ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к соединению как ингибитору пути сигнальной трансдукции WNT, а также к композиции, содержащей его. Кроме того, настоящее изобретение относится к применению указанного соединения и к способу ингибирования пути сигнальной трансдукции WNT.
УРОВЕНЬ ТЕХНИКИ
Передача сигналов WNT важна и для эмбриогенеза, и для гомеостаза взрослых животных. Путь WNT состоит, в общем, из сети белков, которые регулируют следующие процессы: 1: выработка и секреция белков WNT; 2: связывание WNT с клеточными рецепторами; и 3: внутриклеточная трансдукция биохимических ответов, запускаемых этим взаимодействием (Mikels and Nusse, 2006; MacDonald, 2009; Moon, 2005).
Так называемый канонический путь WNT, запускаемый связыванием белков WNT с корецепторами клеточной поверхности Frizzled LRP5/6, приводит к изменению количества β-катенина, достигающего ядра, где он взаимодействует с факторами транскрипции семейства TCF/LEF для ускорения транскрипции определенных генов.
Неканонический путь WNT, преобразованный другим набором внутриклеточных белков, контролирует планарную клеточную полярность у насекомых и некоторые процессы, такие как гаструляция, - у позвоночных.
Передача сигналов WNT известна также своей ролью в контролировании плюрипотентности и дифференцировки эмбриональных и взрослых стволовых клеток (Nusse, 2008). Например, образование первичной полоски при гаструляции связано с локализованной активацией WNT в эмбриоидных тельцах (ten Berge, 2008). На образование множества типов клеток, таких как клетки сердца, панкреатические бета-клетки, допаминергические нейроны и гепатоциты печени, из эмбриональных стволовых клеток или клеток iPS, влияет модулирование WNT (Yang, 2008; D'Amour, 2006; Inestrosa and Arenas, 2010; Sullivan, 2010). Путь WNT играет особенно важную роль в развитии ткани скелета, таком как в остеогенез и хондрогенез (Hoeppner, 2009; Chun, 2008).
Передача сигналов WNT также связана с нейрорегенерацией центральной нервной системы взрослых (Lie, 2005).
В результате измененной активности пути WNT могут возникать различные заболевания. Например, гиперактивация канонического пути WNT может приводить к аберрантному росту клеток (Reya and Clevers, 2005). А именно, 90% случаев рака толстой и прямой кишок обусловлены потерей аденоматозного коли-полипозного (АРС) гена, супрессора пути WNT/β-катенина (Kinzler and Vogelstein, 1996). Повышенная экспрессия белков WNT и потеря внеклеточных ингибиторов, которые обычно подавляют функцию белков WNT, могут вызывать образование WNT-зависимых опухолей (Polakis, 2007). С другой стороны, было показано, что неканонический путь WNT также играет роль в развитии некоторых типов рака (Camilli and Weeraratna, 2010). Недавно было описано также, что передача сигналов WNT непосредственно связана со стволовыми раковыми клетками (Takahashi-Yanaga and Kahn, 2010).
Данные позволяют предположить, что воздействие на Wnt-опосредованный путь сигнальной трансдукции будет терапевтически пригодным в широком ряде заболеваний (Barker and Clevers, 2006). Мутации АРС, бета-катенина и аксина-1, приводящие к конститутивной активации канонического пути Wnt, представляют собой критические события в различных типах рака человека, включая рак толстой и прямой кишок, меланому, гепатоцеллюлярную карциному, рак желудка, рак яичников и прочие (Polakis, 2007). Было показано, что блокада пути Wnt в различных типах рака с помощью генетических или химических подходов прекращает аберрантный рост клеток (Herbst and Kolligs, 2007). Кроме того, ингибирование этого пути может напрямую влиять на клетки, которые выдерживают рост раковых клеток и дают возможность метастаза, и которые предположительно являются устойчивыми к традиционным химиотерапевтическим агентам.
Помимо активации, обусловленной мутациями генных продуктов, следующими за этими рецепторами, аберрантная активность пути Wnt, обусловленная другими механизмами, связана с широким рядом раковых заболеваний. Эти раковые заболевания включают, но не ограничиваясь этим: рак легких (мелкоклеточный и немелкоклеточный), молочной железы, предстательной железы, карциноидные опухоли, рак мочевого пузыря, карциному, рак пищевода, яичников, шейки матки, эндометрия, мезотелиому, меланому, саркому, остеосаркому, липосаркому, рак щитовидной железы, десмоиды, хронический миелоцитарный лейкоз (AML) и хронический миелоцитарный лейкоз (CML). В настоящее время существует множество примеров раковых клеток, зависящих от активированной аутокринной или паракринной передачи сигналов Wnt, а также было показано, что клеточные линии из остеосаркомы, рака молочной железы, головы и шеи, и рака яичников приобретают защиту в результате апоптоза под действием аутокринной или паракринной передачи сигналов Wnt (Kansara, 2009; Bafico, 2004; Akiri, 2009; DeAlmeida, 2007; Chan, 2007; Chen, 2009; Rhee, 2002).
Более того, аберрантный путь Wnt участвует в развитии фиброза, включая, но не ограничиваясь этим: фиброз легких, такой как идиопатический легочный фиброз и радиационно-стимулированный фиброз, фиброз почек и фиброз печени (Morrisey, 2003; Hwang, 2009; Cheng, 2008).
Другие расстройства, связанные с аберрантной передачей сигналов WNT, включают, но не ограничиваясь этим, костные и хрящевые расстройства, такие как остеопороз и остеоартрит, связанный с ожирением диабет II типа и нейродегенеративные заболевания, такие как болезнь Альцгеймера (Hoeppner, 2009; Ouchi, 2010; Blom, 2010; Boonen, 2009). Передача сигналов WNT способствует также самообновлению и сохранению ГСК, а неправильная передача сигналов WNT отвечает за различные расстройства из-за ГСК, такие как лейкозы и различные другие раковые заболевания крови (Reya, 2005).
Соответственно, определение способов и соединений, которые модулируют WNT-зависимые клеточные ответы, может обеспечить направление для регуляции физиологических функций и терапевтического лечения заболеваний, связанных с аберрантной активностью этих путей.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении представлено, в основном, соединение и его фармацевтическая композиция, при этом указанное соединение используют в качестве ингибитора передачи сигналов WNT, а также применение этого соединения для ингибирования сигнального пути WNT.
Определение
При использовании в настоящем документе, «сигнальный путь WNT» или «путь WNT» относится к пути, по которому связывание белка WNT с клеточными рецепторами приводит к изменениям поведения клетки. Путь WNT включает различные белки, в том числе Frizzled, Disheveled, Axin, АРС, GSK3β, β-катенин, факторы транскрипции LEF/TCF и молекулы, участвующие в синтезе и секреции белков WNT. Примеры белков, участвующих в секреции функциональных WNT, включают, но не ограничиваясь этим, белки, прерывающиеся отсутствием wnt/ровностью (Wls/Evi), поркупин (Porcn) и Vps35p.Wls/Evi представляет собой 7-проходный трансмембранный белок, который находится в аппарате Гольджи и необходим для секреции Wg (дрозофила) МОМ-2 (с. elegans) и Wnt3A. Он содержит консервативный структурный мотив, структура и функции которого неизвестны. Поркупин (Рогсп) представляет собой члена мембраносвязанного семейства О-ацилтрансферазы (МВОАТ) пальмитоил-трансфераз. Жирнокислотная модификация Wnt является необходимой для их функции. Wnt являются пальмитоилированными в одном или двух высококонсервативных сайтах. Поэтому ингибиторы Porcn могут полностью блокировать передачу сигналов функционального Wnt. Vps35p представляет собой субъединицу мультибелкового комплекса, называемого ретромерным комплексом, который участвует во внутриклеточном перемещении белков. Действие Vps35p заключается в связывании белков-мишеней, таких как WNT, для их вовлечения в везикулы.
«Ингибитор пути WNT» или «ингибитор передачи сигналов WNT» представляет собой небольшую органическую молекулу, которая подавляет активность передачи сигналов WNT и, как правило, имеет молекулярный вес около 800 г/моль или менее.
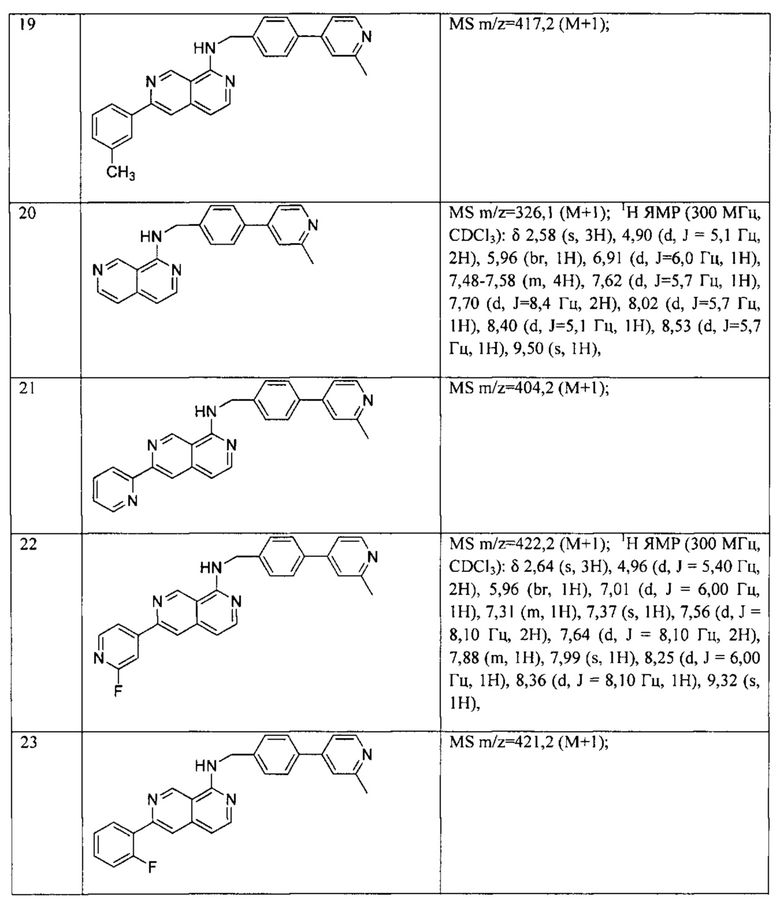
Термин «способ ингибирования пути WNT» относится к способам ингибирования известных биохимических событий, связанных с выработкой функциональных белков WNT или с клеточными реакциями на белки WNT. Как рассмотрено в настоящем документе, небольшие органические молекулы могут ингибировать ответ WNT в соответствии с этим определением.
«Белок WNT» представляет собой белок, который связывается с корецепторами Frizzled и LRP5/6 так, что происходит активация канонической или неканонической передачи сигналов WNT. Конкретные примеры белков WNT включают: WNT-1 (NM005430), WNT-2 (NM003391), WNT-2B/WNT-13 (NM004185), WNT-3 (NM030753), WNT3a (NM033131), WNT-4 (NM030761), WNT-5A (NM003392), WNT-5B (NM032642), WNT-6 (NM006522), WNT-7A (NM004625), WNT-7B (NM058238), WNT-8A (NM058244), WNT-8B (NM003393), WNT-9A/WNT-14) (NM003395), WNT-9B/WNT-15 (NM003396), WNT-10A (NM025216), WNT-10В (NM003394), WNT-11 (NM004626), WNT-16 (NMO16087).
«Расстройство пути WNT» представляет собой состояние или болезненное состояние с аберрантной передачей сигналов WNT. В одном аспекте аберрантная передача сигналов WNT представляет собой уровень передачи сигналов WNT в клетке или ткани, которая предположительно подвержена заболеванию, при этом указанный уровень превышает уровень передачи сигналов WNT в нормальной клетке или ткани. В одном конкретном аспекте WNT-опосредованное расстройство включает рак или фиброз.
Термин «рак» относится к патологическому состоянию у людей, которое характеризуется нерегулируемой клеточной пролиферацией. Примеры включают, но не ограничиваясь этим: карциному, лимфому, бластому и лейкоз. Более конкретные примеры раковых заболеваний включают, но не ограничиваясь этим: рак легких (мелкоклеточный и немелкоклеточный), молочной железы, предстательной железы, карциноидные опухоли, рак мочевого пузыря, рак желудка, поджелудочной железы, печени (гепатоцеллюлярный), гепатобластому, толстой и прямой кишок, плоскоклеточную карциному головы и шеи, рак пищевода, яичников, шейки матки, эндометрия, мезотелиому, меланому, саркому, остеосаркому, липосаркому, рак щитовидной железы, десмоиды, хронический миелоцитарный лейкоз (AML) и хронический миелоцитарный лейкоз (CML).
Термин «фиброз» относится к патологическому состоянию у людей, которое, как правило, характеризуется неконтролируемой пролиферацией фибробластных клеток и уплотнения ткани. Конкретные примеры включают, но не ограничиваясь этим: фиброз легких (идиопатический легочный фиброз и радиационно-стимулированный фиброз), фиброз почек и фиброз печени, включая цирроз печени.
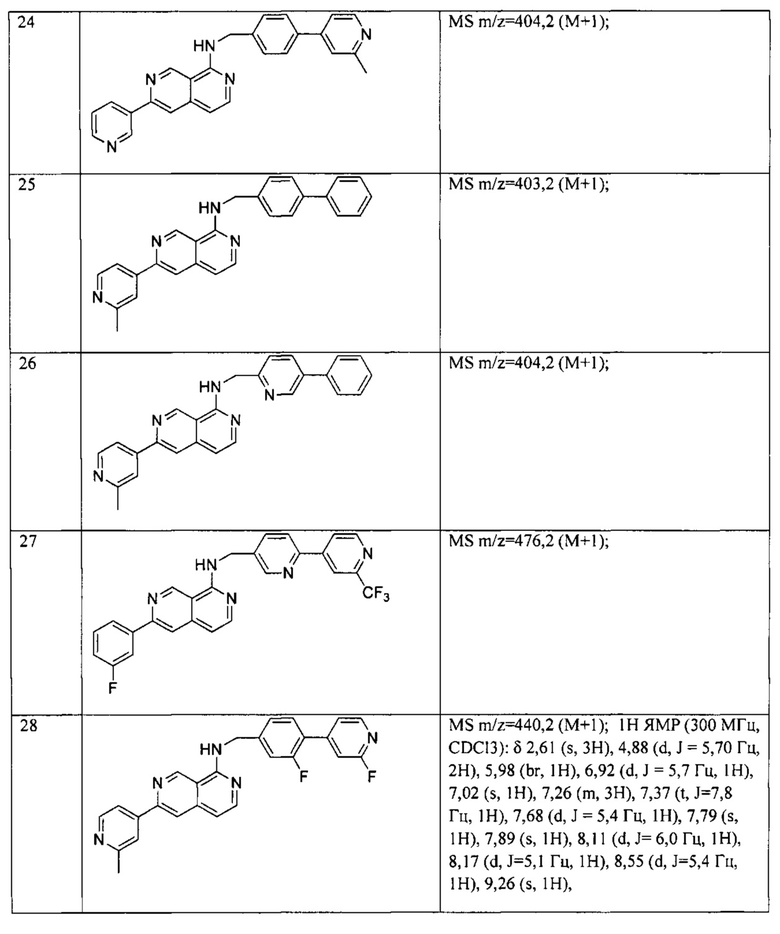
«Ингибирование» или «лечение» или «терапия» относится к уменьшению, терапевтическому лечению и профилактическому или превентивному лечению, при этом задача заключается в снижении или предупреждении заданного патологического расстройства или состояния. В одном примере, после введения ингибитора передачи сигналов WNT онкологический пациент может испытывать снижение размера опухоли. «Лечение» или «терапия» включает (1) подавление заболевания у субъекта, испытывающего или проявляющего патологию или симптомы заболевания, (2) улучшение заболевания у субъекта, который испытывает или проявляет патологию или симптомы заболевания, и/или (3) влияние на любое измеримое уменьшение заболевания у субъекта или пациента, который испытывает или проявляет патологию или симптомы заболевания. В той степени, в которой ингибитор пути WNT может предотвращать рост и/или уничтожать раковые клетки, он может быть цитостатическим и/или цитотоксическим.
Термин «терапевтически эффективное количество» относится к количеству ингибитора пути WNT, эффективному для «лечения» расстройства пути WNT у субъекта или млекопитающего. В случае рака терапевтически эффективное количество лекарства может снижать количество раковых клеток или снижать размер опухоли, ингибировать инфильтрацию раковых клеток в периферийные органы, подавлять метастазы опухоли, замедлять рост опухоли до некоторой степени и/или до некоторой степени облегчать один или более симптомов, связанных с раком.
Введение «в комбинации с» одним или более дополнительными терапевтическими агентами включает одновременное (параллельное) и последовательное введение в любом порядке. При использовании в настоящем документе, термин «фармацевтическая комбинация» относится к продукту, полученному смешиванием или комбинированием активных ингредиентов, и включает как фиксированные, так и нефиксированные комбинации активных ингредиентов. Термин «фиксированная комбинация» означает, что активные ингредиенты, например, соединение Формулы (1) и совместный агент вводят пациенту одновременно в форме единого целого или единой дозы. Термин «нефиксированная комбинация» означает, что активные ингредиенты, например, соединение Формулы (1) и совместный агент вводят пациенту в виде отдельных единиц, одновременно, параллельно или последовательно, без каких-либо конкретных временных ограничений, при этом такое введение обеспечивает терапевтически эффективные концентрации активных ингредиентов в организме пациента. Последнее относится также к терапии смесями, например, к введению трех или более активных ингредиентов.
«Химиотерапевтический агент» представляет собой химическое соединение, применимое для лечения рака. Примеры включают, но не ограничиваясь этим: гемцитабин, иринотекан, доксорубицин, 5-фторурацил, цитозина арабинозид («Ara-C»), циклофосфамид, тиотепа, бусульфан, цитоксин, таксол, метотрексат, цисплатин, мелфалан, винбластин и карбоплатин.
Описание изобретения
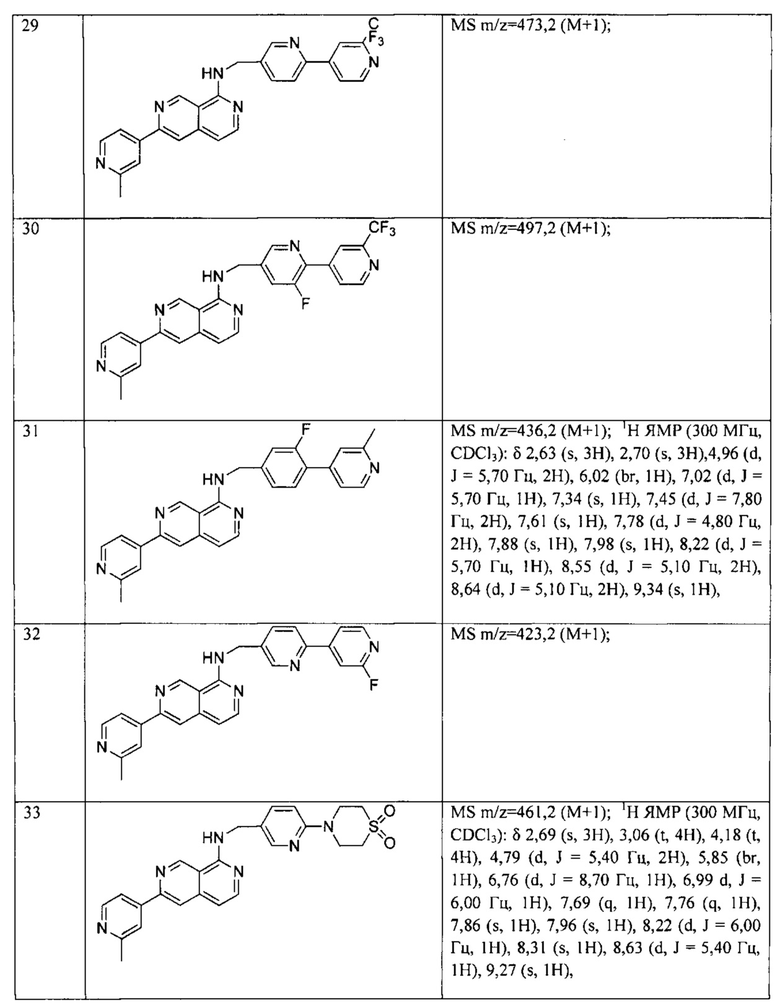
В одном аспекте настоящего изобретения представлено соединение как ингибитор передачи сигналов WNT, которое имеет структуру Формулы I:
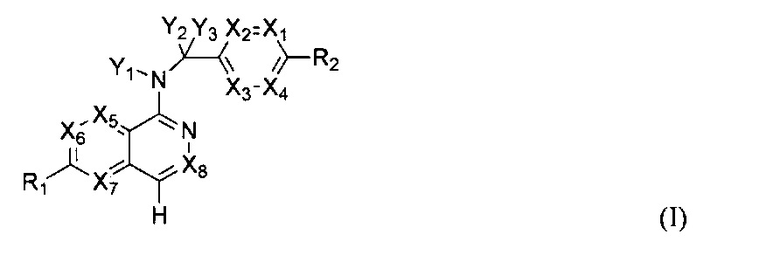
или его физиологически приемлемая соль,
где
X1, X2, X3, X4, X5, Х6, Х7 и X8 независимо представляют собой CR4 или N;
Y1 представляет собой водород или -C(R4)3, каждый R4 является одинаковым или различным;
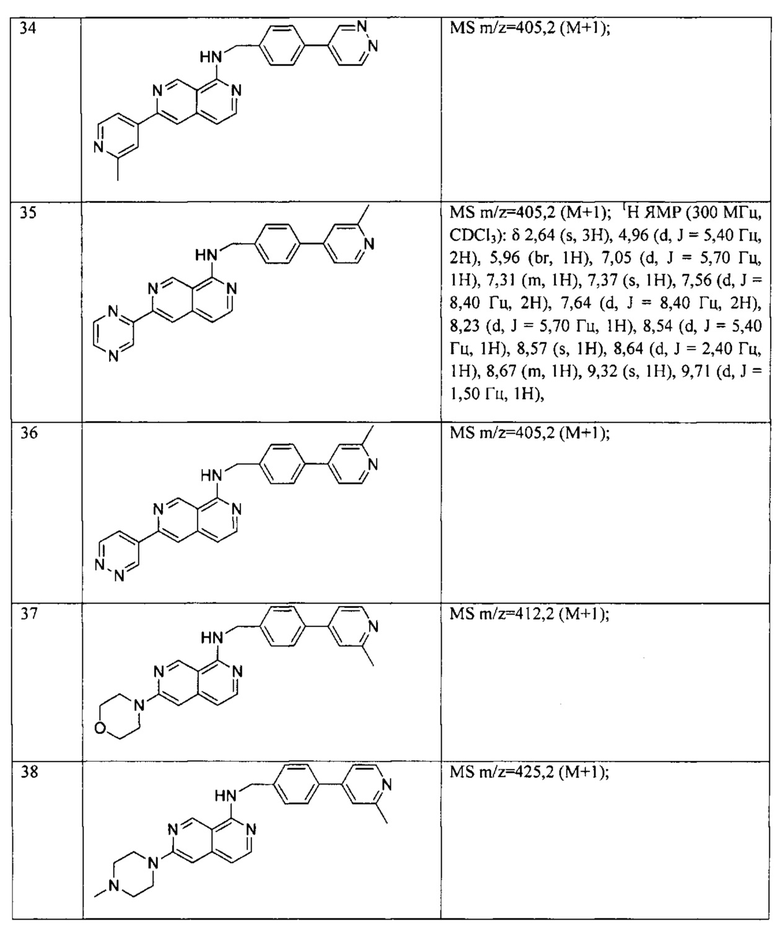
Y2 и Y3 независимо представляют собой водород, галоген или -C(R3)3, каждый R3 является одинаковым или различным;
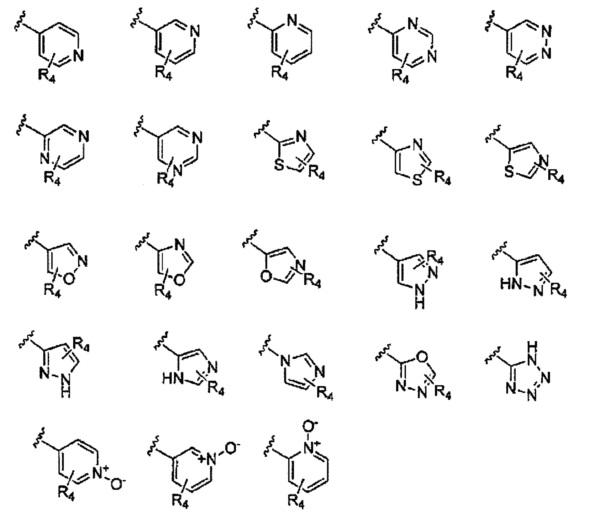
R1 и R2 независимо выбраны из водорода, галогена, С1-6алкила, хинолинила,  , C6-30арила, 3-6-членного гетероциклоалкила, содержащего 1-2 гетероатома, выбранных из N, О и S, и 5- или 6-членного гетероарила, содержащего 1-4 гетероатома, выбранных из N, О и S, где каждый из хинолинила,
, C6-30арила, 3-6-членного гетероциклоалкила, содержащего 1-2 гетероатома, выбранных из N, О и S, и 5- или 6-членного гетероарила, содержащего 1-4 гетероатома, выбранных из N, О и S, где каждый из хинолинила,  , C6-30арила, 3-6-членного гетероциклоалкила и 5- или 6-членного гетероарила может быть необязательно замещен одним или двумя и одинаковыми или различными R4;
, C6-30арила, 3-6-членного гетероциклоалкила и 5- или 6-членного гетероарила может быть необязательно замещен одним или двумя и одинаковыми или различными R4;
каждый R3 независимо выбран из водорода, галогена, циано, C1-6алкила и C1-6алкокси, где каждый из C1-6алкила и C1-6алкокси может быть необязательно замещен галогеном, амино, гидроксилом, C1-6алкокси или циано;
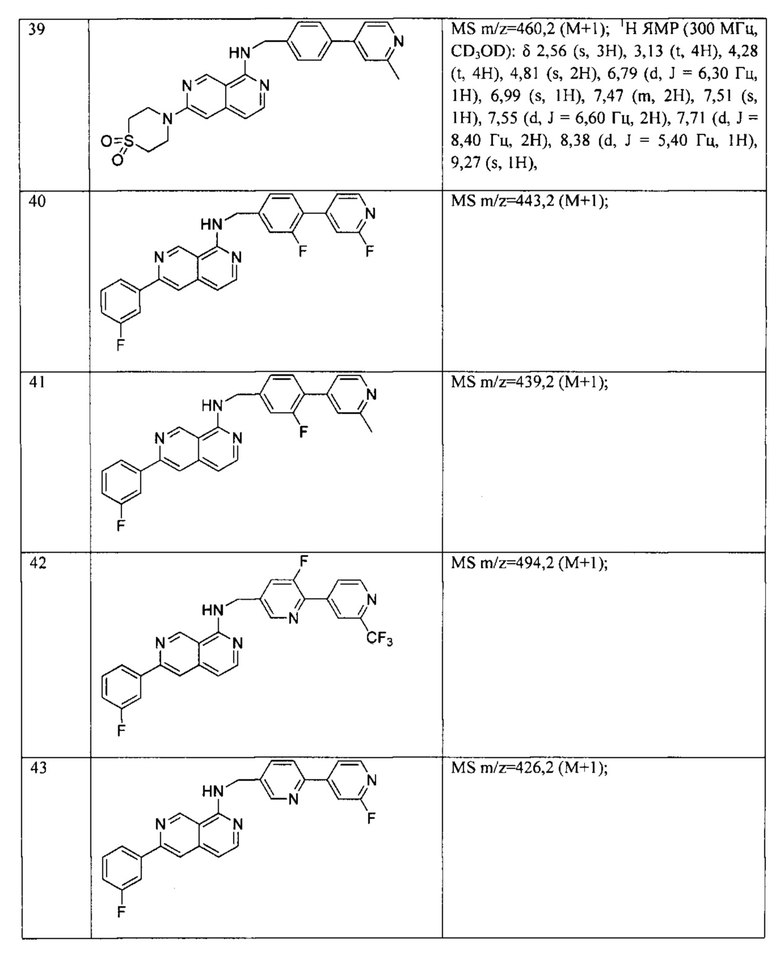
каждый R4 независимо выбран из водорода, галогена, циано, C1-6алкокси, -S(O)2R5, -C(O)OR5, -C(O)R5, -C(O)NR6R7, C1-6алкила, C2-6алкенила и C2-6алкинила, при этом каждый из C1-6алкокси, -S(O)2R5, -C(O)OR5, -C(O)R5, -C(O)NR6R7, C1-6алкила, C2-6алкенила и C2-6алкинила может быть необязательно замещен галогеном, амино, гидроксилом, C1-6алкокси или циано; и
R5, R6 и R7 независимо выбраны из водорода, C1-6алкила, С2-6алкенила и С2-6алкинила, где каждый из C1-6алкила, C2-6алкенила и С2-6алкинила может быть необязательно замещен галогеном, амино, гидроксилом, C1-6алкокси или циано.
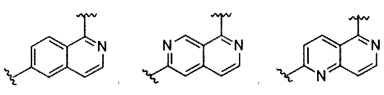
В частности, Формула (I) представляет собой следующие центральные структуры, но не ограничиваясь этим:
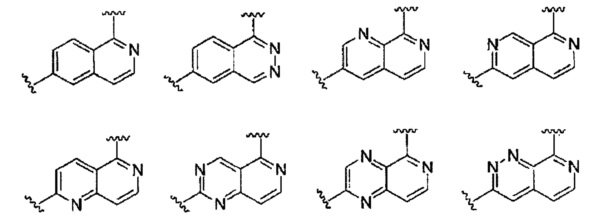
В Формуле I кольцо, обозначенное X1, X2, Х3 и Х4, может быть любой из следующих групп, но не ограничиваясь этим:
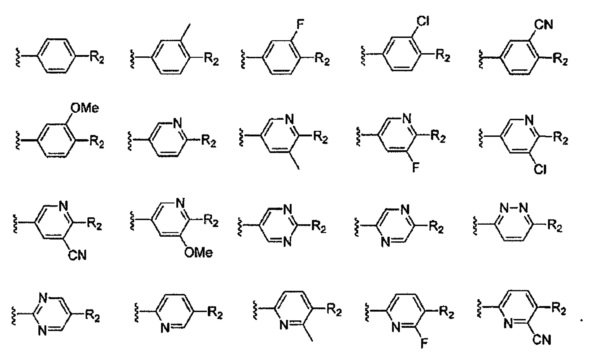
Предпочтительно, R1 и R2 в Формуле I могут быть независимо выбраны из водорода, фтора, хлора, метила, 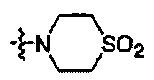 , фенила, морфолинила, пиперазинила и 5- или 6-членного гетероарила, выбранного из:
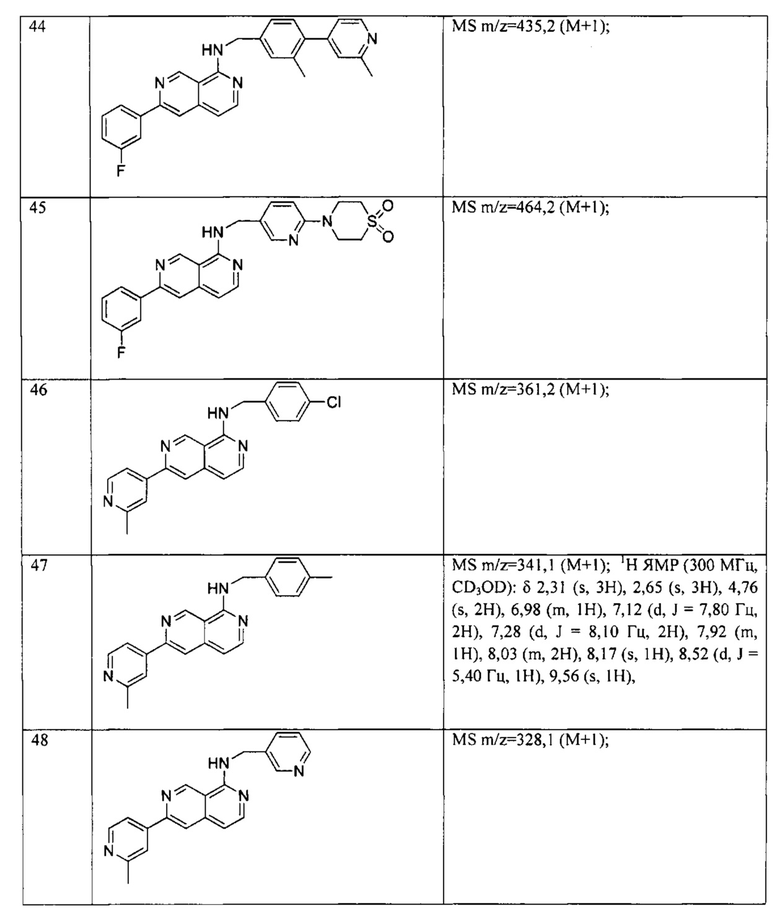
, фенила, морфолинила, пиперазинила и 5- или 6-членного гетероарила, выбранного из:
Предпочтительно, R4 может быть одинаковым или различным, и каждый независимо выбран из водорода, хлора, фтора, циано, -CH3,-CHF2, -CF3, -OCH3, -СООСН3.
В одном варианте реализации по меньшей мере один атом в Формуле I представляет собой по меньшей мере один из соответствующих изотопов(-а), выбранных из 2Н, 3Н, 11С, 13С, 14С, 15N, 17O, 18O, 35S, 18F, 36Cl и 123I.
При использовании в настоящем документе, атом Н, например, в любых группах заместителей (например, СН2), охватывает все подходящие изотопные вариации, например, Н, 2Н и 3Н.
При использовании в настоящем документе, другие атомы, например, в любых группах заместителей, охватывают все подходящие изотопные вариации, включая, но не ограничиваясь этим, 11C, 13С, 14С, 15N, 17O, 18O, 35S, 18F, 36Cl и/или 123I.
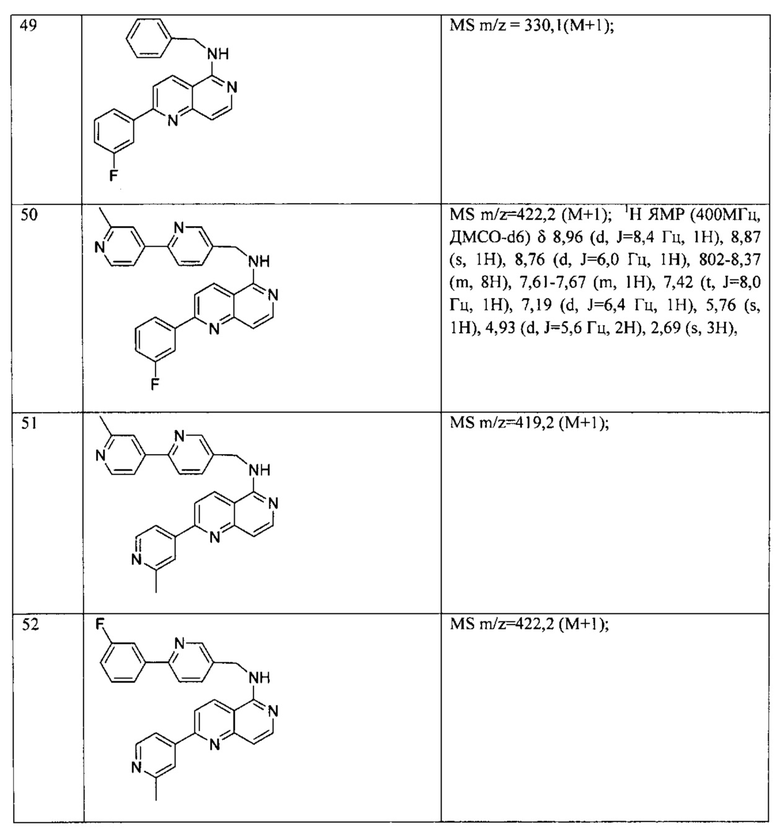
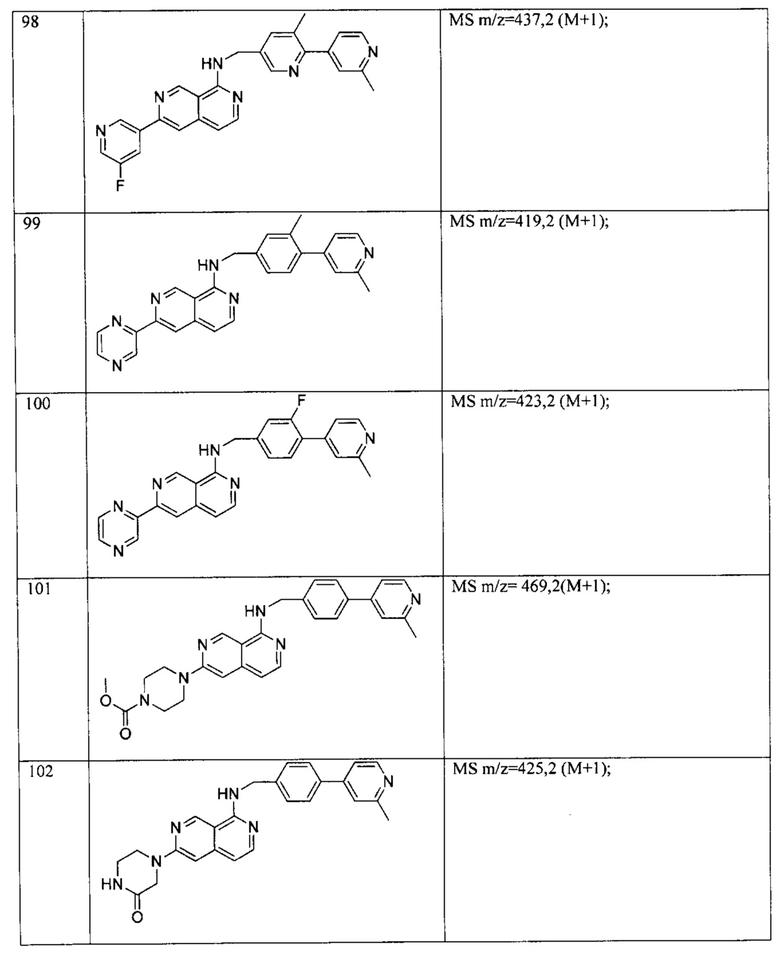
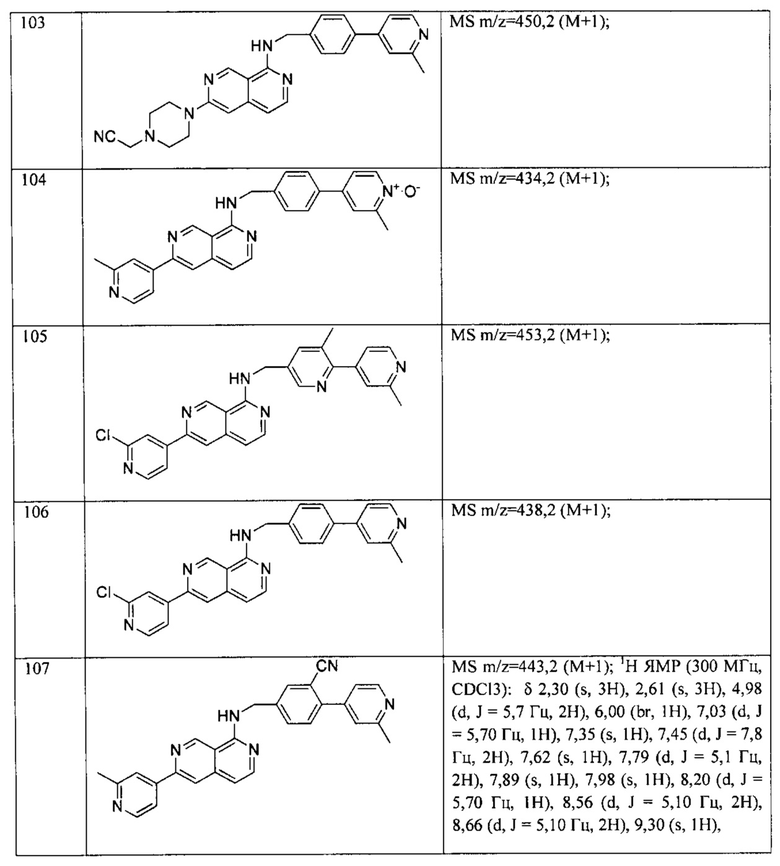
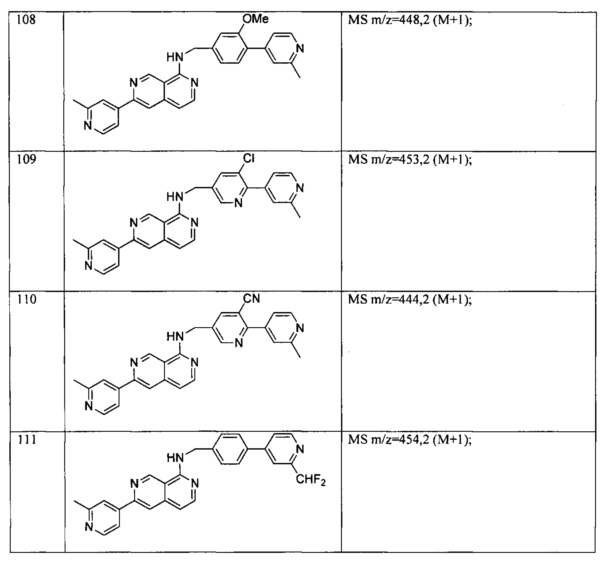
В предпочтительном варианте реализации пример соединения настоящего изобретения включает, но не ограничиваясь этим:
N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)-7-фенилхиназолин-4-амин;
N-((5-(2-метилпиридин-4-ил)пиридин-2-ил)метил)-7-фенилхиназолин-4-амин;
N-(4-морфолинобензил)-7-фенилхиназолин-4-амин;
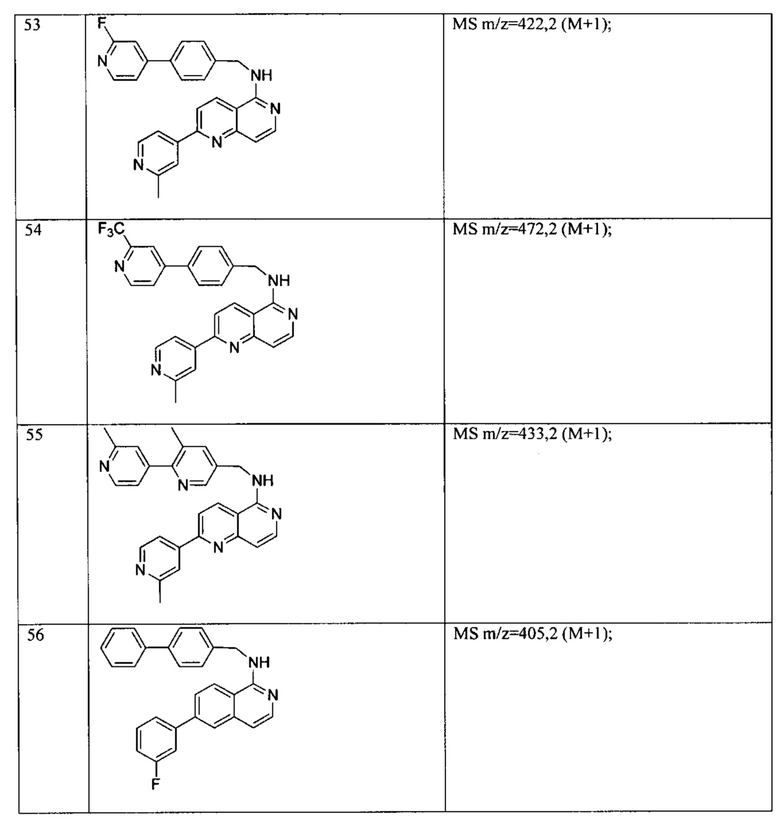
N((6-морфолинопиридин-3-ил)метил)-7-фенилхиназолин-4-амин;
N-((6-(2-метилморфолино)пиридин-3-ил)метил)-7-фенилхиназолин-4-амин;
N-((6-(4-метилпиперазин-1-ил)пиридин-3-ил)метил)-7-фенилхиназолин-4-амин;
4-(5-(((7-фенилхиназолин-4-ил)амино)метил)пиридин-2-ил)тиоморфолина 1,1-диоксид;
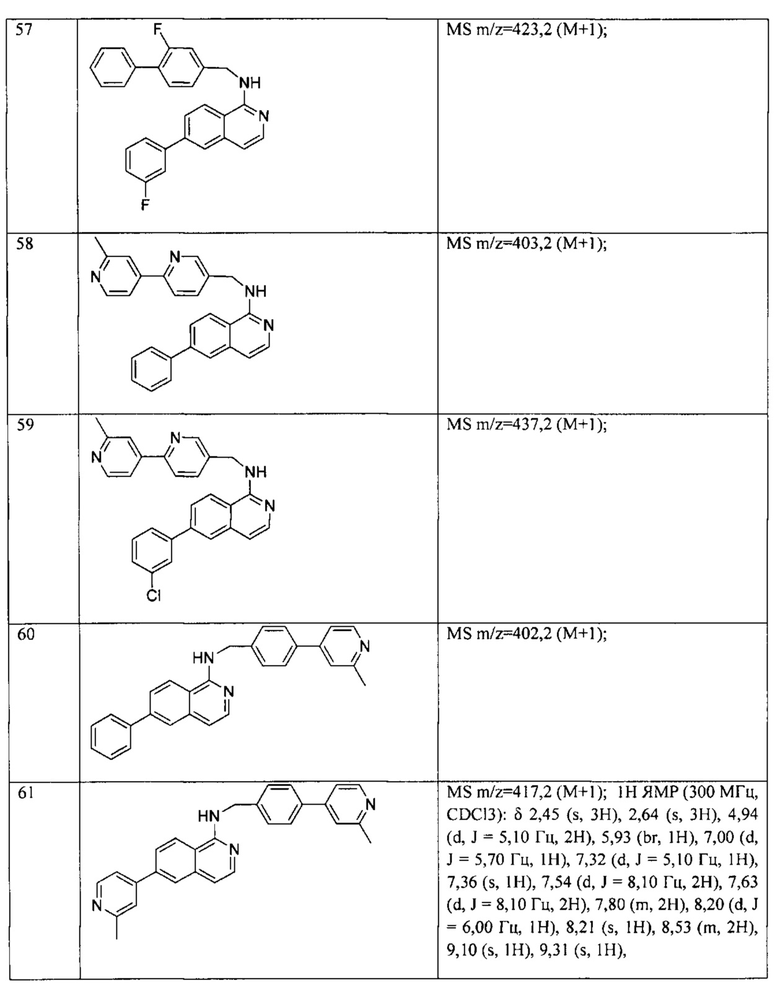
N-((6-(6-метилпиридин-3-ил)пиридин-3-ил)метил)-7-фенилхиназолин-4-амин;
N-((6-(5-метилпиридин-3-ил)пиридин-3-ил)метил)-7-фенилхиназолин-4-амин;
7-фенил-N-((6-(пиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-фенил-N-((6-(пиридин-3-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-фенил-N-((6-(пиридин-2-ил)пиридин-3-ил)метил)хиназолин-4-амин;
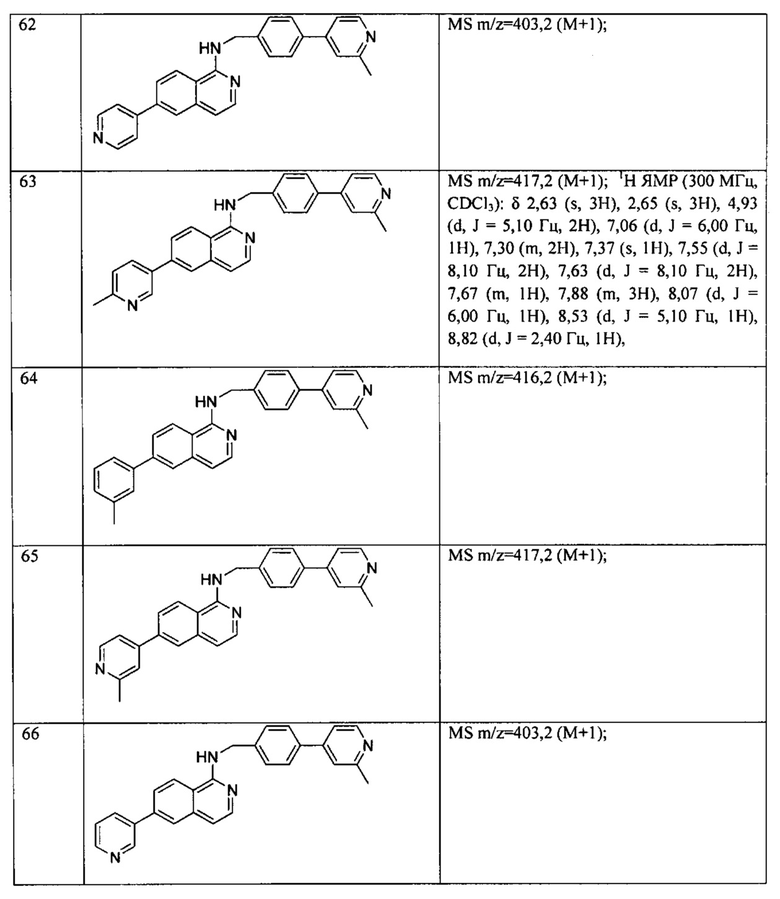
7-фенил-N-((6-(пиридазин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-фенил-N-((6-(пиразин-2-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-фенил-N-((6-(пиримидин-5-ил)пиридин-3-ил)метил)хиназолин-4-амин;
N-((6-(2-фторпиридин-4-ил)пиридин-3-ил)метил)-7-фенилхиназолин-4-амин;
N-((6-(4-метил-1Н-имидазол-1-ил)пиридин-3-ил)метил)-7-фенилхиназолин-4-амин;
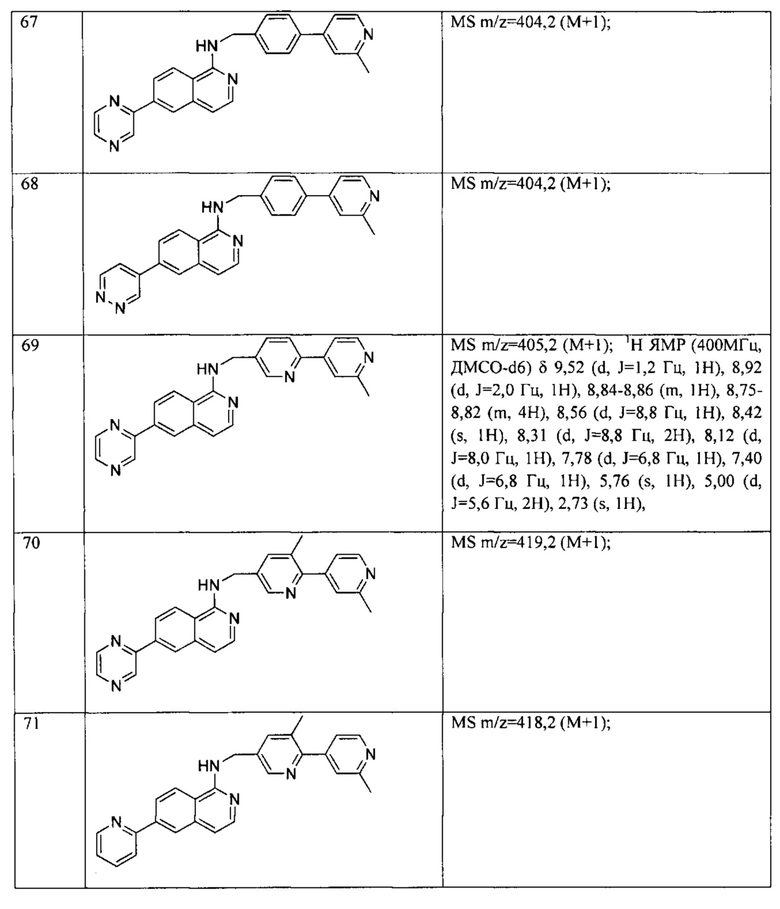
N-((6-(1-метил-1Н-пиразол-4-ил)пиридин-3-ил)метил)-7-фенилхиназолин-4-амин;
N-((5-(6-метилпиридин-3-ил)пиридин-2-ил)метил)-7-фенилхиназолин-4-амин;
N-(4-(2-метилпиридин-4-ил)бензил)-7-фенилхиназолин-4-амин;
N-(4-(2-фторпиридин-4-ил)бензил)-7-фенилхиназолин-4-амин;
N-бензил-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
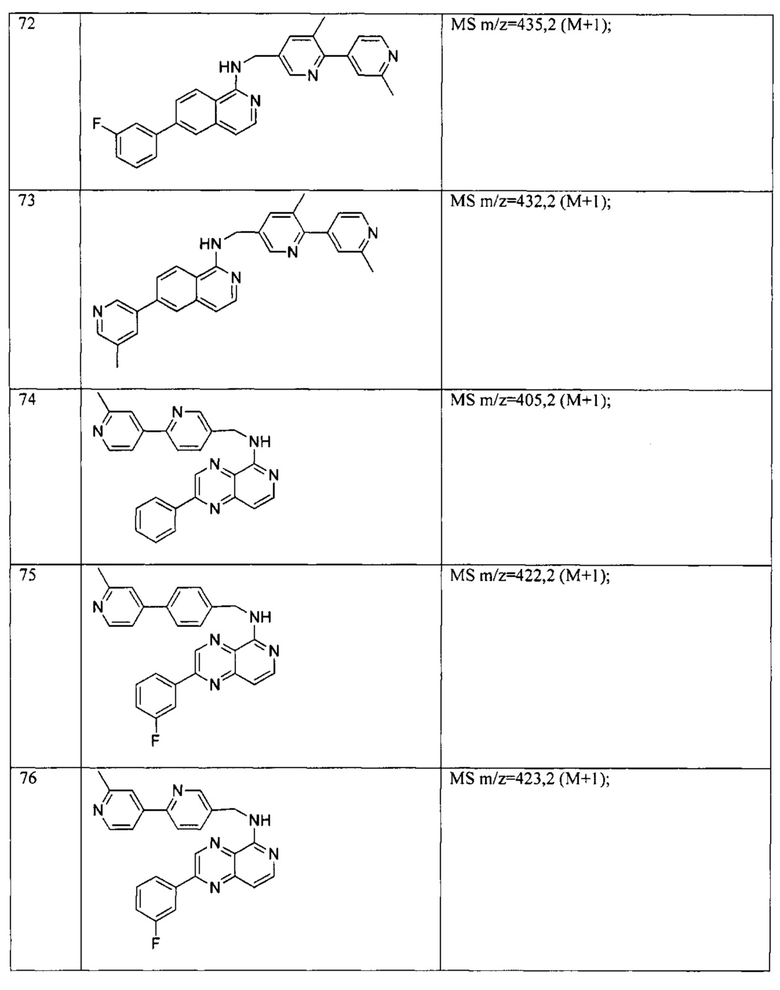
N-(4-метилбензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
N-(4-метоксибензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
N-(4-фторбензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
N-(4-хлорбензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
N-(4-бромбензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
N-(4-(трифторметил)бензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
4-((7-(2-метилпиридин-4-ил)хиназолин-4-иламино)метил)бензонитрил;
N-(4-морфолинобензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
N-(4-фенилбензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
N-(3-фтор-4-фенилбензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
N-(4-(3-фторфенил)бензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
7-(3-фторфенил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-(3-хлорфенил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)-7-м-толилхиназолин-4-амин;
3-(4-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метиламино)хиназолин-7-ил)бензонитрил;
4-(4-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метиламино)хиназолин-7-ил)бензонитрил;
7-(2-метилпиридин-4-ил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-(6-метилпиридин-3-ил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-(5-метилпиридин-3-ил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)-7-(пиридин-2-ил)хиназолин-4-амин;
N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)-7-(пиридин-3-ил)хиназолин-4-амин;
N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)-7-(пиридин-4-ил)хиназолин-4-амин;
N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)-7-(пиридазин-4-ил)хиназолин-4-амин;
N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)-7-(пиразин-2-ил)хиназолин-4-амин;
N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)-7-(пиримидин-5-ил)хиназолин-4-амин;
7-(2-фторпиридин-4-ил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-(2-(трифторметил)пиридин-4-ил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-(2-метоксипиридин-4-ил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-(3-метилпиридин-4-ил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)-7-морфолинохиназолин-4-амин;
N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)-7-(пиперидин-1-ил)хиназолин-4-амин;
7-(4-метилпиперазин-1-ил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
1-(4-(4-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метиламино)хиназолин-7-ил)пиперазин-1-ил)этанон;
4-(4-(((2'-метил-[2,4'-бипиридин]-5-ил)метил)амино)хиназолин-7-ил)тиоморфолина 1,1-диоксид;
7-(1,2,3,6-тетрагидропиридин-4-ил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-(1,2,3,6-тетрагидропиридин-4-ил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
1-(4-(4-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метиламино)хиназолин-7-ил)пиперидин-1-ил)этанон;
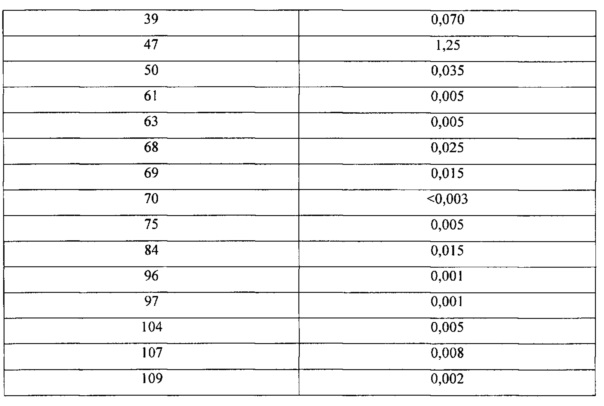
N-((2'-метил-[2,4'-бипиридин]-5-ил)метил)-7-(4-(метилсульфонил)пиперазин-1-ил)хиназолин-4-амин;
7-(1-метил-1Н-пиразол-4-ил)-4-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
7-(изоксазол-4-ил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)хиназолин-4-амин;
N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)-7-(тиазол-2-ил)хиназолин-4-амин;
N-(3-метил-4-(2-метилпиридин-4-ил)бензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
N-(3-фтор-4-(2-метилпиридин-4-ил)бензил)-7-(2-метилпиридин-4-ил)хиназолин-4-амин;
N-(4-(2-метилпиридин-4-ил)бензил)-7-(пиразин-2-ил)хиназолин-4-амин;
N-(4-(2-метилпиридин-4-ил)бензил)-7-(2-фторпиридин-4-ил)хиназолин-4-амин;
N-(4-(2-метилпиридин-4-ил)бензил)-7-морфолинохиназолин-4-амин;
2-(3-фторфенил)-N-(4-(2-метилпиридин-4-ил)бензил)пиридо[3,4-b]пиразин-5-амин;
2-(3-фторфенил)-N-((2'-метил-[2,4'-бипиридин]-5-ил)метил)пиридо[3,4-b]пиразин-5-амин;
2-(3-фторфенил)-N-(3-метил-4-(2-метилпиридин-4-ил)бензил)пиридо[3,4-b]пиразин-5-амин;
N-(3-фтор-4-(2-метилпиридин-4-ил)бензил)-2-(3-фторфенил)пиридо[3,4-b]пиразин-5-амин;
2-(2-метилпиридин-4-ил)-N-(4-(2-метилпиридин-4-ил)бензил)пиридо[3,4-b]пиразин-5-амин;
N-((2'-метил-[2,4'-бипиридин]-5-ил)метил)-2-(2-метилпиридин-4-ил)пиридо[3,4-b]пиразин-5-амин;
N-(3-метил-4-(2-метилпиридин-4-ил)бензил)-2-(2-метилпиридин-4-ил)пиридо[3,4-b]пиразин-5-амин;
N-(3-фтор-4-(2-метилпиридин-4-ил)бензил)-2-(2-метилпиридин-4-ил)пиридо[3,4-b]пиразин-5-амин;
N-((2',3-диметил-[2,4'-бипиридин]-5-ил)метил)-6-(пиразин-2-ил)-2,7-нафтиридин-1-амин;
6-(2-метилморфолино)-N-(4-(2-метилпиридин-4-ил)бензил)-2,7-нафтиридин-1-амин;
(S)-6-(2-метилморфолино)-N-(4-(2-метилпиридин-4-ил)бензил)-2,7-нафтиридин-1-амин;
(R)-6-(2-метилморфолино)-N-(4-(2-метилпиридин-4-ил)бензил)-2,7-нафтиридин-1-амин;
1-(4-(8-((4-(2-метилпиридин-4-ил)бензил)амино)-2,7-нафтиридин-3-ил)пиперазин-1-ил)этанон;
6-(1Н-имидазол-1-ил)-N-(4-(2-метилпиридин-4-ил)бензил)-2,7-нафтиридин-1-амин;
6-(4-метил-1Н-имидазол-1-ил)-N-(4-(2-метилпиридин-4-ил)бензил)-2,7-нафтиридин-1-амин;
N-(4-(2-метилпиридин-4-ил)бензил)-6-(1Н-тетразол-5-ил)-2,7-нафтиридин-1-амин;
6-(5-метил-1,3,4-оксадиазол-2-ил)-N-(4-(2-метилпиридин-4-ил)бензил)-2,7-нафтиридин-1-амин;
6-(1-метил-1Н-пиразол-3-ил)-N-(4-(2-метилпиридин-4-ил)бензил)-2,7-нафтиридин-1-амин;
N-(4-(2-метилпиридин-4-ил)бензил)-6-(тиазол-5-ил)-2,7-нафтиридин-1-амин;
N-(4-(2-метилпиридин-4-ил)бензил)-6-(оксазол-5-ил)-2,7-нафтиридин-1-амин;
N-((2',3-диметил-[2,4'-бипиридин]-5-ил)метил)-6-(5-метилпиридин-3-ил)-2,7-нафтиридин-1-амин;
N-((2',3-диметил-[2,4'-бипиридин]-5-ил)метил)-6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-амин;
N-((3-фтор-2'-метил-[2,4'-бипиридин]-5-ил)метил)-6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-амин;
N-((2',3-диметил-[2,4'-бипиридин]-5-ил)метил)-6-(5-фторпиридин-3-ил)-2,7-нафтиридин-1-амин;
N-(3-метил-4-(2-метилпиридин-4-ил)бензил)-6-(пиразин-2-ил)-2,7-нафтиридин-1-амин;
N-(3-фтор-4-(2-метилпиридин-4-ил)бензил)-6-(пиразин-2-ил)-2,7-нафтиридин-1-амин;
метил-4-(8-((4-(2-метилпиридин-4-ил)бензил)амино)-2,7-нафтиридин-3-ил)пиперазин-1-карбоксилат;
4-(8-((4-(2-метилпиридин-4-ил)бензил)амино)-2,7-нафтиридин-3-ил)пиперазин-2-он;
2-(4-(8-((4-(2-метилпиридин-4-ил)бензил)амино)-2,7-нафтиридин-3-ил)пиперазин-1-ил)ацетонитрил;
2-метил-4-(4-(((6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-ил)амино)метил)фенил)пиридин 1-оксид;
6-(2-хлорпиридин-4-ил)-N-((2',3-диметил-[2,4'-бипиридин]-5-ил)метил)-2,7-нафтиридин-1-амин;
6-(2-хлорпиридин-4-ил)-N-(4-(2-метилпиридин-4-ил)бензил)-2,7-нафтиридин-1-амин;
2'-метил-4-(((6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-ил)амино)метил)-2Н-[1,4'-бипиридин]-2-он;
2-(2-метилпиридин-4-ил)-5-(((6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-ил)амино)метил)бензонитрил;
N-(3-метокси-4-(2-метилпиридин-4-ил)бензил)-6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-амин;
N-((3-хлор-2'-метил-[2,4'-бипиридин]-5-ил)метил)-6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-амин;
N-(4-(2-(дифторметил)пиридин-4-ил)бензил)-6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-амин;
или их физиологически приемлемые соли.
В другом аспекте настоящего изобретения представлена фармацевтическая композиция, содержащая соединение настоящего изобретения, и обычно содержащая по меньшей мере один фармацевтически приемлемый носитель или разбавитель, в котором указанное соединение находится в свободной форме или в форме фармацевтически приемлемой соли. Такая композиция может быть пероральной композицией, композицией для инъекций или суппозиторием. И указанная композиция может быть изготовлена стандартным образом, способами смешивания, гранулирования или нанесения покрытия.
В одном варианте реализации настоящего изобретения композиция представляет собой пероральную композицию, и она может быть таблеткой или желатиновой капсулой. Предпочтительно, пероральная композиция содержит соединение настоящего изобретения вместе с а) разбавителями, например, лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином; b) смазывающими веществами, например, диоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; для таблеток - вместе с c) связывающими веществами, например, силикатом магния-алюминия, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, карбоксиметилцеллюлозой натрия и/или поливинилпирролидоном; и, при необходимости, d) средствами для улучшения распадаемости таблеток, например, крахмалами, агаром, альгиновой кислотой или ее натриевой солью, или шипучими смесями; и/или е) добавками, например, абсорбентами, красителями, ароматизаторами и подсластителями.
В другом варианте реализации настоящего изобретения композиция представляет собой композицию для инъекций и может быть водным изотоническим раствором или суспензией.
В другом варианте реализации настоящего изобретения композиция представляет собой суппозиторий и может быть приготовлена из жирной эмульсии или суспензии.
Предпочтительно, композицию стерилизуют, и/или она содержит адъювант. Такой адъювант может быть консервирующим, стабилизирующим, увлажняющим и/или эмульгирующим агентом, ускорителем растворения, солью для регуляции осмотического давления, буфером и/или любой их комбинацией.
Альтернативно или дополнительно, композиция может дополнительно содержать другие терапевтически ценные вещества для различных применений, такие как солюбилизаторы, стабилизаторы, агенты для усиления тоничности, буферы и/или консерванты.
В одном варианте реализации настоящего изобретения композиция может быть препаратом, пригодным для трансдермального применения. Такой препарат содержит эффективное количество соединения настоящего изобретения и носитель. Предпочтительно, носитель может содержать абсорбируемые фармакологически приемлемые растворители для облегчения прохождения через кожу реципиента. Также может быть использовано трансдермальное устройство, содержащее этот препарат. Трансдермальное устройство может быть в форме бандажа, содержащего подкладочный элемент, резервуар, содержащий соединение, необязательно с носителями; необязательно барьер, контролирующий скорость доставки для обеспечения доставки соединения на кожу реципиента с контролируемой и заранее определенной скоростью в течение продолжительного периода времени, и средства для закрепления устройства на коже. Или может быть использован также матричный трансдермальный препарат.
В другом варианте реализации настоящего изобретения композиция может представлять собой препарат, пригодный для местного применения, например, на кожу и глаза, и может быть водным раствором, мазью, кремом или гелем, общеизвестным в данной области техники.
В другом аспекте настоящего изобретения представлен способ ингибирования секреции WNT из клетки путем контакта этой клетки с эффективным количеством указанного выше соединения или его физиологически приемлемой соли, или указанной выше фармацевтической композиции.
В другом аспекте настоящего изобретения представлен способ ингибирования передачи сигналов WNT в клетке с помощью эффективного количества указанного выше соединения или его физиологически приемлемой соли, или указанной выше фармацевтической композиции. В одном варианте реализации клетка находится в организме млекопитающего, а введенное количество представляет собой терапевтически эффективное количество. В другом варианте реализации ингибирование передачи сигналов WNT дополнительно приводит к ингибированию роста клетки. В дополнительном варианте реализации клетка представляет собой раковую клетку. В другом варианте реализации клетка представляет собой фиброгенную клетку.
Клеточную пролиферацию измеряют с помощью способов, известных специалистам в данной области техники. Например, удобный анализ для измерения клеточной пролиферации представляет собой анализ CellTiter-Glo™, имеющийся в продаже у компании Promega (Мэдисон, штат Висконсин). Этот аналитический прием подразумевает добавление реагента CellTiter-Glo® к клеткам, выращенным на многолуночных планшетах. Люминесцентный сигнал, измеренный люминометром или визуализирующим устройством, пропорционален количеству присутствующего АТФ, которое прямо пропорционально количеству жизнеспособных клеток, содержащихся в культуре. Кроме того, клеточную пролиферацию можно измерить также с помощью анализов образования колоний, известных в данной области техники.
В настоящем изобретении представлен также способ лечения раковых заболеваний или фиброзов, связанных с сигнальным путем WNT, с помощью эффективного количества соединения настоящего изобретения. Специалисты в данной области техники могут легко определить, связан ли рак с путем Wnt, с помощью анализа раковых клеток с применением одного или нескольких приемов, известных в данной области. Например, можно исследовать раковые клетки на аберрации концентраций белков или мРНК, участвующих в передаче сигналов Wnt, с помощью иммунных способов и способов обнаружения нуклеиновых кислот.
Раковые заболевания и фиброзы, связанные с путем Wnt, включают те, в которых активность одного или более компонентов сигнальных путей Wnt повышающе регулирована относительно исходных значений. В одном варианте реализации ингибирование пути Wnt может подразумевать подавление секреции Wnt. В другом примере ингибирование пути Wnt может подразумевать подавление компонентов, следующих за рецепторами клеточной поверхности. В другом варианте реализации подавление секреции Wnt может подразумевать ингибирование активности любых белков, участвующих в секреции функциональных WNT.
Кроме того, в настоящем изобретении представлен способ лечения расстройства пути WNT у субъекта, страдающего от указанного расстройства, путем введения этому субъекту терапевтически эффективного количества ингибитора WNT. В одном варианте реализации расстройство представляет собой расстройство клеточной пролиферации, связанное с аберрантной, например, повышенной активностью передачи сигналов WNT. В другом варианте реализации расстройство обусловлено повышенным количеством белка WNT. В другом варианте реализации расстройство клеточной пролиферации представляет собой рак, включая, но не ограничиваясь этим: рак легких (мелкоклеточный и немелкоклеточный), молочной железы, предстательной железы, карциноидные опухоли, рак мочевого пузыря, рак желудка, поджелудочной железы, печени (гепатоцеллюлярный), гепатобластому, толстой и прямой кишок, рак головы и плоскоклеточную карциному шеи, рак пищевода, яичников, шейки матки, эндометрия, мезотелиому, меланому, саркому, остеосаркому, липосаркому, рак щитовидной железы, десмоиды, хронический миелоцитарный лейкоз (AML) и хронический миелоцитарный лейкоз (CML). В другом варианте реализации расстройство клеточной пролиферации представляет собой фиброз, включая, но не ограничиваясь этим: фиброз легких, такой как идиопатический легочный фиброз и радиационно-стимулированный фиброз, фиброз почек и фиброз печени, включая цирроз печени. В другом варианте реализации расстройство представляет собой остеоартрит, болезнь Паркинсона, ретинопатию, дегенерацию желтого пятна.
Для терапевтического применения соединение настоящего изобретения может быть введено в терапевтически эффективном количестве любым приемлемым способом, известным в данной области техники, в качестве монотерапии. При использовании в настоящем документе, терапевтически эффективное количество может широко варьироваться в зависимости от тяжести заболевания, возраста и относительного состояния здоровья субъекта, эффективности используемого соединения и других факторов. Как правило, удовлетворительный результат достигается при рекомендованном систематическом введении суточной дозы от около 0,03 до 2,5 мг/кг веса тела субъекта. В одном варианте реализации рекомендованная суточная доза для более крупного млекопитающего, такого как человек, находится в диапазоне от около 0,5 мг до около 100 мг. Предпочтительно, соединение вводят раздельными дозами до четырех раз в сутки или в форме с замедленным высвобождением. В другом варианте реализации подходящие разовые лекарственные формы для перорального введения содержат от около 1 до 100 мг активного ингредиента.
Альтернативно, соединение настоящего изобретения может быть введено в терапевтически эффективном количестве в качестве активного ингредиента в комбинации с одним или более терапевтическими агентами, например, в фармацевтических комбинациях. Может наблюдаться синергетический эффект при использовании соединения настоящего изобретения с химиотерапевтическим агентом, известным в данной области техники. Доза введенных совместно соединений может варьироваться в зависимости от типа совместно используемого лекарства, конкретного используемого лекарства, состояния, подлежащего лечению, и так далее.
Соединение настоящего изобретения или его композиция может быть введена любым стандартным способом. В одном варианте реализации его вводят энтерально, например, перорально, и в форме таблеток или капсул. В другом варианте реализации его вводят парентерально и в форме растворов или суспензий для инъекций. В другом варианте реализации его вводят локально и в форме лосьонов, гелей, мазей или кремов, или в назальной форме, или в форме суппозитория.
В другом аспекте настоящего изобретения представлена также фармацевтическая комбинация, предпочтительно набор, содержащий а) первый агент, который представляет собой соединение настоящего изобретения, описанное в настоящем документе, в свободной форме или в форме фармацевтически приемлемой соли, и b) по меньшей мере один совместный агент. Кроме того, набор может содержать инструкции по применению.
Комбинация настоящего изобретения может быть использована in vitro или in vivo. Предпочтительно, заданное терапевтическое преимущество такого введения может быть достигнуто путем контакта клетки, ткани или организма с одной композицией или фармакологическим препаратом, который содержит соединение настоящего изобретения и один или более агентов, или путем контакта клетки с двумя или более отдельными композициями или препаратами, при этом одна композиция содержит один агент, а другая содержит другой. Агенты такой комбинации могут быть введены в одно время или по отдельности через определенный период времени. Предпочтительно, раздельное введение может приводить к заданному терапевтическому преимуществу. Соединение настоящего изобретения может быть введено до, одновременно и/или после других агентов с интервалами, варьирующимися от минут до недель. Специалист в данной области техники, как правило, может соблюсти такой интервал времени для каждой доставки, в течение которого агенты, введенные по отдельности, все еще могут проявлять эффект преимущественного сочетания в отношении клетки, ткани или организма. В одном варианте реализации предусмотрено, что контакт клетки, ткани или организма с двумя, тремя, четырьмя или более средствами в качестве потенциального вещества может быть выполнен практически одновременно, то есть в пределах менее, чем около одной минуты. В другом варианте реализации один или более агентов могут быть введены в интервале от около 1 минуты до 14 дней.
В другом аспекте настоящего изобретения представлено применение представленного соединения или его физиологически приемлемой соли, или представленной фармацевтической композиции для производства лекарственного средства для лечения расстройства, опосредованного путем WNT, как описано выше.
В другом аспекте настоящего изобретения представлен способ получения соединения настоящего изобретения или его солей, или производных.
В одном варианте реализации соединение, имеющее Формулы (I), может быть получено по одному из способов синтеза, описанных в представленных ниже Примерах. В описанных реакциях реакционноспособные функциональные группы, например, гидрокси, амино, имино, тио или карбокси-группы, при необходимости их наличия в конечном продукте, могут быть защищены во избежание их нежелательного участия в реакциях. Стандартные защитные группы могут быть использованы в соответствии со стандартной практикой (см., например, T.W. Greene and P.G.M. Wuts в публикации "Protective Groups in Organic Chemistry", John Wiley and Sons, 1991). Подходящие уходящие группы для применения в описанных способах синтеза включают галогенные уходящие группы и другие стандартные уходящие группы, известные в данной области. Предпочтительно, уходящая группа представляет собой хлор или бром.
В другом варианте реализации соединение настоящего изобретения или его соли могут быть также получены в форме гидратов, или их кристаллы могут содержать, например, растворитель, использованный для кристаллизации (представлены в виде сольватов). Соли, как правило, могут быть преобразованы в соединения в свободной форме с помощью их обработки щелочными агентами, предпочтительно карбонатами щелочных металлов, гидрокарбонатами щелочных металлов или гидроксидами щелочных металлов, более предпочтительно карбонатом калия или гидроксидом натрия. Соединение настоящего изобретения в форме соли присоединения основания может быть преобразовано в соответствующую свободную кислоту с помощью обработки подходящей кислотой, такой как хлороводородная кислота. С учетом тесной взаимосвязи новых соединений в свободной форме и этих соединений в форме их солей, включая те соли, которые могут быть использованы в качестве промежуточных соединений, например, при очистке или идентификации новых соединений, любое упоминание свободных соединений следует понимать как относящееся также к соответствующим солям, если это уместно.
Соли соединения настоящего изобретения с солеобразующей группой могут быть получены по способу, известному в данной области техники. Так, соли присоединения кислот соединения Формулы (I) могут быть получены обработкой кислотой или соответствующим анионообменным реагентом. Фармацевтически приемлемые соли соединения настоящего изобретения могут быть образованы в форме солей присоединения кислот соединения Формулы (I) с основным атомом азота и органическими или неорганическими кислотами.
Предпочтительно, подходящие неорганические соли включают, но не ограничиваясь этим, галоводородные кислоты, такие как хлороводородная кислота, серную кислоту или фосфорную кислоту.
Предпочтительно, подходящие органические кислоты включают, но не ограничиваясь этим, карбоновые, фосфорные, сульфоновые или сульфаминовые кислоты, например, уксусная кислота, пропионовая кислота, октановая кислота, декановая кислота, додекановая кислота, гликолевая кислота, молочная кислота, фумаровая кислота, янтарная кислота, адипиновая кислота, пимелиновая кислота, субериновая кислота, азелаиновая кислота, яблочная кислота, винная кислота, лимонная кислота, аминокислоты, такие как глютаминовая кислота или аспарагиновая кислота, малеиновая кислота, гидроксималеиновая кислота, метилмалеиновая кислота, циклогексанкарбоновая кислота, адамантанкарбоновая кислота, бензойная кислота, салициловая кислота, 4-аминосалициловая кислота, фталевая кислота, фенилуксусная кислота, миндальная кислота, коричная кислота, метан- или этансульфоновая кислота, 2-гидроксиэтансульфоновая кислота, этан-1,2-дисульфоновая кислота, бензолсульфиновая кислота, 2-нафталинсульфиновая кислота, 1,5-нафталиндисульфиновая кислота, 2-, 3- или 4-метилбензолсульфоновая кислота, метилсерная кислота, этилсерная кислота, додецилсерная кислота, N-циклогексилсульфаминовая кислота, N-метил-, N-этил- или N-пропилсульфаминовая кислота или другие органические протонные кислоты, такие как аскорбиновая кислота.
Альтернативно, можно использовать также фармацевтически неприемлемые соли для выделения или очистки, например, пикраты или перхлораты. Но для терапевтического применения используют только фармацевтически приемлемые соли свободных соединений, при необходимости в форме фармацевтических препаратов.
В другом варианте реализации соединение настоящего изобретения в не окисленной форме может быть получено из N-оксидов соединения настоящего изобретения обработкой восстанавливающим агентом в подходящем инертном органическом растворителе при температуре от 0 до 80°C. Предпочтительно, восстанавливающий агент представляет собой серу, диоксид серы, трифенилфосфин, боргидрид лития, боргидрид натрия, трихлорид, трибромид фосфора или тому подобные. Предпочтительно, инертный органический растворитель представляет собой ацетонитрил, этанол, водный диоксан или тому подобные.
В другом варианте реализации пролекарственные производные соединения настоящего изобретения могут быть получены по способам, известным в данной области техники (дополнительные подробности представлены в публикации Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters, Vol. 4, p. 1985). В предпочтительном варианте реализации соответствующее пролекарство может быть получено взаимодействием не дериватизованного соединения настоящего изобретения с подходящим карбамилирующим агентом, таким как 1,1-ацилоксиалкилкарбанохлоридат, паранитрофенилкарбонат или тому подобными.
В другом варианте реализации защищенные производные соединения настоящего изобретения могут быть получены известными в данной области техники способами. Подробное описание приемов, применимых для создания защитных групп и их удаления, представлены в публикации Т.W. Greene, "Protecting Groups in Organic Chemistry", 3rd edition, John Wiley and Sons, Inc., 1999.
В другом варианте реализации соединение настоящего изобретения может быть получено в виде отдельных стереоизомеров. Указанный способ включает взаимодействие рацемической смеси соединения с оптически активным разделяющим агентом с образованием пары диастереоизомерных соединений, разделение диастереомеров и выделение оптически чистых энантиомеров. Разделение энантиомеров может быть выполнено с помощью ковалентных диастереомерных производных соединения настоящего изобретения или с помощью диссоциирующих комплексов, таких как кристаллические диастереомерные соли. Диастереомеры имеют разные физические свойства, представленные температурами плавления, температурами кипения, растворимостью, реакционноспособностью и так далее, и могут быть легко разделены за счет использования преимущества этих различий. Диастереомеры могут быть разделены фракционной кристаллизацией, хроматографией или способами выделения/разделения на основе различий растворимости. Затем выделяют оптически чистый энантиомер вместе с разделяющим агентом с помощью любого практического способа, не приводящего к рацемизации. Более подробное описание приемов, применимых для выделения стереоизомеров соединений из их рацемической смеси, представлено в публикации Jean Jacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions", John Wiley And Sons, Inc., 1981.
В заключение, соединение настоящего изобретения может быть получено по способу, описанному в Примерах;
соединение настоящего изобретения необязательно может быть преобразовано в фармацевтически приемлемую соль;
не окисленная форма соединения настоящего изобретения необязательно может быть преобразована в фармацевтически приемлемый N-оксид;
из смеси изомеров необязательно выделяют отдельный изомер соединения настоящего изобретения; и
не дериватизованное соединение настоящего изобретения необязательно может быть преобразовано в фармацевтически приемлемое пролекарство.
Если получение исходных материалов подробно не описано, то эти соединения являются известными или могут быть получены способами, аналогичными известным в данной области техники способами, или так, как описано далее в Примерах. Специалистам в данной области понятно, что описанные выше преобразования представляют собой лишь примеры способов получения соединений настоящего изобретения, и что точно так же могут быть использованы другие общеизвестные способы.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ РЕАЛИЗАЦИИ
Настоящее изобретение дополнительно иллюстрировано, но не ограничиваясь этим, следующим описанием и Примерами, демонстрирующими получение соединений настоящего изобретения.
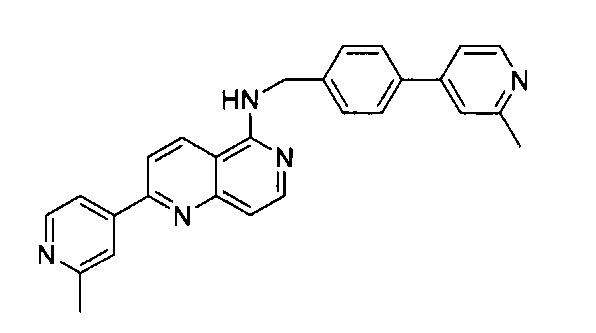
Пример 1: N-(4-(2-метилпиридин-4-ил)бензил)-6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-амин (Соединение №1)
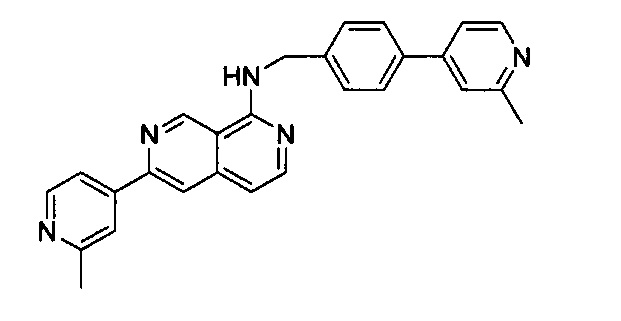
Стадия 1:
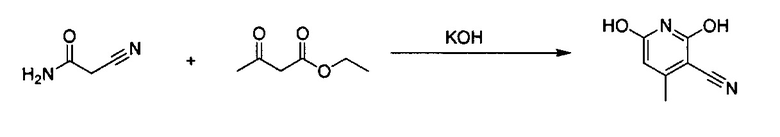
2-Цианоацетамид (50 г, 601,8 ммоль) и этилацетоацетат (75 мл, 601,8 ммоль) растворили в МеОН. КОН (37,0 г, 1,1 экв.) растворили в МеОН и по каплям добавили в смесь, образовалось небольшое количество белого твердого вещества. Смесь нагревали до дефлегмации на масляной бане в течение 8 часов, а затем охладили до комнатной температуры. Твердое вещество отфильтровали, а затем повторно растворили в горячей воде, а затем снова отфильтровали. В фильтрат для нейтрализации добавили 6 н. HCl до pH<7. Снова образовалось белое твердое вещество, которое отфильтровали. Твердое вещество дополнительно промыли МеОН, водой и МеОН, а затем высушили в вакууме с получением конечного продукта, 3-этинил-4-метилпиридин-2,6-диола (выход ~41%).
Стадия 2:
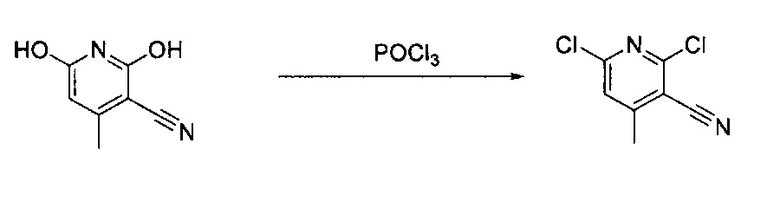
3-Этинил-4-метилпиридин-2,6-диол (28,0 г, 195,2 ммоль) растворили в POCl3 (60,0 мл). Реакционную смесь закрыли в пробирке для работы под давлением и нагревали до 180°C в течение 6 часов. После охлаждения реакционной смеси до комнатной температуры, избыточный POCl3 удалили под вакуумом. В смесь медленно добавили дробленый лед, и образовалось твердое вещество. Твердое вещество отфильтровали и высушили под вакуумом с получением конечного продукта, 2,6-дихлор-4-метилпиридин-3-карбонитрила (выход ~92%) без дополнительной очистки.
Стадия 3:
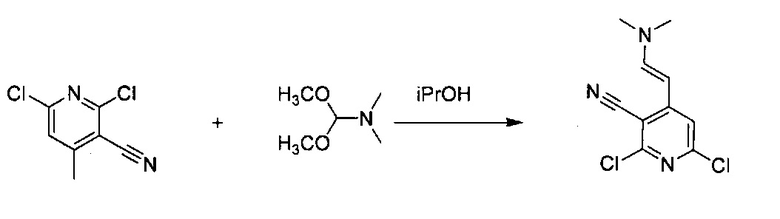
К 2,6-дихлор-4-метилпиридин-3-карбонитрилу (20,0 г, 107,5 ммоль) в 200 мл изопропилового спирта добавили N,N-диметилформамид-диметилацеталь (12,82 г, 107,5 ммоль) и перемешивали реакционную смесь при 65°C в течение 18 часов. После охлаждения реакционной смеси до комнатной температуры осадок собрали фильтрацией и промыли 50 мл изопропилового спирта, и высушили на воздухе с получением продукта, 2,6-дихлор-4-((Е)-2-(диметиламино)винил)пиридин-3-карбонитрила (выход ~26%) без дополнительной очистки.
Стадия 4:
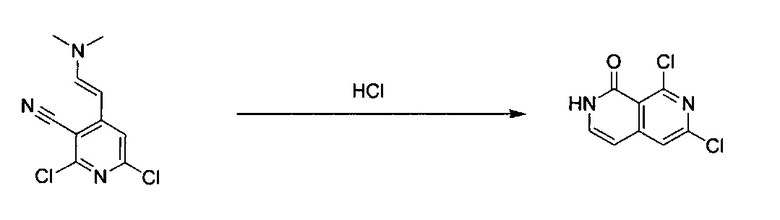
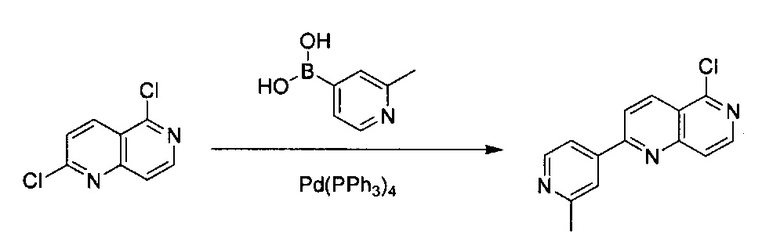
2,6-Дихлор-4-((Е)-2-(диметиламино)винил)пиридин-3-карбонитрил (4,0 г, 16,6 ммоль) добавили с 20 мл концентрированной HCl в закрытую пробирку. Реакционную смесь перемешивали при 45°C в течение 18 часов. После охлаждения реакционной смеси до комнатной температуры к раствору добавили ледяную воду, получив густую желтую взвесь. Осадок собрали фильтрацией, промыли холодной водой, эфиром и этилацетатом и высушили под вакуумом с получением светло-желтого твердого вещества, 6,8-дихлор-2,7-нафтиридин-1(2Н)-она (выход ~80%). MS m/z 215,0 (М+1). 1H ЯМР (300 МГц, ДМСО-d6): δ 11,75 (s, 1Н), 7,76 (s, 1Н), 7,50 (t, J=6,6 Гц, 1H), 6,52 (d, J=6,6 Гц, 1H).
Стадия 5:
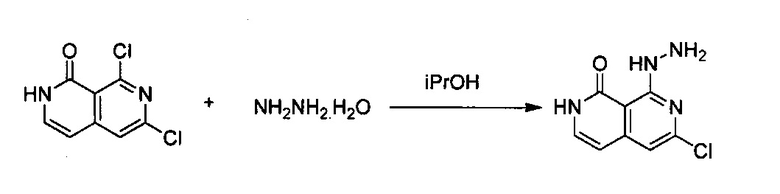
6,8-Дихлор-2,7-нафтиридин-1(2Н)-он (3,0 г, 13,96 ммоль) растворили в iPrOH (120 мл) с получением некоторой суспензии. Раствор охладили до 0°C на ледяной бане, а затем по каплям добавили раствор гидразина (5,6 г, 80%, 10 экв.). Смесь перемешивали при комнатной температуре в течение 15 минут, а затем нагревали на масляной бане при 55°C в течение ночи. После охлаждения реакционной смеси до комнатной температуры, ее отфильтровали с получением сразу твердого вещества, а затем это твердое вещество промыли 70 мл МеОН и высушили под вакуумом. Продукт, 6-хлор-8-гидразинил-2,7-нафтиридин-1(2Н)-он (выход ~98%) использовали на следующей стадии реакции напрямую, без дополнительной очистки.
Стадия 6:
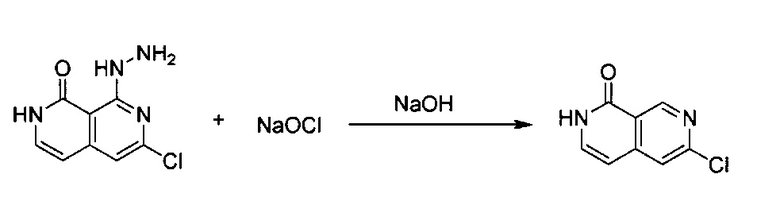
6-Хлор-8-гидразинил-2,7-нафтиридин-1(2Н)-он (1,50 г, 7,12 ммоль) растворили в MeCN (90 мл) с получением некоторой суспензии. Добавили 1 н. NaOH (17,80 мл, 2,5 экв.), а затем в смесь добавили равное количество воды (107,80 мл). Реакционную смесь нагревали при 50°C, перемешивали до образования прозрачного раствора. Раствор снова охладили до 0°C и по каплям добавили NaOCl (11,05 г, 12% раствор, 2,5 экв.), а затем реакционную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции раствор охладили до 0°C, а затем добавили в 1 н. HCl для нейтрализации (pH ~6). Осадок собрали, а фильтрат экстрагировали с использованием 100 мл×2 ЕА. Органический слой объединили и высушили над Na2SO4, и выпарили с получением дополнительного количества неочищенного продукта. Объединенный твердый материал, 6-хлор-2,7-нафтиридин-1(2Н)-он (выход ~93%) использовали на следующей стадии реакции без дополнительной очистки. MS m/z 181,1 (М+1).
Стадия 7:
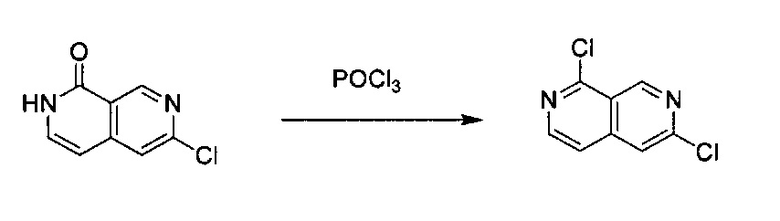
6-Хлор-2,7-нафтиридин-1(2Н)-он (400 мг, 2,2 ммоль) добавили в POCl3 (20,0 мл) в пробирке для работы под давлением. Реакционную смесь нагревали до 160°C в течение 4 часов с получением прозрачного раствора. Раствор охладили до комнатной температуры и вылили в ДХМ, и медленно добавили дробленый лед. В смесь добавили насыщенный раствор NaHCO3 для нейтрализации HCl, образовавшегося в реакции. Использовали вакуум для удаления ДХМ, а оставшийся водный раствор экстрагировали 100 мл×2 ЕА. Объединенные органические слои промыли один раз насыщенным солевым раствором и высушили с помощью Na2SO4, а затем выпарили под вакуумом с получением твердого 1,6-дихлор-2,7-нафтиридина (выход ~73%), который использовали на следующей стадии реакции без дополнительной очистки. MS m/z 199,0 (М+1).
Стадия 8:
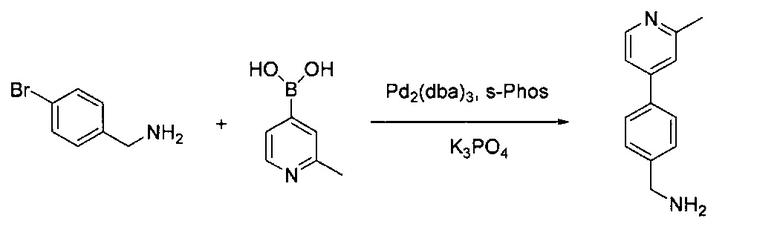
(4-Бромфенил)метанамин (1,00 г, 5,37 ммоль) и 2-метилпиридин-4-ил-4-бороновую кислоту (883,30 мг, 6,45 ммоль) растворили в BuOH (10,0 мл) и воде (2,0 мл). Добавили K3PO4 (2,28 г, 10,75 ммоль), Pd2(dba)3 (120,20 мг, 0,27 ммоль) и S-phos (220,70 мг, 0,54 ммоль) под N2. Реакционную смесь закрыли в пробирке для работы под давлением и нагревали до 125°C в течение 1 часа. После охлаждения реакционной смеси до комнатной температуры смесь вылили в воду и экстрагировали 100 мл×ЕА. Объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4 и концентрировали под вакуумом с получением неочищенного продукта. Твердое вещество очистили силикагелевой колонкой с 10% МеОН (содержащим ~2 н. NH3) в ДХМ с получением чистого (4-(2-метилпиридин-4-ил)фенил)метанамина (выход ~89%). MS m/z 199,1 (М+1).
Стадия 9:
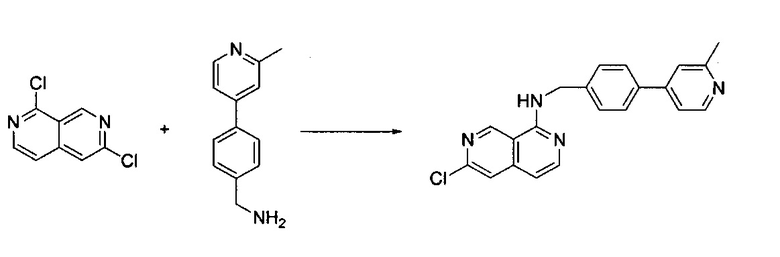
1,6-Дихлор-2,7-нафтиридин (160 мг, 0,80 ммоль) и (4-(2-метилпиридин-4-ил)фенил)метанамин (239,10 мг, 1,21 ммоль) растворили в BuOH (5,0 мл) и нагревали до 115°C в течение ночи. После охлаждения реакционной смеси до комнатной температуры, органический растворитель удалили под вакуумом. Неочищенный продукт очистили силикагелевой флэш-хроматографией с ЕА/гексаном (1:1) с получением твердого N-(4-(2-метилпиридин-4-ил)бензил)-6-хлор-2,7-нафтиридин-1-амина (выход ~90%). MS m/z 361,1 (М+1).
Стадия 10:
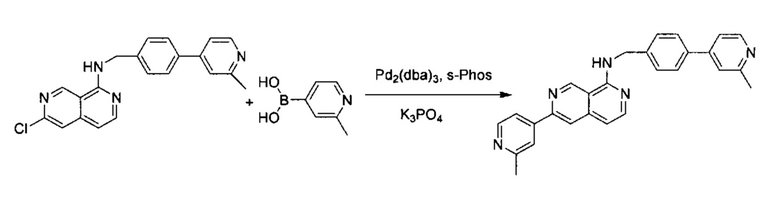
N-(4-(2-Метилпиридин-4-ил)бензил)-6-хлор-2,7-нафтиридин-1-амин (50,00 мг, 0,14 ммоль) и 2-метилпиридин-4-ил-4-бороновую кислоту (56,90 мг, 0,42 ммоль) растворили в BuOH (3,0 мл) и воде (0,6 мл). В смесь добавили K3PO4 (88,20 мг, 0,028 ммоль), Pd2(dba)3 (6,20 мг, 0,014 ммоль) и S-phos (11,40 мг, 0,011 ммоль) под N2. Реакционную смесь закрыли в пробирке для работы под давлением и нагревали до 105°C в течение ночи. После охлаждения реакционной смеси до комнатной температуры смесь вылили в воду и три раза экстрагировали ЕА. Объединенный органический слой промыли насыщенным солевым раствором, высушили с помощью Na2SO4 и концентрировали под вакуумом. Неочищенный продукт дополнительно очистили препаративной ТСХ с 5% МеОН в ДХМ с получением конечного продукта, N-(4-(2-метилпиридин-4-ил)бензил)-6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-амина (выход ~70%). MS m/z 418,2 (М+1). 1Н ЯМР (300 МГц, CDCl3): δ 2,46 (s, 3Н), 2,63 (s, 3Н), 4,94 (d, J=5,10 Гц, 2Н), 5,94 (br, 1H), 6,97 (d, J=5,70 Гц, 1Н), 7,31 (d, J=4,20 Гц, 1Н), 7,36 (s, 1Н), 7,54 (d, J=8,10 Гц, 2Н), 7,63 (d, J=8,40 Гц, 2Н), 7,90 (s, 1Н), 8,19 (d, J=6,00 Гц, 1H), 8,22 (s, 1H), 8,51 (m, 2H), 9,08 (s, 1H), 9,30 (s, 1H).
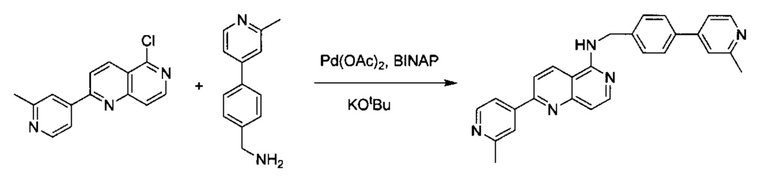
Пример 2: N-(3-метил-4-(2-метилпиридин-4-ил)бензил)-6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-амин (Соединение №2)
Стадия 1:
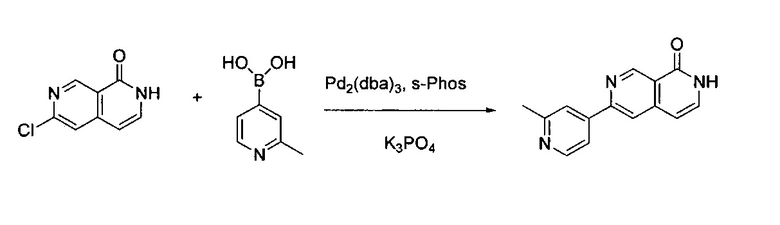
6-Хлор-2,7-нафтиридин-1(2Н)-он (200 мг, 1,10 ммоль) и 2-метилпиридин-4-ил-4-бороновую кислоту (227,60 мг, 1,66 ммоль) растворили в BuOH (5,0 мл) и воде (1,0 мл). Добавили K3PO4 (705,20 г, 3,32 ммоль), Pd2(dba)3 (49,60 мг, 0,22 ммоль) и S-phos (91,00 мг, 0,11 ммоль) под N2. Реакционную смесь в пробирке для работы под давлением нагревали до 130°C в течение 1 часа. После охлаждения реакционной смеси до комнатной температуры смесь вылили в воду, три раза экстрагировали ЕА. Объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4, концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт очистили колонкой с 5% МеОН в ДХМ с получением конечного продукта, 6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1(2Н)-она (выход ~61%). MS m/z 238,1 (М+1).
Стадия 2:
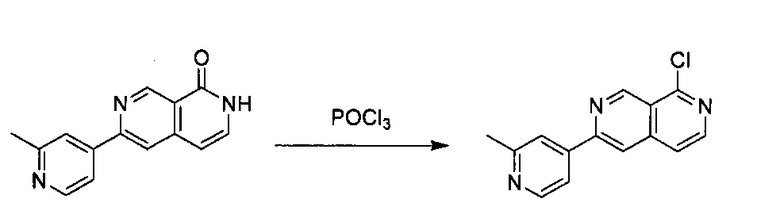
6-(2-Метилпиридин-4-ил)-2,7-нафтиридин-1(2Н)-он (150 мг, 0,63 ммоль) растворили в POCl3 (15,0 мл), пробирку для работы под давлением закрыли и нагревали до 160°C в течение 4 часов. После охлаждения реакционной смеси до комнатной температуры избыточный POCl3 удалили под вакуумом. В смесь медленно добавили дробленый лед, а затем добавили в NaHCO3 для нейтрализации до pH ~7,5. Раствор три раза экстрагировали с помощью ЕА, объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очистили колонкой с ЕА/гексаном (1:1) с получением соединения 1-хлор-6-(2-метилпиридин-4-ил)-2,7-нафтиридина (выход ~55%). MS m/z 256,1 (М+1).
Стадия 3:
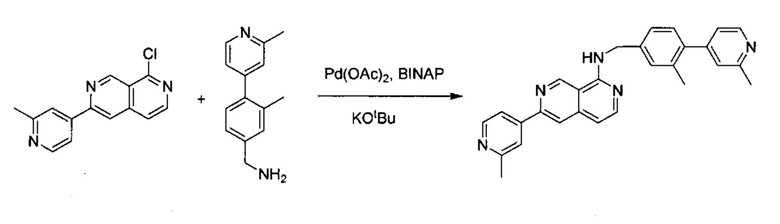
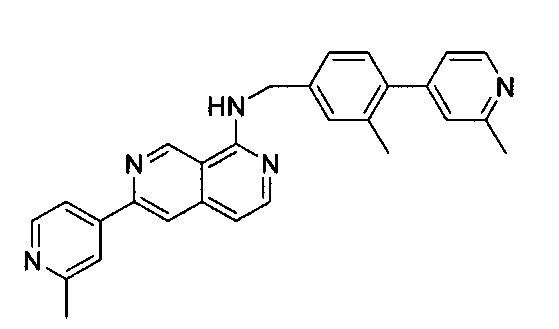
1-Хлор-6-(2-метилпиридин-4-ил)-2,7-нафтиридин (10,00 мг, 0,039 ммоль) и (3-метил-4-(2-метилпиридин-4-ил)фенил)метанамин (10,00 мг, 0,047 ммоль) растворили в толуоле (1,0 мл). В смесь добавили KOtBu (8,80 мг, 0,078 ммоль), Pd(OAc)2 (0,90 мг, 0,0039 ммоль) и BINAP (4,90 мг, 0,0078 ммоль) под N2. Реакционную смесь нагревали до 100°C в течение ночи. После охлаждения реакционной смеси до комнатной температуры смесь вылили в воду, три раза экстрагировали ЕА. Объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4, затем концентрировали под вакуумом. Неочищенный продукт очистили препаративной ТСХ с ЕА/гексаном (4:1) с получением N-(3-метил-4-(2-метилпиридин-4-ил)бензил)-6-(2-метилпиридин-4-ил)-2,7-нафтиридин-1-амина (8,8 мг, выход ~52%). 1Н ЯМР (300 МГц, CDCl3): д 2,31 (s, 3Н), 2,63 (s, 3Н), 2,70 (s, 3Н), 4,91 (d, J=5,10 Гц, 2Н), 5,88 (br, 1Н), 7,00 (d, J=5,40 Гц, 1Н), 7,08 (d, J=5,10 Гц, 1Н), 7,12 (s, 1Н), 7,22 (d, J=7,50 Гц, 1Н), 7,36 (m, 2Н), 7,77 (d, J=4,50 Гц, 1Н), 7,88 (s, 1Н), 7,98 (s, 1Н), 8,24 (d, J=6,00 Гц, 1Н), 8,53 (d, J=4,80 Гц, 1Н), 8,64 (d, J=5,40 Гц, 1Н), 9,31 (s, 1Н). MS m/z 432,2 (М+1).
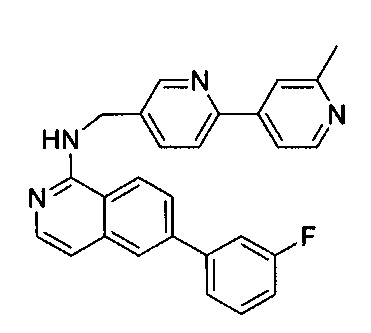
Пример 3: 6-(3-фторфенил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)изохинолин-1-амин (Соединение №3)
Стадия 1:
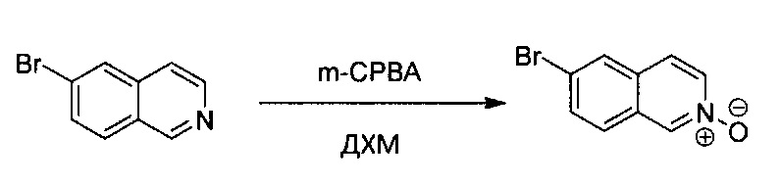
6-Бромизохинолин (1,80 г, 8,66 ммоль) растворили в ДХМ (40 мл), после охлаждения реакционной смеси до 0°C небольшими частями медленно добавили m-СРВА (2,30 г, 1,3 экв., не более 77%). Реакционную смесь нагрели до комнатной температуры с получением некоторой белой суспензии. Через 4 часа в раствор добавили 100 мл ДХМ и промыли насыщенным раствором Na2CO3, водой и насыщенным солевым раствором. Отделенный органический слой высушили над Na2SO4 и удалили под вакуумом с получением желтого твердого N-оксида 6-бромизохинолина без дополнительной очистки (1,82 г, выход ~93%).
Стадия 2:
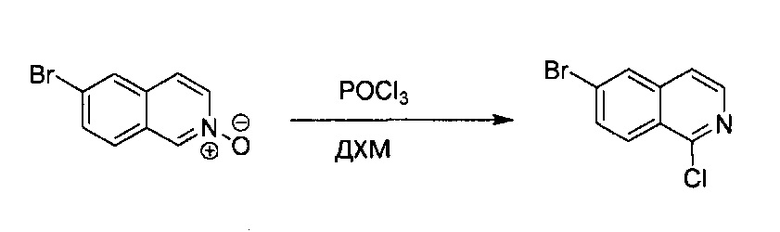
N-оксид 6-бромизохинолина (1,82 г, 8,12 ммоль) растворили в сухом ДХМ (80 мл), по каплям добавили POCl3 (1,12 мл, 1,5 экв.) при комнатной температуре. Реакционную смесь нагревали до 45°C в течение 2 часов. После охлаждения реакционной смеси до комнатной температуры ДХМ и избыточный POCl3 удалили под вакуумом. Неочищенный продукт повторно растворили в 100 мл ДХМ и промыли насыщенным раствором Na2CO3, водой и насыщенным солевым раствором. Отделенный органический слой высушили над Na2SO4 и концентрировали с получением коричневого твердого вещества. Неочищенный материал очистили флэш-колонкой, используя 2% МеОН в ДХМ с получением бледно-желтого твердого 6-бром-1-хлоризохинолина (1,27 г, выход ~65%). MS m/z 242,0 (М+1).
Стадия 3:
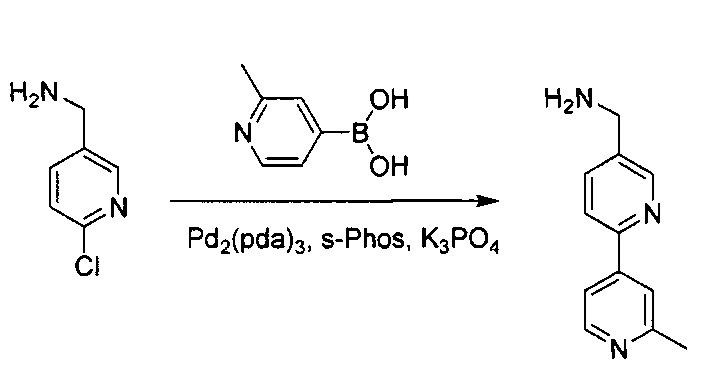
(6-Хлорпиридин-3-ил)метанамин (300 мг, 2,1 ммоль) и 2-метилпиридин-4-илбороновую кислоту (345 мг, 2,52 ммоль) растворили в пробирке для работы под давлением в н-бутаноле (10 мл) и воде (2 мл). Добавили K3PO4 (893 мг, 4,2 ммоль), Pd2(dba)3 (96,3 мг, 0,105 ммоль) и S-phos (86,4 мг, 0,21 ммоль) под защитным слоем азота. Реакционную смесь нагревали до 125°C в течение 30 минут, а затем охладили до комнатной температуры. Раствор вылили в воду и три раза экстрагировали с помощью ЕА. Объединенный органический слой промыли насыщенным солевым раствором и высушили над Na2SO4, и концентрировали под вакуумом. Неочищенный продукт дополнительно очистили флэш-хроматографией с 10% МеОН (содержащим ~2 н. NH3) в ДХМ с получением чистого (6-(2-метилпиридин-4-ил)пиридин-3-ил)метанамина (0,19 г, выход ~45%). MS m/z 200,1 (М+1).
Стадия 4
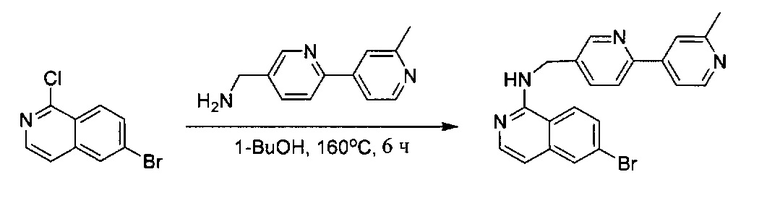
6-Бром-1-хлоризохинолин (100 мг, 0,41 ммоль) и (6-(2-метилпиридин-4-ил)пиридин-3-ил)метанамин (165 мг, 0,82 ммоль) растворили в 0,5 мл n-BuOH в закрытой пробирке. Реакционную смесь нагревали до 160°C в течение 6 часов и охладили до комнатной температуры. Неочищенный продукт очистили флэш-хроматографией, используя 8% МеОН (содержащий ~2 н. NH3) в ДХМ с получением чистого 6-бром-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)изохинолин-1-амина (116 мг, ~70%). MS m/z 405,2 (М+1).
Стадия 5:
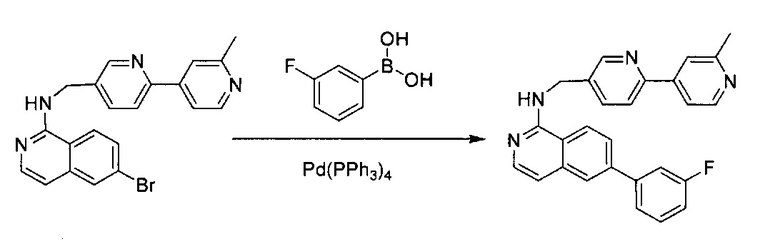
6-Бром-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)изохинолин-1-амин (20 мг, 0,05 ммоль), 3-фторфенилбороновую кислоту (10,5 мг, 0,075 ммоль), Na2CO3 (21 мг, 0,2 ммоль) и тетракис(трифенилфосфин)палладий (5,8 мг, 0,005 ммоль) добавили в пробирку для работы под давлением. В пробирку добавили диоксан/воду (3:1, 2 мл) и нагревали до 125°C в течение 10 минут. После охлаждения реакционной смеси до комнатной температуры раствор разбавили 50 мл воды и 3 раза экстрагировали ЕА. Объединенный органический слой высушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт дополнительно очистили флэш-хроматографией с 10% МеОН (содержащим ~2 н. NH3) в ДХМ с получением чистого 6-(3-фторфенил)-N-((6-(2-метилпиридин-4-ил)пиридин-3-ил)метил)изохинолин-1 -амина (15,8 мг, ~75%). 1Н ЯМР (300 МГц, CDCl3): д 2,71 (s, 3Н), 5,00 (d, J=5,6 Гц, 2Н), 7,32-7,38 (m, 2Н), 7,59-7,65 (m, 1Н), 7,75-7,83 (m, 3Н), 8,10 (d, J=8,4 Гц, 1Н), 8,21 (d, J=8,8 Гц, 1Н), 8,27-8,31 (m, 2Н), 8,39 (s, 2H), 8,72 (d, J=8,8 Гц, 1H), 8,79 (d, J=6,0 Гц, 1H), 8,91 (d, J=1,6 Гц, 1H), 10,02 (s, 1H). MS m/z 421,2 (M+1).
Пример 4: N-(4-(2-метилпиридин-4-ил)бензил)-2-(2-метилпиридин-4-ил)-1,6-нафтиридин-5-амин (Соединение №4)
Стадия 1:
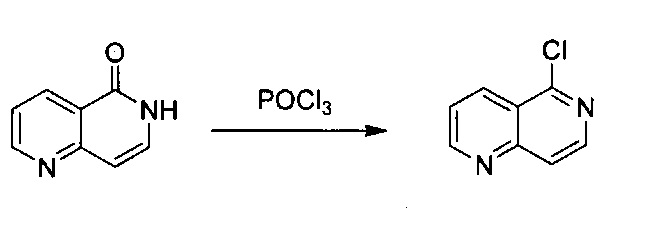
1,6-Нафтиридин-5(6Н)-он (2,9 г, 19,84 ммоль) растворили в POCl3 (40 мл) и нагревали до 100°C в течение 24 часов. После охлаждения реакционной смеси до комнатной температуры избыточный POCl3 удалили под вакуумом. Медленно добавили небольшое количество дробленого льда в насыщенном растворе Na2CO3, выделилось большое количество пузырьков и твердое вещество. Твердое вещество отфильтровали, а раствор 3 раза экстрагировали ЕА. Объединенный органический слой высушили над Na2SO4 и концентрировали под вакуумом. Объединенное твердое вещество дополнительно высушили под вакуумом с получением 5-хлор-1,6-нафтиридина без дополнительной очистки (2,6 г, выход ~80%). MS m/z 165,1 (M+1).
Стадия 2:
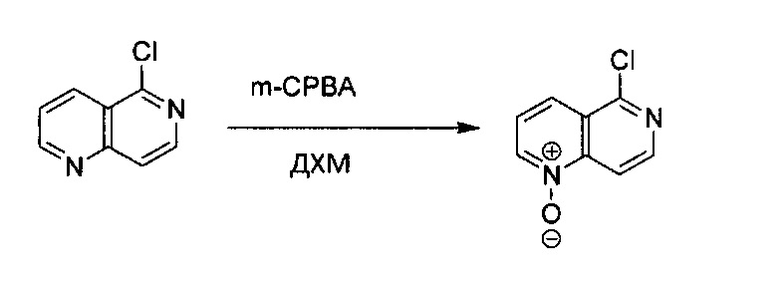
5-Хлор-1,6-нафтиридин (1,5 г, 9,11 ммоль) растворили в ДХМ (45 мл) и охладили на ледяной бане, небольшими частями и медленно добавили m-СРВА (3,7 г, 2 экв., не более 77%). Реакционную смесь нагрели до комнатной температуры и продолжали реакцию в течение 3 часов. В раствор добавили еще 100 мл ДХМ и промыли насыщенным раствором Na2CO3, водой и насыщенным солевым раствором. Органический слой высушили над Na2SO4 и концентрировали под вакуумом с получением желтого твердого N-оксида 5-хлор-1,6-нафтиридина без дополнительной очистки (1,25 г, выход ~76%).
Стадия 3:
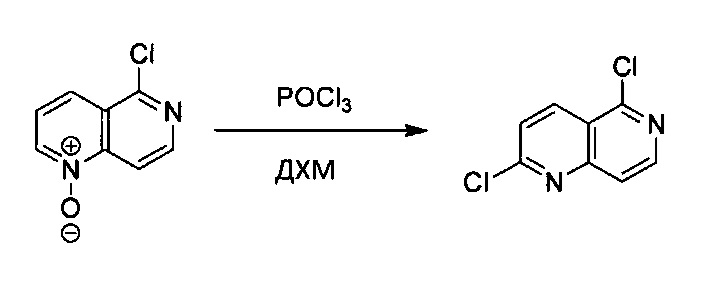
N-Оксид 5-хлор-1,6-нафтиридина (1,2 г, 6,64 ммоль) растворили в сухом ДХМ (30 мл), добавили Et3N (1,85 мл, 13,29 ммоль), затем по каплям добавили POCl3 (0,93 мл, 9,97 ммоль) в 5 мл сухого ДХМ. Реакционную смесь нагревали до 48°C в течение 2 часов. В раствор добавили еще 100 мл ДХМ и промыли насыщенным раствором Na2CO3, водой и насыщенным солевым раствором. Органический слой высушили над Na2SO4 и концентрировали под вакуумом с получением желтого твердого вещества. Неочищенный продукт дополнительно очистили силикагелевой колонкой, используя ЕА/гексан (1:4), с получением белого твердого 2,5-дихлор-1,6-нафтиридина (0,6 г, выход ~45%). MS m/z 199,0 (М+1).
Стадия 4:
2,5-Дихлор-1,6-нафтиридин (200 мг, 1,0 ммоль), 2-метилпиридин-4-ил-4-бороновую кислоту (137 мг, 1,0 ммоль), Na2CO3 (424 мг, 4,0 ммоль) и тетракис(трифенилфосфин)палладий (116 мг, 0,1 ммоль) добавили в колбу, затем добавили 16 мл диоксана и 4 мл воды. Реакционную смесь тщательно перемешивали и нагревали до 90°C в течение 4 часов. После охлаждения реакционной смеси до комнатной температуры раствор разбавили 100 мл воды и 3 раза экстрагировали ЕА. Объединенный органический слой высушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очистили флэш-хроматографией с ЕА/гексаном (1:1) с получением твердого 5-хлор-2-(2-метилпиридин-4-ил)-1,6-нафтиридина (143 мг, выход ~56%). MS m/z 256,1 (М+1).
Стадия 5:
5-Хлор-2-(2-метилпиридин-4-ил)-1,6-нафтиридин (20,00 мг, 0,078 ммоль) и (4-(2-метилпиридин-4-ил)фенил)метанамин (25 мг, 0,118 ммоль) растворили в толуоле (2,0 мл). В смесь добавили KOtBu (13,2 мг, 0,118 ммоль), Pd(OAc)2 (2,7 мг, 0,012 ммоль) и BINAP (15,0 мг, 0,024 ммоль) под N2. Реакционную смесь нагревали до 100°C в течение ночи. После охлаждения реакционной смеси до комнатной температуры смесь вылили в воду, три раза экстрагировали ЕА. Объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4, затем концентрировали под вакуумом. Неочищенный продукт очистили препаративной ТСХ с 8% МеОН в ДХМ с получением N-(4-(2-метилпиридин-4-ил)бензил)-2-(2-метилпиридин-4-ил)-1,6-нафтиридин-5-амина (31 мг, выход -61%). 1Н ЯМР (400 МГц, ДМСО-d6): д 9,12 (d, J=8,8 Гц, 1Н), 8,77-8,83 (m, 2Н), 8,49 (d, J=8,4 Гц, 1Н), 8,40 (s, 1H), 8,31 (d, J=6,4 Гц, 1Н), 8,21 (s, 1Н), 8,11 (d, J=5,6 Гц, 1H), 8,06 (d, J=6,4 Гц, 1H), 7,99 (d, J=8,4 Гц, 2Н), 7,65 (d, J=8,4 Гц, 2Н), 7,23 (d, J=6,4 Гц, 1Н), 5,76 (s, 1Н), 4,93 (d, J=5,6 Гц, 2Н), 2,72 (s, 6Н). MS m/z 432,2 (М+1).
Пример 5: N-(4-(2-метилпиридин-4-ил)бензил)-2-фенилпиридо[4,3-b]пиразин-5-амин (Соединение №5)
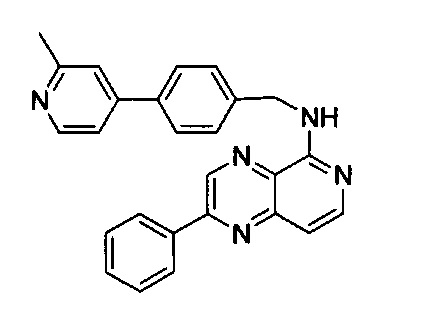
Стадия 1:
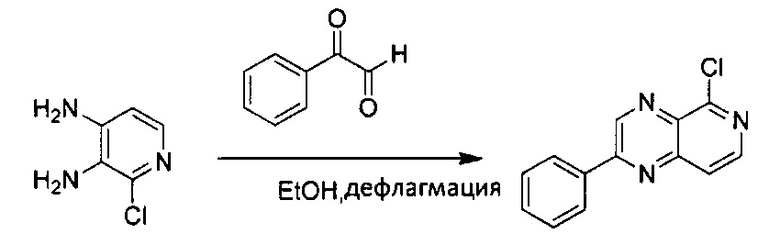
К 20 мл этанола добавили фенилглиоксаля моногидрат (940 мг, 6,99 ммоль) и 2-хлор-3,4-диаминопиридин (1000 мг, 6,99 ммоль). Смесь нагревали с дефлегматором в течение ночи. После охлаждения реакционной смеси неочищенный выпавший в осадок продукт отфильтровали и промыли 15 мл этанола, и высушили под вакуумом с получением 5-хлор-2-фенилпиридо[3,4-b]пиразина без дополнительной очистки (1,28 г, выход ~76%), MS m/z 241,0 (М+1); 1Н ЯМР (300 МГц, ДМСО-d6): δ 9,82 (s, 1H), 8,64 (d, J=6,0 Гц, 1Н), 8,38-8,43 (m, 2Н), 8,07 (d, J=6,0 Гц, 1Н), 7,64-7,68 (m, 3Н).
Стадия 2:
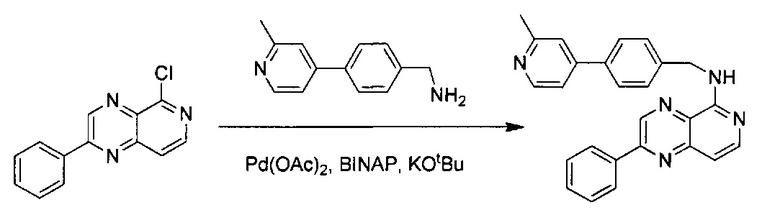
N-(4-(2-Метилпиридин-4-ил)бензил)-2-фенилпиридо[3,4-b]пиразин-5-амин (50 мг, 0,21 ммоль) и (4-(2-метилпиридин-4-ил)фенил)метанамин (42 мг, 0,21 ммоль) растворили в толуоле (4,0 мл). В смесь добавили KOtBu(24 мг, 0,21 ммоль), Pd(OAc)2 (4,5 мг, 0,021 ммоль) и BINAP (26,4 мг, 0,042 ммоль) под N2. Реакционную смесь нагревали до 100°C в течение ночи. После охлаждения реакционной смеси до комнатной температуры смесь вылили в воду, три раза экстрагировали ЕА. Объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4, затем концентрировали под вакуумом. Неочищенный продукт очистили флэш-хроматографией, используя 7% МеОН в ДХМ, с получением N-(4-(2-метилпиридин-4-ил)бензил)-2-фенилпиридо[4,3-b]пиразин-5-амина (61 мг, выход ~72%). MS m/z=404,2 (М+1); 1Н ЯМР (400МГц, ДМСО-d6) δ 9,53 (s, 1Н), 8,77 (d, J=6,4 Гц, 1Н), 8,35-8,39 (m, 2Н), 8,21 (s, 1Н), 8,11 (d, J=6,0 Гц, 1Н), 8,07 (d, J=6,4 Гц, 1Н), 7,96 (d, J=8,4 Гц, 2Н), 7,60-7,65 (m, 5Н), 7,14 (d, J=6,0 Гц, 1Н), 5,76 (s, 1Н), 4,90 (d, J=6,4 Гц, 2Н), 2,71 (s, 3Н).
Специалисты в данной области могут легко понять и узнать, что по такой же стратегии, как в примерах 1-5, могут быть получены другие соединения.
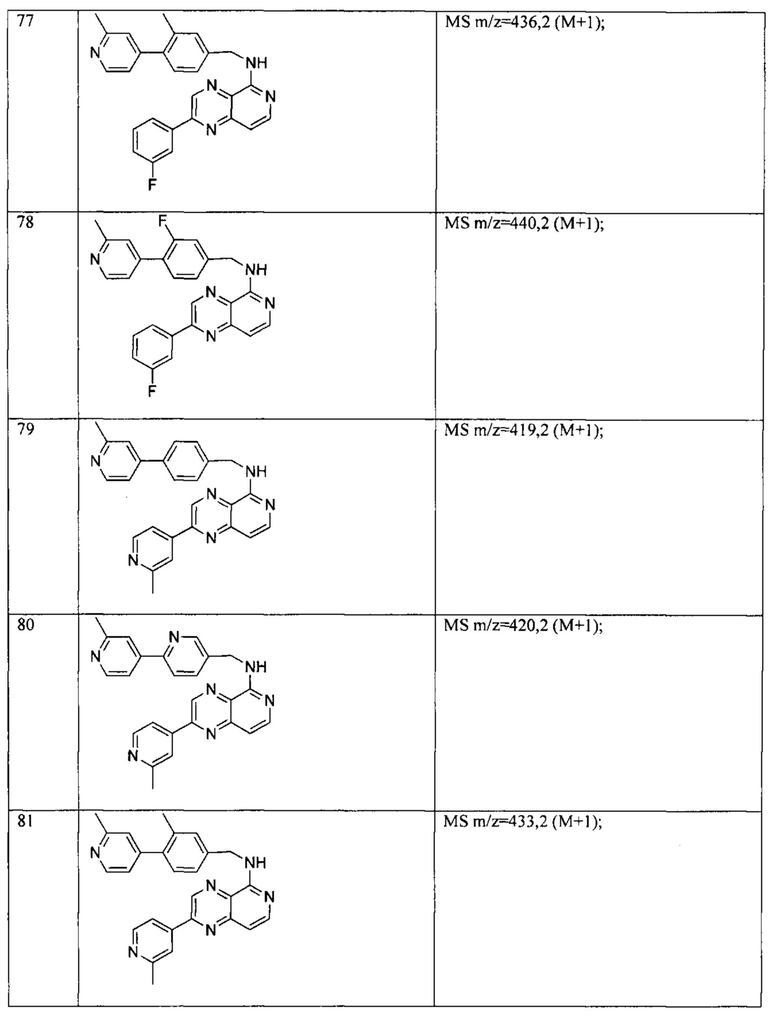
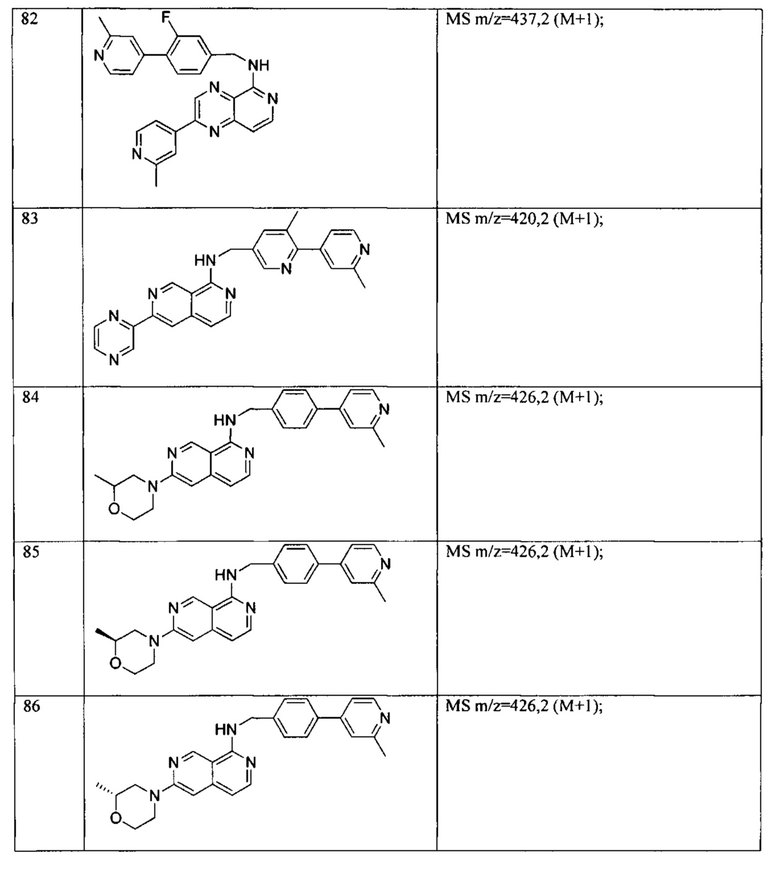
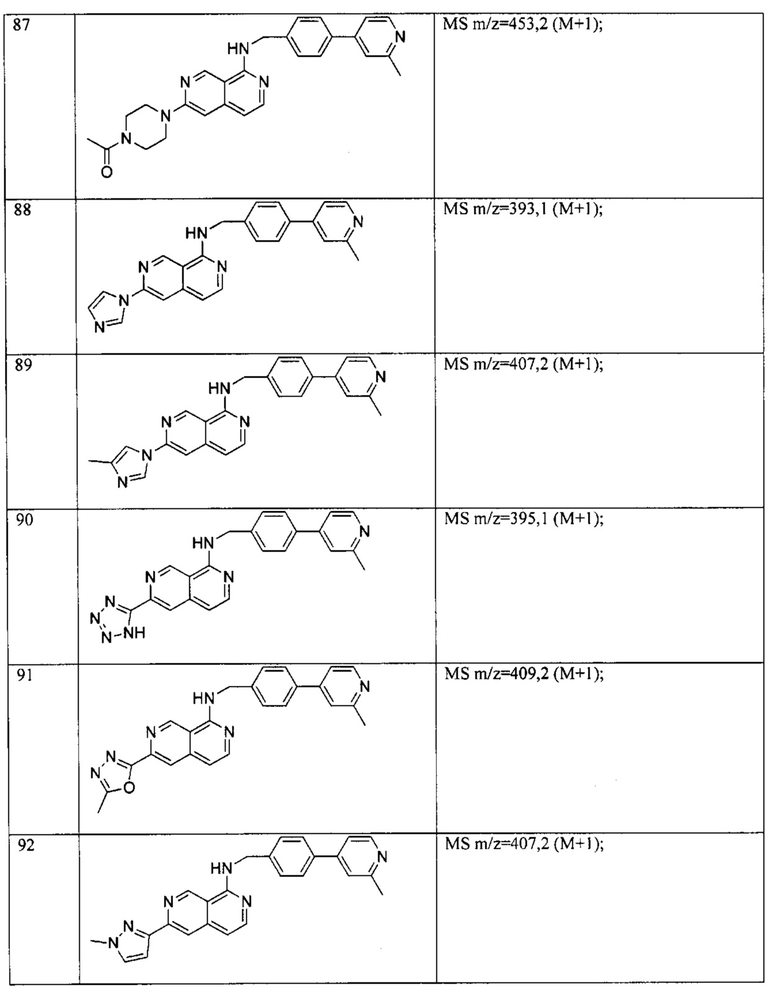
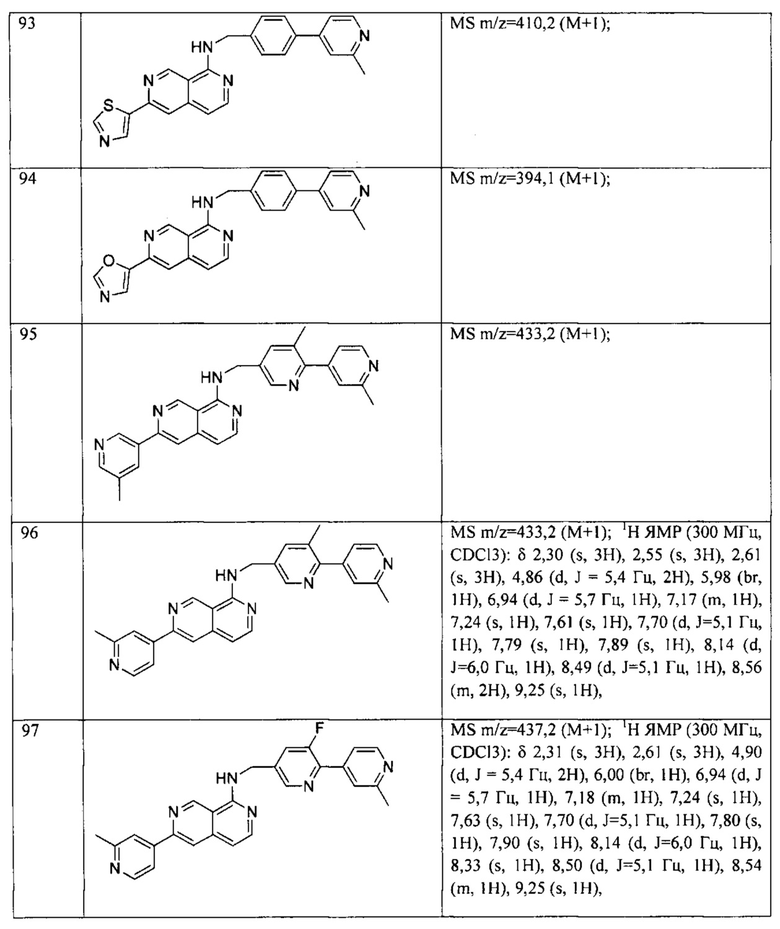
Пример 6: Анализ репортерного гена пути WNT.
Материалы и способы:
Фибробластные клетки мышей NIH3T3 (American Type Culture Collection, Манассас, штат Вирджиния) трансфицировали плазмидой, содержащей ген люциферазы, управляемый 5 копиями элементов TCF. Утратившие свежесть клетки, отобранные с помощью 1 мкг/мл зеоцина (Gibco/Invitrogen, Карлсбад, штат Калифорния), выращивали в среде Игла, модифицированной по способу Дульбекко (Invitrogen, Карлсбад, штат Калифорния) с добавлением 10% FBS (Invitrogen), 50 ед./мл пенициллина и 50 мкг/мл стрептомицина при 37°C с 5% CO2 в атмосфере воздуха. Суспензию клеток НЕК293 (АТСС) трансфицировали плазмидой, содержащей последовательность человеческой кДНК WNT-3а полной длины под управлением промотора CMV, а стабильные клетки отобрали в среде FreeStyle 293 (Invitrogen) с добавлением 100 мкг/мл G418.
Клетки NIH3T3 TCF-Luc и 293 WNT3a выращивали вместе в 96-луночном планшете со средой DMEM с добавлением 0,5% FBS. Через 16 часов активность люциферазы светлячков измерили с помощью люциферазной аналитической системы Steady-Glo™ (Promega). Во время совместного выращивания клетки обрабатывали различными концентрациями соединений настоящего изобретения. Значения IC50 определили как концентрацию, при которой эти соединения снижали интенсивность люминесценции на 50%. Затем для нормализации к количеству и жизнеспособности клеток выполнили анализ CellTiter Glo на планшетах в двух экземплярах.
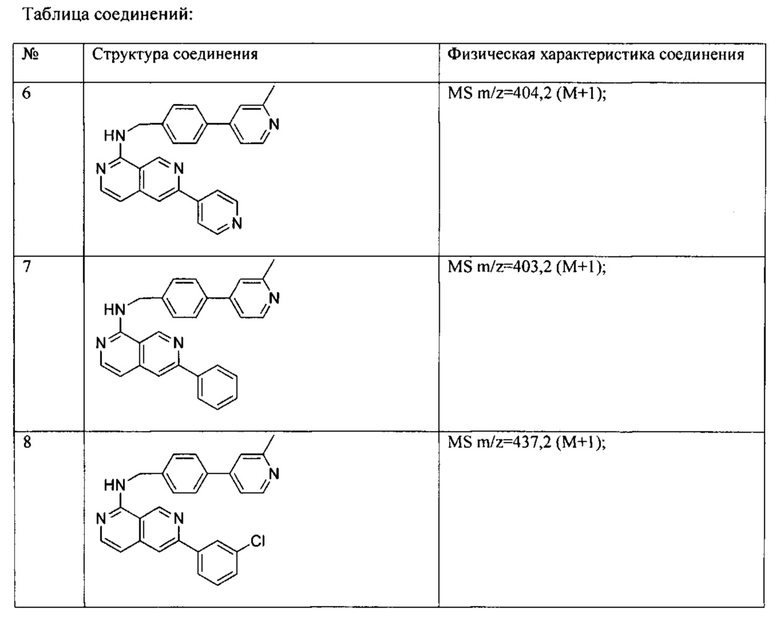
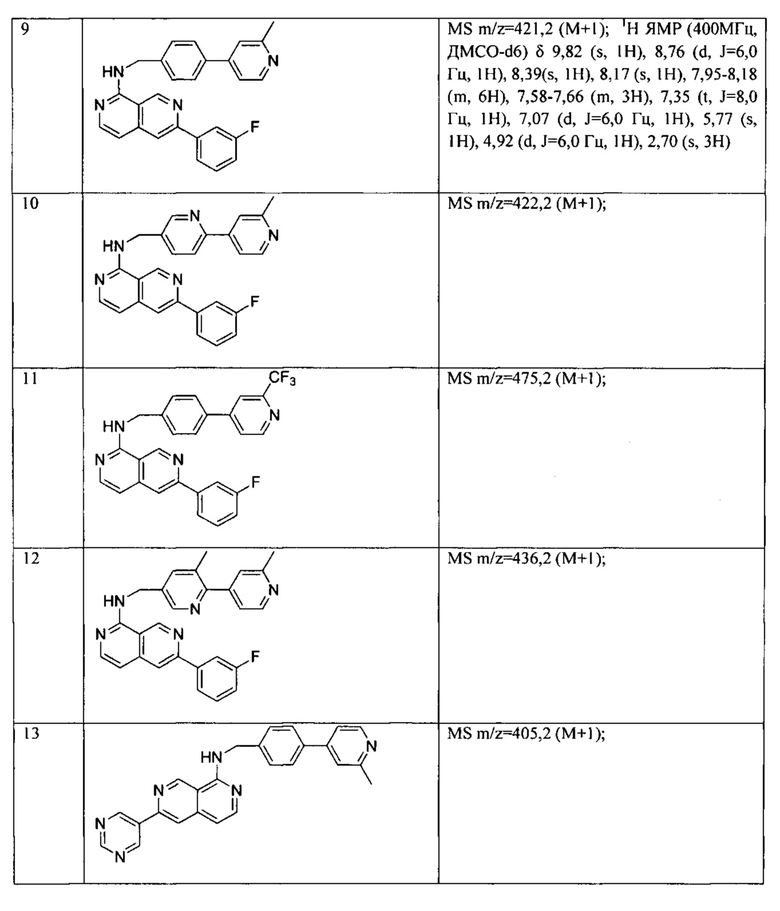
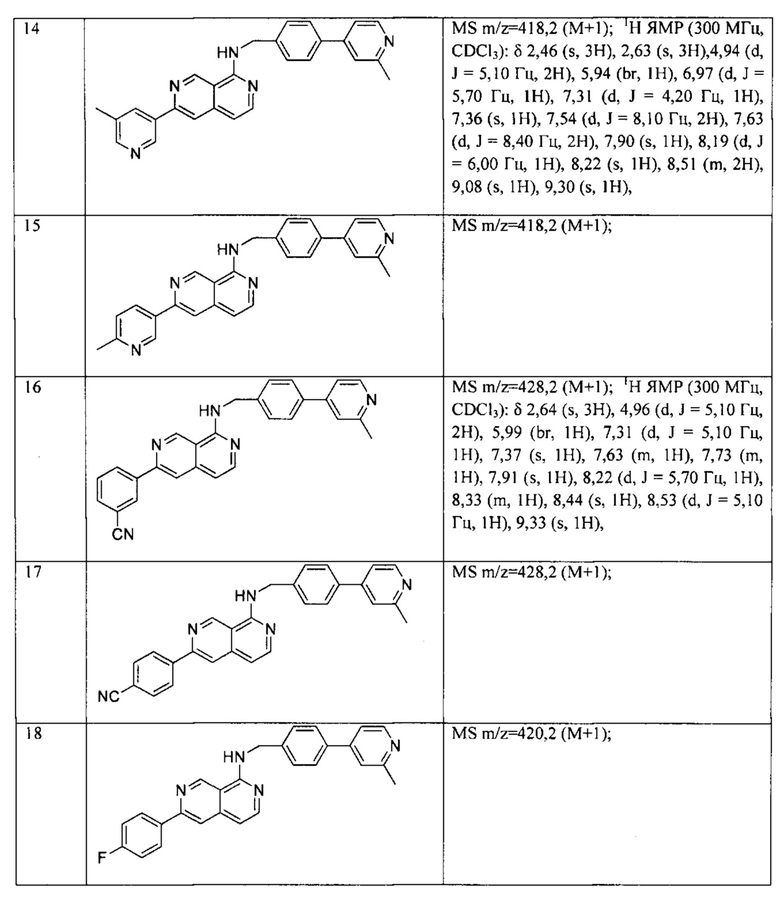
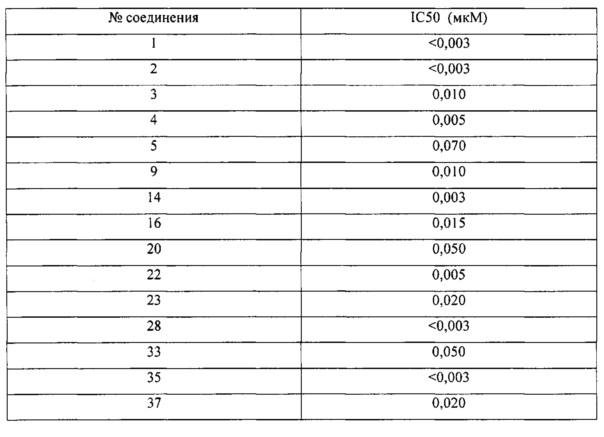
Все соединения, представленные в настоящем патенте, имели значения IC50<5 мкМ в анализе репортерного гена пути WNT. Некоторые примеры перечислены ниже в таблице.
Пример 7: Механистические исследования ингибиторов пути WNT.
Соединения, которые ингибируют активность репортерного гена TCF, индуцированную совместно выращенными клетками Wnt-3а в первичном анализе, затем подвергли механистическому исследованию для определения точки действия этих соединений. Исследовали два различных активатора, один с очищенным рекомбинантным белком Wnt-3а (StemRD Inc., Бурлингейм, штат Калифорния), другой - с ингибитором GSK-3b 6-броминдирубин-3'-оксимом (StemRD Inc., Бурлингейм, штат Калифорния).
Результаты таких механистических исследований показали, что некоторые из активных соединений настоящего изобретения ингибируют активацию пути WNT в точке до взаимодействия WNT-3а с рецепторами, поскольку они не ингибируют активацию репортерного гена TCF рекомбинантным белком WNT-3а. Возможные примеры такого действия включают, но не ограничиваясь этим, белки, прерывающиеся отсутствием wnt/ровностью (Wls/Evi), поркупин (Porcn) и Vps35p.
Пример 5: Влияние ингибиторов пути WNT на раковые клетки
Соединения, которые ингибируют секрецию Wnt и внутриклеточную сигнальную трансдукцию, предположительно подавляют пролиферацию раковых клеток, которые зависят от аутокринной передачи сигналов Wnt. Известно, что для влияния ингибиторов пути Wnt на клеточную пролиферацию в 2-D культуре, «безъякорный» рост клеток и устойчивость к апоптозу в клеточных линиях необходима аутокринная передача сигналов Wnt. Соединения оценивали с помощью стандартных анализов на Wnt-зависимых клеточных линиях, известных в опубликованной литературе: РА-1 (тератокарциномный рак яичников), MDA-MB-157 (рак молочной железы), Saos-2 (остеосаркома) и SNU1076 (плоскоклеточная карцинома головы и шеи). В этих клеточных линиях заметно влияние указанных ингибиторов, что дополнительно подтверждает активность, предполагаемую для этих соединений.
ССЫЛКИ:
Akiri G, Cherian MM, Vijayakumar S, Liu G, Bafico A, Aaronson SA. Wnt pathway aberrations including autocrine Wnt activation occur at high frequency in human non-small-cell lung carcinoma. Oncogene. 28 May, 2009; 28 (21): 2163-72.
Bafico A, Liu G, Goldin L, Harris V, Aaronson SA. An autocrine mechanism for constitutive Wnt pathway activation in human cancer cells. Cancer Cell. 2004 Nov; 6 (5): 497-506.
Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006 Dec; 5 (12): 997-1014.
Blom AB, van Lent PL, van der Kraan PM, van den Berg WB. To seek shelter from the WNT in osteoarthritis? WNT-signaling as a target for osteoarthritis therapy. Curr Drug Targets. 2010 May; 11 (5): 620-9.
Boonen RA, van Tijn P, Zivkovic D. Wnt signaling in Alzheimer's disease: up or down, that is the question. Ageing Res Rev. 2009 Apr; 8 (2): 71-82.
Camilli TC, Weeraratna AT. Striking the target in Wnt-y conditions: intervening in Wnt signaling during cancer progression. Biochem Pharmacol. 2010 Sep 1; 80 (5): 702-11.
Chan SL, Cui Y, van Hasselt A, Li H, Srivastava G, Jin H, Ng KM, Wang Y, Lee KY, Tsao GS, Zhong S, Robertson KD, Rha SY, Chan AT, Tao Q. The tumor suppressor Wnt inhibitory factor 1 is frequently methylated in nasopharyngeal and esophageal carcinomas. Lab Invest. 2007 Jul; 87 (7): 644-50.
Chen В, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, Wei S, Hao W, Kilgore J, Williams NS, Roth MG, Amatruda JF, Chen C, Lum L. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009 Feb; 5 (2): 100-7.
Cheng JH, She H, Han YP, Wang J, Xiong S, Asahina K, Tsukamoto H. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008; 294 (1): G39-49.
Chun JS, Oh H, Yang S, Park M. Wnt signaling in cartilage development and degeneration. BMB Rep.2008 Jul 31; 41 (7): 485-94.
Chien AJ, Moon RT. WNTS and WNT receptors as therapeutic tools and targets in human disease processes. Front Biosci. 2007 Jan 1; 12: 448-57.
DeAlmeida VI, Miao L, Ernst JA, Koeppen H, Polakis P, Rubinfeld B. The soluble wnt receptor Frizzled-8CRD-hFc inhibits the growth of teratocarcinomas in vivo. Cancer Res. 2007 Jun 1; 67 (11): 5371-9
D'Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006 Nov; 24(11):1392-401.
Herbst A, Kolligs FT. Wnt signaling as a therapeutic target for cancer. Method Mol Biol. 2007; 361: 63-91.
Hoeppner LH, Secreto FJ, Westendorf JJ. Wnt signaling as a therapeutic target for bone diseases. Expert Opin Ther Targets. 2009 Apr; 13 (4): 485-96.
Hwang I, Seo EY, Ha H. Wnt/beta-catenin signaling: a novel target for therapeutic intervention of fibrotic kidney disease. Arch Pharm Res. 2009 Dec; 32 (12): 1653-62.
Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci. 2010 Feb; 11(2):77-86.
Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. WNT signalling regulates adult hippocampal neurogenesis. Nature 437 (7063): 1370-5, 2005.
Kansara M, et al. Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J Clin Invest. 2009 Apr; 119 (4): 837-51
MacDonald ВТ, Tamai К, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009 Jul; 17 (1): 9-26.
Mikels AJ, Nusse R. Writs as ligands: processing, secretion and reception. Oncogene. 2006 Dec 4; 25 (57): 7461-8.
Moon RT. Wnt/beta-catenin pathway. Sci STKE.; 2005(271): cm1.
Morrisey EE. Wnt signaling and pulmonary fibrosis. Am J Pathol. 2003 May; 162 (5): 1393-7.
Nusse R. WNT signaling and stem cell control". Cell Res. 18 (5): 523-7, 2008
Ouchi N, Higuchi A, Ohashi K, Oshima Y, Gokce N, Shibata R, Akasaki Y, Shimono A, Walsh K. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science. 2010 Jul 23; 329 (5990): 454-7.
Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005 Apr 14; 434 (7035): 843-50.
Rhee CS, Sen M, Lu D, Wu C, Leoni L, Rubin J, Corr M, Carson DA. Wnt and frizzled receptors as potential targets for immunotherapy in head and neck squamous cell carcinomas. Oncogene. 2002 Sep 26; 21 (43): 6598-605.
Sullivan GJ, et al. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology. 2010 Jan; 51 (1): 329-35.
Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010 Jun 15; 16 (12): 3153-62.
Ten Berge, D. et al. WNT signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell 3, 508-518, 2008.
Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, Henckaerts E, Bonham K, Abbott GW, Linden RM, Field LJ, Keller GM. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008 May 22; 453 (7194): 524-8.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ N-(1H-ИНДАЗОЛ-4-ИЛ) ИМИДАЗОЛ [1,2-А]ПИРИДИН-3- КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНОЙ ТИРОЗИНКИНАЗЫ III ТИПА | 2011 |
|
RU2591195C2 |
| АЛЬФА-СПИРАЛЬНЫЕ МИМЕТИКИ И СПОСОБЫ, СВЯЗАННЫЕ С НИМИ | 2009 |
|
RU2512538C2 |
| 1Н-ПИРАЗОЛО[3,4-B]ПИРИДИНЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2013 |
|
RU2689141C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2013 |
|
RU2679130C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИН КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ ГИПЕРПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2644947C2 |
| СПИРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2016 |
|
RU2745069C2 |
| БИЦИКЛИЧЕСКИЕ АМИНЫ В КАЧЕСТВЕ НОВЫХ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2018 |
|
RU2764980C2 |
| Бициклические производные пиридина, полезные в качестве ингибитора белков, связывающих жирные кислоты (FABP) 4 и/или 5 | 2013 |
|
RU2648247C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗИН-4-АМИНА | 2011 |
|
RU2625791C2 |
| ИНГИБИТОРЫ ИНТЕГРИНА AVB6 | 2018 |
|
RU2769702C2 |
Настоящее изобретение относится к области органической химии, а именно к гетероциклическому соединению общей формулой (I) или его N-оксиду, где X1, Х2, Х3 и Х4 независимо представляют собой CR4 или N, где 0 или 1 из X1-X4 может представлять собой N; Y1, Y2 и Y3 представляют собой водород; R1 выбран из водорода, 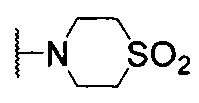 , С6 арила, 6-членного гетероциклоалкила, содержащего 2 гетероатома, выбранных из N и О, и 5- или 6-членного гетероарила, содержащего 1-4 гетероатома, выбранных из N, О и S, где каждый из С6- арила, 6-членного гетероциклоалкила и 5- или 6-членного гетероарила может быть необязательно замещен одним R4; R2 выбран из водорода, галогена, C1-6 алкила,
, С6 арила, 6-членного гетероциклоалкила, содержащего 2 гетероатома, выбранных из N и О, и 5- или 6-членного гетероарила, содержащего 1-4 гетероатома, выбранных из N, О и S, где каждый из С6- арила, 6-членного гетероциклоалкила и 5- или 6-членного гетероарила может быть необязательно замещен одним R4; R2 выбран из водорода, галогена, C1-6 алкила, 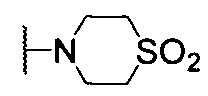 , С6 арила и 6-членного гетероарила, содержащего 1-2 атома N, где каждый из С6 арила и 6-членного гетероарила может быть необязательно замещен одним R4; причем если Х5 представляет собой N, R2 выбирают из галогена, C1-6 алкила,
, С6 арила и 6-членного гетероарила, содержащего 1-2 атома N, где каждый из С6 арила и 6-членного гетероарила может быть необязательно замещен одним R4; причем если Х5 представляет собой N, R2 выбирают из галогена, C1-6 алкила, 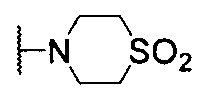 , С6 арила и 6-членного гетероарила, содержащего 1-2 атома N, где каждый из С6 арила, и 6-членного гетероарила может быть необязательно замещен одним R4; каждый R4 независимо выбран из водорода, галогена, циано, оксо, C1-6 алкокси, -C(O)OR5, -C(O)R5, C1-6 алкила, при этом C1-6 алкил может быть необязательно замещен 1-3 заместителями, выбранными из галогена и циано; R5 представляет собой C1-6 алкил; и где центральная структура Формулы I, ограниченная Х5, Х6, Х7 и Х8, представляет собой:
, С6 арила и 6-членного гетероарила, содержащего 1-2 атома N, где каждый из С6 арила, и 6-членного гетероарила может быть необязательно замещен одним R4; каждый R4 независимо выбран из водорода, галогена, циано, оксо, C1-6 алкокси, -C(O)OR5, -C(O)R5, C1-6 алкила, при этом C1-6 алкил может быть необязательно замещен 1-3 заместителями, выбранными из галогена и циано; R5 представляет собой C1-6 алкил; и где центральная структура Формулы I, ограниченная Х5, Х6, Х7 и Х8, представляет собой:
 или
или
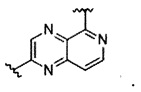
Также изобретение относится к конкретным соединениям, способу ингибирования секреции передачи сигналов WNT в клетке, применению соединения формулы (I), способу лечения расстройства, опосредованного путем WNT. Технический результат: получены новые гетероциклические соединения, полезные при лечении рака, фиброза и остеоартрита. 6 н. и 16 з.п. ф-лы.
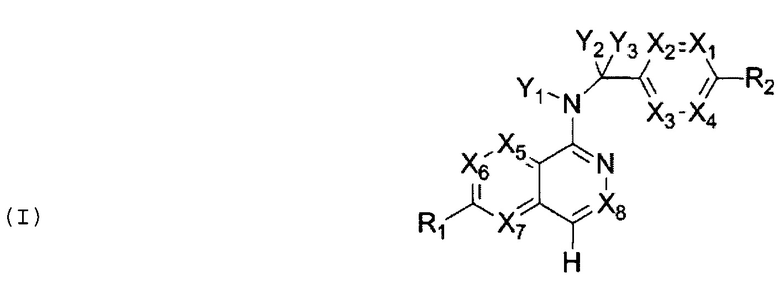
1. Соединение, имеющее структуру следующей Формулы I:
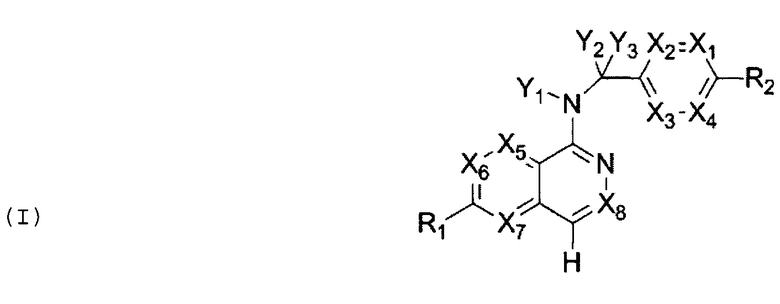
или его N-оксид,
где X1, Х2, Х3 и Х4 независимо представляют собой CR4 или N, где 0 или 1 из X1-X4 может представлять собой N;
Y1 представляет собой водород;
Y2 и Y3 представляют собой водород;
R1 выбран из водорода, 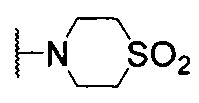 , С6 арила, 6-членного гетероциклоалкила, содержащего 2 гетероатома, выбранных из N и О, и 5- или 6-членного гетероарила, содержащего 1-4 гетероатома, выбранных из N, О и S, где каждый из С6 арила, 6-членного гетероциклоалкила и 5- или 6-членного гетероарила может быть необязательно замещен одним R4;
, С6 арила, 6-членного гетероциклоалкила, содержащего 2 гетероатома, выбранных из N и О, и 5- или 6-членного гетероарила, содержащего 1-4 гетероатома, выбранных из N, О и S, где каждый из С6 арила, 6-членного гетероциклоалкила и 5- или 6-членного гетероарила может быть необязательно замещен одним R4;
R2 выбран из водорода, галогена, C1-6 алкила, 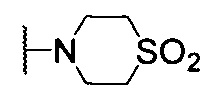 , С6 арила и 6-членного гетероарила, содержащего 1-2 атома N, где каждый из С6 арила и 6-членного гетероарила может быть необязательно замещен одним R4;
, С6 арила и 6-членного гетероарила, содержащего 1-2 атома N, где каждый из С6 арила и 6-членного гетероарила может быть необязательно замещен одним R4;
причем если Х5 представляет собой N, R2 выбирают из галогена, C1-6 алкила, 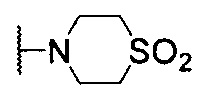 , С6- арила и 6-членного гетероарила, содержащего 1-2 атома N, где каждый из С6 арила и 6-членного гетероарила может быть необязательно замещен одним R4;
, С6- арила и 6-членного гетероарила, содержащего 1-2 атома N, где каждый из С6 арила и 6-членного гетероарила может быть необязательно замещен одним R4;
каждый R4 независимо выбран из водорода, галогена, циано, оксо, C1-6 алкокси, -C(O)OR5, -C(O)R5, C1-6 алкила, при этом C1-6 алкил может быть необязательно замещен 1-3 заместителями, выбранными из галогена и циано;
R5 представляет собой C1-6 алкил; и
где центральная структура Формулы I, ограниченная Х5, Х6, Х7 и Х8, представляет собой
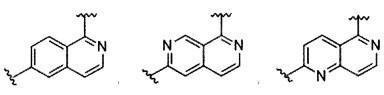 или
или 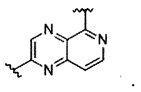
2. Соединение по п.1, отличающееся тем, что центральная структура Формулы I, ограниченная Х5, Х6, Х7 и Х8, выбрана из:
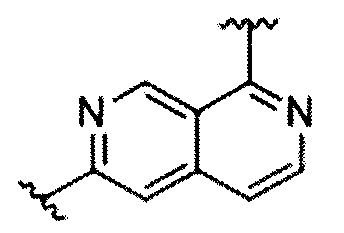 или
или 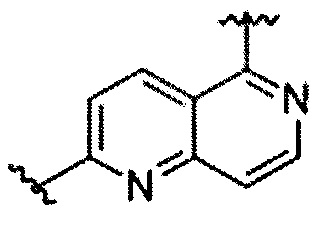 .
.
3. Соединение по п.1 или 2, отличающееся тем, что кольцо в Формуле I, определенное X1, Х2, Х3 и Х4, выбрано из:
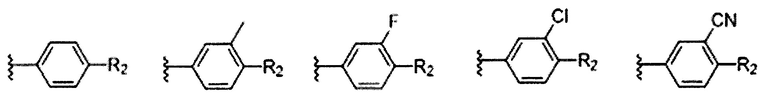
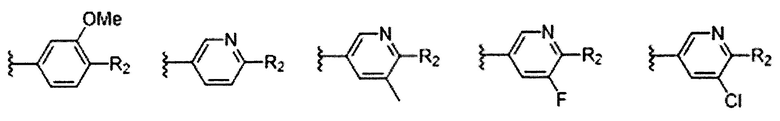
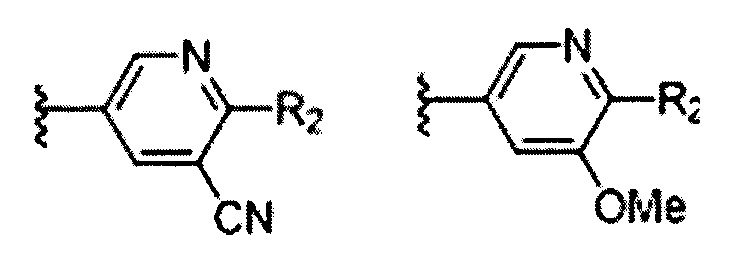
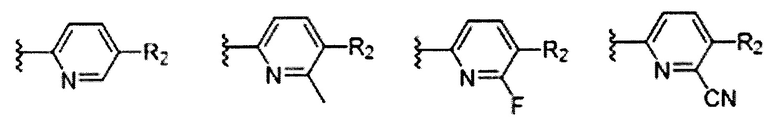
4. Соединение по п.2, отличающееся тем, что R1 выбран из водорода,  , фенила, морфолинила, пиперазинила и 5- или 6-членного гетероарила, выбранного из:
, фенила, морфолинила, пиперазинила и 5- или 6-членного гетероарила, выбранного из:
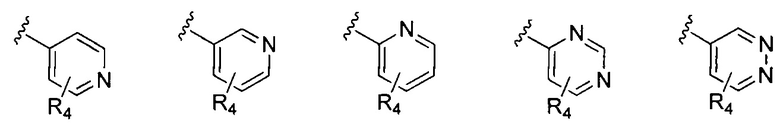
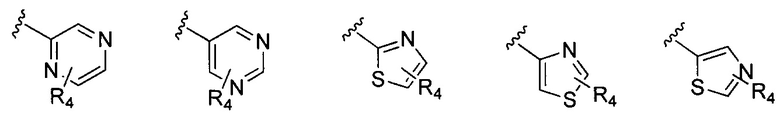
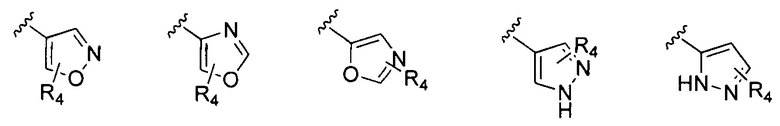
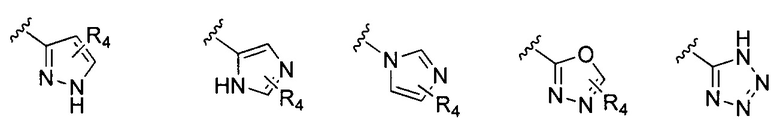
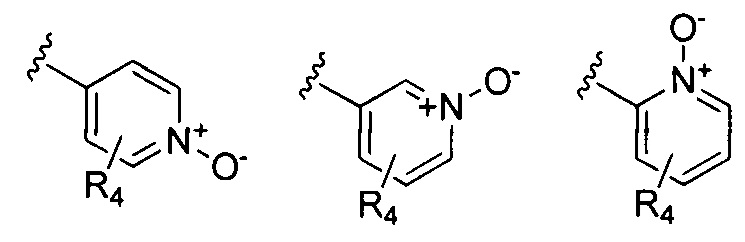
5. Соединение по п.2, где каждый R4 независимо выбран из водорода, хлора, фтора, циано, -СН3, -CHF2, -CF3, -ОСН3, -СООСН3.
6. Соединение по п.2, где R2 выбран из водорода, фтора, хлора, метила, 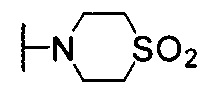 , фенила и 6-ти членного гетероарила, выбранного из:
, фенила и 6-ти членного гетероарила, выбранного из:
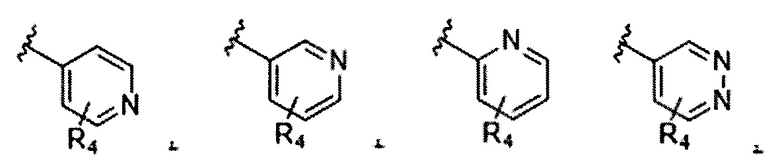
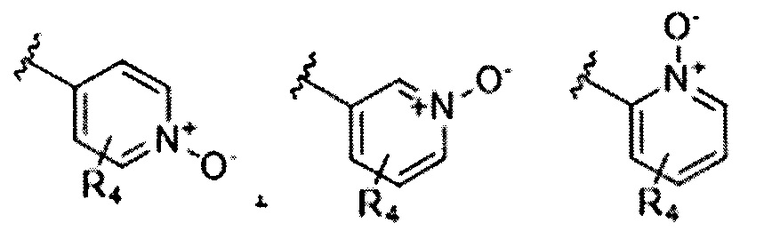
7. Соединение, выбранное из:
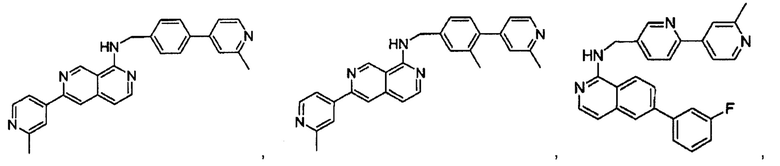
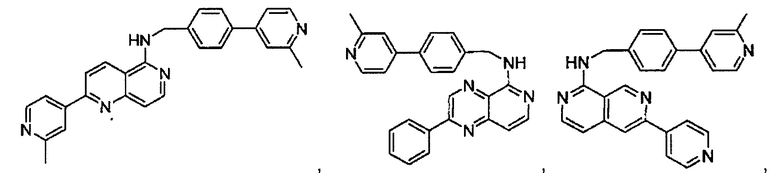
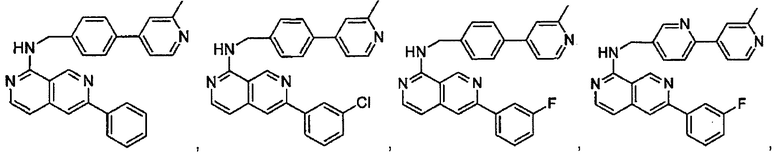
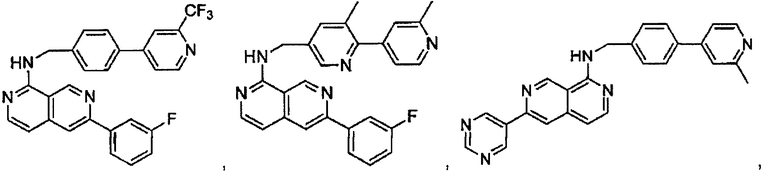
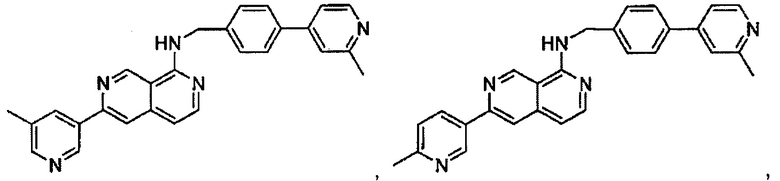
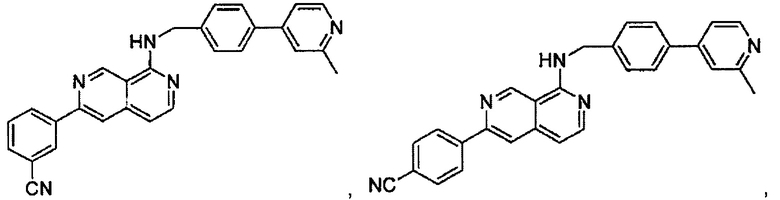
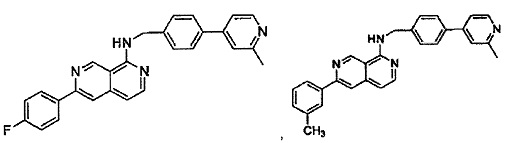
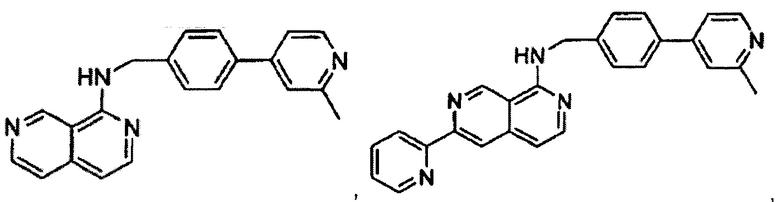
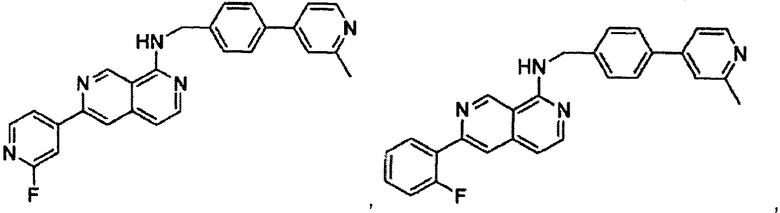
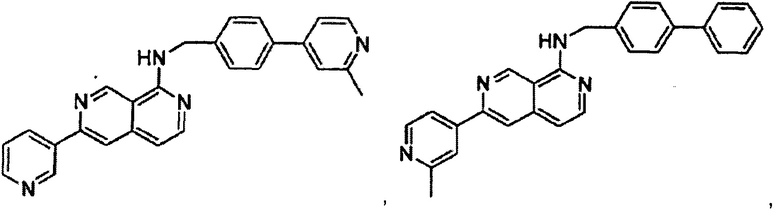
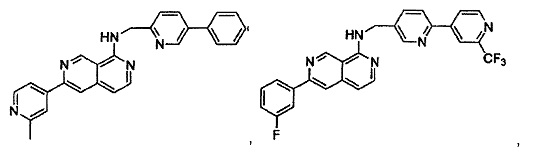
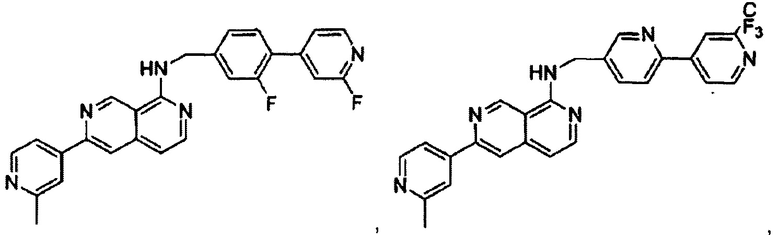
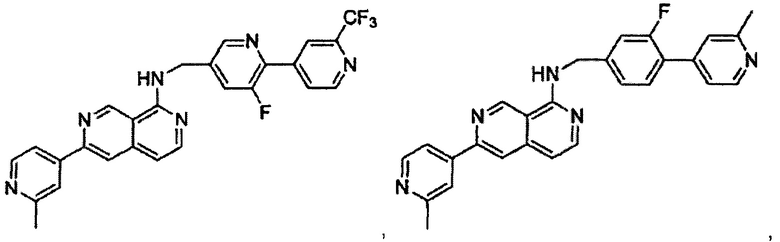
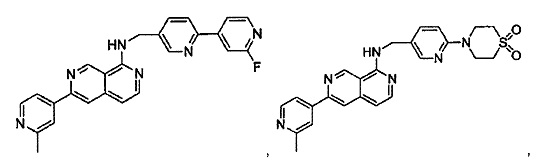
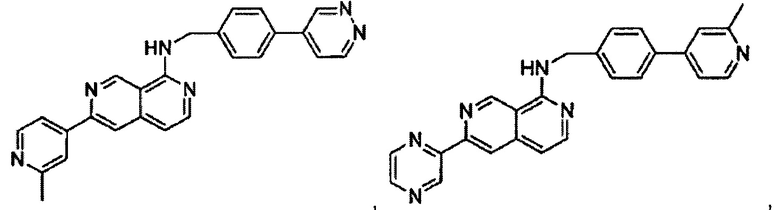
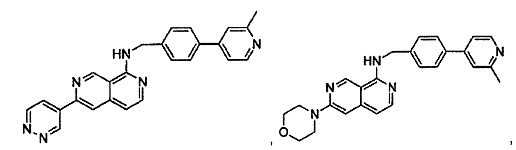
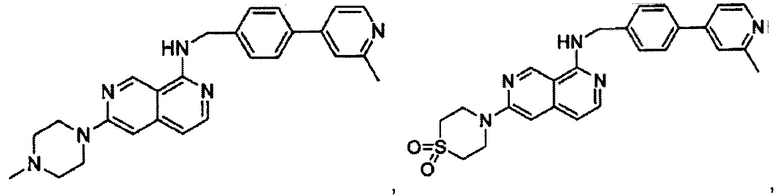
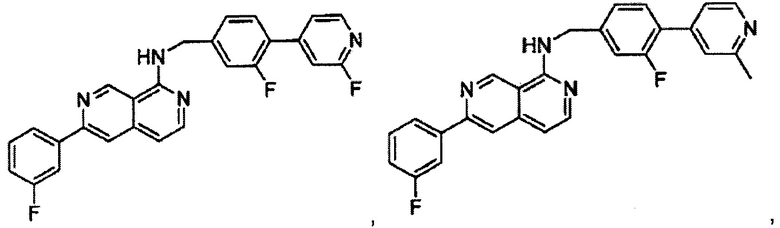
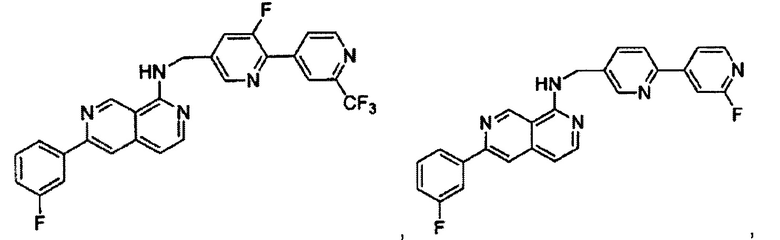
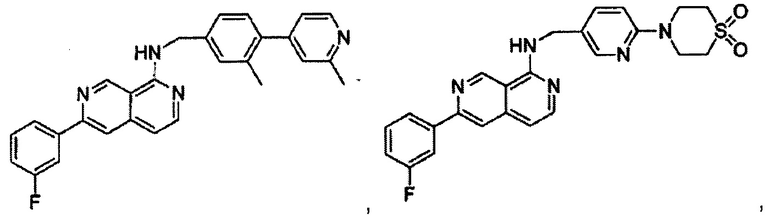
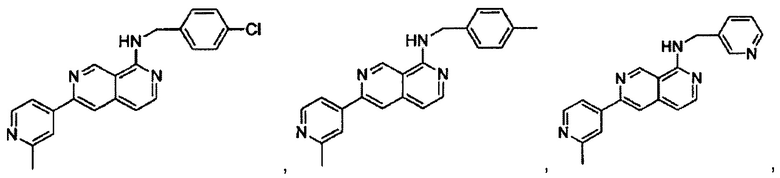
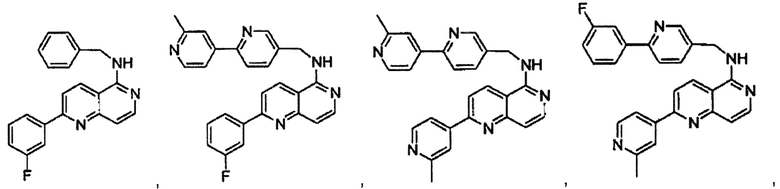
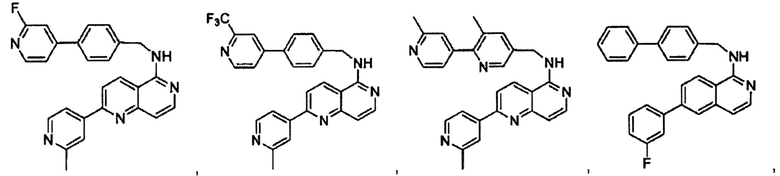
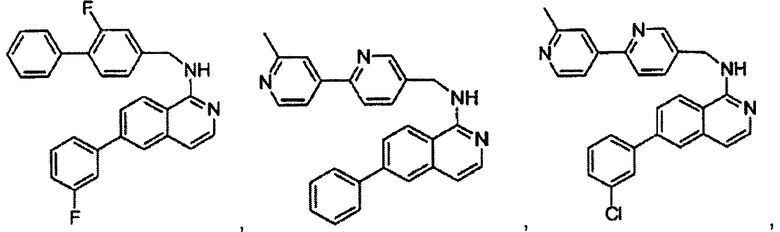
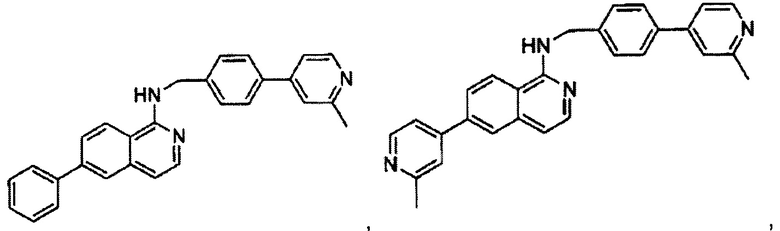
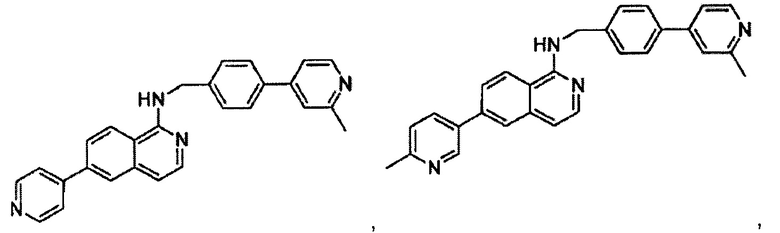
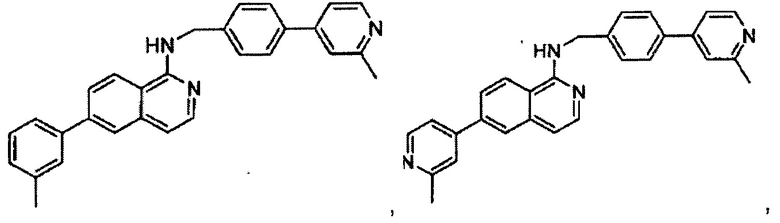
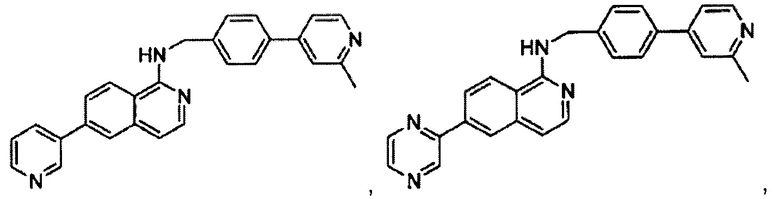
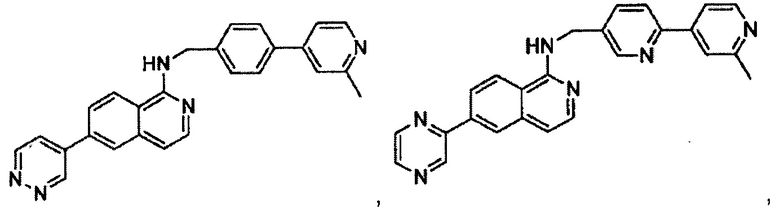
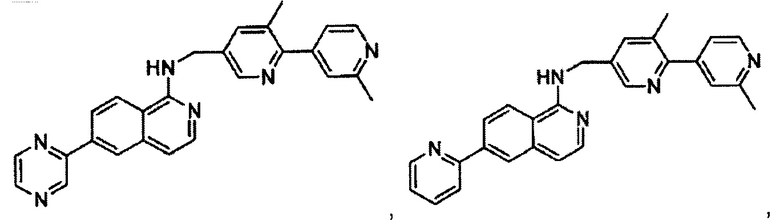
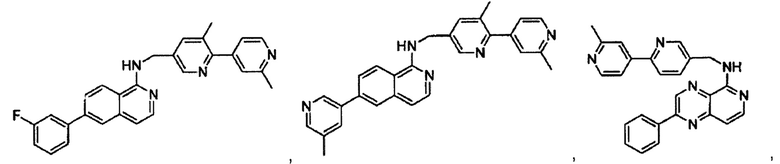
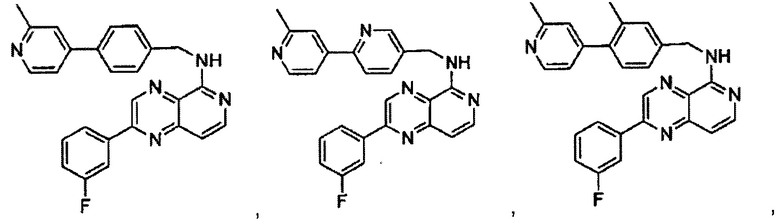
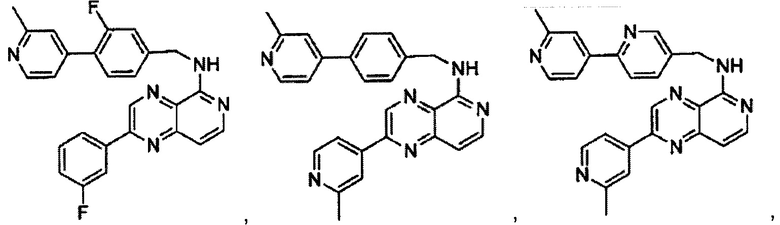
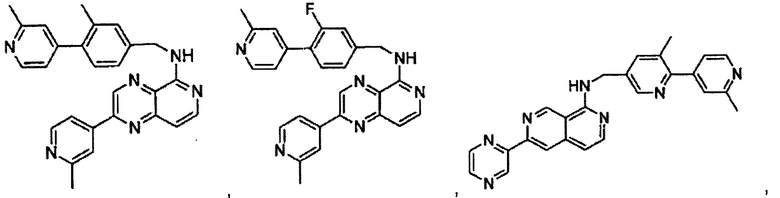
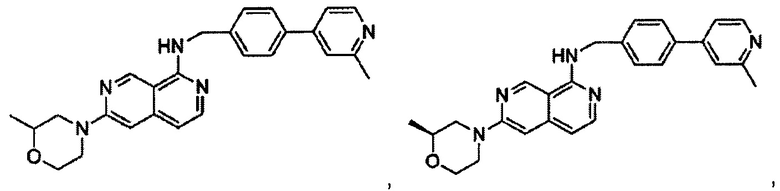
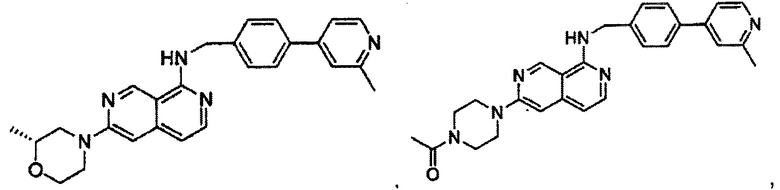
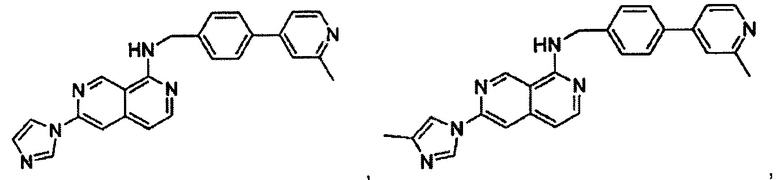
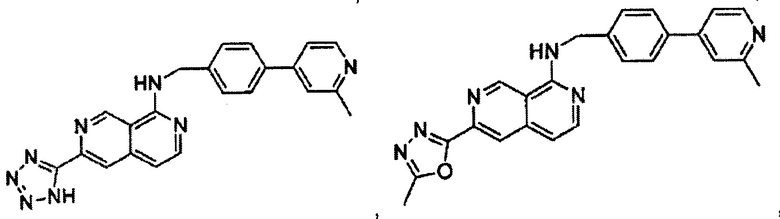
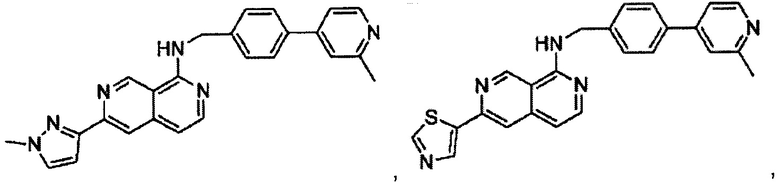
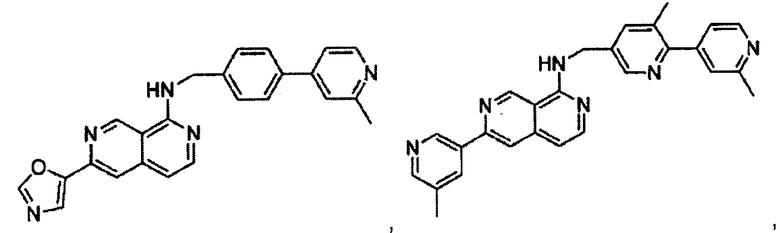
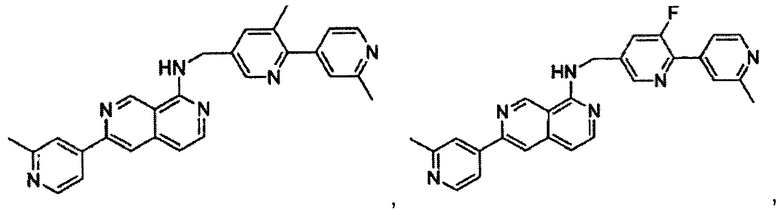
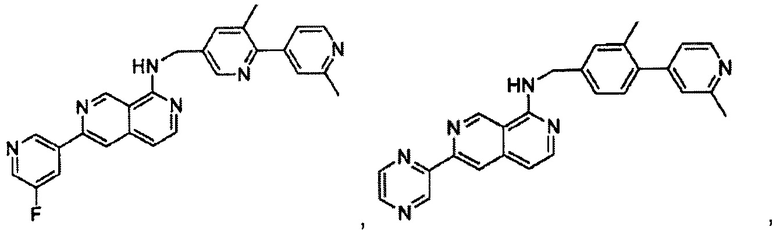
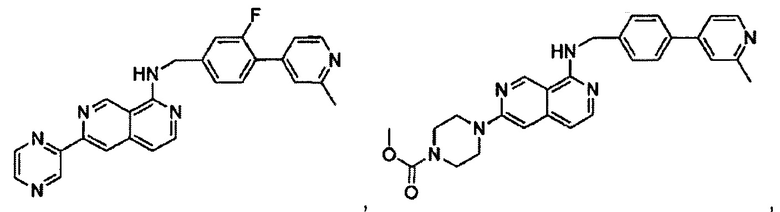
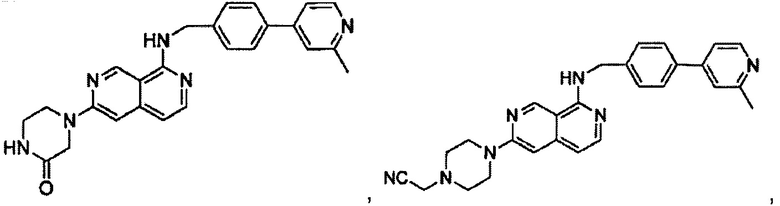
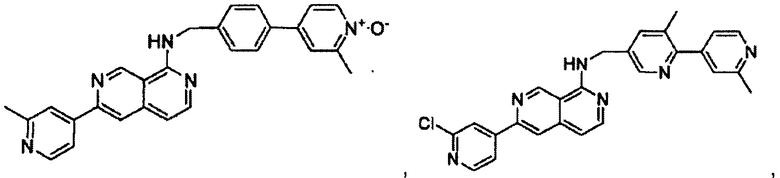
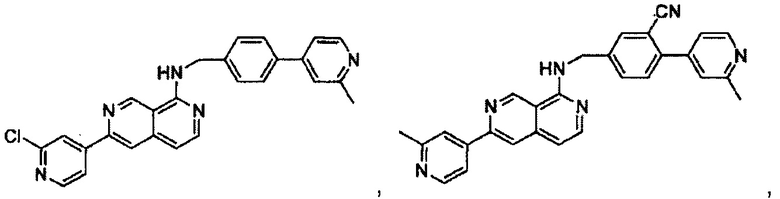
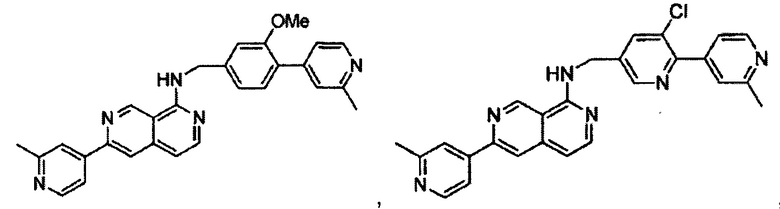
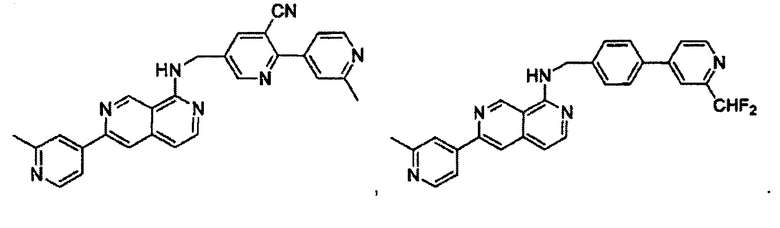
8. Способ ингибирования секреции WNT из клетки, включающий контакт клетки с эффективным количеством соединения по любому из пп.1-7.
9. Способ ингибирования секреции передачи сигналов WNT в клетке, включающий контакт клетки с эффективным количеством соединения по любому из пп.1-7.
10. Применение соединения по любому из пп.1-7 для производства лекарственного средства для лечения расстройства, опосредованного путем WNT.
11. Применение по п.10, отличающееся тем, что указанное расстройство представляет собой рак, фиброз, остеоартрит.
12. Применение по п.11, отличающееся тем, что рак выбран из: рака легких, включая мелкоклеточный и немелкоклеточный рак, рака молочной железы, предстательной железы, карциноидных опухолей, рака мочевого пузыря, рака желудка, поджелудочной железы, рака печени или гепатоцеллюлярного рака, гепатобластомы, рака толстой и прямой кишок, рака почек и плоскоклеточной карциномы головы и шеи, рака пищевода, яичников, шейки матки, эндометрия, мезотелиомы, меланомы, саркомы, остеосаркомы, липосаркомы, рака щитовидной железы, десмоидов, хронического миелоцитарного лейкоза (AML) и хронического миелоцитарного лейкоза (CML).
13. Применение по п.11, отличающееся тем, что фиброз выбран из системного склероза, фиброза кожи, идиопатического легочного фиброза, фиброза почек, фиброза печени, вызванного лекарствами фиброза и вызванного облучением фиброза.
14. Применение по любому из пп.12, 13, отличающееся тем, что терапевтически эффективное количество составляет от около 0,03 до около 2,5 мг/кг веса тела в суточных дозах.
15. Применение по п.14, отличающееся тем, что терапевтически эффективное количество составляет от около 0,5 мг до около 100 мг для людей.
16. Способ лечения расстройства, опосредованного путем WNT у субъекта, страдающего от него, включающий введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-7.
17. Способ по п.16, отличающийся тем, что указанное расстройство представляет собой рак, фиброз, остеоартрит.
18. Способ по п.17, отличающийся тем, что рак выбран из: рака легких, включая мелкоклеточный и немелкоклеточный рак, рака молочной железы, предстательной железы, карциноидных опухолей, рака мочевого пузыря, рака желудка, поджелудочной железы, рака печени или гепатоцеллюлярного рака, гепатобластомы, рака толстой и прямой кишок, рака почек и плоскоклеточной карциномы головы и шеи, рака пищевода, яичников, шейки матки, эндометрия, мезотелиомы, меланомы, саркомы, остеосаркомы, липосаркомы, рака щитовидной железы, десмоидов, хронического миелоцитарного лейкоза (AML) и хронического миелоцитарного лейкоза (CML).
19. Способ по п.17, отличающийся тем, что фиброз выбран из системного склероза, фиброза кожи, идиопатического легочного фиброза, фиброза почек, фиброза печени, вызванного лекарствами фиброза и вызванного облучением фиброза.
20. Способ по любому из пп.16-19, отличающийся тем, что терапевтически эффективное количество составляет от около 0,03 до около 2,5 мг/кг веса тела в суточных дозах.
21. Способ по п.20, отличающийся тем, что терапевтически эффективное количество составляет от около 0,5 мг до около 100 мг для людей.
22. Соединение по любому из пп.1-6, имеющее центральную структуру
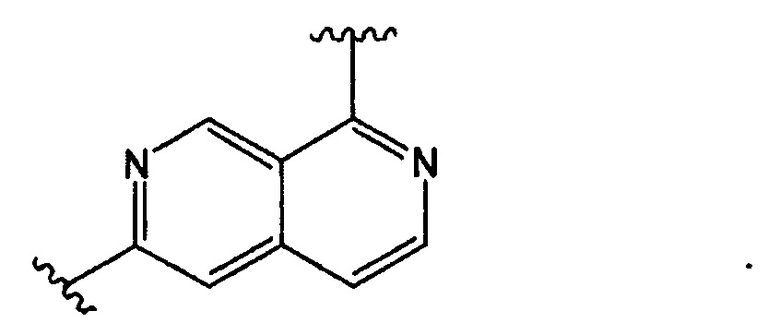
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| RU 2008129807 A, 20.02.2010. | |||

