Область техники
Настоящее изобретение относится к области фармацевтической химии. В частности, настоящее изобретение относится к классу пиридопиримидиновых или пиримидопиримидиновых соединений, изомеров, фармацевтически приемлемых солей, сложных эфиров, пролекарств или сольватов указанных соединений, способу получения указанных соединений, фармацевтической композиции, содержащей указанные соединения, и применению указанных соединений для получения ингибитора мишени рапамицина у млекопитающих (mTOR). Такое соединение или его фармацевтическая композиция в качестве ингибитора mTOR может быть применено для лечения заболевания или состояния, вызванного неправильной работой сигнального пути фосфатидилинозитол-3-киназа-киназа AKT-mTOR (PI3K-AKT-mTOR).
Уровень техники
Исследования последних лет показали, что сигнальный путь PI3K-AKT-mTOR играет ключевую роль в развитии, распространении, инвазии и метастазировании опухолевых клеток, и что блокирование сигнального пути PI3K-AKT-mTOR в клетках может препятствовать пролиферации опухолевых клеток и даже стимулирует апоптоз опухолевых клеток. В различных видах опухолей человека некоторое ключевые белки в сигнальном пути PI3K-AKT-mTOR могут быть чрезмерно активированы в результате мутации или амплификации кодирующих генов, например, мутации или амплификации расположенной выше по каскаду (upstream) рецепторной тирозинкиназы, мутации или амплификации гена PIK3CA, кодирующего каталитическую субъединицу p110α в различных опухолевых клетках, чрезмерной активации Akt и PDK1 и общей делеции в отрицательном регуляторе PTEN.
Мишень рапамицина у млекопитающих, mTOR, которая является одним из наиболее важных субстратов для Akt, представляет собой неклассическую серин/треониновую протеинкиназу семейства связанных с фосфатидидинозитол-3-киназой киназ (PIKK). Сигнальный путь mTOR может регулировать большое количество процессов жизнедеятельности посредством интегрирования сигналов, передаваемых из молекул питания, энергетического статуса и факторов роста и, таким образом, является ключевым путем для регулирования роста и пролиферации клеток. Аномальная активация сигнального пути mTOR является распространенным признаком возникновения и развития различных опухолей, таким образом, она становится важным пунктом в исследовании и разработке противоопухолевых ингибиторов.
Тем не менее, было обнаружено, что существует по меньшей мере два функциональных комплекса, mTORC1 и mTORC2, которые являются посредниками как связанной, так и независимой биологической передачи сигналов. Применяемое в клинической практике рапамициновое лекарственное средство, включая рапамицин и его аналоги, связывается с РКВР12-рапамицинсвязывающим доменом (FRB) около каталитической зоны mTORC1 посредством алостерии для оказания действия, заключающегося в частичном ингибировании белка mTOR. Эти соединения ни непосредственно не ингибируют mTORC2, ни полностью не блокируют все сигналы, посредником для которых является mTORC1. Несмотря на то, что рапамициновое лекарственное средство показало определенную клиническую эффективность в отношении ряда некоторых опухолей, режим действия такого вида лекарственного средства не позволяет обеспечить полный потенциал нацеленных на mTOR лекарственных средств против рака. В частности, в некоторых крупных солидных опухолях гиперфосфорилование (активация) АКТ, опосредованное mTORC2, является крайне необходимым для поддержания и развития опухолей, но рапамициновые лекарственные средства не могут ингибировать mTORC2.
Разработка АТФ-конкурентных и специфичных ингибиторов mTOR в виде малых молекул обеспечивает возможность для лечения различных форм рака. По сравнению с рапамициновыми лекарственными средствами, некоторые АТФ-конкурентные ингибиторы, как недавно сообщалось, демонстрируют лучшее ингибирующее действие на рост и выживаемость, синтез белка и биологический энергетический метаболизм опухолевых клеток. В исследованиях на животных, этот вид лекарственного средства показал сильную противоопухолевую активность при действии одним лекарственным средством на клетки рака молочной железы MDA361, глиомы U87MG, рака легких А549 и Н1975 и рака почки А498 и 786-0.
В итоге, ввиду того, что сигнальный путь mTOR принимает участие в ряде различных опухолей разработка более эффективного ингибитора mTOR обеспечивает новую идею и стратегию для нового противоопухолевого лекарственного средства широкого спектра действия. Недавно несколько ингибиторов mTOR перешли на стадию клинических исследований, которая показала, что АТФ-конкурентный ингибитор mTOR может представлять собой новое поколение лекарственных средств против рака, которые будут применяться в клинической практике.
Авторы настоящего изобретения подтвердили, что ингибитор mTOR является АТФ-конкурентным ингибитором, таким образом, этот механизм действия отличается от механизма действия рапамициновых лекарственных средств. Кроме того, авторы настоящего изобретения получили класс новых пиридопиримидиновых или пиримидопиримидиновых соединений посредством рационального планирования и обширного рассмотрения факторов, таких как растворимость в воде, метаболическая стабильность и т.п. соединений, на основе данных о предварительно описанных соединениях. Такие соединения демонстрируют хорошую ингибирующую активность mTOR на ферментативном и клеточном уровнях. После дальнейшей оптимизации и скрининга эти соединения, как предполагается, должны стать легко получаемыми лекарственными средствами против рака с высокой активностью.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
Одной из задач настоящего изобретения является предложение пиридопиримидинового или пиримидопиримидинового соединения, представленного формулой (I), изомера, фармацевтически приемлемой соли, сложного эфира, пролекарства или сольвата указанных соединений.
Другой задачей настоящего изобретения является предложение способа получения соединения.
Еще одной задачей настоящего изобретения является предложение применения пиридопиримидинового или пиримидопиримидинового соединения, представленного формулой (I), изомера, фармацевтически приемлемых солей, сложного эфира, пролекарства или сольвата указанных соединений в качестве ингибитора mTOR и применение в лечении заболевания или состояния, вызванного дисфункцией сигнального пути PI3K-AKT-mTOR, в частности, опухолевого заболевания.
Другой задачей настоящего изобретения является предложение фармацевтической композиции, содержащей один или более компонентов, выбранных из группы, состоящей из пиридопиримидинового или пиримидопиримидинового соединения, представленного формулой (I), изомера, фармацевтически приемлемой соли, сложного эфира, пролекарств или сольвата указанных соединений.
Другой задачей настоящего изобретения является предложение способа лечения заболевания или состояния, в частности опухолевого заболевания, вызванного дисфункцией сигнального пути PI3K-AKT-mTOR.
ТЕХНИЧЕСКОЕ РЕШЕНИЕ
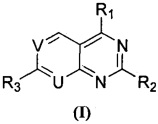
Согласно одному аспекту настоящего изобретения предложены пиридопиримидиновое или пиримидопиримидиновое соединение, представленное формулой (I), изомер, фармацевтически приемлемая соль, сложный эфир, пролекарство или сольват указанных соединений:
 ,
,
где
один из U и V представляет собой N, а другой представляет собой СН, или оба U и V представляют собой N;
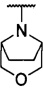
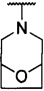
каждый из R1 и R2 независимо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, при этом каждый из RA и RB независимо представляет собой Н, С1-С6 алкил, незамещенный или замещенный С1-С6 алкокси или галогеном, или С1-С6 алкокси, незамещенный или замещенный галогеном, или RA и RB совместно с N, к которому они присоединены, образуют азотсодержащий насыщенный гетероцикл, содержащий от 4 до 8 атомов в кольце, который является незамещенным или замещен С1-С6 алкилом, С1-С6 алкокси или галогеном, причем указанный азотсодержащий насыщенный гетероцикл включает пиперидиновое кольцо, морфолиновое кольцо, пиперазиновое кольцо, N-метилпиперазиновое кольцо, высшее гомоморфолиновое кольцо, гомопиперазиновое кольцо и т.п.,
предпочтительно каждый из R1 и R2 независимо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, при этом каждый из RA и RB независимо представляет собой Н, С1-С3 алкил, незамещенный или замещенный С1-С3 алкокси или галогеном, или С1-С3 алкокси, незамещенный или замещенный галогеном, или RA и RB совместно с N, к которому они присоединены, образуют азотсодержащий насыщенный гетероцикл, содержащий от 6 до 7 атомов в кольце, который является незамещенным или замещен С1-С3 алкилом, С1-С3 алкокси или галогеном, причем указанный азотсодержащий насыщенный гетероцикл предпочтительно представляет собой морфолиновое кольцо,
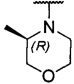
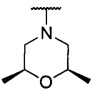
более предпочтительно каждый из R1 и R2 независимо представляет собой 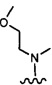 ,
,  ,
,  ,
, 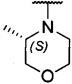 ,
,  ,
, или
или 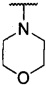 ; и
; и
R3 представляет собой фенил или пиридил, незамещенный или замещенный от 1 до 5 заместителями, при этом заместителем может быть галоген; гидроксил; циано; С1-С7 алкил, незамещенный или замещенный С1-С7 алкокси, галогеном или гидрокси; С1-С7 алкокси; -NHS(=O)2C1-C7 алкил; амино, незамещенный или замещенный С5-С6 арилом, С1-С7 алкилом или ди(С1-С7 алкилом); -С(O)NH2 или -C(O)NHC1-C3 алкилом,
предпочтительно R3 представляет собой фенил, незамещенный или замещенный от 1 до 3 заместителями, при этом заместителем может быть галоген; гидроксил; циано; С1-С4 алкил, незамещенный или замещенный С1-С4 алкокси, галогеном или гидрокси; С1-С4 алкокси; -NHS(=O)2C1-C4 алкил; амино, незамещенный или замещенный С5-С6 арилом, С1-С4 алкилом или ди(С1-С4 алкилом); -C(O)NH2 или -C(O)NHC1-C3 алкилом,
более предпочтительно R3 представляет собой 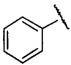 ,
, 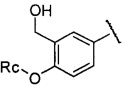 или
или 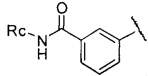 , где Rc представляет собой Н или С1-С3 алкил, предпочтительно Rc представляет собой H или метил.
, где Rc представляет собой Н или С1-С3 алкил, предпочтительно Rc представляет собой H или метил.
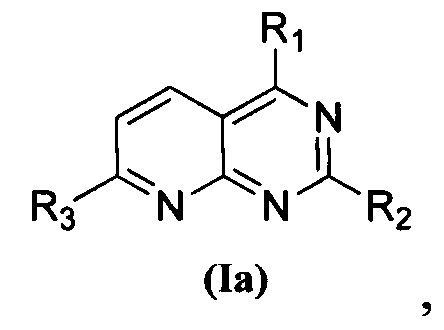
В приведенной выше формуле (I), когда U представляет собой N и V представляет собой CH, соединение, представленное формулой (I), предпочтительно представляет собой соединение, представленное формулой (Iа):
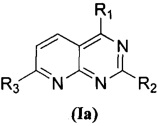 ,
,
где
каждый из R1 и R2 независимо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, при этом каждый из RA и RB независимо представляет собой H, С1-С6 алкил, незамещенный или замещенный С1-С6 алкокси или галогеном, или С1-С6 алкокси, незамещенный или замещенный галогеном, или RA and RB совместно с N, к которому они присоединены, образуют азотсодержащий насыщенный гетероцикл, содержащий от 4 до 8 атомов в кольце, который является незамещенным или замещен С1-С6 алкилом, С1-С6 алкокси или галогеном, причем указанный азотсодержащий насыщенный гетероцикл включает пиперидиновое кольцо, морфолиновое кольцо, пиперазиновое кольцо, N-метилпиперазиновое кольцо, высшее гомоморфолиновое кольцо, гомопиперазиновое кольцо и т.п.,
предпочтительно каждый из R1 и R2 независимо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, при этом каждый из RA и RB независимо представляет собой Н, С1-С3 алкил, незамещенный или замещенный С1-С3 алкокси или галогеном, или С1-С3 алкокси, незамещенный или замещенный галогеном, или RA и RB совместно с N, к которому они присоединены, образуют азотсодержащий насыщенный гетероцикл, содержащий от 6 до 7 атомов в кольце, который является незамещенным или замещен С1-С3 алкилом, С1-С3 алкокси или галогеном, причем указанный азотсодержащий насыщенный гетероцикл предпочтительно представляет собой морфолиновое кольцо,
более предпочтительно каждый из R1 и R2 независимо представляет собой , , , , , или ; и
R3 представляет собой фенил или пиридил, незамещенный или замещенный 1-5 заместителями, при этом заместителем может быть галоген; гидроксил; циано; С1-С7 алкил, незамещенный или замещенный С1-С7 алкокси, галогеном или гидрокси; С1-С7 алкокси; -NHS(=O)2C1-C7 алкил; амино, незамещенный или замещенный С5-С6 арилом, С1-С7 алкилом или ди(С1-С7 алкилом); -C(O)NH2 или -C(O)NH-C1-C3 алкилом,
предпочтительно R3 представляет собой фенил, незамещенный или замещенный 1-3 заместителями, при этом заместителем может быть галоген; гидроксил; циано; С1-С4 алкил, незамещенный или замещенный С1-С4 алкокси, галогеном или гидрокси; С1-С4 алкокси; -NHS(=O)2C1-C4 алкил; амино, незамещенный или замещенный С5-С6 арилом, С1-С4 алкилом или ди(С1-С4 алкилом); -C(O)NH2 или -C(O)NHC1-C3 алкилом,
более предпочтительно R3 представляет собой 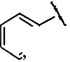 ,
, 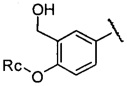 или
или 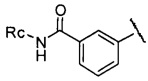 , где Rc представляет собой Н или С1-С3 алкил, предпочтительно Rc представляет собой Н или метил.
, где Rc представляет собой Н или С1-С3 алкил, предпочтительно Rc представляет собой Н или метил.
В приведенной выше формуле (I), когда U представляет собой СН, V представляет собой N, соединение, представленное формулой (I), предпочтительно представляет собой соединение, представленное формулой (Ib):
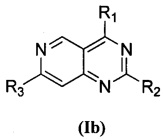 ,
,
где
каждый из R1 и R2 независисмо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, при этом каждый из RA и RB независимо представляет собой Н, С1-С6 алкил, незамещенный или замещенный С1-С6 алкокси или галогеном, или С1-С6 алкокси, незамещенный или замещенный галогеном, или RA и RB совместно с N, к которому они присоединены, образуют азотсодержащий насыщенный гетероцикл, содержащий от 4 до 8 атомов в кольце, который является незамещенным или замещен С1-С6 алкилом, С1-С6 алкокси или галогеном, причем указанный азотсодержащий насыщенный гетероцикл включает пиперидиновое кольцо, морфолиновое кольцо, пиперазиновое кольцо, N-метилпиперазиновое кольцо, высшее гомоморфолиновое кольцо, гомопиперазиновое кольцо и т.п.,
предпочтительно каждый из R1 и R2 независимо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, при этом каждый из RA и RB независимо представляет собой Н, С1-С3 алкил, незамещенный или замещенный С1-С3 алкокси или галогеном, или С1-С3 алкокси, незамещенный или замещенный галогеном, или RA и RB совместно с N, к которому они присоединены, образуют азотсодержащий насыщенный гетероцикл, содержащий от 6 до 7 атомов в кольце, который является незамещенным или замещен С1-С3 алкилом, С1-С3 алкокси или галогеном, причем указанный азотсодержащий насыщенный гетероцикл предпочтительно представляет собой морфолиновое кольцо,
более предпочтительно каждый из R1 и R2 независимо представляет собой , , , , , или ; и
R3 представляет собой фенил или пиридил, незамещенный или замещенный 1-5 заместителями, при этом заместителем может быть галоген; гидроксил; циано; С1-С7 алкил, незамещенный или замещенный С1-С7 алкокси, галогеном или гидрокси; С1-С7 алкокси; -NHS(=O)2C1-C7 алкил; амино, незамещенный или замещенный С5-С6 арилом, С1-С7 алкилом или ди(С1-С7 алкилом); -C(O)NH2 или -C(O)NH-C1-C3 алкилом,
предпочтительно R3 представляет собой фенил, незамещенный или замещенный 1-3 заместителями, при этом заместителем может быть галоген; гидроксил; циано; С1-С4 алкил, незамещенный или замещенный С1-С4 алкокси, галогеном или гидрокси; С1-С4 алкокси; -NHS(=O)2C1-C4 алкил; амино, незамещенный или замещенный С5-С6 арилом, С1-С4 алкилом или ди(С1-С4 алкилом); -C(O)NH2 или -C(O)NHC1-C3 алкилом,
более предпочтительно R3 представляет собой , или , где Rc представляет собой Н или С1-С3 алкил, предпочтительно Rc представляет собой Н или метил.
В приведенной выше формуле (I), когда и U и V представляют собой N, соединение, представленное формулой (I), предпочтительно представляет собой соединение, представленное формулой (Iс):
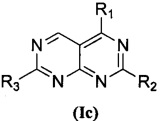 ,
,
где
каждый из R1 и R2 независимо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, при этом каждый из RA и RB независимо представляет собой Н, С1-С6 алкил, незамещенный или замещенный С1-С6 алкокси или галогеном, или С1-С6 алкокси, незамещенный или замещенный галогеном, или RA и RB совместно с N, к которому они присоединены, образуют азотсодержащий насыщенный гетероцикл, содержащий от 4 до 8 атомов в кольце, который является незамещенным или замещен С1-С6 алкилом, С1-С6 алкокси или галогеном, причем указанный азотсодержащий насыщенный гетероцикл включает пиперидиновое кольцо, морфолиновое кольцо, пиперазиновое кольцо, N-метилпиперазиновое кольцо, высшее гомоморфолиновое кольцо, гомопиперазиновое кольцо и т.п.,
предпочтительно каждый из R1 и R2 независимо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, при этом каждый из RA и RB независимо представляет собой Н, С1-С3 алкил, незамещенный или замещенный С1-С3 алкокси или галогеном, или С1-С3 алкокси, незамещенный или замещенный галогеном, или RA и RB совместно с N, к которому они присоединены, образуют азотсодержащий насыщенный гетероцикл, содержащий от 6 до 7 атомов в кольце, который является незамещенным или замещен С1-С3 алкилом, С1-С3 алкокси или галогеном, причем указанный азотсодержащий насыщенный гетероцикл предпочтительно представляет собой морфолиновое кольцо,
более предпочтительно каждый из R1 и R2 независимо представляет собой , , , , , или ; и
R3 представляет собой фенил или пиридил, незамещенный или замещенный 1-5 заместителями, при этом заместителем может быть галоген; гидроксил; циано; С1-С7 алкил, незамещенный или замещенный С1-С7 алкокси, галогеном или гидрокси; С1-С7 алкокси; -NHS(=O)2C1-C7 алкил; амино, незамещенный или замещенный С5-С6 арилом, С1-С7 алкилом или ди(С1-С7 алкилом); -C(O)NH2 или -C(O)NH-C1-C3 алкилом,
предпочтительно R3 представляет собой фенил, незамещенный или замещенный 1-3 заместителями, при этом заместителем может быть галоген; гидроксил; циано; С1-С4 алкил, незамещенный или замещенный С1-С4 алкокси, галогеном или гидрокси; С1-С4 алкокси; -NHS(=O)2C1-C4 алкил; амино, незамещенный или замещенный С5-С6 арилом, С1-С4 алкилом иди ди(С1-С4 алкилом); -C(O)NH2 или -C(O)NHC1-С3 алкилом,
более предпочтительно R3 представляет собой , или , где Rc представляет собой Н или С1-С3 алкил, предпочтительно Rc представляет собой Н или метил.
В настоящем изобретении, особенно предпочтительным конкретным соединением является одно из следующих соединений:
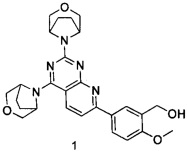
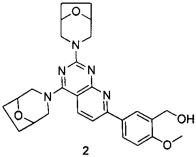
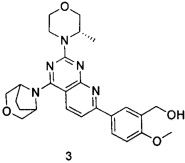
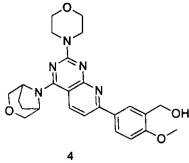
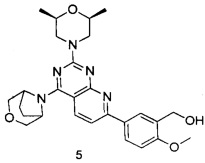
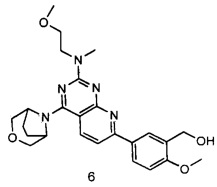
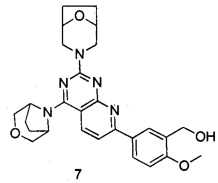
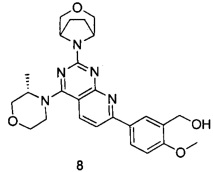
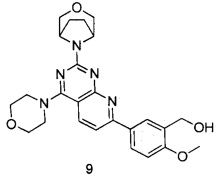
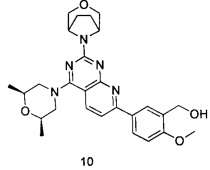
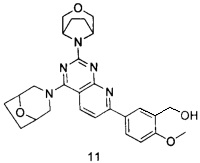
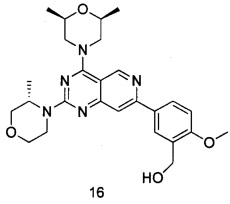
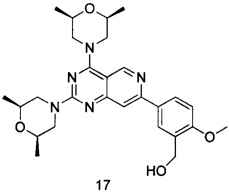
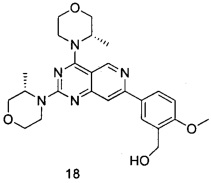
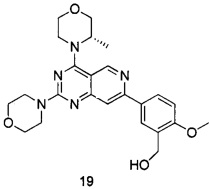
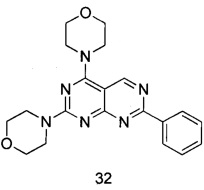
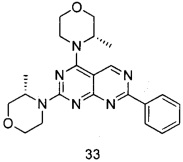
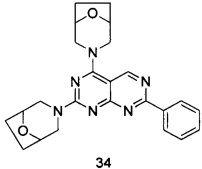
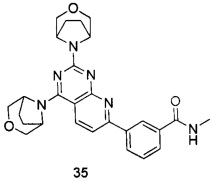
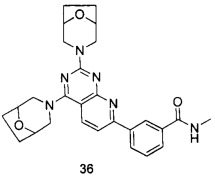
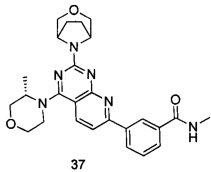
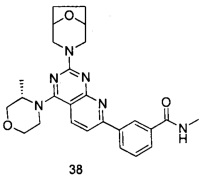
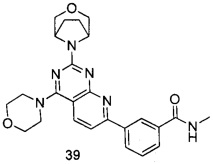
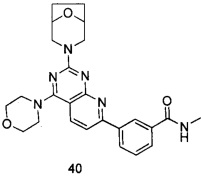
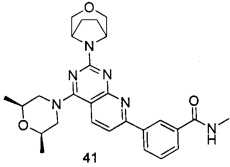
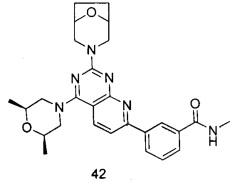
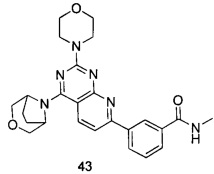
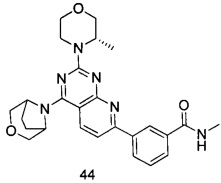
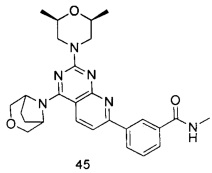
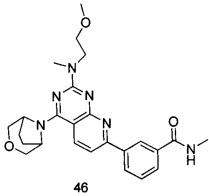
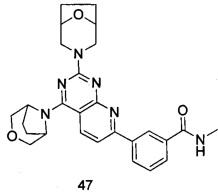
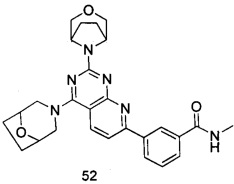 .
.
Фармацевтически приемлемая соль пиридопиримидиновых или пиримидопиримидиновых соединений, представленных формулой (I) в настоящем изобретении, может быть получена путем растворения пиридопиримидинового или пиримидопиримидинового соединения, представленного формулой (I), в спиртовом растворе, насыщенном кислотой, соответствующей соли, необходимой для проведения реакции, например, пиридопиримидиновое или пиримидопиримидиновое соединение, предложенные в настоящем изобретении, могут быть растворены в метанольном растворе, насыщенном HCl, перемешаны при комнатной температуре в течение 30 минут, и затем проводят выпаривание растворителя досуха с получением соответствующего гидрохлорида.
Если не указано иное, в следующих схемах реакций все символы в соединениях имеют то же значение, что и в формуле (I). Соединения в схемах реакций могут включать соли указанных соединений, например, соли соединений, имеющих структуру формулы (I), и т.п.
Для иллюстративных целей, реакционные схемы, представленные ниже, обеспечивают возможные пути синтеза соединений настоящего изобретения, а также ключевых промежуточных соединений. Более подробное описание отдельных реакционных стадий можно найти в следующих примерах. Специалистам в данной области техники будет понятно, что другие синтетические пути могут быть использованы в синтезе соединений настоящего изобретения. Хотя реакционные схемы, показанные и описанные дальше, включают в себя определенные исходные вещества и реагенты, они могут быть легко заменены другими исходными веществами и реагентами, чтобы обеспечить многообразие производных и/или условия реакции. Кроме того, с учетом того, что раскрыто в настоящем изобретении, соединения, полученные посредством данного способа могут быть дополнительно модифицированы путем использования обычных химических методов, хорошо известных специалистам в данной области техники.
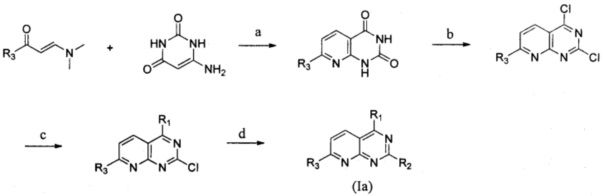
Замещенное пиридо[2,3-d]пиримидиновое соединение, представленное формулой (Ia), или изомер, фармацевтически приемлемая соль, сложный эфир, пролекарство или сольват указанного соединения могут быть получены следующим способом, и в отношении конкретных реагентов и условий реакции ссылка может быть сделана на пример 1.
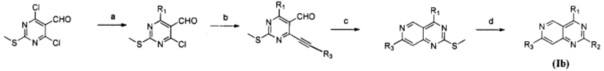
Замещенное пиридо[4,3-d]пиримидиновое соединение, представленное формулой (Ib), изомер, фармацевтически приемлемая соль, сложный эфир, пролекарство или сольват указанного соединения может быть получено следующим способом, и в отношении конкретных реагентов и условий реакции ссылка может быть сделана на пример 13.
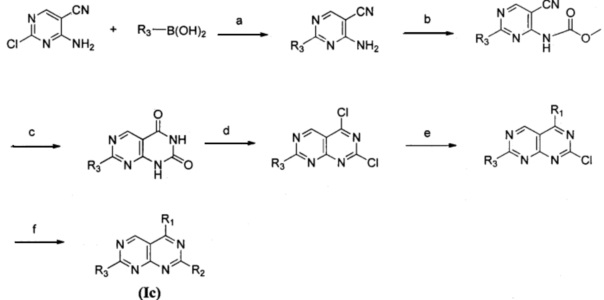
Замещенное пиримидо[4,5-d]пиримидиновое соединение, представленное формулой (Ic), изомер указанного соединения, фармацевтически приемлемая соль, сложный эфир, пролекарство или сольват указанного соединения могут быть получены следующим способом, и в отношении конкретных реагентов и условий реакции ссылка может быть сделана на пример 32.
Согласно другому аспекту настоящего изобретения, предложено применение пиридопиримидинового или пиримидопиримидинового соединения, представленного формулой (I), изомера, фармацевтически приемлемой соли, сложного эфира, пролекарств или сольвата указанных соединений в качестве ингибитора mTOR и применение для лечения заболевания или состояния, вызванного дисфункцией сигнального пути PI3K-AKT-mTOR, в частности опухолевого заболевания. В частности, опухолевое заболевание включает, но не ограничивается ими, меланому, рак печени, рак почки, острый лейкоз, немелкоклеточный рак легкого, рак предстательной железы, рак щитовидной железы, рак кожи, рак толстой кишки, рак прямой кишки, рак поджелудочной железы, рак яичников, рак молочной железы, миелодиспластический синдром, рак пищевода, рак желудочно-кишечного тракта и мезотелиому.
Согласно другому аспекту настоящего изобретения, также предложена фармацевтическая композиция, содержащая один или более компонентов, выбранных из группы, состоящей из пиридопиримидинового или пиримидопиримидинового соединения, представленного формулой (I), изомера, фармацевтически приемлемой соли, сложного эфира, пролекарства или сольвата указанных соединений, которая может быть применена в качестве ингибитора mTOR, и указанная фармацевтическая композиция необязательно может содержать фармацевтически приемлемый носитель или вспомогательное вещество.
Фармацевтически приемлемый носитель относится к обычным для фармацевтической области фармацевтическим носителям, например, растворителю, такому как вода и т.п.; наполнителю, такому как крахмал, сахароза и т.п.; связующему веществу, такому как производное целлюлозы, альгинат, желатин, поливинилпирролидон; смачивающему агенту, такому как глицерин; разрыхлителю, такому как агар, карбонат кальция и бикарбонат натрия; абсорбенту, такому как соединения четвертичного аммония; поверхностно-активному веществу, такому как цетиловый спирт; адсорбционному носителю, такому как каолин и бентонит; смазывающему веществу, такому как тальк, стеарат кальция и стеарат магния, и полиэтиленгликоль и т.п. Кроме того, в фармацевтическую композицию также могут быть добавлены другие адьюванты, такие как ароматизирующий агент, подсластитель и т.д.
Согласно другому аспекту настоящего изобретения также предложен способ лечения заболевания или состояния, вызванного дисфункцией сигнального пути РВК-AKT-mTOR, в частности опухолевого заболевания, при этом указанный способ включает введение пациенту терапевтически эффективного количества одного или более компонентов, выбранных из группы, состоящей из пиридопиримидинового или пиримидопиримидинового соединения, представленного формулой (I), изомера, фармацевтически приемлемой соли, сложного эфира, пролекарства или сольвата указанных соединений, или вышеупомянутой фармацевтической композиции согласно настоящему изобретению.
Соединение или фармацевтическая композиция, предложенные в настоящем изобретении, могут быть введены перорально, ректально или парентерально пациенту, нуждающемуся в таком лечении. Для перорального введения они могут быть приготовлены в виде обычного твердого препарата, такого как таблетка, порошок, гранула, капсула и т.д. или жидкого препарата, такого как водная или масляная суспензия, или другого жидкого препарата, такой как сироп и т.п.; а для парентерального введения они могут быть превращены в инъекционный раствор, водную или масляную суспензию и т.п.
ЭФФЕКТ, ОБЕСПЕЧИВАЮЩИЙ ПРЕИМУЩЕСТВО
Соединения согласно настоящему изобретению демонстрируют хорошую ингибирующую активность в отношении mTOR и также демонстрируют сильный ингибирующий эффект в отношении пролиферации клеток глиомы человека U87MG и клеток рака предстательной железы человека LNCap, при этом соединения имеющие наилучшие активности, такие как соединение 1, соединение 3 и соединение 8, обладают сопоставимой активностью с аналогичным соединением в клинических исследованиях в предшествующем уровне техники, таким как AZD8055.
ПОДРОБНОЕ ОПИСАНИЕ ИЛЛЮСТРАТИВНОГО ВАРИАНТА РЕАЛИЗАЦИИ
Понятно, что специалист в данной области с помощью вышеприведенного описания и без дополнительной информации может найти лучшее применение настоящему изобретению. Таким образом, следующие примеры предназначены исключительно для дополнительной иллюстрации настоящего изобретения и не предназначены для ограничения объема настоящего изобретения каким-либо образом.
Исходные материалы являются коммерчески доступными продуктами, или они могут быть получены способом, известным в данной области техники, или получены в соответствии со способом, описанным в данном документе.
Структуры соединений были идентифицированы посредством спектров ядерно магнитного резонанса (1Н-ЯМР) и/или масс-спектрометрии (МС). Измерения посредством ЯМР проводили на приборе ЯМР типа Varian АМХ-400 с использованием в качестве растворителя для проведения измерений дейтерированного хлороформа (CDCl3) или дейтерированного диметилсульфоксида (DMSO-D6), в качестве внутреннего стандарта использовали тетраметилсилан (ТМС). Измерения посредством МС проводили на жидкостном хроматографе с масс-спектрометром типа Thermo Finnigan LCQ-Deca ХР (ИЭР). Для очистки продуктов путем колоночной хроматографии исрользовали прибор для флэш-хроматографии ISCO CombiFlash® Rf 75, в качестве носителя использовали силикагель 200-300 меш, полученный от компании Qingdao Haiyang Chemical Со Ltd. Инициатор Biotage для получения микроволнового излучения использовали для микроволнового нагрева.
ПРИМЕРЫ
ПРИМЕР 1: получение 5-(2,4-ди(3-окса-8-азабицикло[3.2.1]октан-8-ил)-пиридо[2,3-d]пиримидин-7-ил)-2-метоксифенилметанола (соединение 1)
Реакции проводили согласно следующей схеме:
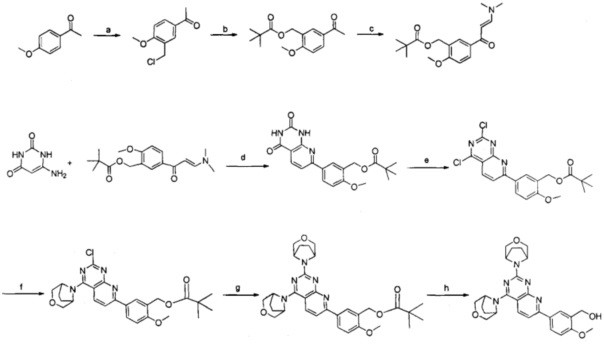
Реагенты и условия: a) параформальдегид, концентрированная соляная кислота, 60°С; b) пивалиновая кислота, карбонат калия, диметилформамид, 65°С; с) m-бутоксибис(диметиламино)метан, 54°С; d) 6-аминоурацил, ледяная уксусная кислота, вода, диметилсульфоксид, 99°С; е) оксихлорид фосфора, N,N-диизопропилэтиламин, анизол, 80°С; f) 3-окса-8-азабицикло[3.2.1]октана гидрохлорид, N,N-диизопропилэтиламин, тетрагидрофуран, комнатная температура; g) 3-окса-8-азабицикло[3.2.1]октана гидрохлорид, N,N-диизопропилэтиламин, изопропанол, микроволновое излучение, 160°С, 80 минут; h) гидроксид калия, тетрагидрофуран, метанол, комнатная температура.
a) 1-(3-(хлорметил)-4-метоксифенил)этанон
К n-метоксиацетофенону (1,0 г, 6,65 ммоль) добавляли параформальдегид (362 мг, 11,97 ммоль) и концентрированную соляную кислоту (10,5 мл), и проводили перемешивание в течение ночи при 60°. Далее полученную реакционную смесь охлаждали до комнатной температуры, наливали на раздробленный лед, экстрагировали этилацетатом, промывали последовательно водой, насыщенным водным раствором бикарбоната натрия, солевым раствором, сушили над безводным сульфатом натрия, и отгоняли органический растворитель при пониженном давлении с получением 1,2 г указанного в заголовке соединения в виде твердого вещества серого цвета с выходом 92%.
1Н ЯМР (400 МГц, CDCl3): δ 8,03-7,91 (m, 2Н), 6,94 (dd, J=8,6, 2,0 Гц, 1H), 4,67 (s, 2Н), 3,95 (s, 3Н), 2,57 (s, 3Н).
b) 5-ацетил-2-метоксибензилпивалат
Пивалиновую кислоту (868,4 мг, 8,51 ммоль), карбонат калия (1,2 г, 8,70 ммоль) растворяли в диметилформамиде и 1-(3-(хлорметил)-4-метоксифенил)этанон (1,2 г, 6,06 ммоль), полученный как описано выше, растворенный в 3 мл диметилформамида добавляли туда же в атмосфере аргона. Смесь нагревали до 65°С и проводили реакцию 4 часа, добавляли воду, экстрагировали этилацетатом, промывали водой, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле с элюированием смесью петролейный эфир/этилацетат (об./об.=8:1) с получением 1,4 г указанного в заголовке соединения в виде масла желтого цвета с выходом 87,5%.
МС(ЭИ): 264.
1Н ЯМР (400 МГц, CDCl3) δ 8,01-7,88 (m, 2Н), 6,92 (d, J=9,1 Гц, 1Н), 5,17 (s, 2Н), 3,91 (s, 3Н), 2,56 (s, 3Н), 1,27 (s, 9Н).
c) (E)-5-(3-(диметиламино)акрил)-2-метоксибензилпивалат
К 5-ацетил-2-метоксибензилпивалату (750 мг, 3,0 ммоль) полученному как описано выше, добавляли трет-бутокси-бис(диметиламино)метан (2 г, 12 ммоль). Смесь подвергали реакции при 54°С 6 часов, добавляли воду, экстрагировали этилацетатом, промывали водой, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 900 мг указанного в заголовке соединения в виде масла желтого цвета с выходом 94%.
МС(ИЭР): 320(М+1), соединение использовали на следующей стадии без дополнительной очистки.
d) 5-(2,4-диoкco-1,2,3,4-тeтpaгидpoпиpидo[2,3-d]пиpимидин-7-ил)-2-мeтoкcибeнзил - пивалат
6-Аминоурацил (726 мг, 5,71 ммоль) добавляли к ледяной уксусной кислоте (7,1 мл) и воду (1,8 мл), наревали до 99°С. (Е)-5-(3-(диметиламино)акрил)-2-метоксибензил- пивалат, полученный как описано выше, растворяли в 2,7 мл диметилсульфоксида и полученный раствор добавляли по каплям в течение 80 минут к вышеуказанной смеси и проводили реакцию при 99°С 3 часа. Реакционную смесь охлаждали до 0°С и добавляли 7 г гидроксида калия растворенного в 14 мл воды на ледяной бане (pH=7), с последующим добавлением водного раствора карбонат калия для доведения смеси до рН=9-10. Смесь перемешивали при комнатной температуре в течение ночи. Выпадал бежевый осадок, его фильтровали, и полученное твердое вещество промывали водным раствором карбоната калия. Затем полученное твердое вещество растворяли в растворе лимонной кислоты при перемешивании в течение 2 часов (рН=4), фильтровали, промывали водой до нейтральной реакции с получением 864 мг указанного в заголовке соединения в виде твердого вещества желтого цвета с выходом 40%.
ИЭР: 384(М+1). 1Н ЯМР (400 МГц, DMSO) δ 11,67 (s, 1Н), 11,42 (s, 1Н), 8,27 (d, J=8,2 Гц, 1H), 8,17 (dd, J=8,7, 2,3 Гц, 1Н), 8,11 (d, J=2,3 Гц, 1H), 7,75 (d, J=8,3 Гц, 1Н), 7,22 (d, J=8,8 Гц, 1Н), 5,13 (s, 2Н), 3,89 (s, 3Н), 1,18 (s, 9Н).
d) 5-(2,4-дихлорпиридо[2,3-d]пиримидин-7-ил)-2-метоксибензилпивалат
5-(2,4-Диoкco-1,2,3,4-тeтpaгидpoпиpидo[2,3-d]пиpимидин-7-ил)-2-мeтoкcибeнзил - пивалат (800 мг, 2,09 ммоль), полученный как описано выше, растворяли в анизоле и добавляли N,N-диизопропилэтиламина (675 мг, 5,22 ммоль), и затем оксихлорид фосфора (963 мг, 6,26 ммоль). После перемешивания при комнатной температуре в течение 1,5 часов, реакционную смесь нагревали до 80°С, и затем проводили реакцию в течение 4,5 часов при 80°С. Затем, оксихлорид фосфора и часть растворителя удаляли при пониженном давлении, и добавляли 4 мл 2М калия гидрокарбоната и этилацетат. Смесь отстаивали в течение ночи и отфильтровывали, и твердое вещество промывали этилацетатом с получением 700 мг указанного в заголовке соединения в виде твердого вещества желтого цвета.
МС(ИЭР): 420(М+1), соединение использовали на следующей стадии без дополнительной очистки.
f) 5-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-2-хлорпиридо[2,3-d]пиримидин-7-ил)-2-метоксибензилпивалат
3-Окса-8-азабицикло[3.2.1]октана гидрохлорид (490 мг, 3,27 ммоль) растворяли в 30 мл тетрагидрофурана, добавляли N,N-диизопропилэтиламин (425 мг, 3,29 ммоль), и проводили реакцию при комнатной температуре в течение 2 часов. Полученную смесь затем добавляли к 5-(2,4-дихлорпиридо[2,3-d]пиримидин-7-ил)-2-метоксибензил- пивалату (580 мг, 1,62 ммоль), полученному как описано выше, который растворяли в 30 мл тетрагидрофурана, добавляли N,N-диизопропилэтиламин (425 мг, 3,29 ммоль), и проводили реакцию при комнатной температуре в течение ночи. Смесь отделяли от растворителя при пониженном давлении, добавляли воду, экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле с элюированием смесью петролейный эфир/этилацетат (об./об.=1:1) с получением 560 мг указанного в заголовке соединения в виде твердого вещества желтого цвета с выходом 82,0%.
МС(ИЭР): 497(М+1).
1H ЯМР (400 МГц, CDCl3) δ 8,36 (dd, J=8,7, 2,4 Гц, 1H), 8,19 (dd, J=17,3, 5,5 Гц, 2Н), 7,78 (d, J=8,7 Гц, 1H), 7,01 (s, 1H), 5,21 (s, 2H), 4,84 (s, 2H), 4,00 (d, J=10,9 Гц, 2H), 3,92 (s, 3H), 3,81 (d, J=10,9 Гц, 2H), 2,21-2,09 (m, 2H), 2,09-1,98 (m, 2H), 1,30-1,15 (m, 9H).
g) 5-(2,4-ди(3-oкca-8-aзaбициклo[3.2.1]oктaн-8-ил)пиpидo[2,3-d]пиpимидин-7-ил)-2-метоксибензилпивалат
5-(4-(3-Oкca-8-aзaбициклo[3.2.1]oктaн-8-ил)-2-xлopпиpидo[2,3-d]пиpимидин-7-ил)-2-метоксибензилпивалат (78 мг, 0,157 ммоль), полученный как описано выше, растворяли в 2 мл изопропанола, добавляли N,N-диизопропилэтиламин (42,9 мг, 0,32 ммоль), и затем, h 3-окса-8-азабицикло[3.2.1]октана гидрохлорид (36 мг, 0,238 ммоль). Смесь подвергали реакции в микроволновом излучении при 160°С в течение 80 минут, выпаривали растворитель с получением 70 мг землистого твердого вещества желтого цвета. Продукт использовали на следующей стадии без дополнительной очистки.
h) 5-(2,4-ди(3-oкca-8-aзaбициклo[3.2.1]oктaн-8-ил)пиpидo[2,3-d]пиpимидин-7-ил)-2-метоксифецилметанол
К 70 мг 5-(2,4-ди(3-oкca-8-aзaбициклo[3.2.1]oктaн-8-ил)-пиpидo[2,3-d]пиpимидин-7-ил)-2-метоксибензилпивалата, полученного как описано выше, добавляли 5 мл метанола, 3 мл тетрагидрофурана и около 80 мг гидроксида калия, и проводили реакцию при комнатной температуре в течение ночи. Смесь отделяли от растворителя при пониженном давлении, добавляли воду, экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле с элюированием смесью петролейный эфир/этилацетат (об./об.=1:2) с получением 30 мг указанного в заголовке соединения в виде твердого вещества желтого цвета с выходом 50,0%.
МС(ИЭР): m/z 490[М+Н]+.
1Н ЯМР (400 МГц, CDCl3) 5 8,21-8,13 (m, 2Н), 8,06 (d, J=8,3 Гц, 1Н), 7,46 (d, J=8,3 Гц, 1Н), 6,99 (d, J=8,4 Гц, 1Н), 4,78 (s, 2Н), 4,58 (s, 2Н), 4,17 (s, 1H), 4,04-3,97 (m, 3Н), 3,94 (s, 3Н), 3,77-3,68 (m, 5Н), 3,49 (s, 1Н), 2,17-1,93 (m, 8Н).
ПРИМЕР 2: получение 5-(2,4-ди(8-окса-3-азабицикло[3.2.1]октан-3-ил)-пиридо[2,3-d]пиримидин-7-ил)-2-метоксифенилметанола (соединение 2)
Указанное в заголовке соединение получали таким же способом, как в примере 1, и стадии a, b, c, d, e и h были такими же, как в примере 1, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадиях f и g был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 38,0%.
МС(ИЭР): m/z 490[М+Н]+.
ПРИМЕР 3: получение 5-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-2-((S)-3-метилморфолин)пиридо[2,3-d]пиримидин-7-ил)-2-метоксифенилметанола (соединение 3)
Указанное в заголовке соединение получали таким же способом, как в примере 1, и стадии a, b, c, d, e, f и h были такими же, как в примере 1, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии g был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 64%.
МС(ИЭР): m/z 478[М+Н]+.
ПРИМЕР 4: получение 5-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-2-морфолинилпиридо[2,3-d]пиримидин-7-ил)-2-метоксифенилметанола (соединение 4)
Указанное в заголовке соединение получали таким же способом, как в примере 1, и стадии a, b, c, d, e, f и h были такими же, как в примере 1, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии g был заменен морфолином. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 47%.
МС(ИЭР): m/z 464[М+Н]+.
ПРИМЕР 5: получение 5-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-2-((2S,6R)-2,6-диметилморфолин)пиридо[2,3-d]пиримидин-7-ил)-2-метоксифенил-метанола (соединение 5)
Указанное в заголовке соединение получали таким же способом, как в примере 1, и стадии a, b, c, d, e, f и h были такими же, как в примере 1, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии g был заменен (2S,6R)-2,6-диметилморфолином. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 79%.
МС(ИЭР): m/z 492[М+Н]+.
ПРИМЕР 6: получение 5-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-2-((2-метоксиэтил)метиламино)пиридо[2,3-d]пиримидин-7-ил)-2-метоксифенил-метанола (соединение 6)
Указанное в заголовке соединение получали таким же способом, как в примере 1, и стадии a, b, c, d, e, f и h были такими же, как в примере 1, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии g был заменен 2-метокси-N-метилэтиламином. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 80%.
МС(ИЭР): m/z 466[М+Н]+.
ПРИМЕР 7: получение 5-(2-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-4-((3-окса-8-азабицикло[3.2.1]октан-8-ил)пиридо[2,3-d]пиримидин-7-ил)-2-метоксифенил-метанола (соединение 7)
Указанное в заголовке соединение получали таким же способом, как в примере 1, и стадии a, b, c, d, e, f и h были такими же, как в примере 1, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии g был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 60%. МС(ИЭР): m/z 490[М+Н]+.
ПРИМЕР 8: получение 5-(2-((1R,5S)-3-окса-8-азабицикло[3.2.1]октан-8-ил)-4-((S)-3-метилморфолин)пиридо[2,3-d]пиримидин-7-ил)-2-метоксифенилметанола (соединение 8)
Указанное в заголовке соединение получали таким же способом, как в примере 1, и стадии a, b, c, d, e, g и h были такими же, как в примере 1, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии f был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 82%.
МС(ИЭР): m/z 478[М+Н]+.
ПРИМЕР 9: получение 5-(2-((1R,5S)-3-окса-8-азабицикло[3.2.1]октан-8-ил)-4-морфолинилпиридо[2,3-d]пиримидин-7-ил)-2-метоксифенилметанола (соединение 9)
Указанное в заголовке соединение получали таким же способом, как в примере 1, и стадии a, b, c, d, e, g и h были такими же, как в примере 1, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии f был заменен морфолином. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 82%.
МС(ИЭР): m/z 464[М+Н]+.
ПРИМЕР 10: получение 5-(2-((1R,5S)-3-окса-8-азабицикло[3.2.1]октан-8-ил)-4-((2S,6R)-2,6-диметилморфолин)пиридо[2,3-d]пиримидин-7-ил)-2-метоксифенил-метанола (соединение 10)
Указанное в заголовке соединение получали таким же способом, как в примере 1, и стадии a, b, c, d, e, g и h были такими же, как в примере 1, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии f был заменен (2S,6R)-2,6-диметилморфолином. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 48%.
МС(ИЭР): m/z 492[М+Н]+.
ПРИМЕР 11: получение 5-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-2-((1R,5S)-3-окса-8-азабицикло[3.2.1]октан-8-ил)пиридо[2,3-d]пиримидин-7-ил)-2-метоксифенилметанола (соединение 11)
Указанное в заголовке соединение получали таким же способом, как в примере 1, и стадий a, b, c, d, e, g и h были такими же, как в примере 1, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии f был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 75%.
МС(ИЭР): m/z 490[М+Н]+.
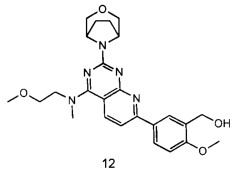
ПРИМЕР 12: получение 5-(2-((1R,5S)-3-окса-8-азабицикло[3.2.1]октан-8-ил)-4-((2-метоксиэтил)амино)пиридо[2,3-d]пиримидин-7-ил)-2-метоксифенилметанола (соединение 12)
Указанное в заголовке соединение получали таким же способом, как в примере 1, и стадии a, b, c, d, e, g и h были такими же, как в примере 1, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии f был заменен 2-метокси-N-метилэтиламином, t. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 75%.
МС(ИЭР): m/z 466[М+Н]+.
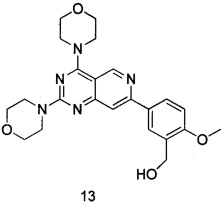
ПРИМЕР 13: получение (5-(2,4-диморфолинилпиридо[4,3-d]пиримидин-7-ил)-2-метоксифенил)метанола (соединение 13)
Реакции проводили согласно следующей схеме:
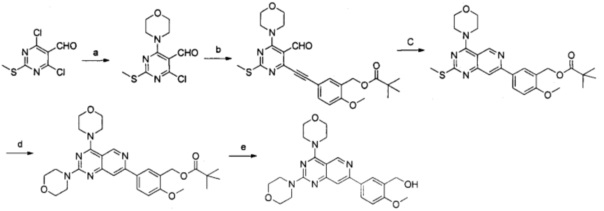
Реагенты и условия: а) морфолин, 0°С, 2 часа; b) 5-этинил-2-метоксибензилпивалат, бис(трифенилфосфин)палладия (II) дихлорид, иодид меди, триэтиламин, диметилформамид, 45°С; c) m-бутиламин, 120°С; d) i. м-хлорпербензойная кислота, дихлорметан, комнатная температура; ii. морфолин, диметилсульфоксид, 75°С; е) гидроксид калия, тетрагидрофуран, комнатная температура.
a) 2-метилтио-4-хлор-6-морфолинилпиримидин-5-карбоксальдегид
2-(метилтио)-4,6-дихлорпиримидин-5-карбоксальдегид (1,5 г, 6,76 ммоль) растворяли в 50 мл метанола, добавляли по каплям морфолин (590 г, 6,76 ммоль) растворенный в 2 мл метанола на ледяной бане, проводили реакцию при комнатной температуре в течение 2 часов и фильтровали. Твердое вещество промывали метанолом с получением 1,24 г указанного в заголовке соединения в виде твердого вещества желтого цвета с выходом 67%.
МС(ИЭР): m/z 274[М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 10,21 (s, 1H), 3,88-3,76 (m, 4Н), 3,72-3,54 (m, 4Н), 2,52 (s, 3Н).
b) 5-((5-формил-2-(метилтио)-6-морфолинилпиримидин-4-ил)ацетенил)-2-метоксибензилпивалат
К 2-метилтио-4-хлор-6-морфолинилпиримидин-5-карбоксальдегиду (602 мг, 2,21 ммоль), полученному как описано выше, добавляли 5 мл диметилформамида, 5-ацетенил-2-метоксибензилпивалат (814 мг, 3,31 ммоль), иодид меди (13 мг, 0,066 ммоль) и триэтиламин (668 мг, 6,61 ммоль). Смесь продували аргоном в течение несколько минут, добавляли бис(трифенилфосфин)палладия (II) дихлорид, проводили реакцию при 45°C в течение ночи, добавляли воду и экстрагировали этилацетатом три раза. Объединенную органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле с элюированием смесью петролейный эфир/этилацетат (об./об.=2:1) с получением 604 мг указанного в заголовке соединения в виде твердого вещества желтого цвета с выходом 57,0%.
1Н ЯМР (400 МГц, CDCl3) δ10,43 (s, 1Н), 7,57 (s, 1Н), 6,90 (s, 1Н), 6,88 (s, 1Н), 5,12 (s, 2Н), 3,88 (s, 3Н), 3,83-3,69 (m, 8Н), 2,54 (s, 3Н), 1,26 (s, 9Н).
с) 2-метокси-5-(2-(метилтио)-4-морфолинилпиридо[4,3-d]пиримидин-7-илбензил-пивалат
К 5-((5-формил-2-(метилтио)-6-морфолинилпиримидин-4-ил)ацетенил)-2-метокси-бензилпивалату (590 мг, 1,22 ммоль), полученному как описано выше, добавляли 40 мл трет-бутидамина и проводили предварительную реакцию в запаянной трубке в течение ночи при 120°C. Реакционную смесь концентрировали при пониженном давлении и остаток очищали посредством колоночной флэш-хроматографии на силикагеле с элюированием смесью петролейный эфир/этилацетат (об./об.=2:1) с получением 389 мг указанного в заголовке соединения в виде твердого вещества желтого цвета с выходом 66,0%.
МС (ИЭР): m/z, 483[М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 8,18-8,04 (m, 3Н), 7,82 (d, J=0,7 Гц, 1Н), 7,01 (d, J=8,6 Гц, 1Н), 5,21 (s, 2Н), 4,00-3,94 (m, 4Н), 3,91 (s, 3Н), 3,91-3,88 (m, 4Н), 2,62 (s, 3Н), 1,25 (s, 9H).
d) 5-(2,4-димopфoлинилпиpидo[4,3-d]пиpимидин-7-ил)-2-мeтoкcибeнзилпивaлaт
К 2-метокси-5-(2-(метилтио)-4-морфолинилпиридо[4,3-d]пиримидин-7-илбензил-пивалату (258 мг, 0,54 ммоль), полученному как описано выше, добавляли 3 мл дихлорметана и мета-хлорпербензойную кислоту (369 мг, 2,14 ммоль). Смесь подвергали реакции при комнатной температуре в течение 5 часов, концентрировали при пониженном давлении, чтобы удалить растворитель, добавляли соответствующее количество насыщенного водного раствора бикарбоната натрия, экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. К остатку без его дополнительной очистки добавляли 10 мл диметилсульфоксида для растворения, а затем 250 мг морфолина, полученную смесь подвергали реакции при 75°С в течение ночи, добавляли воду, экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 85 мг указанного в заголовке соединения в виде твердого вещества желтого цвета с общим выходом 57,0% в две стадии.
МС (ИЭР): m/z, 522[М+Н]+, соединение использовали в последующей стадии без дополнительной очистки.
e) (5-(2,4-димopфoлинилпиpидo[4,3-d]пиpимидин-7-ил)-2-мeтoкcифeнил)мeтaнoл
5-(2,4-димopфoлинилпиpидo[4,3-d]пиpимидин-7-ил)-2-мeтoкcибeнзилпивaлaт (28 мг, 0,048 ммоль), полученный как описано выше, растворяли в 3 мл тетрагидрофурана, добавляли около 30 мг гидроксида калия, подвергали реакции в течение 7 часов при комнатной температуре, удаляли растворитель при пониженном давлении и добавляли воду. Полученную смесь экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток разделяли на пластине с силикагелем, собирали продукт и элюировали смесью петролейный эфир/этилацетат (об./об.=1:3) с получением 5 мг указанного в заголовке соединения в виде твердого вещества желтого цвета с выходом 47,0%.
МС (ИЭР): m/z, 438[М+Н]+.
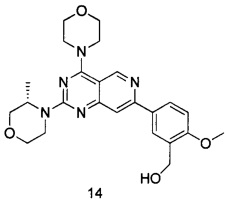
ПРИМЕР 14: получение (S)-(2-метокси-5-(2-(3-метилморфолинил-4-морфолинил)пиридо[4,3-d]пиримидин-7-ил)фенил)метанола (соединение 14)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии a, b, c и e были такими же, как в примере 13, за исключением того, что морфолин на стадии d был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 50%.
МС (ИЭР): m/z, 452[М+Н]+.
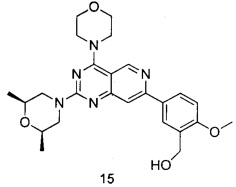
ПРИМЕР 15: получение 5-(2-((2S,6R)-2,6-диметилморфолинил-4-морфолинил)пиридо[4,3-d]пиримидин-7-ил)-2-метоксифенил)метанола (соединение 15)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии a, b, c и e были такими же, как в примере 13, за исключением того, что морфолин на стадии d был заменен (2S,6R)-2,6-диметилморфолином. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 52%.
МС (ИЭР): m/z, 466[М+Н]+.
ПРИМЕР 16: получение 5-(4-((2S,6R)-2,6-диметилморфолинил-2-((S)-3-метилморфолинил)пиридо[4,3-d]пиримидин-7-ил)-2-метоксифенил)метанола (соединение 16)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии b, c и e были такими же, как в примере 13, за исключением того, что морфолин на стадии а был заменен (2S,6R)-2,6-диметилморфолином, а морфолин на стадии d был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали в виде твердого вещества белого цвета с выходом 35%.
МС (ИЭР): m/z, 480[М+Н]+.
ПРИМЕР 17: получение (5-(2,4-ди((2S,6R)-2,6-диметил)пиридо[4,3-d]-пиримидин-7-ил)-2-метоксифенил)метанола (соединение 17)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии b, c и e были такими же, как в примере 13, за исключением того, что морфолин на стадиях a n d был заменен (2S,6R)-2,6-диметилморфолином. Указанное в заголовке соединение получали в виде твердого вещества белого цвета с выходом 55%.
МС (ИЭР): m/z, 494[М+Н]+.
ПРИМЕР 18: получение (5-(2,4-ди((S)-3-метилморфолинил)пиридо[4,3-d]-пиримидин-7-ил)-2-метоксифенил)метанола (соединение 18)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии b, c и e были такими же, как в примере 13, за исключением того, что морфолин на стадиях a и d был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали в виде твердого вещества белого цвета с выходом 56%.
МС (ИЭР): m/z, 466[М+Н]+.
ПРИМЕР 19: получение (S)-(2-метокси-5-(4-(3-метилморфолинил)-2-морфолинилпиридо[4,3-d]пиримидин-7-ил)фенил)метанола (соединения 19)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии b, c, d и e были такими же, как в примере 13, за исключением того, что морфолин на стадии a был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали в виде твердого вещества белого цвета с выходом 30%.
МС (ИЭР): m/z, 452[М+Н]+.
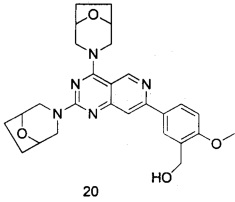
ПРИМЕР 20: получение (5-(2,4-ди(8-окса-3-азабицикло [3.2.1]октан-3-ил)-пиридо[4,3-d]пиримидин-7-ил)-2-метоксифенил)метанола (соединение 20)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии b, c и e были такими же, как в примере 13, за исключением того, что морфолин на стадиях a и d был заменен 8-окса-3-азабицикло[3.2,1]октана гидрохлоридом. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 45%.
МС (ИЭР): m/z, 490[М+Н]+.
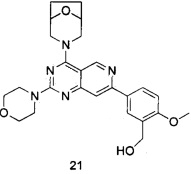
ПРИМЕР 21: получение (5-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-2-морфолинилпиридо[4,3-d]пиримидин-7-ил)-2-метоксифенил)метанола (соединение 21)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии b, c, d и e были такими же, как в примере 13, за исключением того, что морфолин на стадии а был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали в виде твердого вещества белого цвета с выходом 35%.
МС (ИЭР): m/z, 464[М+Н]+.
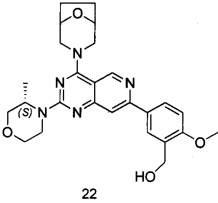
ПРИМЕР 22: получение (5-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-2-((S)-3-метилморфолинил)пиридо[4,3-d]пиримидин-7-ил)-2-метоксифенил)метанола (соединение 22)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии b, c и e были такими же, как в примере 13, за исключением того, что морфолин на стадии а был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом, а морфолин на стадии d был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 35%.
МС (ИЭР): m/z, 478[М+Н]+.
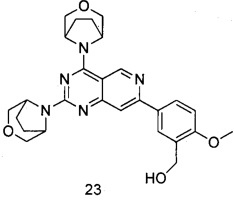
ПРИМЕР 23: получение (5-(2,4-ди(3-окса-8-азабицикло[3.2.1]октан-8-ил)пиридо[4,3-d]пиримидин-7-ил)-2-метоксифенил)метанола (соединение 23)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии b, c и e были такими же, как в примере 13, за исключением того, что морфолин на стадиях a и d был заменен 3-окса-8-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали в виде твердого вещества белого цвета с выходом 50%.
МС (ИЭР): m/z, 490[М+Н]+.
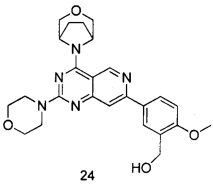
ПРИМЕР 24: получение (5-(4-(1R,5S)-3-окса-8-азабицикло[3.2.1]октан-8-ил)-2-морфолинилпиридо[4,3-d]пиримидин-7-ил)-2-метоксифенил)метанола (соединение 24)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии b, c, d и e были такими же, как в примере 13, за исключением того, что морфолин на стадии а был заменен 3-окса-8-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали в виде твердого вещества белого цвета с выходом 35%.
МС (ИЭР): m/z, 464[М+Н]+.
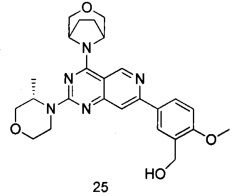
ПРИМЕР 25: получение (5-(4-((1R,5S)-3-окса-8-азабицикло[3.2.1]октан-8-ил)-2-((S)-3-метилморфолинил)пиридо[4,3-d]пиримидин-7-ил)-2-метоксифенил)-метанола (соединение 25)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии b, c и e были такими же, как в примере 13, за исключением того, что морфолин на стадии а был заменен 3-окса-3-азабицикло[3.2.1]октана гидрохлоридом, а морфолин на стадии d был заменен (S)-8-метилморфолином. Указанное в заголовке соединение получали в виде твердого вещества желтого цвета с выходом 35%.
МС (ИЭР): m/z, 478[М+Н]+.
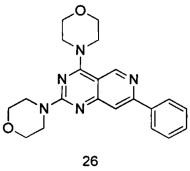
ПРИМЕР 26: получение (4,4'-(7-фенилпиридо[4,3-d]пиримидин-2,4-диил)-диморфолина) (соединение 26)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии a, c, d и e были такими же, как в примере 13, за исключением того, что 5-ацетенил-2-метоксибензилпивалат на стадии b был заменен фенилацетиленом. Указанное в заголовке соединение получали с выходом 52%.
МС (ИЭР): m/z, 378[М+Н]+.
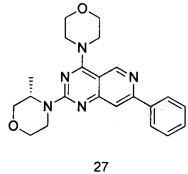
ПРИМЕР 27: получение (S)-3-метил-4-(4-морфолинил-7-фенилпиридо[4,3-d]пиримидин-2-ил)морфолина) (соединение 27)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии a, c и e были такими же, как в примере 13, за исключением того, что 5-ацетенил-2-метоксибензилпивалат на стадии b был заменен фенилацетиленом, а морфолин на стадии d был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали с выходом 42%.
МС (ИЭР): m/z, 391[М+Н]+.
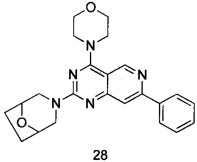
ПРИМЕР 28: получение 3-(4-морфолинил-7-фенилпиридо[4,3-d]пиримидин-2-ил)-8-окса-3-азабицикло[3.2.1]октана (соединение 28)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии a, c и e были такими же, как в примере 13, за исключением того, что 5-ацетенил-2-метоксибензилпивалат на стадии b был заменен фенилацетиленом, а морфолин на стадии d был заменен 8-окса-3-азадицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали с выходом 42%.
МС (ИЭР): m/z, 404[М+Н]+.
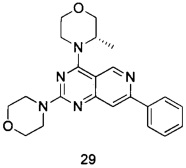
ПРИМЕР 29: получение (S)-3-метил-4-(2-морфолинил-7-фенилпиридо-[4,3-d]пиримидин-4-ил)морфолина (соединение 29)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии c, d и e были такими же, как в примере 13, за исключением того, что 5-ацетенил-2-метоксибензилпивалат на стадии b был заменен фенилацетиленом, а морфолин на стадии а был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали с выходом 42%.
МС (ИЭР): m/z, 392[М+Н]+.
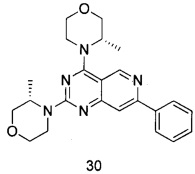
ПРИМЕР 30: получение (3S,3'S)-4,4'-(7-фенилпиридо[4,3-d]пиримидин-2,4-диил)-ди(3-метилморфолина) (соединение 30)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии сие были такими же, как в примере 13, за исключением того, что 5-ацетенил-2-метоксибензилпивалат на стадии b был заменен фенилацетиленом, а морфолин на стадиях a и d был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали с выходом 42%.
МС (ИЭР): m/z, 406[М+Н]+.
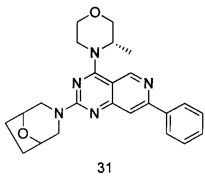
ПРИМЕР 31: получение
3-(4-((S)-3-метилморфолинил)-7-фенилпиридо[4,3-d]-пиримидин-2-ил)-8-окса-3-азабицикло[3.2.1]октана (соединение 31)
Указанное в заголовке соединение получали таким же способом, как в примере 13, и стадии c и e были такими же, как в примере 13, за исключением того, что 5-ацетенил-2-метоксибензилпивалат на стадии b был заменен фенилацетиленом, морфолин на стадии а был заменен (S)-3-метилморфолином, а морфолин на стадии d был заменен на 8-окса-3-азадицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали с выходом 52%.
МС (ИЭР): m/z, 418 [М+Н]+.
ПРИМЕР 32: получение 4,4'-(7-фенилпиримидо[4,5-d]пиримидин-2,4-диил)-диморфолина (соединение 32)
Реакции проводили согласно следующей схеме:
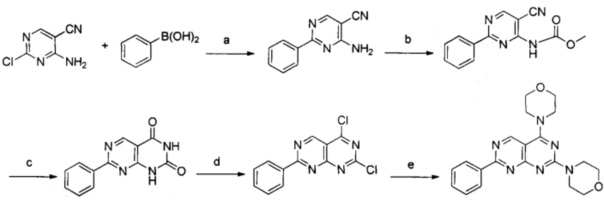 Реагенты и условия: a) фосфат калия, ацетат палладия, 1,1'-бис(ди-m-бутилфосфино)ферроценпалладия дихлорид, диоксан, обратный холодильник; b) метилхлорформиат, гидрид натрия, тетрагидрофуран, комнатная температура; с) 30 мас. % пероксид водорода, гидроксид нактрия, этанол, обратный холодильник; d) оксихлорид фосфора, 1 N,N-диизопропилэтиламин, обратный холодильник; e) морфолин, N,N-диизопропилэтиламин, тетрагидрофуран, комнатная температура.
Реагенты и условия: a) фосфат калия, ацетат палладия, 1,1'-бис(ди-m-бутилфосфино)ферроценпалладия дихлорид, диоксан, обратный холодильник; b) метилхлорформиат, гидрид натрия, тетрагидрофуран, комнатная температура; с) 30 мас. % пероксид водорода, гидроксид нактрия, этанол, обратный холодильник; d) оксихлорид фосфора, 1 N,N-диизопропилэтиламин, обратный холодильник; e) морфолин, N,N-диизопропилэтиламин, тетрагидрофуран, комнатная температура.
a) 4-амино-2-фенилпиримидин-5-карбонитрил
К 4-амино-2-хлорпиримидин-5-карбонитрилу (1 г, 6,47 ммоль) добавляли 30 мл диоксана, фенилбороновую кислоту (1,2 г, 9,70 ммоль), фосфат калия (2,7 г, 12,94 ммоль), ацетат палладия (72,5 мг, 0,32 ммоль) и 1,1'-бис(ди-m-бутил фосфино) ферроцен палладия дихлорид (153 мг, 0,32 ммоль). Полученную смесь дегазировали аргоном три раза, нагревали с обратным холодильником в течение ночи, охлаждали до комнатной температуры и непосредственно помещали на силикагелевую колонку, элюируя смесью петролейный эфир/этилацетат (об./об.=8:1) с получением 500 мг указанного в заголовке соединения в виде твердого вещества белого цвета с выходом 42%.
МС: m/z, 197[М+Н]+.
b) метил-5-циано-2-фенилпиримидин-4-илкарбамат
Гидрид натрия (350 мг, 14,3 ммоль) суспендировали в 60 мл тетрагидрофурана. 4-амино-2-фенилпиримидин-5-карбонитрил (1 г, 5,09 ммоль), полученный как описано выше, растворенный в 40 мл тетрагидрофурана, добавляли по каплям в раствор гидрида натрия на ледяной бане, проводили реакцию при комнатной температуре в течение 3 часов и добавляли по каплям метил хлорформиат (722 мг, 7,65 ммоль) и далее проводили реакцию в течение ночи. После того, как растворитель удалили при пониженном давлении, к полученной смеси добавляли воду, экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии на сйликагеле с элюированием смесью петролейный эфир/этилацетат (об./об=4:1) с получением 1 г указанного в заголовке соединения в виде твердого вещества белого цвета с выходом 78%.
МС (ИЭР): m/z, 255[М+Н]+.
c) 7-фенилпиримидо[4,5-d]пиримидин-2,4(1H,3H)-дион
К метил-5-циано-2-фенилпиримидин-4-илкарбамату (100 мг, 0,39 ммоль) добавляли 8 мл этанола, 30 мг гидроксида натрия и 2 мл 30 масс. % пероксида водорода. Смесь нагревали с обратным холодильником в течение 2 часов, добавляли 8 мл воды, концентрировали при пониженном давленгии и фильтровали с получением твердого вещества белого цвета, которое промывали водой три раза с получением 30 мг указанного в заголовке соединения в виде твердого вещества белого цвета с выходом 32%.
МС (ИЭР): m/z, 241 [М+Н]+.
d) 2,4-дихлор-7-фенилпиримидо[4,5-d]пиримидин
К 7-фенилпиримидо[4,5-d]пиримидин-2,4(1Н,3Н)-диону (30 мг, 0,13 ммоль) добавляли 2 мл оксихлорида фосфора и N,N-диизопропилэтиламин (18 мг, 0,14 ммоль). Смесь нагревали с обратным холодильником в течение ночи, охлаждали до комнатной температуры, наливали на раздробленный лед, нейтрализовали насыщенным водным раствором карбоната натрия до pH of 7-8 и экстрагировали дихлорметаном. Органическую фазу промывали последовательно насыщенным раствором бикарбоната натрия и насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 32 мг указанного в заголовке соединения в виде твердого вещества желтовато-коричневого цвета с выходом 92%. Продукт использовали непосредственно на следующей стадии без дополнительной очистки.
е) 4,4'-(7-фенилпиримидо[4,5-d]пиримидин-2,4-диил)диморфолин
2,4-дихлор-7-фенилпиримидо[4,5-d]пиримидин (32 мг, 0,16 ммоль) растворяли в 2 мл тетрагидрофурана, добавляли N,N-диизопропилэтиламин (35 мг, 0,27 ммоль) и морфолин (50 мг, 0,57 ммоль) и проводили реакцию при комнатной температуре в течение ночи. После того как растворитель удалили при пониженном давлении, к смеси добавляли воду и экстрагировали этилацетатом. Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле с элюированием смесью петролейный эфир/этилацетат (об./об.=2:1) с получением 21 мг указанного в заголовке соединения в виде твердого вещества желтого цвета с выходом 49%.
МС (ИЭР): m/z, 379[М+Н]+.
ПРИМЕР 33: получение (3S,3'S)-4,4'-(7-фенил пиримидо[4,5-d]пиримидин-2,4-диил)-ди(3-метилморфолина) (соединение 33)
Указанное в заголовке соединение получали таким же способом, как в примере 32, и стадии a, b, c и d были такими же, как в примере 32, за исключением того, что морфолин на стадии e был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали с выходом 66%.
МС (ИЭР): m/z, 407[М+Н]+.
ПРИМЕР 34: получение 3,3'-(7-фенилпиримидо[4,5-d]пиримидин-2,4-диил)-ди(8-окса-3-азабицикло[3.2.1]октана) (соединение 34)
Указанное в заголовке соединение получали таким же способом, как в примере 32, и стадии a, b, c и d были такими же, как в примере 32, за исключением того, что морфолин на стадии е был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали с выходом 64%.
МС (ИЭР): m/z, 431[М+Н]+.
ПРИМЕР 35: получение 3-(2,4-ди(3-окса-8-азабицикло[3.2.1]октан-8-ил)-пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 35)
Реакции проводили согласно следующей схеме:
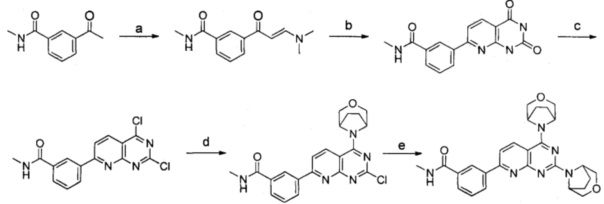
Реагенты и условия: a) N,N-диметилформамид диметил ацеталь, толуол, обратный холодильник; b) 6-аминоурацил, ледяная уксусная кислота, вода, диметилсульфоксид, 99°C; с) оксихлорид фосфора, N,N-диизопропилэтиламин, анизол, 80°С; d) 3-окса-8-азабицикло[3.2.1]октана гидрохлорид, N,N-диизопропилэтиламин, тетрагидрофуран, комнатная температура; е) 3-окса-8-азабицикло[3.2.1]октана гидрохлорид, N,N-диизопропилэтиламин, изопропанол, микроволновое излучение при 160°С, 80 мин.
a) 3-формамидоацетофенон и N,N-диметилформамид диметил ацеталь подвергали реакции конденсации для получения (E)-3-(3-(диметиламино)акрил)-N-метил бензамида; b) (E)-3-(3-(диметиламино)акрил)-N-метилбензамид и 6-аминоурацил подвергали реакции конденсации с получением 3-(2,4-диoкco-1,2,3,4-тeтpaгидpoпиpидo[2,3-d]пиpимидин-ил)-N-бeнзaмидa; c) 3-(2,4-диoкco-1,2,3,4-тeтpaгидpoпиpидo[2,3-d]пиpимидин-ил)-N-бeнзaмид подвергали реакции в присутствии оксихлорида фосфора с получением дихлорированного промежуточного соединения; d) и е) дихлорированное промежуточное соединение подвергали последовательно реакции замещения с 3-окса-8-азабицикло[3.2.1]октана гидрохлоридом с получением указанного в заголовке соединения с выходом 64%.
МС (ИЭР): m/z, 487[М+Н]+.
ПРИМЕР 36: получение 3-(2,4-ди(8-окса-3-азабицйкло[3.2.1]октан-3-ил)-пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 36)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b, и c были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадиях d и e был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали с выходом 64%.
МС (ИЭР): m/z, 487[М+Н]+.
ПРИМЕР 37: получение 3-(2-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-4-((S)-3-метилморфолинил)пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 37)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b, c и e были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии d был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали с выходом 64%.
МС (ИЭР): m/z, 475[М+Н]+.
ПРИМЕР 38: получение 3-(2-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-4-((S)-3-метилморфолинил)пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 38)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b и c были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии d был заменен (S)-3-метилморфолином, а 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии е был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали с выходом 64%.
МС (ИЭР): m/z, 475[М+Н]+.
ПРИМЕР 39: получение 3-(2-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-4-морфолинилпиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 39)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b, c, d и e были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии d был заменен морфолином. Указанное в заголовке соединение получали с выходом 60%.
МС (ИЭР): m/z, 461[М+Н]+.
ПРИМЕР 40: получение 3-(2-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-4-морфолинилпиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 40)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b и c были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии d был заменен морфолином, а 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии е был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали с выходом 60%.
МС (ИЭР): m/z, 461[М+Н]+.
ПРИМЕР 41: получение 3-(2-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-4-((2S,6R)-2,6-диметилморфолинил)пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 41)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b, c и e были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии d был заменен (2S,6R)-2,6-диметилморфолином. Указанное в заголовке соединение получали с выходом 64%.
МС (ИЭР): m/z, 489[М+Н]+.
ПРИМЕР 42: получение 3-(2-(8-окса-3-азабицикло[3.2.1]октан-8-ил)-4-((2S,6R)-2,6-диметилморфолинил)пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 42)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b и c были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии d был заменен (2S,6R)-2,6-диметилморфолином, а 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии e был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали с выходом 66%.
МС (ИЭР): m/z, 489[М+Н]+.
ПРИМЕР 43: получение 3-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-2-морфолинилпиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 43)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b, c и d были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии е был заменен морфолином. Указанное в заголовке соединение получали с выходом 64%. МС (ИЭР): m/z, 461[М+Н]+.
ПРИМЕР 44: получение 3-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-2-((S)-3-метилморфолинил)пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 44)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b, c и d были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии e был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали с выходом 64%.
МС (ИЭР): m/z, 475[М+Н]+.
ПРИМЕР 45: получение 3-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-2-((2S,6R)-2,6-диметилморфолинил)пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 45)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b, c и e были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии е был заменен (2S,6R)-2,6-диметилморфолином. Указанное в заголовке соединение получали с выходом 66%.
МС (ИЭР): m/z, 489[М+Н]+.
ПРИМЕР 46: получение 3-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-2-((2-метоксиэтил)метиламино)пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 46)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b, c и d были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии e был заменен 2-(метоксиэтил)метиламином. Указанное в заголовке соединение получали с выходом 60%.
МС (ИЭР): m/z, 463[М+Н]+.
ПРИМЕР 47: получение 3-(2-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-4-((3-окса-8-азабицикло[3.2.1]октан-8-ил)пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 47)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b, c и d были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии е был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали с выходом 60%.
МС (ИЭР): m/z, 487[М+Н]+.
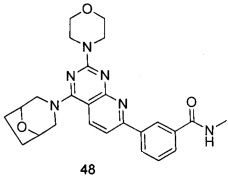
ПРИМЕР 48: получение 3-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-2-морфолинилпиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 48)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b и c были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии d был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом, а 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии e был заменен морфолином. Указанное в заголовке соединение получали с выходом 64%.
МС (ИЭР): m/z, 461[М+Н]+.
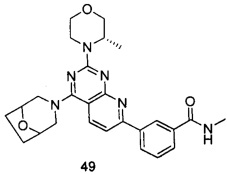
ПРИМЕР 49: получение 3-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-2-((S)-3-метилморфолинил)пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 49)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b и c были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии d был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом, а 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии e был заменен (S)-3-метилморфолином. Указанное в заголовке соединение получали с выходом 64%.
МС (ИЭР): m/z, 475[М+Н]+.
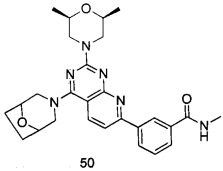
ПРИМЕР 50: получение 3-(4-(8-окса-3-азабицикло [3.2.1] октан-8-ил)-2-((2S,6R)-2,6-диметилморфолинил)пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 50)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b и c были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1] октана гидрохлорид на стадии d был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом, а 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии е был заменен (2S,6R)-2,6-диметилморфолином. Указанное в заголовке соединение получали с выходом 64%.
МС (ИЭР): m/z, 489[М+Н]+.
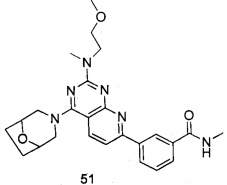
ПРИМЕР 51: получение 3-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-2-((2-метоксиэтил)метиламино)пиридо[2,3-d]пиримидин-7-ил)-N-метилбензамида (соединение 51)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b и с были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии d был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом, а 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии e был заменен (2-метоксиэтил)метиламином. Указанное в заголовке соединение получали с выходом 60%.
МС (ИЭР): m/z, 463[М+Н]+.
ПРИМЕР 52: получение 3-(2-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-4-((8-окса-3-азабицикло[3.2.1]октан-3-ил)пиридо[2,3-d]пиримидин-7-ил)-N-метил-бензамида (соединение 52)
Указанное в заголовке соединение получали таким же способом, как в примере 35, и стадии a, b, c и e были такими же, как в примере 35, за исключением того, что 3-окса-8-азабицикло[3.2.1]октана гидрохлорид на стадии d был заменен 8-окса-3-азабицикло[3.2.1]октана гидрохлоридом. Указанное в заголовке соединение получали с выходом 66%.
МС (ИЭР): m/z, 487[М+Н]+.
Материалы и способы для фармакологического эксперимента
Исследование с киназой mTOR
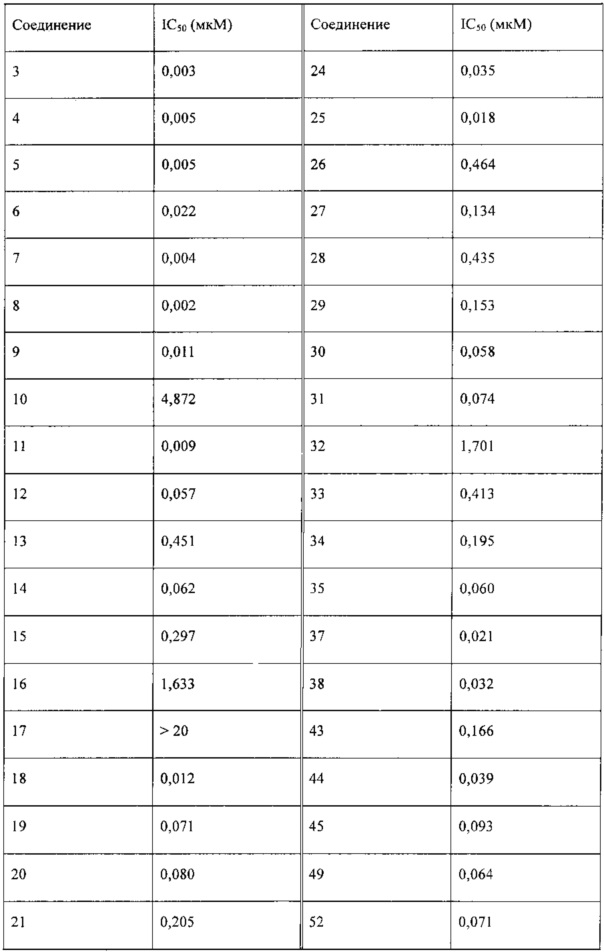
Исследование активности в отношении киназы mTOR проводили с использованием 1,7 нМ mTOR (Millipore, 14-770М), 50 нМ ULight-4EBP1 (Perkin-Elmer, TRF0128M) и 100 мкМ АТФ на системе детектирования LANCE® Ultra (PerkinElmer). Сначала получали соединение в исходном растворе с концентрацией 20 мМ, а затем указанный раствор постепенно разбавляли для добавления в реакционную систему с ферментом mTOR на 384-луночных планшетах. Исследуемые концентрации составляли 20, 4, 0,8, 0,16, 0,032, 0,0064, 0,00128, 0,000256 мкМ (n=3). После проведения ферментативной реакции с mTOR в течение 1,5 часов реакцию прекращали, и проводили анализ с использованием системы LANCE® Ultra (PerkinElmer) в течение 1 часа. Результаты анализа получали с помощью многомодового считывателя микропланшетов Synergy II (BioTek). Относительную активность в отношении mTOR % = (найденное значение света в лунке с лекарством - значение света в контрольной группе (без mТСЖ)) / (значение света в группе с ДМСО - значение света в контрольной группе) × 100%. Экспериментальные данные обрабатывали с помощью Microsoft Office Excel и Graphpad PRISM 5, и рассчитывали IC50. Ингибирующее отношение для mTOR выражали в виде среднего ± стандартное отклонение (CO). Результаты исследования представлены в таблице 1.
Исследование пролиферации клеток
Исследование пролиферации клеток проводили с использованием двух типов клеточных линий: клеток глиомы головного мозга человека U87MG и клеток рака предстательной железы человека LNCap - обе из которых были получены из коллекции АТСС. U87MG и LNCap содержат делецию в гене PTEN и принадлежат к опухолевым клеткам, зависимым от сигнала mTOR. Среда для культур клеток и соответствующие реагенты были получены из GIBCO. Клетки U87MG культивировали в полной минимальной питательной среде (MEM) (содержащей 10% фетальной бычьей сыворотки, 100 Е/мл пенициллина и 100 мкг/мл стрептомицина). Клетки LNCap культивировали в полной среде RPMI-1640 (содержащей 10% фетальной бычьей сыворотки, 100 Е/мл пенициллина и 100 мкг/мл стрептомицина). За один день до эксперимента клетки U87MG и LNCap в фазе роста подвергали ферментативному гидролизу трипсином с получением суспензии клеток, которую добавляли в 96-луночный планшет в количестве 5×103 клеток на лунку (150 мкл на лунку) и культивировали в инкубаторе с 5% CO2 при 37°С, чтобы клетки были слипшимися (adherent) на следующий день и готовыми к применению. Исходный раствор (20 мМ) постепенно разбавляли средой согласно схеме эксперимента, добавляли в исследуемые клетки по 50 мкл на лунку, и конечные концентрации составляли 60, 20, 6,67, 2,22, 0,74, 0,247, 0,0823, 0,027 мкМ (n=3). Клетки/соединение культивировали в течение 3 дней, и использовали метод с метилтетразолсульфатом (MTS) для анализа пролиферации клеток. MTS и метилфеназиния метосульфат (PMS) приобретали в компании Sigma, раствор MTS/PMS (20:1) добавляли в исследуемые клетки в количестве 20 мкл на лунку. По истечение соответствующего времени инкубации получали результаты пролиферации клеток с помощью считывателя микропланшетов для 96-луночного планшета.
Относительная жизнеспособность клеток в % = (значение A490 для группы введения - значение света для контрольной группы)/(значение света для группы с ДМСО - значение света для контрольной группы) × 100%.
Экспериментальные данные обрабатывали с помощью Microsoft Office Excel и Graphpad PRISM 5, и рассчитывали IC50. Значение ингибирования пролиферации клеток выражали в виде среднего ± CO. Результаты исследования приведены в таблице 2.
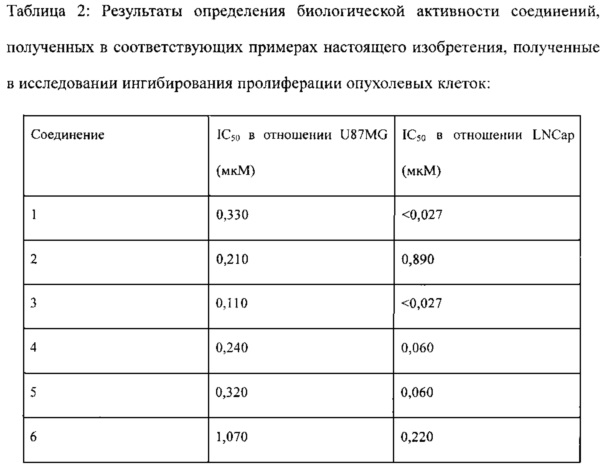
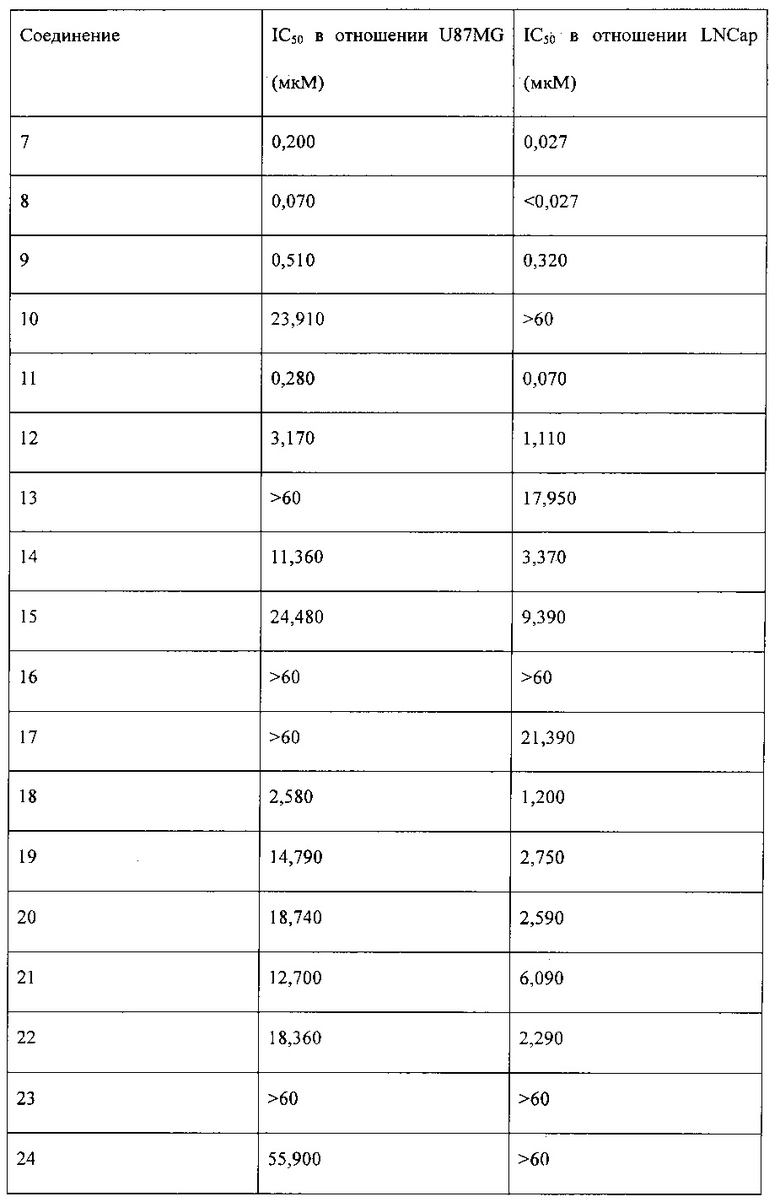
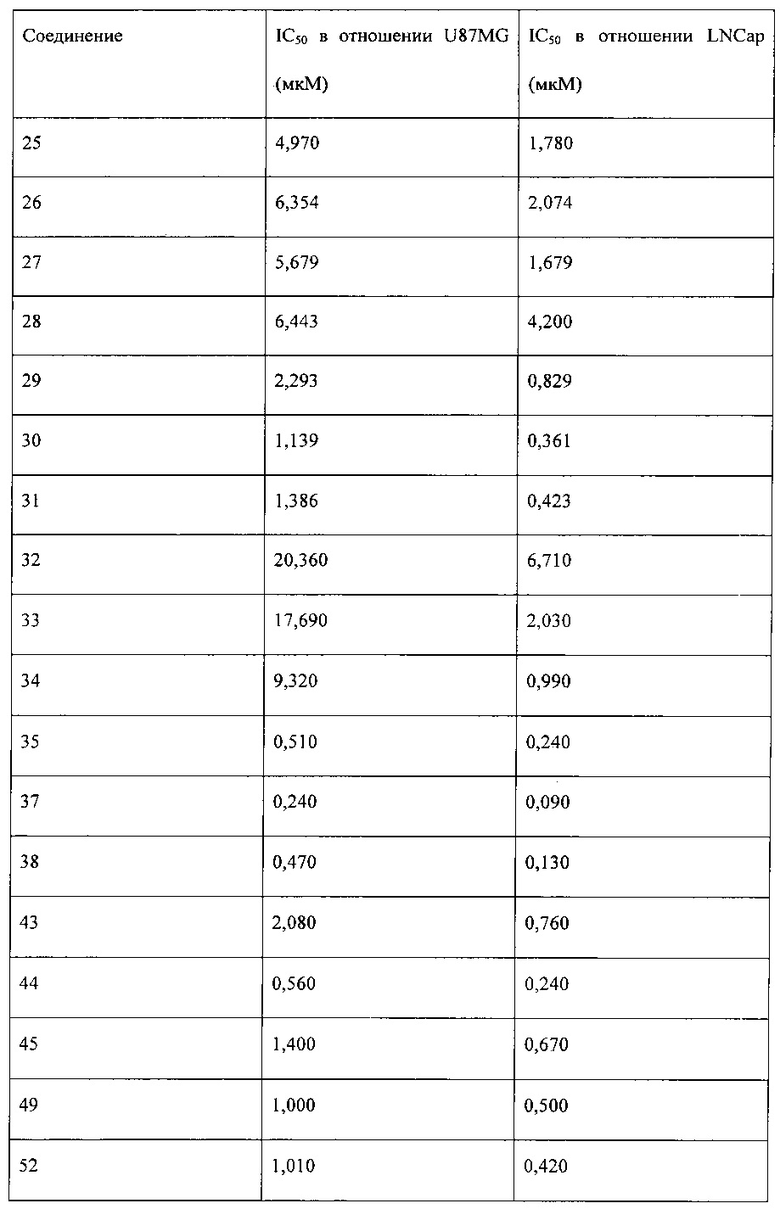
Соединения, перечисленные в таблице 1 и таблице 2, обладают сильной ингибирующей активностью в отношении mTOR, а также mTOR-зависимой противоопухолевой активностью.
Приведенные выше примеры служат исключительно для иллюстративных целей, и объем настоящего изобретения не ограничен ими. Специалисту в данной области техники очевидны различные модификации, и объем настоящего изобретения ограничен только прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ПРОСТОГО ЭФИРА ИЗОКСАЗОЛИЛА В КАЧЕСТВЕ ПАМ ГАМК A АЛЬФА5 | 2019 |
|
RU2800160C2 |
| МОРФОЛИНО-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ БИЦИКЛИЧЕСКИХ ПИРИМИДИНМОЧЕВИНЫ ИЛИ КАРБАМАТА В КАЧЕСТВЕ ИНГИБИТОРОВ mTOR | 2012 |
|
RU2609208C2 |
| ДИФТОРМЕТИЛАМИНОПИРИДИНЫ И ДИФТОРМЕТИЛАМИНОПИРИМИДИНЫ | 2015 |
|
RU2712091C2 |
| ЛЕЧЕНИЕ НЕВРОЛОГИЧЕСКИХ РАССТРОЙСТВ | 2017 |
|
RU2765868C2 |
| 3,3-ДИЗАМЕЩЕННЫЙ-(8-АЗАБИЦИКЛО[3.2.1]ОКТ-8-ИЛ)-[5-(1Н-ПИРАЗОЛ-4-ИЛ)-ТИОФЕН-3-ИЛ]-МЕТАНОН И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2593753C2 |
| ПИРРОЛИДИНОВЫЕ И ПИПЕРИДИНОВЫЕ СОЕДИНЕНИЯ | 2020 |
|
RU2803455C1 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2018 |
|
RU2765152C2 |
| СОЕДИНЕНИЯ 1-(3-АМИНОПРОПИЛ)-ЗАМЕЩЕННОГО ЦИКЛИЧЕСКОГО АМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ПРИМЕНЕНИЯ | 2014 |
|
RU2671179C2 |
| УСИЛИТЕЛЬ ПРОТИВООПУХОЛЕВОГО ЭФФЕКТА С ПРИМЕНЕНИЕМ НОВОГО СОЕДИНЕНИЯ БИФЕНИЛА | 2018 |
|
RU2765153C2 |
| ПРОИЗВОДНОЕ 1,2,4-ТРИАЗОЛОНА | 2011 |
|
RU2566754C2 |
Изобретение относится к новому пиридопиримидиновому соединению общей формулы (Ia), его стереоизомеру или фармацевтически приемлемой соли. Соединения обладают свойствами ингибитора фосфатидилинозитол-3-киназа-киназа AKT-mTOR (PI3K-AKT-mTOR) и могут быть использованы для лечения заболевания или состояния, вызванного дисфункцией сигнального пути PI3K-AKT-mTOR, в частности опухолевого заболевания. В общей формуле (Ia)
где каждый из R1 и R2 независимо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, и по меньшей мере один из R1 и R2 представляет собой 3-окса-8-азабицикло[3.2.1]октанил или 8-окса-3-азабицикло[3.2.1]октанил; при этом каждый из RA и RB независимо представляет собой С1-С6 алкил, незамещенный или замещенный С1-С6 алкокси, или RA и RB совместно с N, к которому они присоединены, образуют морфолиновое кольцо, которое является незамещенным или замещенным С1-С6 алкилом; и R3 представляет собой 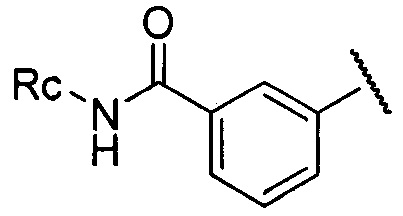 , где Rc представляет собой С1-С3 алкил. 3 н. и 6 з. п. ф-лы, 2 табл., 52 пр.
, где Rc представляет собой С1-С3 алкил. 3 н. и 6 з. п. ф-лы, 2 табл., 52 пр.
1. Пиридопиримидиновое соединение, представленное формулой (Ia), его стереоизомер или фармацевтически приемлемая соль:
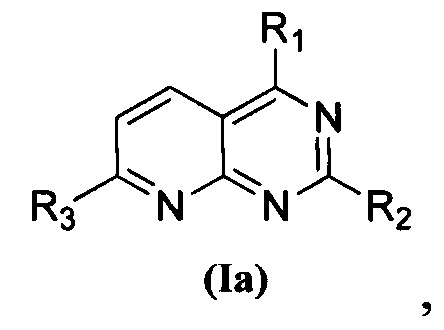
где
каждый из R1 и R2 независимо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, и по меньшей мере один из R1 и R2 представляет собой 3-окса-8-азабицикло[3.2.1]октанил или 8-окса-3-азабицикло[3.2.1]октанил;
при этом каждый из RA и RB независимо представляет собой С1-С6 алкил, незамещенный или замещенный С1-С6 алкокси, или RA и RB совместно с N, к которому они присоединены, образуют морфолиновое кольцо, которое является незамещенным или замещенным С1-С6 алкилом; и
R3 представляет собой 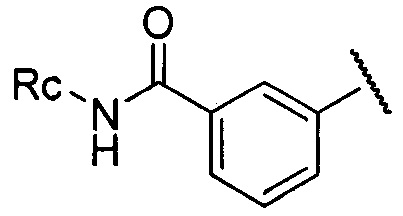 , где Rc представляет собой С1-С3 алкил.
, где Rc представляет собой С1-С3 алкил.
2. Пиридопиримидиновое соединение, его стереоизомер или фармацевтически приемлемая соль по п. 1, где
каждый из R1 и R2 независимо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, и по меньшей мере один из R1 и R2 представляет собой 3-окса-8-азабицикло[3.2.1]октанил или 8-окса-3-азабицикло[3.2.1]октанил;
при этом каждый из RA и RB независимо представляет собой С1-С3 алкил, незамещенный или замещенный С1-С3 алкокси, или RA и RB совместно с N, к которому они присоединены, образуют морфолиновое кольцо, которое является незамещенным или замещенным С1-С3 алкилом.
3. Пиридопиримидиновое соединение, его стереоизомер или фармацевтически приемлемая соль по п. 1, где
каждый из R1 и R2 независимо представляет собой 3-окса-8-азабицикло[3.2.1]октанил, 8-окса-3-азабицикло[3.2.1]октанил или NRARB, и по меньшей мере один из R1 и R2 представляет собой 3-окса-8-азабицикло[3.2.1]октанил или 8-окса-3-азабицикло[3.2.1]октанил;
при этом RA и RB совместно с N, к которому они присоединены, образуют морфолиновое кольцо, незамещенное или замещенное С1-С3 алкилом.
4. Пиридопиримидиновое соединение, его стереоизомер или фармацевтически приемлемая соль по п. 1, где
R1 представляет собой  или
или  , R2 представляет собой
, R2 представляет собой 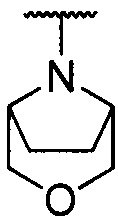 ,
, 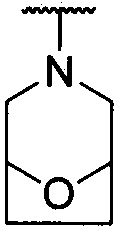 ,
, 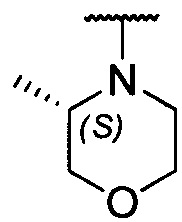 ,
, 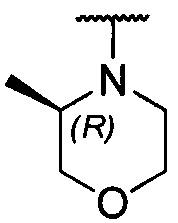 ,
, 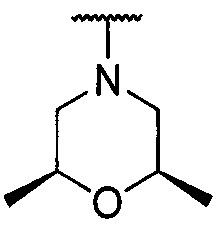 ,
, 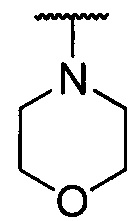 или
или 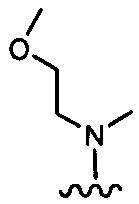 ; или R2 представляет собой
; или R2 представляет собой 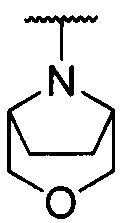 или
или 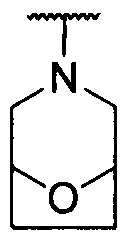 , a R1 представляет собой
, a R1 представляет собой 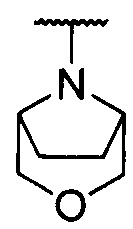 ,
, 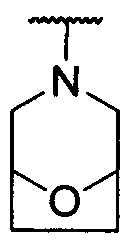 ,
, 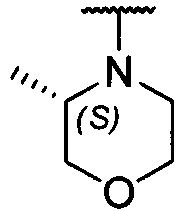 ,
, 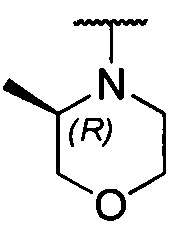 ,
, 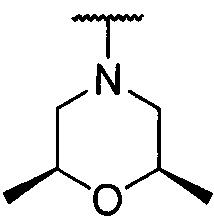 ,
, 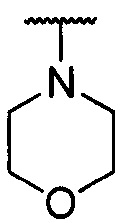 или
или 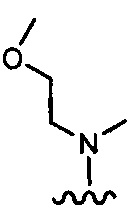 .
.
5. Пиридопиримидиновое соединение, его стереоизомер или фармацевтически приемлемая соль по п. 1, где
Rc представляет собой метил.
6. Пиридопиримидиновое соединение, его стереоизомер или фармацевтически приемлемая соль по п. 1, где соединение, представленное формулой (Ia), выбрано из группы, состоящей из следующих соединений:
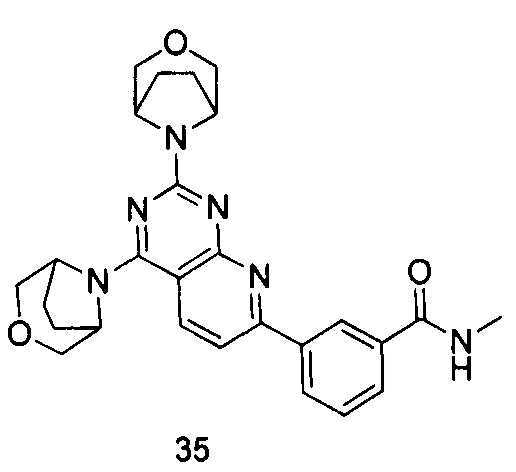
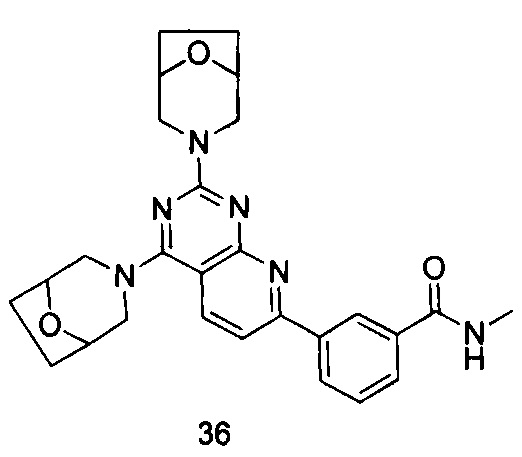
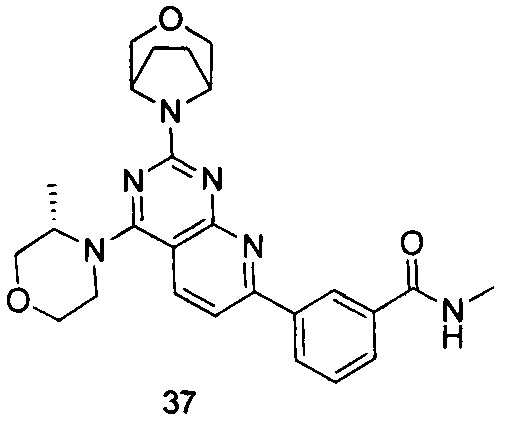
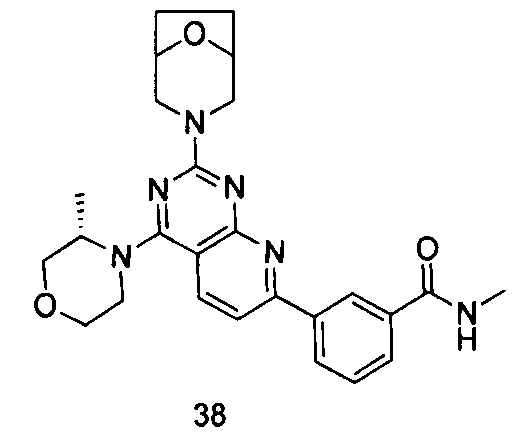
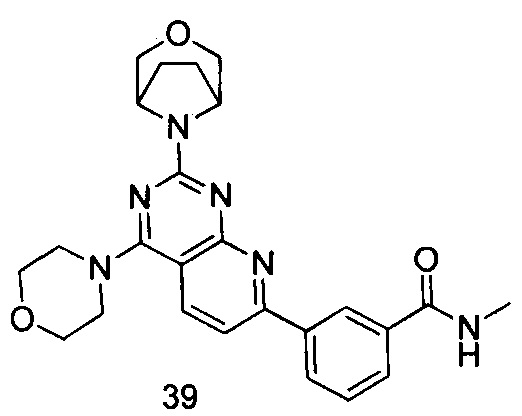
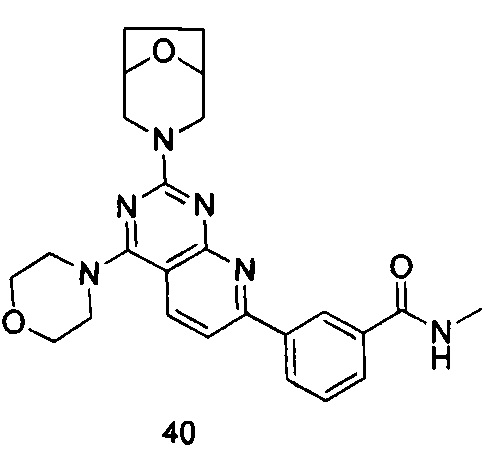
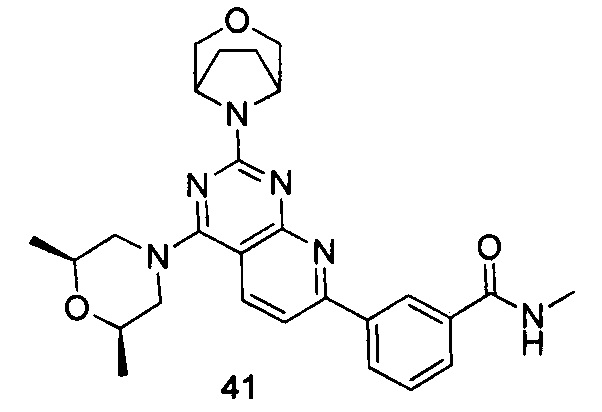
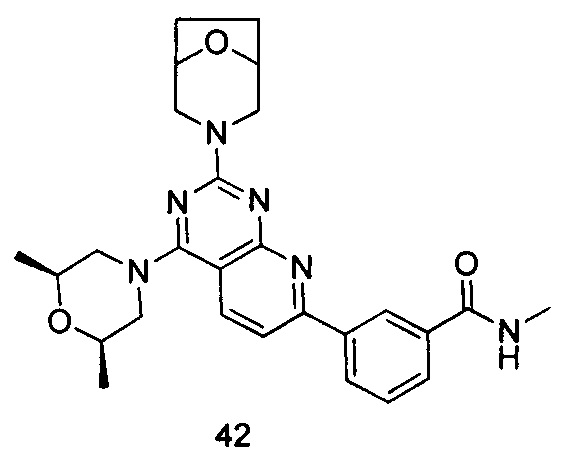
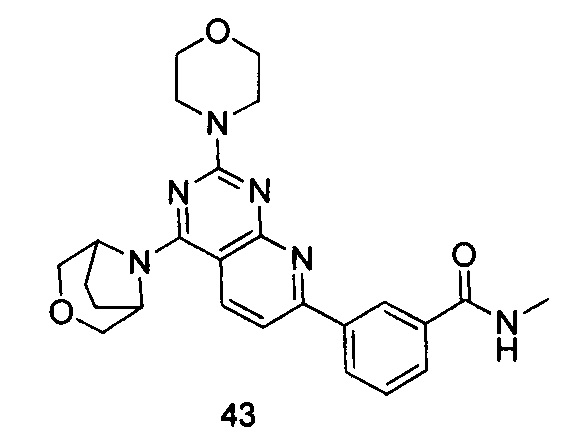
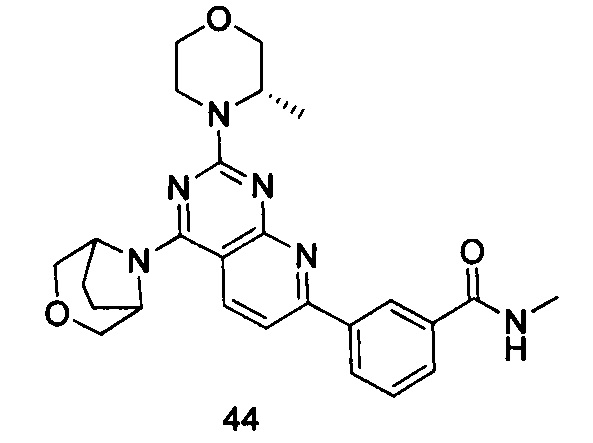
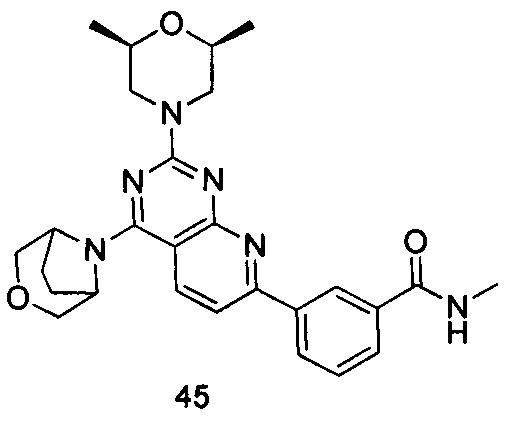
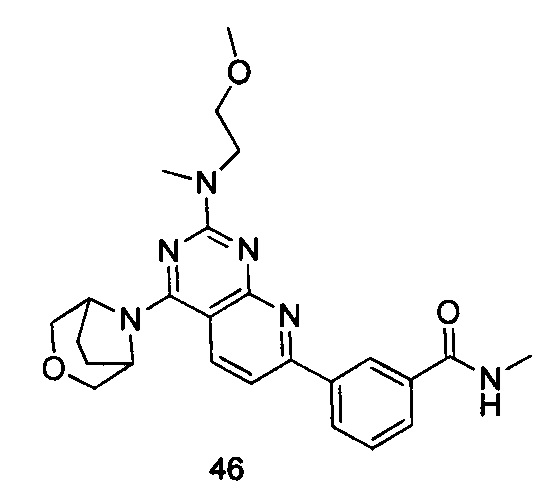
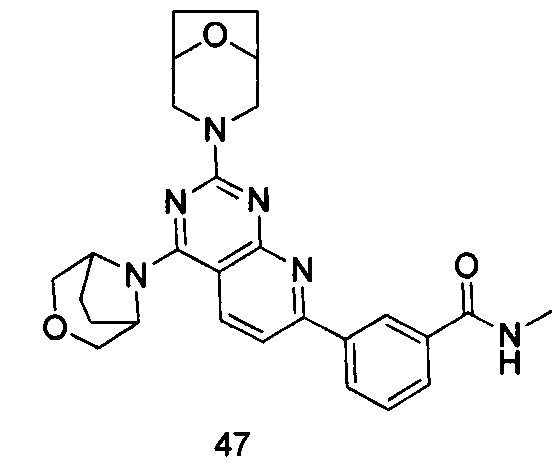
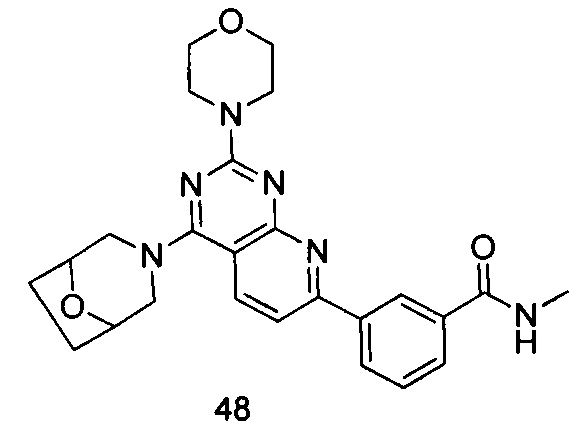
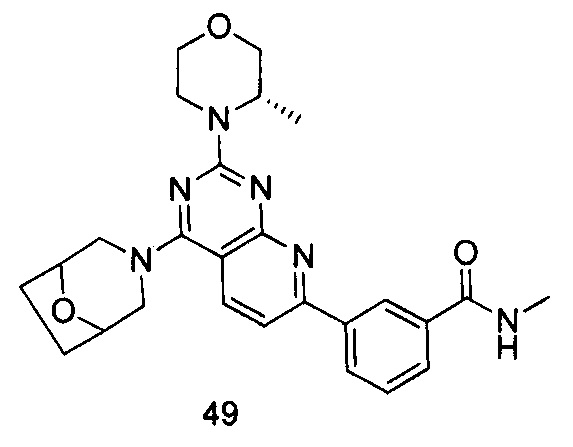
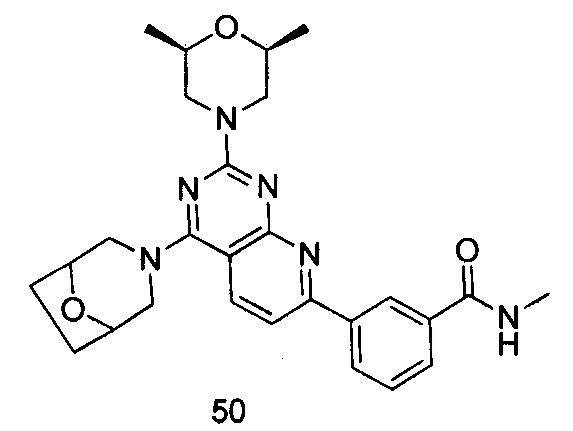
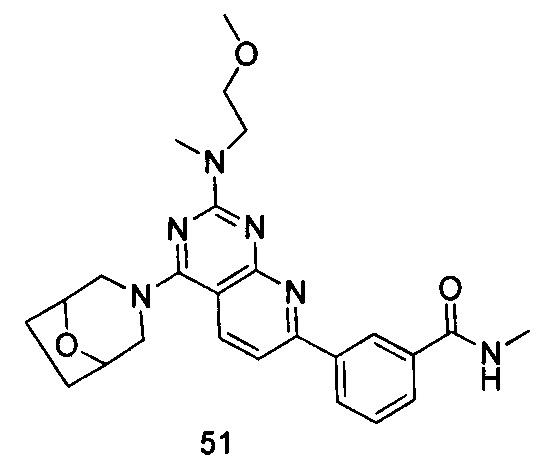
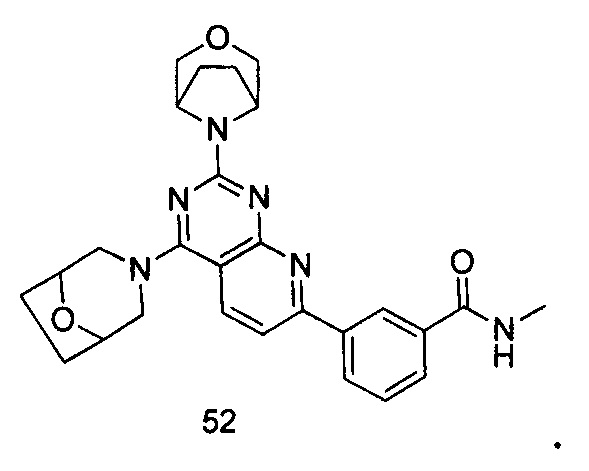
7. Применение пиридопиримидинового соединения, его стереоизомера или фармацевтически приемлемой соли по любому из пп. 1-6 в качестве ингибитора фосфатидилинозитол-3-киназа-киназа AKT-mTOR (PI3K-AKT-mTOR) для лечения заболевания или состояния, вызванного дисфункцией сигнального пути PI3K-AKT-mTOR.
8. Применение по п. 7, в котором указанное заболевание или состояние, вызванное дисфункцией сигнального пути PI3K-AKT-mTOR, представляет собой опухолевое заболевание.
9. Фармацевтическая композиция, обладающая свойствами ингибитора PI3K-AKT-mTOR, содержащая терапевтически эффективное количество пиридопиримидинового соединения, его стереоизомера или фармацевтически приемлемой соли по любому из пп. 1-6 и фармацевтически приемлемый носитель или вспомогательное вещество.
| CN 103588792 A, 19.02.2014 | |||
| CN 102887895 A, 23.01.2013 | |||
| WO 2013016999 А1, 07.02.2013 | |||
| ПРОИЗВОДНОЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРА PI3K И ЕГО ПРИМЕНЕНИЕ | 2007 |
|
RU2448109C2 |

