Область техники
Изобретение относится в общем плане к рецепторам клеточной поверхности и, более конкретно, касается рецепторов глюкагона.
Предпосылки данного изобретения
Глюкагон представляет собой гормон из 29 аминокислот, продуцируемый α-клетками панкреатических островков. Глюкагон ответственен за поддержание нормальных уровней глюкозы во многих животных, в том числе в человеке, действуя как противодействующий инсулину гормон. В частности известно, что в то время как инсулин быстро снижает уровни глюкозы в крови, глюкагон уравновешивает эти эффекты, способствуя повышению уровней глюкозы в крови.
Взаимодействия глюкагона и инсулина очень важны для поддержания уровней глюкозы внутри тела. Считают, что дисбаланс глюкагона или инсулина играет роль в различных заболеваниях, таких как сахарный диабет и диабетический кетоацидоз. Согласно одной теории, гипергликемическое состояние сахарного диабета может быть вызвано не только снижением использования глюкозы (вследствие пониженного уровня инсулина), но также избыточным образованием глюкозы вследствие повышенных концентраций глюкагона (см. Under, "Diabetes and the alpha cell", Diabetes 25:136-151, 1976; Unger and Orci, "Тhе essential role of glucagon in the pathogenesis of diabetes mellitus". Lancet 1:14-16, 1975).
Важным фактором в изучении глюкагона и его роли в таких заболеваниях, как сахарный диабет, является рецептор глюкагона, который при связывании с глюкагоном переносит сигнал к клетке, запуская тем самым гликогенолиз (гидролиз гликогена) и глюконеогенез (синтез глюкозы).
В настоящее время считают, что эффекты глюкагона опосредованы отчасти повышением внутриклеточных уровней циклического аденозинмонофосфата (сАМР). В частности связывание глюкагона с его клеточным рецептором активирует аденилатциклазу для продуцирования сАMР, повышая таким образом уровни внутриклеточного сАМР. Считают, что это повышение внутриклеточных уровней сАМР приводит к гликогенолизу и глюконеогенезу и в конечном счете к повышению образования глюкозы печенью (см. Unson et al., "Biological Activities of des-HisI[Glu]9Glucagon Amide, a Glucagon Antagonist", Peptides 10:1171-1177, 1989).
Однако были предположения также о дополнительных путях стимулирования гликогенолиза и глюконеогенеза. В частности сообщалось, что глюкагон связывается с рецепторами в мембране гепатоцита (печеночной клетки), которые соединены через G-белок с фосфолипазой С. При стимулировании этот белок вызывает распад фосфатидилинозитол-4,5-бифосфата с образованием вторичных мессенджеров (посредников) инозиттрифосфата и 1,2-диацилглицерина (см. Wakelam et al. , "Activation of two signal-transduction systems in hepatocytes by glucagon", Nature 323: 68-71, 1986; Unson et al., Peptides 10:1171-1177, 1989; и Pittner and Fain, Btochem. J. 277:371-378, 1991). Стимулирование глюкагона метаболизмом инозитфофолипида может быть дополнительным путем, при помощи которого глюкагон может стимулировать гликогенолиз и глюконеогенез.
Данное изобретение описывает рецептор (рецепторы) глюкагона и, кроме того, обеспечивает другие связанные с этим объектом преимущества.
Краткое изложение сущности изобретения
Согласно одному аспекту данного изобретения предложены выделенные молекулы ДНК, кодирующие рецептор глюкагона. Применяемый здесь термин "изолированная молекула ДНК относится к молекулам ДНК или последовательностям ДНК, которые отделены, помещены отдельно от других клеточных компонентов. Например, молекула ДНК изолирована, если она отделена от других молекул ДНК, в том числе от других хромосомных последовательностей, с которыми она природно ассоциирована в геноме и, в частности, свободна от других структурных генов. Изолированная молекула ДНК может содержать 5'- и 3'- нетранслируемые последовательности, с которыми она природно ассоциирована. В одном из вариантов изобретения рецептор глюкагона выбран из группы, состоящей из крысиных и человеческих рецепторов глюкагона. В другом варианте молекула ДHК содержит последовательность нуклеотидов SEQ ID 14, от нуклеотида 145 до нуклеотида 1599. В другом варианте молекула ДНК кодирует рецептор глюкагона, содержащий аминокислотную последовательность SEQ ID 15, от метионина, номер аминокислоты 1, до треонина, номер аминокислоты 485. В следующем варианте молекула ДНК содержит последовательность, нуклеотидов SEQ ID 24, от нуклеотида 53 до нуклеотида 1486. Еще в одном варианте молекула ДНК кодирует рецептор глюкагона, содержащий последовательность аминокислот SEQ ID 25, от метионина, номер аминокислоты 1, до фенилаланина, номер аминокислоты 477. Также обеспечены конструкции ДНК, содержащие первый сегмент ДНК, кодирующий рецептор глюкагона, оперативно связанный с дополнительными сегментами ДНК, необходимыми для экспрессии первого сегмента ДНК, клетки-хозяева, содержащие такие конструкции ДНК, а также способы получения рецептора глюкагона, предусматривающие стадию культивирования клетки-хозяина при условиях, способствующих экспрессии сегмента ДНК, кодирующего рецептор глюкагона.
Согласно другому аспекту изобретения предложены изолированные пептиды рецептора глюкагона. Согласно одному варианту предложен изолированный пептид рецептора глюкагона, содержащий последовательность аминокислот SEQ ID 15, от глутамина, аминокислоты 28, до тирозина, номер аминокислоты 142.
Согласно другому аспекту изобретения предложены изолированные антитела, специфически связывающиеся с рецепторами глюкагона. В одном варианте эти антитела представляют собой моноклональные антитела. В дополнительном варианте обеспечены моноклональные антитела, способные блокировать связывание глюкагона с рецептором глюкагона. Также обеспечены гибридомы, продуцирующие описанные выше моноклональные антитела.
Еще в одном аспекте данного изобретения обеспечен способ обнаружения присутствия антагонистов глюкагона, предусматривающий стадии (а) экспонирования соединения в присутствии агониста глюкагона с рекомбинантным рецептором глюкагона, связанным с путем ответной реакции, при условиях и в течение времени, достаточных для того, чтобы произошло связывание соединения с рецептором и произошла ассоциированная ответная реакция через путь метаболизма и (b) детектирования уменьшения в стимулировании ответного пути, вызываемого связыванием испытуемого соединения с рецептором глюкагона, по сравнению со стимулированием ответного пути одним агонистом глюкагона, и определение из этих данных присутствия антагониста глюкагона.
В различных вариантах изобретения ответным путем является ответная реакция мембраносвязанной аденилатциклазы и стадия детектирования предусматривает измерение уменьшения продуцирования циклического АМР в ответном пути, медиируемом мембраносвязанной аденилатциклазой. В другом варианте изобретения ответная реакция включает в себя люциферазную репортерную систему.
Еще в одном аспекте данного изобретения обеспечены зонды по меньшей мере из 12 нуклеотидов, способные гибридизоваться с нуклеиновыми кислотами, кодирующими рецептор глюкагона.
Эти и другие аспекты данного изобретения станут понятными в ходе дальнейшего детального описания со ссылками на прилагаемые чертежи. Кроме того, различные ссылки изложены ниже, описывающие в больших деталях определенные процедуры или композиции (например, плазмиды и т.д.) и поэтому даваемые здесь в виде полных ссылок.
Краткое описание чертежей
Фиг.1 иллюстрирует структуру типичного рецептора глюкагона. Использованы следующие символы: ЕАТD (внеклеточный аминоконцевой домен); ЕD (эффекторный домен), окруженный пунктирной линией; 1ID, первый внутриклеточный петлевой домен; 2lD, второй внутриклеточный петлевой домен; 3ЕlD, третий внутриклеточный петлевой домен; С-ID, карбоксиконцевой внутриклеточный домен; IELD, первый домен внеклеточный петлевой домен; 2ELD, второй внеклеточный петлевой домен; 3ELD, третий внеклеточный петлевой домен; TMDI, первый трансмембранный домен; ТМD2, второй трансмембранный домен; ТМD3, третий трансмембранный домен; ТМD4, четвертый трансмембранный домен; TMD5, пятый трансмембранный домен; ТМD6, шестой трансмембранный домен и ТМD7, седьмой трансмембранный домен.
Фиг.2 графически изображает гидрофобность крысиного рецептора глюкагона.
Фиг. 3 графически изображает связывание 125I-глюкагона с рецепторами глюкагона.
Фиг. 4 представляет собой анализ Скетчарда кажущейся Кd для рецепторов глюкагона.
Фиг. 5 дает аминокислотную последовательность крысиного рецептора глюкагона с линиями над трансмембранными доменами.
Детальное описание изобретения
Как отмечено выше, данное изобретение обеспечивает изолированные молекулы ДНК, кодирующие рецепторы глюкагона. Считают, что в их природной конфигурации рецепторы глюкагона существуют в виде мембраносвязанных белков, состоящих из внеклеточного аминоконцевого домена, а также нескольких наружных и внутренних доменов меньшей величины (см. фиг.1). В контексте данного изобретения "рецептором глюкагона" называют такие белки и подобные им производные. Производные могут быть аллельными разновидностями и генетически сконструированными вариантами, содержащими консервативные аминокислотные замены и (или) минорные добавления, замены или делеции аминокислот. Рецепторы глюкагона данного изобретения способны связывать глюкагон и осуществлять трансдукцию этого сигнала в клетку. Предпочтительно, рецепторы глюкагона данного изобретения способны связывать глюкагон с Кd 100 нМ или менее, более предпочтительно 50 нМ или менее и наиболее предпочтительно 33 нМ или менее. Типичные тесты, которые могут быть использованы для определения связывания глюкагона рецептором глюкагона, описаны более детально в примерах 3 и 6.
Обычно трансдукция сигнала происходит при активации ответного пути метаболизма наружным стимулом, который обычно, но не всегда, связывается с мембраносвязанным рецептором. Отвечающие на стимул пути обычно индуцируют клеточные ответные реакции, такие как внеклеточная матриксная секреция восприимчивых клеточных линий, секреция гормона, хемотаксис, дифференциация, или инициация, или ингибирование клеточного деления восприимчивых клеток. Сопряжением рецепторов с восприимчивыми к исследуемому сигналу путями называют здесь прямую активацию отвечающего на сигнал пути или трансдукцию сигнала через вторичный мессенджер, такой как G-белок, для активации пути клеточного ответа.
Многие пути клеточного ответа могут быть использованы рецепторами глюкагона для трансдукции сигнала связывания глюкагона к клетке, в том числе, например, путь аденилатциклазного ответа и путь ответа внутриклеточного кальция.
Тесты определения аденилатциклазной активности хорошо известны в этой области исследований, например, описанные Lin et al. (Biochemistry 14: 1559-1563, 1975). Данное изобретение также обеспечивает измерение биологической активности рецептора глюкагона на основе концентраций внутриклеточного кальция (см. Grynkiewicz et аl., J.Biol.Сhеm. 260:3440-3450, 1985), а также путем применения люцифразной репортерной системы, описанной в больших деталях ниже. Кроме того, биологические ответные реакции, осуществляющиеся через инозит-трифосфатный путь, могут оцениваться путем измерения метаболизма инозиттрифосфата, как описано в Subers and Nataanson (J.Mol.Cell.Cardiol. 20: 131-140, 1988) или Pittner and Fain. (Biochem.J. 277:371-378, 1991). Следует отметить, что в контексте данного изобретения не все отвечающие на сигнал пути обязательно должны присутствовать для того, чтобы рецептор глюкагона переносил сигнал в клетку. Например, некоторые клеточные ответные реакции, такие как увеличение уровней внутриклеточного кальция, могут инициироваться связыванием глюкагона с его рецептором в отсутствие сАМР- или инозитфосфатных сигналов.
Изолирование (выделение) клонов кДНК рецептора глюкагона
Как отмечено выше, данное изобретение обеспечивает изолированные молекулы ДHK, кодирующие рецепторы глюкагона. Вкратце, геномные или кДНК-молекулы, кодирующие рецепторы глюкагона, могут быть получены из библиотек, приготовленных из клеток и тканей согласно процедурам, описанным ниже и в примерах. Клетки и ткани, которые можно использовать в рамках данного изобретения, можно получать из множества млекопитающих, в том числе, например, из человека, макак, крупного рогатого скота, свиней, лошадей, собак, крыс и мышей. Предпочтительными клетками и тканями являются жировая ткань, почки, поджелудочная железа, сердце и печень.
В одном аспекте изобретения крысиный рецептор глюкагона может быть изолирован и клонирован при помощи описанных здесь процедур. Вкратце, поли(А)+-РНК выделяли из Sрrаguе Dаwlеу крыс и использовали в качестве матрицы для синтеза кДНК в основном, как описано Ноuаmеd еt аl. (Sсiеnсе 252:1318-1321, 1991), для получения кДНК полной длины. Затем библиотеку, содержащую приблизительно 1•106 клонов, конструировали в экспрессирующей плазмиде клеток млекопитающих путем направленного клонирования кДНК, больших, чем 800 н. п. Затем плазмидные ДНK, полученные из пулов, содержащих 5000 клонов, трансфицировали в COS-7 клетки, отбирали и выращивали на предметных стеклах для микроскопа. Трансфицированные клетки анализировали после 72 часов путем связывания с 1251-глюкагоном с последующей эмульсионной радиографией (McMahan et аl., ЕМВО J. 10:2821-2832, 1931). Положительные пулы последовательно разбивались, пока не был изолирован отдельный клон. Плазмида, полученная из этого клона, обозначенная как рLJ4, содержит приблизительно 2,0 т. п. н. инсерцию, которая кодирует белок из 485 аминокислот с предсказанной мол. массой 54962 дальтон (см. SEQ ID 15).
В других аспектах данного изобретения обеспечены способы выделения и клонирования человеческого рецептора глюкагона. Многие технологии можно использовать для обеспеченного здесь способа, в том числе, например, применение полимеразной цепной реакции (РСR) для амплификации последовательностей, кодирующих рецептор глюкагона (пример 4), которые затем могут быть использованы в идентификации библиотек, которые содержат последовательности, кодирующие человеческий рецептор глюкагона с последующим клонированном этого рецептора (пример 5). Особенно предпочтительными стратегиями для клонирования человеческого рецептора глюкагона являются стратегии, изложенные в примерах 4 и 5. Альтернативно экспрессирующая библиотека, содержащая человеческие кДНК, может быть получена из подходящих источников РНК, описанных в примере 1 и скринированных, как описано в примере 3 для клонов, экспрессирующих функциональные рецепторы глюкагона.
Получение рекомбинантных рецепторов глюкагона
Данное изобретение обеспечивает получение рекомбинантных рецепторов глюкагона путем культивирования клеток-хозяев, содержащих конструкцию ДНК, содержащую первый сегмент ДНК, кодирующий рецептор глюкагона, оперативно соединенный с дополнительными сегментами ДНК, необходимыми для экспрессии первого сегмента ДНК. Как отмечено ранее, в контексте данного изобретения под рецепторами глюкагона подразумевают и их производные, в основном сходные с рецепторами. Кроме того, рецепторы глюкагона могут кодироваться последовательностями ДНК, в основном сходными с описанными здесь последовательностями ДНК. Последовательность ДНК считают "в основном сходной", если: (а) эта последовательность ДНК произведена из кодирующего района нативного гена рецептора глюкагона (в том числе, например, аллельные вариации описанных ниже последовательностей); (b) эта последовательность ДНК способна гибридизоваться с последовательностями ДНК данного изобретения при высокой или низкой строгости (см. Sambrook еt аl., Моlесulаr Cloning: A Lаbоrаtоrу Маnuаl, 2d Ed., Соld Sрring Наrbоr Laboratory Рrеss, V, 1989); или (c) последовательности ДНК являются вырожденными в результате вырожденности генетического кода до последовательностей ДНК, описанных в (а) или (b).
Мутации в нуклеотидных последовательностях, сконструированных для экспрессии вариантных рецепторов глюкагона, должны сохранять рамку считывания кодирующих последовательностей. Кроме того, мутации предпочтительно не должны создавать комплементарные районы, которые могли бы гибридизоваться с образованием вторичных структур мРНК, таких как петли или шпильки, которые могут неблагоприятно влиять на трансляцию мРНК рецептора. Хотя сайт мутации может быть заданным, не обязательно, что природа мутации per Se должна быть заранее определена. Например, для отбора оптимальных характеристик мутантов при данном сайте мутагенез можно проводить на кодоне-мишени с последующим скринингом мутантных рецепторов глюкагона на биологическую активность.
Мутации могут быть введены при определенных локусах путем синтеза олигонуклеотидов, содержащих мутантную последовательность, фланкированную сайтами рестрикции, позволяющими лигирование с фрагментами нативной последовательности. После лигирования полученная последовательность кодирует производное, имеющее желаемые аминокислотные инсерцию, замену или делению.
Альтернативно, процедуры олигонуклеотиднаправленного сайтспецифического мутагенеза могут быть использованы для обеспечения измененного гена, имеющего измененные определенные кодоны в соответствии с желаемыми заменой, делецией или инсерцией. Примеры получения изложенных выше изменений описаны Walder еt аl. (Gеnе 42:133-1986); Bauer еt аl. (Gеnе 37:73, 1985); Craik (Вiо Techniques, Jаnuаry 1985, 12-19); Smith еt аl. (Gеnеtiс Еngineering: Рrinсiрlеs and Methods, Plenum Press, 191); and Sаmbrook et аl. (Supra).
Первичная аминокислотная структура рецептора глюкагона также может быть модифицирована путем образования ковалентных или агрегированных конъюгатов с другими химическими частями молекулы, такими как гликозильные группы, липидные, фосфатные, ацетильные группы или с другими белками, или полипептидами. В следующем варианте рецепторы глюкагона могут быть слиты с другими пептидами, облегчающими очистку или идентификацию рецепторов глюкагона. Например, рецептор глюкагона можно получить в виде слитого белка с FLAG-полипептидной последовательностью (см. U. S. Раtеnt 4851341; см. также Норр еt аl., Bio/Technology 6:1204, 1988). FLAG-полипептидная последовательность обладает высокой антигенностью и обеспечивает эпитоп для связывания специфическими моноклональными антителами, делая возможной быструю очистку экспрессируемого рекомбинантного белка. Эта последовательность также специфически расщепляется энтерокиназой бычьей слизистой оболочки при остатке, следующем непосредственно после пары Аsр-Lys. Для удобства могут быть приготовлены многочисленные конструкции ДНК, включающие в себя полные последовательности или части последовательностей нативного или вариантного рецепторов глюкагона, обсужденных выше. В контексте данного изобретения конструкцией ДНК называют молекулу ДНК или клон такой молекулы (одноцепочечной или двухцепочечной), которая была модифицирована таким образом, что она содержит сегменты ДНК, объединенные и помещенные рядом таким образом, что образуется молекула, которая не существовала в природе. Конструкции ДHК данного изобретения содержат первый сегмент ДНК, кодирующий рецептор глюкагона, оперативно соединенный с дополнительными сегментами ДНК, необходимыми для экспрессии первого сегмента ДНК. В контексте данного изобретения дополнительные сегменты ДНК в основном будут включать в себя промоторы и терминаторы транскрипции и могут, кроме того, содержать энхансеры и другие элементы.
Конструкции ДНК, известные также как экспрессирующие векторы, могут также содержать сегменты ДНК, необходимые для управления секрецией целевого полипептида. Такие сегменты ДНК могут содержать по меньшей мере одну секреторную сигнальную последовательность. Предпочтительными секреторными сигналами являются секреторная сигнальная последовательность глюкагона (пре-пропоследовательность), сигнальная последовательность альфа-фактора (пре-пропоследовательность; Kurian аnd Herskowitz, Cell 30:933-943, 1982; Kurian еt аl. , U.S. Раtеnt 4546082; Brake, EP 116 201), сигнальная последовательность РН05 (Весk еt аl., WO 86/00637), секреторная сигнальная последовательность BAR I (MасКаy еt аl., U.S. Раtеnt 4613572; MacKay WO 87/002670), сигнальная последовательность SUC2 (Carlson et al., Mol. Cell. Biol. 3:439-447, 1983), сигнальная последовательность α-1-антитрипсина (Кurасhi еt аl., Рrос. Nаtl. Acad. Sci. USA 78:6826-6830, 1981), сигнальная последовательность ингибитора плазмина α-2 (Тоnе еt аl., J. Biochem. (Tokyo) 102:1033-1042, 1987), сигнальная последовательность тканевого активатора плазминогена (Реnniса еt аl. , Nаture 301:214-221, 1983) сигнальная последовательность PhoA Е. соli (Yuаn еt al. , J.Biol. Сhem. 265:13528-13552, 1990) или любая из бактериальных сигнальных последовательностей, обзор которых дан, например, Оlivеr (Аnn. Rеv. Miсrоbiоl. 39:615-649, 1985). Альтернативно, секреторная сигнальная последовательность может быть синтезирована согласно установленным правилам, например, Heinje (Eur. J.Biochem. 133:17-21, 1983; J. Mol. Biol. 184: 99-105, 1985; Nuc. Acids Res. 14:4683-4690, 1986).
Секреторные сигнальные последовательности могут быть использованы по отдельности или могут быть скомбинированы. Например, первая секреторная сигнальная последовательность может применяться с последовательностью, кодирующей третий домен Ваrriеr (описанный в U.S. Раtеnt 5037243, на который дается полная ссылка). Последовательность, кодирующая третий домен Ваrriеr может быть помещена в подходящей рамке считывания 3' от целевой последовательности ДНК или 5' по отношению к сегменту ДНК и в подходящей рамке считывания как с секреторной сигнальной последовательностью, так и с целевым сегментом ДНК.
Для экспрессии молекулу ДНК, кодирующую рецептор глюкагона, встраивают в подходящую конструкцию ДНК, которую в свою очередь используют для трансформации или трансфекции подходящих клеток-хозяев для экспрессии. Клетки-хозяева для применения в практике данного изобретения представляют собой клетки млекопитающих, птиц, растений, насекомых, бактериальные и грибные клетки. Предпочтительными эукариотическими клетками являются культивируемые линии клеток млеокпитающих (например, клеточные линии грызунов или человека) и грибные клетки, в том числе виды дрожжей (например, Saccharomyces sрр., в частности S. cerevisiae, Schizosaccharomyces spp. или Kluyveromyces spp.) или нитчатые грибы (например, Aspergillus spp., Neurospora spp.). Особенно предпочтительны штаммы дрожжей Saccharomyces cerevisiae. Способы получения рекомбинантных белков во множестве прокариотических и эукариотических клеток-хозяев известны специалистам в этой области (см. "Gеnе Expression Technology", Methods in Еnzymоlоgу, Vol. 185, Goeddel (ed.), Асаdеmic Press, San Diego, Calif., 1990; см. также, "Guide to Yеаst Genetics and Molecular Вiоlоgy", Methodk in Enzyrmology, Guthrie and Fink (ed.) Academic Press, Sаn Diego, Calif., 1991). Как правило, клетку-хозяина выбирают на основе ее способности продуцировать целевой белок на высоком уровне или на основе ее способности проводить по меньшей мере некоторые из стадий процессинга, необходимых для биологической активности этого белка. Таким путем число клонированных последовательностей ДНК, которое должно быть трансфицировано в клетку-хозяин, может быть сведено к минимуму, и общий выход биологически активного белка может быть оптимизирован.
Подходящими дрожжевыми векторами для применения в данном изобретении являются векторы YRр7 (Struhl еt аl., Рrос. Nаtl. Acad. Sci. USA 76:1035-1039, 1978), YЕр13 (Вrоасh еt al., Gene 8:121-133, 1979), POT (Kawasaki et аl. , U. S. Patent 4931373, который включен здесь в виде ссылки) pJDB249 и pJDВ219 (Beggs, Nature 275:104-108, 1978) и их производные. Такие векторы обычно содержат селектируемый маркер, который может быть одним из любого числа генов, проявляющих доминантный фенотип, для которого существует фенотипический тест, позволяющий проводить отбор трансформантов. Предпочтительными являются такие селектируемые маркеры, которые дополняют ауксотрофию клетки-хозяина, обеспечивают устойчивость к антибиотикам или делают клетку способной использовать специфические источники углерода и включают в себя LЕU2 (Broach et al., ibid.), URА 3 (Botstein et al., Gene 8:17, 1979), HIS 3 (Struhl et аl., ibid.) или РОТI (Каwаsакi et al., ibid.). Другим пригодным селектируемым маркером является CAT ген, который сообщает дрожжевым клеткам устойчивость к хлорамфениколу.
Предпочтительными промоторами для применения в дрожжах являются промоторы из дрожжевых гликолитических генов (Нit Zeman et al., J.Biol. Сhеm. 255: 12073-12080, 1980; Аlbеl and Каwаsакi, J.Моl. Аррl. Gеnеt. 1:419-434, 1982; Каwаsакi., U.S. Patent 4599311) или гены алкогольдегидрогеназы (Young еt аl. , in Genetic Engineering of Microorganisms оf Сhеmiсаl, Hollaender et al., (ads. ), p.355, Plenum, Nеw Yоrk, 1982; Ammеrеr, Meth. Еnzymol. 101:192-201, 1983). В этой связи особенно предпочтительными промоторами являются TPLI промотор (Kawasaki, U.S. Раtеnt 4599311, 1986) и ADH2-4C промотор (Pussel et аl. , Nature 304:652-654, 1983; Irani and Kilgore, U.S. Раtеnt Аррliсаtion Serial 07/764653, который включен здесь ссылкой). Единицы экспрессии могут также содержать терминатор транскрипции. Предпочтительным является TPLI терминатор транскрипции (Alber and Kawasaki, ibid.).
Кроме дрожжей белки данного изобретения могут экспрессироваться в нитевидных грибах, например в штаммах гриба Asper-gillus (McKnight et al., U.S. Раtеnt 4935-349, включен в ссылки). Примерами применимых промоторов являются промоторы, произведенные из гликолитических генов Aspergillus nidulans, такие как АDН3 промотор (МсКnight еt аl., ЕМВО J. 4:2093-2099, 1989) и tpiA промотор. Примером пригодного терминатора является АDН3 терминатор (МсKight еt аl., 1985). Единицы экспрессии, использующие такие компоненты, клонированы в векторы, способные к встраиванию в хромосомную ДНК Aspergillus.
Способы трансформации грибов хорошо известны в литературе и были описаны, например, Beggs (ibid.), Нinеnеt et аl., (Рrос.Nаtl. Acad. Sсi. USA 75: 1929-1933, 1978) Yеltоn еt аl., (Рrос. Nаtl. Асаd. Sci USA 81:1740-1747, 1984) и Russel (Nаturе 301:167-169, 1983). Генотип клетки-хозяина обычно содержит генетический дефект, который дополняется селектируемым маркером, присутствующим на экспрессирующем векторе. Выбор определенного хозяина и селектируемого маркера доступны для людей с обычным уровнем квалификации в данной области. Для оптимизации получения гетерологичных белков в дрожжах, например, предпочтительно, чтобы штамм хозяина нес мутацию, такую как рер4 мутация дрожжей (Jones, Genetics 85:23-33, 1977), приводящую к пониженной протеолитической активности.
Кроме грибных клеток в данном изобретении в качестве клеток-хозяев можно использовать культивированные клетки млекопитающих. Предпочтительными культивированными клетками млекопитающих для применения в изобретении являются линии клеток COS-I (АТСС CRL 1650), COS-7 (АТСС CRL 1651), ВНК (АТСС СRL 1632) и 293 (АТСС СRL 1573; Graham et al., J. Gеn. Virol. 36:59-72, 1977). Предпочтительной клеточной линией ВНК является клеточная линия ВНК 570 (депонированная в АТСС под Accession CRL 10314). Кроме того, ряд других клеточных линий млекопитающих может применяться в данном изобретении, в том числе клеточные линии Rat Нер I (АТСС CRL 1600), Rat Hep II (АТСС СRL 1548), ТСMК (АТСС CCL 139), Human lung (клетки легких человека) (АТСС CCL 75.1), Human hepatoma (клетки гепатомы человека) (АТСС НТВ-52), Нер G2 (АТСС НВ 8065). Mouse liver (клетки печени мышей) (АТСС CСL 29.1), N СТС 1469 (АТСС ССL 9.1), SP2/O-Ag-14 (АТСС 1581), HIT-Т15 (АТСС CRL 1777) и RINm 5AHT2B (Orskov and Nielson, FEBS 229 (1): 175-178, 1988).
Экспрессирующие векторы млекопитающих для применения в данном изобретении содержат промотор, способный направлять транскрипцию клонированного гена или кДНК. Предпочтительными промоторами являются вирусные промоторы и клеточные промоторы. Вирусные промоторы представляют собой самый ранний промотор цитомегаловируса (Boshart еt аl., Сеll 41:521-530, 1985) и промотор SV40 (Subramani et аl., Mol. Сеll. Вiol. I: 854-864, 1981). Клеточные промоторы включают мышиный промотор металлотионеина-1 (Palmiter еt аl., U.S. Patent 4579821), мышиный Vk промотор (Веrgmаn еt аl., Рrос. Nаtl. Acad. Sci. USA 81: 7041-7045, 1983; Grаnt еt аl., Nuс. Acids Res. 15:5496, 1987) и мышиный VH промотор (Lоh еt аl., Cell 33:85-93, 1983). Особенно предпочтительным промотором является основной поздний промотор из аденовируса 2 (Каufmаn and Shаrр, Моl. Сеll. Вiоl. 2:1304-1319, 1982). Такие экспрессирующие векторы могут также содержать ряд сайтов сплайсинга РНК, локализованных в направлении 5' --> 3' от промотора и 3' --> 5' от последовательности ДНК, кодирующей целевой пептид или белок. Предпочтительные сайты сплайсинга РНК могут быть получены из генов аденовируса и (или) генов иммуноглобулинов. В экспрессирующих векторах находится также сигнал полиаденили-рования, расположенный в направлении 5' --> 3' от кодирующей целевой последовательности. Пригодными сигналами полиаденилирования являются ранние или поздние сигналы полиаденилирования из SV40 (Каufmаn аnd Sharp, ibid.), сигнал полиаденилирования из ЕIВ района аденовируса и терминатор гена гормона роста человека (DeNoto еt аl., Niс. Acids Res. 9:3719-3730, 1981). Экспрессирующие векторы могут содержать некодирующую вирусную лидерную последовательность, такую как состоящий из трех частей лидер аденовируса 2, расположенный между промотором и сайтами сплайсинга РНК. Предпочтительные векторы могут также содержать энхансерные последовательности, такие как энхансер SV40 и μ энхансер мышей (Gillies, Cеll 33:717-728, 1983). Экспрессирующие векторы могут также содержать последовательности, кодирующие РЕК VA аденовируса. Пригодные векторы могут быть получены из коммерческих источников (например, Stratagene, La Jоllа, СА).
Клонированные последовательности ДНК могут быть введены в культивированные клетки млекопитающих, например, путем медиируемой фосфатом кальция трансфекции (Wigler еt аl., Сеll 14:725, 1978; Corsaro and Pearson, Somatic Cell Genetics 7: 603, 1981; Graham and Vаn der Eb, Virology 52:456, 1973), путем электропорации (Nеumаnn еt аl., ЕМВО J. 1:841-845, 1982) или путем медиированной ДЭАЭ-декстраном трансфекции (Ausubel et al., (eds.), Currеnt Protocols in Моlесulаr Вiоlоgy, Jоhn Wilеy and Sons, Inс., NY, 1987), которые включены в виде ссылки. Для идентификации клеток, стабильно интегрировавших клонированную ДНК, обычно вводят в клетки селектируемый маркер вместе с целевыми геном или кДНК. Предпочтительными селектируемыми маркерами для применения в культивируемых клетках млекопитающих являются гены, придающие клеткам устойчивость к лекарственным средствам, таким как неомицин, гигромицин и метотрексат. Селектируемый маркер может быть амплифицируемым селектируемым маркером. Предпочтительными амплифицируемыми селектируемыми маркерами являются ген DHRR и ген устойчивости к неомицину. Селектируемые маркеры рассмотрены Thily (Mammalian Cell Technology, Butterworth Рuоblishers, Stoneham, МА, который включен в ссылки). Выбор селектируемых маркеров вполне доступен при уровне обычной квалификации в этой области.
Селектируемые маркеры могут быть введены в клетку на отдельном векторе одновременно с последовательностью рецептора глюкагона или они могут быть введены на том же векторе. Если их вводят на одном векторе, то селектируемый маркер и последовательность рецептора глюкагона могут находиться под контролем различных промоторов или одного и того же промотора, причем последний вариант даст дицистронную мРНК. Конструкции этого типа известны специалистам (например, Lеvinsоn аnd Simonsen, U.S. Раtеnt 4713-339). Предпочтительно бывает добавление дополнительной ДНК, известной как "ДНК-носитель", к смеси, которая вводится в клетки.
Трансфицированным клеткам млекопитающих позволяют расти в течение периода времени обычно 1-2 дней, после чего они начинают экспрессировать целевую последовательность (целевые последовательности) ДНК. Затем применяют отбор при помощи лекарственного средства для селекции по росту клеток, которые экспрессируют стабильно селектируемый маркер. Для клеток, трансфицированных амплифицируемым селектируемым маркером, концентрацию лекарственного средства можно увеличивать ступенчато для селекции на повышенное число копий клонированных последовательностей, т. е. на повышение уровня экспрессии. Клетки, экспрессирующие введенные последовательности, отбирают и подвергают скринингу на продуцирование целевого белка в желаемой форме и на желаемом уровне. Клетки, удовлетворяющие этим критериям, можно затем клонировать и оценивать на производительность.
Предпочтительными прокариотическими клетками-хозяевами для применения в проведении данного изобретения являются штаммы бактерии Escheriсhiа соli, хотя также можно использовать Baccillus и другие роды. Техника трансформации этих хозяев и экспрессии чужеродных последовательностей ДНК хорошо известны в этой области исследований (см.например Maniatis et аl., Molecular Сlоning: A Laboratory Manual, Cold Spring Harbor Laboratory, 1982, который включен в ссылки; или Sаmbrооk еt аl., suрrа). Векторы, применяемые для экспрессии клонированных последовательностей ДНК в бактериальных хозяевах, обычно содержат селектируемый маркер, такой как ген устойчивости к антибиотику, и промотор, функционирующий в клетке-хозяине. Предпочтительными промоторами являются промоторные системы trp (Nichols and Yanofsky, Meth. Enzymol. 101: 155-164, 1983), lac (Casadaban et al., J.Bacteriol. 143:971-980, 1980) и фага λ (Quееn, J.Mоl. Аррl. Genet 2:1-10, 1983). Плазмидами, применимыми для трансформации бактерий, являются рВР 322 (Bolivar еt аl., Gene 2:95-113, 1977), рUС (Messing, Meth. Еnzymоl. 101:20-78, 1983; Viеirа аnd Messing, Gеnе 19:259-268, 1982), pCQV2 (Оuееn, ibid.) и их производные. Плазмиды могут содержать как вирусные, так и бактериальные элементы. После данных здесь объяснений промоторы, терминаторы и способы введения экспрессирующих векторов, кодирующих рецепторы глюкагона данного изобретения в клетки растений, птиц, насекомых, будут ясны для специалистов в данной области. Применение бакуловирусов, например, в качестве векторов для экспрессии гетерологичных последовательностей ДНК в клетках насекомых было рассмотрено Atkinson еt аl. (Реstiс. Sci. 28: 215-224, 1990). Кроме того, применение Аgrоbасtеriumrhizogenes в качестве векторов для экспрессии генов в клетках растений было рассмотрено в обзоре Sinkar еt аl. (J.Вiоsсi. (Ваngаlоrе) 11:47-58, 1987).
Клетки-хозяева, содержащие конструкции ДНК данного изобретения, культивируют для экспрессии сегмента ДНК, кодирующего рецептор глюкагона. Клетки культивируют в соответствии со стандартными способами в культуральной среде, содержащей питательные элементы, необходимые для роста выбранных клеток-хозяев. Известно много подходящих сред, которые обычно содержат источник углерода, источник азота, незаменимые аминокислоты, витамины и минеральные соли, а также другие компоненты, например факторы роста или сыворотку, которые могут быть необходимы для определенных клеток-хозяев. Ростовая среда будет селектировать клетки, содержащие конструкцию (конструкции) ДНК, например, при помощи селекции с применением лекарственного средства или селекции при недостаточности основного питательного элемента, что дополняется селектируемым маркером на конструкции ДНК или на другом векторе, котрансфицируемым вместе с конструкцией ДНК.
Подходящие условия роста для дрожжевых клеток предусматривают, например, культивирование в химически определенной среде, содержащей источник азота, который может быть иным, чем аминокислоты, или дрожжевой экстракт, неорганические соли, витамины и добавки незаменимых аминокислот при температуре между 4oС и 37oС, предпочтительно при 30oС. рН среды поддерживают предпочтительно более 2 и менее 8, более предпочтительно 5-6. Способы для поддержания стабильного рН предусматривают применение буферов и постоянного контроля рН. Предпочтительным агентом для контроля рН является гидроксид натрия. Предпочтительными буферными агентами являются янтарная кислота и Bis-Tris (Sigma Chemical Co., St.Louis, МО). Вследствие тенденции дрожжевых клеток-хозяев гипергликозилировать гетерологичные белки, может быть предпочтительно экспрессировать рецепторы глюкагона данного изобретения в дрожжевых клетках, имеющих дефект в гене, необходимом для связанного с аспарагином гликозилирования. Такие клетки предпочтительно растут в среде, содержащей осмотический стабилизатор. Предпочтительным осмотическим стабилизатором является сорбит, добавляемый в среду при концентрации между 0,1 М и 1,5 М, предпочтительно при 0,5 М или 1,0 М. Культивируемые клетки млекопитающих обычно культивируют в коммерчески доступных сывороточных и бессывороточных средах. Выбор среды и условий роста, пригодных для определенной клеточной линии, находится в пределах знаний специалистов обычной квалификации в данной области.
Рецепторы глюкагона могут также экспрессироваться в трансгенных животных (не в человеке), в частности в трансгенных теплокровных животных. Способы получения трансгенных животных, в том числе мышей, крыс, кроликов, овец и свиней, известны специалистам и описаны, например, Hammer еt al. (Nature 315:680-683, 1985), Palmiter et аl. (Science 222:809-814, 1983), Вrinster еt аl. (Рrос. Nаtl. Acad. Sci. USA 82:4438-4442, 1985), Palmiter аnd Brinster (Cell 41:343-345, 1985) и U.S. Patent 4736866, включенных здесь в виде ссылок. Вкратце, единицу экспрессии, содержащую последовательность ДНК, которую требуется экспрессировать вместе с правильно расположенными контролирующими экспрессию последовательностями, вводят в пронуклеусы оплодотворенных яйцеклеток. Введение ДНК обычно выполняют микроинъекцией. Интеграцию инъекцированной ДНК детектируют блоттингом ДНК из проб ткани, обычно проб из ткани хвостов. Предпочтительно, чтобы введенная ДНК была включена в зародышевую линию животного, чтобы она передавалась потомству животного.
В предпочтительном варианте изобретения трансгенное животное, такое как мышь, создавали с помощью нацеленной мутации, чтобы разрушить последовательность рецептора глюкагона (см. Mansour еt аl., "Disruption of thе рrоtооnсоgеnе int-2 in mоusе em bryo-derived stecells: a general strateqy for targeting mutations to non-selectable genes", Nature 336:348-352, 1938). Таких животных можно легко использовать в качестве модели для исследования роли рецептора глюкагона в метаболизме.
Пептиды рецептора глюкагона
Как отмечалось выше, данное изобретение обеспечивает также пептиды рецептора глюкагона. В контексте данного изобретения пептиды рецептора глюкагона содержат части рецептора глюкагона или его производные, обсужденные выше, которые не содержат трансмембранных доменов и имеют длину по меньшей мере 10 аминокислот. Вкратце, структура рецептора глюкагона, а также возможных трансмембранных доменов может быть предсказана из продуктов первичной трансляции с использованием hydrophopicity plot function, например, Р/С Gеnе или Intelligenetics Suite. (Intelligenetics, Мt. Viеw, СА) или в соответствии со способами, описанными Kyte and Dооlittlе (J. Моl. Вiоl. 157:105-132, 1962). График гидрофобности крысиного рецептора глюкагона изображен в фиг.2. Считают, что рецепторы глюкагона имеют структуру, показанную в фиг.1. В частности считают, что эти рецепторы содержат внеклеточный аминоконцевой домен, три внеклеточных домена в виде петель и четыре внутриклеточных домена в виде петель, разделенных трансмембранными доменами.
В одном аспекте данного изобретения обеспечен выделенный пептид рецептора глюкагона, содержащий внеклеточный аминоконцевой домен рецептора. В предпочтительном варианте обеспечен выделенный пептид рецептора глюкагона, содержащий последовательность аминокислот SEQ ID 15, от глутамина, номер аминокислоты 18, до тирозина, номер аминокислоты 142. Также обеспечены другие выделенные пептиды рецептора глюкагона, которые могут быть выбраны из внеклеточных и внутриклеточных доменов в виде петель рецептора глюкагона (см.фиг. 1 и 5). В одном варианте пептиды рецептора глюкагона выбраны из группы, состоящей из 1 ID (SEQ ID 15, от лизина, номер аминокислоты 169, до гистидина, номер аминокислоты 178), 1ELD (SEQ ID 15, от тирозина, номер аминокислоты 203, до изолейцина, номер аминокислоты 231), 2 ID (SEQ ID 15, от фенилаланина, номер аминокислоты 259, до серина, номер аминокислоты 266), 2ЕLD (SEQ ID 15, от валина, номер аминокислоты 293, до изолейцина, номер аминокислоты 307), 3 ID (SEQ ID 15, от лейцина, номер аминокислоты 334, до лизина, номер аминокислоты 345), и 3ELD (SEQ ID 15, от аспарагиновой кислоты, номер аминокислоты 371, до серина, номер аминокислоты 380).
Пептидцы рецептора глюкагона данного изобретения могут быть получены с применением рекомбинантной техники, как обсуждалось выше, или синтетическими способами и могут быть дополнительно очищены, как описано ниже.
Очистка пептидов рецептора глюкагона
Изолированные пептиды рецептора глюкагона можно получать среди других методов, культивированием подходящих систем хозяин/вектор для получения рекомбинантных продуктов трансляции данного изобретения. Супернатанты из таких клеточных линий можно затем обработать при помощи различных процедур очистки для выделения пептида рецептора глюкагона. Например, сначала супернатант можно концентрировать при помощи коммерчески доступных фильтров для концентрирования белков, таких как Amicon или Milliроrе Реllсоn ультрафильтрационная единица. После концентрирования концентрат можно нанести на подходящий очищающий матрикс, такой как, например, антитела против рецептора глюкагона или глюкагон, связанные с подходящим носителем. Альтернативно можно использовать для очистки рецептора или пептида анионо- или катионообменные смолы. Наконец, стадии жидкостной хроматографии высокого разрешения с обращенной фазой (RP-HPLC) могут быть применены для дальнейшей очистки пептида рецептора глюкагона.
Пептид рецептора глюкагона считают "изолированным" или очищенным в контексте данного изобретения, если детектируют только одну полосу при анализе в ПААГ-ДСН с последующим окрашиванием при помощи Кумасси бриллиантового синего.
Антитела к рецепторам глюкагона
В одном аспекте данного изобретения рецепторы глюкагона, в том числе их производные, а также части или фрагменты этих белков, такие как обсужденные выше пептиды рецепторов глюкагона, могут быть использованы для получения антител, специфически связывающихся с рецепторами глюкагона. В контексте данного изобретения термин "антитела" охватывает поликлональные антитела, моноклональные антитела, их фрагменты, такие как F(аb')2 и Fаb, а также полученные рекомбинантной техникой партнеры связывания. Эти партнеры связывания включают в себя вариабзльные районы из гена, кодирующего специфически связывающее моноклональное антитело. Антитела считают специфически связывающимися, если они связываются с рецептором глюкагона с Кa большей или равной 107 М-1. Аффинность моноклинального антитела или партнера связывания может быть легко определена специалистом в этой области (см. Scatcharcd, Ann. N.Y. Acad. Sci. 51:660-672, 1949).
Поликлональные антитела можно легко получить квалифицированному специалисту из многих теплокровных животных, таких как лошади, коровы, козы, овцы, собаки, цыплята, кролики, мыши или крысы. Вкратце, рецептор глюкагона применяют для иммунизации животного при помощи внутрибрюшинной, внутримышечной, внутриглазной или подкожной инъекций. Иммуногенность рецептора глюкагона или пептида рецептора глюкагона может быть увеличена применением адъюванта, такого как адъювант Фрейнда или неполный адъювант. После нескольких повторных иммунизации берут небольшие пробы сыворотки и тестируют их на реактивность с рецептором глюкагона. Можно использовать множество тестов для детектирования антител, специфически связывающихся с рецептором глюкагона. Примеры тестов описаны в деталях в Antibodies: A Laboratory Manual, Harbow and Lane (eds.), Cold Sping Harbor Laboratory Press, 1988. Характерные примеры таких тестов включают: противоточный иммуноэлектрофорез (СIЕР), радиоиммунотесты, радиоиммунопреципитации, твердофазный иммуноферментный анализ (ELISA), дот-блот-тесты, ингибиторные или конкурентные тесты и "сэндвич"-тесты (cм. U.S. Раtеnt 4376110 и 4486530; см. также Antibodies: A Laboratory Manual, supra). Особенно предпочтительные поликлональные антисыворотки будут давать сигнал, который по меньшей мере в три раза больше фона. Как только титр животного достигнет плато с точки зрения реактивности антител с рецептором глюкагона, большие количества поликлональных антител можно легко получать либо еженедельным извлечением крови, либо после забоя животного.
Моноклональные антитела могут быть также легко получены при помощи хорошо известных способов (см. U. S. Parent RE 32001, 4902614, 4543439 и 4411993; см. также Моnосlоnаl Antibodies, Hybritdomas: A New Dimension in Biological Analyses, Plenum Press, Kennet, McKearn and Bechtol (eds.), 1980 и Antibodies: A Laboratory Manual, Harlow and Lane (eds.), Cold Spring Harobor Laboratory Press. 1988). Вкратце, в одном варианте животное, такое как крыса или мышь, инъецируют формой рецептора глюкагона, пригодной для генерирования иммунной ответной реакции против рецептора глюкагона. Типичными примерами пригодных форм, среди других, являются клетки, которые экспрессируют рецептор глюкагона, или пептиды,
основанные на последовательности рецептора глюкагона. Кроме того, известны многие способы для увеличения получаемого иммунного ответа, например, путем соединения рецептора или пептидов рецептора с другим белком, таким как овальбумин или гемоцианин фиссурели (KLH), или путем применения адъювантов, таких как полный или неполный адъювант Фрейнда. Начальная иммунизация может быть внутрибрюшинной, внутримышечной, внутриглазной или подкожной.
Между одной и тремя неделями после исходной иммунизации животное можно реиммунизировать повторной иммунизацией. Затем кровь и сыворотку животного тестируют на связывание с рецептором глюкагона при помощи описанных выше тестов. Дополнительные иммунизации могут проводиться до тех пор, пока животное не выйдет на плато по его реактивности с рецептором глюкагона. Затем животному дают конечную инъекцию рецептора глюкагона или пептида рецептора глюкагона и через 3-4 дня убивают. В это время у животного берут селезенку и лимфатические узлы и измельчают в клеточную суспензию проведением органов через сито или разрушением мембран селезенки или лимфатических узлов, окружающих клетки. В одном из вариантов эритроциты затем лизируют путем добавления гипотонического раствора с последующим моментальным возвратом к изотоничности.
В другом варианте подходящие для получения моноклональных антител клетки получают при помощи техники иммунизации in vitro. Вкратце, животное умерщвляют и клетки селезенки и лимфатических узлов удаляют, как описано выше. Получают суспензию отдельных клеток и эти клетки помещают в культуру, которая содержит форму рецептора глюкагона, пригодную для генерирования иммунной ответной реакции, как описано выше. Затем собирают лимфоциты и сливают, как описано выше.
Клетки, полученные с применением иммунизации или из иммунизированного животного, как описано выше, могут быть иммортализованы трансфекцией вирусом, таким как вирус Эпштейна-Барра (ЕВV) (см. Glasky and Reading, Нybridоmа 8 (4): 377-389, 1989). Альтернативно, в предпочтительном варианте клеточные суспензии собранных селезенки и (или) лимфатических узлов сливали с подходящими миеломными клетками для получения "гибридомы", секретирующей моноклональные антитела. Пригодными миеломными линиями являются предпочтительно дефектные по конструкции или экспрессии антител линии, которые, кроме того, являются сингенными с клетками из иммунизированного животного. Известны многие такие миеломные клеточные линии, и они могут быть получены, например, из American Type Culture Collection (ATCC), Rockville, Maryland (см. Catalogue of Cell Lines and Hybridomas, 6-th ad., ATCC, 1938). Типичными миеломными линиями являются: для человека UС 729-6 (ATCC СRL 8061), MC/CAR-Z2 (ATCC CRL 8147) и SKO-007 (ATCC CRL 8033); для мышей SP2/O-Ag 14 (ATCC CRL 1581) и Р3•63Аg 8 (ATCC TIB 9); и для крыс, Y3-Аg1.2.3 (ATCC CRL 1631) и Y2/0 (ATCC CRL 1662). Особенно предпочтительными слитыми линиями являются NS-1 (ATCC TIB 18) и Р3•63Аg 8,653 (ATCC 1580), которые можно применять для слияний с клеточными линиями мыши, крысы или человека. Слияние между миеломной клеточной линией и клетками из иммунизированных животных можно выполнять различными способами, в том числе с применением полиэтиленгликоля (РЕG) (см. Antibodies: A Laboratory Manual, Наrlоw and Lane (eds.), Cold Spring Наrbоr Laboratory Press, 1988) или электрослияния (см. Zimmerman and Vienken, J. Membrane Biol. 67:165-182, 1982).
После слияния клетки помещают в культуральные чашки, содержащие подходящую среду, такую как RPM1 1640 или DMEM (модифицированная по способу дульбекко среда Игла) (JRH Biosciences, Lenexa, Kan.). Эта среда может также содержать дополнительные ингредиенты, такие как плодная бычья сыворотка (FBS, т. е. из Hyclone, Logan, Utah или JRН Bioscience), тимоциты, которые могут быть взяты у детеныша животного того же вида, который использовали для иммунизации, или агар для отверждения питательной среды. Кроме того, среда должна содержать реагент, избирательно позволяющий расти клеткам селезенки и миеломы. В частности, предпочтительно применять HAT (гипоксантин, аминоптерин и тимидин) (Sigma Chemical Co., St. Louis, Missouri). Приблизительно после 7 дней полученные слитые клетки или гибридомы могут быть подвергнуты скринингу для определения присутствия антител, узнающих рецептор глюкагона. После нескольких клональных разбавлений и повторных тестов можно выделить гибридому, которая связывается с рецептором глюкагона.
Могут быть использованы также и другие способы для конструирования моноклональных антител (см. William D. Hues еt аl., "Generation of а Lаrgе Combinational Library оf the Immunoglobulin Repertoire in Phage Lambda", Sсiеnсе 246: 1275-1281, December 1989; см. также L. Sastry et al., "Cloning оf the Immunological Repertorie in Escherichia соli for Generation of Monoclonal Catalytic Antibodies: Construction of a Heavy Chain Variable Region-Specific с DNA Library" Proc. Natl. Acad. Sci. USA 86:5728-5732, August 1989; см. также Michelle Alting-Mees et al., "Monoclonal Antibody Expression Libraries: A Rapid Alternative to Hybridomas", Strategies in Molecular Biology 3: 1-9, January 1990; эти ссылки описывают коммерческую систему, доступную из Stratacyte, La Jolla, California, которая позволяет получать антитела при помощи рекомбинантных методов). Вкратце, мРНК выделяют из популяции В-клеток и используют для создания экспрессионных библиотек кДНК тяжелой и легкой цепей иммуноглобулина в векторах λIMMUNOZAP(H) и λIMMUNOZAP(L). Эти векторы можно скринировать индивидуально или коэкспрессировать с образованием Fab фрагментов или антител (cм. Huse et al., supra; см.также Sastry et аl., supra). Положительные пятна можно затем превратить в нелитическую плазмиду, делающую возможной высокий уровень экспрессии фрагментов моноклональных антител из Е.соli.
Подобным образом могут быть также сконструированы партнеры связывания при помощи техники рекомбинантных ДНК для включения вариабельных районов гена, кодирующего специфически связывающие антитела. Конструирование этих белков можно легко выполнять при наличии обычной квалификации в данной области (см. например, James W. Larrick еt аl., "Polymerase Chain Rеасtiоn Using Mixed Primers: Cloning of Нumаn Моnосlоnal Antibody Variable Rеgion Genes From Single Hybridoma Cells", Biotechnology 7:934-938, September 1989; Riechmann et al., "Reshaping Human Antibodies for Therapy", Nature 332:323-327, 1988; Roberts et al., "Generation of an Antibody with Enhanced Affinity and Specificity for its Antigen by Protein Engineering", Nature 328:731-734, 1987; Verhoeyen et al., "Reshaping Human Antibodies: Grafting an Antilysozyme Activity", Science 239:1534-1536, 1988; Chaudhary et al., "A Recombinant Immunotoxin Consisting of Two Antibody Variable Domains fused to Preudomonas Exotoxin", Nature 339:394-397, 1989; см. также U.S. Patent 5132405 с названием "Biosynthetic Antibody Binding Sites"). Вкратце, в одном варианте сегменты ДИК, кодирующие антигенсвязывающие домены, специфические для рецептора глюкагона, амплифицируют из гибридом, продуцирующих специфически связывающие рецептор моноклональных антитела, и вводят непосредственно в геном клетки, продуцирующей человеческие антитела (см. Verhoeyen et al., supra; см. также Rеiсhmаn еt аl., supra). Эта техника позволяет переносить антигенсвязывающий сайт обладающих специфическим связыванием крысиных или мышиных антител в антитело человека. Такие антитела предпочтительны для терапевтического применения в человеке, поскольку они имеют меньшую антигенность, чем крысиные или мышиные антитела.
Альтернативно антигенсвязывающие сайты (вариабельный район) могут быть либо соединены с другим, совершенно отличающимся белком или встроены в такой белок, что приводят к образованию нового белка с антигенсвязывающими сайтами антитела, а также с функциональной активностью второго белка. Как понятно специалистам, антигенсвязывающие сайты или домен связывания рецептора глюкагона этих антител могут быть обнаружены в вариабельном районе антител. Кроме того, последовательности ДНК, кодирующие небольшие части этих антител или вариабельные районы, которые специфически связываются с рецептором глюкагона млекопитающих, можно также применять в контексте данного изобретения. Эти части можно легко тестировать на специфичность связывания при помощи описанных ниже тестов.
В предпочтительном варианте гены, кодирующие вариабельный район, из гибридомы, продуцирующей целевое моноклональное антитело, амплифицируют с применением олигонуклеотидных праймеров для вариабельного района. Эти праймеры могут быть синтезированы специалистами или могут быть приобретены из коммерчески доступных источников. Stratacyte (La Jolla, Саlif.) присылает праймеры для мышиных и человеческих вариабельных районов, в том числе праймеры для  VL и CL районов. Эти праймеры могут быть применены для амплификации вариабельных районов тяжелых и легких цепей, которые могут затем быть встроены в векторы, такие как IMMUNOZAP* (Н) или IMMUNOZAP(L)* (Stratacyte) соответственно. Затем эти векторы могут быть введены в Е.соli для экспрессии. При помощи этих способов можно получить большие количества одноцепочечного белка, содержащего слитые VH и VL домены (см. Bird et. аl., Science 242:423-426, 1988).
VL и CL районов. Эти праймеры могут быть применены для амплификации вариабельных районов тяжелых и легких цепей, которые могут затем быть встроены в векторы, такие как IMMUNOZAP* (Н) или IMMUNOZAP(L)* (Stratacyte) соответственно. Затем эти векторы могут быть введены в Е.соli для экспрессии. При помощи этих способов можно получить большие количества одноцепочечного белка, содержащего слитые VH и VL домены (см. Bird et. аl., Science 242:423-426, 1988).
В других вариантах партнер связывания сливают внутри экспрессирующего вектора с другим белком, таким как токсин. Клетки, связанные таким партнером связывания, могут быть убиты в результате включения токсина (см. Chaudhary еt аl., supra). После получения подходящих антител или партнеров связывания их можно изолировать или очистить различными хорошо известными способами (см. Antibodies: A Laboratory Manual, supra). Такими способами являются пептидные или белковые аффинные колонки, HPLC или RP-HRLC, очистка на колонках с А- или G-белками или комбинирование этих способов. В контексте данного изобретения термин "изолированный" относится к антителам или партнерам связывания, в основном не содержащим других компонентов крови.
Антитела и партнеры связывания данного изобретения имеют много применений. Например, их можно использовать в проточной цитометрии для сортинга (сортировки) несущих рецептор глюкагона клеток или для гистохимического окрашивания тканей, несущих рецептор глюкагона. Вкратце, для того, чтобы детектировать рецепторы глюкагона на клетках, клетки инкубируют с меченым моноклональным антителом, которое специфически связывается с рецепторами глюкагона, с последующим детектированием присутствия связанного антитела. Эти стадии можно также проводить с дополнительными стадиями, такими как промывания для удаления несвязавшегося антитела. Метками, пригодными для применения в данном изобретении, являются хорошо известные метки, такие как флуоресцеинизотиоцианат (FIТС), фикоэритрин (РЕ), пероксидаза хрена (HRP) и коллоидное золото. В проточной цитометрии особенно предпочтителен FIТС, который может быть конъюгирован с очищенным антителом по способу Keltkamp в "Conjigation of Fluorescein Isothiocyanate to Antibodies. I. Experiments of the Conditions of Conjuhation, Immunology 18:865-873, 1970. (См. также Ketkamp, "Conjugation of Fluorescein Isothiocyanate to Antibodies. II. A Reproducible Method", Immunology 18:875-881, 1970; и Goding, "Conjugation of Antibodies with Fluorochromes: Modification to the Standart Methods", J. Immunol. Methods 13:215-226, 1970). Для гистохимического окрашивания предпочтительна НRР, которая может быть конъюгирована с очищенным антителом по способу Nакаnе and Kawaoi ("Peroxidase-Labeled Аntibоdу: А Nеw Method оf Соnjugаtion", J.Нistосhem. Cytochem. 22:1084-1091, 1974; см.также Tijssen and Кurstак, "Highly Еffiсiеt and Simple Methods of Рrераrаtiоn of Peroxiodase and Active Peroxidase Antibody Conjugates for Enzyme Immunoassays", Anal. Biochem. 136:451-457, 1984).
Кроме того, очищенные антитела или партнеры связывания могут быть также использованы терапевтически для блокирования связывания глюкагона с рецептором глюкагона in vitro и in vivo. Вкратце, блокирующими антителами являются те антитела, которые связываются с эпитопами рецептора глюкагона таким образом, что они предотвращают связывание глюкагона с его рецептором или предотвращают действие глюкагона на трансдукцию сигнала. Как было отмечено выше, можно использовать множество тестов для обнаружения антител, которые блокируют или ингибируют связывание глюкагона с рецептором глюкагона, в том числе inter alia, ингибиторные и конкурентные тесты, упомянутые выше. В одном варианте моноклональные антитела (полученные, как описано выше) тестируют на связывание с рецептором глюкагона в отсутствие глюкагона, а также в присутствие различных концентраций глюкагона. Блокирующими антителами или партнерами связывания являются те, которые, например, связываются с рецепторами глюкагона и в присутствии глюкагона блокируют или ингибируют связывание глюкагона с рецептором глюкагона.
Антитела или партнеры связывания, которые должны применяться терапевтически, обеспечивают предпочтительно в виде терапевтической композиции, содержащей антитело, или партнер связывания и физиологически приемлемый носитель, или разбавитель. Пригодными носителями или разбавителями среди других являются забуференный до нейтрального рН солевой раствор или солевой раствор, которые могут также содержать дополнительные наполнители или стабилизаторы, такие как буферы, сахара, такие как глюкоза, сахароза или декстроза, хелатирующие агенты, такие как ЭДТА, и различные консерванты.
Антагонисты глюкагона
Как отмечено выше, данное изобретение обеспечивает способы детектирования антагонистов глюкагона. В контексте данного изобретения антагонистом называют молекулу, способную связываться с рецептором, но не стимулирующую или снижающую стимуляцию отвечающего на глюкагон пути внутри клетки. В частности антагонисты глюкагона обычно идентифицируют по их способности связываться с рецептором глюкагона и вследствие этого снижать стимуляцию отвечающего на глюкагон пути метаболизма внутри клетки.
В одном аспекте данного изобретения обеспечены способы для обнаружения присутствия антагонистов глюкагона, предусматривающие стадии (а) экспонирования соединения в присутствии агониста глюкагона с рекомбинантным рецептором глюкагона, сопряженным с отвечающим на глюкагон путем, при условиях и в течение времени, достаточных для того, чтобы произошло связывание испытуемого соединения с рецептором и была получена ответная реакция пути, и (b) детектирование снижения стимулирования отвечающего на глюкагон пути, вызываемого связыванием соединения с рецептором глюкагона, по сравнению со стимулированием этого пути одним агонистом глюкагона, с последующим определением из полученных данных присутствия антагониста глюкагона. В контексте данного изобретения агонистами глюкагона являются соединения (в том числе и сам глюкагон), способные связываться с рецептором глюкагона и стимулировать восприимчивый к глюкагону путь внутри клетки.
При помощи таких способов можно скринировать множество соединений. Типичные примеры включают блокирующие антитела, обсужденные выше, пептиды рецептора глюкагона и аналоги глюкагона (в том числе как пептидные, так и непептидные лиганды). U.S. Serial 07/741931, например, обеспечивает способы получения больших количеств аналогов глюкагона при помощи пулов последовательностей ДНК, кодирующих такие аналоги. Такие пулы последовательностей ДНК, кодирующих аналоги глюкагона, можно получить насыщающим мутагенезом последовательностей ДНК, кодирующих глюкагон (например, Little, Gеnе 88: 113-115, 1990; Hembers et аl., Gеnе 88:143-151, 1989), сегментнаправленным мутагенезом (например, Shortle еt аl., Рrос. Nаtl. Acad. Sci. USA 77:5375-5379, 1980), усиленным неправильным включением нуклеотидов (например, Liao and Wise, Gene 88:107-111, 1990) или путем применения произвольно мутированных олигонуклеотидов (Hutchinson еt al. , Proc. Natl. Acad. Sci. USA 85: 710-714, 1986). Отдельные трансформанты, экспрессирующие аналог глюкагона, можно затем клонировать, как описано выше, или накапливать.
Соединения экспонируют с рекомбинантным рецептором глюкагона в присутствии агониста глюкагона при условиях и в течение времени, достаточных для прохождения связывания соединения с рецептором и ассоциированного с ним ответа через метаболический путь. В данном изобретении условия и время, достаточные для связывания антагониста глюкагона с рецептором, будут вариировать в зависимости от источника рецептора, однако обычно пригодными условиями являются 4oС-55oС в буферном растворе с 0-2 М NаСl, предпочтительно с 0-0,9 м NаСl, наиболее предпочтительно с 0,1 М NаCl, и в диапазоне рН 5-9, предпочтительно 6,8-8. Достаточное время для связывания и получения ответной реакции - 5-15 минут после начала экспозиции.
После экспозиции испытуемого соединения с рекомбинантным рецептором глюкагона в присутствии агониста глюкагона при условиях и времени, достаточных для прохождения связывания соединения с рецептором, снижение стимулирования отвечающего на глюкагон пути можно обнаружить, если это соединение конкурирует с агонистом глюкагона за рекомбинантный рецептор глюкагона. В одном варианте изобретения, отвечающим на глюкагон путем, является ответная аденилатциклазная реакция. В этом случае стадия детектирования предусматривает измерение снижения образования cAMP мембраносвязанной аденилатциклазой по сравнению с образованием циклического АMР в присутствии одного агониста глюкагона. Для целей данного изобретения предпочтительно, чтобы снижение стимулирования ответной реакции было одинаково или больше, чем снижение, вызываемое дез-Нis1-глюкагоном. Тесты аденилатциклазной активности можно выполнять, например, с применением способа, описанного Lin еt аl. (Вiосhеm. 14: 1559-1563, 1975) и в примерах. Эти способы измеряют стимулирование сАMР по сравнению с нативным глюкагоном и обычно предусматривают экспонирование препарата клеток, экспрессируюцих биологически активный рекомбинантный рецептор глюкагона, со смесью глюкагона и тестируемого соединения в присутствии радиоактивно меченого АТР.
Альтернативно образование сАMР может быть легко измерено при помощи способов, хорошо известных в этой области, в том числе, например, способами, описанными Salomon еt аl. (Аnаl. Bioсhem. 58:541-548, 1976) или Krishna еt аl. (J. Phаrmacol. Ехр. Тhеr. 163:379, 1968) или предпочтительно с применением коммерчески доступных китов, таких как Scintillation Proximity Assay Кit из Amersham Соrроrаtiоn. Этот кит измеряет образование сАMР при помощи конкуренции иодинированного сАMР с анти-сАМР антителами. Особенно предпочтительные рецепторы глюкагона имеют биологическую активность в таких тестах ЕD50 (эффективная доза для 50% ответной реакции) менее 1 нМ, более предпочтительно с ED50 менее 0,7 нМ и наиболее предпочтительно с ЕD50 менее 0,25 нМ.
В следующем варианте изобретения ответной реакцией является люциозеразная репортерная система. Вкратце, люцифераза является ферментом, который катализирует высвобождение фотонов люциферином и поэтому может быть легко обнаружена в присутствии люциферина (Аlаm и Сооk, Аnаl. Biochem. 188:245-254, 1990). Как описано более детально ниже, в особенно предпочтительном варианте изобретения обеспечивают конструкцию ДНК, содержащую элемент, отвечающий на циклическую АМР, такой как проэнкефалиновый отвечающий на сАМР элемент, оперативно соединенный с кДНК люциферазы. Эта конструкция ДНК, содержащая кДHK люциферазы, стабильно трансфицируется в клетку-хозяин. Затем эту клетку-хозяин трансфицируют второй конструкцией ДНК, содержащей первый сегмент ДНК, кодирующий рецептор глюкагона, оперативно соединенный с дополнительными сегментами ДНК, необходимыми для экспрессии рецептора. При связывании агониста рецептора глюкагона повышенные уровни сАМР индуцируют экспрессию люциферазы. Люциферазу экспонируют с люциферином и измеряют фотоны, высвобождаемые во время окисления люциферина люциферазой.
В других вариантах изобретения активация ответной реакции приводит к увеличению внутриклеточной концентрации свободного кальция. Для определения концентрации свободного внутриклеточного кальция можно применять различные тесты, например способ Calcim fluor Quinz, описанный Charest et al. (J.Biol. Chem. 259: 8769-8773, 1983) или аеquorin photoprotein - способ, описанный Nakajima- Shimada (Proc. Natl. Acad. Sci. USA 88:6878-8882, 1991). Особенно предпочтителен способ фотоизображения внутриклеточного кальция, который описан более детально в примере 6. Вкратце, в одном варианте клетки трансформируют плазмидой, экспрессирующей рецептор глюкагона, и выращивают в течение трех дней при обычных условиях культивирования. Затем ростовую среду удаляют и заменяют раствором, содержащим 10 мкМ fura-2АМ (см. Grynkiewicz et al. . , J. Biol Chem 260:3440-3450, 1985). Затем клетки инкубируют в течение 30-120 минут. Фотовизуализацию (Формирование изображения) можно проводить при помощи инвертированного флуоресцентного микроскопа Nicon Diарhоt, оборудованного лампой с ртутной дугой. Клетки сначала наблюдают по меньшей мере в течение 60 секунд для установления линии иона, после чего стимулируют буфером, содержащим глюкагон. Изображения обычно записывают по меньшей мере в течение 3 минут после стимулирования. Для обработки и количественного выражения изображений можно использовать программное обеспечение, такое как Inovision (Research Triangle Park, N.С.).
Антагонисты глюкагоны, которые могут быть обнаружены, как описано выше, могут быть очищены ионообменной и распределительной хроматографией, как описано, например, Coy et al. (Peptides Structure and Function, Pierce Сhеmiсаl Company, Rосkfоrоd IL., pp.369-372, 1982), хроматографией с обращенной фазой (Andreu and Меrrifield, Eur. J. Biochem. 164:585-590, 1987) или при помощи HPLC (например, Коfоd еt аl. , Int., J. Peptide Protein Res. 32:436-440, 1988). Дополнительная очистка может быть достигнута с применением обычных способов химической очистки, таких как жидкостная хроматография, градиентное центрифугирование и гель-электрофорез и другие. Способы очистки белков известны специалистам (см. Scopes, R., Protein Purification, Springer-Verlag, NY, 1982) и могут быть использованы для описанных здесь рекомбинантных антагонистов глюкагона. Альтернативно, анатагонисты глюкагона могут быть синтезированы по твердофазному способу Barany and Меrrifield (in The Peptides, Vol. 2A, Gross and Meienhofeer, eds., Academic Press, NY pp.1-284, 1979) или с применением автоматизированного пептидного синтезатора.
Предпочтительны практически чистые антагонисты глюкагона с гомогенностью по меньшей мере 50%, более предпочтительно 70-80% и наиболее предпочтительно 95-99%, в частности для фармацевтического использования. В очищенном до гомогенности виде антагонисты глюкагона могут применяться в терапии. Как правило, антагонисты могут вводиться парентерально или путем инфузии. В типичном случае антагонисты присутствуют в виде свободных оснований или в виде солей с кислотами. Подходящие соли должны быть фармацевтически приемлемыми. Типичными примерами являются соли металлов, соли щелочных и щелочно-земельных металлов, такие как соли калия и натрия. Другими фармацевтически приемлемыми солями являются соли лимонной, янтарной, молочной, соляной и бромистоводородной кислот. Парентеральные композиции могут быть приготовлены в виде водных изотонических растворов с рН между 5,6 и 7,4. Подходящими изотоническими растворами могут быть растворы хлорида натрия, декстрозы, борной кислоты-тартрата натрия и полиэтиленгликоля. Терапевтические дозы антагонистов могут вводиться одновременно с инсулином либо в одной и той же композиции, либо в отдельных композициях.
Диагностическое применение зондов рецептора глюкагона
В другом аспекте данного изобретения обеспечены зонды и праймеры для детектирования рецепторов глюкагона. В одном варианте изобретения обеспечены зонды, которые способны гибридизоваться с ДНК или РНК рецептора глюкагона. Для целей данного изобретения зонды "способны гибридизоваться" с ДНК рецептора глюкагона, если они гибридизуются либо при высокой строгости, либо при низкой строгости (см. Sambrook et аl., supra). Предпочтительно, чтобы зонд мог быть применен для гибридизации с подходящими нуклеотидными последовательностями в присутствии 6•SSC, 1•Denhardt's (Sambrook еt аl., supra), 0,1% додецилсульфата натрия при 65oС и по меньшей мере одной промывке для удаления избытка зонда в присутствии 2•SSC, 1•Denhardt'S, 0,1% ДС Nа при 65oС. Последовательности зондов построены предпочтительно таким образом, чтобы была возможна гибридизация с последовательностями ДНК рецептора глюкагона, но не с последовательностями ДНК секретина, кальцитонина или рецептора паратиреоидного гормона.
Зонды данного изобретения могут состоять либо из ДНК, либо из РНК и могут иметь в длину всего лишь приблизительно 12 нуклеотидов, обычно 14-18 нуклеотидов, но могут быть и такими большими, как вся последовательность рецептора глюкагона. Выбор размера зонда зависит от его применения. Например, для определения присутствия различных полиморфных форм рецептора глюкагона внутри индивидуума предпочтителен зонд, содержащий фактически всю длину кодирующей последовательности рецептора глюкагона. Зонды рецептора глюкагона могут быть применены для идентификации полиморфизмов, связанных с геном рецептора глюкагона (см., например, Wеbеr, Gеnomiсs 7:524-530, 1990; и Weber and May, Аmеr. J. Hum. Gen. 44:388-396, 1989). Такие полиморфизмы могут быть ассоциированы с наследуемыми заболеваниями, такими как диабет.
Зонды могут быть сконструированы и помечены при помощи способов, хорошо известных в этой области знаний. Короткие зонды, например, из 12 оснований могут быть получены синтетическим путем. Более длинные зонды, приблизительно от 75 оснований до менее 1,5 кб (т.п.н.), получают предпочтительно, например, РСR амплификацией в присутствии меченых предшественников, таких как 32Р-dСТР, диоксигенин-dUТР или биотин-dАТР. Зонды длиной более 1,5 кб обычно более легко амплифицировать путем трансфекции клетки плазмидой, содержащей соответствующий зонд, выращивания трансфицированных клеток до больших количеств и очистки соответствующей последовательности из трансфицированных клеток.
Зонды могут быть помечены различными маркерами, в том числе, например, радиоактивными маркерами, флуоресцентными маркерами, ферментными и хромогенными маркерами. Применение 32Р особенно предпочтительно для мечения определенных зондов.
Зонды данного изобретения можно использовать также для обнаружения присутствия мРНК или ДНК рецептора глюкагона внутри пробы. Однако если рецепторы глюкагона присутствуют лишь в ограниченном количестве, или если желательно обнаружить выбранную мутантную последовательность, которая присутствует только в ограниченном количестве, или если желательно клонировать рецептор глюкагона из выбранного теплокровного животного, то может быть выгодно амплифицировать соответствующую последовательность, чтобы ее можно было более легко обнаружить или получить.
Для амплификации выбранной последовательности могут быть применены различные способы, в том числе, например, амплификация РНК (cм. Lizardi et al., Bio/Technology 6: 1197-1202, 1988; Kramer et al., Nature 339:401-402, 1969; Lomeli et al., Clinical Сhеm. 35(9): 1826-1831, 1989; U.S. Patеnt 4786600; и амплификация ДНК с применением полимеразной цепной реакции (PCR) (см. U.S. Раtеnt 4683195, 4683202 и 4800159) (см. также U. S. Раtеnt 4876187 и 5011769, которые описывают альтернативную систему детектирования/амплификации, предусматривающую использование разрезаемых связей).
В особенно предпочтительном варианте РСR амплификацию применяют для обнаружения или получения ДНК рецептора глюкагона. Вкратце, как описано в деталях ниже, пробу ДНК денатурируют при 95oС для генерирования одноцепочечной ДНК. Затем специфические праймеры, как обсуждается ниже, гибридизуют при 37oС-70oС в зависимости от соотношения AT/GC в праймерах. Праймеры удлиняют при 72oС при помощи Таq полимюразы для получения цепи, противоположной матрице. Эти стадии составляют один цикл, который может повторяться для амплификации выбранной последовательности.
Праймеры для амплификации выбранной последовательности должны быть выбраны из последовательностей, которые высокоспецифичны и образуют стабильные дуплексы с последовательностью-мишенью. Праймеры должны быть также некомплементарными, в частности при 3'-конце, не должны образовывать димеров друг с другом или с другими праймерами и не должны образовывать вторичные структуры или дуплексы с другими районами ДНК. Как правило предпочтительны праймеры из 18-20 нуклеотидов, которые можно легко синтезировать при помощи известных способов. Особенно предпочтительные праймеры представлены в таблице 1 и включают в себя вырожденные олигонуклеотиды ZC4715 и ZC4701 (SEQ ID 9 и 8 соответственно), а также олигонуклеотиды ZC4758 и ZC4778 (SEQ ID 10 и 11 соответственно).
Дополнительные применения нуклеотидных последовательностей рецептора глюкагона
Еще в одном аспекте данного изобретения обеспечены вирусные векторы, которые могут быть использованы для лечения заболеваний, при которых рецептор глюкагона (или мутантный рецептор глюкагона) избыточно экспрессируется или при которых не экспрессируется рецептор глюкагона. Вкратце, в одном варианте изобретения обеспечены вирусные векторы, которые направляют образование антисмысловой РНК рецептора глюкагона и тем самым препятствуют избыточному образованию рецепторов глюкагона или экспрессии мутантных рецепторов глюкагона. В другом варианте обеспечены вирусные векторы, которые направляют экспрессию кДНК рецептора глюкагона. Вирусными векторами, пригодными для применения в данном изобретении, среди других являются рекомбинантные векторы вируса осповакцины (U.S. Раtеnt 4603112 и 4769330), рекомбинантные векторы поксвируса (РСТ Publication WO 89/01973) и предпочтительно рекомбинантные ретровирусные векторы ("Recombinant Retroviruses with Anphotropic and Ecoptropic Host Ranges", PCT Publication WO 90/02806; "Rеtrоvirаl Расkаging Сеll Lines and Processes оf Using Sаmе", РСТ Publication WO 89/07150; и "Antisense RNА for Тrеаtmеnt оf Rеtrоvirаl Disease States", PCT Publication WO/03451).
Как отмечалось выше, вирусные векторы данного изобретения могут быть использованы в лечении болезненных состояний, при которых либо избыточно экспрессируется рецептор глюкагона, либо экспрессируется мутантный рецептор глюкагона или в случаях, когда не экспрессируется рецептор глюкагона.
Следующие далее примеры представлены для иллюстрации, но не для ограничения изобретения.
ПРИМЕРЫ
Пример 1
Синтез кДHK и получение библиотек кДНК
А. Синтез кДНК печени крыс
Печени из 30 г - самок крыс Sprague-Dawley (Simonsen Labs, Gilroy, CA) удаляли и сразу же помещали в жидкий азот. Тотальную РНК получали из ткани печени с применением гуанидинизотиоцианата (Сhirgwin et al., Biochemistry 18:52-94, 1979) и центрифугирования в CSCI. Поли(А)+ РНК выделяли с применением олиго d(Т)-целлюлозы хроматографически (Aviv аnd Leqer, Рrос. Nаtl. Acad. Sci. USA 69:1408-1412, 1972).
Первую цепь кДНК синтезировали из поли(А)+ РНК печени, дважды отобранной при помощи поли d(Т). 10 мкл раствора, содержащего 10 мкг поли(А)+ РНК печени, смешивали с 2 мкл 20 рмоль/мкл праймера первой цепи ZC3747 (SEQ ID 7) и 4 мкл обработанной диэтилпирокарбонатом воды. Эту смесь нагревали при 65oC в течение 4 минут и охлаждали на льду.
Синтез первой цепи кДНК инициировали добавлением 8 мкл 5x SUPERSCPIRT буфера (GIBCO BRL, Gaithersburg, Md.), 4 мкл 100 мМ дитиотреитола и 2,0 мкл раствора дезоксинуклеотидтрифосфатов, содержащего 10 мМ каждого из dATP, dGТР, dTTP и 5-метил-dСTP (Pharmacia LKB Вiotесhnоlоgу Inc., Piscataway, N. J.) к РНК-праймерной смеси. Реакционную смесь инкубировали при 42oC в течение 3 минут. После инкубирования добавляли 6,0 мкл 200 Е/мкл обратной транскриптазы SUPERSCRIPT (GIBCO BRL). Эффективность синтеза первой цепи анализировали в параллельной реакции путем добавления 10 мкКи 32P-αdCTP к аликвоте 10 мкл реакционной смеси для мечения продуктов реакции. Реакционные смеси для синтеза первой цепи инкубировали при 45oC в течение 45 минут с последующей 15-минутной инкубацией при 50oC. Реакции останавливали добавлением воды до конечного объема 100 мкл с последующими экстракциями дважды смесью фенол/хлороформ (1:1) и один раз смесью хлороформ/изо-амиловый спирт. Невключенные нуклеотиды удаляли из каждой реакции двойным осаждением кДНК в присутствии 6 мкг гликогена в качестве носителя, 2,5 М ацетата аммония и 2,5 объемов этанола. Немеченую кДНК ресуспендировали в 50 мкл воды и применяли для синтеза второй цепи. Длину первой цепи кДНК оценивали ресуспендированием меченой кДНК в 20 мкл воды и определением размера кДНК при помощи гель-электрофореза в агарозе.
Синтез второй цепи проводили на гибриде РНК-ДНК из реакции синтеза первой цепи при условиях, способствующих использованию первой цепи в качестве праймера в синтезе второй цепи с образованием шпильки ДНК. Реакционную смесь готовили таким образом, что она содержала 20,0 мкл 4х буфера для полимеразы I (100 мМ Трис-HCl, рН 7,4, 500 мМ КСl, 25 мМ MgCl2, 50 мМ (NН4)2SO4), 4,0 мкл 100 мМ дитиотреитола, 1,0 мкл раствора, содержащего 10 мМ каждого из дезоксинуклеотидтрифосфатов, 3,0 мкл β-NAD, 15,0 мкл 3 Е/мкл ДНК-лигазы Е.соli (NВL Enzymes Ltd., Cramlington., Northumbriа, Еnglаnd, 5,0 мкл 10 Е/мкл ДНК-полимеразы I E.coli (GIBCO ВRL) и 50,0 мкл немеченой первой цепи ДНК. Параллельную реакцию, в которой аликвоту 10 мкл смеси для синтеза второй цепи метили путем добавления 10 мкКи 32P-αdCTP, применяли для мониторинга эффективности синтеза второй цепи. Реакционные смеси инкубировали при комнатной температуре в течение 4 минут с последующим добавлением 1,5 мкл 2 Е/мкл РНКазы Е (GIВСО BRL) к каждой реакционной смеси. Реакции инкубировали при 15oC в течение 2 часов с последующей инкубацией в течение 15 минут при комнатной температуре. Каждую реакцию останавливали добавлением 4 мкл 500 мМ ЭДТА с последующей экстракцией смесью фенол/хлороформ и смесью хлороформ/изоамиловый спирт, как описано выше. ДНК из каждой реакции осаждали в присутствии этанола и 2,5 М ацетата аммония. ДНК из немеченой реакции ресуспендировали в 50 мкл воды. Меченую ДНК ресуспендировали и подвергали электроторезу, как описано выше.
Одноцепочечную ДНК в шпилечной структуре расщепляли при помощи нуклеазы маша. Реакционная смесь содержала 10 мкл 10х буфер для нуклеазы маша (Stratagene Сlоning Systems, La Jolla, Calif.), 4 мкл 200 мМ дитиотреитола, 34 мкл воды, 50 мкл второй цепи кДНК и 2 мкл 1:10 разведения нуклеазы маша (Promega Corp. , Madison. , Wis. ) в буфере для разведения Stratagene MB (Stratagene Cloning Systems). Реакцию инкубировали при 37oC в течение 15 минут и останавливали добавлением 20 мкл Трис-HCl, рН 8,0 с последующими последовательными экстракциями смесью фенол/хлороформ и смесью хлороформ/изоамиловый спирт, как описано выше. После экстракций ДНК осаждали в этаноле и ресуспендировали в воде.
Ресуспендированную кДНК "затупляли" при помощи Т4 ДНК-полимеразы. Эту кДНК ресуспендировали в объеме 192 мкл воды, смешивали с 50 мкл 5х буфера для Т4 ДНК-полимеразы (250 мМ Трис-HCl, рН 8,0, 250 мМ КСl, 25 мМ MgCl2), 3 мкл 100 мМ дитиотреитола, 3 мкл раствора, содержащего 10 мМ каждого из дезоксинуклеотидтрифосфатов и 2 мкл 6,7 Е/мкл Т4 ДНК-полимеразы (Pharmacia LKB Biotechnology Inc.). После инкубирования при 15oC в течение 30 минут реакцию останавливали добавлением 2 мкл 500 мМ ЭДТА с последующими последовательными экстракциями смесью фенол/хлороформ и смесью хлороформ/изоамиловый спирт, как описано выше. На основе включения 32P-dCTP было определено, что выход кДНК составлял 4 мкг из исходной матрицы мРНК 10 мкг.
В. Получение библиотеки кДНК печени крысы
Для облегчения клонирования кДНК в экспрессирующий вектор млекопитающих к полученной, как описано выше, кДНК добавляли Есо R1 адаптеры. Аликвоту 10 мкл кДНК и 800 рмолей адаптера (12 мкл) смешивали с 4,0 мкл 10х лигазного буфера (Stratagene Cloning Systems), 4,0 мкл 10 мМ АТР, 6,0 мкл воды и 16 Е Т4 ДНК-лигазы (4,0 мкл; Stratagene Сlоning Systems). Реакцию инкубировали 16 часов при температурном градиенте 4oС-15oC. Реакцию останавливали добавлением 185 мкл воды, 25 мкл RЕАСТ 2 буфера (GIВСО BRL) с последующим инкубированием при 65oС в течение 30-60 минут. После инкубирования реакцию экстрагировали смесью фенол/хлороформ, затем смесью хлороформ/изоамиловый спирт и осаждали этанолом, как описано выше. После центрифугирования осадок ДНК промывали 70% этанолом и высушивали на воздухе. Осадок ресуспендировали в 180 мкл воды.
Для облегчения направленного встраивания кДНК в экспрессирующий вектор млекопитающих кДНК расщепляли при помощи Xho I, получая кДНК, имеющую 5'-Есо R1 липкий конец и 3'-Xho I липкий конец. Сайт рестрикции Xho I при 3' - конце кДНК вводили через праймер ZC3747 (SЕQ ID 7). Рестрикционное расщепление останавливали последовательными экстракциями смесями фенол/хлороформ и хлороформ/изоамиловый спирт. кДНК осаждали этанолом, и полученный осадок промывали 70% этанолом и сушили на воздухе. Осадок ресуспендировали в 1х буфере для нанесения (10 мМ фосфатный буфер, рН 8,8, 5% глицерин, 0,125% бромфенол синий).
Ресуспендированную кДНК нагревали до 65oС в течение 10 минут, охлаждали на льду и подвергали электрофорезу на 0,9% агарозном геле с низкой температурой плавления (Seaplaque GTG Low Melt Agarose, FMC Corp., Rockland, Me.) с применением ВRL I kb ladder(GiBCO BRL) и Pharmacia 100 bp ladder (Pharmacia LKB Biotechnology Inc.) в качестве маркеров размеров. Примесные адаптеры и фрагменты побочных продуктов размером ниже 800 н.п. вырезали из геля. Электроды меняли местами и проводили электрофорез кДНК до тех пор, пока она не концентрировалась вблизи начала дорожки. Эту зону, содержащую концентрированную ДНК, вырезали, помещали в микроцентрифужную пробирку и определяли приблизительный объем отрезка геля. В пробирку добавляли аликвоту ТЕ, эквивалентную половине объема геля, и агарозу расплавляли нагреванием до 65oС в течение 15 минут. После уравновешивания пробы до 42oС добавляли приблизительно 5 единиц β-Agarase I (New Еnglаnd Biolabs, Beverly, Mass.). Пробу инкубировали 90 минут для расщепления агарозы. После инкубирования к пробе добавляли 0,1х объем 3 М ацетата натрия и смесь инкубировали на льду в течение 15 минут. После инкубирования пробу центрифугировали при 14000 g в течение 15 минут при 4oC для удаления нерасщепленной агарозы. Затем осаждали кДНК в супернатанте этанолом. Осадок кДНК промывали 70% этанолом, сушили на воздухе и ресуспендировали в 10 мкл воды.
Полученную кДНК клонировали в векторе p ZCEP E.coli, являющимся производным p CDANAI (invitr ogen), в котором селектируемый маркер SupF заменен β-лактамазной кассетой из pUC18. Плазмиду рZСЕР, которую переводили в линейную форму расщеплением при помощи Есо RI и Xho, лигировали с Есо RI-Xho I-кДНК. Полученные плазмиды вводили электропорацией в клетки штамма Е.соli DH10В ELECTROMAX (GIBCO BRL).
С. Синтез кДНК клеток панкреатических островков человека
Клетки островков выделяли из поджелудочных желез человека, полученных из доноров трансплантируемых органов, для которых не было подходящего реципиента. После перфузии холодным раствором UW (DuPont, Boston, Mass.) каждую поджелудочную железу осторожно вырезали, панкреатический проток канюлировали и вливали раствор коллагеназы 4 мг/мл (Type V, Sigma, St.Louis, Мо.) при постоянной скорости, сначала при 4oС и затем при 39oC. Железу разбирали на кусочки и высвобождаемые фрагменты промывали при помощи центрифугирования, измельчали через иглы с уменьшающимся калибром и очищали центрифугированием с непрерывным градентом плотности фиколла (Warnock Diabeles 35 (Suppl. I): 136-139, 1989). Материал, собранный из верхних интерфаз объединяли и учитывали после определения чистоты островков при помощи окрашивания дитиазоном. Островки, применяемые в конструировании библиотеки, имели чистоту более 65%, тогда как островки, применяемые в блотах Нозерна, имели чистоту более 40%. Средний диаметр островков был 175 мкм. Кроме того, выделенные островки обнаруживали как первую, так и вторую фазы функции секреции инсулина после перфузии либо высокой концентрацией глюкозы, либо изобутилметилксантином (IВМХ).
Поли(А)+РНК выделяли с применением FАSТТRАСК кита для выделения мРНК (Invitrogen) в соответствии с инструкциями изготовителя. Вкратце, 30000 очищенных островков быстро лизировали в буфере для лизиса, гомогенизировали при помощи игл уменьшающегося калибра и расщепляли в присутствии протеиназы К и РНКазина, затем выделяли поли(А)+РНК при помощи хроматографии на олиго-d(Т)-целлюлозе. Концентрацию и чистоту элюированных фракций определяли при OD 260/280.
Приблизительно 2,5 мкг поли(А)+ РНК из человеческих панкреатических островков использовали для создания библиотеки кДНК с применением системы конструирования библиотеки кДНК LIBRAPIAN R11 (Invitrogen) и клеток E.coli DH10B ELSCTROMAX (GIBCO BRL) в соответствии с инструкциями изготовителя. Вкратце, приблизительно 2,5 мкг поли(А)+РНК, выделенной из человеческих островков, превращали в двухцепочечную кДНК с последующим добавлением Вst Х I непалиндромных линкеров (Invitroqen). Эту кДНК фракционировали по размеру и непрореагировавшие линкеры удаляли электрофорезом в агарозном геле и электроэлюцией. Отбирали комплементарные цепи ДНК, большие чем 600 н.п.
Пример 2
Выделение кДНК крысиного рецептора глюкагона при помощи амплификации с применением полимеразной цепной реакции (PCR-амплификации)
КДНК печени крыс использовали в качестве матрицы для амплификации последовательностей нуклеотидов рецептора глюкагона с применением вырожденных олигонуклеотидов (ZC4715 и ZC4701; SEQ ID 9 и 8 соответственно), соответствующих районам высокой консервативности среди членов семейства генов секретина. 50 мкл реакционной смеси содержали 5 нг матрицы кДНК (пример 1А); 100 пмолей каждого из олигонуклеотидов ZC4715 (SEQ ID 9) и ZC4701 (SEQ ID 9); 0,25 мМ каждого из дезоксинуклеотидтрифосфатов (Cetus, Emeryville, СА); 1х Promega 10х буфер (Promega Соrр.); 1,25 единиц Таq полимеразы (Promega). PCR реакцию проводили в виде 40 циклов (1 минута при 95oC, 1 минута при 42oС и 2 минуты при 72oC) с последующим инкубированием в течение 7 минут при 72oС.
PCR продукт 650 н.п. выделяли при помощи гель-электрофореза и лигировали с рСR1000 (Stratagene Cloning Systems). Полученную плазмиду применяли для трансформации клеток ХL-I Е. соli. Плазмидную ДНК получали из отобранных трансформантов, обозначенных G 13/рСR1000, и секвенировали (SEQ ID 14). Секвенирование клона показало, что инсерция кодировала полипептид, родственный рецептору секретина.
Пример 3
Клонирование кДНК полной длины крысиного рецептора глюкагона
КДНК полной длины крысиного рецептора глюкагона получали скринингом библиотеки, описанной в примере 1В, при помощи теста связывания глюкагона. Библиотеку высевали, получая один миллион независимых клонов. Колонии трансформанта из каждой чашки выскребали в 10 мл LB-Amp (Sambrook et al., supra). Клетки откручивали и среду выбрасывали. Осадки клеток ресус-пендировали в 4 мл LВ-Аmр, 15% глицерине, и аликвоты по 1 миллилитру хранили при -80oC. Первый глицерериновый исходный раствор титровали и засевали 100 пулов из 5000 колоний на чашку. После роста колоний каждую чашку выскребали в 10 мл LB-Аmр. Брали аликвоту клеток из каждого пула для применения в приготовлении плазмидной ДНК. Оставшиеся клеточные смеси доводили до конечной концентрации глицерина 15%, делили на аликвоты и замораживали при -80oC. Плазмидную ДНК получали из каждого пула клеток и полученную ДНК расщепляли РНКазои (Boehringer Mannheim, Indianopolis, Ind.) в соответствии с инструкциями изготовителя. РНК-азную реакцию останавливали экстрагированием смесью фенол/хлороформ/изоамиловый спирт (24:24:1) и ДНК осаждали этанолом.
Цлазмидную ДНК из каждого пула трансфицировали в СОS-7 клетки (АТСС СRL 1651) и трансфектанты скринировали на присутствие рецепторов глюкагона при помощи теста связывания 125I-глюкагона. Вкратце, за день до трансфекции приблизительно 2•105 клеток COS-7 высевали на стерильные предметные стекла с одной камерой (NUNC AS, Roskil de, Denmark), которые покрывали 10 мкг/мл человеческого фибронектина (таблица 1) на 30 минут при комнатное температуре и промывали забучеренным фосфатом солевым раствором (PBS, Sigma Chemical Co. , St. Louis, Mo.). Два микрограмма плазмидной ДНК из каждого пула использовали для трансфекции клеток, росших на отдельных стеклах с камерой, при помощи способа, описанного McMahan et al. (EMBO J. 10:2821-2832, 1991, который включен в виде полной ссылки). После трансфекции клетки выращивали в течение 72 часов при 37oС в 5% СО2.
Лиофилизированный порошок растворяли в буферном растворе. Затем добавляли сульфат аммония до концентрации 25% и раствору давали осаждаться в течение 2 часов при 4oС. Фибронектин осаждали центрифугированием при 1000 об/мин в центрифуге Bench Top (Beckman Instruments, Inc. Irvine, Calif.) в течение 15 минут. Супернатант выбрасывали и осадок растворяли в 10 мл NaPO4-буферном растворе (см. выше).
Фибронектин в конечном объеме 16,9 мл диализовали в течение ночи против 1 л NаРО4-буферного раствора (описанного выше). Диализованный материал разбавляли в 3 раза 1 мМ фосфатным буфером, рН 7,4, получая раствор в 1 мМ NаРО4, рН 7,4, 100 мМ NaCl. Затем фибронектин разбавляли в 2 раза дистиллированной водой. Тягучий нерастворимый осадок вынимали стеклянной палочкой.
Фибронектин подвергали FPLC через 50 мл колонку DЕАЕ FF Sepharose (Pharmacia LKB Вiotесhnоlоgу Inc., Рis cataway, N.J.), которая была уравновешена тремя объемами 10 мМ Трис-HCl, рН 8,1, 50 мМ NаСl. После того как колонку промывали 10 мМ Трис, рН 8,1, 50 мМ NаСl до получения пробы фона, фибронектин элюировали градиентом соли до 10 мМ Трис, рН 8,1, 300 мм NaCl. Фракции собирали, аликвоты фракций подвергали электрофорезу на полиакриламидных гелях и гели анализировали окрашиванием Кумасси синим и анализом Вестерна. Фракции пиков объединяли и диализовали против 10 мМ CAPS (3-(циклогексиламино)-1-пропансульфокислота, Sigma), рН 11,0, 10 мМ CaCl2, 150 мМ NaCl. Раствор хранили при -80oC.
Для получения трансфектантов для теста связывания 125I-глюкагона истощенные среды отсасывали из клеток и клетки промывали три раза холодным (4oС) РВS. После конечной промывки клетки покрывали средой для связывания (таблица 2) и инкубировали 10 минут при комнатной температуре. Среды заменяли 0,5 мл среды для связывания, содержащей 0,5 нМ 125I-глюкагон (Аmеrsham receptor grade, удельная активность 2000 Ки/ммоль;
Amersham). Клетки затем качали при 30oС в течение 1 часа. Среды отсасывали из клеток, добавляли холодную (oC) среду для связывания без глюкагона и клетки инкубировали 5 минут при комнатной температуре. Среды отсасывали из клеток и клетки промывали 3 раза холодным (4oC) PBS. После последней промывки клетки фиксировали 1 мл 2,5% глутаровым альдегидом в PBS при комнатной температуре в течение 20 минут. Глутаровый альдегид удаляли и клетки ополаскивали 3 раза PBS. Предметные стекла сушили на воздухе в течение 1 часа при комнатной температуре, погружали в жидкую фотографическую эмульсию (Eаstman Коdak Co., Rochester, N.Y.) в соответствии с инструкциями изготовителя и сушили при комнатной температуре в темноте по меньшей мере 30 минут. Стекла затем помещали в светонепроницаемый бокс на 72 часа при 4oС. Клетки, способные связывать глюкагон, обнаруживали при 2,5Х увеличении при ярком освещении поля. Одни пул, 57, был идентифицирован как пул, содержащий клетки, способные связывать глюкагон.
1 M бикарбоната натрия
8,4 г твердого NaCO3
Бикарбонат натрия высыпали в мерную колбу на 100 мл и добавляли 80 мл дистиллированной воды. Раствор перемешивали до растворения твердого вещества, добавляли дистиллированную воду до 100 мл. Раствор еще раз перемешивали и хранили при 4oС в закрытой пробкой склянке.
Среда
1 миллилитр 1 М бикарбоната натрия добавляли на литр дистиллированной воды. 4 литра готовили заранее и раствор охлаждали в течение ночи или готовили раствор с применением холодной дистиллированной воды.
69% раствор сахарозы
69 г сахарозы
Сахарозу растворяли в 31 мл дистиллированной воды при нагревании. Концентрацию раствора измеряли при помощи рефрактометра. Добавляли твердую сахарозу или воду (по необходимости) для получения концентрации 69±0,5%.
42,3% раствор сахарозы
42 г сахарозы
Сахарозу растворяли в 57 мл дистиллированной воды при нагревании. Концентрацию раствора измеряли при помощи рефрактометра. Добавляли 69% раствор сахарозы или воду (по необходимости) для получения концентрации 42,3±1%.
2х буфер для связывания
100 мМ HEPES, pH 7,3
300 мМ NaCl
2 мМ ЭДТА
2% бычий сывороточный альбумин
1,6 мг/мл бацитрацин
Буфер для получения изображения
140 мМ NаСl
10 мМ HEPES
5,6 мМ глюкоза
5 мМ КСl
1 мМ MgSO4
1 мМ CaCl2
Раствор Fura-2 AM
50 мг fura-2 AM (Molecular Probes)
50 мл DMSO
5 мл буфера для получения изображения
50 мг fura-2 АM растворяли в 50 мл DMSO. После растворения твердого вещества раствор смешивали с 5 мл буфера для получения изображения.
Аликвоту плазмидной дНК из пула 57 подвергали РСR амплификации с применением олигонуклеотидов ZC4701 и ZC4715 (SEQ ID 8 и 9 соответственно). Реакционную смесь 50 мкл готовили таким образом, что она содержала 200-400 нг плазмидной ДНК из пула 57; 100 пмолей каждого из олигонуклеотидов ZC4701 и ZC4715 (SEQ ID 8 и 9 соответственно); 50 мМ KСl; 10 мМ Трис-HCl, рН 9,0 (при 20oС); 1,5 мМ MgCl2; 0,01% желатин; 0,1% Тритон Х-100; 0,2 мМ каждого из дезоксинуклеотидтрифосфатов (Pharmacia LKB Biotechnology Inc.) и 1 единицу Taq полимеразы (Promega). PCR реакцию проводили в виде 30 циклов с последующим 7-минутным инкубированном при 72oC. Реакционную смесь хранили при 4oС. Анализ РСR-продукта при помощи гель-электрофореза обнаружил присутствие полосы 700 н.п., приблизительно того же размера, что и продукт, описанный в примере 2.
Исходный раствор в глицерине из пула 57 титровали и засевали 2% чашек из 500 колоний. Колонии объединяли и исходный раствор и плазмидную ДНК готовили, как описано выше. Плазмидную ДНК трансфицировали в СОS-7 клетки и трансфектанты скринировали с применением теста связывания глюкагона, как описано выше. Один пул, 57-18, был идентифицирован как пул, содержащий клетки, способные связывать глюкагон.
Аликвоту плазмидной ДНК из пула 57-18 подвергали PCR, амплификации с применением олигонуклеотидов ZC4701 и ZC4715 (SEQ ID 8 и 9 соответственно), как описано выше. Анализ PCR продукта при помощи гель-электрофореза обнаружил присутствие полосы 700 н.п., подтвердив присутствие последовательностей ДНК рецептора глюкагона.
Исходный раствор в глицерине из пула 57-18 титровали и засевали 6 чашек из 50 колоний и 47 чашек из 20 колоний. Колонии объединяли и исходные глицериновые растворы и плазмидные ДНК готовили, как описано выше. Аликвоту из каждого пула плазмидной ДНК трансфицировали в COS-7 клетки и трансфектанты скринировали с применением теста связывания глюкагона, описанного выше. Кроме того, аликвоту из каждого пула плазмидной ДНК подвергали PCR амплификации с применением олигонуклеотидов ZС4701 и ZC4715 (SEQ ID 8 и 9 соответственно), как описано выше. 4 положительных пула ( 57-18-16, 57-18-18, 57-18-36 и 57-15-48) были идентифицированы как пулы, содержащие клетки, способные связывать глюкагон, и было показано PCR амплификацией, что они содержат ту же полосу 700 н.п.
Для выделения кДНK две чашки с диаметром 150 мм засевали 2500 колониями (каждую) пула 57-18. Фильтры-лифты готовили по способу, описанному Hanahan and Meselson (Gеnе 10:63, 1980) и Sambrook et al (ibid), на которых даются полные ссылки. Гибридизационный зонд получали путем PCR амплификации плазмидной ДНК из пула 57 с применением олигонуклеотидов и способов, описанных выше. РCR продукт очищали в геле из агарозы с низкой температурой плавления и использовали для него произвольные праймеры из кита MЕGАРRIME (Amersham, Arlingtоn Heights, III.) в соответствии с инструкциями изготовителя. Фильтры гибридизовали в растворе, содержащем 6х SSC, 5х Denhardt's, 5% ДСН, 200 мкг/мл обработанной ультразвуком ДHK спермы лосося и 2•105 имп/мин на мл 32Р-меченого РСR фрагмента. Фильтры гибридизовали в течение ночи при 65oС. Избыток метки удаляли тремя промываниями при помощи 1x SSC, 1% ДСН при 65oС. Фильтры экспонировали на пленке в течение 4 часов при -80oС с двумя экранами. Положительный клон, содержащий плазмиду, pLJ4, был идентифицирован и секвенирован. Плазмида pLJ4 была депонирована с АТСС (12301 Parklawn Dr., Roскvillе, MD 20852) как трансформант E.coli под входным номером 69056 21 августа 1992. Рестрикционнык анализ и секвенирование показали, что pLJ4 содержит инсерцию приблизительно 2 кб (т.п.н.), кодирующую белок из 485 аминокислот с предсказанной мол.массой 54962 дальтон. Эта последовательность нуклеиновой кислоты и выведенная из нее аминокислотная последовательность показаны в SEQ ID 14 и 15. Анализ гидропатии по способу Kyte and Doolittle (J. Mol. Biol 157:105-132, 1982, включая здесь в виде ссылки) выявил 8 кластеров гидрофобных аминокислот, соответствующих амино-концевой сигнальной последовательности и 7 трансмембранным доменам (фиг.2). Кроме того, анализ выведенной аминоксилотной последовательности показал присутствие 4 потенциально N-соединенных сайтов гликозилирования в удлиненной гидрофильной последовательности и присутствие 6 цистеинов в том же районе.
Пример 4
Выделение кДHK человеческого рецептора глюкагона при помощи РСR амплификации
КДНК клеток панкреатических островков человека (пример 1 C) использовали в качестве матрицы для амплификации последовательностей нуклеотидов рецептора глюкагона при помощи вырожденных олигонуклеотидов ZC4715 и ZC4701 (SEQ ID 9 и 8 соответственно). 50 мкл реакционной смеси содержали 5 нг матричной кДНК (пример 1 C); 100 пмолей каждого из олигонуклеотидов ZC4715 (SЕQ ID 9) и ZC4701 (SEQ ID 8); 0,25 мМ каждого из дезоксинуклеотидтрифосфатов (Cetus, Emeryville, Calif); 1x Promega 10x буфер (Рrоmеgа) и 1,25 единиц Таq полимеразы (Рromеgа). РCR реакцию проводили в виде 40 циклов (1 минута при 95oC, 1 минута при 45oC и 2 минуты при 72oС) с последующим инкубированием в течение 7 минут при 72oС.
При помощи гель-электрофореза был выделен РСR продукт размером приблизительно 750 н.п. Одну десятую выделенного РСR продукта применяли в качестве матрицы для другой РCR реакции с применением олигонуклеотидов ZС4758 и ZС4778 (SEQ ID 10 и 11), которые были сконструированы для встраивания сайта рестрикации Bam H1 при 3'-конце и сайта рестрикации Eco R1 при 5'-конце РСR продукта для субклонирования. Реакционная смесь 50 мкл была приготовлена, как описано выше. PCR, реакцию проводили в виде 40 циклов (1 минута при 95oС, 1 минута при 50oC и 1,5 минуты при 72oС) с последующим 7-минутным инкубированием при 72oС.
Для скрининга трансформантов на последовательность нуклеотидов рецептора глюкагона инсерцию ДНК, присутствующую в каждом трансформанте, амплифицировали с применением олигонуклеотидов ZC447 и ZC976 (SEQ ID 1 и 2 соответственно), которые были сконструированы как универсальные pUC праймеры секвенирования и присоединялись к pUC последовательностям, фланкирующим встроенный РСR продукт. 48 трансформантов добавляли (каждый) в 25 мкл реакционной смеси, содержащей 20 пикомолей каждого из олигонуклеотидов; 0,125 мМ каждого из дезоксинуклеотидтрифосфатов (Cetus, Emeryville, Саlif.); 1x Promega 10x буфер (Promega); и 1,25 единиц Таq, полимеразы (Promega). РСR реакцию проводили в виде 30 циклов (1 минута при 95oС, 1 минута при 45oC и 1,5 минуты при 72oC с последующим 7-минутным инкубированием при 72oC.
РСR продукты затем анализировали гибридизацией по Саузерну (Southern, J. Mol. Biol. 98: 503, 1975; и Sambrook et аl., ibid; включенные здесь в виде полных ссылок) с применением 1,9 кб Eco R1-Xho 1 фрагмента pLJ4, который был помечен произвольным включением затравок с использованием кита Amersham MEGAPRIME (Amersham) в качестве зонда. Было показано, что один клон, G30, гибридизовался с полноразмерным кДНК-зондом крыс. Эта нуклеотидная последовательность G30 показана в SEQ ID 16.
Пример 5
Клонирование кДНК полной длины рецептора глюкагона человека
Для идентификации библиотеки, содержащей последовательности, кодирующие человеческий рецептор глюкагона ряд библиотек скринировали при помощи PCR c применением олигонуклеотидных праимеров ZC5433 и ZС5432 (SEQ ID 13 и 12 соответственно), которые были сконструированы таким образом, что они содержат последовательности из клона G30, обсуждаемого выше. Приобретенные и приготовленные человеческие геномные и кДНК библиотеки из печени человека, клеток островков, НерG2 клеток, мозга и плаценты человека подвергали скринингу (таблица 3). Индивидуальные 50 мкл реакционные смеси для PCR реакции готовили таким образом, что они содержали ДHK из каждой библиотеки при объемах, указанных в таблице 4, 20 пмолей/мкл каждого из ZC5433 и ZC5432 (SEQ ID 13 и 12 соответственно), 0,25 мМ каждого из дезоксинуклеотидтрифосфатов, 5 мкл 10x Taq 1 буфера (Promega), 15 мМ MgCl2, 19,5 мкл дистиллированной воды и 0,5 мкл 5 Е/мкл Taq 1 полимеразы (Promega). Кроме того, отдельные реакционные смеси содержали рLJ4 в качестве положительного контроля или не содержали ДНК в качестве отрицательного контроля.
РСR реакции проводили в виде 30 циклов (1 минута при 94oС, 1 минута при 50oС и 1,5 минуты при 72oC) с последующим 10-минутным инкубированием при 72oС. Затем PCR продукты анализировали при помощи агарозного гель-электрофореза. Только NIН библиотека печени генерировала полосу 320-410 н.п., т.е. размера, равного размеру полосы, видимой в случае положительного контроля (pLJ4).
Библиотека KJHK печени человека, клонированная в плазмиду рсD2 (Chen and Okayama, Mol. Сеll. Biol. 7:2745-2752, 1987), полученную от Dr.Rоgеr Bertolloti (Nаtionаl Institutes of Health, Bethesda, Md.), была использована для получения клона кДНК полной длины, кодирующего человеческий рецептор глюкагона. Библиотеку высевали, получая 1 миллион независимых клонов. Колонки трансформантов из каждой чашки выскребали в 10 мл LB-Amp (Sambrook et al. , ibid. ). Клетки откручивали и среду выбрасывали. Осадки клеток ресуспендировали в 4 мл LВ-Аmр, 15% глицерина и 4 аликвоты по 1 мл хранили при -80oС. Первый глицериновый исходный раствор титровали и 100 пулов 5000 колоний высевали на чашку. После роста колонии каждую чашку выскребали в 10 мл LB-Amp. Брали аликвоту клеток из каждого пула для использования в получении плазмидной ДНК. Оставшиеся клеточные смеси доводили до конечной концентрации 15% глицерина, делили на аликвоты и замораживали при -80oC. Плазмидную ДНК готовили из каждого пула клеток и ДНК расщепляли РНКазой (Boehringer Mannheim, Indianapolis, In.) согласно инструкциям изготовителя. РНКазную реакцию останавливали экстракцией смесью фенол/хлороторм/изоамиловый спирт (24:24:1) и ДНК осаждали этанолом.
Аликвоты ресуспендированной плазмидной ДНК из каждого пула объединяли в группы (т.е. 1-10, 11-20, 21-30, 31-40 и т.д.). Плазмидную ДНК разбавляли 1: 20 и 1 мкл ДНК из каждого пула использовали в РСR. реакционной смеси, идентичной реакционной смеси, описанной выше. Реакционную смесь подвергали амплификации при условиях, описанных выше. Анализ продуктов РСR при помощи агарозного гель-электрофореза показал, что пул 31-40 содержал полосу 320-410 того же размера, что и полоса положительного контроля.
Плазмидные ДНК из исходных пулов 31-40 разбавляли 1:20 и 1 мкл каждого пула использовали в реакционной смеси, идентичной смеси, описанной выше. РСR амплификацию реакционных смесей выполняли с применением условий, описанных выше. Анализ РСR продуктов при помощи агарозного гель-электрофореза показал, что пул 40 дал размер полосы приблизительно 310-420 н.п.
Концентрация плазмидной ДНК была грубо оценена как приблизительно 70 нг/мкл при агарозном гель-электрофорезе 1 мкл пула 40, который был разведен 1: 10. 70 нг пула 40 плазмидной ДНК вводили электропорацией в клетки штамма DНIOB E.coli. при 2,3 вольтах, 400 омах, 25 микрофарадах и ресуспендировали в 1 мл SOC (Sаmbrооk еt al., ibid.). Готовили три разведения клеток: 10-2, 10-3 и 10-4. 100 мкл каждого из разведении высевали на 4 чашки. Было определено, что образовалось приблизительно 10000 колоний на чашку для 4 чашек, содержащих клетки из разведения 10-2, и приблизительно 1000 колоний на чашку для 4 чашек, содержащих клетки из разведения 10-3. По 2 фильтра-лифта готовили из каждой из 4 чашек с разведением 10-3 и одной из чашек с разведением 10-2, которые были обозначены как пулы 1 - 5. Один фильтр из каждых двух повторностей помещали на твердую среду и оставляли для роста, пока не образовались колонии. Колонии затем выскребали и использовали для получения ДНK для PСR амплификации. Кроме того, три оставшиеся чашки с разведением 10-2 выскребали и из этих клеток получали плазмидную ДНК.
Оставшиеся фильтры предварительно промывали 3х SSC+0,5% ДСН при 65oС при перемешивании в течение 12 часов для удаления остатков бактерий. Затем фильтры прегибридизовали в буфере Ульриха (Ulrich, EMВО J. 3:361-364, 1984)+50% формамид, 1% ДСН в течение ночи при 37oС. Клон ДНК G30, который метили при помощи набора Аmеrshаm MGАРRIMЕ (Amersham) согласно предлагаемым изготовителем условиям, кипятили и добавляли к раствору для гибридизации (буфер Ульриха + формамид) до конечной концентрации 6•105 имп/млн на мл. Фильтры инкубировали в течение ночи при 37oС с перемешиванием. После инкубирования в течение ночи раствор зонда удаляли и фильтры промывали 5 минут при 65oС в 2х SSC + 0,1% ДСН. После первой промывки фильтры промывали в том же растворе 15 минут при 65oС, затем 5 минут с качанием при комнатной температуре. Протокол последнего промывания повторяли еще 2 раза. Фильтры экспонировали с пленкой в течение ночи при комнатной температуре. Одна колония на чашке 2, которая соответствовала пулу 2, давала положительный результат гибридизации с G 30. Плазмидные ДНК, полученные из отдельных независимых колоний, подвергали анализу секвенирования. Один клон, 40-2-2, подвергали дальнейшему секвенированию и депонировали с АТСС (12301 Parklawn Dr., Rockville, МD 20852) как трансформант E.соli. под входным 69055 21 августа 1992 г. Частичная последовательность ДНК клона 40-2-2 и выведенная из нее аминокислотная последовательность показаны в виде SEQ ID 17 и 18.
Для подтверждения присутствия нуклеотидных последовательностей рецептора глюкагона в гибридизации на фильтрах проводили РСR амплификацию на каждом препарате плазмидной ДНК (плазмидной ДНК, полученной из каждого повторного фильтра, и плазмидной ДНK, полученной из каждой из 3 чашек 10-2). Объединенные плазмидные ДНК разбавляли 1:20 в воде и 1 мкл каждой ДНК применяли в качестве матрицы в РСR реакции, выполняемой, как описано выше. Агарозный гель-электрофорез полученных PСR продуктов показал, что пул 2 и три для 4 пулов из 10000 содержали генерируемые РСR полосы между 310 и 420 н.п. Присутствие генерируемой РСR полосы в пуле 2, соответствующем чашке 2, подтвердило присутствие последовательностей ДНК рецептора глюкагона.
F. Стратегия клонирования 5' -последовательности человеческого рецептора глюкагона
Анализ частичной кДНК последовательности клона 40-2-2 и последовательности кДНК полной длины крысиного рецептора глюкагона показал, что клон 40-2-2 не имел амино-концевой последовательности, соответствующей приблизительно 25 аминокислотам. 5'-последовательность кДНК человеческого рецептора глюкагона получали адаптацией способа, описанного Frohman et al. (Рrос. Nаtl. Acad. Sci. USA 85:8998-9002, 1988). Вкратце, был сконструирован олигонуклеотидный праймер таким образом, что он гибридизовался с последовательностью вблизи 5'-конца кодирующей последовательности 40-2-2. Этот праймер гибридизовали с имеющей G-хвост первой цепью кДНК-матрицы из печени человека и праймер удлиняли в направлении 5'-конца с применением Таq 1 ДНК-полимеразы. Второй поли d(C) праймер гибридизовали с имеющей G-хвост кДНК-матрицей, что делало возможной полимеразную цепную реакцию для амплификации, с последующими клонированном, секвенированием и сплайсингом до образования кодирующей последовательности, присутствующей в клоне 40-2-2.
Получали три кДНК-матрицы. Первая матрица была коммерчески доступной первой цепью кДНК печени человека. Вторую и третью матрицы кДНК получали синтезом первой цепи кДНК из коммерчески доступной мРНК печени человека (Clontech) с использованием либо олигонуклеотида, содержащего последовательности, специфические для человеческого рецептора глюкагона, либо традиционного применения в качестве праймера олиго d(T) соответственно.
Вторую кДНК-матрицу получали синтезом из мРНК печени человека с применением олигонуклеотида ZC5433 (SEQ ID 13), который является специфическим для кодирующей последовательности человеческого рецептора глюкагона. Реакционную смесь, содержащую 2 мкл мРНК печени человека (1 мкг/мкл), 8 мкл 20 пмоль/мкл ZC5453 (SEQ ID 13) и 0,5 мкл 10 мМ Трис, рН 7,4, 0,1 мМ ЭДТА, инкубировали в течение 7 минут при 68oС и выдерживали затем 2 минуты на льду. После инкубирования к реакционной смеси добавляли 4 мкл 5Х SUPERSCRIPT буфера (GIBCO-BRL), 1 мкл 200 мМ дитиотреитола, 1 мкл раствора, содержащего 10 мМ каждого из dNTP, 1 мкл 0,25 мкКи/мкл α32P-dCTP и 5 мкл обратной транскриптазы SUPERSCRIPT. Реакцию инкубировали в течение 1 часа при 45oC. После инкубирования реакцию инкубировали еще в течение 10 минут при 45oС. Реакцию останавливали добавлением 80 мкл ТЕ. РНК гидролизовали добавлением 1 мкл 0,5 М ЭДТА и 1 мкл КОН. Реакцию гидролиза проводили при 65oС в течение 5 минут. После инкубирования пробу разбавляли до 1 мл 50 мМ КОН, 0,1 мМ ЭДTA и проводили через концентратор СENTRICON 100 (Аmicon, Danvers, Mass.). Колонку промывали 1 мл 50 мМ КОН, 0,1 мМ ЭДТА. Концентрированную кДНК собирали и нейтрализовали 1/2 объема 100 мМ НСl. Нейтрализованную пробу кДНК осаждали этанолом и осадок ресуспендировали в 26 мкл дистиллированной воды.
Третью кДНК-матрицу получали синтезом из мРНК печени человека с применением приобретенного олиго d(T) праймера. Готовили реакционную смесь, содержащую 2 мкл 1 мкг/мкл мРНК печени человека, 1 мкл 1 мкг/мкл олиго d(T) 18 (Nеw England Biolabs, Beverly, Mass.) и 5 мкл 10 мМ Триса, рН 7,4, 0,1 мМ ЭДТА. Синтез кДНК проводили с применением условий, описанных для такого синтеза выше.
К кДНК первой цепи присоединяли G-хвост. Готовили реакционные пробирки, содержащие 4 мкл кДНК первой цепи печени человека (QUICK CLONE; Clontech, Palo Alto, Calif. ), 4 мкл кДНК первой цени из печени человека из праймера ZC5433 (SEQ ID 13) или 4 мкл кДНК первой цепи из печени человека из олиго d(T) праймера. Каждая реакционная смесь получала 22 мкл воды, 8 мкл 5X буфера (Рrоmеgа), 4 мкл 20 мМ d GTP и 2 мкл 15 Е/мкл концевой трансферазы, и реакции проводили в течение 30 минут при 37oC с последующим 10-минутным инкубированном при 65oC. Реакционные смеси затем разбавляли до 90 мкл 10 мМ Трисом, рН 7,4, 1 мМ ЭДТА и осаждали этанолом.
Синтез второй цепи всех имеющих G-конец (хвост) кДНК проводили таким же образом. Каждую кДНК сначала суспендировали в 50 мкл дистиллированной воды. Затем к каждой кДНК добавляли 100 пмолей ZC4814 (SEQ ID 20) в 5 мкл. Затем кДНК гибридизовали с праймером путем нагревания каждой смеси до 68oС в течение 5 минут с последующим выдерживанием 2 минуты на льду. После того как праймер гибридизовался с кДНК, каждая смесь получала 20 мкл 5х буфера для полимеразы 1 (пример 1), 1 мкл 100 мМ дитиотреитола, 2 мкл раствора, содержащего 10 мМ каждого из dNTP, 2 мкл 0,5 мкКи/мкл α32P-dCTP, 2 мкл 3 Е/мкл ДНК-лигазы E. coli (New England Вiоlаbs), 5 мкл 7 Е/мкл дНК-полимеразы 1 E. coli (Amersham). Реакцию проводили в течение 5 минут при 22oC с последующим добавлением 1,5 мкл 2 Е/мкл РНКазы Н (GIBCO-BRL), после чего инкубирование продолжали при 16oC в течение 2 часов. Реакцию останавливали добавлением 200 мкл 10 мМ Триса, рH 8,0, 1 мМ ЭдТА с последующей экстракцией 150 мкл водонасыщенного фенола и 150 мкл хлороформа. Смесь перемешивали на вортексе и затем центрифугировали 3 минуты при 22oС для разделения фаз. Водную фазу удаляли и реэкстрагировали фенолом и хлороформом, как описано выше. После второй экстракции фенолом и хлороформом водный слой экстрагировали хлороформом. Затем кДНК в водном слое осаждали добавлением 5 мкг гликогена мидий, 200 мкл 8 М ацетата аммония и 300 мкл изо-пропанола для селективного осаждения больших нуклеиновых кислот, тогда как невключенные олигонуклеотидные праймеры остаются в супернатанте. Затем кДНК осаждали центрифугированием и осадок промывали 70% этанолом и сушили на воздухе. Осадок кдНК ресуспендировали в 15 мкл бидистиллированной воды.
Двухцепочечные кДНК амплифицировали в 5 параллельных реакциях с применением олигонуклеотидного праймера, гомологичного 5'-концу ZС4814 (SEQ ID 20) и содержащего сайты рестриктаз для облегчения субклонирования и олигонуклеотидного праймера, специфического для самого 5'-конца кодирующей последовательности, присутствующей в клоне 40-2-2. Каждая реакционная смесь содержала 5 мкл 10х буфера для Таq, 1,3 мкл 25 мМ MgCl2, 5 мкл раствора, содержащего 2,5 мМ каждого из dNTP, 1 мкл двухцепочечный кДНК и 0,5 мкл Таq 1 полимеразы (Promegа). дистиллированную воду добавляли до конечного объема 50 мкл. Реакцию денатурировали в течение 5 минут при 98oC перед добавлением 1 мкл каждого из 10 рмоль/мкл ZС5624 (SEQ ID 21) и 20 рмоль/мкл ZC4812 (SEQ ID 19). На каждую пробу наслаивали 70 мкл минерального масла и пробы выдерживали при 90oC. Амплификацию проводили в виде 30 циклов (95oC 60 секунд, 57oC 40 секунд, 72oС 60 секунд) с последующим 7-минутным удлинением при 72oC.
Каждый продукт PСR подвергали агарозному гель-электрофорезу и амплифицированные фрагменты вырезали по отдельности и субклонировали в PCR 1000 с применением кита ТА для клонирования (Linvitrogen). Три клона из каждой реакции лигирования отбирали и анализировали на присутствие инсерции с применением олигонуклеотидных праймеров ZC5624 (SEQ ID 21) и ZC4812 (SEQ ID 19) в независимых РСR реакционных смесях, каждая из которых содержала инокулят из клона в качестве матричной ДНК. РСR реакцию проводили, как описано выше, за исключением того, что проводили только 30 циклов амплификации. Один инсерция-положительный клон из каждой исходной PСR реакции подвергали секвенированию. Из двух клонов, показавших, что они содержат не имеющую ошибочных нуклеотидов 5'-кодирующую последовательность рецептора глюкагона, был отобран один клон для получения 5'-кодирующей последовательности человеческого рецептора глюкагона. Клон 9А расщепляли Есо R1 и Крn 1 для получения фрагмента 551 н.п., содержащего 5'-кодирующую последовательность рецептора глюкагона. 3'-кодирующая последовательность рецептора глюкагона была получена в виде Крn 1-Ваm Н1 фрагмента 561 н.п. из 40-2-2. Eсо R1- Крn 1 фрагмент и Крn 1-Bаm Н1 фрагмент 561 н. п. легировали в присутствии Есо R1 и Bаm H1 для предотвращения конкатомеризации. Продукт лигирования, фрагмент 1112 н.п. очищали гель-электрофорезом и обозначали как 9А1. Для удобства фрагмент 9А1 лигировали в расщепленную Eco R1-Bam H1 плазмиду pUC18. Лигировапную смесь трансформировали в клетки штамма DН10b Е.соli. и отобранные клоны анализировали на присутствие инсерции. Один клон, 9А11, содержал инсерцию.
Кодирующая последовательность рецептора глюкагона была встроена в экспрессирующий вектор млекопитающих при помощи плазмиды р9А11 и клона 40-2-2 для получения полнокодирующей последовательности рецептора глюкагона. илазмиду р9А11 расщепляли рестриктазами Рvu 11 и Bam H1 для выделения фрагмента 967 н.п. Клон 40-2-2 расщепляли Ваm H1 и Sac 1 для выделения фрагмента 828 н.п., содержащего 3'-кодирующую последовательность человеческого рецептора глюкагона. Экспрессирующий вектор млекопитающих pHZ1 линеризовали (превращали в линейный) расщеплением с Eco R1 и его концы затупляли при помощи Т4 ДНK-полимеразы. Затем линеаризованный вектор с тупыми концами расщепляли при помощи Sac I. Pvu 11-Batn H1 фрагмент 967 н.п. и Bam H1 - Sac 1 Фрагмент 828 н.п. лигировали с расщеплением Sас 1 вектором с тупыми концами pHZ1.
Плазмида pHZ1 представляет собой экспрессирующий вектор, который можно применять для экспрессии белка в клетках млекопитающих или в системе трансляции социтов лягушки из мРНК, которые транскрибировались in vitro. pHZ1 единица экспрессии содержит мышиный промотор металлотионеина-1, промотор бактериофага Т7, фланкированные множественными клонирующими фрагментами, содержащими уникальные сайты рестрикции для инсерции кодирующих последовательностей, терминатор человеческого гормона роста и терминатор бактериофага Т7. Кроме того, рНZ1 содержит затравку репликации E.coli; бактериальный ген β-лактамазы; единицу экспрессии селектируемого маркера млекопитающих, содержащую промотор SV40 и затравку, ген устойчивости к неомицину и терминатор транскрипции SV40.
Плазмида рНZ1, содержащая кДНК последовательность рецептора глюкагона в корректной ориентации по отношению к промотору, была названа pLJ6'. Соединительные участки инсерции и вектора были секвенированы для подтверждения присутствия корректной последовательности. Плазмида pLJ6' была депонирована в АТСС (12301 Park Lаwn Drive, Rockville, MD 20852) под входным номером 69183 15 января 1993 г. Эта последовательность ДНК и выведенная из нее аминокислотная последовательность рецептора глюкагона показаны в SEQ ID 24 и SEQ ID 25.
Пример 6
Клонирование кДНК рецептора глюкагона клеток панкреатических островков человека
Кроме клонирования рецептора глюкагона из клеток печени человека, кДНК рецептора глюкагона получали из библиотеки кДНК клеток панкреатических островков человека. Аликвоту библиотеки кДНК клеток островков (пример 1 С) подвергали РСR амплификации с применением олигонуклеотида ZC5763 (SEQ ID 22), являющегося смысловым олигонуклеотидом, содержащим сайт Есо R1, фланкированный последовательностями из 5'-нетранслируемой последовательности кДНК человеческого рецептора глюкагона, и олигонуклеотида ZC5849 (SEQ ID 23), который является антисмысловым олигонуклеотидом, содержащим сайт Xho 1, фланкированный последовательностями из 3'-нетранслируемой последовательности кДНК человеческого рецептора глюкагона. Включение сайтов рестрикции в олигонуклеотидные праймеры облегчало направленное клонирование продуктов РСR в подходящий плазмидный вектор. Готовили реакционную смесь для РСR, содержащую 4 мкл библиотеки кДНК клеток островков (пример 1 С), 8 мкл 10х Рrоmеgа PCR буфера (Promega), 20 рмолей ZC5763 (SEQ ID 22), 20 пмолей ZC5849 (SEQ ID 23), 1 мкл раствора, содержащего 20 мМ каждого из дезоксирибонуклеотидов и 46,5 мкл воды. Смесь нагревали до 95oС в течение 3 минут, затем температуру снижали до 80oС в течение 3 минут. Смесь выдерживали при 80oС, пока она не была готовой для применения. Для начала реакции к реакционной смеси добавляли 20 мкл ферментной смеси, содержащей 2 мкл 10х Promega буфера для РСR (Рrоmеgа), 2 мкл 5 E/мкл Taq, 1 полимеразы (Cetus) и 16 мкл воды. На смесь наслаивали 50 мкл минерального масла (Sigma) и реакционную смесь подвергали 30 циклам PCR (95oС в течение 1 минуты, 55oС в течение 1 минуты, 72oС в течение 2 минут и 15 секунд, в течение которых инкубирование при 72oС проводили на 3 секунды дольше в каждом цикле) с последующим 10-минутным инкубированием при 72oС. После этого реакционную смесь выдерживали при 4oС. Агарозный гель-электрофорез 10 мкл-аликвоты продукта РСR показал присутствие фрагмента приблизительно 1,8-1,9 кб. На основании присутствия в pLJ6', кДНК человеческого рецептора глюкагона ожидали обнаружения фрагмента приблизительно 1845 н.п.
РСR реакционную смесь экстрагировали хлороформом с последующими двумя экстракциями смесью фенол/хлороформ и конечной экстракцией хлороформом. После конечной экстракции добавляли 5 мкл 4 мкг/мкл гликогена-носителя (Boerhinger Mannheim Corporation) и смесь осаждали в присутствии ацетата аммония и этанола. ДНK осаждали центрифугированием и осадок промывали 70% этанолом. Осадок ДНК ресуспендировали в воде и расщепляли при помощи Xhо 1 и Eсо R1. ДНК очищали гель-электрофорезом и субклонировали в плазмиду pBLUESCRIPT SK+ (Stratagenе Cloning Systems), которая была линеаризована при помощи Xho 1 и Eсо R1. Смесь для лигирования трансформировали в клетки штамма DH10B E.coli. (GIBCO-BRL). Из отобранных трансформантов получали плазмидную ДНK и подвергали ее действию рестриктазы и блоттингу по Саузерну. Клоны сравнивали с инсерцией кДНК человеческого рецептора глюкагона, присутствующей в pLG6'. Отобранные на основании диагностического расщепления рестриктазами клоны подвергали секвенированию. Секвенирование показало, что один клон, pSLIGR-1, содержал кодирующие последовательности рецептора глюкагона. Кодирующий район рSLIGR-1 содержал три нуклеотидные замены в кодирующем районе рецептора глюкагона по сравнению с кДНК печени, присутствующей в pLJ6'. Одной из нуклеотидных замен была молчащая мутация, две оставшиеся приводили к изменениям в консервативных аминокислотах, как показано в таблице 5. Анализ изменений позволяет предположить, что они могут быть результатом аллельных вариаций, представляющих полиморфные различия.
Пример 7
Клонирование гена рецептора глюкагона человека
Для получения геномного клона рецептора глюкагона дне библиотеки геномной ДНК скринировали с использованием кДНК человеческого рецептора глюкагона в качестве зонда. Амплифицированная геномная библиотека человеческой плаценты λFIX II и амплифицированная геномная библиотека легких человека λFIX (обе библиотеки получены из Stratagene Cloning Systems, номера по каталогу 946203 и 944201 соответственно) были подвергнуты скринингу на ген рецептора глюкагона человека.
Амплифицированную геномную библиотеку легких человека титровали и приблизительно 4•104 бляшкообразующих единиц (pfu) высевали с клетками штамма LE392 Е.соli (Stratagene Cloning Systems) на каждую из 30 чашек с диаметром 150 мм. Дополнительные 10 чашек засевали клетками штамма LE392 E.coli, приблизительно 6•104 pfu на чашку. Чашки инкубировали в течение ночи при 37oC и 30 чашек отбирали для скрининга.
Для каждой из 30 чашек готовили по два фильтра. Каждый фильтр готовили наложением на чашку НYВОND найлоновой мембраны (Amersham) в соответствии с процедурой, рекомендуемой изготовителем. Фильтры поднимали из чашек и клетки лизировали в 1,5 M NaCl, 0,5 М NаОН 5 минут при комнатной температуре. Фильтры нейтрализовали 5 минут в 1 М Трис-НСl (7,5), 1,5 М NaCl и фиксировали при помощи УФ (1200 микроджоулей) в STRATALINKER (Stratagene Сlоning Systems). После фиксации фильтров их предварительно промывали три раза в 0,25х SSC, 0,25% ДСН, 1 мМ ЭДТА при 65oС. После предварительного промывания фильтры делили на 6 серий из 10 фильтров, которые прегибридизовали в прегибридизационном растворе (5х SSC, 5x раствор Dеnhаrdt, 0,2% ДСН, 1 мМ ЭДТА), который фильтровали через фильтр 0,45 мкм и к которому добавляли 100 мкг/мл (конечная концентрация) денатурированной нагреванием ДНК спермы лосося непосредственно перед использованием. Фильтры прегибридизовали при 65oС в течение ночи.
кДНК человеческого рецептора глюкагона из р40-2-2 соединяли с произвольными праймерами с применением кита Amersham MEGAPRiME (Amersham) согласно способу, рекомендуемому изготовителем. Прегибридизационныи раствор из каждой серии фильтров заменяли свежим прегибридизационным раствором, содержащим 28,5•105 имп/мин зонда. Фильтры гибридизовали при 65oC в течение 20 часов. После гибридизации гибридизационный раствор удаляли и фильтры споласкивали 4 или 5 раз в промывном растворе, содержащем 0,25х SSC, 0,2% ДСН, 1 мМ ЭДТА при комнатной температуре. После споласкивания фильтры промывали последовательно 8 раз при 65oC в промывном растворе с последующим конечным промыванием при 70oC. После этого промывания Фильтры экспонировали на пленке для радиоавтографии (ХАR-5; Eastman Коdаk Со.; NY) в течение 4 дней при -70oC с усиливающим экраном.
Исследование радиоавтографа обнаружило присутствие 4 районов гибридизации с радиактивно меченым зондом. Пробку агара из каждого из 4 районов брали для очистки. Каждую агровую пробку вымачивали в течение ночи в 1 мл SM (Maniatis et al., eds., Molecular Cloning: A Laboratory Manual, Cold Spring Наrbоr, NY, 1982; здесь даны полные ссылки), 1% хлороформе. После инкубирования в течение ночи фаг из каждой пробки разводили 1:1000 SМ. Аликвоты 5, 25 и 50 мкл высевали с клетками штамма LE392 E.coli. Чашки инкубировали и по одному фильтру готовили из каждой чашки из посевов 5 и 25 мкл. Фильтры готовили, прегнбридизовали и гибридизовали и промывали, как описано выше. Фильтры экспонировали на пленке для радиоавтографии.
Исследование радиоавтографов обнаружило положительно меченые районы из каждого из 4 клонов. В целом 10 агаровых пробок брали из положительных зон, представляющих по меньшей мере две положительные зоны для каждого из исходных клонов. Агаровые пробки обрабатывали, как описано выше. Фаг из каждой агаровой пробки разводили 1: 10000 в SM. Аликвоты 2,5 и 10 мкл высевали с клетками штамма LE392 E.coli. Чашки инкубировали и по одному фильтру готовили из чашек, имеющих хорошо разделенные бляшки, фильтры готовили и гибридизовали, как описано выше. Радиоавтографы обнаружили зоны, соответствующие отдельным бляшкам. 12 положительных бляшек вынимали, и они представляли по меньшей мере один клон из каждого из 4 исходных положительных клонов. Одну бляшку из каждой чашки отбирали для дальнейшего анализа.
Агаровые пробки из фаговых клонов 2-2-1, 3-3-1, 14-2-1 и 11-2-1 обрабатывали, как описано выше. Фаг разводили 1:1000 в SМ и инокулировали в культуру клеток штамма LE392 E.coli. Двухцепочечную ДНК получали, как описано (С) Grossberger (Nuс. Acids Res. 15:6737, 1987; включается здесь в виде ссылки). Двухцепочечную ДНК расщепляли при помощи Хbа 1 для высвобождения геномной инсерции. Агарозный гель-электрофорез показал, что клоны 2-2-1 и 11-2-1 содержали инсерцию Хbа 1 9 кб (т.п.н.), клон 14-2-1 содержал инсерцию 15 кб и 3-3-1 содержал инсерцию 13 кб. Блоттинг по Саузерну расщепленных Хbа 1 и Xbа-Ваm H1 клонов показал, что кДНК человеческого рецептора глюкагона гибридизовалась с фрагментами, показанными в таблице 6.
Клоны 11-2-1 и 14-2-1 были выбраны для дальнейшего анализа. Для удобства названия клонов были изменены, как указано в таблице 6. Двухцепочечную дНК получали из каждого клона фага для субклонирования в плазмидный вектор по способу, описанному Maniatis. ДНК расщепляли Хbа 1, очищали на геле и субклонировали в плазмиду pBLUESCRUPT SK+ (Stratagene Сlоning Systems), которая была линеаризована расщеплением при помощи Хbа 1 и обработана щелочной фосфатазой теленка для предотвращения замыкания в кольцо. Смеси для лигирования вводили электропорацией в клетки DН10В ЕLЕСТROMАХ (GIBCO-BRL) в Biorad GENEPULSER (Biorad Laboratories; Richmond, CA) при 400 омах, 25 микрофарадах и 2,3 киловольтах. Из отобранных трансоормантов готовили плазмидную ДНК. Клоны, содержащие геномные инсерции, из клонов 6 и 2 были названы pSLHGR 6 и рSLHGR 2 соответственно. Эти клоны были секвенированы. Секвенирование и сравнение с кодирующим районом кДНК человеческого рецептора глюкагона обнаружили присутствие 12 экзонов, содержащих кодирующий район. Анализ локализации в хромосомах проводили на хромосомных нитях в метафазе, как описано Durnamet еt аl. (Mol. Cell. Biol. 8:1863-1867, 1988, включ. полной ссылкой) с применением биотинилированного зонда гена человеческого рецептора глюкагона и специфического для хромосомы 17 центромерного зонда. Денатурацию хромосом, гибридизацию и детектирование цвета проводили в основном, как описано Pinkel et al. (Рrос. Natl. Асаd. Sci. USA 83:2334-2938, 1386, включено в виде полной ссылки) в модификации Kievits еt аl. (Cytogenet. Cell Genet. 53: 134-136, 1990, включено в виде полной ссылки), за исключением того, что гибридизацию проводили в смеси 65% (об./об.) формамид/10% сульфат декстрана/2х SSC и постгибридизационные промывки выполняли со смесью 65% формамид/2х SSC при 42oС и затем 0,1х SSC при 55oC, как описано Palmiter et al. (Proc. Natl. Acad. Sci. 89: 6333-6337, 1932, включенный в виде полной ссылки). Местоположение q25 было подтверждено окрашиванием DАР1 некоторых гибридизованных метафазных нитей, как описано Testa et al. (Cytogenet. Cell. Genet. 60: 247-243, 1932; включ. здесь в виде полной ссылки). Окрашивание DАР1 дало Q-полоса-подобный рисунок.
Скрининг амплифицированной геномной библиотеки человеческой планцеты (Stratagene) на ген рецептора глюкагона при помощи описанного выше способа был неудачным. Вкратце, библиотеку титровали и клетки штампа LE392 E.coli и приблизительно 5•104 рfu высевали на каждую из 30 чашек с диаметром 150 мм. Кроме того, 11 таких же чашек засевали приблизительно 105 pfu и клетками птамма Е. coli LE392. Чашки инкубировали в течение ночи при 37oC и отбирали 38 чашек для скрининга.
Фильтры-лифты готовили, промывали и прегибридизовали, как описано выше. Фрагмент кДНК рецептора глюкагона панкреатических островков человека, G 30 (пример 4) произвольно праимировали с применением кита Amersham MEGAPRIME (Аmеrshаm) по способу, рекомендуемому изготовителем. Прегибридизационный раствор для каждой серии фильтров заменяли свежим прегибридизационным раствором, содержащим 1,1•106 имп/мин зонда.
Фильтры гибридизовали при 65oС в течение ночи. После гибридизации гибридизационный раствор удалили, и фильтры споласкивали 4 или 5 раз в промывном растворе, содержащем 0,25•SSC, 0,25% ДСН, 1 мМ ЭДТА при комнатной температуре. После ополаскивания фильтры промывали последовательно 8 раз при 65oС в промывном растворе. После промывания при 70oС фильтры экспонировали на пленке для радиоавтографии (XAR-5; Eastman Kodak Со.) в течение 4 дней при -70oC с усиливающим экраном.
Исследование радиоавтографов обнаружило отсутствие достоверных положительных сигналов; однако 7 зон, соответствующих слабо меченым участкам, брали для дальнейшего анализа. Эти клоны подвергали вторичному скринингу, как описано выше, но радиоавтографы вторичного скрининга не обнаружили меченых зон. Этими клонами далее не занимались.
Пример 8
Экспрессия кДНК рецептора глюкагона в клетках млекопитающих
А. Экспрессия крысиного рецептора глюкагона в клетках ВНК570
Плазмиду рLJ4 котрансфицировали с плазмидой рLJ1 в клетки BHK570 (депонированные в АТСС под Accession 10314) при помощи кальций-фосфатного способа, в основном описанного Graham and Vander Eb (Virology 52:456, 1973, включ. здесь в виде полной ссылки). Плазмида pLJ1 была произведена из плазмиды р416, которая содержит затравку Adenovirus 5, энхансер SV40, основной поздний промотор Adenovirus 2, состоящий из трех частей лидер Adenovirus 2, 5'- и 3'- сайты сплайсинга, кДHK DНFR, сигнал полиаденилирования SV40 и рМ-1 вектор (Lusky and Botchan, Nature 293:79-81, 1981). Единицу экспрессии Есо R1-Хbа 1 DНFR из р416 лигировали в pUC18, которая была линеаризована расщеплением с Есо R1 и Хbа, для конструирования плазмиды pLJ1. Трансфицирозанные клетки выращивали на ростовой среде (модифицированной по способу Дульбекко среде Игла), содержащей 10% плодную сыворотку теленка, 1x PSN смесь антибиотиков (GIBСО BRL 600-5640) и 2,0 мМ L-глутамина). После нескольких дней в неселективной ростовой среде ростовую среду заменяли селективной средой (ростовой средой, содержащей 250 мМ метотрексат (MTX)). Клетки разрушали и разводили 1:20 и 1:50 в селективных средах на 10-см чашках. После 7-10 дней селекции в 250 мМ MТХ колонии брали при помощи клонирующих цилиндров в ячейки 24-ячеечных планшетов. Полученные колонии тестировали на способность связываться с глюкагоном, как описано выше (пример 3). Связывание глюкагона проводили также на целых клетках путем посева 2•105 клеток каждого клона в ячейки планшета с 24 ячейками. Планшеты инкубировали в течение 72 часов при 37oС в 5% CO2. Связывание глюкагона проводили, как описано в примере 3, за исключением того, что после конечного промывания PBS клетки удаляли из ячеек в пробирку путем трипсинизации. Пробирки считали на радиоактивность в γ-счетчике. Клетки, имеющие наивысший счет импульсов и, следовательно, способные связывать больше всего глюкагона, были выбраны для дальнейшей характеристики. Отобранные трансформанты также тестировались на медиируемую глюкагоном ответную реакцию внутриклеточного кальция, как описано в примере 8Е, и медиированную глюкагоном ответную реакцию сАMР, как описано в примере 8D. Тесты связывания глюкагона проводили также на препаратах мембран из трансфектантов, как описано ниже.
В. Связывание глюкагона препаратами мембран
Мембраны из pLJ4 BНК трансфектантов сравнивали с препаратами мембран печени крыс на способность связывать 125I-глюкагон. Мембраны получали в основном, как описано Rodbell et al. (J.Biol. Chem. 246:1861-1871, 1971, включ. здесь в виде полной ссылки). Две серии мембран печени получали приблизительно из 80 г печени из 12-16 обезглавленных самок крыс. Печени удаляли быстро и помещали в охлажденный льдом химический стакан. Печени делили на части по 10 г. Части измельчали ножницами и соединительные ткани удаляли во время процедуры измельчения.
Части по 10 г отдельно гомогенизировали в гомогенизаторе Dоunсе. Для каждой части добавляли 25 мл среды (таблица 2) к измельченной ткани в гомогенизаторе и ткань гомогенизировали в стакане на льду при помощи 8 энергичных ударов незакрепленным пестиком. После гомогенизации гомогенаты собирали в 450 мл холодной среды (таблица 2). Объединенные гомогенаты перемешивали в течение 3 минут и фильтровали через 2 слоя марли с последующим фильтрованием через 4 слоя марли. Затем гомогенаты центрифугировали в течение 30 минут при 1500 g, при 4oC. Супернатанты выбрасывали, а осадки объединяли в чистый гомогенизатор Dounce. Осадки ресуспендировали тремя мягкими ударами незакрепленного пестика. Ресуспендированные осадки декантировали в 250-мл мерный сосуд, содержащий 62 мл 69% раствора сахарозы (таблица 2). Дистиллированную воду добавляли до конечного объема 110 мл и смесь тщательно смешивали и хранили на холоду. Концентрацию раствора доводили до 44,0%±0,1% при помощи 69% сахарозы или воды в зависимости от измерений рефрактометром (Baush and Lomb, Rochester, N. Y. ). Сахарозную суспензию распределяли поровну в ультрацентрифужные пробирки 25•89 мм. На каждую суспензию наслаивали 20 мл 42,3% раствор сахарозы (таблица 2) и суспензии центрифугировали в течение 150 минут при 24000 об/мин, в роторе SW28 (Sorval, Dupont Company, Wilmington Dеl.) при 4oС.
После центрифугирования плавающий материал из каждой пробирки удаляли отсасыванием в 10-мл шприц через иглу 18 размера. Материал из каждой пробирки соединяли и ресуспендировали приблизительно в 10 мл среды (таблица 2) путем втягивания и выпускания смеси через иглу в центрифужной пробирке. Пробирку заполняли средой (таблица 2) и центрифугировали при 15000 об/мин в роторе SS-34 (Sorval).
Супернатант осторожно декантировали и выбрасывали. Осадок ресуспендировали в среде (таблица 2) и разбавляли 1:1000 дистиллированной водой. Поглощение измеряли в кювете на 1 см для определения концентрации белка. Концентрацию белка определяли по формуле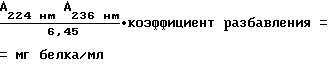
Препарат мембран делили на аликвоты, быстро замораживали в бане сухой лед/этанол и хранили при -80oС.
Мембраны получали из ВНК трансфектантов, которые выращивали до слияния в чашках с диаметром 150 мм в селективной среде. Две чашки слившихся трансфектантов ополаскивали 2 раза холодным забуференным при помощи фосфатного буфера солевым раствором (PBS; Sigma Chemical Co., St.Louis, Md.) и к каждой чашке добавляли 10 мл PBS, содержащего 1 мМ PMSF. Клетки из каждой чашки соскребали в раствор PBS и клетки из каждой чашки переносили в свежие пробирки. Каждую чашку ополаскивали 5 мл PBS, содержащего 1 мМ PMSF и промывки соединяли с соответствующими клетками. Клетки центрифугировали при 2000 об/мин в настольной центрифуге при 4oС. Супернатанты выбрасывали, а клетки ресуспендировали в 30 мл 5 мМ HEPES 1 мМ РМSF, рН 7,5. Клетки инкубировали на льду 15 минут с последующим центрифугированием при 47800 g при 4oС. Каждый осадок ресуспендировали в растворе PBS, содержащем 1 M PMSF, разделяли на аликвоты и замораживали при -80oC.
Конкурентный анализ при помощи тестов связывания глюкагона проводили на препаратах мембран печени крыс и ВНК трансформантов. Вкратце, готовили реакционные пробирки, содержащие 20 мкл глюкагона из его растворов 10-11 М - 10-6 М, разведенных в 10 мМ НОАс, или 20 мкл БСА. К каждой пробирке добавляли 100 мкл 2х буфера для связывания; 20 мкл 125I-глюкагона (Аmеrsham); 20 мкл 1 мМ NaHCO3; 20 мкл 10 мМ HOAc, 0,5% БСA (Novo Nordisk N/A, Bagsvaerod, Dеnmаrk); и 40 мкл дистиллированной воды. Реакцию связывания инициировали добавлением 20 мкл препарата мембран к реакционным пробиркам. Реакции инкубировали 30 минут при 30oС. Мембраны осаждали центрифугированием в микроцентрифуге при высокой скорости в течение 10 минут при 4o0. Супернатанты из каждой пробы отсасывали и осадки считали на радиоактивность. Конкуренция с намеченым глюкагоном дала приблизительно одинаковые сигмоидальные кривые для связывания глюкагона (фиг.3). Анализ Скетчарда (Scatchard, Ann. N.Y. Acad. Sсi. 51: 660-672, 1949, включ. здесь в виде полной ссылки) показал, что видимая Кd равна 50 нМ для клонированного рецептора и 49 нМ для мембран печени крыс (фиг.4).
Специфичность рецептора, кодируемого pLJ4, определяли по способности родственных пептидных гормонов конкурировать за связывание 125I-глюкагона. Микромолярные количества глюкагона и родственных пептидов (таблица 7) добавляли с 125I-глюкагоном к мембранным препаратам pLJ4 трансфектантов в тесте связывания, описанном выше. Только нативный глюкагон и антагонист амид дез-His1[Glu 9] глюкагона были способны конкурировать с 125I-глюкагоном за связывание с мембранами.
С. Экспрессия крысиного рецептора глюкагона в COS-7 клетках
Способность COS-7 клеток, трансфицированных pLJ4, стимулироваться глюкагоном для увеличения уровня сАМР оценивали при помощи Amersham SPA кита (Amersham), как описано нитке (пример 8). Анализ показал, что стимулируемые глюкагоном рLJ4 трансфектанты накапливали приблизительно в 5 раз больше сАМР, чем контрольные, трансфицировянные только вектором COS-7 клетки. Каждый из родственных пептидов, таких как секретин, VIР, РТН, GLP и кальцитонин, добавляли к трансфектантам в концентрациях 100 нМ - 1000 нМ и оценивали их способность повышать уровни сАМР. Результаты этих испытание показали, что ни один из этих пентидов не был способен индуцировать значительно уровни сАМР.
D. Определения люциферазной и аденилатциклазной активности на целых клетках
кДНK крысиного рецептора глюкагона экспрессировали в клеточной линии ВHK570, стабильно трансфицированной ZК6, единицей экспрессии, содержащей промотор, в которое был по меньшей мере один отвечающий на cAМP элемент, кДНК люциферазы и hGH-терминатор. Эта клеточная линия делает возможным измерение люциферазной активности, аденилатциклазной активности и внутриклеточных концентраций кальция в ответ на связывание глюкагона с его рецептором.
Проэнкефалиновый отвечающий на cAМP элемент (СRE) в плазмиде ZК6 получали из Zem233. Zem233 была произведена из плазмид Zem67 и Zem106. Плазмиду Zem106 конструировали из предшественника Zеm93. Для конструирования Zem93 Kрn I-Bam HI фрагмент, содержащий MТ-1 промотор, выделяли из MTRGHIII (Palmiter et al. , Science 222:809-814, 1983) и встраивали в рUС18. Затем плазмиду Zеm93 расщепляли рестриктазой Sst1 и повторно лигировали с образованием плазмиды Zem106, в которой удалялись приблизительно 600 н.п. последовательности в направлении 5' по отношению к MT-1 промотору.
Проэнкефалиновый СRE выстраивали в 5'-конец SV40 промотора в Zem106 путем расщепления сначала Zem106 рестриктазами Eсо R1 и Sst1 для выделения содержащего вектор фрагмента. Олигонуклеотиды ZС982 и ZC983 (SEQ 1D 3 и 4 соответственно) были сконструированы таким образом, чтобы они кодировали при гибридизации проэнкефалиновый СRЕ от нуклеотида - 73 до нуклеотида - 133 (Come et al., Nаturе 323:353-356, 1986), фланкированный 5'-сайтом Eco RI и 3'-сайтом Sst1. Олигонуклеотиды ZC982 и ZC983 (SEQ ID 3 и 4 соответственно) подвергали действию киназы, гибридизовали и лигировали с линеаризованной плазмидой Zеm106 с образованием плазмиды Zеm224.
Плазмиду Zem67 получали расщеплением р1С19R (Marsh еt аl., Gene 32: 481-486, 1984) растриктазами Sma I и Hind III. Затем затравку (оri - район) SV 40 от положения карты 270 (Pvu II) до положения 5171 (Нind III) лигировали с линеаризованной pICI9R, получая плазмиду Zеm67. Затем концевой Hind III-Bam HI фрагмент гена устойчивости к неомицину SV 40 из плазмиды pSV 2-nео (АТСС Accession 37149) встраивали в расщепленную Нind III-Ваm II плазмиду Zem67, получая Zem220.
Экспрессирующую единицу, SV 40 промотор-ген устойчивости к неомицину-SV40 терминатор, из плазмиды Zem220 выделяли в виде фрагмента Есо RI. плазмиду Zem224 расщепляли Есо RI и обрабатывали щелочной фосфатазой теленка для предотвращения замыкания на себя с образованием кольца. Единицу экспрессии неомицина и линеаризованную плазмиду Zem224 лигировали. Плазмида, содержащая SV40 промотор, смежный с СRE, была названа Zem233.
Плазмиду Zem233 модифицировали инсерцией дополнительной СRЕ последовательности, ТАТА-блока и части lacZ кодирующей и поли(А)-последовательностей, соединенных непосредственно 3' с проэнкефалиновой последовательностью СRЕ, так что полученная единица экспрессии была в противоположной ориентации относительно единицы экспрессии устойчивости к неомицину, присутствующей в Zem233. Плазмиду Zem233 линеаризовали расщеплением плазмидами SstI и Ваm HI. Олигонуклеотиды ZC3509 и ZC3510 (SEQ ID 5 и 6 соответственно) конструировали таким образом, что при гибридизации полученный дуплекс кодировал α-гликопротеин СRЕ (Dеlеgеаnе еt al., Mol. Сеll. Вiоl. 7:3994-4002, 1987) с 5' SstI липким концом и 3' Eco RI липким концом. Олигонуклеотиды гибридизовали по стандартным способам. ТАТА-блок тимидинкиназы получали в виде EcoRI-Pst I фрагмента от нуклеотида -79 до нуклеотида +18 гена тимидинкиназы (McKnight, Cell 31:355-366, 1982). 3'-последовательность lасZ гена и связанную с ним поли (А) последовательность получали в виде Рst I-Bam HI фрагмента из плазмиды рLасF (полученной из Jaques Peschon, Immunex Corp., Seattle, wash.), которая содержит lасZ кодирующий район и мышиную протаминовую терминаторную последовательность, клонированную в рUС18 вектор. Линеаризованную рестриктазами Sst I-Bam HI плазмиду Zem233, SstI-Eco RI ZC3509/ZC3510 адаптер, Eco RI-Pst I фрагмент ТАТА-блока и Pst I-Bam HI lacZ последовательность лнгировали. Плазмида, содержащая единицу экспрессии в правильной ориентации по отношению к единице экспрессии гена устойчивости к неомицину Zem233, была названа КZ5.
Ген люцеферазы и терминаторную последовательность человеческого гормона роста (hGH) использовали для замены lасZ кодирующей и поли (А) последовательностей, присутствующих в КZ5. Ген люциферазы исходно получали из плазмиды α-168luc (Dеlеgеаnе еt аl., Mol. Cell. Вiоl. 7:3994-4002, 1987; и de Wet et al. , Mоl. Cell. Biоl. 7:725-737, 1987) в виде Xho I-Xba I фрагмента 1,7 кб. Терминатор hGH получали в виде Хbа I-Sal I фрагмента из Zеm219b (депонированной в виде трансформанта Е.соli в АТСС (Rockville, Md.) под Accessi on АТСС 68079). Последовательности гена люциферазы и терминатора hGH субклонировали в линеаризованную рестриктазами Xho I-Sal I плазмиду pICI9H (Marsh et al., ibid.) для удобства. Полученную плазмиду КZ8 расщепляли Xho I и Sal I для выделения последовательностей гена люциферазы-терминатора hGH. Плазмиду KZ5 расщепляли Sal I для выделения содержащего вектор фрагмента и обрабатывали щелочной фосфатазой теленка для предотвращения повторного образования кольца. Xho I - Sal I лицифераза-терминатор hGН фрагмент лигировали с расщепленной Sal I плазмидой KZ5. Плазмида, содержащая фрагмент люцифераза-терминатор hGН в правильной ориентации по отношению к промотору, была названа КZ6.
Плазмиду KZ6 трансфицировали в ВHK570 клетки (депонированные в АТСС под Accession 10314) с применением способа осаждения фосфатом кальция, описанного в основном Graham and Van dеr Eb (Virology 52:456, 1973, включ. здесь в виде полной ссылки). Трансфицированные клетки выращивали в среде для выращивания (модифицированной по способу Дульбекко среде игла (DMEM), содержащей 10% плодную сыворотку теленка, 1x PSN смесь антибиотиков (GIBCO-BRL) и 2,0 мМ L-глутамин). После нескольких дней в неселективной среде для выращивания эту среду заменяли селективной средой G418 (среда для выращивания, содержащая G418 в количестве 500 мкг/мл). Затем клеткам давали расти до слияния, после чего их трипсинизировали и высевали при лимитирующем разведении в ячейки планшета с 96 ячейками. Клетки росли в течение 1-2 недель в селективной среде G418. Клоны из ячеек, содержащих отдельные колонии, тестировали на способность отвечать на форсколин в описанном ниже люциферазном тесте. Форсколин повышает уровень клеточной cAМР и, следовательно, связанные сАМР-зависимые биологические ответные пути независимым от рецептора образом. Клон, способный отвечать на форсколин, был назван ВНК/КZ 6-19-46.
ВНК/КZ 6-19-46 клетки котрансфицировали с pLJ4 и pLJ1 или с рZСЕP и рLJ1 (рZСЕР трансфектанты использовали в качестве отрицательных контролей), как описано выше при помощи медиированной фосфатом кальция трансфекции. Трансфектанты отбирали в 250 нМ метотрексате, как описано ранее.
Трансфектанты тестировали в трех повторностях на индуцирование СRЕ-люциферазной ответной реакции выбранными агонистами. Тестировали 6 произвольно выбранных рLJ4 трансфектантов и произвольные рZСЕP трансфектанты (отрицательные контроли). Микротитрационные планшеты готовили таким образом, что каждая ячейка содержала 2•104 клеток в 100 мкл селективной среди, и клетки выращивали в течение ночи. Агонисты готовили в селективной среде при 2-конечной концентрации
1 мкМ - глюкагон
200 нМ - глюкагон-подобный пептид (GLР)
20 нМ - вазоактивный кишечный пептид (VIP)
100 нМ - кальцитонин (СТ)
20 мкМ - форсколин (CalBiochem, Sаn Diеgо, Саlif.)
Индукцию инициировали добавлением 100 мкл каждого из 2-х растворов в ячейки с тремя повторностями. Неиндуцированные уровни определяли в трех ячейках, к которым добавляли 100 мкл DMEM, содержащей 10% плодную сыворотку теленка. Планшеты инкубировали в течение 4 часов при 37oС и 5% СО2 для индуцирования образования люциферазы.
После индуцирования среду удаляли и ячейки промывали 1 раз 200 мкл/ячейку PBS. После промывания к каждой ячейке добавляли 25 мкл 1х реагента для лизиса культур клеток (Luciferase Assay System, Promega Corp., Madison, Wis.) и планшеты инкубировали 15 минут при комнатной температуре. Затем планшеты переносили в Labsystems Luminoskan microtiter luminometer (Labsystems Inc. , Morton Grove, Ill.), который добавлял 40 мкл субстракта для определения люциферазы (Luciferase Assay System, Promega), смешивали реакционную смесь 3 секунды и интегрировал люциферазный сигнал за 2 секунды на ячейку. Индукцию в какое-то количество раз люциферазы для каждого агониста рассчитывали следующим образом:
Один клон pLJ4 трансфектанта, КZ6/rGR-DHEFR-2, обнаружил индуцирование в 6-10 раз люциферазы глюкагоном, но он не индуцировался GLP, VIP или СТ.
сАMР-ответ клона трансфектанта KZ6/rGR-DHFr-2 на глюкагон и форсколин тестировали также при помощи радиоиммуноанализа с применением cAМР [125I] сцинтилляционный тест-системы близости (Amersham) согласно инструкциям изготовителя. Вкратце, 100 мкл 2•105 клеток на мл KZ6/rGR-DHFR-2-клеток высевали в ячеки культурального планшета со множеством ячеек и выращивали в течение ночи в селективной среде. Глюкагон и форсколин готовили в DMEM, 10% плодной сыворотке теленка, 10 мкМ IВМХ при 0,0001-1000 нМ и 26 мкМ соответственно.
Среды для выращивания заменяли 50 мкл на ячейку агониста (либо глюкагона, либо форсколина). Клетки инкубировали с агонистами 10 минут при 37oС, 5% СО2. После инкубирования клетки лизировали путем добавления 200 мкл кипящей воды в каждую ячейку. После 25 минут собирали супернатанты и разводили 1: 5 или 1:40 в ацетатном буфере (сАМР [125I] Scintillation Proximity Assay System (Amersham)). Пробы ацетилировали с применением триэтиламина и уксусного ангидрида согласно протоколу, обеспеченному изготовителем.
Аликвоту каждой ацетилированнои пробы соединяли с 75 мкл 125I-cАMР, 75 мкл антнсыворотки против сукцинил-сАMР и 75 мкл SPA бусин, соединенных с ослиными анти-кроличьими IgG (все тест-растворы обеспечены в сАМР [125I] Sсintillаtiоn Prохimity Assay System (Amersham)) в ячейке LKB Т-планшета. Планшеты закрывали герметично и инкубировали в течение ночи с непрерывным качанием на ротационном шейкере при 200 об/мин. Пробы считали на радиоактивность в жидкостном сцинтилляционном счетчике 1205 BETAPLATE (Pharmacia LKB Instruments Inc., Gaithersburg, Md.). Также получали стандартную кривую 2-128 фмолей ацетилированного сАМР. Определяли также общую связанную 125I-сАMР и неспецифическое связывание. КZ6/rGR-DНFR-2 обнаружил индукцию в 140 раз уровней сАМР при насыщающем глюкагоне (10-100 нМ) и ED50 0,25 нМ.
Е. Определение концентрации внутриклеточного кальция
Ответы внутриклеточного кальция pLJ4 трансфектантов на глюкагон тестировали при помощи способа, описанного Grynkiewicz еt al. (J.Biol. Chem. 260: 3440-3450, 1985, включ. здесь в виде полной ссылки). Плазмидные pLJ4 ВHК трансфектанты высевали в 2-луночные камеры покровных стекол (NUNC) при 5•104 клетках на камеру. Клетки выращивали 1-3 дня при обычных условиях культуры в селективной среде с метотрексатом. Среду удаляли отсасыванием и камеры ополаскивали дважды 1 мл буфера для получения изображения (Imaging Buffer) (таблица 3). Клетки инкубировали в течение 30 минут в темноте при комнатной температуре с раствором Fura-2 AM (таблица 3). После инкубирования раствор Fura-2 AM удаляли и клетки споласкивали 1 мл Imaging Buffer трижды. После конечного ополаскивания 0,5 мл буфера оставляли в каждой камере. Клетки держали в темноте при комнатной температуре 30-120 минут.
Получение изображения проводили в обращенном флуоресцентном микроскопе Nicon Diaphot, оборудованном лампой с ртутной дугой и 10х и 40х Nicon Fluor dry objective линзами. Эксперименты контролировали и анализировали с применением SUN SPАRС II Work Station и программным обеспечением Inovision (Rеsеаrсh Triangle Park, N.С.) RATIOTOOL. Чередующиеся возбуждающие длины волн контролировались этим программным обеспечением через автоматизированный фильтр-диск (диск с фильтрами), содержащий фильтры, пропускающие полосы 340 им и 380 нм. Эмиссионные изображения направлялись дихроичным зеркалом (отсекающим 380 нм) к камере Dage-MTI 72 СCD, снабженной усилителем изображения Genesis II, и записывались в цифровом виде программным обеспечением.
Внутриклеточную концентрацию кальция контролировали путем расчета отношения интенсивностей эмиссии при каждой из двух возбуждающих длин волн (340/380) для каждого элемента изображения (пиксела) цифрового микроскопического поля изображения (512•480 пикселов). Grynkiewicz et аl. (ibid.) показали, что это отношение связано с концентрацией кальция, обнаруживаемой при помощи Fura-2-красителя, внутри клеток, которые поглощают и деэтерифицируют ацетоксиметилпроизводное (fura-2 AM), использованное для загрузки клеток. Программные средства RATIOTOOL изображают эту информацию в виде ложного цветного изображения, которое может быть калибровано для концентрации кальция. Были получены изображения и такой расчет выполняли с интервалом 5 секунд во время каждого эксперимента.
Клетки контролировали в течение по меньшей мере 60 секунд (12 изображений) для установления уровня отсчета перед стимулированием. Стимулирование выполняли путем добавления 0,5 мл буфера для изображения, содержащего 200 нМ глюкагона, к 0,5 мл этого буфера в камере, что давало конечную концентрацию 100 нМ. проводили миниторинг клеток и изображения записывали в течение по меньшей мере 3 минут после стимулирования.
Отношения изображений, соответствующие числу клеток в каждом поле, резко изменялись вскоре после добавления глюкагона. Это было выражено в количественном виде при помощи RАТIOТООL программного обеспечения для расчета средней величины для специфических участков отношения изображения, соответствующего каждой из отвечающих клеток. Клетки со средним остановившимся отношением приблизительно 1,4 обнаруживают быстрый рост этого отношения до величины 3-4. Они оставались при этой высокой величине в течение 40-50 секунд, а затем постепенно возвращались обратно к фоновой линии. Независимые калибровочные эксперименты свидетельствуют о том, что это отражает изменения во внутриклеточной концентрации кальция от исходной величины приблизительно 150 нМ до пика приблизительно 400 нМ.
F. Определение инозитфосфата
ВHК 570 клетки, экспрессирующие рецептор глюкагона из pLJ4, или ложно-трансфицированные ВНК 570 клетки высевали в 24-луночные культуральные планшеты для культуры ткани при 200000 клеток на лунку. После 24 часов клетки в каждой лунке метили инкубированием в 0,5 мл селективной среды с метотрексатом (МТХ), содержащей 2,0 мкКи мио-(2-3Н)инозита (удельная активность - 20 Ки/ммоль; Amersham). В конце 24-часовой инкубации клетки промывали 1 мл предварительно нагретой DMEM (модифицированной по способу Дульбекко средой Игла; JRРН Biosciences, Lenexa, Kan.), которая содержала буфер 20 мМ HEPES, рН 7,0 (Sigma Chemical Co.), содержащий 10 мM LiCl. Промывную среду удаляли отсасыванием и заменяли 900 мкл свежей забуференной среды. Клетки инкубировали 5 минут при 37oС. После инкубации каждый агонист или антагонист добавляли к трем лункам (три повторности) и инкубировали в соответствии с объемами и условиями, изложенными в таблице 8.
Реакцию останавливали помещением клеток на лед. После отсасывания сред клетки лизирозали добавлением 1 мл DMЕМ и 1 мл ледяной 10% хлорной кислоты. После 10 минут клеточные лизаты переносили в пробирки нa льду, каждая из которых содержала 500 мкл 10 мМ ЭДТА, рH 7,0. Пробы нейтрализовали добавлением 900 мкл 1,5 М КОН в 60 мМ HEPES-буфере и добавлением по каплям раствора KOH-HEPES до тех пор, пока не был достигнут рН между 7 и 7,5. Нейтрализованные пробы замораживали при -20oС в течение ночи. Замороженные пробы оттаивали и осадкам давали осесть. Супернатанты наносили на микроколонки AМPREP (Amicon), которые последовательно промывали 5 мл метанола и 5 мл 1 M KHCO3 с последующим промыванием 15 мл воды. После нанесения проб собирали проходящий через колонки раствор. Колонки промывали 1 мл воды 4 раза и после каждой промывки собирали фракции по 1 мл. Инозитфосфат элюировали из колонки четырьмя последовательными нанесениями по 1 мл 0,25 М KНCO3, что дало 1 мл-пробу после каждого нанесения КНСО3. 10 мл OPTIFLUOR (packard Instrument Co., Menden, Conn. ) добавляли к каждой пробе и пробы считали на радиактивность. Стимуляция инозитфосфатного пути подтверждалась увеличением уровней меченого инозитсоосфата. Ни в одной пробе не наблюдали увеличения образования инозитфосфата.
G. Экспрессия человеческого рецептора глюкагона в СОS - 7 клетках
Плазмиду pLJ6' трансфицировали в COS 7 клетки по способу с ДEАЭ-декстраном, как описано выше. Клетки выращивали на предметных стеклах с камерами (как описано в примере 3) в течение 72 часов после трансфекции. Анализ связывания глюкагона in situ с применением 125I-глюкагона с последующей эмульсионной радиоавтографией выполняли, как описано в примере 3. Более 50% клеток, трансфицированных плазмидой pLJ6', специфически связывали глюкагон.
Н. Экспрессия человеческого рецептора глюкагона в BНК570 клетках
Клетки BHK570 (депонированные в АТСС под Accession 10314) были трансфицированы плазмидой pLJ6' при помощи способа трансфекции с фосфатом кальция (пример 8). Трансфицированные клетки отбирали в присутствии G418, пока не становились видны отдельные колонии. Колонии клонировали с применением клонирующих цилиндров. Было показано, что эти клоны связывают глюкагон. Связывание in situ с иодинированным глюкагоном проводили, как описано выше.
Трансфицированные плазмидой pLJ6' клетки тестировали на накопление cAMP, как описано в примере 8С. Трансфектанты тестировали после стимуляции глюкагоном, секретином, VIР или GLР-I. Тесты показали, что трансфицированные pLJ6' клетки накапливали повышенные концентрации cAMP после стимулирования глюкагоном, по сравнению с контрольными клетками. Стимулирование трансфектантов секретином, VIP или GLP-I не обнаружило увеличения накопления сАMP. Ответ внутриклеточного кальция определяли в основном, как описано в примере 8E. Стимулируемые глюкагоном трансфицированные рLJ6' клетки показали увеличение уровней внутриклеточного кальция, как показано быстрым увеличением флуоресценции индикатора кальция Furа-2. Эти результаты показывают, что pLJ6' кодирует функциональный человеческий рецептор глюкагона, способный связывать глюкагон и облегчать трансакцию сигнала.
На основании изложенного будет понятно, что хотя специфические варианты данного изобретения описаны здесь в целях иллюстрации, могут быть сделаны многочисленные модификации без отхода от сущности и сферы данного изобретения. Поэтому изобретение не ограничивается описанным, за исключением прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИСПОЛЬЗОВАНИЕ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ, СОДЕРЖАЩЕЙ ПЕПТИД, ПОДАВЛЯЮЩИЙ АППЕТИТ | 1997 |
|
RU2197261C2 |
| НОВЫЙ ЛИГАНД РЕЦЕПТОРА ЦИТОКИНА ZCYTOR17 | 2003 |
|
RU2490276C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЛОГИЧНОГО БЕЛКА | 1994 |
|
RU2143495C1 |
| ТОЛЕРОГЕННАЯ ДНК-ВАКЦИНА | 2017 |
|
RU2752608C2 |
| БЕЛКИ, СВЯЗЫВАЮЩИЕ АНТИГЕН - ЛИГАНД CD30 ЧЕЛОВЕКА | 2013 |
|
RU2650800C2 |
| АНТИТЕЛА К NKG2A И ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2499001C2 |
| ПОЛИПЕПТИД ТРОМБОПОЭТИН (ТРО), ДНК, КОДИРУЮЩАЯ ПОЛИПЕПТИД ТРО (ВАРИАНТЫ), СПОСОБ ПОЛУЧЕНИЯ ПОЛИПЕПТИДА (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ (ВАРИАНТЫ), АНТИТЕЛО К ПОЛИПЕПТИДУ ТРО | 1995 |
|
RU2194074C2 |
| РЕЦЕПТОР ГЛЮКАНОВОГО АКТИВАТОРА И КОДИРУЮЩАЯ ЕГО МОЛЕКУЛА ДНК | 1995 |
|
RU2146705C1 |
| МОДИФИЦИРОВАННЫЙ ФАКТОР VII | 1995 |
|
RU2214833C2 |
| ВЫДЕЛЕННЫЙ РАСТВОРИМЫЙ IL-20 РЕЦЕПТОР (ВАРИАНТЫ) | 2000 |
|
RU2279441C2 |































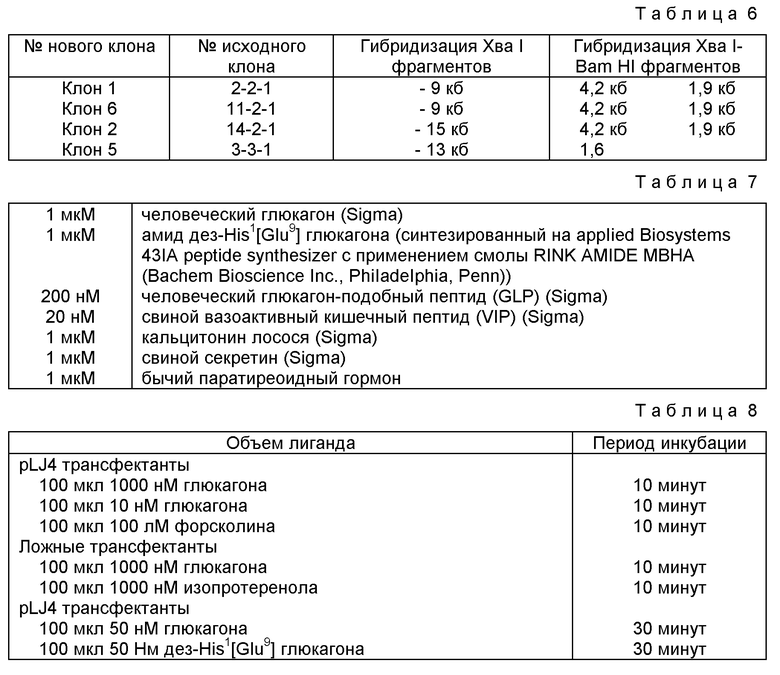
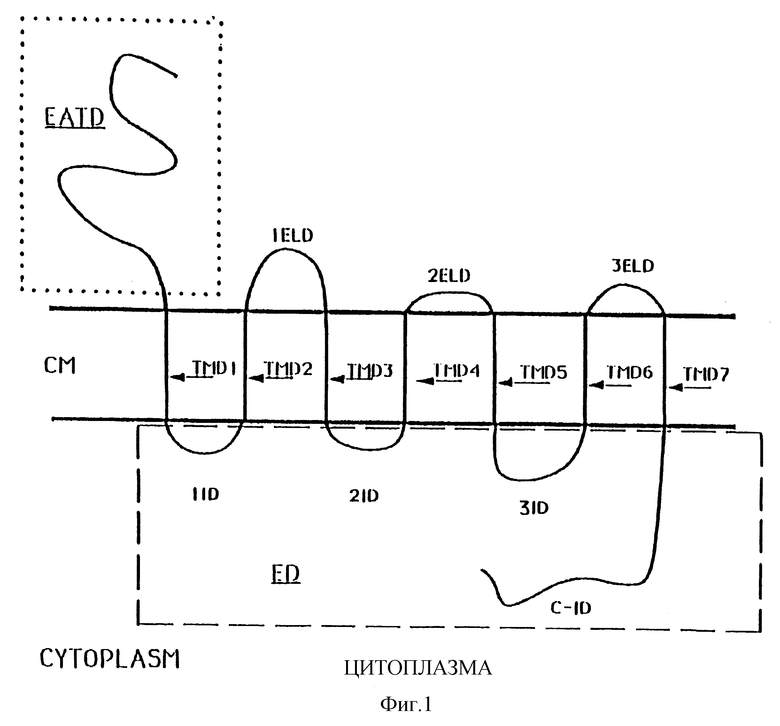
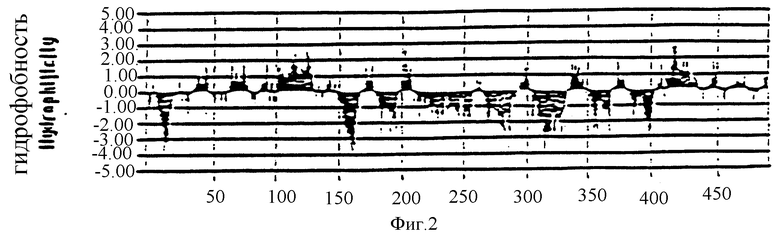
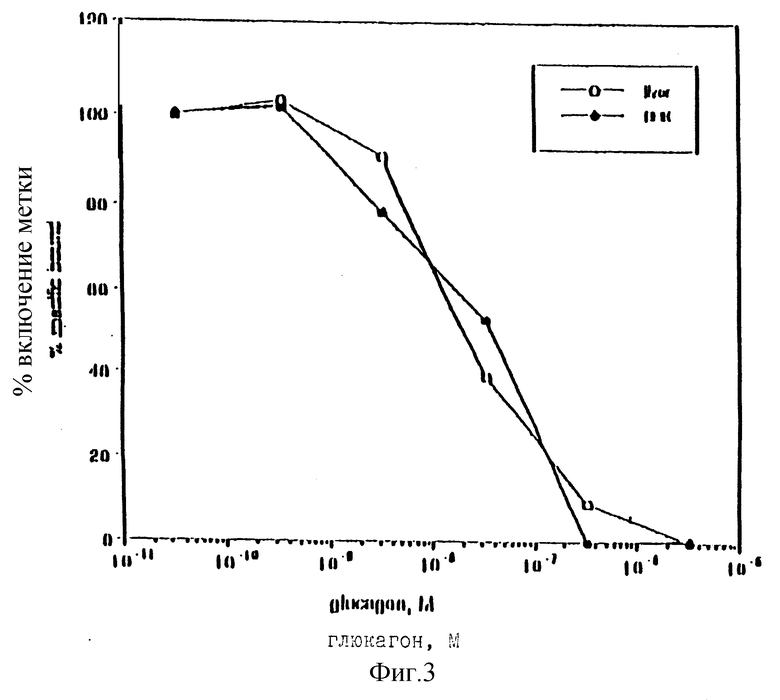
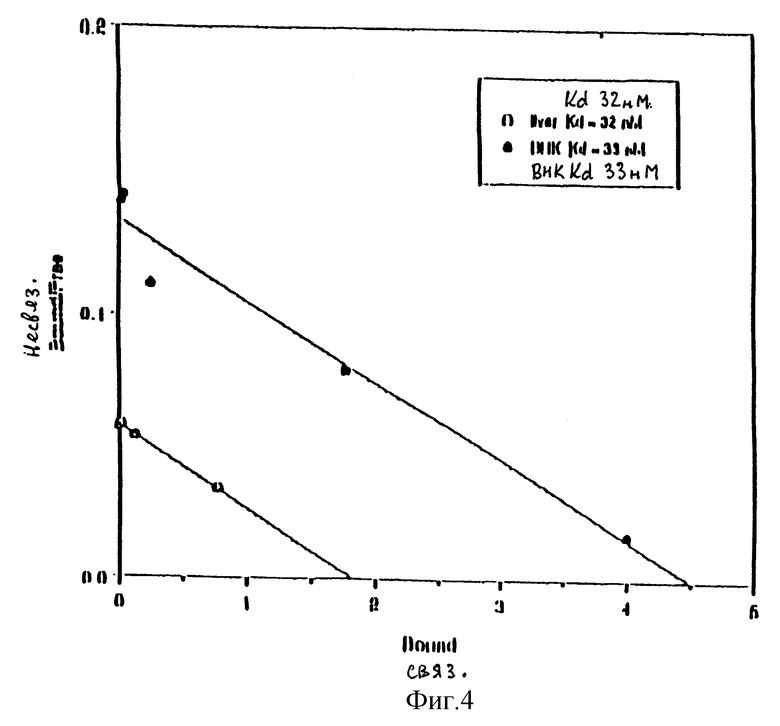



Изобретение касается рецептора глюкагона. Предложена изолированная молекула ДНК, кодирующая рецептор глюкагона или пептид рецептора глюкагона. ДНК-конструкция обеспечивает экспрессию изолированной молекулы ДНК и встраивается в линию клеток COS или ВНК. Также предложен способ получения рецептора глюкагона, предусматривающий культивирование линий клеток COS или ВНК и выделение целевого продукта. В изобретении представлено также изолированное моноклональное антитело, специфически связывающееся с рецептором глюкагона и блокирующее связывание глюкагона с рецептором глюкагона. Другим аспектом изобретения является способ обнаружения антагониста глюкагона, предусматривающий связывание агониста с изолированным рецептором глюкагона. Изобретение может быть использовано в стимулировании выделения глюкагона, что связано со стимулированием гликолиза и гликогенолиза и изучением роли глюкагона в таком заболевании, как сахарный диабет. 9 с. и 4 з.п. ф-лы, 5 ил., 8 табл.
Приоритет по пунктам и признакам:
28.08.1992 по пп. 1, 3, 8, пп. 2, 4-7, 9-13, которые прямо или косвенно ссылаются на п. 1 в части, касающейся рецептора глюкагона крысы (SEQ ID 14 и 15);
01.07.1993 по пп. 2, 4-7, 9-13, которые прямо или косвенно ссылаются на п. 1 в части, касающейся рецептора глюкагона человека (SEQ ID 24).
| А.V | |||
| BINCENT et al | |||
| The characterization of monoclonal antibodies to the hepatic glucagon receptor, THE JOURNAL OF CELL BIOLOGY, vol.107, № 6 (3), december 1988, р.65. |
