Область техники
Настоящее изобретение относится к области фармацевтического синтеза. В частности, настоящее изобретение относится к способу синтеза производных 9-аллилкамптотецина (Ximingtecan гидрохлорид, соединение 1H).
Уровень техники
В 2007 году Шанхайский институт Материа Медика, Китайская академия наук, осуществил ряд модификаций в 9-м положении ядра камптотецина на основе 10-гидроксикамптотецина (публикации международных заявок WO 2005/044821, WO 2007/104214) и, наконец, обнаружил, что 9-аллил-10-гидроксикамптотецин (Jimmytecan, соединение 6), среди прочих, показал превосходную противоопухолевую активность при оценке in vivo и in vitro. Растворимое в воде пролекарство указанного выше Ximingtecan гидрохлорида (соединение 1H) было предметом тщательной оценки, клинические испытания были применены от CFDA в октябре 2010 года, а документ, утверждающий клинические испытания, был получен в мае 2012 года. Согласно указанному документу соединение 1Н является перспективным кандидатом в качестве противоопухолевых лекарственных средств.
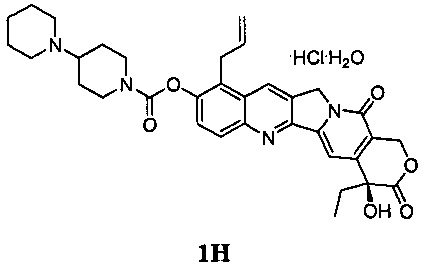
В настоящее время основным способом синтеза Ximingtecan гидрохлорида является конденсация 9-аллил-10-гидроксикамптотецина (соединение 6) и хлорида пиперидинилпиперидина хлормуравьиной кислоты (амид хлормуравьиной кислоты) (соединение 7).
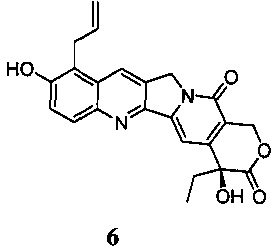
При этом существует два основных способа синтеза соединения 6.
В одном способе 10-гидроксикамптотецин применяют в качестве исходного материала, и продукт получают с помощью двух стадий реакции, алкилирования и перегруппировки Кляйзена (публикация международной заявки WO 2005/044821), а путь указанного способа показан следующим образом:
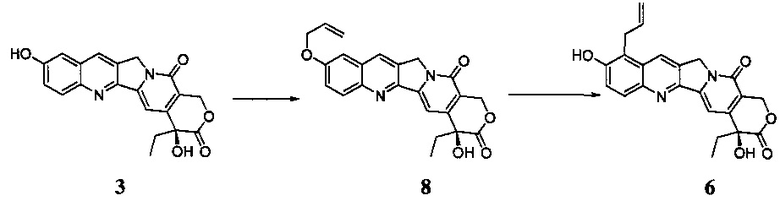
Указанный путь синтеза принят в опытном производстве в настоящее время. Тем не менее существуют очевидные недостатки указанного пути: во-первых, во время процесса перегруппировки была бы получена примесь изомера, который перестроен по 11-му положению (соединение 9). Это соединение трудно полностью удалить даже при использовании колоночной хроматографии; во-вторых, продолжительность реакции перегруппировки составляет до 72 часов, и соединение 8 не может быть полностью превращено в соединение 6, и в реакционной системе присутствует все равно большое количество соединения 8 после завершения реакции. Поскольку продукт похож на примеси в структуре, общий выход значительно ниже, и выделение и очистка продукта затруднены. В процессе реальной работы мы обнаружили, что трудно получить соединение 6 с уровнем чистоты более 95%; поэтому необходима повторная перекристаллизация для очистки конечного продукта 1H, что, таким образом, дополнительно снижает выход. Общий выход по этому пути составляет в настоящее время примерно 16-20%.
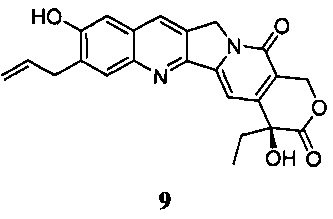
Другой способ синтеза соединения 6, указанный в литературе, представляет собой применение катализируемых металлическим палладием реакций сочетания (сочетание Сузуки или Стилле) для получения соединения 6 (CN 101880285); путь реакции выглядит следующим образом:
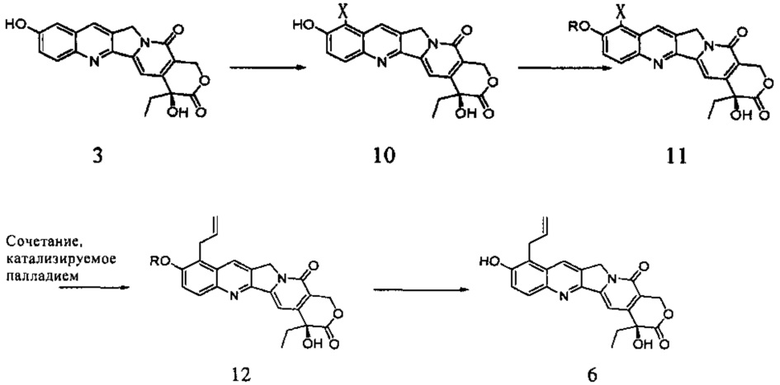
где X представляет собой Cl, Br или I, R представляет собой обычно применяемую защитную группу, в частности, метоксиметил, ацетил, этоксикарбонил и т.д.
Несмотря на то, что при указанном пути может быть получено соединение 6 высокой чистоты и можно избежать перегруппировки изомера 9, все еще существуют значительные проблемы: во-первых, если было использовано сочетание Стилле, то необходимы высокотоксичные реагенты олова, применения которых следует избегать в производстве лекарственного средства; во-вторых, что касается сочетания Сузуки, по сравнению с предыдущим путем синтеза есть еще две стадии в ходе реакции, в то время как общий выход не был значительно увеличен, что приводит к увеличению затрат рабочей силы и операционных расходов; в-третьих, палладиевый катализатор, используемый в реакции, был низкой активности, что делает невозможным достижение хорошего выхода; наконец, в реакции есть две стадии, включающие хроматографическую очистку, которые увеличат сложность эксплуатации и производственные затраты. Этот путь не был использован в реальном производстве.
Таким образом, разработка процесса синтеза для получения соединения 1, которое обладает высокой эффективностью, низкой стоимостью, легкостью масштабирования и хорошей воспроизводимостью, имеет большое значение для промышленного производства лекарственных средств в будущем.
Краткое описание изобретения
Задачей настоящего изобретения является обеспечение способа синтеза для получения моногидрата гидрохлорида 10-((4'-пиперидинилпиперидин)карбонилокси)-9-аллилкамптотецина (соединение 1), который обладает хорошей селективностью, высокой степенью чистоты и высоким общим выходом. При применении этого способа может быть значительно увеличен выход продукта, снижены производственные затраты и сэкономлено время производства, что, таким образом, позволяет непосредственно получить API высокой чистоты для клинического применения.
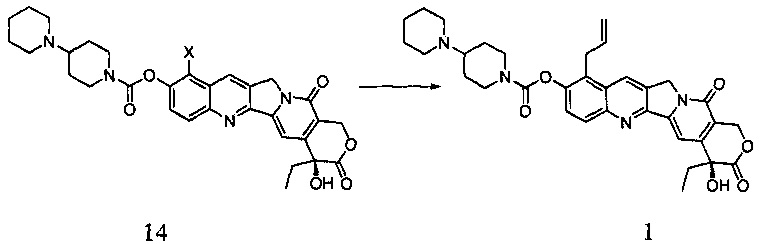
Согласно первому аспекту настоящего изобретения предложено соединение формулы 14

где X представляет собой галоген.
В другом предпочтительном варианте реализации галоген представляет собой хлор, бром или йод.
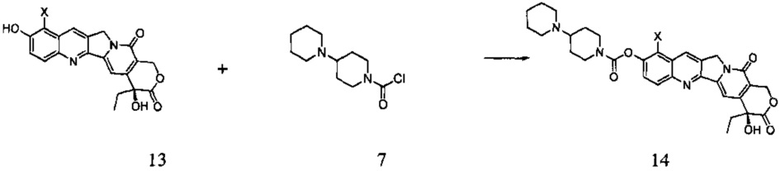
Согласно второму аспекту настоящего изобретения предложен способ получения соединения 14, включающий стадию взаимодействия соединения 13 с соединением 7 в инертном растворителе с получением соединения 14

где в приведенных выше формулах X представляет собой галоген.
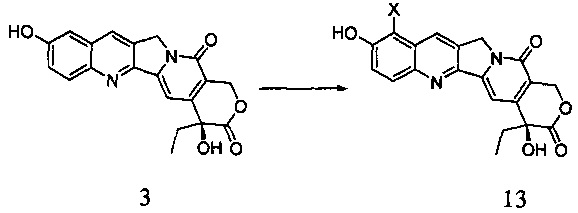
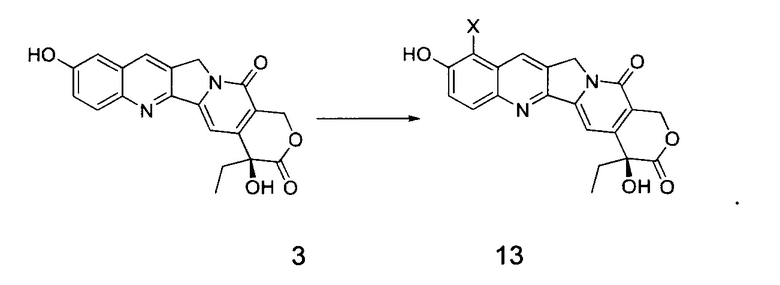
В другом предпочтительном варианте реализации соединение 13 получают способом, включающим следующие стадии:
Взаимодействие соединения 3 с галогенирующим агентом в инертном растворителе с получением соединения 13
 .
.
В другом предпочтительном варианте реализации галогенирующий агент выбран из следующей группы: бром, йод, хлорид йода, N-бромсукцинимид (NBS), N-иодсукцинимид (NIS), хлорид брома, 1,3-дибром-1,3,5-триазин-2,4,6-трион.
Согласно третьему аспекту настоящего изобретения предложен способ получения соединения 1, включающий стадию проведения реакции Сузуки между соединением 14 и аллилборным реагентом с получением соединения 1

где в приведенных выше формулах X представляет собой галоген.
В другом предпочтительном варианте реализации аллилборный реагент выбран из группы, состоящей из пинаколового эфира аллилборной кислоты, комплексной соли фторида аллилбора.
В другом предпочтительном варианте реализации комплексная соль фторида аллилбора выбрана из следующей группы: комплексная соль трифторида аллилбора и фторида калия.
В другом предпочтительном варианте реализации соединение 14 получают способом согласно второму аспекту настоящего изобретения.
В другом предпочтительном варианте реализации реакцию Сузуки проводили в системе, содержащей следующие агенты: палладиевый катализатор, фосфиновый лиганд, щелочь и инертный растворитель.
В другом предпочтительном варианте реализации
Палладиевый катализатор выбран из группы, состоящей из: трис(дибензилиденацетон)дипалладия (Pd2(dba)3), тетракис(трифенилфосфин)палладия (Pd(PPh3)4), ацетата палладия, дихлорбис(трифенилфосфин)палладия, трифторацетата палладия, ацетата трифенилфосфинпалладия, дихлорбис(три-о-бензил-фосфин)палладия, дихлор[1,2-бис(дифенилфосфино)этан]палладия или их комбинаций;
Фосфиновый лиганд выбран из следующей группы: три-трет-бутилфосфин, тетрафторборат три-трет-бутилфосфония, три-н-бутилфосфин, трифенилфосфин, три-п-бензил-фосфин, трициклогексилфосфин, три-о-бензил-фосфин или их комбинации; основание выбрано из следующей группы: фторид калия, фторид цезия, гидратированный фосфат калия, карбонат калия, карбонат натрия, гидрокарбонат натрия, 1,8-диазабицикло[5.4.0]ундец-7-ен, триэтиламин, диизопропилэтиламин, пиридин или их комбинации;
Инертный растворитель выбран из следующей группы: 1,4-диоксан, тетрагидрофуран, ацетонитрил, диметилсульфоксид, N,N-диметилформамид, толуол, метанол, этанол, изопропанол, н-бутанол, трет-бутанол, изо-бутанол, бензиловый спирт, вода или их комбинации.
В другом предпочтительном варианте реализации палладиевый катализатор выбран из группы, состоящей из: трис(дибензилиденацетон)дипалладия (Pd2(dba)3), тетракис(трифенилфосфин)палладия (Pd(PPh3)4) или их комбинаций; и/или
Фосфиновый лиганд выбран из следующей группы: три-трет-бутилфосфин, тетрафторборат три-трет-бутилфосфония или их комбинации; основание выбрано из следующей группы: фторид калия, фторид цезия, гидратированный фосфат калия, диизопропилэтиламин, триэтиламин или их комбинации;
Инертный растворитель выбран из следующей группы: 1,4-диоксан, изопропанол, вода, бензиловый спирт или их комбинации.
В другом предпочтительном варианте реализации реакцию Сузуки проводят в системе, выбранной из следующей группы:
(1) трис(дибензилиденацетон)дипалладий, фторид калия, три-трет-бутилфосфин и 1,4-диоксан;
(2) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, гидратированный фосфат калия и 1,4-диоксан;
(3) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, карбонат калия и 1,4-диоксан;
(4) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, гидратированный фосфат калия, фторид калия и 1,4-диоксан;
(5) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия и 1,4-диоксан;
(6) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид цезия и 1,4-диоксан;
(7) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, триэтиламин, фторид калия и 1,4-диоксан;
(8) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, триэтиламин, фторид калия и тетрагидрофуран;
(9) тетракис(трифенилфосфин)палладий, три-трет-бутилфосфин, фторид калия и 1,4-диоксан;
(10) ацетат палладия, три-трет-бутилфосфин, фторид калия и 1,4-диоксан;
(11) дихлорбис(трифенилфосфин)палладий, три-трет-бутилфосфин, фторид калия и 1,4-диоксан;
(12) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, триэтиламин, фторид калия и метанол;
(13) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия и метанол;
(14) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин и метанол;
(15) тетракис(трифенилфосфин)палладий, три-трет-бутилфосфин, диизопропилэтиламин и метанол;
(16) дихлорбис(трифенилфосфин)палладий, три-трет-бутилфосфин, диизопропилэтиламин и метанол;
(17) трис(дибензилиденацетон)дипалладий, трифенилфосфин, диизопропилэтиламин, фторид калия и изопропанол;
(18) трис(дибензилиденацетон)дипалладий, тетрафторборат три-трет-бутилфосфония, диизопропилэтиламин и изопропанол;
(19) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия, вода и изопропанол;
(20) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия, вода и н-бутиловый спирт;
(21) дихлорбис(трифенилфосфин)палладий, три-трет-бутилфосфин, диизопропилэтиламин, вода и н-бутиловый спирт;
(22) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия и н-бутиловый спирт;
(23) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин и н-бутиловый спирт;
(24) трис(дибензилиденацетон)дипалладий, фторид калия, тетрафторборат три-трет-бутилфосфония, диизопропилэтиламин, 1,4-диоксан и вода;
(25) трис(дибензилиденацетон)дипалладий, фторид калия, тетрафторборат три-трет-бутилфосфония, диизопропилэтиламин, 1,4-диоксан и вода;
(26) трис(дибензилиденацетон)дипалладий, фторид калия, тетрафторборат три-трет-бутилфосфония, диизопропилэтиламин, изопропанол и вода;
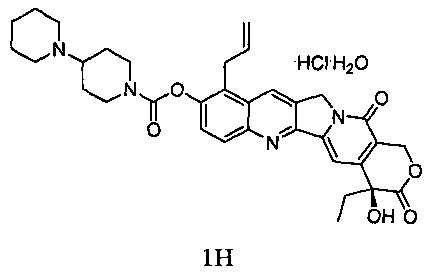
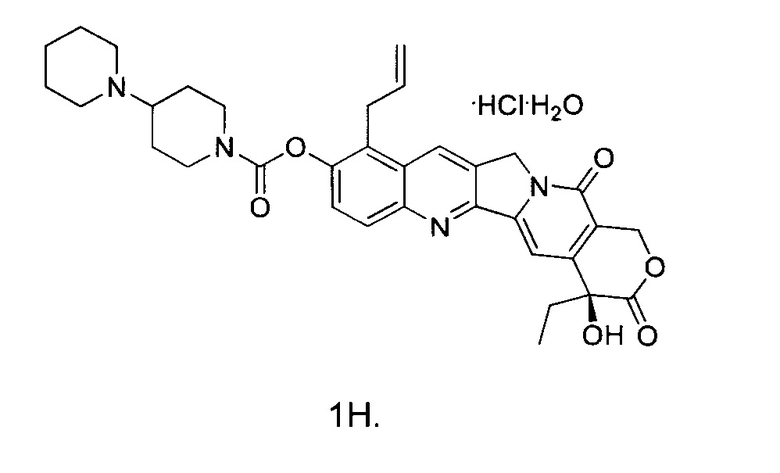
В другом предпочтительном варианте реализации способ дополнительно включает стадию подкисления соединения 1 с получением соединения 1Н.
Следует понимать, что в настоящем изобретении каждую из технических характеристик, в частности, описанных выше и ниже (например, те, которые указаны в примерах), можно комбинировать друг с другом, тем самым составляя новые или предпочтительные технические решения, которые не обязательно еще раз указывать в настоящем описании.
Варианты осуществления изобретения
Проводя исследование, автор настоящего изобретения неожиданно обнаружил способ получения промежуточного продукта для получения моногидрата гидрохлорида 10-((4'-пиперидинилпиперидин)карбонилокси)-9-аллилкамптотецина (соединение 1) и для получения соединения 1. Способ характеризуется хорошей селективностью, высокой степенью чистоты, а также значительно повышенным общим выходом, что значительно снижает стоимость производства, экономит время и позволяет непосредственно получить лекарственное средство с высокой степенью чистоты для клинического применения. Автор изобретения создал настоящее изобретение на основе указанных выше исследований.
В настоящем изобретении предложен способ получения соединения 1, при этом 10-гидроксикамптотецин (соединение 3) применяют в качестве исходного материала, соединение 1 получают путем трех стадий галогенирования, сочетания и реакции Сузуки, и способ включает следующие стадии:
(1) В инертном растворителе (например, ДМФА, CCl4, хлороформ, уксусная кислота и т.д.), при определенной температуре (например, от -20°С до 50°С) соединение 3 галогенировали галогенирующим агентом в течение определенного времени (например, от 0,5 до 6 часов), получив таким образом соединение 13; где X представляет собой галоген (например, хлор, бром, йод);
 .
.
при этом галогенирующий агент выбран из следующей группы: бром, йод, хлорид йода, N-бромсукцинимид (NBS), N-иодсукцинимид (NIS), хлорид брома, 1,3-дибром-1,3,5-триазин-2,4,6-трион.
(2) В инертном растворителе (например, дихлорметан, пиридин, тетрагидрофуран и т.д.), при определенной температуре (например, от -20°С до 25°С) соединение 13 подвергают взаимодействию с соединением 7 в течение определенного времени (например, от 0,5 до 3 часов), получая таким образом соединение 14.
(3) При определенной температуре (например, от 50°С до 120°С) проводят реакцию Сузуки между соединением 14 и аллилборным реагентом в течение определенного времени (например, от 1 до 18 часов), получая таким образом соединение 1;
 .
.
Реакцию Сузуки проводят в системе, выбранной из следующей группы, при этом применение некоторых из реагентов подтверждено экспериментально, в то время как применение других может быть осуществлено с помощью опыта путем простой замены реагентов в соответствии с опытом: (1) трис(дибензилиденацетон)дипалладий, фторид калия, три-трет-бутилфосфин и 1,4-диоксан; (2) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, гидратированный фосфат калия и 1,4-диоксан; (3) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, карбонат калия и 1,4-диоксан; (4) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, гидратированный фосфат калия, фторид калия и 1,4-диоксан; (5) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия и 1,4-диоксан; (6) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид цезия и 1,4-диоксан; (7) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, триэтиламин, фторид калия и 1,4-диоксан; (8) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, триэтиламин, фторид калия и тетрагидрофуран; (9) тетракис(трифенилфосфин)палладий, три-трет-бутилфосфин, фторид калия и 1,4-диоксан; (10) ацетат палладия, три-трет-бутилфосфин, фторид калия и 1,4-диоксан; (11) дихлорбис(трифенилфосфин)палладий, три-трет-бутилфосфин, фторид калия и 1,4-диоксан; (12) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, триэтиламин, фторид калия и метанол; (13) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия и метанол; (14) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин и метанол; (15) тетракис(трифенилфосфин)палладий, три-трет-бутилфосфин, диизопропилэтиламин и метанол; (16) дихлорбис(трифенилфосфин)палладий, три-трет-бутилфосфин, диизопропилэтиламин и метанол; (17) трис(дибензилиденацетон)дипалладий, трифенилфосфин, диизопропилэтиламин, фторид калия и изопропанол; (18) трис(дибензилиденацетон)дипалладий, тетрафторборат три-трет-бутилфосфония, диизопропилэтиламин и изопропанол; (19) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия, вода и изопропанол; (20) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия, вода и н-бутиловый спирт; (21) дихлорбис(трифенилфосфин)палладий, три-трет-бутилфосфин, диизопропилэтиламин, вода и н-бутиловый спирт; (22) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия и н-бутиловый спирт; (23) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин и н-бутиловый спирт; (24) трис(дибензилиденацетон)дипалладий, фторид калия, тетрафторборат три-трет-бутилфосфония, диизопропилэтиламин, 1,4-диоксан и вода; (25) трис(дибензилиденацетон)дипалладий, фторид калия, тетрафторборат три-трет-бутилфосфония, диизопропилэтиламин, 1,4-диоксан и вода; (26) трис(дибензилиденацетон)дипалладий, фторид калия, тетрафторборат три-трет-бутилфосфония, диизопропилэтиламин, изопропанол и вода;
Во время стадии (3), после завершения реакции Сузуки, соединение 1 можно очистить с помощью фильтрации, колоночной хроматографии и перекристаллизации.
Кроме того, способ может дополнительно включать в себя стадию подкисления соединения 1 с получением соединения 1Н.
По сравнению с предшествующим уровнем техники настоящее изобретение, главным образом, обладает следующими преимуществами:
1. Предложен новый способ получения соединения 1. По сравнению с используемыми в настоящее время способами производства время производства одной партии может быть уменьшено на 50% посредством способа синтеза по настоящему изобретению, что значительно повышает эффективность производства. Во-вторых, общий выход увеличен с 16-20% до 70-80%, что значительно повышает эффективность применения исходных материалов и снижает затраты. Наконец, технология позволяет избежать применения высокотоксичного промежуточного продукта 9-аллил-10-гидроксикамптотецина (соединение 6) в соответствии со способом по настоящему изобретению, что значительно повышает безопасность рабочей силы. И способ работы настоящей технологии очень прост.
2. Дополнительно предложен способ получения промежуточного продукта соединения 1.
Настоящее изобретение будет дополнительно проиллюстрировано ниже со ссылкой на конкретные примеры. Следует понимать, что эти примеры приведены только для иллюстрации настоящего изобретения, но не для ограничения объема настоящего изобретения. Экспериментальные способы без конкретных условий, описанные в следующих примерах, как правило, выполняют в обычных условиях или в соответствии с инструкциями производителя. Если не указано иное, части и проценты рассчитаны по массе.
Реакция I:
Пример 1

10-гидроксикамптотецин (соединение 3, 20,0 г, 54,95 ммоль) растворили в ДМФА (480 мл), а затем внутреннюю температуру понизили до 0°С в бане с ледяной водой. Добавили N-бромсукцинимид (9,78 г, 54,95 ммоль) и проводили реакцию при комнатной температуре в течение 2 ч. После завершения реакции реакционную смесь вылили в 800 мл ледяной воды, и значение рН довели до 3-4 с помощью 1H HCl. Смесь тщательно перемешали, отфильтровали путем отсасывания, промыли водой и просушили в сушильном шкафу при температуре 40°С с получением 24 г твердого вещества желтого цвета (соединение 13'), выход которого составил 98%.
1Н-ЯМР (ДМСО-d6): δ 0,87 (t, J=7,2 Гц, 3Н), 1,82-1,89 (m, 2Н), 5,30 (s, 2Н), 5,42 (s, 2Н), 6,51 (s, 1Н), 7,28 (s, 1H), 7,62 (d, J=9,2 Гц, 1H), 8,22 (d, J=9,2, 1H), 8,74 (s, 1H), 11,19 (s, 1Н).
Пример 2

10-гидроксикамптотецин (соединение 3, 500 мг, 1,37 ммоль) растворили в ДМФА (12 мл), а затем внутреннюю температуру снизили до 0°С в бане с ледяной водой. Добавили N-иодсукцинимид (309 мг, 1,37 ммоль) и проводили реакцию при комнатной температуре в течение 2 ч. После завершения реакции реакционную смесь вылили в 20 мл ледяной воды, и значение рН довели до 3-4 с помощью 1H HCl. Смесь тщательно перемешали, отфильтровали путем отсасывания, промыли водой и просушили в сушильном шкафу при температуре 40°С с получением 730 мг твердого вещества желтого цвета (соединение 15), выход которого составил 97%.
1Н-ЯМР (ДМСО-d6): δ 0,87 (t, J=7,2 Гц, 3Н), 1,82-1,89 (m, 2Н), 5,31 (s, 2Н), 5,42 (s, 2Н), 6,53 (s, 1H), 7,28 (s, 1Н), 7,57 (d, J=8,8 Гц, 1H), 8,06 (d, J=9,2 Гц, 1Н), 8,66 (s, 1Н), 11,29 (s, 1H).
Реакция II:
Пример 3
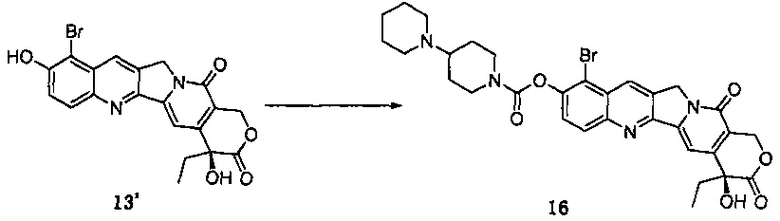
Способ подщелачивания хлористоводородной соли 4-пиперидинилпиперидин-формилхлорида:
10%-ный раствор гидроксида натрия предварительно охладили до комнатной температуры. Хлористоводородную соль 4-пиперидинилпиперидин-формилхлорида (23,5 г, 87,92 ммоль) поместили в реакционную колбу, добавили метиленхлорид (240 мл) и перемешивали до тех пор, пока твердые частицы не стали равномерно рассредоточены и не стало очевидной агломерации. Добавили 10%-ный раствор гидроксида натрия (175 мл) и энергично перемешивали в течение 20 секунд. Слои быстро разделили, водный слой экстрагировали дихлорметаном (120 мл), и комбинированный органический слой промыли насыщенным раствором хлорида натрия, высушили над безводным сульфатом натрия и отфильтровали. Твердые вещества промыли, и фильтрат высушили в вакууме и переместили в пробирку для применения.
Соединение 13' (24 г, 54,17 ммоль) поместили в реакционную колбу, добавили пиридин (300 мл) и медленно перемешивали при комнатной температуре до полного растворения. После полного растворения внутреннюю температуру понизили примерно до -10°С на ледяной бане с солью. Предварительно подщелоченный хлорангидрид 4-пиперидинилпиперидинкарбоновой кислоты (соединение 7, 23,5 г, 87,92 ммоль) растворили в дихлорметане (50 мл) и переместили в капельную воронку постоянного давления, реакционный раствор медленно добавляли по каплям, и температуру поддерживали на уровне менее чем -5°С. После завершения добавления реакционную жидкость перемешивали в течение 2 часов при комнатной температуре. После завершения реакции добавили воду (240 мл) и перемешивали в течение 10 мин, экстрагировали дихлорметаном (240 мл), водный слой промыли насыщенным раствором карбоната натрия (24 мл), а затем экстрагировали дихлорметаном (240 мл), органические слои объединили, промыли насыщенным раствором хлорида натрия (300 мл), высушили над безводным сульфатом натрия и высушили в вакууме, и удалили пиридин для получения продукта в виде твердого вещества. Затем твердое вещество перекристаллизовали в дихлорметане (с 5%-ным раствором изопропанола) (75 мл) и диэтиловом эфире (220 мл) и отфильтровали с получением кристаллов. Кристаллы промыли диэтиловым эфиром и высушили в вакууме при 40°С, получив таким образом примерно 33 г продукта (соединение 16) в виде светло-желтого твердого вещества, выход которого составил 95%.
1Н-ЯМР (ДМСО-d6): δ 0,88 (t, J=9,0 Гц, 3Н), 1,41-1,63 (m, 9Н), 1,81-1,92 (m, 4Н), 2,91-2,97 (m, 1Н), 3,10-3,16 (m, 1H), 4,05-4,08 (m, 1H), 4,31-4,35 (m, 1H), 5,33 (s, 2Н), 5,43 (s, 2Н), 6,56 (s, 1H), 7,35 (s, 1Н), 7,82 (d, J=8,8 Гц, 1H), 8,22 (d, J=8,8 Гц, 1H), 8,90 (s, 1H).
Пример 4
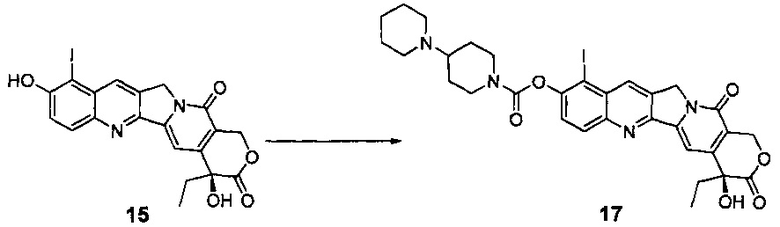
Соединение 15' (730 мг, 54,17 ммоль) поместили в реакционную колбу, добавили пиридин (10 мл) и медленно перемешивали при комнатной температуре до полного растворения. После полного растворения внутреннюю температуру понизили примерно до -10°С на ледяной бане с солью. Предварительно подщелоченный хлорангидрид 4-пиперидинилпиперидинкарбоновой кислоты (366 мг, 1,37 ммоль) растворили в дихлорметане (5 мл) и переместили в капельную воронку, реакционный раствор медленно добавляли по каплям, и внутреннюю температуру поддерживали на уровне менее чем -5°С (что требует медленного добавления по каплям). После завершения добавления реакционную смесь перемешивали в течение 2 часов при комнатной температуре. После завершения реакции добавили воду (20 мл) и перемешивали в течение 10 мин, экстрагировали дихлорметаном (20 мл), водный слой промыли насыщенным раствором карбоната натрия (5 мл), а затем экстрагировали дихлорметаном, и органические слои объединили, промыли насыщенным раствором хлорида натрия (30 мл), высушили над безводным сульфатом натрия и высушили в вакууме, и удалили пиридин. Затем твердые вещества перекристаллизовали в дихлорметане (с 5%-ным раствором изопропанола) (2 мл) и диэтиловом эфире (6 мл), и отфильтровали, кристаллы собрали, промыли диэтиловым эфиром и высушили в вакууме при 40°С. Получили 909 мг продукта в виде светло-желтого твердого вещества (соединение 17), выход которого составил 97%.
1Н-ЯМР (ДМСО-d6): δ 0,88 (t, J=7,2 Гц, 3Н), 1,41-1,66 (m, 9Н), 1,82-1,92 (m, 4Н), 2,90-2,97 (m, 1Н), 3,10-3,16 (m, 1H), 4,06-4,09 (m, 1H), 4,35-4,38 (m, 1H), 5,35 (s, 2Н), 5,44 (s, 2Н), 6,56 (s, 1H), 7,36 (s, 1Н), 7,73-7,76 (d, J=8,8 Гц, 1Н), 8,18-8,21 (d, J=8,8 Гц, 1Н), 8,83 (s, 1Н).
Реакция III:
Пример 5
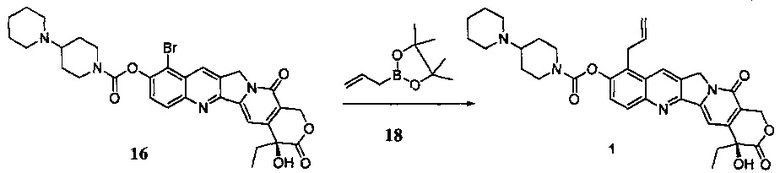
Соединение 16 (2500 мг, 3,925 ммоль), Pd2(dba)3 (359 мг, 0,392 ммоль), тетрафторборат три-трет-бутилфосфония (273 мг, 0,942 ммоль) и KF (6829 мг, 117,739 ммоль) поместили в трехгорлую реакционную колбу объемом 250 мл. В защитной атмосфере азота добавили 1,4-диоксан (150 мл) при комнатной температуре и перемешивали до однородной дисперсии в системе. При комнатной температуре добавили DIPEA (1519 мг, 11,774 ммоль) и H2O (7064 мг, 392,465 ммоль) и перемешали до получения однородной смеси, добавили пинаколовый эфир аллилборной кислоты (соединение 18) (6593 мг, 39,246 ммоль), перемешали до получения однородной смеси, затем нагревали до 60°С в течение 2,5 ч. После завершения реакции добавили 20 мл дихлорметана, отфильтровали через целит и промыли 30 мл дихлорметана, растворитель высушили в вакууме и остаток очистили с помощью колоночной хроматографии (дихлорметан : метанол = 50:1-15:1) с получением 2,2 г желтого твердого вещества (соединение 1), выход которого составил 88%.
1Н-ЯМР (HMCO-d6): δ 0,89 (t, J=7,5 Гц, 3Н), 1,37-1,39 (m, 1Н), 1,67-1,93 (m, 9Н), 2,17-2,23 (m, 2Н), 2,87-2,93 (m, 3Н), 3,09-3,13 (m, 1Н), 3,35-3,41 (m, 3Н), 3,81 (d, J=6,0 Гц, 2Н), 4,16-4,18 (m, 1Н), 4,38-4,40 (m, 1Н), 5,01-5,07 (m, 2Н), 5,27 (s, 2Н), 5,43 (s, 2Н), 5,96-6,04 (m, 1Н), 6,53 (s, 1H), 7,33 (s, 1H), 7,68 (d, J=9,0 Гц, 1H), 8,08 (d, J=9,0 Гц, 1Н), 8,87 (s, 1H), 10,72 (s, 1H).
Пример 6
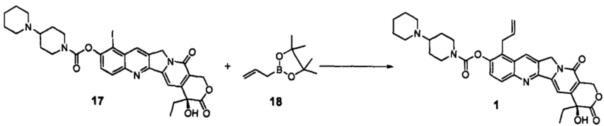
Соединение 17 (100 мг, 0,146 ммоль), Рd2(dba)3 (14 мг, 0,014 ммоль), тетрафторборат три-трет-бутилфосфония (8 мг, 0,015 ммоль), KF (8 мг, 0,146 ммоль) и тригидрат фосфата калия (116 мг, 0,438 ммоль) поместили в трехгорлую реакционную колбу объемом 50 мл. В защитной атмосфере азота добавили 1,4-диоксан (6 мл) при комнатной температуре, перемешивали до однородной дисперсии в системе. При комнатной температуре добавили DIPEA (30 мг, 0,233 ммоль) и H2O (28,3 мг, 1,570 ммоль) и перемешивали до получения однородной смеси, добавили пинаколовый эфир аллилборной кислоты (соединение 18) (28 мг, 0,160 ммоль) и перемешивали до получения однородной смеси, затем нагревали до 60°С в течение 2,5 ч. После завершения реакции добавили 10 мл дихлорметана и отфильтровали через целит, отфильтрованный осадок промыли 10 мл дихлорметана, растворитель высушили в вакууме и остаток очистили с помощью колоночной хроматографии (дихлорметан : метанол = 50:1-15:1) с получением 35 мг желтого твердого вещества (соединение 1), выход которого составил 40%. Данные ЯМР были идентичны приведенным в примере 5.
Пример 7
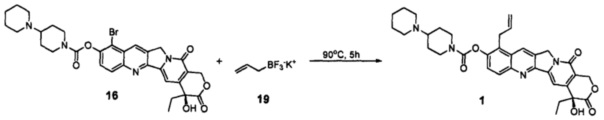
Соединение 16 (1000 мг, 1,57 ммоль), соединение 19 (2320 мг, 15,7 ммоль), Pd2(dba)3 (140 мг, 0,16 ммоль), тетрафторборат три-трет-бутилфосфония (110 мг, 0,24 ммоль) и KF (2731 мг, 47,1 ммоль) поместили в трехгорлую реакционную колбу объемом 100 мл. В защитной атмосфере азота добавили изопропанол (60 мл) при комнатной температуре и перемешали. Затем добавили DIPEA (608 мг, 4,71 ммоль) и H2O (700 мг, 39 ммоль), перемешали и нагревали до 90°С в течение 5 ч. После завершения реакции добавили 100 мл дихлорметана и отфильтровали через целит, промыли 100 мл дихлорметана, растворитель удалили в вакууме, и остаток очистили с помощью колоночной хроматографии (дихлорметан : метанол = 50:1-15:1) с получением 750 мг желтого твердого вещества, выход которого составил 80%. Данные ЯМР были идентичны приведенным в примере 5.
Соединение 1 также может быть получено в соответствии со следующими условиями из соединения 16 или соединения 17:
(1) трис(дибензилиденацетон)дипалладий, фторид калия, три-трет-бутилфосфин и 1,4-диоксан;
(2) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, гидратированный фосфат калия и 1,4-диоксан;
(3) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, карбонат калия и 1,4-диоксан;
(4) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, гидратированный фосфат калия, фторид калия и 1,4-диоксан;
(5) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия и 1,4-диоксан;
(6) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид цезия и 1,4-диоксан;
(7) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, триэтиламин, фторид калия и 1,4-диоксан;
(8) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, триэтиламин, фторид калия и тетрагидрофуран;
(9) тетракис(трифенилфосфин)палладий, три-трет-бутилфосфин, фторид калия и 1,4-диоксан;
(10) ацетат палладия, три-трет-бутилфосфин, фторид калия и 1,4-диоксан;
(11) дихлорбис(трифенилфосфин)палладий, три-трет-бутилфосфин, фторид калия и 1,4-диоксан;
(12) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, триэтиламин, фторид калия и метанол;
(13) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия и метанол;
(14) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин и метанол;
(15) тетракис(трифенилфосфин)палладий, три-трет-бутилфосфин, диизопропилэтиламин и метанол;
(16) дихлорбис(трифенилфосфин)палладий, три-трет-бутилфосфин, диизопропилэтиламин и метанол;
(17) трис(дибензилиденацетон)дипалладий, трифенилфосфин, диизопропилэтиламин, фторид калия и изопропанол;
(18) трис(дибензилиденацетон)дипалладий, тетрафторборат три-трет-бутилфосфония, диизопропилэтиламин и изопропанол;
(19) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия, вода и изопропанол;
(20) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия, вода и н-бутиловый спирт;
(21) дихлорбис(трифенилфосфин)палладий, три-трет-бутилфосфин, диизопропилэтиламин, вода и н-бутиловый спирт;
(22) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин, фторид калия и н-бутиловый спирт;
(23) трис(дибензилиденацетон)дипалладий, три-трет-бутилфосфин, диизопропилэтиламин и н-бутиловый спирт;
Реакция IV:
Пример 8
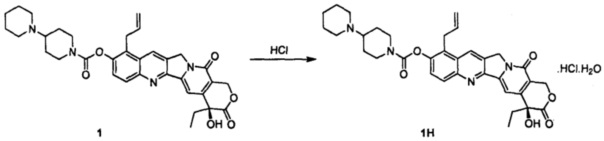
Соединение 1 (5 г) растворили в дихлорметане (с 5%-ным раствором изопропанола), перемешали и охладили до температуры ниже 10°С. 4 М раствора соляной кислоты в изопропаноле добавили по каплям до рН=3~5. Внутреннюю температуру повысили до комнатной температуры и перемешивали в течение 30 минут. Добавили по каплям диэтиловый эфир, и после добавления смесь перемешивали в течение 1 часа с получением твердого вещества желтого цвета. После фильтрования отфильтрованный осадок промыли эфиром и высушили в вакууме при 35°С с получением примерно 5,8 г желтого твердого вещества.
Указанное выше высушенное твердое вещество добавили к 11 мл воды и растворили. Добавили по каплям ацетон (88 мл) при кипячении с обратным холодильником, и осаждение кристаллов происходило при естественном охлаждении и при -10°С в течение ночи. На следующий день кристаллы отфильтровали, промыли ацетоном и высушили с получением 4,8 г твердого вещества. Твердое вещество повторно растворили в воде (10 мл) и добавили по каплям ацетон (85 мл) при кипячении с обратным холодильником. Осаждение кристаллов происходило при естественном охлаждении и при -10°С в течение ночи. На следующий день кристаллы отфильтровали с получением соединения 1Н в виде светло-желтого или белого твердого вещества (3,5 г).
1Н-ЯМР (ДМСО-d6): δ 0,88 (t, J=7,6 Гц, 3Н), 1,40-1,43 (m, 1H), 1,70-1,93 (m, 9Н), 2,18-2,25 (m, 2Н), 2,93-2,99 (m, 3Н), 3,12-3,16 (m, 1H), 3,35-3,42 (m, 3Н), 3,80-3,81 (d, J=5.6 Гц, 2Н), 4,15-4,18 (m, 1H), 4,37-4,40 (m, 1Н), 5,00-5,06 (m, 2Н), 5.27 (s, 2Н), 5,43 (s, 2Н), 5,96-6,03 (m, 1H), 6,55 (s, 1H), 7,33 (s, 1H), 7,66-7,68 (d, J=9.0 Гц, 1H), 8,07-8,09 (d, J=9.0 Гц, 1Н), 8,87 (s, 1H), 10,62 (s, 1H).
Все источники литературы, упомянутые в настоящей заявке, включены в данное описание посредством ссылки, как если бы каждый из них по отдельности был включен посредством ссылки. Кроме того, следует понимать, что после прочтения вышеизложенного специалисты в данной области техники могут сделать различные изменения и модификации в настоящем изобретении. Эти эквиваленты также попадают в рамки, определяемые прилагаемой формулой изобретения.
Изобретение относится к производным 9-аллилкамптотецина, включая соединение формулы 14 в качестве важнейшего промежуточного продукта для получения соединения формулы 1 и его гидрохлорида формулы 1Н, который обладает противоопухолевым действием. Изобретение также относится к способу получения соединения формулы 14 и способу получения соединений 1 и 1Н. Технический результат - высокий общий выход продуктов. 3 н. и 7 з.п. ф-лы, 8 пр.


1. Соединение формулы 14

где X представляет собой галоген.
2. Способ получения соединения 14, включающий стадию: взаимодействие соединения 13 в инертном растворителе с соединением 7 с получением соединения 14
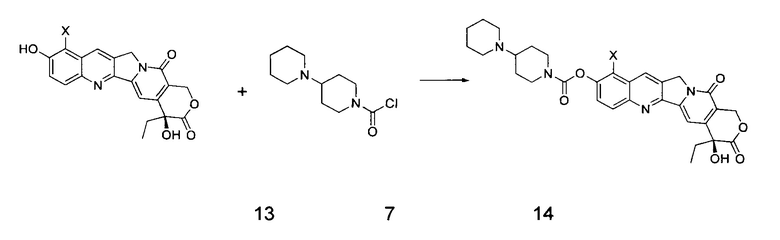
где в приведенных выше формулах X представляет собой галоген.
3. Способ получения по п. 2, отличающийся тем, что соединение формулы 13 получают способом, включающим следующую стадию:
взаимодействие соединения 3 в инертном растворителе с галогенирующим агентом с получением соединения 13
4. Способ получения соединения 1, включающий стадию: проведение реакции Сузуки между соединением 14 и аллилборным реагентом с получением соединения 1
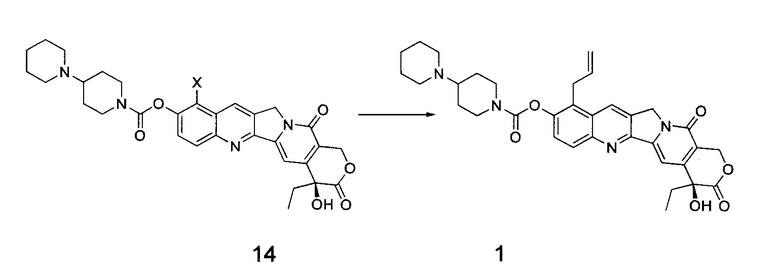
где в приведенных выше формулах X представляет собой галоген.
5. Способ получения по п. 4, отличающийся тем, что аллилборный реагент выбран из группы, состоящей из: пинаколового эфира аллилборной кислоты, комплексной соли фторида аллилбора.
6. Способ получения по п. 4, отличающийся тем, что соединение формулы 14 получено способом по п. 2.
7. Способ получения по п. 4, отличающийся тем, что реакцию Сузуки проводят в системе, содержащей следующие агенты: палладиевый катализатор, фосфиновый лиганд, щелочь и инертный растворитель.
8. Способ получения по п. 7, отличающийся тем, что
палладиевый катализатор выбран из группы, состоящей из:
трис(дибензилиденацетон)дипалладия (Pd2(dba)3),
тетракис(трифенилфосфин)палладия (Pd(PPh3)4), ацетата палладия, дихлорбис(трифенилфосфин)палладия, трифторацетата палладия, ацетата трифенилфосфинпалладия, дихлорбис(три-о-бензил-фосфин)палладия, дихлор[1,2-бис(дифенилфосфино)этан]палладия или их комбинаций;
фосфиновый лиганд выбран из следующей группы: три-трет-бутилфосфин, тетрафторборат три-трет-бутилфосфония, три-н-бутилфосфин, трифенилфосфин, три-п-бензил-фосфин, трициклогексилфосфин, три-о-бензил-фосфин, или их комбинации; основание выбрано из следующей группы: фторид калия, фторид цезия, гидратированный фосфат калия, карбонат калия, карбонат натрия, гидрокарбонат натрия, 1,8-диазабицикло[5.4.0]ундец-7-ен, триэтиламин, диизопропилэтиламин, пиридин или их комбинации; инертный растворитель выбран из следующей группы: 1,4-диоксан, тетрагидрофуран, ацетонитрил, диметилсульфоксид, N,N-диметилформамид, толуол, метанол, этанол, изопропанол, н-бутиловый спирт, т-бутанол, изо-бутанол, бензиловый спирт, вода или их комбинации.
9. Способ получения по п. 7, отличающийся тем, что
палладиевый катализатор выбран из группы, состоящей из:
трис(дибензилиденацетон)дипалладия (Pd2(dba)3),
тетракис(трифенилфосфин)палладия (Pd(PPh3)4), или их комбинаций; фосфиновый лиганд выбран из следующей группы: три-трет-бутилфосфин, тетрафторборат три-трет-бутилфосфония или их комбинации; и/или
основание выбрано из следующей группы: фторид калия, фторид цезия, гидратированный фосфат калия, диизопропилэтиламин, триэтиламин или их комбинации; инертный растворитель выбран из следующей группы: 1,4-диоксан, изопропанол, вода, бензиловый спирт или их комбинации.
10. Способ получения по п. 4, отличающийся тем, что указанный способ дополнительно содержит следующую стадию: кислотная обработка соединения 1 с получением соединения 1Н
| WO 2007104214 A1 20.08.2007 | |||
| CN 101880285 A 10.11.2010 | |||
| WO 2005044821 A1 19.05.2005 | |||
| RU 98117449 A 10.09.2000. |

