ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к лекарственной дозированной форме для предпочтительно орального введения один раз в сутки, которая содержит фармакологически активный агент в соответствии с общей формулой (I)
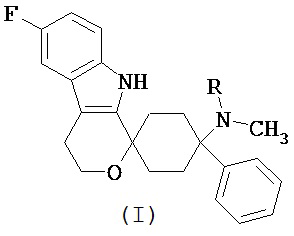
где R представляет собой -H или -CH3, или его физиологически приемлемую соль, для применения для лечения невропатической боли, предпочтительно хронической невропатической боли.
Фармакологически активные агенты в соответствии с общей формулой (I) также могут обозначаться как 6’-фтор-(N-метил- или N,N-диметил-)-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин. Если специально не указано иначе, этот термин также включает физиологически приемлемые соли.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Фармакологически активные агенты в соответствии с изобретением известны из уровня техники и могут вводиться орально, перорально, парентерально, внутривенно, внутрибрюшинно, внутрикожно, внутримышечно, интратекально, эпидурально, интраназально, буккально, ректально или местно, например, на кожу, слизистые оболочки или в глаза. Соединения проявляют анальгезирующие свойства и чрезвычайно пригодны для лечения острой, висцеральной, невропатической или хронической боли (ср., например, WO 2004/043967 и WO 2008/040481).
Общепринятые болеутоляющие средства обычно доступны в виде препаратов, обеспечивающих немедленное высвобождение или в виде препаратов, обеспечивающих пролонгированное высвобождение.
С одной стороны, препараты, обеспечивающие немедленное высвобождение при оральном введении, обладают тем преимуществом, что они приводят к быстрому высвобождению обезболивающего средства в желудочно-кишечный тракт. В результате этого, сравнительно высокая доза обезболивающего средства быстро абсорбируется, что приводит к высоким уровням в плазме в течение короткого периода времени и обеспечивает быстрое начало облегчения боли, то есть обезболивающее действие начинается вскоре после введения. Это чрезвычайно желательно при острой боли.
Тем не менее, наряду с этим, обычно наблюдается быстрое уменьшение обезболивающего действия, поскольку метаболизация и/или распределение и/или экскреция обезболивающего средства вызывает снижение его уровней в плазме крови. По этой причине, препараты, обеспечивающие немедленное высвобождение болеутоляющих средств, обычно необходимо вводить часто, например, восемь раз в сутки. Это не только неблагоприятно по отношению к соблюдению больным режима и схемы лечения, но также может вызывать относительно высокие пики концентрации лекарственного средства в плазме крови и высокие флуктуации между пиком и минимумом концентрации лекарственного средства в плазме крови, что, в свою очередь, может ухудшать переносимость.
С другой стороны, препараты, обеспечивающие пролонгированное высвобождение при оральном введении, обладают тем преимуществом, что их необходимо вводить менее часто, обычно один раз в сутки или два раза в сутки. Это улучшает соблюдение больным режима и схемы лечения и также может уменьшать пик концентрации лекарственного средства в плазме крови и флуктуации между пиком и минимумом концентрации лекарственного средства в плазме крови, что, в свою очередь, может улучшать переносимость.
Тем не менее, наряду с этим, высвобождение обезболивающего средства в желудочно-кишечном тракте пролонгируется. В результате этого, относительно низкая доза обезболивающего средства быстро абсорбируется, что приводит к низким уровням в плазме и обуславливает замедленное начало облегчения боли, то есть обезболивающее действие начинается довольно долго после первого введения.
Кроме того, поскольку препараты, обеспечивающие пролонгированное высвобождение обычно содержат более высокие дозы болеутоляющих средств, по сравнению с препаратами, обеспечивающими немедленное высвобождение, они могут быть связаны с более высоким риском неправильного использования. Пожилые пациенты особенно часто имеют трудности с приемом твердых лекарственных дозированных форм. Кроме того, для наиболее пожилых пациентов необходимы адаптации дозировок вследствие различных ADME (absorption, distribution, metabolism, excretion) характеристик с возрастом, что является другой причиной потребности в распадающихся таблетках. Для решения этой проблемы, были разработаны различные устройства, с помощью которых твердые лекарственные дозированные формы можно измельчить /или пульвелизировать (“дробилки таблеток”). Такие устройства используются, например, обслуживающим персоналом в домах престарелых. Лекарственные дозированные формы после этого вводятся людям, нуждающимся в лечении, уже не в виде таблеток и т.д., но предпочтительно в виде порошка, например, для устранения сложностей, связанных с проглатыванием таблеток. Тем не менее, измельчение лекарственных дозированных форм с помощью таких устройств является проблемным, если лекарственные дозированные формы представляют собой препараты с пролонгированным высвобождением. Как правило, измельчение впоследствии приводит к разрушению внутренней структуры лекарственной дозированной формы, которая отвечает за пролонгированное высвобождение, таким образом аннулируя действие с пролонгированным высвобождением. Следовательно, после введения, зачастую все физиологически активное вещество, которое исходно присутствует в лекарственной дозированной форме, высвобождается в течение относительно короткого периода времени, тем самым относительно очень высокая концентрация в плазме вещества неожиданно достигается в течение относительно короткого периода (дозовая разгрузка). Таким образом, препараты с исходным пролонгированным высвобождением становятся препаратами с быстрым высвобождением. Однако в зависимости от физиологического действия вещества это может вызывать значительные побочные действия, и в чрезвычайных случаях может даже приводить к смерти пациента (ср., например, J. Е. Mitchell, Oral Pharmaceutical dosage forms That Should Not Be Crushed: 2000 Update, Hospital Pharmacy, 2000; H. Miller и др., То Crush or Not - Crush, Nursing 2000; R. Griffith et al., Tablet Crushing and the law: the implications for nursing; Prof. Nurse 2003). Преднамеренное жевание препаратов с пролонгированным высвобождением также может приводить к передозировке вещества, которое в них содержится. Иногда пациенты жуют лекарственные дозированные формы умышленно, хотя часто не зная тип и назначение препарата с пролонгированным высвобождением, поскольку они надеются получить более быстрый эффект.
Также известны препараты, обеспечивающие двойной режим высвобождения, то есть комбинацию немедленного высвобождения с пролонгированным высвобождением (ср., например, СМ. Lopez и др., Compressed Matrix Core Tablet as a Quick/Slow Dual-Component Delivery System Containing Ibuprofen, AAPS PharmSciTech 2007; 8(3), E1-E8). Тем не менее, эти препараты обычно рассчитаны на единицы с быстрым высвобождением и единицы с пролонгированным высвобождением, которые местно разделены друг от друга и, следовательно, такие лекарственные дозированные формы могут быть приготовлены с помощью специфических и дорогостоящих методов.
Лечение хронической боли включает долговременное анальгетическое лечение, для которого часто необходимы более высокие дозы по сравнению с теми, которых достаточно для эпизодов острой боли. Для поддержания побочных явлений на переносимом уровне, может потребоваться титрование дозы обезболивающего средства перед началом лечения, в особенности в случае применения общепринятых μ-опиоидных болеутоляющих средств, таких как морфин. Таким образом, титрованную долговременную терапию опиоидами обычно начинают с субтерапевтических доз, которые постепенно повышают до тех пор, пока не достигается достаточная аналгезия.
Задачей настоящего изобретения является обеспечение лекарственных дозированных форм, содержащих 6’-фтор-N-метил- или N,N-диметил-)-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин, которые обладают преимуществами по сравнению с лекарственными дозированными формами, известными из уровня техники. В особенности, лекарственные дозированные формы будут обеспечивать хорошую биодоступность и достаточность облегчения боли, но также будут иметь высокую переносимость, хорошее соблюдение пациентами инструкций по приему препарата, и безопасность.
Эта задача решается с помощью объектов формулы изобретения.
Было обнаружено, что 6’-фтор-(N-метил- или N,N-диметил-)-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин имеет относительно плохую растворимость в воде. Кроме того, было обнаружено, что несмотря на указанную плохую растворимость в воде, могут быть приготовлены лекарственные дозированные формы, которые обеспечивают немедленное высвобождение 6’-Фтор-(N-метил- или N,N-диметил-)-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амина и обеспечивают хорошую биодоступность. Более того, неожиданно было обнаружено, что 6’-фтор-(N-метил- или N,N-диметил-)-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин имеет относительно большой фармакокинетический конечный период полувыведения (t1/2≈60-90 ч) и следовательно, обеспечивает фармакологическую активность в течение сравнительно продолжительного периода времени после введения (операционный период полувыведения составляет приблизительно 24 ч). Для более подробного понимания терминов конечный период полувыведения и операционный период полувыведения можно привести ссылку, например, на S. Sahin и др., Pharm. Res., 2008, 25(12), 2869-2877.
Таким образом, неожиданно было обнаружено, что при предпочтительно оральном введении лекарственной дозированной формы, содержащей фармакологически активный агент в соответствии с изобретением, может быть достигнуто быстрое начало облегчения боли с последующим пролонгированным обезболивающим эффектом, несмотря на то, или даже если, лекарственная дозированная форма обеспечивает немедленное высвобождение. Следовательно, лекарственная дозированная форма в соответствии с изобретением объединяет благоприятные свойства общепринятых препаратов, обеспечивающих немедленное высвобождение - быстрое облегчение боли благодаря достаточно высокой концентрации активного компонента только вскоре, например, приблизительно один час, после введения фармацевтической композиции - с благоприятными свойствами общепринятых препаратов, обеспечивающих пролонгированное высвобождение - продолжительное обезболивающее действие благодаря достаточно высокому уровню активного компонента в течение пролонгированного времени - и в то же время даже преодолевая недостатки указанных общепринятых препаратов. Принимая фармакологически активный агент в препарате в соответствии с изобретением, пациент может эффективно бороться со своей болью остро и, в то же время, лечить ее эффективно в течение пролонгированного периода без дополнительных измерений и только путем регулярного введения через 24 часовые интервалы.
Является чрезвычайно неожиданным, что лекарственная дозированная форма в соответствии с изобретением не только предоставляет возможность фармакологически активному агенту начинать поступать быстро в плазму при первом введении лекарственной дозированной формы, что приводит к быстрому началу облегчения боли у пациента благодаря немедленному высвобождению, но в то же время обеспечивает продолжительную терапевтическую эффективность в течение относительно длительного периода (по меньшей мере 24 часов). Следовательно, боль, от которой страдает пациент, может быть быстро облегчена при введении лекарственной дозированной формы в соответствии с изобретением без быстрого повторного снижения обезболивающего действия.
Кроме того, неожиданно было обнаружено, что вследствие его большого фармакокинетического периода полувыведения, наибольшие концентрации в плазме (пики концентрации в плазме) 6’-Фтор-(N-метил- или N,N-диметил-)-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амина повышаются после введения один раз в сутки фиксированных доз. Максимальная концентрация (Cmax) неожиданно наблюдается позже, а именно через приблизительно 4-6 часов после введения. Неожиданно было обнаружено, что один раз в сутки введение фиксированных, сравнительно небольших доз 6’-фтор-(N-метил- или N,N-диметил-)-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амина, например, суточных доз 40 мкг, приводит к субтерапевтическим пикам концентрации в плазме в первый день терапии, но к терапевтическим пикам концентрации в плазме в дальнейшем. Например, в случае суточной дозы 40 мкг, существенный обезболивающий эффект наблюдали у пациентов с болезненной диабетической нейропатией на четвертый и пятый день при введении один раз в сутки.
Кроме того, неожиданно было обнаружено, что в таких сравнительно небольших дозах, возникновение побочных эффектов по сравнению с морфином значительно снижается при эквианальгезирующих условиях.
Эти неожиданные эффекты свидетельствуют о том, что в течение долговременной терапии невропатической боли, предпочтительно хронической невропатической боли, доза 6’-Фтор-(N-метил- или N,N-диметил-)-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амина может обычно поддерживаться на низком уровне, не только изначально, но и на всем протяжении полной анальгезирующей терапии и таким образом, нежелательные побочные эффекты могут уменьшаться или даже полностью подавляться. Титрование дозы в течение фазы начального введения возможно, но не является необходимым.
Лекарственная дозированная форма в соответствии с изобретением обеспечивает хорошее соблюдение больным режима и схемы лечения и безопасность. Даже если лекарственная дозированная форма в соответствии с изобретением изменяется, например, с помощью дробилки таблеток, дозовая разгрузка не может произойти - измельчение лекарственной дозированной формы дополнительно не усиливает кривую немедленного высвобождения. Эти данные подтверждаются фармакокинетическими профилями трех различных галеновых препаратов (раствор в макроголе, самоэмульгирующиеся капсулы, заполненные жидкостью, и таблетки.
На фигуре 1 показаны средние значения цифровой рейтинговой шкалы, измеренные в течение 24-х часового периода после введения различных одноразовых доз соединения в соответствии с формулой (I’b) (200, 400, 600 мкг) по сравнению с морфином с замедленным высвобождением и плацебо у пациентов с острой послеоперационной болью после ортопедической хирургии (бурсэктомия).
На фигуре 2 показаны среднесуточные изменения боли (изменения NRS значения) в течение 5-дневного периода после введения суточных доз соединения в соответствии с формулой (I,b) (40 мкг, 120 мкг) по сравнению с плацебо у пациентов с болезненной диабетической нейропатией.
На фигуре 3 показаны среднесуточные изменения боли (изменения NRS значения) в течение 5-дневного периода после введения суточных доз соединения в соответствии с формулой (I’b) (80 мкг, 200 мкг) по сравнению с плацебо у пациентов с болезненной диабетической нейропатией.
На фигуре 4 показаны среднесуточные изменения боли (изменения NRS значения) в течение 5-дневного периода после введения суточных доз соединения в соответствии с формулой (I’b) (100 мкг) по сравнению с плацебо и замедленным высвобождением морфина (60 мг) у пациентов с болезненной диабетической нейропатией.
На фигуре 5 показаны средняя максимальная концентрация в плазме соединения в соответствии с формулой (I,b), измеренная в последний день 5-дневного периода дозирования один раз в сутки по сравнению с концентрацией в плазме, измеренной позже на 8-10 день в конце фазы выведения.
Изобретение относится к лекарственной дозированной форме для введения один раз в сутки и которая содержит фармакологически активный агент в соответствии с общей формулой (I)
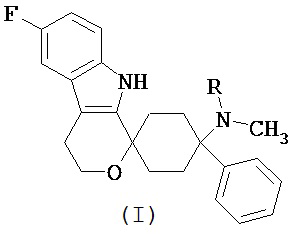 ,
,
где R представляет собой -H или -CH3,
или его физиологически приемлемую соль,
- которая обеспечивает немедленное высвобождение in vitro фармакологически активного агента в соответствии с общей формулой (I) в соответствии с Европейской фармакопеей; и
- которая содержит фармакологически активный агент в соответствии с общей формулой (I) в дозе от 10 мкг до 190 мкг; и
- где фармакокинетический параметр tmax находится в диапазоне от 0,5 до 16 ч,
для применения для лечения невропатической боли, предпочтительно хронической невропатической боли.
Если специально не указано иначе, все дозировки относительно фармакологически активного агента в соответствии с изобретением предпочтительно выражаются в виде эквивалентных весовых дозировок, исходя из свободного основания.
Фармакологически активный агент в соответствии с общей формулой (I) также может обозначаться как “6’-фтор-N-метил-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин”, где R представляет собой -H, и “6’-фтор-N,N-диметил-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин”, где R представляет собой -CH3; для целей настоящей заявки, фармакологически активный агент в соответствии с общей формулой (I) также может обозначаться как “6’-фтор-(N-метил- или N,N-диметил-)-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин”.
В предпочтительном варианте осуществления, фармакологически активный агент в соответствии с общей формулой (I) имеет стереохимию в соответствии с общей формулой (I’)
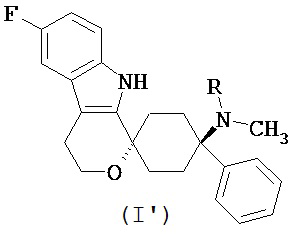
где R представляет собой -H или -CH3, или его физиологически приемлемую соль.
В другом варианте осуществления лекарственной дозированной формы в соответствии с изобретением, соединение формулы (I) выбирают из
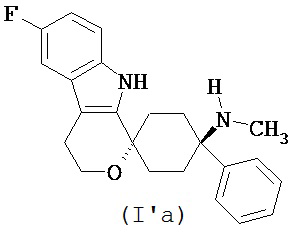
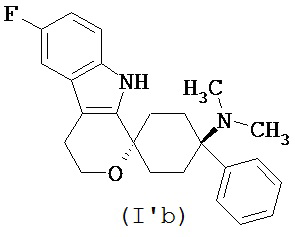
в форме свободного основания или его физиологически приемлемой соли.
Свободное основание в соответствии с общей формулой (I’a) может быть систематически обозначено как “1,1-(3-метиламино-3-фенилпентаметилен)-6-фтор-1,3,4,9-тетрагидропирано[3,4-b]индол (транс)” или как “(1r,4r)-6’-фтор-N-метил-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин”, соответственно.
Свободное основание в соответствии с общей формулой (I’b) может быть систематически обозначено как “1,1-(3-диметиламино-3-фенилпентаметилен)-6-фтор-1,3,4,9-тетрагидропирано[3,4-b]индол (транс)” или как “(1r,4r)-6’-фтор-N,N-диметил-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин”, соответственно.
Определение фармакологически активного агента в соответствии с общей формулой (I), как используется в настоящей заявке, включает 6’-фтор-(N-метил- или N,N-диметил-4-фенил-4,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин, его производные и его стереоизомеры в любой возможной форме, таким образом предпочтительно включая сольваты и полиморфы, соли, в частности соли присоединения кислот и соответствующие сольваты и полиморфы.
В предпочтительном варианте осуществления, фармакологически активный агент в соответствии с общей формулой (I) представлен в виде отдельного диастереомера в соответствии с общей формулой (I’).
В другом предпочтительном варианте осуществления фармакологически активный агент в соответствии с общей формулой (I) представлен в виде смеси диастереомеров. Такая смесь может содержать диастереомеры в любом соотношении. Диастереомерная смесь может, например, содержать диастереомеры в соотношении 60±5:40±5, 70±5:30±5, 80±5:20±5 или 90±5:10±5. Предпочтительно, лекарственная дозированная форма в соответствии с изобретением содержит диастереомер в соответствии с общей формулой (I’) в диастереомерном избытке (de) по меньшей мере 50%de, более предпочтительно по меньшей мере 60%de, еще более предпочтительно по меньшей мере 70%de, также более предпочтительно по меньшей мере 80%de, даже более предпочтительно по меньшей мере 90%de, наиболее предпочтительно по меньшей мере 95%de, и в особенности по меньшей мере 98%de, по отношению к другому диастереомеру (то есть транс отн. цис и анти отн. син, соответственно).
6’-фтор-(N-метил- или N,N-диметил-)-4-фенил-4,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин может присутствовать в лекарственной дозированной форме в соответствии с изобретением в форме свободного основания или в форме соли присоединения кислоты, таким образом можно использовать любую подходящую кислоту, способную образовывать такую соль присоединения.
Превращение 6’-Фтор-(N-метил- или N,N-диметил-)-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амина в соответствующую соль присоединения, например, путем реакции с подходящей кислотой можно осуществлять с помощью способа, хорошо известному специалисту в данной области техники. Подходящие кислоты включают, но не ограничиваясь только ими, соляную кислоту, бромистоводородную кислоту, серную кислоту, метансульфоновую кислоту, муравьиную кислоту, уксусную кислоту, щавелевую кислоту, янтарную кислоту, винную кислоту, миндальную кислоту, фумаровую кислоту, молочную кислоту, лимонную кислоту, глутаминовую кислоту и/или аспарагиновую кислоту. Образование соли предпочтительно осуществляют в растворителе, например, диэтиловом эфире, диизопропиловом эфире, алкил ацетатах, ацетоне и/или 2-бутаноне. Кроме того, триметилхлорсилан в водном растворе также пригоден для приготовления гидрохлоридов.
Фармакологически активный агент в соответствии с общей формулой (I) содержится в лекарственной дозированной форме в терапевтически эффективном количестве, то есть в количестве, которое является терапевтически эффективным по отношению к суточному введению дозированной формы для лечения невропатической боли, предпочтительно хронической невропатической боли. Количество, которое составляет терапевтически эффективное количество, изменяется в зависимости от соединения, состояния, подвергаемого лечению, тяжести указанного состояния, пациента, подвергаемого лечению, и от того, создана ли лекарственная дозированная форма для немедленного или ретардированного высвобождения.
В предпочтительном варианте осуществления, фармакологически активный агент в соответствии с общей формулой (I) содержится в дозированной форме в таком количестве, что однократное введение дозированной формы не приводит к какому-либо обезболивающему эффекту, то есть фармакологически активный агент в соответствии с общей формулой (I) содержится в дозированной форме в количестве, которое является субтерапевтическим по отношению к однократному введению дозированной формы. Тем не менее, предпочтительно, введение дозированной формы один раз в сутки приводит к обезболивающему эффекту, самое позднее, на пятый день, более предпочтительно самое позднее на четвертый день и еще более предпочтительно самое позднее на третий день при введении один раз в сутки.
В особенно предпочтительном варианте осуществления, по отношению к лечению невропатической боли, предпочтительно хронической невропатической боли, введение дозированной формы один раз в сутки приводит к субтерапевтической концентрации в плазме фармакологически активного агента в первый день введения, но к терапевтическим концентрациям в плазме фармакологически активного агента после введения дозированной формы один раз в сутки в течение по меньшей мере 3, или по меньшей мере 4, или по меньшей мере 5 последующих дней.
В предпочтительном варианте осуществления, фармакологически активный агент в соответствии с общей формулой (I) содержится в дозированной форме в таком количестве, что дозированная форма не эффективна для лечения острой боли, то есть фармакологически активный агент содержится в количестве, которое является субтерапевтическим по отношению к лечению острой боли. Предпочтительно, количество является настолько низким, что даже после повторного введения в течение нескольких последовательных дней, например, ≥5 дней, не достигается существенный эффект для лечения острой боли.
Предпочтительно, фармакологически активный агент в соответствии с общей формулой (I) содержится в дозированной форме в таком количестве, что не требуется титрование начальной дозы.
Предпочтительно, фармакологически активный агент в соответствии с общей формулой (I) содержится в дозированной форме в таком количестве, что побочные действия, которые встречаются при введении дозированной формы, снижаются по сравнению с дозированной формой, содержащий чистый агонист μ-опиодного рецептора, такой как морфин, в эквианальгезирующих дозах.
В предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме в соответствии с изобретением составляет самое большее 95 мас. %, более предпочтительно самое большее 50 мас. %, также более предпочтительно самое большее 25 мас. %, еще более предпочтительно самое большее 10 мас. %, даже более предпочтительно самое большее 5 мас. %, наиболее предпочтительно самое большее 1,0 мас. %, и в особенности самое большее 0,5 мас. %.
В другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме в соответствии с изобретением составляет по меньшей мере 0,001 мас. %, более предпочтительно по меньшей мере 0,005 мас. %, также более предпочтительно по меньшей мере 0,01 мас. %, еще более предпочтительно по меньшей мере 0,05 мас. %, даже более предпочтительно по меньшей мере 0,1 мас. %, наиболее предпочтительно по меньшей мере 0,5 мас. %, и в особенности по меньшей мере 1,0 мас. %.
Если четко не указано иначе, в контексте настоящего изобретения указание “мас. %” будет обозначать вес соответствующего ингредиента на общий вес лекарственной дозированной формы. В случае, если лекарственная дозированная форма имеет пленочную оболочку или инкапсулирована с помощью инкапсулирующей среды, которая не содержит какого-либо количества фармакологически активного агента в соответствии с общей формулой (I) и окружена ядром, которое, в свою очередь, содержит общее количество фармакологически активного агента в соответствии с общей формулой (I), указание “мас. %” будет обозначать вес соответствующего ингредиента на общий вес композиции, образующей указанное ядро.
Если лекарственная дозированная форма инкапсулирована или имеет пленочную оболочку, то фармакологически активный агент в соответствии с общей формулой (I) предпочтительно гомогенно распределен в ядре лекарственной дозированной формы. Предпочтительно, инкапсулирующая среда или пленочная оболочка не содержит какого-либо фармакологически активного агента в соответствии с общей формулой (I).
Дозированная форма в соответствии с изобретением адаптирована для введения один раз в сутки и содержит фармакологически активный агент в соответствии с общей формулой (I) в дозе от 10 мкг до 190 мкг, то есть дозированная форма в соответствии с изобретением содержит фармакологически активный агент в соответствии с общей формулой (I) в суточной дозе от 10 мкг до 190 мкг.
В предпочтительном варианте осуществления, доза фармакологически активного агента в соответствии с общей формулой (I) предпочтительно находится в диапазоне от 10 мкг до 180 мкг, предпочтительно в пределах от 12,5 мкг до 150 мкг, более предпочтительно в пределах от 15 мкг до 120 мкг, еще более предпочтительно в пределах от 17,5 мкг до 100 мкг, также более предпочтительно в пределах от 20 мкг до 90 мкг, наиболее предпочтительно в пределах от 25 мкг до 80 мкг, и в особенности в пределах от 30 мкг до 75 мкг.
В предпочтительном варианте осуществления, доза фармакологически активного агента в соответствии с общей формулой (I) находится в диапазоне от 10 мкг до 50 мкг.
В предпочтительном варианте осуществления, доза фармакологически активного агента в соответствии с общей формулой (I) находится в пределах от 1,0 мкг до 100 мкг.
В предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 30±20 мкг, более предпочтительно 30±15 мкг, наиболее предпочтительно 30±10 мкг, и в особенности 30±5 мкг. В другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 35±25 мкг, более предпочтительно 35±20 мкг, еще более предпочтительно 35±15 мкг, наиболее предпочтительно 35±10 мкг, и в особенности 35±5 мкг. В предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 40±30 мкг, более предпочтительно 40±25 мкг, еще более предпочтительно 40±20 мкг, также более предпочтительно 40±15 мкг, наиболее предпочтительно 40±10 мкг, и в особенности 40±5 мкг. В другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 45±35 мкг, более предпочтительно 45±30 мкг, еще более предпочтительно 45±25 мкг, также более предпочтительно 45±20 мкг, даже более предпочтительно 45±15 мкг, наиболее предпочтительно 45±10 мкг, и в особенности 45±5 мкг. В еще другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 50±35 мкг, более предпочтительно 50±30 мкг, еще более предпочтительно 50±25 мкг, также более предпочтительно 50±20 мкг, даже более предпочтительно 50±15 мкг, наиболее предпочтительно 50±10 мкг, и в особенности 50±5 мкг. В другом также предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 55±35 мкг, более предпочтительно 55±30 мкг, еще более предпочтительно 55±25 мкг, также более предпочтительно 55±20 мкг, даже более предпочтительно 55±15 мкг, наиболее предпочтительно 55±10 мкг, и в особенности 55±5 мкг. В предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 60±40 мкг или 60±35 мкг, более предпочтительно 60±30 мкг, еще более предпочтительно 60±25 мкг, также более предпочтительно 60±20 мкг, даже более предпочтительно 60±15 мкг, наиболее предпочтительно 60±10 мкг, и в особенности 60±5 мкг. В другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 65±40 мкг или 65±35 мкг, более предпочтительно 65±30 мкг, еще более предпочтительно 65±25 мкг, также более предпочтительно 65±20 мкг, даже более предпочтительно 65±15 мкг, наиболее предпочтительно 65±10 мкг, и в особенности 65±5 мкг. В еще другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 70±40 мкг или 70±35 мкг, более предпочтительно 70±30 мкг, еще более предпочтительно 70±25 мкг, также более предпочтительно 70±20 мкг, даже более предпочтительно 70±15 мкг, наиболее предпочтительно 70±10 мкг, и в особенности 70±5 мкг. В другом также предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 75±40 мкг или 75±35 мкг, более предпочтительно 75±30 мкг, еще более предпочтительно 75±25 мкг, также более предпочтительно 75±20 мкг, даже более предпочтительно 75±15 мкг, наиболее предпочтительно 75±10 мкг, и в особенности 75±5 мкг. В предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 80±45 мкг или 80±40 мкг, более предпочтительно 80±35 мкг или 80±30 мкг, еще более предпочтительно 80±25 мкг, также более предпочтительно 80±20 мкг, даже более предпочтительно 80±15 мкг, наиболее предпочтительно 80±10 мкг, и в особенности 80±5 мкг. В другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 85±45 мкг или 85±40 мкг, более предпочтительно 85±35 мкг или 85±30 мкг, еще более предпочтительно 85±25 мкг, также более предпочтительно 85±20 мкг, даже более предпочтительно 85±15 мкг, наиболее предпочтительно 85±10 мкг, и в особенности 85±5 мкг. В еще другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 90±45 мкг или 90±40 мкг, более предпочтительно 90±35 мкг или 90±30 мкг, еще более предпочтительно 90±25 мкг, также более предпочтительно 90±20 мкг, даже более предпочтительно 90±15 мкг, наиболее предпочтительно 90±10 мкг, и в особенности 90±5 мкг. В другом также предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 95±35 мкг, более предпочтительно 95±30 мкг, еще более предпочтительно 95±25 мкг, также более предпочтительно 95±20 мкг, даже более предпочтительно 95±15 мкг, наиболее предпочтительно 95±10 мкг, и в особенности 95±5 мкг. В предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 100±80 мкг, более предпочтительно 100±60 мкг, еще более предпочтительно 100±40 мкг, даже более предпочтительно 100±20 мкг, наиболее предпочтительно 100±10 мкг, и в особенности 100±5 мкг. В другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 110±35 мкг, более предпочтительно 110±30 мкг, еще более предпочтительно 110±25 мкг, также более предпочтительно 110±20 мкг, даже более предпочтительно 110±15 мкг, наиболее предпочтительно 110±10 мкг, и в особенности 110±5 мкг. В еще другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 120±60 мкг, более предпочтительно 120±50 мкг, еще более предпочтительно 120±40 мкг, также более предпочтительно 120±30 мкг, даже более предпочтительно 120±20 мкг, наиболее предпочтительно 120±10 мкг, и в особенности 120±5 мкг. В еще другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 130±60 мкг, более предпочтительно 130±50 мкг, еще более предпочтительно 130±40 мкг, также более предпочтительно 130±30 мкг, даже более предпочтительно 130±20 мкг, наиболее предпочтительно 130±10 мкг, и в особенности 130±5 мкг. В другом также предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 140±50 мкг, более предпочтительно 140±50 мкг, еще более предпочтительно 140±40 мкг, также более предпочтительно 140±30 мкг, даже более предпочтительно 140±20 мкг, наиболее предпочтительно 140±10 мкг, и в особенности 140±5 мкг. В еще другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 150±60 мкг, более предпочтительно 150±50 мкг, еще более предпочтительно 150±40 мкг, также более предпочтительно 150±30 мкг, даже более предпочтительно 150±20 мкг, наиболее предпочтительно 150±10 мкг, и в особенности 150±5 мкг. В предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 160±30 мкг, более предпочтительно 160±25 мкг, еще более предпочтительно 160±20 мкг, также более предпочтительно 160±15 мкг, наиболее предпочтительно 160±10 мкг, и в особенности 160±5 мкг. В другом предпочтительном варианте осуществления, содержание фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме находится в диапазоне 170±20 мкг, более предпочтительно 170±10 мкг, и в особенности 170±5 мкг.
В предпочтительном варианте осуществления, лекарственная дозированная форма в соответствии с изобретением адаптирована для орального введения. Подходящие альтернативные пути введения лекарственной дозированной формы в соответствии с изобретением включают, но не ограничиваясь только ими, вагинальное и ректальное введение.
Лекарственная дозированная форма в соответствии с изобретением предназначена для введения один раз в сутки.
Для целей настоящей заявки, “введение один раз в сутки” (sid, OD) предпочтительно обозначает, что лекарственная дозированная форма адаптирована для введения в соответствии со схемой, включающей введение первой лекарственной дозированной формы в соответствии с изобретением и последующее введение второй лекарственной дозированной формы в соответствии с изобретением, где обе, первая и вторая лекарственные дозированные формы вводятся в течение промежутка времени приблизительно 48 часов, но где вторая лекарственная дозированная форма вводится не раньше, чем через 18 часов, предпочтительно не раньше, чем через 20 часов, более предпочтительно не раньше, чем через 22 часа и в особенности, приблизительно через 24 часа после введения первой лекарственной дозированной формы.
Для специалиста в данной области техники совершенно очевидно, что схемы введения “один раз в сутки” могут быть реализованы путем введения одной лекарственной дозированной формы, содержащей суммарное количество фармакологически активного агента в соответствии с общей формулой (I) для введения в конкретный момент времени или, альтернативно, введения множества дозированных единиц, то есть двух, трех или большего количества дозированных единиц, сумма такого множества дозированных единиц содержит суммарное количество фармакологически активного агента в соответствии с общей формулой (I) для введения в указанный конкретный момент времени, где индивидуальные дозированные единицы адаптированы для одновременного введения или введения в течение короткого периода времени, например, в пределах 5, 10 или 15 минут.
Дозированная форма в соответствии с изобретением предназначена для применения для лечения невропатической боли, предпочтительно хронической невропатической боли, такой как болезненная диабетическая нейропатия. Предпочтительно, боль является умеренной, тяжелой или от умеренной до тяжелой.
Для целей настоящей заявки, невропатическая боль представляет собой боль, которая имеет происхождение из поврежденного нерва или нарушения функции нерва. Предпочтительно, невропатическую боль выбирают из острой невропатической боли и хронической невропатической боли. Невропатическая боль может вызываться повреждением или заболеванием, поражающим центральные или периферические части нервной системы, задействованные в телесные ощущения (соматосенсорная система). Предпочтительно, дозированная форма в соответствии с изобретением предназначена для применения для лечения хронической невропатической боли или острой невропатической боли, периферической невропатической боли или центральной невропатической боли, мононевропатической боли или полиневропатической боли. Если невропатическая боль является хронической, то она может быть хронической периферической невропатической болью или хронической центральной невропатической болью, в предпочтительном варианте осуществления хронической периферической мононевропатической болью или хронической центральной мононевропатической болью, в другом предпочтительном варианте осуществления хронической периферической полиневропатической болью или хронической центральной полиневропатической болью. Если невропатическая боль является острой, то она может быть острой периферической невропатической болью или острой центральной невропатической болью, в предпочтительном варианте осуществления острой периферической мононевропатической болью или острой центральной мононевропатической болью, в другом предпочтительном варианте осуществления острой периферической полиневропатической болью или острой центральной полиневропатической болью. Изобретение также относится к фармакологически активному агенту в соответствии с общей формулой (I) или его физиологически приемлемой соли для применения для лечения невропатической боли, как описано выше, предпочтительно путем введения один раз в сутки лекарственной дозированной формы в соответствии с изобретением.
Центральная невропатическая боль обнаруживается при повреждениях спинного мозга, рассеянном склерозе, и некоторых ударах. Фибромиалгия является потенциально нарушением с центральной болью и чувствительна к лекарственным средствам, которые эффективны при невропатической боли. Кроме диабетической нейропатии и других метаболических состояний, обычными причинами болезненных периферических нейропатий являются инфекция, вызванная вирусом простого герпеса, нейропатий, связанные с ВИЧ, недостаточности питания, токсины, отдаленные проявления злокачественных новообразований, генетические, и опосредованные иммунной системой нарушения или физическая травма нервного ствола. Невропатическая боль является обычной при злокачественном новообразовании вследствие прямого действия злокачественного новообразования на периферические нервы (например, компрессия опухолью), или вследствие побочного действия химиотерапии, лучевого поражения или хирургии.
В другом предпочтительном варианте осуществления, боль, подвергаемая лечению, выбирают из группы, включающей боль, являющуюся или ассоциированную с паническим расстройством [эпизодическая пароксизмальная тревожность] [F41.0]; диссоциативными [конверсионными] расстройствами [F44]; устойчивым соматоформным болевым расстройством [F45.4]; паническими нарушениями, исключительно относящимися к психологическим факторам [F45.41]; диспареунией неорганического происхождения [F52.6]; другими стойкими изменениями личности [F62.8]; садомазохизмом [F65.5]; преувеличением физических симптомов по психологическим причинам [F68.0]; мигренью [G43]; другими синдромами головных болей [G44]; невралгией тройничного нерва [G50.0]; атипичной лицевой болью [G50.1]; синдромом фантомной конечности с болью [G54.6]; синдромом фантомной конечности без боли [G54.7]; острой и хронической болью, не классифицированной в других рубриках [G89]; глазной болью [Н57.1]; оталгией [Н92.0]; стенокардией, неопределенной [I20.9]; другими перечисленными нарушениями носа и придаточных пазух носа [J34.8]; другими заболеваниями глотки [J39.2]; височно-нижнечелюстным синдромом [К07.6]; другими перечисленными нарушениями зубов и поддерживающих структур [К08.8]; другими перечисленными заболеваниями челюстей [К10.8]; другими и неклассифицированными поражениями слизистой оболочки рта [К13.7]; синдромом жжения полости рта [К14.6]; другими перечисленными заболеваниями анального отверстия и прямой кишки [К62.8]; болью в суставе [М25.5]; болью в плече [М25.51]; крестцово-копчиковыми нарушениями, не классифицированными в других рубриках [М53.3]; болью в спине [М54.]; радикулопатией [M54.1]; цервикалгией [M54.2]; ишиасом [M54.3]; болью в пояснично-крестцовой области [M54.5]; болью в грудном отделе позвоночника [M54.6]; другой болью в верхних отделах спины [M54.8]; болью в верхних отделах спины, неопределенной [M54.9]; другими поражениями плеча [M75.8]; другими нарушениями мягких тканей, не классифицированными в других рубриках [M79]; миалгией [M79.1]; невралгией и невритом, не классифицированными [M79.2]; болью в конечности [M79.6]; другими перечисленными нарушениями костей [M89.8]; неклассифицированной почечной коликой [N23]; другими перечисленными нарушениями полового члена [N48.8]; другими перечисленными нарушениями мужских половых органов [N50.8]; мастодинией [N64.4]; болью и другими состояниями, ассоциированными с женскими половыми органами и менструальным циклом [N94]; болью, напоминающей менструальную, но возникающей в середине цикла, в период овуляции [N94.0]; другими перечисленными состояниями, ассоциированными с женскими половыми органами и менструальным циклом [N94.8]; болью в горле и грудной клетке [R07]; болью в горле [R07.0]; болью в груди при дыхании [R07.1]; прекордиальной болью [R07.2]; другой болью в груди [R07.3]; болью в груди, неопределенной [R07.4]; абдоминальной болью и болью в области таза [R10]; острой болью в брюшной полости [R10.0]; болью, локализованной в верхнем отделе брюшной полости [R10.1]; болью в области таза и паховой области [R10.2]; болью, локализованной в других частях нижнего отдела брюшной полости [R10.3]; другой и неопределенной болью в области живота [R10.4]; метеоризмом и связанными состояниями [R14]; напряженностью в области живота [R19.3]; другими и неопределенными нарушениями чувствительности кожи [R20.8]; болью, связанной с мочеиспусканием [R30]; другими и неопределенными симптомами и признаками, задействующими мочевыделительную систему [R39.8]; головной болью [R51]; болью, не классифицированной в других рубриках [R52]; острой болью [R52.0]; хронической некупируемой болью [R52.1]; другой хронической болью [R52.2]; болью, неклассифицированной [R52.9]; другими осложнениями сердечных и сосудистых протезных устройств, имплантов и трансплантатов [T82.8]; другими осложнениями мочеполовых протезных устройств, имплантов и трансплантатов [Т83.8]; другими осложнениями внутренних ортопедических протезных устройств, имплантов и трансплантатов [Т84.8]; другими осложнениями внутренних протезных устройств, имплантов и трансплантатов, не классифицированными в других рубриках [Т85.8]; где информация в квадратных скобках относится к классификации в соответствии с ICD-10. Изобретение также относится к фармакологически активному агенту в соответствии с общей формулой (I) или его физиологически приемлемой соли для применения для лечения боли, предпочтительно невропатической боли, как описано выше, предпочтительно путем введения один раз в сутки лекарственной дозированной формы в соответствии с изобретением.
Лекарственная дозированная форма в соответствии с изобретением обеспечивает немедленное высвобождение фармакологически активного агента в соответствии с общей формулой (I). Лекарственная дозированная форма специфически создана для обеспечения немедленного высвобождения фармакологически активного агента в соответствии с общей формулой (I) in vitro в соответствии с Европейской фармакопеей Если лекарственная дозированная форма покрыта, например, оболочкой, которая растворима в желудочном соке, то за кинетикой высвобождения предпочтительно наблюдают после растворения такого покрытия.
В соответствии с описанием, термин “немедленное высвобождение” относится к любой кривой высвобождения, которая соответствует по меньшей мере одному, предпочтительно обоим, следующим требованиям. Во-первых, лекарственная дозированная форма распадается в течение 10 минут или меньше после воздействия дезинтегрирующей среды. Способы определения времени распадаемости известны специалисту в данной области техники. Например, они могут быть определены в соответствии с USP XXIV теста на распадаемость, используя, например, Erweka ZT-71 прибор для определения распадаемости. Во-вторых, лекарственная дозированная форма высвобождает по меньшей мере 70 мас. % лекарственного средства в течение 15 минут после воздействия среды растворения. Предпочтительно, in vitro свойства высвобождения лекарственной дозированной формы в соответствии с изобретением определяют в соответствии со способом с использованием лопастной мешалки с спикером при 50, 75 или 100 об./мин., предпочтительно в условиях in vitro при 37±0,5°C в 900 мл искусственного желудочного сока при pH 1,2, или в идентичных условиях в не-искусственном желудочном соке.
В предпочтительном варианте осуществления, лекарственная дозированная форма высвобождает в условиях in vitro в 900 мл искусственного желудочного сока при pH 1,2 и 37±0,5°С через 30 минут в соответствии со способом с использованием лопастной мешалки с синкером при 100 об./мин. по меньшей мере 50 мас. %, более предпочтительно по меньшей мере 60 мас. %, еще более предпочтительно по меньшей мере 70 мас. %, также более предпочтительно по меньшей мере 80 мас. %, наиболее предпочтительно по меньшей мере 90 мас. %, и в особенности по меньшей мере 95 мас. % фармакологически активного агента в соответствии с общей формулой (I), исходя из общего количества фармакологически активного агента в соответствии с общей формулой (I), которое исходно присутствует в лекарственной дозированной форме.
Лекарственная дозированная форма в соответствии с изобретением проявляет чрезвычайно хорошие срок годности при хранении и стабильность при хранении, то есть ни химический состав, ни физические характеристики, ни профиль растворимости лекарственной дозированной формы существенно не изменяются при хранении.
В предпочтительном варианте осуществления, лекарственная дозированная форма в соответствии с изобретением обеспечивает достаточную стабильность по отношению к фармакологически активному агенту в соответствии с общей формулой (I), которое в них содержится, таким образом, что после хранения лекарственной дозированной формы при 40±2°С при 75% ОВ ±5% в течение минимально периода времени 6 недель, предпочтительно 3 месяца, концентрации нежелательных продуктов разложения и примесей, соответственно, предпочтительно вследствие распада или разложения фармакологически активного агента в соответствии с общей формулой (I) фактически, составляет самое большее 1,0 мас. %, более предпочтительно самое большее 0,8 мас. %, еще более предпочтительно самое большее 0,6 мас. %, также более предпочтительно самое большее 0,4 мас. %, даже более предпочтительно самое большее 0,2 мас. %, наиболее предпочтительно самое большее 0,1 мас. %, и в особенности самое большее 0,05 мас. %, относительно первоначального содержания фармакологически активного агента в соответствии с общей формулой (I) в лекарственной дозированной форме, то есть его содержания перед хранением лекарственной дозированной формы.
Общепринятое ускоренное испытание для определения стабильности лекарственного средства в соответствии с ICH и FDA руководствами относится к хранению фармацевтического препарата, содержащего лекарственное средство (например, в его контейнере и упаковке). В соответствии с ICH руководствами, так называемое определение ускоренного хранения может быть осуществлено для фармацевтических препаратов при 40±2°C при 75% ОВ ±5% в течение минимально периода времени 6 месяцев. Дополнительно, так называемое исследование долговременного хранения может быть осуществлено для фармацевтических препаратов при 25±2°C при не менее чем 60% ОВ ±5% в течение минимально периода времени 12 месяцев. В случае если все критерии удовлетворяются для условий определения ускоренного хранения и исследования долговременного хранения в течение периода 6 месяцев, исследование долговременного хранения может быть сокращено до 6 месяцев и соответствующие данные удвоены для получения предполагаемых данных для периода 12 месяцев.
При хранении, образцы фармацевтического препарата отбирают в указанные промежутки времени и анализируют относительно их содержания лекарственного средства, присутствия примесей, их кривых высвобождения и, в случае необходимости, других параметров. В соответствии с ICH руководствами, во всех образцах чистота лекарственного средства должна составлять ≥98%, содержание лекарственного средства должно составлять 95-105% (FDA руководство: 90-110%). Кроме того, фармацевтический препарат должен высвобождать >80% лекарственного средства в течение 30 минут.
В случае дозированных форм, которые содержат меньше, чем 50 мг лекарственного средства, дополнительно необходимо провести исследование однородности состава для 10 случайно выбранных дозированных форм.
Фармацевтический препарат соответствует требованиям, если ни одно из содержимого индивидуальных единиц не выходит за пределы от 85% до 115% среднего содержимого. В случае если содержимое индивидуальной единицы выходит за эти пределы, следует анализировать другие 30 дозированные формы. Препарат не проходит тест, если более, чем 3 содержимого индивидуальных единиц выходят за пределы от 85 до 115% среднего содержимого или, если одно или больше содержимого индивидуальных единиц выходят за пределы от 75% до 125% среднего содержимого.
В предпочтительном варианте осуществления, после хранения лекарственной дозированной формы в течение 6 месяцев в условиях долговременного хранения (25°C и 60% относительная влажность) в запечатанном стеклянном контейнере, деградация фармакологически активного агента в соответствии с общей формулой (I) не превышает 2,0%, более предпочтительно 1,5%, еще более предпочтительно 1,0%, и наиболее предпочтительно 0,5%.
В другом предпочтительном варианте осуществления, после хранения лекарственной дозированной формы в течение 6 месяцев в условиях ускоренного хранения (40°C и 75% относительная влажность) в запечатанном стеклянном контейнере, деградация фармакологически активного агента в соответствии с общей формулой (I) не превышает 4%, более предпочтительно 3%, еще более предпочтительно 2%, также более предпочтительно 1%, и наиболее предпочтительно 0,5%.
Предпочтительно, после хранения лекарственной дозированной формы в течение 6 месяцев в условиях долговременного хранения (25°C и 60% относительная влажность), лекарственная дозированная форма высвобождает в условиях in vitro в 900 мл искусственного желудочного сока при pH 1,2 и 37±0,5°C через 30 минут в соответствии со способом с использованием лопастной мешалки с спикером при 100 об./мин. по меньшей мере 50 мас. %, более предпочтительно по меньшей мере 60 мас. %, еще более предпочтительно по меньшей мере 70 мас. %, и наиболее предпочтительно по меньшей мере 80 мас. % фармакологически активного агента в соответствии с общей формулой (I), исходя из общего количества фармакологически активного агента в соответствии с общей формулой (I), которое исходно присутствует в лекарственной дозированной форме.
Предпочтительно, после хранения лекарственной дозированной формы в течение 6 месяцев в условиях ускоренного хранения (40°C и 75% относительная влажность), лекарственная дозированная форма высвобождает в условиях in vitro в 900 мл искусственного желудочного сока при pH 1,2 и 37±0,5°C через 30 минут в соответствии со способом с использованием лопастной мешалки с синкером при 100 об./мин. по меньшей мере по меньшей мере 50 мас. %, более предпочтительно по меньшей мере 60 мас. %, еще более предпочтительно по меньшей мере 70 мас. %, и наиболее предпочтительно по меньшей мере 80 мас. % фармакологически активного агента в соответствии с общей формулой (I), исходя из общего количества фармакологически активного агента в соответствии с общей формулой (I), которое исходно присутствует в лекарственной дозированной форме.
Абсорбционные свойства фармакологически активного агента, вводимого с помощью лекарственной дозированной формы, могут быть описаны с помощью фармакокинетических параметров Cmax, tmax и AUC0-t. Определение Cmax и tmax, а также расчет AUC хорошо известны специалисту в данной области техники и описаны, например, в Bauer, Fromming, Fuhrer, “Lehrbuch der Pharmazeutischen Technologie,” 6ое издание (1999). Если специально не указано иначе, все фармакокинетические параметры выражают в виде средних значений для популяции субъектов.
Существуют экспериментально полученные данные, указывающие на то, что AUC0-t и Cmax фармакологически активного агента в соответствии с общей формулой (I) пропорциональны дозе.
Для целей настоящей заявки, Cmax представляет собой наибольшую концентрацию в плазме фармакологически активного агента, достигаемую после однократного введения лекарственной дозированной формы.
Для целей настоящей заявки, tmax представляет собой время, необходимое для достижения Cmax. Предпочтительно, если специально не указано иначе, tmax и Cmax относятся к фармакокинетическим параметрам, которые наблюдаются после однократного введения дозированной формы в соответствии с изобретением субъекту, которого ранее не лечили с помощью фармакологически активного агента в соответствии с общей формулой (I) (то есть Cmax=Cmax, 1 day и tmax=tmax, 1 day).
Для целей настоящей заявки, Cmax, n days представляет собой наибольшую концентрацию в плазме фармакологически активного агента, достигаемую после введения один раз в сутки лекарственной дозированной формы в течение n последовательных дней, где n может представлять собой, например, 1,2, 3, 4, 5, 6, и др.
Для целей настоящей заявки, Cmax, ≥5 days представляет собой наибольшую концентрацию в плазме фармакологически активного агента, достигаемую после введения один раз в сутки лекарственной дозированной формы в течение по меньшей мере 5 последовательных дней. В предпочтительном варианте осуществления, стабильные установившиеся состояния достигаются через 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 или больше дней, то есть непрерывное введение дозированной формы в соответствии с изобретением в дополнительные последовательные дни существенно по повышает как-либо дополнительно Cmax.
Для целей настоящей заявки, tmax, n days представляет собой время, необходимое для достижения Cmax, n days (после использования препарата), где n может представлять собой, например, 1, 2, 3, 4, 5, 6, и т.д.
Для целей настоящей заявки, tmax, ≥5 days представляет собой время, необходимое для достижения Cmax, ≥5 days (после использования препарата).
Для целей настоящей заявки, AUC0-t представляет собой площадь под кривой после однократного введения до времени t последнего образца, который содержит аналитически определяемую концентрацию фармакологически активного агента.
Для целей настоящей заявки, AUC0-72h представляет собой площадь под исходной кривой после однократного введения до 72 часов после этого.
Предпочтительно, Cmax находится в диапазоне от 1 до 250 мкг/м3, более предпочтительно в диапазоне от 5 до 200 мкг/м3, еще более предпочтительно в диапазоне от 10 до 150 мкг/м3, наиболее предпочтительно в диапазоне от 15 до 120 мкг/м3, и в особенности в диапазоне от 20 до 100 мкг/м3.
В предпочтительном варианте осуществления, Cmax находится в диапазоне 20±17,5 мкг/м3, более предпочтительно в пределах от 20±15 мкг/м3, еще более предпочтительно в пределах от 20±12,5 мкг/м3, также более предпочтительно в пределах от 20±10 мкг/м3, и наиболее предпочтительно в пределах от 20±5 мкг/м3. В другом предпочтительном варианте осуществления, Cmax находится в диапазоне 25±20 мкг/м3, более предпочтительно в пределах от 25±17,5 мкг/м3, еще более предпочтительно в пределах от 25±15 мкг/м3, также более предпочтительно в пределах от 25±12,5 мкг/м3, наиболее предпочтительно в пределах от 25±10 мкг/м3, и в особенности в пределах от 25±5 мкг/м3. В еще другом предпочтительном варианте осуществления, Cmax находится в диапазоне 30±20 мкг/м3, более предпочтительно в пределах от 30±17,5 мкг/м3, еще более предпочтительно в пределах от 30±15 мкг/м3, также более предпочтительно в пределах от 30±12,5 мкг/м3, наиболее предпочтительно в пределах от 30±10 мкг/м3, и в особенности в пределах от 30±5 мкг/м3. В другом также предпочтительном варианте осуществления, Cmax находится в диапазоне 35±20 мкг/м3, более предпочтительно в пределах от 35±17,5 мкг/м3, еще более предпочтительно в пределах от 35±15 мкг/м3, также более предпочтительно в пределах от 35±12,5 мкг/м3, наиболее предпочтительно в пределах от 35±10 мкг/м3, и в особенности в пределах от 35±5 мкг/м3. В предпочтительном варианте осуществления, Cmax находится в диапазоне 40±35 мкг/м3, более предпочтительно в пределах от 40±30 мкг/м3, еще более предпочтительно в пределах от 40±25 мкг/м3, также более предпочтительно в пределах от 40±20 мкг/м3, наиболее предпочтительно в пределах от 40±15 мкг/м3, и в особенности в пределах от 40±10 мкг/м3. В другом предпочтительном варианте осуществления, Cmax находится в диапазоне 50±40 мкг/м3, более предпочтительно в пределах от 50±30 мкг/м3, еще более предпочтительно в пределах от 50±25 мкг/м3, также более предпочтительно в пределах от 50±20 мкг/м3, наиболее предпочтительно в пределах от 50±15 мкг/м3, и в особенности в пределах от 50±10 мкг/м3. В еще другом предпочтительном варианте осуществления, Cmax находится в диапазоне 60±40 мкг/м3, более предпочтительно в пределах от 60±30 мкг/м3, еще более предпочтительно в пределах от 60±25 мкг/м3, также более предпочтительно в пределах от 60±20 мкг/м3, наиболее предпочтительно в пределах от 60±15 мкг/м3, и в особенности в пределах от 60±10 мкг/м3. В другом также предпочтительном варианте осуществления, Cmax находится в диапазоне 70±45 мкг/м3, более предпочтительно в пределах от 70±40 мкг/м3, еще более предпочтительно в пределах от 70±30 мкг/м3, также более предпочтительно в пределах от 70±25 мкг/м3, даже более предпочтительно в пределах от 70±20 мкг/м3, наиболее предпочтительно в пределах от 70±15 мкг/м3, и в особенности в пределах от 70±10 мкг/м3. В другом предпочтительном варианте осуществления, Cmax находится в диапазоне 80±50 мкг/м3, более предпочтительно в пределах от 80±40 мкг/м3, еще более предпочтительно в пределах от 80±30 мкг/м3, также более предпочтительно в пределах от 80±25 мкг/м3, даже более предпочтительно в пределах от 80±20 мкг/м3, наиболее предпочтительно в пределах от 80±15 мкг/м3, и в особенности в пределах от 80±10 мкг/м3. В предпочтительном варианте осуществления, Cmax находится в диапазоне 90±50 мкг/м3, более предпочтительно в пределах от 90±30 мкг/м3, еще более предпочтительно в пределах от 90±25 мкг/м3, также более предпочтительно в пределах от 90±20 мкг/м3, наиболее предпочтительно в пределах от 90±15 мкг/м3, и в особенности в пределах от 90±10 мкг/м3. В другом предпочтительном варианте осуществления, Cmax находится в диапазоне 100±50 мкг/м3, более предпочтительно в пределах от 100±30 мкг/м3, еще более предпочтительно в пределах от 100±25 мкг/м3, также более предпочтительно в пределах от 100±20 мкг/м3, наиболее предпочтительно в пределах от 100±15 мкг/м3, и в особенности в пределах от 100±10 мкг/м3. В еще другом предпочтительном варианте осуществления, Cmax находится в диапазоне 120±50 мкг/м3, более предпочтительно в пределах от 120±30 мкг/м3, еще более предпочтительно в пределах от 120±25 мкг/м3, также более предпочтительно в пределах от 120±20 мкг/м3, наиболее предпочтительно в пределах от 120±15 мкг/м3, и в особенности в пределах от 120±10 мкг/м3.
Предпочтительно, соотношение Cmax/дозу находится в диапазоне от 0,01 до 3,00 м-3, также более предпочтительно в диапазоне от 0,02 до 2,50 м-3, более предпочтительно в диапазоне от 0,04 до 2,00 м-3, и наиболее предпочтительно в диапазоне от 0,06 до 1,69 м-3.
В предпочтительном варианте осуществления, соотношение Cmax/дозу находится в диапазоне 0,30±0,25 м-3, более предпочтительно 0,30±0,20 м-3, еще более предпочтительно 0,30±0,15 м-3, наиболее предпочтительно 0,30±0,10 м-3, и в особенности 0,30±0,05 м-3. В другом предпочтительном варианте осуществления, соотношение Cmax/дозу находится в диапазоне 0,40±0,35 м-3, более предпочтительно 0,40±0,30 м-3, еще более предпочтительно 0,40±0,25 м-3, также более предпочтительно 0,40±0,20 м-3, даже более предпочтительно 0,40±0,15 м-3, наиболее предпочтительно 0,40±0,10 м-3, и в особенности 0,40±0,05 м-3. В еще другом предпочтительном варианте осуществления, соотношение Cmax/дозу находится в диапазоне 0,50±0,35 м-3, более предпочтительно 0,50±0,30 м-3, еще более предпочтительно 0,50±0,25 м-3, также более предпочтительно 0,50±0,20 м-3, даже более предпочтительно 0,50±0,15 м-3, наиболее предпочтительно 0,50±0,10 м-3, и в особенности 0,50±0,05 м-3. В другом также предпочтительном варианте осуществления, соотношение Cmax/дозу находится в диапазоне 0,60±0,40 м-3, более предпочтительно 0,60±0,30 м-3, еще более предпочтительно 0,60±0,25 м-3, также более предпочтительно 0,60±0,20 м-3, наиболее предпочтительно 0,60±0,15 м-3, и в особенности 0,60±0,10 м-3. В также другом предпочтительном варианте осуществления, соотношение Cmax/дозу находится в диапазоне 0,70±0,40 м-3, более предпочтительно 0,70±0,35 м-3, еще более предпочтительно 0,70±0,30 м-3, также более предпочтительно 0,70±0,25 м-3, даже более предпочтительно 0,70±0,20 м-3, наиболее предпочтительно 0,70±0,15 м-3, и в особенности 0,70±0,10 м-3. В предпочтительном варианте осуществления, соотношение Cmax/дозу находится в диапазоне 0,80±0,70 м-3, более предпочтительно 0,80±0,60 м-3, еще более предпочтительно 0,80±0,50 м-3, также более предпочтительно 0,80±0,40 м-3, даже более предпочтительно 0,80±0,30 м-3, наиболее предпочтительно 0,80±0,20 м-3, и в особенности 0,80±0,10 м-3. В другом предпочтительном варианте осуществления, соотношение Cmax/дозу находится в диапазоне 0,90±0,70 м-3, более предпочтительно 0,90±0,60 м-3, еще более предпочтительно 0,90±0,50 м-3, также более предпочтительно 0,90±0,40 м-3, даже более предпочтительно 0,90±0,30 м-3, наиболее предпочтительно 0,90±0,20 м-3, и в особенности 0,90±0,10 м-3. В еще другом предпочтительном варианте осуществления, соотношение Cmax/дозу находится в диапазоне 1,00±0,70 м-3, более предпочтительно 1,00±0,60 м-3, еще более предпочтительно 1,00±0,50 м-3, также более предпочтительно 1,00±0,40 м-3, даже более предпочтительно 1,00±0,30 м-3, наиболее предпочтительно 1,00±0,20 м-3, и в особенности 1,00±0,10 м-3. В другом предпочтительном варианте осуществления, соотношение Cmax/дозу находится в диапазоне 1,10±0,70 м-3, более предпочтительно 1,10±0,60 м-3, еще более предпочтительно 1,10±0,50 м-3, также более предпочтительно 1,10±0,40 м-3, даже более предпочтительно 1,10±0,30 м-3, наиболее предпочтительно 1,10±0,20 м-3, и в особенности 1,10±0,10 м-3. В другом также предпочтительном варианте осуществления, соотношение Cmax/дозу находится в диапазоне 1,20±1,05 м-3, более предпочтительно 1,20±0,90 м-3, еще более предпочтительно 1,20±0,75 м-3, также более предпочтительно 1,20±0,60 м-3, даже более предпочтительно 1,20±0,45 м-3, наиболее предпочтительно 1,20±0,30 м-3, и в особенности 1,20±0,15 м-3.
В предпочтительном варианте осуществления, Cmax, 25 days находится в диапазоне от 1 до 150 мкг/м3, более предпочтительно в диапазоне от 10 до 120 мкг/м3, еще более предпочтительно в диапазоне от 15 до 100 мкг/м3, также более предпочтительно в диапазоне от 20 до 80 мкг/м3, наиболее предпочтительно в диапазоне от 20 до 70 мкг/м3, и в особенности в диапазоне от 25 до 60 мкг/м3
В предпочтительном варианте осуществления, Cmax, 25 days находится в диапазоне 25±20 мкг/м3, более предпочтительно в пределах от 25±15 мкг/м3, еще более предпочтительно в пределах от 25±12,5 мкг/м3, также более предпочтительно в пределах от 25±10 мкг/м3, наиболее предпочтительно в пределах от 25±7,5 мкг/м3, и в особенности в пределах от 25±5 мкг/м3. В другом предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 30±20 мкг/м3, более предпочтительно в пределах от 30±15 мкг/м3, еще более предпочтительно в пределах от 30±12,5 мкг/м3, также более предпочтительно в пределах от 30±10 мкг/м3, наиболее предпочтительно в пределах от 30±7,5 мкг/м3, и в особенности в пределах от 30±5 мкг/м3. В еще другом предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 35±20 мкг/м3, более предпочтительно в пределах от 35±15 мкг/м3, еще более предпочтительно в пределах от 35±12,5 мкг/м3, также более предпочтительно в пределах от 35±10 мкг/м3, наиболее предпочтительно в пределах от 35±7,5 мкг/м3, и в особенности в пределах от 35±5 мкг/м3. В другом также предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 40±20 мкг/м3, более предпочтительно в пределах от 40±15 мкг/м3, еще более предпочтительно в пределах от 40±12,5 мкг/м3, также более предпочтительно в пределах от 40±10 мкг/м3, наиболее предпочтительно в пределах от 40±7,5 мкг/м3, и в особенности в пределах от 40±5 мкг/м3. В предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 45±30 мкг/м3, более предпочтительно в пределах от 45±25 мкг/м3, еще более предпочтительно в пределах от 45±20 мкг/м3, также более предпочтительно в пределах от 45±15 мкг/м3, наиболее предпочтительно в пределах от 45±10 мкг/м3, и в особенности в пределах от 45±5 мкг/м3. В другом предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 50±30 мкг/м3, более предпочтительно в пределах от 50±25 мкг/м3, еще более предпочтительно в пределах от 50±20 мкг/м3, также более предпочтительно в пределах от 50±15 мкг/м3, наиболее предпочтительно в пределах от 50±10 мкг/м3, и в особенности в пределах от 50±5 мкг/м3. В еще другом предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 55±30 мкг/м3, более предпочтительно в пределах от 55±25 мкг/м3, еще более предпочтительно в пределах от 55±20 мкг/м3, также более предпочтительно в пределах от 55±15 мкг/м3, наиболее предпочтительно в пределах от 55±10 мкг/м3, и в особенности в пределах от 55±5 мкг/м3. В другом также предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 60±30 мкг/м3, более предпочтительно в пределах от 60±25 мкг/м3, еще более предпочтительно в пределах от 60±20 мкг/м3, также более предпочтительно в пределах от 60±15 мкг/м3, наиболее предпочтительно в пределах от 60±10 мкг/м3, и в особенности в пределах от 60±5 мкг/м3. В предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 65±30 мкг/м3, более предпочтительно в пределах от 65±25 мкг/м3, еще более предпочтительно в пределах от 65±20 мкг/м3, также более предпочтительно в пределах от 65±15 мкг/м3, наиболее предпочтительно в пределах от 65±10 мкг/м3, и в особенности в пределах от 65±5 мкг/м3. В другом предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 70±30 мкг/м3, более предпочтительно в пределах от 70±25 мкг/м3, еще более предпочтительно в пределах от 70±20 мкг/м3, также более предпочтительно в пределах от 70±15 мкг/м3, наиболее предпочтительно в пределах от 70±10 мкг/м3, и в особенности в пределах от 70±5 мкг/м3. В еще другом предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 75±30 мкг/м3, более предпочтительно в пределах от 75±25 мкг/м3, еще более предпочтительно в пределах от 75±20 мкг/м3, также более предпочтительно в пределах от 75±15 мкг/м3, наиболее предпочтительно в пределах от 75±10 мкг/м3, и в особенности в пределах от 75±5 мкг/м3. В другом также предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 80±30 мкг/м3, более предпочтительно в пределах от 80±25 мкг/м3, еще более предпочтительно в пределах от 80±20 мкг/м3, также более предпочтительно в пределах от 80±15 мкг/м3, наиболее предпочтительно в пределах от 80±10 мкг/м3, и в особенности в пределах от 80±5 мкг/м3. В предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 85±50 мкг/м3, более предпочтительно в пределах от 85±40 мкг/м3, еще более предпочтительно в пределах от 85±30 мкг/м3, также более предпочтительно в пределах от 85±20 мкг/м3, наиболее предпочтительно в пределах от 85±10 мкг/м3, и в особенности в пределах от 85±5 мкг/м3. В другом предпочтительном варианте осуществления, Cmax, ≥5days находится в диапазоне 90±50 мкг/м3, более предпочтительно в пределах от 90±40 мкг/м3, еще более предпочтительно в пределах от 90±30 мкг/м3, также более предпочтительно в пределах от 90±20 мкг/м3, наиболее предпочтительно в пределах от 90±10 мкг/м3, и в особенности в пределах от 90±5 мкг/м3. В еще другом предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 100±50 мкг/м3, более предпочтительно в пределах от 100±40 мкг/м3, еще более предпочтительно в пределах от 100±30 мкг/м3, также более предпочтительно в пределах от 100±20 мкг/м3, наиболее предпочтительно в пределах от 100±10 мкг/м3, и в особенности в пределах от 100±5 мкг/м3. В другом также предпочтительном варианте осуществления, Cmax, ≥5 days находится в диапазоне 120±30 мкг/м3, более предпочтительно в пределах от 120±25 мкг/м3, еще более предпочтительно в пределах от 120±20 мкг/м3, также более предпочтительно в пределах от 120±15 мкг/м3, наиболее предпочтительно в пределах от 120±10 мкг/м3, и в особенности в пределах от 120±5 мкг/м3.
Предпочтительно, соотношение Cmax, ≥5 days/дозу находится в диапазоне от 0,25 до 1,50 м-3, более предпочтительно в диапазоне от 0,40 до 1,10 м-3, еще более предпочтительно в диапазоне от 0,45 до 1,00 м-3, также более предпочтительно в диапазоне от 0,50 до 0,95 м-3, наиболее предпочтительно в диапазоне от 0,55 до 0,90 м-3, и в особенности в диапазоне от 0,60 до 0,85 м-3.
Предпочтительно, соотношение Cmax, 5 days/Cmax, 1 day составляет ≥1,00, более предпочтительно >1,00.
В предпочтительном варианте осуществления, соотношение Cmax, 5 days/Cmax, 1 day составляет ≥1,00,. более предпочтительно ≥1,10, еще более предпочтительно ≥1,20, также более предпочтительно ≥1,30, наиболее предпочтительно ≥1,40, и в особенности ≥1,50. В другом предпочтительном варианте осуществления, соотношение Cmac, 5 days/Cmax, 1 day составляет ≥1,60, более предпочтительно ≥1,70, еще более предпочтительно ≥1,80, также более предпочтительно ≥1,90, наиболее предпочтительно ≥2,00, и в особенности ≥2,10.
В предпочтительном варианте осуществления, соотношение Cmax, 5 days/Cmax, 1 day составляет ≤3,10, более предпочтительно ≤3,00, еще более предпочтительно ≤2,90, также более предпочтительно ≤2,80, наиболее предпочтительно ≤2,70, и в особенности ≤2,60. В другом предпочтительном варианте осуществления, соотношение Cmax, 5 days/Cmax, 1 day составляет ≤2,50, более предпочтительно ≤2,40, еще более предпочтительно ≤2,30, также более предпочтительно ≤2,20, наиболее предпочтительно ≤2,10, и в особенности ≤2,00.
Для целей настоящей заявки, C0-3h представляет собой наибольшую концентрацию в плазме фармакологически активного агента, достигаемую после однократного введения лекарственной дозированной формы в течение первых 3 часов после введения. Таким образом, для целей настоящей заявки, C0-3h, 1 day представляет собой наибольшую концентрацию в плазме фармакологически активного агента, достигаемую в течение первых 3 часов после введения один раз в сутки лекарственной дозированной формы в самый первый день интервала введения, в то время как C0-3h, 5 day представляет собой наибольшую концентрацию в плазме фармакологически активного агента, достигаемую в течение первых 3 часов после введения один раз в сутки лекарственной дозированной формы на пятый день указанного интервала введения, включающего 5 последовательных дней, где дозированная форма вводится один раз в сутки.
В предпочтительном варианте осуществления, лекарственная дозированная форма содержит фармакологически активный агент в таком количестве, что C0-3h, 5d ≤ C0-3h, 1d. В другом предпочтительном варианте осуществления, лекарственная дозированная форма содержит фармакологически активный агент в таком количестве, что C0-3h, 5d ≥ С0-3h, 1d. В предпочтительном варианте осуществления, частное (C0-3h, 5d)/(C0-3h, 1d) составляет самое большее 2,5 или 2,4 или 2,3, более предпочтительно самое большее 2,2 или 2,1 или 2,0, еще более предпочтительно самое большее 1,9 или 1,8 или 1,7, также более предпочтительно самое большее 1,6 или 1,5 или 1,4, наиболее предпочтительно самое большее 1,3 или 1,2 или 1,1, и в особенности самое большее 1,0 или 0,9 или 0,8. В другом предпочтительном варианте осуществления, частное (C0-3h, 5d)/(C0-3h, 1d) составляет по меньшей мере 0,8 или 0,9 или 1,0, более предпочтительно по меньшей мере 1,1 или 1,2 или 1,3, еще более предпочтительно по меньшей мере 1,4 или 1,5 или 1,6, также более предпочтительно по меньшей мере 1,7 или 1,8 или 1,9, наиболее предпочтительно по меньшей мере 2,0 или 2,1 или 2,2, и в особенности по меньшей мере 2,3 или 2,4 или 2,5.
В предпочтительном варианте осуществления, наивысшая концентрация в плазме фармакологически активного агента, достигаемая в день 5 5-дневного периода при введении один раз в сутки лекарственной дозированной формы, выше, чем наибольшие концентрации в плазме, достигаемые в первый и/или второй и/или третий и/или четвертый день указанного периода.
В предпочтительном варианте осуществления, суточная средняя концентрация в плазме фармакологически активного агента неуклонно повышается в течение первых 5 дней по меньшей мере 5-дневного периода при введении один раз в сутки лекарственной дозированной формы.
Предпочтительно, концентрация в плазме фармакологически активного агента, измеренная через 10 дней после однократного введения лекарственной дозированной формы, все еще составляет по меньшей мере 0,5 пг/мл, более предпочтительно по меньшей мере 1,0 пг/мл, еще более предпочтительно 1,25 пг/мл, также более предпочтительно по меньшей мере 1,5 пг/мл, наиболее предпочтительно по меньшей мере 1,75 пг/мл, и в особенности по меньшей мере 2,0 пг/мл.
Предпочтительно, концентрация в плазме фармакологически активного агента, измеренная через 10 безмедикаментозных дней после введения один раз в сутки лекарственной дозированной формы в течение по меньшей мере 5 последовательных дней, все еще составляет по меньшей мере 0,5 пг/мл, более предпочтительно по меньшей мере 1,0 пг/мл, еще более предпочтительно 1,25 пг/мл, также более предпочтительно по меньшей мере 1,5 пг/мл, наиболее предпочтительно по меньшей мере 1,75 пг/мл, и в особенности по меньшей мере 2,0 пг/мл.
В соответствии с изобретением, фармакокинетический параметр tmax находится в диапазоне от 0,5 до 16 ч. Предпочтительно, tmax находится в диапазоне от 1 до 12 ч, и в особенности в диапазоне от 2 до 10 ч.
В предпочтительном варианте осуществления, tmax находится в диапазоне 4±3,5 ч, более предпочтительно 4±3 ч, еще более предпочтительно 4±2,5 ч, также более предпочтительно 4±2 ч, даже более предпочтительно 4±1,5 ч, наиболее предпочтительно 4±1 ч, и в особенности 4±0,5 ч. В другом предпочтительном варианте осуществления, tmax находится в диапазоне 5±3,5 ч, более предпочтительно 5±3 ч, еще более предпочтительно 5±2,5 ч, также более предпочтительно 5±2 ч, даже более предпочтительно 5±1,5 ч, наиболее предпочтительно 5±1 ч, и в особенности 5±0,5 ч. В еще другом предпочтительном варианте осуществления, tmax находится в диапазоне 6±4 ч, более предпочтительно 6±3 ч, еще более предпочтительно 6±2,5 ч, также более предпочтительно 6±2 ч, даже более предпочтительно 6±1,5 ч, наиболее предпочтительно 6±1 ч, и в особенности 6±0,5 ч. В другом также предпочтительном варианте осуществления, tmax находится в диапазоне 8±7 ч, более предпочтительно 8±6 ч, еще более предпочтительно 8±5 ч, также более предпочтительно 8±4 ч, даже более предпочтительно 8±3 ч, наиболее предпочтительно 8±2 ч, и в особенности 8±1 ч. В также другом предпочтительном варианте осуществления, tmax находится в диапазоне 12±3 ч, более предпочтительно 12±2 ч, и наиболее предпочтительно 12±1 ч.
В предпочтительном варианте осуществления, tmax, ≥5 days находится в диапазоне от 1 до 12 ч, более предпочтительно в диапазоне от 1,5 до 10 ч, еще более предпочтительно в диапазоне от 2 до 9 ч, также более предпочтительно в диапазоне от 2,5 до 8 ч, наиболее предпочтительно в диапазоне от 3 до 7 ч, и в особенности в диапазоне от 4 to 6 ч.
В предпочтительном варианте осуществления, tmax, ≥5 days находится в диапазоне 4±3,5 ч, более предпочтительно 4±3 ч, еще более предпочтительно 4±2,5 ч, также более предпочтительно 4±2 ч, даже более предпочтительно 4±1,5 ч, наиболее предпочтительно 4±1 ч, и в особенности 4±0,5 ч. В другом предпочтительном варианте осуществления, tmax, ≥5 days находится в диапазоне 5±3,5 ч, более предпочтительно 5±3 ч, еще более предпочтительно 5±2,5 ч, также более предпочтительно 5±2 ч, даже более предпочтительно 5±1,5 ч, наиболее предпочтительно 5±1 ч, и в особенности 5±0,5 ч. В еще другом предпочтительном варианте осуществления, tmax, ≥5 days находится в диапазоне 6±4 ч, более предпочтительно 6±3 ч, еще более предпочтительно 6±2,5 ч, также более предпочтительно 6±2 ч, даже более предпочтительно 6±1,5 ч, наиболее предпочтительно 6±1 ч, и в особенности 6±0,5 ч.
Предпочтительно, соотношение AUC0-t/дозу находится в диапазоне от 0,3 до 20 ч/м3, более предпочтительно в диапазоне от 0,4 до 18 ч/м3, еще более предпочтительно в диапазоне от 0,5 до 16,5 ч/м3 и наиболее предпочтительно в диапазоне от 0,55 до 12,5 ч/м3. В предпочтительном варианте осуществления, соотношение AUC0-t/дозу находится в диапазоне 3±2,5 ч/м3, более предпочтительно 3±2 ч/м3, еще более предпочтительно 3±1,5 ч/м3, также более предпочтительно 3±1 ч/м3, даже более предпочтительно 3±0,75 ч/м3, наиболее предпочтительно 3±0,5 ч/м3, и в особенности 3±0,25 ч/м3. В другом предпочтительном варианте осуществления, соотношение AUC0-t/дозу находится в диапазоне 6±5 ч/м3, более предпочтительно 6±4 ч/м3, еще более предпочтительно 6±3 ч/м3, также более предпочтительно 6±2 ч/м3, даже более предпочтительно 6±1,5 ч/м3, наиболее предпочтительно 6±1 ч/м3, и в особенности 6±0,5 ч/м3. В еще другом предпочтительном варианте осуществления, соотношение AUC0-t/дозу находится в диапазоне 7,5±7 ч/м3, более предпочтительно 7,5±6 ч/м3, еще более предпочтительно 7,5±5 ч/м3, также более предпочтительно 7,5±4 ч/м3, даже более предпочтительно 7,5±3 ч/м3, наиболее предпочтительно 7,5±2 ч/м3, и в особенности 7,5±1 ч/м3. В другом также предпочтительном варианте осуществления, соотношение AUC0-t/дозу находится в диапазоне 9±8 ч/м3, более предпочтительно 9±7 ч/м3, еще более предпочтительно 9±5 ч/м3, также более предпочтительно 9±4 ч/м3, даже более предпочтительно 9±3 ч/м3, наиболее предпочтительно 9±2 ч/м3, и в особенности 9±1 ч/м3. В другом предпочтительном варианте осуществления, соотношение AUC0-72h/дозу находится в диапазоне 10±7 ч/м3, более предпочтительно 10±6 ч/м3, еще более предпочтительно 10±5 ч/м3, также более предпочтительно 10±4 ч/м3, даже более предпочтительно 10±3 ч/м3, наиболее предпочтительно 10±2 ч/м3, и в особенности 10±1 ч/м3.
Предпочтительно, соотношение AUC0-72h/дозу находится в диапазоне от 0,3 до 20 ч/м3, более предпочтительно в диапазоне от 0,4 до 18 ч/м3, еще более предпочтительно в диапазоне от 0,5 до 16,5 ч/м3 и наиболее предпочтительно в диапазоне от 0,55 до 12,5 ч/м3. В предпочтительном варианте осуществления, соотношение AUC0-72h/дозу находится в диапазоне 3±2,5 ч/м3, более предпочтительно 3±2 ч/м3, еще более предпочтительно 3±1,5 ч/м3, также более предпочтительно 3±1 ч/м3, даже более предпочтительно 3±0,75 ч/м3, наиболее предпочтительно 3±0,5 ч/м3, и в особенности 3±0,25 ч/м3. В другом предпочтительном варианте осуществления, соотношение AUC0-72h/дозу находится в диапазоне 6±5 ч/м3, более предпочтительно 6±4 ч/м3, еще более предпочтительно 6±3 ч/м3, также более предпочтительно 6±2 ч/м3, даже более предпочтительно 6±1,5 ч/м3, наиболее предпочтительно 6±1 ч/м3, и в особенности 6±0,5 ч/м3. В еще другом предпочтительном варианте осуществления, соотношение AUC0-72h/дозу находится в диапазоне 7,5±7 ч/м3, более предпочтительно 7,5±6 ч/м3, еще более предпочтительно 7,5±5 ч/м3, также более предпочтительно 7,5±4 ч/м3, даже более предпочтительно 7,5±3 ч/м3, наиболее предпочтительно 7,5±2 ч/м3, и в особенности 7,5±1 ч/м3. В другом также предпочтительном варианте осуществления, соотношение AUC0-72h/дозу находится в диапазоне 9±8 ч/м3, более предпочтительно 9±7 ч/м3, еще более предпочтительно 9±5 ч/м3, также более предпочтительно 9±4 ч/м3, даже более предпочтительно 9±3 ч/м3, наиболее предпочтительно 9±2 ч/м3, и в особенности 9±1 ч/м3.
В предпочтительном варианте осуществления, лекарственная дозированная форма в соответствии с изобретением вводится один раз в сутки в течение интервала введения, включающего начальную фазу, на протяжении которой временная зависимость концентрации в плазме существенно изменяется изо дня в день, и установившуюся равновесную фазу, в течение которой временная зависимость концентрации в плазме существенно не изменяется изо дня в день. В связи с этим, на протяжении установившейся равновесной фазы временная зависимость концентрации в плазме может все еще изменяться в течение дня, то есть концентрация в плазме, измеренная, например, через 1 час после введения, может существенно отличаться от концентрации в плазме, измеренной, например, через 2, 3, 4, 6, 12 или 20 часов после аналогичного введения в тот же самый день. Тем не менее, на протяжении установившейся равновесной фазы, концентрация в плазме, измеренная через X часов после введения в день N, существенно не отличается от концентрации в плазме, измеренной через X часов после введения на следующий день N+1. Предпочтительно, начальная фаза продолжается 1, 2, 3, 4 или 5 последовательных дней, пока не начинается установившаяся равновесная фаза. В предпочтительном варианте осуществления, на протяжении установившейся равновесной фазы, лекарственная дозированная форма обеспечивает и поддерживает при введении один раз в сутки фармакологически эффективную концентрацию в плазме фармакологически активного агента в соответствии с общей формулой (I) в течение по меньшей мере 12 ч, предпочтительно по меньшей мере 18 ч, более предпочтительно по меньшей мере 20 ч, также более предпочтительно по меньшей мере 22 ч, и в особенности все 24 ч по меньшей мере 1,0 пг/мл, по меньшей мере 2,0 пг/мл, или по меньшей мере 3,0 пг/мл, более предпочтительно по меньшей мере 4,0 пг/мл, по меньшей мере 5,0 пг/мл, или по меньшей мере 6,0 пг/мл, еще более предпочтительно по меньшей мере 7,0 пг/мл, по меньшей мере 8,0 пг/мл, или по меньшей мере 9,0 пг/мл, также более предпочтительно по меньшей мере 10 пг/мл, по меньшей мере 12,5 пг/мл, или по меньшей мере 15 пг/мл, даже более предпочтительно по меньшей мере 17,5 пг/мл, по меньшей мере 20 пг/мл, или по меньшей мере 22,5 пг/мл, наиболее предпочтительно по меньшей мере 25 пг/мл, по меньшей мере 27,5 пг/мл, или по меньшей мере 30 пг/мл, и в особенности по меньшей мере 35 пг/мл, по меньшей мере 40 пг/мл, или по меньшей мере 50 пг/мл.
В предпочтительном варианте осуществления, лекарственная дозированная форма в соответствии с изобретением является монолитной.
В другом предпочтительном варианте осуществления, лекарственная дозированная форма в соответствии с изобретением содержит ядро, которое окружено оболочкой или инкапсулирующим материалом. В предпочтительном варианте осуществления, ядро представляет собой жидкость и фармакологически активный агент в соответствии с общей формулой (I) диспергирован, предпочтительно растворен в жидкости.
В предпочтительном варианте осуществления, лекарственная дозированная форма в соответствии с изобретением обеспечивает фармакологически активный агент в соответствии с общей формулой (I) в форме само-(микро) эмульгирующих систем доставки лекарственных средств, твердых растворов, наночастиц, циклодекстриновых комплексов, липосом, мицелл, микронизированных и/или аморфных структур.
В общих чертах, варианты для приготовления препарата плохо растворимых в воде лекарственных средств включают кристаллические твердые, аморфные и липидные препараты.
Скорость растворения фармакологически активного агента из кристаллических препаратов может быть повышена путем уменьшения размера частиц, таким образом повышая площадь поверхности для растворения, например, с помощью общепринятой микронизации фармакологически активного агента до размера частиц приблизительно 2-5 мкм. В некоторых случаях, этого недостаточно и применяют технологию нанокристаллов. Нанокристаллы имеют размер частиц 100-250 нм, который может быть получен с помощью измельчения в шаровой мельнице или с помощью технологии плотного газа.
Твердые растворы обеспечивают фармакологически активный агент в аморфном состоянии, иммобилизированном на полимере. Аморфные растворы могут содержать поверхностно-активные вещества и полимеры, таким образом обеспечивая поверхностную активность в процессе дисперсии при контактировании с водой. Твердые растворы могут быть образованы с использованием различных технологий, таких как распылительная сушка и экструзия расплава.
Липидные препараты, проявляющие различные характеристики, можно использовать для диспергирования и образования мицеллярных растворов, включая простые растворы и самоэмульгирующие системы доставки лекарственных средств (SEDDS). В зависимости наполнителей, для одних требуется переваривание (например, простые маслянистые жидкости), другие легко могут быть абсорбированы без переваривания. Эти наполнители могут быть классифицированы в соответствии с системой классификации липидных препаратов (LFCS) следующим образом:
Другим вариантом является образование циклодекстриновых комплексов, в которых фармакологически активный агент расположен в полости циклодекстрина и поэтому присутствует в более растворимой форме в присутствии водной среды. Успех подгонки существенным образом зависит от качества циклодекстринов, а также от физико-химических свойств и размера фармакологически активного агента.
В предпочтительном варианте осуществления, лекарственная дозированная форма в соответствии с изобретением может рассматриваться как самоэмульгирующаяся система доставки лекарственного средства (SEDDS).
С этой целью, фармакологически активный агент в соответствии с общей формулой (I) предпочтительно заделан в самоэмульгирующийся препарат. Так называемая самоэмульгирующаяся система доставки лекарственного средства (SEDDS) представляет собой систему доставки лекарственного средства, в которой используется эмульсия, получаемая химическими, а не механическими, методами. Другими словами с помощью присущих свойств лекарственного препарата, а не путем специфического смешивания и обработки. Указанный препарат разводят в водной среде и получают эмульсию. В случае если средний размер капли меньше или равен 50 нм, самоэмульгирующаяся система доставки лекарственного средства обозначается как самомикроэмульгирующаяся = система доставки лекарственного средства (SMEDDS). В соответствии с системой классификации липидных препаратов, эти препараты обычно относятся к группе препаратов III типа.
Предпочтительной подгруппой SEDDS являются самоэмульгирующиеся масляные препараты (SEOF). SEOF обычно содержат природное или синтетическое масло, поверхностно-активное вещество и гидрофильный растворитель и иногда сорастворители. Основной характеристикой SEOF является их способность образовывать тонкие эмульсии масло-в-воде или микро эмульсии при слабом перемешивании вслед за разбавлением водными фазами. Эти препараты можно диспергировать в просвет желудочно-кишечного тракта с образованием микро эмульсий или тонкодисперсных эмульсий, при разведении желудочно-кишечными жидкостями.
В другом предпочтительном варианте осуществления, лекарственная дозированная форма содержит фармакологически активный агент в соответствии с общей формулой (I) в форме твердого раствора, то есть молекулярно диспергирован в твердом матриксе. Твердый раствор предпочтительно содержит фармакологически активный агент в соответствии с общей формулой (I) в молекулярно диспергированной форме и аморфный полимерный матрикс, имеющий сравнительно большую удельную поверхность. Фармакологически активный агент в соответствии с общей формулой (I) предпочтительно представлен в молекулярно диспергированной форме, то есть соединение действительно растворено и равномерно распределено в отвержденном растворе. Размер частиц соединения не является ни кристаллическим, ни мелкокристаллическим. Типичный размер частиц предпочтительно составляет 0,1-1 нм.
В еще другом предпочтительном варианте осуществления, фармакологически активный агент в соответствии с общей формулой (I) обеспечен с помощью нанотехнологического препарата со средним размером наночастиц предпочтительно меньше, чем 1 мкм. Фармакологически активный агент в соответствии с общей формулой (I) предпочтительно смешан с указанными наночастицами и таким образом адсорбирован на поверхности частиц. Наночастицы предпочтительно выбирают из органических наночастиц и неорганических наночастиц.
Органические наночастицы предпочтительно содержат небольшие белки, которые присутствуют в виде кластера или агломерата небольших белков, олигопептидов или липидов.
Неорганические наночастицы предпочтительно содержат кристаллические силикаты. Эти силикаты имеют минеральное происхождение или искусственные силикаты, такие как металлосиликаты (например, цеолиты). В предпочтительном варианте осуществления, наночастицы модифицированы таким образом, что они несут электростатический заряд. Наночастицы предпочтительно представляют собой ультра тонкоизмельченные силикаты и фармакологически активный агент в соответствии с общей формулой (I) предпочтительно связан с микропористой поверхностью наночастиц.
Образование наночастиц известно специалисту в данной области техники. Одним методом является получение коллоидных наночастиц в качестве носителей для орального высвобождения лекарственного средства путем распыления фармакологически активного агента в соответствии с общей формулой (I) под давлением при определенной температуре, совместно с подходящим материалом носителя, таким как протамин, через джеты, которые оборудованы фильтровальными плитами, в сильно охлажденные колонны. Результатом быстрого охлаждения является аморфная фаза, состоящая из наночастиц. Другим методом является измельчение фармакологически активного агента в соответствии с общей формулой (I) с подходящими макромолекулами в растворе. При добавлении гидрофобных соединений, молекулы растворителя удаляются из раствора и происходит десольватация. В связи с этим происходит образование очень маленьких частиц, в которые интегрирован фармакологически активный агент в соответствии с общей формулой (I). Для отвердения образованных наночастиц в раствор может быть добавлен сшиватель.
Для получения, например, твердой липидной наночастицы можно использовать метод гомогенизации под высоким давлением и последующего охлаждения распылением. Предпочтительно, фармакологически активный агент в соответствии с общей формулой (I) растворен в подходящем растворителе или в форме субмикрочастиц. При необходимости, в раствор можно добавлять липидный носитель и поверхностно-активное вещество. В завершение тонкоизмельченные пористые заполнители в качестве внешней фазы, а также глиданты и дополнительные поверхностно-активные вещества могут быть добавлены для заполнения полученного препарата, например, в капсулы, такие как твердые желатиновые капсулы.
В другом также предпочтительном варианте осуществления, фармакологически активный агент в соответствии с общей формулой (I) обеспечиваются в виде циклодекстриновых (включение) комплексов.
Циклодекстрины состоят из молекул Сахаров, образующих кольцо, и обычно содержат 5 или больше α-D-гликопиранозидных единиц, которые связаны посредством положения 1-4. Типичное количество связанных мономеров Сахаров находится в диапазоне от 6 до 8 единиц. Шестичленная кольцевая молекула сахара называется α-циклодекстрин. Семичленная кольцевая молекула сахара называется β-циклодекстрин и восьмичленная кольцевая молекула сахара называется γ-циклодекстрин. Форма этих соединений представляет собой тороид с большим и меньшим отверстиями, подверженными воздействию растворителя. Вследствие этого образования внутренняя часть тороида не является гидрофобной, но значительно менее гидрофильна по сравнению с водной окружающей средой и, следовательно, способна принимать гидрофобные молекулы. Наружная часть тороида достаточно гидрофильна для придания циклодекстринам растворимости в воде.
Включение фармакологически активного ингредиента в соответствии с общей формулой (I) в циклодекстрины чрезвычайно модифицирует физические и химические свойства. В большинстве случаев механизм контролированного разложения таких комплексов и получающегося в результате этого высвобождения лекарственного средства основан на изменении pH водных растворов, что приводит к отщеплению водорода или ионных связей между циклодекстринами и включенными молекулами. Альтернативными способами разрушения комплексов является нагревание или воздействие ферментов, способных расщеплять α-1-4 связи между α-D-гликопиранозидами.
В другом предпочтительном варианте осуществления, фармакологически активный агент в соответствии с общей формулой (I) обеспечивается в форме липосом. Липосома предпочтительно состоит из фосфолипидов и предпочтительно имеет сферическую форму. Оболочка этой формы предпочтительно представляет собой ламеллярную или бислойную структуру. Другим типом расположения фосфолипидов является монослой.
Фосфолипиды содержат молекулы с амфифильным свойством, то есть молекулы имеют гидрофобную (липофильную) и гидрофильную (липофобную) часть. В присутствии воды, гидрофильная часть притягивается водой и образует поверхность, обращенную к воде, в то время как гидрофобная часть отталкивается водой и образует поверхность вдали от воды. Поэтому амфифильные молекулы сами по себе упорядочиваются в одном из вышеуказанных типов.
Двухслойные структуры предпочтительно располагаются в сферической конфигурации, где внутренняя часть заполнена водным раствором. Этот тип называется “липосома”. Гидрофобные части молекул обращены друг к другу в середине слоя и гидрофильные части молекул обращены к молекулам воды снаружи липосомы. Водный раствор внутри липосомы является таким же, как и снаружи липосомы. Ингредиенты, растворенные в этом водном растворе, например, фармакологически активные агенты в соответствии с общей формулой (I), таким образом находятся внутри липосомы. Типичный диаметр липосом находится в диапазоне от 25 нм до 1 мкм. Более мелкие липосомы (25 нм - 200 нм) состоят из одного единственного бислоя, тогда как более крупные липосомы (200 нм - 1 мкм) содержат несколько двухслойных оболочек на верхушке друг друга.
Монослойные структуры также упорядочиваются в сферические конфигурации. Вследствие амфифильного характера молекул и сферической конфигурации монослойных структур, внутренняя часть сферических структур заполнена /образована гидрофобными частями молекул. Эти типы называются мицеллами. Внутри структуры отсутствует растворитель. В предпочтительном варианте осуществления, внутренние части мицелл содержат фармакологически активные агенты в соответствии с общей формулой (I).
В другом предпочтительном варианте осуществления фармакологически активный агент в соответствии с общей формулой (I) обеспечивается в микронизированном состоянии. С помощью техники микронизации могут быть получены частицы фармакологически активного агента в соответствии с общей формулой (I) с диаметром в наномерном диапазоне. Такие частицы имеют большое отношение поверхности к объему.
Перемалывание и дробление является пригодным методом для получения частиц в наномерном диапазоне. Сложные техники для микронизации включают RESS (быстрое расширение сверхкритических растворов), SAS (сверхкритический антирастворитель) и PGSS (образование частиц в насыщенных газом растворах).
В RESS методе используют сверхкритическую жидкость, где фармакологически активный агент в соответствии с общей формулой (I) растворен при высоком давлении и температуре, таким образом получая гомогенную сверхкритическую фазу. После распространения раствора через форсунку, образуются небольшие частицы. Вследствие распространения в конце форсунки растворенный фармакологически активный агент в соответствии с общей формулой (I) осаждается в виде кристаллов и включает небольшое количество растворителя. Растворитель изменяет со сверхкритического жидкостного состояния в нормальное состояние, предпочтительно газовую фазу, и кристаллы распадаются изнутри наружу. Таким образом и вследствие того факта, что кристаллы сталкиваются друг с другом, образуются частицы с диаметром в наномерном диапазоне.
В SAS методе фармакологически активный агент в соответствии с общей формулой (I) растворен в предпочтительно органическом растворителе. Сверхкритическую жидкость добавляют в раствор под давлением и таким образом вынуждают также растворяться в растворителе. Вследствие этого, объем полной системы повышается и растворимость фармакологически активного агента в соответствии с общей формулой (I) снижается. Вследствие снижения его растворимости, соединение в соответствии с общей формулой (I) осаждается и образует частицы, имеющие небольшой диаметр.
PGSS метод похож на SAS метод. В этом случае, фармакологически активный агент в соответствии с общей формулой (I) расплавлен и сверхкритическая жидкость растворена в расплаве. Вследствие распространения через форсунку, фармакологически активный агент в соответствии с общей формулой (I) осаждается и образует частицы в наномерном диапазоне.
В предпочтительном варианте осуществления, лекарственная дозированная форма в соответствии с изобретением содержит
- неионное поверхностно-активное вещество (например, Кремофор® EL, Кремофор® ОВ 40, Кремофор® ОВ 60, d-альфа-токоферол полиэтилен гликоль 1000 сукцинат, полисорбат 20, полисорбат 80, Солютол® HS 15, сорбитан моноолеат, полоксамер 407, Лабрафил® M-1944CS, Лабрафил® М-2125CS, Лабразол®, Гелуцип® 44/14, Softigen® 767, и моно- и ди-эфиры жирных кислот PEG 300, 400 или 1750); и/или
- анионное поверхностно-активное вещество, такое как лаурилсульфат натрия (додецилсульфат натрия, например, Texapon® К12), цетилсульфат натрия (например, Lanette Е®), цетилстеарил сульфат натрия, стеарил сульфат натрия, диоктилсульфосукцинат натрия (докузат натрия); и/или
- нерастворимый в воде липид (например, касторовое масло, кукурузное масло хлопковое масло, оливковое масло, арахисовое масло, масло перечной мяты, сафлоровое масло, кунжутное масло, соевое масло, гидрогенизированные растительные масла, гидрогенизированное соевое масло, и среднецепочечные триглицериды кокосового масла и пальмового растительного масла); и/или
- органическая жидкость/полутвердое вещество (например, пчелиный воск, d-альфа-токоферол, олеиновая кислота, среднецепочечные моно- и диглицериды); и/или
- циклодекстрин (например, альфа-циклодекстрин, бета-циклодекстрин, гидроксипропил-бета-циклодекстрин, и сульфобутилэфир-бета-циклодекстрин); и/или
- фосфолипид (например, гидрогенизированные соевый фосфатидилхолин, дистеароилфосфатидилглицерин, L-альфа-димиристоилфосфатидилхолин, и L-альфа-димиристоилфосфатидилглицерин).
Предпочтительно, фармакологически активный агент в соответствии с общей формулой (I) молекулярно диспергирован в матриксе.
В предпочтительном варианте осуществления, фармакологически активный агент в соответствии с общей формулой (I) молекулярно диспергирован в некристаллическом матриксе.
В другом предпочтительном варианте осуществления, фармакологически активный агент в соответствии с общей формулой (I) молекулярно диспергирован в неаморфном матриксе.
Предпочтительно, фармакологически активный агент в соответствии с общей формулой (I) гомогенно распределен в лекарственной дозированной форме в соответствии с изобретением. Содержание фармакологически активного агента в соответствии с общей формулой (I) двух сегментов лекарственной дозированной формы, имеющих объем 1,0 мм3 каждый, отличается друга от друга предпочтительно не более, чем ±10%, более предпочтительно не более, чем ±7,5%, еще более предпочтительно не более, чем ±5,0%, наиболее предпочтительно не более, чем ±2,5%, и в особенности не более, чем ±1,0%. Если лекарственная дозированная форма инкапсулирована или покрыта оболочкой, то указанные два сегмента лекарственной дозированной формы, имеющих объем 1,0 мм3 каждый, предпочтительно представляют собой сегменты ядра, то есть не содержат какой-либо инкапсулирующей среды или пленочной оболочки, соответственно.
Предпочтительно, лекарственная дозированная форма в соответствии с изобретением характеризуется сравнительно гомогенным распределением плотности. Предпочтительно, плотности двух сегментов лекарственной дозированной формы, имеющих объем 1,0 мм3 каждый, отличается друга от друга не более, чем ±10%, более предпочтительно не более, чем ±7,5%, еще более предпочтительно не более, чем ±5,0%, наиболее предпочтительно не более, чем ±2,5%, и в особенности не более, чем ±1,0%. Если лекарственная дозированная форма инкапсулирована, то указанные два сегмента лекарственной дозированной формы, имеющих объем 1,0 мм3 каждый, предпочтительно представляют собой сегменты ядра, то есть не содержат какой-либо инкапсулирующей среды или пленочной оболочки.
В предпочтительном варианте осуществления, лекарственная дозированная форма дополнительно содержит поверхностно-активное вещество. Предпочтительно, поверхностно-активное вещество содержится в матриксе, в котором диспергирован фармакологически активный агент в соответствии с общей формулой (I), предпочтительно молекулярно.
В предпочтительном варианте осуществления, фармакологически активный агент в соответствии с общей формулой (I) и поверхностно-активное вещество равномерно гомогенно распределено в матриксе таким образом, что матрикс не содержит любых сегментов, где либо фармакологически активный агент в соответствии с общей формулой (I) присутствует при отсутствии поверхностно-активного вещества или где поверхностно-активное вещество присутствует при отсутствии фармакологически активного агента в соответствии с общей формулой (I).
В предпочтительном варианте осуществления, лекарственная дозированная форма содержит поверхностно-активное вещество. В другом предпочтительном варианте осуществления, лекарственная дозированная форма содержит смесь двух или более поверхностно-активных веществ.
В предпочтительном варианте осуществления, поверхностно-активное вещество действует в качестве эмульгатора типа масло-вода. В другом предпочтительном варианте осуществления, поверхностно-активное вещество действует в качестве эмульгатора типа вода-масло.
Предпочтительно, лекарственная дозированная форма содержит поверхностно-активное вещество, имеющее гидрофильно-липофильный баланс (HLB) по меньшей мере 10 или по меньшей мере 11. Более предпочтительно, гидрофильно-липофильный баланс (HLB) составляет по меньшей мере 12 или по меньшей мере 13. Наиболее предпочтительно, гидрофильно-липофильный баланс (HLB) находится в диапазоне от 14 до 16.
Предпочтительно, гидрофильно-липофильный баланс (HLB) поверхностно-активного вещества составляет самое большее 30, более предпочтительно самое большее 28, еще более предпочтительно самое большее 26, также более предпочтительно самое большее 24, даже более предпочтительно самое большее 22, наиболее предпочтительно самое большее 20 и в особенности самое большее 18.
В другом предпочтительном варианте осуществления, гидрофильно-липофильный баланс (HLB) поверхностно-активного вещества составляет по меньшей мере 27, более предпочтительно по меньшей мере 29, еще более предпочтительно по меньшей мере 31, также более предпочтительно по меньшей мере 33, даже более предпочтительно по меньшей мере 35, наиболее предпочтительно по меньшей мере 37 и в особенности по меньшей мере 39.
В предпочтительном варианте осуществления, HLB значение поверхностно-активного вещества находится в диапазоне 10±3,5, более предпочтительно 10±3, еще более предпочтительно 10±2,5, также более предпочтительно 10±2, даже более предпочтительно 10±1,5, наиболее предпочтительно 10±1, и в особенности 10±0,5. В другом предпочтительном варианте осуществления, HLB значение поверхностно-активного вещества находится в диапазоне 12±3,5, более предпочтительно 12±3, еще более предпочтительно 12±2,5, также более предпочтительно 12±2, даже более предпочтительно 12±1,5, наиболее предпочтительно 12±1, и в особенности 12±0,5. В еще другом предпочтительном варианте осуществления, HLB значение поверхностно-активного вещества находится в диапазоне 14±3,5, более предпочтительно 14±3, еще более предпочтительно 14±2,5, также более предпочтительно 14±2, даже более предпочтительно 14±1,5, наиболее предпочтительно 14±1, и в особенности 14±0,5. В другом предпочтительном варианте осуществления, HLB значение поверхностно-активного вещества находится в диапазоне 15±3,5, более предпочтительно 15±3, еще более предпочтительно 15±2,5, также более предпочтительно 15±2, даже более предпочтительно 15±1,5, наиболее предпочтительно 15±1, и в особенности 15±0,5. В другом также предпочтительном варианте осуществления, HLB значение поверхностно-активного вещества находится в диапазоне 16±3,5, более предпочтительно 16±3, еще более предпочтительно 16±2,5, также более предпочтительно 16±2, даже более предпочтительно 16±1,5, наиболее предпочтительно 16±1, и в особенности 16±0,5. В другом предпочтительном варианте осуществления, HLB значение поверхностно-активного вещества находится в диапазоне 18±3,5, более предпочтительно 18±3, еще более предпочтительно 18±2,5, также более предпочтительно 18±2, даже более предпочтительно 18±1,5, наиболее предпочтительно 18±1, и в особенности 18±0,5.
Поверхностно-активное вещество может быть ионным, амфотерным или неионным.
В предпочтительном варианте осуществления, лекарственная дозированная форма содержит ионное поверхностно-активное вещество, в особенности анионное поверхностно-активное вещество.
Подходящие анионные поверхностно-активные вещества включают, но не ограничиваясь только ими, сложные эфиры серной кислоты, такие как лаурилсульфат натрия (додецилсульфат натрия, например, Texapon® К12), цетилсульфат натрия (например, Lanette Е®), цетилстеарил сульфат натрия, стеарил сульфат натрия, диоктилсульфосукцинат натрия (докузат натрия); и их соответствующие калиевые или кальциевые соли.
Предпочтительно, анионное поверхностно-активное вещество имеет общую формулу (II-a)
где n представляет собой целое число от 8 до 30, предпочтительно 10 до 24, более предпочтительно 12 до 18; и М выбирают из Li+, Na+, K+, NH4+ 1/2 Mg2+ и 1/2 Са2+.
Другие подходящие анионные поверхностно-активные вещества включают соли холевой кислоты, включая гликохолат натрия (например, Konakion® ММ, Cernevit®), таурохолат натрия и соответствующие калиевые или аммониевые соли.
В другом предпочтительном варианте осуществления, лекарственная дозированная форма содержит неионное поверхностно-активное вещество. Подходящие неионные поверхностно-активные вещества включают, но не ограничиваясь только ими
- жирные спирты, которые могут быть линейными или разветвленными, такие как цетиловый спирт, стеариловый спирт, цетилстеариловый спирт, 2-октилдодекан-1-ол и 2-гексилдекан-1-ол;
- стеролы, такие как холестерин;
- неполные сложные эфиры жирных кислот и сорбитана, такие как сорбитанмонолаурат, сорбитанмонопальмитат, сорбитанмоностеарат, сорбитантристеарат, сорбитанмоноолеат, сорбитанполутораолеат и сорбитантриолеат;
- неполные сложные эфиры полиоксиэтилен сорбитана и жирной кислоты (сложные эфиры полиоксиэтилен-сорбитана и жирной кислоты), предпочтительно моноэфир полиоксиэтилен сорбитана и жирной кислоты, диэфир полиоксиэтилен сорбитана и жирной кислоты, или триэфир полиоксиэтилен сорбитана и жирной кислоты; например, моно- и три- лаурил, пальмитил, стеарил и олеил сложные эфиры, такие как тип, известен под названием “полисорбат” и коммерчески доступен под торговым названием “Твин”, включая Твин® 20 [полиоксиэтилен(20)сорбитан монолаурат], Твин® 21 [полиоксиэтилен(4)сорбитан монолаурат], Твин® 40 [полиоксиэтилен(20)сорбитан монопальмитат], Твин® 60 [полиоксиэтилен(20)сорбитан моностеарат], Твин® 65 [полиоксиэтилен(20)сорбитан тристеарат], Твин® 80 [полиоксиэтилен(20)сорбитан моноолеат], Твин 81 [полиоксиэтилен(5)сорбитан моноолеат], и Твин® 85 [полиоксиэтилен(20)сорбитан триолеат]; предпочтительно моноэфир полиоксиэтиленсорбитана и жирной кислоты в соответствии с общей формулой (II-b)
где (w+x+y+z) находится в диапазоне от 15 до 100, предпочтительно от 16 до 80, более предпочтительно 17 до 60, еще более предпочтительно от 18 до 40 и наиболее предпочтительно 19 до 21;
и алкилен представляет собой необязательно насыщенную алкиленовую группу, содержащую 6 до 30 атомов углерода, более предпочтительно 8 до 24 атома углерода и наиболее предпочтительно 10 до 16 атомов углерода;
- сложные эфиры полиоксиэтиленглицерина и жирных кислот, такие как смеси моно-, ди- и триэфиров глицерина и ди- и моноэфиров макроголей, имеющих молекулярный вес в диапазоне от 200 до 4000 г/моль, например, макроголглицеринкаприлокапрат, макроголглицеринлаурат, макроголглицеринококоат, макроголглицеринлинолеат, макрогол-20-глицеринмоностеарат, макрогол-6-глицеринкаприлокапрат, макроголглицеринолеат; макроголглицеринстеарат, макроголглицерингидроксистеарат (например, Кремофор® ОВ 40), и макроголглицеринрицинолеат (например, Кремофор® EL);
- полиоксиэтиленовые эфиры жирных кислот, жирная кислота предпочтительно имеет от приблизительно 8 до приблизительно 18 атомов углерода, например, макрогололеат, макроголстеарат, макрогол-15-гидроксистеарат, полиоксиэтиленовые сложные эфиры 12-гидроксистеариновой кислоты, такие как тип, известен и коммерчески доступен под торговым названием “Солютол HS 15”; предпочтительно в соответствии с общей формулой (II-c)
где n представляет собой целое число от 6 до 500, предпочтительно 7 до 250, более предпочтительно 8 до 100, еще более предпочтительно от 9 то 75, также более предпочтительно 10 до 50, даже более предпочтительно 11 до 30, наиболее предпочтительно 12 до 25, и в особенности 13 до 20; и где m представляет собой целое число от 6 до 28; более предпочтительно 6 до 26, еще более предпочтительно 8 до 24, также более предпочтительно 10 до 22, даже более предпочтительно 12 до 20, наиболее предпочтительно 14 до 18 и в особенности 16;
- полиоксиэтиленовые эфиры жирного спирта, например, макроголцетилстеариловый эфир, макроголариловый эфир, макрогололеиловый эфир, макроголстеариловый эфир;
- полиоксипропилен-полиоксиэтилен блок-сополимеры (полоксамеры);
- сложные эфиры жирных кислот и сахарозы; например, дистеарат сахарозы, диолеат сахарозы, дипальмитат сахарозы, моностеарат сахарозы, моноолеат сахарозы, монопальмитат сахарозы, мономиристат сахарозы и монолаурат сахарозы;
- сложные эфиры полиглицерина и жирной кислоты, например, полиглицеринолеат;
- полиоксиэтиленовые сложные эфиры альфа-токоферил сукцината, например, D-альфа-токоферил-PEG-1000-сукцинат (TPGS);
- полигликолизированные глицериды, такие как тиры, известные и коммерчески доступные под торговыми названиями “Гелуцип 44/14”, Телуцип 50/13 и “Лабразол”;
- продукты реакции природного или гидрогенизированного касторового масла и этилен оксида, такие как различные жидкие поверхностно-активные вещества, известные и коммерчески доступные под торговым названием “юКремофор”; и
- неполные сложные эфиры многофункциональных спиртов и жирных кислот, такие как сложные эфира глицерина и жирной кислоты, например, моно- и три-лауриловые, пальмитиловые, стеариловые и олеиловые сложные эфиры, например, глицерин моностеарат, глицерин моноолеат, например, глицерил моноолеат 40, известный и коммерчески доступный под торговым названием “Пецеол”; глицерил дибегенат, глицерин дистеарат, глицерин монолинолеат; этиленгликоль моностеарат, этиленгликоль монопальмитостеарат, пентаэритритол моностеарат.
В предпочтительном варианте осуществления, лекарственная дозированная форма в соответствии с изобретением содержит поверхностно-активное вещество или смесь различных поверхностно-активных веществ, получаемую путем
(i) эстерифицирования насыщенных или ненасыщенных C12-C18-жирных кислот, необязательно несущих гидроксильную группу, с полиэтилен гликолем и необязательно, глицерином; полиэтилен гликоль предпочтительно содержит от 10 до 40 этилен оксидных единиц (-CH2CH2O-); и/или
(ii) эстерифицирования триглицеридов насыщенных или ненасыщенных C12-C18-жирных кислот, несущих гидроксильную группу с этилен оксидом таким образом, что полиэтилен гликольный компонент связывается с гидроксильной группой C12-C18-жирных кислот с помощью эфирной связи; полиэтилен гликольный компонент предпочтительно содержит от 30 до 50 этилен оксидных единиц (-CH2CH2O-).
Предпочтительно, поверхностно-активное вещество выбирают из группы, включающей макроголгидроксистеарат, макроголглицерилгидроксистеарат и макроголглицериллаурат, где макрогольный компонент предпочтительно содержит от 15 до 45 этилен оксидных единиц.
Особенно предпочтительные поверхностно-активные вещества этого класса, которые содержатся в лекарственной дозированной форме в соответствии с изобретением, представляют собой неионные поверхностно-активные вещества, имеющие гидрофильно-липофильный баланс (HLB) по меньшей мере 10, в особенности неионные поверхностно-активные вещества, имеющие HLB значение по меньшей мере 12, более предпочтительно неионные поверхностно-активные вещества, имеющие HLB значение в пределах от 14 до 16. Примерами поверхностно-активных веществ этого типа являются вышеперечисленные поверхностно-активные вещества “Твин® 80” и “Солютол® HS 15”.
Солютол® HS-15 представляет собой смесь полиэтиленгликоля 660 12-гидроксистеарата и полиэтилен гликоля. Он представляет собой белую пасту при комнатной температуре, которая становится жидкой приблизительно при 30°C и имеет HLB приблизительно 15.
Твин® 80 [полиоксиэтилен(20)сорбитан моноолеат] представляет собой жидкость при комнатной температуре, имеет вязкость 375-480 мПа. с и имеет HLB приблизительно 15.
В другом предпочтительном варианте осуществления лекарственная дозированная форма в соответствии с изобретением содержит смесь по меньшей мере одного поверхностно-активного вещества, имеющего HLB значение по меньшей мере 10 (гидрофильное поверхностно-активное вещество) и по меньшей мере одного поверхностно-активного вещества, имеющего HLB значение ниже 10 (липофильное поверхностно-активное вещество). Например, дозированная форма может содержать макрогол-глицерингидроксистеарат 40 (например, Кремофор® ОВ 40) в качестве гидрофильного поверхностно-активного компонента и глицерил моноолеат 40 (например, Пецеол®) в качестве липофильного поверхностно-активного компонента.
Предпочтительно, относительное весовое соотношение поверхностно-активного вещества, имеющего HLB значение по меньшей мере 10 (гидрофильное поверхностно-активное вещество) и поверхностно-активного вещества, имеющего HLB значение ниже 10 (липофильное поверхностно-активное вещество) находится в диапазоне 15:1 до 1:20, более предпочтительно 10:1 до 1:15, еще более предпочтительно 8:1 до 1:12, также более предпочтительно 6:1 до 1:10, даже более предпочтительно 5:1 до 1:7, наиболее предпочтительно 4:1 до 1:4 и в особенности 2:1 до 1:2.
В предпочтительном варианте осуществления, содержание поверхностно-активного вещества составляет по меньшей мере 0,001 мас. % или по меньшей мере 0,005 мас. %, более предпочтительно по меньшей мере 0,01 мас. % или по меньшей мере 0,05 мас. %, еще более предпочтительно по меньшей мере 0,1 мас. %, по меньшей мере 0,2 мас. %, или по меньшей мере 0,3 мас. %, также более предпочтительно по меньшей мере 0,4 мас. %, по меньшей мере 0,5 мас. %, или по меньшей мере 0,6 мас. %, и в особенности по меньшей мере 0,7 мас. %, по меньшей мере 0,8 мас. %, по меньшей мере 0,9 мас. %, или по меньшей мере 1,0 мас. %, исходя из общего веса лекарственной дозированной формы.
В другом предпочтительном варианте осуществления, в особенности если лекарственная дозированная форма содержит инкапсулированное ядро, содержание поверхностно-активного вещества составляет по меньшей мере 10 мас. %, более предпочтительно по меньшей мере 15 мас. %, еще более предпочтительно по меньшей мере 20 мас. %, также более предпочтительно по меньшей мере 25 мас. % и в особенности по меньшей мере 30 мас. %, исходя из общего веса композиции, образующей ядро. В предпочтительном варианте осуществления, содержание поверхностно-активного вещества находится в интервале предпочтительно от 0,1 мас. % до 95 мас. %, более предпочтительно от 1 мас. % до 95 мас. %, еще более предпочтительно от 5 мас. % до 90 мас. %, также более предпочтительно от 10 мас. % до 80 мас. %, наиболее предпочтительно от 20 мас. % до 70 мас.%, и в особенности от 30 мас. % до 75 мас. %, исходя из общего веса композиции, образующей ядро.
В предпочтительном варианте осуществления, лекарственная дозированная форма содержит ядро, которое инкапсулировано с помощью инкапсулирующей среды. Ядро может быть жидким, полужидким или твердым.
Предпочтительно, указанная инкапсулирующая среда представляет собой мягкую желатиновую капсулу или твердую желатиновую капсулу, в особенности твердую желатиновую капсулу.
В предпочтительном варианте осуществления, лекарственная дозированная форма содержит жидкое ядро, инкапсулированное твердым материалом, где фармакологически активный агент в соответствии с общей формулой (I) диспергирован в жидком ядре. Предпочтительно, твердый материал представляет собой твердую желатиновую капсулу.
В предпочтительном варианте осуществления, лекарственная дозированная форма в соответствии с изобретением содержит самоэмульгирующийся препарат, в который предпочтительно заделан фармакологически активный агент в соответствии с общей формулой (I). Предпочтительно, фармакологически активный агент в соответствии с общей формулой (I) молекулярно диспергирован в других ингредиентах жидкого ядра. Для целей настоящей заявки, “молекулярно диспергирован в жидком ядре”, например, в других ингредиентах жидкого ядра, обозначает, что существенная часть общего содержания фармакологически активного агента в соответствии с общей формулой (I) представлена в некристаллической форме, то есть не обеспечивает отражения рентгеновских лучей. Предпочтительно, фармакологически активный агент в соответствии с общей формулой (I) растворен в других ингредиентах жидкого ядра. Предпочтительно, содержание некристаллического фармакологически активного агента в соответствии с общей формулой (I) составляет по меньшей мере 60 мас. %, более предпочтительно по меньшей мере 65 мас. %, еще более предпочтительно по меньшей мере 70 мас. %, также более предпочтительно по меньшей мере 75 мас. %, даже более предпочтительно по меньшей мере 80 мас. %, наиболее предпочтительно по меньшей мере 85 мас. %, и в особенности по меньшей мере 90 мас. %, исходя из общего содержания фармакологически активного агента в соответствии с общей формулой (I).
В предпочтительном варианте осуществления, самоэмульгирующийся препарат содержит поверхностно-активное вещество и масло.
В другом предпочтительном варианте осуществления, самоэмульгирующийся препарат представляет собой самоэмульгирующийся маслянистый препарат (SEOF), то есть он содержит поверхностно-активное вещество, масло и дополнительно гидрофильный растворитель.
Для целей настоящей заявки, масло предпочтительно относится к любому веществу, которое представляет собой жидкость при температуре окружающей среды или имеет точку плавления ниже 70°C и является гидрофобным, но растворимо в органических растворителях.
Предпочтительно, масло представляет собой сложный эфир C12-C18-жирной кислоты и моноспирта (например, C1-C12-алкилспирты), сложный эфир ди-C12-C18-жирной кислоты и диспирта (например, этилен гликоль) или сложный эфир три-C12-C18-жирной кислоты и триспирта (например, глицерин).
Предпочтительно, масло имеет точку плавления ниже 60°C, более предпочтительно ниже 55°C, еще более предпочтительно ниже 50°C, также более предпочтительно ниже 45°C, даже более предпочтительно ниже 40°C, наиболее предпочтительно ниже 35°C и в особенности ниже 30°C.
Предпочтительно, чистое масло имеет плотность в пределах от 0,94±0,07 г/см3, более предпочтительно 0,94±0,06 г/см3, еще более предпочтительно 0,94±0,05 г/см3, также более предпочтительно 0,94±0,04 г/см3, даже более предпочтительно 0,94±0,03 г/см3, наиболее предпочтительно 0,94±0,02 г/см3, и в особенности 0,94±0,01 г/см3.
Предпочтительно, чистое масло имеет вязкость при 20°C, измеренную в соответствии с Европейской фармакопеей 2.2.8, в пределах от 30±9 миллипаскаль-секунда, более предпочтительно 30±8 миллипаскаль-секунда, еще более предпочтительно 30±7 миллипаскаль-секунда, также более предпочтительно 30±6 миллипаскаль-секунда, даже более предпочтительно 30±5 миллипаскаль-секунда, наиболее предпочтительно 30±4 миллипаскаль-секунда, и в особенности 30±3 миллипаскаль-секунда.
В предпочтительном варианте осуществления, масло выбирают из группы, включающей
- насыщенные C8-C14 жирные кислоты, такие как миристиновая кислота;
- ненасыщенные C8-C18 жирные кислоты и их сложные эфиры, такие как олеиновая кислота и этил олеат;
- смеси насыщенных и ненасыщенных C8-C18 жирных кислот, такие как соевое масло и арахисовое масло; и
- триглицериды жирных кислот, предпочтительно C6-C12 жирных кислот, более предпочтительно C6-C12 жирных кислот, такие как смеси каприловых/каприновых триглицеридов, наиболее предпочтительно триглицериды со средней цепью в соответствии с Европейской фармакопеей или фармакопеей США, например, известные и коммерчески доступные под торговыми названиями “Каптекс 355” и “Миглиол 812”; и
- сложные эфиры пропилен гликоля и жирной кислоты, такие как пропиленгликоль монокаприлат (известные и коммерчески доступные под торговыми названиями “Каприол 90”);
особенно предпочтительными являются триглицериды со средней цепью в соответствии с Европейской фармакопеей или фармакопеей США, такие как указанные смеси каприловых/каприновых триглицеридов.
В предпочтительном варианте осуществления, содержание масла в лекарственной дозированной форме находится в диапазоне от 1 мас. % до 90 мас. %, предпочтительно от 2 мас. % до 80 мас. %, более предпочтительно от 5 мас. % до 60 мас. %, еще более предпочтительно от 10 мас. % до 50 мас. % и наиболее предпочтительно от 15 мас. % до 30 мас. %, предпочтительно исходя из общего веса ядра.
В предпочтительном варианте осуществления, относительное весовое соотношение поверхностно-активного вещества к маслу находится в диапазоне от 20:1 до 1:20, более предпочтительно 10:1 до 1:10, еще более предпочтительно 7,5:1 до 1:5, также более предпочтительно 7:1 до 1:1, наиболее предпочтительно 5:1 до 1,5:1 и в особенности 4:1 до 2:1.
Предпочтительно, самоэмульгирующийся препарат представлен в виде жидкого ядра, инкапсулированного в твердую желатиновую капсулу.
В предпочтительном варианте осуществления, самоэмульгирующийся препарат дополнительно содержит гидрофильный растворитель.
Предпочтительно, гидрофильный растворитель представляет собой органический спирт, такой как органический моноспирт, органический диспирт или органический триспирт.
Предпочтительно, чистый гидрофильный растворитель имеет точку кипения при атмосферном давлении в пределах от 78±22°C, более предпочтительно 78±18°C, еще более предпочтительно 78±15°C, также более предпочтительно 78±12°C, даже более предпочтительно 78±8°C, наиболее предпочтительно 78±5°C, и в особенности 78±2°C.
Предпочтительно, гидрофильный растворитель выбирают из группы этанола, изопропанола, глицерина и пропилен гликоля; особенно предпочтительно представляет собой этанол. Предпочтительно, содержание гидрофильного растворителя находится в диапазоне от приблизительно 1 мас. % до приблизительно 90 мас. %, предпочтительно от приблизительно 2 мас.% до приблизительно 80 мас. %, более предпочтительно от приблизительно 5 мас. % до приблизительно 60 мас. %, еще более предпочтительно от приблизительно 10 мас. % до приблизительно 50 мас. %, наиболее предпочтительно от приблизительно 15 мас. % до приблизительно 30 мас. %, предпочтительно исходя из общего веса ядра.
В предпочтительном варианте осуществления, лекарственная дозированная форма содержит жидкое ядро, содержащее фармакологически активный агент в соответствии с общей формулой (I), поверхностно-активное вещество, масло и гидрофильный растворитель, где относительное весовое соотношение поверхностно-активное вещество: масло: гидрофильный растворитель находится в диапазоне 60:20±17,5:20±17,5, более предпочтительно 60:20±15:20±15, еще более предпочтительно 60:20±12,5:20±12,5, также более предпочтительно 60:20±10:20±10, даже более предпочтительно 60:20±7,5:20±7,5, наиболее предпочтительно 60:20±5:20±5, и в особенности 60:20±2,5:20±2,5.
В другом предпочтительном варианте осуществления, лекарственная дозированная форма содержит жидкое ядро, содержащее фармакологически активный агент в соответствии с общей формулой (I), поверхностно-активное вещество, имеющее HLB значение по меньшей мере 10 (гидрофильное поверхностно-активное вещество), масло и поверхностно-активное вещество, имеющее HLB значение ниже 10 (липофильное поверхностно-активное вещество), где относительное весовое соотношение гидрофильное: масло: липофильный растворитель находится в диапазоне 60:20±17,5:20±17,5, более предпочтительно 60:20±15:20±15, еще более предпочтительно 60:20±12,5:20±12,5, также более предпочтительно 60:20±10:20±10, даже более предпочтительно 60:20±7,5:20±7,5, наиболее предпочтительно 60:20±5:20±5, и в особенности 60:20±2,5:20±2,5.
В другом предпочтительном варианте осуществления, лекарственная дозированная форма содержит жидкое ядро, содержащее фармакологически активный агент в соответствии с общей формулой (I), поверхностно-активное вещество, имеющее HLB значение по меньшей мере 10 (гидрофильное поверхностно-активное вещество), масло и поверхностно-активное вещество, имеющее HLB значение ниже 10 (липофильное поверхностно-активное вещество), где относительное весовое соотношение гидрофильное: масло: липофильный растворитель находится в диапазоне 40:40±35:20±17,5, более предпочтительно 40:40±30:20±15, еще более предпочтительно 40:40±25:20±12,5, также более предпочтительно 40:40±20:20±10, даже более предпочтительно 40:40±15:20±7,5, наиболее предпочтительно 40:40±10:20±5, и в особенности 40:40±5:20±2,5.
Предпочтительные варианты осуществления A1-A20 жидкого ядра лекарственной дозированной формы в соответствии с изобретением, то есть жидкого ядра, которое инкапсулировано инкапсулирующим материалом, обобщены в таблице, представленной ниже в данной заявке:
где
природа относится к химической природе ингредиента;
сод. относится к содержанию ингредиента в мас. %, исходя из общего веса ядра;
W1 обозначает фармакологически активный агент в соответствии с общей формулой (I) или его физиологически приемлемую соль;
W2 обозначает фармакологически активный агент в соответствии с общей формулой (I’) или его физиологически приемлемую соль;
W3 обозначает (1r,4r)-6’-фтор-N,N-диметил-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин, или (1r,4r)-6’-фтор-N-метил-4-фенил-4’,9’-дигидро-3’H-спиро[циклогексан-1,1’-пирано[3,4,b]индол]-4-амин, или его физиологически приемлемую соль;
X1 обозначает поверхностно-активное вещество, имеющее HLB значение по меньшей мере 10;
X2 обозначает неионное поверхностно-активное вещество, имеющее HLB значение в интервале от 14 до 16;
X3 обозначает полигликолизированный глицерид;
X4 обозначает сложный эфир полиоксиэтилена и жирной кислоты, жирная кислота предпочтительно имеет от приблизительно 8 до приблизительно 18 атомов углерода;
Y1 обозначает моно-, ди- или триэфир C6-С18 жирных кислот;
Y2 обозначает триглицериды C6-C12 жирных кислот (триглицериды со средней цепью);
Y3 обозначает сложный эфир пропилен гликоля и жирной кислоты;
Y4 смесь каприловых/каприновых триглицеридов;
Z1 обозначает гидрофильный растворитель
Z2 обозначает гидрофильный растворитель, выбранный из органического моноспирта, диспирта или триспирта;
Z3 обозначает поверхностно-активное вещество, имеющее HLB значение ниже 10;
Z4 обозначает этанол.
Например, в соответствии с таблицей, представленной выше, вариант осуществления A9 относится к фармацевтическому препарату в соответствии с изобретением, который содержит фармакологически активный агент в соответствии с общей формулой (I’) или его физиологически приемлемую соль в количестве 0,25±0,24 мас. %, неионное поверхностно-активное вещество, имеющее HLB значение в интервале от 14 до 16 в количестве 50±15 мас. %, триглицериды C6-C12 жирных кислот в количестве 25±7,5% и гидрофильный растворитель, выбранный из органического моноспирта, диспирта или триспирта в количестве 25±7,5%, исходя из общего веса жидкого ядра.
Предпочтительно, самоэмульгирующийся препарат представляет собой жидкий препарат типа IIIA или типа IIIB, в соответствии с системой классификации липидных препаратов (LFCS).
Предпочтительно, самоэмульгирующийся препарат образует эмульсии со средним размером капли, меньшим или равным 10 микрометров, более предпочтительно, меньшим или равным 1000 нанометров, наиболее предпочтительно, меньшим или равным 100 нанометров, при подвергании воздействию водной среды.
В другом предпочтительном варианте осуществления, самоэмульгирующийся препарат представляет собой само-микро эмульгирующуюся систему доставки лекарственного средства (SMEDDS), то есть при подвергании воздействию водной среды, препарат образует микроэмульсии со средним размером капли, меньшим или равным 50 нанометров, которые содержат фармакологически активный агент в соответствии с общей формулой (I). В другом предпочтительном варианте осуществления, средний размер капли меньше или равен 10 нанометров
В предпочтительном варианте осуществления, средний размер капли находится в диапазоне 50±70 нм, более предпочтительно 50±60 нм, еще более предпочтительно 50±50 нм, также более предпочтительно 50±40 нм, даже более предпочтительно 50±30 нм, наиболее предпочтительно 50±20 нм, и в особенности 50±10 нм.
В предпочтительном варианте осуществления, средний размер капли находится в диапазоне 75±70 нм, более предпочтительно 75±60 нм, еще более предпочтительно 75±50 нм, также более предпочтительно 75±40 нм, даже более предпочтительно 75±30 нм, наиболее предпочтительно 75±20 нм, и в особенности 75±10 нм.
В предпочтительном варианте осуществления, средний размер капли находится в диапазоне 100±70 нм, более предпочтительно 100±60 нм, еще более предпочтительно 100±50 нм, также более предпочтительно 100±40 нм, даже более предпочтительно 100±30 нм, наиболее предпочтительно 100±20 нм, и в особенности 100±10 нм.
В предпочтительном варианте осуществления, средний размер капли находится в диапазоне 125±70 нм, более предпочтительно 125±60 нм, еще более предпочтительно 125±50 нм, также более предпочтительно 125±40 нм, даже более предпочтительно 125±30 нм, наиболее предпочтительно 125±20 нм, и в особенности 125±10 нм.
В предпочтительном варианте осуществления, средний размер капли находится в диапазоне 150±70 нм, более предпочтительно 150±60 нм, еще более предпочтительно 150±50 нм, также более предпочтительно 150±40 нм, даже более предпочтительно 150±30 нм, наиболее предпочтительно 150±20 нм, и в особенности 150±10 нм.
В особенно предпочтительно варианте осуществления,
- лекарственная дозированная форма содержит от 0,01% до 95% фармакологически активного агента (А); и/или
- лекарственная дозированная форма имеет вес в диапазоне от 0,1 мг до 2,000 мг; и/или
- лекарственная дозированная форма адаптирована для орального введения; и/или
- лекарственная дозированная форма содержит фармакологически активный агент в соответствии с общей формулой (I) в дозе от 10 мкг до 50 мкг или от 25 мкг до 80 мкг; и/или
- фармакологически активный агент в соответствии с общей формулой (I) содержится в дозированной форме в количестве, которое является субтерапевтическим по отношению к однократному введению дозированной формы; и/или
- фармакологически активный агент в соответствии с общей формулой (I) содержится в дозированной форме в количестве, которое является субтерапевтическим по отношению к лечению острой боли; и/или
- фармакологически активный агент в соответствии с общей формулой (I) содержится в дозированной форме в таком количестве, что не требуется титрование начальной дозы; и/или
- фармакологически активный агент в соответствии с общей формулой (I) содержится в дозированной форме в таком количестве, что количество побочных действий, которые встречаются при введении дозированной формы снижается по сравнению с дозированной формой, содержащий чистый агонист μ-опиодного рецептора, такой как морфин, в терапевтически адекватно эффективном количестве; и/или
- tmax находится в диапазоне от 2 до 10 ч, предпочтительно от 2 до 6 ч; и/или
- соотношение AUC0-t/дозу находится в диапазоне от 0,5 до 16,5 ч/м3, предпочтительно от 6 до 12 ч/м3; и/или
- соотношение Cmax/дозу находится в диапазоне от 0,06 до 1,69 м-3, предпочтительно в диапазоне от 0,30 до 1,30 м-3; и/или
- наивысшая концентрация в плазме фармакологического агента, достигаемая после введения один раз в сутки лекарственной дозированной формы в течение по меньшей мере 5 последовательных дней, находится в диапазоне от 10 до 120 мкг/м3, предпочтительно от 20 до 80 мкг/м3.
Дальнейший аспект изобретения относится к способу лечения невропатической боли, предпочтительно хронической невропатической боли, включающей введение фармакологически эффективного количества фармакологически активного агента в соответствии с общей формулой (I) или его физиологически приемлемую соль; предпочтительно один раз в сутки, предпочтительно оральное введение лекарственной дозированной формы в соответствии с изобретением; субъекту, который в этом нуждается.
Предпочтительно, боль представляет собой хроническую невропатическую боль или острую невропатическую боль, периферическую невропатическую боль или центральную невропатическую боль, мононевропатическую боль или полиневропатическую боль. Если невропатическая боль является хронической, то она может быть хронической периферической невропатической болью или хронической центральной невропатической болью, в предпочтительном варианте осуществления хронической периферической мононевропатической болью или хронической центральной мононевропатической болью, в другом предпочтительном варианте осуществления хронической периферической полиневропатической болью или хронической центральной полиневропатической болью. Если невропатическая боль является острой, то она может быть острой периферической невропатической болью или острой центральной невропатической болью, в предпочтительном варианте осуществления острой периферической мононевропатической болью или острой центральной мононевропатической болью, в другом предпочтительном варианте осуществления острой периферической полиневропатической болью или острой центральной полиневропатической болью.
Предпочтительно, боль представляет собой хроническую невропатическую боль. Для целей настоящей заявки, невропатическая боль представляет собой боль, которая имеет происхождение из поврежденных нервов или нарушения функции нервов. Ее классифицируют как хроническую невропатическую боль, если она присутствует в течение более 3 месяцев.
ПРИМЕРЫ
Последующие примеры дополнительно иллюстрируют изобретение, но не должны рассматриваться как ограничивающие его объем.
ПРИМЕР 1:
Клинические исследования проводили для определения анальгезирующей эффективности и переносимости однократных доз соединения в соответствии с формулой (I’b) (200 мкг, 400 мкг и 600 мкг; гемицитрат пероральный раствор соединения (I’b) в Макроголе 400; все дозировки относительно свободного основания лекарственного средства) по сравнению с таковой у морфина (60 мг, форма с контролируемым высвобождением) и плацебо у пациентов с острой послеоперационной болью после ортопедической хирургии (бурсэктомия).
Для этого, 258 пациентов любого пола включали в рандомизированное, плацебо-контролируемое, двойное слепое клиническое исследование в параллельных группах. Леченные группы были хорошо сбалансированы относительно демографических и исходных характеристик с незначительным дисбалансом относительно исходной боли и этнической принадлежности.
После хирургии, всех пациентов изначально лечили с помощью местной послеоперационной анестезии через подколенный блок. Вследствие различных кинетик соединения в соответствии с формулой (I’b) и морфина, пациентов затем лечили с помощью одного из двух лекарственных средств или с помощью плацебо в незначительно отличающееся время:
За один час до прекращения подколенного блока, пациентов рандомизировали и часть из них затем получала однократную дозу соединения в соответствии с формулой (I’b) (200 мкг, 400 мкг или 600 мкг) или плацебо, тогда как другие получали морфин или плацебо 2 часов после прекращения подколенного блока.
Конечной точкой оценки первичной эффективности была абсолютная интенсивность боли в течение 24 часового периода. Интенсивность боли измеряли с использованием 11-бальной количественной шкалы оценки (NRS). В каждый момент времени, пациентов инструктировали для оценки их текущей интенсивности боли относительно 11-бальной количественной шкалы оценки. Оценка ноль соответствовала отсутствию боли и оценка 10 соответствовала наихудшей возможной боли. Пропущенную запланированную оценку боли для пациентов условно начисляли с помощью метода переноса данных последнего наблюдения вперед (LOCF). Полученные средние NRS значения в течение 24 часового периода представлены на фигуре 1.
Сумма отличий интенсивности боли в течение различных периодов времени анализировали с использованием модели ковариационного анализа (ANCOVA) с факторами для оценки лечения и сайта и исходной интенсивности боли (используя оценку интенсивности боли NPRS). Включали только субъектов с непропущенной исходной интенсивностью боли. Обобщенные данные анализа для 2-10-часового периода представлены в таблице 1.
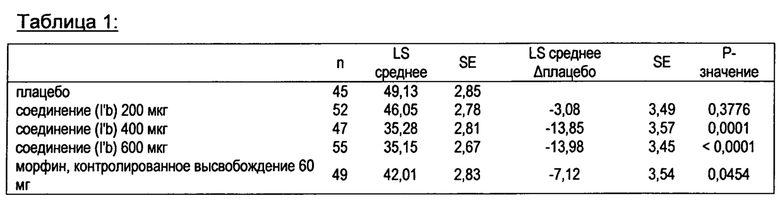
Полученные p-значения анализа большого количества оцененных окон обобщены в Таблице 2.
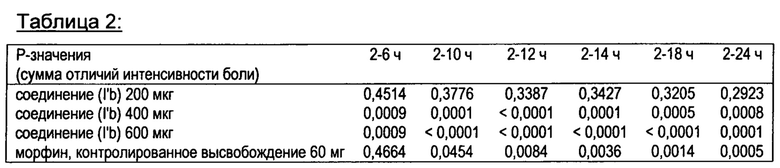
Таким образом, согласно первичному параметру, статистически достоверное различие наблюдали между группами, которые получали дозу 400 мкг или 600 мкг соединения (I’b) и плацебо группами, в то время так не наблюдали статистически достоверного различия для групп, которые получали дозу 200 мкг соединения (I’b).
В таблицах 3 и 4 обобщены вызванные лечением нежелательные явления (ТЕАЕ), которые наблюдали в пяти леченных группах.
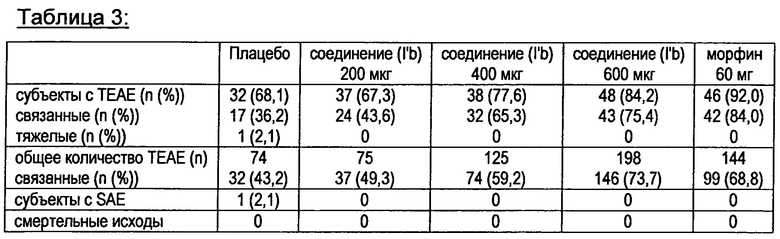
TEAE: вызванное лечением нежелательное явление; SAE: тяжелое нежелательное явление
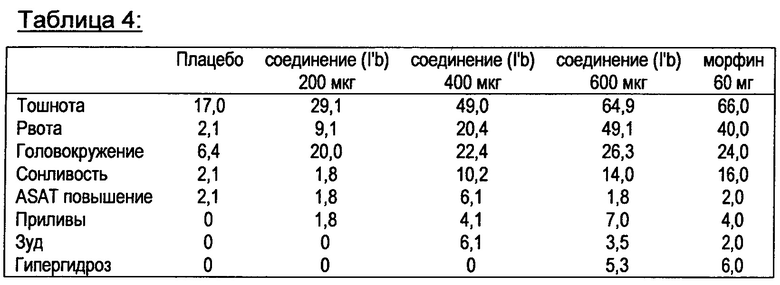
Из таблиц 3 и 4 становится очевидным, что все четыре активных лечения хорошо переносились при таких обстоятельствах и нежелательные явления, которые проявлялись наиболее часто, соответствовали тем, которые можно наблюдать для агонистов μ-опиодного рецептора. Для группы пациентов, которая получала лечение с помощью соединения (I’b), распространенность нежелательных явлений повышалась с дозой, и при дозе 600 мкг распространенность нежелательных явлений была сопоставима с таковой для группы пациентов, получавших морфин, хотя доза 400 мкг была уже сопоставима по эффективности.
ПРИМЕР 2:
Клинические исследования проводили для определения биодоступности препарата капсулы, заполненной жидкостью, содержащего соединение (I’b) с мощностью дозы 400 мкг по сравнению с гемицитратным оральным раствором соединения (I’b) (400 мкг, 400 мкг/мл орального раствора) в препарате Макрогол 400 после однократного перорального введения. 24 здоровые белые особи мужского пола включали в рандомизированное, без контроля плацебо, трехстороннее перекрестное, одноцентровое клиническое исследование. Основными фармакокинетическими параметрами являлись AUC0-t, AUC0-72h и Cmax.
Результаты обобщены в таблицах 5 до 7.

N=24; В таблице представлены средние арифметические +/- среднеквадратическое отклонение (коэффициент вариации).
Таким образом, относительная биодоступность капсулы 400 мкг и 400 мкг/мл орального раствора на основе AUC0-72h составляла 105%, с 90%-Cl в диапазоне обычно используемого для оценки биоэквивалентности.
Однократное оральное введение дозы 400 мкг соединения (ГЬ) было безопасным и хорошо переносилось, независимо от галенового препарата. Не наблюдали тяжелых нежелательных явлений.
Пример 3:
Клинические исследования проводили для определения анальгезирующей эффективности и переносимости многократных доз соединения (I’b) (40 мкг, 80 мкг, 100 мкг, 120 мкг и 200 мкг; все дозировки в виде эквивалентных весовых дозировок относительно свободного основания, в форме гемицитрата.) по сравнению с таковой у морфина (60 мг, контролируемое высвобождение) и плацебо у пациентов с болезненной диабетической нейропатией.
Для этого, 86 пациентов любого пола включали в рандомизированное, плацебо- и дозо-контролируемое, двойное слепое, с тройным перекрытием клиническое исследование в параллельных группах.
Проводили три исследования с рандомизированной, двойной слепой и перекрывающейся схемой:
Вследствие - CA требования вводить ‘высокую дозу’ только после воздействия ‘низкой дозы’, в исследованиях A и B применяли только три из шести возможных последовательностей, тогда как в исследовании C использовали все шесть последовательностей с двойным плацебо контролируемым введением.
В первые 14-18 дней исследований, пациенты не получали никакого лечения для аннулирования любых лекарственных средств из предыдущего лечения. После окончания этой начальной фазы, определяли интенсивности боли и пациентов рандомизировали согласно одной из возможных последовательностей. После этого, каждый пациент получал дозы препаратов, содержащие соответствующую дозу соединения (I’b), морфина или плацебо один раз в сутки в течение 5 дней. За этой фазой следовала фаза выведения продолжительностью 8-10 дней. Оставшиеся две дозы препаратов вводили соответственно, то есть один раз в сутки в течение 5 дней с последующей фазой выведения (8-10 дней).
Все леченные группы были хорошо сбалансированы относительно демографических и исходных характеристик, только в распределении по полу проявлялись релевантные колебания.
Первичным критерием конечной точки было уменьшение от исходного значения средней оценки интенсивности боли в течение последних 24 часов, измеренной с помощью 11-точечной количественной шкалы оценки (NRS) в последний день лечения каждого леченного периода, по сравнению с плацебо. Оценку боли проводили 5 раз в день, начиная с 7 ч. утра почти каждые 4 часа. В каждый момент времени, пациентов инструктировали для оценки их текущей интенсивности боли относительно 11-точечной количественной шкалы оценки посредством электронного дневника. Оценка ноль соответствовала отсутствию боли и оценка 10 соответствовала наихудшей возможной боли. Пропущенную запланированную оценку боли для пациентов условно начисляли с помощью метода переноса данных последнего наблюдения вперед (LOCF). Полученные среднесуточные изменения боли (среднее изменение NRS значения) в течение 5-дневных периодов лечения представлены на Фигурах 2-4.
Изменения интенсивности боли от исходного периода и суммарное исходное анализировали с использованием описательной статистики и модели ковариационного анализа (ANCOVA) с факторами для лечения и побочных действий, влияниями последовательности лечения и периода, и влияниями исходной боли. Для исследований A и C идентифицировали влияние периода. Обобщенный анализ описательной статистики и ANCOVA анализов представлен в Таблицах 8, 9 и 10.



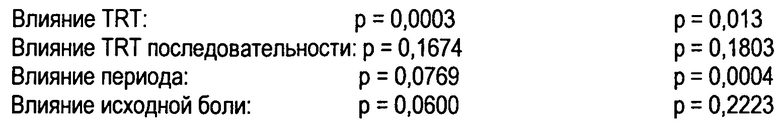
В соответствии с этими результатами, для параметра ‘среднее изменение суточной интенсивности боли с исходного’, все группы, которые получали соединение (I’b) в пределах от 80 мкг до 200 мкг, проявили статистически значимое различие по сравнению с плацебо (за исключением группы 120 мкг, наиболее вероятно, вследствие сильного влияния последовательности лечения). Для этих доз, отрыв от плацебо начинали в день 1 введения с повышающим эффектом в течение 5 последовательных дней суточного введения. Становится очевидным из Таблицы 10 и Фигуры 4, что для дозы 100 мкг соединения (I’b), обезболивающий эффект сходный с таковым для 60 мг морфина. Кроме того, Из фигуры 2 становится очевидным, что влияние дозы 40 мкг соединения (I’b) нельзя отличить от плацебо в день 1 далее, но отличимо от плацебо в день 5.
Эти результаты согласуются с фармакокинетическими параметрами, которые измеряли через регулярные промежутки времени. Арифметическое и геометрическое среднее значение наивысшей концентрации в плазме, наблюдаемое после введения соединения (I’b) в день 5 (Cmax, 5 d), а также арифметическое и геометрическое среднее значение времени, необходимого для ее достижения после введения для этой пятой последовательной суточной дозы (tmax, 5 d). Обобщены в Таблице 11.
Результаты концентраций в плазме, измеренные в 0-3 ч после введения в день 1, то есть C0-3h, 1d, и в 0-3 ч после введения в день 5, то есть С0-3h, 5d, представлены в Таблице 12 (среднее значение ± стандартное отклонение, количество субъектов N):
В Таблице 13 обобщены концентрации в плазме, которые измерены в различные временные точки в течение пятидневной схемы введения:
На фигуре 5 показано сравнение средних значений Cmax измеренных в день 5, по сравнению с концентрацией в плазме, которую наблюдали перед введением следующей дозы (Cnext predose), то есть через 8-10 дней после введения пятой дозы при окончании периода выведения.
Из фигуры 5 становится очевидным, что образцы, отобранные через 8-10 дней после предыдущего лечения с помощью соединения (I’b), все еще содержат это лекарственное средство в обнаружимых (и статистически релевантных) концентрациях. Даже образцы, отобранные через 10-15 дней после предыдущего лечения с помощью соединения I’b), все еще имели концентрации >2,0 пг/мл.
В Таблицах 14 и 15 обобщены вызванные лечением нежелательные явления (ТЕАЕ), наблюдаемые для леченных групп.
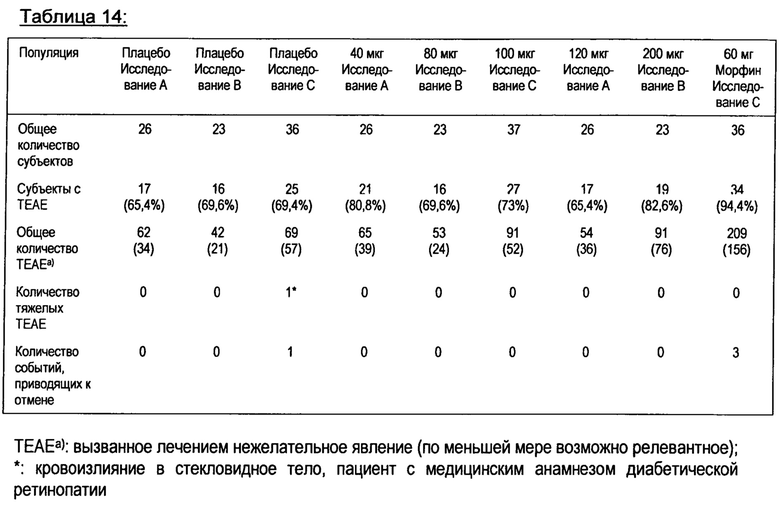
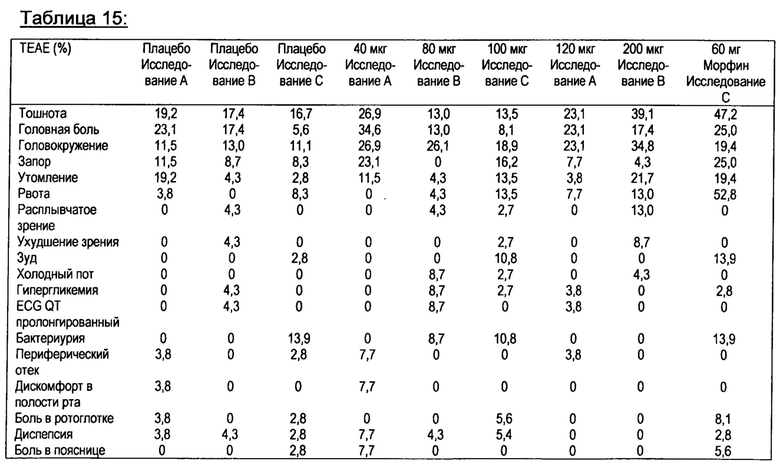
Из таблиц 14 и 15 становится очевидным, что дозы вплоть до 120 мкг соединения (I’b) имели степень вызванных лечением нежелательных явлений (ТЕАЕ), сходную с плацебо, за исключением головокружения, которые описывали более часто по сравнению с плацебо для всех исследуемых доз. Типичные побочные действия, ожидаемые для агонистов μ-опиодного рецептора, начинали проявляться только для верхней дозы 200 мкг. Очевидно больше TEAE, описанное после введения 60 мг морфина по сравнению с 100 мкг соединения (I’b), спарено со сравнимой анальгетической эффективностью (ср. Таблица 10 и Фигура 4).
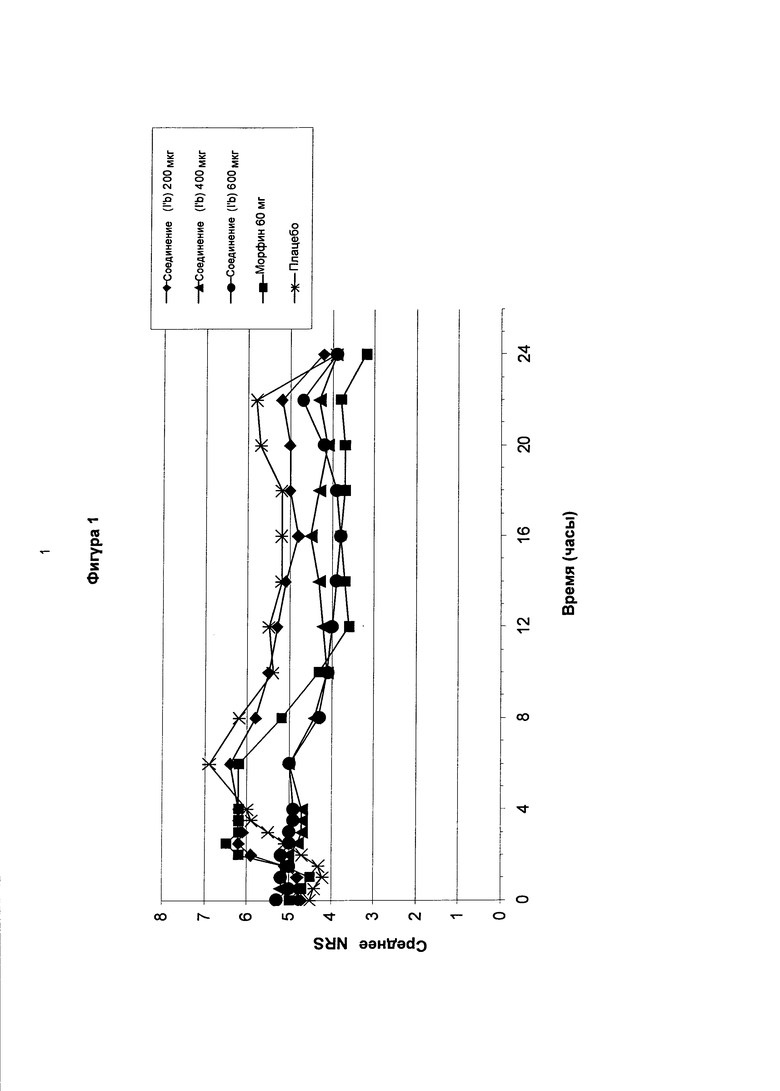
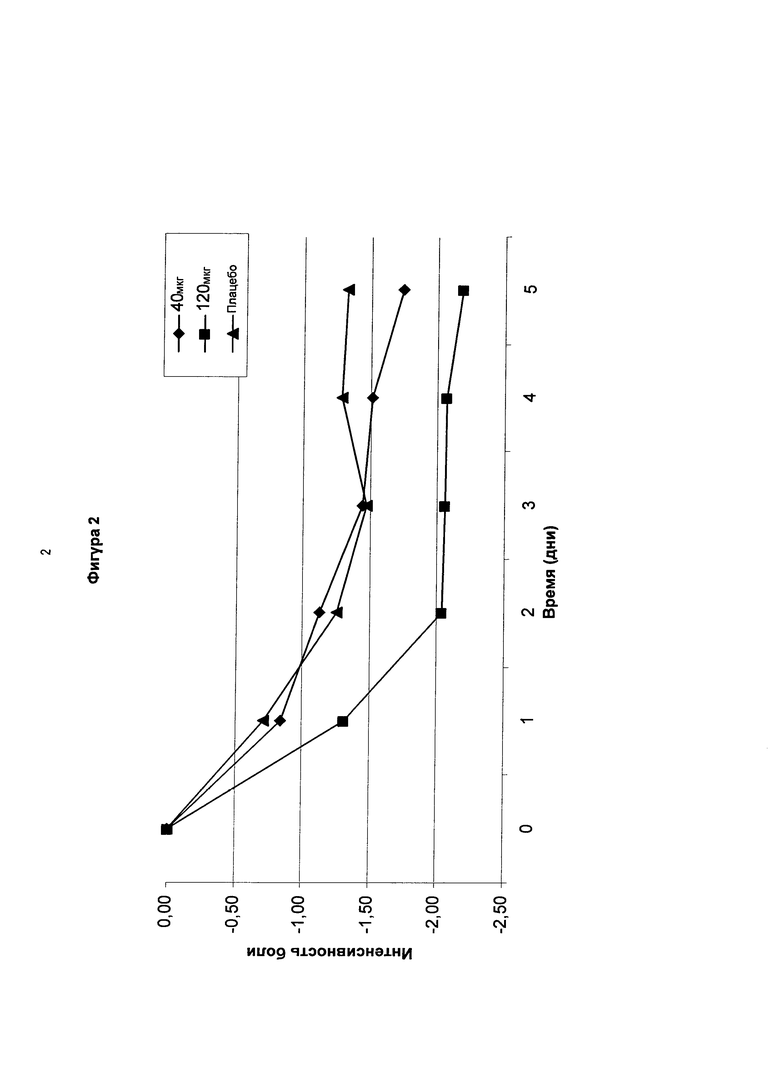
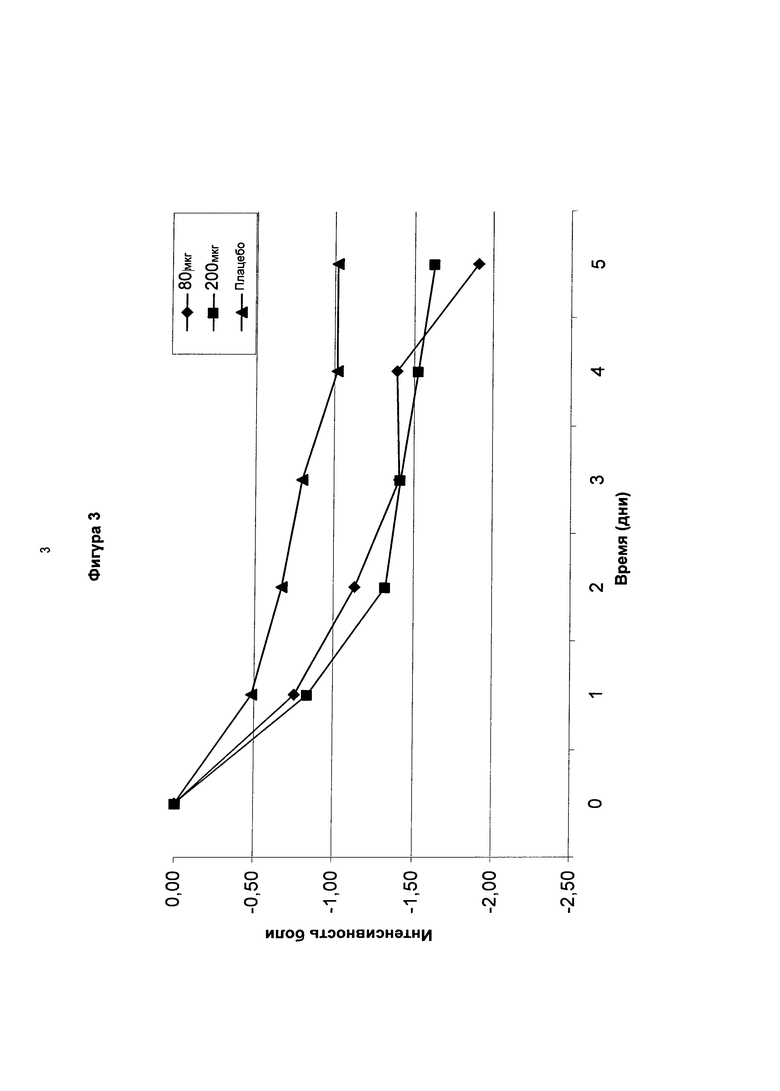
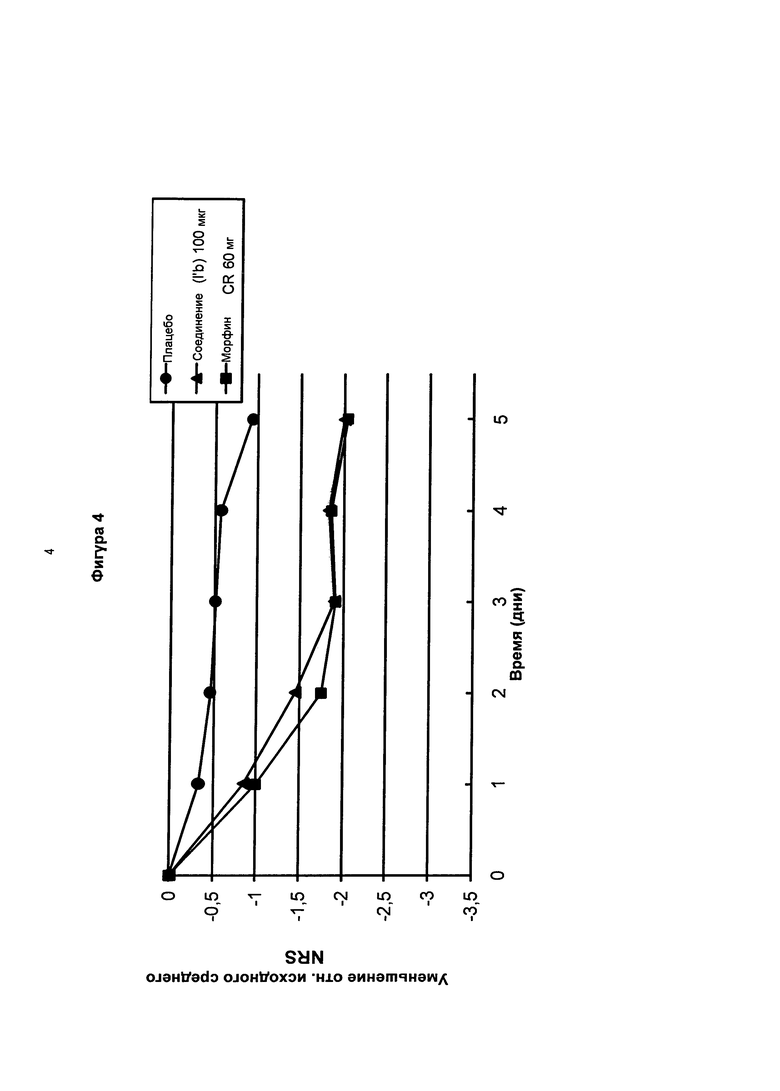
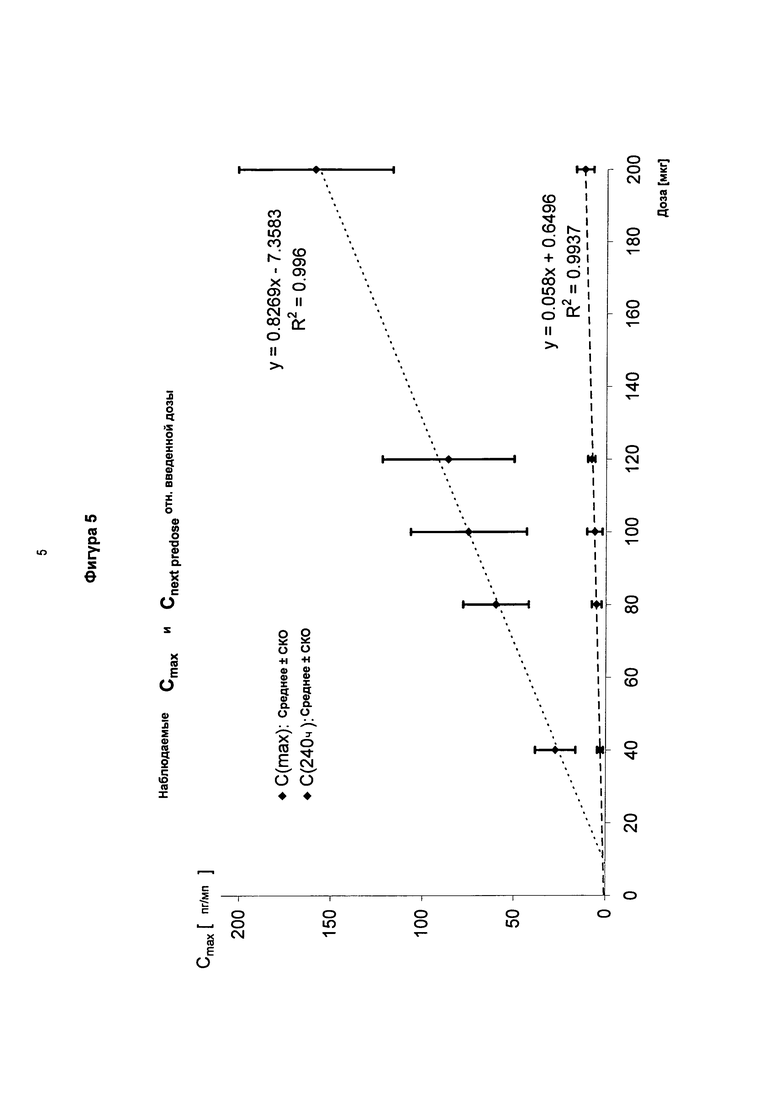
Изобретение относится к фармацевтической промышленности, а именно к способу лечения хронической невропатической боли. Способ включает оральное введение один раз в сутки лекарственной дозированной формы, содержащей фармакологически активный агент в соответствии с общей формулой (I)
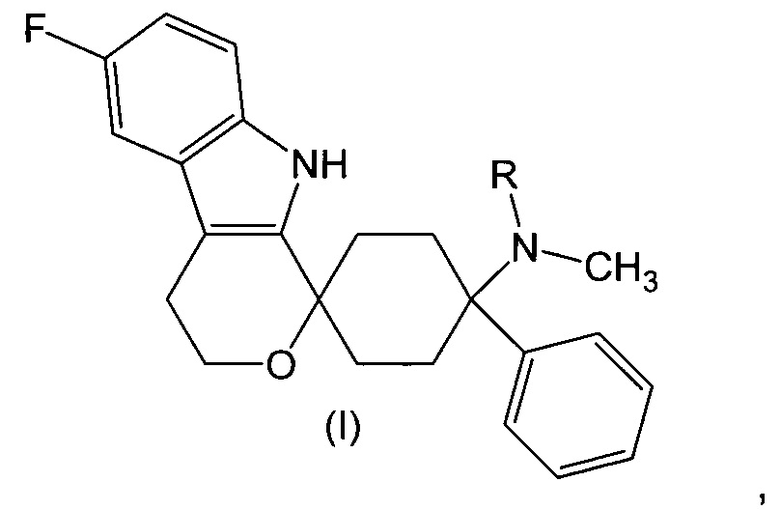
где R представляет собой -Н или -СН3, или его физиологически приемлемую соль, субъекту, который в этом нуждается. Лекарственная дозированная форма обеспечивает немедленное высвобождение in vitro фармакологически активного агента, который содержится в дозе от 40 до 190 мкг, а фармакокинетический параметр tmax находится в диапазоне от 4 до 6 ч. Изобретение обеспечивает хорошую биодоступность и достаточность облегчения боли, а также высокую переносимость и безопасность. 11 з.п. ф-лы, 5 ил., 15 табл., 3 пр.
1. Способ лечения хронической невропатической боли, включающий
оральное введение один раз в сутки лекарственной дозированной формы, содержащей фармакологически активный агент в соответствии с общей формулой (I)
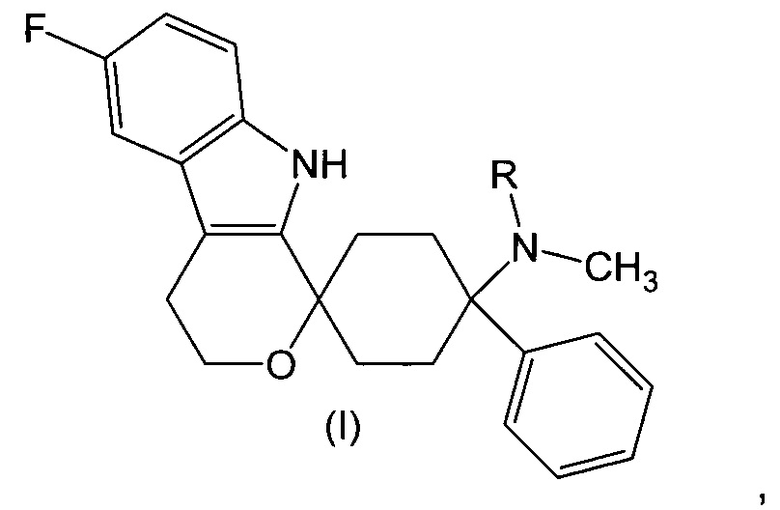
где R представляет собой -Н или -СН3,
или его физиологически приемлемую соль,
которая обеспечивает немедленное высвобождение in vitro фармакологически активного агента, и где фармакологически активный агент содержится в дозе от 40 до 190 мкг, выраженной в виде эквивалентных весовых дозировок, исходя из свободного основания; а фармакокинетический параметр tmax находится в диапазоне от 4 до 6 ч, субъекту, который в этом нуждается.
2. Способ в соответствии с п. 1, где фармакологически активный агент содержится в дозе от 40 до 80 мкг, выраженной в виде эквивалентных весовых дозировок, исходя из свободного основания.
3. Способ в соответствии с п. 1 или 2, где фармакологически активный агент содержится в дозе от 40 до 50 мкг, выраженной в виде эквивалентных весовых дозировок, исходя из свободного основания.
4. Способ в соответствии с п. 1 или 2, где фармакологически активный агент в соответствии с общей формулой (I) имеет стереохимию в соответствии с общей формулой (I')
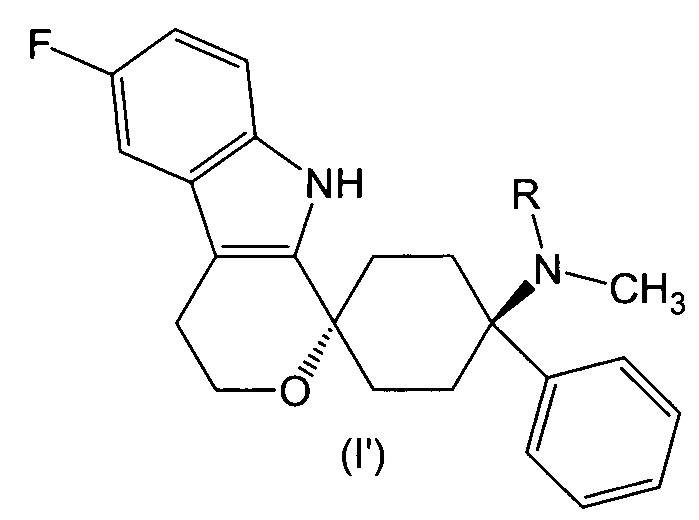
где R имеет значения, как указано в п. 1.
5. Способ в соответствии с п. 1 или 2, где фармакологически активный агент в соответствии с общей формулой (I) представляет собой (1r,4r)-6'-фтор-N,N-диметил-4-фенил-4',9'-дигидро-3'Н-спиро[циклогексан-1,1'-пирано[3,4,b]индол]-4-амин, (1r,4r)-6'-фтор-N-метил-4-фенил-4',9'-дигидро-3'Н-спиро[циклогексан-1,1'-пирано[3,4,b]индол]-4-амин или его физиологически приемлемую соль.
6. Способ в соответствии с п. 1 или 2, где фармакологически активный агент в соответствии с общей формулой (I) содержится в количестве, которое приводит к субтерапевтической концентрации в плазме фармакологически активного агента в первый день введения дозированной формы один раз в сутки, но к терапевтическим концентрациям в плазме фармакологически активного агента после введения дозированной формы один раз в сутки в течение по меньшей мере 3, или по меньшей мере 4, или по меньшей мере 5 последующих дней.
7. Способ в соответствии с п. 1 или 2, где фармакологически активный агент в соответствии с общей формулой (I) содержится в таком количестве, что не требуется титрование начальной дозы.
8. Способ в соответствии с п. 1 или 2, где фармакокинетический параметр AUC0-t / дозу находится в диапазоне от 0,3 до 20 ч/м3.
9. Способ в соответствии с п. 1 или 2, где фармакокинетический параметр Cmax / дозу находится в диапазоне от 0,04 до 2,00 м-3.
10. Способ в соответствии с п. 1 или 2, где наивысшая концентрация в плазме фармакологического агента, достигаемая после введения один раз в сутки лекарственной дозированной формы в течение по меньшей мере 5 последовательных дней, находится в диапазоне от 10 до 120 мкг/м3.
11. Способ в соответствии с п. 1 или 2, где наивысшая концентрация в плазме фармакологического агента, достигаемая после введения один раз в сутки лекарственной дозированной формы в течение по меньшей мере 5 последовательных дней, находится в диапазоне от 20 до 80 мкг/м3.
12. Способ в соответствии с п. 10 или 11, где время до достижения наивысшей концентрации в плазме фармакологического агента, достигаемой после введения один раз в сутки лекарственной дозированной формы в течение по меньшей мере 5 последовательных дней, находится в диапазоне от 4 до 6 ч.
| СПИРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА | 2003 |
|
RU2354656C2 |
| US 20090163716 A1, 25.06.2009 | |||
| US 20080125475 A1, 29.05.2008 | |||
| WO 2008040534 A2, 10.04.2008 | |||
| US 20090209575 A1, 20.08.2009 | |||
| ANTHONY PALMIERI III, Dissolution theory, methodology and testing, 2007, "Introduction", абзац 1 | |||
| В.И | |||
| ЧУЕШОВ, Промышленная технология лекарств, том 1, Харьков, издательство НФАУ, 2002, стр | |||
| Пишущая машина для тюркско-арабского шрифта | 1922 |
|
SU24A1 |

