Область техники, к которой относится изобретение
Перекрестная ссылка на родственные заявки
По данной заявке испрашивается приоритет в соответствии с заявкой на выдачу патента Японии №2012-148850, поданной 2 июля 2012 года, и с заявкой на выдачу патента Японии №2012-215902, поданной 28 сентября 2012 года, полное содержание которых включено в настоящий документ посредством ссылки.
Настоящее изобретение относится к усилителю противоопухолевого эффекта и противоопухолевому лекарственному средству, содержащему комбинацию указанного усилителя и одного или нескольких других противоопухолевых средств.
Предпосылки создания изобретения
Широкий спектр лекарственных средств был определен для использования в качестве противоопухолевых средств. Приближенно они делятся на алкилирующие агенты, соединения на основе платины, антиметаболиты, ингибиторы топоизомеразы, ингибиторы микротрубочек, противоопухолевые антибиотики, лекарственные средства, направленные на молекулярные мишени, и т.п. Кроме того, вместо введения одного противоопухолевого средства в последние годы широко распространено комбинированное применение нескольких лекарственных средств.
AKT представляет собой серин/треонин-специфичную киназу, которая служит в качестве нижележащего эффектора в отношении фосфатидилинозитол-3-киназы (PI3-киназы), которая активируется сигналом от рецепторной тирозинкиназы. AKT часто активирована или высоко экспрессирована при многих злокачественных опухолях (почечно-клеточный рак, рак желудка, рак молочной железы, рак легких, колоректальный рак, рак поджелудочной железы, рак яичников, печеночно-клеточный рак, множественная миелома, лимфома, лейкоз, рак головы и шеи, меланома и т.п.), и при некоторых злокачественных опухолях сообщалось о генетической амплификации или активирующей мутации (непатентный документ 1). Что касается функции, то сообщается, что AKT играет важную роль в образовании опухоли, а именно, в пролиферации клеток, устойчивости клеток к апоптозу, ангиогенезе, метастазировании и инвазии, а также в метаболизме глюкозы и метаболизме липидов (непатентный документ 2). Также сообщается, что AKT высоко экспрессирована при опухолях, невосприимчивых или устойчивых к существующим способам лечения с использованием противоопухолевых средств. Таким образом, можно надеяться на эффективность комбинированного применения ингибитора AKT и существующих противоопухолевых средств, включая противоопухолевые средства, направленные на молекулярную мишень (непатентный документ 3).
Например, имеются сообщения о способах лечения с использованием комбинации MK-2206, который представляет собой ингибитор AKT, и доцетаксела (непатентный документ 4 и патентный документ 1).
Перечень ссылок
Патентный документ
Патентный документ 1: публикация заявки на выдачу патента США №2011-0319354
Непатентные документы
Непатентный документ 1: Annals of Oncology, 21, p. 683-691 (2010)
Непатентный документ 2: Cell, 129, p. 1261-1274 (2007)
Непатентный документ 3: Drug Resistance Updates, 11, p. 32-50 (2008)
Непатентный документ 4: Mol. Cancer Ther., 9, p. 1956-1967 (2010)
Краткое описание сущности изобретения
Техническая задача
При этом, совершенно неизвестно и непредсказуемо, какая комбинация противоопухолевых средств может усиливать их противоопухолевые эффекты, или, в случае усиления противоопухолевых эффектов, происходит ли также усиление токсичности средств, как и усиление противоопухолевых эффектов.
Задача настоящего изобретения заключается в обеспечении комбинированного применения двух противоопухолевых средств, которые могут усиливать их противоопухолевые эффекты без значительного усиления их побочных эффектов.
Решение задачи
В свете данной проблемы, авторы настоящего изобретения сосредоточили внимание на специфическом типе ингибитора AKT и провели исследование эффекта комбинированного применения соединения и других противоопухолевых средств. В результате, авторы изобретения обнаружили, что имидазооксазиновое соединение, представленное ниже формулой (I), или его фармацевтически приемлемая соль, действует как высокоэффективный ингибитор AKT, и что комбинированное применение соединения и противоопухолевого(ых) средства(средств) прекрасно усиливает противоопухолевый эффект, увеличивая тем самым эффективную площадь действия и спектр противоопухолевого эффекта. Полученные результаты были положены в основу настоящего изобретения.
Вариант осуществления настоящего изобретения относится к усилителю противоопухолевого эффекта для усиления одного или нескольких других противоопухолевых средств, содержащих в качестве активного ингредиента имидазооксазиновое соединение, представленное формулой (I), или его фармацевтически приемлемую соль
[Хим. 1]
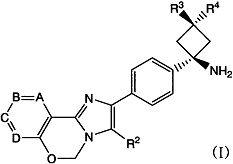 ,
,
где A, B, C и D представляют собой C-R1a, C-R1b, C-R1c и C-R1d соответственно или один или два из A, B, C и D представляют собой атом N;
по меньшей мере два из R1a, R1b, R1c и R1d представляют собой водород, а другой(ие) представляет(ют) собой галоген; циано; C1-6алкил, который может содержать в качестве заместителя(ей) гидроксильную(ые) группу(ы); C1-6алкокси; карбонил, содержащий в качестве заместителя гидроксил, амино, необязательно замещенный моно- или ди-(C1-6алкил)амино или моно- или ди-(C1-6алкокси)амино; или ненасыщенную гетероциклическую группу;
R2 представляет собой фенил, пиридил или тиенил;
R3 представляет собой водород, метил, этил или циклопропил; и
R4 представляет собой водород или гидрокси.
Согласно варианту осуществления настоящего изобретения, A, B, C и D представляют собой C-R1a, C-R1b, C-R1c и C-R1d, соответственно, или один или два из A, B, C и D представляют собой атом N;
по меньшей мере два из R1a, R1b, R1c и R1d представляют собой водород, а другой(ие) по отдельности представляет(ют) собой хлор, фтор, циано, метил, гидроксиметил, метокси, этокси, карбоксил, карбамоил, метиламинокарбонил, этиламинокарбонил, гидроксиэтиламинокарбонил, этоксиаминокарбонил или пиразолил;
R2 представляет собой фенил, пиридил или тиенил;
R3 представляет собой водород, метил, этил или циклопропил; и
R4 представляет собой водород или гидрокси.
Другой вариант осуществления настоящего изобретения относится к усилителю противоопухолевого эффекта для усиления одного или нескольких других противоопухолевых средств, содержащему в качестве активного ингредиента имидазооксазиновое соединение согласно любому из последующих пунктов (a)-(t) или его соль:
(a) транс-3-амино-1-циклопропил-3-(4-(10-фтор-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(b) транс-3-амино-1-циклопропил-3-(4-(10-фтор-3-(пиридин-4-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(c) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(d) транс-3-амино-1-циклопропил-3-(4-(10-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(e) транс-3-амино-1-циклопропил-3-(4-(9-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(f) транс-3-амино-1-циклопропил-3-(4-(8-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(g) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(h) транс-3-амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(i) транс-3-амино-1-этил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(j) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[3,4-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(k) транс-3-амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[3,4-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(l) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[4,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(m) транс-3-амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[4,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(n) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[3,2-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(o) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиразино[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(p) транс-3-амино-3-(4-(9-(гидроксиметил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол,
(q) 2-(4-(транс-1-амино-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбонитрил,
(r) транс-3-амино-1-метил-3-(4-(3-фенил-9-(1H-пиразол-5-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(s) 2-(4-(транс-1-амино-3-гидрокси-3-метилциклобутил)фенил)-N-метил-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбоксамид, и
(t) 2-(4-(транс-1-амино-3-гидрокси-3-метилциклобутил)фенил)-N-этокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбоксамид.
Другой вариант осуществления настоящего изобретения относится к противоопухолевому лекарственному средству, содержащему комбинацию любого из представленных выше имидазооксазиновых соединений или его фармацевтически приемлемой соли и одного или нескольких других противоопухолевых средств.
Другой вариант осуществления настоящего изобретения относится к применению любого из представленных выше имидазооксазиновых соединений или его фармацевтически приемлемой соли для усиления одного или нескольких других противоопухолевых средств.
Другой вариант осуществления настоящего изобретения относится к применению любого из представленных выше имидазооксазиновых соединений или его фармацевтически приемлемой соли для получения усилителя противоопухолевого эффекта для усиления одного или нескольких других противоопухолевых средств.
Другой вариант осуществления настоящего изобретения относится к применению любого из представленных выше имидазооксазиновых соединений или его фармацевтически приемлемой соли для получения противоопухолевого лекарственного средства, содержащего комбинацию имидазооксазинового соединения или его фармацевтически приемлемой соли и одного или нескольких других противоопухолевых средств.
Согласно другому варианту осуществления настоящего изобретения, одно или несколько других противоопухолевых средств представляют собой паклитаксел, карбоплатин, лапатиниб, иринотекан, доксорубицин, эверолимус, бортезомиб, эрлотиниб, трастузумаб (герцептин), метформин, доцетаксел, и комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции для предупреждения и/или лечения опухолей, содержащей любое из представленных выше имидазооксазиновых соединений или его фармацевтически приемлемую соль и одно или несколько других противоопухолевых средств.
Другой вариант осуществления настоящего изобретения относится к способу усиления противоопухолевого эффекта, включающему введение пациенту любого из представленных выше имидазооксазиновых соединений или его фармацевтически приемлемой соли в количестве, эффективном для лечения и/или предупреждения.
Другой вариант осуществления настоящего изобретения относится к способу предупреждения и/или лечения опухолей, включающему введение пациенту комбинации любого из представленных выше имидазооксазиновых соединений или его фармацевтически приемлемой соли и одного или нескольких других противоопухолевых средств в количестве, эффективном для лечения и/или предупреждения.
Другой вариант осуществления настоящего изобретения относится к продукту в виде комбинированного препарата, подлежащего использованию одновременно, последовательно или через некоторый интервал времени для предупреждения и/или лечения опухолей, причем такой продукт содержит комбинацию любого из представленных выше имидазооксазиновых соединений или его фармацевтически приемлемой соли и одного или нескольких других противоопухолевых средств.
Полезные эффекты настоящего изобретения
Будучи использованным с противоопухолевыми средствами, имидазооксазиновое соединение (I) или его фармацевтически приемлемая соль усиливает различные противоопухолевые средства. Более конкретно, при использовании имидазооксазинового соединения (I) с другими противоопухолевыми средствами: 1) оно значимо не усиливает побочные эффекты противоопухолевых средств, позволяя тем самым комбинированное применение нескольких лекарств без снижения их эффективных доз в количествах, эквивалентных количествам конкретных лекарств, дающим максимальный эффект; 2) имидазооксазиновое соединение (I) усиливает противоопухолевые средства независимо от чувствительности к противоопухолевым средствам, подлежащим к комбинированному применению; и 3) такой противоопухолевый эффект наблюдается даже при низких количествах, при которых имидазооксазиновое соединение (I) не может по отдельности проявлять противоопухолевый эффект. Соответственно, настоящее изобретение ведет к обеспечению клинически высоко применимого терапевтического способа увеличения эффективной площади противоракового терапевтического воздействия, улучшения терапевтического эффекта, и т.п.
Краткое описание чертежей
[Фиг. 1] Эффект комбинированного применения соединения I в дозе 8 мг/кг/сутки, 16 мг/кг/сутки и 24 мг/кг/сутки с паклитакселом у бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака яичников A2780.
[Фиг. 2] Изменения массы тела бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака яичников A2780 при комбинированном применении соединения I в дозе 8 мг/кг/сутки, 16 мг/кг/сутки и 24 мг/кг/сутки с паклитакселом.
[Фиг. 3] Эффект комбинированного применения соединения I в дозе 8 мг/кг/сутки, 16 мг/кг/сутки и 24 мг/кг/сутки с паклитакселом у бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака желудка NCI-N87.
[Фиг. 4] Изменения массы тела бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака желудка NCI-N87 при комбинированном применении соединения I в дозе 8 мг/кг/сутки, 16 мг/кг/сутки и 24 мг/кг/сутки с паклитакселом.
[Фиг. 5] Эффект комбинированного применения соединения I в дозе 16 мг/кг/сутки с карбоплатином у бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака яичников A2780.
[Фиг. 6] Изменения массы тела бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака яичников A2780 при комбинированном применении соединения I в дозе 16 мг/кг/сутки с карбоплатином.
[Фиг. 7] Эффект комбинированного применения соединения I в дозе 16 мг/кг/сутки с лапатинибом у бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака желудка NCI-N87.
[Фиг. 8] Изменения массы тела бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака желудка NCI-N87 при комбинированном применении соединения I в дозе 16 мг/кг/сутки с лапатинибом.
[Фиг. 9] Эффект комбинированного применения соединения I в дозе 16 мг/кг/сутки с иринотеканом у бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака яичников A2780.
[Фиг. 10] Изменения массы тела бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака яичников A2780 при комбинированном применении соединения I в дозе 16 мг/кг/сутки с иринотеканом.
[Фиг. 11] Эффект комбинированного применения соединения I в дозе 16 мг/кг/сутки с доксорубицином у бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака яичников A2780.
[Фиг. 12] Изменения массы тела бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака яичников A2780 при комбинированном применении соединения I в дозе 16 мг/кг/сутки с доксорубицином.
[Фиг. 13] Эффект комбинированного применения соединения I в дозе 16 мг/кг/сутки с эверолимусом у бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака желудка NCI-N87.
[Фиг. 14] Изменения массы тела бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака желудка NCI-N87 при комбинированном применении соединения I в дозе 16 мг/кг/сутки с эверолимусом.
[Фиг. 15] Эффект комбинированного применения соединения I в дозе 16 мг/кг/сутки (A) и 24 мг/кг/сутки (B) с TS-1 у бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака желудка 4-1ST.
[Фиг. 16] Изменения массы тела бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака желудка 4-1ST при комбинированном применении соединения I в дозе 16 мг/кг/сутки (A) и 24 мг/кг/сутки (B) с TS-1.
[Фиг. 17] Эффект комбинированного применения соединения I в дозе 16 мг/кг/сутки (A) и 24 мг/кг/сутки (B) с трастузумабом у бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака желудка 4-1ST.
[Фиг. 18] Изменения массы тела бестимусных мышей с подкожно имплантированной человеческой клеточной линией рака желудка 4-1ST при комбинированном применении соединения I в дозе 16 мг/кг/сутки (A) и 24 мг/кг/сутки (B) с трастузумабом.
Описание вариантов осуществления
Используемый в настоящем описании термин «комбинация» означает применение или введение имидазооксазинового соединения согласно настоящему изобретению или его фармацевтически приемлемой соли и одного или нескольких противоопухолевых средств в количестве, достаточном для предупреждения и/или лечения опухоли у пациента, в виде одной композиции или двух различных композиций, как одновременно, последовательно, так и через некоторый интервал времени. Имидазооксазиновое соединение согласно настоящему изобретению, или его фармацевтически приемлемая соль, может быть введено перед, одновременно или после введения других противоопухолевых средств.
Имидазооксазиновое соединение согласно настоящему изобретению или его фармацевтически приемлемая соль представленной ниже формулы (I) усиливает противоопухолевые эффекты противоопухолевых средств.
[Хим. 2]
В формуле (I) A, B, C и D представляют собой C-R1a, C-R1b, C-R1c и C-R1d, соответственно, или один или два из A, B, C и D представляют собой атом N (любой из A, B, C и D, который не представляет собой атом N, представляет собой C-R1a, C-R1b, C-R1c или C-R1d).
По меньшей мере два из R1a, R1b, R1c и R1d представляют собой водород, а другой(ие) представляет(ют) собой галоген, циано, C1-6алкил, который может содержать в качестве заместителя(ей) гидроксильную(ые) группу(ы), C1-6алкокси, карбонил, содержащий гидроксил, амино, необязательно замещенный моно- или ди-(C1-6алкил)амино или моно- или ди-(C1-6алкокси)амино; или ненасыщенную гетероциклическую группу.
Примеры атома галогена, представленного R1a, R1b, R1c или R1d, включают атом хлора, атом брома, атом фтора и атом иода, предпочтительно атом хлора или атом фтора.
C1-6алкил из «C1-6алкила, который может содержать в качестве заместителя(ей) гидроксильную(ые) группу(ы)» предпочтительно представляет собой неразветвленный или разветвленный C1-6алкил; его примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил и гексил. C1-6алкил предпочтительно представляет собой C1-3алкил, более предпочтительно метил. Число гидроксильных групп (заместителей) составляет от 0 до 2, предпочтительно 0 или 1.
«C1-6алкокси», представленный R1a, R1b, R1c или R1d, представляет собой неразветвленный или разветвленный C1-6алкокси, такой как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси или т.п., предпочтительно C1-3алкокси, более предпочтительно метокси или этокси.
Моно- или ди-(C1-6алкил)аминокарбонил из «необязательно замещенного моно- или ди-(C1-6алкил)аминокарбонила» из «карбонила, содержащего в качестве заместителя гидроксил, амино, необязательно замещенный моно- или ди-(C1-6алкил)амино или моно- или ди-(C1-6алкокси)амино», представленный R1a, R1b, R1c или R1d, представляет собой вышеупомянутый аминокарбонил, содержащий 1 или 2 C1-6алкила, предпочтительно моно- или ди-(C1-3алкил)аминокарбонил, более предпочтительно метиламинокарбонил, диметиламинокарбонил или этиламинокарбонил. Заместитель предпочтительно представляет собой гидроксил. Число заместителей предпочтительно составляет 1.
Моно- или ди-(C1-6алкокси)аминокарбонил представляет собой вышеупомянутый аминокарбонил, содержащий 1 или 2 C1-6алкокси, предпочтительно моно- или ди-(C1-3алкокси)аминокарбонил, более предпочтительно этоксиаминокарбонил.
«Карбонил, содержащий в качестве заместителя гидроксил, амино, необязательно замещенный моно- или ди-(C1-6алкил)амино или моно- или ди-(C1-6алкокси)амино», представленный R1a, R1b, R1c или R1d, в частности, предпочтительно представляет собой карбоксил, карбамоил, метиламинокарбонил, этиламинокарбонил, гидроксиэтиламинокарбонил или этоксиаминокарбонил.
Ненасыщенная гетероциклическая группа, представленная R1a, R1b, R1c или R1d, предпочтительно представляет собой 5-10-членную моноциклическую или бициклическую ненасыщенную гетероциклическую группу, содержащую от 1 до 4 гетероатомов, выбранных из группы, состоящей из N, S и O. Ее примеры включают имидазолил, тиенил, фурил, пирролил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиразолил, триазолил, тетразолил, пиридил, пиразил, пиримидинил, пиридазинил, индолил, изоиндолил, индазолил, бензофуранил, бензимидазолил, бензоксазолил, бензотиазолил, пуринил, хинолил, изохинолил, хиназолинил и хиноксалил. Среди них, пиразолил является предпочтительным.
Предпочтительные примеры соединения, представленного формулой (I), включают следующие соединения:
(a) транс-3-амино-1-циклопропил-3-(4-(10-фтор-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(b) транс-3-амино-1-циклопропил-3-(4-(10-фтор-3-(пиридин-4-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(c) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(d) транс-3-амино-1-циклопропил-3-(4-(10-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(e) транс-3-амино-1-циклопропил-3-(4-(9-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(f) транс-3-амино-1-циклопропил-3-(4-(8-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(g) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(h) транс-3-амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(i) транс-3-амино-1-этил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(j) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[3,4-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(k) транс-3-амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[3,4-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(l) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[4,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(m) транс-3-амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[4,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(n) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[3,2-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(o) транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиразино[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол,
(p) транс-3-амино-3-(4-(9-(гидроксиметил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол,
(q) 2-(4-(транс-1-амино-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбонитрил,
(r) транс-3-амино-1-метил-3-(4-(3-фенил-9-(1H-пиразол-5-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол,
(s) 2-(4-(транс-1-амино-3-гидрокси-3-метилциклобутил)фенил)-N-метил-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбоксамид, и
(t) 2-(4-(транс-1-амино-3-гидрокси-3-метилциклобутил)фенил)-N-этокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбоксамид.
Фармацевтически приемлемые соли имидазооксазинового соединения (I) относятся к солям, характеризующимся желаемой фармакологической активностью имидазооксазинового соединения (I), полученным из фармацевтически приемлемого основания или кислоты, включая неорганические/органические основания и неорганические/органические кислоты.
Примеры фармацевтически приемлемых солей имидазооксазинового соединения (I) включают кислотно-аддитивные соли с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, угольная кислота, пикриновая кислота, метансульфоновая кислота, пара-толуолсульфоновая кислота, глутаминовая кислота и т.п.; соли с неорганическими основаниями, такими как натрий, калий, магний, кальций, алюминий и т.п., органическими основаниями, такими как метиламин, этиламин, меглюмин, этаноламин и т.п., или основными аминокислотами, такими как лизин, аргинин, орнитин и т.п.; и соли аммония. Кроме того, имидазооксазиновое соединение (I) включает оптические изомеры и гидраты.
Имидазооксазиновое соединение (I), которое усиливает противоопухолевый эффект согласно настоящему изобретению, может быть получено, например, следующими способами получения или способами, описанными в разделе «Примеры». Однако способ получения соединения согласно настоящему изобретению не ограничивается указанными примерами.
Соединение (I) согласно настоящему изобретению может быть получено с использованием, например, следующего способа получения A и способа получения B.
[Хим. 3]
Способ получения A
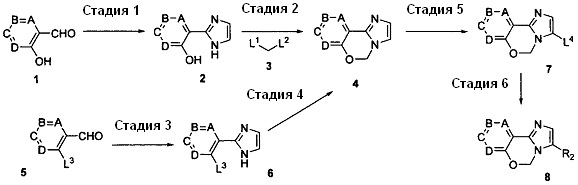
(в формуле L1, L2, L3 и L4 являются одинаковыми или отличаются друг от друга, и каждый представляет собой уходящую группу; а другие символы определены выше).
Стадия 1
Указанная стадия представляет собой способ получения соединения 2 из альдегидного соединения 1.
Исходное соединение 1 представляет собой коммерчески доступный продукт, или оно может быть получено в соответствии с известным способом. Первая стадия может быть проведена способом, описанным в документах (например, J. Med. Chem., Vol. 46, p. 5416, 2003; J. Org. Chem., Vol. 68, p. 5415, 2003), способом, основанным на них, или комбинаций указанных способов с обычными способами.
Например, при использовании в реакции водного раствора аммиака и водного раствора глиоксаля количество подлежащего использованию водного раствора аммиака составляет от 1 до 10 эквивалентов относительно соединения 1. Количество подлежащего использованию водного раствора глиоксаля составляет от 1 до 10 эквивалентов относительно соединения 1.
Примеры пригодных для использования растворителей включают метанол, этанол, тетрагидрофуран, этилацетат, N,N-диметилформамид, уксусную кислоту и воду. Растворители могут быть использованы по отдельности или в комбинации. Время реакции составляет от 0,1 до 100 часов и предпочтительно от 0,5 до 24 часов. Температура реакции составляет от 0°C до температуры кипения растворителя, и предпочтительно от 0 до 100°C.
Полученное таким образом соединение 2 может быть выделено и очищено известными способами разделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстрагирование растворителем, повторное осаждение и хроматография, а затем использовано на следующей стадии; или оно может быть использовано на следующей стадии без выделения и очистки.
Стадия 2
Указанная стадия представляет собой способ получения соединения 4, при котором проводят реакцию алкилирования соединения 2 соединением 3 в присутствии основания.
Соединение 3, в котором в качестве L1 и L2 упомянуты хлор, бром, иод и т.п., представляет собой коммерчески доступный продукт, или оно может быть получено в соответствии с известным способом.
Соединение 3 может быть использовано в количестве от 1 до 100 эквивалентов, и предпочтительно от 1 до 10 эквивалентов, относительно соединения 2.
Примеры основания включают неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия и гидроксид цезия, и органические амины, такие как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин. Основание может быть использовано в количестве от 1 до 100 эквивалентов, и предпочтительно от 2 до 10 эквивалентов.
Примеры пригодных для использования растворителей включают N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, тетрагидрофуран, 1,4-диоксан, N-метилпирролидин-2-он, ацетонитрил и воду. Растворители могут быть использованы по отдельности или в комбинации. Время реакции составляет от 0,1 до 100 часов, и предпочтительно от 0,5 до 24 часов. Температура реакции составляет от 0°C до температуры кипения растворителя, и предпочтительно от 0 до 100°C.
Полученное таким образом соединение 4 может быть выделено и очищено известными способами разделения и очистки, а затем использовано на следующей стадии; или оно может быть использовано на следующей стадии без выделения и очистки.
Стадия 3
Указанная стадия представляет собой способ получения соединения 6 из соединения 5.
Соединение 5, в котором в качестве L3 упомянуты хлор, бром, иод и т.п., представляет собой коммерчески доступный продукт, или оно может быть получено в соответствии с известным способом.
Стадия 3 может быть проведена тем же способом, что и стадия 1.
Стадия 4
Указанная стадия представляет собой способ получения соединения 4, при котором взаимодействие соединения 6 с формальдегидом проводят в присутствии основания.
Формальдегид может быть использован в количестве от 1 до 100 эквивалентов, и предпочтительно от 1 до 10 эквивалентов, относительно соединения 6. Формальдегид может быть использован в форме водного раствора или в форме параформальдегида.
Примеры основания включают гидроксид натрия, карбонат натрия, гидроксид калия, карбонат цезия, трет-бутоксид натрия и трет-бутоксид калия. Основание может быть использовано в количестве от 1 до 100 эквивалентов, и предпочтительно от 2 до 10 эквивалентов.
Примеры пригодных для использования растворителей включают N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, тетрагидрофуран, 1,4-диоксан, N-метилпирролидин-2-он, ацетонитрил и воду. Растворители могут быть использованы по отдельности или в комбинации. Время реакции составляет от 0,1 до 100 часов, и предпочтительно от 0,5 до 24 часов. Температура реакции составляет от 0°C до температуры кипения растворителя, и предпочтительно от 0 до 100°C.
Полученное таким образом соединение 4 может быть выделено и очищено известными способами разделения и очистки, а затем использовано на следующей стадии; или оно может быть использовано на следующей стадии без выделения и очистки.
Стадия 5
Указанная стадия представляет собой способ получения соединения 7 путем проведения галогенирования, например, путем осуществления воздействия галогенирующего агента на соединение 4 (L4 = Cl, Br или I). Галогенирование может быть проведено в соответствии с общеизвестным способом; например, галогенирование может быть проведено в реакционном растворителе, который не оказывает негативное влияние на реакцию.
Полученное таким образом соединение 7 может быть выделено и очищено известными способами разделения и очистки, а затем использовано на следующей стадии; или оно может быть использовано на следующей стадии без выделения и очистки.
Стадия 6
Указанная стадия представляет собой способ получения соединения 8 путем осуществления взаимодействия соединения 7 с арилбороновой кислотой, сложным эфиром арилбороновой кислоты, ненасыщенной гетероциклической бороновой кислотой или сложным эфиром ненасыщенной гетероциклической бороновой кислоты.
Указанная стадия может быть проведена в соответствии с общеизвестным способом (например, Chemical Reviews, Vol. 95, p. 2457, 1995); например, указанная стадия может быть проведена в растворителе, который не оказывает негативное влияние на реакцию, в присутствии катализатора, содержащего металл переменной валентности, и основания.
Арилбороновая кислота, сложный эфир арилбороновой кислоты, ненасыщенная гетероциклическая бороновая кислота или сложный эфир ненасыщенной гетероциклической бороновой кислоты могут быть использованы в количестве от 1 до 10 эквивалентов, и предпочтительно от 1 до 3 эквивалентов, относительно соединения 7.
Примеры пригодных для использования катализаторов, содержащих металл переменной валентности, включают палладиевые катализаторы (например, ацетат палладия, хлорид палладия, тетракис(трифенилфосфин)палладий и т.п.) и никелевые катализаторы (например, хлорид никеля и т.п.). При необходимости, могут быть добавлены лиганды (например, трифенилфосфин, три-трет-бутилфосфин и т.п.), и в качестве сокатализаторов могут быть добавлены оксиды металлов (например, оксид меди, оксид серебра и т.п.), и т.п. Хотя подлежащее использованию количество катализатора, содержащего металл переменной валентности, варьирует в зависимости от типа катализатора, как правило, оно составляет приблизительно от 0,0001 приблизительно до 1 моль, и предпочтительно приблизительно от 0,01 приблизительно до 0,5 моль, относительно соединения 7 (1 моль). Подлежащее использованию количество лиганда, как правило, составляет приблизительно от 0,0001 приблизительно до 4 моль, и предпочтительно приблизительно от 0,01 приблизительно до 2 моль, относительно соединения 7 (1 моль). Подлежащее использованию количество сокатализатора, как правило, составляет приблизительно от 0,0001 приблизительно до 4 моль, и предпочтительно приблизительно от 0,01 приблизительно до 2 моль, относительно соединения 7 (1 моль).
Примеры основания включают органические амины (например, триметиламин, триэтиламин, диизопропилэтиламин, N-метилморфолин, 1,8-диазабицикло[5.4.0]ундец-7-ен, пиридин, N,N-диметиланилин и т.п.), соли щелочных металлов (например, гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия, карбонат цезия, фосфат натрия, фосфат калия, гидроксид натрия, гидроксид калия и т.п.), гидриды металлов (например, гидрид калия, гидрид натрия и т.п.), алкоксиды щелочных металлов (например, метоксид натрия, этоксид натрия, трет-бутоксид натрия, трет-бутоксид калия и т.п.), дисилазиды щелочных металлов (например, дисилазид лития, дисилазид натрия, дисилазид калия и т.п.). Среди них, предпочтительными являются соли щелочных металлов, такие как карбонат калия, карбонат цезия, фосфат натрия и фосфат калия; алкоксиды щелочных металлов, такие как трет-бутоксид натрия и трет-бутоксид калия; органические амины, такие как триэтиламин и диизопропилэтиламин; и т.п. Подлежащее использованию количество основания, как правило, составляет от 0,1 до 10 моль, и предпочтительно приблизительно от 1 приблизительно до 5 моль, относительно соединения 7 (1 моль).
Любые растворители могут быть использованы при условии, если они не оказывают негативное влияние на реакцию. Их примеры включают углеводороды (например, бензол, толуол, ксилол, и т.п.), галогенированные углеводороды (например, хлороформ, 1,2-дихлорэтан, и т.п.), нитрилы (например, ацетонитрил, и т.п.), эфиры (например, диметоксиэтан, тетрагидрофуран, и т.п.), спирты (например, метанол, этанол, и т.п.), апротонные полярные растворители (например, диметилформамид, диметилсульфоксид, гексаметилфосфорамид, и т.п.), воду и их смеси. Время реакции составляет от 0,1 до 100 часов, и предпочтительно от 0,5 до 24 часов. Температура реакции составляет от 0°C до температуры кипения растворителя, и предпочтительно от 0 до 150°C.
Полученное таким образом соединение 8 может быть выделено и очищено известными способами разделения и очистки, а затем использовано на следующей стадии; или оно может быть использовано на следующей стадии без выделения и очистки.
[Хим. 4]
Способ получения B
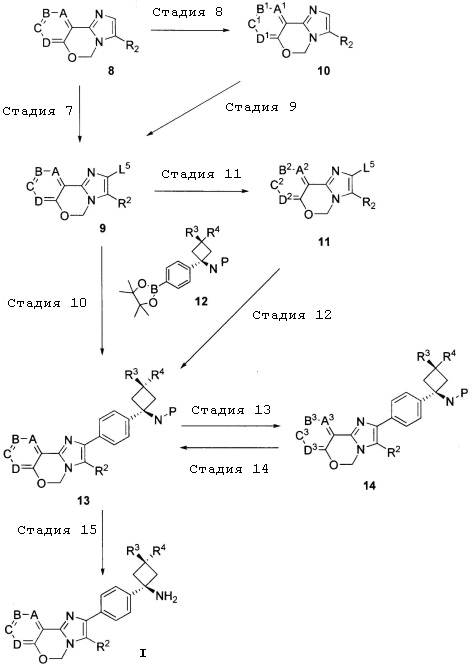
(в формуле L5 являются одинаковыми или отличаются друг от друга, и каждый представляет собой уходящую группу; P представляет собой защитную группу; а другие символы определены выше).
Стадия 7
Стадия 7 может быть проведена тем же способом, что и стадия 5.
Стадия 8
Указанная стадия представляет собой способ преобразования любого из A - D соединения 8 в любой из A1 - D1, соответственно, путем проведения реакции сочетания и т.п. с использованием общеизвестного способа.
Если любой из A - D соединения 8 содержит уходящую группу, такую как галоген, то реакцию сочетания проводят в присутствии катализатора, содержащего металл переменной валентности, с получением соединения 10.
В случае преобразования уходящей группы, такой как галоген, в цианогруппу используют цианид цинка. В случае преобразования в ароматическое кольцо или гетероароматическое кольцо используют коммерчески доступную бороновую кислоту или сложный бороновый эфир, или бороновую кислоту или сложный бороновый эфир, которые могут быть получены в соответствии с известным способом.
В случае преобразования в сложноэфирную группу используют монооксид углерода.
Полученное таким образом соединение 10 может быть выделено и очищено известными способами разделения и очистки, а затем использовано на следующей стадии; или оно может быть использовано на следующей стадии без выделения и очистки.
Стадия 9
Стадия 9 может быть проведена тем же способом, что и стадия 5.
Стадия 10
Указанная стадия представляет собой способ получения соединения 13 путем проведения реакции сочетания соединения 9 и соединения 12.
Соединение 12 может быть получено способом, описанным в документах (например, в WO 2008/070016, WO 2009/148877, WO 2009/148916, WO 2010/088177, WO 2010/114780, WO 2010/104933), или способом, основанным на нем.
Указанная стадия может быть проведена тем же способом, что и стадия 6.
Полученное таким образом соединение 13 может быть выделено и очищено известными способами разделения и очистки, а затем использовано на следующей стадии; или оно может быть использовано на следующей стадии без выделения и очистки.
Стадия 11
Указанная стадия представляет собой способ преобразования любого из A - D соединения 9 в любой из A1 - D1, соответственно, путем проведения реакции преобразования функциональных групп и т.п. с использованием общеизвестного способа.
Если любой из A - D соединения 9 содержит сложноэфирную группу, то соединение 11 получают путем преобразования сложноэфирной группы в спирт с использованием общеизвестной реакции восстановления.
Полученное таким образом соединение 11 может быть выделено и очищено известными способами разделения и очистки, а затем использовано на следующей стадии; или оно может быть использовано на следующей стадии без выделения и очистки.
Стадия 12
Стадия 12 может быть проведена тем же способом, что и стадия 10.
Стадия 13
Указанная стадия представляет собой способ получения соединения 14 путем проведения гидролиза в основных условиях, если любой из A - D соединения 13 содержит сложноэфирную группу.
Основание, такое как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия, гидроксид калия и гидроксид лития, может быть использовано в количестве от 1 до 100 эквивалентов, и предпочтительно от 1 до 30 эквивалентов.
Примеры пригодных для использования растворителей включают воду, метанол, этанол, изопропанол, тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид. Растворители могут быть использованы по отдельности или в комбинации. Время реакции составляет от 0,1 до 100 часов, и предпочтительно от 0,5 до 24 часов. Температура реакции составляет от 0°C до температуры кипения растворителя, и предпочтительно от 0 до 100°C.
Полученное таким образом соединение 14 может быть выделено и очищено известными способами разделения и очистки, а затем использовано на следующей стадии; или оно может быть использовано на следующей стадии без выделения и очистки.
Стадия 14
Указанная стадия представляет собой способ получения соединения 13 путем проведения реакции амидирования соединения 14 амином в органическом растворителе.
Амидирование может быть проведено общеизвестным способом. Примеры такого способа включают способ, при котором реакцию сочетания 14 с соответствующим амином проводят в присутствии агента конденсации (см. «Pepuchido Gosei No Kiso To Jikken [Foundation and Experiments of Peptide Synthesis]», Nobuo Izumiya, et al., опубликовано Maruzen Co. в 1983 г.). Полученное таким образом соединение 13 может быть выделено и очищено известными способами разделения и очистки, а затем использовано на следующей стадии; или оно может быть использовано на следующей стадии без выделения и очистки.
Стадия 15
Указанная стадия представляет собой способ получения соединения (I) путем снятия защитных групп с защищенных аминогрупп соединения 13. Снятие защитных групп может быть проведено общеизвестным способом, например, способом, раскрытым в Protective Groups in Organic Synthesis, T.W. Greene, John Wiley & Sons (1981); или способом, основанным на нем.
Примеры защитной группы включают трет-бутилоксикарбонил и фталимид. Например, при использовании в качестве защитной группы трет-бутилоксикарбонила, снятие защитных групп предпочтительно проводят в кислых условиях. Примеры кислоты включают соляную кислоту, уксусную кислоту, трифторуксусную кислоту, серную кислоту и толуолсульфоновую кислоту.
Подлежащее использованию количество кислоты предпочтительно составляет приблизительно от 1 приблизительно до 100 эквивалентов относительно соединения 13.
В реакции могут быть использованы любые растворители при условии, если они не оказывают негативное влияние на реакцию. Например, могут быть использованы спирты (например, метанол, и т.п.), углеводороды (например, бензол, толуол, ксилол и т.п.), галогенированные углеводороды (например, метиленхлорид, хлороформ, 1,2-дихлорэтан и т.п.), нитрилы (например, ацетонитрил и т.п.), эфиры (например, диметоксиэтан, тетрагидрофуран и т.п.), апротонные полярные растворители (например, N,N-диметилформамид, диметилсульфоксид, гексаметилфосфорамид и т.п.), или их смеси. Время реакции составляет от 0,1 до 100 часов и предпочтительно от 0,5 до 24 часов. Температура реакции составляет от 0 до 100°C и предпочтительно от 0 до 50°C.
При использовании в качестве защитной группы фталимида, может проводиться обработка гидразином. Подлежащее использованию количество гидразина предпочтительно составляет от 1 до 100 эквивалентов относительно соединения 13.
Для проведения синтеза реакция может быть проведена при нагревании с использованием микроволнового реактора и т.п. В реакции могут быть использованы любые растворители при условии, если они не оказывают негативное влияние на реакцию. Например, могут быть использованы спирты (например, метанол, этанол и т.п.), углеводороды (например, бензол, толуол, ксилол и т.п.), галогенированные углеводороды (например, метиленхлорид, хлороформ, 1,2-дихлорэтан и т.п.), нитрилы (например, ацетонитрил и т.п.), эфиры (например, диметоксиэтан, тетрагидрофуран и т.п.), апротонные полярные растворители (например, N,N-диметилформамид, диметилсульфоксид, гексаметилфосфорамид и т.п.) или их смеси. Время реакции составляет от 0,1 до 100 часов и предпочтительно от 0,5 до 24 часов. Температура реакции составляет от 0 до 200°C и предпочтительно от 0 до 150°C.
Полученное таким образом соединение (I) может быть выделено и очищено известными способами разделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстрагирование растворителем, повторное осаждение и хроматография.
Имидазооксазиновое соединение (I) представляет собой противоопухолевое средство, обладающее высокой ингибирующей активностью в отношении AKT и сниженным побочным эффектом. При применении с различными другими противоопухолевыми средствами (здесь и далее в документе называемых противоопухолевым средством A), имидазооксазиновое соединение (I) усиливает противоопухолевый эффект противоопухолевого средства A без значительного увеличения его токсичности.
Примеры противоопухолевого средства A, усиливаемого имидазооксазиновым соединением (I), включают без ограничения противоопухолевые антибиотики, такие как доксорубицин или эпирубицин; алкилирующие агенты, такие как циклофосфамид или нимустин; средства на основе платины, такие как цисплатин, карбоплатин или оксалиплатин; антиметаболиты на основе пиримидина, такие как 5-фторурацил (5-FU), тегафур/гимерацил/отерацил калия (то есть, комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия) (TS-1, обобщенное название «комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия» (наименование препарата: «Ti-Esu-Wan»)), тегафур/урацил (комбинированное лекарственное средство из тегафура и урацила) (UFT, обобщенное название «комбинированное лекарственное средство из тегафура и урацила» (наименование препарата: «Yu-Efu-Ti»)), капецитабин, доксифлуридин, 5-фтор-2’-дезоксиуридин (FdUrd), гемцитабин или цитарабин; антиметаболиты на основе пурина, такие как флударабин, кладрибин или неларабин; антиметаболиты фолиевой кислоты, такие как пеметрексед или метотрексат; растительные алкалоидные противоопухолевые средства, такие как паклитаксел (наименования препарата: «таксол», «абраксан», и т.п.), доцетаксел, иринотекан или винкристин; низкомолекулярные лекарственные средства направленного действия, такие как гефинитиб, эрлотиниб, лапатиниб, эверолимус, темсиролимус, селуметиниб, траметиниб, сорафениб, афатиниб, регорафениб, дабрафениб, вемурафениб, бортезомиб или карфилзомиб; лекарственные средства-антитела направленного действия, такие как трастузумаб (герцептин), цетуксимаб, бевацизумаб, панитумумаб, велтузумаб или ритуксимаб; метформин, дексаметазон, талидомид и леналидомид.
Предпочтительные примеры противоопухолевого средства A, усиливаемого имидазооксазиновым соединением (I), включают паклитаксел, карбоплатин, лапатиниб, иринотекан, доксорубицин, эверолимус, бортезомиб, эрлотиниб, трастузумаб (герцептин), метформин, доцетаксел и комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия.
Злокачественные опухоли, подвергаемые лечению имидазооксазиновым соединением (I) совместно с усиливаемым противоопухолевым средством A, включают без ограничения рак головы и шеи, рак пищевода, рак желудка, рак толстого кишечника, рак прямой кишки, гепатокарциному, рак желчного пузыря, холангиокарциному, рак поджелудочной железы, рак легких, рак молочной железы, рак яичников, рак шейки матки, рак эндометрия, рак почки, рак мочевого пузыря, рак предстательной железы, рак яичка, остеосаркому, саркому мягких тканей, лейкоз, злокачественную лимфому, множественную миелому, рак кожи и опухоль головного мозга.
Согласно одному варианту осуществления, настоящее изобретение относится к усилителю противоопухолевого эффекта для усиления одного или нескольких противоопухолевых средств, содержащему в качестве активного ингредиента имидазооксазиновое соединение (I) или его фармацевтически приемлемую соль.
Согласно другому варианту осуществления, настоящее изобретение относится к противоопухолевому лекарственному средству, содержащему сочетание имидазооксазинового соединения (I) или его фармацевтически приемлемой соли и одного или нескольких других противоопухолевых средств.
Согласно другому варианту осуществления, настоящее изобретение относится к фармацевтической композиции для предупреждения и/или лечения опухолей, содержащей имидазооксазиновое соединение (I) или его фармацевтически приемлемую соль и одно или несколько других противоопухолевых средств.
Согласно другому варианту осуществления, настоящее изобретение относится к применению имидазооксазинового соединения (I) или его фармацевтически приемлемой соли для усиления одного или нескольких других противоопухолевых средств.
Согласно другому варианту осуществления, настоящее изобретение относится к применению имидазооксазинового соединения (I) или его фармацевтически приемлемой соли для получения усилителя для усиления одного или нескольких других противоопухолевых средств.
Согласно другому варианту осуществления, настоящее изобретение относится к способу усиления противоопухолевого эффекта, включающему введение пациенту терапевтически и/или профилактически эффективного количества имидазооксазинового соединения (I) или его фармацевтически приемлемой соли.
Согласно другому варианту осуществления, настоящее изобретение относится к способу предупреждения и/или лечения опухолей, включающему введение терапевтически и/или профилактически эффективного количества сочетания имидазооксазинового соединения (I) или его фармацевтически приемлемой соли и одного или нескольких других противоопухолевых средств.
Согласно другому варианту осуществления, настоящее изобретение относится к продукту, содержащему имидазооксазиновое соединение (I) или его фармацевтически приемлемую соль и одно или нескольких других противоопухолевых средств, в виде комбинированного препарата, подлежащего применению одновременно, последовательно или через некоторый интервал времени для предупреждения и/или лечения опухоли.
Посредством комбинирования имидазооксазинового соединения (I) или его фармацевтически приемлемой соли и противоопухолевого средства A получают усиленное противоопухолевое лекарство. Примеры формы такого нового противоопухолевого лекарства включают отдельный препарат, содержащий имидазооксазиновое соединение (I) или его фармацевтически приемлемую соль и противоопухолевое средство A; и комбинацию отдельных препаратов, т.е. препарат, содержащий имидазооксазиновое соединение (I) или его фармацевтически приемлемую соль, и препарат, содержащий противоопухолевое средство A. Кроме того, средства для введения композиции, содержащей имидазооксазиновое соединение (I), и средства для введения композиции, содержащей противоопухолевое средство A, могут быть одинаковыми или разными (например, пероральное введение и инъекция).
Если имидазооксазиновое соединение (I) или его фармацевтически приемлемая соль содержится в составе фармацевтической композиции, то при необходимости может быть добавлен фармацевтический носитель, образующий тем самым подходящую лекарственную форму, соответствующую целям предупреждения и лечения. Примеры лекарственной формы включают пероральные препараты, инъецируемые препараты, суппозитории, мази, пластыри и т.п. Предпочтительными являются пероральные препараты. Такие лекарственные формы могут быть приготовлены способами, общеизвестными специалистам в данной области техники.
В качестве фармацевтического носителя, различные органические и неорганические вещества-носители, обычно используемые в качестве веществ для приготовления, могут быть смешаны в качестве эксципиента, связующего вещества, разрыхлителя, смазки или красителя в твердых препаратах; или в качестве растворителя, солюбилизатора, суспендирующего агента, изотонического агента, буфера или успокаивающего средства в жидких препаратах. Более того, при необходимости, может быть использована добавка к фармацевтическому препарату, такая как, антисептик, антиоксидант, краситель, подсластитель и стабилизатор.
Пероральные твердые препараты могут быть получены следующим образом. С использованием обычного способа к соединению согласно настоящему изобретению добавляют эксципиент, необязательно вместе со связующим веществом, разрыхлителем, смазкой, красителем, подсластителем/вкусоароматизатором, и т.п., с получением таблеток, таблеток с покрытием, гранул, порошков, капсул или т.п.
Примеры наполнителей включают лактозу, сахарозу, D-маннит, глюкозу, крахмал, карбонат кальция, каолин, микрокристаллическую целлюлозу и ангидрид кремниевой кислоты.
Примеры связующих веществ включают воду, этанол, 1-пропанол, 2-пропанол, простой сироп, жидкую глюкозу, жидкий α-крахмал, жидкий желатин, D-маннит, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилкрахмал, метилцеллюлозу, этилцеллюлозу, шеллак, фосфат кальция и поливинилпирролидон.
Примеры разрыхлителей включают сухой крахмал, альгинат натрия, порошок агара, гидрокарбонат натрия, карбонат кальция, лаурилсульфат натрия, моноглицерид стеариновой кислоты и лактозу.
Примеры смазок включают очищенный тальк, стеарат натрия, стеарат магния, тетраборат натрия и полиэтиленгликоль.
Примеры красителей включают оксид титана и оксид железа.
Примеры подсластителей/вкусоароматизаторов включают сахарозу, кожуру дикого апельсина, лимонную кислоту и винную кислоту, и т.п.
При необходимости, посредством способов, известных для пероральных препаратов, может быть нанесено кишечнорастворимое покрытие или покрытие для увеличения продолжительности эффектов. Примеры таких покровных средств включают гидроксипропилметилцеллюлозу, этилцеллюлозу, гидроксиметилцеллюлозу, гидроксипропилцеллюлозу, полиоксиэтиленгликоль и Твин 80 (зарегистрированная торговая марка).
При приготовлении пероральных жидких препаратов к имидазооксазиновому соединению (I) с использованием обычного способа добавляют подсластитель, буфер, стабилизатор, вкусоароматизатор и т.п. с получением жидкого лекарственного средства для внутреннего применения, сиропа, эликсира и т.п. В этом случае применимы описанные выше подсластители и вкусоароматизаторы. Примеры буферов включают цитрат натрия и т.п., и примеры стабилизаторов включают трагакант, аравийскую камедь и желатин.
Инъецируемые препараты могут быть приготовлены следующим образом. К имидазооксазиновому соединению (I) с использованием обычного способа добавляют регулятор рН, буфер, стабилизатор, изотоническое средство, местный анестетик и т.п. с получением подкожно инъецируемого препарата, внутримышечно инъецируемого препарата или внутривенно инъецируемого препарата. Примеры возможных к использованию в этом случае регуляторов рН и буферов включают цитрат натрия, ацетат натрия и фосфат натрия. Примеры стабилизаторов включают пиросульфит натрия, EDTA, тиогликолевую кислоту и тиомолочную кислоту. Примеры местных анестетиков включают гидрохлорид прокаина и гидрохлорид лидокаина. Примеры изотонических средств включают хлорид натрия, глюкозу, D-маннит и глицерин.
Суппозитории могут быть приготовлены следующим образом. К соединению согласно настоящему изобретению добавляют фармацевтический носитель, известный в данной области техники, такой как полиэтиленгликоль, ланолин, масло какао и триглицерид жирной кислоты, необязательно вместе с Твин 80 (зарегистрированная торговая марка) или подобным поверхностно-активным веществом, с последующим получением с использованием обычного способа.
Мази могут быть приготовлены следующим образом. К соединению согласно настоящему изобретению при необходимости добавляют обычную основу, стабилизатор, увлажнитель, консервант и т.п., смешивают и приготавливают с использованием обычного способа. Примеры основ включают жидкий парафин, белый вазелин, белый пчелиный воск, октилдодециловый спирт и парафин. Примеры консервантов включают метилпарагидроксибензоат, этилпарагидроксибензоат и пропилпарагидроксибензоат.
Пластыри могут быть получены путем покрытия обычной подложки описанными выше мазью, кремом, гелем, пастой и т.п. с использованием обычного способа. Примеры подложек включают тканые и нетканые материалы, изготовленные из хлопка, штапельных волокон и химических волокон; и пленки и пенопластовые листы из мягкого винилхлорида, полиэтилена и полиуретана.
Лекарственное средство согласно настоящему изобретению может быть использовано в отношении млекопитающих, включая людей (например, людей, коров, лошадей, свиней, обезьян, собак, кошек, мышей, крыс, кроликов, коз, овец и т.п.), предпочтительно в отношении людей.
Количество имидазооксазинового соединения (I), которое должно содержаться в такой стандартной лекарственной форме, варьирует в зависимости от состояния пациента или от лекарственной формы. Желаемое количество в одной стандартной лекарственной форме обычно составляет приблизительно от 0,05 до 1000 мг в случае перорального препарата, приблизительно от 0,01 до 500 мг в случае инъецируемого препарата и приблизительно от 1 до 1000 мг в случае суппозитория. «Низкое количество, при котором имидазооксазиновое соединение (I) по отдельности не проявляет противоопухолевый эффект» в одной стандартной лекарственной форме составляет приблизительно от 0,05 до 20 мг в случае перорального препарата.
Суточная доза лекарственного средства в такой лекарственной форме зависит от состояния, массы тела, возраста, пола и т.п. пациента. Например, суточная доза для взрослого (масса тела 50 кг), как правило, может составлять приблизительно от 0,05 до 5000 мг, и предпочтительно от 0,1 до 1000 мг, и предпочтительно вводится одной дозой или в виде двух или трех раздельных доз в сутки.
Если препарат, содержащий имидазооксазиновое соединение (I) или его соль, и препарат, содержащий противоопухолевой средство A, представляют собой отдельные препараты, то они могут быть введены в одно и то же время или так, что один из препаратов вводят с произвольным интервалом во времени до или после введения другого препарата. Два препарата могут быть введены одновременно, последовательно, или с некоторым интервалом во времени.
Соотношение имидазооксазинового соединения (I) или его фармацевтически приемлемой соли и противоопухолевого средства A при введении или смешивании конкретно не ограничено в том случае, если усиление их противоопухолевого эффекта может быть гарантировано; однако количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли, как правило, составляет приблизительно от 0,001 до 100 моль, предпочтительно приблизительно от 0,005 до 50 моль, на моль противоопухолевого средства A.
Например, если противоопухолевое средство A представляет собой паклитаксел, то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 0,05 до 50 моль на моль паклитаксела. Если противоопухолевое средство A представляет собой карбоплатин, то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 0,005 до 5 моль на моль карбоплатина. Если противоопухолевое средство A представляет собой лапатиниб, то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 0,01 до 20 моль на моль лапатиниба. Если противоопухолевое средство A представляет собой иринотекан, то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 0,05 до 30 моль на моль иринотекана. Если противоопухолевое средство A представляет собой доксорубицин, то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 0,05 до 20 моль, на моль доксорубицина. Если противоопухолевое средство A представляет собой эверолимус, то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 0,05 до 20 моль, на моль эверолимуса. Если противоопухолевое средство A представляет собой бортезомиб, то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 1 до 500 моль, на моль бортезомиба. Если противоопухолевое средство A представляет собой эрлотиниб, то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 0,01 до 10 моль, на моль эрлотиниба. Если противоопухолевое средство A представляет собой трастузумаб (герцептин), то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 0,02 до 20 моль, на моль трастузумаба (герцептина). Если противоопухолевое средство A представляет собой метформин, то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 0,00001 до 0,01 моль, на моль метформина. Если противоопухолевое средство A представляет собой доцетаксел, то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 0,001 до 0,3 моль на моль доцетаксела. Если противоопухолевое средство A представляет собой комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия, то количество имидазооксазинового соединения (I) или его фармацевтически приемлемой соли составляет приблизительно от 0,05 до 50 моль на моль тегафура.
Примеры
Настоящее изобретение подробно описано ниже со ссылкой на справочные примеры, примеры и тестовые примеры, которые не предназначены для ограничения объема настоящего изобретения. Кроме того, в представленных ниже тестовых примерах количества различных противоопухолевых средств, усиливаемых соединением согласно настоящему изобретению, определяли исходя из максимально переносимой дозы (MTD), раскрытой в научных работах и т.п., или исходя из максимальной дозы, переносимой в контексте свойства противоопухолевого средства.
Количество, обеспечивающее максимальный эффект противоопухолевого средства, близко к количеству, определяющему его токсичность. Если максимальный противоопухолевый эффект лекарственного средства оценивают с использованием модели на животных, то оценку, как правило, проводят с использованием дозы, близкой к MTD. В представленных ниже тестовых примерах MTD и вызывающее максимальный эффект количество являются одними и теми же.
Если иное не указано особо, то реагенты, использованные в примерах, являются коммерчески доступными продуктами. Для проведения хроматографии на силикагеле использовали Purif-Pack SI производства Shoko Co. или Biotage SNAP Cartridge KP-Sil производства Biotage, и для проведения хроматографии на щелочном силикагеле использовали Purif-Pack NH производства Shoko Co. или Biotage SNAP Cartridge KP-NH производства Biotage.
Для проведения препаративной тонкослойной хроматографии использовали Kieselgel TM60F254, Art. 5744 производства Merck & Co. или NH2 Silica Gel 60 F254 Plate-Wako производства Wako. Для проведения препаративной высокоэффективной жидкостной хроматографии с обращенной фазой использовали CombiPrep Pro C18 (φ 30×50 мм) производства YMC Co.
1H-ЯМР спектры измеряли с использованием спектрометров AL400 (400 МГц) производства JEOL, Mercury (400 МГц) производства Varian; или Inova (400 МГц) производства Varian; используя тетраметилсилан в качестве стандартного вещества. Кроме того, масс-спектры измеряли с использованием спектрометров Micromass ZQ или SQD производства Waters с ионизацией электрораспылением (ESI) или с химической ионизацией при атмосферном давлении (APCI). Реакции с микроволновой обработкой проводили с использованием реактора Initiator производства Biotage.
Ниже определены сокращения:
с: синглет; д: дублет; т: триплет; кв: квартет; дд: двойной дублет; дт: двойной триплет; тд: тройной дублет; тт: тройной триплет; ддд: двойной двойной дублет; ддт: двойной двойной триплет; дтд: двойной тройной дублет; тдд: тройной двойной дублет; м: мультиплет; уш.: уширенный; ДМСО-d6: дейтерированный диметилсульфоксид; CDCl3: дейтерированный хлороформ; ТГФ: тетрагидрофуран; DMF: N,N-диметилформамид; ДМСО: диметилсульфоксид; WSC: гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида; HOBt: моногидрат 1-гидроксибензотриазола; Pd(PPh3)4: тетракис(трифенилфосфин)палладий
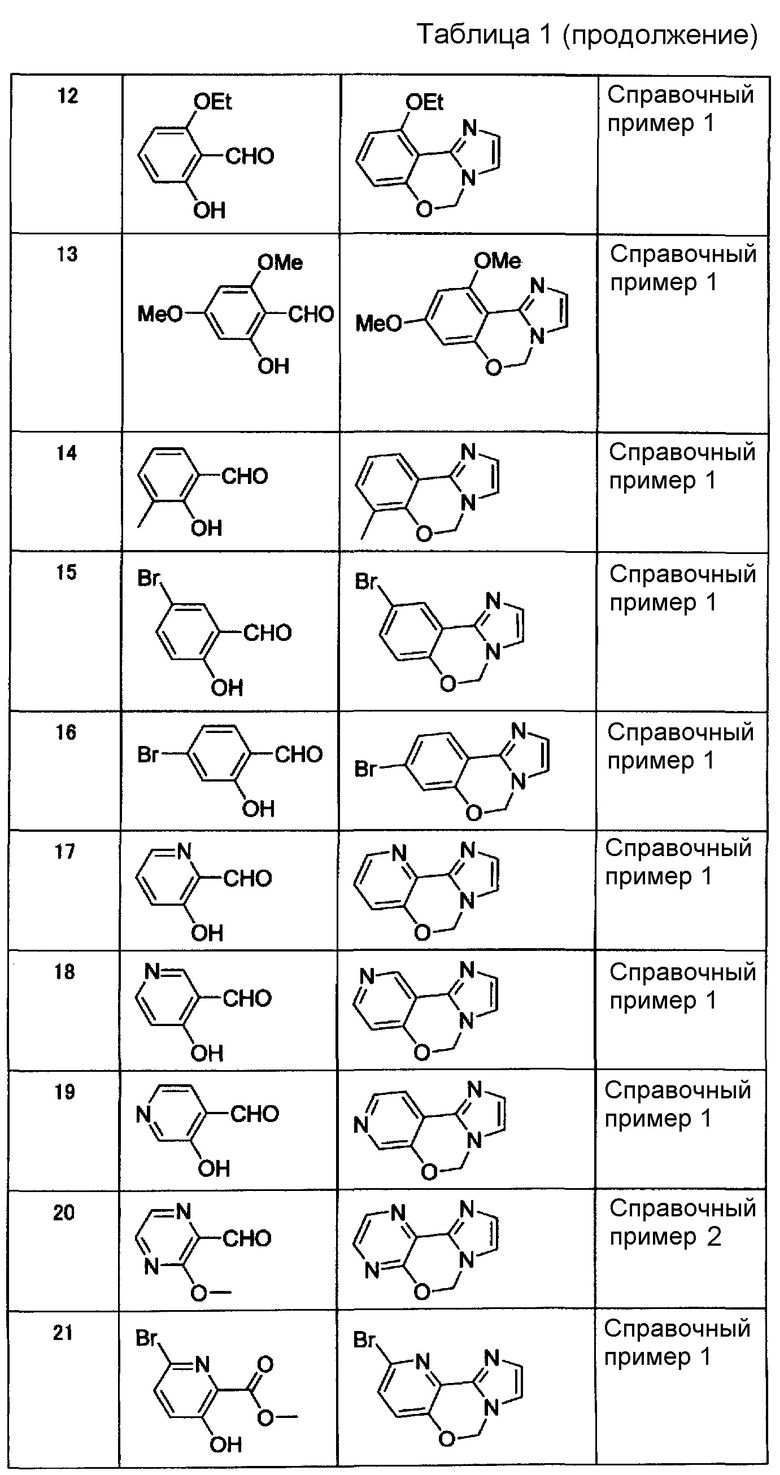
Справочный пример 1
10-Фтор-5H-бензо[e]имидазо[1,2-c][1,3]оксазин
К раствору 2-фтор-6-гидроксибензальдегида (500 мг) в метаноле (7,0 мл) добавляли 28% водный раствор аммиака (2,2 мл) и 40% водный раствор глиоксаля (1,3 мл), и перемешивали смесь при комнатной температуре в течение 5 часов. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением соответствующего имидазофенольного соединения. Полученное имидазофенольное соединение использовали в следующей реакции без дополнительной очистки. К раствору полученного имидазофенольного соединения в DMF (7,2 мл) добавляли карбонат калия (1,98 г) и дииодметан (0,44 мл), и перемешивали смесь при 80°C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой, а затем экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (415 мг, выход: 61%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,32-7,22 (2H, м), 6,98-6,88 (3H, м), 5,82 (2H, c).
ESI-MS m/z 224, 191 (MH+)
Справочный пример 2
Справочный пример 2(1): 2-Бром-3-(1H-имидазол-2-ил)пиридин
К раствору 2-бромникотинальдегида (10 г) в метаноле (90 мл) добавляли 28% водный раствор аммиака (50 мл) и 40% водный раствор глиоксаля (50 мл), и перемешивали смесь при комнатной температуре в течение 14 часов. Реакционную смесь фильтровали, и концентрировали фильтрат в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (4,62 г, выход: 38%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 10,71-10,28 (1H, уш.м), 8,61 (1H, дд, J=7,8, 2,0 Гц), 8,35 (1H, дд, J=4,6, 2,0 Гц), 7,40 (1H, дд, J=7,8, 4,6 Гц), 7,30-7,23 (2H, уш.м).
ESI-MS m/z 224, 226 (MH+)
Справочный пример 2(2): 5H-имидазо[1,2-c]пиридо[3,2-e][1,3]оксазин
К раствору продукта (44,8 мг), полученного в справочном примере 2(1), в 2-пропаноле (2,0 мл) добавляли гидроксид калия (66 мг) и 37% водный раствор формалина (0,20 мл), и перемешивали смесь при 80°C в течение 14 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (16,7 мг, выход: 48%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 8,29-8,24 (2H, м), 7,27 (1H, с), 7,25 (1H, д, J=1,2 Гц), 7,17 (1H, дд, J=7,3, 5,1 Гц), 6,98 (1H, д, J=1,2 Гц), 6,01 (2H, с).
ESI-MS m/z 174 (MH+)
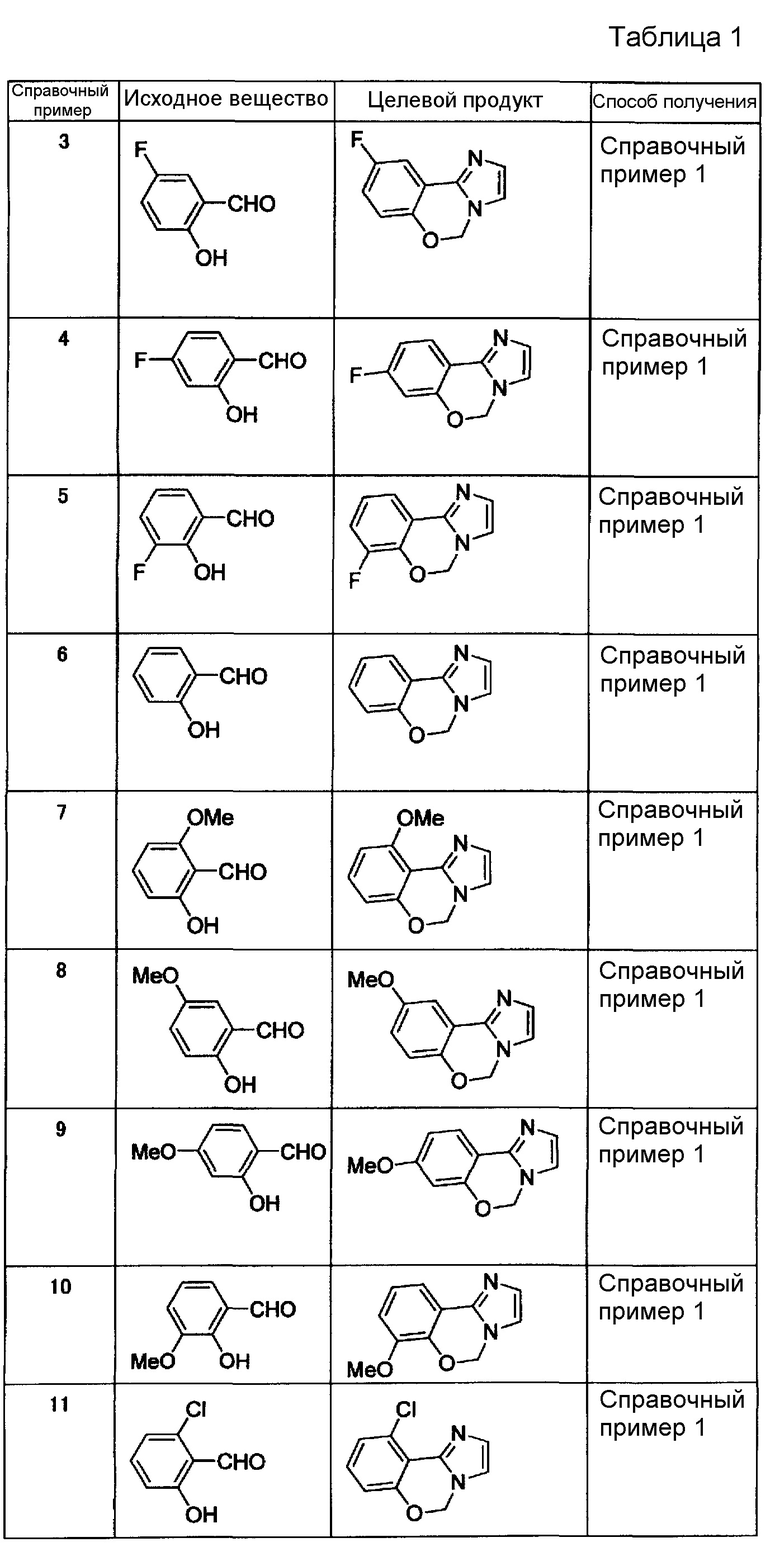
Справочные примеры 3-21
Соединения, представленные ниже в таблице 1, синтезировали в соответствии с любым способом согласно справочному примеру 1 или 2.
[Таблица 1]
[Таблица 2]
Соединения справочных примеров 20 и 21, представленных в таблице 1, синтезировали следующими способами в соответствии со способом, представленным в справочном примере 1, или способом, представленным в справочном примере 2, с использованием коммерчески доступных исходных веществ, представленных в таблице, или исходных веществ, которые могут быть синтезированы известным способом.
Справочный пример 20
Справочный пример 20(1): 2-(1H-имидазол-2-ил)-3-метоксипиразин
К раствору 3-метоксипиразин-2-карбальдегида (480 мг) в метаноле (7,5 мл) добавляли 40% водный раствор глиоксаля (0,80 мл), и медленно по каплям добавляли 28% водный аммиак (1,94 мл) при 8°C. Реакционную смесь перемешивали в течение 10 минут, а затем перемешивали при комнатной температуре в течение 1 часа. Остаток, полученный путем концентрирования реакционной смеси в условиях пониженного давления, очищали методом хроматографии на щелочном силикагеле (хлороформ/метанол) с получением целевого продукта (410 мг, выход: 66%) в виде светло-коричневато-красного аморфного вещества.
1H-ЯМР (CDCl3) δ: 10,52 (1H, уш.с), 8,25 (1H, д, J=2,4 Гц), 8,10 (1H, д, J=2,4 Гц), 7,38 (1H, уш.с), 7,21 (1H, уш.с), 4,20 (3H, с).
ESI-MS m/z 177 (MH+)
Справочный пример 20(2): 5H-имидазо[1,2-c]пиразино[2,3-e][1,3]оксазин
Раствор продукта (460 мг), полученного в справочном примере 20(1), в 5M соляной кислоте (15 мл) перемешивали при 120°C в течение 30 минут с использованием микроволнового реактора. Реакционную смесь охлаждали, подвергали азеотропной перегонке с этанолом и концентрировали в условиях пониженного давления. К раствору полученного остатка в DMF (50 мл) добавляли карбонат калия (1,79 г) и дииодметан (0,42 мл), и перемешивали смесь при 80°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и хлороформом, и экстрагировали хлороформом. Объединенный органический слой промывали водой и насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом препаративной тонкослойной хроматографии на силикагеле (хлороформ/метанол) с получением целевого продукта (36 мг, выход: 8%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 8,43 (1H, д, J=2,8 Гц), 8,19 (1H, д, J=2,8 Гц), 7,41 (1H, д, J=1,2 Гц), 7,06 (1H, д, J=1,2 Гц), 6,11 (2H, с).
ESI-MS m/z 175 (MH+)
Справочный пример 21
Справочный пример 21(1): Метил-6-бром-3-(метоксиметокси)пиколинат
К раствору метил-6-бром-3-гидроксипиридин-2-карбоксилата (970 мг) в хлороформе (20 мл) добавляли диизопропилэтиламин (1,46 мл) и помещали в атмосферу азота. Затем реакционную смесь охлаждали до 0°C, и добавляли к ней хлорметоксиметан (0,38 мл). Реакционную смесь перемешивали при 0°C в течение 5 минут, а затем перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь охлаждали до 0°C, разбавляли водой и экстрагировали хлороформом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (1,22 г, выход: 100%) в виде бесцветного масла.
1H-ЯМР (CDCl3) δ: 7,54 (1H, д, J=8,8 Гц), 7,51 (1H, д, J=8,8 Гц), 5,26 (2H, с), 3,96 (3H, с), 3,51 (3H, с).
ESI-MS m/z 276, 278 (MH+)
Справочный пример 21(2): 6-Бром-3-(метоксиметокси)пиколинальдегид
Раствор продукта (1,22 г), полученного в справочном примере 21(1), в ТГФ (20 мл) помещали в атмосферу азота. Реакционную смесь затем охлаждали до -78°C, и добавляли к ней 0,99M раствор диизобутилалюминийгидрида в толуоле (5,08 мл). Реакционную смесь перемешивали при -78°C в течение 1 часа. Затем к смеси добавляли еще 0,99M раствор диизобутилалюминийгидрида в толуоле (0,51 мл), и перемешивали смесь при -78°C в течение 1 часа. К реакционной смеси добавляли насыщенный водный раствор сегнетовой соли, а затем нагревали смесь до комнатной температуры. Реакционную смесь экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления с получением целевого продукта (1,03 г, выход: 100%) в виде бесцветного масла.
1H-ЯМР (CDCl3) δ: 10,20 (1H, с), 7,61 (1H, д, J=8,8 Гц), 7,58 (1H, д, J=8,8 Гц), 5,33 (2H, с), 3,52 (3H, с).
ESI-MS m/z 246, 248 (MH+)
Справочный пример 21(3): 6-Бром-2-(1H-имидазол-2-ил)-3-(метоксиметокси)пиридин
К раствору продукта (1,03 г), полученного в справочном примере 21(2), в метаноле (16 мл) добавляли 40% водный раствор глиоксаля (0,96 мл), и медленно в условиях охлаждения на льду по каплям добавляли 28% водный аммиак (2,32 мл). После перемешивания при комнатной температуре в течение 4 часов, реакционную смесь концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на щелочном силикагеле (хлороформ/метанол) с получением целевого продукта (0,91 г, выход: 77%) в виде бледно-желтовато-коричневого твердого вещества.
1H-ЯМР (CDCl3) δ: 10,46 (1H, уш.с), 7,53 (1H, д, J=8,8 Гц), 7,35 (1H, д, J=8,8 Гц), 7,33 (1H, уш.с), 7,17 (1H, уш.с), 5,39 (2H, с), 3,54 (3H, с).
ESI-MS m/z 284, 286 (MH+)
Справочный пример 21(4): 9-Бром-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин
К раствору продукта (0,91 г), полученного в справочном примере 21(3), в хлороформе (12 мл) по каплям в условиях охлаждения на льду добавляли трифторуксусную кислоту (6,0 мл). После перемешивания при комнатной температуре в течение 14 часов, реакционную смесь подвергали азеотропной перегонке с толуолом/хлороформом и концентрировали в условиях пониженного давления. К полученному остатку добавляли DMF (20 мл), карбонат калия (2,22 г) и дииодметан (0,52 мл), и перемешивали смесь при 80°C в течение полутора часов. Затем к смеси добавляли еще карбонат калия (0,22 г) и дииодметан (0,052 мл), и перемешивали смесь при 80°C в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и хлороформом и фильтровали через целит. Полученный фильтрат экстрагировали 10% раствором метанола в хлороформе. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия, подвергали азеотропной перегонке с толуолом и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на щелочном силикагеле (хлороформ/метанол) с получением целевого продукта (0,67 г, выход: 82%) в виде светло-коричневого твердого вещества.
1H-ЯМР (CDCl3) δ: 7,38 (1H, д, J=8,8 Гц), 7,34 (1H, д, J=1,2 Гц), 7,24 (1H, д, J=8,8 Гц), 6,99 (1H, д, J=1,2 Гц), 5,89 (2H, с).
ESI-MS m/z 252, 254 (MH+)
Справочный пример 22
Справочный пример 22(1): 3-Бром-10-фтор-5H-бензо[e]имидазо[1,2-c][1,3]оксазин
Раствор продукта (349 мг), полученного в справочном примере 1, в хлороформе (7,0 мл) охлаждали до 0°C. К раствору добавляли N-бромсукцинимид (343 мг), и перемешивали смесь при 0°C в течение 1 часа. Реакционную смесь очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (360 мг, выход: 73%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,32-7,26 (1H, м), 7,25 (1H, с), 6,99-6,91 (2H, м), 5,78 (2H, с).
ESI-MS m/z 269, 271 (MH+)
Справочный пример 22(2): 2-Бром-10-фтор-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин
К раствору продукта (513 мг), полученного в справочном примере 22(1), в 1,4-диоксане (10 мл) и воде (1,3 мл) добавляли фенилбороновую кислоту (349 мг) и карбонат цезия (1,55 г), и помещали смесь в атмосферу азота. К смеси добавляли Pd(PPh3)4 (221 мг), и перемешивали смесь при 100°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением соответствующего продукта сочетания. Полученный продукт сочетания использовали в следующей реакции без дополнительной очистки. Раствор полученного продукта сочетания в хлороформе (5,0 мл) охлаждали до 0°C. К раствору добавляли N-бромсукцинимид (380 мг), и перемешивали смесь при комнатной температуре в течение 1 часа. Реакционную смесь очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (602 мг, выход: 91%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,55-7,42 (5H, м), 7,32-7,27 (1H, м), 6,99-6,94 (1H, м), 6,92-6,89 (1H, м), 5,73 (2H, с).
ESI-MS m/z 345, 347 (MH+).
Справочный пример 23
Справочный пример 23(1): 3,9-Дибром-5H-бензо[e]имидазо[1,2-c][1,3]оксазин
Целевой продукт (389 мг, выход: 98%) получали в виде бесцветного твердого вещества тем же способом, что и описанный в справочном примере 22(1), с использованием в реакции продукта (300 мг), полученного в справочном примере 15.
1H-ЯМР (CDCl3) δ: 8,03 (1H, д, J=2,4 Гц), 7,41 (1H, дд, J=8,8, 2,4 Гц), 7,16 (1H, с), 6,96 (1H, д, J=8,8 Гц), 5,76 (1H, с).
ESI-MS m/z 331 (MH+)
Справочный пример 23(2): 9-Бром-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин
К раствору продукта (9,44 г), полученного в справочном примере 23(1), в 1,4-диоксане (250 мл) и воде (40 мл) добавляли фенилбороновую кислоту (3,35 г) и карбонат цезия (23,3 г), и помещали смесь в атмосферу азота. Затем к смеси добавляли Pd(PPh3)4 (3,30 г) и перемешивали смесь при комнатной температуре в течение 14 часов и при 50°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (7,32 г, выход: 78%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ,: 8,11 (1H, д, J=2,4 Гц), 7,50-7,32 (6H, м), 7,28 (1H, с), 6,95 (1H, д, J=8,5 Гц), 5,84 (2H, с).
ESI-MS m/z 327, 329 (MH+)
Справочный пример 23(3): Метил-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбоксилат
К раствору продукта (5,0 г), полученного в справочном примере 23(2), в DMF (30 мл) и метаноле (30 мл) добавляли диизопропилэтиламин (8,0 мл) и комплекс дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладия(II) и дихлорметана (1,38 г) и помещали смесь в атмосферу монооксида углерода, а затем перемешивали при 70°C в течение 28 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли насыщенным водным раствором гидрокарбоната натрия и экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (2,12 г, 45%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 8,70 (1H, д, J=2,0 Гц), 8,02 (1H, дд, J=8,5, 2,0 Гц), 7,52-7,46 (2H, м), 7,44-7,36 (3H, м), 7,31 (1H, с), 7,13 (1H, д, J=8,5 Гц), 5,93 (2H, с), 3,93 (3H, с).
ESI-MS m/z 307 (MH+).
Справочный пример 23(4): Метил-2-бром-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбоксилат
К раствору продукта (1,0 г), полученного в справочном примере 23(3), в хлороформе (16 мл) добавляли N-бромсукцинимид (754 мг) и перемешивали смесь при комнатной температуре в течение 1 часа. Реакционную смесь фильтровали и промывали остаток хлороформом с получением целевого продукта (800 мг, выход: 64%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 8,71 (1H, д, J=2,0 Гц), 8,04 (1H, дд, J=8,5, 2,0 Гц), 7,56-7,42 (5H, м), 7,12 (1H, д, J=8,5 Гц), 5,80 (2H, с), 3,93 (3H, с).
ESI-MS m/z 385, 387 (MH+)
Справочный пример 24
(2-Бром-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-ил)метанол
Раствор продукта (550 мг), полученного в справочном примере 23(4), в хлористом метилене (14 мл) охлаждали до 0°C. К раствору добавляли 0,99M раствора диизобутилалюминийгидрида в толуоле (4,3 мл) и перемешивали смесь при 0°C в течение 1 часа. К реакционной смеси добавляли насыщенный раствор сегнетовой соли, после чего смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (397 мг, выход: 78%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ,: 7,98 (1H, д, J=2,0 Гц), 7,55-7,42 (5H, м), 7,37 (1H, дд, J=8,3, 2,2 Гц), 7,07 (1H, д, J=8,3 Гц), 5,73 (2H, с), 4,74-4,70 (2H, уш.м)
ESI-MS m/z 357, 359 (MH+)
Справочный пример 25
Синтез 2-Бром-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбонитрила
К раствору продукта (500 мг), полученного в справочном примере 23(2), в 1,4-диоксане (3,0 мл) и DMF (3,0 мл) в атмосфере азота добавляли цианид цинка (360 мг) и ди-трет-бутилпалладий (78,2 мг) и перемешивали смесь при 100°C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали. Фильтрат последовательно промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением соответствующего цианосоединения. Цианосоединение использовали в следующей реакции без дополнительной очистки. К раствору полученного цианосоединения в хлороформе (8,0 мл) добавляли N-бромсукцинимид (352 мг) и перемешивали смесь при комнатной температуре в течение 1 часа. Реакционную смесь фильтровали, и промывали остаток хлороформом с получением целевого продукта (207 мг, выход: 36%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 8,26 (1H, д, J=2,0 Гц), 7,58 (1H, дд, J=8,5, 2,0 Гц), 7,53-7,40 (5H, м), 7,14 (1H, д, J=8,5 Гц), 5,80 (2H, с).
ESI-MS m/z 352, 354 (MH+).
Справочный пример 26
2-Бром-3-фенил-9-(1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин
К раствору продукта (100 мг), полученного в справочном примере 23(2), в 1,4-диоксане (3,0 мл) и воде (0,5 мл) добавляли 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол (198 мг) и карбонат цезия (250 мг), и помещали смесь в атмосферу азота. Затем к смеси добавляли Pd(PPh3)4 (35,4 мг), и перемешивали смесь при 100°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением соответствующего продукта сочетания. Полученный продукт сочетания использовали в следующей реакции без дополнительной очистки. К раствору полученного продукта сочетания в хлороформе (3,0 мл) добавляли N-бромсукцинимид (65,4 мг) и перемешивали смесь при комнатной температуре в течение 1 часа. Реакционную смесь очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (150 мг, выход: 93%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 8,16 (1H, д, J=2,0 Гц), 7,70 (1H, дд, J=8,3, 2,2 Гц), 7,57 (1H, д, J=1,7 Гц), 7,54-7,42 (5H, м), 7,15 (1H, д, J=8,3 Гц), 6,45 (1H, д, J=1,7 Гц), 5,77 (2H, с), 5,45 (2H, с), 3,77-3,71 (2H, м), 0,99-0,94 (2H, м), 0,00 (9H, с).
ESI-MS m/z 523, 525 (MH+).
Справочный пример 27
2-Бром-3-фенил-9-(1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин
К раствору продукта (100 мг), полученного в справочном примере 23(2), в 1,4-диоксане (3,0 мл) и воде (0,5 мл) добавляли 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол (148 мг) и карбонат цезия (250 мг), и помещали смесь в атмосферу азота. Затем к смеси добавляли Pd(PPh3)4 (35,4 мг), и перемешивали смесь при 100°C в течение 1,5 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением соответствующего продукта сочетания. Полученный продукт сочетания использовали в следующей реакции без дополнительной очистки. К раствору полученного продукта сочетания в хлороформе (3,0 мл) добавляли N-бромсукцинимид (60,0 мг) и перемешивали смесь при комнатной температуре в течение 1 часа. Реакционную смесь очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (120 мг, выход: 75%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 8,12 (1H, д, J=2,2 Гц), 7,86-7,85 (2H, м), 7,55-7,43 (6H, м), 7,09 (1H, д, J= 8,5 Гц), 5,75 (2H, с), 5,46 (2H, с), 3,64-3,58 (2H, м), 0,97-0,92 (2H, м), 0,00 (9H, с).
ESI-MS m/z 525, 527 (MH+).
Справочный пример 28
9-Метил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин
К раствору продукта (50 мг), полученного в справочном примере 21(4), в 1,4-диоксане (2,0 мл) и воде (0,32 мл) добавляли метилбороновую кислоту (17,8 мг) и карбонат цезия (162 мг), и помещали смесь в атмосферу азота. Затем к смеси добавляли Pd(PPh3)4 (22,9 мг), и перемешивали смесь при 80°C в течение 4 часов. К реакционной смеси добавляли метилбороновую кислоту (17,8 мг), и перемешивали смесь при 110°C в течение 2 часов. К смеси дополнительно добавляли метилбороновую кислоту (17,8 мг), и перемешивали смесь при 110°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали хлороформом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (15,2 мг, выход: 41%) в виде бесцветного масла.
1H-ЯМР (CDCl3) δ: 7,30 (1H, д, J=1,2 Гц), 7, 26 (1H, д, J=8,4 Гц), 7,08 (1H, д, J=8,4 Гц), 6,97 (1H, д, J=1,2 Гц), 5,84 (2H, с), 2,60 (3H, с).
ESI-MS m/z 188 (MH+)
Справочный пример 29
9-Метокси-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин
К раствору продукта (80 мг), полученного в справочном примере 21(4), в метаноле (2,0 мл) добавляли 25 масс.% раствор метоксида натрия в метаноле (0,36 мл) и перемешивали смесь при 110°C в течение 22 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и хлороформом и экстрагировали хлороформом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом препаративной тонкослойной хроматографии на щелочном силикагеле (хлороформ/метанол) с получением целевого продукта (58,4 мг, выход: 91%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,32 (1H, д, J=8,8 Гц), 7,31-7,30 (1H, м), 6,96 (1H, д, J=0,8 Гц), 6,71 (1H, д, J=8,8 Гц), 5,81 (2H, с), 4,05 (3H, с).
ESI-MS m/z 204 (MH+)
Справочные примеры 30-55
Соединения, представленные ниже в таблице 2, синтезировали в соответствии с любым способом, представленным в справочных примерах 21-25.
[Таблица 3]
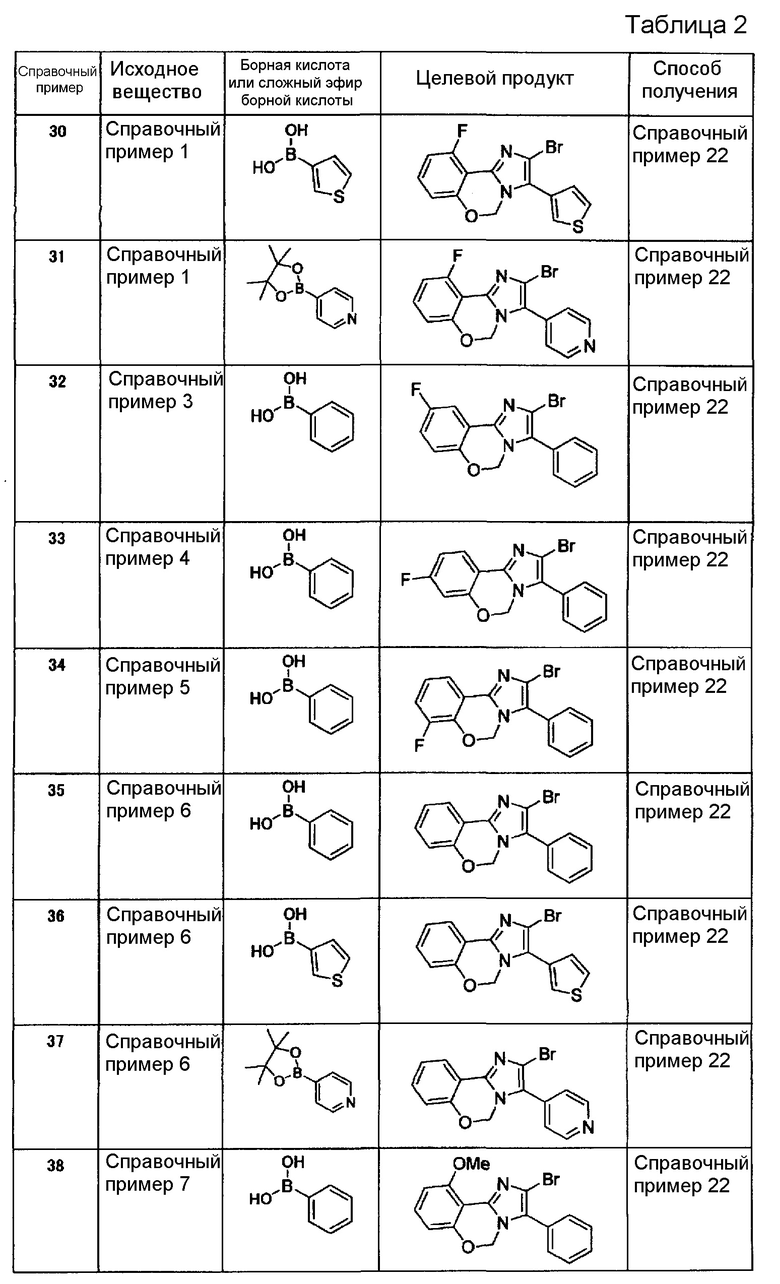 [Таблица 4]
[Таблица 4]
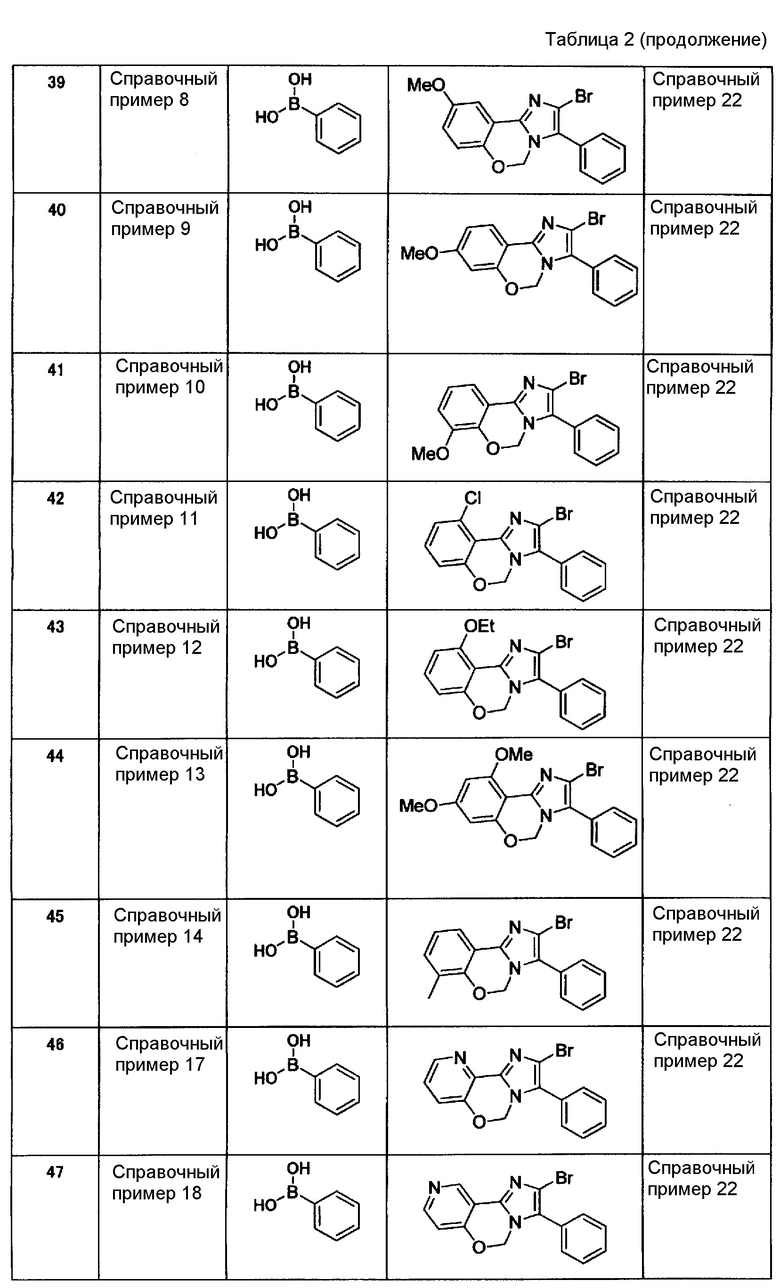
[Таблица 5]
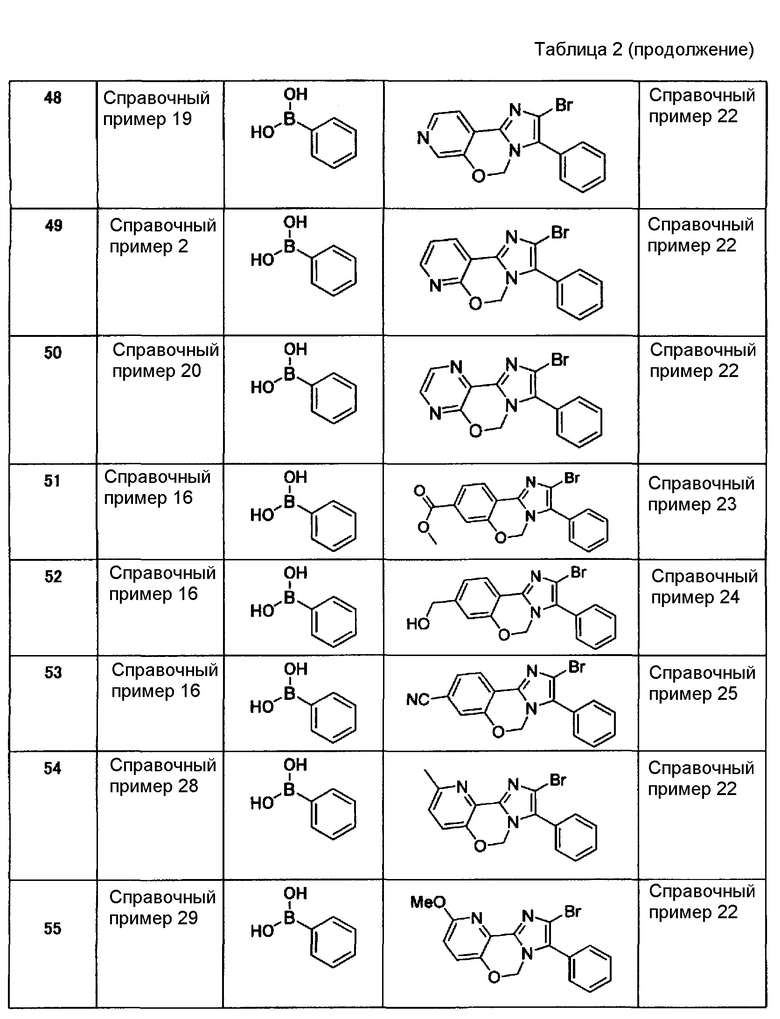
Справочный пример 56
Справочный пример 56(1): 1-(4-бромфенил)циклобутанкарбонитрил
Раствор гидроксида калия (56,5 г) и бромида тетрабутиламмония (2,92 г) в толуоле (400 мл) и воде (30 мл) нагревали до 70°C. Затем к раствору последовательно добавляли 1,3-дибромпропан (39,0 г) и 2-(4-бромфенил)ацетонитрил (35,5 г), и перемешивали смесь при 100°C в течение 3 часов. После охлаждения реакционной смеси до 80°C, к ней добавляли гептан (100 мл), и дополнительно охлаждали смесь до комнатной температуры. Реакционную смесь фильтровали и промывали гексаном и разделяли органический слой. Полученный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (24,0 г, выход: 56%) в виде бесцветного масла.
1H-ЯМР (CDCl3) δ,: 7,53 (2H, д, J=8,8 Гц), 7,29 (2H, д, J=8,8 Гц), 2,87-2,79 (2H, м), 2,63-2,54 (2H, м), 2,50-2,38 (1H, м), 2,13-2,03 (1H, м).
ESI-MS m/z 236, 238 (MH+).
Справочный пример 56(2): 1-(4-Бромфенил)циклобутанкарбоновая кислота
К раствору продукта (24,0 г), полученного в справочном примере 56(1), в бутаноле (100 мл) добавляли 50% водный раствор гидроксида натрия (35 мл) и перемешивали смесь при 120°C в течение 14 часов. После охлаждения до комнатной температуры, к реакционной смеси добавляли воду (100 мл), а затем промывали эфиром. Эфирный слой дополнительно дважды экстрагировали 1н водным раствором гидроксида натрия (50 мл). К объединенному водному слою добавляли 5M соляную кислоту, доводили значение pH до 2, а затем экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Путем добавления к полученному остатку гексана и проведения фильтрования получали целевой продукт (20,4 г, выход: 79%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,45 (2H, д, J=8,5 Гц), 7,17 (2H, д, J=8,5 Гц), 2,88-2,79 (2H, м), 2,53-2,43 (2H, м), 2,15-2,02 (1H, м), 1,93-1,81 (1H, м).
ESI-MS m/z 255, 257 (MH+).
Справочный пример 56(3): трет-Бутил-1-(4-бромфенил)циклобутилкарбамат
К раствору продукта (12,7 г), полученного в справочном примере 56(2), в ТГФ (150 мл) последовательно добавляли ди-трет-бутоксидикарбонат (12,0 г), азид натрия (11,3 г), бромид тетрабутиламмония (2,41 г) и трифторметансульфонат цинка (181 мг) и смесь нагревали с обратным холодильником в течение 14 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и водой и экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (14,7 г, выход: 91%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,45 (2H, д, J=8,5 Гц), 7,30 (2H, д, J=8,5 Гц), 5,08 (1H, уш.с), 2,56-2,43 (4H, м), 2,16-2,04 (1H, м), 1,91-1,79 (1H, м), 1,37 (9H, с).
ESI-MS m/z 326, 327 (MH+).
Справочный пример 56(4): трет-Бутил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутилкарбамат
К раствору продукта (3,21 г), полученного в справочном примере 56(3), в DMF (25 мл) последовательно добавляли ацетат калия (2,41 г) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (6,25 г), и помещали смесь в атмосферу азота. К смеси добавляли комплекс дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладия(II) и дихлорметана (360 мг), и перемешивали смесь при 80°C в течение 10 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли к ней воду, а затем экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (3,20 г, выход: 87%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,79 (2H, д, J=8,0 Гц), 7,43 (2H, д, J=8,0 Гц), 5,07 (1H, уш.с), 2,59-2,31 (4H, м), 2,14-2,03 (1H, м), 1,90-1,78 (1H, м), 1,36 (9H, с), 1,34 (23H, с).
ESI-MS m/z 374 (MH+).
Справочный пример 57
Справочный пример 57(1): цис-1-(4-Бромфенил)-3-гидроксициклобутанкарбоновая кислота
Раствор 4-бромфенилуксусной кислоты (107,8 г) в ТГФ (100 мл) при перемешивании в условиях охлаждения на льду по каплям добавляли к раствору 2M изопропилмагнийхлорида в ТГФ (560 мл), смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. К полученной суспензии при комнатной температуре по каплям добавляли эпихлоргидрин (73 мл), и смесь нагревали от 26°C до температуры реакции, охлаждали, и перемешивали в течение 3 часов с поддержанием указанной температуры. К полученной темно-коричневой реакционной смеси при комнатной температуре по каплям добавляли раствор 2M изопропилмагнийхлорида в ТГФ (560 мл), и перемешивали смесь в течение ночи на водяной бане. К реакционной смеси в условиях охлаждения на льду осторожно добавляли 2M соляную кислоту (900 мл), и экстрагировали этилацетатом. Полученный органический слой промывали 1M соляной кислотой, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток суспендировали в этилацетате, твердое вещество отделяли фильтрованием, а затем промывали этилацетатом и сушили в условиях пониженного давления, с получением целевого продукта (91,46 г, выход: 68%) в виде бесцветного твердого вещества.
1H-ЯМР (CD3ОD) δ: 7,49 (2H, д, J=8,8 Гц), 7,34 (2H, д, J=8,8 Гц), 4,01 (1H, квинтет, J=7,3 Гц), 2,88-2,80 (2H, м), 2,69-2,61 (2H, м).
ESI-MS m/z 269 и 271 (MH-).
Справочный пример 57(2): Метил-цис-1-(4-бромфенил)-3-гидроксициклобутанкарбоксилат
Продукт (116,0 г), полученный в справочном примере 57(1), растворяли в метаноле (500 мл). К нему при комнатной температуре добавляли концентрированную серную кислоту (3,5 мл), и нагревали смесь с обратным холодильником в течение ночи. Реакционную смесь концентрировали в условиях пониженного давления для удаления метанола, разбавляли водой и экстрагировали этилацетатом. Объединенный органический слой промывали 1M водным раствором гидроксида натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления с получением целевого продукта (112,5 г, выход: 99%) в виде твердого вещества светло-желтого цвета.
1H-ЯМР (CDCl3) δ: 7,47 (2H, д, J=8,5 Гц), 7,22 (2H, д, J=8,5 Гц), 4,19 (1H, м), 3,64 (3H, с), 2,93-2,85 (2H, м), 2,76-2,69 (2H, м), 2,21 (1H, д, J=6,3 Гц).
Справочный пример 57(3): Метил-1-(4-бромфенил)-3-оксоциклобутилкарбоксилат
Продукт (112,5 г), полученный в справочном примере 57(2), растворяли в хлороформе (500 мл) и добавляли к нему N-метилморфолин-N-оксид (63,3 г) и измельченные 4А молекулярные сита (120 г). Смесь охлаждали на льду, добавляли к ней тетра-н-пропиламмония перрутенат (2,76 г), и перемешивали смесь в течение 24 часов в процессе нагревания до комнатной температуры. Реакционную смесь разбавляли гексаном, адсорбировали на силикагеле, элюировали смесью гексан/этилацетат (3/1), и концентрировали элюат в условиях пониженного давления. Полученное светло-желтое твердое вещество суспендировали в гексане, твердое вещество отделяли фильтрованием, а затем промывали гексаном и сушили в условиях пониженного давления с получением целевого продукта (83,4 г, выход: 69%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,52 (2H, д, J=8,8 Гц), 7,24 (2H, д, J=8,8 Гц), 3,95-3,87 (2H, м), 3,71 (3H, с), 3,57-3,49 (2H, м).
Справочный пример 57(4): транс-3-Амино-3-(4-бромфенил)-1-циклопропилциклобутанол
Раствор продукта (18,57 г), полученного в справочном примере 57(3), в толуоле (200 мл) охлаждали до -40°C и по каплям добавляли к нему раствор 0,7M циклопропилмагнийбромида в ТГФ (310 мл). После перемешивания при -40°C в течение 15 минут и при 0°C в течение 3 часов к реакционной смеси осторожно добавляли лед, а затем насыщенный водный раствор хлорида аммония и экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток растворяли в 1,4-диоксане (100 мл), при комнатной температуре добавляли к нему 1M водный раствор гидроксида натрия (150 мл), а затем перемешивали в течение ночи. Реакционную смесь концентрировали в условиях пониженного давления, и удаляли 1,4-диоксан. Водный слой промывали толуолом. Полученный водный раствор подкисляли добавлением 2M соляной кислоты и экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток растворяли в 1,4-диоксане (215 мл) и при комнатной температуре добавляли к нему N,N-диизопропилэтиламин (7,60 мл) и дифенилфосфорилазид (8,77 мл). Смесь перемешивали при комнатной температуре в течение 4 часов, а затем при 63°C в течение 4 часов и охлаждали до комнатной температуры. Полученную реакционную смесь по каплям добавляли к энергично перемешиваемой 0,5M соляной кислоте (1000 мл) и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь промывали этилацетатом, и подщелачивали полученный водный раствор добавлением 2M водного раствора гидроксида натрия. После растворения хлорида натрия до насыщения проводили экстрагирование хлороформом. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления с получением целевого продукта (5,52 г, выход: 30%) в виде светло-желтого масла.
1H-ЯМР (CDCl3) δ: 7,46 (2H, д, J=8,8 Гц), 7,34 (2H, д, J=8,8 Гц), 2,60-2,54 (2H, м), 2,31-2,26 (2H, м), 1,36-1,29 (1H, м), 0,61-0,55 (2H, м), 0,47-0,42 (2H, м).
ESI-MS m/z 282 и 284 (MH+).
Справочный пример 57(5): 2-(транс-1-(4-бромфенил)-3-циклопропил-3-гидроксициклобутил)изоиндолин-1,3-дион
К раствору продукта (882 мг), полученного в справочном примере 57(4), в хлороформе (15,6 мл) добавляли триэтиламин (0,52 мл) и N-этоксикарбонилфталимид (683 мг) и перемешивали смесь при 70°C в течение 38 часов. Реакционную смесь охлаждали, разбавляли водой и экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (1,18 г, выход: 92%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,77-7,73 (2H, м), 7,70-7,66 (2H, м), 7,60-7,56 (2H, м), 7,47-7,43 (2H, м), 3,11-2,99 (4H, м), 1,49 (1H, с), 1,16-1,12 (1H, м), 0,51-0,45 (2H, м), 0,32-0,27 (2H, м).
Справочный пример 57(6): 2-(транс-3-цклопропил-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)изоиндолин-1,3-дион
К раствору продукта (1,26 г), полученного в справочном примере 57(5), в 1,4-диоксане (15 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (1,14 г), ацетат калия (883 мг) и комплекс дихлор(1,1'-бис(дифенилфосфино)ферроцен)палладия(II) и дихлорметана (245 мг), и перемешивали смесь в атмосфере азота при 80°C в течение 16 часов. Реакционную смесь охлаждали, разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) и концентрировали в условиях пониженного давления. Полученное твердое вещество промывали этилацетатом/гексаном с получением целевого продукта (1,12 г, выход: 81%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,81-7,63 (8H, м), 3,14-3,05 (4H, м), 1,49 (1H, с), 1,32 (12H, c), 1,16-1,10 (1H, м), 0,50-0,44 (2H, м), 0,33-0,28 (2H, м).
Справочный пример 58
Справочный пример 58(1): транс-1-(4-Бромфенил)-3-гидрокси-3-метилциклобутанкарбоновая кислота
Раствор продукта (11,62 г), полученного в справочном примере 57(3), в ТГФ (210 мл) охлаждали до -40°C и по каплям добавляли раствор 3M метилмагнийхлорида в ТГФ (48 мл). После перемешивания при -40°C в течение 15 минут и при 0°C в течение 2 часов, к реакционной смеси осторожно добавляли лед, а затем насыщенный водный раствор хлорида аммония, и экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток растворяли в 1,4-диоксане (60 мл), к нему при комнатной температуре добавляли 1M водный раствор гидроксида натрия (62 мл), а затем перемешивали в течение ночи. Полученную реакционную смесь концентрировали в условиях пониженного давления для удаления 1,4-диоксана и вливали в 0,5M водный раствор гидроксида натрия, и промывали водный слой этилацетатом. Полученный щелочной водный раствор подкисляли добавлением 2M соляной кислоты и экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток кристаллизовали из смеси хлороформ/гексан с получением целевого продукта (5,92 г, выход: 51%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,45 (2H, д, J=8,5 Гц), 7,17 (2H, д, J=8,5 Гц), 3,09-3,04 (2H, м), 2,62-2,56 (2H, м), 1,43 (3H, с).
ESI-MS m/z 283 и 285 (MH-).
Справочный пример 58(2): транс-3-Амино-3-(4-бромфенил)-1-метилциклобутанол
К раствору продукта (4,28 г), полученного в справочном примере 58(1), в 1,4-диоксане (60 мл) добавляли триэтиламин (2,20 мл) и дифенилфосфорилазид (3,40 мл) и перемешивали смесь при 80°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли к охлажденной на льду 1M соляной кислоте (60 мл) и перемешивали смесь при комнатной температуре в течение 2 часов. К реакционной смеси добавляли воду, смесь промывали диэтиловым эфиром, подщелачивали добавлением 5M раствора гидроксида натрия и экстрагировали хлороформом. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления с получением целевого продукта (3,23 г, выход: 84%) в виде бесцветного масла.
1H-ЯМР (CDCl3) δ: 7,49-7,43 (2H, м), 7,27-7,22 (2H, м), 2,64-2,57 (2H, м), 2,40-2,33 (2H, м), 1,64 (3H, с).
ESI-MS m/z 256, 258 (MH+).
Справочный пример 58(3): трет-Бутил-транс-1-(4-бромфенил)-3-гидрокси-3-метилциклобутилкарбамат
К раствору продукта (3,23 г), полученного в справочном примере 58(2), в 1,4-диоксане (63 мл) добавляли ди-трет-бутил-дикарбонат (3,30 г), и перемешивали смесь при 70°C в течение 3 часов. Реакционную смесь концентрировали в условиях пониженного давления и перекристаллизовывали из гексана/этилацетата с получением целевого продукта (3,50 г, выход: 78%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,47-7,42 (2H, м), 7,28 (2H, д, J=8,5 Гц), 4,96 (1H, уш.с), 2,77-2,47 (4H, м), 1,67 (1H, с), 1,58 (3H, с), 1,38 (9H, уш.с).
ESI-MS m/z 356, 358 (MH+).
Справочный пример 58(4): трет-Бутил-транс-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутилкарбамат
К раствору продукта (3,74 г), полученного в справочном примере 58(3), в DMF (42 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (3,47 г) и ацетат калия (3,09 г) и помещали смесь в атмосферу азота. К раствору добавляли комплекс дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладия(II) и дихлорметана (0,43 г), и перемешивали смесь при 80°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли к ней воду, а затем экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (3,39 г, выход: 80%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,78 (2H, д, J=8,1 Гц), 7,41 (2H, д, J=8,1 Гц), 4,95 (1H, уш.с), 2,78-2,49 (4H, м), 1,65 (1H, с), 1,58 (3H, с), 1,37 (9H, уш.с), 1,34 (12H, с).
ESI-MS m/z 404 (MH+).
Справочный пример 59: трет-Бутил-транс-3-этил-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутилкарбамат
Целевой продукт получали в виде бесцветного твердого вещества с использованием в реакции продукта, полученного в справочном примере 57(3), тем же способом, что и описанный в справочном примере 58, но используя этилмагнийбромид вместо метилмагнийхлорида в справочном примере 58(1).
1H-ЯМР (CDCl3) δ: 7,78 (2H, д, J=7,8 Гц), 7,43 (2H, д, J=7,8 Гц), 4,92 (1H, уш.с), 2,80-2,45 (4H, м), 1,83 (2H, кв, J=7,2 Гц), 1,53 (1H, с), 1,45-1,25 (9H, м), 1,34 (12H, с), 0,97 (3H, т, J=7,2 Гц).
ESI-MS m/z 418 (MH+).
Пример 1
транс-3-Амино-1-циклопропил-3-(4-(10-фтор-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
К раствору продукта (15,0 мг), полученного в справочном примере 22(2) в 1,4-диоксане (1,0 мл) и воде (0,13 мл), добавляли продукт (30,0 мг), полученный в справочном примере 57(6), и карбонат цезия (35,4 мг) и помещали смесь в атмосферу азота. Затем к ней добавляли Pd(PPh3)4 (5,0 мг), и перемешивали смесь при 100°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением соответствующего продукта сочетания. Полученный продукт сочетания использовали в следующей реакции без дополнительной очистки. К раствору полученного продукта сочетания в этаноле (2,0 мл) добавляли моногидрат гидразина (0,5 мл) и перемешивали смесь при 110°C в течение 20 минут с использованием микроволнового реактора. Реакционную смесь охлаждали до комнатной температуры, разбавляли насыщенным гидрокарбонатом натрия и экстрагировали хлороформом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом препаративной высокоэффективной жидкостной хроматографии с обращенной фазой (0,1% трифторуксусная кислота, ацетонитрил/вода) и концентрировали в условиях пониженного давления. Затем проводили процедуру обессоливания с использованием Bond Elut (зарегистрированный товарный знак) (метанол) производства Varian, Inc. с получением указанного в заголовке соединения (16,8 мг, выход: 83%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,60 (2H, д, J=8,3 Гц), 7,48 (1H, дд, J=5,0, 3,0 Гц), 7,38-7,32 (3H, м), 7,28-7,23 (1H, м), 7,08 (1H, дд, J=5,0, 1,3 Гц), 6,99-6,93 (1H, м), 6,92-6,88 (1H, м), 5,69 (2H, с), 2,64-2,58 (2H, м), 2,33-2,27 (2H, м), 1,34 (1H, тт, J=8,3, 5,4 Гц), 0,59-0,53 (2H, м), 0,48-0,43 (2H, м).
ESI-MS m/z 468 (MH+).
Пример 2
транс-3-Амино-3-(4-(10-фтор-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол
К раствору продукта (15,0 мг), полученного в справочном примере 22(2), в 1,4-диоксане (1,0 мл) и воде (0,13 мл) добавляли продукт (28,3 мг), полученный в справочном примере 58(4), и карбонат цезия (35,4 мг) и помещали смесь в атмосферу азота. Затем к ней добавляли Pd(PPh3)4 (5,0 мг) и перемешивали смесь при 100°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением соответствующего продукта сочетания. Полученный продукт сочетания использовали в следующей реакции без дополнительной очистки. К раствору полученного продукта сочетания в хлороформе (1,0 мл) добавляли трифторуксусную кислоту (0,5 мл) и перемешивали смесь при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в условиях пониженного давления, полученный остаток очищали методом препаративной высокоэффективной жидкостной хроматографии с обращенной фазой (0,1% трифторуксусная кислота, ацетонитрил/вода) и концентрировали в условиях пониженного давления. Затем проводили процедуру обессоливания с использованием Bond Elut (зарегистрированный товарный знак) (метанол) производства Varian, Inc. с получением указанного в заголовке соединения (15,2 мг, выход: 79%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 7,57-7,53 (2H, м), 7,50-7,42 (3H, м), 7,38-7,33 (2H, м), 7,28-7,20 (3H, м), 6,99-6,93 (1H, м), 6,91-6,87 (1H, м), 5,65 (2H, с), 2,63-2,58 (2H, м), 2,39-2,32 (2H, м), 1,62 (3H, с).
ESI-MS m/z 442 (MH+).
Пример 3
1-(4-(10-Фтор-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанамин
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 22(2), и того же способа, что и описанный в примере 2, но с использованием продукта, полученного в справочном примере 56(4), вместо продукта, полученного в справочном примере 58(4).
1H-ЯМР (CDCl3) δ: 7,57 (2H, д, J=8,3 Гц), 7,51-7,44 (3H, м), 7,39-7,35 (2H, м), 7,30-7,22 (3H, м), 7,00-6,94 (1H, м), 6,92-6,88 (1H, м), 5,66 (2H, с), 2,57-2,48 (2H, м), 2,17-1,99 (3H, м), 1,78-1,69 (1H, м).
ESI-MS m/z 412 (MH+).
Пример 4
транс-3-Амино-1-циклопропил-3-(4-(10-фтор-3-(тиофен-3-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 30, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 7,60 (2H, д, J=8,3 Гц), 7,48 (1H, дд, J=5,0, 3,0 Гц), 7,38-7,32 (3H, м), 7,28-7,23 (1H, м), 7,08 (1H, дд, J=5,0, 1,3 Гц), 6,99-6,93 (1H, м), 6,92-6,88 (1H, м), 5,69 (2H, с), 2,64-2,58 (2H, м), 2,33-2,27 (2H, м), 1,34 (1H, тт, J=8,3, 5,4 Гц), 0,59-0,53 (2H, м), 0,48-0,43 (2H, м).
ESI-MS m/z 474 (MH+).
Пример 5
транс-3-Амино-1-циклопропил-3-(4-(10-фтор-3-(пиридин-4-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 31, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 8,71 (2H, д, J=6,1 Гц), 7,53 (2H, д, J=8,5 Гц), 7,37 (2H, д, J=8,5 Гц), 7,33-7,24 (3H, м), 7,01-6,91 (2H, м), 5,74 (2H, с), 2,64-2,58 (2H, м), 2,34-2,28 (2H, м), 1,35 (1H, тт, J=8,3, 5,4 Гц), 0,60-0,54 (2H, м), 0,49-0,44 (2H, м).
ESI-MS m/z 469 (MH+).
Пример 6
транс-3-Амино-3-(4-(10-фтор-3-(тиофен-3-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 30, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 7,59 (2H, д, J=8,5 Гц), 7,48 (1H, дд, J=4,9, 2,9 Гц), 7,33 (1H, дд, J=3,0, 1,3 Гц), 7,29-7,23 (3H, м), 7,08 (1H, дд, J=4,9, 1,3 Гц), 6,99-6,93 (1H, м), 6,92-6,88 (1H, м), 5,69 (2H, с), 2,66-2,60 (2H, м), 2,41-2,34 (2H, м), 1,64 (3H, с).
ESI-MS m/z 448 (MH+).
Пример 7
транс-3-Амино-3-(4-(10-фтор-3-(пиридин-4-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 31, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 8,68 (2H, д, J=5,9 Гц), 7,51 (2H, д, J=8,3 Гц), 7,32-7,22 (5H, м), 7,00-6,90 (2H, м), 5,73 (2H, с), 2,66-2,60 (3H, м), 2,41-2,35 (2H, м), 1,64 (3H, с).
ESI-MS m/z 443 (MH+).
Пример 8
транс-3-Амино-3-(4-(9-фтор-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 32, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 7,80-7,76 (1H, м), 7,55-7,43 (5H, м), 7,37-7,33 (2H, м), 7,25-7,21 (2H, м), 7,05-6,96 (2H, м), 5,65 (2H, с), 2,64-2,58 (2H, м), 2,38-2,32 (2H, м), 1,62 (3H, с).
ESI-MS m/z 443 (MH+).
Пример 9
транс-3-Амино-3-(4-(8-фтор-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 33, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 8,07 (1H, дд, J=8,5, 6,1 Гц), 7,55-7,33 (7H, м), 7,24 (2H, д, J=8,3 Гц), 6,92 (1H, тд, J=8,7, 2,4 Гц), 6,84-6,79 (1H, м), 5,68 (2H, с), 2,65-2,59 (2H, м), 2,39-2,34 (2H, м), 1,63 (3H, с).
ESI-MS m/z 443 (MH+).
Пример 10
транс-3-Амино-1-циклопропил-3-(4-(7-фтор-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 34, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 7,88-7,85 (1H, м), 7,55 (2H, д, J=8,5 Гц), 7,51-7,45 (3H, м), 7,38-7,32 (4H, м), 7,15-7,10 (2H, м), 5,73 (2H, с), 2,63-2,57 (2H, м), 2,32-2,27 (2H, м), 1,34 (1H, тт, J=8,3, 5,4 Гц), 0,59-0,53 (2H, м), 0,48-0,43 (2H, м).
ESI-MS m/z 468 (MH+).
Пример 11
транс-3-Амино-1-циклопропил-3-(4-(3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 35, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 8,09 (1H, дд, J=7,7, 1,6 Гц), 7,58-7,29 (10H, м), 7,21-7,16 (1H, м), 7,07 (1H, дд, J=8,0, 1,0 Гц), 5,67 (2H, с), 2,62-2,56 (2H, м), 2,31-2,25 (2H, м), 1,33 (1H, тт, J=8,3, 5,4 Гц), 0,58-0,52 (2H, м), 0,42-0,47 (2H, м).
ESI-MS m/z 450 (MH+).
Пример 12
транс-3-Амино-1-метил-3-(4-(3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 35, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 8,09 (1H, дд, J=7,7, 1,6 Гц), 7,56-7,43 (5H, м), 7,37-7,29 (3H, м), 7,25-7,16 (3H, м), 7,06 (1H, дд, J=8,3, 1,0 Гц), 5,67 (2H, с), 2,64-2,58 (2H, м), 2,38-2,32 (2H, м), 1,62 (3H, с).
ESI-MS m/z 424 (MH+).
Пример 13
1-(4-(3-Фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанамин
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 35, и того же способа, что и описанный в примере 2, но с использованием продукта, полученного в справочном примере 56(4), вместо продукта, полученного в справочном примере 58(4).
1H-ЯМР (CDCl3) δ: 8,09 (1H, дд, J=7,7, 1,6 Гц), 7,55 (2H, д, J=8,5 Гц), 7,50-7,43 (3H, м), 7,39-7,27 (5H, м), 7,22-7,17 (1H, м), 7,07 (1H, дд, J=8,0, 1,0 Гц), 5,68 (2H, с), 2,58-2,49 (2H, м), 2,19-2,00 (3H, м), 1,79-1,71 (1H, м).
ESI-MS m/z 394 (MH+).
Пример 14
1-(4-(3-(Тиофен-3-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанамин
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 36, и того же способа, что и описанный в примере 2, но с использованием продукта, полученного в справочном примере 56(4), вместо продукта, полученного в справочном примере 58(4).
1H-ЯМР (CDCl3) δ: 8,07 (1H, дд, J=8,0, 1,6 Гц), 7,59-7,57 (2H, м), 7,47 (1H, дд, J=4,8, 2,8 Гц), 7,34-7,30 (4H, м), 7,21 (1H, ддд, J=7,6, 7,6, 1,2 Гц), 7,10-7,06 (2H, м), 5,71 (2H, с), 2,58-2,51 (2H, м), 2,17-2,03 (3H, м), 1,79-1,70 (1H, м).
ESI-MS m/z 400 (MH+).
Пример 15
1-(4-(3-(Пиридин-4-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанамин
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 37, и того же способа, что и описанный в примере 2, но с использованием продукта, полученного в справочном примере 56(4), вместо продукта, полученного в справочном примере 58(4).
1H-ЯМР (CDCl3) δ: 8,69 (2H, дд, J=4,4, 1,6 Гц), 8,09 (1H, дд, J=7,6, 1,6 Гц), 7,52 (2H, д, J=7,6 Гц), 7,38-7,34 (3H, м), 7,27-7,26 (2H, м), 7,21 (1H, ддд, J=7,6, 7,6, 1,2 Гц), 7,10 (1H, дд, J=7,6, 1,2 Гц), 5,76 (2H, с), 2,58-2,52 (2H, м), 2,19-2,02 (3H, м), 1,79-1,70 (1H, м).
ESI-MS m/z 395 (MH+).
Пример 16
транс-3-Амино-1-циклопропил-3-(4-(10-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 36, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 7,59 (2H, д, J=8,3 Гц), 7,49-7,43 (3H, м), 7,38-7,35 (2H, м), 7,32-7,23 (3H, м), 6,77 (1H, дд, J=8,5, 0,7 Гц), 6,72 (1H, дд, J=8,0, 0,7 Гц), 5,58 (2H, с), 4,06 (3H, с), 2,62-2,56 (2H, м), 2,31-2,25 (2H, м), 1,33 (1H, тт, J=8,3, 5,4 Гц), 0,58-0,42 (4H, м).
ESI-MS m/z 480 (MH+).
Пример 17
транс-3-Амино-1-циклопропил-3-(4-(9-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 39, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 7,61-7,53 (3H, м), 7,50-7,41 (3H, м), 7,39-7,31 (4H, м), 7,00 (1H, д, J=8,8 Гц), 6,68 (1H, дд, J=9,0, 2,9 Гц), 5,63 (2H, с), 3,89 (3H, с), 2,64-2,57 (2H, м), 2,33-2,26 (2H, м), 1,38-1,29 (1H, м), 0,60-0,42 (4H, м).
ESI-MS m/z 480 (MH+).
Пример 18
транс-3-амино-1-циклопропил-3-(4-(8-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 40, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 8,00 (1H, д, J=8,5 Гц), 7,54 (2H, д, J=8,3 Гц), 7,49-7,40 (3H, м), 7,38-7,30 (4H, м), 6,76 (1H, дд, J=8,7, 2,3 Гц), 6,62 (1H, д, J=2,2 Гц), 5,64 (2H, с), 3,85 (3H, с), 2,63-2,57 (2H, м), 2,32-2,26 (2H, м), 1,38-1,28 (1H, м), 0,59-0,42 (4H, м).
ESI-MS m/z 480 (MH+).
Пример 19
транс-3-Амино-1-циклопропил-3-(4-(7-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 41, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 7,71 (1H, дд, J=7,8, 1,0 Гц), 7,55 (2H, д, J=8,3 Гц), 7,50-7,41 (3H, м), 7,38-7,30 (4H, м), 7,18-7,11 (1H, м), 6,95 (1H, дд, J=8,3, 1,2 Гц), 5,71 (2H, с), 3,93 (3H, с), 2,63-2,57 (2H, м), 2,32-2,26 (2H, м), 1,38-1,29 (1H, м), 0,59-0,42 (4H, м).
ESI-MS m/z 480 (MH+).
Пример 20
транс-3-Амино-3-(4-(9-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 39, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 7,59 (1H, д, J=2,9 Гц), 7,53 (2H, д, J=8,5 Гц), 7,49-7,42 (3H, м), 7,37-7,32 (2H, м), 7,25 (2H, д, J=8,5 Гц), 6,99 (1H, д, J=9,0 Гц), 6,87 (1H, дд, J=9,0, 2,9 Гц), 5,62 (2H, с), 3,88 (3H, с), 2,65-2,59 (2H, м), 2,41-2,35 (2H, м), 1,62 (3H, с).
ESI-MS m/z 454 (MH+).
Пример 21
транс-3-Амино-3-(4-(8-метокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 40, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 8,00 (1H, д, J=8,5 Гц), 7,52 (2H, д, J=8,3 Гц), 7,48-7,40 (3H, м), 7,36-7,32 (2H, м), 7,24 (2H, д, J=8,3 Гц), 6,76 (1H, дд, J=8,5, 2,4 Гц), 6,61 (1H, д, J=2,4 Гц), 5,63 (2H, с), 3,84 (3H, с), 2,64-2,58 (2H, м), 2,39-2,33 (2H, м), 1,62 (3H, с).
ESI-MS m/z 454 (MH+).
Пример 22
транс-3-Амино-3-(4-(10-хлор-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-циклопропилциклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 42, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 7,60 (2H, д, J=8,3 Гц), 7,51-7,44 (3H, м), 7,39-7,30 (4H, м), 7,27-7,18 (2H, м), 7,00 (1H, дд, J=7,9, 1,3 Гц), 5,62 (2H, с), 2,63-2,57 (2H, м), 2,33-2,27 (2H, м), 1,33 (1H, тт, J=8,5, 5,6 Гц), 0,58-0,52 (2H, м), 0,47-0,42 (2H, м).
ESI-MS m/z 484 (MH+).
Пример 23
транс-3-Амино-1-циклопропил-3-(4-(10-этокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 43, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 7,60 (2H, д, J=8,4 Гц), 7,50-7,41 (3H, м), 7,39-7,35 (2H, м), 7,30 (2H, д, J=8,4 Гц), 7,22 (1H, дд, J=8,4, 8,2 Гц), 6,75 (1H, д, J=8,4 Гц), 6,70 (1H, д, J=8,2 Гц), 5,55 (2H, с), 4,28 (2H, кв, J=7,0 Гц), 2,62-2,56 (2H, м), 2,32-2,26 (2H, м), 1,63 (3H, т, J=7,0 Гц), 1,37-1,28 (1H, м), 0,57-0,42 (4H, м).
ESI-MS m/z 494 (MH+).
Пример 24
транс-3-Амино-3-(4-(10-этокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 43, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 7,60 (2H, д, J=8,6 Гц), 7,50-7,42 (3H, м), 7,39-7,35 (2H, м), 7,25-7,19 (3H, м), 6,76 (1H, дд, J=8,4, 0,8 Гц), 6,70 (1H, дд, J=8,2, 0,8 Гц), 5,55 (2H, с), 4,28 (2H, д, J=7,0 Гц), 2,64-2,59 (2H, м), 2,38-2,32 (2H, м), 1,63 (3H, т, J=7,0 Гц), 1,62 (3H, с).
ESI-MS m/z 468 (MH+).
Пример 25
транс-3-Амино-1-циклопропил-3-(4-(8,10-диметокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 44, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 7,56-7,52 (2H, м), 7,46-7,41 (3H, м), 7,35-7,24 (4H, м), 6,32 (1H, д, J=2,2 Гц), 6,27 (1H, д, J=2,2 Гц), 5,56 (2H, с), 3,98 (3H, с), 3,84 (3H, с), 2,64-2,56 (2H, м), 2,38-2,30 (2H, м), 1,35-1,25 (1H, м), 0,55-0,49 (2H, м), 0,46-0,40 (2H, м).
ESI-MS m/z 510 (MH+).
Пример 26
транс-3-Амино-1-циклопропил-3-(4-(7-метил-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 45, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 7,93 (1H, д, J=6,6 Гц), 7,56 (2H, д, J=8,5 Гц), 7,49-7,42 (3H, м), 7,39-7,35 (2H, м), 7,33 (2H, д, J=8,5 Гц), 7,19-7,16 (1H, м), 7,11-7,06 (1H, м), 5,69 (2H, с), 2,62-2,57 (2H, м), 2,32-2,27 (2H, м), 1,34 (1H, тт, J=8,0, 5,4 Гц), 0,59-0,53 (2H, м), 0,48-0,43 (2H, м).
ESI-MS m/z 464 (MH+).
Пример 27
транс-3-Амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 46, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 8,49 (1H, дд, J=4,8, 1,6 Гц), 7,68 (2H, д, J=8,0 Гц), 7,52-7,48 (3H, м), 7,41-7,38 (3H, м), 7,30 (2H, д, J=8,0 Гц), 7,25 (1H, дд, J=8,0, 4,8 Гц), 5,71 (2H, с), 2,62-2,58 (2H, м), 2,31-2,28 (2H, м), 1,38-1,31 (1H, м), 0,58-0,53 (2H, м), 0,47-0,44 (2H, м).
ESI-MS m/z 451 (MH+).
Пример 28
транс-3-Амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 46, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 8,49 (1H, дд, J=4,8, 1,6 Гц), 7,67 (2H, д, J=8,0 Гц), 7,52-7,48 (3H, м), 7,42-7,37 (3H, м), 7,26-7,20 (3H, м), 5,71 (2H, с), 2,63-2,60 (2H, м), 2,38-2,34 (2H, м), 1,64 (3H, с).
ESI-MS m/z 425 (MH+).
Пример 29
1-(4-(3-Фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанамин
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 46, и того же способа, что и описанный в примере 2, но с использованием продукта, полученного в справочном примере 56(4), вместо продукта, полученного в справочном примере 58(4).
1H-ЯМР (CDCl3) δ: 8,49 (1H, дд, J=4,8, 1,3 Гц), 7,67 (2H, д, J=8,5 Гц), 7,52-7,47 (3H, м), 7,43-7,36 (3H, м), 7,28-7,22 (3H, м), 5,71 (2H, с), 2,57-2,49 (2H, м), 2,16-2,00 (3H, м), 1,79-1,69 (1H, м).
ESI-MS m/z 395 (MH+).
Пример 30
транс-3-Амино-1-этил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 46, и того же способа, что и описанный в примере 2, но с использованием продукта, полученного в справочном примере 59, вместо продукта, полученного в справочном примере 58(4).
1H-ЯМР (CDCl3) δ: 8,49 (1H, дд, J=4,4, 1,6 Гц), 7,67 (2H, д, J=8,0 Гц), 7,52-7,48 (3H, м), 7,41-7,36 (3H, м), 7,29-7,21 (3H, м), 5,71 (2H, с), 2,57-2,54 (2H, м), 2,37-2,34 (2H, м), 1,91 (2H, кв, J=7,2 Гц), 0,97 (3H, т, J=7,2 Гц).
ESI-MS m/z 439 (MH+).
Пример 31
транс-3-Амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 47, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 8,49 (1H, дд, J=4,8, 1,6 Гц), 7,68 (2H, д, J=8,0 Гц), 7,52-7,48 (3H, м), 7,41-7,38 (3H, м), 7,30 (2H, д, J=8,0 Гц), 7,25 (1H, дд, J=8,0, 4,8 Гц), 5,71 (2H, с), 2,62-2,58 (2H, м), 2,31-2,28 (2H, м), 1,38-1,31 (1H, м), 0,58-0,53 (2H, м), 0,47-0,44 (2H, м).
ESI-MS m/z 451 (MH+).
Пример 32
транс-3-Амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[3,4-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 47, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ 9,26 (1H, с), 8,49 (1H, д, J=5,6 Гц) 7,58-7,44 (5H, м), 7,40-7,33 (2H, м), 7,29-7,22 (2H, м), 6,99 (1H, д, J=5,6 Гц), 5,76 (2H, с), 2,66-2,58 (2H, м), 2,40-2,33 (2H, м), 1,64 (3H, с), 1,61 (3H, уш.с).
ESI-MS m/z 425 (MH+).
Пример 33
1-(4-(3-Фенил-5H-имидазо[1,2-c]пиридо[3,4-e][1,3]оксазин-2-ил)фенил)циклобутанамин
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 47, и того же способа, что и описанный в примере 2, но с использованием продукта, полученного в справочном примере 56(4), вместо продукта, полученного в справочном примере 58(4).
1H-ЯМР (CDCl3) δ: 9,27 (1H, с), 8,50 (1H, д, J=5,6 Гц), 7,58 (5H, м), 7,40-7,25 (4H, м), 7,00 (1H, д, J=5,6 Гц), 5,76 (2H, с), 2,59-2,48 (2H, м), 2,20-1,98 (3H, м), 1,82-1,69 (1H, м).
ESI-MS m/z 395 (MH+).
Пример 34
транс-3-Амино-1-этил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[3,4-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 47, и того же способа, что и описанный в примере 2, но с использованием продукта, полученного в справочном примере 59, вместо продукта, полученного в справочном примере 58(4).
1H-ЯМР (CDCl3) δ: 9,27 (1H, с), 8,49 (1H, д, J=5,6 Гц), 7,59-7,44 (5H, м), 7,40-7,34 (2H, м), 7,29 (2H, д, J=8,3 Гц), 7,00 (1H, д, J=5,6 Гц), 5,76 (2H, с), 2,60-2,53 (2H, м), 2,40-2,33 (2H, м), 1,90 (2H, кв, J=7,3 Гц), 1,62 (3H, уш.с), 0,97 (3H, т, J= 7,3 Гц).
ESI-MS m/z 439 (MH+).
Пример 35
транс-3-Амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[4,3-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 48, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 9,26 (1H, с), 8,49 (1H, д, J=5,6 Гц), 7,59-7,43 (5H, м), 7,40-7,32 (4H, м), 6,99 (1H, д, J=5,6 Гц), 5,76 (2H, с), 2,63-2,57 (2H, м), 2,33-2,26 (2H, м), 1,61 (3H, уш.с), 1,34 (1H, тт, J=8,3, 5,4 Гц), 0,61-0,41 (4H, м).
ESI-MS m/z 451 (MH+).
Пример 36
транс-3-Амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[4,3-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 48, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 8,45-8,45 (1H, м), 8,42 (1H, дд, J=4,9, 1,0 Гц), 7,91 (1H, дд, J=4,9, 0,7 Гц), 7,54-7,47 (5H, м), 7,38-7,34 (2H, м), 7,27-7,23 (2H, м), 5,74 (2H, с), 2,64-2,59 (2H, м), 2,39-2,32 (2H, м), 1,63 (3H, с).
ESI-MS m/z 425 (MH+).
Пример 37
1-(4-(3-Фенил-5H-имидазо[1,2-c]пиридо[4,3-e][1,3]оксазин-2-ил)фенил)циклобутанамин
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 48, и того же способа, что и описанный в примере 2, но с использованием продукта, полученного в справочном примере 56(4), вместо продукта, полученного в справочном примере 58(4).
1H-ЯМР (CDCl3) δ: 8,47 (1H, д, J=0,6 Гц), 8,45 (1H, д, J=5,1 Гц), 7,92 (1H, дд, J=5,1, 0,6 Гц), 7,56-7,47 (5H, м), 7,40-7,36 (2H, м), 7,31 (2H, д, J=8,5 Гц), 5,75 (2H, с), 2,57-2,49 (2H, м), 2,18-2,00 (3H, м), 1,79-1,70 (1H, м).
ESI-MS m/z 395 (MH+).
Пример 38
транс-3-Амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[3,2-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 49, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 8,40 (1H, дд, J=7,6, 2,0 Гц), 8,26 (1H, дд, J=5,0, 2,0 Гц), 7,56-7,45 (5H, м), 7,39-7,32 (4H, м), 7,20 (1H, дд, J =7,6, 5,0 Гц), 5,85 (2H, с), 2,63-2,57 (2H, м), 2,32-2,26 (2H, м), 1,33 (1H, тт, J=8,3, 5,4 Гц), 0,59-0,53 (2H, м), 0,48-0,42 (2H, м).
ESI-MS m/z 451 (MH+).
Пример 39
транс-3-Амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[3,2-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 49, и того же способа, что и описанный в примере 2.
1H-ЯМР (ДМСО-D6) δ: 8,30 (1H, дд, J=7,3, 2,0 Гц), 8,26 (1H, дд, J=4,9, 2,0 Гц), 7,56-7,49 (3H, м), 7,45-7,39 (4H, м), 7,33-7,28 (3H, м), 5,96 (2H, с), 4,74 (1H, с), 2,39-2,33 (2H, м), 2,18-2,13 (2H, м), 1,48 (3H, с).
ESI-MS m/z 425 (MH+).
Пример 40
1-(4-(3-Фенил-5H-имидазо[1,2-c]пиридо[3,2-e][1,3]оксазин-2-ил)фенил)циклобутанамин
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 49, и того же способа, что и описанный в примере 2, но с использованием продукта, полученного в справочном примере 56(4), вместо продукта, полученного в справочном примере 58(4).
1H-ЯМР (CDCl3) δ: 8,41 (1H, дд, J=7,6, 2,0 Гц), 8,27 (1H, дд, J=4,9, 2,0 Гц), 7,55-7,45 (5H, м), 7,40-7,36 (2H, м), 7,33-7,29 (2H, м), 7,20 (1H, дд, J=7,6, 4,9 Гц), 5,86 (2H, с), 2,58-2,48 (2H, м), 2,18-1,99 (4H, м), 1,79-1,69 (1H, м).
ESI-MS m/z 395 (MH+).
Пример 41
транс-3-Амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиразино[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде твердого вещества светло-желтого цвета с использованием продукта, полученного в справочном примере 50, и того же способа, что и описанный в примере 1.
1H-ЯМР (CDCl3) δ: 8,46 (1H, д, J=2,8 Гц), 8,19 (1H, д, J=2,8 Гц), 7,65 (2H, д, J=8,4 Гц), 7,53-7,50 (3H, м), 7,42-7,40 (2H, м), 7,32 (2H, д, J=8,4 Гц), 5,92 (2H, с), 2,62-2,58 (2H, м), 2,31-2,28 (2H, м), 1,38-1,30 (1H, м), 0,58-0,54 (2H, м), 0,47-0,43 (2H, м).
ESI-MS m/z 452 (MH+).
Пример 42
транс-3-Амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиразо[3,2-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде твердого вещества светло-желтого цвета с использованием продукта, полученного в справочном примере 50, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 8,46 (1H, д, J=2,8 Гц), 8,19 (1H, д, J=2,8 Гц), 7,64 (2H, д, J=8,4 Гц), 7,54-7,52 (3H, м), 7,42-7,40 (2H, м), 7,23 (2H, д, J=8,4 Гц), 5,92 (2H, с), 2,63-2,60 (2H, м), 2,37-2,34 (2H, м), 1,64 (3H, с).
ESI-MS m/z 426 (MH+).
Пример 43
транс-3-Амино-1-этил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиразо[3,2-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 50, и того же способа, что и описанный в примере 2, но с использованием продукта, полученного в справочном примере 59, вместо продукта, полученного в справочном примере 58(4).
1H-ЯМР (CDCl3) δ: 8,47 (1H, д, J=2,4 Гц), 8,20 (1H, д, J=2,4 Гц), 7,65 (2H, д, J=8,4 Гц), 7,54-7,52 (3H, м), 7,43-7,40 (2H, м), 7,30-7,23 (2H, м), 5,92 (2H, с), 2,57-2,54 (2H, м), 2,37-2,34 (2H, м), 1,91 (2H, кв, J=7,6 Гц), 0,97 (3H, т, J=7,6 Гц).
ESI-MS m/z 440 (MH+).
Пример 44
транс-3-Амино-3-(4-(9-(гидроксиметил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 24, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 8,10 (1H, д, J=1,7 Гц), 7,56-7,44 (5H, м), 7,38-7,23 (5H, м), 7,06 (1H, д, J=8,0 Гц), 5,66 (2H, с), 4,72 (2H, с), 2,65-2,60 (2H, м), 2,39-2,33 (2H, м), 1,63 (3H, с).
ESI-MS m/z 454 (MH+).
Пример 45
транс-3-Амино-3-(4-(8-(гидроксиметил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 52, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 8,06 (1H, д, J=7,8 Гц), 7,54 (2H, д, J=8,3 Гц), 7,50-7,42 (3H, м), 7,38-7,34 (2H, м), 7,24 (2H, д, J=8,3 Гц), 7,17 (1H, дд, J=7,8, 1,5 Гц), 7,10 (1H, д, J=1,5 Гц), 5,66 (2H, с), 4,72 (2H, с), 3,49 (1H, с), 2,65-2,60 (2H, м), 2,39-2,33 (2H, м), 1,63 (3H, с).
ESI-MS m/z 454 (MH+).
Пример 46
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбонитрил
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 25, и того же способа, что и описанный в примере 2.
1H-ЯМР (ДМСО-D6) δ: 8,28 (1H, д, J=2,0 Гц), 7,85 (1H, дд, J=8,5, 2,0 Гц), 7,57-7,29 (10H, м), 5,94 (2H, с), 2,39-2,32 (2H, м), 2,17-2,11 (2H, м), 1,50 (3H, с).
ESI-MS m/z 449 (MH+).
Пример 47
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбонитрил
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 53, и того же способа, что и описанный в примере 2.
1H-ЯМР (ДМСО-D6) δ: 8,03 (1H, д, J=8,0 Гц), 7,72 (1H, д, J=1,5 Гц), 7,66 (1H, дд, J=8,0, 1,5 Гц), 7,55-7,49 (3H, м), 7,45-7,38 (4H, м), 7,30 (2H, д, J=8,5 Гц), 5,90 (2H, с), 2,36-2,30 (2H, м), 2,15-2,10 (2H, м), 1,48 (3H, с).
ESI-MS m/z 449 (MH+).
Пример 48
транс-3-Амино-1-метил-3-(4-(3-фенил-9-(1H-пиразол-5-ил)-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 26, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 8,47 (1H, д, J=2,2 Гц), 7,77 (1H, дд, J=8,5, 2,2 Гц), 7,62 (1H, д, J=2,4 Гц), 7,55 (2H, д, J=8,5 Гц), 7,50-7,43 (3H, м), 7,39-7,35 (2H, м), 7,27-7,23 (2H, м), 7,12 (1H, д, J=8,5 Гц), 6,70 (1H, д, J=2,2 Гц), 5,70 (2H, с), 2,66-2,60 (2H, м), 2,41-2,35 (2H, м), 1,62 (3H, с).
ESI-MS m/z 490 (MH+).
Пример 49
2-(4-(транс-1-амино-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбонитрил
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 27, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 8,23 (1H, д, J=2,2 Гц), 7,94-7,93 (2H, м), 7,56 (2H, д, J=8,3 Гц), 7,50-7,44 (4H, м), 7,39-7,36 (2H, м), 7,28-7,24 (2H, м), 7,09 (1H, д, J=8,5 Гц), 5,69 (2H, с), 2,66-2,60 (2H, м), 2,40-2,34 (2H, м), 1,64 (3H, с).
ESI-MS m/z 490 (MH+).
Пример 50
транс-3-Амино-1-метил-3-(4-(9-метил-3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)циклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 54, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 7,65 (2H, д, J=8,0 Гц), 7,52-7,48 (3H, м), 7,40-7,38 (2H, м), 7,28-7,26 (1H, м), 7,21 (2H, д, J=8,0 Гц), 7,10 (1H, д, J=8,4 Гц), 5,67 (2H, с), 2,65 (3H, с), 2,63-2,60 (2H, м), 2,38-2,34 (2H, м), 1,64 (3H, с).
ESI-MS m/z 439 (MH+).
Пример 51
транс-3-Амино-3-(4-(9-метокси-3-фенил-5H-имидазо[1,2-c]пиридо[2,3-e][1,3]оксазин-2-ил)фенил)-1-метилциклобутанол
Указанное в заголовке соединение получали в виде бесцветного твердого вещества с использованием продукта, полученного в справочном примере 55, и того же способа, что и описанный в примере 2.
1H-ЯМР (CDCl3) δ: 7,60 (2H, д, J=8,0 Гц), 7,52-7,46 (3H, м), 7,38-7,36 (2H, м), 7,32 (1H, д, J=8,8 Гц), 7,22 (2H, д, J=8,0 Гц), 6,72 (1H, д, J=8,8 Гц), 5,65 (2H, с), 4,11 (3H, с), 2,63-2,60 (2H, м), 2,38-2,34 (2H, м), 1,64 (3H, с).
ESI-MS m/z 455 (MH+).
Пример 52
Пример 52(1): Метил-2-(4-(транс-1-(трет-бутоксикарбониламин)-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбоксилат
К раствору продукта (148 мг), полученного в справочном примере 23(4), в 1,4-диоксане (2,4 мл) и воде (0,4 мл) добавляли трет-бутил-транс-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутилкарбамат (125 мг) и карбонат цезия (194 мг), и помещали смесь в атмосферу азота. Затем к ней добавляли Pd(PPh3)4 (27,5 мг), и перемешивали смесь при 100°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением целевого продукта (191 мг, выход: 71%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 8,79 (1H, д, J=2,2 Гц), 8,02 (1H, дд, J=8,5, 2,2 Гц), 7,57-7,19 (7H, м), 7,11 (1H, д, J=8,5 Гц), 6,75 (2H, д, J=8,8 Гц), 5,73 (2H, с), 5,23-5,13 (1H, уш.м), 3,94 (4H, с), 2,79-2,60 (4H, м), 1,56 (3H, с), 1,44-1,29 (9H, уш.м).
ESI-MS m/z 582 (MH+).
Пример 52(2): 2-(4-(транс-1-(трет-бутоксикарбониламино)-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбоновая кислота
К раствору продукта (140 мг), полученного в примере 52(1), в метаноле (2,5 мл) добавляли 2M водный раствор гидроксида калия (0,6 мл) и перемешивали смесь при комнатной температуре в течение 5 часов. К реакционной смеси добавляли 0,5M водный раствор гидросульфата калия и экстрагировали хлороформом. Объединенный органический слой промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления с получением целевого продукта (120 мг, выход: 88%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 9,14 (1H, д, J=1,8 Гц), 8,03 (1H, дд, J=8,4, 1,8 Гц), 7,60 (2H, д, J=8,3 Гц), 7,47-7,06 (7H, м), 6,71 (1H, д, J=8,4 Гц), 5,71 (2H, с), 5,11-4,89 (1H, уш.м), 2,76-2,45 (4H, м), 1,53 (3H, с), 1,45-1,24 (9H, уш.м).
ESI-MS m/z 568 (MH+).
Пример 52(3): 2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-N-метил-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбоксамид
К раствору продукта (20 мг), полученного в примере 52(2), в DMF (0,5 мл) добавляли гидрохлорид метиламина (5,0 мг), триэтиламин (0,025 мл), WSC гидрохлорид (13,5 мг) и HOBt (10,8 мг), и перемешивали смесь при комнатной температуре в течение 2 часов и при 90°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (гексан/этилацетат) с получением соответствующего соединения. Полученное соединение использовали в следующей реакции без дополнительной очистки. К раствору полученного соединения в хлороформе (1,0 мл) добавляли трифторуксусную кислоту (0,5 мл) и перемешивали смесь при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в условиях пониженного давления и очищали полученный остаток методом хроматографии на силикагеле (хлороформ/метанол) с получением указанного в заголовке соединения (14,8 мг, выход: 87%) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 8,37 (1H, д, J=2,2 Гц), 7,91 (1H, дд, J=8,5, 2,2 Гц), 7,51-7,44 (5H, м), 7,37-7,32 (2H, м), 7,23-7,18 (2H, м), 7,11 (1H, д, J=8,5 Гц), 6,67-6,57 (1H, уш.м), 5,70 (2H, с), 2,99-2,94 (3H, м), 2,63-2,56 (2H, м), 2,37-2,30 (2H, м), 1,62 (3H, с).
ESI-MS m/z 481 (MH+).
Пример 53
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбоксамид
Указанное в заголовке соединение получали в виде бесцветного твердого вещества тем же способом, что и описанный в примере 52, но с использованием 28% водного аммиака вместо гидрохлорида метиламина в примере 52(3).
1H-ЯМР (CDCl3) δ: 8,45 (1H, д, J=2,0 Гц), 7,96 (1H, дд, J=8,5, 2,0 Гц), 7,55-7,44 (5H, м), 7,39-7,34 (2H, м), 7,28-7,24 (2H, м), 7,16 (1H, д, J=8,5 Гц), 5,74 (2H, с), 2,66-2,60 (2H, м), 2,39-2,33 (2H, м), 1,63 (3H, с).
ESI-MS m/z 467 (MH+).
Пример 54
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-N,N-диметил-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбоксамид
Указанное в заголовке соединение получали в виде бесцветного твердого вещества тем же способом, что и описанный в примере 52, но с использованием гидрохлорида диметиламина вместо метиламина гидрохлорида в примере 52(3).
1H-ЯМР (CDCl3) δ: 8,14 (1H, д, J=2,2 Гц), 7,55-7,42 (6H, м), 7,38-7,34 (2H, м), 7,23 (2H, д, J=8,5 Гц), 7,10 (1H, д, J=8,3 Гц), 5,69 (2H, с), 3,16-3,02 (6H, м), 2,64-2,58 (2H, м), 2,38-2,32 (2H, м), 1,62 (3H, с).
ESI-MS m/z 495 (MH+).
Пример 55
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-N-этил-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбоксамид
Указанное в заголовке соединение получали в виде бесцветного твердого вещества тем же способом, что и описанный в примере 52, но с использованием этиламина гидрохлорида вместо гидрохлорида метиламина в примере 52(3).
1H-ЯМР (CDCl3) δ: 8,38 (1H, д, J=2,2 Гц), 7,93 (1H, дд, J=8,5, 2,2 Гц), 7,52-7,44 (5H, м), 7,38-7,33 (2H, м), 7,25-7,21 (2H, м), 7,13 (1H, д, J=8,5 Гц), 6,60-6,50 (1H, м), 5,71 (2H, с), 3,54-3,45 (2H, м), 2,64-2,58 (2H, м), 2,38-2,32 (2H, м), 1,62 (3H, с), 1,27 (3H, т, J=7,3 Гц).
ESI-MS m/z 495 (MH+).
Пример 56
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-N-метил-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбоксамид
Указанное в заголовке соединение получали в виде бесцветного твердого вещества тем же способом, что и описанный в примере 52, но с использованием в реакции продукта, полученного в справочном примере 51, вместо продукта, полученного в справочном примере 23(4).
1H-ЯМР (CDCl3) δ: 8,12 (1H, д, J=8,0 Гц), 7,56-7,45 (7H, м), 7,39-7,35 (2H, м), 7,28-7,23 (2H, м), 6,18-6,11 (1H, м), 5,71 (2H, с), 3,04 (3H, д, J=4,9 Гц), 2,65-2,60 (2H, м), 2,39-2,34 (2H, м), 1,64 (3H, с).
ESI-MS m/z 481 (MH+).
Пример 57
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-N,N-диметил-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбоксамид
Указанное в заголовке соединение получали в виде бесцветного твердого вещества тем же способом, что и описанный в примере 52, но с использованием гидрохлорида диметиламина вместо гидрохлорида метиламина в примере 52(3), а также с использованием в реакции продукта, полученного в справочном примере 51, вместо продукта, полученного в справочном примере 23(4).
1H-ЯМР (CDCl3) δ: 8,10 (1H, д, J=8,0 Гц), 7,56-7,44 (5H, м), 7,38-7,35 (2H, м), 7,28-7,21 (3H, м), 7,16 (1H, д, J=1,5 Гц), 5,69 (2H, с), 3,13 (3H, с), 3,03 (3H, с), 2,65-2,60 (2H, м), 2,40-2,35 (2H, м), 1,64 (3H, с).
ESI-MS m/z 495 (MH+).
Пример 58
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-N-(2-гидроксиэтил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбоксамид
Указанное в заголовке соединение получали в виде бесцветного твердого вещества тем же способом, что и описанный в примере 52, но с использованием 2-аминоэтанола вместо гидрохлорида метиламина в примере 52(3), а также с использованием в реакции продукта, полученного в справочном примере 51, вместо продукта, полученного в справочном примере 23(4).
1H-ЯМР (CDCl3-CD3ОD) δ: 8,09 (1H, д, J=8,0 Гц), 7,62-7,57 (2H, м), 7,52-7,44 (5H, м), 7,36-7,31 (2H, м), 7,29-7,25 (2H, м), 5,70 (2H, с), 3,81-3,75 (2H, м), 3,61-3,54 (2H, м), 2,69-2,64 (2H, м), 2,43-2,37 (2H, м), 1,60 (3H, с).
ESI-MS m/z 511 (MH+).
Пример 59
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-N-этокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбоксамид
Указанное в заголовке соединение получали в виде бесцветного твердого вещества тем же способом, что и описанный в примере 52, но с использованием гидрохлорида O-этилгидроксиламина вместо гидрохлорида метиламина в примере 52(3), а также с использованием в реакции продукта, полученного в справочном примере 51, вместо продукта, полученного в справочном примере 23(4).
1H-ЯМР (CDCl3-CD3ОD) δ: 8,04-8,00 (1H, м), 7,52-7,43 (7H, м), 7,33-7,23 (4H, м), 5,66 (2H, с), 4,08 (2H, кв, J=7,1 Гц), 2,69-2,63 (2H, м), 2,42-2,35 (2H, м), 1,59 (3H, с), 1,34 (3H, т, J=7,1 Гц).
ESI-MS m/z 511 (MH+).
Пример 60
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-N-(2-гидроксиэтил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбоксамид
Указанное в заголовке соединение получали в виде бесцветного твердого вещества тем же способом, что и описанный в примере 52, но с использованием 2-аминоэтанола вместо гидрохлорида метиламина в примере 52(3).
1H-ЯМР (CDCl3-CD3ОD) δ: 8,35 (1H, д, J=2,0 Гц), 7,99-7,94 (1H, м), 7,50-7,43 (5H, м), 7,35-7,26 (4H, м), 7,15 (1H, д, J=8,5 Гц), 5,72 (2H, с), 3,83-3,78 (2H, м), 3,63-3,58 (2H, м), 2,70-2,65 (2H, м), 2,43-2,37 (2H, м), 1,60 (3H, с).
ESI-MS m/z 511 (MH+).
Пример 61
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-N-этокси-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбоксамид
Указанное в заголовке соединение получали в виде бесцветного твердого вещества тем же способом, что и описанный в примере 52, но с использованием гидрохлорида O-этилгидроксиламина вместо метиламина гидрохлорида в примере 52(3).
1H-ЯМР (CDCl3-CD3ОD) δ: 8,19 (1H, д, J=2,0 Гц), 7,96 (1H, дд, J=8,8, 2,0 Гц), 7,50-7,42 (5H, м), 7,35-7,27 (4H, м), 7,17 (1H, д, J=8,8 Гц), 5,72 (2H, с), 4,11 (2H, кв, J=7,1 Гц), 2,71-2,65 (2H, м), 2,44-2,37 (2H, м), 1,61 (3H, с), 1,38 (3H, т, J=7,1 Гц).
ESI-MS m/z 511 (MH+).
Пример 62
2-(4-(транс-1-Амино-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбоксамид
Указанное в заголовке соединение получали в виде бесцветного твердого вещества тем же способом, что и описанный в примере 52, но с использованием 28% водного аммиака вместо гидрохлорида метиламина в примере 52(3), а также с использованием в реакции продукта, полученного в справочном примере 51, вместо продукта, полученного в справочном примере 23(4).
1H-ЯМР (CDCl3) δ: 8,05 (1H, д, J=8,5 Гц), 7,55-7,42 (7H, м), 7,34-7,29 (2H, м), 7,22 (2H, д, J=8,3 Гц), 6,62-6,35 (1H, уш.м), 6,14-5,82 (1H, уш.м), 5,65 (2H, с), 2,64-2,58 (2H, м), 2,36-2,31 (2H, м), 1,61 (3H, с).
ESI-MS m/z 467 (MH+).
Пример 63
Гидрохлорид 2-(4-(транс-1-амино-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-9-карбоновой кислоты
К раствору продукта (19,5 мг), полученного в примере 52(2), в этилацетате (1,0 мл) добавляли раствор (0,5 мл) 4M соляной кислоты в этилацетате и перемешивали смесь при комнатной температуре в течение 1 часа. Реакционную смесь фильтровали и промывали остаток этилацетатом с получением указанного в заголовке соединения (6,0 мг, выход: 35%) в виде бесцветного твердого вещества.
1H-ЯМР (ДМСО-D6) δ: 8,69-8,59 (3H, уш.м), 8,58 (1H, д, J=2,2 Гц), 8,00 (1H, дд, J=8,5, 2,2 Гц), 7,59-7,47 (9H, м), 7,31 (1H, д, J=8,5 Гц), 5,94 (2H, с), 2,71-2,59 (4H, м), 1,42 (3H, с).
ESI-MS m/z 468 (MH+).
Пример 64
Гидрохлорид 2-(4-(транс-1-амино-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбоновой кислоты
2-(4-(транс-1-(трет-бутоксикарбониламино)-3-гидрокси-3-метилциклобутил)фенил)-3-фенил-5H-бензо[e]имидазо[1,2-c][1,3]оксазин-8-карбоновой кислоты получали тем же способом, что и описанный в примере 52, но с использованием продукта, полученного в справочном примере 51, вместо продукта, полученного в справочном примере 23(4). Затем указанное в заголовке соединение получали в виде бесцветного твердого вещества тем же способом, что и описанный в примере 63.
1H-ЯМР (ДМСО-D6) δ: 8,68-8,57 (3H, уш.м), 8,12 (1H, д, J=8,0 Гц),7,82 (1H, дд, J=8,0, 1,5 Гц), 7,62 (1H, д, J=1,5 Гц), 7,58-7,45 (9H, м), 5,90 (2H, с), 2,69-2,58 (4H, м), 1,41 (3H, с).
ESI-MS m/z 468 (MH+).
Перечень соединений представлен ниже в таблице 3.
[Таблица 6]
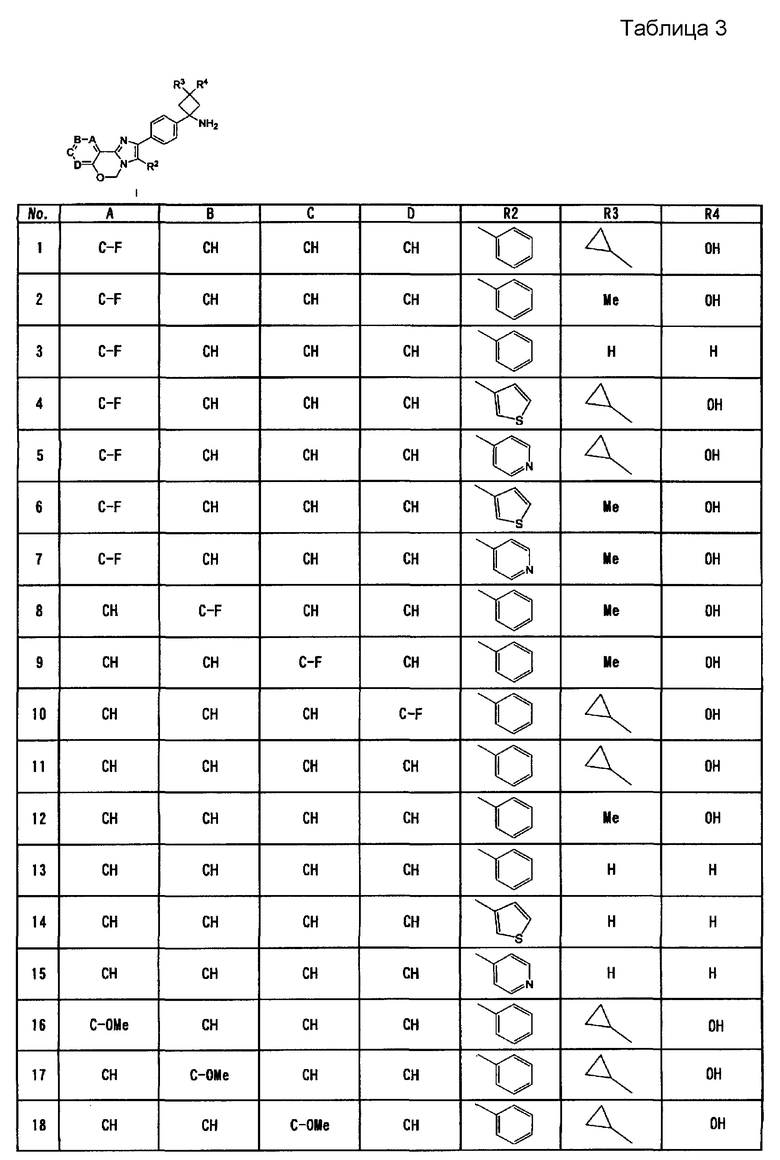
[Таблица 7]
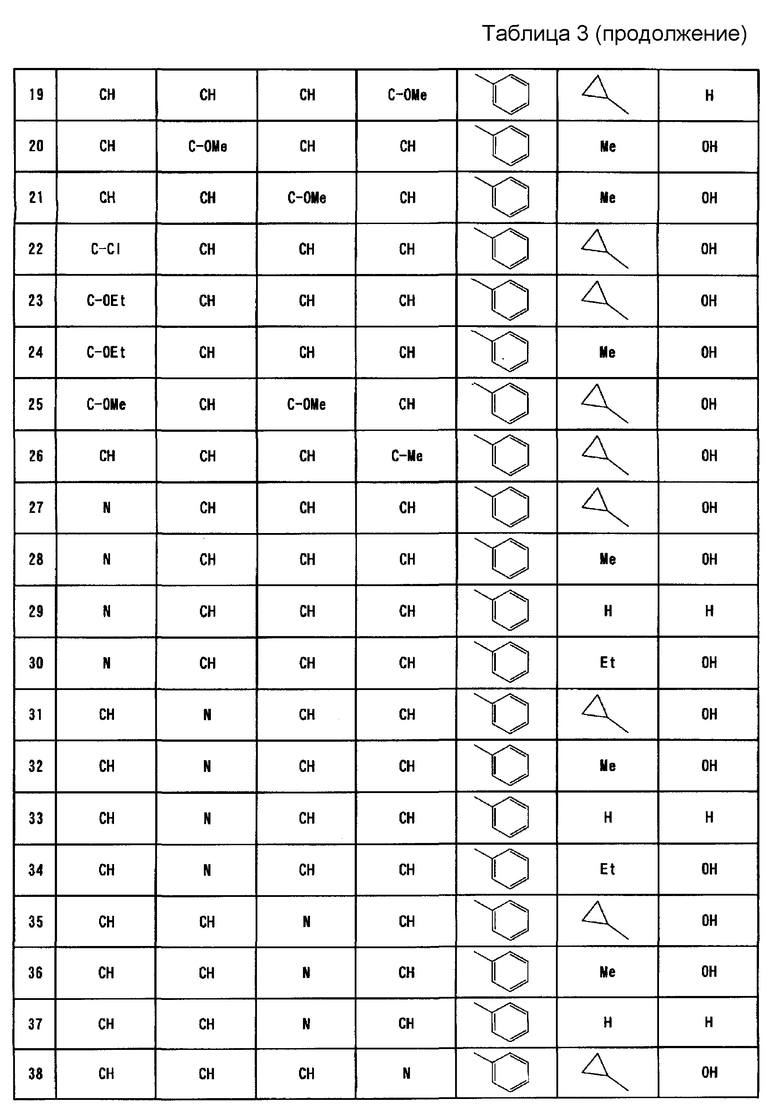
[Таблица 8]
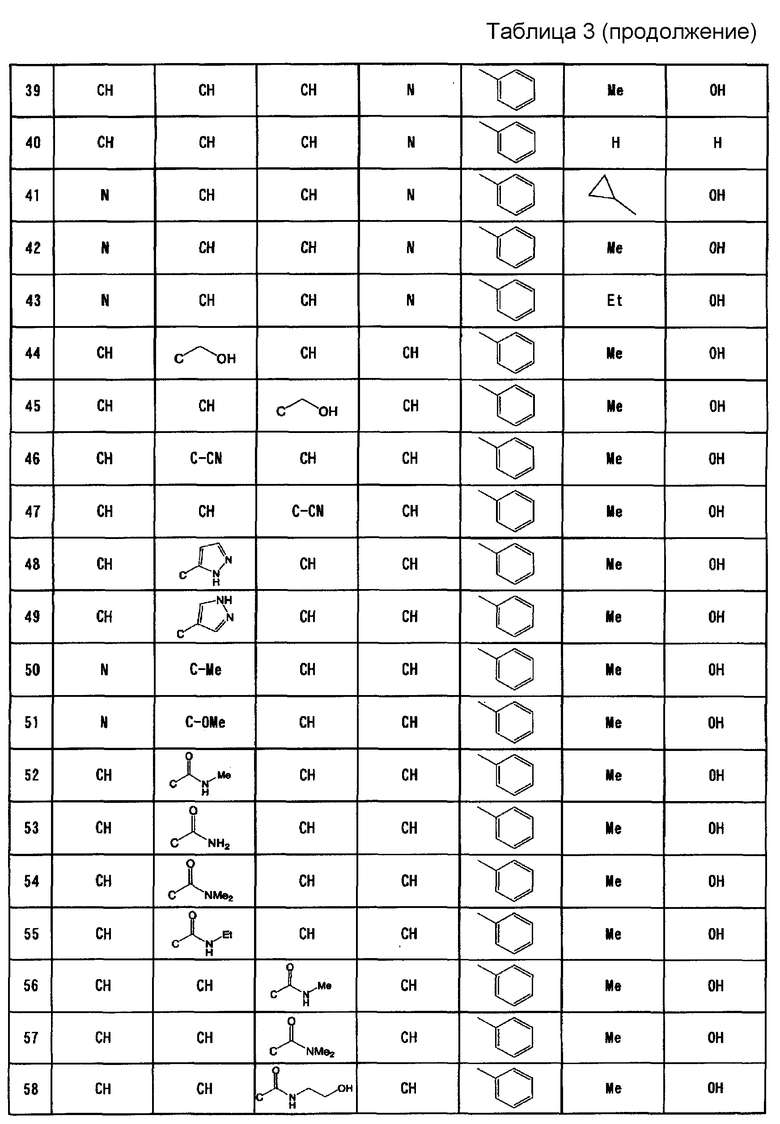
[Таблица 9]
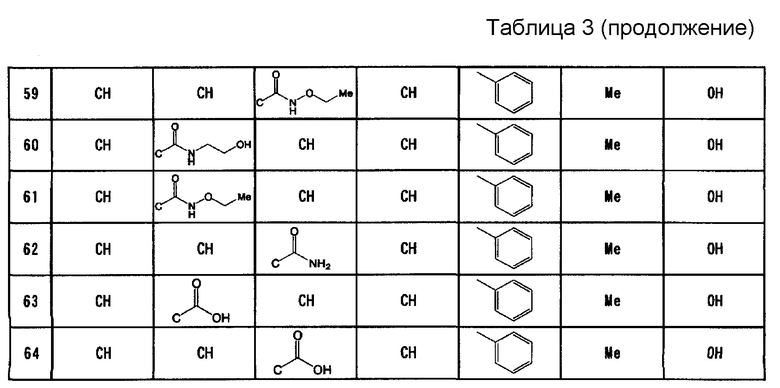
Тестовые примеры
В представленных тестовых примерах соединение I представляет собой транс-3-амино-1-метил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[3,4-e][1,3]оксазин-2-ил)фенил)циклобутанол, полученный в представленном выше примере 32, и соединение II представляет собой транс-3-амино-1-циклопропил-3-(4-(3-фенил-5H-имидазо[1,2-c]пиридо[4,3-e][1,3]оксазин-2-ил)фенил)циклобутанол, полученный в представленном выше примере 35.
Тестовый пример 1
Усиление противоопухолевого эффекта паклитаксела
Клеточную линию рака яичников человека A2780 подкожно имплантировали в правый бок самцам мышей BALB/cA Jcl-nu/nu в возрасте 7 недель. После вживления импланта измеряли длину (мм) и ширину (мм) опухоли и вычисляли объем опухоли (TV). Затем мышей разделяли на две группы так, чтобы среднее значение TV стало одинаковым в каждой группе. День разделения на группы (n=5) обозначали как 0 сутки.
Приготавливали тестовый раствор для группы, получавшей только паклитаксел (Wako Pure Chemical Ind. Ltd.), растворяя паклитаксел растворителем, содержащим 10% этиловый спирт/10% кремофор/80% физиологический раствор, для обеспечения введения паклитаксела в дозе 60 мг/кг/сутки. Приготавливали тестовые растворы для групп, получавших только соединение I, растворяя соединение I 0,5% гидроксипропилметилцеллюлозой (HPMC), для обеспечения введения соединения I в дозе 8 мг/кг/сутки, 16 мг/кг/сутки и 24 мг/кг/сутки. Соединение I вводили один раз в сутки при пероральном введении в течение 14 суток, начиная с 1 суток, и паклитаксел вводили через хвостовую вену каждой мыши в 1 сутки и 8 сутки. Группе, получавшей только одно из двух лекарственных средств, вводили носитель 10% этиловый спирт/10% кремофор/80% физиологический раствор или носитель 0,5% HPMC, вместо паклитаксела или соединения I. В группах, получавших два лекарственных средства, соединение I вводили в дозах 8 мг/кг/сутки, 16 мг/кг/сутки и 24 мг/кг/сутки и паклитаксел вводили в дозе 60 мг/кг/сутки.
В качестве показателя противоопухолевого эффекта измеряли TV на 15 сутки для каждой из групп введения лекарственного средства, и вычисляли относительный объем опухоли (RTV) по сравнению с 1 сутками и T/C(%) согласно следующим ниже формулам для оценки противоопухолевого эффекта. Эффект комбинированного введения оценивали так, что если среднее значение RTV группы комбинированного введения было статистически значимо меньше, чем среднее значение RTV группы введения только лекарственного средства (тест пересечения-объединения Уэлша), то комбинированное введение определяли эффективным. На фигуре звездочка представляет собой результат, обладающий статистически значимым различием по сравнению с группой введения только лекарственного средства.
Результаты представлены на Фиг. 1 и в таблице 10.
TV(мм3) = (длина × ширина2)/2
RTV = (TV на 15 сутки)/(TV на 1 сутки)
T/C(%) = (среднее значение RTV для группы введения тестового раствора)/(среднее значение RTV для контрольной группы)×100
Кроме того, в качестве показателя токсичности с течением времени измеряли массу тела, и вычисляли изменение средней массы тела [BWC(%)] на 15 сутки относительно 1 суток согласно представленной ниже формуле (n представляет собой сутки измерения массы тела, которое происходило два раза в неделю; сутками последнего измерения являлись 15 сутки, которые являлись сутками последней оценки).
Результаты представлены на Фиг. 2.
BWC(%) = [(BW на n сутки)-(BW на 1 сутки)]/(BW на 1 сутки)×100
Сутки 15
Среднее значение + SD
p<0,05 (в сравнении с 8, 16, 24 мг/кг соединения I)
p<0,01 (в сравнении с 60 мг/кг паклитаксела)
Тестовый пример 2
Усиление противоопухолевого эффекта паклитаксела
Человеческую клеточную линию рака желудка (NCI-N87) подкожно имплантировали в правый бок мышей BALB/cA Jcl-nu/nu в возрасте 7 недель и использовали тем же способом, что и в тестовом примере 1.
Приготавливали тестовый раствор для группы, получавшей только паклитаксел (Wako Pure Chemical Ind. Ltd.), для обеспечения введения паклитаксела в дозе 60 мг/кг/сутки. Кроме того, приготавливали тестовые растворы для групп, получавших только соединение I, для обеспечения введения соединения I в дозе 8 мг/кг/сутки, 16 мг/кг/сутки и 24 мг/кг/сутки. Соединение I вводили один раз в сутки при пероральном введении в течение 14 суток, начиная с 1 суток, и паклитаксел вводили через хвостовую вену каждой мыши в 1 сутки и 8 сутки. Как и в тестовом примере 1, группе, получавшей только одно из двух лекарственных средств, вводили носитель вместо паклитаксела или соединения I. В группах, получавших два лекарственных средства, соединение I вводили в дозах 8 мг/кг/сутки, 16 мг/кг/сутки и 24 мг/кг/сутки, и паклитаксел вводили в дозе 60 мг/кг/сутки. Оценку проводили тем же способом, что и в тестовом примере 1. Эффект комбинированного введения статистически определяли тем же способом, что и в тестовом примере 1. На фигуре и в таблице звездочка представляет собой результат, обладающий статистически значимым различием по сравнению с группой введения только лекарственного средства.
Результаты представлены на Фиг. 3 и в таблице 11.
Кроме того, с течением времени оценивали изменение массы тела в качестве показателя токсичности тем же способом, что и в тестовом примере 1. Результаты представлены на Фиг. 4.
p<0,05 (в сравнении с группой, получавшей паклитаксел/носитель, и группой, получавшей носитель/соединение I)
Тестовый пример 3
Усиление противоопухолевого эффекта карбоплатина
Человеческую клеточную линию рака яичников (A2780) подкожно имплантировали в правый бок самцам бестимусных крыс в возрасте 7 недель и использовали тем же способом, что и в тестовом примере 1. Карбоплатин приготавливали, двукратно растворяя инъекционный раствор параплатина (Bristol-Myers Squibb, 50 мг/5 мл) физиологическим раствором, для обеспечения введения карбоплатина в дозе 50 мг/кг/сутки. Кроме того, приготавливали тестовый раствор соединения I, корректируя его дозу до 16 мг/кг/сутки.
Соединение I вводили один раз в сутки в виде перорального введения в течение 14 суток, начиная с 1 суток, и карбоплатин вводили через хвостовую вену каждой мыши в 1 сутки и 8 сутки. Группе введения только лекарственного средства вводили 0,5% HPMC или физиологический раствор в качестве носителя вместо соединения I или карбоплатина.
В группе комбинированного введения соединение I и карбоплатин вводили в дозах 16 мг/кг/сутки и 50 мг/кг/сутки, соответственно, и проводили оценку тем же способом, что и в тестовом примере 1. Результаты представлены на Фиг. 5 и в таблице 12. Кроме того, с течением времени оценивали изменение массы тела в качестве показателя токсичности тем же способом, что и в тестовом примере 1. Эффект комбинированного введения статистически определяли тем же способом, что и в тестовом примере 1. На фигуре и в таблице звездочка представляет собой результат, обладающий статистически значимым различием по сравнению с группой введения только лекарственного средства.
Результаты представлены на Фиг. 6.
Тестовый пример 4
Усиление противоопухолевого эффекта лапатиниба
Человеческую клеточную линию рака желудка (NCI-N87) подкожно имплантировали в правый бок самцам мышей BALB/cA Jcl-nu/nu в возрасте 7 недель и использовали тем же способом, что и в тестовом примере 1. Лапатиниб приготавливали, корректируя его дозу до 100 мг/кг/сутки. Кроме того, приготавливали тестовый раствор соединения I, корректируя его дозу до 16 мг/кг/сутки. Носитель 0,5% HPMC/0,1% Tween 80 и носитель 0,5% HPMC использовали для лапатиниба и соединения I, соответственно.
В группе введения только лекарственного средства соединение I в дозе 16 мг/кг/сутки или лапатиниб в дозе 100 мг/кг/сутки вводили перорально один раз в сутки в течение 14 суток, начиная с 1 суток. В группе комбинированного введения соединение I и лапатиниб вводили в дозе 16 мг/кг/сутки и 100 мг/кг/сутки соответственно и проводили оценку тем же способом, что и в тестовом примере 1. Эффект комбинированного введения статистически определяли тем же способом, что и в тестовом примере 1. На фигуре и в таблице звездочка представляет собой результат, обладающий статистически значимым различием по сравнению с группой введения только лекарственного средства. Результаты представлены на Фиг. 7 и в таблице 13. Кроме того, с течением времени оценивали изменение массы тела в качестве показателя токсичности тем же способом, что и в тестовом примере 1. Результаты представлены на Фиг. 8.
Тестовый пример 5
Усиление противоопухолевого эффекта иринотекана
Человеческую клеточную линию рака яичников (A2780) подкожно имплантировали в правый бок самцам мышей BALB/cA Jcl-nu/nu в возрасте 7 недель и использовали тем же способом, что и в тестовом примере 1. Иринотекан приготавливали, разбавляя раствор для внутривенных инфузий Campto (Yakult Co., Ltd., 100 мг/5 мл) физиологическим раствором, для обеспечения введения иринотекана в дозе 75 мг/кг/сутки. Кроме того, приготавливали тестовый раствор соединения I, корректируя его дозу до 16 мг/кг/сутки. Физиологический раствор и 0,5% HPMC использовали в качестве носителя для иринотекана и соединения I соответственно.
В группе введения только лекарственного средства соединение I перорально вводили один раз в сутки в течение 14 суток, начиная с 1 суток при дозировке 16 мг/кг/сутки, и иринотекан вводили в хвостовую вену каждой мыши в дозе 75 мг/кг/сутки в 1 сутки и 8 сутки. В группе комбинированного введения соединение I и иринотекан вводили в дозах 16 мг/кг/сутки и 75 мг/кг/сутки, соответственно, и проводили оценку тем же способом, что и в тестовом примере 1. Эффект комбинированного введения статистически определяли тем же способом, что и в тестовом примере 1. На фигуре и в таблице звездочка представляет собой результат, обладающий статистически значимым различием по сравнению с группой введения только лекарственного средства. Результаты представлены на Фиг. 9 и в таблице 14. Кроме того, с течением времени оценивали изменение массы тела в качестве показателя токсичности тем же способом, что и в тестовом примере 1. Результаты представлены на Фиг. 10.
Тестовый пример 6
Усиление противоопухолевого эффекта доксорубицина
Человеческую клеточную линию рака яичников (A2780) подкожно имплантировали в правый бок самцам мышей BALB/cA Jcl-nu/nu в возрасте 7 недель и использовали тем же способом, что и в тестовом примере 1.
В группе, получавшей только доксорубицин, приготавливали тестовый раствор, разбавляя доксорубицин (раствор для инъекций Adriacin, Kyowa Hakko Kogyo Co., Ltd.) физиологическим раствором, для обеспечения введения доксорубицина в дозе 7 мг/кг/сутки. В группе, получавшей только соединение I, приготавливали тестовый раствор, разбавляя соединение I 0,5% HPMC, для обеспечения введения соединения I в дозе 16 мг/кг/сутки. В группе комбинированного введения соединение I вводили в дозе 16 мг/кг/сутки, и доксорубицин вводили в дозе 7 мг/кг/сутки. Соединение I перорально вводили в течение 14 следующих подряд суток, начиная с 1 суток, доксорубицин перорально вводили в 1 сутки и 8 сутки, и проводили оценку тем же способом, что и в тестовом примере 1. Эффект комбинированного введения статистически определяли тем же способом, что и в тестовом примере 1. На фигуре и в таблице звездочка представляет собой результат, обладающий статистически значимым различием по сравнению с группой введения только лекарственного средства. Результаты представлены на Фиг. 11 и в таблице 15.
С течением времени оценивали изменение массы тела в качестве показателя токсичности тем же способом, что и в тестовом примере 1. Результаты представлены на Фиг. 12.
Тестовый пример 7
Усиление противоопухолевого эффекта эверолимуса
Человеческую клеточную линию рака желудка (NCI-N87) подкожно имплантировали в правый бок самцам мышей BALB/cA Jcl-nu/nu в возрасте 7 недель и использовали тем же способом, что и в тестовом примере 1.
В группе, получавшей только эверолимус (IS Chemical Technology), приготавливали тестовый раствор для обеспечения введения эверолимуса в дозе 5 мг/кг/сутки. Использовали носитель 5% этиловый спирт/5% PEG400/4% Tween 20/86% дистиллированная вода. В группе, получавшей только соединение I, приготавливали тестовый раствор, разбавляя соединение I 0,5% HPMC, для обеспечения введения соединения I в дозе 16 мг/кг/сутки. В группе комбинированного введения соединение I вводили в дозе 16 мг/кг/сутки, и эверолимус вводили в дозе 5 мг/кг/сутки. И соединение I, и эверолимус вводили перорально один раз в сутки в течение 14 суток, начиная с 1 суток, и проводили оценку тем же способом, что и в тестовом примере 1. Результаты представлены на Фиг. 13 и в таблице 16.
Оценку эффекта комбинированного введения проводили так, что если среднее значение RTV группы комбинированного введения было статистически значимо меньше, чем среднее значение RTV группы введения только лекарственного средства (тест пересечения-объединения Уэлша, общее максимальное значение p<0,05), то комбинированное введение определяли эффективным. На фигуре и в таблице звездочка представляет собой результат, обладающий статистически значимым различием по сравнению с группой введения только лекарственного средства.
С течением времени оценивали изменение массы тела в качестве показателя токсичности тем же способом, что и в тестовом примере 1. Результаты представлены на Фиг. 14.
Тестовый пример 8
Усиление противоопухолевого эффекта TS-1
Человеческую клеточную линию рака желудка (4-1ST) подкожно имплантировали в правый бок самцам мышей BALB/cA Jcl-nu/nu в возрасте 6 недель и использовали тем же способом, что и в тестовом примере 1.
Приготавливали TS-1, смешивая FT (тегафур; Taiho Pharmaceutical Co., Ltd.), CDHP (гимерацил; Taiho Pharmaceutical Co., Ltd.) и Oxo (отерацил калия; Taiho Pharmaceutical Co., Ltd.) в молярном соотношении 1/0,4/1, добавляя водный раствор 0,5% (масс./об.) HPMC к смеси при концентрации FT 1,66 мг/мл и подвергая смесь ультразвуковой обработке для получения однородной суспензии (раствор для введения, обладающий двойной концентрацией раствора в группе с дозой 8,3 мг/кг/сутки). Суспензию разбавляли в два раза водным раствором 0,5% (масс./об.) HPMC до концентрации 0,83 мг/мл, приводя тем самым к получению раствора для введения, который будет использоваться в группе с дозой 8,3 мг/кг/сутки. Кроме того, приготавливали тестовый раствор соединения I, корректируя его дозу до 16 и 24 мг/кг/сутки.
Соединение I и TS-1 перорально вводили один раз в сутки в течение 14 суток, начиная с 1 суток. В группе комбинированного введения вводили соединение I в дозе 16 мг/кг/сутки и 24 мг/кг/сутки и TS-1 в дозе 8,3 мг/кг/сутки, и проводили оценку тем же способом, что и в тестовом примере 1. Результаты представлены на Фиг. 15A и 15B и в таблице 17.
Тем же способом, что и в тестовом примере 7, статистически определяли эффект комбинированного введения. На фигуре и в таблице звездочка представляет собой результат, обладающий статистически значимым различием по сравнению с группой введения только лекарственного средства.
С течением времени оценивали изменение массы тела в качестве показателя токсичности тем же способом, что и в тестовом примере 1. Результаты представлены на Фиг. 16A и 16B.
Тестовый пример 9
Усиление ингибирующего пролиферацию злокачественных клеток эффекта бортезомиба
Человеческую происходящую из множественной миеломы клеточную линию MM.1S или MM.1R субкультивировали в среде RPMI-1640, содержащей 10% эмбриональную телячью сыворотку. Клетки MM.1S или MM.1R высевали в 384-луночный микропланшет (1500 клеток/20 мкл/лунка) и культивировали в течение ночи в инкубаторе при 37°C, 5% CO2 и влажности 100%. Планшет забирали из инкубатора, и добавляли в лунки соединение II после разбавления средой RPMI-1640, содержащей растворитель ДМСО и 10% эмбриональную телячью сыворотку, до конечных концентраций 7,8, 15,6, 31,3, 62,5, 125, 250, 500, 1000 и 2000 нМ; каждую концентрацию добавляли в 4 каждые лунки (5 мкл в лунку). Бортезомиб добавляли в лунки после разведения средой RPMI-1640, содержащей растворитель ДМСО и 10% эмбриональную телячью сыворотку до конечных концентраций 7,8, 15,6, 31,3, 62,5, 125, 250, 500, 1000 и 2000 нМ; каждую концентрацию добавляли в 4 лунки (5 мкл на лунку). В группах комбинированного введения соединение II и бортезомиб разбавляли для получения всех комбинаций указанных выше конечных концентраций, и каждую комбинацию добавляли в 4 лунки (5 мкл на лунку). В качестве контрольной группы ДМСО, разбавленный средой RPMI-1640, содержащей 10% эмбриональную телячью сыворотку, добавляли в 16 лунок для контрольных измерений (5 мкл на лунку). Планшет помещали обратно в инкубатор и культивировали еще в течение 3 суток. Через 3 суток в лунки добавляли CellTiter-Gloтм (Promega) (25 мкл на лунку), и подсчитывали количество клеток. Результаты для ингибирующего клеточную пролиферацию эффекта, полученные при обработке только соединением II, при обработке с использованием только бортезомиба и при обработке с использованием двух лекарственных средств, анализировали согласно способу медиана-эффект, описанному Chou и Talalay в Adv. Enzyme Regul. 1984; 22:27-55, используя CalcuSyn версия 2.1 (Biosoft), получая тем самым показатель аддитивности (CI) в качестве показателя эффекта комбинированного введения. Эффект определяли конкурентным, если величина CI составляла 1,2 или более, аддитивным, если величина CI составляла менее чем 1,2 и не менее чем 0,85, и синергическим, если величина CI составляла менее чем 0,85. Ниже в таблице представлен показатель аддитивности. Величина CI составляла от 0,71 до 1,0 для MM.1S и от 0,60 до 1,13 для MM.1R, демонстрируя, что соединение II проявляло аддитивный и синергический эффект ингибирования клеточной пролиферации при применении с бортезомибом.
Тестовый пример 10
Усиление ингибирующего эффекта эрлотиниба в отношении пролиферации раковой клетки
Человеческую клеточную линию NCI-N87, происходящую из рака желудка, субкультивировали в среде RPMI-1640, содержащей 10% эмбриональную телячью сыворотку. Клетки NCI-N87 высевали в 96-луночный микропланшет (3750 клеток/100 мкл/лунка), и культивировали в течение ночи в инкубаторе при 37°C, 5% CO2 и 100% влажности. Серии разведений соединения II получали при разбавлении его раствором ДМСО с кратностью разведения 200 до конечных концентраций 0 (только ДМСО), 10, 30, 100, 300, 1000, 3000 и 10000 нМ, и серии разведений эрлотиниба, получаемые при разбавлении его до конечных концентраций в 0,1, 0,3, 1, 3 и 10 раз от конечных концентраций соединения II. Планшет забирали из инкубатора и разбавляли каждый из указанных выше разбавленных ДМСО растворов средой RPMI-1640, содержащей 10% эмбриональную телячью сыворотку, до концентрации, четырехкратной от конечной концентрации. В группах комбинированного введения соединение II и эрлотиниб добавляли к лункам во всех комбинациях указанных выше конечных концентраций (50 мкл на лунку). На данной стадии общее количество среды в лунках составляло 200 мкл. В качестве контрольной группы в лунки добавляли ДМСО (100 мкл на лунку), разбавленный средой RPMI-1640, содержащей 10% эмбриональную телячью сыворотку. Планшет помещали обратно в инкубатор и культивировали еще в течение 3 суток. Через 3 суток планшет забирали при комнатной температуре и удаляли 100 мкл надосадочной жидкости. В лунки добавляли CellTiter-GloTM (Promega) (50 мкл на лунку) и подсчитывали количество клеток. Результаты ингибирующего пролиферацию клеток эффекта, полученные при схеме лечения с использованием только соединения II, при обработке с использованием только эрлотиниба и при обработке с использованием двух лекарственных средств, анализировали тем же способом, что и в тестовом примере 9, получая тем самым показатель аддитивности (CI) в качестве показателя эффекта комбинированного введения. Эффект определяли конкурентным, если величина CI составляла 1,2 или более, аддитивным, если величина CI составляла менее чем 1,2 и не менее чем 0,85, и синергическим, если величина CI составляла менее чем 0,85. Показатель аддитивности представлен ниже в таблице 19. Величина CI для клеточной линии NCI-N87 составляла от 0,25 до 0,52, демонстрируя, что соединение II проявляло синергический эффект ингибирования клеточной пролиферации при применении с эрлотинибом.
Тестовый пример 11
Усиление ингибирующего пролиферацию раковых клеток эффекта трастузумаба (герцептина)
Человеческую клеточную линию рака желудка (4-1ST) подкожно имплантировали в правый бок самцам мышей BALB/cA Jcl-nu/nu в возрасте 6 недель и использовали тем же способом, что и в тестовом примере 1.
Согласно вкладышу-инструкции по применению, в ампулу, содержащую 150 мг трастузумаба (Roche Pharma), добавляли 7,2 мл воды для инъекции с использованием стерилизованного шприца для инъекций. Полученный раствор в концентрации 21 мг/мл хранили в морозильной камере. Во время использования замороженный раствор разбавляли в 10,5 раз 4,75 мл физиологического раствора при концентрации трастузумаба 2,0 мг/мл, с получением в результате раствора для введения с дозой трастузумаба 20 мг/кг/сутки. Приготавливали тестовые растворы соединения I для обеспечения введения в дозах 16 и 24 мг/кг/сутки.
Соединение I и трастузумаб вводили один раз в сутки с 1 по 14 сутки. В группах комбинированного введения соединение I вводили в дозе 16 и 24 мг/кг/сутки, трастузумаб вводили в дозе 20 мг/кг/сутки, и проводили оценку тем же способом, что и в тестовом примере 1. Результаты представлены на Фиг. 17A и 17B и в таблице 20. В группе введения среды/соединения I тестовый раствор вводили перорально один раз в сутки в течение последующих суток. В группе введения трастузумаба тестовый раствор вводили интраперитонеально один раз в сутки в течение последующих суток.
Эффект комбинированного введения статистически определяли тем же способом, что и в тестовом примере 7. На фигуре и в таблице звездочка представляет собой результат, обладающий статистически значимым различием по сравнению с группой введения только лекарственного средства.
С течением времени оценивали изменение массы тела в качестве показателя токсичности тем же способом, что и в тестовом примере 1. Результаты представлены на Фиг. 18A и 18B.
Как наглядно представлено на Фиг. 1, 3, 5, 7, 9, 11, 13, 15 и 17, имидазооксазиновое соединение (I) или его фармацевтически приемлемая соль значительно усиливают различные противоопухолевые средства. Эффект наблюдали даже при низкой дозе имидазооксазинового соединения (I), то есть, 8 мг/кг/сутки (количество, при котором имидазооксазиновое соединение (I) не проявляло противоопухолевого эффекта по отдельности; Фиг. 1 и таблица 11). В группе бестимусных мышей, получавших высокую дозу (24 мг/кг/сутки) (доза для получения максимального эффекта), наблюдали значительное уменьшение размеров опухоли при комбинированном введении (таблицы 11 и 13). Кроме того, снижение веса не ухудшалось в группе комбинированного введения по сравнению со снижением веса при введении для каждого противоопухолевого средства в отдельности (Фиг. 2, 4, 12 и 14). Такие данные показывают, что соединение согласно настоящему изобретению усиливает различные противоопухолевые средства без усиления их токсичности.
Кроме того, например, как показано в сравнении Фиг. 1 и 3, хотя противоопухолевый эффект (чувствительность к лекарственному средству) паклитаксела варьирует в зависимости от типа опухоли, усиление эффекта наблюдали во всех случаях его комбинированного применения с имидазооксазиновым соединением (I) даже в той же самой дозировке. Более конкретно, считается, что даже для опухолей, обладающих низкой чувствительностью к паклитакселу, комбинированное применение паклитаксела и имидазооксазинового соединения (I) усиливает ингибирующий пролиферацию опухоли эффект паклитаксела. Это указывает на то, что при комбинированном применении с имидазооксазиновым соединением (I) спектр его противоопухолевого эффекта противоопухолевого средства увеличивается.
Более конкретно, комбинации имидазооксазинового соединения (I) или его фармацевтически приемлемой соли и различных противоопухолевых средств продемонстрировали усиление противоопухолевого эффекта и увеличение спектра противоопухолевого эффекта без проявления значительного усиления токсичности.
Тестовый пример 12
Усиление ингибирующего пролиферацию раковых клеток эффекта метформина
Человеческую клеточную линию рака яичников A2780 субкультивировали в среде RPMI-1640, содержащей 10% эмбриональную телячью сыворотку. Клетки A2780 высевали в 96-луночный микропланшет (2000 клеток/100 мкл/лунка), и культивировали в течение ночи в инкубаторе при 37°C, 5% CO2 и 100% влажности. Соединение I разбавляли раствором ДМСО с кратностью разведения 200 до конечных концентраций 0 (только ДМСО), 10, 30, 100, 300, 1000, 3000 и 10000 нМ и разбавляли метформин средой RPMI-1640, содержащей 10% эмбриональную телячью сыворотку, до конечных концентраций в 1000, 3333 и 10000 раз от конечных концентраций соединения I. Планшет забирали из инкубатора. В группах комбинированного введения соединения I и метформина, соединение I и метформин разбавляли для получения всех комбинаций из указанных выше конечных концентраций и добавляли в лунки. На данной стадии общее количество среды в лунках составляло 200 мкл. В качестве контрольной группы в лунки добавляли ДМСО (100 мкл на лунку), разбавленный средой RPMI-1640, содержащей 10% эмбриональную телячью сыворотку, до конечной концентрации 0,5%. Планшет помещали обратно в инкубатор и культивировали еще в течение 3 суток. Через 3 суток планшет забирали при комнатной температуре и удаляли 150 мкл надосадочной жидкости. В лунки добавляли CellTiter-GloTM (Promega) (50 мкл на лунку), и подсчитывали количество клеток. Результаты ингибирующего пролиферацию клеток эффекта, полученные при схеме лечения с использованием только соединения I, при схеме лечения с использованием только эрлотиниба и при обработке с использованием двух лекарственных средств, анализировали тем же способом, что и в тестовом примере 9, получая тем самым показатель аддитивности (CI) в качестве показателя эффекта комбинированного введения. Эффект определяли конкурентным, если величина CI составляла 1,2 или более, аддитивным, если величина CI составляла менее чем 1,2 и не менее чем 0,85, и синергическим, если величина CI составляла менее чем 0,85. Показатель аддитивности показан ниже в таблице. Величина CI составляла от 0,08 до 0,96 для клеточной линии A2780, демонстрируя, что при применении с метформином соединение I проявляло аддитивный или синергический ингибирующий клеточную пролиферацию эффект.
Тестовый пример 13
Усиление ингибирующего пролиферацию злокачественных клеток эффекта доцетаксела
Человеческую клеточную линию рака яичников A2780 субкультивировали в среде RPMI-1640, содержащей 10% эмбриональную телячью сыворотку. Клетки A2780 высевали в 96-луночный микропланшет (2000 клеток/150 мкл/лунка) и культивировали в течение ночи в инкубаторе при 37°C, 5% CO2 и 100% влажности. Соединение I разбавляли раствором ДМСО с кратностью разведения 200 до конечных концентраций 0 (только ДМСО), 10, 30, 100, 300, 1000, 3000 и 10000 нМ, и доцетаксел разбавляли средой RPMI-1640, содержащей 10% эмбриональную телячью сыворотку, до конечных концентраций в 0,01 и 0,03 раз от конечных концентраций соединения I. Планшет забирали из инкубатора и, как и для группы комбинированного введения соединения I и доцетаксела, соединение I и доцетаксел разбавляли для получения все комбинаций из указанных выше конечных концентраций и добавляли в лунки. На данной стадии общее количество среды в лунке составляло 200 мкл. В качестве контрольной группы в лунки добавляли ДМСО (50 мкл на лунку), разбавленный средой RPMI-1640, содержащей 10% эмбриональную телячью сыворотку, до конечной концентрации 0,5%. Планшет помещали обратно в инкубатор и культивировали еще в течение 3 суток. Через 3 суток планшет забирали при комнатной температуре и удаляли 150 мкл надосадочной жидкости. В лунки добавляли CellTiter-Gloтм (Promega) (50 мкл на лунку) и подсчитывали количество клеток. Результаты ингибирующего пролиферацию клеток эффекта, полученные при схеме лечения с использованием только соединения I, при схеме лечения с использованием только доцетаксела и при схеме лечения с использованием двух лекарственных средств, анализировали тем же способом, что и в тестовом примере 9, получая тем самым показатель аддитивности (CI) в качестве показателя эффекта комбинированного введения. Эффект определяли конкурентным, если величина CI составляла 1,2 или более, аддитивным, если величина CI составляла менее чем 1,2 и не менее чем 0,85, и синергическим, если величина CI составляла менее чем 0,85. Показатель аддитивности показан ниже в таблице. Величина CI составляла от 0,33 до 0,67 для клеточной линии A2780, демонстрируя, что при применении с доцетакселом соединение I проявляло аддитивный или синергический ингибирующий клеточную пролиферацию эффект.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ИМИДАЗООКСАЗИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2012 |
|
RU2578608C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| ИСПОЛЬЗОВАНИЕ ЛИГАНДОВ РЕЦЕПТОРА ЕР4 ДЛЯ ЛЕЧЕНИЯ ОПОСРЕДОВАННЫХ IL-6 ЗАБОЛЕВАНИЙ | 2003 |
|
RU2285527C2 |
| ДИСПИРОПИРРОЛИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2012 |
|
RU2612534C2 |
| 6, 6-БИЦИКЛИЧЕСКИЕ КОЛЬЦЕВЫЕ ЗАМЕЩЕННЫЕ ГЕТЕРОБИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2005 |
|
RU2379308C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИН-2-ОНА, ОБЛАДАЮЩИЕ mTOR ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 2009 |
|
RU2478636C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ SUV39H2 | 2016 |
|
RU2729187C1 |
| КОМБИНАЦИЯ, СОДЕРЖАЩАЯ ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ ОПУХОЛЕЙ | 2021 |
|
RU2827100C1 |
| ПРОИЗВОДНОЕ 1,2,3,4-ТЕТРАГИДРОХИНОКСАЛИНА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2804127C2 |
| ПИРРОЛО[2,3-D]ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2712269C2 |

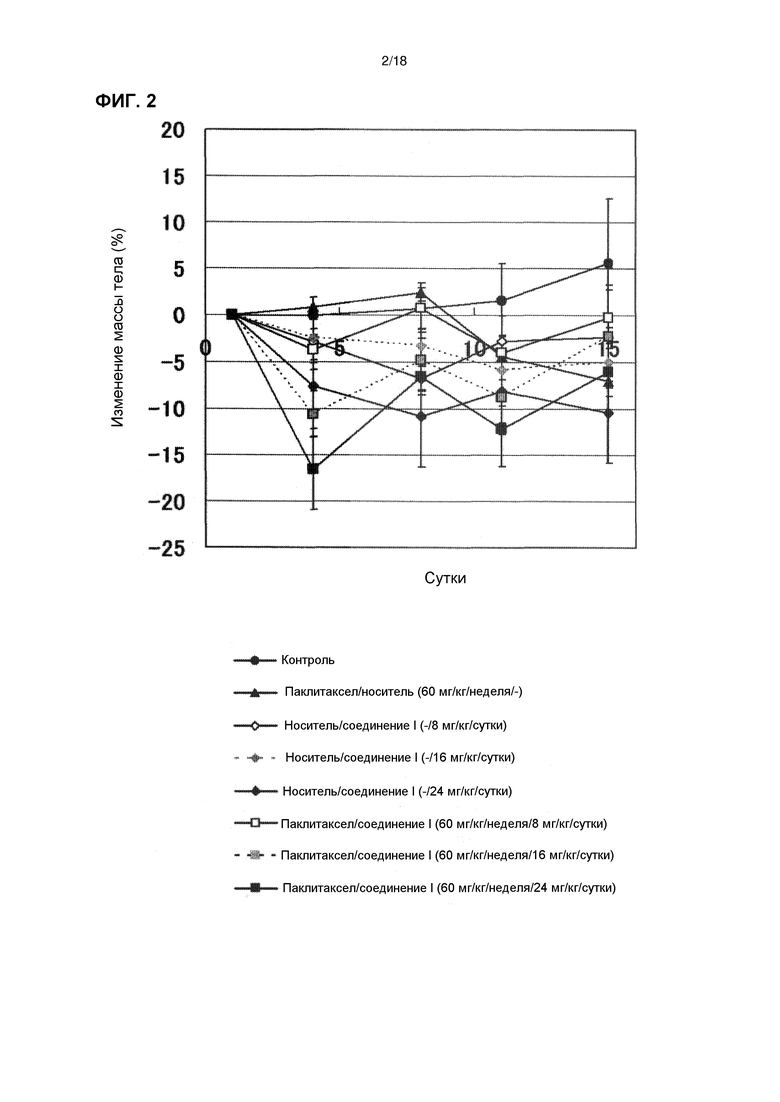
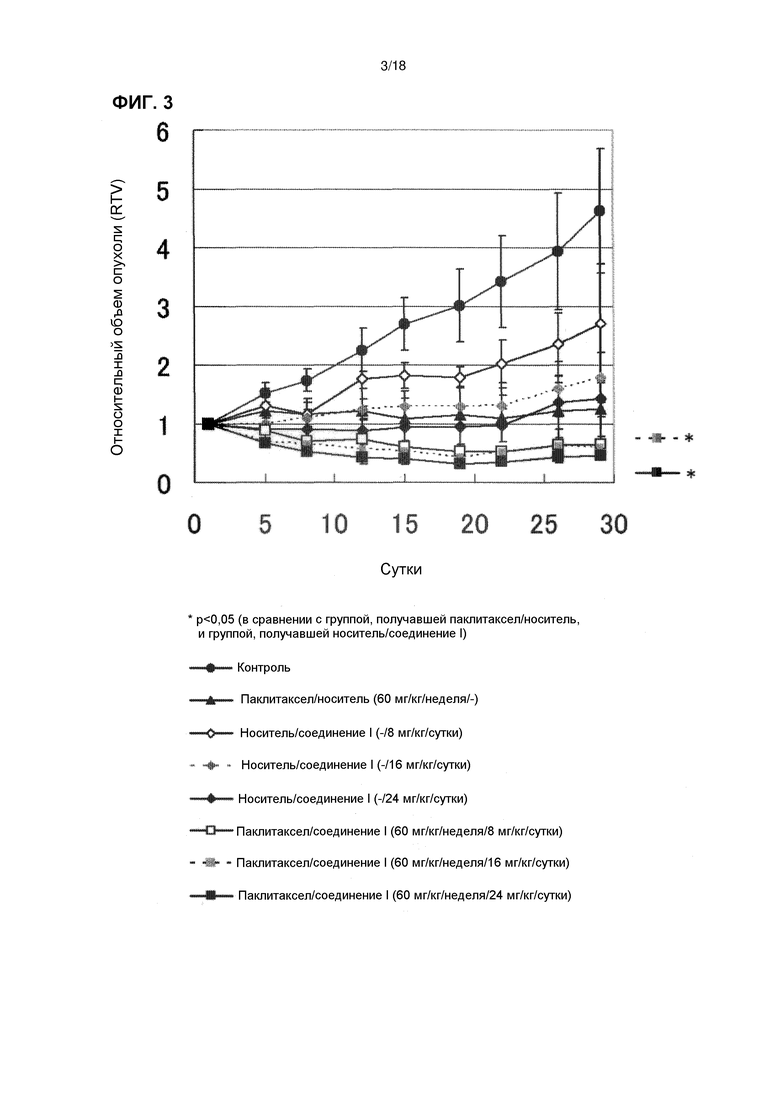
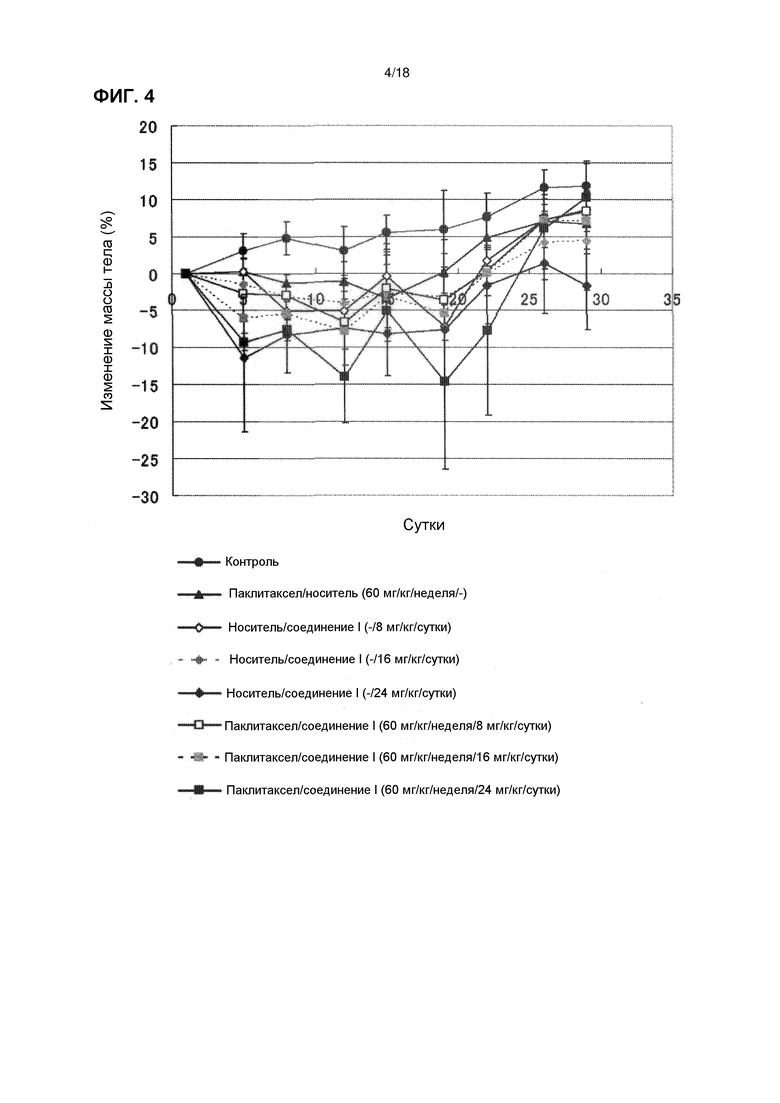
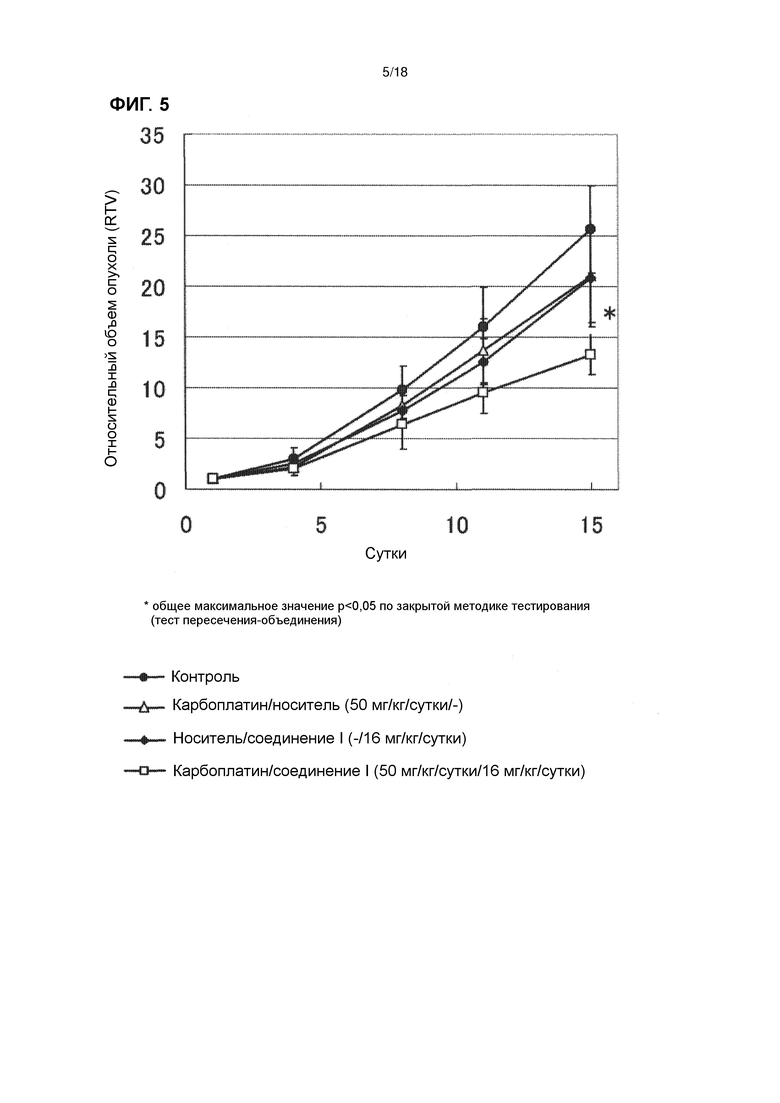
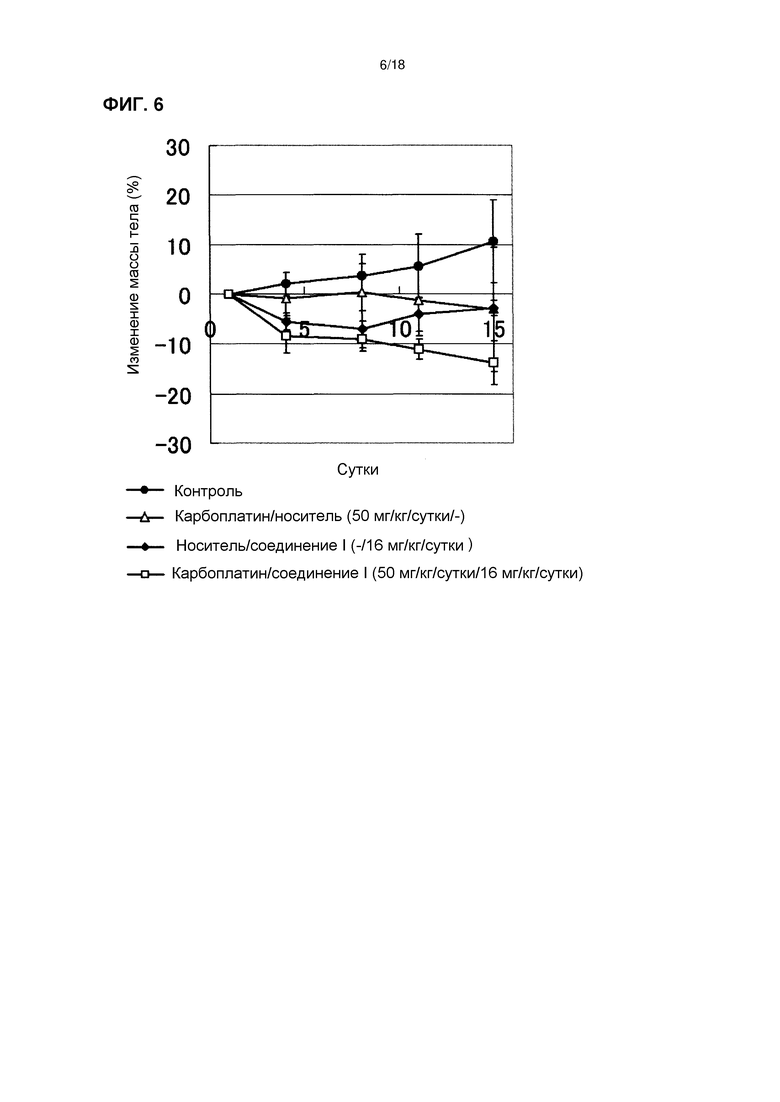
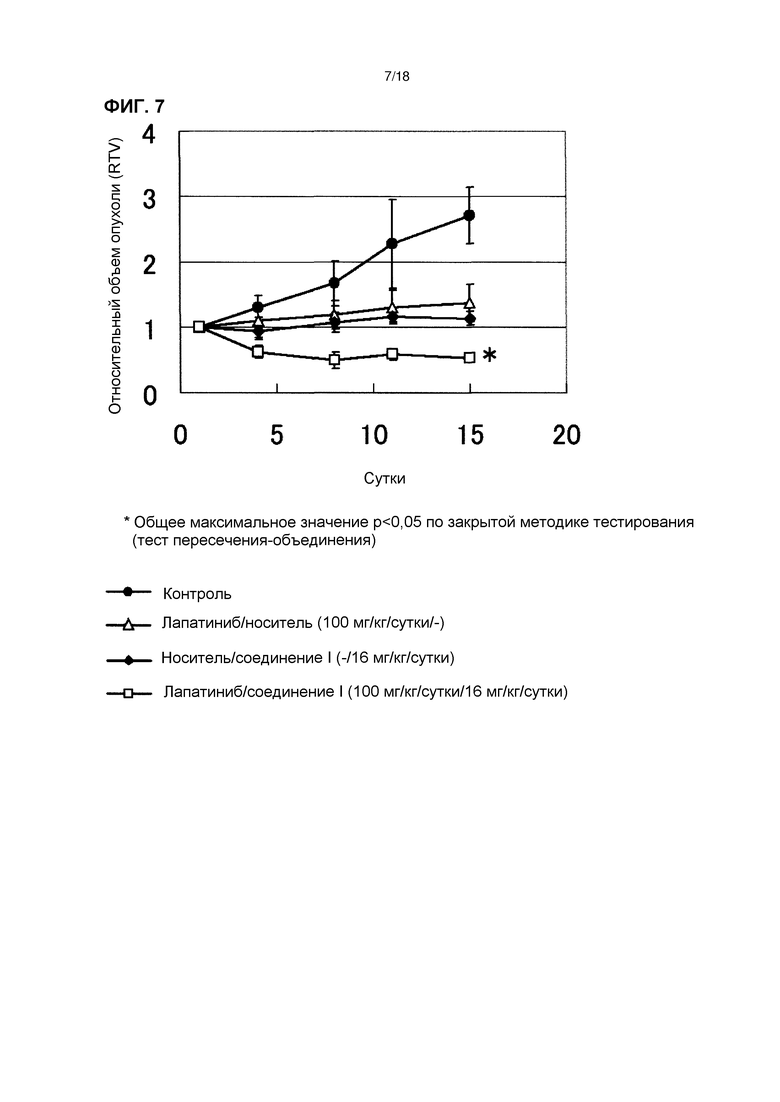

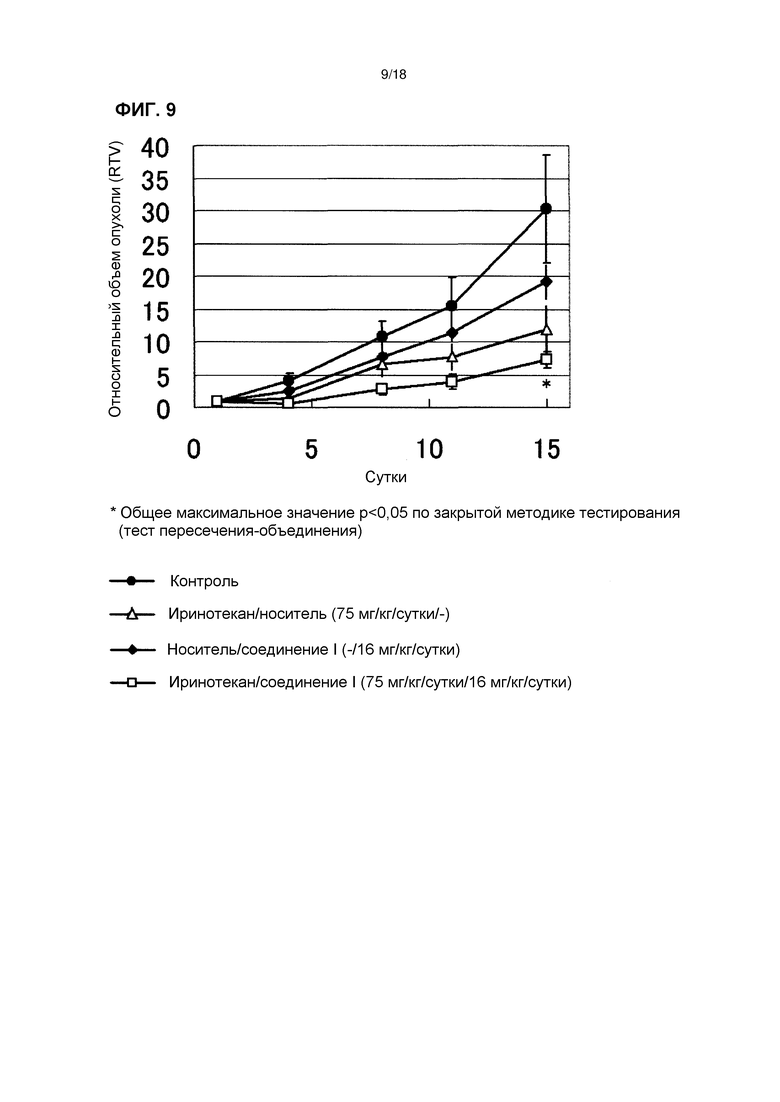
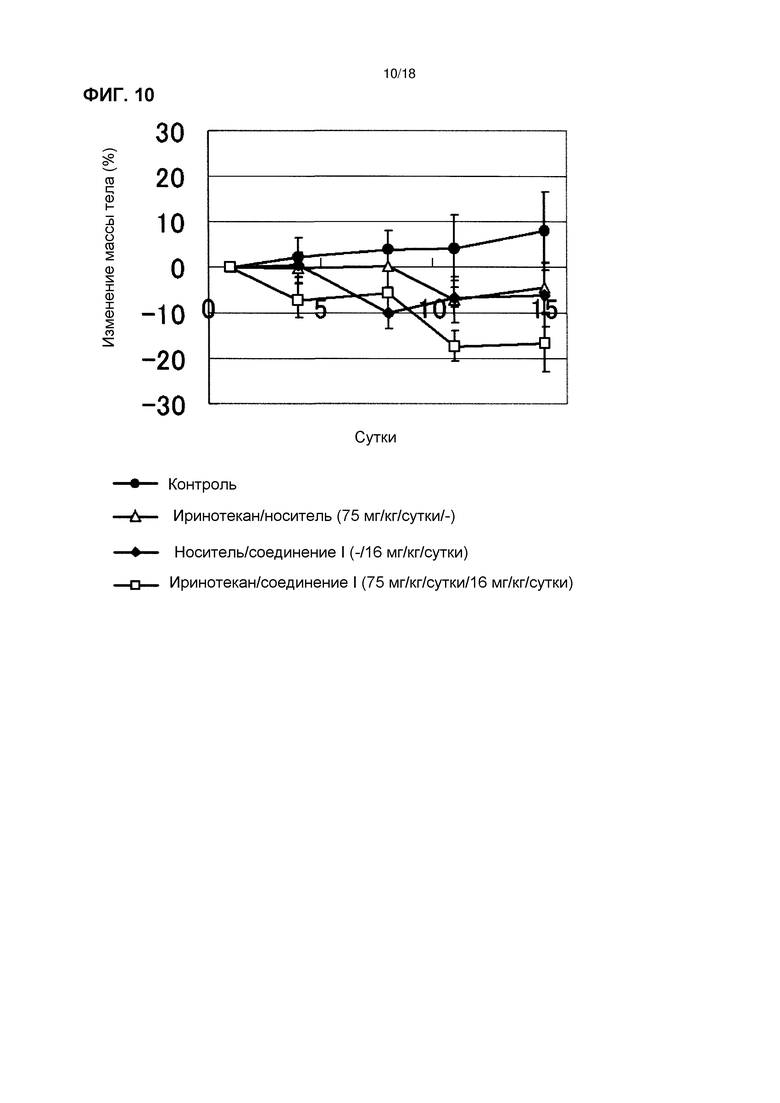
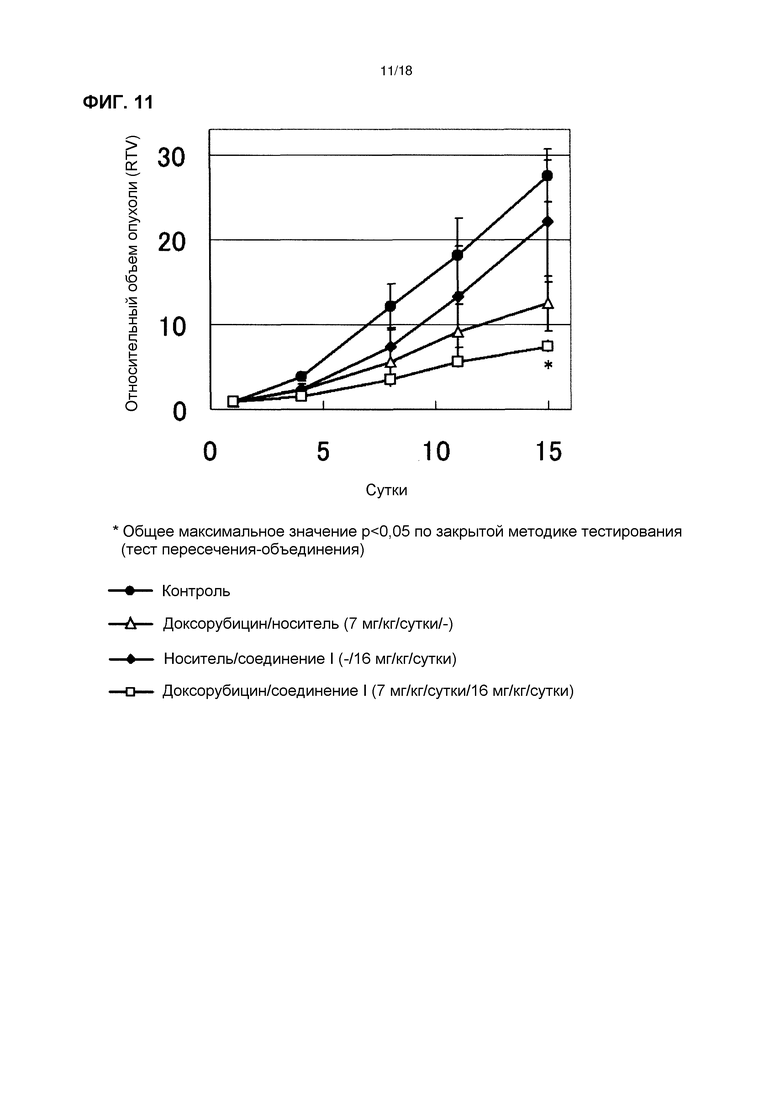
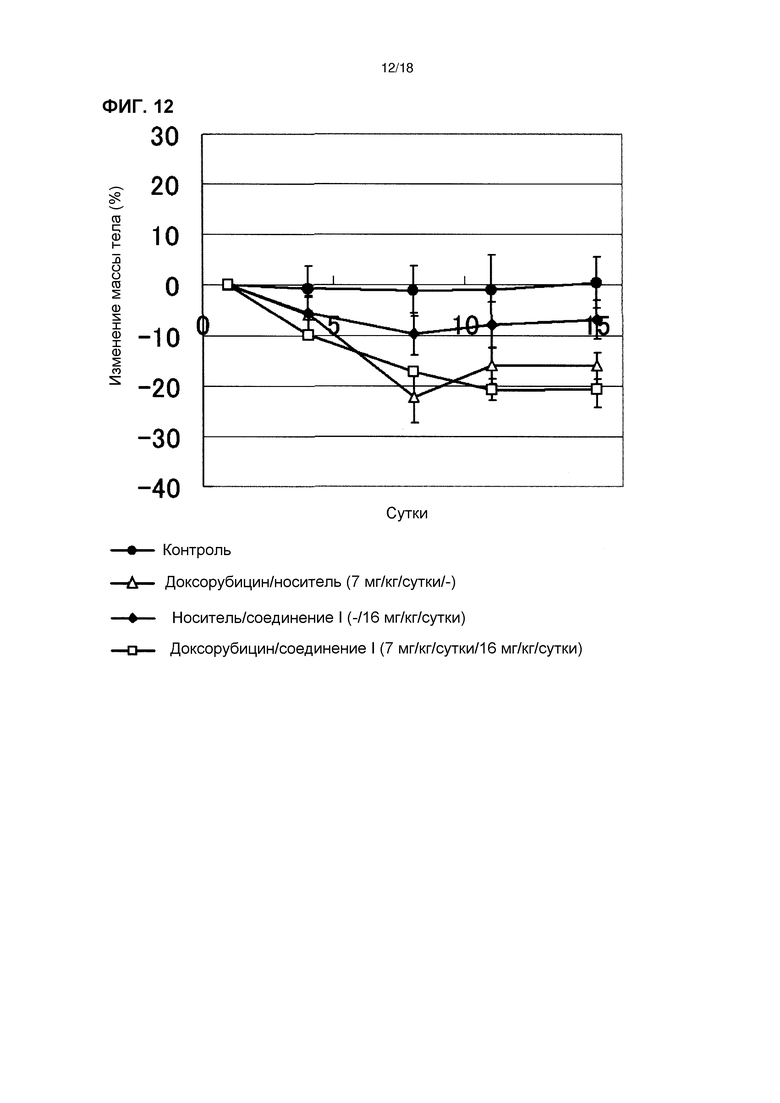
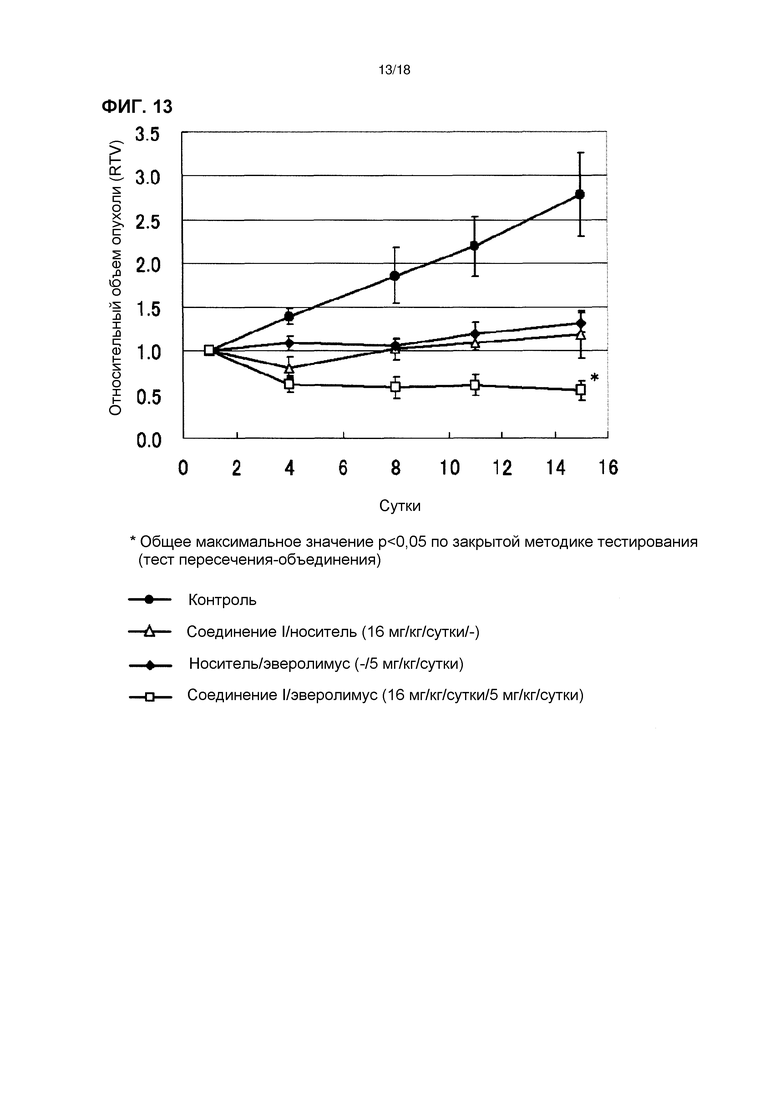
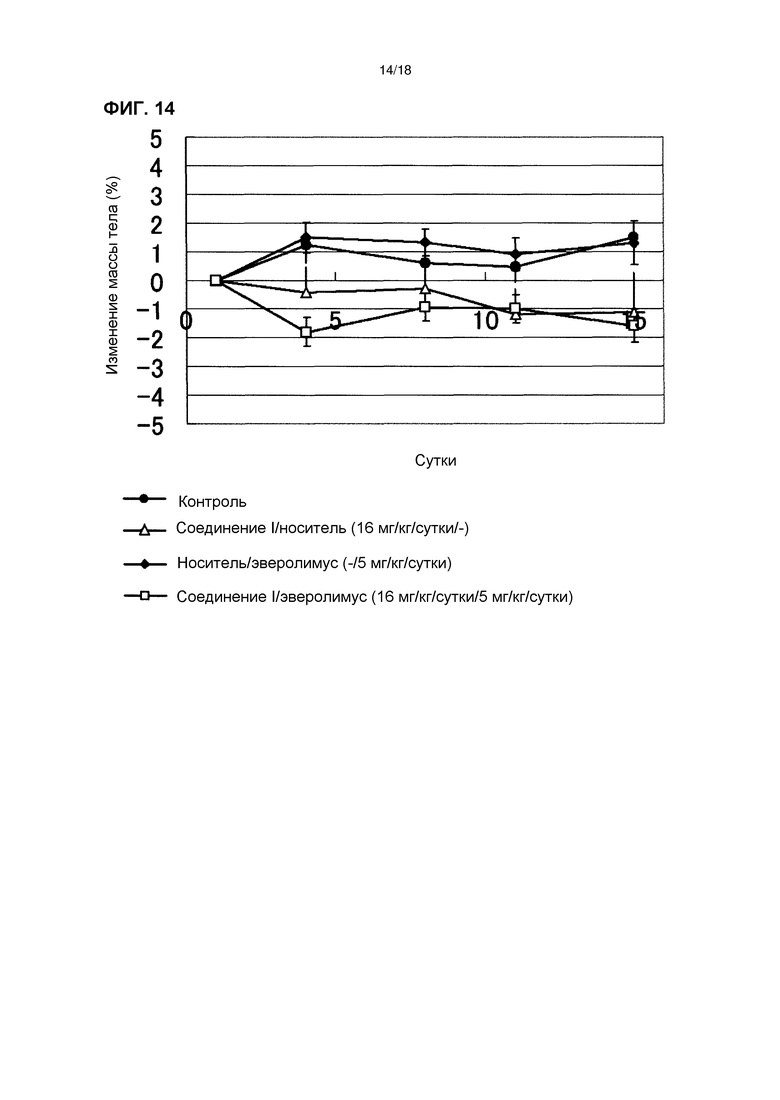

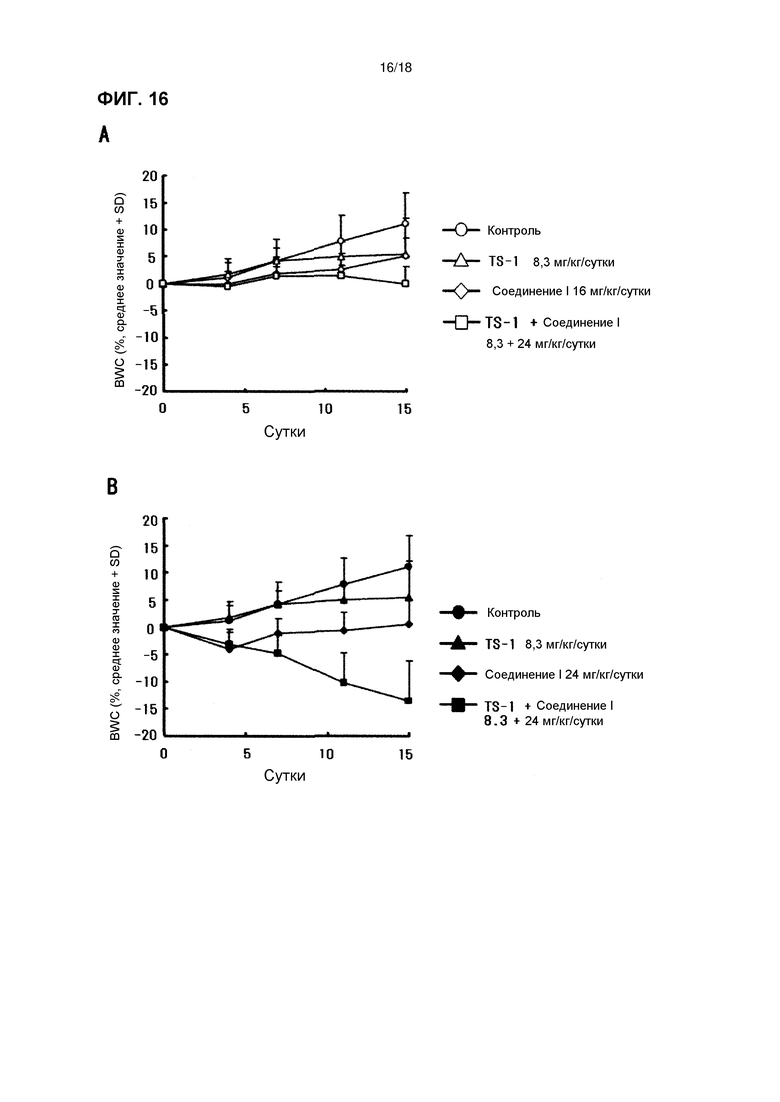
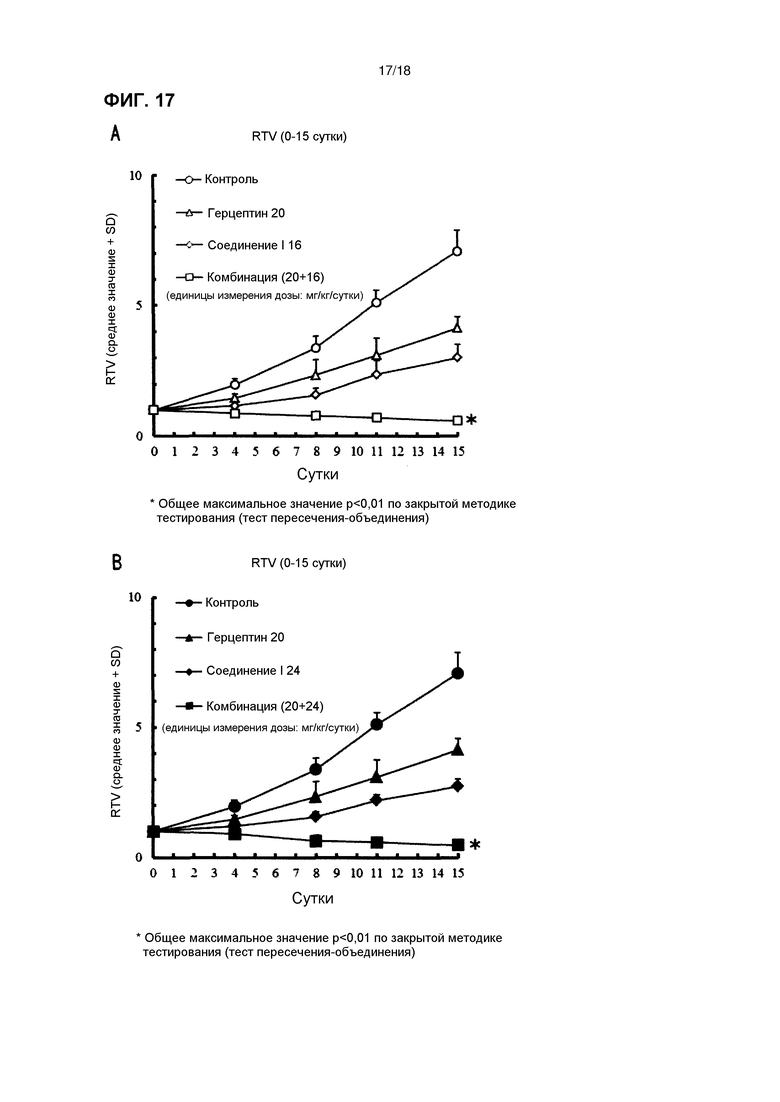
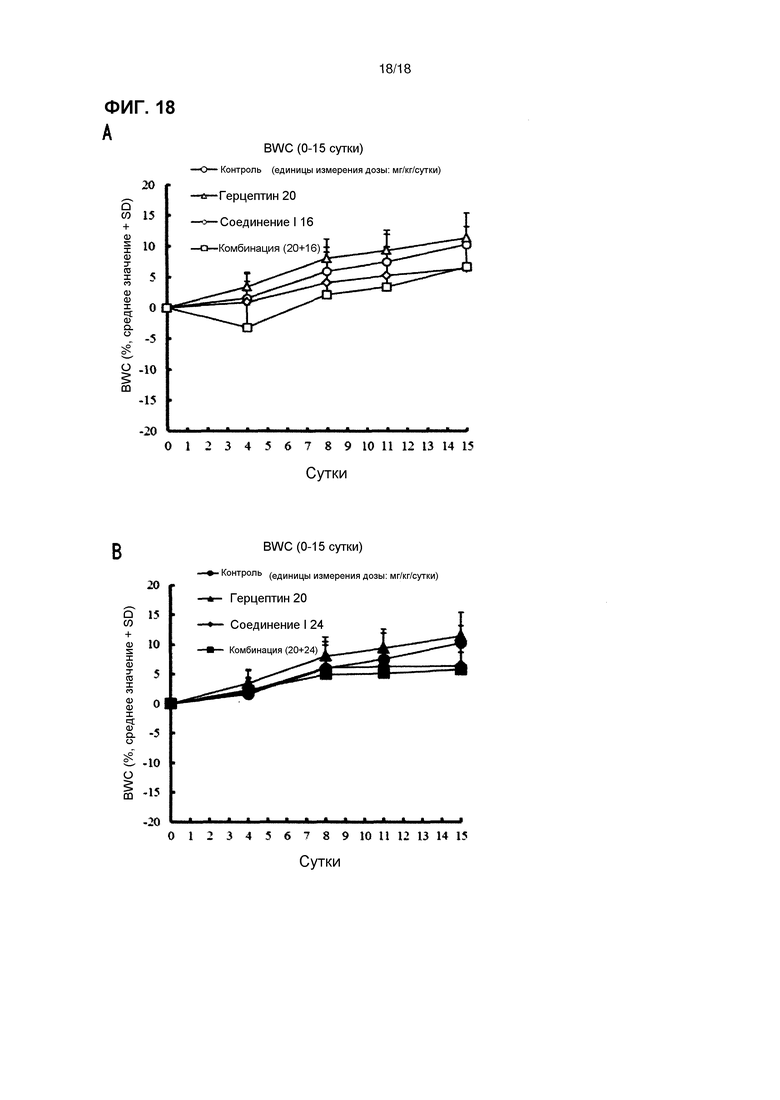
Изобретение относится к химико-фармацевтической промышленности и представляет собой усилитель противоопухолевого эффекта для усиления противоопухолевого средства, содержащий в качестве активного ингредиента имидазооксазиновое соединение, которое представляет собой транс-3-амино-1-метил-3-(4-(3-фенил-5Н-имидазо[1,2-с]пиридо[3,4-е][1,3]оксазин-2-ил)фенил)циклобутанол или транс-3-амино-1-циклопропил-3-(4-(3-фенил-5Н-имидазо[1,2-c]пиридо[4,3-е][1,3]оксазин-2-ил)фенил)циклобутанол, или его фармацевтически приемлемую соль. Также изобретение относится к фармацевтической композиции, противоопухолевому лекарственному средству, содержащим указанные соединения, их применениям, способам усиления противоопухолевого эффекта и лечения опухолей. Изобретение обеспечивает комбинированную противоопухолевую терапию, позволяющую усилить противоопухолевой эффект противоопухолевых лекарственных средств без значительного увеличения их токсичности. 9 н. и 9 з.п. ф-лы, 77 пр., 22 табл., 18 ил.
1. Усилитель противоопухолевого эффекта для усиления противоопухолевого средства, содержащий в качестве активного ингредиента имидазооксазиновое соединение, которое представляет собой транс-3-амино-1-метил-3-(4-(3-фенил-5Н-имидазо[1,2-с]пиридо[3,4-е][1,3]оксазин-2-ил)фенил)циклобутанол, или транс-3-амино-1-циклопропил-3-(4-(3-фенил-5Н-имидазо[1,2-c]пиридо[4,3-е][1,3]оксазин-2-ил)фенил)циклобутанол, или его фармацевтически приемлемую соль.
2. Усилитель противоопухолевого эффекта по п. 1, где противоопухолевое средство представляет собой паклитаксел, карбоплатин, лапатиниб, иринотекан, доксорубицин, эверолимус, бортезомиб, эрлотиниб, трастузумаб (герцептин), метформин, доцетаксел и комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия.
3. Противоопухолевое лекарственное средство, содержащее комбинацию имидазооксазинового соединения, которое представляет собой транс-3-амино-1-метил-3-(4-(3-фенил-5Н-имидазо[1,2-с]пиридо[3,4-е][1,3]оксазин-2-ил)фенил)циклобутанол, или транс-3-амино-1-циклопропил-3-(4-(3-фенил-5Н-имидазо[1,2-c]пиридо[4,3-е][1,3]оксазин-2-ил)фенил)циклобутанол, или его фармацевтически приемлемую соль, и противоопухолевого средства.
4. Противоопухолевое лекарственное средство по п. 3, где противоопухолевое средство представляет собой паклитаксел, карбоплатин, лапатиниб, иринотекан, доксорубицин, эверолимус, бортезомиб, эрлотиниб, трастузумаб (герцептин), метформин, доцетаксел и комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия.
5. Применение имидазооксазинового соединения, которое представляет собой транс-3-амино-1-метил-3-(4-(3-фенил-5Н-имидазо[1,2-c]пиридо[3,4-е][1,3]оксазин-2-ил)фенил)циклобутанол, или транс-3-амино-1-циклопропил-3-(4-(3-фенил-5Н-имидазо[1,2-с]пиридо[4,3-е][1,3]оксазин-2-ил)фенил)циклобутанол, или его фармацевтически приемлемую соль, для усиления противоопухолевого эффекта противоопухолевого средства.
6. Применение по п. 5, где противоопухолевое средство представляет собой паклитаксел, карбоплатин, лапатиниб, иринотекан, доксорубицин, эверолимус, бортезомиб, эрлотиниб, трастузумаб (герцептин), метформин, доцетаксел и комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия.
7. Применение имидазооксазинового соединения, которое представляет собой транс-3-амино-1-метил-3-(4-(3-фенил-5Н-имидазо[1,2-c]пиридо[3,4-е][1,3]оксазин-2-ил)фенил)циклобутанол, или транс-3-амино-1-циклопропил-3-(4-(3-фенил-5Н-имидазо[1,2-с]пиридо[4,3-е][1,3]оксазин-2-ил)фенил)циклобутанол, или его фармацевтически приемлемую соль, для получения усилителя противоопухолевого эффекта для усиления противоопухолевого эффекта противоопухолевого средства.
8. Применение по п. 7, где противоопухолевое средство представляет собой паклитаксел, карбоплатин, лапатиниб, иринотекан, доксорубицин, эверолимус, бортезомиб, эрлотиниб, трастузумаб (герцептин), метформин, доцетаксел и комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия.
9. Применение имидазооксазинового соединения, которое представляет собой транс-3-амино-1-метил-3-(4-(3-фенил-5Н-имидазо[1,2-c]пиридо[3,4-е][1,3]оксазин-2-ил)фенил)циклобутанол, или транс-3-амино-1-циклопропил-3-(4-(3-фенил-5Н-имидазо[1,2-с]пиридо[4,3-е][1,3]оксазин-2-ил)фенил)циклобутанол, или его фармацевтически приемлемую соль, для получения противоопухолевого лекарственного средства, содержащего комбинацию имидазооксазинового соединения или его фармацевтически приемлемой соли и противоопухолевое средство.
10. Применение по п. 9, где противоопухолевое средство представляет собой паклитаксел, карбоплатин, лапатиниб, иринотекан, доксорубицин, эверолимус, бортезомиб, эрлотиниб, трастузумаб (герцептин), метформин, доцетаксел и комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия.
11. Фармацевтическая композиция для лечения опухолей, содержащая имидазооксазиновое соединение, которое представляет собой транс-3-амино-1-метил-3-(4-(3-фенил-5Н-имидазо[1,2-с]пиридо[3,4-е][1,3]оксазин-2-ил)фенил)циклобутанол, или транс-3-амино-1-циклопропил-3-(4-(3-фенил-5Н-имидазо[1,2-c]пиридо[4,3-е][1,3]оксазин-2-ил)фенил)циклобутанол, или его фармацевтически приемлемую соль, и противоопухолевое средство.
12. Фармацевтическая композиция по п. 11, где противоопухолевое средство представляет собой паклитаксел, карбоплатин, лапатиниб, иринотекан, доксорубицин, эверолимус, бортезомиб, эрлотиниб, трастузумаб (герцептин), метформин, доцетаксел и комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия.
13. Способ усиления противоопухолевого эффекта, включающий введение пациенту имидазооксазинового соединения, которое представляет собой транс-3-амино-1-метил-3-(4-(3-фенил-5Н-имидазо[1,2-c]пиридо[3,4-е][1,3]оксазин-2-ил)фенил)циклобутанол, или транс-3-амино-1-циклопропил-3-(4-(3-фенил-5Н-имидазо[1,2-с]пиридо[4,3-е][1,3]оксазин-2-ил)фенил)циклобутанол, или его фармацевтически приемлемую соль, в количестве, эффективном для лечения опухолей.
14. Способ по п. 13, где противоопухолевое средство представляет собой паклитаксел, карбоплатин, лапатиниб, иринотекан, доксорубицин, эверолимус, бортезомиб, эрлотиниб, трастузумаб (герцептин), метформин, доцетаксел и комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия.
15. Способ лечения опухолей для усиления противоопухолевого средства, включающий введение пациенту комбинации из имидазооксазинового соединения, которое представляет собой транс-3-амино-1-метил-3-(4-(3-фенил-5Н-имидазо[1,2-c]пиридо[3,4-е][1,3]оксазин-2-ил)фенил)циклобутанол, или транс-3-амино-1-циклопропил-3-(4-(3-фенил-5Н-имидазо[1,2-c]пиридо[4,3-е][1,3]оксазин-2-ил)фенил)циклобутанол, или его фармацевтически приемлемую соль, и противоопухолевого средства.
16. Способ по п. 15, где противоопухолевое средство представляет собой паклитаксел, карбоплатин, лапатиниб, иринотекан, доксорубицин, эверолимус, бортезомиб, эрлотиниб, трастузумаб (герцептин), метформин, доцетаксел и комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия.
17. Продукт в виде комбинированного препарата, подлежащий использованию одновременно, последовательно или с некоторым интервалом во времени для лечения опухолей, причем продукт включает комбинацию из имидазооксазинового соединения, которое представляет собой транс-3-амино-1-метил-3-(4-(3-фенил-5Н-имидазо[1,2-c]пиридо[3,4-е][1,3]оксазин-2-ил)фенил)циклобутанол, или транс-3-амино-1-циклопропил-3-(4-(3-фенил-5Н-имидазо[1,2-с]пиридо[4,3-е][1,3]оксазин-2-ил)фенил)циклобутанол, или его фармацевтически приемлемую соль, и противоопухолевого средства.
18. Продукт по п. 17, где противоопухолевое средство представляет собой паклитаксел, карбоплатин, лапатиниб, иринотекан, доксорубицин, эверолимус, бортезомиб, эрлотиниб, трастузумаб (герцептин), метформин, доцетаксел и комбинированное лекарственное средство из тегафура, гимерацила и отерацила калия.
| US 20090156604 A1, 18.06.2009 | |||
| US 20110319354 A1, 29.12.2011 | |||
| WO 2009148916 A1, 10.12.2009 | |||
| ПРОИЗВОДНОЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРА PI3K И ЕГО ПРИМЕНЕНИЕ | 2007 |
|
RU2448109C2 |

