ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области синтеза лекарственных средств и, в частности, относится к ингибитору трициклического производного, способу его получения и его применению.
УРОВЕНЬ ТЕХНИКИ
Семейство белков фосфатидилинозитол-3-киназы (PI3K) делится на четыре типа: I, II, III и IV, и участвует в регуляции множества функций клеток, таких как клеточный рост, пролиферация, дифференцировка, выживание и метаболизм глюкозы. Четыре типа белков PI3K имеют разные структуры и функции, и наиболее широко изучен тип I PI3K. Этот тип I PI3K делится на четыре подтипа: PI3Kα, PI3Kβ, PI3Kδ и PI3Kγ. Среди них PI3Kα демонстрирует активирующие мутации и амплификацию в различных опухолях и тесно связана с возникновением и развитием опухоли. Сообщается, что PI3Kβ может активировать тромбоциты и играть важную роль в развитии таких заболеваний, как тромбоз. PI3Kδ и PI3Kγ в основном экспрессируются в кровеносной системе и тесно связаны с иммунной системой и воспалением. PI3Kγ также тесно связан со стабильностью артериального давления и сокращением гладких мышц.
PI3Kα демонстрирует активирующую мутацию и амплификацию при различных опухолях и является движущим фактором, ведущим к онкогенезу. PI3Kα представляет собой гетеродимер, состоящий из каталитической субъединицы p110 и регуляторной субъединицы p85. PI3Kα активируется рецепторными тирозинкиназами (RTK) и рецепторами, связанными с G-белком (GPCR). После активации она катализирует образование фосфатидилинозитол-3-фосфата (PIP3) из фосфатидилинозитол-2-фосфата (PIP2). PIP3 может дополнительно активировать протеинкиназу B (PKB, также известную как AKT) и ее нижерасположенные сигнальные пути. Различные факторы роста клеток, такие как эпидермальный фактор роста (EGF), фактор роста фибробластов (FGF), фактор роста эндотелия сосудов (VEGF), фактор роста гепатоцитов (HGF) и инсулин, могут активировать PI3Kα, тем самым активируя нижерсположенные сигнальные пути, связанные с пролиферацией клеток. Патологическая активация PI3Kα может привести к быстрой пролиферации клеток, тем самым приводя к онкогенезу.
PI3Kα всегда была важной мишенью при разработке противоопухолевых лекарственных средств. Однако большинство соединений являются ингибиторами PI3K широкого спектра действия и могут вызывать очевидные побочные действия в клинических исследованиях, которые серьезно ограничивают дальнейшую разработку ингибитора PI3K. В текущих исследования было установлено, что большинство побочных действий ингибиторов PI3K широкого спектра действия вызвано ингибированием подтипов PI3Kβ, PI3Kδ и PI3Kγ. PI3Kβ играет важную роль в побочных действиях тромбоцитопении и тромбозе. Ингибирование PI3Kδ может вызывать нарушения в иммунной системе, а аутоиммунные и вирусные инфекции, такие как пневмония, гепатит и диарея/энтерит, тесно связаны с ингибированием мишени PI3Kδ. PI3Kγ тесно связана со стабильностью артериального давления и сокращением гладких мышц и является основной мишенью, вызывающей побочное действие гипертензию. Следовательно, необходимо разработать ингибитор PI3Kα с высокой активностью и высокой селективностью, который может дополнительно улучшить противоопухолевое действие ингибитора PI3Kα и уменьшить или устранить серьезные побочные действия, такие как различные воспаления, тромбоцитопения и гипертония, вызываемые ингибированием других подтипов.
PI3Kα-селективный ингибитор BYL-719, разработанный Novartis, в настоящее время проходит III фазу клинических исследований. PI3Kα-селективный ингибитор MLN1117, разработанный Takeda, вошел во II фазу клинических исследований. Селективный ингибитор GDC-0077, разработанный Genentech, проходит фазу I клинических исследований.
В международных патентных заявках WO2010029082 (A1) и WO2011022439 (A1) раскрыты соединения, родственные PI3Kα-селективному ингибитору. Однако последующие исследования показывают, что активность данных соединений в клетках невысока, что влияет на их клиническое противоопухолевое действие. Поэтому существует острая необходимость в разработке селективного ингибитора PI3Kα с высокой активностью и высокой селективностью. Селективные ингибиторы PI3Kα могут применяться для лечения множества различных опухолей с помощью активирующей мутации или амплификации PI3Kα и имеют важное клиническое значение.
Исследования показывают, что соединения согласно примерам настоящего изобретения обладают более высокой активностью и селективностью в отношении фермента PI3Kα, лучшей активностью в клетках, лучшей степенью ингибирования опухоли в фармакодинамической модели у мышей и более высокой безопасностью.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является обеспечение соединения формулы (I), его стереоизомера или его фармацевтически приемлемой соли, где структура соединения формулы (I) представляет собой следующую:

где:
каждый из Q, Y и Z независимо выбран из группы, состоящей из N и -CRaa;
кольцо A выбрано из группы, состоящей из циклоалкила, гетероциклила, арила и гетероарила;
R1 выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, галогеналкокси, галогена, амино, нитро, гидрокси, циано, алкенила, алкинила, циклоалкила, гетероциклила, оксогетероциклила, тиоксогетероциклила, арила, гетероарила, -(CH2)n1Rbb, -(CH2)n1ORbb, -NRaaC(O)(CH2)n1ORbb, -NRaaC(S)(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc, где каждый из указанных алкила, дейтерированного алкила, галогеналкила, алкокси, галогеналкокси, алкенила, алкинила, циклоалкила, гетероциклила, оксогетероциклила, тиоксогетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, замещенного или незамещенного циклоалкилалкила, замещенного или незамещенного циклоалкилгалогеналкила, галогена, замещенного или незамещенного циклоалкиламино, оксо, тиоксо, нитро, циано, гидрокси, замещенного или незамещенного циклоалкилалкенила, замещенного или незамещенного циклоалкилалкинила, замещенного или незамещенного циклоалкилалкокси, замещенного или незамещенного циклоалкилгалогеналкокси, замещенного или незамещенного циклоалкилгидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1Rdd, -(CH2)n1ORdd, -(CH2)n1SRdd, -(CH2)n1C(O)Rdd, -(CH2)n1C(O)ORdd, -(CH2)n1S(O)m1Rdd, -(CH2)n1NRddRee, -(CH2)n1C(O)NRddRee, -(CH2)n1C(O)NHRdd, -(CH2)n1NRddC(O)Ree и -(CH2)n1NRddS(O)m1Ree;
каждый из Rx и Ry независимо выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, галогеналкокси, галогена, амино, тиола, нитро, гидрокси, циано, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc, где каждый из указанных алкила, дейтерированного алкила, галогеналкила, алкокси, галогеналкокси, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, замещенного или незамещенного циклоалкилалкила, замещенного или незамещенного циклоалкилгалогеналкила, галогена, замещенного или незамещенного циклоалкиламино, тиола, оксо, нитро, циано, гидрокси, замещенного или незамещенного алкенила, замещенного или незамещенного алкинила, замещенного или незамещенного циклоалкилалкокси, замещенного или незамещенного циклоалкилгалогеналкокси, замещенного или незамещенного циклоалкилгидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1Rdd, -(CH2)n1ORdd, -(CH2)n1SRdd, -(CH2)n1C(O)Rdd, -(CH2)n1C(O)ORdd, -(CH2)n1S(O)m1Rdd, -(CH2)n1NRddRee, -(CH2)n1C(O)NRddRee, -(CH2)n1C(O)NHRdd, -(CH2)n1NRddC(O)Ree и -(CH2)n1NRddS(O)m1Ree;
или любые два смежных или несмежных Rx связаны с образованием циклоалкила, гетероциклила, арила или гетероарила, где указанный циклоалкил, гетероциклил, арил или гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, замещенного или незамещенного алкила, замещенного или незамещенного галогеналкила, галогена, замещенного или незамещенного амино, оксо, нитро, циано, гидрокси, замещенного или незамещенного алкенила, замещенного или незамещенного алкинила, замещенного или незамещенного алкокси, замещенного или незамещенного галогеналкокси, замещенного или незамещенного гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc;
или любые два смежных или несмежных Ry связаны с образованием циклоалкила, гетероциклила, арила или гетероарила, где указанный циклоалкил, гетероциклил, арил или гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, замещенного или незамещенного алкила, замещенного или незамещенного галогеналкила, галогена, замещенного или незамещенного амино, оксо, нитро, циано, гидрокси, замещенного или незамещенного алкенила, замещенного или незамещенного алкинила, замещенного или незамещенного алкокси, замещенного или незамещенного галогеналкокси, замещенного или незамещенного гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc;
Raa выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, гидроксиалкила, галогеналкокси, галогена, циано, нитро, гидрокси, амино, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из указанных алкила, дейтерированного алкила, галогеналкила, алкокси, гидроксиалкила, галогеналкокси, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, замещенного или незамещенного алкила, галогена, гидрокси, замещенного или незамещенного амино, оксо, нитро, циано, замещенного или незамещенного алкенила, замещенного или незамещенного алкинила, замещенного или незамещенного алкокси, замещенного или незамещенного гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила и замещенного или незамещенного гетероарила;
каждый из Rbb, Rcc, Rdd и Ree независимо выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, гидроксиалкила, галогеналкокси, галогена, циано, нитро, гидрокси, амино, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из указанных алкила, дейтерированного алкила, галогеналкила, алкокси, гидроксиалкила, галогеналкокси, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, замещенного или незамещенного алкила, галогена, гидрокси, замещенного или незамещенного амино, оксо, нитро, циано, замещенного или незамещенного алкенила, замещенного или незамещенного алкинила, замещенного или незамещенного алкокси, замещенного или незамещенного гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила и замещенного или незамещенного гетероарила;
n равно 0, 1, 2 или 3;
p равно 0, 1, 2, 3, 4, 5 или 6;
q равно 0, 1, 2, 3, 4, 5 или 6;
m1 равно 0, 1 или 2; и
n1 равно 0, 1, 2, 3, 4 или 5.
В предпочтительном варианте осуществления Rx представляет собой -(CH2)n1NRbbC(RffRgg)C(O)Rcc;
каждый из Rff и Rgg независимо выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, гидроксиалкила, галогеналкокси, галогена, циано, нитро, гидрокси, амино, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из указанных алкила, дейтерированного алкила, галогеналкила, алкокси, гидроксиалкила, галогеналкокси, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, замещенного или незамещенного алкила, галогена, гидрокси, замещенного или незамещенного амино, оксо, нитро, циано, замещенного или незамещенного алкенила, замещенного или незамещенного алкинила, замещенного или незамещенного алкокси, замещенного или незамещенного гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила и замещенного или незамещенного гетероарила; и
n1, Rbb и Rcc являются такими, как определено в формуле (I).
В еще одном предпочтительном варианте осуществления, когда кольцо A представляет собой бензольное кольцо, Ry представляет собой водород, Q и Y представляют собой N, Z представляет собой -CRaa, Raa представляет собой водород, n равно 1, и R1 представляет собой  Rx не является -NHCHRffC(O)NH2, где Rff представляет собой CH3-, циклопропил- или -CH2CH3;
Rx не является -NHCHRffC(O)NH2, где Rff представляет собой CH3-, циклопропил- или -CH2CH3;
когда кольцо A представляет собой бензольное кольцо, Ry представляет собой водород, Q и Y представляют собой N, Z представляет собой -CRaa, Raa представляет собой водород, n равно 1, и R1 представляет собой 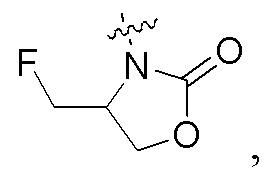 Rx не явялется -NCHRffC(O)NH2, где Rff представляет собой CH3- или циклопропил-; и
Rx не явялется -NCHRffC(O)NH2, где Rff представляет собой CH3- или циклопропил-; и
когда кольцо A представляет собой бензольное кольцо, Ry представляет собой водород, Q и Y представляют собой N, Z представляет собой -CRaa, Raa представляет собой водород, n равно 1, и R1 представляет собой 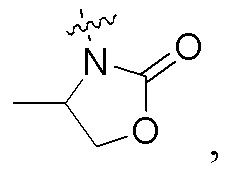 Rx не является -NHCHRffC(O)NH2, где Rff представляет собой циклопропил- или циклобутил-.
Rx не является -NHCHRffC(O)NH2, где Rff представляет собой циклопропил- или циклобутил-.
В предпочтительнм варианте осуществления настоящего изобретения обеспечены соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения является такой, как показано в формуле (II):

где:
W выбран из группы, состоящей из кислорода и серы, и предпочтительно кислорода;
каждый из R9 и R10 независимо выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, галогеналкокси, галогена, амино, тиола, нитро, гидрокси, циано, алкенила, алкинила, циклоалкила, галогенциклоалкила, гетероциклила, арила, гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc, где каждый из указанных алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогеналкила, галогена, амино, тиола, оксо, нитро, циано, гидрокси, алкенила, алкинила, алкокси, галогеналкокси, гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1Rdd, -(CH2)n1ORdd, -(CH2)n1SRdd, -(CH2)n1C(O)Rdd, -(CH2)n1C(O)ORdd, -(CH2)n1S(O)m1Rdd, -(CH2)n1NRddRee, -(CH2)n1C(O)NRddRee, -(CH2)n1C(O)NHRdd, -(CH2)n1NRddC(O)Ree и -(CH2)n1NRddS(O)m1Ree;
или R9 и R10 могут быть связаны с образованием гетероциклила или гетероарила, где указанный гетероциклил или гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогеналкила, галогена, амино, оксо, нитро, циано, гидрокси, алкенила, алкинила, алкокси, галогеналкокси, гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc; и
кольцо A, H, Y, Z, R23-R26, Rx, Ry, n, p, q, m1 и n1 являются такими, как определено в формуле (II).
В предпочтительном варианте осуществления настоящего изобретения обеспечены соединение формулы (II), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения показана в формуле (II-A) или (II-B):
 ,
,  ,
,
где:
G выбран из группы, состоящей из кислорода и серы;
L выбран из группы, состоящей из азота, кислорода, серы и -CRaa;
кольцо В выбрано из группы, состоящей из гетероциклила и гетероарила, и предпочтительно тиоксогетероциклила или оксогетероциклила;
Rz выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, галогеналкокси, галогена, амино, тиола, нитро, гидрокси, циано, оксо, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc, где каждый из указанных алкила, галогеналкила, алкокси, галогеналкокси, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогеналкила, галогена, амино, тиола, оксо, нитро, циано, гидрокси, алкенила, алкинила, алкокси, галогеналкокси, гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1Rdd, -(CH2)n1ORdd, -(CH2)n1SRdd, -(CH2)n1C(O)Rdd, -(CH2)n1C(O)ORdd, -(CH2)n1S(O)m1Rdd, -(CH2)n1NRddRee, -(CH2)n1C(O)NRddRee, -(CH2)n1C(O)NHRdd, -(CH2)n1NRddC(O)Ree и -(CH2)n1NRddS(O)m1Ree;
или любые два смежных или несмежных Rz могут быть связаны с образованием циклоалкила, гетероциклила, арила или гетероарила, где указанный циклоалкил, гетероциклил, арил или гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогеналкила, галогена, амино, оксо, нитро, циано, гидрокси, алкенила, алкинила, алкокси, галогеналкокси, гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc;
R2 присутствует или отсутствует, когда L представляет собой азот или -CRaa, R2 выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, галогеналкокси, галогена, амино, тиола, нитро, гидрокси, циано, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, гидроксиалкила, галогеналкокси, галогена, амино, тиола, нитро, гидрокси, циано, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc, где каждый из указанных алкила, дейтерированного алкила, галогеналкила, алкокси, галогеналкокси, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогеналкила, галогена, амино, тиола, оксо, нитро, циано, гидрокси, алкенила, алкинила, алкокси, галогеналкокси, гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1Rdd, -(CH2)n1ORdd, -(CH2)n1SRdd, -(CH2)n1C(O)Rdd, -(CH2)n1C(O)ORdd, -(CH2)n1S(O)m1Rdd, -(CH2)n1NRddRee, -(CH2)n1C(O)NRddRee, -(CH2)n1C(O)NHRdd, -(CH2)n1NRddC(O)Ree и -(CH2)n1NRddS(O)m1Ree;
или любые две группы из R2, R3, R4 и Raa связаны с образованием циклоалкила, гетероциклила, арила или гетероарила, где указанный циклоалкил, гетероциклил, арил или гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогеналкила, галогена, амино, оксо, нитро, циано, гидрокси, алкенила, алкинила, алкокси, галогеналкокси, гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc;
m равно 0, 1, 2, 3, 4, 5 или 6;
t равно 0, 1, 2, 3, 4, 5 или 6;
q равно 0, 1, 2, 3, 4, 5 или 6; и
кольцо A, Q, Y, Z, Rbb, Rcc, Rdd, Ree, Rx, Ry, n, p, q, m1 и n1 являются такими, как определено в формуле (I).
В предпочтительном варианте осуществления настоящего изобретения обеспечены соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения является такой, как показано в формуле (III):
 ,
,
где:
каждый из R5, R6 и R14 независимо выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, галогеналкокси, галогена, амино, тиола, нитро, гидрокси, циано, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc, где каждый из указанных алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогеналкила, галогена, амино, тиола, оксо, нитро, циано, гидрокси, алкенила, алкинила, алкокси, галогеналкокси, гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила и замещенного или незамещенного гетероарила;
или R5 и R6 связаны с образованием циклоалкила, гетероциклила, арила или гетероарила, где указанный циклоалкил, гетероциклил, арил или гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогеналкила, галогена, амино, оксо, нитро, циано, гидрокси, алкенила, алкинила, алкокси, галогеналкокси, гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc;
Q, Y, Z, Rbb, Rcc, R1, R2, Ry, n, p, q, m1 и n1 являются такими, как определено в формуле (I); и
G, m, R3 и R4 являются такими, как определено в формуле (II-A).
В предпочтительном варианте осуществления настоящего изобретения обеспечено соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения является такой, как показано в формуле (IV):
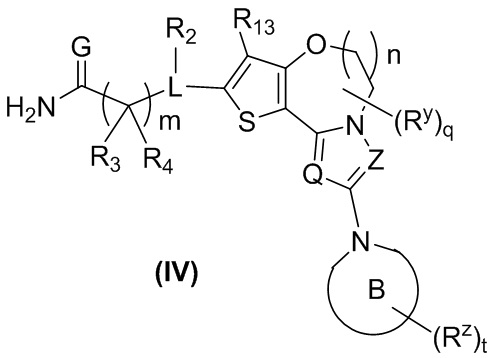 ,
,
где:
R13 выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогена, циано, нитро, галогеналкила, гидрокси, амино, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из указанных алкила, циклоалкила, гетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогена, гидрокси, амино, оксо, нитро, циано, алкенила, алкинила, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила, и предпочтительно галогена, амино, нитро, циано, алкила, галогеналкила или циклоалкила; и
кольцо B, Q, Z, G, R2-R4, Ry, Rz, m, n, q и t являются такими, как определено в формуле (III).
В предпочтительном варианте осуществления настоящего изобретения обеспечено соединение формулы (III), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения показана в формуле (III-A) или (III-B):
 ,
, ,
,
где:
каждый из R7, R8, R11 и R12 независимо выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, галогеналкокси, галогена, амино, тиола, нитро, гидрокси, циано, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc, где каждый из указанных алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогеналкила, галогена, амино, тиола, оксо, нитро, циано, гидрокси, алкенила, алкинила, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
или любые две группы из R7, R8, R11 и R12 могут быть связаны с образованием циклоалкила, гетероциклила, арила или гетероарила, где указанный циклоалкил, гетероциклил, арил или гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогеналкила, галогена, амино, оксо, нитро, циано, гидрокси, алкенила, алкинила, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R9 и R10 являются такими, как определено в формуле (II);
Q, Z, G, R2-R6, Rbb, Rcc, Ry, m, n, q, m1 и n1 являются такими, как определено в формуле (III); и
R14 является таким, как определено в формуле (III-A).
В предпочтительном варианте осуществления настоящего изобретения обеспечено соединение формулы (III-A), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения показана в формуле (V):
 ,
,
где:
кольцо B является таким, как определено в формуле (II-A); и
Q, Z, G, L, R2-R8, R11, R12, R14, Rz, m и t являются такими, как определено в формуле (III-A).
В предпочтительном варианте осуществления настоящего изобретения обеспечено соединение формулы (III-A), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения показана в формуле (VI):
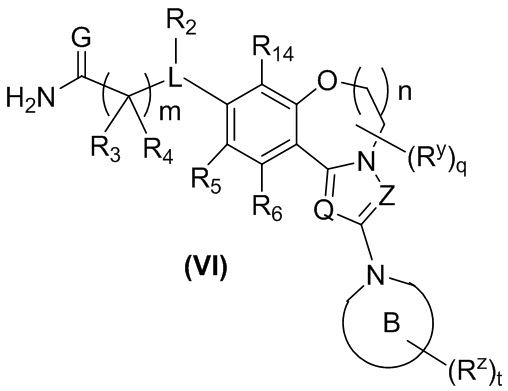 ,
,
где:
кольцо B является таким, как определено в формуле (II-A); и
Q, Z, G, L, R2-R6, R14, Ry, Rz, q, m и t являются такими, как определено в формуле (III-A).
В предпочтительном варианте осуществления настоящего изобретения любое из соединения формулы (II-A), (II-B), (IV), (V) или (VI), его стереоизомер или его фармацевтически приемлемая соль характеризуется тем, что
кольцо B выбрано из группы, состоящей из:
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,
 ,
, ,
,  ,
,  ,
, 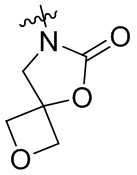 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 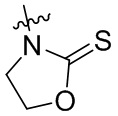 ,
,  ,
, 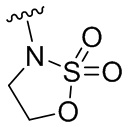 ,
,  ,
,  ,
,  и
и 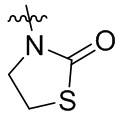 .
.
В предпочтительном варианте осуществления настоящего изобретения обеспечено соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения является такой, как показано в формуле (VII):
 ,
,
где:
Raa выбран из группы, состоящей из водорода, дейтерия, алкила, дейтерированного алкила, галогеналкила, алкокси, гидроксиалкила, галогеналкокси, галогена, циано, нитро, гидрокси, амино, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из указанных алкила, дейтерированного алкила, галогеналкила, алкокси, гидроксиалкила, галогеналкокси, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогена, гидрокси, амино, оксо, нитро, циано, алкенила, алкинила, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила; и
L, R2-R8, R11, R12, R14 и m являются такими, как определено в формуле (V).
Предпочтительно, когда Raa, R2, R5-R8, R11, R12 и R14 одновременно не являются водородом, R3 и R4 являются такими, как определено в формуле (VII).
Также предпочтительно, когда Raa, R2, R5-R8, R11, R12 и R14 одновременно представляют собой водород, любые две группы из R2, R3 и R4 связаны с образованием циклоалкила, гетероциклила, арила или гетероарила, где указанный циклоалкил, гетероциклил, арил или гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, галогеналкила, галогена, амино, оксо, нитро, циано, гидрокси, алкенила, алкинила, алкокси, галогеналкокси, гидроксиалкила, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероциклила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -(CH2)n1-, -(CH2)n1Rbb, -(CH2)n1ORbb, -(CH2)n1SRbb, -(CH2)n1C(O)Rbb, -(CH2)n1C(O)ORbb, -(CH2)n1S(O)m1Rbb, -(CH2)n1NRbbRcc, -(CH2)n1C(O)NRbbRcc, -(CH2)n1NRbbC(O)Rcc и -(CH2)n1NRbbS(O)m1Rcc.
В предпочтительном варианте осуществления настоящего изобретения обеспечено соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения является такой, как показано в формуле (VIII-A):
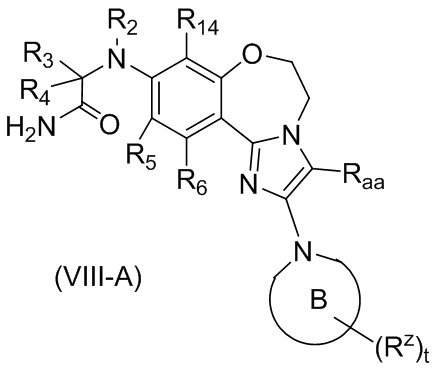 ,
,
где:
кольцо B выбрано из группы, состоящей из:
, , , , , и ;
R2 выбран из группы, состоящей из водорода, C1-6 алкила и C1-6 галогеналкила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси и -(CH2)n1ORbb;
или R3 и R4 связаны с образованием C3-8 циклоалкила или 3-8-членного гетероциклила, и предпочтительно оксетанила;
или R2 и R3 или R2 и R4 связаны с образованием 3-8-членного гетероциклила, и предпочтительно пирролидинила или азетидинила;
каждый из R5, R6 и R14 независимо выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила, C1-6 алкокси и C1-6 галогеналкила;
Raa выбран из группы, состоящей из водорода, C1-6 алкила, галогена и циано;
Rz выбран из группы, состоящей из водорода, оксо, C1-6 алкила, C1-6 галогеналкила и -(CH2)n1Rbb;
Rbb выбран из группы, состоящей из водорода, C1-6 алкила, галогена и циано; и
t равно 0, 1, 2 или 3.
Предпочтительно,
R2 выбран из группы, состоящей из водорода, C1-3 алкила и C1-3 галогеналкила, и более предпочтительно водорода, метила, этила или пропила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, C1-3 алкила, C1-3 алкокси и C1-3 алкила, замещенного C1-3 алкокси, и более предпочтительно водорода, метила, этила, пропила, метокси, этокси, CH3OCH2- или CH3CH2OCH2-;
или R3 и R4 связаны с образованием C4-6 циклоалкила или 4-6-членного гетероциклила, предпочтительно 4-6-членного гетероциклила, содержащего один атом кислорода или азота, и более предпочтительно оксетанила;
R2 и R3 или R2 и R4 связаны с образованием 3-8-членного гетероциклила, предпочтительно 4-6-членного гетероциклила, содержащего азот или кислород, где число гетероатомов равно одному или двум, и более предпочтительно тетрагидропирролила, тетрагидрофуранила, пиперидинила или азетидинила;
каждый из R5 и R6 независимо выбран из группы, состоящей из водорода, галогена, циано, C1-3 алкила, C1-3 алкокси и C1-3 галогеналкила, и предпочтительно водорода;
R14 выбран из группы, состоящей из водорода и галогена, и предпочтительно водорода, фтора или хлора;
Rz выбран из группы, состоящей из водорода, галогена, оксо, C1-3 алкила, C1-3 алкила, замещенного галогеном и -(CH2)n1Rbb, предпочтительно водорода, фтора, хлора, брома, иода, циано, ацетонитрилила, пропионитрилила или C1-3 алкила, замещенного фтором, и более предпочтительно фтора, метила, ацетонитрилила, -CHF2, -CF2CH3 или CHF2CH2-;
Raa выбран из группы, состоящей из водорода и галогена, и предпочтительно водорода;
Rbb представляет собой циано;
n1 равно 0, 1, 2 или 3; и
t равно 0, 1, 2 или 3.
При условии, что
когда Raa представляет собой водород, кольцо B представляет собой 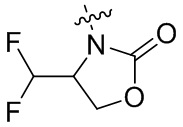 , и все из R2, R4, R5, R6, R14 и Raa представляют собой водород, R3 не является -CH(CH3), циклопропилом или CH3CH2-;
, и все из R2, R4, R5, R6, R14 и Raa представляют собой водород, R3 не является -CH(CH3), циклопропилом или CH3CH2-;
когда Raa представляет собой водород, кольцо B представляет собой , и все из R2, R3, R5, R6, R14 и Raa представляют собой водород, R4 не является -CH(CH3), циклопропилом или CH3CH2-;
когда Raa представляет собой водород, кольцо B представляет собой  , и все из R2, R4, R5, R6, R14 и Raa представляют собой водород, R3 не является -CH3 или циклопропилом;
, и все из R2, R4, R5, R6, R14 и Raa представляют собой водород, R3 не является -CH3 или циклопропилом;
когда Raa представляет собой водород, кольцо B представляет собой , и все из R2, R3, R5, R6, R14 и Raa представляют собой водород, R4 не является -CH3 или циклопропилом;
когда Raa представляет собой водород, кольцо B представляет собой  , и все из R2, R4, R5, R6, R14 и Raa представляют собой водород, R3 не является циклопропилом или циклобутилом; и
, и все из R2, R4, R5, R6, R14 и Raa представляют собой водород, R3 не является циклопропилом или циклобутилом; и
когда Raa представляет собой водород, кольцо B представляет собой , и все из R2, R3, R5, R6, R14 и Raa представляют собой водород, R4 не является циклопропилом или циклобутилом.
В предпочтительном варианте осуществления настоящего изобретения обеспечено соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения является такой, как показано в формуле (VIII):
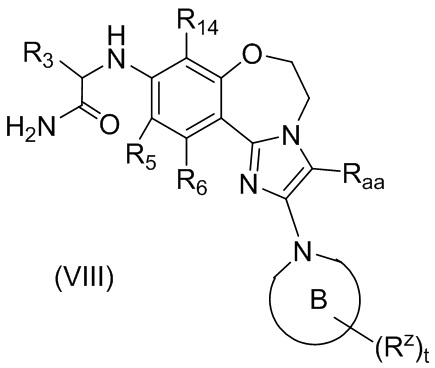 ,
,
где: кольцо B, R3, R5, R6, R14, Rz, Raa и t являются такими, как определено в формуле (III-A); и
Rz и t являются такими, как определено в формуле (V).
В предпочтительном варианте осуществления кольцо B выбрано из группы, состоящей из
, , , , , и ;
R2 выбран из группы, состоящей из водорода и C1-6 алкила, предпочтительно водорода или C1-3 алкила, и более предпочтительно водорода, метила, этила или пропила;
R3 выбран из группы, состоящей из C1-6 алкила, C1-6 алкокси и C1-6 алкокси, замещенного алкилом, предпочтительно C1-3 алкила, C1-3 алкокси или C1-3 алкила, замещенного C1-3 алкокси, и более предпочтительно метила, этила, пропила, метокси, этокси, CH3OCH2- или CH3CH2OCH2-;
каждый из R5 и R6 независимо выбран из группы, состоящей из водорода и галогена и предпочтительно водорода;
R14 выбран из группы, состоящей из водорода и галогена, и предпочтительно водорода, фтора или хлора; и
Rz выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила и C1-6 алкила, замещенного галогеном, предпочтительно водорода, фтора, хлора, брома, иода, циано, ацетонитрилила, пропионитрилила или C1-3 алкила, замещенного галогеном, и более предпочтительно фтора, метила, ацетонитрилила, -CHF2, -CF2CH3 или CHF2CH2-.
В предпочтительном варианте осуществления настоящего изобретения обеспечено соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения является такой, как показано в формуле (IX):
 ,
,
где:
каждый из R15 и R16 независимо выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 галогеналкила и -(CH2)n1Rbb; и
R2-R4, R6, R14, Raa и Rbb являются такими, как определено в формуле (VIII-A).
Предпочтительно,
R2 выбран из группы, состоящей из водорода и C1-6 алкила, предпочтительно водорода или C1-3 алкила, и более предпочтительно водорода, метила, этила или пропила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 алкокси и C1-6 алкокси, замещенного алкилом, предпочтительно водорода, C1-3 алкила, C1-3 алкокси или C1-3 алкила, замещенного C1-3 алкокси, и более предпочтительно водорода, метила, этила, пропила, метокси, этокси, CH3OCH2- или CH3CH2OCH2-;
R3 и R4 связаны с образованием C4-6 циклоалкила или 4-6-членного гетероциклила, предпочтительно 4-6-членного гетероциклила, содержащего один атом кислорода или азота, и более предпочтительно оксетанила;
R2 и R3 или R2 и R4 связаны с образованием 4-6-членного гетероциклила, предпочтительно 4-6-членного гетероциклила, содержащего азот или кислород, где число гетероатомов равно одному или двум, и более предпочтительно тетрагидропирролила, тетрагидрофуранила, пиперидинила или азетидинила;
R6 выбран из группы, состоящей из водорода и галогена, и предпочтительно водорода;
R14 выбран из группы, состоящей из водорода и галогена, и предпочтительно водорода, фтора или хлора;
Raa выбран из группы, состоящей из водорода и галогена, и предпочтительно водорода; и
каждый из R15 и R16 независимо выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила и C1-6 алкила, замещенного галогеном, предпочтительно водорода, фтора, хлора, брома, иода, циано, ацетонитрилила, пропионитрилила или C1-3 алкила, замещенного галогеном, и более предпочтительно водорода, фтора, метила, ацетонитрилила, -CHF2, -CF2CH3 или CHF2CH2-.
В предпочтительном варианте осуществления настоящего изобретения обеспечено соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль, где структура соединения является такой, как показано в формуле (X):
 ,
,
где:
каждый из R15 и R16 независимо выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 галогеналкила и -(CH2)n1Rbb; и
R2-R4, R6, R14, Raa и Rbb являются такими, как определено в формуле (VIII-A).
Предпочтительно,
R2 выбран из группы, состоящей из водорода и C1-6 алкила, предпочтительно водорода или C1-3 алкила, и более предпочтительно водорода, метила, этила или пропила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 алкокси и C1-6 алкокси, замещенного алкилом, предпочтительно водорода, C1-3 алкила, C1-3 алкокси или C1-3 алкила, замещенного C1-3 алкокси, и более предпочтительно водорода, метила, этила, пропила, метокси, этокси, CH3OCH2- или CH3CH2OCH2-;
R3 и R4 связаны с образованием C4-6 циклоалкила или 4-6-членного гетероциклила, предпочтительно 4-6-членного гетероциклила, содержащего один атом кислорода или азота, и более предпочтительно оксетанила;
R2 и R3 или R2 и R4 связаны с образованием 4-6-членного гетероциклила, предпочтительно 4-6-членного гетероциклила, содержащего азот или кислород, где число гетероатомов равно одному или двум, и более предпочтительно тетрагидропирролила, тетрагидрофуранила, пиперидинила или азетидинила;
R3 и R4 связаны с образованием C4-6 циклоалкила или 4-6-членного гетероциклила, предпочтительно 4-6-членного гетероциклила, содержащего один атом кислорода или азота, и более предпочтительно оксетанила;
R6 выбран из группы, состоящей из водорода и галогена, и предпочтительно водорода;
R14 выбран из группы, состоящей из водорода и галогена, и предпочтительно водорода, фтора или хлора;
Raa выбран из группы, состоящей из водорода и галогена, и предпочтительно водорода; и
каждый из R15 и R16 независимо выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила и C1-6 алкила, замещенного галогеном, предпочтительно водорода, фтора, хлора, брома, иода, циано, ацетонитрилила, пропионитрилила или C1-3 алкила, замещенного галогеном, и более предпочтительно водорода, фтора, метила, ацетонитрилила, -CHF2, -CF2CH3 или CHF2CH2-.
В предпочтительном варианте осуществления настоящего изобретения обеспечено любое из соединений формулы (I), его стереоизомер или его фармацевтически приемлемая соль, где
R2 присутствует или отсутствует, когда он присутствует, R2 выбран из группы, состоящей из водорода, метокси, C1-6 алкила и C1-6 галогеналкила;
или R2 и R3 или R2 и R4 связаны с образованием 3-8-членного гетероциклила, и предпочтительно пирролидинила или азетидинила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси и 3-8-членного гетероциклила;
или R3 и R4 связаны с образованием C3-8 циклоалкила или 3-8-членного гетероциклила, и предпочтительно оксетанила;
каждый из R5 и R6 независимо выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила, C1-6 алкокси и C1-6 галогеналкила;
или R5 и R6 связаны с образованием C3-8 циклоалкила или 3-8-членного гетероциклила, и предпочтительно циклобутанила, циклопентила или 1,3-диоксоланила;
R14 выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси и C3-8 циклоалкила;
Ry выбран из группы, состоящей из водорода, C1-6 алкила, галогена, C1-6 алкокси, C1-6 галогеналкила и -(CH2)n1-, предпочтительно водорода, C1-3 алкила или C1-3 галогеналкила, и более предпочтительно водорода, метила или -(CH2)n1-; и
Raa выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила, C1-6 галогеналкила, C1-6 алкокси и C3-8 циклоалкила.
В предпочтительном варианте осуществления, R2 выбран из группы, состоящей из водорода, C1-3 алкила, C1-3 гидроксиалкила и C1-3 галогеналкила, и предпочтительно метила, этила, пропила, гидроксиметила, гидроксиэтила, гидроксипропила, галогенметила, галогенэтила или галогенпропила;
каждый из R3 и R4 независимо выбран из группы, состоящей из C1-3 алкила, C1-3 гидроксиалкила, C1-3 галогеналкила и C1-3 алкокси, и предпочтительно метила, этила, пропила, гидроксиметила, гидроксиэтила, гидроксипропила, галогенметила, галогенэтила, галогенпропила, метокси, этокси или пропокси;
каждый из R5 и R6 независимо выбран из группы, состоящей из водорода, C1-3 алкила, C1-3 алкокси и C1-3 галогеналкила, и предпочтительно метила, этила, пропила, галогенметила, галогенэтила, галогенпропила, метокси, этокси или пропокси;
R14 выбран из группы, состоящей из водорода, C1-3 алкила, C1-3 алкокси и C1-3 галогеналкила, и предпочтительно метила, этила, пропила, галогенметила, галогенэтила, галогенпропила, метокси, этокси или пропокси;
Ry выбран из группы, состоящей из водорода, метила и -(CH2)n1-; и
Raa выбран из группы, состоящей из галогена, циано, C1-3 алкила, C1-3 алкокси и C1-3 галогеналкила, и предпочтительно метила, этила, пропила, галогенметила, галогенэтила, галогенпропила, метокси, этокси или пропокси.
В предпочтительном варианте осуществления настоящего изобретения в любом из соединений формулы (I), его стереоизомере или его фармацевтически приемлемой соли, где Rz выбран из группы, состоящей из водорода, галогена, оксо, тиоксо, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила и -(CH2)n1-, где каждый из указанных C1-6 алкила, C1-6 алкокси и C1-6 галогеналкила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из водорода, галогена, оксо, тиоксо, C1-6 алкила, C1-6 алкокси и C1-6 галогеналкила, Rz предпочтительно представляет собой галоген, C1-6 алкил, C1-6 галогеналкил или оксо, и более предпочтительно галоген, C1-3 алкил, C1-3 галогеналкил или оксо.
Настоящее изобретение также относится к способу получения соединения формулы (IV), его стереоизомера или его фармацевтически приемлемой соли, включающему следующую стадию

реакции соединения формулы (IV-1) и соединения формулы (IV-2) с получением соединения формулы (IV), его стереоизомера или его фармацевтически приемлемой соли;
где:
Х представляет собой галоген; и
кольцо B, Q, Z, G, L, R2-R4, Ry, Rz, q, m, n и t являются такими, как определено в формуле (IV).
Настоящее изобретение также относится к способу получения соединения формулы (VI), его стереоизомера или его фармацевтически приемлемой соли, включающему слудющую стадию

реакции соединения формулы (VI-1) и соединения формулы (IV-2) с получением соединения формулы (VI), его стереоизомера или его фармацевтически приемлемой соли;
где:
Х представляет собой галоген; и
кольцо B, Q, Z, G, L, R2-R6, R14, Ry, Rz, q, m, n и t являются такими, как определено в формуле (VI).
Настоящее изобретение также относится к способу получения соединения формулы (IV), его стереоизомера или его фармацевтически приемлемой соли, включающему следующие стадии:

где:
Х представляет собой галоген; и
кольцо B, Q, Z, G, L, R2-R6, Ry, Rz, q, m, n и t являются такими, как определено в формуле (VI).
Настоящее изобретение также относится к способу получения соединения формулы (VI), его стереоизомера или его фармацевтически приемлемой соли, включающему следующие стадии:
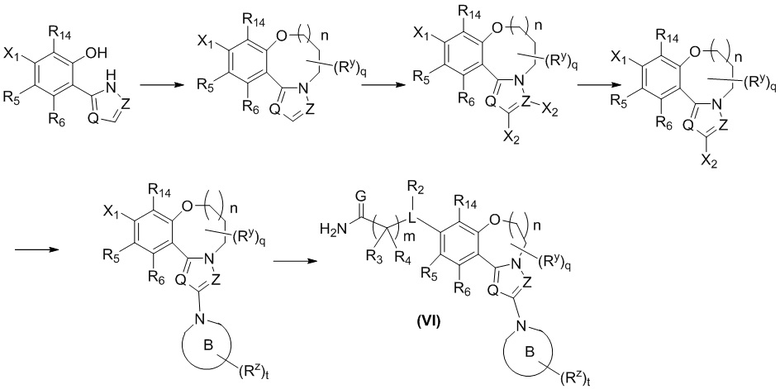
где:
X1 и X2 представляют собой галоген; и
кольцо B, Q, Z, G, L, R2-R6, Ry, Rz, q, m, n и t являются такими, как определено в формуле (VI).
Настоящее изобретение также относится к способу получения соединения формулы (X), его стереоизомера или его фармацевтически приемлемой соли, включающему следующую стадию

реакции соединения формулы (IX) и реагента Лоуссона с получением соединения формулы (IX-A), его стереоизомера или его фармацевтически приемлемой соли;
где:
R2-R4, R6, R14-R16 и Raa являются такими, как определено в формуле (IX).
Настоящее изобретение также относится к способу получения соединения формулы (X), его стереоизомера или его фармацевтически приемлемой соли, включающему следующую стадию

реакции соединения формулы (IX-A) с комплексом переходного металла и его лигандом с получением соединения формулы (X), его стереоизомера или его фармацевтически приемлемой соли;
где:
комплекс переходного металла и его лиганд предпочтительно представляют собой димер дихлор(п-цимен)рутения (II) и 2-бициклогексилфосфино-2',6'-диметоксибифенил; и
R2-R4, R6, R14-R16 и Raa являются такими, как определено в формуле (X).
Настоящее изобретение дополнительно относится к фармацевтической композиции, содержащей терапевтически эффективное количество любого из соединения формулы (I), его стереоизомера или его фармацевтически приемлемой соли и один или более фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ.
Настоящее изобретение, кроме того, относится к применению любого из соединения формулы (I), его стереоизомера или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для получения лекарственного средства, регулирующего PI3K, и предпочтительно лекарственного средства-ингибитора PI3Kα.
Настоящее изобретение также относится к применению соединения формулы (I), его стереоизомера или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции, для получения лекарственного средства для лечения рака, заболевания костей, воспалительного заболевания, иммунного заболевания, заболевания нервной системы, метаболического заболевания, заболевания органов дыхательной системы и заболевания сердца, где указанный рак выбран из группы, состоящей из рака молочной железы, рака поджелудочной железы, немелкоклеточного рака легкого (НМРЛ), рака щитовидной железы, семиномы, меланомы, рака мочевого пузыря, рака печени, рака почки, миелодиспластического синдрома (МДС), острого миелоидного лейкоза (ОМЛ) и колоректального рака.
Настоящее изобретение также относится к применению соединения формулы (I), его стереоизомера или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции, для получения лекарственного средства для лечения рака, заболевания костей, воспалительного заболевания, иммунного заболевания, заболевания нервной системы, метаболического заболевания, заболевания органов дыхательной системы и заболевания сердца.
Настоящее изобретение, кроме того, относится к способу предупреждения и/или лечения рака, включающему введение пациенту терапевтически эффективного количества соединения формулы (I), его стереоизомера или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции.
В настоящем изобретении также предложен способ лечения патологического состояния с помощью соединения или фармацевтической композиции согласно настоящему изобретению, где указанное патологическое состояние включает, не ограничиваясь перечисленным, состояния, связанные с нарушением функции киназ PI3Kα, PI3Kβ, PI3Kδ и PI3Kγ.
Настоящее изобретение также относится к способу лечения гиперпролиферативного заболевания у млекопитающего, включающему введение указанному млекопитающему терапевтически эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, сложного эфира, пролекарства, сольвата, гидрата или производного.
В некоторых вариантах осуществления способ включает лечение заболевания, такого как рак, заболевание костей, воспалительное заболевание, иммунное заболевание, заболевание нервной системы, метаболического заболевания, заболевание органов дыхательной системы и заболевание сердца.
В некоторых вариантах осуществления способ включает лечение рака, такого как острый миелоидный лейкоз, миелодиспластический синдром (МДС), рак тимуса, рак головного мозга, рак легкого (НМРЛ и МРЛ), плоскоклеточная карцинома, семинома, меланома, рак кожи, рак глаза, ретинобластома, внутриглазная меланома, рак полости рта и ротоглотки, рак мочевого пузыря, рак желудочно-кишечного тракта, рак желудка, рак поджелудочной железы, рак мочевого пузыря, рак молочной железы, рак шейки матки, рак головы, рак шеи, рак почек, рак почки, рак печени, рак яичника, рак предстательной железы, рак эндометрия, колоректальный рак, рак пищевода, рак яичка, гинекологический рак, рак щитовидной железы, рак ЦНС (центральная нервная система), рак ПНС (паранеопластический неврологический синдром), СПИД-ассоциированный (синдром иммунодефицита) рак (например, лимфома и саркома Капоши) и вирусиндуцируемый рак. В некоторых вариантах осуществления способ относится к лечению незлокачественного гиперпролиферативного заболевания, такого как кожное заболевание (например, псориаз), рестеноз и доброкачественная гиперплазия предстательной железы (например, доброкачественная гиперплазия предстательной железы (ДГПЖ)). В некоторых вариантах осуществления рак представляет собой меланому или колоректальный рак.
Предложенный в настоящем документе способ лечения включает введение субъекту терапевтически эффективного количества соединения согласно настоящему изобретению. В одном из вариантов осуществления настоящего изобретения предложен способ лечения воспалительного заболевания, включая аутоиммунное заболевание, у млекопитающего. Способ включает введение терапевтически эффективного количества соединения млекопитающему согласно настоящему изобретению или его фармацевтически приемлемой соли, сложного эфира, пролекарства, сольвата, гидрата или производного. Заболевание, связанное с одним или более типами нарушения функции ERK, включает, не ограничиваясь перечисленным, острый рассеянный энцефаломиелит (ОРЭМ), болезнь Аддисона, антифосфолипидный синдром (АФС), апластическую анемию, аутоиммунный гепатит, целиакию, болезнь Крона, диабет (1 типа), синдром Гудпасчера, болезнь Грейвса, синдром Гийена-Барре (СГБ), болезнь Хашимото, системная красная волчанка, рассеянный склероз, миастению гравис, опсо-миоклональный синдром (ОМС), неврит зрительного нерва, тиреоидит Орда, пузырчатку, полиартрит, первичный билиарный цирроз, псориаз, ревматоидный артрит, синдром Литла, артериит Такаясу, височный артериит (также известный как «гигантоклеточный артериит»), аутоиммунную гемолитическую анемию с тепловыми антителами, гранулематоз Вегенера, универсальную алопецию, болезнь Чагаса, синдром хронической усталости, вегетативную дисфункцию, эндометриоз, гнойный гидраденит, интерстициальный цистит, нервно-мышечную ригидность, саркоидоз, склеродермию, язвенный колит, витилиго и вульводинию. Другие заболевания включают нарушение, связанное с резорбцией кости, и тромбоз.
ОПРЕДЕЛЕНИЯ
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют приведенные ниже значения:
Термин «алкил» относится к насыщенной алифатической углеводородной группе, которая представляет собой группу с прямой или разветвленной цепью, содержащую от 1 до 20 атомов углерода, предпочтительно алкил, содержащий от 1 до 8 атомов углерода, более предпочтительно алкил, содержащий от 1 до 6 атомов углерода, и наиболее предпочтительно алкил, содержащий от 1 до 3 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их различные разветвленные изомеры. Более предпочтительно алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и тому подобное. Алкильная группа может быть замещенной или незамещенной. В случае, если она является замещенной, группа (группы) заместителя может быть замещена(ны) в любой доступной соединительной точке. Группа (группы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила. Алкил согласно настоящему изобретению предпочтительно выбран из группы, состоящей из метила, этила, изопропила, трет-бутила, галогеналкила, дейтерированного алкила, алкоксизамещенного алкила и гидроксизамещенного алкила.
Термин «алкилен» относится к алкилу, атом водорода которого дополнительно замещен, например, «метилен» относится к -CH2-, «этилен» относится к -(CH2)2-, «пропилен» относится к -(CH2)3-, «бутилен» относится к -(CH2)4- и т.п. Термин «алкенил» относится к алкилу, как определено выше, который состоит по меньшей мере из двух атомов углерода и по меньшей мере из одной двойной углерод-углеродной связи, например, этенилу, 1-пропенилу, 2-пропенилу, 1-, 2- или 3-бутенилу и т.п. Алкенильная группа может быть замещенной или незамещенной. Когда она является замещенной, группа (группы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио и гетероциклилтио.
Термин «циклоалкил» относится к насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 8 атомов углерода и наиболее предпочтительно от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и тому подобное. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо. Циклоалкил предпочтительно представляет собой циклопропил, циклобутил, циклогексил, циклопентил или циклогептил.
Термин «спироциклоалкил» относится к 5-20-членной полициклической группе с отдельными кольцами, соединенными через один общий атом углерода (называемый спироатомом), где кольца могут содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Спироциклоалкил предпочтительно представляет собой 6-14-членный спироциклоалкил и более предпочтительно 7-10-членный спироциклоалкил. По количеству спироатомов, общих между кольцами, спироциклоалкил можно подразделить на моноспироциклоалкил, диспироциклоалкил или полиспироциклоалкил, и спироциклоалкил предпочтительно представляет собой моноспироциклоалкил или диспироциклоалкил, и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспироциклоалкил. Неограничивающие примеры спироциклоалкила включают:
 ;
;
и также включают спироциклоалкил, в котором циклоалкил и гетероциклил связаны посредством одного спироатома, их неограничивающие примеры включают:
 и
и  .
.
Термин «конденсированный циклоалкил» относится к 5-20-членной полициклической группе, состоящей только из атомов углерода, где каждое кольцо в системе имеет общую смежную пару атомов углерода с другим кольцом, одно или более колец могут содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Конденсированный циклоалкил предпочтительно представляет собой 6-14-членный конденсированный циклоалкил и более предпочтительно 7-10-членный конденсированный циклоалкил. По количеству входящих в него колец конденсированный циклоалкил можно подразделить на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, и конденсированный циклоалкил предпочтительно представляет собой бициклический или трициклический конденсированный циклоалкил, и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный циклоалкил. Неограничивающие примеры конденсированного циклоалкила включают:
 .
.
Термин «мостиковый циклоалкил» относится к 5-20-членной полициклической группе, состоящей только из атомов углерода, в которой каждые два кольца в системе имеют два общих несвязанных атома углерода, кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Мостиковый циклоалкил предпочтительно представляет собой 6-14-членный мостиковый циклоалкил и более предпочтительно 7-10-членный мостиковый циклоалкил. По количеству входящих в него колец мостиковый циклоалкил можно подразделить на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, и мостиковый циклоалкил предпочтительно представляет собой бициклический, трициклический или тетрациклический мостиковый циклоалкил, и более предпочтительно бициклический или трициклический мостиковый циклоалкил. Неограничивающие примеры мостикового циклоалкила включают:

Циклоалкильное кольцо может быть конденсировано с кольцом арила, гетероарила или гетероциклила, где кольцо, связанное с исходной структурой, представляет собой циклоалкил. Неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептил и тому подобное. Циклоалкил может быть необязательно замещенным или незамещенным. Когда он является замещенным, группа (группы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин «гетероциклил» относится к 3-20-членной насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, где один или более атомов кольца представляют собой гетероатомы, выбранные из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2), но исключая -O-O-, -O-S- или -S-S- в кольце, при этом оставшиеся атомы кольца являются атомами углерода. Предпочтительно гетероциклил имеет от 3 до 12 атомов кольца, где от 1 до 4 атомов представляют собой гетероатомы; более предпочтительно от 3 до 8 атомов кольца; более предпочтительно от 3 до 8 атомов кольца; и наиболее предпочтительно от 4 до 6 атомов кольца. Неограничивающие примеры моноциклического гетероциклила включают оксетанил, пирролидинил, пирролидонил, имидазолидинил, тетрагидрофуранил, тетрагидротиенил, дигидроимидазолил, дигидрофуранил, дигидропиразолил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил, пиранил и т.п., и предпочтительно оксетанил, пирролидинил, пирролидонил, тетрагидрофуранил, пиразолидинил, морфолинил, пиперазинил и пиранил. Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо. Гетероциклил, содержащий спирокольцо, конденсированное кольцо или мостиковое кольцо, необязательно связан с другой группой посредством одинарной связи или дополнительно связан с другими циклоалкилом, гетероциклилом, арилом и гетероарилом посредством любых двух или более атомов кольца.
Термин «спирогетероциклил» относится к 3-20-членной полициклической гетероциклильной группе с отдельными кольцами, соединенными посредством одного общего атома (называемого спироатомом), где один или более атомов кольца представляют собой гетероатомы, выбранные из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2), причем оставшиеся атомы кольца являются атомами углерода, и кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Спирогетероциклил предпочтительно представляет собой 6-14-членный спирогетероциклил и более предпочтительно 7-10-членный спирогетероциклил. По количеству спироатомов, общих между кольцами, спирогетероциклил можно подразделить на моноспирогетероциклил, диспирогетероциклил или полиспирогетероциклил, и спирогетероциклил предпочтительно представляет собой моноспирогетероциклил или диспирогетероциклил, и более предпочтительно 3-членный/5-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспирогетероциклил. Неограничивающие примеры спирогетероциклила включают:

Термин «конденсированный гетероциклил» относится к 5-20-членной полициклической гетероциклильной группе, где каждое кольцо в системе имеет общую смежную пару атомов с другим кольцом, одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы, и один или более атомов кольца представляют собой гетероатомы, выбранные из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2), причем оставшиеся атомы кольца являются атомами углерода. Конденсированный гетероциклил предпочтительно представляет собой 6-14-членный конденсированный гетероциклил и более предпочтительно 7-10-членный конденсированный гетероциклил. По количеству входящих в него колец конденсированный гетероциклил можно подразделить на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, и предпочтительно бициклический или трициклический конденсированный гетероциклил, и более предпочтительно 3-членный/5-членный, 4-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Неограничивающие примеры конденсированного гетероциклила включают:
Термин «мостиковый гетероциклил» относится к 5-14-членной полициклической гетероциклильной группе, где каждые два кольца в системе имеет два общих несвязанных атома, где кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы, и один или более атомов кольца представляют собой гетероатомы, выбранные из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2), причем оставшиеся атомы кольца являются атомами углерода. Мостиковый гетероциклил предпочтительно представляет собой 6-14-членный мостиковый гетероциклил и более предпочтительно 7-10-членный мостиковый гетероциклил. По количеству входящих в него колец мостиковый гетероциклил можно подразделить на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил, и мостиковый гетероциклил предпочтительно представляет собой бициклический, трициклический или тетрациклический мостиковый гетероциклил, и более предпочтительно бициклический или трициклический мостиковый гетероциклил. Неограничивающие примеры мостикового гетероциклила включают:
 .
.
Кольцо гетероциклила может быть конденсировано с кольцом арила, гетероарила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероциклил. Неограничивающие примеры включают:


 и т.п.
и т.п.
Гетероциклил может быть необязательно замещенным или незамещенным. Когда он является замещенным, группа (группы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин «арил» относится к 6-14-членному моноциклическому или полициклическому конденсированному кольцу (т. е. каждое кольцо в системе имеет общую пару атомов углерода с другим кольцом в системе), состоящему только из атомов углерода, имеющему сопряженную π-электронную систему, предпочтительно 6-10-членному арилу, например, фенилу и нафтилу. Арил более предпочтительно представляет собой фенил. Кольцо арила может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой кольцо арила. Неограничивающие примеры включают:
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и  .
.
Арил может быть замещенным или незамещенным. Когда он является замещенным, группа (группы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила.
Термин «гетероарил» относится к 5-14-членной гетероароматической системе, имеющей от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, S и N. Гетероарил предпочтительно представляет собой 5-10-членный гетероарил и более предпочтительно 5- или 6-членный гетероарил, например, имидазолил, фурил, тиенил, тиазолил, пиразолил, оксазолил, пирролил, триазолил, тетразолил, пиридил, пиримидинил, тиадиазолил, пиразинил и т.п., предпочтительно триазолил, тиенил, имидазолил, пиразолил, пиридил, пиримидинил и тиазолил, и более предпочтительно триазолил, пирролил, тиенил, тиазолил, пиридил и пиримидинил. Кольцо гетероарила может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой кольцо гетероарила. Неограничивающие примеры включают:
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и  .
.
Гетероарил может быть необязательно замещенным или незамещенным. Когда он является замещенным, группа (группы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила.
Термин «алкокси» относится к группе -O-(алкил) или -O-(незамещенный циклоалкил), где алкил представляет собой такой, как определено выше. Неограничивающие примеры алкокси включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси. Алкокси может быть необязательно замещенным или незамещенным. Когда он является замещенным, группа (группы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила.
«Галогеналкил» относится к алкильной группе, замещенной одним или более атомами галогена, где алкил представляет собой такой, как определено выше.
«Галогеналкокси» относится к алкоксигруппе, замещенной одним или более атомами галогена, где алкокси представляет собой такой, как определено выше.
«Гидроксиалкил» относится к алкильной группе, замещенной гидроксигруппой(ами), где алкил представляет собой такой, как определено выше.
«Алкенил» относится к алкенилу цепочечной структуры, также известному как алкеновая группа. Алкенил может быть дополнительно замещен другой родственной группой, например, алкилом, алкенилом, алкинилом, алкокси, алкилтио, алкиламино, галогеном, тиолом, гидрокси, нитро, циано, циклоалкилом, гетероциклилом, арилом, гетероарилом, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси или алкоксикарбонилом.
«Алкинил» относится к (CH≡C-). Алкинил может быть дополнительно замещен другой родственной группой, например алкилом, алкенилом, алкинилом, алкокси, алкилтио, алкиламино, галогеном, тиолом, гидрокси, нитро, циано, циклоалкилом, гетероциклилом, арилом, гетероарилом, циклоалкокси, гетероциклоалкокси, циклоалкилтиотио, карбокси или алкоксикарбонилом.
«Гидрокси» относится к группе -ОН.
«Галоген» относится к фтору, хлору, брому или иоду.
«Амино» относится к группе -NH2.
«Циано» относится к группе -CN.
«Нитро» относится к группе -NO2.
«Карбокси» относится к группе -C(O)OH.
«ТГФ» относится к тетрагидрофурану.
«EtOAc» относится к этилацетату.
«EA» относится к этилацетату.
«МеОН» относится к метанолу.
«ДМФА» относится к N,N-диметилформамиду.
«DIPEA» относится к диизопропилэтиламину.
«TFA» относится к трифторуксусной кислоте.
«MeCN» относится к ацетонитрилу.
«DMA» относится к N,N-диметилацетамиду.
«Et2O» относится к диэтиловому эфиру.
«DCE» относится к 1,2-дихлорэтану.
«DIPEA» относится к N,N-диизопропилэтиламину.
«NBS» относится к N-бромсукцинимиду.
«NIS» относится к N-иодосукцинимиду.
«Cbz-Cl» относится к бензилхлорформиату.
«Pd2(dba)3» относится к трис(дибензилиденацетон)дипалладию.
«Dppf» относится к 1,1'-бисдифенилфосфиноферроцену.
«HATU» относится к 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфату.
«KHMDS» относится к гексаметилдисилазиду калия.
«LiHMDS» относится к бис(триметилсилил)амиду лития.
«MeLi» относится к метиллитию.
«n-BuLi» относится к н-бутиллитию.
«NaBH(OAc)3» относится к триацетоксиборгидриду натрия.
«ДХМ» относится к дихлорметану.
Различные выражения, такие как «X выбран из группы, состоящей из A, B или C», «X выбран из группы, состоящей из A, B и C», «X представляет собой A, B или C», «X представляет собой A, B и C» и т.п. имеют один и тот же смысл, то есть X может представлять собой одно или более из A, B и C.
Атом водорода согласно настоящему изобретению может быть замещен его изотопом дейтерием. Любой из атомов водорода в соединениях из примеров настоящего изобретения также может быть замещен атомом дейтерия.
«Необязательный» или «необязательно» означает, что описанное далее явление или обстоятельство может, но не обязательно должно, происходить, и такое описание включает ситуацию, в которой явление или обстоятельство происходит или не происходит. Например, «гетероциклил, необязательно замещенн алкилом» означает, что алкильная группа может, но не обязательно должна, присутствовать, и такое описание включает ситуацию, когда гетероциклил замещен алкилом, и когда гетероциклил не замещен алкилом.
«Замещенный» относится к одному или более атомам водорода в группе, предпочтительно до 5 и более предпочтительно от 1 до 3 атомов водорода, независимо замещенных соответствующим числом заместителей. Разумеется, заместители существуют только в возможных для них химических положениях. Специалист в данной области техники способен определить, возможно или невозможно провести замещение с помощью экспериментов или теоретических знаний, не прилагая чрезмерных усилий. Например, комбинация амино- или гидроксигрупп, имеющих свободные атомы водорода, и атомов углерода, имеющих ненасыщенные связи (такие как олефиновые), может быть нестабильной.
«Фармацевтическая композиция» относится к смеси одного или более описанных соединений согласно настоящему изобретению или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами и другими компонентами, такими как физиологически/фармацевтически приемлемые носители и вспомогательные вещества. Назначение фармацевтической композиции состоит в том, чтобы облегчить введение соединения в организм, что способствует абсорбции активного ингредиента, необходимой для проявления биологической активности.
«Фармацевтически приемлемая соль» относится к соли соединения согласно настоящему изобретению, которая является безопасной и эффективной при применении у млекопитающих и обладает желаемой биологической активностью.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение будет дополнительно описано со ссылкой на следующие примеры, которые не следует рассматривать как ограничивающие объем настоящего изобретения.
ПРИМЕРЫ
Структуры соединений согласно настоящему изобретению идентифицировали с помощью ядерного магнитного резонанса (ЯМР) и/или жидкостной хроматографии и тандемной масс-спектрометрии (ЖХ-МС). Сдвиги ЯМР (δ) приведены в миллионных долях (м.д.). Спектры ЯМР определяли на приборе Bruker AVANCE-400. Растворители для определения представляли собой дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный метанол (CDCl3) и дейтерированный хлороформ (CD3OD), а внутренний стандарт представлял собой тетраметилсилан (ТМС).
Жидкостную хроматографию и тандемную масс-спектрометрию (ЖХ-МС) проводили на масс-спектрометре Agilent 1200 Infinity Series. Высокоэффективную жидкостную хроматографию (ВЭЖХ) проводили на жидкостном хроматографе высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire C18 150×4,6 мм) и жидкостном хроматографе высокого давления Waters 2695-2996 (хроматографическая колонка Gimini C18 150×4,6 мм).
Силикагелевые пластины Yantai Huanghai HSGF254 или Qingdao GF254 использовали в качестве пластины для тонкослойной хроматографии на силикагеле (ТСХ). Размер силикагелевой пластины, используемой в ТСХ, составлял от 0,15 до 0,2 мм, а размер силикагелевой пластины, используемой при очистке продукта, составлял от 0,4 мм до 0,5 мм. В качестве носителя для колоночной хроматографии обычно использовали силикагель Yantai Huanghai фракции от 200 до 300 меш.
Исходные вещества, используемые в примерах настоящего изобретения, известны и коммерчески доступны, или могут быть синтезированы путем принятия или в соответствии с методами, известными в данной области техники.
Если не указано иное, все реакции согласно настоящему изобретению проводили при непрерывном перемешивании магнитной мешалкой в атмосфере сухого азота или аргона, растворитель был сухим, а температура реакции была в градусах Цельсия.
Промежуточное соединение 1
(S)-4-(дифторметил)оксазолидин-2-он

Стадия 1: Получение (R)-3-бензил-4-(гидроксиметил)оксазолидин-2-она

(R)-оксиран-2-илметанол (3,7 г, 50,0 ммоль) и (изоцианатометил)бензол (6,66 г, 50,0 ммоль) смешивали в дихлорметане (50 мл). В атмосфере азота реакционный раствор нагревали до 45°C и перемешивали в течение ночи. После охлаждения добавляли 100 мл насыщенного водного раствора бикарбоната натрия, затем экстрагировали реакционный раствор дихлорметаном (100 мл×2). Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (R)-3-бензил-4-(гидроксиметил)оксазолидин-2-она (4,14 г, 40%).
MS m/z (ESI): 208,2 [M+H]+.
Стадия 2: Получение (S)-3-бензил-4-(дигидроксиметил)оксазолидин-2-она

(R)-3-бензил-4-(гидроксиметил)оксазолидин-2-он (4,14 г, 20,0 ммоль) и IBX (16,8 г, 60,0 ммоль) смешивали в этилацетате (100 мл). В атмосфере азота реакционный раствор перемешивали при 85°C в течение 3 часов. Реакционный раствор охлаждали и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта (S)-3-бензил-4-(дигидроксиметил)оксазолидин-2-она (4,46 г), который сразу же использовали на следующей стадии.
MS m/z (ESI): 224,2 [M+H]+.
Стадия 3: Получение (S)-3-бензил-4-(дифторметил)оксазолидин-2-она
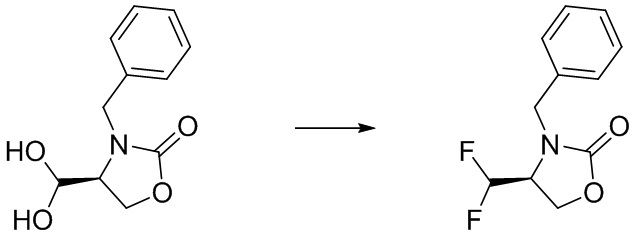
(S)-3-бензил-4-(дигидроксиметил)оксазолидин-2-он (4,46 г, 20,0 ммоль) растворяли в дихлорметане (100 мл). В атмосфере азота по каплям добавляли DAST (6,45 г, 40,0 ммоль) на ледяной бане, а затем реакционный раствор естественным образом нагревали до комнатной температуры и оставляли реагировать в течение 3 часов. Реакционный раствор медленно по каплям добавляли к предварительно охлажденному насыщенному водному раствору бикарбоната натрия, а затем экстрагировали дихлорметаном (200 мл×2). Органические фазы объединяли и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-3-бензил-4-(дифторметил)оксазолидин-2-она (1,82 г, выход для двух стадий 40%).
MS m/z (ESI): 228,2 [M + H]+.
Стадия 4: Получение (S)-4-(дифторметил)оксазолидин-2-она

(S)-3-бензил-4-(дифторметил)оксазолидин-2-он (1,82 г, 8 ммоль) растворяли в этаноле (100 мл) с последующим добавлением Pd(OH)2/C (300 мг). В атмосфере водорода реакционный раствор перемешивали при 70°C в течение ночи. Реакционный раствор охлаждали и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (S)-4-(дифторметил)оксазолидин-2-он (0,88 г, 80%).
1H ЯМР (400 МГц, CDCl3) δ 4,05-4,18 (m, 1H), 4,39-4,45 (m, 1H), 4,54 (t, J = 9,3 Гц, 1H), 5,78 (td, J = 55,3, 4,7 Гц, 1H), 6,07 (s, 1H);
MS m/z (ESI): 138,1 [M+H]+.
Промежуточное соединение 2
9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин
Стадия 1: Получение 5-бром-2-(1H-имидазол-2-ил)фенола
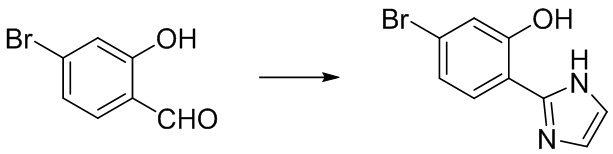
Водный раствор глиоксаля (40 мас.%, 87 г, 597 ммоль) добавляли к раствору 4-бром-2-гидроксибензальдегида (24,0 г, 119 ммоль) в метаноле (250 мл). На водяной бане медленно по каплям добавляли аммиак (28 мас.%, 121 г, 860 ммоль) медленно по каплям добавляли к реакционному раствору при перемешивании, процесс добавления длился 30 минут, и температуру реакционного раствора контролировали так, чтобы она не превышала 40°C. Смесь перемешивали при 35°C в течение двух дней, охлаждали и концентрировали при пониженном давлении, чтобы удалить органический растворитель и получить неочищенный продукт 5-бром-2-(1H-имидазол-2-ил)фенол, который сразу же использовали на следующей стадии.
MS m/z (ESI): 239,0 [M+H]+.
Стадия 2: Получение 9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин

Неочищенный продукт 5-бром-2-(1H-имидазол-2-ил)фенол (приблизительно 29 г, 119 ммоль), карбонат цезия (158 г, 485 ммоль) и 1,2-дибромэтан (42 мл, 485 ммоль) смешивали в ДМФА (250 мл), и перемешивали реакционный раствор при 85°C в течение ночи. Реакционный раствор охлаждали и разбавляли большим количеством этилацетата. Органическую фазу несколько раз промывали насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (12,5 г, выход за две стадии: 38%).
MS m/z (ESI): 265,0 [M+H]+.
Стадия 3: Получение 9-бром-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина

NIS (29,8 г, 132 ммоль) порциями добавляли к раствору 9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (11,7 г, 44,1 ммоль) в ДМФА (150 мл) при комнатной температуре и перемешивали реакционный раствор при 60°C в течение ночи. Реакционный раствор охлаждали и добавляли воду для осаждения твердого вещества. После фильтрации твердое вещество растворяли в этилацетате. Раствор последовательно промывали 1 M водным раствором NaOH и насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали с получением указанного в заголовке соединения 9-бром-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (22,5 г, выход: 98,7%).
MS m/z (ESI): 516,7 [M+H]+.
Стадия 4: Получение 9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина
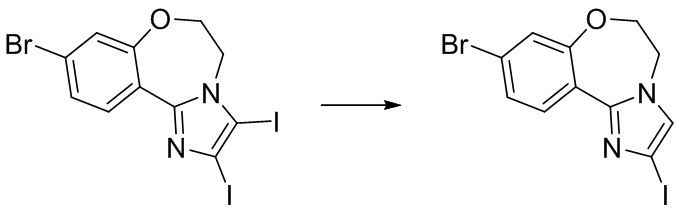
EtMgBr (1,0 M, раствор ТГФ, 60,9 мл, 60,9 ммоль) медленно по каплям добавляли к раствору 9-бром-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (21,0 г, 40,6 ммоль) в ТГФ (140 мл) при минус 20°C. После завершения добавления реакционный раствор перемешивали при минус 15°C в течение 3 часов. Реакционный раствор медленно нагревали до комнатной температуры и по каплям добавляли насыщенный водный раствор хлорида аммония. После перемешивания в течение 15 минут реакционный раствор несколько раз экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (12,5 г, выход за две стадии: 79%).
MS m/z (ESI): 390,9 [M+H]+.
Стадия 5: Получение (S)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-она

9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (300 мг, 0,77 ммоль), (S)-4-(дифторметил)оксазолидин-2-он (105 мг, 0,77 ммоль), (1R,2R)-N1,N2-диметилциклогексан-1,2-диамин (43 мг, 0,30 ммоль), ацетат меди (27 мг, 0,15 ммоль) и карбонат цезия (489 мг, 1,5 ммоль) смешивали с 2-метилтетрагидрофураном (6 мл). Реакционную систему трижды продували азотом и оставляли реагировать при 78°C в течение 22 часов. Реакционный раствор охлаждали до комнатной температуры и добавляли 15% аммиак. Реакционный раствор перемешивали в течение 5 минут и трижды экстрагировали EtOAc. Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-она (186 мг, 61%).
1H ЯМР (400 МГц, CDCl3) δ 4,35-4,41 (m, 2H), 4,44-4,52 (m, 2H), 4,53-4,55 (m, 1H), 4,73-4,76 (m, 1H), 4,89-4,91 (m, 1H), 6,62-6,71 (m, 1H), 7,19-7,28 (m, 2H), 7,30 (s, 1H), 8,21 (d, J = 8,6 Гц, 1H);
MS m/z (ESI): 400,1 [M+H]+.
Пример 1
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин-10-ил)амино)пропанамида
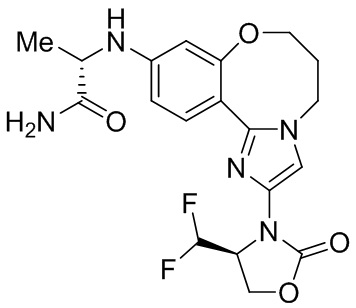
Стадия 1: Получение 10-бром-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцина

5-бром-2-(1H-имидазол-2-ил)фенол (1,0 г, 4,2 ммоль), 1,3-дибромпропан (3,2 г, 15,9 ммоль) и карбонат цезия (5,2 г, 15,9 ммоль) смешивали в N,N-диметилформамиде (20 мл) и перемешивали реакционный раствор при комнатной температуре в течение 1,5 часов. Добавляли воду и перемешивали реакционный раствор в течение 5 минут и трижды экстрагировали EtOAc. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 10-бром-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцина (1,1 г, 94%).
1H ЯМР (400 МГц, ДМСО-d6) δ 1,89-1,94 (m, 2H), 4,03-4,09 (m, 4H), 7,03 (d, J = 0,8 Гц, 1H), 7,28-7,33 (m, 3H), 7,95 (s, 1H);
MS m/z (ESI): 279,0 [M+H]+.
Стадия 2: Получение 10-бром-2,3-дииодо-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцина

Смешивали 10-бром-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин (1,1 г, 3,96 ммоль) и N-иодсукцинимид (2,5 г, 11,08 ммоль) в N, N-диметилформамиде (15 мл). Реакционную систему трижды продували азотом и перемешивали при 80°C в течение ночи. Реакционный раствор охлаждали до комнатной температуры, а затем в реакционную колбу добавляли ледяную воду. Реакционный раствор перемешивали в течение 5 минут и трижды экстрагировали EtOAc. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 10-бром-2,3-дииодо-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцина (720 г, 34%).
MS m/z (ESI): 530,8 [M+H]+.
Стадия 3: Получение 10-бром-2-иодо-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцина
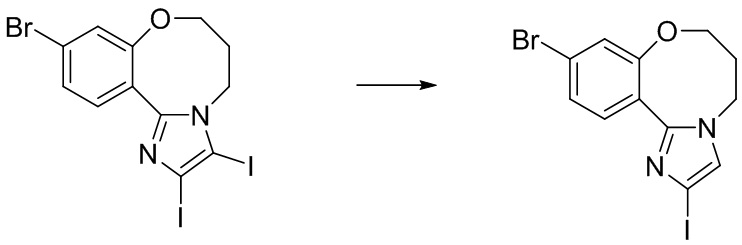
10-бром-2,3-дииодо-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин (500 мг, 0,94 ммоль) растворяли в тетрагидрофуране (10 мл). Реакционную систему охлаждали до минус 20°C и трижды продували азотом. По каплям добавляли этилмагнийбромид (0,35 мл, 1,05 ммоль), и реакционный раствор оставляли реагировать при минус 20°C в течение 3 часов. Добавляли насыщенный раствор хлорида аммония для остановки реакции и экстрагировали реакционный раствор EtOAc три раза. Органическую фазу промывали насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 10-бром-2-иодо-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцина (365 г, 96%).
1H ЯМР (400 МГц, ДМСО-d6) δ 1,87-1,96 (m, 2H), 4,00-4,11 (m, 4H), 7,30-7,37 (m, 2H), 7,47-7,52 (m, 2H);
MS m/z (ESI): 404,9 [M+H]+.
Стадия 4: Получение (S)-3-(10-бром-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин-2-ил)-4-(дифторметил)оксазолидин-2-она

10-бром-2-иодо-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин (300 мг, 0,75 ммоль), (S)-4-(дифторметил)оксазолидин-2-он (103 мг, 0,75 ммоль), (1R,2R)-N1,N2-диметилциклогексан-1,2-диамин (43 мг, 0,30 ммоль), иодид меди (29 мг, 0,15 ммоль) и карбонат калия (205 мг, 1,5 ммоль) смешивали в 1,4-диоксане (6 мл). Реакционную систему трижды продували азотом и оставляли реагировать при 100°C в течение 5 часов. Реакционный раствор охлаждали до комнатной температуры и добавляли 15% аммиак. Реакционный раствор перемешивали в течение 5 минут и трижды экстрагировали EtOAc. Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-3-(10-бром-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин-2-ил)-4-(дифторметил)оксазолидин-2-она (187 мг, 60%).
MS m/z (ESI): 414,0 [M+H]+.
Стадия 5: Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин-10-ил)амино)пропанамида

(S)-3-(10-бром-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин-2-ил)-4-(дифторметил)оксазолидин-2-он (100 мг, 0,24 ммоль), L-аланин (43 мг, 0,48 ммоль), иодид меди (9 мг, 0,048 ммоль) и фосфат калия (103 мг, 0,48 ммоль) смешивали с диметилсульфоксидом (5 мл). Реакционную систему трижды продували азотом и оставляли реагировать при 100°C в течение 5 часов. Реакционный раствор охлаждали до комнатной температуры, затем добавляли хлорид аммония (78 мг, 1,45 ммоль) и триэтиламин (367 мг, 3,63 ммоль) и перемешивали реакционный раствор в течение 5 минут. Добавляли O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфат (830 мг, 2,18 ммоль) и перемешивали реакционный раствор при комнатной температуре в течение 2 часов и фильтровали. К фильтрату добавляли насыщенный водный раствор бикарбоната натрия, который затем трижды экстрагировали этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин-10-ил)амино)пропанамида (34 мг, 34%).
1H ЯМР (400 МГц, ДМСО-d6) δ 1,31 (d, J = 6,9 Гц, 3H), 1,82-1,96 (m, 2H), 3,71-3,82 (m, 1H), 3,87-4,06 (m, 4H), 4,51-4,62 (m, 2H), 4,89-4,95 (m, 1H), 6,11-6,17 (m, 2H), 6,35-6,40 (m, 1H), 6,54-6,82 (m, 1H), 7,01 (s, 1H), 7,20 (s, 1H), 7,32 (d, J = 8,6 Гц, 1H), 7,39 (s, 1H);
MS m/z (ESI): 422,1 [M+H]+.
Пример 2 и Пример 3
Получение (S)-2-(((S)-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6-метил-5,6-дигидробензо[f] имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида и (S)-2-(((R)-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
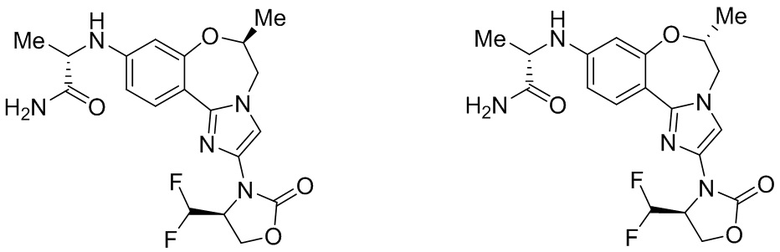
Стадия 1: Получение 2-(5-бром-2-фторфенил)-1H-имидазола

5-бром-2-фторбензальдегид (5,0 г, 24,6 ммоль) растворяли в изопропаноле/воде (25 мл/25 мл) при комнатной температуре с последующим добавлением ацетата аммония (17,6 г, 221,7 ммоль) и добавлением по каплям глиоксаля (4,5 мл, 221,7 ммоль), и реакционный раствор перемешивали в течение ночи. Реакционный раствор разбавляли изопропанолом, фильтровали и концентрировали при пониженном давлении. К концентрату добавляли дихлорметан и воду и разделяли две фазы. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 2-(5-бром-2-фторфенил)-1H-имидазола (3,3 г, 56%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,18-7,27 (m, 2H), 7,33-7,38 (m, 1H), 7,56-7,60 (m, 1H), 8,10-8,16 (m, 1H);
MS m/z (ESI): 241,0[M+H]+.
Стадия 2: Получение 9-бром-6-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина

2-(5-бром-2-фторфенил)-1H-имидазол (1,0 г, 4,2 ммоль) растворяли в N,N-диметилформамиде (5 мл) с последующим добавлением гидрида натрия (221 мг, 4,6 ммоль) в бане с ледяной водой и перемешивали реакционный раствор в течение 10 минут. Добавляли 1,2-пропиленоксид (292 мг, 5,1 ммоль) и нагревали реакционный раствор до 95°C и перемешивали в течение 6 часов. Реакционный раствор охлаждали до комнатной температуры, а затем в реакционную колбу добавляли насыщенный водный раствор хлорида аммония. Реакционный раствор трижды экстрагировали дихлорметаном. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 9-бром-6-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (1,1 г, 94%).
1H ЯМР (400 МГц, ДМСО-d6) δ 1,40 (d, J = 6,4 Гц, 3H), 4,19-4,25 (m, 1H), 4,42-4,53 (m, 2H), 6,96 (d, J = 8,7 Гц, 1H), 7,07 (s, 1H), 7,31 (s, 1H), 7,38-7,42 (m, 1H), 8,41 (d, J = 2,5 Гц, 1H);
MS m/z (ESI): 279,0 [M+H]+.
Стадия 3: Получение 9-бром-2,3-дииод-6-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина

9-бром-2,3-дииод-6-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин получали способом, описанным в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ 1,42 (d, J = 6,4 Гц, 3H), 4,14-4,20 (m, 1H), 4,31-4,36 (m, 1H), 4,44-4,56 (m, 1H), 6,99 (d, J = 8,7 Гц, 1H), 7,43-7,48 (m, 1H), 8,26 (d, J = 2,5 Гц, 1H);
MS m/z (ESI): 530,8 [M+H]+.
Стадия 4: Получение 9-бром-2-иод-6-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина

9-бром-2-иод-6-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин получали способом, описанным в примере 1.
MS m/z (ESI): 404,9 [M+H]+.
Стадия 5: Получение (4S)-3-(9-бром-6-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-она

(4S)-3-(9-бром-6-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-он получали способом, описанным в примере 1.
MS m/z (ESI): 414,0 [M+H]+.
Стадия 6: Получение (S)-2-(((S)-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6-метил-5,6-дигидробензо[f] имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида и (S)-2-(((R)-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-(((S)-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6-метил-5,6-дигидробензо[f] имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид и (S)-2-(((R)-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1, и хиральным разделением.
ЯМР и масс-спектрометрические данные смешанной пары эпимеров представляют собой следующие:
1H ЯМР (400 МГц, ДМСО-d6) δ 1,24-1,41 (m, 6H), 3,65-3,77 (m, 1H), 4,01-4,13 (m, 1H), 4,35-4,41 (m, 2H), 4,55-4,65 (m, 2H), 4,91-4,97 (m, 1H), 5,69 (d, J = 7,0 Гц, 1H), 6,50-6,53 (m, 1H), 6,61-6,94 (m, 1H), 6,76-6,81 (m, 1H), 6,97 (s, 1H), 7,29 (s, 1H), 7,33 (s, 2H);
MS m/z (ESI): 422,1 [M+H]+.
Пример 4
Получение (S)-2-(((R)-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-(((R)-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид способом, описанным в примере 2 и примере 3.
1H ЯМР (400 МГц, CD3OD) δ 1,47 (t, J = 7,4 Гц, 6H), 3,78-3,87 (m, 1H), 4,16-4,23 (m, 1H), 4,40-4,45 (m, 1H), 4,55-4,65 (m, 3H), 4,91-4,97 (m, 1H), 6,17-6,21 (m, 1H), 6,41-6,45 (m, 1H), 6,43-6,73 (m, 1H), 7,20 (s, 1H), 8,06-8,09 (m, 1H);
MS m/z (ESI): 422,1 [M+H]+.
Пример 5
Получение (S)-2-(((S)-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-(((S)-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 2 и примере 3.
1H ЯМР (400 МГц, CD3OD) δ 1,47 (t, J = 7,4 Гц, 6H), 3,78-3,87 (m, 1H), 4,16-4,23 (m, 1H), 4,40-4,45 (m, 1H), 4,55-4,65 (m, 3H), 4,91-4,97 (m, 1H), 6,17-6,21 (m, 1H), 6,41-6,45 (m, 1H), 6,43-6,73 (m, 1H), 7,20 (s, 1H), 8,06-8,09 (m, 1H);
MS m/z (ESI): 422,1 [M+H]+.
Пример 6
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6,6-диметил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6,6-диметил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 2 и примере 3.
1H ЯМР (400 МГц, ДМСО-d6) δ 1,26-1,33 (m, 9H), 3,69-3,75 (m, 1H), 3,90-3,98 (m, 2H), 4,55-4,63 (m, 2H), 4,90-5,00 (m, 1H), 5,78 (d, J = 7,1 Гц, 1H), 6,54-6,58 (m, 1H), 6,78-6,83 (m, 1H), 6,55-6,86 (m, 1H), 6,94 (d, J = 2,8 Гц, 1H), 6,98 (s, 1H), 7,35 (s, 1H), 7,39 (s, 1H);
MS m/z (ESI): 436,1 [M+H]+.
Пример 7
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,5-диметил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,5-диметил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 2 и примере 3.
MS m/z (ESI): 436,1 [M+H]+.
Пример 8
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5H-спиро[бензо[f]имидазо[1,2-d][1,4]оксазепин-6,1'-циклопропан]-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5H-спиро[бензо[f]имидазо[1,2-d][1,4]оксазепин-6,1'-циклопропан]-9-ил)амино)пропанамид получали способом, описанным в примере 2 и примере 3.
MS m/z (ESI): 434,1 [M+H]+.
Пример 9
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6H-спиро[бензо[f]имидазо[1,2-d][1,4]оксазепин-5,1'-циклопропан]-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6H-спиро[бензо[f]имидазо[1,2-d][1,4]оксазепин-5,1'-циклопропан]-9-ил)амино)пропанамид получали способом, описанным в примере 2 и примере 3.
MS m/z (ESI): 434,1 [M+H]+.
Пример 10
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

Стадия 1: Получение 9-бром-3-фтор-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина
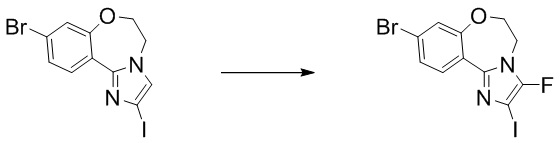
К раствору 9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (500 мг, 1,28 ммоль) в тетрагидрофуране (10 мл) по каплям добавляли раствор LDA (1,28 мл, 2,56 ммоль) в тетрагидрофуране (10 мл) при минус 78°C. После завершения добавления реакционный раствор перемешивали при минус 78°C в течение 30 минут. По каплям добавляли раствор N-фторбензолсульфонамида (806 мг, 2,56 ммоль) в тетрагидрофуране (9 мл) и перемешивали реакционный раствор при минус 78°C в течение 30 минут. Реакцию останавливали насыщенным водным раствором хлорида аммония и экстрагировали реакционный раствор дихлорметаном (100 мл×2). Органическую фазу промывали насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 9-бром-3-фтор-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (150 мг, 29%).
1H ЯМР (400 МГц, ДМСО-d6) δ 4,31-4,34 (m, 2H), 4,43-4,48 (m, 2H), 7,19-7,34 (m, 2H), 8,17 (d, J = 8,6 Гц, 1H);
MS m/z (ESI): 408,9 [M+H]+.
Стадия 2: Получение (S)-3-(9-бром-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-она

9-бром-3-фтор-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (100 мг, 0,24 ммоль), (S)-4-(дифторметил)оксазолидин-2-он (33,5 мг, 0,24 ммоль), (1R,2R)-N1,N2-диметилциклогексан-1,2-диамин (35 мг, 0,24 ммоль), иодид меди (46 мг, 0,24 ммоль) и фосфат калия (155 мг, 0,73 ммоль) смешивали с диметилсульфоксидом (10 мл), и реакционный раствор оставляли реагировать при 130°C в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и добавляли 15% аммиак. Реакционный раствор перемешивали в течение 5 минут и трижды экстрагировали EtOAc. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-3-(9-бром-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-она (21 мг, 20%).
1H ЯМР (400 МГц, CDCl3) δ 4,25-4,29 (m, 1H), 4,42-4,50 (m, 2H), 4,56-4,69 (m, 4H), 6,16-6,35 (m, 1H) , 7,20-7,25 (m, 2H), 8,15 (d, J = 8,4 Гц, 1H);
MS m/z (ESI): 417,9 [M+H]+.
Стадия 3: Получение (2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-L-аланина

(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-L-аланин получали способом, описанным в примере 1.
MS m/z (ESI): 427,1 [M+H]+.
Стадия 4: Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CDCl3) δ 1,55 (d, J = 7,0 Гц, 3H), 3,70-3,87 (m, 1H), 4,21 (d, J = 3,6 Гц, 2H), 4,43 (d, J = 5,2 Гц, 2H), 4,57-4,66 (m, 2H), 5,35 (s, 1H), 6,10-6,27 (m, 2H), 6,37-6,50 (m, 2H), 8,07 (d, J = 8,6 Гц, 1H).
MS m/z (ESI): 426,1 [M+H]+.
Пример 11
Получение (S)-1-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамида

(S)-1-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид получали способом, описанным в примере 10.
MS m/z (ESI): 452,1 [M+H]+.
Пример 12
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)окси)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)окси)пропанамид получали способом, описанным в примере 10.
MS m/z (ESI): 427,1 [M+H]+.
Пример 13
Получение (S)-2-((3-фтор-2-((S)-2-оксо-4-(трифторметил)оксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
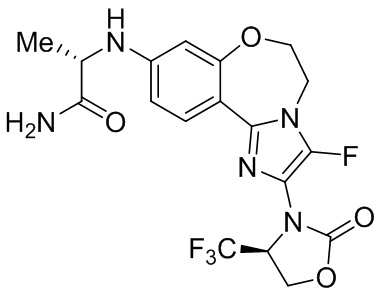
(S)-2-((3-фтор-2-((S)-2-оксо-4-(трифторметил)оксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 10.
MS m/z (ESI): 444,1 [M+H]+.
Пример 14
Получение (S)-2-((3-хлор-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
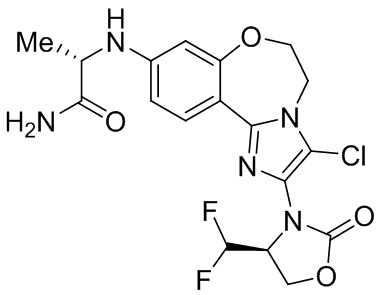
(S)-2-((3-хлор-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 10.
1H ЯМР (400 МГц, CD3OD) δ 1,46 (d, J = 7,0 Гц, 3H), 3,80-3,86 (m, 1H), 4,29-4,32 (m, 2H), 4,43-4,46 (m, 2H), 4,57-4,67 (m, 3H), 6,07-6,31 (m, 2H), 6,43-6,46 (m, 1H), 7,98 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 442,1 [M+H]+.
Пример 15
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
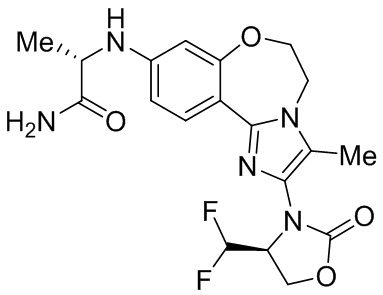
Стадия 1: Получение 5-бром-2-(5-метил-1H-имидазол-2-ил)фенола

Водный раствор пирувальдегида (40 мас.%, 80 мл) добавляли к раствору 4-бром-2-гидроксибензальдегида (5 г, 119 ммоль) в метаноле (100 мл). На водяной бане медленно по каплям добавляли аммиак (28 мас.%, 40 г) к реакционному раствору при перемешивании, процесс добавления длился 30 минут, и температуру реакционного раствора контролировали так, чтобы она не превышала 40°C. Реакционный раствор перемешивали при 75°C в течение 2 часов, затем охлаждали до комнатной температуры для осаждения твердого вещества, которое отфильтровывали с получением указанного в заголовке соединения 5-бром-2-(5-метил-1H-имидазол-2-ил)фенола (3,6 г, 57%).
MS m/z (ESI): 253,0 [M+H]+.
Стадия 2: Получение 9-бром-3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина

5-бром-2-(5-метил-1H-имидазол-2-ил)фенол (2,5 г, 9,8 ммоль), карбонат цезия (12,2 г, 37,5 ммоль) и 1,2-дибромэтан (42,0 мл, 37,5 ммоль) смешивали в ДМФА (30 мл) и перемешивали реакционный раствор при 85°C в течение ночи. Реакционный раствор охлаждали до комнатной температуры и разбавляли большим количеством этилацетата. Органическую фазу несколько раз промывали насыщенным рассолом, сушили над сульфатом натрия и концентрировали. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 9-бром-3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (0,92 г, 33%).
1H ЯМР (400 МГц, CDCl3) δ 2,25 (s, 3H), 4,12-4,29 (m, 2H), 4,40-4,53 (m, 2H), 6,94 (s, 1H), 7,14-7,18 (m, 1H), 7,20-7,22 (m, 1H), 8,37 (d, J = 8,6 Гц, 1H);
MS m/z (ESI): 279,1 [M+H]+.
Стадия 3: Получение 9-бром-2-иод-3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина

9-бром-2-иод-3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин получали способом, описанным в примере 1.
MS m/z (ESI): 404,9 [M+H]+.
Стадия 4: Получение (S)-3-(9-бром-3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-она

(S)-3-(9-бром-3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-он получали способом, описанным в примере 1.
MS m/z (ESI): 414,0 [M+H]+.
Стадия 5: Получение (2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-L-аланина

(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-L-аланин получали способом, описанным в примере 1.
MS m/z (ESI): 423,1 [M+H]+.
Стадия 6: Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 1,37 (d, J = 7,0 Гц, 3H), 2,08 (s, 3H), 3,68-3,75 (m, 1H), 4,18-4,24 (m, 2H), 4,32-4,35 (m, 2H), 4,45-4,61 (m, 3H), 6,10 (m, 2H), 6,34 (d, J = 8,8 Гц, 1H), 7,83 (d, J = 8,8 Гц, 1H).
MS m/z (ESI): 422,2 [M+H]+.
Пример 16
Получение (S)-1-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамида

(S)-1-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид получали способом, описанным в примере 15.
MS m/z (ESI): 448,2 [M+H]+.
Пример 17
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)окси)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)3-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)окси)пропанамид получали способом, описанным в примере 15.
MS m/z (ESI): 423,1 [M+H]+.
Пример 18
Получение (S)-2-((3-метил-2-((S)-2-оксо-4-(трифторметил)оксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
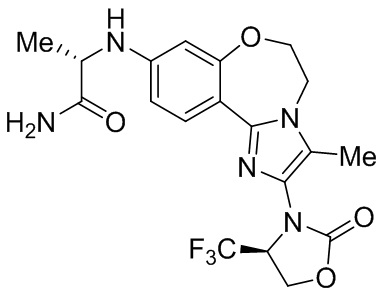
(S)-2-((3-метил-2-((S)-2-оксо-4-(трифторметил)оксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 15.
MS m/z (ESI): 440,1 [M+H]+.
Пример 19
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)3-(трифторметил)5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)3-(трифторметил)5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 15.
MS m/z (ESI): 476,1 [M+H]+.
Пример 20
Получение (S)-2-((3-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((3-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 15.
MS m/z (ESI): 433,1 [M+H]+.
Пример 21
Получение (S)-1-(3-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамида

(S)-1-(3-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид получали способом, описанным в примере 15.
MS m/z (ESI): 459,2 [M+H]+.
Пример 22
Получение (S)-2-((3-циано-2-((S)-2-оксо-4-(трифторметил)оксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((3-циано-2-((S)-2-оксо-4-(трифторметил)оксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 15.
MS m/z (ESI): 451,1 [M+H]+.
Пример 23
Получение (S)-2-((3-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)окси)пропанамида
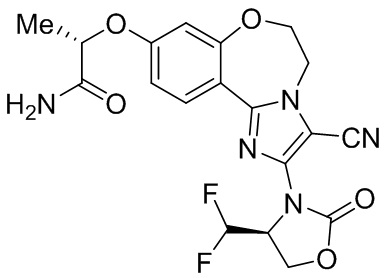
((S)-2-((3-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)окси)пропанамид получали способом, описанным в примере 15.
MS m/z (ESI): 434,1 [M+H]+.
Пример 24
Получение (S)-2-((3-циклопропил-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((3-циклопропил-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 15.
MS m/z (ESI): 448,2 [M+H]+.
Пример 25
Получение (S)-2-((9-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-2-ил)амино)пропанамида
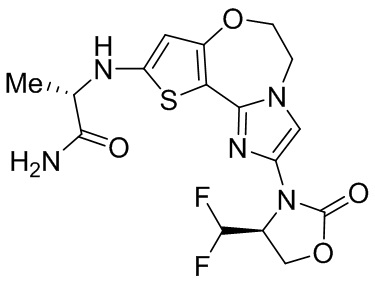
Стадия 1: Получение метил-3-(цианометокси)тиофен-2-карбоксилата

Метил-3-гидрокситиофен-2-карбоксилат (1,58 г, 10 ммоль), бромацетонитрил (2,4 г, 20 ммоль) и карбонат цезия (9,77 г, 30 ммоль) добавляли к ДМФА (40 мл). Реакционный раствор нагревали до 60°C и оставляли реагировать в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли воду (200 мл) и экстрагировали реакционный раствор ЕА (200 мл×3). Органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия (200 мл×3). Органические фазы собирали и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения метил-3-(цианометокси)тиофен-2-карбоксилата (1,58 г, 80%).
Стадия 2: Получение 3,4-дигидротиено[2,3-f][1,4]оксазепин-5(2H)-она

Метил-3-(цианометокси)тиофен-2-карбоксилат (1,58 г, 8 ммоль), никель Ренея (400 мг) и аммиак (2 мл) добавляли к этанолу (100 мл). В атмосфере водорода (50 psi (фунтов на квадратный дюйм)) реакционный раствор нагревали до кипения с обратным холодильником и оставляли реагировать в течение 5 часов. Реакционный раствор охлаждали до комнатной температуры и фильтровали, и концентрировали фильтрат. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 3,4-дигидротиено[2,3-f][1,4]оксазепин-5(2H)-она (1 г, 74%).
MS m/z (ESI): 170,2 [M+H]+.
Стадия 3: Получение метил-2-(5-оксо-2,3-дигидротиено[2,3-f][1,4]оксазепин-4(5H)-ил)ацетата

3,4-дигидротиено[2,3-f][1,4]оксазепин-5(2H)-он (1 г, 5,91 ммоль), метилбромацетат (1,09 г, 7,09 ммоль) и карбонат калия (1,63 г, 11,8 ммоль) добавляли к ацетону (20 мл). Реакционный раствор нагревали до кипения с обратным холодильником и оставляли реагировать в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали. К концентрату добавляли ДХМ и воду и разделяли две фазы. Органическую фазу концентрировали и подвергали остаток колоночной хроматографии с получением указанного в заголовке соединения метил-2-(5-оксо-2,3-дигидротиено[2,3-f][1,4]оксазепин-4(5H)-ил)ацетата (1,21 г, 85%).
MS m/z (ESI): 242,2 [M+H]+.
Стадия 4: Получение 5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-9(8H)-она

Метил-2-(5-оксо-2,3-дигидротиено[2,3-f][1,4]оксазепин-4(5H)-ил)ацетат (1,21 г, 5,02 ммоль) и аммиак (5 мл) добавляли в трет-амиловый спирт (25 мл). Реакционный раствор нагревали до 120°C и оставляли реагировать в течение 5 часов в запаянной пробирке. Реакционный раствор охлаждали до комнатной температуры и концентрировали. К концентрату добавляли ДХМ и воду и разделяли две фазы. Органическую фазу концентрировали, и остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-9(8H)-она (521 мг, 50%).
MS m/z (ESI): 209,2 [M+H]+.
Стадия 5: Получение 9-бром-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4] оксазепина

5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-9(8H)-он (521 мг, 2,50 ммоль) растворяли в 1,2-дихлорэтане (15 мл) с последующим добавлением оксибромида фосфора (2,15 г, 7,50 ммоль). Реакционный раствор нагревали до кипения с обратным холодильником и оставляли реагировать в течение ночи. Реакционный раствор охлаждали до комнатной температуры и доводили его pH до нейтрального насыщенным водным раствором бикарбоната натрия. Реакционный раствор экстрагировали ДХМ и концентрировали органическую фазу. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 9-бром-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепина (407 мг, 60%).
MS m/z (ESI): 271,1 [M+H]+.
Стадия 6: Получение (S)-4-(дифторметил)-3-(5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-9-ил)оксазолидин-2-она

(S)-4-(дифторметил)-3-(5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-9-ил)оксазолидин-2-он получали согласно примеру 21.
MS m/z (ESI): 328,1 [M+H]+.
Стадия 7: Получение (S)-4-(дифторметил)-3-(2-иод-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-9-ил)оксазолидин-2-она

(S)-4-(дифторметил)-3-(5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-9-ил)оксазолидин-2-он (327 мг, 1,0 ммоль) растворяли в дихлорметане (5 мл) и уксусной кислоте (5 мл) с последующим добавлением NIS (248 мг, 1,1 ммоль). Реакционный раствор оставляли реагировать при комнатной температуре в течение ночи. pH реакционного раствора доводили до нейтрального насыщенным водным раствором бикарбоната натрия. Реакционный раствор экстрагировали ДХМ и концентрировали органическую фазу. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-4-(дифторметил)-3-(2-иод-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-9-ил)оксазолидин-2-она (363 мг, 80%).
MS m/z (ESI): 454,1 [M+H]+.
Стадия 8: Получение (S)-2-((9-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-2-ил)амино)пропанамида

(S)-2-((9-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-2-ил)амино)пропанамид получали согласно примеру 1.
MS m/z (ESI): 414,1 [M+H]+.
Пример 26
Получение (S)-2-((9-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-3-фтор-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-2-ил)амино)пропанамида

(S)-2-((9-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-3-фтор-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-2-ил)амино)пропанамид получали согласно примеру 25.
MS m/z (ESI): 432,1 [M+H]+.
Пример 27
Получение (S)-2-((9-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-2-ил)окси)пропанамида

(S)-2-((9-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-2-ил)окси)пропанамид получали согласно примеру 25.
MS m/z (ESI): 415,1 [M+H]+.
Пример 28
Получение (S)-1-(9-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-2-ил)пирролидин-2-карбоксамида

(S)-1-(9-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-2-ил)пирролидин-2-карбоксамид получали согласно примеру 25.
MS m/z (ESI): 440,1 [M+H]+.
Пример 29
Получение (S)-2-((9-((S)-2-оксо-4-(трифторметил)оксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-2-ил)амино)пропанамида

(S)-2-((9-((S)-2-оксо-4-(трифторметил)оксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]тиено[2,3-f][1,4]оксазепин-2-ил)амино)пропанамид получали согласно примеру 25.
MS m/z (ESI): 432,1 [M+H]+.
Пример 30
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6,10,11-тетрагидроциклобута[5,6]бензо[1,2-f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

Стадия 1: Получение 1-(бицикло[4.2.0]окта-1(6),2,4-триен-3-ил)этан-1-она
AlCl3 (3,33 г, 25 ммоль) суспендировали в нитрометане (25 мл) с последующим добавлением по каплям раствора бицикло[4.2.0]окта-1(6),2,4-триена (2,08 г, 20 ммоль) и ацетилхлорида (1,73 г, 22 ммоль) в нитрометане (25 мл) на ледяной бане в атмосфере N2. Реакционный раствор нагревали естественным образом до комнатной температуры и оставляли реагировать в течение ночи. Реакционный раствор добавляли к 200 мл ледяной воды и экстрагировали ДХМ (200 мл×2). Органические фазы объединяли и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 1-(бицикло[4.2.0]окта-1(6),2,4-триен-3-ил)этан-1-она (800 мг, 27%).
Стадия 2: Получение 1-(5-бромбицикло[4.2.0]окта-1(6),2,4-триен-3-ил)этан-1-она

1-(бицикло[4.2.0]окта-1(6),2,4-триен-3-ил)этан-1-он (731 мг, 5 ммоль) растворяли в уксусной кислоте (20 мл). В атмосфере N2 по каплям добавляли бром (878,9 мг, 5,5 ммоль), и реакционный раствор оставляли реагировать при комнатной температуре в течение 3 часов. Реакционный раствор концентрировали, к концентрату добавляли ДХМ и насыщенный водный раствор бикарбоната натрия, и разделяли две фазы. Органическую фазу концентрировали при пониженном давлении и остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 1-(5-бромбицикло[4.2.0]окта-1(6),2,4-триен-3-ил)этан-1-она (900 мг, 80%).
Стадия 3: Получение 5-бромбицикло[4.2.0]окта-1(6),2,4-триен-3-ила ацетата

1-(5-бромбицикло[4.2.0]окта-1(6),2,4-триен-3-ил)этан-1-он (900 мг, 4 ммоль) и m-CPBA (75%, 2,30 г, 10 ммоль) смешивали в ДХМ (20 мл). В атмосфере N2 реакционный раствор нагревали до кипения с обратным холодильником и оставляли реагировать в течение ночи. Реакционный раствор охлаждали до комнатной температуры и фильтровали для удаления нерастворимых веществ. Реакционный раствор промывали насыщенным водным раствором бикарбоната натрия, и органическую фазу концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 5-бромбицикло[4.2.0]окта-1(6),2,4-триен-3-ила ацетата (723 мг, 75%).
Стадия 4: Получение 5-бромбицикло[4.2.0]окта-1(6),2,4-триен-3-ола

5-бромбицикло[4.2.0]окта-1(6),2,4-триен-3-ила ацетат (723 мг, 3 ммоль) растворяли в метаноле (20 мл) с последующим добавлением 5 н. водного раствора гидроксида натрия (3 мл). Реакционный раствор оставляли реагировать при комнатной температуре в течение ночи. К реакционному раствору добавляли 50 мл воды и доводили его pH до 5 с помощью 1 н. соляной кислоты. Реакционный раствор экстрагировали ДХМ (50 мл×2), органические фазы объединяли и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 5-бромбицикло[4.2.0]окта-1(6),2,4-триен-3-ола (567 мг, 95%).
Стадия 5: Получение 5-бром-3-гидроксибицикло[4.2.0]окта-1(6),2,4-триен-2-карбальдегида
5-бромбицикло[4.2.0]окта-1(6),2,4-триен-3-ол (567,2 мг, 2,85 ммоль), хлорид магния (407 мг, 4,28 ммоль) и TEA (1,15 г, 11,4 ммоль) добавляли к ацетонитрилу (5 мл). Реакционный раствор нагревали до 40°C и оставляли реагировать в течение 30 минут. Добавляли параформальдегид (770 мг, 8,55 ммоль), и реакционный раствор оставляли реагировать при 80°C в течение ночи. Реакционный раствор охлаждали до комнатной температуры, добавляли 50 мл воды и pH реакционного раствора доводили до 5 с помощью 4 н. соляной кислоты. Реакционный раствор экстрагировали ДХМ (50 мл×2), органические фазы объединяли и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 5-бром-3-гидроксибицикло[4.2.0]окта-1(6),2,4-триен-2-карбальдегида (517,6 мг, 80%).
Стадия 6: Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6,10,11-тетрагидроциклобута[5,6]бензо[1,2-f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6,10,11-тетрагидроциклобута[5,6]бензо[1,2-f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали согласно примеру 1.
MS m/z (ESI): 434,2 [M+H]+.
Пример 31
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6,10,11-тетрагидроциклобута[5,6]бензо[1,2-f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-2-метоксиацетомида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6,10,11-тетрагидроциклобута[5,6]бензо[1,2-f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-2-метоксиацетомид получали согласно примеру 30.
MS m/z (ESI): 450,1 [M+H]+.
Пример 32
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6,11,12-тетрагидро-10H-имидазо[1,2-d]индено[4,5-f][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6,11,12-тетрагидро-10H-имидазо[1,2-d]индено[4,5-f][1,4]оксазепин-9-ил)амино)пропанамид получали согласно примеру 30.
MS m/z (ESI): 448,1 [M+H]+.
Пример 33
Получение (S)-2-((11-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-7,8-дигидро-[1,3]диоксоло[4',5':5,6]бензо[1,2-f]имидазо[1,2-d][1,4]оксазепин-4-ил)амино)пропанамида
(S)-2-((11-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-7,8-дигидро-[1,3]диоксоло[4',5':5,6]бензо[1,2-f]имидазо[1,2-d][1,4]оксазепин-4-ил)амино)пропанамид получали согласно примеру 30.
MS m/z (ESI): 452,1 [M+H]+.
Пример 34
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-9-ил)амино)пропанамида

Стадия 1: Получение 1-(4-бром-2-метоксифенил)этан-1-она

1-(4-бром-2-гидроксифенил)этан-1-он (5,00 г, 23,3 ммоль), карбонат калия (4,82 г, 35,0 ммоль) и метилиодид (2,94 мл, 46,5 ммоль) смешивали в ДМФА (60 мл) и перемешивали реакционный раствор при комнатной температуре в течение 3 часов. Твердое вещество осаждали добавлением воды к реакционному раствору и сушили с получением указанного в заголовке соединения 1-(4-бром-2-метоксифенил)этан-1-она (5,3 г, 99%).
Стадия 2: Получение метил-3-(4-бром-2-метоксифенил)-3-оксопропаноата

В атмосфере азота к суспензии NaH (1,75 г, 43,7 ммоль) в ТГФ (40 мл) добавляли диметилкарбонат (2,76 мл, 32,7 ммоль). Реакционный раствор нагревали до 70°C, после чего медленно по каплям добавляли раствор 1-(4-бром-2-метоксифенил)этан-1-она (2,50 г, 10,9 ммоль) в ТГФ (10 мл). После завершения добавления реакционный раствор перемешивали при 70°C в течение 3 часов. Реакционный раствор охлаждали и добавляли 1 М раствор HCl для подкисления системы. Реакционный раствор несколько раз экстрагировали этилацетатом. Органические фракции объединяли, сушили над безводным натрия сульфатом и концентрировали. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения метил-3-(4-бром-2-метоксифенил)-3-оксопропаноата (2,2 г, 70%).
MS m/z (ESI): 227,0 [M+H]+.
Стадия 3: Получение 5-(4-бром-2-метоксифенил)-1,2-дигидро-3H-пиразол-3-она
Раствор гидразингидрата (80 мас.%, 3 мл) добавляли к раствору метил-3-(4-бром-2-метоксифенил)-3-оксопропаноата (1,20 г, 4,18 ммоль) в этаноле (50 мл). Реакционный раствор перемешивали при кипении с обратным холодильником в течение 1 часа. Реакционный раствор охлаждали и добавляли воду для осаждения твердого 5-(4-бром-2-метоксифенил)-1,2-дигидро-3H-пиразол-3-она (600 мг). Фильтрат концентрировали и остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 5-(4-бром-2-метоксифенил)-1,2-дигидро-3H-пиразол-3-она (400 мг). После объединения двух веществ было получено в общей сложности 1,0 г указанного в заголовке соединения 5-(4-бром-2-метоксифенил)-1,2-дигидро-3H-пиразол-3-она (89%).
MS m/z (ESI): 269,0 [M+H]+.
Стадия 4: Получение 5-(4-бром-2-гидроксифенил)-1,2-дигидро-3H-пиразол-3-она
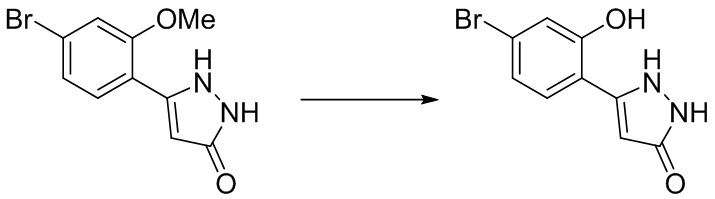
5-(4-бром-2-метоксифенил)-1,2-дигидро-3H-пиразол-3-он (100 мг, 0,372 ммоль) смешивали с раствором BBr3 в ДХМ (1 M, 4 мл) и перемешивали реакционный раствор при комнатной температуре в течение ночи. Реакционный раствор концентрировали при пониженном давлении, чтобы удалить органический растворитель и получить неочищенный продукт 5-(4-бром-2-гидроксифенил)-1,2-дигидро-3H-пиразол-3-он, который сразу же использовали на следующей стадии.
MS m/z (ESI): 255,0 [M+H]+.
Стадия 5: Получение 9-бром-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-2(3H)-она
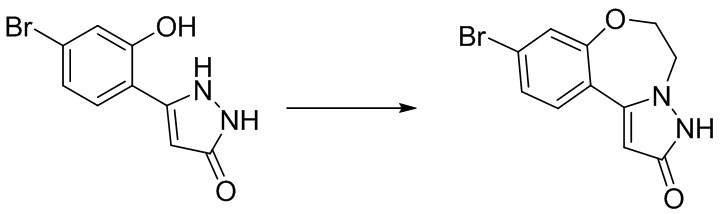
Неочищенный продукт с описанной выше стадии растворяли в ДМФА (4 мл) с последующим последовательным добавлением 1,2-дибромэтана (70 мг, 0,372 ммоль) и карбоната калия (515 мг, 3,72 ммоль). Реакционный раствор перемешивали при 60°C в течение 1 часа, перемешивали при 75°C в течение 1 часа и перемешивали при 90°C в течение 1 часа. Реакционный раствор охлаждали, разбавляли этилацетатом, несколько раз промывали насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 9-бром-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-2(3H)-она (44 мг, выход для двух стадий:42%).
MS m/z (ESI): 281,0 [M+H]+.
Стадия 6: Получение 9-бром-5,6-дигидробензо[f]пиразоло[1,5-d][1,4] оксазепин-2-ил-трифторметансульфоната

Трифторметансульфоновый ангидрид (48 мг, 0,171 ммоль) добавляли по каплям к раствору 9-бром-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-2(3H)-она (40 мг, 0,142 ммоль) в пиридине (1 мл) на бане с ледяной водой. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 9-бром-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-2-ил-трифторметансульфоната (42 мг, 71%).
MS m/z (ESI): 412,9 [M+H]+.
Стадия 7: Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-9-ил)амино)пропанамид получали согласно примеру 1.
MS m/z (ESI): 408,1 [M+H]+.
Пример 35
(S)-1-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид
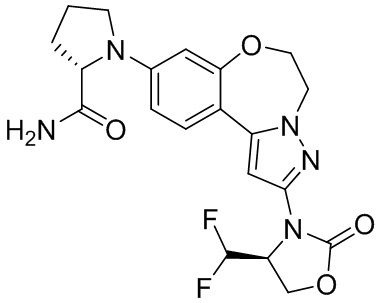
(S)-1-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид получали способом, описанным в примере 34.
MS m/z (ESI): 434,2 [M+H]+.
Пример 36
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-9-ил)окси)пропанамид

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-9-ил)окси)пропанамид получали способом, описанным в примере 34.
MS m/z (ESI): 409,1 [M+H]+.
Пример 37
(S)-2-((2-((S)-2-оксо-4-(трифторметил)оксазолидин-3-ил)-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-9-ил)амино)пропанамид

((S)-2-((2-((S)-2-оксо-4-(трифторметил)оксазолидин-3-ил)-5,6-дигидробензо[f]пиразоло[1,5-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 34.
MS m/z (ESI): 426,1 [M+H]+.
Пример 38
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-3-метилбутанамида
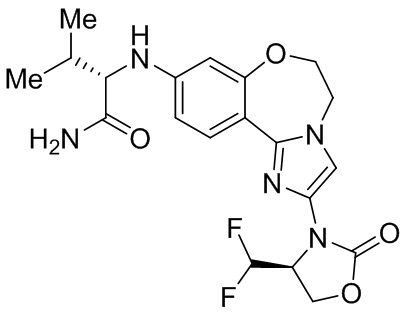
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-3-метилбутанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 1,09 (t, J = 6,1 Гц, 6H), 2,13 (d, J = 7,0 Гц, 1H), 3,60 (d, J = 6,4 Гц, 1H), 4,38 (d, J = 19,3 Гц, 4H), 4,68-4,60 (m, 3H), 6,27 (s, 1H), 6,43-6,78 (m, 2H), 7,17 (s, 1H), 8,06 (d, J = 8,7 Гц, 1H);
MS m/z (ESI): 436,1 [M+H]+.
Пример 39
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-2-метоксиацетамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-2-метоксиацетамид получали способом, описанным в примере 1.
MS m/z (ESI): 424,1 [M+H]+.
Пример 40
Получение (R)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-3-фторпропанамида

(R)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-3-фторпропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 426,1 [M+H]+.
Пример 41
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-2-(оксетан-3-ил)ацетамида
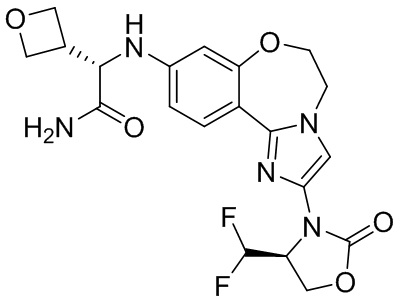
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-2-(оксетан-3-ил)ацетамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 3,26-3,33 (m, 2H), 4,08 (d, J = 9,6 Гц, 1H), 4,22-4,25 (m, 2H), 4,29-4,31 (m, 2H), 4,40-4,50 (m, 5H), 4,61-4,69 (m, 1H), 6,18 (d, J = 2,2 Гц, 1H), 6,44-6,50 (m, 2H), 7,06 (s, 1H), 7,97 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 450,1 [M+H]+.
Пример 42
Получение (S)-2-((2-(4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-2-метилпропанамида

(S)-2-((2-(4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-2-метилпропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 1,50 (s, 6H), 4,31-4,36 (m, 2H), 4,38-4,43 (m, 2H), 4,61-4,65 (m, 2H), 4,95 (d, J = 10,6 Гц, 1H), 6,19 (d, J = 2,2 Гц, 1H), 6,64-6,81 (m, 2H), 7,17 (s, 1H), 8,05 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 422,1 [M+H]+.
Пример 43
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)(метил)амино)пропанамида
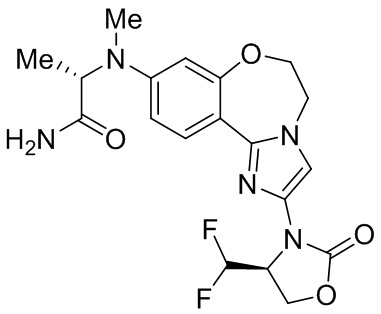
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)(метил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD): δ 1,40 (d, J = 6,8 Гц, 3H), 2,90 (s, 3H), 4,37-4,64 (m, 7H), 4,96 (m, 1H), 6,41 (s, 1H), 6,46-6,74 (m, 2H), 7,16 (s, 1H), 8,13 (d, J = 9,2 Гц, 1H);
MS m/z (ESI): 422,1 [M+H]+.
Пример 44
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропантиоамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропантиоамид получали способом, описанным в примере 1.
MS m/z (ESI): 424,1 [M+H]+.
Пример 45
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)тио)пропанамида
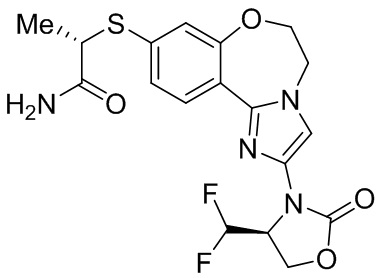
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)тио)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 425,1 [M+H]+.
Пример 46
Получение (S)-3-((2-(4-дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)оксетан-3-карбоксамида

(S)-3-((2-(4-дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)оксетан-3-карбоксамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 4,35 (m, 4H), 4,63 (m, 4H), 4,90 (m, 1H), 5,10 (d, J = 8,0 Гц, 2H), 5,90 (s, 1H), 6,29 (d, J = 8,0 Гц, 1H), 6,59 (t, J = 56 Гц, 1H), 7,16 (s, 1H), 8,10 (d, J = 8,0 Гц, 1H);
MS m/z (ESI): 436,1 [M+H]+.
Пример 47
Получение (S)-3-(9-(((3-аминооксетан-3-ил)метил)амино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-она

(S)-3-(9-(((3-аминооксетан-3-ил)метил)амино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-он получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD): δ 3,35 (s, 2H), 4,24 (d, J = 4,7 Гц, 2H), 4,30 (d, J = 4,7 Гц, 2H), 4,41 (d, J = 6,4 Гц, 2H), 4,45-4,60 (m, 5H), 6,22 (d, J = 2,3 Гц, 1H), 6,27-6,71 (m, 2H), 7,05 (s, 1H), 7,94 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 422,1 [M+H]+.
Пример 48
Получение (S)-1-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)азетидин-2-карбоксамида

(S)-1-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)азетидин-2-карбоксамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 2,30-2,40 (m, 1H), 2,52-2,58 (m, 1H), 3,66-3,72 (m, 1H), 3,91-3,96 (m, 1H), 4,22-4,27 (m, 2H), 4,28-4,34 (m, 2H), 4,48-4,59 (m, 2H), 4,79-4,85 (m, 2H), 6,00 (d, J = 2,2 Гц, 1H), 6,20-6,22 (m, 1H), 6,37-6,65 (m, 1H), 7,08 (s, 1H), 8,06 (d, J = 8,7 Гц, 1H).
MS m/z (ESI): 420,1 [M+H]+.
Пример 49
Получение (S)-1-(2-(4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)азетидин-3-карбоксамида

(S)-1-(2-(4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)азетидин-3-карбоксамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 3,42-3,49 (m, 1H), 3,87 (t, J = 6,7 Гц, 2H), 3,98 (t, J = 7,9 Гц, 2H), 4,23-4,27 (m, 2H), 4,29-4,33 (m, 2H), 4,50-4,58 (m, 3H), 5,97 (d, J = 2,2 Гц, 1H), 6,17-6,20 (m, 1H), 6,36-6,64 (m, 1H), 7,07 (s, 1H), 8,02 (d, J = 8,7 Гц, 1H);
MS m/z (ESI): 420,1 [M+H]+.
Пример 50
Получение (S)-2-((2-((S)-4-(дифторметил)-2-тиоксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-тиоксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 424,1 [M+H]+.
Пример 51
Получение (S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

Стадия 1: Получение (S)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-тиона

Реагент Лоуссона (1,01 г, 2,5 ммоль) добавляли к раствору (S)-3-(10-бром-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин-2-ил)-4-(дифторметил)оксазолидин-2-она (100 мг, 0,25 ммоль) в толуоле (10 мл). Реакционный раствор оставляли реагировать в микроволновой печи при 140°C в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и фильтровали, и промывали фильтровальный осадок EtOAc (20 мл). Фильтрат сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-тиона (42 мг, 40%).
1H ЯМР (400 МГц, ДМСО-d6) δ 4,43-4,52 (m, 4H), 4,79-4,86 (m, 2H), 5,24-5,35 (m, 1H), 6,57-6,85 (m, 1H), 7,23-7,38 (m, 2H), 8,10 (s, 1H), 8,26 (d, J = 8,6 Гц, 1H);
MS m/z (ESI): 416,1 [M+H]+.
Стадия 2: Получение (R)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)тиазолидин-2-она

К раствору (S)-3-(10-бром-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-тиона (33 мг, 0,079 ммоль) в толуоле (1 мл) добавляли димер дихлор(п-цимен)рутения (II) (14,7 мг, 0,024 ммоль) и 2-бисциклогексилфосфино-2',6'-диметоксибифенил (9,7 мг, 0,024 ммоль). Реакционный раствор реагировал в атмосфере воздуха при 110°C в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры и разбавляли EtOAc. Органические фазы промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (R)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)тиазолидин-2-она (26 мг, 79%).
1H ЯМР (400 МГц, CDCl3) δ 3,57-3,72 (m, 2H), 4,28-4,41 (m, 2H),4,44-4,47 (m, 2H) 5,14-5,24 (m, 1H), 6,29-6,67 (m, 1H), 7,14-7,25 (m, 2H), 7,42 (s, 1H), 8,21 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 416,1 [M+H]+.
Стадия 3: Получение (S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(R)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)тиазолидин-2-он (26 мг, 0,062 ммоль), L-аланин (19,5 мг, 0,22 ммоль), иодид меди (6 мг, 0,03 ммоль) и фосфат калия (40 мг, 0,19 ммоль) смешивали в диметилсульфоксиде (3 мл). Реакционную систему трижды продували азотом и оставляли реагировать при 100°C в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли хлорид аммония (20 мг, 0,37 ммоль) и триэтиламин (95 мг, 0,94 ммоль) и перемешивали реакционный раствор в течение 5 минут. Добавляли O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфат (212 мг, 0,56 ммоль) и перемешивали реакционный раствор при комнатной температуре в течение 2 часов. Реакционный раствор фильтровали, к фильтрату добавляли насыщенный водный раствор бикарбоната натрия, который затем трижды экстрагировали этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида (15 мг, 56%).
1H ЯМР (400 МГц, CD3OD) δ 1,37 (d, J = 7,2 Гц, 3H), 3,57-3,61 (m, 1H), 3,83-3,87 (m, 2H), 4,33-4,41 (m, 4H), 5,12-5,19 (m, 1H), 6,15-6,17 (m, 1H), 6,47-6,52 (m, 2H), 7,28 (s, 1H), 8,10 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 424,1 [M+H]+.
Пример 52
Получение (S)-2-((2-((S)-5-(дифторметил)-2-оксоимидазолидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-5-(дифторметил)-2-оксоимидазолидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 51.
MS m/z (ESI): 407,2 [M+H]+.
Пример 53
Получение (S)-2-((2-((S)-5-(дифторметил)-3-метил-2-оксоимидазолидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
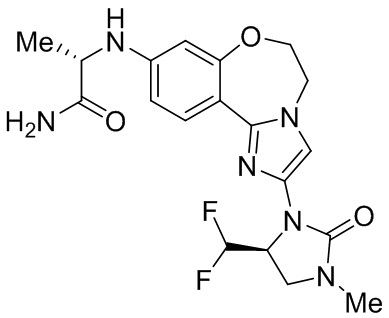
(S)-2-((2-((S)-5-(дифторметил)-3-метил-2-оксоимидазолидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 51.
1H ЯМР (400 МГц, CD3OD) δ 1,46 (d, J = 7,0 Гц, 3H), 2,85 (s, 3H), 3,62-3,68 (m, 2H), 3,79-3,85 (m, 1H), 4,27-4,30 (m, 2H), 4,35-4,37 (m, 2H), 4,63-4,69 (m, 1H), 6,17 (d, J = 2,0 Гц, 1H), 6,34-6,62 (m, 2H), 7,05 (s, 1H), 8,01 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 421,2 [M+H]+.
Пример 54
Получение (S)-2-((2-((4S,5R)-4-(дифторметил)-5-метил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

Стадия 1: Получение метил-(4S,5R)-5-метил-2-оксооксазолидин-4-карбоксилата

Гидрохлорид метил-L-треонината (500 мг, 2,95 ммоль) растворяли в дихлорметане (15 мл), и полученный раствор охлаждали до 0°C на бане с ледяной водой. Добавляли трифосген (289 мг, 0,97 ммоль), затем по каплям добавляли раствор триэтиламина (895 мг, 8,84 ммоль) в дихлорметане (2 мл). После завершения добавления реакционный раствор оставляли реагировать при 0°C в течение 1 часа. Добавляли воду и экстрагировали реакционный раствор дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением указанного в заголовке соединения метил-(4S,5R)-5-метил-2-оксооксазолидин-4-карбоксилата (251 мг, 53%).
MS m/z (ESI): 160,1 [M+H]+.
Стадия 2: Получение метил-(4S,5R)-3-бензил-5-метил-2-оксооксазолидин-4-карбоксилата

Метил-(4S,5R)-5-метил-2-оксооксазолидин-4-карбоксилат (200 мг, 1,26 ммоль) растворяли в ДМФА (5 мл), и полученный раствор охлаждали до минус 15°C. Добавляли NaH (60% в керосине, 50 мг, 1,26 ммоль) и перемешивали реакционный раствор при минус 15°C в течение 1 часа. Добавляли бензилбромид (322 мг, 1,89 ммоль) и перемешивали реакционный раствор в течение 2 часов. Добавляли воду для остановки реакции и экстрагировали реакционный раствор дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением указанного в заголовке соединения метил-(4S,5R)-3-бензил-5-метил-2-оксооксазолидин-4-карбоксилата (260 мг, 83%).
MS m/z (ESI): 250,1 [M+H]+.
Стадия 3: Получение (4R,5R)-3-бензил-4-(гидроксиметил)-5-метилоксазолидин-2-она

Метил-(4S,5R)-3-бензил-5-метил-2-оксооксазолидин-4-карбоксилат (260 мг, 1,0 ммоль) растворяли в метаноле (5 мл), и полученный раствор охлаждали до 0°C на бане с ледяной водой. Порциями добавляли боргидрид натрия (11 мг, 3,1 ммоль), реакционный раствор постепенно нагревали до комнатной температуры и оставляли реагировать в течение 2 часов. Реакционный раствор концентрировали, и полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением указанного в заголовке соединения (4R,5R)-3-бензил-4-(гидроксиметил)-5-метилоксазолидин-2-она (180 мг, 78%).
MS m/z (ESI): 222,1 [M+H]+.
Стадия 4: Получение (4S,5R)-3-бензил-5-метил-2-оксооксазолидин-4-карбальдегида

(4R,5R)-3-бензил-4-(гидроксиметил)-5-метилоксазолидин-2-она (180 мг, 0,81 ммоль) и IBX (683 мг, 2,44 ммоль) смешивали в этилацетате (10 мл). В атмосфере азота реакционный раствор оставляли реагировать при 85°C в течение 3 часов. Реакционный раствор охлаждали и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта (4S,5R)-3-бензил-5-метил-2-оксооксазолидин-4-карбальдегида (178 мг), который сразу же использовали на следующей стадии.
MS m/z (ESI): 220,2 [M+H]+.
Стадия 5: Получение (4S,5R)-3-бензил-4-(дифторметил)-5-метилоксазолидин-2-она

(4S,5R)-3-бензил-5-метил-2-оксооксазолидин-4-карбальдегид (178 мг, 0,81 ммоль) растворяли в дихлорметане (10 мл). В атмосфере азота раствор охлаждали до 0°C на бане с ледяной водой и по каплям добавляли DAST (262 мг, 1,62 ммоль). Реакционный раствор нагревали естественным образом до комнатной температуры и оставляли реагировать в течение 3 часов. Реакционный раствор медленно по каплям добавляли к предварительно охлажденному насыщенному водному раствору бикарбоната натрия, а затем экстрагировали дихлорметаном (20 мл×2). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (4S,5R)-3-бензил-4-(дифторметил)-5-метилоксазолидин-2-она (110 мг, выход для двух стадий:56%).
1H ЯМР (400 МГц, CDCl3) δ 1,33 (d, J = 6,4 Гц, 3H), 3,27-3,33 (m, 1H), 4,16-4,20 (m, 1H), 4,41-4,64 (m, 1H), 4,91 (d, J = 15,0 Гц, 1H), 5,56-5,88 (m, 1H), 7,27-7,44 (m, 5H);
MS m/z (ESI): 242,1 [M+H]+.
Стадия 6: Получение (4S,5R)-4-(дифторметил)-5-метилоксазолидин-2-она

(4S,5R)-3-бензил-4-(дифторметил)-5-метилоксазолидин-2-он (110 мг, 0,46 ммоль) растворяли в мезитилене (2 мл) с последующим добавлением метансульфоновой кислоты (438 мг, 4,56 ммоль). Реакционный раствор нагревали до 135°C и оставляли реагировать в течение 5 часов. После охлаждения до комнатной температуры реакционный раствор медленно по каплям добавляли к предварительно охлажденному насыщенному водному раствору бикарбоната натрия, а затем экстрагировали дихлорметаном (20 мл×2). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением неочищенного указанного в заголовке соединения (4S,5R)-4-(дифторметил)-5-метилоксазолидин-2-она (68 мг), которое сразу же использовали на следующей стадии.
MS m/z (ESI): 152,1 [M+H]+.
Стадия 7: Получение (4S,5R)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)-5-метилоксазолидин-2-она

9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (100 мг, 0,25 ммоль), (4S,5R)-4-(дифторметил)-5-метилоксазолидин-2-он (38,5 мг, 0,25 ммоль), (1R,2R)-N1,N2-диметилциклогексан-1,2-диамин (22 мг, 0,15 ммоль), иодид меди (14 мг, 0,08 ммоль) и фосфат калия (108 мг, 0,51 ммоль) смешивали с диметилсульфоксидом (3 мл), и реакционный раствор оставляли реагировать при 130°C в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и добавляли 15% аммиак (5 мл). Реакционный раствор перемешивали в течение 5 минут и трижды экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-3-(9-бром-3-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-она (61 мг, 57%).
MS m/z (ESI): 414,2 [M+H]+.
Стадия 8: Получение (S)-2-((2-((4S,5R)-4-(дифторметил)-5-метил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(4S,5R)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)-5-метилоксазолидин-2-он (61 мг, 0,15 ммоль), L-аланин (39 мг, 0,44 ммоль), иодид меди (14 мг, 0,07 ммоль) и фосфат калия (94 мг, 0,44 ммоль) смешивали с диметилсульфоксидом (5 мл). Реакционную систему трижды продували азотом и оставляли реагировать при 100°C в течение 5 часов. Реакционный раствор охлаждали до комнатной температуры, затем добавляли хлорид аммония (47 мг, 0,88 ммоль) и триэтиламин (223 мг, 2,21 ммоль) и перемешивали реакционный раствор в течение 5 минут. Добавляли O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфат (505 мг, 1,33 ммоль) и перемешивали реакционный раствор при комнатной температуре в течение 2 часов. Реакционный раствор фильтровали, к фильтрату добавляли насыщенный водный раствор бикарбоната натрия, который затем трижды экстрагировали этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин-10-ил)амино)пропанамида (33 мг, 53%).
1H ЯМР (400 МГц, CD3OD) δ 1,46 (d, J = 6,8 Гц, 3H), 1,53 (d, J = 6,2 Гц, 3H), 3,79-3,85 (m, 1H), 4,32-4,39 (m, 4H), 4,46-4,55 (m, 1H), 4,93-4,95 (m, 1H), 6,17 (s, 1H), 6,39-6,72 (m, 2H), 7,14 (s, 1H), 8,03 (d, J = 8,6 Гц, 1H);
MS m/z (ESI): 422,1 [M+H]+.
Пример 55
Получение (R)-2-((2-((4S,5R)-4-(дифторметил)-5-метил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(R)-2-((2-((4S,5R)-4-(дифторметил)-5-метил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 54.
MS m/z (ESI): 422,2 [M+H]+.
Пример 56
Получение (S)-2-((2-((S)-4-(дифторметил)-5,5-диметил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
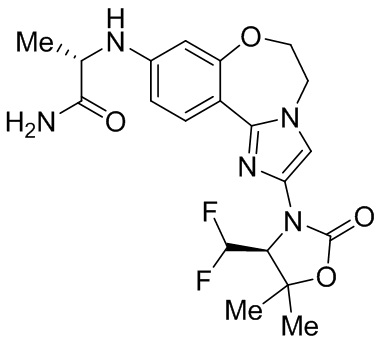
(S)-2-((2-((S)-4-(дифторметил)-5,5-диметил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 54.
MS m/z (ESI): 436,2 [M+H]+.
Пример 57
Получение (S)-2-((2-((S)-7-(дифторметил)-5-оксо-4-окса-6-азаспиро[2,4]гептан-6-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-7-(дифторметил)-5-оксо-4-окса-6-азаспиро[2,4]гептан-6-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 54.
MS m/z (ESI): 434,2 [M+H]+.
Пример 58
Получение (S)-2-((2-((S)-8-(дифторметил)-6-оксо-2,5-диокса-7-азаспиро[3,4]октан-7-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-8-(дифторметил)-6-оксо-2,5-диокса-7-азаспиро[3,4]октан-7-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 54.
MS m/z (ESI): 450,2 [M+H]+.
Пример 59
Получение (S)-2-((2-(6-оксо-2,7-диокса-5-азаспиро[3,4]октан-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
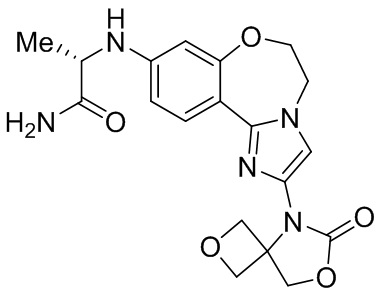
(S)-2-((2-(6-оксо-2,7-диокса-5-азаспиро[3,4]октан-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 1,37 (d, J = 7,0 Гц, 3H), 3,69-3,77 (m, 1H), 4,27-4,38 (m, 4H), 4,62 (d, J = 7,4 Гц, 2H), 4,70 (s, 2H), 5,12 (d, J = 7,4 Гц, 2H), 6,10 (d, J = 2,3 Гц, 1H), 6,33-6,38 (m, 1H), 7,18 (s, 1H), 7,96 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 400,2 [M+H]+.
Пример 60
Получение (S)-2-((2-((R)-4-(метоксиметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((R)-4-(метоксиметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 1,46 (d, J = 7,0 Гц, 3H), 3,34 (s, 3H), 3,57-3,62 (m, 1H), 3,77-3,85 (m, 2H), 4,31-4,35 (m, 2H), 4,37-4,41 (m, 2H), 4,42-4,45 (m, 1H), 4,53-4,55 (m, 1H), 4,63-4,69 (m, 1H), 6,16-6,19 (m, 1H), 6,40-6,45 (m, 1H), 7,12 (s, 1H), 8,01 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 402,2 [M+H]+.
Пример 61
Получение (2S)-2-((2-((5S)-5-(дифторметил)-3-оксо-2-окса-4-азабицикло[3,1.0]гексан-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(2S)-2-((2-((5S)-5-(дифторметил)-3-оксо-2-окса-4-азабицикло[3,1.0]гексан-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 420,1 [M+H]+.
Пример 62
Получение (S)-2-((2-((1R,5S)-6,6-дифтор-3-оксо-2-окса-4-азабицикло[3,1.0]гексан-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((1R,5S)-6,6-дифтор-3-оксо-2-окса-4-азабицикло[3,1.0]гексан-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 406,1 [M+H]+.
Пример 63
Получение (S)-2-((2-((S)-1,1-дифтор-5-оксо-6-окса-4-азаспиро[2,4]гептан-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-1,1-дифтор-5-оксо-6-окса-4-азаспиро[2,4]гептан-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 420,1 [M+H]+.
Пример 64
Получение (S)-2-((2-((S)-5-(дифторметил)-3-метил-2,4-диоксоимидазолидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-5-(дифторметил)-3-метил-2,4-диоксоимидазолидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 435,2 [M+H]+.
Пример 65
Получение метил-(9-(((S)-1-амино-1-оксопропан-2-ил)амино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)((S)-2,2-дифторциклопропил)карбамата

Метил-(9-(((S)-1-амино-1-оксопропан-2-ил)амино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)((S)-2,2-дифторциклопропил)карбамат получали способом, описанным в примере 1.
MS m/z (ESI): 422,2 [M+H]+.
Пример 66
Получение (S)-2-((2-(5,5-дифтор-2-оксо-1,3-оксазинан-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
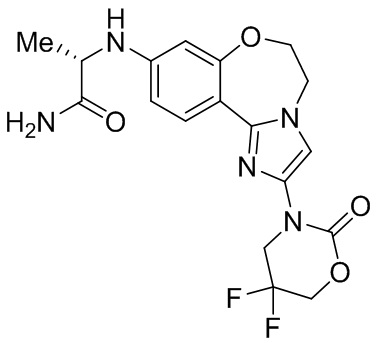
((S)-2-((2-(5,5-дифтор-2-оксо-1,3-оксазинан-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 1,37 (d, J = 7,0 Гц, 3H), 3,70-3,75 (m, 1H), 4,21-4,25 (m, 2H), 4,26-4,32 (m, 3H), 4,44-4,52 (m, 3H), 6,08 (d, J = 2,2 Гц, 1H), 6,36-6,33 (m, 1H), 7,13 (s, 1H), 7,96 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 408,1 [M+H]+.
Пример 67
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксо-1,3-оксазинан-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

Стадия 1: Получение метил-((бензилокси)карбонил)-L-гомосерината

Карбонат калия (545 мг, 3,95 ммоль) и метилиодид (617 мг, 4,35 ммоль) последовательно добавляли к раствору ((бензилокси)карбонил)-L-гомосерина (1,0 г, 3,95 ммоль) в N,N-диметилформамиде (6 мл) и перемешивали реакционный раствор при комнатной температуре в течение ночи. Добавляли насыщенный раствор бикарбоната натрия для остановки реакции и экстрагировали реакционный раствор EtOAc. Органические фазы собирали, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения метил-((бензилокси)карбонил)-L-гомосерината (920 мг, 87%).
MS m/z (ESI): 268,1 [M+H]+.
Стадия 2: Получение метил-L-гомосерината

Метил-((бензилокси)карбонил)-L-гомосеринат (920 мг, 3,4 ммоль) растворяли в метаноле (10 мл) с последующим добавлением Pd/C (50 мг). В атмосфере водорода реакционный раствор перемешивали при комнатной температуре в течение ночи. Реакционный раствор фильтровали и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения метил-L-гомосерината (288 мг, 64%).
MS m/z (ESI): 134,1 [M+H]+.
Стадия 3: Получение метил-(S)-2-оксо-1,3-оксазинан-4-карбоксилата
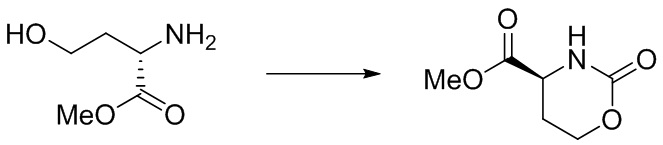
Метил-L-гомосеринат (288 мг, 2,2 ммоль) растворяли в дихлорметане (15 мл) и охлаждали полученный раствор на ледяной бане. Добавляли трифосген (258 мг, 0,87 ммоль), затем по каплям добавляли раствор триэтиламина (658 мг, 6,51 ммоль) в дихлорметане (2 мл). После завершения добавления реакционный раствор оставляли реагировать на ледяной бане в течение 1 часа. Добавляли воду и экстрагировали реакционный раствор дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением чистого продукта метил-(S)-2-оксо-1,3-оксазинан-4-карбоксилата (110 мг, 32%).
MS m/z (ESI): 160,1 [M+H]+.
Стадия 4: Получение метил-(S)-3-бензил-2-оксо-1,3-оксазинан-4-карбоксилата

Метил-(S)-2-оксо-1,3-оксазинан-4-карбоксилат (110 мг, 0,7 ммоль) растворяли в тетрагидрофуране (12 мл) и охлаждали полученный раствор на ледяной бане. Добавляли гидрид натрия (42 мг, 1,06 ммоль), и реакционный раствор перемешивали в течение 10 минут, затем по каплям добавляли раствор бензилбромида (142 мг, 0,84 ммоль) в тетрагидрофуране (2 мл). После завершения добавления реакционный раствор постепенно нагревали до комнатной температуры и оставляли реагировать в течение 2 часов. Добавляли насыщенный раствор хлорида аммония и экстрагировали реакционный раствор этилацетатом. Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением чистого продукта метил-(S)-3-бензил-2-оксо-1,3-оксазинан-4-карбоксилата (100 мг, 57%).
MS m/z (ESI): 250,1 [M+H]+.
Стадия 5: Получение (S)-3-бензил-4-(гидроксиметил)-1,3-оксазинан-2-она

Метил-(S)-3-бензил-2-оксо-1,3-оксазинан-4-карбоксилат (100 мг, 0,4 ммоль) растворяли в метаноле (5 мл) и охлаждали полученный раствор на ледяной бане. Порциями добавляли боргидрид натрия (30 мг, 0,8 ммоль) и постепенно нагревали реакционный раствор до комнатной температуры и оставляли реагировать в течение 2 часов. Реакционный раствор концентрировали, и полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением чистого продукта (S)-3-бензил-4-(гидроксиметил)-1,3-оксазинан-2-она (70 мг, 79%).
MS m/z (ESI): 222,1 [M+H]+.
Стадия 6: Получение (S)-3-бензил-2-оксо-1,3-оксазинан-4-карбальдегида

(S)-3-бензил-4-(гидроксиметил)-1,3-оксазинан-2-она (70 мг, 0,32 ммоль) и IBX (269 мг, 0,96 ммоль) смешивали в этилацетате (5 мл). В атмосфере азота реакционный раствор перемешивали при 85°C в течение 3 часов. Реакционный раствор охлаждали и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта (S)-3-бензил-2-оксо-1,3-оксазинан-4-карбальдегида (68 мг), который сразу же использовали на следующей стадии.
MS m/z (ESI): 220,1[M+H]+.
Стадия 7: Получение (S)-3-бензил-4-(дифторметил)-1,3-оксазинан-2-она

(S)-3-бензил-2-оксо-1,3-оксазинан-4-карбальдегид (68 мг, 0,31 ммоль) растворяли в дихлорметане (5 мл). В атмосфере азота к реакционному раствору на ледяной бане по каплям добавляли DAST (100 мг, 0,62 ммоль). Реакционный раствор нагревали естественным образом до комнатной температуры и оставляли реагировать в течение 3 часов. Реакционный раствор медленно по каплям добавляли к предварительно охлажденному насыщенному водному раствору бикарбоната натрия и экстрагировали дихлорметаном (10 мл×2). Органические фазы объединяли и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-3-бензил-4-(дифторметил)-1,3-оксазинан-2-она (55 мг, 73%).
MS m/z (ESI): 242,1 [M+H]+.
Стадия 8: Получение (S)-4-(дифторметил)-1,3-оксазинан-2-она

(S)-3-бензил-4-(дифторметил)-1,3-оксазинан-2-он (55 мг, 0,23 ммоль) растворяли в этаноле (5 мл) с последующим добавлением Pd(OH)2/C (10 мг). В атмосфере водорода реакционный раствор перемешивали при 70°C в течение ночи. Реакционный раствор охлаждали и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (S)-4-(дифторметил)-1,3-оксазинан-2-она (28 мг, 81%).
MS m/z (ESI): 152,1 [M+H]+.

(S)-2-((2-((S)-4-(дифторметил)-2-оксо-1,3-оксазинан-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 422,2 [M+H]+.
Пример 68
Получение (S)-2-((2-((S)-3-(дифторметил)-5-оксоморфолино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

Стадия 1: Получение (S)-2-(бензиламино)-3,3-дифторпропан-1-ола

5 моль/л водный раствор гидроксида натрия (1,5 мл, 7,5 ммоль) добавляли к раствору (S)-3-бензил-4-(дифторметил)оксазолидин-2-она (340 мг, 1,5 ммоль) в метаноле (5 мл) при комнатной температуре. После завершения добавления реакционный раствор нагревали до 55°C и перемешивали при этой температуре в течение 3 часов. Реакционный раствор охлаждали и концентрировали при пониженном давлении для удаления органического растворителя. В реакционную колбу добавляли воду и трижды экстрагировали реакционный раствор этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя и получения неочищенного указанного в заголовке соединения (S)-2-(бензиламино)-3,3-дифторпропан-1-ола, которое сразу же использовали на следующей стадии.
MS m/z (ESI): 202,1 [M+H]+.
Стадия 2: Получение (S)-N-бензил-2-хлор-N-(1,1-дифтор-3-гидроксипропан-2-ил)ацетамида

Раствор хлорацетилхлорида (186 мг, 1,65 ммоль) в тетрагидрофуране (2 мл) по каплям добавляли к раствору (S)-2-(бензиламино)-3,3-дифторпропан-1-ола (301 мг, 1,5 ммоль) и триэтиламина (379 мг, 3,75 ммоль) в тетрагидрофуране (10 мл) на ледяной бане. После завершения добавления реакционный раствор перемешивали при этой температуре в течение 2 часов. В реакционную колбу добавляли воду для остановки реакции и трижды экстрагировали реакционный раствор этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя и получения неочищенного указанного в заголовке соединения (S)-N-бензил-2-хлор-N-(1,1-дифтор-3-гидроксипропан-2-ил)ацетамида, которое сразу же использовали на следующей стадии.
MS m/z (ESI): 278,1 [M+H]+.
Стадия 3: Получение (S)-4-бензил-5-(дифторметил)морфолин-3-она

Гидрид натрия (72 мг, 1,8 ммоль) добавляли к раствору (S)-N-бензил-2-хлор-N-(1,1-дифтор-3-гидроксипропан-2-ил)ацетамида (415 мг, 1,5 ммоль) в тетрагидрофуране (8 мл) на ледяной бане. После завершения добавления реакционный раствор постепенно нагревали до комнатной температуры и перемешивали в течение 3 часов. В реакционную колбу добавляли насыщенный раствор хлорида аммония для остановки реакции и трижды экстрагировали реакционный раствор этилацетатом. Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-4-бензил-5-(дифторметил)морфолин-3-она (240 мг, 66%).
MS m/z (ESI): 242,1 [M+H]+.
Стадия 4: Получение (S)-5-(дифторметил)морфолин-3-она
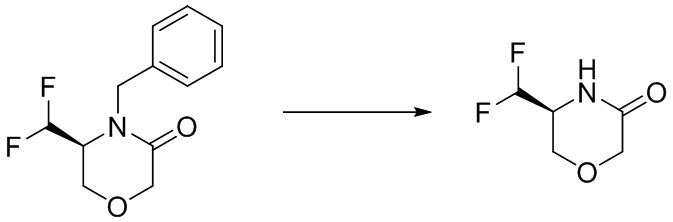
(S)-4-бензил-5-(дифторметил)морфолин-3-он (240 мг, 1,0 ммоль) растворяли в растворе метансульфоновой кислоты (0,5 мл) и мезитилена (2,5 мл) при комнатной температуре. Реакционный раствор оставляли реагировать в микроволновой печи при 135°C в течение 1,5 часов. Реакционный раствор концентрировали, добавляли насыщенный водный раствор карбоната натрия и трижды экстрагировали реакционный раствор этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя и получения неочищенного указанного в заголовке соединения (S)-5-(дифторметил)морфолин-3-она.
MS m/z (ESI): 152,1 [M+H]+.
Стадия 5: Получение (S)-4-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-5-(дифторметил)морфолин-3-она

10-бром-2-иод-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин (258 мг, 0,66 ммоль), (S)-5-(дифторметил)морфолин-3-он (100 мг, 0,66 ммоль), (1R,2R)-N1,N2-диметилциклогексан-1,2-диамин (38 мг, 0,27 ммоль), иодид меди (25 мг, 0,13 ммоль) и карбонат калия (183 мг, 1,32 ммоль) смешивали в 1,4-диоксане (4 мл). Реакционную систему трижды продували азотом и оставляли реагировать при 125°C в течение 5 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли 15% аммиак (5 мл) и перемешивали реакционный раствор в течение 5 минут и трижды экстрагировали EtOAc. Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-4-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-5-(дифторметил)морфолин-3-она (110 мг, 40%).
MS m/z (ESI): 414,0 [M+H]+.
Стадия 6: Получение (S)-2-((2-((S)-3-(дифторметил)-5-оксоморфолино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-4-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-5-(дифторметил)морфолин-3-он (50 мг, 0,12 ммоль), L-аланин (22 мг, 0,24 ммоль), иодид меди (5 мг, 0,025 ммоль) и фосфат калия (51 мг, 0,24 ммоль) смешивали в диметилсульфоксиде (3 мл). Реакционную систему трижды продували азотом и оставляли реагировать при 105°C в течение 2,5 часов. Реакционный раствор охлаждали до комнатной температуры, затем добавляли хлорид аммония (39 мг, 0,73 ммоль) и триэтиламин (183 мг, 1,82 ммоль) и перемешивали реакционный раствор в течение 5 минут. Добавляли O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфат (414 мг, 1,09 ммоль) и перемешивали реакционный раствор при комнатной температуре в течение 2 часов. Реакционный раствор фильтровали, к фильтрату добавляли насыщенный водный раствор бикарбоната натрия, который затем трижды экстрагировали этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-2-((2-((S)-3-(дифторметил)-5-оксоморфолино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида (8,6 мг, 17%).
1H ЯМР (400 МГц, CD3OD) δ 1,37 (d, J = 7,0 Гц, 3H), 3,75-3,77 (m, 1H), 3,92-3,97 (m, 1H), 4,19-4,21 (m, 1H), 4,22-4,25 (m, 2H), 4,28-4,33 (m, 3H), 4,45-4,53 (m, 2H), 6,06-6,10 (m, 1H), 6,22-6,37 (m, 2H), 7,26 (s, 1H), 7,95 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 422,1 [M+H]+.
Пример 69
Получение (S)-2-((2-((S)-2-(дифторметил)-4-метил-6-оксопиперазин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

Стадия 1: Получение (S)-2-(бензиламино)-3,3-дифторпропан-1-ола
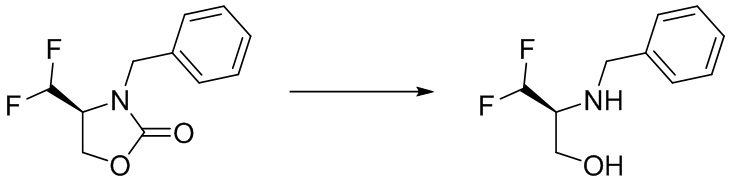
5 моль/л водный раствор гидроксида натрия (8,8 мл, 44,0 ммоль) добавляли к раствору (S)-3-бензил-4-(дифторметил)оксазолидин-2-она (2,0 г, 8,8 ммоль) в метаноле (30 мл) при комнатной температуре. После завершения добавления реакционный раствор нагревали до 55°C и перемешивали при этой температуре в течение 3 часов. Реакционный раствор охлаждали и концентрировали при пониженном давлении для удаления органического растворителя. В реакционную колбу добавляли воду и трижды экстрагировали реакционный раствор этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя и получения неочищенного указанного в заголовке соединения (S)-2-(бензиламино)-3,3-дифторпропан-1-ола, которое сразу же использовали на следующей стадии.
MS m/z (ESI): 202,1 [M+H]+.
Стадия 2: Получение (S)-N-бензил-2-хлор-N-(1,1-дифтор-3-гидроксипропан-2-ил)ацетамида
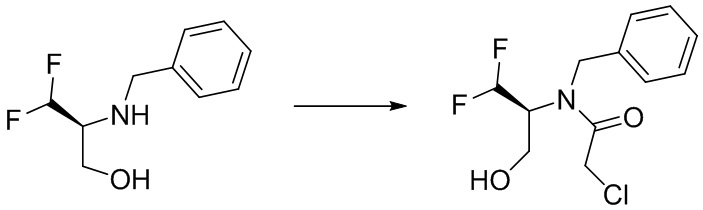
Раствор хлорацетилхлорида (1,2 г, 10,6 ммоль) в тетрагидрофуране (5 мл) по каплям добавляли к раствору (S)-2-(бензиламино)-3,3-дифторпропан-1-ола (1,8 г, 8,8 ммоль) и триэтиламина (1,8 г, 17,6 ммоль) в тетрагидрофуране (30 мл) на ледяной бане. После завершения добавления реакционный раствор перемешивали при этой температуре в течение 2 часов. В реакционную колбу добавляли воду для остановки реакции и трижды экстрагировали реакционный раствор этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя и получения неочищенного указанного в заголовке соединения (S)-N-бензил-2-хлор-N-(1,1-дифтор-3-гидроксипропан-2-ил)ацетамида, которое сразу же использовали на следующей стадии.
MS m/z (ESI): 278,1 [M+H]+.
Стадия 3: Получение (S)-N-бензил-N-(1,1-дифтор-3-гидроксипропан-2-ил)-2-(метиламино)ацетамида
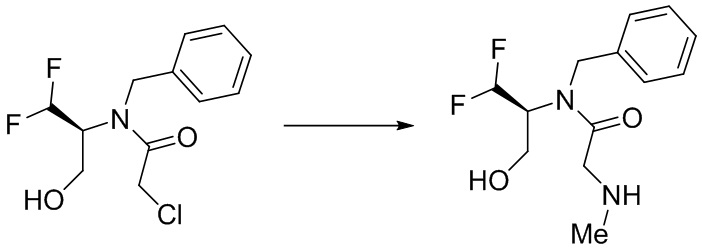
(S)-N-бензил-2-хлор-N-(1,1-дифтор-3-гидроксипропан-2-ил)ацетамид (1,9 г, 6,9 ммоль), гидрохлорид метиламина (2,3 г, 34,5 ммоль) и триэтиламин (4,2 г, 41,4 ммоль) растворяли в тетрагидрофуране (25 мл). Реакционный раствор подвергали взаимодействию при комнатной температуре в течение 1 часа, нагревали до 60°C и оставляли реагировать в течение 2 часов. Реакционный раствор концентрировали, и полученный остаток очищали с помощью колоночной хроматографии с получением чистого продукта (S)-N-бензил-N-(1,1-дифтор-3-гидроксипропан-2-ил)-2-(метиламино)ацетамида (1,0 г, 53%).
MS m/z (ESI): 273,1 [M+H]+.
Стадия 4: Получение (S)-1-бензил-6-(дифторметил)-4-метилпиперазин-2-она

(S)-N-бензил-N-(1,1-дифтор-3-гидроксипропан-2-ил)-2-(метиламино)ацетамид (272 мг, 1,0 ммоль), трифенилфосфор (341 мг, 1,3 ммоль), диизопропилазодикарбоксилат (263 мг, 1,3 ммоль) и N,N-диизопропилэтиламин (194 мг, 1,5 ммоль) растворяли в тетрагидрофуране (12 мл) и оставляли реагировать при комнатной температуре в течение ночи. Реакционный раствор концентрировали, и полученный остаток очищали с помощью колоночной хроматографии с получением чистого продукта (S)-1-бензил-6-(дифторметил)-4-метилпиперазин-2-она (78 мг, 30%).
MS m/z (ESI): 255,1 [M+H]+.
Стадия 5: Получение (S)-6-(дифторметил)-4-метилпиперазин-2-она
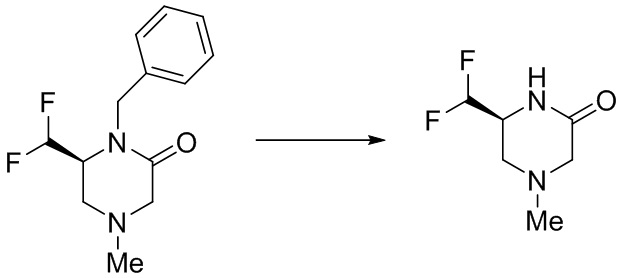
(S)-1-бензил-6-(дифторметил)-4-метилпиперазин-2-он (70 мг, 0,28 ммоль) растворяли в 0,5 мл метансульфоновой кислоты при комнатной температуре. Реакционный раствор оставляли реагировать в микроволновой печи при 150°C в течение 1,5 часов. Реакционный раствор концентрировали, добавляли насыщенный водный раствор карбоната натрия и трижды экстрагировали реакционный раствор этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя и получения неочищенного указанного в заголовке соединения (S)-6-(дифторметил)-4-метилпиперазин-2-она.
MS m/z (ESI): 152,1 [M+H]+.
Стадия 6: Получение (S)-1-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-6-(дифторметил)-4-метилпиперазин-2-она

10-бром-2-иод-6,7-дигидро-5H-бензо[b]имидазо[2,1-d][1,5]оксазоцин (71 мг, 0,18 ммоль), (S)-6-(диформетил)-4-метилпиперазин-2-он (30 мг, 0,18 ммоль), (1R,2R)-N1,N2-диметилциклогексан-1,2-диамин (7 мг, 0,04 ммоль), иодид меди (10 мг, 0,07 ммоль) и карбонат калия (51 мг, 0,37 ммоль) смешивали в 1,4-диоксане (4 мл). Реакционную систему трижды продували азотом и оставляли реагировать при 125°C в течение 5 часов. Реакционный раствор охлаждали до комнатной температуры и добавляли 15% аммиак. Реакционный раствор перемешивали в течение 5 минут и трижды экстрагировали EtOAc. Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-1-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-6-(дифторметил)-4-метилпиперазин-2-она (63 мг, 82%).
MS m/z (ESI): 427,1 [M+H]+.
Стадия 7: Получение (S)-2-((2-((S)-2-(дифторметил)-4-метил-6-оксопиперазин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-1-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-6-(дифторметил)-4-метилпиперазин-2-он (63 мг, 0,15 ммоль), L-аланин (53 мг, 0,6 ммоль), иодид меди (11 мг, 0,06 ммоль) и фосфат калия (191 мг, 0,9 ммоль) смешивали с диметилсульфоксидом (3 мл). Реакционную систему трижды продували азотом и оставляли реагировать при 105°C в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли хлорид аммония (49 мг, 0,9 ммоль) и N,N-диизопропилэтиламин (290 мг, 2,25 ммоль), и перемешивали реакционный раствор в течение 5 минут. Добавляли O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфат (513 мг, 1,35 ммоль) и перемешивали реакционный раствор при комнатной температуре в течение 1 часа. Реакционный раствор фильтровали, к фильтрату добавляли насыщенный водный раствор бикарбоната натрия, который затем трижды экстрагировали этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-2-((2-((S)-2-(дифторметил)-4-метил-6-оксопиперазин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида (4,6 мг, 7%).
1H ЯМР (400 МГц, CD3OD) δ = 1,46 (d, J = 7,0, 3H), 2,36 (s, 3H), 2,76-2,81 (m, 1H), 3,02 (s, 1H), 3,15-3,21 (m, 1H), 3,50-3,55 (m, 1H), 3,79-3,85 (m, 1H), 4,30-4,35 (m, 2H), 4,38-4,42 (m, 2H), 4,72-4,79 (m, 1H), 6,07-6,36 (m, 1H), 6,15-6,19 (m, 1H), 6,40-6,46 (m, 1H), 7,26 (s, 1H), 8,02 (d, J = 8,8, 1H);
MS m/z (ESI): 435,1 [M+H]+.
Пример 70
Получение (S)-2-((2-((S)-2-(дифторметил)-6-оксопиперазин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
(S)-2-((2-((S)-2-(дифторметил)-6-оксопиперазин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 69.
MS m/z (ESI): 421,2 [M+H]+.
Пример 71
Получение (S)-2-((2-((S)-2-(дифторметил)-4-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
(S)-2-((2-((S)-2-(дифторметил)-4-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 408,1 [M+H]+.
Пример 72
Получение (S)-2-((2-((S)-4-(хлордифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
(S)-2-((2-((S)-4-(хлордифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 442,1 [M+H]+.
Пример 73
Получение (2S)-2-((2-((4S)-4-(дифторметил)-2-оксидо-1,2,3-оксатиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(2S)-2-((2-((4S)-4-(дифторметил)-2-оксидо-1,2,3-оксатиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 428,1 [M+H]+.
Пример 74
Получение (S)-2-((2-((S)-4-(дифторметил)-2,2-диоксидо-1,2,3-оксатиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
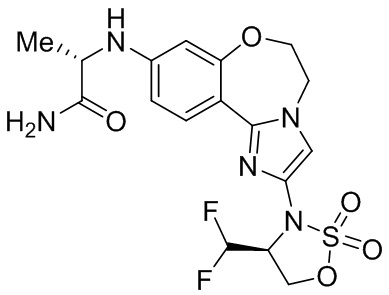
(S)-2-((2-((S)-4-(дифторметил)-2,2-диоксидо-1,2,3-оксатиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 444,1 [M+H]+.
Пример 75
Получение (S)-2-((2-((S)-3-(дифторметил)-1,1-диоксидоизотиазолидин-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-3-(дифторметил)-1,1-диоксидоизотиазолидин-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 442,1 [M+H]+.
Пример 76
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-N-гидроксипропанамида
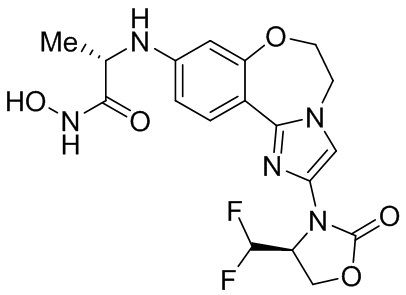
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-N-гидроксипропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD): δ 1,45 (d, J = 6,9 Гц, 3H), 3,79-3,94 (m, 1H), 4,31-4,41 (m, 4H), 4,50-4,70 (m, 3H), 6,18-6,22 (m, 1H), 6,42-6,73 (m, 2H), 7,15 (s, 1H), 8,04 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 424,1 [M+H]+.
Пример 77
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

Стадия 1: Получение 4-бром-3-фтор-2-метоксибензальдегида

Метилат натрия (733 мг, 13,56 ммоль) добавляли к раствору 4-бром-2,3-дифторбензальдегида (2,0 г, 9,05 ммоль) в метаноле (25 мл) при комнатной температуре. Реакционный раствор нагревали до 65°C и оставляли реагировать в течение 2 часов. Реакционный раствор концентрировали, и полученный остаток очищали с помощью колоночной хроматографии с получением 4-бром-3-фтор-2-метоксибензальдегида (1,78 г, 85%).
MS m/z (ESI): 233,0 [M+H]+.
Стадия 2: Получение 4-бром-3-фтор-2-гидроксибензальдегида

Бромистоводородную кислоту (8,7 мл, 48%) добавляли к раствору 4-бром-3-фтор-2-метоксибензальдегида (1,78 г, 7,67 ммоль) в уксусной кислоте (15 мл) при комнатной температуре. Реакционный раствор нагревали до 120°C и оставляли реагировать в течение 16 часов. Реакционный раствор охлаждали и концентрировали при пониженном давлении. В реакционную колбу добавляли воду и этилацетат, а затем разделяли две фазы. Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Полученный остаток очищали с помощью колоночной хроматографии с получением 4-бром-3-фтор-2-гидроксибензальдегида (1,12 г, 67%).
MS m/z (ESI): 219,0 [M+H]+.
Стадия 3: Получение 3-бром-2-фтор-6-(1H-имидазол-2-ил)фенола

Водный раствор глиоксаля (40 мас.%, 3,73 г, 25,7 ммоль) добавляли к раствору 4-бром-3-фтор-2-гидроксибензальдегида (1,12 г, 5,14 ммоль) в метаноле (12 мл). На водяной бане медленно по каплям добавляли аммиак (28 мас.%, 5,14 г, 51,4 ммоль) реакционному раствору при перемешивании, процесс добавления длился 30 минут, и температуру реакционного раствора контролировали так, чтобы она не превышала 40°C. Смесь перемешивали при 35°C в течение двух дней, охлаждали и концентрировали при пониженном давлении для удаления органического растворителя. Полученный остаток очищали с помощью колоночной хроматографии с получением 3-бром-2-фтор-6-(1H-имидазол-2-ил)фенола (1,31 г, 100%).
MS m/z (ESI): 257,0 [M+H]+.
Стадия 4: Получение 9-бром-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина

3-бром-2-фтор-6-(1H-имидазол-2-ил)фенол (1,31 г, 5,14 ммоль), карбонат цезия (6,3 г, 19,53 ммоль) и 1,2-дибромэтан (3,6 г, 19,12 ммоль) смешивали в ДМФА (12 мл) и перемешивали реакционный раствор при 85°C в течение ночи. Реакционный раствор охлаждали и разбавляли этилацетатом. Органическую фазу несколько раз промывали насыщенным рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Полученный остаток очищали с помощью колоночной хроматографии с получением указанного в заголовке соединения 9-бром-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (995 мг, 69%).
MS m/z (ESI): 283,0 [M+H]+.
Стадия 5: Получение 9-бром-8-фтор-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина
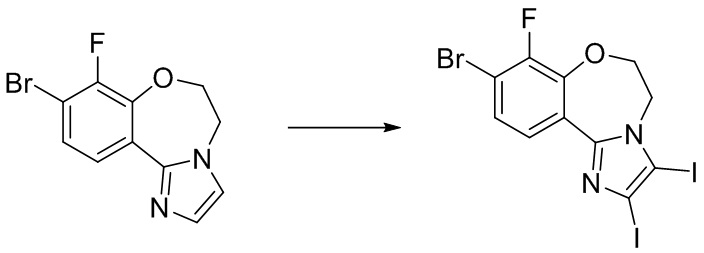
NIS (2,23 г, 9,88 ммоль) добавляли к раствору 9-бром-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (995 мг, 3,53 ммоль) в ДМФА (8 мл) при комнатной температуре, и перемешивали реакционный раствор при 60°C в течение ночи. Реакционный раствор охлаждали и добавляли воду для осаждения твердого вещества. После фильтрации твердое вещество растворяли в этилацетате. Раствор последовательно промывали 1 M водным раствором NaOH и насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали с получением указанного в заголовке соединения 9-бром-8-фтор-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (1,79 г, 94%).
MS m/z (ESI): 534,7 [M+H]+.
Стадия 6: Получение 9-бром-8-фтор-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина

EtMgBr (1,0 M, раствор в ТГФ, 1,23 мл, 3,69 ммоль) медленно по каплям добавляли к раствору 9-бром-8-фтор-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (1,79 г, 3,35 ммоль) в ТГФ (10 мл) при минус 20°C. После завершения добавления реакционный раствор перемешивали при минус 15°C в течение 3 часов. Реакционный раствор медленно нагревали до комнатной температуры, по каплям добавляли насыщенный водный раствор хлорида аммония и перемешивали реакционный раствор в течение 15 минут. Реакционный раствор несколько раз экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 9-бром-8-фтор-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (610 мг, 45%).
MS m/z (ESI): 408,9 [M+H]+.
Стадия 7: Получение (S)-4-(дифторметил)-3-(8-фтор-9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)оксазолидин-2-она

9-бром-8-фтор-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (300 мг, 0,74 ммоль), (S)-4-(дифторметил)оксазолидин-2-он (102 мг, 0,74 ммоль), (1R,2R)-N1,N2-диметилциклогексан-1,2-диамин (42 мг, 0,30 ммоль), иодид меди (28 мг, 0,15 ммоль) и карбонат калия (205 мг, 1,5 ммоль) смешивали с 1,4-диоксаном (6 мл). Реакционную систему трижды продували азотом и проводили реакцию при 105°C в течение 5 часов. Реакционный раствор охлаждали до комнатной температуры и добавляли 15% аммиак. Раствор перемешивали в течение 5 минут и трижды экстрагировали EtOAc. Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-4-(дифторметил)-3-(8-фтор-9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)оксазолидин-2-она (225 мг, 65%).
MS m/z (ESI): 466,0 [M+H]+.
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.

1H ЯМР (400 МГц, CD3OD) δ 1,50 (d, J = 7,0 Гц, 3H), 3,95-4,01 (m, 1H), 4,36-4,41 (m, 2H), 4,47-4,53 (m, 2H), 4,57-4,67 (m, 2H), 4,93-4,98 (m, 1H), 6,37-6,42 (m, 1H), 6,44-6,73 (m, 1H), 7,20 (s, 1H),7,87-7,91 (m, 1H);
MS m/z (ESI): 426,1 [M+H]+.
Пример 78
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-11-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-11-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 1,46 (d, J = 4,0 Гц, 3H), 3,84 (m, 1H), 4,24 (m, 2H), 4,49 (m, 2H), 4,60 (m, 3H), 6,19 (s, 1H), 6,28 (d, J = 8,0 Гц, 1H), 6,49 (t, J = 56 Гц, 1H), 7,30 (s, 1H);
MS m/z (ESI): 426,1 [M+H]+.
Пример 79
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-10-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
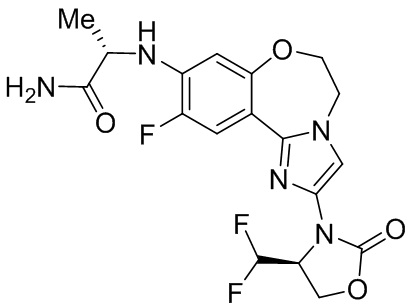
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-10-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD): δ 1,52 (d, J = 6,8 Гц, 3H), 3,86-3,96 (m, 1H), 4,30-4,42 (m, 4H), 4,60-4,69 (m, 3H), 4,91-5,00 (m, 1H), 6,19-6,25 (m, 1H), 6,46-6,76 (m, 1H), 7,18 (s, 1H), 8,04 (d, J = 13,4 Гц, 1H);
MS m/z (ESI): 426,1 [M+H]+.
Пример 80
Получение (S)-3-(9-(4-амино-5-метил-1H-имидазол-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-она

(S)-3-(9-(4-амино-5-метил-1H-имидазол-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-он получали способом, описанным в примере 1.
MS m/z (ESI): 417,1 [M+H]+.
Пример 81
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-8-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-8-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 1,51 (d, J = 6,9 Гц, 3H), 2,15 (s, 3H), 3,99-4,02 (m, 1H), 4,33-4,37 (m, 2H), 4,43-4,47 (m, 2H), 4,55-4,68 (m, 2H), 4,93-4,97 (m, 1H), 6,36 (d, J = 8,9 Гц, 1H), 6,43-6,71 (m, 1H), 7,19 (s, 1H),7,94 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 422,1 [M+H]+.
Пример 82
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-11-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
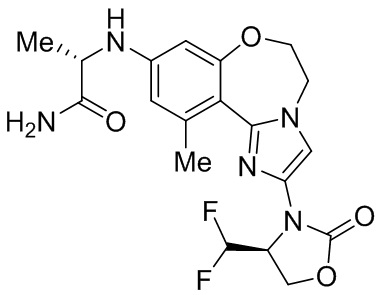
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-11-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 422,1 [M+H]+.
Пример 83
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-10-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-10-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD): δ 1,52 (d, J = 6,9 Гц, 3H), 2,19 (s, 3H), 3,85-3,93 (m, 1H), 4,25-4,36 (m, 4H), 4,55-4,67 (m, 2H), 4,92-4,96 (m, 1H), 6,09 (s, 1H), 6,43-6,71 (m, 1H), 7,12 (s, 1H), 7,90 (s, 1H);
MS m/z (ESI): 422,1 [M+H]+.
Пример 84
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-8-метокси-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
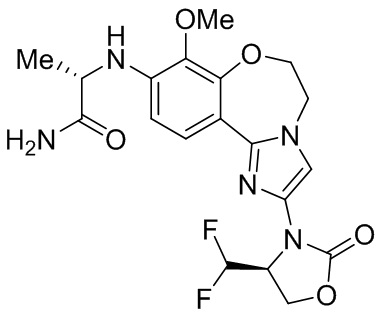
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-8-метокси-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 438,1 [M+H]+.
Пример 85
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-11-метокси-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-11-метокси-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 438,1 [M+H]+.
Пример 86
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-10-метокси-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-10-метокси-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 438,1 [M+H]+.
Пример 87
Получение (S)-2-((8-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((8-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 433,1 [M+H]+.
Пример 88
Получение (S)-2-((11-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((11-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 433,1 [M+H]+.
Пример 89
Получение (S)-2-((10-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((10-циано-2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 433,1 [M+H]+.
Пример 90
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-3-метоксипропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-3-метоксипропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 3,39 (s, 3H), 3,67-3,76 (m, 2H), 3,94-3,98 (m, 1H), 4,30-4,34 (m, 2H), 4,37-4,41 (m, 2H), 4,57-4,66 (m, 2H), 4,91-4,96 (m, 1H), 6,21-6,25 (m, 1H), 6,43-6,46 (m, 1H), 6,48-6,73 (m, 1H), 7,15 (s, 1H), 8,06 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 438,2 [M+H]+.
Пример 91
Получение (2S,3R)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксибутанамида

(2S,3R)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксибутанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD): δ 1,23-1,27 (d, J = 6,9 Гц, 3H), 3,39 (s, 3H), 3,75-3,80 (m, 1H), 3,88-3,93 (m, 1H), 4,29-4,43 (m, 4H), 4,56-4,68 (m, 2H), 4,89-4,98 (m, 1H), 6,22-6,25 (m, 1H), 6,43-6,74 (m, 2H), 7,15 (s, 1H), 8,03-8,08 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 452,2 [M+H]+.
Пример 92
Получение (2S,3S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-3-метоксибутанамида

(2S,3S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-3-метоксибутанамид получали способом, описанным в примере 1.
MS m/z (ESI): 452,2 [M+H]+.
Пример 93
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]пиридо[2,3-f][1,4]оксазепин-9-ил)амино)пропанамида
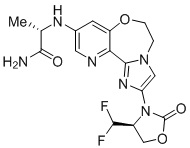
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]пиридо[2,3-f][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 409,2 [M+H]+.
Пример 94
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 409,2 [M+H]+.
Пример 95
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 409,2 [M+H]+.
Пример 96
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f][1,2,4]триазоло[1,5-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f][1,2,4]триазоло[1,5-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 409,2 [M+H]+.
Пример 97
Получение (S)-2-((2-((S)-2-(дифторметил)-5-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-2-(дифторметил)-5-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ 1,30 (d, J = 8,0 Гц, 3H), 2,20-2,45 (m, 3H), 3,31 (d, J = 8,0 Гц, 1H), 3,76 (t, J = 7,6 Гц, 1H), 4,32-4,36 (m, 4H), 4,69-4,78 (m, 1H), 6,08 (s, 1H), 6,15 (d, J = 8,0 Гц, 1H), 6,41 (d, J = 8,0 Гц, 1H), 6,66 (t, J = 56 Гц, 1H), 7,00 (s, 1H), 7,38 (d, J = 8,0 Гц, 1H), 7,40 (s, 1H), 8,00 (d, J = 8,0 Гц, 1H);
MS m/z (ESI): 406,2 [M+H]+.
Пример 98
Получение (S)-2-((2-((S)-7-(дифторметил)-2,5-диокса-8-азаспиро[3,4]октан-8-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
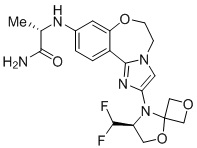
(S)-2-((2-((S)-7-(дифторметил)-2,5-диокса-8-азаспиро[3,4]октан-8-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 436,2 [M+H]+.
Пример 99
Получение (S)-2-((2-(N-((S)-2,2-дифтор-1-(оксетан-3-ил)этил)ацетамидо)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-(N-((S)-2,2-дифтор-1-(оксетан-3-ил)этил)ацетамидо)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 450,2 [M+H]+.
Пример 100
Получение (S)-N-(9-((1-амино-1-оксопропан-2-ил)амино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-N-(2,2-дифторэтил)оксетан-3-карбоксамида

(S)-N-(9-((1-амино-1-оксопропан-2-ил)амино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-N-(2,2-дифторэтил)оксетан-3-карбоксамид получали способом, описанным в примере 1.
MS m/z (ESI): 436,2 [M+H]+.
Пример 101
Получение (S)-N-(9-(((S)-1-амино-1-оксопропан-2-ил)амино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-N-(2,2-дифторэтил)оксетан-2-карбоксамида

(S)-N-(9-(((S)-1-амино-1-оксопропан-2-ил)амино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-N-(2,2-дифторэтил)оксетан-2-карбоксамид получали способом, описанным в примере 1.
MS m/z (ESI): 436,2 [M+H]+.
Пример 102
Получение (R)-N-(9-(((S)-1-амино-1-оксопропан-2-ил)амино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-N-(2,2-дифторэтил)оксетан-2-карбоксамида

(R)-N-(9-(((S)-1-амино-1-оксопропан-2-ил)амино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-N-(2,2-дифторэтил)оксетан-2-карбоксамид получали способом, описанным в примере 1.
MS m/z (ESI): 436,2 [M+H]+.
Пример 103
Получение (S)-2-((2-(N-(2,2-дифторэтил)-2-метоксиацетамидо)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-(N-(2,2-дифторэтил)-2-метоксиацетамидо)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 424,2 [M+H]+.
Пример 104
Получение (S)-2-((2-((3S,5S)-5-(дифторметил)-3-метокси-2-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
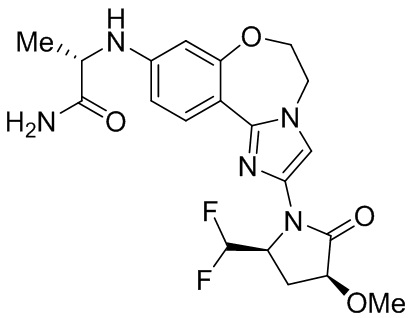
(S)-2-((2-((3S,5S)-5-(дифторметил)-3-метокси-2-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 1,46 (d, J = 7,0 Гц, 3H), 2,10-2,20 (m, 1H), 2,74-2,84 (m, 1H), 3,57 (s, 3H), 3,81 (q, J = 7,0 Гц, 1H), 4,25-4,40 (m, 5H), 4,71-4,84 (m, 1H), 6,13-6,18 (m, 1H), 6,37-6,70 (m, 2H), 7,38 (s, 1H), 8,04 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 436,2 [M+H]+.
Пример 105
Получение (S)-2-((2-((3R,5S)-5-(дифторметил)-3-метокси-2-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
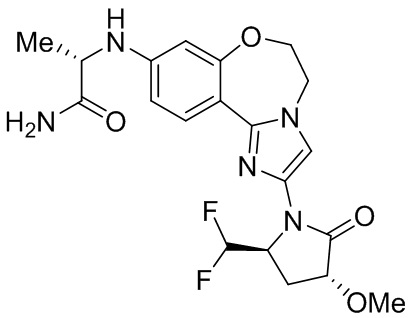
(S)-2-((2-((3R,5S)-5-(дифторметил)-3-метокси-2-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 436,2 [M+H]+.
Пример 106
Получение (1S,5R)-2-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-2-азабицикло[3,1.0]гексан-1-карбоксамида

(1S,5R)-2-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-2-азабицикло[3,1.0]гексан-1-карбоксамид получали способом, описанным в примере 1.
MS m/z (ESI): 446,2 [M+H]+.
Пример 107
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-гидроксипропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-гидроксипропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 3,87 (s, 2H), 4,34 (d, J = 4,3 Гц, 2H), 4,37-4,43 (m, 2H), 4,62 (m, 4H), 6,23 (d, J = 2,6 Гц, 1H), 6,41-6,62 (m, 2H), 7,16 (s, 1H), 8,06 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 424,1[M+H]+.
Пример 108
Получение (S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
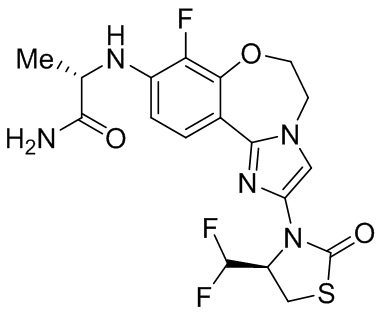
Стадия 1: Получение 4-бром-3-фтор-2-метоксибензальдегида
Метилат натрия (733 мг, 13,56 ммоль) добавляли к раствору 4-бром-2,3-дифторбензальдегида (2,0 г, 9,05 ммоль) в метаноле (25 мл) при комнатной температуре. Реакционный раствор нагревали до 65°C и оставляли реагировать в течение 2 часов. Реакционный раствор концентрировали, и полученный остаток очищали с помощью колоночной хроматографии с получением 4-бром-3-фтор-2-метоксибензальдегида (1,78 г, 85%).
MS m/z (ESI): 233,0 [M+H]+.
Стадия 2: Получение 4-бром-3-фтор-2-гидроксибензальдегида
Бромистоводородную кислоту (8,7 мл, 48%) добавляли к раствору 4-бром-3-фтор-2-метоксибензальдегида (1,78 г, 7,67 ммоль) в уксусной кислоте (15 мл) при комнатной температуре. Реакционный раствор нагревали до 120°C и оставляли реагировать в течение 16 часов. Реакционный раствор охлаждали и концентрировали при пониженном давлении. В реакционную колбу добавляли воду и этилацетат, а затем разделяли две фазы. Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Полученный подвергали колоночной хроматографии с получением 4-бром-3-фтор-2-гидроксибензальдегида (1,12 г, 67%).
MS m/z (ESI): 219,0 [M+H]+.
Стадия 3: Получение 3-бром-2-фтор-6-(1H-имидазол-2-ил)фенола
Водный раствор глиоксаля (40 мас.%, 3,73 г, 25,7 ммоль) добавляли к раствору 4-бром-3-фтор-2-гидроксибензальдегида (1,12 г, 5,14 ммоль) в метаноле (12 мл). На водяной бане медленно по каплям добавляли аммиак (28 мас.%, 5,14 г, 51,4 ммоль) реакционному раствору при перемешивании, процесс добавления длился 30 минут, и температуру реакционного раствора контролировали так, чтобы она не превышала 40°C. Смесь перемешивали при 35°C в течение двух дней, охлаждали и концентрировали при пониженном давлении для удаления органического растворителя. Полученный остаток очищали с помощью колоночной хроматографии с получением 3-бром-2-фтор-6-(1H-имидазол-2-ил)фенола (1,31 г, 100%).
MS m/z (ESI): 257,0 [M+H]+.
Стадия 4: Получение 9-бром-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина
3-бром-2-фтор-6-(1H-имидазол-2-ил)фенол (1,31 г, 5,14 ммоль), карбонат цезия (6,3 г, 19,53 ммоль) и 1,2-дибромэтан (3,6 г, 19,12 ммоль) смешивали в ДМФА (12 мл) и перемешивали реакционный раствор при 85°C в течение ночи. Реакционный раствор охлаждали и разбавляли этилацетатом. Органическую фазу несколько раз промывали насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Полученный остаток очищали с помощью колоночной хроматографии с получением указанного в заголовке соединения 9-бром-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (995 мг, 69%).
MS m/z (ESI): 283,0 [M+H]+.
Стадия 5: Получение 9-бром-8-фтор-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина
NIS (2,23 г, 9,88 ммоль) добавляли к раствору 9-бром-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (995 мг, 3,53 ммоль) в ДМФА (8 мл) при комнатной температуре, и перемешивали реакционный раствор при 60°C в течение ночи. Реакционный раствор охлаждали и добавляли воду для осаждения твердого вещества. После фильтрации твердое вещество растворяли в этилацетате. Раствор последовательно промывали 1 M водным раствором NaOH и насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали с получением указанного в заголовке соединения 9-бром-8-фтор-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (1,79 г, 94%).
MS m/z (ESI): 534,7 [M+H]+.
Стадия 6: Получение 9-бром-8-фтор-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина
EtMgBr (1,0 M, раствор в ТГФ, 1,23 мл, 3,69 ммоль) медленно по каплям добавляли к раствору 9-бром-8-фтор-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (1,79 г, 3,35 ммоль) в ТГФ (10 мл) при минус 20°C. После завершения добавления реакционный раствор перемешивали при минус 15°C в течение 3 часов. Реакционный раствор медленно нагревали до комнатной температуры и по каплям добавляли насыщенный водный раствор хлорида аммония. Реакционный раствор перемешивали в течение 15 минут и несколько раз экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения 9-бром-8-фтор-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (610 мг, 45%).
MS m/z (ESI): 408,9 [M+H]+.
Стадия 7: Получение (S)-4-(дифторметил)-3-(8-фтор-9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)оксазолидин-2-она
9-бром-8-фтор-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (300 мг, 0,74 ммоль), (S)-4-(дифторметил)оксазолидин-2-он (102 мг, 0,74 ммоль), (1R,2R)-N1,N2-диметилциклогексан-1,2-диамин (42 мг, 0,30 ммоль), иодид меди (28 мг, 0,15 ммоль) и карбонат калия (205 мг, 1,5 ммоль) смешивали с 1,4-диоксаном (6 мл). Реакционную систему трижды продували азотом и проводили реакцию при 105°C в течение 5 часов. Реакционный раствор охлаждали до комнатной температуры и добавляли 15% аммиак. Реакционный раствор перемешивали в течение 5 минут и трижды экстрагировали EtOAc. Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-4-(дифторметил)-3-(8-фтор-9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)оксазолидин-2-она (225 мг, 65%).
MS m/z (ESI): 466,0 [M+H]+.
Стадия 8: Получение (S)-4-(дифторметил)-3-(8-фтор-9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)оксазолидин-2-тиона

Реагент Лоуссона (1,92 г, 4,73 ммоль) добавляли к раствору (S)-4-(дифторметил)-3-(8-фтор-9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)оксазолидин-2-она (220 мг, 0,47 ммоль) в толуоле (20 мл). Реакционный раствор нагревали до 145°C и оставляли реагировать в течение 6 часов. Реакционный раствор охлаждали до комнатной температуры и фильтровали, и промывали фильтровальный осадок EtOAc (20 мл). Фильтрат сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-4-(дифторметил)-3-(8-фтор-9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)оксазолидин-2-тиона (105 мг, 46%).
MS m/z (ESI): 482,1[M+H]+.
Стадия 9: Получение (R)-4-(дифторметил)-3-(8-фтор-9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)тиазолидин-2-она
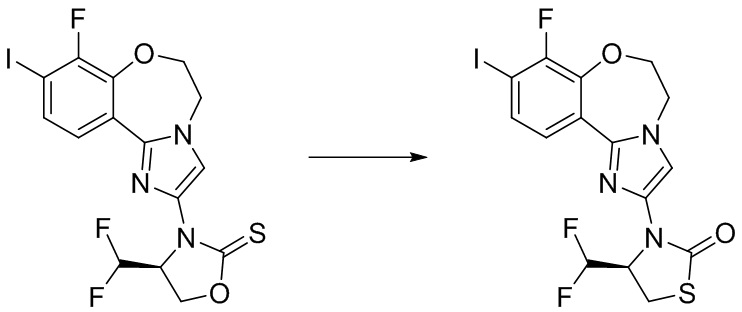
Димер дихлор(п-цимен)рутения (II) (27 мг, 0,045 ммоль) и 2-бициклогексилфосфино-2',6'-диметоксибифенил (27 мг, 0,065 ммоль) добавляли к раствору (S)-3-(9-бром-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-дифторметил)оксазолидин-2-тиона (105 мг, 0,22 ммоль) в толуоле (3 мл). Реакционный раствор оставляли реагировать на воздухе при 115°C в течение 16 часов. Реакционный раствор охлаждали до комнатной температуры и разбавляли EtOAc. Органические фазы промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (R)-4-(дифторметил)-3-(8-фтор-9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)тиазолидин-2-она (55 мг, 52%).
MS m/z (ESI): 482,1 [M+H]+.
Стадия 10: Получение (S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(R)-4-(дифторметил)-3-(8-фтор-9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)тиазолидин-2-он (40 мг, 0,083 ммоль), L-аланин (15 мг, 0,17 ммоль), иодид меди (6,3 мг, 0,033 ммоль) и фосфат калия (53 мг, 0,25 ммоль) смешивали с диметилсульфоксидом (3 мл). Реакционную систему трижды продували азотом и оставляли реагировать при 125°C в течение 1,5 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли хлорид аммония (27 мг, 0,5 ммоль) и DMAP (161 мг, 1,25 ммоль) и перемешивали реакционный раствор в течение 5 минут. Добавляли O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфат (284 мг, 0,75 ммоль) и перемешивали реакционный раствор при комнатной температуре в течение 2 часов. Реакционный раствор фильтровали, к фильтрату добавляли насыщенный водный раствор бикарбоната натрия, который затем трижды экстрагировали этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида (7,9 мг, 22%).
1H ЯМР (400 МГц, CD3OD) δ 1,49 (d, J = 7,0 Гц, 3H), 3,54-3,60 (m, 1H), 3,76-3,93 (m, 1H), 3,95-4,00 (m, 1H), 4,36-4,40 (m, 2H), 4,47-4,52 (m, 2H), 5,10-5,20 (m, 1H), 6,32-6,62 (m, 2H), 7,32 (s, 1H), 7,85-7,91 (m, 1H);
MS m/z (ESI): 442,1 [M+H]+.
Пример 109
Получение (S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксипропанамида
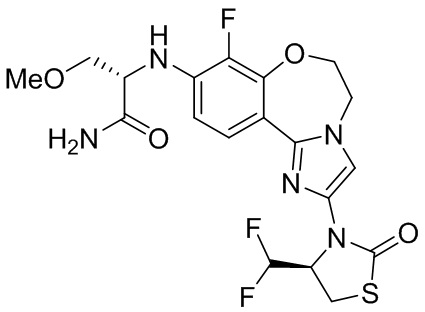
(S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-3-метоксипропанамид получали согласно примеру 108.
1H ЯМР (400 МГц, CD3OD) δ 3,40 (s, 3H), 3,53-3,60 (m, 1H), 3,69-3,83 (m, 3H), 4,06-4,13 (m, 1H), 4,35-4,41 (m, 2H), 4,47-4,52 (m, 2H), 5,10-5,21 (m, 1H), 6,30-6,60 (m, 2H), 7,32 (s, 1H), 7,89 (d, J = 8,5 Гц, 1H);
MS m/z (ESI): 472,1 [M+H]+.
Пример 110
Получение (S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксипропанамида
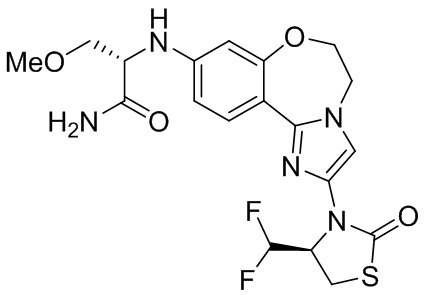
Стадия 1: Получение (S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксипропанамида

(R)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)тиазолидин-2-он (26 мг, 0,062 ммоль), O-метил-L-серин (22 мг, 0,18 ммоль), иодид меди (6,0 мг, 0,03 ммоль) и фосфат калия (40 мг, 0,19 ммоль) смешивали в диметилсульфоксиде (3 мл). Реакционную систему трижды продували азотом и оставляли реагировать при 100°C в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли хлорид аммония (20 мг, 0,37 ммоль) и триэтиламин (95 мг, 0,94 ммоль) и перемешивали реакционный раствор в течение 5 минут. Добавляли O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфат (212 мг, 0,56 ммоль) и перемешивали реакционный раствор при комнатной температуре в течение 2 часов. Реакционный раствор фильтровали, к фильтрату добавляли насыщенный водный раствор бикарбоната натрия, который затем трижды экстрагировали этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении для удаления органического растворителя. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксипропанамида (13 мг, 46%).
1H ЯМР (400 МГц, CD3OD) δ 3,39 (s, 3H), 3,53-3,57 (m, 1H), 3,62-3,76 (m, 3H), 3,93-3,98 (m, 1H), 4,16-4,30 (m, 4H), 5,06-5,16 (m, 1H), 6,21-6,23 (m, 1H), 6,28-6,52 (m, 2H), 7,23 (s, 1H), 8,02 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 454,1 [M+H]+.
Пример 111
Получение (S)-1-(2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамида

(S)-1-(2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид получали способом, описанным в примере 51.
1H ЯМР (400 МГц, ДМСО-d6) δ 1,83-1,92 (m, 2H), 2,09-2,15 (m, 1H), 3,72-3,81 (m, 4H), 4,25-4,32 (m, 4H), 5,07-5,15 (m, 1H), 5,93-5,97 (m, 1H), 6,22-6,28 (m, 1H), 6,35-6,65 (s, 1H), 7,00 (s, 1H), 7,26 (s, 1H), 7,35 (s, 1H), 7,99 (d, J = 8,6 Гц, 1H);
MS m/z (ESI): 450,1 [M+H]+.
Пример 112
Получение (S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)(метил)амино)пропанамида

(S)-2-((2-((R)-4-(дифторметил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)(метил)амино)пропанамид получали способом, описанным в примере 51.
1H ЯМР (400 МГц, CD3OD) δ 1,40 (d, J = 7,0 Гц, 3H), 2,90 (s, 3H), 3,53-3,58 (m, 1H), 3,75-3,80 (m, 1H), 4,30-4,44 (m, 4H), 4,46-4,51 (m, 1H), 5,08-5,18 (m, 1H), 6,22-6,41 (m, 2H), 6,51-6,73 (m, 1H),7,28 (s, 1H), 8,11 (d, J = 9,0 Гц, 1H);
MS m/z (ESI): 438,1[M+H]+.
Пример 113
Получение (S)-2-((2-((R)-5-(метоксиметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((R)-5-(метоксиметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 1,48 (d, J = 7,0 Гц, 3H), 3,44 (s, 3H), 3,59-3,72 (m, 2H), 3,83 (q, J = 7,0 Гц, 1H), 3,95-4,02 (m, 6,5 Гц, 1H), 4,20 (t, J = 9,3 Гц, 1H), 4,28-4,33 (m, 2H), 4,35-4,42 (m, 2H), 4,81-4,86 (m, 1H), 6,17-6,21 (m, 1H), 6,43 (dd, J = 8,8, 2,3 Гц, 1H), 7,12 (s, 1H),8,01 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 402,2 [M+H]+.
Пример 114
Получение (S)-2-((2-((R)-4-метокси-2-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((R)-4-метокси-2-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CDCl3) δ 1,52 (d, J = 6,9 Гц, 3H), 2,57-2,67 (m, 1H), 2,78-2,88 (m, 1H), 3,40 (s, 3H), 3,79-3,88 (m, 1H), 4,10-4,43 (m, 7H), 6,19 (s, 1H), 6,42 (d, J = 8,8 Гц, 1H), 7,32 (s, 1H),8,16 (d, J = 8,7 Гц, 1H);
MS m/z (ESI): 386,2 [M+H]+.
Пример 115
Получение (2S)-2-((2-(4-(цианометил)-2-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
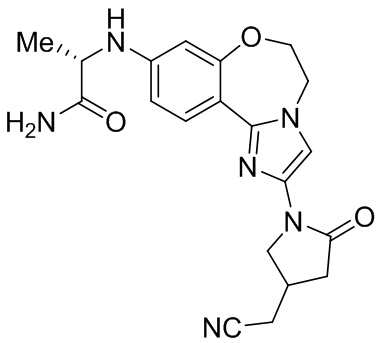
(2S)-2-((2-(4-(цианометил)-2-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ 1,30 (d, J = 6,9 Гц, 3H), 2,29 (dd, J = 16,9, 6,2 Гц, 1H), 2,70 (dd, J = 16,5, 8,0 Гц, 1H), 2,77-2,88 (m, 3H), 3,64 (dd, J = 10,8, 4,4 Гц, 1H), 3,76 (dt, J = 13,7, 6,9 Гц, 1H), 4,13 (dd, J = 10,6, 7,1 Гц, 1H), 4,33 (dd, J = 8,5, 5,8 Гц, 4H), 6,08 (d, J = 2,0 Гц, 1H), 6,12 (d, J = 7,0 Гц, 1H), 6,40 (dd, J = 8,8, 2,2 Гц, 1H), 6,99 (d, J = 0,7 Гц, 1H), 7,37 (s, 2H), 7,99 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 395,1 [M+H]+.
Пример 116
Получение (2S)-2-((2-(4циано-2-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
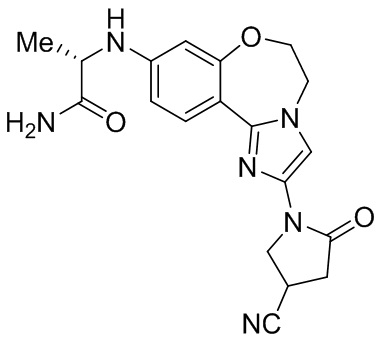
(2S)-2-((2-(4-циано-2-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, CD3OD) δ 1,49 (d, J = 7,0 Гц, 3H), 2,88 (dd, J = 17,0, 6,7 Гц, 1H), 3,01 (dd, J = 17,0, 9,3 Гц, 1H), 3,64-3,76 (m, 1H), 3,83 (q, J = 7,1 Гц, 1H), 4,25 (dd, J = 10,8, 5,9 Гц, 1H), 4,30-4,46 (m, 5H), 6,19 (d, J = 1,8 Гц, 1H), 6,44 (dd, J = 8,8, 2,3 Гц, 1H), 7,38 (s, 1H), 7,75 (s, 1H), 8,06 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 381,1 [M+H]+.
Пример 117
Получение (S)-2-((2-((S)-2-(цианометил)-5-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида
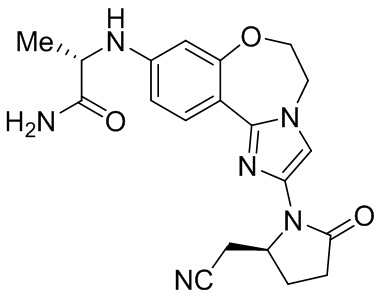
(S)-2-((2-((S)-2-(цианометил)-5-оксопирролидин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ 1,30 (d, J = 6,8 Гц, 3H), 1,99 (t, J = 10,8 Гц, 1H), 2,42 (dd, J = 22,2, 12,2 Гц, 3H), 2,62-2,71 (m, 1H), 3,13 (dd, J = 16,8, 2,8 Гц, 1H), 3,69-3,80 (m, 1H), 4,17-4,48 (m, 4H), 4,53-4,66 (m, 1H), 6,08 (d, J = 1,7 Гц, 1H), 6,15 (d, J = 7,0 Гц, 1H), 6,40 (dd, J = 8,8, 1,8 Гц, 1H), 7,01 (s, 1H), 7,38 (s, 1H), 7,41 (s, 1H), 7,98 (d, J = 8,9 Гц, 1H);
MS m/z (ESI): 395,2 [M+H]+.
Пример 118
Получение (S)-2-((2-((R)-4-(цианометил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((R)-4-(цианометил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ 1,30 (d, J = 6,9 Гц, 3H), 3,14 (dd, J = 17,1, 2,4 Гц, 1H), 3,45 (dd, J = 17,1, 5,0 Гц, 1H), 3,72-3,80 (m, 1H), 4,33 (ddd, J = 13,3, 8,4, 4,7 Гц, 5H), 4,66 (t, J = 8,9 Гц, 1H), 4,77 (dt, J = 8,4, 4,9 Гц, 1H), 6,08 (d, J = 2,1 Гц, 1H), 6,17 (d, J = 7,0 Гц, 1H), 6,40 (dd, J = 8,8, 2,2 Гц, 1H), 7,02 (s, 1H), 7,20 (s, 1H), 7,39 (d, J = 1,0 Гц, 1H), 7,98 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 397,1 [M+H]+.
Пример 119
Получение (S)-2-((2-((R)-4-(цианометил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((R)-4-(цианометил)-2-оксотиазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали способом, описанным в примере 1.
MS m/z (ESI): 436,2 [M+H]+.
Пример 120
Получение (S)-2-((2-((R)-4-(цианометил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксипропанамида

(S)-2-((2-((R)-4-(цианометил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксипропанамид получали способом, описанным в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ 3,14 (dd, J = 17,6, 2,8 Гц, 1H), 3,28 (s, 3H), 3,46 (dd, J = 17,3, 4,4 Гц, 1H), 3,56 (d, J = 5,3 Гц, 2H), 3,92-4,01 (m, 1H), 4,24-4,48 (m, 5H), 4,66 (t, J = 9,0 Гц, 1H), 4,74-4,81 (m, 1H), 6,09-6,21 (m, 2H), 6,46 (dd, J = 8,7, 1,7 Гц, 1H), 7,16 (s, 1H), 7,20 (s, 1H), 7,46 (s, 1H), 7,99 (d, J = 8,8 Гц, 1H);
MS m/z (ESI): 427,1 [M+H]+.
Пример 121
Получение (S)-2-((2-((R)-4-(2-метоксиэтил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((R)-4-(2-метоксиэтил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали согласно примеру 1.
MS m/z (ESI): 416,1 [M+H]+.
Пример 122
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксо-1,3-тиазинан-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(тиазинан)-2-оксо-1,3-оксазинан-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали согласно примеру 51.
MS m/z (ESI): 438,1 [M+H]+.
Пример 123
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксо-1,3-тиазинан-3-ил)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксо-1,3-тиазинан-3-ил)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали согласно примеру 51.
MS m/z (ESI): 456,1 [M+H]+.
Пример 124
Получение (S)-2-((2-((S)-4-(дифторметил)-2-оксо-1,3-тиазинан-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксипропанамида

(S)-2-((2-((S)-4-(дифторметил)-2-оксо-1,3-тиазинан-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксипропанамид получали согласно примеру 51.
MS m/z (ESI): 468,1 [M+H]+.
Пример 125
Получение (S)-2-((2-((S)-3-(дифторметил)-5-оксоморфолино)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

((S)-2-((2-((S)-3-(дифторметил)-5-оксоморфолино)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали согласно примеру 68.
MS m/z (ESI): 440,1 [M+H]+.
Пример 126
Получение (S)-2-((2-((S)-3-(дифторметил)-5-оксоморфолино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)3-метоксипропанамида

(S)-2-((2-((S)-3-(дифторметил)-5-оксоморфолино)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)3-метоксипропанамид получали согласно примеру 68.
MS m/z (ESI): 452,1 [M+H]+.
Пример 127
Получение (S)-2-((2-((S)-2-(дифторметил)-4-метил-6-оксопиперазин-1-ил)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида

(S)-2-((2-((S)-2-(дифторметил)-4-метил-6-оксопиперазин-1-ил)-8-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид получали согласно примеру 69.
MS m/z (ESI): 453,1 [M+H]+.
Пример 128
Получение (S)-2-((2-((S)-2-(дифторметил)-4-метил-6-оксопиперазин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксипропанамида
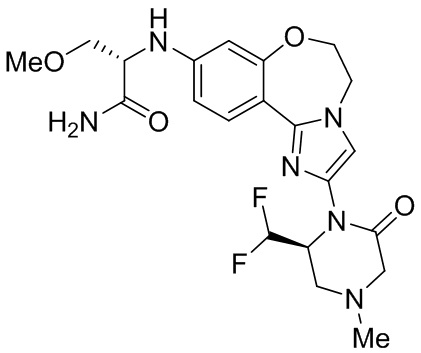
(S)-2-((2-((S)-2-(дифторметил)-4-метил-6-оксопиперазин-1-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)-3-метоксипропанамид получали согласно примеру 69.
MS m/z (ESI): 465,1 [M+H]+.
Пример 129
Получение (2S,3R)-1-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-3-метилпирролидин-2-карбоксамида

(2S,3R)-1-(2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-3-метилпирролидин-2-карбоксамид получали согласно примеру 1.
MS m/z (ESI): 448,1 [M+H]+.
Биологический анализ и оценка
Настоящее изобретение дополнительно описано ниже в сочетании со следующими тестовыми примерами, которые не предназначены для ограничения объема настоящего изобретения.
Тестовый пример 1. Определение ингибирующего действия соединений согласно примерам настоящего изобретения на активность киназы PI3Kα/β/γ/δ
Цель эксперимента: Целью этого тестового примера является определение ингибирующего действия соединений согласно примерам на активность киназы PI3Kα/β/γ/δ.
Оборудование для эксперимента: центрифуга (5810R, приобретена у Eppendorf), пипетка (приобретена у Eppendorf или Rainin) и считыватель для микропланшетов (модель: полнофункциональный считыватель для микропланшетов SynergyH1, приобретен у BioTek, США).
Методика эксперимента: в этом эксперименте применялся способ анализа липидкиназы Promega ADP-Glo (Promega № V9102). Липидкиназа PI3Kα/β/γ/δ катализировала образование АДФ из АТФ в присутствии субстрата PIP2:3PS и АТФ. Активность липидкиназы характеризовали путем измерения содержания АДФ в реакции, и таким образом получали полуингибирующую концентрацию IC50 соединения в отношении ингибирования активности киназы PI3Kα/β/γ/δ.
Конкретный процесс эксперимента выглядит следующим образом:
Киназную реакцию проводили в белом 384-луночном планшете (Perkin Elmer № 6007299). В каждую лунку добавляли 2 мкл раствора соединения различных концентраций, разбавленного ddH2O, содержащей 1% ДМСО. 2 мкл ddH2O, содержащей 1% ДМСО, добавляли в лунку положительного контроля. В каждую лунку добавляли 2 мкл 0,1-2 нМ раствора киназы PI3K, разведенного 5× киназным буфером (HEPES 250 мМ, MgCl2 15 мМ, NaCl 250 мМ, BSA 0,05%). 2 мкл 5× киназного буфера добавляли в лунку отрицательного контроля. 4 мкл 50 мкМ субстрата PIP2: 3PS (Promega # V1701), приготовленного с 10× буфером для разведения и ddH2O, добавляли во все лунки. Наконец, для начала реакции добавляли 2 мкл 50-100 мкМ раствора АТФ, разбавленного водой. После проведения реакции при комнатной температуре в течение 90-120 минут в каждую лунку добавляли 10 мкл реагента ADP-Glo (содержащего 10 мМ MgCl2), и он реагировал при комнатной температуре в течение 60 минут для удаления избытка АТФ в реакции. В каждую лунку добавляли 20 мкл реагента для обнаружения киназ, и он реагировал при комнатной температуре в темноте в течение 20 минут. Величину хемилюминесценции измеряли с помощью считывателя для микропланшетов BioTek Synergy H1.
Название
Методика обработки экспериментальных данных:
Данные процентного ингибирования лунки, обработанной соединением, рассчитывали с помощью лунки положительного контроля (лунки с контролем ДМСО) и лунки отрицательного контроля (без добавления киназы) на планшете {% степени ингибирования = 100 - [(значение для тестируемого соединения - значение для отрицательного контроля)] / (значение для положительного контроля - значение для отрицательного контроля) × 100}. Значение IC50 рассчитывали путем аппроксимации данных для различных концентраций и соответствующих процентных степеней ингибирования четырехпараметрическим нелинейным логическим уравнением с помощью GraphPad prism.
Результаты эксперимента:
Согласно приведенной выше схеме, соединения согласно примерам настоящего изобретения в тесте активности в отношении киназы PI3Kα/β/γ/δ продемонстрировали биологические активности, приведенные в таблице 1.
Таблица 1
Приведенные выше данные показывают, что соединения согласно примерам настоящего изобретения обладают хорошей активностью и селективностью в отношении ингибирования активности киназы PI3Kα/β/γ/δ.
Тестовый пример 2. Определение эффекта ингибирования пролиферации соединений согласно примерам настоящего изобретения на мутантные по PI3Kα раковые клетки
Цель эксперимента: Целью этого тестового примера является определение активности ингибирования пролиферации соединений согласно примерам в отношении мутантных по PI3Kα раковых клеток HCC1954 (H1047R), HGC-27 (E542K) и MKN1 (E545K).
Оборудование для эксперимента: центрифуга (5702R, приобретена у Eppendorf), инкубатор с диоксидом углерода (приобретен у Thermo), бокс биологической безопасности (приобретен у Shanghai Boxun Company), пипетка (приобретена у Eppendorf или Rainin) и считыватель для микропланшетов (модель: полнофункциональный считыватель для микропланшетов SynergyH1, приобретен у BioTek, США).
Методика эксперимента: Эффект ингибирования пролиферации соединений согласно примерам в отношении мутантных по PI3Kα линий раковых клеток HCC1954, HGC-27 и MKN1 определяли методом Cell Titer-Glo. Линии клеток культивировали в среде RPMI 1640 (Gibco № 22400089), содержащей 10% FBS (Gibco № 10091148) и 1% P/S (Hyclone № SV30010) при 37°C и 5% CO2. Перед экспериментом клетки собирали и подсчитывали, а затем регулировали плотность клеток. Клетки инокулировали в белый 96-луночный планшет (Corning № 3610) при плотности от 1000 до 10000 клеток/лунку и инкубировали в инкубаторе при 37°C, 5% CO2 в течение ночи. Добавляли раствор соединения различных концентраций и одновременно устанавливали соответствующий контроль носителем. Планшет с клетками дополнительно инкубировали в инкубаторе при 37°C, 5% CO2 в течение 48-96 часов. Планшет с клетками и его содержимое уравновешивали до комнатной температуры. В каждую лунку добавляли от 20 до 100 мкл раствора Cell Titer-Glo (Promega № G7573), планшет хорошо встряхивали и инкубировали при комнатной температуре в темноте в течение 5-30 минут. Величину хемилюминесценции измеряли с помощью считывателя для микропланшетов BioTek Synergy H1.
Методика обработки экспериментальных данных:
Данные процентного ингибирования лунки, обработанной соединениями согласно примерам, рассчитывали с помощью лунки с контролем носителем на планшете {% степени ингибирования = 100 - (значение для тестируемого соединения / значение для контроля носителем) × 100}. Значение IC50 рассчитывали путем аппроксимации данных для различных концентраций и соответствующих процентных степеней ингибирования четырехпараметрическим нелинейным логическим уравнением с помощью GraphPad prism.
Результаты эксперимента:
Согласно приведенной выше схеме, соединения согласно примерам настоящего изобретения в тесте ингибирования пролиферации мутантных по PI3Kα раковых клеток HCC1954 (H1047R), HGC-27 (E542K) и MKN1 (E545K) продемонстрировали биологические активности, приведенные в таблице 2.
Таблица 2
Приведенные выше данные показывают, что соединения согласно примерам настоящего изобретения обладают хорошей активностью в отношении ингибирования пролиферации мутантных по PI3Kα раковых клеток HCC1954 (H1047R), HGC-27 (E542K) и MKN1 (E545K).
Тестовый пример 3. Тест на токсичность 7-дневного многократного внутрижелудочного введения крысам СД
3.1 Цель эксперимента
Целью этого исследования является изучение возможной токсичности GDC-0077 и соединения примера 51 у крыс SD (Спрег-Доули) после 7-дневного многократного внутрижелудочного введения, а также сравнение различий в токсичности между GDC-0077 и соединением примера 51.
3.2 Материалы и оборудование для эксперимента
3.2.1 Тестируемое соединение
Тестируемое соединение 1: GDC-0077
Тестируемое соединение 2: соединение примера 51
3.2.2 Носитель
Наименование: 20% водный раствор SBE-β-CD (каптизол)
3.2.3 Информация о животных
Виды и линии: Крысы Спрег-Доули (СД)
Класс животного: свободные от специфической патогенной микрофлоры (класс SPF)
Количество и пол животных: 112 крыс, половина самцов и половина самок
3.2.4 Оборудование
Автоматический анализатор крови ADVIA®2120, который использовался для подсчета клеток крови;
Коагулометр SYSMEX CA-500, который использовался для определения показателей функции свертывания;
Автоматический биохимический анализатор TBA-120FR, который использовался для определения биохимических показателей крови;
Анализатор электролитов Easylyte, который использовался для определения электролитов; и
Жидкостная хроматография - масс-спектрометрия (детектор: API4000, источник электроспрей (ESI) в режиме положительных ионов, колонка: Agilent ZORBAX XDB-C18 (3,5 мкм, 2,1×50 мкм)), которая использовалась для биологического анализа образца плазмы.
3.3 Методика эксперимента
1) В эксперименте 112 крыс (56 крыс/пол) были разделены на 14 групп по полу и массе тела, из которых 70 крыс были использованы для токсикологического исследования (группы с 1 по 7, 5 крыс/пол/группа) и 42 крысы были использованы для токсикологического исследования (группы с 8 по 14, 3 крысы/пол/группа).
2) В качестве контрольной группы носителя животным в группах 1 и 8 внутрижелудочно вводили 20%-й водный раствор SBE-β-CD (каптизола).
3) Животным в группах 2 и 9, 3 и 10 и 4 и 11 внутрижелудочно вводили GDC-0077 в дозах 10, 30 и 60 мг/кг, соответственно.
4) Животным в группах 5 и 12, 6 и 13 и 7 и 14 внутрижелудочно вводили соединение примера 51 в дозах 10, 30 и 60 мг/кг, соответственно.
5) Животным осуществляли введение раз в сутки в течение 7 дней подряд (животным из групп 7 и 14 осуществляли введение в течение 6 дней подряд).
6) Объем введения составлял 10 мл/кг.
7) В ходе эксперимента изучались такие параметры, как клинические наблюдения, масса тела, прием пищи, клинико-патологические показатели (общий анализ крови, функция свертывания, биохимия крови), токсикокинетика и т.п.
8) Все животные были умерщвлены на 8 день (животные в группах 7 и 14 были умерщвлены после введения на 6 день).
9) Во время эксперимента проводили патологоанатомическое исследование животных в группах с 1 по 7, животных в группе 14 и мертвых животных (включая животных, участвовавших в токсикокинетическом исследовании), и проводили гистопатологическое исследование патологических тканей, тканей желудочно-кишечного тракта (таких как ободочная и слепая кишка) и иммунных тканей (таких как вилочковая железа).
3.4 Перечень данных теста
3.4.1 Предсмертное состояние/смерть
В случае GDC-0077 или соединения примера 51 в дозе 60 мг/кг наблюдались животные в предсмертном состоянии/мертвые. В случае других доз, мертвых животных/животных в предсмертном состоянии не наблюдалось.
3.4.2 Токсикокинетика
В дозе 30 мг/кг среднее системное воздействие AUC соединения примера 51 после последнего введения (самцы: 11400 ч*нг/мл, самки: 15900 ч*нг/мл) было приблизительно в 2,4-3,8 раза выше, чем у GDC-0077 в той же дозе (самцы: 3000 ч*нг/мл, самки: 6510 ч*нг/мл) и аналогично GDC-0077 в дозе 60 мг/кг после первого введения (самцы: 15400 ч*нг/мл, самки: 22800 ч*нг/мл).
В дозе 10 мг/кг среднее системное воздействие AUC (площадь под кривой) соединения примера 51 после последнего введения (самцы: 2110 ч*нг/мл, самки: 3170 ч*нг/мл) было приблизительно в 1,4-2,5 раза выше, чем у GDC-0077 (самцы: 845 ч*нг/мл, самки: 2250 ч*нг/мл).
Следовательно, системное воздействие соединения примера 51 было значительно выше, чем воздействие GDC-0077 в той же дозе.
3.4.3 Клинические наблюдения
GDC-0077: В группе, получавшей высокую дозу, наблюдались патологические симптомы, такие как выгнутая спина, жидкий стул, загрязнение околоанальной области и пушистая шерсть.
Соединение примера 51: В группах, получавших среднюю и высокую дозу, наблюдались патологические симптомы, такие как выгнутая спина, жидкий стул, загрязнение околоанальной области и пушистая шерсть.
3.4.4 Масса тела и потребление пищи
GDC-0077: В группах, получавших среднюю и высокую дозу, конечная масса тела всех животных снизилась, и потребление ими пищи также снизилось за тот же период.
Соединение примера 51: В группах, получавших низкую, среднюю и высокую дозу, конечная масса тела снизилась, и соответствующее потребление пищи также снизилось.
3.4.5 Подсчет клеток крови и функция свертывания
GDC-0077: В каждой группе введения дозы ретикулоциты снизились как у самцов, так и у самок.
Соединение примера 51: В каждой группе введения дозы ретикулоциты снизились как у самцов, так и у самок; и в группе, получавшей 30 мг/кг, и группе, получавшей 60 мг/кг, нейтрофилы и тромбоциты снизились как у самцов, так и у самок.
3.4.6 Биохимия крови
GDC-0077: В каждой группе, получавшей дозу, глюкоза и холестерин повысились у самцов животных; в группе, получавшей 60 мг/кг, АСТ (аспартатаминотрансфераза) и мочевина повысились как у самцов, так и у самок, A/G (альбумин-глобулиновый индкс) снизились как у самцов, так и у самок, а АЛТ (аланинаминотрансфераза) повысилась у самцов; и в группе, получавшей 30 мг/кг, АСТ и мочевина повысились как у самцов, так и у самок, а АЛТ повысилась у самок.
Соединение примера 51: В группе, получавшей 60 мг/кг, АСТ, глюкоза и мочевина повысились как у самцов, так и у самок; в группе, получавшей 30 мг/кг, АСТ и мочевина повысились как у самцов, так и у самок, а глюкоза повысилась у самцов; и в группе, получавшей 10 мг/кг, АСТ повысилась как у самцов, так и у самок.
3.4.7 Патологическая анатомия
GDC-0077: Патологические изменения под микроскопом в основном включали атрофию бокаловидных клеток слизистой оболочки слепой кишки, увеличение количества окрашивающихся телец макрофагов в вилочковой железе, и эрозию, кровотечение и отек слизистой оболочки желудка.
Соединение примера 51: Патологические изменения под микроскопом в основном включали кровотечение и атрофию железистой ткани слизистой желудка; кровотечение слизистой оболочки слепой кишки, атрофию бокаловидных клеток, атрофия бокаловидных клеток слизистой оболочки ободочной кишки; атрофию белой пульпы селезенки; и атрофию коры или коры и мозгового вещества вилочковой железы, увеличение количества окрашивающихся телец макрофагов и тому подобное.
Основными органами-мишенями токсичности GDC-0077 и соединения примера 51 являются ткани желудочно-кишечного тракта (такие как желудок и слепая кишка) и иммунные ткани (такие как вилочковая железа).
3.5 Вывод после эксперимента
Во время данного эксперимента тестируемое соединение GDC-0077 и соединение примера 51 вводили внутрижелудочно крысам SD в течение 7 дней в дозе 10, 30 и 60 мг/кг (раз в сутки). Летальная доза GDC-0077 и соединения примера 51 составляет 60 мг/кг, а максимальная переносимая доза (MTD) составляет 30 мг/кг. В дозе 30 мг/кг Cmax и AUC(0-24 ч) соединения примера 51 значительно выше, чем у GDC-0077. Таким образом, переносимость соединения примера 51 лучше, чем переносимость GDC-0077.
Тестовый пример 4. Тест на эффективность соединений согласно примерам настоящего изобретения in vivo
4.1 Цель эксперимента
Цель состоит в том, чтобы отобрать соединения со значительной эффективностью и меньшими токсичными и побочными действиями с помощью теста эффективности in vivo.
4.2 Основное оборудование и материалы для эксперимента
4.2.1 Оборудование:
1. Бокс биологической безопасности (BSC-1300II A2, Shanghai Boxun Medical Biological Instrument Corp.)
2. Сверхчистый рабочий стол (CJ-2F, Suzhou Fengshi Laboratory Animal Equipment Co., Ltd.)
3. CO2-инкубатор (Thermo-311)
4. Центрифуга (Centrifuge 5702R, Eppendorf)
5. Автоматический счетчик клеток (Countess II, Life)
6. Пипетка (10-20 мкл, Eppendorf)
7. Микроскоп (TS2, Nikon)
8. Штангенциркуль с нониусом (CD-6''AX, Mitutoyo, Япония)
9. Колба для клеточных культур (T75/T225, Corning)
10.Электронные весы (CPA2202S, Sartorius)
4.2.2 Реагенты:
1. Среда RPMI-1640 (22400-089, Gibco)
2. Фетальная бычья сыворотка (FBS) (10091-148, Gibco)
3.0,25% трипсин (25200-056, Gibco)
4. Двойные антибиотики пенициллин-стрептомицин (15140-122, Gibco)
5. Натрий-фосфатный буфер (PBS) (10010-023, Gibco)
6. Matrigel Matrix (356234, Corning)
4.2.3 Животное:
Голые мыши BALB/c в возрасте от 6 до 8 недель, самцы, приобретенные в Shanghai Sippr-BK laboratory animal Co. Ltd.
4.3 Процесс эксперимента
4.3.1 Культура клеток и получение клеточной суспензии
a. Клетки HCC1954 были взяты из банка клеток и реанимированы в среде RPMI-1640 (RPMI-1640 + 10% FBS (фетальная бычья сыворотка) + 1% SP). Реанимированные клетки помещали в колбу для культивирования клеток (тип клетки, дата, имя оператора и т.п. были отмечены на стенке колбы) и культивировали в CO2-инкубаторе (температура инкубатора составляла 37°C, а концентрация СО2 составляла 5%).
b. Пассаж проводили после того, как клетки покрывали от 80 до 90% дна культуральной колбы. После проведения пассажа клетки продолжали культивировать в CO2-инкубаторе. Этот процесс повторяли до тех пор, пока количество клеток не соответствовало требованиям для теста эффективности in vivo.
c. Культивированные клетки собирали и подсчитывали с помощью автоматического счетчика клеток. Клетки повторно суспендировали в PBS и matrigel в соответствии с результатами подсчета для получения клеточной суспензии (плотность: 5×107/ мл), которую помещали в холодильную камеру для последующего использования.
4.3.2 Инокуляция клеток
a. Перед инокуляцией голых мышей метили одноразовыми универсальными ушными бирками для крыс и мышей.
b. Во время инокуляции суспензию клеток хорошо перемешивали. От 0,1 до 1 мл клеточной суспензии отбирали шприцем на 1 мл, пузырьки воздуха удаляли и шприц помещали на пузырь со льдом для последующего использования.
c. Голую мышь держали левой рукой. Место в правой части спины рядом с правым плечом голой мыши (место инокуляции) дезинфицировали 75% спиртом и через 30 секунд осуществляли инокуляцию.
d. Тестируемых голых мышей последовательно подвергали инокуляции (каждой мыши инокулировали 0,1 мл клеточной суспензии).
4.3.3 Измерение объема опухоли, распределение на группы и введение мышам с опухолью
a. В соответствии с ростом опухоли опухоль измеряли через 14-18 дней после инокуляции и рассчитывали размер опухоли.
Расчет объема опухоли: объем опухоли (мм3) = длина (мм) × ширина (мм) × ширина (мм)/2
b. В соответствии с массой тела и размером опухоли мыши с опухолью осуществляли распределение на группы случайным образом.
c. Согласно результатам распределения на группы вводили тестируемые соединения (способ введения: пероральное введение; вводимая доза: 10 мг/кг; вводимый объем: 10 мл/кг; частота введения: раз в сутки; цикл введения: 21 день; носитель: 0,5% КМЦ (карбоксиметилцеллюлоза)/1% Твин 80).
d. Объем опухоли и массу тела измеряли дважды в неделю после начала введения тестируемых соединений.
e. После окончания эксперимента животных умерщвляли.
f. Данные обрабатывали с помощью программного обеспечения, такого как Excel. Расчет степени ингибирования роста опухоли TGI (%) соединения: когда опухоль не регрессировала, TGI (%) = [(1-(средний объем опухоли в группе лечения в конце введения - средний объем опухоли группа лечения в начале введения))/(средний объем опухоли в группе контроля носителем в конце лечения - средний объем опухоли в группе контроля носителем в начале лечения)]×100%. Когда опухоль регрессировала, TGI (%) = [1-(средний объем опухоли в группе лечения в конце введения - средний объем опухоли в группе лечения в начале введения)/средний объем опухоли в группе лечения в начале введения]×100%.
4.4 Данные тестов представлены в таблице 3:
Таблица 3
4.5 Результаты эксперимента
Из приведенных выше результатов можно видеть, что указанные выше соединения согласно настоящей заявке показали хорошую степень ингибирования роста опухоли.
Тестовый пример 5. Фармакокинетический (ФК) анализ соединений согласно примерам настоящего изобретения на мышах
Фармакокинетический анализ предпочтительных соединений согласно примерам настоящего изобретения на мышах проводили на самцах мышей Balb/c (Shanghai Jiesijie Laboratory Animal Co., LTD).
- Способ введения: однократное внутрижелудочное введение.
- Вводимая доза: 5 мг/10 мл/кг (массы тела).
- Препарат: соединение растворяли в 0,5% Na-КМЦ с помощью ультразвука для получения прозрачного раствора или однородной суспензии.
-Точки взятия образцов: 0,5, 1, 2, 4, 6, 8 и 24 часа после введения.
-Пример процесса:
1) 0,1 мл крови брали из глазной вены, помещали в пробирку с K2-ЭДТА (этилендиаминтетрауксусная кислота) и центрифугировали от 5 до 20 минут при комнатной температуре при 1000-3000×g для отделения плазмы, которую затем хранили при минус 80°C.
2) 160 мкл ацетонитрила добавляли к 40 мкл образца плазмы для осаждения, а затем смесь центрифугировали в течение 5-20 минут при 500-2000×g.
3) отбирали 100 мкл обработанного супернатанта и анализировали с помощью анализа ЖХ/МС/МС для определения концентрации тестируемого соединения.
Анализ ЖХ-МС/МС:
- Условия проведения жидкостной хроматографии: Насос Shimadzu LC-20AD
- Условия проведения масс-спектрометрии: Масс-спектрометр AB Sciex API 4000
- Хроматографическая колонка: phenomenex Gemiu 5 мкм C18 50×4,6 мм
- Подвижная фаза: раствор A представлял собой 0,1% водный раствор муравьиной кислоты, а раствор B представлял собой ацетонитрил
- Скорость потока: 0,8 мл/мин
- Время элюирования: 0-4 минуты градиентного элюирования
Фармакокинетика:
Основные параметры рассчитывали с помощью WinNonlin 6.1. Результаты эксперимента по проведению фармакокинетического анализа на мышах приведены в таблице 4 ниже:
Таблица 4
Из приведенных в таблице результатов фармакокинетического анализа видно, что соединения согласно примерам настоящего изобретения показали хорошие метаболические свойства, и как уровни в плазме AUC, так и максимальная концентрация в плазме Cmax были хорошими.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ С ТРЕМЯ КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ СОЛЬ ИЛИ КРИСТАЛЛИЧЕСКАЯ ФОРМА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2020 |
|
RU2835105C1 |
| ИНГИБИТОР, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ ПРОИЗВОДНОЕ ПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2807611C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| СОЕДИНЕНИЯ БЕНЗОКСАЗЕПИНОКСАЗОЛИДИНОНОВ И СПОСОБЫ ПРИМЕНЕНИЯ | 2016 |
|
RU2730529C2 |
| РЕГУЛЯТОРЫ ПРОИЗВОДНЫХ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2803499C1 |
| МОДИФИКАТОР ПРОИЗВОДНОГО ЧЕТЫРЕХЧЛЕННОГО КОЛЬЦА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2833027C1 |
| ИНГИБИТОР, СОДЕРЖАЩИЙ БИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2820948C2 |
| РЕГУЛЯТОРЫ-ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2797408C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| ИНГИБИТОР EGFR И ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2702631C2 |
Изобретение относится к соединению формулы (VI), его стереоизомеру или его фармацевтически приемлемой соли, где кольцо В выбрано из группы, состоящей из:
 L представляет собой азот; G представляет собой кислород; Q представляет собой N; Z представляет собой -CRaa; Raa выбран из группы, состоящей из водорода и дейтерия; каждый Ry независимо выбран из группы, состоящей из водорода и дейтерия; каждый из R5, R6 и R14 независимо выбран из группы, состоящей из водорода, дейтерия и галогена; каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, дейтерия, C1-6 алкила, C1-6 дейтерированного алкила и -(CH2)n1ORbb, где каждый из указанных C1-6 алкила и C1-6 дейтерированного алкила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия и C1-6 алкокси; R2 выбран из группы, состоящей из водорода, дейтерия и C1-6 алкила; или R2 и R3 связаны с образованием 4-6-членного гетероциклила, содержащего азот, где число атомов азота равно одному или двум; Rz выбран из группы, состоящей из водорода, дейтерия, C1-6 алкила, C1-6 дейтерированного алкила и C1-6 галогеналкила, где указанный C1-6 алкил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано; Rbb выбран из группы, состоящей из водорода, C1-6 алкила и циано; n1 равно 0,1, 2 или 3; m равно 1; t равно 1; n равно 1; и q равно 0, 1, 2, 3, 4, 5 или 6. Изобретение также к способам получения соединения формулы (VI), фармацевтической композиции для ингибирования активности фосфатидилинозитол-3-киназы (PI3K) на его основе. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине при лечении рака и заболеваний или состояний, опосредованных или зависящих от дисбаланса PI3K. 7 н. и 17 з.п. ф-лы, 4 табл., 129 пр.
L представляет собой азот; G представляет собой кислород; Q представляет собой N; Z представляет собой -CRaa; Raa выбран из группы, состоящей из водорода и дейтерия; каждый Ry независимо выбран из группы, состоящей из водорода и дейтерия; каждый из R5, R6 и R14 независимо выбран из группы, состоящей из водорода, дейтерия и галогена; каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, дейтерия, C1-6 алкила, C1-6 дейтерированного алкила и -(CH2)n1ORbb, где каждый из указанных C1-6 алкила и C1-6 дейтерированного алкила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия и C1-6 алкокси; R2 выбран из группы, состоящей из водорода, дейтерия и C1-6 алкила; или R2 и R3 связаны с образованием 4-6-членного гетероциклила, содержащего азот, где число атомов азота равно одному или двум; Rz выбран из группы, состоящей из водорода, дейтерия, C1-6 алкила, C1-6 дейтерированного алкила и C1-6 галогеналкила, где указанный C1-6 алкил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано; Rbb выбран из группы, состоящей из водорода, C1-6 алкила и циано; n1 равно 0,1, 2 или 3; m равно 1; t равно 1; n равно 1; и q равно 0, 1, 2, 3, 4, 5 или 6. Изобретение также к способам получения соединения формулы (VI), фармацевтической композиции для ингибирования активности фосфатидилинозитол-3-киназы (PI3K) на его основе. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине при лечении рака и заболеваний или состояний, опосредованных или зависящих от дисбаланса PI3K. 7 н. и 17 з.п. ф-лы, 4 табл., 129 пр.

1. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль:
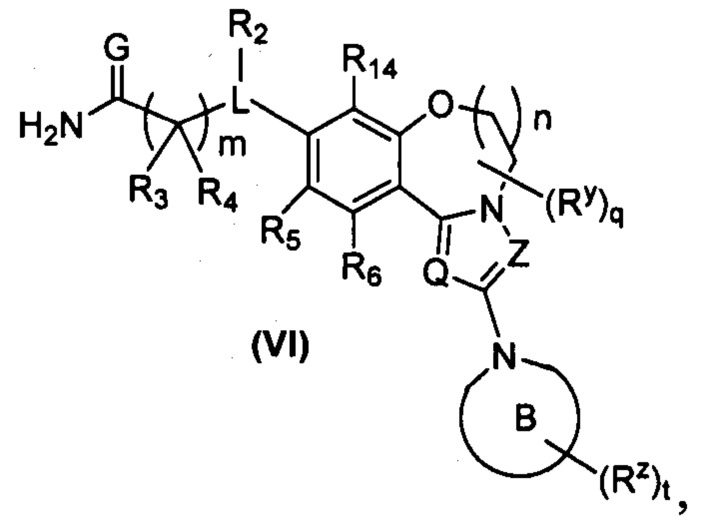
где:
кольцо В выбрано из группы, состоящей из:

L представляет собой азот;
G представляет собой кислород;
Q представляет собой N;
Z представляет собой -CRaa;
Raa выбран из группы, состоящей из водорода и дейтерия;
каждый Ry независимо выбран из группы, состоящей из водорода и дейтерия;
каждый из R5, R6 и R14 независимо выбран из группы, состоящей из водорода, дейтерия и галогена;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, дейтерия, C1-6 алкила, C1-6 дейтерированного алкила и -(CH2)n1ORbb, где каждый из указанных C1-6 алкила и C1-6 дейтерированного алкила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия и C1-6 алкокси;
R2 выбран из группы, состоящей из водорода, дейтерия и C1-6 алкила;
или R2 и R3 связаны с образованием 4-6-членного гетероциклила, содержащего азот, где число атомов азота равно одному или двум;
Rz выбран из группы, состоящей из водорода, дейтерия, C1-6 алкила, C1-6 дейтерированного алкила и C1-6 галогеналкила, где указанный C1-6 алкил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, галогена, циано;
Rbb выбран из группы, состоящей из водорода, C1-6 алкила и циано;
n1 равно 0, 1, 2 или 3;
m равно 1;
t равно 1;
n равно 1; и
q равно 0, 1, 2, 3, 4, 5 или 6.
2. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 1, характеризующееся тем, что оно дополнительно представляет собой такое, как показано в формуле (VIII-А):
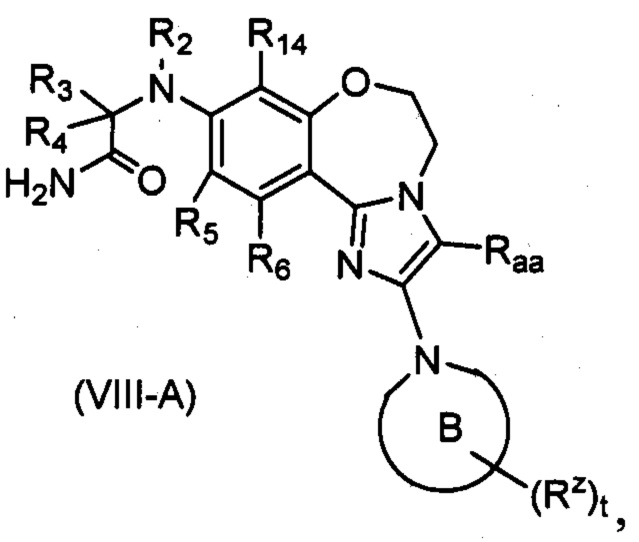
где:
кольцо В выбрано из группы, состоящей из:

R2 выбран из группы, состоящей из водорода и C1-6 алкила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, C1-6 алкила и -(CH2)n1ORbb;
или R2 и R3 связаны с образованием 4-6-членного гетероциклила, содержащего азот, где число атомов азота равно одному или двум;
каждый из R5, R6 и R14 независимо выбран из группы, состоящей из водорода, галогена;
Raa представляет собой водород;
Rz выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 галогеналкила и -(CH2)n1Rbb;
Rbb выбран из группы, состоящей из водорода, C1-6 алкила и циано;
n1 равно 0, 1, 2 или 3; и
t равно 1.
3. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 2, характеризующееся тем, что R2 и R3 связаны с образованием пирролидинила или азетидинила.
4. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 2, характеризующееся тем, что
R2 выбран из группы, состоящей из водорода и С1-3 алкила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, С1-3 алкила и С1-3 алкила, замещенного С1-3 алкокси;
или R2 и R3 связаны с образованием 4-6-членного гетероциклила, содержащего азот, где число атомов азота равно одному или двум;
каждый из R5 и R6 независимо выбран из группы, состоящей из водорода и галогена;
R14 выбран из группы, состоящей из водорода, фтора и хлора;
Rz выбран из группы, состоящей из водорода, С1-3 алкила, С1-3 алкила, замещенного галогеном, и -(СН2)n1Rbb;
Raa представляет собой водород;
Rbb представляет собой циано;
n1 равно 0, 1, 2 или 3; и
t равно 1.
5. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 4, характеризующееся тем, что
R2 выбран из группы, состоящей из водорода и метила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, метила, этила, пропила, СН3ОСН2- и СН3СН2ОСН2-;
R2 и R3 связаны с образованием пирролидинила, пиперидинила или азетидинила;
Rz выбран из группы, состоящей из водорода, ацетонитрилила, пропионитрилила и С1-3 алкила, замещенного фтором.
6. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 4 или 5, характеризующееся тем, что
Rz выбран из группы, состоящей из ацетонитрилила, -CHF2, -CF2CH3 и CHF2CH2-.
7. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 1, характеризующееся тем, что оно дополнительно представляет собой такое, как показано в формуле (VIII):

где:
кольцо В, R3, R5, R6, R14, Rz, Raa и t являются такими, как определено в п. 1.
8. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 7, характеризующееся тем, что кольцо В выбрано из группы, состоящей из
R2 представляет собой водород;
R3 выбран из группы, состоящей из С1-3 алкила и С1-3 алкила, замещенного С1-3 алкокси;
каждый из R5 и R6 независимо выбран из группы, состоящей из водорода и галогена;
R14 выбран из группы, состоящей из водорода, фтора и хлора;
Rz выбран из группы, состоящей из водорода, С1-6 алкила и С1-6 алкила, замещенного галогеном.
9. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 7, характеризующееся тем, что
R3 выбран из группы, состоящей из метила, этила, пропила, СН3ОСН2- и СН3СН2ОСН2-;
каждый из R5 и R6 независимо выбран из группы, состоящей из водорода и галогена;
Rz выбран из группы, состоящей из водорода, ацетонитрилила, пропионитрилила и С1-3 алкила, замещенного галогеном.
10. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 7, характеризующееся тем, что
Rz выбран из группы, состоящей из метила, ацетонитрилила, -CHF2, -CF2CH3 и CHF2CH2-.
11. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 1, характеризующееся тем, что оно дополнительно представляет собой такое, как показано в формуле (X):

где:
R15 выбран из группы, состоящей из водорода, С1-6 алкила, С1-6 галогеналкила и -(CH2)n1Rbb;
R16 представляет собой водород;
R2-R4, R6, R14, n1, Raa и Rbb являются такими, как определено в п. 2.
12. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 11, характеризующееся тем, что
R2 выбран из группы, состоящей из водорода и C1-6 алкила;
или R2 и R3 связаны с образованием 4-6-членного гетероциклила, содержащего азот, где число атомов азота равно одному или двум;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода и С1-3 алкила;
каждый из R5 и R6 независимо выбран из группы, состоящей из водорода и галогена;
R14 выбран из группы, состоящей из водорода и галогена;
Raa представляет собой водород.
13. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 12, характеризующееся тем, что R2 и R3 связаны с образованием пирролидинила или азетидинила.
14. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 12, характеризующееся тем, что
R2 выбран из группы, состоящей из водорода и С1-3 алкила;
каждый из R3 и R4 независимо выбран из группы, состоящей из метила, этила и пропила;
R5 и R6 представляют собой водород;
R14 представляет собой водород.
15. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 1, 2 или 7, характеризующееся тем, что Rz представляет собой С1-3 алкил или С1-3 галогеналкил.
16. Соединение формулы (VI), его стереоизомер или его фармацевтически приемлемая соль по п. 1, характеризующееся тем, что структура соединения представляет собой следующую:
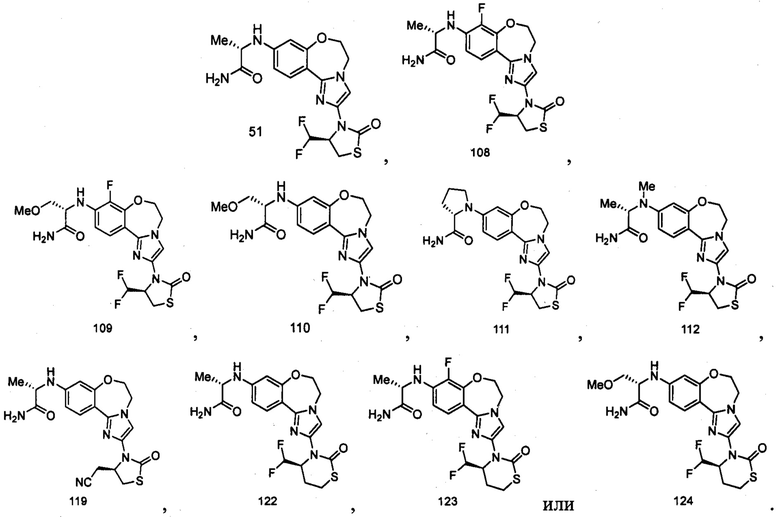
17. Способ получения соединения формулы (VI), его стереоизомера или его фармацевтически приемлемой соли, характеризующийся тем, что он включает следующую стадию

взаимодействия соединения формулы (VI-1) и соединения формулы (IV-2) с получением соединения формулы (VI), его стереоизомера или его фармацевтически приемлемой соли;
где:
X представляет собой галоген; и
кольцо В, Q, Z, G, L, R2-R6, R14, Ry, Rz, q, m, n и t являются такими, как определено в п. 1.
18. Способ получения соединения формулы (X), его стереоизомера или его фармацевтически приемлемой соли, характеризующийся тем, что он включает следующие стадии

взаимодействия соединения формулы (IX) и реагента Лоуссона с получением соединения формулы (IX-A), его стереоизомера или его фармацевтически приемлемой соли, и взаимодействия соединения формулы (IX-А) с комплексом переходного металла и его лигандом с получением соединения формулы (X), его стереоизомера или его фармацевтически приемлемой соли;
где:
R2-R4, R6, R14-R16 и Raa являются такими, как определено в п. 11.
19. Способ получения соединения формулы (X), его стереоизомера или его фармацевтически приемлемой соли, характеризующийся тем, что он включает следующую стадию

взаимодействия соединения формулы (IX-А) с комплексом переходного металла и его лигандом с получением соединения формулы (X), его стереоизомера или его фармацевтически приемлемой соли;
где:
R2-R4, R6, R14-R16 и Raa являются такими, как определено в п. 11.
20. Способ по п. 19, характеризующийся тем, что комплекс переходного металла и его лиганд представляют собой димер дихлор(п-цимен)рутения (II) и 2-бициклогексилфосфино-2',6'-диметоксибифенил.
21. Фармацевтическая композиция для ингибирования активности фосфатидилинозитол-3-киназы (PI3K), содержащая терапевтически эффективное количество соединения формулы (VI), его стереоизомера или его фармацевтически приемлемой соли по любому из пп. 1-16 и один или более фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ.
22. Применение соединения формулы (VI), его стереоизомера или его фармацевтически приемлемой соли по любому из пп. 1-16 или фармацевтической композиции по п. 21 для получения лекарственного средства-ингибитора PI3K.
23. Применение по п. 22, характеризующееся тем, что лекарственное средство-ингибитор PI3K представляет собой лекарственное средство-ингибитор P13Kα.
24. Применение соединения формулы (VI), его стереоизомера или его фармацевтически приемлемой соли по любому из пп. 1-16 или фармацевтической композиции по п. 21 для получения лекарственного средства для лечения рака, заболевания костей, воспалительного заболевания, иммунного заболевания, заболевания нервной системы, метаболического заболевания, заболевания органов дыхательной системы и заболевания сердца, опосредованных активностью PI3K, где указанный рак выбран из группы, состоящей из рака молочной железы, рака поджелудочной железы, немелкоклеточного рака легкого, рака щитовидной железы, семиномы, меланомы, рака мочевого пузыря, рака печени, рака почки, миелодиспластического синдрома, острого миелоидного лейкоза и колоректального рака.
| WO 2017001645 A1, 05.01.2017 | |||
| WO 2017001658 A1, 05.01.2017 | |||
| WO 2011036280 A1, 31.03.2011 | |||
| WO 2012126901 A1, 27.09.2012 | |||
| TIMOPHY P | |||
| HEFFRON et al., J | |||
| Med | |||
| Chem., vol.59, no | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Аппарат для сжигания нефти | 1920 |
|
SU985A1 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗОКСАЗЕПИНОВЫХ СОЕДИНЕНИЙ | 2014 |
|
RU2649976C2 |

