Область техники
Настоящее изобретение относится к адсорбенту десульфуризации. В частности, настоящее изобретение относится к адсорбенту десульфуризации для углеводородного масла, его получению и использованию.
Уровень техники
Китайская патентная заявка № CN1355727A относится к адсорбирующей композиции, содержащей оксид цинка, диоксид кремния, оксид алюминия и никель или кобальт, и к способу получения этой адсорбирующей композиции. В соответствии с данным способом сначала получают носитель, содержащий оксид цинка, диоксид кремния и оксид алюминия, и затем в него вводят никель способом пропитки. Эта адсорбирующая композиция может использоваться при удалении серы из бензина флюид-каталитического крекинга (FCC) или дизельного топлива.
Согласно выложенному китайскому патенту № CN1208124C, носитель адсорбента, содержащий оксид цинка, вспученный перлит и оксид алюминия, пропитывают металлом-промотором, таким как кобальт и никель, и далее при подходящей температуре промотор восстанавливают, чтобы получить адсорбент для использования при удалении сульфидов из FCC-бензина.
При использовании адсорбентов известного уровня техники в атмосфере водорода для удаления серы из бензина неизбежно происходит уменьшение октанового числа из-за насыщения олефинов.
В китайской патентной заявке № CN101433821A упоминается адсорбент для удаления серы из углеводородных масел, содержащий редкоземельный фожазит, оксид активного металла и носитель, в котором носитель содержит оксид алюминия и оксид цинка, в котором редкоземельный фожазит предварительно смешивают с носителем и формуют в пористые твердые частицы, и далее вводят компоненты активного металла в пористые твердые частицы для получения адсорбента.
В китайской патентной заявке № CN101434854A упоминается адсорбент для удаления серы из легких углеводородных масел, содержащий модифицированный фосфором редкоземельный фожазит, оксид активного металла и носитель, содержащий оксид алюминия и оксид цинка, в котором редкоземельный фожазит предварительно модифицируют фосфором и далее смешивают с носителем, и далее формуют в пористые твердые частицы, и далее вводят компоненты активного металла в пористые твердые частицы для получения адсорбента.
Адсорбенты известного уровня техники предполагают увеличение октанового числа бензина благодаря содержанию формоселективного молекулярного сита для изомеризации, однако страдают от проблем, связанных с недостатком подходящего количества металлов-промоторов и приемников серы, приводящих к тому, что адсорбент имеет недостаточную активность в десульфуризации.
В связи с этим в известном уровне техники по-прежнему сохраняется потребность в адсорбенте десульфуризации, который проявляет высокую адсорбирующую активность в десульфуризации и одновременно повышает октановое число продукта десульфуризации.
Сущность изобретения
Настоящее изобретение было разработано специально для решения этих проблем известного уровня техники, и относится к адсорбенту десульфуризации для углеводородного масла, его получению и использованию.
В частности, настоящее изобретение относится к следующим аспектам.
1. Адсорбент десульфуризации для углеводородного масла, содержащий следующие компоненты в расчете на общую массу адсорбента:
1) Si-Al молекулярное сито со структурой A-FAU, где А представляет собой одновалентный катион, в количестве 1-20% масс.,
2) по меньшей мере одно связующее, выбранное из группы, состоящей из диоксида титана, диоксида олова, диоксида циркония и оксида алюминия, в количестве 3-35% масс.,
3) источник диоксида кремния в количестве 5-40% масс.,
4) оксид цинка в количестве 10-80% масс., и
5) по меньшей мере один металл-промотор, выбранный из группы, состоящей из кобальта, никеля, железа и марганца, в зависимости от металла в количестве 5-30% масс., где по меньшей мере 10% масс. металла-промотора присутствует в состоянии пониженной валентности.
2. Адсорбент согласно любому из вышеперечисленных аспектов, в котором Si-Al молекулярное сито со структурой A-FAU находится в количестве 2-15% масс., связующее находится в количестве 5-25% масс., источник диоксида кремния находится в количестве 10-30% масс., оксид цинка находится в количестве 25-70% масс., металл-промотор находится в количестве 8-25% масс.
3. Адсорбент согласно любому из вышеперечисленных аспектов, в котором Si-Al молекулярное сито со структурой A-FAU находится в количестве 2-10% масс., связующее находится в количестве 8-15% масс., источник диоксида кремния находится в количестве 12-25% масс., оксид цинка находится в количестве 40-60% масс., металл-промотор находится в количестве 12-20% масс.
4. Адсорбент согласно любому из вышеперечисленных аспектов, в котором источник диоксида кремния выбирают из группы, состоящей из диоксида кремния или природного минерала, имеющего содержание диоксида кремния более 45% масс.
5. Адсорбент согласно любому из вышеперечисленных аспектов, в котором Si-Al молекулярное сито со структурой A-FAU является по меньшей мере молекулярным ситом, выбранным из молекулярного сита Х и молекулярного сита Y.
6. Способ получения адсорбента десульфуризации для углеводородного масла согласно любому из вышеперечисленных аспектов, включающий:
(1) контактирование по меньшей мере одного связующего, выбранного из группы, состоящей из диоксида титана, диоксида олова, диоксида циркония и оксида алюминия, с источником диоксида кремния, Si-Al молекулярным ситом со структурой A-FAU (где А представляет собой одновалентный катион) и оксидом цинка для получения носителя,
(2) контактирование носителя с соединением, содержащим металл-промотор, для получения предшественника адсорбента, и
(3) при условии, достаточном для присутствия по меньшей мере 10% масс. металла-промотора в состоянии пониженной валентности (или превращения по меньшей мере 10% масс. металла-промотора в состояние пониженной валентности), обработку предшественника адсорбента для получения адсорбента десульфуризации для углеводородного масла.
7. Способ по любому из вышеперечисленных аспектов, в котором этап (1) включает следующие стадии:
(1a) связующее или предшественник связующего смешивают с кислотой для образования суспензии, и
(1b) суспензию, источник диоксида кремния, Si-Al молекулярное сито со структурой A-FAU и оксид цинка смешивают, формуют, высушивают и прокаливают для получения носителя.
8. Способ по любому из вышеперечисленных аспектов, в котором этап (1) включает следующие стадии:
(1a') по меньшей мере часть источника диоксида кремния и связующего или предшественника связующего смешивают с кислотой для образования суспензии, и
(1b') суспензию, оставшуюся часть источника диоксида кремния, Si-Al молекулярное сито со структурой A-FAU и оксид цинка смешивают, формуют, высушивают и прокаливают для получения носителя.
9. Способ по любому из вышеперечисленных аспектов, в котором предшественник связующего является по меньшей мере предшественником, выбранным из группы, состоящей из галогенидов, алкоксилатов, солей карбоновой кислоты, гидратированных оксидов, гидроксидов, гидратированных гидроксидов и оксигалогенидов титана, олова, циркония и/или алюминия.
10. Способ по любому из вышеперечисленных аспектов, в котором кислота является по меньшей мере кислотой, выбранной из группы, состоящей из водорастворимой неорганической кислоты и водорастворимой органической кислоты, и количество используемой кислоты таково, что значения pH суспензии достигают 0,5-6.
11. Способ десульфуризации углеводородного масла, включающий контактирование серосодержащего углеводородного масла с адсорбентом десульфуризации для углеводородного масла по любому из вышеперечисленных аспектов, при условии, достаточном для удаления по меньшей мере части серы из серосодержащего углеводородного масла.
Технические эффекты
В соответствии с настоящим изобретением адсорбент десульфуризации для углеводородного масла проявляет высокую адсорбирующую активность в десульфуризации и одновременно способен существенно повысить октановое число продукта десульфуризации (например, бензина).
Лучший вариант осуществления настоящего изобретения
В контексте настоящего изобретения, если не указано иное, любая реакция или стадия проводятся при давлении, обычно применяемом в данной области (например, нормальном давлении), и/или при температуре, обычно применяемой в данной области (например, нормальной температуре).
Настоящее изобретение относится к адсорбенту десульфуризации для углеводородного масла, содержащему следующие компоненты в расчете на общую массу адсорбента: 1) Si-Al молекулярное сито со структурой A-FAU, где А представляет собой одновалентный катион, в количестве 1-20% масс., 2) по меньшей мере одно связующее, выбранное из группы, состоящей из диоксида титана, диоксида олова, диоксида циркония и оксида алюминия, в количестве 3-35% масс., 3) источник диоксида кремния в количестве 5-40% масс., 4) оксид цинка в количестве 10-80% масс., и 5) по меньшей мере один металл-промотор, выбранный из группы, состоящей из кобальта, никеля, железа и марганца, в зависимости от металла в количестве 5-30% масс., где по меньшей мере 10% масс. металла-промотора присутствует в состоянии пониженной валентности (предпочтительно, в металлическом состоянии).
Согласно одному варианту осуществления настоящего изобретения Si-Al молекулярное сито со структурой A-FAU присутствует в количестве 2-15% масс., связующее присутствует в количестве 5-25% масс., источник диоксида кремния присутствует в количестве 10-30% масс., оксид цинка присутствует в количестве 25-70% масс., металл-промотор присутствует в количестве 8-25% масс.
Согласно одному варианту осуществления настоящего изобретения Si-Al молекулярное сито со структурой A-FAU присутствует в количестве 2-10% масс., связующее присутствует в количестве 8-15% масс., источник диоксида кремния присутствует в количестве 12-25% масс., оксид цинка присутствует в количестве 40-60% масс., металл-промотор присутствует в количестве 12-20% масс.
Согласно дополнительному варианту осуществления настоящего изобретения адсорбент десульфуризации для углеводородного масла может быть получен в соответствии со способом, включающим следующие этапы:
(1) контактирование по меньшей мере одного связующего, выбранного из группы, состоящей из диоксида титана, диоксида олова, диоксида циркония и оксида алюминия, с источником диоксида кремния, Si-Al молекулярным ситом со структурой A-FAU (где А представляет собой одновалентный катион) и оксидом цинка для получения носителя,
(2) контактирование носителя с соединением, содержащим металл-промотор, для получения предшественника адсорбента, и
(3) при условии, достаточном для присутствия по меньшей мере 10% масс. металла-промотора в состоянии пониженной валентности, обработку предшественника адсорбента для получения адсорбента десульфуризации для углеводородного масла.
В соответствии с настоящим изобретением в качестве Si-Al молекулярного сита со структурой A-FAU может использоваться любое Si-Al молекулярное сито со структурой A-FAU (где А представляет собой одновалентный катион), известное в данной области, без каких-либо определенных ограничений. Можно использовать молекулярное сито одного типа или смесь из двух или более типов.
Si-Al молекулярное сито со структурой A-FAU является молекулярным ситом типа фожазита, которое имеет трехмерные каналы с двенадцатичленным кольцом, и размер пор 7,4 Α×7,4 Α. В качестве примера Si-Al молекулярного сита со структурой A-FAU могут быть приведены молекулярное сито Х и молекулярное сито Y. В общем случае, молекулярное сито Х имеет молярное отношение SiO2/Al2O3, равное 2,2-3,0, молекулярное сито Y имеет молярное отношение SiO2/Al2O3 более 3,0. И молекулярное сито Х и молекулярное сито Y имеют каркасную структуру гексагональной системы и пространственную группу Fd3m. Молекулярное сито Х имеет параметр ячейки а=24,86-25,02 Α, молекулярное сито Y имеет параметр ячейки а=24,6-24,85 Α.
В качестве примера Si-Al молекулярного сита со структурой A-FAU могут быть приведены модифицированное молекулярное сито и немодифицированное молекулярное сито. В качестве примера способа модификации может быть приведен гидротермический способ, способ химической обработки (например, химическая обработка неорганической кислотой, изоморфное замещение кремнефтористоводородной кислотой, или газофазный метод SiCl4) или любое их сочетание. В качестве примера модифицированного молекулярного сита может быть приведено, среди прочего, ультрастабильное молекулярное сито Y (USY).
В соответствии с настоящим изобретением в Si-Al молекулярном сите со структурой A-FAU, замещающий ион А должен быть одновалентным катионом, например, по меньшей мере одновалентным катионом, выбранным из группы, состоящей из NH4+, H+, Na+ и Ag+.
В соответствии с настоящим изобретением не существует определенных ограничений в отношении размера частиц Si-Al молекулярного сита со структурой A-FAU, который обычно составляет 10-1000 нм, предпочтительно 100-500 нм, но этим не ограничивается. Не существует определенных ограничений в отношении величины удельной поверхности Si-Al молекулярного сита со структурой A-FAU, которая обычно составляет 150 м2/г или более, но не этим ограничивается.
В соответствии с настоящим изобретением источник диоксида кремния относится к материалу, содержащему диоксид кремния, который может быть диоксидом кремния per se, также может быть природным минералом, имеющим содержание диоксида кремния более 45% масс. Природный минерал может содержать другие компоненты, такие как Al2O3, K2O, CaO, MgO, Fe2O3, TiO2 и тому подобное. Источником диоксида кремния, например, может быть по меньшей мере источник, выбранный из группы, состоящей из диатомита, вспученного перлита, каолина, силикалита, коллоидного кремнезема, осажденного кремнезема, пирогенного кремнезема, силикатного золя, крупнопористого кремнезема и силикагеля. Можно использовать источник диоксида кремния одного типа или смесь из двух или более типов.
В соответствии с настоящим изобретением оксид цинка обычно имеет чистоту более 99%.
В соответствии с настоящим изобретением связующее является по меньшей мере связующим, выбранным из группы, состоящей из диоксида титана, диоксида олова, диоксида циркония и оксида алюминия. Связующее может использоваться в кристаллической форме (например, диоксид титана анатазной формы), либо в аморфной форме или в коллоидной форме (например, золь или коллоид), или в форме дисперсии (например, водная суспензия) без какого-либо определенного ограничения. Можно использовать связующее одного типа или смесь из двух или более типов.
В соответствии с настоящим изобретением термин «металл-промотор» относится к металлическому компоненту, который помогает обеспечивать удаление серы из углеводородного масла после введения в адсорбент по настоящему изобретению. Металл-промотор предпочтительно является по меньшей мере металлом-промотором, выбранным из группы, состоящей из кобальта, никеля, железа и марганца, более предпочтительно - никелем. Можно использовать металл-промотор одного типа или смесь из двух или более типов.
В соответствии с настоящим изобретением по меньшей мере часть металла-промотора в адсорбенте по настоящему изобретению должна находиться в состоянии пониженной валентности. При этом без намерения связывать себя какой-либо теорией, полагают, что промотор в состоянии пониженной валентности способен химически адсорбировать, отщеплять или удалять серу. Для этого необходимо уменьшить количество атомов кислорода, связанных с металлом, или сделать степень окисления металла равной 0 (т.е. как в металлическом или элементарном состоянии). Например, если в качестве металла-промотора используется никель, известно, что оксид никеля (NiO) представляет нормальную степень окисления, и тогда никель (металл-промотор) в состоянии пониженной валентности может быть металлическим никелем (Ni0) или оксидом никеля, имеющим формулу NiO(1-х) с нестехиометрической степенью окисления, где 0<x<1. Если в качестве металла-промотора используется кобальт, известно, что оксид кобальта (CoO) представляет нормальную степень окисления, и тогда кобальт (металл-промотор) в состоянии пониженной валентности может быть металлическим кобальтом (Co0) или оксидом кобальта, имеющим формулу CoO(1-y) с нестехиометрической степенью окисления, где 0<y<1. Если в качестве металла-промотора используется железо, известно, что оксид железа (Fe2O3) представляет нормальную степень окисления, и тогда железо (металл-промотор) в состоянии пониженной валентности может быть металлическим железом (Fe0) или оксидом железа, имеющим формулу Fe2O(3-a) с нестехиометрической степенью окисления, где 0<a<3. Если в качестве металла-промотора используется марганец, известно, что оксид марганца (MnO2) представляет нормальную степень окисления, и тогда марганец (металл-промотор) в состоянии пониженной валентности может быть металлическим марганцем (Mn0) или оксидом марганца, имеющим формулу MnO(2-b) с нестехиометрической степенью окисления, где 0<b<2.
Предпочтительно, чтобы содержание металла-промотора в адсорбенте десульфуризации для углеводородного масла данного изобретения было таким, что когда адсорбент приводится в контакт с серосодержащим углеводородным маслом в условиях десульфуризации, описанных в данном описании, он может эффективно удалять серу из указанного углеводородного масла. Для оптимальной активности десульфуризации в адсорбенте по настоящему изобретению, относительно общего содержания металла-промотора, предпочтительно по меньшей мере примерно 10% масс. металла-промотора находится в состоянии пониженной валентности (предпочтительно в металлическом состоянии), более предпочтительно по меньшей мере примерно 40% масс. металла-промотора находится в состоянии пониженной валентности (предпочтительно в металлическом состоянии), и наиболее предпочтительно по меньшей мере 80% масс., по меньшей мере 90% масс. или более металла-промотора находится в состоянии пониженной валентности (предпочтительно в металлическом состоянии).
Согласно способу получения адсорбента десульфуризации для углеводородного масла по настоящему изобретению на этапе (1) (в дальнейшем в этом документе называется этапом контактирования) исходные материалы (т.е. по меньшей мере одно связующее, выбранное из группы, состоящей из диоксида титана, диоксида олова, диоксида циркония и оксида алюминия, источник диоксида кремния, Si-Al молекулярное сито со структурой A-FAU и оксид цинка) приводятся в контакт друг с другом (например, смешиваются) в соответствующем вышеуказанном заранее определенном количестве, для получения продукта контактирования по настоящему изобретению (т.е. носителя). В данном этапе не существует определенных ограничений в отношении порядка контактирования или последовательности контактирования исходных материалов.
В соответствии с настоящим изобретением не существует определенных ограничений относительно того, как осуществлять этап контактирования, при условии, что исходные материалы могут в достаточной степени контактировать друг с другом, чтобы образовать гомогенный продукт контактирования. Например, исходные материалы могут быть смешаны до гомогенного состояния любыми способами, известными в данной области.
При необходимости, для более полного и гомогенного контактирования, или для облегчения контактирования, этап контактирования можно осуществлять в присутствии жидкой или газовой среды. При этом полученный продукт контактирования может быть в виде суспензии, тестообразной или кремообразной смеси.
Этап контактирования можно осуществлять при любой температуре, находящейся в диапазоне от 0°С до 70°С. С точки зрения удобства нормальная температура является предпочтительной, но этим не ограничивается. Контактирование может длиться в течение периода времени, достаточного для получения гомогенного продукта контактирования, обычно от 0,5 до 5 часов, но этим не ограничивается.
В соответствии с настоящим изобретением продукт контактирования после получения при необходимости может дополнительно формоваться, высушиваться и прокаливаться любыми способами, известными в данной области.
В частности, полученный таким образом продукт контактирования может быть сформован в экструдаты, гранулы, таблетки, сферы или микросферы любыми способами, известными в данной области. Например, когда продукт контактирования присутствует в виде тестообразной или кремообразной смеси, продукт контактирования может быть непосредственно сформован (предпочтительно экструдированием) в цилиндрический экструдат, имеющий диаметр, как правило, 1,0-8,0 мм и длину, как правило, 2,0-5,0 мм. Затем полученный таким образом экструдат высушивают и прокаливают. Когда продукт контактирования присутствует в виде суспензии, продукт контактирования может быть высушен до тестообразной или кремообразной консистенции и далее сформован, как описано выше. Или, в качестве альтернативы, суспензия может быть непосредственно сформована с помощью способа распылительной сушки в микросферы с размером частицы примерно 20-200 мкм. При этом для удобства осуществления распылительной сушки предпочтительно скорректировать содержание твердого вещества в суспензии перед сушкой до уровня 10-50% масс., предпочтительно 20-50% масс.
В соответствии с настоящим изобретением сушку можно осуществлять любым общепринятым способом, без каких-либо определенных ограничений. Например, сушка может быть сушкой на воздухе, в печи или активным вентилированием. Температура в процессе сушки может быть от комнатной температуры до 400°С, предпочтительно 100-350°С, но этим не ограничивается.
В соответствии с настоящим изобретением прокаливание можно осуществлять любым общепринятым способом, без каких-либо определенных ограничений. В общем случае температура при прокаливании может быть 400-700°С, предпочтительно 450-650°С, в то время как продолжительность прокаливания обычно может быть по меньшей мере 0,5 ч, предпочтительно 0,5-100 ч, более предпочтительно 0,5-10 ч, но этим не ограничивается.
Учитывая данные варианты осуществления, предпочтительно, чтобы этап (1) способа получения адсорбента десульфуризации для углеводородного масла по настоящему изобретению дополнительно включал стадию формования (как описано выше), сушки (как описано выше) и прокаливания (как описано выше) продукта контактирования (названную стадией (1-1)). Продукт, полученный на этой стадии (1-1), в контексте настоящего изобретения также называется носителем.
Согласно одному варианту осуществления настоящего изобретения этап контактирования предпочтительно осуществляют в соответствии со следующим способом А или способом B.
Способ А включает следующие стадии:
(1a) связующее или предшественник связующего смешивают с кислотой с образованием суспензии, и
(1b) суспензию, источник диоксида кремния, Si-Al молекулярное сито со структурой A-FAU и оксид цинка смешивают, формуют, высушивают и прокаливают для получения носителя.
Способ B включает следующие стадии:
(1a') по меньшей мере часть источника диоксида кремния и связующего или предшественника связующего смешивают с кислотой для образования суспензии, и
(1b') суспензию, оставшуюся часть источника диоксида кремния, Si-Al молекулярное сито со структурой A-FAU и оксид цинка смешивают, формуют, высушивают и прокаливают для получения носителя.
В соответствии с настоящим изобретением используемая кислота является по меньшей мере кислотой, выбранной из группы, состоящей из водорастворимой неорганической кислоты и водорастворимой органической кислоты, и содержание используемой кислоты (если используется более одной кислоты, - общее содержание используемых кислот) является таким, что в конечном счете полученная суспензия имеет значение pH в диапазоне 0,5-6, предпочтительно 1-6, более предпочтительно 1-5, еще более предпочтительно 1-4, наиболее предпочтительно 1,5-4. Можно использовать кислоту одного типа или смесь из двух или более типов.
Кислота предпочтительно является по меньшей мере кислотой, выбранной из группы, состоящей из соляной кислоты, азотной кислоты, фосфорной кислоты и уксусной кислоты. При необходимости или для облегчения работы кислота может использоваться в виде водного раствора.
В соответствии с настоящим изобретением выражение «предшественник связующего» относится к соединению, которое может быть превращено в связующее с помощью соответствующей обработки (например, после прохождения стадий (1a) и (1b), или после прохождения стадий (1a') и (1b')). Примером предшественника могут быть галогениды, алкоксилаты, соли карбоновой кислоты, гидратированные оксиды, гидроксиды, гидратированные гидроксиды и оксигалогениды металла M, где M является по меньшей мере металлом, выбранным из группы, состоящей из титана, олова, циркония и алюминия. Можно использовать предшественник одного типа или смесь из двух или более типов.
В частности, примером предшественника диоксида титана может служить тетрахлорид титана, этилтитанат, изопропилтитанат, ацетат титана и гидратированный оксид титана. Если начинать с диоксида титана анатазной формы, можно повторно получать диоксид титана после стадий (1a) и (1b) или стадий (1а') и (1b'), и поэтому диоксид титана анатазной формы также иногда называют предшественником диоксида титана. Можно использовать предшественник диоксида титана одного типа или смесь из двух или более типов. Примером предшественника оксида циркония могут служить тетрахлорид циркония, оксихлорид циркония, ацетат циркония и гидратированный диоксид циркония. Если начинать с аморфного диоксида циркония, после стадий (1a) и (1b) или стадий (1а') и (1b') можно повторно получать диоксид циркония, и поэтому аморфный диоксид циркония также иногда называют предшественником диоксида циркония. Можно использовать предшественник диоксида циркония одного типа или смесь из двух или более типов. Примером предшественника диоксида олова могут служить тетрахлорид олова, тетраизопропаноат олова, ацетат олова и гидратированный оксид олова. Можно использовать предшественник диоксида олова одного типа или смесь из двух или более типов. Предшественником оксида алюминия могут служить гидратированный оксид алюминия, золь алюминия, бемит, псевдобемит, тригидратированный оксид алюминия и аморфный гидроксид алюминия. Можно использовать предшественник оксида алюминия одного типа или смесь из двух или более типов. Оксид алюминия в конечном счете образуется из предшественника оксида алюминия, как правило, представленного в виде γ-Al2O3.
В соответствии с настоящим изобретением не существует определенных ограничений относительно того, как осуществлять стадию (1а), при условии, что связующее или предшественник и кислота (предпочтительно водный раствор кислоты) могут быть тщательно перемешаны для получения гомогенной суспензии. Аналогичным образом, не существует определенных ограничений относительно того, как осуществлять стадию (1а'), при условии, что связующее или предшественник, кислота (предпочтительно водный раствор кислоты) и по меньшей мере часть источника диоксида кремния могут быть тщательно перемешаны для получения гомогенной суспензии. В обоих случаях перемешивание может проводиться при температуре в диапазоне от 0°С до 70°С, с точки зрения удобства предпочтительно от 5°C до 40°C, но этим не ограничивается. Продолжительность перемешивания такова, чтобы можно было получить гомогенную суспензию: обычно от 0,5 до 5 ч, но этим не ограничивается.
В соответствии с настоящим изобретением выражение «по меньшей мере часть источника диоксида кремния» означает, что содержание источника диоксида кремния, используемого на стадии (1a'), представляет по меньшей мере часть от общего содержания источника диоксида кремния, используемого для получения адсорбента десульфуризации для углеводородного масла по настоящему изобретению. Например, содержание, представленное выражением «по меньшей мере часть», в данном документе может быть 30% масс. или более, предпочтительно 50% масс. или более, более предпочтительно 70% масс. или более, более предпочтительно 90% масс. или более от общего содержания источника диоксида кремния, или, например, 100% масс. Кроме того, выражение «оставшаяся часть источника диоксида кремния» относится к остальному содержанию источника диоксида кремния, полученному вычитанием вышеуказанного содержания, представленного выражением «по меньшей мере часть», из общего содержания. Например, содержание, представленное выражением «оставшаяся часть», в данном документе может быть 70% масс. или менее, предпочтительно 50% масс. или менее, более предпочтительно 30% масс. или менее, еще более предпочтительно 10% масс. или менее от общего содержания источника диоксида кремния, или, например, 0% масс.; в этом случае на стадии (1b') не будет использоваться источник диоксида кремния.
Если предшественник используется на стадии (1a), - предшественник взаимодействует с кислотой с образованием адгезионного гидролизатного раствора, т.е. гель-раствора. Гель-раствор в настоящем документе также называется суспензией. Аналогичным образом, если предшественник используется на стадии (1a'), - предшественник взаимодействует с кислотой с образованием адгезионного гидролизатного раствора, т.е. гель-раствора. Гель-раствор и по меньшей мере часть источника диоксида кремния образуют так называемую в данном документе суспензию. В этом случае длительность перемешивания на стадии (1а) или стадии (1а') является такой же, как описано выше. Тем не менее, для более полного протекания реакции гидролиза и более гомогенной системы, в данной области техники известно, что при необходимости полученный гель-раствор или суспензия могут быть подвергнуты выдерживанию. Выдерживание можно осуществлять любым способом, известным в данной области, например, при температуре 60-90°С гель-раствор или суспензию оставляют стоять от 0,5 до 3 ч, но этим не ограничиваются. В соответствии с настоящим изобретением гель-раствор или суспензия после этого выдерживания также называется суспензией.
Согласно стадии (1b) по настоящему изобретению полученная суспензия, источник диоксида кремния, Si-Al молекулярное сито со структурой A-FAU и оксид цинка смешивают, формуют, высушивают и прокаливают, чтобы получить носитель. Или, в соответствии со стадией (1b'), суспензию, оставшуюся часть источника диоксида кремния, Si-Al молекулярное сито со структурой A-FAU и оксид цинка смешивают, формуют, высушивают и прокаливают для получения носителя. В обоих случаях суспензия, источник диоксида кремния (или его оставшаяся часть), Si-Al молекулярное сито со структурой A-FAU и оксид цинка могут быть смешаны в любой последовательности и любым способом, при условии, что может быть получена гомогенная смесь (т.е. продукт контактирования). Например, допустимо введение в суспензию источника диоксида кремния (или его оставшейся части), и после этого или одновременно оксида цинка и Si-Al молекулярного сита со структурой A-FAU, или введение в суспензию этих трех компонентов одновременно. Или, в качестве альтернативы, допустимо непосредственно вводить в суспензию источник диоксида кремния (или его оставшуюся часть), оксид цинка и/или Si-Al молекулярное сито со структурой A-FAU. Очевидно, также допустимо превращать источник диоксида кремния (или его оставшуюся часть), оксид цинка и/или Si-Al молекулярное сито со структурой A-FAU в водную суспензию, и затем осуществлять вышеуказанное введение.
Стадия (1b) или стадия (1b') может проводиться при температуре в диапазоне от 0°С до 70°С, с точки зрения удобства предпочтительно от 5°C до 40°C, но этим не ограничивается. Продолжительность перемешивания такова, чтобы можно было получить гомогенную суспензию: обычно от 0,5 до 5 ч, но этим не ограничивается.
После получения смеси или продукта контактирования допустимо проводить формование, сушку и прокаливание таким же способом, как описано применительно к этапу (1) или стадии (1-1) для продукта контактирования, чтобы получить носитель.
Далее, в соответствии с этапом (2) носитель контактирует с соединением, содержащим металл-промотор, для получения предшественника адсорбента.
В соответствии с настоящим изобретением соединение, содержащее металл-промотор, является соединением, которое можно превратить в металл-промотор прокаливанием. Примером соединения, содержащего металл-промотор, могут быть ацетаты, карбонаты, нитраты, сульфаты, тиоцианаты, гидроксиды, гидратированные оксиды, гидратированные гидроксиды или оксиды металла M', где M' представляет собой по меньшей мере металл из кобальта, никеля, железа и марганца, предпочтительно никель. Можно использовать соединение, содержащее металл-промотор, одного типа или смесь из двух или более типов.
В соответствии с настоящим изобретением не существует определенных ограничений относительно способа контактирования носителя с соединением, содержащим металл-промотор, при условии, что может быть достигнуто полное контактирование с образованием гомогенного продукта контактирования (т.е. предшественника адсорбента). В данном случае не существует определенных ограничений относительно порядка контактирования или последовательности контактирования между носителем и соединением, содержащим металл-промотор.
При необходимости, для более полного и гомогенного контактирования, или для облегчения контактирования, допустимо проводить контактирование в присутствии жидкой или газовой среды. В этом случае полученный продукт контактирования может быть в виде суспензии.
Этап контактирования можно осуществлять при температуре в диапазоне от 0°С до 70°С, с точки зрения удобства предпочтительно от 5°C до 40°C, но этим не ограничиваясь. Продолжительность перемешивания такова, чтобы можно было получить гомогенную суспензию: обычно от 1 до 20 мин, но этим не ограничивается.
В соответствии с вариантом осуществления настоящего изобретения контактирование между носителем и соединением, содержащим металл-промотор, может осуществляться способом пропитки или способом осаждения, известным в данной области. В соответствии со способом пропитки получают водный раствор или водную суспензию соединения, содержащего металл-промотор, и далее носитель пропитывают раствором или суспензией, чтобы получить продукт контактирования наподобие суспензии. В соответствии со способом осаждения получают водный раствор или водную суспензию соединения, содержащего металл-промотор, и далее носитель пропитывают раствором или суспензией, и затем в него вводят водный раствор аммиака, в результате чего соединение, содержащее металл-промотор, осаждается на/в носителе, с получением влажного продукта контактирования или продукта контактирования наподобие суспензии.
В соответствии с настоящим изобретением после получения продукта контактирования, особенно если продукт контактирования находится во влажной форме или в форме суспензии, допустимо иногда высушивать продукт любым способом, известным в данной области, для удаления любой жидкой среды (например, воды) или газовой среды, которая могла вводиться туда в ходе получения. Например, продукт контактирования может быть высушен при температуре примерно 50-300°C, предпочтительно 100-250°C, при продолжительности высушивания примерно 0,5-8 ч, предпочтительно примерно 1-5 ч.
В соответствии с настоящим изобретением после получения продукта контактирования (предпочтительно после вышеуказанной сушки), допустимо иногда прокаливать продукт любым способом, известным в данной области, чтобы превратить металл-промотор, содержащийся в продукте контактирования, в соответствующий оксид металла. Например, прокаливание может осуществляться в присутствии кислорода или в кислородной атмосфере (с содержанием кислорода, например, более 20 об.%, предпочтительно более 40 об.%), при температуре примерно 300-800°С, более предпочтительно 450-750°С. Продолжительность прокаливания обычно составляет около 0,5-4 ч, предпочтительно 1-3 ч. Продукт контактирования после данного прокаливания также называется в данном документе предшественником адсорбента или иногда прокаленным предшественником адсорбента.
В связи с этим, способ получения адсорбента десульфуризации для углеводородного масла по настоящему изобретению предпочтительно включает на этапе (2) стадию высушивания (как описано выше) и прокаливания (как описано выше) предшественника адсорбента (названную стадией (2-1)).
Далее, в соответствии с этапом (3) данного изобретения предшественник адсорбента (предпочтительно прокаленный предшественник адсорбента или предшественник адсорбента, полученный на стадии (2-1)), обрабатывают в условиях, достаточных для присутствия по меньшей мере 10% масс. металла-промотора в состоянии пониженной валентности (например, восстановленного в водородной атмосфере), для получения адсорбента десульфуризации для углеводородного масла по настоящему изобретению.
В соответствии с настоящим изобретением предшественник адсорбента может быть восстановлен при температуре 300-600°C и в водородной атмосфере, чтобы безусловно получить по меньшей мере 10% масс. металла-промотора, находящегося в состоянии пониженной валентности (например, в металлическом состоянии). При этом температура более предпочтительно составляет 400-500°C, содержание водорода в водородной атмосфере составляет обычно 10-60 об.%, продолжительность восстановления обычно составляет 0,5-6 ч, предпочтительно 1-3 ч.
Настоящее изобретение также относится к способу десульфуризации углеводородного масла, включающему этап контактирования серосодержащего углеводородного масла с адсорбентом десульфуризации для углеводородного масла по настоящему изобретению, при условии, достаточном для удаления по меньшей мере части серы из серосодержащего углеводородного масла.
В соответствии с настоящим изобретением углеводородное масло может быть FCC-бензином или дизельным топливом, при этом термин «FCC-бензин» относится к углеводородной смеси, имеющей температурный интервал кипения от 40 до 210°C, или любой ее фракции, которая представляет собой продукт способа термического или каталитического крекинга. Приемлемый способ термического крекинга включает, среди прочего, коксование, термический крекинг, висбрекинг или любое их сочетание. Приемлемый способ каталитического крекинга включает, среди прочего, каталитический крекинг на псевдоожиженном слое, каталитический крекинг тяжелого масла или любое их сочетание. В данных обстоятельствах приемлемый FCC-бензин включает, среди прочего, бензин коксования, бензин термического крекинга, бензин висбрекинга, бензин каталитического крекинга на псевдоожиженном слое, бензин каталитического крекинга тяжелого масла или любое их сочетание. В некоторых случаях, при необходимости, FCC-бензин может быть разделен на фракции и/или гидрогенизирован перед использованием в способе десульфуризации по настоящему изобретению в качестве углеводородного масла. В соответствии с настоящим изобретением термин «дизельное топливо» относится к углеводородной смеси, имеющей температурный интервал кипения от 170°C до 450°C, или любой ее фракции. Углеводородное масло данного типа включает, среди прочего, легкий рецикловый газойль, керосин, дизельное масло прямой перегонки, гидроочищенное дизельное масло или любое их сочетание.
В контексте настоящего изобретения термин «сера» включает в себя серу любой природы, например, органические соединения серы, обычно находящиеся в углеводородных маслах (например, в FCC-бензине или дизельном топливе). Сера, которая может быть найдена в серосодержащем углеводородном масле согласно настоящему изобретению, может являться, среди прочего, карбонилсульфидом (COS), дисульфидом углерода (CS2), меркаптаном или другими тиофеновыми соединениями или любым их сочетанием, особенно тиофенами, бензотиофенами, алкилтиофенами, алкилбензотиофенами и алкилдибензотиофенами, и тиофеновыми соединениями с большей молекулярной массой, которые могут обычно находиться в дизельном топливе.
В соответствии с настоящим изобретением «условия, достаточные для удаления по меньшей мере части серы из серосодержащего углеводородного масла» включают: присутствие водорода, молярное отношение водорода к углеводородному маслу 0,1-3, более предпочтительно 0,3-0,8, WHSV (массовая часовая объемная скорость) 1-15 ч-1, более предпочтительно 3-8 ч-1, температуру реакции 350-500°C, предпочтительно 400-450°C, давление реакции 0,5-4 МПа, предпочтительно 1,0-2,0 МПа.
При необходимости адсорбент десульфуризации для углеводородного масла по настоящему изобретению можно использовать повторно (например, после потери по меньшей мере части активности десульфуризации) с помощью процедуры регенерации и восстановления. Условия регенерации включают: в присутствии кислорода или в кислородной атмосфере (с содержанием кислорода, например, более 20 об.%, предпочтительно более 40 об.%), при нормальном давлении, при температуре 400-700°C, предпочтительно 500-600°C, продолжительность регенерации 0,5-2 ч. Условия восстановления включают: в присутствии водорода или в водородной атмосфере, при температуре 350-500°С, предпочтительно 370-450°С, при давлении 0,2-2 МПа, предпочтительно 0,2-1,5 МПа, продолжительность восстановления 0,2-2 ч.
Примеры
Следующие примеры более подробно иллюстрируют, но не ограничивают настоящее изобретение.
В следующих примерах и сравнительных примерах состав адсорбента десульфуризации для углеводородного масла определяли рентгеноструктурным анализом (XRD).
Пример I-1
Адсорбент получали следующим образом: 2,42 кг тетрахлорида титана (от Beijing Chemical Plant, AR, 99% масс.) медленно добавляли к 3,2 кг кислой воды, и затем осторожно перемешивали, чтобы избежать осаждения кристаллов оксида титана, при этом получали бесцветный и прозрачный коллоидный раствор, названный золем титана. Затем в титановый золь добавляли 2,10 кг вспученного перлита (от компании Global Mineral, 2,06 кг в пересчете на сухое вещество) и далее перемешивали до гомогенного состояния, для получения суспензии А.
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.), 0,84 кг бета-молекулярного сита (от Nanjing Catalyst Division, Sinopec, 0,70 кг в пересчете на сухое вещество) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Суспензию носителя подвергали распылительной сушке с помощью распылительной сушилки модели Bowen Nozzle Tower™, при давлении распылительной сушки от 8,5 до 9,5 МПа, температуре на входе 500°С или менее, температуре на выходе примерно 150°С. Микросферы, полученные при распылительной сушке, сначала высушивали при 180°С в течение 1 ч, затем прокаливали при 635°С в течение 1 ч для получения носителя адсорбента.
3,2 кг носителя адсорбента пропитывали в растворе 3,51 кг гексагидрата нитрата никеля (от Beijing Chemical Reagent Company, имеющего чистоту более 98,5% масс.) в 0,6 кг деионизированной воды, полученную смесь высушивали при 180°C в течение 4 ч, и затем прокаливали в воздушной атмосфере при 635°С в течение 1 ч для получения предшественника адсорбента.
Предшественник адсорбента восстанавливали при 425°C в водородной атмосфере в течение 2 ч для получения адсорбента. Адсорбент назвали адсорбентом I-A1. Состав адсорбента I-A1: содержание оксида цинка 44,3% масс., содержание вспученного перлита 20,6% масс., содержание бета-молекулярного сита 7,0% масс., содержание диоксида титана 10,0% масс., содержание никеля 18,1% масс.
Пример I-2
1,26 кг диоксида титана (анатазной формы, содержащего 1,17 кг диоксида титана в пересчете на сухое вещество) добавляли к 2,6 кг 10% соляной кислоты (СР, от Beijing Chemical Plant), после этого перемешивали и подкисляли в течение 1 ч, при этом оксид титана полностью растворялся в бесцветный и прозрачный коллоидный раствор, названный золем титана. Затем в золь титана добавляли 1,54 кг диатомита (от компании Global Mineral, 1,50 кг в пересчете на сухое вещество) и далее смешивали при перемешивании для получения суспензии А.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 0,36 кг бета-молекулярного сита (от Nanjing Catalyst Division, Sinopec, 0,30 кг в пересчете на сухое вещество) и 5,0 кг деионизированной воды, и перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера I-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента I-А2. Состав адсорбента I-A2: содержание оксида цинка 55,2% масс., содержание диоксида титана 11,7% масс., содержание диатомита 15,0% масс., содержание бета-молекулярного сита 3,0% масс., содержание никеля 15,1% масс.
Пример I-3
Адсорбент получали следующим образом: 3,87 кг этилтитаната (от компании Aldrich, AR, 99%) при перемешивании медленно добавляли к 3,2 кг 10% азотной кислоты (коммерчески чистой (CP), от Beijing Chemical Plant), после этого перемешивали в течение 1 ч, при этом получали светло-желтый и прозрачный коллоидный раствор, названный золем титана.
Смешивали 4,93 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 1,64 кг диатомита (от компании Global Mineral, 1,60 кг в пересчете на сухое вещество), 0,56 кг молекулярного сита USY (от Qilu Catalyst Division, Sinopec, 0,50 кг в пересчете на сухое вещество) и 6,40 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии. Суспензию добавляли в золь титана и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера I-1, суспензию носителя высушивали распылением и формовали, и далее вводили в нее активные компоненты никеля и кобальта для получения адсорбента I-А3. Состав адсорбента I-A3: содержание оксида цинка 49,3% масс., содержание диоксида титана 13,5% масс., содержание диатомита 16,0% масс., содержание USY 5,0% масс., содержание никеля 8,1% масс., содержание кобальта 8,1% масс.
Пример I-4
Адсорбент получали следующим образом: 3,36 кг этилтитаната (от компании Aldrich, AR, 99%) при перемешивании медленно добавляли к 3,2 кг 10% азотной кислоты (CP, от Beijing Chemical Plant), после этого перемешивали в течение 1 ч, при этом получали светло-желтый и прозрачный коллоидный раствор, названный золем титана.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 2,03 кг каолина (от Suzhou Kaolin Factory, 1,50 кг в пересчете на сухое вещество), 0,36 кг молекулярного сита Х (от Qilu Catalyst Division, Sinopec, 0,30 кг в пересчете на сухое вещество) и 6,40 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии. Суспензию добавляли в золь титана и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера I-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента I-А4. Состав адсорбента I-A4: содержание оксида цинка 55,2% масс., содержание диоксида титана 11,7% масс., содержание каолина 15,0% масс., содержание молекулярного сита Х 3,0% масс., содержание никеля 15,1% масс.
Пример I-5
Адсорбент получали следующим образом: 2,42 кг тетрахлорида титана (от Beijing Chemical Plant, AR, 99% масс.) медленно добавляли к 3,2 кг кислой воды, и затем осторожно перемешивали, чтобы избежать осаждения кристаллов оксида титана, при этом получали бесцветный и прозрачный коллоидный раствор, названный золем титана. Затем в титановый золь добавляли 2,10 кг вспученного перлита (от компании Global Mineral, 2,06 кг в пересчете на сухое вещество) и далее перемешивали до гомогенного состояния, для получения суспензии А.
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.), 0,78 кг молекулярного сита USY (от Qilu Catalyst Division, Sinopec, 0,70 кг в пересчете на сухое вещество) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера I-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента I-А4. Состав адсорбента I-A4: содержание оксида цинка 44,3% масс., содержание вспученного перлита 20,6% масс., содержание молекулярного сита USY 7,0% масс., содержание диоксида титана 10,0% масс., содержание никеля 18,1% масс.
Сравнительный пример I-1
Адсорбент получали следующим образом: 3,31 кг тетрахлорида титана (от Beijing Chemical Plant, AR, 99% масс.) медленно добавляли к 5,0 кг кислой воды, и затем осторожно перемешивали, чтобы избежать осаждения кристаллов оксида титана, при этом получали бесцветный и прозрачный коллоидный раствор, названный золем титана. Затем в золь титана добавляли 2,45 кг вспученного перлита (от компании Global Mineral, 2,40 кг в пересчете на сухое вещество) и далее перемешивали до гомогенного состояния, для получения суспензии А.
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера I-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента I-B1. Состав адсорбента I-B1: содержание оксида цинка 44,3% масс., содержание вспученного перлита 24,0% масс., содержание диоксида титана 13,6% масс., содержание никеля 18,1% масс.
Сравнительный пример I-2
1,26 кг диоксида титана (анатазной формы, содержащего 1,17 кг диоксида титана в пересчете на сухое вещество) добавляли к 2,6 кг 10% соляной кислоты (СР, от Beijing Chemical Plant), после этого перемешивали и подкисляли в течение 1 ч, при этом оксид титана полностью растворялся в бесцветный и прозрачный коллоидный раствор, названный золем титана. Затем в золь титана добавляли 1,85 кг диатомита (от компании Global Mineral, 1,80 кг в пересчете на сухое вещество) и далее смешивали при перемешивании для получения суспензии А.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%) и 5,0 кг деионизированной воды, и перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера I-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента I-B2. Состав адсорбента I-B2: содержание оксида цинка 55,2% масс., содержание диоксида титана 11,7% масс., содержание диатомита 18,0% масс., содержание никеля 15,1% масс.
Сравнительный пример I-3
Адсорбент получали следующим образом: 3,87 кг этилтитаната (от компании Aldrich, AR, 99%) при перемешивании медленно добавляли к 3,2 кг 10% азотной кислоты (CP, от Beijing Chemical Plant), после этого перемешивали в течение 1 ч, при этом получали светло-желтый и прозрачный коллоидный раствор, названный золем титана.
Смешивали 4,93 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 2,15 кг диатомита (от компании Global Mineral, 2,10 кг в пересчете на сухое вещество) и 6,80 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии. Суспензию добавляли в золь титана и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера I-1, суспензию носителя высушивали распылением и формовали, и далее вводили в нее активные компоненты никеля и кобальта для получения адсорбента I-B3. Состав адсорбента I-B3: содержание оксида цинка 49,3% масс., содержание диоксида титана 13,5% масс., содержание диатомита 21,0% масс., содержание никеля 8,1% масс., содержание кобальта 8,1% масс.
Сравнительный пример I-4
Адсорбент получали следующим образом: 3,36 кг этилтитаната (от компании Aldrich, AR, 99%) при перемешивании медленно добавляли к 3,2 кг 10% азотной кислоты (CP, от Beijing Chemical Plant), после этого перемешивали в течение 1 ч, при этом получали светло-желтый и прозрачный коллоидный раствор, названный золем титана.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, с чистотой 99,7%), 2,44 кг каолина (от Suzhou Kaolin Factory, 1,80 кг в пересчете на сухое вещество) и 7,00 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии. Суспензию добавляли в золь титана и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера I-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента I-B4. Состав адсорбента I-B4: содержание оксида цинка 55,2% масс., содержание оксида титана 11,7% масс., содержание каолина 18,0% масс., содержание никеля 15,1% масс.
Пример I-6
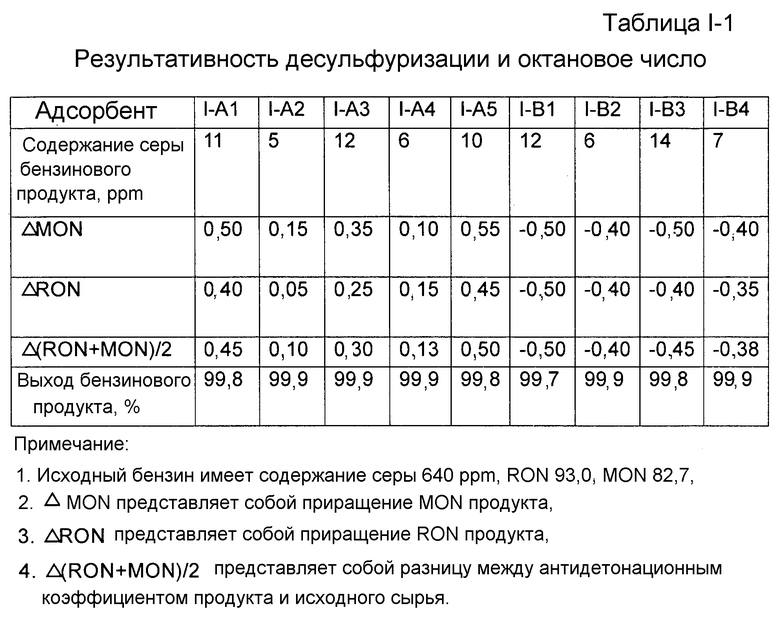
Каждый адсорбент, полученный как описано выше, исследовали на результативность десульфуризации и октановое число. Результативность десульфуризации характеризовали по содержанию серы продукта десульфуризации. Содержание серы продукта десульфуризации определяли с помощью автономно работающего хроматографа после микрореактора с неподвижным слоем. Исходным маслом для испытания реакции десульфуризации был бензин каталитического крекинга, имеющий содержание серы 640 ppm (частей на миллион). Условиями испытания реакции десульфуризации были: водородная атмосфера, температура реакции 410°С, давление реакции 1,38 МПа, молярное отношение водорода к углеводородному маслу 0,35, WHSV 4 ч-1. Для точного отражения активности адсорбента, применяемого в промышленных масштабах, после завершения реакции адсорбент регенерировали при 550°C в воздушной атмосфере. Испытания повторяли в течение шести циклов реакции и регенерации, и после этого адсорбент проявлял практически стабильную активность. Результативность десульфуризации адсорбента здесь характеризуется содержанием серы бензинового продукта, измеренным с данным адсорбентом, проявляющим практически стабильную активность, что представлено в таблице I-1. Бензиновый продукт взвешивали для расчета выхода.
В соответствии с китайскими национальными стандартами GB/T 503-1995 и GB/T 5487-1995 определяли соответственно октановое число бензина по моторному методу (MON) и по исследовательскому методу (RON) до и после испытания реакции десульфуризации. Результаты приведены в таблице I-1. Как можно видеть из таблице I-1, при использовании адсорбента, содержащего молекулярное сито со структурой A-FAU, октановое число бензинового продукта может быть повышено в каждом случае.
Таблица I-1. Результативность десульфуризации и октановое число
Пример II-1
2,33 кг кристаллического тетрахлорида олова (SnCl4 .5H2O, от компании Alfa Aesar, имеющего чистоту 99% масс.) медленно добавляли к 3,2 кг кислой воды, и затем осторожно перемешивали, чтобы избежать осаждения кристаллов оксида олова, для получения бесцветного и прозрачного золя олова. Затем в золь олова добавляли 2,10 кг вспученного перлита (от компании Global Mineral, 2,06 кг в пересчете на сухое вещество) и далее перемешивали до гомогенного состояния для получения суспензии А.
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.), 0,84 кг бета-молекулярного сита (от Nanjing Catalyst Division, Sinopec, 0,70 кг в пересчете на сухое вещество) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Суспензию носителя подвергали распылительной сушке с помощью распылительной сушилки модели Bowen Nozzle Tower™, при давлении распылительной сушки от 8,5 до 9,5 МПа, температуре на входе 500°С или менее, температуре на выходе примерно 150°С. Микросферы, полученные при распылительной сушке, сначала высушивали при 180°С в течение 1 ч, затем прокаливали при 635°С в течение 1 ч для получения носителя адсорбента.
3,2 кг носителя адсорбента пропитывали в растворе 3,51 кг гексагидрата нитрата никеля (от Beijing Chemical Reagent Company, имеющего чистоту более 98,5% масс.) в 0,6 кг деионизированной воды, полученную смесь высушивали при 180°C в течение 4 ч, и затем прокаливали в воздушной атмосфере при 635°С в течение 1 ч для получения предшественника адсорбента.
Предшественник адсорбента восстанавливали при 425°C в водородной атмосфере в течение 2 ч для получения адсорбента. Адсорбент назвали адсорбентом II-A1. Состав адсорбента II-A1: содержание оксида цинка 44,3% масс., содержание вспученного перлита 20,6% масс., содержание бета-молекулярного сита 7,0% масс., содержание диоксида олова 10,0% масс., содержание никеля 18,1% масс.
Пример II-2
1,26 кг оксида дибутилолова (от компании Aldrich, AR, 99% масс.) добавляли в 2,6 кг 10% соляной кислоты (СР, от Beijing Chemical Plant) и после этого нагревали до 80°C и выдерживали в течение 1 ч, при этом оксид олова полностью растворялся в бесцветный и прозрачный коллоидный раствор, названный золем олова. Затем в золь олова добавляли 1,54 кг диатомита (от компании Global Mineral, 1,50 кг в пересчете на сухое вещество) и далее смешивали при перемешивании для получения суспензии А.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 0,36 кг бета-молекулярного сита (от Nanjing Catalyst Division, Sinopec, 0,30 кг в пересчете на сухое вещество) и 5,0 кг деионизированной воды, и перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера II-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента II-А2. Состав адсорбента II-A2: содержание оксида цинка 55,2% масс., содержание диоксида олова 11,7% масс., содержание диатомита 15,0% масс., содержание бета-молекулярного сита 3,0% масс., содержание никеля 15,1% масс.
Пример II-3
3,19 кг ацетата олова (от компании Aldrich, AR, 99%) при перемешивании медленно добавляли к 3,5 кг 5% соляной кислоты (CP, от Beijing Chemical Plant), после этого перемешивали в течение 1 ч, при этом получали раствор в виде белого золя.
Смешивали 4,93 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 1,64 кг диатомита (от компании Global Mineral, 1,60 кг в пересчете на сухое вещество), 0,56 кг молекулярного сита USY (от Qilu Catalyst Division, Sinopec, 0,50 кг в пересчете на сухое вещество) и 6,40 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к белому золю и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера II-1, суспензию носителя высушивали распылением и формовали, и далее вводили в нее активные компоненты никеля и кобальта для получения адсорбента II-A3. Состав адсорбента II-A3: содержание оксида цинка 49,3% масс., содержание диоксида олова 13,5% масс., содержание диатомита 16,0% масс., содержание USY 5,0% масс., содержание никеля 8,1% масс., содержание кобальта 8,1% масс.
Пример II-4
2,64 кг ацетата олова (от компании Aldrich, AR, 99%) при перемешивании медленно добавляли к 3,5 кг 5% соляной кислоты (CP, от Beijing Chemical Plant), после этого перемешивали в течение 1 ч, при этом получали раствор в виде белого золя.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 2,03 кг каолина (от Suzhou Kaolin Factory, 1,50 кг в пересчете на сухое вещество), 0,36 кг молекулярного сита Х (от Qilu Catalyst Division, Sinopec, 0,30 кг в пересчете на сухое вещество) и 6,40 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии В. Суспензию В добавляли к белому золю и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера II-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента II-А4. Состав адсорбента II-A4: содержание оксида цинка 55,2% масс., содержание диоксида олова 11,7% масс., содержание каолина 15,0% масс., содержание молекулярного сита Х 3,0% масс., содержание никеля 15,1% масс.
Пример II-5
2,33 кг кристаллического тетрахлорида олова (SnCl4 .5H2O, от компании Alfa Aesar, имеющего чистоту 99% масс.) медленно добавляли к 3,2 кг кислой воды, и затем осторожно перемешивали, чтобы избежать осаждения кристаллов оксида олова, для получения бесцветного и прозрачного золя олова. Затем в золь олова добавляли 2,10 кг вспученного перлита (от компании Global Mineral, 2,06 кг в пересчете на сухое вещество) и далее перемешивали до гомогенного состояния для получения суспензии А.
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.), 0,78 кг молекулярного сита USY (от Qilu Catalyst Division, Sinopec, 0,70 кг в пересчете на сухое вещество) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера II-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента II-А5. Состав адсорбента II-A5: содержание оксида цинка 44,3% масс., содержание вспученного перлита 20,6% масс., содержание молекулярного сита USY 7,0% масс., содержание диоксида олова 10,0% масс., содержание никеля 18,1% масс.
Сравнительный пример II-1
3,17 кг кристаллического тетрахлорида олова (SnCl4 .5H2O, от компании Alfa Aesar, имеющего чистоту 99% масс.) медленно добавляли к 4,2 кг кислой воды, и затем осторожно перемешивали, чтобы избежать осаждения кристаллов оксида олова, для получения бесцветного и прозрачного золя олова. Затем в золь олова добавляли 2,45 кг вспученного перлита (от компании Global Mineral, 2,40 кг в пересчете на сухое вещество) и далее перемешивали до гомогенного состояния для получения суспензии А.
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера II-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента II-B1. Состав адсорбента II-B1: содержание оксида цинка 44,3% масс., содержание вспученного перлита 24,0% масс., содержание диоксида олова 13,6% масс., содержание никеля 18,1% масс.
Сравнительный пример II-2
1,26 кг оксида дибутилолова (от компании Aldrich, AR, 99% масс.) добавляли в 2,6 кг 10% соляной кислоты (СР, от Beijing Chemical Plant) и после этого нагревали до 80°C и выдерживали в течение 1 ч, при этом оксид олова полностью растворялся в бесцветный и прозрачный коллоидный раствор, названный золем олова. Затем в золь олова добавляли 1,85 кг диатомита (от компании Global Mineral, 1,80 кг в пересчете на сухое вещество) и далее смешивали при перемешивании для получения суспензии А.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%) и 5,0 кг деионизированной воды, и перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера II-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента II-B2. Состав адсорбента II-B2: содержание оксида цинка 55,2% масс., содержание диоксида олова 11,7% масс., содержание диатомита 18,0% масс., содержание никеля 15,1% масс.
Сравнительный пример II-3
3,19 кг ацетата олова (от компании Aldrich, AR, 99%) при перемешивании медленно добавляли к 3,5 кг 5% соляной кислоты (CP, от Beijing Chemical Plant), после этого перемешивали в течение 1 ч, при этом получали раствор в виде белого золя.
Смешивали 4,93 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 2,15 кг диатомита (от компании Global Mineral, 2,10 кг в пересчете на сухое вещество) и 6,80 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к белому золю и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера II-1, суспензию носителя высушивали распылением и формовали, и далее вводили в нее активные компоненты никеля и кобальта для получения адсорбента II-B3. Состав адсорбента II-B3: содержание оксида цинка 49,3% масс., содержание диоксида олова 13,5% масс., содержание диатомита 21,0% масс., содержание никеля 8,1% масс., содержание кобальта 8,1% масс.
Сравнительный пример II-4
2,64 кг ацетата олова (от компании Aldrich, AR, 99%) при перемешивании медленно добавляли к 3,5 кг 5% соляной кислоты (CP, от Beijing Chemical Plant), после этого перемешивали в течение 1 ч, при этом получали раствор в виде белого золя.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 2,44 кг каолина (от Suzhou Kaolin Factory, 1,80 кг в пересчете на сухое вещество) и 6,80 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии В. Суспензию В добавляли к белому золю и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера II-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента II-B4. Состав адсорбента II-B4: содержание оксида цинка 55,2% масс., содержание диоксида олова 11,7% масс., содержание каолина 18,0% масс., содержание никеля 15,1% масс.
Пример II-6
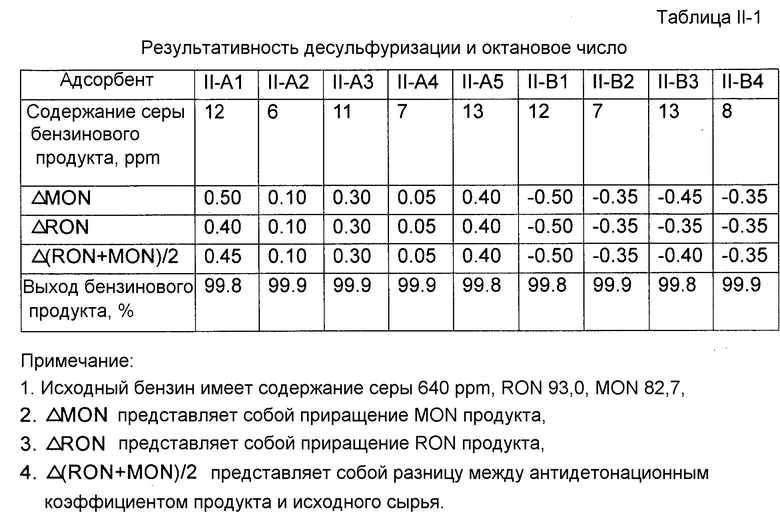
Каждый адсорбент, полученный как описано выше, исследовали на результативность десульфуризации и октановое число. Результативность десульфуризации характеризовали по содержанию серы продукта десульфуризации. Содержание серы продукта десульфуризации определяли с помощью автономно работающего хроматографа после микрореактора с неподвижным слоем. Исходным маслом для испытания реакции десульфуризации был бензин каталитического крекинга, имеющий содержание серы 640 ppm. Условиями испытания реакции десульфуризации были: водородная атмосфера, температура реакции 410°С, давление реакции 1,38 МПа, молярное отношение водорода к углеводородному маслу 0,35, WHSV 4 ч-1. Для точного отражения активности адсорбента, применяемого в промышленных масштабах, после завершения реакции адсорбент регенерировали при 550°C в воздушной атмосфере. Испытания повторяли в течение шести циклов реакции и регенерации, и после этого адсорбент проявлял практически стабильную активность. Результативность десульфуризации адсорбента здесь характеризуется содержанием серы бензинового продукта, измеренным с данным адсорбентом, проявляющим практически стабильную активность, что представлено в табл.II-1. Бензиновый продукт взвешивали для расчета выхода.
В соответствии с китайскими национальными стандартами GB/T 503-1995 и GB/T 5487-1995 определяли соответственно октановое число бензина по моторному методу (MON) и по исследовательскому методу (RON) до и после испытания реакции десульфуризации. Результаты приводятся в табл.II-1. Как можно видеть из табл.II-1, при использовании адсорбента, содержащего молекулярное сито со структурой A-FAU, октановое число бензинового продукта может быть повышено в каждом случае.
Таблица II-1. Результативность десульфуризации и октановое число
Пример III-1
Адсорбент получали следующим образом: 1,90 кг тетрахлорида циркония (от Beijing Chemical Plant, AR, 99% масс.) медленно добавляли к 2,7 кг 5% масс. раствора азотной кислоты, и затем осторожно перемешивали, чтобы избежать осаждения кристаллов диоксида циркония, для получения бесцветного и прозрачного коллоидного раствора, названного золем циркония. Затем в золь циркония добавляли 2,10 кг вспученного перлита (от компании Global Mineral, 2,06 кг в пересчете на сухое вещество) и далее перемешивали до гомогенного состояния для получения суспензии А.
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.), 0,84 кг бета-молекулярного сита (от Nanjing Catalyst Division, Sinopec, 0,70 кг в пересчете на сухое вещество) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Суспензию носителя подвергали распылительной сушке с помощью распылительной сушилки модели Bowen Nozzle Tower™, при давлении распылительной сушки от 8,5 до 9,5 МПа, температуре на входе 500°С или менее, температуре на выходе примерно 150°С. Микросферы, полученные при распылительной сушке, сначала высушивали при 180°С в течение 1 ч, затем прокаливали при 635°С в течение 1 ч для получения носителя адсорбента.
3,2 кг носителя адсорбента пропитывали в растворе 3,51 кг гексагидрата нитрата никеля (от Beijing Chemical Reagent Company, имеющего чистоту более 98,5% масс.) в 0,6 кг деионизированной воды, полученную смесь высушивали при 180°C в течение 4 ч, и затем прокаливали в воздушной атмосфере при 635°С в течение 1 ч для получения предшественника адсорбента.
Предшественник адсорбента восстанавливали при 425°C в водородной атмосфере в течение 2 ч для получения адсорбента. Адсорбент назвали адсорбентом III-A1. Состав адсорбента III-A1: содержание оксида цинка 44,3% масс., содержание вспученного перлита 20,6% масс., содержание бета-молекулярного сита 7,0% масс., содержание связующего диоксида циркония 10,0% масс., содержание никеля 18,1% масс.
Пример III-2
1,71 кг оксихлорида циркония (от компании Aldrich, AR, 98,5% масс.) при перемешивании медленно добавляли к 3,2 кг 15% масс. соляной кислоты (CP, от Beijing Chemical Plant) и после этого перемешивали и подкисляли в течение 1 ч, при этом получали бесцветный и прозрачный коллоидный раствор, названный золем циркония. Затем в золь циркония добавляли 1,54 кг диатомита (от компании Global Mineral, 1,50 кг в пересчете на сухое вещество) и далее смешивали при перемешивании для получения суспензии А.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 0,36 кг бета-молекулярного сита (от Nanjing Catalyst Division, Sinopec, 0,30 кг в пересчете на сухое вещество) и 5,0 кг деионизированной воды, и перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера III-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента III-A2. Состав адсорбента III-A2: содержание оксида цинка 55,2% масс., содержание связующего диоксида циркония 11,7% масс., содержание диатомита 15,0% масс., содержание бета-молекулярного сита 3,0% масс., содержание никеля 15,1% масс.
Пример III-3
Адсорбент получали следующим образом: 1,76 кг гидроксида циркония (от компании Aldrich, AR, 99%) добавляли к 3,1 кг 30% масс. соляной кислоты (CP, от Beijing Chemical Plant) и после этого перемешивали и подкисляли в течение 1 ч для получения прозрачного коллоидного раствора, названного золем циркония.
Смешивали 4,93 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 1,64 кг диатомита (от компании Global Mineral, 1,60 кг в пересчете на сухое вещество), 0,56 кг молекулярного сита USY (от Qilu Catalyst Division, Sinopec, 0,50 кг в пересчете на сухое вещество) и 6,40 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к золю циркония и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера III-1, суспензию носителя высушивали распылением и формовали, и далее вводили в нее активные компоненты никеля и кобальта для получения адсорбента III-A3. Состав адсорбента III-A3: содержание оксида цинка 49,3% масс., содержание связующего диоксида циркония 13,5% масс., содержание диатомита 16,0% масс., содержание USY 5,0% масс., содержание никеля 8,1% масс., содержание кобальта 8,1% масс.
Пример III-4
Адсорбент получали следующим образом: 1,53 кг гидроксида циркония (от компании Aldrich, AR, 99%) добавляли к 3,1 кг 30% масс. соляной кислоты (CP, от Beijing Chemical Plant) и после этого перемешивали и подкисляли в течение 1 ч для получения прозрачного коллоидного раствора, названного золем циркония.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 2,03 кг каолина (от Suzhou Kaolin Factory, 1,50 кг в пересчете на сухое вещество), 0,36 кг молекулярного сита Х (от Qilu Catalyst Division, Sinopec, 0,30 кг в пересчете на сухое вещество) и 7,40 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии В. Суспензию В добавляли к золю циркония и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера III-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента III-A4. Состав адсорбента III-A4: содержание оксида цинка 55,2% масс., содержание связующего диоксида циркония 11,7% масс., содержание каолина 15,0% масс., содержание молекулярного сита Х 3,0% масс., содержание никеля 15,1% масс.
Пример III-5
Адсорбент получали следующим образом: 1,90 кг тетрахлорида циркония (от Beijing Chemical Plant, AR, 99% масс.) медленно добавляли к 2,7 кг 5% масс. раствора азотной кислоты, и затем осторожно перемешивали, чтобы избежать осаждения кристаллов диоксида циркония, для получения бесцветного и прозрачного коллоидного раствора, названного золем циркония. Затем в золь циркония добавляли 2,10 кг вспученного перлита (от компании Global Mineral, 2,06 кг в пересчете на сухое вещество) и далее перемешивали до гомогенного состояния для получения суспензии А.
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.), 0,78 кг молекулярного сита USY (от Qilu Catalyst Division, Sinopec, 0,70 кг в пересчете на сухое вещество) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера III-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента III-A5. Состав адсорбента III-A5: содержание оксида цинка 44,3% масс., содержание вспученного перлита 20,6% масс., содержание молекулярного сита USY 7,0% масс., содержание связующего диоксида циркония 10,0% масс., содержание никеля 18,1% масс.
Сравнительный пример III-1
Адсорбент получали следующим образом: 2,58 кг тетрахлорида циркония (от Beijing Chemical Plant, AR, 99% масс.) медленно добавляли к 4,2 кг 5% масс. раствора азотной кислоты, и затем осторожно перемешивали, чтобы избежать осаждения кристаллов диоксида циркония, для получения бесцветного и прозрачного коллоидного раствора, названного золем циркония. Затем в золь циркония добавляли 2,45 кг вспученного перлита (от компании Global Mineral, 2,40 кг в пересчете на сухое вещество) и далее перемешивали до гомогенного состояния для получения суспензии А.
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера III-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента III-B1. Состав адсорбента III-B1: содержание оксида цинка 44,3% масс., содержание вспученного перлита 24,0% масс., содержание связующего диоксида циркония 13,6% масс., содержание никеля 18,1% масс.
Сравнительный пример III-2
1,71 кг оксихлорида циркония (от компании Aldrich, AR, 98,5% масс.) при перемешивании медленно добавляли к 3,2 кг 15% масс. соляной кислоты (CP, от Beijing Chemical Plant) и после этого перемешивали и подкисляли в течение 1 ч, при этом получали бесцветный и прозрачный коллоидный раствор, названный золем циркония. Затем в золь циркония добавляли 1,85 кг диатомита (от компании Global Mineral, 1,80 кг в пересчете на сухое вещество) и далее смешивали при перемешивании для получения суспензии А.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%) и 5,0 кг деионизированной воды, и перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера III-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента III-B2. Состав адсорбента III-B2: содержание оксида цинка 55,2% масс., содержание связующего диоксида циркония 11,7% масс., содержание диатомита 18,0% масс., содержание никеля 15,1% масс.
Сравнительный пример III-3
Адсорбент получали следующим образом: 1,76 кг гидроксида циркония (от компании Aldrich, AR, 99%) добавляли к 3,1 кг 30% масс. соляной кислоты (CP, от Beijing Chemical Plant) и после этого перемешивали и подкисляли в течение 1 ч для получения прозрачного коллоидного раствора, названного золем циркония.
Смешивали 4,93 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 2,15 кг диатомита (от компании Global Mineral, 2,10 кг в пересчете на сухое вещество) и 6,90 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к золю циркония и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера III-1, суспензию носителя высушивали распылением и формовали, и далее вводили в нее активные компоненты никеля и кобальта для получения адсорбента III-B3. Состав адсорбента III-B3: содержание оксида цинка 49,3% масс., содержание связующего диоксида циркония 13,5% масс., содержание диатомита 21,0% масс., содержание никеля 8,1% масс., содержание кобальта 8,1% масс.
Сравнительный пример III-4
Адсорбент получали следующим образом: 1,53 кг гидроксида циркония (от компании Aldrich, AR, 99%) добавляли к 3,1 кг 30% масс. соляной кислоты (CP, от Beijing Chemical Plant) и после этого перемешивали и подкисляли в течение 1 ч для получения прозрачного коллоидного раствора, названного золем циркония.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 2,44 кг каолина (от Suzhou Kaolin Factory, 1,80 кг в пересчете на сухое вещество) и 7,40 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии В. Суспензию В добавляли к золю циркония и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера III-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента III-B4. Состав адсорбента III-B4: содержание оксида цинка 55,2% масс., содержание связующего диоксида циркония 11,7% масс., содержание каолина 18,0% масс., содержание никеля 15,1% масс.
Пример III-6
Каждый адсорбент, полученный как описано выше, исследовали на результативность десульфуризации и октановое число. Результативность десульфуризации характеризовали по содержанию серы продукта десульфуризации. Содержание серы продукта десульфуризации определяли с помощью автономно работающего хроматографа после микрореактора с неподвижным слоем. Исходным маслом для испытания реакции десульфуризации был бензин каталитического крекинга, имеющий содержание серы 640 ppm. Условиями испытания реакции десульфуризации были: водородная атмосфера, температура реакции 410°С, давление реакции 1,38 МПа, молярное отношение водорода к углеводородному маслу 0,35, WHSV 4 ч-1. Для точного отражения активности адсорбента, применяемого в промышленных масштабах, после завершения реакции адсорбент регенерировали при 550°C в воздушной атмосфере. Испытания повторяли в течение шести циклов реакции и регенерации, и после этого адсорбент проявлял практически стабильную активность. Результативность десульфуризации адсорбента здесь характеризуется содержанием серы бензинового продукта, измеренным с данным адсорбентом, проявляющим практически стабильную активность, что представлено в табл.III-1. Бензиновый продукт взвешивали для расчета выхода.
В соответствии с китайскими национальными стандартами GB/T 503-1995 и GB/T 5487-1995 определяли соответственно октановое число бензина по моторному методу (MON) и по исследовательскому методу (RON) до и после испытания реакции десульфуризации. Результаты приведены в табл.III-1. Как можно видеть из табл.III-1, при использовании адсорбента, содержащего молекулярное сито со структурой A-FAU, октановое число бензинового продукта может быть повышено в каждом случае.
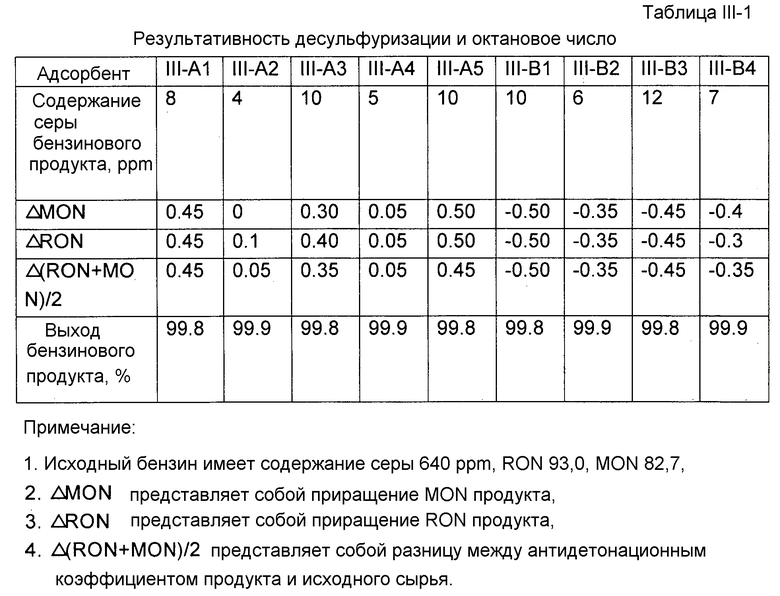
Пример IV-1
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.), 0,84 кг бета-молекулярного сита (от Nanjing Catalyst Division, Sinopec, 0,70 кг в пересчете на сухое вещество) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии A.
1,37 кг оксида алюминия (от Shandong Aluminium Plant, 1,00 кг в пересчете на сухое вещество), и 2,10 кг вспученного перлита (от компании Global Mineral, 2,06 кг в пересчете на сухое вещество) смешивали при перемешивании, и далее вводили туда 4,6 кг деионизированной воды, после этого смешивали до гомогенного состояния, вводили туда 360 мл 30% масс. соляной кислоты (CP, от Beijing Chemical Plant), после этого перемешивали и подкисляли в течение 1 ч, нагревали до 80°C и выдерживали в течение 2 ч, далее вводили туда суспензию А, и затем перемешивали в течение 1 ч для получения суспензии носителя.
Суспензию носителя подвергали распылительной сушке с помощью распылительной сушилки модели Bowen Nozzle Tower™, при давлении распылительной сушки от 8,5 до 9,5 МПа, температуре на входе 500°С или менее, температуре на выходе примерно 150°С. Микросферы, полученные при распылительной сушке, сначала высушивали при 180°С в течение 1 ч, затем прокаливали при 635°С в течение 1 ч для получения носителя адсорбента.
3,2 кг носителя адсорбента пропитывали в растворе 3,51 кг гексагидрата нитрата никеля (от Beijing Chemical Reagent Company, имеющего чистоту более 98,5% масс.) в 0,6 кг деионизированной воды, полученную смесь высушивали при 180°C в течение 4 ч, и затем прокаливали в воздушной атмосфере при 635°С в течение 1 ч для получения предшественника адсорбента.
Предшественник адсорбента восстанавливали при 425°C в водородной атмосфере в течение 2 ч для получения адсорбента. Адсорбент назвали адсорбентом IV-A1. Состав адсорбента IV-A1: содержание оксида цинка 44,3% масс., содержание вспученного перлита 20,6% масс., содержание бета-молекулярного сита 7,0% масс., содержание связующего оксида алюминия 10,0% масс., содержание никеля 18,1% масс.
Сравнительный пример IV-1
Адсорбент получали следующим образом: смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии оксида цинка.
1,87 кг оксида алюминия (от Shandong Aluminium Plant, 1,36 кг в пересчете на сухое вещество), и 2,46 кг вспученного перлита (от компании Global Mineral, 2,40 кг в пересчете на сухое вещество) смешивали при перемешивании, и далее вводили туда 4,6 кг деионизированной воды, после этого смешивали до гомогенного состояния, вводили туда 360 мл 30% соляной кислоты (CP, от Beijing Chemical Plant), после этого подкисляли при перемешивании в течение 1 ч, нагревали до 80°C и выдерживали в течение 2 ч, далее вводили туда суспензию оксида цинка, и затем перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера IV-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента IV-B1. Состав адсорбента IV-B1: содержание оксида цинка 44,3% масс., содержание связующего оксида алюминия 13,6% масс., содержание вспученного перлита 24,0% масс., содержание никеля 18,1% масс.
Пример IV-2
1,61 кг псевдобемита (от Shandong Aluminium Plant, 1,17 кг в пересчете на сухое вещество), и 1,54 кг диатомита (от компании Global Mineral, 1,50 кг в пересчете на сухое вещество) смешивали при перемешивании, и далее вводили туда 8,2 кг деионизированной воды, после этого смешивали до гомогенного состояния, вводили туда 260 мл 30% соляной кислоты (CP, от Beijing Chemical Plant), после этого подкисляли при перемешивании в течение 1 ч, нагревали до 80°C и выдерживали в течение 2 ч, после этого охлаждали, далее вводили туда 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%) и 0,36 кг бета-молекулярного сита (от Nanjing Catalyst Division, Sinopec, 0,30 кг в пересчете на сухое вещество), после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера IV-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента IV-A2. Состав адсорбента IV-A2: содержание оксида цинка 55,2% масс., содержание связующего оксида алюминия 11,7% масс., содержание диатомита 15,0% масс., содержание бета-молекулярного сита 3,0% масс., содержание никеля 15,1% масс.
Сравнительный пример IV-2
1,61 кг псевдобемита (от Shandong Aluminium Plant, 1,17 кг в пересчете на сухое вещество) и 1,85 кг диатомита (от компании Global Mineral, 1,80 кг в пересчете на сухое вещество) смешивали при перемешивании, и далее вводили туда 8,2 кг деионизированной воды, после этого смешивали до гомогенного состояния, вводили туда 260 мл 30% соляной кислоты (CP, от Beijing Chemical Plant), после этого подкисляли при перемешивании в течение 1 ч, нагревали до 80°C и выдерживали в течение 2 ч, после этого охлаждали, далее вводили туда 5,52 кг порошка оксида цинка (от компании Headhorse, с чистотой 99,7%) и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера IV-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента IV-B2. Состав адсорбента IV-B2: содержание оксида цинка 55,2% масс., содержание связующего оксида алюминия 11,7% масс., содержание диатомита 18,0% масс., содержание никеля 15,1% масс.
Пример IV-3
Адсорбент получали следующим образом: смешивали 4,93 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 0,56 кг молекулярного сита USY (от Qilu Catalyst Division, Sinopec, 0,50 кг в пересчете на сухое вещество) и 5,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии A.
1,85 кг псевдобемита (от Shandong Aluminium Plant, 1,35 кг в пересчете на сухое вещество) и 1,64 кг диатомита (от компании Global Mineral, 1,60 кг в пересчете на сухое вещество) смешивали при перемешивании, и далее вводили туда 4,6 кг деионизированной воды, после этого смешивали до гомогенного состояния, вводили туда 300 мл 30% соляной кислоты (CP, от Beijing Chemical Plant) для получения суспензии с pH 2,5, после этого подкисляли при перемешивании в течение 1 ч, нагревали до 80°C и выдерживали в течение 2 ч, далее вводили туда суспензию А, и затем перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера IV-1, суспензию носителя высушивали распылением и формовали, и далее вводили в нее активные компоненты никеля и кобальта для получения адсорбента IV-A3. Состав адсорбента IV-A3: содержание оксида цинка 49,3% масс., содержание связующего оксида алюминия 13,5% масс., содержание диатомита 16,0% масс., содержание USY 5,0% масс., содержание никеля 8,1% масс., содержание кобальта 8,1% масс.
Сравнительный пример IV-3
Адсорбент получали следующим образом: смешивали 4,93 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%) и 5,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии оксида цинка.
1,85 кг псевдобемита (от Shandong Aluminium Plant, 1,35 кг в пересчете на сухое вещество) и 2,16 кг диатомита (от компании Global Mineral, 2,10 кг в пересчете на сухое вещество) смешивали при перемешивании, и далее вводили туда 4,6 кг деионизированной воды, после этого смешивали до гомогенного состояния, вводили туда 300 мл 30% соляной кислоты (CP, от Beijing Chemical Plant) для получения суспензии с pH 2,5, после этого подкисляли при перемешивании в течение 1 ч, нагревали до 80°C и выдерживали в течение 2 ч, далее вводили туда суспензию оксида цинка, и затем перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера IV-1, суспензию носителя высушивали распылением и формовали, и далее вводили в нее активные компоненты никеля и кобальта для получения адсорбента IV-B3. Состав адсорбента IV-B3: содержание оксида цинка 49,3% масс., содержание связующего оксида алюминия 13,5% масс., содержание диатомита 21,0% масс., содержание никеля 8,1% масс., содержание кобальта 8,1% масс.
Пример IV-4
Адсорбент получали следующим образом: смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 0,36 кг молекулярного сита USY (от Qilu Catalyst Division, Sinopec, 0,30 кг в пересчете на сухое вещество) и 5,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии A.
1,61 кг псевдобемита (от Shandong Aluminium Plant, 1,17 кг в пересчете на сухое вещество) и 2,03 кг каолина (от Suzhou Kaolin Factory, 1,50 кг в пересчете на сухое вещество) смешивали при перемешивании, и далее вводили туда 4,6 кг деионизированной воды, после этого смешивали до гомогенного состояния, вводили туда 300 мл 30% соляной кислоты (CP, от Beijing Chemical Plant) для получения суспензии с pH 2,5, после этого подкисляли при перемешивании в течение 1 ч, нагревали до 80°C и выдерживали в течение 2 ч, далее вводили туда суспензию А, и затем перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера IV-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента IV-A4. Состав адсорбента IV-A4: содержание оксида цинка 55,2% масс., содержание связующего оксида алюминия 11,7% масс., содержание каолина 15,0% масс., содержание молекулярного сита Х 3,0% масс., содержание никеля 15,1% масс.
Сравнительный пример IV-4
Адсорбент получали следующим образом: смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%) и 5,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии оксида цинка.
1,61 кг псевдобемита (от Shandong Aluminium Plant, 1,17 кг в пересчете на сухое вещество) и 2,44 кг каолина (от Suzhou Kaolin Factory, 1,80 кг в пересчете на сухое вещество) смешивали при перемешивании, и далее вводили туда 3,6 кг деионизированной воды, после этого смешивали до гомогенного состояния, вводили туда 300 мл 30% соляной кислоты (CP, от Beijing Chemical Plant) для получения суспензии с pH 2,5, после этого подкисляли при перемешивании в течение 1 ч, нагревали до 80°C и выдерживали в течение 2 ч, далее вводили туда суспензию оксида цинка, и затем перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера IV-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента IV-B4. Состав адсорбента IV-В4: содержание оксида цинка 55,2% масс., содержание связующего оксида алюминия 11,7% масс., содержание каолина 18,0% масс., содержание никеля 15,1% масс.
Пример IV-5
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.), 0,78 кг молекулярного сита USY (от Qilu Catalyst Division, Sinopec, 0,70 кг в пересчете на сухое вещество) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии А.
1,37 кг оксида алюминия (от Shandong Aluminium Plant, 1,00 кг в пересчете на сухое вещество), и 2,10 кг вспученного перлита (от компании Global Mineral, 2,06 кг в пересчете на сухое вещество) смешивали при перемешивании, и далее вводили туда 4,6 кг деионизированной воды, после этого смешивали до гомогенного состояния, вводили туда 360 мл 30% масс. соляной кислоты (CP, от Beijing Chemical Plant), после этого подкисляли при перемешивании в течение 1 ч, нагревали до 80°C и выдерживали в течение 2 ч, далее вводили туда суспензию А, и затем перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера IV-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента IV-A5. Состав адсорбента IV-A5: содержание оксида цинка 44,3% масс., содержание вспученного перлита 20,6% масс., содержание молекулярного сита USY 7,0% масс., содержание связующего оксида алюминия 10,0% масс., содержание никеля 18,1% масс.
Пример IV-6
Каждый адсорбент, полученный как описано выше, исследовали на результативность десульфуризации и октановое число. Результативность десульфуризации характеризовали по содержанию серы продукта десульфуризации. Содержание серы продукта десульфуризации определяли с помощью автономно работающего хроматографа после микрореактора с неподвижным слоем. Исходным маслом для испытания реакции десульфуризации был бензин каталитического крекинга, имеющий содержание серы 640 ppm (частей на миллион). Условиями испытания реакции десульфуризации были: водородная атмосфера, температура реакции 410°С, давление реакции 1,38 МПа, молярное отношение водорода к углеводородному маслу 0,35, WHSV 4 ч-1. Для точного отражения активности адсорбента, применяемого в промышленных масштабах, после завершения реакции адсорбент регенерировали при 550°C в воздушной атмосфере. Испытания повторяли в течение шести циклов реакции и регенерации, и после этого адсорбент проявлял практически стабильную активность. Результативность десульфуризации адсорбента здесь характеризуется содержанием серы бензинового продукта, измеренным с данным адсорбентом, проявляющим практически стабильную активность, что представлено в табл.IV-1. Бензиновый продукт взвешивали для расчета выхода.
В соответствии с китайскими национальными стандартами GB/T 503-1995 и GB/T 5487-1995 определяли соответственно октановое число бензина по моторному методу (MON) и по исследовательскому методу (RON) до и после испытания реакции десульфуризации. Результаты приведены в табл.IV-1. Как можно видеть из табл.IV-1, при использовании адсорбента, содержащего молекулярное сито со структурой A-FAU, октановое число бензинового продукта может
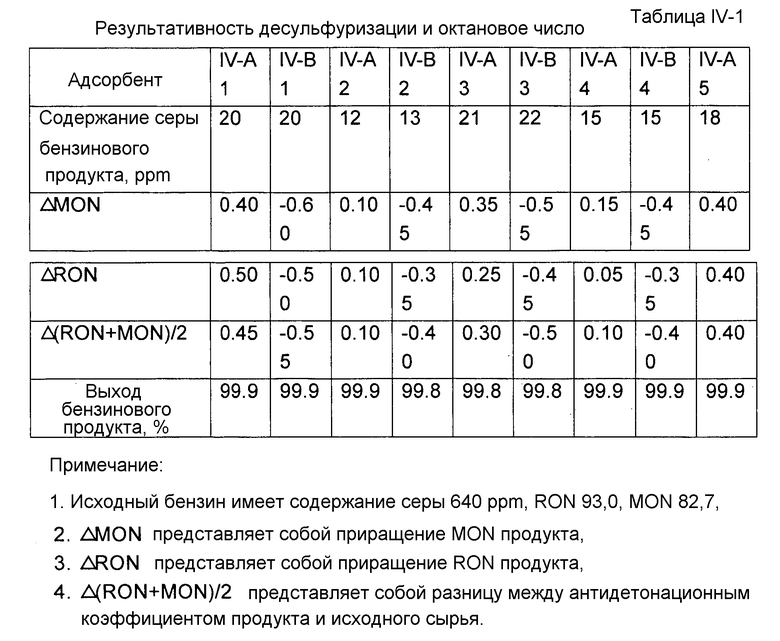
Пример V-1
Адсорбент получали следующим образом: 1,90 кг тетрахлорида циркония (от Beijing Chemical Plant, AR, 99% масс.) медленно добавляли к 2,7 кг 5% масс. раствора азотной кислоты, и затем осторожно перемешивали, чтобы избежать осаждения кристаллов диоксида циркония, для получения бесцветного и прозрачного коллоидного раствора, названного золем циркония. Затем в золь циркония добавляли 2,10 кг вспученного перлита (от компании Global Mineral, 2,06 кг в пересчете на сухое вещество) и далее перемешивали до гомогенного состояния для получения суспензии А.
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.), 0,78 кг молекулярного сита USY (от Qilu Catalyst Division, Sinopec, 0,70 кг в пересчете на сухое вещество) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Суспензию носителя подвергали распылительной сушке с помощью распылительной сушилки модели Bowen Nozzle Tower™, при давлении распылительной сушки от 8,5 до 9,5 МПа, температуре на входе 500°С или менее, температуре на выходе примерно 150°С. Микросферы, полученные при распылительной сушке, сначала высушивали при 180°С в течение 1 ч, затем прокаливали при 635°С в течение 1 ч для получения носителя адсорбента.
3,2 кг носителя адсорбента пропитывали в растворе 3,51 кг гексагидрата нитрата никеля (от Beijing Chemical Reagent Company, имеющего чистоту более 98,5% масс.) в 0,6 кг деионизированной воды, полученную смесь высушивали при 180°C в течение 4 ч, и затем прокаливали в воздушной атмосфере при 635°С в течение 1 ч для получения предшественника адсорбента.
Предшественник адсорбента восстанавливали при 425°C в водородной атмосфере в течение 2 ч для получения адсорбента. Адсорбент назвали адсорбентом V-A1. Состав адсорбента V-A1: содержание оксида цинка 44,3% масс., содержание вспученного перлита 20,6% масс., содержание молекулярного сита USY 7,0% масс., содержание связующего диоксида циркония 10,0% масс., содержание никеля 18,1% масс.
Пример V-2
1,26 кг оксида дибутилолова (от компании Aldrich, AR, 99% масс.) добавляли в 2,6 кг 10% соляной кислоты (СР, от Beijing Chemical Plant) и после этого нагревали до 80°C и выдерживали в течение 1 ч, при этом оксид олова полностью растворялся в бесцветный и прозрачный коллоидный раствор, названный золем олова. Затем в золь олова добавляли 1,54 кг диатомита (от компании Global Mineral, 1,50 кг в пересчете на сухое вещество) и далее смешивали при перемешивании для получения суспензии А.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 0,38 кг молекулярного сита NaY (от Qilu Catalyst Division, Sinopec, 0,30 кг в пересчете на сухое вещество) и 5,0 кг деионизированной воды, и перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера V-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента V-A2. Состав адсорбента V-A2: содержание оксида цинка 55,2% масс., содержание диоксида олова 11,7% масс., содержание диатомита 15,0% масс., содержание молекулярного сита NaY 3,0% масс., содержание никеля 15,1% масс.
Пример V-3
Адсорбент получали следующим образом: 3,87 кг этилтитаната (от компании Aldrich, AR, 99%) при перемешивании медленно добавляли к 3,2 кг 10% азотной кислоты (CP, от Beijing Chemical Plant), после этого перемешивали в течение 1 ч, при этом получали светло-желтый и прозрачный коллоидный раствор, названный золем титана.
Смешивали 4,93 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 1,64 кг диатомита (от компании Global Mineral, 1,60 кг в пересчете на сухое вещество), 0,59 кг молекулярного сита HY (от Qilu Catalyst Division, Sinopec, 0,50 кг в пересчете на сухое вещество) и 6,40 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии. Суспензию добавляли в золь титана и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера V-1, суспензию носителя высушивали распылением и формовали, и далее вводили в нее активные компоненты никеля и кобальта для получения адсорбента V-A3. Состав адсорбента V-A3: содержание оксида цинка 49,3% масс., содержание диоксида титана 13,5% масс., содержание диатомита 16,0% масс., содержание HY 5,0% масс., содержание никеля 8,1% масс., содержание кобальта 8,1% масс.
Пример V-4
1,61 кг псевдобемита (от Shandong Aluminium Plant, 1,17 кг в пересчете на сухое вещество), и 1,54 кг диатомита (от компании Global Mineral, 1,50 кг в пересчете на сухое вещество) смешивали при перемешивании, и далее вводили туда 8,2 кг деионизированной воды, после этого смешивали до гомогенного состояния, вводили туда 260 мл 30% соляной кислоты (CP, от Beijing Chemical Plant), после этого подкисляли при перемешивании в течение 1 ч, нагревали до 80°C и выдерживали в течение 2 ч, после этого охлаждали, далее вводили туда 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%) и 0,36 кг молекулярного сита X (от Nanjing Catalyst Division, Sinopec, 0,30 кг в пересчете на сухое вещество), после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера V-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента V-A2. Состав адсорбента V-A2: содержание оксида цинка 55,2% масс., содержание связующего оксида алюминия 11,7% масс., содержание диатомита 15,0% масс., содержание молекулярного сита X 3,0% масс., содержание никеля 15,1% масс.
Сравнительный пример V-1
Адсорбент получали следующим образом: 2,58 кг тетрахлорида циркония (от Beijing Chemical Plant, AR, 99% масс.) медленно добавляли к 4,2 кг 5% масс. раствора азотной кислоты, и затем осторожно перемешивали, чтобы избежать осаждения кристаллов диоксида циркония, для получения бесцветного и прозрачного коллоидного раствора, названного золем циркония. Затем в золь циркония добавляли 2,45 кг вспученного перлита (от компании Global Mineral, 2,40 кг в пересчете на сухое вещество) и далее перемешивали до гомогенного состояния для получения суспензии А.
Смешивали 4,43 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7% масс.) и 4,57 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера V-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента V-B1. Состав адсорбента V-B1: содержание оксида цинка 44,3% масс., содержание вспученного перлита 24,0% масс., содержание связующего диоксида циркония 13,6% масс., содержание никеля 18,1% масс.
Сравнительный пример V-2
1,26 кг оксида дибутилолова (от компании Aldrich, AR, 99% масс.) добавляли в 2,6 кг 10% соляной кислоты (СР, от Beijing Chemical Plant) и после этого нагревали до 80°C и выдерживали в течение 1 ч, при этом оксид олова полностью растворялся в бесцветный и прозрачный коллоидный раствор, названный золем олова. Затем в золь олова добавляли 1,85 кг диатомита (от компании Global Mineral, 1,80 кг в пересчете на сухое вещество) и далее смешивали при перемешивании для получения суспензии А.
Смешивали 5,52 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%) и 5,0 кг деионизированной воды, и перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к суспензии А и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера V-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента V-B2. Состав адсорбента V-B2: содержание оксида цинка 55,2% масс., содержание диоксида олова 11,7% масс., содержание диатомита 18,0% масс., содержание никеля 15,1% масс.
Сравнительный пример V-3
Адсорбент получали следующим образом: 1,76 кг гидроксида циркония (от компании Aldrich, AR, 99%) добавляли к 3,1 кг 30% масс. соляной кислоты (CP, от Beijing Chemical Plant) и после этого перемешивали и подкисляли в течение 1 ч для получения прозрачного коллоидного раствора, названного золем циркония.
Смешивали 4,93 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 2,15 кг диатомита (от компании Global Mineral, 2,10 кг в пересчете на сухое вещество) и 6,90 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии B. Суспензию В добавляли к золю циркония и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера V-1, суспензию носителя высушивали распылением и формовали, и далее вводили в нее активные компоненты никеля и кобальта для получения адсорбента V-B3. Состав адсорбента V-B3: содержание оксида цинка 49,3% масс., содержание связующего диоксида циркония 13,5% масс., содержание диатомита 21,0% масс., содержание никеля 8,1% масс., содержание кобальта 8,1% масс.
Сравнительный пример V-4
1,61 кг псевдобемита (от Shandong Aluminium Plant, 1,17 кг в пересчете на сухое вещество) и 1,85 кг диатомита (от компании Global Mineral, 1,80 кг в пересчете на сухое вещество) смешивали при перемешивании, и далее вводили туда 8,2 кг деионизированной воды, после этого смешивали до гомогенного состояния, вводили туда 260 мл 30% соляной кислоты (CP, от Beijing Chemical Plant), после этого подкисляли при перемешивании в течение 1 ч, нагревали до 80°C и выдерживали в течение 2 ч, после этого охлаждали, далее вводили туда 5,52 кг порошка оксида цинка (от компании Headhorse, с чистотой 99,7%) и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера V-1, суспензию носителя высушивали распылением и формовали, и затем вводили в нее активный компонент никеля для получения адсорбента V-B2. Состав адсорбента V-B2: содержание оксида цинка 55,2% масс., содержание связующего оксида алюминия 11,7% масс., содержание диатомита 18,0% масс., содержание никеля 15,1% масс.
Сравнительный пример V-5
Адсорбент получали следующим образом: 3,87 кг этилтитаната (от компании Aldrich, AR, 99%) при перемешивании медленно добавляли к 3,2 кг 10% азотной кислоты (коммерчески чистой (CP), от Beijing Chemical Plant), после этого перемешивали в течение 1 ч, при этом получали светло-желтый и прозрачный коллоидный раствор, названный золем титана.
Смешивали 4,93 кг порошка оксида цинка (от компании Headhorse, имеющего чистоту 99,7%), 1,64 кг диатомита (от компании Global Mineral, 1,60 кг в пересчете на сухое вещество), 0,72 кг молекулярного сита ReY (от Qilu Catalyst Division, Sinopec, 0,50 кг в пересчете на сухое вещество, с содержанием Re2O3 16,2% масс.) и 6,40 кг деионизированной воды, и после этого перемешивали в течение 30 мин для получения суспензии. Суспензию добавляли в золь титана и после этого перемешивали в течение 1 ч для получения суспензии носителя.
Следуя процедуре примера V-1, суспензию носителя высушивали распылением и формовали, и далее вводили в нее активные компоненты никеля и кобальта для получения адсорбента V-B3. Состав адсорбента V-B3: содержание оксида цинка 49,3% масс., содержание диоксида титана 13,5% масс., содержание диатомита 16,0% масс., содержание молекулярного сита ReY 5,0% масс., содержание никеля 8,1% масс., содержание кобальта 8,1% масс.
Пример V-5
Каждый адсорбент, полученный как описано выше, исследовали на результативность десульфуризации и октановое число. Результативность десульфуризации характеризовали по содержанию серы продукта десульфуризации. Содержание серы продукта десульфуризации определяли с помощью автономно работающего хроматографа после микрореактора с неподвижным слоем. Исходным маслом для испытания реакции десульфуризации был бензин каталитического крекинга, имеющий содержание серы 640 ppm. Условиями испытания реакции десульфуризации были: водородная атмосфера, температура реакции 410°С, давление реакции 1,38 МПа, молярное отношение водорода к углеводородному маслу 0,35, WHSV 4 ч-1. Для точного отражения активности адсорбента, применяемого в промышленных масштабах, после завершения реакции адсорбент регенерировали при 550°C в воздушной атмосфере. Испытания повторяли в течение шести циклов реакции и регенерации, и после этого адсорбент проявлял практически стабильную активность. Результативность десульфуризации адсорбента здесь характеризуется содержанием серы бензинового продукта, измеренным с данным адсорбентом, проявляющим практически стабильную активность, что представлено в табл.V-1. Бензиновый продукт взвешивали для расчета выхода.
В соответствии с китайскими национальными стандартами GB/T 503-1995 и GB/T 5487-1995 определяли соответственно октановое число бензина по моторному методу (MON) и по исследовательскому методу (RON) до и после испытания реакции десульфуризации. Результаты приведены в табл.V-1. Как можно видеть из табл.V-1, при использовании адсорбента, содержащего молекулярное сито со структурой A-FAU, октановое число бензинового продукта может быть повышено в каждом случае.
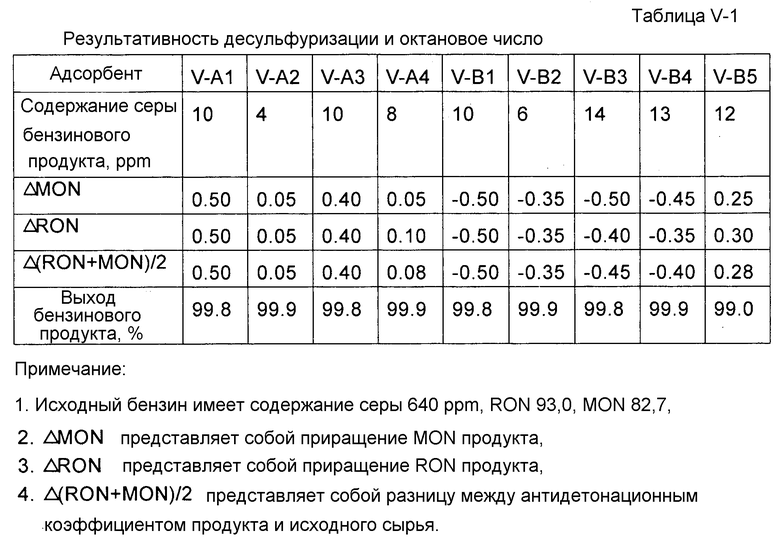
Специалистам будет понятно, что различные модификации и варианты могут быть внесены в настоящее изобретение без отступления от сущности или объема настоящего изобретения. Таким образом, подразумевается, что настоящее изобретение охватывает модификации и варианты по настоящему изобретению, при условии, что они попадают в пределы объема прилагаемой формулы изобретения и ее эквивалентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| АДСОРБИРУЮЩЕЕ ВЕЩЕСТВО ДЛЯ ДЕСУЛЬФУРИЗАЦИИ УГЛЕВОДОРОДНОГО МАСЛА, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2012 |
|
RU2585633C2 |
| КАТАЛИЗАТОР ДЕСУЛЬФУРИЗАЦИИ, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2017 |
|
RU2749402C1 |
| Катализатор для обессеривания жидких нефтепродуктов, его получение и применение | 2017 |
|
RU2745372C2 |
| ОБЕССЕРИВАЮЩИЙ АДСОРБЕНТ, СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ И ИСПОЛЬЗОВАНИЯ | 2009 |
|
RU2498849C2 |
| КАТАЛИЗАТОР КАТАЛИТИЧЕСКОГО КРЕКИНГА И ЕГО ПОЛУЧЕНИЕ | 2018 |
|
RU2755891C2 |
| АДСОРБЕНТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ УДАЛЕНИЯ СЕРЫ ИЗ КРЕКИНГ-БЕНЗИНА ИЛИ ДИЗЕЛЬНОГО ТОПЛИВА | 2009 |
|
RU2517639C2 |
| КАТАЛИЗАТОР КРЕКИНГА И СПОСОБ ДЛЯ ЕГО ПРИГОТОВЛЕНИЯ | 2021 |
|
RU2832734C1 |
| СПОСОБ ОЧИСТКИ БЕНЗИНА | 2017 |
|
RU2742646C2 |
| ФОСФОРСОДЕРЖАЩЕЕ МОЛЕКУЛЯРНОЕ СИТО, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2018 |
|
RU2782564C2 |
| МОДИФИЦИРОВАННОЕ МАГНИЕМ МОЛЕКУЛЯРНОЕ СИТО ТИПА Y, ЕГО ПОЛУЧЕНИЕ И СОДЕРЖАЩИЙ ЕГО КАТАЛИЗАТОР | 2018 |
|
RU2770421C2 |
Настоящее изобретение относится к адсорбенту для десульфуризации углеводородного масла, его получению и использованию. Адсорбент десульфуризации для каталитического крекинг-бензина содержит следующие компоненты в расчете на общую массу адсорбента: Si-Al молекулярное сито со структурой A-FAU, где А представляет собой одновалентный катион, в количестве 3-20% масс., связующее, выбранное из диоксида титана, диоксида олова, диоксида циркония и оксида алюминия, в количестве 3-35% масс., источник диоксида кремния в количестве 5-40% масс., оксид цинка в количестве 10-80% масс., металл-промотор, выбранный из кобальта, никеля, железа и марганца, в количестве 5-30% масс., где по меньшей мере 10% масс. металла-промотора присутствует в состоянии пониженной валентности. Адсорбент по изобретению проявляет повышенную активность и стабильность и одновременно способен существенно повысить октановое число бензинового продукта. 3 н. и 8 з.п. ф-лы, 5 табл.
1. Адсорбент десульфуризации для каталитического крекинг-бензина, содержащий следующие компоненты в расчете на общую массу адсорбента:
1) Si-Al молекулярное сито со структурой A-FAU, где А представляет собой одновалентный катион, в количестве 3-20% масс.,
2) по меньшей мере одно связующее, выбранное из группы, состоящей из диоксида титана, диоксида олова, диоксида циркония и оксида алюминия, в количестве 3-35% масс.,
3) источник диоксида кремния в количестве 5-40% масс.,
4) оксид цинка в количестве 10-80% масс., и
5) по меньшей мере один металл-промотор, выбранный из группы, состоящей из кобальта, никеля, железа и марганца, в зависимости от металла в количестве 5-30% масс., где по меньшей мере 10% масс. металла-промотора присутствует в состоянии пониженной валентности.
2. Адсорбент по п. 1, в котором Si-Al молекулярное сито со структурой A-FAU присутствует в количестве 3-15% масс., связующее присутствует в количестве 5-25% масс., источник диоксида кремния присутствует в количестве 10-30% масс., оксид цинка присутствует в количестве 25-70% масс., металл-промотор присутствует в количестве 8-25% масс.
3. Адсорбент по п. 1, в котором Si-Al молекулярное сито со структурой A-FAU присутствует в количестве 3-10% масс., связующее присутствует в количестве 8-15% масс., источник диоксида кремния присутствует в количестве 12-25% масс., оксид цинка присутствует в количестве 40-60% масс., металл-промотор присутствует в количестве 12-20% масс.
4. Адсорбент по п. 1, в котором источник диоксида кремния выбирают из группы, состоящей из диоксида кремния или природного минерала, имеющего содержание диоксида кремния более 45% масс.
5. Адсорбент по п. 1, в котором Si-Al молекулярное сито со структурой A-FAU является по меньшей мере молекулярным ситом, выбранным из молекулярного сита X и молекулярного сита Y.
6. Способ получения адсорбента десульфуризации для каталитического крекинг-бензина по любому из пп. 1-5, включающий:
(1) контактирование по меньшей мере одного связующего, выбранного из группы, состоящей из диоксида титана, диоксида олова, диоксида циркония и оксида алюминия, с источником диоксида кремния, Si-Al молекулярным ситом со структурой A-FAU (где А представляет собой одновалентный катион) и оксидом цинка для получения носителя,
(2) контактирование носителя с соединением, содержащим металл-промотор, для получения предшественника адсорбента, и
(3) при условии, достаточном для присутствия по меньшей мере 10% масс. металла-промотора в состоянии пониженной валентности, обработку предшественника адсорбента для получения адсорбента десульфуризации для каталитического крекинг-бензина.
7. Способ по п. 6, в котором этап (1) включает следующие стадии:
(1а) связующее или предшественник связующего смешивают с кислотой для образования суспензии, и
(1b) суспензию, источник диоксида кремния, Si-Al молекулярное сито со структурой A-FAU и оксид цинка смешивают, формуют, высушивают и прокаливают для получения носителя.
8. Способ по п. 6, в котором этап (1) включает следующие стадии:
(1а′) по меньшей мере часть источника диоксида кремния и связующего или предшественника связующего смешивают с кислотой для образования суспензии, и
(1b′) суспензию, оставшуюся часть источника диоксида кремния, Si-Al молекулярное сито со структурой A-FAU и оксид цинка смешивают, формуют, высушивают и прокаливают для получения носителя.
9. Способ по п. 7 или 8, в котором предшественник связующего является по меньшей мере предшественником, выбранным из группы, состоящей из галогенидов, алкоксилатов, солей карбоновой кислоты, гидратированных оксидов, гидроксидов, гидратированных гидроксидов и оксигалогенидов титана, олова, циркония и/или алюминия.
10. Способ по п. 7 или 8, в котором кислота является по меньшей мере кислотой, выбранной из группы, состоящей из водорастворимой неорганической кислоты и водорастворимой органической кислоты, и количество используемой кислоты таково, что значения рН суспензии достигают 0,5-6.
11. Способ десульфуризации каталитического крекинг-бензина, включающий контактирование серосодержащего каталитического крекинг-бензина с адсорбентом десульфуризации для каталитического крекинг-бензина по любому из пп. 1-5, при условии, достаточном для удаления по меньшей мере части серы из серосодержащего каталитического крекинг-бензина.
| КОМПОЗИЦИЯ СОРБЕНТА, СПОСОБ ЕЁ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ ПРИ ДЕСУЛЬФУРИЗАЦИИ | 2000 |
|
RU2230608C2 |
| CN 101481627 A, 15.07.2009 | |||
| ТВЕРДЫЙ КОМПОЗИЦИОННЫЙ АДСОРБЕНТ ДЛЯ ОЧИСТКИ УГЛЕВОДОРОДНЫХ ПОТОКОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ УДАЛЕНИЯ ПРИМЕСЕЙ | 2001 |
|
RU2264855C2 |
| US 6150300 A, 21.11.2000 | |||
| CN 102114407 B, 12.12.2012 | |||
| CN 102114406 A, 06.07.2011 | |||
| СПОСОБ ДЕСУЛЬФУРИЗАЦИИ УГЛЕВОДОРОДНОЙ НЕФТИ | 2004 |
|
RU2335528C2 |

