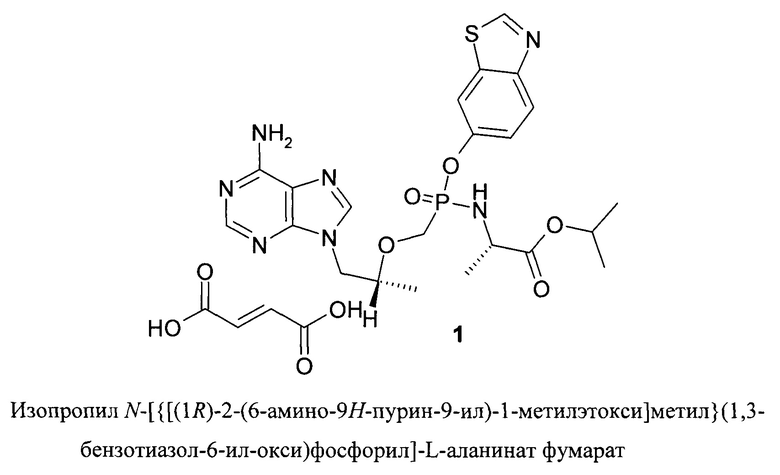
Настоящее изобретение относится к изопропил N-[{[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}(1,3-бензотиазол-6-ил-окси)фосфорил]-L-аланинат фумарату, а также его композициям, способам применения и получения. Это соединение и его композиции являются ингибиторами обратной транскиптазы и может использоваться при лечении вызываемых вирусами заболеваний, в частности гепатита Б.
Известно, что соединение 9-(2-фосфонил-метоксипропил)аденин, содержащее нуклеозидный остаток, соединенный с остатком форсфорной кислоты, Тенофовир, обладает сильным ингибирующим действием на обратную транскриптазу.
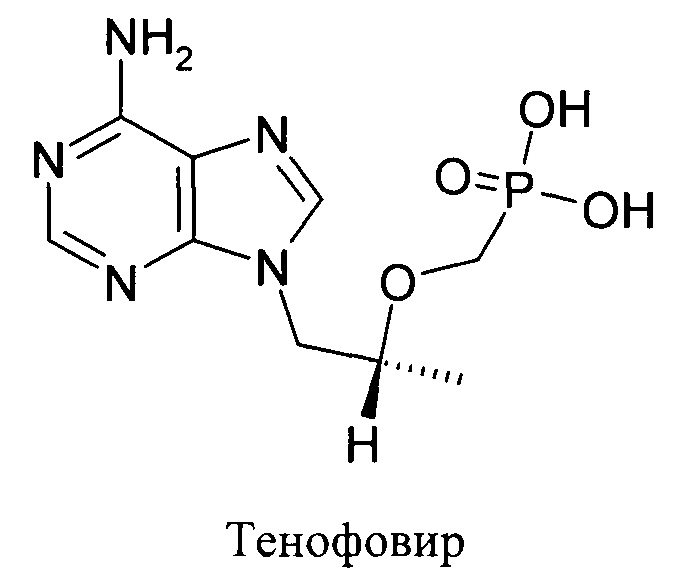
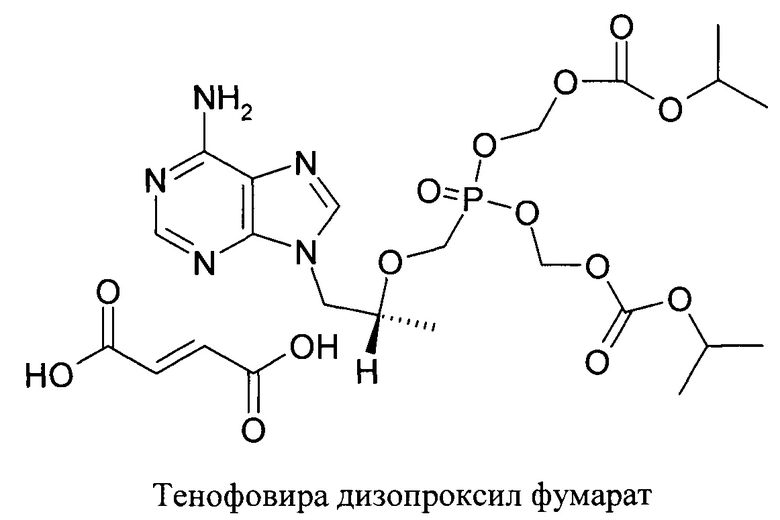
В то же время, в форме двузамещенной фосфорной кислоты Тенофовир обладает нулевой биодоступностью при пероральном применении. Были предложены различные пролекарственные формы Тенофовира, в частности Тенофовир дизопроксил фумарат, обладающие хорошей биодоступностью при пероральном применении и высвобождающие незамещенный 9-(2-фосфонил-метоксипропил)аденин в результате метаболизма, преимущественно в печени. 9-(2-Фосфонил-метоксипропил)аденин после высвобождения подвергается фосфорилированию и переходит в активные формы ди- и трифосфата Тенофовира.
Заявители внезапно обнаружили, что смешанный эфиро-амид Тенофовира - изопропил N-[{[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}(1,3-бензотиазол-6-ил-окси)фосфорил]-L-аланинат в виде соли - фумарата, соединение формулы 1, является эффективным пролекарством Тенофовира и способен подавлять вирус гепатита Б в ин-виво модели заболевания гепатита Б в утках.
Ниже приведены определения терминов, которые использованы в описании этого изобретения.
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и других готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Фармацевтическая композиция» обозначает композицию, включающую в себя активный компонент и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлимых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные такие как мази и кремы, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, дихлорацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей можно найти в справочнике фармацевтичнеских солей [Р.Н. Stahl, C.G. Wermuth (Eds.). Handbook of Pharmaceutical Salts, Properties, Selection, and Use. VHCA, Verlag Helvetica Chimica Acta,  , Switzerland, and Wiley-VCH, Weinheim, Germany. 2002.]. Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
, Switzerland, and Wiley-VCH, Weinheim, Germany. 2002.]. Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Цель настоящего изобретения заключается в создании новой пролекарственной формы Тенофовира, активной при пероральном приеме.
Поставленная цель достигается изопропил N-[{[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}(1,3-бензотиазол-6-ил-окси)фосфорил]-L-аланинат фумаратом формулы 1
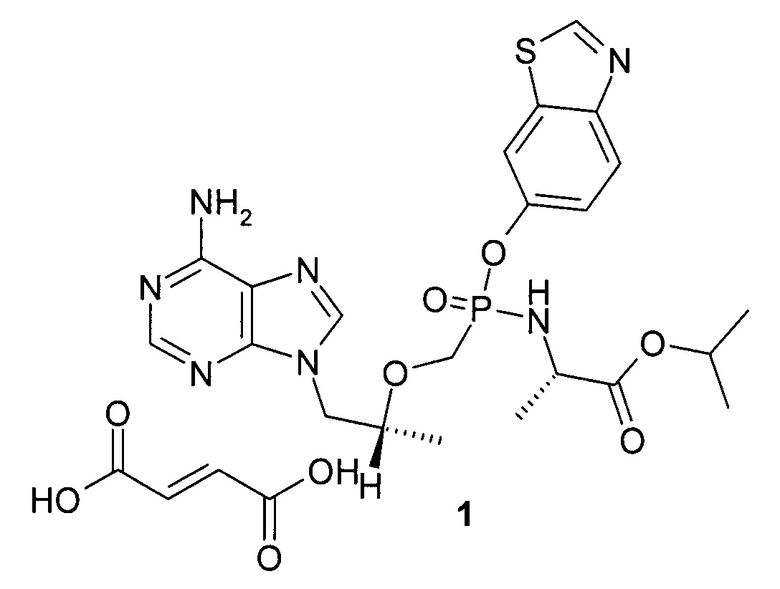
Предметом настоящего изобретения является новый ингибитор обратной транскриптазы формулы 1.
Соединение формулы 1 по настоящему изобретению может также включать все изотопы атомов, присутствующих в соединении 1. Изотопы включают атомы, имеющие одинаковый атомный номер, но разные массовые числа. Например, изотопы водорода включают тритий и дейтерий.
Соединение 1 по настоящему изобретению может быть получено с использованием известных методов органического синтеза и может быть синтезировано в соответствии с любым из многочисленных возможных путей синтеза. Реакция для получения соединения по изобретению может быть осуществлена в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Каждая реакция может быть осуществлена в одном растворителе или в смеси растворителей. В зависимости от конкретной стадии реакции, подходящие растворители для конкретной стадии реакции могут быть выбраны специалистом в данной области.
Преимуществом нового соединения формулы 1 является более высокая активность в ин-виво модели гепатита Б в утках по сравнению с Тенофовира дизопроксид фумаратом. Такая более высокая активность может быть объяснена более удачной формой пролекартсва, позволяющей достичь лучшей биодоступности при пероральном применении и большей концентрации незамещенного Тенофовира и его активных метаболитов в пораженном органе (печени). Исследовалось вещество формулы 1 (ZB98-0002) в сравнении со стандартным пролекарством Тенофовира - TDF (тенофовир дизопроксил фумарат). Как видно из таблицы 1 - к двадцатому дню терапии средние уровни вирусной нагрузки снизились в группе 1 (доза TDF 10 мг/кг) - на 3,40 lg; в группе 2 (доза ZB98-0002 10 мг/кг) - на 3,82 lg, в группе 3 (доза ZB98-0002 1,0 мг/кг) - на 3,36 lg Таким образом, препарат ZB98-0002 продемонстрировал способность подавлять репликацию модельного гепаднавируса DHBV, сопоставимую с таковой референсного препарата TDF, при этом применение обеих доз препарата ZB98-0002, 10 мг/кг и 1 мг/кг, на протяжении трех недель приводило к значительному подавлению вирусной репликации, о чем свидетельствовало снижение вирусной нагрузки на 3,36-3,82 lg/мл.


Настоящее изобретение относится к способу получения соединения формулы 1. Способ получения соединения формулы 1 включает взаимодействие соединения формулы 1а [(1R)-2-(6-амино-9Н-пурин-9-ил)-1-метилэтокси]метил фосфорной кислоты (Тенофововир) последовательно с оксалил-хлоридом в диметилформамиде и потом с 1,3-бензотиазол-6-олом 2а в сульфолане с образованием моно-1,3-бензотиазолил {[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}фосфоната 3, переводом его в хлорангидрид моно-1,3-бензотиазолил {[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}фосфоната гидрохлорид 4 при помощи реакции с тионилхлоридом, взаимодействие последнего с изопропиловым эфиром L-аланина с получением изопропил N-[{[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}(1,3-бензотиазол-6-ил-окси)фосфорил]-L-аланината 5 и реакцией последнего с фумаровой кислотой с получением Изопропил N-[{[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}(1,3-бензотиазол-6-ил-окси)фосфорил]-L-аланинат фумарата 1.
Соединение формулы 1 по настоящему изобретению может являться пролекарством противовирусного препарата Тенофовир, обеспечивая высвобождение последнего после всасывания в желудочно-кишечном тракте, преимущетсвенно в печени под воздействием печеночных энзимов, а также в крови.
Предметом настоящего изобретения является способ лечения гепатита Б путем терапевтически эффективного количества соединения формулы 1 по настоящему изобретению или его фармацевтической композиции.
В соответствии с настоящим изобретением соединение формулы 1 может быть введено пациенту в виде фармацевтических композиций. Эти композиции могут быть получены способом, хорошо известным в фармацевтической области, и могут быть введены различными способами в зависимости от того, требуется местное или системное лечение и на площади, подлежащей обработке. Композиции могут быть в форме таблеток, пилюль, порошков, лепешек, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (как твердое вещество или в жидкой среде), мазей, содержащих, например, до 10% по весу активного соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных инъекционных растворов и стерильно упакованных порошков. Композиции могут быть приготовлены в виде единичной дозы, причем каждая доза содержит от примерно 5 мг до примерно 1000 мг, обычно предпочтительно от 100 мг до 500 мг, активного ингредиента. Термин "единичные дозы" относится к физически дискретным единицам, пригодным в качестве единичных доз для человека и других млекопитающих, причем каждая единица содержит заданное количество активного материала, рассчитанное на получение желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим наполнителем.
Активное соединение может быть эффективным в широком диапазоне доз и обычно вводится в фармацевтически эффективном количестве. Следует понимать, однако, что количество фактически принимаемого соединения обычно будет определяться лечащим врачом в соответствии с состоянием пациента, подлежащего лечению.
Ниже изобретение будет описано более подробно с помощью конкретных примеров, которые представлены с целью иллюстрации, и не предназначены для ограничения изобретения каким-либо образом. Специалисты в данной области техники легко поймут различие некритических параметров, которые могут быть изменены или модифицированы, чтобы получить те же результаты.
Пример 1. Метод синтеза изопропил N-[{[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}(1,3-бензотиазол-6-ил-окси)фосфорил]-L-аланинат фумарата (1)
Изопропил N-[{[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}(1,3-бензотиазол-6-ил-окси)фосфорил]-L-аланинат фумарат получали в соответствии со схемой 1.
Схема 1
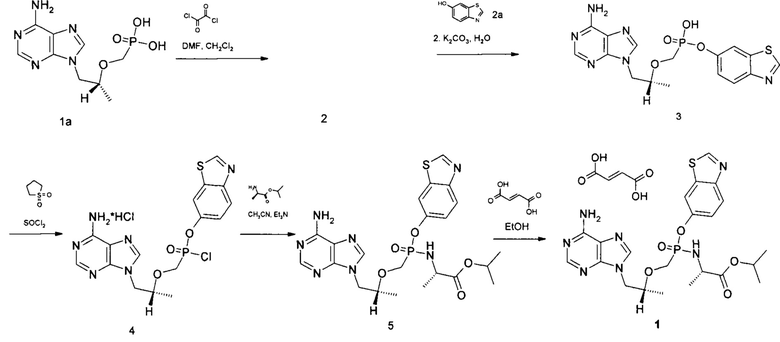
где 2 это хлорангидрид {[(1R)-2-(6-{[(1E)-(диметиламино)метилен]амино}-9H-пурин-9-ил)-1-метилэтокси]метил}фосфорной кислоты дигидрохлорид
Синтез хлорангидрида {[(1R)-2-(6-{[(1E)-(диметиламино)метилен]амино}-9H-пурин-9-ил)-1-метилэтокси]метил}фосфорной кислоты дигидрохлорид 2. К суспензии [(1R)-2-(6-амино-9Н-пурин-9-ил)-1-метилэтокси]метил фосфорной кислоты 1а (50.00 г., 0.174 моль) в абсолютном хлористом метилене (1000 мл), охлажденной до 10°С прибавили абсолютный диметилформамид (14.00 г., 0.191 моль), затем по каплям прибавили оксалил хлорид (88.34 г., 0.696 моль). По окончании прибавления реакционную смесь кипятили 16 часов, затем растворитель отогнали. Полученное белое кристаллическое вещество без дополнительной очистки использовали в следующей стадии, выход количественный. Спектр ЯМР 1Н (400 МГц, ДСМО-д6, δ, м.д., J/Гц): 1.07 (д, 3Н, J 5.9 Гц), 3.28 (с, 3Н), 3.40 (с, 3Н), 3.51-3.69 (м, 2Н), 3.74-3.89 (м, 1Н), 3.92-4.05 (м, 1Н), 4.25-4.36 (м, 1Н), 4.42-4.51 (м, 1Н), 6.72-7.788 (м, 2Н), 8.66 (с, 1Н), 9.59 (с, 1Н).
Синтез моно-1,3-бензотиазолил {[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}фосфоната 3. В 3-х горлую колбу, снабженную магнитной мешалкой, термометром и системой подачи аргона поместили хлорангидрид {[(1R)-2-(6-{[(1E)-(диметиламино)метилен]амино}-9H-пурин-9-ил)-1-метилэтокси]метил}фосфорной кислоты дигидрохлорид 2 (78.67 г., 0.174 моль) в абсолютном сульфолане (200 мл) и прибавили 1,3-
бензотиазол-6-ол 2а (31.57 г., 0.209 моль). Реакционную смесь перемешивали при 65-75°С на глицериновой бане в токе аргона в течение 12-16 часов. Полноту прохождения реакции контролировали методом ЛС-МС пробы. Затем реакционную смесь охладили до комнатной температуры и прибавили ацетон (400 мл), выпавший осадок отфильтровали, промыли на фильтре несколько раз ацетоном (по 100 мл). Отфильтрованный осадок перенесли в 2-х литровый стакан и растворили в воде (500 мл). Полученный темный раствор перемешивали при 60°С в течение 1 часа, затем прибавили 20% раствор гидроксида калия до рН 7, после этого прибавили к реакционной массе карбонат калия (48.02 г., 0.348 моль). Реакционную массу перемешивали при 70°С в течение 16-20 часов, контроль полноты прохождения реакции осуществляли методом ЛС-МС пробы. К охлажденной реакционной массе прибавляли соляную кислоту (конц) до рН 7, после этого отогнали при пониженном давлении воду и переупарили несколько раз реакционную смесь с сухим бензолом (2×100 мл).
К полученному густому темному остатку прибавили абсолютный метанол (200 мл) и отфильтровали выпавший неорганический осадок. Фильтраты собрали, концентрировали и нанесли на слой силикагеля (толщина 20 см), собирали фракцию с Rf 0.2 в системе хлороформ/метанол 1/1. Выделенные фракции упарили досуха, соединение 3 перекристаллизовали в системе ацетонитрил/метанол 10/1. Сушили при температуре 60°С на воздухе. Получили 51.7 г.(70.7%) белого кристаллического вещества. Спектр ЯМР 1Н (400 МГц, ДСМО-д6, δ, м.д., J/Гц): 0.94 (д, 3Н, J 6.2 Гц), 3.38-3.46 (м, 1H), 3.47-3.56 (м, 1Н), 3.85-3.94 (м, 1H), 4.08-4.16 (м, 1Н), 4.21-4.29 (м, 1Н), 7.07 (с, 2Н), 7.23 (д, 1Н, J 9.1 Гц), 7.82-7.87 (м, 2Н), 8.12 (с, 1Н), 8.26 (с, 1Н), 9.15 (с, 1Н).
Синтез хлорангидрида моно-1,3-бензотиазолил {[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}фосфоната гидрохлорида 4. В 3-х горлую колбу, снабженную магнитной мешалкой, термометром и системой подачи аргона поместили моно-1,3-бензотиазолил {[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}фосфонат 3 (51.7 г., 0.123 моль), прибавили абсолютный сульфолан (150 мл) и медленно по каплям прибавляли предварительно перегнанный хлористый тионил (58.54 г., 0.49 моль) с такой скоростью, чтобы температура реакционной смеси не превышала 60°С. По окончании прибавления всего количества хлористого тионила реакционную смесь перемешивали при 60°С в течение 10-12 часов (контроль за прохождением реакции осуществляли методом ЛС-МС обработанной абсолютным метанолом пробы). Избыток хлористого тионила отогнали при пониженном давлении. Полученное соединение 4 без дополнительной очистки использовали в следующей стадии.
Синтез изопропил N-[{[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}(1,3-бензотиазол-6-ил-окси)фосфорил]-L-аланинат 5. В 3-х горлую колбу, снабженную магнитной мешалкой, термометром и системой подачи аргона поместили суспензию хлорангидрида моно-1,3-бензотиазолил {[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}фосфоната гидрохлорида 4 (58.47 г., 0.123 моль) в абсолютном ацетонитриле (500 мл), свежеперегнанный изопропиловый эфир L-аланина (16.94 г., 0.129 моль). Охладили реакционную смесь до -40°С и в токе аргона прибавили триэтиламин (24.85 г., 0.246 моль), после этого довели температуру реакционной смеси до комнатной. Перемешивали реакционную смесь при комнатной температуре 30 минут, отфильтровали выпавший осадок, промыли его на фильтре тетрагидрофураном (3×100 мл). Фильтрат упарили при пониженном давлении, температура не должна превышать 30-35°С. Вещество выделяли методом колоночной хроматографии (силикагель, ТГФ-ТГФ/МеОН 10/1). Выделенные фракции объединили, упарили растворитель при пониженном давлении и температуре 20°С. Получили 34.12 г.(52%) бежевого кристаллического вещества. Спектр ЯМР 1Н (400 МГц, ДСМО-д6, δ, м.д., J/Гц): 1.00-1.18 (м, 12Н), 3.78-4.05 (м, 3Н), 4.13-4.35 (м, 2Н), 4.73-4.84 (м, 1Н), 5.55-5.76 (м, 1H), 7.20-7.32 (м, 3Н), 7.87 (д, 1Н, J 11.8 Гц), 8.04 (дд, 1Н, J1 8.9 Гц, J2 19.0 Гц), 8.13 (д, 1Н, J 1.6 Гц), 8.15 (д, 1H, J 3.4 Гц), 9.33 (с, 1Н).
Синтез изопропил N-[{[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}(1,3-бензотиазол-6-ил-окси)фосфорил]-L-аланинат фумарат 1. К раствору соединения изопропил N-[{[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}(1,3-бензотиазол-6-ил-окси)фосфорил]-L-аланинат 5 (34.12 г., 0.064 моль) в абсолютном этаноле (100 мл) при комнатной температуре прибавили фумаровую кислоту (7.42 г., 0.064 моль). Упарили растворитель при пониженном давлении и температуре 42°С. К твердому остатку прибавили холодный абсолютный этанол (25 мл), осадок отфильтровали, сушили в вакуумном шкафу. Получили 35.85 г. (86.3%) светло-бежевого кристаллического вещества. Спектр ЯМР 1Н (400 МГц, ДСМО-д6, δ, м.д., J/Гц): 1.00-1.18 (м, 12Н), 3.78-4.05 (м, 3Н), 4.13-4.35 (м, 2Н), 4.73-4.84 (м, 1Н), 5.55-5.76 (м, 1Н), 6.63 (с, 2Н), 7.20-7.32 (м, ЗН), 7.87 (д, 1Н, J 11.8 Гц), 8.04 (дд, 1Н, J1 8.9 Гц, J2 19.0 Гц), 8.13 (д, 1Н, J 1.6 Гц), 8.15 (д, 1Н, J 3.4 Гц), 9.33 (с, 1Н).
Пример 2. Ин-виво активность соединения формулы 1 в модели гепатита Б в утках.
Моделирование DHBV-инфекции.
Были сформированы 4 группы уток, по 10 животных в каждой. Все животные были заражены в 3-дневном возрасте внутрибрюшинно вирусом DHBV, штамм «Уфа-04», в дозе 0,2×106 ID50/животное. Через 3 недели у каждого животного была подтверждена DHBV-инфекция на основании выявления ДНК DHBV сыворотке крови методом полимеразной цепной реакции (ПЦР).
Моделирование курса противовирусной терапии в эксперименте Каждая из четырех групп животных соответствовала препарату/дозе: группа 1 - TDF в дозе 10 мг/кг веса животного; группа 2 - ZB98-0002 (вещество формулы 1) в дозе 10 мг/кг веса животного; группа 3 - ZB98-0002 (вещество формулы 1) в дозе 1,0 мг/кг животного, группа 4 - плацебо.
Водную суспензию препаратов ZB98-0002 (вещество формулы 1) и TDF (тенофовир дизопроксил фумарат - перпарат сравнения) готовили с использованием стерильной бидистиллированной воды. В качестве плацебо использовали воду, применявшуюся для приготовления суспензий ZB98-0002 и TDF (1 мл на животное).
Поскольку на момент начала эксперимента средняя масса животных составляла 1,0 кг, концентрация суспензии TDF составила 10 мг/мл, для достижения дозы 10 мг/кг при однократном введении 1 мл препарата в группе 1. Концентрация суспензии препарата ZB98-0002 составила 10 мг/мл для достижения дозы 10 мг/кг при однократном введении 1 мл препарата в группе 2; концентрация суспензии препарата ZB98-0002 составила 1 мг/мл для достижения дозы 1 мг/кг при однократном введении 1 мл препарата в группе 3.
Препараты вводили ежедневно на протяжении 20 дней перорально инсулиновым шприцем без иглы.
На протяжении эксперимента проводилась корректировка дозы с учетом массы тела каждого животного. Для этого с интервалом в семь дней взвешивали уток. С учетом увеличения массы тела животных меняли концентрацию препаратов в суспензии. Данные по изменению массы тела экспериментальных животных и корректировке дозы препаратов приведены в таблице 2.

В ходе эксперимента каждые 3 дня, начиная с дня, предшествующего началу курса приема препаратов, у уток проводили забор образцов крови - дни 0, 3, 6, 10, 13, 17 и 20. В образцах сыворотки крови определяли концентрацию ДНК DHBV методом ПЦР в реальном времени. Кровь у животных отбирали из яремной вены в объеме примерно 0,5 мл индивидуально в одноразовые шприцы и переносили кровь из шприцов в индивидуальные стерильные пробирки объемом 1,5 мл. Для отделения плазмы пробирки откручивали в центрифуге (4000 об./мин., 10 минут), отбирали сыворотку в криопробирки с завинчивающейся крышкой, маркировали пробирки и помещали образцы плазмы на хранение при -70°С. Затем из образцов сыворотки крови выделяли тотальные нуклеиновые кислоты и определяли концентрацию ДНК DHBV методом ПЦР.
Определение вирусной нагрузки DHBV.
Тотальную ДНК выделяли из 50 мкл сыворотки крови с помощью набора для выделения ДНК/РНК (НПФ Литех, Россия) по протоколу производителя. Количественное определение ДНК DHBV проводили методом ПЦР в реальном времени на анализаторе TaqMan 48 (Roche) с комплектом реагентов для ПЦР в реальном времени производства НПО «Синтол» по протоколу производителя. Для ПЦР в реальном времени использовали праймеры: DHBVf (5'- agcaatcactagaccaatcc -3'), DHBVr (5'- ccctaagtgatgcctagc -3') и зонд к консервативному участку вирусного генома, DHBVp (5'-FAM- ctctcccacgtagttcgagca-RTQ1-3'). В качестве стандарта для количественного определения ДНК DHBV использовали разведения плазмидной ДНК, несущей последовательность DHBV. Диапазон разведений стандарта составлял 103-1012 копий ДНК/мл.
Статистическая обработка данных.
Полученные значения вирусной нагрузки ДНК DHBV выражали в десятичных логарифмах копий ДНК в миллилитре (lg копий ДНК DHBV/мл). Для каждой группы в каждой точке (день терапии) определяли среднее значение вирусной нагрузки и стандартное отклонение (SD). Для оценки изменения вирусной нагрузки DHBV в ходе эксперимента полученные средние значения сравнивали с исходными значениями до начала терапии (день 0) для каждой группы, соответственно. Степень достоверности различий оценивали с помощью критерия Уилкоксона (Wilcoxon mathhed pairs test), рассчитанного с помощью программы Statistica 8.0. Статистически достоверными признавались различия при значении вероятности Р<0.05.
Изобретение относится к изопропил N-[{[(1R)-2-(6-амино-9H-пурин-9-ил)-1-метилэтокси]метил}(1,3-бензотиазол-6-ил-окси)фосфорил]-L-аланинат фумарату формулы 1. Соединение по изобретению обладает противовирусными свойствами по отношению к вирусам гепатита Б и предназначено для лечения вирусного гепатита Б. 4 н. и 1 з.п. ф-лы, 2 табл., 2 пр.
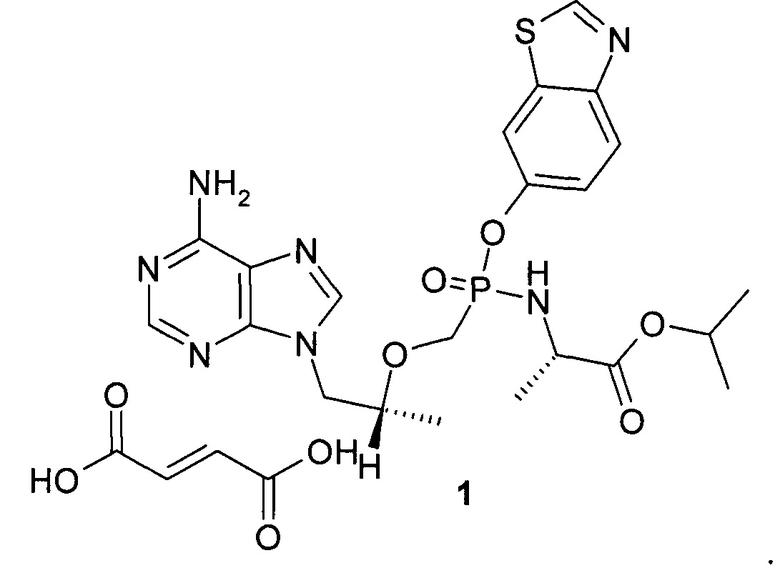
1. Соединение общей формулы 1
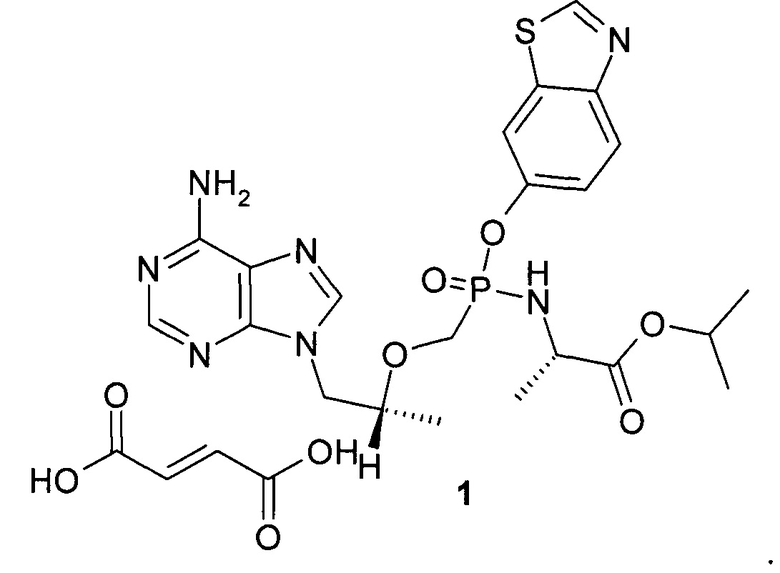
2. Фармацевтическая композиция, обладающая противовирусными свойствами по отношению к вирусам гепатита Б, содержащая в качестве активного компонента соединение общей формулы 1 по пп. 1 в терапевтически эффективном количестве.
3. Фармацевтическая композиция по п. 2 для использования в форме таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку.
4. Способ лечения вирусного гепатита Б, включающий введение указанному пациенту терапевтически эффективного количества соединения по пп. 1 или фармацевтической композиции по пп. 2 или 3.
5. Способ получения соединения общей формулы 1 по п. 1, включающий постадийное проведение реакций по схеме 1 в подходящем растворителе или смеси растворителей
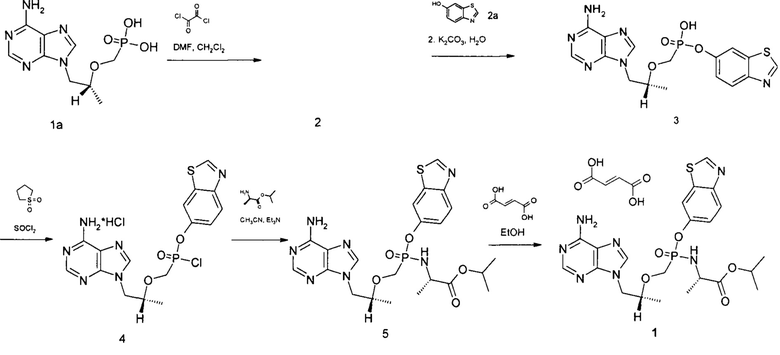
где 2 - это хлорангидрид {[(1R)-2-(6-{[(1E)-(диметиламино)метилен]амино}-9H-пурин-9-ил)-1-метилэтокси]метил}фосфорной кислоты дигидрохлорид.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Электрод-инструмент | 1949 |
|
SU81797A1 |

