Изобретение относится к новым физиологически активным веществам, новым ингибиторам рПОб - дельта-изоформы фосфоинозитид-3-киназы (PI3K), активным компонентам для фармацевтических композиций, к фармацевтическим композициям и лекарственным средствам, содержащим (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетат в качестве ингибитора р110δ - дельта-изоформы фосфоинозитид-3-киназы (PI3K), который может применяться при лечении фолликулярной лимфомы и других злокачественных лимфопролиферативных заболеваний из ряда неходжкинских лимфом.
Лимфомы неходжкинского типа обусловлены раковыми новообразованиями В- и Т- клеточного типа. Источник такой патологии способен зародиться в любом лимфатическом узле, либо другом органе, а затем «заразить» лимфу. В дальнейшем метастазирование происходит тремя путями: гематогенным, лимфогенным, гематогенно-лимфогенным. Название «Фолликулярная лимфома» возникло потому, что в большинстве случаев злокачественное новообразование начинало формироваться и развиваться в покрывных клетках эпидермиса, патологически изменяя волосяные луковицы, иначе называемые фолликулами. Группа риска - возрастная категория до сорока лет.
По типу локализации фолликулярные лимфомы делятся на нодальные - лимфатические саркомы, локализованные в лимфоузлах, и экстранодальные - в случае другого места локализации (слюнные железы, миндалины, щитовидная железа, эпидермис, головной мозг, легкие и так далее). Фолликулярными (нодулярными) или диффузными их делает структурная составляющая новообразования.
На сегодняшний день фолликулярная лимфома - наиболее часто встречающаяся форма индолентных (вялотекущих) лимфом. Каждый год в мире регистрируется более 75 тысяч человек, страдающих от этого злокачественного заболевания. В России с диагнозом «фолликулярная лимфома» ежегодно сталкиваются 3500 пациентов.
Опухоль при фолликулярной лимфоме состоит из зрелых В-лимфоцитов, которые образуются в фолликулярном центре лимфатического узла, в котором происходит рост и развитие лимфоцитов - основных клеток иммунной защиты человека.
Один из универсальных сигнальных путей, характерных для большинства клеток человека, отвечающий за уход от апоптоза, рост, пролиферацию клеток, метаболизм, представляет собой группу ферментов, которые являются компонентами внутриклеточного сигнального пути PI3K/AKT/mTOR, Активация данного пути наблюдается в подавляющем большинстве опухолей человека, с ним часто связано развитие устойчивости клеток опухоли, поэтому PI3K представляет собой перспективную мишень для противоопухолевой терапии..
Различают 4 изоформы PI3K: α, β, γ, and δ. Изоформы α и β распостранены во многих тканях организма, в то время как δ и γ более специфически экспрессированы в лейкоцитах. Таким образом, ингибирование этих изоформ представляет собой перспективный специфичный метод терапии различных типов гемобластозов, в том числе и фолликулярной лимфомы.
В последние годы показана высокая эффективность ингибиторов тирозинкиназ Btk (ибрутиниб и др.) и PI3K δ (иделалисиб и др.). Иделалисиб, послуживший прототипом данного изобретения, разрабатывался как препарат второй линии терапии, обычно для использования в комбинации с ритуксимабом, но также планировался к самостоятельному использованию также у пациентов, у которых терапия ритуксимабом противопоказана или неэффективна в связи с сопутствующими заболеваниями или непереносимостью ритуксимаба, а также для применения для лечения пациентов с рецидивом фолликулярной формы. К сожалению, в данный момент Иделалисиб отозван с рынка в связи высокой токсичностью. Поэтому превосходство безопасности соединения формулы I над прототипом является ее неоспоримым преимуществом.
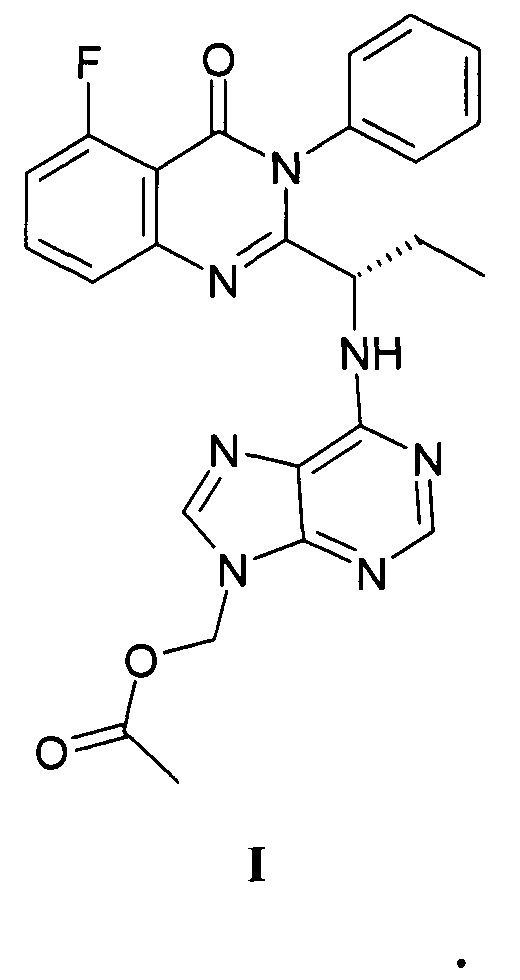
Было неожиданно обнаружено, что (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидро-хиназолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетат - соединение формулы I
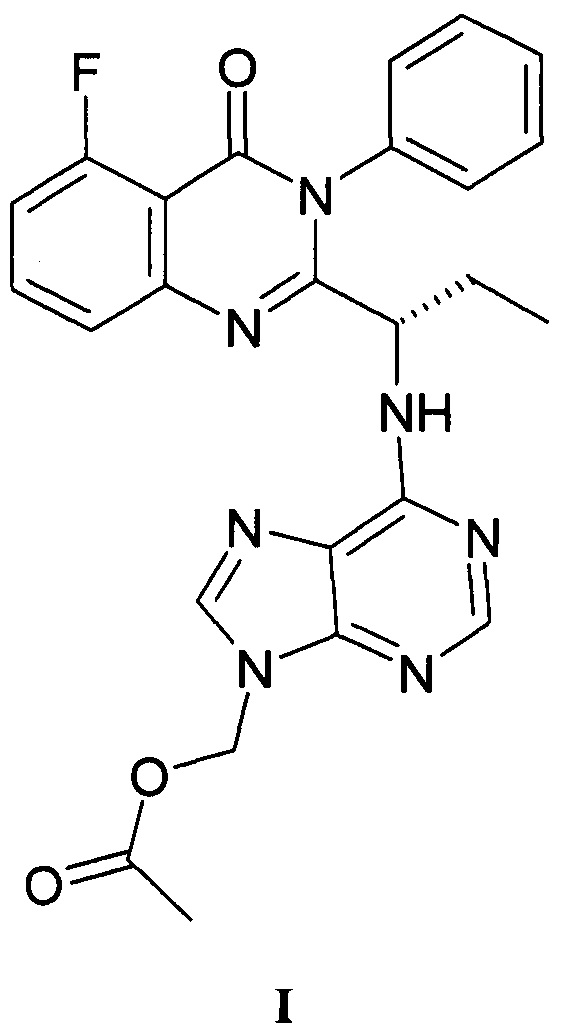
является эффективны ингибитором р110δ - дельта-изоформы фосфоинозитид-3-киназы (РБК) и может быть использовано для разработки лекарственных средств для лечения фолликулярной лимфомы и других неходжкинских лимфом.
Ниже приведены определения терминов, которые использованы в описании этого изобретения.
«Агонисты» означают соединения, которые, связываясь с рецепторами определенного типа, активно способствуют передаче этими рецепторами свойственного им специфического сигнала и тем самым вызывают биологический ответ клетки.
«Активный компонент» (лекарственное вещество, лекарственная субстанция, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Действующее вещество» (лекарственное вещество, лекарственная субстанция, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного средства.
«Ингибиторы» (лат. inhibere - задерживать) - общее название веществ, подавляющих или задерживающих течение физиологических и физико-химических (главным образом ферментативных) процессов, изучение ингибирования ферментов играет важную роль в создании лекарств, в изучении механизма действия и структуры ферментов,
«Лекарственное комбинированное средство (препарат)» - комбинация нескольких лекарственных веществ для одновременного использования в виде таблеток, капсул, инъекций, мазей, ректальных суспензий и гелей и др. готовых форм, предназначенный для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего. Лекарственные вещества в одном комплекте могут быть представлены в виде различных готовых форм, предназначенных для введения в организм животного или человека различными способами, например перорально и ректально.
«Рецепторы» (от латинского recipere - получать, узнавать) представляют собой биологические макромолекулы, расположенные на цитоплазматической мембране клетки или внутриклеточно, способные специфически взаимодействовать с ограниченным набором физиологически активных веществ (лигандов) и трансформировать сигнал об этом взаимодействии в определенный клеточный ответ.
«PI3K» - семейство ферментов, фосфорилирующих фосфатидилинозитол в положении 3D инозитольного кольца. Являются ключевым элементом PI3K сигнального пути. Фосфоинозитид-3-киназный путь является одним из универсальных сигнальных путей, характерных для большинства клеток человека. Он контролирует такие процессы, как: апоптоз, рост и пролиферация клеток, метаболизм. Гиперактивация фосфоинозитид-3-киназного пути в большинстве случаев приводит к развитию онкологических патологий. В этой связи, фосфоинозитид-3-киназы вызывают повышенный интерес как объекты противораковой терапии.
«Сигнальный каскад» (сигнальная система, каскад передачи сигнала) означает совокупность взаимосвязанных последовательных и параллельных молекулярных процессов регуляции клеточного метаболизма внешними (первичными) сигналами, несущими в клетку информацию, что принципиально отличает их от других поступающих в клетку химических соединений, служащих для нее источником материи и энергии. Молекулярные механизмы передачи (трансдукции) внешних сигналов в клетку подразумевают не только передачу сигналов как таковую, но и весь комплекс событий, с ней сопряженных, в том числе усиление, ослабление и подавление (или выключение) сигналов.
«Фармацевтическая композиция» обозначает композицию, включающую в себя соединение общей формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как, консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, алгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения. Фармацевтические композиции, как правило, получают с помощью стандартных процедур, предусматривающих смешение активного соединения с жидким или тонко измельченным твердым носителем. Для изготовления суппозиториев помимо активных компонентов используют также масло какао, сплавы его с парафином и гидрогенизированными жирами, растительные и животные гидрогенизированные жиры, твердый жир, ланоль, сплавы гидрогенизированных жиров с воском, твердым парафином и другие основы, разрешенные для медицинского применения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные. (Подробное описание свойств таких солей дано в Berge S.М., et а1., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как, холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
«Фармацевтически приемлемые эксципиенты» под фармацевтически приемлимыми эксципиентами подразумеваются применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или носители.
Цель настоящего изобретения заключается в создании новых ингибиторов р110δ - дельта-изоформы фосфоинозитид-3-киназы как потенциальных препаратов для лечения фолликулярной лимфомы.
Поставленная цель достигается (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохин-азолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетатом формулы I
Предметом настоящего изобретения является новый ингибитор р110δ - дельта-изоформы фосфоинозитид-3-киназы, представляющий собой соединение формулы I.
Предметом данного изобретения является способ получения (6-{[(1S)-1(5-фтор-4-оксо-З -фенил-3,4-дигидрохин-азолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетата формулы I исходя из 2-фтор-6-нитробензойной кислоты (2), (2S)-2-[(трет-бутоксикарбонил)амино]бутановой кислоты (4) и 6-хлор-9H-пурина (8). (схема 1).
Схема 1
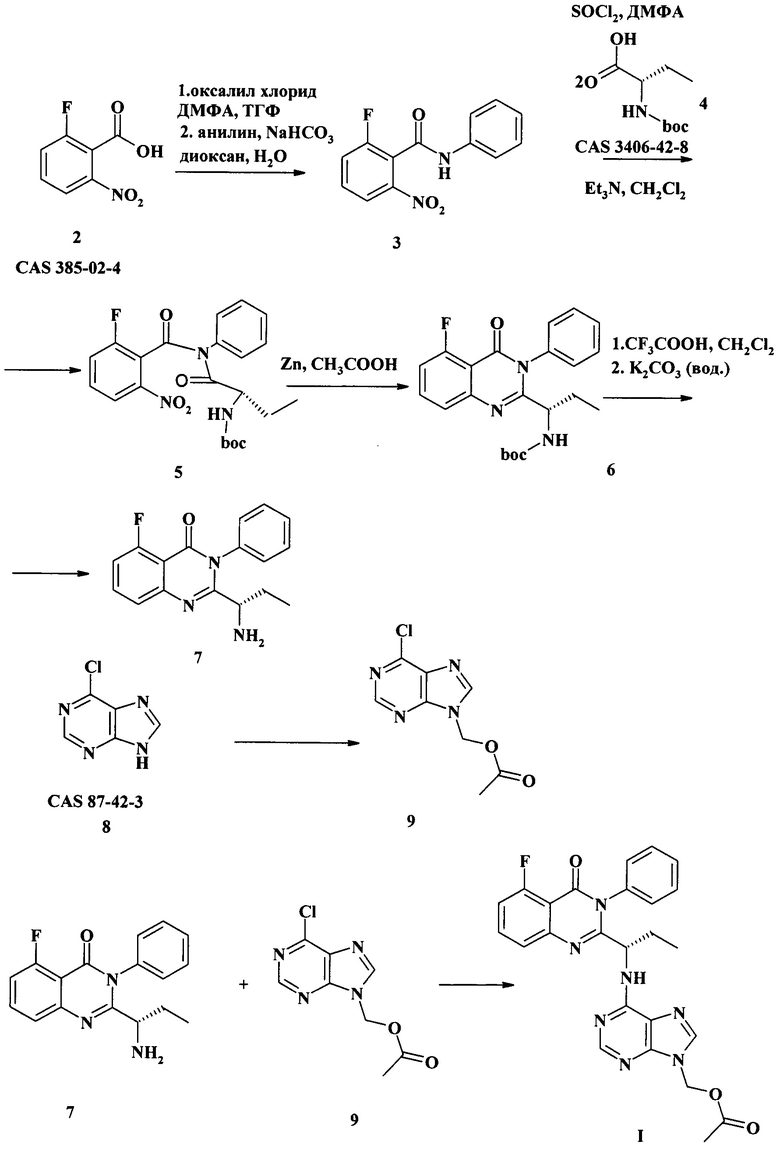
Схема включает получение вторичного анилида фторнитробензойной кислоты, который в условиях активации вторично амидируется (2S)-2-[(трет-бутоксикарбонил)амино]-бутановой кислотой (4). Полученый интермедиат циклизуется с обрзованием ключевого интермедиата - амин-замещенного хиназолинонового цикла (7). 6-хлор-9H-пурин (8) модифицируется по имидазольному азоту последовательным воздействием ацетилхлорида и формальдегида с образованием хлорида (9) - второго ключевого интермедиата. Каплинг интермедиатов 7 и 9 приводит к образованию (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохин-азолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетата формулы I.
Предпочтительный вариант способа получения предусматривает следующие этапы:
а) к раствору 2-фтор-6-нитробензойной кислоты в смеси дихлорметана и диметилформамида добавляют раствор оксалил хлорида в дихлорметане. Затем реакционную массу перерастворяют в диоксане и при охлаждении добавляют суспензию анилина и K2CO3 с образованием осадка 2-фтор-6-нитро-N-фенилбензамида;
б) раствор полученного на предыдущей стадии 2-фтор-6-нитро-N-фенилбензамида в диметилформамиде обрабатывают тионилхлоридом при нагревании, а затем (2S)-2-[(трет-бутоксикарбонил)амино]бутановой кислотой в дихлорметане при 10°С. Реакционную массу отфильтроваывают, и маточный раствор хроматографируют с образованием третбутилового эфира (S)-[1-(2-фтор-6-нитробензоил)-фенил-аминокарбонил]-пропил-карбаминовой кислоты
в) третбутиловый эфир (S)-[1-(2-фтор-6-нитробензоил)-фенил-аминокарбонил]-пропилкарбаминовой кислоты с предыдущей стадии растворяют в уксусной кислоте в присутствии порошка цинка. Реакционную массу отфильтровывают, концентрируют, продукт - третбутиловый эфир (S)-[1-(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)-пропил]-карбаминовой кислоты - экстрагируют и очищают хроматографически.
г) третбутиловый эфир (S)-[1-(5-фтор-4-оксо-3-фенил-3,4-дигидро-хиназолин-2-ил)-пропил]-карбаминовой кислоты с предыдущей стадии расторяют в дихлорметане и циклизуют под воздействием трифторуксусной кислоты. Реакционную массу нейтрализуют и концентрируют с образованием (S)-2-(1-аминопропил)-5-фтор-3-фенил-3H-хиназолин-4-она.
д) Смесь 6-хлор-9H-пурина параформа, уксусной кислоты и уксусного ангидрида нагревают в толуоле, затем отфильтровывают и концентрируют с образованием кристаллического 6-хлор-9H-пурин-9-ил)метил ацетата.
е) (S)-2-(1-Аминопропил)-5-фтор-3-фенил-3H-хиназолин-4-он и (6-Хлор-9H-пурин-9-ил)метил ацетат растворяют в третбутаноле в присутствии диизопропилэтиламин (ДИПЭА) при нагревании. Реакционную смесь концентрируют и выделяют продукт - (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохин-азолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетат хроматографически.
Схема 2
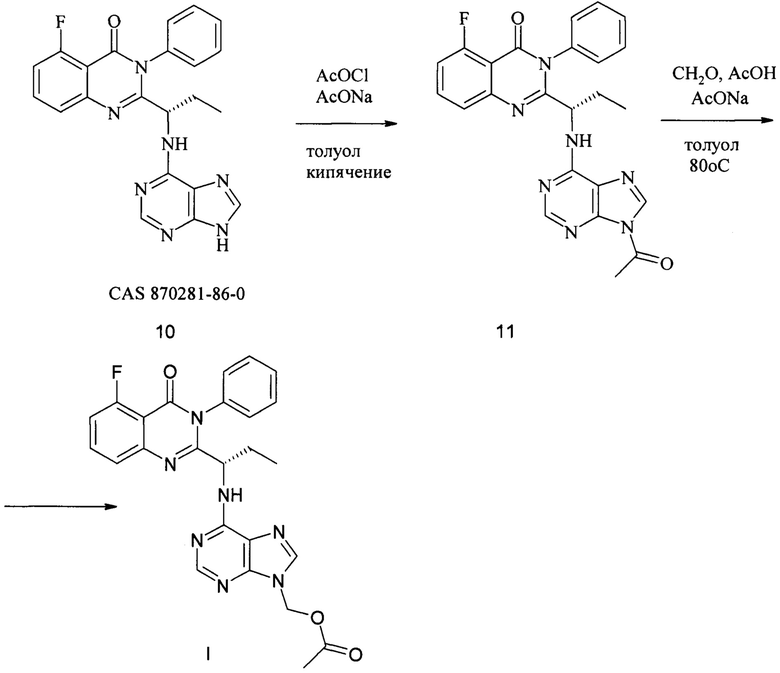
а) к раствору ацетилхлорида в ацетонитриле добавляют 5-фтор-3-фенил-2-[(1S)-1-(9Н-пурин-6-иламино)пропил]хиназолин-4(3H)-он с образованием осадка 2-{(1S)-1-[(9-ацетил-9Н-пурин-6-ил)амино]пропил}-5-фтор-3-фенилхиназолин-4(3H)-она;
б) к толуольному раствору 2-{(1S)-1-[(9-ацетил-9Н-пурин-6-ил)амино]пропил}-5-фтор-3-фенилхиназолин-4(3H)-она, полученного на этапе а) добавляют параформ. По окончании взаимодействия продукт - (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохин-азолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетата формулы I - выделяют последовательным концентрированием, хроматографией и рядом перекристаллизаций из подходящих растворителей.
Конкретные режимы/ условия осуществления способа получения могут быть подобраны специалистом эксперементальным путем.
Предметом настоящего изобретения является активный компонент обладающий свойством ингибитора р110δ - дельта-изоформы фосфоинозитид-3-киназы (PI3K), представляющий собой соединение (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохин-азолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетат формулы I.
Предметом данного изобретения является фармацевтическая композиция, предназначенная для лечения фолликулярной лимфомы и других злокачественных лимфопролиферативных заболеваний из ряда неходжкинских лимфом, содержащая в эффективном количестве активный компонент обладающий свойством ингибитора р110δ - дельта-изоформы фосфоинозитид-3-киназы (PI3K). Фармацевтическая композиция может быть выполнена в форме таблеток, капсул и инъекций, помещенных в фармацевтически приемлемую упаковку.
Фармацевтическая композиция может включать фармацевтически приемлемые эксципиенты. Под фармацевтически приемлемыми эксципиентами подразумеваются применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или носители. Фармацевтическая композиция наряду с активным компонентом по настоящему изобретению может включать и другие активные ингредиенты, при условии, что они не вызывают нежелательных эффектов, например, аллергических реакций.
При необходимости использования фармацевтических композиций по настоящему изобретению в клинической практике они могут смешиваться для изготовления различных форм, при этом они могут включать в свой состав традиционные фармацевтические носители; например, пероральные формы (такие как, таблетки, желатиновые капсулы, пилюли, растворы или суспензии); формы для инъекций (такие как, растворы или суспензии для инъекций, или сухой порошок для инъекций, который требует лишь добавления воды для инъекций перед использованием); местные формы (такие как, мази или растворы).
Носители, используемые в фармацевтических композициях по настоящему изобретению, представляют собой носители, которые применяются в сфере фармацевтики для получения распространенных форм, в том числе: в пероральных формах используются связующие вещества, смазывающие агенты, дезинтеграторы, растворители, разбавители, стабилизаторы, суспендирующие агенты, бесцветные агенты, корригенты вкуса; в формах для инъекций используются антисептические агенты, солюбилизаторы, стабилизаторы; в местных формах используются основы, разбавители, смазывающие агенты, антисептические агенты.
Предпочтительно фармацевтическая композиция, содержит в эффективном количестве соединение формулы I, в количестве 25 мг, и фармацевтически приемлемые добавки: целлюлоза микрокристаллическая в количестве 35,0 мг, лактозы моногидрат 26,0 мг, гидроксипропилцеллюлоза в количестве 5 мг, натрия карбоксиметилкрахмал в количестве 4 мг, натрия кроскармелоза в количестве 4 мг, магния стеарат, в количестве 1 мг, а также Опадрай II белый (поливиниловый спирт - 60%, титана диоксид - 25%, полиэтиленгликоль -10%, тальк - 5%) в количестве 3 мг.
Новая фармацевтическая композиции может быть получена смешением с инертным наполнителем и/или растворителем активного компонента, представляющего собой, соединение формулы I.
Предметом данного изобретения является лекарственное средство, содержащее соединение (6-{[(1S)-1(5-фтор-4-оксо-3 -фенил-3,4-дигидрохиназолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетат формулы I в качестве активного компонента.
Лекарственные средства могут вводиться перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно, местно или ректально). Клиническая дозировка активного компонента (субстанции), фармацевтической композиции или лекарственного комбинированного средства, включающих фармацевтически эффективное количество активного компонента, у пациентов может корректироваться в зависимости от терапевтической эффективности и биодоступности активных ингредиентов в организме, скорости их обмена и выведения из организма, а также в зависимости от возраста, пола и стадии заболевания пациента, при этом суточная доза у взрослых обычно составляет 1-300 мг, предпочтительно 50-100 мг. Поэтому во время приготовления фармацевтических композиций по настоящему изобретению в виде единиц дозировки необходимо учитывать вышеназванную эффективную дозировку, при этом каждая единица дозировки препарата должна содержать 1-300 мг, предпочтительно - 5-100 мг. В соответствии с указаниями врача или фармацевта данные препараты могут приниматься несколько раз в течение определенных промежутков времени (предпочтительно - от одного до шести раз).
Предметом данного изобретения является способ лечения фолликулярной лимфомы и других злокачественных лимфопролиферативных заболеваний из ряда неходжкинских лимфом введением в эффективном количестве нового активного компонента или новой фармацевтической композиции или нового лекарственного средства.
Предметом данного изобретения является применение соединения формулы I для получения лекарственного средства для лечения фолликулярной лимфомы и других злокачественных лимфопролиферативных заболеваний из ряда неходжкинских лимфом.
Предметом данного изобретения является способ ингибирования р110δ - дельта-изоформы фосфоинозитид-3-киназы (PI3K) путем введения в эффективном количестве соединения формулы I нуждающемуся в этом реципиенту
Также заявленное изобретение относится к соединению формулы I для применения в способе лечения фолликулярной лимфомы и других злокачественных лимфопролиферативных заболеваний из ряда неходжкинских лимфом.
Представленные ниже примеры иллюстрируют, но не ограничивают изобретение.
Структуры полученных соединений подтверждались данными химического, хроматографического и спектрального анализа.
Пример 1. Получение 2-фтор-6-нитро-N-фенилбензамида (3)
К раствору 2-фтор-6-нитробензойной кислоты (10 г, 54 ммоль) (2) в смеси дихлорметана (60 мл) и диметилформамида (0,5 мл) с помощью капельной воронки добавляют раствор оксалил хлорида в дихлорметане (2М, 41 мл, 80 ммоль, 1,5 экв.). Реакционную массу перемешивают в течение 2 часов при комнатной температуре, после чего растворители отгоняют на роторном испарителе. Остаток растворяют в диоксане (8 мл), охлаждают до 6°С и медленно добавляют суспендированные в водном растворе диоксана (25 мл:25 мл) анилин (4,9 мл, 54 ммоль, 1 экв) и K2CO3 (9 г, 108 ммоль, 2 экв.). Температура реакционной массы при этом поднимается до 27°С. Реакционную массу перемешивают в течение 30 минут, после чего добавляют 120 мл воды. Образовавшийся осадок отфильтровывают, промывают водой, гексаном, высушивают и получают 2-фтор-6-нитро-N-фенилбензамид с выходом 93% (13 г.)
Спектр ЯМР 1Н (400.13 МГц, ДМСО-d6, δ, м.д., J/Гц): 10.82 (с 1Н), 8.12 (д, J=7.7 Hz, 1Н), 7.91-7.77 (м, 2Н), 7.64 (д, J=7.7 Hz, 2Н), 7.38 (т, J=7.9 Hz, 2Н), 7.15 (т, J=7.4 Hz, 1Н);
LCMS: один пик с временем удерживания 4.36 мин. (М+Н+=261) с содержанием вещества 99.3…99.9% по УФ-детекторам и 96.2% по ELSD.
Пример 2 Получение третбутилового эфира (S)-[Т-(2-фтор-6-нитробензоил)-фенил-аминокарбонил]-пропилкарбаминовой кислоты (5)
К 2-Фтор-6-нитро-М-фенилбензамиду (3) (50 ммоль) добавляют диметилформамид (0,5 мл), тионилхлорид (25,6 мл, 250 ммоль, 5 экв) и перемешивают реакционную массу в течение 5 часов при 85°С, после чего избыток тионилхлорида отгоняют на роторном испарителе. К остатку, растворенному в дихлорметане (20 мл), медленно прибавляют раствор (2S)-2-[(трет-бутоксикарбонил)амино]бутановой кислоты (4) (11,2 г, 55 ммоль, 1,1 экв) и триэтиламина (7,7 мл, 0,55 моль, 1,1 экв) в дихлорметане (60 мл) при 10°С. Реакционную массу перемешивают при комнатной температуре в течение 3 часов, нерастворимый осадок отфильтровывают, маточный раствор промывают водой, раствором бикарбоната натрия, 5% раствором лимонной кислоты и раствором хлорида натрия. Органический слой подсушивают с помощью сульфата магния и растворитель отгоняют на роторном испарителе. Продукт выделяют методом колоночной хроматографии, используя раствор гексан : этилацетат (10%-25%) в качестве элюента. Выход продукта составляет 66% (14,7 г).
Спектр ЯМР 1Н (400.13 МГц, ДМСО-d6, δ, м.д., J/Гц): 8.13 (д, J=8.0 Hz, 1Н), 7.84 (т, J=8.6 Hz, 1Н), 7.78-7.67 (м, 1Н), 7.65-7.49 (м, 3H), 7.40-7.28 (м, 2Н), 7.19 (д, J=7.5 Hz, 1Н), 4.05 (шс 1Н), 1.75-1.30 (м, 2Н), 1.34 (с 9Н), 0.93 (шс 3H).
LCMS: один пик с временем удерживания 6.96 мин. (М+Н+=446.3) с содержанием вещества 99.3…99.9% по УФ-детекторам и 96.2% по ELSD.
Пример 3. Получение третбутилового эфира (S)-[1-(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)-пропил]-карбаминовой кислоты (6)
К раствору третбутилового эфира (S)-[1-(2-фтор-6-нитробензоил)-фенил-аминокарбонил]-пропилкарбаминовой кислоты (5) (12,5 ммоль, 1 экв) в уксусной кислоте (50 мл) добавляют порошок цинка (4,8 г, 74 ммоль, 6 экв). Реакционную массу перемешивают в течение 2 часов при комнатной температуре, осадок отфильтровывают и промывают уксусной кислотой. Маточный раствор упаривают на роторном испарителе, остаток растворяют в этилацетате (400 мл), раствор промывают водой и водный слой экстрагируют с помощью этилацетата. Органические слои объединяют, промывают водой, раствором бикарбоната натрия, раствором хлорида натрия и подсушивают с помощью сульфата магния. Растворитель отгоняют на роторном испарителе, а продукт выделяют из остатка методом колоночной хроматографии, используя раствор гексан : этилацетат (10%-25%) в качестве элюента. Выход продукта составляет 69% (3,3 г).
Спектр ЯМР 1Н (400.13 МГц, CDCl3, δ, м.д., J/Гц): 7.66-7.75 (м, 1H), 7.50-7.64 (м, 4Н), 7.38 (м, 1Н), 7.29 (м, 1H), 7.13 (т, J=9 Hz, 1Н), 5.46 (м, 1Н), 4.42 (м, 1Н), 1.68-1.8 (м, 1Н), 1.52-1.62 (м, 1Н), 1.41 (с 9Н), 0.77 (т, J=7.3 Hz, 3H).
LCMS один пик с временем удерживания 13.00 мин. (М+Н+=398.3) с содержанием вещества 96.4…99.3% по УФ-детекторам и 83.9% по ELSD.
Пример 4. Получение (S)-2-(1 -аминопропил)-5-фтор-3-фенил-3H-хиназолин-4-она (7)
К раствору третбутилового эфира (S)-[1-(5-фтор-4-оксо-3-фенил-3,4-дигидро-хиназолин-2-ил)-пропил]-карбаминовой кислоты (6) (8,5 ммоль) в дихлорметане (6 мл) добавляют трифторуксусную кислоту (6 мл). Реакционную массу перемешивают в течение 1 часа при комнатной температуре, после чего разбавляют дихлорметаном (15 мл), и органический слой промывают 10%-ным водным раствором бикарбоната калия. Водный слой экстрагируют дихлорметаном и объединенные органические слои промывают водой, раствором хлорида натрия и сушат с помощью сульфата магния. Растворитель отгоняют на роторном испарителе и получают продукт с выходом 88% (2,2 г).
Спектр ЯМР 1Н (400.13 МГц, CDCl3, δ, м.д., J/Гц): 7.73-7.65 (м, 1Н), 7.62-7.49 (м, 4Н), 7.32-7.22 (м, 2Н), 7.13-7.06 (м, 1Н), 3.42 (дд, J=7.5, 5.2 Hz, 1Н), 1.87-1.70 (м, 1Н), 1.58-1.43 (м, 1H), 0.80 (т, J=7.4 Hz, 3H).
LCMS один пик с временем удерживания 7.00 мин. (М+Н+=298.2) с содержанием вещества 96.4…99.3% по УФ-детекторам и 83.9% по ELSD.
Пример 5. Получение (6-Хлор-9H-пурин-9-ил)метил ацетата (9)
Смесь 6-хлор-9H-пурина 8 (300 мг, 2 ммоль), параформа (120 мг, 4 ммоль, 2 экв), уксусной кислоты (0,12 г, 2 ммоль, 1 экв) и уксусного ангидрида (400 мг, 4 ммоль, 2 экв) растворяют в толуоле (10 мл). Реакционную массу перемешивают при 80°С в течение 5 часов, после чего охлаждают до комнатной температуры. Выпавший осадок отфильтровывают, маточный раствор упаривают на роторном испарителе, получая светло-желтые кристаллы продукта с выходом 34% (150 мг).
Спектр ЯМР 1Н (400.13 МГц, CDCl3, δ, м.д., J/Гц): 2.13 (с 3H), 6.21 (с 2Н), 8.4 (с 1Н), 8.84 (с 1Н).
Пример 6. Получение (6-{[(1S)-1-(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)пропил]амино}-9H-пурин-9-ил)метил ацетата (1).
(S)-2-(1-Аминопропил)-5-фтор-3-фенил-3H-хиназолин-4-он 7 (300 мг, 1 ммоль) растворяют в третбутаноле (4 мл), добавляют (6-Хлор-9H-пурин-9-ил)метил ацетат 9 (1,1 ммоль, 1,1 экв), а также диизопропилэтиламин (ДИПЭА) (260 мг, 2 ммоль, 2 экв). Реакционную массу нагревают до 80°С и перемешивают при этой температуре в течение 24 часов, после чего охлаждают, а растворитель отгоняют на роторном испарителе. Технический продукт растворяют в 5 мл хлористого метилена и наносят на 40 г силикагеля, разведенного и уплотненного с 90 мл хлористого метилена. Смывают смесью хлористый метилен - тетрагидрофуран (10:1) в количестве 0,3 л. Первую фракцию объемом 100 мл отбрасывают. Затем собирают фракцию объемом 200 мл, содержащую продукт. Фракции, содержащие продукт, упаривают на роторном испарителе и получают бесцветное масло. К продукту прибавляют 5 мл этилового спирта и перемешивают в течение часа при комнатной температуре. Выпавший продукт отфильтровывают. Продукт сушат в вакуумном сушильном шкафу при 45°С и остаточном давлении 10±5 мм рт.ст. в течение 4 часов. Выход продукта (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)пропил]амино}-9H-пурин-9-ил)метил ацетата 8 составлял 13% (12 мг).
Спектр ЯМР 1Н (400.13 МГц, CDCl3, δ, м.д., J/Гц): 0.84 (т, 3H), 1.75-1.82 (м, 1Н), 1.9-1.398 (м, 1Н), 2.08 (с 3H), 5.18 (шс, 1Н), 6.09 (з, 2Н), 6.74 (шс, 1H), 7.1 (т, 1Н), 7.34 (д, 1H), 7.44-7.61 (м, 5Н), 7.65 (м, 1Н), 8.04 (шс 1Н), 8.33 (с 1Н).
LCMS один пик с временем удерживания 5.50 мин. (М+Н+=487.9) с содержанием вещества 96.4…99.3% по УФ-детекторам и 99.9% по ELSD.
Пример 7. Получение 2-{(1S)-1-[(9-ацетил-9Н-пурин-6-ил)амино]пропил}-5-фтор-3-фенилхиназолин-4(3H)-она (11)
Смесь 45 мл толуола, 5-фтор-3-фенил-2-[(1S)-1-(9Н-пурин-6-иламино)пропил]-хиназолин-4(3H)-она 10 (1, 5 г, 3,6 ммоль), уксусного ангидрида (0.55 г, 5,4 ммоль, 1,5 экв) и безводного ацетата натрия (0.59 г, 7,2 ммоль, 2 экв) кипятят с обратным холодильником в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры и промывают дважды водой (порциями по 10 мл). Растворитель отгоняют на роторном испарителе и образовавшийся осадок отфильтровывают, промывают эфиром и сушат на воздухе до постоянной массы. Выход продукта 2-{(1S)-1-[(9-ацетил-9Н-пурин-6-ил)амино]пропил}-5-фтор-3-фенилхиназолин-4(3H)-он 11 составлял 92% (1,5 г).
Спектр ЯМР 1Н (400.13 МГц, ДМСО-d6, δ, м.д., m/Гц): 0.75 (т, 3H), 1.92 (м, 2Н), 2.86 (с 3H), 4.68 (м, 1Н), 7.28 (т, 1H), 7.42-7.45 (м, 3H), 7.5-7.6 (м, 3H), 8.24-8.30 (м, 1Н), 8.31 (с 1Н), 8.66 (с 1Н).
Пример 8. Получение (6-{[(1S)-1-(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)пропил]амино}-9H-пурин-9-ил)метил ацетата (1).
2-{(1S)-1-[(9-ацетил-9Н-пурин-6-ил)амино]пропил}-5-фтор-3-фенилхиназолин-4(3H)-он 11 (1,4 г, 3 ммоль), уксусную кислоту (0,18 г, 3 ммоль, 1 экв), параформ (0,18 г, 6 ммоль, 2 экв) и безводный ацетат натрия (0,25 г, ммоль, 1 экв) растворяют в 30 мл толуола. Реакционную массу перемешивают при 80°С в течение 5 часов, после чего охлаждают, а растворитель отгоняют на роторном испарителе. Технический продукт растворяют в 5 мл хлористого метилена и наносят на 40 г силикагеля, разведенного и уплотненного с 90 мл хлористого метилена. Смывают смесью хлористый метилен - тетрагидрофуран (10:1) в количестве 0,3 л. Первую фракцию объемом 100 мл отбрасывают. Затем собирают фракцию объемом 200 мл, содержащую продукт. Фракции, содержащие продукт, упаривают на роторном испарителе и получают бесцветное масло. К продукту прибавляют 5 мл этилового спирта и перемешивают в течение часа при комнатной температуре. Выпавший продукт отфильтровывают. Продукт сушат в вакуумном сушильном шкафу при 45°С и остаточном давлении 10±5 мм рт.ст. в течение 4 часов. Выход продукта (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)пропил]амино}-9H-пурин-9-ил)метил ацетата 8 составляет 73% (1,1 г).
Спектр ЯМР 1Н (400.13 МГц, CDCl3, δ, м.д., J/Гц): 0.84 (т, 3H), 1.75-1.82 (м, 1Н), 1.9-1.398 (м, 1Н), 2.08 (с 3H), 5.18 (шс, 1Н), 6.09 (з, 2Н), 6.74 (шс, 1Н), 7.1 (т, 1H), 7.34 (д, 1Н), 7.44-7.61 (м, 5Н), 7.65 (м, 1Н), 8.04 (шс 1Н), 8.33 (с 1Н).
LCMS один пик с временем удерживания 5.50 мин. (М+Н+=487.9) с содержанием вещества 96.4…99.3% по УФ-детекторам и 99.9% по ELSD.
Пример 9. Исследование активности in vivo.
Противоопухолевая активность соединения формулы 1 была изучена ин виво в модели В-клеточной лимфомы человека. В качестве препарата сравнения использован Иделалисиб. Для формирования модели лимфомы иммунодефицитным мышам линии SCID подкожно имплантировали клетки В-клеточной лимфомы линии Ramos. После формирования опухолей животных рандомизировали по группам (9 мышей в группе) и начинали лечение. Соединение формулы 1 вводили внутрижелудочно в дозах 10, 25, 50 и 100 мг/кг, Иделалисиб - в дозе 50 мг/кг, контрольной группе животных вводили растворитель, объем введения - 10 мг/кг. Наблюдение за животными продолжалось в течение 28 дней после инъекции клеток. В процессе исследования следили за поведением и состоянием животных, весом и смертностью животных.
Показано, что соединение формулы 1 дозозависимо замедляло рост опухолей. Максимального эффекта удалось достичь в дозе 100 мг/кг. При этом противоопухолевый эффект соединения формулы 1 в дозе 50 мг/кг был сравним с эффектом Иделалисиба в эквивалентной дозе.
Пример 10. Исследование субхронической токсичности in vivo
Целью исследования являлась оценка хронической токсичности соединения формулы I при его ежедневном внутрижелудочном введении в дозе, аналогичной терапевтической для Иделалисиба (ТД), 5-ти и 10-тикратной ТД крысам обоего пола - а именно в дозах 25 мг/кг, 50 мг/кг и 100 мг/кг в течение 3-х месяцев с последующим периодом наблюдения в течение 1 месяца после окончания введения.
Основная задача доклинического изучения хронической токсичности состояла в том, чтобы установить выраженность токсического действия и переносимость препарата при его хроническом введении крысам по сравнению с Иделалисибом.
В Pharmacology review of Idelalisib (Мау 1, 2014) сообщается, что в исследовании 28-дневной токсичности на крысах внутрижелудочное введение Иделалисиба в дозах 100 и 150 мг/кг/сутки приводило к гибели животных (3 смерти в дозе 100 и 5 смертей в дозе 150 мг/кг). Таргетными органами токсичности были зафиксированы костный мозг, сердце, печень и семенники.
В 3-мес исследовании токсичности на крысах использование Иделалисиба в дозе 90 мг/кг вело к увеличению веса сердца. Таргетными органами токсичности были сердце, поджелудочная железа, язык и семенники
Внутрижелудочное введение соединения формулы I в дозе 100 мг/кг в течение трех месяцев не вызывало токсических эффектов и гибели животных, что говорит о меньшей токсичности соединения формулы I по сравнению с Иделалисибом при одинаковом пути введения в одинаковых дозах.
Детализированные результаты исследования токсичности соединения формулы I
Ниже приводятся результаты макроскопических и микроскопических исследований внутренних органов крыс, полученных в эксперименте.
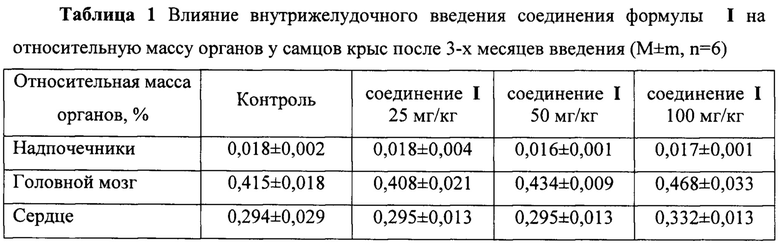
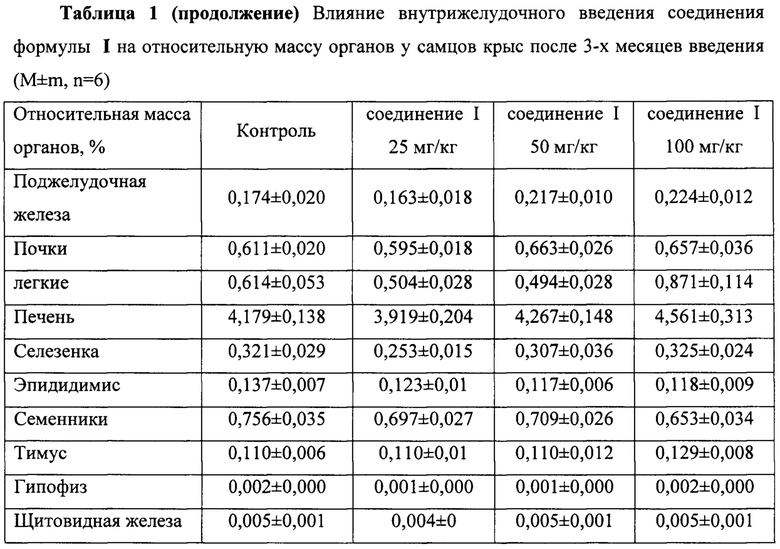
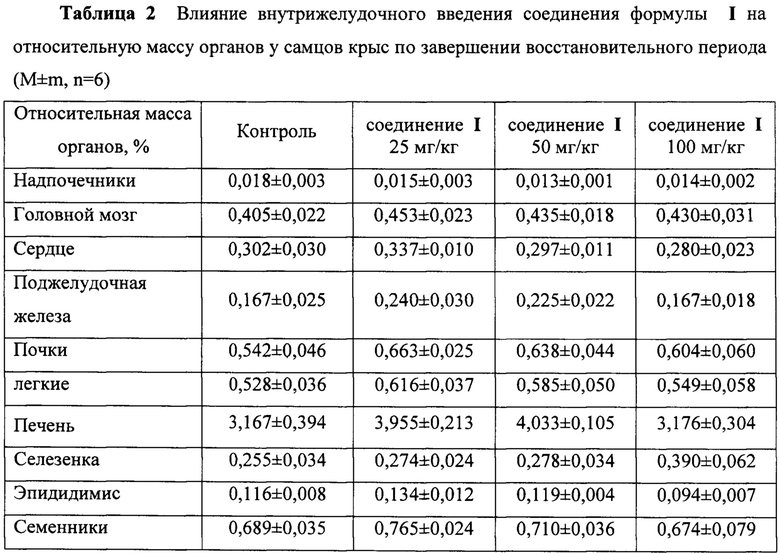
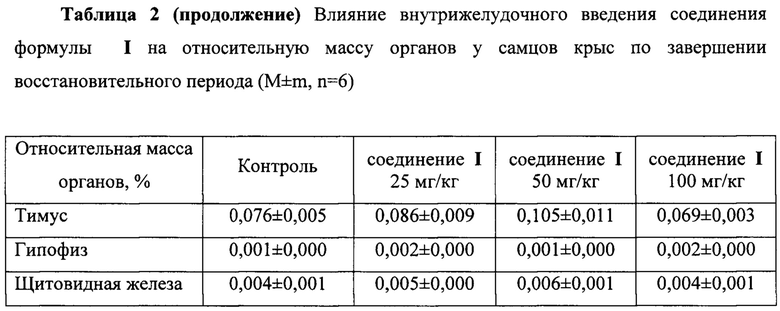
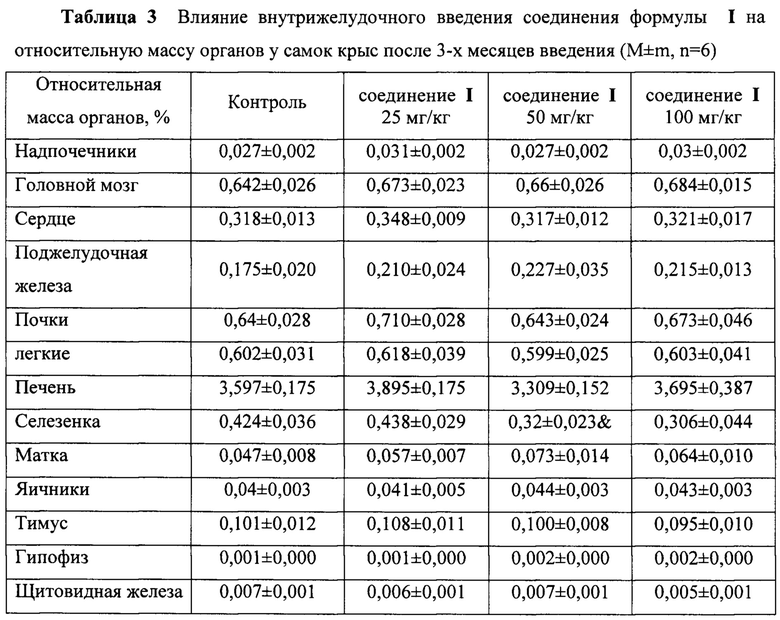
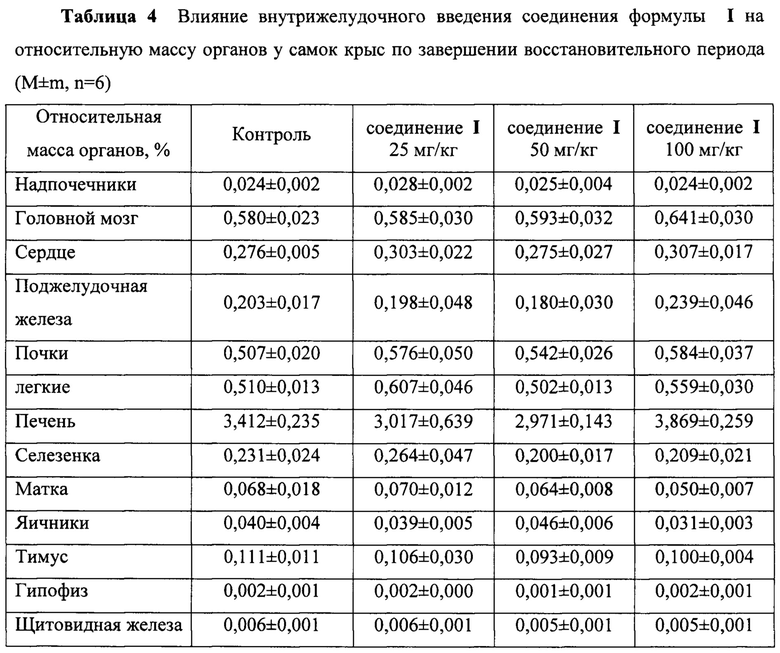
Масса органов
Результаты представлены в таблицах 2-5.
При патоморфологическом исследовании не выявлено отличий в относительной массе органов самцов и самок крыс, получавших ФС препарата ZC065-0001 в ТД, 2ТД и 4ТД в течение 3-х месяцев, по сравнению с параллельным контролем. Различия не отмечены и по завершении восстановительного периода.
Гистологическое исследование
Крысы, органы которых представлены для исследования, были поделены на две экспериментальные серии (А и Б) по восемь групп в каждой. Серию «А» составили самцы и самки, получавшие соединение I 3 месяца. Серию «Б» - составили самцы и самки, которые в течение 1 месяца находились в восстановительном периоде после трехмесячного применения соединение I.
СЕРИЯ "А"
- Группа 1А - контроль, самцы, получавшие растворитель;
- Группа 2А - самцы, получавшие соединение I в дозе 25 мг/кг;
- Группа 3А - самцы, получавшие соединение I в дозе 50 мг/кг;
- Группа 4А - самцы, получавшие соединение I в дозе 100 мг/кг;
- Группа 5А - контроль, самки, получавшие растворитель;
- Группа 6А - самки, получавшие соединение I в дозе 25 мг/кг;
- Группа 7А - самки, получавшие соединение I в дозе 25 мг/кг;
- Группа 8А - самки, получавшие соединение I в дозе 100 мг/кг;
СЕРИЯ "Б"
- Группа 1Б - контроль самцы, получавшие растворитель и проходившие восстановительный период;
- Группа 2Б - самцы, получавшие соединение I в дозе 25 мг/кг и проходившие восстановительный период;
- Группа 3Б - самцы, получавшие соединение I в дозе 50 мг/кг и проходившие восстановительный период;
- Группа 4Б - самцы, получавшие соединение I в дозе 100 мг/кг и проходившие восстановительный период;
- Группа 5Б - контроль самки, получавшие растворитель и проходившие восстановительный период;
- Группа 6Б - самки, получавшие соединение I в дозе 25 мг/кг и проходившие восстановительный период;
- Группа 7Б - самки, получавшие соединение I в дозе 50 мг/кг и проходившие восстановительный период;
- Группа 8Б - самки, получавшие соединение I в дозе 100 мг/кг и проходившие восстановительный период.
На исследование были взяты следующие органы: печень, почки, сердце, легкие, селезенка, тимус, надпочечники, семенники, яичники, головной мозг, щитовидная железа, желудок, тонкая и толстая кишка, поджелудочная железа.
Результаты исследования. Гистологическое исследование тимуса, селезенки, сердца, надпочечников, печени, почек, щитовидной железы, семенников, яичников, легких, головного мозга, желудка, тонкого и толстого кишечника, поджелудочной железы животных контрольных групп, как в период введения растворителя (группы 1А и 5А), так и в период восстановления (группы 1Б и 5Б), показало отсутствие особенностей строения, свидетельствующих о наличии патологии.
В месте введения - желудок, тонкая кишка, толстая кишка, поджелудочная железа -признаков раздражающего действия, патологических изменений у контрольных животных не выявлено.
При изучении структуры органов опытных животных, получавших соединение I, получены следующие результаты:
Печень - при применении всех изученных доз препарата выраженных патологических нарушений в органе не выявлено. Печеночные трабекулы у животных хорошо выражены. Степень развития междольковой соединительной ткани у опытных животных по сравнению с контролем не различалась.
Почки - применение препарата во всех изученных дозах не вызывало существенных нарушений строения почек в опытных группах. Почечные тельца не имеют повреждений. Кровенаполнение капилляров клубочков, перитубулярной сети и вен не отличалось от контроля. Эпителий дистальных и проксимальных канальцев в корковом веществе не имел повреждений.
Миокард - строение миокарда типично, существенно не отличалось от миокарда контрольных животных. Дистрофических изменений ядер и кардиомиоцитов, а также повреждения миофибрилл не найдено.
Легкие - у крыс опытных групп строение легких соответствовало норме. Патологические изменения в структуре органа отсутствовали.
Селезенка - фолликулярная структура была сохранена у опытных животных. Степень кровенаполнения красной пульпы у опытных животных не отличалась от кровенаполнения у животных контрольной группы. По завершении восстановительного периода патологические изменения в структуре органа отсутствовали.
Тимус (вилочковая железа) - тимус у животных опытных групп при применении препарата не имел нарушений структуры. Дольки тимуса типичной формы, варьировались по размеру. Граница между корковым и мозговым веществом отчетливая. По завершении восстановительного периода патологические изменения в структуре органа отсутствовали.
Надпочечники - выраженность клубочковой, сетчатой и пучковой зон коры в опытных группах не отличались от животных контрольных групп. Изменений в строении эндокриноцитов коркового и мозгового вещества не обнаружено. Кровенаполнение сосудов было идентично животным контрольной группы. По завершении восстановительного периода патологические изменения в структуре органа отсутствовали.
Семенники - во всех опытных группах сперматогенные клетки базальной и адлюминальной части извитых семенных канальцев не имели повреждений. Клетки Лейдига без изменений. Отличий в строении от контрольной группы животных не выявлено. После восстановительного периода патологических изменений в структуре органа не было обнаружено.
Яичники - во всех опытных группах при применении изученных доз препарата отличий от контрольной группы животных в строении органа не обнаружено: корковое вещество обычного строения, представлено в основном фолликулами на различных стадиях развития. Мозговое вещество было сохранено, в небольшом количестве, содержало довольно крупные извитые кровеносные сосуды, строма обычного микроскопического строения. После восстановительного периода патологических изменений в структуре органа не выявлено.
Головной мозг - при применении препарата во всех изученных дозах строение головного мозга было сохранено. Белое и серое вещество в пределах нормы. Признаков дистрофии нейронов, участков глиоза, признаков нейронофагии, воспалительной инфильтрации, фокусов деструкции, патологической гиперемии не было обнаружено. После восстановительного периода патологические изменения в структуре органа минимальны или полностью отсутствовали.
Щитовидная железа - у животных всех опытных групп строение органа типичное. Фолликулы различного размера. Коллоид гомогенного вида во всех наблюдаемых фолликулах. Тиреоциты кубической формы. Различий с группой контроля не выявлено. По завершении восстановительного периода патологические изменения в структуре органа отсутствовали.
Желудок - во всех опытных группах строение стенки желудка сохранено. В строении собственных желез у опытных животных не имелось признаков дистрофии или гипертрофии железистых клеток, не наблюдалось эрозивных или язвенных повреждений слизистой оболочки, воспалительных изменений не выявлено. По завершении восстановительного периода патологические изменения в структуре органа отсутствовали.
Тонкая кишка - стенка тонкой кишки во всех опытных группах имеет обычное строение. Эпителий ворсинок сохранен, воспалительной инфильтрации во всех слоях стенки не было найдено. Эрозий и язв не найдено. По завершении восстановительного периода патологические изменения в структуре органа отсутствовали.
Толстая кишка - строение стенки кишки соответствовала норме во всех опытных группах. Крипты сохранны. Количество секрета бокаловидных клеток между опытными группами не отличается. Воспалительной инфильтрации ни одного слоя стенки кишки, эрозий и язв не было. После восстановительного периода патологических изменений в структуре органа не обнаружено
Поджелудочная железа - строение органа у животных опытных групп существенно не различалось и не имело отличий от группы контроля. В ацинусах и выводных протоках не наблюдалось признаков атрофии или склероза. Признаки деструкции островков Лангерганса отсутствовали. По завершении восстановительного периода патологические изменения в структуре органа отсутствовали.
Заключение. В результате гистологического исследования органов крыс самцов и самок, получавших соединение I, показано, что при применении ТД препарата (25 мг/кг), а также доз 50 мг/кг (двухкратная ТД) и 100 мг/кг (четрырехкратная ТД) признаки интоксикации у крыс отсутствовали. По завершении восстановительного периода признаки интоксикации в опытных группах не выявлены.
Результаты проведенного исследования свидетельствуют, что ежедневное внутрижелудочное введение соединения формулы I в течение 3-х месяцев в ТД, 2ТД и 4ТД не вызывало гибели животных и не оказывало влияния на интегральные показатели их жизнедеятельности. Общее состояние животных, внешний вид, подвижность, отношение к корму и воде на протяжении всего эксперимента были удовлетворительны и не различались в опытных и контрольной группах.
Введение соединения формулы I не оказывало существенного влияния на динамику массы тела крыс обоего пола по сравнению с контролем в течение всего периода эксперимента.
У самок крыс введение соединения формулы I в течение 3-х месяцев в дозах 50 мг/кг и 100 мг/кг вызвало достоверное увеличение амплитуды R относительно контрольной группы; при введении в дозе 25 мг/кг наблюдалось достоверное снижение амплитуды Т и увеличение интервала QRS. Коме того, при введении в течение трех месяцев соединения формулы I в дозе 50 мг/кг наблюдалось достоверное увеличение R-R интервала при одновременном снижении частоты сердечных сокращений. По окончании восстановительного периода амплитуда К также была достоверно повышена группах самок, получавших соединение формулы I в дозах 50 и 100 мг/кг. Кроме того, наблюдалось достоверное увеличение амплитуды Т в группах самок получавших соединения формулы I в дозах 25 и 50 мг/кг.
При внутрижелудочном введении соединения формулы I самцам крыс в течение трех месяцев выявлены достоверные изменения лейкоцитарной формулы в группах самцов, получавших препарат в дозах 50 мг/кг и 100 мг/кг. Также наблюдалось достоверное снижение среднего содержания гемоглобина в эритроцитах и увеличение ширины распределения эритроцитов в группе самцов, получавших соединение формулы I в дозе 100 мг/кг. По окончании восстановительного периода достоверных отличий от контрольной группы не наблюдалось.
При внутрижелудочном введении соединения формулы I самкам крыс были выявлены изменения в лейкоцитарной формуле а также в других показателях периферической крови во всех исследуемых дозах. По окончании восстановительного периода достоверные изменения в лейкоцитарной формуле обнаружены в группе самок крыс, получавших соединение формулы I в дозе 50 мг/кг - снижение количества лимфоцитов и увеличение количества гранулоцитов.
Препарат соединения формулы I при внутрижелудочном введении в течение 3-х месяцев вызывал изменения биохимических показателей сыворотки крови крыс обоего пола, которые по окончании восстановительного периода возвращались к показателям контрольной группы.
Показано, что соединение формулы I при внутрижелудочном введении в течение 3-х месяцев в ТД, 2ТД и 4ТД не оказывало влияния на показатели мочевыделительной системы самцов крыс; статистически значимых отличий между опытными группами и контролем не наблюдалось. У самок крыс в группах, получавших соединение формулы I в течение 3 месяцев в дозах 25 мг/кг и 100 мг/кг наблюдалось достоверное увеличение удельной плотности мочи. По окончании восстановительного периода достоверных отличий от контрольной группы в показателях мочи экспериментальных групп не выявлено
Соединение формулы I при 3-х месячном внутрижелудочном введении и после восстановительного периода не вызывал изменения массовых коэффициентов внутренних органов.
В результате гистологического исследования органов крыс, получавших соединение формулы I, показано, что при применении в течении 3-х месяцев в ТД (25 мг/кг), а также в дозах 50 мг/кг (2ТД) и 100 мг/кг (4ТД) признаки интоксикации у крыс отсутствовали. По завершении восстановительного периода признаков интоксикации и реактивных изменений со стороны органов иммунной системы в опытных группах не выявлено.
При макроскопическом исследовании ЖКТ (местно-раздражающее действие) - желудок, тонкая кишка, толстая кишка, поджелудочная железа - признаков раздражающего действия не обнаружено. Отсутствие местно-раздражающего действия подтвердилось и при гистологическом исследовании.
Суммируя полученные результаты, можно заключить, что 3-х месячное введение препарата соединения формулы I в ТД, а также в дозах, равных 2 ТД и 4ТД вызывает умеренно выраженные изменения, полностью или частично нормализующиеся в восстановительном периоде. Гибели животных ни в одной из исследованных групп не наблюдалось.
В исследовании 28-дневной токсичности Иделалисиба на крысах внутрижелудочное введение препарата в дозах 100 и 150 мг/кг/сутки приводило к гибели животных (3 смерти в дозе 100 и 5 смертей в дозе 150 мг/кг). Таргетными органами токсичности были зафиксированы костный мозг, сердце, печень и семенники.
В 3-мес исследовании токсичности на крысах использование Иделалисиба в дозе 90 мг/кг вело к увеличению веса сердца. Таргетными органами токсичности были сердце, поджелудочная железа, язык и семенники
Внутрижелудочное введение соединения формулы I в дозе 100 мг/кг в течение трех месяцев не вызывало токсических эффектов и гибели животных, что говорит о меньшей токсичности соединения формулы I по сравнению с Иделалисибом при одинаковом пути введения в одинаковых дозах, что говорит о меньшей токсичности соединения формулы I по сравнению с известным аналогом.
| название | год | авторы | номер документа |
|---|---|---|---|
| Производные хромена в качестве ингибиторов фосфоинозитид-3-киназ | 2016 |
|
RU2722383C2 |
| ДЕЙТЕРИРОВАННЫЕ СОЕДИНЕНИЯ ХИНАЗОЛИНОНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2015 |
|
RU2656485C2 |
| НОВЫЕ ХИНАЗОЛИОНОВЫЕ ПРОИЗВОДНЫЕ, ИНГИБИРУЮЩИЕ PI3K, И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2017 |
|
RU2702904C1 |
| ПУРИНОВЫЕ ИНГИБИТОРЫ ЧЕЛОВЕЧЕСКОЙ ФОСФАТИДИЛИНОЗИТ 3-КИНАЗЫ ДЕЛЬТА | 2013 |
|
RU2658006C2 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
| ПИРИДАЗИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОИНОЗИТИД-3-КИНАЗ | 2015 |
|
RU2695374C2 |
| ПУРИНОВЫЕ ИНГИБИТОРЫ ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ ДЕЛЬТА ЧЕЛОВЕКА | 2013 |
|
RU2661896C2 |
| СОЕДИНЕНИЯ 1-(3-АМИНОПРОПИЛ)-ЗАМЕЩЕННОГО ЦИКЛИЧЕСКОГО АМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ПРИМЕНЕНИЯ | 2014 |
|
RU2671179C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ - 759 | 2008 |
|
RU2481348C2 |
| ПИРИМИДИНЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ПУРИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ИНГИБИРОВАНИЯ ПРОТЕИНКИНАЗ, СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ЗАБОЛЕВАНИЙ, ЧУВСТВИТЕЛЬНЫХ К ИНГИБИРОВАНИЮ ПРОТЕИНКИНАЗ И СПОСОБ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2008 |
|
RU2518098C2 |
Изобретение относится к (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетату (соединение формулы I), обладающему свойством ингибитора p110δ - дельта-изоформы фосфоинозитид-3-киназы (PI3K). Кроме того, изобретение относится к способу получения соединения формулы I, активному компоненту, фармацевтической композиции и лекарственному средству на его основе, а также его применению для лечения и способу лечения фолликулярной лимфомы и других злокачественных лимфопролиферативных заболеваний из ряда неходжикинских лимфом. 8 н. и 2 з.п. ф-лы, 4 табл., 10 пр.
1. Соединение (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетат формулы I
2. Способ получения соединения формулы I, заключающийся во взаимодействии 2-фтор-6-нитробензойной кислоты в смеси дихлорметана и диметилформамида с оксалил хлоридом в дихлорметане с получением раствора 2-фтор-6-нитро-N-фенилбензамида с последующей обработкой последовательно сульфонил хлоридом в диметилформамиде и (2S)-2-[(трет-бутоксикарбонил)амино]бутановой кислотой в дихлорметане, приводящей к получению третбутилового эфира (S)-[1-(2-фтор-6-нитробензоил)-фенил-аминокарбонил]-пропил-карбаминовой кислоты, и последующим восстановлением ее под действием порошка цинка в уксусной кислоте и циклизацией полученного третбутилового эфира (S)-[1-(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)-пропил]-карбаминовой кислоты под воздействием трифторуксусной кислоты с образованием (S)-2-(1-аминопропил)-5-фтор-3-фенил-3H-хиназолин-4-она и последующим взаимодействием его с (6-хлор-9H-пурин-9-ил)метил ацетатом с получением и выделением продукта - (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохин-азолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетата формулы I хроматографически.
3. Способ по п. 2, включающий следующие этапы:
а) к раствору ацетилхлорида в ацетонитриле добавляют 5-фтор-3-фенил-2-[(1S)-1-(9Н-пурин-6-иламино)пропил]хиназолин-4(3Н)-он с образованием осадка 2-{(1S)-l-[(9-ацетил-9Н-пурин-6-ил)амино]пропил}-5-фтор-3-фенилхиназолин-4(3Н)-она;
б) к толуольному раствору 2-{(1S)-1-[(9-ацетил-9Н-пурин-6-ил)амино]пропил}-5-фтор-3-фенилхиназолин-4(3Н)-она, полученного на этапе а), добавляют параформ. По окончании взаимодействия продукт - (6-{[(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохин-азолин-2-ил)пропил]амино}-9Н-пурин-9-ил)метилацетата формулы I - выделяют последовательным концентрированием, хроматографией и рядом перекристаллизаций из подходящих растворителей.
4. Активный компонент, обладающий свойством ингибитора p110δ - дельта-изоформы фосфоинозитид-3-киназы (PI3K), включающий в свой состав соединение формулы I в фармацевтически эффективном количестве.
5. Фармацевтическая композиция, обладающая свойством ингибитора р110δ - дельта-изоформы фосфоинозитид-3-киназы (PI3K) для лечения фолликулярной лимфомы и других злокачественных лимфопролиферативных заболеваний из ряда неходжкинских лимфом, содержащая в фармацевтически эффективном количестве соединение формулы I по п. 1 или активный компонент по п. 4 и фармацевтически приемлемые добавки.
6. Фармацевтическая композиция по п. 5, содержащая в эффективном количестве соединение формулы I по п. 1 в количестве 25 мг и фармацевтически приемлемые добавки: целлюлоза микрокристаллическая в количестве 35,0 мг, лактозы моногидрат 26,0 мг, гидроксипропилцеллюлоза в количестве 5 мг, натрия карбоксиметилкрахмал в количестве 4 мг, натрия кроскармелоза в количестве 4 мг, магния стеарат в количестве 1 мг, а также Опадрай II белый (поливиниловый спирт - 60%, титана диоксид - 25%, полиэтиленгликоль - 10%, тальк - 5%) в количестве 3 мг.
7. Лекарственное средство, содержащее соединение формулы I в эффективном количестве в качестве активного компонента.
8. Способ лечения фолликулярной лимфомы и других злокачественных лимфопролиферативных заболеваний из ряда неходжкинских лимфом посредством введения в эффективном количестве соединения формулы I по п. 1, или активного компонента по п. 4, или фармацевтической композиции по пп. 5-6, или лекарственного средства по п. 7 нуждающемуся в этом реципиенту.
9. Применение соединения формулы I по п. 1 для получения лекарственного средства для лечения фолликулярной лимфомы и других злокачественных лимфопролиферативных заболеваний из ряда неходжкинских лимфом.
10. Способ ингибирования р110δ - дельта-изоформы фосфоинозитид-3-киназы (PI3K) введением в эффективном количестве соединения формулы I по п. 1 нуждающемуся в этом реципиенту.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ использования делительного аппарата ровничных (чесальных) машин, предназначенных для мериносовой шерсти, с целью переработки на них грубых шерстей | 1921 |
|
SU18A1 |
| Устройство для подъема и перемещения грузов | 1917 |
|
SU2674A1 |
| WO 2017079558 A1, 11.05.2017 | |||
| НЕКОТОРЫЕ ХИМИЧЕСКИЕ СТРУКТУРЫ, КОМПОЗИЦИИ И СПОСОБЫ | 2009 |
|
RU2513636C2 |

