Изобретение относится к способу получения иодированных рентгеноконтрастных агентов и, в частности, к способу получения иоформинола - контрастного агента, пригодного для проведения рентгенологических исследований. Более конкретно, изобретение относится к получению иоформинола из смеси соединений, содержащей 1-формиламино-3,5-бис(2,3-бис(формилокси)пропан-1-илкарбамоил)-2,4,6-трииодбензол, ключевое промежуточное соединение в процессе получения иоформинола.
За последние 50 лет в качестве рентгеноконтрастных агентов использовали главным образом растворимые иодсодержащие соединения. Имеющиеся в продаже контрастные среды, содержащие иодированные контрастные агенты, обычно классифицируют как ионные мономеры, например, диатризоат (Gastrografen™); ионные димеры, например иоксаглат (Hexabrix™); неионные мономеры, например иогексол (Omnipaque™), иопамидол (Isovue™), иомепрол (lomeron™) и неионный димер иодиксанол (Visipaque™). Наиболее широко применяемые коммерческие неионные рентгеноконтрастные агенты, такие как агенты, упомянутые выше, считаются безопасными. Контрастные среды, содержащие иодированные контрастные агенты, ежегодно применяют в США при более чем 20 млн рентгенологических исследований, и количество побочных эффектов считают приемлемым. Однако так как рентгенологическое исследование с контрастированием может требовать примерно до 200 мл контрастных сред в суммарной дозе, непрерывно продолжаются поиски усовершенствованных контрастных сред.
Часть контингента больных, рассматриваемая как пациенты высокого риска, увеличивается. Для того чтобы удовлетворить требования постоянного усовершенствования in vivo рентгенологических диагностических агентов для всего контингента больных, непрерывно продолжают исследования для обнаружения рентгеноконтрастных агентов, которые обладают улучшенными свойствами, в том числе с точки зрения контраст-индуцированной нефротоксичности (КИН).
Рентгеноконтрастные среды, содержащие в качестве активного фармацевтического ингредиента (ингредиентов) химическое соединение, имеющее две трииодированные фенильные группы, связанные соединительной группой, обычно называют димерными контрастными агентами, или димерами. В течение многих лет был предложен широкий спектр иодированных димеров. В настоящее время на рынке имеется одна контрастная среда, содержащая иодированный неионный димер в качестве активного фармацевтического ингредиента - продукт Visipaque™, содержащий соединение иодиксанол.
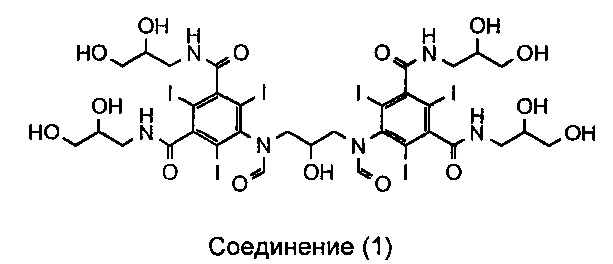
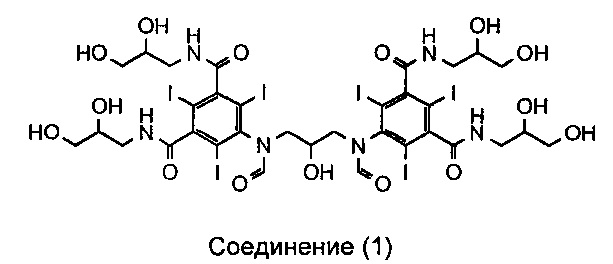
В публикации WO 2009/008734 автора данной заявки раскрыт новый димерный контрастный агент, называемый иоформинолом. Свойства этого агента описаны более подробно в публикациях Chai et al. «Predicting cardiotoxicity propensity of the novel iodinated contrast medium GE-145: ventricular fibrillation during left coronary arteriography in pigs» (Прогнозирование склонности к кардиотоксичности новой иодированной контрастной среды GE-145: фибрилляция желудочков при артериографии левых коронарных сосудов у свиней), Acta Radiol, 2010; и Wistrand, L.G. et al. «GE-145, a new low-osmolar dimeric radiographic contrast medium» (GE-145 - новое низкоосмотическое димерное рентгеноконтрастное вещество), Acta Radiol, 2010. Иоформинол (GE-145) в тексте данного описания называют соединением 1, и он имеет следующую структуру:
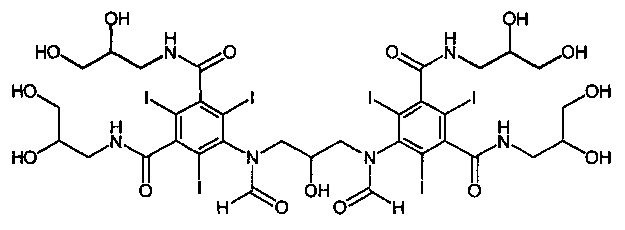
Соединение 1:
5,5'-(2-гидроксипропан-1,3-диил)бис(формилазанедиил)бис(N1,N3-бис (2,3-дигидроксипропил)-2,4,6-трииодизофталамид)
Изготовление неионных рентгеноконтрастных сред включает получение химического лекарственного препарата, активного фармацевтического ингредиента (АФИ), то есть контрастного агента, с последующим приготовлением готовой лекарственной формы, которая в тексте данного описания именуется рентгеноконтрастной композицией. Публикация WO 2009/008734 автора данной заявки обеспечивает ход синтеза для получения АФИ иоформинола. Иоформинол, например, можно синтезировать, согласно общему описанию получения и Примеру 1 из WO 2009/008734, из 5-амино-N,N'-бис-(2,3-дигидроксипропил)-2,4,6-трииодоизофталамида (соединение (4)), который имеется в продаже. Получение этого соединения известно из синтеза как иогексола, так и иодиксанола, и его можно также получить из 5-нитроизофталевой кислоты, например, как описано в WO 2006/016815, с участием гидрогенизации и последующего иодирования, например, хлоридом иода, ICl. В качестве альтернативы можно использовать 5-амино-2,4,6-трииодизофталевую кислоту, которая является имеющимся в продаже предшественником, например, от Sigma-Aldrich. Затем ацилируют свободную аминогруппу изофталамидного соединения (соединение (4)), и гидроксильные группы в заместителях также можно защитить посредством ацилирования. Защитные группы можно удалить, например, гидролизом, с получением N1,N3-бис(2,3-дигидроксипропил)-5-формиламино-2,4,6-трииодизофталамида. На стадии димеризации он участвует в реакции, например, бисалкилирования с эпихлоргидрином, с образованием соединения иоформинола - контрастного агента.
Существующий уровень техники в отношении синтеза иоформинола, раскрытый в примерах 1 и 2 WO 2009/008734, приведен на Схеме 1 ниже.
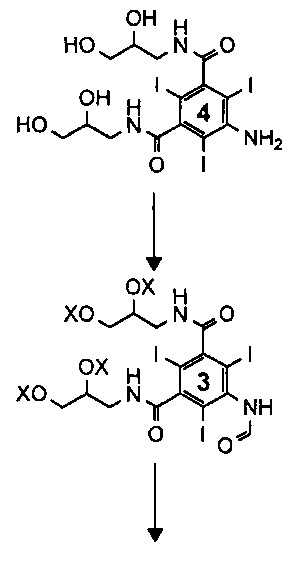
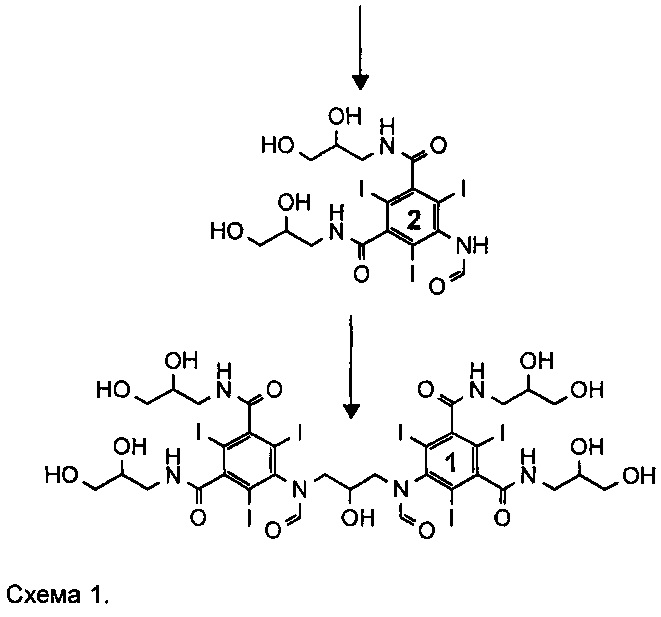
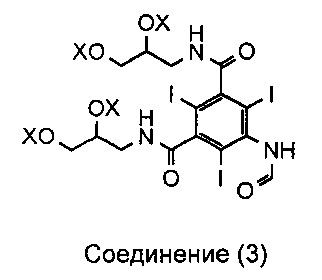
Как описано в WO 2009/008734, соединение 3 представляет собой смесь, включающую 1-формиламино-3,5-бис(2,3-бис(формилокси)пропан-1-илкарбамоил)-2,4,6-трииодбензол, а X, соответственно, является формильной группой.
На каждой стадии синтеза важно оптимизировать выход, минимизировать получение примесей, но при этом свести к минимуму затраченное время и средства. Задачу, которую должно решить настоящее изобретение, можно рассматривать как проведение оптимизации процесса получения иоформинола из соединения (3) схемы 1, то есть смеси, содержащей 1-формиламино-3,5-бис(2,3-бис(формилокси)пропан-1-илкарбамоил)-2,4,6-трииодбензол. Таким образом, изобретение направлено на способ, включающий гидролиз соединения (3) и реакцию димеризации с получением иоформинола.
На существующем уровне техники способ, раскрытый в WO 2009/008734, Пример 2, процедура В, получение соединения (1) (иоформинола) из 1-формиламино-3,5-бис(2,3-бис(формилокси)пропан-1-илкарбамоил)-2,4,6-трииодбензола (соединение (3), в котором X представляет собой формильную группу) осуществляют в ходе процесса, в котором соединение (3) растворено в смеси воды и метанола, дополнительно содержащей борную кислоту, при pH от 11,6 до 11,7, установленном путем добавления гидроксида калия. После получения раствора несколькими порциями добавляли эпихлоргидрин. Смесь оставляли при перемешивании, и несколько раз корректировали pH путем добавления гидроксида калия, одновременно регулируя температуру. Как сообщали, в целом процесс получения соединения (1) из соединения (3) занимает по меньшей мере 48 часов.
Был обнаружен более эффективный в экономическом отношении способ получения иоформинола (соединения (1)), в котором время протекания реакции сокращено, а выход соединения (1) увеличен. Заявители обнаружили, что соединение (1) можно получить с помощью эффективного в экономическом отношении и не причиняющего вреда окружающей среде способа, включающего гидролиз in situ защитных групп соединения (3) с последующим бис-алкилированием, также указанным как димеризация, при использовании воды в качестве единственного растворителя. Был обнаружен способ, занимающий значительно меньше времени, чем способ существующего уровня техники.
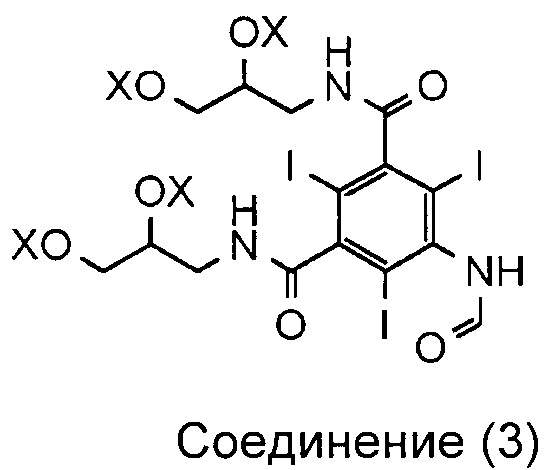
Соответственно, в первом аспекте данное изобретение обеспечивает способ получения соединения (1)
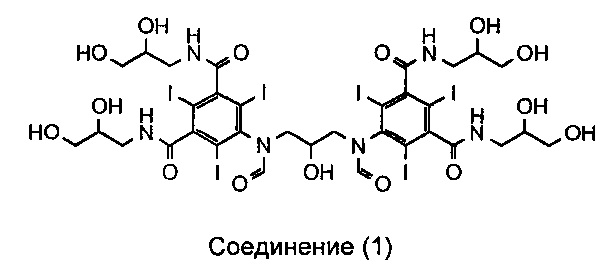
из соединения (3),
в котором каждый X по отдельности представляет собой водород, формильную группу (-CO-H) или ацетильную группу (-CO-CH3);
при этом способ включает стадию гидролиза in situ защитных групп (-OX) соединения (3), а соединение (3) суспендировано только в воде.
Соединение (3) представляет собой смесь различных соединений как с формильными, так и с ацетильными защитными группами. Это является результатом предшествующей стадии формилирования, на которой соединение (3) получают из соединения (4), предпочтительно с использованием смешанных ангидридов. В одном примере воплощения соединение 3 содержит смесь соединений, в которых все группы X по отдельности являются формильными или ацетильными. Основным компонентом соединения (3) является 1-формиламино-3,5-бис(2,3-бис(формилокси)пропан-1-илкарбамоил)-2,4,6-трииодбензол. Следовательно, в одном воплощении все группы X представляют собой формил.
В одном воплощении изобретения способ включает, перед реакцией бис-алкилирования, следующие последовательные стадии:
i) суспендирование соединения (3) в воде;
ii) регулирование pH раствора, полученного на стадии i), до 10,0-12,5.
После суспендирования соединения (3) и регулирования pH происходит гидролиз in situ формильных и ацетильных защитных групп соединения (3). В этой реакции получают соединение (2), но его не выделяют. В то же время в качестве побочных продуктов получают формильные и ацетильные соли. Это образование солей может оказывать ускоряющее воздействие на скорость реакции бис-алкилирования, протекающей после гидролиза in situ, обеспечивая быстрое и полное бис-алкилирование. При суспендировании исходного материала - соединения (3) - только в воде и при использовании данной процедуры гидролиза in situ наблюдали неожиданное увеличение скорости реакции бис-алкилирования. Полагают, что полученные соли увеличивают скорость реакции путем координации агента диалкилирования, применяемого при бис-алкилировании, и/или стабилизации переходного комплекса реакции. В то же время этот способ обеспечивает высокий выход. Было обнаружено, что реакцию превращения соединения (3) в соединение (1) можно провести менее чем за 24 часа, например, менее чем за 20 часов, с обеспечением выхода 90% или выше, например 93% или выше, или выше 95%. Что является неожиданным, проводимая только в воде стадия способа по данному изобретению обеспечивает выход примерно на 20% выше и экономит примерно один день времени получения по сравнению со способом существующего уровня техники, описанным в Примере 2 WO 2009/008734.
На стадии i) в качестве единственного растворителя используют воду, и неожиданно было обнаружено, что нет необходимости добавлять другие растворители или добавки, такие как, например, борная кислота. В способе по изобретению исходный материал - соединение (3) - предпочтительно представляет собой мелкий порошок с низким содержанием кислоты. Смесь (3) соединений может, в одном из примеров воплощения, содержать некоторые остатки антирастворителей, например, спиртов; но нет необходимости добавлять спирт. В способе получения соединения (3), для оптимизации получения его в форме порошка, можно применять спирт с короткой цепью; и было обнаружено, что полезно, когда соединение (3), в том виде, как его используют в качестве исходного материала в заявленном способе, не высушено полностью, но содержит от 0 до 15% спирта; и приемлемым является остаточное содержание спирта от 0 до 7%, наиболее предпочтительно от 2 до 5%. Остаточное содержание спирта в соединении (3) обычно представляет собой спирт с короткой цепью и является C1-C6 спиртом с прямой или разветвленной цепью, или смесью таких спиртов. Спирт может быть одноатомным или двухатомным. Предпочтительными спиртами являются метанол, этанол и пропанолы; при этом наиболее предпочтительным являются пропанолы, особенно изо-пропанол. Количество воды, необходимое для создания оптимальных реакционных условий, зависит от таких факторов, как содержание остаточного спирта и температура. Было обнаружено, что приемлемым количеством воды является примерно 0,5-2,0 литров воды на килограмм соединения (3), например, примерно 1 литр воды на килограмм соединения (3).
На стадии ii), следовательно перед реакцией бис-алкилирования, pH регулируют до 10,0-12,5, а более предпочтительно до 11,0-11,8, а наиболее предпочтительно до 11,0-11,2, путем добавления основания к суспензии соединения (3). Предпочтительно pH регулируют ступенчато, чтобы нейтрализовать кислоты и избежать слишком резкого выделения тепла. Основание выбирают из сильных водорастворимых оснований, например гидроксида натрия и гидроксида калия; предпочтительным является раствор (например, 50%) гидроксида натрия. Добавление основания обеспечивает приводимый в действие основанием гидролиз защитных сложноэфирных групп соединения (3); при этом в качестве побочных продуктов образуются формильные и ацетильные соли, например формильные соли натрия и ацетильные соли натрия. Кроме того, такое регулирование pH обеспечивает оптимальные условия по pH для бис-алкилирования. В одном из примеров воплощения регулирование pH проводят с использованием системы pH-стата (pH-stat), чтобы гарантировать поддержание стационарного pH. Такая pH-система содержит как кислоту, так и основание, например растворы HCl и NaOH.
Бис-алкилирование (стадия димеризации) посредством 2-гидроксипропанового мостика предпочтительно происходит путем добавления соответствующего количества диалкилирующего агента к щелочному раствору стадии ii). Такой агент выбирают из дигалогензамещенного алканола или галогензамещенного гетероциклоалкила, такого как 1,3-дихлор-2-пропанол, 1-хлор-2,3-пропанол, 1,3-дибром-2-гидроксипропан и эпихлоргидрин (ЭХГ), при этом особенно предпочтительным является ЭХГ. Соответственно, в еще одном примере воплощения данного изобретения способ дополнительно включает стадию добавления к щелочному раствору стадии ii) диалкилирующего агента. Диалкилирующий агент добавляют к щелочному водному раствору одной или большим количеством порций, например, 1-5 порциями, предпочтительно 3 равными порциями. Примерно 2 мольных эквивалента соединения (3) реагируют и образуют мостик с одним мольным эквивалентом диалкилирующего агента. Можно применять небольшой мольный избыток диалкилирующего агента ввиду небольшого потребления диалкилирующего агента основанием. В ходе добавления и после него реакционную смесь поддерживают при перемешивании в течение периода, необходимого для завершения реакции бис-алкилирования. Это может потребовать от 5 до 20 часов, предпочтительно от 10 до 15 часов.
Перед, на протяжении и/или после гидролиза можно также регулировать температуру, например, снижая ее ниже комнатной, до 12-16°C или ниже. В предпочтительном примере воплощения перед добавлением диалкилирующего агента температуру регулируют примерно до 15°C. Особенно предпочтительно перед добавлением pH регулируют до 11,0-11,2, а температуру регулируют примерно до 15°C, и эти условия поддерживают до завершения бис-алкилирования.
Полученные соединения, например соединение (1), можно очистить любым удобным образом, например промывкой, препаративной хроматографией, перекристаллизацией или ультра/нанофильтрацией. Таким образом, возможными дополнительными стадиями являются очистка и сушка.
В том виде, как они получены заявленным способом, смесь (3) соединений и соединение (1) содержат оптически активные изомеры и существуют в нескольких изомерных формах, из-за хиральных атомов углерода. В дополнение эти соединения проявляют экзо/эндо изомерию из-за ограниченного вращения связи N-CO в формильной функциональной группе, обусловленного близостью объемного атома йода. Способ по данному изобретению охватывает получение как энантиомерно чистых продуктов, так и смесей оптических изомеров.
Соединения, полученные по данному изобретению, можно использовать в качестве контрастных агентов, и их можно объединять с традиционными носителями и вспомогательными веществами, чтобы получить диагностические контрастные среды. Таким образом, с точки зрения дополнительных аспектов данное изобретение обеспечивает иоформинол (соединение (1)) и диагностическую композицию, содержащую иоформинол, полученный способом по данному изобретению, где указанная композиция включает по меньшей мере один физиологически приемлемый носитель или вспомогательное вещество, например водный раствор для инъекций, возможно совместно с добавленными ионами плазмы или растворенным кислородом. Композиция контрастного агента по данному изобретению может быть в концентрации, готовой для применения, или она может быть в форме концентрата, предназначенного для разбавления перед введением. Таким образом, данное изобретение дополнительно охватывает применение при рентгеноконтрастных исследованиях иоформинола, полученного в соответствии с данным способом получения, и содержащей его диагностической композиции.
Изобретение проиллюстрировано со ссылкой на следующие неограничивающие примеры.
Примеры
Пример 1: Получение соединения (1) из соединения (3)
Соединение (3) (1103 кг, 890 моль) было суспендировано в воде (1213 л) с использованием реактора с введенной сверху механической мешалкой. Суспензию охлаждали до 10 градусов и в течение 12 часов добавляли водный раствор NaOH (50%), поддерживая pH и температуру ниже 12,5 и 20 градусов, соответственно. Раствор охлаждали до 16 градусов, и pH устанавливали на 11,1, используя систему pH-стата, заправленную HCl (30%) и NaOH (50%). Систему оставили работающей до тех пор, пока не была достигнута температура <18 градусов. ЭХГ (41 кг, 445 моль) непрерывно добавляли в течение 2,5 часов, поддерживая температуру между 15 и 18°C. После перемешивания в течение 38 часов реакционную смесь подавляли, регулируя pH на 7 с использованием HCl (30%). Высокоэффективная жидкостная хроматография показала ~95,5% (УФ) выход соединения (1).
В этом примере время реакции диалкилирования было более длительным, чтобы гарантировать завершение реакции и максимизировать выход. Реакцию можно прервать значительно раньше, например на 10-12 часов, без существенной потери по выходу.
| название | год | авторы | номер документа |
|---|---|---|---|
| Получение промежуточного соединения синтеза иоформинола | 2013 |
|
RU2654461C2 |
| ОЧИСТКА РЕНТГЕНОКОНТРАСТНЫХ ВЕЩЕСТВ | 2013 |
|
RU2662941C2 |
| КОНТРАСТНЫЕ АГЕНТЫ | 2008 |
|
RU2469021C2 |
| Получение промежуточных соединений для получения рентгеноконтрастных веществ | 2013 |
|
RU2659214C2 |
| СПОСОБ И РЕАГЕНТ ДЛЯ ПОЛУЧЕНИЯ ДИАГНОСТИЧЕСКОЙ КОМПОЗИЦИИ | 2014 |
|
RU2662319C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЙОПАМИДОЛА | 2014 |
|
RU2657238C2 |
| СПОСОБ ЙОДИРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2009 |
|
RU2469997C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИОГЕКСОЛА | 1997 |
|
RU2173315C2 |
| ИОДИРОВАННЫЕ АРИЛЬНЫЕ СОЕДИНЕНИЯ И ДИАГНОСТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2145955C1 |
| Способ получения инъектируемой контрастной среды | 1990 |
|
SU1829940A3 |
Изобретение относится к способу получения соединения (1), используемого в качестве контрастного агента для проведения рентгенологических исследований, из соединения (3). В соединении (3) каждый X по отдельности обозначает водород, формильную группу (-СО-Н) или ацетильную группу (-СО-СН3). При этом способ включает приводимый в действие основанием in situ гидролиз защитных групп (-ОХ) соединения (3), включающий следующие последовательные стадии: i) суспендирование соединения (3) в воде; ii) регулирование рН раствора, полученного на стадии i), до величины от 10,0 до 12,5. Предлагаемый способ позволяет сократить время протекания реакции и увеличить выход соединения (1). 5 з.п. ф-лы, 1 пр.
1. Способ получения соединения (1)
из соединения (3)
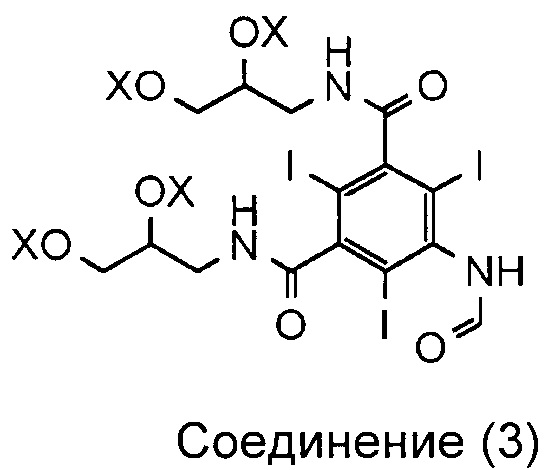 ,
,
в котором каждый X по отдельности обозначает водород, формильную группу (-СО-Н) или ацетильную группу (-СО-СН3);
причем указанный способ включает приводимый в действие основанием in situ гидролиз защитных групп (-ОХ) соединения (3), включающий следующие последовательные стадии:
i) суспендирование соединения (3) в воде;
ii) регулирование рН раствора, полученного на стадии i), до величины от 10,0 до 12,5.
2. Способ по п.1, в котором исходный материал - соединение (3) - представляет собой мелкий порошок с низким содержанием кислоты.
3. Способ по п.1, в котором исходный материал - соединение (3) - содержит от 0 до 15% спирта.
4. Способ по п.1, дополнительно включающий реакцию бис-алкилирования, которую проводят путем добавления к щелочному раствору стадии (ii) соответствующего количества диалкилирующего агента.
5. Способ по п.4, в котором диалкилирующий агент представляет собой эпихлоргидрин.
6. Способ по п.1, включающий стадию регулирования температуры раствора, полученного на стадии i) или ii), до 12-16°С.
| Колосоуборка | 1923 |
|
SU2009A1 |
| ПОЛУЧЕНИЕ ИОДИКСАНОЛА | 2005 |
|
RU2385316C2 |

