Изобретение относится к способам обработки соединений на основе карбида урана общей формулы UCx, которые могут применяться в качестве мишеней в любых устройствах, использующих карбиды урана, где их необходимо стабилизировать после использования, и особенно в исследовательских ускорителях (где такие мишени после использования считаются отходами), которые должны удовлетворять установленным управлениями по ядерной безопасности критериям приемлемости, основанным, в частности, на их химической стабильности при нормальных условиях хранения (при температуре и давлении окружающей среды).
Действительно, в исследовательских ускорителях мишени из UCx используются в качестве источника тяжелых ионов, в частности, в ускорителе GANIL (French National Heavy Ion Large Accelerator, ускорительный комплекс тяжелых ионов) в г. Кан, Франция, с установкой SPIRAL 2 (2-nd Generation In-Line Radioactive Ion Production System, система второго поколения для поточного производства радиоактивных ионов).
Вещество мишени UCx, которое может использоваться в работе исследовательских ускорителей, обычно синтезируют карботермическим восстановлением сверхстехиометрической смеси графита и порошка UO2 и затем прессуют для формирования гранул сантиметрового размера. Его структура обычно содержит две фазы: фазу дикарбида урана UC2, составляющую подавляющую часть материала мишени (порядка 90% по весу) и другую фазу, состоящую из свободного углерода, обозначенного CL, находящегося в форме графита. Что касается объемного распределения, то графит, объемная доля которого обычно составляет 70%, в конечном веществе UCx может иметь объемную долю, варьирующуюся от 0% до более чем 75%. В исходном веществе может также в очень небольших количествах (обычно менее 1%) обнаруживаться UC, который локально синтезируется на этапе карботермического восстановления.
Следует отметить, что реальное стабилизируемое вещество после облучения может содержать продукты распада/активации, такие как Co, Cs, B, Br, Kr, Zr, Rh и т.п.
Вообще говоря, способы стабилизации должны позволить удовлетворить следующим требованиям:
- конверсия вещества UCx в форму стабилизированного продукта типа UOx (U3O8, UO2, UO3 и т.п.) должна быть совместима с требованиями зон вывода/захоронения, предусмотренными управлениями по ядерной безопасности и ANDRA (Французское национальное агентство по обращению с радиоактивными отходами);
- применение способа стабилизации веществ UCx посредством особой окислительной термообработки должно дать возможность управлять реакцией окисления, являющейся сильно экзотермической, и устранять любые явления неконтролируемого нагрева во время реакции;
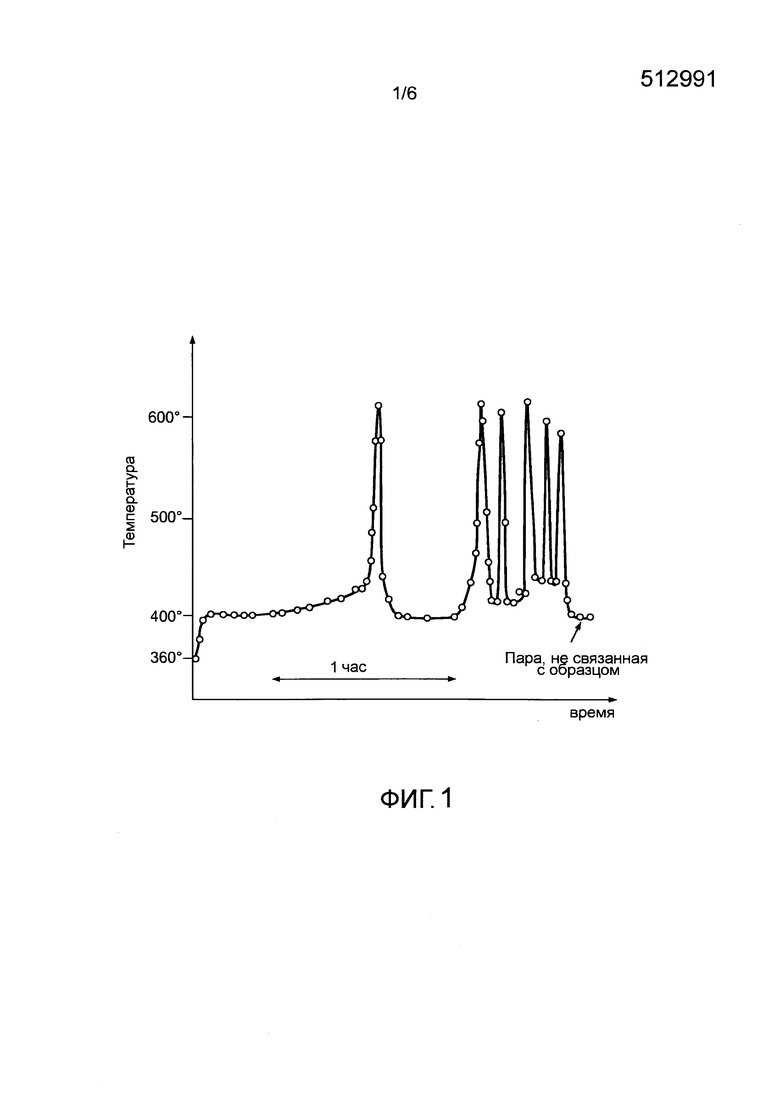
- удержание химической активности вещества в пределах параметрического и ограниченного диапазона (ограничение неконтролируемого повышения температуры, селективность реакции окисления, контроль температуры воспламенения в ходе окисления веществ UCx для предотвращения неравномерной работы. Так, фигура 1 иллюстрирует неожиданное и неконтролируемое восстановление реакционной способности и более конкретно - пример неконтролируемого нагрева, характеризующийся псевдопериодическим перегревом при окислении образца металлического урана при 390°C (Yves Adda, Kinetic study of the oxidation, nitridation and hydridation of uranium, отчет Французской комиссии по атомной энергии № 757, 1958);
- возможность минимизировать образование газообразных выбросов и отходов, которые всегда дорого устранять и присутствие которых в ядерных средах должно быть ограничено, посредством использования оптимального рабочего диапазона в процессе, что дает возможность полностью стабилизировать одну только фазу UC2, в то же время предотвращая окисление избытком свободного углерода, присутствующего в исходном веществе UCx. Конечной целью является осуществление способа в радиационной обстановке (экранированной ячейке) посредством простого способа обработки, при котором не образуются жидкие отходы;
- подтверждение отсутствия реакционной способности продуктов после их стабилизации в окисленной форме, причем полученное вещество должно быть стабильно в отношении химических реакций с воздухом в условиях температуры и давления внешней среды;
- применение процесса стабилизации, совместимого с полупромышленными рабочими режимами: уменьшенное время обработки, надежность способа, в частности, в отношении изменения входных параметров (масса вещества, плотности, пористости, фазы) и управляемый мониторинг индикаторов во время процесса.
В настоящее время использованные UCx-мишени хранятся в ожидании подходящей выгрузки и/или процесса обработки; так, например, происходит в разделителе изотопов ISOLDE (Isotope Separator On Line Detector) в Женеве.
Химические способы переработки уже были описаны, в частности, в международной заявке на патент WO/2004/012206, которая описывает способ электрохимического окисления путем растворения. Предложенная обработка делает этот способ полностью несовместимым с UCx-веществом мишеней, поскольку при использовании способа образуется значительное количество жидких отходов (из-за химического растворения), что не соответствует целям настоящего изобретения.
Имеются также научные публикации, относящиеся к окислению содержащих уран карбидов типа UC/UC2, которые можно отнести к трем основным категориям в соответствии с природой используемого окислителя: диоксид углерода, жидкая вода или вода в форме пара и молекулярный кислород, в различных концентрациях.
Что касается реакций окисления карбидов актинидов с CO2, авторы Peakall K.A., Antill J.E. в статье Oxydation of Uranium Monocarbide, J. Less-Common Metals, 4 (1961), 426-435, сообщают об исследованиях окисления UC, проведенного в атмосфере диоксида кремния в качестве окислительного газа. Полученные результаты говорят о том, что способность химического взаимодействия карбидов с CO2 относительно мала и несовместима с промышленным способом (в частности по критерию времени обработки). Кроме того, Murbach et al., E.W. и G.E. Brand, 1965, в статье «Pyrochemical reprocessing of uranium carbide», Summary Report, Atomics International, p.38, наблюдали крайне изменчивую химическую активность в зависимости от морфологической структуры UC, которые приводят к незаконченным и неполным циклам окисления, что неприемлемо для намеченного приложения. В целом эти наблюдения, относящиеся к значительному замедлению кинетики окисления карбидов в присутствии CO2, несовместимы с указанными выше требованиями, предъявляемыми к переработке вещества, образованного из мишеней UCx, которые накладываются в целях ускорения конверсии.
Что касается реакций окисления карбидов актинидов водой в виде жидкости и в виде пара, несколько исследований, приведенных ниже в качестве примера, включая упомянутые в следующих документах: Bradley M., «Hydrolysis of Uranium Carbides between 25 and 100°C», II Uranium Dicarbide, Uranium Metal Monocarbide Mixtures and Uranium Monocarbide-Dicarbide Mixtures, Inorganic Chemistry, 3 (1964), 189-195, Herrmann B., Herrmann, FJ., Kinetics of oxidation of uranium monocarbide by dry or humid oxygen, отчет Комиссии по атомной энергии Франции, 19 (1968), показывают, что карбиды реагируют с водой и водяным паром. Результаты сообщают, что водяной пар является важным фактором механизма окисления, и что предварительное воздействие воздуха или слабо окислительной влажной среды значительно повышает их реакционную способность. Следует отметить, что окислительная обработка карбидов водой в жидкой форме совершенно не пригодна для способа, предусмотренного для материалов, образованных из мишеней UCx, с точки зрения основных ограничений, связанных, в частности, с обработкой отходов, которые образуются в ходе обработки. Хотя присутствие водяного пара оказывает эффект повышения реакционной способности карбидов, а именно сверхстехиометрически, вследствие увеличения скорости превращения в оксидную фазу, представленные в указанных документах исследования окисления в неизотермической среде и в присутствии только водяного пара выявляют два главных недостатка в разработке способа, предназначенного для веществ на основе карбида урана, являющихся предметом способа стабилизации по настоящему изобретению, из-за:
- медленной конверсии карбидов в оксидную фазу в присутствии только водяного пара и в присутствии молекулярного кислорода в аналогичных условиях окисления;
- образования новых газообразных продуктов, как описано Litz, M. в Uranium Carbides: «Their Preparation, Structure and Hydrolysis», PhD Thesis, Ohio State University, NP-1453 (1945): CH4 (для UC), C2H6 (для UC2), и, в частности, образования в потенциально больших количествах молекулярного водорода H2 (или из UC, или из UC2), взрывоопасная природа которого угрожает безопасности способа. Отсюда следует, что результаты, полученные в присутствии водяного пара, преимущественно с UC, нельзя напрямую переносить на требования, установленные для вещества UCx, из-за описанных выше ограничений, а также из-за изменчивости входных характеристик (высокое содержание избыточного углерода, которое приводит к дополнительному увеличению концентрации H2 гидролизом/газификацией, если не будут приняты особые предосторожности).
Наконец, что касается реакций окисления UC и UC2 кислородом O2, было опубликовано много исследований об окислении урансодержащих карбидов в атмосфере молекулярного кислорода при различной концентрации. Однако следует обратить внимание, что эти исследования, за исключением работы Nawada H.P. et al., Thermogravimetrical study of oxidation behaviour of uranium dicarbide, Journal of Thermal Analysis, 35 (1989), 1145-1155, относятся к веществу UC стехиометрического состава, которое, следовательно, существенно отличается по своей природе и свойствам от многофазного вещества UCx, рассматриваемого в настоящем изобретении, состоящего из двух основных фаз (карбид урана и освободный углерод в форме графита). Единственные доступные данные, касающиеся стехиометрического UC2, также показывают другой тип поведения в отношении окисления из-за отсутствия свободного углерода, содержание которого также изменяется во время стабилизационной обработки в зависимости от параметрического диапазона и используемых рабочих условий.
Вообще говоря, при осуществлении окислительной термообработки можно выделить два основных пути:
- окислительная обработка карбидов проводится при переменных температурах (неизотермические условия);
- окислительная обработка карбидов проводится при фиксированной температуре (изотермические условия).
Неизотермические условия окисления несовместимы с применением способа стабилизации согласно настоящему изобретению, так как они не позволяют гарантировать стабильные, безопасные и воспроизводимые условия окисления. Действительно, постепенное увеличение температуры, применяемое во время обработки и, следовательно, поступление в систему энергии в форме тепла вызывает риск неконтролируемого нагрева и нестабильные условия окисления карбидов, что приводит к:
- неожиданному увеличению локальной температуры и кинетики окисления (как показано на фигуре 1);
- неконтролируемому нагреву в реакции и потенциальному спонтанному самовозгоранию вещества UCx (в особенности в форме порошка), сопровождающемуся выраженным экзотермическим пиком из-за высокой энтальпии реакции окисления порядка -1450 кДж/моль.
Для предотвращения этих явлений и осуществления процесса при уменьшении подачи окислителя, начиная с заранее заданного горючего сырья и с заранее заданной температуры активации (принцип безопасной работы, кроме того, важен для демонстрации управления процессом), должна быть предусмотрена окислительная обработка в изотермических условиях.
Кроме того, на поведение урансодержащих карбидов в отношении окисления значительно влияют такие структурные и морфологические различия, как:
- исходная природа вещества: UC при окислении ведет себя иначе, чем UC2 (есть отличие в прибавке массы), что справедливо также для UCx, богатого избыточным углеродом;
- морфология: у порошка существенно иная температура воспламенения, чем у одной или более гранул, имеющих заданные объемы и плотности (влияет, например, высота слоя порошка, обрабатываемая масса и т.п.).
Известные технологии окисления основаны, в частности, на нескольких исследованиях, проведенных с UC, полученным карботермическим восстановлением в порошковой форме в изотермических условиях, в частности, на исследовании, описанном в Ohmichi, T. (1968), «Oxidation of UC and UN Powder in Air», Journal of Nuclear Science and Technology, 5, 600-602. Подробный анализ результатов показывает, что эти данные не могут быть перенесены на вещества мишени типа UCx из-за нескольких ограничений: ограниченное исходное количество вещества (масса UC менее 30 мг), слишком высокий диапазон используемых температур окисления (вплоть до 1400°C), а также состав и геометрическая форма исходного карбида, которые отличаются: UC не дает при окислении такой прибавки массы, как UC2, и геометрия UC (порошок с размером частиц 150 мкм) нетипична для стабилизируемых мишеней из UCx, о которых идет речь в настоящем изобретении (сравнимая для большинства из них с геометрией пористых гранул сантиметрового размера).
В других исследованиях, проведенных с монолитным UC, как исследование Herrmann, Kinetics of oxidation of uranium monocarbide by dry or humid oxygen», French Atomic Energy Commission Report, 19 (1968), также показаны профили изменения массы, существенно отличающиеся от полученных для вещества мишени UCx из-за разницы в исходном содержании углерода в фазе карбида (на 60% большее увеличение массы при образовании одного и того же оксида U3O8 в случае UC, чем в случае UC2, окисленного в близких условиях).
В исследовании S.K. Mukerjee, G.A. R. Rao, J.V. Dehadraya, V.N. Vaidya, V. Venugopal, D.D. Sood (1994), «Oxidation of Uranium Monocarbide Microspheres», Journal of Nuclear Materials, 1, 97-106, и E.W. Murbach, G.E. Brand, 1965, «Pyrochemical Reprocessing of Uranium Carbide», Summary Report, p. 38, Atomics International, было также проанализировано влияние начальной массы UC (от 30 до 200 мг у Mukerjee и до 10 кг у Murbach) на кинетику окисления. Представленные результаты показывают, что они не могут быть перенесены на вещества на основе карбида согласно настоящему изобретению, так как исследования проводились не в изотермических условиях (Mukerjee), и образцы UC изначально были синтезированы методом дуговой плавки (Murbach), следовательно, они проявляли структурные свойства, в частности, удельную плотность, которые радикально отличаются от таковых у веществ на основе карбида урана по настоящему изобретению.
Некоторые факты, относящиеся к окислению UC2, и, следовательно, наиболее характерные для исследуемого способа, в отношении структурного состава мишеней из UCx, имеются в исследовании Nawada et al., Thermogravimetrical Study of Oxidation Behaviour of Uranium Dicarbide, Journal of Thermal Analysis, 35 (1989), 1145-1155. Окислительная обработка UC2 проводилась в неизотермических условиях, за которыми следовали длительные стационарные условия окисления продолжительностью от 4 до более 100 часов. Полный цикл окисления, следовательно, растягивался в целом на 118 часов. В полученных результатах можно выделить 4 этапа, чтобы было проще понять реакцию окисления UC2 до U3O8:
- первый этап, характеризующийся постепенным и очень медленным окислением UC2 до промежуточного оксида α-UO3 с увеличением массы более чем на порядка 19% для температур, меняющихся от 25 до 260°C;
- второй этап, характеризующийся окислением углерода, содержащегося в исходной фазе UC2, что приводит к потере массы примерно в два раза, при температурах от 260 до 410°C;
- третий этап, соответствующий окислению фазы α-UO3 до окисленной фазы U3O8, что также приводит к потере массы, в диапазоне температур от 410 до 560°C;
- четвертый и последний этап, который определяется окислением остаточного свободного углерода, который, как предполагается, присутствует в исходном веществе, при температурах окисления от 560 до 690°C, также сопровождающийся регистрацией потери массы.
Хотя в упомянутом исследовании приводятся данные для понимания окисления UC2, оно описывает факты, несовместимые с использованием способа конверсии UCx в U3O8 по нескольким причинам:
- неподходящий программируемый тепловой режим (сочетание неизотермических условий окисления, за которыми следуют длительные стационарные условия окисления), не удовлетворяющий применению окислительной термообработки с контролируемыми потенциальными изменениями реакционной способности, необходимыми для гарантии безопасности способа;
- чрезмерно высокое время окисления: полная продолжительность окисления UC2 в данном исследовании оценивается более чем в 118 часов, что делает его не совместимым с полупромышленной обработкой, которая требует применения способа для быстрой конверсии карбидных фаз в U3O8;
- недостаток входных данных, например, таких как масса исходного UC2 (не упоминается), или отсутствие описания физических свойств исходного вещества UC2 (плотности, пористости, геометрии гранул), что не гарантирует гибкости описываемой окислительной обработки. Приведенные в упомянутом исследовании данные показывают, кроме того, что вещество значительно отличается от указанного выше вещества UCx (а именно отличается стехиометрией свободного углерода в исходном веществе, что значительно меняет поведение в отношении окисления);
- отсутствие сравнительных данных, относящихся к химической активности UC2 на различных этапах окисления (энтальпия каждой промежуточной реакции окисления), а также к изменению измеренных выходных значений (полученная масса, полученный газ CO2) как функции входных параметров (массы, концентрации O2).
Отсутствие таких данных показывает, что это исследование, важное, в частности, для понимания механизма окисления UC2, не позволяет создать способ, поскольку оно несовместимо с требованиями безопасности способа стабилизации в отношении контроля термической нестабильности и контроля реакции окисления посредством управляемого введения парциального давления O2, управляемой скорости потока и подходящей массы. Кроме того, не описаны критерии, позволяющие гарантировать окончание реакции, за исключением обработки полностью при высокой температуре, что не является совместимым с целями/ограничениями настоящего изобретения.
Исходя из всех этих библиографических данных представлялется очевидным, что никакая окислительная термообработка не может быть применима к материалам, состоящим из мишеней из карбида урана, имеющим сверхстехиометрическую концентрацию углерода, так, чтобы гарантировать, что конверсия UCx в UOx будет проведена быстрым, безопасным и надежным способом окисления, соответствующим желаемым функциям, указанным выше.
Поэтому в настоящем изобретении предлагается решение комплексной проблемы стабилизации безопасным, управляемым, надежным и ускоренным способом композиционного материала, имеющего формулу UCx+yC, где действительное число x может быть больше или равно 1, а действительное число y больше 0.
Решение, предлагаемое в настоящем изобретении, позволяет разработать промышленный способ, учитывающий требования, связанные с необходимостью ограничить образование газообразных или жидких отходов (работа в радиационной обстановке внутри экранированных оболочек) и отвечающий требованиям, налагаемым управлениями по безопасности относительно конверсии UCx в отходы типа UOx (в основном U3O8), что достигается стабилизирующей обработкой, контролируемой в каждый момент реакции.
Более конкретно объектом настоящего изобретения является способ химической стабилизации соединения карбида урана, соответствующего формуле
UCx+yC, где x, y являются действительными числами, x≥1 и y>0, помещенного в стабилизационную камеру,
отличающийся тем, что он содержит следующие этапы:
- этап повышения температуры внутри указанной камеры до температуры «окисления» указанного соединения на основе карбида урана примерно от 380°C до 550°C, причем указанная камера заполнена инертным газом;
- этап изотермической окислительной обработки при указанной температуре окисления, причем указанная камера находится под парциальным давлением O2;
- этап контроля завершения стабилизации указанного соединения, включающий отслеживание количества израсходованного молекулярного кислорода и/или выделенного диоксида углерода, или выделенных диоксида углерода и моноксида по меньшей мере до достижения заданного входного значения молекулярного кислорода, или минимального порогового значения для указанного количества диоксида углерода, или пороговых значений для диоксида углерода и моноксида углерода.
Согласно одному альтернативному варианту осуществления изобретения, этап контроля завершения стабилизации дополнительно содержит отслеживание изменения массы твердых соединений на основе углерода и урана в камере, причем увеличение массы в текущий момент коррелирует с окислением карбида урана.
Согласно одному альтернативному варианту осуществления изобретения, этап контроля завершения стабилизации осуществляется путем повышения температуры внутри указанной камеры в интервале от указанной температуры окисления до температуры окисления углерода (не включая) и отслеживанием количества выделенного CO2.
Согласно одному альтернативному варианту осуществления изобретения, способ содержит введение парциального давления водяного пара в указанную камеру перед и/или во время этапа окисления. Предпочтительно дальше он может включать определение H2 как маркера для отслеживания окончания окисления в указанной камере.
Согласно одному альтернативному варианту осуществления изобретения, этап контроля окончания стабилизации проводится введением в указанную камеру (в предполагаемом конце обработки, то есть при снижении уровня CO2 ниже порогового значения) некоторого количества водяного пара при температуре окисления и отслеживанием содержания H2 в камере, при значении ниже порогового это позволяет установить окончание реакции окисления UCx.
Согласно одному альтернативному варианту осуществления изобретения, этап контроля завершения стабилизации содержит операцию создания избыточного давления реакционных газов, имеющихся в указанной камере, чтобы ускорить окончание реакции окисления указанного соединения.
Согласно одному альтернативному варианту осуществления изобретения, этап контроля завершения стабилизации дополнительно содержит цикл операции создания избыточного давления и операции снятия избыточного давления реакционных газов, имеющихся в указанной камере.
Согласно одному альтернативному варианту осуществления изобретения, способ содержит предварительный этап определения оптимальной температуры окисления посредством термогравиметрического анализа образца соединения UCx+yC.
Согласно одному альтернативному варианту осуществления изобретения, оптимальная температура окисления, которая меняется в зависимости от кондиционирования указанного карбида урана, находится между примерно 380°C и 550°C.
В способе по изобретению указанное соединение может иметь морфологию порошкового типа или типа пористой или сплошной гранулы.
Другим объектом настоящего изобретения является устройство химической стабилизации соединения карбида урана, имеющее камеру с печью для окисления, отличающееся тем, что оно содержит:
- модуль подачи газа, который позволяет создавать инертную атмосферу аргона, или азота, или частично окислительную атмосферу O2 и/или H2O с помощью внешнего контура подачи, причем потоки газа подаются в указанную печь для окисления;
- модуль электропитания, питающий печь для окисления, который подает к ней заданный поток, что делает возможным повышение температуры;
- указанную камеру, подающую выходящий поток газа к модулю управления и автоматического регулирования;
- указанный модуль управления и автоматического регулирования, содержащий первый модуль измерения температуры и тепловой мощности и второй модуль анализа различных количеств газа, имеющихся в печи для окисления, подающий заданный поток на указанное устройство подачи газа и на указанное устройство электропитания.
Согласно одному альтернативному варианту осуществления изобретения, модуль подачи газа содержит контур создания водяного пара, соединенный с регулятором давления водяного пара, устройство подачи аргона/азота, устройством подачи аргона/молекулярного кислорода, соединенное с регулятором давления молекулярного кислорода.
Модуль управления и автоматического регулирования, который, таким образом, содержит модуль, позволяющий измерять температуру и тепловую мощность, и модуль анализа концентрация различных газов, таких как O2, CO2, CO, H2O и H2, позволяет производить непрерывное регулирование с обратной связью параметров протекания процесса, таких как парциальное давление кислорода, температура стабилизации, путем отслеживания в реальном времени температуры и тепловой мощности печи для термического окисления.
Согласно альтернативному варианту осуществления изобретения, указанная камера дополнительно оснащена устройством взвешивания твердых соединений на основе углерода и урана.
Изобретение станет более понятным, и другие его преимущества выявятся при изучении следующего неограничивающего описания и прилагаемых чертежей, на которых:
- фигура 1 иллюстрирует пример неравномерного нагрева, характеризующегося неконтролируемым псевдопериодическим перегревом, во время окисления образца металлического урана при 390°C;
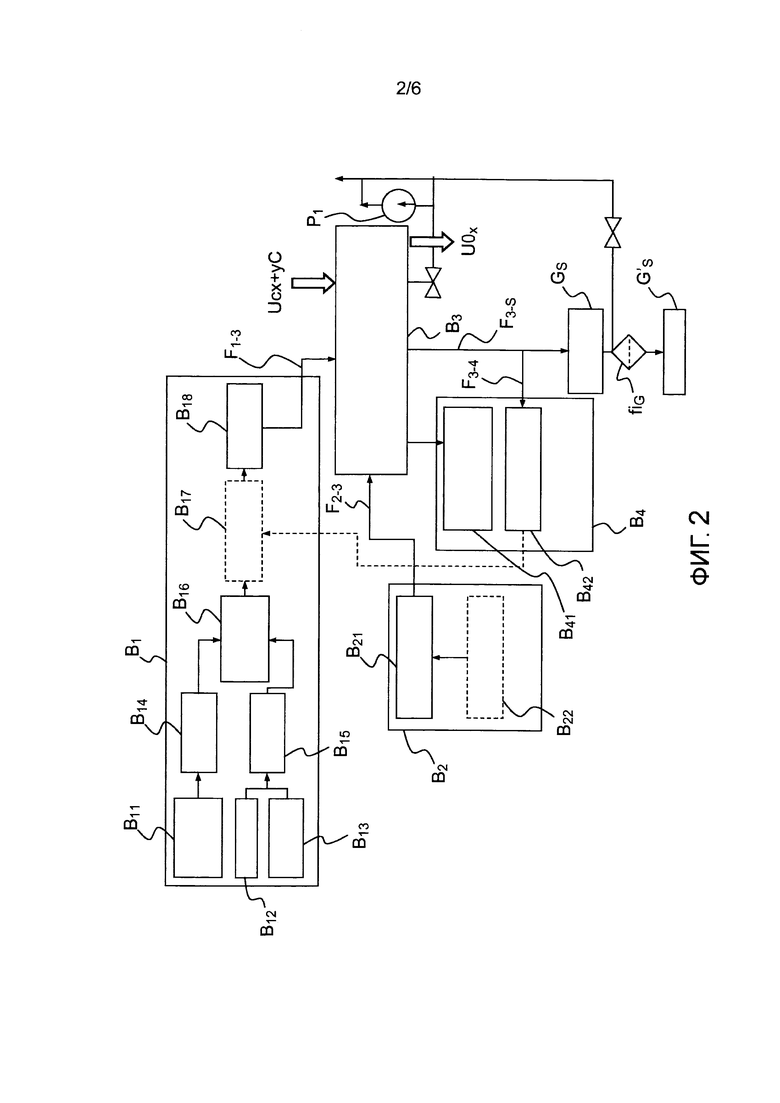
- фигура 2 представляет собой блок-схему, показывающую различные устройства, предназначенные для осуществления способа по настоящему изобретению;
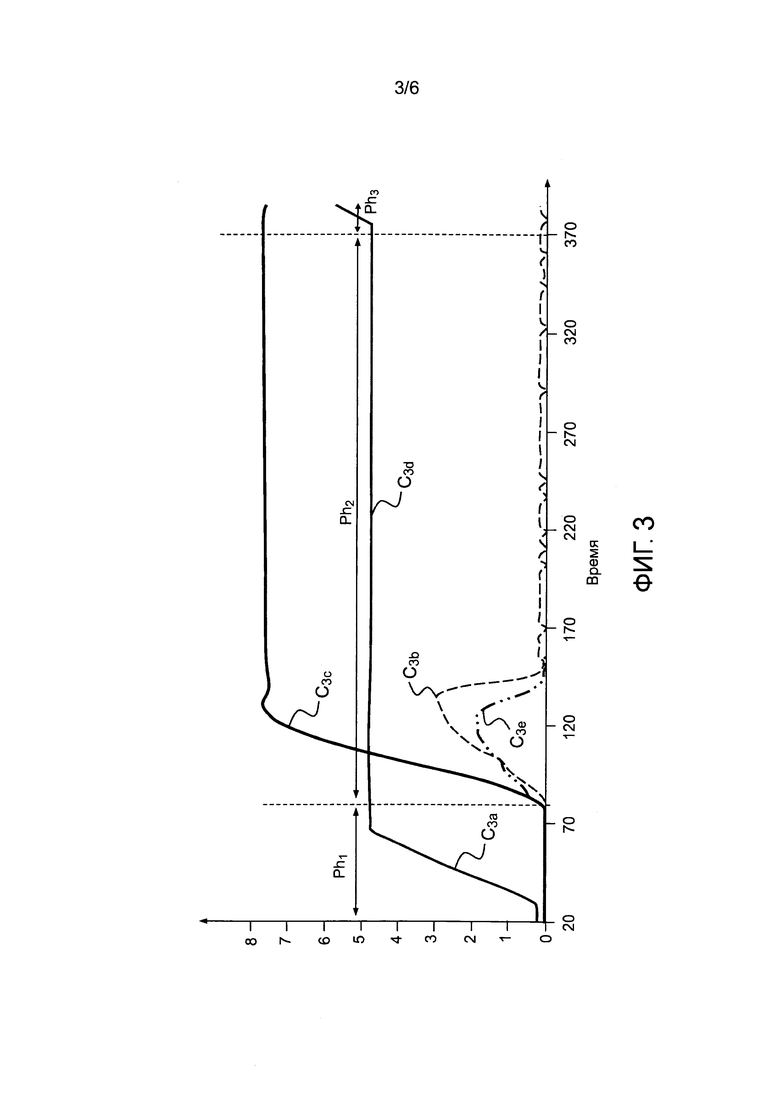
- фигура 3 иллюстрирует различные фазы операций согласно способу по изобретению;
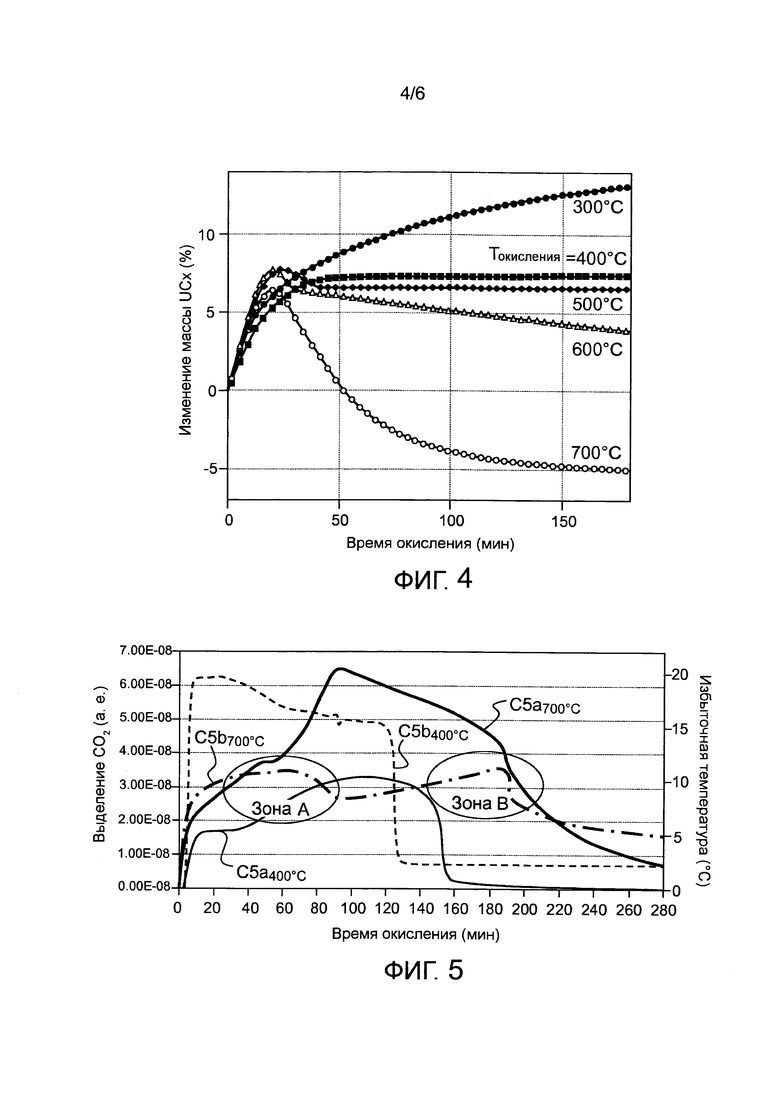
- фигура 4 иллюстрирует изменение массы соединения UCx как функцию времени для разных изотермических температур окисления;
- фигура 5 иллюстрирует изменения высвобождения CO2 в случаях локального перегрева, обнаруженных во время осуществления способа, соответственно для двух разных температур окисления (Tокисления=400 и затем 700°C);
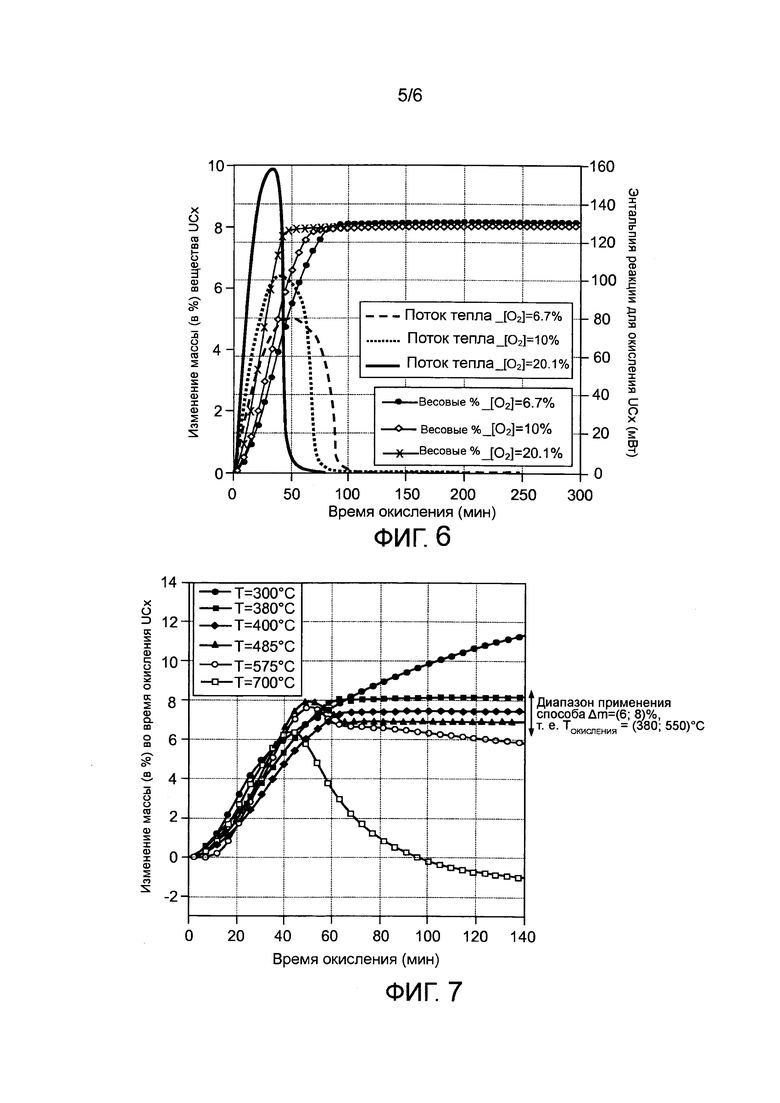
- фигура 6 иллюстрирует изменения массы в процентах и выделившийся тепловой поток при изотермическом окислении UCx для трех разных концентраций молекулярного кислорода;
- фигура 7 иллюстрирует профили изменения массы, полученные при изотермическом окислении UCx в окислительной атмосфере для разных температур окисления;
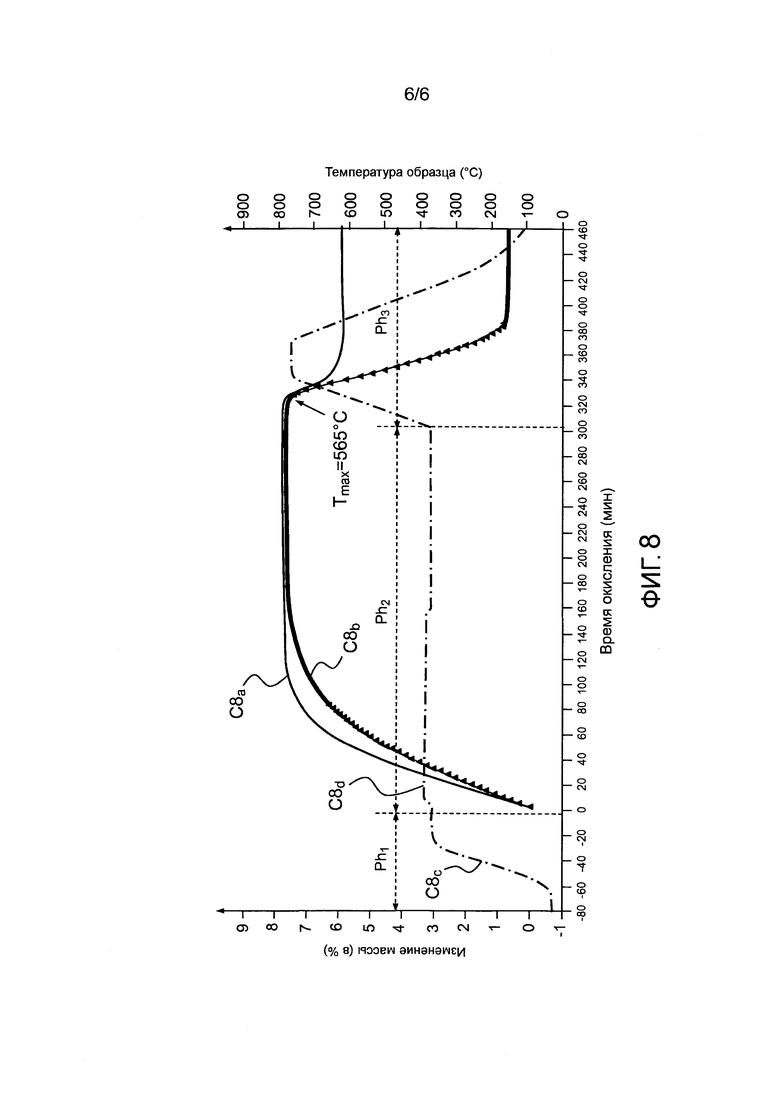
- фигура 8 иллюстрирует термогравиметрические кривые, показывающие влияние геометрического строения на способ стабилизации вещества UCx при средней температуре Tокисления=400°C.
В целом, способ по настоящему изобретению включает:
- доведение температуры вещества в инертной среде до подходящей для дальнейшего окисления;
- операцию контролируемой стабилизации фазы UCx+yC изотермической окислительной обработкой в оптимальном диапазоне температур (380°C; 550°C), в частности, как функции природы, количества, структуры и состава (величины x и y в исходном веществе) при парциальном давлении O2 (от 5% до 25% O2) (предпочтительно 10% O2). На этом этапе условия обработки выбирают, в частности, так, чтобы убедиться, что продукты являются реакционноспособными, и что эта реакционная способность регулируется только подачей кислорода. Подтверждение удовлетворительного протекания окислительной обработки осуществляется отслеживанием в реальном времени израсходованного молекулярного кислорода O2 и выделенного диоксида углерода CO2/моноксида углерода CO;
- операцию подтверждения завершения стабилизации композитного материала. Этот последний этап может осуществляться, в частности, путем значительного, но контролируемого повышения температуры окисления, или последовательным введением парциального давления водяного пара для стимулирования окисления полученных фрагментов UC2, которые, возможно, не окислились на первой стадии окисления, или изменением давления реакционных газов в процессе (положительное изменение (ограничено величиной максимум 1 бар) или отрицательное изменение (ограничено величиной минимум 1 мбар)), или иначе комбинацией двух или трех альтернативных способов.
Определение реакционной способности присутствующих реакционных газов (CO, CO2, H2) из этого изменения условий позволяет установить окончание реакции стабилизации, не опасаясь высокой реакционной способности части отходов, которые потенциально еще не были стабилизированы на предыдущем этапе. Если реакционная способность этих газов отсутствует, обработку останавливают.
Подробное описание ниже имеет целью показать, что с точки зрения химической природы обрабатываемого вещества UCx+yC, количества и объемы, которые могут использоваться для достижения цели настоящего изобретения, то есть большие количества подлежащих переработке отходов, обычно более нескольких килограммов, способ по настоящему изобретению позволяет обеспечить полную стабилизацию вещества в окисленной форме, который стабилен на воздухе при температуре и давлении внешней среды, если использовать подходящую температуру обработки при оптимальной скорости потока газа и оптимальной концентрации O2.
Действительно, особенности структуры указанного композитного вещества (двухфазное соединение, в частности, например, из UC2 и свободного углерода в форме графита, гетерогенное, высокопористое) приводят к противоречиям в том, что касается цели, и даже к физическим ограничениям, что делает особенно выгодными различные оптимизации способа по настоящему изобретению, раскрытые дальше в описании.
Эти трудности основаны, в частности, на следующих противоречиях:
- необходимо гарантировать стабилизацию отходов вида UCx+yC, однако без конверсии всего углерода (yC), изначально имеющегося в веществе UCx+yC или являющегося промежуточным продуктом различных реакций окисления. Это происходит потому, что при полной конверсии этих углеродсодержащих форм выделяется значительное количество газа (в основном CO2, CO), что может нанести большой вред и поэтому недопустимо с точки зрения переработки газа (значительное удаление) и длительности применения способа на полупромышленном масштабе. Кроме того, выбор полной стабилизации всех компонентов вещества UCx+yC (UC2, UC и углерод) заставляет работать при более высоких температурах окисления, что значительно способствует выделению радиоактивных веществ в месте выхода газообразных отходов;
- стабилизация, адаптированная к конкретной части вещества UCx+yC (фазы UC2, UC), становится все более проблематичной, поскольку стабилизирующая окислительная реакция является сильно экзотермической (трудно контролировать реакционную способность), что противоречит поставленной цели;
- контроль реакционной способности все более сложным, поскольку помимо экзотермичности, он обусловлен доступом окислителя в зоны реакции и зависит от образующихся побочных продуктов (UOx), способных создавать ограничения для протекания реакции, которая во время обработки может прерваться более или менее внезапно.
Способ по настоящему изобретению должен, таким образом, позволить преодолеть перечисленные выше физические ограничения, используя оптимальный рабочий диапазон, для того, чтобы:
- полностью окислить только фазу UCx, не сжигая полностью избыточный графит, присутствующий в исходном веществе (yC), а также, возможно, в контейнере мишени, также состоящем из графита, и по оценкам обычно имеющий вес более 1 кг;
- ограничить время обработки для стабилизации/конверсии вещества UCx благодаря использованию исследованного диапазона температур окисления, что дает возможность ускорить окисление UCx до UOx;
- ограничить только получение CO2 в результате окисления только UCx до U3O8/UO2 путем ингибирования сильного выделения CO2, образующегося при окислении избыточного углерода/графита, объемы которых, вводимые с веществом UCx+yC и графитовым контейнером для мишеней UCx, требуют длительного процесса обработки;
- ограничить летучесть и распространение потенциальных продуктов деления или активации, локализуя их по возможности внутри мишеней из UCx, намеченных для обработки при подходящей и умеренной температуре окисления;
- дать систему для проведения способа, которая позволит контролировать химическую реакционную способность и подтвердить удовлетворительную стабилизацию вещества после его окисления данным способом;
- предотвратить появление любых нестабильных форм окисления вещества UCx, в частности, в отношении разнообразия геометрии (гранулы, порошок, сферические бусины) и природы сырья на основе карбидов урана.
Пример устройства, позволяющего осуществить способ стабилизации соединения UCx+yC:
фигура 2 показывает в виде диаграммы пример устройства, позволяющего проводить изотермическую окислительную обработку соединения под парциальным давлением O2 в печи для окисления:
- первый модуль B1 используется для подачи газа и позволяет создавать инертную атмосферу аргона, или азота, или же частично окислительную атмосферу O2 и/или H2O с использованием внешнего контура подачи. Эти атмосферы непрерывно регулируются расходомерами и манометрами и затем вводятся в печь для окисления для стабилизации композитного материала из UCx+yC. Более конкретно данный модуль B1 может содержать, в частности, контур B11 создания водяного пара, соединенный с регулятором B14 давления водяного пара, устройство B12 подачи аргона/азота, устройство B13 подачи аргона/молекулярного кислорода, соединенное с регулятором B15 давления молекулярного кислорода, два регулятора, подающие смесителю B16 O2 и/или H2O в направлении регулятора B17 входного давления, соединенного с регулятором B18 скорости потока выхода газов для подачи потока F1-3 камере, соответствующей третьему модулю B3 стабилизации термообработки, содержащему печь для окисления, в котором происходит стабилизация соединения;
- второй модуль B2 электропитания предназначен для подачи на блок B3 заданного потока F2-3 и содержит модуль электропитания B21 и модуль B22 программирования теплового цикла стабилизации, приспособленного к разнообразным исходным композитным материалам;
- третий модуль B3 содержит печь для окисления с контролируемой атмосферой; он также позволяет загружать исходное вещество оптимально распределенным по подвесному контейнеру с точки зрения разнообразия природы и структуры исходного вещества и затем удалять стабилизированные отходы для потенциального анализа после аварии (в частности, взвешивания полученного остатка и взятия пробы остатка для определения характеристик) перед их последующей упаковкой и помещением на хранение;
- четвертый модуль B4 предназначен для управления и автоматического регулирования; он содержит модуль B41, позволяющий измерять температуру и тепловую мощность, и модуль B42 анализа концентраций различных газов, таких как O2, CO2, CO, H2O или H2. Этот четвертый модуль позволяет осуществлять непрерывное регулирование с обратной связью параметров протекания процесса, а именно парциального давления окислителя и температуры стабилизации, путем отслеживания в реальном времени температуры и тепловой мощности печи для окисления, расхода газа (O2, N2, Ar, H2O) и получения газообразных реагентов (CO2, CO, H2, CH4, C2H6). Опционально может быть также записано изменение массы UCx в процессе его окисления, чтобы идентифицировать различные реакции окисления, распознать противоположные фазы и отслеживать степень конверсии стабилизируемого вещества.
Потоки F3-S газа, выходящие из камеры, с одной стороны, перед удалением фильтруются насосом P1 и фильтром fiG и, с другой стороны, анализируются с помощью взятия пробы указанных газов F3-4.
Подробное описание на примере различных этапов осуществления способа по изобретению:
1) Этап повышения температуры до температуры окисления предпочтительно в интервале примерно от 380°C до 550°C может проводиться в камере в инертной атмосфере.
Чтобы соблюсти условия изотермического окисления, вещество UCx+yC постепенно нагревают в атмосфере инертного газа до температуры окисления, подходящей для применения способа. Выбор данной температуры окисления зависит, в частности, от типа печи и ее характеристик, от природы и структуры исходного вещества, от геометрии подвесного контейнера для загрузки и расположения окисляемого вещества внутри этого контейнера. Для наилучшего регулирования температуры обработки, возможно, необходимы предварительные тесты с меньшими количествами вещества (они будут позже раскрыты в данном описании). Продолжительность этого первого этапа обычно может составлять порядка 60 мин.
2) После периода стабилизации в инертной атмосфере (средняя продолжительность 60 мин) газ, содержащий O2, вводят в печь для окисления. Обычно после применения способа при температурах Tокисления, меняющихся от 380 до 550°C, вещество UCx, изначально имевшее химический состав UC2 + углерод в форме графита CF и геометрию в виде «гранул», окисляется и образует много гомогенного порошка с химическим составом U3O8 + углерод в форме графита CF. Увеличение объема вещества UCx после процесса составляет порядка 50%. Окисление вещества UCx отслеживают в реальном времени газоанализатором на выходе печи для окисления. Обработку окислением останавливают, когда концентрация O2 достигает установленного входного значения и когда концентрация CO2, выделенного во время окисления мишеней из UCx, меньше порогового значения, которое обычно может составлять порядка 100 ч/млн.
3) Окисление UCx+yC можно предпочтительно отслеживать анализом изменения массы (если это позволяет измерительное устройство) и измерением в реальном времени полученных в процессе газов, в частности: отслеживание израсходованного молекулярного кислорода O2, CO2, полученного при окислении UCx до оксида UOx, опционально оксида углерода CO и молекулярного H2, выделенного при последующем программированном добавлении водяного пара во время реакции. Действительно, может быть выгодным использовать водяной пар также для более мягкой стабилизации через контролируемое окисление.
4) Стабилизация вещества UCx считается завершенной, когда:
- исходная масса обрабатываемого вещества увеличивается на прибавку Δm, соответствующую образованию структуры вида UOx, преимущественно U3O8 (изменение массы Δm обычно может составлять от 6% до 10%);
- концентрация O2 на выходе достигает предписанного для способа значения (предпочтительно концентрации 10 об.%);
- концентрации полученных газов CO, CO2, H2 опускаются ниже порогового значения (обычно ниже 100 ч/млн);
- окисленное вещество UCx больше не вступает в реакции (отсутствует химическая активность) под воздействием изменения температуры ΔT, концентрации (Δ(O2), например), влажности среды (Δ(H2O)) или давления ΔP.
Следует отметить, что воздействия могут быть такими:
- резкое, но контролируемое увеличение ΔT температуры окислительной обработки, так что Tокисления+ΔT<Tmax, где Tокисления является температурой проведения окислительной обработки (Tокисления лежит между 300 и 550°C), а Tmax - максимальная температура, допустимая перед окислением избыточного свободного углерода (Tmax составляет около 560°C); отсутствие расхода O2 и выделения CO2 во время такого воздействия говорит об окончании процесса;
- изменение давления в печи. Изменение давления способствует проникновению газов в центр окисляемого вещества и ускоряет кинетику реакции. Для этого может быть проведен цикл снижения давления (Pmin около 1 мбар)-компрессии (Pmax около 1 бар) посредством системы насосов и соленоидального клапана, соединенных с печью для окисления;
- добавление оставшегося количества водяного пара до, во время или после обработки, чтобы способствовать предпочтительному окислению веществ UCx, в частности, имеющих высокую удельную плотность, с предпочтительным окислением шариков UC2 в окислительной атмосфере, содержащей воду. Добавление водяного пара ограничено максимум 5% об., чтобы исключить заполнение атмосферы избытком H2 (максимально допустимым безопасным значением является объемная концентрация 5% H2), причем газообразный H2 образуется при окислении UCx водяным паром. Введение H2O в конце цикла является выгодным, поскольку позволяет использовать H2 в качестве нового индикаторного газа для окисления только UCx и поскольку он полностью безопасен в случае восстановления реакционной способности вещества UCx, так как при этом получаются значительно меньшие количества благодаря тому, что материал UCx уже по большей части стабилизирован в окисленную форму благодаря как ограничению температуры (реакция газификации невозможна при Tокисления<Tmax, так и в результате ограничения концентрации (H2O));
- также можно использовать одновременно несколько описанных выше воздействий.
Фигура 3 иллюстрирует все эти этапы, схематически представленные в виде фаз Ph1, Ph2 и Ph3. Кривая C3a показывает изменение температуры в зависимости от времени, кривая C3b показывает количество выделенного CO2, кривая C3c показывает изменение массы твердых соединений, кривая C3d показывает количество O2, и кривая C3e показывает количество H2 в водяном паре.
Обычно возможно использовать парциальное давление кислорода, равное 10%.
Чтобы удовлетворить этим критериям удовлетворительного протекания процесса, было показано, что предпочтительно возможно заранее определить оптимальные температуры стабилизации в диапазоне от 300 до 550°C. Эти температуры тщательно выбраны, чтобы способствовать окислению только фазы UC2 до UOx, не влияя на избыточный графит, содержащийся в исходном веществе UCx, причем целью является окисление как можно меньшего количества графита из вещества и контейнера, в котором он содержится.
Этот этап оптимизации температуры окисления ниже проиллюстрирован более подробно для случая вещества, имеющего состав UC2+2C. Так как это вещество UCx является многофазным и гетерогенным, то его окисление в изотермических условиях стало в настоящей заявке предметом углубленного анализа. Чтобы показать, что желаемые характеристики вещества после осуществления стабилизации зависят от многих параметров и, в частности, от оптимального диапазона температур окисления, на фигуре 4 показан конкретный пример изотермических сеток, полученных термогравиметрически и дифференциальным термоанализом при парциальном давлении O2, равном 10%. Каждая кривая показывает изменение массы вещества UCx как функцию времени для различных температур окисления, обозначенных Tокисления. Обнаруженное увеличение массы отражает тот факт, что вещество UCx, имевшее изначально химическую формулу UC2+yCF (CF обозначает избыточный графит, присутствующий в исходном веществе UCx), окислилось до твердого химического соединения вида UOz+yCF и/или UOz. Когда обнаруживается уменьшение массы, это отражает тот факт, что произошло окисление твердого реагента до газа, что в данном случае соответствует окислению углеродсодержащей формы до CO/CO2.
Таким образом, ясно, что для температуры Tокисления около 300°C кинетика окисления вещества UCx до фазы UOz (в этом случае, для примера, до U3O8) является постепенной и довольно медленной.
Следует помнить, что основная реакция в процессе окисления такова:
UC2+2CF+4/3O2→1/3U3O8+2CUCx+2CF
и она приводит к теоретическому увеличению массы Δmтеор=15%. Полученное увеличение массы нужно поэтому сравнить с теоретическими значениями увеличения массы.
При этой температуре не должно происходить выделения газообразного CO2, что и было подтверждено с использованием соединенного газоанализатора на выходе термогравиметрического устройства.
При температуре Tокисления около 400°C увеличение массы проходит быстрее и приводит к получению хорошо определяемого постоянного значения, показывающего, что окисленное вещество UCx больше не меняется, хотя оно все еще находится в окислительной среде. Эта оптимальная температура окисления, таким образом, делает возможной быструю и стабильную конверсию вещества UCx до фазы оксида (а особенно U3O8), которая в этом примере задается следующей реакцией:
UC2+2CF+10/3O2→1/3U3O8+2CF+2CO2 Δmтеор=7,2%
Для температуры Tокисления, равной 500°C, профиль изменения массы во время окисления UCx показывает увеличение, за которым следует временная потеря массы, которая затем возвращается к стабилизированному постоянному значению Δm. Увеличение массы соответствует окислению фазы UC2 до U3O8, и потеря массы говорит об окислении оставшегося в небольшом количестве углерода из UC2, которое сопровождается выделением незначительного количества CO2. В конце стационарного состояния окисления остаются химические фазы U3O8 и CF, так что полностью реакцию окисления можно записать в виде:
UC2+2CF+(10/3+α)O2→1/3U3O8+(2-α)CF+(2+α)CO2 Δmтеор=<7%.
Для температур, больших или равных 600°C, профили изменения массы одновременно выявляют увеличение, за которым следует постепенная потеря массы, амплитуда которого пропорциональна используемой температуре окисления. Профили Δm, таким образом, проходят через максимум, также известный как выброс, амплитуда и положение которого для одного и того же вещества меняются в зависимости от используемой температуры окисления. С этой точки зрения эта потеря массы сопровождается значительным выделением CO2, что означает, помимо окисления UCx до формы U3O8, окисление всего избыточного графита. Отношение окисления двух фаз (UC2 и CF), составляющих вещество UCx, таким образом, зависит строго от используемой температуры окисления Tокисления.
Такое определение кинетики окисления вещества UCx и влияния выбранной температуры в изотермических условиях показывает, таким образом, что диапазон оптимальных температур для применения способа по настоящему изобретению лежит вблизи 400°C +/-100°C. Эти температуры позволяют убедиться, что достигнуто полное окисление фазы UC2, и в то же время:
- не происходит полного окисления оставшегося углерода (как от окисления UCx (CUCx), так и изначально имевшегося (CF)), содержащегося в мишенях;
- не требуется продолжительность обработки, не приемлемая для процесса: термогравиметрические кривые на фигуре 4 показывают, что окончательная стабилизация вещества UCx (другими словами, изменение массы, которое больше не меняется в процессе окисления) при температуре Tокисления=400°C происходит в четыре раза быстрее, чем при температуре окисления 700°C, и в то же время предотвращается окисление оставшегося графита;
- не происходит избыточного перегрева стабилизируемого загруженного вещества, так что предотвращается неконтролируемый нагрев, а также окисления других элементов, которое не нужно и может даже быть вредным для обработки газов.
Например, фигура 5 показывает изменения выделения CO2 (C5a 400°C и C5a 700°C) и случаи перегрева, соответствующие локальным превышениям температур (C5b 400°C и C5b 700°C), обнаруженным при применении способа. Полученные данные показывают, в частности, феномены восстановления реакционной способности, наиболее конкретно при температуре 700°C (на фигуре 5 обозначены как Зона A и Зона B), что свидетельствует об экзотермичности протекающих реакций. Дополнительно при той же температуре окисления 700°C выделение CO2, все еще происходящее после окислительного воздействия продолжительностью 280 минут, показывает, что процесс стабилизации еще не окончен. С другой стороны, для более умеренных температур около 400°C выделение CO2 становится меньше порогового значения (100 ч/млн) после окислительного воздействия продолжительностью только 200 минут, что означает превращение почти всего вещества UCx в UOx. Аналогично феномены восстановления термической реакционной способности при этих «умеренных» температурах гораздо слабее, фактически они не имеют места.
Протекающие во время процесса реакции схематически можно записать так (по очереди относительно реакции (1)):
UCx+yCF+(x+4/3)O2→1/3U3O8+yCF+xCO2 x=1 до 2, y=1 до n (1)
UCx+yCF+4/3O2→1/3U3O8+xCUCx+yCF x=1 до 2, y=1 до n (2)
UCx+yCF+(x+z/2)O2→1UOz+yCF+xCO2 x=1 до 2, y=1 до n, z=2 до 3 (3).
Напротив, реакциями, нежелательными для вещества UCx, являются те, в которых происходит окисление углерода одновременно с окислением фазы UC2 и более конкретно свободного углерода, обозначенного CF, который в форме графита присутствует в исходном веществе UCx в больших количествах (70% об.). Например, ниже приведены несколько нежелательных реакций, которые не показывают больше присутствия углерода CF и/или CUCx среди продуктов реакции окисления.
UCx+yCF+(4/3+x+y)O2→1/3U3O8+(x+y)CO2 x=1 до 2, y=1 до n (4)
UCx+yCF+(z/2+x+y)O2→1UOz+(x+y)CO2 x=1 до 2, y=1 до n, z=2 до 3 (5).
Оптимизация окислительного парциального давления и выделяемого тепла:
В заявке также показано, что можно стабилизировать окислительное парциальное давление и выделенное тепло в зависимости от времени. С этой целью было изучено воздействие парциального давления O2 на ход окисления UCx. Конкретный пример показан на фигуре 6, где представлены изменения массы в % (сплошные линии) и выделенного потока тепла (пунктирные линии) во время изотермического окисления UCx при использовании температуры процесса 400°C при 3 различных парциальных концентрациях (O2) ((O2)=6,7%, 10% и 21%).
Полученные результаты показывают, что парциальное давление O2 не влияет на диапазон применимости способа: колебания прибавки массы идентичны и лежат вблизи среднего конечного значения Δm=+8%, какое бы парциальное давление O2 не использовалось. Результатом является окисление только фазы UC2 до оксида типа U3O8. Избыточный графит CF, в свою очередь, все еще присутствует в окисленном веществе, ограничивая таким образом образование диоксида углерода CO2, вредного для контроля газовых потоков в процессе после термообработки. Парциальное давление важно для кинетики окисления UCx и, следовательно, для времени обработки в способе: при высоких концентрациях ((O2)=21%) парциальное давление (это O2) позволяет стабилизировать фазу UC2 вещества UCx только после применения способа в течение 40 мин, тогда как при низкой концентрации ((O2)=10%) стабилизация UCx достигает порогового значения Δm=+8% после 70 мин.
Парциальное давление O2 также важно для измеренных значений теплового потока, которые характеризуют экзотермичность реакции окисления UCx до U3O8; максимальное количество выделенного тепла в два раза больше, когда способ стабилизации UCx проводится при парциальных концентрациях O2, меняющихся от 6,7% до 21%. Поскольку такое количество единовременно выделенного тепла способно отрицательно повлиять на способ в том случае, когда увеличение локально слишком высокой температуры может привести к увеличению полной температуры окисления, в особенности до значений, больших Tmax (которая определяется как та температура, при которой начинается окисление избыточного углерода), необходимо установить оптимальные экспериментальные условия, которые позволят найти компромисс между скоростью превращения и контролем тепловыделения, которое может изменить реакционную способность.
Следовательно, парциальная концентрация O2 около 10% позволяет, таким образом, оптимизировать время конверсии UCx в форму оксида, при этом ограничивая выделение тепла в данной реакции окисления.
Оптимизация температуры стабилизационной термообработки:
Прибавки массы, полученные при температурах окисления, меняющихся от 380 до 550°C, и стабилизация этих значений вблизи порогового значения Δm=(6;8)% определяют надежность способа в отношении температуры, которая применяется при обработке способом по изобретению, дают возможность проводить контролируемую конверсию только фазы UC2 вещества UCx в форму оксида типа U3O8 (возможны следы UO2).
Фигура 7 показывает изотермическую сетку, полученную вблизи оптимальной температуры осуществления способа, равной 400°C. Полученные профили (они получены в тех же изотермических условиях, что и профили с фигуры 3) позволяют проверить надежность способа, определяя максимальную температуру, ведущую к окислению избыточного углерода, в показанных термогравиметрических кривых.
Следует отметить, что термогравиметрические кривые, полученные для температур окисления, больших 550°C (2 термогравиметрические кривые, полученные при Tоксиления=575°C и затем при 700°C, представленные, например, на фигуре 7) показывают потерю массы, которая значительно возрастает и линейно затухает: они подчеркивают постепенность окисления избыточного углерода CF, которое становится все более выраженным с повышением температуры окисления.
Оптимизация способа по изобретению добавлением водяного пара:
Было также исследовано добавление водяного пара до и во время изотермического цикла обработки по способу, и оказалось возможным сделать следующие выводы о:
- влиянии водяного пара на скорость конверсии вещества UCx в UOx в окислительной среде вне зависимости от времени добавления водяного пара (до или во время окислительной обработки);
- возможности использования нового индикаторного газа H2, связанного с реакцией между UCx и H2O согласно формуле
UC2+yCF+xH2O→UOx+xH2+yCF (6).
Присутствие H2 в концентрации, которая, согласно измерению, более чем в 100 раз ниже концентрации CO2, выделившегося во время окисления UCx, может использоваться, аналогично последнему, в качестве критерия прекращения удовлетворительного протекания процесса, причем этот критерий достигается, когда выделение H2 меньше минимального порогового значения;
- ускорении химического распада очень плотных веществ и скорости окисления UCx до оксида (например, было измерено увеличение времени, равное 10 мин, при проведении окислительной обработки в изотермических условиях при 420°C);
- снижении количества выделенного тепла и, следовательно, избыточной температуры ΔT, полученных во время способа и особенно в начале экзотермической реакции окисления UCx до U3O8 (уменьшение ΔT составило 8% в присутствии водяного пара).
Было также исследовано влияние водяного пара на стабилизацию UCx методом средовой сканирующей электронной микроскопии. Результаты окисления in situ, полученные средовой электронной микроскопией при различных температурах окисления и давлениях водяного пара, позволили продемонстрировать появление локализованных трещин на поверхности UCx. Эти трещины облегчают взаимодействие между молекулами O2 и кластерами UC2, которые, находясь в массе вещества UCx, не являются очень доступными. Эти трещины позволяют молекулам O2 более эффективно проникать в массу вещества и поэтому значительно улучшают полную скорость конверсии UC2 в фазу оксида. Рентгеновский анализ после аварии при испытаниях на окисление UCx методом электронной микроскопии при различных парциальных давлениях водяного пара P(H2O) выявил наличие UO2, U3O8 и избыточного углерода в окисленном веществе.
Использование реагента, являющегося комбинацией O2/H2O, в обработке по способу также позволяет использовать два типа реакций (коррозии и окисления) с изменением молярного объема, происходящего из-за продуктов окисления фазы UC2 (среди них UO2 и U3O8). Наличие этих двух оксидов способствует изменению объема окисленного продукта и появлению напряжений на границах раздела фаз), что приводит к появлению трещин, обеспечивающих лучший доступ O2 к неокисленным поверхностям, и значительному улучшению кинетики окисления.
Добавление водяного пара в способ становится еще более важным для сыпучих и плотных исходных веществ, доступ молекулярного кислорода в центральную часть которых затруднен. Водяной пар, таким образом, влияет на структуру исходного стабилизируемого вещества.
Проверка способа по изобретению для различных типов структуры соединения карбида урана:
Стабилизация мишеней из UCx проводилась при температуре стабилизации 400°C с использованием двух различных геометрических форм: порошка UCx (размер частиц 150 мкм) и сборки нескольких склеенных друг с другом гранул UCx (гранулы Φ=15 мм, t=1 мм, гидростатическая плотность = 8, пористость >50%). Программирование изотермического цикла окисления и изменения массы двух данных геометрических форм UCx во время окислительной обработки показаны на фигуре 8. Более конкретно кривая C8a показывает изменение массы в случае гранул, кривая C8b показывает изменение массы в случае порошка, кривая C8c показывает изменение температуры в случае гранул и кривая C8d показывает изменение температуры в случае порошка.
Во время данных испытаний был также запрограммирован цикл окисления при неизотермических условиях (подъем температуры до Tокисления=800°C с градиентом 10°C/мин) после проведения процесса в течение 300 мин, чтобы:
- определить максимальную температуру Tmax, соответствующую запуску окисления избыточного углерода вещества UCx;
- проанализировать различия в потере массы избыточного углерода как функции структуры исходного вещества.
Итак, полученные результаты показывают, что способ по настоящему изобретению:
- применим для различных веществ UCx в виде порошка или гранул, поскольку прирост массы вещества UCx (формованного или в виде порошка) во время окисления стремится к постоянному значению, равному Δm=7,6%, в соответствии с получением стабилизированного конечного продукта, определяемого формулой U3O8+CF и подтвержденного рентгеновским методом;
- оптимален для исходного вещества UCx, имеющего геометрию типа «гранулы», поскольку кинетика реакции, связанная с реакцией окисления UCx до U3O8 быстрее (постоянное значение достигается быстрее) и менее экзотермична, чем в случае геометрии типа «порошка» (постоянное значение Δm достигается более стремительно, и локально избыточная температура ΔT ниже и имеет меньшую амплитуду);
- может подстраиваться в отношении температуры обработки в процессе, вне зависимости от геометрической природы UCx. Действительно, в обоих случаях температура Tmax, соответствующая запуску окисления избыточного углерода, идентична и при измерениях получается равной 565°C. Опыт, таким образом, показывает, что ΔT=Tmax-Tокисления подходит для проверки в конце реакции удовлетворительного протекания способа стабилизации UCx до формы UOx. Способ может также регулироваться в обоих случаях, так как увеличение массы такое же, как и при окислительной обработке в изотермических условиях.
Зарегистрированные изменения потери массы во время окисления избыточного углерода показывают, что, кроме как при температуре Tmax, применение способа не позволяет полностью окислить избыточный углерод CF, присутствующий в исходном веществе UCx, в частности, если последнее имеет геометрию типа «гранулы». Тем не менее, на основе строго геометрических факторов сравнения, если температура применения способа должна быть больше температуры Tmax (в частности, для проверки окончания реакции), использование вещества UCx типа «гранулы», а не типа «порошка», оказывается выгодным в том смысле, что окисление избыточного углерода является только частичным, ограничивая, таким образом, получение значимых количеств CO/CO2, которое будет иметься в наличии после осуществления способа.
Пример условий работы для способа стабилизации согласно изобретению:
Исходное вещество UCx в форме порошка или гранул сантиметрового размера помещают внутрь подвесного контейнера, который, в свою очередь, помещают внутрь печи для окисления.
Затем в печь вводят инертный газ, например аргон, и запускают цикл нагрева 10°C/мин до тех пор, пока не будет достигнута заданная температура, обозначенная Tокисления, составляющая примерно 400°C.
Как только данная температура Tокисления получена, программируют стационарное состояние стабилизации в течение 30 мин в атмосфере аргона.
После такого стационарного состояния стабилизации восстановленный воздух, в чистом виде или разбавленный аргоном, при содержании O2 10% неожиданно вводят в измерительное устройство при скорости потока газа, пропорциональной исходному количеству UCx.
Затем начинают окисление UCx в изотермических условиях при температуре стабилизации Tокисления=400°C в течение в среднем 5 ч, и система анализа газов позволяет отслеживать в реальном времени, в частности, расход O2 и выделение CO2, полученного во время окисления UCx до U3O8.
Когда концентрация O2 достигает заданного значения, установленного перед началом процесса (предпочтительно 10% об.), и когда концентрация CO2 достигает значения менее 100 ч/млн, проводят испытания на подтверждение восстановления реакционной способности. Эти испытания состоят, например, в резком, но контролируемом повышении температуры выше заданного значения, обычно на ΔT=+50°C, и измерении изменения концентрации O2 и CO2 во время этого изменения температуры. В качестве критериев испытаний может быть также предусмотрено изменение давления и/или введение парциального давления водяного пара.
При отсутствии выделения CO2 выше порогового значения (100 ч/млн) и/или расхода O2 во время такой проверки программируют охлаждение печи воздухом (охлаждение несколько десятков °C/мин).
Если выделение CO2 и/или расход O2 во время такого испытания имеют место до конца реакции, то стабилизацию UCx продолжают при новой температуре Tокисления+ΔT до тех пор, пока количества CO2 не станут меньше порогового значения (100 ч/млн). Может быть предусмотрено добавление водяного пара к окислительной среде для существенного ускорения полной стабилизации UCx в форму UOx. Присутствие водяного пара позволит также отслеживать новый индикатор, H2, который появляется во время оставшегося окисления UCx до формы оксида. Эти температурные испытания проводят до тех пор, пока применяемая температура в целом не превысит максимальное значение, соответствующее окислению избыточного углерода, присутствующего в веществе UCx (Tmax составляет около 560°C). Если не происходит нового выделения газа, печь охлаждают в тех же условиях, что и в случае отрицательного ответа на проверку окончания реакции.
Окисленный остаток, имеющий состав U3O8+CF и окончательную структуру в виде порошка, затем собирают и упаковывают согласно стандартам, предусмотренным для выгрузки. Также берут пробу для рентгеновского анализа.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПЕРЕРАБОТКИ УРАНСОДЕРЖАЩИХ КОМПОЗИЦИЙ | 1999 |
|
RU2158973C2 |
| СПОСОБ ПЕРЕРАБОТКИ ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА | 2015 |
|
RU2591215C1 |
| КАТАЛИЗАТОР, СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА ИЗ МЕТАНА | 2007 |
|
RU2350386C1 |
| УСТОЙЧИВЫЙ К ВОЗДЕЙСТВИЮ ТЕМПЕРАТУРЫ КАТАЛИЗАТОР ДЛЯ ОКИСЛЕНИЯ ХЛОРОВОДОРОДА В ГАЗОВОЙ ФАЗЕ | 2008 |
|
RU2486006C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИГАЛОГЕНИДА, И/ИЛИ ОКСИДА АКТИНИДА(ОВ), И/ИЛИ ЛАНТАНИДА(ОВ) ИЗ СРЕДЫ, СОДЕРЖАЩЕЙ ПО КРАЙНЕЙ МЕРЕ ОДНУ РАСПЛАВЛЕННУЮ СОЛЬ | 2012 |
|
RU2610067C2 |
| КАТАЛИЗАТОР И СПОСОБ ИЗГОТОВЛЕНИЯ ХЛОРА ПУТЕМ ОКИСЛЕНИЯ ХЛОРОВОДОРОДА В ГАЗОВОЙ ФАЗЕ | 2008 |
|
RU2469790C2 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА | 2011 |
|
RU2548410C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ПЕРФТОРУГЛЕРОДНЫХ СОЕДИНЕНИЙ В ГЕКСАФТОРИДЕ УРАНА | 1999 |
|
RU2154028C1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ ТАБЛЕТОК ЯДЕРНОГО ТОПЛИВА | 2009 |
|
RU2396611C1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА ДЛЯ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА ИЗ МЕТАНА, КАТАЛИЗАТОР, ПРИГОТОВЛЕННЫЙ ПО ЭТОМУ СПОСОБУ, И СПОСОБ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА ИЗ МЕТАНА С ЕГО ИСПОЛЬЗОВАНИЕМ | 2016 |
|
RU2638831C1 |
Изобретение относится к способу химической стабилизации соединения карбида урана и устройству для осуществления способа. Способ включает следующие этапы: этап повышения температуры внутри указанной камеры до температуры окисления указанного соединения на основе карбида урана в интервале приблизительно от 380°C до 550°C, причем в указанную камеру поступает инертный газ; этап изотермической окислительной обработки при указанной температуре окисления, причем указанная камера находится под парциальным давлением O2; этап контроля завершения стабилизации указанного соединения, который содержит отслеживание количества поглощенного молекулярного кислорода и/или диоксида углерода или выделенных диоксида или моноксида углерода до достижения входного заданного значения указанного количества молекулярного кислорода, минимального порогового значения указанного количества диоксида углерода или минимальных пороговых значений диоксида углерода и моноксида углерода. Техническим результатом является возможность безопасного, надежного управляемого и ускоренного решения комплексной проблемы стабилизации соединений карбида урана с формулой UCx + yC, где число x может быть больше или равно 1, а действительное число y больше нуля. 2 н. и 11 з.п. ф-лы, 8 ил.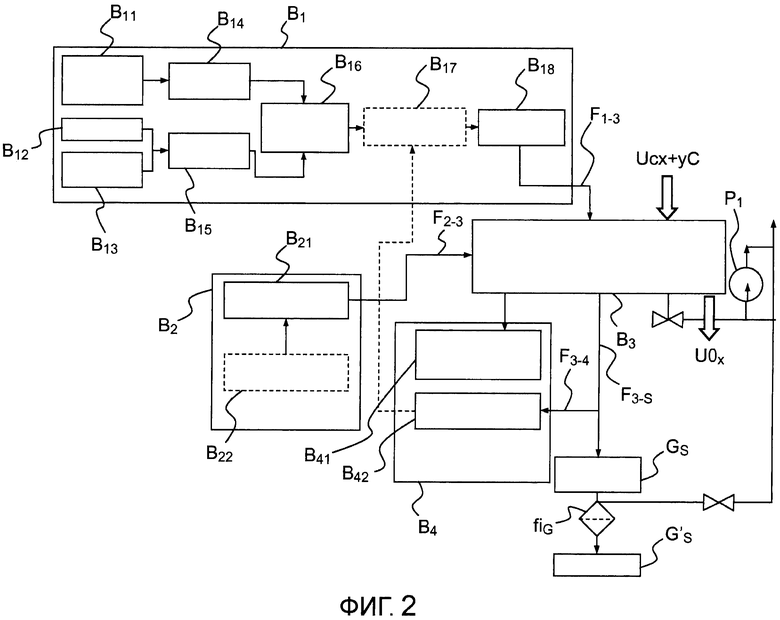
1. Способ химической стабилизации соединения карбида урана, соответствующего формуле
UCx+yC,
где x≥1 и y>0, причем x и y являются действительными числами,
помещенного в стабилизационную камеру, отличающийся тем, что он включает следующие стадии:
- стадию повышения температуры внутри указанной камеры до температуры окисления указанного соединения на основе карбида урана в интервале приблизительно от 380°C до 550°C, причем в указанную камеру поступает инертный газ;
- стадию изотермической окислительной обработки при указанной температуре окисления, причем указанная камера находится под парциальным давлением O2;
- стадию контроля завершения стабилизации указанного соединения, которая включает отслеживание количества поглощенного молекулярного кислорода и/или диоксида углерода или выделенных диоксида или моноксида углерода до достижения входного заданного значения указанного количества молекулярного кислорода, минимального порогового значения указанного количества диоксида углерода или минимальных пороговых значений диоксида углерода и моноксида углерода.
2. Способ химической стабилизации соединения карбида урана по п. 1, отличающийся тем, что стадия контроля завершения стабилизации дополнительно включает отслеживание изменения массы твердых соединений на основе углерода и урана в камере, причем увеличение массы коррелирует с постепенным окислением карбида урана.
3. Способ химической стабилизации соединения карбида урана по п. 1, отличающийся тем, что стадию контроля завершения стабилизации осуществляют с повышением температуры внутри указанной камеры в интервале от указанной температуры окисления до температуры окисления углерода (причем эта температура исключается из интервала) и отслеживания количества выделенного CO2.
4. Способ химической стабилизации соединения карбида урана по любому из пп. 1 и 2, отличающийся тем, что он включает введение парциального давления водяного пара в указанную камеру до, и/или во время, и/или после этапа окисления.
5. Способ химической стабилизации соединения карбида урана по п. 4, отличающийся тем, что стадия контроля и завершения стабилизации включает определение H2 в качестве маркера контроля окончания окисления в указанной камере.
6. Способ химической стабилизации соединения карбида урана по п. 1, отличающийся тем, что стадия контроля завершения стабилизации включает операцию создания избыточного давления реакционных газов, имеющихся в указанной камере так, чтобы ускорить завершение реакции окисления указанного соединения.
7. Способ химической стабилизации соединения карбида урана по п. 6, отличающийся тем, что стадия контроля завершения стабилизации дополнительно включает цикл операций создания избыточного давления и операцию снятия избыточного давления реакционных газов, имеющихся в указанной камере.
8. Способ химической стабилизации соединения карбида урана по п. 1, в котором указанное соединение присутствует в виде порошка или пористой или сплошной гранулы.
9. Способ химической стабилизации соединения карбида урана по п. 1, отличающийся тем, что он включает предварительную стадию определения оптимальной температуры окисления методом термогравиметрического анализа образца соединения UCx+yC.
10. Способ химической стабилизации соединения карбида урана по п. 9, отличающийся тем, что оптимальная температура окисления, которая меняется в зависимости от кондиционирования указанного карбида урана, составляет примерно от 380°C до 550°C.
11. Устройство химической стабилизации соединения карбида урана, содержащее камеру с печью для окисления (B3) и использующее способ по любому из пп. 1-10, отличающееся тем, что оно включает:
- модуль подачи газа (В1), который позволяет создавать инертную атмосферу аргона, или азота, или частично окислительную атмосферу O2 и/или H2O с помощью внешнего контура подачи, причем потоки газа подаются в указанную печь для окисления;
- модуль (B2) электропитания, питающий печь для окисления, который подает к ней заданный поток, что делает возможным повышение температуры;
- указанную камеру, подающую выходящий поток газа к модулю (B4) управления и автоматического регулирования;
- указанный модуль управления и автоматического регулирования, содержащий первый модуль (B41) измерения температуры и тепловой мощности и второй модуль (B42) анализа различных количеств газа, имеющихся в печи для окисления, подающий заданный поток указанному устройству подачи газа и указанному устройству электропитания.
12. Устройство химической стабилизации соединения карбида урана по п. 11, отличающееся тем, что модуль (B1) подачи газа содержит контур (B11) создания водяного пара, соединенный с регулятором (B14) давления водяного пара, устройство (B12) подачи аргона/азота, устройство (B13) подачи аргона/молекулярного кислорода, соединенное с регулятором (B15) давления молекулярного кислорода.
13. Устройство химической стабилизации соединения карбида урана по любому из пп. 11 и 12, отличающееся тем, что указанная камера оснащена устройством для взвешивания твердых соединений на основе углерода и урана.
| АВТОМАТИЧЕСКОЕ УСТРОЙСТВО ДЛЯ ПОДАЧИ УГЛЯ К ТОПКАМ | 1920 |
|
SU297A1 |
| US 20020038070 A1, 28.03.2002 A1 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПЕКТИНА | 1995 |
|
RU2095372C1 |
| СПОСОБ ПЕРЕРАБОТКИ ОТХОДОВ РЕАКТОРНОГО ГРАФИТА | 2003 |
|
RU2242814C1 |

