Протеинкиназы являются одним из самых крупных семейств ферментов человека и регулируют множество различных сигнальных процессов посредством присоединения фосфатных групп к белкам, в частности тирозинкиназы фосфорилируют белки по спиртовой группе тирозина. Семейство тирозинкиназ включает в себя ферменты, которые контролируют клеточный рост, миграцию и дифференцировку клеток. Аномальная активность киназ вовлечена в различные заболевания человека, включая рак, аутоиммунные и воспалительные заболевания. Так, протеинкиназы являются одними из ключевых регуляторов клеточной сигнализации, они являются модуляторами клеточной функции вместе с низкомолекулярными ингибиторами киназной активности и, тем самым, представляют собой хорошие цели при разработке лекарственных препаратов. В дополнение к лечению опосредованных киназами процессов заболевания, селективные и эффективные ингибиторы киназ также полезны для изучения клеточных сигнальных процессов и выявления других клеточных мишеней, интересных с терапевтической точки зрения.
SYK (тирозинкиназа селезенки) представляет собой безрецепторную тирозинкиназу, которая важна для В-клеточной активации через BCR сигналинг. SYK активируется при связывании с фосфорилированным BCR и, таким образом, инициирует ранние сигналинговые события после BCR активации. Мыши, дефицитные по SYK проявляют раннюю блокировку развития В-клеток. Поэтому, в качестве средства для лечения аутоиммунных заболеваний предлагается ингибирование ферментативной активности SYK в клетках посредством его воздействия на продукцию аутоантител.
В дополнение к роли SYK в сигналинге BCR и активации В-клеток, она также играет ключевую роль в FcεRI-опосредованной дегрануляции тучных клеток и активации эозинофилов. Так, SYK вовлечена в аллергические заболевания, включая астму. SYK связывает фосфорилированную гамма-цепь FcεRI через ее SH2-домены и является существенной для нисходящего сигналинга. Дефицитные по SYK тучные клетки показали нарушенную дегрануляцию и секрецию арахидоновой кислоты и цитокинов. Это также было показано для фармацевтических агентов, ингибирующих активность SYK в тучных клетках. Обработка антисмысловыми олигонуклеотидами SYK ингибирует антиген-индуцированную инфильтрацию эозинофилами и нейтрофилами в модели астмы на животных. Эозинофилы, дефицитные по SYK также показали нарушенную активацию в ответ на стимуляцию FcεR. Таким образом, низкомолекулярные ингибиторы SYK будут полезными для лечения вызванных аллергией воспалительных заболеваний, включая астму.
Ввиду многочисленных состояний, для которых ожидается польза при лечении, включающем модуляцию сигнального пути SYK, сразу становится очевидным, что новые соединения, которые модулируют сигнальный путь SYK и способы применения этих соединений должны обеспечить существенные терапевтические улучшения для широкого спектра пациентов. В настоящем изобретении предложены новые соединения для применения при терапевтическом лечении аутоиммунных и воспалительных заболеваний посредством воздействия на сигнальный путь SYK или посредством ингибирования SYK киназы.
В настоящем изобретении предложено соединение Формулы I, II или III
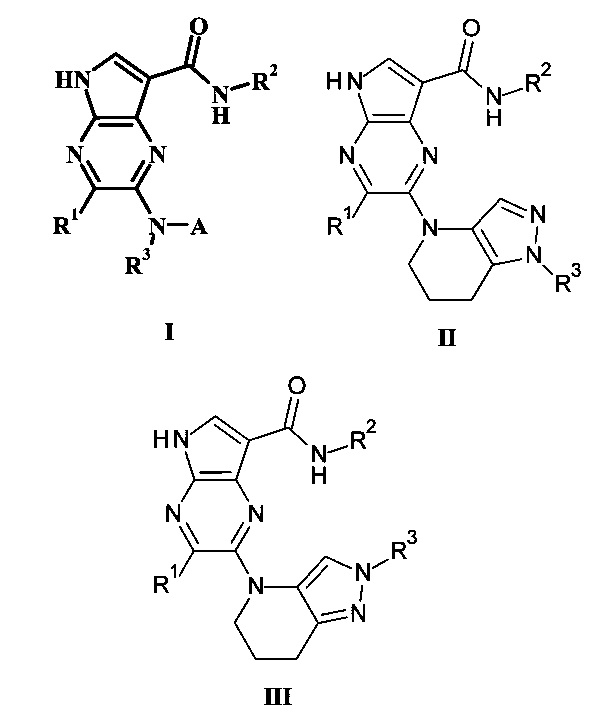
где:
R1 представляет собой Н, гало, или низший алкил;
R2 представляет собой низший алкил или низший гидроксиалкил;
R3 представляет собой Н или низший алкил;
А представляет собой моноциклический или бициклический гетероарил (предпочтительно пиридинил, пиримидил, пиразолил, изоксазолил, тиадиазолил, тиазолил или пиразинил) или фенил, возможно замещенный одним или более А'; и
каждый А' представляет собой независимо низший алкил, гало, низший алкилсульфонил, низший алкиламид, амидо, низший гидроксиалкил, низший алкокси, амино-низший алкил или дейтерий;
или его фармацевтически приемлемая соль.
В настоящем изобретении предложен способ лечения воспалительных или аутоиммунных заболеваний, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложена фармацевтическая композиция, содержащая соединение Формулы I, в смеси с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
Определения
Указанное в настоящем изобретении единственное число относится также и к множественному числу, например, соединение обозначает одно или более соединений или по меньшей мере одно соединение. Таким образом, единственное число, «один или более» и «по меньшей мере один» могут использоваться в настоящем изобретении взаимозаменяемо.
Как используется в настоящем изобретении, в переходной фразе или в пункте формулы изобретения, термин «содержит» или «содержащий» подразумевает неограничивающее значение. Другими словами, указанный термин является синонимом фраз «имеющий по меньшей мере» или «включающий по меньшей мере». Когда термин «содержащий» используется в контексте способа, он означает, что способ включает по меньшей мере перечисленные стадии, но может также включать дополнительные стадии. Когда термин «содержащий» используется в контексте соединения или композиции, он означает, что соединение или композиция включают по меньшей мере перечисленные признаки или компоненты, но также могут содержать дополнительные признаки или компоненты.
Как используется в настоящем изобретении, если специально не указано другого, слово «или» используется во «включительном» смысле «и/или», а не в альтернативном смысле «тот или иной».
Термин «независимо» используется в настоящем изобретении для обозначения, что варианты применимы к любому отдельному случаю безотносительно к присутствию или отсутствию переменной с таким же значением или разных определений внутри одного и того же соединения. Так, соединение, у которого R" показано дважды и определено как «независимо углерод или азот», оба R" могут быть углеродами, оба R" могут быть азотами, или один R" может быть углеродом, а другой - азотом.
Когда любые переменные встречаются более одного раза в любом остатке или формуле, обозначающим или описывающим применяемые или охватываемые настоящим изобретением соединения, их определения в каждом случае являются независимыми от определений в любых других случаях. Также, комбинации заместителей и/или переменных являются возможными только в случае, если такие соединения являются стабильными.
Символы "*" на конце связи или "-----" прорисованные через связь, каждый обозначает точку присоединения функциональной группы или другого химического остатка к остову молекулы, частью которой он является. Так, например:
Связь, изображенная внутри кольцевой системы (в отличие от присоединенных к отдельным вершинам) обозначает, что связь может быть присоединена к любому подходящему кольцевому атому.
Термин "необязательный" или "необязательно" используемый в настоящем изобретении означает, что далее описанные события или обстоятельства могут, но не обязательно, иметь место, при этом описание включает примеры, где события или обстоятельства имеют место, и примеры, где не имеют. Например, «необязательно замещенный» означает, что необязательно замещенный остаток может включать атом водорода или заместитель.
Фраза «необязательная связь» означает, что связь может присутствовать или не присутствовать, и что описание включает простую, двойную или тройную связь. Если заместитель определен как «связь» или «отсутствует», атомы, связанные с данным заместителем тогда являются связанными напрямую.
Термин «около» используется в настоящем изобретении для обозначения приблизительно, в области, примерно или вблизи. Когда термин «около» используется вместе с числовым диапазоном, он изменяет этот диапазон расширением верхней и нижней границы указанных числовых значений. В общем, термин «около» используется в настоящем изобретении для изменения числового значения выше и ниже указанного значения на отклонение в 20%.
Некоторые соединения могут проявлять таутомеризм. Таутомерные соединения могут существовать в виде двух или более взаимопереходящих видов. Прототропные таутомеры образуются в результате миграции ковалентно связанного атома водорода между двумя атомами. Таутомеры обычно существуют в равновесии, и попытки изолировать отдельные таутомеры обычно приводят к получению смеси, химические и физические свойства которой соответствуют смеси соединений. Положение равновесия зависит от химических свойств внутри молекулы. Например, у многих алифатических альдегидов и кетонов, таких как ацетальдегид, преобладает кето-форма, в то время как у фенолов преобладает енольная форма. Часто встречающиеся прототропные таутомеры включают кето/енол (-С(=O)-СН-↔-С(-ОН)=СН-), амид/имидовая кислота (-C(=O)-NH-↔-С(-OH)=N-) и амидиновые (-C(=NR)-NH-↔-C(-NHR)=N-) таутомеры. Последние два особенно распространены в гетероарильных и гетероциклических кольцах, и настоящее изобретение охватывает все таутомерные формы заявленных соединений.
Технические и научные термины, используемые в настоящем изобретении, имеют значение, обычно понимаемое квалифицированным специалистом в данной области техники, к которой относится настоящее изобретение, если не указано иного. Различные способы и материалы, на которые ссылается настоящее изобретение, известны специалистам в данной области техники. Стандартные справочные издания, в которых излагаются общие принципы фармакологии включают Goodman and Gilman's The Pharmacological Basis of Therapeutics, 10th Ed., McGraw Hill Companies Inc., New York (2001). Любые подходящие материалы и/или способы, известные квалифицированным специалистам могут быть использованы при осуществлении настоящего изобретения. Однако, предпочтительные материалы и способы описаны. Материалы, реагенты и т.п., на которые ссылается следующее описание и примеры могут быть получены из коммерческих источников, если не указано иного.
Определения, описанные в настоящем изобретении, могут соединяться с образованием химически релевантных комбинаций, таких как "гетероалкиларил", "галоалкилгетероарил", "арилалкилгетероциклил", "алкилкарбонил", "алкоксиалкил" и т.п. Когда термин "алкил" используется в качестве суффикса после очередного термина, как в "фенилалкиле" или "гидроксиалкиле", это предназначено для обозначения алкильной группы, как определено выше, замещенной одним или двумя заместителями, выбранными из другой специально обозначенной группы. Так, например, "фенилалкил" относится к алкильной группе, содержащей от одного до двух фенильных заместителей, и, таким образом, включает бензил, фенилэтил и бифенил. "Алкиламиноалкил" представляет собой алкильную группу, имеющую от одного до двух алкиламино заместителей. "Гидроксиалкил" включает в себя 2-гидроксиэтил, 2-гидроксипропил, 1-(гидроксиметил)-2-метилпропил, 2-гидроксибутил, 2,3-дигидроксибутил, 2-(гидроксиметил), 3-гидроксипропил и т.д. Соответственно, используемый в настоящем изобретении термин «гидроксиалкил» используется для обозначения подгруппы гетероалкильных групп, определенных ниже. Термин -(ар)алкил относится как к незамещенным алкильным или аралкильным группам. Термин (гетеро)алкил или (гет)арил относится как к арильным, так и к гетероарильным группам.
Термин "спироциклоалкил", как используется в настоящем изобретении, означает спироциклическую циклоалкильную группу, таку как, например, спиро[3.3]гептан. Термин спирогетероциклоалкил, как используется в настоящем изобретении, означает спироциклический гетероциклоалкил, такой как, например, 2,6-диазаспиро[3.3]гептан.
Термин "ацил", используемый в настоящем изобретении, означает группу формулы -C(=O)R, где R представляет собой водород или низший алкил как определено в настоящем изобретении. Термин "алкилкарбонил", используемый в настоящем изобретении, означает группу формулы C(=O)R, где R представляет собой алкил, как определено в настоящем изобретении. Термин С1-6 ацил относится к группе -C(=O)R, содержащей 6 атомов углерода. Термин "арилкарбонил", используемый в настоящем изобретении, означает группу формулы C(=O)R, где R представляет собой арильную группу; термин "бензоил", используемый в настоящем изобретении, означает арилкарбонильную группу, где R представляет собой фенил.
Термин «эфир», как используется в настоящем изобретении, обозначает группу формулы -C(=O)OR, где R представляет собой низший алкил, как определено в настоящем изобретении.
Термин "алкил", используемый в настоящем изобретении, означает насыщенный, моновалентный, углеводородный остаток с неразветвленной или разветвленной цепочкой, содержащий от 1 до 10 атомов углерода. Термин "низший алкил" означает углеводородный остаток с неразветвленной или разветвленной цепочкой, содержащий от 1 до 6 атомов углерода. "С1-10 алкил", используемый в настоящем изобретении, относится к алкилу, состоящему из 1-10 углеродных атомов. Примеры алкильных групп включают, без ограничения, низшие алкильные группы, включая метил, этил, пропил, i-пропил, n-бутил, изобутил, трет-бутил или пентил, изопентил, неопентил, гексил, гептил и октил.
Когда термин "алкил" используется в качестве суффикса со следующим далее термином, как, например, в терминах "фенилалкил" или "гидроксиалкил", он предназначен для обозначения алкильной группы, как определено выше, замещенной от одного до двух заместителей, выбранных из другой специфично названной группы. Так, например, "фенилалкил" обозначает радикал R'R"-, где R' представляет собой фенильный радикал, a R" представляет собой алкиленовый радикал, как определено в настоящем изобретении, с пониманием того, что точка присоединения фенилалкильного остатка будет на алкиленовом радикале. Примеры арилалкильных радикалов включают, без ограничения, бензил, фенилэтил, 3-фенилпропил. Термины "арилалкил" или "аралкил" интерпретируются одинаково, исключая R' представляет собой арильный радикал. Термины "(гет)арилалкил" или "(гет)аралкил" интерпретируются одинаково, исключая R' представляет собой арильный или гетероарильный радикал.
Термин «галоалкил» или «гало-низший алкил» или «низший алкил» относится к углеводородному остатку с прямой или разветвленной цепочкой, содержащему 1-6 атомов углерода, где один или более атомов замещены одним или более атомами галогена.
Термин "алкилен" или "алкиленил", используемый в настоящем изобретении, означает бивалентный насыщенный линейный углеводородный радикал из от 1 до 10 атомов углерода (например, (СН2)n) или разветвленный насыщенный бивалентный углеводородный радикал из 2-10 атомов углерода (например, -СНМе- или -СН2СН(i-Pr)СН2-), если не указано иного. За исключением случая с метиленом, открытые валентности алкиленовой группы не присоединены к одному атому. Примеры алкиленовых радикалов включают, без ограничения, метилен, этилен, пропилен, 2-метил-пропилен, 1,1-диметил-этилен, бутилен, 2-этилбутилен.
Термин "алкокси", используемый в настоящем изобретении, означает -О-алкил группу, где алкил является таким, как определено выше, такую как метокси, этокси, n-пропилокси, i-пропилокси, n-бутилокси, i-бутилокси, t-бутилокси, пентилокси, гексилокси, включая их изомеры. "Низший алкокси", используемый в настоящем изобретении, означает алкокси группу с "низшей алкильной" группой, как определено выше. "С1-10 алкокси", используемый в настоящем изобретении, относится к -О-алкилу, где алкил представляет собой С1-10.
Термин "галоалкокси" или "гало-низший алкокси" или "низший галоалкокси" относится к низшей алкоксигруппе, где один или более атомов углерода замещены одним или более атомами галогена.
Термин "гидроксиалкил", используемый в настоящем изобретении, означает алкильный радикал, как определено выше, где от одного до трех атомов водорода на разных атомах углерода заменен(ы) гидроксильной(ыми) группой(ами).
Термин "алкилсульфонил" и "арилсульфонил", как используется в настоящем изобретении, относится к группе формулы -S(=O)2R, где R представляет собой алкил или арил соответственно, и алкил и арил являются акими, как определено в настоящем изобретении. Термин "гетероалкилсульфонил", как используется в настоящем изобретении, относится к группе формулы -S(=O)2R, где R представляет собой "гетероалкил", как определено в настоящем изобретении.
Термин «низший алкиламид», используемый в настоящем изобретении, относится к группе формулы CONRaRb, где Ra и Rb независимо представляют собой Н и низший алкил, как определено в настоящем изобретении.
Термины «алкилсульфониламино» и "арилсульфониламино", как используется в настоящем изобретении, относится к группе формулы -NR'S(=O)2R, где R представляет собой алкил или арил соответственно, R1 представляет собой водород или С1-3 алкил, и алкил и арил являются такими, как определено в настоящем изобретении.
Термин "циклоалкил", используемый в настоящем изобретении, относится к насыщенному карбоциклическому кольцу, содержащему от 3 до 8 атомов углерода, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил. "С3-7 циклоалкил", используемый в настоящем изобретении, относится к циклоалкилу, содержащему 3-7 атомов углерода в карбоциклическом кольце.
Термин карбоксиалкил, как используется в настоящем изобретении, относится к алкильному остатку, в котором один атом водорода заменен карбоксилом, с понимание того, что точка присоединения гетероалкильного радикала находится на атоме углерода. Термин "карбокси" или "карбоксил" относится к остатку -CO2H.
Термины "гетероарил" или «гетероароматический», используемые в настоящем изобретении, означают моноциклический или бициклический радикал из 5-12 кольцевых атомов, имеющий по меньшей мере одно ароматическое или частично ненасыщенное кольцо, содержащее от четырех до восьми атомов на кольцо, включающих один или более гетероатомов N, О или S, остальные кольцевые атомы представляют собой углерод, принимая во внимание, что точка присоединения гетероарильного радикала будет находиться на ароматическом или частично ненасыщенном кольце. Как хорошо известно специалистам в области техники, гетероарильные кольца обладают меньшим ароматичеким характером, чем их полностью углеродные аналоги. Таким образом, для целей изобретения, гетероарильная группа должна обладать только некоторой степенью ароматического характера. Примеры гетероарильных остатков включают моноциклические ароматические гетероциклы с 5-6 кольцевыми атомами и 1-3 гетероатомами, включая, без ограничения, пиридинил, пиримидинил, пиразинил, оксазинил, пирролил, пиразолил, имидазолил, оксазолил, 4,5-дигидро-оксазолил, 5,6-дигидро-4Н-[1,3]оксазолил, изоксазол, тиазол, изотиазола, триазолин, тиадиазол и оксадиаксолин, которые могут быть возможно замещены одним или более, предпочтительно одним или двумя заместителями, выбранными из гидрокси, циано, алкил, алкокси, тио, низший галогеналкокси, алкилтио, гало, низший галогеналкил, алкилсульфинил, алкилсульфонил, галоген, амино, алкиламино, диалкиламино, аминоалкил, алкиламиноалкил и диалкиламиноалкил, нитро, алкоксикарбонил и карбамоил, алкилкарбамоил, диалкилкарбамоил, арилкарбамоил, алкилкарбониламино и арикарбониламино. Примеры бициклических остатков включают, без ограничения, хинолинил, изохинолинил, бензофурил, бензотиофенил, бензоксазол, бензизоксазол, бензотиазол, нафтиридинил, 5,6,7,8-тетрагидро-[1,6]нафтиридинил и бензизотиазол. Бициклические остатки могут быть возможно замещены в любом кольце, однако точки присоединения находятся на кольце, содержащем гетероатом.
Термины «гетероциклил», «гетероциклоалкил» или «гетероцикл», как используется в настоящем изобретении обозначет моновалентный насыщенный циклический радикал, состоящий из одного или более колец, предпочтительно одного или двух колец, включая спироциклические кольцевые системы, из 3-8 атомов на кольцо, включающих один или более кольцевых гетероатомов (выбранных из N,O или S(O)0-2), и которые возможно могут быть независимо замещены одним или более, предпочтительно одним или двумя заместителями, выбранными из гидрокси, оксо, циано, низший алкил, низший алкокси, низший галогеналкокси, алкилтио, галоген, низший галогеналкил, гидроксиалкил, нитро, алкоксикарбонил, амино, алкиламино, алкилсульфонил, арилсульфонил, алкиламиносульфонил, ариламиносульфонил, алкилсульфониламино, арилсульфониламино, алкиламинокарбонил, ариламинокарбонил, алкилкарбониламино, арилкарбониламино и их ионных форм, если не указано иное. Примеры гетероциклических радикалов включают, без ограничения, морфолинил, пиперазинил, пиперидинил, азетидинил, пирролидинил, гексагидроазепинил, оксетанил, тетрагидрофуранил, тетрагидротиофенил, оксазолидинил, тиазолидинил, изоксазолидинил, тетрагидропиранил, тиоморфолинил, хинуклидинил и имидазолинил и их ионные формы. Примерами могут быть также бициклические, такие как, например, 3,8-диазабицикло[3.2.1]октан, 2,5-диазабицикло[2.2.2]октан или октагидро-пиразино[2,1-с][1,4]оксазин.
Ингибиторы SYK
В настоящем изобретении предложено соединение Формулы I, II или III
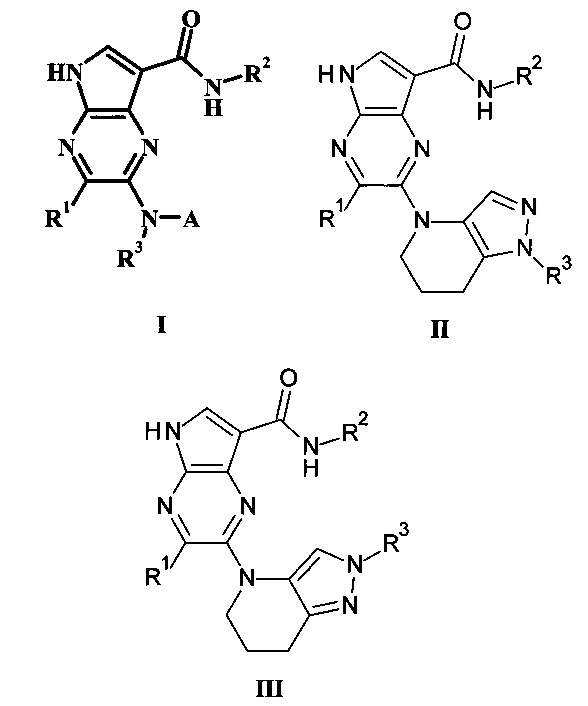
где:
R1 представляет собой Н, гало, или низший алкил;
R2 представляет собой низший алкил или низший гидроксиалкил;
R3 представляет собой Н или низший алкил;
А представляет собой моноциклический или бициклический гетероарил (предпочтительно пиридинил, пиримидил, пиразолил, изоксазолил, тиадиазолил, тиазолил или пиразинил) или фенил, возможно замещенный одним или более А'; и
каждый А' представляет собой независимо низший алкил, гало, низший алкилсульфонил, низший алкиламид, амидо, низший гидроксиалкил, низший алкокси, амино-низший алкил или дейтерий;
или его фармацевтически приемлемая соль.
В частности, в настоящем изобретении предложено соединение Формулы I
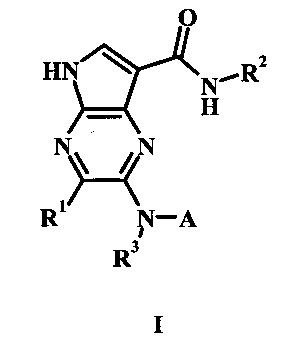
где:
R1 представляет собой Н, гало, или низший алкил;
R2 представляет собой низший алкил или низший гидроксиалкил;
R3 представляет собой Н или низший алкил;
А представляет собой моноциклический или бициклический гетероарил (предпочтительно пиридинил, пиримидил, пиразолил, изоксазолил, тиадиазолил, тиазолил или пиразинил) или фенил, возможно замещенный одним или более А'; и
каждый А' представляет собой независимо низший алкил, гало, низший алкилсульфонил, амидо, низший гидроксиалкил, низший алкокси, амино-низший алкил или дейтерий;
или его фармацевтически приемлемая соль.
В настоящем изобретении предложено соединение Формулы I, II или III, где R3 представляет собой низший алкил.
В настоящем изобретении предложено соединение Формулы I, II или III, где R3 представляет собой Н.
В настоящем изобретении предложено соединение Формулы I, II или III, где R2 представляет собой низший алкил.
В настоящем изобретении предложено соединение Формулы I, II или III, где R3 представляет собой Н, a R2 представляет собой низший алкил.
В настоящем изобретении предложено соединение Формулы I, II или III, где R1 представляет собой Н.
В настоящем изобретении предложено соединение Формулы I, II или III, где R1 представляет собой Н, a R3 представляет собой Н.
В настоящем изобретении предложено соединение Формулы I, II или III, где R1 представляет собой Н, a R2 представляет собой низший алкил.
В настоящем изобретении предложено соединение Формулы I, II или III, где R1 представляет собой Н, R3 представляет собой Н, a R2 представляет собой низший алкил.
В настоящем изобретении предложено соединение Формулы I, где А представляет собой моноциклический гетероарил, возможно замещенный одним или более А'.
В настоящем изобретении предложено соединение Формулы I, где А представляет собой моноциклический гетероарил, возможно замещенный одним или более A', R1 представляет собой Н, R3 представляет собой Н, a R2 представляет собой низший алкил.
В настоящем изобретении предложено соединение Формулы I, где А' представляет собой низший алкил.
В настоящем изобретении предложено соединение Формулы I, где А' представляет собой дейтерий.
В настоящем изобретении предложено соединение Формулы I, где А представляет собой моноциклический гетероарил, возможно замещенный одним или более A', R1 представляет собой Н, R3 представляет собой Н, R2 представляет собой низший алкил, а А' представляет собой низший алкил.
В настоящем изобретении предложено соединение Формулы I, где А' представляет собой низший алкилсульфонил.
В настоящем изобретении предложено соединение Формулы I, где А представляет собой моноциклический гетероарил, возможно замещенный одним или более A', R1 представляет собой Н, R3 представляет собой Н, R2 представляет собой низший алкил, а А' представляет собой низший алкилсульфонил.
В настоящем изобретении предложено соединение Формулы I, где А представляет собой фенил возможно замещенный одним или более А'.
В настоящем изобретении предложено соединение Формулы I, где А представляет собой фенил, возможно замещенный одним или более A', R1 представляет собой Н, R3 представляет собой Н, a R2 представляет собой низший алкил.
В настоящем изобретении предложено соединение Формулы I, где А' представляет собой низший алкил или низший алкилсульфонил.
В настоящем изобретении предложено соединение Формулы I, где А' представляет собой низший алкил или низший алкилсульфонил, А представляет собой фенил, возможно замещенный одним или более A', R1 представляет собой Н, R3 представляет собой Н, a R2 представляет собой низший алкил.
В настоящем изобретении предложено соединение Формулы I, где А представляет собой бициклический гетероарил, возможно замещенный одним или более А'.
В настоящем изобретении предложено соединение Формулы I, где А представляет собой бициклический гетероарил, возможно замещенный одним или более A', R1 представляет собой Н, R3 представляет собой Н, a R2 представляет собой низший алкил.
В предпочтительном воплощении, в настоящем изобретении предложено соединение Формулы I, где R1 представляет собой Н; R2 представляет собой низший алкил или низший гидроксиалкил; R3 представляет собой Н; А представляет собой пиридинил, пиримидил, пиразолил, изоксазолил, тиадиазолил, тиазолил, пиразинил или фенил, возможно замещенный одним или более А'; и
каждый А' представляет собой независимо низший алкил, гало, низший алкилсульфонил, низший гидроксиалкил, низший алкокси, амино-низший алкил или дейтерий;
или его фармацевтически приемлемая соль.
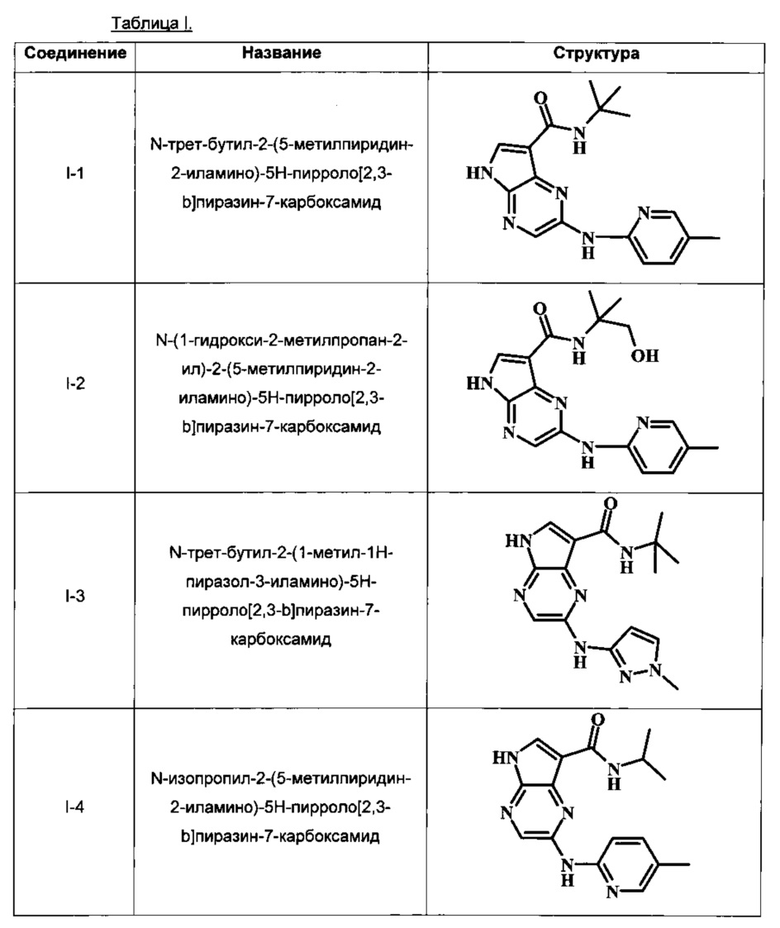
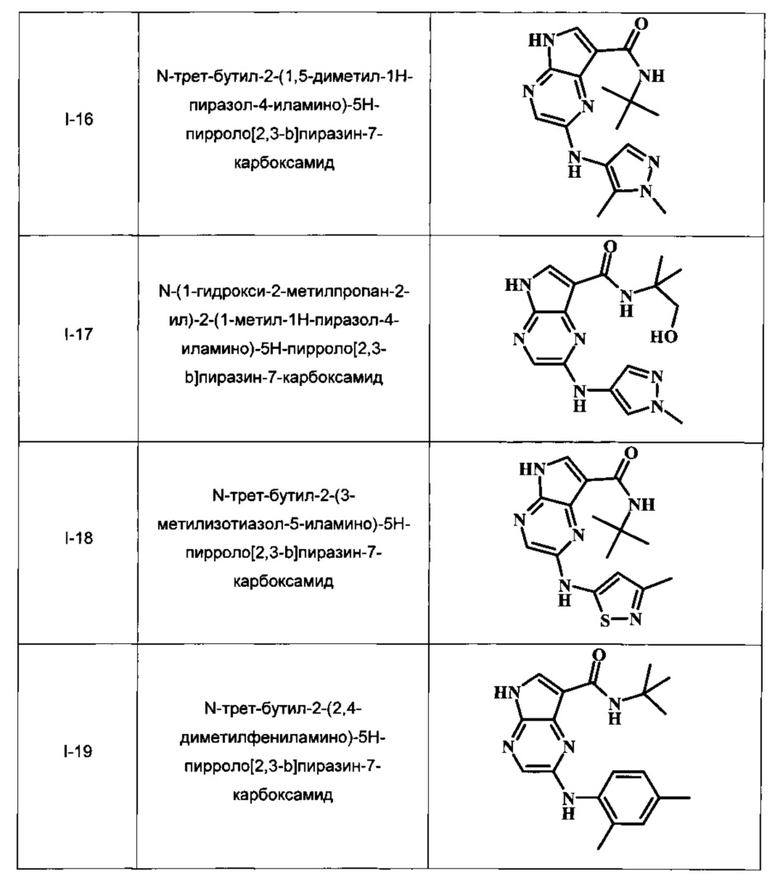
В настоящем изобретении предложено соединение, выбранное из группы, состоящей из:
N-трет-бутил-2-(5-метилпиридин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-(1-гидрокси-2-метилпропан-2-ил)-2-(5-метилпиридин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(1-метил-1Н-пиразол-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-изопропил-2-(5-метилпиридин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
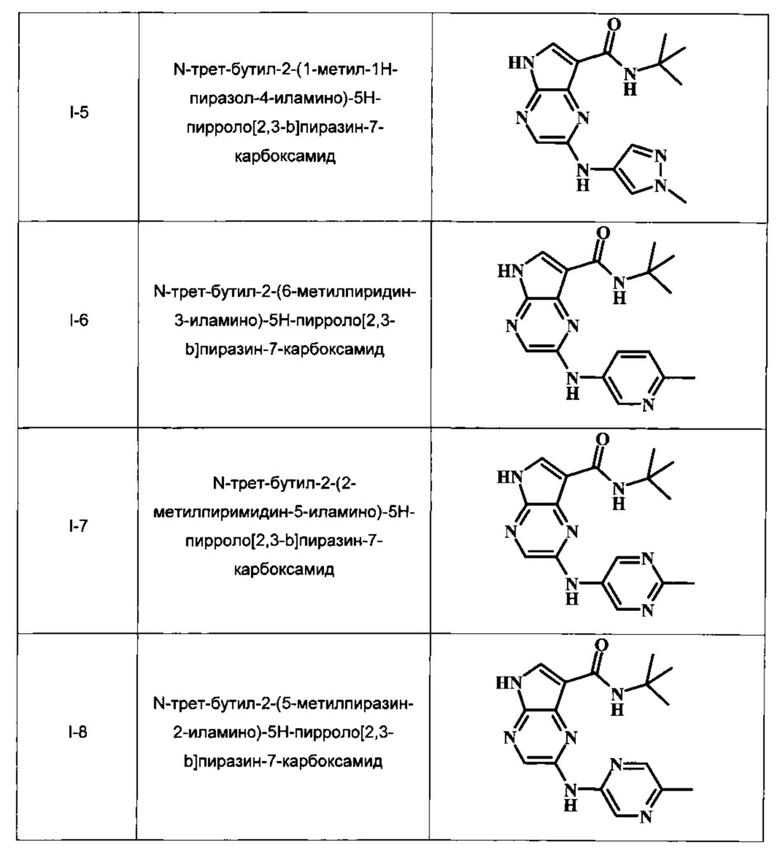
N-трет-бутил-2-(1-метил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(6-метилпиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(2-метилпиримидин-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(5-метилпиразин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
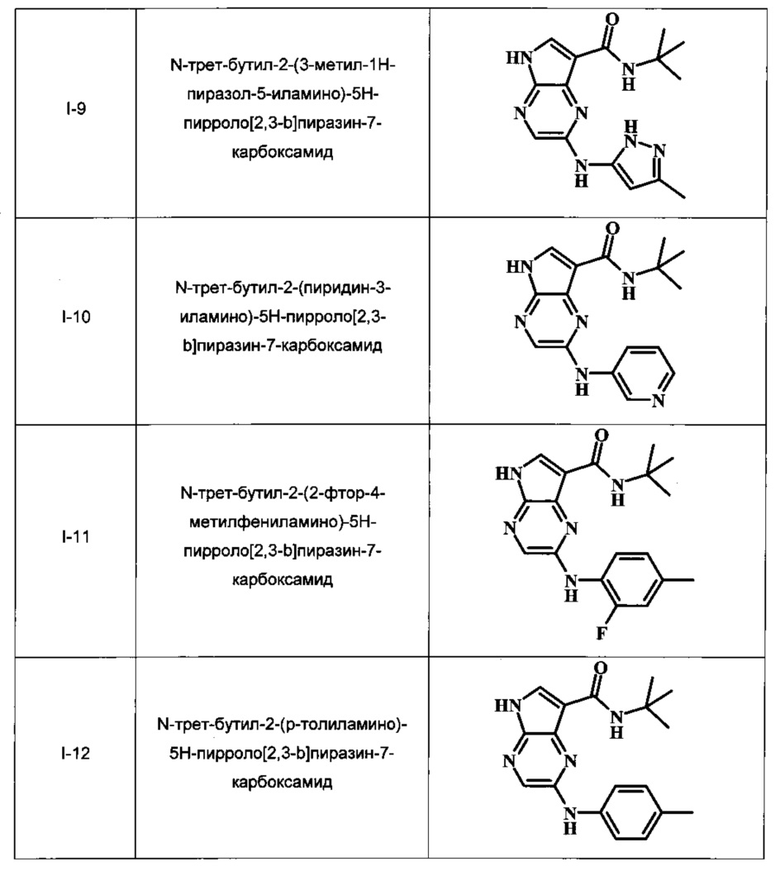
N-трет-бутил-2-(3-метил-1Н-пиразол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(2-фтор-4-метилфениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(р-толиламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
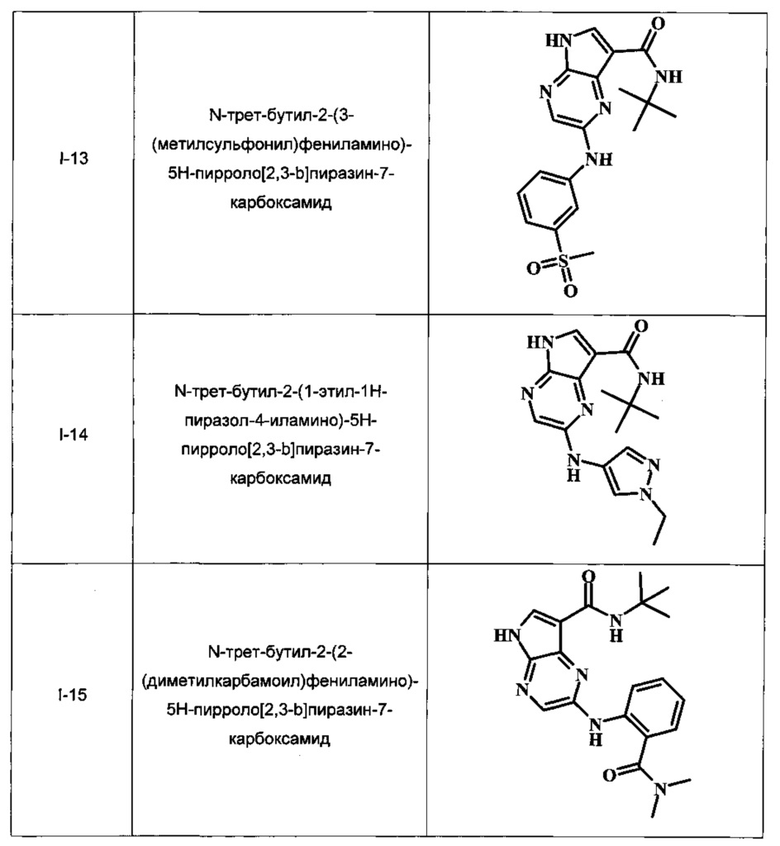
N-трет-бутил-2-(3-(метилсульфонил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(1-этил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(2-(диметилкарбамоил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(1,5-диметил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-(1-гидрокси-2-метилпропан-2-ил)-2-(1-метил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(3-метилизотиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(2,4-диметилфениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
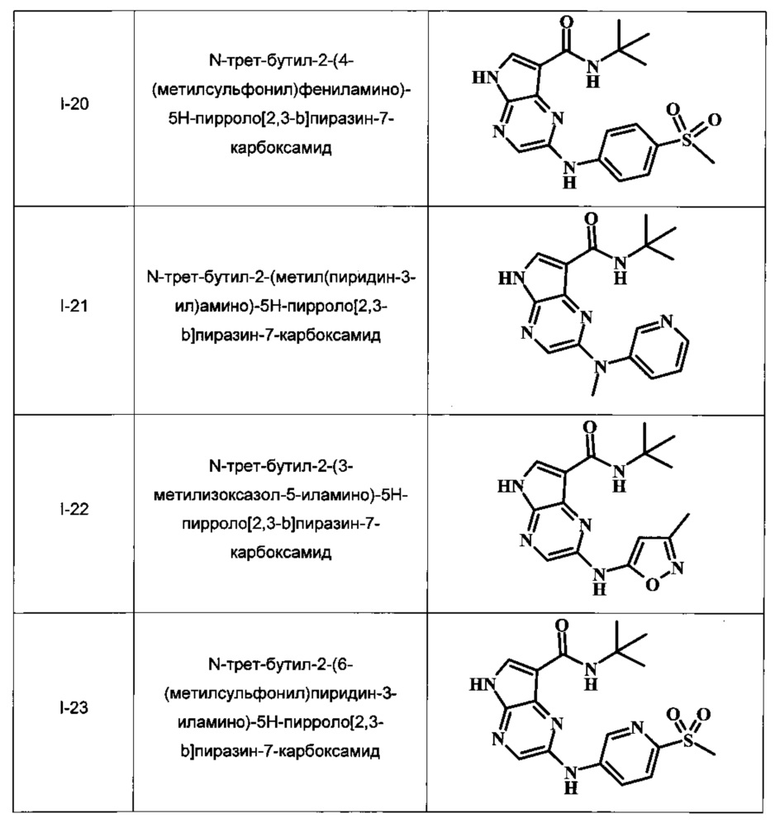
N-трет-бутил-2-(4-(метилсульфонил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(метил(пиридин-3-ил)амино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(3-метилизоксазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(6-(метилсульфонил)пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
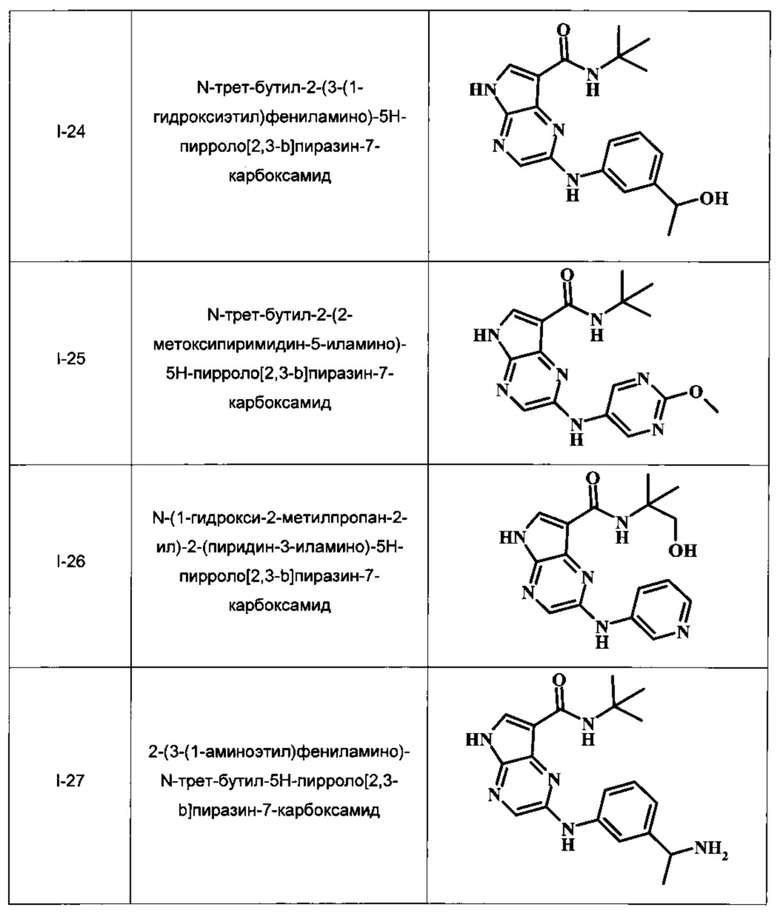
N-трет-бутил-2-(3-(1-гидроксиэтил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(2-метоксипиримидин-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-(1-гидрокси-2-метилпропан-2-ил)-2-(пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
2-(3-(1-аминоэтил)фениламино)-N-трет-бутил-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
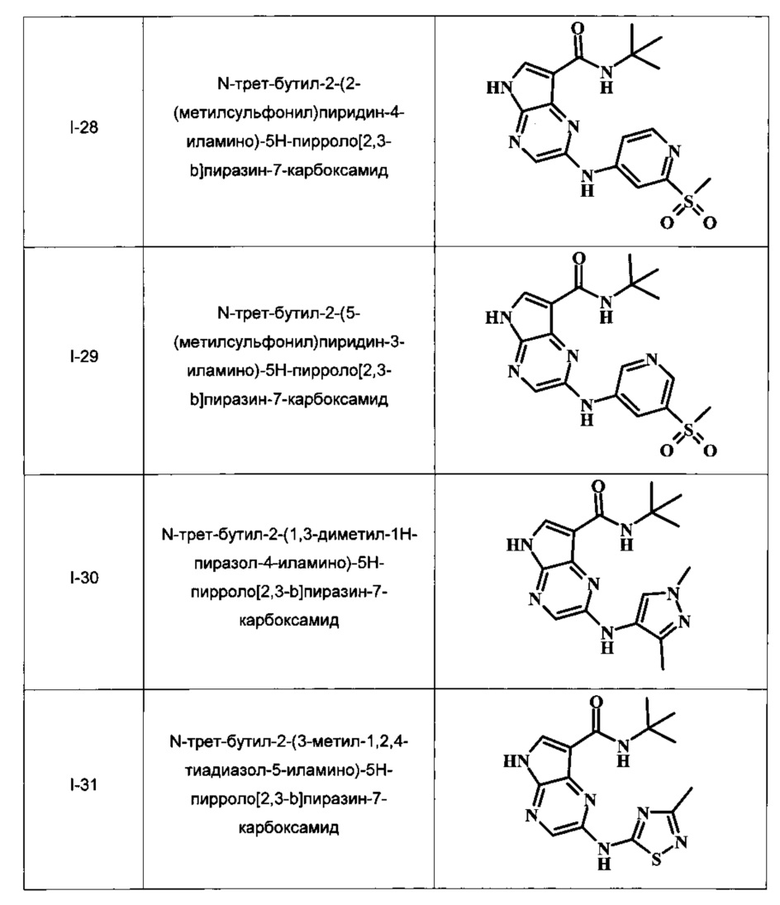
N-трет-бутил-2-(2-(метилсульфонил)пиридин-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(5-(метилсульфонил)пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(1,3-диметил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(3-метил-1,2,4-тиадиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(2-метил-6,7-дигидро-2H-пиразоло[4,3-b]пиридин-4(5H)-ил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(1-метил-6,7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
3-метил-2-(1-метил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид;
2-(3-метансульфонил-фениламино)-3-метил-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид;
3-метил-2-(3-метил-изотиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид;
2-фениламино(D5)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид; and
N-трет-бутил-3-хлор-2-(3-метилизотиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид.
В настоящем изобретении предложен способ лечения воспалительный или аутоиммунных заболеваний, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен вышеуказанный способ, также содержащий введение дополнительного терапевтического средства выбранного из химиотерапевтического или антипролиферативного средства, противовоспалительного средства, иммуномодулятора или иммуносупрессивного средства, нейротрофического фактора, средства для лечения сердечнососудистых заболеваний, средства для лечения диабета или средства для лечения иммунодефицитных состояний.
В настоящем изобретении предложен способ лечения воспалительных заболеваний, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения ревматоидного артрита, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения астмы, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения иммунных нарушений, включая волчанку, рассеянный склероз, ревматоидный артрит, псориаз, сахарный диабет I типа, осложнения трансплантации органов, ксенотрансплантацию, диабет, рак, бронхиальную астму, атопический дерматит, аутоиммунные заболевания щитовидной железы, язвенный колит, болезнь Крона, болезнь Альцгеймера и лейкемию, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения воспалительных заболеваний содержащий совместное введение пациенту, нуждающемуся в этом, терапевтически эффективное количество противовоспалительного соединения в комбинации с соединением Формулы I.
В настоящем изобретении предложен способ лечения иммунного заболевания, содержащий совместное введение пациенту, нуждающемуся в этом, терапевтически эффективное количество иммуносупрессора в комбинации с соединением Формулы I.
В настоящем изобретении предложена фармацевтическая композиция, содержащая соединение Формулы I, смешанное с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
В настоящем изобретении предложена вышеуказанная фармацевтическая композиция, также содержащее дополнительное терапевтическое средство, выбранное из химиотерапевтического или антипролиферативного средства, противовоспалительного средства, иммуномодулятора или иммуносупрессивного средства, нейротрофического фактора, средства для лечения сердечно-сосудистых заболеваний, средства для лечения диабета и средства для лечения иммунодефицитных состояний.
В настоящем изобретении предложено применение соединения Формулы I для изготовления лекарственного средства, полезного при лечении заболеваний, ассоциированных с Syk.
В настоящем изобретении предложено применение соединения Формулы I для изготовления лекарственного средства, полезного при лечении ревматоидного артрита.
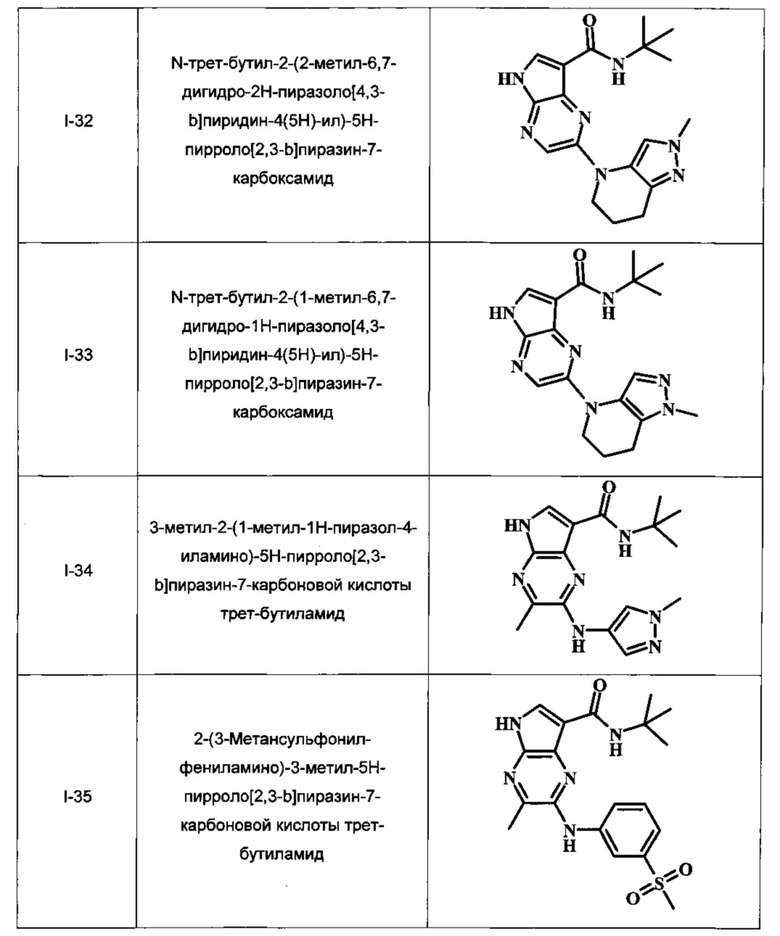
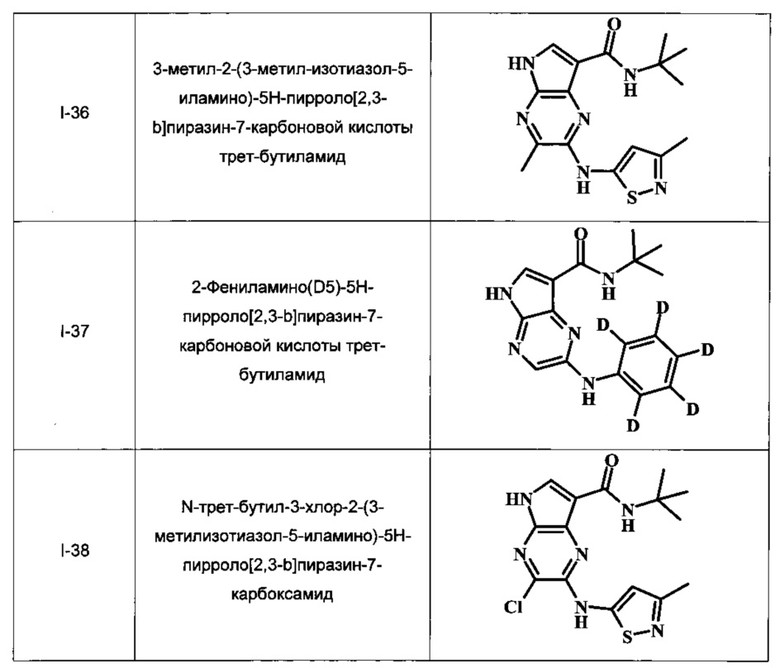
Примеры индивидуальных соединений, охватываемых настоящим изобретением и находящихся в его объеме, представлены в следующей таблице. Эти примеры и способы приведены далее для более четкого понимания и осуществления настоящего изобретения квалифицированными специалистами. Они не должны рассматриваться как ограничивающие объем изобретения, а лишь в качестве его иллюстрирования и воплощения.
В основном, номенклатура, использованная в настоящем изобретении основана на AUTONOMTM v.4.0, компьютеризованной системе института Бельштейн, или Struct=Name, приложения CambridgeSoft® для генерации названий химических соединений по ИЮПАК. Если наблюдается несоответствие между изображенной структурой и названием, данным для этой структуры, изображенная структура обладает более высоким весом. Кроме того, если стереохимия структуры или части структуры не обозначена, например, связью или пунктирной линией, структура или часть структуры должны интерпретироваться как охватывающие все их стереоизомеры.
В Таблице 1 изображены примеры соединений в соответствии с формулой I.
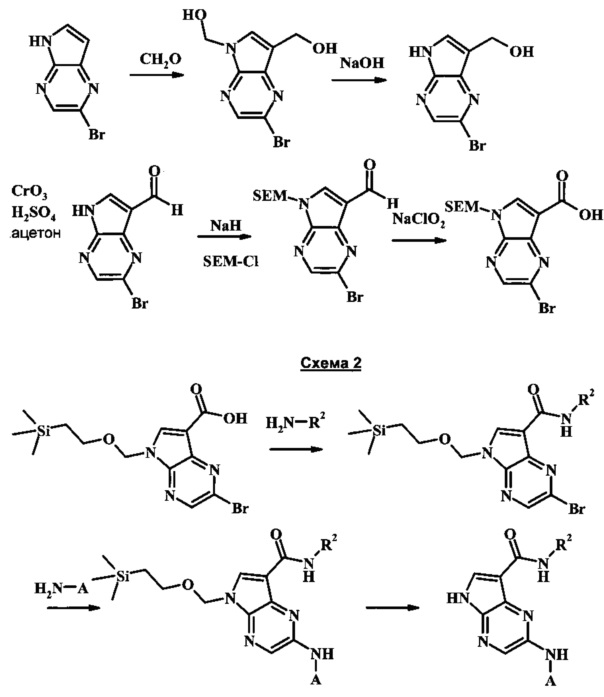
Синтез
Общие схемы
На Общей Схеме, данной ниже, R2 может быть низшим алкилом или низшим гидроксиалкилом; А может быть моноциклическим или бициклическим гетероарилом или фенилом, возможно замещенным одним или более А'; и каждый А' может быть независимо низшим алкилом, гало, низшим алкилсульфонилом, амидо, низшим гидроксиалкилом, низшим алкокси, амино-низшим алкилом, или дейтерием.
Схема 1
Фармацевтические композиции и пути введения
Соединения согласно настоящему изобретению могут быть приготовлены в виде различных лекарственных форм и носителей для перорального введения. Соединения для перорального введения могут быть в форме таблеток, покрытых оболочкой таблеток, драже, твердых и мягких желатиновых капсул, растворов, эмульсий, сиропов или суспензий. Соединения настоящего изобретения являются эффективными при введении другими способами введения, включая непрерывное (внутривенное вливание), местное, парентеральное, внутримышечное, внутривенное, подкожное, трансдермальное (которое может включать средство для улучшения проницаемости), буккальное, назальное введение, ингаляцию и с помощью суппозиториев среди других путей введения. Предпочтительным способом введения, в основном, является пероральное с использованием удобного ежедневного режима дозирования, который может быть скорректирован в зависимости от тяжести заболевания и реакции пациента на активный ингредиент.
Соединение или соединения по настоящему изобретению, а также их фармацевтически полезные соли, вместе с одним или более традиционными эксципиентами, носителями или разбавителями, могут быть в форме фармацевтических композиций и единичных дозированных форм. Фармацевтические композиции и единичные дозированные формы могут состоять из обычных ингредиентов в обычных пропорциях, с или без дополнительных активных соединений или основ и единичные дозированные формы могут содержать любое подходящее эффективное количество активного ингредиента пропорционально предполагаемому применяемому дневному диапазону дозировки. Фармацевтические композиции могут быть использованы в виде твердых веществ, таких как таблетки или капсулы, полутвердых, порошков, препаратов с замедленным высвобождением или жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или наполненные капсулы для перорального применения, или в виде суппозиториев для ректального или вагинального введения, или в виде стерильных растворов для инъекций для парентерального применения. Типичный препарат будет содержать примерно от 5% до примерно 95% активного соединения или соединений (масс./масс.). Термин «препарат» или «дозированная форма» предназначен для обозначения твердых и жидких лекарственных форм активного соединения и квалифицированному специалисту в данной области техники будет понятно, что активный ингредиент может существовать в различных препаратах в зависимости от органа-мишени или ткани и от желаемого дозы и фармакокинетических параметров.
Термин "эксципиент", используемый в настоящем изобретении, относится к соединению, которое является полезным в приготовлении фармацевтической композиции, обычно безопасному, нетоксичному и не являющемуся ни биологически, ни как либо еще нежелательным, и включает в себя эксципиенты, которые являются приемлемыми для ветеринарного использования, а также для фармацевтического применения для человека. Соединения настоящего изобретения могут быть введены сами по себе, но обычно их вводят в смеси с одним или несколькими подходящими фармацевтическими эксципиентами, разбавителями или носителями, выбранными с учетом предполагаемого способа введения и стандартной фармацевтической практики.
"Фармацевтически приемлемый" означает такой, который полезен в приготовлении фармацевтической композиции, который обычно является безопасным, нетоксичным и не является ни биологически, ни как либо еще нежелательным и включает такие, которые являются приемлемыми для ветеринарного использования, а также для фармацевтического применения для человека.
"Фармацевтически приемлемая соль" является формой активного ингредиента, которая может также изначально придать желательные фармакокинетические свойства активному ингредиенту, которые отсутствовали в несолевой форме, и которая может положительно влиять на фармакодинамику активного ингредиента в отношении его терапевтической активности в организме. Фраза «фармацевтически приемлемая соль" соединения означает соль, которая является фармацевтически приемлемой и которая обладает требуемой фармакологической активностью исходного соединения. Такие соли включают: (1) кислотно-аддитивные соли, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или образованные с органическими кислотами, такими как уксусная кислота, пропионовая, капроновая кислоты, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этан-дисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфокислота, 4-толуолсульфокислота, камфорсульфокислота, 4-метилбицикло[2.2.2]окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, третбутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислоты, гидроксинафтоевая кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.п., или (2) соли, образованные, когда протон кислоты присутствует в исходном соединении, либо заменен на ион металла, например, ион щелочного металла, ион щелочноземельного металла или ион алюминия, или связан с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п.
Твердые лекарственные препараты включают порошки, таблетки, пилюли, капсулы, саше, суппозитории и диспергируемые гранулы. Твердый носитель может быть одним или несколькими веществами, которые также могут действовать как разбавители, вкусовые добавки, растворители, любриканты, суспендирующие агенты, связующие вещества, консерванты, разрыхлители для таблеток или инкапсулирующий материал. В случае порошков носитель обычно представляет собой мелко измельченное твердое вещество, которое смешано с мелко измельченным активным компонентом. В случае таблеток активный компонент обычно смешан с носителем, имеющим необходимую связывающую способность, в подходящих пропорциях и спрессован в нужную форму нужного размера. Подходящие носители включают, без ограничения, карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлоза, натрий-карбоксиметилцеллюлоза, легкоплавкий воск, масло какао и т.п. Твердые лекарственные препараты могут содержать, в дополнение к активному компоненту, красители, ароматизаторы, стабилизаторы, буферы, искусственные и натуральные подсластители, диспергаторы, загустители, солюбилизирующие агенты и т.п.
Жидкие препараты также являются пригодными для перорального введения включая жидкие композиции, включая эмульсии, сиропы, эликсиры, водные растворы, водные суспензии. Они включают в себя твердые формы препаратов, которые предназначены для превращения в жидкую форму незадолго до использования. Эмульсии могут быть получены в растворах, например, в водном растворе пропиленгликоля или могут содержать эмульгаторы, такие как лецитин, сорбитанмоноолеат или камедь. Водные растворы могут быть получены растворением активного компонента в воде и добавлением подходящих красителей, ароматизаторов, стабилизаторов и загустителей. Водные суспензии могут быть получены диспергированием тонкоизмельченного активного компонента в воде с вязким веществом, таким как природная или синтетическая камедь, смолы, метилцеллюлоза, натрий-карбоксиметилцеллюлоза и другие хорошо известные суспендирующие агенты.
Соединения по настоящему изобретению могут быть приготовлены для парентерального введения (например, путем инъекции, в частности болюсной инъекции или непрерывной инфузии) и могут быть представлены в единичной дозированной форме в ампулах, предварительно заполненных шприцах, инфузионных растворах небольшого объема или в многодозовых контейнерах с добавлением консерванта. Композиции могут быть в таких формах как суспензии, растворы или эмульсии на масляных или водных основах, например, растворы в водном полиэтиленгликоле. Примеры масляных или неводных носителей, разбавителей, растворителей или основ включают пропиленгликоль, полиэтиленгликоль, растительные масла (например, оливковое масло) и инъекционные органические сложные эфиры (например, этилолеат), и могут содержать вспомогательные вещества, такие как консерванты, увлажнители, эмульгаторы или суспендирующие средства, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может быть в форме порошка, полученного путем асептического выделения стерильного твердого вещества или путем лиофилизации из раствора для разбавления перед использованием с подходящей основой, например, стерильной, апирогенной водой.
Соединения по настоящему изобретению могут быть приготовлены для местного введения в верхний слой кожи в виде мазей, кремов или лосьонов или в виде трансдермального пластыря. Мази и кремы могут, например, быть приготовлены на водной или масляной основе с добавлением подходящих загустителей и/или желатинирующих агентов. Лосьоны могут быть приготовлены на водной или масляной основе и обычно также содержат один или более эмульгаторов, стабилизаторов, диспергирующих агентов, суспендирующих агентов, загустителей или красителей. Препараты, подходящие для местного применения в полости рта, включают лепешки, содержащие активные вещества на ароматизированной основе, как правило, сахарозе и камеди или трагаканте; пастилки, содержащие активный ингредиент на инертной основе, такой как желатин и глицерин или сахароза и камедь, и жидкости для полоскания рта, содержащие активный ингредиент в подходящем жидком носителе.
Соединения по настоящему изобретению могут быть приготовлены для введения в виде суппозиториев. Легкоплавкий воск, такой как смесь глицеридов жирных кислот, или масло какао сначала расплавляют и активный компонент равномерно диспергируют, например, путем перемешивания. Затем расплавленную гомогенную смесь выливают в формы подходящего размера, дают остыть и затвердеть.
Соединения по настоящему изобретению могут быть приготовлены для вагинального введения. Из уровня техники известно, что вагинальные суппозитории, тампоны, кремы, гели, пасты, пены или спреев, содержащие в дополнение к активному ингредиенту такие носители являются подходящими.
Соединения по настоящему изобретению могут быть приготовлены для назального введения. Растворы или суспензии вводят непосредственно в полость носа с помощью обычных средств, например, капельницы, пипетки или спрея. Композиции могут быть представлены в виде одиночной или многодозовой формы. В последнем случае для капельницы или пипетки, это может быть достигнуто путем введения пациенту необходимого, предварительно заданного объема раствора или суспензии. В случае спрея, это может быть достигнуто, например, путем регулирования распыления спреем.
Соединения по настоящему изобретению могут быть приготовлены для аэрозольного введения, в частности, в дыхательные пути, включая интраназальное введение. Соединение обычно имеет небольшой размер частиц, например, порядка пяти (5) микрон или меньше. Такой размер частиц может быть получен с помощью известных в данной области способов, например, путем тонкого измельчения. Активный ингредиент содержится в герметичной упаковке с подходящим пропеллентом, таким как хлорфторуглерод (ХФУ), например, дихлордифторметаном, трихлорфторметаном или дихлортетрафторэтаом или диоксидом углерода или другим подходящим газом. Для удобства, аэрозоль может содержать поверхностно-активные вещества, такие как лецитин. Доза препарата может контролироваться дозирующим клапаном. Кроме того, активные ингредиенты могут быть предоставлены в виде сухого порошка, например порошковой смеси соединения в подходящей порошковой основе, такой как лактоза, крахмал, производные крахмала, такие как гидроксипропилметилцеллюлоза и поливинилпирролидон (ПВП). Порошок-носитель образует гель в носовой полости. Порошковая композиция может быть представлена в виде единичной дозированной формы, например, в капсулах или картриджах, например, желатиновой или блистерной упаковке, из которой порошок может быть введен с помощью ингалятора.
Когда необходимо, композиции могут быть приготовлены с кишечнорастворимой оболочкой, приспособленной для задержанного или контролируемого высвобождения активного ингредиента. Например, соединения настоящего изобретения могут быть приготовлены для устройств трансдермальной или подкожной доставки лекарственных средств. Эти системы доставки обладают преимуществом, когда необходимо задержанное высвобождение соединения и когда соблюдение пациентом режима лечения имеет решающее значение. Соединения в трансдермальных системах доставки часто соединяют с адгезивной по отношению к коже твердой подложкой. Интересующее соединение также может быть объединено с усилителями проницаемости, например, с Azone (1-додецилаза-циклогептан-2-он). Системы доставки с замедленным высвобождением вводятся подкожно в субдермальный слой хирургическим путем или инъекций. Подкожные имплантаты инкапсулируют соединения в жирорастворимой мембране, например, силиконовом каучуке или биоразлагаемом полимере, например, полимолочной кислоте.
Подходящие фармацевтические композиции наряду с фармацевтическими носителями, разбавителями и эксципиентами описаны у Remington: The Science and Practice of Pharmacy 1995, под редакцией E.W. Martin, Mack Publishing Company, 19th edition, Easton, Pennsylvania. Квалифицированный в фармацевтике специалист может изменить композиции в рамках раскрытия изобретения с получением многочисленных препаратов для конкретного пути введения без потери стабильности композициями настоящего изобретения или ущерба для их терапевтической активности.
Модификация соединений настоящего изобретения с целью сделать их более растворимыми в воде или другом носителе, например, может быть легко достигнута путем незначительных изменений (образование соли, этерификация и т.д.), которые известны специалистам в данной области. Также квалифицированным специалистам хорошо известны изменения способа введения и режима дозирования конкретного соединения с целью контроля фармакокинетики соединений настоящего изобретения для максимального положительного эффекта у пациентов.
Термин "терапевтически эффективное количество", используемый в настоящем изобретении, означает количество, требуемое для уменьшения симптомов заболевания у пациента. Доза будет скорректирована с учетом индивидуальных требований в каждом конкретном случае. Это доза может варьироваться в широких пределах в зависимости от многих факторов, таких как тяжесть заболевания, подлежащего лечению, возраста и общего состояния здоровья пациента, других лекарственных средств, которыми пациент лечится, путей и форм введения и предпочтений и опыта лечащего врача. Для перорального введения подходящей должна быть суточная доза примерно от 0,01 до примерно 1000 мг/кг веса тела в день при монотерапии и/или в комбинированной терапии. Предпочтительная суточная доза составляет от приблизительно 0,1 до приблизительно 500 мг/кг массы тела, более предпочтительно от 0,1 до 100 мг/кг массы тела и наиболее предпочтительно 1,0 и 10 мг/кг массы тела в сутки. Таким образом, для введения пациенту с весом 70 кг, диапазон доз будет приблизительно от 7 мг до 0,7 г в день. Суточная доза может быть введена в виде однократной дозы или в виде нескольких доз, как правило, от 1 до 5 доз в день. Как правило, лечение начинают с меньших доз, которые меньше оптимальной дозы соединения. После этого доза увеличивается на малую величину, пока не будет достигнут оптимальный эффект для конкретного пациента. Специалисты в лечении описанных здесь заболеваний смогут без лишних экспериментов на основе личных знаний, опыта и раскрытия настоящего изобретения установить терапевтически эффективное количество соединения по настоящему изобретению для данного заболевания и пациента.
Фармацевтические препараты предпочтительно находятся в единичных дозированных формах. В такой форме препарат разделен на стандартные дозы, содержащие соответствующие количества активного компонента. Единичная дозированная форма может представлять собой упакованный препарат, эта упаковка содержит дискретные количества препарата, такие как упакованные таблетки, капсулы и порошки во флаконах или ампулах. Кроме того, единичная дозированная форма может представлять собой капсулу, таблетку, саше или лепешку сама по себе или может быть соответствующим числом каждого из указанных в упакованном виде.
Композиции
Фармацевтические композиции для доставки различными путями составлены как показано в следующих далее Таблицах. "Активный ингредиент" или "Активное вещество", как используется в Таблицах, означает одно или более соединений Формулы I.
Композиция для перорального введения

Ингредиенты смешали и заполнили ими капсулы, каждая содержащая примерно 100 мг; одна капсула приблизительно соответствует общей суточной дозе.
Композиция для перорального введения

Ингредиенты объединили и гранулировали с использованием растворителя, такого как метанол. Затем композицию высушили и сформировали в таблетки (содержащие около 20 мг активного вещества) с использованием подходящего устройства для формирования таблеток.
Композиция для перорального введения

Ингредиенты смешали с получением суспензии для перорального введения.
Парентеральная Композиция

Активный ингредиент растворили в части воды для инъекций. Затем добавили достаточное количество хлорида натрия при перемешивании, чтобы сделать раствор изотоническим. Раствор довели до необходимого веса остатком воды для инъекций, профильтровали через мембранный фильтр 0,2 мкм и расфасовали в стерильных условиях.
Композиция для суппозиториев

Ингредиенты расплавили вместе и смешали на паровой бане, и разлили по формам, содержащим общий вес 2.5 г.
Композиции для местного применения
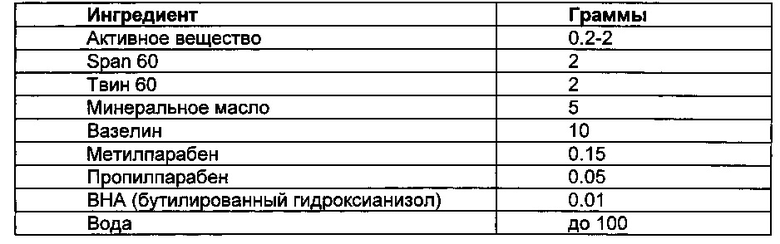
Все ингредиенты, за исключением воды, смешивали и нагревали до приблизительно 60°С при перемешивании. Затем добавили достаточное количество воды при температуре около 60°С при интенсивном перемешивании для эмульгирования ингредиентов, и затем добавили воду, необходимую до приблизительно 100 г.
Композиции назального спрея
Несколько водных суспензий, содержащих приблизительно от 0.025-0.5 процентов активного вещества приготовили в виде препаратов назальных спреев. Препараты возможно содержат неактивные ингредиенты, такие как, например, микрокристаллическая целлюлоза, натрийкарбоксиметилцеллюлоза, декстроза и т.п. Соляная кислота может быть добавлена для выравнивания рН. Препараты назального спрея могут быть доставлены посредством дозированного насоса назального спрея, обычно доставляющего около 50-100 мкл препарата на одно срабатывание. Типичный режим дозирования составляет 2-4 применения спрея каждые 4-12 ч.
Показания и способы лечения
Соединения, описанные здесь, являются ингибиторами киназы, в частности ингибиторами SYK. Эти ингибиторы могут быть полезными для лечения одного или более заболеваний, чувствительных к ингибированию киназы, в том числе заболеваний, чувствительных к ингибированию SYK и/или ингибированию пролиферации В-клеток у млекопитающих. Безотносительно какой-либо конкретной теории, полагают, что взаимодействие соединений по изобретению с SYK приводит к ингибированию активности SYK и, таким образом, к фармацевтической полезности этих соединений. Соответственно, настоящее изобретение включает способ лечения млекопитающего, например, человека, имеющего заболевание, чувствительное к ингибированию активности SYK и/или ингибированию пролиферации В-клеток, включающий введение млекопитающему, имеющему такое заболевание, эффективного количества по меньшей мере одного химического соединения, предложенного здесь. Эффективная концентрация может быть установлена экспериментально, например, путем анализа концентрации соединения в крови, или теоретически путем расчета биодоступности. Другие киназы, которые могут быть затронуты в дополнение к SYK включают, без ограничения, другие тирозинкиназы и серин/треонин киназы.
В настоящем изобретении предложен способ лечения воспалительных или аутоиммунных заболеваний, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен вышеуказанный способ, также содержащий введение дополнительного терапевтического средства выбранного из химиотерапевтического или антипролиферативного средства,
противовоспалительного средства, иммуномодулятора или иммуносупрессивного средства, нейротрофического фактора, средства для лечения сердечнососудистых заболеваний, средства для лечения диабета или средства для лечения иммунодефицитных состояний.
В настоящем изобретении предложен способ лечения воспалительных заболеваний, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения ревматоидного артрита, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения астмы, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения иммунных заболеваний, включая волчанку, рассеянный склероз, ревматоидный артрит, псориаз, сахарный диабет I типа, осложнений трансплантации органов, ксенотрансплантации, диабет, рак, бронхиальную астму, атопический дерматит, аутоиммунные заболевания щитовидной железы, язвенный колит, болезнь Крона, болезнь Альцгеймера, а также лейкемию, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения воспалительных заболеваний, содержащий совместное введение пациенту, нуждающемуся в этом, терапевтически эффективного количества противовоспалительного соединения в комбинации с соединением Формулы I.
В настоящем изобретении предложен способ лечения иммунного заболевания, содержащий совместное введение пациенту, нуждающемуся в этом, терапевтически эффективного количества иммуносупрессора в комбинации с соединением Формулы I.
ПРИМЕРЫ
Аббревиатуры
Обычно используемые сокращения включают: ацетил (Ас), азо-бис-изобутирилнитрил (AIBN), атмосферы (атм), 9-борабицикло[3.3.1]нонан (9-BBN или BBN), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP), трет-бутоксикарбонил (Boc), ди-трет-бутилпирокарбонат или boc-ангидрид (BOC2O), бензил (Bn), бутил (Bu), регистрационный номер в системе кодирования в реферативном журнале "Chemical Abstracts" (CASRN), бензилоксикарбонил (CBZ или Z), карбонилдиимидазол (CDI), 1,4-диазабицикло[2.2.2]октан (DABCO), диэтиламиносеры трифторид (DAST), дибензилиденацетон (DBA), 1,5-диазабицикло[4.3.0]нон-5-ен, (DBN), 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), N,N'-дициклогексилкарбодиимид (DCC), 1,2-дихлорэтан (DCE), дихлорметан (ДХМ), 2,3-дихлор-5,6-дициано-1,4-бензохинон (DDQ), диэтилазодикарбоксилат (DEAD), ди-изо-пропилазодикарбоксилат (DIAD), ди-изо-бутилалюминия гидрид (DIBAL или DIBAL-H), ди-изо-пропилэтиламин (DIPEA), N,N-диметилацетамид (DMA), 4-N,N-диметиламинопиридин (DMAP), N,N-диметилформамид (ДМФ), диметилсульфоксид (ДМСО), 1,1'-бис-(дифенилфосфино)этан (DPPE), 1,1'-бис-(дифенилфосфино)ферроцен (DPPF), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDCI), 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (EEDQ), этил (Et), этилацетат (EtOAc), этанол, (EtOH), 2-этокси-2Н-хинолин-1-карбоновой кислоты этиловый эфир (EEDQ), диэтиловый эфир (Et2O), этилизопропиловый эфир (EtOiPr), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфатуксусная кислота (HATU), уксусная кислота (НОАс), 1-N-гидроксибензотриазол (HOBt), высокоэффективная жидкостная хроматография (ВЭЖХ), изопропанол (IPA), изопропилмагнийхлорид (iPrMgCl), гексаметилдисилазан (HMDS), жидкостная хроматография с масс-спектрометрией (ЖХ-МС), лития гексаметилдисилазан (LiHMDS), мета-хлорпербензойная кислота (m-СРВА), метанол (МеОН), температура плавления (т.пл. или MP), MeSO2-(мезил или Ms), метил (Me), ацетонитрил (MeCN), м-хлорпербензойная кислота (МСРВА), масс-спектр (ms или MS), метил-трет-бутиловый эфир (МТВЕ), метил-тетрагидрофуран (МеТГФ), N-бромсукцинимид (NBS), н-бутиллитий (nBuLi), N-карбоксиангидрид (NCA), N-хлорсукцинимид (NCS), N-метилморфолин (НММ), N-метилпирролидон (NMP), хлорхромат (РСС), дихлор-((бис-дифенилфосфино)ферроценил)палладий(II) (Pd(dppf)Cl2), палладия(11)ацетат (Pd(OAc)2), трис(дибензилиденацетон)дипалладий (0) (Pd2(DBA)3), дихромат пиридиния (PDC), фенил (Ph), пропил (Pr), изо-пропил (i-Pr), фунт на квадратный дюйм (PSI), пиридин (pyr), 1,2,3,4,5-пентафенил-1-(ди-трет-бутилфосфино)ферроцен (Q-Phos), комнатная температура (собственная температура, кт или КТ), втор-бутиллитий (sBuLi), трет-бутилдиметилсилил или трет-BuMe2Si (TBDMS), тетра-н-бутиламмония фторид (TBAF), триэтиламин (TEA или Et3N), 2,2,6,6-тетраметилпиперидин-1-оксил (TEMPO), триметилсилилэтоксиметил (SEM), трифлат или CF3SO2-(Tf), трифторуксусная кислота (ТФУ), 1,1'-бис-2,2,6,6-тетраметилгептан-2,6-дион (TMHD), О-бензотриазол-1-ил-N,N,N',N'-тетраметилурония тетрафторборат (TBTU), тонкослойная хроматография (ТСХ), тетрагидрофуран (ТГФ), триметилсилил или Me3Si (TMS), р-толуолсульфокислота (TsOH или pTsOH), 4-Me-C6H4SO2 или тозил (Ts) и N-уретан-N-карбоксиангидрид (UNCA). Обычные номенклатуры, включая префиксы нормальный (N), изо (I-), вторичный (втор-), третичный (трет-) и нео имеют свои обычные значения при использовании их с алкильной частью. (J. Rigaudy и D.P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford).
Общие условия.
Если не указано иное, все температуры, включая температуру плавления (т.е. MP) приведены в градусах Цельсия (°С). Следует понимать, что реакция, с помощью которой получают указанный и/или желаемый продукт, не обязательно непосредственно приводит к нему из комбинации двух реагентов, которые были добавлены первоначально, т.е. могут существовать одно или более промежуточных соединений, которые образуются в смеси, что в конечном итоге приводит к образованию указанного и/или желаемого продукта. Данные выше сокращения могут быть использованы в способах получения и примерах. Все названия были генерированы с помощью Autonom или ChemDraw.
Следующие способы получения и примеры приведены с целью более четкого понимания и осуществления на практике настоящего изобретения специалистами в данной области техники. Их следует рассматривать не как ограничивающие объем изобретение, а только как иллюстрирующие и являющиеся типичными для настоящего изобретения.
Примеры Получения
Пример 1
Методика 1
2-бромо-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбальдегид
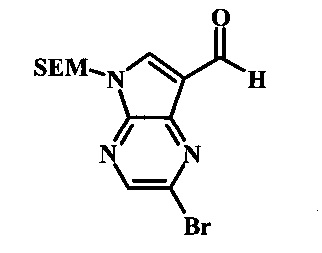
Способ А
Стадия 1
(2-бромо-7-гидроксиметил-пирроло[2,3-b]пиразин-5-ил)-метанол 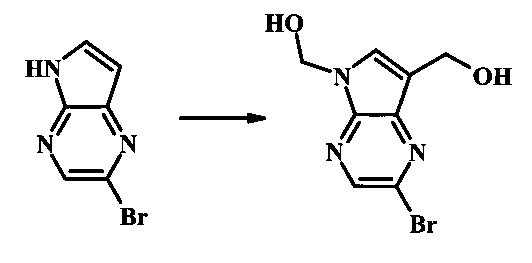
К частичной суспензии 2-бромо-5Н-пирроло[2,3-b]пиразина (5.0 г, 25.2 ммоль) в 1,4-диоксане (100 мл) добавили 2.0 М водный NaOH (25 мл, 50.0 ммоль) и 37% водный формальдегид (19 мл, 252 ммоль). Темную гомогенную реакционную смесь перемешивали при комнатной температуре в течение ночи. Органические фазы эвапорировали при пониженном давлении. Водный слой нейтрализовали 1.0 М HCl и экстрагировали EtOAc (2х). Объединенные органические фазы сконцентрировали с получением 2.6 г оранжевого осадка. После отстаивания в водном слое образовался густой коричневый преципитат. Преципитат собрали посредством фильтрации и высушили. Коричневый осадок экстрагировали горячей смесью 10% МеОН/EtOAc (3×200 мл). Экстракты объединили и эвапорировали с получением дополнительного количества 3.05 г оранжевого осадка. Общий выход составил 5.65 г (87%) (2-бромо-7-гидроксиметил-пирроло[2,3-b]пиразин-5-ил)-метанола. 1Н NMR (DMSO-d6, 400 MHz): δ (ppm) 8.43 (s, 1H), 7.96 (s, 1H), 6.71 (t, J=7.3 Hz, 1H), 5.59 (d, J=7.6 Hz, 2H), 5.10 (t, J=5.3 Hz, 1H), 4.66 (d, J=5.6 Hz, 2H).
Стадия 2
(2-бромо-5Н-пирроло[2.3-b]пиразин-7-ил)-метанол
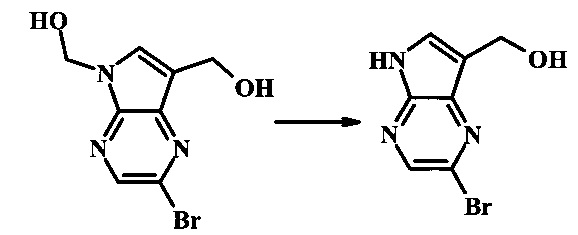
К суспензии (2-бромо-7-гидроксиметил-пирроло[2,3-b]пиразин-5-ил)-метанола (5.65 г, 21.9 ммоль) в ТГФ (150 мл) добавили раствор 2.0 М водного NaOH (33 мл, 66 ммоль). Гомогенную реакционную смесь перемешивали в течение ночи, затем органическую фазу удалили при пониженном давлении. Водный остаток довели до рН 4 с помощью 1.0 М водной HCl. Получившийся преципитат собрали посредством фильтрации и промыли H2O с получением 3.68 г желтого осадка. Фильтрат экстрагировали EtOAc (2х) и органические фазы сконцентрировали при пониженном давлении с получением дополнительных 0.92 г желтого осадка. Общий выход составил 4.60 г (92%) (2-бромо-5Н-пирроло[2,3-b]пиразин-7-ил)-метанола. 1Н NMR (DMSO-d6, 300 MHz): δ (ppm) 12.19 (br. s., 1H), 8.33 (s, 1H), 7.85 (s, 1H), 4.96 (t, J=5.3 Hz, 1H), 4.62 (d, J=4.9 Hz, 2H).
Стадия 3
2-бромо-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбальдегид
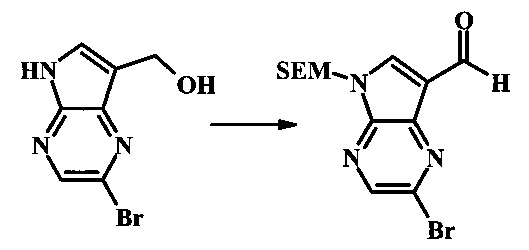
Стоковый раствор реактива Джонса (2.67 М) получили посредством осторожного добавления концентрированной H2SO4 (2.3 мл) к CrO3 (2.67 г), затем разбавили до 10 мл с помощью H2O. К частичной суспензии (2-бромо-5Н-пирроло[2,3-b]пиразин-7-ил)-метанола (4.6 г, 20.1 ммоль) в ацетоне (300 мл) медленно добавили реактив Джонса (9 мл, 24.0 ммоль). По мере добавления исходное соединение постепенно растворилось и образовался вязкий зеленый преципитат. Реакционную смесь перемешивали в течение 15 мин, затем погасили с помощью i-PrOH (2 мл) и отфильтровали через целит, промывая ацетоном. Фильтрат сконцентрировали с получением 4.76 г 2-бромо-5Н-пирроло[2,3-b]пиразин-7-карбальдегида в виде желто-оранжевого осадка, который использовали без дополнительной очистки. В раствор этого осадка в ДМФ (50 мл) при 0°С добавили NaH (60% в минеральном масле, 1.2 г, 30.1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, затем снова охладили до 0°С и медленно добавили 2-(триметилсилил)этоксиметилхлорид (4.3 мл, 24.1 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч, затем погасили с помощью H2O и экстрагировали EtOAc (3х). Объединенные органические фазы промыли H2O (3х) и солевым раствором, затем высушили над MgSO4 и сконцентрировали. Остаток очистили посредством SiO2 хроматографии (20%-30% EtOAc/гексаны) для выделения 3.82 г (53%) 2-бромо-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбальдегида в виде желтого осадка. 1Н NMR (CDCl3, 300 MHz): δ (ppm) 10.37 (s, 1Н), 8.50 (s, 1Н), 8.33 (s, 1Н), 5.73 (s, 2Н), 3.53-3.70 (m, 2Н), 0.90-1.05 (m, 2Н), 0.00 (s, 9Н).
Способ В
Стадия 1
2-бромо-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин
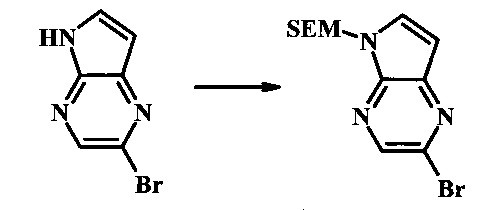
В сухой круглодонной колбе 2-бромо-5Н-пирроло[2,3-b]пиразин (5.0 г, 25.2 ммоль) растворили в ДМФ (50 мл). Реакционную смесь охладили до 0°С и добавили гидрид натрия (60% дисперсия в минеральном масле, 1.22 г, 30.6 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 15 мин, затем снова охладили до 0°С и медленно добавили SEM-Cl (5.4 мл, 30.4 ммоль). После завершения добавления, ледяную ванну убрали и реакционную смесь перемешивали при комнатной температуре в течение 1.5 ч. Реакционную смесь погасили с помощью 50 мл воды и экстрагировали 150 мл диэтилового эфира (2х). Объединенные органические слои промыли дважды 30 мл воды и один раз 30 мл солевого раствора, затем высушили над сульфатом натрия, отфильтровали и сконцентрировали. Остаток абсорбировали на -20 г SiO2 и хроматографировали через 200 г SiO2 с помощью ЕtOАс/гексаны (градиент: 0-15% EtOAc). Все фракции, содержащие продукт, объединили и сконцентрировали с получением 2-бромо-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразина (6.61 г, 80%) в виде бледно-желтого масла, которое постепенно затвердело. 1Н NMR (CDCl3, 300 MHz): δ (ppm) 8.38 (s, 1Н), 7.70 (d, J=3.8 Hz, 1H), 6.76 (d, J=3.8 Hz, 1H), 5.68 (s, 2H), 3.50-3.65 (m, 2H), 0.88-1.03 (m, 2H), 0.00 (s, 9H).
Стадия 2
2-бромо-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбальдегид
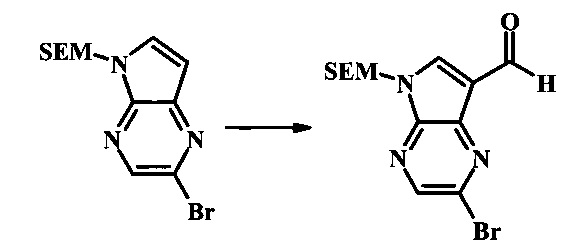
В круглодонной колбе 2-бромо-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин (6.58 г, 20.0 ммоль) растворили в хлороформе (стабилизированном пентеном, 120 мл) и добавили хлорметилендиметилиминия хлорид (10.3 г, 80.2 ммоль) добавили. Реакционную смесь перемешивали при кипении с обратным холодильником в течение 8 ч при барботировании стационарного потока азота через реакционную смесь. Темно-коричневый раствор охладили до комнатной температуры и перемешивали в течение ночи. Реакционную смесь осторожно погасили с помощью ~100 мл насыщенного раствора NaHCO3 (осторожно: экзотермично) и затем дважды экстрагировали 200 мл диэтилового эфира. Органические слои объединили, высушили над сульфатом натрия, отфильтровали и сконцентрировали. Остаток абсорбировали на ~20 г SiO2 и хроматографировали через 200 г SiO2 с помощью EtOAc/гексаны (градиент: 0-25% EtOAc). Все фракции, содержащие продукт, объединили и сконцентрировали с получением 5.92 г (83%) приблизительно 3:1 смеси 2-бромо-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбальдегида и 2-хлор-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбальдегида в виде желтого осадка. Бромид: 1Н NMR (CDCl3, 300 MHz): δ (ppm) 10.37 (s, 1Н), 8.50 (s, 1Н), 8.33 (s, 1Н), 5.73 (s, 2Н), 3.56-3.67 (m, 2Н), 0.91-1.02 (m, 2Н), 0.00 (s, 9Н); Хлорид: 1Н NMR (CDCl3, 300 MHz): 6 (ppm) 10.36 (s, 1Н), 8.41 (s, 1Н), 8.35 (s, 1Н), 5.74 (s, 2Н), 3.56-3.67 (m, 2Н), 0.91-1.02 (m, 2Н), 0.00 (s, 9Н).
Методика 2
2-бромо-5-(2-триметилсиланилэтоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновая кислота
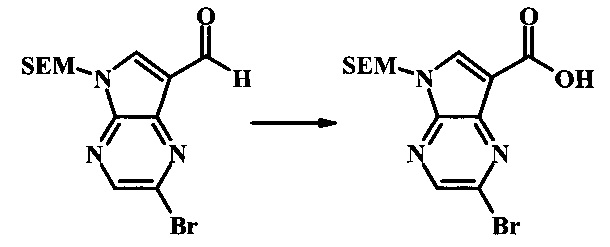
В колбе 2-бромо-5-(2-триметилсиланилэтоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбальдегид (3.11 г, 8.74 ммоль) растворили в диоксане (120 мл) и H2O (30 мл) и смесь охладили до 0°С. Сульфаминовую кислоту (5.09 г, 52.4 ммоль) добавили, с последующим раствором хлорита натрия (1.28 г, 11.4 ммоль) и дигидрофосфатом калия (14.3 г, 104.9 ммоль) в H2O (75 мл) через капельную воронку в течение 15 мин. Смеси дали нагреться до комнатной температуры в течение 2 ч. Получившийся желтый осадок отфильтровали, промыли H2O и гексаном и высушили. Фильтрат затем экстрагировали EtOAc, и объединенные органические фазы промыли солевым раствором, высушили над MgSO4 и сконцентрировали с получением дополнительного продукта. В суме получили 3.71 г 2-бромо-5-(2-триметилсиланилэтоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты в виде желтого осадка. 1Н NMR (CDCl3, 400 MHz): δ (ppm) 8.52 (s, 1Н), 8.42 (s, 1H), 5.73 (s, 2H), 3.56-3.65 (m, 2H), 0.90-1.02 (m, 2H), 0.00 (s, 9H).
Методика 3
2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
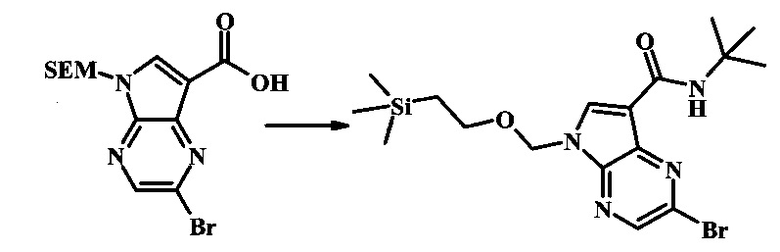
В 100 мл круглодонной колбе, 2-бромо-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновую кислоту (1.00 г, 2.69 ммоль) растворили в ДМФ (6 мл). Трет-бутиламин (1.7 мл, 16.2 ммоль) добавили с последующим HATU (1.12 г, 2.95 ммоль). Желтую суспензию перемешивали при комнатной температуре в течение 72 ч, затем погасили водой и экстрагировали смесью диэтилового эфира и EtOAc. Органические слои промыли дважды водой и один раз солевым раствором, затем объединили, высушили над сульфатом натрия, отфильтровали и сконцентрировали. Остаток тритурировали с петролейным эфиром с получением 989 мг (86%) 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида в виде серо-белого порошка. 1Н NMR (CDCl3, 300 MHz): δ (ppm) 8.40 (s, 1H), 8.31 (s, 1H), 7.86 (s, 1H), 5.65 (s, 2H), 3.46-3.58 (m, 2H), 1.54 (s, 9H), 0.84-0.98 (m, 2H), -0.04 (s, 9H).
Методика 4
2-бромо-N-(1-гидрокси-2-метилпропан-2-ил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
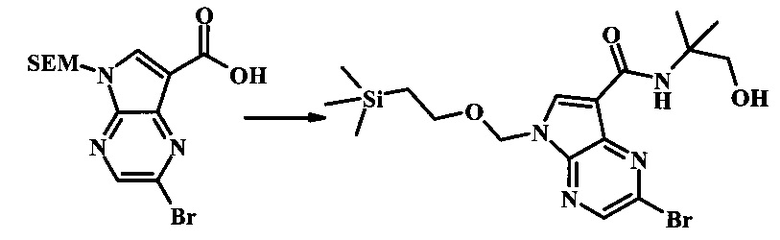
К смеси 2-бромо-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты (2 г, 5.37 ммоль), 2-амино-2-метилпропан-1-ола (575 мг, 6.45 ммоль) и HATU (2.25 г, 5.91 ммоль) добавили DIPEA (2.08 г, 2.81 мл, 16.1 ммоль) и ДМФ (18 мл). Смесь перемешивали при комнатной температуре в течение 16 ч, затем разбавили EtOAc и 10% лимонной кислотой. Фазы разделили и органическая фаза была затем промыта последовательно 10% лимонной кислотой, NaHCO3 и солевым раствором. Органическую фазу высушили (MgSO4), отфильтровали и очистили посредством хроматографии (силикагель, 20-60% этилацетат в гексанах) с получением 2-бромо-N-(1-гидрокси-2-метилпропан-2-ил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (1.95 г, 4.4 ммоль, 82%) в виде белого осадка. 1Н NMR (400 MHz, ХЛОРОФОРМ-d) δ ppm: 8.47 (s, 1Н), 8.34 (s, 1Н), 8.13 (s, 1Н), 5.69 (s, 2Н), 3.78 (s, 2Н), 3.57 (t, J=8.4 Hz, 3H), 1.52 (s, 60.96 (t, J=8.4 Hz, 3H),0.00 (s, 9H).
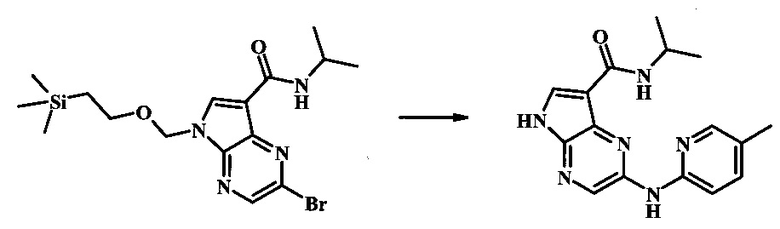
Пример 1
N-трет-бутил-2-(5-метилпиридин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
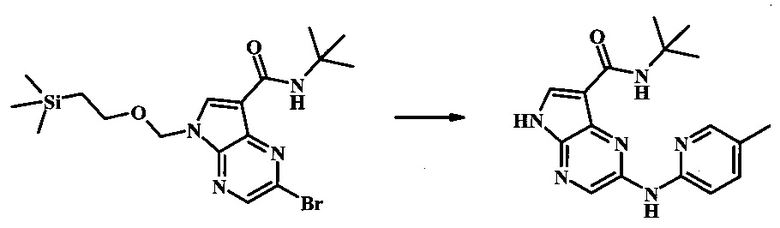
Стадия 1
К смеси 5-метилпиридин-2-амина (38.0 мг, 351 мкмоль), 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (100 мг, 234 мкмоль), (R)-(+)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (7.28 мг, 11.7 мкмоль), ацетата палладия (II) (13.1 мг, 58.5 мкмоль) и трет-бутоксида натрия (56.2 мг, 585 мкмоль) добавили ДМФ (1 мл) и толуол (500 мкл) и смесь нагревали в микроволновой печи при 140°С в течение 20 мин. Образовавшуюся темную смесь влили в воду и экстрагировали этилацетатом. Органические слои объединили, промыли солевым раствором, высушили (сульфат магния), отфильтровали и очистили посредством хроматографии (силикагель, 30-70% этилацетат в гексанах) с получением N-трет-бутил-2-(5-метилпиридин-2-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (62 мг, 136 мкмоль, 58%) в виде серо-белого осадка. MS (EI/CI) m/z: 455.3 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(5-метилпиридин-2-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (162 мг, 356 мкмоль) в дихлорметане (3 мл) добавили трифторуксусную кислоту (813 мг, 549 мкл, 7.13 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь затем сконцентрировали под вакуумом и полученный остаток растворили в дихлорметане (3 мл), метаноле (1 мл) и гидроксиде аммония (0.6 мл) и смесь перемешивали при комнатной температуре в течение 30 мин. Образовавшийся желтый преципитат собрали посредством фильтрации и высушили. Маточную жидкость очистили посредством хроматографии (силикагель, 5-25% 1:4 метанол:дихлорметан раствор в дихлорметане) и объединили с осадком с получением N-трет-бутил-2-(5-метилпиридин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (77 мг, 237 мкмоль, 67%) в виде серо-белого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.37 (s, 1 Н), 9.88 (s, 1 Н), 8.38 (s, 1 Н), 8.13 (d, J=2.5 Hz, 1 H), 8.04 (d, J=3.2 Hz, 1 H), 7.88 (d, J=8.5 Hz, 1 H), 7.74 (s, 1 H), 7.51 (dd, J=8.2, 2.3 Hz, 1 H), 2.25 (s, 3 H), 1.47 (s, 9 H); MS (EI/CI) m/z: 325.1 [M+Н].
Пример 2
N-(1-гидрокси-2-метилпропан-2-ил)-2-(5-метилпиридин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
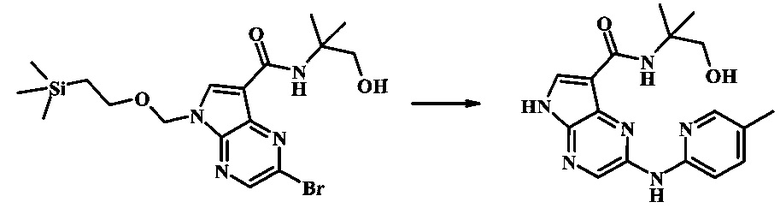
Стадия 1
К смеси 5-метилпиридин-2-амина (72.1 мг, 666 мкмоль), 2-бромо-N-(1-гидрокси-2-метилпропан-2-ил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (197 мг, 444 мкмоль), (R)-(+)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (13.8 мг, 22.2 мкмоль), ацетата палладия (II) (24.9 мг, 111 мкмоль) и трет-бутоксида натрия (107 мг, 1.11 ммоль) добавили ДМФ (2 мл) и толуол (1 мл), затем нагревали в микроволновой печи при 140°С в течение 20 мин. Образовавшуюся темную смесь влили в воду и экстрагировали этилацетатом (3х). Органические слои объединили, промыли солевым раствором (2х), высушили над сульфатом магния и очистили посредством хроматографии (силикагель, 65-100% этилацетат в гексанах) с получением N-(1-гидрокси-2-метилпропан-2-ил)-2-(5-метилпиридин-2-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (66 мг, 140 мкмоль, 32%) в виде светло-желтого осадка. MS (EI/CI) m/z: 471.4.
Стадия 2
К раствору N-(1-гидрокси-2-метилпропан-2-ил)-2-(5-метилпиридин-2-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (66 мг, 140 мкмоль) в дихлорметане (1.5 мл) добавили трифторуксусную кислоту (320 мг, 216 мкл, 2.8 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом, затем остаток растворили в дихлорметане (2 мл), метаноле (0.5 мл) и гидроксиде аммония (0.3 мл). После перемешивания при комнатной температуре в течение 30 мин, смесь сконцентрировали под вакуумом и тритурировали с эфиром и водой. Желтый преципитат собрали посредством фильтрации и промыли водой, затем эфиром с получением N-(1-гидрокси-2-метилпропан-2-ил)-2-(5-метилпиридин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (31 мг, 91.1 мкмоль, 65%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.38 (s, 1 Н), 9.86 (s, 1 Н), 8.39 (s, 1 Н), 8.12 (d, J=2.5 Hz, 1 H), 8.04 (d, J=3.3 Hz, 1 H), 7.95 (d, J=8.4 Hz, 1 H), 7.74 (s, 1 H), 7.54 (dd, J=8.6, 2.3 Hz, 1 H), 5.06 (s, 1 H), 3.58 (s, 2 H), 2.25 (s, 3 H), 1.39 (s, 6 H); MS (EI/CI) m/z: 341.1 [M+Н].
Пример 3
N-трет-бутил-2-(1-метил-1Н-пиразол-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
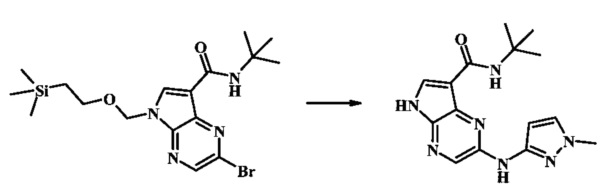
Стадия 1
К смеси 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (200 мг, 468 мкмоль), 1-метил-1Н-пиразол-3-амина (68.2 мг, 702 мкмоль), BINAP (14.6 мг, 23.4 мкмоль), ацетата палладия (II) (26.3 мг, 117 мкмоль) и трет-бутоксида натрия (112 мг, 1.17 ммоль) добавили ДМФ (1.01 мл) и толуол (503 мкл), нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь охладили, затем разбавили водой и экстрагировали этилацетатом. Органические слои объединили и промыли солевым раствором и затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 20-100% этилацетат в гексанах) дала N-трет-бутил-2-(1-метил-1Н-пиразол-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (140 мг, 316 мкмоль, 67%) в виде желтого осадка. MS (EI/CI) m/z: 444.3.
Стадия 2
К раствору N-трет-бутил-2-(1-метил-1Н-пиразол-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (140 мг, 316 мкмоль) в дихлорметане (3.5 мл) добавили трифторуксусную кислоту (720 мг, 486 мкл, 6.31 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток растворили в дихлорметане (3 мл), метаноле (1.5 мл), и гидроксиде аммония (0.65 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали под вакуумом, затем тритурировали эфиром и водой. Осадок собрали посредством фильтрации, затем высушили под вакуумом с получением N-трет-бутил-2-(1-метил-1Н-пиразол-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (44 мг, 140 мкмоль, 45%) в виде коричневого осадка. 1Н NMR (400 MHz, метанол-d) δ ppm: 8.07 (s, 1 Н), 7.93 (s, 1 Н), 7.81 (s, 1 Н), 7.35 (d, J=2.2 Hz, 1 H), 6.32 (d, J=2.2 Hz, 1 H), 3.67 (s, 3 H), 1.35 (s, 9 H); MS (EI/CI) m/z: 314.1 [M+Н].
Пример 4
N-Isopropyl-2-(5-метилпиридин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
Стадия 1
К смеси 2-бромо-N-изопропил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 363 мкмоль), 5-метилпиридин-2-амина (58.9 мг, 544 мкмоль), BINAP (11.3 мг, 18.1 мкмоль), ацетата палладия (II) (20.4 мг, 90.7 мкмоль) и трет-бутоксида натрия (87.2 мг, 907 мкмоль) добавили ДМФ (780 мкл) и толуол (390 мкл), нагревали в микроволновой печи при 140°С в течение 20 мин. Раствор охладили, затем разбавили водой и этил ацетатом. Смесь экстрагировали этилацетатом (3х), затем органические экстракты объединили и промыли солевым раствором, высушили над сульфатом магния и очистили посредством хроматографии (силикагель, 30-70% этилацетат в гексанах) с получением N-изолролил-2-(5-метилпиридин-2-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (90 мг, 204 мкмоль, 56%) в виде серо-белого осадка. MS (EI/CI) m/z: 441.3 [М+Н].
Стадия 2
К раствору N-изолролил-2-(5-метилпиридин-2-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (90 мг, 204 мкмоль) в дихлорметане (2.5 мл) добавили трифторуксусную кислоту (466 мг, 315 мкл, 4.09 ммоль) и перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом, затем полученный остаток растворили в дихлорметане (3 мл), метаноле (1.5 мл) и гидроксиде аммония (0.45 мл). Через 3 ч при комнатной температуре, смесь сконцентрировали под вакуумом затем тритурировали с водой и эфиром. Осадок собрали посредством фильтрации с получением N-изолролил-2-(5-метилпиридин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (23 мг, 74.1 мкмоль, 36%) в виде серо-белого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.40 (s, 1 Н), 9.92 (s, 1 Н), 8.43 (s, 1 Н), 8.14 (s, 1 Н), 8.09 (d, J=3.2, 1 H), 7.86 (s, 1 H), 7.85 (d, J=15.1 Hz, 1 H), 7.55 (dd, J=8.5, 2.3 Hz, 1 H), 4.16 (m, 1 H), 2.26 (s, 3 H), 1.25 (d, J=6.5, 6 H); MS (EI/CI) m/z: 311.1 [M+Н].
Пример 5
N-трет-бутил-2-(1-метил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
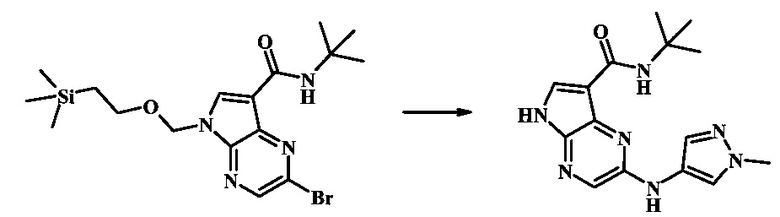
Стадия 1
К смеси 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 1-метил-1Н-пиразол-4-амина гидрохлорида (70.3 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (84.3 мг, 877 мкмоль) добавили ДМФ (1 мл) и толуол (500 мкл). После нагревания в микроволновой печи при 140°С в течение 20 мин, реакционную смесь охладили, разбавили водой и экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния и очистили посредством хроматографии (силикагель, 30-70% этилацетат в гексанах) с получением N-трет-бутил-2-(1-метил-1Н-пиразол-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (89 мг, 201 мкмоль, 57%) в виде желтого осадка. MS (EI/CI) m/z: 314.1 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(1-метил-1Н-пиразол-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (89 мг, 201 мкмоль) в дихлорметане (3 мл) добавили трифторуксусную кислоту (458 мг, 309 мкл, 4.01 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали под вакуумом, затем растворили в дихлорметане (3 мл), метаноле (1.5 мл) и гидроксиде аммония (0.45 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. После концентрирования под вакуумом, смесь тритурировали водой и отфильтровали для сбора осадка. Осадок промыли эфиром и высушили с получением N-трет-бутил-2-(1-метил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (58 мг, 185 мкмоль, 92%) в виде коричневого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.18 (s, 1 Н), 9.11 (s, 1 Н), 7.90 (d, J=3.2, 1 H), 7.86 (s, 1 H), 7.85 (s, 1 H), 7.72 (s, 1 H), 7.55 (s, 1 H), 3.80 (s, 3 H), 1.43 (s, 9 H); MS (EI/CI) m/z: 314.1 [M+Н].
Пример 6
N-трет-бутил-2-(6-метилпиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
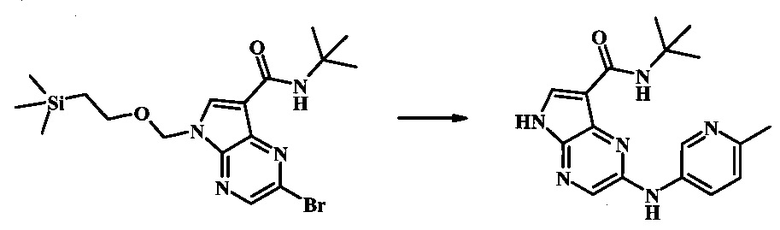
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 6-метилпиридин-3-амина (56.9 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (84.3 мг, 877 мкмоль) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь охладили, затем разбавили водой и экстрагировали в этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 30-70% этилацетат в гексанах) дала N-трет-бутил-2-(6-метилпиридин-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (57 мг, 125 мкмоль, 36%) в виде коричневого осадка. MS (EI/CI) m/z: 455.2 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(6-метилпиридин-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (55 мг, 121 мкмоль) в дихлорметане (1.9 мл) добавили трифторуксусную кислоту (276 мг, 186 мкл, 2.42 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и затем полученный остаток растворили в дихлорметане (2 мл), метаноле (1 мл) и гидроксиде аммония (0.25 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали и тритурировали водой, затем отфильтровали с получением осадка, который промыли эфиром и высушили с получением N-трет-бутил-2-(6-метилпиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (33 мг, 102 мкмоль, 84%) в виде коричневого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.23 (s, 1 Н), 9.41 (s, 1 Н), 8.60 (d, J=2.3, 1 H), 7.91 (m, 3 H), 7.59 (s, 1 H), 7.06 (d, J=8.5, 1 H), 2.31 (s, 3 H), 1.32 (s, 9 H); MS (EI/CI) m/z. 325.1 [M+Н].
Пример 7
N-трет-бутил-2-(2-метилпиримидин-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
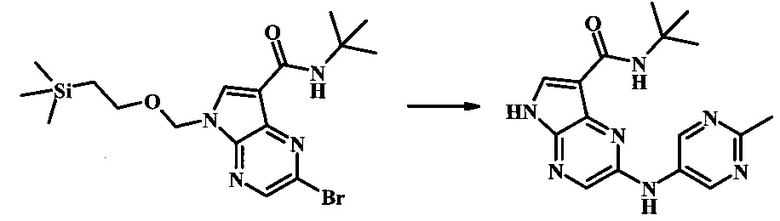
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 2-метилпиримидин-5-амина (57.4 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (84.3 мг, 877 мкмоль) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 40-80% этилацетат в гексанах) дала N-трет-бутил-2-(2-метилпиримидин-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (78 мг, 171 мкмоль, 49%) в виде коричневого осадка. MS (EI/CI) m/z: 456.2 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(2-метилпиримидин-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (78 мг, 171 мкмоль) в дихлорметане (2.5 мл) добавили трифторуксусную кислоту (390 мг, 264 мкл, 3.42 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (2.5 мл), метанолом (1.2 мл) и гидроксидом аммония (0.35 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь сконцентрировали лоб вакуумом затем тритурировали водой. Осадок собрали посредством фильтрации и промыли водой и эфиром с получением N-трет-бутил-2-(2-метилпиримидин-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (43 мг, 132 мкмоль, 77%) в виде коричневого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.31 (s, 1 Н), 9.57 (s, 1 Н), 8.90 (s, 2 Н), 7.96 (d, J=3.3, 1 H), 7.92 (s, 1 H), 7.53 (s, 1 H), 2.46 (s, 3 H), 1.32 (s, 9 H); MS (EI/CI) m/z: 326.1 [M+Н].
Пример 8
N-трет-бутил-2-(5-метилпиразин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
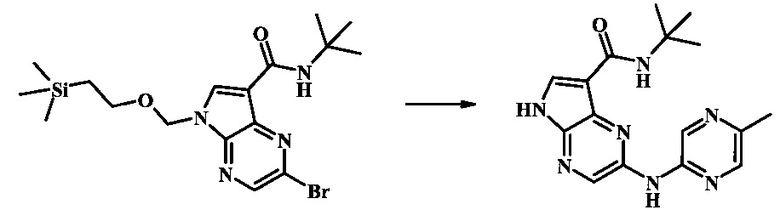
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 5-метилпиразин-2-амина (57.4 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (84.3 мг, 877 мкмоль) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 30-70% этилацетат в гексанах) дала N-трет-бутил-2-(5-метилпиразин-2-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (72 мг, 158 мкмоль, 45%) в виде серо-белого осадка. MS (EI/CI) m/z: 456.2 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(5-метилпиразин-2-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (72 мг, 158 мкмоль) в дихлорметане (2.5 мл) добавили трифторуксусную кислоту (360 мг, 243 мкл, 3.16 ммоль) и перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали лоб вакуумом и полученный остаток разбавили дихлорметаном (2.5 мл), метанолом (1.2 мл) и гидроксидом аммония (0.35 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали и тритурировали водой. Осадок собрали посредством фильтрации и промыли водой и эфиром с получением N-трет-бутил-2-(5-метилпиразин-2-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (26 мг, 79.9 мкмоль, 51%) в виде светло-зеленого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.45 (s, 1 Н), 10.12 (s, 1 Н), 9.17 (d, J=1.5, 1 H), 8.40 (s, 1 H), 8.21 (s, 1 H), 8.10 (d, J=3.2, 1 H), 7.73 (s, 1 H), 2.42 (s, 3 H), 1.46 (s, 9 H); MS (EI/CI) m/z: 326.1 [M+Н].
Пример 9
N-трет-бутил-2-(3-метил-1Н-пиразол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид 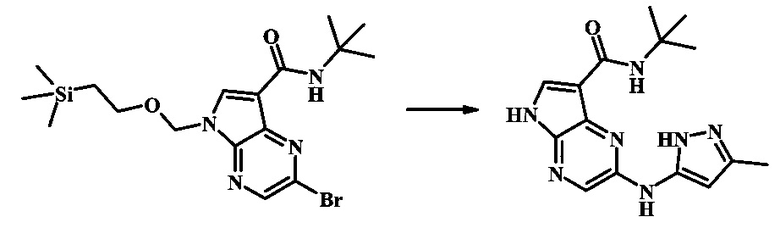
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 3-метил-1Н-пиразол-5-амина (51.1 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (84.3 мг, 877 мкмоль) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 50-100% этилацетат в гексанах) дала N-трет-бутил-2-(3-метил-1Н-пиразол-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (27 мг, 60.9 мкмоль, 17.3%) в виде коричневого осадка. MS (EI/CI) m/z: 444.3 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(3-метил-1Н-пиразол-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (25 мг, 56.4 мкмоль) в дихлорметане (1 мл) добавили трифторуксусную кислоту (129 мг, 86.8 мкл, 1.13 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (1 мл), метанолом (0.5 мл) и гидроксидом аммония (0.15 мл) и перемешивали при комнатной температуре в течение 1 ч. Очистка посредством хроматографии (силикагель, 20-50% [10% метанол/ дихлорметан/ 0.5% гидроксид аммония]/ дихлорметан) дала N-трет-бутил-2-(3-метил-1Н-пиразол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (10 мг, 31.9 мкмоль, 57%) в виде светло-коричневого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.19 (s, 1 Н), 10.12 (s, 1 Н), 9.53 (s, 1 Н), 8.07 (s, 1 Н), 7.93 (d, J=3.0, 1 H), 7.81 (s, 1 H), 6.33 (s, 1 H), 2.19 (s, 3 H), 1.45 (s, 9 H); MS (EI/CI) m/z: 314.1 [M+Н].
Пример 10
N-трет-бутил-2-(пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
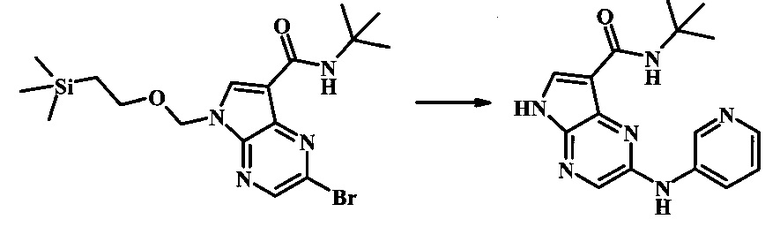
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), пиридин-3-амина (49.5 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (84.3 мг, 877 мкмоль) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 30-70% этилацетат в гексанах) дала N-трет-бутил-2-(пиридин-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (44 мг, 99.9 мкмоль, 28.5%) в виде желтого осадка. MS (EI/CI) m/z: 311.1 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(пиридин-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (44 мг, 99.9 мкмоль) в дихлорметане (1.6 мл) добавили трифторуксусную кислоту (228 мг, 154 мкл, 2.00 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (1.6 мл), метанолом (0.8 мл) и гидроксидом аммония (0.2 мл) и перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали и тритурировали водой. Осадок собрали посредством фильтрации и промыли водой и эфиром с получением N-mpem-бутил-2-(пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (24 мг, 77.3 мкмоль, 77%) в виде коричневого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.26 (s, 1 Н), 9.54 (s, 1 Н), 8.77 (d, J=2.5, 1 H), 8.O4 (d, J=4.6, 1 H), 7.98 (d, J=8.5, 1 H), 7.92 (d, J=3.3, 1 H), 7.91 (s, 1 H), 7.57 (s, 1 H), 7.19 (m, 1 H), 1.31 (s, 9 H); MS (EI/CI) m/z: 311.1 [M+Н].
Пример 11
N-трет-бутил-2-(2-фтор-4-метилфениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид

Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 2-фтор-4-метиланилина (65.9 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (84.3 мг, 877 мкмоль) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 20-50% этилацетат/гексаны) дала N-трет-бутил-2-(2-фтор-4-метилфениламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (87 мг, 184 мкмоль, 53%) в виде серо-белого осадка. MS (EI/CI) m/z: 470.1 [М-Н].
Стадия 2
К раствору N-трет-бутил-2-(2-фтор-4-метилфениламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (87 мг, 184 мкмоль) в дихлорметане (3 мл) добавили трифторуксусную кислоту (421 мг, 284 мкл, 3.69 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (3 мл), метанолом (1.5 мл) и гидроксидом аммония (0.4 мл) и перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали и тритурировали водой. Осадок собрали посредством фильтрации и промыли водой и эфиром с получением N-трет-бутил-2-(2-фтор-4-метилфениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (58 мг, 170 мкмоль, 92%) в виде серо-белого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.27 (s, 1 Н), 8.97 (s, 1 Н), 8.10 (s, 1 Н), 7.96 (d, J=2.3, 1 H), 7.81 (t, J=8.6, 1 H), 7.70 (s, 1 H), 7.10 (d, J=12.4, 1 H), 6.95 (d, J=8.0, 1 H), 2.29 (s, 3 H), 1.32 (s, 9 H); MS (EI/CI) m/z: 342.1 [M+Н].
Пример 12
N-трет-бутил-2-(р-толиламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
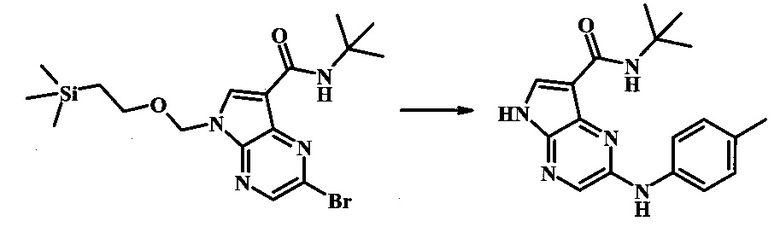
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), п-толуидина (56.4 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (84.3 мг, 877 мкмоль) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 20-45% этилацетат в гексанах) дала N-трет-бутил-2-(р-толиламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (80 мг, 116 мкмоль, 33%) в виде коричневой смолы. MS (EI/CI) m/z: 454.4 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(р-толиламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (80 мг, 176 мкмоль) в дихлорметане (3 мл) добавили трифторуксусную кислоту (402 мг, 272 мкл, 3.53 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (3 мл), метанолом (1.5 мл) и гидроксидом аммония (0.4 мл) и перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали и тритурировали водой. Осадок собрали посредством фильтрации и промыли водой и эфиром. Очистка посредством хроматографии (силикагель, 20-40% раствор 10:89.5:0.5 метанол:дихлорметан:гидроксид аммония в дихлорметане) дала N-трет-бутил-2-(р-толиламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (17 мг, 52.6 мкмоль, 30%) в виде светло-желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.26 (s, 1 Н), 9.34 (s, 1 Н), 7.97 (s, 1 Н), 7.96 (d, J=3.3, 1 H), 7.76 (s, 1H), 7.57 (t, J=8.5, 2 H), 7.09 (d, J=8.1, 2 H), 2.26 (s, 3 H), 1.44 (s, 9 H); MS (EI/CI) m/z: 324.1 [M+Н].
Пример 13
N-трет-бутил-2-(3-(метилсульфонил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
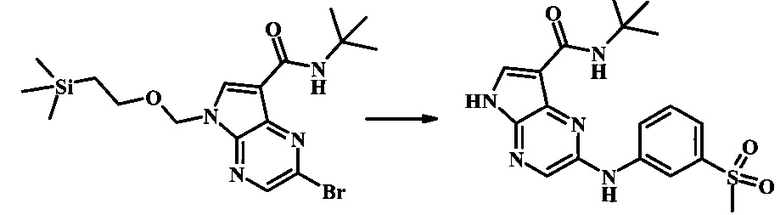
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 3-(метилсульфонил)анилина гидрохлорида (109 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (101 мг, 1.05 ммоль, Экв: 3) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 20-50% этилацетат в гексанах) дала N-трет-бутил-2-(3-(метилсульфонил)фениламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (61 мг, 118 мкмоль, 34%) в виде коричневой смолы. (EI/CI) m/z: 518.3 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(3-(метилсульфонил)фениламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (61 мг, 118 мкмоль) в дихлорметане (1.8 мл) добавили трифторуксусную кислоту (269 мг, 182 мкл, 2.36 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (1.8 мл), метанолом (0.9 мл) и гидроксидом аммония (0.25 мл) и перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали и тритурировали водой. Осадок собрали посредством фильтрации и промыли водой и эфиром. Очистка посредством хроматографии (силикагель, неизвестный градиент дихлорметан/ метанол/ 0.5% гидроксид аммония) дала N-трет-бутил-2-(3-(метилсульфонил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (26 мг, 67.1 мкмоль, 57%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.42 (s, 1 Н), 9.87 (s, 1 Н), 8.33 (d, J=8.2, 1 H), 8.05 (d, J=3.4, 1 H), 8.03 (s, 1H), 7.79 (t, J=2.0, 1 H), 7.68 (s, 1 H), 7.54 (t, J=8.0, 1 H), 7.47 (d, J=7.7, 1 H), 3.21 (s, 3 H), 1.42 (s, 9 H); MS (EI/CI) m/z: 388.2 [M+Н].
Пример 14
N-трет-бутил-2-(1-этил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид

Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 1-этил-1Н-пиразол-4-амина гидрохлорида (51.8 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (101 мг, 1.05 ммоль, Экв: 3) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 40-80% этилацетат в гексанах) дала N-трет-бутил-2-(1-этил-1Н-пиразол-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (82 мг, 179 мкмоль, 51%) в виде желтой смолы. (EI/CI) m/z.458.4 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(1-этил-1Н-пиразол-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (82 мг, 179 мкмоль) в дихлорметане (2.7 мл) добавили трифторуксусную кислоту (409 мг, 276 мкл, 3.58 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (3 мл), метанолом (1.5 мл) и гидроксидом аммония (0.4 мл) и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь сконцентрировали, затем очистили посредством хроматографии (силикагель, 15-65% дихлорметан/ [10% дихлорметан/ метанол/ 0.5% гидроксид аммония]) с получением N-трет-бутил-2-(1-этил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (33 мг, 101 мкмоль, 56%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.16 (s, 1 Н), 9.06 (s, 1 Н), 7.90 (d, J=3.3, 1 H), 7.85 (s, 1 H), 7.84 (s, 1H), 7.73 (s, 1 H), 7.59 (s, 1 H), 4.08 (q, J=7.3, 2 H), 1.42 (s, 9 H), 1.36 (t, J=7.3, 3 H); MS (EI/CI) m/z: 328.1 [M+Н].
Пример 15
N-трет-бутил-2-(2-(диметилкарбамоил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
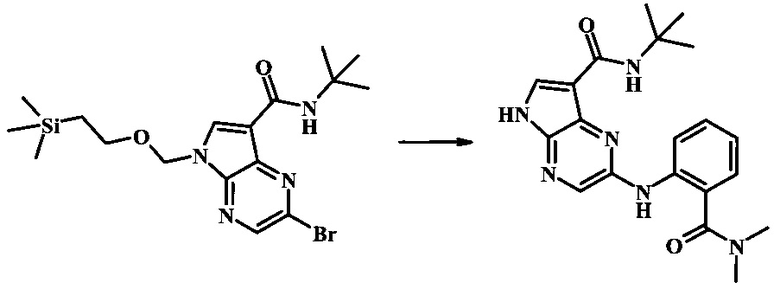
Стадия 1
К раствору 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль) в дихлорметане (5.5 мл) добавили трифторуксусную кислоту (800 мг, 541 мкл, 7.02 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (5 мл), метанолом (2.5 мл) и гидроксидом аммония (0.7 мл) и перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали и тритурировали водой. Осадок собрали посредством фильтрации и промыли водой и эфиром с получением 2-бромо-N-трет-бутил-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (80 мг, 269 мкмоль, 77%) в виде серо-белого осадка.
Стадия 2
Смесь 2-бромо-N-трет-бутил-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (80 мг, 269 мкмоль), 2-амино-N,N-диметилбензамида гидрохлорида (81.0 мг, 404 мкмоль), BINAP (8.38 мг, 13.5 мкмоль), ацетата палладия (II) (15.1 мг, 67.3 мкмоль) и трет-бутоксида натрия (77.6 мг, 808 мкмоль, Экв: 3) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (15-45% дихлорметан/ [10% дихлорметан/ метанол/ 0.5% гидроксид аммония]) дала N-трет-бутил-2(2(диметилкарбамоил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (16 мг, 42.1 мкмоль, 16%) в виде серо-белого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.25 (s, 1 Н), 8.77 (s, 1 Н), 8.06 (s, 1 Н), 7.96 (d, J=3.0, 1 H), 7.91 (d, J=7.9, 1 H), 7.78 (s, 1H), 7.38 (t, J=7.7, 1 H), 7.30 (d, J=7.5, 1 H), 7.11 (t, J=7.5, 1 H), 2.88 (s, 3 H), 2.80 (s, 3 H), 1.27 (s, 9 H); MS (EI/CI) m/z: 381.2 [M+Н].
Пример 16
N-трет-бутил-2-(1,5-диметил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
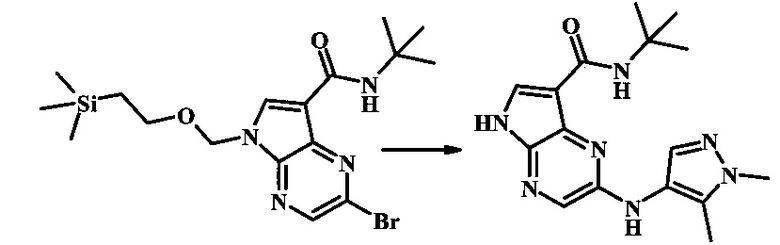
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 1,5-диметил-1Н-пиразол-4-амина дигидрохлорида (96.9 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (118 мг, 1.23 ммоль) в ДМФ (1 мл) и толуол (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (50-100% этилацетат в гексанах) дала N-трет-бутил-2-(1,5-диметил-1Н-пиразол-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (45 мг, 98.3 мкмоль, 28%) в виде желтой смолы. (EI/CI) m/z: 458.3 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(1,5-диметил-1Н-пиразол-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (45 мг, 98.3 мкмоль) в дихлорметане (1.5 мл) добавили трифторуксусную кислоту (224 мг, 152 мкл, 1.97 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (1.5 мл), метанолом (0.75 мл) и гидроксидом аммония (0.2 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь сконцентрировали, затем очистили посредством хроматографии (силикагель, 10-45% раствора 1:4 метанол:дихлорметан в дихлорметане) с получением N-трет-бутил-2-(1,5-диметил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (18 мг, 55.0 мкмоль, 56%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.10 (s, 1 H), 8.39 (s, 1 H), 7.87 (s, 1 H), 7.86 (s, 1 H), 7.85 (d, J=3.3, 1 H), 3.71 (s, 3 H), 2.16 (s, 3 H), 1.30 (s, 9 H); MS (EI/CI) m/z: 328.2 [M+Н].
Пример 17
N-(1-гидрокси-2-метилпропан-2-ил)-2-(1-метил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
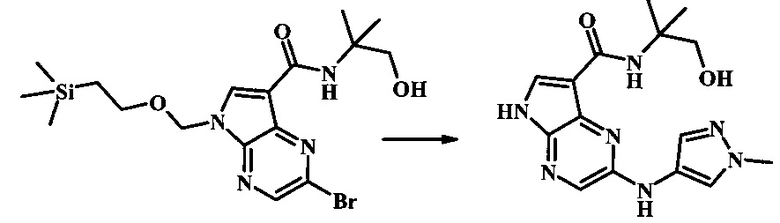
Стадия 1
Смесь 2-бромо-N-(1 -гид рокси-2-метил пропан-2-ил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 338 мкмоль), 1-метил-1Н-пиразол-4-амина гидрохлорида (67.8 мг, 526 мкмоль), BINAP (10.5 мг, 16.9 мкмоль), ацетата палладия (II) (19.0 мг, 84.6 мкмоль) и трет-бутоксида натрия (81.3 мг, 846 мкмоль) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 20-65% раствор 10:89.5:0.5 метанол:дихлорметан:гидроксид аммония в дихлорметане) дала N-(1-гидрокси-2-метил пропан-2-ил)-2-(1-метил-1Н-пиразол-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (28 мг, 60.9 мкмоль, 18%) в виде светло-желтого осадка. (EI/CI) m/z: 460.3 [М+Н].
Стадия 2
К раствору N-(1-гидрокси-2-метилпропан-2-ил)-2-(1-метил-1Н-пиразол-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (26 мг, 56.6 мкмоль) в дихлорметане (1 мл) добавили трифторуксусную кислоту (129 мг, 87.2 мкл, 1.13 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (1 мл), метанолом (0.5 мл) и гидроксидом аммония (0.15 мл) и перемешивали при комнатной температуре в течение 2 ч. Смесь сконцентрировали и тритурировали водой. Осадок собрали посредством фильтрации и промыли водой и эфиром с получением N-(1-гидрокси-2-метилпропан-2-ил)-2-(1-метил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (12 мг, 36.4 мкмоль, 64%) в виде коричневого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.19 (s, 1 Н), 9.16 (s, 1 Н), 8.01 (s, 1 Н), 7.91 (d, J=3.0, 1 H), 7.86 (s, 1H), 7.70 (s, 1 H), 7.49 (s, 1 H), 5.15 (t, J=5.7, 1 H), 3.82 (s, 3 H), 3.57 (d, J=5.9, 2 H), 1.37 (s, 9 H); MS (EI/CI) m/z: 330.2 [M+Н].
Пример 18
N-трет-бутил-2-(3-метилизотиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид

Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 3-метилизотиазол-5-амина гидрохлорида (79.3 мг, 526 мкмоль), BINAP (10.9 мг, 17.5 мкмоль), ацетата палладия (II) (19.7 мг, 87.7 мкмоль) и трет-бутоксида натрия (101 мг, 1.05 ммоль, Экв: 3) в ДМФ (1.2 мл) и толуоле (600 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 50-100% этилацетат в гексанах) дала N-трет-бутил-2-(3-метилизотиазол-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (32 мг, 69.5 мкмоль, 20%) в виде желтого осадка. (EI/CI) m/z. 461.3 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(3-метилизотиазол-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (32 мг, 69.5 мкмоль) в дихлорметане (1.2 мл) добавили трифторуксусную кислоту (158 мг, 107 мкл, 1.39 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (1.2 мл), метанолом (0.6 мл) и гидроксидом аммония (0.15 мл) и перемешивали при комнатной температуре в течение 2 ч. Смесь сконцентрировали и тритурировали водой. Осадок собрали посредством фильтрации и промыли водой и эфиром с получением N-трет-бутил-2-(3-метилизотиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (18 мг, 54.5 мкмоль, 78.4%) в виде коричневого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.55 (s, 1 Н), 11.39 (s, 1 Н), 8.15 (d, J=3.2, 1 H), 8.11 (s, 1 H), 7.37 (s, 1H), 6.70 (s, 1 H), 2.33 (s, 3 H), 1.55 (s, 9 H); MS (EI/CI) m/z. 331.1 [M+Н].
Пример 19
N-трет-бутил-2-(2,4-диметилфениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид

Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (200 мг, 468 мкмоль), 2,4-диметиланилина (85.1 мг, 526 мкмоль), BINAP (14.6 мг, 23.4 мкмоль), ацетата палладия (II) (26.3 мг, 117 мкмоль) и трет-бутоксида натрия (112 мг, 1.17 ммоль) в ДМФ (1 мл) и толуоле (500 мкл) нагревали в микроволновой печи при 140°С в течение 20 мин. Реакционную смесь разбавили водой, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промыли солевым раствором, затем высушили над сульфатом магния. Очистка посредством хроматографии (силикагель, 20-60% этилацетат в гексанах) дала N-трет-бутил-2-(2,4-диметилфениламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (82 мг, 175 мкмоль, 37.5%) в виде коричневой смолы. (EI/CI) m/z: 468.4 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(2,4-диметилфениламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (82 мг, 175 мкмоль) в дихлорметане (2.7 мл) добавили трифторуксусную кислоту (400 мг, 270 мкл, 3.51 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток разбавили дихлорметаном (3 мл), метанолом (1.5 мл) и гидроксидом аммония (0.4 мл) и перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали и тритурировали водой. Осадок собрали посредством фильтрации и промыли водой и эфиром с получением N-трет-бутил-2-(2,4-диметилфениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (7 мг, 20.7 мкмоль, 12%) в виде желтого осадка. 1Н NMR (400 MHz, CDCl3-d) δ ppm: 9.19 (s, 1 Н), 8.12 (d, J=3.0, 1 H), 7.95 (s, 1 H), 7.86 (s, 1H), 7.50 (d, J=7.8, 1 H), 7.15 (s, 1 H), 7.08 (d, J=8.2, 1 H), 6.33 (s, 1 H), 2.38 (s, 3 H), 2.34 (s, 1 H), 1.49 (s, 9 H); MS (EI/CI) m/z: 338.2 [M+Н].
Пример 20
N-трет-бутил-2-(4-(метилсульфонил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
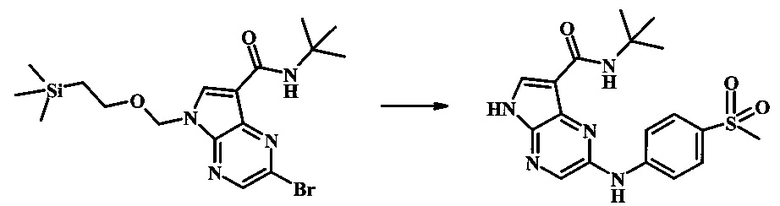
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (125 мг, 292 мкмоль), 4-(метилсульфонил)анилина (75.1 мг, 439 мкмоль), ксантфоса (50.8 мг, 87.7 мкмоль), Pd2(dba)3 (26.8 мг, 29.2 мкмоль) и карбоната цезия (191 мг, 585 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Раствор охладили, отфильтровали через целит, и фильтрат сконцентрировали под вакуумом. Очистка посредством хроматографии (силикагель, 20-75% этилацетат в гексанах) дала N-трет-бутил-2-(4-(метилсульфонил)фениламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (90 мг, 174 мкмоль, 59%) в виде светло-желтого осадка. (EI/CI) m/z. 518.3 [М+Н].
Стадия 2
N-трет-бутил-2-(4-(метилсульфонил)фениламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (90 мг, 174 мкмоль) растворили в дихлорметане (3 мл) и обработали трифторуксусной кислотой (396 мг, 268 мкл, 3.48 ммоль). После перемешивания при комнатной температуре в течение 16 ч, смесь сконцентрировали под вакуумом, затем перерастворили в дихлорметане (3 мл), метаноле (1.5 мл) и гидроксиде аммония (0.35 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали под вакуумом, затем разбавили водой, тритурировали и отфильтровали. Полученный осадок промыли эфиром, затем высушили с получением N-трет-бутил-2-(4-(метилсульфонил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (65 мг, 168 мкмоль, 97%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.55 (s, 1 H), 10.12 (s, 1 H), 8.19 (d, J=2.8, 1 H), 8.16 (s, 1H), 7.95 (d, J=8.8, 2 H), 7.88 (d, J=8.8, 2 H), 7.75 (s, 1 H), 3.22 (s, 3 H), 1.54 (s, 9 H); MS (EI/CI) m/z. 388.2 [M+Н].
Пример 21
N-трет-бутил-2-(метил(пиридин-3-ил)амино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид

Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (125 мг, 292 мкмоль), N-метилпиридин-3-амина (47.4 мг, 439 мкмоль), ксантфоса (50.8 мг, 87.7 мкмоль), Pd2(dba)3 (26.8 мг, 29.2 мкмоль) и карбоната цезия (191 мг, 585 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Раствор охладили и затем отфильтровали через слои целита. Фильтрат сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, этилацетат в гексанах) с получением N-трет-бутил-2-(метил(пиридин-3-ил)амино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (38 мг, 83.6 мкмоль, 29%) в виде желтой смолы. (EI/CI) m/z.455.4 [М+Н].
Стадия 2
N-трет-бутил-2-(метил(пиридин-3-ил)амино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (38 мг, 83.6 мкмоль) растворили в дихлорметане (1.5 мл) и обработали трифторуксусной кислотой (191 мг, 129 мкл, 1.67 ммоль). Через 16 ч, смесь сконцентрировали и остаток перерастворили в дихлорметане (1.5 мл), метаноле (0.75 мл) и гидроксиде аммония (0.2 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали, затем тритурировали водой и отфильтровали с получением осадка. Его промыли эфиром, затем высушили с получением N-трет-бутил-2-(метил(пиридин-3-ил)амино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (16 мг, 49.3 мкмоль, 59%) в виде светло-желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.33 (s, 1 Н), 8.64 (d, J=2.5, 1 H), 8.40 (dd, J=4.7, 1.2, 1 H), 8.05 (d, J=3.1, 1 H), 7.96 (s, 1H), 7.92 (s, 1 H), 7.82 (m, 1 H), 7.46 (m, 1 H), 3.50 (s, 3 H), 1.32 (s, 9 H); MS (EI/CI) m/z: 325.1 [M+Н].
Пример 22
N-трет-бутил-2-(3-метилизоксазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
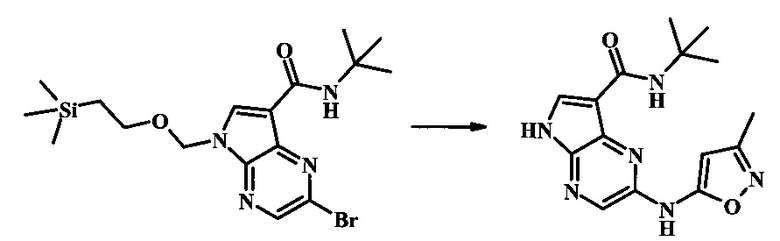
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (125 мг, 292 мкмоль), 3-метилизоксазол-5-амина (43.0 мг, 439 мкмоль), ксантфоса (50.8 мг, 87.7 мкмоль), Pd2(dba)3 (26.8 мг, 29.2 мкмоль) и карбоната цезия (191 мг, 585 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Раствор был охлажден, затем отфильтрован через слои целита. Фильтрат сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, 20 - 75% этилацетат в гексанах) с получением N-трет-бутил-2-(3-метилизоксазол-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (33 мг, 74.2 мкмоль, 25.4%) в виде желтого осадка. (EI/CI) m/z: 455.3 [М+Н].
Стадия 2
N-трет-бутил-2-(3-метилизоксазол-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (33 мг, 74.2 мкмоль) растворили в дихлорметане (1.3 мл) и обработали трифторуксусной кислотой (169 мг, 114 мкл, 1.48 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч, затем сконцентрировали и перерастворили в дихлорметане (1.3 мл), метаноле (0.65 мл) и гидроксиде аммония (0.2 мл) и перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали и разбавили водой, тритурировали и отфильтровали. Полученный осадок промыли эфиром и высушили с получением N-трет-бутил-2-(3-метилизоксазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (16 мг, 45.8 мкмоль, 62%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.53 (s, 1 Н), 11.06 (s, 1 Н), 8.14 (d, J=3.0, 1 H), 8.09 (s, 1 H), 7.57 (s, 1H), 6.18 (s, 1 H), 2.17 (s, 3 H), 1.48 (s, 9 H); MS (EI/CI) m/z: 315.1 [M+Н].
Пример 23
N-трет-бутил-2-(6-(метилсульфонил)пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
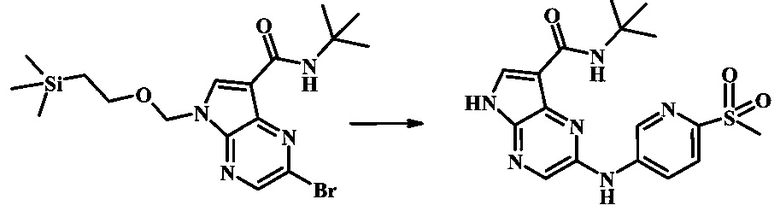
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 6-(метилсульфонил)пиридин-3-амина (90.7 мг, 526 мкмоль), ксантфоса (60.9 мг, 105 мкмоль), Pd2(dba)3 (32.1 мг, 35.1 мкмоль) и карбоната цезия (229 мг, 702 мкмоль) в диоксане (2.4 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Раствор был охлажден, затем отфильтрован через слои целита. Фильтрат сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, 20-95% этилацетат в гексанах) с получением N-трет-бутил-2-(6-(метилсульфонил)пиридин-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (97 мг, 187 мкмоль, 53%) в виде желтого осадка. (EI/CI) m/z: 458.3 [М+Н].
Стадия 2
N-трет-бутил-2-(6-(метилсульфонил)пиридин-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (97 мг, 187 мкмоль) растворили в дихлорметане (2.88 мл) и обработали трифторуксусной кислотой (426 мг, 288 мкл, 3.74 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали затем перерастворили в дихлорметане (3 мл), метаноле (1.5 мл) и гидроксиде аммония (0.5 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали под вакуумом, затем тритурировали водой и отфильтровали. Полученный осадок промыли эфиром, затем высушили с получением N-трет-бутил-2-(6-(метилсульфонил)пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (71 мг, 183 мкмоль, 98%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.51 (s, 1 Н), 10.26 (s, 1 Н), 8.99 (d, J=2.5, 1 H), 8.37 (dd, J=8.4, 2.7, 1 H), 8.14 (s, 1H), 8.11 (s, 1 H), 7.92 (d, J=8.7, 1 H), 7.62 (s, 1 H), 3.21 (s, 3 H), 1.45 (s, 9 H); MS (EI/CI) m/z: 389.2 [M+Н].
Пример 24
N-трет-бутил-2-(3-(1-гидроксиэтил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
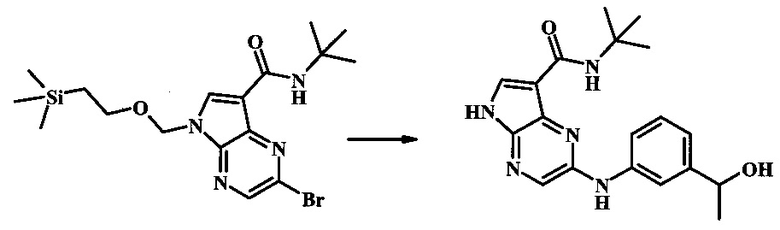
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 1-(3-аминофенил)этанола (72.2 мг, 526 мкмоль), ксантфоса (60.9 мг, 105 мкмоль), Pd2(dba)3 (32.1 мг, 35.1 мкмоль) и карбоната цезия (229 мг, 702 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Раствор был охлажден, затем отфильтрован через слои целита. Фильтрат сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, 40-70% этилацетат в гексанах) с получением N-трет-бутил-2-(3-(1-гидроксиэтил)фениламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (143 мг, 148 мкмоль, 42%) в виде желтого осадка. (EI/CI) m/z: 519.3 [М+Н].
Стадия 2
N-трет-бутил-2-(3-(1-гидроксиэтил)фениламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (143 мг, 296 мкмоль) растворили в дихлорметане (4.5 мл) и обработали трифторуксусной кислотой (674 мг, 456 мкл, 5.91 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали, затем перерастворили в дихлорметане (4 мл), метаноле (3 мл) и гидроксиде аммония (0.7 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь сконцентрировали, затем очистили посредством хроматографии (силикагель, метанол в дихлорметане) с получением N-трет-бутил-2-(3-(1-гидроксиэтил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (55 мг, 156 мкмоль, 53%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.29 (s, 1 Н), 9.44 (s, 1 Н), 8.00 (s, 1 Н), 7.99 (s, 1 Н), 7.98 (d, J=2.8, 1 H), 7.78 (s, 1 H), 7.19 (m, 2 H), 6.91 (d, J=7.5, 1 H), 5.17 (d, J=4.0, 1 H), 4.70 (m, 1 H), 3.38 (q, J=7.2, 1 H), 1.43 (s, 9 H), 1.34 (d. J=6.3, 3 H), 1.09 (t, J=7.2, 1 H); MS (EI/CI) m/z: 354.2 [M+Н].
Пример 25
N-трет-бутил-2-(2-метоксипиримидин-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид

Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 2-метоксипиримидин-5-амина (65.9 мг, 526 мкмоль), ксантфоса (60.9 мг, 105 мкмоль), Pd2(dba)3 (32.1 мг, 35.1 мкмоль) и карбоната цезия (229 мг, 702 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Раствор был охлажден, затем отфильтрован через слои целита. Фильтрат сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, 40-80% этилацетат в гексанах) с получением N-трет-бутил-2-(2-метоксипиримидин-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (99 мг, 210 мкмоль, 60%) в виде желтого осадка. (EI/CI) m/z: 484.3 [М+Н].
Стадия 2
N-трет-бутил-2-(2-метоксипиримидин-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (100 мг, 212 мкмоль) растворили в дихлорметане и обработали трифторуксусной кислотой (484 мг, 327 мкл, 4.24 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали, затем перерастворили в дихлорметане (3 мл), метаноле (1.5 мл) и гидроксиде аммония (0.45 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали под вакуумом, затем тритурировали водой и отфильтровали. Полученный осадок промыли эфиром, затем высушили с получением N-трет-бутил-2-(2-метоксипиримидин-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (64 мг, 187 мкмоль, 88%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.35 (s, 1 Н), 9.48 (s, 1 Н), 8.86 (s, 2 Н), 8.01 (s, 1 Н), 7.97 (s, 1Н), 7.63 (s, 1 Н), 3.88 (s, 3 Н), 1.38 (s, 9 Н); MS (EI/CI) m/z: 342.1 [M+Н].
Пример 26
N-(1-гидрокси-2-метилпропан-2-ил)-2-(пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
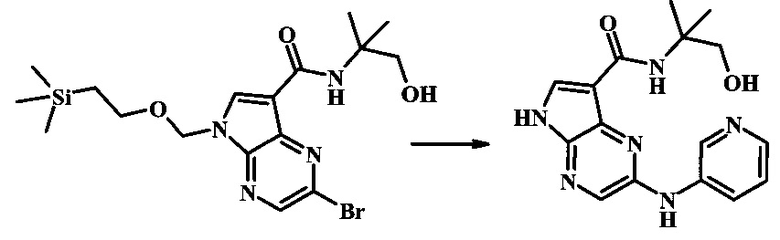
Стадия 1
Смесь 2-бромо-N-(1-гидрокси-2-метил пропан-2-ил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 338 мкмоль), пиридин-3-амина (47.8 мг, 507 мкмоль), ксантфоса (58.7 мг, 101 мкмоль), Pd2(dba)3 (31.0 мг, 33.8 мкмоль) и карбоната цезия (220 мг, 677 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Раствор был охлажден, затем отфильтрован через слои целита. Фильтрат сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, 40-100% градиент этилацетата в гексанах, затем дихлорметан/ метанол) с получением N-(1-гидрокси-2-метилпропан-2-ил)-2-(пиридин-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (32 мг, 59.6 мкмоль, 18%) в виде светло-желтого осадка. (EI/CI) m/z: 472.3 [М+Н].
Стадия 2
N-(1-гидрокси-2-метилпропан-2-ил)-2-(пиридин-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (32 мг, 70.1 мкмоль) растворили в дихлорметане (1.2 мл) и обработали трифторуксусной кислотой (160 мг, 108 мкл, 1.4 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали, затем перерастворили в дихлорметане (1 мл), метаноле (0.5 мл) и гидроксиде аммония (0.2 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали под вакуумом, затем тритурировали водой и отфильтровали. Полученный осадок промыли эфиром, затем высушили с получением N-(1-гидрокси-2-метилпропан-2-ил)-2-(пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (21 мг, 64.3 мкмоль, 92%) в виде светло-желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.39 (s, 1 Н), 9.66 (s, 1 Н), 8.85 (d, J=2.5, 1 H), 8.21 (m, 1 H), 8.16 (dd, J=4.7, 1.6, 1 H), 8.O4 (s, 1 H), 8.03 (s, 1 H), 7.67 (s, 1 H), 7.33 (m, 1 H), 5.05 (t, J=5.6, 1 H), 3.58 (d, J=5.9, 2 H), 1.36 (s, 6 H); MS (EI/CI) m/z: 327.1 [M+Н].
Пример 27
2-(3-(1-Aminoethyl)фениламино)-N-трет-бутил-5Н-пирроло[2,3-b]пиразин-7-карбоксамид

Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), трет-бутил 1-(3-аминофенил)этилкарбамата (124 мг, 526 мкмоль), ксантфоса (60.9 мг, 105 мкмоль), Pd2(dba)3 (32.1 мг, 35.1 мкмоль) и карбоната цезия (229 мг, 702 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Раствор был охлажден, затем отфильтрован через слои целита. Фильтрат сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, 20-40% этилацетат в гексанах) с получением трет-бутил 1-(3-(7-(трет-бутилкарбамоил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-2-иламино)фенил)этилкарбамата (150 мг, 257 мкмоль, 73%) в виде желтой смолы. (EI/CI) m/z. 583.5 [М+Н].
Стадия 2
К раствору трет-бутил 1-(3-(7-(трет-бутилкарбамоил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-2-иламино)фенил)этилкарбамата (150 мг, 257 мкмоль) в дихлорметане (4 мл) добавили трифторуксусную кислоту (587 мг, 397 мкл, 5.15 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали, затем перерастворили в дихлорметане (4 мл), метаноле (2 мл) и гидроксиде аммония (0.7 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь сконцентрировали, затем очистили посредством хроматографии (силикагель, 6-10% метанол/ дихлорметан) с получением 2-(3-(1-аминоэтил)фениламино)-N-трет-бутил-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (64 мг, 182 мкмоль, 71%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 9.44 (s, 1 Н), 8.01 (s, 1 Н), 7.99 (d, J=7.9, 1 H), 7.98 (s, 1 H), 7.77 (s, 1H), 7.22 (t, J=7.8, 1 H), 7.20 (s, 1 H), 6.98 (d, J=7.8, 1 H), 4.05 (q, J=6.1, 1 H), 1.43 (s, 9 H), 1.32 (d, J=6.5, 3 H); MS (EI/CI) m/z. 353.2 [M+Н].
Пример 28
N-трет-бутил-2-(2-(метилсульфонил)пиридин-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
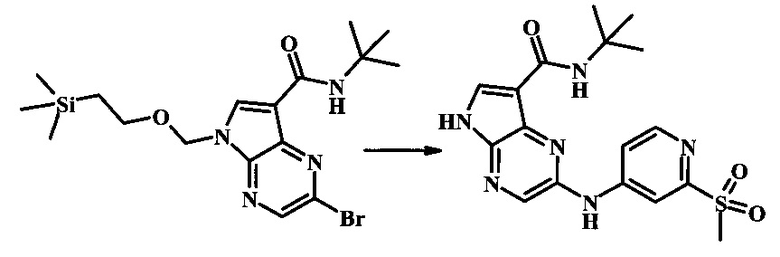
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (120 мг, 281 мкмоль), 2-(метилсульфонил)пиридин-4-амина (55 мг, 319 мкмоль), ксантфоса (48.7 мг, 84.2 мкмоль), Pd2(dba)3 (25.7 мг, 28.1 мкмоль) и карбоната цезия (183 мг, 562 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Раствор был охлажден, затем отфильтрован через слои целита. Фильтрат сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, 35-70% этилацетат в гексанах) с получением N-трет-бутил-2-(2-(метилсульфонил)пиридин-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (127 мг, 245 мкмоль, 87%) в виде светло-желтой смолы. (EI/CI) m/z: 519.1 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(2-(метилсульфонил)пиридин-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (127 мг, 245 мкмоль) в дихлорметане добавили трифторуксусную кислоту (558 мг, 377 мкл, 4.9 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали, затем перерастворили в дихлорметане (4 мл), метаноле (2 мл) и гидроксиде аммония (0.7 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали под вакуумом, затем тритурировали водой и отфильтровали. Полученный осадок промыли эфиром, затем высушили с получением N-трет-бутил-2-(2-(метилсульфонил)пиридин-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (94 мг, 242 мкмоль, 99%) в виде серо-белого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.51 (s, 1 Н), 10.36 (s, 1 Н), 8.39 (d, J=5.9, 1 H), 8.10 (s, 1 H), 8.01 (s, 1H), 7.99 (dd, J=5.3, 2.3, 1 H), 7.83 (d, J=2.1, 1 H), 7.52 (s, 1 H), 3.15 (s, 3 H), 1.35 (s, 9 H); MS (EI/CI) m/z: 389.1 [M+Н].
Пример 29
N-трет-бутил-2-(5-(метилсульфонил)пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид 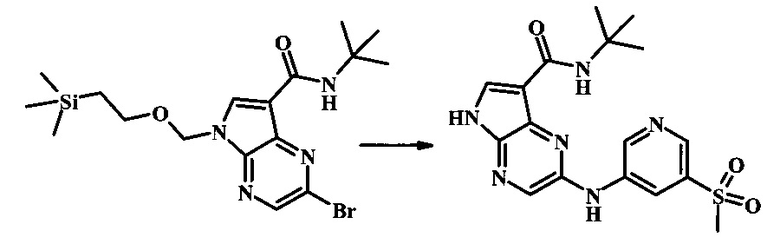
Стадия 1
Смесь 2-амино-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (92 мг, 253 мкмоль), 3-бромо-5-(метилсульфонил)пиридина (77.7 мг, 329 мкмоль), ксантфоса (43.9 мг, 75.9 мкмоль), Pd2(dba)3 (23.2 мг, 25.3 мкмоль) и карбоната цезия (165 мг, 506 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Раствор был охлажден, затем отфильтрован через слои целита. Фильтрат сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, 30-70% этилацетат в гексанах) с получением N-трет-бутил-2-(5-(метилсульфонил)пиридин-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (125 мг, 241 мкмоль, 95%) в виде желтой смолы. (EI/CI) m/z: 519.3 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(5-(метилсульфонил)пиридин-3-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (125 мг, 241 мкмоль) в дихлорметане добавили трифторуксусную кислоту (550 мг, 371 мкл, 4.82 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали, затем перерастворили в дихлорметане (3.7 мл), метаноле (1.6 мл) и гидроксиде аммония (0.5 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали под вакуумом, затем тритурировали водой и отфильтровали. Полученный осадок промыли эфиром, затем высушили с получением N-трет-бутил-2-(5-(метилсульфонил)пиридин-3-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (92 мг, 237 мкмоль, 98%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.38 (s, 1 Н), 9.90 (s, 1 Н), 9.40 (d, J=2.5, 1 H), 8.55 (d, J=1.9, 1 H), 8.06 (t, J=2.2, 1H), 8.01 (s, 1 H), 7.96 (s, 1 H), 7.54 (s, 1 H), 3.22 (s, 3 H), 1.31 (s, 9 H); MS (EI/CI) m/z: 389.1 [M+Н].
Пример 30
N-трет-бутил-2-(1,3-диметил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
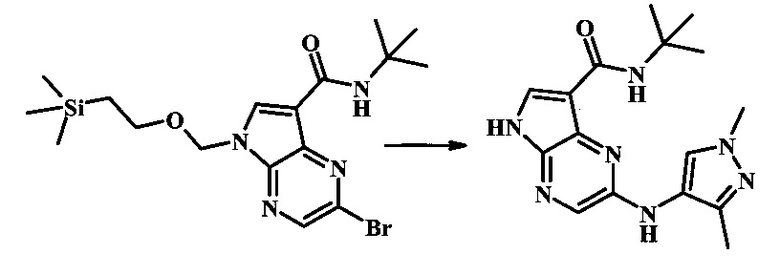
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 1,3-диметил-1Н-пиразол-4-амина (58.5 мг, 526 мкмоль), ксантфоса (60.9 мг, 105 мкмоль), Pd2(dba)3 (32.1 мг, 35.1 мкмоль) и карбоната цезия (229 мг, 702 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Раствор был охлажден, затем отфильтрован через слои целита. Фильтрат сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, 25-100% этилацетат в гексанах) с получением N-трет-бутил-2-(1,3-диметил-1Н-пиразол-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (89 мг, 194 мкмоль, 55%) в виде желтой смолы. (EI/CI) m/z: 458.3 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(1,3-диметил-1 Н-пиразол-4-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (89 мг, 194 мкмоль) в дихлорметане (3 мл) добавили трифторуксусную кислоту (443 мг, 300 мкл, 3.89 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали, затем перерастворили в дихлорметане (3 мл), метаноле (1.5 мл) и гидроксиде аммония (0.45 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь сконцентрировали, затем очистили посредством хроматографии (силикагель, 5-8% метанол в дихлорметане) с получением N-трет-бутил-2-(1,3-диметил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (40 мг, 122 мкмоль, 63%) в виде светло-коричневого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.13 (s, 1 Н), 8.44 (s, 1 Н), 7.91 (s, 1 Н), 7.87 (d, J=3.2, 1 H), 7.82 (s, 1 H), 7.81 (s, 1 H), 3.71 (s, 3 H), 2.09 (s, 3 H), 1.35 (s, 9 H); MS (EI/CI) m/z: 328.1 [M+Н].
Пример 31
N-трет-бутил-2-(3-метил-1,2,4-тиадиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
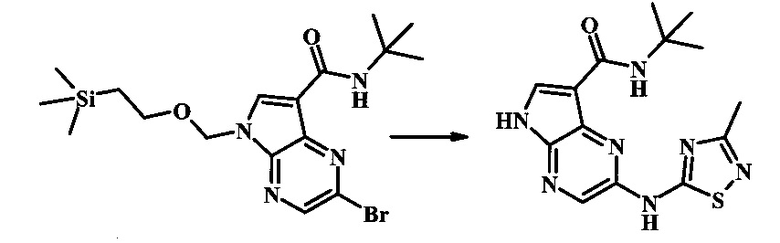
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (150 мг, 351 мкмоль), 3-метил-1,2,4-тиадиазол-5-амина (60.6 мг, 526 мкмоль), ксантфоса (60.9 мг, 105 мкмоль), Pd2(dba)3 (32.1 мг, 35.1 мкмоль) и карбоната цезия (229 мг, 702 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Очистка посредством хроматографии (силикагель, 30-70% этилацетат в гексанах) дала N-трет-бутил-2-(3-метил-1,2,4-тиадиазол-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид (128 мг, 277 мкмоль, 79%) в виде оранжевого осадка. (EI/CI) m/z: 462.3 [М+Н].
Стадия 2
К раствору N-трет-бутил-2-(3-метил-1,2,4-тиадиазол-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (128 мг, 277 мкмоль) в дихлорметане (4.3 мл) добавили трифторуксусную кислоту (632 мг, 427 мкл, 5.55 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали затем перерастворили в дихлорметане (4 мл), метаноле (2 мл) и гидроксиде аммония (0.6 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали под вакуумом, затем тритурировали водой и отфильтровали. Полученный осадок промыли эфиром, затем высушили с получением N-трет-бутил-2-(3-метил-1,2,4-тиадиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (90 мг, 272 мкмоль, 98%) в виде светло-желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.80 (s, 1 Н), 12.57 (s, 1 Н), 8.35 (s, 1 Н), 8.34 (s, 1 Н), 7.34 (s, 1 Н), 2.53 (s, 3 Н), 1.62 (s, 9 Н); MS (EI/CI) m/z: 332.1 [M+Н].
Пример 32
N-трет-бутил-2-(2-метил-6,7-дигидро-2Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
Стадия 1
4,5,6,7-тетрагидро-1Н-пиразоло[4,3-b]пиридин
250 мл бутыль Парра заполнили раствором 1Н-пиразоло[4,3-b]пиридина (2 г, 16.8 ммоль, полученного от J&W PharmLab, LLC) в ТФУ (20.0 мл) и оксидом платины (IV) (381 мг, 1.68 ммоль). Реакционную смесь дважды продули водородом и встряхивали под давлением водорода 55 фунт/кв. дюйм в течение 24 ч. Реакционную смесь отфильтровали через целит, и фильтрационный остаток промыли 5% метанолом в CH2Cl2. Фильтрат сконцентрировали под вакуумом с получением светло-коричневого масла, которое растворили в метаноле (20 мл). Концентрированный раствор гидроксида аммония добавили по каплям до основного рН (рН 8). Смесь сконцентрировали под вакуумом, затем остаток абсорбировали на силикагеле и очистили посредством хроматографии (силикагель, 150 г колонка, 50 мкм от компании Analogix, 0-5% раствор 9:1 метанолтидроксид аммония в дихлорметане, 15 мин) с получением желтого масла. Это масло затвердело после сушки в глубоком вакууме с получением 4,5,6,7-тетрагидро-1Н-пиразоло[4,3-b]пиридина (1.712 г, 13.9 ммоль, 83%) в виде желтого осадка. 1Н NMR (ХЛОРОФОРМ-d) δ: 7.09 (s, 1Н), 3.14-3.25 (m, 2Н), 2.76 (t, J=6.6 Hz, 2H), 1.88-2.02 (m, 2H). MS (EI/CI) m/z: 124.1 [M+H]+.
Стадия 2
трет-бутил 6,7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-карбоксилат

К бесцветному раствору 4,5,6,7-тетрагидро-1Н-пиразоло[4,3-b]пиридина (168 мг, 1.36 ммоль) в метаноле (10 мл) добавили ди-трет-бутилдикарбонат (327 мг, 348 мкл, 1.5 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Растворителя эвапорировали под вакуумом и неочищенный остаток очистили посредством хроматографии (40 г колонка, 50 мкм от компании Analogix, 0-5% 9:1 метанол:гидроксид аммония в CH2Cl2, 20 мин) с получением трет-бутил 6,7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-карбоксилата (188 мг, 62%) в виде бесцветного масла. 1Н NMR (ХЛОРОФОРМ-d) δ: 11.38 (br. s., 1Н), 7.43-8.06 (m, 1Н), 3.69 (br. s., 2Н), 2.75 (t, J=6.2 Hz, 2H), 1.89-2.O2 (m, 2H), 1.43-1.65 (m, 9H). MS (EI/CI) m/z: 224.1 [M+H]+.
Стадия 3
трет-бутил 2-метил-6.7-дигидро-2Н-пиразоло[4,3-b]пиридин-4(5Н)-карбоксилат и трет-бутил 1-метил-6,7-дигидро-1H-пиразоло[4.3-b]пиридин-4(5Н)-карбоксилат
Раствор трет-бутил 6,7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-карбоксилата (188 мг, 842 мкмоль) в N,N-диметилформамиде (5.0 мл) охладили при 0°С при перемешивании, затем медленно добавили раствор трет-бутоксида калия (1M в ТГФ, 1.18 мл, 1.18 ммоль). После перемешивания в течение 30 мин добавили метилйодид (167 мг, 73.7 мкл, 1.18 ммоль). Через 30 мин смесь нагревали до комнатной температуры и перемешивали в течение дополнительных 18 ч. Смесь погасили насыщенным раствором NH4Cl в воде и продукт экстрагировали CH2Cl2 (3×30 мл). Объединенные органические фазы высушили над сульфатом магния, затем сконцентрировали под вакуумом до серо-белого осадка. Осадок растворили в толуоле и очистили с помощью флеш-хроматографии (115 г колонка, 50 мкм силикагель от компании Analogix, 0-20% этилацетат в гексанах, 15 мин) с получением двух продуктов алкилирования. Мене полярный, N-2 продукт алкилирования, трет-бутил 2-метил-6,7-дигидро-2Н-пиразоло[4,3-b]пиридин-4(5Н)-карбоксилат (56 мг, 28%) получили в виде светло-желтого осадка 1Н NMR (ХЛОРОФОРМ-d) δ: 7.22-7.78 (m, 1Н), 3.78 (s, 3Н), 3.56-3.70 (m, 2Н), 2.71 (t, J=6.4 Hz, 2Н), 1.87-2.00 (m, 2Н), 1.46-1.59 (m, 9Н). MS (EI/CI) m/z: 238.1 [М+Н]+. Более полярный, N-1 алкилированный продукт, трет-бутил 1-метил-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5Н)-карбоксилат (55 мг, 28%) получили в виде светло-желтого осадка. 1Н NMR (ХЛОРОФОРМ-d) δ: 7.52 (br. s., 1Н), 3.73 (s, 3Н), 3.66 (br. s., 2Н), 2.65 (t, J=6.4 Hz, 2H), 1.91-2.06 (m, 2H), 1.57 (br. s., 9H). MS (EI/CI) m/z: 238.1 [M+H]+.
Стадия 4
2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-b]пиридин
К перемешанному раствору трет-бутил 2-метил-6,7-дигидро-2Н-пиразоло[4,3-b]пиридин-4(5Н)-карбоксилата (56 мг, 236 мкмоль) в CH2Cl2 (4.00 мл) добавили ТФУ (740 мг, 500 мкл, 6.49 ммоль) при комнатной температуре. Через 18 ч, реакционную смесь сконцентрировали под вакуумом и полученный остаток очистили посредством хроматографии (40 г колонка, 50 мкм силикагель от компании Analogix, 0-5% 9:1 метанол:гидроксид аммония в CH2Cl2, 20 мин) с получением 2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-b]пиридина (25 мг, 77%). 1Н NMR (ХЛОРОФОРМ-d) δ: 6.81 (s, 1Н), 3.75 (s, 3Н), 3.11-3.18(m, 2Н), 3.04 (s, 1Н), 2.73 (t, J=6.4 Hz, 2Н), 1.86-2.00 (m, 2Н).
Стадия 5
N-трет-бутил-2-(2-метил-6,7-дигидро-2Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
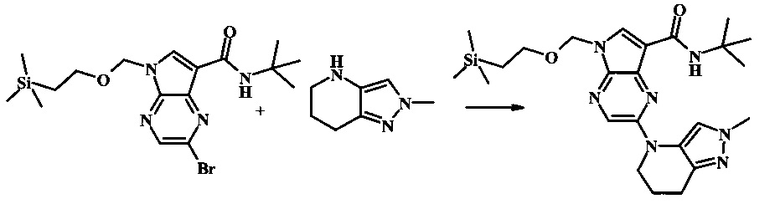
Пробирку для микроволновой печи заполнили смесью 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (77.9 мг, 182 мкмоль), 2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-b]пиридина (25 мг, 182 мкмоль), ксантфоса (31.6 мг, 54.7 мкмоль), Pd2(dba)3 (16.7 мг, 18.2 мкмоль) и Cs2CO3 (119 мг, 364 мкмоль) в диоксане (2 мл). Смесь продули аргоном и затем нагревали при 150°С в течение 20 мин под микроволновым излучением. Смесь разбавили CH2Cl2 и затем отфильтровали через слои целита. Фильтрат сконцентрировали под вакуумом и остаток очистили посредством хроматографии (40 г колонка, 50 мкм силикагель от компании Analogix, 0-5% 9:1 метанолтидроксид аммония в CH2Cl2, 20 мин) с получением N-трет-бутил-2-(2-метил-6,7-дигидро-2Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (87 мг, 99%) в виде желтой пены. 1Н NMR (ХЛОРОФОРМ-d) δ: 8.20 (s, 1Н), 8.15 (s, 1Н), 7.99 (s 1Н), 7.85 (s, 1Н), 5.64 (s, 2Н), 3.91 (s, 3H), 3.50-3.63 (m, 2Н), 2.93 (t, J=6.6 Hz, 2Н), 2.15-2.30 (m, 2Н), 1.53-1.62 (m, 11Н), 0.86-1.05 (m, 2Н), 0.00 (s, 9Н). MS (EI/CI) m/z: 484.0 [М+Н]+.
Стадия 6
N-трет-бутил-2-(2-метил-6.7-дигидро-2Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид

К перемешанному раствору N-трет-бутил-2-(2-метил-6,7-дигидро-2Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (87 мг, 180 мкмоль) в CH2Cl2 (4 мл) добавили ТФУ (740 мг, 500 мкл, 6.49 ммоль). Через 24 ч, смесь сконцентрировали под вакуумом и остаток перерастворили в 5 мл смеси (CH2Cl2/метанол/гидроксид аммония; 60:10:1) и смесь перемешивали при комнатной температуре в течение 24 ч, затем эвапорировали до желтого осадка, который очистили посредством хроматографии (сферический силикагель 20-45 мкм, 23 г, Versaflash Supelco, 0-5% 9:1 метанолтидроксид аммония в CH2Cl2, 30 мин) с получением остатка, который растворили в CH2Cl2, затем преципитировали с помощью циклогексана. Желтый осадок получили декантированием маточной жидкости и высушили его в глубоком вакууме с получением N-трет-бутил-2-(2-метил-6,7-дигидро-2Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (23 мг, 36%). 1Н NMR (ХЛОРОФОРМ-d) δ: 9.46 (br. s., 1Н), 8.08-8.21 (m, 2Н), 7.97 (br. s., 1Н), 7.82 (s, 1Н), 3.88 (m, 5Н), 2.89 (t, J=6.2 Hz, 2H), 2.14-2.28 (m, 2H), 1.56 (s, 9H). MS (EI/CI) m/z: 354.1 [M+H]+.
Пример 33
N-трет-бутил-2-(1-метил-6,7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
Стадия 1
1-метил-4,5,6,7-тетрагидро-1Н-пиразоло[4.3-b]пиридин
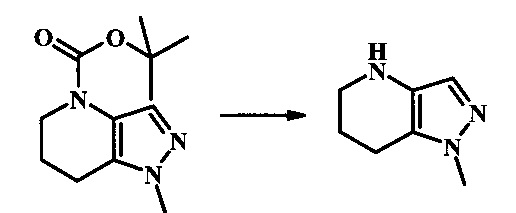
К перемешанному раствору трет-бутил 1-метил-6,7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-карбоксилата (55 мг, 232 мкмоль) в CH2Cl2 (4 мл) добавили ТФУ (740 мг, 500 мкл, 6.49 ммоль). Через 18 ч, реакционную смесь сконцентрировали под вакуумом и полученный остаток очистили посредством хроматографии (40 г колонка, 50 мкм силикагель от компании Analogix, 0-5% 9:1 метанол:гидроксид аммония в CH2Cl2, 20 мин) с получением 1-метил-4,5,6,7-тетрагидро-1Н-пиразоло[4,3-b]пиридина (22 мг, 69%). 1Н NMR (ХЛОРОФОРМ-d) δ: 7.05 (s, 1Н), 3.70 (s, 3H), 3.08-3.21 (m, 2Н), 2.88 (br. s., 1Н), 2.64 (t, J=6.4 Hz, 2H), 1.84-2.01 (m, 2H). MS (EI/CI) m/z: 138.2 [M+H]+.
Стадия 2
N-трет-бутил-2-(1-метил-6.7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид

Пробирку для микроволновой печи заполнили смесью 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (68.5 мг, 160 мкмоль), 1-метил-4,5,6,7-тетрагидро-1Н-пиразоло[4,3-b]пиридина (22 мг, 160 мкмоль), ксантфоса (27.8 мг, 48.1 мкмоль), Pd2(dba)3 (14.7 мг, 16.0 мкмоль), Cs2CO3 (105 мг, 321 мкмоль) и диоксана (2 мл). Реакционную смесь продули аргоном и затем нагревали до 150°С в течение 20 мин под микроволновым излучением. Смесь разбавили CH2Cl2 и затем отфильтровали через слои целита. Фильтрат сконцентрировали под вакуумом и остаток очистили посредством хроматографии (40 г колонка, 50 мкм силикагель от компании Analogix, 0-5% 9:1 метанол хидроксид аммония в CH2Cl2, 20 мин) с получением N-трет-бутил-2-(1-метил-6,7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (77 мг, 99%) в виде желтой пены. 1Н NMR (ХЛОРОФОРМ-d) δ: 8.29 (s, 1Н), 8.14 (s, 1Н), 8.10 (s 1Н), 7.88 (s, 1Н), 5.63 (s, 2Н), 3.93-4.01 (m, 2Н), 3.87 (s, 3H), 3.52-3.63 (m, 2Н), 2.80-2.91 (m, 2Н), 2.16-2.29 (m, 2Н), 1.55-1.61 (m, 11Н), 0.87-1.02 (m, 2Н), 0.00 (s, 9Н) MS (EI/CI) m/z: 484.0 [М+Н]+.
Стадия 3
N-трет-бутил-2-(1-метил-6.7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5Н-пирроло[2.3-b]пиразин-7-карбоксамид
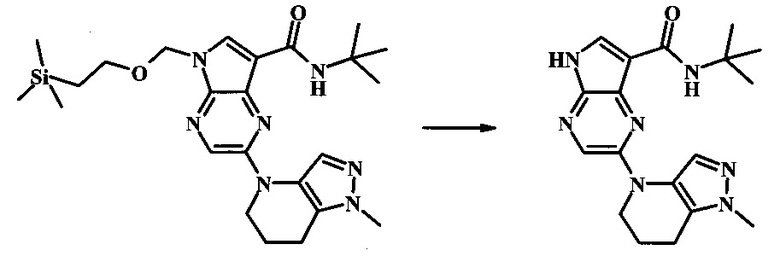
К перемешанному раствору N-трет-бутил-2-(1-метил-6,7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5-((2-(триметилсилил)этокси)метил)-5H-пирроло[2,3-b]пиразин-7-карбоксамида (77 мг, 159 мкмоль) в CH2Cl2 (4 мл) добавили ТФУ (740 мг, 500 мкл, 6.49 ммоль). Через 24 ч растворители эвапорировали и остаток перерастворили в 5 мл смеси (CH2Cl2/метанол/гидроксид аммония; 60:10:1). После перемешивания при комнатной температуре в течение 24 ч, смесь сконцентрировали под вакуумом до желтого осадка, который очистили посредством хроматографии (сферический силикагель 20-45 мкм, 23 г, Versaflash Supelco, 0-5% 9:1 метанол хидроксид аммония в CH2Cl2, 30 мин) с получением продукта, который растворили в CH2Cl2 и преципитировали посредством добавления циклогексана. Образовавшийся желтый осадок разделили посредством декантирования маточной жидкости и затем высушили в глубоком вакууме с получением N-трет-бутил-2-(1-метил-6,7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (33 мг, 59%) в виде желтого осадка. 1Н NMR (ХЛОРОФОРМ-d) δ: 8.95 (br. s., 1Н), 8.24 (s, 1Н), 8.02-8.14 (m, 2Н), 7.85 (s, 1Н), 3.88-3.99 (m, 2Н), 3.83 (s, 3H), 2.81 (t, J=6.6 Hz, 2H), 2.08-2.24 (m, 2H), 1.55 (s, 9H). MS (EI/CI) m/z: 354.1 [M+H]+.
Пример 34
3-метил-2-(1-метил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид
Стадия 1
3,5-Дибром-6-метил-пиразин-2-иламин
3,5-Дибром-6-метил-пиразин-2-иламин получили посредством бромирования 6-метил-пиразин-2-иламина, в соответствии со способом, описанным у Sato (Sato, N., J. Het. Chem., 1980, 17, 143-147). 1H NMR (400 MHz, DMSO-d6) δ ppm 2.34 (s, 3 H), 6.86 (br s, 2 H, -NH2); MS (EI/CI) m/z: 266 [M+Н].
Стадия 2
5-бромо-6-метил-3-триметилсиланилэтинил-пиразин-2-иламин
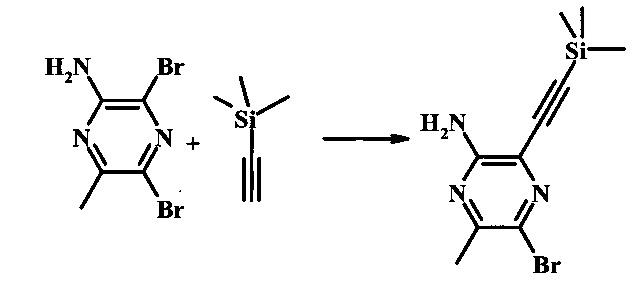
Раствор триметилсилилацетилена (1.03 г, 29.4 ммоль) в тетрагидрофуране (2 мл) добавили по каплям к охлажденной (водяная баня при приблизительно 15°С) смеси 3,5-дибром-6-метил-пиразин-2-иламина (7.48 г, 28 ммоль), йодида меди (I) (1.07 г, 5.6 ммоль), бис-трифенилфосфинпалладия дихлорида (1.97 г, 2.3 ммоль), триэтиламина (3.542 г, 35 ммоль) и тетрагидрофурана (75 мл). Смесь перемешивали в атмосфере аргона в течение 16 часов, в течение этого времени реакционной смеси дали нагреться до комнатной температуры. Реакционную смесь влили в воду и экстрагировали гексанами-этилацетатом (2:1). Органический слой промыли последовательно насыщенным раствором хлорида аммония, водой, и затем снова насыщенным раствором хлорида аммония, водой, затем высушили над безводным сульфатом магния, обработали активированным улем, отфильтровали через целит и сконцентрировали при пониженном давлении. Очистка посредством хроматографии (силикагель, 20% этилацетат в гексанах) дала (6.19 г, 77%) 5-бромо-6-метил-3-триметилсиланилэтинил-пиразин-2-иламин в виде серо-белого осадка. 1Н NMR (400 MHz, DMSO-d6) δ ppm 0.26 (s, 9 Н), 2.39 (s, 3 Н), 6.71 (br s, 2Н, -NH2).
Стадия 3
2-бромо-3-метил-5Н-пирроло[2,3-b]пиразин
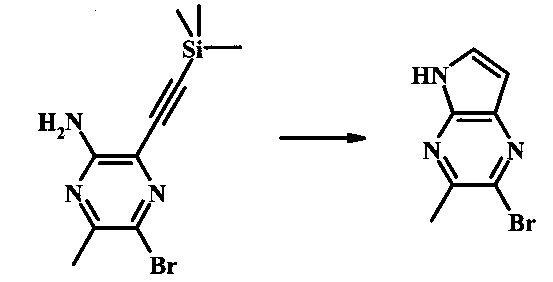
Раствор 5-бромо-6-метил-3-триметилсиланилэтинил-пиразин-2-иламина (6.19 г, 21.8 ммоль) в тетрагидрофуране (15 мл) добавили по каплям в атмосфере аргона в раствор тетрагидрофурана (24 мл) и 1 М трет-бутоксида калия в тетрагидрофуране (24 мл, 24 ммоль), который охладили в ванной при -30°С. Смесь перемешивали в течение 1 ч, охлаждающую ванну удалили и смесь перемешивали при комнатной температуре. Через 19 ч смесь охладили в ванне со льдом и погасили посредством добавления 0.5 М соляной кислоты (48 мл). Затем смесь влили в воду и экстрагировали дважды этилацетатом. Объединенные этилацетатные слои промыли водой, высушили над безводным сульфатом магния, обработали активированным углем, отфильтровали через целит и сконцентрировали при пониженном давлении с получением оранжевого осадка (3.83 г), которые представлял собой в основном 2-бромо-3-метил-5Н-пирроло[2,3-b]пиразин вместе с небольшим количеством дасиалированного исходного соединения. Этот материал использовали на следующей стадии без дополнительной очистки. ЯМР данные получили от вещества, полученного в отдельной реакции и очищенного посредством хроматографии (силикагель, гексаны-этилацетат 70:30). 1Н NMR (400 MHz, DMSO-d6) δ ppm 2.64 (s, 3 Н), 6.57 (d, d, J=1.5, 3.3 Hz, 1 H), 7.84 (t, J=3.3 Hz, 1 H), 12.12 (brs, 1 H, NH)
MS (EI/CI) m/z: 212 [M+Н].
Стадии 4
2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин
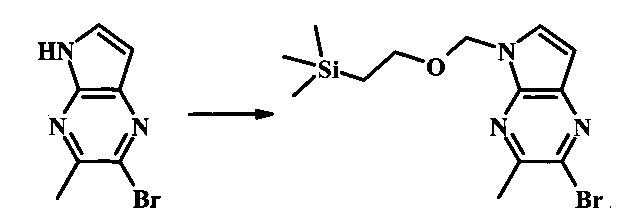
К смеси неочищенного 2-бромо-3-метил-5Н-пирроло[2,3-b]пиразина (3.83 г, 18 ммоль), полученного выше, и N, N-диметилформамида (25 мл), в атмосфере аргона и охлаждении в ванне со льдом, добавили 95% масляную дисперсию гидрида натрия (0.912 г, 36 ммоль) (выделение газа). Смесь перемешивали в течение 15 минут, и затем добавили по каплям (2-хлорметокси-этил)-триметилсилан (3.61 г, 22 ммоль) в N,N-диметилформамиде (3 мл). Через 10 минут охлаждающую ванну удалили и смесь перемешивали в течение дополнительных 3 ½ часов при комнатной температуре. Смесь охладили в ванне со льдом, погасили посредством добавления воды, и затем влили в воду и экстрагировали три раза эфиром. Объединенные эфирные экстракты промыли дважды водой, высушили над безводным сульфатом магния, отфильтровали и сконцентрировали при пониженном давлении. Очистка посредством хроматографии (силикагель, 15% этилацетат в гексанах) дала 2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b] пиразин (3.47 г, 56%) в виде янтарного осадка.
1Н NMR (400 MHz, DMSO-d6) δ ppm -0.11 (s, 9 Н), 0.82 (m, 2 Н), 2.67 (s, 3 Н), 3.51 (m, 2 Н), 5.59 (s, 2 Н), 6.67 (d, J=3.5 Hz, 1 H), 8.01 (d, J=3.5 Hz, 1 H).
Стадия 5
2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбальдегид
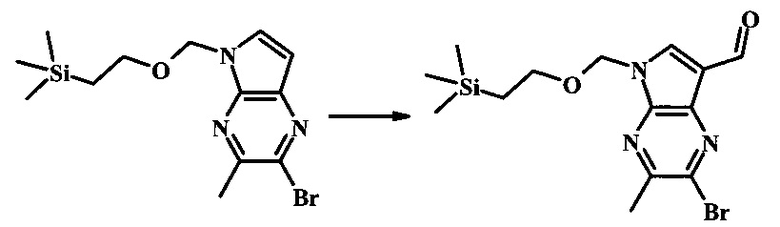
Трифторуксусный ангидрид (10.2 мл, 60.8 ммоль) добавили по каплям, с охлаждением (ледяная ванна) к N, N- диметилформамиду (24 мл) в атмосфере аргона. Охлаждающую ванну удалили в течение 15 мин для облегчения перемешивания и затем возвратили обратно. Раствор 2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b] пиразина (3.47 г, 10.1 ммоль) в дихлорметане (5 мл) добавили по каплям. Охлаждающую ванну удалили; смесь перемешивали в течение 15 мин, затем поместили в ванну при 45°С и перемешивали в течение 16 ч. Смесь охладили, затем влили в воду со льдом water (200 мл), содержащую карбонат натрия (13 г) и экстрагировали четыре раза эфиром. Объединенные эфирные экстракты дважды промыли водой, высушили над безводным сульфатом магния, отфильтровали и сконцентрировали при пониженном давлении. Очистка посредством хроматографии (силикагель, 20% этилацетат в гексанах) дала 2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b] пиразин-7-карбальдегид (3.17 г, 84%) в виде светло-желтого осадка. 1Н NMR (400 MHz, DMSO-d6) δ ppm -0.09 (s, 9 Н), 0.85 (t, J=8.0 Hz, 2 H), 2.71 (s, 3 H), 3.58 (t, J=8.0 Hz, 2 H), 5.68 (s, 2 H), 8.87 (s, 1 H), 10.04 (s, 1 H).
Стадия 6
2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновая кислота
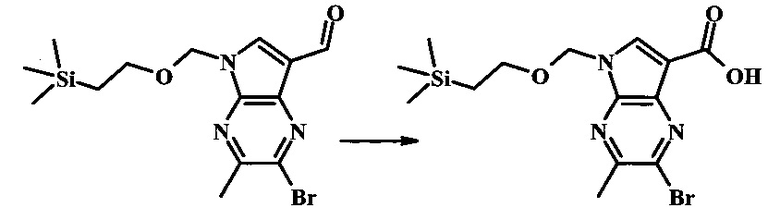
К перемешанной смеси 2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b] пиразин-7-карбальдегида (3.17 г, 8.55 ммоль), трет-бутанола (10 мл), тетрагидрофурана (10 мл), и воды (10 мл) добавили сульфаминовую кислоту (3.32 г, 34 ммоль) и затем раствор дигидрофосфата калия (8.85 г, 65 ммоль) и хлорида натрия (1.93 г, 21 ммоль) в воде (42 мл). Через 1 ч смесь разбавили этилацетатом, промыли один раз солевым раствором, высушили над безводным сульфатом магния, отфильтровали и сконцентрировали при пониженном давлении с получением 2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b] пиразин-7-карбоновой кислоты (3.02 г, 91%) в виде светло-желтого осадка. Этот материал использовали на следующей стадии без дополнительной очистки. 1Н NMR (400 MHz, DMSO-d6) δ ppm -0.10 (s, 9 Н), 0.83 (t, J=8.0 Hz, 2 H), 2.69 (s, 3 H), 3.55 (t, J=8.0 Hz, 2 H), 5.64 (s, 2 H), 8.64 (s, 1 H), 12.46 (br s, J=9.06 Hz, 1 H, -COOH).
Стадия 7
2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид
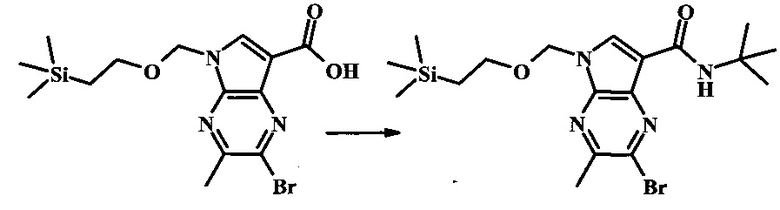
Смесь 2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b] пиразин-7-карбоновой кислоты (1.16 г, 3 ммоль), HBTU [1-[бис(диметиламино)метилен]-1Н-бензотриазола 3-оксида, гексафторфосфата (1-) (1:1) (1.48 г, 3.9 ммоль), N,N-диметилформамида (12 мл) и триэтиламина (0.61 г, 6 ммоль) перемешивали при комнатной температуре в течение 1 ч, и затем добавили трет-бутиламин (0.63 мл, 6 ммоль). Смесь перемешивали дополнительный час и затем разбавили этилацетатом, промыли один раз 0.5 М карбонатом натрия, дважды водой, один раз солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и сконцентрировали при пониженном давлении. Очистка посредством хроматографии (силикагель, 20% этилацетат в гексанах) дала 2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b] пиразин-7-карбоновой кислоты трет-бутиламид (1.22 г, 92%) в виде белого кристаллического осадка. 1Н NMR (400 MHz, DMSO-d6) δ ppm -0.09 (s, 9 Н), 0.83 (t, J=8.2 Hz, 2 H), 1.43 (s, 9 H), 2.71 (s, 3 H), 3.54 (t, J=8.2 Hz, 2 H), 5.64 (s, 2 H), 7.64 (s, 1 H), 8.49 (s, 1 H).
MS (EI/CI) m/z: 441 [M+H]
Стадия 8
3-метил-2-(1-метил-1Н-пиразол-4-иламино)-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид
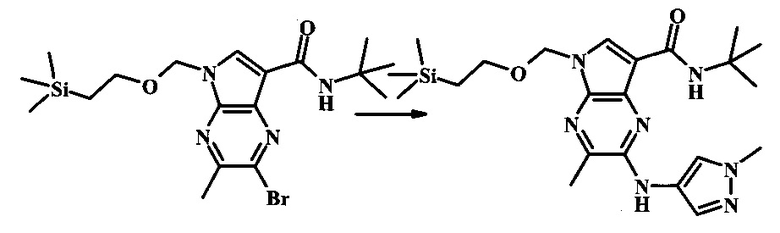
Смесь 2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b] пиразин-7-карбоновой кислоты трет-бутиламида (0.110 г, 0.25 ммоль), 1-метил-1Н-пиразол-4-иламина 1.35-HCl соли (0.073 г, 0.5 ммоль), трис(дибензилиденацетон)дипалладия (0.011 г, 0.0125 ммоль), рацемического BINAP [rac-2,2'-бис(дифенилфосфино)-1,1'-бинафтила] (0.023 г, 0.038 ммоль) и трет-бутоксида натрия (0.094 г, 0.98 ммоль) в толуоле (2 ½ мл), в атмосфере аргона нагревали и перемешивали в закрытом сосуде при 110°С в течение 18.5 ч. Охлажденную реакционную смесь разбавили этилацетатом, промыли один раз солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и сконцентрировали при пониженном давлении. Очистка посредством хроматографии (силикагель, 70% этилацетат в гексанах) дала 0.104 g слегка загрязненного 3-метил-2-(1-метил-1Н-пиразол-4-иламино)-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида в виде желто-коричневого осадка. Этот материал объединили с 0.022 г частично очищенного продукта, полученного в отдельной реакции связывания с низким выходом (ацетат палладия использовался вместо трис(дибензилиденацетон)дипалладия), и очистили посредством хроматографии (силикагель, этилацетат) с получением 3-метил-2-(1-метил-1Н-пиразол-4-иламино)-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида (0.112 г) в виде светло-желтого осадка. 1Н NMR (400 MHz, DMSO-d6) δ ppm -0.08 (s, 9 Н), 0.83 (t, J=8.2 Hz, 2 H), 1.38 (s, 9 H), 2.54 (s, 3 H), 3.51 (t, J=8.2 Hz, 2 H), 3.80 (s, 3 H), 5.55 (s, 2 H), 7.61 (s, 1 H), 7.68 (s, 1 H), 7.85 (s, 1 H), 8.01 (s, 1 H), 8.19 (s, 1 H); MS (EI/CI) m/z: 458 [M+Н].
Стадия 9
3-метил-2-(1-метил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид
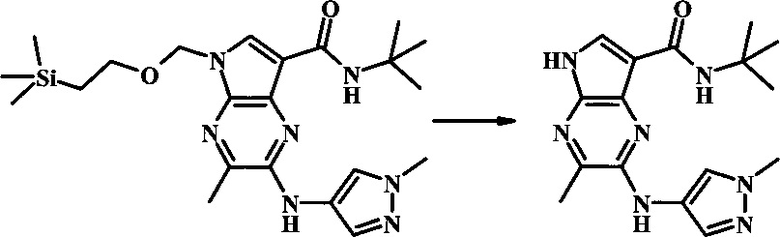
Смесь 3-метил-2-(1-метил-1Н-пиразол-4-иламино)-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида (0.110 г, 0.24 ммоль), дихлорметана (2.5 мл) и трифторуксусной кислоты (0.5 мл) оставили при комнатной температуре на 13 ч и затем сконцентрировали при пониженном давлении. Желтый остаток перемешивали с дихлорметаном-метанол-гидроксид аммония (50:10:1, 10 мл) в течение 1 ½ ч и затем сконцентрировали при пониженном давлении. Остаток растворили в этилацетате и промыли последовательно 0.5 М карбонатом натрия, водой и затем солевым раствором. Этилацетатный слой высушили над безводным сульфатом натрия, отфильтровали и сконцентрировали при пониженном давлении с получением 0.082 г светло-желтого осадка. По данным ЯМР конверсия прошла не полностью, поэтому смесь перемешивали дополнительные 2 часа с дихлорметаном-метанол-гидроксид аммония (5:5:1) в течение 1.5 часов и затем разбавили этилацетатом и промыли один раз водой и один раз солевым раствором. Этилацетатный слой высушили над безводным сульфатом натрия, отфильтровали и сконцентрировали при пониженном давлении с получением 0.075 г светло-желтого осадка. Перекристаллизация из ацетонитрил-метанола дала 3-метил-2-(1-метил-1Н-пиразол-4-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид (0.047 г, 41%) в виде светло-желтого кристаллического осадка. 1Н NMR (400 MHz, DMSO-d6) δ ppm 1.38 (s, 9 Н), 2.52 (s, 3 Н), 3.80 (s, 3 Н), 7.60 (s, 1 Н), 7.68 (s, 1 Н), 7.79 (s, 1 Н), 7.84 (s, 1 Н), 8.07 (s, 1 Н, -NH), 11.97 (br s, 1 H, -NH). MS (EI/CI) m/z: 328 [M+Н].
Пример 35
2-(3-Метансульфонил-фениламино)-3-метил-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид
Стадия 1
2-(3-Метансульфонил-фениламино)-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид

Смесь 2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b] пиразин-7-карбоновой кислоты трет-бутиламида (0.110 г, 0.25 ммоль), 3-метансульфонил-фениламина (0.086 г, 0.5 ммоль), трис(дибензилиденацетон)дипалладия (0.011 г, 0.0125 ммоль), рацемического BINAP [rac-2,2'-бис(дифенилфосфино)-1,1'-бинафтила] (0.023 г, 0.038 ммоль) и трет-бутоксида натрия (0.060 г, 0.63 ммоль) в толуоле (2.5 мл), в атмосфере аргона, нагревали и перемешивали в закрытом сосуде при 110°С в течение 21.5 ч. Охлажденную реакционную смесь разбавили этилацетатом, промыли один раз солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и сконцентрировали при пониженном давлении. Очистка посредством хроматографии (силикагель, 75% этилацетат в гексанах) дала 0.107 г слегка загрязненного 2-(3-метансульфонил-фениламино)-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида в виде желтого стеклообразного вещества. Реакцию повторили, как указано выше, и объединенные слегка загрязненные продукты очистили дополнительно посредством хроматографии (силикагель, 33% этилацетат в гексанах) с получением 2-(3-метансульфонил-фениламино)-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида (0.169 г, 64%) в виде светло-желтого осадка. 1Н NMR (400 MHz, DMSO-d6) δ ppm -0.07 (s, 9 H), 0.85 (t, J=8.2 Hz, 2 H), 1.30 (s, 9 H), 2.64 (s, 3 H), 3.19 (s, 3 H), 3.54 (t, J=8.2 Hz, 2 H), 5.59 (s, 2 H), 7.49 (m, 1 H), 7.56 (m, 1 H), 7.62 (s, 1 H), 7.96 (m, 1 H), 8.05 (m, 1 H), 8.16 (s, 1 H), 8.74 (s, 1 H). MS (EI/CI) m/z. 532 [M+Н].
Стадия 2
2-(3-Метансульфонил-фениламино)-3-метил-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид

Смесь 2-(3-метансульфонил-фениламино)-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида (0.167 г, 0.31 ммоль), дихлорметана (2.5 мл) и трифторуксусной кислоты (0.5 мл) оставили при комнатной температуре на 14 ч и затем сконцентрировали при пониженном давлении. Желтый остаток перемешивали с дихлорметаном-метанол-гидроксид аммония (3:3:1, 7 мл) в течение 3 ч. Реакционную смесь затем разбавили этилацетатом, промыли один раз солевым раствором, высушили над безводным сульфатом натрия и сконцентрировали при пониженном давлении, с получением 0.126 г светло-желтого осадка. Продукт перекристаллизовали из ацетонитрил-метанола с получением 2-(3-метансульфонил-фениламино)-3-метил-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида (0.063 г, 50%) в виде светло-желтого кристаллического осадка. 1Н NMR (400 MHz, DMSO-d6) δ ppm 1.30 (s, 9 Н), 2.61 (s, 3 Н), 3.19 (s, 3 Н), 7.46 (m, 1 Н), 7.54 (m, 1 Н), 7.64 (s, 1 Н), 7.94 (m, 1 Н), 7.96 (s, 1 Н), 8.04 (m, 1 Н), 8.66 (s, 1 Н), 12.21 (br s, 1 H). MS (EI/CI) m/z: 402 [M+Н].
Пример 36
3-метил-2-(3-метил-изотиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид
Стадия 1
3-метил-2-(3-метил-изотиазол-5-иламино)-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид
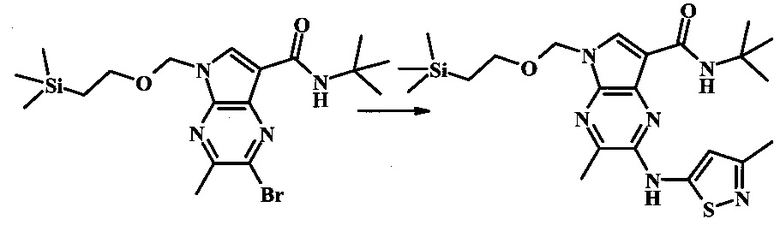
Смесь 2-бромо-3-метил-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b] пиразин-7-карбоновой кислоты трет-бутиламида (0.221 г, 0.5 ммоль), 3-метил-изотиазол-5-иламина гидрохлорида (0.151 г, 1 ммоль), трис(дибензилиденацетон)дипалладия (0.023 г, 0.025 ммоль), рацемического BINAP [rас-2,2'-бис(дифенилфосфино)-1,1'-бинафтила] (0.047 г, 0.075 ммоль) и трет-бутоксида натрия (0.144 г, 1.5 ммоль) в толуоле (3.5 мл), в атмосфере аргона, нагревали и перемешивали в закрытом сосуде при 110°С в течение 19 ч. Охлажденную реакционную смесь разбавили этилацетатом, промыли один раз солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и сконцентрировали при пониженном давлении. Очистка посредством хроматографии (силикагель, 75% этилацетат в гексанах, затем повторно 50% этилацетат в гексанах, и снова 40% этилацетат в гексанах) дала 3-метил-2-(3-метил-изотиазол-5-иламино)-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид (0.101 г, 43%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d6) δ ppm -0.08 (s, 9 Н), 0.84 (t, J=8.0 Hz, 2 H), 1.55 (s, 9 H), 2.35 (s, 3 H), 2.67 (s, 3 H), 3.53 (t, J=8.0 Hz, 2 H), 5.61 (s, 2 H), 6.94 (s, 1 H), 7.34 (s, 1 H), 8.25 (s, 1 H), 10.49 (s, 1 H). MS (EI/CI) m/z: 475 [M+Н].
Стадия 2
3-метил-2-(3-метил-изотиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид
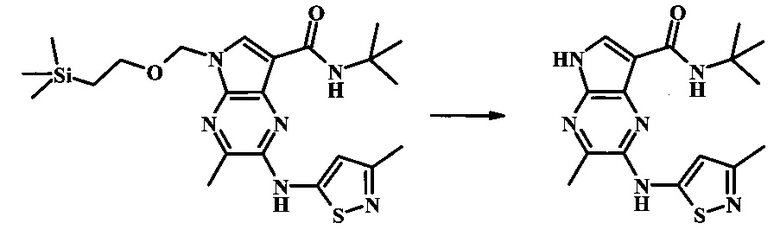
Смесь 3-метил-2-(3-метил-изотиазол-5-иламино)-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида (0.099 г, 0.21 ммоль), дихлорметана (2.5 мл) и трифторуксусной кислоты (0.5 мл) оставили при комнатной температуре на 14 ч и затем сконцентрировали при пониженном давлении. Желтый остаток перемешивали с дихлорметаном-метанол-гидроксид аммония (3:3:1, 7 мл) в течение 3 ч. Реакционную смесь затем разбавили этилацетатом, промыли один раз солевым раствором, высушили над безводным сульфатом натрия и сконцентрировали при пониженном давлении с получением 0.079 г светло-желтого осадка. Продукт перекристаллизовали из ацетонитрил-метанола с получением 3-метил-2-(3-метил-изотиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида (0.046 г, 64%) в виде светло-желтого кристаллического осадка. 1Н NMR (400 MHz, DMSO-d6) δ ppm 1.55 (s, 9 Н), 2.34 (s, 3 Н), 2.65 (s, 3 Н), 6.92 (s, 1 Н), 7.32 (s, 1 Н), 8.04 (d, J=1.5 Hz, 1 H), 10.41 (s, 1 H), 12.36 (s, 1 H). MS (EI/CI) m/z: 345 [M+Н].
Пример 37
2-Фениламино(D5)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид
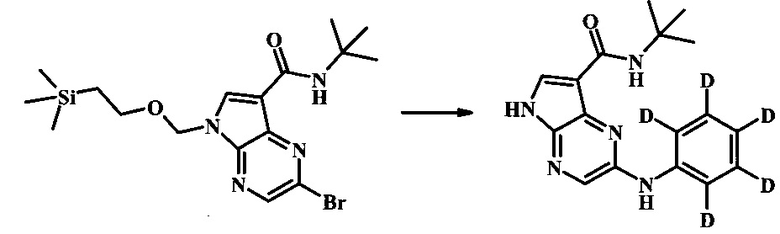
Стадия 1
Смесь 2-бромо-N-трет-бутил-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (125 мг, 292 мкмоль), анилина-D5 (43.1 мг, 439 мкмоль), ксантфоса (50.8 мг, 87.7 мкмоль), Pd2(dba)3 (26.8 мг, 29.2 мкмоль) и карбоната цезия (191 мг, 585 мкмоль) в диоксане (2 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Реакционную смесь отфильтровали через слои целита, затем фильтрат сконцентрировали под вакуумом и очистили посредством хроматографии (силикагель, 20-60% этилацетат в гексанах) с получением 2-фениламино(D5)-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида (40 мг, 90.0 мкмоль, 31%) в виде светло-желтого осадка. 1Н NMR (400 MHz, CDCl3-d) δ ppm: 8.15 (s, 1 Н), 7.99 (s, 1 Н), 7.93 (s, 1 Н), 6.70 (s, 1 Н), 5.63 (s, 2 Н), 3.57 (t, J=8.2, 2 H), 1.57 (s, 9 H), 0.96 (t, J=8.1, 2 H), 0.01 (s, 9 H).
Стадия 2
К раствору 2-фениламино(05)-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида (40 мг, 90.0 мкмоль) в дихлорметане (1.5 мл) добавили трифторуксусную кислоту (205 мг, 139 мкл, 1.8 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали, затем перерастворили в дихлорметане (1.5 мл), метаноле (0.75 мл) и гидроксиде аммония (0.2 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали под вакуумом, затем тритурировали водой и отфильтровали. Полученный осадок промыли эфиром, затем высушили с получением 2-фениламино(D5)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламида (24 мг, 76.3 мкмоль, 85%) в виде желтого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.31 (s, 1 Н), 9.47 (s, 1 Н), 8.00 (s, 1 Н), 7.99 (s, 1 Н), 7.76 (s, 1 Н), 1.44 (s, 9 Н); MS (EI/CI) m/z: 315.1 [M+Н].
Пример 38
N-трет-бутил-3-хлор-2-(3-метилизотиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
2-бромо-N-трет-бутил-3-хлор-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
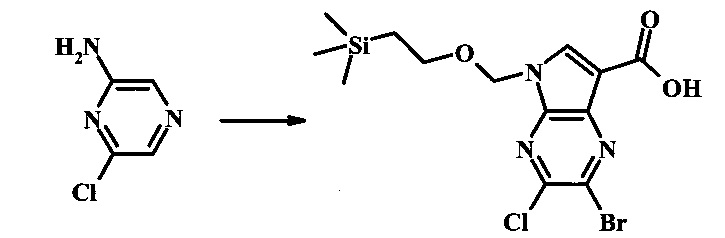
Стадия 1
3,5-Дибром-6-хлор-пиразин-2-иламин

К раствору 6-хлор-пиразин-2-иламина (5.0 г, 38.8 ммоль) в хлороформе (194 мл) добавили N-бромсукцинимид (20.7 г, 116 ммоль) в атмосфере азота и реакционную смесь перемешивали при комнатной температуре в течение 5 ч. Получившуюся смесь влили в водный раствор K2CO3 (300 мл) и экстрагировали дихлорметаном (200 мл × 4). Объединенные органические экстракты высушили и очистили посредством хроматографии (силикагель, 10-20% этилацетат в гексанах) с получением 3,5-дибром-6-хлор-пиразин-2-иламина (4.2 г, 38%) в виде желтого осадка. LC-MS: 286.0 (М-Н).
Стадия 2
5-бромо-6-хлор-3-триметилсиланилэтинил-пиразин-2-иламин
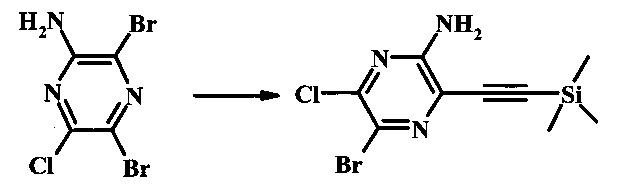
К перемешанному раствору 3,5-дибром-6-хлор-пиразин-2-иламина (5.0 г, 17.42 ммоль) в безводном ТГФ (40 мл) добавили триэтиламин (3.52 г, 34.84 ммоль) и Cul (0.333 г, 1.74 ммоль). Смесь дегазировали с помощью барботирования аргона в течение 10 мин, затем откачали и снова заполнили аргоном. Ее основательно продули аргоном в течение 20 мин и добавили Pd(PPh3)2Cl2 (0.355 г, 0.52 ммоль). Реакционную смесь охладили до -5°С, очень медленно добавили TMS-ацетилен (1.90 г, 19.16 ммоль) и температуру медленно повысили до 15°С в течение 3 ч. Реакционную смесь разбавили водой (30 мл) и экстрагировали этилацетатом (100 мл × 3). Объединенные органические слои сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, 5% этилацетат в гексанах) с получением 5-бромо-6-хлор-3-триметилсиланилэтинил-пиразин-2-иламина (4.0 г, 75%) в виде желтого осадка. LC-MS: 304.0 (М+Н).
Стадия 3
2-бромо-3-хлор-5Н-пирроло[2,3-b]пиразин
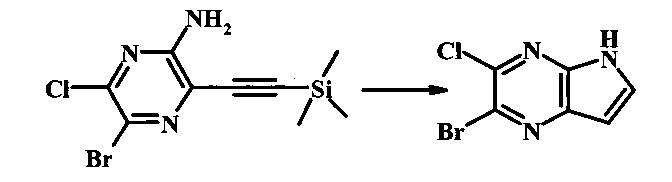
К перемешанному раствору 5-бромо-6-хлор-3-триметилсиланилэтинил-пиразин-2-иламина (2.0 г, 6.58 ммоль) в ТГФ (28 мл) медленно добавили 1 М раствор KOtBu в ТГФ (7.24 мл, 7.24 ммоль) при 0°С в атмосфере азота. Реакционную смесь перемешивали при 0°С в течение 10 мин, и затем нагрели до комнатной температуры. Через 2 ч смесь сконцентрировали под вакуумом, и полученный остаток суспендировали в воде (7.5 мл) и насыщенном водном NaHCO3 (7.5 мл) и перемешивали в течение ночи при комнатной температуре. Осадок отфильтровали, промыли водой и гексанами, затем высушили в печи при 80°С с получением 2-бромо-3-хлор-5Н-пирроло[2,3-b]пиразина (1.64 г) в виде коричневого осадка. LC-MS: 232 (М-Н). Его использовали сразу без дополнительной очистки.
Стадия 4
2-бромо-3-хлор-пирроло[2,3-b]пиразин-5-карбоновой кислоты трет-бутиловый эфир

К перемешанной суспензии 2-бромо-3-хлор-5Н-пирроло[2,3-b]пиразина (1.52 г, 6.55 ммоль) в ацетонитриле (25 мл) добавили DMAP (0.008 г, 0.07 ммоль). Через 5 мин, добавили ди-трет-бутилдикарбонат (1.82 мл, 8.52 ммоль) и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь сконцентрировали под вакуумом и полученный остаток затем очистили посредством хроматографии (силикагель, 7% этилацетат в гексанах) с получением 2-бромо-3-хлор-пирроло[2,3-b]пиразин-5-карбоновой кислоты трет-бутилового эфира (1.0 г, 46% за две стадии) в виде желтого осадка. LC-MS: 334.2 (М+Н).
Стадия 5
(2-бромо-3-хлор-7-гидроксиметил-пирроло[2,3-b]пиразин-5-ил)-метанол
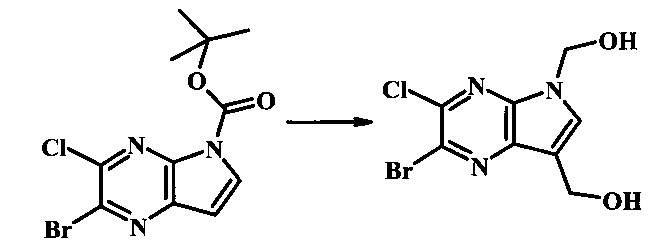
К перемешанному раствору 2-бромо-3-хлор-пирроло[2,3-b]пиразин-5-карбоновой кислоты трет-бутилового эфира (6.0 г, 18.0 ммоль) в 1,4-диоксане (100 мл) добавили метаналь (5.4 г, 180.4 ммоль) и 1 М раствор NaOH (54 мл) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. 1.2 н HCl добавили в реакционную смесь до достижения смесью рН 7. Реакционную смесь сконцентрировали под вакуумом, и полученный остаток разбавили водой (50 мл) и экстрагировали этилацетатом (150 мл × 3). Органические слои объединили и сконцентрировали под вакуумом с получением (2-бромо-3-хлор-7-гидроксиметил-пирроло[2,3-b]пиразин-5-ил)-метанола (4.4 г, 83%) в виде желтого осадка. LC-MS: 292.2 (М-Н). Его использовали сразу без дополнительной очистки.
Стадия 6
2-бромо-3-хлор-5Н-пирроло[2,3-b]пиразин-7-карбальдегид
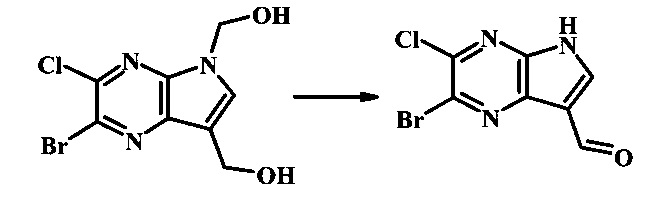
К перемешанной суспензии (2-бромо-3-хлор-7-гидроксиметил-пирроло[2,3-b]пиразин-5-ил)-метанола (2.0 г, 6.84 ммоль) в ацетоне (67 мл) добавили реактив Джонса (2.12 мл) и смесь перемешивали при комнатной температуре в течение 20 мин. Раствор был охлажден, затем отфильтрован через слои целита и фильтрат сконцентрировали под вакуумом. Остаток разбавили ТГФ (35 мл) и 1 н раствором NaOH (18 мл). Получившуюся смесь перемешивали при комнатной температуре в течение 16 ч, затем сконцентрировали под вакуумом до ~20 мл и довели до рН7 посредством добавления 1 н HCl. Смесь экстрагировали этилацетатом (100 мл × 3), высушили и сконцентрировали с получением 2-бромо-3-хлор-5Н-пирроло[2,3-b]пиразин-7-карбальдегида (1.4 г, 80%) в виде коричневого осадка. LC-MS: 260.2 (М-Н). Его использовали сразу без дополнительной очистки.
Замечание: Получение реактива Джонса: Раствор CrO3 (13.66 г) в воде (20 мл) охладили до 0°С и медленно добавили конц.H2SO4 (11.5 мл). Объем получившегося раствора довели до 50 мл посредством добавления воды с получением реактива Джонса.
Стадия 7
2-бромо-3-хлор-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2.3-b]пиразин-7-карбальдегид

К перемешанной суспензии 2-бромо-3-хлор-5Н-пирроло[2,3-b]пиразин-7-карбальдегида (2.0 г, 7.69 ммоль) добавили LiHMDS (1Mb ТГФ) (9.46 мл, 9.46 ммоль) при 0°С. Через 1 ч добавили триметилсилил этоксиметоксихлорид (1.5 мл, 8.46 ммоль) и реакционную смесь нагрели до комнатной температуры за 30 мин. Реакционную смесь разбавили водным раствором Na2CO3 (1.56 г в 35 мл воды) и затем экстрагировали этилацетатом (100 мл × 3). Объединенные органические слои промыли солевым раствором, затем очистили посредством хроматографии (силикагель, 5-10% этилацетат в гексанах) с получением 2-бромо-3-хлор-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбальдегида (1.96 г, 65%) в виде коричневого осадка. LC-MS: 392.2 (М+Н).
Стадия 8
2-бромо-3-хлор-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновая кислота
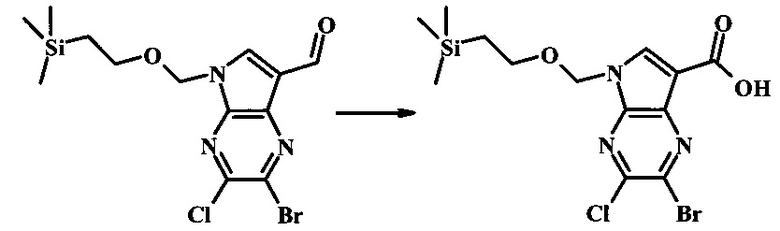
К перемешанному раствору 2-бромо-3-хлор-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбальдегида (1.96 г, 5.03 ммоль) в смешанной системе растворителей (1:1) 1,4-диоксан:вода (105 мл), добавили сульфаминовую кислоту (2.93 г, 30.15 ммоль), хлорид натрия (0.591 г, 6.53 ммоль) и дигидрофосфат калия (8.20 г, 60.3 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Получившуюся смесь разбавили водой (50 мл) и экстрагировали этилацетатом (150 мл × 3). Объединенные органические фазы промыли солевым раствором, высушили и сконцентрировали под вакуумом до желтого осадка, который тщательно промыли гексаном с получением 2-бромо-3-хлор-5-(2-триметилсиланил-этоксиметил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты (1.62 г, 79%) в виде серо-белого осадка. LC-MS: 406.2 (М-Н).
Стадия 9
2-бромо-N-трет-бутил-3-хлор-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид 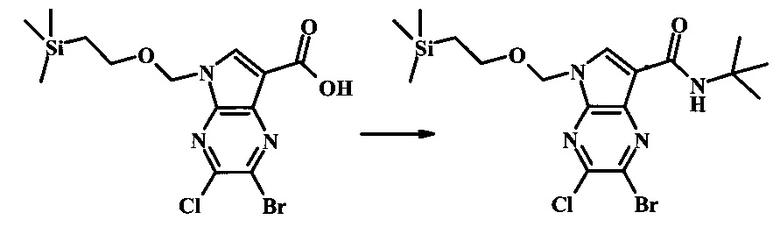
К раствору 2-бромо-3-хлор-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты (600 мг, 1.48 ммоль) в ДМФ (5 мл) добавили 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (650 мг, 3.39 ммоль) и НОВТ (520 мг, 3.39 ммоль) и смесь перемешивали в течение 10 мин. 2-метилпропан-2-амин (432 мг, 5.9 ммоль) добавили и смесь перемешивали при комнатной температуре в течение 16 ч, затем разбавили 10% лимонной кислотой и этилацетатом. Органический слой разделили и промыли 10% лимонной кислотой, насыщенным водным NaHCO3 и солевым раствором, затем сконцентрировали под вакуумом и очистили посредством хроматографии (силикагель, 5-20% этилацетат в гексанах) с получением 2-бромо-N-трет-бутил-3-хлор-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (530 мг, 1.15 ммоль, 78%) в виде белого осадка. 1Н NMR (400 MHz, ХЛОРОФОРМ-d) δ ppm: 8.30 (s, 1 Н), 7.72 (s, 1 Н), 5.64 (s, 2 Н), 3.57 (t, J=8.3 Hz, 2 Н), 1.56 (s, 9 Н), 0.96 (t, J=8.3 Hz, 2 H), 0.01 (s, 9 H).
Стадия 10
N-трет-бутил-3-хлор-2-(3-метилизотиазол-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид

Смесь 2-бромо-N-трет-бутил-3-хлор-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (125 мг, 271 мкмоль), 3-метилизотиазол-5-амина гидрохлорида (61.2 мг, 406 мкмоль), Pd2(dba)3 (24.8 мг, 27.1 мкмоль), ксантфоса (47.0 мг, 81.2 мкмоль) и карбоната цезия (265 мг, 812 мкмоль) в диоксане (1.5 мл) нагревали в микроволновой печи при 150°С в течение 20 мин. Pd2(dba)3 (24.8 мг, 27.1 мкмоль) и ксантфос (47.0 мг, 81.2 мкмоль) добавили и продолжили нагревание в микроволновой печи при 150°С в течение 30 мин. Смесь охладили, затем отфильтровали через слои целита. Фильтрат сконцентрировали под вакуумом, затем очистили посредством хроматографии (силикагель, 10-50% этилацетат в гексанах) с получением N-трет-бутил-3-хлор-2-(3-метилизотиазол-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (40 мг, 48.5 мкмоль, 17.9%) в виде коричневой смолы. (EI/CI) m/z: 495.1 [М+Н].
Стадия 11
N-трет-бутил-3-хлор-2-(3-метилизотиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид
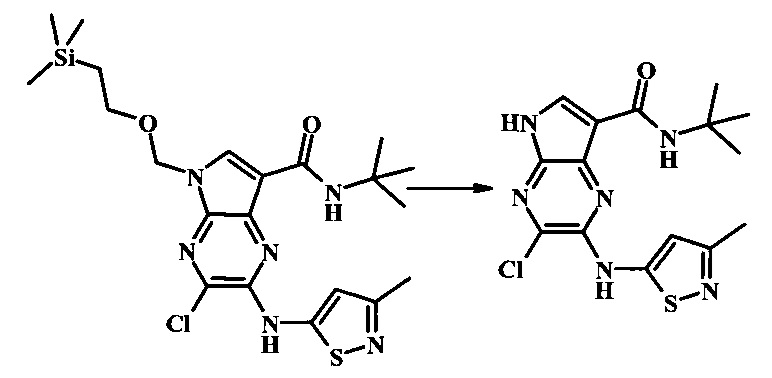
К раствору N-трет-бутил-3-хлор-2-(3-метилизотиазол-5-иламино)-5-((2-(триметилсилил)этокси)метил)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (40 мг, 80.8 мкмоль) в дихлорметане (1.5 мл) добавили трифторуксусную кислоту (184 мг, 124 мкл, 1.62 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь сконцентрировали, затем перерастворили в дихлорметане (1.5 мл), метаноле (0.75 мл) и гидроксиде аммония (0.2 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь сконцентрировали под вакуумом, затем тритурировали водой и отфильтровали. Полученный осадок промыли эфиром, затем высушили с получением N-трет-бутил-3-хлор-2-(3-метилизотиазол-5-иламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамида (15 мг, 37.0 мкмоль, 46%) в виде темно-оранжевого осадка. 1Н NMR (400 MHz, DMSO-d) δ ppm: 12.76 (s, 1 Н), 10.91 (s, 1 Н), 8.30 (d, J=3.2, 1 H), 7.33 (s, 1 H), 7.13 (s, 1 H), 2.42 (s, 3 H), 1.62 (s, 9 H); MS (EI/CI) m/z.365.0 [M+Н].
Биологические Примеры
Данные SYK анализа
Определение значений IC50 ингибирования тирозинкиназы селезенки (SYK):
Анализ SYK киназы представляет собой стандартный киназный анализ, адаптированный для использования в 96 луночных планшетах. Этот анализ проводился в 96-луночных планшетах для определения значений IC50 с 8 образцами, которые представляют собой десятичные полулогарифмические разведения в 40 мкл реакционном объеме. В анализе измеряется включение радиоактивно меченного 33Р γАТФ в биотинилированный на N-конце пептидный субстрат, полученный из природной фосфоакцепторной консенсусной последовательности (Biotin-11аа DY*E). Фосфорилированный продукт определяли сразу после завершения реакции посредством ЭДТА и добавления покрытых стрептавидином шариков. Результаты представлены в Таблице II выше.
Планшеты для анализа: 96-луночные планшеты Multiscreen с 0.65 мкм фильтром (Millipore Cat. No.: MADVNOB10).
Покрытые стрептавидином шарики: Streptavidin Sepharose ТМ, суспензия 5.0 мл, в 50 мМ ЭДТА/PBS разведенная (1:100), (Amersham, Cat. No.: 17-5113-01).
Соединения: 10 мМ в 100% диметилсульфоксиде (ДМСО), конечная конц.: соединения 0.003-100 мкМ в 10% ДМСО.
Фермент: SYK RPA очищенная, усеченная конструкция тирозинкиназы селезенки аа 360-635, стоковый раствор 1 мг/мл, MW: 31.2 кДа, конечная конц.:0.0005 мкМ.
Пептид 1: биотинилированный пептид получили из природной фосфор-акцепторной консенсусной последовательности (Биотин-EPEGDYEEVLE), специальный заказ для QCB, стоковый раствор 20 мМ, конечная конц.: 5.0 мкМ.
АТФ: Аденозин-5'-трифосфат 20 мМ, (ROCHE Cat. No.: 93202720), конечная концентрация: 20 мкМ
Буфер: HEPES: 2-Гидроксиэтил пиперазин-2-этансульфоновая кислота (Sigma, Cat. No.: Н-3375) конечная концентрация: 50 мМ HEPES рН7.5
БСА: Бычий сывороточный альбумин, фракция V, свободный от жирных кислот (Roche Diagnostics GmbH, Cat. No. 9100221), разведенный до конечной концентрации 0.1%
ЭДТА: ЭДТА стоковый раствор 500 мМ, (GIBCO, Cat. No.: 15575-038) конечная концентрация: 0.1 мМ
ДТТ: 1,4-дитиотрейтол (Roche Diagnostics GmbH, Cat. No.: 197777), конечная конц.: 1 мМ
MgCl2 × 6H2O: MERCK, Cat. No.: 105833.1000, конечная концентрация: 10 мМ
Буфер для разведения (ADB): 50 мМ HEPES, 0.1 мМ EGTA, 0.1 мМ ванадат Na, 0.1 мМ β-глицерофосфат, 10 мМ MgCl2, 1 мМ ДТТ, 0,1% БСА, рН 7.5
Буфер для промывки шариков: 10 г/л PBS (фосфатно-солевой буферный раствор) с 2М NaCl+ 1% фосфорной кислоты.
Экспериментальный способ:
В объеме 40 мкл, 26 мкл разведенного в ADB, очищенного рекомбинантного человеческого SYK360-635 [0.5 нм] смешали с 4 мкл 10Х концентрациями тестируемых соединений, [обычно 100 мкМ- 0.003 мкМ] в [10%] ДМСО и смесь инкубировали в течение 10 мин при комнатной температуре.
Киназную реакцию инициировали посредством добавления 10 мкл 4х субстратной смеси, содержащей DYE пептидный субстрат [0 или 5 мкМ], АТФ [20 мкМ] и 33РγАТФ [2 мКю/реакция]. После инкубации при 30°С в течение 15 мин, реакцию останавливали переносом 25 мкл реакционного образца в 96 луночные планшеты с мембранами 0.65 мкм Millipore MADVNOB, содержащие 200 мкл 5 мМ ЭДТА и 20% покрытых стрептавидином шариков в PBS.
Не связавшиеся радионуклеотиды промыли под вакуумом 3×250 мкл 2М NaCl; 2×250 мкл 2М NaCl+1% фосфорной кислотой; 1×250 мкл H2O. После последней промывки мембраны/планшеты поместили на адаптерный планшет, высушили нагреванием в течение 15 мин при 60°С, и 50 мкл сцинтиляционной смеси добавили в каждую лунку и спустя 4 ч радиоактивность была количественно подсчитана на сцинтиляционном счетчике.
Процент ингибирования подсчитывали на основе не ингибированного уровня фермента:
% ингибирования=100/(1+(IC50/конц. ингибитора)")
Значения IC50 рассчитывали с помощью нелинейной аппроксимации кривой, используя программное обеспечение XLfit (ID Business Solution Ltd., Guilford, Surrey, UK).
FLIPR анализ выхода кальция в клетках Ramos
Клеточная линия В-клеточной лимфомы человека Ramos культивировалась в RPMI-1640 с 10% фетальной бычьей сывороткой (Invitrogen). Клетки посеяли в планшеты для анализа и загрузили кальциевым красителем посредством добавления 30 мкл загрузочного буфера красителя в соответствии с инструкцией производителя (Beckton Dickinson Calcium Kit, каталожный №# 80500). Клетки обработали серийно разведенными соединениями в течение 30 мин перед стимуляцией. Базовую линию флуоресценции записывали в течение приблизительно 20 секунд с последующей стимуляцией мышиными антителами против человеческого IgM (10 мкг/мл, клон М2Е6, Antibody Solutions Inc.) для клеток Ramos, и записывали максимальные значения флуоресценции в каждой лунке. Максимальные значения, отделенные от значений базовой линии, определенные как степенные изменения от базовой линии для каждой лунки, использовались для расчета IC50.
Анализ увеличения CD69 в В-клетках цельной крови человека
Кровь была собрана у здоровых добровольцев в вакутайнеры (BD Biosciences, San Jose, СА), содержащих гепарин натрия. Тестируемые соединения суспендировали в ДМСО и сделали девять полулогарифмических серийных разведений. Концентрация соединения в анализе составляла 0.5%. 100 мкл цельной крови предварительно инкубировали с соединением в течение 30 мин и затем стимулировали козьими IgM антителами к F(ab')2 человека (50 мкг/мл, Southern Biotech) в течение 20 ч. В конце 20 часовой инкубации образцы инкубировали с флуорохром-конъюгированными антителами, РЕ мышиными против CD20 человека и АРС мышиными против CD69 человека (BD Biosciences), в течение 30 минут. Затем образцы лизировали с помощью лизирующего раствора (BD) и промыли ФСБ, содержащим 2% фетальной бычьей сыворотки (FBS). Флуоресцентные сигналы измеряли на проточном цитометре LSR II (BD) и данные анализировали с помощью Flow Jo. Процент активированных (CD69hi) В-лимфоцитов (CD20+) определяли с использованием не стимулированных (отрицательный контроль) и стимулированных (положительный контроль) лунок в качестве референсных значений. Процент ингибирования рассчитывался и строилась кривая IC50 с использованием программного обеспечения GraphPad Prism с аппроксимацией сигмоидальной кривой.
Данные показаны ниже в мкМ в Таблице II.

Изложенное выше изобретение было описано в некоторых деталях с целью иллюстрирования и в качестве примера для наглядности и понимания. Для специалиста в данной области очевидно, что в рамках формулы изобретения могут быть осуществлены изменения и модификации. Поэтому следует понимать, что приведенное выше описание предназначено для иллюстрации, а не ограничения. Объем изобретения, следовательно, не должен быть ограничен приведенным выше описанием, но должен определяться с учетом формулы изобретения, наряду с полным объемом эквивалентов, к которым применима указанная формула изобретения.
Все патенты, патентные заявки и публикации, цитируемые в данной заявке, включены в качестве ссылки во всей своей полноте для всех целей в той же степени, как если бы каждый отдельный патент, заявка на патент или публикация были отдельно описаны.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ JAK | 2010 |
|
RU2538204C2 |
| СОЕДИНЕНИЯ ПИРИДАЗИНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ СЕЛЕЗЕНКИ (SYK) | 2013 |
|
RU2627661C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2821941C2 |
| ПРОИЗВОДНЫЕ 6-КАРБОНИТРИЛ ДИПИРИДОПИРРОЛЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ РАКА | 2010 |
|
RU2575635C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ, АКТИВНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2013 |
|
RU2666538C2 |
| ИМИДАЗОПИРИДАЗИНЫ | 2012 |
|
RU2662443C2 |
| СОЕДИНЕНИЕ-ИНГИБИТОР JAK И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2820445C2 |
| ПИРРОЛОПИРАЗИНОВЫЕ ИНГИБИТОРЫ КИНАЗЫ | 2012 |
|
RU2673064C2 |
| 6,7-ДИГИДРО-5H-ПИРИДО[2,3-C]ПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛКОВ BCLXL И ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2020 |
|
RU2832191C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
Изобретение относится соединениям формулы I
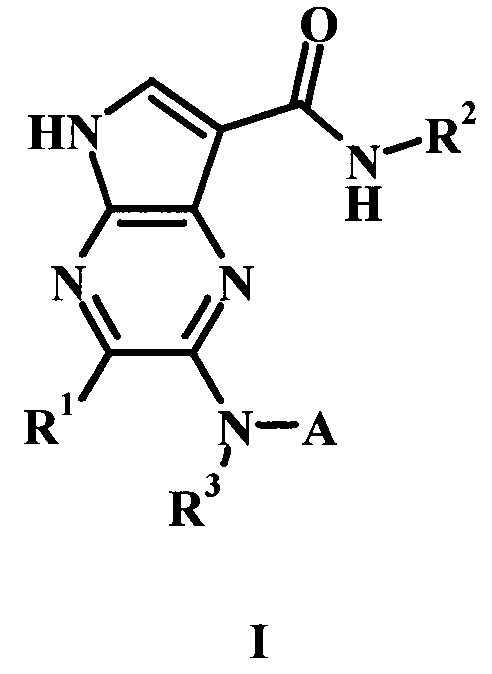
Технический результат: получены новые соединения, которые являются ингибиторами SYK и полезны для лечения аутоиммунных и воспалительных заболеваний. 3 н. и 6 з.п. ф-лы, 2 табл., 38 пр.
1. Соединение формулы I
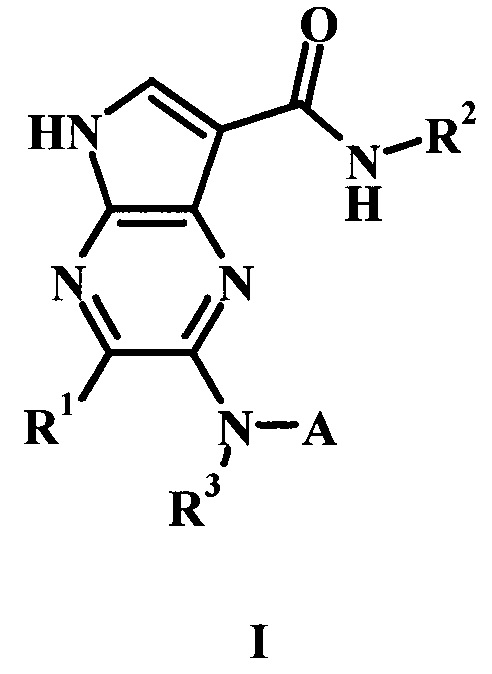
где R1 представляет собой Н;
R2 представляет собой трет-бутил;
R3 представляет собой Н или С1-С6-алкил;
А представляет собой фенил, возможно замещенный одним или более А', и каждый А' представляет собой независимо С1-С6-алкил, гало, C1-C6-алкилсульфонил, амидо, С1-С6-гидроксиалкил, С1-С6-алкокси, амино-С1-С6-алкил или дейтерий;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, где R3 представляет собой С1-С6-алкил.
3. Соединение по п. 1, где R3 представляет собой Н.
4. Соединение по пп. 1-3, где каждый А' представляет собой независимо С1-С6-алкил, гало, C1-С6-алкилсульфонил, С1-С6-гидроксиалкил, С1-С6-алкокси, амино-С1-С6-алкил или дейтерий.
5. Соединение по пп. 1-4, где А' представляет собой С1-С6-алкил.
6. Соединение по пп. 1-5, где А' представляет собой С1-С6-алкилсульфонил.
7. Соединение по п. 1, где А' представляет собой С1-С6-алкил или С1-С6-алкилсульфонил.
8. Соединение, выбранное из группы, состоящей из:
N-трет-бутил-2-(2-фтор-4-метилфениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(р-толиламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(3-(метилсульфонил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(2-(диметилкарбамоил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(2,4-диметилфениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(4-(метилсульфонил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
N-трет-бутил-2-(3-(1-гидроксиэтил)фениламино)-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
2-(3-(1-аминоэтил)фениламино)-N-трет-бутил-5Н-пирроло[2,3-b]пиразин-7-карбоксамид;
2-(3-метансульфонил-фениламино)-3-метил-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид;
2-фениламино(D5)-5Н-пирроло[2,3-b]пиразин-7-карбоновой кислоты трет-бутиламид.
9. Фармацевтическая композиция для лечения нарушений, ассоциированных с Syk, содержащая соединение по любому из пп. 1-8 в эффективном количестве, смешанное с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
| WO 2011144585 A1, 24.11.2011 | |||
| RU 2010139566 A, 10.04.2012 | |||
| ИНГИБИТОР КИНАЗЫ | 2007 |
|
RU2440352C2 |

