Техническая область изобретения
Настоящее изобретение относится к области синтеза органических макромолекул. Более конкретно, настоящее изобретение относится к способу получения представляющих интерес макромолекул из моносахаридных или олигосахаридных звеньев или производных таких звеньев. Этот способ осуществляется в жидкой фазе (растворе) и в нем используются якорные молекулы, растворимые в органической среде и, в частности, в неполярных растворителях. Эти якорные молекулы (или жидкая подложка) придают интересующим макромолекулам (или промежуточным продуктам), когда они связаны с ними, следующее: временную защиту по меньшей мере одной химической функциональной группы и превосходную растворимость в неполярных органических растворителях и, следовательно, очистку путем простой экстракции или фильтрации на силикагеле. Макромолекулы-мишени могут, в частности, представлять собой макромолекулы, представляющие биологический интерес, такие как олигонуклеотиды и олигосахариды, и они могут содержать звенья, которые не являются моно- или олигосахаридами.
Уровень техники
Настоящее изобретение относится к новому способу синтеза макромолекул, содержащих звенья моносахаридов, и/или олигосахаридов, и/или их производных; эти звенья могут быть или могут содержать, в частности, пентозы или гексозы. Таким образом, и более конкретно, настоящее изобретение относится к синтезу олигонуклеотидов (или их производных) и синтезу олигосахаридов (или их производных).
Обычно олигонуклеотиды получают в лабораторных масштабах путем синтеза на твердой подложке с помощью автоматов. Многочисленные усилия были предприняты для улучшения этого оборудования на промышленной стадии для более крупных производств (>1 кг). Однако с конца прошлого века число терапевтических олигонуклеотидов значительно расширилось, вызвав настоящий интерес к этим молекулярным видам. На сегодняшний день более 100 олигонуклеотидов проходят клинические испытания, и восемь препаратов были одобрены Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) (см. Y.S. Sanghvi, «А Status Update for Modified Oligonucleotides for Chemotherapeutics Applications», Curr. Protoc. Nucleic Acid Chem. 2011, 46:4.1.1-4.1.22.).
Первым одобренным олигонуклеотидом был фомивирсен или Vitravene™ (см. М.D. de Smet et al, «Fomovirsen - a phosphorothioate oligonucleotide for the treatment of CMV retinitis»r Ocul. Immunol. Inflamm. 1999, 7, 189-198, и S.T. Cooke et al, «RNA-Targeted Therapeutics», Cell Metabolism, 2018, 27, 714-739), олигодезоксинуклеотид, состоящий из 21 звена, связанных фосфоротиоатными связями. Применяется при лечении цитомегаловирусного ретинита.
Позже в 2004 и 2013 годах были последовательно одобрены Macugen™ (пегаптаниб натрия) и Kynamro™ (мипомерсен натрия), показанные для лечения неоваскулярной (влажной) формы возрастной дегенерации желтого пятна (AMD) и гомозиготной семейной гиперхолестеринемии, соответственно (см. Sanghvi and Schulte, Curr. Opin. Drug Discovery Dev. 2004, 6, 765).
До сих пор твердофазный синтез олигонуклеотидов обычно производил олигонуклеотиды в килограммовом масштабе с хорошими качествами (см. Sanghvi, Y.S. (2019) «Large-scale automated synthesis of therapeutic oligonucleotides: A status update», published in S. Agrwal & M.J. Gait (Eds.) Advances in nucleic acid therapeutic (pp. 453-473)). Несмотря на то, что было предпринято много усилий по оптимизации, еще есть возможности для улучшения методов получения олигонуклеотидов. Синтез олигонуклеотидов в жидкой фазе все чаще позиционируется как эффективный ответ на производство олигонуклеотидов.
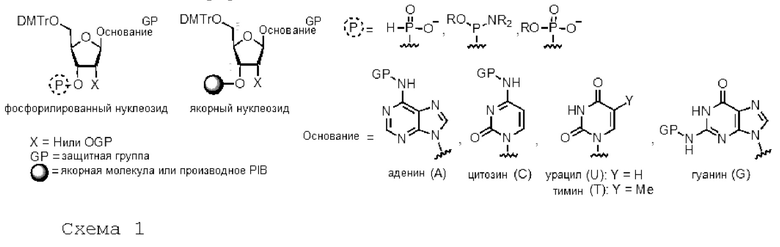
Независимо от того, в какой фазе (твердая (гетерогенная) или жидкая (раствор)), в которой протекает реакция, синтез олигонуклеотидов сводится к образованию фосфорно-эфирных связей между нуклеозидами в определенном порядке (см. Н. Lonneberg, «Synthesis of oligonucleotides on a soluble support», Beilstein J. Org. Chem., 2017, 13, 1368-1387, и A. Molina & Y.S. Sanghvi, «Liquid-Phase Oligonucleotide Synthesis: Past, Present, and Future Predictions», Current Protocols in Nucleic Acid Chemistry, 2019, 77 (1)). Часто жидкофазный синтез олигонуклеотидов (дезоксирибонуклеиновой кислоты (ДНК) или рибонуклеиновой кислоты (РНК)) начинается с присоединения якорной молекулы к 3'-концу нуклеозида; удлинение происходит от 3' до 5'. На этой стадии начинается первый цикл синтеза производного нуклеозида, фосфорилированного в 3'-положении (фосфорамидит, или Н-фосфинат, или фосфотриэфир), который будет взаимодействовать со спиртом в 5'-положении нуклеозида, заякоренного на носителе синтеза.
За исключением реагирующего спирта, все остальные нуклеофильные функциональные группы должны быть заранее защищены. Так, амины нуклеиновых оснований предпочтительно защищены ацилированием (бензоил для цитозина и аденина, изобутил для гуанина), тогда как спиртовые функциональные группы дезоксирибозы и рибозы соответственно маскируются тритиловыми (диметокситритиловыми или монометокситритиловыми) эфирами в 5'-положении и/или силиловыми (трет-бутилдиметилсилиловыми или триизопропилсилилоксиметиловыми) эфирами в положении 2'.
В зависимости от природы выбранного фосфорилированного нуклеозида рабочие условия реакции сочетания различны. В случае нуклеозидных фосфорамидитов (см. S.L. Beaucage & М.Н. Caruthers, «Deoxynucleoside Phosphoramidites - A new Class of Key Intermediates for Deoxypolynucleotide Synthesis», Tetrahedron Letters, 1981, 22, 1859-1862) и нуклеозидных H-фосфонатов за реакциями сочетания следует реакция окисления с получением соответствующих фосфотриэфиров. В случае нуклеозидфосфатов соответствующие фосфотриэфиры получают путем этерификации с предшествующим нуклеозидом (см. С.В. Reese & Z. Pei-Zhuo, «Phosphotriester Approach to the Synthesis of Oligonucleotides: A Reappraisal», J. Chem. Soc, Perkin Trans. 1, 1993, 2291-2301, V.A. Efimov et al, Application of new catalytic phosphate protecting groups for the highly efficient phosphotriester oligonucleotide synthesis», Nucleic Acids Res. 1986, 14, 6525-6540, G. van der Marel et al, «A New Approach to the Synthesis of Phosphotriester Intermediates of Nucleosides and Nucleic Acids,» Tetrahedron Lett. 1981, 22, 3887-3890, и E. de Vroom et al, «Use of a 1-hydroxybenzotriazole activated phosphorylating reagent towards the synthesis of short RNA fragments in solution», Nucleic Acids Res. 1986, 14, 5885-5900).
Различные якорные молекулы были описаны в литературе в связи с синтезом олигонуклеотидов в жидкой фазе. Исторически основа для синтеза олигонуклеотидов была заложена в работах Hayatsu и Khorana (см. «Deoxyribooligonucleotide Synythesis on a Polymer Support», J. Am. Chem. Soc. 1966, 88, 13, 3182-3183). Этот подход, вдохновленный методологией синтеза пептидов, описанной Меррифилдом, использовал растворимый полистирол в качестве привитой якорной молекулы (5'-О-монометокситритил (MMTr)). Однако этот полистирол становится нерастворимым в органических растворителях за пределами трех нуклеотидов, и в результате выделение продукта становится плохим.
Были изучены и другие полимеры, такие как целлюлоза (Biochemistry 1968, 7, 8, 2809-2813) или поливиниловый спирт (Н. Schott et al., «Liquid-Phase-Synthese von Oligothymidylphosphaten», Die Makromolekulare Chemie, 1973, 173, 247-251). В обоих случаях при включении первого нуклеозида в носитель необходима так называемая реакция копирования свободных (нефункционализированных) гидроксильных групп во избежание появления укороченных олигонуклеотидов в конце синтеза.
В 1990-х годах в качестве якорной молекулы широко изучался другой класс полимеров: полиэтиленгликоль (или ПЭГ). Используя этот тип растворимых носителей, группа Боноры достигла синтеза олигонуклеотидов в масштабе грамма при значительном сокращении времени обработки (см. «HELP (High Efficiency Liquid Phase) new oligonucleotide synthesis on soluble polymeric support», Nucleic Acids Research, 1990, 18, 3155-3159, и «Large scale, liquid phase synthesis of oligonucleotides by the phosphoramidite approach», Nucleic Acids Research, 1993, 21, 1213-1217).
Совсем недавно группа Ливингстона разработала и использовала мембраны для молекулярного разделения с помощью нанофильтрации (см. P. R. J. Gaffney et al, «Liquid-Phase Synthesis of 2'-Methyl-РИА on a Homos tar Support through Organic-Solvent Nanofiltration», Chem. Eur. J., 2015, 21, 9535-9543, и J.F. Kim et al, «Organic Solvent Nanofimltration (OSN): A New Technology Platform for Liquid-Phase Oligonucleotide Synthesis (LPOS)», Org. Proc. Res. Dev., 2016, 20, 1439-1452). Они облегчают и ускоряют стадии очистки растущих олигонуклеотидов.
В общем, использование полиэтиленгликолей (ПЭГ) в качестве якорной матрицы имеет несколько преимуществ, таких как: использование ацетонитрила в качестве растворителя; получение олигонуклеотидов, имеющих хорошую чистоту; упрощенные очистки (на каждой стадии); возможность получения разумных количеств олигонуклеотидов; универсальность на уровне реакций связывания (фосфорамидиты, Н-фосфонат (см. K.J. Padiya et al, «Large Scale, Liquid Phase Oligonucleotide Synthesis by Alkyl H-phosphonate Approach», Bioorg. Med. Chem., 2000, 8, 337-342) и фосфаты) и уменьшение количества вовлеченных мономеров.
Основными препятствиями для использования ПЭГ в промышленных масштабах являются: большое количество стадий осаждения; перерасход растворителей и стоимость нанофильтрационных мембран.
В 2017 году было описано использование производного адамантана в качестве якорной молекулы для жидкофазного синтеза олигонуклеотидов (A. Schwenger et al., «Solution-Phase Synthesis of Branched Oligonucleotides with up to 32 Nucleotides and the Reversible Formation of Materials», Eur. J. Org. Chem., 2017, 5852-5864). Одним из преимуществ этой подложки является очистка простой экстракцией. Основным недостатком этой якорной молекулы является ее низкий Е-фактор (который выражает желаемое массовое отношение отходов к продукту), поскольку для различных стадий экстракционной очистки требуется большой объем растворителя. Та же исследовательская группа описала тетразамещенное производное адамантана в качестве якорной молекулы для жидкофазного синтеза олигонуклеотидов. Опять же, большой объем растворителя, необходимого для очистки, и избыток фосфорамидитных мономеров сдерживают его промышленное развитие.
В 2006 г. была описана первая молекула ионного якоря, в данном случае имидазолиевого типа (R. A. Donga et al., «А Novel Approach to Oligonucleotide Synthesis Using an Imidazolium Ion Tag as a Soluble Support», J. Org. Chem., 2006, 71, 7907-7910). Очистку удлиненного олигонуклеотида осуществляют осаждением и экстракцией. С промышленной точки зрения эта жидкая подложка утяжеляется большим объемом растворителя, необходимого для очистки растущего олигонуклеотида.
Было показано, что якорные молекулы растворимого типа [3-циклодекстрина на основе D-глюкопиранозы эффективны для синтеза олигонуклеотидов в жидкой фазе (A. Gimenez Molina et al., «Acetylated and Methylated β-Cyclodextrins as Viable Soluble Supports for the Synthesis of Short 2'-Oligodeoxyribonucleotides in Solution», Molecules, 2012, 17, 12102-12120). Преимущества, связанные с этими растворимыми носителями, заключаются в их стоимости и уменьшении количества мономеров, необходимых для стадий связывания.
Слабые стороны этих матриц заключаются в способе очистки. Действительно, для каждого цикла обязательна очистка на хроматографической колонке.
Описан другой класс якорных молекул, состоящий из функционализированных производных пентаэритрита. Они были использованы для синтеза ДНК (V. Kungurtsev et al., «Solution-Phase Synthesis of Short O1igo-2'-deoxyribonucleotides by Using Clustered Nucleosides as a Soluble Support», (Eur. J. Org. Chem., 2013, 6687-6693) и РНК (A. Gimenez Molina et al., «Solution phase synthesis of short oligoribonucleotides on a precipitative tetrapodal support», Beilstein J. Org. Chem., 2014, 10, 2279-2285 and Current Organic Synthesis, 2014, 12, 202-207). Преимущества этой жидкой подложки: возможность проведения реакций удлинения в химии фосфорамидитов или фосфотриэфиров; стабильность матрицы; наличие четырех анкерных площадок; и очистки осаждением.
Основными препятствиями для индустриализации этой жидкой подложки являются использование двух гидрофобных защитных групп (в 2' и 5') при синтезе РНК, а также использование некоммерческих мономеров и многочисленные очистки путем осаждения.
Производные галловой кислоты в качестве растворимого носителя также использовали для получения олигонуклеотидов в растворе (см. JP 2010-275254 and WO 2013/179412, а также публикации S. Kim et al, «Liquid-Phase RNA Synthesis by Using Alkyl-Chain-Soluble Support», Chem. Eur. J., 2013, 19, 8615-8 620, and T. Shoji et al, «Synthesis of Conjugated Oligonucleotide in Solution Phase Using Alkyl-chain-soluble Support», Chem. - Lett. 2014, 43, 1251-1253). При использовании последнего в сочетании с фосфорамидитной химией был достигнут синтез (21-мерной) РНК с отличным выходом (26%) и хорошей чистотой (78%). Однако многочисленные стадии очистки (>50) делают эти якорные молекулы непригодными для промышленного использования.
В том же ключе были описаны якорные молекулы, названные Ajiphase™, для получения олигонуклеотидов (см. S. Katayama & K. Hirai, «Liquid-phase synthesis of oligonucleotides», published in S. Obika & M. Sekine (Eds.), «Synthesis of therapeutic oligonucleotides» (pp. 83-95), Springer Singapore, 2018). Использование Ajiphase™ в качестве якорной молекулы для получения олигонуклеотидов имеет некоторые преимущества: заякоренные олигомеры можно очистить путем простого осаждения в ацетонитриле или метаноле; количество эквивалентов фосфорамидита невелико (<2 эквивалентов); общий выход улучшается, а потребление растворителя снижается. Однако основным ограничением этого метода остается очистка (одно осаждение за цикл).
Настоящее изобретение также относится к олигосахаридам, ранее называемым «углеводами». Они участвуют в нескольких биологических процессах и играют важную роль в живом мире. Действительно, сахара участвуют в структуре основных молекул, таких как нуклеиновые кислоты (рибоза и дезоксирибоза). Олигосахариды и полисахариды состоят из моносахаридов, связанных между собой гликозидной связью. С точки зрения понимания роли Сахаров в живом мире химики добились концептуальных и практических достижений, которые открывают доступ ко все более сложным олигосахаридам (М. Panza et al., «Automated Chemical Oligosaccharide Synthesis: Novel Approach to Traditional Challenges», Chem. Rev. 2018, 118, 17, 8105-8150, и M. Guberman & P.H. Seeberger, «Automated Glycan Assembly: A Perspective», J. Am. Chem. Soc. 2019, 141, 14, 5581-5592).
После работы Меррифилда в 1960-х годах по синтезу пептидов на твердой подложке многие химики разработали аналогичные методы, адаптированные к синтезу олигосахаридов, чтобы извлечь выгоду из преимуществ этой методологии, а именно: использование избыточных реагентов и очистки путем простой промывки твердой подложки. Однако этот подход к синтезу олигосахаридов связан с некоторыми проблемами, такими как: выбор твердой подложки, дизайн спейсера, выбор защитных групп, мониторинг реакций, выбор донорных или акцепторных мономеров и окончательное отцепление целевого олигосахарида от матрицы (см. P.Н. Seeberger & S.J. Danishefsky, «Solid-Phase Synthesis of Oligosaccharides and Glycoconj ugates by the Glycal Assembly Method: A Five Year Retrospective», Acc. Chem. Res. 1998, 31, 685-695; P.H. Seeberger & W.-C. Haase, «Solid-Phase Oligosaccharide Synthesis and Combinatorial Carbohydrate Libraries», Chem. Rev. 2000, 100, 4349-4393, и P.H. Seeberger, «Automated oligosaccharide synthesis», Chem. Soc. Rev. 2008, 37, 19-28).
В 1973 г. был описан первый твердофазный синтез трисахарида (см. J.М. Frechet & С. Schuerch, «Solid-Phase Synthesis of Oligosaccharides. I. Prepapration of the Solid Support. Poly[p-(1-propen-3-ol-l-yl)styrene]», J. Am. Chem. Soc. 1971, 93, 492-496; Frechet, J.M. in «Polymer-Supported Reactions in Organic Synthesis», Hodge, P., Sherrington, D.C., Eds.; John Wiley & Sons Ltd: Chichester, UK, 1980, pp 407-434, и Carbohydr. Res. 1972, 399-412).
Вдохновленные этой работой, Seeberger et al. разработали машину для автоматизированного синтеза олигосахаридов (Р.Н. Seeberger, «Solid Phase Oligosaccharide Synthesis», J. Carbohydr. Chem, 2002, 21, 613-643, и M. Weishaupt et al, «Solid Phase Synthesis of Oligosaccharides», published in Methods in Enzymology, 2010, 478, 463-484). Так, они описали синтез нескольких сложных олигосахаридов, таких как производные додекамера β-глюкана.
Использование гликаля для целей гликозидного связывания изучалось на твердой подложке (J.Т. Randolph et al, «Major Simplifications in Oligosaccharide Synthesis Arising from a Solid-Phase Based Method: An Application to the Synthesis of the Lewis b Antigen», J. Am. Chem. Soc. 1995, 117, 5712-5719; C. Zheng et al, «Solid Support Oligosaccharide Synthesis: Construction of [3-Linked Oligosaccharides by Coupling of Glycal Derived Thioethyl Glycosyl Donors», J. Org. Chem, 1998, 63, 1126-1130, и K.A. Savin et al, «A New Polymer Support Silylene Linking Method for Hindered Hydroxy1-Bearing Systems», J. Org. Chem. 1999, 64, 4183-4186). Эта концепция гликозидного сочетания основана на активации гликаля электрофилом (например, диметилдиоксираном).
В результате только что изложенного основными проблемами твердофазного синтеза олигосахаридов являются: использование избыточных мономерных звеньев, невозможность контролировать аномерность каждого вводимого мономера, экстраполяция на любой тип гликозидных связей, низкая кинетика расщепления спейсеров, сложный синтез спейсеров, потребность в специальном оборудовании (способном работать до -20°С) и небольшие производственные мощности.
Короче говоря, производство макромолекул, олигонуклеотидов и олигосахаридов в промышленных масштабах с низкими затратами и с низким воздействием на окружающую среду является иллюзорным при существующих методологиях. Именно это и является целью настоящего изобретения. Действительно, авторы изобретения разработали ряд защитных групп (или якорных молекул, или солюбилизирующих молекул, или якорных матриц), способных обеспечить синтез олигонуклеотидов и олигосахаридов с низкой стоимостью, низким воздействием на окружающую среду и низкой сложностью. Другими словами, использование этих якорных молекул позволяет сочетать преимущества жидкофазного синтеза и твердофазного синтеза (гомогенность реакционной среды за счет растворимости носителей, что обеспечивает линейную кинетику; возможность уменьшить количество дорогие реагенты, а также растворители; возможность проведения крупномасштабных реакций (периодическое); упрощение очистки удлиняющихся макромолекул, избытка реагентов и побочных продуктов, основанное именно на физико-химических свойствах и природе якорной молекулы, их чаще всего проводят жидкостно-жидкостной экстракцией, основанной на селективном распределении (или коэффициенте распределения, или коэффициенте распределения) растворенных веществ в двух практически несмешивающихся жидкостях.
Объект изобретения
В настоящем изобретении предлагается решить трудности, обнаруженные в предшествующем уровне техники, в частности, в отношении техники очистки, с одной стороны, путем дериватизации защитных групп, которые могут быть растворимы в неполярных растворителях, и, с другой стороны, с использованием полиолефинов или олигомеров полиолефинов или полиалкенов, способных вызывать избирательную растворимость при получении биологических макромолекул (олигонуклеотидов и олигосахаридов) в жидкой фазе (или растворе). Действительно, авторы изобретения обнаружили, что использование полиолефинов и, в частности, производных полиизобутена (PIB), в качестве защитных групп, или якорных молекул, или жидких носителей, или якорных матриц позволяет синтезировать эти макромолекулы в растворе (галогенированном и/или негалогенированном растворителе), облегчая их очистку жидкостно-жидкостной экстракцией.
Таким образом, настоящее изобретение относится к синтезу макромолекул путем последовательного удлинения различных звеньев, которые в основном являются производными углеводов, которые могут быть идентичными или разными. Указанные макромолекулы могут, в частности, представлять собой олигонуклеотиды или олигосахариды.
Первой целью настоящего изобретения является способ синтеза макромолекул, состоящих из звеньев U, которые в основном представляют собой моносахариды или производные моносахаридов, которые могут быть идентичными или разными, указанные макромолекулы имеют первый и второй концы, где указанный способ синтеза осуществляется путем последовательного удлинения указанного второго конца мономером или олигомером М, имеющим по меньшей мере две функциональные группы, и указанный способ характеризуется следующим:
- на первой так называемом стадии заякоривания первое звено U1 указанной макромолекулы, соответствующее мономеру M1 или концевому звену олигомера M1, присоединяется посредством ковалентной связи (эфирной, сложноэфирной или любой другой функциональной связи, совместимой с настоящим способом) к якорной молекуле, растворимой в органических растворителях, указанная ковалентная связь является результатом взаимодействия первой из функциональных групп указанного мономера M1 или указанного олигомера M1 с функциональной группой указанной якорной молекулы, второй конец, возможно, представляет собой другую функциональную группу указанного мономера M1 или указанного олигомера M1, защищенной до указанной реакции по меньшей мере первой защитной группой GP1 и, возможно, второй защитной группой GP2 и n-й защитной группой GPn;
- на второй так называемой стадии удаления защиты удаляют одну из указанных защитных групп GP1, или GP2, или GPn, оставляя по меньшей мере одну свободную химическую функциональную группу на указанном мономере M1 или указанном олигомере M1;
- на третьей так называемой стадии связывания второй мономер М2 или олигомер М2, несущий по меньшей мере одну свободную химическую функциональную группу и по меньшей мере одну химическую функциональную группу, защищенную защитной группой GP3, вступает в реакцию таким образом, что его свободная химическая функциональная группа образует в результате взаимодействия с указанной свободной функциональной группой указанного первого мономера M1 или олигомера M1 ковалентную связь, таким образом создавая новую молекулу, образованную указанным мономером M1 или олигомером M1, присоединенным своим первым концом к указанной якорной молекуле, и указанным мономером М2 или олигомером М2, присоединенным к другому из ее концов, и указанный способ характеризуется тем, что указанная якорная молекула содержит полиолефиновую цепь, или полиолефиновый олигомер, или полиалкен по меньшей мере с 5 мономерными звеньями, предпочтительно, от 10 до 50 мономерными звеньями, при этом указанная полиолефиновая цепь представляет собой разветвленную цепь и, предпочтительно, полиизобутеновую цепь.
Таким образом, путем сочетания можно добавить n-й мономер Mn или олигомер Mn; этот способ позволяет синтезировать макромолекулы.
Способ включает по меньшей мере одну стадию, на которой указанную макромолекулу, присоединенную к указанной якорной молекуле, выделяют из реакционной среды методом жидкостной экстракции в неполярном органическом растворителе с несмешивающимся полярным растворителем (или в смеси полярных растворителей). Таким образом, реальной задачей является сохранение селективной растворимости заякоренного мономера или олигомера M1 после реакций удаления защиты и связывания по отношению к побочным продуктам указанных реакций.
Именно разветвленный характер полиолефиновой цепи якорной молекулы обеспечивает ей превосходную растворимость в неполярных растворителях и очень низкую растворимость в полярных растворителях, что необходимо для выделения якорной молекулы из реакционной среды методом жидкостной экстракции.
Способ может включать стадию полного удаления защиты у указанной макромолекулы. После связывания последнего мономера или олигомера защитные группы макромолекулы удаляются.
Указанные моносахаридные звенья или производные моносахаридов представляют собой, в частности, производные пентоз (и, в частности, нуклеозидов) или гексоз. Среди пентоз можно назвать, например, нуклеозиды, такие как С-нуклеозиды, ациклические нуклеозиды, карбоксильные нуклеозиды, имино-С-нуклеозиды, нуклеозиды с модифицированными основаниями; все эти молекулы должны использоваться в соответственно защищенной форме. Другими пригодными для использования производными моносахаридов являются озамины (аминосахара), такие как глюкозамин, галактозамин, маннозамин, меглумин.
Связь между двумя последовательными звеньями представляет собой озидную (предпочтительно, гликозидную) или фосфорилированную (особенно озо-1-фосфатную) или углеводную, или N-гетерозидную, или S-гетерозидную связь.
В одном варианте осуществления способа некоторые из звеньев U могут не быть моносахаридами или производными моносахаридов. В частности, такие звенья U могут быть выбраны из аминокислот и липидов (производных изопрена).
Упомянутая якорная молекула, преимущественно, имеет среднемассовую молекулярную массу от 300 до 20000, предпочтительно, от 500 до 15000. Ее полиолефиновая цепь представляет собой олигомер или полимер, построенный из мономеров, которые предпочтительно являются идентичными.
В одном предпочтительном варианте осуществления изобретения указанную полиолефиновую цепь или полиолефиновый или полиалкеновый олигомер получают полимеризацией мономера. Этот мономер преимущественно получают из биоресурсов.
В одном конкретном варианте осуществления изобретения указанная полиолефиновая или полиалкеновая олигомерная цепь содержит ненасыщенные углерод-углеродные связи в количестве, не превышающем 5% и, предпочтительно, не превышающем 3%.
Второй целью изобретения является применение способа согласно любому из вариантов осуществления изобретения для синтеза олигонуклеотидов или олигосахаридов.
В случае олигонуклеотидов мономерное звено обычно состоит из соответствующим образом защищенного фосфорилированного нуклеозида (или олигонуклеотидной цепи) (схема 1). Это звено будет взаимодействовать со спиртом в 5'-положении защищенного соответствующим образом нуклеозида (или олигонуклеотидной цепи) на нуклеиновой основе и/или во 2'-положении сахара и заякориваться в 3'-положении на жидком носителе.
[Химические формулы 1]
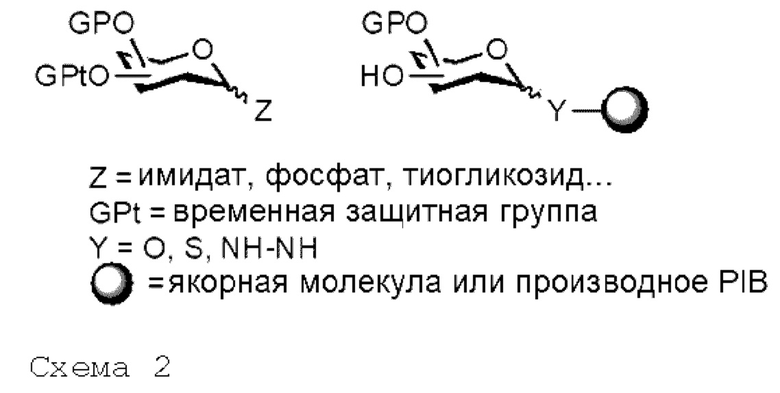
В случае олигосахаридов мономерная единица обычно состоит из соответствующим образом защищенного или незащищенного донорного гликозила (или олигосахаридной цепи), несущего группу Z в аномерном положении, которая может быть, но не ограничивается этим, циклическим полуацеталем, ацетатом, тиогликозидом, фосфатом или имидатом. Спиртовая функциональная группа соответствующим образом защищенного или незащищенного акцепторного гликозида (или олигосахаридной цепи), заякоренного на жидком носителе, будет взаимодействовать с указанным мономерным звеном (схема 2).
[Химические формулы 2]
Вообще говоря, способ по изобретению можно осуществлять с использованием одинаковых или различных устройств. Таким образом, например, может быть синтезирован олигосахарид, который представляет собой гомополимер, а именно: состоит из идентичных моносахаридных звеньев. Олигосахарид также может быть синтезирован из различных моносахаридных звеньев. Также можно использовать звенья, которые представляют собой дисахариды, трисахариды или более длинные олигосахариды.
Точно так же олигонуклеотиды могут быть синтезированы из одинаковых или разных фосфорилированных нуклеозидных (или олигонуклеотидных) звеньев.
Например, для получения олигонуклеотидов можно использовать нуклеозиды 3'-(2-цианоэтил-N,N-диалкилфосфорамидит) или нуклеозиды 3'-(Н-фосфонаты) или нуклеозидфосфаты (ди- или триэфиры). В синтезе олигосахаридов можно использовать соответствующим образом защищенные или незащищенные донорные гликозилы. В обоих случаях применяют реакции связывания, известные специалистам в данной области техники.
Способ по изобретению также позволяет синтезировать смешанные полисахариды, включающие моносахаридные звенья и нуклеотидные звенья.
Способ по изобретению также позволяет вводить в макромолекулу звенья, которые не являются или не содержат ни олигосахаридов, ни олигонуклеотидов.
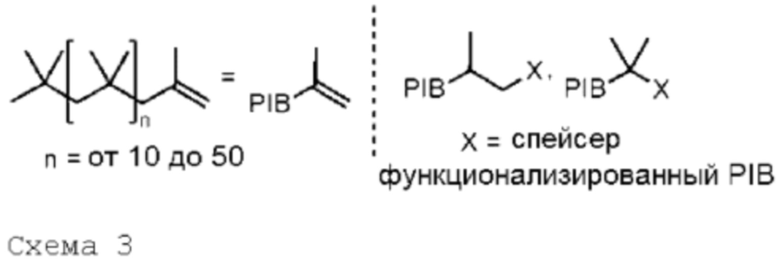
Одним существенным признаком способа по изобретению является использование якорных молекул, или солюбилизирующих молекул, или якорных матриц. Они должны быть растворимы в неполярном растворителе. Они состоят из полиолефиновой цепи, содержащей не менее 5 мономерных звеньев, предпочтительно, от 10 до 50 звеньев; они представляют собой полиолефины или олигомеры полиолефинов или полиалкенов. Преимущественно, якорные молекулы функционализированы, по меньшей мере, на одном из своих концов, чтобы обеспечить защиту или присоединение исходного мономерного (или олигомерного) звена.
Указанная якорная молекула может содержать в каждом из своих звеньев идентичные или неидентичные алкильные группы, которые, предпочтительно, выбраны из группы, включающей метил и этил. Указанная полиолефиновая цепь, преимущественно, имеет среднемассовую молекулярную массу от 300 до 20000, предпочтительно, от 500 до 15000. Указанная полиолефиновая цепь может содержать ненасыщенные углерод-углеродные связи в количестве, не превышающем 5% и, предпочтительно, не превышающем 3%. Предпочтительно, она представляет собой полиизобутеновую (сокращенно PIB) цепь. На схеме 3 показаны некоторые PIB, применимые в рамках изобретения; в этих формулах X представляет собой спейсер, который выполняет функцию, предназначенную для взаимодействия с первым звеном U1 макромолекулы для заякоривания на жидком носителе.
[Химические формулы 3]
Способ по изобретению позволяет получить макромолекулы высокой чистоты. Этот способ индуцирует выгодный Е-фактор (фактор окружающей среды), поскольку очистка выполняется методом жидкостной экстракции с разумными количествами растворителя, что позволяет свести к минимуму стадии очистки с помощью хроматографических колонок; генерируя, следовательно, финансовую экономию. Таким образом, реакции дериватизации побочных продуктов, растворимых в неполярных растворителях, можно проводить до жидкостно-жидкостной экстракции.
В одном конкретном варианте осуществления изобретения указанная якорная молекула содержит полиолефиновую цепь (или представляет собой полиолефиновую цепь), оканчивающуюся по меньшей мере одной группой, выбранной из группы (спейсер), состоящей из алифатической цепи (разветвленной или неразветвленной, ненасыщенной или ненасыщенной), (гетеро)кольца и/или (гетеро)арила (замещенного или незамещенного).
В одном преимущественном осуществлении изобретения среднемассовая молекулярная масса якорных молекул, за исключением их концевой функциональной группы, составляет от 300 до 20000, предпочтительно, от 500 до 15000. При средней молекулярной массе около 20000 эти молекулы могут проявлять чрезмерную вязкость, что ограничивает их растворимость в органических растворителях.
Некоторые производные PIB, используемые в рамках настоящего изобретения, коммерчески доступны в качестве лигандов для гомогенного катализа. Например, могут быть использованы 2-метил-3-[полиизобутил(12)]пропанол (среднемассовая молекулярная масса 7 57, включая концевую функциональную группу) или 4-[полиизобутил(18)]фенол (среднемассовая молекулярная масса 1104, включая концевую функциональную группу), которые распространяются, соответственно, под номерами ссылок 06-1037 и 06-104 8 компанией Strem Chemicals. Эти две молекулы являются производными полиизобутена, цепь которого оканчивается, соответственно, группой -СН2-С(СН3) (Н)-СН2-ОН (то есть изопропанолом) и группой -СН2-С(СН3)2-С6Н4-ОН (то есть фенолом).
В соответствии с одним признаком изобретения якорные молекулы действуют как защитные группы, спиртовые функциональные группы и/или любые другие функциональные группы, которые должны быть инертными в условиях способа, являющегося предметом настоящего изобретения.
В случае синтеза олигонуклеотидов спиртовая функциональная группа в 3'-положении рибозы/дезоксирибозы и/или амина нуклеооснования может быть замаскирована, одновременно или нет, по меньшей мере одной солюбилизирующей защитной группой.
В случае синтеза олигосахаридов исходная аномерная функциональная группа заякоривается (защищается) солюбилизирующей защитной группой.
Эти дериватизации позволяют солюбилизировать мономеры и олигомеры в неполярных растворителях, таких как циклогексан, гептан(ы) или гексан(ы).
Согласно другому признаку изобретения использование якорных молекул, растворимых в органических растворителях, описанных выше (и, более конкретно, использование полиолефинов), и нерастворимых в некоторых полярных растворителях (таких как вода, и/или этанол, и/или ацетонитрил), облегчает очистку заякоренных мономеров и олигомеров простой жидкостной экстракцией или простой фильтрацией на силикагеле.
В соответствии с еще одним признаком изобретения используются коммерчески доступные якорные молекулы или якорные молекулы, которые могут быть синтезированы простым и прямым способом из коммерчески доступных предшественников, включая некоторые производные полиизобутена. Другими словами, к PIB могут быть присоединены обычные защитные группы, такие как бензил, силил или производные карбоновой кислоты.
На схеме 4 ниже представлены некоторые не исчерпывающие структуры, способные играть роль защитной группы в синтезе олигонуклеотидов.
[Химические формулы 4]
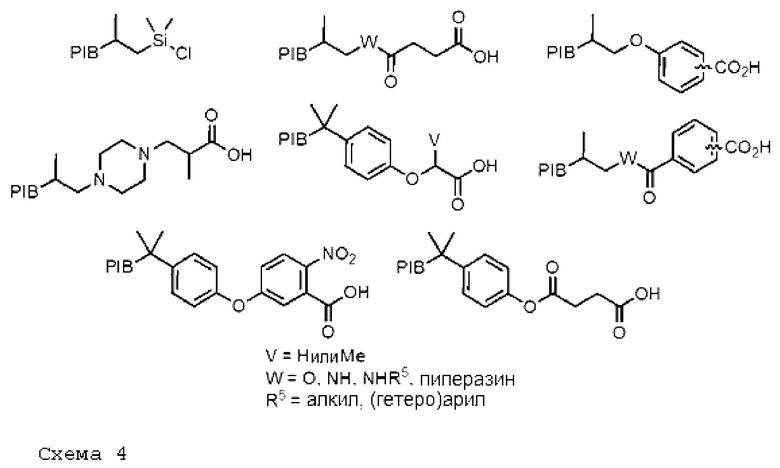
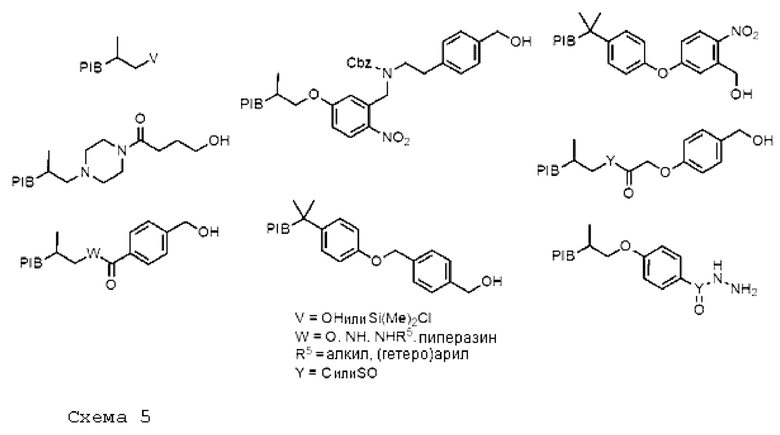
На схеме 5 ниже приведены некоторые не исчерпывающие структуры, способные играть роль защитной группы в синтезе олигосахаридов.
[Химические формулы 5]
Согласно еще одному признаку изобретения исходный мономер (или олигомер) присоединяется к якорной молекуле в соответствии с известными реакциями, зависящими от химический функциональных групп, в соответствующем растворителе, таком как (галогенированные или негалогенированные) смеси растворителей, и при температура от -50°С до 150°С.
Согласно еще одному признаку изобретения химические функциональные группы мономера (или олигомера), которые несовместимы с этим способом, могут быть временно замаскированы соответствующими защитными группами, такими как трет-бутилдиметилсилил (сокращенно TBDMS или TBS), диметокситритил (сокращенно DMTr) или любой другой защитной группой, совместимой с данным способом.
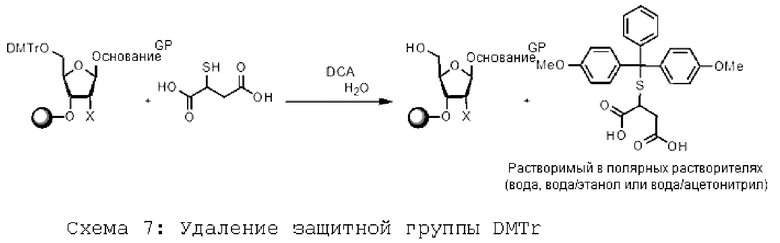
Согласно еще одному признаку изобретения после стадии удаления защиты защитные группы могут быть дериватизированы, предпочтительно, in situ с образованием соединений, растворимых в полярном растворителе (или смеси полярных растворителей). Таким образом, мономер (или олигомер), связанный с производным PIB, первичный спирт которого защищен тритильной группой, расщепляется в кислой среде в присутствии соответствующего акцептора карбокатиона (тритила), что делает его растворимым в водной (или полярной) фазе. Поглотители тритилкарбокатионов включают, но не ограничиваются ими, тиогликолевую кислоту, 3-меркаптопропроновую кислоту, 3-меркапто-1-пропансульфоновую кислоту, цистеин или тиомалиновую кислоту (или меркаптоянтарную кислоту).
В соответствии с еще одним признаком изобретения указанная якорная молекула взаимодействует с надлежащим образом защищенным первым мономером (или олигомером), что приводит к ковалентной связи, такой как сложноэфирная или О-гликозидная связь, между этими двумя видами молекул.
Согласно еще одному признаку изобретения мономеры, имеющие химические функциональные группы, несовместимые с условиями реакции цикла удлинения (или итерации), могут быть временно замаскированы солюбилизирующей защитной группой. В этом случае, в зависимости от длины макромолекулы-мишени, химические функциональные группы могут быть замаскированы одной или несколькими солюбилизирующими защитными группами и одной или несколькими обычными защитными группами, такими как: бензоил, ацетил, трет-бутилдиметилсилил.
В соответствии с еще одним признаком изобретения способ синтеза макромолекул осуществляется с использованием в качестве отправной точки мономерных звеньев (или олигомеров), связанных с якорной молекулой.
В случае синтеза олигонуклеотидов первый цикл элонгации начинается с селективного удаления защиты с 5'-защитной группы заякоренного нуклеозида в кислой среде и в присутствии поглотителя, если это тритильное производное.
В случае синтеза олигосахаридов первый цикл элонгации начинается с реакции между подходящим образом защищенным или незащищенным донорным гликозилом и гликозидом, заякоренным и лишенным защиты на спиртовой функциональной группе, которая будет участвовать в реакции. Опять же, если защитная группа представляет собой производное тритила, с нее снимают защиту в кислой среде в присутствии поглотителя.
В соответствии с еще одним признаком изобретения интеграция звена (или повторения) мономера требует нескольких стадий.
В случае синтеза олигонуклеотидов для нуклеозидфосфатов необходимы две стадии (присоединение и удаление защиты со спирта в 5'-положении) и три стадии для нуклеозидфосфорамидитов или Н-фосфонатов (связывание, окисление и удаление защиты со спирта в 5'-положении).
В случае синтеза олигосахаридов необходимы две стадии (связывание и удаление защиты).
Согласно еще одному признаку изобретения указанная макромолекула образована n мономерными звеньями с химической функциональной группой, связанной с якорной молекулой. В ходе осуществления способа олигомерная цепь растет за счет последовательного удлинения (или цикла) или итерации, и на каждой из этих стадий к свободному спиртовому концу добавляется мономерное или олигомерное звено.
Согласно еще одному признаку изобретения в указанной олигомерной цепи могут быть использованы природные и/или неприродные, и/или синтетические мономеры (или олигомеры).
В соответствии с еще одним признаком изобретения в указанной олигомерной цепи можно использовать одно или несколько подходящим образом защищенных аналоговых звеньев мономера (или олигомера), таких как мономеры морфолинофосфора или мономеры N-ацетилмоносахарида.
В соответствии с еще одним признаком изобретения по меньшей мере одна стадия, на которой указанную олигомерную цепь выделяют из реакционной среды путем экстракции в не смешивающемся с водой аполярном органическом растворителе (или не смешивающемся со смесью вода/этанол или смесью вода/ацетонитрил) или фильтрованием на силикагеле. В качестве неполярного органического растворителя предпочтительными являются углеводороды, содержащие только атомы углерода и водорода, такие как линейные алканы, разветвленные алканы и циклоалканы, и особенно предпочтительными являются циклогексан, гептаны и гексаны.
В соответствии с еще одним признаком изобретения способ позволяет получать олигомеры высокой чистоты после полного удаления защиты и расцепления якорных молекул для их предполагаемого использования, например, в качестве активного начала для доклинических испытаний, клинической помощи или любых других применений.
Согласно еще одному признаку изобретения в способе согласно изобретению якорные молекулы могут быть использованы повторно (рециклированы).
В соответствии с еще одним признаком изобретения экстракционные растворители могут быть использованы повторно (рециклированы) в способе по изобретению.
Способ по изобретению может быть автоматизирован и/или реализован в проточной химии.
Подробное описание
В настоящем изобретении приведенные ниже термины используются в следующем значении, которое соответствует терминологии Международного союза теоретической и прикладной химии (IUPAC), и любые другие используемые термины также следует понимать в соответствии с определением IUPAC.
Термин «углевод» включает моносахариды, олигосахариды и полисахариды, а также соединения, полученные из моносахаридов путем восстановления карбонильной функциональной группы (особенно альдегидной или кетоновой функциональной группы), путем окисления по меньшей мере одной функциональной группы на конце цепи до карбоксильной группы, кислотой или заменой одной или нескольких гидроксильных групп атомом водорода, аминогруппой, тиольной группой или любым подобным атомом. Этот термин также включает производные таких соединений.
Термин «моносахарид» относится к мономеру углеводов.
Термин «гликозиламин» означает соединение, в котором углевод связан с аминогруппой в аномерном положении.
Термин «нуклеозид» относится к рибозил- или дезоксирибозилпроизводным некоторых пиримидиновых или пуриновых оснований, точнее гликозиламинов, состоящих из нуклеооснования, связанного с аномерным атомом углерода пентозного остатка, обычно рибозы (рибонуклеозида) или дезоксирибозы (дезоксирибонуклеозида), посредством гликозидная связь от атома N1 пиримидина или атома N9 пурина.
Термин «нуклеотид» относится к органической молекуле, которая является строительным блоком нуклеиновой кислоты, такой как ДНК или РНК; эта молекула состоит из нуклеинового основания, пятиуглеродной азы и, наконец, от одной до трех фосфатных групп.
Термин «защитная группа» относится к молекуле, используемой для обратимой защиты функциональной группы во время химической реакции, чтобы сделать указанную функциональную группу нереакционноспособной в указанном процессе химической реакции, который преобразовал бы указанную незащищенную функциональную группу.
Термин «функциональная группа» означает атом или группу атомов, которые могут реагировать с другими функциональными группами. Примерами функциональных групп являются следующие: альдегид, карбоновая кислота, фосфорная кислота, фосфоновая кислота, сульфоновая кислота, первичный или вторичный амин, кетон, алкилгалогенид, гидразин, гидроксиламин, гидроксил, изоцианат, изотиоцианат, тиол.
Термин «макромолекула» относится к молекуле с высокой молекулярной массой, созданной из последовательности единиц с низкой молекулярной массой. Эти единицы могут быть связаны индивидуально и последовательно, чтобы сформировать цепочку; олигомер, содержащий несколько таких звеньев, также может быть связан. Вообще говоря, эти единицы могут быть одинаковыми или разными. Макромолекулы могут иметь синтетическое или биологическое происхождение или их комбинацию. Они могут включать одну или несколько функциональных групп. Используемый в настоящем документе термин «макромолекула» включает полимеры; в случае полимера указанное звено называется мономером.
Термин «полимер» относится к макромолекуле, включающей повторяющиеся структурные единицы, а именно мономеры, соединенные химическими связями линейным, кольцевым, разветвленным, сшитым, дендримерным образом или их комбинацией. Понятно, что полимер может, например, также содержать по меньшей мере одну функциональную группу. Полимер называется «гомополимером», если полимер состоит из одних и тех же мономеров, и называется «сополимером», если полимер состоит из разных мономеров.
В соответствии с одним признаком изобретения, который более подробно описан ниже, якорные молекулы или солюбилизирующие защитные группы или якорная матрица представляют собой полиолефины или, точнее, полиолефиновые олигомеры (полиолефины также называются полиалкенами) и их производные, то есть они несут хотя бы одну функциональную группу.
В одном предпочтительном варианте осуществления в способ по изобретению используются полиолефины или, точнее, олигомеры полиолефинов (полиолефины также называют полиалкенами) и их производные в качестве якорной молекулы или защитной группы или якорной матрицы из нескольких функциональных групп различных мономеров, по меньшей мере, монофункциональных, связанных ковалентной связью (сложный эфир, амид, простой эфир, тиоэфир или любые другие подходящие химические функциональные группы), образуя новое мономерное производное растворим в неполярной жидкой фазе. Молекулы полиолефинов состоят из цепочки атомов углерода, связанных одинарными связями. Они могут включать разветвления, состоящие из одинаковых или разных, но предпочтительно одинаковых алкильных групп. Предпочтительно, полимеры состоят из мономерных звеньев в количестве по меньшей мере 10 и, предпочтительно, от 15 до 50. Предпочтительны гомополимеры, но можно использовать и сополимеры (насыщенные или ненасыщенные). В случае ненасыщенных полимеров или сополимеров количество ненасыщенных связей в цепи атомов углерода, предпочтительно, не превышает 5% и, предпочтительно, не превышает 3%.
В одном предпочтительном варианте осуществления они представляют собой производные полиизобутенов (PIB), класс полимеров, известный с 1930-х годов, но также могут быть использованы производные полипропиленов.
Эти якорные молекулы, используемые в способе согласно изобретению, предпочтительно, находятся в форме функционализированных производных. Предыдущие схемы 4 и 5 показывают ряд производных PIB.
В соответствии с одним признаком изобретения эти якорные молекулы связаны с мономерным (или олигомерным) звеном ковалентной связью, такой как амидная, простая эфирная, тиоэфирная, тиоэфирная, сульфонилгидразидная или ацилгидразидная (неполный список). Это предполагает, что задействованные производные PIB соответствующим образом функционализированы. Эта функционализация якорных молекул, как правило, происходит в концевом положении, а именно, предпочтительно, на одном из концов цепи атомов углерода.
Согласно изобретению многофункциональные мономеры (или олигомеры) могут быть функционализированы производными PIB посредством ковалентной связи в виде сложного эфира, простого эфира, простого тиоэфира, сложного тиоэфира или любой другой химической функциональной группы, совместимой с настоящим способом. Это действует как солюбилизирующая защитная группа для мономеров (или олигомеров).
Полиолефиновые олигомеры, используемые в качестве якорных молекул, обычно характеризуются среднемассовой молекулярной массой, но также можно использовать «чистые» олигомеры, которые имеют идентичные молекулы с заданной длиной цепи.
Взаимодействие между якорной молекулой и мономером (или олигомером) приводит к образованию новой молекулы с низкой растворимостью в воде (<30 мг/мл).
В соответствии с другим признаком изобретения функционализация PIB в результате химических реакций приводит к различным молекулярным структурам, способным действовать в качестве солюбилизирующих защитных групп по меньшей мере интересующей бифункциональной молекулы (или промежуточного продукта) в процессе многостадийного синтеза. Подразумевается, что защитная группа также является по меньшей мере монофункциональной. В противном случае химическая функциональная группа, не участвующая в связывании между производным PIB и интересующей молекулой (или промежуточным продуктом), должна быть инертной или должным образом защищена, чтобы избежать любых ложных продуктов или побочных реакций.
В соответствии с другим признаком изобретения химические функциональные группы мономера (или олигомера), не вовлеченные в ковалентную связь с производным PIB, прямо или нет, должны быть пассивными или должным образом защищены, чтобы избежать образования нежелательных продуктов.
В другом аспекте изобретения молекула, полученная в результате реакции между производным PIB и мономером (или олигомером) посредством образования ковалентной связи, прямо или косвенно, характеризуется тем, что она имеет низкую растворимость в воде (<30 мг/мл). Другими словами, производное PIB действует как солюбилизирующая молекула.
Согласно другому признаку изобретения молекула, образующаяся в результате взаимодействия между производным PIB и мономером (или олигомером) посредством образования ковалентной связи, прямо или косвенно, характеризуется тем, что производное PIB значительно повышает растворимость мономера (или олигомера) в неполярных растворителях (циклогексан, гептан(ы), гексан(ы) или ароматические растворители) или в любом другом подходящем растворителе. Таким образом, новое мономерное (или олигомерное) производное обладает селективной растворимостью (высоким коэффициентом распределения) для неполярного растворителя при жидкостно-жидкостной экстракции (в присутствии воды или смеси вода/этанол или вода/ацетонитрил), что делает процесс очистки простым, быстрым и дешевым.
Реакцию введения защитных групп между производным PIB и мономером (или олигомером) посредством образования ковалентной связи, прямо или косвенно (спейсер), проводят в любом растворителе или инертной жидкости, которые могут растворять реагенты, при соответствующей температуре. Применимые растворители, чистые или в виде смесей, включают, но не ограничиваются ими, галогенированные или негалогенированные углеводороды. Предпочтительными растворителями являются дихлорметан и толуол (отдельно или в присутствии N-N-диметилформамида).
В зависимости от свободных химических функциональных групп мономера (или олигомера) и якорной молекулы возможны различные химические реакции. Таким образом, образование нового мономера или производного олигомера можно проводить всеми способами, известными специалисту в данной области. В качестве неисчерпывающих примеров применимые реакции включают реакции этерификации, реакции амидирования или реакции этерификации. Следовательно, условия реакции (растворитель(и), температура(и), концентрация(и), продолжительность(и)) должны быть адаптированы для каждой реакции введения защитных групп.
В зависимости от химической природы связи между мономером (или олигомером) и якорной молекулой стадии удаления защитных групп можно проводить с использованием условий реакции, известных специалисту в данной области. Не будучи исчерпывающим, можно упомянуть омыление, гидролиз и гидрирование. Точнее, способ растворения подходящим образом защищенного и заякоренного мономера (или олигомера) посредством ковалентной связи согласно изобретению характеризуется тем, что его растворяют в органическом растворителе. Другими словами, якорная молекула действует как солюбилизирующая молекула и защитная группа химической функциональной группы мономера (или олигомера).
Мономер (или олигомер), надлежащим образом защищенный и ковалентно связанный с якорной молекулой (различного типа), характеризуется низкой растворимостью в воде (<30 мг/мл). Другими словами, якорная матрица действует как солюбилизирующее вещество. Эта дериватизация значительно увеличивает растворимость новой молекулы до такой степени, что она становится растворимой в неполярных органических растворителях. Следовательно, мономеры (или олигомеры), связанные с производным PIB, имеют высокий коэффициент распределения (селективную растворимость (или селективное распределение)) для неполярной органической фазы во время жидкостно-жидкостной экстракции в присутствии циклогексана, или гептана(ов), или гексана(ов) и воды, или смеси вода/этанол или вода/ацетонитрил, что обеспечивает простую и быструю очистку.
Настоящее изобретение открывает возможность конвергентного синтеза длинных олигомеров, который может быть достигнут с использованием по меньшей мере двух подходящим образом защищенных олигомерных фрагментов, по меньшей мере один из которых связан с якорной молекулой.
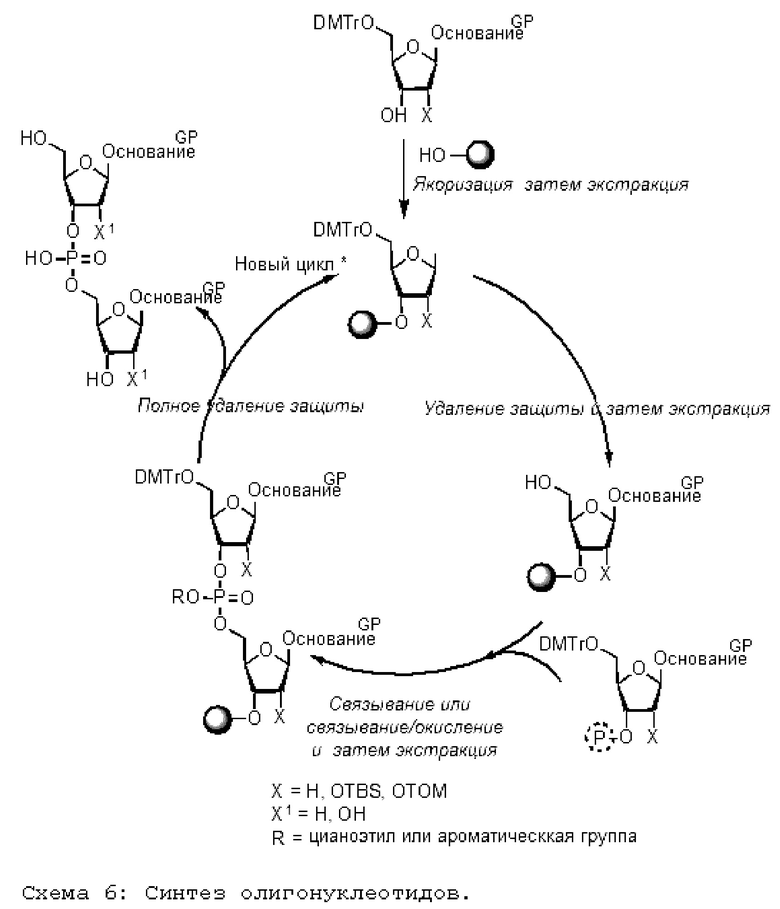
На приведенной ниже схеме реакции 6 показана полная последовательность получения олигонуклеотида. Точнее, на первой так называемой стадии заякоривания первое мономерное звено (в данном случае производное дезоксирибозы), защищенное защитной группой (в данном случае DMTr), присоединяется к молекуле жидкого носителя согласно изобретению; полученный продукт очищают жидкостно-жидкостной экстракцией. На второй так называемой стадии удаления защиты защитную группу (DMTr) удаляют и убирают в кислой среде в присутствии поглотителя, а полученный продукт очищают жидкостно-жидкостной экстракцией.
Предпочтительно выполнять заякоривание и удаление защиты (с очисткой) без выделения промежуточного защищенного спирта.
[Химические формулы 6]
После стадии удаления защиты защитные группы могут быть дериват из ированы, предпочтительно, in situ с образованием соединений, растворимых в полярном растворителе (или их смеси). Таким образом, мономер (или олигомер), связанный с производным PIB, первичный спирт которого защищен тритильной группой, расщепляется в кислой среде в присутствии поглотителя соответствующего карбокатиона (тритила) для придания ему растворимости в водной (или полярной) фазе. Поглотители тритилкарбокатиона могут быть предпочтительно выбраны из группы, включающей: тиогликолевая кислота, 3-меркаптопропионовая кислота, 3-меркапто-1-пропансульфокислота, цистеин, тиомалиновая кислота, меркаптоянтарная кислота. Пример такого удаления защиты представлен на схеме 7 ниже.
[Химические формулы 7]
На третьей так называемом стадии связывания/окисления (сульфуризации) вводится второе мономерное звено (в данном случае фосфорилированное производное рибозы), защищенное защитной группой (в данном случае DMTr). Б случае химии фосфорамидитов реакцию сочетания проводят в присутствии тетразола или любых других подходящих реагентов (бензилтио-1Н-тетразол (ВТТ), 4,5-дицианимидазол) с последующей реакцией окисления (метахлорпербензойная кислота (мХПБК), йод, пероксид 2-бутанона). Таким образом получают динуклеотид; реагенты и побочные продукты разделяют экстракцией.
На четвертой стадии, называемой общим снятием защиты, с этого динуклеотида, который все еще связан с молекулой жидкого носителя согласно изобретению, можно удалить защиту, а затем отсоединить от этого носителя. В качестве альтернативы он может войти в новый цикл для добавления третьего звена и так далее.
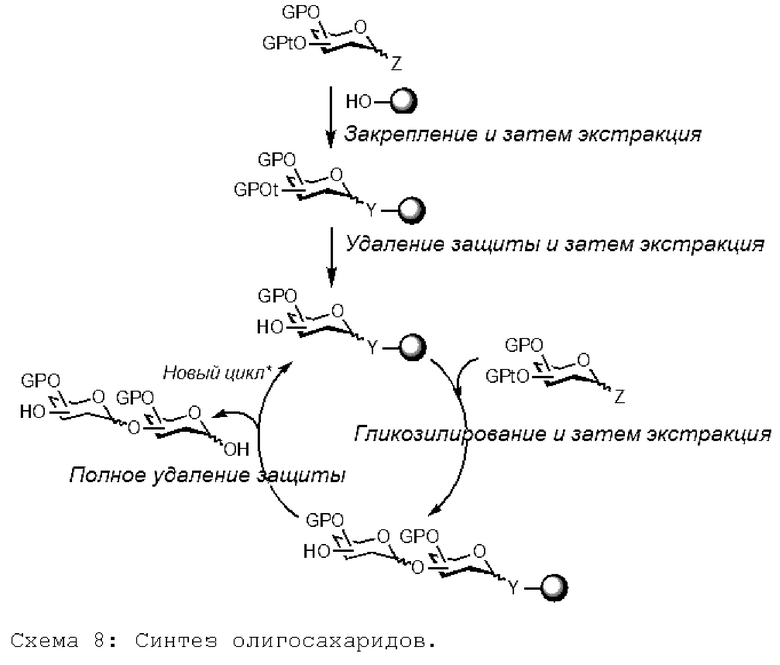
На схеме реакции 8 ниже показана полная последовательность получения олигосахарида. Точнее, на первой так называемой стадии заякоривания первое мономерное звено (в данном случае производное гексозы), защищенное или не защищенное первой, более сильной защитной группой (GP) и второй, более лабильной защитной или незащитной группой, называемой временной (GPt), присоединено к молекуле жидкого носителя в аномерном положении согласно изобретению; это соединение очищают экстракцией. На второй так называемой стадии селективного удаления защиты указанную вторую защитную группу (GPt) удаляют, и соединение очищают экстракцией. На третьей так называемой стадии гликозилирования добавляют второе мономерное звено (в данном случае защищенное или незащищенное донорное гликозилпроизводное) и связывают с указанным первым звеном с образованием заякоренного дисахарида; реагенты и побочные продукты удаляются жидкостно-жидкостной экстракцией. На четвертой так называемом стадии полного удаления защиты с этого дисахарида удаляют защиту, а затем отделяют от жидкой подложки. Альтернативно, он может вступить в новый цикл, после селективного удаления защиты функциональной группы, при необходимости инициировать новый цикл и так далее.
[Химические формулы 8]
Среди якорных молекул предпочтительны те, которые могут быть получены методом полимеризации из простых мономеров. Это относится к полиизобутенам (PIB), которые представляют собой один особенно предпочтительный тип молекул-якорей. Мономер полиизобутенов, а именно изобутен, может быть изготовлен в промышленных масштабах из биосырья, и PIB могут быть получены из изобутена биологического происхождения путем простой полимеризации. Таким образом, настоящее изобретение может быть реализовано с использованием якорных молекул из биологического источника и, в частности, из биоисточника PIB.
Концепция содержания биоресурсов определена в стандарте ISO 16620-1:2015 «Plastics - Biosourced Content - Part 1: General Principles», включая определение терминов «синтетический полимер, полученный из биоресурсов», «содержание синтетического полимера, полученного из биоресурсов», «содержание» и «массовое содержание биосырья», а также в ISO 16620-2:2015 «Plastlcs Biobased Content - Part 2: Determination of Blobased Carbon Content», и ISO 16620-3:2015 «Plastics - Biosourced Content Part 3: Determination of Biobased Synthetic Polymer Content», для методов определения и количественной оценки биоресурсов.
Преимущественно якорные молекулы, используемые в настоящем изобретении, имеют содержание углерода из биологического источника более 90%, предпочтительно, более 93% и, еще более предпочтительно, более 95%.
Способ по изобретению имеет много преимуществ.
Первое преимущество заключается в том, что он позволяет получать олигомеры, связанные с матрицей, с хорошей чистотой путем простой жидкостной экстракции в неполярном органическом растворителе и воде или в смеси вода/этанол или вода/ацетонитрил, или путем фильтрации на силикагеле, что приводит к удалению побочных продуктов (солей, избыточных реагентов или любых других молекулярных частиц), которые не связаны с полиолефиновыми или полиалкеновыми олигомерными производными. Неполярные органические растворители, такие как циклогексан, гептан(ы), гексан(ы), которые имеют температуру воспламенения <15°С, подходят для солюбилизации полиолефина или олигомера полиолефина или производных полиалкена в процессе экстракции или промывки. Таким образом, способ по изобретению упрощает стадии очистки и дает меньше отходов (эффлюенты и стационарная фаза).
Вторым особенно интересным преимуществом является возможность автоматизации способа по изобретению.
Третьим преимуществом является возможность повторного использования растворителей для экстракции, а также якорных молекул (полиолефинов или олигомеров полиолефинов или полиалкенов), особенно в промышленных масштабах. Действительно, эти защитные группы могут быть легко удалены в конце синтеза с помощью реакций, обычно используемых в органическом синтезе (таких как гидролиз, омыление, гидрирование или любая другая реакция, совместимая с настоящим способом) и использованы повторно. Это доказывает, что способ по изобретению соответствует экологически чистой или устойчивой химии, в отличие от существующих способов производства.
Четвертое преимущество изобретения заключается в возможности получения больших олигомеров либо путем модулирования размера молекулы-якоря, либо путем перехода к конвергентному синтезу, либо путем введения одной или нескольких молекул-якорей в мономерные звенья.
Пятым преимуществом является возможность контролировать чистоту олигомера в процессе синтеза на каждой стадии с помощью различных аналитических методов, таких как масс-спектрометрия, высокоэффективная жидкостная хроматография, протонный или углерод-13 ядерный магнитный резонанс.
Шестым преимуществом является возможность реализации в промышленных масштабах без дорогостоящего оборудования.
Благодаря своей высокой чистоте макромолекулы, полученные этим способом, могут быть использованы в качестве фармацевтических препаратов, косметических средств,
фитосанитарных продуктов или сельскохозяйственных пищевых продуктов или для получения доступа к любому из этих продуктов.
Еще одно преимущество заключается в том, что предпочтительные якорные молекулы, а именно производные полиизобутена, могут быть получены из изобутена биологического происхождения, как объяснялось выше.
Примеры:
Следующие примеры иллюстрируют синтез некоторых функционализированных якорных молекул, которые можно использовать для реализации способа по изобретению.
Если не указано иное, известные производные PIB были получены из предшественников и описанными способами (Tetrahedron, 2005, 61, 12081) и коммерческих реагентов.
Пример 1: Общий способ О-арилирования
[Химические формулы 9]
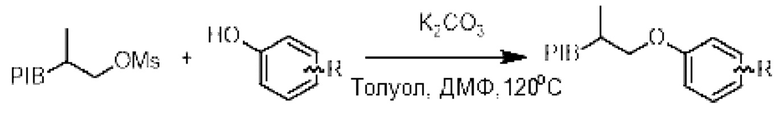
К смеси производного PIB-CH£-CH (СН3)-CH2-OMs (1 эквивалент) и фенола (3 эквивалента) в смеси толуол/N-N-диметилформамид (1/1) (0,1 М) добавляли карбонат калия (5 эквивалентов), а затем реакционную среду нагревали при температуре 120°С в течение 16 ч, а затем охлаждали до комнатной температуры. Реакционную среду трижды экстрагировали циклогексаном и смесью ацетонитрила/воды или этанола/воды (90/10), промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении и остаток очищали фильтрованием на силикагеле, если это необходимо, с получением соответствующего О-арильного производного.
Пример 2:
[Химические формулы 10]
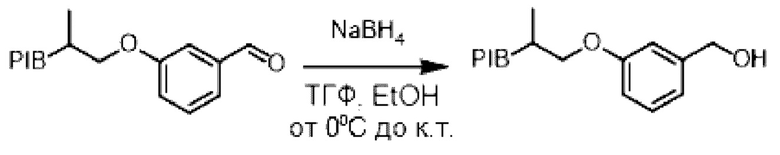
Закрепленный альдегид (1 эквивалент) растворяли в смеси (0,1 М) тетрагидрофуран/этанол (1/1) и затем охлаждали до температуры 0°С в течение 5 минут. В реакционную смесь (небольшими частями) добавляли боргидрид натрия (3 эквивалента) и затем реакционную смесь перемешивали при комнатной температуре в течение 3 0 минут. Реакционную среду концентрировали при пониженном давлении, и затем к остатку последовательно добавляли 1н раствор гидроксида натрия и циклогексан. Органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением соответствующего бензилового спирта.
Пример 3:
[Химические формулы 11]
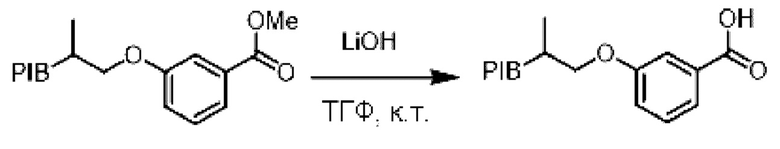
Закрепленный метиловый эфир (1 эквивалент) растворяли в смеси (0,1 М.) тетрагидрофуран/ДМСО/вода (8/1/1). В реакционную среду добавляли гидроксид лития (3 эквивалента) и затем реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь три раза экстрагировали циклогексаном и последовательно промывали раствором соляной кислоты (1 н), смесью этанол/вода (90/10), насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении и остаток очищали фильтрованием на силикагеле, если необходимо, с получением соответствующей карбоновой кислоты.
Пример 4:
[Химические формулы 12]
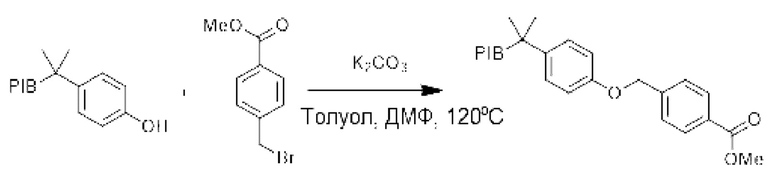
К смеси производного PIB-фенола (1 экв.) и метил-4-(бромметил)бензоата (3 экв.) в смеси (0,1 М) толуол/N-N-диметилформамид (1/1) добавляли карбонат калия (5 эквивалентов), затем реакционную смесь нагревали при температуре 120°С в течение 16 ч, затем охлаждали до комнатной температуры. Реакционную смесь три раза экстрагировали циклогексаном и последовательно промывали смесью ацетонитрил/вода или этанол/вода, насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении, и остаток очищали фильтрованием на силикагеле, если необходимо, с получением соответствующего О-арильного производного.
Пример 5:
[Химические формулы 13]
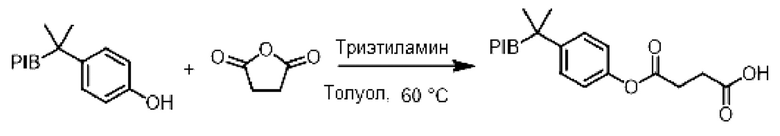
К раствору производного PIB-фенола (1 эквивалент) в толуоле (0,1 М.) при перемешивании и при комнатной температуре добавляли янтарный ангидрид (2 эквивалента), а затем триэтиламин (3 эквивалента). Реакционную смесь нагревали при температуре 60°С в течение 16 ч и затем охлаждали до комнатной температуры. После добавления раствора (1 н) соляной кислоты реакционную смесь три раза экстрагировали циклогексаном и органическую фазу последовательно промывали смесью этанол/вода (90/10), насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении и остаток очищали фильтрованием на силикагеле, если необходимо, с получением соответствующего сложного эфира.
Пример 6:
[Химические формулы 14]
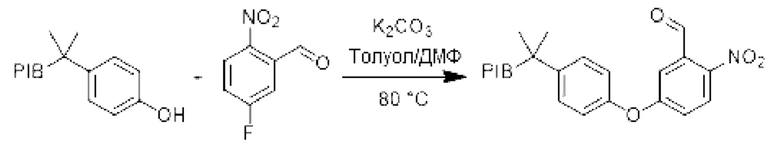
К раствору производного PIB-фенола (1 эквивалент) в толуоле/ДМФ (1/1) (0,1 М) при перемешивании и при комнатной температуре добавляли 5-фтор-2-нитробензальдегид (3 эквивалента) и затем карбонат калия (3 эквивалента). Реакционную смесь нагревали при температуре 80°С в течение 4 В ч и затем охлаждали до комнатной температуры. Реакционную смесь три раза экстрагировали циклогексаном и промывали смесью этанол/вода (90/10), насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении и остаток очищали фильтрованием на силикагеле, если необходимо, с получением соответствующего арилового эфира.
Пример 7:
[Химические формулы 15]
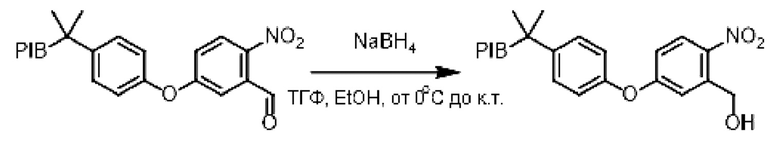
Заякоренный альдегид (1 эквивалент) растворяли в смеси (0,1 М) тетрагидрофуран/этанол (1/1) и затем в течение 5 минут охлаждали до температуры 0°С. В реакционную среду (небольшими частями) добавляли боргидрид натрия (3 эквивалента) и затем реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную среду концентрировали при пониженном давлении, и затем 1н раствор гидроксида натрия и циклогексан к остатку последовательно добавляли. Органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением соответствующего бензилового спирта.
Пример 8:
[Химические формулы 16]
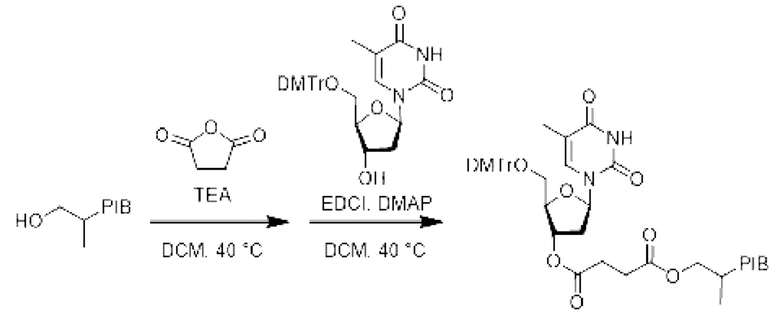
К раствору производного PIB-спирта (1 эквивалент) в дихлорметане (0,1 М) при перемешивании, в инертной атмосфере и при комнатной температуре добавляли янтарный ангидрид (1,1 эквивалент) и затем триэтиламин (1.2 эквивалент). Реакционную смесь нагревали при температуре 40°С в течение 1 В ч и затем охлаждали до комнатной температуры.
На этой стадии в реакционную среду последовательно добавляли 5'-О-(4,4'-диметокситритил)тимидин (1,1 эквивалент), гидрохлорид этил-(N,N-диметиламино) пропилкарбодиимида (EDCI) (1,1 эквивалент) и 4-(N,N-диметиламино) пиридин (DMAP) (0,5 эквивалента); затем реакционную смесь нагревали при температуре 40°С в течение 18 ч. Реакционную смесь упаривали и затем экстрагировали циклогексаном, промывали 3 раза смесью этанол/вода (90/10), насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении и остаток очищали фильтрованием на силикагеле, если необходимо, с получением соответствующего заякоренного тимидина.
Пример 9:
[Химические формулы 17]
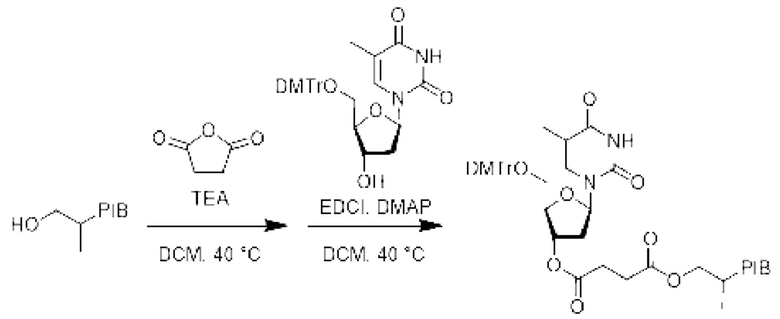
К раствору производного PIB-карбоновой кислоты (1 эквивалент) в дихлорметане при перемешивании в инертной атмосфере и при комнатной температуре добавляли 5'-О-(4,4'-диметокситритил)тимидин (1,1 эквивалент), гидрохлорид этил-(N,N-диметиламино) пропилкарбрдиимида (EDCI) (1,1 эквивалента) и 4-(N,N-диметиламино) пиридин (DMAP) (0,5 эквивалента), и затем реакционную смесь нагревали при температуре 40°С в течение 18 ч. Реакционную смесь упаривали и затем экстрагировали циклогексаном, промывали 3 раза смесью этанол/вода (90/10), насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении и остаток очищали фильтрованием на силикагеле, если необходимо, с получением соответствующего заякоренного тимидина.
Пример 10:
[Химические формулы 18]
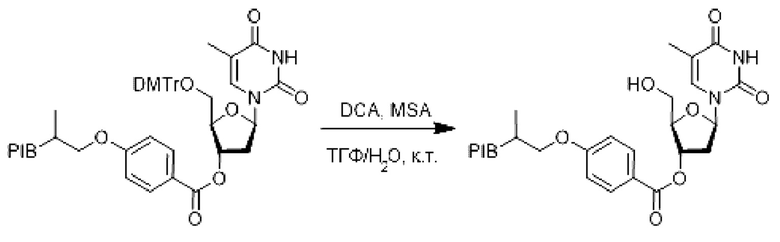
К раствору производного заякоренного производного 5'-O-(4,4'-диметокситритил) тимидина (1 эквивалент) в смеси (0,1 М) ТГФ/Н2О при комнатной температуре добавляли меркаптоянтарную кислоту (5 эквивалентов), затем дихлоруксусную кислоту (5 об. %), и затем реакционную смесь перемешивали в течение 1 ч. Реакционную смесь упаривали и затем экстрагировали циклогексаном, промывали 3 раза смесью этанол/вода (90/10), насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением производного соответствующего связанного тимидина с удаленной защитой.
Пример 11:
[Химические формулы 19]
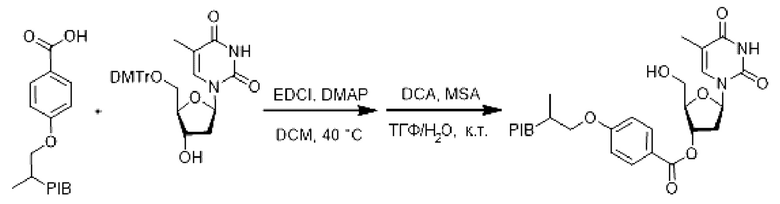
К раствору производного PIB-карбоновой кислоты (1 эквивалент) в дихлорметане (0,1 М) при перемешивании в инертной атмосфере и при комнатной температуре добавляли 5'-О-(4,4'-диметокситритил)тимидин (1,.1 эквивалент), гидрохлорид этил-(N,N-диметиламино) пропилкарбодиимида (EDCI) (1,1 эквивалент) и 4-(N,N-диметиламино) пиридин (DMAP) (0,5 эквивалент). Затем реакционную смесь нагревали при температуре 45°С в течение 18 ч и затем упаривали.
Остаток солюбилизировали в смеси (0,1 М) ТГФ/H2O (9/1) при комнатной температуре и затем последовательно добавляли меркаптоянтарную кислоту (5 эквивалентов), дихлоруксусную кислоту (5 об. %), затем реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь упаривали и затем экстрагировали циклогексаном, промывали 3 раза смесью этанол/вода (90/10), насыщенным раствором бикарбоната натрия, насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением производного соответствующего заякоренного тимидина с удаленной защитой.
Пример 12:
[Химические формулы 20]
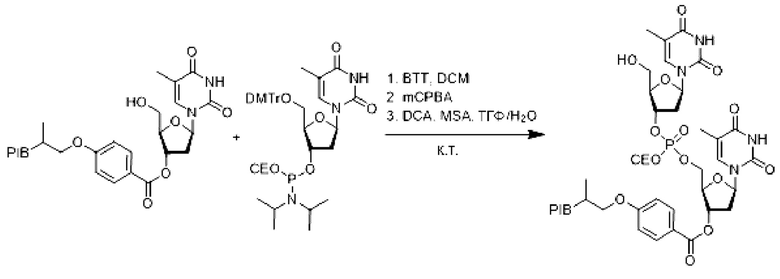
Производное заякоренного тимидина С удаленной защитой (1 эквивалент) и DMT-dT фосфорамидит (2 эквивалента) выпаривали 3 раза совместно с безводным толуолом, а затем сушили в вакууме. К остатку в инертной атмосфере добавляли дихлорметан (0,1 М), затем раствор бензилтио-1Н-тетразола (ВТТ) (4,5 эквивалента) в ацетонитриле (0,01 М) и реакционную смесь перемешивали в течение 16 ч при комнатной температуре. К реакционной среде добавляли mCPBA (3 эквивалента), затем перемешивали в течение 1 часа и затем упаривали.
На этой стадии при комнатной температуре остаток растворяли в смеси (0,1 М) ТГФ/Н2О (9/1), затем последовательно добавляли меркаптоянтарную кислоту (5 эквивалентов), дихлоруксусную кислоту (5 об. %) и затем реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь упаривали и затем экстрагировали циклогексаном, промывали 3 раза смесью этанол/вода (90/10), водным раствором бикарбоната натрия (10%), насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением димерного производного соответствующего заякоренного тимидина с удаленной защитой.
Пример 13:
[Химические формулы 21]
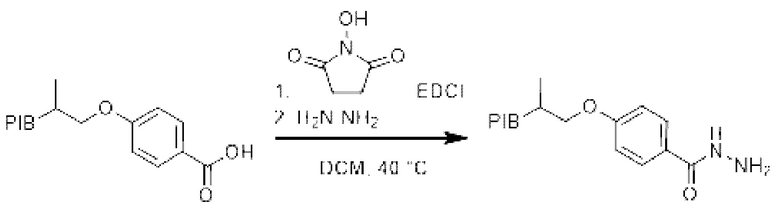
К раствору производного PIB-бензойной кислоты (1 эквивалент) в дихлорметане (0,1 М) при перемешивании в инертной атмосфере и при комнатной температуре добавляли N-гидроксисукцинимид (1,1 эквивалент), гидрохлорид этил-(N,N-диметиламино) пропилкарбодиимида (EDCI) (1,1 эквивалент) и 4-(N,N-диметиламино) пиридин (DMAP) (0,1 эквивалент), и затем реакционную смесь нагревали при температуре 40°С в течение 30 мин и затем охлаждали до комнатной температуры. В реакционную среду добавляли гидразин гидрат и затем нагревали при температуре 40°С в течение 30 мин. Реакционную смесь упаривали и затем экстрагировали циклогексаном, промывали 3 раза смесью этанол/вода (90/10), насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением соответствующего производного PIB-ацилгидразида.
Пример 14:
[Химические формулы 22]
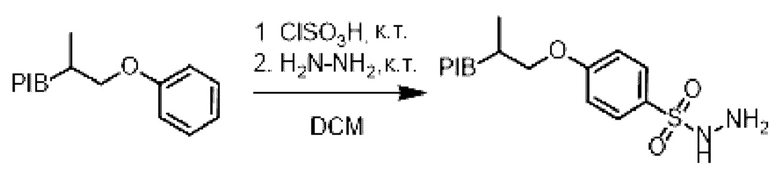
К раствору производного PIB-арила (1 эквивалент) в дихлорметане (0,1 М) при перемешивании в инертной атмосфере и при комнатной температуре добавляли по каплям хлористый сульфурин (1,1 эквивалент), затем реакционную смесь перемешивали при комнатной температуре в течение 30 мин. В реакционную среду добавляли гидразин гидрат (3 эквивалента) и затем перемешивали в течение 30 мин. Реакционную смесь упаривали и затем экстрагировали циклогексаном, промывали 3 раза смесью этанол/вода (90/10), насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением соответствующего производного PIB-сульфонилгидразида.
Пример 15:
[Химические формулы 23]
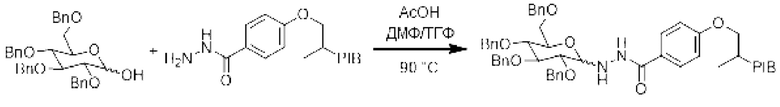
К смеси РIB-ацилгидразида (1 эквивалент) и 2,3,4,6-тетра-О-бензил-D-глюкопиранозы (1,5 эквивалента) в смеси (0,1 М) ДМФ/ТГФ (9/1) добавляли уксусной кислоту (0,01 эквивалента) и затем реакционную смесь нагревали при температуре 90°С в течение 18 часов. Реакционную среду выпаривали и затем экстрагировали циклогексаном, промывали 3 раза смесью этанол/вода (90/10), насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением соответствующего производного PIB-гликозилгидразида.
Пример 16:
[Химические формулы 24]
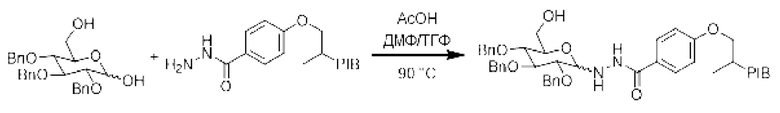
К смеси PIB-ацилгидразида (1 эквивалент) и 2,3,4-три-О-бензил-D-глюкопиранозы (1,5 эквивалента) в смеси (0,1 М) ДМФА/ТГФ (9/1) добавляли уксусную кислоту (0,01 эквивалента), затем реакционную смесь нагревали при температуре 90°С в течение 18 часов. Реакционную среду выпаривали и затем экстрагировали циклогексаном, промывали 3 раза смесью этанол/вода (90/10), насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении с получением соответствующего производного PIB-гликозилгидразида.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДОВ, ИЛИ БЕЛКОВ, ИЛИ ПЕПТИДОМИМЕТИКОВ | 2020 |
|
RU2828026C2 |
| АНАЛОГИ L-РИБО-ЗНК | 2000 |
|
RU2258708C2 |
| МЕТАЛЛИРОВАННЫЙ ЩЕЛОЧНЫМ МЕТАЛЛОМ ПОЛИОЛЕФИН И СПОСОБ ПОЛУЧЕНИЯ БЛОКСОПОЛИМЕРА | 1992 |
|
RU2107697C1 |
| КОНЪЮГАТЫ ОЛИГОНУКЛЕОТИДОВ | 2013 |
|
RU2653438C2 |
| НУКЛЕИНОВЫЕ КИСЛОТЫ, МОДИФИЦИРОВАННЫЕ АМИНОКИСЛОТАМИ | 1995 |
|
RU2154638C2 |
| КОНЪЮГАТЫ УГЛЕВОДА И LNA-ОЛИГОНУКЛЕОТИДА | 2014 |
|
RU2649367C2 |
| БИЦИКЛИЧЕСКИЕ АНАЛОГИ НУКЛЕОЗИДОВ, НУКЛЕОТИДОВ И ОЛИГОНУКЛЕОТИДОВ | 1998 |
|
RU2243231C2 |
| 3' И/ИЛИ 2'-АМИНО- ИЛИ ТИОЛМОДИФИЦИРОВАННЫЕ НУКЛЕОЗИДЫ, НУКЛЕОТИДЫ ИЛИ ОЛИГОНУКЛЕОТИДЫ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ | 1991 |
|
RU2073682C1 |
| РЕЗИСТЕНТНЫЕ К НУКЛЕАЗЕ ОЛИГОНУКЛЕОЗИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И РЕЗИСТЕНТНЫЙ К НУКЛЕАЗЕ НУКЛЕОЗИДНЫЙ ДИМЕР | 1991 |
|
RU2131436C1 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОКИСЛОТНЫХ ИЛИ НУКЛЕИНОВОКИСЛОТНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ НА СУБСТРАТЕ, СПОСОБ СКРИНИНГА БОЛЬШОГО КОЛИЧЕСТВА АМИНОКИСЛОТНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ, СУБСТРАТ ДЛЯ СКРИНИНГА | 1990 |
|
RU2107072C1 |
Изобретение относится к синтезу макромолекул, состоящих из моносахаридных или олигосахаридных звеньев. Предложен способ синтеза макромолекул, состоящих из звеньев, представляющих собой моносахариды или производные моносахаридов, путём последовательного удлинения цепи на мономер или олигомер, обладающий по меньшей мере двумя функциональными группами, где на первой стадии происходит заякоривание мономера или концевого звена олигомера якорной молекулой с последующим удалением защитной группы, оставляя свободную функциональную группу, к которой затем присоединяется второй мономер или олигомер, который может содержать свободную функциональную группу, защищённую защитной группой, при этом указанная якорная молекула содержит полиолефиновую цепь, или полиолефиновый олигомер, или полиалкен с по меньшей мере от 10 до 50 мономерными звеньями, указанная полиолефиновая цепь представляет собой разветвленную цепь и предпочтительно полиизобутеновую цепь; а также применение указанного способа для синтеза олигонуклеотидов или олигосахаридов. Технический результат – упрощение способа синтеза макромолекул, состоящих из моносахаридных или олигосахаридных звеньев, с уменьшением воздействия на окружающую среду. 2 н. и 10 з.п. ф-лы, 16 пр.
1. Способ синтеза макромолекул, состоящих из звеньев U, которые в основном представляют собой моносахариды или производные моносахаридов, которые могут быть идентичными или разными, при этом указанные макромолекулы имеют первый и второй конец, где указанный способ синтеза заключается в последовательном удлинении указанного второго конца на мономер или олигомер М, обладающий по меньшей мере двумя функциональными группами, причем указанный способ характеризуется следующим:
- на первой так называемой стадии заякоривания первое звено U1 указанной макромолекулы, соответствующее мономеру M1 или концевому звену олигомера M1, присоединяется ковалентной связью к якорной молекуле, растворимой в органических растворителях, указанная ковалентная связь является результатом взаимодействия первой из функциональных групп указанного мономера M1 или указанного олигомера M1 с функциональной группой указанной якорной молекулы, второй конец, возможно, представляет собой другую функциональную группу указанного мономера M1 или указанного олигомера M1, которая была защищена до указанного взаимодействия по меньшей мере с первой защитной группой GP1 и, возможно, со второй защитной группой GP2;
- на второй так называемой стадии удаления защиты удаляют одну из указанных защитных групп GP1 или GP2, оставляя так называемую свободную функциональную группу на указанном мономере M1 или олигомере M1;
- на третьей так называемой стадии связывания второй мономер М2 или олигомер М2, несущий по меньшей мере одну свободную функциональную группу и по меньшей мере одну функциональную группу, защищенную защитной группой GP3, вводят в реакцию, так что свободная функциональная группа образует в результате реакции с указанной свободной функциональной группой указанного первого мономера M1 или олигомера M1 ковалентную связь, создавая таким образом новую молекулу, образованную указанным мономером M1 или олигомером M1, присоединенным своим первым концом к указанной якорной молекуле, и указанным мономером М2 или олигомером М2, присоединенным к другому ее концу, и указанный способ отличается тем, что указанная якорная молекула содержит полиолефиновую цепь, или полиолефиновый олигомер, или полиалкен с по меньшей мере от 10 до 50 мономерными звеньями, указанная полиолефиновая цепь представляет собой разветвленную цепь.
2. Способ по п. 1, отличающийся тем, что указанная полиолефиновая цепь представляет собой полиизобутеновую цепь.
3. Способ по п. 1, отличающийся тем, что n-й мономер Mn или олигомер Mn добавляют путем связывания.
4. Способ по любому из пп. 1-3, отличающийся тем, что после связывания последнего мономера или олигомера проводят удаление защитных групп макромолекулы.
5. Способ по любому из пп. 1-3, включающий по меньшей мере одну стадию, на которой указанную макромолекулу, присоединенную к указанной якорной молекуле, выделяют из реакционной среды путем экстракции в неполярном органическом растворителе и/или путем экстракции или промывания полярным растворителем и/или фильтрованием.
6. Способ по любому из пп. 1-3, включающий стадию полного удаления защиты у указанной макромолекулы.
7. Способ по любому из пп. 1-3, отличающийся тем, что указанные моносахаридные звенья или моносахаридные производные представляют собой производные пентозы и, в частности, нуклеозида или гексозы.
8. Способ по любому из пп. 1-3, отличающийся тем, что связь между двумя последовательными звеньями представляет собой связь озидного типа и, предпочтительно, гликозидного типа или фосфорилированного типа, в частности оза-1-фосфатного типа, или углеводного типа, или N-гетерозидного типа, или S-гетерозидного типа.
9. Способ по любому из пп. 1-3, отличающийся тем, что указанная якорная молекула имеет среднемассовую молекулярную массу от 300 до 20000, предпочтительно, от 500 до 15000.
10. Способ по любому из пп. 1-3, отличающийся тем, что указанная полиолефиновая или полиолефиновая олигомерная или полиалкеновая цепь содержит ненасыщенные углерод-углеродные связи в количестве, не превышающем 5% и предпочтительно не превышающем 3%.
11. Способ по любому из пп. 1-3, отличающийся тем, что указанную полиолефиновую цепь, или полиолефиновый олигомер, или полиалкен получают путем полимеризации мономера предпочтительно биологического происхождения.
12. Применение способа по любому из пп. 1-11 для синтеза олигонуклеотидов или олигосахаридов.
| Kim S | |||
| et al | |||
| Liquid‐Phase RNA Synthesis by Using Alkyl‐Chain‐Soluble Support | |||
| Chemistry-A European Journal | |||
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| EP 2857412 B1, 11.01.2017 | |||
| Gravert D | |||
| J | |||
| et al | |||
| Organic synthesis on soluble polymer supports: liquid-phase methodologies | |||
| Chemical reviews | |||
| Электрическое сопротивление для нагревательных приборов и нагревательный элемент для этих приборов | 1922 |
|
SU1997A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Takahashi D | |||
| et al | |||
| AJIPHASE®: a | |||

