ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к области фармакологии и медицины и касается способа синтеза фторклозапина и его производных, которые могут быть использованы для изготовления лекарственных средств для лечения психических заболеваний или психических расстройств.
УРОВЕНЬ ТЕХНИКИ
В настоящее время психическое здоровье принадлежит к числу наиболее серьезных проблем, стоящих перед всеми странами, поскольку в тот или иной период жизни такие проблемы возникают, по крайней мере, у каждого четвертого человека. Разработка новых антипсихотических препаратов является крайне важной задачей, стоящей перед современной медициной, поскольку единственный препарат, эффективно устраняющий позитивные и негативные симптомы шизофрении - клозапин - обладает также опасными побочными эффектами, ограничивающими его применение в медицинской практике.
Фторированные аналоги клозапина являются эффективными антипсихотическими препаратами, в которых сравнимая с клозапином высокая эффективность сочетается со относительной безопасностью применения. Получение фторпроизводных клозапина высокой чистоты важно, поскольку даже небольшие количества примесей могут существенно изменить фармакологический эффект и профиль безопасности субстанции.
Из уровня техники известен способ синтеза одного из производных фторклозапина (патент РФ №2441867, МПК C07B 403/04, ООО «Валентек», опубл. 10.02.2012), общая схема которого выглядит следующим образом:

Одной из стадий указанного способа является стадия получения соединения формулы (b). Для этого растворяют 89 г соединения формулы (а) в 1335 мл абсолютного этанола. К смеси прибавляют 112,2 мл концентрированного раствора соляной кислоты, присыпают 206,5 г хлорида олова и нагревают реакционную массу до кипения. Смесь кипятят 12 ч. Затем смесь охлаждают до комнатной температуры, упаривают растворитель на роторно-пленочном испарителе (РПИ). В кубовый остаток прибавляют 1 л воды и подщелачивают водным раствором NaOH до pH 9. Полученную суспензию экстрагируют этилацетатом три раза по 250 мл. Получают продукт в виде светло - желтого или светло - зеленого порошка. Выход 64,9 г, 81% от теоретического.
Однако полученный по известной методике конечный продукт является загрязненным, и содержит не менее 0.15% единичной примеси, предположительно дихлорпроизводного следующей формулы:

Появление примеси может быть отслежено на стадии восстановления хлоридом олова.
Хотя дихлорпроизводное по своей структуре близко к целевому фторхлорпроизводному, известно, что атом хлора по своей природе является более объемным заместителем, поэтому рецепторный профиль соединений может существенно отличаться. Кроме того связь С-Cl более подвержена гидролизу по сравнению со связью С-F, и образование реактивного метаболита дихлорпроизводного более вероятно. Поэтому наличие примеси дихлорпроизводного в конечном продукте крайне нежелательно.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Задачей изобретения является создание нового способа синтеза фторклозапина и его производных высокой степени чистоты, где указанные соединения в совокупности представлены общей формулой (1):

где цикл А представляет собой фенил, 5- или 6-членный гетероарил, содержащий 1 атом О, или 1 атом O и 1-2 атома Ν, или 1 атом S, или 1 атом S и 1-2 атома Ν, или 1-3 атома Ν, при этом цикл А опционально замещен 1-3 заместителями R1;
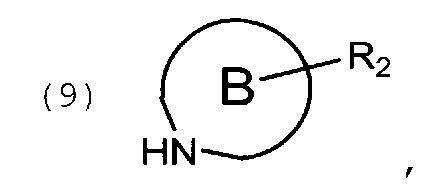
цикл В представляет собой 5-7 членный насыщенный или частично ненасыщенный гетероцикл, содержащий 1 атом О, или 1 атом О и 1-2 атома N, или 1 атом S, или 1 атом S и 1-2 атома N, или 1-3 атома N, при этом цикл В опционально замещен 1-3 заместителями R2; где
R1 представляет собой водород, C1-3 алкил, опционально замещенный 1-3 R3, O-C1-3-алкил, опционально замещенный 1-3 R3, С1-3-алкил-O-С1-3-алкил, опционально замещенный 1-3 R3 или галоген,
R2 представляет собой водород, галоген или C1-3-алкил, опционально замещенный 1-3 R3,
R3 представляет собой галоген или ОН.
Под упомянутом выше термином «галоген» следует понимать фтор, хлор, бром, иод.
Техническим результатом, достигаемым при использовании изобретения, является повышение степени чистоты фторклозапина и его производных, в совокупности представленных указанной выше общей формулой (1), за счет понижения содержания примесей.
Поставленная задача и требуемый технический результат достигаются за счет нового способа синтеза соединений указанной выше формулы, который включает:
взаимодействие 2,4-дифторо-5-нитро-1-хлорбензола (2) с соединением общей формулы (3)

в которой цикл А и R1 определены выше, с получением соединения общей формулы (4)
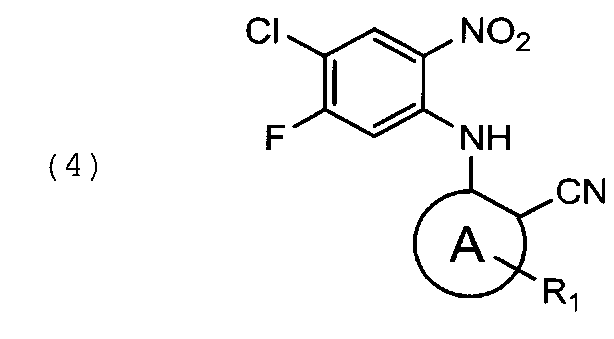
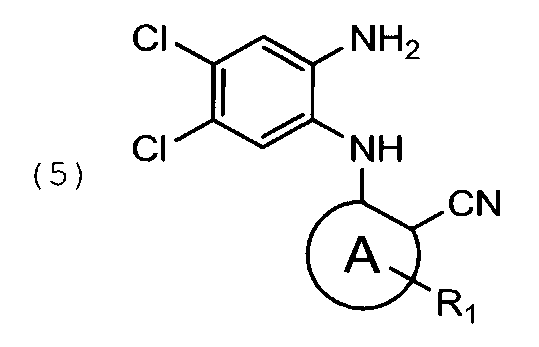
восстановление указанного соединения общей формулы (4) с помощью дитионита натрия в подходящем растворителе с получением соединения общей формулы (5)
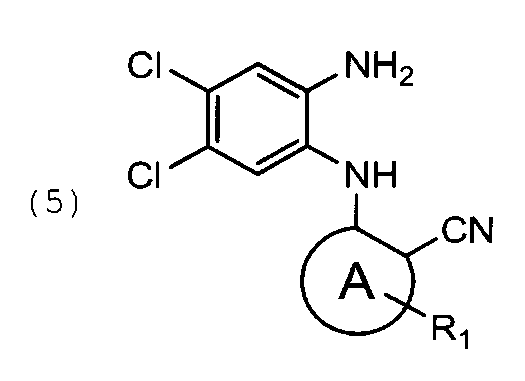
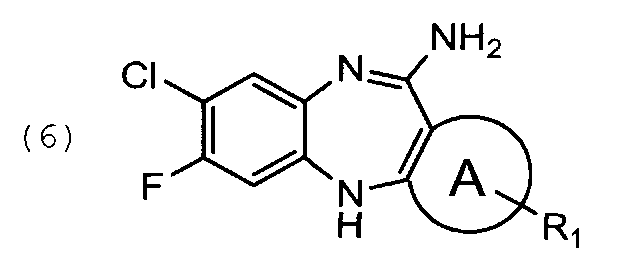
циклизацию указанного соединения общей формулы (5) с образованием соединения общей формулы (6)
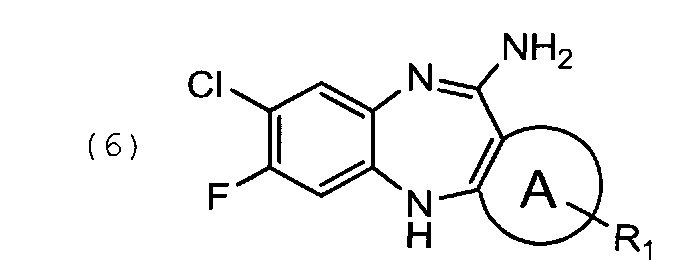
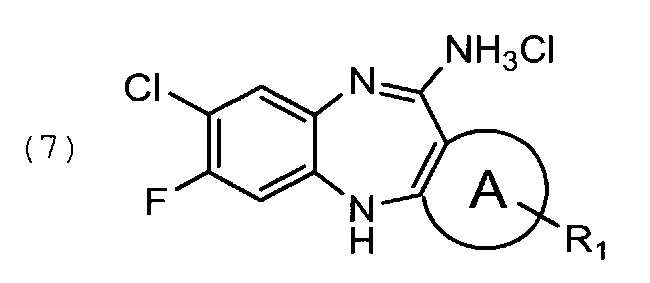
кристаллизацию из указанного соединения общей формулы (6) соединения общей формулы (7)
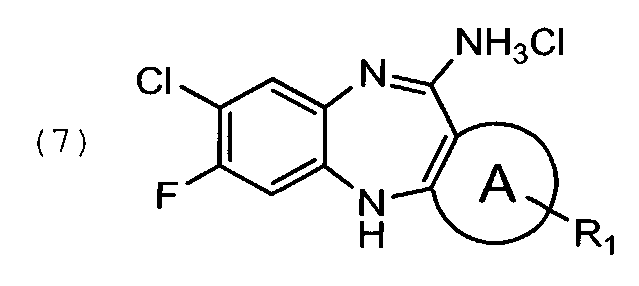
в подходящем растворителе;
взаимодействие указанного соединения общей формулы (7) с аминопроизводным соединением общей формулы (9)

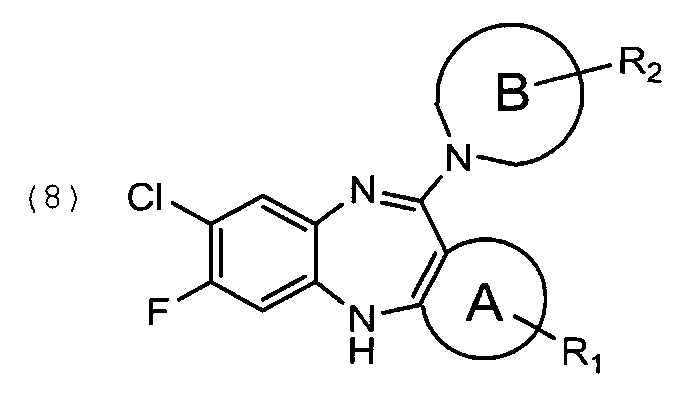
в которой цикл В и R2 определены выше, с получением соединения общей формулы (8)
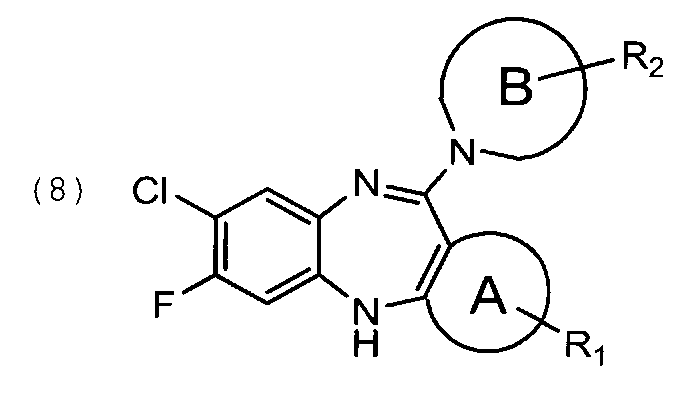
при этом взаимодействие соединения формулы (2) с соединением формулы (3) осуществляют в присутствии KF, нагревая реакционную смесь 5-6 суток при 140-180°С, в основных условиях в полярном апротонном растворителе, где в качестве основания используют NaH, KH, t-BuOK, LiOH или CsCO3, а в качестве растворителя используют диметилформамид, N-метилпирролидон, ДМСО или ТГФ, кроме того взаимодействие соединения формулы (7) с соединением формулы (9) осуществляют в апротонном растворителе, например, ДМСО или толуоле, стадию циклизацию осуществляют в кислых условиях, а на стадии кристаллизации в качестве растворителя используют диэтиловый эфир, метил-терт-бутиловый эфир, диоксан, и на стадии восстановления используют ТГФ, 2-метилтетрагидрофуран, циклопентилметиловый эфир.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
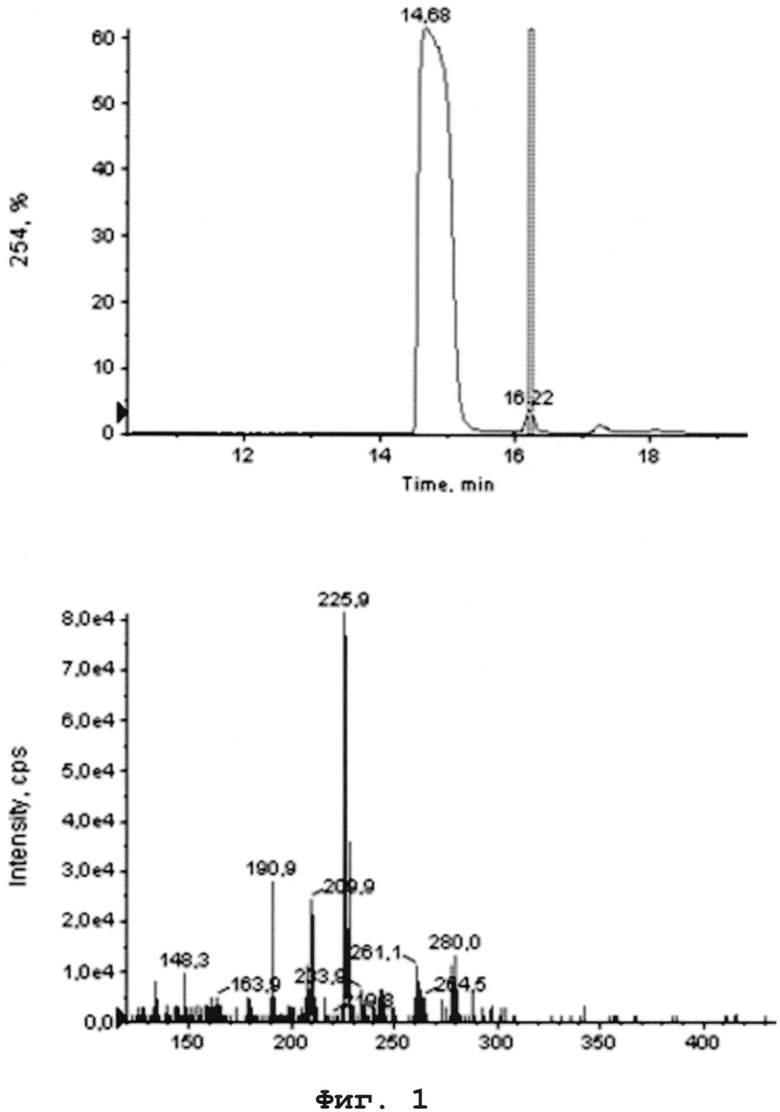
На Фиг. 1 - изображена ЖХ МС продукта восстановления-циклизации (соединения формулы (6′), полученного предложенным способом.
На Фиг. 2 - изображена ЖХ МС соединения А-1.
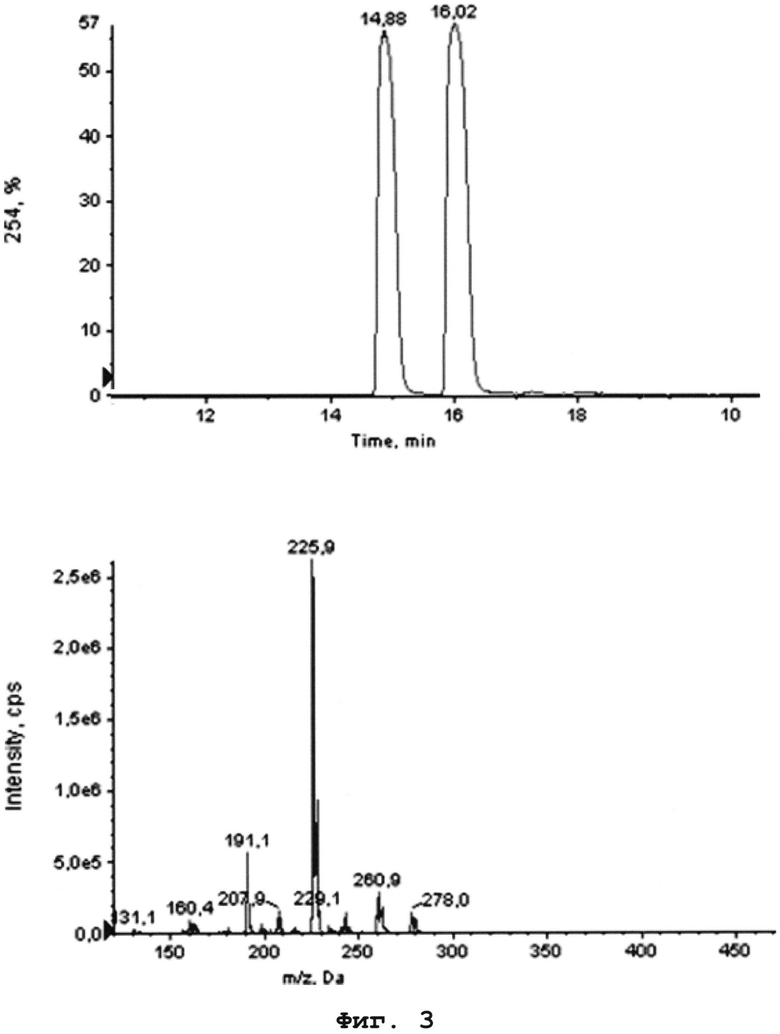
На Фиг. 3 - изображена ЖХ МС смеси интермедиата 6′ и примеси А-1.
На Фиг. 4 - изображена хроматограмма образца фторклозапина.
ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
Ниже с целью пояснения существенных отличий предложенного изобретения представлена общая схема синтеза соединений указанной выше формулы (1).

Соединение указанной выше формулы (4), может быть получено из 2,4-дифторо-5-нитро-1-хлорбензола 2 и соответствующего нитрила (3) в присутствии KF при нагревании реакционной смеси 5-6 суток при 140-180°С.
Альтернативно сочетание соединений формулы (2) и (3) может быть проведено в основных условиях в полярном апротонном растворителе. Подходящие основания включают NaH, КН, t-BuOK, LiOH, CsCO3. В качестве растворителей могут быть использованы диметилформамид, Ν-метилпирролидон, ДМСО, ТГФ. 2,4-дифторо-5-нитро-1-хлорбензол (2) может быть получен из 2,4-дифторо-5-нитро-1-хлорбензола по стандартной методике.
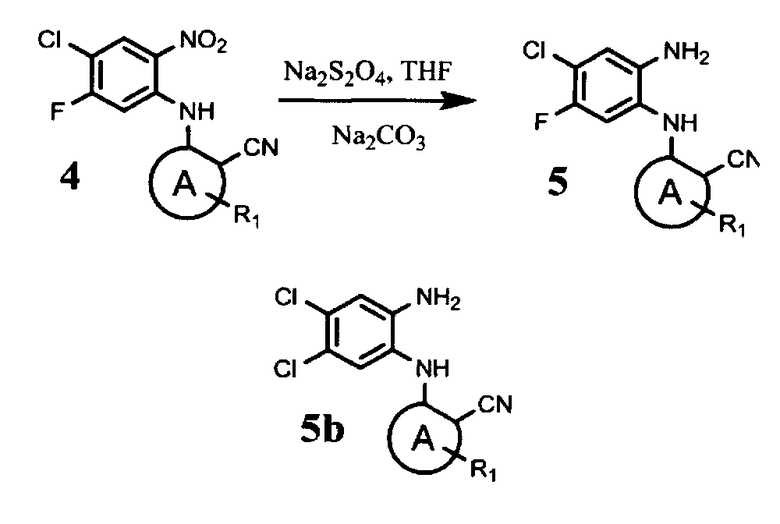
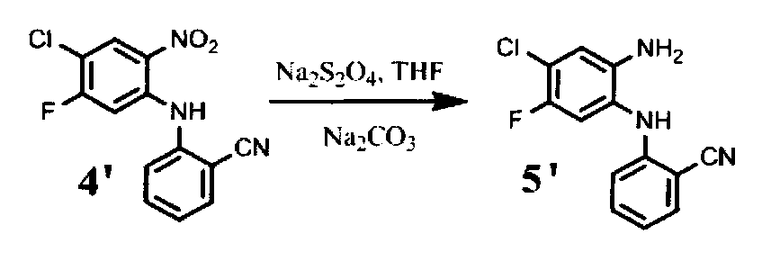
Соединение формулы (5) может быть получено из соединения формулы (4) восстановлением с помощью дитионита натрия, например, в ТГФ. Известный из уровня техники способ получения аналогов соединения формулы (5) включает восстановление нитрогруппы с помощью хлорида олова. Однако ионы хлора способны заместить атом фтора соединений формулы (4) и (5), что приводит к образованию нежелательного побочного продукта - соединения формулы (5b), которое очень сложно отделить от целевого соединения формулы (5). При использовании Na2S2O4 в качестве восстанавливающего агента образование побочного продукта формулы (5b) исключается, и реакция протекает с образованием только соединения формулы (5).

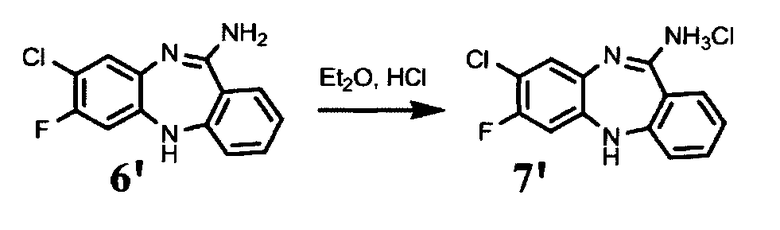
Циклизацию соединения формулы (5) с образованием соединения формулы (6) проводят в кислых условиях. Хлорид формулы (7) кристаллизуют из свободного основания формулы (6) в подходящем растворителе, например, дизтиловом эфире.
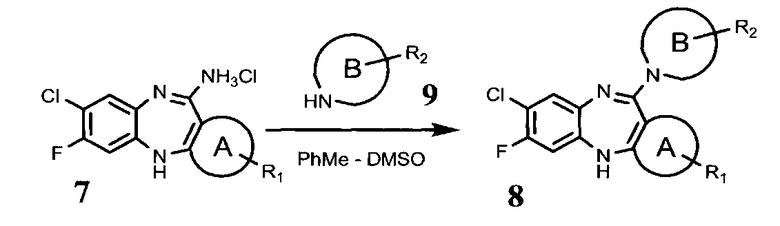
Соединение формулы (8) может быть получено из соединения формулы (7) и соответствующего аминопроизводного формулы (9). Реакцию проводят в апротонном растворителе, например, ДМСО или толуоле.
Ниже с целью иллюстрации отдельных аспектов осуществления изобретения приведены примеры осуществления предлагаемого способа, а именно получения фторклозапина (8′) и его производных (9′), (10) и (16). Приведенные ниже примеры не предназначены для того, чтобы каким-либо образом ограничивать объем настоящего изобретения. 

Пример. 1. Способ синтеза фторклозапина
1.1. 2,4-дифторо-5-нитро-1-хлоробензол, соединение формулы (2)
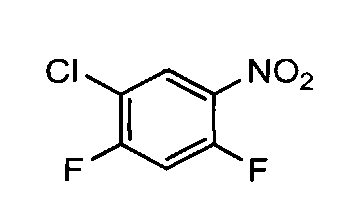
К охлажденной до 0°С азотной кислоте (1.5 л) прибавляют по каплям 0.5 л соединения формулы (2а). Смесь перемешивают 15 минут и выливают в 25 л воды, охлажденной до +5°С. Реакционную массу перемешивают 2 минуты и дают отстояться 1 час. Продукт формируется на дне в виде тяжелого масла. Водную фазу декантируют, экстрагируют 1 л CH2Cl2. Экстракт объединяют с продуктом, промывают 4 раза водой до pH 7. Органический слой сушат сульфатом натрия, растворитель удаляют на РПИ. Выход соединения формулы (2) 90-95%.

1.2. 2-(2-нитро-5-фторо-4-хлорофениламино)
бензонитрил, соединение общей формулы (4)

В 500 мл колбу, снабженную песчаной баней, термопарой и магнитной мешалкой, помещают 115 г соединения формулы (2), 34,5 г KF и 84,2 г соединения формулы (3′). Реакционную массу прогревают при перемешивании при 150-160°С 5-6 суток, контролируя протекание реакции по ЯМР. Реакцию считают завершенной, когда содержание исходного соединения формулы (2) не более 15% по отношению к продукту.
Реакционную смесь охлаждают до 60°С и прибавляют, при перемешивании, 500 мл спирта этилового ректификованного. Смесь охлаждают при перемешивании до 20°С, фильтруют, отжимают. Вещество промывают на фильтре тремя порциями этанола по 150 мл и двумя порциями воды по 200 мл. Выход 87 г, 50% от теоретического.
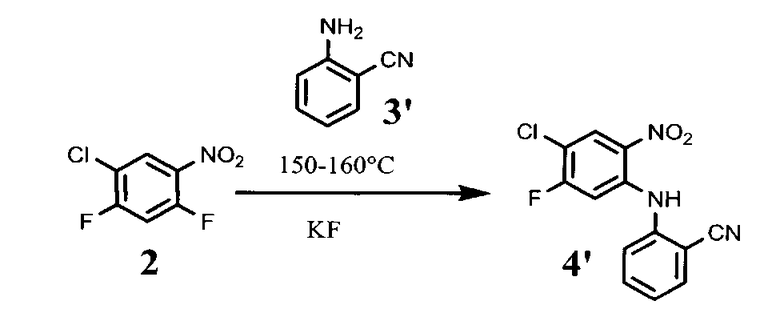
1.3. 2-(2-амино-5-фторо-4-хлорофениламино)
бензонитрил, соединение общей формулы (5)

48.5 г соединения формулы (4′) растворяют в 670 мл тетрагидрофурана. Затем прибавляют раствор 144,7 г дитионита натрия в 560 мл воды и 45 мл метанола. Реакционную массу выдерживают при перемешивании 1 час. После полного прохождения реакции в реакционную смесь прибавляют раствор 70,5 г бикарбоната натрия в 750 мл воды. Перемешивают 15 минут. Получают продукт в виде светло-серого порошка. Выход 60 г, 65% от теоретического.
1.4. 11-амино-7-фторо-8-хлоро-5Н-дибензо[b,e][1,4] диазепин, соединение общей формулы (6)
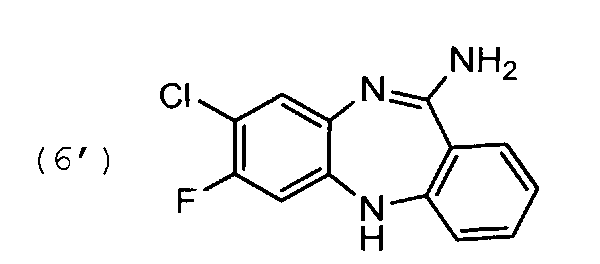
105 г соединения формулы (5′) растворяют в 1 л спирта этилового ректификованного. К смеси прибавляют 68 мл концентрированного водного раствора HCl. Реакционную массу нагревают до кипения при перемешивании и продолжают кипячение 4-5 часов. Затем смесь охлаждают до комнатной температуры и удаляют растворитель на РПИ. В кубовый остаток прибавляют 800 мл ацетонитрила. Смесь перемешивают 15 мин. Образовавшиеся кристаллы отфильтровывают, отжимают и промывают на фильтре 200 мл ацетонитрила. Выход 85 г, 81% от теоретического.

1.5. 11-амино-7-фторо-8-хлоро-5Н-дибензо[b,е][1,4]диазепина гидрохлорид, соединение общей формулы (7)

К суспензии 64,9 г соединения формулы (6′) в 45С мл диэтилового эфира прибавляют 124 мл 3М HCl в диоксане. Реакционную массу перемешивают 30 мин. Образовавшиеся кристаллы отфильтровывают, отжимают и промывают на фильтре двумя порциями по 150 мл диэтилового эфира. Выход 73 г, 98% от теоретического.
1.6. 11-(4-метилпиперазин-1-ил)-7-фторо-8-хлоро-5Н-дибензо[b,е][1,4]диазепин, соединение общей формулы (8)

В 2 л плоскодонной колбе, снабженной магнитной мешалкой и обратным холодильником, растворяют 98 г соединения формулы (7′) в смеси толуол:ДМСО=490:490 мл, прибавляют 291,7 мл метилпиперазина. Прибор продувают аргоном, убирают подачу инертного газа с баллона, (оставляя шар заполненный аргоном) и кипятят 36 ч. Выливают реакционную массу в воду и трижды экстрагируют этилацетатом порциями по 150 мл. Объединенный органический слой промывают тремя порциями воды по 150 мл. Сушат над сульфатом натрия и отгоняют растворитель на РПИ.
В кубовый остаток прибавляют 500 мл хлороформа, перемешивают на МПУ 10 мин. Выпавшие кристаллы отфильтровывают, отжимают на фильтре и промывают 100 мл метанола на фильтре. Продукт формулы (8′) чистят перекристаллизацией из метанола с активированные углем, рассчитывая объем растворителя на 1 г продукта - 15 мл метанола и 10% массы соединения формулы (8′) активированного угля. Выход 59 г 61%.

Пример 2. Синтез 11-(пиперазин-1-ил)-7-фторо-8-хлоро-5Н-дибензо[b,е][1,4]диазепина
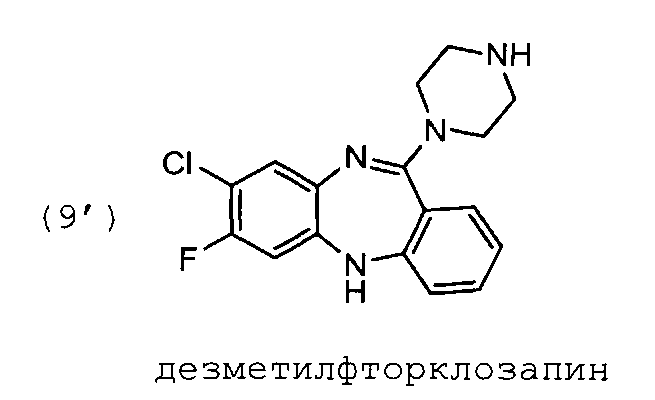
В 2 л плоскодонной колбе, снабженной магнитной мешалкой и обратным холодильником, растворяют 63 г соединения формулы (7′) в смеси толуол:ДМСО=315:315 мл, прибавляют 145,6 г пиперазина. Прибор продувают аргоном, убирают подачу инертного газа с баллона, (оставляя шар заполненный аргоном) и кипятят реакционную массу 36 ч. ТСХ-контроль (дихлорметан:метанол =20:1).
Смесь охлаждают до комнатной температуры, и выливают в 2 л воды. Экстрагируют тремя порциями по 200 мл этилацетата. Объединенные органические слои промывают тремя порциями воды по 100 мл. Затем органический слой сушат над сульфатом натрия.
Органическую фазу отфильтровывают через слой силикагеля 10 мм и промывают на фильтре 2 литрами этилацетата. Маточный раствор экстрагируют 4 Μ раствором соляной кислоты в воде. После этого водную фазу промывают тремя порциями этилацетата по 150 мл. Затем, при охлаждении, подщелачивают водную фазу предварительно охлажденным 4 Μ раствором NaOH до pH 10. Выпавшие кристаллы отфильтровывают, отжимают и промывают на фильтре тремя порциями по 150 мл воды.
Полученный продукт формулы (8") перекристаллизовывают из толуола (этанола, метилэтилкетона), рассчитывая объем растворителя на 1 г продукта 3 мл растворителя. Выход 45 г, 50% от теоретического.
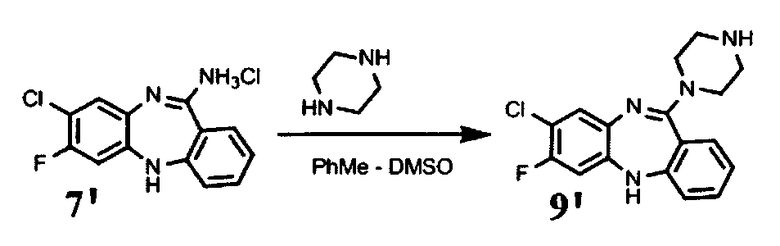
Пример 3. Синтез 11-(4-(2-(2-гидроксиэтокси)этил) пиперазин-1-ил)-7-фторо-8-хлоро-5Н-дибензо[b,е][1,4]диазепина
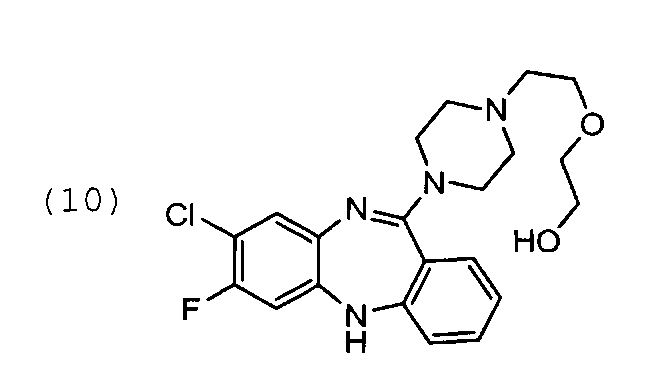
В 2 л плоскодонной колбе, снабженной магнитной мешалкой и обратным холодильником, растворяют 75 г соединения формулы (7′) в смеси толуол:ДМСО=375:375 мл, прибавляют 413 мл 1-[2-(2-гидроксизтокси) этил]пиперазина. Прибор продувают аргоном, убирают подачу инертного газа с баллона, (оставляя шар заполненный аргоном) и кипятят 36 ч. Выход 48 г 61%.

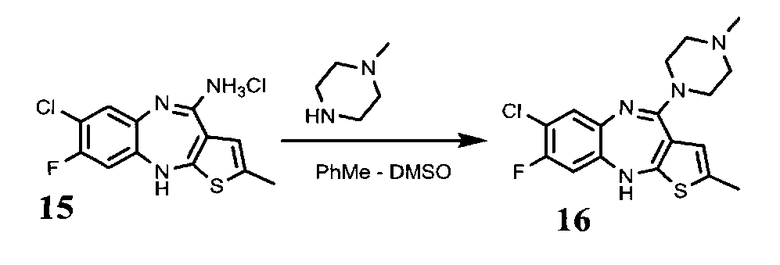
Пример 4. 2-метил-4-(4-метилпиперазин-1-ил)-8-фторо-7-хлоро-10Н-бензо[b]тиено[2,3-е][1,4]диазепин, соединение формулы (16)

4.1. 2-амино-5-метилтиофена-3-карбонитрил, соединение формулы (11)
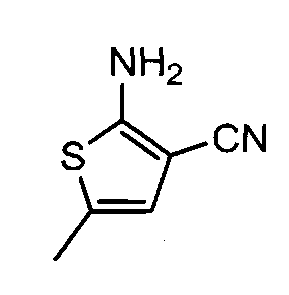
Сера (S8, 0,9 г), пропионовый альдегид (2 мл) и диметилформамид (6 мл) переносятся в трехгорлую круглодонную колбу, снабженную капельной воронкой и холодильником. Смесь охлаждается до 0°С, и по каплям добавляется триэтиламин (2,3 мл). Образующийся темный раствор нагревается до комнатной температуры в течение 1 часа. Раствор малононитрила (1,71 мл) в ДМФ (3,2 мл) переносится в воронку и добавляется по каплям. Образующаяся коричневая жидкость перемешивается в течение ночи при комнатной температуре.
Выливают реакционную массу в 80 мл воды и льда, образовавшийся оранжевый осадок отфильтровывают, промывают холодной водой и высушивают в вакууме. Выход 78%.
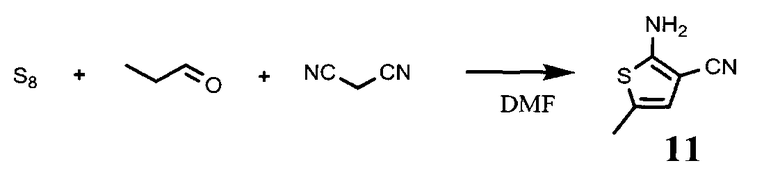
4.2. 5-метил-2-(2-нитро-5-фторо-4-хлорофенил-амино)тиофена-3-карбонитрил, соединение формулы (12)

К 0.26 г NaH (полученного из 55% суспензии в масле промыванием в гексане) добавляют 1 мл безводного ТГФ. 0,5 г соединения формулы (11) и 0,7 г соединения формулы (2) растворяются в 1,5 мл безводного ТГФ и добавляются к суспензии NaH по каплям, поддерживая температуру ниже 30°С. Реакционная смесь перемешивается в течение ночи в атмосфере азота.
Смесь выливают в 11 мл смеси воды со льдом, нейтрализуют концентрированной HCl, и экстрагируют 36 мл дихлорометана. Органический слой сушат над сульфатом магния и выпаривают досуха. Остаток очищается хроматографически на силикагеле (элюент этилацетат:гексан 1:9). Выход 0,68 г, 60%.
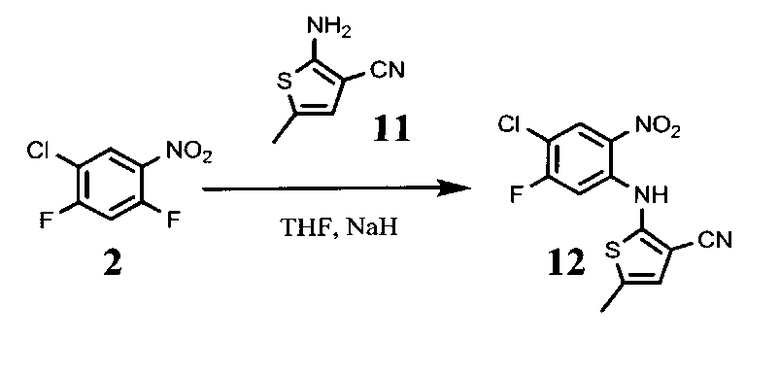
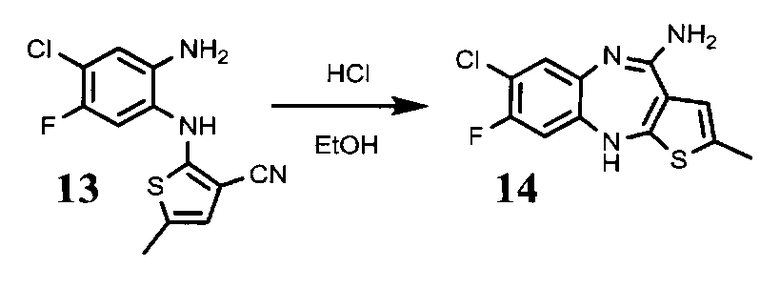
4.3. 2-(2-амино-5-фторо-4-хлорофениламино)-5-метилтиофена-3-карбонитрил, соединение формулы (13)

0,52 г соединения формулы (12) растворяют в 6,7 мл тетрагидрофурана. Затем прибавляют раствор 1,45 г дитионита натрия в 5,6 мл воды и 0,4 5 мл метанола. Реакционную массу выдерживают при перемешивании 1 час. После полного прохождения реакции в реакционную смесь прибавляют раствор 0,70 г бикарбоната натрия в 7,5 мл воды. Перемешивают 15 минут. Получают продукт в виде светло-серого порошка. Выход 0.25 г, 53% от теоретического.

4.4. 4-амино-2-метил-8-фторо-7-хлоро-10Н-бензо[b]тиено[2,3-е][1,4]диазепин, соединение формулы (14)
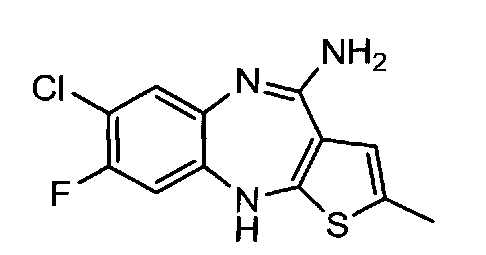
1,13 г соединения формулы (13) растворяют в 10 мл 96% этанола. К смеси прибавляют 0,68 мл концентрированного водного раствора HCl. Реакционную массу нагревают до кипения при перемешивании и продолжают кипячение 4-5 часов. Затем смесь охлаждают до комнатной температуры и удаляют растворитель на РПИ. В кубовый остаток прибавляют 8 мл ацетонитрила. Смесь перемешивают 15 мин. Образовавшиеся кристаллы отфильтровывают, отжимают и промывают на фильтре 2 мл ацетонитрила. Выход 1 г, 89% от теоретического.
4.5. 4-амино-2-метил-8-фторо-7-хлоро-10Н-бензо[b]тиено[2,3-е][1,4]диазепина гидрохлорид, соединение формулы (15)

К суспензии 0,7 г соединения 14 в 4,5 мл дизтилового эфира прибавляют 1,24 мл 3М HCl в диоксане. Реакционную массу перемешивают 30 мин. Образовавшиеся кристаллы отфильтровывают, отжимают и промывают на фильтре двумя порциями по 1,5 мл диэтилового эфира. Выход 0,78 г, 98% от теоретического.

4.6. 2-метил-4-(4-метилпиперазин-1-ил)-8-фторо-7-хлоро-10Н-бензо[b]тиено[2,3-е][1,4]диазепин, соединение формулы (16)
1,2 соединения 15 растворяют в смеси толуол:ДМСО=4,9:4,9 мл, прибавляют 2,9 мл метилпиперазина. Смесь кипятят в атмосфере инертного газа в течение 36 ч. Выливают реакционную массу в воду и трижды экстрагируют этилацетатом порциями по 1,5 мл. Объединенный органический слой промывают тремя порциями воды по 1,5 мл. Сушат над сульфатом натрия и отгоняют растворитель на РПИ. Выход 1,1 г, 77% от теоретического.
Ниже приведены данные, подтверждающие повышения чистоты продукта, полученного предложенным способом.
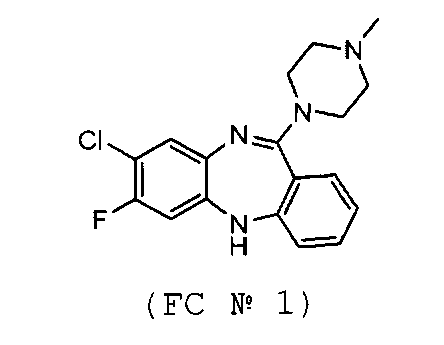
Образец FC №1 был синтезирован по стандартной методике с использованием SnCl2. Анализ образца №1 показал, что содержание единичной примеси не превышает 0,332%, а суммарное содержание примесей -1.248%. Исследование интермедиатов показало, что образование примесей происходит на стадии восстановления SnCl2.
На Фиг. 1 приведена ЖХ МС продукта восстановления-циклизации (соединения формулы (6′)), полученного предложенным способом. Было сделано предположение, что основная примесь, образующаяся на стадии восстановления представляет собой производное дихлордибензодиазепина А-1.

Гипотеза была подтверждена сравнением ЖХ МС спектра продукта восстановления и дихлордибензодиазепина. На Фиг. 2 изображена ЖХ МС соединения А-1. На Фиг. 3 приведена ЖХ МС смеси интермедиата 6′ и примеси А-1.

Образец фторклозапина FC №2 был синтезирован согласно предложенному способу, в котором восстановление проводилось с помощью дитионита натрия. На Фиг. 4 изображена хроматограмма указанного соединения. Площадь пика на Фиг. 4 соответствует количеству примеси. Видно, что содержание основного вещества (пик №8)>99,85%, суммарное содержание примесей в новом образце 0,15%, максимальное содержание единичной примеси 0,047% (пик №6). Таким образом, сравнивая данные для образца FC №1, полученного по прототипу, и данные для образца FC №2, полученного согласно изобретению, видно, что степень чистоты фторклозапина FC №2, полученного способом согласно изобретению, выше, что подтверждает достижение поставленного технического результата.
Предложенное изобретение не ограничено описанными выше вариантами осуществления, а наоборот оно охватывает различные модификации и варианты в рамках сущности и объема предлагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛИ ТРИАЗОЛИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PAR1, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2009 |
|
RU2494100C2 |
| ПРОИЗВОДНЫЕ 1-ПИПЕРАЗИН- И 1-ГОМОПИПЕРАЗИНКАРБОКСИЛАТОВ, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2356890C2 |
| [1,4] ДИАЗЕПИНО [6,7,1-IJ] ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ АНТИПСИХОТИЧЕСКИХ СРЕДСТВ И СРЕДСТВ ПРОТИВ ОЖИРЕНИЯ | 2003 |
|
RU2312866C2 |
| ПРОИЗВОДНЫЕ 1,5-БЕНЗОДИАЗЕПИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЙ ИХ ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1994 |
|
RU2135486C1 |
| ПРОИЗВОДНЫЕ ДИГИДРОБЕНЗО[b][1,4]ДИАЗЕПИН-2-ОНА В КАЧЕСТВЕ АНТАГОНИСТОВ I mGluR2 | 2002 |
|
RU2270197C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3-ХИНОЛОНКАРБОНОВОЙ КИСЛОТЫ | 1994 |
|
RU2138483C1 |
| Способ получения замещенных -6-арил4н- -триазоло/3,4-с/тиено /2,3-с/-1,4диазепинов или их солей | 1976 |
|
SU622406A3 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ ИМИДАЗОЛА И ИХ НОВЫЕ КРИСТАЛЛИЧЕСКИЕ СТРУКТУРЫ | 2002 |
|
RU2286342C2 |
| ЗАМЕЩЕННЫЕ АЦИЛАМИНОБЕНЗАМИДЫ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2034829C1 |
| Способ получения производных 1,4-диазепина | 1975 |
|
SU648104A3 |


Изобретение способу синтеза фторклозапина и его производных формулы (1), которые могут быть использованы для изготовления лекарственных средств для лечения психических заболеваний или психических расстройств. Способ включает взаимодействие 2,4-дифторо-5-нитро-1-хлорбензола (2) с соединением формулы (3) с получением соединения формулы (4), восстановление соединения формулы (4) с помощью дитионита натрия в подходящем растворителе с получением соединения формулы (5), циклизацию соединения формулы (5) с образованием соединения формулы (6), кристаллизацию из соединения формулы (6) соединения формулы (7), взаимодействие указанного соединения формулы (7) с аминопроизводным соединением формулы (9). Новый способ синтеза позволяет получить соединения общей формулы (1), обладающие повышенной степенью чистоты. 9 з.п. ф-лы, 4 ил., 4 пр.



1.Способ получения соединения формулы (1):
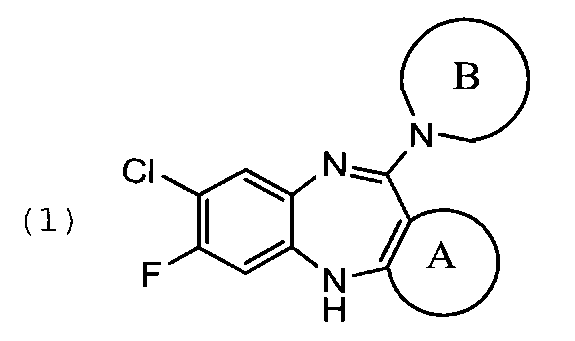
где цикл А представляет собой фенил, 5- или 6-членный гетероарил, содержащий 1 атом О, или 1 атом O и 1-2 атома N, или 1 атом S, или 1 атом S и 1-2 атома N, или 1-3 атома N, при этом цикл А опционально замещен 1-3 заместителями R1;
цикл В представляет собой 5-7-членный насыщенный или частично ненасыщенный гетероцикл, содержащий 1 атом О, или 1 атом О и 1-2 атома N, или 1 атом S, или 1 атом S и 1-2 атома N, или 1-3 атома N, при этом цикл В опционально замещен 1-3 заместителями R2; где
R1 представляет собой водород, C1-3 алкил, опционально замещенный 1-3 R3, O-С1-3-алкил, опционально замещенный 1-3 R3, С1-3-алкил-O-С1-3-алкил, опционально замещенный 1-3 R3, или галоген,
R2 представляет собой водород, галоген или С1-3-алкил, опционально замещенный 1-3 R3,
R3 представляет собой галоген или ОН,
который включает:
взаимодействие 2,4-дифторо-5-нитро-1-хлорбензола (2) с соединением формулы (3)
в которой цикл А и R1 определены выше, с получением соединения формулы (4)
восстановление указанного соединения формулы (4) с помощью дитионита натрия в подходящем растворителе с получением соединения формулы (5)
циклизацию указанного соединения формулы (5) с образованием соединения формулы (6)
кристаллизацию из указанного соединения формулы (6) соединения формулы (7)
в подходящем растворителе;
взаимодействие указанного соединения формулы (7) с аминопроизводным соединением формулы (9)
в которой цикл В и R2 определены выше, с получением соединения формулы (8)
2. Способ по п. 1, в котором взаимодействие соединения формулы (2) с соединением формулы (3) осуществляют в присутствии KF, нагревая реакционную смесь 5-6 суток при 140-180°С.
3. Способ по п. 1, в котором взаимодействие соединения формулы (2) с соединением формулы (3) осуществляют в основных условиях в полярном апротонном растворителе.
4. Способ по п. 3, в котором в качестве основания используют NaH, КН, t-BuOK, LiOH или CsCO3.
5. Способ по п. 3, в котором в качестве растворителя используют диметилформамид, Ν-метилпирролидон, ДМСО или ТГФ.
6. Способ по п. 1, в котором взаимодействие соединения формулы (7) с соединением формулы (9) осуществляют в апротонном растворителе.
7. Способ по п. 6, в котором в качестве растворителя используют ДМСО или толуол.
8. Способ по п. 1, в котором циклизацию осуществляют в кислых условиях.
9. Способ по п. 1, в котором на стадии кристаллизации в качестве растворителя используют диэтиловый эфир, метил-терт-бутиловый эфир, диоксан.
10. Способ по п. 1, в котором в качестве растворителя на стадии восстановления используют ТГФ, 2-метилтетрагидрофуран, циклопентилметиловый эфир.
| ПРОИЗВОДНЫЕ 5H-ДИБЕНЗО[b, e][1, 4]ДИАЗЕПИНА И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2441867C2 |
| WO 1991010661 A1, 25.07.1991 | |||
| WO 2006088541 A2, 24.08.2006 | |||

