Область техники
[0001] Настоящее изобретение относится к способу получения производных феноксипиридина, применимых в качестве противоопухолевого средства и ингибитора метастаза рака, обладающих ингибирующей активностью в отношении рецептора фактора роста гепатоцитов (даже упоминают как ″HGFR″), противоопухолевой активностью, ингибирующей активностью в отношении ангиогенеза, ингибирующей активностью в отношении метастазирования рака или им подобной.
Уровень техники
[0002] Патентный источник 1 раскрывает производное феноксипиридина, обладающего ингибирующей активностью в отношении к HGFR, и которое применимо в качестве противоопухолевого средства, ингибитора ангиогенеза или ингибитора метастазирования рака,
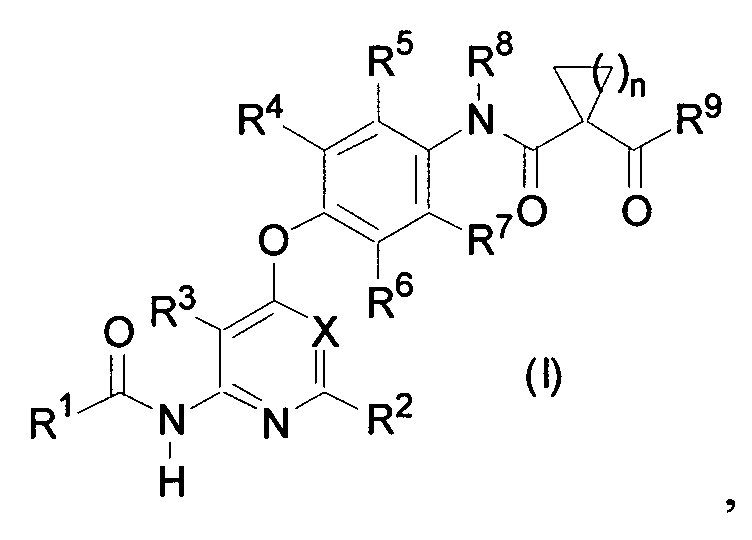
где R1 представляет собой 3-10-членную неароматическую гетероциклическую группу или ей подобную, R2 и R3 каждый представляет собой атом водорода, R4, R5, R6 и R7 могут быть одинаковыми или различными и каждый представляет собой атом водорода, атом галогена, C1-6 алкильную группу или подобные им, R8 представляет собой атом водорода или подобный ему, R9 представляет собой 3-10-членную неароматическую гетероциклическую группу или подобную ей, n представляет собой целое число 1 или 2 и X представляет собой группу, представленную формулой -CH= или атомом азота.
[0003] В качестве способа получения такого производного феноксипиридина патентный источник 1 раскрывает в примере 48 следующую реакцию в N,N-диметилформамиде в присутствии триэтиламина и бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфата:
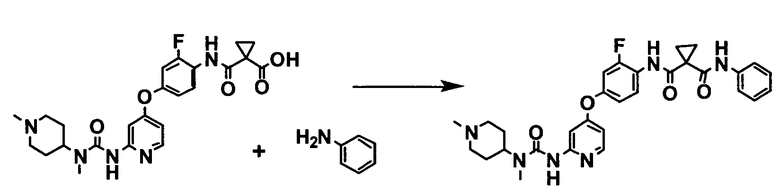
[0004] Патентный источник 2 описывает способ получения, в котором производное анилина и производное карбоновой кислоты реагируют в присутствии конденсирующего средства, как способ получения производного феноксипиридина, подходящий для промышленного крупномасштабного синтеза,
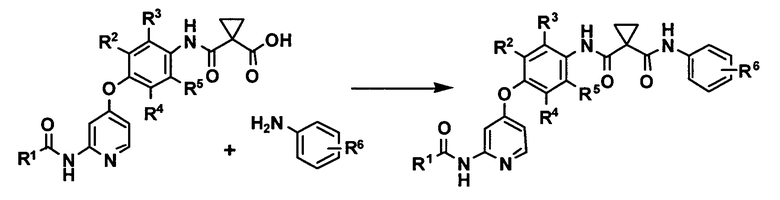
где R1 представляет собой азетидин-1-ильную группу, необязательно замещенную каким-либо заместителем или подобным ему, R2, R3, R4 и R5 могут быть одинаковыми или различными и каждый представляет собой атом водорода или атом фтора, и R6 представляет собой атом водорода или атом фтора.
[0005] Патентный источник 3 раскрывает способ получения, изображенный на следующей схеме, в качестве другого способа получения производного феноксипиридина:
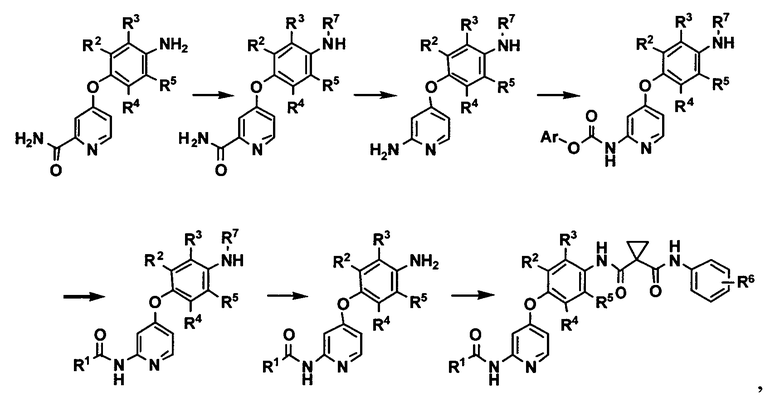
где R1 представляет собой 4-(4-метилпиперазин-1-ил)пиперидин-1-ильную группу или 3-гидроксиазетидин-1-ильную группу, R2, R3, R4 и R5 могут быть одинаковыми или различными и каждый представляет собой атом водорода или атом фтора при условии, что два или три из R2, R3, R4 и R5 представляют собой атом водорода, R6 представляет собой атом водорода, R6 представляет собой атом водорода или атом фтора, R7 представляет собой защитную группу для аминогруппы, а Ar представляет собой фенильную группу или ей подобную.
Перечень ссылок
Патентный источник
[0006] Патентный источник 1: WO 2007/023768.
Патентный источник 2: WO 2008/026577.
Патентный источник 3: WO 2009/104520.
Краткое описание изобретения
Техническая проблема
[0007] Однако способы получения, описанные выше в патентных источниках 2 и 3, применяемые в качестве промышленных способов получения, никогда не являлись полностью удовлетворительными при достижении более высокого выхода, более высокой чистоты и более короткого времени реакции. Кроме того, в последние годы внимание привлекали экологически чистые технологии, так называемые технологии зеленой химии, как, например, применение минимально возможного количества органического растворителя, однако способы получения, описанные в приведенных выше патентных источниках 2 и 3, не являлись в достаточном объеме экологически безопасными.
Решение проблемы
[0008] Таким образом, настоящие авторы предприняли значительные усилия и обнаружили, что изобретение по настоящей заявке, которое представляет собой способ получения, более пригодно для способа промышленного производства и экологически безопасно.
[0009] Более конкретно, настоящее изобретение обеспечивает:
(1) способ получения соединения или его соли, представленного формулой (I)
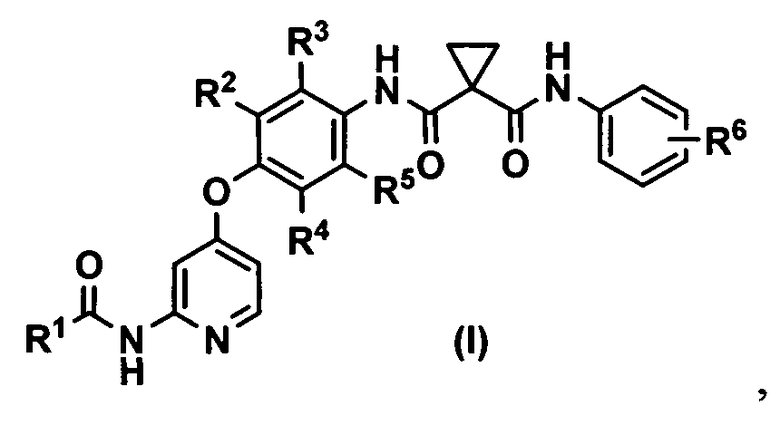
где R1 представляет собой 4-[3-(диметиламино)азетидин-1-ил]пиперидин-1-ильную группу, 4-(4-метилпиперазин-1-ил)пиперидин-1-ильную группу, 3-гидроксиазетидин-1-ильную группу или метил(1-метилпиперидин-4-ил)аминогруппу, R2, R3, R4 и R5 могут быть одинаковыми или различными и каждый представляет собой атом водорода или атом фтора, R6 представляет собой атом водорода или атом галогена, включающий реакцию соединения или его соли, представленного формулой (II)
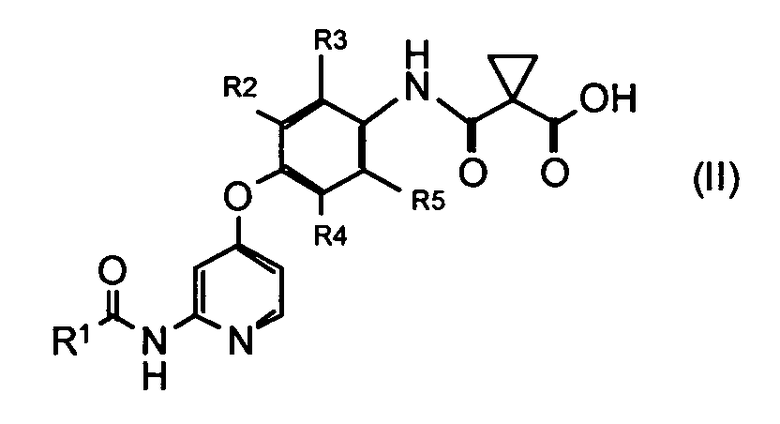
и производного анилина, представленного формулой (III)
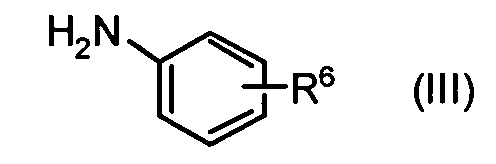
в воде или смешанном растворителе из воды и органического растворителя в присутствии 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида и, главным образом, при отсутствии основания.
(2) Способ получения согласно (1), где R1 представляет собой 4-(4-метилпиперазин-1-ил)пиперидин-1-ильную группу.
(3) Способ получения согласно (1) или (2), где каждый R2, R4 и R5 представляет собой атом водорода и R3 представляет собой атом фтора.
(4) Способ получения по любому из (1)-(3), где производное анилина, представленное формулой (III), представляет собой 4-фторанилин.
Полезные эффекты изобретения
[0010] Применяют способ получения производного феноксипиридина согласно настоящему изобретению, который в сравнении с общепринятыми способами получения обеспечивает более высокий выход и требует меньшее количество органического растворителя.
Описание вариантов осуществления
[0011] Будут определены символы и термины, используемые в данном документе, и настоящее изобретение будет подробно описано ниже.
[0012] Структурные формулы соединений в настоящем описании для удобства могут представлять только определенные изомерные формы, но изобретение охватывает все изомеры, такие как геометрические изомеры, оптические изомеры на основе ассиметричных атомов углерода, стереоизомеры, таутомеры и смеси этих изомеров, которые образуются из структур соединений, не будучи ограниченными любой из формул, показанных для удобства, и могут быть либо одним из изомеров, либо их смесью.
[0013] Поэтому соединения настоящего изобретения, представленные в молекулярной и в оптически активной или рацемической форме, иногда могут содержать асимметричные атомы углерода, однако настоящее изобретение не ограничено какой-либо одной, а включает обе. Также не существует ограничений при существовании их полиморфных кристаллических форм, и соединения могут находиться в одной кристаллической форме или в смеси различных кристаллических форм. Кроме того, также включены ангидриды и гидраты соединений настоящего изобретения.
[0014] Способ получения согласно настоящему изобретению подробно описан ниже.
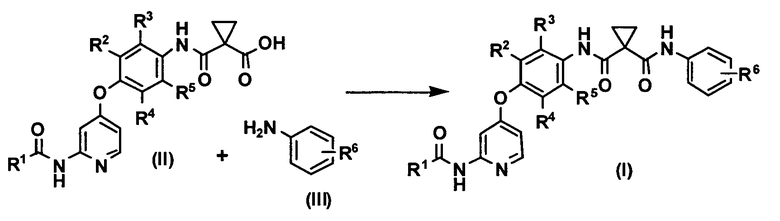
[0015] Настоящая реакция представляет собой реакцию получения соединения или его соли, представленного формулой (I), путем реакции соединения, представленного формулой (II), или его соли и производного анилина, представленного формулой (III), в воде или смешанном растворителе из воды и органического растворителя в присутствии 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида и, главным образом, при отсутствии основания.
[0016] Соединения или их соли, представленные формулой (II), могут быть получены из общеизвестных соединений, коммерчески доступных соединений или соединений, которые обычно легко получают при помощи способов, применяемых специалистами в данной области техники, из коммерчески доступных соединений.
[0017] Производные анилина, представленные формулой (III), могут быть получены из известных соединений, коммерчески доступных соединений или соединений, которые обычно легко получают при помощи способов, применяемых специалистами в данной области техники, из коммерчески доступных соединений.
[0018] Растворитель, применяемый для данного этапа, может представлять собой, как описано выше, воду в чистом виде или смешанный растворитель из воды и водорастворимого органического растворителя (например, тетрагидрофуран (THF), или ацетонитрил, или им подобные). Предпочтительно, чтобы растворителем была вода, используемая в чистом виде.
[0019] Температура реакции, в целом, будет различной, в зависимости от исходных материалов, растворителя и других реагентов, применяемых в реакции, и составляет предпочтительно 0-50°C, более предпочтительно 0-30°C.
[0020] Время реакции будет, как правило, отличаться, в зависимости от исходных материалов, растворителя и других реагентов, применяемых в реакции, и составляет предпочтительно от 10 минут до 12 часов, более предпочтительно от 30 минут до 6 часов.
[0021] Производное анилина, представленное формулой (III), может быть использовано в количестве 1,0-3,0-кратного молярного эквивалента, предпочтительно 1,0-1,5-кратного молярного эквивалента по отношению к соединению или его соли, представленного формулой (II).
[0022] 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (EDC) может быть использован в количестве 1,0-3,0-кратного молярного эквивалента, предпочтительно 1,0-1,5-кратного молярного эквивалента, по отношению к соединению или его соли, представленного формулой (II).
[0023] Как правило, если амин и карбоновая кислота вступают в реакцию с применением EDC для образования амидной связи, тогда должно присутствовать основание. Однако неожиданно было обнаружено, что настоящая реакция эффективно протекает, главным образом, при отсутствии основания, без снижения выхода и увеличения времени реакции. Выражение ″главным образом, при отсутствии основания″, как указано в настоящем изобретении, означает, что основание отсутствует при реакции или основание может содержаться в таком объеме, в котором оно не оказывает существенного влияния на реакцию настоящего изобретения. Примеры ″основания″ в данном документе включают неорганические основания, такие как гидроксид лития, гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, карбонат цезия, гидрокарбонат натрия, гидрокарбонат калия и карбонат цезия; металлорганические реагенты, такие как бутиллитий, метиллитий, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия и бис(триметилсилил)амид калия; гидриды, такие как гидрид лития, гидрид натрия и гидрид калия; гетероциклы, такие как имидазол, пиридин и 4-диметиламинопиридин; и органические амины, такие как триэтиламин и N,N-диизопропилэтиламин.
[0024] Кроме того, настоящая реакцию может быть проведена с учетам условий реакции, послереакционных стадий, способов очистки и т.п., описанных примерах, которые описывают далее.
[0025] Соли соединения, представленного формулой (I), и соли соединения, представленного формулой (II), в частности, не ограничены, и примеры включают соль неорганической кислоты, соль органической кислоты, соль неорганического основания, соль органического основания и соль с кислой или основной аминокислотой.
[0026] Предпочтительно соль неорганической кислоты включает, например, соль соляной кислоты, бромоводородной кислоты, серной кислоты, азотной кислоты или фосфорной кислоты.
Предпочтительно соль органической кислоты включает, например, соль уксусной кислоты, янтарной кислоты, фумаровой кислоты, малеиновой кислоты, винной кислоты, лимонной кислоты, молочной кислоты, стеариновой кислоты, бензойной кислоты, метансульфоновой кислоты, этансульфоновой кислоты или п-толуолсульфоновой кислоты.
[0027] Предпочтительно соль неорганического основания включает, например, соль щелочного металла, такую как соль натрия и соль калия, соль щелочноземельного металла, такую как соль кальция и соль магния, соль алюминия или соль аммония. Предпочтительно соль органического основания включает, например, соль диэтиламина, диэтаноламина, меглумина или N,N-дибензилэтилендиамина.
[0028] Предпочтительно соль с кислой аминокислотой включает, например, соль аспарагиновой кислоты или глутаминовой кислоты. Предпочтительно соль основной аминокислоты включает в себя, например, соль аргинина, лизина или орнитина.
[0029] Соответствующие заместители соединения, представленного формулой (I), формулой (II) или формулой (III), будут описаны ниже.
[Определение R1]
[0030] R1 представляет собой 4-[3-(диметиламино)азетидин-1-ил]пиперидин-1-ильную группу, 4-(4-метилпиперазин-1-ил)пиперидин-1-ильную группу, 3-гидроксиазетидин-1-ильную группу или метил(1-метилпиперидин-4-ил)аминогруппу. Структурная формула каждой группы показана ниже:
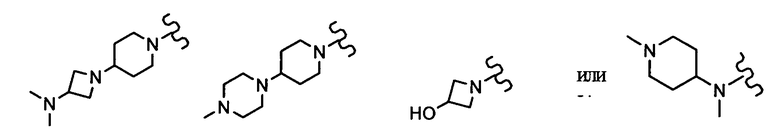
Предпочтительно, чтобы R1 был 4-(4-метилпиперазин-1-ил)пиперидин-1-ильной группой.
[Определение R2, R4 и R5]
[0031] R2, R3, R4 и R5 могут быть одинаковыми или различными и каждый представляет собой атом водорода или атом фтора. Все из R2, R3, R4 и R5 могут быть атомом водорода, все они могут быть атомом фтора или некоторые из них могут быть атомом водорода, а остальные могут быть атомом фтора, но предпочтительно, чтобы каждый R2, R3, R4 и R5 был атомом водорода и R3 был атомом фтора.
[Определение R6]
[0032] R6 представляет собой атом водорода или атом галогена. Атом галогена означает атом фтора, атом хлора, атом брома или атом йода. Положение R6 в производном анилина, представленного формулой (III), может быть любым из орто-положений, мета-положений и пара-положений по отношению к аминогруппе. R6 предпочтительно представляет собой атом галогена, более предпочтительно атом фтора, более предпочтительно атом фтора, присоединенный в пара-положении относительно аминогруппы.
Пример
[0033] Примеры настоящего изобретения пояснены ниже, но настоящее изобретение не ограничено данными примерами.
Пример 1: Синтез N-(2-фтор-4-{[2-({[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]карбонил}амино)пиридин-4-ил]окси}фенил)-N′-(4-фторфенил)циклопропан-1,1-дикарбоксиамида
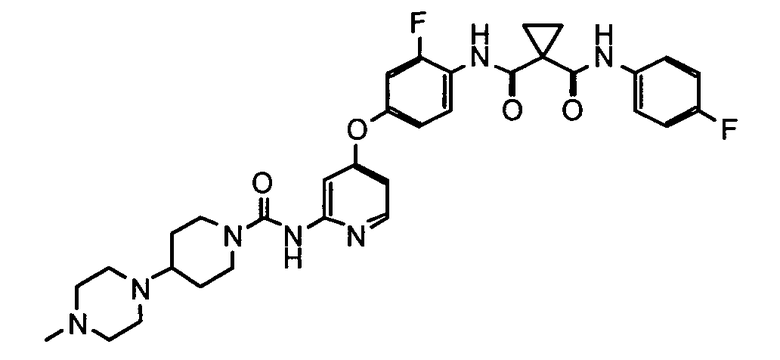
1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида (493,2 г) добавляли при 15°C к водному (7,5 кг) раствору дигидрата тригидрохлорида 1-[2-фтор-4-(2-{[4-(4-метилпиперазин-1-ил)пиперидин-1-карбонил]амино}пиридин-4-илокси)фенилкарбамоил]циклопропанкарбоновой кислоты (1495,5 г) и 4-фторанилина (250 г) и перемешивали в течение 1 часа. Ацетон (6,95 кг) и водный раствор 5N гидроксида натрия (1,92 кг) добавляли к реакционной смеси, pH доводили до щелочного и добавляли к ней затравку кристаллов (1,5 г) N-(2-фтор-4-{[2-({[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]карбонил}амино)пиридин-4-ил]окси}фенил)-N′-(4-фторфенил)циклопропан-1,1-дикарбоксиамид, полученную по способу, описанному в WO 2008/026577. Реакционную смесь перемешивали в течение 16,5 часов при 15°C и после подтверждения осаждения кристаллов добавляли вод (8,82 кг), и затем смесь перемешивали в течение 23 часов. Полученное твердое вещество собирали путем фильтрования и сушили для получения 1,29 кг титульного соединения.
[0034] Применяя способ, аналогичный примеру 9 (способ 3), описанный в WO 2008026577, как сравнительный пример, время реакции конденсации, ее выход и чистоту сравнивали с таковыми способа, описанного в примере 1, и оба они приведены в таблице ниже. Чистота означает числовую величину, полученную путем деления площади пика целевого соединения на сумму всех площадей пиков в анализе высокоэффективной жидкостной хроматографией (ВЭЖХ).
[0035] Соединение, представленное формулой (II), используемое в изобретении настоящей заявки в качестве исходного вещества, может быть альтернативно синтезировано с применением следующих справочных примеров.
[0036] (Справочный пример 1)
Сложный бензиновый эфир
1[4-(2-карбамоилпиридин-4-илокси)-2-фторфенилкарбамоил]циклопропанкарбоновой кислоты
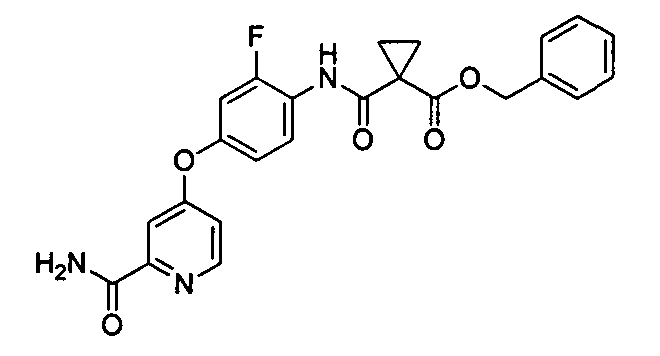
Тионил хлорид добавляли при -20°C к раствору 1-(бензилоксикарбонил)циклопропанкарбоновой кислоты (97,98 г) в тетрагидрофуране (200 мл) и N-метилпирролидоне (200 мл) и перемешивали в течение 30 минут. Раствор 4(4-амино-3-фторфенокси)пиридин-2-карбоксиамида (100 г) и N-металморфолина(40,91 г) в N-метилпирролидоне (600 мл) добавляли к реакционной смеси при такой же температуре и перемешивали в течение 1 часа при 10°C. Воду (100 г) добавляли к реакционной смеси и перемешивали в течение 1 часа при 20°C. После подтверждения осаждения кристаллов добавляли водный раствор (1 л) 1N гидроксида натрия и затем перемешивали в течение 19 часов. Осажденное твердое вещество собирали путем фильтрования и промывали водой (500 г). Полученное твердое вещество сушили в течение 20 часов при 60°C, чтобы получить 176,7 г титульного соединения.
[0037] (Справочный пример 2)
Дигидрат тригидрохлорида
1-[2-фтор-4-(2-{[4-(4-метилпиперазин-1-ил)пиперидин-1-карбонил]амино}пиридин-4-илокси)фенилкарбамоил]циклопропанкарбоновой кислоты
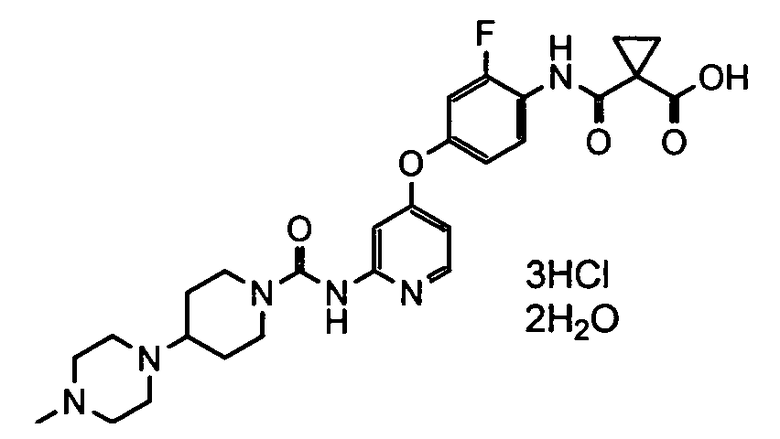
Водный раствор (40 мл) 5N гидроксида натрия добавляли при комнатной температуре к раствору тригидрохлорида сложного бензинового эфира 1-[2-фтор-4-(2-{[4-(4-метилпиперазин-1-ил)пиперидин-1-карбонил]амино}пиридин-4-илокси)фенилкарбамоил]циклопропанкарбоновой кислоты (20 г) в тетрагидрофуране (200 мл) и перемешивали в течение 2 часов и 42 минут. После окончания реакции реакционную смесь отстаивали и разделяли слои. Полученный органический слой добавляли по каплям в течение 1 часа и 5 минут при комнатной температуре к суспензии раствора ацетона (200 мл), воды (20 мл), 35% соляной кислоты (16 мл) и затравки кристаллов (0,2 г) дигидрата тригидрохлорида 1-[2-фтор-4-(2-{[4-(4-метилпиперазин-1-ил)пиперидин-1-карбонил]амино}пиридин-4-илокси)фенилкарбамоил]циклопропанкарбоновой кислоты, полученных по способу, описанному в примере 8-2 в WO 2008/026577. Смесь перемешивали в течение 2 часов и 28 минут при комнатной температуре и после подтверждения осаждения кристаллов добавляли ацетон (100 мл) и затем смесь перемешивали в течение 1 часа и 25 минут Полученное твердое вещество собирали путем фильтрования и сушили, чтобы получить 19,2 г титульного соединения.
[0038] (Справочный пример 3)
Сложный бензиновый эфир 1-[2-фтор-4-(2-феноксикарбониламинопиридин-4-илокси)фенилкарбамоил]циклопропанкарбоновой кислоты
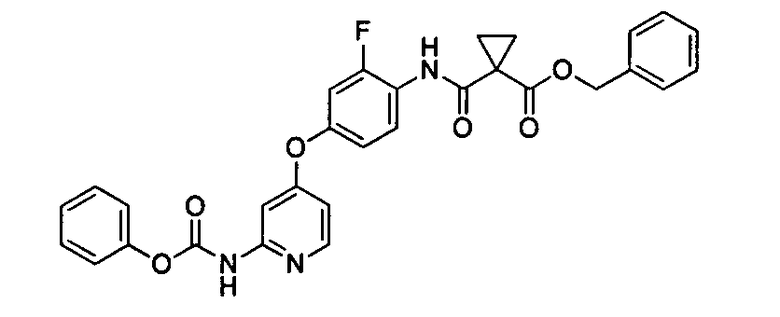
Пиридин (50 мл) смешивали с тетрагидрофураном (40 мл) и охлаждали до 10°C. Раствор фенилхлорформиата (4,83 г) в ацетонитриле (50 мл) и раствор бензинового эфира 1-[4-(2-аминопиридин-4-илокси)-2-фторфенилкарбамоил]циклопропанкарбоновой кислоты (10,00 г) в тетрагидрофуране (80 мл) добавляли по каплям к раствору тетрагидрофуран-пиридина в течение 2 часов при 10°C. На данном этапе количества раствора фенилхлорформиат-ацетонитрил и раствора тетрагидрофуран/сложный бензиновый эфир 1-[4-(2-аминопиридин-4-илокси)-2-фторфенилкарбамоил]циклопропанкарбоновой кислоты, добавляемого по каплям, доводили до постоянной скорости так, чтобы каждый раствор добавляли по каплям в течение 2 часов. В то время как растворы смешивали по каплям, кристаллы выпадали в осадок. После завершения добавления по каплям окончание реакции подтверждали с применением устройства ВЭЖХ. Затем добавляли смешанный раствор воды (10 мл) и ацетонитрила (50 мл) к реакционной смеси. Кристаллы собирали путем фильтрования и промывали смешанным растворам тетрагидрофурана (15 мл) и ацетонитрила (15 мл) и сушили при пониженном давлении, чтобы получить титульное соединение (12,74 г). Наблюдаемый выход 98,0%, чистота 94,6% (процентное отношение площади ВЭЖХ).
[0039] (Справочный пример 4)
Сложный бензиновый эфир 1-[2-фтор-4-(2-феноксикарбониламинопиридин-4-илокси)фенилкарбамоил]циклопропанкарбоновой кислоты
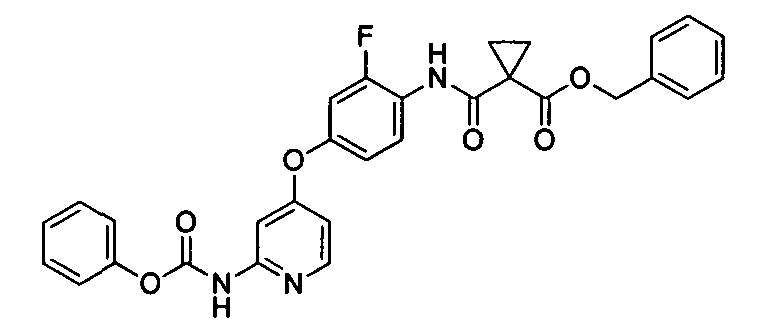
Сложный бензиновый эфир 1-[4-(2-аминопиридин-4-илокси)-2-фторфенилкарбамоил]циклопропанкарбоновой кислоты (176,0 кг), тетрагидрофуран (1876 кг), ацетонитрил (692 кг) и пиридин (860 кг) смешивали и охлаждали до 10°C. Фенилхлорформиат (105 кг) добавляли по каплям к смешанному раствору в течение 1 часа при 10°C. В то время как раствор добавляли по каплям, кристаллы выпадали в осадок. После завершения добавления по каплям окончание реакции подтверждали используя устройство ВЭЖХ. Затем к реакционной смеси добавляли ацетонитрил (692 кг). Кристаллы собирали путем фильтрования, промывали смешанным раствором тетрагидрофурана (234 кг) и ацетонитрила (207 кг) и сушили при пониженном давлении, чтобы получить неочищенные кристаллы бензилового эфира 1-[2-фтор-4-(2-феноксикарбониламинопиридин-4-илокси)фенилкарбамоил]циклопропанкарбоновой кислоты (202,9 кг). Неочищенные кристаллы (202,9 кг), ацетонитрил (638 кг) и воду (812 л) перемешивали в течение 1 часа при 20°C. Кристаллы собирали с помощью фильтрования, промывали ацетонитрилом (319 кг) и сушили при пониженном давлении, чтобы получить указанное соединение (174,8 кг). Наблюдаемый выход 77,1%, чистота 93,9% (процентное отношение площади ВЭЖХ).
[0040] (Справочный пример 5)
Сложный бензиловый эфир 1-[4-(2-карбамоилпиридин-4-илокси)-2-фторфенилкарбамоил]циклопропанкарбоновой кислоты
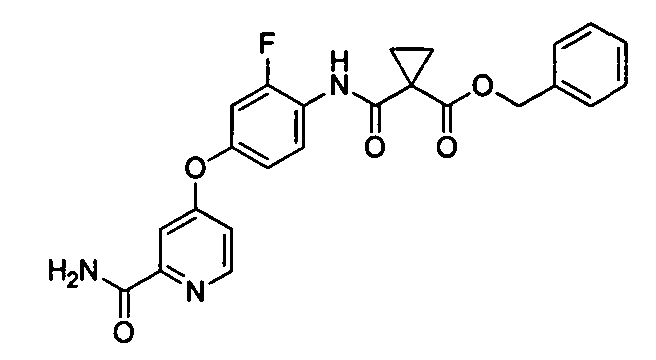
1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида (1,16 г) добавляли при комнатной температуре к раствору 4-(4-амино-3-фторфенокси)пиридин-2-карбоксамида (1,0 г) и 1-(бензилоксикарбонил)циклопропанкарбоновой кислоты (1,07 г) в ацетоне (10 мл), воде (10 мл) и 5N растворе соляной кислоты (1,4 мл) и перемешивали в течение 5 минут. После подтверждения протекания реакции полученное твердое вещество собирали путем фильтрования, промывали водой (5 мл) и сушили для получения 1,52 г титульного соединения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ АМИДЫ, СОДЕРЖАЩИЕ НАСЫЩЕННУЮ СВЯЗЫВАЮЩУЮ ГРУППУ, И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2006 |
|
RU2412181C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2267489C2 |
| ПИРРОЛИДИНИЛАЛКИЛАМИДНЫЕ ПРОИЗВОДНЫЕ, ИХ ПОЛУЧЕНИЕ И ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛИГАНДОВ РЕЦЕПТОРА CCR3 | 2009 |
|
RU2514824C2 |
| ПРОИЗВОДНОЕ ПИРИМИДИН-4,6-ДИАМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2834353C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2020 |
|
RU2835082C2 |
| ПРОИЗВОДНЫЕ 2-СУЛЬФАНИЛБЕНЗИМИДАЗОЛ-1-ИЛУКСУСНОЙ КИСЛОТЫ В КАЧЕСТВЕ АНТАГОНИСТОВ CRTH2 | 2005 |
|
RU2409569C2 |
| ПРОИЗВОДНЫЕ [1,2,4]ТРИАЗОЛО[1,5-a]ПИРИМИДИН-2-ИЛМОЧЕВИНЫ И ЕГО ПРИМЕНЕНИЕ | 2004 |
|
RU2348636C2 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ И АНАЛОГИ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2019 |
|
RU2797316C2 |
| ПРОИЗВОДНОЕ ПИРИМИДИНА С ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ПРОТЕИНКИНАЗ И СОДЕРЖАЩАЯ ЕГО ТЕРАПЕВТИЧЕСКАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2022 |
|
RU2833427C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ИНГИБИРОВАНИЯ ТИРОЗИНКИНАЗНОЙ АКТИВНОСТИ | 2011 |
|
RU2598852C2 |
Изобретение относится к способу получения соединения или его соли, представленного формулой (I), который включает реакцию соединения или его соли, представленного формулой (II), и производного анилина, представленного формулой (III), в воде или смешанном растворителе из воды и органического растворителя в присутствии 1-этил-3-(3-диметиламинопропил)карбодиимида и, главным образом, при отсутствии основания:
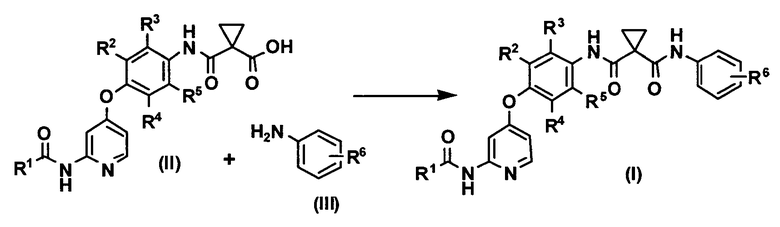 .
.
Технический результат: разработан новый улучшенный способ получения соединения или его соли, представленного формулой (I). 1 табл., 5 пр., 2 з.п. ф-лы.
1. Способ получения соединения или его соли, представленного формулой (I)
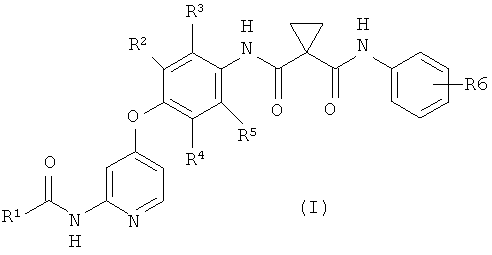 ,
,
где R1 представляет собой 4-(4-метилпиперазин-1-ил)пиперидин-1-ильную группу,
R2, R3, R4 и R5 могут быть одинаковыми или различными, и каждый представляет собой атом водорода или атом фтора, и
R6 представляет собой атом водорода или атом галогена,
который включает введение в реакцию соединения или его соли, представленного формулой (II)
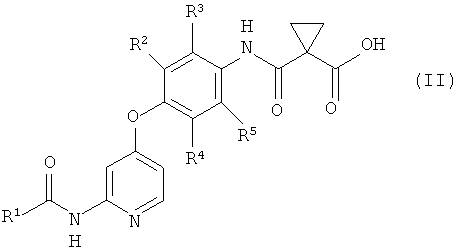 ,
,
и производного анилина, представленного формулой (III)
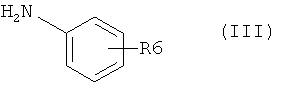 ,
,
в воде или смешанном растворителе из воды и органического растворителя в присутствии 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида и, по сути, при отсутствии основания.
2. Способ получения по п.1, где каждый из R2, R4 и R5 представляет собой атом водорода и R3 представляет собой атом фтора.
3. Способ получения по п.1, где производное анилина, представленное формулой (III), представляет собой 4-фторанилин.
| WO2008026577 A1, 06.03.2008 ? | |||
| НОВОЕ ПРОИЗВОДНОЕ ПИРИДИНА И ПРОИЗВОДНОЕ ПИРИМИДИНА (3) | 2006 |
|
RU2362771C1 |
| WO2008102870 A1, 28.08.2008 | |||
| НОВЫЕ ПИРИДИНОВОЕ ПРОИЗВОДНОЕ И ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ (1) | 2005 |
|
RU2330021C2 |
| WO2009104520 A1, 27.08.2009 | |||
| МОНОЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛЫ, ИНГИБИРУЮЩИЕ КИНАЗУ | 2005 |
|
RU2350603C2 |
| WO2010051373 A1, 06.05.2010. | |||

