СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к адсорбенту для удаления серы из жидкого тока, такого как крекинг-бензин и дизельное топливо.
УРОВЕНЬ ТЕХНИКИ
С повышением внимания к окружающей среде природоохранное законодательство становится все более строгим. Считается, что понижение содержания серы в бензине или дизельном топливе становится одной из наиболее важных мер для повышения качества воздуха, поскольку сера, содержащаяся в топливе, неблагоприятно влияет на функционирование каталитического конвертера автомобилей и транспортных средств. Оксид серы, присутствующий в отработанных газах, выходящих из автомобильного двигателя, снижает активность катализатора, основанного на благородных металлах, в конвертере и необратимо отравляет его. Газы, испускаемые из неэффективно работающего конвертера или конвертера, содержащего отравленный катализатор, содержат несгораемые неметановые углеводороды, оксид азота и монооксид углерода, все из которых легко образуют фотохимический смог, при катализе солнечным светом.
В Китае большая часть серы, содержащейся в бензинах, происходит из бензина при термообработке, который, как правило, представляет собой каталитический крекинг-бензин. Поэтому понижение содержания серы в крекинг-бензине может облегчить снижение содержания серы в этих бензинах. Существующий стандарт для бензинового продукта - GB 17930-2006 «Бензин для автомобильной промышленности», который ограничивает содержание серы в бензине с 31 декабря 2009 г. Существует требование, чтобы содержание серы в бензине было снижено до 50 промилле. Это обстоятельство означает, что крекинг-бензин должен быть максимально обессерен, чтобы соответствовать экологическим требованиям.
При снижении содержания серы в топливе автомобильного двигателя, изменений в содержании олефинов, которые приводят к снижению октанового числа (включая октановое число бензина по исследовательскому (ROM) и моторному методам (MON)) можно избежать, сохраняя характеристики сгорания топлива автомобильного двигателя. Как правило, негативные изменения содержания олефинов бывают вызваны реакцией гидрирования, при удалении соединений тиофена (включая тиофен, бензотиофен, алкилтиофен, алкилбензотиофен и алкилдибензотиофен). Кроме того, можно также избежать потерь ароматических углеводородов в крекинг-бензине из-за насыщения при условиях гидрирования. Поэтому большинство желаемых подходов состоит в обессеривании бензина при сохранении его октанового числа.
С другой стороны, как при гидрообессеривании, так и при гидрировании углеводородов потребляется водород, что приводит к повышению эксплуатационных расходов на обессеривание. Следовательно, существует необходимость в способе обессеривания без потребления водорода в больших объемах и в обеспечении, таким образом, более экономичного способа обработки крекинг-бензина или дизельного топлива.
Традиционно процесс производства бензина с фиксированным слоем катализатора обычно используют для обессеривания в жидкой фазе. Однако данный процесс является неблагоприятным с точки зрения гомогенности реакции и регенерации материала. По сравнению с процессом с фиксированным слоем процесс с использованием псевдоожиженного слоя является благоприятным, с учетом более широких перспектив применения в будущем, вследствие лучшего теплопереноса и перепада давлений. В этой связи, реактор с псевдоожиженным слоем обычно снабжают гранулированными реагентами; а используемые частицы катализатора должны быть достаточно стойкими к истиранию. Следовательно, большое значение придается поиску адсорбента, обладающего как высокой стойкостью к истиранию, так и кпд обессеривания.
В китайских патентах CN 1110931 A и CN 1151333 A описан новый адсорбционный состав, содержащий оксид цинка, оксид кремния, коллоидный оксид и катализатор; а также способ изготовления такого состава. В данном способе псевдоожиженные частицы получают штамповкой, и объемы пор частиц повышаются за счет добавления к коллоидному веществу порообразующего агента, который становится воспламеняемым при нагреве.
В Патенте США №6150300, китайских патентных публикациях CN 1355727А и CN 1382071 A раскрыт состав гранулированного адсорбента, содержащего смесь оксида цинка, кремнезема, глинозема, никеля или кобальта в восстановленном состоянии. Адсорбент изготавливают путем предварительного смешивания кремнезема, глинозема и оксида цинка под действием сдвигающей силы и получения твердых частиц с помощью гранулятора. В этих способах активный компонент, никель, вводят пропиткой.
В китайских патентных публикациях CN 1422177 A и CN 1627988 A раскрыт устойчивый к истиранию состав адсорбента, приготовленный пропиткой носителя, содержащего оксид цинка, вспученный перлит и глинозем, в таком катализаторе, как никель, оксид никеля, или в соединении - «предшественнике» оксида никеля, и последующего снижения валентности металла-катализатора в полученном составе носителя, содержащего металлический адсорбент. Состав адсорбента применяют для удаления элементарной серы и сульфида, например сероводорода и органического сульфида, из крекинг-бензина и дизельного топлива. В соответствии с этими патентами активный компонент можно вводить пропиткой носителя адсорбента в соединении-«предшественнике», содержащем металл-катализатор, и данный способ пропитки является благоприятным вследствие простоты приготовления и внедрения. Однако состав, содержащий металл-катализатор, вводимый указанным способом, часто не обладает достаточной однородностью, что может привести к недостаточной обессеривающей активности в адсорбенте. В данной ситуации количество используемого металла-катализатора обычно повышают для усиления активности адсорбента, но это также повышает себестоимость изготовления адсорбента.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем изобретении обеспечен адсорбент для удаления серы из крекинг-бензина или дизельного топлива, причем адсорбент обладает достаточно высокой активностью. В одном варианте воплощения адсорбент имеет активные компоненты, равномерно распределенные по носителю. В другом варианте воплощения активные компоненты распределены по носителю практически в виде монослоя.
В настоящем изобретении также обеспечен способ получения такого адсорбента.
В настоящем изобретении дополнительно обеспечено применение такого адсорбента, например, при удалении элементарной серы и сульфидов из крекинг-бензина и/или дизельного топлива.
На практике, адсорбент согласно изобретению содержит:
1) носитель, содержащий источник кремнезема, неорганических оксидных связок и, по меньшей мере, одного оксида металла, выбранного из групп IIB, VB и VIB;
2) по меньшей мере, один металл-катализатор, пригодный для восстановления серы, находящейся в окисленном состоянии, до сероводорода, и который имеет значение η<0,5.
Параметр «η» задан следующим образом: η=(количество металла-катализатора в кристаллической фазе, в процентах /(количество металла-катализатора в адсорбенте, в процентах).
Методики определения процентных долей, из которых вычисляют η, разъяснены ниже.
Адсорбент, обеспеченный в настоящем документе, содержит примерно 1-40 мас.% источника диоксида кремния, и примерно 10-80 мас.% оксида металла, и примерно 3-35 мас.% связующего вещества на основе неорганического оксида в пересчете на оксиды, и примерно 3-30 мас.% металла-катализатора в пересчете на оксиды исходя из общей массы адсорбента. Является предпочтительным, чтобы адсорбент содержал примерно 10-25 мас.% источника диоксида кремния, примерно 40-60 мас.% оксида металла, примерно 10-18 мас.% связующего вещества на основе неорганического оксида в пересчете на оксиды и примерно 5-20 мас.% металла-катализатора в пересчете на оксиды исходя из общей массы катализатора. Является более предпочтительным, чтобы содержание металла-катализатора составляло в диапазоне примерно 8-20 мас.%.
В соответствии с описываемым здесь адсорбентом источник кремнезема может представлять собой либо чистый кремнезем, либо кремнеземсодержащую смесь, например, одно или несколько веществ из следующих: глина (например, каолин, столбчатая глина, и т.д.), диатомит, вспученный перлит, кремнистая порода, гидролизный кремнезем, крупнопористый кремнезем и силикагель. Примеры такой столбчатой глины включают в себя (но не ограничены): ректорит, глину Юмменга, бентонит, монтмориллонит и смектит; предпочтительным является ректорит.
Связующее вещество на основе неорганического оксида представляет собой оксидное связующее вещество на основе кремнеземно-глиноземной смеси, которое можно выбрать из одного или нескольких веществ: глинозем, кремнезем и аморфная смесь кремнезем-глинозем; глинозем является предпочтительным, более предпочтительным - глинозем, выбранный из одного или нескольких веществ: γ-глинозем, η-глинозем, θ-глинозем и χ-глинозем.
Приведенный здесь оксид металла представляет собой оксид одного или нескольких металлов, выбранных из групп IIB, VB и VIB, или любой другой оксид металла, обладающий свойствами сохранения серы; предпочтительными являются оксиды ванадия, цинка или молибдена; а наиболее предпочтительным - оксид цинка.
Металл-катализатор может представлять собой любой металл, способный восстанавливать окисленную серу до сероводорода. Предпочтительными являются один или несколько металлов, выбранных из никеля, кобальта, марганца, железа, меди, серебра, молибдена, хрома, ванадия, вольфрама и лантаноидов; наиболее предпочтительным является металл-катализатор, содержащий никель.
В некоторых вариантах воплощения согласно настоящему изобретению η<0,5, в других вариантах воплощения η=0, т.е., по меньшей мере, один металл-катализатор, который способен восстанавливать серу, находящуюся в окисленном состоянии, до сероводорода, распределен по поверхности носителя практически в виде монослоя.
Термин «крекинг-бензин», используемый в настоящем документе, относится к углеводородам с диапазоном температур кипения 40-210°С или к любым их фракциям, получаемым путем термического или каталитического крекинга с преобразованием крупных молекул углеводорода в мелкие молекулы. Подходящие процессы термического крекинга включают в себя (но не ограничены) пиролиз, термический крекинг, легкий крекинг или их сочетания. Примеры подходящих процессов каталитического крекинга включают в себя (но не ограничены) крекинг на псевдоожиженном слое катализатора и каталитический крекинг тяжелого топлива и их сочетания. Поэтому подходящий каталитический крекинг-бензин включает в себя (но не ограничен) бензин коксования, бензин, полученный путем термокрекинга, бензин, полученный путем крекинга на псевдоожиженном слое катализатора, бензин, полученный путем крекинга тяжелой нефти, и их сочетания. Согласно процессу, описываемому в изобретении, в некоторых случаях крекинг-бензин при его использовании в качестве углеводородсодержащего флюида можно разгонять на фракции и/или гидрогенизировать перед обессериванием.
Термин «дизельное топливо», используемый в настоящем изобретении, означает любую углеводородную смесь или любые ее фракции с диапазоном температур выкипания 170-450°С. Такие углеводородсодержащие флюиды включают в себя (но не ограничены) легкий рецикловый газойль, керосин, дизельное масло прямой перегонки, гидрогенизированное дизельное масло и их сочетания.
Термин «сера», используемый в настоящем изобретении, означает элемент серу в любой форме, такой как органический сульфид, существующий в углеводородсодержащих жидкостях, таких как крекинг-бензин или дизельное топливо. Сера, содержащаяся в углеводородсодержащей жидкости согласно настоящему изобретению, включает в себя (но не ограничена) сернистый карбонил (COS), сероуглерод (CS2), меркаптаны или другие соединения тиофена и их сочетания, в частности тиофен, бензотиофен, алкилтиофен, алкилбензотиофен и алкилдибензотиофен, и соединения тиофена, обладающие молекулярной массой большей, чем у соединений, обычно содержащихся в дизельном топливе.
Настоящее изобретение также относится к способу получения адсорбента, включающему в себя стадии:
(1) приведение в контакт источника кремнезема, неорганического оксидного соединения-«предшественника» и оксида одного или нескольких металлов, выбранных из групп IIB, VB и VIB или их «предшественников», с последующим формованием и высушиванием смеси с образованием носителя;
(2) загрузки носителя в псевдоожиженную подложку и пропускания органического соединения, содержащего металл-катализатор, пригодный для восстановления окисленной серы до сероводорода, причем упомянутое органическое соединение переносится в газовой фазе, для получения «предшественника» адсорбента;
(3) высушивания и обжига «предшественника» абсорбента, полученного на предыдущей стадии (2) для преобразования органического соединения, содержащего металл-катализатор, в оксид металла;
(4) восстановления «предшественника» абсорбента, обработанного на стадии (3) в восстановительной атмосфере таким образом, чтобы металл-катализатор по существу сам находился бы в восстановленном состоянии, а адсорбент имел бы значение η<0,5, где η=(количество, в процентах, металла-катализатора в кристаллической фазе)/(количество, в процентах, металла-катализатора в адсорбенте).
На стадии (1), носитель можно приготавливать в соответствии со способами, известными из уровня техники. Например, микросферический носитель можно изготавливать, перемешивая и измельчая источник кремнезема, неорганическое оксидное связующее соединение-«предшественник», оксид одного или нескольких металлов, выбранных из групп IIВ, VB и VIB или их «предшественник», и воду, в кислотной среде, получая, таким образом, суспензию примерно с 10-40 мас.% твердых частиц, с последующей распылительной сушкой.
На стадии (1), источник диоксида кремния выбирают из одной или нескольких глин (например, каолина, столбчатой глины, и т.д.), диатомита, вспученного перлита, кремнистой породы, гидролизного диоксида кремния, макропористого диоксида кремния и силикагеля, предпочтительно одного или нескольких веществ: вспученного перлита, диатомита и глины. Примеры такой столбчатой глины включают в себя (но не ограничены): ректорит, глину Юнменга, бентонит, монтмориллонит и смектит; ректорит является предпочтительным.
На стадии (1) оксид одного или нескольких металлов, выбранных из групп IIB, VB и VIB, или его «предшественники» включает в себя оксиды как таковые или их «предшественники», причем «предшественниками» оксидов являются соединения, которые можно преобразовывать в оксиды при вышеупомянутых условиях приготовления. Примеры этих соединений представляют собой, например, сульфиды, сульфаты, гидроксиды, карбонаты, ацетаты и нитраты одного или нескольких металлов групп IIB, VB и VIB. Предпочтительными оксидами металлов являются оксиды ванадия, цинка или молибдена, а наиболее предпочтительно - оксид цинка.
На стадии (1) неорганическое оксидное связующее соединение-«предшественник» представляет собой вещество, которое может образовывать термостойкий неорганический оксид в ходе процесса образования адсорбента, включая, например, одно или несколько веществ: глинозем, кремнезем и аморфный «предшественник» смеси кремнезема и глинозема. Например, «предшественник» глинозема можно выбрать из гидратированной окиси алюминия и/или золя алюминия; гидратированную окись алюминия выбирают из одного или нескольких веществ: бемита, псевдобемита, тригидрата окиси алюминия, аморфного гидроксида алюминия. «Предшественник» кремнезема можно выбрать из одного или нескольких веществ: силикатного золя, силикагеля и водомерного стекла. «Предшественник» аморфной кремнеземно-глиноземной смеси можно выбрать из одного или нескольких соединений: кремнеземно-глиноземного золя, смеси силикатного золя и глиноземного золя и кремнеземно-глиноземного геля. Эти «предшественники» термостойких неорганических оксидов являются известными для специалистов в данной области техники.
Согласно процессу по настоящему изобретению является предпочтительным осуществление обжига высушенного носителя после стадии (1). Температура обжига составляет примерно 350-700°С, предпочтительно примерно 450-650°С, а период обжига составляет примерно 1-10 часов, предпочтительно примерно 1-4 часа.
На стадии (2), газ является неокислительным газом, предпочтительно инертным газом, таким как азот, в частности предпочтительным является безводный инертный газ. Давление газа составляет примерно 1-5 атм, предпочтительно примерно атмосферное давление. Температура газа составляет примерно 50-200°С, предпочтительно примерно 50-130°С.
На стадии (2), металл-катализатор может представлять собой любой металл, пригодный для восстановления окисленной серы до сероводорода. Предпочтительными являются один или несколько металлов, выбранных из следующих металлов: никель, кобальт, марганец, железо, медь, серебро, молибден, хром, ванадий, вольфрам и лантаноиды, более предпочтительно - металл-катализатор, содержащий никель. Органическое соединение металла-катализатора выбирают из одного или нескольких соединений: формиатов, ацетатов, карбонилов металлов и нафтенатов с давлением пара примерно 0,5-100 кПа при комнатной температуре (298К). Согласно одному варианту воплощения настоящего изобретения металл-катализатор представлен карбонилом никеля, и, в частности, тетракарбонилом никеля.
На стадии (3) «предшественник» адсорбента обжигают примерно при 300-800°С, предпочтительно при 450 - примерно 750°С в течение примерно 0,5-4 часов, предпочтительно в течение примерно 1-3 часов в присутствии кислорода или кислородсодержащей атмосферы до удаления летучих материалов и преобразования металлов-катализаторов в оксиды металлов.
На стадии (4) является предпочтительным восстанавливать металлы-катализаторы в восстановительной атмосфере перед использованием адсорбента с получением, таким образом, металлов-катализаторов в практически восстановленном состоянии (предпочтительно с нулевой валентностью) и с получением, в свою очередь, адсорбента по настоящему изобретению. Температура восстановления составляет примерно 300-600°С, предпочтительно примерно 400-500°С. Время восстановления составляет примерно 0,5-6 часов, предпочтительно примерно 1-3 часов. Количество восстанавливающего газа составляет примерно 10-100 по объему в восстанавливающей атмосфере; предпочтительный восстанавливающий газ представляет собой газообразный водород, а остаток может представлять собой инертный газ, предпочтительно газообразный водород или газообразный аргон.
Адсорбент, полученный на стадии (4), имеет η<0,5; при η=0, по меньшей мере, один металл-катализатор, который пригоден для восстановления серы, находящейся в окисленном состоянии, до сероводорода, распределен по поверхности носителя практически в виде монослоя.
Настоящее изобретение дополнительно относится к способу обессеривания крекинг-бензина или дизельного топлива, включающему в себя приведение в контакт серосодержащего вещества с адсорбентом согласно изобретению, при котором сера, содержащаяся в веществе, адсорбируется адсорбентом, и, таким образом, получается продукт с низким содержанием серы.
Условия приведения в контакт в этом отношении следующие:
- температура: примерно 350-500°С, предпочтительно примерно 400-425°С;
- объемно-весовая скорость: примерно 2-8 ч-1, предпочтительно примерно 4-6 ч-1;
- давление: примерно 1000-5000 кПа, предпочтительно примерно 1500-3000 кПа;
- атмосфера: восстановительная атмосфера, предпочтительно атмосфера водорода;
в которых адсорбент может рециркулировать после того, как он был подвергнут окислительно-восстановительному процессу регенерации.
Обессеривающие свойства адсорбента сильно зависят от эффективного содержания металла-катализатора, которое связано с количеством металла-катализатора, способного контактировать и адсорбировать серу на поверхности адсорбента. Обессеривающие свойства адсорбента повышаются с повышением количества металла-катализатора перед достижением содержания металла-катализатора определенного значения η. Однако обессеривающие свойства адсорбента невозможно повысить даже путем повышения количества металла-катализатора после того как эффективное количество металла-катализатора достигло определенного значения η.
Определено, что η=(количество, в процентах, металла-катализатора в кристаллической фазе)/(количество, в процентах, металла-катализатора в адсорбенте). Если металл-катализатор в основном распределен по поверхности адсорбента, то максимум, соответствующий содержанию металла-катализатора в адсорбенте при анализе кристаллической фазы, отсутствует и тогда количество, в процентах, металла-катализатора в кристаллической фазе равно нулю, таким образом, η=0. Чем ниже значение η, тем меньше количество металла-катализатора, пригодного для контактирования с серой и ее поглощения. Поэтому η свидетельствует о распределении металла-катализатора по поверхности адсорбента. То есть для случая однослойного распределения или распределения, близкого к монослою, возникает ситуация, когда η=0; металл-катализатор начинает скапливаться, а некоторые металлы-катализаторы никогда не присутствуют на поверхности, когда η>0. Чем выше значение η, тем больше количество металла-катализатора в агломерированном состоянии. Обычно адсорбент, приготовленный в соответствии с настоящим изобретением, имеет η<0,5, тогда как адсорбент, приготовленный в соответствии с уровнем техники, имеет η>0,5.
Способ кипящего слоя, сжижаемого газом, используемый в настоящем изобретении, может обеспечить однослойное распределение органического соединения металла-катализатора по сжижаемой поверхности носителя. Кроме того, металл-катализатор также может быть распределен по поверхности носителя в виде монослоя при обжиге и восстановлении до восстановленного состояния (в основном до нулевой валентности). Адсорбент проявляет максимальную адсорбирующую активность при обессеривании, когда количество используемого металла-катализатора таково, что дисперсионный объем металла-катализатра является объемом самого крупного дисперсионного слоя. Содержание металла-катализатора в адсорбенте согласно настоящему изобретению много ниже, чем содержание металла-катализатора, вводимого в адсорбент пропиткой в соответствии с уровнем техники, демонстрируя те же характеристики, и поэтому стоимость изготовления адсорбента может быть значительно снижена.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
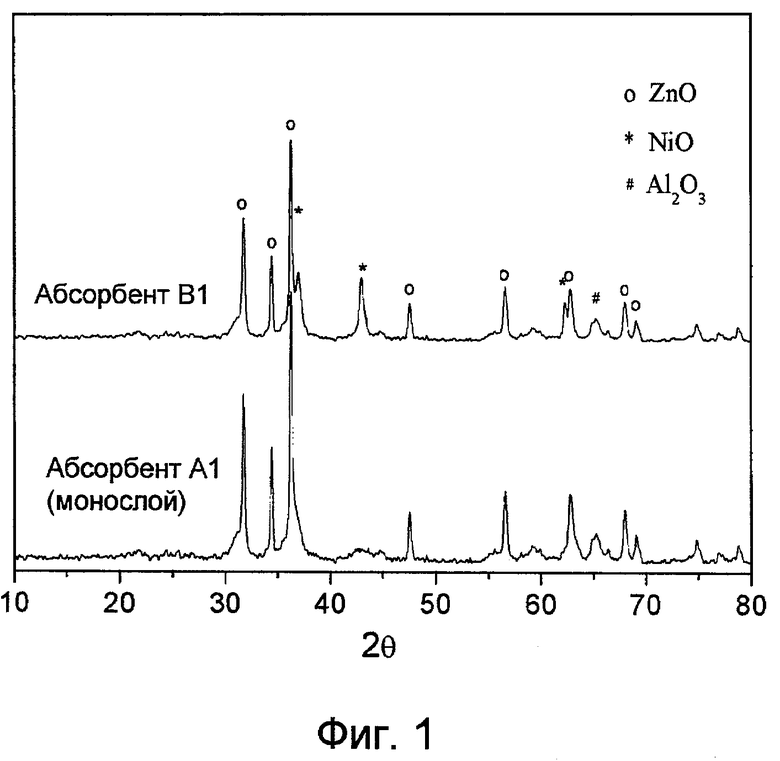
Фиг.1 представляет собой рентгенограммы адсорбента А1 (изобретение) и В1 (уровень техники). Металлический Ni, диспергированный в виде монослоя в адсорбенте А1, в основном показывает отсутствие пиков кристаллизации оксида никеля, тогда как адсорбент В1, в который введен металл-катализатор никель способом согласно уровню техники, демонстрирует четкий пик кристаллизации оксида никеля (характеристический пик кристаллизации виден при 42,9°).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Тогда как варианты воплощения настоящего раскрытия описаны в соответствии с вышеописанными вариантами воплощения и соответствующим текстом и фигурой, это не должно рассматриваться как ограничение формулы изобретения частными вариантами воплощения, приведенными в этих описаниях. Напротив, описание охватывает все альтернативы, модификации и эквиваленты, находящиеся в пределах сущности настоящего изобретения. Настоящее изобретение далее будет проиллюстрировано со ссылкой на следующие примеры, но оно не ограничено ими.
Пример 1
0,80 кг вспученного перлита (выпускаемого компанией Worldminerals, M27, обладающего сухим весом 0,79 кг), 0,71 кг каолина (выпускаемого компанией Suzhou Kaolin CO., 31, обладающего сухим весом 0,59 кг), 1,15 кг глинозема (выпускаемого компанией Shandong Aluminum Corporation, обладающего сухим весом 0,78 кг) и 9,0 кг кислой воды (вода, очищенная от катионов, рН 3), были смешаны при интенсивном перемешивании, а затем к смеси было добавлено 110 мл 30% НС1 (химически чистой марки, выпускаемой компанией Beijing Chemical Works) при интенсивном перемешивании, в целях подкисления, в течение 1 часа. Затем было добавлено 3,5 кг порошкового оксида цинка, перемешано и взболтано в течение 1 часа для получения взвеси-носителя. Взвесь была подвергнута распылительной сушке с использованием Niro Bowen Nozzle Tower™ распылительной сушилки, при давлении 8,5-9,5 МПа, температуре на входе ниже 500°С и температуре на выходе примерно 150°С. Полученная таким образом микросфера была высушена при 180°С в течение 1 часа, а затем обожжена при 635°С в течение 1 часа до получения носителя адсорбента.
Носитель адсорбента был загружен в псевдоожиженный слой, и через него был пропущен высокочистый газообразный азот, содержащий тетракарбонил никеля (50°С) в течение 6 часов. Затем носитель адсорбента, заполненный никелем, был вынут и обожжен на воздухе при 650°С в течение 1 часа с получением «предшественника» адсорбента. «Предшественник» адсорбента был восстановлен в атмосфере водорода при 425°С в течение 2 часов с получением адсорбента, который упоминался как адсорбент A1.
Адсорбент A1 состоял из 57,9 мас.% оксида цинка, 12,7 мас.% глиноземного связующего вещества, 12,8 мас.% вспученного перлита, 9,6 мас.% каолина, и 8,0 мас.% никеля (в виде оксида никеля).
Пример 2
После замены носителя адсорбента на «предшественник» адсорбента A1, процесс внедрения никеля путем осаждения адсорбента из паровой фазы был повторен. Затем «предшественник» адсорбента был обожжен при 635°С на воздухе в течение 1 часа, с получением «предшественника» адсорбента, который был восстановлен в атмосфере водорода при 410°С в течение 4 часов, с получением адсорбента, который упоминался как адсорбент А2.
Адсорбент А2 состоял из 54,5 мас.% оксида цинка, 12,1 мас.% глиноземного связующего вещества, 12,3 мас.% вспученного перлита, 9,2 мас.% каолина и 11,9 мас.% никеля (в виде оксида никеля).
Пример 3
Адсорбент был приготовлен следующим образом: были перемешны 3,5 кг порошкового оксида цинка (выпускаемого компанией Beijing Chemical Works) и 4,97 кг деонизованной воды, а затем взболтаны в течение 30 минут для получения взвеси оксида цинка.
0,61 кг диатомита (выпускаемого компанией Beijing Chemical Reagent Co., Ltd, обладающего сухим весом 0,58 кг), 0,96 кг ректорита (выпускаемого компанией Qilu Petrochemical Catalyst Company, обладающего сухим весом 0,80 кг), глинозема (выпускаемого компанией Shandong Aluminum Corporation, обладающего сухим весом 0,82 кг), и 5,0 кг кислой воды (декатионизованной воды, рН 3) были смешаны при встряхивании, а затем к ним было добавлено 115 мл 30% НСl (химически чистой марки, выпускаемой компанией Beijing Chemical Works) при взбалтывании в течение 1 часа, для подкисления. Затем был добавлен вышеупомянутый порошковый оксид цинка, а смесь была перемешана и взболтана в течение 1 часа для получения взвеси-носителя. Взвесь была подвергнута распылительной сушке с использованием распылительной сушилки Niro Bowen Nozzle Tower™, при давлении 8,5-9,5 МПа, температуре на входе ниже 500°С и температуре на выходе примерно 150°С. Полученные, таким образом, микросферы были высушены при 180°С в течение 1 часа, а затем обожжены при 635°С в течение 1 часа для получения носителя адсорбента.
Носитель адсорбента был загружен в псевдоожиженный слой, и через него в течение 6 часов был пропущен высокочистый газообразный азот с растворенным тетракарбонилом никеля (50°С). Затем носитель адсорбента был обожжен на воздухе при 630°С в течение 1 часа, с получением «предшественника» адсорбента. «Предшественник» адсорбента был восстановлен в атмосфере водорода при 425°С в течение 2 часов, с получением адсорбента, который упоминался, как адсорбент A3.
Адсорбент A3 состоял из 57,9 мас.% диоксида цинка, 12,7 мас.% глиноземного связующего вещества, 9,5 мас.% диатомита, 12,9 мас.% ректорита и 8,0 мас.% никеля (в виде диоксида никеля).
Пример 4
Носитель адсорбента был приготовлен согласно способу по Примеру 1.
Носитель адсорбента был загружен в псевдоожиженный слой, и через него в течение 4 часов был пропущен высокочистый газообразный азот с растворенным тетракарбонилом кобальта (50°С). Затем носитель адсорбента, заполненный никелем, был вынут и обожжен на воздухе при 650°С в течение 1 часа с получением предшественника адсорбента. Предшественник адсорбента был восстановлен в атмосфере водорода при 425°С в течение 2 часов, с получением адсорбента, который упоминался как адсорбент А4.
Адсорбент А4 состоял из 57,9 мас.% оксида цинка, 12,7 мас.% глиноземного связующего вещества, 12,8 мас.% вспученного перлита, 9,6 мас.% каолина, и 8,0 мас.% кобальта (в виде оксида кобальта).
Сравнительный пример 1
Носитель адсорбента был приготовлен согласно способу по Примеру 1 со следующими отличиями. 3,24 кг носителя адсорбента (обладающего сухим весом 3,0 кг) было пропитано в 1,03 кг гексагидрата нитрата никеля и 0,2 кг деионизованной воды. Смесь, полученная указанным способом, была высушена при 180°С в течение 4 часов и обожжена на воздухе при 635°С в течение 1 часов до получения предшественника адсорбента. Предшественник адсорбента был восстановлен в атмосфере водорода при 425°С в течение 2 часов, с получением адсорбента, который упоминался как адсорбент В1.
Адсорбент В1 состоял из 57,9 мас.% оксида цинка, 12,7 мас.% глиноземного связующего вещества, 12,8 мас.% вспученного перлита, 9,6 мас.% каолина и 8,0 мас.% никеля (в виде оксида никеля).
Сравнительный пример 2
Носитель адсорбента был приготовлен в соответствии со способом по Примеру 2 со следующими отличиями. 3,24 кг носителя адсорбента (обладающего сухим весом 3,0 кг) было пропитано в 1,03 кг гексагидрата нитрата никеля и 0,2 кг деионизованной воды. Полученная таким образом смесь была высушена при 180°С в течение 4 часов и обожжена на воздухе при 635°С в течение 1 часа для получения предшественника адсорбента. Предшественник адсорбента был восстановлен в атмосфере водорода при 425°С в течение 2 часов, с получением адсорбента, который упоминался как адсорбент В2.
Адсорбент В2 состоял из 54,5 мас.% оксида цинка, 12,1 мас.% глиноземного связующего вещества, 12,3 мас.% вспученного перлита, 9,2 мас.% каолина и 11,9 мас.% никеля (в виде оксида никеля).
Сравнительный пример 3
Носитель адсорбента был приготовлен в соответствии со способом по Примеру 3 со следующими отличиями. 3,24 кг носителя адсорбента (обладающего сухим весом 3,0 кг) было пропитано в 1,61 кг гексагидрата нитрата никеля и 0,3 кг деионизованной воды. Полученная таким образом смесь была высушена при 180°С в течение 4 часов и обожжена на воздухе при 635°С в течение 1 часа для получения предшественника адсорбента. Предшественник адсорбента был восстановлен в атмосфере водорода при 425°С в течение 2 часов, с получением адсорбента, который упоминался как адсорбент В3.
Адсорбент В3 состоял из 57,9 мас.% оксида цинка, 12,7 мас.% глиноземного связующего вещества, 9,5 мас.% диатомита, 12,9 мас.% ректорита и 8,0 мас.% никеля (в виде оксида никеля).
Сравнительный пример 4
Носитель адсорбента был приготовлен согласно способу по Примеру 1 со следующими отличиями. Адсорбент В4 был приготовлен согласно способу по Сравнительному примеру 1, с пропиткой нитратом кобальта и последующим отжигом и восстановлением.
Адсорбент В4 состоял из 57,9 мас.% оксида цинка, 12,7 мас.% глиноземного связующего вещества, 12,8 мас.% вспученного перлита, 9,6 мас.% каолина и 8,0 мас.% кобальта (в виде оксида кобальта).
Пример 5
Для различения вышеупомянутых различных адсорбентов были измерены находящиеся в кристаллической фазе составы предварительно восстановленных адсорбентов А1-А3 и В1-В3 и были вычислены значения η. Кристаллическая фаза была проанализирована с использованием дифракции рентгеновских лучей и фазовой фильтрации (R.V.Siriwardane, J.A.Poston, G. Evans, Jr. Ind. Eng. Chem. Res.33 (1994) 2810-2818), адаптированная форма работы «Rietveld modeling (RIQAS rietveld analysis, Operators Manual, Material Data, Inc., Berkley, CA (1999)») и вычисления составов кристаллической фазы с помощью методологии пригонки. Все рентгенографические измерения были проведены с использованием генератора Philips XRG 3100, снабженного источником рентгеновского излучения в виде длинной тонкой фокусирующей медной пластины, снабженной электроэнергией при 40 кВ и 30 мА; цифрового гониометра Philips 3020 и управляющего гониометра Philips 3710 MPD; а также охлажденного кремниевого детектора Kevex PSI Peltier. Детектор Kevex был использован с контроллером ионного насоса 4601, источником тока Kevex 4608 Peltier, детектором тока подмагничивания Kevex 4621, импульсным процессором Kevex 4561 А и одноканальным анализатором Kevex 4911-А.
Рентгенограммы были получены с использованием программного обеспечения Philips APD версии 4.1 с. Все вычисления Rietveld были осуществлены с использованием программного обеспечения Material Data, Inc. Riqas версии 3.1 с (Outokumpu HSC Chemistry for Windows: Users Guide, Outokumpo Research Oy, Pori. Finland (1999)). Программы были запущены под операционную систему MS Windows® 2000 с использованием персонального компьютера класса Intel Pentium® IV 2,0 ГГц, снабженного оперативной памятью 512 Мбайт. Составы различных образцов, находящиеся в кристаллической фазе, были показаны в Таблице 1.
Определение
ηNi = (процентное содержание Ni в кристаллической фазе/процентное содержание Ni в адсорбенте).
Эффективное содержание Ni было определено с помощью Метода H2-TPD, состоящего в измерении на адсорбционном инструменте Autochem II 2920 компании Micromeritics Co. Образцы адсорбента были восстановлены газообразным водородом при 450°С в течение 1 часа, охлаждены до температуры окружающей среды и подвергнуты продувке в течение 30 мин, а затем были подвергнуты продувке высокочистым азотом до достижения стабильного состояния и, наконец, нагреты до 650°С. Сигналы были записаны с помощью термокондуктометрического детектора (TCD), а общее количество адсорбированного водорода было вычислено по отношению площадей х-х пиков. Затем адсорбированное количество Ni на атом водорода, т.е. эффективное содержание никеля, было вычислено исходя из зависимости одного атома водорода, адсорбированного атомом никеля, от количества атомов никеля. Указанные результаты показаны в таблице 1.
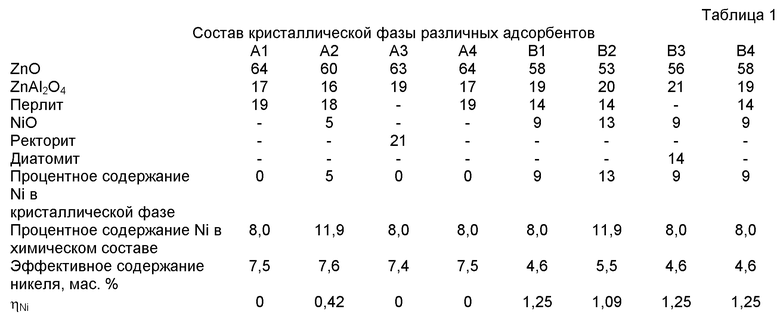
Как видно из таблицы 1, Со или NiO в адсорбентах А1, A3 и А4 присутствует в виде монослойной дисперсии и, как таковой, не может быть отражен в составе кристаллической фазы. То есть процентное содержание Ni/Co в кристаллической фазе равно 0, и, таким образом, η=0. Адсорбент А2 имеет ηNi=0,42, и это означает, что эффективное содержание никеля выше, чем в прототипе (ηNi в сравнительных вариантах воплощения B1, B2 и В3 выше, чем 1). Также из эффективного содержания можно видеть, что адсорбенты, приготовленные согласно настоящему изобретению, обладают эффективным содержанием никеля примерно 7,5 мас.%, тогда как адсорбенты, приготовленные в соответствии с уровнем техники, обладали эффективным содержанием никеля не более 5,5 мас.%.
Пример 6
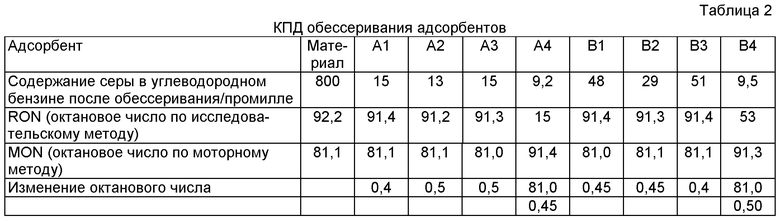
Силу адсорбентов, приготовленных различными способами, оценивают с помощью метода вертикального истирания трубы, и данный метод может относиться к методу RIPP 29-90, описываемому в работе «Анализ нефтепродуктов, методы испытаний RIPP». Упомянутые результаты приведены в таблице 2.
Следующие методы были использованы для оценки кпд обессеривания этих адсорбентов. Старение: адсорбент был восстановлен в атмосфере водорода при давлении 0,1 МПа и температуре 420°С в течение 3 часов, до восстановления всего никеля, содержащегося в адсорбенте до нулевой валентности. Затем газовая смесь 10 об.% H2S+10 об.% Н2 + 80 об.% N2 была пропущена через адсорбент, который выдерживали при 420°С в течение 2 часов. Затем была осуществлена продувка азотом для удаления водорода из системы в течение 0,5 часа, с последующим внедрения воздуха в адсорбент и поддержанием адсорбента при 510°С в течение 2 часов для регенерации. Данный процесс восстановления-осеривания-окисления-регенерации повторяли три раза.
Для оценки кпд обессеривания было использовано устройство для микрореакции в стационарном слое. Материал для реакции адсорбции представляет собой каталитический крекинг-бензин, обладающий содержанием серы 800 промилле. Испытание на адсорбцию было осуществлено в атмосфере водорода при объемно-массовой скорости 4 ч-1. Содержание серы в бензине после реакции представлено в таблице 2.
Изобретение относится к области сероочистки. Адсорбент для удаления серы из крекинг-бензина или дизельного топлива содержит носитель, состоящий из источника кремнезема, связующее вещество на основе неорганического оксида, оксид металла, выбранный из группы IIB, и металл-катализатор, который пригоден для восстановления серы, находящейся в окисленном состоянии, до сероводорода. Адсорбент характеризуется значением η<0,5, где η является отношением количества металла-катализатора в кристаллической фазе (в процентах) к количеству металла-катализатора в адсорбенте (в процентах). Металл-катализатор, находящийся в адсорбенте в окисленном состоянии, предпочтительно равномерно диспергирован по поверхности носителя, образуя монослой. Предложен также способ приготовления адсорбента и способ его использования для удаления серы из жидкого потока. Изобретение обеспечивает повышенную активность адсорбента. 3 н. и 17 з.п. ф-лы, 1 ил., 2 табл., 5 пр.
1. Обессеривающий адсорбент, содержащий:
1) носитель, состоящий из источника кремнезема, связующего вещества на основе неорганического оксида и, по меньшей мере, одного оксида металла, выбранного из группы IIB;
2) по меньшей мере, один металл-катализатор, который пригоден для восстановления серы, находящейся в окисленном состоянии, до сероводорода и имеет значение η<0,5, где η=(количество металла-катализатора в кристаллической фазе, в процентах)/(количество металла-катализатора в адсорбенте, в процентах), в котором металл-катализатор представляет собой один или несколько металлов: никель, кобальт, марганец, железо, медь, серебро, молибден, хром, ванадий, вольфрам и лантаноиды;
причем адсорбент содержит примерно 1-40 мас.% источника кремнезема, примерно 10-80 мас.% оксидов металла, примерно 3-35 мас.% связующего вещества на основе неорганического оксида, в пересчете на оксиды, и примерно 3-30 мас.% металла-катализатора, в пересчете на оксиды, исходя из общей массы адсорбента.
2. Адсорбент по п.1, причем адсорбент содержит примерно 10-25 мас.% источника кремнезема, примерно 40-60 мас.% оксидов металла, примерно 10-18 мас.% связующего вещества на основе неорганического оксида, в пересчете на оксиды, и примерно 5-20 мас.% металла-катализатора, в пересчете на оксиды, исходя из общей массы адсорбента.
3. Адсорбент по п.1, в котором, по меньшей мере, один металл-катализатор, который пригоден для восстановления серы, находящейся в окисленном состоянии, до сероводорода, диспергирован по поверхности носителя практически в виде монослоя.
4. Адсорбент по п.1, в котором источник кремния выбран из одного или нескольких веществ: глины, диатомита, вспученного перлита, кремнистой породы, гидролизного кремнезема, макропористого кремнезема и силикагеля.
5. Адсорбент по п.1, в котором связующее вещество на основе неорганического оксида представляет собой оксидное связующее вещество на основе кремнеземно-глиноземной смеси.
6. Адсорбент по п.1 или п.4, в котором связующее вещество на основе неорганического оксида выбрано из одного или нескольких веществ: глинозема, кремнезема и аморфной кремнеземно-глиноземной смеси.
7. Адсорбент по п.1, в котором оксид металла выбран из оксида ванадия, оксида цинка или оксида молибдена.
8. Адсорбент по п.1, в котором металл-катализатор включает в себя никель.
9. Способ получения адсорбента по п.1, включающий в себя стадии:
(1) приведения в контакт источника кремнезема, «предшественника» связующего вещества на основе неорганического оксида и оксида одного или нескольких металлов, выбранных из группы IIB, или их «предшественников», с последующим формованием и высушиванием смеси с образованием носителя;
(2) обработки носителя в псевдоожиженном слое с органическим соединением, содержащим металл-катализатор, пригодный для восстановления окисленной серы до сероводорода, который растворен в безводном инертном газе, причем металл-катализатор представляет собой один или несколько из следующих металлов: никель, кобальт, марганец, железо, медь, серебро, молибден, хром, ванадий, вольфрам и лантаноиды;
(3) высушивания и обжига «предшественника» адсорбента, полученного на стадии (2), для преобразования органического соединения, содержащего металл-катализатор, в оксид металла;
(4) восстановления «предшественника» адсорбента, обработанного на стадии (3), в восстановительной атмосфере, вследствие чего металл-катализатор по существу сам по себе присутствует в восстановленном состоянии, и адсорбент имеет значение η<0,5, где η= (количество в процентах металла-катализатора в кристаллической фазе, в процентах)/(количество металла-катализатора в адсорбенте, в процентах).
10. Способ по п.9, в котором источник кремнезема по этапу (1) выбирают из одного или нескольких веществ: глины, диатомита, вспученного перлита, кремнистой породы, гидролизного кремнезема, макропористого кремнезема и силикагеля.
11. Способ по п.9, в котором оксид металла выбран из оксида ванадия, оксида цинка или оксида молибдена, а «предшественники» оксидов металлов выбраны из сульфидов, сульфатов, гидроксидов, карбонатов, ацетатов и нитратов.
12. Способ по п.9, в котором «предшественник» связующего вещества на основе неорганического оксида по этапу (1) выбран из одного или нескольких веществ: глинозема, кремнезема и «предшественника» аморфной кремнеземно-глиноземной смеси.
13. Способ по п.9, в котором температура газа составляет примерно 50°C-200°C, а давление газа составляет примерно 1-5 атм.
14. Способ по п.9, в котором органическое соединение, содержащее металл-катализатор, представляет собой органическое соединение с давлением пара примерно 0,5-100 кПа при 298 К.
15. Способ по п.14, в котором органическое соединение, содержащее металл-катализатор, выбрано из одного или нескольких соединений: формиатов, ацетатов, карбонилов металла и нафтенатов.
16. Способ по п.9, в котором органическое соединение, содержащее металл-катализатор, представляет собой тетракарбонил никеля.
17. Способ по п.9, в котором металл-катализатор по этапу (4) восстанавливают в восстановительной атмосфере, получая, таким образом, компонент в виде металла-катализатора по существу в состоянии нулевой степени валентности.
18. Способ по п.9, в котором, по меньшей мере, один металл-катализатор, который пригоден для восстановления серы, находящейся в окисленном состоянии, до сероводорода, присутствующего в адсорбенте, полученном на стадии (4), диспергирован по поверхности носителя практически в виде монослоя.
19. Способ обессеривания крекинг-бензина или дизельного топлива, включающий в себя приведение в контакт серы, содержащей материал, с адсорбентом по п.1, в ходе которого сера, содержащаяся в веществе, адсорбируется в адсорбент, и, таким образом, получается продукт, обладающий низким содержанием серы.
20. Способ по п.19, в котором условия контактирования включают в себя: температуру примерно 350-500°C; объемно-массовую скорость примерно 2-8 ч-1; и давление примерно 1000-5000 кПа.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| СПОСОБ СНИЖЕНИЯ ОБЩЕГО СОДЕРЖАНИЯ СЕРЫ В ГАЗАХ, ВКЛЮЧАЮЩИХ СЕРОВОДОРОД И ДРУГИЕ СОДЕРЖАЩИЕ СЕРУ КОМПОНЕНТЫ | 1997 |
|
RU2177361C2 |
| ДЕСУЛЬФИРОВАНИЕ И СОРБЕНТЫ ДЛЯ ЭТОГО | 2002 |
|
RU2309795C2 |

