Изобретение относится к способу получения йодированных рентгеноконтрастных веществ и, в частности, к способу получения ключевых промежуточных соединений, используемых для получения йодированных рентгеноконтрастных веществ. Изобретение дополнительно относится к усовершенствованному способу получения 5-ацетамидо-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодоизофталамида (Соединения А) или N,N'-бис(2,3-дигидроксипропил)-5-формамидо-2,4,6-трийодоизофталамида (Соединения С), которые являются промежуточными соединениями при промышленном получении неионных рентгеноконтрастных веществ. В частности, изобретение относится к способу деацилирования ацилированных гидроксильных групп промежуточного соединения, образующегося при получении названных соединений. Дополнительно, изобретение относится к способу получения таких контрастных веществ, как йодиксанол, йогексол и йоформинол, применяемых для получения рентгеновских изображений.
В течение последних пяти десятилетий наиболее широко используемыми среди рентгеноконтрастных веществ являются растворимые йодсодержащие соединения. Коммерчески доступные контрастные препараты, содержащие йодированные контрастные вещества, обычно разделяют на ионные мономеры, такие как диатризоат (Gastrografen™), ионные димеры, такие как йоксаглат (Hexabrix™), неионные мономеры, такие как йогексол (Omnipaque™), йопамидол (Isovue™), йомепрол (Iomeron™) и неионный димер йодиксанол (Visipaque™). Наиболее широко используемые коммерчески доступные неионные рентгеноконтрастные вещества, например перечисленные выше, считаются безопасными. Ежегодно в США проводят более 20 миллионов рентгенологических исследований с использованием контрастных препаратов, содержащих йодированные контрастные вещества, и количество побочных явлений считается приемлемым. Неионные рентгеноконтрастные вещества составляют очень важный класс выпускаемых в больших количествах фармацевтических соединений. Важными примерами таких соединений являются 5-[N-(2,3-дигидроксипропил)-ацетамидо]-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодоизофталамид (йогексол) и 1,3-бис(ацетамидо)-N,N'-бис[3,5-бис(2,3-дигидроксипропил-аминокарбонил)-2,4,6-трийодфенил]-2-гидроксипропан (йодиксанол). Например, йодиксанол, выпускаемый под торговым наименованием Visipaque ®, является одним из наиболее широко используемых в диагностических рентгенологических процедурах веществом. Его в больших количествах производит GE Healthcare, Норвегия.
Производство неионных рентгеноконтрастных препаратов включает получение химического препарата, активного фармацевтического ингредиента (АФИ), т.е. контрастного вещества, которое затем вводят в лекарственный препарат, далее называемый рентгенологической композицией.
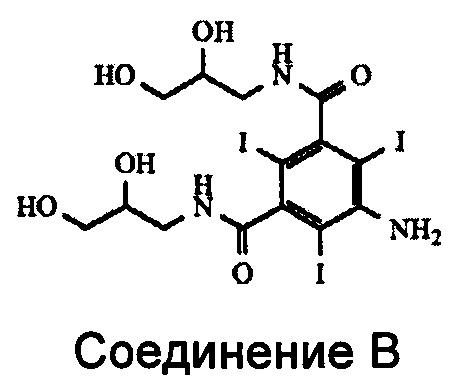
Промышленное получение йодиксанола и йогексола включает многостадийный химический синтез, последние стадии которого, начиная с 5-амино-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодоизофталамида, называемого Соединением В, представлены ниже на Схеме 1. Получение Соединения В подробно описано в предшествующем уровне техники. См. также патент US 6974882.
Схема 1
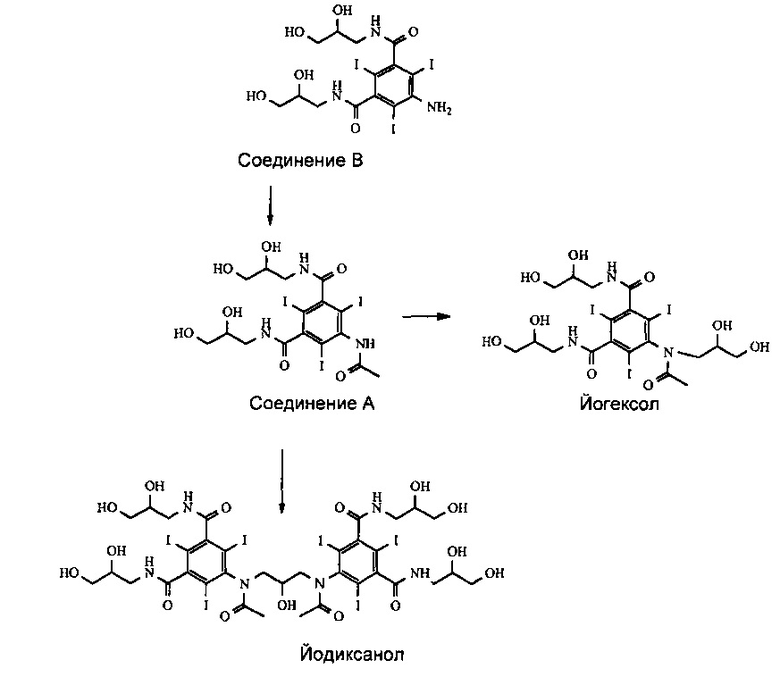
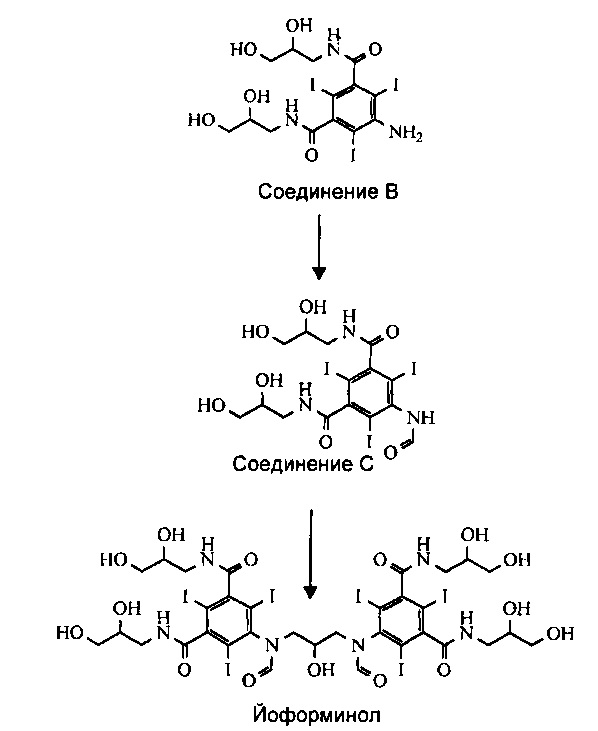
Также, как показано ниже на Схеме 2, из Соединения В может быть получено соединение, называемое йоформинолом, 5,5'-(2-гидроксипропан-1,3-диил)бис(формилазандиил)бис(N1,N3-бис(2,3-дигидроксипропил)-2,4,6-трийодоизофталамид).
Схема 2
Для снижения стоимости контрастного вещества и готового препарата необходимо оптимизировать каждую из стадий синтеза с целью получения оптимального выхода и снижения образования загрязняющих веществ. При крупномасштабном производстве даже небольшое усовершенствование способа проведения реакции может привести к значительной экономии.
Для получения N-ацилированных мономерных соединений из соединения В, например, N-ацетилированного соединения 5-ацетамидо-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодоизофталамида, в настоящем описании называемого Соединением А, и N-формилированного N,N'-бис(2,3-дигидроксипропил)-5-формамидо-2,4,6-трийодоизофталамида, в настоящем описании называемого Соединением С, был разработан усовершенствованный способ.
На стадии ацетилирования при синтезе йогексола и йодиксанола в производственных масштабах для получения 5-ацетиламино-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодоизофталамида (Соединения А) производят ацетилирование 5-амино-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодоизофталамида (Соединения В) таким ацетилирующим реагентом, как уксусный ангидрид. Авторами изобретения было обнаружено, что Соединение А может быть получено из Соединения В оптимизированным способом, в котором, кроме того, значительно снижено образование некоторых загрязняющих веществ.
Задача, решаемая настоящим изобретением, может рассматриваться как оптимизация способа получения N-ацилированных мономерных соединений, которые являются промежуточными соединениями в синтезе контрастных веществ для рентгенологического исследования, в частности в получении Соединения А и Соединения С.
В реакции ацилирования Соединения В происходит ацилирование функциональной аминогруппы, т.е. либо ее ацетилирование, либо формилирование в зависимости от типа получаемого конечного продукта. Но при этом также происходит ацилирование всех четырех гидроксильных групп Соединения В (см. Схему 3). Наличие в промежуточном соединении, называемом Соединением В2, ацилированных гидроксильных групп нежелательно, и после проведения реакции ацилирования эти группы необходимо деацилировать. Настоящее изобретение включает способ деацилирования тех ацилированных групп Соединения В2, ацилирование которых нежелательно, которые получают на стадии ацилирования.
Схема 3
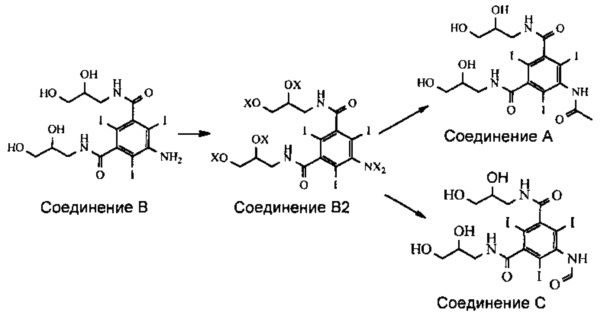
В Соединении В2 X означает водород или ацильную группу.
Способ получения Соединения А из Соединения В, включающий деацетилирование О-ацетилированных групп Соединения В2, известен, например, из патентной публикации ЕР 2281811 А1, GE Healthcare AS, в которой рассмотрен непрерывный способ, включающий деацетилирование ацетилированных гидроксильных групп в первом реакторе при рН, составляющем от 11 до 12, при добавлении подходящего основания, например, водного раствора гидроксида натрия.
Первая стадия способа получения Соединения А из Соединения В представляет собой ацетилирование. Следующая стадия состоит в деацетилировании с целью отщепления О-ацетильных групп, образованных в результате реакции ацетилирования. В настоящее время было обнаружено, что одним из основных веществ, загрязняющих неочищенное Соединение А, полученное способом согласно предшествующему уровню техники, является Соединение 1, представленное ниже:
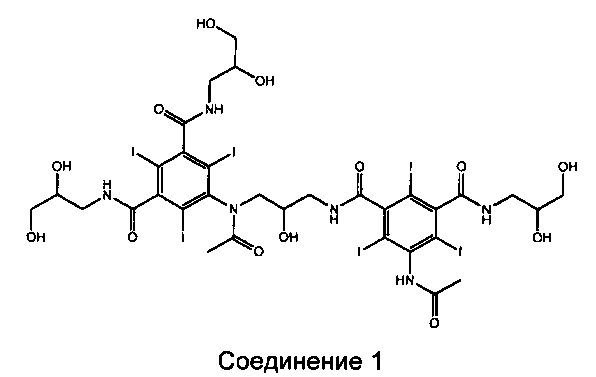
Соединение 1 представляет собой 5-ацетамидо-N1-(3-(N-(3,5-бис((2,3-дигидроксипропил)карбамоил)-2,4,6-трийодфенил)ацетамидо)-2-гидроксипропил)-N3-(2,3-дигидроксипропил)-2,4,6-трийодоизофталамид.
Тщательное исследование механизма реакции показало, что Соединение 1 образуется в несколько стадий при осуществлении способа согласно предшествующему уровню техники. Обычно при проведении стадии деацилирования гидроксид натрия (50%, водн.) добавляют в раствор, содержащий избыточно ацилированное Соединение В, т.е. к соединению, обозначенному на Схеме 2 как Соединение В2. Неожиданно было обнаружено, что деацилирование может быть произведено альтернативным образом, при котором значительно снижается количество образующегося загрязняющего вещества, называемого Соединением 1. Обнаруженная альтернатива заключается в добавлении раствора Соединения В2 к деацилирующему агенту, например к основанию.
Соответственно, первый аспект изобретения относится к способу получения N-ацилированного мономерного соединения, выбранного из Соединения А и Соединения С, который включает стадию, в которой Соединение В2,
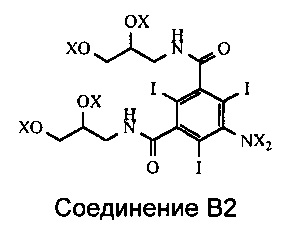
где каждый из X независимо означает водород или ацильную группу,
подвергают деацилированию ацилированных гидроксильных групп путем добавления Соединения В2 к водному раствору деацилирующего агента.
В одном из воплощений Соединение В2 добавляют к деацилирующему агенту в виде раствора. В другом воплощении при добавлении в деацилирующий агент Соединение В2 представляет собой твердое вещество. Предпочтительно применяют раствор Соединения В2.
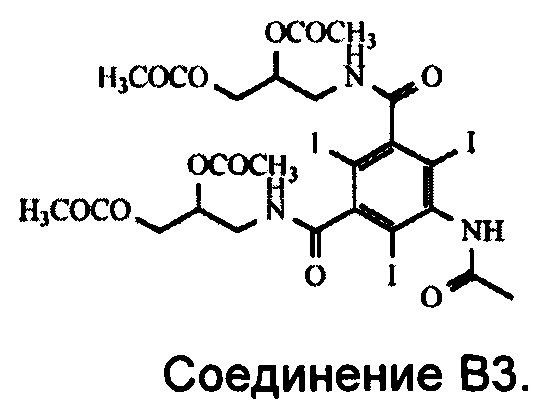
Группа X представляет собой либо водород, либо ацил при условии, что по меньшей мере одна из групп X, присоединенных к атому азота, представляет собой ацильную группу. Ацильная группа выбрана из формильной и ацетильной группы и предпочтительно представляет собой ацетильную группу. Основным компонентом Соединения В2 предпочтительно является Соединение B3.
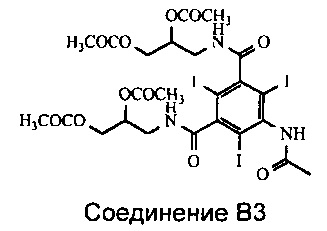
В одном из воплощений Соединение В2 представляет собой Соединение B3.
По сравнению со способом согласно предшествующему уровню техники, вместо добавления деацилирующего агента, обычно основания, к Соединению В2, Соединение В2 добавляют к деацилирующему агенту. Основным преимуществом альтернативного способа деацилирования согласно изобретению является уменьшение получаемого количества определенных загрязняющих веществ. В частности, при получении Соединения А согласно изобретению достигают снижения количества образующегося загрязняющего Соединения 1 и снижения теплоты, выделяемой при проведении деацилирования. Соединение 1 представляет собой загрязняющее вещество, трудноудаляемое из неочищенного продукта - Соединения А. Например, это загрязняющее вещество нельзя удалить кристаллизацией. Соответственно, значительное снижение количества образующегося Соединения 1 в способе согласно изобретению оказывает влияние в особенности на выход при использовании полученного Соединения А на последующих стадиях синтеза, например, при получении контрастного вещества, такого как йогексол или йодиксанол.
Было обнаружено, что образование загрязняющего Соединения 1 в способе согласно предшествующему уровню техники происходит в несколько стадий. Две последние стадии протекают на стадии деацилирования. Сначала образуется эпоксид - Соединение 2.
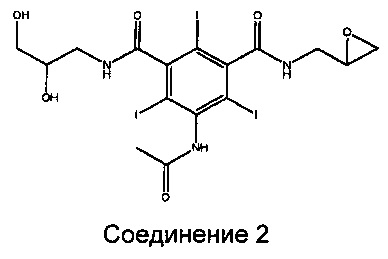
Этот эпоксид отличается реакционной способностью и, во-вторых, реагирует в щелочных условиях с получаемой молекулой анионного Соединения А, как показано ниже, с образованием загрязняющего Соединения 1.
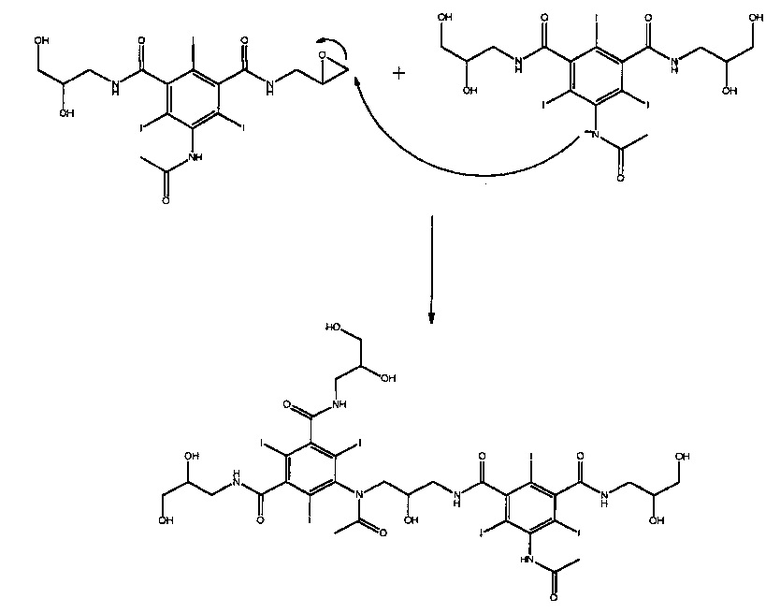
Эта стадия образования Соединения 1 представляет собой реакцию димеризации, и, поскольку эта реакция протекает между двумя молекулами, было показано, что, таким образом, протекание этой стадии зависит от концентрации. Соответственно, можно предположить, что при проведении стадии деацилирования при большем разбавлении образование Соединения 1 будет подавляться. В способе согласно предшествующему уровню техники гидроксид натрия (50%, водный) добавляют в кислый раствор, содержащий избыточно ацилированное Соединение А (т.е. соединение В2).
Определив загрязняющее Соединения 1 и путь его образования и предположив о необходимости большего разбавления, очевидное решение проблемы заключается в том, что разбавление кислого раствора Соединения В2 может быть осуществлено значительно большим количеством растворителя, таким как метанол с водой, перед проведением стадии деацилирования для подавления образования Соединения 1. Однако было обнаружено, что недостатком такого решения является снижение эффективности производства.
Как было указано при рассмотрении первого аспекта изобретения, было неожиданно обнаружено удобное и экономически эффективное альтернативное решение этой проблемы, которое состоит в добавлении Соединения В2, такого как кислый раствор Соединения В2, к водному раствору деацилирующего агента. В таком альтернативном способе снижается количество теплоты, выделяющейся на стадии деацилирования, по сравнению со способом, включающим добавление деацилирующего агента в раствор Соединения В2, поскольку большую часть теплоты, выделяющейся при разбавлении, удаляют еще до начала деацилирования. Таким образом, деацилирование можно проводить в более мягких температурных условиях. Наиболее важное отличие от способа деацилирования согласно предшествующему уровню техники состоит в получении иного профиля концентрации субстрата соединения В2 во время деацилирования. В способе деацилирования согласно изобретению концентрация будет гораздо ниже по сравнению с концентрацией, получаемой в способе согласно предшествующему уровню техники, вследствие чего значительно снижается количество образующегося Соединения 1.
Перед деацилированием ацилированных гидроксильных групп Соединения В2 производят ацилирование Соединения В. Таким образом, в этом воплощении способ включает указанную стадию, то есть изобретение относится к способу получения N-ацилированного мономерного соединения, выбранного из Соединения А и Соединения С, включающему:
первую стадию ацилирования Соединения В с образованием Соединения В2;
с последующим проведением стадии, в которой осуществляют деацилирование ацилированных гидроксильных групп Соединения В2 посредством добавления Соединения В2 к водному раствору деацилирующего агента.
Ацилирование Соединения В может быть выполнено любым подходящим способом, например, включающим применение ангидридов и кислот. Если стадия ацилирования представляет собой формилирование с целью получения Соединения С, то может быть применен любой подходящий способ формилирования, например, включающий применение в качестве формилирующего агента активированной муравьиной кислоты, например смешанных ангидридов. Смешанные ангидриды могут быть получены множеством способов, рассмотренных в литературе. Один из подходящих способов получения смешанных ангидридов состоит в добавлении ангидрида карбоновой кислоты к избытку уксусной кислоты при поддержании регулируемой температуры. Предпочтительно на стадии формилирования применяют смесь муравьиной кислоты и уксусного ангидрида. В результате проведения стадии формилирования при использовании смешанных ангидридов, Соединение В2 представляет собой смесь различных соединений, содержащих как формильные, так и ацетильные защитные группы. Могут быть достигнуты различные степени О-формилирования, но при этом степень N-формилирования высока, что обеспечивает высокий выход N-формилированного Соединения С после деацилирования. Если стадия ацилирования представляет собой стадию ацетилирования с образованием Соединения А, то предпочтительно применяют уксусную кислоту и уксусный ангидрид. В этом воплощении основным компонентом Соединения В2 является Соединение B3, в котором все ацилированные ОН группы являются ацетилированными.
Выбор типа деацилирующего агента не имеет особенных ограничений, и для осуществления настоящего изобретения может быть применен любой деацилирующий агент, обычно используемый для традиционного проведения подобных реакций. Примеры подходящих деацилирующих агентов включают водные растворы неорганических оснований, включающие карбонаты щелочных металлов, например карбонат натрия, карбонат калия или карбонат лития; и гидроксиды щелочных металлов, например гидроксид натрия, гидроксид калия или гидроксид лития. Предпочтительными среди перечисленных соединений являются гидроксиды щелочных металлов, в частности гидроксид натрия или гидроксид калия, и наиболее предпочтительным является гидроксид натрия.
В одном из воплощений перед добавлением деацилирующего агента в раствор Соединения В2 деацилирующий агент разбавляют водой. В одном из предпочтительных воплощений деацилирующий агент представляет собой гидроксид натрия, и если применяют этот агент, то, например, 50% водный раствор гидроксида натрия добавляют в воду в соотношении от 1:1 до 1:5, например в соотношении от 1:1 до 1:2. Предпочтительно концентрация применяемого основания, например NaOH, составляет приблизительно от 10 до 50% масс., более предпочтительно от 20 до 25%. В начале добавления раствора Соединения В2 к деацилирующему агенту рН смешиваемого раствора составляет приблизительно 14; затем, по мере добавления раствора Соединения В2 и к завершению добавления, величина рН снижается приблизительно до 11-13. Продолжительность добавления составляет, например, от 0,5 до 2 часов. Раствор Соединения В2 добавляют к деацилирующему агенту либо несколькими небольшими порциями в течение указанного времени, либо более предпочтительно раствор Соединения В2 добавляют непрерывно медленно и осторожно при перемешивании. В этих условиях не подвергается гидролизу и остается ацилированной только необходимая NH-ацильная группа. При этом количество образующегося загрязняющего Соединения 1 оказывается минимальным.
В этом способе на стадии деацилирования выделяется малое количество теплоты, поскольку большую часть теплоты, выделяемой при разбавлении, удаляют еще до начала деацилирования. Таким образом, в целом, температура повышается на меньшую величину по сравнению со способом согласно предшествующему уровню техники. Таким образом, деацилирование может быть проведено в более мягких температурных условиях. Во время добавления раствора Соединения В2 к раствору деацилирующего агента, температура, в зависимости от, например, типа применяемого оборудования, повышается, например, от начальной температуры, составляющей от 20 до 35°C, до конечной температуры, составляющей от 50 до 60°C. По окончании добавления, перед началом кристаллизации, раствор может быть дополнительно разбавлен, предпочтительно водой для достижения требуемой концентрации.
Реакцию деацилирования предпочтительно проводят в присутствии растворителя, и перед добавлением раствора Соединения В2 к деацилирующему агенту Соединение В2 растворяют в растворителе. Выбор типа растворителя не имеет особенных ограничений при условии, что растворитель не оказывает негативного воздействия на ход реакции или на реагенты, участвующие в реакции, и может растворять эти реагенты по меньшей мере до некоторой степени. Примеры подходящих растворителей включают: простые эфиры, например простой диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; спирты, например метанол или этанол; и воду. Предпочтительными из перечисленных растворителей являются спирты, в частности метанол или смесь воды и одного или более спиртов. В частности, в одном из воплощений для получения Соединения С, Соединение В2 не растворяют в растворителе перед добавлением к деацилирующему агенту, и, таким образом, его добавляют в виде твердого вещества.
После проведения стадии деацилирования, т.е. после добавления всего Соединения В2 в деацилирующий агент, к смеси для снижения рН добавляют кислоту для понижения растворимости полученного Соединения А или Соединения С, в результате чего образуется более концентрированная суспензия и, таким образом, начинается осаждение Соединения А или Соединения С. Добавляемая кислота представляет собой растворимую в воде сильную неорганическую кислоту, которая предпочтительно выбрана из группы, включающей серную кислоту, азотную кислоту и соляную кислоту; наиболее предпочтительной является соляная кислота. Величину рН снижают до диапазона от 2,0 до 8,0, например от 5,0 до 8,0, и предпочтительно до приблизительно 7 при перемешивании в течение времени, достаточного для полного осаждения, например в течение по меньшей мере получаса. В одном из воплощений до, во время или после регулирования рН в реакционный раствор добавляют затравочный кристалл Соединения А или Соединения С, в зависимости от типа получаемого соединения. Полученную суспензию предпочтительно оставляют перемешиваться при пониженной температуре, например от 10 до 25°C, предпочтительно приблизительно при 20°C в течение времени, составляющего от 5 до 20 часов, например приблизительно 10 часов. Затем полученное Соединение А собирают и факультативно очищают. В одном из воплощений способ включает дополнительную стадию сбора продукта, предпочтительно фильтрованием, например, с помощью нутч-фильтра, например вакуумного нутч-фильтра или нутч-фильтра, работающего под давлением, или их комбинации, также факультативно при одновременном нагревании. Затем продукт предпочтительно промывают одной или более порциями растворителя, например применяют от 1 до 5 порций и предпочтительно 3 порции, предпочтительно тем же растворителем, который применяли на стадии деацилирования, например метанолом, и затем факультативно сушат в подходящем оборудовании для сушки. Соответственно, в дополнительном воплощении изобретения получаемое после деацилирования Соединение А или Соединение С собирают и факультативно очищают, например кристаллизацией.
Способ согласно изобретению позволяет получать Соединение А и Соединение С с постоянным качеством и выходом. Процедуру повторяли множество раз, каждый раз получая чистоту более 99,5%, как в мелкомасштабных, так и в крупномасштабных экспериментах, например, при получении порядка 100 кг вещества. В частности, количество загрязняющего Соединения 1, образующегося при получении Соединения А способом согласно изобретению, минимально, и высушенное готовое Соединение А включает 0,08% или менее, предпочтительно менее 0,05% и более предпочтительно менее 0,025% Соединения 1. Дополнительный аспект изобретения относится к получению высушенного готового Соединения А, которое содержит 0,08% или предпочтительно менее 0,05% масс. и более предпочтительно менее 0,025% масс. загрязняющего Соединения 1. Такой продукт может быть получен способом согласно изобретению. При осуществлении способа согласно изобретению образуется значительно меньшее количество Соединения 1 по сравнению со способом согласно предшествующему уровню техники, что, в частности, важно при использовании полученного Соединения А в дальнейшем синтезе контрастных веществ. Например, снижение содержания образующегося Соединения 1 в очищенном Соединении А с 0,09% масс. до 0,02% масс. повышает общий выход получаемого йодиксанола на 1-5%, то есть приводит к значительному экономическому выигрышу.
Таким образом, способы дальнейшего синтеза, например получения таких контрастных веществ, как йодиксанол, йогексол или йоформинол, в которых используют Соединение А или Соединение С, полученные способом согласно настоящему изобретению, также включены в объем настоящего изобретения. Таким образом, дополнительный аспект изобретения относится к способу получения контрастного вещества, который включает способ получения Соединения А или Соединения С, рассмотренный в первом аспекте. Такой способ может включать дополнительные стадии алкилирования или бис-алкилирования (димеризации) с образованием йогексола или йодиксанола, соответственно, или с образованием йоформинола, если производят бис-алкилирование Соединения С. Например, последняя стадия получения йодиксанола включает бис-алкилирование с участием 2-гидроксипропанового мостика. Эта стадия может быть выполнена, как описано в европейском патенте 108638 и международной патентной заявке WO 98/23296, например, с использованием в качестве агента димеризации эпихлоргидрина, 1,3-дихлор-2-гидроксипропана или 1,3-дибром-2-гидроксипропана. Димеризацию предпочтительно проводят в присутствии агента, связывающего кислоту, например, органического или неорганического основания; в качестве основания может быть использован алкоголят щелочного металла, например, метилат натрия, или гидроксид щелочного металла, например гидроксид натрия и гидроксид калия.
Соединения, полученные способом согласно изобретению, то есть Соединения А и С, и готовые контрастные вещества содержат оптически активные изомеры, и из-за наличия в них хиральных атомов углерода существуют в нескольких изомерных формах. Кроме того, из-за ограничения вращения вокруг связи N-CO функциональной ацильной группы, обусловленного близостью объемного атома йода, указанные соединения имеют экзо-/эндо-изомеры. Способ согласно изобретению включает как получение энантиомерно чистых продуктов, так и смесей оптических изомеров.
Соединения, получаемые согласно изобретению, могут быть применены в качестве контрастных веществ, и для получения диагностического контрастного препарата могут быть включены в композиции, содержащие традиционные носители и вспомогательные вещества. Таким образом, дополнительный аспект изобретения относится к диагностической композиции, включающей контрастное вещество, полученное способом согласно изобретению, и по меньшей мере один физиологически приемлемый носитель или вспомогательное вещество, например водный раствор для инъекций, в который факультативно добавлены ионы плазмы или растворенный кислород. Композиция, включающая контрастное вещество согласно изобретению, может иметь концентрацию препарата, готового к употреблению, или может представлять собой концентрированный препарат, который перед введением должен быть разбавлен. Таким образом, изобретение дополнительно включает применение контрастного вещества, полученного способом получения, и содержащей его диагностической композиции для рентгеновских исследований с применением контрастных веществ.
Ниже для иллюстрации изобретения приведены некоторые неограничивающие примеры.
ПРИМЕРЫ
Пример 1
Ацетилирование Соединения В с последующим деацетилированием способом согласно предшествующему уровню техники и способом согласно изобретению с целью получения Соединения А
Ацетилирование Соединения В проводили в смеси уксусного ангидрида и уксусной кислоты. В качестве катализатора применяли пара-толуолсульфокислоту (ПТСК). После ацетилирования раствор концентрировали под уменьшенным давлением, и перед стадией деацетилирования добавляли метанол и воду. Этот раствор (раствор Соединения В2) делили на две равные части. Одну часть подвергали деацетилированию согласно предшествующему уровню техники, а другую часть подвергали деацетилированию способом согласно изобретению.
Сравнительный способ согласно предшествующему уровню техники
Гидроксид натрия (50%, 150 мл) добавляли в раствор, содержащий Соединение В2. Перед добавлением гидроксида натрия в раствор Соединения В2 температура раствора составляла 30°C. Продолжительность добавления гидроксида натрия составляла приблизительно 20 минут, и во время добавления температура поднималась до приблизительно 55°C. Затем раствор разбавляли водой до достижения общего объема, составляющего 850 мл. Затем добавляли соляную кислоту (17,5%) до незначительного помутнения раствора, и в раствор помещали затравочный кристалл Соединения А (0,9 г). Суспензию перемешивали в течение 45 минут, а затем добавляли соляную кислоту (17,5%) до достижения рН, приблизительно составляющего 7. Затем суспензию охлаждали до 15°C в течение ночи. На следующие сутки суспензию отфильтровывали, и осадок на фильтре промывали метанолом и сушили в вакуумном шкафу. Высушенный продукт анализировали с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ), уровень загрязняющего Соединения 1 составил 0,09%.
Способ согласно изобретению
Гидроксид натрия (50%, 145 мл) добавляли в воду (250 мл). Температура разбавленного раствора гидроксида натрия перед добавлением раствора Соединения В2 составляла 40°C. Продолжительность добавления раствора избыточно ацилированного Соединения В составляла приблизительно 30 минуты, и во время добавления температура повысилась приблизительно до 55°C. Затем раствор разбавляли небольшим количеством воды до общего объема, составляющего 850 мл. Затем добавляли соляную кислоту (17,5%) до незначительного помутнения раствора, и в раствор помещали затравочный кристалл Соединения А (0,9 г). Суспензию перемешивали в течение 45 минут, а затем добавляли соляную кислоту (17,5%) до достижения рН, приблизительно составляющего 7. Затем суспензию охлаждали до 15°C в течение ночи. На следующий день суспензию отфильтровывали, и осадок на фильтре промывали метанолом и сушили в вакуумном шкафу. Высушенный продукт анализировали с помощью ВЭЖХ, уровень загрязняющего Соединения 1 составил лишь 0,02%.
Пример 2
Сравнение количества образующегося в растворе загрязняющего
Соединения 1
Аналогично процедуре, описанной в Примере 1, были проведены две реакции ацетилирования. В процедуру Примера 1 были внесены некоторые изменения с целью получения более высокого содержания предшественников эпоксидного Соединения 2.
Сравнительный способ согласно предшествующему уровню техники
Гидроксид натрия (50%, 200 мл) добавляли в раствор, содержащий избыточно ацилированное Соединение В (раствор Соединения В2). Затем раствор разбавляли водой до достижения общего объема, составляющего 870-880 мл. Анализ раствора способом ВЭЖХ, проведенный перед кристаллизацией, показал, что уровень Соединения 1 составил 5,8%.
Способ согласно изобретению
Гидроксид натрия (50%, 170 мл) добавляли в воду (280 мл). Затем в разбавленный раствор гидроксида натрия добавляли раствор Соединения В2. Затем полученный раствор разбавляли водой до достижения общего объема, составляющего 870-880 мл. Анализ раствора способом ВЭЖХ, проведенный перед кристаллизацией, показал, что уровень Соединения 1 составил 0,3%, что показывает, что при применении способа согласно изобретению образуется значительно меньшее количество Соединения 1, чем в способе согласно предшествующему уровню техники, что позволяет повысить общий выход.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОЧИСТКА РЕНТГЕНОКОНТРАСТНЫХ ВЕЩЕСТВ | 2013 |
|
RU2662941C2 |
| Получение промежуточного соединения синтеза иоформинола | 2013 |
|
RU2654461C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОНТРАСТНОГО АГЕНТА ЙОМЕПРОЛА | 2019 |
|
RU2795091C2 |
| КОНТРАСТНЫЕ АГЕНТЫ | 2008 |
|
RU2469021C2 |
| Получение иоформинола - рентгеноконтрастного агента | 2013 |
|
RU2642436C2 |
| Способ получения фармацевтической субстанции на основе йогексола | 2017 |
|
RU2655619C1 |
| ДИАГНОСТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ КАТИОНЫ ПЛАЗМЫ КРОВИ, ОБЛАДАЮЩАЯ ПРЕВОСХОДНЫМ ПРОФИЛЕМ БЕЗОПАСНОСТИ | 2010 |
|
RU2544113C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИОГЕКСОЛА | 1997 |
|
RU2173315C2 |
| КОМПОЗИЦИИ, КОРРИГИРУЮЩИЕ ВКУС КОНТРАСТНЫХ СРЕДСТВ | 2014 |
|
RU2683568C1 |
| ПРЕПАРАТЫ И СПОСОБ ДЛЯ РЕНТГЕНОКОНТРАСТНОГО ИССЛЕДОВАНИЯ ЛИМФАТИЧЕСКОЙ СИСТЕМЫ | 2000 |
|
RU2171690C1 |
Изобретение относится к способу получения йодированных рентгеноконтрастных веществ и, в частности, к получению ключевых промежуточных соединений, используемых для получения йодированных рентгеноконтрастных веществ. Способ получения N-ацилированного мономерного соединения, выбранного из 5-ацетамидо-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодоизофталамида (Соединения А) и N,N'-бис(2,3-дигидроксипропил)-5-формамидо-2,4,6-трийодоизофталамида (Соединения С), включает стадию деацилирования ацилированных гидроксильных групп путем добавления Соединения В2 к водному раствору деацилирующего агента, который представляет собой неорганическое основание. При этом у соединения В2 каждый из X независимо обозначает водород или ацильную группу, при условии, что по меньшей мере одна из групп X, присоединенных к атому азота, представляет собой ацильную группу. Способ позволяет получить оптимальный выход и снизить образование загрязняющих веществ. 5 з.п. ф-лы, 2 пр.
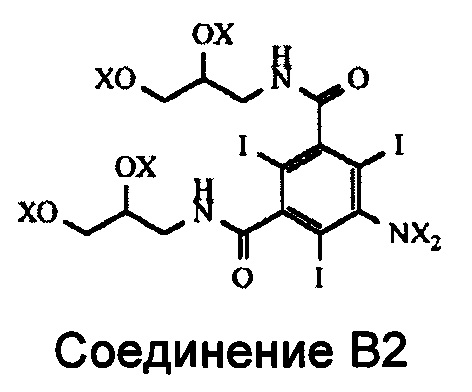
1. Способ получения N-ацилированного мономерного соединения, выбранного из 5-ацетамидо-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодоизофталамида (Соединения А) и N,N'-бис(2,3-дигидроксипропил)-5-формамидо-2,4,6-трийодоизофталамида (Соединения С), включающий стадию, на которой Соединение В2,
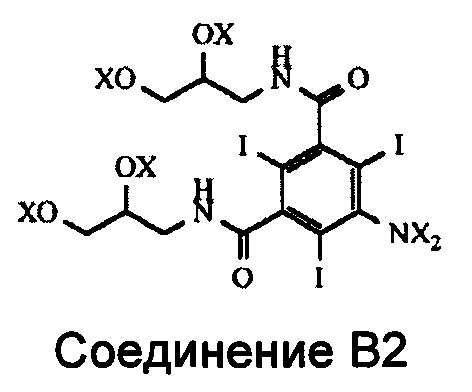
в котором каждый из X независимо обозначает водород или ацильную группу, при условии, что по меньшей мере одна из групп X, присоединенных к атому азота, представляет собой ацильную группу;
подвергают деацилированию ацилированных гидроксильных групп путем добавления Соединения В2 к водному раствору деацилирующего агента, где деацилирующий агент представляет собой неорганическое основание.
2. Способ по п. 1, в котором Соединение В2 добавляют к деацилирующему агенту в виде раствора.
3. Способ по п. 1, в котором Соединение В2 представляет собой Соединение В3
4. Способ по п. 1, в котором рН смешанного раствора Соединения В2 и деацилирующего агента снижают со значения, приблизительно равного 14, до значения, составляющего от 11 до 13, во время добавления к деацилирующему агенту.
5. Способ по п. 1, в котором Соединение В2 добавляют в водный раствор деацилирующего агента в течение времени, составляющего от 0,5 до 2 часов, либо в несколько небольших порций, либо непрерывно.
6. Способ по п. 1, включающий:
первую стадию ацилирования Соединения В,
в результате которой получают Соединение В2;
с последующим проведением стадии, на которой раствор Соединения В2 подвергают деацилированию ацилированных гидроксильных групп путем добавления Соединения В2 к водному раствору деацилирующего агента, где деацилирующий агент представляет собой неорганическое основание.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| ИОДИРОВАННЫЕ АРИЛЬНЫЕ СОЕДИНЕНИЯ И ДИАГНОСТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2145955C1 |
| ПОЛУЧЕНИЕ ИОДИКСАНОЛА | 2005 |
|
RU2385316C2 |

