Настоящее изобретение относится к способам получения декаборана B10H14, который является единственным бороводородным предшественником в синтезе и производстве незамещенных и замещенных карборанов общей формулы RCB10H10CR, используемых в качестве компонентов специальных составов с уникальными свойствами, высокотермостойких полимеров и суперклеев. Декаборан также является исходным продуктом в синтезе солей стабильных анионов клозо-декабората [B10H10]2- и клозо-додекабората [В12Н12]2-, применяемых в медицине при нейтронозахватной терапии злокачественных опухолей.
Известен способ получения декаборана пиролизом диборана В2Н6 при температуре 180-250°C при пропускании газообразного диборана над нагретой поверхностью. При этом наряду с декабораном образуются другие гидриды бора - тетраборан B4H10, пентаборан В5Н9 и твердые высшие гидриды бора (ВН)n, в количествах, сравнимых с количеством декаборана (40-60%). Кристаллический декаборан осаждается на холодных стенках трубы аппарата пиролиза, а непрореагировавший диборан и другие гидриды бора возвращаются в реакционную зону путем рециркуляции в токе водорода. Получаемый декаборан растворяют в органическом растворителе (гексан, бензол, толуол) и хранят в растворе (Патент США №3078530, МПК С01В 35/18, 1963).
Недостатками способа являются использование ядовитых легковоспламеняющихся газов и образование твердых высших бороводородов, что снижает выход и затрудняет выделение декаборана, кроме того, этот способ требует высоких энергетических затрат.
Известен способ получения декаборана пиролизом диборана под давлением 28 атм и температуре 82°C в присутствии растворителя 2,3-диметилбутана. Полученный раствор смеси бороводородов охлаждают, кристаллический декаборан отфильтровывают, а маточник возвращают в процесс пиролиза (Патент США №3169829, МПК С01В 35/18, 1965).
Недостатками способа являются использование высокотоксичного и взрывоопасного газа диборана, необходимость применения высокого давления и образование смеси более низких гидридов бора.
Известен способ синтеза декаборана с использованием излучения химического лазера (DF) из диборана. Диборан перекачивают в реактор, охлаждая до -126°C. Сам реактор представляет собой металлический цилиндр длиной 10 см, снабженный окошком диаметром 3 см из NaCl или KCl. При давление в реакторе 10 Торр происходит облучение лазером диборана в течении 13 сек. При этом наблюдается незначительный нагрев реактора. Продукт в виде чисто-белых кристаллов осаждается на стенках реакционной камеры, после чего из камеры удаляются остатки газа, а продукт смывается метилциклогексаном для его последующей перекристаллизации в сухой камере.
Недостатками данного способа является малый выход и невозможность получения продуктов в промышленных масштабах, а также работа с высокотоксичным и взрывоопасным дибораном и образование не менее высокоопасного пентаборана(9) (Патент США №4144151, МПК С01В 35/02, 1979).
Известен непиролитический метод получения декаборана через промежуточную соль ундекаборат натрия NaB11H14. Ундекаборат натрия NaB11H14 получают путем конденсации боргидрида натрия NaBH4 под действием кислот Льюиса. В качестве кислоты Льюиса используют трехфтористый бор BF3, который пропускают прямо в реакционную смесь (суспензия боргидрида натрия в диглиме). Газообразный трехфтористый бор поглощают диглимом, полученный диглимат трехфтористого бора реагирует с боргидридом натрия, образуя раствор ундекабората натрия и суспензию борфторида натрия NaBF4. Путем азеотропной отгонки с водой диглим заменяют на воду, водный раствор ундекабората натрия подкисляют серной кислотой и окисляют диоксидом марганца MnO2, или хромовым ангидридом CrO3, или солью трехвалентного железа, а декаборан, образовавшийся в процессе, экстрагируют из реакционной смеси неполярным органическим растворителем и после отгонки растворителя подвергают вакуумной возгонке (Патент РФ №2346890, МПК С01В 35/18, 2009).
Недостатками способа являются образование больших объемов нерастворимых и токсичных отходов (NaBF4, MnO2, MnSO4, Cr2(SO4)3), что сильно затрудняет экстракцию декаборана и приводит к потери целевого продукта, азеотропная отгонка диглима с водой способствует удорожанию декаборана, т.к. возникает необходимость в регенерации растворителя, а также сложность работы с газообразным трехфтористым бором.
Наиболее близким по технической сущности и принятым нами в качестве прототипа является способ получения декаборана из боргидрида натрия NaBH4 и алкилгалогенидов (C5H11Br и др.) в качестве кислот Льюиса в диглиме при температуре 100-110°C с образованием раствора ундекабората натрия NaB11H14. Получаемый ундекаборат натрия, освобожденный от диглима азеотропной отгонкой с водой, окисляют в водной среде в присутствии разбавленной серной кислоты. В качестве окислителя используют перманганат калия (KMnO4), бихромат натрия (Na2Cr2O7) или перекись водорода (Н2О2) в присутствии соли трехвалентного железа. Образующийся декаборан экстрагируют из реакционной смеси бензолом, или гексаном, или диэтиловым эфиром (Патент США №4115521, кл. С01В 35/18, 1978; G.B. Dunks, K. Barker et al. Inorg. Chem., 20, 1692, 1981).
Недостатками способа являются:
а) низкий выход целевого продукта;
б) необходимость отделения бромида натрия, так как он реагирует с вышеупомянутыми окислителями в присутствии серной кислоты с образованием газообразного брома, что приводит к бромированию декаборана;
в) образование значительных количеств токсичных побочных продуктов;
г) невозможность использования толуола в качестве экстрагента, так как используемые окислители реагируют с ним в условиях реакции.
Задачей изобретения является повышение выхода декаборана за счет создания эффективного, технологически простого экологически чистого и безопасного способа его получения.
Для решения поставленной задачи предложен способ получения декаборана из боргидрида натрия и алкилгалогенида с промежуточным образованием ундекабората натрия, который осуществляют путем нагревания раствора диглима и боргидрида натрия до температуры 105°С, при этом к раствору прикапывают алкилгалогенид, после добавления всего алкилгалогенида продолжают перемешивание реакционной массы в течение 60 минут при той же температуре, полученный продукт продувают и охлаждают в токе азота до комнатной температуры, при этом диглим и непрореагировший алкилгалогенид отгоняют при остаточном давлении 10-15 мм рт. ст. и температуре 60-90°С до получения твердого остатка, отличающийся тем, что к полученному охлажденному твердому остатку прибавляют углеводородный растворитель и перемешивают, при этом в реакционную массу при комнатной температуре по каплям добавляют 40% водный раствор серной кислоты, далее реакционную смесь выдерживают при температуре 50°С до полного прекращения газовыделения, полученный кислый раствор диглимата ундекабората натрия охлаждают до температуры 35°С, причем в реакционную массу прикапывают окислитель в течение 15-185 минут, а в качестве окислителя используют органические соединения, содержащие карбонильную группу (альдегиды и кетоны), при этом процесс окисления происходит в присутствии образующегося в результате реакции конденсации галогенида натрия, после окончания подачи окислителя останавливают перемешивание и выдерживают реакционную массу до ее расслоения, нижний водный слой удаляют, верхний органический - промывают дистиллированной водой и отгоняют углеводородный растворитель до концентрации декаборана 17-20%.
Предлагаемый способ в сравнении с прототипом позволяет повысить выход декаборана на 15-25%, упростить стадию окисления ундекабората натрия, существенно сократить время ведения процесса, уменьшить количество отходов и снизить стоимость продукта за счет использования в качестве окислителя ундекабората натрия широкодоступных и дешевых органических соединений класса альдегидов и кетонов. При этом процесс окисления-восстановления может проходить в присутствии побочного продукта реакции конденсации бромида натрия, что позволяет исключить стадию фильтрации осадка и экстракцию из него целевого продукта. Водные отходы процесса не содержат токсичных солей тяжелых металлов хрома и марганца, а низкокипящие органические соединения легко удаляются с помощью азеотропной отгонки. Кроме того, возможно осуществлять окисление ундекабората натрия в ароматических растворителях, включая толуол как наиболее дешевый и распространенный растворитель, так как заявленные органические окислители хорошо растворяются в таких растворителях и реакция окисления проходит в гомогенной среде.
Процесс окисления ундекабората натрия протекает по следующим химическим реакциям:
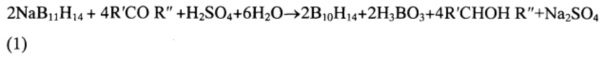
где R'=Rʺ=-Н, -СН3, -С6Н5;
R'=-СН3, Rʺ=-С2Н5;
R'=-Н, Rʺ=-С6Н5;
R'=-СН3, Rʺ=-СН2СОСН3;


Способ осуществляют следующим образом. В продутую азотом четырехгорлую круглодонную колбу, снабженную мешалкой, прямым холодильником, термометром, капельной воронкой, загружают осушенный диглим, боргидрид натрия, нагревают до температуры 100-105°С и затем медленно прикапывают галоидный алкил. По окончании прикапывания галоидного алкила реакционную смесь выдерживают 1 час при температуре 100-105°С, охлаждают в токе азота, после чего отгоняют диглим и непрореагировавший алкилгалогенид при остаточном давлении 10-15 мм рт. ст. и температуре 60-90°С. Далее к полученному маслянистому твердому остатку, содержащему ундекаборат натрия и бромид натрия, прибавляют толуол и разбавленную серную кислоту. Затем при перемешивании и охлаждении прикапывают окислитель - соединение, содержащее карбонильную группу. Органический слой отделяют, промывают водой, растворитель упаривают до концентрации декаборана 17-20%. Продукт анализируют методами хроматографического анализа и 11В, 1Н ЯМР-спектроскопией.
Сущность предлагаемого изобретения иллюстрируется следующими примерами.
Пример 1. Получение ундекабората натрия.
В четырехгорлую реакционную колбу объемом 2 литра, предварительно продутую азотом, снабженную мешалкой, прямым холодильником, капельной воронкой и термометром, загружают 1000 мл осушенного диглима и 120 г (3,17 моля) боргидрида натрия. В полученный раствор, нагретый до 105°C, медленно прикапывают 480 мл (3,85 моля) бромпентана. Выделение водорода контролируют по барботеру, заполненному водным раствором этанола в соотношении 1:1. После добавления всего бромпентана продолжают перемешивание реакционной массы в течение 1 часа при температуре 105°C, после чего полученный продукт продувают и охлаждают в токе азота до комнатной температуры. Для количественной оценки содержания ундекабората натрия, который находится в форме диглимата, из реакционной массы отфильтровывают осадок бромистого натрия, а из маточника отбирают аликвоту и обрабатывают тетраэтиламинбромидом. По количеству выпавшей в осадок соли тетраэтиламинундекабората после фильтрования и сушки судят о выходе реакции, который составляет 30,17 г (0,19 моля) (67%).
Пример 2. Окисление диглимата ундекабората натрия формальдегидом.
Стадию получения диглимата ундекабората натрия ведут, как указано в примере 1, исключая стадию фильтрования бромида натрия. К полученному охлажденному твердому остатку, оставшемуся после отгонки растворителя и алкилгалогенида, прибавляют 320 мл толуола и включают перемешивание. При комнатной температуре в реакционную массу по каплям добавляют 1600 г 40% водного раствора серной кислоты. При этом наблюдается обильное пенообразование, газовыделение, разогрев реакционной смеси. Реакционную смесь выдерживают при температуре 50°C до полного прекращения газовыделения. Полученный кислый раствор диглимата ундекабората натрия охлаждают до температуры 35°C и медленно прикапывают 60 мл (0,21 моль) формальдегида в течение 100 минут. После окончания подачи водного раствора формальдегида выключают перемешивание и выдерживают до расслоения реакционной массы. Нижний водный слой удаляют, а верхний органический - промывают дистиллированной водой, из толуольного раствора отгоняют растворитель, а полученный декаборан возгоняют в вакууме. Выход декаборана 18,3 г (0,15 моля) (78,9% в расчете на ундекаборат натрия).
Пример 3. Окисление диглимата ундекабората натрия ацетоном.
Стадию получения диглимата ундекабората натрия ведут, как указано в примере 1, исключая стадию фильтрования бромида натрия. К полученному охлажденному твердому остатку, оставшемуся после отгонки растворителя и алкилгалогенида, прибавляют 320 мл толуола и включают перемешивание. При комнатной температуре в реакционную массу по каплям добавляют 1600 г 40% водного раствора серной кислоты. При этом наблюдается обильное пенообразование, газовыделение, разогрев реакционной смеси. Затем выдерживают реакционную смесь при температуре 50°C до полного прекращения газовыделения. Полученный кислый раствор диглимата ундекабората натрия охлаждают до температуры 35°C и медленно прикапывают 100 мл (1,36 моль) ацетона в течении 105 минут. После окончания подачи окислителя выключают перемешивание и выдерживают до расслоения реакционной массы. Нижний водный слой удаляют, а верхний органический - промывают дистиллированной водой, затем из толуольного раствора отгоняют растворитель, а полученный декаборан возгоняют в вакууме. Выход декаборана - 19,8 г (0,16 моль) (84,2% в расчете на ундекаборат натрия).
Пример 4. Окисление диглимата ундекабората натрия ацетилацетоном.
Стадию получения диглимата ундекабората натрия ведут, как указано в примере 1, исключая стадию фильтрования бромида натрия. К полученному охлажденному твердому остатку, оставшемуся после отгонки растворителя и алкилгалогенида, прибавляют 320 мл толуола и включают перемешивание. При комнатной температуре в реакционную массу по каплям добавляют 1600 г 40% водного раствора серной кислоты. При этом наблюдается обильное пенообразование, газовыделение, разогрев реакционной смеси. Реакционную смесь выдерживают при температуре 50°C до полного прекращения газовыделения. Полученный кислый раствор диглимата ундекабората натрия охлаждают до температуры 35°C и медленно прикапывают 153 мл (1,50 моль) ацетиацетона в течение 100 минут. После окончания подачи ацетилацетона выключают перемешивание и выдерживают до расслоения реакционной массы. Нижний водный слой удаляют, а верхний органический - промывают дистиллированной водой, из толуольного раствора отгоняют растворитель, а полученный декаборан возгоняют в вакууме. Выход декаборана 11,0 г (0,09 моля) (47,4% в расчете на ундекаборат натрия).
Пример 5. Окисление диглимата ундекабората натрия бензальдегидом.
Стадию получения диглимата ундекабората натрия ведут, как указано в примере 1, исключая стадию фильтрования бромида натрия. К полученному охлажденному твердому остатку, оставшемуся после отгонки растворителя и алкилгалогенида, прибавляют 320 мл толуола и включают перемешивание. При комнатной температуре в реакционную массу по каплям добавляют 1600 г 40% водного раствора серной кислоты. При этом наблюдается обильное пенообразование, газовыделение, разогрев реакционной смеси. Реакционную смесь выдерживают при температуре 50°C до полного прекращения газовыделения. Полученный кислый раствор диглимата ундекабората натрия охлаждают до температуры 35°C и медленно прикапывают 54 мл (0,53 моль) бензальдегида в течение 50 минут. После окончания подачи окислителя выключают перемешивание и выдерживают до расслоения реакционной массы. Нижний водный слой удаляют, а верхний органический - промывают дистиллированной водой, толуольный слой концентрируют и анализируют на хроматографе на содержание основного вещества. Выход декаборана 13,6 г (0,11 моля) (57,9% в расчете на ундекаборат натрия).
Пример 6. Окисление диглимата ундекабората натрия циклогексаноном.
Стадию получения диглимата ундекабората натрия ведут, как указано в примере 1, исключая стадию фильтрования бромида натрия. К полученному охлажденному твердому остатку, оставшемуся после отгонки растворителя и алкилгалогенида, прибавляют 320 мл толуола и включают перемешивание. При комнатной температуре в реакционную массу по каплям добавляют 1600 г 40% водного раствора серной кислоты. При этом наблюдается обильное пенообразование, газовыделение, разогрев реакционной смеси. Реакционную смесь выдерживают при температуре 50°C до полного прекращения газовыделения. Полученный кислый раствор диглимата ундекабората натрия охлаждают до температуры 35°C и медленно прикапывают 45,6 мл (0,44 моль) циклогексанона в течение 15 минут. После окончания подачи циклогексанона выключают перемешивание и выдерживают до расслоения реакционной массы. Нижний водный слой удаляют, а верхний органический - промывают дистиллированной водой, из толуольного раствора отгоняют растворитель, а полученный декаборан возгоняют в вакууме. Выход декаборана 10,8 г (0,09 моля) (47,4% в расчете на ундекаборат натрия).
Пример 7. Окисление диглимата ундекабората натрия циклогександионом.
Стадию получения диглимата ундекабората натрия ведут, как указано в примерах 1, исключая стадию фильтрования бромида натрия. К полученному охлажденному твердому остатку, оставшемуся после отгонки растворителя и алкилгалогенида, прибавляют 320 мл толуола и включают перемешивание. При комнатной температуре в реакционную массу по каплям добавляют 1600 г 40% водного раствора серной кислоты. При этом наблюдается обильное пенообразование, газовыделение, разогрев реакционной смеси. Реакционную смесь выдерживают при температуре 50°C до полного прекращения газовыделения. Полученный кислый раствор диглимата ундекабората натрия охлаждают до температуры 35°C и порциями добавляют 79,9 г (0,71 моль) циклогександиона в течение 185 минут. После окончания подачи циклогександиона выключают перемешивание и выдерживают до расслоения реакционной массы. Нижний водный слой удаляют, а верхний органический - промывают дистиллированной водой, из толуольного раствора отгоняют растворитель, а полученный декаборан возгоняют в вакууме. Выход декаборана 13,8 г (0,11 моля) (57,9% в расчете на ундекаборат натрия).
Выход декаборана по предлагаемому способу определяется выбором окислителя и может достигать до 80% при использовании формальдегида и более 84% при использовании ацетона. Эти окислители являются наиболее предпочтительными, учитывая также их доступность, невысокую стоимость и простоту удаления из готового продукта.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДЕКАБОРАНА | 2006 |
|
RU2346890C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1,2-БИС(ГИДРОКСИМЕТИЛ)-О-КАРБОРАНА | 2011 |
|
RU2486191C1 |
| СПОСОБ ПОЛУЧЕНИЯ КЛОЗО-БОРАТНЫХ КЛАСТЕРОВ | 2007 |
|
RU2344070C1 |
| Способ получения солей додекагидрододекабората BH | 2016 |
|
RU2647733C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИМЕТИЛАМИНБОРАНА | 2004 |
|
RU2259373C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИЭТИЛАМИДА НИКОТИНОВОЙ КИСЛОТЫ | 2000 |
|
RU2182903C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-N-МЕТИЛАМИНО-1-ФЕНИЛ-1-ПРОПАНОЛА | 2007 |
|
RU2331632C1 |
| Способ получения замещенных гетероциклом производных 5сульфамоилбензойной кислоты | 1975 |
|
SU634664A3 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ГИДРОКСИАДАМАНТАН-4-ОНА (ЛЕКАРСТВЕННОЕ СРЕДСТВО "КЕМАНТАН") | 1994 |
|
RU2104994C1 |
| СПОСОБ ПОЛУЧЕНИЯ α,ω-БИС(МЕТИЛДИФЕНИЛСИЛИЛ)ОЛИГОДИОРГАНОСИЛОКСАНОВ | 2011 |
|
RU2471818C1 |
Изобретение относится к химической промышленности и может быть использовано в синтезе и производстве незамещенных и замещенных карборанов общей формулы RCB10H10CR. Сначала нагревают раствор диглима и боргидрида натрия до 105°С, прикапывая алкилгалогенид. После добавления всего алкилгалогенида реакционную массу перемешивают в 60 минут при той же температуре. Полученный продукт продувают и охлаждают в токе азота до комнатной температуры. Диглим и непрореагировавший алкилгалогенид отгоняют при остаточном давлении 10-15 мм рт. ст. и температуре 60-90°С до получения твердого остатка. К охлажденному твердому остатку прибавляют углеводородный растворитель, перемешивают, добавляют по каплям 40% водный раствор серной кислоты при комнатной температуре. Затем реакционную смесь выдерживают при 50°С до полного прекращения газовыделения. Полученный кислый раствор диглимата ундекабората натрия охлаждают до 35°С. После этого в течение 15-185 минут прикапывают окислитель - органические соединения, содержащие карбонильную группу (альдегиды и кетоны). Окисление происходит в присутствии галогенида натрия, образующегося в результате реакции конденсации. После окончания подачи окислителя останавливают перемешивание и выдерживают реакционную массу до расслоения, затем нижний водный слой удаляют, верхний органический промывают дистиллированной водой и отгоняют углеводородный растворитель до концентрации декаборана В10Н14 17-20%. Изобретение позволяет повысить выход декаборана на 15-25%, упростить стадию окисления ундекабората натрия, сократить время процесса и уменьшить количество отходов. 7 пр.
Способ получения декаборана из боргидрида натрия и алкилгалогенида с промежуточным образованием ундекабората натрия осуществляют путем нагревания раствора диглима и боргидрида натрия до температуры 105°С, при этом к раствору прикапывают алкилгалогенид, после добавления всего алкилгалогенида продолжают перемешивание реакционной массы в течение 60 минут при той же температуре, полученный продукт продувают и охлаждают в токе азота до комнатной температуры, при этом диглим и непрореагировавший алкилгалогенид отгоняют при остаточном давлении 10-15 мм рт. ст. и температуре 60-90°С до получения твердого остатка, отличающийся тем, что к полученному охлажденному твердому остатку прибавляют углеводородный растворитель и перемешивают, при этом в реакционную массу при комнатной температуре по каплям добавляют 40% водный раствор серной кислоты, далее реакционную смесь выдерживают при температуре 50°С до полного прекращения газовыделения, полученный кислый раствор диглимата ундекабората натрия охлаждают до температуры 35°С, причем в реакционную массу прикапывают окислитель в течение 15-185 минут, причем в качестве окислителя используют органические соединения, содержащие карбонильную группу (альдегиды и кетоны), при этом процесс окисления происходит в присутствии образующегося в результате реакции конденсации галогенида натрия, после окончания подачи окислителя останавливают перемешивание и выдерживают реакционную массу до расслоения, затем нижний водный слой удаляют, верхний органический промывают дистиллированной водой и отгоняют углеводородный растворитель до концентрации декаборана 17-20%.
| US 4115521 A, 19.09.1978 | |||
| СПОСОБ ПОЛУЧЕНИЯ ДЕКАБОРАНА | 2006 |
|
RU2346890C2 |
| US 3078530 A, 26.02.1963 | |||
| US 3169829 A, 16.02.1965 | |||
| US 4144151 A, 13.03.1979 | |||
| В.В.СУХОВЕЙ, Перспективы промышленного производства соединений додекагидро-клозо-додекаборатного аниона, ХI Международная конференция студентов и молодых ученых "Перспективы развития фундаментальных наук", Томск, 2014, стр.503-505. | |||

