Эта заявка является продолжением части заявки США No. 08/362652, поданной 21 декабря 1994 г., которая ожидает решения, которая является продолжением части заявки США N 08/286889, поданной 5 августа 1994, которая опубликована как патент США 5470953 от 28 ноября 1995, которая в свою очередь является продолжением части заявки США N 08/173497, поданной 23 декабря 1993 г, которая опубликована как патент США 5437958 от 1 августа 1995.
Настоящее изобретение относится к клонированию и экспрессии полинуклеотидов, кодирующих новую человеческую β2 интегриновую α субъединицу, обозначенную αd, которая является структурно связанной с известными человеческими β2 интегриновыми α субъединицами, CD11a, CD11b, и CD11c. Настоящее изобретение также относится к полинуклеотидам, выделенным из других видов, которые проявляют гомологию с человеческими α кодирующими последовательностями.
Интегрины представляют класс мембранно-ассоциированных молекул, которые активно участвуют в клеточной адгезии. Интегрины представляют трансмембранные гетеродимеры, включающие α субъединицу в нековалентной связи с β субъединицей. До сего времени были идентифицированы по крайней мере четырнадцать α субъединиц и восемь β субъединиц [Обзор в Springer, Nature 346:425-434 (1990)] . Обычно β субъединицы являются способными ассоциировать с более чем одной α субъединицей, и гетеродимеры обменивающие общую α субъединицу, были класифицированы как субсемейства внутри интегриновой популяции.
Один класс человеческих интегринов, затрудняющих экспрессию в белых кровяных клетках, характеризуется общей β2 субъединицей, В результате этой клеточно-специфической экспрессии на эти интегрины обычно ссылаются как на лейкоцитовые интегрины, Leu-CAMs или лейкоинтегрины. Из-за общей β2 субъединицы альтернативное обозначение этого класса представляет β2 интегрины. β2 субъединица (CD18) была выделена ранее в ассоциации с одной из трех четких α субъединиц, CD11a, CD11b, и CD11c. Выделение с-ДНК, кодирующей человеческую CD18, описано у Kashimoto et.al., Cell 48: 681-690 (1987). В официальной WHO номенклатуре гетеродимерные белки относятся к CD11a/CD18, CD11b/CD18 или CD11C/CD18; в обычной номенклатуре они относятся к LFA-1, Мас-1, или Mol и р15095 или Leu M5, соответственно [Cobbold, относятся к LFA-1, Mac-1, или Mol и р15095 или Leu M5 соответственно [Cobbold, et.al., in Leukocyte Tiping 111, McMichael (ed), Oxford Press, p.788 (1987)]. Было продемонстрировано, что человеческие β2 интегриновые α субъединицы, CD11a, CD11b, и CDHc, мигрируют в условиях восстановления в электрофорезе с кажущимися молекулярными весами приблизительно 180 kD, 155 kD и 150 kD соответственно, и было проведено клонирование ДНК-т, кодирующих эти субъединицы [CD11a, Larson, et.al., J. Cell Biol. 108: 703-712 (1989); CD11b, Corbi, et.al., J.Biol.Chem. 263: 12403-12411 (1988) и CD11c Corbi, et.al. EMBO J. 6:4023-4028 (1987)]. Предполагаемые гомологи человеческих β2 интегриновых α и β цепей, определенные с помощью приблизительной аналогии в молекулярном весе, были ранее идентифицированы в других видах, включающих обезьян и других приматов [Letvin, et.al., Blood 61:408-410 (1983)], мышей [Sanchez-Madrid, et.al., J.Exp.Med. 154:1517 (1981)] и собак [Moore, et.al., Rissue Antigens 36:211-220(1990)],
Было показано, что абсолютные молекулярные веса предполагаемых гомологов из других видов значительно изменялись [смотри, например, Danilenko et.al., Tissue Antigens 40:13-21 (1992)], и отсутствие последовательной информации делает невозможным определенную корреляцию между человеческими интегриновыми субъединицами и субъединицами, идентифицированными в других видах. Однако между различными видами наблюдали изменение в количестве элементов в белковом семействе. Полагают, например, что было выделено больше IgA изотопов у кроликов, чем у людей [Burnett, et. al., EMBO J. 8:4041-4047 (1989) and Schneiderman, et. al. , Proc.Natl.Acad.Sci (USA) 86:7561-7565 (1989)]. Аналогично у людей были ранее идентифицированы по крайней мере шесть вариантов металлотионинового белка [Karin and Richards, Nature 299: 797-802 (1982) and Varshney, et. al., Mol.Cell.Biol, 6:26-37, (1986)], тогда как у мышей находятся в наличии только два таких варианта [Serle, et.al,, Mol.Cell.Biol. 4: 1221-1230 (1984)]. Поэтому существование множества элементов белкового семейства в одном виде не обязательно подразумевает, что соответствующие элементы семейства существуют в других видах.
В конкретном контексте β2 интегринов у собак наблюдали, что преимущественно собачья β2 часть, противоположная человеческой CD18, является способной образовывать димер с таким большим числом субъединиц, как четыре потенциально четких α субъединицы [Danilenko, et.al., в печати]. Антитела, генерированные иммунизованной мышью с собачьими спленоцитами, приводили к моноклональным антителам, которые иммунопреципитировали белки, временно обозначенные как собачьи гомологи в человеческих CD18, CD11a, CD11b и CD11c в основном на основании подобия, но не идентичности молекулярных весов. Другое собачье спленоцитовое антитело, СаI18Н2, узнавало и иммунопреципитировало четвертую α связь собачьей субъединицы, также способную к ассоцициации с β2 субъединицей, но имеющую уникальный молекулярный вес и рестриктированное в экспрессии в субнаборе дифференцированных тканевых макрофагов.
Антитела, генерированные иммунизацией хомячков мышиными дендритными клетками, приводили к двум антиинтегриновым антителам [Metlay, et.al., J. Exp. Med. 171: 1753-1771 (1990)], Одно антитело, 2Е6, иммунопреципитировало преимущественно гетеродимер с субъединицами, имеющими приблизительные молекулярные веса 180 kD и 90 kD, дополнительно к небольшим полосам в области молекулярного веса 150-160 kD. Второе антитело, N418, преципитировало другой наблюдаемый гетеродимер, имеющий приблизительные молекулярные веса 150 kD и 90 kD. На основании исследований блокирования клеточной адгезии была высказана гипотеза, что антитело 2Е6 узнает мышиную часть, противоположную человеческой CD18. В то время как молекулярный вес N418 антигена подтвердил узнавание мышиного гомолога в человеческой CD11c/CD18, дальнейший анализ показал, что мышиный антиген обнаруживает картину распределения ткани, которая не согласуется с той, которая наблюдается для человеческой CD11с/CD18.
Антигены, узнанные собачьим СаI1.8Н2 антителом и мышиным N418 антителом, могут представлять вариант видов (например, гликозилирования или сплайс вариант) ранее идентифицированной собачьей или мышиной α субъединицы. Или же эти антигены могут представлять уникальные собачьи и мышиные интегриновые α субъединицы. При отсутствии конкретной информации, касающейся первичной структуры, не может быть проведено различие среди этих альтернативных вариантов.
У людей CD11a/CD18, экспрессируется на всех лейкоцитах. CD11b/CD18 и CD11с/CD18 ограничиваются по существу в экспрессии на моноцитах, гранулоцитах, макрофагах и естественных клетках-убийцах (NK), но CD11с/CD18 обнаруживается также на некоторых типах В-клеток. Вообще, CD11a/CD18 преобладает на лимфоцитах, CD11b/CD18 на гранулоцитах и CD11с/CD18 на макрофагах [смотри обзор Arnaout, Blood 75:1037-1050 (1990)]. Экспрессия α цепей, однако, является изменяемой относительно состояния активации и дифференциации индивидуальных клеточных типов [смотри обзор, Larson and Springer, Immunol. Rev. 114:181-217(1990)].
Включение β интегринов в человеческую иммунную и воспалительную реакции было продемонстрировано с использованием моноклинальных антител, которые являются способными к блокированию адгезии β2 интегрин-ассоциированной клетки. Например, CD11a/CD18, CD11b/CD18 и CD11с/CD18 активно участвуют в связывании естественной клетки-убийцы (NK) в клетках лимфомы и аденокарциномы [Patarroyo, et.al., Immunol.Rev. 114:67-108 (1990)], аккумуляции гранулоцитов [Nourshargh, et.al., J.Immunol. 142:3192-3198 (1990)], гранулоцит-независимой утечки плазмы [Arford, et.al., в печати] и адгезии лейкоцитов в сосудистом эндотелии [Price, et.al., J.Immunol 139:4174-4177 (1987) и Smith, et.al., J.Clin.Invest. 83: 2008-2017 (1989)]. Фундаментальная роль β2 интегринов в иммунных и воспалительных реакциях становится очевидной в клиническом синдроме, на который ссылаются как на дефицит адгезии лейкоцитов (LAD), где клинические проявления включают повторяющиеся и часто угрожающие жизни бактериальные инфекции. LAD является следствием гетерогенных мутаций в β субъединице [Kishimoto, et.al., Cell, 50:193-202 (1987)] и тяжесть состояния заболевания является пропорциональной степени дефицита в экспрессии β2 субъединицы. Образование полного интегринового гетеродимера ослабляется β2 мутацией [Kishimoto, et.al., выше].
Интересно было показано, что по крайней мере одно антитело, специфичное для CD18, ингибирует человеческий вирус иммунодефицита типа 1 (HIV-1) образования синцития in vitro, хотя точный механизм этого ингибирования является неясным [Hildreth and Orentas, Science 244:1075-1078 (1989)]. Это наблюдение согласуется с открытием, что принципиальный противорецептор CD11a/CD18, IСАМ-1, является также поверхностным рецептором для большой группы риновирусных серотипов [Greve, et.al., Cell 56:839 (1989)].
Значение связывающей активности β2 интегрина в человеческой иммунной и воспалительной реакциях подчеркивает необходимость развития более полного понимания этого класса поверхностных белков. Идентификация пока неизвестных элементов этого субсемейства, а также их противорецепторов и генерирование моноклональных антител или других растворимых факторов, которые могут изменять биологическую активность β2 интегринов, будет обеспечивать практический путь для терапевтического вмешательства в β2 интегрин-связанные иммунные и воспалительные реакции.
В одном из аспектов настоящее изобретение обеспечивает новые очищенные и выделенные полинуклеотиды (например, ДНК и РНК транскрипты, обоих типов кодируемых и некодируемых нитей), кодирующих новую человеческую β2 интегриновую α субъединицу, αd, и их варианты (т.е. делеции, аналоги присоединения или замещения), которые обладают связывающими и/или иммунологическими свойствами, присущими αd. Предпочтительные молекулы ДНК изобретения включают сДНК, геномную ДНК и полностью или частично синтезированные молекулы ДНК. Теперь предпочтительный нуклеотид представляет ДНК впредь в виде SEQ ID NO: 1, кодирующего полипептид SEQ ID NO:2, Обеспечивается также рекомбинантная плазмида и вирусные ДНК конструкции (экспрессионные конструкции), которые включают αd кодирующие последовательности, где αd кодирующая последовательность является оперативно связанной с гомологами или гетеролагами транскрипционального регуляторного элемента или элементов.
Настоящим изобретением обеспечивают также выделение и очистку мышиных и крысиных полинуклеотидов, которые проявляют гомологию к полинуклеотидам, кодирующим человеческую αd. Предпочтительный мышиный полинуклеотид представляется впредь в SEQ ID NO:52, а предпочтительный крысиный полинуклеотид представляет впредь в SEQ ID NO:54.
В качестве другого объекта изобретения обеспечивают прокариотические или эукариотические клетки-хозяева, транформированные или трансфекцированные ДНК последовательностями изобретения, которые экпрессируют αd полинуклеотид или его варианты. Клетки-хозяева изобретения являются особенно полезными для крупномасштабного производства αd полипептида, который может быть выделен либо из самой клетки-хозяина, либо из среды, в которой выращивается клетка-хозяин. Клетки-хозяева, которые экпрессируют αd полипептид на их внутриклеточной мембранной поверхности также являются полезными в качестве иммуногенов в производстве αd - специфических антител. Предпочтительно, чтобы клетки-хозяева, трансфекцированные αd, являлись бы сотрансфекцированными с экпрессией β2 интегриновой субъединицы для того, чтобы позволить поверхностную экспрессию гетеродимера.
Настоящим изобретением обеспечивают также выделение и очистку αd полипептидов, фрагментов и их вариантов. Предпочтительный αd полипептид представляется впредь в SEQ ID NO:2. Новые αd продукты изобретения могут быть получены в виде выделенных из натуральных источников, но аналог с продуктами αd варианта предпочтительно производится с помощью рекомбинантных процедур, включающих клетки-хозяева изобретения. Полностью гликозилированные, частично гликозилированные и полностью дегликозилированные формы αd полипептида могут быть генерированы с помощью изменения клетки-хозяина, выбранной для рекомбинантного производства и/или обработки после выделения. Варианты αd полипептидов изобретения могут включать водорастворимые и не растворимые в воде αd полипептиды, включающие аналоги, где одна или больше аминокислот делегируются или заменяются: 1) без потери и предпочтительно с усилением одной или большего количества биологических активностей или иммунологических характеристик специфических для αd; или 2) со специфической неспособностью конкретного лиганда/рецептора к функции связывания или сигналирования. Обеспечиваются также составные полипептиды, где αd аминокислотные последовательности экспрессируются со смежными аминокислотными последовательностями из других полипептидов. Такие составные полипептиды могут обладать модифицированными биологическими, биохимическими и/или иммунологическими свойствами по сравнению с αd дикого типа. Рассматривается аналог полипептидов, включающий дополнительный аминокислотный (например, лизин или цистеин) остаток, который облегчает образование мультимера.
Настоящим изобретением рассматриваются также полипептиды и другие непептидные молекулы, которые специфически связываются с αd. Предпочтительные связывающие молекулы включают антитела (например, моноклональные или поликлональные антитела), противорецепторы (например, мембранно-ассоциированные и растворимые формы) и другие лиганды (например, молекулы естественного происхождения или синтетические молекулы), включающие те лиганды, которые конкурентно связывают αd в присутствии αd моноклональных антител и/или специфических противорецепторов. Связывающие молекулы являются полезными для очистки αd полипетидов и идентификации типов клеток, которые экспрессируют αd. Связывающие молекулы являются также полезными для модуляции (т.е. ингибирования, блокирования или стимулирования) in vitro связывающей или сигнальной трансдукционной активностей αd.
Обеспечиваются также анализы для идентификации αd. связывающих молекул, включающие анализы иммобилизованных связывающих лигандов, анализы связывающих растворов, сцинтилляционные проксимальные анализы, ди-гибридные скринирующие анализы и им подобные.
Анализы in vitro для идентификации антител или других соединений, которые модулируют активность αd, могут включать, например, иммобилизацию αd или естественного лиганда, с которым связывается αd, обнаружимое мечение неиммобилизованного связывающего партнера, инкубирование связывающих партнеров вместе и определение влияния испытываемого соединения на количество связанных меток, где уменьшение в связанной метке в присутствии испытываемого соединения по сравнению с количеством связанной метки в отсутствие испытываемого соединения указывает на то, что испытываемый агент является ингибитором αd связывания.
Другой тип анализа для идентификации соединений, которые модулируют вазимодействие между αd и лигандом, включает иммобилизацию αd или ее фрагмента на твердую подложку (или импрегнированную агентом), покрытую флуоресцирующим агентом, мечение лиганда соединением, способным возбуждать флуоресцирующий агент, контактирование иммобилизованной αd с меченным лигандом в присутствии и отсутствие предполагаемого модулирующего соединения, обнаружение излучения света флуоресцирующим агентом и идентификацию модулирующего соединения как такого соединения, которое оказывает влияние на излучение света флуоресцирующим агентом по сравнению с излучением света флуоресцирующим агентом в отсутствие модулирующего соединения. Или же αd лиганд может быть иммобилизованным, и αd может быть меченным в анализе.
Изобретением рассматривается еще один способ для идентификации соединений, которые модулируют взаимодействие между αd и лигандом, включая трансформацию или трансфекцию соответствующих клеток-хозяев конструкцией ДНК, содержащей сообщаемый ген, под контролем промотора, регулируемого фактором транскрипции, имеющим ДНК связывающий домен и активирующий домен, экспрессируют в клетки-хозяева первую гибридную ДНК последовательность, кодирующую первую составную часть или все αd и либо ДНК связывающий домен, либо активирующий домен фактора транскрипции, который не вводится в первый гибрид, оценивают влияние предполагаемого модулирующего соединения на взаимодействие между αd и лигандом, за счет обнаружения связывания лиганда со второй гибридной ДНК последовательностью αd в конкретной клетке-хозяине за счет измерения продуцирования сообщаемого генного продукта в отсутствие модулирующего соединения. Теперь предпочтительными для использования в анализе являются промотор Lex A, Lex A ДНК связывающий домен, GaLI4 домен трансактивации, LacZ сообщаемый ген и дрожжевая клетка-хозяин.
Модифицированная версия вышеуказанного анализа может быть использована в выделении полинуклеотида, кодирующего белок, который связывает αd за счет трансформирования или трансфекцирования соответствующих клеток-хозяев конструкцией ДНК, включающей сообщаемый ген под контролем промотора, регулируемого фактором транскрипции, имеющим ДНК связывающий домен и активирующий домен, экспрессии в клетки-хозяева первой гибридной ДНК последовательности, кодирующей первую составную часть или все αd и либо ДНК связывающий домен, либо активирующий домен фактора транскрипции, экспрессии в клетки-хозяева библиотеки вторых гибридных ДНК последовательностей, кодирующих вторые гибриды части или всех предполагаемых αd связывающих белков и ДНК связывающего домена, или активирующего домена фактора транскрипции, который не вводится в первый гибрид, обнаружения связывания αd связывающего белка в αd в конкретной клетке-хозяине за счет обнаружения продуцирования сообщаемого генного продукта в клетке-хозяине, и выделении вторых гибридных ДНК последовательностей, кодирующих αd связывающий белок из конкретной клетки-хозяина.
Гибридомные линии клеток, которые продуцируют антитела, специфичные для αd, также рассматриваются изобретением. Приемы продуцирования гибридом, которые секретируют моноклональные антитела, являются хорошо известными специалистам в этой области. Гибридомные линии клеток могут быть генерированы после иммунизации животного очищенной αd, вариантами αd, или клетками, которые экспрессируют αd или ее варианты на экстраклеточной мембранной поверхности. Иммуногенные клеточные типы включают клетки, которые экспрессируют αd in vivo, или трансфекцированные прокариотические или эукариотические линии клеток, которые обычно не подвергаются нормальной экспрессии αd in vivo. Предпочтительные антитела изобретения секретируются теперь гибридомами, обозначенными 169А, 169В, 170D, 170F, 170Т, 170Х, 170Н, 188А, 188В, 188С, 188Е, 188F, 188G, 188I, 188К, 188L, 188М, 188N, 188Р, 188R, 188Т, 195А, 195С, 195D, 195Е, 195Н, 197А-1, 197А-2, 197А-3, 197А-4, 199А и 199М.
Выявляется значение информации, внесенной за счет раскрытия ДНК и аминокислотных последовательностей αd. В одной из серий примеров раскрытая αd СДНК последовательность делает возможным выделение человеческой αd геномной ДНК последовательности, включающей транскрипциональные элементы контроля для геномной последовательности. Рассматривается также идентификация αd аллельных вариантов и гетерологичных видов (например, крысы или мыши) ДНК-т. Выделение человеческой αd геномной ДНК и ДНК-т гетерологичных видов может достигаться за счет стандартных приемов ДНК/ДНК гибридизации при соответственно строгих условиях, с использованием всех или части αd сДНК последовательности в качестве пробы для скринирования всей библиотеки. Или же реакция полимеразной цепи (PCR) с использованием олигонуклеотидных праймеров, которые конструируются на основе известной сДНК последовательности, может быть использована для усиления и идентификации геномных αd сДНК последовательностей. Синтетические ДНК-ты, кодирующие αd полипептид, включающие фрагменты и другие их варианты, могут быть получены обычными синтетическими методами.
Информация о ДНК последовательности изобретения также делает возможным развитие за счет гомологичной рекомбинации или стратегий "нокаута" [смотри, например, Kapecchi, Science 244:1288-1292 (1989)], получать грызунов, которые не в состоянии экспрессировать функциональный αd полипептид или которые экспрессируют вариант αd пептида. Такие грызуны являются полезными в качестве моделей для изучения активностей αd и αd модуляторов in vivo.
ДНК и аминокислотные последовательности изобретения делают также возможным анализ αd эпитопов, которые активно участвуют в противорецепторном связывании, так же как и эпитопов, которые могут скорее регулировать, чем активно участвовать в связывании. Идентификация эпитопов, которые могут участвовать в трансмембранной сигнальной трансдукции, также рассматривается изобретением.
ДНК изобретения является также полезной для обнаружения клеточных типов, которые экпрессируют αd полипептид. Стандартные приемы ДНК/РНК гибридизации, которые используют αd ДНК для обнаружения αd РНК, могут быть использованы для определения представленного уровня αd транскрипции внутри клетки, так же как и изменений в уровне транскрипции в ответ на действие внутренних или внешних агентов. Идентификация агентов, которые модифицируют транскрипцию и/или трансляцию αd, может быть в свою очередь оценена для потенциальной терапевтической или профилактической цели. ДНК изобретения делает также возможной гибридизацию in situ αd ДНК в клеточной РНК для определения клеточной локализации αd специфических переносов внутри комплексных клеточных популяций и тканей.
ДНК изобретения является также полезной для идентификации нечеловеческих полинуклеотидных последовательностей, которые проявляют гомологию к человеческим αd последовательностям. Обладание нечеловеческими αd ДНК последовательностями позволяет проводить развитие животных моделей (включающих, например, трансгенные модели) человеческой системы.
В качестве другого аспекта изобретения рассматриваются моноклональные или поликлональные антитела, специфические для αd, которые могут быть применены в иммуногистохимическом анализе для локализации αd в субклеточных положениях или индивидуальных клетках внутри тканей. Иммуногистохимические анализы этого типа являются особенно полезными, когда используются в комбинации с гибридизацией in situ для локализации и αd mРНК и полинуклеотидных продуктов αd гена.
Идентификация клеточных типов, которые экспрессируют αd, может иметь значительные ответвления для развития терапевтических или профилактических агентов. Предполагается, что продукты изобретения, отнесенные к αd, могут быть применены в лечении заболеваний, где макрофаги являются существенным элементом процесса заболевания. Животные модели для многих патологических состояний, ассоциированные с макрофаговой активностью, были описаны в литературе. Например, у мышей макрофаговое пополнение в сайты хронических и острых воспалений сообщалось Jutila, et. al., J.Leucocyte Biol. 54:30-39 (1993). У крыс Adams, et.al.[Transplantation 53:1115-1119 (1992) and Transplantation 56: 794-799 (1993)] описывает модель для трансплантированных артериосклерозов после гетеротропной абдоминальной сердечной аллографической трансплантации. Rosenfeld, et.al., [Arteriosclerosis 7:9-23 (1987) and Arteriosclerosis 7:24-34 (1987)] описывает индуцированные атеросклерозы у кроликов, в рационе которых содержался дополнительной холестерин. Henenberg, et. al., [Diabetologia 32:126-134 (1989)] сообщал о спонтанном развитиии инсулинзависимого диабета у ВВ крыс. Yamada, et.al. [Gastroenterology 104: 759-771 (1993)] описали индуцированное воспалительное заболевание кишечника, хронические грануломатозные колиты, у крыс после инъекций стрептококковых пептидогликан-полисахаридных полимеров. Cromartie, et.al. [J.Exp. Med. 146: 1585-1602 (1977] and Schwab, et.al., [Infection and Immunity 59:4436-4442 (1991)] сообщали, что инъекция крысам белка стрептококковой клеточной стенки приводила к артритному состоянию, характеризующемуся воспалением периферийных суставов и последующему разрушению сустава. Наконец, Huitinga, et.al. [Eur. J.Immunol., 23:709-715, (1993)] описал экспериментальные аллергические энцефаломиелиты, модель для множественных склерозов у крыс Льюиса. В каждой из этих моделей αd антитела, другие αd связывающие белки или растворимые формы αd используются для ослабления состояния заболевания, преимущественно за счет инактивации макрофаговой активности.
Изобретением обеспечиваются фармацевтические композиции для лечения этих и других состояний заболевания. Фармацевтические композиции составляются с целью ингибирования взаимодействия между αd и его лигандом(ми) и включают различные растворимые и мембранно-ассоциированные формы αd связывающих белков (включающих антитела, лиганды и им подобные), внутриклеточные или внеклеточные модуляторы αd связывающей активности, и/или модуляторы αd и/или αd-лиганд полипептидной экспрессии, включающей модуляторы транскрипции, трансляции, посттрансляционного процессинга и/или внутриклеточного транспорта.
Изобретение рассматривает также способы лечения состояний заболевания, в которых подразумевается αd связывание, или локализованная аккумуляция клеток, которые экпрессируют αd, где пациент, страдающий от указанного состояния заболевания, обеспечивается количеством фармацевтической композиции изобретения, достаточной для модулирования уровней αd, связывания или для модулирования аккумуляции типов клеток, которые экспрессируют αd. Способ лечения согласно изобретению является подходящим для состояний заболевания, таких как, но не ограничивается ими, диабеты типа 1, атеросклерозы, множественные склерозы, астма, псориаз, воспаление легких, синдром острого респираторного растройства и ревматоидные артриты.
Многочисленные другие аспекты и преимущества настоящего изобретения будут очевидны при рассмотрении следующего описания, со ссылками на чертежи, где:
Фиг. 1А - 1D включают расстановку человеческих аминокислотных последовательностей CD11b (SEQ ID NO:3), CD-He (SEQ ID NO:4) и αd (SEQ ID NO:2).
Настоящее изобретение иллюстрируется следующими примерами, касающимися выделения сДНК клона, кодирующего αd из человеческой селезенки сДНК библиотеки. Более конкретно Пример 1 иллюстрирует применение собачьего αTM1 антитела в попытке обнаружения гомологичного человеческого белка. Пример 2 детализирует очистку собачьего αTM1 и N-терминального секвенирования полипептида для конструирования олигонуклеотидных праймеров для PCR амплификации собачьего αTM1 гена. Пример 3 направлен на крупномасштабную очистку собачьего αTM1 для внутреннего секвенирования для того, чтобы сконструировать дополнительные PCR праймеры. Пример 4 описывает применение PCR и праймеров внутренней последовательности для амплификации фрагмента собачьего αTM1 гена. Пример 5 направлен на клонирование человеческой αd-кодирующей сДНК последовательности. Пример 6 описывает гибридизационный анализ Нозерн блоттинга человеческой ткани и клеток для экспрессии αd mРНК. Пример 7 детализирует конструкцию человеческих αd плазмид экспрессии и трансфекции COS клеток с получением плазмид. Пример 8 описывает анализ ELISA αd экспрессии в трансфекцированных COS клетках. Пример 9 описывает анализ FACS клеток COS, трансфекцированных человеческими αd плазмидами экспрессии. Пример 10 направлен на иммунопреципитацию CD18 совместно с αd в со-трансфекцированных COS клетках. Пример 11 относится к устойчивой трансфекции αd конструкций экспрессии в клетках яичников китайских хомячков. Пример 12 направлен на CDI8-зависимое связывание αd с внутриклеточной адгезией молекулы ICAM-R, с ICAM-R мутантным белком и комплементным фактом iC3b. Пример 13 описывает сцинтиляционные скринирующие анализы для идентификации ингибиторов или энхансеров (т.е. модуляторов) αd лиганд/антилиганд связывающих взаимодействий. Пример 14 направлен на конструирование плазмид экспрессии, которые кодируют растворимые формы αd, и анализы связывания продуктов экспрессии. Пример 15 относится к продуцированию αd-специфических поликлональных сывороток и моноклональных антител. Пример 16 описывает анализ αd распределения ткани, экспрессию αd в периферийных лейкоцитах крови и αd экспрессию в воспалительной и невоспалительной синовиальной оболочках с использованием анти-αd поликлональной синовиальной оболочки. Пример 17 описывает выделение крысиных сДНК последовательностей, которые проявляют гомологию к человеческим αd генным последовательностям. Пример 18 относится к конструкции крысиных αd плазмид экспрессии полной длины, крысиных αd I доменовых экспрессионных плазмид, включающих I домен/IgG составные белки, и продуцированию моноклональных антител полной длины и I доменовых составных белков. Пример 19 направлен на выделение мышиных сДНК последовательностей, которые проявляют гомологию к человеческим αd генным последовательностям. Пример 20 описывает выделение дополнительных мышиных αd сДНК клонов, используемых для подтверждения анализа последовательности. Пример 21 относится к гибридизационному анализу in situ различных мышиных тканей для определения тканевой и клеточной специфической экспрессии предполагаемого мышиного гомолога в человеческой αd. Пример 22 описывает генерирование экспрессионных конструкций, которые кодируют предполагаемый мышиный гомолог человеческой αd. Пример 23 направлен на конструирование "нокаут" мыши, где ген, кодирующий предполагаемый мышиный гомолог человеческой αd, разрушается. Пример 24 описывает выделение кроличьих сДНК клонов, которые проявляют гомологию к человеческим αd кодирующим последовательностям. Пример 25 описывает животные модели человеческих состояний заболевания, где анализируется модуляция αd на терапевтическую восприимчивость. Пример 26 описывает экспрессию αd в животной модели состояний заболевания.
Пример 1
Попытки обнаружения человеческой гомологии собачьего αTM1
Моноклональное антитело Са11.8Н2 [Moore, et.al., выше], специфичное для собачьей αTM1 было испытано на перекрестную реактивность в лейкоцитах человеческой периферической крови в попытке идентификации человеческого гомолога собачьей αTM1. Клеточные препараты (обычно 1•106 клеток) были проинкубированы неразбавленным супернатантом гибридомы или очищенным мышиным IgG-отрицательным контрольным антителом (10 мкг/мл) во льду в присутствии 0.1% азида натрия. Связывание моноклонального антитела было обнаружено за счет последующей инкубации FIТС-сопряженным лошадиным анти-мышиным IgG (Vector Laboratories, Burlingame, CA) при 6 мкл/мл. Окрашенные клетки были зафиксированы 2% вес/об, параформальдегидом в фосфатном буферном рассоле (PBS) и проанализированы с помощью Facstar Plus флуоресцентно-активированной клетки sorter (Becton Dickenson, Mountain View, CA). Обычно анализировали 10000 клеток с использованием логарифмической амплификации для интенсивности флуоресценции.
Результаты указывают на то, что СаI1.8Н2 не подвергается перекрестному взаимодействию с поверхностью белков, экспрессированных на лейкоцитах человеческой периферической крови, в то время как контрольные клетки неопластических собачьих лимфоцитов переферической крови по-существу все проявляли положительную реакцию на αTM1.
Так как моноклональное антитело СаI1.8Н2, специфическое для собачьей α субъединицы, не подвергается перекрестному взаимодействию с человеческим гомологом, выделение собачьей αTM1. ДНК считалось необходимым предварительным требованием для выделения противочасти человеческого гена, если он существовал.
Пример 2
Аффинная очистка собачьей αTM1 для М-теоминального секвенирования.
Собачья αTM1. была подвергнута аффинной очистке для того, чтобы определить N-терминальные аминокислотные последовательности для конструирования олигонуклеотидной пробы/праймера. Короче, антиαTM1. моноклональное антитело СаI1.8Н2 было подвергнуто сочетанию с Affigel 10 хромографической смолой (Bio Rad, Hercules, CA), и белок был выделен за счет специфического взаимодействия антитело-белок. Антитело было подвергнуто конъюгированию со смолой согласно протоколу Bio Rad при концентрации приблизительно 5 мг антитела на мл смолы. После проведения реакции конъюгирования избыток антитела удаляли и смолу блокировали тремя объемами 0.1 М этаноламина. Затем смолу промывали раствором фосфатного солевого буфера (PBS) объемом, равным тридцати объемам колонки.
Двадцать пять граммов одной собачьей селезенки были гомогенезированы в 250 мл буфера, содержащего 0.32 М сахарозы в 25 мМ Трис-HCl, рН 8.0, протеазными ингибиторами. Ядерный и клеточный дебрисы были подвергнуты осаждению центрифугированием при 1000 g в течение 15 минут. Мембраны собирали в виде комочков из супернатанта центрифугированием при 100000 g в течение 30 минут. Мембранный комочек был ресуспендирован в 200 мл лизисного буфера (50 mM NaCl, 50 mМ бората, рН 8.0 с 2% NP-40) и проинкубирован в течение 1 часа во льду. Затем нерастворимый материал был сконцентрирован в виде комочка центрифугированием при 100000 g в течение 60 минут, 10 мл очищенного лизата переносили в 15 мл полипропиленовую пробирку с 0.5 мл СаI1.8Н2-конъюгированного с Affagel 10 смолой, описанной выше. Пробирка была проинкубирована в течение ночи при 4oС вращением, и смола последовательно промыта раствором D-PBS с объемом, равным 50 объемам колонки. Затем смола была перенесена в пробирку для микроцентрифугирования и подвергнута кипячению в течение 10 минут в 1 мл Laemmli образца буфера (не восстановленного), содержащего 0.1 М Трис-HCl, рН 6.8, 2% SDS, 20% глицерина и 0.002% бромфенолового голубого. Смола была сконцентрирована в комочек центрифугированием и выгружена; супернатант обрабатан 1/15 объема β-меркаптоэтанола (Sigma, St.Louis, МО) и нанесен на 7% полиакриламидный гель. Выделенные белки были перенесены на Immobilon PVDF мембрану (Millipore, Bedford, MA) следующим образом.
Гели промывали один раз в деионизованной воде, Millipore-отфильтрованной воде и уравновешивали в течение 15-45 минут в 10 мМ буфера переноса 3[циклогексиламино]-1-пропансульфоновой кислоты (CASP), рН 10.5 с 10% метанола. Immobilon мембраны увлажняли метанолом, промывали профильтрованной водой и уравновешивали в течение 15-30 минут CASP буфере переноса. Начальный перенос проводили с использованием аппарата переноса Biorad при 70 В в течение 3 часов. Immobilon мембрану удаляли после переноса и окрашивали в профильтрованном 0.1% R250 кумазине в течение 10 минут. Мембраны обесцвечивали в 50% растворе метанола/10% уксусной кислоте три раза, каждый раз в течение 10 минут. После обесцвечивания мембраны промывали в профильтрованной воде и сушили на воздухе.
Обнаруживали полосы белков приблизительно 150 kD, 95 kD, 50 kD и 30 kD. Вероятно, полосы 50 kD и 30 kD появлялись из-за загрязнения антителом. N-терминальное секвенирование пробовали затем на обеих полосах 150 kD и 95 kD, но 95 kD белок был блокирован, предотвращая секвенирование. Белковая полоса 150 kD была вырезана из мембраны и непосредственно секвенирована Applied Biosystems (Forster City, CA) Model 573A белковым секвенатором в соответствии с инструкциями производителя. Получающаяся аминокислотная последовательность представляется в SEQ ID NO:5, использующим единственное написание аминокислотных обозначений.
FNLDVEEPMVFQ (SEQ ID NO:5)
Идентифицированная последовательность включала FNLD характеристику последовательности α субъединиц интегринового семейства [Tamura, et.al., J. Cell.Biol. 111:1593-1604(1990)].
Конструирование праймера и попытка амплификациии собачьих αTM1 последовательностей.
Из информации об N-терминальной последовательности были сконструированы три олигонуклеотидных пробы для гибридизации: a)"Tommer" полностью дегенерированный олигонуклеотид; 6)"Patmer" частично дегенерированный олигонуклеотид и c)"Guessmer" недегенерированный олигонуклеотид на основании использования кодона млекопитающего. Эти пробы приводятся ниже в виде SEQ ID NO: 6, 7 и 8 соответственно. Символы нуклеиновых кислот в соответствии с 37 C.F. R. п. 1.882 для этих и всех других нуклеотидных последовательностей, приведенных здесь.

На основании данных секвенирования соответствующие клоны не были обнаружены с использованием этих олигонуклеотидов в некоторых не очень строгих условиях гибридизации (in sevral low stringency hybridizations) в сДНК библиотеке собачьей селезенки/макрофаге периферической крови, клонированной в λZAP (Stratagene, La Jolla, CA).
Четыре других олигонуклеотидных праймера, обозначенных 5' Deg, 5' Spec, 3' Deg и 3'Spec (представленных в SEQ ID NO: 9, 10, 11 и 12 соответственно, где Deg указывает на дегенерированный и Spec указывает на недегенерированный) были впоследствии сконструированы на основе установленной N-терминальной последовательности для попыток экстракопирования собачьих αTM1 последовательностей с помощью PCR из фаговой библиотеки ДНК, очищенной от пластинок лизата Stratagene библиотеки, описанной выше.

αTM1 олигонуклеотидные праймеры были подвергнуты спариванию с Т3 или Т7 векторными праймерами, как представляются в SEQ ID NO: 13 и 14 соответственно, которые гибридизовали с последовательностями, фланкирующими полилинкерную область в Bluescript фагмиде, найденном в λZAP.
PCR экстракопирование было проведено в Taq буфере (Boehringer Mannheim, Indianapolis, IN), содержащем магний, с 150 нг библиотеки ДНК, 1 мкг каждого праймера, 200 мкМ dNTPs и 2.5 единицами Taq полимеразы (Boehringer Mannheim) и продукты были выделены электрофорезом в 1% агарозном геле в буфере Трис-ацетат-ЭДТА (ТАЕ) с 0.25 мкг/мл этидиний бромида. ДНК была перенесена на Hybond (Amersham, Arlington Heights, IL) мембрану тампоном в течение ночи в 10Х SSPE. После переноса иммобилизованную ДНК денатурировали 0.5 М NaOH, 0.6 М NaCl, нейтрализовали 1.0 М Трис-HCl, рН 8.0, в 1.5 М NaCl, и промывали 2Х SSPE до УФ сшивания с помощью Stratalinker (Stratagene) сшивающего устройства. Мембрана была проинкубирована в предварительно гибридизованном буфере (5Х SSPE, 4X Denhardts, 0.8 SDS, 30% формамиде) в течение 2 часов при 50oС с перемешиванием.
Олигонуклеотидные пробы 5' Deg, 5' Spec, 3' Deg и 3'Spec (в SEQ ID NO: 9, 10, 11 и 12 соответственно) были мечены с использованием Boehringer Mannheim киназного буфера с 100-300 мкСi γP32-dATP и 1-3 единиц полинуклеотидкиназы в течение 1-3 часов при 37oС. Не вошедшую метку удаляли с помощью Sephadex G-25 (Pharmacia, Piscataway, NJ) хроматографии с использованием 10 нМ Трис-HCl, рН 8.0, 1 mM ЭДТА (ТЕ) буфера и вышедший из колонки раствор непосредственно добавляли к раствору прегибридизации. Мембраны подвергали зондированию в течение 16 часов при 42oС с перемешиванием и промыванием повторно, с конечным строгим промыванием 1Х SSPE/0.1% SDS при 50oС в течение 15 минут. Затем блот подвергали экспозиции на Kodak X-Omat AR пленку в течение 1-4 часов при -80oС.
Олигонуклеотиды 5' Deg, 5' Spec, 3' Deg и 3'Spec гибридизовались только с PCR продуктами из реакций, в которых они были использованы в качестве праймеров, и были неспособны к гибридизации, как ожидалось, с PCR продуктами из реакций, в которых они не были использованы в качестве праймеров. Таким образом, было сделано заключение, что не один из PCR продуктов не был специфичен для αTM1, потому что не было продукта, гибридизованного со всеми соответствующими пробами.
Пример 3
Крупномасштабная аффинная очистка собачьих αTM1 для внутреннего секвениоования.
Для того чтобы обеспечить дополнительную аминокислотную последовательность для праймерного конструирования собачья αTM1 была очищена для внутреннего секвенирования. Три среза замороженной селезенки (приблизительно 50 г каждый) и замороженные клетки из двух частичных селезенок от взрослых собак были использованы для генерирования белка для внутреннего секвенирования. Пятьдесят граммов селезенки были гомогенезированы в 200-300 мл боратного буфера с помощью смесителя Waring. Гомогенизованный материал был разбавлен 1 объемом буфера, содержащего 4% NP-40, и смесь затем осторожно перемешивали в течение по крайней мере одного часа. Полученный лизат был очищен от крупного дебриса центрифугированием при 2000 g в течение 20 минут и затем отфильтрован либо через Corning (Corning NY) префильтр, либо через Corning 0.8 микронный фильтр. Далее лизат был очищен фильтрованием через Corning 0.4 микронную фильтрующую систему.
Селезеночный лизат и антитело, конъюгированное с Affigel 10 смолой, описанной в Примере 2, были объединены при объемном отношении 150:1 в 100 мл аликвотах и проинкубированы в течение ночи при 4oС встряхиванием. Лизат был удален после центрифугирования при 1000 g в течение 5 минут, объединен с большей частью антитела, конъюгированного с Affigel 10 смолой, и проинкубирован в течение ночи, как описано выше. Аликвоты поглощенной смолы затем объединяли и промывали 50 объемами D-PBS/0.1% Твин 20 и смолу переносили в 50 мл Biorad колонку. Поглощенный белок элюировали из смолы 3-5 объемами 0.1 М глицина (рН 2.5); собирали фракции приблизительно по 900 мкл и нейтрализовали 100 мкл 1 М Трис буфера, рН 8.0. Из каждой фракции удаляли 15 мкл аликвоты и кипятили в равном объеме 2Х Laemmli буфера с 1/15 объема 1 М дитиотрейтола (DDT). Эти образцы были подвергнуты электрофорезу на 8% Novex (San Diego, CA) полиакриламидных гелях и визуализованы либо с помощью окрашивания кумазином, либо с помощью серебрения, используя набор Daiichi (Enprotech, Natick, MA) в соответствии с протоколом, предложенным производителем. Фракции, которые содержали наибольшие количества белка, были объедены и подвергнуты концентрированию в вакууме. Остающийся раствор был разбавлен 50% восстанавливающим Laemmli буфером и испытание проведено на 1,5 мм 7% полиакриламидных гелях в Трис-глицин/SDS буфере. Белок был перенесен из гелей на Immobilon мембрану с помощью процедуры, описанной в Примере 2, с использованием для переноса устройства Hoefer.
Полосы белка, соответствующие собачьей αTM1, были вырезаны из 10 PVDF мембран, и было получено приблизительно 47 мкг общего белка. Полосы были обесцвечены в 4 мл 50% метанола в течение 5 минут, высушены на воздухе и разрезаны на куски 1 х 2 мм. Куски мембраны были погружены в 2 мл 95% ацетона при 4oС в течение 30 минут с периодическим переворачиванием и затем высушены на воздухе.
До протеолитического расщепления белка, связанного с мембраной, 3 мг цианоген бромида (CNBr) (Pierce, Rockford, IL) были растворены в 1.25 мл 70% муравьиной кислоты. Затем этот раствор был добавлен в пробирку, содержащую PVDF куски мембраны, и пробирка проинкубирована в темноте при комнатной температуре в течение 24 часов. Затем супернатант (S1) удаляли в другую пробирку и куски мембран промывали 0.25 мл 70% муравьиной кислоты. Этот супернатант (S2) был удален и добавлен к ранее полученному супернатанту (S1). К объединенным супернатантам (S1 и S2) было добавлено два миллилитра Milli Q воды и раствор лиофилизован. PVDF куски мембраны были подвергнуты сушке в атмосфере азота и вновь проэкстрагированы 1.25 мл 60% ацетонитрила, 0.1% тетрафторуксусной кислоты (ТФУК) при 42oС в течение 17 часов. Этот супернатант (S3) был удален и куски мембран проэкстрагированы вновь 1.0 мл 80% ацетонитрила, 0.1% тетрафторуксусной кислоты (ТФУК) при 42oС в течение 1 часа. Этот супернатант (S4) был объединен с ранее полученными супернатантами (S1, S2 и S3) и высушен в вакууме.
Высушенные CNBr фрагменты были затем растворены в 63 мкл 8 М мочевины, 0.4 М NH4НСО3. Фрагменты были восстановлены в 5 мкл 45 мМ дитиотрейтола (DDT) и впоследствии проинкубированы при 50oС в течение 15 минут. Раствор затем был охлажден до комнатной температуры и фрагменты проалкилированы добавлением 5 мкл 100 мМ иодацетамида (Sigma, St. Louis, МО). После 15 минут инкубации при комнатной температуре образец разбавляли 187 мкл Milli Q воды до конечной концентрации мочевины 2.0 М. Затем был добавлен Трипсин (Worthington, Freehold, NJ) при отношении 1:25 (вес:вес) фермента к белку и белок подвергнут расщеплению в течение 24 часов при 37oС. Расщепление прерывали добавлением 30 мкл ТФУК.
Затем были выделены белковые фрагменты с помощью гельпроникающей жидкостной хроматографии (HPLC) на системе Waters 625 LC (Millipore, Milford, MA) с использованием 2.1 х 250 мм, 5 микронной Vydac С-18 колонки (Vydac, Hespria, CA), уравновешенной в 0.05% растворе ТФУК и воде для гельпроникающей хроматографии (буфер А). Белки элюировали 80% ацетонитрилом с повышающейся концентрацией в 0.04% ТФУК (буфер В) с градиентом 38-75% буфера В в течение 65-95 минут и 75-98% буфером В в течение 95-105 минут. Белки были расфракционированы при скорости потока 0.2 мл/мин и обнаружены при 210 нм.
После фракционирования аминокислотная последовательность белков анализировалась за счет проведения автоматической Edman деструкции на Applied Biosystems Model 437A секвинаторе с использованием стандартных циклов производителя и программы Model 610 A Data Analysis, версии 1.2.1. Все секвинирующие реагенты были поставлены Applied Biosystems. Аминокислотные последовательности семи из восьми внутренних фрагментов приводятся ниже, где "X" указывает на то, что идентичность аминокислоты была неопределенной.

Конструирование праймера
Одна полученная внутренняя аминокислотная последовательность (представленная в SEQ ID NO:22) была затем использована для конструирования полностью перерожденного олигонуклеотидного праймера, обозначенного p4(R) в виде представленного в SEQ ID NO:23.

Пример 4
PCR клониоование собачьего αTM1 фрагмента.
5'часть собачьего αTM1 гена была экстракопирована из двутяжной сДНК собачьей селезенки с помощью PCR.
А. Генерирование двутяжной сДНК собачьей селезенки.
Один грамм замороженного материала из селезенки молодой особи собаки был измельчен в жидком азоте в сухом льду и гомогенизирован в 20 мМ PHK-Stat 60 буфера (Tel- Nest В, Inc. Friendswood, TX). Было добавлено 4 мл хлороформа и раствор проэкстрагирован центрифугированием при 12000 g в течение 15 минут. РНК была осаждена из водного слоя 10 мл этанола. Затем была выбрана поли А+ РНК на Dynal Oligo dT Dynabeads (Dynal, Oslo, Norwey). Пять аликвот 100 мкг общей РНК были объединены и разбавлены равным объемом 2Х связывающего буфера (20 мМ Трис-HCl, рН 7.5, 1.0 М LiCl, 1 mM ЭДТА, 0.1% SDS), в соответствии с протоколом, предложенным производителем до элюирования поли A+ mPHK 2 мМ ЭДТА, рН 7.5. Затем была генерирована двутяжная сДНК с использованием элюированной поли А+ РНК и сДНК набора Boehringer Mannheim для синтеза в соответствии с протоколом, предложенным производителем.
Б. Выделение частичной собачьей αTM1 сДНК.
Олигонуклеотидные праймеры 5' Deg (SEQ ID NO:9) и p4(R) (SEQ ID NO:23) были использованы в стандартной PCR реакции с использованием 150 нг двутяжной сДНК, 500 нг каждого праймера, 200 мкМ dNTPs и 1.5 единиц Taq полимеразы (Boehringer Mannheim) в Taq буфере (Boehringer Mannheim) с магнием. Полученные продукты (1 мкл продукта исходной реакции) были подвергнуты второму кругу PCR реакции с теми же самыми праймерами для увеличения выхода продукта. Эта полоса была подвергнута элюированию из 1% агарозного геля на Schleicher and Schuell (Keene.NH) NA 45 бумагу в буфере, содержащем 10 мМ Трис-HCl, рН 8, 1мМ ЭДТА, 1.5 М NaCl при 65oС, осаждена и лигирована в pCRtmII вектор (Invitrogen, San Diego, CA) с использованием ТА клонирующего набора (Invitrogen) и протокола, предложенного производителем. Лигированная смесь с помощью электропорации была перенесана в XL-1 Blue бактерии (Stratagene). Был определен один клон 2.7 с содержанием последовательностей, соответствующих αTM1 пептидным последовательностям, которые не были использованы при конструировании праймеров.
Секвенирование было проведено с Applied Biosystems 373A ДНК секвестором (Foster City, CA) с набором Dye-deoxy терминаторной циклической последовательности (ABI), в которую был введен флуоресцентно-меченный dNTPs при асимметрической PCR реакции [McCabe, "Получение однонитевой ДНК с помощью асимметрической PCR реакции", в PCR Protocol: A Guide to Methods and Applications. Innis. et.al.(eds), с. 76-83 Academic Press: New York (1990)] следующим образом. Образцы выдерживали при 96oС в течение 4 минут и подвергали 25 циклам постадийной последовательной обработки: 96oС в течение 15 секунд, 50oС в течение 1 секунды; 60oС в течение 4 минут. Последовательные данные автоматически загружались в файлы образца на компьютере, которые включали хроматограмму и текстовые файлы. Последовательность полного инсерта клона 2.7 представлялась в SEQ ID NO:24.
Попытки выделить собачью αTM1 сДНК полной длины из Stratagene бибилиотеки (как описано в Примере 2) не увенчались успехом. Были скринированы приблизительно 1•106 фаговых бляшек с помощью гибридизации в нестрогих условиях с использованием 30% формамида с клоном 2.7 в качестве пробы, но не было получено положительных клонов. Попытки эстракопирования соответствующих последовательностей нижнего потока из тех, которые представлены в клоне 2.7 с использованием специфических олигонуклеотидов, полученных из клона 2.7, или вырожденных праймеров на основании аминокислотной последовательности из других пептидных фрагментов, спаренных с вырожденным олигонуклеотидом на основе сохраненной αTM1 субъединицы аминокислотного мотива GFFKR [Tamura et. al., выше], также были безуспешными.
Пример 5
Клонирование предполагаемого человеческого гомолога собачьей α сДНК.
В попытке выделить человеческую последовательность, гомологичную собачьей αTM1 в качестве пробы, был использован приблизительно 1 кб собачий αTM1 фрагмент из клона 2.7. Проба была генерирована с помощью PCR реакции в условиях, описанных в Примере 2, с использованием NT2 (представленного в SEQ ID NO:25) праймеров.
5′-GTNTTYCARGARGAYGG-3′ (SEQ ID NO:25)
PCR продукт был очищен с использованием Qiagen (Chatsworth, GA) Quick Spin набора и протокола, предложенного производителем. Очищенная ДНК (200 нг) была мечена 200 мк Ci α32 PdCTR с использованием Boehringer Mannheim Random Prime Labelling набора и протокола, предложенного производителем. Невошедший изотоп был удален с помощью хроматографии под действием силы тяжести с Сефадексом G25 (мелкозернистым). Проба до использования была подвергнута денатурированию 0.2 N раствором NaOH и нейтрализации 0.4 М Трис-HCl, рН 8.0.
Были приготовлены колонии клеток (colony lifts) на Hybon фильтрах (Amersham) с сДНК библиотекой человеческой селезенки в pCDNA/Amp (Invitrogen, San Diego, CA). Фильтры были сначала денатурированы и нейтрализованы, как описано в Примере 2, и впоследствии денатурированы в растворе прегибридизации (8 мл/фильтр) с 30% формамида при 50oС при осторожном перемешивании в течение 2 часов. К этому раствору была добавлена меченая проба, как описано выше, и проинкубирована с фильтрами в течение 14 часов при 42oС. Фильтры были два раза промыты в 2Х SSC/0.1% SDS при 37oС и два раза в 2Х SSC/0.1% SDS при 50oС. Окончательное тщательное промывание было проведено 1Х SSC/0.1% SDS дважды при 65oС (1Х SSC представляет 150 мМ NaCl, 15 мМ цитрата натрия, рН 7.0). Фильтры подвергали экспозиции на пленке Kodak X-Omat AR в течение 6 часов с интенсифицирующим экраном. Колонии, дающие сигналы на дупликатных подъемах (lifts), были посеяны штрихом в LB среду в чашки с магний (LВМ)/карбеницилином и проинкубированы в течение ночи при 37oС. Полученные посеянные штрихом колонии были подняты Hybond фильтрами и эти фильтры были обработаны, как указано выше. Фильтры были прогибридизованы при более строгих условиях с 1 кб пробой из клона 2.7, мечены, как описано выше, в 50% формамидном гибридизационном растворе при 50oС в течение 3 часов. Фильтры, в которые были введены пробы, были промыты с окончательной тщательностью 0.1Х SSC/0.1% SDS при 65oС и подвергнуты экспозиции на пленку Kodak X-Omat AR в течение 2.5 часов при -80oС с интенсифицирующим экраном. Положительные колонии были идентифицированы и подвергнуты культивированию в (LВМ)/карбеницилиновой среде в течение ночи. ДНК из культур была приготовлена с использованием минипрепаративного набора Promega Wizard в соответствии с протоколом, предложенным производителем, и получающаяся ДНК была подвергнута секвенированию.
Начальный скрининг привел к 18 положительным клонам, в то время как вторичное скринирование при более строгих условиях гибридизации давало один положительный клон, который был обозначен 19А2. ДНК и полученные аминокислотные последовательности человеческого αd клона 19А2 представляются в SEQ ID NO:1 и 2 соответственно.
Характеристики человеческой αd сДНК и предсказанного полипептида.
Клон 19А2 включает в себя полную кодирующую область для зрелого белка, плюс 48 оснований (16 аминокислотных остатков) сигнальной последовательности 5'верхнего потока и 214 оснований 3' нетранслированной последовательности, которая не обрывается в полиаденилированной последовательности. Молекулярный вес ядра зрелого белка предсказывается равным около 125 kD. Экстраклеточный (межклеточный) домен предсказывается включающим аминокислотные остатки приблизительно от 17 до 1108 SEQ ID NO:2. Эта экстраклеточная область является областью последовательностей с около 20 аминокислотами гомологичной с человеческой CD11c трансмембранной областью (остатки 1109 до 1128 SEQ ID NO:2). Цитоплазматический домен включает приблизительно 30 аминокислот (остатки от 1129 до 1161 SEQ ID NO:2). Белок содержит также область (вокруг 150-352 остатков) приблизительно из 202 аминокислот, гомологичных I (инсертированному) домену, общему для CD11a, CD11b и CD11c [Larson and Springer, выше], αE [Shaw, et. al. , J.Biol.Chem. 269:6016-6025 (1994)] и в VLA-1 VLA-2 [Tamura et. al. , выше]. Было показано, что I домен в других интегринах участвует в ICAM связывании [Landis, et.al., J.Cell.Biol. 120:1519-1527 (1993)]; Diamond, et. al. , J.Cell.Biol. 120:1031-1043(1993)], подтверждая, что αd может также связывать элементы семейства ICAM поверхностных молекул. Не было продемонстрировано, что эта область существует в любых других интегриновых субъединицах.
Выведенная аминокислотная последоваительность αd показывает приблизительно 36% идентичности аминокислотной последовательности CD11a, приблизительно 60% идентичности CD11b и приблизительно 66% идентичности CD11c. Упорядочение аминокислотных последовательностей для (CD11b SEQ ID NO:3), CD11с (SEQ ID NO:4) и αd (SEQ ID NO:2) представляется на Фиг.1.
Цитоплазматические домены α субъединиц в β2 интегринах являются обычно отличными друг от друга в пределах одних и тех видов, в то время как индивидуальные α субъединицы показывают высокие степени гомологии по видовым микроокружениям.
В соответствии с этими наблюдениями цитоплазматическая область αd заметно отличается от CD11a, CD11b, и CD11c за исключением мембранной проксимальной GFFKR аминокислотной последовательности, которая, как показано, сохраняется среди всех α интегринов [Rojiani, et.al., Biochemistry 30: 9859-9866 (1991)] . Так как цитоплазматическая хвостовая область интегринов была вовлечена в "изнаночное" ("inside out") сигналирование и авидность регулирования [Landis et. al., выше], возможно, что αd взаимодействует с цитоплазматическими молекулами, отличными от молекул, взаимодействующих с CD11a, CD11b и CD11c, и в результате участвует в путях сигналирования, отличных от путей, включающих другие β2 интегрины.
Экстраклеточный домен αd содержит сохраненную DGSGS аминокислотную последовательность, смежную с 1-доменом; в CD11b, DGSGS последовательность представляет область, связывающую металл, необходимую для взаимодействия лиганда [Michishita, et. al., 72:857-867 (1993)]. Три дополнительных предполагаемых сайта, связывающих катион в CD11b и CD11c, являются сохраненными в αd последовательности в аминокислотах 465-474, 518-527 и 592-600 в клоне 19А2 (SEQ ID NO: 1). αd I-домен представляет 36%, 62% и 57% идентичность соответствующим областям в CD11a, CD11b, и CD11c соответственно, и относительно низкая последовательная гомология в этой области подтверждает, что αd может взаимодействовать с набором экстраклеточных белков, отличных от белков, с которыми взаимодействуют другие известные β2 интегрины. Или же сродство αd для известных β2 интегриновых лигандов, например, ICAM-1, ICAM-2 и/или ICAM-R, может быть отличным от того, которое демонстрируется для других β2 интегриновых/ICAM взаимодействий [смотри Пример 12].
Выделение дополнительных человеческих αd сДНК клонов для секвенируюшего контроля.
Для того чтобы подтвердить ДНК последовательность, кодирующую человеческую αd, были выделены дополнительные человеческие сДНК путем гибридизации человеческой селезеночной олиго-dт-примированной сДНК библиотеки (Invitrogen) в рсДНК/Аmр, (описанной в Примере 5), размер которой был выбран с помощью электрофореза в агарозном геле для сДНК больше чем 3 кб в длину. Проба для гибридизации была получена из 5' области αd, как описано ниже. Условия гибридизации были такими же, как описано выше для выделения начального человеческого αd клона за исключением того, что после гибридизации фильтры были дважды промыты в 2Х SSC/0.1% SDS при комнатной температуре и один раз 2Х SSC/0.1% SDS при 42oС. Фильтры подвергали экспозиции на пленке Kodak X-Omat AR в течение ночи.
5' αd проба гибридизации была генерирована PCR реакцией из 19А2 клона с использованием праймеров CD11c 5' For (SEQ ID NO:94) и CD11c 5' для Rev(SEQ ID NO: 95) при следующих условиях. Образцы выдерживали при 94oС в течение 4 минут и подвергали 30 циклам обработки последовательными температурными стадиями i) 94oC в течение 15 секунд; ii)5oC в течение 30 секунд; iii)72oС в течение 1 минуты в аппарате Perkin-Elmer 9600 thermoceycler.
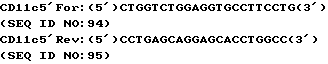
Продукт амплификации был очищен с использованием Bio Pad (Hercules, СА) Prep-A-Gene набора в соответствии с протоколом, предложенным производителем. Получающаяся 5' αd проба включала в длину приблизительно 720 пар оснований, соответствующих области от нуклеотида 1121 до нуклеотида 1839 в SEQ ID NO:1. Очищенная ДНК (приблизительно 50 нг) была мечена 32P-dCTR с использованием Boehringer Mannheim (Indianapolis, Indiana) Random Prime Labelling набора согласно протоколу, предложеному производителем. Не вошедший изотоп был удален с использованием Centrisep Spin колонок (Princeton Separetion, Adelphia, NJ) в соответствии с протоколом, предложенным производителем. Меченая проба была добавлена к фильтрам в раствор прегибридизации, содержащий 45% формамида и инкубированию позволяли протекать в течение ночи при 50oС. После инкубирования фильтры были промыты, как описано выше.
Тринадцать колоний давали сигналы на дупликатных подъемах. Положительные колонии были отобраны из основных чашек, разбавлены в LBM и карбецилине (100 мкг/мл) и культивированы при различных разбавлениях на Hybon (Amersham) фильтры. Удвоенные фильтры были прогибридизованы тем же самым раствором от первичной гибридизации, и после гибридизации фильтры были промыты с окончательной тщательностью 2Х SSC/0.1% SDS при 42oС и подвергнуты экспозиции на пленку.
Десять из первоначально идентифицированных тринадцати положительных колоний были подтверждены при вторичном скринировании. Из этих десяти клонов два (обозначенных A7.Q и A8.Q) были подвергнуты секвенированию и определены для кодирования человеческой αd. Было найдено, что клон A7.Q представляет в длину приблизительно 2.5 кб, включая 5'лидерную часть, часть, кодирующую область, и дополнительно 60 оснований 5' нетранслированной последовательности. Было определено, что неполная кодирующая область является следствием отклонения от сплайсинга интронной области при соответствующем нуклеотиде 2152 SEQ ID NO:1. Было определено, что клон A8.Q представляет в длину приблизительно 4 кб, перекрывая всю αd кодирующую область и включающую также интронную последовательность при соответствующем основании 305 SEQ ID NO:1. По сравнению с первоначально выделенным αd клоном (SEQ ID NO:1) наблюдалось одно отличие в том, что, как было определено, оба клона A7.Q и A8.Q имеют три инсерции основного CAG кодона, происходящие в основании 1495. Последовательности для клонов A7.Q и A8.Q представляются в SEQ ID NO:96 и 97 соответственно, и композитная человеческая последовательность, полученная из клонов A7. Q и A8.Q и ее соответствующая выведенная аминокислотная последовательность представляется в SEQ ID NO:98 и 99, соответственно.
Пример 6
Анализ Нозерн-блоттинга человеческой αd экспрессии в тканях.
Для того чтобы определить относительный уровень экспрессии и тканевую специфичность αd, был выполнен анализ Нозерн-блоттинга с использованием фрагментов в качестве проб из клона 19А2. Приблизительно 10 мкг полной РНК из каждой из нескольких человеческих тканей или культивированных линий клеток загружали на формальдегид агарозный гель в присутствии 1 мкг этидиний бромида. После электрофореза при 100 B в течение 4 часов, РНК переносили на нитроцеллюлозную мембрану (Schleicher and Schuell) с помощью тампона в 10Х SSC в течение ночи. Мембрану отжигали в течение 1.5 часов при 80oС в вакууме. Для блокирования мембраны в течение 3 часов при 42oС был использован раствор прегибридизации, содержащий 50% формамида в буфере 3-(N-морфолин)пропан сульфоновой кислоты (MOPS). Фрагменты клона 19А2 были мечены Boehringer Mannheim Random Prime Labelling набором согласно инструкциям производителя, включая αP32 dCTR и a α
Гибридизация с использованием BstX1 фрагмента из клона 19А2 (соответствующего нуклеотидам 2011-3388 в SEQ ID NO:1) приводила к обнаружению слабого сигнала в области приблизительно 5 кб в образцах печени, планцеты, тимуса и миндалин полной РНК. Сигнал не был обнаружен в образцах почки, мозга или сердца. Количество РНК, присутствующей в почечной полосе, было минимальным, как определено с помощью окрашивания этидиний бромидом.
Когда использовали второй фрагмент клона 19А2 (включающий область от оснований 500 до 2100 в SEQ ID NO:1), были обнаружены РНК транскрипты двух различных размеров в человеской мульти-ткани Нозерн-блоттинга (MTN) с использованием полиА+ РНК (Clontech). Полосу приблизительно 6.5 кб наблюдали в селезенке и скелетной мышце, в то время как полосу 4.5 кб обнаруживали в легком и лейкоцитах периферической крови. Изменения в наблюдающихся размерах могут быть вызваны специфическим аденилированием ткани, перекрестной реактивностью пробы с другими членами семейства интегринов или гибридизацией с мРНК, подвергнутой альтернативному сплайсингу.
Нозерн анализ с использованием третьего фрагмента из клона 19А2, перекрывающего нуклеотиды 2000-3100 в SEQ ID NO:1, давал результат, согласующийся с результатом, использующим другие фрагменты клона 19А2.
РНК из трех лианезированных линий клеток спинного мозга также была испытана с использованием фрагментов, соответствующих нуклеотидам 500-2100 и 2000-3100 в SEQ ID NO:1. ТНР-1 линия клеток, стимулированная ранее РМА, давала диффузный сигнал в той же самой области размеров (приблизительно 5.0 кб) со слегка более сильной интесивностью, чем в случае тканевых сигналов. РНК из нестимулированных и ДМСО-стимулированных HL-60 клеток гибридизовали αd пробой при той же интенсивности, как и в случае тканевых образцов, однако РМА обработка, по видимому, увеличивала интенсивность сигнала. Так как РМА и ДМСО управляли HL-60 клеточной дифференциацией по отношению к моноцит/макрофагу и гранулоцитовым путям соответственно, этот результат подтверждал усиленную αd экспрессию в моноцит/макрофаговых клеточных типах. U937 клетки экпрессировали αd перенос, и этот сигнал не увеличивался стимулированием РМА. Не было обнаружено полосы в Molt, Daudi, H9, JY или клетках Jukart.
Пример 7
Неустойчивая экспрессия человеческих αd конструкций.
А. Генерирование экспрессионных конструкций.
Человеческий клон 19А2 испытывал недостаток в инициации метионинового кодона и, возможно, некоторой 5' сигнальной последовательности. Поэтому для того чтобы генерировать человеческую плазмиду экспрессии, содержащую 19А2 последовательности, были использованы два различных подхода. Во-первых, были сконструированы две плазмиды, в которых сигнальные пептидные последовательности, полученные из генов, кодирующих либо CD11b, либо CD11c, были подвергнуты сплайсингу в клон 19А2 для генерирования химерной αd последовательности. Во втором подходе была сконструирована третья плазмида, в которую было добавлено аденозиновое основание в положение 0 в клоне 19А2 для кодирования инициации метионина.
Три плазмиды содержали различные области, которые кодировали 5'часть αd последовательности или химерной αd последовательности, αd область была подвергнута PCR амплификации (смотри условия в Примере 2) специфическим 3' праймером Bam Rev (представленном ниже в SEQ ID NO:26) и одним из трех 5' праймеров. Три 5' праймера содержали в последовательности: (1) Идентичные неспецифические основания в положениях 1-6, предусматривающие гидролиз, EcoRI сайт из положений 7-12 и согласованную Kazak последовательность из положений 13-18; (2)часть CD11b (праймер ERIB) или CD11c (праймер ERIC) сигнальной последовательности или аденозин (праймер ERID); и (3)дополнительные 15-17 оснований, специфически перекрывающих 5' последовательности из клона 19А2 для того, чтобы предусмотреть праймерный отжиг. Праймеры ERIB, ERIC и ERID представляются в SEQ ID NO:27, 28 или 29 соответственно, где метиониновый кодон инициации является 6 подслоенным и EcoRI сайт является дважды подслоенным.

Получающийся PCR продукт был гидролизован EcoRI и BamHI.
Все три плазмиды содержали общую вторую αd область (которая немедленно инсертируется в нижний поток из 5' области, описанной в предыдущем параграфе), включающую 3' конец αd клона. Вторая αd область, которая простирается от нуклеотида 625 в Xbal сайт в векторе 3' полилинкерной области клона 19А2, была выделена гидролизом клона 19А2 BamHI и Xbal.
Были проведены три реакции лигирования, в которых 3' BamHI/Xbal фрагмент был лигирован в один из трех 5' αd EcoRI/BamHI фрагментов с использованием Boehringer Mannheim лизатного буфера и Т4 лигазы (1 единица на реакцию). После 4 часов инкубирования при 14oС соответствующее количество вектора рсДНК. 3 (Invirogen), прогидролизованного EcoRI и Xbal, было добавлено к каждой реакции с дополнительной единицей лигазы. Реакциям позволяли протекать еще 14 часов. Затем одну десятую реакционной смеси превращали в XL-1 Blue компетентные клетки. Получающиеся колонии были культивированы, и ДНК выделена, как в Примере 5. Гидролиз EcoRI идентифицировал три клона, которые были положительными для указанного сайта рестрикции и, таким образом, конструировали сигнальную последовательность. Клоны были обозначены рАТМ.В1 (CD11b/αd, из праймера ER1B), рАТМ.С10 (CD11b/αd, из праймера ER1C) pATM.D12 (аденозин/αd из праймера ER1D). Присутствие соответствующих сигнальных последовательностей в каждом клоне было подтверждено с помощью секвенирования нуклеиновой кислотой.
Б. Трансфекция COS клеток.
Экспрессия из αd плазмид, обсуждавшаяся выше, была осуществлена с помощью сотрансфекции COS клеток с индивидуальными плазмидами и CD18 экспрессионной плазмидой, pRC. CD18. В качестве положительного контроля COS клетки были также подвергнуты со-трансфекции с плазмидой pRC.CD18 и CD11 экспрессионной плазмидой, pDC.CD11A.
Клетки были пассированы в культурную среду (DMEM/10%FBS/penstrep) в 10 см чашки Петри с обработанной Corning тканевой культурой при 50% слиянии за 16 часов до трансфекции. Клетки были удалены из чашек буфером Versene (0.5 мМ NаЭДТА в PBS) без трипсина для всех процедур. До трансфекции чашки промывали один раз свободным от сыворотки DMEM. Пятнадцать микрограммов каждой плазмиды были добавлены к 5 мл буфера трансфекции (DMEM с 20 мкг/мл DEAE-декстран и 0.5 мМ хлорокина) в каждую чашку. После 1.5 часов инкубирования при 37oС клетки были подвергнуты шоку в течение 1 минуты 5 мл DMEM/мл ДМСО. Этот раствор ДМСО был затем заменен 10 мл/чашку культурной среды.
Полученные трансфектанты были проанализированы с помощью ELISA, FACS и иммунопреципитации, как описано в Примерах 8, 9 и 10.
Пример 8
ELISA анализ COS трансфектантов.
Для того чтобы определить, являются ли COS клетки со-трансфекцированными с CD18 плазмидой экспрессии pRC.CD18 и αd плазмидой экспрессированной αd на клеточной поверхности вместе с CD18, был проведен анализ ELISA с использованием первичных антител, возникающих против CD18 (например, TS1/18 очищенных из АТСС НВ203). В качестве положительного контроля были выполнены также анализы ELISA на клетках, со-трансфекцированных с CD18 плазмидой экспрессии и CD11a плазмидой экспрессии pDC.CD11A. Первичные антитела в этом контроле индуцировали CD18 антителами и анти-CD11a антителами (например, TS1/22, очищенными из АТСС НВ202).
Для анализа ELISA клетки из каждой чашки были удалены буфером Versene и перенесены в лунки 96-луночного планшета, содержащего на дне тканевую культуру Corning. Клеткам позволяли инкубироваться в культурной среде в течение 2 суток до анализа. Затем чашки дважды промывали 150 мкл/лунку D-PBS/0.5% раствором желатина кожи teleost (Sigma). Этот буфер был использован во всех стадиях за исключением стадии роста. Все промывки и инкубирования были проведены при комнатной температуре. Лунки были блокированы раствором желатина в течение 1 часа. Первичные антитела были разбавлены до 10 мкг/мл в растворе желатина и затем 50 мкл были добавлены в каждую лунку. Для каждого первичного антитела были использованы тройные лунки. После 1 часа инкубирования чашки промывали 3Х раствором желатина с концентрацией 150 мкл/ячейку. Вторичное антитело (специфическое овечье антимышиное Ig/HRP-Fc [Jackson, West Grove, PA]) при разбавлении 1:3500 было добавлено при концентрации 50 мкл/лунку, и чашки были проинкубированы в течение 1 часа. После трех промывок чашки были проявлены в течение 20 минут 100 мкл/лунку раствора о-фенилдиамина (OPD)(Slgma) (с концентрацией 1 мг/мл OPD в цитратном буфере) до добавления 50 мкл/лунку 15% серной кислоты.
Анализ трансфектантов в формате ELISA со специфическими антителами анти-СD18 показывал незначительную экспрессию выше фона в клетках, трансфекцированных только плазмидой, кодирующей CD18. Клетки, со-трансфекцированные с плазмидой, содержащей CD11a и CD18, показывали увеличение в экспрессии против фона, когда анализировались со специфическими антителами CD18 или реагентами, специфическими для CD11a. Дальнейший анализ клеток, со-трансфекцированных с плазмидами, кодирующими CD18, и одной из конструкций αd экспрессии (рАТМ.С10 или рATM.D12), показывал, что клеточная поверхностная экспрессия CD18 исключается сопутствующей экспрессией αd. Увеличение в обнаруживаемой CD18 экспрессии в COS клетках, трансфекцированных рАТМ.С10 или рATM. D12, было сравнимо с экспрессией, наблюдаемой в со-трансфекцированных CD11a/CD18 положительных контрольных клетках.
Пример 9
FACS анализ COS трансфектантов.
Для FACS анализа клетки в чашках Петри были загружены свежей культурной средой спустя сутки после трансфекции, и им было позволено инкубироваться в течение 2 суток до анализа. Трансфектантные клетки были удалены из чашек 3 мл Versene, промыты один раз 5 мл FACS буфера (DMEM/2% FBS/0.2% азида натрия) и разбавлены до 500000 клеток/образец в 0,1 мл FACS буфера. Десять микролитров либо 1 мг/мл FITC-конъюгированных CD18, CD11a либо CD11b (Becton Dickinson) специфических антител или 800 мкг/мл CFSE-конъюгированных мышиных 23F2G (aнти-CD18)(ATCC HB11081) были добавлены к каждому образцу. Затем образцы были проинкубированы во льду в течение 45 минут, промыты 3 раза 5мл/промывку FACS буфера и ресуспендированы в 0.2 мл FACS буфера. Затем образцы обрабатывали на Becton Dickinson FACscan и данные анализировали с помощью Lysys II software (Becton Dickinson).
COS клетки, трансфекцированные только CD18 последовательностями, не окрашивались в CD18, CD11a или CD11b. Если со-трансфекцировали с CD11a/CD18, около 15% клеток окрашивались антителами в CD11а или CD18. Все клетки, трансфекцированные с CD18 и любой конструкцией αd, не приводили к обнаруживаемому окрашиванию в CD11a и CD11b. рАТМ.В1, рАТМ.С10 и рATM.D12 группы окрашивались 4%, 13% и 8% положительными в CD18 соответственно. Флуоресценция в положительной популяции в CD11a/CD18 группе была в 4 раза выше, чем фон. По сравнению с со-трансфекцированными αd конструкциями CD18 конструкция продуцировала положительную популяцию, которая показывала 4-7-кратное увеличение в интенсивности флуоресценции по сравнению с фоном.
Пример 10
Биотин-меченная иммунопреципитация человеческих αd/СD18 комплексов из со-трансфекцированных COS клеток.
Была предпринята попытка иммунопреципитации на клетках, со-трансфекцированных с CD18 и каждой из плазмид αd экспрессии, отдельно описанных в Примере 7 для того, чтобы определить, может ли быть αd выделена в виде части αβ гетеродимерного комплексного параметра интегринов.
Трансфекцированные клетки (1-3•108 клеток/группу) были удалены из чашек Петри буфером Versene и промыты три раза в 50 мл/группу D-PBS. Каждый образец был мечен 2 мг Сульфо-NHS Biotin (Pierce, Rockford, IL) в течение 15 минут при комнатной температуре. Реакцию гасили промыванием 3 раза в 50 мл/группу холодным D-PBS. Промытые клетки ресуспендировали в 1 мл лизисного буфера (1% NP40, 50 мМ Трис-HCl, рН 8.0, 0.2 М NaCl, 2 мМ Са++, 2 мМ Мg++, протеазные ингибиторы) и проинкубированы в течение 15 минут во льду. Нерастворимое вещество было отделено центрифугированием при 10000 g в течение 5 минут, и супернатант удален в свежие пробирки. Для того чтобы удалить неспецифически реактивное вещество с мышиным иммуноглобулином, была проведена стадия предварительного просветления. Двадцать пять граммов мышиного иммуноглобулина (Cappel, West Chester, PA) были проинкубированы супернатантами при 4oС. После 2.5 часов 100 мкл (25 мкг) кроличьего антимышиного Ig, конъюгированного с сефарозой (приготовленного из Protein A Sepharose 4B и кроличьего антимышиного IgG, оба из Zymed, San Francisco, СА), были добавлены в каждый образец; инкубирование было продолжено при 4oС с встряхиванием в течение 16 часов. Сефарозные гранулы были удалены из супернатантов центрифугированием. После предварительного просветления супернатанты были затем обработаны 20 мкг антитела анти-СD18 (TS1.18) в течение 2 часов при 4oС. Антитело/антигенные комплексы были выделены из супернатантов инкубированием 100 мкл/образец кроличьего антимышиного/белок А сефарозного препарата, описанного выше. Гранулы были 4 раза промыты 10 мМ HEPES, 0.2 М NaCl и 1% Тритон-Х 100. Промытые гранулы были собраны и подвергнуты кипячению в течение 10 минут в 20 мкл 2Х Laemmli буферного образца с 2% β- меркаптоэтанола. Образцы были отцентрифугированы и загружены на 8% предварительно налитый полиакриламидный гель Novex (Novex) при 100 B в течение 30 минут. Белок был перенесен на нитроцеллюлозную мембрану (Schleicher and Schuell) в TBS-T буфере при 200 мА в течение 1 часа. Мембраны были блокированы в течение 2 часов 3% BSA в TBS-T. Мембраны были обработаны 1:6000 разбавлением степавидиновой пероксидазой хрена (POD) (Boehringer Mannheim) в течение 1 часа с последующими тремя промывками в TBS-T. Затем для проявления блота был использован набор Amersham Enhanced Chemiluminescence в соответствии с инструкциями производителя. Мембрана была подвергнута экспозиции на Hyperfilm MP (Amersham) в течение 0.5-2 минут.
Иммунопреципитация CD18 комплексов из клеток, трансфекцированных PRC. CD18 и либо рАТМ.В1, рАТМ.С10, либо pATM.D12, обнаруживающим поверхностную экспрессию гетеродимерных образцов, состоящих из приблизительно 100 kD β цепей, согласовывалась с предсказанным размером CD18 и α цепи приблизительно 150 kD, соответствующей αd.
Пример 11
Устойчивая трансфекция человеческой αd в клетках яичника китайского хомячка.
Для определения того, экспрессируется ли αd на клеточную поверхность в виде гетеродимера вместе с CD18, сДНК-ты, кодирующие каждую цепь, были обе кратковременно и устойчиво трансфекцированы в линию клетки, которой недоставало обеих составляющих αd и CD18.
Для этих экспериментов αd сДНК была подвергнута стимуляции дополнительными лидерными последовательностями и Kozak согласованной последовательностью, как описано в Примере 7, и субклонирована в векторе экспрессии рсДНК3. Конечная конструкция, обозначенная pATM.D12, была со-трансфекцирована с модифицированным коммерческим вектором, pDCl.CD18, кодирующим человеческую CD18 в дигидрофолат редуктазных (DHFR)-клетках яичника (СНО) китайского хомячка. Плазмида pDC1.CD18 кодировала DHFR+ маркер и трансфектант мог быть выбран с использованием соответствующей нуклеозид-дефицитной среды. Модификации, которые давали pDC1.CD18, являются следующими.
Плазмида pRC/CMV (Invitrogen) является вектором экспрессии млекопитающего с цитомегаловирусным промотором и с сопротивлением гена маркера ампициллину. DHFR ген из плазмиды pSC1190-DHFR был инсертирован в pRC/CMV 5' SV40 источника репликации. Кроме того, полилинкер из 5' области плазмиды pHF2G-DHF был лигирован в pRC/CMV/DHFR конструкцию, 3' в DHFR гене. CD18 кодирующие последовательности впоследствии клонировались в получающуюся плазмиду между 5' фланкирующей полилинкерной областью и бычьим гормоном роста поли А кодирующей области.
Поверхностная экспрессия CD18 была проанализирована с помощью цитометрии в потоке с использованием моноклонального антитела TS1/18. Образование гетеродимера, обнаруженное между αd и CD18, в этой клеточной линии согласовывалось с иммунопреципитацией, описанной в Примере 10, с кратковременной экспрессией в COS клетках.
Пример 12
Связывание человеческой αd с ICAM-R в СD18-зависимом гибриде.
В свете того, что сообщалось, демонстрируется взаимодействие между лейкоцитовыми интегринами и молекулами с межклеточной адгезией (ICAMs), которые опосредуют контакт клетка-клетка [Hynes, Cell 69:11-25 (1992)], способность СНО клеток, экспрессирующих αd/CD18, к связыванию IСАМ-1, ICAM-R или VCAM-1, определенную двумя способами.
В репликационных анализах растворимые IСАМ-1, ICAM-R или VCAM-1, IgG1 составные белки были иммобилизованы в пластик и была определена способность αd/CD18 СНО трансфекцированных клеток к связыванию иммобилизованных лигандов. Трансфекцированные клетки были подвергнуты мочению кальцеином, промыты в связывающем буфере (PRMI с 1% BSA), и проинкубированы либо только в буфере (с или без 10 нг/мл РМА), либо в буфере с aнти-CD18 моноклональными антителами с концентрацией 10 мкг/мл. Трансфекцированные клетки были добавлены в 96-луночные Immulon 4 микротитрические планшеты, заполненные ранее растворимым ICAM-1/IgGt, ICAM-R/IgGI, VCAM-1/IgG1 составным белком или бычьим сывороточным альбумином (BSA) в качестве отрицательного контроля. Конструирование растворимых форм этих адгезивных молекул описывается и полностью раскрывается в совместной заявке США No. 08/102852, поданной 5 августа 1993 г. Лунки были блокированы 1% BSA в PBS до добавления меченых клеток. После промывания планшетов погружением в PBS с 1% BSA в течение 20 минут была измерена полная флуоресценция, остающаяся в каждой лунке, с использованием Cytofluor 2300 (Millipore, Milford, MA).
В экспериментах с иммобилизованными ICAMs, αd/CD18 co-трансфектантами, последовательно показано 3-5-кратное увеличение в связывании с ICAM-R/IgG1 лунками по сравнению с лунками, покрытыми BSA. Была продемонстрирована специфичность СD18-зависимости этого связывания с помощью ингибирующих эффектов анти-СD18 антитела TS1/18. Связывание клеток, трансфекцированных CD11a/CD18 с ICAM-1/IgG1 лунками, было сравнимым со связыванием, наблюдаемым с лунками, заполненными BSA. CD11a/CD18 трансфекцированные клетки показывали 2-3 кратное увеличение в связывании с ICAM-1/IgG1 лунками, только после предварительной обработки РМА. РМА обработка αd/CD18 трансфектантов не влияла на связывание с ICAM-1/IgG1 или ICAM-R/IgG1 лунками. Не наблюдали обнаруживаемого связывания αd/CD18 трансфектантов с VCAM-1/IgG1 лунками.
Связывание αd/СD18-трансфекцированных клеток с растворимыми ICAM-1/IgG1, ICAM-R/IgG1 или VCAM-1/IgG1 составными белками было определено с помощью проточной цитометрии. Приблизительно один миллион αd/CD18-трансфекцированных СНО клеток (выращенных в центрифужных пробирках до более высокой экспрессии) на измерение было суспендировано в 100 мкл связывающего буфера (PRMI и 1% BSA) с или без добавления 10 мкг/мл анти-CD18 антитела. После 20 минут инкубации при комнатной температуре клетки были промыты в связывающем буфере и был добавлен растворимый ICAM-1/IgG1 или ICAM-R/IgG1 составной белок с конечной концентрацией 5 мкг/мл. Связыванию было позволено протекать в течение 30 минут при 37oС, после чего клетки были три раза промыты и ресуспендированы в 100 мкл связывающего буфера, содержащего FITC-конъюгированный овечий анти-человеческий IgG1 при рабавлении 1:100. После 30 минут инкубирования образцы были три раза промыты и суспендированы в 200 мкл связывающего буфера для анализа Becton Dickinson FaCScan.
Приблизительно 40-50% αd/CD18 трансфектантов указывали на связывание с ICAM-R/IgG1, но не на связывание с ICAM-1/IgG1 или VCAM-1/IgG1 белками.
Предварительная обработка трансфекцированных клеток РМА не оказывала влияния на αd/CD18 связывание либо с ICAM-1/IgG1, ICAM-R/IgG1, либо VCAM-1/IgG1, которое согласовывалось с иммобилизованным адгезионным анализом. Связывание с помощью ICAM-R было уменьшенным относительно уровней фона после обработки αd/CD18 трансфектантов анти-СD18 антителом TS1/18.
Общие данные из этих двух анализов по связыванию иллюстрируют, что αd/CD18 связывается с ICAM-R и связывается предпочтительно по сравнению с IСАМ-1 и VCAM-1. αd/CD18 связывание, предпочтительное для ICAM-R, по сравнению с IСАМ-1 является противоположным тому, что наблюдается с CD11a/CD18 и CD11b/CD18. Таким образом, модуляция αd/CD18 связывания может селективно оказывать влияние на нормальную и патологическую иммунную функцию, где ICAM-R играет заметную роль. Кроме того, результаты аналогичных анализов, в которых были испытаны антитела, иммуноспецифические для различных экстраклеточных доменов ICAM-R на их способность к ингибированию связывания ICAM-R с αd/CD18 трансфектантами, указывают, что αd/CD18 и CD11a/CD18 взаимодействуют с различными доменами ICAM-R.
Неспособность CD11a/CD18 к связыванию ICAM-1/IgG1 в растворе подтверждает, что способность связывания между CD11a/CD18 и IСАМ-1 или ICAM-R является слишком низкой для того, чтобы позволить связывание в растворе. Обнаружение αd/CD18 связывания с ICAM-R/IgG1, однако, подтверждает необычно высокую связывающую способность.
Был использован адгезионный анализ FACS, описанный выше, для изучения связывания ICAM-R мутанта E37A/Ig с СНО клетками, экспрессирующими αd/CD18.. Было показано, что E37A/Ig устраняет связывание в LFA-1/Ig химере [Sadhu, et. al. , Cell Adgesion and Communication 2:429-440 (1994)]. Мутантный белок был экспрессирован в растворимой форме из устойчиво трансфекцированной СНО линии клетки и очищен на колонке Prosep А, как описано Sadhu, et.al.,выше.
E37A/Ig связывание с αd/CD18 трансфектантами не было обнаружено в повторных анализах. Значение интенсивности флуоресценции (MFI) E37A/Ig химеры, обнаруженное с помощью FITC-конъюгированного античеловеческого антитела, было идентичным MFI одного обнаруживаемого антитела, указывая на то, что не было обнаруживаемого сигнала выше фона с использованием в анализе E37A/Ig мутантного белка. Аналогично в ELISA анализе, выполненном, как описано в Примере 14, E37A/Ig мутант не вызывает связывания иммобилизованного αd/CD18.
αd Связывание с iC3b.
Комплементный компонент С3 может быть протеолитически расщеплен с образованием комплекса iC3b, который инициирует альтернативный путь комплементной активации и приводит одновременно к клетка-опосредованной деструкции мишени. Обе субъединицы CD11b и CD11c были вовлечены в iC3b связывание и последующий фагоцитоз iC3b-покрытых частиц. Белковый фрагмент в CD11b I домене был недавно идентифицирован в виде сайта iC3b взаимодействия [Ueda, et.al., Proc.Natl.Acad.Sci., (USA) 91:10680-10684 (1994]. Область iC3b связывания является высоко консервативной областью в CD11b и CD11c, и αd, предполагая αd/iC3b связывающее взаимодействие.
Связывание αd с iC3b проводилось с использованием трансфектантов или линий клеток, естественно экспрессирующих αd (например, РМА-стимулированных HL60 клеток) и iC3b-покрытых овечьих красных кровяных клеток (sRBC) в анализе розеткообразной структуры [Dana, et. al., J.CIin. Invest. 73: 153-159 (1984)] . Способности αd/CD18 CHO трансфектантов, VLA4-CHO трансфектантов (отрицательный контроль) и РМА-стимулированных HL60 клеток (положительный контроль) к образованию розеткообразных структур сравниваются в присутствии и отсутствии aнти-CD18 моноклонального антитела (например, TS1/18).
Пример 13
Скринирование с помощью анализа, близкого к сцинтилляционному.
Специфические ингибиторы связывания между αd лигандами настоящего изобретения и их связывающими партнерами (пара αd лиганд/антилиганд) могут быть определены с помощью различных способов, таких как техника анализа, близкая к сцинтилляционной, как в общем описано в патенте США 4271139, Hart and Greenwald, Mol.Immunol. 12:265-267 (1979) and Hart and Greenwald, J.Nuc.Med. 20:1062-1065 (1979), каждый из которых представлен здесь в виде ссылки.
Один элемент пары αd лиганд/антилиганд связывается с твердой подложкой либо непосредственно, либо не непосредственно. Непрямой захват будет включать моноклональное антитело, непосредственно связанное с подложкой, которая узнает специфический эпитоп при С-терминусе растворимой интегриновой β цепи белка. Этот эпитоп будет либо гемаглутиновым белком, либо митобактериальным IIIE9 эпитопом [Anderson, et.al., J.Immunol. 141:607-613 (1988)]. Флуоресцирующий агент также связывается с подложкой. Или же флуоресцирующий агент мог быть интегрирован в твердую подложку, как описано в патенте США 4568649, представленном здесь в виде ссылки. Не связанный с подложкой элемент пары αd лиганд/антилиганд метится радиоактивным соединением таким образом, что испускает излучение, способное возбуждать флуоресцирующий агент. Когда лиганд связывает радиоактивномеченый антилиганд, метка вводится достаточно близко к флуоресцирующему агенту, связанному с подложкой для возбуждения флуоресцирующего агента и для излучения света. Когда лиганд не связан, метка является обычно слишком удаленной от твердой подложки для возбуждения флуоресцирующего агента, и излучение света является слишком слабым. Излученный свет измеряется и коррелируется со связыванием между лигандом и антилигандом. Добавление ингибитора связывания к образцу будет уменьшать излучение флуоресценции за счет предохранения радиоактивной метки от того, чтобы быть захваченной вблизи твердой подложки. Поэтому ингибиторы связывания могут быть идентифицированы за счет их влияния на излучение флуоресценции из образца. Потенциальные антилиганды к αd также могут быть идентифицированы с помощью аналогичных методов.
Растворимая рекомбинанта αd/CD18 лейциновой конструкции типа "застежка молния" (смотри Пример 14) используется в анализе, близком к сцинтилляционному в скринировании в модуляторах САМ связывания с помощью следующего способа. Рекомбинантный интегрин иммобилизуется неблокирующей анти-α субъединицей или анти-β субъединицей антитела, ранее нанесенного на плашку с введенным сцинтиллянтом. Химическая библиотека соединений и специфическая биотинилированная CAM/Ig химера одновременно добавляются в чашку. Связывание CAM/Ig химеры обнаруживается с помощью меченого стрептовидина. В анализе ICAM-1/Ig и ICAM-3/Ig биотинилируются NHS-сульфобиотин-LC (длинноцепочечным, Pierce) в соответствии с протоколом, предложенным производителем. Меченые белки являются еще реактивными с САМ специфическими антителами и могут быть показаны в реакции с иммобилизованным LFA-1 с помощью анализа ELISA, с детектированием с помощью Стрептавидин-HRP и последующим проявлением OPD.
Или же рекомбинантный лейциновый белок типа "застежка молния" очищается или частично очищается и запролняется непосредственно в чашку с внедренным сцинтиллянтом. Немеченая CAM/Ig химера и химическая библиотека соединений добавляются одновременно. Связывание CAM/Ig обнаруживается с помощью 125I-меченного античеловеческого Ig. Связывание рекомбинантного интегрина обнаруживается меченой неблокирующей α или β субъединицей антитела.
Пример 14
Растворимые человеческие αd экспрессионные конструкции.
Экспрессию растворимого человеческого αd/CD18 гетеродимерного белка полной длины обеспечивает легко очищаемый материал для иммунизации и анализов по связыванию. Преимущество генерирования растворимого белка состоит в том, что он может быть скорее очищен из супернатантов, чем из клеточных лизатов (как с мембранно-связанным αd/CD18 полной длины); поэтому с улучшенной регенерацией и уменьшенным количеством примесей.
Растворимая αd экспрессионная плазмида была сконструирована следующим образом. Нуклеотидный фрагмент, соответствующий области из оснований от 0 до 3161 в SEQ ID NO:1, клонированный в плазмиду pATM.D12, был выделен гидролизом Hind III и AatII. PCR фрагмент, соответствующий основаниям 3130-3390 в SEQ ID NO: 1, перекрывающий Hind III/AatII фрагмент и содержащий дополнительный MIuI рестриктный сайт при 3' терминусе, был амплифицирован из pATM. D12 с праймерами sHAD.5 sHAD.3, представленными в SEQ ID NO:30 и 31 соответственно.
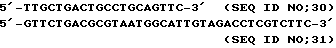
Продукт PCR амплификации был гидролизован AatII и MIuI и лигирован в Hind III/AatII фрагмент. Полученный продукт был лигирован в Hind III/MIuI гидролизованную плазмиду pDC1.s.
Эта конструкция являлась соэкспрессированной с растворимой CD18 в стабильно трансфекцированные СНО клетки и экспрессия обнаруживалась с помощью авторадиографической визуализации иммунопреципитированных CD18 комплексов, полученных из 35S-меченных метионином клеток. Конструкция являлась также соэкспрессированной с растворимой CD18 в 293 клетках [Berman, et.al., J.Cell Biochem. 52:183-195 (1993)].
Растворимая αd конструкция полной длины.
Изобретением рассматриваются также альтернативные αd экспрессионные конструкции. Для того чтобы облегчить экспрессию и очистку интактного αd/CD18 гетеродимера, будут сконструированы растворимые αd и CD18 экспрессионные плазмиды для включения в составную последовательность типа "застежка молния", которая должна стабилизировать гетеродимер в процессе очистки [Chang, et. al. , Proc.Natl.Acad.Sci (USA), 91:11408-11412 (1994)]. ДНК, кодирующая кислотные и основные аминокислотные цепи типа "застежка молния", была генерирована с помощью отжига праймера с использованием олигонуклеотидов, описанных Chang, et. al. ДНК последовательности были далее модифицированы с включением дополнительных MIuI и Хbа1 рестриктных сайтов при 3' и 5' концах соответственно ДНК для облегчения субклонирования в αd или CD18 экспрессионные конструкции, описанные ранее. Кроме того, были добавлены последовательности, представляющие либо гемаглутининовый белок, либо полигистидиновую последовательность, так же как и стоп-кодон, инсертированный после Хbа1 сайта. Гемаглутининовые или полигистидиновые последовательности вводятся для облегчения аффинной очистки экспрессированного белка. Последовательности, кодирующие основную цепь типа "застежка молния", вводятся в плазмидный вектор, экспрессирующий CD18; кислотная цепь вводится в конструкцию α цепи. При экспрессии модифицированных αd и CD18 белков в клетках хозяевах, вероятно, взаимодействие между кислотными и основными цепями структуры типа "застежка молния", будет стабилизировать гетеродимер и позволит выделение интактной αd/CD18 молекулы за счет аффинной очистки, как описано выше.
Для экспрессии растворимых αd и CD18 последовательностей с кислотными и основными элементами типа "застежка молния" были сконструированы плазмиды и трансфекцированы в COS клетки с помощью способа DEAE/Dextran, описанного в Примере 7. На полученный белок ссылались как на αd/CD18LZ. Гемаглутининовые и полигистидиновые мишени не вводились в αd/CD18LZ. Трансфекцированные клетки были выращены в течение 14 дней в условиях, содержащих уменьшенное количество сыворотки (2%). Супернатанты из трансфекцированных клеток, собираемые каждые пять суток, были проанализированы на продуцирование белка с помощью анализа ELISA, как описано в Примере 8. Короче, αd/CD18LZ гетеродимер был иммобилизован в чашки, заполненные анти-αd моноклинальным антителом 169В (смотри Пример 15). αd/CD18LZ комплекс обнаруживали путем добавления биотинилированного aнти-CD18 моноклонального антитела, TS1/18.1 (смотри Пример 8), с последующим добавлением конъюгата стрептавидин/пероксидазы хрена (HRP) и о-фенилдиамина (OPD). Белок был четко обнаруживаемым в супернатантах.
Анализы по связыванию с использованием растворимых αd продуктов экспрессии полной длины.
Функциональные анализы по связыванию с использованием гетеродимера αd/CD18LZ полной длины, описанного выше, были проведены за счет иммобилизации гетеродимера на планшеты, заполненные моноклональным антителом 169В или неблокирующим анти-CD18 моноклональным антителом (смотри Пример 15). Лунки были блокированы желатином из рыбьей кожи для предотвращения неспецифического связывания до добавления CAM/Ig химер (смотри Пример 12) при начальной концентрации 10 мкг/мл. Связывание химер с αd/CD18 было обнаружено с овечьим античеловеческим Ig HRP конъюгатом (Jackson Labs) и с последующим проявлением OPD.
VCAM-1/Ig наблюдали в связывании с захваченным αd/CD18LZ при уровне, в 3-5 раз превышающем уровень с захваченным CD11a/CD18. ICAM-1/Ig и ICAM-2/Ig связывали растворимый гетеродимер CD11a/CD18 приблизительно с уровнем, 15 и 10-кратно превышающим фон соответственно, но не связывали αd/CD18. VCAM-1 связывание снижалось приблизительно на 50% в присутствии VCAM-1 специфических антител 130К и 130Р, использованных в комбинации.
Анализ по связыванию проводили также с ICAM-1/Ig белком, иммобилизованным в 96-луночныые планшеты с последующим добавлением рекомбинантного растворимого интегрина в клеточный супернатант. Связывание растворимых интегринов было обнаружено с немечеными неблокирующими α и β субъединицами специфического мышиного антитела с последующим инкубированием с HPR-конъюгированным овечьим антимышиным антителом и проявлением OPD.
Результаты указывают на то, что неблокирующее антитело обнаруживало αd/CD18LZ связывание с ICAM-R/Ig, 10-кратно превышающее связывание, обнаруживаемое в контрольной лунке, не содержащей антитела. Связывание растворимой αd/CD18 не было обнаружено с иммобилизованным ICAM-1/Ig, однако связывание было обнаружено между αd/CD18 и иммобилизованными CD11b/CD18 и CD11a/CD18 в 15 и 5 раз соответственно выше фонового связывания.
Так как прежние исследования продемонстрировали, что CD11b и CD11c связывают липополисахарид (LPS) [Wright, Curr. Opin.lmmunol. 3:83-90 (1991); Inhalls and Golenbock, J. Exp. Med. 181:1473-1479 (1995)], связывание LPS также было проанализировано с использованием проточной цитометрии и анализов на плашках. Результаты указывают на то, что FIТС-меченные LPS выделенные из S. Minnesota and S. typhosa (обе полученных из Sigma) при концентрации 20 мкг/мл были способны к слабому связыванию αd/CD18 трансфекцированных СНО клеток. Связывания не наблюдали с нетрансфекцированными контрольными СНО клетками. В формате анализов ELISA биотинилированные LPS [Luk, et.al., Alan. Biochem. 232:217-224 (1995)] при концентрации 0.5-3.0 мкг связывали иммобилизованную αd/CD18LZ с сигналом, в четыре раза превышающим сигнал захваченного антитела и самого блокирующего антитела. Кажущееся связывание LPS с CD11a/CD18 было поправлено на превышение за счет извлечения из каждой экспериментальной величины фонового связывания с анти-CD11а и антителом TS2/4.
Для того чтобы идентифицировать другие лиганды для αd/CD18, рекомбинантный αd/CD18LZ белок использовался в друхярусном исследовании. Связывание различных клеточных типов с иммобилизованным белком использовалось для определения, какие клетки экспрессируют αd лиганды на клеточной поверхности. Затем использовалось ингибирование антитела для определения, будет ли наблюдающееся клеточное связывание возникать из взаимодействия с известными молекулами с поверхностной адгезией. Если в результате не происходит ингибирования, со-иммунопреципитация с αd/CD18LZ, связанным с белками из лизатов клеток, которые будут связывать αd, используется в попытке идентификации лиганда.
Экспрессионные конструкции растворимого человеческого αd I домена.
Ранее сообщалось, что I домен в CD11а может быть экспрессирован в виде независимой структурной единицы, которая сохраняет способности лиганд связывания лиганда и узнавания антитела [Randi and Hogg, J.Biol.Chem. 269: 12395-12398 (1994); Zhout, et. al., J.Biol.Chem. 269: 17075-17079 (1994); Michshita, et. al., Cell 72:857-867 (1993)]. Для генерирования растворимого составного белка, включающего αd I домен и человеческий lgG4, αd I домен амплифицировался с помощью PCR реакции с использованием праймеров, сконструированных для присоединения фланкирующих BamHI и XhoI рестриктных сайтов для облегчения субклонирования. Эти праймеры представляются в SEQ ID NO:32 и 33 с рестриктными сайгами, приведенными ниже.
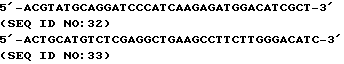
С нуклеотид непосредственно 3' в BamHI сайте в SEQ ID NO:32 соответствует нуклеотиду 435 в SEQ ID NO:1; G нуклеотид 3' в XhoI сайте в SEQ ID NO: 33 является комплементарным нуклеотиду 1067 в SEQ ID NO:1. Амплифицированный I домен гидролизуется соответствующими ферментами, очищенный фрагмент лигируется в векторе экспрессии pDCs и прокариотическом векторе экспрессии pGEX-4T-3 (Pharamacia) и фрагмент I домена секвенируется. Составной белок затем экспрессировался в COS, CHO или E.coli клетках, трансфекцированных или трансформированных соответствующей экспрессионной конструкцией.
Данное сродство αd для ICAM-R, экспрессии αd I домена могут иметь достаточное сродство для того, чтобы быть полезным ингибитором клеточной адгезии, в которой участвует αd.
Анализ составных белков человеческого αd I домена/IgG4.
Белок был разделен с помощью SDS-PAGE в условиях восстановления и не восстановления и визуализован либо за счет серебряного окрашивания, либо кумазинового окрашивания. Затем белок был перенесен на Immobilon PVDF мембрану и подвергнут анализу Вестерн-блоттинга с использованием античеловеческих IgG моноклональных антител или антибычьих IgG моноклональных антител.
Было определено, что обнаруженный белок мигрирует при около 120 кD при восстанавливающих условиях. Небольшие полосы были также обнаружены в невосстанавливающих гелях при приблизительно 40-45 кD, которые были реактивными с античеловеческими, но не антибычьими антителами. Было определено анализом Вестерн-блоттинга, что 200 kD небольшая полоса является бычьей Ig.
Анализы по связыванию с использованием экспрессионных продуктов I домена.
Способность I домена к специфическому узнаванию ICAM-R/IgG химерного белка была испытана в формате ELISA. Серийные разбавления αd I домена IgG4 составного белка (Iαd/IgG4) в TBS были проинкубированы ICAM-1/IgG, ICAM-R/IgG или неотносящимся к IgG1 миеломным белком, иммобилизованным на Immulon IV RIA/EIA плашки. CD11a I домен/IgG химерный белок и человеческий IgG4/каппа миеломный белок были использованы в качестве негативных контролей. Связывание IgG4 было обнаружено с биотинилированным анти-IgG4 моноклональным антителом НР6023 с последующим добавлением стрептовидин-пероксидазного конъюгата и проявлено субстратом о-фенилдиамина.
В повторных анализах не было обнаружено связывания CD11a/IgG4 белка или IgG4 миеломного белка с любыми иммобилизованными белками. Iαd/IgG4 белок не связывался с желатином рыбьей кожи или блокирующими агентами бычьего сывороточного альбумина, человеческого IgG1 или ICAM-1/IgG. Двух-трехкратное увеличение в связывающем сигнале по сравнению с фоном было обнаружено в лунках, заполненных ICAM-R/IgG белком с использованием концентраций 1-5 мкг/мл Iαd/IgG4 белка. Сигнал в лунках, заполненных VCAM-1/IgG белком, был в 7-10 раз выше, чем уровень фона. В предыдущих анализах αd/CD18 трансфекцированные СНО клетки не связывали VCAM-1/IgG белок, подтверждая, что VCAM-1 связывание может быть характеристикой выделенного I домена аминокислотных последовательностей.
Дополнительные конструкции αd I домена.
Дополнительные конструкции αd I домена генерировались таким же образом, как предыдущая конструкция, но с введением большего количества аминокислот вокруг αd I домена. Специфические конструкции включали: i) Последовательности из экзона 5 (аминокислоты 127-353 в SEQ ID NO:2), предшествующего текущей конструкции, ii) относящиеся к EF повторы (EF-hand repeats) (аминокислоты 17-603 в SEQ ID NO:2), после I домена и iii)альфа цепь, усеченную в трансмембранной области (аминокислоты 17-1029 в SEQ ID NO:2) с IgG4 хвостом для целей очистки и обнаружения. Эти конструкции лигировались либо в векторе экспрессии PDCS1 млекопитающего, либо в прокариотическом векторе экспрессии pGEX-4T-3 (Pharmacia) и I домен секвенировался. Затем составные белки экспрессировались в COS, СНО, или E.coli клетках, трансформированных или трансфекцированных соответствующей экспрессионной конструкцией. Белок очищался на ProSepA колонке (Bioprocessing Limited, Durham, England), испытывался на реактивность с анти-IgG4 моноклональным антителом НР6023 и визуализировался на полиакриламидных гелях кумазиновым окрашиванием.
Для того чтобы сконструировать экспрессионную плазмиду для полного αd полипетида, pATM. D12, описанная выше, модифицировалась для экспрессии αd-IgG4 составного белка следующим образом. IgG4, кодирующий ДНК, выделялся из вектора pDCS1 за счет PCR с использованием праймеров, которые индивидуально вводили 5' AatII рестриктный сайт (SEQ ID NO:89) и 3' XbaI рестриктный сайт (SEQ ID NO:90).

Плазмида pATM. D12 гидролизовапась AatII и XbaI и соответственно гидролизованный и очищенный lgG4 PCR продукт лигировался в линкерном векторе.
Пример 15
Продуцирование человеческих αd специфических антител.
А. Продуцирование моноклональных антител.
1. Неустойчиво трансфекцированные клетки из Примера 7 были три раза промыты в Dulbecco фосфатном забуференном рассоле (D-PBS) и введены при концентрации 5•106 клеток/мышь Balb/c мышам с 50 мкг/мышь мурамил дипептидазы (Sigma) в PBS. Инъекции мышам были проведены более чем два раза в том же виде с двухнедельными интервалами. Предварительно обескровленная и иммунизованная сыворотка от мыши была проанализирована с помощью FACS анализа, как подчеркнуто в Примере 9, и селезенка из мыши с самой высокой реактивностью к клеткам, трансфекцированным αd/CD18, была слита. Гибридомные супернатанты культуры были затем испытаны отдельно на отсутствие реактивности по отношению к клеткам COS, трансфекцированным CD11a/CD18, и реактивности с клетками, сотрансфекцированными с αd экспрессионной плазмидой и CD18.
Этот метод не приводил к моноклональным антителам.
2. В качестве альтернативного способа для продуцирования моноклональных антител, растворимый αd I домен/IgG4 составной белок был аффинно очищен от супернатанта устойчиво трансфекцированных СНО клеток и использован для иммунизации Balb/c мышей, как описано выше. Гибридомы были подтверждены и супернатанты из этих гибридом были подвергнуты скринированию с помощью анализа ELISA на реактивность по отношению к αd I домен составному белку. Затем положительные культуры были проанализированы на реактивность с αd/CD18 комплексами, экспрессированными на СНО трансфектантах.
Мыши 1908 получали три первоначальные иммунизации αd/CD18 трансфекцированных СНО клеток и две последующие повторные иммунизации растворимым αd/CD18 гетеродимером. Две конечные иммунизации включали 50 мкг/мышь αd I домена/IgG4 составного белка. Гибрид продуцировал 270 IgG-продуцирующих лунок. Супернатант из 45 лунок показывал с помощью анализа ELISA по крайней мере в 7 раз большее связывание с Iαd/IgG4 составным белком, чем с человеческим IgG4. Ни один из супернатантов не взаимодействовал с αd/CD18, трансфекцированными СНО клетками, как определено с помощью FACS анализа.
Для определения того, способны ли супернатанты узнавать интегриновые альфа субъединицы белков в другом контексте, свежезамороженные селезеночные кусочки были окрашены супернатантами из 24 лунок из общего количества 45 лунок. Было определено, что три супернатанта являются положительными: один окрашивал большие клетки в красной пульпе, в то время как два других окрашивали рассеянные клетки в красной пульпе, а также в перекладине.
Эти супернатанты были далее проанализированы за счет их способности к иммунопреципитации биотинилированными CD18 комплексами либо из αd/CD18, трансфекцированных СНО клеток, либо из РМА-стимулированных HL60 клеток. Гибридные лунки с супернатантами, которые узнавали белок в детергентных лизатах (которые не должны быть в виде конформационно ограниченных как белок экспрессированный в виде гетеродимеров), были выбраны для дальнейшего субклонирования. Моноклональные антитела, которые узнают белок в детергенте, могут быть более полезными в иммунопреципитации гетеродимерных комплексов из трансфектантов, тканей и клеточных линий.
3. В качестве другого альтернативного варианта для продуцирования моноклонального антитела CD18 комплексы были иммунопреципитированы из лизатов человеческой селезнки анти-CD18 моноклональным антителом 23F2G после просветления CD11a/CD18 (с использованием моноклонального антитела TS2/4) и CD11b/CD18 (с использованием моноклонального антитела Мо-1). Пять Balb/c мышей в возрасте от десяти до двенадцати недель были иммунизованы подкожной инъекцией приблизительно 30 мкг полученного белка в полном адъюванте Freund на 0 день с последующими двумя повторными инъекциями 30 унг иммуногена/мышь на 28 и 43 сутки в неполном адъюванте Freund. Были отобраны десять сывороток на десятый день после последней повторной инъекции и реактивность была проанализирована с использованием 1:500 разбавления каждой сыворотки для обнаружения 1 мкг/дорожку иммуногена в анализе Вестерн-блоттинга. Сыворотки от трех мышей обнаруживали полосы приблизительно 95 и 150 kD; сигнала не было видно в полосах, обработанных 1:50 разбавлением предварительно иммунизованных сывороток. Было высказано предположение, что полоса 150 kD представляет αd в in vivo гликозилированном состоянии. Кроме того, все постиммунные сывротки иммунопреципитированного белка из лизатов биотинилированных αd/CD18 CHO клеток, которые мигрируют с соответствующими молекулярными весами на SDS-PAGE, представляли гетеродимер. По данным этих результатов была выбрана мышь # 2212 и была далее иммунизована с помощью внутрибрюшинной инъекции на 64 день 30 мкг иммуногена в PBS. Мышь была умерщвлена спустя четыре дня и селезенка удалена в стерильных условиях.
Была получена одноклеточная суспензия путем измельчения селезенки между двумя концами замороженных стеклянных слайдов микроскопа, погруженных в свободный от сыворотки RPMI 1640, дополненный 2 мМ L-глутамина, 1 мМ пирувата натрия, 100 единицами/мл пенициллина и 100 мкг/мл стрептомицина (RPMI) (Gibco Canada). Клеточная суспензия была профильтрована через стерильное 70 меш Nitex cell сито (Becton Dickinson, Parsippany, New Jersey), и фильтрат дважды промыт центрифугированием при 200 g в течение 5 минут. Полученный комечек был ресуспендирован в 20 мл свободного от сыворотки RPMI. Тимоциты, взятые от трех мышей Balb/c, были приготовлены аналогичным образом.
До гибридизации NS-1 миеломные клетки, хранившиеся в log фазе в RPMI с 10% сыворотки Fetalclone (FBS) (Hyclone Laboratories, Inc., Logan, Utha) в течение 3 дней до гибридизации, были сконцентрированы центрифугированием при 200 g в течение 5 минут, дважды промыты, как описано в вышеприведенном параграфе, и подсчитаны. Приблизительно 2•10е клеток селезенки были объединены с 4•107 NS-1 клеток, и полученная смесь сконцентрирована центрифугированием при 200 g. Супернатант был выгружен. Клеточный шарик удаляли постукиванием пробирки, и 2 мл 50% PEG 1500 в 75 мМ Hopes (pH 8.0, 37oС) (Boehhnger Mannheim) было добавлено в течение одной минуты при перемешивании. Дополнительные 14 мл свободного от сыворотки RPMI было последовательно добавлено в течение следующих семи минут с последующим немедленным добавлением 16 мл RPMI. Полученная смесь была отцентрифугирована при 200 g в течение 10 минут, и супернатант выгружен. Шарик был ресуспендирован в 200 мл RPMI, содержащего 16 мМ FBS, 100 мМ гидроксантина натрия, 0.4 мМ аминоптерина, 16 мМ тимидина (HAT) (Gibco), 25 единиц/мл IL-6 (Boehringer Mannheim) и 1.5•106 тимоцитов/мл и распределен в 96-луночные плоскодонные планшеты тканевой культуры (Coming, United Kingdom) с концентрацией 200 мкл/лунку. Клетки были загружены на 2, 4 и 6 сутки после гибридизации всасыванием приблизительно 100 мкл из каждой лунки иглой 18G (Becton Dickinson) и добавлением 100 мкл/лунку микробиологической среды, описанной выше, за исключением содержания 10 единиц/лунку IL-6 и недостающих тимоцитов.
На 7-10 день после гибридизации супернатант из каждой лунки был испытан с помощью ELISA на захват антитела,испытанием на присутствие мышиного IgG. Immunolon 4 плашки (Dynatech, Cambridge, Massachusetts) были заполнены 50 мкл/лунку овечьего антимышиного IgA, IgG или IgM (Organon Teknicka), разбавленного 1: 5000 в 50 мМ карбонатного буфера, pH 9.6, при 4oС. Плашки были промыты 3Х PBS, содержащим 0.5% Твин 20 (PBST) и было добавлено 50 мкл супернатанта культуры из каждой лунки. После инкубирования при 37oС в течение 30 минут лунки были промыты PBST, как описано выше, и 50 мкл пероксидазы хрена, конъюгированной с овечьим антимышиным IgG (fc) (Jackson ImmunoRasearch, West Grove, Prnnsylvaia), разбавленной 1:3500, в PBST, было добавлено в каждую лунку. Планшеты были проинкубированы, как описано выше, промыты 4Х PBST, и было добавлено 100 мкл субстрата, содержащего 1 мг/мл о-фенилендиамина (Sigma) и 0.1 мкл/мл 30% перекиси водорода в 100 мМ Цитрата, рН 4.5. Окрашивание реакционной смеси было остановлено через 5 минут добавлением 50 мкл 15% серной кислоты. Поглощение при 490 нм регистрировали для каждой лунки с использованием аппарата для прочтения планшета (Dynatech).
Гибридомы были далее охарактеризованы следующим образом. Супернатанты из IgG-продуцирующих культур были проанализированы с помощью проточной цитометрии на реактивность к αd/CD18 трансформированным СНО клеткам, но не клеткам JY (В-клеточная линия положительная на LFA-1, но не на другие β2 интегрины, как наблюдалось в предыдущих экспериментах окрашивания в лунке). Короче, 5•105 αd/CD18 трансформированных СНО или αd/CD18-JY клеток были суспендированы в 50 мкл RPMI, содержащего 2% PBS и 10 мМ NaN3 (FACS буфера). Индивидуальные клеточные суспензии были добавлены к 50 мкл IgG супернатанта положительной гибридомной культуры в лунки 96-луночных круглодонных планшетов (Corning). После 30 минут инкубирования во льду клетки были дважды промыты концентрированием в клинической центрифуге, супернатант из каждой лунки был выгружен, и комочки ресуспендированы в 200-300 мкл FACS буфера. Последнюю промывку заменяли 50 мкл/лунку разбавлением 1:100 F(ab')2 фрагмента овечьего антимышиного IgG (H+L)-FITC конъюгата (Sigma, St.Louis, Mussouri), приготовленного в FACS буфере. После инкубирования, как описано выше, клетки дважды промывали Dulbecco's PBS (D-PBS) с дополнением 10 мМ NaN3 и, наконец, рессупендировали в D-PBS, содержащий 1% параформальдегида. Затем образцы были перенесены в полистирольные пробирки для анализа проточной цитометрией (FACS) анализатором Becton Dickinson FACsan.
Гибридизация давала четыре культуры, считающиеся положительными по обоим критериям. Когда вторичное скринирование было повторено на увеличенных в объеме супернатантах спустя приблизительно четыре дня, три из четырех культур оставались положительными. Три лунки, обозначенные 169А, 169В, 169D, были успешно клонированы от двух до трех раз путем двойного разбавления в RPMI, 15% FBS, 100 мМ гипоксантина натрия, 16 мМ тимидина и 10 единицах/мл IL-6. Лунки планшетов с клонами были подсчитаны визуально после четырех дней, и было зрегистрировано число колоний в лунке с наименьшей плотностью. Выбранные лунки каждого клонирования были проанализированы с помощью FACS спустя 7-10 суток. Активность была обнаружена в двух культурах 169А и 169В. При окончательном клонировании положительные лунки, содержащие единичные колонии, были увеличены в объеме в RPMI с 11% FBS. Антитела из клональных супернатантов 169А и 169В были изотипированы с использованием набора IsoStrip (Boehringer Mannheim) согласно методике, предложенной производителем, и было найдено, что они являются IgG1 изотипом.
Иммунопреципитация αd/CD18 комплексов из СНО трансфектантов и РМА-стимулированных HL-60 клеток была использована в качестве третичного скринирования на специфичность. Гибридомы 169А и 169В осаждали соответствующие полосы из СНО линий и образцов единичной α цепи 150-160 kD из HL60 клеток, как определено с помощью SDS-PAGE. Гибридомы 169А и 169В были осаждены 31 мая 1995 г American Type Culture Collection, 12301 Parklawn Drive, Rockville, Meryland 20852 и обозначены Accession Number HB11907 и НВ 11906 соответственно.
Для того чтобы более полно охарактеризовать связывающие свойства 169А и 169В, была изучена способность каждого антитела ингибировать связывание другого антитела или анти-CD18 антитела TS1/18 с растворимой αd/CD18. Растворимая αd/CD18 полной длины была иммобилизована каждым немеченым антителом отдельно в формате 96-луночного планшета, и были использованы биотинилированные антитела для обнаружения белка, связанного теми же или другими немечеными антителами. Связывание было обнаружено с использованием овечьего антимышиного Ig/HRP конъюгата с последующим добавлением OPD субстрата. Результаты указывают на то, что антитело 169А было способно к блокированию связывания биотинилированного 169А и TS1/18, в то время как антитело 169В блокировало связывание только самой себя.
4. Была выбрана другая мышь (#2214), иммунизованная с таким же протоколом, как и мышь #2212, и далее иммунизована с помощью предварительно гибридизованной повторной иммунизации на 70 сутки 30 мкг очищенной αd из селезеночных лизатов в PBS. Мышь была умерщвлена спустя четыре дня. и селезенка удалена в стерильных условиях.
Гибридизация и клонирование положительных клеток были проведены, как описано выше. Гибридизация продуцировала пять анти-αd моноклональных гибридом, обозначенных 170D, 170F, 170Т, 170Х и 170Н, которые были изотипированы в виде IgG1 с использованием набора IsoStrip (Boehringer Mannheim) согласно методикам, предложенным производителем.
5. Была выбрана еще другая мышь (#2211), иммунизованная с таким же первоначальным протоколом, как и мышь #2212 и мышь #2214, и далее иммунизована на 88 сутки 30 мкг иммуногена и предварительно гибридизованной повторной иммунизацией 30 мкг иммуногена на 203 сутки. Мышь была умерщвлена спустя четыре дня, и селезенка удалена и проведена гибридизация, как описано выше. Гибридомный супернатант был подвергнут скринированию с помощью анализа ELISA на захват антитела и с помощью проточной цитометрии, как детально описано в приведенном выше параграфе.
Были идентифицированы пятнадцать положительных гибридом, обзначенных 188А, 188В, 188С, 188Е, 188F, 188G, 188I, 188J, 188К, 188L, 188М, 188N 188Р, 188R и 188Т и изотипированы в анализе ELISA. Короче, Immunolon 4 планшета (Dynatech, Cambridge, Massachusetts) были заполнены при 4oС 50 мкл/лунку овечьим антимышиным IgA, G, М (Organon Teknicka), разбавленным 1:5000 в 50 мМ карбонатного буфера, рН 9.6. Планшеты были подвергнуты блокированию в течение 30 минут при 37oС 1% BSA в PBS, промыты 3 раза PBS, содержащим 0.05% Твин 20 (PBST) и было добавлено 50 мкл супернатанта культуры (разбавленного 1: 10 в PBST). После инкубирования и промывания, как описано выше, было добавлено 50 мкл пероксидазы хрена, конъюгированной с кроличьим антимышиным IgG1, G2a или G3 (Zymed, San Francisco, California) разбавленной 1:1000 в PBST с 1% нормальной овечьей сыворотки. Планшеты были проинкубированы, как описано выше, промыты 4 раза PBST, после чего было добавлено 100 мкл субстрата, содержащего 1 мг/мл о-фенилендиамина (Sigma) и 0.1 мкл/мл 30% перекиси водорода в 100 мМ цитрата, рН 4.5. Окрашивание реакционной смеси было остановлено через 5 минут добавлением 50 мкл 15% серной кислоты. А490 была зарегистрирована использованием аппарата для прочтения планшета (Dynatech) для всех пятнадцати антител, и были определены антитела, являющиеся IgG1.
Избыток селезеночных клеток из мыши #2211 был заморожен в криостате и хранился в жидком азоте. Криостат подвергали быстрому оттаиванию, помещая в водяную баню при 37oС, и помешивали ее круговыми движениями до тех пор, пока содержимое не оттает. Клетки были перенесены в 15 мл центрифужные пробирки, куда медленно добавляли 1 мл теплого RPMI, содержащего 11% FBS, с интервалами между добавлениями от трех до пяти минут. Добавляли еще 5 мл теплого RPMI и после пяти минут ожидания пробирки центрифугировали при 200 g в течение пяти минут и супернатант отсасывали. Клетки были ресуспендированы в RPMI и была проведена гибридизация, как описано выше. Гибридомный супернатант был охарактеризован с помощью захвата антитела и проточной цитометрии, как описано выше.
Гибридизация давала пять клонов, обозначенных 195А, 195С, 195D, 195Е и 195Н. Клоны были изотипированы с помощью анализа ELISA, как описано выше; моноклональные антитела 195А, 195С, 195D, 195Е, и 195Н были определены как являющиеся IgG2a.
6. Для того чтобы идентифицировать способность антител ингибировать функциональное αd связывание, для иммунизации использовалась растворимая αd/CD18LZ (см. Пример 14). Белок выделялся на смолу, использованную в аффиннной хроматографии, из супернатанта неустойчиво трансфекцированных COS клеток и αd, связанная со смолой, использовалась в качестве иммуногена. Выбранная мышь иммунизировалась, как описано выше, и повторную конечную иммунизацию проводили спустя две недели после начальной иммунизации. Иммунизация с использованием этого приема предотвращает возможные изменения в конформации белка, часто связанные с детергентным лизисом клеток. Дополнительная мышь иммунизовалась рекомбинантным белком, также связанным со смолой, но не была первоначально иммунизована рекомбинантным белком, очищенным из клеточного лизата.
Гибридомы, приготовленные, как описано выше, которые возникали после иммунизации, скринировались с помощью анализа ELISA на рекомбинантном белке, иммобилизованном из клеточного супернатанта с использованием Fab фрагмента не блокирующего антитела. Или же использовалась проточная цитометрия для анализа на реактивность к JY клеткам, предварительно трансфекцированным αd сДНК.
7. В качестве другого альтернативного варианта моноклональные антитела генерировались следующим образом. Аффинно очищенный αd/CD18 гетеродимерный белок из детергентных лизатов устойчиво трасфекцированных СНО клеток использовался с 50 мкг/мл мурамил дипептидазы для иммунизации Balb/c мышей, как описано выше. Мыши получали три иммунизации до реактивности сывортоки по отношению к αd/CD18, определяемой иммунопреципитацией биотинилированных комплексов в СНО трансфектантах. Гибридомы от положительных животных устанавливались согласно стандартным протоколам, после чего выбирались гибридомные культуры с помощью проточной цитометрии с использованием αd/CD18 трансфектантов. CD11a/CD18 трансфектанты использовались в контроле на реактивность только для CD18.
8. В качестве другого альтернативного варианта для продуцирования моноклонального антитела Balb/c мыши подвергались протоколу иммунизации/иммуносуппрессии, сконструированному для снижения реактивности СНО клеточных детерминант на трасфектантах, использованных для иммунизации. Этот протокол включал иммунизацию с нетрансфекцированными СНО клетками и последующее уничтожение СНО-реактивных В-клеточных бластов циклофосфамидной обработкой. После трех кругов иммунизации проводилась и циклофосфамидная обработка, и мыши иммунизовались αd/CD18 СНО трансфекцированными клетками, как описано выше.
9. В качестве другого альтернативного варианта CD18 комплексы из детергентных лизатов РМА стимулированных HL60 клеток обогащались за счет предварительного просветления, как описано выше. Другие β2 интегрины просветлялись на тех же колонках. Иммунизация полученными комплексами, гибридомное продуцирование и протоколы скринирования выполнялись, как описано выше.
В. Продуцирование поликлональной сыворотки.
Очищенная αd I домен/IgG4 химера (Пример 14) была использована для генерирования поликлональной антисыворотки у кроликов. Была проведена инъекция αd I домен/IgG4 антигена с концентрацией 100 мкг/кролика сначала в полном адъюванте Freund с последующими тремя повторными иммунизациями с тем же количеством белка в неполном адъюванте Freund. Испытываемые спуски крови были проанализированы после третьей и четвертой инъекций. Кроличий иммуноглобулин (Ig) был очищен от сыворотки на белок А-сефарозной колонке, а предварительно просветленный античеловеческий IgG на человеческой IgG/Affigel колонке. Реактивность, установленная с помощью ELISA в I доменной химере, но не человеческом IgG, была использована для подтверждения полного предварительного просветления.
Предварительно просветленная поликлональная сыворотка была использована для иммунопреципитации белка из детергентных лизатов поверхностно-биотинилированных СНО клеток, предварительно трансфекцированных αd и CD18 экспрессионными векторами. Иммунопреципитация была проведена с помощью метода, предварительно описанного в Примере 10. Предварительно просветленная сыворотка распознавала белковый комплекс такого же молекулярного веса, как молекулярный вес, преципитированный с помощью анти-CDIS моноклинального антитела TS1.18. Кроме того, сыворотка распознавала единственную полосу соответствующего размера в анализе Вестерн-блоттинга, CD18 комплексов из αd/CD18 трансфекцированных СНО клеток. Аффинно очищенные интегрины CD11a/CD18, CD11d/CD18 и VLA4 из человеческой селезенки не были распознаны с помощью кроличьей поликлональной сыворотки. Сыворотка была не в состоянии взаимодействовать с αd-трансфекцированными СНО клетками в растворе, как определено с помощью проточной цитометрии. Поэтому было сделано заключение о том, что поликлональная кроличья сыворотка способна узнавать только денатурированные αd I домен/IgG4 белки.
При попытке продуцирования поликлональной антисыворотки против αd/CD18, мышь была иммунизована 3 раза αd- трансфекцированными СНО клетками (D6.CHO, αd/CD18) с адъювантным белком и один раз очищенным αd/CD18 гетеродимером. Конечная повторная иммунизация включала только αd/CD18 гетеродимер. Приблизительно 100 мкл иммунизованной сыворотки было предварительно просветлено добавлением приблизительно 108 LFA-1-трансфекцированных СНО клеток в течение 2 часов при 4oС. Полученная сыворотка была проанализирована на αd реактивность при разбавлениях 1/5000, 1/10000, 1/20000 и 1/40000 на нормальную человеческую селезенку. Поликлональное антитело было реактивным при разбавлении 1/20000, в то время, как разбавление 1/40000 давало очень слабое окрашивание.
Пример 16
Анализ αd распределения.
Тканевое распределение αd/CD18 было определено с использованием антисыворотки, генерированной, как описано в Примере 15.
Очищенное кроличье поликлональное антитело было использовано при концентрациях в области между 120 нг/мл и 60 нг/мл для иммуноцитохимических анализов замороженных срезов человеческой селезенки. Срезы 6-микронной толщины распределяли в виде слоев в Superfrost Plus Slides (VWR) и хранили при -70oС. Перед использованием слайды извлекали из морозильника при -70oС и помещали при 55oС на 5 минут. Затем срезы фиксировали в холодном ацетоне в течение 2 минут и сушили на воздухе. Срезы блокировали в растворе, содержащем 1% BSA, 30% нормальной человеческой сыворотки и 5% нормальной кроличьей сыворотки в течение 30 минут при комнатной температуре. Первичное антитело было добавлено к каждому срезу в течение 1 часа при комнатной температуре. Несвязанное антитело было удалено промыванием слайдов 3 раза в TBS буфере в течение 5 минут на промывку. Затем к каждому срезу в том же TBS буфере добавляли кроличье антимышиное IgG связанное антитело. Антитело мышиной щелочной фосфатазы антищелочной фосфатазы (АРААР), проинкубированное в течение 30 минут при комнатной температуре, было использовано для обнаружения вторичного антитела. Затем срезы промывали 3 раза в TBS буфере. Был добавлен субстрат Fast Blue (Vector Labs) и развитие окраски останавливали погружением в воду. Срезы были подвергнуты противоокрашиванию в Nuclear Fast Red(Sigma) и промыты до помещения на предметное стекло Aqua Mount (Baxter). Окрашивание было обнаружено в селезеночной красной пульпе с этим реагентом, но не с не относящимся к препарату кроличьим поликлональным Ig или неочищенной предварительно иммунизованной сывороткой от тех же животных.
Было определено, что одна мышиная сыворотка обладает специфичной αd реактивностью, она была использована для окрашивания различных лимфоидных и нелимфоидных тканей. Моноклональные антитела, узнающие CD18, CD11a, CD11b и CD11c, были использованы в тех же экспериментах в качестве контролей. Окрашивание нормальных селезеночных срезов αd поклональной сывороткой и моноклональными антителами CD11a, CD11b, CD11c и CD18 показало следующие результаты. Картина, наблюдавшаяся с αd поликлональной сывороткой, не обнаруживала такой же картины мечения как CD11a, CD11b, CD11c и CD18. Имело место четкая картина мечения некоторыми клетками, расположенными в краевой зоне белой пульпы, и четкое мечение клеток периферийных с краевой зоной. Это картина не наблюдалась с другими антителами. Индивидуальные клетки, рассеянные по всей красной пульпе, которые могли быть или могли не быть такой же популяцией или субнабором, видным с CD11a и CD18, также были мечеными.
Мечение CD11c не обнаруживалось некоторыми клетками, окрашиваемыми в краевой зоне, но антитело не показывало четкой кольцевой картины вокруг белой пульпы, когда сравнивалось с αd поликлональной сывороткой, только мечение в красной пульпе давало такую же картину окрашивания, как αd поликлональная сыворотка.
Поэтому картина мечения, наблюдаемая с αd поликлональной сывороткой, была уникальной по сравнению с той, которая наблюдалось с использованием антител с другими β2 интегринами (CD11a, CD11b, CD11c и CD18), и подтверждала, что распределение αd у человека in vivo отличается от распределения других β2 интегринов.
Характеристика человеческой αd экспрессии моноклональными антителами.
Антитела, секретированные гибридомами 169А и 169В, были использованы для анализа человеской αd экспрессии в замороженных срезах ткани с помощью иммуноцитохимии и на линиях клеток лейкоцитов периферической крови с помощью проточной цитометрии. Гибридомные супернатанты, использованные в обоих наборах экспериментов, были неразбавленными.
Окрашивание ткани.
Все процедуры окрашивания были выполнены, как описано выше, за исключением печеночных срезов, которые были окрашены следующим образом. После фиксации ацетоном срезы блокировали в 1% раствором перекиси водорода и 1% растворе азида натрия в TBS в течение 15 минут при комнатной температуре. После первичного антительного окрашивания было добавлено кроличье антимышиное антитело, непосредственно конъюгированное с пероксидазой, в течение 30 минут при комнатной температуре. Срезы три раза промывали в TBS буфере. Свиное антикроличье антитело, непосредственно конъюгированное с пероксидазой, было проинкубировано в течение 30 минут при комнатной температуре для обнаружения вторичного антитела. Затем срезы промывали 3 раза в TBS буфере и был добавлен АЕС субстрат (Vector Labs), и окраске было позволено проявиться. Срезы подвергали противоокрашиванию Hematoxylin Gill's No. 2 (Sigma) и последовательно промывали в воде до дегидратации и помещения на предметное стекло.
В селезеночных срезах большая часть экспрессии локализовалась в селезеночной красной пульпе на клетках, идентифицированных морфологическими данными как гранулоциты и макрофаги. Большое количество гранулоцитов было окрашено, в то время как небольшой субнабор макрофагов давал сигнал. Небольшое количество фоликулярных дендритных клеток в белой пульпе также были слабо окрашенны αd антителами. CD11a и CD18 окрашивание было обнаружено по всей красной и белой пульпе. CD11c окрашивание было более заметно в больших клетках, преимущественно являющихся макрофагами в селезеночной белой пульпе и краевой зоне, окружающей белую пульпу; диффузное окрашивание в красной пульпе также было заметно, Оказывалось, что CD11b имело распределение, перекрывающееся, но не идентичное αd в красной пульпе, без включения белой пульпы.
Было проведено сравнение интегриновой экспрессии в нормальных и (ревматоидных) артритных синовиальных тканях. Минимальное окрашивание со всеми антиинтегриновыми антителами (включая антитела специфически иммунореактивные с CD11a, CD11b, CD11c и CD18, а также с αd) было замечено в нормальных тканях с широким распределением на живущих клетках, преимущественно макрофагах. В воспаленном синовиуме экспрессия всех интегринов была более локализованной в клетках, кластеризованных вокруг лимфатических сосудов. В то время как экспрессионные партнеры αd и CD11b были подобными, CD11c не проявляла себя как сильно экспрессированная единица и была рестректированной в субъединице лейкоцитов.
У собак, CD11b, но не αd, экспрессия была обнаружена на печеночных макрофагах, или клетках Kuppfer. Окрашивание нормальных человеческих печеночных срезов (как ранее описано для окрашивания собачьего печеночного среза, выше) подтверждает сохранение этого окрашивающего партнера в людях. Кроме того, CD11c была обнаружена с низкими уровнями. В срезах от пациента с гепатитом окрашивание всех лейкоинтегринов было выше, чем наблюдали на нормальной печенке, в то время как αd экспрессия в этих образцах была обнаружена на макрофагах и гранулоцитах.
Минимальное окрашивание нормальных человеческих колоновых срезов наблюдали с анти-αd антителами; наблюдали слабое окрашивание гладкой мышцы и окрашивание лейкоцитов. Все лейкоинтегрины были обнаружены с более высокими уровнями в срезах от пациентов с заболеванием Crohb's.
Нормальное легкое показывало ограниченное количество слабо αd-положительных клеток; было определено морфологически, что эти клетки являются макрофагами и нейтрофилами. В легочной ткани от пациента с эмфиземой αd окрашивание наблюдали на нейтрофилах и на макрофагах, содержащих гемосидерин, железосодержащий пигмент, указывающий на поглощение красных клеток этими клетками.
Были исследованы срезы нормального мозга и поражений в виде бляшек от пациентов с множественным склерозом (MS) на интегриновую экспрессию. В нормальном мозге αd окрашивание было менее интенсивным, чем окрашивание CD11a, CD11b, CD11c, и рестриктированных в клетках, морфологически типично представленных в виде микроглиальных клеток и CD68 окрашивание. CD11b положительные клетки были обнаружены окружающими сосуды и по всей ткани. CD11c+ клетки оказывались расположенными внутри сосудов, тогда как α
Оба среза грудной аорты и брюшной аорты из PDAY (Патологические детерминантны атеросклероза в юности, LSU Medical Center) образцов ткани были проанализированы с антилейкоинтегринами и анти-САМ антителами. Исследованные повреждения согласовывались с жировыми прожилками аорты (aortic fatty streaks), состоящими из находящихся под внутренней оболочкой сосуда агрегатов больших ксантомных клеток (большей частью макрофагов с абсорбированными липидами) и инфильтратов меньших лейкоцитов. Исследования по единичному мечению моноклональными антителами, специфичными для αd и других β2 интегринов α цепей (CD11a, CD11b, и CD11c), плюс макрофаговый маркер (CD68), показали, что большая часть макрофагов, нагруженных липидами, экспрессировали умеренный уровень αd и CD18, в то время как экспрессия CD11a и CD11c была на слабом или от слабого до умеренного уровнях соответственно. CD11b была слабо экспрессирована и к тому же только за счет субнабора макрофагов.
Исследования по двойному мечению были проведены для определения относительной локализации αd и ICAM-R антигенов в срезах аорты. Так как ксантомные клетки в этих срезах окрашивались антителом Ham 56, специфичным для макрофагового маркера, но не антителами актина гладкой мышцы, было определено, что ксантомные клетки не образовывались из находящихся под внутренней оболочкой сосуда клеток гладкой мышцы. CD68 положительные макрофаги, экспрессирующие αd, были окруженными и разбросанными небольшими ICAM-R положительными лейкоцитами. Это, оказывается, является ограничением количества небольших лейкоцитов, которые были CD68 отрицательными, но окрашенными обеими антителами αd и ICAM-R.
Оказывается, распределение αd в нормальных тканях находилось на постоянных лейкоцитах в картине перекрывания CD11b и CD11c, но не являясь идентичным распределению CD11b и CD11c, двух других лейкоинтегринов α цепи, которые были ранее охарактеризованы как имеющие рестриктное распределение лейкоцитов. Клеточная морфология указывает на то, что αd окрашивание, главным образом, ограничивается в макрофагах и гранулоцитах с ограниченным окрашиванием лимфоцитов. Вообще, воспаление ткани, оказывается, увеличивало количество и типы лейкоцитов, наблюдавшихся в конкретной ткани, вместе с увеличенным окрашиванием лейкоинтегринов, включая αd. Так как клеточное и пространственное распределение лейкоинтегринов было не идентичным в патологичных тканях, было сделано заключение, что отчетливые функции и лиганды существуют для каждого члена семейства, включая αd, в конкретных контекстах.
Оказывается, что αd экспрессия при ранних атеросклеротических повреждениях являлась более заметной, чем экспрессия CD11a, CD11b и CD11c, подтверждая, что αd может играть центральную роль в установлении этих повреждений. Приложенное распределение αd и ICAM-R положительных клеток поддерживается данными, предполагающими взаимодействие между αd и ICAM-R, предполагая, что αd может быть включена в восстановление лейкоцитов или активацию на ранних стадиях в этих повреждениях.
Линия клеток и окрашивание лейкоцитов периферической крови.
Антитела 169А и 169В окрашивали промиелмоноцитическую линию клеток, HL60, с помощью FACS. Поверхностная экспрессия αd в этих клетках отрицательно затрагивается РМА стимуляцией, которая, как сообщается, индуцирует дифференциацию вдоль пути макрофага, но не затрагивается ДМСО, который индуцирует дифференциацию гранулоцита [Cjllins, et.al., Blood 70:1233-1244 1987)]. FACS профили 169А и 169В были антитетическими с РМА стимуляцией в отношении профилей, наблюдавшихся с анти-CD11b и CD11c моноклинальными антителами. Моноцитная линия клеток ТНР-1 также показывала слабое окрашивание 169А и 169В. Кроме того, субнабор клеток в лимфоците и моноцитных путях лейкоцитов периферической крови, оказывается, является слабо положительным по данным FACS. Субнабор моноцитов периферической крови слабо окрашивается 169А и 169В, в то время как было найдено, что В лимфоциты не имеют поверхностной экспрессии αd. CD8+ субнабор Т лимфоцитов был α
В свете полученных результатов с HL60 клетками гранулоциты были выделены из периферической крови центрифугированием с фикол/гипаковым ингредиентом и последующим лизисом красных кровяных клеток. Было найдено, что все препараты являются больше чем на 90% РМА, с помощью визуализации ядерной морфологии в уксусной кислоте. Отдельные популяции стимулировали в течение 30 минут 50 г/мл РМА или 10-8 М формилпептида (fMLP) для высвобождения потенциальных внутриклеточных интегриновых пулов. Нестимулированные популяции показывали низкую, но значительную экспрессию 169А и 169В антигенов по сравнению с IgG1 контролем с обнаруживаемым увеличением при стимуляции. В PMNs, уровни αd и CD11c поверхностной экспрессии были более похожими, чем уровни, которые наблюдали в HL60 клетках. Антитело 169В было использовано впоследствии для осаждения гетеродимерной молекулы из детергентного лизата биотинилированных PMNs субъединицей с размером приблизительно 150 и 95 kD соответствующих αd и CD18 соответственно.
Присутствие αd в PMNs нельзя предположить из информации, известной о сабачьей αd экспрессии. Собачьи нейтрофилы, непохожие на их человеческие контрчасти, экспрессируют Т хелперный клеточный маркер CD4, а также интегриновый VLA-4, и поэтому могут иметь другие лиганды и функции у собак, чем у человека.
Окрашивание PBL субгрупп.
Настоящее исследование предпринято для определения распределения этого β2 интегрина в человеческих лейкоцитах периферической крови. Кроме того, было проведено сравнение плотности клеточной поверхности αd относительно других β2 интегринов. Наконец, сильная регуляция αd экспрессии в очищенных человеческих эозинофилах также была оценена.
Лейкоциты человеческой периферической крови были выделены центрифугированием с градиентом плотности в моноядерной клеточной фракции (содержащей моноциты, лимфоциты и базофилы) и гранулоцитах (нейтрофилы и эозинофилы) [Warner, et.al., J.Immunol.Meth. 105:107-110 1987)]. Для некоторых экспериментов эозинофилы были очищены с использованием CD16 иммуномагнитной селекции с примесями более чем 95% [Hansel, et.al., J.lmmunol.Meth., 122:97-103 (1989)] . Клетки кожи молочной железы (skin mast cells) были ферментативно диспергированными из кожи и обогащены, как ранее описано [Lawrence, et.al., J.lmmunol. 139:3062-3069 (1987)].
Клетки были мечены соответствующими разбавлениями моноклонального антитела, специфичного либо для CD11a(MHM24), Cd11b (Н5А4), CD11c (BU-15), либо для αd(169A). Был использован также мышиный контроль IgG1. Клетки были промыты и затем проинкубированы избытком мышиного IgG и FITC-меченного мышиного моноклонального антитела или овечьего поликлонального антитела, специфичного для конкретной клетки (например, CD3, CD4, или CD8 для Т-клеток; CD16+ лимфоцитов для NK клеток; анти-IgE для базофилов [Bochner, et.al. , J.lmmunol.Meth. 125:265-271 (1989)]. Затем образцы были исследованы с помощью проточной цитометрии (Coulter EPICS Profile) с использованием открытия мембранного канала для идентификации клеточных субнаборов.
Для изучения с человеческими эозинофилами, в которых была исследована сильная сверхрегуляция αd экспрессии, клетки были стимулированы в течение 15 минут при 37oС форболовым эфиром (10 нг/мл), RANTES (100 нг/мл) [Shall, Cytokine, 3: 165-183 (1991)], или IL-5 (10 нг/мл) до мечения различными моноклональными антителами, как описано выше.
Результаты показали, что αd присутствовала во всех эозинофилах, базофилах, нейтрофилах, моноцитах периферической крови и NK клетках. Небольшой субнабор (приблизительно 30%) CD8+ лимфоцитов был также найден в экспрессии αd. Клетки кожи молочной железы и CD4+ лимфоциты не экпрессировали αd. В общем, CD11a и CD11b присутствуют при более высоких плотностях на лейкоцитах, чем αd, последняя является экспрессированной при относительно низких уровнях подобно CD11c. Среди лейкоцитов моноциты и CD8+ клетки имели самую высокую плотность αd, в то время как эозинофилы имели самый низкий уровень αd экспрессии. Экспрессия на нейтрофилах, базофилах и NK клетках была промежуточной.
Стимулирование периферических эозинофилов СС хемокином RANTES не вызывала изменений в экспрессии любых β2 интегринов. Обработка форболовым эфиром, однако, продуцировала двух-трехкратное увеличение в экспрессии и CD11b и αd, но не оказывала влияния на экспрессию CD11a или CD11c. IL-5 обработка приводила к селективному сверхрегулированию CD11b экспрессии без влияния на уровни других интегриновых субъединиц.
В совокупности эти результаты указывают на то, что в лейкоцитах периферической крови αd обычно экспрессируется с уровнем, сравнимым с СD11с. Самые высокие уровни были найдены в моноцитах и субнаборе СD8+ лимфоцитов. Человеческие клетки кожи молочной железы не экспрессировали αd. Очищенные эозинофилы, по видимому, имели предварительно образованные внутрицитопластические пулы хранения CD11c и αd. Однако дифференциальное сверхрегулирование, показанное IL-5 в сравнении с HVF, подтверждает, что эти пулы хранения разделяются друг от друга.
Картины окрашивания для субгрупп лейкоцитов периферической крови (PBL) также были определены с помощью проточной цитометрии с использованием комбинации открытия мембранного канала и поверхностных маркеров, как описано выше, в попытке более точно определить 169А/В негативную лимфоцитную группу. PBL были выделены на Ficoll, как описано ранее, и окрашены отдельно 169А, 169В и моноклональными антителами к CD14 (моноцит/макрофаговый маркер), CD20 (В клетка), CD56 (NK клетка), Т клеточном рецепторе α/β (Т клетка), CD16 (нейтрофилах, Nks), и α4 (отрицательный маркер для нейтрофилов). Пути были определены размером и распределением маркера.
Результаты указывают на то, что клетки в CD14+ моноцитном канале показывают низкие уровни 169А и 169В окрашивания. Бимодальная картина экспрессии, наблюдавшаяся в ранних экспериментах в лимфоцитном канале, была разрешенной за счет увеличения переднего рассеивания. Смешанные TCR/CD20+ популяции, по-видимому, имели низкие, но гомогенные уровни 169А/В экспрессии, тогда как популяция, картированная при слегка более высоком боковом рассеивании (клеточной кмплексности), которая окрашивала 50% положительных популяций для CD56, по-видимому, имела четкую 169А/В отрицательную популяцию. Отрицательная популяция не узнавалась также TCP, CD20, CD14 или CD16 антителами.
Синовиальное распределение αd.
Для того чтобы определить клеточное распределение αd, других β2 интегринов и их контррецепторов в воспалительных и невоспалительных синовиумах, были использованы моноклональные антитела в различных β2 интегриновых и иммуноглобулиновых супергенных семействах в иммуногистологических исследованиях. Белковая экспрессия была определена в образцах нормальных, остеоартритных и ревматоидных синовиальных тканях.
Результаты указывают на то, что слой синовиальной выстилающей клетки экпрессировал высокие уровни VCAM-1, CD11b/CD18 и αd/CD18. В этих клетках CD11c/CD18 экспрессия ограничивалась, и CD11a/CD18 обычно не обнаруживалась. В ревматоидных артритных синовитах экспрессия β2 интегринов в синовиальном клеточном слое увеличивалась пропорционально степени гиперплазии. Коэффициент клеток, которые экспрессировали CD11c, значительно увеличивался, приближаясь к коэффициенту CD11b и αd, но не наблюдалось увеличения в CD11a экпрессии.
В субвыстилающем пространстве ткани, агрегаты и диффузные инфильтраты CD3/CD11a/ICAM-R+ лимфоцитов перераспределялись среди CD68/CD11b/αd макрофагов. Значительное количество агрегатов демонстрировало интенсивное αd окрашивание, особенно в зонах, обогащенных Т клетками.
Синовиальный эндотелий изменчиво экспрессировал IСАМ-1 и ICAM-2 с минимальным присутствием ICAM-R экспрессии.
Вместе взятые, эти результаты указывают на то, что синовиальные макрофаги и макрофагоподобные синовиальные клетки экспрессируют высокие уровни β2 интегринов CD11b и αd. В синовитах имеет место расширение этого субнабора клеток в обеих областях в выстланной и субвыстланной вместе с кажущимся увеличением в экспрессии CD11c. Специфические популяции ревматоидных синовиальных Т лимфоцитов дополнительно к экспрессии CD11a и ICAM-R также экспрессировали высокие уровни αd, последняя молекула, которая была показана выше, являлась экспрессированной при низких уровнях лимфоцитами периферической крови.
Пример 17.
Выделение крысиных сДНК клонов.
В свете существования обеих αd субъединиц, собачьей и человеческой, были предприняты попытки выделения гомологичных генов в других видах, включая крыс (этот пример) и мышей (Пример 17, ниже).
Частичная последовательность крысиной сДНК, показывающая гомологию с человеческим αd геном, была получена из крысиной селезенки λgt10 библиотеки (Clontech). Библиотека была посеяна в планшет при 2•104 pfu/чашку в 150 мм LBM/агаровые чашки. Библиотека была поднята на Hybon мембранах (Amersham), денатурирована в течение 3 минут, нейтрализована в течение 3 минут и промыта в течение 5 минут буфером, как описано в стандартных протоколах [Sambrook, et. al. , Molecular Cloning: a laboratory manual. 0.2.110]. Мембраны были немедленно помещены в Stratalinker (Stratagene), и ДНК сшита с использованием автосшивающего набора. Мембраны были прегибридизованы и гибридизованы в 30% или 50% формамиде, в слабожестких и высокожестких условиях соответственно. Мембраны первоначально были скринированы Р-меченной пробой, генерированной из человеской αd сДНК, соответствующей основаниям 500 до 2100 в клоне 19А2 (SEQ ID NО:1). Пробы были мечены с использованием Boehringer Mannheim's Random Prime набора в соответствии с протоколом, предложенным производителем. Фильтры были промыты 2Х SSC при 55oС.
Были идентифицированы два клона, обозначенные 648.3 и 705,1, которые показывали последовательность, гомологичную человеческому αd гену, человеческой CD11b и человеческой CD11c. Оба клона встраивались
в человеческий αd ген в 3'область гена, начиная от основания 1871 и простираясь до основания 3012 для клона 648.3 и от основания 1551 до 3367 для клона 705.1.
Для того чтобы выделить более полную крысиную последовательность, которая включала 5' область, та же библиотека была повторно скринирована с использованием того же протокола, который применяли для первоначального скринирования, но используя мышиную пробу, генерированную из клона АН 60 (см. Пример 17 ниже). Были выбраны единичные выделенные бляшки из второго скринирования и поддержаны в виде единичных клонов на LBM/агаровых чашках. Секвенированные праймеры 434FL и 434FR (SEQ ID NO: 34 и 35 соответственно) были использованы в стандартном протоколе с генерированием ДНК для секвенирования.
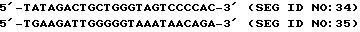
ДНК из PCR была очищена с использованием Qick Spin колонки (Quagen) в соответствии с протоколом, предложенным производителем.
Были идентифицированы два клона, обозначенные 741.4 и 741.11, которые перекрывали клоны 648.3 и 705.1; в областях перекрывания клоны 741.1 и 741.11 были на 100% гомологичными клонам 648.3 и 705.1. Состав крысиной сДНК, обладающей гомологией к человеческому αd гену, представляется в SEQ ID NO: 36; предсказанная аминокислотная последовательность представляется в SEQ ID NО:37.
Клонирование 5'конца крысиной αd.
Был получен 5' сДНК фрагмент из крысиного αd гена с использованием клонирующего набора Clonetech rat spleen RAGE в соответствии с протоколом, предложенным производителем. Использованные ген специфические олигонуклеотиды были обозначены 741.11#2R и 741.2#1R (SEQ ID NO:58 и 59 соответственно).

Олиго 741.11# 2R включает пары оснований 131-152 в SEQ ID NO:36, в обратной ориентации и 741.2#1R включает пары оснований 696-715 в SEQ ID NO:36, также в обратной ориентации. Первичная PCR также была выполнена с использованием самого большого 3' олиго, 741.2# 1R. Вторая PCR следовала, используя олиго 741.11# 2R и ДНК, генерированную в первичной реакции. Полоса приблизительно 300 пар оснований была обнаружена на 1% агарозном геле.
Вторичный PCR продукт был лигирован в плазмиду pCRTAII (Invirogen) в соответствии с протоколом, предложенным производителем. Были отобраны белые положительные колонии и добавлены к 100 мкл LBM, содержащего 1 мкл 50 мг/мл готового раствора карбеницилина и 1 мкл М13К07 фаговой культуры в индивидуальных лунках в круглодонных 96-луночных планшетах культуры ткани. Смесь была проинкубирована при 37oС в течение от 30 минут до 1 часа. После начального периода инкубации были добавлены 100 мкл LBM (содержащего 1 мкл 50 мг/мл карбеницилина и 1:250 разбавление 10 мг/мл готового раствора канамицина), и инкубирование было продолжено в течение ночи при 37oС.
Используя стерильную 96-луночную металлическую гребенку для переноса, супернатант из 96-луночного планшета был перенесен на четыре нейлоновых фильтра Amerscham Hybond. Фильтры были денатурированы, нейтрализованы и сшиты с помощью стандартного протокола. Фильтры были прегибридизованы в 20 мл буфера прегибридизации (5Х SSPE; 5X Denhardts; 1% SDS; 50 унг/мл ДНК денатурированной спермы лосося) при 50oС в течение нескольких часов с потряхиванием.
Олигопробы 741.11# 1 и 741.111 R (SEQ ID NO:56 и 57, соответственно), включающие пары оснований 86-105 (SEQ ID NO:36) в прямой и обратной ориентации соответственно были мечены следующим образом.

Приблизительно 65 нг олиго ДНК в 12 мкл dH2O было нагрето до 65oС в течение 2 минут. 3 мкл 10 мСi/мл γ-32P-ATP были добавлены в пробирку вместе с 4 мкл 5х киназного буфера (Gibco) и 1 мкл Т4 ДНК Киназы (Gibco). Смесь была проинкубирована при 37oС в течение 30 минут. После инкубирования было добавлено 16 мкл каждой меченой олигопробы к прегибридизованному буферу и фильтрам, и гибридизация была продолжена в течение ночи при 42oС. Фильтры были промыты три раза в 5х SSPE; 0.1% SDS в течение 5 минут на каждую промывку при комнатной температуре, авторадиографированы в течение 6 часов. Положительные клоны были размножены, и ДНК очищена с использованием Mag Mini Prep набора (Promega) в соответствии с протоколом, предложенным производителем. Для секвенирования был выбран клон 2F7, показывающий 100% гомологию к клону 741.11 в перекрывающей области. Полная крысиная последовательность αd нуклеиновой кислоты представлена в SEQ ID NO:54; аминокислотная последовательность представлена в SEQ ID NO:55.
Характеристики крысиной сДНК и аминокислотных последовательностей.
Ни нуклеиновая кислота, ни аминокислотная последовательности не сообщались ранее для крысиных α субъединиц в β2 интегринах. Однако сравнение последовательности с сообщавшейся человеческой β2 интегриновой α субъединицей подтвердило, что выделенный крысиный клон и его предсказанная аминокислотная последовательность являются наиболее близко связанными с αd нуклеотидом и аминокислотными последовательностями.
На нуклеиновокислотном уровне выделение крысиного сДНК клона показало 80% идентичности, по сравнению с человеческой αd сДНК; 68% идентичности по сравнению с человеческой CD11b; 70% идентичности по сравнению с человеческой CD11c; и 65% идентичности по сравнению с мышиной CD11b. Значительной идентичности не наблюдается по сравнению с человеческой CD11а и мышиной CD11а.
На аминокислотном уровне предсказанный крысиный полипептид, кодированный выделенной сДНК, показывал 70% идентичности по сравнению с человеческим αd полипептидом; 28% идентичности по сравнению с человеческой CD11a; 58% идентичности по сравнению с человеческой CD11b; 61% идентичности по сравнению с человеческой CD11c; 28% идентичности по сравнению с мышиной CD11a и 55% идентичности по сравнению с мышиной CD11Ь.
Пример 18.
Продуцирование и характеристика αd специфичных антител грызунов.
А. Антитела против крысиных α I домен/НuIgG4 составных белков.
В свете того, что I домен человеческих β2 интегринов, как было продемонстрировано, участвовал в лигандном связывании, было высказано предположение, что то же самое будет иметь место и для крысиного αd белка. Моноклональные антитела, иммуноспецифические для крысиного αd I домена, могут быть поэтому полезными в крысиных моделях человеческих состояний заболения, где подразумевается αd связывание.
Олигопроизводные "крысиной альфа-D15" (SEQ ID NO:87) и "крысиной альфа-D13" (SEQ ID NO:88) были генерированы из крысиной αd последовательности, соответствующей парам оснований 469-493 и парам оснований 1101-1125 (в обратной ориентации) соответственно в SEQ ID NO:54. Олигопроизводные были использованы в стандартной PCR реакции для генерирования крысиного αd ДНК фрагмента, содержащего I домен, включающий диапазон пар оснований 459-1125 в SEQ ID NO:54. PCR продукт был лигирован в вектор pCRTAII (Invirogen) в соответствии с протоколом, предложенным производителем. Была выбрана положительная колония и размножена для ДНК очистки с использованием Qiagen (Chatswoth, GA) Midi Prep набора в соответствии с протоколом, предложенным производителем. ДНК была гидролизована XhoI и ВgI11 в стандартном рестриктном ферменте гидролиза и полоса 600 пар оснований была очищена на геле, который был впоследствии лигирован в pDCS1/HuIgG4 вектор экспрессии. Была выбрана положительная колония, размножена, и ДНК очищена с помощью Qiagen Маху Prep набора.
COS клетки были посеяны при половинном слиянии на 100 мм чашки с культурой и выращены в течение ночи при 37oС в 7% CO2. Клетки были промыты один раз 5 мл DMEM. К 5 мл DMEM было добавлено 50 мкл DEAE-декстрана, 2 мкл хлорокина и 15 мкг крысиной αd I домен/НuIgG4 ДНК, описанной выше. Смесь была добавлена к COS клеткам и проинкубирована при 37oС в течение 3 часов. Затем среда была удалена и было добавлено 5 мл 10% ДМСО в CMF-PBS точно в течение 1 минуты. Клетки осторожно были промыты один раз DMEM. Десять мл DMEM, содержащего 10% FBS, было добавлено к клеткам, и инкубирование продолжено в течение ночи при 37oС в 7% CO2. На следующий день среда была заменена свежей средой и инкубирование продолжено в течение трех дополнительных суток. Среда была собрана, и свежая среда была добавлена в чашки. После трех суток среда была собрана вновь и чашки опорожнены. Процедура была повторена до тех пор, пока не было собрано 2 л супенатанта культуры.
Супенатант, собранный, как описано выше, был загружен в Prosep-A колонку (Bioprocessing Limited), и белок очищен, как описано ниже.
Колонка первоначально была промыта промывным буфером в объеме, равном 15 объемам колонки, содержащем 35 мМ Трис и 150 мМ NaCl, pH 7.5. Супернатант был загружен с низкой скоростью с объемом меньше, чем объем, приблизительно равный 60 объемам колонки в час. После загрузки колонка была промыта промывным буфером в объеме, равном 15 объемам колонки, 0.55 М диэтаноламином, pH 8.5, в объеме, равном 15 объемам колонки, 50 мМ лимонной кислоты, pH 5.0, в объеме, равном 15 объемам колонки. Белок был элюирован 50 мМ лимонной кислоты, pH 3.0. Белок был нейтрализован 1.0 М Трис, pH 8.0 и диализован в стерильном PBS.
Крысиный αd I доменовый белок был проанализирован, как описано в Примере 14. Обнаруженный белок мигрировал таким же образом, как описано с человеческим I доменовым белком.
В. Продуцирование моноклональных антител с крысиными αd I доменовыми/НuIgG4 составными белками.
Мыши были индивидуально иммунизованы 50 мкг очищенного крысиного αd I доменового/НuIgG4 составного белка, предварительно эмульгированного в равном объеме полного адъюванта Freunds (FCA) (Sigma). Приблизительно 200 мкл антиген/адъювантного препарата было инъецировано в 4 точки спины и в бока каждой мыши. Четыре недели спустя мыши были повторно иммунизованы инъекцией 100 мкл крысиного αd I доменового/НuIgG антигена (50 мкг/мышь), предварительно эмульгированного в равном объеме полного адъюванта Freunds (FCA). Дополнительно через две недели мыши были повторно иммунизованы 100 мкг антигена в 200 мкл PBS в виде внутривенной инъекции.
Для оценки сывороточных титров у иммунизованных мышей было проведено ретро-орбитальное кровопускание животным спустя десять дней после третьей иммунизации. Крови было позволено свернуться, и сыворотка выделена центрифугированием. Сыворотка была использована в иммунопреципитации на биотинилированных (BIP) крысиных спленоцитах. Сыворотка от каждой мыши иммунопреципитировала полосы белка ожидаемого молекулярного веса для крысиной αd и крысиной CD18. Для получения гибрида была выбрана одна мышь и подвергнута повторной иммунизации четвертый раз, как описано выше для третьей повторной иммунизации.
Гибридомные супернатанты были подвергнуты скринированию с помощью захвата антитела, описываемого следующим образом. Immunolon 4 плашки (Dynatech, Cambridge, Massachusetts) были нагружены при 4oС 50 мкл/лунку овечьим антимышиным IgA, IgG или IgM (Organon Teknika), разбавленным 1:5000 в 50 мМ карбонатного буфера, рН 9.6. Плашки были промыты 5х PBS, содержащим 0.05% Твин 20 (PBST), и были добавлены 50 мкл супернатанта культуры. После инкубирования при 37oС в течение 30 минут и промывания, как описано выше, было добавлено 50 мкл пероксидазы хрена, конъюгированной с овечьим антимышиным IgG9(Fc) (Jackson ImmunoReserch, West Grove, Pensylvania), разбавленным 1: 3500 в PBST. Немедленно после этого было добавлено 100 мкл субстрата, содержащего 1 мг/мл о-фенилендиамина (Sigma) и 0.1 мкл/мл 30% H2O2 в 100 мМ цитрата, рН 4.5. Окрашивание продуктов реакции было остановлено через 5 минут добавлением 50 мкл 15% H2SO4. Поглощение при 490 нм регистрировали на Dynatech plate reader.
Супернатант из лунок, содержащих антитело, также был проанализирован с помощью анализа ELISA с иммунизованным крысиным αd I домен/НuIgG4 составным белком. В анализе ELISA плашки, нагруженные HuIgG4 антителом, служили в качестве контроля на реактивность по отношению к IgG гибридному партнеру. Положительные лунки были выбраны для дальнейшей проверки с помощью ВIР на крысином спленоцитном лизате с использованием приемов, описанных ниже.
С. Продуцирование поликлональной сыворотки в крысином αd I доменовом/НuIgG4 составном белке.
Двум кроликам было предварительно проведено кровопускание до иммунизации 100 мкг очищенного крысиного αd I доменового/НuIgG4 составного белка в полном адъюванте Freund's. Инъекции повторяли с теми же дозами каждые три недели в неполном адъюванте Freund's (IFA). После трех инъекций кроликам было проведено кровопускание и собранную сыворотку использовали в стандартной иммунопреципитации в крысиных спленоцитовых лизатах. Было определено, что сыворотка от обоих кроликов была иммунореактивной с крысиным αd. Кролики были вновь повторно иммунизованы 100 унг антигена в IFA, и собранная сыворотка проанализирована на увеличенную иммунореактивность с крысиным αd с помощью иммунопреципитации. Животным была проведена конечная повторная иммунизации, и спустя 10 дней было проведено кровопускание и собрана сыворотка.
Крысиная αd гистология.
Кроличья поликлональная сыворотка, генерированная против крысиного αd "I" домена, была использована для иммуногистохимического окрашивания срезов крысиных тканей с помощью приемов, описанных в Примере 16. Картина окрашивания, обнаруженная на замороженных и на внедренных в парафин крысиных селезеночных срезах, была по существу идентичной той, которую наблюдали с антителами против человеческой αd, с окрашиванием индивидуальных клеток по всей красной пульпе. Картина окрашивания отличалась от картины, наблюдавшейся с моноклональными антителами против крысиных CD11a, CD11b и CD11c. Кроме того, положительная картина окрашивания была видна в тимусе на индивидуальных клетках по всему кортексу. Ни одна из этих тканей не давала никакого сигнала, когда окрашивалась кроличьей преиммунной сывороткой.
Д. Анализ специфичности антитела.
Крысы были умерщвлены с помощью асфиксии СО2, и селезенки были удалены с использованием стандартных хирургических приемов. Спленоциты были собраны осторожным продавливанием селезенки через проволочное сито 3 см3 шприцем в 20 мл RPMI. Клетки были собраны в 50 мл конические пробирки и промыты в соответствующем буфере.
Клетки были три раза промыты в холодном D-PBS и ресуспендированы при плотности 108-109 клеток в 40 мл PBS. Четыре мг NHS-Biotin (Pierce) были добавлены к клеточной суспензии, и реакции было позволено продолжаться в течение точно 15 минут при комнатной температуре. Клетки были собраны в комочки и промыты три раза в холодном D-PBS.
Клетки были ресуспендированы при плотности 108 клеток/мл в холодном лизисном буфере (1% NP40, 50 mM Трис-HCl, рН 8.0; 150 mM NaCl; 2 mM CaCl; 2 mM MgCl; 1: 100 растворе пепстатина, лейпептина и апротинина, добавленном до добавления к клеткам; и 0.0001 г PMSF кристаллов, добавленных до добавления к клеткам). Лизаты были перемешаны покручиванием в течение приблизительно 30 секунд, проинкубированы в течение 5 минут при комнатной температуре и далее проинкубированы в течение 15 минут во льду, Лизаты были подвергнуты центрифугированию в течение 10 минут при 10000 g до образования нерастворимого материала в виде комочков. Супернатант был собран в новые пробирки и хранился при температуре между 4oС и -20oС.
Один мл клеточного лизата был предварительно просветлен с помощью инкубирования 200 мкл А сефарозной суспензии, содержащей белок (Zymed), в течение ночи при 4oС. Аликвоты предварительно просветленного лизата были помещены в аппендорфовские пробирки при 50 мкл/пробирку для каждого антитела, которое испытывалось. Двадцать пять мкл поликлональной сыворотки или от 100 до 500 мкл супернатанта моноклонального антитела были добавлены к предварительно просветленным лизатам и полученная смесь проинкубирована в течение 2 часов при 4oС с вращением. Затем была добавлена одна сотая мкл кроличьего антимышиного IgG (Jackson), связанного с белком А сефарозными гранулами в суспензии PBS, и инкубирование продолжено в течение 30 минут при комнатной температуре с вращением. Гранулы были собраны осторожным центрифугированием и три раза промыты холодным промывным буфером (10 мМ HEPES; 0.2 М NaCl; 1% Тритон Х-100). Супернатант был удален отсасыванием, и было добавлено 20 мкл 2Х SDS буфера, содержащего 10% β-меркаптоэтанола. Образец был подвергнут кипячению в течение 2 минут на водяной бане и образец был загружен в 5% SDS PAGE гель. После разделения белки были перенесены на нитроцеллюлозу при постоянном токе в течение ночи. Нитроцеллюлозные фильтры были блокированы 3% BSA в TBS-T в течение 1 часа при комнатной температуре, и блокирующий буфер был удален. Стрептавидин-HRP конъюгат (Jackson) в 0.1% BSA TBS-T был добавлен, и инкубирование продолжено в течение 30 минут при комнатной температуре. Фильтры были промыты три раза в течение 15 минут каждый TBS-T и авторадиографированы с использованием Amerscham's ECL набора в соответствии с протоколом, предложенным производителем.
Е. Продуцирование моноклональных антител в крысином αd белке полной длины.
Очистка αd крысиного белка.
Крысиный αd был очищен от крысиных спленоцитов для приготовления иммуногена для генерирования антикрысиных αd моноклональных антител. Селезенки из соответственно 50 нормальных крысиных самок Lewis 12-20 недельного возраста были собраны, и была приготовлена одноклеточная суспензия из ткани путем продавливания ее через тонкое проволочное сито. Красные кровяные клетки были удалены за счет лизиса в буфере, содержащем 150 мМ NH4Cl, 10 mM КНСО3, 0.1 мМ ЭДТА, рН 7.4, и оставшиеся лейкоциты были промыты два раза фосфатным буферным рассолом (PBS). Спленоциты были переведены в комочки центрифугированием и лизированы в буфере, содержащем 50 мМ Трис, 150 mM NaCl; 2 mM CaCl2; 2 mM MgCl2; 10 мМ PMSF, лейпептина пепстатина и 1% Тритон Х-100 на 5•108 спленоцитов. Нерастворимый материал был удален центрифугированием.
CD11a, CD11b и CD11c были удалены из селезеночного лизата с помощью иммунопреципитации следующим образом. 750 мкл объем А сефарозной суспензии белка был проинкубирован 2 мг кроличьего антимышиного иммуноглобулина при 4oС в течение 30 минут. Кроличий антимышиный А сефарозный белок был три раза промыт лизисным буфером и суспендирован в конечный объем 1.5 мл лизисного буфера. Приблизительно 200 мкг каждого из крысиных β2 интегриновых моноклональных антител, 515 F (специфичных для крысиной CD11a), ОХ-42 (специфичных для крысиной CD11b) и 100 g (специфичных для крысиной CD11c) были добавлены к 50 мл крысиного селезеночного лизата. После 30 минут инкубирования при 4oС было добавлено 500 мкл кроличьего антимышиного А сефарозного белка к селезеночным лизатам и смешано с вращением снова и снова в течение 30 минут при 4oС. Лизат был отцентрифугирован при 2500 g в течение 10 минут до образования шариков CD11a, CD11b и CD11c, связанных с кроличьим антимышиным А сефарозным белком, а супернатант перенесен в чистые 50 мл центрифужные пробирки. Иммунопреципитация антителами 515 F, ОХ-42 и 100 g была повторена дополнительно два раза для обеспечения полного удаления CD11a, CD11b и CD11c.
β2 интегрины, оставшиеся в лизате, были выделены с использованием аффинной очистки. Приблизительно 250 мкл суспензии антикрысиного CD18 моноклонального антитела 20С5В конъюгированного с CNBr-сефарозой было добавлено к лизатам и смешано с вращением снова и снова в течение 30 минут при 4oС. Комплексы антитело/антиген были собраны в комочки центрифугированием при 2500 g в течение 10 минут, и комочки три раза промыты лизисным буфером до того, как поместить на хранение при 4oС.
Иммунизация армянских хомячков.
Армянские хомячки шести-восьми недельного возраста были первоначально иммунизованы приблизительно 50 мкг рекомбинантного белка, состоящего из I домен крысиного αd гибридизованного с человеческой IgG4 тяжелой цепью, эмульгированной в полном адъюванте Freund's. Первичная иммунизация, за которой следовали последующие иммунизации крысиным I доменом/НuIgG4 эмульгировали в неполном адъюванте Freund's на 14, 33 и 95 сутки. Впоследствии было осуществлено получение двух раздельных гибридов, обозначенных 197 и 199.
За четыре дня до гибридизации 197 (306 сутки) одному хомячку была введена комбинация крысиного белка, очищенного от спленоцитов и СНО клеток, трансфекцированных крысиным αd. Повторная гибридная иммунизация была проведена за три дня до гибридизации (307 сутки) очищенным крысиным αd белком и αd трансфекцированными СНО клетками. Крысиные αd трансфекцированные СНО клетки были приготовлены, как описано ниже.
Генный сегмент, кодирующий крысиный αd белок полной длины, был инсертирован в pDCl вектор и трансфекцирован электропорацией в СНО клетки вместе с человеческой CD18-pRC конструкцией. Трансфекцированные клетки были выращены в присутствии гипоксантина для того, чтобы провести выбор клеток, успешно трансфекцированных pCR конструкцией и в присутствии g418 для того, чтобы провести выбор клеток, трансфекцированных pDCl конструкцией. После трех недель клетки были окрашены крысиной αd специфической кроличьей поликлональной сывороткой и подвергнуты сортировке с помощью FACS. Небольшое количество клеток, которые экспрессировали самые высокие уровни поверхностного αd (приблизительно 3% общей популяции) были собраны и далее размножены. FACS селекция была повторена несколько раз для обеспечения популяций клеток с самыми высокими уровнями αd поверхностной экспрессии.
αd трансфекцированные клетки были охарактеризованы также с помощью проточной цитометрии с использованием крысиной αd специфичной поликлональной сыворотки и человеческого CD18 специфичного моноклонального антитела, TS1.18.1. Результаты подтвердили, что трансфекцированные СНО клетки экспрессировали высокие уровни и крысиной αd и человеческого CD18.
Наконец, αd и CD18 экспрессия в клетки была оценена с помощью иммунопреципитации. Было найдено, что крысиная αd специфическая кроличья поликлональная сыворотка иммунопреципитирует белки с двумя четкими молекулярными весами: самым высокомолекулярным белком(ми), который имеет 170 кD, и самым низкомолекулярным белком(ми) 95 kD. Эти найденные результаты согласовывались с экспрессией крысиного αd/человеческого CD18 гетеродимерного комплекса на поверхности трансфекцированных СНО клеток.
На день гибридизации селезенка была удалена и получена одноклеточная суспензия за счет измельчения ткани между замороженными концами двух стеклянных микроскопических слайдов, погруженных в сыворотку, свободную от RPMI 1640, дополненную 2 мМ L-глутамина, 1 мМ пирувата натрия, 100 единицами/мл пенициллина и 100 мкг/мл стрептомицина (RPMI)(Gibco, Canada). Клеточная суспензия была профильтрована через стерильное сито с ячейками 70 меш Nitex (Becton Dickinson, Persyppany, New Jersey), промыта два раза центрифугированием при 200 g в течение 5 минут, и шарики ресуспендированы в 20 мл сыворотки, свободной от RPMI. Тимоциты, взятые от трех нативных Balb/c мышей, были приготовлены аналогичным образом. NS-1 миеломные клетки, хранившиеся в log фазе в RPMI с 10% сыворотки Fetaclone (FBS) (Hyclone Laboratories, Inc. Logan, Utha) в течение трех суток до гибридизации, были центрифугированы при 200 g в течение пяти минут, шарики были два раза промыты, как описано ранее.
Приблизительно 1.15•108 селезеночных клеток были объединены с 5.8•107 NS-1 клеток, отцентрифугированы, и супернатант удален отсасыванием. Клеточный шарик был удален с помощью конической трубки, и семь мл ПЭГ 1500 при 37oС (50% в 75 мл Hopes, pH 8.0)(Boehringer Mannheim) было добавлено в течение более чем одной минуты с последующим добавлением 14 мл сыворотки, свободной от RPMI, в течение более чем семи минут. Было добавлено дополнительно восемь мл RPMI, и клетки были отцентрифугированы при 200 g в течение 10 минут. Супернатант был удален, и шарик ресуспендирован в 200 мл RPMI, содержащего 15% FBS, 100 мМ гипоксантина натрия, 0.4 мМ аминоптерина, 16 мМ тимидина (HAT) (Gibco), 25 единиц/мл IL-6 (Boehringer Mannheim) и 1.5•106 тимоцитов/мл. Суспензия была распределена в десять 96-луночных плоскодонных планшетов с тканевой культурой (Corning, United Kingdom) при 200 мкл/лунку, и клетки были выгружены на 4, 5, 6 и 7 дни после гибридизации отсасыванием приблизительно 100 мкл из каждой лунки иглой 18G (Becton Dickinson) и добавлением 100 мкл культивированной среды, описанной выше, за исключением недостающих тимоцитов.
На 10 день супернатанты из гибридизованных лунок были определены на реактивность с помощью проточной цитометрии к крысиным αd/человеским CD18 трансфекцированным СНО клеткам. Приблизительно 5•105 крысиных αd трансфекцированных СНО клеток были суспендированы в 50 мкл RPMI, содержащего 2.0% FBS и 0.05% азида натрия, и добавлены в приблизительно 100 мкл супернатанта гибридомной культуры в 96-луночные круглодонные планшеты. Положительные контроли для окрашивания включали кроличью анти-αd- поликлональную сыворотку и TS1/18 (античеловеческую CD18). Клетки были проинкубированы в течение 30 минут во льду, три раза промыты в FACS буфере (RPMI, 2.0% FBS, 0.05% азида Na) и проинкубированы в течение 30 минут во льду с FITC-конъюгированным овечьим антихомячковым антителом (Jackson Immunol Reserch Labs) с конечным разбавлением 1:200 в FACS буфере. Клетки были три раза промыты в FACS буфере и ресуспендированы в 200 мл FACS буфера. Образцы были проанализированы на Becton Dickinson FACscan анализаторе. Для того чтобы убедиться, что лунки с положительными клонами были специфичными для крысиного αd, определение было повторено с нетрансфекцированными СНО клетками, Лунки, которые отвечали критерию реактивности с крысиными αd СНО трансфектантами и не отвечали критерию реактивности с нетрасфекцированными СНО клетками, были клонированы.
После первичного скринирования клетки из лунок с положительной реакцией были клонированы первоначально двойным разбавлением и впоследствии с помощью ограничивающего разбавления в RPMI, 15% FBS 100 мМ гипоксантина натрия, 16 мМ тимидина и 10 единиц/мл IL-6. В стадии ограничивающего разбавления было определено количество лунок, в которых наблюдали рост, и предсказана клональность с использованием Poisson анализа распределения. Лунки, в которых наблюдали рост, были проанализированы с помощью FACS после 10-12 суток. После конечного клонирования положительные лунки были размножены в RPMI и 11% FBS. Клонирование давало одну культуру, считающуюся положительной по этому критерию, из которой были размножены четыре отдельных субклона, обозначенных 197А-1, 197А-2, 197А-3 и 197А-4.
До гибридизации 199 второй хомячок был повторно иммунизован на 307 сутки 2.3•106 крысиных αd трансфекцированных СНО клеток. Две конечных иммунизации проводили за 4 дня до гибридизации (334 сутки) и вновь за три дня до гибридизации (335 сутки). Повторная иммунизация на 334 сутки состояла из 2•106 крысиных αd трансфекцированных СНО клеток и 200 мкл очищенного крысиного αd связанного с сефарозой (предварительно описано), введенных внутривенной инъекцией. Повторная иммунизация на 335 сутки состояла из 5•106 крысиных αd трансфекцированных СНО клеток, также введенных внутривенной инъекцией. Гибриды и протоколы скринирования для гибридизации 199 были идентичными с гибридизацией 197, и были идентифицированы и клонированы три гибридомы, обозначенные 199А, 199Н и 199М, супернатантом, реактивным с крысиным αd. Гибридома, обзначенная 199М, была осаждена 1 марта 1996 Аmerical Type Culture Collection, 12301 Parklawn Drive, Rockville, Maryland 20852, и приписана к No. HB-12058.
Характеристика моноклональных антител в крысином αd.
Для того чтобы охарактеризовать антикрысиные αd антитела, были приготовлены печеночные лизаты, меченные биотином, как описано в Примере 18, раздел Д, приведенном выше. Лизаты были предварительно осветлены до использования в опытах по иммунопреципитации. Первоначально 50 мкг/мл нормального мышиного иммуноглобулина было добавлено к лизату, и полученный раствор смешан вращением вновь и вновь в течение 30 минут при 4oС. 75 мкл суспензии А-сефарозного белка, нагруженной кроличьим антимышиным иммуноглобулином, было добавлено и перемешано с непрерывным вращением вновь и вновь в течение 30 минут. Гранулы, покрытые кроличьим антимышиным белком А, были собраны в комочки центрифугированием при 15000 об/мин в настольной микроцентрифуге в течение пяти минут при 4oС, и супернатант собран. Материал в виде комочков был выгружен.
Для каждой клонированной гибридомы приблизительно 300 мкл суспентатнта было помещено в эппендорфовскую центрифужную пробирку, в которую было добавлено 30 мкл 10% Тритона Х-100, 30 мкл 100Х заранее приготовленного раствора пепстатина, лейпептина и апротинина, 100 мкг PMSF кристаллов и 50 мкл предварительно осветленного биотинилированного лизата кроличьей селезенки. Образцы осторожно покручивали и помещали во вращающееся устройство при 4oС в течение 30 минут. Контрольный образец был приготовлен добавлением 10 мг/мл кроличьего антикрысиного αd специфичного поликлонального антитела к 50 мкл лизата крысиной селезенки.
После 30 минут инкубирования было добавлено 75 мкл гранул А сефарозного белка в суспензии PBS к каждому образцу и проинкубировано с вращением вновь и вновь при 4oС в течение 30 минут. А-спаренные гранулы белка были собраны в комочки центрифугированием при 15000 об/мин в настольной микроцентрифуге в течение 5 минут при 4oС, и супернатант собран. Гранулы в виде комочков были последовательно промыты сериями 1 мл детергентных промывок следующим образом. Буфер # 1, содержащий 10 мМ Трис, 400 мМ NaCl, 1.0% Тритон Х-100, рН 8.0; буфер #2, содержащий 10 мМ Трис, 400 мМ NaCl, 0.5 Тритон Х-100, рН 8.0; буфер #3, содержащий 10 мМ Трис, 400 мМ NaCl, 1,0% Тритон X-100, 0.1% деоксихолата, рН 8.0 и буфер #4, содержащий 10 мМ Трис, 400 мМ NaCl, 0.5 М LiCl2, рН 8.0. Конечная промывка была проведена промывным буфером #1. Гранулы осторожно покручивали после каждой промывки и собирали в комочки с помощью настольной микроцентрифуги. Супернатанты удаляли с помощью переносной пипетки и после конечной промывки весь оставшийся буфер удаляли из гранул сифоном Hamilton. 50 мкл аликвоты SDS буферного образца, содержащего бромфеноловый голубой и Пирониновый Y красители и β-меркаптоэтанол с конечной концентрацией 10% были добавлены к каждому шарику. Смесь интенсивно покручивали в течение 1-2 минут и инкубировали при комнатной температуре в течение 5-10 минут. Образцы центрифугировали в течение 5 минут при 15000 об/мин в настольной микроцентрифуге при 4oС и высвободившийся белок собирали и переносили в новые микроцентрифужные пробирки. Аликвоты из каждого образца кипятили в течение 4 минут на водяной бане до загрузки в 7.5% SDS-PAGE гели. После разделения с помощью PAGE белки были перенесены на нитроцеллюлозные фильтры в течение 1 часа при 200 мА, и фильтры были блокированы в растворе 3.0% BSA/TBS-T в течение ночи при 4oС. Раствор 0.1% BSA/TBS-T, содержащий 1:6000 разбавление стрептавидин-OPD, был добавлен к каждому фильтру, и инкубирование продолжалось в течение 1 часа при комнатной температуре. Фильтры были пять раз промыты в течение 5 минут каждый в TBS-T, проявлены с использованием Amerscham's ECL набора в соответствии с протоколом, предложенным производителем.
Было найдено, что клон 199М иммунопреципитирует гетеродимерный белок. Большая часть белковой субъединицы имела приблизительно молекулярный вес 170-175 kD, который согласовывался с размером белка, иммунопреципитированного кроличьим антикрысиным αd поликлональным контролем. Второй белок также был осажден с молекулярным весом приблизительно 95 kD, согласующимся с весом CD18.
Пример 19
Выделение мышиных сДНК клонов.
Была предпринята попытка выделения мышиного αd гомолога.
Были проведены перекрестные виды гибридизации с использованием PCR-генерированных проб: 1.5 kb фрагмента, соответствующего основаниям 522-2047 из человеческого клона 19А2 (SEQ ID NO:1), и 1.0 kb крысиного фрагмента, который соответствовал основаниям 1900-2900 в человеческом клоне 19А2 (SEQ ID NO: 1). Человеческая проба была генерирована с помощью PCR с использованием первичных праймерных пар, обозначенных АТМ-2 и 9-10.1, представленных в SEQ ID NO:38 и 39 соответственно; крысиная проба была генерирована с помощью PCR с использованием первичных праймерных пар 434L и 434R, представленных в SEQ ID NO:34 и 35
соответственно. Образцы были проинкубированы при 94oС в течение 4 минут и подвергнуты 30 циклам обработки с последовательностью температурных стадий: 94oС; 50oС в течение 2 минут; 72oС в течение 4 минут.
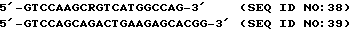
PCR продукты были очищены с использованием Quagen Quick Spin набора в соответствии с протоколом, предложенным производителем, и приблизительно 180 нг ДНК были мечены 200 мкCi[32P]-dCTP с использованием Boehringer Mannheim Random Primer Labelling набора в соответствии с протоколом, предложенным производителем. Невошедший изотоп был удален с использованием Centri-sep Spin колонки (Princeton Separation, Adelphia, NJ) в соответствии с протоколом, предложенным производителем. Пробы до использования были денатурированы 0.2 N NaOH и нейтрализованы 0.4 М Трис-HCl, рН 8.0,
Мышиная тимусная олиго dT-примированная сДНК библиотека в ламбда ZAP II (Stratageen) была помещена с количеством приблизительно 30000 бляшек на 15 см чашку. Бляшковые подъемы на нитроцеллюлозных фильтрах (Schleicher & Schuell, Keene, NH) были проинкубированы при 50oС с перемешиванием в течение 1 часа в растворе прегибридизации (8 мл/подъем), содержащем 30% формамида. Меченые человеческие и крысиные пробы были добавлены к раствору прегибридизации, и инкубирование продолжено в течение ночи при 50oС. Фильтры были дважды промыты в 2Х SSC/0.1% при комнатной температуре, один раз в 2Х SSC/0.1% SDS при 37oС и один раз в 2Х SSC/0.1% SDS при 42oС. Фильтры были подвергнуты экспозиции на пленке Kodak X-Omat AR при -80oС в течение 27 часов с интенсифицирующим экраном.
Четыре бляшки, дававшие положительные сигналы на дупликатных подъемах, пересеяны штрихом в LB среде с магний (LВМ)/карбеницилином (100 мг/мл) в чашках и проинкубированы в течение ночи при 37oС. Фаговые бляшки были подняты Hybon фильтрами (Amerscham), прозондированы в первоначальном экране и подвергнуты экспозиции на пленке Kodak X-Omat AR при -80oС в течение 27 часов с интенсифицирующим экраном.
Двенадцать бляшек, дававшие положительные сигналы, были перенесены в фаговый разбавитель с низким содержанием Mg++, содержащий 10 мМ Трис-НСl и 1 мМ MgCl2. Размер инсерта был определен с помощью PCR амплификации с использованием Т3 и Т7 праймеров (SEQ ID NO: 13 и 14 соответственно) и следующих условий реакции. Образцы были проинкубированы при 94oС в течение 4 минут и подвергнуты 30 температурным циклам обработки с последовательностью стадий: 94oС в течение 15 секунд; 50oС в течение 30 секунд и 72oС в течение 1 минуты.
Шесть образцов продуцировали четкие полосы, которые находилсь в области размеров от 300 оснований до 1 кb. Фагмиды были выделены через ко-инфицирование с хелперным фагом и рециркулиризованы с генерированием Bluescript SK- (Stratagen). Полученные колонии были культивированы в LBM/карбеницилине (100 мг/мл) в течение ночи. ДНК была выделена с использованием Promega Wizard минипрераративного набора (Madison, WI) в соответствии с протоколом, предложенным производителем. EcoRI рестрикционный анализ очищенной ДНК подтвердил молекулярные веса, которые были обнаружены с использованием PCR. Инсертированная ДНК была секвенирована М13 и М13 обратным 1 праймерами, представленными в SEQ ID NO:40 и 41 соответственно.

Секвенирование было проведено, как описано в Примере 4.
Из шести клонов только два, обозначенные 10.3-1 и 10.5-2, обеспечивали последовательную информацию и были идентичны 600 bp фрагментам. Последовательность 600 bp представляла 68% идентичности соответствующей области человеческой αd, 40% идентичности человеческой CD11a, 58% идентичности человеческой CD11c и 54% идентичности мышиной CD11b. Этот фрагмент 600 bp был затем использован для выделения более полной сДНК, кодирующего предполагаемого мышиного αd гомолога.
Мышиная селезеночная сДНК библиотека (олиго dT- и статистически примированная) в ламбда Zapll(Stratagen) была культивирована с концентрацией 2.5•104 фага/15 см LBM чашку. Бляшки были приподняты на Hybond нейлоновых переносных мембранах (Amerscham), денатурированы 0.5 М NaOH/1.5 M NaCl, нейтрализованы 0,5 М Трис основанием/1.5 М NaCl/11.6 HCl, и промыты в 2Х SSC. ДНК была перекрестно связана с фильтрами за счет ультрафиолетового облучения.
Приблизительно 500000 бляшек были испытаны с использованием проб 10.3-1 и 10.5-2, предварительно меченных, как описано выше. Пробы были добавлены к раствору прегибридизации и проинкубированы в течение ночи при 50oС. Фильтры были дважды промыты в 2Х SSC/0.1% SDS при комнатной температуре, один раз в 2Х SSC/0.1% SDS при 37oС и один раз в 2Х SSC/0.1% SDS при 42oС, Фильтры были подвергнуты экспозиции на пленку Kodak X-Omat AR при -80oС в течение 13 часов с интенсифицирующим экраном.
Восемнадцать положительных бляшек были перенесены в фаговый разбавитель с низким содержанием Mg++ и размер инсерта определен с помощью PCR амплификации, как описано выше. Семь образцов давали сигнальные полосы, которые имели размер в области от 600 bp до 4 kb. EcoRI рестрикционный анализ очищенной ДНК подтвердил размеры, наблюдавшиеся из PCR, и ДНК была секвенирована праймерами М13 и М13 обратным 1 (SEQ ID NO:40 и 41 соответственно).
Один клон, обозначенный В3800, содержал 4 kb инсерт, который соответствовал области 200 оснований нижнего потока 5' конца человеческого αd 19А2 клона и включал 533 основания 3' нетранслированной области. Клон В3800 показывал 77% идентичности соответствующей области человеческой αd, 44% идентичности соответствующей области человеческой CD11a, 59% идентичности соответствующей области человеческой CD11c и 51% идентичности соответствующей области мышиной CD11b. Второй клон А1160 представлял 1.2 kb инсерт, который пристраивал к 5'концу кодирующей области человеческой αd, 12 нуклеиновых кислот нижнего потока инициирующего метионина. Клон А1160 показывал 75% идентичности соответствующей области человеческой αd, 46% идентичности соответствующей области человеческой CD11a, 62% идентичности соответствующей области человеческой CD11с и 66% идентичности соответствующей области мышиной CD11b.
Фрагмент клона А1160 ближе к 5' концу человеческого клона 19А2 составлял 1160 оснований в длину и разделял область перекрывания с клоном В3800, начиная от основания 205 и продолжаясь до основания 1134. Клон А1160 содержал инсерции 110 оснований (основания 704-814 клона А1160), не присутствующих в области перекрывания клона В3800. Эта инсерция имела место, вероятно при эксон-интронном связывании [Fleming, et.al., J.Immunol. 150:480-490 (1993)] и была удалена до последующего лигирования клонов А1160 и В3800.
Быстрое экстракопирование 5' конца сДНК предполагаемого мышиного αd клона.
RACE PCR [Frohman, "RACE: Быстрая амплификация сДНК концов" в PCR протоколах: A Guide Methoda and Applications. Innis, et.al. (eds) pp.28-38, Academic Press: New York (1990)] была использована для получения утерянных 5' последовательностей предполагаемого мышиного αd клона, включающего 5' нетранслированную последовательность и инициирующий метионин. Мышиный селезеночный RACE-Ready набор (Clonech, Palo Alto, СА) был использован в соотвествии с протоколом, предложенным производителем. Два античувствительных ген-специфических праймера, А1160 RACE1-первичный и А1160 РАСЕ2-внутригенный (SEQ ID NO: 42 и 43) были сконструированы для выполнения первичной и внутригенной PCR реакций.

Праймеры SEQ ID NO:42 и 43, соответствуют областям, начинающимся 302 и 247 основаниями от 5" конца соответственно. PCR была проведена, как описано выше, с использованием 5' якорного праймера (SEQ ID NO:44) и мышиной селезеночной сДНК, снабжаемой набором,
Электрофорез PCR продукта обнаруживает полосу приблизительно размером 280 оснований, которая была субклонирована с использованием ТА клонирующего набора (Invitrogen) в соответствии с протоколом, предложенным производителем. Десять получающихся колоний были культивированы и ДНК выделена и секвинирована. Дополнительные 60 оснований 5' последовательности были идентифицированы этим способом, которые соответствовали основаниям 1-60 в SEQ ID NO: 45.
Характеристики мышиной сДНК и предсказанной аминокислотной последовательности.
Композитная последовательность мышиной сДНК, кодирующая предполагаемый гомолог человеческой αd, представлена в SEQ ID NO:45. Хотя гомология между внешними доменами человеческих и мышиных клонов является высокой, гомология между цитоплазматическими доменами составляет только 30%. Наблюдаемое изменение может указывать на С-терминальное функциональное различие между человеческими и мышиными белками. Или же изменение в цитоплазматических доменах может возникать из сплайс вариаций, или может указывать на существование дополнительного β интегринового гена(нов).
На аминокислотном уровне мышиная сДНК предсказывает белок (SEQ ID NO:46) с 28% идентичностью мышиной CD11a, 53% идентичностью мышиной CD11b, 28% идентичностью человеческой CD11a, 55% идентичностью человеческой CD11b, 59% идентичностью человеческой CD11c и 70% идентичностью человеческой αd, Сравнение аминокислотных последовательностей цитоплазматических доменов человеческой αd и предполаемого мышиного гомолога указывает на области той же длины, но имеющие дивергентную первичную структуру. Аналогичная длина последовательности в этих областях подтверждает скорее изменение образца, чем сплайс форм варианта. Если сравнивать с предсказанным крысиным полипетидом Примера 16, выше, мышиные и крысиные цитоплазматические домены показывают больше, чем 60% идентичности.
Пример 20
Выделение дополнительных мышиных сДНК клонов для последующего подтверждения.
Для того, чтобы подтвердить нуклеиновые и аминокислотные последовательности, описанные в Примере 19 для мышиной αd, были выделены дополнительные мышиные последовательности с целью подтверждения.
Выделение мышиной сДНК гибридизацией двумя гомологичными αd пробами (3' и 5') было выполнено с использованием обеих библиотек и мышиной селезеночной статистической примированной библиотеки и олиго dT-примированной сДНК библиотеки в лабда ZAPll(Stratagen). Библиотка была культивирована при 5•105 фагов на 15 см LBM чашку. Бляшки были подняты на Hybond нейлоновой мембране (Amerscham) и мембраны были денатурированы (0.5 М NaOH/1.5 M NaCl), нейтрализованы (0.5 Трис основание/1.5 М NaCl/11.6 М HCl) и промыты (2Х SSC солевым раствором). ДНК была сшита с фильтрами с помощью ультрафиолетового излучения.
Пробы были генерированы с использованием праймеров, описанных ниже в PCR реакции при следующих условиях. Образцы выдерживали при 94oС в течение 4 минут и затем пропускали через 30 температурных циклов с последовательностью стадий (94oС в течение 15 секунд, 50oС в течение 30 секунд; 72oС в течение 1 минуты в аппарате Perkin-Elmer 9600 thermocycler).
3' проба имела длину приблизительно 900 оснований и простиралась в области от нуклеотидов 2752 до 3651 (в SEQ ID NO:1)(5'-3') и была продуцирована праймерами 11. b-1/2FOR11 и 11.b-1/2REV2, как показано в SEQ ID NO:69 и 74 соответственно. Эта проба была использована в первом наборе подъемов.
5' проба имела длину приблизительно 800 оснований и простиралась в области от нуклеотидов 149 до 946 (в SEQ ID NO:1)(5'-3') и была продуцирована праймерами 11.b-1/2FOR1 и 11.a-1/1REV1, как показано в SEQ ID NO:50 и 85 соответственно. Эта проба была использована во втором наборе подъемов.
В третьем наборе подъемов обе пробы, описанные выше, были использованы вместе на тех же самых чашках.
Скринированию были подвергнуты приблизительно 500000 бляшек с использованием двух приведенных выше проб, которые были мечены таким же образом, как описано в Примере 17. Меченые пробы были добавлены к раствору прегибридизации, содержащему 45% формамида, и проинкубированы в течение ночи при 50oС. Фильтры были дважды промыты 2Х SSC/0.1% SDS при комнатной температуре (22oС). Конечное промывание было проведено в 2Х SSC/0.1% SDS при 50oС. Авторадиография была проведена в течение 19 часов при -80oС на пленку Kodak X-Omat AR с интенсифицирующим экраном.
Тринадцать бляшек, дающие положительные сигналы на по крайней мере дупликатных подъемах, были подвергнуты вторичному скринированию, выполненному, как описано для первоначального скринирования за исключением того, что обе 3'и 5' меченые пробы были использованы для гибридизации и было введено дополнительное конечное промывание с использованием 2Х SSC/0.1% SDS при 65oС. Авторадиография была проведена, как описано выше, в течение 2.5 часов.
Тринадцать бляшек (обозначенных от MS2P1 до MS2P13), дающие положительные сигналы, были перенесены в фаговый разбавитель с низким содержанием Мg++. Размер инсерта был определен с помощью PCR амплификации (Perkin-Elmer 9600 thermocycler) с использованием праймеров Т3 и Т7, которые отжигали в Bluescript фагмиде в ZAP II (последовательность, описанную ранее) при тех же условиях, как показано выше. Размеры полос находились в области от 500 оснований до 4 kb. Т































































































































Изобретение относится к биотехнологии и иммунологии и может быть использовано в иммуногистохимическом анализе для определения локализации αd. Гибридому получают путем иммунизации хомячка очищенным крысиным αd и трансфектированными крысиным αd СНО клетками с последующим слиянием силеноцитов с NS-1 миеломными клетками и скринингом целевых клеток. Гибридома продуцирует моноклональное антитело, реактивное с α-субъединицей крысиного интегрина (αd). Изобретение позволяет определить локализацию αd в тканях. 2 с.п.ф-лы, 4 ил., 1 табл.
2. Моноклональное антитело, реактивное с α-субъединицей крысиного интегрина (αd), характеризующееся тем свойством, что оно секретируется гибридомой по п. 1.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |

