Настоящее изобретение относится к улучшенным способам скрининга и, в частности, к способам скрининга библиотек антилигандов для идентификации антилигандов, специфичных для дифференциально и/или нечасто экспрессирующихся лигандов.
Библиотеки на основе белков или пептидов часто используются для селекции молекул антилигандов со специфичностью к определенным лигандам.
Такие библиотеки сконструированы таким образом, что молекула белка каким-то образом оказывается физически сопряженной с молекулой, содержащей генетическую информацию, кодирующую эту определенную белковую молекулу. Белковая молекула, таким образом, оказывается представлена вместе с кодирующим ее геном.
Обычно используемые форматы дисплеев основаны на клеточных или вирусных частицах-хозяевах, использующихся для представления белковой молекулы, и включают в себя бактериальный дисплей (Francisco et al., 1993) и фаговый дисплей (Smith, 1985; Smith and Scott, 1993; Winter et al.,). Такие системы экспонируют потенциальный антилиганд на поверхности частицы-хозяина, в то время как генетическая информация для отображаемой молекулы спрятана внутри частицы, и упомянутые способы были успешно использованы для селекции специфических антилигандов на белковой основе.
Существуют другие форматы дисплеев, основанные на системах трансляции in vitro; включая различные формы рибосомных дисплеев (Mattheakis et al., 1994; Hanes and Pluckthun, 1997; He and Taussig, 1997)), которые основаны на нековалентном связывании содержащей генетическую информацию молекулы с белковой молекулой, а также другие форматы дисплеев, также основанные на системах трансляции in vitro, причем молекула, содержащая генетическую информацию, связана ковалентной связью с белковой молекулой потенциального антилиганда, например Profusion (Weng et al., 2002) или Covalent Display Technology (Gao et al., 1997).
Библиотеки экспонируемых пептидных или белковых антилигандов могут быть полностью рандомизированными, например, когда используются пептидные библиотеки, или они могут быть основаны на каркасной (скаффолд, scaffold) структуре постоянной области и включать в себя дополнительную структуру, придающую изменчивость.
Часто используемые каркасные структуры имеют в основе на вариабельных доменах тяжелой и легкой цепей антител (McCafferty et al., 1990), но могут также иметь в основе и другие скаффолды, такие как фибронектин (Jacobsson and Frykberg, 1995; Koide et al., 1998), домены белка A (Stahl et al., 1989) или небольшие стабильные белковые домены, например, BPTI (Markland et al., 1991) 1991).
Селекция антилигандов, демонстрирующих определенную специфичность связывания, из библиотек дисплеев, часто осуществляется с помощью способов так называемого «биопэннинга».
Целевой лиганд может быть иммобилизован на твердой поверхности, и специфические антилиганды из библиотеки подвергают воздействию иммобилизованного целевого лиганда, чтобы дать возможность антилигандам, представляющим интерес, связаться с целевым лигандом. Не связавшиеся элементы библиотеки впоследствии смывают, а антилиганды, представляющие интерес, извлекают и амплифицируют.
Белковые частицы, иные чем элементы библиотеки антилигандов, например, экспрессируемые фагом фрагменты антител, могут «прилипать», приводя к связыванию и выделению некоторых нецелевых специфических молекул. Неспецифическое связывание может быть сведено к минимуму добавлением некоторых соединений, например, молока, бычьего сывороточного альбумина, сыворотки (человеческой/фетальной телячьей), желатина и для некоторых (не клеточных) приложений, детергента, к конструкции дисплея антилигандов/смеси лигандов для того, чтобы служить в качестве блокирующих агентов с целью уменьшения такого фонового связывания неспецифических антилигандов.
Был разработан ряд процедур промывки, чтобы уменьшить неспецифическое связывание входящих в библиотеку антилигандов с клетками и помочь отделению клеток от загрязняющих и/или неспецифически связавшихся элементов библиотеки.
Такие способы включают в себя промывку клеток, фиксированных магнитным образом на колонке (Siegel et al., 1997), с тем чтобы свести к минимуму сдвигающие воздействия и позволить повторное связывание диссоциированного фага. Другой способ промывки клеток заключается в центрифугировании в среде более высокой плотности, такой как Ficoll или Percoll, чтобы селективно удалить неспецифические антилиганды и антилиганды с низкой аффинностью и дополнительно пространственно отделить клетки и связанные с клетками антилиганды от свободных антилигандов и неспецифически связавшихся антилигандов (Carlsson et al., 1988; Williams and Sharon, 2002).
В зависимости от эффективности процесса селекции может потребоваться несколько раундов пэннинга, чтобы устранить или, по меньшей мере, значительно уменьшить содержание неспецифически связавшихся антилигандов до желаемого уровня (Dower et al., 1991).
В другом способе селекции целевой лиганд(ы) связывается со специфическими элементами библиотеки антилигандов, находясь в растворе. Связавшиеся антилиганды затем выделяют с использованием, например, извлекаемого тега, прикрепленного к целевому лиганду. Наиболее широко используемый тег представляет собой биотин, что позволяет извлечь комплекс между целевой молекулой и отображаемым специфическим элементом библиотеки с использованием авидина, соединенного с твердой подложкой, например, магнитным шариком (Siegel et al., 1997).
Данные способы используются, когда целевой лиганд хорошо известен и доступен в очищенной форме. Селекции по одному целевому лиганду за один раз являются обычными. Селекция по нескольким заданным целевым лигандам может быть выполнена одновременно. Целевые лиганды могут представлять собой один или несколько из небольших гаптенов, белков, углеводов, ДНК и липидов.
Специфические антилиганды против дифференциально экспрессирующихся лигандов представляют интерес для многих приложений. Например, белки могут по-разному экспрессироваться на клетках и тканях, происходящих от пациентов с болезнью, по сравнению с таковыми, полученными в качестве контролей от здоровых людей. К таким заболеваниям относятся микробные, вирусные или паразитарные инфекции, астма, хронические воспалительные и аутоиммунные расстройства, рак, неврологические, сердечно-сосудистые или желудочно-кишечные заболевания. Сходным образом, белковый состав жидкостей организма, например, плазмы, спинномозговой жидкости, мочи, спермы, слюны и слизи, может отличаться у пациентов с болезнью по сравнению с контрольной здоровой группой.
Следовательно, помимо их общей применимости в качестве исследовательских инструментов для идентификации дифференциально экспрессирующихся лигандов, антилиганды, специфичные для дифференциально экспрессирующихся лигандов, могут быть использованы в качестве инструментов для применения в диагностике, профилактике и/или лечении заболевания.
Последние достижения в областях геномики и протеомики указали на наличие множества еще не определенных дифференциально экспрессирующихся молекул, что подчеркивает важность способов генерации специфических антилигандов для таких потенциальных целевых лигандов.
Ожидается, что многие из таких дифференциально экспрессирующихся молекул присутствуют на клеточных поверхностях и, тем самым, представляют собой потенциальные мишени для целевых терапий с использованием, например, специфических антител, которые могут быть конъюгированы с биоактивными (например, цитотоксическими) агентами.
Большие и высокодиверсифицированные библиотеки дисплеев антилигандов предоставляют способы выделения антилигандов со специфичностью к неизвестным клеточным лигандам углеводного, белкового, липидного или объединенного происхождения.
Способы биопэннинга, имеющиеся в настоящее время, включают в себя способы, основанные на целых клетках, участках клеток и клеточной мембране, что, в принципе, позволяет выделить конструкции дисплеев, экспонирующие антилиганды, специфические к лигандам на клеточных мембранах в их нативной конфигурации.
Человеческие и гуманизированные терапевтические антитела все чаще используется для лечения различных заболеваний, включая острые и хронические воспалительные заболевания, иммунологические нарушения и расстройства центральной нервной системы и рак. Человеческие терапевтические антитела считаются наиболее привлекательными формами для лечения заболеваний человека в силу их полностью человеческой природы и связанного с этим отсутствия иммуногенности, оптимальной способности осуществлять эффекторные механизмы иммунного ответа хозяина, опосредованные Fc-фрагментом антител, и длительного in vivo времени полураспада по сравнению с их мышиными, химерными и гуманизированными дубликатами. На сегодняшний день человеческие антитела получают рутинным образом различными технологиями, включающими библиотеки антител гуманизированных мышей и высоко диверсифицированные фаговые библиотеки антител.
Большие (более 105 уникальных клонов антител) библиотеки человеческих антител являются достаточно диверсифицированными, чтобы содержать высокоаффинные антитела, специфические к значительному числу антигенов, включая практически все виды аутоантигенов. Аутоантигены представляют собой антигены, которые, несмотря на то, что они являются составляющей нормальной ткани, служат мишенью гуморального или клеточного иммунного ответа, как и в аутоиммунном заболевании, и представляют категорию антигена, вызывающего особый терапевтический интерес.
Дополнительно полагают, что библиотеки человеческих антител обеспечивают преимущества по сравнению с трансгенными мышами, несущими гены человеческого иммуноглобулина, при селекции на антитела, которые связываются с рецепторными эпитопами, которые являются структурно консервативными у человека и мыши, поскольку данная категория антител отрицательно селектирована в in vivo условиях механизмами аутотолерантности. Такие консервативные области вызывают особый терапевтический интерес, поскольку консервативные области являются часто функционально связанными (например, лиганд-связывающие домены, необходимые для связывания и присвоения индуцированных лигандом/рецептором клеточных ответов), и антитела, нацеленные на такие консервативные эпигоны, могут быть подвергнуты скринингу на in vivo терапевтическую активность в сингенных системах экспериментальной модели болезни.
Высокоаффинные антитела, специфичные для практически всех видов растворимых антигенов человека (таких как цитокины, хемокины, факторы роста, липиды, углеводы и конъюгированные молекулы и т.д.), а также рецепторов клеточной поверхности (например, охватывающие рецепторы 1ТМ, 4ТМ, 7ТМ и мульти-ТМ и т.д.) были успешно выделены из высокодиверсифицированных библиотек человеческих антител.
Рецепторы клеточной поверхности составляют одну категорию мишеней, вызывающих особый терапевтический интерес, и несколько антител, которые связываются с рецепторами, ассоциированными с различными раковыми клетками, были одобрены для лечения рака, включая ритуксимаб (анти-CD20), трастузумаб (анти-Her2) и цетуксимаб (анти-EGFR).
Однако терапевтическая эффективность не так легко предсказывается, исходя из специфичности антител к рецепторам; антитела к одному и тому же целевому рецептору могут значительно различаться по терапевтической эффективности независимо от их связывающей аффинности (Beers et al., 2008; Cragg and Glermie, 2004), и антитела против альтернативных молекулярных мишеней могут проявить перспективный, а иногда неожиданно терапевтический потенциал (Beck et al., 2010; Cheson and Leonard, 2008). Например, различные клоны специфических антител CD20, которые связываются со сходной аффинностью к антигену CD20 и несут константные области, идентичные мышиному IgG2a, принципиально отличаются в способности истощать В-клетки in vivo (Beers et al., 2008; Cragg and Glennie, 2004), и антитела против других опухолеассоциированных рецепторов клеточной поверхности, отличные от CD20, могут обладать значительной противоопухолевой активностью против раковых В-клеток (для обзора см. (Cheson and Leonard, 2008)).
Таким образом, в высокодиверсифицированной библиотеке антител наиболее терапевтически эффективные, сильнодействующие и наиболее переносимые антитела по отношению к любому данному типу рака, вероятно, являются специфичными для любого из нескольких различных рецепторов, и для определения клонов терапевтически оптимальных антител в высоко диверсифицированной библиотеке требуется функциональный скрининг многих, а в идеале всех, элементов библиотеки, которые являются специфичными для различных рецепторов, связанных с больными клетками.
Ранее заявитель разработал технологию скрининга (способ биопэннинга), позволяющую выявлять клоны связывающихся с различными поверхностными рецепторами антител, которые дифференциально экспрессируются на одной клеточной популяции (целевые клетки) по сравнению с другой (нецелевые клетки) из фаговых библиотек человеческих антител (WO 2004/023140, Fransson et al., 2006; Frendeus, 2006) (далее упоминается как дифференциальный биопэннинг). Раскрытие заявки WO 2004/023140 (и все национальные заявки, вытекающие из нее) включено в данный документ посредством ссылки в полном объеме.
Данный способ скрининга состоит из по существу шести стадий, как указано на фигуре 1. Важно отметить, что данный способ содержит стадии скрининга в следующем порядке:
1) дифференциальный биопэннинг с последующим
2) скринингом на мишень в сравнении с нецелевой специфичностью, после чего следует
3) обычное секвенирование способом Sanger меньшего числа клонов.
Используя данную технологию, было возможно генерировать пул антител, которые показали высокую специфичность для дифференциально экспрессирующихся поверхностных рецепторов целевых клеток в сравнении с дифференциально экспрессирующимися поверхностными рецепторами нецелевых клеток.
Секвенирование способом Sanger представляет собой пример способа, который в настоящее время используется для идентификации уникальных «связывателей» (binders) в режиме «низкой пропускающей способности» («low throughput»). Другие примеры включают в себя прогон ДНК генов антител в гелях до и после расщепления ферментами рестрикции для выявления уникальных размеров и, через различную чувствительность к различным ферментам рестрикции, непрямым путем, различных последовательностей.
Когда данный способ применили для выделения антител, направленных против дифференциально экспрессирующихся поверхностных рецепторов раковых В-клеток (целевые) в сравнении с дифференциально экспрессирующимися поверхностными рецепторами Т-клеток (нецелевые) (дифференциальный биопэннинг «BnonT»), то были идентифицированы антитела, специфичные для различных дифференциально экспрессирующихся поверхностных рецепторов клеток-мишеней, включая HLA-DR, поверхностный Ig и ICAM-1 (таблица 1).
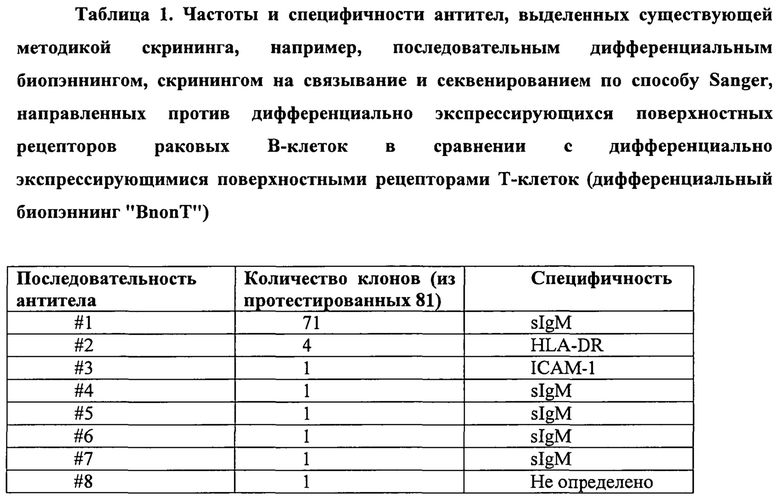
Однако, все целевые рецепторы экспрессировались на относительно высоком уровне (от 50000 до 400000 рецепторов на клетку), и число уникальных последовательностей антител, определенных (скринировано 8 из 81) данным способом, было ограничено.
В то время как было секвенировано лишь ограниченное количество клонов, специфичных для дифференциально экспрессирующихся поверхностных рецепторов целевых клеток, высокая частота одного клона антитела указывала на ограниченное разнообразие антител в пуле антител, извлеченном способом «BnonT». Таким образом, в то время как данный способ обеспечил значительное улучшение по сравнению с предыдущими способами пэннинга, основанными на клеточном подходе, в том смысле, что способом ограниченного скрининга (limited screening) (Fransson et al., 2006) были идентифицированы антитела с терапевтическим потенциалом к нескольким различным дифференциально экспрессирующимся рецепторам, данное наблюдение показало, что требуются дополнительные улучшения, поскольку, в соответствии с преобладающей общей точкой зрения, пэннинг только генерировал пул антител ограниченного разнообразия, состоящий из антител против относительно высокоэкспрессирующихся и строго дифференциально экспрессирующихся поверхностных рецепторов (Hoogenboom, 2002) (Liu et al., 2004; Mutubema et al., 1999; Osboum et al., 1998).
Расчеты in silico, выполненные, как описано в более раннем способе биопэннинга (WO 2004/023140 и Frendeus, 2006), выявили, что пул дифференциально селектированных антител «BnonT» должен содержать гораздо большее количество антител против каждого из нескольких дифференциально экспрессирующихся поверхностных рецепторов (фигура 2).
Возможности секвенирования того времени делали секвенирование значительно большего числа клонов антител в пуле крайне тяжелым (практически невозможным), поэтому гипотеза о том, что пул дифференциально селектированных антител должен быть гораздо более диверсифицированным, чем явствует из начальных скринингов, была проверена с использованием непрямого подхода. Таким образом, используя иммуногранулы, конъюгированные с рекомбинантным белком ICAM-1 (ICAM-1 представляет собой рецептор клеточной поверхности и является мишенью для одного клона антител из первоначально 81 секвенированных клонов в пуле дифференциально селектированных антител из таблицы 1), пул антитела, дифференциально селектированного способом «BnonT», был проверен на наличие дополнительных клонов антител, специфичных для ICAM-1. Скрининг выявленных 1260 клонов антител, с последующим пэннингом пула дифференциально селектированных антител против рекомбинантного ICAM-1, идентифицировал двадцать одну (21) дополнительные последовательности/клоны антител, специфичных для ICAM-1.
Данные наблюдения показали, что первоначальным способом дифференциального биопэннинга было возможно идентифицировать клоны антител к дифференциально экспрессирующимся антигенам, но пул дифференциально селектированных антител был гораздо более диверсифицированным, чем явствует из таких первоначальных скриннингов, а также значительно больше, чем если бы это было определено традиционными подходами скрининга.
В настоящее время заявитель разработал способ повышения точности способа дифференциального биопэннинга для обнаружения множества различных антилигандов к лиганду, представляющему интерес. Таким образом, настоящее изобретение описывает методику, позволяющую извлекать из библиотеки человеческих антител (и других молекулярных библиотек) пул высокоаффинных антилигандов, таких как человеческие антитела, которые являются специфичными для различных лигандов (например, рецепторов), дифференциально экспрессирующихся в нативной конфигурации, которую они имеют на поверхности клеток, с уровнями экспрессии от низкого до высокого в популяции целевых клеток, по сравнению с другой клеточной популяцией(ми).
Настоящее изобретение отличается от ранее разработанных методик скрининга в нескольких отношениях (фигура 8). Во-первых, объединяя уникально мощную методику дифференциального биопэннинга с глубоким секвенированием (deep sequencing) нового поколения и последующим подтверждающим скринингом в отношении специфичности антитела для дифференциально экспрессирующихся поверхностных рецепторов целевых клеток – «обратным скринингом», настоящее изобретение позволяет получать пул антител, который является 1) качественно и 2) количественно уникальным.
Важно отметить, что антилиганды, такие как клоны антител, идентифицированные с помощью такого подхода, могут все иметь терапевтический потенциал, основанный, во-первых, на их высокоаффинном связывании с рецепторами, которые a) дифференциально экспрессируются на целевых клетках по сравнению с нецелевыми клетками и b) экспрессируются на целевых клетках в нативной конфигурации, которую они имеют на поверхности клеток, и, во-вторых, на задокументированной способности антител с данными свойствами опосредовать терапевтические эффекты в соответствующих in vitro и in vivo системах экспериментальных моделей болезни (Beck et al., 2010; Fransson et al., 2006).
Таким образом, следовательно, настоящее изобретение позволяет:
1. Генерацию дифференциальным биопэннингом пула антител, содержащего антитела, специфические к дифференциально экспрессирующимся поверхностным рецепторам, экспрессируемым на высоком, промежуточном и низком уровнях.
2. Существование более низкого порогового значения для числа клонов секвенированных антител, которое должно быть превышено в целях выявления антител, специфичных для поверхностных рецепторов, экспрессирующихся на промежуточном и низком уровне.
3. Секвенирование увеличивающего числа клонов антител, превышающего данное более низкое пороговое значение, увеличивает количество выявленных антител, специфичных для поверхностных рецепторов, экспрессирующихся на промежуточном и низком уровне.
4. Комплексную идентификацию антител, специфичных для поверхностных рецепторов, экспрессирующихся на промежуточном и низком уровне, в пуле дифференциально экспрессирующихся антител, которая требует глубокого секвенирования.
Таким образом, первый объект настоящего изобретения относится к способу выделения по меньшей мере одного антилиганда к по меньшей мере одному дифференциально экспрессирующемуся целевому лиганду, содержащему следующие стадии:
(a) выполнение дифференциального биопэннинга на библиотеке антилигандов с тем, чтобы выделить по меньшей мере один антилиганд; и
(b) выполнение секвенирования с высокой пропускной способностью на антилигандах, выделенных на стадии (a).
Данный способ может дополнительно содержать стадию:
(c) выполнения подтверждающего скрининга на специфичность антитела для дифференциально экспрессирующегося лиганда.
Стадия дифференциального биопэннинга способа по изобретению может содержать подстадии:
(i) получение библиотеки антилигандов;
(ii) получение первой популяции лигандов, содержащей лиганд, фиксированный к или встроенный в конструкцию субтрактивного лиганда;
(iii) получение второй популяции лигандов, содержащей тот же лиганд со стадии (ii), фиксированный к или встроенный в конструкцию целевого лиганда;
(iv) определение количества конструкций субтрактивного лиганда и конструкций целевого лиганда в популяциях с использованием одного или более уравнений, полученных из всеобщего закона действующих масс 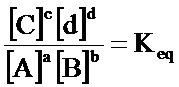
где:
A, B, C&D=представляют собой участники реакции (реагенты и продукты)
a, b, c&d=представляют собой коэффициенты, необходимые для сбалансированного химического уравнения
так, чтобы обеспечить выделение антилиганда к дифференциально экспрессирующемуся целевому лиганду;
(v) предоставление конструкции субтрактивного лиганда в количестве, как определено на стадии (iv);
(vi) предоставление конструкции целевого лиганда в количестве, как определено на стадии (iv);
(vii) предоставление средств разделения для отделения антилиганда, связанного с конструкцией целевого лиганда, от антилиганда, связанного с конструкцией субтрактивного лиганда;
(viii) подвергание библиотеки (i) воздействию конструкций лигандов, являющихся продуктами стадий (v) и (vi), с тем, чтобы сделать возможным связывание антилигандов с лигандами; и
(ix) использование средств разделения для выделения антилиганда, связавшегося с лигандом, фиксированным к или включенным в конструкцию целевого лиганда.
Не предполагается, что стадии по изобретению обязательно должны быть выполнены в любом определенном порядке.
Под «предоставлением определенного количества» авторы понимают предоставление количества лиганда, которое уже было известно, так что уравнения по изобретению были использованы для проверки того, что предоставляемое известное количество является подходящим для выделения желаемого антилиганда(ов).
Параметры реакции, которые используются для данного способа селекции, могут быть оптимизированы в соответствии с настоящим изобретением расчетами, применяющими закон действующих масс и уравнения, полученные из него, а также с учетом таких параметров как разнообразие молекулярной библиотеки, число копий антилиганда, желаемый предел обнаружения позитивной регуляции, аффинность желаемого антилиганда и концентрация лиганда.
Стадия секвенирования с высокой пропускной способностью способа по первому объекту изобретения может проводиться секвенированием 454 (454 sequencing), Illumina, способами SOLiD, системой Helicos system или таковыми от Complete Genomics и Pacific Biosciences.
Появление секвенирования нового поколения («next generation sequencing») позволило проводить секвенирование большого числа (от 1000 до 1000000) генов-кандидатов в режиме высокой пропускной способности (здесь и далее упоминается как «глубокое секвенирование»).
Способ секвенирования 454 описан (и включен в настоящее описание посредством ссылки) у Margulies et al. (2005). При способе 454 ДНК, подлежащая секвенированию, либо фракционируется и снабжается адаптерами, либо сегменты ДНК могут быть амплифицированы посредством ПЦР с использованием праймеров, содержащих адаптеры. Адаптеры представляют собой последовательности из 25 нуклеотидов, необходимые для связывания с шариками DNA Capture Beads и для отжига эмульсии PCR Amplification Primers и Sequencing Primer. Фрагменты ДНК делают одноцепочечными и прикрепляют к шарикам захвата ДНК (DNA Capture Beads) таким образом, чтобы к одному шарику был присоединен только один фрагмент ДНК. Далее шарики, содержащие ДНК, эмульгируют в смеси вода-в-масле, в результате чего получаются «микрореакторы», содержащие только один шарик.
Фрагмент ПЦР-амплифицируется внутри микрореактора, в результате чего число копий достигает нескольких миллионов на шарик. После выполнения ПЦР эмульсию разрушают и шарики загружают в пико-титровальный планшет (pico titer plate). Каждая лунка пико-титровального планшета может содержать только один шарик. В лунки добавляют секвенирующие ферменты и нуклеотиды пропускают через лунки в определенном порядке. Встраивание нуклеотида приводит к высвобождению пирофосфата, который катализирует реакцию, приводящую к появлению хемилюминесцентного сигнала. Данный сигнал регистрируется с помощью ПЗС(ССВ)-камеры, и программное обеспечение используется для перевода сигналов в последовательность ДНК.
При способе Illumina (Bentley (2008)) одноцепочечные, снабженные адаптерами фрагменты прикрепляют к оптически прозрачной поверхности и подвергают «мостовой амплификации» (bridge amplification). Данная процедура приводит к появлению нескольких миллионов кластеров, каждый из которых содержит копии уникального фрагмента ДНК. Добавляют ДНК-полимеразу, праймеры и четыре меченых нуклеотида, представляющих собой обратимые терминаторы, и поверхность визуализируется с помощью лазерной флуоресценции для определения местоположения и характера меток. Защитные группы затем удаляют и способ повторяют в течение нескольких циклов.
Способ SOLiD (Shendure (2005)) аналогичен способу 454 sequencing, фрагменты ДНК амплифицируются на поверхности шариков. Секвенирование включает в себя циклы лигирования и обнаружения меченых зондов.
В настоящее время разрабатываются несколько других способов для секвенирования с высокой пропускной способностью. Примерами таких способов являются Helicos system (Харрис (2008)), Complete Genomics (Drmanac (2010)) и Pacific Biosciences (Lundquist (2008)). Поскольку это чрезвычайно быстро развивающаяся техническая область, возможность приложения к настоящему изобретению способов секвенирования с высокой пропускной способностью будет очевидна для специалиста в данной области техники.
В то время как приборы, способные секвенировать длинные участки ДНК, такие как участки, кодирующие последовательности вариабельных доменов антител (Fv), scFv или Fab, находятся только на стадии прототипа, имеющиеся в настоящее время приборы в действительности позволяют проводить секвенирование более коротких участков ДНК, таких как последовательности, кодирующие и охватывающие от CDRH1- до CDRH3-домены scFv. Однако, поскольку технология секвенирования улучшается с тем, чтобы позволить секвенирование длинных участков ДНК, данные способы будут также хорошо работать в способах по изобретению.
Стадия подтверждающего скрининга способа по изобретению может проводиться детектированием специфического лигандного связывания выделенного пула антилигандов и/или отдельных клонов антилигандов с целевой конструкцией в сравнении с субтрактивной конструкцией с помощью любого анализа, предназначенного для детектирования связывания лиганд/антилиганд, такого как проточная цитометрия (Flow-cytometry), FMAT (Fluorescent Microvolumetric Assay Technology), твердофазный иммуноферментный анализ ELISA (Enzyme-linked immunosorbent assay), MSD (Meso Scale Discovery) and CBA (Cytometric Bead Array).
В одном варианте осуществления лиганд по настоящему способу не экспрессируется как на целевой конструкции так и на субтрактивной конструкции, т.е. он экспрессируется либо на целевой конструкции, либо на субтрактивной конструкции.
В другом варианте осуществления лиганд по настоящему способу экспрессируется на более высоких уровнях как на целевой конструкции, так и на субтрактивной конструкции.
Способ дифференциального биопэннинга может содержать дополнительную подстадию высвобождения антилиганда от лиганда.
Предпочтительно, стадии от (ii) до (ix) стадии дифференциального биопэннинга проводят параллельно, чтобы выделить множество антилигандов к множеству различных лигандов.
Стадии от (ii) до (ix) стадии дифференциального биопэннинга повторяют один или более раз.
Предпочтительно, количество одной субтрактивной конструкции или целевой конструкции на стадии дифференциального биопэннинга предоставляется в избытке по сравнению с количеством другой субтрактивной конструкции или целевой конструкции. Превышение лиганда может составлять от 10 до 1000 раз, но также может составлять от 2 до 10 раз, или 1000 и 100000 раз.
Степень избытка популяции субтрактивных лигандов определяет максимально возможное «разрешение» (то есть, насколько хорошо вы сможете различить антилиганды со специфичностью для лигандов, активность которых позитивно регулируется на низком уровне, на умеренном уровне, на высоком уровне, или которые являются уникально экспрессирующимися), которое вы сможете детектировать, и насколько хорошо вы сможете различать дифференциально экспрессирующиеся лиганды. Например, если вы используете библиотеку со 100 антилигандами, специфическими к целевым лигандам, и вы добавляете достаточно большие концентрации позитивного лиганда, так что весь антилиганд будет связан с лигандом в равновесных условиях, то 10-кратный избыток популяции субтрактивных лигандов позволит вам уменьшить частоту антилигандов со специфичностью к обычно экспрессирующимся лигандам на 90%, в то время как 200-кратный избыток (удвоенное количество специфических связывателей антилигандов) позволит вам удалить общие связыватели (см. WO 2004/023140, фигура 5 и самый последний абзац примера 4 для данных, показывающих это).
В одном варианте осуществления уравнение на стадии (iv) стадии дифференциального биопэннинга представляет собой:
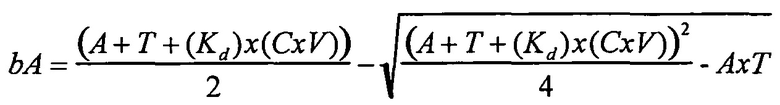
где
bA представляет собой связанный антилиганд (Bound anti-ligand)
А представляет собой общее число антилиганда
T представляет собой общее число лигандов
C представляет собой постоянную Авогадро (6,022×1023 частиц/моль)
V представляет собой реакционный объем (литры)
Kd представляет собой равновесную константу диссоциации
И в альтернативном варианте осуществления уравнение на стадии (iv) стадии дифференциального биопэннинга представляет собой:
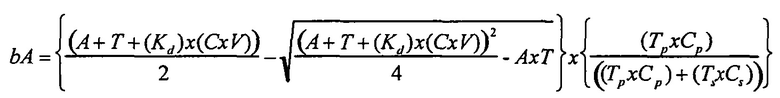
где
bAp представляет собой связанный антилиганд (Bound anti-ligand)
Tp представляет собой число лигандов на Cp
Ts представляет собой число лигандов на Cs
Cp представляет собой число конструкций целевых лигандов
Cs представляет собой число конструкций субтрактивных лигандов
A представляет собой общее число антилиганда
T представляет собой общее число лигандов
C представляет собой постоянную Авогадро (6,022×1023 частиц/моль)
V представляет собой реакционный объем (литры)
Kd представляет собой равновесную константу диссоциации
Средство разделения со стадии дифференциального биопэннинга может быть выбрано из по меньшей мере одного из твердой подложки, клеточной мембраны и/или ее участков, синтетической мембраны, шариков, химических меток (тегов) и свободного лиганда. Средство разделения субтрактивных и целевых конструкций может иметь различную плотность. Средство отделения субтрактивной конструкции может представлять собой предпочтительно мембранную везикулу или мембрану целой клетки.
Стадия (ix) способа дифференциального биопэннинга может быть выполнена по меньшей мере одним из способов разделения, причем данный способ представляет собой одно из: плотностное центрифугирование (Williams and Sharon, 2002), секвестрация на твердой подложке, секвестрация на магнитном шарике (Siegel et al., 1997), связывание через химическую метку (тег) и разделение в водной фазе.
Более предпочтительно, способ разделения представляет собой плотностное центрифугирование, проводимое в градиенте плотности, например, Ficoll; Percoll; йодированной градиентной среды, где во время центрифугирования первый и второй целевые лиганды перемещаются в разной степени через градиент Ficoll, в результате чего первый и второй целевые лиганды могут быть выделены из их различных конечных точек.
Наиболее предпочтительно способ разделения использует градиент полисахарида, например, Ficoll.
Библиотека со стадии (а) предпочтительно представляет собой библиотеку дисплеев, содержащую множество элементов библиотек, которые экспонируют антилиганды. Примером такой библиотеки может служить библиотека фагового дисплея, где антилиганд экспонируется на поверхность бактериофага.
Экспонирование белков и полипептидов, слитых с одним из белков фаговой оболочки, на поверхности бактериофага (фага) служит мощным инструментом для селекции специфических лигандов. Такой способ «фаговового дисплея» первоначально был использован Смитом (Smith) в 1985 году, чтобы создать большие библиотеки антител для целей селекции антител с высокой аффинностью к определенному антигену. Совсем недавно данный способ был использован для экспонирования пептидов, доменов белков и интактных белков на поверхности фага с тем, чтобы идентифицировать лиганды, имеющие желаемые свойства.
Принципы, лежащие в основе технологии фагового дисплея, таковы:
(i) Нуклеиновую кислоту, кодирующую белок или полипептид для дисплея, клонируют в фаг;
(ii) Клонированная нуклеиновая кислота экспрессируется слитно с крепящейся к оболочке «якорной» частью одного из белков фаговой оболочки (обычно белки Р3 или Р8 оболочки в случае нитчатых фагов), так что чужеродный белок или полипептид экспонируется на поверхности фага;
(iii) Фаг, экспонирующий белок или полипептид с желаемыми свойствами, затем селектируют (например, аффинной хроматографией), тем самым получая генотип (связанный с фенотипом), который может быть секвенирован, умножен и перенесен в другие системы экспрессии.
Альтернативно, чужеродный белок или полипептид можно экспрессировать, используя фагмидный вектор (то есть вектор, содержащий точки начала (ориджины) репликации, полученные из фага и плазмиды), который может быть упакован в виде одноцепочечной нуклеиновой кислоты в оболочке бактериофага. При использовании фагмидных векторов используется «фаг-помощник» для обеспечения функций репликации и упаковки фагмидной нуклеиновой кислоты. Полученный фаг будет экспрессировать как белок оболочки дикого типа (кодируется фагом-помощником), так и модифицированный белок оболочки (кодируемый фагмидом), тогда как при использовании фагового вектора экспрессируется только модифицированный белок оболочки.
Использование фагового дисплея для выделения лигандов, которые связываются с биологически релевантными молекулами, было рассмотрено у Felici et al. (1995), Katz (1997) и Hoogenboom et al. (1998). Было сконструировано несколько рандомизированных комбинаторных пептидных библиотек, чтобы селектировать полипептиды, которые связывают различные цели, например, рецепторы клеточной поверхности или ДНК (Kay and Paul, (1996)).
Белки и мультимерные белки были успешно экспонированы в фаге в качестве функциональных молекул (см. Chiswell and McCafferty, (1992)). Кроме того, были экспрессированы функциональные фрагменты антител (например, Fab, одноцепочечный Fv [scFv]) (McCafferty et al. (1990); Barbas et al. (1991); Clackson et al. (1991)), и некоторые из недостатков технологии получения человеческих моноклональных антител были вытеснены после того, как были выделены высоко-аффинные фрагменты человеческих антител (Marks et al. (1991) и Hoogenboom and Winter (1992)).
Дальнейшая информация о принципах и практике фагового дисплея приведена в Phage display of peptides and proteins: a laboratory manual Ed Kay, Winter and McCafferty (1996), раскрытие которого включено в данный документ посредством ссылки.
Библиотека антилигандов может быть построена из по меньшей мере одного, выбранного из антител и антиген-связывающих вариантов, их производных или фрагментов; каркасных молекул с разработанными вариабельными поверхностями; рецепторов и ферментов.
Дифференциально экспрессирующийся лиганд может представлять собой по меньшей мере одно, выбранное из антигенов; рецепторов лигандов; и ферментов-мишеней, которые содержат по меньшей мере одно из углевода; белка; пептида; липида; полинуклеотида; неорганических молекул и конъюгированных молекул.
Способ по изобретению может также содержать дополнительную стадию подвергания лиганда и средства разделения (на стадии дифференциального биопэннинга) воздействию побудительного фактора, влияющего на экспрессию целевых лигандов на упомянутых конструкциях лигандов.
Селектированные антилиганды, описанные в изобретении, могут быть впоследствии использованы при производстве фармацевтической композиции для применения в медицине для лечения, визуализации, диагностики или прогнозирования заболевания. Антилиганды на основе антител и, важнее всего, на основе человеческих антител, имеют большой терапевтический потенциал.
Таким образом, второй объект изобретения относится к способу получения фармацевтической композиции, который содержит, после идентификации антилиганда с желаемыми характеристиками способом по любому из предшествующих пунктов, добавление упомянутого антилиганда к фармацевтически приемлемому носителю.
Третий объект данного изобретения относится к фармацевтической композиции, полученной способом по второму объекту для применения в медицине. Фармацевтическая композиция также может быть использована в производстве лекарственного средства для профилактики, лечения, визуализации, диагностики или прогнозирования заболевания.
Определения
Под «биопэннингом» авторы имеют в виду способ селекции одного элемента из желаемой связывающейся пары антилиганд-лиганд, основанный на его способности связываться с высокой аффинностью с другим элементом.
По «дифференциальным биопэннингом» авторы имеют в виду способ биопэннинга для селекции одного элемента из желаемой связывающейся пары антилиганд-лиганд, который экспрессируется в различных количествах в или на двух разных источниках (например, субтрактор/контроль и мишень), на основе его способности связываться с высокой аффинностью с другим элементом.
Под «секвенированием с высокой пропускной способностью» авторы имеют в виду, что большое количество последовательностей секвенируют параллельно (до нескольких миллионов), так что скоростное секвенирование большого числа молекул оказывается практически осуществимым и выполняется значительно быстрее и дешевле.
Под «подтверждающим скринингом» авторы имеют в виду детектирование специфического лигандного связывания пула выделенных антилигандов и/или клонов отдельных антилигандов с целевой конструкцией в сравнении с субтрактивной конструкцией с помощью любого анализа, предназначенного для детектирования связывания лиганд/антилиганд, например проточная цитометрия, FMAT, ELISA, MSD и СВА. Данный термин дополнительно означает, что как только антилиганд идентифицируется как связывающийся с дифференциально экспрессирующимся лигандом, то изучаются природа и идентичность лиганда, а также связывающие взаимодействия между антилигандом и лигандом.
В термин «лиганд» авторы включают значение одного элемента связывающейся пары лиганд/антилиганд. Лиганд может представлять собой, например, одну из нитей нуклеиновой кислоты в связывающейся паре дуплекса комплементарных, гибридизированных нуклеиновых кислот; молекулу эффектора в связывающейся паре эффектор/рецептор; или антиген в связывающейся паре антиген/антитело или антиген/фрагмент антитела.
В термин «антилиганд» авторы включают значение противоположного элемента связывающейся пары лиганд/антилиганд. Антилиганд может представлять собой другую из нитей нуклеиновой кислоты в связывающейся паре дуплекса комплементарных, гибридизированных нуклеиновых кислот; молекулу рецептора в связывающейся паре эффектор/рецептор; или молекулу антитела или фрагмента антитела в связывающейся паре антиген/антитело или антиген/фрагмент антитела, соответственно.
В термин «антиген» авторы включают значение молекулы или химического соединения, которое способно взаимодействовать с антителами, но не обязательно производить иммунный ответ. Такие антигены включают, но без ограничения, молекулы белка, пептида, нуклеотида, углевода, липида или их конъюгата.
Под «дифференциально экспрессирущимися лигандами» авторы имеют в виду лиганды, которые либо экспрессируются на уровнях, различающихся у целевых и субтрактивных источников, включая лиганды, экспрессирующиеся только в определенных условиях/местах, а не в других; либо случаи, где либо целевой либо субтрактивный лиганд представляет собой модифицированную версию другого целевого или субтрактивного лигандов. Например, некоторые антигены экспрессируются на высоком уровне на клеточной поверхности пораженных клеток (например, раковых клеток) и на низких уровнях или вообще не экспрессируются на эквивалентных здоровых клетках (например, не раковых клетках).
Под «лигандами с низкой экспрессией» авторы имеют в виду те лиганды, которые экспрессируются на низких уровнях, то есть менее чем 20000 копий на клетку, например, от 5000 до 20000 (сюда относится большинство экспрессирующихся рецепторов клеточной поверхности дикого типа), или лиганды, появляющиеся при частоте, составляющей менее 1% от любого другого, экспрессирующегося на более высоком уровне лиганда в образце популяции, содержащей позитивные лиганды.
Под «конструкцией лиганда» авторы имеют в виду систему, которая содержит целевой и/или субтрактивный лиганд, связанный со средством разделения.
Термин «вариант антитела» принят для обозначения любых синтетических антител, рекомбинантных антител или гибридов антител, таких как, но не ограничиваясь ими, одноцепочечная молекула антитела, вырабатываемая фагом, экспонирующим вариабельные и/или константные области легкой и/или тяжелой цепи иммуноглобулина, или другая иммунореактивная молекула, способная связываться с антигеном в формате иммуноанализа, известном специалистам в данной области техники.
Термин «производное антитела» относится к любой модифицированной молекуле антитела, способной связываться с антигеном в формате иммуноанализа, который известен специалистам в данной области техники, такой как фрагмент антитела (например, фрагмент Fab или Fv), или к молекуле антитела, которая модифицирована добавлением одной или более аминокислот или других молекул для облегчения связывания антител с другим пептидом или полипептидом, с большим белком-носителем или с твердой подложкой (например, аминокислотами тирозином, лизином, глутаминовой кислотой, аспарагиновой кислотой, цистеином и их производными, NH2-ацетильными группами или COOH-концевыми амидными группами, среди прочего).
Под «плотностным центрифугированием» авторы имеют в виду разделение элементов, например клеток, органелл и макромолекул, в соответствии с их различиями в плотности. Такое разделение достигается центрифугированием с использованием градиента плотности подходящего раствора, через который элементы, разделяясь, двигаются на основе их плотности.
«Закон действующих масс» является универсальным законом природы, который применим при любых обстоятельствах. Данный закон гласит, что для реакции:
aA+bB→cC+dD
и если данная система находится в равновесии при заданной температуре, то следующее соотношение является постоянным:
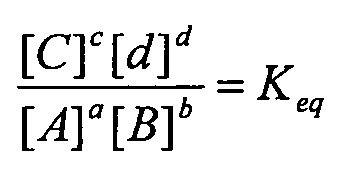
где:
A, B, C&D=представляют собой участники реакции (реагенты и продукты)
a, b, c, & d=представляют собой коэффициенты, необходимые для сбалансированного химического уравнения
И где константа равновесия рассчитывается для значений концентраций (обозначается []) и К имеет единицы Mc+d-(a+b).
Примеры вариантов осуществления некоторых объектов изобретения будут теперь описаны со ссылкой на следующие чертежи, на которых:
Фигура 1. Схема способа дифференциального биопэннинга, описанного в (Fransson et al. Int J Cane 2006). Числа ниже стрелок указывают на количество клонов фага-abs или антител, которые удерживаются в способе скрининга после каждой из пяти стадий скрининга (от 1 до 5) и двух последующих стадий синтеза и верификации (6) и (7). Данные стадии представляют собой следующее: (1) дифференциальный биопэннинг; (2) отбор колоний scFv-преобразованных клонов; (3) экспрессия scFv-клонов на уровне единичного клона; (4) проверка scFv-клонов на специфичность к дифференциально экспрессирующимся поверхностным рецепторам целевых клеток; (5) секвенирование для ID последовательностей уникальных антител; (6) синтез IgG, и (7) in vitro I in vivo функциональный тест.
Фигура 2. Расчеты in silico, показывающие, что пул антител, полученный из дифференциального биопэннинга раковых В-клеток в сравнении с Т-клетками Jurkat («BnonT»), должен содержать гораздо большее количество антител против каждого из нескольких дифференциально экспрессирующихся поверхностных рецепторов, чем экспериментально идентифицируется с использованием обычных способов.
(1) Указывает, что расчеты проводились, как описано в WO 2004/023140.
Фигура 3. Пул фаг-антитело, полученный из дифференциального биопэннинга клеток DU-145 рака простаты в сравнении с Т-клетками Jurkat («DnonT»), обладает высокой специфичностью к популяции целевых клеток (DU-145).
A) На графике представлено зависимое от дозы специфическое связывание фагового пула, полученного после двух циклов дифференциального биопэннинга к целевым клеткам DU145, как проанализировано с помощью FACS. Следует отметить, что связывание с нецелевьми клетками Jurkat не детектируется.
B) На графике представлено специфическое связывание каждого из 1408 отдельных, случайно выбранных клонов из пула, полученного после двух циклов дифференциального биопэннинга клеток DU145 в сравнении с клетками Jurkat, как проанализировано с помощью FMAT.
Фигура 4. Целевые клетки DU-145 экспрессируют несколько поверхностных рецепторов, которые могут быть классифицированы как «высоко дифференциально экспрессирующиеся!, «промежуточно дифференциально экспрессирующиеся», или «низко дифференциально экспрессирующиеся» на основе их абсолютного уровня экспрессии целевыми клетками и их относительного уровня экспрессии на поверхностях целевых клеток в сравнении с нецелевыми клетками.
Целевые (DU145) и нецелевые (Jurkat) клетки подвергали скринингу на экспрессию трех антигенов HER2, CD24 и CD130 проточной цитометрией с использованием антител, меченных Zenon Alexa Fluor 647.
A) На фигуре показана величина средней интенсивности флуоресценции целевых и нецелевых клеток, окрашенных Zenon-мечеными антителами.
B) На фигуре показан процент клеток, которые экспрессируют HER2, CD24 и CD130.
Фигура 5. Анализы графика Скэтчарда выявляют, что уровни экспрессии дифференциально экспрессирующихся поверхностных рецепторов, являющихся мишенью для антител, выделенных дифференциальным биопэннингом и глубоким секвенированием, составляют от 6000 до 400000 рецепторов на клетку.
А. Кривые насыщения.
Б. Графики Розенталя. Аффинность (KD) анти-CD130 антитела, как было оценено, составляет 0,8 нМ, и количество поверхностных рецепторов CD130 составляет 6,300/клетку. Аффинность анти-CD24 антитела, как было оценено, составляет 5,6 нМ, и количество эпитопов составляет 8,400/клетку. Аффинность анти-HER2 антитела, как было оценено, составляет 47 нМ, и число эпитопов составляет 110000/клетку. Оценка была выполнена с графиками Розенталя 125I-меченых антител, связывающихся с клетками DU145, стимулированными INF-гамма.
Фигура 6. Пул антител, полученный из дифференциального биопэннинга клеток DU-145 рака простаты в сравнении с Т-клетками Jurkat («DnonT»), содержит клоны антител, специфичных для сильно экспрессирующихся, промежуточно экспрессирующихся и низко экспрессирующихся дифференциально экспрессирующихся поверхностных рецепторов.
На основании анализов Скэтчарда и FACS (фигуры 4 и 5), HER2, CD24 и CD130 были охарактеризованы как рецепторы, экспрессирующиеся на разных уровнях.
A) На фигуре показано, что клоны антител, специфичные для всех антигенов, присутствуют в пуле антител, генерированном двумя раундами дифференциального биопэннинга клеток DU-145 в сравнении с клетками Jurkat, как проанализировано с помощью ELISA.
B) На фигуре показано, что когда целевые специфические клоны в А были селектированы и повторно протестированы на связывание, большинство из данных клонов были еще позитивными для целевого антигена. Секвенирование полученных клонов антител, специфичных к поверхностным рецепторам-мишеням, показало наличие (по меньшей мере) 12 уникальных антител в пуле дифференциально селектированных антител; восемь (8) анти-HER2, одно (1) анти-CD24 и три (3) анти-CD130 антитела.
Фигура 7. Секвенирование увеличивающего числа дифференциально селектированных клонов антител приводит к идентификации увеличивающего числа клонов антител, специфичных для низко экспрессирующихся дифференциально экспрессирующихся поверхностных рецепторов целевых клеток.
Были определены последовательности антител клонов в трех случайным образом селектированных пулах связывателей увеличивающегося размера (91 клонов, 255 клонов и 813 клонов, соответственно). После этого клоны, которые были найдены во всех трех пулах («часто встречающиеся клоны»), только в двух пулах большего размера («менее часто встречающиеся клоны») или только в самом большом пуле («редкие клоны»), были проанализированы на связывание с клетками DU145 способом FACS, и величины средней интенсивности флуоресценции изобильных, менее частых и редких клонов были подвергнуты сравнению.
Данные ясно показывают, что средние уровни экспрессии рецепторов для часто встречающихся клонов больше, чем для клонов промежуточной частоты, которые, в свою очередь, больше, чем для редких клонов. Таким образом, секвенирование увеличивающего числа дифференциально селектированных клонов антител привело к идентификации увеличивающего числа клонов антител, специфичных для низко экспрессирующихся дифференциально экспрессирующихся поверхностных рецепторов целевых клеток.
A) На фигуре показана величина средней интенсивности флуоресценции клеток DU-145, окрашенных соответствующими клонами антител.
B) Фигура показывает процент целевых клеток, с которыми связываются отдельные клоны антител.
Так как клоны были выбраны случайным образом, некоторые из них не являлись связывателями и они были убраны перед анализом (связыватель был определен либо как клон, дающий сигнал по меньшей мере в два раза превышающий отрицательный контроль, выраженный как процент позитивно окрашенных клеток, так и среднее геометрическое, либо, альтернативно, в три раза превышающий сигнал, выраженный как среднее геометрическое).
*=p<0,05, **=p<0,01 по подсчетам ANOVA с помощью коррекции Бонферрони для нескольких анализов.
Фигура 8. Схематически показано сравнение стадий скрининга описанного ранее способа скрининга дифференциального биопэннинга (А., верхняя панель) и нового улучшенного способа (В., нижняя панель).
Данные два способа отличаются в нескольких отношениях; Во-первых, в способе обратного скрининга были опущены стадии дифференциального биопэннинга преобразования фага-ab в scFv, экспрессии scFv и проверки scFv на специфичность связывания, содержащиеся в WO 2004/023140. Во-вторых, в способе обратного скрининга непосредственно после дифференциального биопэннинга следует (глубокое) секвенирование, в то время как в способе скрининга в WO 2004/023140 секвенирование клонов антител в пуле предшествует скринингу отдельных клонов scFv на специфичность дифференциально экспрессирующихся поверхностных рецепторов целевых клеток. В-третьих, и что наиболее важно, количество клонов антител (10000) и качество клонов антител (включая всеобъемлющую генерацию антител, специфичных для низко экспрессирующихся дифференциально экспрессирующихся рецепторов), достигаемые с помощью способа обратного скрининга, практически не может быть достигнуто с помощью ранее описанного способа дифференциального биопэннинга из А.
Данные стадии представляют собой: (1) дифференциальный биопэннинг; (2) отбор колоний scFv-преобразованных клонов; (3) экспрессия scFv-клонов на уровне единичного клона; (4) проверка scFv-клонов на специфичность к дифференциально экспрессирущимся поверхностным рецепторам целевых клеток; (5) секвенирование для ID последовательностей уникальных Ab; (6) синтез IgG, и (7) in vitro I in vivo функциональный тест,
(2') глубокое секвенирование нового поколения; (3'), синтез IgG HT; (4') проверка клонов IgG на специфичность к дифференциально экспрессирущимся поверхностным рецепторам целевых клеток; (5') in vitro/in vivo функциональный тест
Фигура 9. Расчеты in silico извлеченного пула фаг-антитело, полученного из двух раундов дифференциального биопэннинга клеток DU-145 рака простаты в сравнении с Т-клетками Jurkat
Пример 1. Объединение дифференциального биопэннинга с последующим секвенированием с высокой пропускной способностью генерирует беспрецедентное количество уникальных клонов антител, специфичных для дифференциально экспрессирующихся поверхностных рецепторов целевых клеток.
Генерация пула антител дифференциальным биопэннингом раковых В-клеток в сравнении с Т-клетками Jurkat («BnonT»).
В данном эксперименте 2×1013 фаговых частиц из высоко диверсифицированной библиотеки n-CoDeR, содержащей 101 генотипов уникальных связывателей, смешивают с целыми клетками лимфомы В клеточной линии Ramos (позитивная селекция) и везикулами из плазматической мембраны или неочищенной мембраны из Т-лейкозных клеток линии Jurkat (негативная селекция). Связыватели, специфичные для антигенов, которые уникально экспрессируются на В-клетках лимфомы клеточной линии Ramos, по сравнению с Т-лейкозными клетками клеточной линии Jurkat, должны быть селективно выделены.
Расчет количества позитивных и негативных клеток для селекции
Количество клеток, которое использовалось в различных селекционных раундах, было рассчитано, как описано в (WO 2004/023140). Реакционные параметры, используемые для расчетов, представлены на фигуре 2.
Количества позитивных и негативных клеток были выбраны таким образом, что после трех раундов селекции происходит обогащение в 10000 раз связывателей со специфичностью к антигенам, экспрессирующимся однозначно на В-клетках, относительно количества антигена, экспрессирующегося при равной плотности на В- и Т-клетках.
Исходное количество фаговых связывателей, специфичных для различных категорий антигена (позитивная клетка обогащенная, позитивная клетка уникальная или стандартно экспрессирующийся антиген позитивной/негативной клетки), в 2-м и 3-м селекционных раундах было рассчитано умножением расчитанного количества элюированного фага, специфичного для различных категорий антигена после 1-го и 2-го селекционных раундов, с коэффициентом амплификации (AF).
Коэффициент амплификации получали делением общего количества амплифицированного фага после соответствующего раунда селекции на общее число элюированного фага из того же раунда селекции.
Экспериментальные способы
Клеточные культуры
Клеточную линию Т-клеток Jurkat, клон Е6-1 и клеточную линию В-клеточной лимфомы Ramos культивировали в среде RPMI 1640, дополненной 10% FCS (инактивированной нагреванием только для клеток Ramos), 10 мМ HEPES и 1 мМ пирувата натрия, в увлажненной атмосфере при 37°C. Клетки поддерживали на уровне 1-2×106 клеток/мл (менее 1×106 клеток/мл для Jurkat).
Получение плазматической мембраны Т-клеток Jurkat
Клеточная культура Jurkat
Клетки Jurkat E6-1 поддерживали в среде RPMI-1640 с Glutamax I (Gibco, #61870-010), дополненной 10% фетальной телячьей сывороткой (Gibco, Lot no 1128016), 1 мМ пирувата натрия (Gibco) и 10 мМ Hepes-буфера (Gibco) в увлажненной атмосфере с 5% CO2 при 37°C, и при клеточных плотностях от 1×105 до 1×106 клеток/мл. В конечном пассаже клетки оставляли для достижения максимальной плотности, составляющей 2×106, после чего их собирали.
Разрушение клеток
1. Клетки собирали из культуры центрифугированием в центрифужных пробирках объемом 500 мл (Coming, #431123), помещенных в пробирочные адаптеры, 1500 оборотов в минуту, 15 минут при 4°C.
2. Супернатант отбрасывали и промывали в 0,145 М NaCl. Клеточные суспензии объединяли, подсчитывали клетки (всего 5×109 клеток) и повторяли центрифугирование.
3. Разрушение клеток проводили гипоосмотическим шоком в 1 мМ NaHCO3 1,5 мМ MgAc pH 7,4 на льду в течение от 10 до 30 мин, и последующую кавитацию в азоте проводили в прессе Yeda, 40 бар (4000 кПа) в течение 15 мин при 0°C. Концентрация клеток не превышала 5×107 клеток/мл.
4. После разрушения к суспензии гомогената добавляли 150 мкл 0,5М ЭДТА с получением конечной концентрации ЭДТА, равной 1 мМ (добавление EDTA предотвращает агрегацию мембранных везикул).
5. А) Выделение неочищенной мембраны: гомогенат (50 мл) центрифугировали в течение 10 мин при 1900g (4000 оборотов в минуту в роторе SS34), чтобы удалить неразрушенные клетки и ядра, и собирали супернатант. Промывку и повторное центрифугирование осадка избегали, так как хрупкие ядра, как правило, разрушались, вызывая утечку ДНК и агрегацию; или
В) Выделение плазматической мембраны: 10 мл 37,2%-ной сахарозы наслаивали на дне 6×38,5 мл ультрацентрифужной пробирки Beckman, и 6×27 мл клеточного гомогената с вышеупомянутой стадии 2 аккуратно наносили поверх. Пробирку центрифугировали при 27000 оборотов в минуту в поворотно-откидном роторе SW28 (номинальная мощность 6×39 мл) в течение 2 часов 45 минут при 4°C. Плазматические мембраны выделяли из пробирок в виде белой полосы интерфазы между сахарозной подушкой и фазой образца, и плазматическую мембрану (РМ) собирали, разделяли между 4×35 мл пробирками и разводили в ТЕ-буфере (1 мМ Трис/0,25 М сахароза/0,25 М ЭДТА) до общего объема, составляющего 35 мл.
6. Ультрацентрифугирование проводили в роторе Beckman Type 45.Ti (номинальная емкость 6×94 мл пробирки Nalgene) при 40000 оборотах в минуту (около 200000×g) в течение 1 часа при 4°C.
7. Супернатанты отбрасывали и оставшийся буфер удаляли с использованием пипетки Finn объемом 1 мл. Осадки плазматической мембраны соскабливали в нижней части пробирок металлической палочкой и переносили в небольшой гомогенизатор Даунса. Осажденные мембраны ресуспендировали гомогенизацией в общем объеме, составляющем 2,5 мл, ТЕ-буфера, содержащего 10 мМ Hepes (буфер 10 мМ Hepes/1 мМ Трис/0,25 М сахароза/0,25 М ЭДТА), с от 5 до 10 проходами свободно проходящего стеклянного поршня Даунса. Приблизительно, мембраны, полученные из 2×109 клеток Jurkat, могли быть ресуспендированы в 1 мл ресуспензионного (ТЕ) буфера.
Определение концентрации белка
Определение концентрации белка проводили с использованием набора ВСА в соответствии с инструкциями изготовителя. Вкратце, двойной стандарт BSA готовили 2-кратными разведениями (10 мкл образца+10 мкл буфера) исходного раствора 2 мг/мл BSA в PBS. Строили стандартную кривую и использовали ее для определения концентрации общего белка в образцах мембран.
Активность плазматической мембраны (анализ со щелочной фосфатазой)
Растворы щелочной фосфатазы
Раствор субстрата:
1 таблетка p-NPP на 10 мл боратного буфера (конечная концентрация 1,5 мг/мл) в 50 мМ натрий-боратном буфере (pH 9,8), 1,0 мМ MgCl2
Образец в трех повторах разводили в буфере борат/MgCl2 переносом 50 мкл образца к 50 мкл буфера разбавления (50 мМ натрий-боратный буфер (pH 9,8), 1,0 мМ MgCl2. 200 мкл раствора субстрата (1 таблетка p-NPP на 10 мл боратного буфера до конечной концентрации 1,5 мг/мл в 50 мМ натрий-боратном буфере, pH 9,8, 1,0 мМ MgCl2) добавляли к двум из трех образцов для каждого разведения. Затем образцы инкубировали при 37°C в течение 60 минут и более. Оптическую плотность надосадочной жидкости измеряли при длине волны 410 нм и вычитали значения, полученные из соответствующей контрольной лунки(ок) (например, общий клеточный гомогенат, приготовленный азотной кавитацией, за исключением ядер и тяжелых митохондрий), куда субстрат не был добавлен. Результаты были нанесены на график и проанализированы.
Процедура селекции: Протокол дифференциального биопэннинга
Реакционные параметры
1-й раунд селекции
Исходная фаговая культура n-CoDeR Lib2000, содержащая 1010 фагмидных частиц с уникальными генотипами (Ampr), амплифицированная до общей БОЕ, равной 2х1013, в 1,6 мл 2% молока-PBS (с Ca и Mg).
Общий объем реакционной смеси составлял 2,5 мл
Позитивные - 5×107 клеток B-клеточной лимфомы линии Ramos
Негативные - неочищенные мембраны Т-клеток Jurkat, полученные из 2×10 клеток
2-й раунд селекции
1,5×1012 фаговых частиц, элюированных из предыдущего раунда селекции, затем амплифицировали, осаждали и ресуспендировали в 100 мкл 2% молока-PBS (с Ca и Mg). Общий объем реакционной смеси составлял 0,5 мл Позитивные - 5×106 клеток В-клеточной лимфомы линии Ramos Негативные - везикулы неочищенных мембран Т-клеток Jurkat, полученные из 1×109 клеток
3-й раунд селекции
1×1012 фаговых частиц, элюированных и амплифицированных из предыдущего раунда селекции, ресуспендировали в 100 мкл 2% молока-PBS (с Ca и Mg).
Общий объем реакционной смеси составлял 0,5 мл
Позитивные - 5×106 клеток В-клеточной лимфомы линии Ramos
Негативные - везикулы плазматических мембран Т-клеток Jurkat, полученные из 1×109 клеток
Способ
Исходную фаговую культуру предварительно нагревали при 37°C в течение 15 минут и перемешивали встряхиванием с перерывами. Исходную фаговую культуру центрифугировали в течение 15 минут на полной скорости в центрифуге Эппендорф. После образования осадка супернатант переносили в новую пробирку Эппендорф и ресуспендировали в обезжиренном молоке до конечной концентрации, равной 2%.
Контрольные препараты плазматических мембран клеток Jurkat из 2×109 клеток (1×109 клеток 2-го и 3-го раундов биопэннинга) оттаивали на льду. (10 мкл также сохраняли для определения концентрации белка.) Размороженные препараты плазматических мембран ресуспендировали добавлением исходной фаговой культуры и смешиванием с помощью пипетки, а затем инкубировали в течение 15 минут на льду.
5×107 (5×106 клеток 2-го и 3-го раундов биопэннинга) клеток Ramos центрифугировали при 1200 оборотов в минуту, 6 мин, 4°C.
Надосадочную жидкость отбрасывали и клетки Ramos ресуспендировали в исходном растворе молоко-фаг-негативные клеточные мембраны и инкубировали при 10°C и подвергали медленному (end-over-end) вращению в течение 4 часов.
Инкубируемую смесь клетка/клеточная мембрана/фаг переносили в пробирку Фалькон объемом 15 мл, содержащую 1 мл 100% (окрашенного трипановым синим) Ficoll на дне, и 9 мл наслоенного 40% Ficoll-Paque Plus в 2% BSA/PBS (с Са и Mg). Пробирку центрифугировали при 1500 оборотах в минуту в течение 10 мин, 4°C, поворачивали на 180° и центрифугировали в течение дополнительной 1 минуты для того, чтобы вытеснить клетки от стенки пробирки.
Интерфейс, содержащий целые клетки Ramos и связанный фаг, аккуратно отсасывали с помощью шприца и большей иглы (например Microlance 3 - 19GA11/2 1,1×40 TW РМ). Иглу вводили непосредственно под интерфейс, содержащий клетки, со скошенным концом иглы вверх. Клеточный слой собирали (примерно 150 мкл), и иглу проталкивали через пластиковый материал пробирки в противоположную сторону от входного отверстия. Содержимое шприца переливали в чистую пробирку и промывали дважды, засасывая чистую порцию PBS в иглу (все еще находящуюся в положении проколовшей пробирку). Собранную суспензию клеток ресуспендировали в 500 мкл PBS-2% BSA и повторяли промывку, сохраняя супернатант для титрования.
Клетки ресуспендировали в 1 мл PBS и переносили в новую пробирку Эппендорф объемом 15 мл, в которой их центрифугировали при 1260 оборотах в минуту в течение 10 мин, 4°C. Супернатант удаляли с помощью пипетки, сохраняя супернатант для титрования.
Фаг элюировали из клеток добавлением 150 мкл 76 мМ лимонной кислоты (pH 2,5) в PBS с последующей инкубацией при комнатной температуре в течение 5 мин. Смесь нейтрализовали добавлением 200 мкл 1М Трис-HCl, pH 7,4. Затем клетки центрифугировали и сохраняли элюированный фаг.
Клетки ресуспендировали в 1 мл трипсина и переносили в новую пробирку и инкубировали в течение 10 мин перед инактивацией с 40 мкл 1 мг/мл апротинина. Клетки центрифугировали, супернатант сохраняли для титрования.
Амплификация на больших планшетах после 1-го и 2-го раундов селекции
1. Инициировали рост 10 мл культур HB101F' E. coli (по одной для каждой селекции с целью амплификации+одна для измерения оптической плотности при 600 нм (OD 600)) за от 2,5 до 3 часов перед использованием добавлением 50 мкл ночной культуры в 10 мл среды LB (лизогении бульон), содержащей 15 мкг/мл тетрациклина. OD проверяли на одной культуре приблизительно через 2,5 часа.
2. Пробирки инфицировали половиной элюированного фага при OD600, равной 0,5.
3. Пробирки инкубировали в течение 30 минут при 37°C и 50 оборотов в минуту, и для правильного фенотипирования дополнительно 30 мин при 37°C, 200 оборотов в минуту.
4. Бактерии концентрировали (10 мл) центрифугированием в течение 10 минут при 2060×g (3000 оборотов в минуту Beckman GS-6).
5. Бактерии ресуспендировали в части надосадочной жидкости (приблизительно 3 мл) и распределяли на большие планшеты площадью 500 см с LA (лурия агар), содержащим 100 мкг/мл ампициллина+15 мкг/мл тетрациклина+1% глюкозы.
6. Планшеты инкубировали в течение ночи при 30°C.
7. Бактерии собирали из планшетов добавлением 5 мл LB, содержащей 100 мкг/мл ампициллина и 15 мкг/мл тетрациклина в расчете на планшет и соскоб. Планшеты наклоняли и отсасывали раствор.
8. Планшеты промывали с дополнительными 3 мл среды LB, как указано выше, и объединяли с первой бактериальной суспензией в пробирки Фалкон объемом 50 мл.
9. Бактерии концентрировали центрифугированием в течение 10 минут при 2100×g/3000 оборотов в минуту, Beckman GS6, при комнатной температуре и ресуспендировали в 1 мл LB, содержащей 100 мкг/мл ампициллина и 15 мкг/мл тетрациклина.
10. 500 мкл 50% глицерина добавляли к 1 мл бактериальной суспензии, и исходный раствор в глицерине замораживали при -80°C.
11. 2×10 мл LB, содержащей 100 мкг/мл ампициллина и 15 мкг/мл тетрациклина, инфицировали 2,5 мкл (5 мкл) исходного раствора в глицерине со стадии 10 и выращивали до OD600, равной 0,5.
12. 6×109 БОЕ вспомогательного фага R408 добавляли в расчете на 1 мл культуры, и культуры инкубировали в течение 30 минут при 37°C и 50 оборотов в минуту.
13. Добавляли раствор IPTG до конечной концентрации 100 мкМ (т.е. 2 мкл из 0,5 М исходного раствора на 10 мл культуры), и культуры инкубировали в течение ночи при 25°C и 175 оборотах в минуту.
Сбор и преципитация амплифицируемых исходных фаговых культур
1. Бактерии осаждали центрифугированием в течение 10 минут при комнатной температуре при 2100×g (3000 оборотов в минуту, в Beckman GS-6), и супернатант стерильно фильтровали через 0,2 мкм стерильный фильтр.
2. Пробирки, происходящие из той же селекции, объединяли, и фаг преципитировали добавлением 1/4 объема буфера преципитации фага и инкубировали в течение по меньшей мере 4 часов при 4°C.
3. Пробирки центрифугировали в течение 30 минут при 4°C и 13000×g.
4. Осадок ресуспендировали полностью в 100 мкл PBS в течение ночи при 4°C. Амплификация на планшетах для исходных растворов в глицерине, и ночная культура для минипрепаратов (после раунда селекции 3).
1. Инициировали рост 10 мл культур HB101F' E.coli (по одной для каждой селекции с целью амплификации+одна для измерения оптической плотности при 600 нм (OD 600)) за от 2,5 до 3 часов перед использованием добавлением 50 мкл ночной культуры в 10 мл LB, содержащей 15 мкг/мл тетрациклина. OD проверяли на одной культуре приблизительно через 2,5 часа.
2. Пробирки инфицировали половиной элюированного фага при OD600, равной 0,5.
3. Пробирки инкубировали в течение 30 минут при 37°C и 50 оборотов в минуту, и для правильного фенотипирования дополнительно 30 мин при 37°C, 200 оборотов в минуту.
4. Добавляли 10 мл теплой среды LB, содержащей 200 мкг/мл ампициллина, и инфицированные бактерии разделяли на 2 части по 10 мл каждая.
5. В одной из двух пробирок бактерии концентрировали (10 мл) центрифугированием в течение 10 минут при 2100×g/3000 оборотов в минуту, Beckman GS-6 при комнатной температуре, ресуспендировали в небольшом объеме и распределяли на планшет площадью 500 см2 с LA (100 мкг/мл ампициллина+15 мкг/мл тетрациклина+1% глюкозы) и инкубировали в течение ночи при 30°C.
6. Минипрепарат: другие 10 мл центрифугировали и ресуспендировали в 6 мл LB, содержащей 0,1% глюкозы и 100 мкг/мл ампициллина, и инкубировали в течение ночи при 30°C, 175 оборотов в минуту.
7. Бактерии собирали из планшетов добавлением 5 мл LB, содержащей 100 мкг/мл ампициллина и 15 мкг/мл тетрациклина, в расчете на планшет и соскоб. Планшеты наклоняли и отсасывали раствор.
8. Планшеты промывали с дополнительными 3 мл среды LB, как указано выше, и объединяли с первой бактериальной суспензией в пробирки Фалкон объемом 50 мл.
9. Бактерии концентрировали центрифугированием в течение 10 минут при 2100×g/3000 оборотов в минуту, Beckman GS6, при комнатной температуре и ресуспендировали в 1 мл LB, содержащей 100 мкг/мл ампициллина и 15 мкг/мл тетрациклина.
10. 500 мкл 50% глицерина добавляли к 1 мл бактериальной суспензии, и исходный раствор в глицерине замораживали при -80°C.
11. Очищенную ДНК фаг-антитело получали приготовлением минипрепаратов из 3 мл культуры в соответствии с протоколом из набора производителей (BioRad).
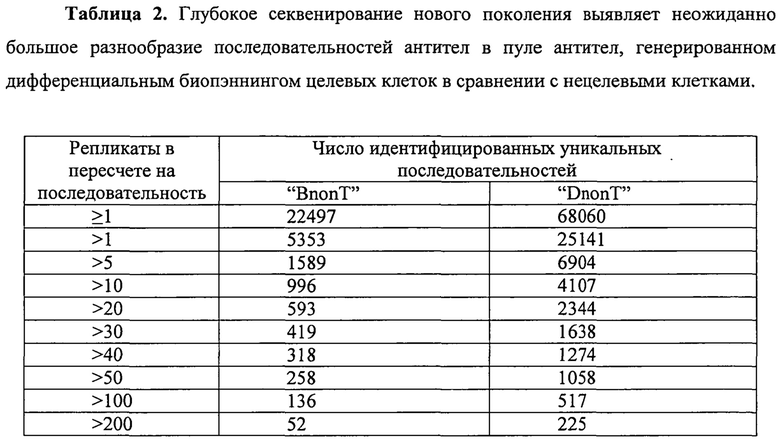
Чтобы оценить непосредственно разнообразие антител в пуле, генерируемым дифференциальным биопэннингом BnonT, авторы использовали технологию 4-5-4 (Margulies et al., 2005) и оценивали разнообразие антител определением количества уникальных вариантов CDRH3 в пуле дифференциально селектированных антител.
Глубокое секвенирование по технологии 4-5-4 проводили на очищенной ДНК фаг-антитело, полученной после трех раундов дифференциального биопэннинга («BnonT»), выявив, в общей сложности, 22497 уникальных последовательностей (таблица 2). Для сравнения, традиционный скрининг с секвенированием Sanger ДНК фаг-антитело из тех же дифференциальных биопэннингов BnonT идентифицировал только восемь уникальных клонов антител (таблица 1).
Данное наблюдение, наряду с ранним сообщением авторов (Fransson et al., 2006) о том, что подавляющее большинство (более 99%) дифференциально селектированных способом BnonT клонов было специфично по отношению к дифференциально экспрессирующимся поверхностным рецепторам раковых В-клеток, свидетельствует о том, что
a) пул дифференциально селектированных антител был на самом деле высоко диверсифицированным, и
b) что при объединении дифференциального биопэннинга и секвенирования с высокой пропускной способностью количество уникальных клонов антител, специфичных для дифференциально экспрессирующихся поверхностных рецепторов, которое можно идентифицировать, превышает на несколько порядков количество, достигаемое при использовании традиционных подходов скрининга.
Для того чтобы продемонстрировать, что дифференциальный биопэннинг с последующим секвенированием с высокой пропускной способностью могут быть воспроизводимо использованы для генерации большого количества антител, специфичных для различных поверхностных рецепторов, дифференциально экспрессирующихся различными типами целевой клетки, проводили новую реакцию дифференциальный биопэннинг/глубокое секвенирование - на этот раз используя клетки рака простаты DU-145 в качестве целевых клеток и Т-клетки в качестве нецелевых клеток в реакции пэннинга «DnonT».
Опять же, дифференциальный биопэннинг генерировал пул антител с высокой специфичностью к клетке-мишени (фигура 3). Глубокое секвенирование по технологии 4-5-4 проводили на очищенной ДНК фаг-антитело, полученной после двух раундов (DU-145 против Т) дифференциального биопэннинга, выявив, в общей сложности, 68060 уникальных последовательностей, соответственно (таблица 2).
Подавляющее большинство из данных последовательностей антител, вероятно, будут специфичны для дифференциально экспрессирующихся антигенов целевых клеток, на что указывает скрининг более 1400 случайно выбранных клонов антител на связывание с клетками DU-145 в сравнении с Т-клетками (фигура 3b) и расчетами in silico (фигура 9).
Авторы пришли к выводу, что объединенное а) приложение дифференциального биопэннинга к высоко диверсифицированной библиотеке человеческих антител с последующим b) глубоким секвенированием пула антител, генерированного дифференциальным биопэннингом, воспроизводимо генерирует гораздо большее количество клонов антител, специфичных для дифференциально экспрессирующихся поверхностных рецепторов целевых клеток, чем это возможно традиционными подходами скрининга.
Пример 2 - Объединение дифференциального биопэннинга с последующим секвенированием с высокой пропускной способностью генерирует качественно уникальный пул клонов антител - включая таковые, специфичные для экспрессирующихся на более низком уровне дифференциально экспрессирующихся поверхностных рецепторов
Расчеты in silico, выполненные, как описано в WO2004/023140 и у Frendeus, (2006), применяя закон действующих масс, свидетельствуют о том, что при применении дифференциального биопэннинга, как выполнено в настоящей заявке (как проиллюстрировано в примере 1, используя раковые В-клетки или клетки рака простаты в качестве мишеней), частота полученных клонов антител в пуле селектированных антител будет находиться в прямой зависимости от а) абсолютной и относительной экспрессии их целевых рецепторов на поверхности целевой клетки в сравнении с нецелевой клеткой и b) их соответственных аффинностей для целевых поверхностных рецепторов.
Данные расчеты дополнительно выявляют, что, вопреки преобладающему общему мнению (Hoogenboom, 2002) (Лю et al., 2004; Mutubema et al., 1999; Osboum et al., 1998), клоны антител, специфичные для поверхностных рецепторов, экспрессирующихся на более низком уровне (например, менее чем 20000 на клетку), а также те клоны антител, которые специфичны для поверхностных рецепторов, экспрессирующихся на промежуточном уровне (например, экспрессирующихся на уровне от 20000 до 50000 рецепторов на клетку), могут и будут селектированы дифференциальным биопэннингом, как описано в настоящем документе, и будут присутствовать в пуле элюированных антител, хотя и со значительно более низкой частотой по сравнению с клонами антител, специфичными к дифференциально экспрессирующимся поверхностным рецепторам с высоким уровнем экспрессии (фигура 2 и 9).
Некоторые подходы были теперь использованы, чтобы продемонстрировать, что пул антител, генерируемый последовательным дифференциальным биопэннингом и глубоким секвенированием, уникален тем, что он содержит клоны антител, специфичных для дифференциально экспрессирующихся поверхностных рецепторов, экспрессирующихся на низких и промежуточных уровнях, и что увеличение глубины секвенирования (т.е. количество анализируемых последовательностей антител клонов) приводит к идентификации антител, специфичных к дифференциально экспрессирующимся поверхностным рецепторам, экспрессирующимся на уменьшающихся (более низких) уровнях на целевых (в сравнении с нецелевыми) клетках.
Во-первых, авторы показали, что пул антител, селектированный дифференциальным биопэннингом, действительно содержит клоны антител, которые являются специфичными для дифференциально экспрессирующихся на низком и промежуточном уровне поверхностных рецепторов (фигуры 4, 5 и 6).
Подвергая пул дифференциально селектированных антител одной дополнительной селекции с внеклеточными доменами (extracellular domains (ECD)) поверхностных рецепторов, проверенных на то, что они дифференциально экспрессируются целевыми клетками в сравнении с нецелевыми клетками, и проверенных графиком Скэтчарда и анализом FACS на то, что они экспрессируются на от низкого до промежуточного уровнях на целевых клетках (фигура 4 и 5 TBG), авторы выделили несколько (двенадцать) клонов антител, специфичных для различных экспрессирующихся на низком и промежуточном уровне поверхностных рецепторов, из пула дифференциально экспрессирующихся антител, включая таковые, специфичные для поверхностных рецепторов CD24, CD130 и HER2 (фигура 6).
Одно выделенное антитело против каждого рецептора было преобразовано в формат IgG и использовано для анализа Скэтчарда и FACS, показывая уровни экспрессии, составляющие от 6000 до 100000 рецепторов/клетка (таблица 3, фигуры 4 и 5).


Экспериментальные способы
Селекция с внеклеточными доменами (ECD) поверхностных рецепторов
Реакционные параметры
Фаги, элюированные и амплифицированные после двух раундов дифференциального биопэннинга (DU-145 против Т, «DnonT») осаждали и ресуспендировали в PBS. 100 мкл (соответствуют 2.4×1011 фагов) были использованы в третьем раунде селекции.
Общий объем реакционной смеси составлял 1,0 мл
При селекции фаги были обогащены связывателями к трем локализованным на поверхности белкам: CD24, CD 130 и HER2.
Способ - селекции ECD
50 пмоль каждого белка (см. выше) использовали для покрытия 4 полистирольных шариков (Polysciences, cat no 17175-100) в реакционном объеме 0,1 М натрий-карбонатного буфера, pH 9,5, равном 1 мл. Покрытие осуществлялось в пробирке Эппендорф при комнатной температуре в течение 1 ч с вращением end-over-end и последующей стадией инкубации в течение ночи при 4°C без вращения.
Шарики с покрытием промывали один раз 1 мл TPBSB-3% (PBS, содержащем 3% BSA, 0,05% Tween-20 и 0,02% NaN3), и блокировали инкубацией в течение 1 ч с 1 мл TPBSB-5% (PBS, содержащий 5% BSA, 0,05% Tween-20 и 0,02% NaN3) при комнатной температуре в течение 1 часа с вращением end-over-end. После промывки с 1 мл TPBSB-3% шарики переносили в чистую пробирку Эппендорф, и фаги добавляли к заблокированным шарикам (в общем объеме TPBSB-3%, равном 1 мл). Смесь инкубировали в течение ночи при 4°C с вращением end-over-end.
Для удаления несвязанных фагов шарики промывали три раза 1 мл TPBSB-3% с последующими тремя промывками с 10 мл TPBS (PBS, содержащий 0,05% Tween-20 и 0,02% NaN3) и тремя промывками с 10 мл PBS. До промывки TPBS шарики собирали, используя фильтр, и переносили в чистую пробирку объемом 50 мл, в которой были выполнены все последующие промывочные стадии. Для облегчения процедуры промывки за каждой стадией промывки следовала трехминутная стадия инкубации при комнатной температуре с вращением end-over-end.
Промытые шарики собирали с использованием сетчатого фильтра и переносили в чистую пробирку Эппендорф. Связанные фаги элюировали, инкубируя шарики с 400 мкл 0,5% трипсина при комнатной температуре в течение 30 минут с вращением end-over-end, с последующим добавлением 40 мкл апротинина (2 мг/мл) для инактивации трипсина. Смесь трипсин/апротинин, содержащую элюированные фаги, переносили в пробирку Эппендорф и промывали шарики с 200 мкл PBS, который впоследствии объединяли с элюированными фагами, в результате чего общий объем достигал 640 мкл.
Амплификация на планшетах для исходных растворов в глицерине и ночная культура для минипрепаратов
Данные объекты способа осуществляли, как описано в примере 1.
Преобразование из формата «связанный с фагом scFv» в формат «растворимый scFv»
Это способ преобразования, когда фрагмент scFv вырезают из фагмида (Amp) и встраивают в вектор pKscFv-3×FH, который несет ген, кодирующий устойчивость к канамицину.
Материал:
Avrll (4 ед/мкл, New England Biolabs Cat No R0174)
10×NEBuffer2 (New England Biolabs Cat No B7002S)
Вода MQ
SfiI (20 ед/мкл. New England Biolabs Cat No R0123)
10×BSA (100 x BSA New England Biolabs Cat No B9001S разбавляют 10× в воде MQ)
ПЦР-комплект очистки QIAquick PCR purification kit (Qiagen Cat No 28104)
pKscFv 3xFH, расщепленный SfiI/AvrII
ДНК-лигаза фага Т4 (1 ед/мкл, Invitrogen Cat No 15224-017)
5× буфер ДНК-лигазы фага Т4 (Invitrogen, Cat No P/N Y90001)
Расщепление фагмидной ДНК
- Подготовить реакции расщепления с использованием фагмидных минипрепаратов:
2 мкг фагмидного минипрепарата
1 мкл ⇒4ед AvrII
2 мкл ⇒1×10× NEBuffer2
MQ H2O до 20 мкл общего объема
- Инкубировать при 37°C в течение 2 часов.
- Продолжать с расщеплением SfiI. К смеси расщепления из вышеприведенного добавить:
1 мкл ⇒20 ед Sfil
5 мкл MQ H2O ⇒Общий объем составлял 30 мкл
- Инкубировать при 50°C в течение 2 часов.
Очищать расщепленную ДНК с использованием ПЦР-набора для очистки QIAquick PCR Purification Kit в соответствии с инструкциями поставщика. Использовать 50 мкл воды для элюции. Будет получено приблизительно 50 мкл с общей концентрацией, составляющей 40 нг/мкл (при условии 100%-го выхода).
Лигирование
- Составление смеси для последующего лигирования:
0,6 мкл ⇒60 нг pKscFv 3xFH, расщепленного SfiI/AvrII.
6 мкл ⇒240 нг минипрепарата расщепленный SfiI/AvrII/очищенный фагмид
5 мкл ⇒1× 5× лигазного буфера
1 мкл ⇒1ед ДНК-лигазы фага Т4
12,4 мкл MQ H2O ⇒Общий объем составлял 25 мкл
- Инкубировать при 16°C в течение ночи.
- Хранить при -20°C до использования.
Трансформация
Лигат, произведенный выше, трансформировали в ТОР10 E. coli.
Одну пробирку (100 мкл/пробирка)/лигат химически компетентных клеток ТОР10 оттаивали на льду. 10 нг лигата добавляли на одну пробирку, и пробирки инкубировали на льду в течение 30 мин.
Затем пробирки инкубировали при 42°C (водяная баня) в течение 90 с, и дополнительно инкубировали на льду в течение 5 мин.
900 мкл LB добавляли в каждую пробирку, и пробирки инкубировали при 37°C, 1 ч, 200 оборотов в минуту.
Содержание каждой пробирки распределяли на одном планшете площадью 500 см2 с LA (100 мкг/мл канамицина+1% глюкозы) и инкубировали в течение ночи при 37°C.
Сбор колоний
В общей сложности 1152 клонов было отобрано из каждой селекции (CD24, CD130 и HER2) из больших LA-планшетов в 3×384 луночные микропланшеты/селекция (всего 9 планшетов) с помощью системы сбора колоний Genetix «Q-bot».
1. Смешать LB среды, 20 мкг/мл канамицина и 1% глюкозы.
2. Заполнить планшеты (Greiner Flat 384 well 781101) средой, 60 мкл/лунка.
3. Собрать колонии с Q-Bot в соответствии с протоколом.
Экспрессия клонов в 384-луночном формате
1. Заполнить экспрессионный планшет (Greiner Flat 384 well 781101) средой, 50 мкл/лунка, включая 20 мкг/мл канамицина.
2. Инокулировать экспрессионный планшет с 5 мкл/лунка из основного планшета.
3. Инкубировать экспрессионный планшет при 37°C, 600 оборотов в минуту в течение 3,5 часов.
4. Индуцировать продукцию scFv с 10 мкл/лунка среды, включая 20 мкг/мл канамицина и 2,5 мМ IPTG.
5. Инкубировать экспрессионный планшет в течение 10 ч при 37°C, 600 оборотов в минуту.
6. Хранить экспрессионный планшет при 4°C.
Скрининг клонов 384 лунок
Материал:
CD24-GST (0,11 мг/мл), Abnova Cat No H00000943-H01
HER-2-Fc (0,1 мг/мл в PBS), R&D Systems Cat No 1129-ER
CD130 (0,2 мг/мл), R&D Systems Cat No 228-GP
человеческий IgG (6,2 мг/мл), Sigma Cat No 12511
Анти His-HRP, R&D Systems Cat No MAB050H
Анти FLAG-AP, Sigma Cat No A9469
Рыбий желатин, Sigma Cat No G7765
Supersignal ELISA Pico, Thermo Scientific Cat No 37069
Tropix CDP Star Emerald II, Applied Biosystems Cat No T2388C
Процедура:
День 1 Покрытие
Покрывают планшеты (Greiner 384 well plate white HB 781074) с:
CD24 0,4 пмоль/лунку
HER2 0,8 пмоль/лунку
CD130 0,9 пмоль/лунку
hIgG (нецелевой) 1 пмоль/лунку
разбавляют в 0,1 М карбонате натрия, pH 9,5, 50 мкл/лунка. Инкубировать планшеты в течение ночи при 4°C.
День 2
2. Промыть планшеты три раза с 1×PBS, 0,05% (об./об.) Твин-20.
3. Добавить PBS+0,05% Твин-20+0,45% рыбьего желатина, 40 мкл/лунка.
4. Добавить экспрессированный scFv, 10 мкл/лунку и инкубировать планшет при комнатной температуре в течение 1 часа.
5. Промыть планшет, как описано выше.
6. Добавить вторичное антитело, разбавленное в PBS+0,05% Tween-20+0,45% рыбьего желатина; α-HIS-HRP, разбавленный 1:4000, в планшеты, покрытые CD24, CD 130 и hIgG, α-FLAG-AP, разбавленный 1:25000, в планшеты, покрытые HER2 и hIgG, 50 мкл/лунку, и инкубировать планшет в течение 1 часа при комнатной температуре. Примечание: Продублировать планшеты для hIgG.
7. Промыть планшеты, как описано выше. Промыть планшеты три раза с 20 мМ Трис-HCl, 10 мМ MgCk, pH 9,8.
8. Добавить субстрат;
Для планшетов с анти-His-HRP добавить Pierce SuperSignal ELISA Pico, разведенный 1:20 в 20 мМ Трис-HCl, 10 мМ MgCl2, pH 9,8, 50 мкл/лунка, и инкубировать планшет в течение 10 мин при комнатной температуре.
К планшетам с анти-Flag добавить Tropix CDP Star Emerald II, разведенный 1:20 в 20 мМ Трис-HCl, 10 мМ MgCl2, pH 9,8, 50 мкл/лунка, и инкубировать планшет в течение 30 мин при комнатной температуре.
9. Считать показания с планшетов.
Анализ FACS экспрессии рецептора DU145 после стимуляции IFN-γ
Материал
Клетки Jurkat
Клетки DU145
Трипсин-ЭДТА (Invitrogen Cat No 25300-054)
rh-IFN-γ (R&D Systems Cat No 285-IF)
Блок мышиный IgG (Jackson Immunoresearch Cat No 015-000-002)
Буфер FACS (Gibco Cat No 14040 PBS w/o Ca и Mg+0,5% BSA)
Zenon Alexa Fluor 647 Human IgG Labeling Kit (Invitrogen Cat No Z25408)
Стимуляция INF-γ
Клетки DU145 стимулировали 2501Е INF-y в течение 24 часов. Клетки собирали с буфером клеточной диссоциации.
Способ проточной цитометрии
Клетки промывали один раз в FACS-буфере и проводили блокирование с использованием 50 мкг/мл мышиного IgG в FACS-буфере на льду в течение 10 минут. Между тем, антитела метили Zenon AF 647 в соответствии с инструкциями изготовителя.
50 мкл клеточной суспензии (приблизительно 500000 клеток) переносили в пробирки FACS и добавляли 50 мкл меченого антитела перед инкубацией в течение 1 ч на льду.
FACS-буфер затем использовали, чтобы промыть клетки, и полученную суспензию анализировали в проточном цитометре (BD Bioscience, FACS Calibur).
Анализ Скэтчарда экспрессии рецептора DU145 после стимуляции INF-γ
Стимуляция inf-γ
Клетки DU145 стимулировали 250IE INF-γ течение 24 часов. Клетки собирали с буфером клеточной диссоциации.
Анализ Скэтчарда
Антитела были помечены 125I с использованием свободного йода и тестируемых пробирок, предварительно покрытых окислительным реагентом lodogen (1,3,4,6-тетрахлор-3α,6α-дифенилглюколурил, Thermo Scientific), в соответствии с инструкцией производителя. Вкратце, 200 мкг антитела метили в течение 10 минут в PBS, и свободный йод удаляли с помощью небольшой одноразовой колонки для обессоливания (NAP 5, GE Healthcare Life Science). Меченые антитела имели удельную активность, составляющую приблизительно 2,5 мкКи/мкг антитела, и содержали менее, чем 1% свободного йода по оценкам с бумажной хроматографией.
0,5×106 клеток инкубировали в течение 2,5 ч (на ледяной бане) с различными концентрациями 125I - меченого антитела. Свободное несвязавшееся антитело (F) отделяли от связанного с клетками антитела (В) центрифугированием через 40% Ficoll-подушку и образцы анализировали в гамма-счетчике.
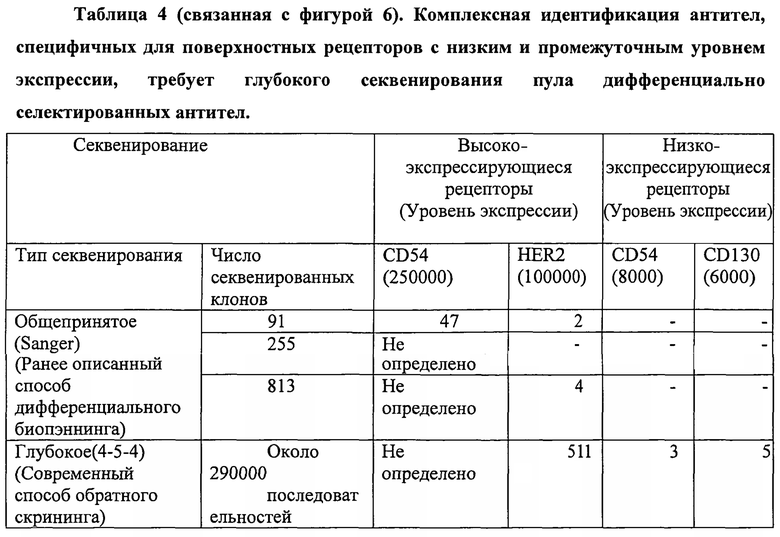
В таблице приведено число извлекаемых клонов антител, специфичных для каждого экспрессирующегося поверхностного рецептора в зависимости от числа секвенированных клонов антител. Рецепторные особенности отдельных клонов антител определяли, как описано на фигуре 6.
Аналогичный скрининг 96 клонов из пула дифференциально селектированных антител идентифицировал 47 последовательностей, специфичных для экспрессирующегося на высоком уровне поверхностного рецептора ICAM-1 (таблица 4).
Затем авторы задались вопросом, были ли идентифицированы антитела (последовательности), специфичные для поверхностных рецепторов, экспрессирующихся на высоком, промежуточном и низком уровнях, соответственно, секвенированием 91, 255 или 813 случайно отобранных клонов антител или последующим глубоким секвенированием около 290000 случайно отобранных клонов антител.
CD54 был идентифицирован анализом Скэтчарда как высоко экспрессирующийся поверхностный рецептор с 250000 копий/клетку (данные не показаны). Скрининг 96 клонов из пула дифференциально селектированных антител «DnonT» идентифицировал 47 клонов, специфичных для CD54 (таблица 4). HER2 с 100000 копий/клетку представляет собой высоко экспрессирующийся поверхностный рецептор, и CD24 с 8000 и CD130 с 6000 копий/клетку представляют собой низко экспрессирующиеся поверхностные рецепторы (фигура 4 и 5).
Антитела (последовательности), специфичные для высоко экспрессирующегося поверхностного рецептора ICAM-1, были определены всеми способами скрининга (таблица 4 и не показанные данные). В противоположность этому, никакие антитела, специфичные для низко экспрессирующихся поверхностных рецепторов, не были определены с помощью обычного скрининга вплоть до 813 случайно отобранных клонов (таблица 4). Поразительно, и в противоположность этому, антитела, специфичные для низко экспрессирующихся поверхностных рецепторов CD24 и CD130, были определены глубоким секвенированием.
Влияние увеличения глубины секвенирования на специфичность извлеченных антител к дифференциально экспрессирующимся поверхностным рецепторам с высоким, промежуточным и низким уровнем экспрессии исследовали следующим образом:
Были определены последовательности антител клонов в трех случайным образом селектированных пулах связывателей увеличивающегося размера (91 клонов, 255 клонов и 813 клонов, соответственно). После этого клоны, которые были найдены во всех трех пулах («изобильные клоны»), только в двух пулах увеличивающего размера («менее частые клоны») или только в самом большом пуле («редкие клоны») были проанализированы на связывание с клетками DU145 способом FACS.
Средние уровни целевой экспрессии рецепторов, служащих мишенью для изобильных, менее частых и редких клонов, уменьшаются с понижением распространенности в пуле дифференциально селектированных антител (фигура 7), свидетельствуя о том, что увеличение глубины секвенирования приводит к идентификации антител, специфичных к дифференциально экспрессирующимся поверхностным рецепторам, экспрессирующимся на уменьшающихся (более низких) уровнях на целевых (в сравнении с нецелевыми) клетках.
Пример 3 - Получение уравнений дифференциального биопэннинга
Применяя всеобщий закон действующих масс (Law of Mass Action (LMA)), можно рассчитать число лигандов, необходимых для выделения антилигандов к лигандам с низкой экспрессией и/или дифференциально экспрессирующимся лигандам из дисплейных библиотек высокой диверсификации.
LMA утверждает, что нековалентное (водородные связи, электростатические, Ван-дер-Ваальса или гидрофобные силы), обратимое связывание между антилигандом А и его целевым лигандом Т и их комплексом AT дается равновесным взаимодействием А+Т⇔AT с константой равновесной диссоциации или аффинности Kd=[А][Т]/[АТ].
Равновесное взаимодействие между антилигандами с идентичной специфичностью (А) для целевого лиганда (Т) может быть описано как
Связавшийся А (bА)⇔свободный A (fA)+свободный Т (fT)
с

Известно, что величина суммарного А или Т представляет собой сумму свободного и связанного А или Т
т.е. [А] (Суммарный А)=[fA]+[bA], и [Т] (Суммарный Т)=[fT]+[bA]
Поэтому, в (I), заменив [fA] на [А]-[bA] и [fT] на [T]-[bA]

Которое преобразовывается с образованием
(Kdx[bA])=([A][T]-[A][bA])-([T][bA]-[bA]2)
0=[bA]2-([A]+[T]+Kd[bA]+[A][T]
Данное одновременное уравнение имеет решение
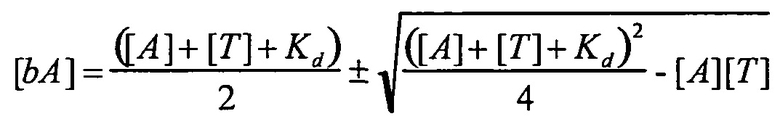
где негативный корень тот, который надо учитывать:
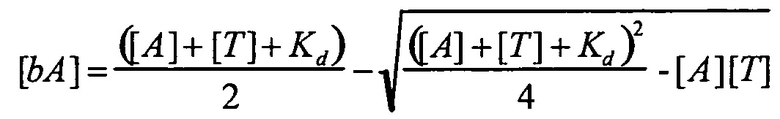
Подставление значений концентраций для числа частиц/ числа частиц на моль (С)/единица объема (V) дает
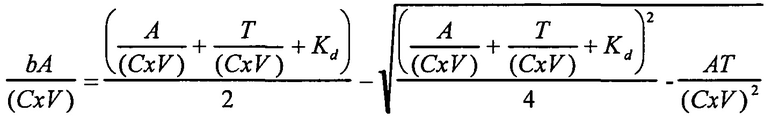
и упрощается до
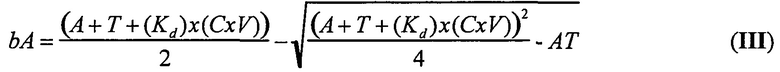
где A представляет собой общее число антилигандов А
T представляет собой общее число лигандов T
V представляет собой реакционный объем (литры)
C представляет собой постоянную Авогадро (6,022×1023 частиц/моль)
Учитывая, что LMA применяется к каждой реакции между различными антилигандами с заданной аффинностью и специфичностью в отношении их соответствующих целевых лигандов, число антилигандов, связанных с лигандами после выполнения способа селекции, может быть рассчитано с применением LMA и уравнения (III).
Кроме того, если качественное отличие между антилигандами, связанными с популяциями субтрактивных или целевых лигандов, отсутствует, то есть не имеется никаких изменений в физико-химических свойствах лиганда во время выполнения способа, то число антилигандов, которые связаны с целевыми лигандами при равновесии, будет равно общему числу связанных антилигандов, умноженному на отношение числа целевых лигандов на конструкциях целевых лигандов к общему числу лигандов (субтрактивный и целевой лиганд):
Вводя
Cp представляет собой число конструкций целевых лигандов
Cs представляет собой число конструкций субтрактивных лигандов
Тр представляет собой число Т лигандов на Ср
TS представляет собой число Т лигандов на Cs
Если целевые и субтрактивные конструкции смешиваются, то общее число лигандов будет составлять:
TTot=(ТP×Ср+TS×CS)
И число антилигандов (А), связанных с позитивными конструкциями при равновесии (bAP), определяется по формуле:

Дополнительно, объединение уравнений (III) и (IV) дает
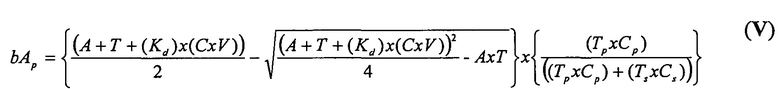
Пример 4 - Оптимизация концентраций лиганда
Уравнения, проиллюстрированные в примере 1, показывают, что использование высоких концентраций как первого субтрактивного лиганда, так и второго целевого лиганда способствует эффективному извлечению антилигандов со специфичностью к дифференциально экспрессирующимся лигандам с низким уровнем экспрессии, а также для уменьшения количества антилигандов со специфичностью к обычно экспрессирующимся лигандам.
Концентрация лиганда может быть увеличена несколькими способами. Во всех случаях концентрация лиганда увеличивается при переходе от двумерного связывания лиганда (связывания с двумерной твердой фазой) к применению свободного лиганда в суспензии или растворе (трехмерная твердая фаза).
В случаях, когда связывание зависит от лиганда, используемого в своей нативной конфигурации, например, для лигандов клеточной поверхности, концентрация лиганда максимизируется увеличением отношения площади поверхности конструкции лиганда к объему конструкции лиганда.
Например, антигены клеточной поверхности могут быть использованы в виде небольших везикул из плазматической мембраны, находящихся свободно в суспензии, в отличие от использования целых клеток, фиксированных к 2-мерной поверхности. Это имеет дополнительное преимущество, увеличивая стабильность лиганда в суспензии или растворе, способствуя тем самым равновесному взаимодействию лиганд-антилиганд.
Если источник лиганда имеет сферическую (или по существу сферическую) форму, это описывается математически следующим уравнением:
Ap/Vp=(4πr2)/(4πr3/3)=π/3r
Где Ар представляет собой площадь сферы
Vp представляет собой объем сферы
т.е. чем меньше радиус сферы, тем больше отношение лиганды/объем и источник лиганда более похож на частицы (похож на суспензию).
Пример 5 - Предпочтительный вариант осуществления
Предпочтительный объект данного изобретения относится к выделению антилигандов со специфичностью к антигенам клеточной поверхности в их нативной конфигурации и независимо от их природы (белок, углевод, липид, комплекс). Кроме того, связавшиеся антигены представляют собой те антигены, которые позитивно регулируются или экспрессируются исключительно на одном типе клеток по сравнению с другим (например, трансформированная раковая клетка, клетка, инфицированная вирусом/микробом/паразитом/грибком, или клетка, стимулированная другим агонистом или активированная другим инфекционным агентом, в сравнении с контрольными клетками).
Когда для селекции используются антилиганды, полученные из антител (например, антилиганды, кодирующие scFv, Fab или Fv), данный способ, одновременно со способом скрининга, генерирует кандидаты в терапевтические антитела, которые взаимодействуют с целевым антигеном в его нативной конфигурации на клеточной мембране.
Поскольку необходимы столь большие концентрации антигена, антиген используется в форме, которая не сдвигает равновесия реакции. Таким образом, антиген используется в формах, которые занимают минимум места и только незначительно увеличивают вязкость и силу сдвига.
Например, когда ищут антилиганды к антигенам клеточной поверхности, может быть использован способ конкурентного биопэннинга с использованием целых клеток-мишеней и избытка мембран субтрактивных клеток, смешанных с элементами высоко диверсифицированной молекулярной библиотеки антилигандов, с последующим разделением по плотности в градиенте Ficoll или Percoll/бычий сывороточный альбумин и избирательным выделением целевых клеток и антилигандов, специфичных для позитивно регулируемых целевых клеток и уникальных антигенов.
В данной методике популяция целевого лиганда (антиген) находится в виде целых клеток (высокая плотность) и субтрактивный лиганд (антиген) находится в виде везикул плазматической мембраны или энуклеированных клеток (низкая плотность).
Популяции целевого и субтрактивного антигенов смешивают с элементами высоко диверсифицированной молекулярной библиотеки контролируемым образом, основываясь на уравнениях, описанных в данном документе.
Например, когда 5×107 целых клеток-мишеней смешивают с везикулами клеточной мембраны 1×1010 субтрактивных клеток и смешивают с элементами высоко диверсифицированной молекулярной библиотеки с числом копий специфического антилиганда, равным 200 (обычно получаемые антилиганды с Kd=10-8 М при отборе на чистом антигене), то можно ожидать выделения антилигандов, специфичных для 10-кратного или большего количества позитивно регулируемых антигенов, включая те, которые экспрессируются при таких низких плотностях как 10000 на клетку-мишень.
Реакционную смесь инкубируют для достижения равновесия. После конкурентного биопэннинга элементы библиотеки, связанные с целевой популяцией, отделяют от несвязавшихся антилигандов и тех антилигандов, которые связались с контрольным субтрактивным антигеном, разделением плотностным центрифугированием, что приводит к обогащению фага, специфичного для высоко экспрессирующихся антигенов, присутствующих среди исследуемой популяции.
Если экспрессия желаемого целевого антигена выше в субтрактивной популяции, процесс идет в обратном направлении, так что популяция субтрактивного лиганда становится популяцией целевого антигена и наоборот.
Помимо создания антилигандов со специфичностью для дифференциально экспрессирующихся и уникальных лигандов, применение способа разделения по различной плотности в градиенте плотности имеет ряд преимуществ, включая:
- Физическое и пространственное отделение антилигандов, находящихся в комплексе с позитивным лигандом, от несвязавшихся антилигандов и антилигандов со специфичностью к лиганду, найденному в контрольной популяции.
- Промывка Ficoll увеличивает силу сдвига. Следовательно, такая промывка является более эффективной и необходимо меньше повторных промывок (раунды пэннинга), и наблюдается минимальная диссоциация специфически связавшихся (с более высокой аффинностью) антилигандов, представляющих интерес.
- Не требуется навешивание тега или химическая модификация клеток (сравните конкурентный биопэннинг на основе FACS (сортировка клеток с активированной флуоресценцией (fluorescence activated cell sorter)) или MACS (магнитная сортировка активированных клеток (magnetic activated cell sorter))), которые могли бы изменить конфигурацию/конформацию и/или композицию лиганда клеточной поверхности.
Пример 6 - Все мембранные везикулы как средство разделения
Целые клетки могут быть заменены мембранными везикулами, полученными в среде более высокой плотности, что позволяет использовать даже более высокие концентрации лиганда без ущерба для равновесной реакции.
Пример 7 - Тестирование эффектов стимулов на позитивную/негативную регуляцию лиганда.
Дополнительный вариант осуществления изобретения может быть использован для выделения антилигандов со специфичностью к клеточным лигандам, которые экспрессируются при очень низкой плотности лишь в небольшом числе клеток в изучаемой клеточной популяции.
Например, можно заподозрить, что некоторый стимул вызывает позитивную регуляцию или негативную регуляцию антигена клеточной поверхности, находящегося на неизвестной клеточной субпопуляции, присутствующей в крови.
Клетки, полученные из цельной крови, подвергнувшейся воздействию данного стимула, могут быть смешаны с плазматическими мембранами, полученными из цельной крови перед воздействием стимула, и может быть проведена реакция конкурентного биопэннинга, аналогичная описанной выше.
Пример - 8 - Диагностическое применение способа скрининга
Дополнительный вариант осуществления изобретения позволяет выделять из высоко диверсифицированных молекулярных библиотек антилиганды против лигандов, присутствующих в различных количествах в биологических образцах (например, плазма, моча, спинномозговая жидкость). Такие антилиганды могут впоследствии быть использованы, например, для анализа экспрессии белка и идентификации потенциальных биомаркеров.
Если используются достаточно высокие концентрации лигандов, способ по изобретению позволяет проводить селективное выделение антилигандов против активированных или уникальных лигандов при сравнении белковой композиции в двух различных образцах. В конечном итоге такой подход позволяет выделить антилиганды, специфичные для лигандов, которые более распространены в одной популяции по сравнению с другой популяцией, таким образом, что результат не зависит от относительных концентраций лигандов внутри популяции позитивного лиганда.
Из-за экстремальных концентраций лиганда, необходимых для осуществления действия последнего, лиганд предпочтительно должен быть использован в суспензии или растворе. Например, лиганд целевой популяции можно разделить и пометить тегом в нескольких различных положениях, чтобы минимизировать разрушение и уничтожение соответствующих лигандов, в то время как лиганды субтрактивной популяции могут быть использованы без тегов или фиктивной обработки (mock treatment). Мечение тегами популяции позитивного лиганда обеспечивает средство для последующего поиска лигандов позитивной популяции и связывателей, связавшихся с лигандами позитивной популяции только за счет использования помеченного лиганда, находящегося в комплексе, например, с помеченными счетчиком магнитными шариками.
Применение данного способа заключается в том, чтобы объединить образцы плазмы, полученные от популяции пациентов с некоторой болезнью, и сравнить с образцами плазмы из контрольной популяции. В данном случае образцы плазмы пациентов будут разделены и помечены тегами, а контрольная популяция не будет помечена тегами.
Источники информации
- Barbas CF, Kang As, Lemer RA and Benkovic SJ, (1991), Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc. Natl. Acad. Sci. USA 88, 7978-7982.
- Beck et al. (2010). Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 10, 345-352.
- Beers et al. (2008). Type II (tositumomab) anti-CD20 monoclonal antibody out performs type I (rimximab-like) reagents in В-cell depletion regardless of complement activation. Blood 112, 4170-4177.
- Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008; 456: 53-59.
- Carlsson, R. et al. (1988) Binding of staphylococcal enterotoxin A to accessory cells is a requirement for its ability to activate human Т cells. J Immunol 140, 2484.
- Chiswell DJ and McCafferty J, (1992), Phage antibodies: will new 'coliclonal' antibodies replace monoclonal antibodies? Trends Biotechnol. 10, 80-84.
- Clackson Т, Hoogenboom HR, Griffiths AD and Winter G, (1991), Making antibody fragments using phage display libraries. Nature 352, 624-628.
- Cragg, M.S., and Glennie, M.J. (2004). Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood 103, 2738-2743.
- Dower, W.J., CWIRLA, S.E. and N.V., A.T. (1991) Recombinant Library Screening Methods. In: World Intellectual Property Organization. SMITH, W.M., United States.
- Drmanac R, et al. Human Genome Sequencing Using Unchained Base Reads on Self-assembing DNA Nanoarrays. Science 2010; 327: 78-81.
- Felici F, Luzzago A, Manaci P, Nicosia A, Sollazzo M and Traboni C, (1995), Peptide and protein display on the surface of filamentous bacteriophage. Biotechnol. Annual Rev. 1, 149-183.
- Francisco, J.A., Stathopoulos, C., Warren, R.A., Kilbum, D.G. and Georgiou, G. (1993) Specific adhesion and hydrolysis of cellulose by intact Escherichia coli expressing surface anchored cellulase or cellulose binding domains. Biotechnology (N Y) 11, 491.
- Fransson, J., Tomberg, U.C., Borrebaeck, C.A., Carlsson, R., and Frendeus, B. (2006). Rapid induction of apoptosis in В-cell lymphoma by functionally isolated human antibodies. Int J Cancer 119, 349-358.
- Gao, C., Lin, C.H., Lo, C.H., Мао, S., Wirsching, P., Lemer, R.A. and Janda, K.D. (1997) Making chemistry selectable by linking it to infectivity. Proc Nati Acad Sci USA 94, 11777.
- Hanes, J. and Pluckthun, A. (1997) In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci USA 94, 4937.
- Harris TD, et al. Single-molecule DNA sequencing of a viral genome. Science 2008; 320: 106-109.
- He, M. and Taussig, M.J. (1997) Antibody-ribosome-mRNA (ARM) complexes as efficient selection particles for in vitro display and evolution of antibody combining sites. Nucleic Acids Res 25, 5132.
- Hoogenboom HR and Winter G, (1992), By-passing immunisation. Human antibodies from synthetic repertoires of germline VH gene segments rearranged in vitro. J. Mol. Biol. 227, 381-388).
- Hoogenboom HR, deBruine AP, Hutton SE, Hoet RM, Arends JW and Rooves RC, (1998), Antibody phage display technology and its application. Immunotechnology 4(1), 1-20.
- Hoogenboom, H.R. (2002). Overview of antibody phage display technology and its applications. In Methods in Molecular Biology P.M. O'Brien, and R. Aitken, eds. (Totowa, NJ: Humana Press Inc.), pp.1-37.
- Jacobsson, K. and Frykberg, L. (1995) Cloning of ligand-binding domains of bacterial receptors by phage display. Biotechniques 18, 878.
- Katz BA, (1997), Structural and mechanistic determinants of affinity and specificity of ligands discovered or engineered by peptide display. Annual Rev. Biophys. Biomol. Struct. 26, 27-45
- Kay BK and Paul JI, (1996), High-throughput screening strategies to identify inhibitors of protein-protein interactions. Mol. Divers. 1, 139-140).
- Koide, A., Bailey, C.W., Huang, X. and Koide, S. (1998) The fibronectin type III domain as a scaffold for novel binding proteins. JMol Biol 284, 1141.
- Liu et al. (2004). Mapping tumor epitope space by direct selection of single-chain Fv antibody libraries on prostate cancer cells. Cancer Res 64, 704-710.
- Lundquist PM, et al. Parallel confocal detection of single molecules in real time. Optics Letters 2008; 33: 1026-1028.
- Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005; 437: 376-380.
- Markland, W., Roberts, B.L., Saxena, M.J., Guterman, S.K. and Ladner, R.C. (1991) Design, construction and function of a multicopy display vector using fusions to the major coat protein of bacteriophage M13. Gene 109, 13.
- Marks JD, Hoogenboom HR, Borinert TP, McCafferty J, Griffiths AD and Winter G, (1991), By-passing immunisation. Human antibodies from V-gene libraries displayed on phage. J. Mol. Biol. 222, 581-597.
- Mattheakis, L.C., Bhatt, R.R. and Dower, W.J. (1994) An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc NatlAcadSci USA 91, 9022.
- McCafferty, J., Griffiths, A.D., Winter, G. and Chiswell, D.J. (1990) Phage antibodies: filamentous phage displaying antibody variable domains. Nature 348, 552.
- Mutubema et al. (1999). Model systems to study the parameters determining the success of phage antibody selections on complex antigens. J Immunol Methods 237, 65-81.
- Osboum et al. (1998). Pathfinder selection: in situ isolation of novel antibodies. Immunotechnology 3, 293-302.
- Shendure J, et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 2005; 309: 1728-1732.
- Siegel et al. (1997). Isolation of cell surface-specific human monoclonal antibodies using phage display and magnetically-activated cell sorting: applications in immunohematology. J Immunol Methods 206, 73-85.
- Smith, G.P. (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315.
- Smith, G.P. and Scott, J.K. (1993) Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol 217, 228.
- Stahl et al. (1989) A dual expression system for the generation, analysis and purification of antibodies to a repeated sequence of the Plasmodium falciparum antigen PH55/RESA. J Immunol Methods 124, 43.
- Weng, S., Gu, K., Hammond, P.W., Lohse, P., Rise, C., Wagner, R.W., Wright, M.C. and Kuimelis, R.G. (2002) Generating addressable protein microarrays with PROfusion covalent mRNA-protein fusion technology. Proteomics 2, 48.
- Williams, B.R. and Sharon, J. (2002) Polyclonal anti-colorectal cancer Fab phage display library selected in one round using density gradient centrifugation to separate antigen-bound and free phage. Immunol Lett 81, 141.
- Winter and McCafferty (1996) Phage display ofpeptides and proteins: a laboratory manual Ed Kay, Academic Press, Inc ISBN 0-12-402380-0.
- Winter, G., Griffiths, A.D., Hawkins, R.E. and Hoogenboom, H.R. (1994) Making antibodies by phage display technology. Annu Rev Immunol 12, 433.
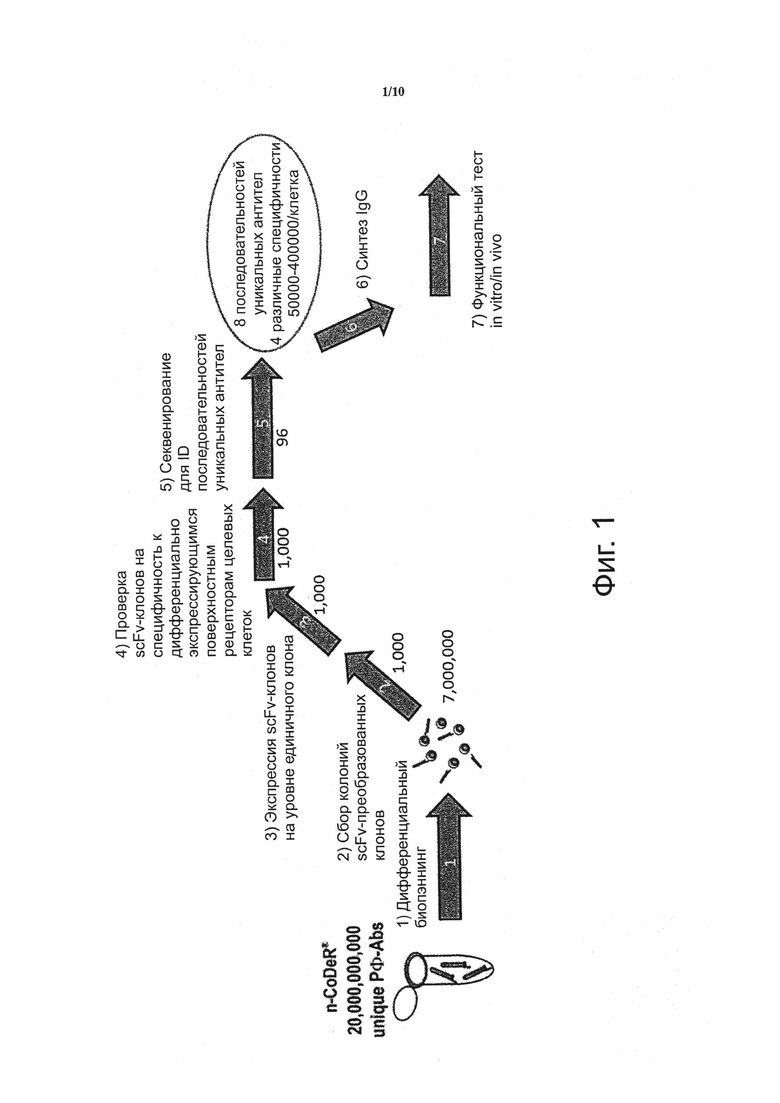

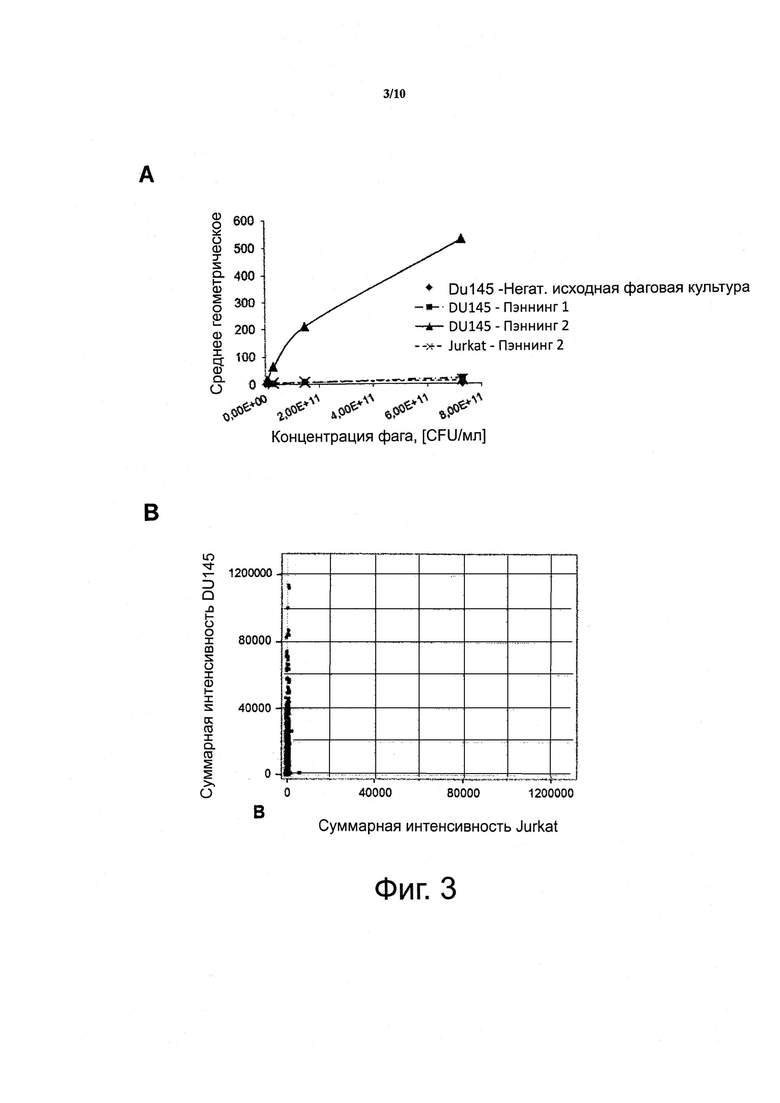
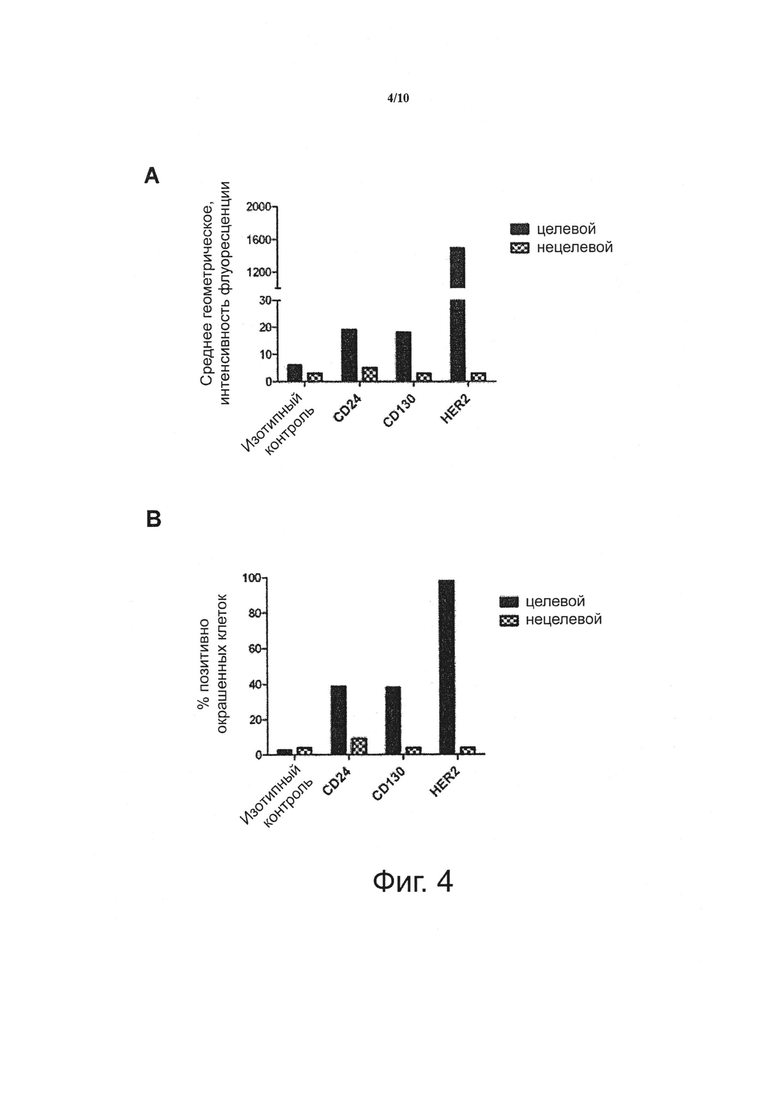
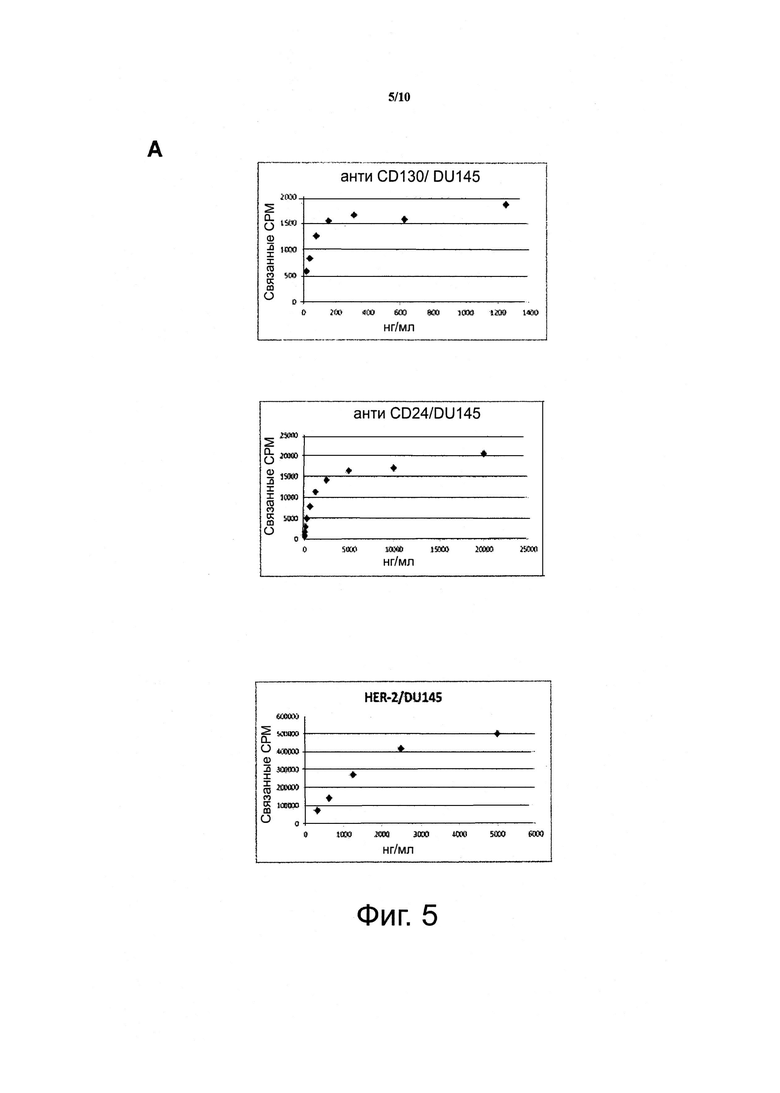

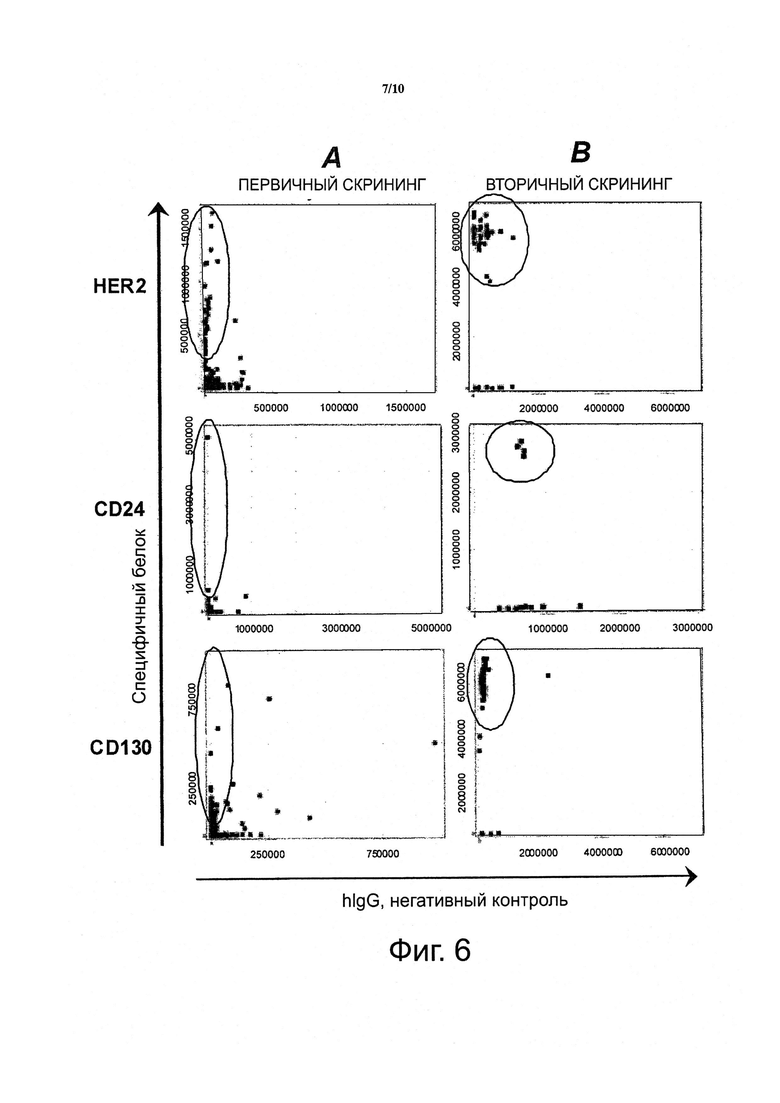
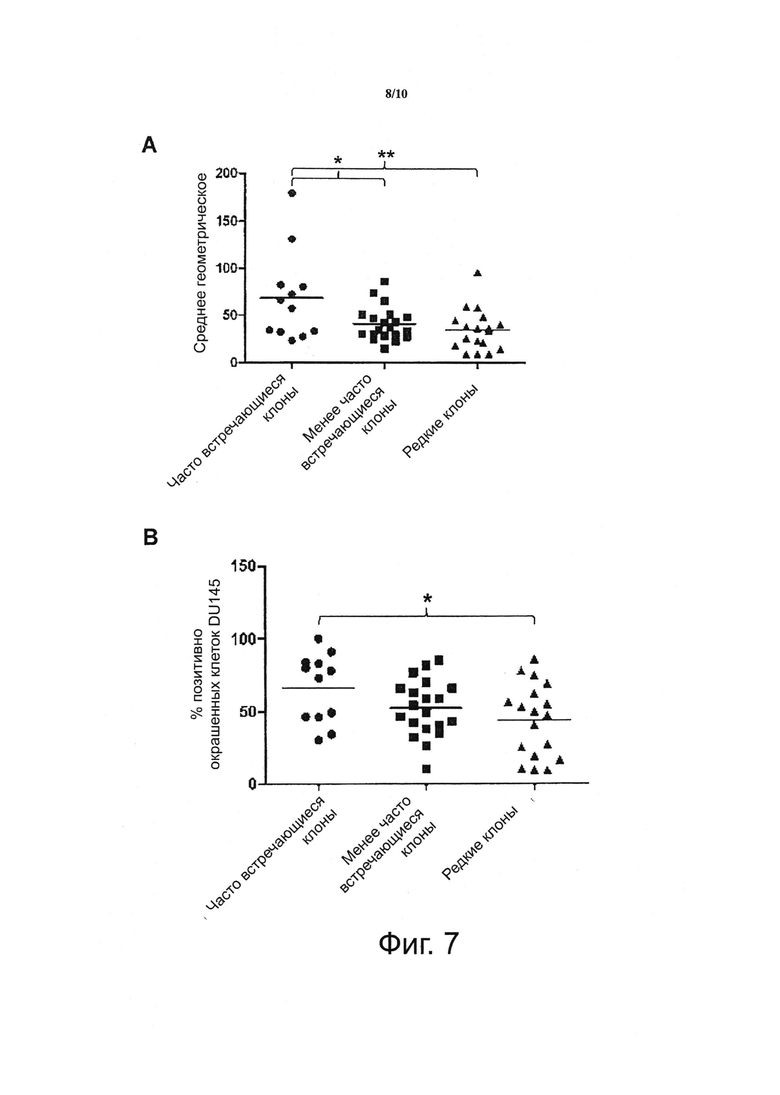
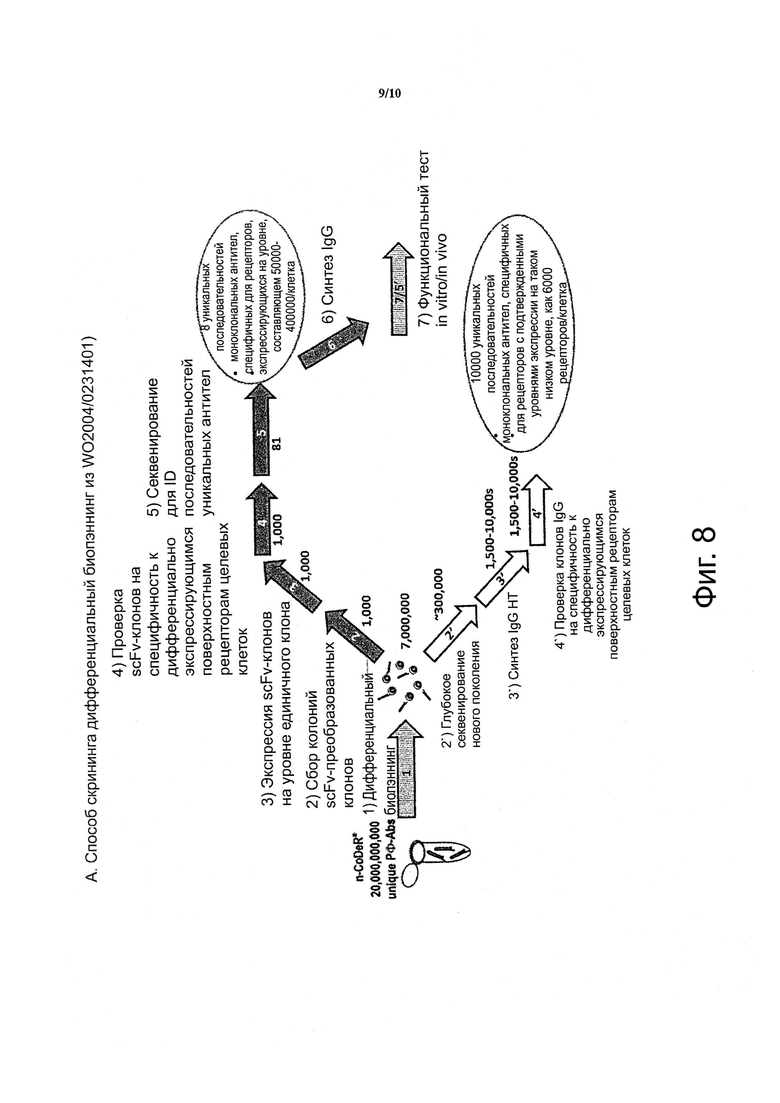
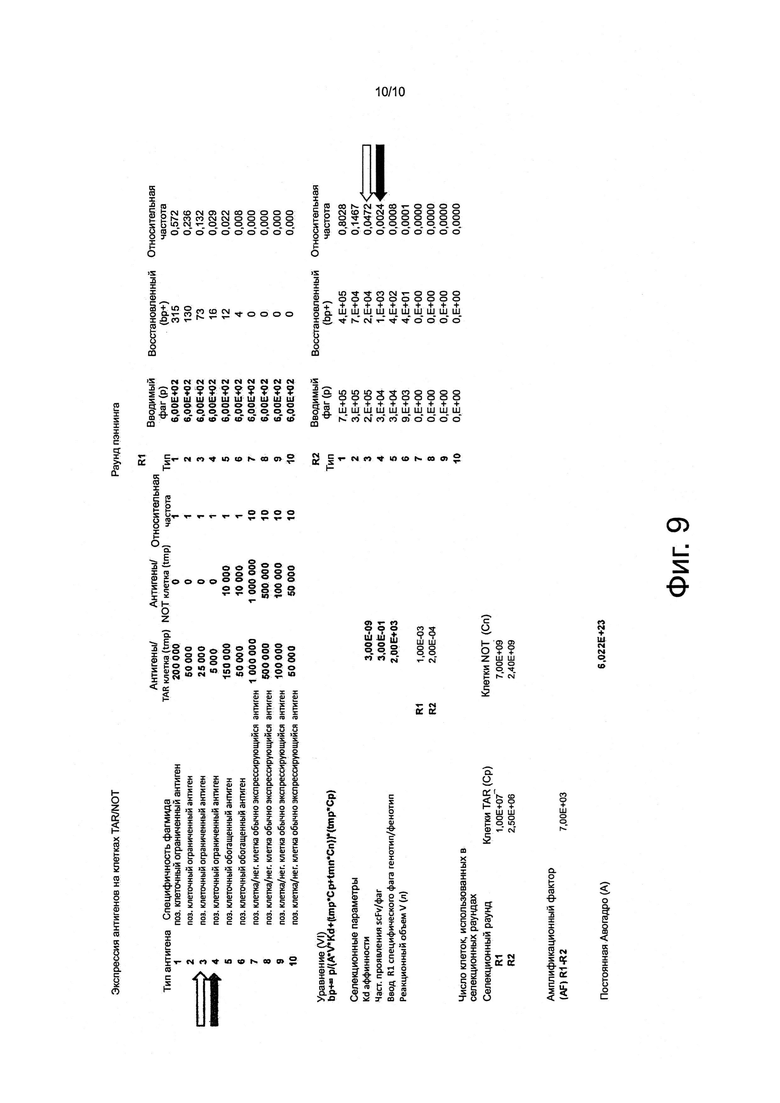
Настоящее изобретение относится к области биотехнологии и касается способа выделения антилиганда, представляющего собой антитело или его антигенсвязывающий вариант, производное или фрагмент, каркасную молекулу со сконструированными вариабельными поверхностями; рецептор или фермент, к дифференциально-экспрессирующемуся целевому лиганду. Представленный способ включает стадии: (а) выполнение дифференциального биопэннинга на библиотеке антилигандов с тем, чтобы выделить по меньшей мере один антилиганд; (b) выполнение секвенирования с высокой пропускной способностью на антилигандах, выделенных на стадии (а), и (c) выполнения подтверждающего скрининга на специфичность к антилиганду для дифференциально-экспрессирующегося лиганда. Представленное изобретение позволяет проводить скрининг библиотек антилигандов для идентификации антилигандов, специфичных для дифференциально и/или не часто экспрессирующихся лигандов. 17 з.п. ф-лы, 9 ил., 4 табл., 8 пр.
1. Способ выделения по меньшей мере одного антилиганда к по меньшей мере одному дифференциально-экспрессирующемуся целевому лиганду, включающий следующие стадии:
(а) выполнение дифференциального биопэннинга на библиотеке антилигандов с тем, чтобы выделить по меньшей мере один антилиганд; где указанные антилиганды выбирают из антител и их антигенсвязывающих вариантов, производных или фрагментов; каркасных молекул со сконструированными вариабельными поверхностями; рецепторов и ферментов, и
стадия дифференциального биопэннинга включает в себя подстадии:
(i) получение библиотеки антилигандов;
(ii) получение первой популяции лигандов, содержащей лиганд, фиксированный на или встроенный в конструкцию субтрактивного лиганда;
(iii) получение второй популяции лигандов, содержащей тот же лиганд, что и на стадии (ii), фиксированный на или встроенный в конструкцию целевого лиганда;
(iv) определение количества конструкций субтрактивного лиганда и конструкций целевого лиганда в популяциях с использованием одного или более уравнений, полученных из всеобщего закона действующих масс 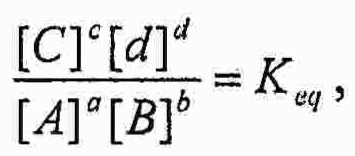
где:
А, В, С & D = представляют собой участников реакции (реагенты и продукты)
а, b, с & d = представляют собой коэффициенты, необходимые для сбалансированного химического уравнения
так, чтобы обеспечить выделение антилиганда к дифференциально экспрессирующемуся целевому лиганду;
(v) обеспечение количества конструкций субтрактивного лиганда, определенного в стадии (iv);
(vi) обеспечение количества конструкций целевого лиганда, определенного в стадии (iv);
(vii) обеспечение средств разделения для выделения анти-лиганда связанного с конструкцией целевого лиганда из конструкции анти-лиганда, связанного с конструкцией субтрактивного лиганда;
(viii) экспонирование библиотеки из (i) конструкциям лигандов, обеспеченных (v) и (vi), для осуществления связывания анти-лигандов с лигандами; и (ix) применения средств разделения для выделения антилиганда, связанного с лигандом, зафиксированным или включенным в конструкцию целевого лиганда
(b) выполнение секвенирования с высокой пропускной способностью на антилигандах, выделенных на стадии (а), и
(c) выполнения подтверждающего скрининга на специфичность к антилиганду для дифференциально-экспрессирующегося лиганда.
2. Способ по п. 1, в котором стадию секвенирования с высокой пропускной способностью проводят секвенированием 454, Illumina, способами SOLiD или системой Helicos.
3. Способ по п. 1, в котором стадию подтверждающего скрининга проводят проточной цитометрией, FMAT, ELISA, MSD или СВА.
4. Способ по п. 1, в котором лиганд не экспрессируется ни на одном из либо целевой конструкции или субтрактивной конструкции.
5. Способ по п. 1, включающий дополнительную стадию высвобождения антилиганда от лиганда.
6. Способ по п. 1, в котором стадии от (ii) до (ix) осуществляют параллельно, чтобы выделить множество антилигандов ко множеству различных лигандов.
7. Способ по п. 1, в котором стадии от (ii) до (ix) повторяют один или более раз.
8. Способ по п. 1, в котором количество одного из субтрактивной конструкции или целевой конструкции предоставляется в избытке относительно количества другого из субтрактивной конструкции или целевой конструкции.
9. Способ по п. 8, в котором избыток лиганда составляет от 10 до 1000 раз, или от 2 до 10 раз, или до 1000 до 1000000 раз.
10. Способ по п. 1, в котором уравнение стадии (iv) представляет собой или:
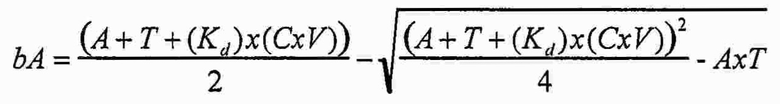
где
bA представляет собой связанный антилиганд (Bound anti-ligand)
А представляет собой общее число антилиганда
Т представляет собой общее число лигандов
С представляет собой постоянную Авогадро (6,022×1023 частиц/моль)
V представляет собой реакционный объем (литры)
Kd представляет собой равновесную константу диссоциации
или
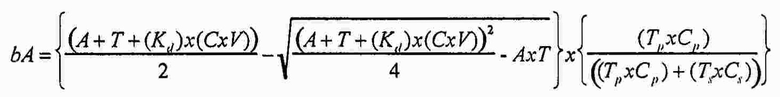
где
bAp представляет собой связанный антилиганд
Тр представляет собой число лигандов на Ср
Ts представляет собой число лигандов на Cs
Ср представляет собой число конструкций целевых лигандов
Cs представляет собой число конструкций субтрактивных лигандов
А представляет собой общее число антилиганда
Т представляет собой общее число лигандов
С представляет собой постоянную Авогадро (6,022×1023 частиц/моль)
V представляет собой реакционный объем (литры)
Kd представляет собой равновесную константу диссоциации
11. Способ по п. 1, в котором средство разделения выбирают из по меньшей мере одного из твердой подложки, клеточной мембраны и/или их участков, синтетической мембраны, шариков, химических меток и свободного лиганда.
12. Способ по п. 1, в котором средства разделения субтрактивной и целевой конструкций имеют различную плотность.
13. Способ по п. 1, в котором средство разделения субтрактивной конструкции представляет собой мембранную везикулу или целую клеточную мембрану.
14. Способ по п. 1, в котором стадию (ix) выполняют по меньшей мере одним из плотностного центрифугирования, секвестрации на твердой подложке, секвестрации на магнитном шарике, связыванием через химическую метку и разделением в водной фазе.
15. Способ по п. 1, в котором библиотека со стадии (а) представляет собой библиотеку дисплеев, содержащую множество элементов библиотеки, которые экспонируют антилиганды.
16. Способ по п. 1, в котором библиотека представляет собой библиотеку фагового дисплея.
17. Способ по п. 1, в котором лиганд представляет собой по меньшей мере одно, выбранное из антигенов; лигандов рецепторов; и мишеней ферментов, которые содержат по меньшей мере одно из углевода; белка; пептида; липида; полинуклеотида; неорганических молекул и конъюгированных молекул.
18. Способ по п. 1, включающий дополнительную стадию подвергания лиганда и средства разделения воздействию побудительного фактора, влияющего на экспрессию целевых лигандов на упомянутых конструкциях лигандов.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| СПОСОБ СКРИНИНГА БИБЛИОТЕКИ ФАГОВОГО ДИСПЛЕЯ | 2005 |
|
RU2402777C2 |

