ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новому способу получения рентгеноконтрастного агента йомепрола.
Точнее, настоящее изобретение относится к новому способу получения рентгеноконтрастного агента йомепрола, который может обеспечить легкое отделение и удаление неорганических солей, образовавшихся во время реакции, без обработки ионообменной смолой при сокращении имеющегося времени получения с помощью одностадийного способа синтеза путем проведения реакции N-метилирования путем добавления неорганического основания, неорганического хлорида и растворителя к 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамиду.
С помощью настоящего изобретения можно экономично получить йомепрол, контрастное соединение, с высоким выходом, равным 99% или более, с помощью одностадийного способа синтеза и перекристаллизации, как описано выше.
УРОВЕНЬ ТЕХНИКИ
Йомепрол является неионогенным контрастным агентом третьего поколения, который представляет собой N,N'-бис(2,3-дигидроксипропил)-5-(2-гидрокси-N-метилацетамидо)-2,4,6-трийодизофталамид, обладающий следующей формулы 1a. Его получают метилированием соединения следующей формулы 1b. Он является рентгеноконтрастным и CT-контрастным агентом трийодизофталамидом, разработанным фирмой Bracco Imaging S.p.A, Italy, и его используют в разных случаях в ангиографии, и он впервые описан в Европейском патенте № EP 0026281.
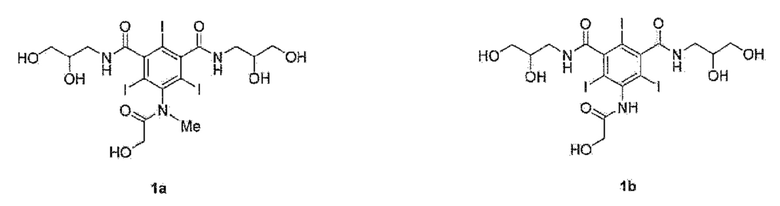
Кроме того, пути получения 5-(гидроксиацил)аминопроизводных на основании реакции перегруппировки Смайлса описан в Международных публикациях №№ WO 88/09328 и WO 00/32651.
Преимущество способа синтеза, описанного в Международной публикации № WO 00/32561, в основном состоит в том, что не используются некоторые реагенты и растворители, такие как тионилхлорид, уксусный ангидрид, метилйодид, метиленхлорид и хлороформ, или исключается реакция с водородом с использованием катализаторов и т. п.
Способ синтеза, указанный выше, является следующим (схема 1).
Схема 1
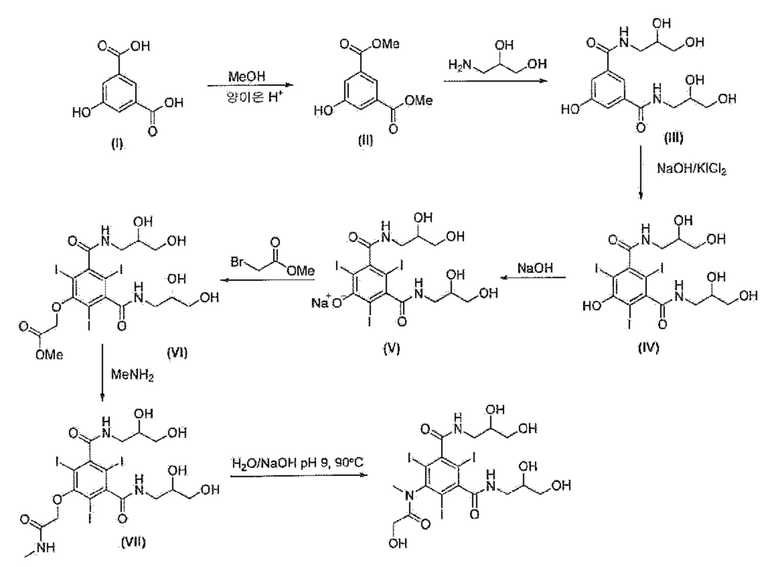
В способе синтеза, приведенном в Международной публикации № WO 00/32561, в условиях промышленного производства не используются вредные вещества. Однако стадия синтеза является длительной и при переходе от соединения формулы (III) к соединению последней стадии в концевой группе содержится оксигруппа, что указывает на растворимость в воде. Таким образом, имеется трудная задача удаления неорганического вещества, использующегося на стадиях 4 и 7.
Поскольку йомепрол растворим в воде, его очень трудно отделить и удалить, если во время получения образуются неорганические соли.
Для решения этой задачи и получения целевого продукта высокой чистоты не только йомепрол, но и другие контрастные агенты на основе йода очищают путем использования ионообменной смолы на заключительной стадии удаления неорганических веществ.
С точки зрения условий промышленного производства устройство с ионообменной смолой обладает теми экономическими недостатками, что его стоимость не является низкой, для его установки требуется пространство и через некоторое время требуется замена смолы.
Для способа синтеза, описанного в Международной публикации № WO 88/09328, требуется более короткое время получения, чем для описанного в Международной публикации № WO 00/32561. Однако, поскольку неорганические соли также используются на конечной стадии способа синтеза, описанного в Международной публикации № WO 88/09328, для разделения полученного йомепрола и неорганических веществ необходимо использовать устройство с ионообменной смолой.
Способ получения без обработки ионообменной смолой, который использовали для подавления образования неорганических веществ, что являлось затруднением в указанных выше патентах, зарегистрировать авторами настоящего изобретения, как патент Кореи 101833334.
В указанном выше патенте каждую гидроксигруппу исходного вещества ацетилируют с образованием защитной группы, так что оно хорошо растворимо в органическом растворителе.
После этого предложен способ синтеза йомепрола, в котором неорганические вещества удаляли с помощью реакции удаления защитной группы после реакции метилирования синтезированного нового вещества. N,N'-бис(2,3-диацетилатпропил)-5-(2-ацетокси-N-метилацетамидо)-2,4,6-трийодизофталамида (схема 2).
Схема 2
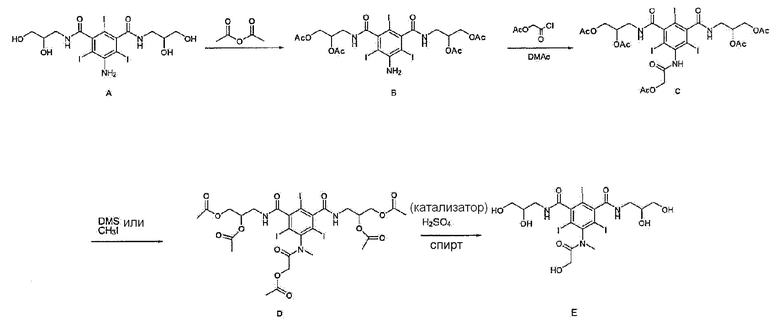
Хотя указанный выше способ получения обладает тем преимуществом, что неорганические вещества удаляются в процессе экстракции с использованием нового растворимого в масле промежуточного продукта, он обладает тем недостатком, что получение является длительным, поскольку способ получения состоит 4 стадий, что увеличивает стоимость производства в реальных условиях.
Поэтому для компенсации недостатков указанных выше способов производства необходима разработка способа производства, в котором неорганические соли удаляют без использования ионообменной смолы, и технология производства, способная гарантировать экономическую эффективность при сокращении времени получения и сведении к минимуму образования примесей при реакции.
ПУБЛИКАЦИЯ ПРЕДШЕСТВУЮЩЕГО УРОВНЯ ТЕХНИКИ
ПАТЕНТНАЯ ПУБЛИКАЦИЯ
Патентная публикация 1: WO 0032561 A
Патентная публикация 2: WO 8809328 A
Патентная публикация 3: Korean Patent No. 101833334 A
СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
РЕШАЕМАЯ ЗАДАЧА
Задачей настоящего изобретения является разработка способа получения рентгеноконтрастного агента йомепрола, в котором можно свести к минимуму образования примесей при реакции и легко удалить неорганические соли при значительном сокращении времени получения по сравнению с имеющимся путем проведения реакции N-метилирования путем добавления неорганического основания, неорганического хлорида и растворителя, который растворяет неорганический хлорид с получением имеющегося в продаже 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида.
ТЕХНИЧЕСКИЕ СРЕДСТВА
Для решения этой технической задачи в первом объекте настоящего изобретения предложен новый способ получения контрастного агента йомепрола, описывающегося формулой 1a, включающий стадию быстрого и стабильного проведения реакции N-метилирования путем добавления N-метилирующего реагента, неорганического основания, неорганического хлорида и растворителя, который растворяет неорганический хлорид, такого как метанол, DMSO, DMAc и DMF, к 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамиду формулы 1b.
ЭФФЕКТ ИЗОБРЕТЕНИЯ
Способ получения йомепрола, предлагаемый в настоящем изобретении, может привести к сокращению времени получения, сведению к минимуму образования примесей при реакции и получению йомепрола высокой чистоты, равной 99% или более, путем обеспечения растворения неорганического хлорида в растворителе для проведения реакции и растворителе для кристаллизации при эффективном отделении и удалении неорганической соли без использования отдельной ионообменной смолы путем проведения реакции N-метилирования при особой температуре путем добавления N-метилирующего реагента, неорганического основания, неорганического хлорида и растворителя, который растворяет неорганический хлорид с образованием 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида (формула 1b).
КОНКРЕТНЫЙ ПУТЬ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Ниже настоящее изобретение описано подробнее.
Способ получения йомепрола, предлагаемый в настоящем изобретении, включает стадию проведения реакции N-метилирования путем добавления N-метилирующего реагента, неорганического основания, неорганического хлорида и растворителя к 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид (формула 1b).
Способ получения, предлагаемый в настоящем изобретении, в частности, представляет собой способ, представленный ниже на схеме 3.
Схема 3
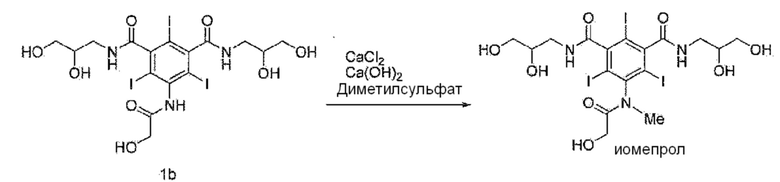
В способе получения, предлагаемом в настоящем изобретении, контрастный агент йомепрол (формула 1a) получают путем добавления N-метилирующего реагента, неорганического основания, неорганического хлорида и растворителя, который растворяет неорганический хлорид , к 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамиду (формула 1b).
5-(2-Гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид, описывающийся формулой 1b, содержит пять гидроксигрупп, но не обладает хорошей растворимостью в воде или спирте.
Для проведения реакции N-метилирования соединение формулы 1b необходимо сначала растворить. Для этого добавляют неорганический хлорид, т. е. хлорид кальция, для образования ионной связи с гидроксигруппой для создания условий для растворения в воде или метаноле.
Неорганическое основание действует, как основание Бренстеда, для удаления водорода из амидной группы для реакции N-метилирования.
В частности, как показано на схеме 3, после добавления 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида (формула 1b) и неорганического хлорида к растворителю, такому как диметилсульфоксид (DMSO) или метанол, и перемешивания для растворения, добавляют основание Бренстеда, т. е. гидроксид натрия, и N-метилирующий реагент, диметилсульфат, растворяется в растворителе для проведения реакции.
N-Метилирующий реагент, использующийся в способе получения, предлагаемом в настоящем изобретении, выбран из группы, состоящей из: диметилсульфата, метилйодида и их комбинация, и предпочтительно представляет собой диметилсульфат.
В способе получения, предлагаемом в настоящем изобретении, используют 1 или большее количество эквивалентов, предпочтительно от 3 до 4 экв. N-метилирующего реагента в пересчете на 1 экв. 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида.
Если содержание N-метилирующего реагента меньше указанного в приведенном выше числовом диапазоне, реакция может протекать в недостаточной степени и время реакции может увеличиться. Если содержание N-метилирующего реагента больше указанного в приведенном выше числовом диапазоне, может увеличиться количество примесей.
Неорганическое основание, использующееся в способе получения, предлагаемом в настоящем изобретении, выбрано из группы, состоящей из: гидроксида лития, гидроксида кальция, гидроксида магния, гидроксида бериллия и их комбинация, и предпочтительно представляет собой гидроксид кальция.
Неорганические основания, такие как гидроксид кальция, после завершения реакции образуют неорганические соли, такие как хлорид кальция, и воду, и хлорид кальция хорошо растворим в метаноле, использующемся в качестве растворителя для проведения реакции, и затем в растворителе для кристаллизации, так что неорганические соли можно эффективно удалить без использования ионообменной смолы. В результате можно получить йомепрол высокой чистоты (формула 1a).
В способе получения, предлагаемом в настоящем изобретении, можно использовать от 0,5 до 2 экв., предпочтительно от 0,6 до 1,2 экв., более предпочтительно от 0,6 до 0,7 экв. неорганического основания в пересчете на 1 экв. 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида (формула 1b).
Если содержание неорганического основания меньше указанного в приведенном выше числовом диапазоне, может снизится скорость реакции. Если содержание больше указанного в приведенном выше числовом диапазоне, может увеличиться количество примесей.
В качестве неорганического хлорида, использующегося в способе получения, предлагаемом в настоящем изобретении, можно использовать хлорид кальция, хлорид лития, хлорид бериллия, хлорид магния и т. п., которые могут растворяться в метаноле, являющемся растворителем для проведения реакции
В случае получения йомепрола без добавления неорганического хлорида, реагент 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид (формула 1b) не растворяется в метаноле, который является растворителем для проведения реакции. Реакция не протекает легко и даже если реакция частично протекает, реакция может не закончиться и может остаться реагент.
Поскольку оставшийся реагент, соединение формулы 1b, обладает структурой, сходной со структурой йомепрола, его очень трудно удалить после завершения реакции.
В способе получения, предлагаемом в настоящем изобретении, можно использовать от 2 до 10 экв., предпочтительно от 2 до 4 экв., более предпочтительно 3 экв. неорганического хлорида в пересчете на 1 экв. 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида (формула 1b).
Если содержание неорганического хлорида слишком велико, велика концентрация раствора реакционной смеси, так что растворение или перемешивание может быть затруднительным. Если его содержание слишком мало или он отсутствует, реакция может не завершиться, поскольку не растворяется соединение формулы 1b.
Растворитель, использующийся в способе получения, предлагаемом в настоящем изобретении, можно выбрать из группы, состоящей из: метанола, диметилформамида (DMF), диметилацетамида (DMAc), диметилсульфоксида (DMSO) и их комбинация. Предпочтительно можно использовать метанол.
В способе получения, предлагаемом в настоящем изобретении, отношение массы растворителя к массе 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида (формула 1b) может равняться от 4 до 10, предпочтительно от 5 до 7.
Если содержание растворителя меньше указанного в приведенном выше числовом диапазоне, растворение может быть затруднительным. Если содержание больше указанного в приведенном выше числовом диапазоне, может не быть обеспечен выход на стадии кристаллизации.
Способ получения, предлагаемый в настоящем изобретении, дополнительно включает стадию кристаллизации продукта путем добавления растворителя для кристаллизации к продукту, полученному по реакции N-метилирования.
Растворителем для кристаллизации может быть любой, выбранный из группы, состоящей из: метанола, этанола, изопропанола, нормального бутанола, 2-бутанола или их комбинация, предпочтительно метанол.
Растворитель для кристаллизации используется для эффективного удаления неорганической соли и увеличения выхода.
На стадии кристаллизации после завершения реакции N-метилирования, кислоту (HCl и т. п.) добавляют для подкисления, и затем растворитель для кристаллизации добавляют для кипячения и перемешивания при температуре от комнатной температуры до 80°C, предпочтительно от 70 до 80°C, в течение от 2 до 24 ч, предпочтительно 3 ч.
Затем кристаллы отфильтровывают, в достаточной степени промывают растворителем для кристаллизации и сушат при пониженном давлении при температуре от 50 до 90°C.
Настоящее изобретение подробнее описано с помощью следующих примеров и сравнительных примеров. Однако они никоим образом не ограничивают объем настоящего изобретения.
ПРИМЕРЫ
Пример 1.
Получение йомепрола
5 г (1 экв.) 5-(2-Гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида (формула 1b) и 3,6 г (5 экв.) хлорида кальция вместе добавляли к 25 г метанола и затем растворяли путем нагревания при комнатной температуре или 70°C в течение 60 мин.
После снижения температуры раствора на 10-15°C добавляли 0,3 г (0,62 экв.) гидроксида кальция, затем перемешивали при такой же температуре в течение 1 ч.
2,48 г (3 экв.) Диметилсульфата добавляли к раствору реакционной смеси и перемешивали при такой же температуре в течение 3 ч до завершения реакции.
После завершения реакции добавляли 1 мл HCl (35%) для подкисления и добавляли 25 мг 2-бутанола и перемешивали при температуре, равной от 70 до 80°C в течение 2 ч. Затем смесь охлаждали, фильтровали и промывали 2-бутанолом и получали неочищенный йомепрол.
После добавления полученного выше неочищенного йомепрола к смеси 25 мл метанола и 10 мл воды температуру повышали до 50°C для их растворения, затем добавляли 20 мл 2-бутанола и кипятили с обратным холодильником при 90°C в течение 3 ч. Затем смесь охлаждали до комнатной температуры и отфильтровывали полученные кристаллы.
После промывки 2-бутанолом их сушили при пониженном давлении при 90°C в течение 12 ч и получали 4,17 г йомепрола (HPLC: 99,3%).
Пример 2.
Получение йомепрола
5 г (1 экв.) 5-(2-Гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида (формула 1b) и 3,6 г (5 экв.) хлорида кальция вместе добавляли к 25 г метанола, и затем растворяли путем нагревания при комнатной температуре или 70°C в течение 30 мин.
После снижения температуры раствора на 0-5°C добавляли 0,3 г (0,62 экв.) гидроксида кальция, затем перемешивали при такой же температуре в течение 1 ч.
2,48 г (3 экв.) Диметилсульфата добавляли к раствору реакционной смеси и перемешивали при такой же температуре в течение 7 ч до завершения реакции.
После завершения реакции добавляли 1 мл HCl (35%) для подкисления и добавляли 25 мг 2-бутанола и перемешивали при температуре, равной от 70 до 80°C в течение 2 ч. Затем смесь охлаждали, фильтровали и промывали 2-бутанолом и получали неочищенный йомепрол.
После добавления полученного выше неочищенного йомепрола к смеси 25 мл метанола и 10 мл воды температуру повышали до 50°C для их растворения, затем добавляли 20 мл 2-бутанола и кипятили с обратным холодильником при 90°C в течение 3 ч. Затем смесь охлаждали до комнатной температуры и отфильтровывали полученные кристаллы.
После промывки 2-бутанолом их сушили при пониженном давлении при 90°C в течение 12 ч и получали 4,21 г йомепрола (HPLC: 99,1%).
Сравнительный пример 1 (без добавления неорганического хлорида)
5-(2-Гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид (формула 1b) добавляли к 25 г метанола и затем кипятили с обратным холодильником в течение 30 мин.
После снижения температуры мутного раствора на 10-15°C добавляли 0,3 г (0,62 экв.) гидроксида кальция, затем перемешивали при такой же температуре в течение 1 ч.
2,48 г (3 экв.) Диметилсульфата добавляли к раствору реакционной смеси и перемешивали при такой же температуре в течение 5 ч.
По данным исследования реакционной способности с помощью HPLC установлено, что синтез йомепрола протекал на 5% и установлено, что реакционная способность была очень низкой, поскольку осталось более 90% 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида (формула 1b).
Сравнительный пример 2 (без добавления неорганического основания)
5-(2-Гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамид (формула 1b) и 3,6 г (5 экв.) хлорида кальция вместе добавляли к 25 г метанола и затем кипятили с обратным холодильником в течение 30 мин.
После снижения температуры раствора на 10-15°C к раствору реакционной смеси добавляли 2,48 г (3 экв.) диметилсульфата и перемешивали при такой же температуре в течение 5 ч.
По данным исследования реакционной способности с помощью HPLC установлено, что синтез йомепрола протекал на 5% и установлено, что реакционная способность была очень низкой, поскольку осталось более 99% 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида (формула 1b).
Как показано выше, поскольку в настоящем изобретении соединение формулы получают 1a метилированием соединения формулы 1b без дополнительных промежуточных стадий путем использования неорганических оснований, неорганических хлоридов и растворителя, который растворяет неорганические хлориды, настоящее изобретение может привести к сокращению имеющегося способа получения и времени получения йомепрола высокой чистоты без необходимости проведения очистки с помощью отдельного устройства с ионообменной смолой.
Подробное описание настоящего изобретения является просто иллюстрацией настоящего изобретения и используется только для описания настоящего изобретения без ограничения сущности или объема настоящего изобретения, описывающихся формулой изобретения.
Поэтому специалисты с общей подготовкой в данной области техники должны понимать, что возможны различные модификации и другие эквивалентные варианты осуществления.
Поэтому истинный технический объем защиты для настоящего изобретения следует определять по объему прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНТРАСТНЫЕ АГЕНТЫ | 2008 |
|
RU2469021C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИОГЕКСОЛА | 1997 |
|
RU2173315C2 |
| Получение промежуточных соединений для получения рентгеноконтрастных веществ | 2013 |
|
RU2659214C2 |
| ОЧИСТКА РЕНТГЕНОКОНТРАСТНЫХ ВЕЩЕСТВ | 2013 |
|
RU2662941C2 |
| Способ получения соединения 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(S-метилсульфонимидоил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида | 2019 |
|
RU2765144C1 |
| СИНТЕЗ 2-МЕТИЛ-4-(4-МЕТИЛ-1-ПИПЕРАЗИНИЛ)-10H-ТИЕНО[2,3-b] [1,5]БЕНЗОДИАЗЕПИНА И ЕГО СОЛЕЙ | 2005 |
|
RU2435775C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-[(3R)-3-МЕТИЛМОРФОЛИН-4-ИЛ]-4-(1-МЕТИЛ-1H-ПИРАЗОЛ-5-ИЛ)-8-(1H-ПИРАЗОЛ-5-ИЛ)-1,7-НАФТИРИДИНА | 2019 |
|
RU2802512C2 |
| Способ получения фармацевтической субстанции на основе йогексола | 2017 |
|
RU2655619C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 5-АМИНО-2,4,6-ТРИИОДО- 1,3-БЕНЗОЛКАРБОНОВОЙ КИСЛОТЫ | 1990 |
|
RU2046795C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТИОБАРБИТУРОВОЙ КИСЛОТЫ | 2000 |
|
RU2242467C2 |
Изобретение относится к способу получения рентгеноконтрастного агента йомепрола. Способ заключается в N-метилировании 5-(2-гидроксиацетамидо)-N,N'-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида путем добавления N-метилирующего реагента, неорганического основания, неорганического хлорида и растворителя, который растворяет неорганический хлорид. В качестве неорганического основания выбирают любое соединение из группы, состоящей из: гидроксида лития, гидроксида кальция, гидроксида магния, гидроксида бериллия или их комбинации. Способ позволяет легко отделять и удалять неорганические соли, образующиеся во время реакции, без обработки ионообменной смолой, с сокращением времени получения продукта и сведением к минимуму образования примесей при реакции. 9 з.п. ф-лы, 2 пр.
1. Способ получения йомепрола, включающий стадию проведения реакции N-метилирования путем добавления N-метилирующего реагента, неорганического основания, неорганического хлорида и растворителя, который растворяет неорганический хлорид, к 5-(2-гидроксиацетамидо)-N,N’-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамиду,
где неорганическим основанием является любое, выбранное из группы, состоящей из: гидроксида лития, гидроксида кальция, гидроксида магния, гидроксида бериллия или их комбинации.
2. Способ получения йомепрола по п.1, в котором N-метилирующим реагентом является любой, выбранный из группы, состоящей из: диметилсульфата, метилйодида или их комбинации.
3. Способ получения йомепрола по п.1, в котором используют от 1 до 10 экв. N-метилирующего реагента в пересчете на 1 экв. 5-(2-гидроксиацетамидо)-N,N’-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида.
4. Способ получения йомепрола по п.1, в котором используют от 1 до 10 экв. неорганического основания в пересчете на 1 экв. 5-(2-гидроксиацетамидо)-N,N’-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида.
5. Способ получения йомепрола по п.1, в котором неорганическим хлоридом является любой, выбранный из: хлорида кальция, хлорида лития, хлорида бериллия и хлорида магния.
6. Способ получения йомепрола по п.1, в котором используют от 2 до 10 экв. неорганического хлорида в пересчете на 1 экв. 5-(2-гидроксиацетамидо)-N,N’-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида.
7. Способ получения йомепрола по п.1, в котором растворителем является любой, выбранный из группы, состоящей из: диметилформамида (DMF), диметилацетамида (DMAc), диметилсульфоксида (DMSO), метанола или их комбинации.
8. Способ получения йомепрола по п.1, в котором отношение массы растворителя к массе 5-(2-гидроксиацетамидо)-N,N’-бис(2,3-дигидроксипропил)-2,4,6-трийодизофталамида равно от 4 до 10.
9. Способ получения йомепрола по п.1, дополнительно включающий стадию кристаллизации продукта путем добавления растворителя для кристаллизации к продукту, полученному по реакции N-метилирования.
10. Способ получения йомепрола по п.9, в котором растворителем для кристаллизации является любой, выбранный из группы, состоящей из: метанола, этанола, изопропанола, нормального бутанола, 2-бутанола или их комбинации.
| US 4352788 A, 05.10.1982 | |||
| KR 20170123748 A, 09.11.2017 | |||
| CN 107253918 A, 17.10.2017 | |||
| KR 20150082293 A, 15.07.2015 | |||
| KR 20130090408 A, 13.08.2013 | |||
| Рама для поддержки и направления двух бесконечных лент на валиках второй пары вытяжного аппарата бумагопрядильных машин | 1929 |
|
SU26281A1 |
| АППАРАТ ДЛЯ ЛЕЧЕНИЯ ГНАТИЧЕСКОЙ ФОРМЫ МЕЗИАЛЬНОЙ ОККЛЮЗИИ У ПОДРОСТКОВ | 2014 |
|
RU2547789C1 |
| KR 20180073981 A, 03.07.2018 | |||
| Способ получения фармацевтической субстанции на основе йогексола | 2017 |
|
RU2655619C1 |

