ССЫЛКА НА РАНЕЕ ПОДАННУЮ ЗАЯВКУ
По этой заявке испрашивается приоритет в соответствии с 35 статьей Кодекса США § 119(е) согласно предварительной заявке на патент US 61/604,998, поданной 29 февраля 2012 и имеющей название "АЗА-АРИЛ-1Н-ПИРАЗОЛ-1-ИЛ-БЕНЗОЛ-СУЛЬФОНАМИДЫ", которая включена полностью этой ссылкой.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Данное изобретение обеспечивает соединения и фармацевтические композиции, содержащие одно или более этих соединений или их фармацевтически приемлемые соли, которые эффективны в ингибировании связывания или функции различных хемокинов к рецепторам хемокинов. Как антагонисты или модуляторы рецепторов хемокинов соединения и композиции используются при лечении различных иммунных нарушения и заболеваний.
Хемокины, также известные как хемотаксисные цитокины, являются группой белков малой молекулярной массы, которые высвобождаются большим разнообразием клеток и имеют множество биологических действий. Хемокины привлекают различные типы клеток иммунной системы, такие как макрофаги, Т-клетки, эозинофилы, базофилы и нейтрофилы, и заставляют их мигрировать из крови в различные лимфоидные и нелимфоидные ткани. Они способствуют проникновению воспалительных клеток к местам воспаления и ответственны за инициирование и сохранение многих воспалительных заболеваний (см. Schall, Cytokine, 3:165-183 (1991), Schall et al., Curr. Opin. lmmunol, 6:865-873 (1994).
В дополнение к стимулированию хемотаксиса хемокины могут вызвать другие изменения в реагирующих клетках, включая изменения в форме клетки, экзоцитоз гранул, интегрин-регулирование, формирование биологически активных липидов (например, лейкотриенов), респираторный бурсит, связанный с активацией лейкоцитов, пролиферацию клеток, сопротивление индукции апоптоза и ангиогенеза.
Таким образом, хемокины - ранние пусковые механизмы воспалительного ответа, вызывающие высвобождение воспалительного медиатора, хемотаксис и кровоизлияние в местах инфекции или воспаления. Они также стимуляторы множества клеточных процессов, которые имеют важные физиологические функции, а также патологические последствия.
Хемокины проявляют свое влияние, активизируя рецепторы хемокинов, выраженные реагирующими клетками. Рецепторы хемокинов - это класс рецепторов, связанных с G-протеинами, также известные как семитрансмембранные рецепторы, обнаруженные на поверхности широкого разнообразия типов клеток, таких как лейкоциты, эндотелиальные клетки, гладкие мышечные клетки и опухолевые клетки.
Хемокины и рецепторы хемокинов выражаются присущими ренальными клетками и инфильтрующими клетками во время почечного воспаления (Segerer et al., J. Am. Soc. Nephrol., 11:152-76 (2000); Morii et al., J. Duabetes Complications, 17:11-5 (2003); Lloyd et al. J. Exp. Med., 185:1371-80 (1997); Gonzalez-Cuadrado et al. Clin. Exp. Immunol., 106:518-22 (1996); Eddy& Giachelli, Kidney Int., 47:1546-57 (1995); Diamond et al., Am. J. Physiol., 266: F926-33 (1994).
Т-лимфоцитная (Т-клеточная) инфильтрация в тонкую кишку и толстую кишку была связана с патогенезом брюшнополостных заболеваний, пищевых аллергий, ревматоидного артрита, воспалительных заболеваний кишечника (IBD) человека, которые включают заболевание Крона и язвенный колит. Блокирование транспорта соответствующих популяций Т-клеток в кишечник может привести к эффективному подходу к лечению IBD человека. Позже было отмечено, что рецептор-9 хемокина (CCR (9)) выражен на циркулирующих Т-клетках в периферической крови, взятой у пациентов с воспалениями тонкой кишки, такими как заболевание Крона и глютеиновое заболевание. Единственный CCR (9) лиганд, идентифицированный до настоящего времени, ТЕСК (тимус-выраженный хемокин), выражен и в тонкой и в толстой кишках, а пара рецептор-лиганд, как теперь полагают, играет основную роль в развитии IBD. В частности, эта пара обуславливает перемещения вызывающих заболевание воспалительных клеток в кишечник. (См. например, Zaballos et al., 5. Immunol., 162(10):5671-5675 (1999); Kunkel et al., J. Exp. Med, 192(5):761-768 (2000); Papadakis et al., J. Immunol., 165(9):5069-5076 (2000); Papadakis et al., Gastroenterology, 121(2):246-254 (2001); Campbell et al., J. Exp. Med., 195(1): 135-141 (2002); Wurbel et al., Blood, 98(9):2626-2632 (2001); and Uehara et al., J. Immunol, 168(6):2811-2819 (2002); Rivera-Nieves et al., Gastroenterology, 2006 Nov; 131(5): 1518-29; and Kontoyiannis et al., J. Exp. Med, Vol. 196, Number 12, Dec. 16, 2002).
Кроме того, CCR (9) несущие лимфоциты, как было показано, способствуют патологии филяриатоза (лимфатическое филяриозное заболевание), и ингибирование CCR (9) было коррелировано с уменьшением патологии, связанной с такими состояниями. (См. например, Babu et al., Journal of Infectious Diseases, 191:1018-26, 2005).
Идентификация соединений, которые модулируют функцию CCR (9), представляет привлекательную новую семью терапевтических агентов для лечения воспалительных и других состояний и заболеваний, связанных с CCR (9) активацией, таких как воспалительное заболевание кишечника.
US 2011/0130426 раскрывает соединения формулы I и их использование в медицинской терапии, такой как модуляция глюкокортикоидного рецептора у теплокровных животных:
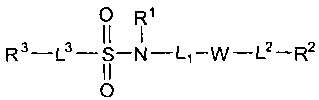
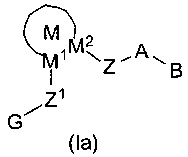
WO 02/00651 раскрывает соединения формулы (Ia) как ингибиторы подобных трипсину ферментов протеазы серина и способы их использования в качестве антикоагулянтных агентов для лечения и предотвращения тромбоэмболических нарушений:
КРАТКОЕ РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Данное изобретение направлено на соединения и их фармацевтически приемлемые соли, композиции и способы, используемые в реагировании активности хемокинов и активности рецепторов хемокинов. Соединения и их соли, композиции и способы, описанные здесь, полезны в лечении или предотвращении хемокин-обусловленных состояний или заболеваний, включая определенные воспалительные и иммунорегуляторные расстройства и болезни.
Соединения данного изобретения, как показано, модулируют один или более из CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR(9), CCR10, CCR11, CCR12, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CXCR6, CXCR7, CX3CR1, C5aR, chemR23, FPRL1, FPR1 и FPRL2. В частности, различные соединения данного изобретения регулируют CCR (9) как показано в примерах.
В одном воплощении представленные соединения могут быть представлены формулой (I) или ее солями:
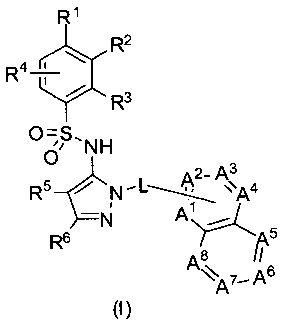
где R1, R2, R3, R4, R5, R6, L, А1, А2, А3, А4, А5, А6, А7 и А8 - как определены ниже.
В другом аспекте данное изобретение обеспечивает композиции, полезные в регулировании активности хемокинов. В одном воплощении композиция по данному изобретению включает соединение по изобретению и фармацевтически приемлемый носитель или наполнитель.
В еще одном аспекте данное изобретение обеспечивает способы регулирования функции хемокинов в клетке, включающие контакт клетки с терапевтически эффективным количеством соединения или композиции по изобретению.
В еще одном аспекте данное изобретение обеспечивает способы для регулирования функции хемокинов, включающие контакт рецептора хемокина с терапевтически эффективным количеством соединения или композиции по изобретению.
В еще одном аспекте данное изобретение обеспечивает способы лечения хемокин-опосредованных состояния или заболевания, включающие введение субъекту безопасного и эффективного количества соединения или композиции по изобретению. Введение может быть оральным, парентеральным, ректальным, трансдермальным, подъязычным, назальным или местным. В некоторых аспектах соединение может вводиться в комбинации с противовоспалительным или болеутоляющим агентом.
В дополнение к описанным здесь соединениям данное изобретение далее обеспечивает фармацевтические композиции, содержащие одно или более этих соединений, а также способы использования этих соединений в терапевтических способах, прежде всего для лечения заболеваний, связанных с сигнальной активностью хемокинов.
CCR (9) обусловленное заболевание или состояние является воспалительным заболеванием кишечника, аллергическим заболеванием, псориазом, атопическим дерматитом, астмой, фиброзными заболеваниями, отторжением трансплантатов, иммунно обусловленными пищевыми аллергиями, аутоиммунными заболеваниями, Глютеиновым заболеванием, ревматоидным артритом, тимомой, тимомной карциномой, лейкемией, твердой опухолью, острой лимфоцитарной лейкемией, меланомой, первичным склерозирующим холангитом, гепатитом, воспалительным заболеванием печени или постоперационной кишечной непроходимостью.
ПОДРОБНОЕ ОПИСАНИЕ
Общее
Данное изобретение направлено на соединения и их соли, композиции и способы, используемые в регулировании функции рецептора хемокина, особенно CCR (9) функции. Регулирование активности рецептора хемокина, как здесь используется в различных формах, охватывает антагонизм, агонизм, частичный антагонизм, инверсный агонизм и/или частичный агонизм активности, связанной с особым рецептором хемокина, предпочтительно CCR (9) рецептором. Соответственно, соединения данного изобретения - это соединения, которые регулируют, по крайней мере, одну функцию или признак CCR (9) млекопитающих, например, человеческий CCR (9) белок.
Способность соединения регулировать функцию CCR (9) может быть продемонстрирована в анализе связывания (например, связывания лиганда или связывания агониста), хемотаксисе (анализе перемещения), сигнальном анализе (например, активации G-белка млекопитающих, индукции быстрого и переходного увеличения концентрации цитозольного свободного кальция) и/или анализе клеточного ответа (например, стимуляции хемотаксиса, экзоцитоза или высвобождения воспалительного медиатора лейкоцитами).
Сокращения и Определения
При описании соединений, композиций, способов и процессов этого изобретения следующие термины имеют следующие значения, если не указано иначе.
Термин "алкил" отдельно или как часть другого заместителя относится к группе углеводородов, которая может быть линейной, циклической или разветвленной или их комбинацией, имеющей определенное число атомов углерода (то есть, С1-8 означает один-восемь атомов углерода).
Термин "циклоалкил" отдельно или как часть другого заместителя относится к циклической алкил-группе, имеющей определенное число углерода, и является подмножеством термина "алкильный". Другие подмножества термина "алкильный" включают "линейные" и "разветвленные" алкильные группы, которые относятся к двум различным типам нециклических алкильных групп. Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, sec-бутил, циклогексил, циклопентил, (циклогексил) метил, циклопропилметил, бицикло [2.2.1] гептан, бицикло-[2.2.2] октан и т.д. В этом списке примеров алкильные примеры метила, этила, н-пропила и н-бутила - также примеры линейных алкил-групп. Точно также изопропил и трет-бутил также являются примерами разветвленных алкил-групп. Циклопентил, циклогексил, (циклогексил) метил, циклопропилметил, бицикло [2.2.1] гептан, бицикло [2.2.2] октан являются примерами циклоалкил-групп. В некоторых воплощениях циклопропил может использоваться в качестве связывающей группы между двумя другими частями и представлен как -СН(СН2)СН-.
Алкил-группы могут быть замещенными или незамещенными, если иначе не обозначено.
Примеры замещенного алкила включают галоалкил, тиоалкил, аминоалкил и т.п. Дополнительные примеры подходящих алкил заместителей содержат, но не ограничиваются, гидроксиизопропил, -С(СН3)2-O, аминометил, 2-нитрометил, 4-цианобутил, 2,3-дихлорпентил, 3-гидрокси-5-карбоксигексил, 2-аминоэтил, пентахлорэтил, трифторметил, 2-диэтиламиноэтил, 2-диметиламинопропил, этоксикарбонилметил, метанилсульфанилметил, метоксиметил, 3-гидроксипентил, 2-карбоксибутил, 4-хлорбутил и пентафторэтил.
"Алкокси" относится к -О-алкилу. Примеры алкокси-группы включают метокси, этокси, н-пропокси и т.д. Алкильная часть алкокси может быть алкилом из от 1 до 16 углерода и в некоторых воплощениях от 1 до 8 углерода.
"Алкенил" относится к ненасыщенной группе углеводорода, которая может быть линейной, цикличной или разветвленной или их комбинацией. Группы алкенила с 2-8 атомами углерода предпочтительны. Алкенил-группа может содержать 1, 2 или 3 двойные связи углерод-углерод. Примеры алкенил-групп включают этенил, н-пропенил, изопропенил, н-бут-2-энил, н-гекс-3-энил, циклогексенил, циклопентенил и т.п. Группы алкенила могут быть замещенными или незамещенными, если иначе не указано.
"Алкинил" относится к ненасыщенной углеводородной группе, которая может быть линейной, цикличной или разветвленной или их комбинацией. Группы алкинила с 2-8 атомами углерода предпочтительны. Алкинил-группа может содержать 1, 2 или 3 углерод-углерод тройные связи. Примеры алкинил-групп включают этинил, н-пропинил, н-бут-2-инил, н-гекс-3-инил и т.п. Группы алкинила могут быть замещенными или незамещенными, если иначе не указано.
"Алкиламино" относится к -N (алкил)2 или -NH (алкил). Когда алкиламино-группа содержит две алкил-группы, они могут быть объединены вместе, чтобы сформировать карбоциклическое или гетероциклическое кольцо. Понятно, что алкил-группы, входящие в алкиламино-группу, могут быть замещенными или незамещенными. Примеры алкиламино-группы включают метиламино, трет-бутиламино, диметиламино, ди-изопропиламино, морфолино и т.п.
"Аминоалкил" как замещенная алкил-группа относится к группе моноаминоалкила или группе полиаминоалкила, которые наиболее типично замещены 1-2 амино-группами. Примеры включают аминометил, 2-аминоэтил, 2-диэтиламиноэтил и т.п.
"Арил" относится к полиненасыщенной ароматической группе углеводорода, имеющей единственное кольцо (моноциклический) или множественные кольца (бициклический), которые могут быть сплавлены вместе или связаны ковалентно. Арил-группы с 6-10 атомами углерода предпочтительны, при этом, число атомов углерода может указываться как С6-10, например. Примеры арил-групп включают фенил и нафтален-1-ил, нафтален-2-ил, бифенил и т.п. Арил-группы могут быть замещенными или незамещенными, если иначе не указано.
Замещенный арил может быть замещен одним или более заместителями. Подходящие заместители для арила включают замещенный или незамещенный C1-8 алкил и заместители, как обсуждалось выше для замещенного алкила.
"Гало" или "галоген", отдельно или как часть заместителя, относится к атому хлора, брома, иода или фтора.
"Галоалкил" как группа замещенного алкила относится к моногалоалкил- или полигалоалкил-группе, наиболее типично замещенной 1-3 атомами галогена. Примеры включают 1-хлорэтил, 3-бромпропил, трифторметил и т.п.
"Гетероциклил" относится к насыщенному или ненасыщенному неароматическому кольцу, содержащему, по крайней мере, один гетероатом (обычно 1-5 гетероатомов), выбранных из азота, кислорода или серы. Гетероциклическое кольцо может быть моноцикличным или бицикличным. Предпочтительно эти группы содержат 0-5 атомов азота, 0-2 атома серы и 0-2 атома кислорода, при условии, что присутствует, по крайней мере, один гетероатом. В некоторых воплощениях эти группы содержат 0-3 атома азота, 0-1 атом серы и 0-1 атом кислорода.
Примеры гетероциклических групп включают пирролидин, пиперидин, имидазолидин, пиразолидин, бутиролактам, валероактам, имидазолидинон, гидантоин, диоксолан, фталимид, пиперидин, 1,4-диоксан, морфолин, тиоморфолин, тиоморфолин-S-оксид, тиоморфолин-S,S-диоксид, пиперазин, пиран, пиридон, 3-пирролин, тиопиран, пирон, тетрагидрофуран, тетрагидротиофен, хинуклидин и т.п.Предпочтенные гетероциклические группы моноцикличны, хотя они могут быть сплавлены или связаны ковалентно с арилом или кольцевой системой гетероарила.
В определениях, приведенных выше, подходящие заместители для замещенного алкила, алкенила и алкинила включают: галоген, -CN, -CO2R', -C(O)R', -C(O)NR'R", оксо (=O или -O"), -OR', -OC(O)R', -OC(O)NR'R"-NO2, -NR'C(O)R", - NR"'C(O)NR'R", -NR'R",-NR'CO2R", -NR(O)R", -NR(O)2R"', -NR"'S(O)NR'R",-NR"'S(O)2NR'R", -SR', -S(O)R', -S(O)2R', -S(O)2NR'R", - NR'-C(NHR")=NR"', -SiR'R"R"', -OSiR'R"R"', -N3, замещенный или незамещенный С6-10арил, замещенный или незамещенный 5-10-членный гетероарил и замещенный или незамещенный 3-10-членный гетероциклил. Число возможных заместителей колеблется от ноля до (2m'+1), где m' является общим количеством атомов углерода в таком радикале. Относительно замещенного алкила R', R" и R'" - каждый независимо относится к множеству групп, включая водород, замещенному или незамещенному С1-8 алкилу, замещенному или незамещенному С2-8 алкенилу, замещенному или незамещенному С2-8 алкинилу, замещенному или незамещенному арилу, замещенному или незамещенному гетероарилу, замещенному или незамещенному гетероциклилу, замещенному или незамещенному арилалилу, замещенному или незамещенному арилоксиалкилу.
Когда R' и R" присоединены к тому же самому атому азота, они могут быть объединены с атомом азота, чтобы сформировать 3-, 4-, 5-, 6- или 7-членное кольцо (например, -NR'R" включает 1-пирролидинил и 4-морфолинил). Кроме того, R' и R", R" и R" ' или R' и R"' могут вместе с атомом (ами), к которому они присоединены, формировать замещенное или незамещенное 5-, 6- или 7-членное кольцо.
В одном предпочтительном воплощении гетероциклические группы могут быть представлены формулой (АА) ниже:
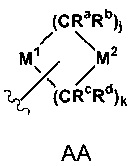
где формула (АА) присоединена через свободную валентность или на М1 или на М2;
М1 представляет О, NRe или S(О)1;
М2 представляет CRfRg, О, S (О)1 или NRe; где может быть необходимо опустить один Rf,
Rg или Ре, чтобы создать свободную валентность на М1 или М2 такую как, например, CRf,
CRg или N;
l - это 0, 1 или 2;
j - 1, 2 или 3, и
k - 1, 2 или 3, при условии, что j+k - 3, 4 или 5; и Ra, Rb, Rc, Rd, Re, Rf и Rg независимо выбраны из группы, состоящей из водорода, галогена, замещенного или незамещенного С1-8 алкила, замещенного или незамещенного С2-8 алкенила, замещенного или незамещенного С2-8 алкинила,
-CORh, -CO2Rh, -CONRhRi, -NRhCORi, -SO2Rh, -SO2NRhRi, -NRhSO2R',
-NRhRi, -ORh, -SiRhRiRj, -OSiRhRiRj, -Q1CORh, -Q1CO2Rh, -Q1CONRhRi,
-Q1NRhCORi, -Q1SO2Rh, -Q1SO2NRhRi, -Q1NRhSO2Ri, -Q1NRhRi, -Q1ORh,
где Q1 - член, выбранный из группы, состоящей из С1-4 алкилена, С2-4 алкенилена и С2-4 алкинилена, и
Rh, Ri и Rj независимо выбраны из группы, состоящей из водорода и С1-8 алкила, и где алифатические части каждого Ra, Rb, Rc, Rd, Re, Rf, Rg Rh, Ri и R заместителя произвольно заменены от одного до трех членами, выбранными из группы, состоящей из галогена, - О, -ORn, -OC(O)NHRn, -OC(O)NRnRo, -SH, -SRn, -S(O)Rn, -S(O)2Rn, -SO2NH2, -S(O)2NHRn, -S(O)2NRnRo, -NHS(O)2Rn, -NRnS(O)2Ro, -С(O)NH2, -С(O)NHRn, -С(O)NRnRo, -С(O)Rn, -NHC(O)Ro, -NRnC(O)Ro, -NHC(O)NH2, -NRnC(O)NH2, -NRnC(O)NHRo, -NHC(O)NHRn, -NRnC(O)NRoRp, -NHC(O)NRnRo, -CO2H, -CO2Rn, -NHCO2Rn, -NRnCO2Ro, -CN, -NO2, -NH2, -NHRn, -NRnRo, -NRnS(O)NH2 и -NRnS(O)2NHRo,
где Rn, Ro и Rp являются независимо незамещенным C1-8 алкилом. Дополнительно любые два из Ra, Rb, Rc, Rd, Re, Rf и Rg могут быть объединены, чтобы сформировать мостиковую или спироциклическую кольцевую систему.
В другом предпочтительном воплощении некоторые из Ra+Rb+Rc+Rd групп, которые отличны от водорода, это 0, 1 или 2.
В более предпочтительном воплощении Ra, Rb, Rc, Rd, Re, Rf и Rg независимо выбраны из группы, состоящей из водорода, галогена, незамещенного или замещенного С1-8 алкила, -CORh, -CO2Rh, -CONRhRh, -NRhCORh, -SO2Rh, -SO2NRhRi, -NSO2RhRi, -NRhRi и -ORh, где Rh и Ri независимо выбраны из группы, состоящей из водорода и незамещенного C1-8 алкила, и где алифатические части каждого из Ra, Rb, Rc, Rd, Re, Rf и Rg заместителей произвольно заменены от одного до трех членами, выбранными из группы, состоящей из галогена, - ОН, -ORn, -OC(O)NHRn, -OC(O)NRnRo, -SH, -SRn, -S(O)Ro, -S(O)2Rn, -SO2NH2, -S(O)2NHRn, -S(O)2NRnRo, -NHS(O)2Rn, -NRnS(O)2Ro, -C(O)NH2, -C(O)NHRn, -C(O)NRnRo, -C(O)Rn, -NHC(O)Rn, -NRnC(O)Ro, -NHC(O)NH2, -NRnC(O)NH2, -NRnC(O)NHRo, -NHC(O)NHRn, -NRnC(O)NRoRp, -NHC(O)NRnRo, -CO2H, -CO2Rn, -NHCO2Rn, -NRnCO2Ro, -CN, -NO2, -NH2, -NHRn, -NRnRo, -NRnS(O)NH2 и -NRnS(O)2NHRo, где Rn, Ro и Rp являются независимо незамещенным C1-8 алкилом.
В более предпочтительном воплощении Ra, Rb, Rc, Rd, Re, Rf и Rg - это независимо водород или C1-4алкил.
В другом предпочтительном воплощении, по крайней мере, три из Ra, Rb, Rc, Rd, Re, Rf и Rg - это водород.
"Гетероарил" относится к ароматической группе, содержащей, по крайней мере, один гетероатом, где гетероарил-группа может быть моноциклической или бициклической. Примеры включают пиридил, пиридазинил, пиразинил, пиримидинил, триазинил, хинолинил, хиноксалинил, хиназолинил, циннолинил, фталазинил, бензотриазинил, пуринил, бензопиразолил, бензотриазолил, бензизоксазолил, изобензоофурил, изоиндолил, индолизинил, бензотриазинил, тиенопиридинил, тиенопиримидинил, пиразолопиримидинил, имидазопиридины, бензотиазолил, бензофуранил, бензотиенил, индолил, азаиндолил, азаиндазолил, хинолил, изохинолил, изотиазолил, пиразолил, индазолил, птеридинил, имидазолил, триазолил, тетразолил, оксазолил, изоксазолил, оксадиазолил, тиадиазолил, пирролил, тиазолил, фурил или тиенил. Предпочтенные гетероарил группы - это те, которые имеют, по крайней мере, один атом азота с арильным кольцом, такие как хинолинил, хиноксалинил, пуринил, бензимидазолил, бензопиразолил, бензотриазолил, бензотиазолил, индолил, хинолил, изохинолил и т.п.
Предпочтительные гетероарил-системы с 6 кольцами включают пиридил, пиридазинил, пиразинил, пиримидинил, триазинил и т.п.
Предпочтительные гетероарил-системы с 5 кольцами включают изотиазолил, пиразолил, имидазолил, тиенил, фурил, триазолил, тетразолил, оксазолил, изоксазолил, оксадиазолил, тиадиазолил, пирролил, тиазолил и т.п.
Гетероциклил и гетероарил могут быть присоединены в любом доступном кольцевом углероде или гетероатоме. Каждый гетероциклил и гетероарил могут иметь одно или более колец. Когда присутствуют многократные кольца, они могут быть сплавлены вместе или связаны ковалентно. Каждый гетероциклил и гетероарил должны содержать, по крайней мере, один гетероатом (как правило, 1-5 гетероатомов), выбранный из азота, кислорода или серы. Предпочтительно эти группы содержат 0-5 атомов азота, 0-2 атома серы и 0-2 атома кислорода. Более предпочтительно эти группы содержат 0-3 атома азота, 0-1 атом серы и 0-1 атом кислорода. Гетероциклил и гетероарил группы могут быть замещенными или незамещенными, если иначе не указано. Для замещенных групп замена может быть на углероде или гетероатоме. Например, когда замена - оксо(=O или -О-), полученная группа может иметь как карбонил (-С(О)-), так и N-оксид (-N+-O-).
Подходящие заместители для замещенного алкила, замещенного алкенила и замещенного алкинила включают галоген, -CN, -CO2R', -С(О)R', -С(О)NR'R", оксо(=O или -О-), -OR', -OSiR'R "R"', -OC(O)R', -OC(O)NR'R' -NO2, -NR'C(O)R", - NR' "C(O)NR'R", -NR'R", -NR'CO2R", - NR(O)R", -NR(O)2R"', -NR"'S(O)NR'R", -NR"'S(O)2NR'R", -SR', -S(O)R', -S(O)2R', -S(O)2NR'R", -NR'-C(NHR")=NR"', -SiR'R"R"', -N3, замещенный или незамещенный С6-10 арил, замещенный или незамещенный 5- 10-членный гетероарил и замещенный или незамещенный 3-10-членный гетероциклил. Число возможных заместителей колеблется от ноля до (2m'+1), где m' является общим количеством атомов углерода в таком радикале.
Подходящие заместители для замещенного арила, замещенного гетероарила и замещенного гетероциклила включают галоген, -CN, -CO2R', -С(О)R', -С(О)NR'R", охо(=O или -О"), -OR', -OSiR'R"R"', -OC(O)R', -OC(O)NR'R", -NO2, -NR'C(O)R", -NR"'C(O)NR'R", -NR'R", -NR'CO2R", -NR(O)R", -NR(O)2R", -NR"'S(O)NR'R", -NR"'S(O)2NR'R", -SR', -S(O)R', -S(O)2R', -S(O)2NR'R", -NR'-C(NHR")=NR"', -SiR'R"R"', -N3, замещенный или незамещенный C1-8алкил, замещенный или незамещенный С2-8 алкенил, замещенный или незамещенный С2-8, алкинил, замещенный или незамещенный С6-10 алкил, замещенный или незамещенный 5-10-членный гетероарил и замещенный или незамещенный 3-10-членный гетероциклил. Число возможных заместителей колеблется от ноля до общего количества открытых валентностей на ароматической кольцевой системе.
Как использовалось выше, R', R" и R'" каждый независимо относится ко множеству групп, включая водород, замещенный или С1-8 алкил, замещенный или незамещенный С2-8 алкенил, замещенный или незамещенный С2-8 алкинил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный гетероциклил, замещенный или незамещенный арилалкил, замещенный или незамещенный арилоксиалкил. Когда R' и R" присоединены к тому же самому атому азота, они могут быть объединены с атомом азота, чтобы сформировать 3-, 4-, 5-, 6- или 7-членное кольцо (например, -NR'R" включает 1-пирролилинил и 4-морфолинил). Кроме того, R' и R", R" и R"' или R' и R'" могут вместе с атомом (ами), к которому они присоединены, формировать замещенное или незамещенное 5-, 6- или 7-членное кольцо.
Два из заместителей на смежных атомах арила или кольца гетероарила могут быть произвольно заменены заместителем формулы -T-C(O)-(CH2)q-U-, где Т и U независимо -NR""-, -О-, -СН2- или единственная связь, и q - целое число от 0 до 2. Альтернативно два из заместителей на смежных атомах арила или кольца гетероарила могут быть произвольно заменены заместителем формулы -А'-(СН2)r-В'-, где А' и В' независимо -СН2-, -О-, - NR""-, -S-, -S(О) -, S(O)2-, -S(O)2NR""- или единственная связь, и r - целое число от 1 до 3. Одна из единственных связей нового кольца, таким образом сформированного, может быть произвольно заменена двойной связью. Альтернативно два из заместителей на смежных атомах арила или кольца гетероарила могут быть произвольно заменены заместителем формулы - (CH2)s-X-(CH2)r, где s и t - независимо целые числа от 0 до 3, и XIV это -О-, - NR""-, -S-, -S(O)-, S(O)2- или -S(O)2NR'-.
R"" выбран из водорода или незамещенного C1-8 алкила.
"Гетероатом" означает включающий кислород (О), азот (N), серу (S) и кремний (Cи).
"Фармацевтически приемлемый" носитель, разжижитель или наполнитель - это носитель, разжижитель или наполнитель, совместимый с другими компонентами состава и не вредный для его потребителя.
"Фармацевтически приемлемая соль" относится к соли, которая является приемлемой для введения пациенту, такому как млекопитающее (например, соли, имеющие приемлемую безопасность для млекопитающих для данного режима дозировки). Такие соли могут быть получены из фармацевтически приемлемых неорганических или органических оснований и из фармацевтически приемлемых неорганических или органических кислот, в зависимости от особых заместителей, обнаруженных на соединениях, описанных здесь. Когда соединения данного изобретения содержат относительно кислотные функциональности, основные дополнительные соли могут быть получены путем соединения с нейтральной формой таких соединений с достаточным количеством желаемой основы, или неразбавленной или в подходящем инертном растворителе. Соли, полученные из фармацевтически приемлемых неорганических оснований, включают соли алюминия, аммония, кальция, меди, соединений окиси железа, железистую, лития, магния, марганцовистую, калия, натрия, цинка и т.п. Соли, полученные из фармацевтически приемлемых органических оснований, включают соли основных, вторичных, третичных и четвертичных аминов, включая замещенные амины, циклические амины, естественные амины и т.п., такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, смолы полиамина, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, тромэтамин и т.п. Когда соединения данного изобретения содержат относительно основные функциональности, кислотные дополнительные соли могут быть получены путем соединения с нейтральной формой таких соединений с достаточным количеством желаемой кислоты, или нерастворенной или в подходящем инертном растворителе. Соли, полученные из фармацевтически приемлемых кислот, включают уксусную, аскорбиновую, бензолсульфоновую, пуринилойную, камфосульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глюкороновую, глутаминовую, гиппуриновую, гидробромистую, хлористоводородную, изотионовую, молочную, лактобионовую, малеиновую, яблочную, манделиновую, метансульфоновую, слизевую, нафталенсульфоновую, никотиновую, азотную, пальминовую, пантотеническую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую и т.п. В некоторых воплощениях соединения включают дополнительную соль натрия.
Также включены соли аминокислот, такие как аргинат и т.п. и соли органических кислот как глюкуроновой или галактуроновой и т.п. (см., например, Berge, S.M. et al, "Pharmaceutical Salts", J. Pharmaceutical Science, 1977, 66:1-19). Определенные специфические соединения данного изобретения содержат и основные и кислые функциональности, которые позволяют соединениям преобразовываться или в основные или в кислотные дополнительные соли.
Нейтральные формы соединений могут быть восстановлены путем соединения с солью с основанием или кислотой и путем изоляции исходного соединения обычным образом. Исходная форма соединения отличается от различных солевых форм определенными физическими свойствами, такими как растворимость в полярных растворителях, но в других отношениях соли эквивалентны исходной форме соединения в целях данного изобретения.
"Его соль " относится к соединению, полученному, когда водород кислоты заменяется катионом, таким как катион металла или органический катион и т.п. Предпочтительно, соль - фармацевтически приемлемая соль, хотя это не требуется для солей промежуточных соединений, которые не предназначаются для введения пациенту.
В дополнение к солевым формам данное изобретение обеспечивает соединения, которые находятся в форме пролекарства. Пролекарства часто полезны, потому что в некоторых ситуациях их легче вводить, чем исходный препарат. Они могут, например, быть биодоступными пероральным приемом, тогда как исходный препарат нет. Пролекарство может также улучшать растворимость в фармацевтических композициях по сравнению с исходным препаратом. Большое разнообразие производных пролекарств известно в уровне техники, такие как те, которые отвечают на гидролитическое расщепление или окислительную активацию пролекарства. Примером пролекарства, без ограничения, могло бы быть соединение данного изобретения, которое вводится как сложный эфир ("пролекарство"), но затем он метаболически гидролизуется в карбоксильную кислоту, активный элемент. Дополнительные примеры включают пептидил производные соединения изобретения.
Пролекарства соединений, описанных здесь, являются теми соединениями, которые легко претерпевают химические изменения при физиологических условиях для обеспечения соединений данного изобретения. Дополнительно пролекарства могут быть преобразованы в соединения данного изобретения химическими или биохимическими способами in или ех vivo окружающих условиях. Например, пролекарства могут медленно преобразовываться в соединения данного изобретения, когда помещены в резервуар трансдермального пластыря с подходящим ферментом или химическим реагентом.
Пролекарства могут быть получены путем изменения функциональных групп, присутствующих в соединениях таким образом, что модификации расщепляются, или в обычном манипулировании или in vivo, в исходные соединения. Пролекарства включают соединения, где гидроксил, амино, сульфидил или группы карбоксила связаны с любой группой, которая при введении млекопитающему субъекту расщепляется, чтобы сформировать свободный гидроксил, амино, сульфидил или группу карбоксила, соответственно. Примеры пролекарств включают, но без ограничения, ацетат, формиат и бензоат производные спирта и функциональные группы амина в соединениях изобретения. Получение, выбор и использование пролекарств обсуждаются в Т. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the A.C.S. Symposium Series; "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985; and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, каждый из которых включен здесь ссылкой во всей своей полноте.
Соединения изобретения могут присутствовать в форме их фармацевтически приемлемых метаболитов. Термин "метаболит" означает фармацевтически приемлемую форму метаболической производной соединения изобретения (или его соли). В некоторых аспектах метаболит может быть функциональной производной соединения, которая легко преобразуется in vivo в активное соединение. В других аспектах метаболит может быть активным соединением.
Термин "кислотные изостеры" означает, если иначе не указано, группу, которая может заменить карбоксильную кислоту, имеющую кислотную функциональность и стерические и электронные характеристики, которые обеспечивают уровень активности (или другую характеристику соединения, такую как растворимость), подобный карбоксильной кислоте. Представительные кислотные изостеры включают: гидроксаминовые кислоты, сульфоновые кислоты, сульфиновые кислоты, сульфонамиды, асцил-сульфонамиды, фосфоновые кислоты, фосфиновые кислоты, фосфорные кислоты, тетразол и оксо-оксадиазолы.
"Терапевтически эффективное количество" относится к количеству, достаточному для эффективного лечения, когда вводится нуждающемуся в лечении пациенту.
"Лечащий" или "лечение", как здесь используются, относятся к лечению болезни или заболеванию (такому как вирусная, бактериальная или грибковая инфекция или другие инфекционные заболевания, а также такому как аутоиммунные или воспалительные заболевания) у пациента, такого как млекопитающее (особенно человек или домашнее животное), которое включает улучшение болезни или медицинского состояния, т.е. устраняет или вызывает регресс болезни или медицинского состояния у пациента; подавление болезни или медицинского состояния, т.е. замедление или прекращение развития болезни или медицинского состояния у пациента; или облегчение симптомов болезни или медицинского состояния у пациента.
Определенные соединения данного изобретения могут существовать в несольватных формах, а также в сольватных формах, включая гидратированные формы. Вообще, и сольватные и несольватные формы охвачены объемом данного изобретения. Определенные соединения данного изобретения могут существовать во множественных кристаллических или аморфных формах (т.е. как полиморфы). Вообще, все физические формы являются эквивалентными для использования, рассмотренного данным изобретением, и охвачены рамками данного изобретения.
Для специалиста будет очевидно, что определенные соединения данного изобретения могут существовать в таутомерных формах; все такие таутомерные формы соединений охвачены рамками изобретения. Например, некоторые соединения, имеющие гетероарил, могут быть заменены одной или более гидроксильными группами. Таутомерные формы поэтому будут включать оксо-заместители. Определенные соединения данного изобретения обладают асимметричными углеродными атомами (оптические центры) или двойными связями; рацематы, диастереомеры, геометрические изомеры и отдельные изомеры (например, отдельные энантиомеры) все охвачены рамками данного изобретения. Соединения данного изобретения могут также содержать неестественные пропорции атомных изотопов на одном или более атомах, которые составляют такие соединения. Например, соединения могут быть радиомеченными радиоактивными изотопами, такими как, например, тритием (3Н), иодом 125 (125I) или углеродом 14 (14С). Все изотопные варианты соединений данного изобретения, радиоактивные или нет, охвачены рамками данного изобретения.
Соединения данного изобретения могут включать обнаружимую метку. Обнаружимая метка - это группа, которая обнаружима при низких концентрациях, обычно меньше чем микромолярная, возможно меньше чем наномолярная и возможно меньше чем пикомолярная, и она может быть легко отличима от других молекул из-за различий в молекулярных свойствах (например, молекулярной массе, отношения массы к заряду, радиоактивности, окислительно-восстановительного потенциала, люминесценции, флюоресценции, электромагнитных свойств, свойств связывания и т.п.). Обнаружимые метки могут быть обнаружены спектроскопическими, фотохимическими, биохимическими, иммунохимическими, электрическими, магнитными, электромагнитными, оптическими или химическими средствами и т.п.
Большое разнообразие обнаружимых меток в рамках данного изобретения, включая гаптеновые метки (например, биотин или метки, используемые в соединении с обнаружимыми антителами, такими как антитела пероксидазы конской редьки); метки массового признака (например, устойчивые метки изотопа); радиоизотопные метки (включая 3Н, 125I, 35, 14С или 32Р); металлические хелатные метки; люминесцентные метки, включая флуоресцентные метки (такие как флюоресцеин, изотиоцианат, Техасский красный, родамин, зеленый флуоресцентный белок и т.п.), фосфоресцирующие метки и хемилюминесцентные метки, как правило, имеющие квантовый выход более чем 0.1; электроактивные и метки передачи электрона; метки модулятора фермента, включающие коэнзимы, металлоорганическую каталитическую пероксидазу конской редьки, щелочную фосфатазу и другие, обычно используемые в ELISA; фотосенсибилизаторы; магнитные шариковые метки, включая Dynabeads; колориметрические метки, такие как коллоидное золото, серебро, селен или другие металлы и метки металлической соли (см. патент US 5 120 643, который здесь включается ссылкой полностью для всех целей), или шариковые метки из окрашенного стекла или пластмассы (например, пенопласта, полипропилена, латекса и т.д.) и сажевые метки. Патенты, раскрывающие использование таких обнаружимых меток, включают патенты US N№3 817 837; 3 850 752; 3 939 350; 3 996 345; 4 277 437; 4 275 149; 4 366 241; 6 312 914; 5 990 479; 6 207 392; 6 423 551; 6 251 303; 6 306 610; 6 322 901; 6 319 426; 6 326 144 и 6 444 143, которые здесь включаются ссылкой в их полноте для всех целей.
Обнаружимые метки коммерчески доступны или могут быть получены как известно специалистам области. Обнаружимые метки могут быть ковалентно присоединены к соединениям, используя реактивную функциональную группу, которая может быть расположена в любом соответствующем положении. Способы присоединения обнаружимой метки известны специалистам области. Когда реактивная группа присоединена к алкилу или замещенной алкильной связи, ограниченной арилзамещенным ядром, реактивная группа может быть расположена в предельном положении алкильной цепи.
Соединения
Данное изобретение обеспечивает соединения, которые регулируют деятельность CCR (9). Рецепторы хемокина - встроенные мембранные белки, которые взаимодействуют с внеклеточным лигандом, таким как хемокин, и добиваются клеточного ответа на лиганд, например, хемотаксис, увеличенную внутриклеточную концентрацию ионов кальция, и т.д. Поэтому модуляция функции рецептора хемокина, например, вмешательство во взаимодействие рецептора хемокина с лигандом, будет модулировать опосредованный ответ рецептора хемокина и лечить или предотвращать обусловленное рецептором хемокина состояние или заболевание. Модуляция функции рецептора хемокина включает как стимулирование, так и ингибирование функции. Тип достигнутой модуляции будет зависеть от особенностей соединения, т.е. антагонист или полный, частичный или обратный агонист.
Например, соединения этого изобретения действуют как мощные CCR (9) антагонисты, и эта антагонистическая активность была далее подтверждена в экспериментах на животных для воспаления, одного из болезненных состояний признака для CCR (9). Соответственно, соединения обеспеченные здесь, используются в фармацевтических соединениях, способах лечения CCR (9) - опосредованных заболеваний и как средства контроля в анализе для идентификации конкурентных CCR (9) антагонистов.
В формулах, приведенных ниже, когда переменная появляется более чем однажды в одной формуле, она может быть той же самой или отличающейся. Например, в формуле (I) один R8 может быть -NH2, а остаток может быть водородом.
В одном воплощении соединения данного изобретения представляются формулой (I), или ее солями:
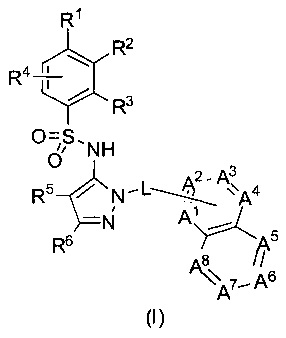
где R1 выбран из группы, состоящей из замещенного или незамещенного С2-8 алкила, замещенного или незамещенного С1-8 алкокси, замещенного или незамещенного C1-8 алкиламино и замещенного или незамещенного С3-10гетероциклила;
R2 - это Н, F, Cl, замещенный или незамещенный С1-8 алкокси; или R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют неароматическое карбоциклическоке кольцо или гетероциклическое кольцо;
R3 - это H, замещенный или незамещенный C1-8 алкил; замещенный или незамещенный C1-8 алкокси или гало; R4 - это Н или F; R5-Н, F, Cl, или -СН3; R6 - это Н, гало, -CN,
-CO2Ra, -CONH2, -NH2, замещенный или незамещенный C1-8 алкил, замещенный или незамещенный C1-8 алкокси, замещенный или незамещенный C1-8 аминоалкил;
Ra - это Н или замещенный или незамещенный С1-8 алкил;
R5 и R6 могут вместе формировать карбоциклическое кольцо;
L - связь или -CH2-, или -СН(СН3)-;
каждый из А1, А2, А3, А4, А5, А6, А7, и А8 независимо выбирается из группы, состоящей из N, N-O, и -CR8-; где, по крайней мере, один и не более чем два из А1, А2, А3, А4, А5, А6, А7 и А8 - это N или N-O;
R8 каждый независимо выбирается из группы, состоящей из Н, гало, -CN, -ОН, оксо, замещенного или незамещенного С1-8 алкила, замещенного или незамещенного C1-8 алкокси, и -NR20R21, замещенного или незамещенного арила, замещенного или незамещенного гетероарила и замещенного или незамещенного гетероциклила, и R20 и R21 - каждый независимо - Н или замещенный или незамещенный C1-8 алкил.
В одном воплощении формулы (I) R1 - замещенный или незамещенный С2-8 алкил; предпочтительно R1 - трет-бутил; R2, R3, R4 и R5 - Н; R6 замещенный или незамещенный С1-8 алкил, замещенный или незамещенный С1-8 алкокси, -CN,-CONH2, -NH2 или С1-8 аминоалкил; предпочтительно R6 - незамещенный С1-8 алкил или С1-8 галоалкил; более предпочтительно R6- это СН3, -CH2F, -CHF2 или -CF3; L - связь; а А1, А2, А3, А4, А3, А6, А7, А8 и R8 - как определяет формула (I).
В другом воплощении формулы (I), R1 замещенный или незамещенный С2-8 алкил; предпочтительно R1 - трет-бутил; R2 - F; R3, R4 и R5 - Н; R6 замещенный или незамещенный C1-8 алкил, замещенный или незамещенный С1-8 алкокси, -CN, -CONH2, -NH2, или замещенный или незамещенный C1-8 аминоалкил; предпочтительно R6 незамещенный C1-8 алкил или С1-8 галоалкил; более предпочтительно R6 - СН3, -CH2F, -CHF2 или -CF3; L - связь; а А1, А2, А3, А4, А5, А6, А7, А8 и R8 как определяет формула (I).
В одном воплощении соединения формулы (I) данного изобретения представляются формулой (II) или ее солями:
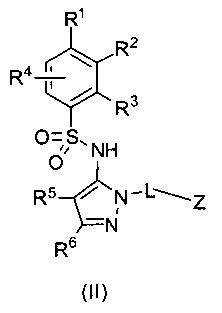
где R1 выбирается из группы, состоящей из замещенного или незамещенного С2-8 алкила, замещенного или незамещенного C1-8 алкокси, замещенного или незамещенного С1-8 алкиламино и замещенного или незамещенного С3-10 гетероциклила;
R2 - Н, F, Cl, или замещенный или незамещенный C1-8 алкокси; или R1 и R2 вместе с атомами углерода, к которым они присоединены, формируют неароматическое карбоциклическое кольцо или гетероциклическое кольцо;
R3 - это Н, замещенный или незамещенный C1-8 алкил, замещенный или незамещенный алкокси или гало;
R4 - Н или F;
R5 - Н, F, Cl или -СН3;
R6 - Н, гало, -CN,-CO2Ra, -CONH2, -NH2, замещенный или незамещенный С1-8 аминоалкил, замещенный или незамещенный C1-8 алкокси;
Ra - Н или замещенный или незамещенный C1-8 алкил; где R5 и R6 могут вместе формировать карбоциклическое кольцо;
L - связь, -СН2-, или -СН(СН3)-;
Z выбирается из группы, состоящей из
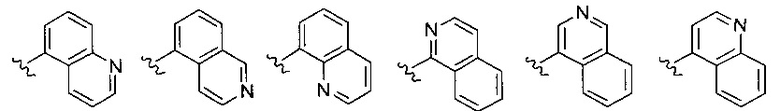
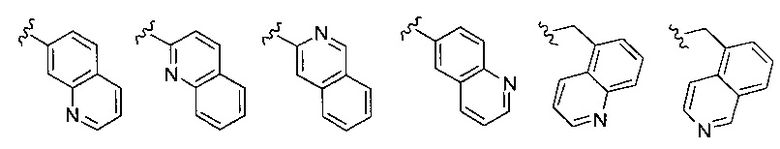
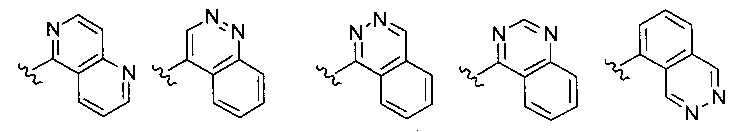
и их N-оксидов; где Z группа может быть незамещена или замещена 1-3 независимо выбранными R8 заместителями; каждый R8 независимо выбирается из группы, состоящей из Н, гало, -CN, -ОН, оксо, замещенного или незамещенного С1-8 алкила, замещенного или незамещенного C1-8 алкокси, -NR20R21, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, замещенного или незамещенного гетероциклила, a R20 и R21 - каждый независимо Н или замещенный или незамещенный C1-8 алкил.
В одном воплощении формулы II Z выбирается из группы, состоящей из: замещенного или незамещенного хинолинила, замещенного или незамещенного изохинолинила, замещенного или незамещенного 1,6-нафтиридинила, замещенного или незамещенного циннолинила, замещенного или незамещенного фталазинила, замещенного или незамещенного хиназолинила.
В одном воплощении формулы (II) R1 - замещенный или незамещенный С2-8 алкил; предпочтительно R1 - трет-бутил; R2, R3, R4 и R5 - Н; и R6 замещенный или незамещенный C1-8 алкил, замещенный или незамещенный C1-8 алкокси, -CN, -CONH2, -NH2, или замещенный или незамещенный C1-8 аминоалкил; предпочтительно R6 незамещенный C1-8 алкил или C1-8 галоалкил; более предпочтительно R6 - -СН3, -CH2F, -CHF2 или -CF3.
В другом воплощении формулы (II) R1 - замещенный или незамещенный С2-8 алкил; предпочтительно R1 - трет-бутил; R2 - F; R3, R4 и R5 - Н; и R6 замещенный или незамещенный C1-8 алкил, замещенный или незамещенный С1-8 алкокси, - CN, -CONH2, -NH2, или замещенный или незамещенный C1-8 аминоалкил; предпочтительно R6 - незамещенный C1-8 алкил, или С1-8 галоалкил; более предпочтительно R6- -СН3, -CH2F, -CHF2 или -CF3.
В одном воплощении соединения формулы (I) настоящего изобретения представлены формулой (IIIa) или (IIIb) или их солями:
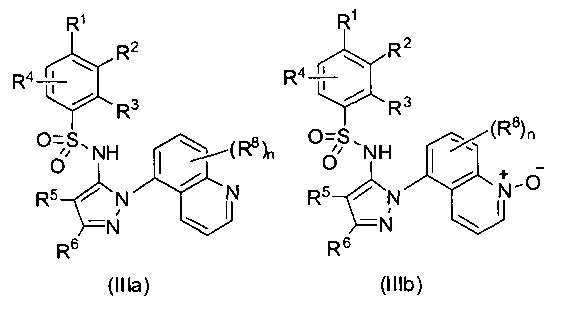
где R1 выбран из группы, состоящей из замещенного или незамещенного С2-8 алкила, замещенного или незамещенного C1-8 алкокси, замещенного или незамещенного С1-8 алкиламино и замещенного или незамещенного С3-10 гетероциклила; предпочтительно замещенного или незамещенного С2-8 алкила; более предпочтительно трет-бутила;
R2 - Н, F, Cl или замещенный или незамещенный C1-8 алкокси; предпочтительно Н или F; более предпочтительно Н; или R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют неароматическое карбоциклическое кольцо или гетероциклическое кольцо;
R3 - Н, замещенный или незамещенный C1-8 алкил, замещенный или незамещенный C1-8 алкокси или гало; предпочтительно Н или гало; более предпочтительно Н;
R4 - Н или F; предпочтительно Н;
R5 - Н, F, Cl или -СН3; предпочтительно Н;
R6 - Н, гало, - CN, -CO2R3, -CONH2, -NH2, замещенный или незамещенный C1-8 аминоалкил, замещенный или незамещенный C1-8 алкил или замещенный или незамещенный C1-8 алкокси; предпочтительно незамещенный C1-8 алкил или С1-8 галоалкил; более предпочтительно -СН3, -CH2F, -CHF2, или -CF3;
Ra - Н или замещенный или незамещенный С1-8 алкил; или где R5 и R6 вместе с углеродными атомами, к которым они присоединены, формируют карбоциклическое кольцо;
каждый R8 независимо выбран из группы, состоящей из Н, гало, - CN, - О, оксо, замещенного или незамещенный C1-8 алкила, замещенного или незамещенный C1-8 алкокси, и -NR20R21, замещенного или незамещенного арила, замещенного или незамещенного гетероарила и замещенного или незамещенного гетероциклила;
R20 и R21 - каждый независимо Н или замещенный или незамещенный С1-8 алкил;
n - 0, 1, 2 или 3.
В одном воплощении формулы (IIIa) или (IIIb) R1 - замещенный или незамещенный С2-8 алкил; предпочтительно R1 - трет-бутил; R2, R3, R4 и R5 - Н; R6 - замещенный или незамещенный С1-8 алкил, замещенный или незамещенный С1-8 алкокси, - CN, -CONH2, -NH2 или замещенный или незамещенный С1-8 аминоалкил; предпочтительно R6 - незамещенный С1-8 алкил или С1-8 галоалкил; более предпочтительно R6- -СН3, -CH2F, -CHF2, или -CF3; L - связь; и А1, А2, А3, А4, А5, А6, А7, А8 и R8 - как определяет формула (I).
В одном воплощении формулы (IIIa) или (IIIb) R1- замещенный или незамещенный С2-8 алкил; предпочтительно R1 - трет-бутил; R2 - F; R3, R4 и R5 - Н; R6 - замещенный или незамещенный С1-8 алкил, замещенный или незамещенный С1-8 алкокси, - CN, -CONH2, -NH2 или замещенный или незамещенный C1-8 аминоалкил; предпочтительно R6 - незамещенный С1-8 алкил или С1-8 галоалкил; более предпочтительно R6- -2СН3, -CH2F, -CHF2 или -CF3, L - связь; и А1, А2, А3, А4, А5, А6, А7, А8 и R8 - как определяет формула (I).
В одном воплощении формулы (IIIa) или (IIIb) R1 выбран из группы, состоящей из -СН2СН3, -СН(СН3)2, -С(СН3)3, -С(СН3)22Н2СН3, C(CH2CH2)CN, -С(O)(СН3)2, -ОСН3, -ОСН2СН3, -ОСН(СН3)2, -ОСН(СН3)3, -ОСН2СН(СН3)2, -OCF3, и морфолино; предпочтительно R1- -С(СН3)3; R2 - Н, F или Cl; предпочтительно R2 - Н или F; R1 и R2 могут вместе формировать - ОСН(СН3)2СН2- или -С(СН3)2СН2СН2-; R3 - Н, -СН3 или -ОСН3; предпочтительно R3 - Н; R4 - Н или F; предпочтительно R4 - Н; R5 - Н; R6 - Н, -СН3, -СН2СН3, -СН(СН3)2, С3Н7, -CH2F, -CHF2, -CF2CH3, -CF3, -CH2OCH3, -CH2OH, -CH2CN, -CN или -CONH2; предпочтительно R6- -CH3, -CH2F, -CHF2 или -CF3; и R8 каждый независимо выбран из группы, состоящей из Н, F, Cl, Br, СН3, -О, -ОСН3, -ОСН2СН3, -NH2, -N(CH3)2 и - CN; предпочтительно R8-Н или -NH2
В некоторых воплощениях R2 - это Н. В некоторых воплощениях R2 - F.
В одном воплощении соединения формулы (IIIa) или (IIIb), или их соли выбраны из группы, состоящей из:
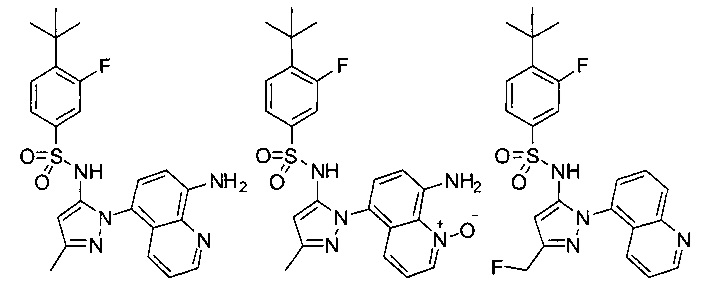
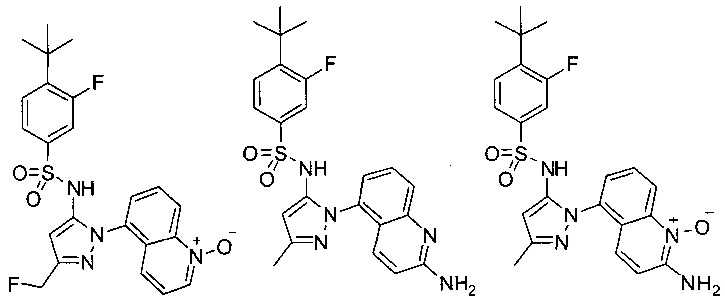
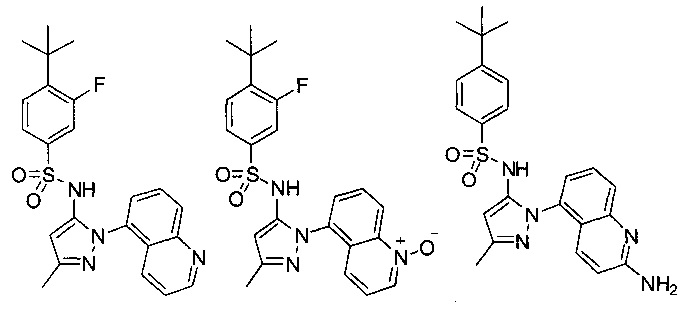
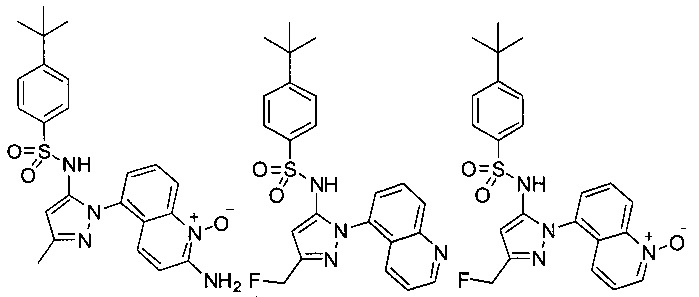
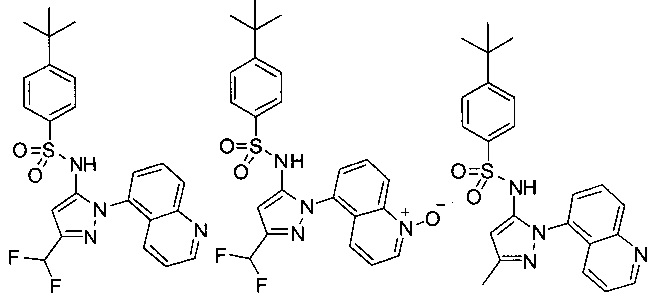
и 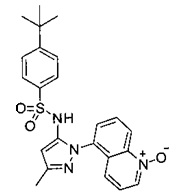
Предпочтительные R1 заместители
В формулах (I, II, IIIa и IIIb) R1 выбран из группы, состоящей из замещенного или незамещенного C2-8 алкила, замещенного или незамещенного C1-8 алкокси, замещенного или незамещенного C1-8 алкиламино и замещенного или незамещенного С3-10 гетероциклила. Когда R1 - замещенный алкил, группа алкила предпочтительно замещена гало или гидрокси. Когда R1 - замещенный алкокси, группа алкокси предпочтительно замещена гало. Предпочтительно R1 - незамещенный С2-8 алкил, включая C3-8 циклоалкил, С2-8 галоалкил, С1-8 гидроксиалкил, незамещенный С1-8 алкокси, С1-8 галоалкокси и С1-8 алкиламино; более предпочтительно незамещенный C2-8 алкил, C2-8 галоалкил, незамещенный С1-8 алкокси и С1-8 алкиламино; еще более предпочтительно незамещенный С2-8 алкил, незамещенный C1-8 алкокси и морфолино; еще более предпочтительно незамещенный С2-8; и наиболее предпочтительно трет-бутил.
Предпочтительные R6 заместители
В формулах (I, II, IIIa и IIIb) R6 - Н, гало, - CN, -CO2Ra, -CONH2, -NH2, замещенный или незамещенный C1-8 алкил, замещенный или незамещенный C1-8 алкокси или замещенный или незамещенный C1-8 аминоалкил. Когда R6 замещенный алкил, группа алкила предпочтительно замещена гало, гидрокси, алкокси или циано. Предпочтительно R6- - CN, -CONH2, -NH2, незамещенный C1-8 алкил, незамещенный C1-8 галоалкил и незамещенный C1-8 алкокси; более предпочтительно незамещенный C1-8 алкил или незамещенный C1-8 галоалкил, еще более предпочтительно незамещенный C1-8 алкил; наиболее предпочтительно метил.
Композиции, которые модулируют активность Хемокина
В другом аспекте настоящее изобретение обеспечивает композиции, которые модулируют активность хемокина, в частности, CCR(9) активность. Вообще композиции для модулирования активности рецептора хемокина в людях и животных будут включать фармацевтически приемлемый инертный наполнитель или растворитель и соединение, имеющее любую из формул I-III.
Термин "композиция", как здесь используется, охватывает продукт, включающий указанные ингредиенты в указанных количествах, а также любой продукт, который получается прямо или косвенно из комбинации указанных ингредиентов в указанных количествах. Под "фармацевтически приемлемым" имеется в виду носитель, растворитель или инертный наполнитель, который должен быть совместим с другими ингредиентами состава и не вредить его получателю.
Фармацевтические композиции для введения соединений этого изобретения могут быть просто представлены в форме единичной дозировки и могут быть получены любым из методов, известных в технологии фармацевтики. Все способы включают этап введения активного ингредиента в соединении с наполнителем, который составляет один или более вспомогательных ингредиентов. В целом, фармацевтические композиции получают путем однородного и глубокого введения активного ингредиента в соединении с жидким носителем или тонко раздробленным твердым носителем или обоими, и затем, в случае необходимости, преобразования продукта в требуемый состав. В фармацевтической композиции активное целевое соединение включено в количестве, достаточном, чтобы произвести желательный эффект на процесс или состояние заболеваний.
Фармацевтические композиции, содержащие активный ингредиент, могут быть представлены в форме, подходящей для орального использования, такие, например, как таблетки, ромбы, водные или масляные суспензии, дисперсные порошки или гранулы, эмульсии и самоэмульгаторы, как описано в патенте US №6451339, твердые или мягкие капсулы, или сиропы или эликсиры. Композиции, предназначенные для орального использования, могут быть получены согласно любому методу, известному в технологии для производства фармацевтических композиций. Такие композиции могут содержать один или более агентов, выбранных из подслащивающих агентов, вкусовых агентов, пигментов и консервантов, чтобы обеспечить фармацевтически изящные и приемлемые составы. Таблетки содержат активный ингредиент в смеси с другими нетоксичными фармацевтически приемлемыми инертными наполнителями, которые являются подходящими для производства таблеток. Эти инертные наполнители могут быть, например, инертными разбавителями, типа целлюлозы, диоксида кремния, окиси алюминия, углекислого кальция, углекислого натрия, глюкозы, маннита, сорбита, лактозы, фосфорнокислого кальция или фосфорнокислого натрия; гранулирующими и дезинтегрирующими агентами, например, кукурузным крахмалом или альгиновой кислотой; связующими веществами, например, PVP, целлюлозой, PEG, крахмалом, желатином или акацией, и смазывающими агентами, например, стеаратом магния, стеариновой кислотой или тальком. Таблетки могут быть неокрашенными, или они могут быть кишечно покрыты или иначе известными способами, чтобы задержать дезинтеграцию и поглощение в желудочно-кишечном трактате и таким образом обеспечить длительное действие за более длительный период. Например, может использоваться материал временной задержки типа глицерил моностеарат или глицерил дистеарат. Они могут также быть покрыты способами, описанными в патентах US 4256108; 4166452 и 4265874, чтобы сформировать осмотические терапевтические таблетки для контроля высвобождения.
Составы для орального использования могут также быть представлены как твердые желатиновые капсулы, в которых активный ингредиент смешан с инертным твердым разбавителем, например, углекислым кальцием, фосфорнокислым кальцием или каолином, или как мягкие желатиновые капсулы, в которых активный ингредиент смешан с водой или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом. Дополнительно эмульсии могут быть получены с неводным смешивающимся ингредиентом, таким как масла, и стабилизированы поверхностно-активными веществами типа монодиглицеридов, сложных эфиров PEG и т.п.
Водные суспензии содержат активные вещества в смеси с инертными наполнителями, подходящими для получения водных суспензий. Такие инертные наполнители - суспендирующие агенты, например, натрий карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрия альгинат, поливинилпирролидон, трагант и аравийская камедь; диспергаторы или смачивающие агенты могут быть естественно встречающимся фосфатидом, например, лецитином или продуктами конденсации оксида алкилена с жирными кислотами, например, стеаратом полиэтиленоксида или продуктами конденсации окиси этилена с длинноцепочечными алифатическими спиртами, например, гептадекаэтиленоксиэтанолом, или продуктами конденсации окиси этилена с частичными эфирами, полученными из жирных кислот и гекситола, такими как полиоксиэтилен-сорбитолмоноолеат, или продукты конденсации окиси этилена с частичными эфирами, полученными из жирных кислот и ангидридов гекситола, например полиэтиленсорбитан-моноолеат. Водные суспензии могут также содержать один или более консервантов, например, этил или н-пропил, п-гидроксибензоат, один или более пигментов, один или более вкусовых агентов и один или более подслащивающих агентов типа сахарозы или сахарина.
Масляные суспензии могут быть составлены путем суспендирования активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, типа жидкого парафина. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Подслащивающие агенты, такие как сформулированные выше, и вкусовые агенты могут быть добавлены, чтобы обеспечить приемлемый оральный состав. Эти композиции могут быть защищены дополнением антиоксиданта, типа аскорбиновой кислоты.
Дисперсные порошки и гранулы, подходящие для получения водной суспензии дополнением воды, обеспечивают активный ингредиент в смеси с дисперсным или смачивающим агентом, суспендирующим агентом и одним или более консервирующими средствами. Подходящие диспергирующие или смачивающие агенты и суспендирующие агенты приведены выше. Дополнительные инертные наполнители, например, подсластители, вкусовые агенты и пигменты, также могут присутствовать.
Фармацевтические композиции изобретения могут также быть в форме масла в водных эмульсиях. Масляная фаза может быть растительным маслом, например, оливковым маслом или арахисовым маслом, или минеральным маслом, например, жидким парафином, или их смесями. Подходящие эмульгаторы могут быть естественно встречающимися смолами, например, аравийская камедь или трагант, естественно встречающимися фосфатидами, например, соевым лецитином и сложными эфирами или частичными эфирами многоатомного спирта, полученными из жирных кислот и ангидридов гекситола, например сорбитанмоноолеат, и продуктами конденсации упомянутых частичных эфиров многоатомного спирта с окисью этилена, например полиэтиленоксидсорбитан-моноолеат. Эмульсии могут также содержать подсластители и вкусовые агенты.
Сиропы и эликсиры могут быть составлены с подсластителями, например глицерином, пропиленгликолем, сорбитом или сахарозой. Такие составы могут также содержать смягчители, консерванты и вкусовые агенты и пигменты. Оральные растворы могут быть получены в комбинации, например, с циклодекстрином, PEG и поверхностно-активными веществами.
Фармацевтические композиции могут быть в форме стерильной инъекционной водной или масляной суспензии. Эта суспензия может быть получена согласно известной технологии, используя подходящие диспергирующие или смачивающие агенты и суспендирующие агенты, которые были упомянуты выше. Стерильный инъекционный состав может также быть стерильным вводимым раствором или суспензией в нетоксичном парентерально доставляемом разбавителе или растворителе, например, таком как раствор в 1,3-бутан-диоле. Среди приемлемых связующих и растворителей, которые могут использоваться, вода, раствор Рингера и изотонический раствор хлористого натрия. Кроме того, стерильные, исключенные масла, традиционно используются в качестве растворителя или суспендирующей среды. С этой целью может использоваться любое мягкое нелетучее масло, включая синтетические моно - или диглицериды. Кроме того, жирные кислоты, типа олеиновой кислоты используются при получении инжектируемых сред.
Соединения настоящего изобретения могут также вводиться в форме свечей для ректальной доставки препарата. Эти композиции могут быть получены путем смешивания препарата с подходящим нераздражающим инертным наполнителем, который является твердым при обычных температурах, но жидким при ректальной температуре и будет поэтому плавиться в прямой кишке, чтобы высвободить препарат. Такие материалы - это масло какао и полиэтиленгликоли. Дополнительно, соединения могут вводиться посредством глазной доставки растворами или мазями. Еще один способ - трансдермальная доставка упомянутых соединений может быть достигнута посредством ионтофоретических пластырей и т.п.
Для местного использования применяются кремы, мази, гели, растворы или суспензии, содержащие соединения настоящего изобретения. Как здесь используется, местное применение также включает использование жидкостей для рта и полосканий.
Фармацевтические композиции и способы настоящего изобретения могут далее включать другие терапевтически активные соединения, как отмечено здесь, такие как примененные при лечении вышеупомянутых патологических состояний.
В одном воплощении настоящее изобретение обеспечивает композицию, состоящую из фармацевтически приемлемого носителя и соединения изобретения.
Способы Лечения
В зависимости от заболевания, которое будут лечить, и состояния субъекта соединения и композиции настоящего изобретения можно вводить оральным, парентеральным (например, внутримышечной, внутрибрюшинной, внутривенной, ICV, внутриполостной инъекцией или инфузией, подкожной инъекцией или имплантированием), ингаляцией, носовым, влагалищным, ректальным, подъязыковым или местным способом введения и они могут быть получены, один или вместе, в подходящих составах единичной дозировки, содержащих обычные нетоксичные фармацевтически приемлемые носители, присадки и связующие средства, соответствующие для каждого способа введения. Настоящее изобретение также рассматривает введение соединений и композиций настоящего изобретения в депо - составе.
При лечении или предотвращении состояний, которое требует модуляцию рецептора хемокина, соответствующий уровень дозировки в целом будет приблизительно 0.001-100 мг на кг массы тела пациента в сутки, которую можно вводить в единственной или многократных дозах. Предпочтительно уровень дозировки будет приблизительно 0.01 - приблизительно 25 мг/кг в сутки; более предпочтительно приблизительно 0.05 - приблизительно 10 мг/кг в сутки. Подходящий уровень дозировки может быть приблизительно 0.01-25 мг/кг в сутки, приблизительно 0.05-10 мг/кг в сутки, или приблизительно 0.1-5 мг/кг в сутки. В пределах этого диапазона дозировка может быть 0.005-0.05, 0.05-0.5, 0.5-5.0 или 5.0-50 мг/кг в сутки. Для орального введения соединения предпочтительно обеспечиваются в форме таблеток, содержащих 1.0-1000 миллиграммов активного ингредиента, в частности, 1.0, 5.0, 10.0, 15.0, 20.0, 25.0, 50.0, 75.0, 100.0, 150.0, 200.0, 250.0, 300.0, 400.0, 500.0, 600.0, 750.0, 800.0, 900.0 и 1000.0 миллиграммов активного ингредиента для симптоматического регулирования дозировки пациенту, которого лечат. Соединения можно вводить при режиме 1-4 раз в сутки, предпочтительно несколько раз в сутки.
Понятно, однако, что определенный уровень дозы и частота дозировки для любого специфического пациента могут быть различны и будут зависеть от разнообразия факторов, включая активность определенного применяемого соединения, метаболической стабильности и времени действия этого соединения, возраста, массы тела, наследственных особенностей, общего здоровья, пола, диеты, способа и времени введения, скорости выведения, комбинации препаратов, серьезности специфического состояния и хозяина, подвергающегося терапии.
В некоторых воплощениях соединения настоящего изобретения вводят как часть комбинированной терапии. Например, количество химиотерапевтического агента или радиации вводят субъекту до, после или в комбинации с соединениями настоящего изобретения. В некоторых воплощениях количество является суб-терапевтическим, когда химиотерапевтический агент или радиация вводится одни. Специалистам отрасли понятно, что "комбинации" могут включить комбинации в лечении (т.е. два или более лекарства можно вводить как смесь, или, по крайней мере, одновременно или, по крайней мере, вводить субъекту в разное время, но так, чтобы оба находились в кровотоке субъекта в одно время). Дополнительно, композиции настоящего изобретения можно вводить до или после второго терапевтического режима, например до или после дозы химиотерапии или радиации.
В еще одних воплощениях представленные способы направлены на лечение аллергических заболеваний, при этом соединение или композиция изобретения вводится или одно или в комбинации со вторым терапевтическим агентом, где упомянутый второй терапевтический агент - это антигистамин или противовоспалительное средство. Когда используется комбинация, практик может вводить комбинацию соединения или композиции настоящего изобретения и второго терапевтического агента. Кроме того, соединение или композиция и второй терапевтический агент можно вводить последовательно, в любом порядке.
Соединения и композиции настоящего изобретения могут комбинироваться с другими соединениями и композициями, полезными для предотвращения и лечения состояния или заболевания интереса, такого как воспалительное состояние и заболевание, включая воспалительное заболевание кишечника (включая заболевание Крона и язвенный колит), аллергические заболевания, псориаз, атопический дерматит и астму, и патологии, отмеченные выше. Выбор соответствующих агентов для использования в комбинированных терапиях может быть сделан обычным специалистом области. Комбинация терапевтических агентов может действовать синергитически на эффект лечения или предотвращения различных нарушений. Используя этот подход, можно достигнуть терапевтической эффективности с более низкими дозировками каждого агента, таким образом снижая возможности неблагоприятных побочных эффектов.
При лечении, предотвращении, улучшении, контроле или снижении риска воспаления соединения настоящего изобретения могут использоваться вместе с противовоспалительным или болеутоляющим агентом, таким как опиатный агонист, ингибитор липоксигеназы, типа ингибитора 5-липоксигеназы, ингибитора циклооксигеназы, типа ингибитора циклооксигеназы-2, ингибитора интерлейкина, типа ингибитора интерлейкина 1, NMDA антагониста, ингибитора окиси азота или ингибитора синтеза окиси азота, аминосалицилаты, кортикостероидов и других иммунодепрессивных препаратов, нестероидного противовоспалительного агента, или подавляющего цитокин противовоспалительного агента, например, с соединением таким как ацетаминофен, аспирин, кодеин, биологические TNF секвестранты, биологические агенты, которые имеют целью a4b7, АСЕ2 ингибиторы, С ингибиторы протеиновой линазы, фентанил, ибупрофен, метиндол, кеторолак, морфин, напроксен, фенакцетин, пироксикам, стероидный анальгетик, суфентанил, сунлиндак, тенидап и т.п.
Точно также соединения настоящего изобретения можно вводить с противоболевым средством; усилителем действия, типа кофеина, Н2-антагониста, симтикона, гидроокиси алюминия или магния; противозастойным средством, типа псевдофедрина; средством против кашля, типа кодеина; мочегонным средством; седативным или неседативным антигистамином; антагонистом самого последнего антигена (VLA-4); иммунодепрессантом, типа циклоспорина, такролимуса, рапамицина, агониста EDG рецептора или других иммунодепрессантов типа FK-506; стероидом; нестероидным антиастматическим агентом, таким как β2-агонист, антагонист лейкотриена или ингибитором биосинтеза лейкотриена; ингибитором фосфодиэстеразы типа IV (PDE-IV); понижающим холестерин агентом, типа ингибитора редуктазы HMG-CoA, секвестанта или ингибитора поглощения холестерина; и антидиабетическим агентом, типа инсулина, ингибиторов α-глюкозидазы или глитазонов.
Весовое соотношение соединения настоящего изобретения ко второму активному ингредиенту может быть различно и будет зависеть от эффективной дозы каждого ингредиента. В целом, будет использоваться эффективная доза каждого. Таким образом, например, когда соединение настоящего изобретения объединено с NSAID, весовое соотношение соединения настоящего изобретения к NSAID будет в общем случае составлять от приблизительно 1000:1 до приблизительно 1:1000, предпочтительно от 200:1 до приблизительно 1:200. Комбинации соединения настоящего изобретения и других активных ингредиентов в общем случае также будут в пределах вышеупомянутого диапазона, но в каждом случае должна использоваться эффективная доза каждого активного ингредиента.
Способы лечения или Предотвращения ССК(9)-опосредованных Состояний или Заболеваний
В еще одном аспекте настоящее изобретение обеспечивает способы лечения или предотвращения CCR(9) - опосредованного состояния или заболевания путем введения субъекту, имеющему такое состояние или заболевание, терапевтически эффективного количества любого соединения формул, приведенных выше. Соединения для использования в представленных методах включают соединения согласно вышеупомянутым формулам, обеспеченные выше как варианты, в частности, иллюстрируемые Примерами, приведенными ниже, и те, которые представлены здесь специфичными составами.
"Субъект" как здесь определено, включает животных, типа млекопитающих, включая, но без ограничения, приматов (например, людей), коров, овец, коз, лошадей, собак, кошек, кроликов, крыс, мышей и т.п. В предпочтительных воплощениях субъект - это человек.
Как здесь используется, фраза "CCR(9) - опосредованное состояние или заболевание" и связанные фразы и термины относятся к состоянию или заболеванию, характеризующемуся несоответствующим, т.е. меньше чем или больше чем нормальная CCR(9) функциональная активность. Несоответствующая CCR(9) функциональная активность могла бы возникнуть как результат CCR(9) экспрессии в клетках, которые обычно не выражают CCR(9), увеличенной CCR(9) экспрессии (приводящей, например, к воспалительным и иммунорегуляторным расстройствам и заболеваниям) или уменьшенной CCR(9) экспрессии. Несоответствующая CCR(9) функциональная активность могла бы также возникнуть как результат ТЕСК секреции клетками, которые обычно не секретируют ТЕСК, увеличенной ТЕСК экспрессии (приводящей, например, к воспалительным и иммунорегуляторным расстройствам и заболеваниям) или уменьшенной ТЕСК экспрессии. CCR(9) - опосредованное состояние или заболевание может быть полностью или частично обусловлено несоответствующей CCR(9) функциональной активностью. Однако, CCR(9) - опосредованное состояние или заболевание - то, в котором модуляция CCR(9) приводит к некоторому влиянию на основное состояние или заболевание (например, CCR(9) антагонист приводит к некоторому улучшению состояния пациента по крайней мере в некоторых пациентах).
Термин "терапевтически эффективное количество" означает количество соответствующего соединения, которое проявляет биологический или медицинский ответ клетки, ткани, системы или животного, такого как человек, который исследуется исследователем, ветеринаром, медицинским доктором или другим лицом, обеспечивающим лечение.
Заболевания и состояния, связанные с воспалением, иммунными расстройствами, инфекцией и раком, можно лечить или предотвращать представленными соединениями, композициями и способами. В одной группе воплощений заболевания или состояния, включая хронические заболевания людей или других разновидностей, можно лечить ингибиторами CCR(9) функции. Эти заболевания или состояния включают: (1) аллергические заболевания, типа системной анафилаксии или ответов гиперчувствительности, аллергии на препараты, аллергии ужаливания насекомого и пищевые аллергии, (2) воспалительные заболевания кишечника, типа заболевания Крона, язвенного колита, микроскопического колита, илеита и энтерита, и постоперационной кишечной непроходимости, (3) вагиниты, (4) псориаз и воспалительный дерматоз, типа дерматита, экземы, атопического дерматита, аллергического контактного дерматита, крапивницы и зуда, (5) васкулиты, (6) спондилоартропатии, (7) склеродерму, (8) астму и дыхательные аллергические заболевания, типа аллергической астмы, аллергического ринита, заболевания гиперчувствительности легких и т.п., (9) аутоиммунные заболевания, типа фибромиалгии, анкилозного спондилита, ювенильного RA, заболевания Стилла, полисуставного ювенильного А, паусиартикулярного ювенильного RA, полимиальгии ревматической, ревматического артрита, псориатического артрита, остеоартрита, полисуставного артрита, рассеянного склероза, системной волчанки, диабета типа I, диабета типа II, гломерулонефритов и т.п., (10) отторжения трансплантата (включая отторжение аллотрансплантата), (11) заболевание трансплантат-против-хозяина (включая и острое и хроническое), (12) другие заболевания, в которых нежелательные воспалительным ответы должны замедляться, типа атеросклероза, миозитов, нейродегенеративных заболеваний (например, болезнь Альцгеймера), энцефалита, менингита, гепатита, нефрита, сепсиса, саркоидоза, аллергического конъюнктивита, отита, хронического обструктивного заболевания легких, синусита, синдрома Бехсета и подагры, (13) иммунные аллергии, обусловленные пищей, типа заболевания брюшной полости, (14) легочный фиброз и другие фиброзные заболевания, (15) синдром раздраженного кишечника, (16) первичное склерозирующее воспаление желчных путей, (17) рак (включая и первичный и метастатический), (18) связанные с бактериями синдромы, типа геморрагического колита и гемолитического уремического синдрома, (19) меланому, (20) первичное склерозирующее воспаление желчных путей, (21) постоперационную кишечную непроходимость, (22) гепатит и (23) воспалительные заболевания печени.
В другой группе воплощений заболевания или состояния можно лечить модуляторами и агонистами CCR (9) функции. Примеры заболеваний, которые лечат, модулируя CCR (9) функцию, включают раковые образования, сердечнососудистые заболевания, заболевания, в которых ангиогенез или неоваскуляризация играют роль (неопластические заболевания, ретинопатия и макулярная дегенерация), инфекционные заболевания (вирусные инфекции, например, ВИЧ инфекция, и бактериальные инфекции) и иммунодепрессивные заболевания, типа состояний пересадки органа и состояний пересадки кожи.
Термин "состояния пересадки органа" включает состояния трансплантата костного мозга и состояния пересадки твердого органа (например, почки, печени, легкого, сердца, поджелудочной железы или их комбинации).
Предпочтительно, представленные способы направлены на лечение заболеваний или состояний, выбранных из воспалительного заболевания кишечника, включая заболевание Крона и Язвенный Колит, аллергические заболевания, псориаз, атипический дерматит и астму, аутоиммунное заболевание, типа ревматического артрита и имунно-обусловленных пищевых аллергий, типа брюшных заболеваний.
В еще одних воплощениях представленные способы направлены на лечение псориаза, где соединение или композиция изобретения используются одни или в комбинации со вторым терапевтическим агентом, типа кортикостероида, смазки, кератолитического агента, производной витамина D3, PUVA и антралина.
В других воплощениях представленные способы направлены на лечение атопического дерматита за счет использования соединения или композиции изобретения или одного или в комбинации со вторым терапевтическим агентом, типа смазки и кортикостероида.
В дальнейших воплощениях представленные способы направлены на лечение астмы за счет использования соединения или композиции изобретения или одного или в комбинации со вторым терапевтическим агентом, типа b2-агониста и кортикостероида.
Получение модуляторов
Следующие примеры предлагаются, чтобы иллюстрировать, но не ограничить, заявленное изобретение.
Дополнительно специалисты в области оценят, что молекулы, заявленные в этом патенте, могут быть синтезированы, используя многообразие стандартных превращений органической химии.
Определенные общие типы реакций, используемые широко, чтобы синтезировать целевые соединения в этом изобретении, суммированы в примерах. В частности, родовые процедуры для образования сульфамида и аза-арил N-оксида описаны и использовались обычным образом.
Если не предназначаются чтобы быть исчерпывающими, представленные синтетические органические превращения, которые могут использоваться для получения соединений изобретения, включены ниже.
Эти представленные превращения включают: стандартные манипуляции с функциональными группами; восстановление, типа нитро в амино; окисления функциональных групп, включая спирты и аза-арилы; замещения арила через IPSO или другие механизмы для введения разнообразия групп, включая нитрил, метил и галоген; введения и удаления защитной группы; формирование Гриньяра и реакция с электрофилами; металл-обусловленные перекрестные связи, включая, но без ограничения реакции Buckwald, Suzuki и Sonigashira; галоидирования и другие электрофильные ароматические реакции замещения; формирования соли диазония и реакции этих разновидностей; этерификации; циклативные конденсации, дегидратации, окисления и восстановления, приводящие к гетероарилгруппам; арилметаллизации и трансметаллизации и реакция следующих разновидностей металл-арил с электрофилом, типа хлорида кислоты или Weinreb амида; амидирования; этерификации; реакции нуклеофильного замещения; алкилирования; ацилирования; формирование сульфамида; хлорсульфониляции; гидролизы сложных эфиров и связанные гидролизы, и т.п.
Определенные молекулы, заявленные в этом патенте, могут существовать в различных энантиометрических и диастереометрических формах, и все такие варианты этих соединений лежат в рамках изобретения. В частности, когда R8 это ОН и орто к азоту, хотя формулой иллюстрируется как -N=C(O)-, должно быть понятно, что таутомерная форма -NH-C(O) - также находится в рамках этой формулы.
В описаниях синтезов, которые далее следуют, некоторые предшественники были получены из коммерческих источников. Эти коммерческие источники включают Aldrich Chemical Co., Acros Organics, Ryan Scientific Incorporated, Oakwood Products Incorporated, Lancaster Chemical CO., Sigma Chemical Co., Lancaster Chemical Co., TCI-America, Alfa Aesar, Davos Chemicals и GFS Chemicals.
Соединения изобретения, включая перечисленные в таблице активности, могут быть получены способами и подходами, описанными в следующем экспериментальном разделе, и при помощи стандартных превращений органической химии, которые известны специалистам отрасли.
Примеры
Типичные соединения, используемые в способе изобретения и в фармацевтических композициях изобретения, включают, но без ограничения, соединения, перечисленные в следующей таблице. Фармацевтически приемлемые соли соединений, перечисленных в этой таблице, также используются в способе по изобретению и в фармацевтических композициях по изобретению. Эти соединения находятся в рамках этого изобретения и были проверены на CCR (9) активность, как описано ниже.
Соединения изобретения были испытаны на активность в пробирочном анализе хемотаксиса, описанном здесь ниже в разделе, названном "Пример n vitro анализа", где описан "хемотаксисный анализ". Все соединения, перечисленные в Таблице 1, имеют IC50<1000 nM в хемотаксисном анализе.

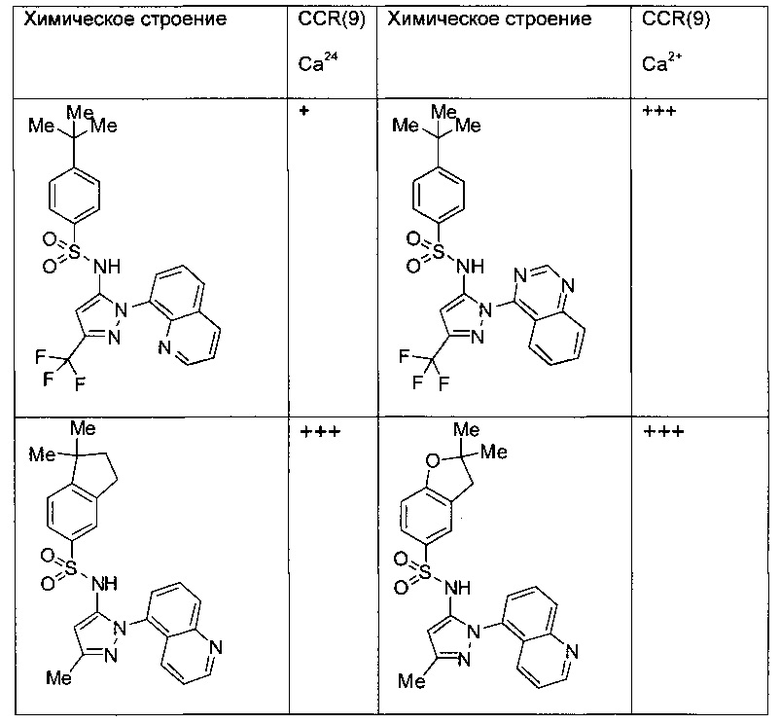
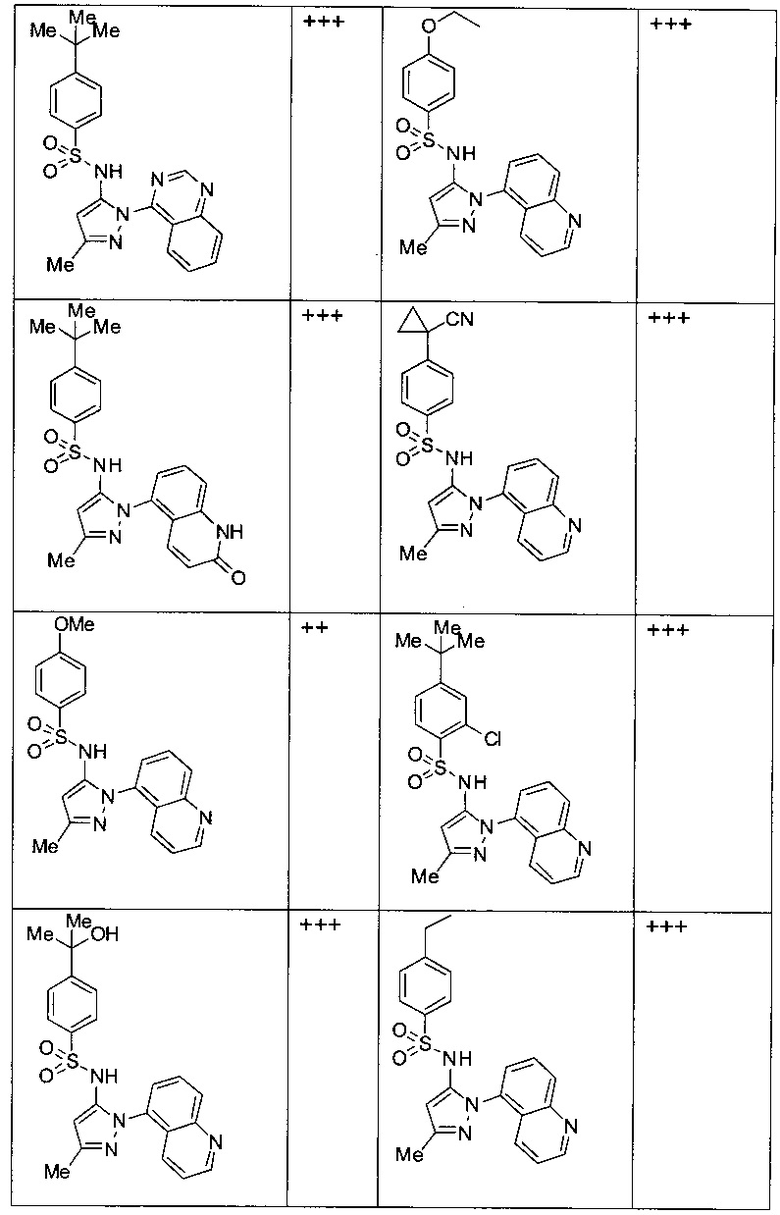
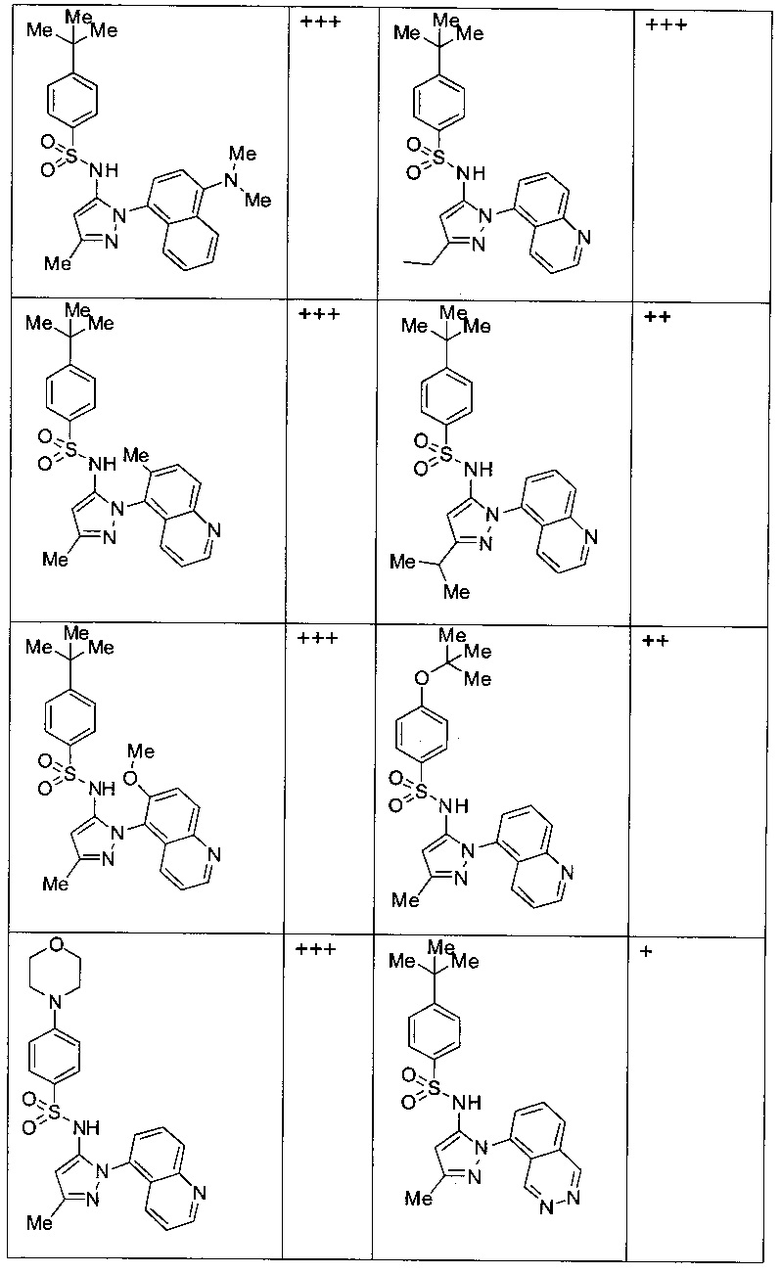
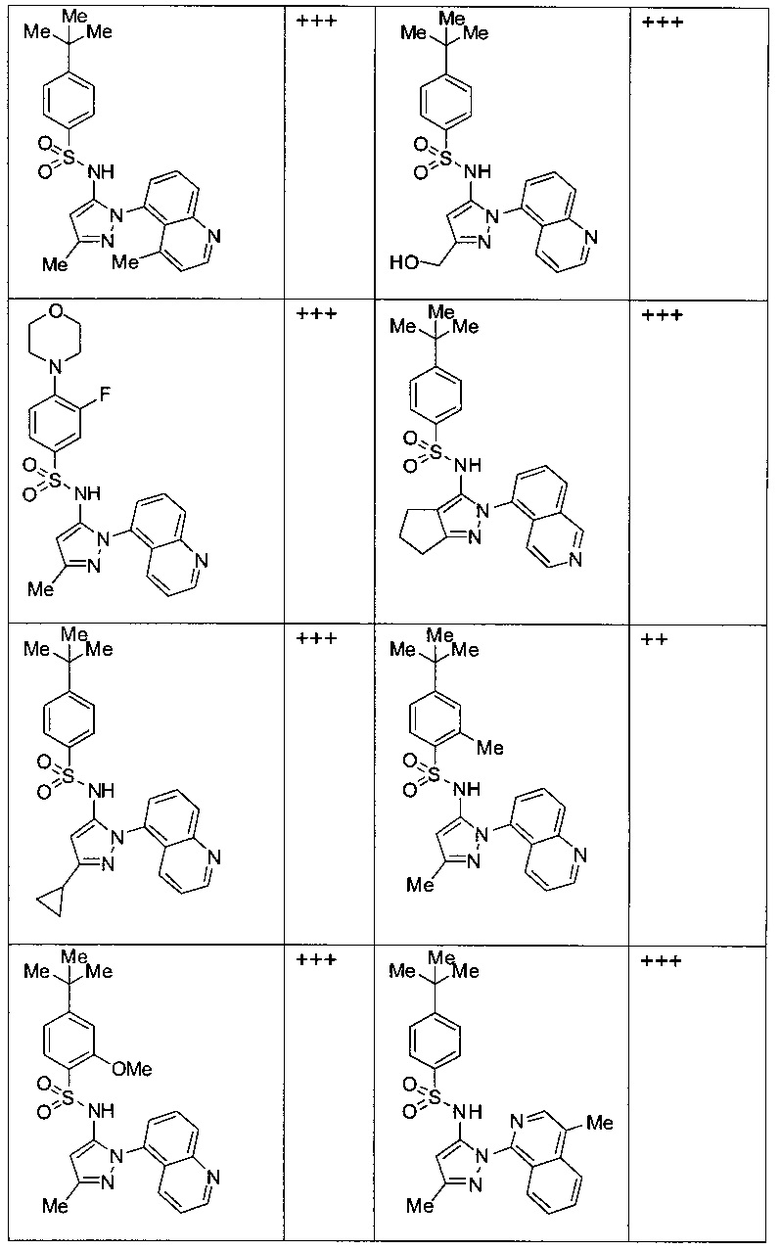
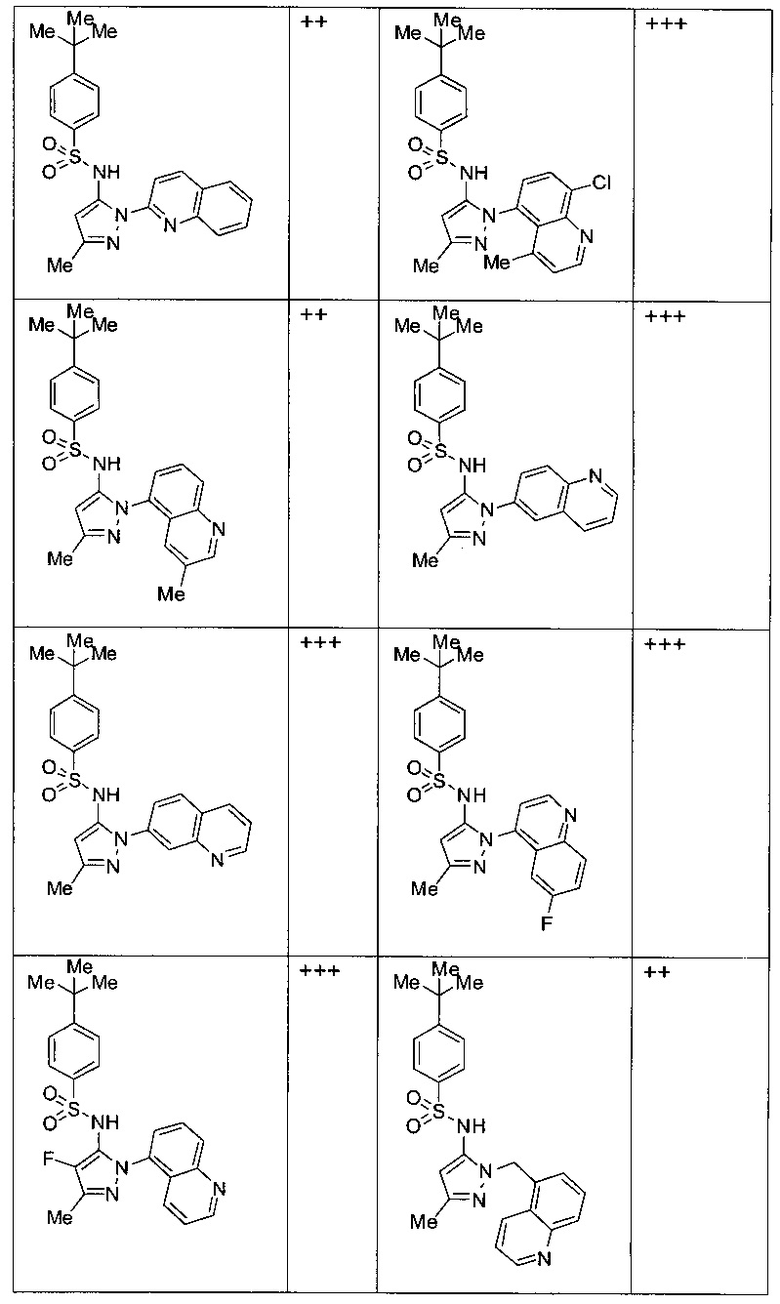
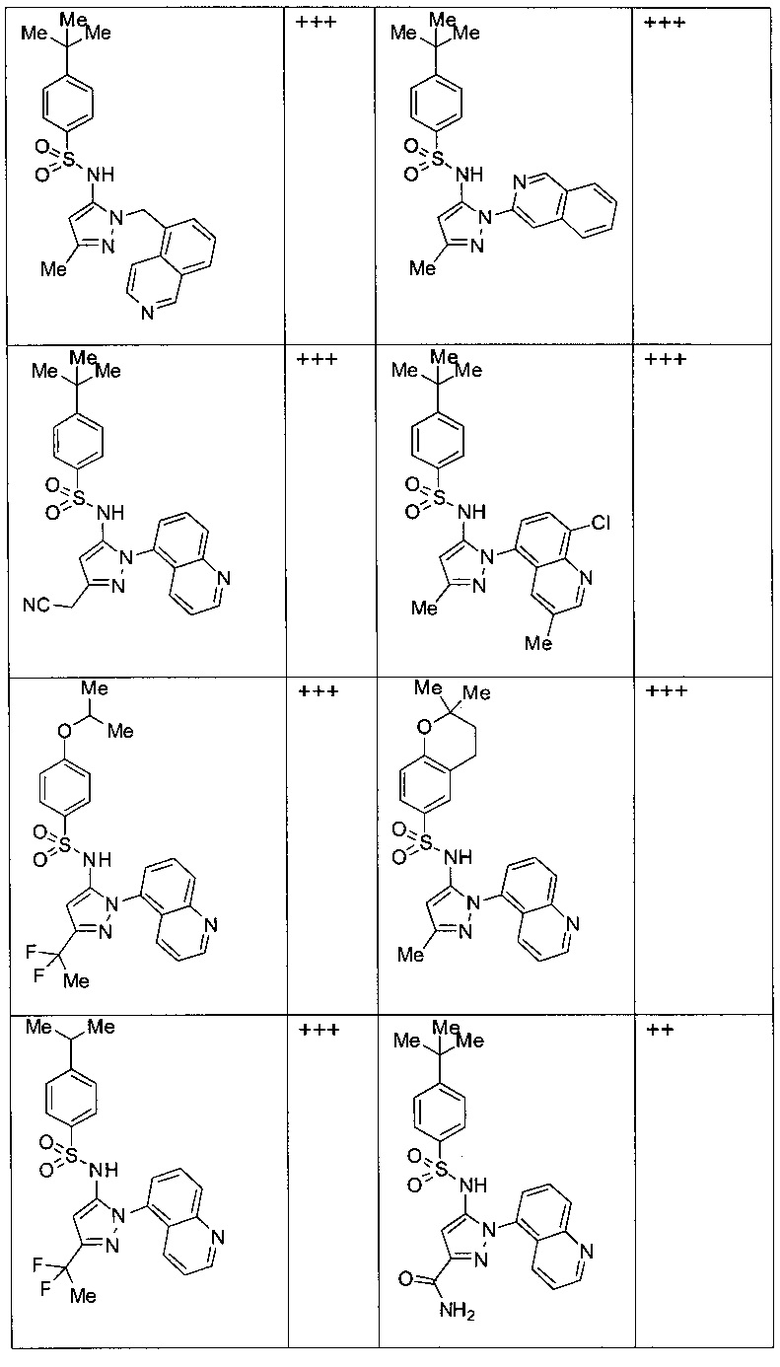
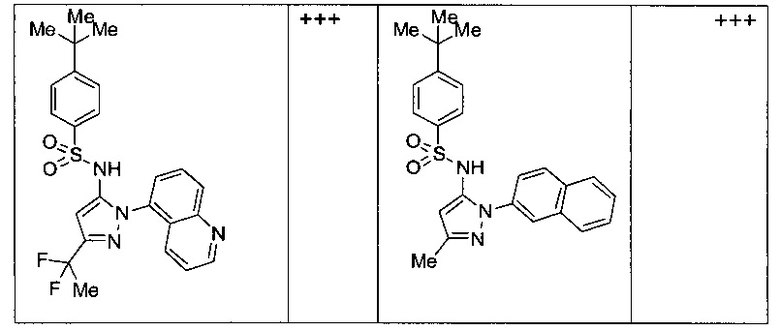
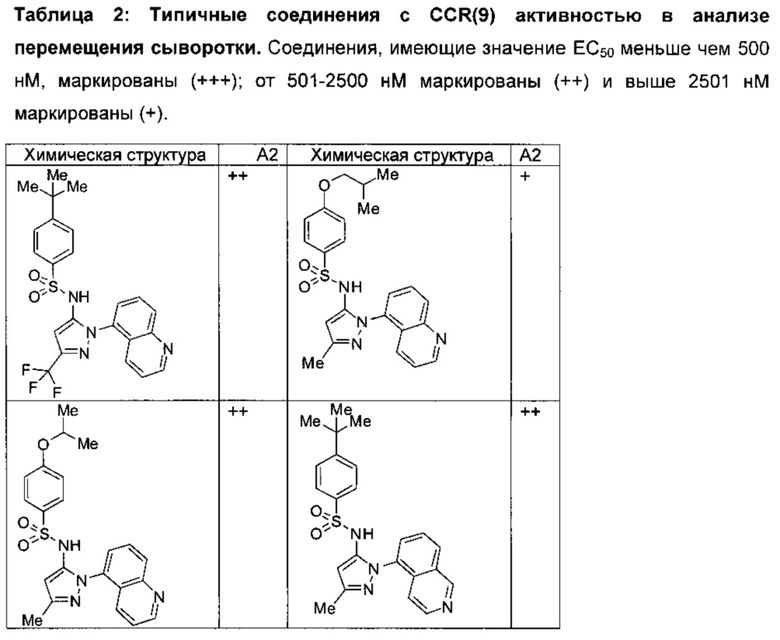
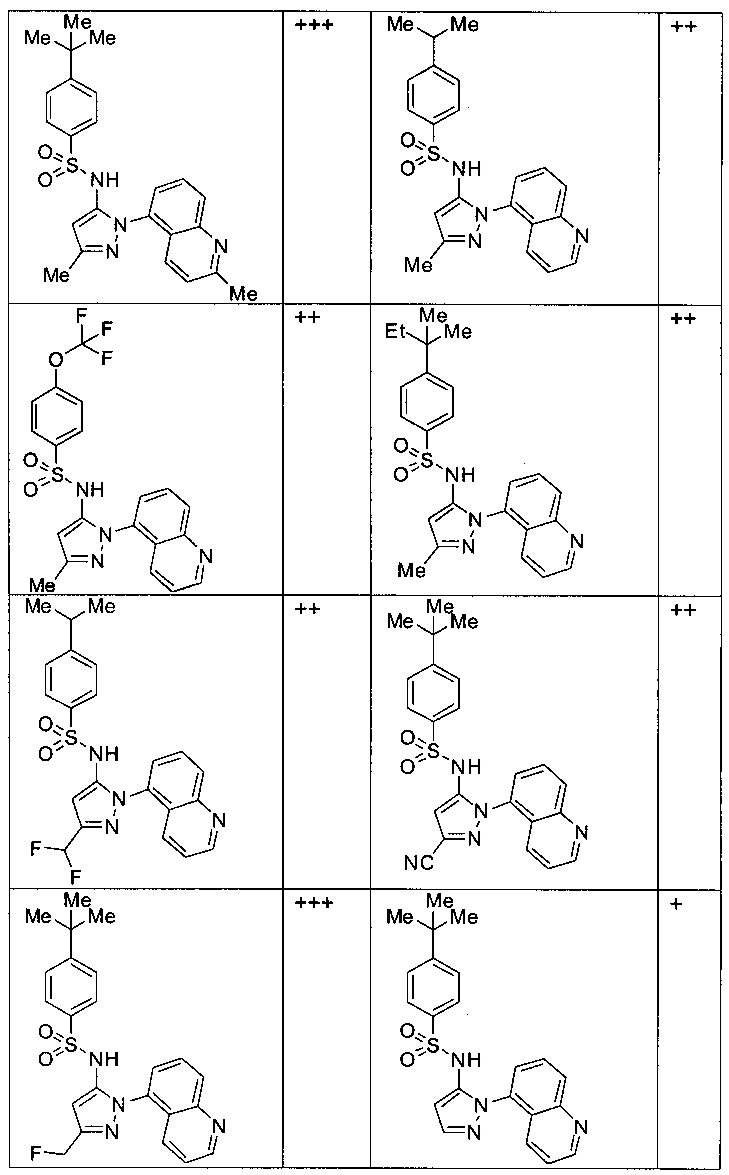
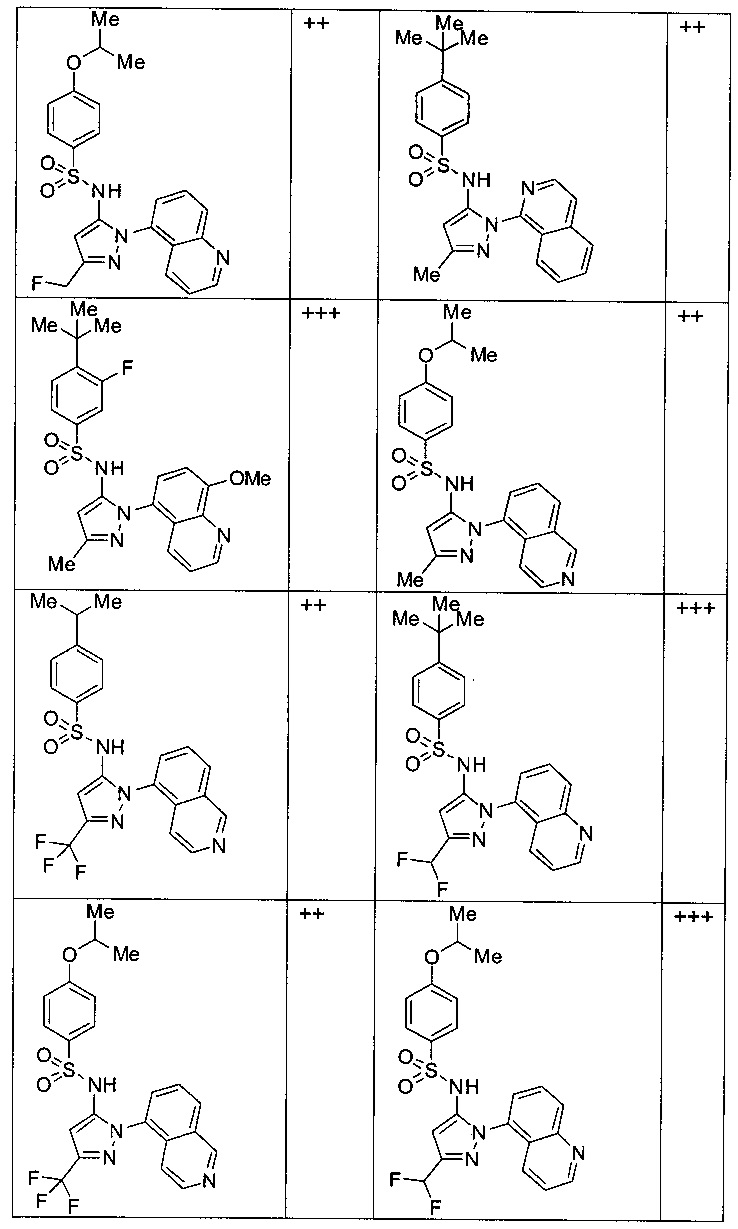
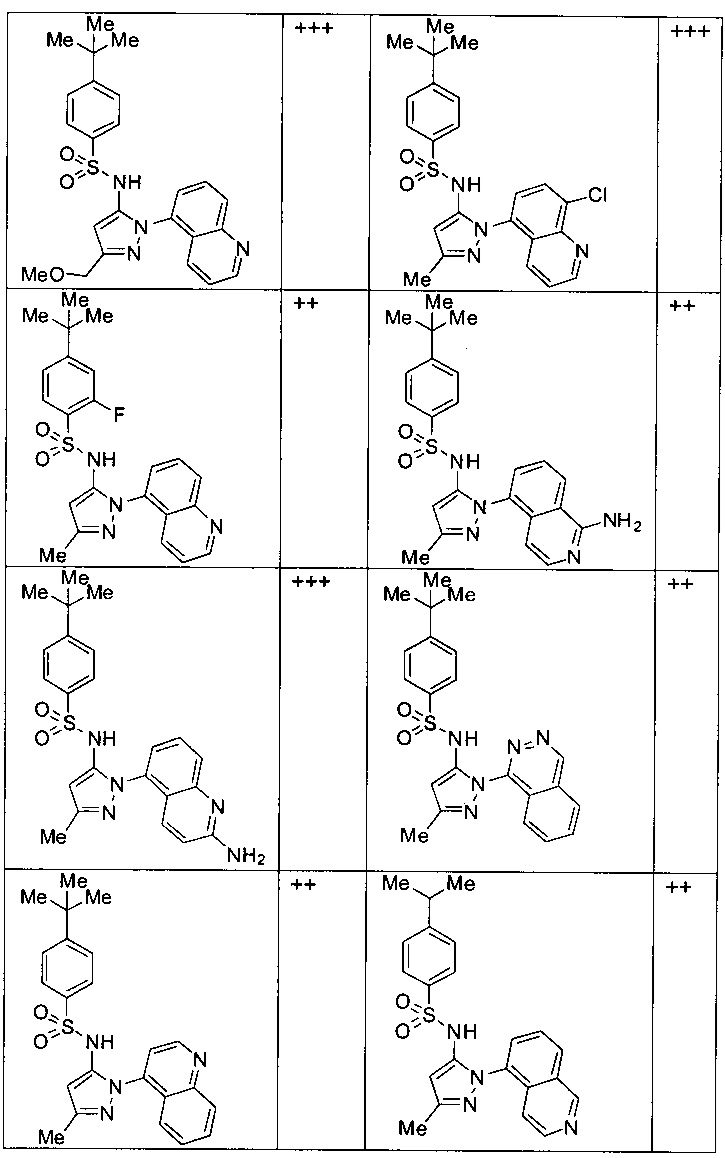
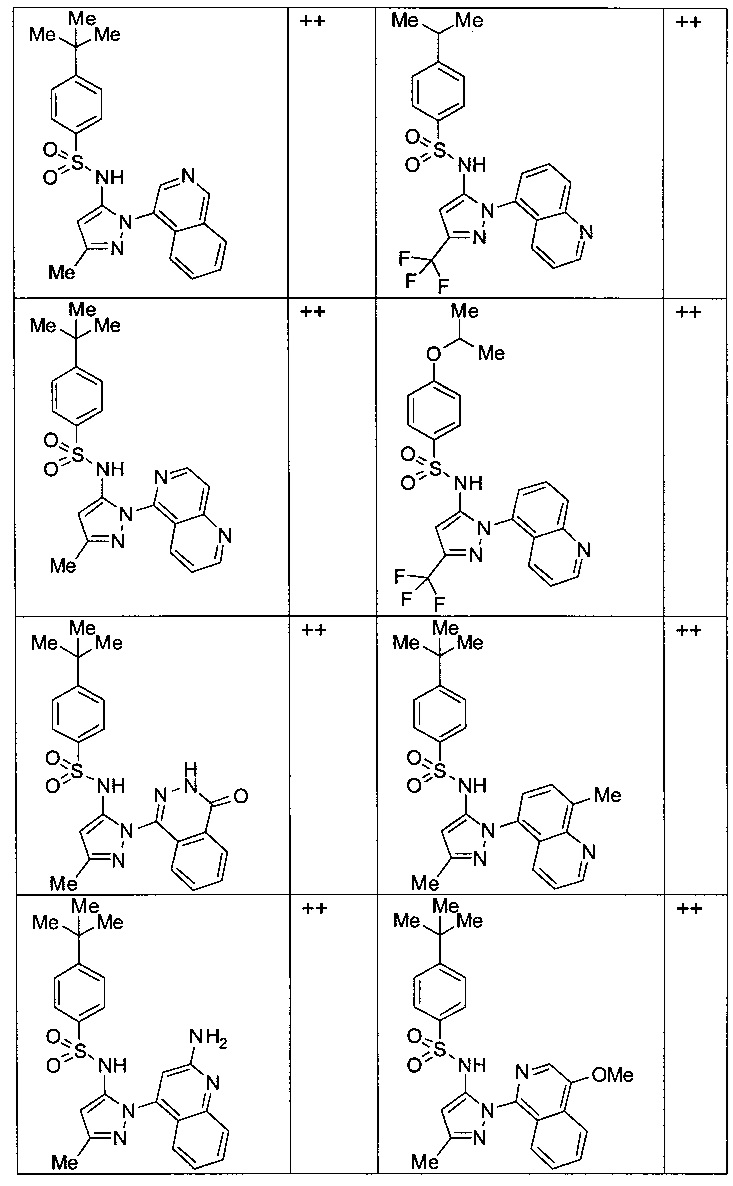
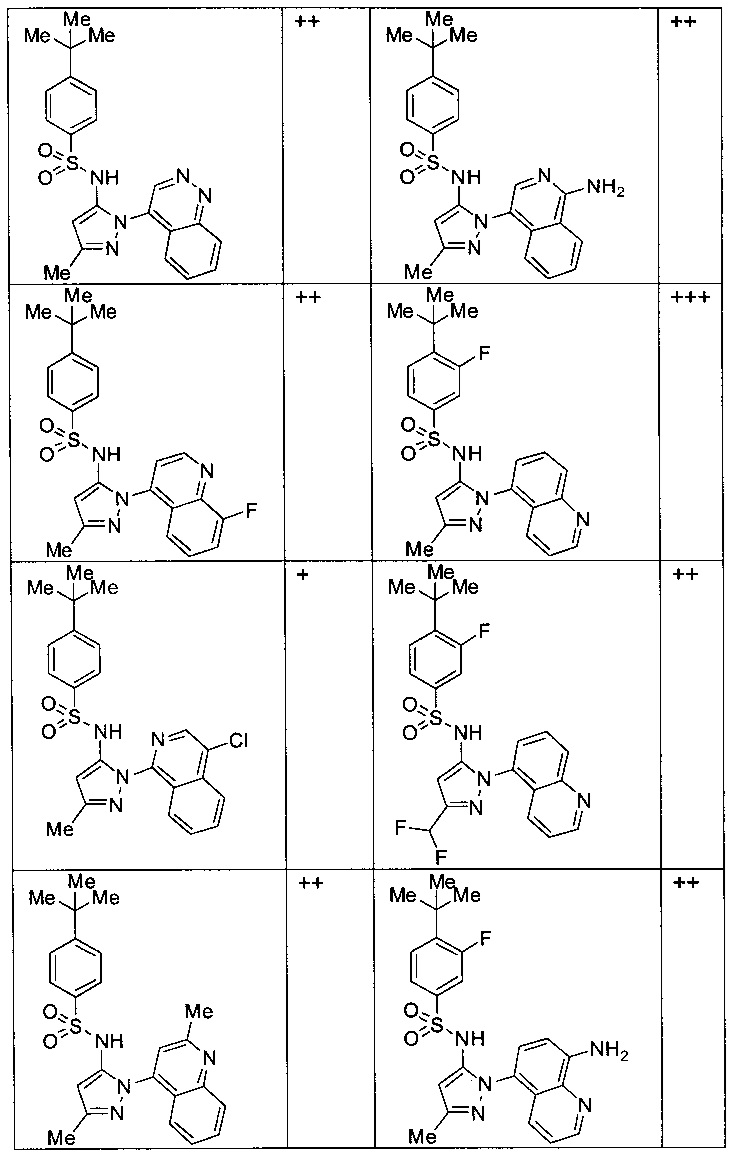
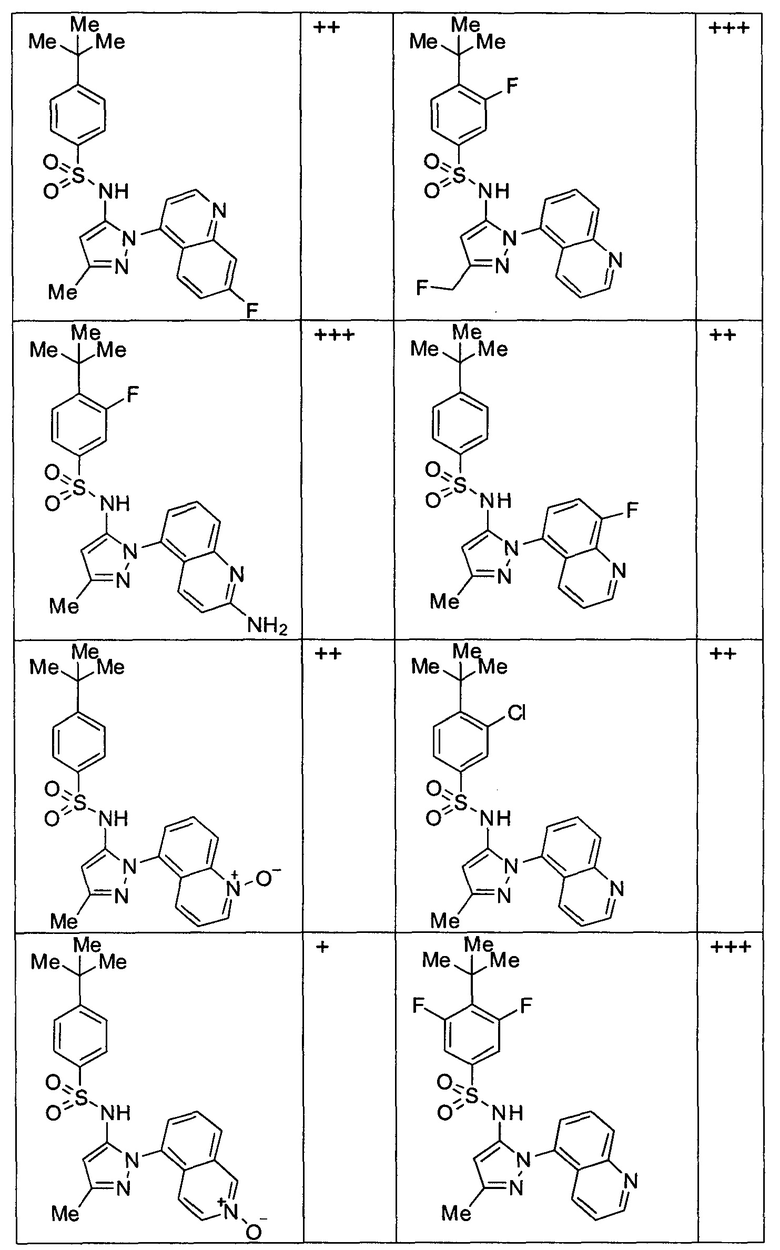
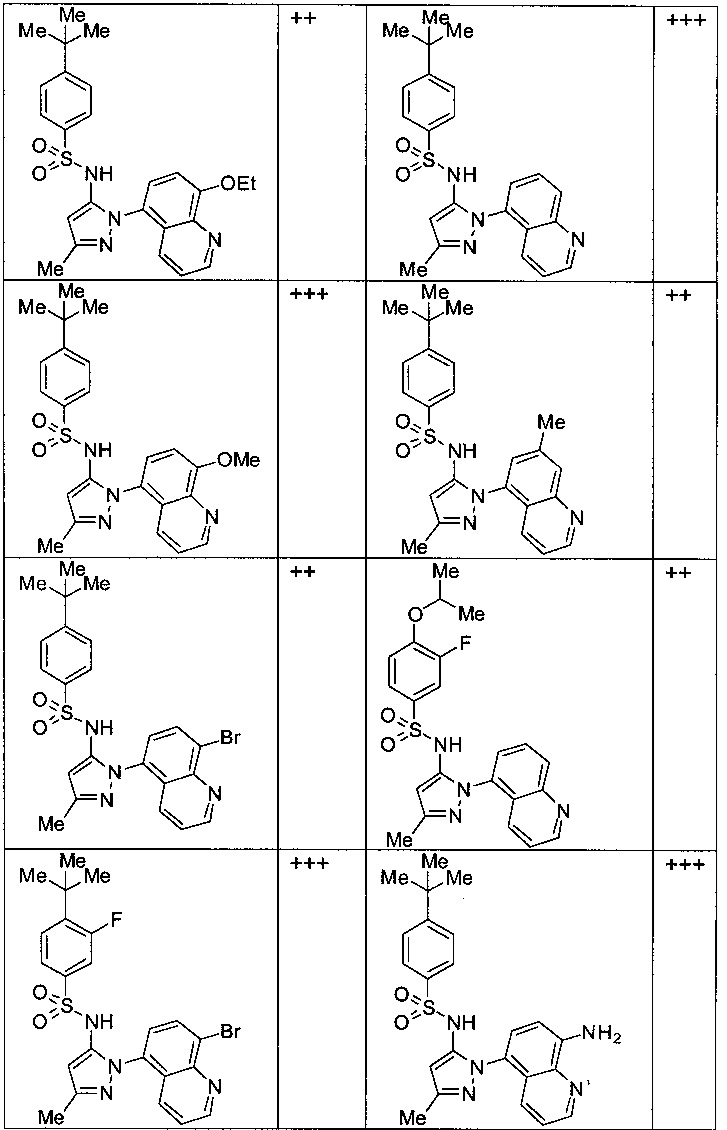
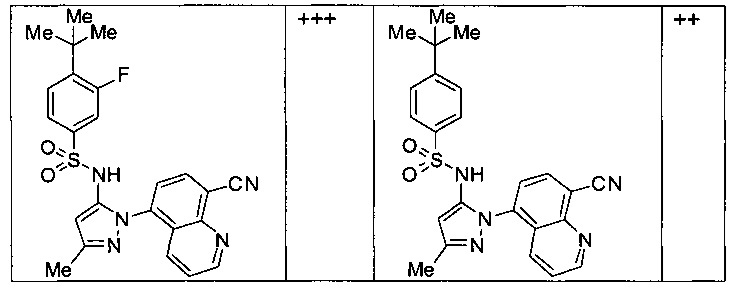
Реактивы и растворители, используемые ниже, могут быть получены из коммерческих источников, такого как Aldrich Chemical Co. (Milwaukee, Wisconsin, USA). 1H-NMR был зарегистрирован на Varian Ртутном NMR спектрометре на 400 МГц. Показательные пики сведены в таблицу в порядке: множественность (br, широкое; s, синглет; d, диполь; t, триплет; q, квартет; м, мультиплет) и число протонов. О результатах масс-спектрометрии говорит отношение массы к заряду, за которым следует относительная распространенность каждого иона (в круглой скобке). В таблицах о единственном значении т/е сообщено для М+Н (или, как отмечено, М-Н, M+Na, и т.д.) иона, содержащего самые общие атомные изотопы. Изотопные характеристики соответствуют ожидаемой формуле во всех случаях. Анализ Электроспрейной Ионизационной (ESI) масс-спектрометрии проводился на Hewlett-Packard MSD электроспрейном масс-спектрометре с использованием HP 1100 HPLC для подачи образца. Обычно аналит растворяли в метаноле при 0.1 мг/мл, и 1 микролитр вводили с доставляющем растворителем в масс-спектрометр, который сканировал от 100 до 1500 дальтон. Все соединения могли быть проанализированы в позитивном ESI режиме, используя ацетонитрил / воду с 1%-ной муравьиной кислотой в качестве доставляющего растворителя. Соединения, обеспеченные ниже, могли также быть проанализированы в отрицательном ESI режиме с использованием 2 мм NH4OAC в ацетонитриле / воде как системе доставки.
Соединения настоящего изобретения могут быть синтезированы общим Синтезом А, показанным ниже. Обработка арилсульфонилхлорида формулы А пиразоламином В в присутствии основы, типа пиридина при соответствующей температуре, например, 80°С, предоставляет арилсульфамиды формулы С. Пиразоламины В могут быть синтезированы обработкой гидразина D нитрилом С при подходящей повышенной температуре в растворителе, таком как этиловый спирт. Специалисту понятно, что заместители, включая, например, R1, R2, IV, R4 и R6, могут нуждаться в защите, как известно специалистам в области, стандартными защитными группами во время синтеза в зависимости от их реактивности к условиям реакции.
Общий Синтез А
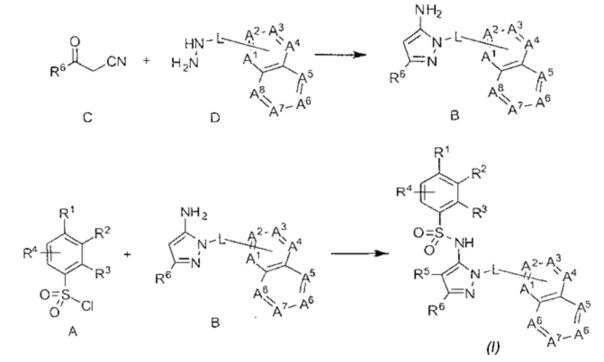
Пример I: Синтез 4-трет-бутил-N-(3-метил-1-(хиназолин-4-ил)-1H-пиразол-5-ил)бензолсульфонамида
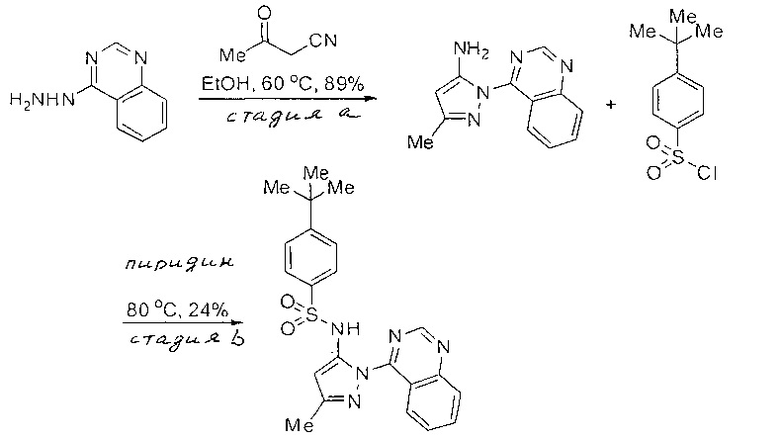
а) Для этого перемешанный раствор 4-гидразиноксиназолина (0.16 г, 1.0 ммоль) и 3-оксо-бутиронитрила (0.083 г, 1.0 ммоль) в этиловом спирте (10 мл) нагрет при 60°С в течение 18 часов. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 30%-ный этилацетат в гексане) с получением требуемого соединения (0.20 г, 0.089 ммоль, 89%).
b) Смесь 4-трет-бутилбензолсульфонилхлорида (0.084 г, 0.36 ммоль) и 3-метил-1-(хиназолин-4-ил)-1H-пиразол-5-амина (0.067 г, 0.30 ммоль) в пиридине (0.6 мл) нагрета при 80°С в течение 15 часов при перемешивании. После охлаждения до комнатной температуры к реакционной смеси добавляют дихлорметан и промывают 1 М водным гидросульфатом натрия (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 5-60% этилацетат в гексане) для получения названного соединения в виде белого твердого (0.30 г, 0.071 ммоль, 24%). 1Н NMR (400 МГц, CDCl3) δ 10.92 (s, 1Н), 9.27 (dd, J=1.2, 8.8 Гц, 1Н), 9.98 (s, 1Н), 7.99 (dd, J=0.8, 8.4 Гц, 1Н), 7.89 (ddd, J-1.2, 6.8, 8.4 Гц, 1Н), 7.66-7.61 (м, 2Н), 7.28-7.25 (м, 2Н), 6.34 (s, 1Н), 2.37 (s, 3Н), 1.13 (s, 9Н); MS: (ES) m/z вычислено для C22H24F3N5O2S [М+Н]+ 422.2, обнаружено 422.3.
Пример 2: Синтез 4-трет-бутил-N-(1-(изохинолин-4-ил)-3-метил-1H-пиразол-5-ил)бензенсульфонамида
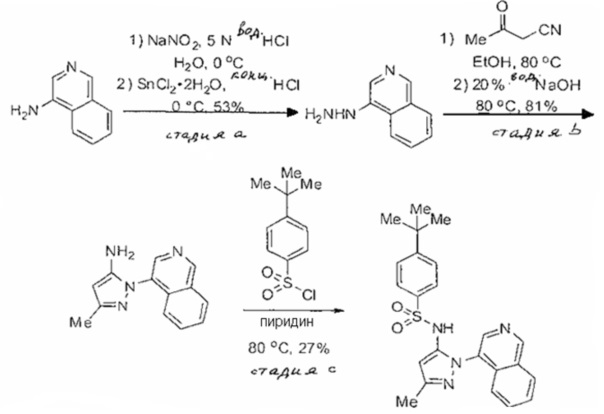
a) К перемешиваемому раствору 4-аминоизохинолина (1.4 г, 10.0 ммоль) в 5 N водной соляной кислоте (12 мл) при 0°С добавлен раствор азотистокислого натрия (NaNO2, 0.069 г, 10.0 ммоль) в деионизированной воде (1 мл) при поддержании внутренней температуры ниже 0°С. Реакционная смесь перемешана при 0°С в течение 30 минут и по каплям добавлен раствор дигидрата хлорида олова (II) (SnCl2⋅2Н2О, 5.6 г, 25.0 ммоль), растворенного в концентрированной соляной кислоте (5 мл). Смесь перемешана при комнатной температуре в течение 2 часов, и раствор отрегулирован до рН~12-14 20%-ной водной гидроокисью натрия. Смесь извлечена 2:1 CHCl3/iPrOH. Органический слой высушен (Na2SO4), профильтрован и сконцентрирован в вакууме. Полученный неочищенный продукт очищен флэш-хроматографией (SiO2, 50% этилацетат в гексане) с получением требуемого соединения (0.84 г, 5.3 ммоль, 53%).
b) Перемешиваемая суспензия 4-гидразинилизохинолина (0.32 г, 2.0 ммоль) и 3-оксо-бутиронитрила (0.16 г, 0.19 ммоль) в этиловом спирте (10 мл) нагрета при 80°С в течение 3 часов. После охлаждения до комнатной температуры 20% водная гидроокись натрия (1 мл) добавлена к реакционной смеси и далее нагрета при 80°С в течение 1 часа. Реакционная смесь охлаждена до комнатной температуры и сконцентрирована в вакууме. Неочищенный остаток растворен в 1:1 дихлорметан/метаноле (40 мл) и фазы разделены. Органический слой высушен (Na2SO4) и отфильтрован через фильтр Целита. Фильтрат сконцентрирован в вакууме, и неочищенный остаток очищен флэш-хроматографией (SiO2, 50-100% этилацетат в гексанах) с получением требуемого продукта (0.36 г, 1.6 ммоль, 81%).
с) Смесь 4-трет-бутилбензолсульфонилхлорида (0.10 г, 0.43 ммоль) и 1-(изохинолин-4-ил)-3-метил-1H-пиразол-5-амина (0.080 г, 0.36 ммоль) в пиридине (5 мл) нагрета при 80°С в течение 15 часов с перемешиванием. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 50-100% этилацетат в гексанах) с получением названного соединения как белого твердого (0.18 г, 0.12 ммоль, 27%). 1H NMR (400 МГц, CDCl3) δ 8.89 (s, 1Н), 8.02 (s 1Н), 8.87 (dd, J=1.6, 6.8 Гц, 1H), 7.58-7.53 (м, 2Н), 7.58 (s, 1Н), 7.56 (s, 1Н), 7.39-7.35 (м, 3Н), 6.25 (s, 1Н), 2.34 (s, 3Н), 1.32 (s, 9Н); MS: (ES) m/z вычислено для C23H25N4O2S [М+H]+ 421.2, обнаружено 422.2.
Пример 3: Синтез 4-трет-бутил-N-(1-(8-фторхинолин-4-ил)-3-метил-1H-пиразол-5-ил)бензолсульфонамида
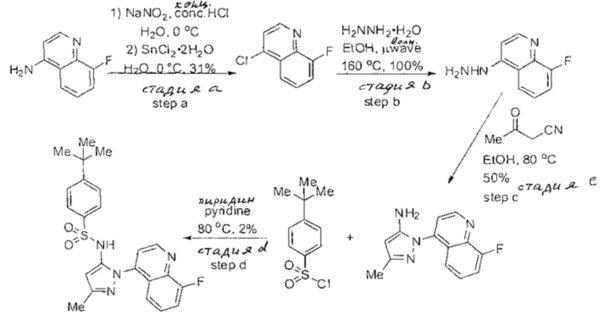
а) К перемешиваемому раствору 8-фторхинолин-4-амина (1.0 г, 6.2 ммоль) в концентрированной соляной кислоте (6.2 мл) и деионизированной воде (6.0 мл) при 0°С добавлен раствор NaNO2 (0.51 г, 7.4 ммоль) в деионизированной воде (3 мл). Реакционная смесь размешана при 0°С в течение 30 минут, и затем добавлен по каплям раствор SnCl2⋅2H2O (2.8 г, 12.4 ммоль), растворенного в деионизированной воде (3 мл). Смесь размешана при комнатной температуре в течение 2 часов, и суспензия превращена в основание водным бикарбонатом натрия. Смесь извлечена 2:1 CHCl3/iPrOH. Органический слой промыт водным бисульфатом натрия, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Полученный неочищенный продукт использован непосредственно без дальнейшей очистки (0.34 г, 1.9 ммоль, 31%).
b) Раствор неочищенного 4-хлор-8-фторхинолина (0.27 г, 1.5 ммоль) и NH2NH2⋅Н2О (1.5 мл, 16.6 ммоль) в этиловом спирте (1.6 мл) нагрет при 160°С в микроволнах в течение 1 часа с перемешиванием. После охлаждения смеси до комнатной температуры водный насыщенный бикарбонат натрия добавлен к реакционной смеси, и водный слой извлечен с 2:1 CHCl3/iPrOH (2×5 мл). Объединенные органические слои промыты водой, высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток использовался непосредственно без дальнейшей очистки (0.27 г, 1.5 ммоль, 100%).
c) Перемешиваемый раствор неочищенного 8-фтор-4-гидразинилхинолина (0.27 г, 1.5 ммоль) и 3-оксо-бутиронитрила (0.15 г, 0.19 ммоль) в пиридине (3 мл) нагрет при 80°С в течение 15 часов. После охлаждения до комнатной температуры водный насыщенный бикарбонат натрия добавлен к реакционной смеси и извлечен дихлорметаном (3×10 мл). Объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток использовался непосредственно без дальнейшей очистки (0.14 г, 0.76 ммоль, 50%).
d) Смесь 4-трет-бутилбензолсульфонилхлорида (0.13 г, 0.55 ммоль) и неочищенного 1-(8-фторхинолин-4-ил)-3-метил-1H-пиразол-5-амина (0.14 г, 0.55 ммоль) в пиридине (1 мл) нагрета при 80°С в течение 15 часов с перемешиванием. После охлаждения до комнатной температуры дихлорметан добавлен к реакционной смеси и промыт 1 М водного гидросульфата натрия (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен обратной фазой HPLC (С18колонка, ацетонитрил-H2O с 0.1% TFA как элюентом) с получением названного соединения как белого твердого (0.005 г, 0.011 ммоль, 2%). 1Н NMR (400 МГц, CDCl3) δ 8.80 (d, J=4.8 Гц, 1Н), 7.61 (d, J=1.6 Гц, 1Н), 7.59 (d, J=2.0 Гц, 1Н), 7.47-7.41 (м, 5Н), 7.13 (d, J=4.8 Гц, 1Н), 6.20 (s, 1Н), 2.35 (s 3Н), 1.35 (s, 9Н); MS: (ES) m/z вычислен для C23H24FN4O2S [М.+Н]+ 439.5, обнаружен 439.3.
Пример 4: Синтез 4-трет-бутил-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
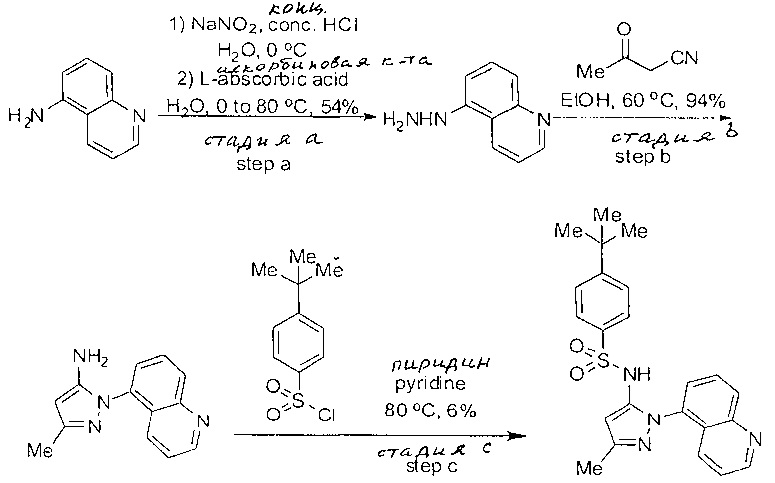
a) Раствор 5-аминохинолина (0.75 г, 5.2 ммоль) в концентрированной соляной кислоте (3.8 мл) смешивают при 0°С в течение 10 мин. Раствор азотистокислого натрия (0.43 г, 6.2 ммоль) в деионизированной воде (0.5 мл) добавлен к холодной реакционной смеси в течение более чем 10 минут и размешан при 0°С в течение 1 часа с формированием гетерогенной смеси. L-аскорбиновая кислота (0.95 г, 5.4 ммоль) затем добавлена к реакционной смеси в течение более чем 10 мин. Реакционная смесь нагрета до комнатной температуры и размешана в течение 45 мин. Жидкая реакционная суспензия затем нагрета при 80°С в течение 20 минут, и добавлена деионизированная вода (4 мл). Суспензия повторно охлаждена до 0°С и размешана в течение 2 часов. Твердое вещество собрано фильтрацией и промыто метанолом с получением требуемого продукта (0.45 г, 2.8 ммоль, 54%).
b) К перемешиваемой суспензии хинолин-5-ил-гидразина (0.25 г, 1.6 ммоль) в 3:1 этиловом спирте/деионизированной воде (2.5 мл) добавлен 3-оксо-бутиронитрил (0.13 г, 1.6 ммоль). Реакционная смесь затем нагрета при 60°С в течение 2 часов. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме, и полученный неочищенный продукт использован непосредственно без дальнейшей очистки (0.21 г, 1.5 ммоль, 94%)
с) Смесь 4-трет-бутилбензолсульфонилхлорида (0.59 г, 2.5 ммоль) и неочищенного 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амииа (0.47 г, 2.1 ммоль) в пиридине (0.6 мл) нагрета при 80°С в течение 15 часов с перемешиванием. После охлаждения до комнатной температуры дихлорметан добавлен к реакционной смеси и промыт 1 М водного годросульфата натрия (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 1-10% метанол, содержащий 10% гидроокиси аммония в дихлорметане) с получением названного соединения как белого твердого (0.18 г, 0.12 ммоль, 6%). 1H NMR (400 МГц, CDCl3) δ 8.87 (dd, J=1.2, 4.0 Гц, 1Н), 8.11 (d, J=8.4 Гц, 1Н), 7.58-7.53 (м., 4Н), 7.42 (s 1Н), 7.40 (s, 1Н), 7.29-7.23 (м, 1H), 6.94 (d, J=7.2 Гц, 1H), 6.28 (s, 1Н), 2.34 (s, 3H), 1.36 (s, 9Н); MS: (ES) m/z вычислено для C23H25N4O2S [Ml+Н]+ 421.2, обнаружено 421.3.
Пример 5: Синтез N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)-4-(трифторметокси)бензолсульфонамида
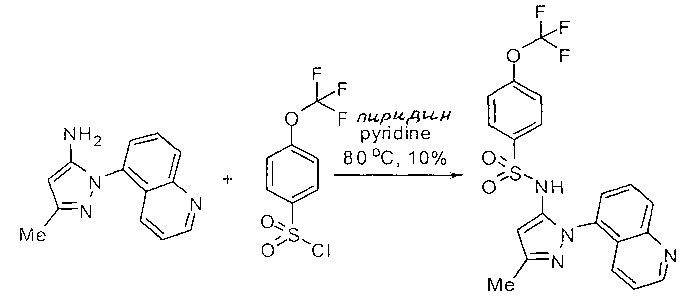
Перемешиваемая смесь 4-(трифторметокси)бензол-1-сульфонилхлорида (0.060 г, 0.23 ммоль) и 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап b, 0.050 г, 0.22 ммоль) в пиридине (1.0 мл) нагрета при 80°С в течение 1 часа. После охлаждения до комнатной температуры дихлорметан добавлен к реакционной смеси и промыт 1 М водного гидросульфата натрия (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 2-10% метанол в дихлорметане) и очищен обратной фазой HPLC (С 18 колонка, ацетонитрил-H2O с 0.1% TFA как элюеита) с получением названного соединения как белого твердого (0.010 г, 0.022 ммоль, 10%). 1Н NMR (400 МГц, CDCl3) δ 8.75 (d, J=4.0 Гц, 1H), 8.01 (d, J=8.8 Гц, 1Н), 7.71 (s, 1H), 7.69 (s, 1H), 7.56 (t, J=8.4 Гц, 1Н), 7.49 (d, J=8.4 Гц, 1H), 7.21-7.17 (м, 4Н), 6.12 (s, 1Н), 2.34 (s, 3H); MS: (ES) m/z вычислено для C20H16F3N4O3S [М+H]+ 449.1, обнаружено 449.7.
Пример 6: Синтез 4-этил-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
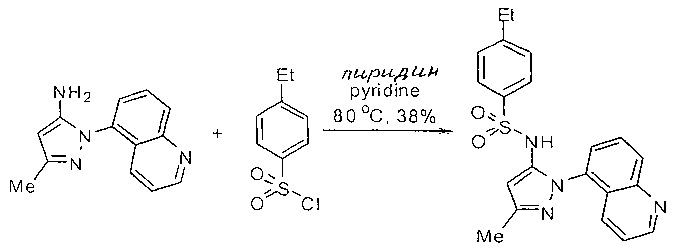
Смесь 4-этилбензолсульфонилхлорида (0.033 г, 0.16 ммоль) и 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получена из Примера 4 этап b, 0.030 г, 0.13 ммоль) в пиридине (1.0 мл) нагрета при 80°С в течение 2 часов с перемешиванием. После охлаждения до комнатной температуры дихлорметан добавлен к реакционной смеси и промыт I М водного гидросульфата натрия (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как элюэнт) с получением названного соединения как белого твердого (0.019 г, 0.049 ммоль, 38%). 1H NMR. (400 МГц, CDCl3) δ 8.86 (dd,J=2.0, 4.0 Гц, 1Н), 8.12 (d., J=8.8 Гц, 1Н), 7.60 (dd, J=7.2, 8.4 Гц, 1Н), 7.51-7.46 (м., 3Н), 7.27-7.23 (м, 1Н), 7.15 (s, 1Н), 7.13 (s, 1Н), 7.06 (d, J=7.6 Гц, 1Н), 6.27 (s, 1Н), 2.68 (q, J=7.6 Гц, 2Н), 2.34 (s, 3Н), 1.27 (t, J=7.6 Гц, 3Н); MS: (ES) m/z вычислено для C21H21N4O2S [М+Н]+ 393.2, обнаружено 393.2
Пример 7: Синтез 4-изопропил-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
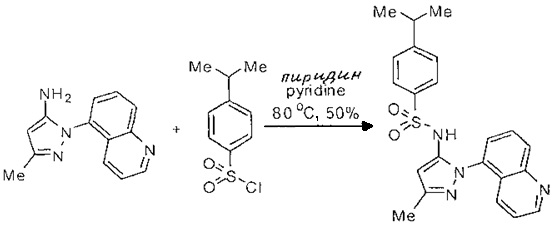
Смесь 4-трет-пентилбензолсульфонилхлорида (0.028 г, 0.13 ммоль) и 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап b, 0.023 г, 0.10 ммоль) в пиридине (1.0 мл) нагрета при 80°С в течение 18 часов с перемешиванием. После охлаждения до комнатной температуры дихлорметан добавлен к реакционной смеси и промыт 1 М водного гидросульфата натрия (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетоыитрил-H2O с 0.1% TFA как элюент) с получением названного соединения как белого твердого (0.020 г, 0.05 ммоль, 50%). 1H NMR (400 МГц, CDCl3) δ 9.02 (d, J=4.4 Гц, 1Н), 8.43 (d, J=8.8 Гц, 1Н), 8.30 (d, J=8.4 Гц, 1Н), 7.87-7.33 (м, 2Н), 7.72-7.70 (м, 3Н), 7.35 (s, 1Н), 7.33 (s, 1Н), 5.94 (s, 1Н), 3.04-2.98 (м, 1Н), 2.32 (s, 3Н), 1.29 (s, 6Н); MS: (ES) m/z вычислено для C22H23N4O2S [М+Н]+ 407.2, обнаружено 407.0.
Пример 8: Синтез 4-изопропокси-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
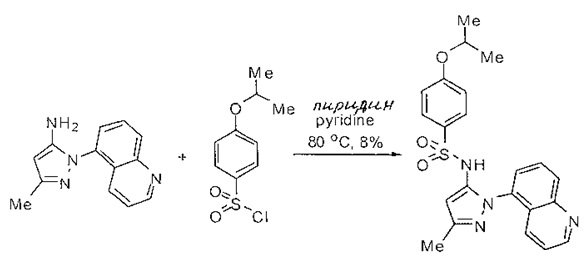
Перемешиваемая смесь 4-изопропоксибензол-1-сульфонилхлорида (0.10 г, 0.52 ммоль) и 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из от Примера 4 этап о, 0.10 г, 0.44 ммоль) в пиридине (2 мл) нагрета при 80°С в течение 1 часа. После охлаждения до комнатной температуры водный насыщенный бикарбонат натрия добавлен к реакционной смеси и извлечен дихлорметаном. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 2-10% метанол в дихлорметане), с последующей обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA на элюэнт) с получением названного соединении как белого твердого (0.015 г, 0.036 ммоль, 8%). 1Н NMR (400 МГц, CDCl3) δ 8.58 (dd, J=2.0, 4.0 Гц, 1Н), 8.09 (d, J=8.4 Гц, 1Н), 7.61 (dd, J=7.6, 8.4 Гц, 1Н), 7.50 (d, J=2.0 Гц, 1Н), 7.48 (d, J=1.6 Гц, 1H), 7.25-7.21 (м, 2Н), 7.14 (dd, J=0.8, 7.2 Гц, 1Н), 6.74 (d, J=2.4 Гц, 1H), 6.72 (d, J=2.0 Гц, 1Н), 6.25 (s, 1H), 4.57 (hept, J=6.0 Гц, 1H), 2.33 (s, 3H), 1.39 (d, J=6.4 Гц, 6H); MS: (ES) m/z вычислено для C23H23N4O3S [M+H]+ 423.2, обнаружено 423.0.
Пример 9: Синтез 4-изобутокси-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
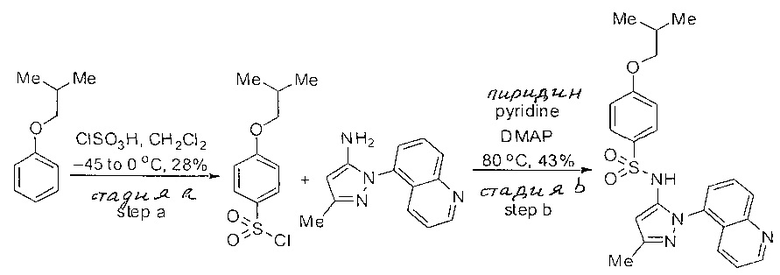
a) К перемешиваемому раствору изобутоксибензола (0.60 г, 4.0 ммоль) в дихлорметане (5 мл) при -45°С добавлена хлорсульфоновая кислота (0.6 мл, 9.1 ммоль) по каплям, и реакционная смесь размешана при -45°С течение 1 часа. Реакционная смесь затем нагрета при 0°С, и дополнительно хлорсульфоновая кислота (0.6 мл, 9.1 ммоль) добавлена по каплям. Реакционная смесь затем размешана при 0°С в течение 1 часа и влита в лед. Водный слой извлечен этилацетатом, и органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 5-10% этилацетат в гексанах) с получением 4-изобутоксибензол-1-сульфонилхлорида (0.32 г, 1.1 ммоль, 28%).
b) Размешиваемая смесь 4-изобутоксибензол-1-сульфонилхлорида (0.060 г, 0.24 ммоль), 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап b, 0.050 г, 0.22 ммоль), и 4-(диметиламино) пиридина (DMAP, 0.025 г, 0.20 ммоль) в пиридине (2 мл) нагрета при 80°С в течение 2 часов. После охлаждения до комнатной температуры водный насыщенный бикарбонат натрия добавлен к реакционной смеси и извлечен дихлорметаном. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 2-5% метанол в дихлорметане), с последующей обратной фазой HPLC (колонка С18, ацетон итрил-H2O с 0.1% TFA как элюентом) с получением названного соединении как белого твердого (0.04 1 г, 0.094 ммоль, 43%). 1H NMR (400 МГц, CDCl3) δ 8.84 (d, 7=4.0 Гц, 1H), 8.11 (d, J=8.0 Гц, 1H), 7.63 (t, J=8.0 Гц, 1H), 7.48-7.44 (м, 2H), 7.23 (d,.7=4.0, Гц, 1H), 7.21 (d, J=4,0, Гц, 1H), 7.15 (dd, J=1.2, 7.2 Гц, 1H), 6.72 (d, J=2.4 Гц, 1Н), 6.70 (d, J=2.4 Гц, 1Н), 6.25 (s, 1Н), 3.72 (dd, J=2.0, 6.4 Гц, 2Н), 2.33 (s, 3Н), 2.13 (hept, J=6.4 Гц, 1H), 1.08 (dd, J=2.4, 6.4 Гц, 6Н); MS: (ES) m/z вычислено для C23H25N4O2S [М+H]+ 437.2, обнаружено 437.0.
Пример 10: Синтез N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)-4-трет-пентилбензолсульфонамида
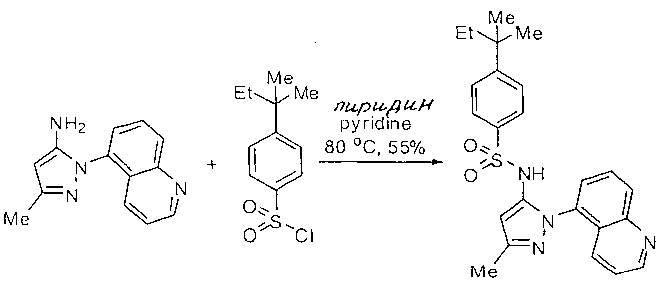
Смесь 4-трет-пентилбензолсульфонилхлорида (0.13 г, 0.53 ммоль) и 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап b, 0.10 г, 0.44 ммоль) в пиридине (1.0 мл) нагрета при 80°С в течение 3 часов с перемешиванием. После охлаждения до комнатной температуры дихлорметан добавлен к реакционной смеси и промыт 1М водного гидросульфата натрия (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы а вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как элюентом) с получением названного соединения как белого твердого (0.11 г, 0.24 ммоль, 55%). 1H NMR (400 МГц, CDCl3) δ 8.96 (dd, J=1.6, 4.8 Гц, 1Н), 8.20 (d, J=8.8 Гц, 1Н), 8.13 (d, J=8.4 Гц, 1H), 7.75 (d, J=7.6 Гц, 1Н), 7.71 (s, 1H), 7.69 (s, 1H), 7.54 (dd, J=4.8, 8.4 Гц, 1Н), 7.46-7.43 (м, 3H), 6.02 (s, 1Н), 2.32 (s, 3Н), 1.70 (q, J=7.2 Гц, 2Н), 1.34 (s, 6Н), 0.70 (t, J=7.2 Гц, 3Н); MS: (ES) m/z вычислено для C24H27N4O2S [М+H]+ 435.2, обнаружено 435.1.
Пример 11: Синтез 4-(2-гидроксипропан-2-ил)-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
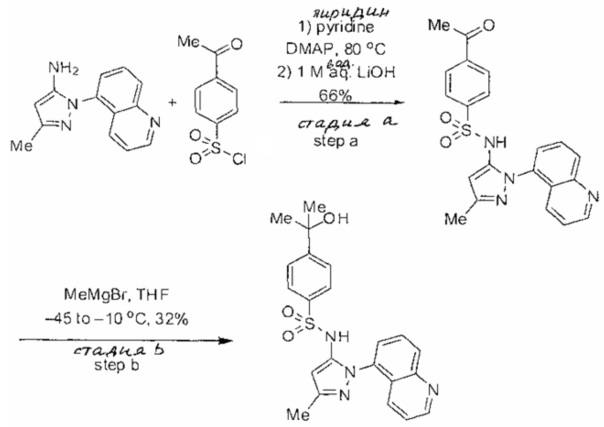
a) Смесь 4-ацетилбензол-1-сульфонилхлорида (0.050 г, 0.22 ммоль), 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап b, 0.060 г, 0.27 ммоль), и DMAP (0.027 г, 0.22 ммоль) в пиридине (2 мл) нагрета при 80°С в течение 2.5 часов с перемешиванием. После охлаждения до комнатной температуры к реакционной смеси добавлен 1 М водной гидроокиси лития (2 мл) и размешана в течение 2 часа. Раствор дополнен 4:1 дихлорметаном/метанолом и промыт 1 М водного хлористого аммония (5 мл). Раствор отрегулирован до pH~8-9 гидроокисью аммония, и фазы разделены. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 2-5% метанол в дихлорметане). Продукт затем рекристаллизован в минимальном количестве 4:1, дихлорметан/метанола, и твердое собрано фильтрацией с получением требуемого твердого (0.059 г, 0.1 5 ммоль, 66 %).
b) Раствор метилмагнийбромида (1.4 М раствора в 3:1 толуол/THF, 1.4 мл, 2.0 ммоль) добавлен в колбу, содержащую 4-ацетил-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамид (0.059 г, 0.15 ммоль) в THF (6 мл) при -45°С с перемешиванием. Реакционная смесь медленно нагрета до -10°С в течение более чем 1 часа и добавлен дихлорметан/метанол 4:1 (2 мл). Органический слой промыт водным насыщенным хлористым аммонием, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как элюентом) с получением названного соединения как белого твердого (0.020 г, 0.047 ммоль, 32%). 1H NMR (400 МГц, CD3OD) δ 8.81 (d, J=4.4 Гц, 1Н), 8.05 (d, J=8.8 Гц, 1Н), 7.54 (t, J=8.0 Гц, 1H), 7.69 (d, J=8.0 Гц, 1Н), 7.67 (s, 1Н), 7.65 (s, 1Н), 7.51-7.47 (м, 3Н), 7.36 (dd, J=4.4, 8.4 Гц, 1Н), 5.73 (s, 1Н), 2.15 (s, 3Н), 1.56 (s, 6H); MS: (ES) m/z вычислено для C22H23N4O3S [М+H]+ 433.2, обнаружено 433.0.
Пример 12: Синтез 2,2-диметил-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)-2,3-дигидробензофуран-5-сульфонамида
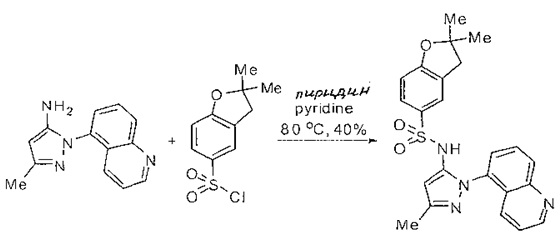
При перемешивании смесь 2,2-диметил-2,3-дигидро-1-бензолфуран-5-сульфонилхлорида (0.10 г, 0.41 ммоль) и 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап и, 0.11 г, 0.49 ммоль) в пиридине (0.41 мл) нагрета при 80°С в течение 1 часа. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 0-20% этилацетат в гексанах) с последующим обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как элюент) с получением названного соединения как белого твердого (0.070 г, 0.16 ммоль, 40%). 1Н NMR (400 МГц, CDCl3) δ 8.82 (s, 1Н), 8.06 (d, J=8.0 Гц, 1Н), 7.60 (1, J=7.6 Гц, 1Н), 7.54 (d, J=8.0 Гц, 1Н), 7.42 (d, J=8.4 Гц, 1H), 7.31 (s, 1H), 7.24 (d, J=3.2 Гц, 1Н), 7.19 (d, J=6.8 Гц, 1Н), 6.61 (d, J=8.8 Гц, 1Н), 6.24 (s, 1Н), 2.89 (s, 2Н), (ES) m/z вычислено для C23H23N4O3S [М+Н]+ 435.2, найдено 435.3.
Пример 13: Синтез 2,2-диметил-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-мл)хроман-6-сульфонамида
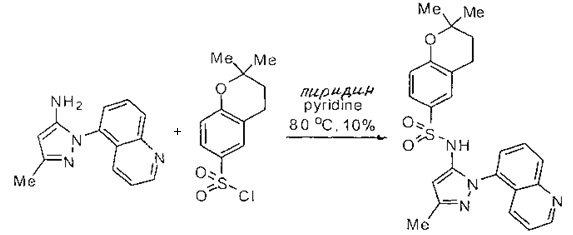
При перемешивании смесь 2,2-диметилхроман-6-сульфонилхлорида (0.050 г, 0.22 ммоль) и 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап b, 0.068 г, 0.26 ммоль) в пиридине (1.0 мл) нагрета при 80°С в течение 15 часов. После охлаждения до комнатной температуры дихлорметан добавлен к реакционной смеси и промыт 1М водного насыщенного бисульфата (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Полученный неочищенный остаток очищен флэш-хроматографией (SiO2, 0-20% этилацетат в гексанах) с последующим обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.010 г, 0.022 ммоль, 10%). 1Н NMR (400 МГц, CDCl3) δ 9.09 (d, J=4.8 Гц, 1H), 8.38 (d, J=8.4 Гц, 1Н), 8.29 (d, J=8.4 Гц, 1Н), 7.87 (t, J=7.6 Гц, 1H), 7.66 (dd, J=4.8, 8.8 Гц, 1Н), 7.61 (d, J=7.6 Гц, 1Н), 7.40-7.38 (м, 2Н), 6.72 (d, J=9.2 Гц, 1H), 6.09 (s, 1H), 2.71 (t, J=6.4 Гц, 2Н), 2.34 (s, 3H), 1.83 (1, J=6.4 Гц, 2Н), 1.36 (s, 6H); MS: (ES) m/z вычислено для C24H25N4O3S [М+Н]+ 449.2, найдено 449.1
Пример 14: Синтез 1,1-диметил-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)-2,3-дигидро-1H-инден-5-сульфонаммда
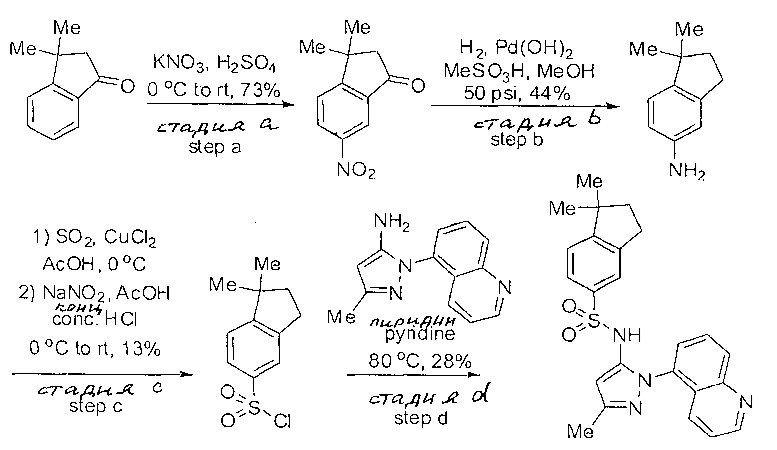
a) К перемешиваемому раствору 3,3-диметил-1-инданона (0.10 г, 0.64 ммоль) в серной кислоте (0.63 мл) при 0°С добавлен азотнокислый калий (KNO3, 0.063 г, 0.63 ммоль) в серной кислоте (0.2 мл). Реакционная смесь перемешана при 0°С в течение I часа, затем нагрета до комнатной температуры и перемешана в течение 15 часов. Реакционная смесь охлаждена льдом, и водный слой извлечен этилацетатом. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 20% этилацетат в гексанах) с получением требуемого продукта (0.096 г, 0.47 ммоль, 73%).
b) В колбе шейкера Парра 3,3-диметил-6-нитро-1-инданон (1.0 г, 4.8 ммоль) и гидроокись палладия на углероде (Pd(OH)2, 20% по весу, 0.52 г) в метаноле (2 мл) и метановой сульфокислоте (MeSO3H, 0.4 мл, 6.2 ммоль) гидрирована при 50 psi в течение 1.5 часа. Реакционная смесь разбавлена метанолом и профильтрована через фильтр Целита. Фильтрат сконцентрирован с вакууме, и полученный неочищенный остаток очищен флэш-хроматографией (SiO2, 100% этилацетат) с получением требуемого продукта (0.34 г, 2.1 ммоль, 44%).
c) Раствор ледяной уксусной кислоты (8 мл) при 0°С барботирован в газе двуокиси серы (SO2) в течение 30 мин. Хлорид меди (II) (CuCl2, 0.29 г, 2.16 ммоль) добавлен к реакционной смеси и размешан в течение 30 минут при 0°С с получением синего/зеленого раствора. В другую колбу, содержащую 1,1-диметмлиндап-5-амин (0.34 г, 2.13 ммоль) в концентрированной соляной кислоте (4.2 мл) при 0°С добавлен NaNO2 (0.22 г, 3.2 ммоль) и размешан в течение 30 мин. Этот раствор диазония затем медленно добавлен к готовому медному раствору и размешан при 0°С в течение 30 мин. Реакционная смесь затем медленно нагрета до 70°С в течение более чем 2 часов. После охлаждения до комнатной температуры реакционная смесь охлаждена деионизированной водой, и водный слой извлечен этилацетатом (2×10 мл). Объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 0-10% этилацетат в гексанах) с получением требуемого продукта (0.067 г, 0.27 ммоль, 13%).
d) Смесь 1,1-диметилиндан-5-сульфонилхлорида (0.030 г, 0.13 ммоль) и 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получен из Примера 4 этап b, 0.028 г, 0.12 ммоль) в пиридине (0.12 мл) нагрета при 80°С в течение 1 часа с перемешиванием. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме. Полученный неочищенный остаток очищен флэш-хроматографией (SiO2, 20% этилацетат в гексанах) с последующим обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.015 г, 0.036 ммоль, 28%). 1Н NMR (400 МГц, DMSO-d6) δ 10.30 (s, 1Н), 8.90 (d, J=3.2 Гц, 1Н), 8.06 (d, J=8.4 Гц, 1Н), 7.73 (t, J=8.0 Гц, 1Н), 7.57 (d, J=9.2 Гц, 1Н), 7.46 (dd, J=4.0, 8.4 Гц, 1Н), 7.30-7.26 (м., 2Н), 7.18 (s, 1Н), 7.11 (d, J=8.0 Гц, 1Н), 6.05 (s, 1Н), 2.71 (d, J=7.6 Гц, 2Н), 2.20 (s, 3Н), 1.84 (d, J=7.2 Гц, 2Н), 1.18 (s, 6Н); MS: (ES) m/z вычислено для C24H25N4O2S [М+Н]+ 433.2, найдено 433.1.
Пример 15: Синтез 3-фтор-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)-4-морфолинобензолсульфонамида
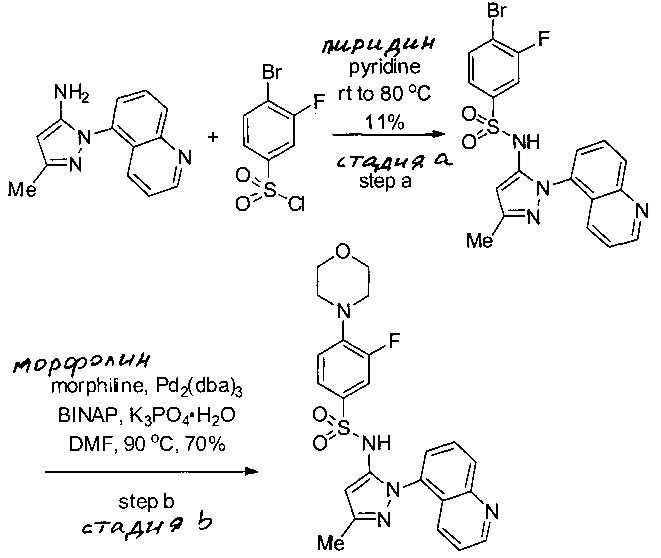
a) Смесь 4-бром-3-фторбензол-1-сульфонилхлорида (1.4 г, 5.2 ммоль) и 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап b, 0.90 г, 4.0 ммоль) в пиридине (10 мл) перемешана при комнатной температуре в течение 2 часов, затем нагрета при 80°С в течение 2 часов. После охлаждения до комнатной температуры 1 N водной соляной кислоты (1 мл) добавлен к реакционной смеси и извлечен дихлорметаном (2×5 мл). Объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 0-10% метанол в этилацетате) с получением требуемого продукта (0.20 г, 0.43 ммоль, 11%).
b) При перемешивании смесь 4-бром-3-фтор-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида (0.07 г, 0.15 ммоль), морфолина (0.066 г, 0.75 ммоль), трис-(дибензилиденацетон)дипалладия (0) (Ра2(dba)3, 0.007 г, 0.008 ммоль), 2,2'-бис-(дифенилфосфино)-1,1'-бинафтила (BINAP, 0.014 г, 0.023 ммоль) и одноосновного фосфорнокислого калия (K3PO4⋅Н2О, 0.21 г, 0.90 ммоль) в безводном N,N-диметилформамиде (DMF, 6 мл) очищена азотом в течение 5 мин. Реакционная смесь нагрета при 90°С в течение 4 часов. После охлаждения до комнатной температуры этилацетат (10 мл) добавлен к реакционной смеси и промыт водным насыщенным бикарбонатом натрия. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Получающийся неочищенный материал очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.049 г, 0.11 ммоль, 70%). 1Н NMR (400 МГц, CDCl3) δ 9.06 (dd, J=1.6, 4.8 Гц, 1Н), 8.36 (d, J=8.4 Гц, 1Н), 8.31 (d, J=8.0 Гц, 1Н), 7.88 (dd, J=8.4, 9.6 Гц, 1Н), 7.68 (d, J=4.8 Гц, 1Н), 7.66 (d, J=6.4 Гц, 1Н), 7.37 (dd, J=1.6, 8.4 Гц, 1Н), 7.31 (dd, J=2.4, 12.4 Гц, 1Н), 6.80 (t, J=6.8 Гц, 1Н), 6.09 (s, 1Н), 3.90-3.86 (м, 4Н), 3.21-3.18 (м, 4Н), 2.35 (s, 3Н); MS: (ES) m/z вычислено для C23H23FN5O3S [М+Н+ 468.2, найдено 468.2.
Пример 16: Синтез 4-трет-бутил-N-(1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
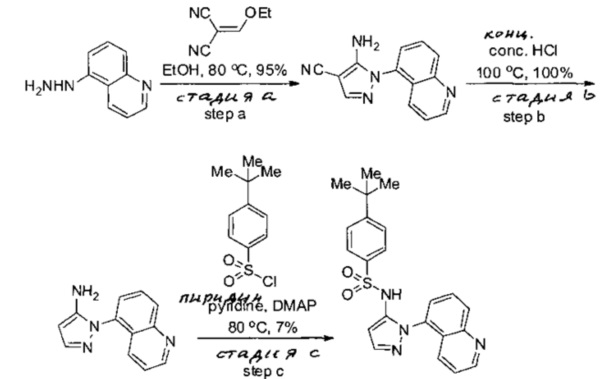
a) Перемешиваемый раствор (этоксиметилен)малононитрила (0.38 г, 3.2 ммоль) и хинолин-5-ил-гидразина (получено из Примера 4 этап а, 0.5 г, 3.2 ммоль) в этиловом спирте (5 мл) нагрет при 80°С в течение* 15 часов. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме, и неочищенное твердое использовано непосредственно без дальнейшей очистки (0.70 г, 3.0 ммоль, 95%).
b) Раствор неочищенного 5-амино-1-(хинолин-5-ил)-1H-пиразол-4-карбонитрила (0.40 г, 1.7 ммоль) в концентрированной соляной кислоте (5 мл) нагрет при 100°С в течение часов с перемешиванием. Реакционная смесь охлаждена до комнатной температуры и превращена в основание водным насыщенным бикарбонатом натрия. Водный слой извлечен 2:1 хлороформ/iPrOH, и органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток использован непосредственно без дальнейшей очистки (0.36 г, 1.7 ммоль, 100%).
с) Размешиваемая смесь сырья 1-(хинолин-5-ил)-1H-пиразол-5-амина (0.080 г, 0.38 ммоль), 4-трет-бутилбензолсульфонилхлорида (0.13 г, 0.57 ммоль) и DM АР (0.068 г, 0.57 ммоль) в пиридине (1.5 мл) нагрета при 80°С в течение 2 часов. После охлаждения до комнатной температуры водный насыщенный бикарбонат натрия добавлен к реакционной смеси и извлечен 2:1 хлороформ/iPrOH. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% NFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.010 г, 0.025 ммоль, 7%). 1H NMR (400 МГц, CD3OD) δ 8.87 (dd, J=2.0, 4.4 Гц, 1Н), 8.13 (d, J=8.4 Гц, 1H), 7.76 (dd, J=7.2, 8.8 Гц, 1Н), 7.69 (d, J=2.4 Гц, 1Н), 7.65-7.62 (м, 1Н), 7.52-7.42 (м, 3Н), 7.51 (d, J=8.8 Гц, 1Н), 7.43 (d, J=8.8 Гц, 1Н), 7.28 (dd, J=1.2, 7.2 Гц, 1H), 6.25 (s, 1H), 1.35 (s, 9Н); MS: (ES) m/z вычислено для C22H23N4O2S [М+Н]+ 407.2, найдено 407.0.
Пример 17: Синтез 4-трет-бутил-N-(3-этил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
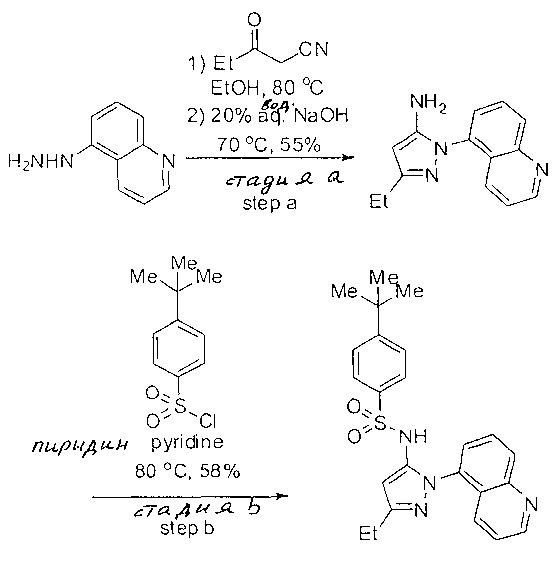
a) Раствор 3-оксопентаннитрила (0.74 г, 7.6 ммоль) и хинолин-5-ил-гидразина (получено из Примера 4 этап а, 1.0 г, 6.3 ммоль) в этиловом спирте (5 мл) нагрет при 80°С в течение 3 часов с перемешиванием. После охлаждения до комнатной температуры 20% водная гидроокись натрия (1.5 мл) добавлена к реакционной смеси и затем нагрета при 70°С в течение 3 часов. Реакционная смесь охлаждена до комнатной температуры и сконцентрирована в вакууме. Неочищенный остаток растворен в 1:1 дихлорметан/метаноле (40 мл) и фазы разделены. Органический слой высушен (Na2SO4) и профильтрован через фильтр Целита. Фильтрат сконцентрирован в вакууме, и неочищенный остаток очищен флэш-хроматографией (SiO2, 1-10% метанол, содержащий 10% гидроокись аммония в дихлорметане) с получением требуемого продукта (0.83 г, 3.5 ммоль, 55%).
b) Смесь 4-трет-бутилбензолсульфонилхлорида (0.064 г, 0.27 ммоль) и 3-этил-1-(хинолин-5-ил)-1H-пиразол-5-амина (0.05 г, 0.21 ммоль) в пиридине (0.5 мл) нагрета при 80°С в течение 15 часов с перемешиванием. После охлаждения до комнатной температуры дихлорметан добавлен к реакционной смеси и промыт 1 М водного гидросульфата натрия (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и концентрированы в вакууме. Неочищенный остаток растворен в метаноле (3 мл) и 1 М водной гидроокиси натрия (1.0 мл, 1.0 ммоль). Реакционная смесь размешана при комнатной температуре в течение 1 часа, и растворитель удален в вакууме. Получающийся остаток разделен между дихлорметаном (3 мл) и 1 М водного гидросульфата натрия (3 мл) и фазы разделены. Водный слой извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.053 г, 0.12 ммоль, 58%). 1Н NMR (400 МГц, CDCl3) δ 9.00 (d, J=3.6 Гц, 1Н), 8.23 (d, J=8.8 Гц, 1Н), 8.19 (d, J=8.0 Гц, 1Н), 7.79 (dd, J=8.0, 8.4 Гц, 1Н), 7.69-7.65 (м, 2Н), 7.59 (dd, J=4.4, 8.8 Гц, 1Н), 7.51-7.47 (м, 3Н), 6.05 (s, 1Н), 2.69 (q, 7=7.6 Гц, 2Н), 1.37 (s, 9Н), 1.29 (t, J=7.6 Гц, 3Н); MS: (ES) m/z вычислено для C24H27N4O2S [М+Н]+ 435.2, найдено 435.2.
Пример 18: Синтез 4-трет-бутил-N-(3-циклопропил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
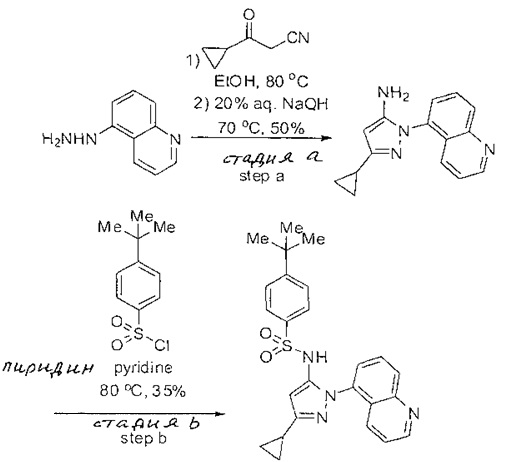
a) Раствор 3-циклопропил-3-оксопропаннитрила (0.74 г, 7.6 ммоль) и хинолин-5-ил-гидразина (получено из Примера 4 этап а, 1.0 г, 6.3 ммоль) в этиловом спирте (5 мл) нагрет при 80°С в течение 3 часов с перемешиванием. После охлаждения до комнатной температуры 20% водная гидроокись натрия (1.5 мл) добавлена к реакционной смеси и нагрета при 70°С в течение 3 часов. Смесь охлаждена до комнатной температуры и сконцентрирована в вакууме. Неочищенный остаток растворен в 1:1 дихлорметан/метаноле (40 мл) и фазы разделены. Органический слой высушен (Na2SO4) и профильтрован через фильтр Целита. Фильтрат сконцентрирован в вакууме, и неочищенный материал очищен флэш-хроматографией (SiO2, 1-10% метанол, содержащий 10% гидроокись аммония в дихлорметане) с получением требуемого продукта (0.80 г, 3.2 ммоль, 50%).
b) Смесь 4-трет-бутилбензолсульфонилхлорида (0.061 г, 0.26 ммоль) и 3-циклопропил-1-(хинолин-5-ил)-1H-пиразол-5-амина (0.05 г, 0.20 ммоль) в пиридине (1 мл) нагрета при 80°С в течение 15 часов с перемешиванием. После охлаждения до комнатной температуры дихлорметан добавлен к реакционной смеси и промыт 1 М водного гидросульфата натрия (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Получающийся неочищенный остаток растворен в метаноле (3 мл) и 1 М водной гидроокиси натрия (1.0 мл, 1.0 ммоль) и размешан при комнатной температуре в течение 1 часа. Реакционная смесь сконцентрирована в вакууме, и получающийся остаток разделен между дихлорметаном (3 мл) и 1 М водного гидросульфата натрия (3 мл). Фазы разделены, и водный слой извлечен дихлорметаном (2×5 мл). Объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-Н2О с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.031 г, 0.070 ммоль, 35%). 1Н NMR (400 МГц, CDCl3) δ 8.84 (d, J=4.0 Гц, 1Н), 8.08 (d, J=8.0 Гц, 1Н), 7.55-7.51 (м, 5Н), 7.41 (s, 1Н), 7.39 (s, 1Н), 6.94 (d, J=7.2 Гц, 1Н), 6.12 (s, 1Н), 1.99-1.93 (м, 1Н), 1.36 (s, 9Н), 1.01-0.92 (м, 2Н), 0.84-0.79 (м, 2Н); MS: (ES) m/z вычислено для C25H27N4O2S [М+Н]+ 447.2, найдено 447.2.
Пример 19: Синтез 4-трет-бутил-N-(3-(цианометил)-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
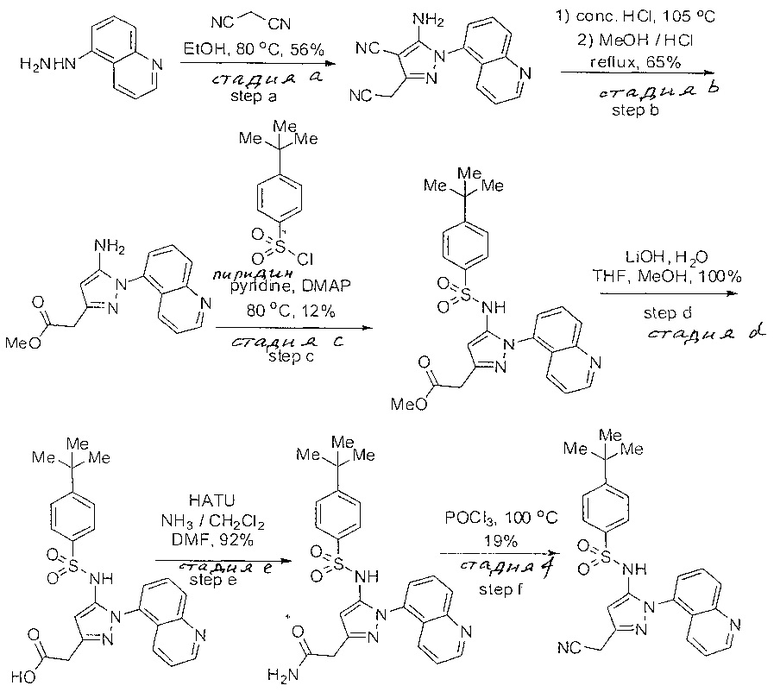
a) К перемешиваемому раствору хинолин-5-ил-гидразина (получено из Примера 4 этап а, 0.62 г, 3.9 ммоль) в этиловом спирте (4 мл) добавлен малононитрил (0.51 г, 7.7 ммоль). Реакционная смесь нагрета при 80°С в течение 15 часов. После охлаждения до комнатной температуры водный насыщенный хлористый аммоний (1.5 мл) добавлен к реакционной смеси и извлечен 2:1 хлороформ/iPrOH. Органический слой промыт соляным раствором, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 0-5% метанол в дихлорметане) с получением требуемого продукта как светло-коричневого твердого (0.60 г, 2.2 ммоль, 56%).
b) Раствор 5-амино-3-(цианометил)-1-(хинолин-5-ил)-1H-пиразол-4-карбонитрила (0.60 г, 2.2 ммоль) в концентрированной соляной кислоте (50 мл) нагрет при 105°С в течение 22 часов с перемешиванием. Реакционная смесь охлаждена до комнатной температуры, и раствор сконцентрирован в вакууме. Метанол (50 мл) и концентрированная соляная кислота (0.5 мл) добавлены к получающемуся остатку, и реакционная смесь нагрета с обратным холодильником в течение 3 часов. После охлаждения до комнатной температуры реакционная смесь превращена в основание водным насыщенным бикарбонатом натрия. Водный слой извлечен 2:1 хлороформом/iPrOH, и органический слой извлечен водным насыщенным хлористым аммонием, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток использован непосредственно без дальнейшей очистки (0.40 г, 1.7 ммоль, 65%).
c) Смесь неочищенного метил-2-(5-амино-1-(хинолин-5-ил)-1H-пиразол-3-ил) ацетата (0.55 г, 2.0 ммоль), 4-треот-бутилбензолсульфонилхлорида (0.30 г, 1.3 ммоль) и DMAP (0.12 г, 1.0 ммоль) в пиридине (5 мл) нагрета при 80°С в течение 2 часов с перемешиванием. После охлаждения до комнатной температуры к реакционной смеси добавлен дихлорметан и промыт 1 М водного гидросульфата натрия (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и комбинированные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-Н2О с 0.1% TFA как растворителем для элюирования) с получением белого твердого (0.075 г, 0.16 ммоль, 12%).
d) К суспензии метил-2-(5-(4-трет-бутилфенилсульфонамидо)-1-(хинолин-5-ил)-1H-пиразол-3-ил)ацетата (0.066 г, 0.14 ммоль) в THF (1 мл) и метаноле (1 мл) добавлен раствор гидроокиси лития (0.05 г, 2.1 ммоль) в деионизированной воде (0.5 мл). Реакционная смесь перемешана при комнатной температуре в течение 1 часа, и получающийся раствор отрегулирован до рН~5,5 N водной соляной кислотой. Водный слой извлечен 2:1 хлороформом/iPrOH, и органический слой промыт соляным раствором, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный материал использован непосредственно без дальнейшей очистки (0.065 г, 0.14 ммоль, 100%).
e) К размешиваемому раствору неочищенной 2-(5-(4-трет-бутилфенилсульфонамидо)-1-(хинолин-5-ил)-7H-пиразол-3-ил) уксусной кислоты (0.065 г, 0.14 ммоль) и N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил) уроний гексафторфосфата (HATU, 0.11 г, 0.28 ммоль) в DMF (1.5 мл) добавлен раствор насыщенного аммония в дихлорметане (0.5 мл). Реакционная смесь размешана при комнатной температуре в течение 1 часа, и добавлен соляной раствор. Водный слой извлечен 2:1 хлороформом/iPrOH, и органический слой промыт соляным раствором, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный материал использован непосредственно без дальнейшей очистки (0.060 г, 0.14 ммоль, 92%).
f) Перемешиваемый раствор неочищенного 2-(5-4-трет-бутилфенилсульфонамидо)-1-(хинолин-5-ил)-1H-пиразол-3-ил) ацетамида (0.03 г, 0.07 ммоль) и фосфорного (V) оксид хлорида (POCl3, 0.5 мл, 5.4 ммоль) нагрет при 100°С в течение 30 мин. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме. Водный насыщенный бикарбонат натрия добавлен к получающемуся остатку и извлечен 2:1 хлороформом/iPrOH. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный продукт очищен обратили фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.006 г, 0.013 ммоль, 19%). 1Н NMR (400 МГц, CD3OD) δ 8.86 (dd, J=2.0, 4.4 Гц, 1Н), 8.12 (d, J=8.4 Гц, 1Н), 7.77 (dd, J=7.2, 8.4 Гц, 1H), 7.68 (d, J=8.4 Гц, 1Н), 7.55-7.53 (м, 2Н), 7.45-7.42 (м, 3Н), 7.34 (dd, J=1.2, 7.2 Гц, 1Н), 6.21 (s, 1Н), 3.88 (s, 2Н), 1.35 (s, 9Н); MS: (ES) m/z вычислено для C24H24N5O2S [М+Н]+ 446.2, найдено 446.3.
Пример 20: Синтез 4-трет-бутил-N-(1-(хинолин-5-ил)-3-(трифторметил)-1H-пиразол-5-ил)бензолсульфонамида
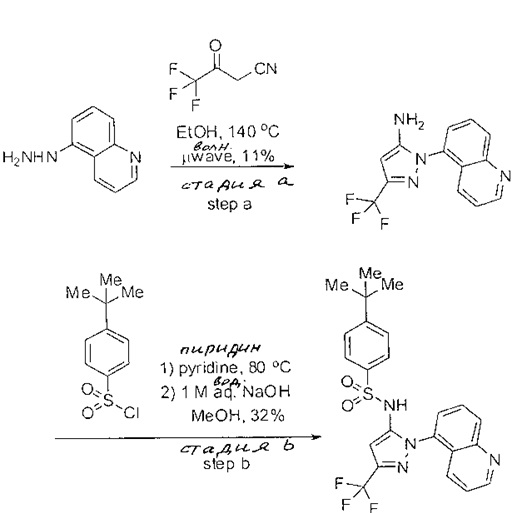
a) Перемешиваемый раствор 4,4,4-трифтор-3-оксобутаннитрила (0.40 г, 2.9 ммоль) и хинолин-5-ил-гидразина (получено из Примера 4 этап а, 0.46 г, 2.9 ммоль) в этиловом спирте (3 мл) нагрет при 140°С в микроволнах в течение 40 мин. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме. Получающийся неочищенный остаток очищен флэш-хроматографией (SiO2, 5-60% этилацетат в гексанах) с получением требуемого продукта как светло-коричневого твердого (0.087 г, 0.31 ммоль, 11%).
b) Смесь 4-трет-бутилбензолсульфонилхлорида (0.067 г, 0.29 ммоль) и 1-(хинолин-5-ил)-3-(трифторметил)-1H-пиразол-5-амина (0.011 г, 0.04 ммоль) в пиридине (0.5 мл) нагрета при 80°С в течение 2 часов с перемешиванием. После охлаждения до комнатной температуры дихлорметан добавлен к реакционной смеси и промыт 1 М водного гидросульфата натрия (1 мл). Водный слой далее извлечен дихлорметаном (2×5 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток растворен в метаноле (3 мл) и 1 М водной гидроокиси натрия (1.0 мл, 1.0 ммоль) и размешан при комнатной температуре в течение 1 часа. Реакционная смесь сконцентрирована в вакууме и разделена между дихлорметаном (3 мл) и 1 М водного гидросульфата натрия (3 мл). Фазы разделены, и водный слой далее извлечен дихлорметаном (2×5 мл). Объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.006 г, 0.013 ммоль, 32%). 1Н NMR (400 МГц, CDCl3) δ 8.78 (dd, J=1.6, 4.4 Гц, 1Н), 8.07 (d, J=8.4 Гц, 1Н), 7.66 (s, 1Н), 7.63 (s, 1Н), 7.55 (t, J=8.4 Гц, 1Н), 7.45 (s, 1Н), 7.48 (s, 1Н), 7.43 (d, J=6.0 Гц, 1Н), 7.27-7.24 (м, 1Н), 7.09 (d, J=6.0 Гц, 1Н), 6.70 (s, 1Н), 1.38 (s, 9Н); MS: (ES) m/z вычислено для C23H22F3N4O2S [М+Н]+ 475.2, найдено 475.3.
Пример 21: Синтез 4-изопропокси-N-(1-(хинолин-5-ил)-3-(трифторметил)-1H-пиразол-5-ил)бензолсульфонамида
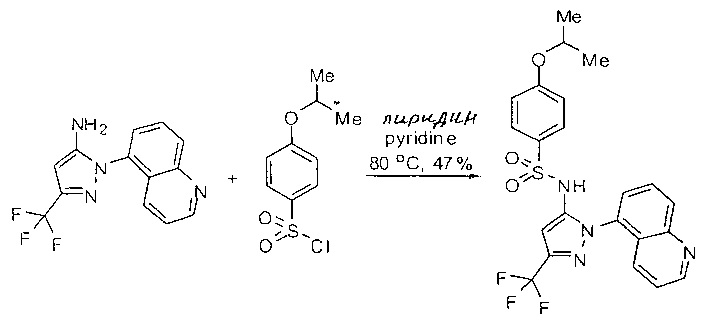
Смесь 4-изопропоксибензолсульфонилхлорида (0.080 г, 0.35 ммоль) и 1-(хинолин-5-ил)-3-(трифторметил)-1H-пиразол-5-амина (получено из Примера 21 этап а, 0.032 г, 0.11 ммоль) в пиридине (0.5 мл) нагрета при 80°С в течение 4 часов с перемешиванием. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме, и неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с ТФК на 0.1% как растворителем для элюирования) с получением названного соединения как белого твердого (0.025 г, 0.052 ммоль, 47%). 1Н NMR (400 МГц, DMSO-d6) δ 8.94 (dd, J=1.6, 4.0 Гц, 1Н), 8.19 (d, J=8.4 Гц, 1Н), 7.82 (t, J=8.4 Гц, 1Н), 7.50-7.46 (м, 2Н), 7.40 (s, 1H), 7.38 (s, 1Н), 7.35 (d, J=8.4 Гц, 1Н), 6.83 (s, 1Н), 6.81 (s, 1Н), 6.64 (s, 1Н), 4.63 (pent, J=6.0 Гц, 1Н), 1.28 (d, J=6.0 Гц, 6Н); MS: (ES) m/z вычислено для C27H20F3N4O2S [М+Н]+ 477.2, найдено 477.3.
Пример 22: Синтез 4-трет-бутил-N-(3-(1,1-дифторэтил)-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
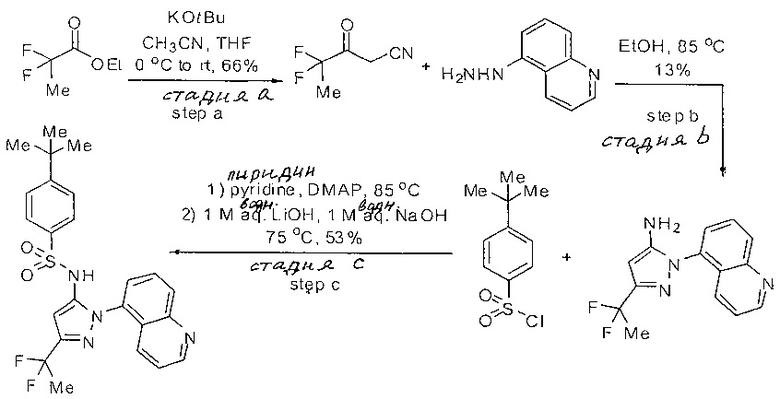
a) К перемешиваемому раствору калия трет-бутоксида (KOtBu, 1.7 М раствор в ТНК, 32 мл, 54.4 ммоль) в THF (10 мл) при 0°С добавлен этил-2,2-дифторпронаноат (5.0 г, 36.2 ммоль) и ацетонитрил (2.8 мл, 54.3 ммоль). Реакционная смесь нагрета до комнатной температуры и размешана в течение 18 часов. Водный насыщенный бисульфат калия добавлен к реакционной смеси, и рН отрегулирован до значения ниже 2. Водный слой извлечен этилацетатом (2×50 мл), и объединенные органические слои высушены (Na2SO4), профильтрованы и сконцентрированы в вакууме. Получающееся коричневое неочищенное масло использовано непосредственно без дальнейшей очистки (3.2 г, 24.1 ммоль, 66%).
b) К перемешивающемуся раствору 5-гидразинилхинолина (получено из Примера 4 этап а, 0.90 г, 5.6 ммоль) в этиловом спирте (10 мл) добавлен неочищенный 4,4-дифтор-3-оксопентаннитрил (0.75 г, 5.6 ммоль) и нагрет при 85°С в течение 6 часов. После охлаждения до комнатной температуры этилацетат добавлен к реакционной смеси и промыт 5 М водной гидроокиси натрия и соляным раствором. Органический слой высушен (Na2SO4.), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 50-100% этилацетат в гексанах) с получением требуемого продукта (0.20 г, 0.73 ммоль, 13%).
c) Смесь 4-трет-бутилбензол-1-сульфонилхлорида (0.080 г, 0.34 ммоль), 3-(1,1-дифторэтил)-1-(хинолин-5-ил)-1H-пиразол-5-амина (0.045 г, 0.16 ммоль) и DMAP (0.020 г, 0.16 ммоль) в пиридине (1 мл) нагрета при 85°С в течение 5 часов с перемешиванием. После охлаждения до комнатной температуры 1М водной гидроокиси лития (1 мл) и 1М водной гидроокиси натрия (1 мл) добавлены к реакционной смеси. Получающаяся смесь нагрета при 75°С в течение 1.5 часов. После охлаждения до комнатной температуры реакционная смесь нейтрализована 1 N водной соляной кислотой. Водный слой извлечен эти л ацетатом (2×10 мл). Объединенные органические слои промыты водным насыщенным бикарбонатом натрия, высушены (Na2SO4), отфильтровнаы и сконцентрированы в вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.040 г, 0.085 ммоль, 53%). 1Н NMR (400 МГц, CD3OD) δ 10.70 (s, 1Н), 8.88 (dd, J=1.6, 4.4 Гц, 1H), 8.14 (dd, J=1.2, 8.4 Гц, 1Н), 7.77 (dd, J=7.2, 8.8 Гц, 1Н), 7.64-7.61 (м, 2Н), 7.52-7.42 (м, 5Н), 7.31 (dd, J=1.2, 7.2 Гц, 1Н), 1.96 (t, J=15.4 Гц, 3Н), 1.35 (s, 9Н); MS: (ES) m/z вычислено для C24H25F2N4O2S [М+H]+ 471.2, найдено 471.2.
Пример 23: Синтез 4-трет-бутил-N-(3-(дифторметил)-1-(хинолин-5-ил)-N-пиразол-5-ил)бензолсульфонамида
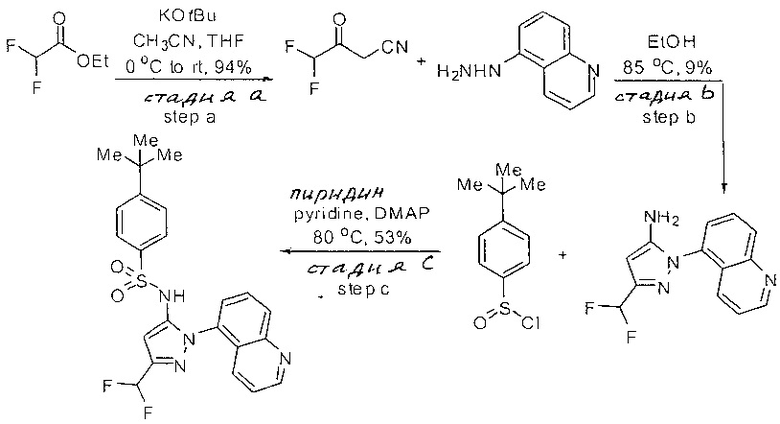
а) К перемешиваемому раствору KOtBu (1.0 М раствор в THF, 121 мл, 121 ммоль) при 0°С добавлен 2,2-дифторацетатэтил (10.0 г, 80.6 ммоль) и ацетонитрил (6.3 мл, 121 ммоль). Реакционная смесь нагрета до комнатной температуры и размешана в течение 18 часов. Водный насыщенный бисульфат калия добавлен к реакционной смеси, и pH отрегулирован до значения ниже 2. Водный слой извлечен этилацетатом (2×50 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме.
Коричневое неочищенное масло использовано непосредственно без дальнейшей очистки (9.0 г, 75 ммоль, 94%).
b) Перемешиваемый раствор неочищенного 4,4-дифтор-3-оксобутаннитрила (0.65 г, 4.0 ммоль) и 5-гидразинилхинолина (получено из Примера 4 этап а, 0.50 г, 4.2 ммоль) в этиловом спирте (8 мл) нагрет при 85°С в течение 6 часов. После охлаждения до комнатной температуры этилацетат добавлен к реакционной смеси и промыт водным насыщенным бикарбонатом натрия и соляным раствором. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 2-10% метанол в этилацетате) с получением требуемого продукта (0.095 г, 0.36 ммоль, 9%).
c) Смесь 4-трет-бутилбензол-1-сульфонилхлорида (0.070 г, 0.30 ммоль), 3-(дифторметил)-1-(хинолин-5-ил)-1H-пиразол-5-амина (0.045 г, 0.17 ммоль) и DMAP (0.020 г, 0.17 ммоль) в пиридине (2 мл) нагрета при 80°С в течение 5 часов с перемешиванием. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 5% метанол в этилацетате), с последующим обратной фазой HPLC (колонка С18, ацетонитрил-Н2О с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.040 г, 0.085 ммоль, 53%). 1Н NMR (400 МГц, CD3OD) δ 8.88 (dd, J=2.0, 4.0 Гц, 1Н), 8.15 (d, J=8.0 Гц, 1Н), 7.77 (dd, J=7.2, 8.4 Гц, 1Н), 7.63 (dddd, J=0.8, 1.6, 1.2, 7.2 Гц, 1Н), 7.53-7.43 (м, 5Н), 7.31 (dd, J=0.8, 7.2 Гц, 1Н), 6.71 (t, J=54.8 Гц, 1Н), 6.42 (s, 1Н), 1.35 (s, 9Н); (ES) m/z вычислено для C23H23F2N4O2S [М+Н]+457.2, найдено 457.2.
Пример 24: Синтез 4-трет-бутил-N-(2-(изохинолин-5-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)бензолсульфонамида
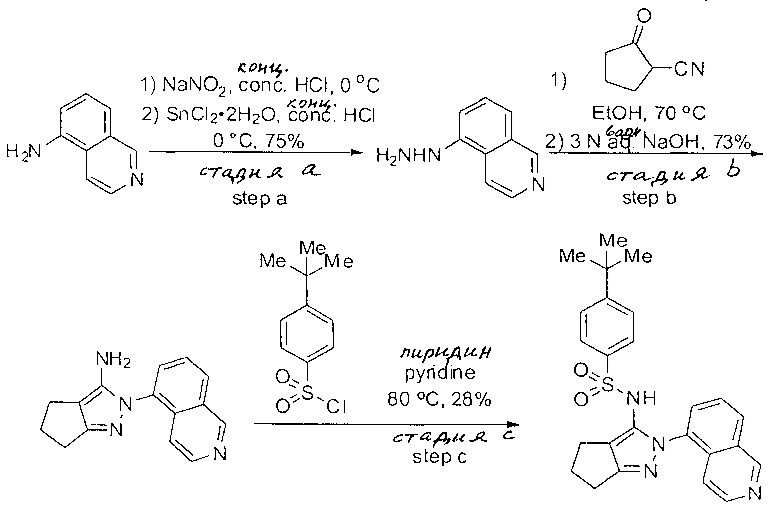
а) В колбу с круглым основанием, оборудованную перемешивающим устройством, содержащую изохинолин-5-амин (15.4 г, 10.0 ммоль) медленно добавлена концентрированная соляная кислота (90 мл). Жидкий реакционный раствор размешан при 0°С в течение 30 минут, и раствор NaNO2 (7.3 г, 105.8 ммоль) в минимальном количестве деионизированной воды добавлен по каплям. Реакционная смесь размешана при 0°С в течение 30 минут и затем при комнатной температуре в течение 30 минут с образованием темно-красного раствора. Реакционная смесь повторно охлаждена до 0°С, и затем добавлен по каплям раствор SnCl2⋅2H2O (47.4, 210.0 ммоль), растворенный в минимальном количестве концентрированной соляной кислоты. Густая коричневая смесь размешана при 0°С в течение 30 минут и затем при комнатной температуре в течение 4 часов. Твердое собрано фильтрацией и промыто холодным этиловым спиртом (200 мл). Желтое твердое суспендировано в 2:1 CHCl3/iPrOH (300 мл), и раствор отрегулирован до рН~12-14 с 2 М водной гидроокиси натрия (300 мл). Фазы отделены, и водный слой далее извлечен с CHCl3/iPrOH (2×300 мл). Объединенные органические слои высушены безводным сульфатом магния (MgSO4, профильтрованы и сконцентрированы в вакууме. Полученный неочищенный продукт использован непосредственно без дальнейшей очистки (12.7 г, 79.8 ммоль, 75%).
b) Перемешиваемая суспензия неочищенных 5-гидразинилизохинолина (0.45 г, 2.2 ммоль) и 2-оксоциклопентанкарбонитрила (получен как в Fleming, et al. J. Org. Chem., 2007, 72, 1431-1436, 0.24 г, 2.2 ммоль) в этаноле (10 мл) нагрета при 70°С в течение 2 часов. После охлаждения до комнатной температуры 3 М водной гидроокиси натрия (0.5 мл) добавлены к реакционной смеси и размешаны при комнатной температуре в течение 1 часа. Смесь затем сконцентрирована в вакууме, и получающийся остаток извлечен этилацетатом. Органический слой высушен (Na2SO4), профильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 5-10% этилацетат в гексанах) с получением требуемого продукта (0.48 г, 1.6 ммоль, 73%).
с) Смесь 4-трет-бутилбензолсульфонилхлорида (0.075 г, 0.32 ммоль) и 2-(изохинолин-5-ил) -2,4,5,6-тетрагидроциклопентапиразол-3-амина (0.050 г, 0.20 ммоль) в пиридине (1 мл) размешана при комнатной температуре в течение 1 часа. В реакционную смесь добавлен раствор 1 N водной соляной кислоты и извлечена в 2:1 CH2Cl2/iPrOH (2×10 мл). Объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Получающийся материал очищен обратной фазой HPLC (колонка С18, ацетонитрил-Н2О с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.025 г, 0.056 ммоль, 28%). 1Н NMR (400 МГц, DMSO-d6) δ 10.10 (s, 1Н), 9.35 (s, 1Н), 8.42 (d, J=6.0 Гц, 1Н), 8.17 (d, J=8.8 Гц, 1Н), 7.68 (t,J=8.0 Гц, 1Н), 7.49 (d, J=7.2 Гц, 1Н), 7.41-7.37 (м, 3Н), 7.18 (d, J=4.8 Гц, 1Н), 2.64 (t, J=7.2 Гц, 2Н), 2.21 (pent, J=7.2 Гц, 2Н), 2.06 (t, J=7,5 Hz, 2Н), 1.25(s, 9H); MS: (ES) m/z вычислено для C25H27N4O2S [M+H]+ 447.2, найдено 447.1.
Пример 25: Синтез N-(1-(1-аминоизохинолин-5-ил)-5-метил-1H-пиразол-5-ил)-4-трет-бутилбензолсульфонамида
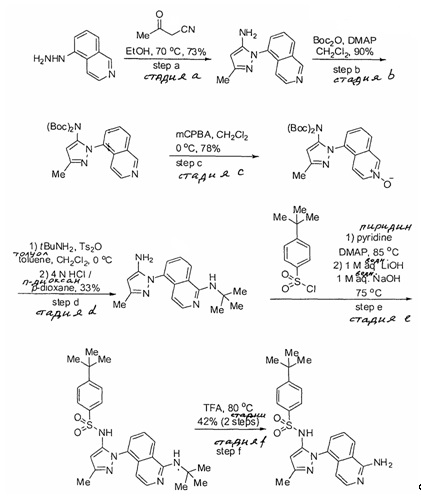
a) Перемешиваемая суспензия 5-гидразинилизохинолииа (получено из Примера 25 этап а, 0.60 г, 3.8 ммоль) и 3-оксобутаннитрила (0.3 I г, 3.8 ммоль) в этиловом спирте (3 мл) нагрета при 80°С в течение 2 часов. После охлаждения до комнатной температуры реакционная смесь аакуумирована, и получающийся неочищенный остаток очищен флэш-хроматографией (SiCh, 0-20% метанол в этилацетате) с получением требуемого продукта (0.067 г, 2.8 ммоль, 73%).
b) К раствору 1-(изохинолин-5-ил)-3-метил-1H-пиразол-5-амина (0.45 г, 2.0 ммоль) в дихлорметане (10 мл) добавлен DMAP (0.30 г, 2.5 ммоль) и ди-трет-бутилбикарбонат (Boc2O, 1.2 г, 5.5 ммоль). Реакционная смесь размешана при комнатной температуре в течение 15 часов, и добавлен этилацетат. Получающийся раствор промыт 2 N водной гидроокисью натрия, 2 N водной соляной кислотой и соляным расвором. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Сырой продукт очищен флэш-хроматографией (SiO2, 20-50% этилацетат в гексанах) с получением требуемого продукта (0.76 г, 1.8 ммоль, 90%).
c) К перемешиваемому раствору ди-трет-бутил-1-(изохинолин-5-ил)-3-метил-1H-пиразол-5-илиминодикарбоната (0.15 г, 0.35 ммоль) в дихлорметане (10 мл) при 0°С добавлена 3-хлорпербензойная кислота (mCPBA, 0.2 г, 0.90 ммоль). Реакционная смесь медленно нагрета до комнатной температуры и размешана при той же самой температуре в течение 4 часа. Раствор 15% iPrOH в дихлорметане добавлен к реакционной смеси и промыт водным насыщенным бикарбонатом натрия. Органический слой высушен (Na2SO4), профильтрован и сконцентрирован в вакууме. Сырой продукт очищен флэш-хроматографией (SiO2, 5-10% метанол в дихлорметане) с получением требуемого продукта (0.12 г, 0.27 ммоль, 78%).
d) Смесь 5-(5-(бис-(трет-бутоксикарбонил) амино)-3-метил-1H-пиразол-1-ил) изохинолин-2-оксида (0.12 г, 0.27 ммоль) в толуоле (3 мл) и дихлорметане (3 мл) при 0°С дополнена трет-бутиламином (0.3 мл, 2.86 ммоль) и n-толуолсульфоновым ангидридом (Ts2O, 0.30 г, 0.93 ммоль) в трех частях. Реакционная смесь медленно нагрета до комнатной температуры в течение более 2 часов и добавлен этилацетат. Органический слой промыт водным насыщенным бикарбонатом натрия, 1 N водной соляной кислотой и соляным раствором. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Получающийся сырой продукт затем растворен в дихлорметане (5 мл), и раствор соляной кислоты в n-диоксане (4.0 N раствора в n-диоксане, 5.0 мл, 20 ммоль) добавлен. Реакционная смесь размешана при комнатной температуре в течение 2 часов, и добавлен этилацетат. Органический слой промыт водным насыщенным бикарбонатом натрия, высушен (Na2SO4), профильтрован и сконцентрирован в вакууме. Сырой продукт использован непосредственно без дальнейшей очистки (0.026 г, 0.090 ммоль, 33%).
e) Смесь сырья 5-(5-амино-3-метил-1H-пиразол-1-ил)-N-трет-бутилизохинолин-1-амина (0.055 г, 0.30 ммоль), 4-ацетилбензол-1-сульфонилхлорида (0.090 г, 0.39 ммоль) и DMAP (0.037 г, 0.30 ммоль) в пиридине (2 мл) нагрета при 85°С в течение 1 часа с перемешиванием. После охлаждения до комнатной температуры 1 М водной гидроокиси лития и 1 М водной гидроокиси натрия (на 1 мл) (1 мл) добавлены к реакционной смеси. Полученная смесь нагрета при 75°С в течение 30 мин. После охлаждения до комнатной температуры реакционная смесь нейтрализована 1 N водной соляной кислотой. Водный слой извлечен этилацетатом, и органический слой промыт водным насыщенным бикарбонатом натрия, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный материал использован непосредственно для следующего этапа.
f) Неочищенный остаток растворен в TFA (8 мл) и нагрет при 80°С в течение 1.5 часов с перемешиванием. Реакционная смесь охлаждена до комнатной температуры и сконцентрирована в вакууме. Сырой продукт затем растворен в 15%-ом метаноле в дихлорметане и промыт водным насыщенным бикарбонатом натрия. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Получающийся сырой продукт очищен обратной фазой HPLC (колонка С18, ацетонитрил-Н2О с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.035 г, 0.080 ммоль, 42% для 2 этапов). 1Н NMR (400 МГц, CD3OD) δ 8.23 (dd, J=0.8, 8.8 Гц, 1Н), 7.60-7.54 (м, 3Н), 7.50-7.42 (м, 4Н), 6.31 (dd, J=0.8, 6.4 Гц, 1Н), 5.93 (s, 1Н), 2.22 (s, 3Н), 1.36 (s, 9Н); MS: (ES) m/z вычислено для C23H26N5O2S [М+Н]+ 436.2, обнаружено 436.1.
Пример 26: Синтез 4-трет-бутил-3-фтор-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
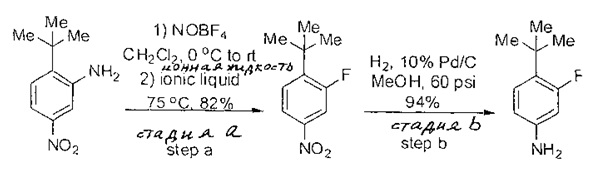
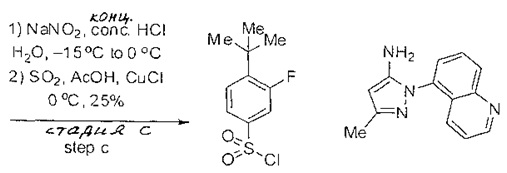
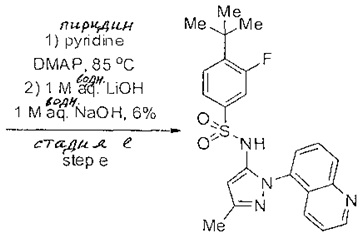
а) К размешиваемой суспензии нитрозилтетрафторбората (8.4 г, 71.9 ммоль) в дихлорметане при 0°С добавлен хинолин-5-ил-гидразин (получен как в Laali, et al. J. Fluorine Chem., 2001, 107, 31-34, 12.0 г, 61.8 ммоль) малыми частями через 5 мин. После завершения добавления реакционная смесь размешана при 0°С в течение 1 часа и медленно нагрета до комнатной температуры в течение более чем I часа с получением тонкой суспензии. К этой суспензии медленно добавлен I-этил-3-метил-имидазолийтетрафторборат (ионная жидкость, 50 г, 252.6 ммоль) и получающаяся смесь нагрета при 75°С в течение 2 часов. Органическое летучее удалено дистилляцией через Dean-Stark конденсатор. После охлаждения смеси до комнатной температуры диизопропилэтиламин (iPr2NEt, 10 мл) добавлен к реакционной смеси и размешан в течение 10 мин. Диэтиловый эфир (300 мл) добавлен к реакционной смеси и промыт 1 N водной соляной кислотой и соляным раствором, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Сырой продукт использован непосредственно без дальнейшей очистки (10.0 г, 50.8 ммоль, 82%).
b) В колбе шейкера Парра, содержащей сырье 1-трет-бутил-2-фтор-4-нитробензола (1.0 г, 5.1 ммоль) и Pd/C (10% по весу, 0.040 g) в метаноле (60 мл), проведена гидрогенизация при 60 psi в течение 2 часов. Реакционная смесь разбавлена метанолом и пропущена через фильтр Целита. Фильтрат сконцентрирован в вакууме, и получающийся остаток использован непосредственно без дальнейшей очистки (0.80 г, 4.8 ммоль, 94%).
c) Колба, содержащая ледяную уксусную кислоту (2 мл), при 0°С барботирована газом двуокиси серы (SO2) в течение 30 мин. Хлорид меди (I) (CuCl, 0.10 г, 1.0 ммоль) добавлен к реакционной смеси и смесь перемешана 30 минут при 0°С с получением сине-зеленого раствора. В отдельной колбе раствор сырья 4-трет-бутил-3-фторанилина (0.20 г, 1.2 ммоль) в концентрированной соляной кислоте (3 мл) при -15°С дополнен раствором NaNO2 (0.12 г, 1.7 ммоль) в деионизированной воде (1 мл). Реакционная смесь размешана при той же самой температуре в течение 30 мин. Этот раствор диазония медленно добавлен к готовому медному раствору, и получающийся раствор барботирован SO2 в течение еще 5 мин. Реакционная смесь размешана при -15°С в течение 1 часа и нагрета до 0°С в течение более чем 1 часа. Диэтиловый эфир затем добавлен к реакционной смеси, и содержимое лили по льду. Получающаяся смесь была извлечена диэтиловым эфиром, и органический слой далее промыт водой со льдом, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Темное сырое масло очищено флэш-хроматографией (SiO2, 1-3% этилацетат в гексанах) с получением требуемого продукта (0.075 г, 0.30 ммоль, 25%).
d) Смесь 4-трет-бутил-3-фторбензол-1-сульфонилхлорида (0.075 г, 0.30 ммоль), 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап b, 0.050 г, 0.22 ммоль) и DMAP (0.027 г, 0.22 ммоль) в пиридине (1 мл) нагрета при 85°С в течение 2.5 часов с перемешиванием. После охлаждения до комнатной температуры 1 М водной гидроокиси лития (1 мл) и 1 М водной гидроокиси натрия (1 мл) добавлены к реакционной смеси. Получающаяся смесь размешана при комнатной температуре в течение 15 часов и нейтрализована 1 N водной соляной кислотой. Водный слой извлечен этилацетатом, и органический слой промыт водным насыщенным бикарбонатом натрия, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Получающийся неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-Н20 с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.006 г, 0.014 ммоль, 6%). 1Н NMR (400 МГц, CD3OD) δ 8.83 (dd, J=1.6, 4.4 Гц, 1Н), 8.08 (dd, J=1.2, 8.4 Гц, 1Н), 7.79 (dd, J=7.2, 8.4 Гц, 1Н), 7.69 (ddd,.7=0.8, 1.6, 7.6 Гц, 1Н), 7.44 (dd, J=1.2, 7.2 Гц, 1Н), 7.40-7.31 (м, 3Н), 7.20 (dd, J=1.6, 7.2 Гц, 1H), 5.88 (s, 1Н), 2.21 (s, 3H), 1.39 (s, 9H); MS: (ES) m/z вычислено для C23H24FN4O2S [M+H]+ 439.2, обнаружено 439.2.
Пример 27: Синтез N-(1-(2-аминохинолин-5-ил)-3-метил-1H-пиразол-5-ил)-4-трет-бутил-3-фторбензолсульфонамида.
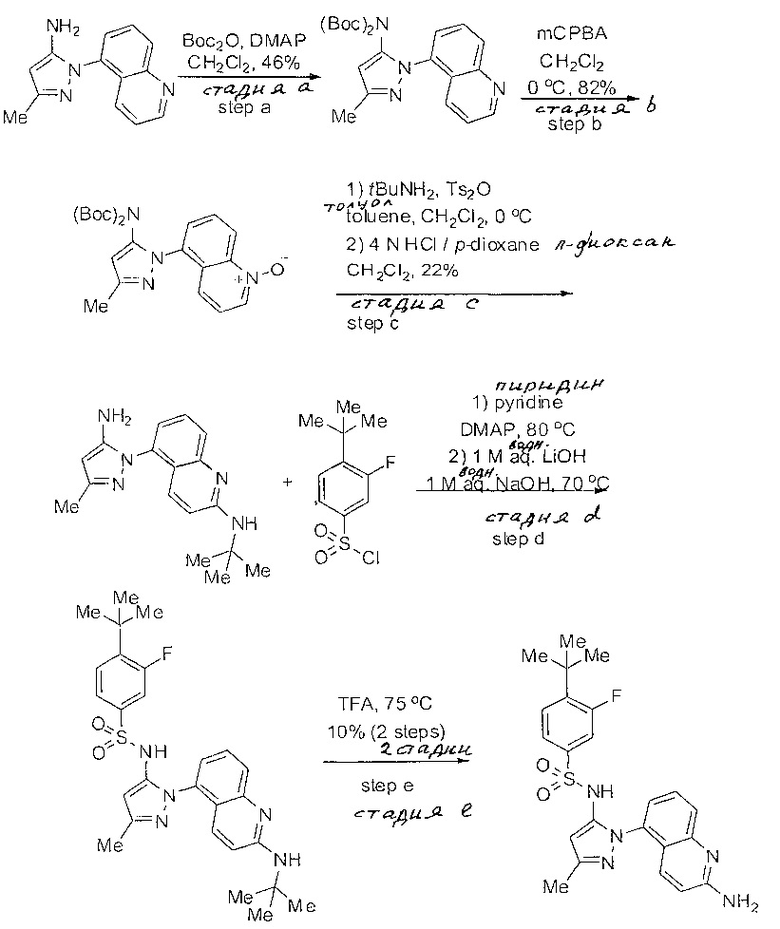
а) К раствору 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап b, 4.0 г, 17.9 ммоль) в дихлорметане (50 мл) добавлен DMAP (2.2 г, 17.9 ммоль) и Boc2O (7.8 г, 35.9 ммоль). Реакционная смесь размешана при комнатной температуре в течение 3 часов, и добавлен этилацетат. Органический слой промыт 1 N водной соляной кислотой, водным насыщенным бикарбонатом натрия и соляным раствором. Органический слой высушен (Na2SO4), профильтрован и сконцентрирован в вакууме, и сырой продукт использован непосредственно без дальнейшей очистки (3.5 г, 8.3 ммоль, 46%).
b) К перемешиваемому раствору сырья ди-трет-бутил-3-метил-1-(хинолин-5-ил)-1H-пиразол-5-илиминодикарбоната (3.5 г, 8.3 ммоль) в дихлорметане (50 мл) при 0°С медленно добавлен тСРВА (4.0 г, 17.8 ммоль). Реакционная смесь медленно нагрета до комнатной температуры и размешана в течение 2 часов. Раствор 15% iPrOH дихлорметане добавлен к реакционной смеси и промыт водным насыщенным бикарбонатом натрия. Органический слой высушен (Na2SO4), профильтрован и сконцентрирован в вакууме. Сырой продукт очищен флэш- хроматографией (SiO2, 2-5% метанол в дихлорметане) с получением требуемого продукта (3.0 г, 6.8 ммоль, 82%).
c) Перемешиваемая смесь 5-(5-(бис (трет-бутоксикарбонил) амино)-3-метил-1H-пиразол-1-ил) хинолин-1-оксида (3.0 г, 6.8 ммоль) в толуоле (40 мл) и дихлорметане (40 мл) при 0°С дополнена трет-бутиламином (5.5 мл, 52.3 ммоль) и Ts2O (5.0 г, 15.3 ммоль) в двух частях. Реакционная смесь медленно нагрета до комнатной температуры в течение более чем 2 часов, и добавлен этилацетат. Органический слой промыт водным насыщенным бикарбонатом натрия, 1 N водной соляной кислотой и соляным раствором. Органический слой высушен (Na2SO4), профильтрован и сконцентрирован в вакууме. Сырой продукт затем растворен в дихлорметане (10 мл) и добавлен раствор соляной кислоты в n-диоксане (4.0 N раствора в n-диоксане, 20 мл, 80 ммоль). Реакционная смесь размешана при комнатной температуре в течение 2 часов, и добавлен этилацетат. Органический слой промыт водным насыщенным бикарбонатом натрия, высушен (Na2SO4), профильтрован и сконцентрирован в вакууме. Сырой продукт использован непосредственно без дальнейшей очистки(рафинации) (0.45 г, 1.52 ммоль, 22%)
d) Смесь сырья 5-(5-амино-3-метил-1H-пиразол-1-ил)-N-трет-бутилхинолин-2-амина (0.075 г, 0.25 ммоль), 4-трет-бутил-3-фторбензол-1-сульфонилхлорида (получено из Примера 27 этап с, 0.11 г, 0.44 ммоль, и DMAP (0.031 г, 0.25 ммоль) в пиридине (2 мл) нагрета при 80°С в течение 2 часов с перемешиванием. После охлаждения до комнатной температуры 1 М водной гидроокиси лития (1 мл) и 1 М водной гидроокиси натрия (1 мл) добавлены к реакционной смеси и нагреты при 70°С в течение 30 мин. После охлаждения до комнатной температуры реакционная смесь нейтрализована 1 N водной соляной кислотой. Водный слой извлечен этилацетатом, и органический слой промыт водным насыщенным бикарбонатом натрия, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный материал использован непосредственно для следующего этапа.
е) Неочищенный остаток растворен в TFA (6 мл) и нагрет при 75°С в течение 2 часов с перемешиванием. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме. Сырой продукт затем растворен в 10% метаноле в дихлорметане и промыт водным насыщенным раствором бикарбоната натрия. Органический слой высушен (Na2SO4) профильтрован и сконцентрирован в вакууме. Получающийся сырой продукт очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.011 г, 0.024 ммоль, 10% для 2 этапов). 1Н NMR (400 МГц DMSO-d6) 11.01 (br s, 1Н), 7.47 (м, 2Н), 7.31 (d, J=8.0 Гц, 1Н), 7.27 (d, J=8.0 Гц, 1Н), 7.25 (dd, J=2.0, 8.0 Гц, 1Н), 7.16 (dd, J=2.0, 8.0 Гц, 1Н), 6.91 (м, 3Н), 5.86 (s, 1Н), 2.14 (s, 3Н), 1.32 (s, 9Н); MS: (ES) m/z вычислено для C23H25FN5O2S [М+Н]+454.2, обнаружено 454.1.
Пример 28: Синтез 4-трет-бутил-3-хлор-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
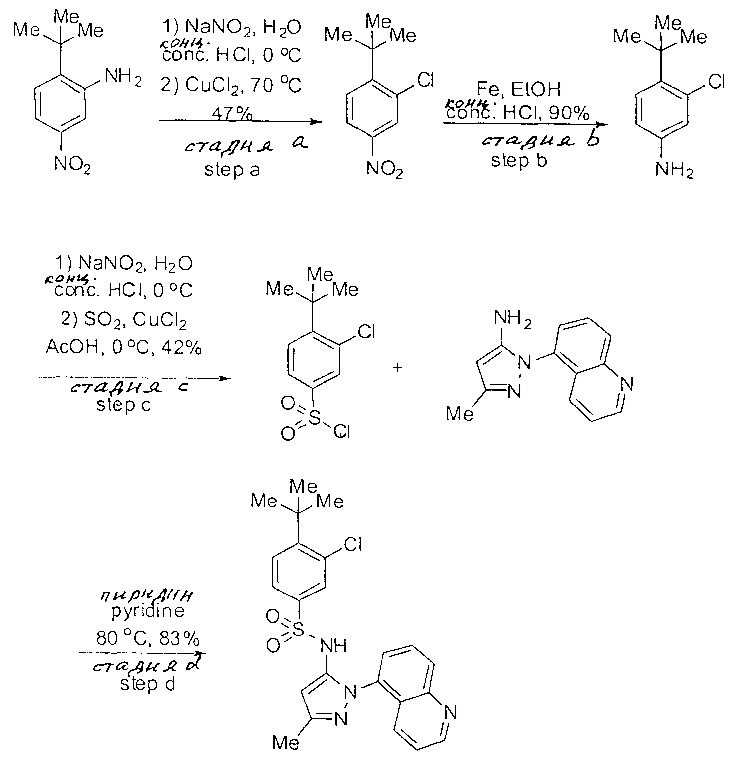
a) К перемешиваемому раствору хинолин-5-ил-гидразина (получен как в Laali, el.al. J. Fluorine Chem., 2001, 107, 31-34, 0.25 г, 1.29 ммоль) в концентрированной соляной кислоте (1.3 мл) при 0°С добавлен раствор NaNO2 (0.13 г, 1.9 ммоль) в деионизированной воде (0.64 мл). Реакционная смесь размешана в течение 30 минут при 0°С и нагрета при 70°С в течение 30 мин. Хлорид меди (II) (0.22 г, 1.6 ммоль) добавлен к горячей смеси и размешан при 70°С в течение 30 мин. После охлаждения реакционной смеси до комнатной температуры получен осадок, и твердое собрано фильтрацией. Твердое промыто холодной деионизированной водой и высушено под вакуумом с получением требуемого продукта (0.13 г, 0.61 ммоль, 47%).
b) Концентрированная соляная кислота (0.25 мл) добавлена медленно к раствору 1-трет-бутил-2-хлор-4-нитробензола (0.13 г, 0.61 ммоль) и железного порошка (0.17 г, 3.0 ммоль) в этиловом спирте (1.2 мл). Реакционная смесь размешана при комнатной температуре в течение I часа, и жидкий раствор разбавлен этанолом. Полученная смесь затем профильтрована через фильтр Целита и промыта дополнительным этанолом (30 мл).
Фильтрат сконцентрирован в вакууме, и получающийся неочищенный материал очищен флэш-хроматографией (SiO2, 0-80% этилацетата в гексанах) с получением требуемого продукта (0.10 г, 0.55 ммоль, 90%).
c) Раствор ледяной уксусной кислоты (2 мл) при 0°С барботирован в газ двуокиси серы (SO2) в течение 30 мин. Хлорид Меди (II) (0.073 г, 0.54 ммоль) добавлен к реакционной смеси и размешан в течение дополнительных 30 минут при 0°С. В другую колбу, содержащую 4-трет-бутил-3-хлоранилин (0.10 г, 0.54 ммоль) в концентрированной соляной кислоте (0.5 мл) добавлен раствор NaNO2 (0.06 г, 0.87 ммоль) в деионизированной воде (0.1 мл) при 0°С с перемешиванием. Этот раствор диазония медленно добавлен к готовому медному раствору и размешан при 0°С в течение 30 мин. Диэтиловый эфир добавлен к реакционной смеси, и фазы отделены. Водный слой далее извлечен этилацетатом (2×10 мл), и объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 0-20% этилацетат в гексанах) с получением требуемого продукта (0.061 г, 0.23 ммоль, 42%).
d) Смесь 4-трет-бутил-3-хлорбензол-1-сульфонилхлорида (0.050 г, 0.19 ммоль) и 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап b, 0.042 г, 0.095 ммоль) в пиридине (0.2 мл) нагрета при 80°С в течение 1 часа с перемешиванием. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме и неочищенный остаток очищен флэш-хроматографией (SiO2, 20%-ный этилацетат в гексанах) с получением названного соединения как белого твердого (0.066 г, 0.16 ммоль, 83%). 1Н NMR (400 МГц, DMSO-d6) δ 8.86 (dd, J=2.0, 4.0 Гц, 1Н), 7.98 (d, J=8.4 Гц, 1Н), 7.76 (t, J=8.4 Гц, 1Н), 7.70 (d, J=8.4 Гц, 1Н), 7.44 (s, 3Н), 7.40 (d, J=7.6 Гц, 1Н), 7.34 (dd, J=4.0, 8.0 Гц, 1Н), 5.48 (s, 1Н), 2.02 (s, 3Н), 1.44 (s, 9Н); MS: (ES) m/z вычислено для C23H24ClN4O2S [М+Н]+ 455.2, обнаружено 455.2.
Пример 29: Синтез 3-фтор-4-изопропокси-N-(3-метил-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
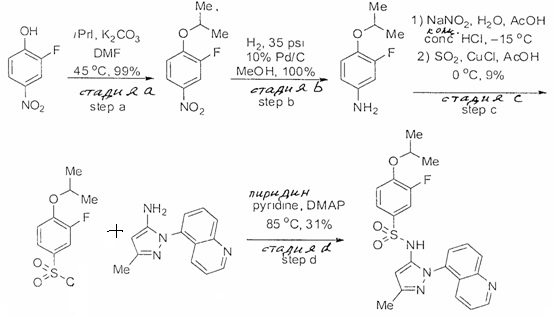
a) Перемешиваемый раствор 2-фтор-4-нитрофенола (1.6 г, 10.2 ммоль) и карбоната калия (K2CO3, 2.5 г, 18.1 ммоль) в DMF (10 мл) размешан при комнатной температуре в течение 5 мин. Изопропиловый йодид (iPrI, 2 мл, 20.0 ммоль) затем добавлен к реакции, и получающаяся смесь размешана при комнатной температуре в течение 2 часов и затем нагрета при 45°С в течение 6 часов. Реакционная смесь охлаждена до комнатной температуры, и добавлен диэтиловый эфир. Смесь промыта деионизированной водой и водным насыщенным бикарбонатом натрия. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Сырой продукт использован непосредственно без дальнейшей очистки (2.0 г, 10.1 ммоль, 99%).
b) В колбе шейкера Парра сырье 2-фтор-1-изопропокси-4-нитробензола (2.0 г, 10.1 ммоль) и Pd/C (10% по весу, 0.50 g) в метаноле (100 мл) гидрировано при 35 psi в течение 1 часа. Реакционная смесь разбавлена метанолом и профильтрована через фильтр Целита. Фильтрат сконцентрирован в вакууме, и получающийся остаток использован непосредственно без дальнейшей очистки (1.7 г, 10.1 ммоль, 100%).
c) Раствор ледяной уксусной кислоты (40 мл) барботирован в SO2. После 15 минут добавлен хлорид меди (1) (0.50 г, 5.1 ммоль) и барботирование газа SO2 продолжено, пока раствор не приобрел зеленый/синий цвет. К другой колбе, содержащей сырье 3-фтор-4-изопропоксианилина (1.5 г, 8.9 ммоль) в 1:1 ледяной уксусной кислоте и концентрированную соляную кислоту (10 мл) при -15°С добавлен раствор NaNO2 (0.75 г, 10.8 ммоль) в деионизированной воде (3 мл) и размешан при -15 0 С в течение 30 мин. Этот раствор диазония затем медленно добавлен в готовый медный раствор и размешан при -15°С в течение 30 мин. Диэтиловый эфир (30 мл) добавлен к реакционной смеси и размешан при -15°С в течение 1 часа. Реакционную смесь лили на лед, и добавлен дополнительный диэтиловый эфир (30 мл). Фазы отделены, и водный слой далее извлечен этилацетатом (2×10 мл). Объединенные органические слои высушены (Na2SO4) отфильтрованы и сконцентрированы в вакууме. Неочищенный остаток очищен флэш-хроматографией (SiO2, 5-10% этилацетат в гексанах с получением требуемого продукта (0.20 г, 0.79 ммоль, 9%).
d) Смесь 3-фтор-4-изопропоксибензол-1-сульфонилхлорида (0.050 г, 0.19 ммоль), 3-метил-1-(хинолин-5-ил)-1H-пиразол-5-амина (получено из Примера 4 этап b, 0.025 г, 0.11 ммоль, и DMAP (0.020 г, 0.16 ммоль) в пиридине (2 мл) нагрета при 85°С в течение 2 часов с перемешиванием. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме, и неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.015 г, 0.034 ммоль, 31%). 1Н NMR (400 МГц, CD3OD) δ 8.85 (d, J=4.4 Гц, 1Н), 8.11 (d, J=8.4 Гц, 1Н), 7.80 (t, J=8.0 Гц, 1Н), 7.62 (8.8 Гц, 1Н), 7.45-7.40 (м, 2Н), 7.25 (d, J=8.8 Гц, 1Н), 7.12 (dd, J=2.4, 10.4 Гц, 1Н), 6.92 (t, J=8.4 Гц, 1Н), 6.10 (s, 1Н), 4.63 (hept, J=6.4 Гц, 1Н), 2.27 (s, 3H), 1.37 (d, J=6.4 Hz, 6H); MS: (ES) m/z вычислено для C22H22FN4O3S [M+H]+ 441.2, обнаружено 441.2.
Пример 30: Синтез N-(1-(8-аминохинолин-5-ил)-3-метил-1H-пиразол-5-ил)-4-трет-бутил-3-фторбензолсульфонамида
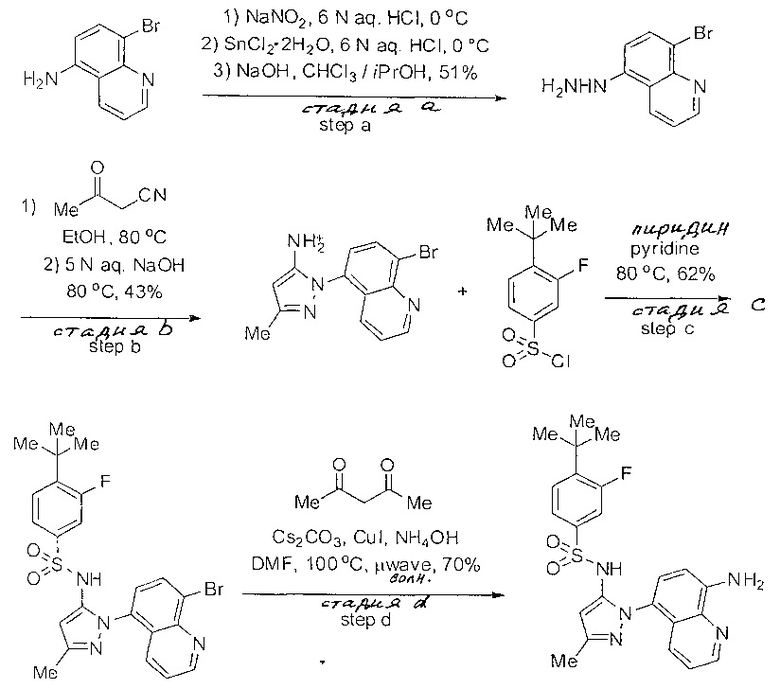
a) К перемешиваемому раствору 5-амино-8-бромхинолина (1.1 г, 5.0 ммоль) в 6 N водной соляной кислоте (10 мл) при 0°С медленно добавлен твердый NaNO2 (1.0 г, 14.5 ммоль), при поддержании внутренней температуры ниже 0°С. Реакционная смесь размешана при 0°С в течение 1 часа, и раствор SnCl2⋅2H2O (3.2 г, 12.5 ммоль), растворенный в 6 N водной соляной кислоте (3 мл), добавлен затем по каплям. Смесь размешана при комнатной температуре в течение 2 часов, и раствор нейтрализован до рН~7 1 М водной гидроокиси натрия. Смесь извлечена 2:1 CHCl3/iPrOH и органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Получающийся сырой продукт очищен флэш-хроматографией (SiCl2, 50% этилацетат в гексанах) с получением требуемого соединения как желтого твердого (0.60 г, 2.6 ммоль, 51 %).
b) Перемешиваемая суспензия 8-бром-5-гидразинилхинолина (2.0 г, 8.4 ммоль) и 3-оксо-бутиронитрила (0.70 г, 8.4 ммоль) в этаноле (20 мл) нагрета при 80°С в течение 3 часов. После охлаждения до комнатной температуры 5 М водной гидроокиси натрия (1 мл) добавлены к реакционной смеси и нагреты при 80°С в течение 1 часа. Получающаяся смесь охлаждена до комнатной температуры и сконцентрирована в вакууме. Неочищенный остаток растворен в 1:1 дихлорметан/метаноле (40 мл) и фазы отделены. Органический слой высушен (Na2SO4) отфильтрован и сконцентрирован в вакууме. Получающееся сырье очищено флэш- хроматографией (SiO2, 50-100% этилацетат в гексанах) с получением коричневого продукта как требуемого продукта (1.1 г, 3.6 ммоль, 43%).
c) Перемешиваемая смесь 4-трет-бутил-3-фторбензол-1-сульфонилхлорида (получен из Примера 27 этап с, 1.1 г, 4.2 ммоль) и 1-(8-бромхинолин-5-ил)-3-метил-1H-пиразол-5-амина (0.97 г, 3.2 ммоль) в пиридине (5 мл) нагрета при 80°С в течение 15 часов. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме. Сырое твердое перекристаллизовано из горячего этанола (5 мл), и полученное твердое собрано фильтрацией с получением требуемого соединения (0.10 г, 0.19 ммоль, 62%).
d) К перемешиваемому раствору N-(1-(8-бромхинолин-5-ил)-3-метил-1H-пиразол-5-ил)-4-трет-бутил-3-фторбензолсульфонамида (0.052 г, 0.10 ммоль) в гидроокиси аммония (1 мл) и DMF (1 мл) добавлен 2,4-пентандион (0.006 г, 0.06 ммоль), карбонат цезия (Cs2CO3, 0.064 г, 0.20 ммоль) и йодид меди (I) (CuI, 0.0095 г, 0.050 ммоль). Реакционная смесь нагрета при 120°С в микроволнах в течение 2 часов. После охлаждения до комнатной температуры к реакционной смеси добавлен этилацетат (100 мл) и промыт деионизированной водой (20 мл) и солевым раствором (2×20 мл). Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Получающееся сырье очищено обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как растворителем для элюирования) с получением названного соединения как коричневого твердого (0.032 г, 0.070 ммоль, 70%). 1Н NMR (400 МГц, CD3OD) δ 8.80 (d, J=3.2 Гц, 1Н), 7.53 (dd, 7=1.2, 8.4 Гц, 1Н), 7.45 (dd, J=4.0, 8.4 Гц, 1Н), 7.40 (t, J=8.0 Гц, 1Н), 7.32 (dd, J=2.0, 8.4 Гц, 1Н), 7.22 (dd, J=2.0, 12.0 Гц, 1Н), 7.08 (s, 2Н), 6.12 (s, 1Н), 2.28 (s, 3Н), 1.41 (s, 9Н); MS: (ES) m/z вычислено для C23H25FN5O2S [М+Н]+ 454.2, обнаружено 454.2.
Пример 31: Синтез N-(1-(2-аминохинолин-5-ил)-5-метил-1H-пиразол-5-ил)-4-трет-бутилбензолсульфонамида
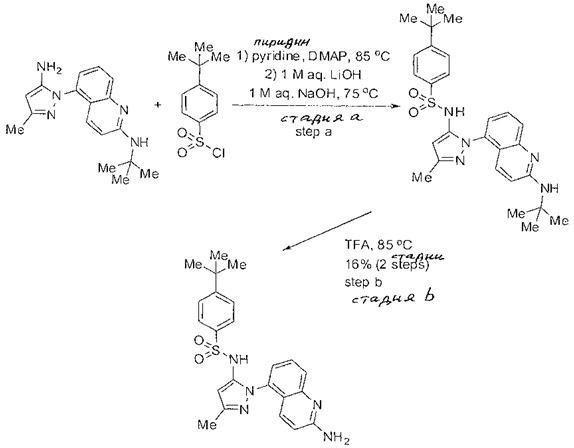
а) Смесь сырья 5-(5-амино-3-метил-1H-пиразол-1-ил)-N-трет-бутилхинолин-2-амина (получено из Примера 28 этап с, 0.090 г, 0.31 ммоль), 4-трет-бутилбензолсульфонил-хлорида (0.15 г, 0.65 ммоль) и DMAP (0.022 г, 0.1.8 ммоль) в пиридине (3 мл) нагрета при 85°С в течение 2 часов с перемешиванием. После охлаждения до комнатной температуры 1 М водной гидроокиси лития (1 мл) и 1 М водной гидроокиси натрия (1 мл) добавлены к реакционной смеси и нагреты при 75°С в течение 1 часа. После охлаждения до комнатной температуры реакционная смесь нейтрализована 1 N водной соляной кислотой. Водный слой извлечен этилацетатом, и органический слой промыт водным насыщенным бикарбонатом натрия, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный материал использован непосредственно для следующего этапа.
b) Неочищенный остаток растворен в TFA (8 мл) и нагрет при 85°С в течение 6 часов с перемешиванием. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме. Сырой продукт затем суспендирован в водном насыщенном бикарбонате натрия и извлечен этилацетатом. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Получающийся сырой продукт очищен обратной фазой HPLC (колонка С18, ацетонитрил-Н2О с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.021 г, 0.048 ммоль, 16% для 2 этапов). 1H NMR (400 МГц, CD3OD) 7.55 (d, J=6.4 Гц, 1Н), 7.55-7.54 (м., 2Н), 7.49 (dd, J=7.2, 8.4 Гц, 1Н), 7.43 (d, J=2.0 Гц, 1Н), 7.41 (s, 1Н), 7.28 (d, J=9.2 Гц, 1Н), 6.90 (d, J=7.2 Гц, 1Н), 6.71 (d, J=9.2 Гц, 1Н), 5.93 (s, 1Н), 2.21 (s, 3Н), 1.35 (s, 9Н); MS: (ES) m/z вычислено для C23H26N5O2S.
Пример 32: Синтез 4-трет-бутил-N-(3-(фторметил)-1-(хинолин-5-ил)-1H-лиразол-5-ил)бензолсульфонамида
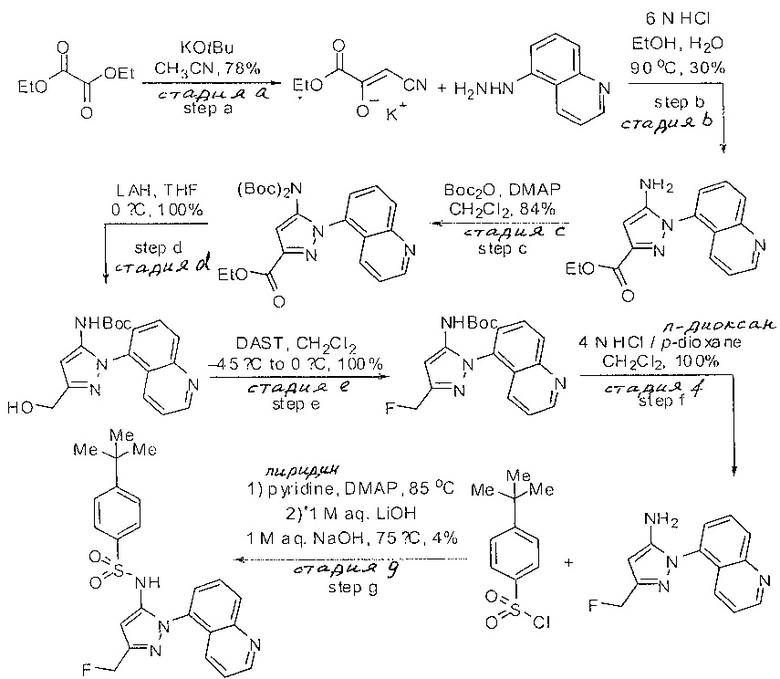
а) К раствору диэтилового оксалата (25.3 г, 173 ммоль) в ацетонитриле (100 мл) добавлен калий трет-бутоксид. (19.5 г, 173 ммоль) более чем тремя частями. Оранжевая суспензия размешана при комнатной температуре в течение 1.5 часов, и твердое собрано фильтрацией с получением желтого порошка как требуемого продукта (24.3 г, 135.8 ммоль, 78%).
b) К перемешиваемой суспензии сырых 5-гидразинилизохинолина (получен из Примера 25 этап а, 6.0 г, 37.7 ммоль) и калия 1-циано-3-этокси-3-оксопроп-1-ен-2-олата (8.1 г, 45.2 ммоль) в этаноле (36 мл) добавлен раствор 6 N водной соляной кислоты (7.7 мл, 45.2 ммоль) и деионизированная вода (10 мл). Реакционная смесь нагрета при 90°С в течение 5 часов. После охлаждения до комнатной температуры реакционная смесь сконцентрирована в вакууме, и получающийся остаток извлечен с 2:1 хлороформом/iPrOH. Органический слой промыт водным насыщенным бикарбонатом натрия, и органический слой высушен (Na2SO4) отфильтрован и сконцентрирован в вакууме. Получающееся твердое суспендировано в дихлорметан/диэтиловом эфире, и желтое твердое собрано фильтрацией с получением требуемого продукта (3.14 г, 11.1 ммоль, 30%).
c) К раствору этил-5-амино-1-(хинолин-5-ил)-1H-пиразол-3-карбоксилата (0.25 г, 0.89 ммоль) в дихлорметане (5 мл) добавлены DMAP (0.15 г, 1.2 ммоль) и Boc2O (0.5 г, 2.3 ммоль). Реакционная смесь размешана при комнатной температуре в течение 6 часов, и добавлен этилацетат. Получающийся раствор промыт водным насыщенным бикарбонатом натрия и соляным раствором. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Сырой продукт очищен флэш-хроматографией (SiO2, 20-50% этилацетат в гексанах) с предоставлением требуемого продукта (0.36 г, 0.75 ммоль, 84%).
d) К раствору этил-5-(бис(трет-бутоксикарбонил)амино)-1-(хинолин-5-ил)-1H-пиразол-3-карбоксилата (0.20 г, 0.41 ммоль) в TFA (6 мл) при 0°С добавлен раствор литий-алюминий гидрида (LAH, 2.0 М раствора в TFA, 0.48 мл, 0.96 ммоль). Реакционная смесь размешана при 0°С в течение 5 минут, и водный насыщенный калий-натрий тартрат добавлен к реакционной смеси. Получающийся раствор извлечен этилацетатом (2×5 мл). Объединенные органические слои высушены (Na2SO4), отфильтрованы и сконцентрированы в вакууме, с получением сырого продукта (0.14 г, 0.41 ммоль, 100%).
e) К перемешиваемому раствору трет-бутил-3-(гидроксиметил)-1-(хинолин-5-ил)-1H-пиразол-5-ил-карбамата (0.20 г, 0.59 ммоль) в дихлорметане (6 мл) при -45°С по каплям добавлен N,N-диэтиламиносульфуртрифторид (DAST, 0.15 мл, 1.2 ммоль), и реакционная смесь размешана при 0°С в течение 15 мин. Реакционную смесь затем влили в лед и добавили водный бикарбонат натрия. Водный слой извлечен этилацетатом, и органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Получающийся неочищенный остаток использован без дальнейшей очистки (0.20 г, 0.59 ммоль, 100%).
f) К перемешиваемому раствору трет-бутил 3-(фторметил)-1-(хинолин-5-ил)-1H-пиразол-5-илкарбамата (0.20 г, 0.59 ммоль) в дихлорметане (5 мл), и метаноле (1 мл) добавлен раствор соляной кислоты в n-диоксане (4 N раствор в n-диоксане, 10 мл, 40 ммоль). Реакционная смесь размешана при комнатной температуре в течение 1 часа, и органическое летучее удалено в вакууме. Полученный остаток растворен в 2:1 хлорметан/iPrOH и промыт водным раствором 1 М гидроокиси натрия и водным насыщенным бикарбонатом натрия. Органический слой высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток использован без дальнейшей очистки (0.14 г, 0.59 ммоль, 100%).
g) Смесь 4-трет-бутилбензолсульфонилхлорида (0.085 г, 0.37 ммоль), 3-(фторметил)-1-(хинолин-5-ил)-1H-пиразол-5-амина (0.05 г, 0.21 ммоль) и DMAP (0.025 г, 0.19 ммоль) в пиридине (1.0 мл) нагрета при 85°С в течение 2 часов с перемешиванием. После охлаждения до комнатной температуры к реакционной смеси добавлены 1 М водной гидроокиси лития (1 мл) и 1 М водной гидроокиси натрия (1 мл) и смесь нагрета при 75°С в течение 1 часа. После охлаждения до комнатной температуры реакционная смесь нейтрализована 1 N раствором водной соляной кислоты. Водный слой извлечен этилацетатом, и органический слой промыт водным насыщенным бикарбонатом натрия, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-Н2О с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.004 г, 0.009 ммоль, 4%). 1Н NMR (400 МГц, CD3OD) δ 8.86 (dd, J=1.2, 4.0 Гц, 1Н), 8.12 (d, J=8.8 Гц, 1Н), 7.79 (dd, J=8.4, 8.4 Гц, 1Н), 7.65 (d, J=8.4 Гц, 1Н), 7.56 (s, 1Н), 7.54 (s, 1Н), 7.43-7.37 (м, 4Н), 6.24 (s, 1Н), 5.34 (с, 1Н), 5.22 (s, 1Н), 1.35 (s, 9Н); MS: (ES) m/z вычислено для C23H24FN4O2S [М+Н]+ 439.2, обнаружено 439.1.
Пример 33: Синтез 4-трет-бутил-3-фтор-N-(3-(фторметил)-1-(хинолин-5-ил)-1H-пиразол-5-ил)бензолсульфонамида
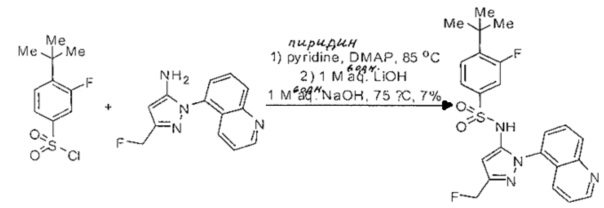
Смесь 4-трет-бутил-3-фторбензол-1-сульфонилхлорида (получено из Примера 27 этан с, 0.050 г, 0.20 ммоль), 3-(фторметил)-1-(хинолин-5-ил)-1H-пиразол-5-амина (0.025 г, 0.10 ммоль) и DMAP (0.012 г, 0.095 ммоль) в пиридине (3 мл) нагрета при 85°С в течение 2 часов с перемешиванием. После охлаждения до комнатной температуры к реакционной смеси добавлены 1 М водной гидроокиси лития (1 мл) и 1 М водной гидроокиси натрия (1 мл) и смесь нагрета при 75°С в течение 1 часа. После охлаждения до комнатной температуры реакционная смесь нейтрализована 1 N водным раствором соляной кислоты. Водный слой извлечен этилацетатом, и органический слой промыт водным насыщенным бикарбонатом натрия, высушен (Na2SO4), отфильтрован и сконцентрирован в вакууме. Неочищенный остаток очищен обратной фазой HPLC (колонка С18, ацетонитрил-H2O с 0.1% TFA как растворителем для элюирования) с получением названного соединения как белого твердого (0.003 г, 0.007 ммоль, 7%). 1H NMR (400 МГц, CD3OD) 8.83 (dd, J=4.4, 11.6 Гц, 1H), 8.10 (dd, J=1.2, 8.8 Гц, 1H), 7.83 (ddd, J=1.6, 7.6, 8.8 Гц, 1H), 7.63 (dd, J=0.8, 8.8 Гц, 1H), 7.53 (dd, J=0.8, 7.6 Гц, 1H), 7.41 (dd, J=1.6, 8.4 Гц, 1Н), 7.38-7.31 (м, 2Н), 7.24 (dd, J=1.6, 12.4 Гц, 1Н), 6.05 (d, J=1.6 Гц, 1Н), 5.28 (s, 1Н), 5.16 (s, 1Н), 1.40 (d, J=0.8 Гц, 9Н); MS: (ES) m/z вычислено для C23H26N5O2S [М+H]+ 457.2, обнаружено 457.2.
Измерение Эффективности Модуляторов Хемокина
In Vitro тестирование
Разнообразие тестирования может использоваться, чтобы оценить обеспеченные здесь соединения, включая сигнальное тестирование, хемотаксис (миграционное тестирование), тестирование связывания лиганда и другие виды тестирования клеточного ответа.
Сигнальное тестирование Рецептора Хемокина может использоваться, чтобы измерить способность соединения, такого как потенциальный CCR (9) антагонист, блокировать CCR (9) лиганд, (например, ТЕСК) - индуцированное сигнализирование. Блокирование такой передачи сигналов может быть полезным в лечении различных заболеваний типа воспалительных заболеваний кишечника, аллергических заболеваний, псориаза, атопического дерматита, астмы, фиброзных заболеваний, отторжения трансплантата, иммунно обусловленных пищевых аллергий, аутоиммунных заболеваний, брюшнополостных заболеваний, ревматического артрита, тимомы, тимьяновой карциномы, лейкемии, твердой опухоли, острой лимфоцитарной лейкемии, меланомы, первичного склерозирования желчных путей, гепатита, воспалительного заболевания печени или постоперативной кишечной непроходимости. Тестирование хемотаксиса может использоваться, чтобы измерить способность интересующего соединения, такого как возможный антагонист хемокина, блокировать хемокин-опосредованное перемещение клетки в пробирке. Последнее, как полагают, напоминает хемокин-вызванное перемещение клетки в естественных условиях. Тестирование связывания лиганда может также использоваться, чтобы измерить способность соединения, такого как потенциальный CCR (9) антагонист, блокировать взаимодействие ТЕСК или других CCR (9) лигандов с их рецептором.
В подходящем тестировании используется белок хемокина (или изолированный или рекомбинантный ген) или другой лиганд, чтобы иметь, по крайней мере, одно свойство, активность или функциональную характеристику хемокин белка млекопитающего. Свойство может быть связующей способностью (с, например, лигандом или ингибитором), сигнализирующей активностью (например, активация G белка млекопитающего, индукцией быстрого и переходного увеличения в концентрации цитозольного свободного иона кальция), клеточной функцией ответа (например, возбуждение хемотаксиса или высвобождения воспалительного медиатора лейкоцитами) и т.п.
Тестирование может быть тестированием на основе клетки, которое использует устойчивость клетки или кратковременно трансформируемость вектором или кассетой экспрессии, имеющей последовательность нуклеиновой кислоты, которая кодирует рецептор хемокина. Линии клеток или изолированные первичные клетки, естественно выражающие хемокин, могут также использоваться. Клетки поддерживаются в условиях, соответствующих экспрессии рецептора, и контактируют с предполагаемым агентом при соответствующих условиях для того, чтобы имело место связывание. Связывание может быть обнаружено с использованием стандартных методик. Например, степень связывания может быть определена относительно подходящего контроля (например, относительно фона в отсутствии предполагаемого агента или относительно известного лиганда). Предпочтительно, клеточная фракция, такая как мембранная фракция, содержащая рецептор, может использоваться вместо целых клеток.
Обнаружение связывания или сложного формирования может быть проведено прямо или косвенно. Например, предполагаемый агент может быть маркирован подходящей меткой (например, флуоресцентная метка, хемилюминесцентная метка, изотопная метка, ферментная метка и т.п.), и связывание может быть определено обнаружением метки. Определенное и/или конкурентное связывание может быть оценено исследованиями конкуренции или перемещения с использованием немаркированного агента или лиганда (например, ТЕСК) как конкурента.
Тестирование ингибирования связывания может использоваться, чтобы оценить присутствующие соединения. В этих тестированиях соединения оценивают как ингибиторы связывания лиганда, используя, например, ТЕСК или лиганды малых молекул. CCR (9) рецептор контактирует с лигандом в присутствии или отсутствии тестового агента, и осуществляется измерение связывания лиганда. Сокращение степени связывания лиганда является показателем ингибирования связывания тестовым агентом. Тестирование ингибирования связывания может быть выполнено с использованием целых клеток, которые выражают рецептор, или мембранной фракции из клеток, которые выражают рецептор.
Далее связывание рецептора, соединенного с G белком, например, агонистом, может привести к событию сигнализирования рецептором. Соответственно, тестирование сигнализирования может также использоваться, чтобы оценить соединения настоящего изобретения, и индукция сигнализирующей функции агентом может быть проверена, используя любой подходящий метод. Например, активность G белка, такая как гидролиз GTP к GDP, или более поздние события сигнализирования, вызванные связыванием рецептора, могут быть тестированы известными способами (см., например, PCT/US 97/15915; Neote et al., Cell, 72:415425 (1993); Van Riper et al., J. Exp. Med., 177:851-856 (1993) и Dahinden et al., J. Exp. Med., 179:751-756 (1994). Тестирование сигнализирования кальция также измеряет GPCR активность, определяя внутриклеточную концентрацию кальция в течение долгого времени, предпочтительно до и после связывания рецептора/лиганда, в присутствии или отсутствии тестового агента. Это тестирование используется при определении способности соединения, такого как соединения настоящего изобретения, производить медиатор сигнализирования рецептора связыванием к интересующему рецептору. Кроме того, это тестирование используется при определении способности соединения типа соединения настоящего изобретения замедлять медиатор сигнализирования рецептора путем интерферирования со связыванием между интересующим рецептором и лигандом.
В кальций сигнализирующем тестировании, используемом для определения способности соединения интерферировать со связыванием между рецептором хемокина и известным лигандом хемокина, выражающие рецептор хемокин клетки (CCR (9) - выражающие клетки, такие как MOLT 4 клетки Т клеточной линии) сначала инкубируют с интересующим соединением, таким как потенциальный антагонист хемокина, при увеличении концентраций. Число клеток может быть от 105 до 5×106 клеток на лунку в 96 - луночной микротитровальной пластине. Концентрация тестируемого соединения может располагаться в диапазоне от 0 до 100 тМ. После периода инкубации (который может составлять от 5 до 60 минут), обработанные клетки помещают во Флюорометрический считыватель изображения пластины (FLIPR®) (доступный от Molecular Devoces Corp., Sunnyvale, CA) согласно инструкции изготовителя. Система FLIPR® известна специалистам области как стандартный метод выполнения тестирования. Клетки затем стимулируют соответствующим количеством лиганда хемокина (ТЕСК для CCR (9) при финальных концентрациях 5-100 нМ, и регистрируют сигнал внутриклеточного увеличения кальция (также называемый поток кальция). Эффективность соединения как ингибитора связывания между хемокином и лигандом может быть вычислена как IC50 (концентрация, необходимая для того, чтобы вызвать 50%-ое ингибирование сигнализирования), или IC90 (при 90%-ом ингибировании).
Хемотаксисное тестирование может также использоваться, чтобы оценить функцию рецептора и оценить представленные здесь соединения. Это тестирование основано на функциональном перемещении клеток in vitro или in vivo, индуцированном агентом, и может использоваться, чтобы оценить связывание и/или влияние на хемотаксис лигандов, ингибиторов или агонистов. Разнообразие хемотаксисного тестирования известно в технологии, и любое подходящее тестирование может использоваться, чтобы оценить соединения настоящего изобретения. Примеры подходящих тестов включают описанные в PCT/US 97/15915; Springer et al., WO 94/20142; Berman et al., Immunol. Invest., 17: 625-677 (1988); и Kavanaugh et al., J. Immunol., 146: 4149-4156 (1991).
In vitro клеточный хемотаксисный анализ может быть выполнен (но без ограничения этим форматом) с использованием 96 - луночной микрокамеры (называемой ChemoTXTM). Система ChemoTXTM известна специалистам в области техники как тип прибора хемотаксисного/клеточного перемещения. В этом анализе CCR (9) - выражающие клетки (такие как MOLT-4) сначала инкубированы с соединением, представляющим интерес, таким как возможный CCR (9) антагонист при увеличивающихся концентрациях. Как правило, используются пятьдесят тысяч клеток в лунке, но количество может составлять от 103 до 106 клеток в лунке. Лиганд хемокина (например, CCR (9) лиганд ТЕСК, обычно при 50 нМ (но может составлять от 5 до 100 нМ)), помещают в нижнюю камеру и собирают прибор перемещения. Двадцать микролитров обработанных тестовым соединением клеток затем помещают на мембрану. Перемещению позволяют иметь место при 37°С в течение периода времени обычно 2.5 часа для CCR (9). В конце инкубации, подсчитывают число клеток, которые мигрировали через мембрану в нижнюю камеру. Эффективность соединения как ингибитора хемокин-опосредованного перемещения клетки может быть вычислена как IC50 (концентрация, необходимая для уменьшения перемещения клеток на 50%), или IC90 (для 90%-ого ингибирования).
In vivo модели эффективности для человеческого IBD
Инфильтрация Т-клеток в тонкую кишку и толстую кишку связана с патогенезом человеческих воспалительных заболеваний кишечника, которые включают брюшнополостные заболевания, заболевание Крона и язвенный колит. Блокирование перемещения релевантных Т-клеток в кишечник, как полагают, является эффективным подходом к лечению человеческого IBD. CCR (9) выражен на кишечно хоуминговых Т-клетках в периферийной крови, взятой у пациентов с небольшим воспалением кишечника, типа заболевания Крона и брюшнополостным заболеванием. ТЕСК CCR (9) лиганда выражается в тонком кишечнике. Таким образом, полагается, что эта пара лиганд-рецептор играет роль в развитии IBD, обуславливая перемещение Т-клеток к кишечнику. Несколько животных моделей существуют и могут использоваться для того, чтобы оценить соединения, представляющие интерес, такие как потенциальные CCR (9) антагонисты, на способность влиять на такое перемещение Т-клетки и/или состояние или заболевание, которая могла бы позволить предсказывать эффективность антагонистов в людях.
Животные модели с патологией, схожей с человеческим язвенным колитом
Крысиная модель, описанная Panwala и сотрудниками (Panwala et al., J Immunol., 161 (10): 5733-44 (1998)), включает генетическое удаление крысиного множественного стойкого к лекарствам гена (MDR). Мыши с удалением MDR (MDR-/-) - склонны к проявлению серьезного, непосредственного кишечного воспаления, когда поддерживаются при определенных условиях без патогена. Кишечное воспаление, отмеченное в мышах MDR-/-, имеет патологию, подобную человеческому воспалительному заболеванию кишечника (IBD) и определено инфильтрацией Т-клеток ТЫ типа в lamina propria толстой кишки.
Другая крысиная модель была описана Davidson и др., J Exp Медиана, 184 (1): 241-51 (1986). В этой модели крысиный ген IL-10 был удален и мыши представлены несовершенными в производстве интерлейкина 10 (IL-10-/-). Эти мыши проявляют хроническое воспалительное заболевание кишечника (IBD), которое преобладает в толстой кишке и разделяет гистопатологические особенности с человеческим IBD. Другая крысиная модель для IBD была описана Powrie et al, Int. Immunol, 5 (11): 1461-71 (1993), в которой подмножество CD4+Т клеток (названных CD45RB (высокая)) от иммунокомпетентных мышей очищено и адаптивно перемещено в иммунодефицитных мышей (таких как С. В-17 scid мыши). Животное, восстановленное с CD45RBhighCD4 + популяцией Т-клеток, развивало смертельное заболевание с серьезными инфильтратами одноядерных клеток в толстом кишечнике, патологически сходное с человеческим IBD.
TNF ARE (-/-) модель. Роль TNF в заболевании Крона в человеке демонстрировалась совсем недавно успехом лечения с использованием anti-TNF-альфа антитела, проведенного Targan et al., N. Engl. J Med., 337 (15): 1029-35 (1997). Мыши с аберрантным производством TNF-альфа из-за генетического изменения в гене TNF (ARE-/-) развивали Крон-подобные воспалительные заболевания кишечника (см. Kontoyiannis et al., Immunity, 10 (3): 387-98 (1999)).
Модель SAMP/yit. Эта модель описана Kosiewicz et al, J Clin. Invest., 107 (6): 695-702 (2001). Штамм мыши, SAMP/Yit, спонтанно развивает хроническое воспаление, локализованное в подвздошной кишке. Результирующий илеит характеризуется массивной инфильтрацией активированных Т-лимфоцитов в lamina propria и имеет замечательное сходство с заболеванием Крона человека.
Примеры in vitro тестов
Реактивы
MOLT-4 клетки получены из американской коллекции Типов Культур (Manassas, VA) и культивированы в питательной среде ткани RPMI с добавленной 10%-ной эмбриональной сывороткой теленка (FCS) в увлажненном 5%-ом инкубаторе CO2 при 37°С. Рекомбинантные человеческие хемокин белки ТЕСК были получены от R&D Systems (Minneapolis, MN). ChemoTX® хемотаксисные микрокамеры куплены у Neuro Probe (Gaithersburg, MD). Наборы клеточной пролиферации CyQUANT® куплены у Molecular Probes (Eugene, Oregon). Fluo-4 краски индикатора кальция куплены у Molecular Devices (Mountain View, CA).
Оценка тестового модулятора в анализе Мобилизации Кальция
Цитоплазматический анализ мобилизации кальция использовался для определения эффективности потенциальных антагонистов рецептора при блокировании сигналов, опосредованных через рецепторы хемокина, такие как CCR (9). Этот анализ обычно выполняется с использованием Флуоресцентного считывателя изображений (FLIPR, Molecular Devices). MOLT-4 клетки маркированы Fluo-4 красками индикатора флуоресценции (Molecular Devices) согласно указаниям изготовителя. После введения меченых атомов клетки собраны центрифугированием (400× г в течение 5 минут при комнатной температуре) и повторно суспендированы в HBSS до плотности клеток 2.5×106 клеток/мл. Испытательные соединения получены в 100%-ном диметилсульфоксиде при 100Х заключительной концентрации; в целом, диапазоны концентраций каждого соединения проверены, с заключительными концентрациями от 0.1 нМ до 10 000 нМ. Маркированные клетки (300 μL) смешаны с соединением или равным объемом диметилсульфоксида (3 мкL) в 96 - луночной пластине; после полного смешивания 50 мкL этой смеси клетки/соединение добавлено в каждую из четырех лунок 384 - луночной пластины FLIPR. Агонист хемокина (т.е. hTECK), полученный в HBSS при 5Х концентрациях предварительно определенной ЕС50 концентрации, добавлен в каждую лунку, и получающиеся изменения во флуоресцентной интенсивности, показательной для установленного рецептором сигнализирования хемокина зарегистрированы на FLIPR. IC50 значения соединения вычислены по этим данным, используя программное обеспечение Graphpad Prism(Graphpad Software) и нелинейную регрессию, модель конкуренции с одним участником.
Оценка тестового модулятора в анализе Сывороточного Хемотаксиса
Анализ Сывороточного хемотаксиса использовался для определения эффективности потенциальных антагонистов рецептора при блокировании перемещения, установленного через рецепторы хемокина, типа CCR (9). Этот анализ выполнен с использованием микрокамерной системы ChemoTX® с поликарбонатной мембраной с размером пор 5-цт.MOLT-4 клетки собраны центрифугированием в 400× г при комнатной температуре, затем суспендированы при 50 миллион/мл в человеческой сыворотке, содержащей 50 мм HEPES (заключительный рН 7.2). Тестируемое соединение или эквивалентный объем его растворителя (DMSO) затем добавлены к смеси клетки/сыворотка при заключительной концентрации DMSO 0.125% (v/v), и эта смесь затем инкубирована при 37°С в течение одного часа. Отдельно рекомбинантный человеческий ТЕСК разбавлен хемотаксисным буфером (HBSS +0.1% BSA), в целом охватывая диапазон от 0.1 нМ до 500 нМ, после чего 29 μl разбавленного хемокина помещено в более низкие лунки пластины ChemoTX®. 5 μl (размер пор) поликарбонатная мембрана помещена на пластину, и 20 μL смеси клетки/соединение перемещено на каждую лунку мембраны. Пластины были инкубированы при 37°С в течение 90 минут, после чего поликарбонатные мембраны удалены, и 5 μl ДНК-интеркалярного агента CeQUANT (Invitrogen, Carlsbad, СА) добавлены в более низкие лунки. Количество флюоресценции, соответствующее числу мигрировавших клеток, измерено с использованием Spectrafluor Plus считывателя пластины (TECAN, San Joce, СА).
Значения А2 вычислены из следующего уравнения, сравнивающего эффективность тестового соединения с DMSO контролем при экви-активных уровнях хемокина:
Log (А2)=log [drug (М)] - log [(A'/A)-1]
где А отражает потенцию агониста в отсутствии антагониста и А' отражает потенцию агониста в присутствии антагониста при данной концентрации препарата ([препарат (М)]).
Примеры In vivo анализов Эффективности
Оценка тестового модулятора в CCR (9) зависимой модели перемещения Т клеток Единичные клеточные суспензии получены из селезенки и лимфатических узлов OT-I Tg CD45.1 мыши. 15×106 в целом клеток (приблизительно 3×106 CD8 Т-клеток) введены в конгенных CD45.2 C57BL/6n мышей одного пола (8-10 недель). 24 часа спустя животные иммунизированы оральным кормлением 25 мг белка Яичного альбумина (Sigma-Aldrich, St. Louis, МО) + 10 μg Токсина Холеры (Calbiochem, San Diego, СА). CCR (9) антагонисты дозированы и введены до орального яичного альбумина во временном интервале, продиктованном фармакокинетикой мышей. Через пять дней после иммунизации животные подвергнуты эвтаназии, и тонкий кишечник изъят.Участки Пейера удалены и после воздействия PBS кишечник вскрыт на влажном квадрате ткани Optima (Allegiance Healthcare). Слизистая оболочка очищена скальпелем и затем диссоциирована перемешиваем в 50 мл среды, содержащей 10%-ую сыворотку новорожденного теленка и DTT (1 мМ) в течение 15 минут при комнатной температуре. После центрифугирования гранулы повторно суспендированы в PBS, содержащем 10%-ую сыворотку новорожденного теленка, закручены в течение 3 минут и быстро пропущены через колонку стекловаты (1.6 г, упакованную в 20 мл шприце; Fisher Scientific). IEL далее очищены на Ficoll-Paque градиенте и окрашены mAds для анализа поточной цитометрии. Перемещенные ОТ-1 Tg CD45.1 Т-клетки обнаружены и определены количественно поточной цитометрией. В этой модели обработка соединением изобретения привела к значительному сокращению частоты ОТ-1 Tg CD45.1 Т-клеток, которые перемещались к тонкому кишечнику в ответ на антиген.
Оценка тестового модулятора в модели клеточной передачи колита
Клеточные суспензии отдельных очищенных CD4+CD25-T клеток получены из селезенки и лимфоузлов мышей Balb/c. 1×106 CD4+CD25-T клетки затем переданы в подобранных по полу и возрасту СВ17 SCID мышей. CD4+CD25-мыши-реципиенты получили или носитель или соединение изобретения за 2 часа до перемещения. Массы тела мышей проверялись еженедельно, поскольку мыши проявляют заболевание, они похудели. Мыши, в которых прогрессия заболевания замедлена или замедлялась, будут иметь отмеченное различие в их массе тела относительно мышей, получающих носитель. В конце исследования толстые кишечники мышей взвешены и измерены, чтобы оценить изменения целевой ткани. Изменения в цитокинах также измерены в толстокишечной ткани гомогенатов. Обработка соединением изобретения приводит к значительной защите от ущерба, связанного с заболеванием, а также нормализации толстокишечного изменения и провоспалительных уровней цитокина.
Оценка тестового модулятора в Модели Ингибирования распространения ВИЧ
В костный мозг/печень/тимус, или "BLT" мыши нетучных диабетических (NOD)/SCID мышей (которые испытывают недостаток в эндогенных Т и В клетках) хирургическим путем внедрены фетальные тимьяновые и печеночные органоиды, как в системе SCID-hu. Мыши затем сублетально облучены и трансплантированы взятыми у той же особи CD34+ стволовыми клетками, полученными от эмбриональной печени, которые занимали место в крысином костном мозге, эффективно принимая человеческий трансплантат костного мозга и приводя к диапазону человеческих клеток в периферийной крови, включая зрелые Т и В лимфоциты, моноциты, макрофаги и дендритные клетки, все из которых показывают обширную инфильтрацию органов и тканей, включая печень, легкое и желудочно-кишечный трактат. Вслед за трансплантацией соединение изобретения введено трансплантированным мышам, чтобы ингибировать передачу человеческих клеток к желудочно-кишечному трактату, главному источнику взаимодействия Т-клеток/ВИЧ. Эффективность соединения измерена как снижение в крови вирусной нагрузки стандартными методиками.
Оценка тестового модулятора в модели артрита
17-дневное исследование вызванного коллагеном артрита типа II проводится для оценки влияния модулятора на вызванное артритом клиническое набухание лодыжки. Вызванный коллагеном артрит крысы - экспериментальная модель полиартрита, которая широко используется для преклинического тестирования многочисленных антиподагрических агентов (см. Trentham et al., J. Exp. Med. 146 (3): 857-868 (1977), Bendele et al., Toxicologic Pathol. 27: 134-142 (1999), Bendele et al., Arthritis Rheum. 42: 498-506 (1999)). Пробы этой модели - надежное начало и прогрессия сильного, легко измеримого полисуставного воспаления, отмеченная деструкция хряща в ассоциации с формированием паннуса и умеренного уменьшения резорбции кости и костной пролиферации надкостницы.
Женские крысы Льюиса (приблизительно 0.2 килограмма) обезболены изофлюраном и инжектированы неполным адъювантом Фреунда, содержащим бычий коллаген типа II при 2 мг/мл в основание хвоста и два участка на задней части в 0 и 6-й дни этого 17-дневного исследования. Тестовый модулятор дозирован ежедневно подкожной инъекцией с 9 дня до 17 дня в дозе 100 мг/кг и объеме 1 мл/кг в следующем носителе (24.5% Cremaphore EL, 24.5% обычное масло, 1%-й бензиловый спирт и 50%-ая Дистиллированная вода). Измерения толщиномером диаметра сустава лодыжки предприняты ежедневно, и снижение набухания сустава принято как мера эффективности.
Оценка тестового модулятора в Модели Язвенного Колита
MDR1a-выбывшие мыши, которые испытывают недостаток в гене Р-гликопротеина, спонтанно проявляют колит при определенных условиях без патогена. Патология у этих животных характеризуется как Th1-тип воспаления, обусловленного Т-клетками, подобного язвенному колиту в людях. Заболевание обычно начинает развиваться в пределах 8-10 недель после рождения. Однако возрасты, в которых заболевание появляется, и окончательный уровень проникновения часто разнятся значительно среди различных животных.
В исследовании, использующем MDR1a выбывших мышей, CCR (9) антагонист изобретения был оценен профилактическим введением его способности задерживать начало заболевания. Женские мыши (n=34) дозированы 10-100 mg/kg один раз в день подкожными инъекциями в течение 14 последовательных недель, начиная с возраста в 10 недель. Исследование было оценено для IBD-связанного запаздывания роста, и тестовое соединение показало эффективность в этой модели.
Оценка тестового модулятора в мышиной модели астмы
Этот пример описывает процедуру оценки эффективности антагонистов для лечения астмы. Животная модель астмы может быть индуцирована путем сенсибилизации грызунов к экспериментальному антигену (например, OVA) стандартной иммунизацией и впоследствии введением того же самого антигена в легкое грызуна аэрозолизацией. Три серии групп грызунов, включая 10 грызунов в группе, активно сенсибилизированы в День 0 единственной i.p. инъекцией 100 ug OVA в фосфат-буферизованном соляном растворе (PBS), вместе с адъювантом, например, гидроксидом алюминия. В 11-й день после сенсибилизации животные размещены в Плексигласовую камеру и опрыснуты аэрозолем OVA (1%) в течение 30 минут, используя сверхзвуковой распылитель (De Vilbliss). Одни серии мышей дополнительно получают PBS и Твин 0.5% i.p. в начальной сенсибилизации и при различных режимах дозирования после этого, вплоть до вызова опрыскиванием аэрозоля OVA. Вторые серии состоят из групп мышей, получающих различные дозы CCR4 антагониста, данного интраперитонеально, внутривенно, подкожно, внутримышечно, орально или через любой другой способ введения в начальном повышении чувствительности и в различных режимах дозирования после этого, вплоть до вызова опрыскивания аэрозолем OVA. Третьи серии мышей, служащие как позитивный контроль, состоят из групп, которые обрабатывают мышиным IL-10 i.p., анти-Ib4 i.p антителами или анти-Ib5 i.p. антителами в начальном повышении чувствительности и при различном режиме дозирования после этого, вплоть до вызова опрыскивания аэрозолем OVA. Животные впоследствии анализированы в различные моменты времени после вызова опрыскивания аэрозолем OVA на легочные функции, клеточные инфильтраты в бронхоальвеолярном лаваже (BAL), гистологическое исследование легких и измерение определенных титров IgE сыворотки OVA.
Это, следовательно, означает, что предшествующее детализированное описание расценивается как иллюстративное вместо ограничивающего, и понятно, что следующая формула изобретения, включая все эквиваленты, предназначена, чтобы определить сущность и объем этого изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-АЦИЛ-4-ФЕНИЛСУЛЬФОНИЛПРОЛИНАМИДА И НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2615997C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИМИДИНА | 2013 |
|
RU2637925C2 |
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
| 6-АРИЛ-4-МОРФОЛИН-1-ИЛПИРИДОНЫ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА ИЛИ ДИАБЕТА | 2017 |
|
RU2754664C2 |
| МОДУЛЯТОРЫ ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ | 2017 |
|
RU2738646C2 |
| СОЕДИНЕНИЯ ЗАМЕЩЕННЫХ ПИРАЗОЛОНОВ И СПОСОБЫ ИСПОЛЬЗОВАНИЯ | 2013 |
|
RU2650895C2 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИЧЕСКИХ ПРЕПАРАТАХ | 2013 |
|
RU2701156C2 |
| АНТАГОНИСТЫ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2013 |
|
RU2646762C2 |
| АНТАГОНИСТЫ CXCR7 | 2013 |
|
RU2649004C2 |
| ПРОИЗВОДНЫЕ 5-ЧЛЕННЫХ ГЕТЕРОЦИКЛОВ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ p38 | 2004 |
|
RU2381219C2 |
Изобретение относится к соединениям формулы (I), в которой радикалы и символы имеют значения, указанные в формуле изобретения, и к их вариантам. Предложенные соединения действуют как мощные антагонисты CCR (9) рецептора. Тестирование животных показывает, что эти соединения полезны для лечения воспаления, заболевания с отличительным признаком для CCR (9). Соединения в целом являются производными арилсульфамида и используются в фармацевтических композициях, способах лечения CCR (9) опосредованных заболеваний и как контроль в анализах для идентификации CCR (9) антагонистов. 9 н. и 17 з.п. ф-лы, 2 табл., 33 пр.
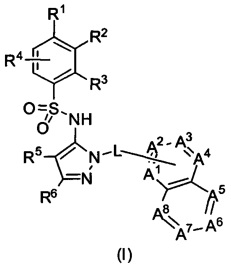
1. Соединение или его фармацевтически приемлемая соль формулы (I):
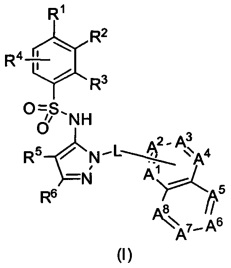
где
R1 выбран из группы, состоящей из замещенного или незамещенного линейного или разветвленного С2-8 алкила, замещенного или незамещенного C1-8 алкокси, цианоциклопропила, или морфолино, где замещенный С2-8 алкил является замещенным гало или гидрокси и где замещенный C1-8 алкокси является замещенным гало;
R2 - это Н, F, Cl или незамещенный C1-8 алкокси; или
R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют
-ОСН(СН3)2СН2-, -ОСН(СН3)2СН2СН2- или -С(СН3)2СН2СН2-;
R3 является Н, незамещенным линейным или разветвленным C1-8 алкилом, незамещенным C1-8 алкокси или гало;
R4 является Н или F;
R5 является Н или F;
R6 является Н, гало, -CN, -CONH2, циклопропилом, замещенным или незамещенным линейным или разветвленным С1-8 алкилом, где замещенный C1-8 алкил замещен гало, гидрокси, -CN или -ОСН3;
где R5 и R6 могут вместе образовать циклопентановое кольцо;
L является связью или -СН2-;
каждый из А1, А2, А3, А4, А5, А6, А7 и А8 независимо выбран из группы, состоящей из N, N-O и -CR8-; где по крайней мере один и не более чем два из А1, А2, А3, А4, А5, А6, А7 и A8 - это -N или N-O;
R8 каждый независимо выбран из группы, состоящей из Н, гало, -CN, - ОН, оксо, незамещенного линейного или разветвленного С1-8 алкила, незамещенного C1-8 алкокси, и -NR20R21; и
R20 и R21 - каждый является независимо Н или незамещенным линейным или разветвленным C1-8 алкилом; при этом соединение не является 3,4-диметокси-N-(3-метил-1-(4,6,8 триметилхинолин 2 ил)-1Н-пиразол-5-ил) бензенсульфонамидом, N-(1-(4,8-диметилхинолин-2-ил)-3-метил-1Н-пиразол-5-ил)-3,4-диметоксибензенсульфамидом, 3,4-диметокси-N-(3-метил-1-(4-метилхинолин-2-ил)-1Н-пиразол-5-ил) бензенсульфонамидом, 3,4-диметокси-N-(3-метил-1-(хинолин-2-ил)-1Н-пиразол-5-ил) бензенсульфонамидом и 3,4-диметокси-N-(1-(8 метокси-4-метилхинолин 2 ил)-3-метил-1Н-пиразол-5-ил) бензенсульфонамидом.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, в котором один из А1 или А2 является N или N-О, и остальные из А1, А2, А3, А4, А5, А6, А7 и А8 - -CR8, где каждый R8 выбран независимо.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, в котором два из А2, А3, А4, А5 являются N или N-О и остальные из А1, А2, А3, А4, А5, А6, А7 и А8 являются -R8-, где каждый R8 выбран независимо.
4. Соединение или его фармацевтически приемлемая соль формулы (II):
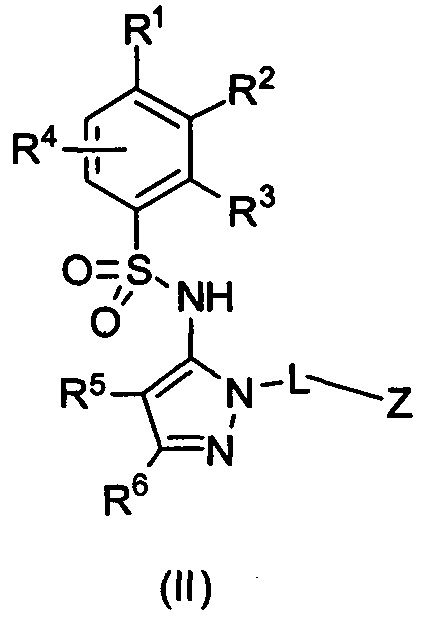
где
R1 выбран из группы, содержащей замещенный или незамещенный линейный или разветвленный С2-8 алкил, замещенный или незамещенный C1-8 алкокси, цианоциклопропил, или морфолино, где замещенный С2-8 алкил замещен гало или гидрокси и где замещенный С1-8 алкокси замещен гало;
R2 является Н, F, Cl или незамещенным C1-8 алкокси; или
R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют
-ОСН(СН3)2СН2-, -ОСН(СН3)2СН2СН2-, или -С(СН3)2СН2СН2-;
R3 является Н, незамещенным линейным или разветвленным C1-8 алкилом, незамещенным C1-8 алкокси или гало;
R4 является Н или F;
R5 является Н или F;
R6 является Н, гало, -CN, -CONH2, циклопропилом, замещенным или незамещенным линейным или разветвленным C1-8 алкилом, где замещенный C1-8 алкил замещен гало, гидрокси, -CN или -ОСН3;
где R5 и R6 могут вместе образовать циклопентановое кольцо;
L является связью или -СН2-; и
Z выбран из группы, состоящей из
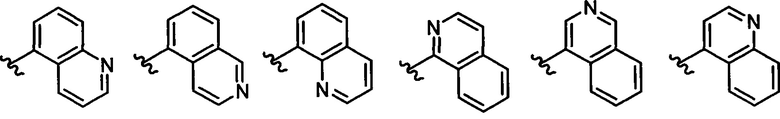
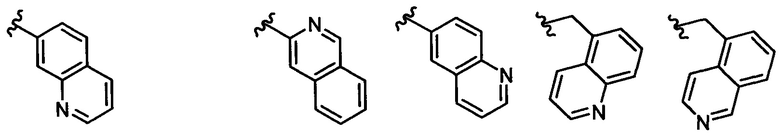
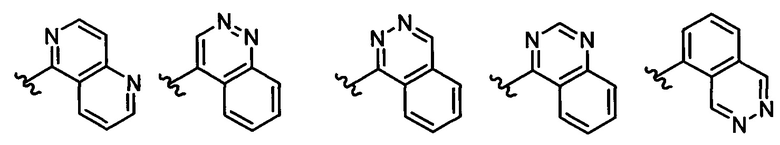
где Z-группа может быть незамещенной или замещенной от 1 до 3 независимо выбранными R8 заместителями;
каждый R8 независимо выбран из группы, содержащей Н, гало, - CN, -ОН, оксо, незамещенный линейный или разветвленный C1-8 алкил, незамещенный C1-8 алкокси, и -NR20R21 и
R20 и R21 - каждый независимо Н или незамещенный линейный или разветвленный С1-8 алкил.
5. Соединение или фармацевтически приемлемая его соль формулы (II):
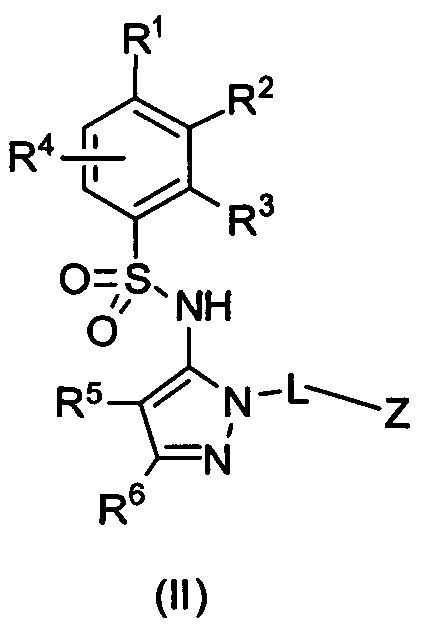
где
R1 выбран из группы, состоящей из замещенного или незамещенного линейного или разветвленного С2-8 алкила, замещенного или незамещенного C1-8 алкокси, цианоциклопропила, или морфолино, где замещенный С2-8 алкил замещен гало или гидрокси и где замещенный C1-8 алкокси замещен гало;
R2 является Н, F, С или незамещенным C1-8 алкокси; или
R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют
-ОСН(СН3)2СН2-, -ОСН(СН3)2СН2СН2- или -С(СН3)2СН2СН2-;
R3 является Н, незамещенным линейным или разветвленным C1-8 алкилом, незамещенным C1-8 алкокси или гало;
R4 является Н или F;
R5 является Н или F;
R6 является Н, гало, -CN, -CONH2, циклопропаном, замещенным или незамещенным линейным или разветвленным C1-8 алкилом, где замещенный C1-8 алкил замещен гало, гидрокси, -CN или -ОСН3;
L является связью; и
Z-это 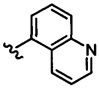 ,
,
где Z группа может быть незамещенной или замещенной от 1 до 3 независимо выбранными R8 заместителями;
каждый R8 независимо выбран из группы, состоящей из Н, гало, -CN, - ОН, оксо, незамещенного линейного или разветвленного C1-8 алкила, незамещенного C1-8 алкокси и -NR20R21; и
R20 и R21 - каждый независимо является Н или незамещенным линейным или разветвленным C1-8 алкилом.
6. Соединение по п. 5 или фармацевтически приемлемая его соль, где
R1 выбран из группы, состоящей из: -СН2СН3, -СН(СН3)2, -С(СН3)3,
-С(СН3)2СН2СН3, -C(CH2CH2)CN, -С(O)(СН3)2, -ОСН3, -ОСН2СН3, -ОСН(СН3)2,
-ОС(СН3)3, -ОСН2СН(СН3)2, -OCF3 и морфолино;
R2 является Н, F, или Cl; или
R3 является Н, -СН3, или -ОСН3;
R4 является Н или F;
R5 является Н;
R6 является Н, -СН3, -СН2СН3, -СН(СН3)2, -С3Н7, -CH2F, -CHF2, -CF2CH3,
-CF3, -CH2OCH3, -CH2OH, -CH2CN, -CN или -CONH2; и
каждый R8 независимо выбран из группы, состоящей из Н, F, Cl, Br,
-СН3, - ОН, -ОСН3, -ОСН2СН3, -NH2, -N(CH3)2 и -CN.
7. Соединение по п. 5 или фармацевтически приемлемая его соль, где
R1 является -С(СН3)3;
R2 является Н или F;
R3 является Н;
R4 является Н;
R5 является Н; и
R6 является -СН3, -CH2F, -CHF2 или -CF3.
8. Соединение по п. 4 или фармацевтически приемлемая его соль формулы (IIIa) или (IIIb):
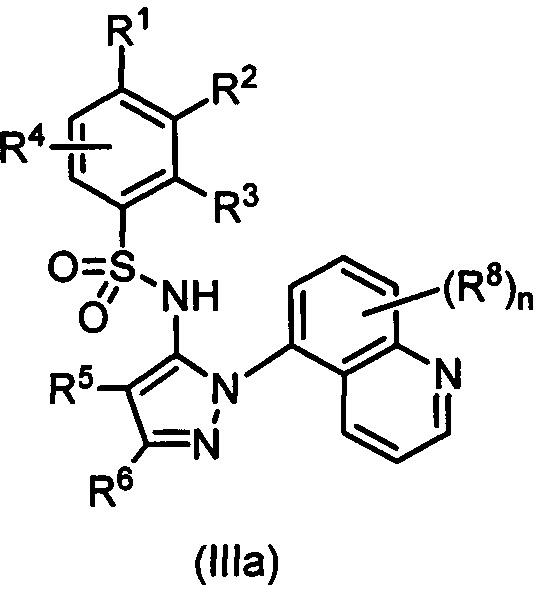
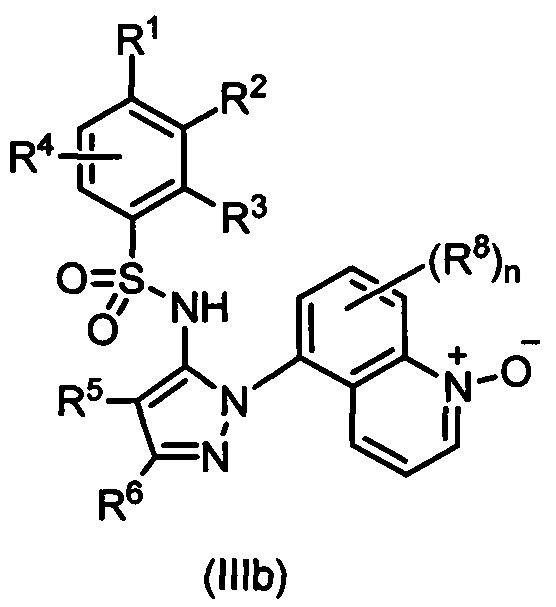
где
R1 выбран из группы, состоящей из замещенного или незамещенного линейного или разветвленного C2-8 алкила, замещенного или незамещенного C1-8 алкокси, цианоциклопропила, или морфолино, где замещенный C2-8 алкил замещен гало или гидрокси и где замещенный С1-8 алкокси замещен гало;
R2 является H, F, Cl или незамещенным C1-8 алкокси; или
R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют
-ОСН(СН3)2СН2-, -ОСН(СН3)2СН2СН2- или -С(СН3)2СН2СН2-;
R3 является Н, незамещенным линейным или разветвленным C1-8 алкилом, незамещенным C1-8 алкокси, или гало;
R4 является Н или F;
R5 является Н или F;
R6 является Н, гало, -CN, -CONH2, циклопропилом, замещенным или незамещенным линейным или разветвленным C1-8 алкилом, где замещенный C1-8 алкил замещают гало, гидрокси, -CN или -ОСН3;
или где R5 и R6 вместе с атомами углерода, к которым они присоединены, образуют циклопентановое кольцо;
каждый R8 независимо выбран из группы, состоящей из Н, гало, -CN, -ОН, оксо, незамещенного линейного или разветвленного C1-8 алкила, незамещенного С1-8 алкокси и -NR20R21;
R20 и R21 - каждый независимо Н, незамещенный линейный или разветвленный C1-8 алкил; и
n - это 0, 1, 2 или 3.
9. Соединение по п. 8 или его фармацевтически приемлемая соль, где
R1 выбран из группы, состоящей из: -СН2СН3, -СН(СН3)2, -С(СН3)3, -С(СН3)2СН2СН3,
-C(CH2CH2)CN, -С(O)(СН3)2, -ОСН3, -ОСН2СН3, -ОСН(СН3)2, -ОС(СН3)3,
-ОСН2СН(СН3)2, -OCF3 и морфолино;
R2 является Н, F или Cl; или
R1 и R2 могут вместе образовать -ОС(СН3)2СН2- или -С(СН3)2СН2СН2-;
R3 является Н, -СН3, или -ОСН3;
R4 является Н или F;
R5 является Н;
R6 является Н, -СН3, -СН2СН3, -СН(СН3)2, -С3Н7, -CH2F, -CHF2, -CF2CH3, -CF3, -CH2OCH3, -CH2OH, -CH2CN, -CN или -CONH2; и
каждый R8 независимо выбран из группы, состоящей из Н, F, Cl, Br, -СН3, - ОН, -ОСН3, -ОСН2СН3, -NH2, -N(СН3)2 и -CN.
10. Соединение по п. 9 или его фармацевтически приемлемая соль, где R1 является -С(СН3)3.
11. Соединение по п. 10 или его фармацевтически приемлемая соль, где
R2 является Н или F;
R3 является Н;
R4 является Н; и
R6 является -СН3, -CH2F, -CHF2 или -CF3.
12. Соединение или его фармацевтически приемлемая соль, выбранные из группы, состоящей из:
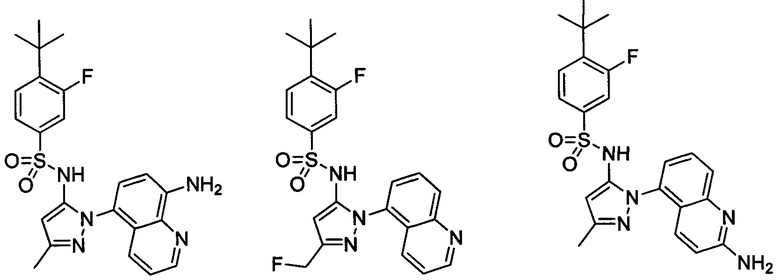
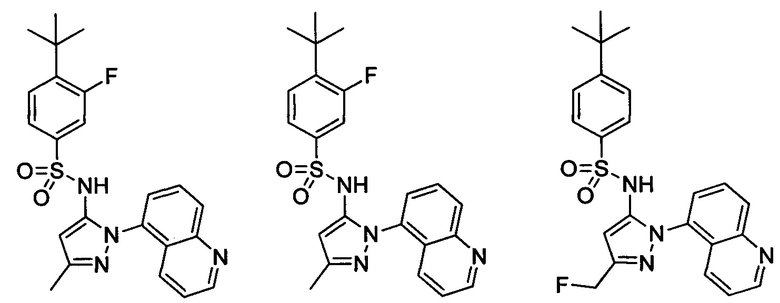
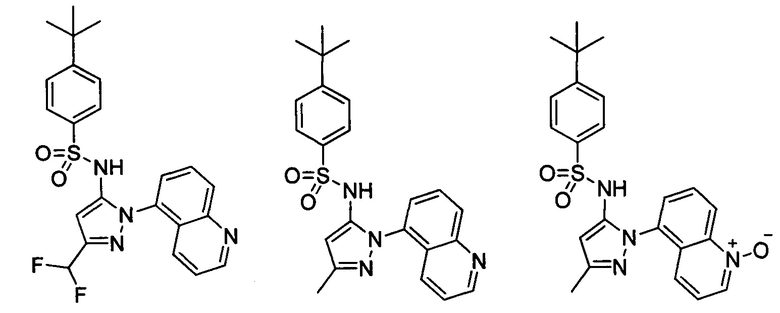
13. Соединение или его фармацевтически приемлемая соль, выбранные из группы, состоящей из
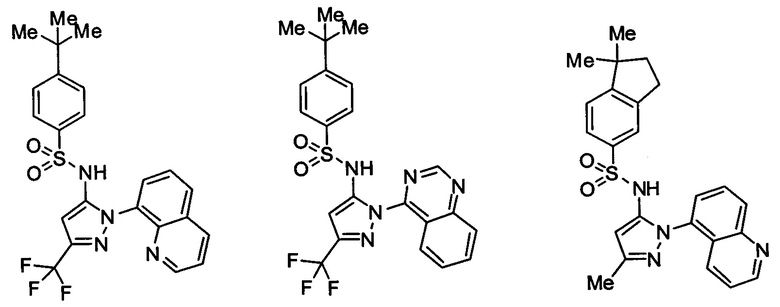
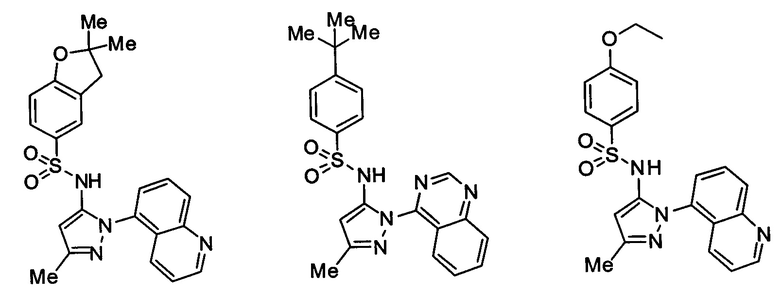
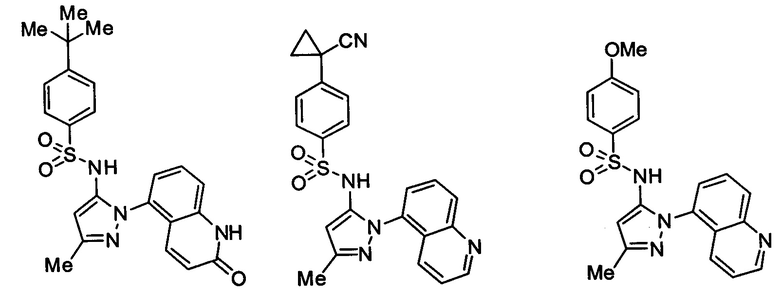
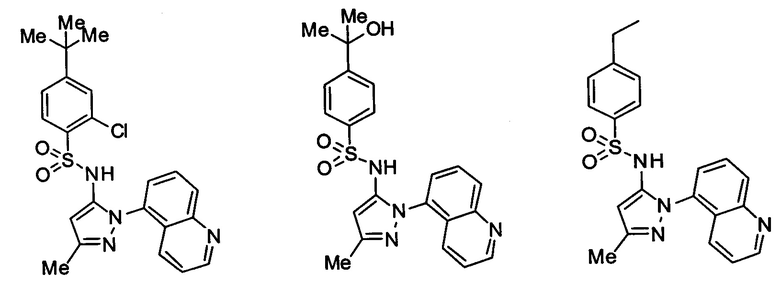
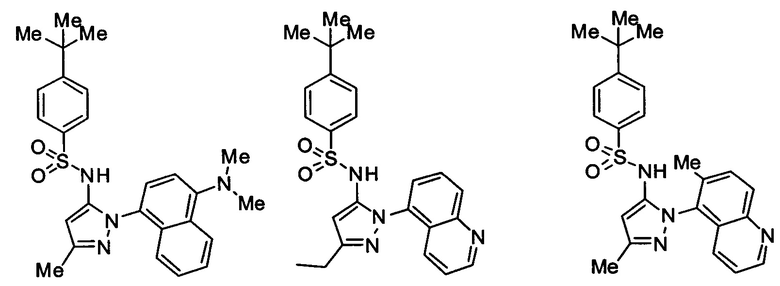
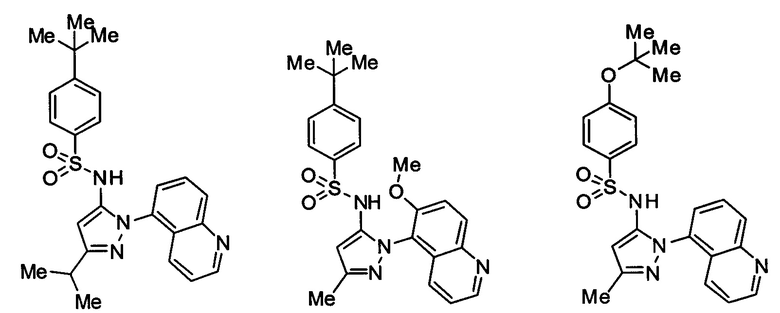
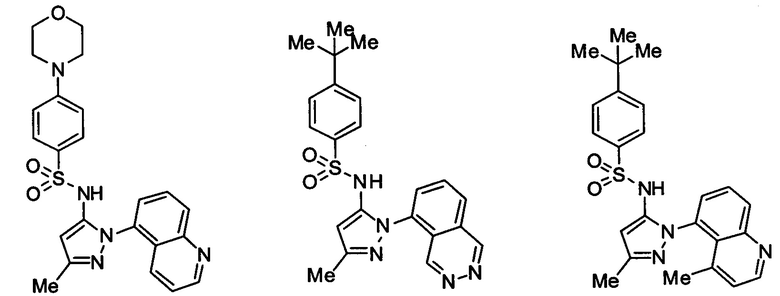
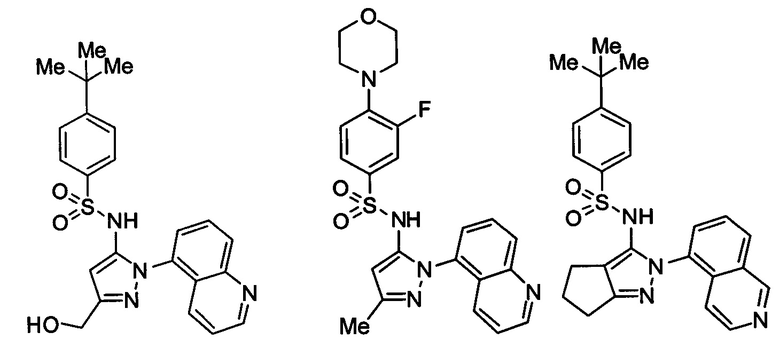
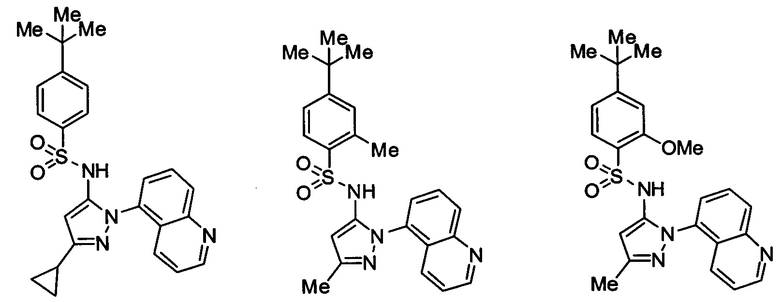
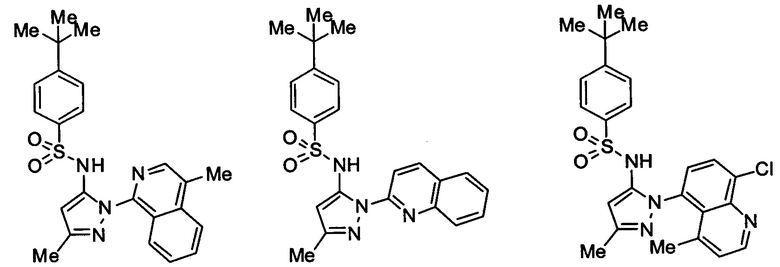
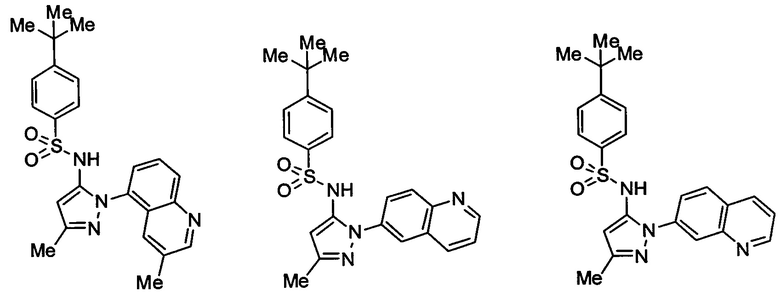
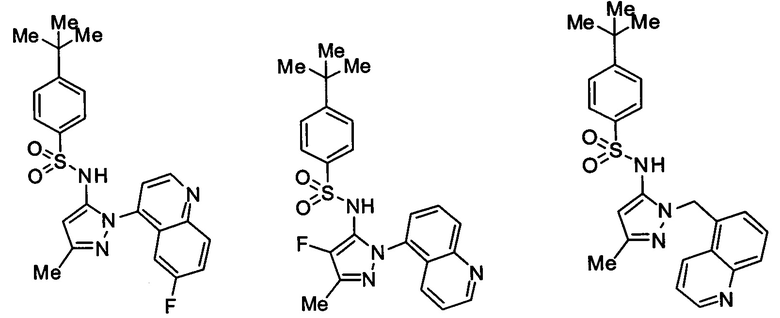
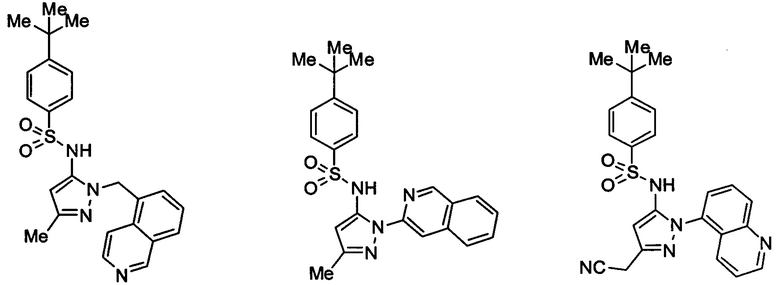
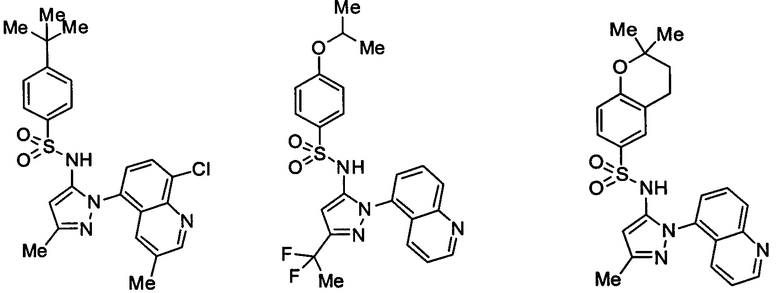
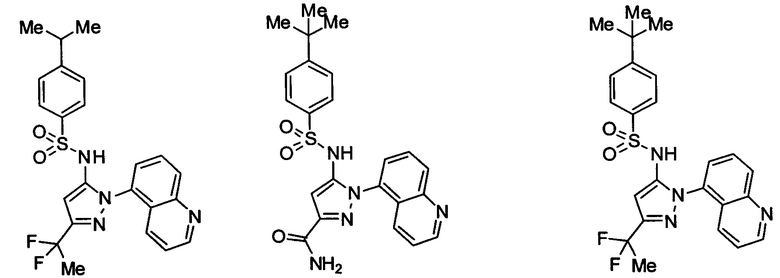
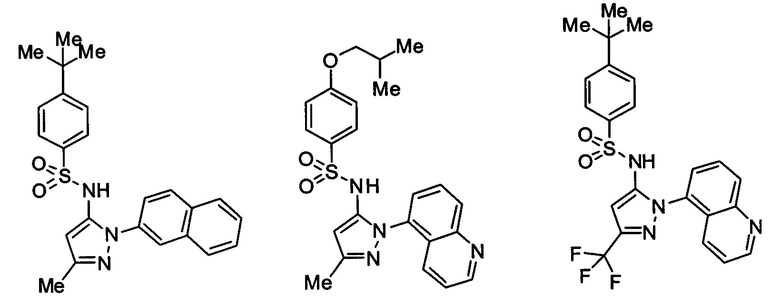
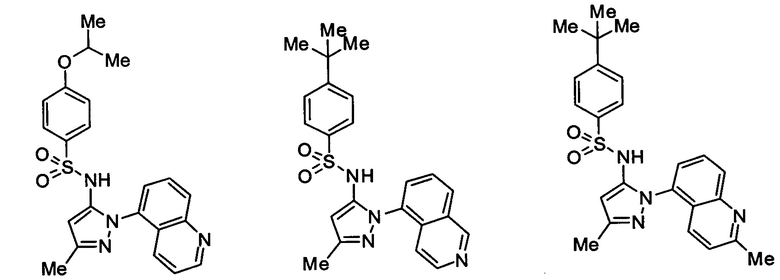
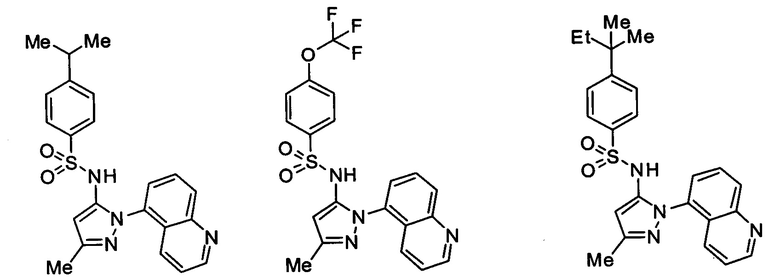
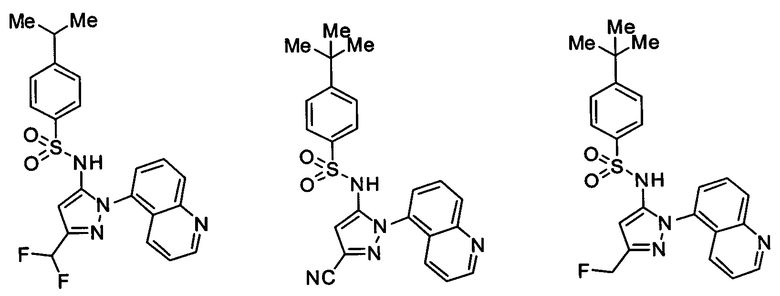
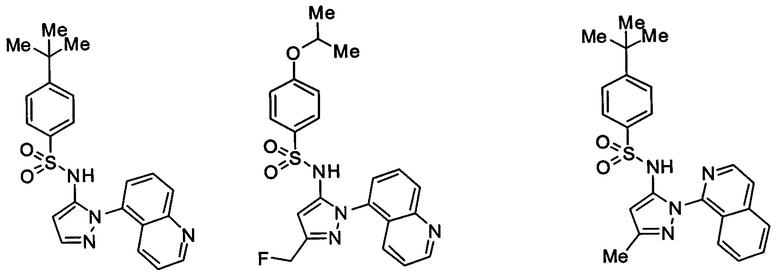
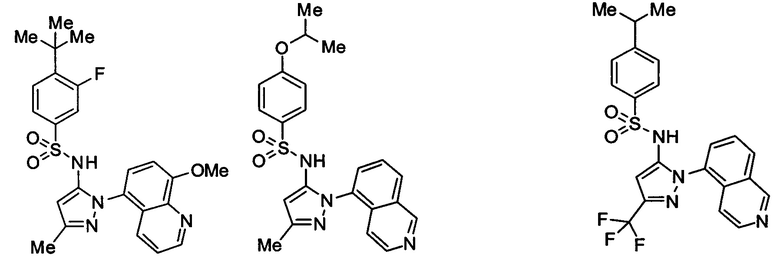
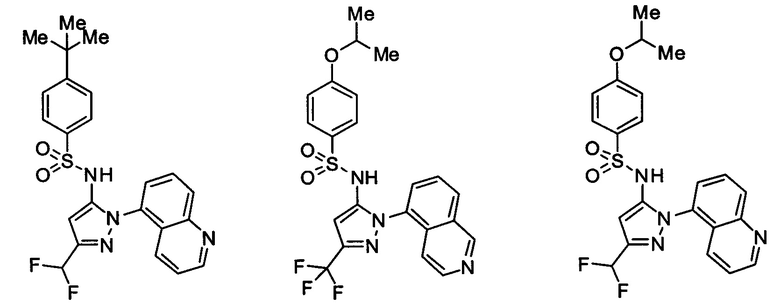
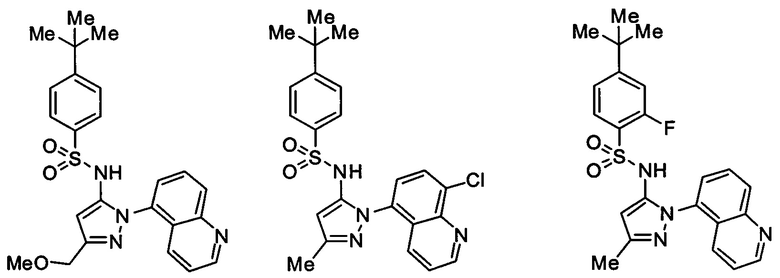
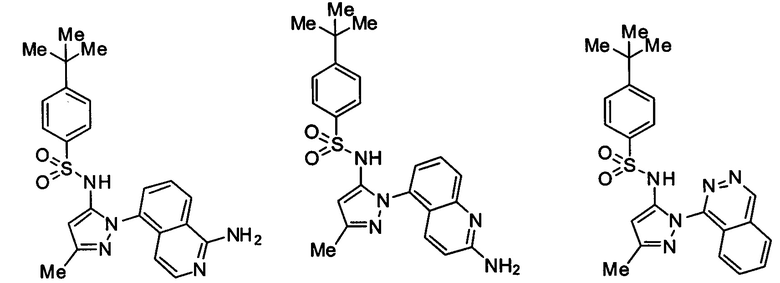
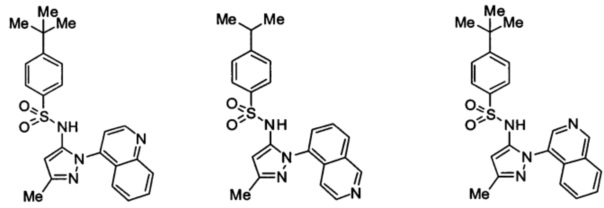
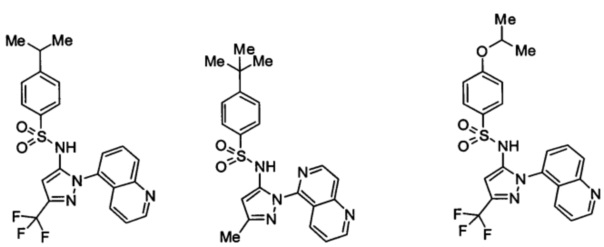
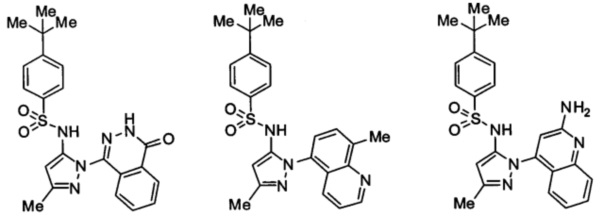
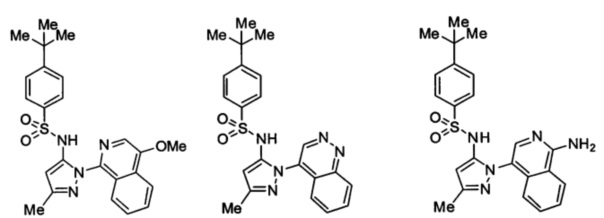
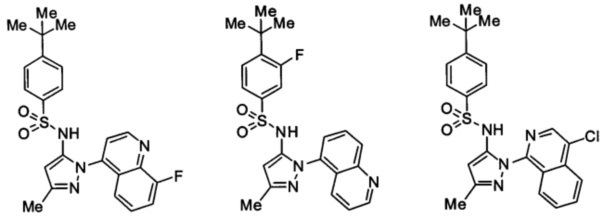
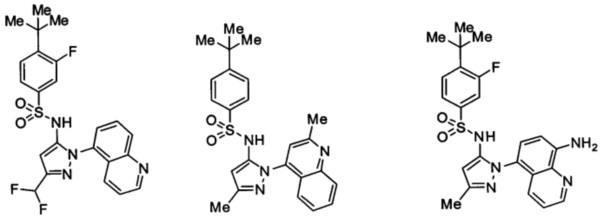
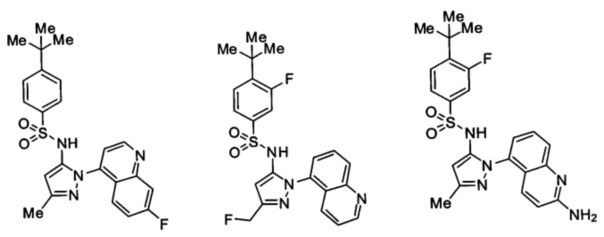
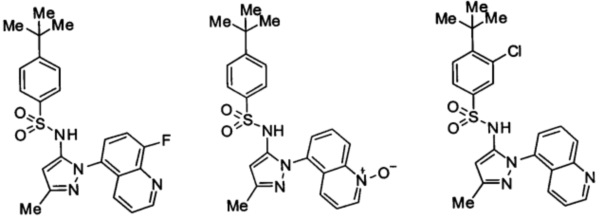
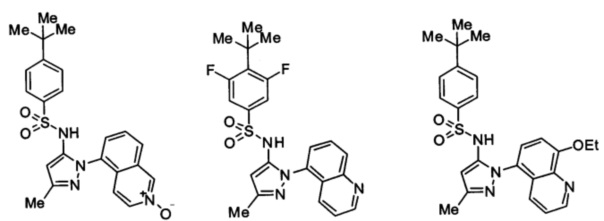
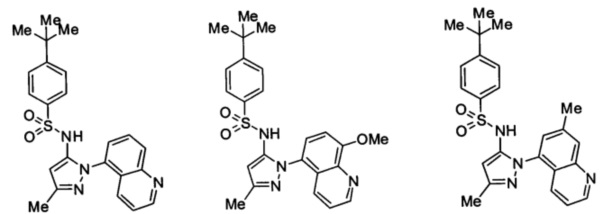
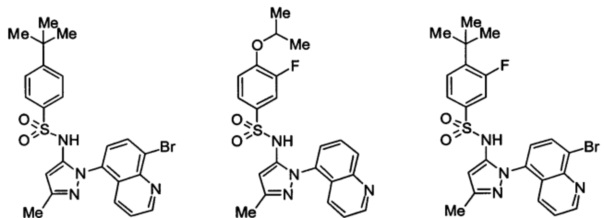
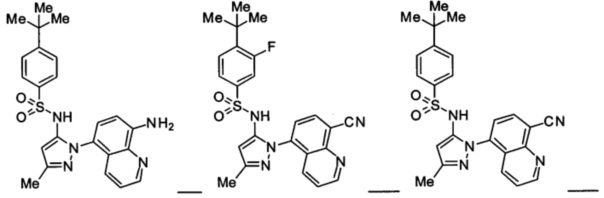
14. Соединение или его фармацевтически приемлемая соль, где соединение - это
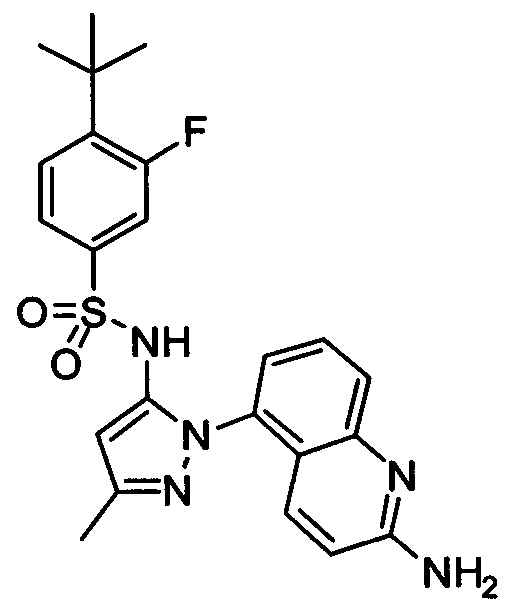
15. Композиция, которая ингибирует CCR (9) функцию, включающая фармацевтически приемлемый носитель и соединение или его фармацевтически приемлемую соль формулы (I):
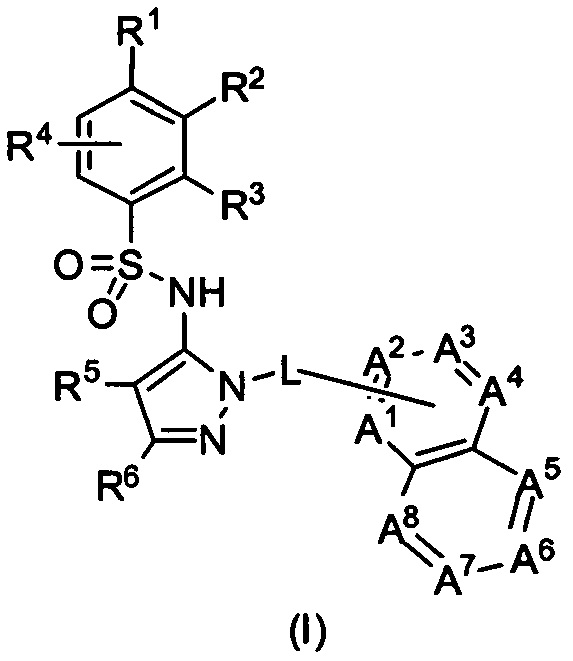
где
R1 выбран из группы, состоящей из замещенного или незамещенного линейного или разветвленного С2-8 алкила, замещенного или незамещенного C1-8 алкокси, цианоциклопропила, или морфолино, где замещенный С2-8 алкил замещен гало или гидрокси и где замещенный С1-8 алкокси замещен гало;
R2 является Н, F, Cl или незамещенным C1-8 алкокси; или
R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют
-ОСН(СН3)2СН2-, -ОСН(СН3)2СН2СН2- или -С(СН3)2СН2СН2-;
R3 является Н, линейным или разветвленным алкилом, незамещенным C1-8 алкокси, или гало;
R4 является Н или F;
R5 является Н или F;
R6 является Н, гало, -CN, -CONH2, циклопропилом, замещенным или незамещенным линейным или разветвленным C1-8 алкилом, где замещенный C1-8 алкил замещен гало, гидрокси, -CN или -ОСН3;
где R5 и R6 могут вместе формировать циклопентановое кольцо;
L является связью или -СН2-;
каждый из А1, А2, А3, А4, А5, А6, А7 и А8 независимо выбран из группы, состоящей из N, N-О и -CR8-; где по крайней мере один и не более чем два из А1, А2, А3, А4, А5, А6, А7 и А8 - это - N или N-O;
R8 каждый независимо выбран из группы, состоящей из Н, гало, -CN, -ОН, оксо, линейного или разветвленного C1-8 алкила, незамещенного C1-8 алкокси и -NR20R21; и
R20 и R21 - каждый независимо Н или незамещенный линейный или разветвленный С1-8 алкил.
16. Композиция по п. 15, где соединение или его фармацевтически приемлемая соль имеют формулу (II):
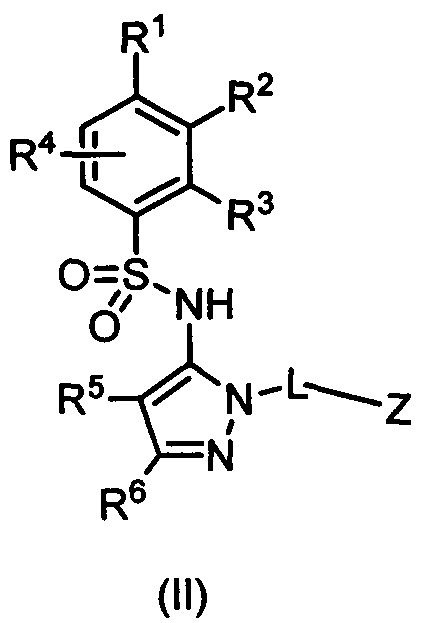
где
R1 выбран из группы, состоящей из замещенного или незамещенного линейного или разветвленного С2-8 алкила, замещенного или незамещенного C1-8 алкокси, цианоциклопропила, или морфолино, где замещенный С2-8 алкил замещен гало или гидрокси и где замещенный C1-8 алкокси замещен гало;
R2 - Н, F, Cl или незамещенный С1-8 алкокси; или
R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют
-ОСН(СН3)2СН2-, -ОСН(СН3)2СН2СН2- или -С(СН3)2СН2СН2-;
R3 является Н, незамещенным линейным или разветвленным C1-8 алкилом, незамещенным C1-8 алкокси или гало;
R4 является Н или F;
R5 является Н или F;
R6 является Н, гало, -CN, -CONH2, циклопропилом, замещенным или незамещенным линейным или разветвленным C1-8 алкилом, где замещенный C1-8 алкил замещен гало, гидрокси, -CN или -ОСН3;
L является связью; и
Z - это 
где Z группа может быть незамещенной или замещенной от 1 до 3 независимо выбранными R8 заместителями;
каждый R8 независимо выбран из группы, состоящей из Н, гало, -CN, -ОН, оксо, незамещенного линейного или разветвленного C1-8 алкила, незамещенного C1-8 алкокси и -NR20R21; и
R20 и R21 - каждый независимо Н или незамещенный линейный или разветвленный C1-8 алкил.
17. Композиция по п. 15, в которой соединение или его фармацевтически приемлемая соль - это
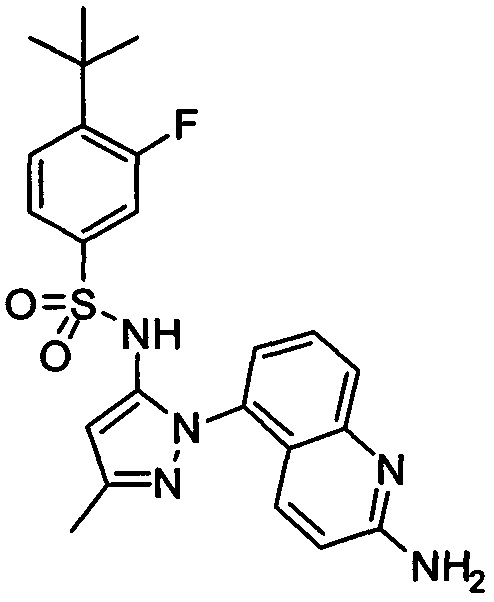
18. Способ ингибирования CCR (9) функции в клетке, включающий контактирование клетки с CCR (9) ингибирующим количеством соединения по любому одному из пп. 1-14 или композиции по любому одному из пп. 15-17.
19. Способ для лечения CCR (9) опосредованного состояния или заболевания, включающий введение субъекту эффективного количества соединения по любому одному из пп. 1-14 или композиции по любому одному из пп. 15-17.
20. Способ по п. 19, в котором субъектом является человек.
21. Способ по п. 19, где введение является оральным, парентеральным, ректальным, трансдермальным, подъязыковым, назальным или местным.
22. Способ по п. 19, где CCR (9) опосредованное заболевание или состояние является воспалительными заболеваниями кишечника, аллергическим заболеванием, псориазом, атопическим дерматитом, астмой, фиброзными заболеваниями, отторжением трансплантата, заболеванием трансплантат против хозяина, иммунно-опосредованными пищевыми аллергиями, аутоиммунными заболеваниями, кишечным заболеванием, ревматоидным артритом, тимомой, относящаяся к зобной железе карциномой, лейкемией, твердой опухолью, острым лимфолейкозом, меланомой, первичным склерозирующим холангитом, гепатитом, воспалительным заболеванием печени или послеоперационной кишечной непроходимостью.
23. Способ по п. 19, где CCR (9) опосредованное заболевание или состояние является воспалительным заболеванием кишечника, выбранным из группы, состоящей из болезни Крона или язвенного колита.
24. Способ по п. 19, где CCR (9) опосредованное заболевание или состояние является астмой.
25. Способ по п. 19, где CCR (9) опосредованное заболевание является заболеванием трансплантат против хозяина.
26. Способ по любому из пп. 19-25, далее включающий введение противовоспалительного или болеутоляющего агента.
| EA 200901583 A1, 29.10.2010 | |||
| Способ парообразования в котлах непрерывной циркуляцией | 1928 |
|
SU15710A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

