Данное изобретение относится к способам синтеза морфолиновых олигомеров с фосфородиамидатным мостиком сочетанием звеньев морфолинового мономера и, в частности, к улучшенным методикам снятия защиты с атома азота морфолинового кольца на каждой стадии связывания и к применению гуаниновых морфолиновых звеньев (MoG) с защитой в обеих N2 и O6/N1 группах гуанинового основания. Морфолиновые олигомеры, синтезированные с использованием данных модификаций, получены с более высокой чистотой и выходом по сравнению с морфолиновыми олигомерами, синтезированными с использованием монозащищенных гуаниновых звеньев и/или обычных методик по снятию защиты с кольцевого атома азота.
Ссылки
Albert, A., Physical Methods in Heterocyclic Chemistry, Vol.I, A.R. Katritzky, Ed., Academic Press, pp.44 (1963).
Fisher, A., Galloway, W.J. and Vaughan, J., J. Chem. Soc. 3591 (1964).
Garrison, A.W. and Boozer, C.E., J. Chem. Soc. 90(13):3486-3494 (1968).
Gough et al. (1979) Nucleic Acids Research 7:1955-1964.
Hata et al. (1983) Tetrahedron Lett. 24:2775-2778.
Jones et al. (1982A) Tetrahedron Lett. 23:2253-2256.
Jones et al. (1982B) Tetrahedron Lett. 23:2257-2260.
Mitsunobu, O. (1981) Synthesis 1:1-28.
Ravikumar, V. et al., U.S. Patent № 5510476.
Reese et al. (1981) Tetrahedron Lett. 22:4755-4758.
Reese et al. (1984) J. Chem Soc., Perkin Trans. I 1263-1270.
Rogne, O., J. Chem Soc., 727 (1970).
Summerton, J.E. and Weller, D.D. (1993) U.S. Patent № 5185444.
Summerton, J.E. and Weller, D.D. Antisense Nucl. Acid Drug Dev. 7(3):187-195 (1997).
Summerton, J.E. and Weller, D.D. U.S. Patent № 5185444 (1993).
Уровень техники
Морфолиновые олигомеры с фосфордиамидатным мостиком, или PMO, представляют собой аналоги нуклеиновой кислоты, которые прочно присоединяются и в определенной последовательности к комплиментарной РНК, и применимы в модулировании синтеза белка и, таким образом, экспрессии гена. Данные олигомеры составлены из известных группировок спаренных оснований (гетероциклических оснований), поддерживаемых системой морфолиновой основы. Морфолиновые звенья для применения в синтезе таких олигомеров могут быть легко приготовлены из соответствующих рибонуклеозидов, которые представляют собой без труда доступные и недорогие предшественники (см., например, Summerton, J.E. and Weller, 1993, 1997).
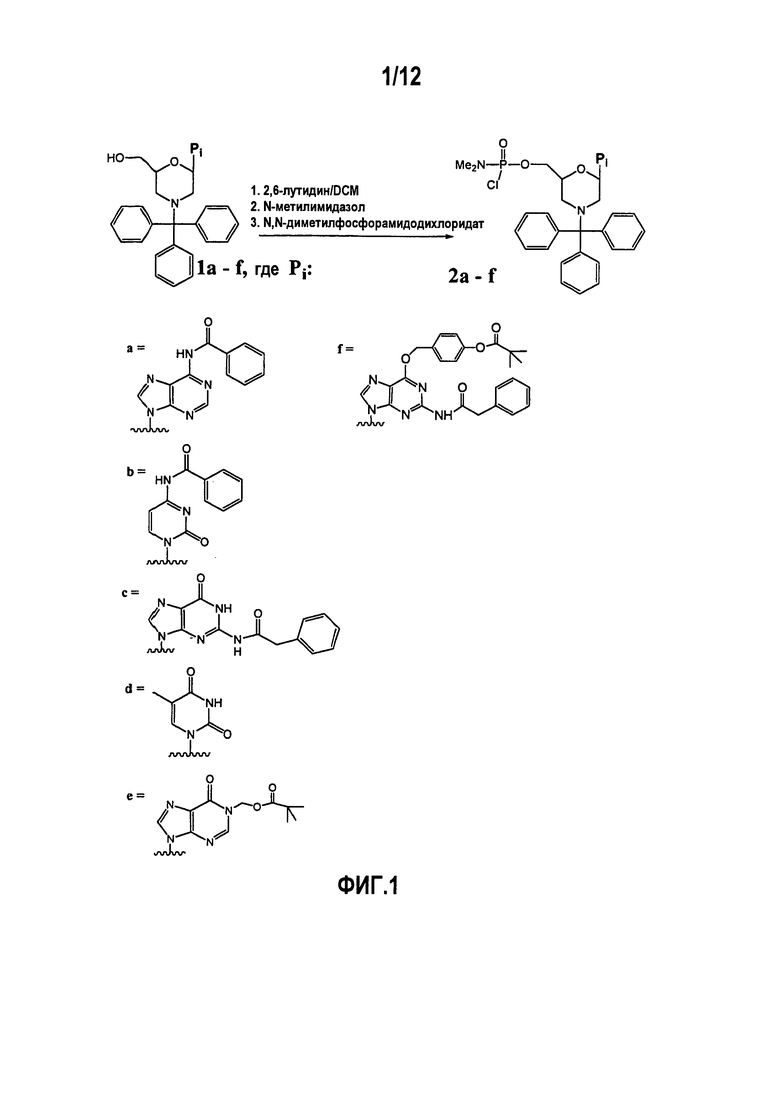
В течение такого синтеза, как, например, в обычном олигонуклеотидном синтезе, функциональные группы на гетероциклических основаниях обычно защищают, чтобы предотвратить взаимные влияния при синтетических превращениях. Например, активация N-тритилированного морфолинового мономера (1a-f; фиг.1) вызывает взаимодействие 5’-гидроксила с подходящим фосфорамидодихлоридатом с образованием активированного звена 2а-f. При большом масштабе (50-100 галлоновый (190-380 л) реактор) неочищенное активированное звено обычно загрязнено большим количеством промежуточных продуктов. После хроматографической очистки активированное звено выделяется примерно с 50% выходом для А, С, I, T, U и их защищенных форм, но только примерно с 5% выходом для активированного однократно защищенного G звена, что, как полагают, происходит из-за присутствия незащищенного О6 кислорода.
О6-незащищенное гуаниновое звено также вызывает подъем побочных реакций на стадии олигомера. Например, О6 кислород может реагировать с активированным звеном при конденсации с образованием О6-фосфорилированных представителей или производных, и в течение окончательного отщепления основных защитных групп аммиаком аммиак может реагировать в С6, чтобы вытеснить данные частицы, образуя диаминопуриновое производное. Такие примеси трудны для удаления хроматографией и вызывают большие потери выхода продукта.
В данной области были предложены различные схемы защиты, чтобы снизить побочные реакции незащищенных гуаниновых О6 положений в обычном олигонуклеотидном синтезе (см., например, Gough et al. 1979; Reese et al. 1981, 1984; Jones et al. 1982А, 1982B). Однако данные протоколы были большей частью неуспешными при применении к синтезу РМО. В соответствии с этим, предприняты попытки для повышения выхода и чистоты синтеза РМО, особенно с использованием G морфолиновых звеньев.
Морфолиновый азот морфолинового звена также защищают перед использованием, обычно с помощью тритильных или замещенных тритильных частиц. По ходу олигомерного синтеза данная группа должна быть удалена в течение каждого цикла, чтобы предоставить возможность для включения следующего звена. Неполное удаление защитной группы приводит к последовательностям с N-1 делецией, что загрязняет требуемый олигомерный продукт.
Тритильные группы обычно удаляются кислотой, и реагенты для снятия защиты, использованные в синтезе РМО, традиционно представляли собой карбоновые кислоты (Summerton, J.E. et al. 1993, 1997). Однако фосфородиамидатные группы являются также чувствительными к кислоте, и карбоновые кислоты, применяемые для удаления тритильной группы, также способны к стимулированию гидролиза фосфородиамидатных мостиков до амидатных типов, как показано на фиг.1, с возможностью более интенсивного разложения основы. Например, цианоуксусная кислота в 20% смеси ацетонитрил/DCM является эффективным реагентом для снятия защиты, но, как установлено, вызывает значительный гидролиз (5-10%) фосфородиамидатных мостиков в РМО продукте.
Карбоновые кислоты также должны быть полностью удалены из полимера-носителя для синтеза перед реакцией связывания; иначе, образуются побочные продукты, которые состоят из усеченных олигомеров, содержащих 3’-ацилированные частицы.
По этим причинам требуются улучшенные реагенты для снятия защиты с морфолинового азота в синтезе РМО.
Сущность изобретения
В одном аспекте данное изобретение относится к морфолиновому соединению, представляющему собой структуру I:
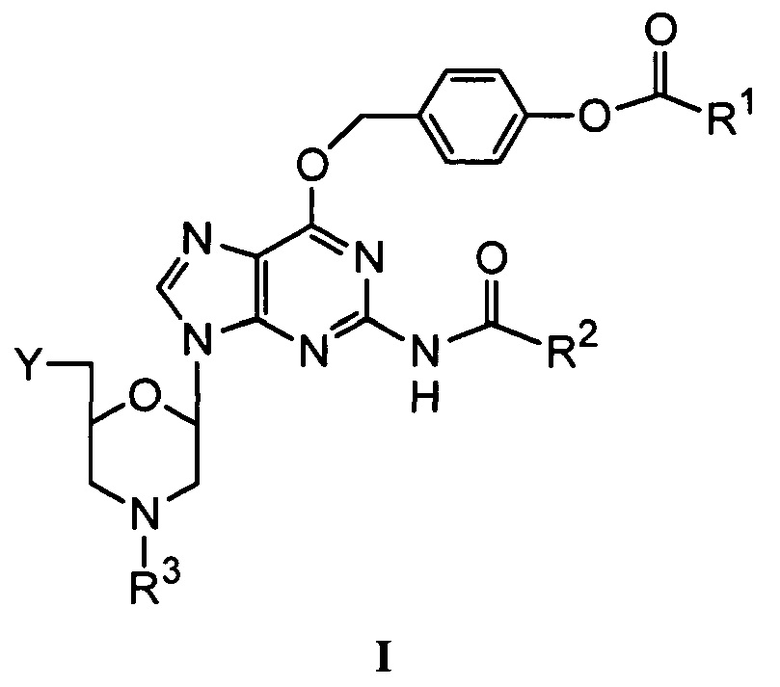
в которой
R1 выбран из группы, состоящей из низшего алкила, ди(низший алкил)амино и фенила;
R2 выбран из группы, состоящей из низшего алкила, моноциклического арилметила и моноциклического(арилокси)метила;
R3 выбран из группы, состоящей из триарилметила и водорода; и
Y выбран из группы, состоящей из: защищенной или незащищенной гидроксильной или аминогруппы; хлорфосфорамидатной группы; и фосфородиамидатного мостика к кольцевому атому азота следующего морфолинового соединения или морфолинового олигомера.
В отдельных вариантах осуществления Y выбран из группы, состоящей из защищенной или незащищенной гидроксильной или аминогруппы; хлорфосфорамидатной группы; например, хлорфосфорамидатной группы вида –О-Р(=О)-N(CH3)2Cl. Когда Y представляет собой защищенную гидроксильную группу, она предпочтительно является гидроксильной группой, защищенной триалкилсилилом.
Группа R3 предпочтительно выбрана из тритил(трифенилметил)а, 4-метокситритила, 4-метилтритила, 4,4’-диметилтритила и 4,4’,4’’-триметилтритила. Группа R1 предпочтительно представляет собой низший алкил, особенно С1-С4-алкил, и наиболее предпочтительно –С(СН3)3 (трет-бутил). Группа R2 предпочтительно выбрана из бензила и –СН(СН3)2 (изо-пропил).
В соответствующем аспекте данное изобретение относится к улучшенному способу синтеза морфолинового олигомера, причем способ включает:
(а) взаимодействие морфолиновых звеньев с носителем в твердой фазе, имеющей незащищенный кольцевой атом азота, с мономером морфолинового звена с защищенным основанием, имеющим кольцевой атом азота, защищенный триарилметилом, и активированную фосфорамидатную группу на 5’-экзоциклическом углероде,
при этом образуется фосфородиамидатный мостик между 5’-экзоциклическим углеродом и незащищенным кольцевым атомом азота;
(b) снятие защиты с защищенного кольцевого атома азота для образования незащищенного кольцевого атома азота; и
(с) повторение стадий (а) и (b) один или несколько раз со следующими мономерами морфолинового звена с защищенным основанием;
где, по меньшей мере, один из мономеров морфолинового звена с защищенным основанием представляет собой дважды защищенное гуаниновое морфолиновое соединение, имеющее структуру I:
в которой
R1 выбран из группы, состоящей из низшего алкила, ди(низший алкил)амино и фенила;
R2 выбран из группы, состоящей из низшего алкила, моноциклического арилметила и моноциклического(арилокси)метила;
R3 выбран из группы, состоящей из триарилметила и водорода; и
Y представляет собой хлорфосфорамидатную группу.
Отдельные варианты осуществления разных структур, представленных в вышеприведенной структуре, включают варианты, описанные выше.
В следующем аспекте данное изобретение относится к улучшенному способу синтеза морфолинового олигомера, причем способ включает:
(а) взаимодействие морфолинового звена с носителем в твердой фазе, имеющей незащищенный кольцевой атом азота, с мономером морфолинового звена с защищенным основанием, имеющим кольцевой атом азота, защищенный триарилметилом, и активированную фосфорамидатную группу на 5’-экзоциклическом углероде,
при этом образуется фосфородиамидатный мостик между 5’-экзоциклическим углеродом и незащищенным кольцевым атомом азота;
(b) снятие защиты с защищенного кольцевого атома азота для образования незащищенного кольцевого атома азота; и
(с) повторение стадий (а) и (b) один или несколько раз со следующими мономерами морфолинового звена с защищенным основанием;
где указанное снятие защиты включает воздействие на кольцевой атом азота, защищенный триарилметилом, раствора реагента, содержащего гетероциклическую аминную соль, в растворителе, содержащем трифторэтанол, при этом соль является солью гетероциклического амина с рКа в интервале 1-4 в протонированной форме, с кислотой, выбранной из сульфоновой кислоты, трифторуксусной кислоты и хлористоводородной кислоты.
Гетероциклический амин предпочтительно выбран из группы, состоящей из: замещенного электронно-акцепторной группой пиридина, триазола, пиридазина, пиразола, триазола и замещенных электронно-акцепторной группой замещенных производных перечисленных соединений. Такие электронно-акцепторные группы (EWG) включают галоген, циано, альдегидную, кето, карбоксисложноэфирную и карбоксамидную группу.
Предпочтительно, гетероциклический амин представляет собой замещенный электронно-акцепторной группой пиридин, такой как хлор- или цианозамещенный пиридин. Аминная соль предпочтительно представляет собой соль сульфоновой кислоты, такую как алкилсульфонат, (фторалкил)сульфонат или п-толуолсульфонат или трифторацетат. В отдельных вариантах осуществления соль выбрана из метансульфоната 3-хлорпиридиния (СРМ) и трифторацетата 4-цианопиридиния (CYTFA).
Растворитель, содержащий TFE, предпочтительно содержит дихлорметан и трифторэтанол в объемном соотношении от примерно 90:10 до 25:75, и предпочтительнее в объемном соотношении примерно 80:20 DCM:TFE.
Триарилметилзащитная группа выбрана из группы, состоящей из тритильной (трифенилметильной), 4-метокситритильной, 4-метилтритильной, 4,4’-диметилтритильной и 4,4’,4’’-триметилтритильной группы.
Модификации и усовершенствования, представленные в настоящем описании, могут быть объединены таким образом, что выполняются вышеприведенные стадии (а)–(с), где:
(i) по меньшей мере, один из звеньев морфолинового мономера с защищенным основанием представляет собой дважды защищенное гуаниновое морфолиновое соединение, имеющее структуру I, представленную выше; и
(ii) снятие защиты c защищенного кольцевого атома азота включает воздействие на кольцевой атом азота, защищенный триарилметилом, раствора реагента, содержащего гетероциклическую аминную соль, в растворителе, содержащем трифторэтанол, при этом соль является солью гетероциклического амина с рКа в интервале 1-4 в протонированной форме, с кислотой, выбранной из сульфоновой кислоты, трифторуксусной кислоты и хлористоводородной кислоты.
Обычно данный синтез еще включает отщепление морфолинового олигомера от твердой фазы и снятие защиты с оснований в соответствии со стандартными методиками.
Данные и другие задачи и особенности изобретения станут более четкими, когда прочитано последующее подробное описание изобретения в сочетании с сопровождающими чертежами.
Краткое описание чертежей
Фиг.1 показывает образование активированного морфолинового звена.
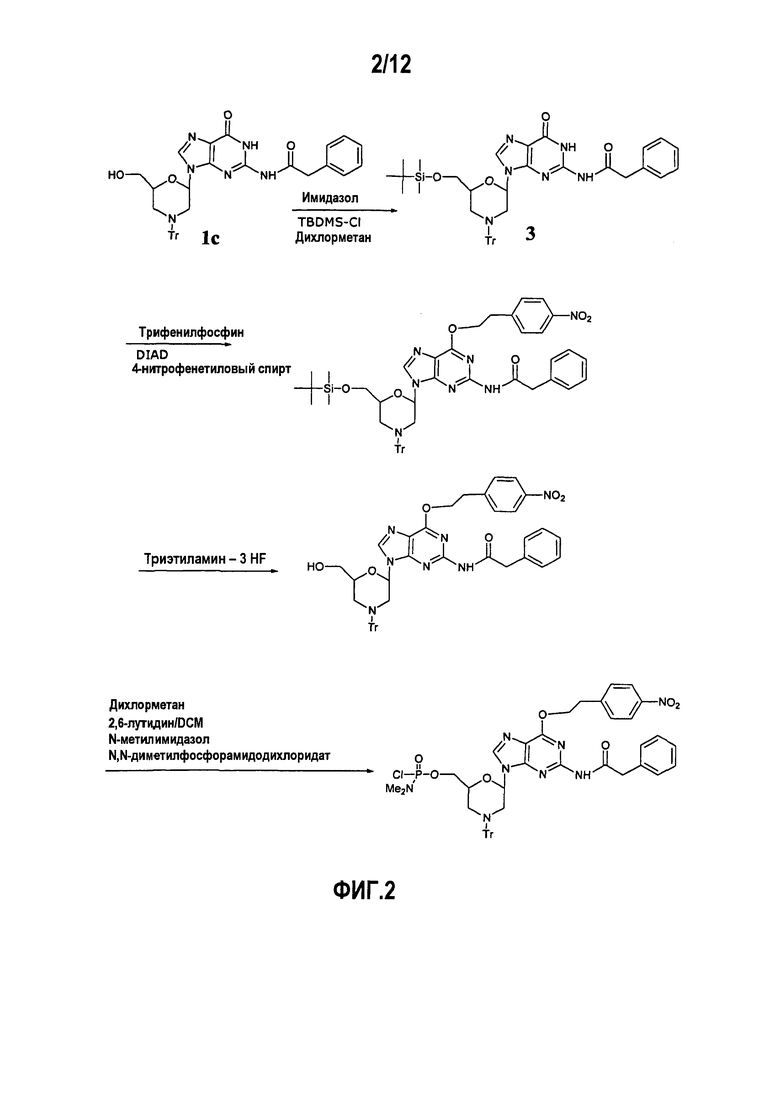
Фиг.2 показывает схему образования производного дважды защищенного морфолино G звена (DPG), в котором N2 положение является фенилацетилированным и О6 положение защищено 4-нитрофенетильной группой (NPE).
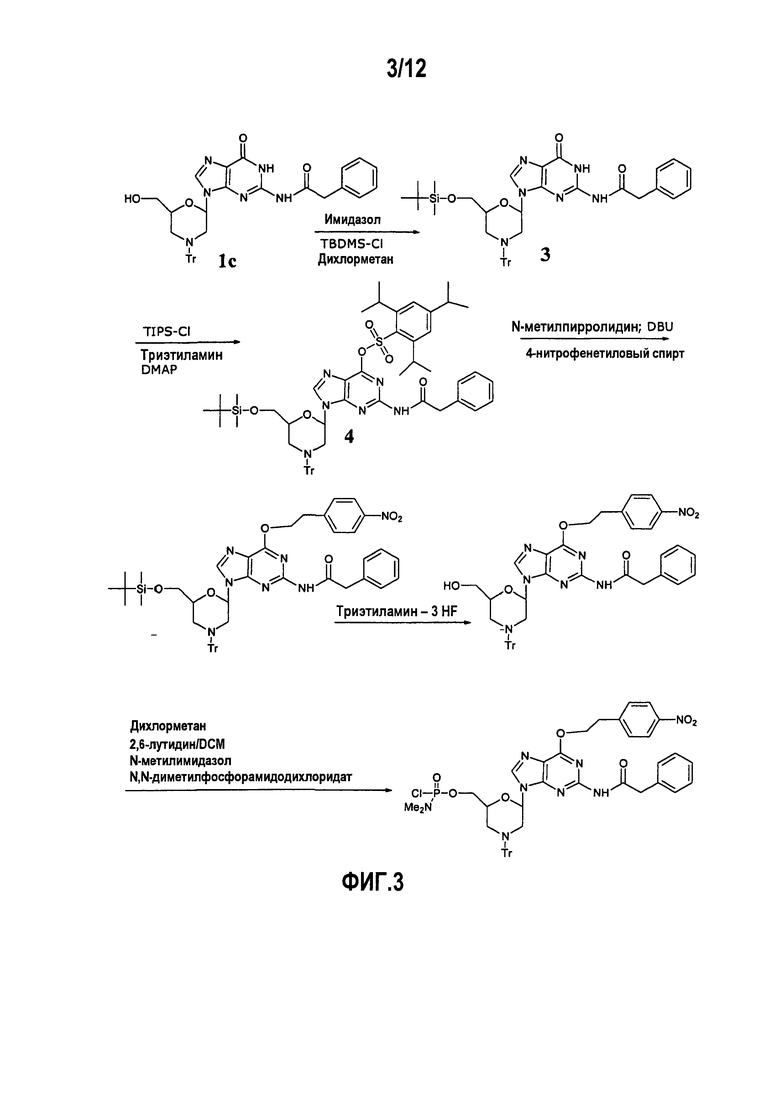
Фиг.3 показывает альтернативную схему образования производного дважды защищенного морфолино G звена (DPG), в котором N2 положение является фенилацетилированным и О6 положение защищено 4-нитрофенетильной группой (NPE).
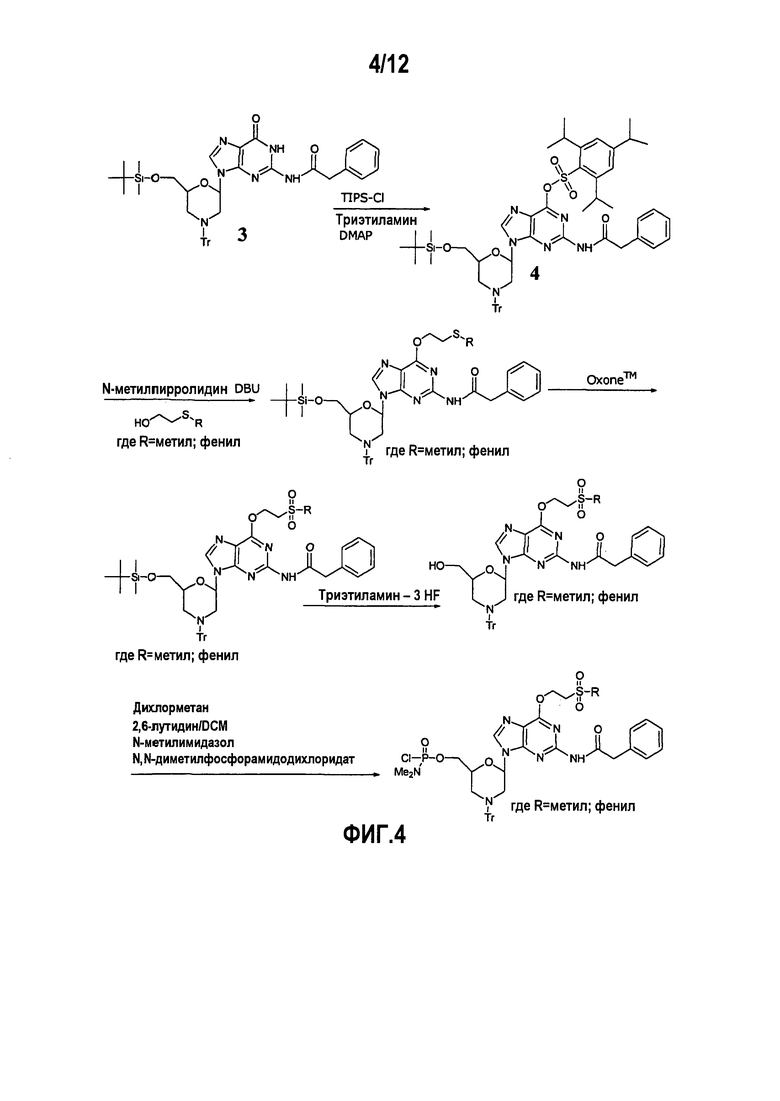
Фиг.4 показывает образование DPG производного, в котором N2 положение является фенилацетилированным и О6 положение защищено либо фенилсульфонилэтильной (PSE), либо метилсульфонилэтильной (MSE) группой.
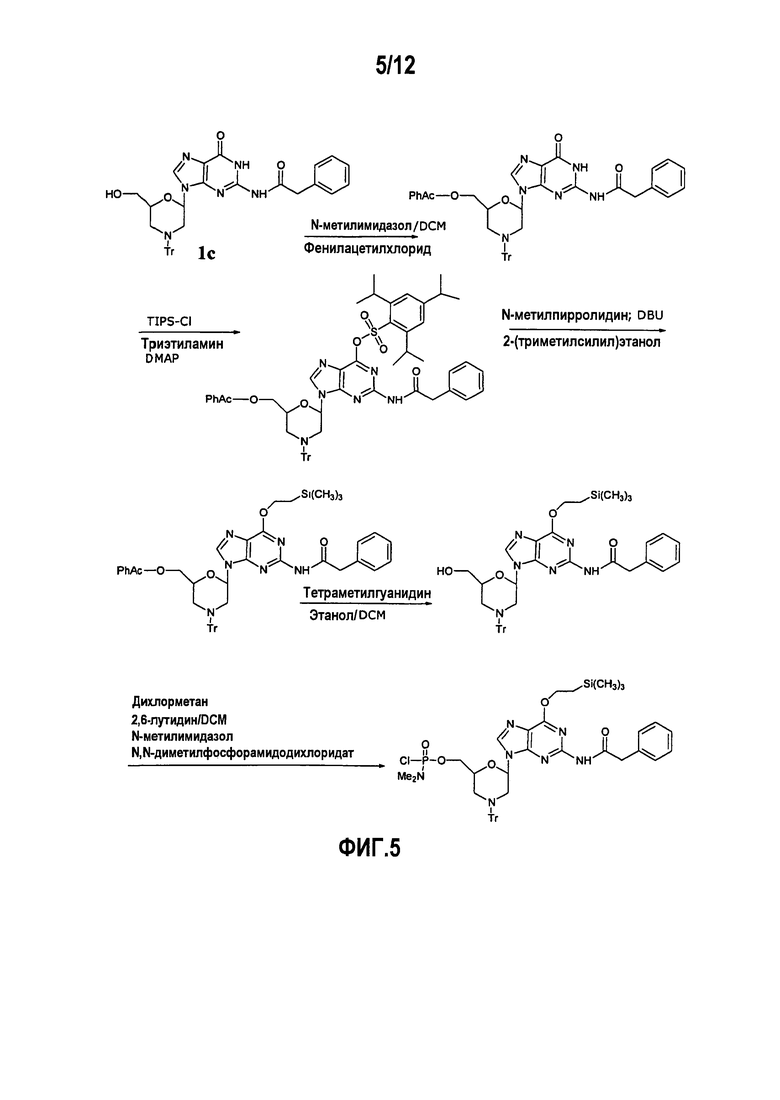
Фиг.5 показывает образование DPG производного, в котором N2 положение является фенилацетилированным и О6 положение защищено триметилсилилэтильной (TMSE) группой.
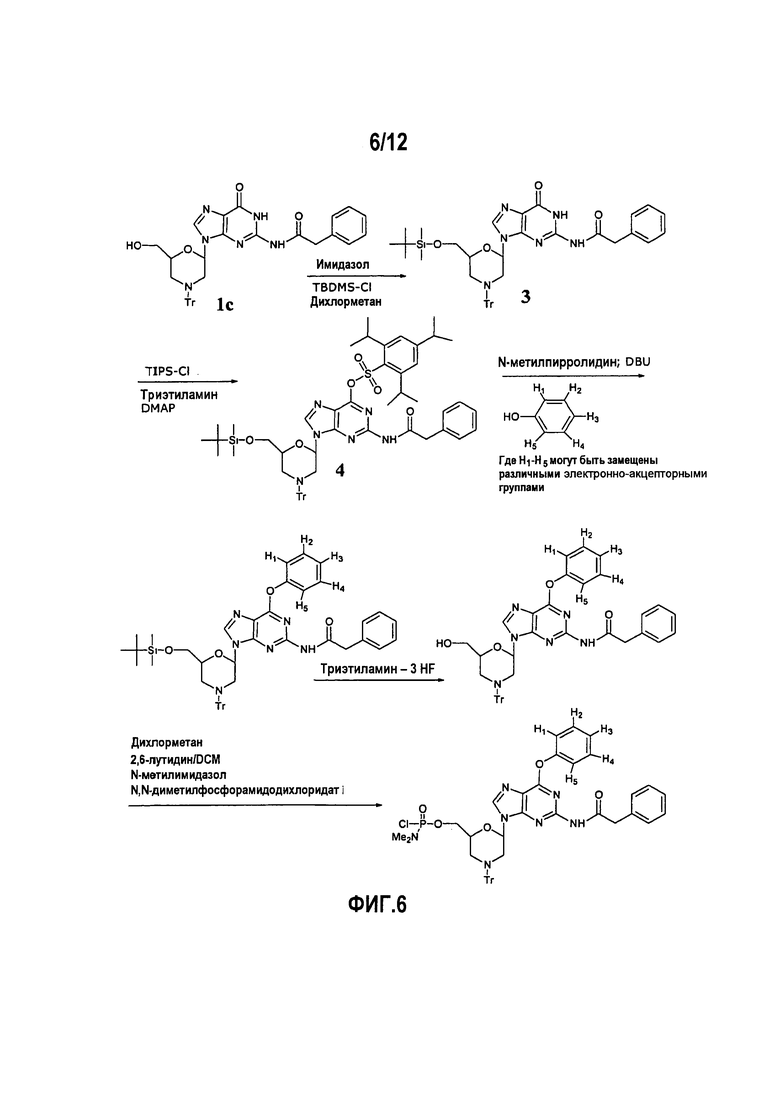
Фиг.6 показывает образование DPG производного, в котором N2 положение является фенилацетилированным и О6 положение защищено рядами арильных производных.
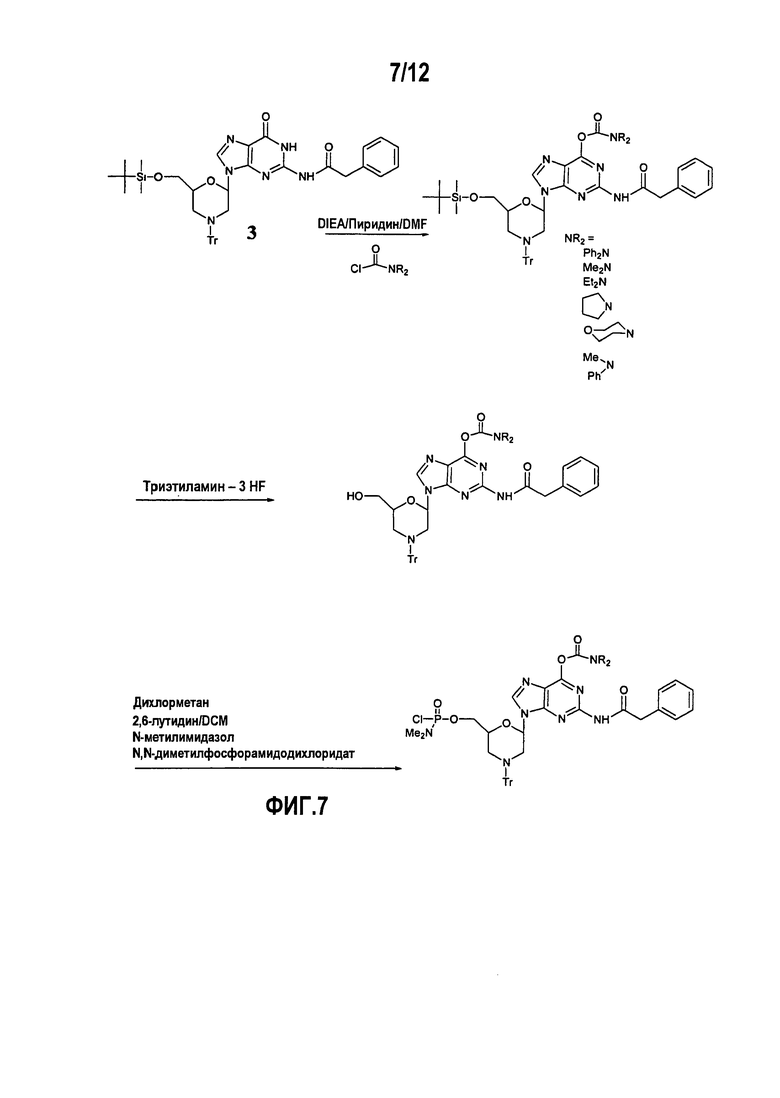
Фиг.7 показывает образование DPG производного, в котором N2 положение является фенилацетилированным и О6 положение защищено рядами карбамоильных производных.
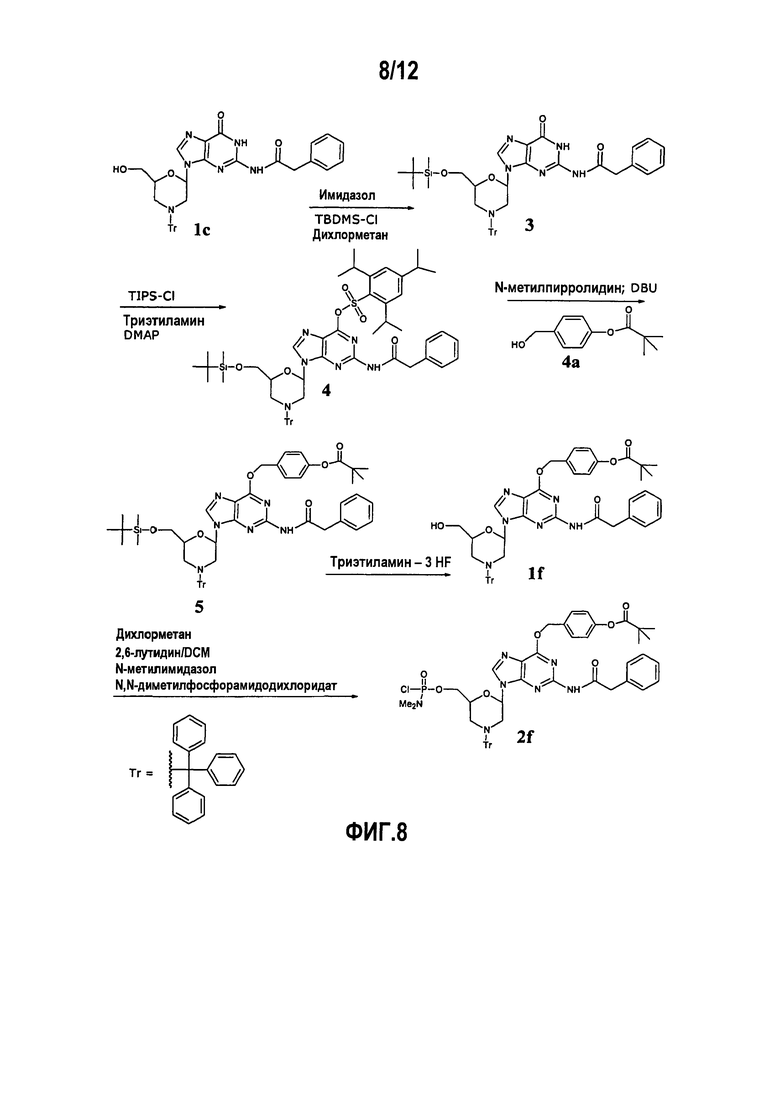
Фиг.8 показывает образование DPG производного, в котором N2 положение является фенилацетилированным и О6 положение защищено 4-(пивалоилокси)бензилокси (РОВ) группой.
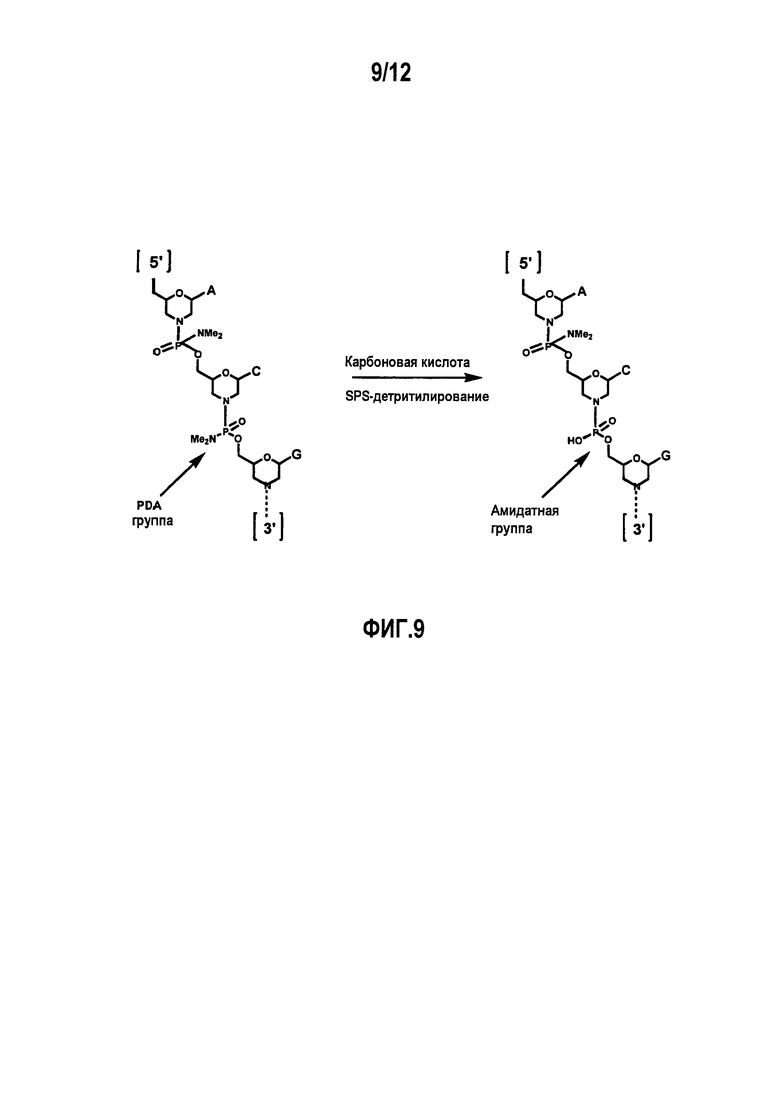
Фиг.9 показывает превращение фосфородиамидатного (PDA) мостика в фосфорамидатные (амидатные) мостики по побочной реакции, которая может иметь место при обработке морфолиновых олигомеров, связанных с фосфородиамидатом (РМО), карбоновыми кислотами.
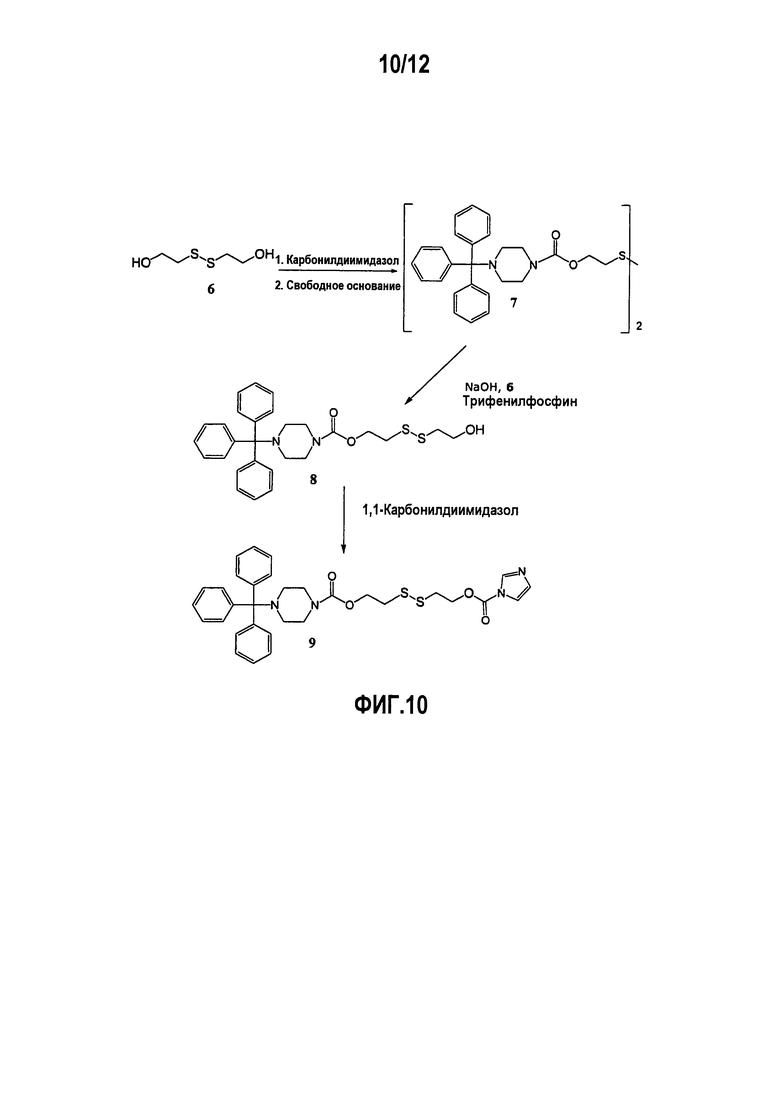
Фиг.10 показывает приготовление дисульфидного анкера для применения в модификации синтетического полимера, используемого для ступенчатого получения морфолинового олигомера, при этом создается возможность для легкого высвобождения олигомера обработкой тиолом.
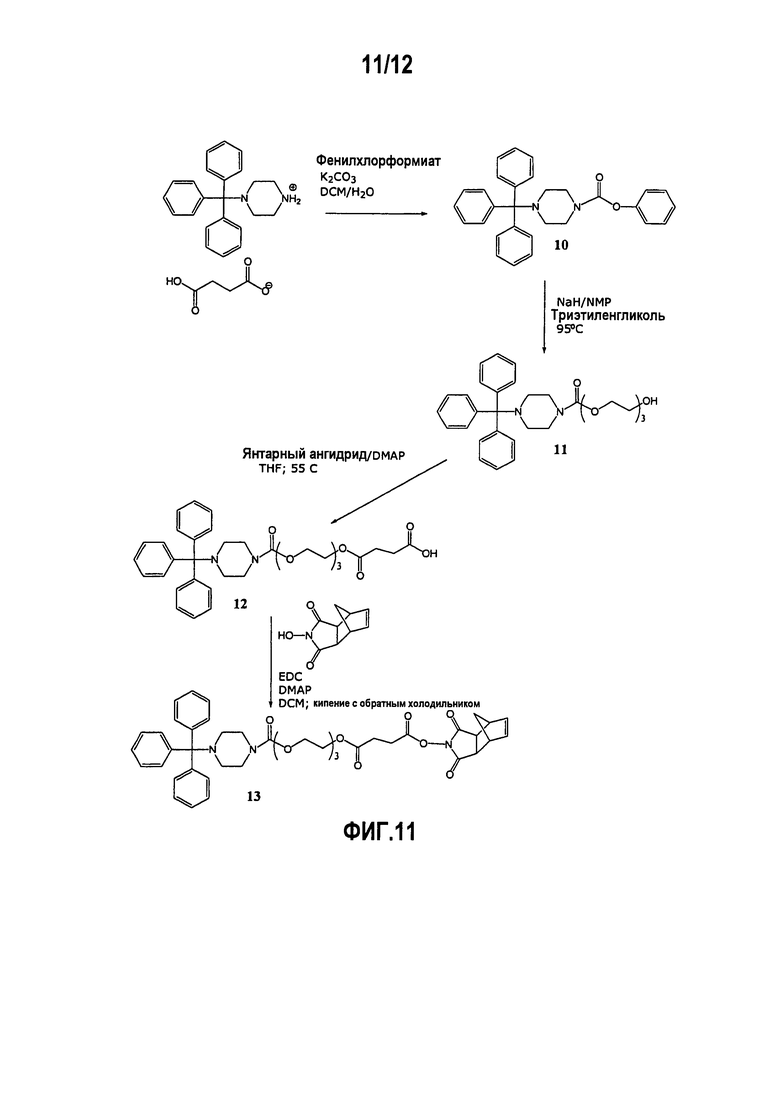
Фиг.11 показывает приготовление фрагмента, содержащего триэтиленгликоль (“хвоста”/“Tail”), который повышает растворимость в воде синтетических антисмысловых олигомеров.
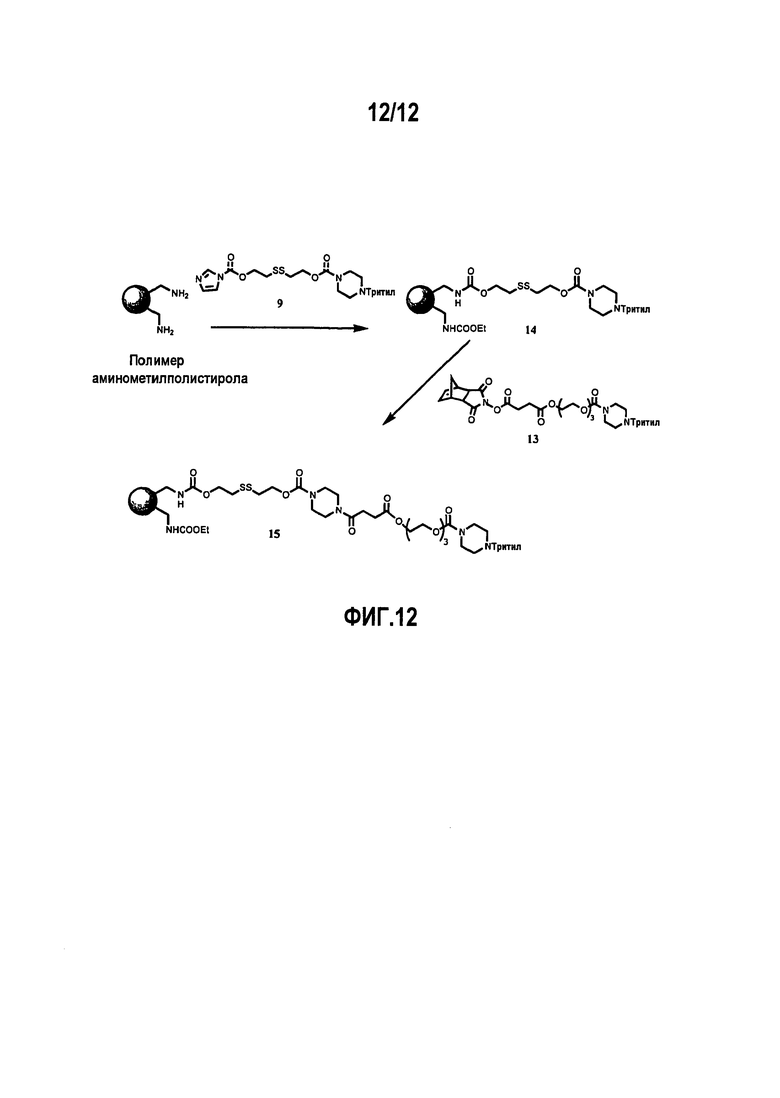
Фиг.12 показывает приготовление полимеров, применимых для твердофазного синтеза морфолиновых олигомеров.
Подробное описание изобретения
I. Определения
Нижеприведенные термины, использованные в настоящем описании, имеют следующие значения, если не оговорено специально.
“Морфолиновый олигомер” относится к полимерной молекуле, содержащей основу, которая обеспечивает основания, способные к образованию водородной связи с обычными полинуклеотидами, где полимер испытывает недостаток в группировке основы из пентозного сахара, а точнее рибозной основы, присоединенной фосфодисложноэфирными связями, которая обычно состоит из нуклеотидов и нуклеозидов, но вместо этого содержит кольцевой атом азота со связыванием через кольцевой атом азота. Предпочтительный морфолиновый олигомер составлен из структур “морфолиновых звеньев”, таких как показано ниже, которые в олигомере предпочтительно связаны вместе (тио)фосфородиамидатными мостиками, причем морфолиноазот одного звена присоединяется к 5’-экзоциклическому углероду соседнего звена. Каждое звено включает спаренную группировку Pi пуринового или пиримидинового основания, которая является эффективной, чтобы присоединяться, посредством водородного связывания, к основанию в полинуклеотиде.
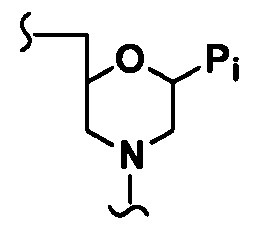
Морфолиновые олигомеры детализированы, например, в патентах США (включая совместные) №№ 5698685, 5217866, 5142047, 5034506, 5166315, 5185444, 5521063 и 5506337, которые все специально включены ссылкой в настоящем описании.
“Фосфородиамидатная” группа включает фосфор с двумя присоединенными атомами кислорода и двумя присоединенными атомами азота, и в настоящем описании также может относиться к фосфору с одним присоединенным атомом кислорода и тремя присоединенными атомами азота. В мостиках внутри звеньев олигомеров, представленных в настоящем описании, один азот обычно является боковой цепью к основной цепи, и второй азот представляет собой кольцевой атом азота в структуре морфолинового кольца, как показано в формуле II ниже. В альтернативном случае или в дополнение, азот может присутствовать при 5’-экзоциклическом углероде, как в формулах III и IV ниже.
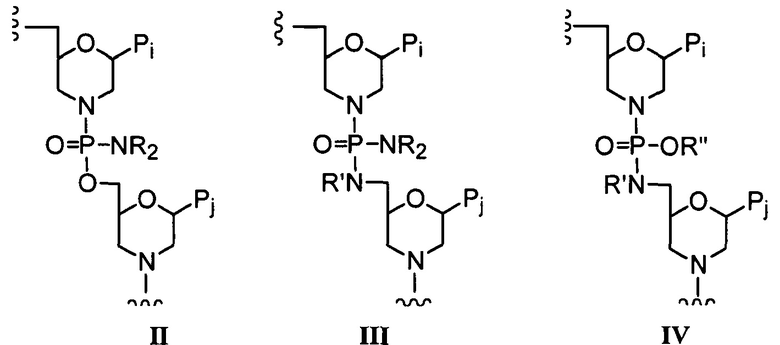
В тиофосфородиамидатном мостике один атом кислорода, обычно бокового кислорода по отношению к основе в олигомерах, представленных в настоящем описании, замещен серой.
“Морфолиновое звено с носителем в твердой фазе” может представлять собой первый или любой последующий мономер морфолинового звена, включенный в морфолиновый олигомер постадийным твердофазовым синтезом, раскрытым в настоящем описании. Данное звено присоединено к твердому носителю или к растущей цепи олигомера на твердом носителе, посредством 5’-экзоциклического углерода. “Защищенное основание” относится к защите групп спаренных оснований, например, пуриновых или пиримидиновых оснований, на морфолиновых звеньях с помощью подходящих защитных групп, чтобы предотвращать взаимодействие или интерференцию групп спаренных оснований в течение постадийного синтеза олигомера.
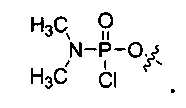
“Активированная фосфорамидатная группа” обычно представляет собой хлорфосфорамидатную группу, имеющую замещение при азоте, которое требуется в окончательном фосфорамидатном мостике в олигомере. Примером является (диметиламино)хлорфосфорамидат, т.е. -О-Р(=О)(NMe2)Cl.
Термины “заряженный”, “незаряженный”, “катионный” и “анионный”, как используются в настоящем описании, относятся к преобладающему состоянию химического фрагмента при рН, близком к нейтральному, например, приблизительно от 6 до 8. Предпочтительно, данный термин относится к преобладающему состоянию химического фрагмента при физиологическом рН, т.е. примерно 7,4.
“Низший алкил” относится к алкильному радикалу из одного до шести атомов углерода, который иллюстрирован метилом, этилом, н-бутилом, изо-бутилом, трет-бутилом, изоамилом, н-пентилом и изопентилом. В отдельных вариантах осуществления “низшая алкильная” группа содержит от одного до четырех атомов углерода, или 1-2 атома углерода; т.е. является метилом или этилом. Аналогично, “низший алкенил” относится к алкенильному радикалу из двух до шести, предпочтительно трех или четырех атомов углерода, который иллюстрирован аллилом или бутенилом.
“Немешающий” заместитель представляет собой заместитель, который не оказывает негативного влияния на способность антисмыслового олигомера, описанного в настоящем документе, присоединяться к предполагаемой мишени. Такие заместители включают небольшие и предпочтительно неполярные группы, такие как метил, этил, метокси, этокси, гидрокси или фтор.
II. Защита основания в синтезе РМО
Из-за определенных проблем в химии морфолина защитная группа для основания должна отвечать нескольким требованиям. Защитная группа должна легко вводиться в/на гетероциклический фрагмент и после этого быть устойчивой к активации звена и условиям очистки и твердофазному синтезу. Защитная группа не должна быть реакционно-способной по отношению к морфолиновому аминному фрагменту растущей цепи и должна создавать возможность для того, чтобы чисто связывать активированное морфолиновое звено с растущей цепью олигомера. Защитная группа должна отщепляться, предпочтительно аммиаком, без введения новых примесей. Наконец, она должна приводить к кристаллическим производным звена, чтобы не создавать необходимости для хроматографической очистки перед активацией.
Как описано ниже и в сравнительных примерах, защитные группы, опубликованные в литературе для дважды защищенных гуанозинов, используемых для синтеза нуклеиновых кислот, адекватно не отвечают данным критериям. Таким образом, потребовалась новая стратегия защиты для морфолино G звеньев. Как описано ниже, применение 4-(пивалоилокси)бензилоксигруппы в О6, как установлено, отвечает всем вышеприведенным критериям.
А. Защитные группы для О6: сравнительные данные
А1. 4-Нитрофенетиловый простой эфир (NPE)
Данное производное готовили, как показано на фиг.2 (Mitsunobu 1981) или фиг.3 (Jones et al. 1982B). Хотя неочищенное звено, защищенное по О6, могло быть приготовлено с рациональным выходом, данное соединение не было четко кристаллическим и могло быть адекватно очищено только хроматографией на силикагеле, что нежелательно для широкомасштабного производства. После тестирования широкого спектра условий ресуспендирования и/или перекристаллизации было установлено, что комбинации бутоксиэтанолсодержащих растворителей могли, с некоторой трудностью, закристаллизовать данный продукт. Однако избыток бутоксиэтанола невозможно удалить из конечного продукта, так как данное соединение, вероятно, кристаллизуется как сольват. Присутствие избытка спиртового растворителя было бы неприемлемым для реакции активации.
NPE группа отщепляется сильным основанием через механизм β-элиминирования. Данные условия приводят к получению реакционноспособного побочного продукта 4-нитростирола, который затем может взаимодействовать с реакционноспособными сайтами олигомера. Хотя разные отщепляющие средства (например, тиолы и 1,3-дикарбонильные соединения) вводили в смесь для снятия защиты в попытке предотвратить улавливание побочного продукта олигомером, никакие из них не были полностью удачными в устранении данной проблемы внутреннего возврата. Даже после очистки олигомеры, приготовленные с данным звеном, имели оттенок желтого цвета.
А2. Фенилсульфонилэтил (PSE) и метилсульфонилэтил (MSE)
Данные группы вводили посредством соответствующих производных 2-тиоэтанола (Jones et al. 1982A, 1982B), как показано на фиг.4. Однако не могла быть найдена методика успешной кристаллизации для полученных звеньев.
Подобно вышеприведенной NPE группе, данные группы отщепляются через механизм β-элиминирования. После включения в олигомер, данные производные создают те же проблемы, что с NPE группой; т.е. внутренний возврат реакционно-способного алкенового побочного продукта, образованного в течение снятия защиты.
А3. Триметилсилилэтиловый простой эфир
Как опубликовано в работе Jones (Jones et al. 1982B), O6-TMSE-модифицированное морфолиногуаниновое звено готовили, как показано на фиг.5, но оно не было устойчивым в течение синтеза олигомера. Олигомеры, сделанные с данным звеном, показали спектр побочных продуктов, подобных побочным продуктам, сделанным из О6-незащищенных G звеньев.
А4. Фениловый простой эфир
Морфолиногуаниновые звенья с О6-фенильным замещением (фиг.6) готовили согласно методике Reese et al. (1981, 1984). Производные включали незамещенный фенил, 2,5-дихлорфенил, пентафторфенил и 3-фторфенил. Такие звенья могли быть включены в PMO, но снятие защиты обычными реагентами, такими как оксим 2-нитробензальдегида и сильное основание, не могли быть выполнены полностью без разложения олигомера.
А5. Карбамат
Некоторые О6-карбаматные производные синтезировали согласно методике Hata et al. 1983 (фиг.7). Использование данных производных в синтезе олигомера давало различные результаты в зависимости от использованных производных. Для более лабильных соединений, таких как дифенилкарбамоильный аналог, отмечен перенос защитной группы к 3’-азоту растущей цепи в течение стадии конденсации твердофазного синтеза, приводящего к укороченным олигомерам, содержащим 3’-дифенилкарбамоильный фрагмент. Кроме того, О6-карбаматы имеют два возможных сайта для взаимодействия с аммиаком. В то время как более реакционно-способные фрагменты, такие как дифенилкарбамоильная группа, проявляли относительно селективное действие на карбонил, более устойчивые диметил и пирролидинилкарбаматы показали значительное конкурирующее взаимодействие аммиака в С6 положении с превращением в диаминопурин.
В. 4-(Пивалоилокси)бензилоксизащитная группа
4-(Пивалоилокси)бензилоксиспирт (4а, фиг.8) вводили в морфолиногуаниновое звено путем эффективного синтеза с высоким выходом. Звено перед активацией (соединение 1f на фиг.1 и 8) может быть синтезировано и воспроизводимо выделено в большом масштабе без хроматографической очистки, и его можно кристаллизовать из множества растворителей (например, ТГФ/вода ТГФ/гептан, ацетонитрил, различные смеси сложный эфир/углеводород). Десять загрузок данного звена, произведенных в масштабе 50-200 галлонов (190-760 литров) (размер загрузки: 8-27 кг соединения 1с), давали средний выход продукта в 65%, имеющего чистоту (по данным ВЭЖХ) от 97,6% до 99,2%.
Звено превращается в активированное звено (т.е. конверсия в 5’-хлорфосфорамидатное соединение) намного более чисто, чем монозащищенный G, и она может быть легче очищена хроматографией на силикагеле. При промышленном изготовлении общий выход от соединения 1f до соединения 2f (фиг.1) приблизительно составляет 50%.
РОВ защитная группа может быть использована с другими комбинациями защитных групп для N2 и атомов азота морфолинового кольца. Подходящие защитные группы для N2 включают фенилацетил (показанный на фиг.8), а также ацетил, пропионил, изобутирил и феноксиацетил. Тритильные частицы, подходящие для защиты азота морфолинового кольца между стадиями связывания, включают незамещенный тритил, 4-метил-, 4,4’диметил- и 4,4’,4’’-триметилтритил, и 4-метокситритил.
Другие ацильные защитные группы также могут быть использованы вместо пивалоила для фенольного фрагмента РОВ группы. Подходящие альтернативы включают N,N-диметилкарбамоил и бензоил.
В течение синтеза РМО не отмечены никакие продукты, в которых пивалоильная группа присоединена к 3’-концу более мелких фрагментов РМО по всей длине РМО. Только определенный заметный побочный продукт представлял собой РМО, содержащий фенольный остаток, полученный от взаимодействия с побочным продуктом хинонметилом для снятия защиты. Однако данный побочный продукт мог быть снижен до следовых количеств достаточным разбавлением аммиачного раствора при снятии защиты. Кроме того, его легко удалить с помощью эффективного связывания фенольного остатка c полимерными смолами, применяемыми для эффективной анионообменной хроматографии. В целом, общий выход очищенного РМО значительно повышен, как видно в таблице 1.
Усовершенствование в получении РМО, достигаемое с помощью гуаниновой группы, защищенной РОВ, наиболее очевидно в очистке после твердофазного синтеза РМО, где трудность в удалении диаминопурина и родственных побочных продуктов может приводить к большой потере в течение эффективной анионообменной (SAX) хроматографии. Например, сырые примеси для AVI-4126, приготовленного с CPM и MPG (монозащищенное гуаниновое звено), находятся в интервале 68-73%, который вычисляет приблизительно 58% сырой выход РМО. В течение очисток продукта на тритильном компоненте и очисток от тритильного компонента значительное количество продукта теряется при получении чистого продукта, и общее выделение от хроматографии составляет 52%. Для AVI-4126, полученного с использованием CYTFA и DPG (дизащищенное гуаниновое звено), сырые примеси составляют 70-75%, сопоставимые с N-1 уровнями по данным масс-спектроскопии (при этом указывается, что эффективности детритилирования реагентов CYTFA и CPM приблизительно эквивалентны) и сырыми выходами примерно в 61%. Однако применение обычных методов очистки открывает 80% РМО из сырой смеси.
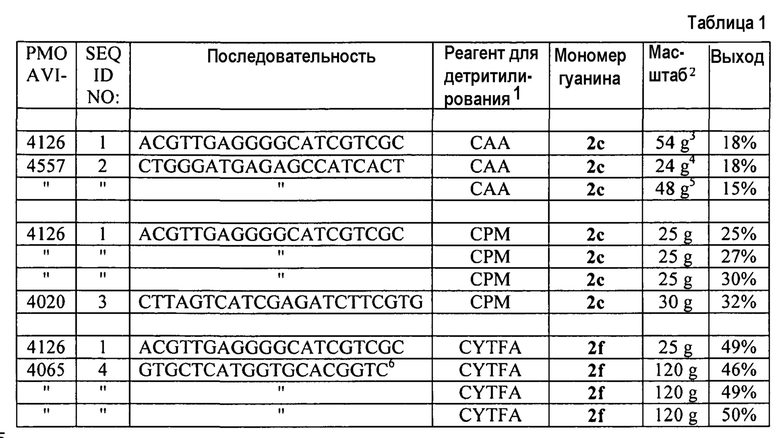
Синтезы выполняли в соответствии с методами, описанными в совместной заявке США 11/801885, используя модификации, указанные в таблице; см. примеры 3-6 ниже. Все РМО имеют 5’-“хвост” и не замещены на 3’-конце.
1САА=11% цианоуксусная кислота (масс./масс.) в смеси 20% ацетонитрил/DCM (об./об.);
СРМ=2% метансульфонат 3-хлорпиридиния (масс./об.) и 0,9% этанол (об./об.) в 20% трифторэтанол/DCM (об./об.); CYTFA=2% трифторацетат 3-цианопиридиния (масс./об.) и 0,9% этанол (об./об.) в 20% трифторэтанол/DCM (об./об.).
2Масштаб означает массу исходного полимера в граммах. Нагрузка полимера равна 480-520 микромоли/г.
3Объединенный выход из 4×12 г и 1×8 г циклов.
4Объединенный выход из 2×12 г циклов.
5Объединенный выход из 4×12 г циклов.
6Добавление конечной С звена выполняли активированным морфолиновым звеном с 4-метокситритильной защитой на морфолиноазоте.
Таким образом, применение дважды защищенного MoG мономера изобретения предоставляет способ синтеза морфолинового олигомера повышенного выхода и чистоты по отношению к ранее известным в данной области способам, и особенно в сравнении с выходами и чистотой, наблюдаемыми, когда использован монозащищенный MoG мономер или другой защищенный MoG мономер, подпадающий под настоящее изобретение. В частности, данный способ предпочтительно дает сниженный уровень диаминопуриновых соединений, то могло бы получаться при использовании MoG мономера, подпадающего под настоящее изобретение.
III. Дважды защищенные гуаниновые морфолиновые звенья
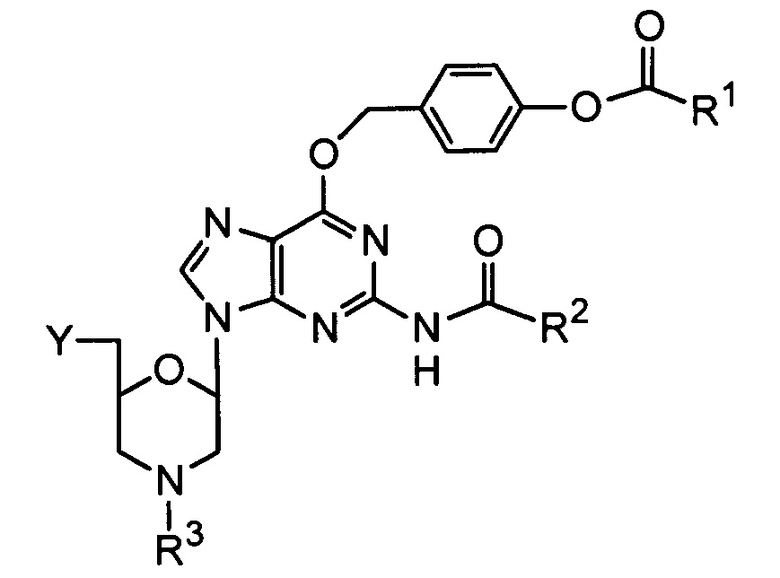
Дважды защищенные гуаниновые (DPG) морфолиновые звенья данного изобретения имеют структуру:
где
R1 выбран из группы, состоящей из низшего алкила, ди(низший алкил)амино и фенила;
R2 выбран из группы, состоящей из низшего алкила, моноциклического арилметила и моноциклического(арилокси)метила;
R3 выбран из группы, состоящей из триарилметила и водорода; и
Y выбран из группы, состоящей из: защищенной или незащищенной гидроксильной или аминогруппы; хлорфосфорамидатной группы; и фосфородиамидатного мостика к кольцевому атому азота следующего морфолинового соединения или морфолинового олигомера.
В отдельных вариантах осуществления Y представляет собой защищенную или незащищенную гидроксильную группу (как в предварительно активированном мономере) или хлорфосфорамидатную группу (как в активированном мономере). Предпочтительные защитные группы для гидроксильной группы включают триалкилсилильные группы, такие как трет-бутилдиметилсилил (TBDMS).
Варианты осуществления, в которых Y представляет собой фосфородиамидатный мостик к кольцевому атому азота следующего морфолинового соединения, или фосфородиамидатный мостик к морфолиновому олигомеру, относятся к соединениям, образованным в течение синтеза морфолинового олигомера, перед снятием защиты с основания.
Как обсуждено ниже, заместители на хлорфосфорамидатной группе (в активированном мономере) могут различаться в зависимости от определенного желаемого фосфордиамидатного мостика.
Данное изобретение также относится, соответственно, к способу синтеза морфолинового олигомера, причем способ включает:
(а) взаимодействие морфолинового звена с носителем твердой фазы, имеющей незащищенный кольцевой атом азота, с мономером морфолинового звена с защищенным основанием, имеющим кольцевой атом азота, защищенный триарилметилом, и активированную фосфорамидатную группу на 5’-экзоциклическом углероде,
при этом образуется фосфородиамидатная связь между 5’-экзоциклическим углеродом и незащищенным кольцевым атомом азота;
(b) снятие защиты с указанного защищенного кольцевого атома азота для образования незащищенного кольцевого атома азота; и
(с) повторение стадий (а) и (b) один или несколько раз со следующими мономерами морфолинового звена с защищенным основанием;
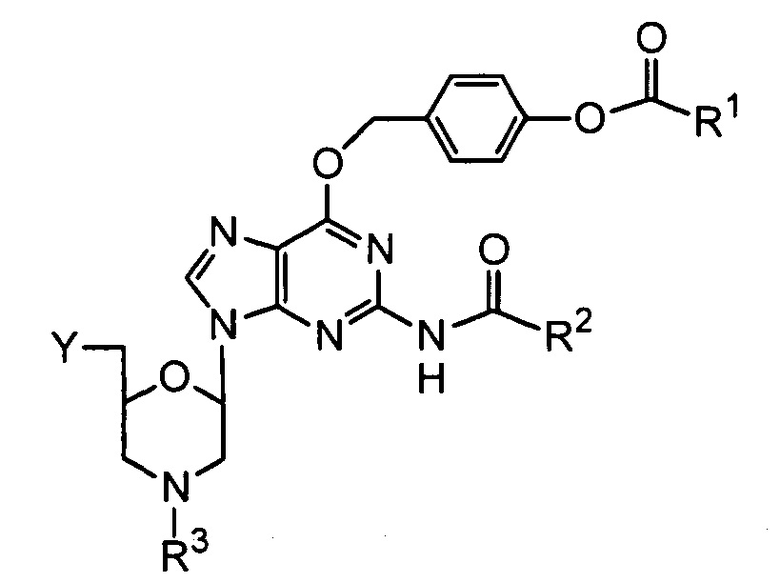
где, по меньшей мере, один из указанных мономеров морфолинового звена с защищенным основанием представляет собой дважды защищенное гуаниновое морфолиновое соединение, имеющее структуру:
в которой
R1 выбран из группы, состоящей из низшего алкила, ди(низший алкил)амино и фенила;
R2 выбран из группы, состоящей из низшего алкила, моноциклического арилметила и моноциклического(арилокси)метила;
R3 выбран из группы, состоящей из триарилметила и водорода; и
Y представляет собой хлорфосфорамидатную группу.
Предпочтительные триарилметильные защитные группы для азота морфолинового кольца (R3) включают тритил(трифенилметил), 4-метокситритил, 4-метилтритил, 4,4’-диметилтритил и 4,4’,4’’-триметилтритил.
R1 заместитель на О6 защитной группе предпочтительно представляет собой С1-С4алкил, особенно –С(СН3)3 (трет-бутил), как в 4-(пивалоилокси)бензилокси (РОВ) группе. Однако R1 может также означать ди(низший алкил)амино, а именно, диметиламино или фенил.
Как отмечено выше, замещение хлорфосфорамидатной группы Y в “активированных” мономерах изменяется в зависимости от структуры желаемого фосфородиамидатного мостика. Для приготовления “стандартного” незаряженного РМО мостика 5’-O-P(=O)(-N(CH3)2)-3’ (как показано в формуле II выше, где R означает метил), хлорфосфорамидатная группа Y представляет собой 5’-O-P(=O)Cl-NR2 (см., например, соединение 2f, фиг.8).
Как описано в совместной заявке, имеющей USSN 11/801885, с регистрацией от 10 мая 2007 года, которая включена в настоящее описание ссылкой, полезные свойства могут быть получены за счет приготовления РМО, имеющих катионные, а также нейтральные мостики между звеньями. В таких олигомерах, по меньшей мере, один межзвенный мостик между двумя последующими структурами морфолиноколец содержит боковую катионную группу. Боковая группа несет дистальный атом азота, который может нести положительный заряд при нейтральном или близком к нейтральному (например, при физиологическом) рН.
Для приготовления таких мостиков хлорфосфорамидатная группа Y в звенных мономерах данного изобретения может иметь одну из следующих структур:
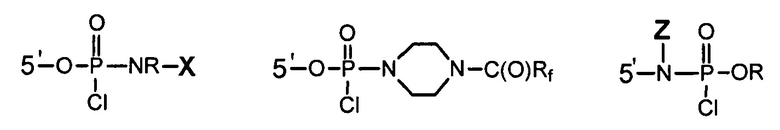
где R представляет собой низший алкил, такой как метил или этил;
Х=-R4-NHC(=O)Rf, где R4 представляет собой бивалентный алкил или олиго-PEG, и Rf представляет собой полностью или частично фторированный метил, этил или изопропил; и
Z=X, определенный выше, или низший алкил. Отметим, что Z-содержащая группа дает 5’-аминсодержащий мостик.
Термин “олиго-PEG” относится к группе, такой как –(СН2-СН2-О)n-CH2-CH2-, где n обычно означает 1-3, и “бивалентный алкил” обычно означает С2-С8алкил.
После приготовления олигомеров при использовании мономеров, содержащих такие активированные хлорфосфорамидатные группы, С(=О)Rf защитные группы удаляют с терминальных атомов азота, которые затем могут быть модифицированы, например, для образования терминальных гуанидильных групп, как описано в совместной заявке USSN 11/801885.
IV. Улучшенные условия для снятия защиты с азота морфолинового кольца в синтезе РМО
Как указано выше, снятие защиты с азота морфолинового кольца, который обычно защищают триарилметильной группой, такой как тритильная, в РМО синтезе, должно завершаться в достаточной степени на каждой стадии, чтобы свести к минимуму N-1 делецию частиц. Однако изучения в поддержку данного изобретения показали, что реагенты, использованные ранее в данной области для данной цели вызывали нежелательное количество гидролиза основы (см. фиг.1) и деградацию. Поэтому предприняты попытки в части эффективных реагентов для снятия защиты, которые одновременно минимизируют такой гидролиз.
Простой анализ использован для тестирования эффективности различных реагентов в снятии защиты (обычно снятии тритильной группы) с N-защищенных морфолиновых звеньев. Модельное соединение, тритилированную moCBz (т.е. бензоилзащищенную цитозинморфолино) звено, показанное ниже, растворяют в растворе для детритилирования, предназначенном для исследования. В разные моменты времени (например, 1, 2, 4 мин) аликвоту гасили и анализировали с помощью ТСХ или ВЭЖХ для завершения снятия защиты с азота морфолина. Обычно для прогноза эффективного детритилирования в течение твердофазного синтеза РМО данная модельная реакция должна быть завершена в течение примерно 2 минут при комнатной температуре.
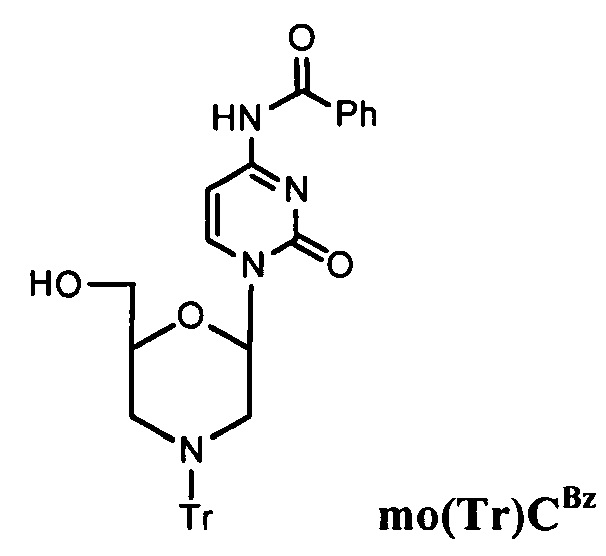
При использовании данного анализа и последующего экспериментирования было обнаружено, что различные пиридиниевые соли сильных кислот в смесях трифторэтанола (TFE) и дихлорметана (DCM) являются отличными катализаторами для удаления триарилметильной защитной группы, например, тритильной группы от атома азота морфолина в ходе твердофазного синтеза РМО.
Для значительных скоростей реакции и солюбилизации пиридиниевых солей предпочитают минимальное количество TFE (~10% об./об. или больше). Поскольку один TFE не вызывает набухания незащищенного полистирола, предпочитают смеси с DCM (дихлорметаном), особенно в ранних циклах синтеза РМО. Предпочтительные композиции растворителей включают от 10 до 75% TFE.
Применение TFE растворителя, как полагают, усиливает селективность реакции детритилирования сверх образования амидата (гидролиз) и расщепления фосфородиамидата (PDA), описанного выше, привлечением различных механизмов расщепления PDA и детритилирования. TFE является потенциальным растворителем для образования водородной связи и снижает реакционную способность нуклеофилов в растворе; поэтому, как считают, он замедляет атаку на фосфор, необходимую для расщепления P-N связи. TFE также ускоряет реакции сольволиза SN1 типа. Характер расщепления растворителем реакций детритилирования амина посредством TFE очевиден по желтой окраске смесей реакции детритилирования и оранжевой окраске смесей реакции деметокситритилирования. Поэтому повышение концентрации TFE полагается как для подавления нуклеофильной атаки на PDA мостик, так и для ускорения детритилирования.
Незамещенные пиридиниевые соли являются недостаточно кислыми для оптимального снятия защиты, но применение пиридиниевых соединений, содержащих электронно-акцепторные группы (EWG) (например, галоген, карбонил, циано), дает быстрое отщепление защитных групп. Обычно, по меньшей мере, 2% (масс./об.) такой соли в растворителе TFE:DCM достаточны для быстрого детритилирования. Предпочтительные уровни пиридиниевых солей составляют от 2 до 10% (масс./об.).
Кислоты, применимые для образования пиридиниевых солей, включают сульфоновые кислоты, такие как метансульфоновая, трифторметансульфоновая и п-толуолсульфоновая кислота, трифторуксусная кислота и хлористоводородная кислота. Хотя карбоновая кислота, трифторуксусная кислота не закрывают растущую цепь РМО, если присутствуют в течение реакции связывания, их карбоксилат является недостаточно нуклеофильным, чтобы ускорять образование амидата. В частности, предпочтительными являются трифторуксусная и особенно метансульфоновая кислота.
Пиридины, применимые для образования пиридиниевых солей, включают галогензамещенные пиридины, особенно менее дорогие хлорпиридины, из которых предпочитают 3-хлорпиридин, и цианопиридины, из которых предпочитают 4-цианопиридин. 3- и 4-цианопиридины являются легко доступными, дешевыми химикатами в больших количествах. В общем, эффективность данных солей связана обратнопропорционально с рКа пиридиниевых соединений. Пиридины с электронно акцепторными группами находятся в интервале рКа от примерно 1 до 4 (Fisher et al. 1964, Rogne 1970).
Также применимыми являются производные никотиновой кислоты (т.е. сложные эфиры, такие как этилникотинат и никотинамид), а также представители их кетонов и альдегидов. Обычно, однако, данные соединения представляют собой менее потенциальные реагенты, чем цианопиридиниевые соли.
Будет оценено, что соли гетероциклов, иных, чем пиридины, могут функционировать как селективные реагенты детритилирования при описанных состояниях при условии, что рКа протонированной формы одинакова с рКа замещенных пиридинов данного изобретения. Примеры можно найти во многих таблицах рКа для гетероциклов, найденных в литературе (например, Albert 1963). Примеры включают тиазол (рКа 2,53), пиридазин (рКа 2,33), пиразол (рКа 2,47), триазол (рКа 2,30) и их замещенные производные, особенно производные, замещенные с помощью EWG, описанных выше.
Две особенно предпочтительные соли представляют собой метансульфонат 3-хлорпиридиния (СРМ) и трифторацетат 4-цианопиридиния (CYTFA), и особенно предпочтительные варианты осуществления реагентов для детритилирования включают растворы 2% (масс./об.) СРМ или CYTFA в 20% смеси трифторэтанол/DCM (об./об.), содержащей 0,9% этанола (об./об.). Как показано в таблице 1 выше, использование данных реагентов приводит к значительному повышению выхода по сравнению с обычными реагентами на основе цианоуксусной кислоты.
Установлено, что более кислый CYTFA является слабо более эффективным, чем СРМ. Однако большое повышение в выходе при сравнении между СРМ и CYTFA реагентами в таблице 1 может быть связано с использованием дважды защищенного гуанинового мономера (DPG), в котором О6 положение защищено 4-(пивалоилокси)бензилоксигруппой, как раскрыто в совместной и конкурентно поданной предварительной заявке, озаглавленной “Improved Synthesis of Morpholino Oligomers using Doubly Protected Guanine Mopholino Subunits”. В целом, использование DPG мономера снижает количество побочных продуктов, содержащих диаминопурин, в то время как улучшенные реагенты для детритилирования снижают количество гидролизующейся основы или укороченных побочных продуктов.
Таким образом, данная модификация обеспечивает способ синтеза морфолинового олигомера с пониженным гидролизом фосфородиамидатного мостика в основе, и предпочтительно со сниженным или эквивалентным уровнем соединений с N-1 делецией по сравнению с предшествующими способами в данной области. В другом аспекте данное изобретение относится к способу снятия защиты с морфолинового кольцевого атома азота, защищенного триарилметилом, в течение синтеза морфолинового олигомера с пониженным гидролизом фосфородиамидатного мостика в основе морфолинового олигомера в сравнении со способом, когда цианоуксусную кислоту используют в качестве реагента для снятия защиты. Предпочтительно, данный способ также обеспечивает сниженный или эквивалентный уровень соединений с N-1 делецией, которые были бы в случае, когда цианоуксусная кислота использована в качестве реагента для снятия защиты.
Следующая полезная модификация представляет собой использование агента для захвата тритила, такого как тиол, чтобы сдвигать равновесие реакции в отношении продуктов. Применение агентов по захвату тритила использовалось для синтеза нуклеиновых кислот (Ravikumar et al., U.S. Patent № 5510476). Меркаптоэтанол является легко доступным, дешевым агентом, применимым для данной цели. Присутствие гидроксильной группы не является важным для захвата, так как простые тиолы, такие как бензилмеркаптан, проявляют себя одинаково хорошо. Спирты, такие как этанол и бутанол, и даже вода, также служат в качестве агентов для захвата тритильного катиона.
Примеры
Пример 1
Синтез N2-PhAc, O6-POB дважды защищенной морфолино G (DPG) звена (см. фиг.8)
Приготовление соединения 3 (исходя из 35 кг 1с): 100 галлоновый (380 л) реактор загружают соединением 1с (35 кг; 1,0 экв.), имидазолом (5,0 кг; 1,3 экв.) и дихлорметаном (279 кг). Загрузку охлаждают до 3°С. 50 галлоновый (190 л) реактор охлаждают до 3°С и загружают трет-бутилхлордиметилсиланом (10,1 кг; 1,2 экв.) и дихлорметаном (93 кг). Раствор в 50 галлоновом (190 л) реакторе переносят в 100 галлоновый (380 л) реактор и данную порцию термостатируют при 20°С. После окончания реакции (1-3 часа) в 100 галлоновый (380 л) реактор загружают метанол (1,8 кг; 1,0 экв.). Спустя 30 минут раствор в 100 галлоновом (380 л) реакторе загружают в 200 галлоновый (760 л) реактор, содержащий цитратный буфер с рН 3 (376 кг 1М лимонной кислоты, отрегулированной до рН 3 твердым NaOH). Данную загрузку перемешивают в течение 30 минут и слои отделяют. Нижний органический слой промывают еще раз цитратным буфером с рН 3 и один раз насыщенным солевым раствором (287 кг 2,5% NaCl/вода (масс./масс.)). Полученный органический раствор перегоняют при <35°С до тех пор, пока анализ загрузки по Карлу Фишеру не показывает <0,05% воды. Данный раствор охлаждают до 3°С в 100 галлоновом (380 л) реакторе и используют непосредственно в приготовлении соединения 4.
Приготовление соединения 4 : 100 галлоновый (380 л) реактор, содержащий раствор соединения 3, загружают триэтиламином (6,8 кг; 1,2 экв.), 4-диметиламинопиридином (0,68 кг; 0,1 экв.) и триизопропилбензолсульфонилхлоридом (18,6 кг; 1,1 экв.). Загрузку нагревают при 20°С. После окончания реакции (3-9 часов) данный раствор загружают в 200 галлоновый (760 л) реактор, содержащий фосфатный буфер с рН 4,5 (228 кг 1М КН2РО4). Данную загрузку перемешивают в течение 30 минут и слои отделяют. Нижний органический слой промывают насыщенным солевым раствором (212 кг 2,5% NaCl/вода (масс./масс.)). Полученный органический раствор перегоняют при <35°С до тех пор, пока анализ загрузки по Карлу Фишеру не показывает <0,01% воды. Данный раствор охлаждают до 3°С в 100 галлоновом (380 л) реакторе и используют непосредственно в приготовлении соединения 5.
Приготовление соединения 4a (исходя из 60 кг 4-гидроксибензальдегида): 750 галлоновый (2850 л) реактор загружают 4-гидроксибензальдегидом (60 кг; 1,0 экв.), толуолом (260 кг) и 1-метилимидазолом (8,1 кг; 0,2 экв.). В данный раствор загружают раствор бикарбоната калия (100 кг; 2,0 экв.) в воде (400 кг), затем - триметилацетилхлорид (83 кг; 1,4 экв.). Двухфазную смесь перемешивают при 20°С. После окончания реакции (1-5 часов) в данную порцию загружают метанол (15,7 кг; 1,0 экв.). Данную загрузку перемешивают при 20°С в течение 1 часа. Слои отделяют. К верхнему органическому слою добавляют воду (200 кг). Данную загрузку перемешивают в течение 30 минут и слои отделяют. В верхний органический слой загружают фосфатный буфер с рН 4,5 (16,5 кг КН2РО4 в 242 кг воды). Данную порцию перемешивают в течение 30 минут и слои отделяют. В верхний органический слой загружают воду (200 кг). Данную порцию перемешивают в течение 30 минут и слои отделяют. Верхний органический слой перегоняют в вакууме при <35°С до достижения объема загрузки в 200 л. В данную порцию загружают ТГФ (70 кг), и порцию переносят в 500 галлоновый (380 л) (1900 л) реактор, содержащий Pd/C (9,6 кг; 0,004 экв.; 5% Pd/C, 50% влажность Johnson Matthey Type A405028-5 или A570129-5). Реактор первоначально поддерживают под давлением 5 psi (0,0344 МПа) H2 c устройством для перемешивания при 50 об/м. Как давление, так и скорость перемешивания медленно повышают по мере протекания реакции до максимума в 25 psi (0,1720 МПа) H2 и 90 об/м. После окончания реакции (8-48 часов) данную порцию фильтруют через слой целлита, затем через 0,1 микронный совмещенный фильтр. Целит промывают толуолом (20 кг). В данную загрузку вносят раствор фосфатного буфера с рН 6,5 (2,7 кг КН2РО4 и 2,3 кг фосфата калия, двухосновного, тригидрата в 200 кг воды). Данную порцию перемешивают в течение 30 минут и слои отделяют. Верхний органический слой перегоняют в вакууме при <35°С до достижения объема загрузки в 140 л. В данную порцию загружают толуол (126 кг) и загрузку перегоняют в вакууме при <35°С до достижения объема загрузки в 140 л. Порцию устанавливают при 20°С и переносят в 500 галлоновый (1900 л) реактор, содержащий н-гептан (821 кг) и затравочные кристаллы соединения 4а (100 г), хранящиеся при 0°С. Загрузку хранят при 0°С в течение 1-2 часов. Добавляют вторую порцию кристаллов (100 грамм), и загрузку хранят при 0°С в течение 1-2 часов. Соединение 4а выделяют фильтрованием. Выход=70-80% от 4-гидроксибензальдегида.
Производное, в котором фенольная группировка защищена в виде ее N,N-диметилкарбамата вместо пивалатного сложного эфира, получают в условиях, одинаковых с условиями получения 4а. Чтобы подтолкнуть к завершению реакции между 4-гидроксибензальдегидом и диметилкарбамоилхлоридом, данную реакцию выполняют в дихлорметане при кипении с обратным холодильником в присутствии N-метилимидазола в качестве основания и 0,2 экв. DMAP в качестве катализатора.
Приготовление соединения 5 : 100 галлоновый (380 л) реактор, содержащий раствор соединения 4, загружают N-метилпирролидином (9,5 кг; 2,0 экв., растворенные в 23 кг дихлорметана). Спустя 10 минут добавляют соединение 4а (14,0 кг; 1,2 экв.), затем 1,8-диазабицикло[5.4.0]ундец-7-ен (10,2 кг; 1,2 экв. в 23 кг дихлорметана). Загрузку нагревают при 20°С. После окончания реакции (1-9 часов) данный раствор разбавляют 327 кг дихлорметана и загружают в 200 галлоновый (760 л) реактор, содержащий фосфатный буфер с рН 4,5 (334 кг 1 М КН2РО4). Данную загрузку перемешивают в течение 30 минут и слои отделяют. Нижний органический слой промывают еще раз фосфатным буфером с рН 4,5 (111 кг 1 М КН2РО4), затем один раз насыщенным солевым раствором (212 кг 2,5% NaCl/вода (масс./масс.). Полученный органический раствор перегоняют при <35°С до тех пор, пока анализ загрузки по методу Карла Фишера не показывает <0,05% воды. Данный раствор используют непосредственно в приготовлении соединения 1f.
Приготовление соединения 1f: 100 галлоновый (380 л) реактор, содержащий раствор соединения 5, загружают триэтиламин тригидрофторидом (18,0 кг; 2,0 экв.). Загрузку нагревают при 20°С. После окончания реакции (4-20 часов) данную загрузку помещают в 200 галлоновый (760 л) реактор. 200 галлоновый (760 л) реактор загружают раствором NaHCO3 (230 кг 5% (масс./масс.) раствора). Данную загрузку перемешивают в течение 30 минут и слои отделяют. Нижний органический слой промывают еще раз раствором NaHCO3 (230 кг 5% (масс./масс.) раствора), затем один раз фосфатным буфером с рН 6,5 (9,3 кг КН2РО4 и 14,0 кг К2НРО4 в 215 кг воды). Полученный органический раствор претерпевает обмен растворителей на ТГФ (чтобы получить <1% DCM масс. в загрузке). Раствор разбавляют с помощью ТГФ (124 кг) и нагревают до 60°С. В ТГФ раствор медленно загружают воду (8 кг на кг соединения 1f в растворе на основе LOE анализа; предварительно нагретые до 60°С). Раствор медленно охлаждают до 3°С и выдерживают в течение >4 часов. Сырое соединение 1f отделяют фильтрованием. Если желательно, может быть произведена вторая перекристаллизация для дополнительной очистки соединения 1f. Выход=53-73% от 1c.
Приготовление соединения 2f (исходя из 12 кг 1f): 50 галлоновый (190 л) реактор загружают соединением 1f (12 кг; 1,0 экв.), дихлорметаном (159 кг), 2,6-лутидином (2,5 кг; 1,6 экв.) и 1-метилимидазолом (0,36 кг; 0,3 экв.). Данный раствор перегоняют для достижения объема загрузки в 69 л и охлаждают до 5°С. Данную порцию загружают N,N-диметилфосфорамидодихлоридатом (3,8 кг; 1,6 экв.). Данную порцию термостатируют при 20°С. После окончания реакции (6-16 часов) данную порцию загружают толуолом. Полученную смесь перегоняют для достижения объема загрузки в 126 л (ГХ анализ загрузки должен показывать 30-45% DCM масс.) и переносят в 100 галлоновый (380 л) реактор, содержащий цитратный буфер с рН 3 (15,4 кг моногидрата лимонной кислоты, 1,4 кг NaOH, 80 кг воды). Данную загрузку перемешивают в течение 30 минут и слои отделяют. Нижний водный слой отбрасывают. Верхний органический слой переносят в 50 галлоновый (190 л) реактор, содержащий сульфат натрия (8,0 кг). Данную загрузку перемешивают в течение 30 минут и осадок отходов от сульфата натрия удаляют фильтрованием. Осадок сульфата натрия промывают дихлорметаном (16 кг). Раствор полученного продукта перегоняют в 50 галлоновом (190 л) реакторе для достижения объема загрузки в 53 л (ГХ анализ загрузки должен показывать 11-15% DCM масс.). 100 галлоновый (380 л) реактор загружают гептаном (238 кг). Загрузку в 50 галлоновом (190 л) реакторе переносят в 100 галлоновый (380 л) реактор в продолжение 2 часов. По окончании переноса загрузку выдерживают при 20°С в течение 4-16 часов. Сырое соединение 6 собирают фильтрованием. Сырой продукт загружают в 100 галлоновый (380 л) реактор. К неочищенным твердым веществам добавляют раствор толуола (16 кг) и гептана (50 кг). Данную смесь перемешивают в течение 3 часов и фильтруют. Ресуспендирование повторяют один или несколько раз. Выход очищенного 2f=80% от 1f.
Очистка соединения 2f хроматографией на силикагеле (исходя из ~6,5 кг сырого соединения 2f): “концентрация” сырого соединения 2f вычислена корректированием массы сырого продукта по ВЭЖХ чистоте и летучим компонентам. Для данной стадии очистки 5,75 кг продукта (исправленного по концентрации) используют на ввод на 50 см хроматографическую колонку. 50 см хроматографическую колонку наполняют взвесью гептан/силикагель (51,8 кг силикагеля). Сырой продукт загружают в колонку в виде раствора в смеси дихлорметан/2,6-лутидин (15 кг дихлорметана, 0,16 кг 2,6-лутидина). Продукт элюируют при двухступенчатом градиенте 4-метил-2-пентанон (MIBK)/2,6-лутидин (первая ступень представляет собой 827 л 39:61 MIBK:гептан (масс.:масс.) с 0,06% 2,6-лутидина (масс.:масс.); вторая ступень представляет собой 1343 л 73:27 MIBK:гептан (масс.:масс.) с 0,06% 2,6-лутидина (масс.:масс.)). Апробированную совокупность фракций концентрировали тонкопленочным упариванием до концентрации в 150 г/л. Данную концентрированную массу осаждали 6 объемами гептана. Очищенное соединение 2f выделяли фильтрованием. Выход очищенного 2f=50% от 1f; 65% от сырого 2f.
Пример 2
Приготовление раствора CYTFA пиридиниевой соли для снятия тритильной группы
К раствору 4-цианопиридина (10,1 г; 1,055 экв.) в дихлорметане (790 мл) добавляют трифторуксусную кислоту (10,5 г; 1,0 экв.), затем 2,2,2-трифторэтанол (198 мл) и этанол (10 мл), и раствор перемешивают в течение 10-30 мин.
Пример 3
Приготовление дисульфидного анкера (см. фиг.10)
Приготовление сукцинатной соли N-тритилпиперазина (NTP): К охлаждаемому раствору пиперазина (10 экв.) в толуоле/метаноле (5:1 толуол/метанол (об.:об.); 5 мл/г пиперазина) медленно добавляли раствор трифенилметил(тритил)хлорида (1,0 экв.) в толуоле (5 мл/г тритилхлорида). После завершения реакции (1-2 ч) данный раствор промывали четыре раза водой. К полученному органическому раствору добавляли водный раствор янтарной кислоты (01,1 экв.; 13 мл воды/г янтарной кислоты). Данную смесь перемешивали в течение 90 мин и твердый продукт собирали фильтрованием. Сырой NTP очищали двойным ресуспендированием в ацетоне. Выход=70%.
Приготовление симметричного дисульфида 7: 1,1’-карбонилдиимидазол (CDI) (12,402 г; 2,2 экв.) суспендировали в дихлорметане (5,25 мл/г) и охлаждали на водяной бане. Гидроксиэтилдисульфид 6 (5,36 г; 1 экв.) растворяли в дихлорметане (10 мл/г) и тетрагидрофуране (1 мл/г). Раствор диола медленно добавляли к CDI, так что температура смеси оставалась ниже 4°С в течение реакции. После завершения реакции (как только заканчивалось добавление) вносили деионизированную воду, чтобы гасить реакцию. Независимым образом сукцинатную соль N-тритилпиперазина (NTP) (32,59 г; 2,1 экв.) растворяли в толуоле (8 мл/г NTP), дихлорметане (2 мл/г NTP) и метаноле (2 мл/г NTP). К2СО3 (22,09 г; 4,6 экв.) растворяли в деионизированной воде (10 мл/г). Раствор К2СО3 добавляли к раствору NTP; смесь перемешивали и затем разделяли на два слоя. Мутный органический слой перегоняли для удаления 90 граммов; образовавшиеся капельки воды отделяли и к органическому слою добавляли ацетон (8 мл/г NTP). Раствор дисульфидного диола, активированного c помощью CDI, добавляли к раствору свободного основания и концентрировали до 225 мл. Добавляли ацетон (10 мл/г NTP) и смесь концентрировали до 225 мл. Смесь нагревали при кипении с обратным холодильником и твердое вещество начинало выкристаллизовываться из раствора. После завершения реакционную смесь охлаждали и твердое вещество (7) выделяли фильтрованием. Выход: 27,92 г; 93,1% (в расчете на анализ массы).
Приготовление дисульфидного спирта 8 : Соединение 7 (36,00г; 32,1 ммоль; 1 экв.) суспендировали в ацетоне (2,8 мл/г 7). Добавляли гидроксиэтилдисульфид (78,51 мл; 20 экв.), затем ацетон (1,7 мл/г 7). Добавляли 5% NaOH/метанол (2,85 мл; 0,1 экв.); рН смеси составлял 10 по рН бумажке. Добавляли трифенилфосфин (8,42 г; 1 экв.), затем ацетон (1,1 мл/г 7). Все твердые вещества переходили в раствор и затем продукт начинал кристаллизоваться. Через шестнадцать часов реакционную смесь нейтрализовали уксусной кислотой (2,4 г; 0,2 экв.). Сырой продукт выделяли фильтрованием. Сырое твердое вещество подвергали двум ресуспендированиям в ацетоне при кипячении с обратным холодильником (5 мл/г 7).
После фильтрования сырой продукт суспендировали в дихлорметане (7,25 мл/г 7). Смесь нагревали до образования прозрачного раствора (35°С). Раствор экстрагировали пять раз равным объемом деионизированной воды и конечный органический раствор концентрировали до 155 мл. Добавляли дихлорметан (4,3 мл/г 7) и раствор снова концентрировали до 155 мл. Добавляли CDI (9,17 г; 1,1 экв.) и смесь перемешивали при комнатной температуре. После завершения реакции (~20 мин) реакционную смесь дважды промывали равным объемом деионизированной воды, затем этилбензолом (2,1 мл/г 7). Раствор концентрировали до 65,2 г, при этом дихлорметан в растворе снижается до 0,17%, и перемешивали на ледяной бане для кристаллизации продукта. Продукт 9 выделяли фильтрованием. Выход: 44%.
Пример 4
Триэтиленгликолевый хвост (см. фиг.11)
Приготовление тритилпиперазинфенилкарбамата 10 : К охлаждаемой суспензии NTP в дихлорметане (6 мл/г NTP) добавляли раствор карбоната калия (3,2 экв.) в воде (4 мл/г карбоната калия). К данной двухфазной смеси медленно добавляли раствор фенилхлорформиата (1,03 экв.) в дихлорметане (2 г/г фенилхлорформиата). Реакционную смесь нагревали до 20°С. После завершения реакции (1-2 ч) слои разделяли. Органический слой промывали водой и сушили над безводным карбонатом калия. Продукт 10 выделяли кристаллизацией из ацетонитрила. Выход=80%.
Приготовление карбаматного спирта 11: Гидрид натрия (1,2 экв.) суспендировали в 1-метил-2-пирролидиноне (32 мл/г гидрида натрия). К данной суспензии добавляли триэтиленгликоль (10,0 экв.) и соединение 10 (1,0 экв.). Полученную взвесь нагревали до 95°С. После завершения реакции (1-2 ч) смесь охлаждали до 20°С. К данной смеси добавляли 30% дихлорметан/метил-трет-бутиловый эфир (об./об.) и воду. Органический слой, содержащий продукт, промывали последовательно водным NaOH, водной янтарной кислотой и насыщенным водным хлоридом натрия. Продукт 11 выделяли кристаллизацией из смеси дихлорметан/метил-трет-бутиловый эфир/гептан. Выход=90%.
Приготовление хвостовой кислоты 12: К раствору соединения 11 в тетрагидрофуране (7 мл/г 11) добавляли янтарный ангидрид (2 экв.) и DMAP (0,5 экв.). Смесь нагревали до 50°С. После завершения реакции (5 ч) смесь охлаждали до 20°С и устанавливали рН 8,5 водным NaHCO3. Добавляли метил-трет-бутиловый эфир и продукт экстрагировали в водный слой. Добавляли дихлорметан и смесь устанавливали при рН 3 водной лимонной кислотой. Органический слой, содержащий продукт, промывали смесью с рН=3 из цитратного буфера и насыщенного водного хлорида натрия. DCM- раствор соединения 12 использовали без выделения в приготовлении соединения 13.
Приготовление соединения 13: К раствору соединения 12 добавляли имид N-гидрокси-5-норборнен-2,3-дикарбоновой кислоты (HONB) (1,02 экв.), 4-диметиламинопиридин (DMAP) (0,34 экв.) и затем гидрохлорид 1-(3-диметиламинопропил)-N’-этилкарбодиимида (EDC) (1,1 экв.). Смесь нагревали до 55°С. После завершения реакции (4-5 ч) смесь охлаждали до 20°С и промывали последовательно смесью 1:1 0,2 М лимонная кислота/насыщенный солевой раствор и насыщенным солевым раствором. Дихлорметановый раствор претерпевал обмен растворителя на ацетон и затем на N,N-диметилформамид, и продукт выделяли осаждением из ацетон/N,N-диметилформамида в насыщенный водный хлорид натрия. Сырой продукт ресуспендировали несколько раз в воде для удаления остаточного N,N-диметилформамида и солей. Выход=70% соединения 13 от соединения 11. Введение активированного “хвоста” на дисульфидный анкерный полимер проводили в NMP методикой, использованной для включения звеньев в течение твердофазного синтеза.
Пример 5
Приготовление твердого носителя для синтеза морфолиновых олигомеров
Пример 5а: Приготовление аминометилполистирол-дисульфидного полимера
Данную методику выполняли в силанированном, снабженном рубашкой пептидном сосуде (закупка сделана фирмой ChemGlass, NJ USA) со стеклянной фриттой грубой пористости (40-60 мкм), верхней мешалкой и 3-ходовым тефлоновым запорным краном, чтобы позволить N2 барботировать через фритту или экстракцию в вакууме. Контроль температуры достигался в реакционном сосуде баней с циркулирующей водой.
Обработка полимера/стадии промывки в последующей методике состоит из двух основных операций: флюидизации полимера и экстракции растворитель/раствор. Для флюидизации полимера запорный кран располагали так, чтобы создавать возможность для потока N2 через фритту, и определенную обработку/промывку полимера добавляли в реактор и создавали возможность для проницаемости и полного смачивания полимера. Затем начинали смачивание и полимерную смесь перемешивали в течение определенного периода времени. Для экстракции растворитель/раствор смешивали и поток N2 останавливали и запускали вакуумный насос, и затем запорный кран устанавливали для создания возможности по удалению отходов обработки/промывке полимера. Все объемы по обработке/промывке полимера составляли 15 мл/г полимера, если не оговорено специально.
К полимеру аминометилполистирола (100-200 меш; ~1,0 ммоль/г N2 замещение; 75 г 1 экв., Polymer Labs, UK, part # 1464-X799) в силанированный, снабженный рубашкой пептидный сосуд добавляли 1-метил-2-пирролидинон (NMP; 20 мл/г полимера), и полимеру позволяли набухать с перемешиванием в течение 1-2 ч. После отделения растворителя для набухания полимер промывали дихлорметаном (2×1-2 мин), 5% диизопропилэтиламином в 25% изопропаноле/дихлорметане (2×3-4 мин) и дихлорметане (2×1-2 мин). После отделения окончательной промывки полимер флюидизировали раствором дисульфидного анкера 9 в 1-метил-2-пирролидиноне (0,17 М; 15 мл/г полимера, ~2,5 экв.), и смесь полимера/реагента нагревали при 45°С в течение 60 ч. По завершении реакции нагревание прекращали и раствор анкера удаляли и полимер промывали 1-метил-2-пирролидоном (4×3-4 мин) и дихлорметаном (6×1-2 мин). Данный полимер обрабатывали раствором 10% (об./об.) диэтилкарбоната в дихлорметане (16 мл; 2×5-6 мин) и затем промывали дихлорметаном (6×1-2 мин.). Полимер 14 сушили в потоке N2 в течение 1-3 ч и затем в вакууме до постоянной массы (±2%). Выход: 110-150% первоначальной массы полимера.
Пример 5b: Определение нагрузки аминометилполистирол-дисульфидного полимера
Нагрузку полимера (число потенциально доступных реакционно-способных сайтов) определяют спектрометрическим анализом по числу трифенилметильных(тритильных) групп на грамм полимера.
Известную массу высушенного полимера (25±3 мг) переносят в силанированную 25 мл волюметрическую колбу и добавляют ~5 мл 2% (об./об.) трифторуксусной кислоты в дихлорметане. Содержимое колбы перемешивают интенсивным вращением и затем позволяют стоять в течение 30 мин. Данный объем доводят до 25 мл дополнительной 2% (об/об.) трифторуксусной кислотой в дихлорметане и содержимое тщательно перемешивают. Используя пипетку для положительного вытеснения, аликвоту тритилсодержащего раствора (500 мкл) переносят в 10 мл волюметрическую колбу и объем доводят до 10 мл метансульфоновой кислотой.
Содержание тритильного катиона в конечном растворе измеряют УФ-поглощением при 431,7 нм, и нагрузку полимера вычисляют по тритильным группам на грамм полимера (мкмоль/г), используя соответствующие объемы, разбавления, коэффициент экстинкции (ε: 41 мкмоль-1см-1) и массу полимера. Оценку проводят в тройной повторности и вычисляют среднюю нагрузку.
Методика для определения нагрузки полимера в данном примере предоставит полимер с нагрузкой приблизительно 500 мкмоль/г. Нагрузку в 300-400 мкмоль/г получали, если стадию включения дисульфидного анкера проводят в течение 24 ч при комнатной температуре.
Пример 5с: Хвостовая нагрузка (см. фиг.12)
Используя ту же самую схему и объемы, как для приготовления аминометилполистирол-дисульфидного полимера, можно ввести хвост в молекулу. Для стадии конденсации использовали раствор соединения 13 (0,2М) в NMP, содержащий 4-этилморфолин (NEM, 0,4 М) вместо раствора дисульфидного анкера. Спустя 2 ч при 45°С полимер 15 промывали дважды 5% диизопропилэтиламином в 25% изопропаноле/дихлорметане и один раз с помощью DCM. К данному полимеру добавляли раствор бензойного ангидрида (0,4М) и NEM (0,4M). Через 25 мин реактор, снабженный рубашкой, охлаждали до комнатной температуры, и полимер промывали дважды 5% диизопропилэтиламином в 25% изопропаноле/дихлорметане и восемь раз с помощью DCM. Полимер 15 фильтровали и сушили в высоком вакууме. Нагрузку для полимера 15 определяют как нагрузку начального аминометилполистирол-дисульфидного полимера 14, использованного в хвостовой нагрузке.
Пример 6
Синтез морфолиновых олигомеров
Пример 6а: Твердофазный синтез
Защищенные олигомеры готовили вручную твердофазным синтезом олигомера на аминометилполистирол-дисульфидном полимере (~500 мкмоль/г нагрузки) в 10 г количестве (исходная масса полимера). Использованные растворы представляли собой следующее:
Растворы для снятия тритильной группы (детритилирования): САА=11% цианоуксусная кислота (масс./масс.) в смеси 20% ацетонитрил/DCM (об./об.);
СРМ=2% метансульфонат 3-хлорпиридиния (масс./об.) и 0,9 этанол (об./об.) в 20% трифторэтаноле/DCM (об./об.);
CYTFA=2% трифторацетат 3-цианопиридиния (масс./об.) и 0,9 этанол (об./об.) в 20% трифторэтаноле/DCM (об./об.);
Раствор для нейтрализации: 5% диизопропилэтиламин в 25% изопропаноле/дихлорметане;
Растворы для связывания: 0,165М (для 2f (DPG), 2c и 2d или других Т звеньев) или 0,18M (для 2а и 2b или других А/С звеньев) активированного морфолинового звена и 0,4 М N-этилморфолин в 1,3-диметилимидазолидиноне (DMI).
Активированную MPG (2c) готовили по Summerton et al. (1993).
После переноса полимера в реактор для синтеза и перед началом синтетических циклов 1-метил-2-пирролидинон (NMP, 20 мл/г полимера) добавляли и выдерживали при стоянии в течение 1-2 ч. После 2 промывок дихлорметаном (10 мл/г полимера) использовали следующие циклы синтеза с добавлением соответствующего конденсирующего раствора активированного морфолинового звена желаемого основания и желаемого типа мостика в каждом цикле с получением специфической последовательности.
** Объемы для связывания достаточны для поддерживания хорошего смешивания и увеличены с учетом набухания полимера.
После включения конечного звена конечный цикл (метокситритилирование) выполняли с 0,32М 4-метокситрифенилметилхлоридом и 0,4М N-этилморфолином в DMI. После метокситритилирования полимер промывали 8 раз с помощью NMP и затем обрабатывали раствором для отщепления, состоящим из 0,1М 1,4-дитиотрейтола (DTT) и 0,73М триэтиламина в NMP (27 мл/г исходного полимера в течение 30 мин). После сбора раствора защищенного олигомера полимер (значительно сниженный в объеме) промывали двумя дополнительными порциями раствора для отщепления (13 мл/г исходного полимера в течение каждых 15 мин) и промывки объединяли с основным раствором. К раствору защищенного олигомера в сосуде с соответствующей величиной давления с тефлоновой пробкой (Ace Glass, NJ, USA) добавляли концентрированный водный аммиак (106 мл/г исходного полимера, предварительно охлажденного до -20°С), сосуд герметизировали и содержимое перемешивали кручением. Сосуд помещали в печь при 45°С на 16-20 ч, чтобы удалить защитные группы на основании и каркасе.
После аммонолиза раствор сырого олигомера охлаждают до комнатной температуры и затем подвергают диафильтрации на фоне 0,28% водного аммиака с использованием PLBC 3kd Regenerated Cellulose мембраны (Millipore), чтобы удалить растворители и небольшие молекулы перед ионообменной хроматографией.
Пример 6b: Очистка морфолиновых олигомеров анионообменной хроматографией
Раствор сырого олигомера, полученный от диафильтрации, устанавливают при рН 11-11,5 и загружают в колонку ToyoPearl Super-Q 650S c анионообменной смолой (Tosoh Bioscience). Метокситритилированный олигомер элюируют при градиенте 5-35 В в пределах 17 объемов колонки (буфер А: 10 мМ гидроксид натрия; буфер В: 1М хлорид натрия в 10 мМ гидроксиде натрия) и накапливают фракции приемлемой чистоты (анионообменная ВЭЖХ и масс-спектроскопия).
Пример 6с: Деметокситритилирование морфолиновых олигомеров
К собранным фракциям от анионообменной хроматографии добавляют ацетонитрил (10% об.), затем 2М Н3РО4 для установления рН 3. Данный раствор перемешивают в течение 45 мин и затем нейтрализуют концентрированным водным аммиаком до рН 7. Раствор олигомера подвергают диафильтрации на фоне 20 мМ ацетата натрия, используя PLBC 3kd Regenerated Cellulose мембрану (Millipore), чтобы обменять буферы перед катионообменной хроматографией.
Пример 6d: Очистка морфолиновых олигомеров катионообменной хроматографией
Раствор олигомера устанавливают при рН 4,5 уксусной кислотой и загружают в колонку c Source 30S катионообменной смолой (GE Healthcare). Олигомер элюируют при градиенте 0-35% в пределах 17 объемов колонки (буфер А: 20 мМ ацетат натрия, 25% ацетонитрил, рН 4,5; буфер В: 0,5М хлорид натрия, 20 мМ ацетат натрия, 25% ацетонитрил, рН 4,5) и накапливают фракции приемлемой чистоты (катионообменная ВЭЖХ и масс-спектроскопия).
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИРАЛЬНЫЕ РЕАГЕНТЫ ДЛЯ ПОЛУЧЕНИЯ ГОМОГЕННЫХ ОЛИГОМЕРОВ | 2016 |
|
RU2791532C2 |
| СПОСОБ СОЕДИНЕНИЯ НУКЛЕОЗИДОВ 3'-5'-МЕЖНУКЛЕОТИДНЫМ СИЛИЛЬНЫМ ЗВЕНОМ | 1991 |
|
RU2079508C1 |
| МОДИФИЦИРОВАННЫЕ ОЛИГОНУКЛЕОТИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2708237C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТИОФОСФАТНЫХ ОЛИГОНУКЛЕОТИДОВ ДЛЯ ТЕРАПЕВТИЧЕСКОГО ПРИМЕНЕНИЯ | 2024 |
|
RU2836132C1 |
| СПОСОБ СИНТЕЗА В РАСТВОРЕ МАКРОМОЛЕКУЛ ИЗ ЗВЕНЬЕВ ПРОИЗВОДНЫХ УГЛЕВОДОВ | 2021 |
|
RU2819631C1 |
| АНТИСМЫСЛОВЫЕ НУКЛЕИНОВЫЕ КИСЛОТЫ | 2015 |
|
RU2702424C2 |
| КАТИОННЫЕ ОЛИГОНУКЛЕОТИДЫ, АВТОМАТИЗИРОВАННЫЕ СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2006 |
|
RU2451022C2 |
| РЕЗИСТЕНТНЫЕ К НУКЛЕАЗЕ ОЛИГОНУКЛЕОЗИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И РЕЗИСТЕНТНЫЙ К НУКЛЕАЗЕ НУКЛЕОЗИДНЫЙ ДИМЕР | 1991 |
|
RU2131436C1 |
| СИНТЕЗ ФОСФИТИЛИРОВАННЫХ СОЕДИНЕНИЙ С ИСПОЛЬЗОВАНИЕМ ЧЕТВЕРТИЧНОГО ГЕТЕРОЦИКЛИЧЕСКОГО АКТИВАТОРА | 2005 |
|
RU2440364C2 |
| АНТИСМЫСЛОВЫЕ НУКЛЕИНОВЫЕ КИСЛОТЫ | 2015 |
|
RU2730681C2 |

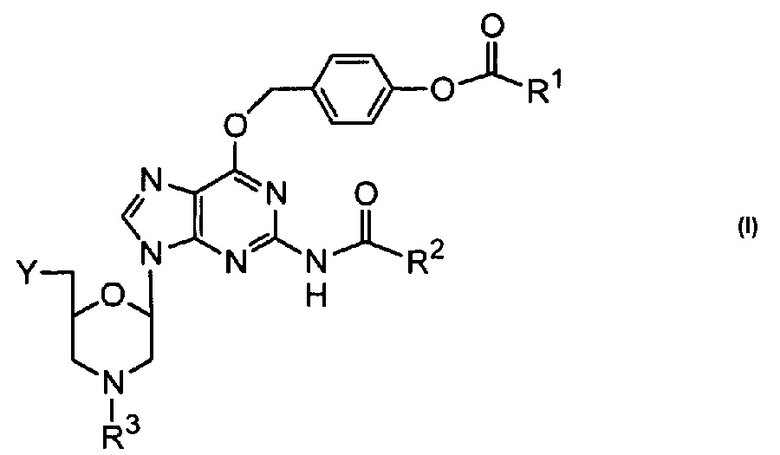
Изобретение относится к применимым в химической промышленности способу снятия защиты с азота морфолинового кольца в морфолиновом олигомере и соединениям формулы (I):
 ,
,
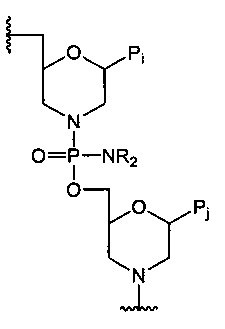
где R1 представляет собой C1-С6 алкил, ди(С1-С6 алкил)амино или фенил; R2 представляет собой C1-С6 алкил или бензил; R3 представляет собой водород, тритил (трифенилметил), 4-метокситритил, 4-метилтритил, 4,4'-диметилтритил и 4,4',4ʺ-триметилтритил; Y представляет собой необязательно защищенный триалкилсилилом гидроксил или группу 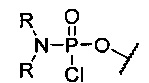 , R представляет собой C1-С6 алкил. В предложенном способе снимают защиту с азота морфолинового олигомера, который состоит из морфолиновых звеньев, связанных фосфородиамидатным мостиком, формулы:
, R представляет собой C1-С6 алкил. В предложенном способе снимают защиту с азота морфолинового олигомера, который состоит из морфолиновых звеньев, связанных фосфородиамидатным мостиком, формулы:
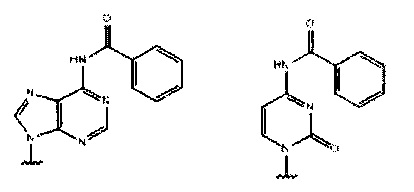
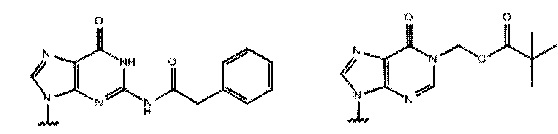
 , где Pi и Pj выбраны из:
, где Pi и Pj выбраны из:
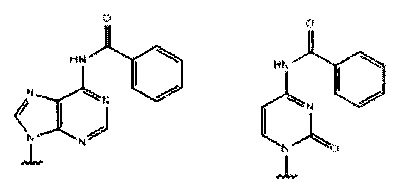
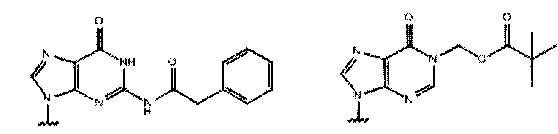 или
или 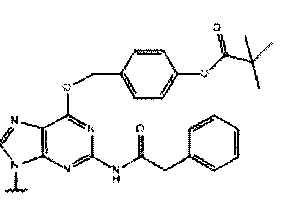 ,
,
R представляет собой C1-С6 алкил, защитная группа выбрана из 4-метокситритила, 4-метилтритила, 4,4'-диметилтритила или 4,4',4ʺ-триметилтритила, при этом способ включает воздействие на защищенный кольцевой атом азота раствора реагента, содержащего соль гетероциклического амина, выбранного из пиридина, тиазола, пиридазина, пиразола или триазола, замещенного электронно-акцепторной группой, выбранной из галогена, циано, альдегидной, кето, карбоксисложноэфирной или карбоксамидной групп, с сульфоновой кислотой, выбранной из алкилсульфоната, (фторалкил)сульфоната или п-толуолсульфоната, трифторуксусной кислотой или хлористоводородной кислотой, с рКа в интервале 1-4 в протонированной форме, в растворителе, содержащем дихлорметан и трифторэтанол. Предложены новые соединения, эффективные в синтезе морфолиновых олигомеров, и улучшенный метод снятия защитных групп с кольцевых атомов азота в морфолиновых олигомерах. 2 н. и 12 з.п. ф-лы, 6 пр., 2 табл., 12 ил.
1. Морфолиновое соединение, имеющее следующую структуру (I):
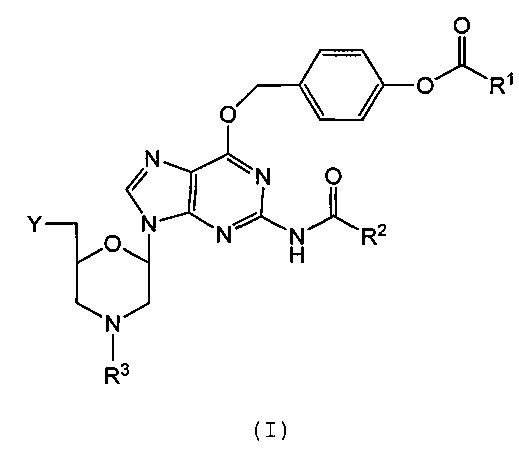 ,
,
в которой
R1 представляет собой C1-С6 алкил, ди(С1-С6 алкил)амино или фенил;
R2 представляет собой C1-С6 алкил или бензил;
R3 представляет собой водород, тритил (трифенилметил), 4-метокситритил, 4-метилтритил, 4,4'-диметилтритил и 4,4',4ʺ-триметилтритил; и
Y представляет собой защищенную триалкилсилилом гидроксильную группу или незащищенную гидроксильную группу, или хлорфосфорамидатную группу структуры:
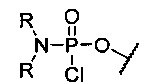
при этом каждый R независимо представляет собой C1-С6 алкил.
2. Соединение по п. 1, в котором Y представляет собой защищенную триалкилсилилом гидроксильную группу или незащищенную гидроксильную группу, при этом каждый алкил содержит от 1 до 6 атомов углерода.
3. Соединение по п. 1, в котором Y представляет собой хлорфосфорамидатную группу
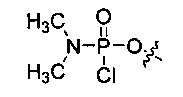
4. Соединение по п. 1, в котором R3 представляет собой тритил, 4-метокситритил, 4-метилтритил, 4,4'-диметилтритил и 4,4',4ʺ-триметилтритил.
5. Соединение по п. 1, в котором R1 представляет собой C1-С6 алкил.
6. Соединение по п. 5, в котором R1 представляет собой C(CH3)3.
7. Соединение по п. 1, в котором R2 представляет собой бензил или -CH(CH3)2.
8. Соединение по п. 1, при этом соединение имеет следующую структуру:
 ,
,
в которой Y представляет собой хлорфосфорамидатную группу, имеющую следующую структуру:
9. Способ снятия защиты с азота морфолинового кольца, защищенного триарилметильной группой, в морфолиновом олигомере с образованием незащищенного азота морфолинового кольца, при этом морфолиновый олигомер включает по меньшей мере два морфолиновых звена, связанных фосфородиамидатным мостиком, формулы:
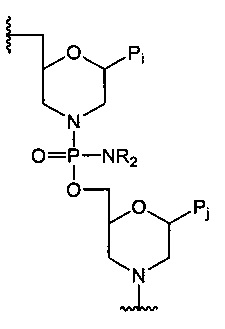 ,
,
где каждый Pi и Pj независимо представляет собой пуриновое или пиримидиновое спариваемое основание, выбранное из аденина (А), цитозина (С), тимина (Т), инозина (I) и гуанина (G):
 или
или 
каждый R независимо представляет собой C1-С6 алкил, и
триарилметильная защитная группа выбрана из 4-метокситритила, 4-метилтритила, 4,4'-диметилтритила или 4,4',4ʺ-триметилтритила,
при этом способ включает воздействие на кольцевой атом азота, защищенного триарилметильной группой, раствора реагента, содержащего гетероциклическую аминную соль, в растворителе, содержащем трифторэтанол, при этом указанная соль является солью гетероциклического амина, выбранного из пиридина, тиазола, пиридазина, пиразола или триазола, замещенного электронно-акцепторной группой, выбранной из галогена, циано, альдегидной, кето, карбоксисложноэфирной или карбоксамидной групп, с сульфоновой кислотой, выбранной из алкилсульфоната,
(фторалкил)сульфоната или п-толуолсульфоната, трифторуксусной кислотой или хлористоводородной кислотой, с рКа в интервале 1-4 в протонированной форме, при этом растворитель, содержащий трифторэтанол, содержит дихлорметан и трифторэнатол.
10. Способ по п. 9, в котором гетероциклический амин представляет собой пиридин.
11. Способ по п. 9, в котором гетероциклический амин представляет собой пиридин, замещенный хлором или циано.
12. Способ по п. 9, в котором соль выбрана из метансульфоната 3-хлорпиридиния (СРМ) или трифторацетата 4-цианопиридиния (CYTFA).
13. Способ по п. 9, в котором растворитель содержит дихлорметан и трифторэтанол в объемном соотношении в диапазоне от 90:10 до 25:75.
14. Способ по п. 13, в котором указанное объемное соотношение равно 80:20.
| НУКЛЕИНОВЫЕ КИСЛОТЫ, МОДИФИЦИРОВАННЫЕ АМИНОКИСЛОТАМИ | 1995 |
|
RU2154638C2 |
| US 5185444 A, 09.02.1993 | |||
| US 2007135333 A1, 14.06.2007 | |||
| James Summerton et al, Antisense&nucleic acid development, 1997, 7, 187-195 | |||
| Tatiana V | |||
| Abramova et al, Bioorganic chemistry, 2007, 35, 258-275 | |||
| Mariano J | |||
| L | |||
| et al, Tetrahedron Letters, 1997, 38, 23, 3995-3998 | |||
| Tatiana V | |||
| Abramova et al, Tetrahedron Letters, 2004, 45, 4361-4364. | |||

