Изобретение относится к способам оценки качества, для идентификации и количественного определения таурина и аллантоина при совместном присутствии в различных лекарственных препаратах, биологически активных добавках, косметической и пищевой продукции.
Известен способ количественного определения таурина в продукции, выпускаемой фармацевтическими предприятиями и аптеками (RU 2167410, G01N 21/78, 20.05.2001 г.), где определение проводят путем обработки исследуемого образца спиртовым раствором нингидрина в среде фосфатного буферного раствора с рН от 6,4 до 7,6 в присутствии аскорбиновой кислоты. Полученный окрашенный раствор разбавляют водой и измеряют оптическую плотность на спектрофотометре.
Известен способ количественного определения субстанции таурина с помощью формолового титрования (ФС.2.1.0039.15, ГФ XIII), включающий растворение субстанции в воде, прибавление формалина и титрование раствором натрия гидроксида до появления слаборозового окрашивания (индикатор-фенолфталеин) или определение точки эквивалентности потенциометрически. Параллельно проводят контрольный опыт.
Известен способ количественного определения субстанции аллантоина с фенилгидразином спектрофотометрическим методом (Chen, Х.В. and M.J. Gomes, 1992. Estimation of microbial protein supply to sheep and cattle based on urinary excretion of purine derivatives An overview of the technical details, International Feed Resources Unit, Rowett Research Institute, Bucksburn Aberdeen AB2 9SB, UK Occasional Publication).
Известен способ количественного определения аллантоина в биологических, косметических и фармацевтических образцах спектрофотометричеким методом: готовят раствор аллантоина с использованием гидроксида натрия и измеряют спектр полученного раствора (Determination of allantoin in biological, cosmetic, and pharmaceutical samples. JAOAC Int. 1996 May-Jun; 79(3):628-35).
Все вышеперечисленные методы с указанными пробоподготовками, не предоставляют возможности количественного определения таурина и аллантоина при совместном присутствии в лекарственных препаратах, биологически активных добавках и косметических средствах. При проведении патентного поиска и анализа литературных источников не выявлено разработанной методики количественного определения таурина и аллантоина при совместном присутствии.
Задача изобретения - разработка методики определения подлинности и количественного содержания таурина и аллантоина при совместном присутствии в различных лекарственных препаратах, биологически активных добавках, косметической и пищевой продукции.
Технический результат заключается в разработке способа на основе метода ВЭЖХ для количественного определения таурина и аллантоина при совместном присутствии таурина и аллантоина в различных лекарственных формах или смесях.
Технический результат достигается тем, что способ количественного определения таурина и аллантоина при совместном присутствии методом ВЭЖХвключает растворение навески исследуемого вещества в подвижной фазе (ПФ), разделение раствора на хроматографической колонке, измерение оптической плотности полученного раствора и определение концентрации исследуемых веществ по калибровочным графикам, причем в качестве подвижной фазы для таурина использовали 30 мкмоль/л раствора ацетата натрия рН 6.0, а растворение проводили из расчета 250 мг исследуемого вещества на 10 мл и доведение до объема раствором ПФ; в качестве подвижной фазы для аллантоина использовали смесь раствора гидрофосфат аммония с рН 7.78 и ацетонитрила 9:1, а растворение проводили из расчета 1000 мг исследуемого вещества на 10 мл и доведение до объема раствором ПФ; для таурина перед разделением раствора с ПФ на хроматографической колонке к нему добавляли боратный буферный раствор с рН 9.0 и раствор 0,1% 2,4-динитрохлорбензола в растворе ацетонитрил - вода (2:1) при соотношении 1:1:1, нагревали полученную смесь, охлаждали до комнатной температуры и на 6 об. ч. полученной смеси добавляли 1 об. ч. 10% раствора уксусной кислоты и 13 об. ч. воды; детектирование проводили при длинах волн 360±2 нм и 218±2 нм для таурина и аллантоина соответственно.
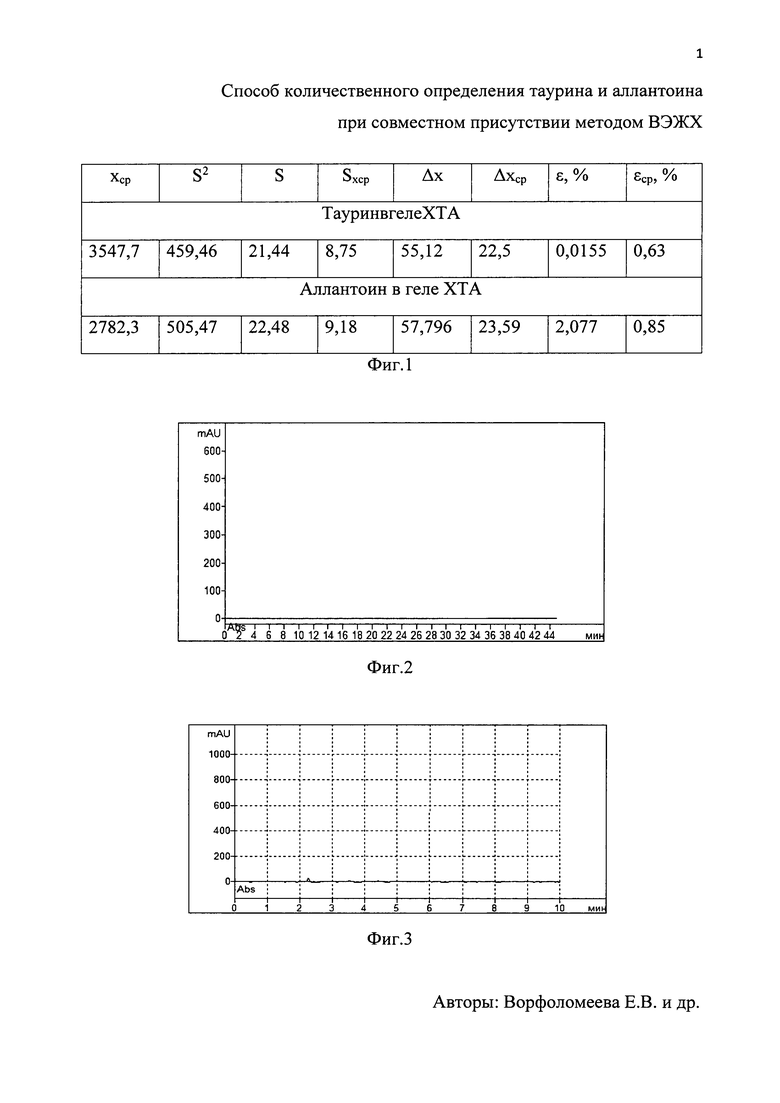
На фиг. 1 приведена метрологическая оценка результатов количественного определения таурина и аллантоина при совместном присутствии в геле на основе хитозана с таурином и аллантоином. На фиг. 2 и фиг. 3 представлены хроматограммы растворителей образца геля ХТА для определения таурина и аллантоина соответственно.
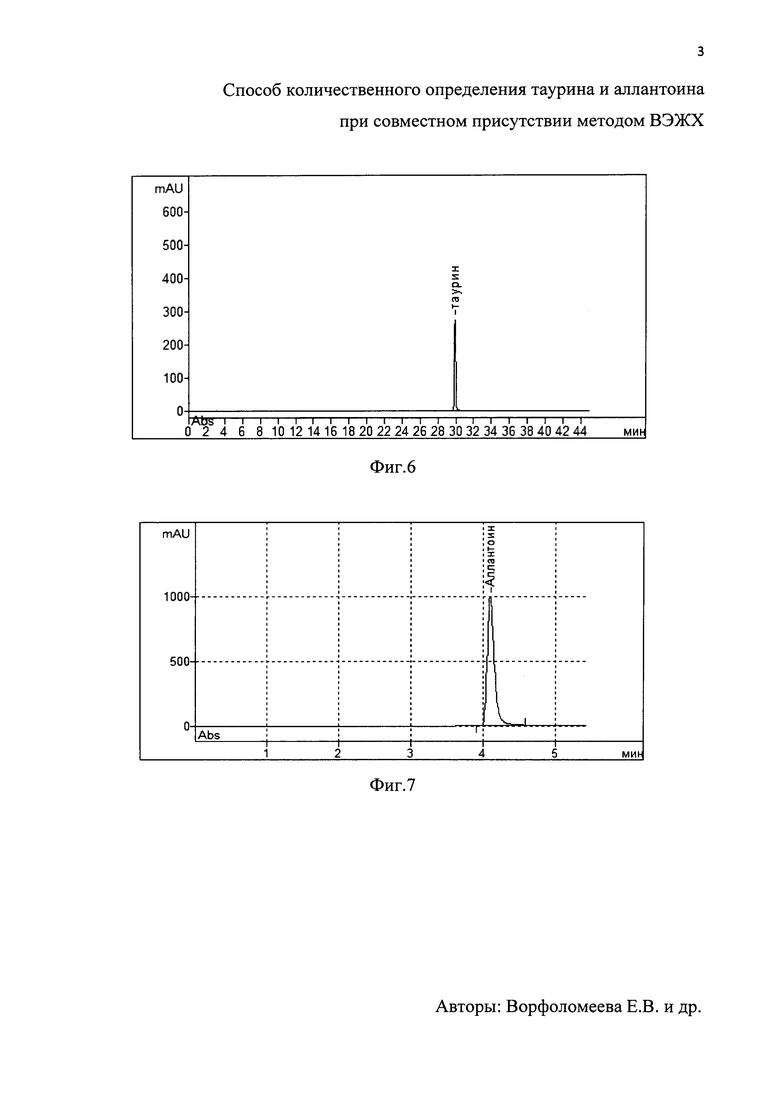
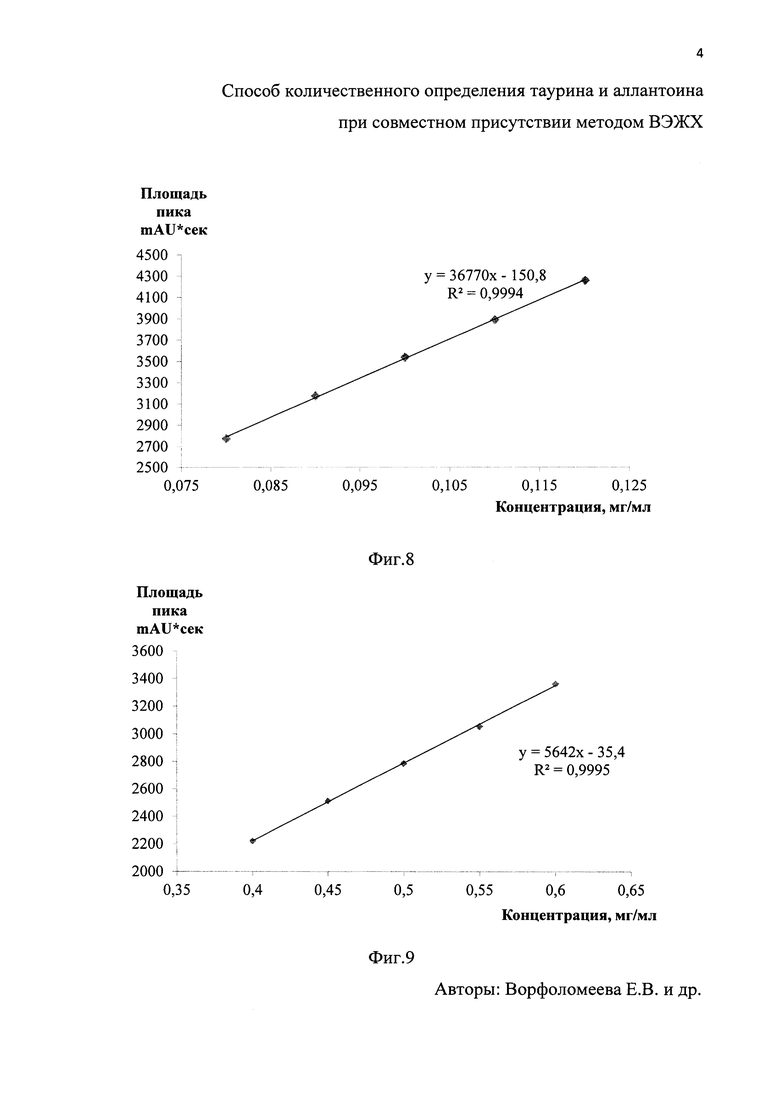
На фиг. 4 - хроматограммараствора стандартного образца таурина и на фиг. 5 - хроматограмма раствора стандартного образца аллантоина. На фиг. 6 - хроматограмма испытуемого раствора таурина в растворе геля ХТА и на фиг. 7 - хроматограмма испытуемого раствора аллантоина в растворе геля ХТА. На фиг. 8 и фиг. 9 приведены калибровочные графики таурина и аллантоина соответственно
Использованные в способе растворы получали следующим образом.
Подвижная фаза для количественного определения таурина. В мерную колбу на 1000 мл помещали 2,46 г СН3СООNа⋅3Н2О, приливали около 900,0 мл воды для хроматографии и полностью растворяли соли. Устанавливали значение рН 6,0 потенциометрически при добавлении 5% триэтиламина или 10% ледяной уксусной кислоты, доводили водой для хроматографии до метки.
Боратный буферный раствор с рН 9.0 0,62 г тетрабората натрия помещали в мерную колбу на 100 мл, доводили значение рН 9.0, при помощи 1М раствора NaOH.
Раствор 0,1% 2,4-динитрохлорбензола. Растворяли 10 мг 2,4-динитрохлорбензолапомещали в 10 мл раствора ацетонитрил - вода (2:1).
Подвижная фаза для количественного определения аллантоина. В мерную колбу на 1000 мл помещали 6,61 г (NH4)2HPO4, приливали 300,0 мл воды для хроматографии и полностью растворяли соль, доводили объем в колбе до метки водой для хроматографии и еще раз тщательно перемешивали, рН 7,78 устанавливали потенциометрически. В мерную колбу вместимостью 1000 мл помещали 900 мл полученного раствора, прибавляли 100 мл ацетонитрила и тщательно перемешивали. Полученную подвижную фазу фильтровали через фильтр с диаметром пор 0,45 мкм и дегазировали.
Пример.
Анализ геля на основе хитозана с таурином и аллантоином.
Пробоподготовка 1.
Навеску геля ХТА массой 250 мг помещали в мерную колбу на 10 мл, растворяли навеску в ПФА. 100 мкл полученного раствора помещали в эппендорф вместимостью 1,5 мл. Добавляли 100 мкл боратного буферного раствора с рН 9.0 и 100 мкл 0,1% раствора 2,4-динитрохлорбензола, закрывали эппендорф и помещали в водяную баню с температурой 90°С на 90 мин, охлаждали до комнатной температуры, добавляли 50 мкл 10% раствора уксусной кислоты и доводили до 1 мл водой для хроматографии. Фильтровали через фильтр с размером пор 0,45 мкм, полученные пробы перемешивали и вводили в хроматограф. Использовали колонку LunaC18 размером 4,6×250 мм, заполненную сферическими частицами силикагеля размером 5 мкм. Детекцию осуществляли спектрофотометрически при длине волны 360 нм. На хроматограммах идентифицировали пики таурина и определяли их площади. Время удерживания составило 30 мин. Количественное определение таурина в геле ХТА проводили методом калибровочного графика. Содержание таурина составляло 100,33±0,82 мкг/мл.
Для подтверждения точности метода был получен стандартный раствор с точно известной концентрацией таурина. 10,0 мг стандартного образца таурина помещали в мерную колбу на 10 мл, растворяли навеску в ПФА. 100 мкл полученного раствора помещали в эппендорф вместимостью 1,5 мл и добавляли 100 мкл боратного буферного раствора с рН 9.0 и 100 мкл 0,1% раствора 2,4-динитрохлорбензола. Закрывали эппендорф и помещали в водяную баню с температурой 90°С на 90 мин. Охлаждали до комнатной температуры, добавляли 50 мкл 10% раствора уксусной кислоты и доводили раствор до 1 мл водой для хроматографии, с последующим фильтрованием через фильтр размером пор 0,45 мкм. Полученные пробы перемешивали и вводили в хроматограф. Использовали колонку LunaC18 размером 4,6×250 мм, заполненную сферическими частицами силикагеля размером 5 мкм
Статистическая обработка данных показала, что средняя ошибка определения количественного содержания таурина заявленным методом не превышала 0,63%.
Пробоподготовка 2.
1000 мг геля помещали в мерную колбу на 10 мл и растворяли в 5 мл раствора подвижной фазы, использовали ультразвуковую ванну при комнатной температуре в течение 5 минут, затем доводили объем до метки раствором подвижной фазы и еще раз тщательно перемешивали, с последующей фильтрацией через бумажный складчатый фильтр. Детекцию осуществляли УФ спектрофотометрически при длине волны 218 нм. На хроматограммах идентифицировали пики аллантоина и определяли их площади. Время удерживания равнялось 4 мин. Количественное определение аллантоина в геле ХТА проводили методом калибровочного графика. Содержание аллантоина составляло 0,5 мг/мл.
Для подтверждения точности метода был получен стандартный раствор с точно известной концентрацией аллантоина. В мерную колбу вместимостью 100 мл помещали 50,0±0,5 мг аллантоина, приливали 50 мл подвижной фазы, растворяли полностью и доводили объем подвижной фазой до метки. Использовали колонку LunaC18 размером 4,6×250 мм, заполненную сферическими частицами силикагеля размером 5 мкм. Концентрация аллантоина 0,500±0,0005 мг/мл. Статистическая обработка данных показала, что средняя ошибка определения количественного содержания аллантоина заявленным методом не превышала 0,85%.
Методика позволяет идентифицировать и количественно определять таурин и аллантоин при совместном присутствии в различных лекарственных формах или смесях, что может служить для стандартизации геля ХТА.
Изобретение относится к способу количественного определения методом ВЭЖХ таурина и аллантоина при их совместном присутствии в различных лекарственных препаратах, биологически активных добавках, косметической и пищевой продукции. Способ включает растворение навески исследуемого вещества в подвижной фазе (ПФ), разделение раствора на хроматографической колонке, измерение оптической плотности полученного раствора и определение концентрации исследуемых веществ по калибровочным графикам. В качестве подвижной фазы для таурина использовали 30 мкмоль/л раствора ацетата натрия рН 6.0, а растворение проводили из расчета 250 мг исследуемого вещества на 10 мл и доведение до объема раствором ПФ. В качестве подвижной фазы для аллантоина использовали смесь раствора гидрофосфат аммония с рН 7.78 и ацетонитрила 9:1, а растворение проводили из расчета 1000 мг исследуемого вещества на 10 мл и доведение до объема раствором ПФ. Для таурина перед разделением раствора с ПФ на хроматографической колонке к нему добавляли боратный буферный раствор с рН 9.0 и 0,1% раствор 2,4-динитрохлорбензола в растворе ацетонитрил - вода (2:1) при соотношении 1:1:1, нагревали полученную смесь, охлаждали до комнатной температуры и на 6 об. ч. полученной смеси добавляли 1 об. ч. 10% раствора уксусной кислоты и 13 об. ч. воды. Детектирование проводили при длинах волн 360±2 нм и 218±2 нм для таурина и аллантоина, соответственно. Способ позволяет идентифицировать и количественно определять таурин и аллантоин при их совместном присутствии в различных лекарственных препаратах. 9 ил.
Способ количественного определения таурина и аллантоина при совместном присутствии методом ВЭЖХ, включающий растворение навески исследуемого вещества в подвижной фазе, разделение раствора на хроматографической колонке, измерение оптической плотности полученного раствора и определение концентрации исследуемых веществ по калибровочным графикам, причем в качестве подвижной фазы для таурина использовали 30 мкмоль/л раствора ацетата натрия рН 6.0, а растворение проводили из расчета 250 мг исследуемого вещества на 10 мл и доведение до объема раствором подвижной фазы; в качестве подвижной фазы для аллантоина использовали смесь раствора гидрофосфат аммония с рН 7.78 и ацетонитрила 9:1, а растворение проводили из расчета 1000 мг исследуемого вещества на 10 мл и доведение до объема раствором подвижной фазы; для таурина перед разделением раствора с подвижной фазой на хроматографической колонке к нему добавляли боратный буферный раствор с рН 9.0 и 0,1% раствор 2,4-динитрохлорбензола в растворе ацетонитрил - вода (2:1) при соотношении 1:1:1, нагревали полученную смесь, охлаждали до комнатной температуры, и на 6 об. частей полученной смеси добавляли 1 об. часть 10% раствора уксусной кислоты и 13 об. частей воды; детектирование проводили при длинах волн 360±2 нм и 218±2 нм для таурина и аллантоина соответственно.
| Т.И | |||
| Ярыгина, Разработка методики количественного определения тауфона (таурина), Журнал Вестник Российского университета дружбы народов, Серия: Медицина, 2010, N4, стр.522-525 | |||
| Kovalenko Svetlana et al., Development and validation of HPLC metod for determining ALLANTOIN in the compound drug for external use to treat diabetic ulcers, Журнал Наука и Технология, Изд-во SCIEURO (Лондон), том 1, N3 с.7-13 | |||
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ АЛИФАТИЧЕСКИХ АМИНОКИСЛОТ | 1999 |
|
RU2167410C2 |
| Ранозаживляющий гель для наружного применения | 2015 |
|
RU2611400C2 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ПРОИЗВОДНЫХ ИМИДАЗОЛА (ГРУППЫ ИМИДАЗОЛИНА) | 2014 |
|
RU2597787C2 |

