ОБЛАСТЬ ИЗОБРЕТЕНИЯ
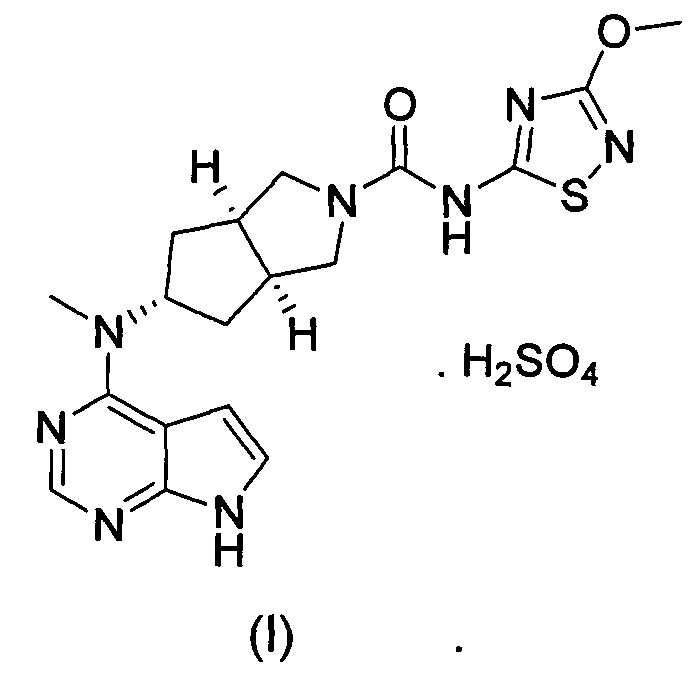
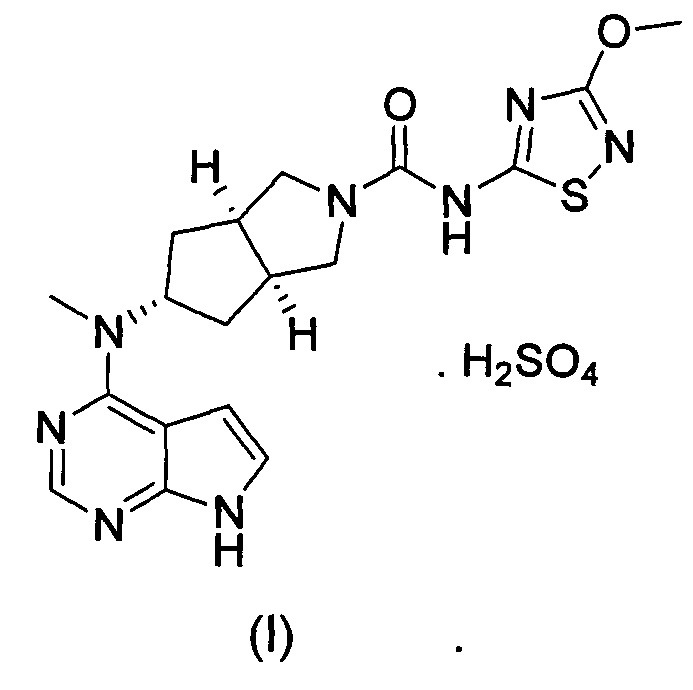
Настоящее изобретение относится к бисульфату ингибитора Янус-киназы (JAK) и к способу его получения. Более конкретно настоящее изобретение относится к бисульфату (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1Н)-карбоксамида и к способу его получения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Артрит является наиболее распространенным хроническим заболеванием в мире, существует много причин, приводящих к артриту, и повреждения суставов, вызванные ими, различны. В настоящее время основные лекарственные средства для лечения ревматоидного артрита включают адалимумаб (Humira) компании America Abbott Laboratories, этанерцепт (Enbrel), совместно разработанный компаниями Pfizer и Amgen, и инфликсимаб (Remicade) фармацевтической компании Janssen. Эти лекарственные средства в настоящее время являются наиболее продаваемыми лекарственными средствами на фармацевтическом рынке, но стоит отметить, что эти наиболее продаваемые лекарственные средства являются только инъекционными лекарственными средствами. Хотя МТХ (метотрексат), который чаще всего вводят перорально, обладает значительной эффективностью, его токсичность очень высока.
Исследования показали, что расстройства биохимического пути передачи сигнала множеством цитокинов играют важную роль в патофизиологическом процессе ревматоидного артрита (PA). Воспалительный каскад, опосредованный серией неконтролируемых цитокинов, ведет к множеству клеток, связанных с РА, включая T-клетки, B-клетки, моноциты, макрофаги и остеокласты в долговременном активированном состоянии, вызывая посредством этого устойчивое воспаление и структурное повреждение суставов. Биохимический путь передачи сигнала Янус-киназы (JAK) может регулировать провоспалительную активность клеток, связанных с РА, где JAK является хаб-белком при преобразовании сигнала в сети воспалительных цитокинов, и уровень JAK значительно увеличивается в синовиальных тканях сустава, пораженного РА. В настоящее время тофацитиниб (СР-690550), разработанный компанией Pfizer, является ингибитором JAK1. Результаты фазы III клинического испытания показали, что эффективность тофацитиниба компании Pfizer значительно лучше, чем метотрексата. В данном испытании исследователи рандомизировали пациентов в группы, одной группе пациентов вводили 5 мг/10 мг тофацитиниба в виде монотерапии, а другой группе пациентов вводили 5 мг/10 мг метотрексата. Результаты показали, что эффективность тофацитиниба при ингибировании внутреннего структурного повреждения человека была несколько лучше, чем метотрексата в течение периода в шесть месяцев, он может эффективно улучшать различные симптомы у пациентов с ревматоидным артритом.
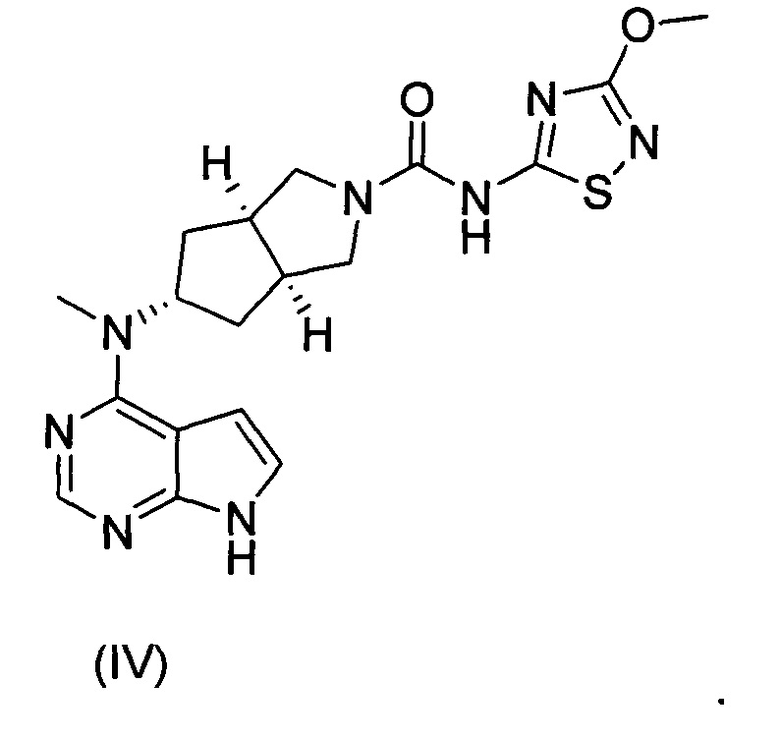
Авторы изобретения исходили из идеи разработки ингибиторов киназы JAK и следили за направлением разработки аналогичных международных лекарственных средств. Основываясь на структуре тофацитиниба, авторы изобретения разработали серию лекарственных средств, обладающих активностью in vitro и in vivo и высокой абсорбцией, и соединение формулы (IV) было успешно получено как ингибитор киназы JAK. Что касается соединения формулы (IV), информация о нем была полностью описана в заявке на патент РСТ № PCT/CN2012/086922, поданной заявителем совместно с другим лицом, полностью включенной в настоящий документ посредством ссылки.
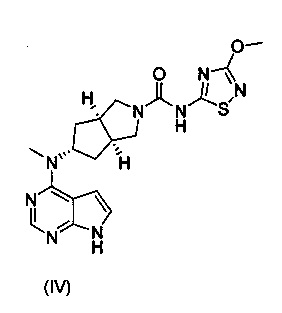
С учетом более низкой растворимости соединения формулы (IV) авторы изобретения изучили образование им солей для улучшения его растворимости и биодоступности, где исследуемые кислоты включали лимонную кислоту, соляную кислоту и серную кислоту. Основываясь на данных растворимости полученных в результате солей и фармакокинетических результатах экспериментов на животных, авторы изобретения неожиданно обнаружили, что соединение формулы (I) должно являться предпочтительным соединением в качестве ингибитора киназы JAK, которое обладает важной научной значимостью при лечении ревматизма и ревматоидного артрита.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В изобретении предложена соль ингибитора киназы JAK, обладающая лучшей растворимостью в воде и улучшенной фармакокинетической активностью. Более конкретно в изобретении предложен бисульфат (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1Н)-карбоксамида формулы (I) и способ его получения.
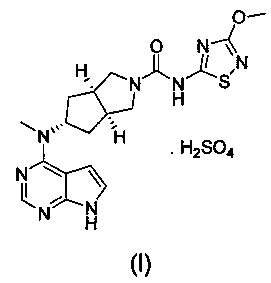
Стехиометрическое отношение (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1Н)-карбоксамида к серной кислоте составляет 1:1.
В другом аспекте, в изобретении предложен способ получения бисульфата (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1Н)-карбоксамида, включающий стадию реакции образования соли соединения формулы (IV) и серной кислоты.
 .
.
Описанную выше реакцию можно проводить в растворителе, где растворитель реакции представляет собой смешанный растворитель из галогеналканов и спиртов, имеющих количество атомов углерода, меньшее или равное 3, предпочтительно смешанный растворитель из дихлорметана и метанола.
Предпочтительная температура описанной выше реакции составляет 10-30°C, и предпочтительное время реакции составляет 0,5-4 часа.
В другом аспекте в изобретении предложена фармацевтическая композиция, содержащая бисульфат (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксамида формулы (I) и фармацевтически приемлемый носитель.
В другом аспекте изобретение относится к применению бисульфат (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1Н)-карбоксамида формулы (I) или содержащих его фармацевтических композиций для получения лекарственного средства для лечения ревматизма и ревматоидного артрита.
Бисульфит формулы (I), полученный в соответствии со способом по изобретению, не содержал остаточный растворитель или содержал лишь небольшое количество остаточного растворителя и удовлетворял требованиям по ограничению содержания остаточного растворителя релевантных фармацевтических препаратов, указанных в Китайской фармакопее, следовательно, бисульфат формулы (I) согласно изобретению можно надлежащим образом применять в качестве активной фармацевтической субстанции.
ПРЕДПОЧТИТЕЛЬНЫЕ ВОПЛОЩЕНИЯ ИЗОБРЕТЕНИЯ
Приведенные ниже примеры служат для подробной иллюстрации изобретения и описания технических решений настоящего изобретения. Следует понимать, что приведенные ниже примеры не ограничивают сущность и объем настоящего изобретения.
ПРИМЕР 1: Получение соединения формулы (IV) (описанного в PCT/CN2012/086922)
Соединение формулы (IV) может быть получено в соответствии со следующим путем:

где получение соединения d было предложено, как описано ниже:
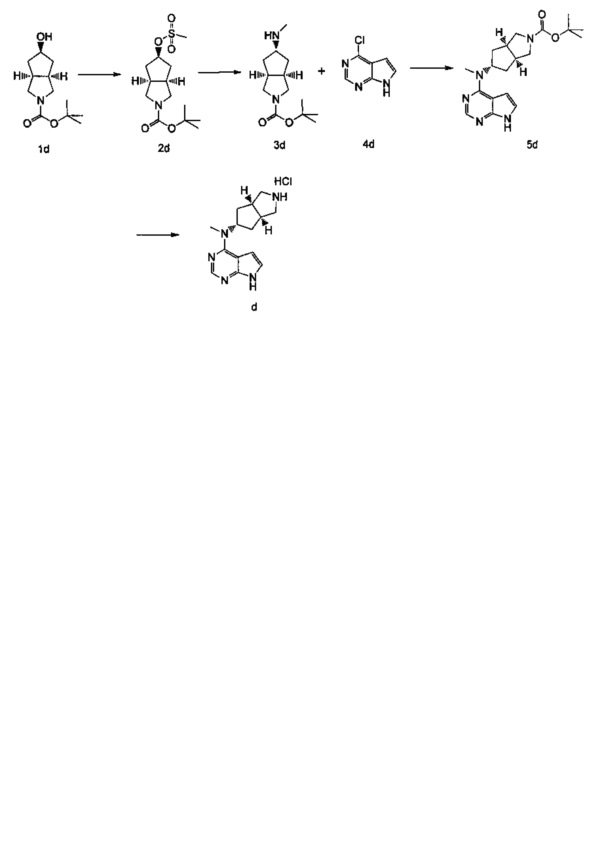
Конкретно получение соединения формулы (IV) включает следующие две части:
Часть I: Получение соединения d
Стадия 1
(3aR,5r,6aS)-трет-бутил-5-((метилсульфонил)окси)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат
(3aR,5r,6aS)-трет-бутил-5-гидроксигексагидроциклопента[c]пиррол-2(1H)-карбоксилат 1d (9 г, 40 ммоль) растворяли в 150 мл дихлорметана с последующим добавлением метилсульфонилхлорида (4,70 мл, 60 ммоль) и триэтиламина (11,20 мл, 80 ммоль) при 0°C. После взаимодействия в течение 2 часов при комнатной температуре, к реакционной смеси добавляли 200 мл насыщенного раствора бикарбоната натрия. Разделяли водную фазу и органическую фазу. Органическую фазу промывали насыщенным раствором хлорида натрия (200 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением продукта, указанного в заголовке, (3aR,5r,6aS)-трет-бутил-5-((метилсульфонил)окси)гексагидроциклопента[c]пиррол-2(1Н)-карбоксилат 2d (12,00 г, выход 98,4%) в виде желтой жидкости.
Стадия 2
(3aR,5s,6aS)-трет-бутил-5-(метиламино)гексагидроциклопента[c]пиррол-2(1H)-карбоксилат
(3aR,5r,6aS)-трет-бутил-5-((метилсульфонил)окси)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат 2d (60 мг, 0,2 ммоль) растворяли в 10 мл метанола с последующим добавлением 5 мл метиламина. После взаимодействия в течение 16 часов при 40°C реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта, указанного в заголовке, (3aR,5s,6aS)-трет-бутил-5-(метиламино)гексагидроциклопента[c]пиррол-2(1Н)-карбоксилата 3d (60 мг, коричневое масло), который непосредственно использовали в следующей стадии без дополнительной очистки.
Масс-спектрометрия (МС) m/z (ионизация электрораспылением (ИЭР)): 241,5 [М+1].
Стадия 3
(3aR,5s,6aS)-трет-бутил-5-(метил(7Н-пиррол[2,3-d]пиримидин-4-ил)амино) гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат
(3aR,5s,6aS)-трет-бутил-5-(метиламино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилат 3d (200 мг, 0,8 ммоль) и 4-хлор-7Н-пирроло[2,3-d]пиримидин 4d (127 мг, 0,8 ммоль) растворяли в 5 мл н-бутанола с последующим добавлением триэтиламина (168 мг, 1,6 ммоль). После взаимодействия в течение 48 часов при 100°C реакционную смесь концентрировали при пониженном давлении с последующим добавлением 10 мл H2O и 10 мл этилацетата. Водную фазу и органическую фазу разделяли. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали высокоэффективной жидкостной хроматографией (ВЭЖХ) с получением продукта, указанного в заголовке, (3aR,5s,6aS)-трет-бутил-5-(метил(7Н-пиррол[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксилата 5d (5 мг, выход 5,0%) в виде белого твердого вещества.
MC m/z (ИЭР): 358,5 [М+1]
1H ЯМР (ядерный магнитный резонанс) (400 МГц, CDCl3): δ 10,07 (s, 1Н), 8,31 (s, 1Н), 7,50 (s, 1Н), 6,55 (s, 1Н), 5,58-5,54 (m, 1Н), 3,65-3,62 (m, 2Н), 3,27-3,23 (m, 5Н), 2,86-2,81 (m, 2Н), 2,06-2,02 (m, 2Н), 1,93-1,91 (m, 2Н), 1,49 (s, 6Н).
Стадия 4
Гидрохлорид N-метил-N-((3aR,5s,6aS)-октагидроциклопента[c]пиррол-5-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина
(3aR,5s,6aS)-трет-бутил-5-(метил(7Н-пиррол[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1Н)-карбоксилат 5d (1,5 г, 4,2 ммоль) растворяли в 20 мл раствора 1 М хлорида водорода в метаноле. После взаимодействия в течение 16 часов реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта, указанного в заголовке, гидрохлорида N-метил-N-((3aR,5s,6aS)-октагидроциклопента[c]пиррол-5-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина d (1,5 г, коричневое твердое вещество).
МС m/z (ИЭР): 258,1 [М+1].
Часть II: Получение соединения формулы (IV)
Стадия 1
Фенил-(3-метокси-1,2,4-тиадиазол-5-ил)карбамат
3-Метокси-1,2,4-тиадиазол-5-амин a (500 мг, 3,82 ммоль) и фенилкарбонохлоридат b (600 мг, 3,82 ммоль) растворяли в 20 мл дихлорметана с последующим добавлением триэтиламина (0,8 мл, 5,73 ммоль). После взаимодействия в течение 16 часов к реакционной смеси добавляли 30 мл H2O для разбавления раствора. Водную фазу и органическую фазу разделяли, водную фазу экстрагировали дихлорметаном  , и органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением продукта, указанного в заголовке, фенил-(3-метокси-1,2,4-тиадиазол-5-ил)карбамата с (200 мг, выход 20,8%) в виде белого твердого вещества.
, и органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением продукта, указанного в заголовке, фенил-(3-метокси-1,2,4-тиадиазол-5-ил)карбамата с (200 мг, выход 20,8%) в виде белого твердого вещества.
МС m/z (ИЭР): 252,0 [М+1].
Стадия 2
(3aS,5s,6aS)-N-(3-Метокси-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамид
Гидрохлорид N-метил-N-((3aR,5s,6aS)-октагидроциклопента[c]пиррол-5-ил)-7Н-пирроло[2,3-d]пиримидин-4-амина d (120 мг, 0,47 ммоль) растворяли в 15 мл тетрагидрофурана с последующим добавлением фенил(3-метокси-1,2,4-тиадиазол-5-ил)карбамата с (117 мг, 0,47 ммоль) и триэтиламина (0,13 мл, 0,94 ммоль). После взаимодействия в течение 5 часов при 60°C реакционную смесь смешивали с 30 мл H2O и экстрагировали дихлорметаном  . Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия , высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования A с получением продукта, указанного в заголовке, (3aR,5s,6aS)-N-(3-метокси-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-(1]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида (IV) (50 мг, выход 25,9%) в виде белого твердого вещества.
. Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия , высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования A с получением продукта, указанного в заголовке, (3aR,5s,6aS)-N-(3-метокси-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-(1]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида (IV) (50 мг, выход 25,9%) в виде белого твердого вещества.
МС m/z (ИЭР): 412,9 [М-1].
1H ЯМР (400 МГц, ДМСО-d6): δ 11,60 (m, 2Н), 8,08 (s, 1Н), 7,06-7,05 (m, 1Н), 6,53-6,51 (m, 1Н), 5,48-5,44 (m, 1Н), 3,90 (s, 3H), 3,69-3,65 (m, 2Н), 3,37-3,32 (m, 2Н), 3,16 (s, 3H), 2,90-2,88 (m, 2Н), 2,02-1,99 (m, 2Н), 1,80-1,77 (m, 2Н).
ПРИМЕР 2: Получение бисульфата (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида (соединения
формулы (I))
(3aR,5s,6aS)-N-(3-метокси-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[c]пиррол-2(1Н)-карбоксамид (IV) (140 г, 0,34 моль), безводный метанол (350 г) и дихлорметан (2,0 кг) добавляли в реакционную колбу на 10 л при перемешивании. Серную кислоту (34,8 г, 0,36 моль) медленно добавляли при комнатной температуре, и реакционный раствор становился прозрачным. После перемешивания в течение 30 мин нерастворимое вещество удаляли фильтрованием, фильтрат концентрировали при пониженном давлении и высушивали с получением продукта, указанного в заголовке, 135 г - 168 г, выход: 80-90%.
МС m/z (ИЭР): 415,1651 [М+1].
1Н ЯМР (400 МГц, ДМСО-d6): δ 12,75 (s, 1Н), 11,04 (s, 1Н), 8,37 (s, 1Н), 7,41-7,42 (t, 1Н), 6,89 (s, 1Н), 5,15-5,19 (m, 1Н), 3,89 (s, 3H), 3,68-3,70 (m, 2Н), 3,38-3,40 (m, 2Н), 3,29 (s, 3H), 2,95 (s, 2Н), 2,09-2,16 (m, 2Н), 1,92-1,97 (m, 2Н)
ПРИМЕР 3
Проводили сравнительные эксперименты по растворимости (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1H)-карбоксамида (соединение формулы (IV)) и соответствующего цитрата, гидрохлорида, сульфата, бисульфата в воде и 0,1 н HCl. Результаты показали, что растворимость его бисульфата была значительно повышена, а также была значительно лучше, чем других солей. Подробные результаты показаны в таблице 1.
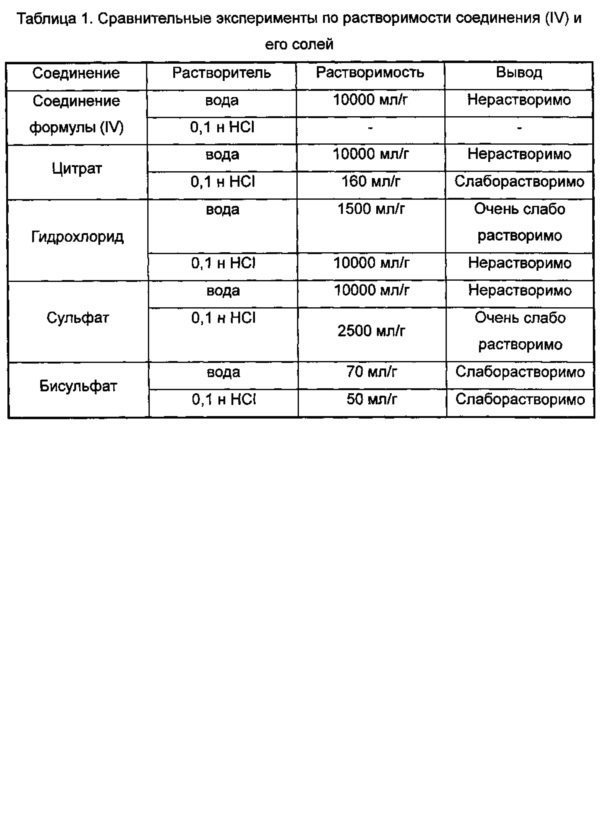
ПРИМЕР 4
Фармакокинетические свойства соединения формулы (IV) и его различных форм солей были исследованы у макак-резусов, и природу различных форм соединений подробно оценивали. В качестве тестируемых животных использовали четырех макак-резусов, половина самцов и половина самок. Макакам-резусам вводили в однократной дозе 50 мг/кг; для введения тестируемым животным различных лекарственных средств был адаптирован план исследования с перекрестным сравнением множества случаев, и период реконвалесценции каждого цикла составлял трое суток; образцы крови (0,5 мл) брали из бедренной вены до введения (0 ч) и через 0,25 ч, 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч и 24 ч после введения, хранили в гепаринизированных пробирках, тщательно перемешивали и центрифугировали в течение 10 минут при 3500 об/мин для отделения плазмы крови. Образцы плазмы хранили при низкой температуре, для измерения концентрации лекарственного средства в плазме и печени использовали жидкостную хроматографию с масс-спектрометрией (ЖХ-МС/МС), и фармакокинетические параметры анализировали с помощью программы WinNonlin 5.3. Результаты экспериментов представлены ниже:

где AUC - площадь под кривой зависимости концентрации от времени, от англ. Area Under the Curve; MRT - среднее время удержания препарата в организме, от англ. mean residence time.
Выводы: Приведенные выше фармакокинетические результаты для обезьян показали, что воздействие in vivo соединения формулы (IV) составляет 41111 нг/мл*ч, но имеются значительные индивидуальные различия; воздействие in vivo цитрата аналогично воздействию основания; воздействие in vivo бисульфата возрастало на 50% относительно воздействия основания, и индивидуальные различия были небольшими, как можно видеть, бисульфат обладал значительным воздействием in vivo и небольшими индивидуальными различиями, следовательно, он подходит для медицинских целей.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИСУЛЬФАТА ИНГИБИТОРА JAK И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2015 |
|
RU2704795C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ИНГИБИТОР ЯНУС-КИНАЗЫ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМУЮ СОЛЬ | 2017 |
|
RU2744432C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2012 |
|
RU2618673C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИСУЛЬФАТНОГО ИНГИБИТОРА JAK-КИНАЗЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2015 |
|
RU2716260C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| ИНГИБИТОРЫ MAGL НА ОСНОВЕ ПИРАЗОЛА | 2018 |
|
RU2789157C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ОПОСРЕДОВАНИЯ АКТИВНОСТИ ТИРОЗИНКИНАЗЫ 2 | 2020 |
|
RU2826012C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ, АКТИВНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2013 |
|
RU2666538C2 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
Изобретение относится к бисульфату (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида формулы (I). Стехиометрическое соотношение (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)-гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида (соединение IV) к серной кислоте составляет 1:1. Способ получения соединения по изобретению включает стадию реакции образования соли соединения формулы (IV) и серной кислоты, где реакцию образования соли проводят в растворителе, а растворитель представляет собой смешанный растворитель из галогеналканов и спиртов, имеющих количество атомов углерода, меньшее или равное 3, предпочтительно смешанный растворитель из дихлорметана и метанола. Температура реакции при реакции образования соли составляет от 10 до 30°С, а время реакции составляет от 0,5 до 4 часов. Бисульфат (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида формулы (I) обладает активностью ингибирования Янус-киназы и предназначен для получения лекарственного средства для лечения ревматизма и ревматоидного артрита. 4 н. и 3 з.п. ф-лы, 2 табл., 4 пр.
 ,
, 
1. Бисульфат (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида формулы (I)
2. Бисульфат (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида формулы (I) по п. 1, где стехиометрическое отношение соединения формулы (IV) к серной кислоте составляет 1:1,

3. Способ получения бисульфата (3aR,5S,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида формулы (I) по п.1, включающий стадию реакции образования соли соединения формулы (IV) и серной кислоты

4. Способ получения по п. 3, где реакцию образования соли проводят в растворителе, а растворитель представляет собой смешанный растворитель из галогеналканов и спиртов, имеющих количество атомов углерода, меньшее или равное 3, предпочтительно смешанный растворитель из дихлорметана и метанола.
5. Способ получения по п. 4, где температура реакции при реакции образования соли составляет от 10 до 30°С, а время реакции составляет от 0,5 до 4 часов.
6. Фармацевтическая композиция, обладающая активностью ингибирования Янус-киназы, содержащая бисульфат (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида формулы (I) по п. 1 и фармацевтически приемлемый носитель.
7. Применение бисульфата (3aR,5s,6aS)-N-(3-метоксил-1,2,4-тиадиазол-5-ил)-5-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)гексагидроциклопента[с]пиррол-2(1Н)-карбоксамида формулы (I) по п. 1 или фармацевтической композиции по п. 6 при получении лекарственного средства для лечения ревматизма и ревматоидного артрита.
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2012 |
|
RU2618673C2 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Устройство для ограждения зоны работ на станционных путях | 1987 |
|
SU1439010A1 |
| Муфта | 1990 |
|
SU1798559A1 |
| WO 2012171863 A1, 20.12.2012 | |||
| ПИРРОЛОПИРИМИДИНЫ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2006 |
|
RU2434871C2 |

