Настоящее изобретение относится к внеклеточному домену, связывающему аллостерический ингибитор, где указанный связывающий домен происходит из тирозинкиназного рецептора, один раз пронизывающего мембрану. Более конкретно, изобретение относится к внеклеточному домену, происходящему из тирозинкиназного рецептора, т.е. рецептора фактора роста фибробластов (FGFR), рецептора фактора роста сосудистого эндотелия (VEGFR) или рецептора фактора роста из тромбоцитов (PDGFR). Изобретение дополнительно относится к применению этого домена для идентификации сходных доменов во внеклеточной части других тирозинкиназных рецепторов и к способу скрининга для идентификации аллостерического ингибитора.
Рецепторы клеточной поверхности представляют собой мишени для большинства имеющихся лекарств (Overington, et al., 2006). Исторически в программах поиска лекарств доминировали попытки разработать антагонисты, которые конкурируют за связывание с эндогенными лигандами в ортостерических сайтах. Напротив, труднее идентифицировать лекарства, которые связываются с аллостерическими сайтами, т.е. доменами, топографически отличными от доменов, используемых ортостерическими лигандами (если мишень представляет собой рецептор) или субстратами (если мишень представляет собой фермент), и модулируют активность белка. Однако в последние годы наблюдается рост количества аллостерических модуляторов, идентифицированных для управляемых лигандами каналов и рецепторов, сопряженных с G-белками (GPCRs) (Christopoulos, 2002; Kenakin, 2010). Неожиданно оказалось, что до сих пор не идентифицированы аллостерические низкомолекулярные вещества, являющиеся модуляторами рецепторных тирозинкиназ (RTKs), узнающих ростовые факторы, несмотря на тот факт, что это надсемейство рецепторов имеет огромные биологическую важность и медицинское значение и, несмотря на тот факт, что аллостерические лекарства могут предоставить особые терапевтические преимущества по сравнению с традиционными ортостерическими лигандами, включая более высокую безопасность и/или избирательность. К настоящему времени большинство способов лечения, направленных на RTKs, сфокусировано либо на моноклональных антителах, узнающих лиганды в виде ростовых факторов, либо на низкомолекулярных химических соединениях, прямо ингибирующих тирозинкиназную активность рецепторов.
Среди других одной областью, которая может получить существенные преимущества от более эффективных и/или селективных низкомолекулярных ингибиторов RTK, является область лечения с помощью антиангиогенных лекарств. Антиангиогенные агенты, направленные на VEGF, продлевают выживаемость онкологических больных, но их общий успех ограничен внутренней резистентностью, избеганием за счет приобретенной устойчивости и, по меньшей мере, в доклинических моделях, стимуляцией метастазирования. Было высказано предположение, что сочетанная терапия с дополнительными антиангиогенными агентами может помочь преодолеть эти проблемы. Фактор 2 роста фибробластов (FGF)-2, первый идентифицированный ангиогенный фактор, является привлекательным кандидатом для лекарства. Действительно, было показано, что сигнализация через FGFR связана с раковыми и воспалительными заболеваниями (Shin et al., 2006; Eswarakumar et al., 2005; Malemud et al., 2007; Carmeliet, 2005), вносит вклад в изменение опухолевого ангиогенеза (Presta et al., 2005; Kubo et al., 2002; Shine et al., 2006; Lavine et al., 2006) и оберегает васкуляризацию опухолей, а также способствует ее восстановлению после лечения ингибиторами VEGF (Casanovas et al., 2005). Несмотря на это, семейство FGF не привлекало существенного внимания при разработке антиангиогенных лекарств частично из-за обилия членов этого семейства из 18 лигандов и 4 FGFRs (Eswarakumar et al., 2005; Beenken and Mohammadi, 2009; Cenni et al., 2005; Bossard et al., 2004; Compagni et al., 2000). Также селективные ингибиторы тирозинкиназы FGFR не были одобрены для клинического использования (Dimitroff et al., 1999; McDermott et al., 2005).
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
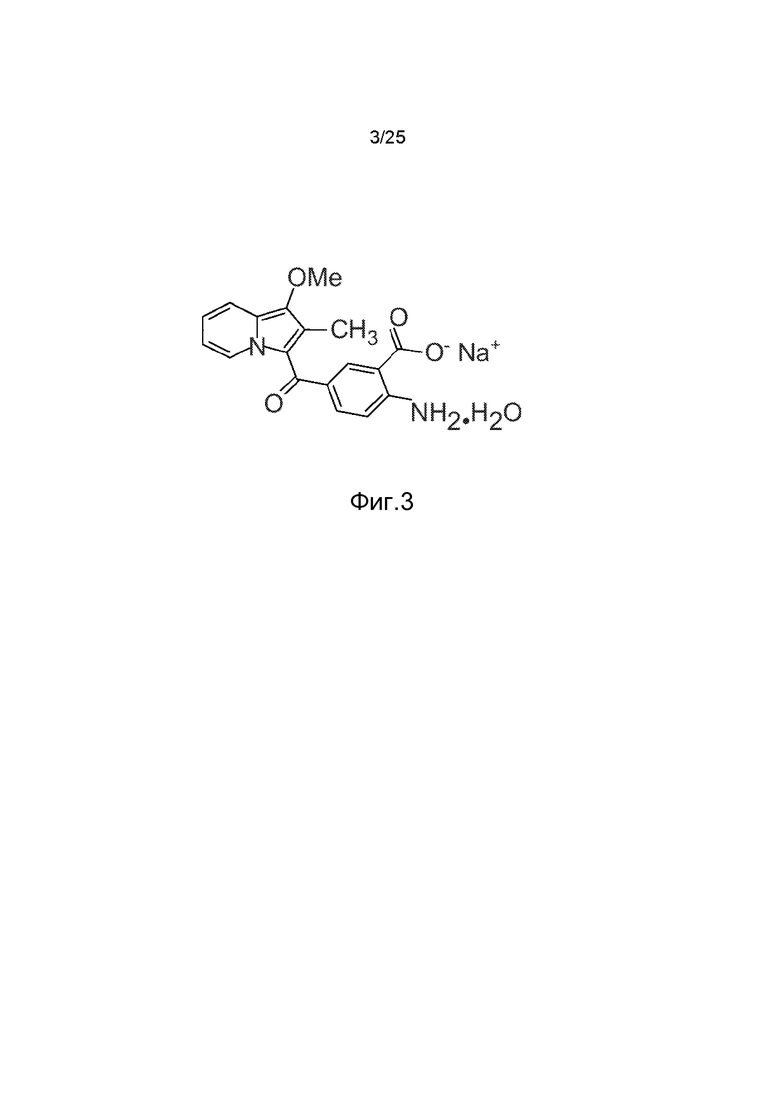
Неожиданно изобретатели нашли, что с помощью высокопроизводительного скрининга в сочетании с химической оптимизацией может быть идентифицирован первый перорально активный низкомолекулярный аллостерический ингибитор RTK, а именно FGFR. Это соединение названо SSR128129 (аббревиатура «SSR») (фиг.3).
Как проиллюстрировано с помощью подробного исследования на основании активности SSR, SSR обладает способностью ингибировать все члены одного и того же семейства, представляющего семейство FGFRs. Как представлено в последующих примерах, SSR способен ингибировать активность FGFR1 (фиг.4 и 6), активность FGFR2 (фиг.8), активность FGFR3 (фиг.9) и активность FGFR4 (фиг.7). Таким образом, этот аллостерический ингибитор связывается с эволюционно консервативным аллостерическим сайтом FGFR, расположенным во внеклеточном домене рецептора, который является общим для различных членов TRKs. Этот консервативный сайт расположен в домене III FGFR (фиг.11). Связывание SSR со своим сайтом связывания индуцирует «избирательный антагонизм». Этот эффект подтверждается тем фактом, что связывание SSR с аллостерическим сайтом связывания ведет к конформационному изменению рецептора, особенно в выявленном фрустрированном домене. Вследствие избирательного антагонизма предлагается путь идентификации аллостерического ингибитора с помощью использования тестирующего скрининга на основе измерений путей фосфо-сигнализации, как описано ниже. С этого момента SSR является первым примером аллостерического ингибитора RTK.
Подтверждение направленности на такой сайт в FGFR и направленности на сходные сайты в других RTKs, таких как VEGFR2 и PDGFRβ, связано с важным практическим применением и должно привести к существенному терапевтическому преимуществу.
Различные аспекты этого изобретения проиллюстрированы в подробном описании изобретения и в последующих примерах.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В первом аспекте в изобретении предлагается аллостерический сайт связывания, происходящий из внеклеточного домена тирозинкиназного рецептора. Как применяется в настоящем описании, аллостерический сайт связывания обозначает сайт, с которым ингибитор, предпочтительно низкомолекулярное вещество, может связываться, не вызывая конкурентного ингибирования связывания лиганда с лигандсвязывающим сайтом рецептора. Как применяется в настоящем описании, «происходящий из» обозначает, что аллостерический сайт связывания состоит из части внеклеточного домена, но не включает полный внеклеточный домен. Предпочтительно, аллостерический сайт связывания составляет между 10 и 200 аминокислотами в длину, более предпочтительно между 10 и 100 аминокислотами, даже более предпочтительно между 20 и 50 аминокислотами, где указанные аминокислоты являются частью внеклеточного домена рецептора.
Как применяется в настоящем описании, низкомолекулярное вещество представляет собой вещество неполимерной природы, предпочтительно с молекулярной массой менее 1000 Да, более предпочтительно менее 900 Да, более предпочтительно менее 800 Да, более предпочтительно менее 700 Да, более предпочтительно менее 600 Да, даже более предпочтительно менее 500 Да.
Тирозинкиназный рецептор и рецепторная тирозинкиназа (RTK) применяются в объеме данного патента как эквивалентные формы. «Тирозинкиназный рецептор» используется для обозначения рецептора, в то время как «рецепторная тирозинкиназа» используется для более конкретного указания на киназную активность рецептора. Тирозинкиназные рецепторы известны специалисту в данной области техники и включают, но не ограничиваются этим, рецепторы EGF, инсулиноподобного фактора роста, PDGF, FGF, VEGF, HGF, Trk, AXL, LTK, TIE, ROR, DDR, PKT7, RYK, CCK4, семейства рецепторов Eph и MuSK. Предпочтительно указанный аллостерический сайт связывания происходит из внеклеточного домена TRK с Ig-доменом, включая AXL, FGFR, MuSK, PDGFR, PTK7, ROR, TIE и VEGFR…; даже более предпочтительно указанный аллостерический сайт связывания происходит из TRK с прерывистым киназным доменом в цитоплазматическом домене; в предпочтительном варианте осуществления TRKs по изобретению представляют собой рецепторы фактора роста фибробластов или их гомологи, ортологи или паралоги.
«Гомологи» белка охватывают пептиды, олигопептиды, полипептиды, белки и ферменты, имеющие аминокислотные замены, делеции и/или вставки относительно интересующего немодифицированного белка и имеющие биологическую и функциональную активность, сходную с немодифицированным белком, из которого они произошли. «Ортологи и паралоги» охватываются эволюционными концепциями, используемыми для описания взаимоотношений предковых генов. Паралоги представляют собой гены тех же видов, которые произошли путем дупликации предкового гена; ортологи представляют собой гены различных организмов, которые произошли при видообразовании, и также происходят от общего предкового гена.
Такое аллостерическое связывание относится к SEQ ID NO: 1, принадлежащей к семейству FGFRs, более конкретно к FGFR2. Предпочтительно указанный аллостерический ингибиторный сайт включает SEQ ID № 1, даже более предпочтительно он состоит из SEQ ID № 1.
В другом аспекте в изобретении предлагается гомолог, паралог или ортолог аллостерического сайта связывания. Предпочтительно полипептидная последовательность этих гомологов, паралогов или ортологов имеет гомологию с SEQ ID NO.1, по меньшей мере, 70%, 80%, 90%, 95% или более.
В качестве примера, такие паралоги аллостерического сайта связывания присутствуют в семействе FGFRs.
Главным образом аллостерический сайт связывания по изобретению локализован в домене III FGFRs.
Таким же образом аллостерический сайт связывания VEGFR2 локализован в Ig-домене 6 рецептора в области, включающей лизин 609 и лизин 648.
Аллостерический сайт связывания присутствует также в PDGFRβ и локализован в области, расположенной вблизи трансмембранного участка, главным образом в Ig-домене 3, в области, включающей лейцин 383 и лизин 387. Предпочтительно связывание аллостерического ингибитора с аллостерическим сайтом связывания индуцирует избирательный антагонизм.
Применяемый в настоящем описании термин «избирательный антагонизм» обозначает, что для рецептора с несколькими нижележащими путями проведения сигнала не все пути задействованы или не все пути задействованы в одинаковой степени при связывании аллостерического ингибитора с аллостерическим ингибиторным сайтом связывания. В предпочтительном варианте осуществления, по меньшей мере, один нижележащий путь ингибирован, тогда как, по меньшей мере, один другой нижележащий путь не затронут.
Предпочтительно аллостерический сайт связывания по изобретению включает, предпочтительно по существу состоит, даже более предпочтительно состоит из фрустрированного домена.
Применяемый в настоящем описании термин «фрустрированный домен» обозначает домен белка или его фрагмента, который неоднозначно ориентирован на одну структурную конформацию; фрустрированные домены известны специалисту в данной области техники, и присутствие фрустрированных доменов определяется либо по неоднозначному ответу в одной программе предсказания вторичной структуры белка; либо по противоречию в предсказании между двумя различными программами предсказания вторичной структуры белка. Предпочтительно он определяется по противоречию в предсказании программой предсказания вторичной структуры белка и реальной структурой, определенной методом определения структуры белка, таким как кристаллизация и дифракция рентгеновских лучей. В качестве неограничивающего примера, противоречие может быть в указании на α-спираль в одном методе и на β-складку в другом методе. Белки являются минимально фрустрированными; однако, некоторые домены (называемые «фрустрированными доменами») индуцируют некоторую фрустрацию, и эти домены склонны индуцировать конформационные изменения белка.
В предпочтительном варианте осуществления указанный фрустрированный домен включает SEQ ID № 2, предпочтительно он состоит из SEQ ID № 2. Этот фрустрированный домен принадлежит к семейству FGFRs, особенно к FGFR2.
Другие фрустрированные домены могут быть идентифицированы, как указано выше.
В другом аспекте в изобретении предлагается использование аллостерического сайта связывания по изобретению для индукции избирательного антагонизма при связывании лиганда с сайтом связывания тирозинкиназного рецептора, в котором локализован аллостерический сайт связывания. В еще одном аспекте в изобретении предлагается использование аллостерического сайта связывания по изобретению для скрининга низкомолекулярных ингибиторов, полученных из случайной библиотеки, связывающихся с указанным сайтом.
В еще одном аспекте в изобретении предлагается способ идентификации аллостерического ингибиторного сайта связывания во внеклеточном домене тирозинкиназного рецептора, включающий скрининг на присутствие фрустрированных доменов в указанном внеклеточном домене. Способы скрининга фрустрированных доменов известны специалисту в данной области техники и пример такого способа описан в примере 8. В качестве неограничивающего примера фрустрированные домены определяют по неоднозначному ответу в одной программе предсказания вторичной структуры белка; предпочтительно по противоречию в предсказании между двумя различными программами предсказания вторичной структуры белка, даже более предпочтительно по противоречию в предсказании программой предсказания вторичной структуры белка и реальной структурой, определенной методом определения структуры белка, таким как кристаллизация и дифракция рентгеновских лучей. Программы предсказания вторичной структуры белка известны специалисту в данной области техники; в качестве неограничивающего примера такие программы описаны Rost (2003). Предпочтительно указанный фрустрированный домен расположен в непосредственной близости от указанного аллостерического сайта связывания; более предпочтительно он локализован не более чем на 20 аминокислот от границы сайта связывания, даже более предпочтительно не более чем на 10 аминокислот, даже более предпочтительно он примыкает к указанному сайту связывания, даже более предпочтительно он перекрывается с указанным сайтом связывания, наиболее предпочтительно он включен в сайт связывания. После идентификации возможных ингибиторных сайтов скрининг может быть завершен подтверждением функции возможного ингибиторного сайта путем создания соединений, таких как малые молекулы, малые пептиды, пептидомиметики, антитела или нанотела, которые связываются с сайтом и для которых может быть протестирована аллостерическая ингибиторная функция.
В другом аспекте в изобретении предлагается способ идентификации низкомолекулярного аллостерического ингибитора, связывающегося с аллостерическим сайтом связывания внеклеточного домена тирозинкиназного рецептора по изобретению, включающий сравнение двух различных репортеров, индуцируемых двумя различными нижележащими путями сигнализации в зависимости от активации указанного тирозинкиназного рецептора. Репортер представляет собой любой ген, белок или соединение, которое дает определяемый сигнал, и в качестве неограничивающего примера может представлять собой ген устойчивости к антибиотикам, ген токсина, приводящего к клеточной гибели, ген, кодирующий флуоресцентный белок, такой как GFR, или ген, кодирующий белок с ферментативной активностью, такой как бета-галактозидаза, или белок, который фосфорилируется или дефосфорилируется, ацетилируется или дезацетилируется или конформация которого изменяется. В случае репортерного гена кодирующую последовательность помещают под контроль подходящего промотора, т.е. промотора, который индуцируется в результате связывания лиганда с рецептором и последующей индукции пути для репортера; в случае двойного пути сигнализации требуется два различных промотора. В качестве неограничивающего примера при сравнении фосфорилирования белков в присутствии или в отсутствие аллостерического ингибитора можно получить различия в фосфорилировании из-за избирательного антагонизма, и эти различия в фосфорилировании могут быть использованы в качестве репортерных.
В предпочтительном варианте осуществления идентификация аллостерического ингибитора RTK может быть выполнена путем осуществления метода скрининга, включающего следующие стадии:
a) введения в контакт аллостерического сайта связывания RTK с кандидатным веществом для аллостерического ингибирования
b) измерения изменений, по меньшей мере, в двух нижележащих путях в зависимости от активации/ингибирования указанного тирозинкиназного рецептора,
c) сравнения изменений в состоянии, по меньшей мере, одного репортера, у которого, по меньшей мере, два различных нижележащих пути зависят от активации/ингибирования указанного тирозинкиназного рецептора,
где аллостерический ингибитор является идентифицированным, когда при наличии связывания лиганда с лигандсвязывающим доменом рецептора, по меньшей мере, один нижележащий путь ингибирован, тогда как, по меньшей мере, один другой нижележащий путь не затронут. Изменение в состоянии репортера зависит от используемого репортера и может представлять собой в качестве неограничивающего примера изменение фосфорилирования репортерного белка или переключение от отсутствия индукции к индукции гена (и наоборот). Предпочтительно указанное изменение в состоянии представляет собой изменение в состоянии фосфорилирования.
Предпочтительно изменения в нижележащих путях прослеживаются путем измерения изменений в путях фосфо-сигнализации, включая сигнальные пути ERK1/2 и PLCγ.
В другом варианте осуществления аллостерический модулятор FGF-Rs может быть идентифицирован при использовании аффинного скрининга на основе SEC-LC/MS, как описано ниже:
Метод SEC-LC/MS представляет собой аналитический метод, используемый для аффинного скрининга, состоящий из 2-компонентной системы, соединенной в режиме реального времени: хроматографии, исключающей по размеру, в сочетании с высокоэффективной жидкостной хроматографией для выделения с последующей ионизацией электрораспылением с использованием для детекции время-пролетной масс-спектрометрии.
Метод основан на способности некоторых соединений взаимодействовать с растворимыми полипептидами (включая пептиды, домены белков или полноразмерные белки). После смешивания пула низкомолекулярных соединений с интересующим пептидом комплекс пептид-лиганд индуцирует сдвиг массы, позволяющий разделить несвязанные и связанные низкомолекулярные соединения с помощью хроматографии, исключающей по размеру. Затем комплекс диссоциируют, и связываемые соединения отделяют от пептида и определяют с использованием LC/ESI-TOF высокого разрешения для точного измерения массы (например, с помощью масс-спектрометра Waters LCT Premier). Данные деконволюционного алгоритма позволяют идентифицировать связанные молекулы из данных анализа определения масс.
Для идентификации низкомолекулярных веществ аллостерических модуляторов FGFRs этот метод может быть применен к внеклеточному домену различных FGFRs, либо нативных, либо мутантных. Нативная форма позволяет определить всех участников связывания с внеклеточным доменом. Альтернативно, можно провести скрининг аллостерических модуляторов путем использования «открытой» формы спирали FGF-R2 в непосредственной близости к сайту связывания SSR. Указанная «открытая» форма может быть получена с помощью мутации Tyr328Arg-Ile329Lys, которая стабилизирует альфа-спираль, позволяя тем самым повысить чувствительность к связыванию SSR. Мутантный FGF-R2 затем используют для скрининга вместо WT FGF-R2. Сходная стратегия может быть использована для скрининга FGF-R1, -R3 или -R4 с мутациями аминокислот, соответствующими Tyr328 и Ile329 в FGF-R2. Мутантная форма с Tyr328Asp (FGF-R2) или других FGF-Rs с мутацией в соответствующих положениях может быть использована в качестве контроля. Действительно, SSR не способен связываться с FGF-R2, который мутирован с Tyr328Asp вблизи гидрофобного кармана. Следовательно, эта мутантная форма может быть использована для отбрасывания части соединений, которые не взаимодействуют с карманом FGF-R2, являющимся мишенью.
Во всех этих случаях данная стратегия ведет к идентификации низкомолекулярных соединений, способных связываться с карманом интересующего пептида, являющимся мишенью. На второй стадии оценивается влияние на сигнализацию в клетке. На основе путей фосфо-сигнализации, идентифицированных матрицей Proteome Profiler™ «набор с матрицей фосфокиназ человека» от R&D Systems, аллостерические модуляторы могут быть проверены с помощью ELISA (на белковых клеточных экстрактах или прямо на клетках) на их способность ингибировать действие FGF-2 на HUVEC на уровне фосфорилирования киназ (на PYK2, eNOS, p53, c-jun, AKT, CREB, Erk1/2) в отсутствие ингибирования не подверженных влиянию киназ, определенных по протеомному профилю.
Сходный подход может быть использован для других RTKs: после идентификации одного или более фрустрированных доменов во внеклеточном домене рецептора, указанный фрустрированный домен может быть использован в подходе SEC-LC/MS для идентификации связывающихся соединений в области фрустрированного домена. Затем может быть протестировано действие связывающегося соединения на путь сигнализации с использованием подхода картирования фосфо-белков, как описано выше, или любой другой репортерной системы пути.
В еще одном аспекте в изобретении предлагается низкомолекулярное соединение, связывающееся с аллостерическим сайтом связывания, называемое также «аллостерическим ингибитором» по изобретению, и/или идентифицированное способом по изобретению.
«Соединение» обозначает любое химическое или биологическое соединение, включая простые или сложные, органические или неорганические молекулы, пептиды, пептидомиметики, белки, антитела, углеводы, нуклеиновые кислоты или их производные.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг.1: A/Эксперимент с количественной ПЦР на HUVEC выявил экспрессию только FGFR1 и FGFR4. B/Анализом ОТ-ПЦР идентифицированы изоформы FGFR1β и FGFR4. FGFR1 представлен в формате варианта IIIc (C).
Фиг.2: Клетки BaF/3, трансфецированные FGFR1βIIIc-hMpI, способны пролиферировать, когда вставленный FGFR активирован. Только FGF4 (A) способен индуцировать FGFR1βIIIc, тогда как FGF19 не способен (B).
Фиг.3: Демонстрация соединения SSR
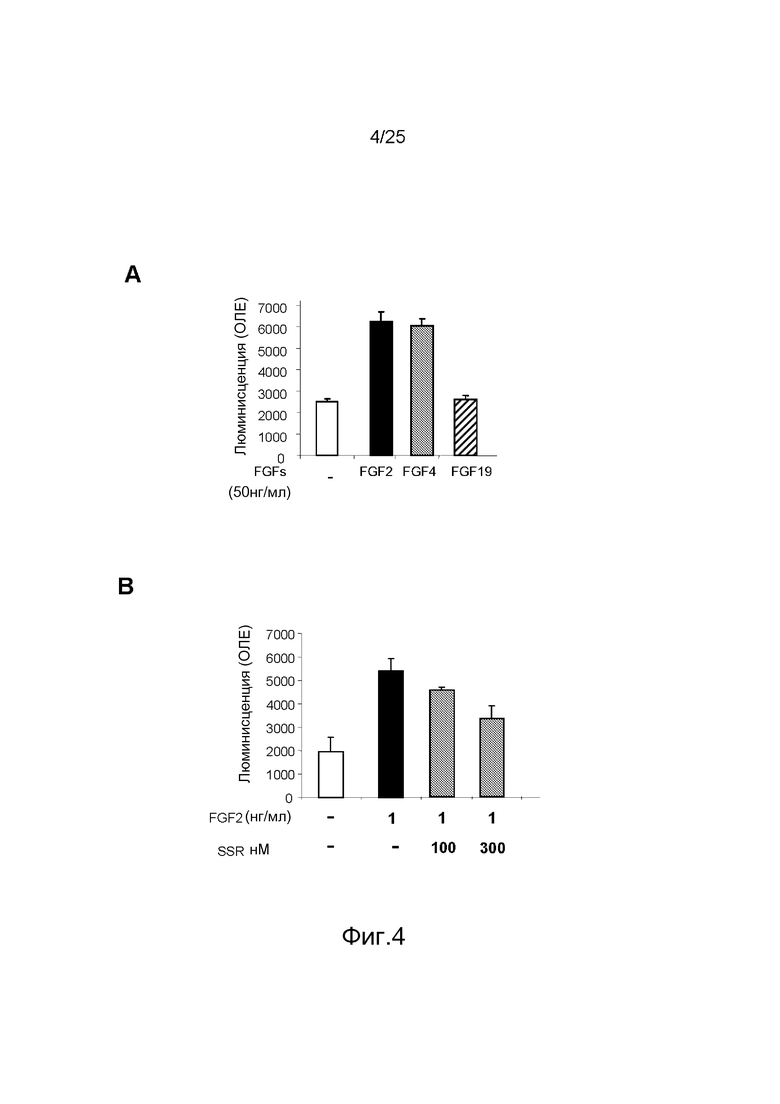
Фиг.4: Исследование активности SSR по пролиферации клеток эндотелия. A/Только FGF2 и FGF4 стимулируют пролиферацию HUVEC, демонстрируя, что FGFR1 управляет пролиферацией в этих клетках. B/SSR ингибирует индуцируемую FGF2 пролиферацию HUVEC, демонстрируя антагонистическое действие SSR на FGFR1. C/В эндотелиальных клетках жировых телец крыс (REPEC), трансфецированных FGFR1, FGF2 индуцировал аутофосфорилирование FGFR1, которое только частично ингибировалось SSR даже при высоких дозах. D/Это ингибирование не обусловлено конкурентным действием SSR на связывание FGF2 в PAEC, трансфецированных FGFR1, или в HUVEC.
Фиг.5: A/Связывание FGF1, флуоресцентно меченного lumio (FGF1-lumio), с очищенным ECD FGFR2 без Fc-метки (FGFR2∂123) по измерению скорости ротации в качестве параметра анизотропии. Не наблюдается прямой конкуренции между SSR и FGF1-lumio. B/SSR не способен ингибировать мультимеризацию FGFR2 или C/димеризацию FGF2.
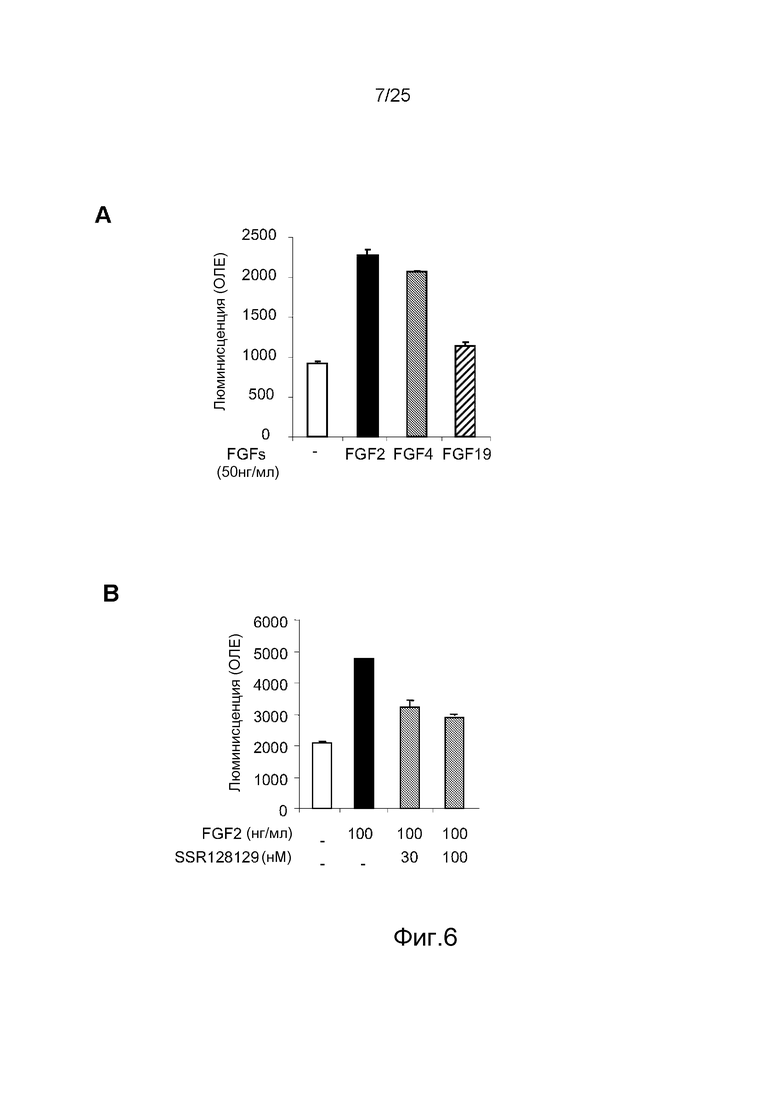
Фиг.6: Исследование активности SSR в отношении хемотаксиса миграции эндотелиальных клеток. A/Только FGF2 и FGF4 стимулируют миграцию HUVEC, указывая на то, что FGFR1 управляет пролиферацией этих клеток. B/SSR ингибирует индуцируемую FGF2 хемотаксическую миграцию HUVEC в соответствии со своим антагонистическим действием на FGFR1.
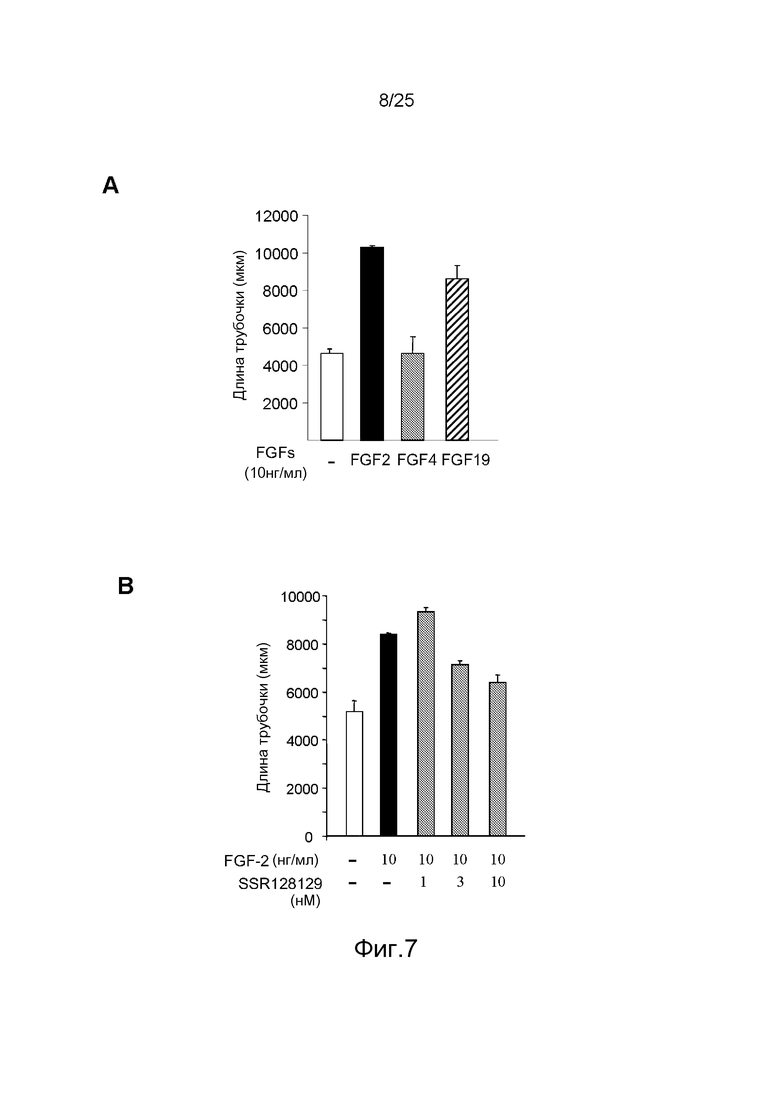
Фиг.7: Исследование активности SSR на эндотелиальных клетках в отношении ангиогенеза in vitro. A/Только FGF2 и FGF19 стимулируют ангиогенез HUVEC, указывая на то, что FGFR4 контролирует данную стадию дифференцировки этих клеток. B/SSR ингибирует индуцируемый FGF2 ангиогенез HUVEC in vitro в соответствии со своим антагонистическим действием на FGFR4.
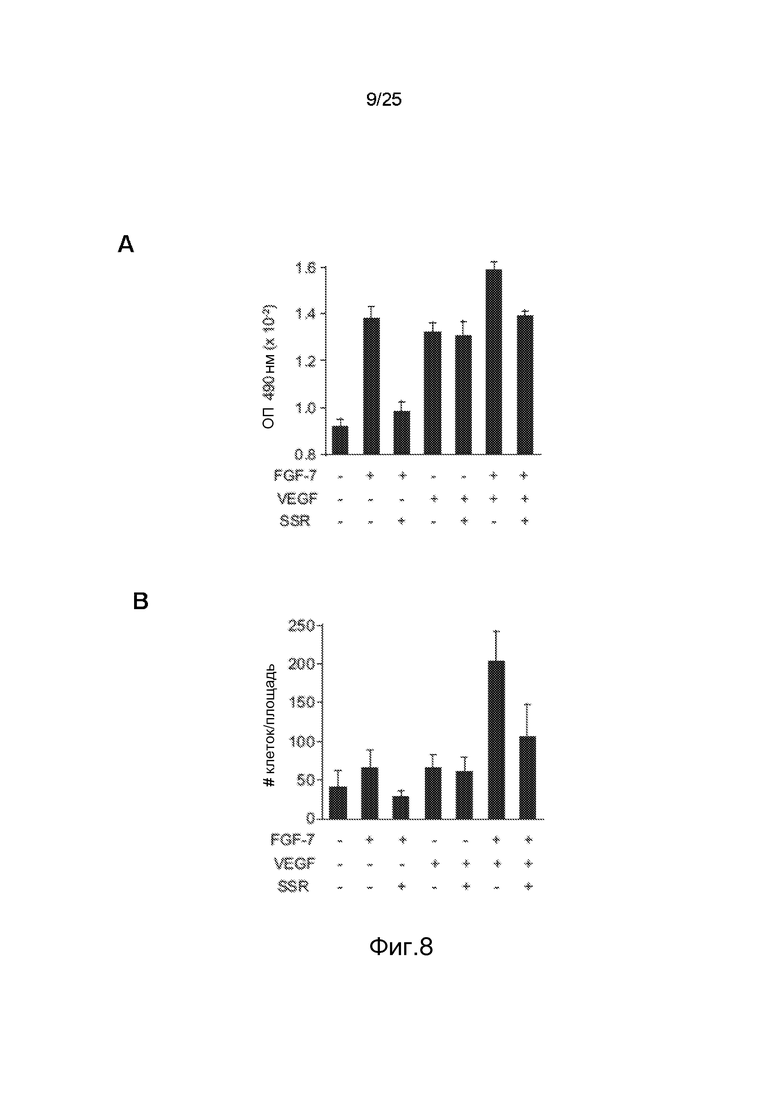
Фиг.8: Исследование активности SSR в отношении пролиферации и миграции PANC02. Клетки PANC02 пролиферируют (A) или мигрируют (B) при стимуляции FGF7 с VEGF или без него, предполагая зависимость системы от FGFR2IIIb. SSR ингибирует индуцируемую FGF7 пролиферацию PANC02 и индуцируемую FGF7+VEGF миграцию клеток, что демонстрирует его способность ингибировать FGFR2IIIb.
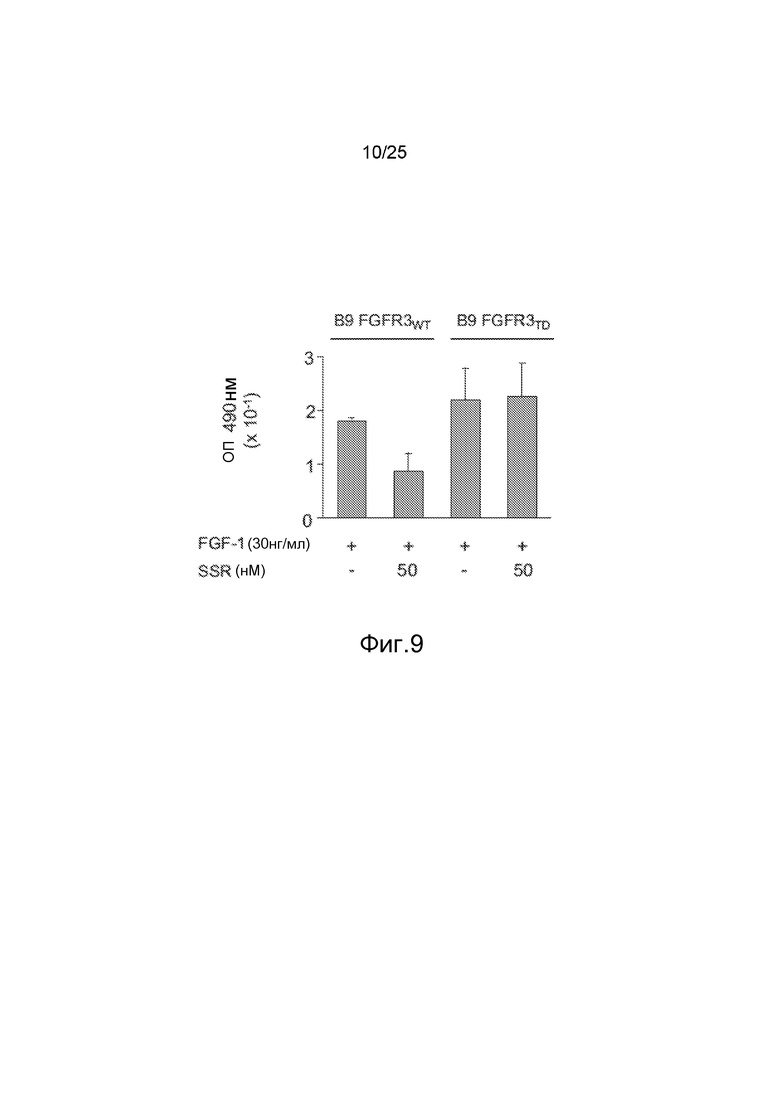
Фиг.9: Исследование активности SSR в отношении пролиферации клеток B9 миеломы. A/FGF1 индуцирует пролиферацию клеток B9 миеломы через FGFR3, а SSR ингибирует эту стимуляцию, указывая на то, что SSR способен блокировать FGFR3. B/Тем не менее, SSR не способен ингибировать пролиферацию клеток B9, трансфецированных аутоактивированным мутантным FGFR3 (с конститутивно фосфорилированным киназным доменом). Эти результаты демонстрируют внеклеточное действие SSR.
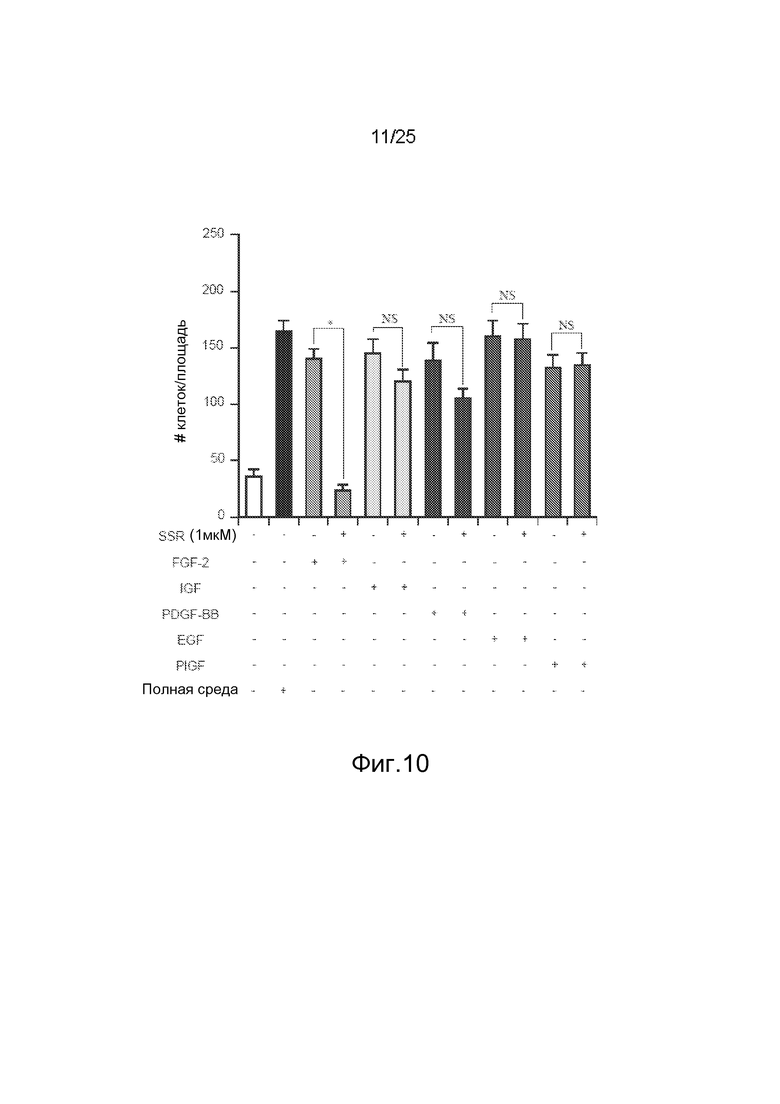
Фиг.10: Тест на миграцию HUVEC, в котором SSR не способен существенно ингибировать клеточную миграцию, индуцируемую различными факторами роста, такими как IGF, PDGF-BB, EGF или PIGF. SSR является специфичным для FGFR и блокирует только индуцируемую FGF миграцию HUVEC.
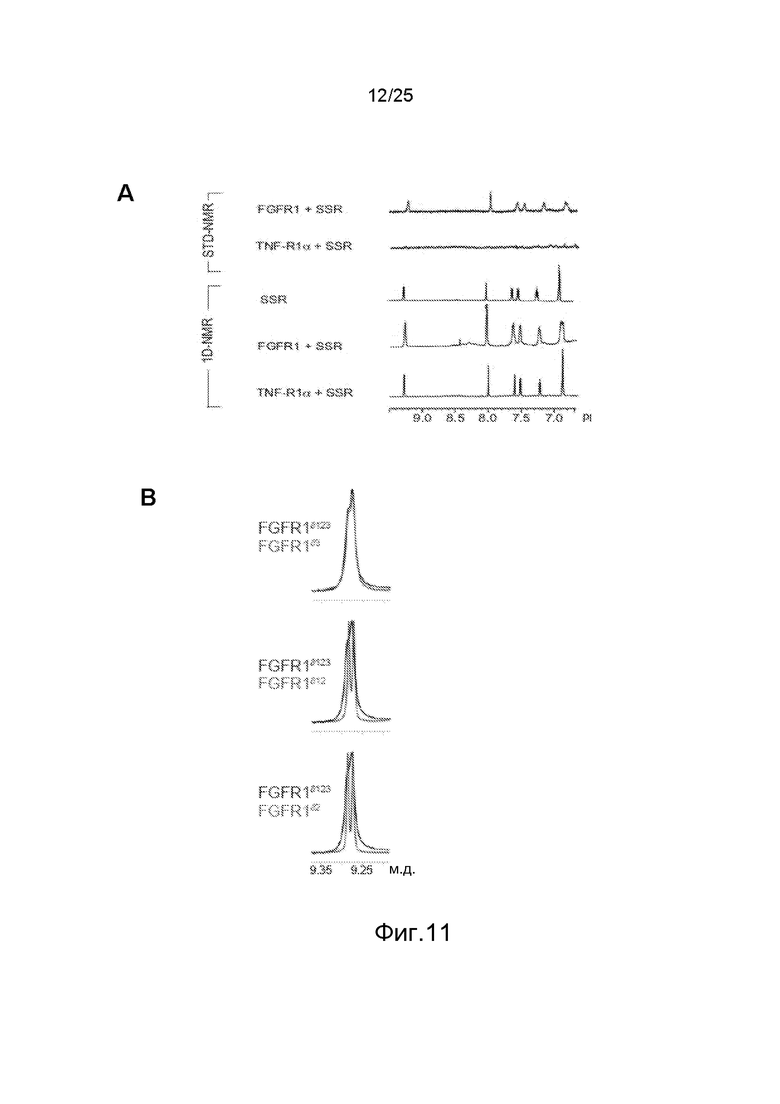
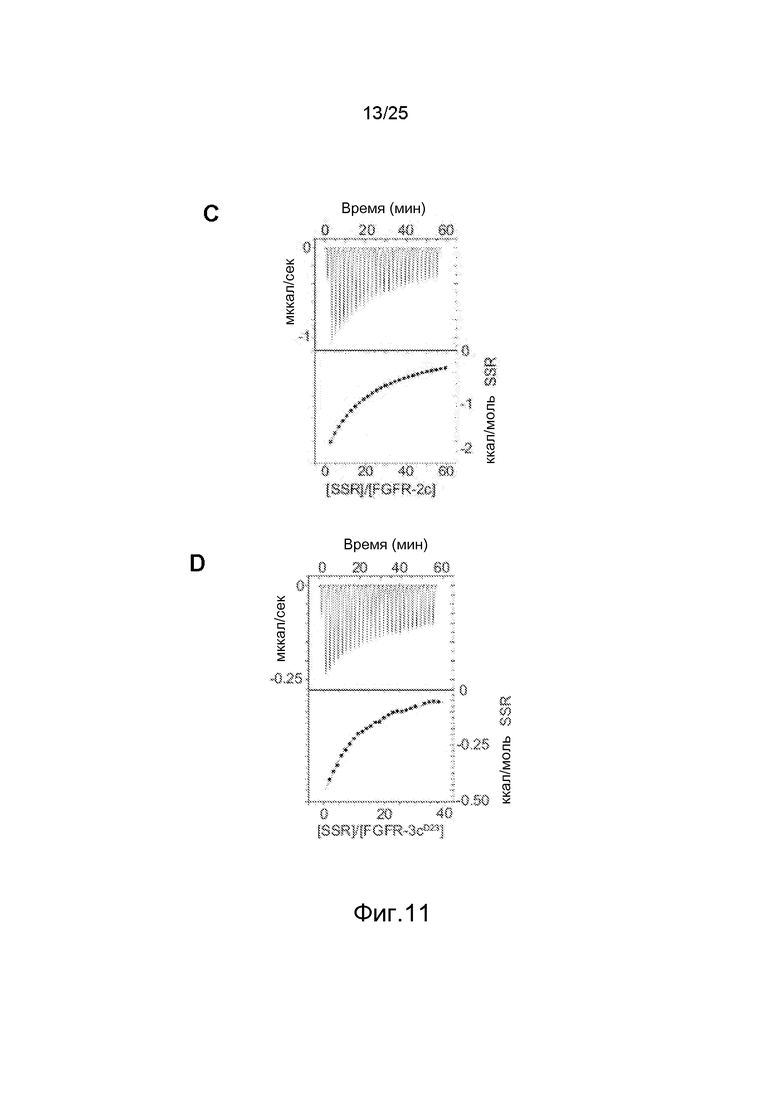
Фиг.11: Исследования с помощью ЯМР, демонстрирующие связывание SSR с доменом III FGFR. A/1D- и STD-NMR анализ связывания SSR с внеклеточным доменом FGFR1. Насыщения не наблюдается с контрольным TNFR1α. B/1D-NMR анализ связывания SSR с различными доменами FGFR1 показывает, что спектры, получаемые с полноразмерным FGFR1 и с доменом II FGFR1 сходны, что предполагает сайт взаимодействия в домене III. C/Изотермическая калориметрия титрования, показывающая способность SSR связываться с FGFR2 и FGFR3. (D) Внеклеточный домен.
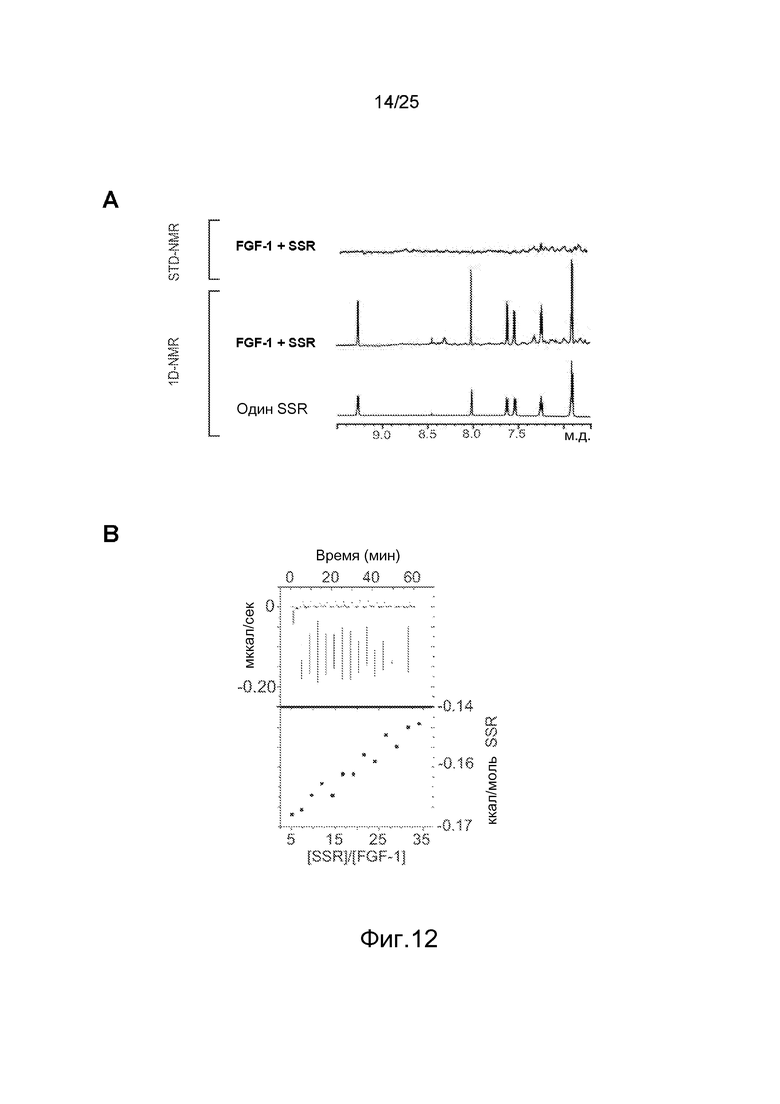
Фиг.12: Исследования с помощью ЯМР (A, C) и ITC (B), демонстрирующие, что SSR не способен взаимодействовать с FGF1 (A, B) и FGF2 (C). D/Не наблюдается вмешательства в связывание SSR с FGFR1 после добавления октасульфата сахарозы (SOS), миметиков гепарина, подтверждая, что SSR не взаимодействует с гепаринсвязывающим сайтом FGFR1.
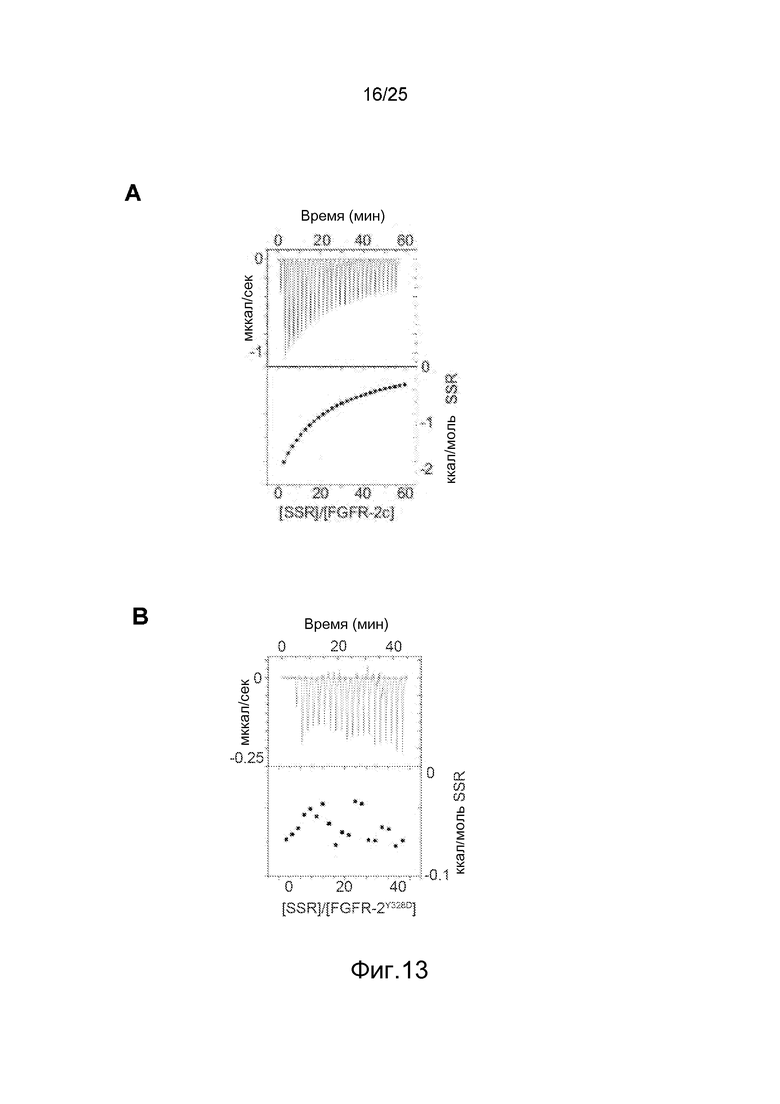
Фиг.13: Моделирование in silico и мутагенез идентифицируют аллостерический сайт связывания для SSR в области аминокислоты Y328. Эксперименты ITC с внеклеточным доменом WT FGFR2 (A) демонстрируют взаимодействие между SSR и FGFR2. Это связывание не происходит, когда измерения выполняются с мутантом Y328D (B), это подтверждает, что мутация Y328D вызывает потерю чувствительности FGFR2 к связыванию SSR.
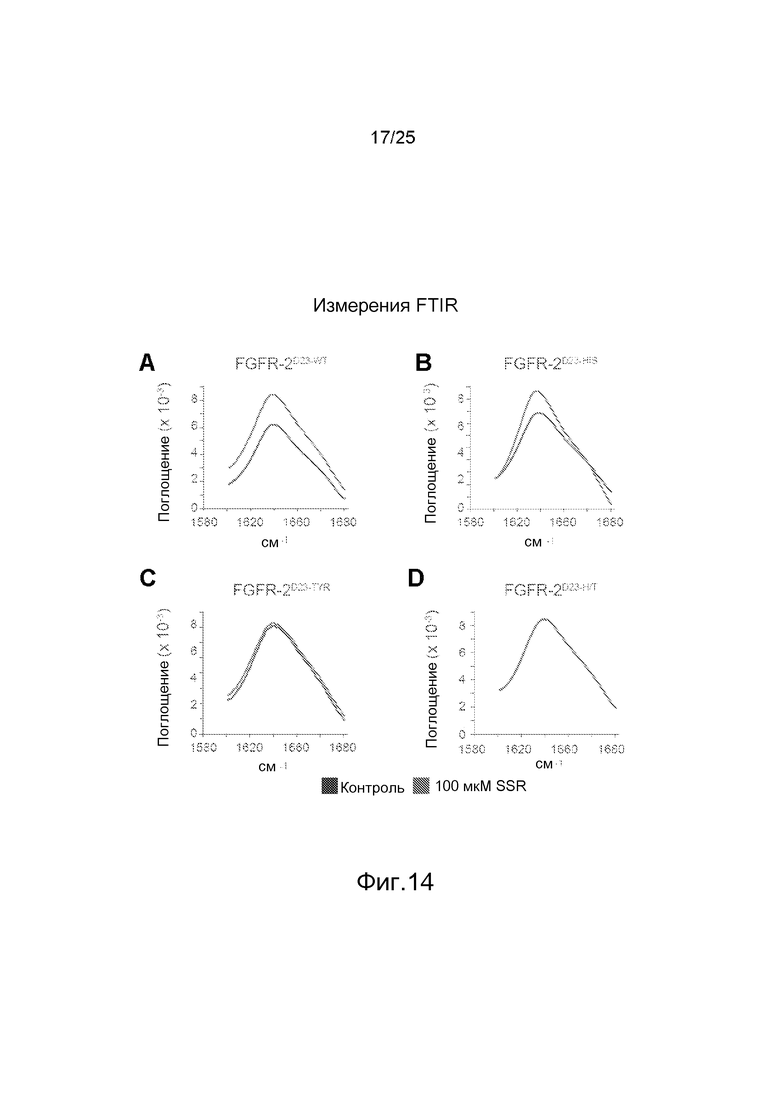
Фиг.14: Измерения с помощью инфракрасной спектроскопии на основе преобразования Фурье (FTIR) очищенного (A) FGFRδ23-WT, (B) FGFR2-δ23-His, (C) FGFR2-δ23-Tyr, (D) FGFR2-δ23-H/T (двойного мутанта H295L/Y328D) без или с 100 мкМ SSR (черная и серая линии, соответственно) идентифицировали конформационное изменение как в WT, так и в His295L мутанте, тогда как мутация Y328D вызывает потерю чувствительности FGFR2 к этому изменению.
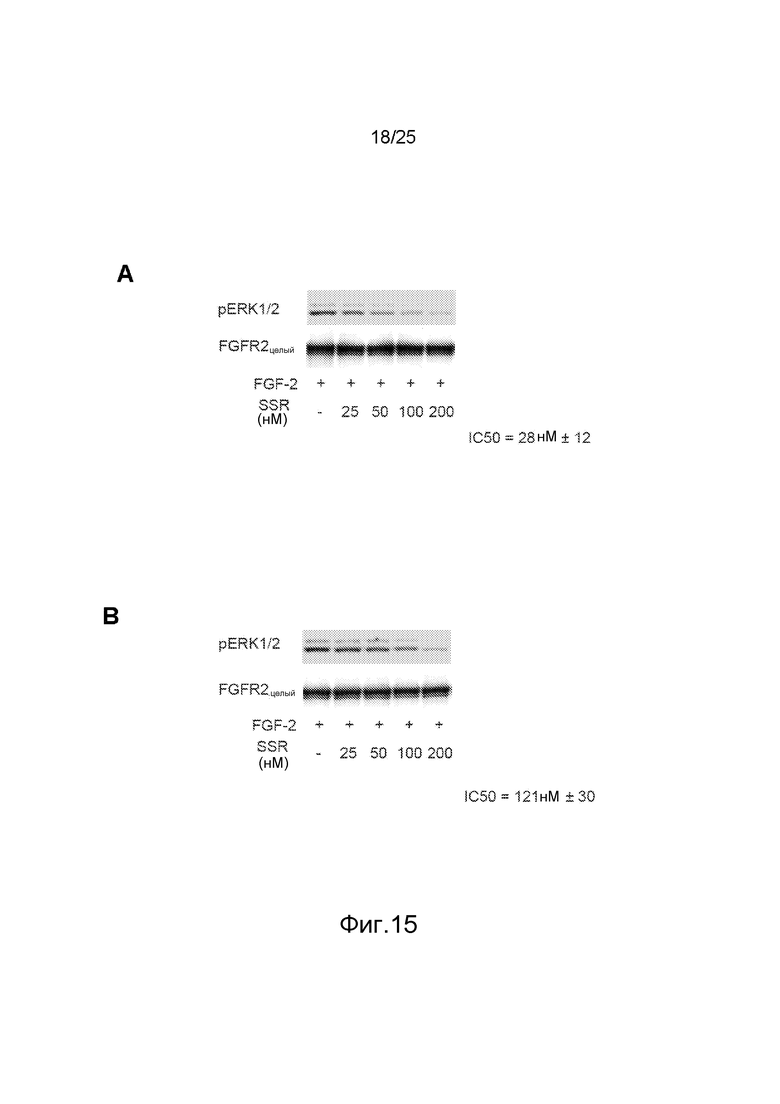
Фиг.15: Анализ иммуноблоттингом активированной Erk1/2 после стимуляции FGF2 (05, нг/мл в течение 5 мин) в клетках HEK293, стабильно трансфецированных полноразмерным FGFR2-WT или -Y328D. Определенная денситометрией величина IC50 (среднее + ст.ош.ср.; три независимых эксперимента) показала, что мутантный рецептор FGFR2-Y328D (B) приблизительно в 5 раз менее чувствителен к ингибированию SSR по сравнению с FGFR2-WT (A).
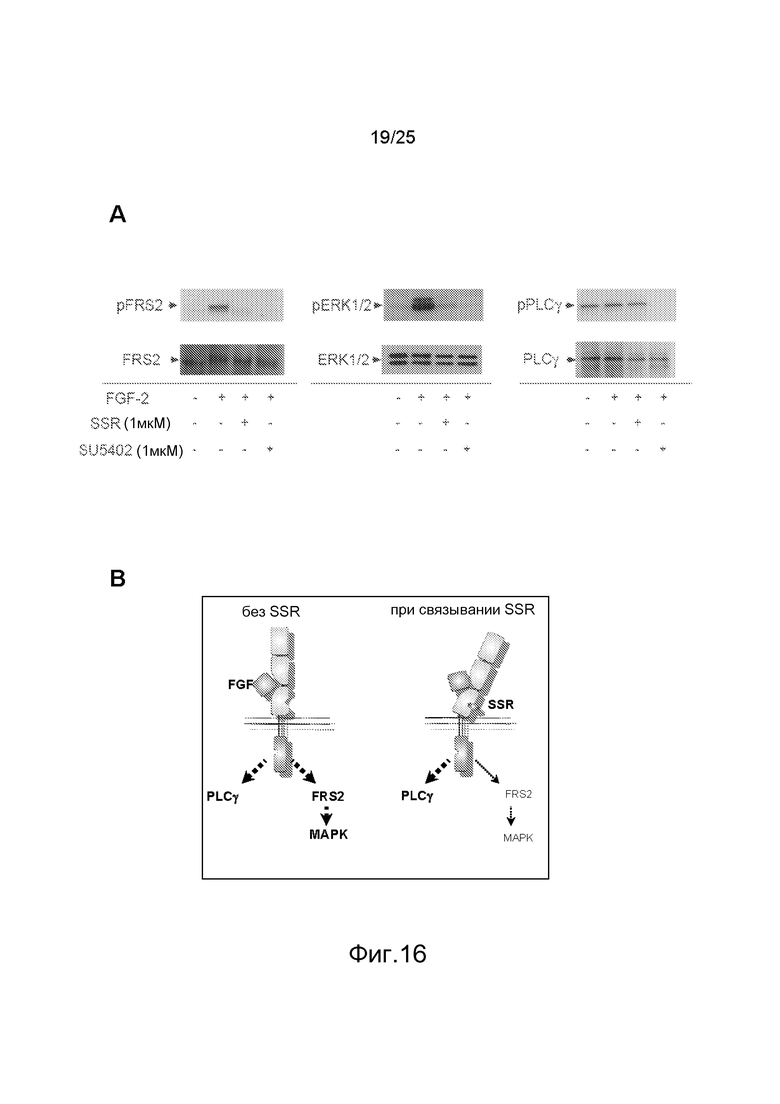
Фиг.16: A/Анализ иммуноблоттингом действия SSR по сравнению с ингибитором SU5402 тирозинкиназы FGFR на клетки HEK, трансфецированные FGFR2, стимулированные FGF2. SU5402 ингибирует фосфорилирование PLCγ, FRS2 и Erk1/2, тогда как SSR не ингибирует путь PLCγ, что указывает на избирательный антагонизм SSR (B).
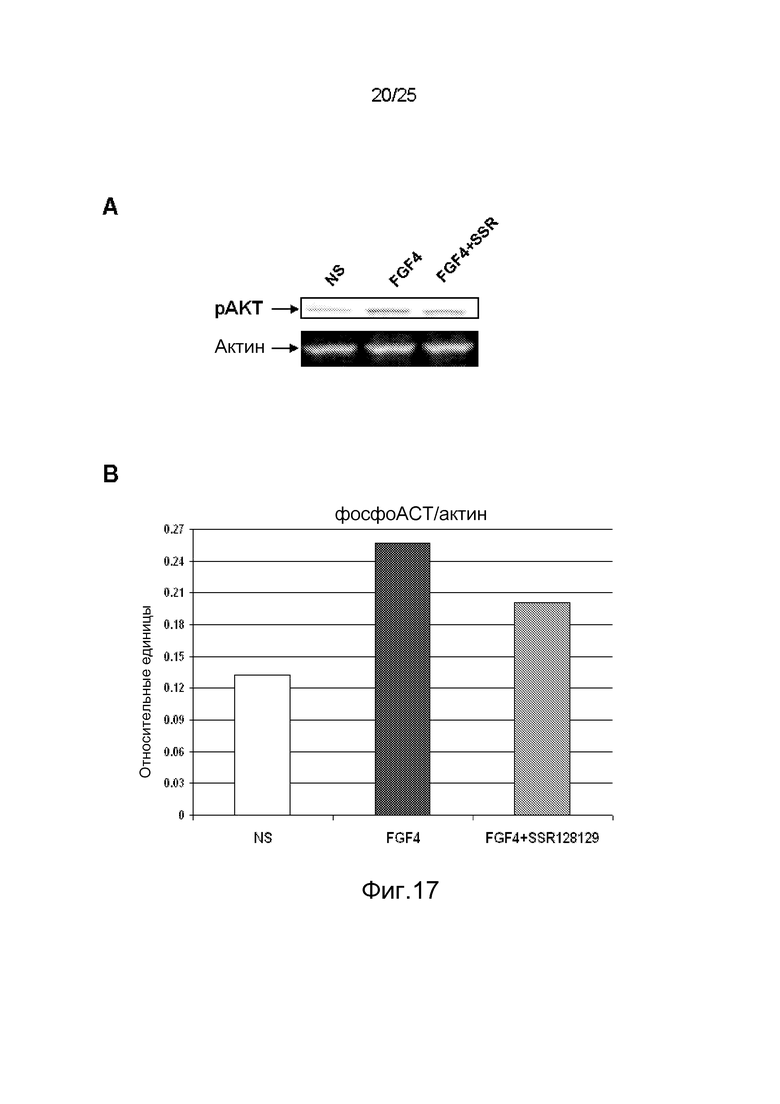
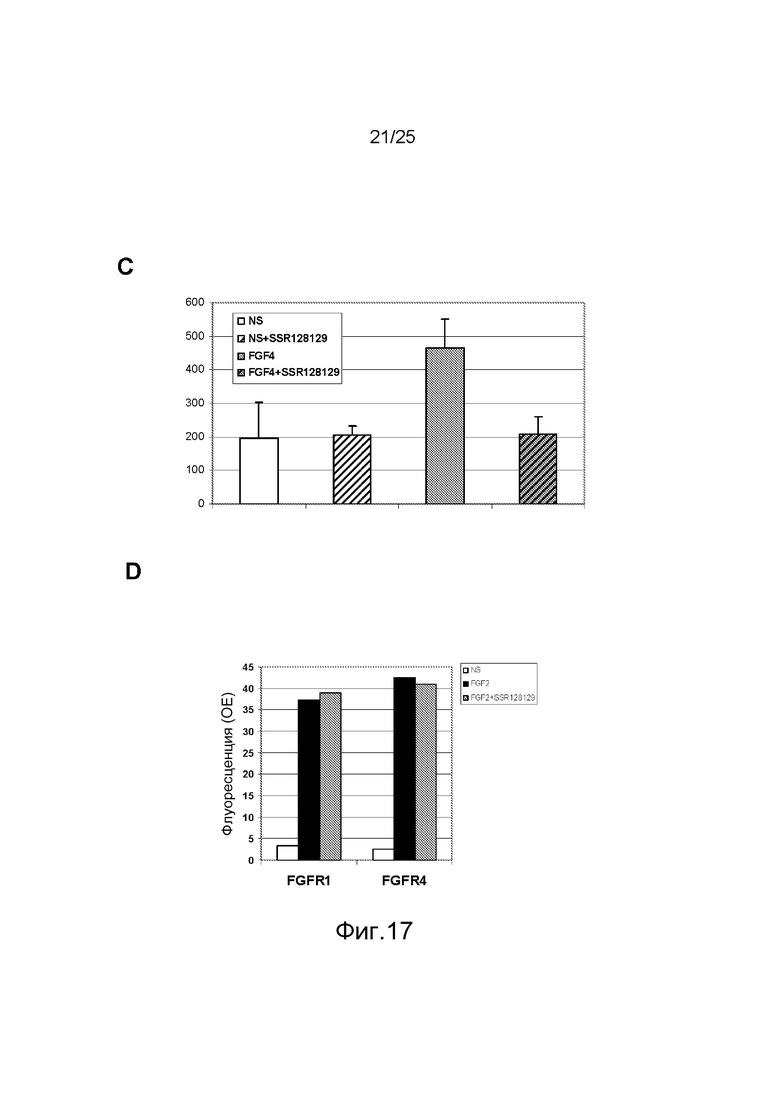
Фиг.17: Анализ иммуноблоттингом (A) действия SSR на индуцируемое FGF2 фосфорилирование AKT в HUVEC с соответствующей диаграммой количественной оценки (B). C/Этот эффект также количественно оценен с помощью ELISA на клетках, с антителами направленными против фосфо-AKT (Ser473). D/Этот эффект не зависит от отсутствия способности SSR конкурировать с FGF за связывание с FGFR.
Фиг.18: Идентификация предполагаемых фрустрированных зон в рецепторе VEGF-R2 с использованием компьютерной программы AGADIR и мутационного анализа. (A) Идентифицированы некоторые области, которые склонны подвергаться структурным изменениям (например, переходам из β-складки в α-спираль). (B) Два лизина (K609 и K648) выбраны для мутации в аспартат из-за их непосредственной близости к трансмембранному домену и из-за их наиболее отрицательного вклада в свойство образовывать спираль после мутации.
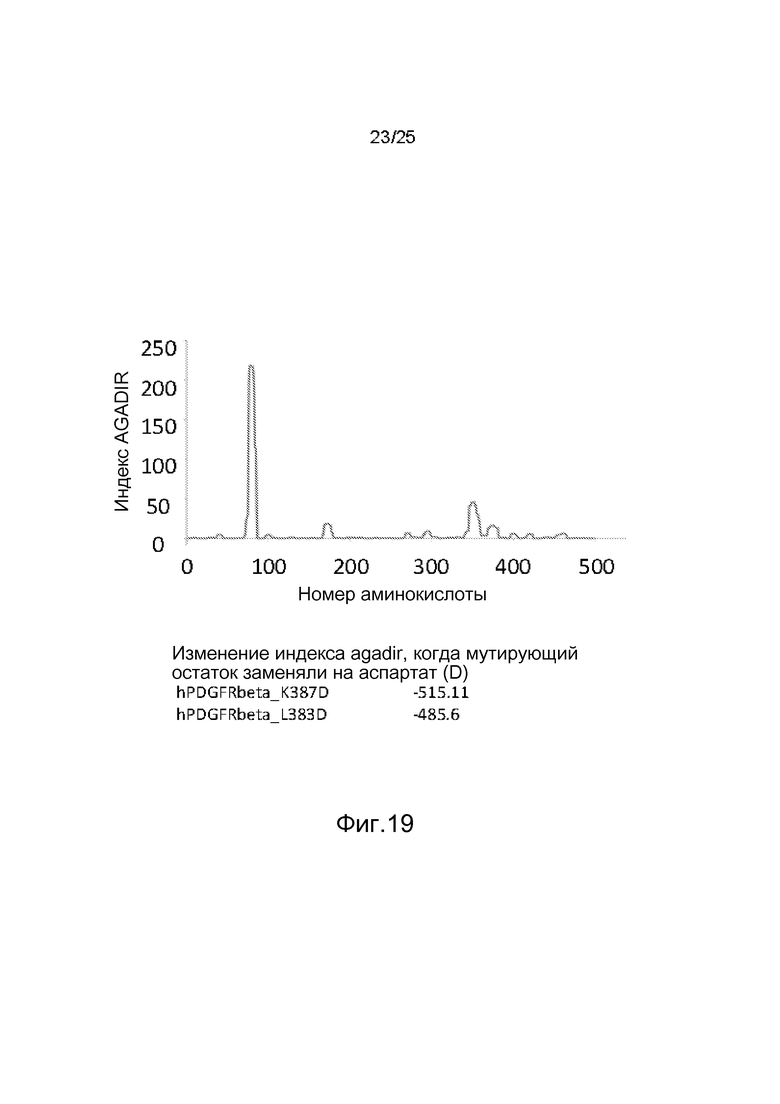
Фиг.19: Идентификация предполагаемых фрустрированных зон в рецепторе PDGF-Rβ с использованием компьютерной программы AGADIR и мутационного анализа. Мутации лизин387 в аспартат и лейцин383 в аспартат, очевидно, характеризуются наибольшим отрицательным вкладом в свойство образовывать спираль.
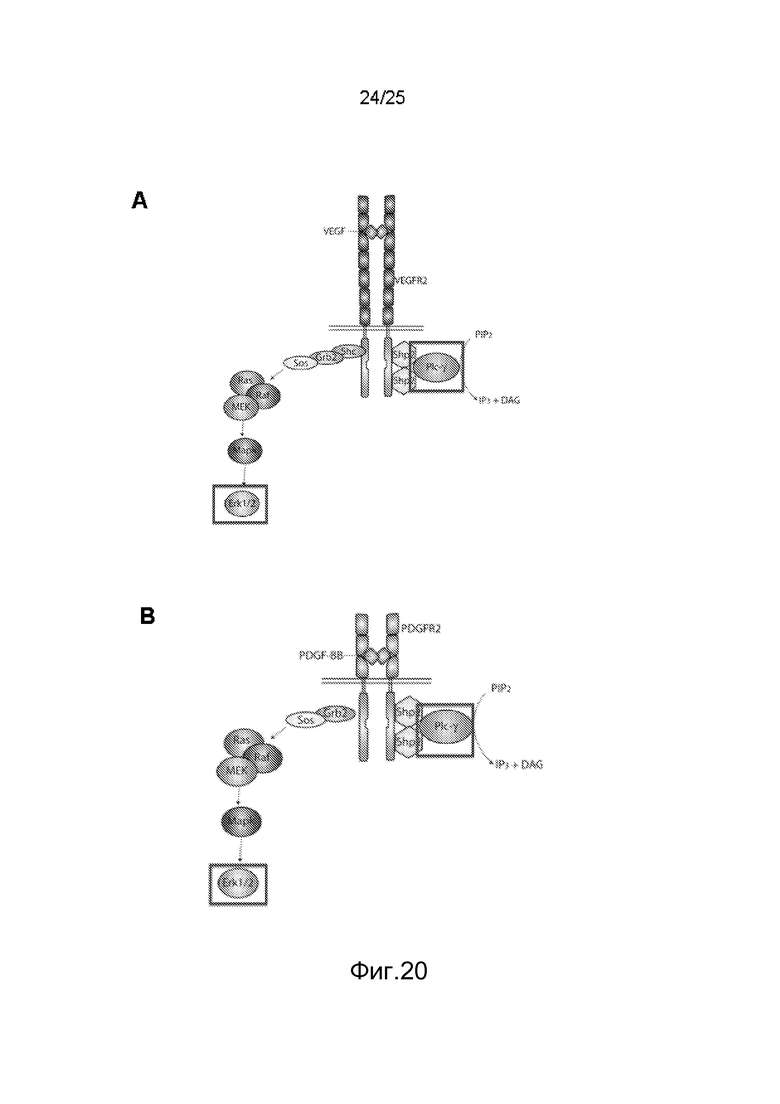
Фиг.20: Схематическое представление активации сигнальных путей с рецепторов VEGF-R2 и PDGF-Rβ через Erk1/2 и PLCγ. (A) Сигнализация через VEGFR2 после стимуляции VEGF и (B) сигнализация через PDGF-Rβ после стимуляции PDGF.
Фиг.21: Определение фосфорилирования Erk1/2 в клетках HEK293, которые гиперэкспрессируют дикий тип или мутантные формы рецепторов VEGF-R2 или PDGF-Rβ c использованием иммуноблоттинга или теста SureFire. (A) Клетки HEK293 стабильно трансфецировали мышиным VEGF-R2 дикого типа и мутированным в K609D или K648D. После выращивания на минимальной среде и стимуляции без (0) или с (+) мышиным VEGF определяли фосфорилирование с помощью иммуноблоттинга. (B) Схематическое представление теста SureFire для определения фосфорилирования Erk1/2 (левая схема) или PLCγ (правая схема) в белковых экстрактах. (C) Данные AlphaScreen SureFire для суммарной или фосфорилированной Erk1/2 в клетках HEK293, трансфецированных PDGFRβ, после стимуляции 10% FBS, 1 или 50 нг/мл PDGF-BB или без стимуляции в качестве контроля (0).
ПРИМЕРЫ
МАТЕРИАЛЫ И МЕТОДЫ К ПРИМЕРАМ
Определение связывания с помощью STD-NMR
Внеклеточный домен (ECD; аминокислоты: 39-358) гена FGFR1 человека (P11362) амплифицировали с помощью ПЦР и клонировали в вектор pETTEV E. coli (с N-концевой His-меткой, за которой следует сайт расщепления протеазой TEV) с использованием сайтов рестрикции Ndel и BamHI. Для продукции белка полученную плазмиду (pET FGFR1 D1D2D3) трансформировали в BL21(DE3) (Novagene) E. coli. Клетки выращивали при 37°С до достижения ОП600 равной 0,6, и продукцию рекомбинантного белка индуцировали добавлением 1 мМ IPTG (изопропил-b-D-тиогалактопиранозида). После индукции в течение 4 часов клетки собирали и хранили при -80°С до использования. Осадок клеток (1 л культуры) размораживали и ресуспендировали в 50 мл буфера 1 (20 мМ трис/HCl, pH 7,5, 200 мМ NaCl), содержащего лизоцим (2 мг) и 40 Е бензоназы (Merck). Клетки разрушали обработкой ультразвуком, тельца включения (IB) осаждали центрифугированием (15000 g, 20 мин, 4°C), и полученный осадок промывали дважды буфером 1. Осадок IB растворяли в 20 мл денатурирующего буфера (6 М гуанидин-HCl, 20 мМ трис/HCl, pH 8,0, 200 мМ NaCl) в течение 40 минут при комнатной температуре. Нерастворимые остатки клеток удаляли центрифугированием (30000 g, 30 мин) и супернатант наносили на колонку Ni-NTA (Qiagen), предварительно уравновешенную буфером А, следуя рекомендациям производителя. ECD FGFR1 элюировали с колонки с использованием денатурирующего буфера с 500 мМ имидазолом. Фракции, содержащие ECD, объединяли и осуществляли повторную укладку с помощью моментального разведения солюбилизированного белка (фактор разведения 1:30) в 50 трис/HCl, pH 8,0, 250 мМ NaCl, 0,5 М L-аргинина, 2 мМ ЭДТА, 0,02% азида, с последующей инкубацией при осторожном перемешивании в течение 24 час при 4°С. Смесь с повторной укладкой центрифугировали при 30000 g в течение 20 мин, концентрировали с помощью мембраны YM10 (конечная концентрация белка 1 мг/мл) в ячейке с перемешиванием Amicon, диализовали против 25 мМ трис/HCl, pH 8,0, 2 мМ ЭДТА, 0,02% азида, наносили на колонку HiTrap Heparin HP 5 мл (GE Healthcare) и элюировали линейным градиентом от 0 до 2 М NaCl. Конечная очистка ECD FGFR1 достигалась хроматографией, исключающей по размеру, с использованием колонки Hi Load 26/60 75 pG (GE Healthcare), уравновешенной 25 мМ трис/HCl, pH 8,0, 200 мМ NaCl, 25 мМ L-аргинина, 2 мМ ЭДТА, 0,02% азида. FGF1 (аминокислоты: 16-155) и FGF2 (аминокислоты 9-155) и TNF-R1α экспрессировали и очищали. Структурная целостность ECD FGFR1 была продемонстрирована по его способности связываться с колонкой гепарина (смотри выше) и по образованию комплекса с FGF1. Образование комплекса анализировали с помощью хроматографии, исключающей по размеру, и последующего анализа на SDS-PAGE.
Все эксперименты по STD- и 1D-NMR осуществляли на BRUKER трехканальном DRX600 и на BRUKER четырехканальном DRX800 спектрометре при стандартной температуре 24,9°C и нормировали на внутренний стандарт 3-триметил-2,2,3,3-тетрадейтеропропионат-натриевую соль (TSP). Обычно образцы для ЯМР содержали 0,5 мл белка (20-300 мМ) в 25 мМ трис/HCl, pH 8,0, 200 мМ NaCl, 25 мМ L-аргинина, 2 мМ ЭДТА, 0,02% азида (в 95% H2O/5% D2O). Для белкового лиганда измерения спектров 1D STD NMR регистрировали с 1 мМ лиганда SSR128129E (100 мМ исходного раствора ДМСО) и 40 мМ белка со слабым 2s RF облучением отдельных резонансных метильных групп белка. Подавление воды осуществляли с использованием стандартной Bruker WATERGATE 3-9-19 последовательности. Данные ЯМР обрабатывали с использованием программы Bruker программного обеспечения xWin для ЯМР.
Измерения с помощью изотермической калориметрии титрования (ITC)
Все калориметрические эксперименты осуществляли при 30°С с помощью калориметра титрования VP-ITC (MicroCal Inc., Northampton, MA), как описано ранее 45. Титрования включали добавление 10 мкл аликвот 1,25 мМ SSR через вращающийся с перемешиванием шприц к ячейке с раствором, содержащим 1,407 мл 10-20 мкМ взаимодействующего белка (т.е. FGFR2∂123, FGFR2∂23 и их описанных мутантов и субдоменов, FGFR3∂123, FGF1, FGF2 и фоллистатина (в качестве отрицательного контроля) с 4 мин интервалами. Поддерживалась постоянная скорость перемешивания 300 об/мин, и данные приводили в соответствие со стандартной моделью одного невзаимодействующего сайта с фиксированным n в виде 1,0, предоставленной MicroCal. Все измерения осуществлялись в 10 мМ HEPES pH 7,2, 150 мМ NaCl, и белки очищали, как описано ранее (Pellegrini et al., 2000). Мутагенез осуществляли, используя «набор для сайт-направленного мутагенеза» (Stratagene).
Измерения с помощью инфракрасной спектроскопии на основе преобразования Фурье
Измерения с помощью инфракрасной спектроскопии на основе преобразования Фурье осуществляли с использованием спектрометра Bruker Tensor 37 FT-IR, оснащенного проточной кюветой AquaSpec. Камеру для образцов термостатировали до 25°С, 100 спектров усредняли для хорошего отношения сигнала к шуму. Белки очищали, как описано выше. Сразу после гель-фильтрации белки диализовали в течение ночи в том же приготовленном буфере (10 мМ Hepes pH 7,2, 150 мМ NaCl) в присутствии или в отсутствие SSR. Образцы буфера для диализа использовали для вычитания фонового сигнала. Анализ осуществляли с использованием пакета компьютерных программ OPUS, предлагаемых Bruker. Интерпретацию результатов осуществляли, как описано {Barth, 2002 #60}.
Трансфекция HEK293 и исследования фосфорилирования Erk1/2,
PLCγ и FRS2
Клетки HEK293 были временно или стабильно трансфецированы (с использованием FuGENE 6, Roche) hFGFR2IIIca или hFGFR2IIIca-Y328D, клонированными в pcDNA3 (Invitrogen). Стабильно трансфецированные клетки выращивали в среде, содержащей G418 (400 мкг/мл). Перед стимуляцией клеткам давали голодать в DMEM в течение ночи (0% сыворотки), и их преинкубировали с SSR128128E в требуемой концентрации. Клетки затем стимулировали FGF2 (концентрация между 0,5-10 нг/мл) в течение 5 мин при 37°С с или без SSR или SU5402 в концентрации 1 мкМ. После промывания в ледяном забуференном фосфатом физиологическом растворе, содержащем ингибиторы фосфатазы (Roche), клетки лизировали в буфере RIPA (трис 30 мМ HCl pH 7,5, 150 мМ NaCl, 1 мМ ЭДТА, 1% тритон-X, 0,5% масс./об. дезоксихолат, содержащем ингибиторы фосфатаз и протеаз, как описано производителем (Roche)). Лизаты клеток центрифугировали при 12000 g в течение 10 мин, и супернатанты собирали. Белки разделяли на полиакриламидных гелях Novex (Invitrogen, Carlsbad, CA) и затем переносили на нитроцеллюлозные мембраны Hybond ECL (Amersham Pharmacia). После инкубации с 5% порошком нежирного молока в ЗФР мембраны инкубировали в течение ночи при 4°С со следующими антителами: фосфо-ERK1/2 (CST:9101), фосфо-FRS2 (CST:3861), фосфо-PLCγ (CST:2821) и FGFR2 (F0300, Sigma).
Пролиферация клеток BaF/3, трансфецированных FGFR:
Конструкты клеток BaF/3, используемые в этом эксперименте, подробно описаны в заявке WO2007/080325.
Количественная ПЦР в реальном времени
Суммарную РНК выделяли из HUVEC с использованием реагента тризола (Invitrogen, USA) и набора RNeasy (Qiagen, Germany), из которой затем получали кДНК с использованием набора Quantitect Reverse Transcription (Qiagen, Germany). Наборы праймеров и меченные красителем зонды FAM™ TaqMan® MGB (Eurogentec, Belgium) конструировали для человеческих FGFR1, FGFR2, FGFR3, FGFR4 и TBP, и реакции ПЦР осуществляли в системе быстрой ПЦР в реальном времени 7500 (ABI, Germany). Каждый образец анализировали в трех параллельных вместе с конкретными стандартами и без матричных контролей. Амплификации осуществляли при использовании смеси 2X TaqMan® Universal PCR Master Mix, 20X Assays-on-demand™ Gene Expression Assay Mix. Расчеты количества копий исходной мРНК в каждом образце проводили в соответствии с методом порогового цикла (CT). Количество копий мРНК FGFR1, FGFR2, FGFR3, FGFR4 нормировали, используя уровни мРНК TBP.
Измерения фосфорилирования FGFR1
Эндотелиальные клетки жировых телец крыс, стабильно трансфецированные hFGFR1IIIcα-гемагглютинином, выращивали до 80-90% конфлуентности и давали голодать без сыворотки (0,5% FBS) в течение 24 час. Стимуляцию осуществляли в течение 5 минут FGF2 в концентрации 2 нг/мл в сочетании с SSR или ДМСО (в качестве контроля). Клеточные лизаты центрифугировали при 12000 g в течение 10 мин, и супернатанты собирали. Белки, меченные HA, иммунопреципитировали путем инкубации клеточных лизатов в течение ночи при 4°С в присутствии агарозы, конъюгированной с антителами против HA. Иммунные комплексы промывали три раза 1 мл буфера для лизиса; белки элюировали путем инкубации с 50 мкл 2x SDS буфера для образцов и кипячения. Белки разделяли на полиакриламидных гелях Novex (Invitrogen, Carlsbad, CA) и затем переносили на нитроцеллюлозные мембраны Hybond ECL (Amersham Pharmacia). После инкубации с 5% порошком нежирного молока в ЗФР мембраны инкубировали в течение ночи при 4°С со следующими антителами: pFGFR (CST: 3471) и FGFR1 (CST: 3472).
Измерения анизотропии
Для оценки ингибирует ли SSR связывание FGF1 со своим связывающим карманом заявители очистили полный внеклеточный домен FGFR2 без Fc-метки (FGFR2∂123) и измерили скорость ротации (в качестве параметра анизотропии) флуоресцентно меченного lumio FGF1 (FGF1-lumio; постоянная концентрация 1 мкМ) в присутствии различных концентраций FGFR2∂123 без (синие) и с (красные) SSR (1 мМ). Когда FGFR2∂123 добавляли к FGF1-lumio, скорость ротации комплекса лиганд-рецептор была более медленной, чем у одного FGF1-lumio из-за его более крупного размера. Большой молярный избыток (1000-кратный) SSR не смог изменить скорость ротации комплекса, подтверждая, что SSR не замещает FGF в FGFR.
Пролиферация HUVEC
Собирают конфлуентные клетки HUVEC, и 5 104 клеток в 100 мкл среды RPMI 1640 (Invitrogen, 32404-014) с 0,5% FCS (Hyclone, SH30070.03), 2 мМ глутамина, заменимыми аминокислотами MEM 1x (Gibco, 11140-035), пируватом натрия MEM 1x (Gibco, 11360-039) высевают в лунки 96-луночных покрытых коллагеном I планшетов (Beckton Dickinson, 354650) на ночь. После этого среду удаляют и заменяют 50 мкл среды, которая содержит 2x FGF2 (R&D, 234-FSE-025), FGF4 (R&D, 235-F4-025) или FGF-19 (полученного на месте), и 50 мкл 2x SSR (200 или 600 нМ). Клетки инкубировали в CO2-инкубаторе при 37°С в течение 3 дней, и пролиферацию оценивали путем количественного определения содержания АТФ с использованием 100 мкл из набора «Cell Titer Glo Luminescent cell viability» (Promega, G7571).
Хемотаксическая миграция HUVEC
Собирают конфлуентные клетки HUVEC и ресуспендируют в среде RPMI 1640 (Invitrogen, 32404-014) без FCS, с 2 мМ глутамина, заменимыми аминокислотами MEM 1x (Gibco, 11140-035), пируватом натрия MEM 1x (Gibco, 11360-039) при концентрации 0,8 106 клеток/мл. 250 мкл раствора клеток вносят с 4x SSR в верхнюю камеру 24-луночной системы ангиогенеза для миграции клеток эндотелия BD Biocoat (BD Biocoat, 354144), и 750 мкл среды с FGF2 (R&D, 234-FSE-025), FGF4 (R&D, 235-F4-025) или FGF-19 (полученного на месте) в концентрации 67 нг/мл вносят в нижнюю камеру. Планшеты инкубируют 2 час при 37°С в CO2-инкубаторе. После этого вставку планшета удаляют и помещают в новый 24-луночный планшет (Falcon, 353504), который содержит 500 мкл кальцеина (Molecular probes, C-3100) на 90 мин. После этого мигрировавшие клетки становятся флуоресцирующими, и миграцию измеряют с помощью люменометра по нижнему показателю при возбуждении 485 нм и испускании 535 нм.
In vitro ангиогенез HUVEC
Гели из коллагена/матригеля готовят помещением в каждую лунку камеры с лунками (с биопокрытием лунок коллагеном, тип I, 8-луночные культуральные камеры: Becton dickinson 354630) 160 мкл разбавленного 1/6 матригеля (матригель с пониженным содержанием ростовых факторов: Becton dickinson 356230) в коллагене I (коллаген из хвоста крысы, тип I: Becton dickinson 354236). Полимеризация происходит при 37°С в течение 1 час. Затем добавляют 15.103 HUVEC на лунку в 400 мкл среды EBM (Clonetics C3121)+2% FCS+hEGF 10 мкг/мл. Эндотелиальные клетки стимулируют 10 нг/мл FGF2 (R&D, 133-FB-025), FGF4 (R&D, 235-F4-025) или FGF19 (R&D, 969-FG-025) в течение 24 час при 37°С в CO2-инкубаторе. Затем измеряют суммарную длину псевдотрубочек с помощью системы биоизображений (Imagenia Biocom, Courtaboeuf, France).
Анализ фосфорилирования AKT в HUVEC с помощью иммуноблотинга
Клетки HUVE (Promocell, C-12200) высевают в 35-мм покрытую коллагеном I чашку (BD Biocoat, 354456) в количестве 0,5.106 клеток в 2 мл среды EBM (Clonetics, CC-3121), содержащей 2% FBS (Clonetics, CC-4101), 10 мкг/мл hEGF (Clonetics, CC-4017) из набора EGM singlequots (Clonetics, CC-4133), 1250 нг/мл гепарина (Sigma, H3149) и 375 нг/мл ECGS (BD Biosciences, 356006). При 90% конфлуентности клетки оставляют голодать в течение ночи в 1,8 мл среды RPMI 1640 (Invitrogen, 32404-014) с 0,5% FCS, 2 мМ глутамина, 1 мМ заменимых аминокислот (Invitrogen, 11140-050), пируватом натрия (Invitrogen, 11360-070). На следующий день клетки стимулируют в течение 10 мин 200 мкл уравновешенной среды голодания, содержащей 10x FGF-4 (30 нг/мл; R&D, 235-F4-025) в присутствии или в отсутствие 10x SSR (3 мкМ). После этого клетки промывают холодным ЗФР и лизируют 75 мкл RIPA, содержащим 2,5 мМ ортованадат и смесь ингибиторов протеаз (Sigma, P8340). Клеточные лизаты центрифугировали при 12000g в течение 10 мин и собирали супернатанты. Белки разделяли на 4-20% трис-глициновых полиакриламидных гелях Novex (Invitrogen) и затем переносили на нитроцеллюлозные мембраны (Invitrogen, IB3010-01). После инкубации с 5% порошком обезжиренного молока в TBS-0,05% твине 80 мембраны инкубировали в течение ночи при 4°С с антителом против фосфо-AKT (Ser473, CST, 4058), разбавленным 1000x в TBS, твине, 1% БСА. После хемилюминесцентного выявления с помощью SuperSignal® West Dura Extended Duration Substrate (Thermo Scientific, 34076) получают сигнал каждой точки, и плотность точек количественно определяют с помощью системы биоизображений Chemigenius2 (Syngene).
Анализ ELISA фосфорилирования AKT в целых клетках
Собирают конфлуентные клетки HUVEC, и 5 104 клеток в 50 мкл среды RPMI 1640 (Invitrogen, 32404-014) с 0,5% FCS (Hyclone, SH30070.03), 2 мМ глутамина, заменимыми аминокислотами MEM 1x (Gibco, 11140-035), пируватом натрия MEM 1x (Gibco, 11360-039) высевают в лунки 96-луночных покрытых коллагеном I планшетов (Beckton Dickinson, 354650) на ночь. Клетки стимулируют 5 мин 100 мкл уравновешенной среды голодания без FCS, содержащей 20 нг/мл FGF4 и 600 нМ SSR. Затем добавляют 50 мкл PFA 8% в ЗФР (Polysciences, 18814) на 15 мин при комнатной температуре, и клетки промывают 3 раза 200 мкл ЗФР в течение 2 мин. Неспецифические сайты блокируют в течение 1 час при комнатной температуре с помощью ЗФР, 0,3% тритона, 0,1% нормальной козьей сыворотки (Zymed, 50-062Z), и блокирующий буфер удаляют и заменяют антителом против фосфо-AKT (Ser473) (CST, 4058), разбавленным 1/500 в ЗФР, 0,3% тритоне, на ночь. Первое антитело затем удаляли, и производили промывку 3 раза 200 мкл ЗФР в течение 2 мин. Для выявления фосфорилирования AKT используют второе антикроличье антитело, конъюгированное с HRP (CST, 7074), после разбавления 1/2000 в ЗФР, 0,3% тритоне в течение 2 час при комнатной температуре. После этого клетки промывают ЗФР, и добавляют 100 мкл субстрата HRP (Uptima, UP664781) на 20 мин в темной комнате. Ферментативную реакцию останавливают с помощью 100 мкл стоп-буфера (Uptima, UPS29590) и измеряют ОП при 450 нм.
Связывание FGF2 на трансфецированных FGFR клетках 300-19
FGF2 метили Alexa Fluor 488 C5-малеимидом (Invitrogen, A10254) в соответствии с рекомендациями продавца.
Этот AF488-FGF2 использовали в концентрации 10 нг/мл в экспериментах по связыванию с мышиными пре-B клетками 300-19, трансфецированными конструктами FGFR1 или FGFR4 в плазмидах pEF6-V5/His Topo (Invitrogen). SSR (конечная концентрация 300 нМ) предварительно инкубировали 20 мин с клетками в среде RPMI 1640 (Invitrogen, 32404-014) с 10% FCS (Hyclone, SH30070.03), 2 мМ глутамина, заменимыми аминокислотами MEM 1x (Gibco, 11140-035), пируватом натрия MEM 1x (Gibco, 11360-039) и 150 мМ монотиоглицерина (Sigma, M6145) при 4°С и встряхивании при 150 об/мин. После этого добавляют FGF2 (конечная концентрация 10 нг/мл) на 30 мин и измеряют связывание с помощью проточного цитометра FACS Calibur (Beckton Dickinson). Анализируют также медиану флуоресценции для каждого состояния.
Миграция клеток с разными факторами роста
Миграцию клеток оценивали с помощью теста с модифицированной камерой Бойдена, используя 24-луночные вставки, содержащие проницаемые подложки между лунками с размером пор 8 мкм с поликарбонатной мембраной (Costar, Corning Inc.). Клетки экспоненциальной фазы роста оставляли голодать в среде, содержащей 0,2% FBS, в течение 16 часов и ресуспендировали при концентрации 5×105 клеток/мл в той же среде с низким содержанием сыворотки. 100 мкл клеточной суспензии высевали в верхнюю камеру, а хемоаттрактанты и/или SSR помещали в нижнюю камеру. Тестированные хемоаттрактанты включают: человеческие PDGF-BB, IGF-I, PIGF, EGF, все в концентрации 100 нг/мл, в присутствии или в отсутствие SSR (1 мкМ). В качестве положительного контроля использовали среду, содержащую 10% FBS. После 6 часов инкубации при 37°С клетки, находящиеся на верхней стороне мембраны, соскабливали с помощью ватного тампона, а мигрировавшие клетки на нижней поверхности фиксировали 1% параформальдегидом в ЗФР, и окрашивали ядра с помощью DAPI для количественного определения с помощью флуоресцентного микроскопа. Количественное определение проводят получением 5 случайных изображений при увеличении 10x и подсчетом количества ядер.
Пролиферация и миграция PANC02:
Пролиферацию клеток анализировали в экспоненциальную фазу роста клеток, которые оставляли голодать в течение 16 часов в 100 мкл среды RPMI 1640 (Invitrogen, 32404-014) с 0,2% FBS (Hyclone, SH30070.03), 2 мМ глутамина, заменимыми аминокислотами MEM 1x (Gibco, 11140-035), пируватом натрия MEM 1x (Gibco, 11360-039) и высевали в количестве 4000 клеток/лунку в 96-луночные микропланшеты. После экспозиции с митогенами и/или SSR в течение 72 часов оценивали пролиферацию клеток с помощью теста CellTiter 96 AQueous One Solution Cell Proliferation (Promega, Madison, Wisconsin, USA) в соответствии с инструкциями производителя. Миграцию клеток оценивали с помощью теста с модифицированной камерой Бойдена, используя 24-луночные вставки, содержащие проницаемые подложки между лунками с размером пор 8 мкм с поликарбонатной мембраной (Costar, Corning Inc.). Клетки экспоненциальной фазы роста оставляли голодать в среде, содержащей 0,2% FBS, в течение 16 часов и ресуспендировали при концентрации 5×105 клеток/мл в той же среде с низким содержанием сыворотки. 100 мкл клеточной суспензии высевали в верхнюю камеру, а хемоаттрактанты и/или SSR помещали в нижнюю камеру. В качестве положительного контроля использовали среду, содержащую 10% FBS. После 6 часов инкубации при 37°С клетки, находящиеся на верхней стороне мембраны, соскабливали с помощью ватного тампона, а мигрировавшие клетки на нижней поверхности фиксировали 4% параформальдегидом, и окрашивали ядра с помощью DAPI для количественного определения.
Пролиферация миеломных клеток B9:
Пролиферацию клеток анализировали в экспоненциальную фазу роста клеток, которые оставляли голодать в течение 16 часов в среде IMDM (Invitrogen, 31980048), содержащей 0,2% FBS, 2 мМ глутамина, и высевали в количестве 4000 клеток/лунку в 96-луночные микропланшеты. После экспозиции с митогенами и/или SSR в течение 72 часов оценивали пролиферацию клеток с помощью теста CellTiter 96 AQueous One Solution Cell Proliferation (Promega, Madison, Wisconsin, USA) в соответствии с инструкциями производителя.
Тест AlphaScreen SureFire
День 0: посеять клетки HEK293:mVEGFR2wt или HEK293:PDGFRβ в количестве 10000 клеток/лунку (96-луночный планшет для связывания клетками Costar) и дать им прикрепиться в течение ночи.
День 1: голодающие, как минимум, на протяжении 3 час в среде DMEM (0% сыворотки) клетки; готовят смесь из 50 нг/мл VEGF164 или PDGF-BB в DMEM (0% сыворотки) и стимулируют клетки в течение 5 или 15 минут; лизируют клетки в буфере для лизиса из набора SureFire (Perkin Elmer): лизируют клетки в 50 мкл буфера, встряхивают планшет в течение 10 мин при комнатной температуре и затем замораживают при -20°С до последующего использования; готовят смесь буфера для лизиса с белком и анализируют на предмет pERK1/2, суммарной ERK1/2 и по заказу pPLCγ и суммарной PLCγ в соответствии с инструкциями производителей.
Пример 1: Идентификация SSR128129E в качестве аллостерического ингибитора многих FGFR
Целью данного исследования было создание низкомолекулярных химических соединений, которые связываются с внеклеточным доменом (ECD) FGFR и ингибируют сигнализацию FGFR. Принимая во внимание то, как это сложно представить себе, чтобы небольшое соединение могло взаимодействовать с гораздо более крупным полипептидом (т.е. FGF) путем создания простого стерического препятствия для ортостерического сайта, для определения того, действуют ли какие-либо выявленные соединения ортостерически или посредством аллостерического механизма, было использовано множество форматов тестирования связывания лигандов. Заявители сначала разработали высокопродуктивный сцинтилляционный тест связывания на основе близости (SPA) для выявления соединений, которые ингибируют связывание 125I-FGF2 с FGFR1-ECD, состоящим из трех Ig-подобных доменов D1-3 и соединенным с Fc-фрагментом (FGFR1d123/Fc). После скрининга >20000 соединений и химической оптимизации одно соединение, SSR128129E (обозначаемое далее как «SSR»), ингибировало связывание 125I-FGF2. В дополнительных тестах SPA SSR действовал как ингибитор многих FGFR, блокируя связывание различных лигандов FGF с различными FGFRs, не ингибируя при этом связывание >100 разных лигандов с родственной структурной гомологией или с совершенно иным химическим составом с их собственными рецепторами; эти наблюдения позволяли предполагать или конкурентный (ортостерический) механизм, или же аллостерическое взаимодействие, отличающееся высокой отрицательной кооперативностью (Christopoulos and Kenakin, 2002).
Одним из отличительных признаков аллостерических взаимодействий является феномен «зависимости от зонда», т.е. вариации величины и направленности аллостерического взаимодействия в зависимости от природы ортостерического лиганд-рецепторного комплекса, с которым взаимодействует модулятор (May et al., 2007). Для определения того, зависит ли действие SSR на связывание 125I-FGF2 в SPA от конфигурации сконструированного гибридного белка FGFR/Fc, помещенного на искусственный субстрат, заявители затем исследовали, ингибирует ли SSR связывание флуоресцентного FGF1, меченного lumio (FGF1-lumio), с очищенным ECD FGFR2 без метки Fc (FGFR2d123) путем измерения скорости ротации в качестве показателя анизотропии. При добавлении FGFR2d123 к FGF1-lumio скорость ротации лиганд-рецепторного комплекса была ниже скорости ротации одного FGF-lumio из-за большего размера. Если бы SSR ингибировал связывание лиганда, скорость ротации должна была бы вновь возрасти. Однако даже при >1000-кратном молярном избытке SSR не влиял на скорость ротации комплекса, что указывает на отсутствие прямой конкуренции между SSR и FGF1-lumio (фиг.5A). Наконец, тестирование связывания I125-FGF2 на клетках эндотелия пупочной вены человека (HUVECs) или клетках эндотелия аорты свиньи с гиперэкспрессией FGFR1 (PAE-FGFR1) также показало, что SSR (даже при высоких мкМ концентрациях) не способен подавлять связывание I125-FGF2 со своим рецептором, когда последний экспрессируется в своей более естественной конформации в интактных клетках; в то же время нейтрализующее αFGF2 антитело было эффективно. В соответствии с этой последней экспериментальной парадигмой и в отличие от SPA SSR также не был способен препятствовать связыванию дополнительных лигандов FGF с другими FGFRs (т.е. FGF2 или FGF4 с FGFR2; FGF2 с FGFR4).
В совокупности эти результаты показали, что ингибирующая активность SSR в отношении связывания лигандов FGF в высокой степени зависит от конформации FGFR и не соответствует простому конкурентному механизму, связанному со стерическим препятствием для перекрывающегося связывающего домена. О способности низкомолекулярных аллостерических модуляторов дифференцированно изменять связывание ортостерических лигандов в зависимости от условий анализа, как это отмечено в настоящем описании в отношении FGFR, ранее сообщалось в отношении GPRCs (Litschig et al., 1999; Price et al., 2005). Предположительно FGFR1d123/Fc находится в конформации, которая обеспечивает передачу отрицательного аллостерического эффекта на сродство 125I-FGF2 путем связывания SSR, тогда как отсутствие метки Fc или экспрессия полностью интактного рецептора в его естественном окружении не обеспечивает передачу эффекта.
Пример 2: SSR является аллостерическим ингибитором многих FGFR
Поскольку анализ экспрессии FGFR на клетках HUVEC (C-12200, Promocell) с помощью количественной ПЦР (фиг.1A) и ОТ-ПЦР с использованием специфических праймеров для выявления экспрессии генов FGFR (фиг.1B) и вариантов FGFR1 (фиг.1C) выявил экспрессию лишь FGF-R4 и FGF-R1β3c, заявители сначала использовали клетки HUVE для изучения антагонистической активности SSR на разных FGFR. Известно, что FGF19 специфически стимулирует FGFR4, а FGF4 (но не FGF19) активирует лишь FGF-R1 в клетках BaF/3, трансфецированных гибридным белком FGFR1-hMpI (фиг.2A), а FGF19 не способен этого делать (фиг.2B). Таким образом, в клетках HUVECs FGFR1 и FGFR4 могут быть стимулированы, соответственно, FGF4 и FGF19.
Пролиферация HUVEC стимулируется FGF2 и FGF4, но не FGF19 (фиг.4A), что позволяет предполагать, что пролиферация HUVEC находится под контролем FGFR1. SSR способен подавлять индуцированную FGF2 пролиферацию HUVEC, что указывает на то, что SSR ингибирует рецептор FGF-R1β3c (фиг.4B). Тестирование связывания I125-FGF2 на клетках эндотелия пупочной вены человека (HUVECs) или клетках эндотелия аорты свиньи с гиперэкспрессией FGFR1 (PAE-FGFR1) дополнительно показало, что SSR (даже при высоких мкМ концентрациях) не способен подавлять связывание I125-FGF2 со своим рецептором. В то же время нейтрализующее αFGF2 антитело было эффективно (фиг.4D). Заявители также проанализировали, ингибирует ли SSR аутофосфорилирование FGFR, являющееся критической стадией сигнализации FGFR. Иммунопреципитация FGFR1, экспрессированного в клетках эндотелия жирового тельца крысы, с последующим иммуноблоттингом фосфорилированного FGFR1 показали, что индуцированное FGF2 фосфорилирование тирозина FGFR1 сильно подавляется SSR в наномолярном диапазоне концентраций (фиг.4C). Примечательно, что даже в высоких дозах SSR не полностью подавляет фосфорилирование тирозина FGFR1 с сохранением низкого остаточного уровня (фиг.4C). Было проанализировано влияние SSR на связывание меченного lumio FGF1 с внеклеточным доменом FGFR2, и SSR не ингибировал взаимодействие FGF1/FGFR2 (фиг.5A). Таким же образом показано, что SSR не способен подавлять димеризацию FGFR2 или FGF2 (фиг.5B и 5C).
Миграция HUVECs также стимулировалась FGF2 и FGF4, но не FGF19 (фиг.6A). В этом контексте SSR также способен ингибировать активность FGFR1, что ведет к снижению индуцированной FGF2 хемотаксической миграции HUVEC (фиг.6B).
Напротив, ангиогенез in vitro стимулируется FGF2 и FGF19, тогда как FGF4 неактивен, что позволяет предполагать, что FGFR4 контролирует ангиогенез in vitro в этом тесте (фиг.7A). В низком наномолярном диапазоне SSR блокирует индуцированный FGF2 ангиогенез HUVEC, что демонстрирует, что SSR способен ингибировать контролируемый FGFR4 клеточный процесс (фиг.7B).
Для оценки активности SSR в отношении FGFR2 и варианта FGFR2-IIIb использовали пролиферацию и миграцию клеток PANC02, поскольку эти клеточные ответы могут быть стимулированы 100 нг/мл FGF7 (фиг.8A и 8B), специфическим лигандом FGFR2-IIIb. Индуцирующее действие FGF7 в присутствии или в отсутствие VEGF блокируется добавлением 100 нМ SSR, что указывает на то, что SSR способен ингибировать рецептор FGFR2 и вариант 3b (фиг.8A и 8B).
Для изучения влияния SSR на FGFR3 оценивали пролиферацию миеломных клеток B9, экспрессирующих либо FGFR3WT, либо FGFR3TD (конститутивно активированный вариант даже в отсутствие лиганда, вызванный мутацией K650E; Truedel et al; blood 2006), в ответ на стимуляцию FGF1 (25 нг/мл). В то время как клетки линии B9-FGFR3WT могли быть индуцированы FGF1 и ингибированы 0,1 мкМ SSR (фиг.9), клетки линии FGFR3TD были нечувствительны к SSR (фиг.9), что указывает на то, что SSR способен ингибировать рецептор FGFR3, и подтверждает, что SSR не действует на киназный домен FGFR.
Все вместе эти результаты указывают на то, что SSR способен ингибировать изоформы FGFR (FGFR1, R2, R3 и R4) и варианты FGFR.
Пример 3: SSR не способен ингибировать клеточные ответы, индуцированные другими ростовыми факторами
Поскольку SSR дифференциально ингибировал эффективность зависимой от FGF сигнализации, заявители затем исследовали, влияет ли он также на зависимые от FGF ответы клеток in vitro. При использовании HUVECs SSR подавлял хемотаксические эффекты FGF2.
SSR не влиял на клеточные ответы, индуцированные PIGF, EGF, PDGF-BB и IGF, которые, как известно, активируют члены семейства тирозинкиназных рецепторов (фиг.10).
Пример 4: SSR128129E связывается с аллостерическим сайтом в Ig-подобном домене D3 внеклеточной области FGFRs:
Поскольку SSR был ингибитором многих FGFR, заявители использовали полипептидные фрагменты из разных подтипов FGFR (человека). Спектры ЯМР разности насыщающего переноса (STD-NMR) SSR показали, что SSR связывается с ECD FGFR1 (FGFR1d123) (фиг.11A). Это было подтверждено анализом профиля одномерного (1D)-ЯМР, который показал уширение пика сигнала FGFR1d123 после добавления SSR (фиг.11A). Это связывание является специфичным, поскольку связывание не наблюдается с внеклеточным белком TNF-R1 (фиг.11A). Заявители затем использовали фрагменты ECD FGFRs для картирования сайта связывания SSR с одним из трех Ig-доменов. Измерения 1D-ЯМР фрагмента, содержащего лишь домен D3 (FGFR1d3), выявили сайт связывания SSR в этом примембранном домене (фиг.11B). Действительно, FGFR1d3 и FGFR1d123 дают идентичные сигналы (широкую линию), что означает, что было получено одинаковое сродство к этим белкам, тогда как FGFR1d12 и FGFR1d2 дают тонкие линии (фиг.11B). Изотермическая калориметрия титрования (ITC) с использованием двух фрагментов ECD, FGFR2d23 (состоящего из доменов D2 и 3) и FGFR3d23 показала, что SSR связывается с FGFR2 и FGFR3 (фиг.11C и 11D).
Связывание SSR с доменом D3 FGFRs было специфичным, поскольку соединение не связывалось с FGF-лигандами (FGF1 и FGF2; фиг.12A, 12B и 12C) при анализе с помощью ITC или STD-NMR. Более того, гепарин не влиял на связывание SSR с FGFR, поскольку STD-NMR выявил сопоставимый сигнал SSR с FGFR или в присутствии, или в отсутствие аналога гепарина октасульфата сахарозы (SOS, фиг.12D).
Пример 5: Аллостерическое связывание SSR индуцирует конформационное изменение в FGFR
Затем заявители исследовали, можно ли получить прямое доказательство конформационного изменения FGFR, опосредуемого связыванием SSR с областью, идентифицированной в предшествующих экспериментах. Поэтому заявители произвели измерения инфракрасной спектроскопией на основе преобразования Фурье (FTIR) фрагментов ECD FGFR2, состоящих из доменов D2-3 (FGFR2d23). Добавление SSR к любому варианту приводило к повышению амплитуды амидной I полосы спектра FTIR с максимумом около 1640 см-1, что соответствует глобальному конформационному изменению (фиг.12A).
Затем заявители исследовали, связывается ли SSR с аминокислотными остатками, которые образуют часть ортостерического сайта в D3, или же с альтернативным аллостерическим сайтом. Вначале заявители использовали алгоритмы молекулярного докинга пакетов программ MOLEGRO, Autodock и YASARA и доступные кристаллографические данные. Циклы докинга SSR на FGFR2d3 с использованием обоих методов выявили два предполагаемых сайта связывания, один с центром около His293 и другой около Tyr328; эти предполагаемые сайты связывания находятся на противоположной относительно FGF-лигандсвязывающего сайта поверхности рецептора и образуют гидрофобный карман на расстоянии ~25 Е от ортостерического сайта связывания. Примечательно, что оба остатка не перекрываются с остатками ортостерического FGF-связывающего кармана. Для оценки функциональной значимости обоих предполагаемых сайтов связывания SSR заявители использовали программу FoldX молекулярного силового поля (Schymkowitz et al., 2005) для конструирования мутаций, которые должны были бы снизить или снять связывание аллостерического лиганда при том, однако, чтобы не нарушить общую конформационную стабильность структуры: (i) FGFR2d23-Y328D, которая снимает гидрофобное взаимодействие с SSR путем замены ароматического остатка отрицательно заряженным аспартатом; (ii) FGFR2d23-H293L, которая удаляет важнейший остаток из другого предполагаемого связывающего сайта для SSR; и (iii) FGFR2d23-Y328D/H293L, двойная мутация (обозначаемая как FGFR2d23-YH). Эксперименты по связыванию с использованием ITC показали, что SSR не связывается с FGFR2d23-Y328D (фиг.13B и 13C). Эти данные соответствуют модели, согласно которой SSR связывается с аллостерическим сайтом, образованным гидрофобным карманом рядом с ортостерическим сайтом связывания лиганда, и в которой остаток Tyr328, по-видимому, является решающим для передачи взаимодействия между SSR и FGFR2. Заявители также проанализировали спектр FTIR упомянутых выше мутантных фрагментов FGFR2. Ни одна из этих одиночных или двойных мутаций не индуцировала существенного сдвига в спектре FTIR, что указывает на то, что общая трехмерная конфигурация этих вариантов FGFR сопоставима. SSR индуцировал сопоставимый сдвиг спектра FTIR FGFR2d23-H293L и нативного фрагмента FGFR2d23 (фиг.14A и 14B), что указывает на то, что мутация заменой His293 на Leu293 не является достаточно радикальной или что His293 не так сильно вовлечен во взаимодействие с SSR. Напротив, SSR не индуцировал это изменение в спектре FTIR мутантных фрагментов FGFR2d23-Y328D или FGFR2d23-YH (фиг.14C и 14D), что указывает на то, что остаток Tyr328, действительно, играл решающую роль в передаче аллостерического конформационного изменения FGFR2 после связывания SSR.
Пример 6: Мутация аллостерического сайта связывания SSR подавляет ингибирующее действие SSR на сигнализацию FGFR
Для оценки функциональной значимости аллостерического сайта для регуляции сигнализации FGFR заявители создали стабильные линии клеток HEK293, экспрессирующие либо функциональный FGFR2WT, либо вариант FGFR2d23-Y328D, и проанализировали, ингибирует ли SSR активацию ERK1/2 под действием FGF2 в этих клеточных линиях. Иммуноблотинг показал, что ингибирование индуцированного FGF2 фосфорилирования ERK1/2 в клетках FGFR2d23-Y328D под действием SSR было снижено (величина IC50: 121±30 нМ) по сравнению с его ингибирующим действием в клетках FGFR2WT (величина IC50: 28±12 нМ) (фиг.15A и 15B), что указывает на то, что этот аллостерический сайт имеет отношение не только к связыванию SSR с очищенными фрагментами FGFR2 in vitro, но и к его ингибирующей активности в отношении сигнализации FGFR2 в физиологических условиях in cellulo. Тот факт, что мутация Y328D не полностью подавляла ингибирующую активность SSR, позволяет предполагать, что кроме Tyr328 в связывание SSR вносят вклад также другие соседние остатки при экспрессии FGFR2 в физиологическом контексте.
Пример 7: SSR представляет собой «избирательный» ингибитор зависимой от FGFR сигнализации фосфорилированием
Для оценки влияния SSR на контролируемую FGFR сигнализацию фосфорилированием клетки HEK293 трансфецировали FGFR2, и исследовали два основных пути после аутофосфорилирования FGFR, PLCγ и FRS2, с помощью иммуноблотинга, в сопоставлении с ингибитором тирозинкиназы FGFR SU5402, который, как описано в публикации, ингибирует зависимые от FGFR каскады FRS2 и PLCγ (Zhen et al., Oncogene 2007). В таких клетках FGF индуцирует фосфорилирование FRS2, ERK1/2 и PLCγ. SU5402 блокирует все эти индуцированные эффекты, тогда как SSR ингибирует лишь путь FRS2 (фиг.16A и 16B), что указывает на избирательный антагонизм, развиваемый SSR. В более общем плане эти различия в фосфорилировании могут быть использованы в качестве репортерного показателя для оценки аллостерических модуляторов.
Пример 8: Методология поиска фрустрированного домена во внеклеточном домене тирозинкиназных рецепторов
Один из возможных молекулярных механизмов описанного в примере 5 конформационного изменения включает наличие фрустрированного домена (определение смотри выше). При анализе доменов D2 и D3 FGFR2 человека с помощью AGADIR (алгоритма предсказания стабильности спирали; Mucoz, V. & Serrano, L. 1994) заявители выявили последовательность остатков между Tyr319 и Arg330 (включающую, таким образом, важнейший остаток Tyr328) в качестве единственной области, склонной подвергаться переходу из β-складки в α-спираль. В соответствии с этой теоретической моделью замена Tyr328 аспартатом (FGFR2d23-Y328D), которая по предсказанию AGADIR должна снижать альфа-спирализацию и тем самым снижать фрустрацию домена, действительно, предотвращала наблюдаемое конформационное изменение, как показано с помощью анализа FTIR. Был проведен сходный анализ последовательности дополнительных рецепторов факторов роста, включая VEGFR1, -2 и -3 и PDGFRβ, наряду с другими TKRs, содержащими области с относительно высокими индексами AGADIR, которые могли бы быть обращены путем мутации важнейшего остатка этой области в аспартат. Заявители располагают некоторыми предварительными данными для VEGFR2, где мутации K609D и K648D приводили к снижению сигнализации ERK1/2 после стимуляции VEGF.
Пример 9: Аффинный скрининг аллостерических модуляторов FGF-Rs с помощью SEC-LC/MS и оценка активности выявленных соединений
Методология SEC-LC/MS представляет собой аналитический метод, используемый для аффинного скрининга на основе 2-компонентной системы, соединенной в оперативном режиме: исключающая по размеру хроматография в соединении с высокоэффективной жидкостной хроматографией с последующей ионизацией электрораспылением - детекция время-пролетной масс-спектрометрией.
Метод основан на способности некоторых соединений взаимодействовать с растворимыми полипептидами (пептидом, белковым доменом или полноразмерным белком). После смешивания пула низкомолекулярных соединений с интересующим пептидом комплекс пептид-лиганд индуцирует изменение массы, обеспечивающее разделение несвязанных и связанных низкомолекулярных соединений с помощью исключающей по размеру хроматографии. После этого вызывают диссоциацию комплексов, и связываемые вещества выявляют с помощью LC/ESI-TOF высокого разрешения для точного измерения массы (например, с помощью масс-спектрометра Waters LCT Premier). Алгоритм деконволюции данных обеспечивает идентификацию связанных молекул по результатам анализа измерения массы.
Для идентификации аллостерических модуляторов FGF-Rs этот метод можно применить к внеклеточному домену разных FGF-Rs, нативных или мутантных. Нативная форма обеспечивает выявление всех соединений, связывающихся с внеклеточным доменом. Другой путь осуществления скрининга аллостерических модуляторов мог бы быть осуществлен с использованием «открытой» формы спирали FGF-R2 рядом с сайтом связывания SSR128129, полученным с помощью мутаций Tyr328Arg-Ile329Lys, которые стабилизируют альфа-спираль, что обеспечивает сенсибилизацию к связыванию SSR128129. В этом случае этот мутантный FGF-R2 может заменять WT FGF-R2. Сходная стратегия может быть использована для проведения скрининга на FGF-R1, -R3 или -R4 с мутациями аминокислот, соответствующих Tyr328 и Ile329 в FGF-R2. Мутантная форма по Tyr328 (FGF-R2) или соответствующие мутантные аминокислоты в других FGF-Rs могут быть использованы для проведения негативного скрининга. Поскольку SSR128129 не связывается с FGF-R2, содержащим мутацию Tyr328Asp рядом с гидрофобным карманом, эта мутантная форма может быть использована для отбраковки части соединений, которые не взаимодействуют с интересующим карманом на FGF-R2.
Во всех случаях эта стратегия ведет к идентификации молекул, способных связываться с целевым карманом в интересующем пептиде. На следующей стадии следует оценить клеточный эффект. Во-первых, отобранные соединения должны ингибировать индуцированный FGF путь, такой как фосфорилирование AKT в клетках HUVEC, как это наблюдалось в экспериментах по иммуноблоттингу с SSR (фиг.17A и 17B). Состояние фосфорилирования AKT в клетках HUVEC может быть измерено с помощью методологии ELISA на клетках. Этот формат анализа был разработан заявителями для прямого определения фосфорилирования AKT в разных клетках, таких как HUVEC, и обеспечивает выявление влияния SSR на стимулированные FGF4 клетки HUVEC (фиг.17C). Типичной особенностью аллостерических модуляторов FGFR является их неспособность конкурировать за связывание FGF. Для оценки этого был разработан тест связывания на мышиных пре-B клетках 300-19, трансфецированных FGFRs. Меченный AlexaFluor488 FGF2 в концентрации 10 нг/мл связывается с FGFR1 или FGFR4, экспрессированными на клетках 300-19, которые в естественных условиях не экспрессируют никакого FGFR, и SSR в концентрации 300 нМ не способен подавлять это связывание на FGFR1 или FGFR4 по данным анализа проточной цитометрией (фиг.17D).
Пример 10: Идентификация предполагаемых фрустрированных зон в рецепторе VEGF-R2 и мутационный анализ предполагаемых фрустрированных зон
Стратегия, разработанная для FGF-Rs, применима для другой рецепторной TK: VEGF-R2, или KDR. В качестве первоначального подхода к идентификации областей, которые могли бы укрывать предполагаемые аллостерические целевые сайты, заявители использовали компьютерную программу AGADIR1 для идентификации областей, которые склонны подвергаться структурным изменениям (например, переходам β-складки в α-спираль), с использованием доступных первичных аминокислотных последовательностей из рецептора VEGF-R2 мыши (регистрационный номер Entrez NP_034742.2). Это дало несколько областей с повышенной склонностью к спирализации, хотя в структуре Ig-домена следует ожидать главным образом β-складчатые структуры. Эти результаты анализа Agadir показаны на фиг.18A.
Затем после последовательных мутаций каждой аминокислоты из VEGF-R2 in silico путем замены остатком аспартата (D) заявители вновь проанализировали показатель AGADIR и отобрали те мутации, (i) в результате которых изменение склонности к спирализации было наибольшим (самые негативные) и (ii) которые локализовались в тех Ig-доменах, которые были ближайшими к трансмембранному домену. Из этих мутаций мутации K609D и K648D, оба остатка которых расположены в домене IgD6 mVEGF-R2 (фиг.18B), дали наибольшее снижение склонности к спирализации и были в дальнейшем использованы для анализа in cellulo (смотри дополнительно пример 12).
Пример 11: Идентификация предполагаемых фрустрированных зон в рецепторе PDGF-Rβ и мутационный анализ предполагаемых фрустрированных зон
В качестве первоначального подхода к идентификации областей, которые могли бы укрывать предполагаемые аллостерические целевые сайты, заявители использовали компьютерную программу AGADIR1 для идентификации областей, которые склонны подвергаться структурным изменениям (например, переходам β-складки в α-спираль), с использованием доступных первичных аминокислотных последовательностей из рецептора PDGFRβ человека (регистрационный номер Entrez NP_002600.1). Это дало несколько областей с повышенной склонностью к спирализации, хотя в структуре Ig-домена следует ожидать главным образом β-складчатые структуры (фиг.19). Затем после последовательных мутаций каждой аминокислоты из PDGFRβ in silico путем замены остатком аспартата (D) заявители вновь проанализировали индекс AGADIR и отобрали те мутации, (i) в результате которых изменение склонности к спирализации было наибольшим (самые негативные) и (ii) которые локализовались в тех Ig-доменах, которые были ближайшими к трансмембранному домену. Из этих мутаций мутации L383D и K387D, оба остатка которых расположены в домене IgD3 hPDGFRβ, дали наибольшее снижение склонности к спирализации (фиг.19).
Пример 12: Метод скрининга для идентификации соединений, индуцирующих «избирательную» сигнализацию
Связывание VEGF или PDGF-BB индуцирует димеризацию соответствующих рецепторов, которая в свою очередь индуцирует фосфорилирование внутриклеточных киназных доменов. Затем активируются два основные пути (интересующие в связи с избирательным антагонистом FGF-Rs, SSR), включая путь ERK1/2 (фиг.21A и 1C) и путь PLCγ (схематически представленный на фиг.20). Измерение активации каждого из путей в присутствии или в отсутствие возможного ингибитора ведет к идентификации соединений, индуцирующих «избирательную» сигнализацию путем ингибирования лишь одного из путей сигнализации.
Для VEGF-R2 два мутантных рецептора VEGFR2 (VEGFR2K609D и VEGFR2K648D), идентифицированных в примере 10, и форму VEGF-R2 дикого типа стабильно экспрессировали в клетках HEK293. Рецептор VEGFR2WT четко отвечает активацией фосфорилирования ERK1/2. В то время как мутант VEGFR2K609D обладает сниженной способностью к сигнализации через ERK1/2 (но достаточной для теста негативного скрининга), у мутанта VEGFR2K648D она утрачена. Результаты суммированы на фиг.21A.
Для PDGF-Rβ клетки HEK293, гиперэкспрессирующие hPDGFRβ, стимулировали либо средой, не содержащей добавок («0»), либо 10% фегальной бычьей сывороткой (10% FBS), 50 нг/мл PDGF-BB (50 нг/мл) или 1 нг/мл PDGF-BB (1 нг/мл) в соответствии с процедурой AlphaScreen SureFire (фиг.21B). На фиг.21C на левой панели вновь схематически представлены смеси для выявления активированного или суммарного белка ERK1/2 после стимуляции. На правой панели показаны результаты измерений после стимуляции клеток PDGF-BB, измеренные методом детекции AlphaScreen SureFire. Результаты показывают, что при стимуляции клеток либо 10% FBS, либо 50 нг/мл PDGF-BB может быть выявлен четкий сигнал, но в отсутствие стимуляции или при стимуляции 1 нг/мл PDGF-BB сигнал намного слабее. Ответ PLCγ измеряют сходным образом.
Метод скрининга для идентификации соединений, индуцирующих «избирательную» сигнализацию VEGF-R2 или PDGF-Rβ, подобно SSR в отношении FGF-Rs, может основываться на ответе Erk1/2 и PLCγ. Сравнение ответа ERK1/2 и PLCγ в присутствии и в отсутствие кандидатных аллостерических ингибиторов обеспечивает идентификацию соединений, которые действуют в качестве избирательного ингибитора: ингибирование лишь одного из двух путей сигнализации. Мутантные конструкты VEGF-R2 или PDGF-Rβ могут использоваться в тесте негативного скрининга для подтверждения механизма действия идентифицированных аллостерических модуляторов. На мутантных рецепторах соединения должны терять свою способность модулировать рецептор.
ССЫЛКИ







| название | год | авторы | номер документа |
|---|---|---|---|
| МОНОКЛОНАЛЬНЫЕ АНТИТЕЛА К РЕЦЕПТОРУ 2 ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2009 |
|
RU2546254C9 |
| СЛИТНЫЙ БЕЛОК АНТИАНГИОГЕННОГО ИНДУЦИРУЮЩЕГО ФАКТОРА И ЕГО ПРИМЕНЕНИЕ | 2012 |
|
RU2560589C2 |
| FGF-R-Fc СЛИТЫЙ БЕЛОК И ЕГО ИСПОЛЬЗОВАНИЕ | 2012 |
|
RU2560573C2 |
| КОМБИНАЦИИ | 2015 |
|
RU2715236C2 |
| КОМБИНАЦИИ ИНГИБИТОРА FGFR И ИНГИБИТОРА IGF1R | 2015 |
|
RU2715893C2 |
| НОВЫЕ АНТИТЕЛА ПРОТИВ FGFR2B | 2020 |
|
RU2840536C1 |
| БИГЕТЕРОАРИЛ-ЗАМЕЩЕННЫЕ 1,4-БЕНЗОДИАЗЕПИНЫ И ПУТИ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2016 |
|
RU2747644C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2016 |
|
RU2747645C2 |
| Новые антитела против FGFR2b | 2020 |
|
RU2841589C1 |
| СПОСОБ ТЕРАПИИ И ПРОФИЛАКТИКИ ОПУХОЛИ, ПОДДАЮЩЕЙСЯ ЛЕЧЕНИЮ ЭНДОКРИННОЙ ТЕРАПИЕЙ, С ПОМОЩЬЮ КОМБИНИРОВАННОГО ПРИМЕНЕНИЯ ИНГИБИТОРА РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ И ЭНДОКРИННОЙ ТЕРАПИИ | 2019 |
|
RU2825636C2 |







Настоящее изобретение относится к области биотехнологии, конкретно к внеклеточному связывающему домену для аллостерического ингибитора, и может быть использовано для идентификации таких ингибиторов. Изобретение позволяет идентифицировать низкомолекулярный аллостерический ингибитор FGFR при сравнении изменений в состоянии, предпочтительно, в индукции гена или фосфорилировании, по меньшей мере, одного репортера для, по меньшей мере, двух разных сигнальных нижележащих путей, зависимых от активации/ингибирования указанного тирозинкиназного рецептора, предпочтительно выбранных из сигнального пути ERK1/2, сигнального пути PLCγ и сигнального пути АКТ. При этом аллостерический ингибитор FGFR идентифицируется тогда, когда при наличии связывания лиганда с лигандсвязывающим доменом рецептора ингибируется, по меньшей мере, один нижележащий путь FGFR, в то время как, по меньшей мере, один другой нижележащий путь остается без изменений. 2 з.п. ф-лы, 21 ил., 12 пр.
1. Способ идентификации низкомолекулярного аллостерического ингибитора FGFR, включающий стадии:
a) приведения аллостерического сайта связывания FGFR, включающего SEQ ID NO: 1 или последовательность, имеющую по меньшей мере 70%, 80%, 90%, 95% или более гомологии с SEQ ID NO: 1, в контакт с низкомолекулярным соединением-кандидатом на роль аллостерического ингибитора,
b) измерения изменений в ингибировании, по меньшей мере, в двух нижележащих сигнальных путях, зависимых от активации/ингибирования указанного тирозинкиназного рецептора,
c) сравнения изменений в состоянии, предпочтительно, в индукции гена или фосфорилировании, по меньшей мере, одного репортера для, по меньшей мере, двух разных сигнальных нижележащих путей, зависимых от активации/ингибирования указанного тирозинкиназного рецептора,
где аллостерический ингибитор идентифицируется тогда, когда при наличии связывания лиганда с лигандсвязывающим доменом рецептора ингибируется, по меньшей мере, один нижележащий путь FGFR, в то время как, по меньшей мере, один другой нижележащий путь остается без изменений.
2. Способ по п. 1, где указанные два разных нижележащих пути FGFR выбраны из сигнального пути ERK1/2, сигнального пути PLCγ и сигнального пути АКТ.
3. Способ по п. 1, где указанные два разных нижележащих пути соответствуют сигнальному пути ERK1/2 и сигнальному пути PLCγ.
| DOERN A | |||
| et al, Characterization of Inhibitory Anti-insulin-like Growth Factor Receptor Antibodies with Different Epitope Specificity and Ligand-blocking Properties, Journal of Biological Chemistry, 2009, v | |||
| СЧЕТНЫЙ ДИСК ДЛЯ РАСЧЕТА СОСТАВНЫХ ЧАСТЕЙ ПИЩИ | 1919 |
|
SU284A1 |
| Прибор для нагревания перетягиваемых бандажей подвижного состава | 1917 |
|
SU15A1 |
| Станок для насечки напильников и ножовочных полотен | 1928 |
|
SU10254A1 |
| WO2005066211 A2, 21.07.2005 | |||
| WO2008012690 A2, 31.01.2008 | |||
| BOGOYEVITCH M.A | |||
| et al, A new paradigm for protein | |||

