механизм его действия до конца не ясен, по-видимому, он воздействует на натриевые каналы нейронов, снижая их активность. Также считается, что лакосамид участвует в восстановлении поврежденных нейронов.
Продукт описан в патенте США RE 38,551. Также в этом документе указаны три различных пути синтеза с использованием D-серина в качестве исходного материала, а также йодистого метила и оксида серебра для метилирования ОН.
Альтернативный способ синтеза лакосамида описан в заявке на патент WO 2006/037574, согласно которой используя в качестве исходного материала N-Boc-защищенный D-серин, осуществляют реакцию метилирования гидроксила с использованием бутиллития и алкилирующего агента.
Еще один альтернативный путь синтеза описан в заявке на патент ЕР 2067765, в которой перед метилированием гидроксила осуществляют защиту амино-группы блокирующей группой, такой как тритил.
Во всех способах получения, описанных до настоящего момента, в качестве исходного материала используется D-серин, а также используются дорогие реагенты, такие как бутиллитий или блокирующие защитные группы, задача которых состоит в минимизации рацемизации продукта.
Причина состоит в том, что до настоящего момента не были разработаны способы разделения рацемических смесей лакосамида и очистка R-энантиомера считается чрезвычайно трудной.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
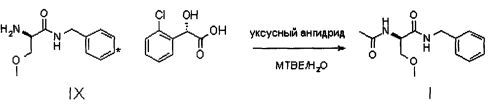
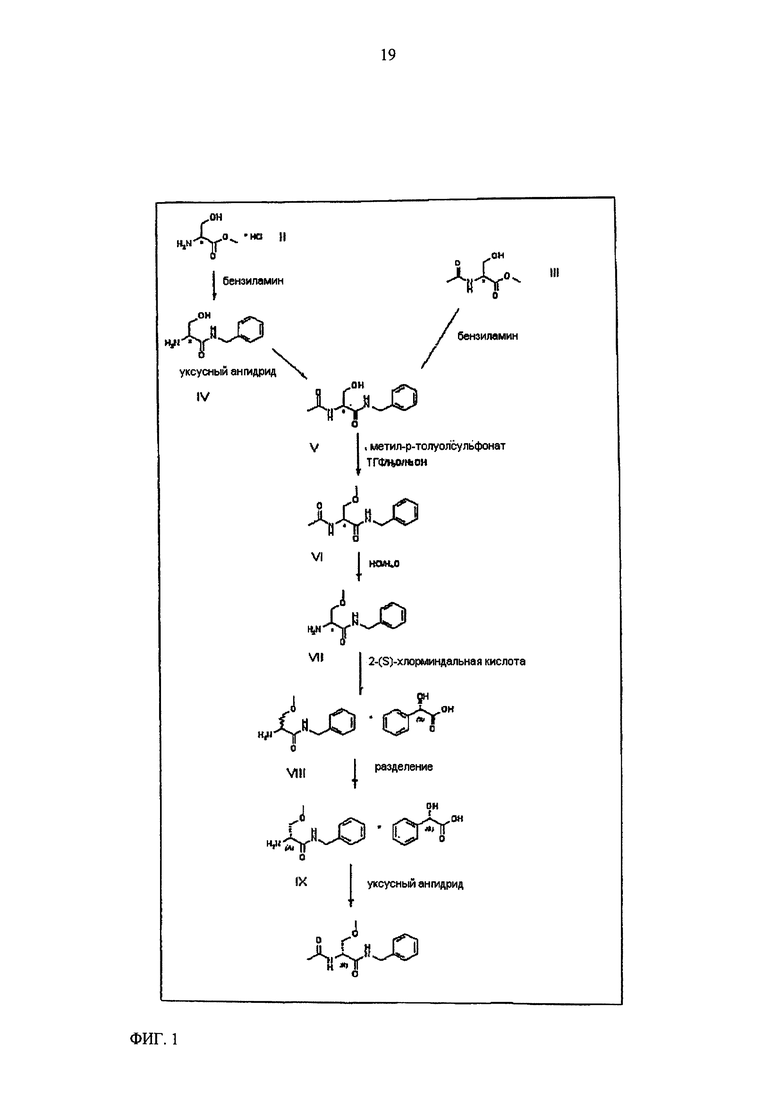
Настоящее изобретение относится к новому способу синтеза лакосамида, в котором в качестве исходного материала используют D,L-серин и в котором реакцию метилирования гидроксила осуществляют с использованием недорогого основания, такого как NaOH, и недорогого алкилирующего агента, нетоксичного и неонкогенного, такого как метил-p-толуолсульфонат; R-энантиомер выделяют из рацемической смеси лакосамида после селективного гидролиза ацетамида, реакции солеобразования рацемической смеси с хиральной кислотой (НХ*) в органическом растворителе, разделения смеси диастереоизомеров, предпочтительно путем преципитации R-энантиомера, и последующего ацетилирования оптически чистого промежуточного соединения.
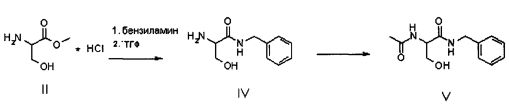
Настоящее изобретение относится к новому способу синтеза лакосамида, в котором в качестве исходного материала используют метиловый эфир D,L-лакосамида формулы II, предпочтительно в форме гидрохлорида, который можно легко получить из D,L-серина широко описанными способами.
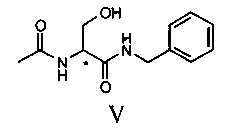
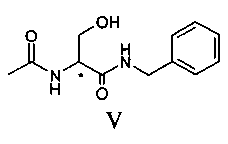
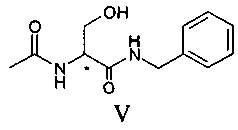
Продукт формулы II превращают в продукт формулы V путем осуществления реакции, в которой вначале используют бензиламин, а затем уксуснокислый ангидрид (или галогенангидрид уксусной кислоты, предпочтительно ацетилхлорид или в смеси с ангидридом), в соответствии со следующей схемой.
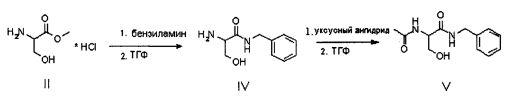
Первую реакцию предпочтительно осуществляют непосредственно в бензиламине (от 2 до 10 эквивалентов) или в апротонном полярном растворителе, таком как, например, ТГФ или ацетон, при температуре в диапазоне от 0°C до температуры флегмы, предпочтительно при 30-40°C; вторую реакцию осуществляют путем проведения реакции соединения IV с уксусным ангидридом (или с ацилгалидом, предпочтительно ацетилхлоридом) в апротонном полярном растворителе, таком как, например, ТГФ, при температуре предпочтительно в диапазоне от 10 до 40°C, предпочтительно от 15 до 30°C, еще более предпочтительно от 20 до 25°C.
В альтернативном варианте соединение V может быть приготовлено из ацетамида метилового эфира D,L-серина формулы III, доступного на рынке, путем осуществления реакции с бензиламидом; в этом случае также реакцию предпочтительно осуществляют непосредственно в бензиламине (от 2 до 10 эквивалентов) или в подходящем органическом растворителе, предпочтительно апротонном полярном растворителе, таком как, например ТГФ, при температуре в диапазоне от 0°C до температуры флегмы растворителя, предпочтительно при примерно 65°C.
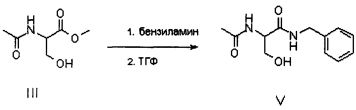
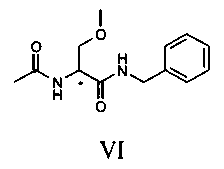
Соединение V превращают в соединение VI путем метилирования гидроксила, присутствующего в молекуле.

Реакцию метилирования можно осуществить путем растворения соединения V в подходящем органическом растворителе и приведения в контакт с алкилирующим агентом в присутствии органического или неорганического основания при температуре предпочтительно в диапазоне от 20 до 40°C, предпочтительно от 30 до 35°C. Растворитель предпочтительно представляет собой апротонный полярный растворитель, такой как, например, ТГФ. Алкилирующий агент предпочтительно выбирают из следующих агентов: йодистый метил, диметилсульфат, метилмезилат, метилпаратолуосульфонат и наиболее предпочтительно он представляет собой метил-пара-толуолсульфонат.
Органическое основание предпочтительно выбирают из третичных аминов NR1R2R3, где R1, R2 или R3, одинаковые или отличающие друг от друга, представляют собой линейные или разветвленные C1-C4 алкильные цепи, предпочтительным третичным амином является триэтиламин. Неорганическое основание предпочтительно представляет собой гидроксид щелочноземельного или щелочного металла, более предпочтительно КОН или NaOH.
Неорганическое основание может быть использовано в виде водного раствора. В случае использования неорганического основания предпочтительно использовать катализатор межфазного переноса для ускорения реакции; указанный катализатор межфазного переноса предпочтительно представляет собой соль тетрабутиламмония, имеющую в качестве противоиона гидросульфат, хлор, бром или иод.
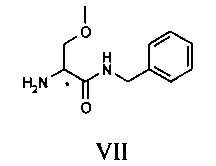
Соединение VI, рацемический лакосамид, превращают в соединение VII путем гидролиза в водном растворе с неорганической минеральной кислотой, предпочтительно HCl, при pH предпочтительно в диапазоне от 0 до 2; такую реакцию гидролиза предпочтительно осуществляют при температуре флегмы. Затем после нейтрализации кислоты экстрагируют соединение VII в органическом растворителе, предпочтительно апротонном неполярном растворителе, таком как, например, CH2Cl2, CHCl3 или C2H4Cl2.
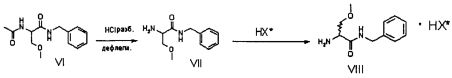
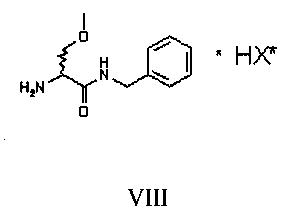
Затем соединение VII подвергают преципитации в виде соли VIII с хиральной кислотой (НХ*), предпочтительно D, такой как, например, дибензоил-винная кислота, камфорсульфокислота, миндальная, 2-хлорминдальная, 3-хлорминдальная, 4-хлорминдальная кислота. Для целей настоящего изобретения кислота предпочтительно представляет собой 2-хлорминдальную кислоту и еще более предпочтительно 2-(S)-хлорминдальную кислоту. Хиральную кислоту используют в количестве предпочтительно от 0.5 до 1.5 эквивалентов. Осаждение предпочтительно осуществляют в по меньшей мере одном апротонном полярном органическом растворителе, таком как, например, этилацетат, ацетон или изопропилацетат, еще более предпочтительно изопропилацетат. Соль VIII количественно осаждают из этого растворителя в виде смеси диастереоизомеров. Реакция солеобразования (с) соединения формулы VII с 2-(S)-хлорминдальной кислотой может быть осуществлена в органическом растворителе, таком как этилацетат, изопропилацетат, ацетон, тетрагидрофуран, метил-тетрагидрофуран, с получением смеси диастереоизомеров VIII.
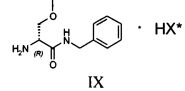
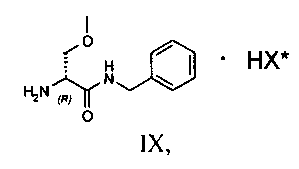
Затем полученную таким образом соль VIII растворяют в подходящей смеси растворителей, способной обеспечить селективную преципитацию только одного энантиомера, предпочтительно, только целевого энантиомера IX. Такая смесь растворителей состоит из по меньшей мере одного апротонного органического растворителя и по меньшей мере одного протонного растворителя. Апротонный органический растворитель предпочтительно выбирают из ТГФ, метил-ТГФ, этилацетата и изопропилацетата; однако предпочтительными являются такие апротонные полярные органические растворители как этилацетат или изопропилацетат. Протонный растворитель предпочтительно выбирают из C1-C4 спиртов (таких, например, как метанол, этанол, изопропанол, n-бутанол, i-бутанол и s-бутанол) и воды; в предпочтительном варианте используют смесь этилацетата и воды.
В соответствии еще с одним предпочтительным аспектом настоящего изобретения используют от 10 до 40 объемов апротонного органического растворителя на объем протонного растворителя.

Превращение соли IX в лакосамид может быть осуществлено в присутствии ацилирующего агента по меньшей мере в одном органическом растворителе, предпочтительно неполярном растворителе, необязательно смешанном с водой. В частности, соединение IX ацетилируют в присутствии ацилирующего агента в подходящем органическом растворителе, предпочтительно, неполярном апротонном растворителе, еще более предпочтительно, в эфире, в присутствии количества воды, лежащего в диапазоне от 0 до 50% масс. по отношению к соединению IX, предпочтительно от 5 до 20%, с получением лакосамида. Для целей настоящего изобретения особенно предпочтительны C2-C8 эфиры, такие как, например, метил-трет-бутиловый эфир; предпочтительным ацилирующим агентом является уксуснокислый ангидрид (может быть также использован галогенангидрид уксусной кислоты, предпочтительно ацилхлорид); реакцию ацилирования предпочтительно осуществляют при температуре от 0 до 40°C, предпочтительно от 20 до 25°C.
Дополнительными объектами настоящего изобретения являются соли формул VIII и IX
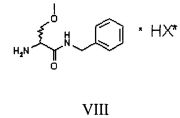
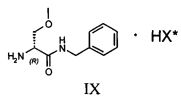
где НХ* имеет перечисленные выше значения и в предпочтительном аспекте изобретения представляет собой 2-(S)-хлорминдальную кислоту.
Полная схема способа согласно настоящему изобретению показана на фигуре 1 применительно к случаю, в котором хиральной кислотой НХ* является 2-(S)-хлорминдальная кислота.
ПРИМЕР 1 (Синтез V из III)
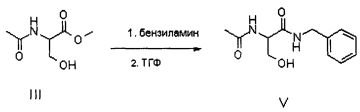
100 г соединения III и 332 г бензиламина помещают в реактор объемом 1 литр, оборудованный механической мешалкой, дефлегматором и термометром и помещенный в атмосферу азота. Смесь нагревают до 65°C и выдерживают при этой температуре с перемешиванием в течение 12 часов. Смесь дистиллируют под вакуумом для удаления избытка бензиламина. Смесь охлаждают примерно до 55-60°C и добавляют 50 мл ТГФ. Затем охлаждают до 20°C и полученное твердое вещество фильтруют, а после этого сушат под вакуумом при 40°C. Получают 123.2 г соединения V.
Молярный выход 84%
ПРИМЕР 2 (Синтез VI из V)

148 г соединения V, 7.4 г тетрабутиламмония сульфата, 740 мл ТГФ, 350 г метил-пара-толуолсульфоната и 440 г 20%-го гидроксида натрия помещают в реактор объемом 3 литра, оборудованный механической мешалкой, дефлегматором и термометром и помещенный в атмосферу азота. Полученную таким образом смесь нагревают до 35°C и выдерживают при указанной температуре с перемешиванием в течение 4 часов. Затем смесь охлаждают до 20-25°C и добавляют 165 г 28%-го гидроксида аммония. Смесь охлаждают до 5°C и доводят pH до 7 с использованием соляной кислоты.
Смесь дистиллируют под вакуумом для удаления ТГФ и затем разделяют с использованием 1 литра воды.
Смесь экстрагируют 4 раза с использованием 500 мл дихлорметана, затем органические фазы объединяют и дистиллируют до небольшого объема. Дихлорметан заменяют на 600 мл изопропилацетата. Полученную суспензию фильтруют, полученное твердое вещество промывают изопропилацетатом и сушат под вакуумом при 40°C.
Получают 115 г соединения VI. Молярный выход 73%.
ПРИМЕР 3 (Синтез VIII из VI)
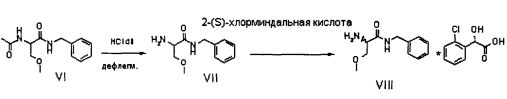
63.5 г соединения VI, 850 мл воды и 65 г 37%-ой HCl помещают в реактор объемом 2 литра, оборудованный механической мешалкой, дефлегматором и термометром и помещенный в инертную атмосферу азота. Смесь нагревают до температуры дефлегмирования и выдерживают с перемешиванием в течение 6 часов, затем смесь охлаждают до 20-25°C. pH доводят до 11.5±0.5 с использованием 30%-го гидроксида натрия. Полученную смесь экстрагируют 2 раза с использованием 300 мл дихлорметана. Объединенные органические фазы концентрируют до небольшого объема, добавляют 300 мл этилацетата и 35 г 2-(S)-хлорминдальной кислоты. Половину растворителя отгоняют и смесь оставляют с перемешиванием при комнатной температуре до завершения преципитации. Твердые вещества отфильтровывают и сушат под вакуумом при 40°C. Получают 84.3 г смеси диастереоизомеров VIII.
Молярный выход 84.1%
ПРИМЕР 4 (Разделение рацемической смеси VIII с получением IX)
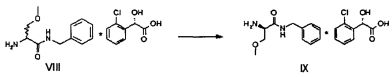
120 г рацемической смеси диастереоизомеров VIII, 3.5 литра этилацетата и 300 мл этанола помещают в реактор объемом 5 литров, оборудованный механической мешалкой, дефлегматором и термометром и помещенный в атмосферу азота. Смесь нагревают до завершения растворения и медленно охлаждают до 20°C, после чего выдерживают с перемешиванием при этой температуре в течение 5 часов. Полученное твердое вещество фильтруют и сушат.
Получают 61 г соединения IX с 37% молярным выходом относительно рацемата.
ПРИМЕР 5 (Синтез I из IX)
35 г соединения X, 700 мл трет-бутилового эфира метила и 5 мл воды помещают в реактор объемом 1 литр, оборудованный механической мешалкой, дефлегматором и термометром и помещенный в инертную атмосферу азота. Смесь охлаждают до 10-15°C и капают в реакционную смесь 10 г уксуснокислого ангидрида. Реакционную смесь выдерживают с перемешиванием в течение 2 часов при комнатной температуре, затем полученное твердое вещество, которое сушат под вакуумом при 40°C фильтруют. Получают 19.5 г лакосамида. Молярный выход 88%.
ПРИМЕР 6 (Синтез V из II)
100 г соединения II и 330 г бензиламина помещают в реактор объемом 1 литр, оборудованный механической мешалкой, дефлегматором и термометром и помещенный в инертную атмосферу азота. Смесь нагревают до 35°C и выдерживают при этой температуре с перемешиванием в течение 22 часов. Смесь дистиллируют под вакуумом до удаления бензиламина и добавляют 1.2 литра ТГФ. Смесь нагревают до получения прозрачного раствора и медленно охлаждают до комнатной температуры. Твердое вещество фильтруют и к полученному раствору медленно добавляют 65 г уксуснокислого ангидрида. Раствор концентрируют до половины объема и затем медленно охлаждают до 0-5°C. Полученное твердое вещество фильтруют и сушат под вакуумом при 40°C. Получают 106 г соединения V. Молярный выход 70%.
Изобретение относится к способу синтеза лакосамида, активного ингредиента, используемого для лечения нейфропатий. Способ осуществляют путем (а) гидроксиметилирования соединения формулы V с получением соединения формулы формулы VI; (b) гидролиза соединения формулы VI с получением соединения формулы VII; (c) реакции солеобразования соединения формулы VII с 2-(S)-хлорминдальной кислотой (HX*) в органическом растворителе с получением смеси диастереоизомеров VIII; (d) разделения смеси диастереоизомеров VIII с получением соли IX; и (е) превращения соли IX в лакосамид. Гидроксиметилирование (а) осуществляют путем проведения реакции соединения формулы V с алкилирующим агентом в присутствии основания, при температуре в диапазоне от 20°C до 40°C. Гидролиз (b) осуществляют в присутствии неорганической кислоты, предпочтительно соляной кислоты. Разделение (d) смеси диастереоизомеров VIII осуществляют путем преципитации из смеси, по меньшей мере, одного апротонного органического растворителя и, по меньшей мере, одного протонного растворителя. Стадию (е) осуществляют в присутствии ацилирующего агента, по меньшей мере, в одном органическом растворителе, предпочтительно неполярном растворителе, необязательно смешанном с водой. Также изобретение относится к промежуточным соединениям, представленным формулами VIII и XI. Технический результат - получение лакосамида за счет разделения рацемической смеси лакосамида и энантиомерной очистки R-энантиомера. 3 н. и 22 з.п. ф-лы, 6 пр.
1. Способ синтеза лакосамида, включающий следующие этапы:
(a) гидроксиметилирование соединения формулы V
с получением соединения формулы VI
(b) гидролиз соединения формулы VI с получением соединения формулы VII
(c) реакция солеобразования соединения формулы VII с 2-(S)-хлорминдальной кислотой (HX*) в органическом растворителе с получением смеси диастереоизомеров VIII;
(d) разделение смеси диастереоизомеров VIII с получением соли IX;
(е) превращение соли IX в лакосамид.
2. Способ по п.1, отличающийся тем, что гидроксиметилирование (а) осуществляют путем проведения реакции соединения формулы V с алкилирующим агентом в присутствии основания.
3. Способ по п.2, отличающийся тем, что указанный алкилирующий агент выбирают из таких агентов, как йодистый метил, диметилсульфат, метилмезилат, метилпаратолуолсульфонат, и, в предпочтительном случае, алкилирующий агент представляет собой метилпаратолуолсульфонат.
4. Способ по п.2, отличающийся тем, что указанное основание представляет собой органическое основание, предпочтительно имеющее формулу NR1R2R3, где R1, R2 или R3, одинаковые или отличающиеся друг от друга, представляют собой линейные или разветвленные С1-С4 алкильные цепи, более предпочтительно представляет собой триэтиламин, и/или неорганическое основание, предпочтительно гидроксид щелочноземельного или щелочного металла, более предпочтительно KOH или NaOH.
5. Способ по п.1, отличающийся тем, что гидроксиметилирование (а) осуществляют при температуре в диапазоне от 20°C до 40°C, предпочтительно от 30°C до 35°C.
6. Способ по п.1, отличающийся тем, что гидроксиметилирование (а) осуществляют в апротонном полярном растворителе, предпочтительно ТГФ.
7. Способ по п.1, отличающийся тем, что гидролиз (b) осуществляют в присутствии неорганической кислоты, предпочтительно соляной кислоты.
8. Способ по п.1, отличающийся тем, что указанный органический растворитель представляет собой апротонный полярный органический растворитель.
9. Способ по п.1, отличающийся тем, что указанную кислоту используют в количестве от 0,5 до 1,5 эквивалентов.
10. Способ по п.8, отличающийся тем, что указанный апротонный полярный органический растворитель выбирают из следующих растворителей: этилацетат, изопропилацетат, ацетон, тетрагидрофуран, метил-тетрагидрофуран, и предпочтительно он представляет собой изопропилацетат.
11. Способ по п.1, отличающийся тем, что разделение (d) смеси диастереоизомеров VIII осуществляют путем преципитации из смеси по меньшей мере одного апротонного органического растворителя и по меньшей мере одного протонного растворителя.
12. Способ по п.11, отличающийся тем, что указанный по меньшей мере один апротонный органический растворитель выбирают из следующих растворителей: ТГФ, метил-ТГФ, этилацетат, изопропилацетат, более предпочтительно этилацетат или изопропилацетат, и/или указанный по меньшей мере один протонный растворитель выбирают из следующих растворителей: вода и С1-С4 спирты, предпочтительно метанол, этанол, изопропанол, n-бутанол, i-бутанол и s-бутанол.
13. Способ по п.11, отличающийся тем, что указанная смесь состоит из этилацетата и этанола.
14. Способ по п.11, отличающийся тем, что используют от 10 до 40 объемов апротонного растворителя на объем протонного растворителя.
15. Способ по п.1, отличающийся тем, что этап (е) осуществляют в присутствии ацилирующего агента по меньшей мере в одном органическом растворителе, предпочтительно неполярном растворителе, необязательно смешанном с водой.
16. Способ по п.15, отличающийся тем, что указанный ацилирующий агент представляет собой уксуснокислый ангидрид или галогенангидрид уксусной кислоты.
17. Способ по п.15, отличающийся тем, что этап (е) осуществляют при температуре в диапазоне от 0 до 40°C, предпочтительно от 20 до 25°C.
18. Способ по п.15, отличающийся тем, что указанный по меньшей мере один органический растворитель представляет собой эфир, предпочтительно С2-С8 эфир, еще более предпочтительно метил-трет-бутиловый эфир.
19. Способ по п.15, отличающийся тем, что вода присутствует в количестве в диапазоне от 0 до 50% масс. по отношению к соединению IX, предпочтительно в диапазоне от 5 до 20%.
20. Способ по любому из предшествующих пунктов, отличающийся тем, что соединение V получают путем:
(аа) амидирования соединения формулы II бензиламином
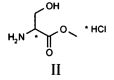
с получением соединения IV
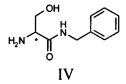
(bb) ацилирования соединения формулы IV с получением соединения V;
или
(сс) амидирования соединения формулы III
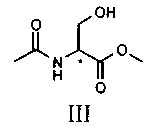
с получением соединения формулы V.
21. Способ по п.20, отличающийся тем, что бензиламин присутствует в количестве от 2 до 10 эквивалентов.
22. Способ по п.20, отличающийся тем, что этап (аа) осуществляют при температуре в диапазоне от 0°C до температуры флегмы растворителя, предпочтительно от 30°C до 40°C; этап (bb) осуществляют при температуре в диапазоне от 10°C до 40°C, предпочтительно от 15 до 30°C, более предпочтительно от 20 до 25°C; и/или этап (сс) осуществляют при температуре в диапазоне от 0°C до температуры флегмы растворителя, предпочтительно при температуре 65°C.
23. Способ по п.20, отличающийся тем, что этап (аа) осуществляют в бензиламине и в апротонном полярном растворителе, предпочтительно ТГФ; этап (bb) осуществляют в присутствии уксуснокислого ангидрида, смеси ангидрида и/или галогенангидрида уксусной кислоты, предпочтительно ацетилхлорида, в апротонном полярном растворителе, предпочтительно ТГФ; и/или этап (сс) осуществляют в бензиламине и в апротонном полярном растворителе, предпочтительно ТГФ.
24. Соль формулы VIII
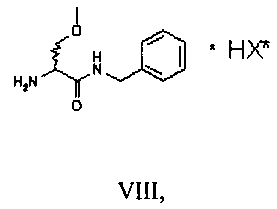
где НХ* представляет собой 2-(S)-хлорминдальную кислоту.
25. Соль формулы XI
где НХ* представляет собой 2-(S)-хлорминдальную кислоту.
| Выбрасывающий аппарат для картофелесажалок | 1926 |
|
SU12588A1 |
| CHOI D | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ANDURKAR S.V | |||
| et al.: "Synthesis and anticonvulsant activities of (R)-(O)-methyleneserine derivatives", TETRAHEDRON ASYMMETRY, 1998, vol.9, p.3841-3854 | |||
| US 5773475 A1, 30.06.1998 | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |

