Настоящее изобретение описывает производные фосфорилхолина по общей формуле (I), которые подходят для иммобилизации на твердой подложке для обеспечения сепарационного материала по общей формуле (II), которые с высокой активностью и высокой специфичностью связываются c белком, точнее, с С-реактивным белком и антифосфорилхолиновыми антителами. Указанные сепарационные материалы являются особенно применимыми в экстракорпоральном удалении С-реактивного белка и антифосфорилхолиновых антител из биологической жидкости пациента для профилактики и/или лечения иммунных дисфункций и сердечно-сосудистых заболеваний. Также обеспечивают колонку, которая включает сепарационный материал по общей формуле (II), а также устройство, содержащее колонку.
Предшествующий уровень техники
C-реактивный белок (СРБ (CRP)) является компонентом острой фазы, продуцируемым печенью в ответ на высвобождение цитокинов при воспалении. Его длительно используют в клинической практике для наблюдения за системным воспалением, особенно бактериальной инфекцией. Позднее, эпидемиологические свидетельства показали, что базальные уровни СРБ в отсутствие очевидных воспалительных заболеваний могут быть информативными в предсказании будущих миокардиальных или цереброваскулярных событий (Ridker et al. Circulation, 2001 103, 1813). Также интересен тот факт, что СРБ является потенциальным воспалительным маркером, предположительно важным в предсказании коронарных событий (Danesh et al. N. Engl. J. Med. 2004 350, 1387), и что СРБ является причинным фактором деструктивных процессов, наблюдаемых в течение недели после инфаркта миокарда (Slagman et al., Blood Purif. 2011, 31, 9).
Некоторые исследования показали аффинность связывания фосфорилхолина (ФХ) (PC)) в отношении СРБ. Следовательно, производные ФХ, иммобилизованные на твердой подложке, широко используют для изоляции белков, связывающих ФХ, из различных биологических источников. Однако, наиболее важное клиническое применение вышеупомянутых производных ФХ, иммобилизованных на твердой подложке, представляет собой экстракорпоральное удаление С-реактивного белка и антифосфорилхолиновых антител из биологических жидкостей.
В WO 90/12632 описан способ удаления СРБ и антифосфорилхолиновых антител из биологических жидкостей для улучшения их клеточных иммунных ответов, и способ удаления СРБ и антифосфорилхолиновых антител из циркуляции пациентов с раком путем проведения экстракорпоральной перфузии плазмы крови пациента через матрицу, адсорбирующую фосфорилхолин, так, чтобы улучшить клеточный иммунный ответ против рака.
В WO 2007/076844 описан способ лечения риска накопления СРБ путем проведения экстракорпоральной перфузии плазмы крови пациентов, имеющих риск сердечно-сосудистых заболеваний или иммунные дисфункции, такие как аутоиммунные заболевания, посредством колонки, которая содержит абсорбирующий матриксный материал, включающий производные ФХ, для удаления СРБ из биологических жидкостей пациента так, чтобы предотвратить и/или лечить аутоиммунные заболевания, сердечно-сосудистые заболевания, такие как инфаркт миокарда, инсульт, диабет, ревматизм и почечная недостаточность.
Также описаны синтез ФХ-производного бычьего альбумина сыворотки (PC-BSA), его иммобилизация на Toyopearl® HW 65 и его применение для аффинной очистки СРБ (Stults et al. Anal. Biochem. 1987, 161, 567). В проведенном исследовании иммобилизацию ФХ производного на твердой подложке осуществляли посредством образования фосфодиэфирных сшивок. Другие производные ФХ сходным образом иммобилизовали на твердой подложке (Spande T.F. J. Org. Chem. 1980, 45, 3081; Martin, L. M. Tetrahedron Lett. 1996 37, 7921).
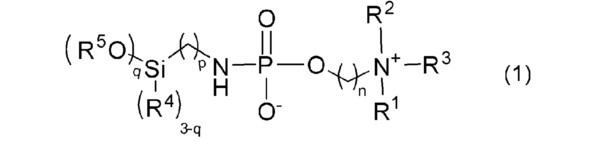
В WO 2013/176084 A1 также описано соединение силилалкилфосфорамидата по общей формуле (1) для покрытия оболочкой медицинского устройства с целью подавления адгезии биологического материала, такого как тромбоциты.
WO 2012/160187 A1 обеспечивает производные фосфоновой кислоты, содержащие аммоний, для применения в лечении воспалительных, аутоиммунных и/или аллергических расстройств. Установлено, что описанные соединения проявляют свою фармакологическую активность посредством ингибирования киназного пути фосфоинозитид 3-киназы (PI3K)/Akt.
Как известно, до настоящего времени иммобилизация ФХ на твердой подложке достигалась только посредством образования фосфодиэфирной связи. Иммобилизация производных ФХ посредством образования фосфодиэфирной связи имеет некоторые преимущества, но также некоторые недостатки. Основным недостатком настоящего подхода является то, что образование фосфодиэфирной связи изменяет общий заряд (суммарный заряд) молекулы ФХ и посредством этого изменяет ориентацию модифицированного таким образом ФХ в связывающем кармане. Кроме того, фосфодиэфирная связь может быть легко расщеплена неспецифическими фосфодиэстеразами, присутствующими в биологических жидкостях, приводя к проблемам нестабильности матриц, которые были заменены таким образом.
Для преодоления вышеупомянутых недостатков, задачей настоящего изобретения является обеспечение соединений, которые могут быть иммобилизованы на твердой подложке для обеспечения сепарационного материала, который с высокой аффинностью и высокой специфичностью связывает СРБ, для профилактики и/или лечения иммунных дисфункций и сердечно-сосудистых заболеваний путем экстракорпорального удаления СРБ и антифосфорилхолиновых антител.
Указанную задачу решали при помощи соединений по общей формуле (I) в соответствии с независимым пунктом формулы изобретения 1, сепарационного материала по независимому пункту формулы изобретения 5 для применения в экстракорпоральном удалении СРБ и антифосфорилхолиновых антител из биологической жидкости пациента для профилактики и/или лечения иммунных дисфункций и сердечно-сосудистых заболеваний.
Задачу настоящего изобретения решают с использованием содержания независимой формулы изобретения. Дополнительные преимущественные характеристики, аспекты и подробности изобретения очевидны из зависимой формулы изобретения, описания, чертежей и примеров настоящей заявки.
Описание изобретения
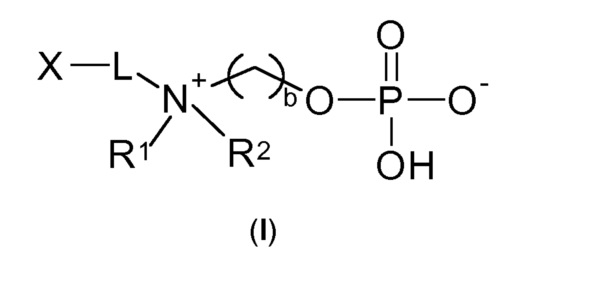
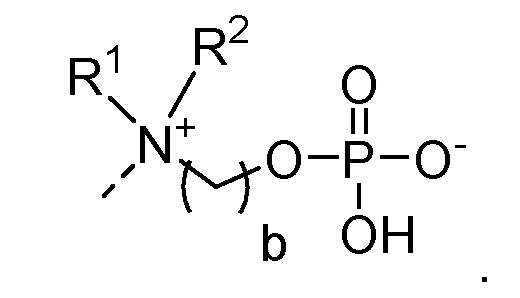
Следовательно, настоящее изобретение относится к соединению по общей формуле (I)
где
b выбирают из 2 и 3;
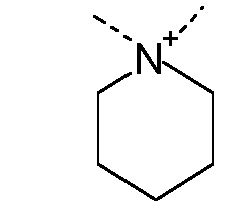
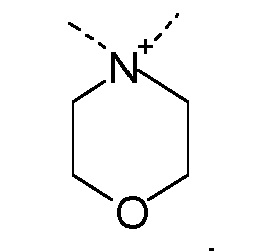
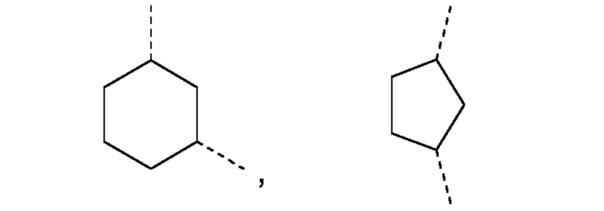
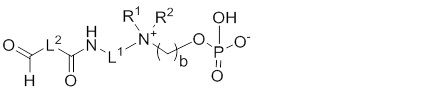
R1 и R2 независимо друг от друга выбирают из: -CH3, -C2H5, -C3H7, -C4H9, -C5H11 и -C6H13, или R1 и R2 могут образовывать вместе с атомом азота, к которому они присоединены, гетероцикл, выбираемый из:
 и
и 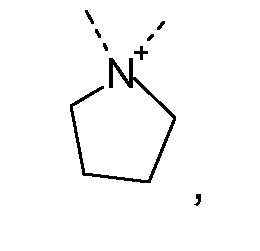
где один или несколько атома(ов) водорода могут быть заменены атомом(ами) фтора;
X выбирают из: -SH, -NHR3, -C≡CH, -CH=CH2, -N3 и -CHO;
R3 выбирают из: -H, -CH3, -C2H5 и -C3H7;
-Lа- выбирают из: -La-, -La-Le-, -La-Lb-Le- и -La-Lb-Ld-Lc-Le-,
где
-La- выбирают из: -(CH2)m-, -(CH2-CH2-O)m-CH2-,
 и
и 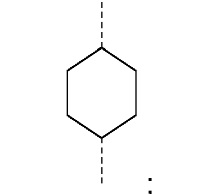 ;
;
-Lb- и -Lc- независимо друг от друга выбирают из: -O-, -NH-C(O)-, -C(O)- NH-, -O-C(O)- NH- и -SO2-;
-Ld- выбирают из: -(CH2)n-, -(CH2-CH2-O)n-CH2-,
и 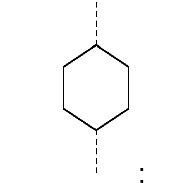 ;
;
-Le- выбирают из: -(CH2)p1-, -(CH2)p1-O- (CH2)p2-,
и  ;
;
m, n, p1 и p2 независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10;
и энантиомеры, смеси энантиомеров, таутомеры, гидраты, сольваты, рацематы, протонированные и депротонированные формы вышеупомянутых соединений.
Специалисту очевидно, что левый конец фрагмента -L- соединен с X и правый конец фрагмента -L- соединен с
Некоторые применения фрагментов: -La-, -Lb-, -Lc-, -Ld- и -Le-.
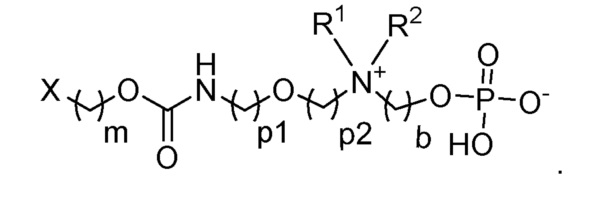
Следовательно, например, если -L- представляет собой -La-Lb-Le- с -La-, являющимся -(CH2)m-, -Lb-, являющимся -O-C(O)- NH-, и -Le-, являющимся -(CH2)p1-O- (CH2)p2-, тогда соединение по общей формуле (I) является следующим:
Экспрессирующийся таутомер, как используется в настоящем описании, относится к органическому соединению, которое взаимопревращается путем химической реакции, называемой таутомеризацией. Таутомеризация катализируется основаниями, кислотами или другими соединениями.
Предпочтительными соединениями по настоящему изобретению являются соединения по общей формуле (I):
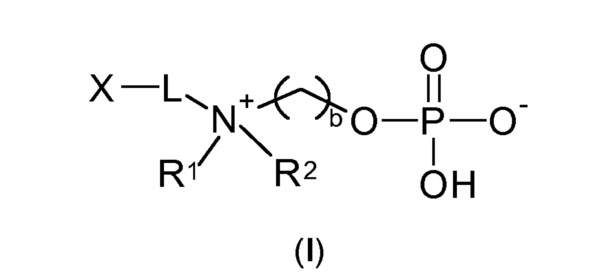
где b выбирают из 2 и 3;
R1 и R2 независимо друг от друга выбирают из: -CH3 и -C2H5, или R1 и R2 могут образовывать вместе с атомом азота, с которым они соединены, гетероцикл, выбираемый из:
и ,
где один или несколько атома(ов) водорода могут быть заменены (a) атомом(ами) фтора;
X выбирают из: -SH, -NHR3, -C≡CH, -CH=CH2, -N3 и -CHO;
R3 выбирают из: -H, -CH3, -C2H5 и -C3H7;
-L- выбирают из: -La-, -La-Le-, -La-Lb-Le- и -La-Lb-Ld-Lc-Le-, где
-La- представляет собой: -(CH2)m- или -(CH2-CH2-O)m-CH2-;
-Lb- и -Lc- независимо друг от друга выбирают из: -O-, -NH-C(O)-, -C(O)- NH-,-O-C(O)- NH- и -SO2-;
-Ld- представляет собой: -(CH2)n- или -(CH2-CH2-O)n-CH2-;
-Le- выбирают из: -(CH2)p1- и -(CH2)p1-O- (CH2)p2-; и
m, n, p1 и p2 независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10.
Предпочтительный вариант осуществления в соответствии с настоящем изобретением относится к соединениям по общей формуле (I), где X выбирают из: -SH, -NHR3, и -CHO, и более предпочтительно его выбирают из: -SH и -NHR3.
Другой предпочтительный вариант осуществления настоящего изобретения относится к соединениям по общей формуле (I), где X представляет собой: -C≡CH, -CH=CH2, или -N3.
Предпочтительные соединения по настоящему изобретению представляют собой соединения по следующей общей формуле (III), (IV), (V) и (VI):
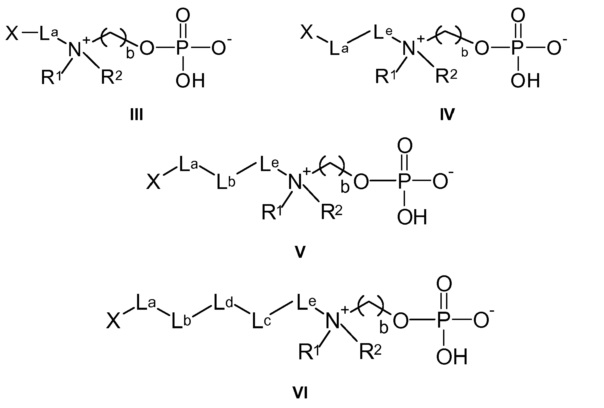
где
-La- представляет собой: -(CH2)m- или -(CH2-CH2-O)m-CH2-;
-Lb- и -Lc- независимо друг от друга выбирают из: -O-, -NH-C(O)-, -C(O)- NH-, -O-C(O)- NH- и -SO2-;
-Ld- представляет собой: -(CH2)n- или -(CH2-CH2-O)n-CH2-;
-Le- выбирают из: -(CH2)p1- и -(CH2)p1-O- (CH2)p2-,
m, n, p1 и p2 независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10;
и b, X, R1 и R2 имеют значения, определяемые выше.
Особенно предпочтительными соединениями являются соединения по общей формуле (I), (III), (IV), (V) и (VI), где
R1 и R2 независимо друг от друга выбирают из: -CH3 и -C2H5;
X выбирают из: -SH и -NHR3;
R3 выбирают из: -H, -CH3, -C2H5 и -C3H7; и
-La- представляет собой: -(CH2)m-.
Еще более предпочтительный вариант осуществления изобретения относится к соединениям по общей формуле (I)
где b выбирают из 2 и 3;
R1 и R2 независимо друг от друга выбирают из: -CH3, -C2H5, -C3H7, -C4H9, -C5H11, -C6H13, или R1 и R2 могут образовывать вместе с атомом азота, к которому они присоединены, гетероцикл, выбираемый из:
и ,
где один или несколько атома(ов) водорода, может быть заменен атомом(ами) фтора;
X выбирают из: -SH, -NHR3, -C≡CH, -CH=CH2, -N3 и -CHO;
R3 выбирают из: -H, -CH3, -C2H5, -C3H7;
-L- выбирают из: -(CH2)m-, -(CH2)m-O-C(O)- NH-- (CH2)p1-, -(CH2)m-O- (CH2)p1-, -(CH2)m-C(O)- NH- (CH2)p1-, -(CH2)m-NH-C(O)- (CH2)p1-,
- (CH2)m-C(O)- NH- (CH2)n-O- (CH2)p1-,
- (CH2)m-O-C(O)- NH- (CH2)n-O- (CH2)p1-,
- (CH2)m-C(O)- NH- (CH2)n-C(O)- NH- (CH2)p1-O- (CH2)p2-,
- (CH2)m-O-C(O)- NH- (CH2)n-C(O)- NH- (CH2)p1-O- (CH2)p2-;
и m, n, p1 и p2 независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10.
Предпочтительно соединения по общей формуле (I) выбирают из:
1. 2-[2-(2-аминоэтокси)этилдиэтиламмонио]этил кислого фосфата
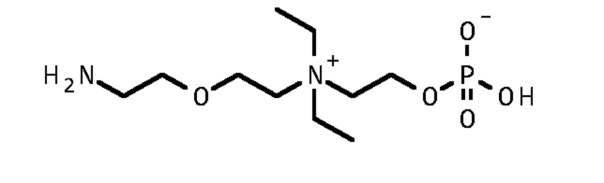
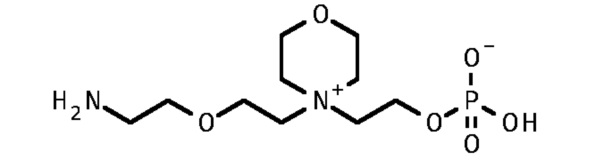
2. 2-[4-[2-(2-аминоэтокси)этил]морфолин-4-иум-4-ил]этил кислого фосфата
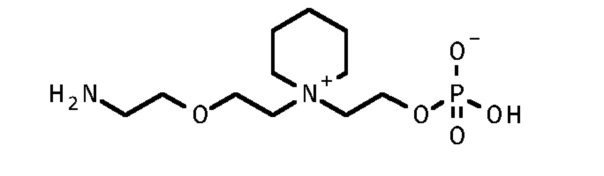
3. 2-[1-[2-(2-аминоэтокси)этил]пиперидин-1-иум-1-ил]этил кислого фосфата
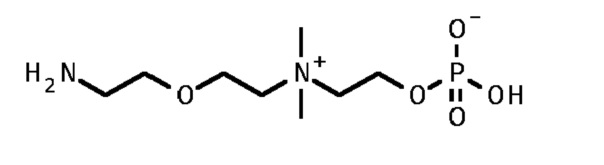
4. 2-[2-(2-аминоэтокси)этилдиметиламмонио]этил кислого фосфата
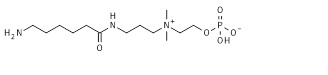
5. 2-[3-аминопропил(диметил)аммонио]этил кислого фосфата
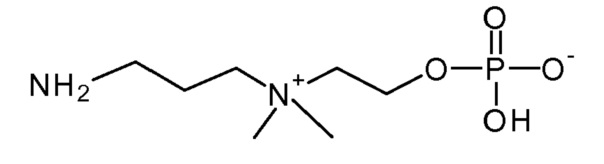
6. 2-[диметил(4-сульфанилбутил)аммонио]этил кислого фосфата
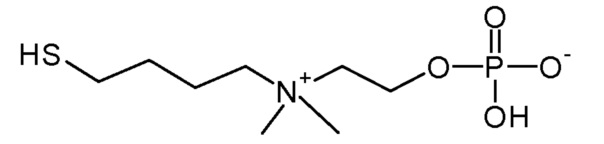
7. 2-[4-азидобутил(диметил)аммонио]этил кислого фосфата
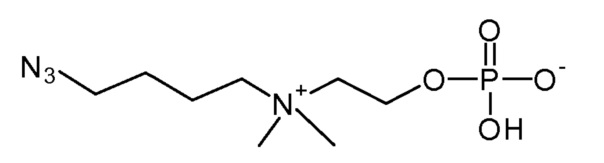
8. 2-[диметил(пент-4-инил)аммонио]этил кислого фосфата
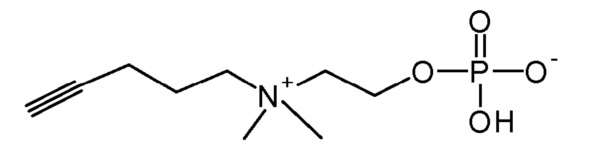
9. 2-[3-(6-аминогексаноиламино)пропилдиэтиламмонио]этил кислого фосфата
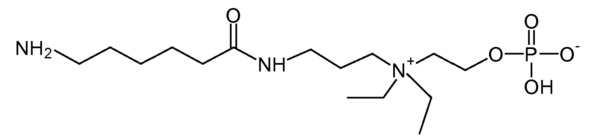
10. 2-[1-[2-[2-(6-аминогексаноиламино)этокси]этил]пиперидин-1-иум-1-ил]этил кислого фосфата
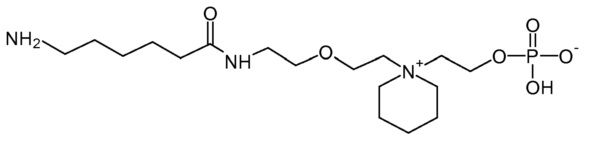
11. 2-[4-[2-[2-[3-(6-аминогексаноиламино)пропаноиламино]этокси]этил]морфолин-4-иум-4-ил]этил кислого фосфата

12. 2-[1-[2-[2-[6-(6-аминогексаноиламино)гексаноиламино]этокси]этил]пирролидин-1-иум-1-ил]этил кислого фосфата

13. 2-[2-аллилоксиэтил(диметил)аммонио]этил кислого фосфата
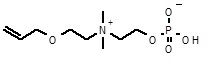
14. 2-[2-аллилоксиэтил(диэтил)аммонио]этил кислого фосфата
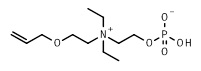
15. 2-[4-(2-аллилоксиэтил)морфолин-4-иум-4-ил]этил кислого фосфата
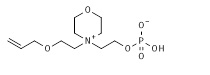
16. 2-[1-(2-аллилоксиэтил)пиперидин-1-иум-1-ил]этил кислого фосфата
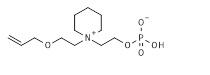
17. 2-[2-[2-(6-аминогексаноиламино)этокси]этилдиметиламмонио]этил кислого фосфата
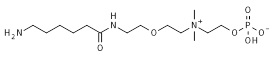
18. 2-[2-[2-[3-(6-аминогексаноиламино)пропаноиламино]этокси]этилдиметиламмонио]этил кислого фосфата
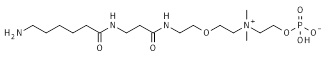
19. 2-[3-азидопропил(диметил)аммонио]этил кислого фосфата
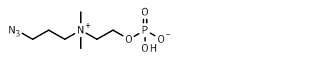
20. 2-[диметил-[2-[2-(проп-2-иноксикарбониламино)этокси]этил]аммонио]этил кислого фосфата
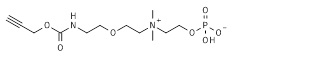
21. 2-[2-[2-(аллилоксикарбониламино)этокси]этилдиметиламмонио]этил кислого фосфата
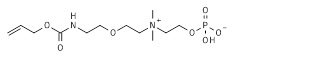
22. 2-[2-[2-[6-(аллилоксикарбониламино)гексаноиламино]этокси]этилдиметиламмонио]этил кислого фосфата
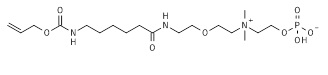
23. 2-[2-(6-аминогексаноиламино)этилдиметиламмонио]этил кислого фосфата
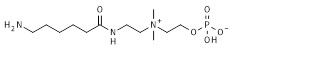
24. 2-[диметил-[3-[6-(проп-2-иноксикарбониламино)гексаноиламино]пропил]аммонио]этил кислого фосфата
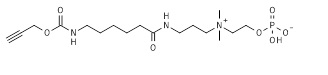
25. 2-[3-(6-аминогексаноиламино)пропилдиметиламмонио]этил кислого фосфата
Соединения по настоящему изобретению были созданы так, что они могут быть иммобилизованы на твердых подложках для получения сепарационных материалов, которые демонстрируют большую стабильность и повышенную селективность к СРБ по сравнению с сепарационными материалами, описанными в предшествующей области техники. Это было достигнуто путем замены одного из алкильных заместителей триалкиламмониевой группы фосфорилхолина фрагментом -L-X. Фрагмент -L-X позволяет иммобилизацию соединений по общей формуле (I) на твердой подложке без модификации общего заряда молекулы фосфорилхолина и путем избегания присутствия нестабильной фосфодиэфирной связи в конструкте.
Следовательно, настоящее изобретение дополнительно обеспечивает сепарационный материал по общей формуле (II):
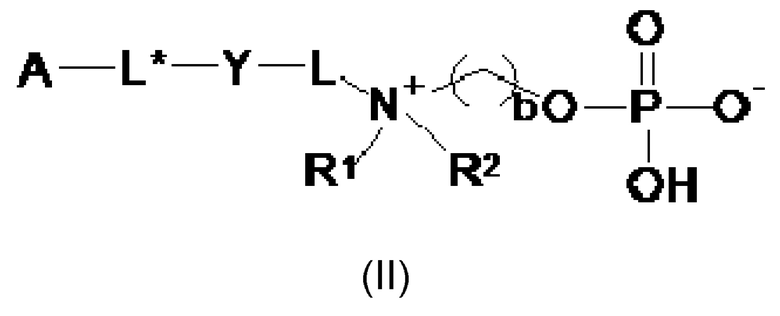
где
b выбирают из 2 и 3;
R1 и R2 независимо друг от друга выбирают из: -CH3, -C2H5, -C3H7, -C4H9, -C5H11 и -C6H13, или R1 и R2 могут образовывать вместе с атомом азота, с которым они соединены, гетероцикл, выбираемый из
и
где один или несколько атома(ов) водорода, могут быть заменены (a) атомом(ами) фтора;
Y выбирают из: -CH(OH)- CH2-N(R4)-, -CH(OH)- CH2-S-, -CH2-NH-, -NH-CH2-, -CH2-CH2-S-, -S-CH2-CH2-,

R4 выбирают из: -H, CH3, -C2H5, -C3H7 и -C(O)- CH3;
-L- выбирают из: -La-, -La-Le-,-La-Lb-Le- и -La-Lb-Ld-Lc-Le-,
где
-La- выбирают из: -(CH2)m-, -(CH2-CH2-O)m-CH2-,
, и  ;
;
-Lb- и -Lc- независимо друг от друга выбирают из: -O-
-NH-C(O)-, -C(O)- NH-, -O-C(O)- NH- и -SO2-;
-Ld- выбирают из: -(CH2)n-, -(CH2-CH2-O)n-CH2-,
, и ;
-Le- выбирают из: -(CH2)p1-, -(CH2)p1-O- (CH2)p2-,
, и ;
-L*- выбирают из: -L*a-, -L*a-L*e- и -L*a-L*b-L*e-, где
-L*a- выбирают из: -(CH2)o-, -(CH2-CH2-O)o-C2H4-, -(CH2-CH2-O)o-CH2- и -CH2-CH(OH)- CH2-;
-L*e- выбирают из: -(CH2)q-, -C2H4- (O-CH2-CH2)q-, и -CH2- (O-CH2-CH2)q-;
-L*b- выбирают из: -O- (CH2)r-O-, -S- (CH2)r-S-, -SO2-, -S-, -O-, -NH-C(O), -C(O)- NH- и -S-S-; и
m, n, p1, p2, o, r, q независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10;
A представляет собой твердую подложку, выбираемую из группы, состоящей из: поливинилпирролидона (PVP), полисульфона (PS), полиэфирсульфона (PES), полиакрилэфирсульфона (PAES), полиакрилата, поли(метилметакрилата) (PMMA), поли(глицидилметакрилата) (PGMA), поли(гидроксиметакрилата), полистирола (PS), политетрафторэтилена (PTFE), полиакриламида, полиакролеина, акрилонитрилбутадиеностирола (ABS), полиакрилонитрила (PAN), полиуретана (PU), eupergit®, полиэтиленгликоля (PEG), гиперфторкарбона, агарозы, альгината, каррагенана, хитина, крахмала, целлюлозы, нитроцеллюлозы, сефарозы®, стекла, диоксида кремния, диатомита, циркония, глинозема, оксида железа и смеси и/или производных указанных твердых подложек;
и протонированных и депротонированных форм указанного сепарационного материала.
Специалисту очевидно, что левый конец фрагмента -L*- соединен с A, правый конец фрагмента -L*- соединен с левым концом фрагмента Y, правый конец фрагмента Y соединен с левым концом фрагмента-L-, чей правый конец соединен с
Некоторые применимы к фрагментам: -La-, -Lb-, -Lc-, -Ld-, -Le-, -L*a-, -L*b- и -L*e-.
Следовательно, например, если и -L*- представляет собой -L*a-L*b-L*e- с -L*a, являющимся -(CH2)q-, Y представляет собой -CH(OH)- CH2-N(R4)-, -L- представляет собой -La-Lb-Le- с -La-, являющимся -(CH2)m-, -Lb-, являющимся -O-C(O)- NH-, и -Le- являющимся -(CH2)p1-O- (CH2)p2-, тогда сепарационный материал по общей формуле (II) является следующим:
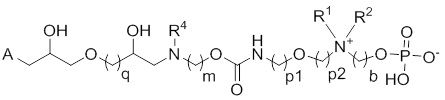
Как используется в настоящем описании, твердая подложка A относится к инертной твердой подложке, которая в преддверии ковалентной иммобилизации соединений по изобретению по общей формуле (I), (III), (IV), (V) и (VI) функционализирована с помощью фрагмента-L*-FG, где FG представляет собой реакционно-способную функциональную группу, которая подходит для реакции с функциональной группой X. Примеры FG групп включают, без ограничения: эпоксид, -CHO, -C≡CH, -CH=CH2, -N3, -CH(OH)- CH2-N3, -NH2, -SH, тресил. Функционализацию твердой подложки достигают посредством способов, хорошо известных специалисту в области техники (Chin. J. Chem. 2012, 30, 2473; Polym. Int. 2013, 62, 991). Кроме того, функционализированные твердые подложки уже коммерчески доступны: Toyopearl® AF-эпокси, Toyopearl® AF-амино, Toyopearl® AF-тресил, TSKgel® тресил, эпокси-активированная Sepharose® 6B (GE Healthcare Life Sciences), CNBr-активированная Sepharose® 4 быстрого тока (GE Healthcare Life Sciences), ECH Sepharose® 4B (GE Healthcare Life Sciences), NHS-активированная Sepharose® 4 быстрого тока (GE Healthcare Life Sciences), концевая винилсульфон активированная Sepharose® 4 быстрого тока (Affiland), альдегид Separopore® (Agarose) 4B, ECH Separopore® (Agarose) 4B (Separopore).
Твердая подложка может быть подвергнута перекрестным сшивкам или другой обработке для улучшения физической или химической стабильности, и может быть преобразована в различные формы, включая, без ограничения, волокна, листы, стержни, шарики и мембраны.
Предпочтительно, твердую подложку A выбирают из полисульфона (PS), полиэфирсульфона (PES), полиакрилэфирсульфона (PAES), их смесей и/или производных.
Твердые подложки на основе PS, PES и PAES могут быть модифицированы так, чтобы представлять на их поверхности фрагмент -L*-FG, где FG представляет собой реакционно-способную функциональную группу, которая способна реагировать с функциональной группой X (Chin. J. Chem. 2012, 30, 2473; Polym. Int. 2013, 62, 991). Такая модификация приводит к увеличению противообрастающих свойств, биосовместимости и других специфических функций.
Предпочтительные сепарационные материалы по настоящему изобретению представляют собой сепарационные материалы по общей формуле (II):
и их протонированные и депротонированные формы,
где
b выбирают из 2 и 3;
R1 и R2 независимо друг от друга выбирают из: -CH3, -C2H5, или R1 и R2 могут образовывать вместе с атомом азота, с которым они соединены, гетероцикл, выбираемый из:
,и
где один или несколько атома(ов) водорода могут быть заменены атомом(ами) фтора;
Y выбирают из: -CH(OH)- CH2-N(R4)-, -CH(OH)- CH2-S-, -CH2-NH-, -NH-CH2-, -CH2-CH2-S-, -S-CH2-CH2-,
R4 выбирают из: -H, CH3, -C2H5, -C3H7 и -C(O)- CH3;
-L- выбирают из: -La-, -La-Le-, -La-Lb-Le- и -La-Lb-Ld-Lc-Le-, где
-La- представляет собой: -(CH2)m- или -(CH2-CH2-O)m-CH2-;
-Lb- и -Lc- независимо друг от друга выбирают из: -O-, -NH-C(O)-, -C(O)- NH-, -O-C(O)- NH- и -SO2-;
-Ld- выбирают из: -(CH2)n- и -(CH2-CH2-O)n-CH2-,
-Le- выбирают из: -(CH2)p1- и -(CH2)p1-O- (CH2)p2-;
-L*- выбирают из: -L*a-, -L*a-L*e- и -L*a-L*b-L*e-, где
-L*a- выбирают из: -(CH2)o-, -(CH2-CH2-O)o-C2H4-, -(CH2-CH2-O)o-CH2- и -CH2-CH(OH)- CH2-;
-L*e- выбирают из: -(CH2)q-, -C2H4- (O-CH2-CH2)q-, и -CH2- (O-CH2-CH2)q-;
-L*b- выбирают из: -O- (CH2)r-O-, -S- (CH2)r-S-, -SO2-, -S-, -O-, -NH-C(O)-, -C(O)- NH- и -S-S-;
m, n, p1, p2, o, r, q независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10;
и A имеет значение, определенное выше, и более предпочтительно A выбирают из полисульфона (PS), полиэфирсульфона (PES), полиакрилэфирсульфона (PAES), их смесей и/или производных.
Предпочтительный вариант осуществления по настоящему изобретению относится к сепарационным материалам по общей формуле (II), где Y выбирают из: -CH(OH)- CH2-N(R4)-, -CH(OH)- CH2-S-, -CH2-NH-, -NH-CH2-, -CH2-CH2-S-, -S-CH2-CH2-,
и более предпочтительно выбирают из: -CH(OH)- CH2-N(R4)-, и -CH(OH)- CH2-S-.
Также предпочтительными являются сепарационные материалы по общей формуле (II), где Y представляет собой:
Предпочтительные соединения по настоящему изобретению представляют собой соединения по следующей общей формуле (VII), (VIII), (IX) и (X)
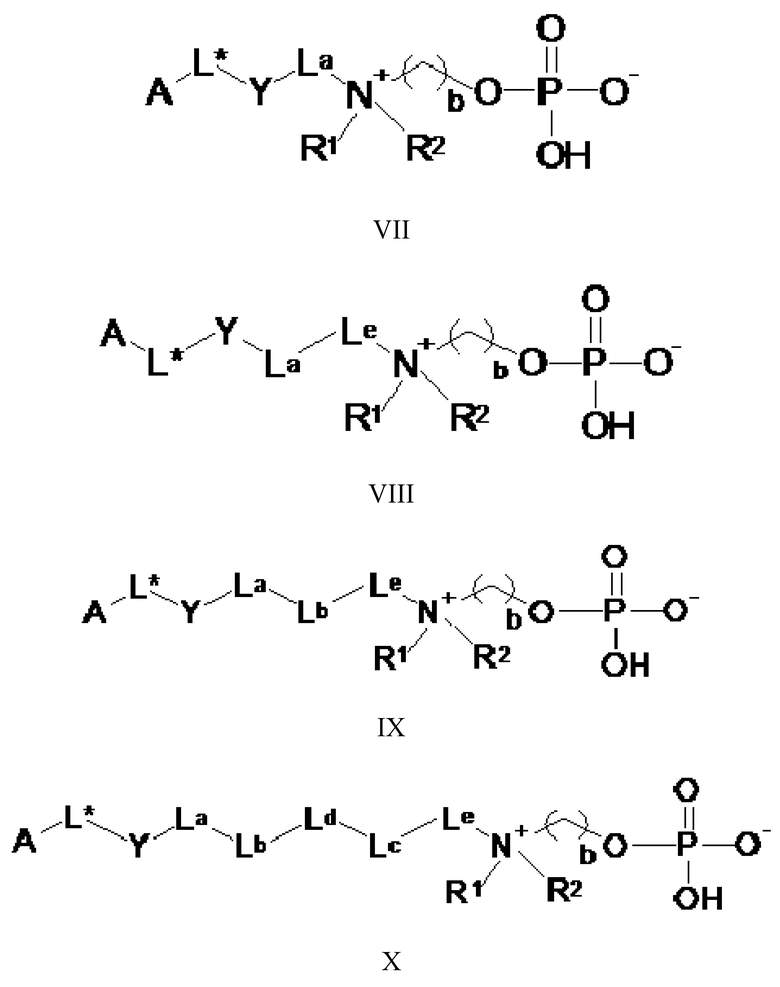
где
-La- выбирают из: -(CH2)m- и -(CH2-CH2-O)m-CH2-;
-Lb- и -Lc- независимо друг от друга выбирают из: -O-, -NH-C(O)-, -C(O)- NH-, -O-C(O)- NH- и -SO2-;
-Ld- выбирают из: -(CH2)n- и -(CH2-CH2-O)n-CH2-;
-Le- выбирают из: -(CH2)p1- и -(CH2)p1-O- (CH2)p2-;
m, n, p1, p2, o, r, q независимо друг от друга выбирают из: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10; и b, A, L*, Y, R1 и R2 имеют значения, определенные выше.
Особенно предпочтительными сепарационными материалами являются сепарационные материалы по общей формуле (II), (VII), (VIII), (IX) и (X), где
-L*- выбирают из: -L*a-, -L*a-L*e- и -L*a-L*b-L*e-; и
-L*a- выбирают из: -(CH2)o- и -CH2-CH(OH)- CH2-;
-L*e- представляет собой -(CH2)q-;
-L*b- выбирают из: -O- (CH2)r-O-, -S- (CH2)r-S-, -S- и -O-; и
o, q и r независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10.
В общих формулах (II), (VII), (VIII), (IX) и (X), L* предпочтительно представляет собой -L*a-L*b-L*e- и A предпочтительно выбирают из: полисульфона (PS), полиэфирсульфона (PES), полиакрилэфирсульфона (PAES), их смесей и/или производных.
Другой предпочтительный вариант осуществления настоящего изобретения относится к сепарационным материалам по общей формуле (II), (VII), (VIII), (IX) и (X), где
R1 и R2 независимо друг от друга выбирают из: -CH3 и -C2H5;
Y выбирают из: -CH(OH)- CH2-N(R4)-, -CH(OH)- CH2-S-, -CH(OH)- CH2-O-, -CH2-NH-, -S-CH2-CH2- и -CH2-CH2-S-;
R4 выбирают из: -H, -CH3, -C2H5, -C3H7 и -C(O)- CH3; и
-La- представляет собой -(CH2)m-.
Еще более предпочтительный вариант осуществления изобретения относится к сепарационному материалу по общей формуле (II)
где
R1 и R2 независимо друг от друга выбирают из: -CH3, -C2H5, -C3H7, -C4H9, -C5H11 и -C6H13, или R1 и R2 могут образовывать вместе с атомом азота, с которым они соединены, гетероцикл, выбираемый из:
, и
где один или несколько атома(ов) водорода может быть заменен атомом(ами) фтора;
Y выбирают из: -CH(OH)- CH2-N(R4)-, -CH(OH)- CH2-S-, -CH2-NH-, -NH-CH2-, -S-CH2-CH2- и -CH2-CH2-S-,
R4 выбирают из: -H, CH3, -C2H5, -C3H7 и -C(O)- CH3;
-L- выбирают из: -(CH2)m-, -(CH2)m-O-C(O)- NH- (CH2)p1-, -(CH2)m-O- (CH2)p1-, -(CH2)m-C(O)- NH- (CH2)p1-, -(CH2)m-NH-C(O)- (CH2)p1-, -(CH2)m-C(O)- NH- (CH2)n-O- (CH2)p1-, -(CH2)m-O-C(O)- NH- (CH2)n-O- (CH2)p1-, -(CH2)m-C(O)- NH- (CH2)n-C(O)- NH- (CH2)p1-O- (CH2)p2- и -(CH2)m-O-C(O)- NH- (CH2)n-C(O)- NH- (CH2)p1-O- (CH2)p2-;
m, n, p1 и p2 независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 и b, A и L* имеют значения, определенные выше, и более предпочтительно A выбирают из: полисульфона (PS), полиэфирсульфона (PES), полиакрилэфирсульфона (PAES), их смесей и/или производных, и протонированных и депротонированных форм указанного сепарационного материала.
Другой предпочтительный вариант осуществления изобретения относится к сепарационному материалу по общей формуле (II)
где b выбирают из 2 и 3;
R1 и R2 независимо друг от друга выбирают из: -CH3, -C2H5, -C3H7, -C4H9, -C5H11 и -C6H13, или R1 и R2 могут образовывать вместе с атомом азота, с которым они соединены, гетероцикл, выбираемый из:
, и
где один или несколько атома(ов) водорода может быть заменен атомом(ами) фтора;
Y выбирают из: -CH(OH)- CH2-N(R4)-, -CH(OH)- CH2-S-, -S-CH2-CH2- и -CH2-CH2-S-;
R4 выбирают из: -H, -CH3, -C2H5, -C3H7 и -C(O)- CH3;
-L- выбирают из: -(CH2)m-, -(CH2)m-O-C(O)- NH- (CH2)p1-, -(CH2)m-O- (CH2)p1-, -(CH2)m-C(O)- NH- (CH2)p1-, -(CH2)m-NH-C(O)- (CH2)p1-, -(CH2)m-C(O)- NH- (CH2)n-O- (CH2)p1-, -(CH2)m-O-C(O)- NH- (CH2)n-O- (CH2)p1-, -(CH2)m-C(O)- NH- (CH2)n-C(O)- NH- (CH2)p1-O- (CH2)p2- и -(CH2)m-O-C(O)- NH- (CH2)n-C(O)- NH- (CH2)p1-O- (CH2)p2-; и m, n, p1 и p2 независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10;
-L*- выбирают из: -(CH2)o-, -(CH2-CH2-O)o-C2H4-, (CH2-CH2-O)o-CH2-C(O)- NH- (CH2)q-, -(CH2)o-SO2- (CH2)q-, -CH2-CH(OH)- CH2-O-CH2-, -CH2-CH(OH)- CH2-O- (CH2)r-O-CH2-, и -(CH2-CH2-O)o-C2H4-O-CH2-;
o, r и q независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10; и A имеет значение, определенное выше, и более предпочтительно выбирают из: полисульфона (PS), полиэфирсульфона (PES), полиакрилэфирсульфона (PAES), их смесей и/или производных.
Другой предпочтительный вариант осуществления изобретения относится к сепарационному материалу по общей формуле (II)
и его пронированным и депротонированным формам
где b выбирают из 2 и 3;
R1 и R2 независимо друг от друга выбирают из: -CH3, -C2H5, -C3H7, -C4H9, -C5H11 и -C6H13, или R1 и R2 могут вместе с атомом азота, с которым они соединены, гетероцикл, выбираемый из:
, и
где один или несколько атома(ов) водорода может быть заменен атомом(ами) фтора;
Y выбирают из:
общей формуле (II), где Y представляет собой:

-L- выбирают из: -(CH2)m-, -(CH2)m-O-C(O)- NH-- (CH2)p1-, -(CH2)m-O- (CH2)p1-, -(CH2)m-C(O)- NH- (CH2)p1-, -(CH2)m-NH-C(O)- (CH2)p1-, -(CH2)m-C(O)- NH- (CH2)n-O- (CH2)p1-, -(CH2)m-O-C(O)- NH- (CH2)n-O- (CH2)p1-, -(CH2)m-C(O)- NH- (CH2)n-C(O)- NH- (CH2)p1-O- (CH2)p2- и -(CH2)m-O-C(O)- NH- (CH2)n-C(O)- NH- (CH2)p1-O- (CH2)p2-; и m, n, p1 и p2 независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10;
-L*- выбирают из: -(CH2)o-, -(CH2-CH2-O)o-C2H4-, -(CH2-CH2-O)o-CH2-C(O)- NH- (CH2)q-, -(CH2)o-SO2- (CH2)q-, -CH2-CH(OH)- CH2-O-CH2-, -CH2-CH(OH)- CH2-O- (CH2)r-O-CH2-, и -(CH2-CH2-O)o-C2H4-O-CH2-;
o, r, и q независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10; и A имеет значение, определенное выше, и более предпочтительно его выбирают из полисульфона (PS), полиэфирсульфона (PES), полиакрилэфирсульфона (PAES), их смесей и/или производных.
Указания:
Неожиданно было обнаружено, что вышеупомянутые соединения по общей формуле (I), (III), (IV), (V) и (VI), а также сепарационные материалы по общей формуле (II), (VII), (VIII), (IX) и (X) связываются с высокой аффинностью и высокой селективностью с СРБ и антифосфорилхолиновыми антителами. Следовательно, соединения по изобретению являются применимыми для удаления СРБ и антифосфорилхолиновых антител из биологических жидкостей.
Один вариант осуществления настоящего изобретения относится к способу экстракорпорального удаления СРБ из биологической жидкости пациента для профилактики и/или лечения иммунных дисфункций и сердечно-сосудистых заболеваний, включающему стадии:
a) обеспечения сепарационного материал по общей формуле (II), (VII), (VIII), (IX) или (X); и
b) контакта биологической жидкости пациента с сепарационным материалом.
Для этой цели сепарационный материал по общей формуле (II), (VII), (VIII), (IX) и (X) загружают в колонку, указанная колонка является применимой для экстракорпорального удаления СРБ из биологической жидкости пациента для профилактики и/или лечения иммунных дисфункций и сердечно-сосудистых заболеваний.
Как используется в настоящем описании термин биологическая жидкость охватывает кровь, плазму крови, перитонеальную жидкость и лимфатическую жидкость.
Размеры колонки не являются критическими, и их необходимо выбирать так, чтобы они были адаптированы к скорости тока биологической жидкости, удаляемой у пациента, что очень ассоциировано с массой тела пациента. Колонка, включающая сепарационный материал по настоящему изобретению, должна быть стерилизована, например, стерилизующим газом, таким как этиленоксид, и или использована немедленно или герметизирована и храниться для последующего применения. Перед использованием колонку необходимо промыть нормальным солевым раствором с последующей промывкой нормальным солевым раствором, содержащим любые другие подходящие подготовленные ингредиенты. Колонка может иметь обычный дизайн картриджа, псевдоожиженный слой, раздвижное ложе или монолит. Получение таких колонок хорошо известно специалисту. Например, получение колонки с обычным дизайном картриджа может быть осуществлено, как описано в патентной заявке EP 0237659 A1.
Регенерация колонки также возможна. С целью регенерации, например, для промывки колонки может быть использован 0,9% солевой раствор. После этого значение pH снижали с использованием буфера глицин/HCl (pH 2,8) с целью разрыва связи между СРБ и сепарационным материалом, с последующей стадией промывки PBS (фосфатный буферный раствор) при значении pH 7,4, и окончательной стадией промывки 0,9% солевым раствором.
Необязательно, колонка, включающая сепарационный материал по настоящему изобретению, содержит дополнительные сепарационные материалы, подходящие для удаления других интересующих веществ из биологической жидкости пациента. Другими веществами предпочтительно являются белки. Примерами других веществ являются интерлейкин-6, интерлейкин-1, TNFα, P-компонент амилоида сыворотки (SAP), PTX3, фибриноген, антитела (такие как IgG, IgE), антифосфолипидные антитела и компоненты системы комплемента, такие как C1q, C3a или C5a.
Колонка по настоящему изобретению может быть включена в устройство для обеспечения устройства, подходящего для экстракорпорального удаления СРБ из биологической жидкости пациента для профилактики и/или лечения иммунных дисфункций и сердечно-сосудистых заболеваний.
Следовательно, устройство по настоящему изобретению включает:
a) колонку, включающую сепарационный материал по общей формуле (II), (VII), (VIII), (IX) и (X); и
b) прибор для содержания колонки ex vivo, включающей сепарационный материал, в контакте с биологической жидкостью пациента, посредством этого снижая уровень СРБ в указанной биологической жидкости, и для возврата биологической жидкости пациенту.
Из-за высокой аффинности и селективности соединений по общей формуле (I), сепарационный материал по общей формуле (II), (VII), (VIII), (IX) и (X), колонка включает сепарационный материал по общей формуле (II), (VII), (VIII), (IX) или (X), и устройство включает колонку, которая включает сепарационный материал по общей формуле (II), (VII), (VIII), (IX) или (X), является особенно применимым в экстракорпоральном удалении СРБ из биологической жидкости пациента для профилактики и/или лечения иммунной дисфункции и сердечно-сосудистых заболеваний.
Предпочтительно, биологическую жидкость выбирают из: крови, плазмы крови, перитонеальной жидкости и лимфатической жидкости.
Термин ʺсердечно-сосудистые заболеванияʺ, как используется в настоящем описании, включает, без ограничения, инфаркт, инсульт, диабет, терминальную стадию заболевания почек, почечную недостаточность из-за гипертонии, эндотелиальные повреждения, эндотелиальную деструкцию, артериосклероз, тромбоз, атеросклероз, стеноз, рестеноз, атеросклеротические или тромботические заболевания, недостаточность кровотока, ишемические события, эмболию легких, стабильную и нестабильную стенокардию напряжения, ишемическую болезнь сердца, инфаркт миокарда, а также патологические результаты артериосклеротических или тромботических заболеваний.
Термин ʺиммунные дисфункцииʺ как используется в настоящем описании, включает, без ограничения, иммунные заболевания, аутоиммунные заболевания, реакции отторжения трансплантата, отторжение аллотрансплантата, отторжение ксенотрансплантата, отторжение типа трансплантат против хозяина, отторжение типа хозяин против трансплантата, сахарный диабет, ревматизм, ревматоидный артрит, псориатический артрит, анкилозирующий спондилит, рассеянный склероз, миастению gravis, вульгарный псориаз, болезнь Грейвса, синдром Гудпасчера, идиопатическую тромбоцитопеническую пурпуру (ITP), апластическую анемию, воспалительные заболевания кишечника (IBD), болезнь Крона (также известную как синдром Крона), язвенный колит, дилатационную кардиомиопатию (DCM), аутоиммунный тиреоидит, тиреоидит Хашимото, заместительную гормональную терапию (HRT), остеоартрит и подагру.
Химический синтез
A. Синтез соединений по общей формуле (I)
A.1 Соединения по изобретению по общей формуле I, где X представляет собой -NHR3, R3 выбирают из: -H, -CH3, -C2H5, -C3H7 и -L- представляет собой -La-, -La-O-Le-, -La-Le- (т.е. соединения по общей формуле 6), или X представляет собой -C≡CH или -CH=CH2 и -L- представляет собой -La-O-C(O)- NH-Le- или -La-O-C(O)- NH-Ld-O-Le- (т.е. соединения по общей формуле 5, где PG1 представляет собой аллилоксикарбонил или пропаргилоксикарбонил и R3 представляет собой -H) могут быть получены, начиная с коммерчески доступных аминоспиртов по общей формуле NHR3-L1-OH (1), где L1 представляет собой -La-, -La-O-Le-, -La-Le-, -Le-, -Ld-O-Le-, или -Ld-Le- в соответствии со схемой 1.
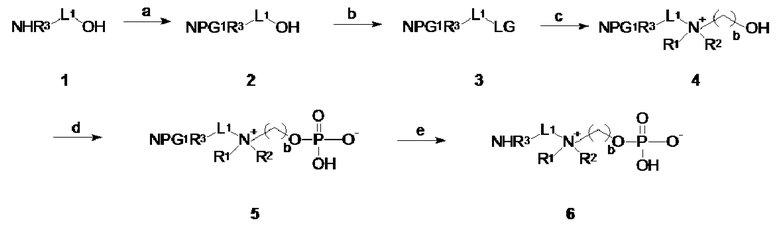
Схема 1: Синтез соединений по общей формуле 5 и 6: a. протекция аминогруппы; b. конверсия гидроксильной группы в уходящую группу LG; c. кватернизация; d. фосфорилирование; e. депротектирование.
Синтез включает защиту концевой аминогруппы аминоспирта 1 с помощью защитной группы PG1, с последующей конверсией концевой гидроксильной группы в уходящую группу LG для обеспечения промежуточного соединения 3. Подходящая аминозащитная группа PG1 хорошо известна специалисту в области техники и включает карбаматы, фталоиловую группу и замещенные фталоиловые группы. Примеры карбаматов включают, без ограничения: бензилоксикарбонил (Cbz или Z), 4-нитробензилоксикарбонил, 4-хлорбензилоксикарбонил, 4-метоксибензилоксикарбонил, 4-метилбензилоксикарбонил, 4-азидобензилоксикарбонил, 9-флуоренилметоксикарбонил (Fmoc), аллилоксикарбонил (Aloc), пропаргилоксикарбонил, трет-бутилоксикарбонил (Boc). Фталоиловые и замещенные фталоиловые защитные группы являются особенно применимыми для защиты первичных аминов (R3 представляет собой H). Пример замещенных фталоиловых групп включает, без ограничения, 4-нитрофталоил, 3-нитрофталоил и тетрахлорфталоил. Предпочтительно, защитную группу PG1 выбирают из аллилоксикарбонила, пропаргилоксикарбонила, бензилоксикарбонила и фталоила. Уходящую LG группу, вставляемую на стадии b, можно выбирать из группы, включающей хлорид, бромид, йодид, тозилат, бензолсульфонат, п-нитробензолсульфонат, мезилат или трифлатную группу. Предпочтительно уходящей группой LG является сложный эфир сульфоновой кислоты (т.е. сульфонат), и более предпочтительно LG представляет собой мезилатную группу. Конверсия спиртовой функциональной группы в группу LG может быть осуществлена, например, путем обработки спирта 2 с помощью MsCl или TsCl в присутствии триэтиламина, с использованием дихлорметана в качестве растворителя при 0°C. Промежуточное соединение 3 дополнительно реагирует с аминоспиртом по общей формуле NHR1R2- (CH2)b-OH, таким как диметилэтаноламин, диметилпропаноламин, N,N-диэтилэтаноламин, N-(2-гидроксиэтил)морфолин, или N-(2-гидроксиэтил)пиперидин для получения аминоспирта 4 посредством реакции кватернизации. Последующая обработка спирта 4 POCl3 и триэтаноламином в ацетонитриле при 0°C приводит к внесению в молекулу фосфатной группы. Депротектирование концевой аминогруппы обеспечивает целевые соединения по общей формуле 6 в соответствии с настоящим изобретением, которые готовы к иммобилизации на твердой подложке посредством концевой аминогруппы. Если защитная группа PG1 представляет собой аллилоксикарбонил или пропаргилоксикарбонил и R3 представляет собой -H, тогда стадия депротектирования является необязательной, пока соединения по общей формуле 5 могут быть непосредственно иммобилизованы на твердой подложке посредством терминальной алкеновой (тиол-еновой химии, быстрой химии, метатеза) или алкиновой группы (клик-химия).
Множество коммерчески доступных аминоспиртов по общей формуле NHR3-L1-OH (1) могут действовать как исходный материал для пути синтеза, описанного на схеме 1. Такие коммерчески доступные аминоспирты включают, без ограничения, 2-аминоэтанол, 3-аминопропан-1-ол, 4-аминобутан-1-ол, 5-аминопентан-1-ол, 6-аминогексан-1-ол, 2-(2-аминоэтокси)этанол, 2-[2-(2-аминоэтокси)этокси]этанол, 2-(3-аминопропокси)этанол. 3-(3-аминопропокси)пропан-1-ол, 4-(3-аминопропокси)бутан-1-ол, 2-(метиламино)этанол, 3-(метиламино)пропан-1-ол, 4-(метиламино)бутан-1-ол, 5-(метиламино)гексан-1-ол, 6-(метиламино)гексан-1-ол, 7-(метиламино)гептан-1-ол, 2-[2-(метиламино)этокси]этанол, 2-[2-[2-(метиламино)этокси]этокси]этанол, 2-[3-(метиламино)пропокси]этанол, 3-[3-(метиламино)пропокси]этанол, 3-[3-(метиламино)пропокси]пропан-1-ол, 4-[3-(метиламино)пропокси]бутан-1-ол.
Специалист в области техники понимает, что применяя стадии b, c и d методики синтеза, описанной в схеме 1, и если необходимо, соответствующую стадию депротектирования до спирта по общей формуле PG2S-L1-OH, HC≡C-L1-OH, CH2=CH-L1-OH, N3-L1-OH или PG3=C-L1-OH, где PG2 является сульфгидрильной защитной группой (например, бензил) и PG3 представляет собой альдегидную защитную группу (например, дитиан или ацеталь), аналоги 6a, 6b, 6c, 6d и 6e будут получены.
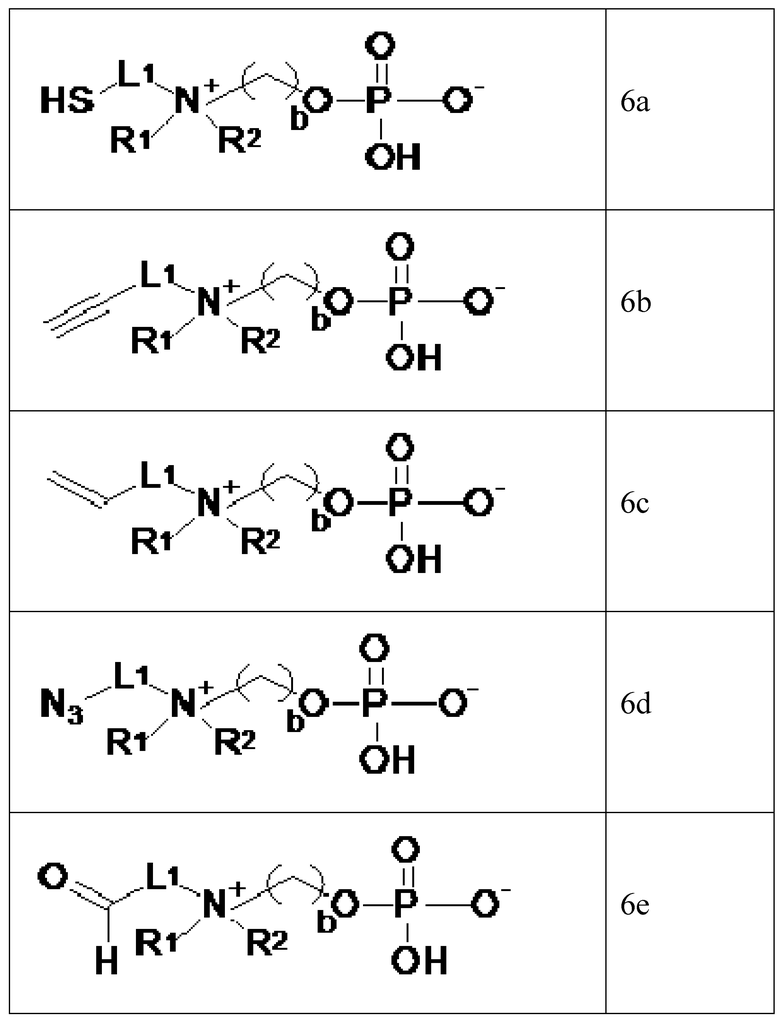
В таком случае пропиоловый спирт, 3-азидопропан-1-ол, 2-азидоэтанол, 4-азидобутан-1-ол, 2-(2-азидоэтокси)этанол, 2-[2-(2-азидоэтокси)этокси]этанол, 2-[2-[2-(2-азидоэтокси)этокси]этокси]этанол, 2-(2-азидоэтилсульфонил)этанол, бут-3-ин-1-ол, пент-4-ин-1-ол, 2-проп-2-иноксиэтанол, 2-(2-проп-2-иноксиэтокси)этанол, 2-[2-(2-проп-2-иноксиэтокси)этокси]этанол могут быть использованы в качестве исходных материалов.
A.2 Соединения по изобретению по общей формуле I, где X представляет собой -NHR3, R3 выбирают из: -H, -CH3, -C2H5, -C3H7 и -L- представляет собой -La-C(O)- NH-Le- (т.е. соединения по общей формуле 7 и 19) или X представляет собой -C≡CH или -CH=CH2 и -L- представляет собой -La-O-C(O)- NH-Ld-C(O)- NH-Le- (т.е. соединения по общей формуле 8 и 18, где PG1 представляет собой аллилоксикарбонил или пропаргилоксикарбонил R3 представляет собой -H) могут быть получены, начиная с аминокислот 9 по общей формуле NHR3-L2-CO2H, где -L2- представляет собой -La- или -Ld-, аминоспиртов 10 по общей формуле NH2-L1-OH (1), где L1 представляет собой -Le-, или диаминов 11 по общей формуле NH2-L3-NR1R2, где L3 представляет собой -Le- в соответствии со схемой 2.
Синтез начинается с защиты аминогруппы на аминокислоте 9 защитной группой PG1, где PG1 имеет значение, определенное в параграфе A.1 для получения карбоновой кислоты 12. Связывание карбоновой кислоты 12 с аминоспиртом 10 дает первичный спирт 13, на котором гидроксильная группа дополнительно преобразуется в уходящую группу LG, где уходящая группа LG определена в параграфе A.1 для обеспечения промежуточного соединения 14. Обработка промежуточного соединения 14 аминоспиртом 10 по общей формуле NHR1R2- (CH2)b-OH (например, диметилэтаноламином, диметилпропанoламином, N,N-диэтилэтаноламином, N-(2-гидроксиэтил)морфолином, или N-(2-гидроксиэтил)пиперидином) приводит к образованию спирта 15, который подвергается реакции кватернизации, с последующим депротектированием, включающим отщепление защитной группы PG1 для получения целевого соединения 7. Как ранее, предшественник 8 с PG1, являющейся аллилоксикарбонилом или пропаргилоксикарбонилом, может быть непосредственно иммобилизован на твердой подложке.
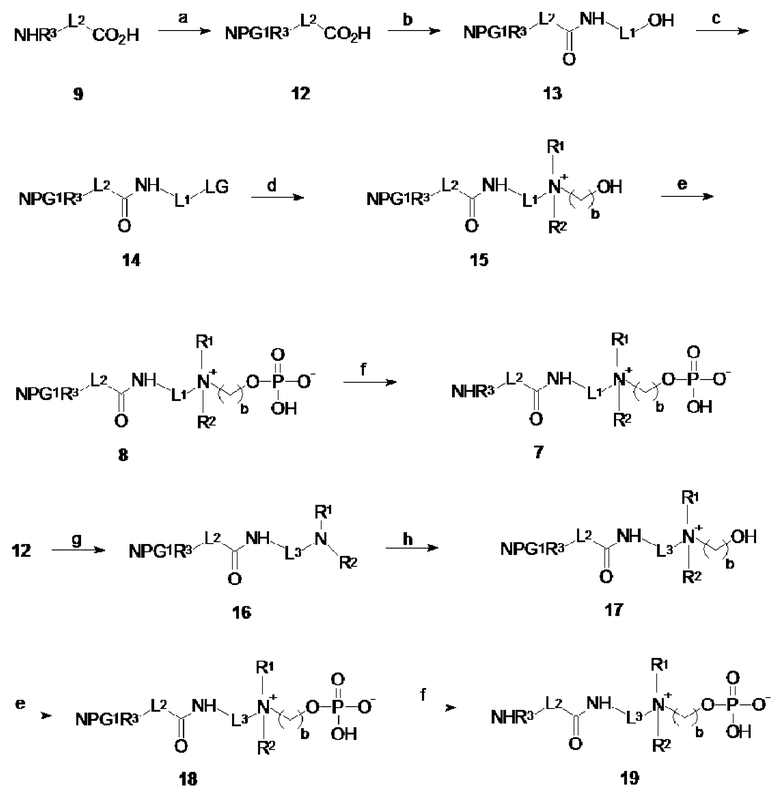
Схема 2: Синтез соединений по общей формуле 7, 8, 18 и 19: a. защита аминогруппы; b. 10, связывание; c. конверсия гидроксильной группы в уходящую гидроксильную группу в уходящую группу LG; d. кватернизация; e. фосфорилирование; f. депротектирование; g. 11, связывание; h. кватернизация.
Альтернативно, карбоновый спирт 12 может быть связан с диамином 11 для обеспечения амида 16, который впоследствии подвергается реакции кватернизации путем обработки производным 2-гидроксиэтила или 3-гидроксипропила. Подходящие производные 2-гидроксиэтила и 3-гидроксипропила включают 2-бром- и 2-йод-этанол и 3-бром и 3-йодпропанол. Реакцию кватернизации предпочтительно проводят в ацетонитриле, при комнатной температуре. Наконец, реакцию фосфорилирования, с последующей реакцией депротектирования, применямой к спирту 18, дает целевое соединение 19.
Аминокислоты 9 по общей формуле NHR3-L2-CO2H включают, без ограничения 7-аминогептанoвую кислоту, 6-аминогексанoвую кислоту, 5-аминопентановую кислоту, 4-аминобутановую кислоту, 3-аминопропановую кислоту, 2-аминоуксусную кислоту, 2-[2-(2-аминоэтокси)этокси]уксусную кислоту, 2-[2-[2-(2-аминоэтокси)этокси]этокси]уксусную кислоту, 2-(2-аминоэтокси)уксусную кислоту, 2-(3-аминопропокси)уксусную кислоту, 3-(3-аминопропокси)пропановую кислоту, 4-(3-аминопропокси)бутановую кислоту, 2-(метиламино)уксусную кислоту, 3-(метиламино)пропановую кислоту, 4-(метиламино)бутановую кислоту, 5-(метиламино)пентановую кислоту, 6-(метиламино)гексановую кислоту, 7-(метиламино)гептановую кислоту, 2-[2-(метиламино)этокси]уксусную кислоту, 2-[2-[2-(метиламино)этокси]этокси] уксусную кислоту, 2-[3-(метиламино)пропокси] уксусную кислоту, 3-[3-(метиламино)пропокси]пропанoвую кислоту, 4-[3-(метиламино)пропокси]бутановую кислоту, 2-[2-[2-[2-(метиламино)этокси]этокси]этокси] уксусную кислоту.
Аминоспирты 10 по общей формуле NHR1R2- (CH2)b-OH, подходящие для использования в схеме синтеза, отображаемой схемой 2, включают, без ограничения: 2-аминоэтанол, 3-аминопропан-1-ол, 4-аминобутан-1-ол, 5-аминопентан-1-ол, 6-аминогексан-1-ол, 2-(2-аминоэтокси)этанол, 2-[2-(2-аминоэтокси)этокси]этанол, 2-(3-аминопропокси)этанол, 3-(3-аминопропокси)пропан-1-ол, 4-(3-аминопропокси)бутан-1-ол.
Примеры диаминов 11 по общей формуле H2N-Le-NR1R2, которые являются коммерчески доступными, включают, без ограничения этилендиамин; 1,3 диаминопропан; N-метилэтилендиамин; 1,4-диаминобутан; 3-(метиламино)пропиламин; N,N′-диметилэтилендиамин; N-этилэтилендиамин; 3-(диметиламино)-1-пропиламин; N-изопропилэтилендиамин; N-пропилэтилендиамин; гексаметилендиамин; 1,2-диаминоциклогексин; 1,4-диаминоциклогексан; и N-гексилэтилендиамин.
Специалист в области техники понимает, что путем применения стадий b-e методики синтеза, описанной в схеме 2, и при необходимости соответствующей стадии депротектирования к карбоновой кислоте по общей формуле PG2S-L2-CO2H, HC≡C-L2-CO2H, CH2=CH-L2-CO2H, N3-L2-CO2H или PG3=C-L2-CO2H, где PG2 представляет собой сульфгидрильную защитную группу (например, бензил) и PG3 представляет собой альдегидную защитную группу (например, дитиан или ацеталь) могут быть получены аналоги 7a, 7b, 7c, 7d и 7e.
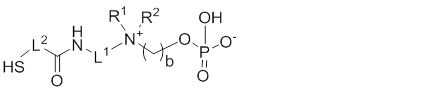
7а
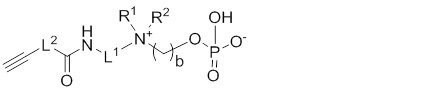
7b
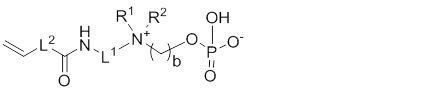
7c
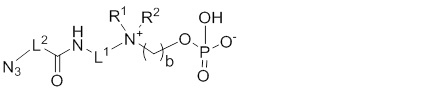
7d
7e
Примеры подходящих карбоновых кислот для использования в качестве исходного материала для получения аналогов 7a-7e включают, без ограничения: 6-тиогексанoвую кислоту, 6-азидогексанoвую кислоту, 5-тиопентановую кислоту, 5-азидопентановую кислоту, 4-тиобутанoвую кислоту, 4-азидобутановую кислоту, 3-тиопропанoвую кислоту, 3-азидопропановую кислоту, тиоуксусную кислоту, 3-бутиноевую кислоту, 4-пентиноевую кислоту.
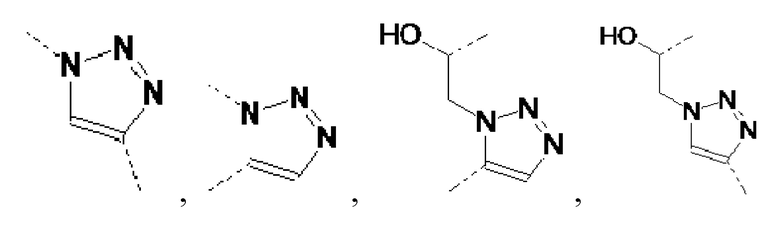
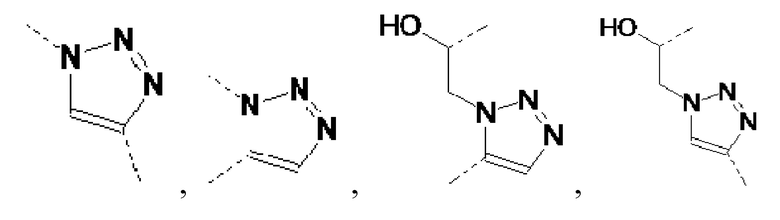
A.3 Альтернативно, диамины по общей формуле R3HN-L4-NR1R2 (20) могут быть успешно использованы для оценки соединений по общей формуле I, где X представляет собой -NHR3, R3 выбирают из: -H, -CH3, -C2H5, -C3H7 и -L- представляет собой -La- (см. схему 3). Диамины 20 могут быть коммерчески доступными (например, N,N-диметил-1,3-пропанeдиамин) или могут быть легко получены, например, начиная с соответствующих производных галогенов посредством двухстадийного синтеза, включающего конверсию производных галогена в азидопроизводные и восстановление азидопроизводных до соответствующих первичных аминов.
Начиная с диаминов 20 по общей формуле R3HN-L4-NR1R2, соединения по общей формуле 24 могут быть получены в четыре стадии, включая защиту первичного или вторичного амина защитной группой PG1, где PG1 имеет значение, определенное в параграфе A.1, кватернизацию третичного амина путем обработки соответствующим производным 2-гидроксиэтила или 3-гидроксипропила с последующим фосфорилированием и депротектированием первичного или вторичного амина.
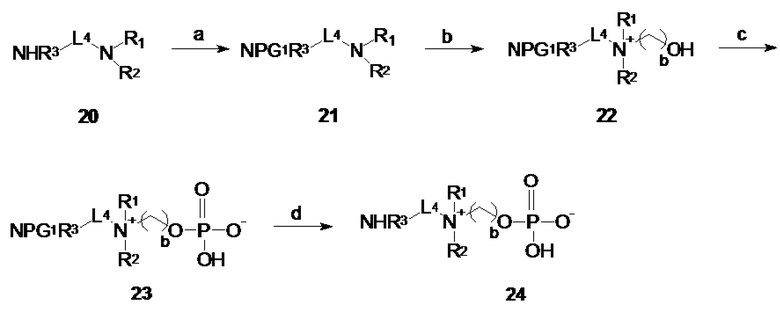
Схема 3: Синтез соединений по общей формуле 24: a. защита аминогруппы; b. кватернизация; c. фосфорилирование; d. депротектирование.
B. Получение сепарационного материала по общей формуле (II)
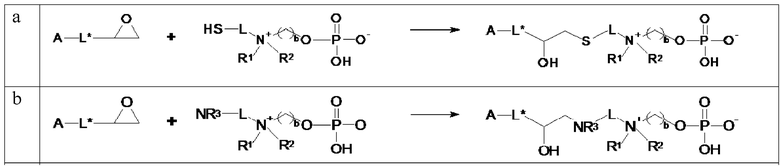
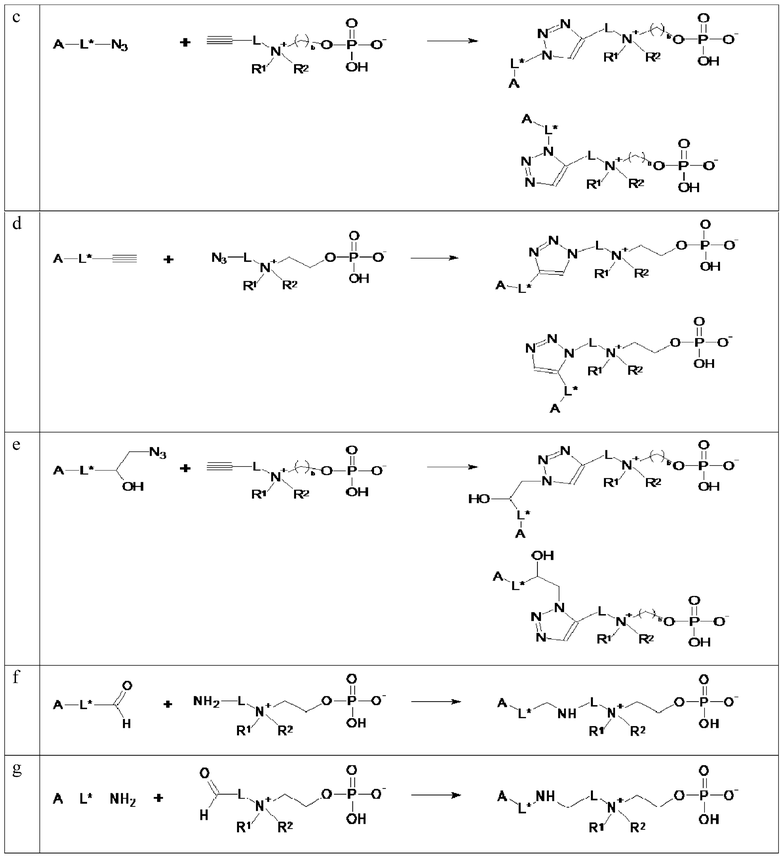
Сепарационный материал по общей формуле (II) может быть получен путем связывания соединений по общей формуле (I) с твердой подложкой A, функционализированной фрагментом-L*-FG, где FG представляет собой функциональную группу, которая подходит для реакции с функциональной группой X. Примеры групп FG включают -CHO, -C≡CH, -N3, -CH=CH2, -NH2, -SH, эпоксид, тресил. На схеме 4 суммированы различные методики связывания, которые могут быть использованы для получения сепарационных материалов по общей формуле (II). Предпочтительно, соединения по общей формуле (I), где X выбирают из -SH, -OH, и -NHR3, связываются с твердой подложкой A посредством открытия эпоксидного цикла (см. схему 5 a, b).
В одном варианте осуществления изобретения связывание соединений по общей формуле (I), где X=-C≡CH, -N3, или -CH=CH2 и твердой подложки имеет место с образованием триазолового фрагмента (см. схему 5 c, d, e). Образование триазолов из азида и алкина, также известное как алкин-азид циклоприсоединение Хисгена, проводили как 1,3-циклоприсоединение. Определенным вариантом 1,3-биполярного циклоприсоединения Хисгена является вариант, катализируемый медью(I), в котором органические азиды и терминальные алкины объединяют с получением 1,4-региоизомеров 1,2,3-триазолов, как единственных продуктов. Такую реакцию называют как катализируемое медью(I) Азид-Алкин циклоприсоединение (CuAAC). Реакция может быть проведена с использованием коммерческих источников меди (I), таких как бромид или йодид меди. Однако, реакция работает существенно лучше с использованием смеси меди (II) (например, сульфата меди(II)) и восстанавливающего агента (например, аскорбата натрия) для получения на месте Cu(l). Так как Cu(l) является нестабильной в водных растворителях, стабилизирующие лиганды являются эффективными для улучшения исходов реакции, особенно если используют трис-(бензилтриазолилметил)амин (TBTA). Реакция может быть запущена во множестве растворителей и смесей воды и множества (частично) смешиваемых органических растворителей, включая спирты, DMSO, DMF, t-BuOH, диоксан, ацетон и их смеси. Кроме того, реакция может быть катализирована рутением. Катализируемое рутением 1,3-биполярное азид-алкин циклоприсоединение (RuAAC) дает 1,5-триазолы. Следовательно, с использованием азид-алкин циклоприсоединения легко могут быть получены сепарационные материалы по общей формуле (II), где Y выбирают из:
Таким же образом, сепарационные материалы по общей формуле (II), где Y выбирают из:
могут быть получены, начиная с азидов и алкенов.
Сепарационные материалы по общей формуле (II), где Y выбирают из -CH2-NH-, и -NH-CH2-, могут быть получены путем восстановительного аминирования с использованием, например, NaBH4 в качестве восстановителя, начиная с соединений по общей формуле (I), где X представляет собой -NH2 или -CH(O), и твердой подложки, функционализированной A с помощью фрагмента -L*-FG, где функциональная группа FG представляет собой -CH(O) и -NH2, соответственно (см. схему 4 f, g).
Соединения по общей формуле (I), где X=-CH=CH2, могут быть иммобилизованы к твердой подложке посредством реакции обмена или реакции Виттига для обеспечения сепарационных материалов по общей формуле (II), где Y выбирают из:
Соединения по общей формуле (I), где X=-CH=CH2 или -SH может быть иммобилизован к твердой подложке посредством тиол-еновой химии для обеспечения сепарационных материалов по общей формуле (II), где Y представляет собой -S-CH2-CH2- или -CH2-CH2-S-.
Схема 5. Методики связывания соединений по общей формуле (I) с твердой подложкой.
Следующие примеры включены для демонстрации предпочтительных вариантов осуществления изобретения. Специалист в области техники понимает, что методики, описанные в следующих примерах, представляют собой методики, предложенные авторами изобретения для благоприятного осуществления изобретения, следовательно, составляют предпочтительные варианты его осуществления. Однако специалист в области техники должен в свете настоящего описания понимать, что множество изменений могут быть осуществлены в специфических вариантах осуществления изобретения, которые описаны, все еще получая подобный или сходный результат, без отклонения от рамок и тенденции изобретения.
Дополнительные модификации и альтернативные варианты осуществления различных аспектов по изобретению очевидны специалисту в области техники в свете описания. Соответственно, указанное описание расценивают, как только иллюстративное с целью указания специалисту в области техники общего варианта осуществления изобретения. Необходимо понимать, что формы изобретения, показанные и описанные в настоящем описании, понимают, как примеры вариантов осуществления изобретения. Элементы и материалы могут быть заменены таковыми, проиллюстрированными и описанными в настоящем описании, части и процессы могут быть переставлены и определенные характеристики изобретения могут быть использованы независимо, все как очевидно специалисту в области техники после получения преимущества настоящего описания изобретения. Изменения могут быть осуществлены в элементах, описанных в настоящем описании, без отклонения от рамок и тенденции изобретения, как описано в следующей формуле изобретения.
ПРИМЕРЫ
Пример 1: Синтез бензил N-[3-(диметиламино)пропил]карбамата (1*).

К перемешиваемому раствору N-(бензилоксикарбонилокси)сукцинимида (68,8 г, 0,276 моль) в хлороформе (300 мл) в атмосфере аргона и при 0°C добавляли по каплям N',N'-диметилпропан-1,3-диамин (24,52 г, 0,24 моль), растворенный в хлороформе (150 мл), при поддержании температуры реакционной смеси на 0°C. Реакционную смесь перемешивали при 0°C в течение 2 ч, затем разводили хлороформом (400 мл) и промывали насыщенным водным раствором гидрокарбоната натрия (3×200 мл) и насыщенным хлоридом натрия (2×200 мл). Органическую фазу сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме для получения 56 г (98,7%) вязкого масла в качестве желаемого продукта 1*, которое использовали как таковое для следующей реакции. MS м/з 236,31 (M+H)+ (236,15 рассчитанная).
Пример 2: Синтез бромида 3-(бензилоксикарбониламино)пропил-(2-гидроксиэтил)диметиламмония (2*).
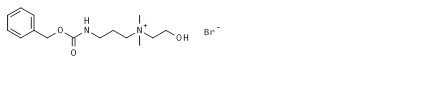
К приблизительно 1 M раствору бензил N-[3-(диметиламино)пропил]карбамата (1*, 56 г, 0,237 моль, пример 1) в ацетонитриле добавляли 2-бромэтанол (32,58 г, 0,261 моль) и реакционную смесь инкубировали при комнатной температуре до того, как весь бензил N-[3-(диметиламино)пропил]карбамат потреблялся (мониторируемый по ЖХ/МС и ТЖХ). К полученному раствору бромида 3-(бензилоксикарбониламино)пропил-(2-гидроксиэтил)диметиламмония добавляли пять объемов метил трет-бутилового эфира. Мутные смешанные растворы перемешивали до того, как обнаруживалось разделение фаз. Верхнюю органическую фазу отделяли от бромида 3-(бензилоксикарбониламино)пропил-(2-гидроксиэтил)диметиламмония путем сливания, и осаждение повторяли снова еще три раза. Наконец, целевое соединение получали после откачивания следового растворителя в вакууме в виде вязкого масла (76,7 г, 89,6% в виде соли бромида). Целевой продукт 2* был гомогенным по ЖХ/МС и ТЖХ, и его использовали на стадии фосфорилирования без последующей очистки. МС м/з 281,33 M+ (281,19 рассчитанная).
Пример 3: Синтез 6-(бензилоксикарбониламино)гексанoвой кислоты (3*).
К перемешиваемой суспензии мелко измельченной 6-аминокапроевой кислоты (32,79 г, 0,25 моль) в дихлорметане (500 мл) добавляли триэтиламин (63,24 г, 0,625 моль). Затем, триметилхлорсилан (54,32 г, 0,5 моль) добавляли по каплям при тщательном перемешивании и полученную смесь дефлегмировали в течение 2 ч. Затем реакционную смесь охлаждали на ледяной бане и добавляли бензилхлорформат (95%, 53,31 г, 0,313 моль). Полученную смесь перемешивали на льду в течение 60 мин и при комнатной температуре в течение 4 ч. После удаления растворителя остаток распределяли между 1000 мл 4% (об./масс.) раствора гидрокарбоната натрия и 500 мл метил трет-бутилового эфира. Водный слой разделяли, подкисляли до pH 2,0 с помощью 2Н соляной кислоты и экстрагировали три раза этилацетатом. Комбинированные органические слои сушили над безводным сульфатом магния, фильтровали и концентрировали для получения прозрачного масла (62,4 г, 94,1%), соответствующего целевой карбоновой кислоте 3*, которая была гомогенной по ЖХ/МС и ТЖХ, и использовали на следующей стадии без дополнительной очистки. МС м/з 265,33 (M+1)+ (265,13 рассчитанная).
Пример 4: Синтез бензил N-[6-[3-(диметиламино)пропиламино]-6-оксогексил]карбамата (4*).
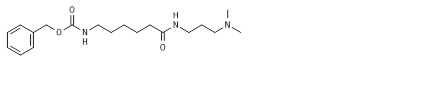
К раствору 6-(бензилоксикарбониламино)гексанoвой кислоты (3*, 66,3 г, 0,25 моль, пример 3) в сухом дихлорметане (500 мл), добавляли каталитическое количество диметилформамида (1,25 мл) и реакционную смесь охлаждали до 0°C на ледяной/солевой бане. Раствор оксалилхлорида (38,08 г, 0,3 моль) в дихлорметане (150 мл) медленно добавляли при поддержании температуры реакционной смеси на 0°C. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Летучие вещества выпаривали в вакууме и остаток растворяли в сухом дихлорметане (500 мл). Полученный раствор охлаждали до 0°C на ледяной/солевой бане и смесь N,N-диметил-1,3-пропандиамина (25,55 г, 0,25 моль) и триэтиламина (55,65 г, 0,55 моль) добавляли по каплям при перемешивании и охлаждении в течение 2 ч. Реакционную смесь перемешивали в течение 6 ч и прогресс реакции отслеживали посредством ТЖХ и ЖХ-МС. Осадок фильтровали, промывали небольшим объемом дихлорметана. Затем раствор дихлорметана промывали последовательно насыщенным гидрокарбонатом натрия, насыщенным хлоридом натрия, сушили над безводным сульфатом магния и концентрировали для получения бензил N-[6-[3-(диметиламино)пропиламино]-6-оксогексил]карбамата (4*, 76,4 г, 87,4%) в виде прозрачного желтоватого масла. Сырой материал очищали ЖХ/МС и ТЖХ и использовали на следующей стадии без дополнительной очистки. МС м/з 249,13 (M+1)+ (349,24 рассчитанная).
Пример 5: Синтез бромида 3-[6-(бензилоксикарбониламино)гексаноиламино]пропил-(2-гидроксиэтил)-диметиламмония (5*).
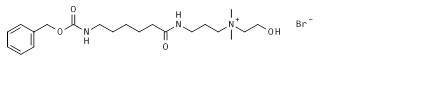
К приблизительно 1 M раствору бензил N-[6-[3-(диметиламино)пропиламино]-6-оксогексил]карбамата (4*, 56 г, 0,16 моль, пример 4) в ацетонитриле, добавляли 2-бромэтанол (22,03 г, 0,176 моль) и реакционную смесь перемешивали при комнатной температуре до потребления всего бензил N-[6-[3-(диметиламино)пропиламино]-6-оксогексил]карбамата (отслеживаемого по ЖХ/МС и ТЖХ). К полученному раствору бромида 3-[6-(бензилоксикарбониламино)гексаноиламино]пропил-(2-гидроксиэтил)-диметиламмония добавляли пять объемов метил трет-бутилового эфира. Непрозрачные смешанные растворы перемешивали до образования разделения фаз. Верхнюю органическую фазу отделяли от бромида 3-[6-(бензилоксикарбониламино)гексаноиламино]пропил-(2-гидроксиэтил)диметиламмония путем сливания и осаждение повторяли снова еще три раза. Наконец, целевое соединение получали после отсасывания следового растворителя в вакууме в виде вязкого масла (5*, 66,3 г, 87,2% в виде соли бромида). Продукт был гомогенным по ЖХ/МС и ТЖХ, и его использовали на стадии фосфорилирования без последующей очистки. МС м/з 394,1 M+ (394,27 рассчитанная).
Пример 6: Синтез бензил N-[2-(2-гидроксиэтокси)этил]карбамата (6*).

2-(2-аминоэтокси)этанол (25,23 г, 0,240 моль) растворяли в 530 мл 10% триэтиламина в MeOH (48 мл, 0,345 моль триэтиламина, 480 мл метанола) и охлаждали до 0°C. Бензилхлорформат (технической чистоты, 95%, 34,4 мл, 0,229 моль) добавляли одномоментно, и реакционной смеси позволяли нагреться до комнатной температуры. Через 12 ч растворитель удаляли путем выпаривания, и продукт очищали с использованием хроматографии на силикагелевой колонке (этилацетат/гексан), получая бесцветное масло (6*, 52 г, 0,163 моль, 95% на основе бензилхлорформата). Сырое вещество было аналитически чистым по ЖХ/МС и ТЖХ и его использовали на следующей стадии без дополнительной очистки. МС м/з 239,17 (M+1)+ (239,12 рассчитанная).
Пример 7: Синтез бензил N-[6-[2-(2-гидроксиэтокси)этиламино]-6-оксогексил]карбамата (7*).
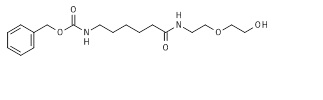
Раствор триметилфосфита (12,5 г, 0,101 моль) в дихлорметане (290 мл) охлаждали на ледяной/солевой бане. Затем порошкообразный йод (25,6 г, 0,101 моль) добавляли по частям при поддержании температуры реакционной смеси на 0°C. После полного растворения твердого йода, 6-(бензилоксикарбониламино)гексановую кислоту (3*, 25,5 г, 0,096 моль, пример 3) и триэтиламин (23,3 г, 0,23 моль) добавляли последовательно и полученный раствор перемешивали в течение 20 мин в охлаждающей бане при 0°C. Добавляли 2-(2-аминоэтокси)этанол (10,6 г, 0,101 моль), растворенный в дихлорметане (50 мл), и реакционную смесь перемешивали при 0°C в течение 30 мин. После удаления охлаждающей бани, реакционную смесь перемешивали в течение ночи при комнатной температуре. Прогресс реакции отслеживали посредством ТЖХ и ЖХ/МС. Затем, реакционную смесь промывали последовательно 10% (об./масс.) гидросульфатом натрия, насыщенным хлоридом натрия, насыщенным гидрокарбонатом натрия и сушили над безводным сульфатом магния. Растворитель удаляли выпариванием и продукт очищали с использованием хроматографии с силикагелевой колонкой (этилацетат/гексан), получая бесцветное масло (34,7 г, 0,091 моль, 95,1% на основании 6-(бензилоксикарбониламино)гексанoвой кислоты). Сырой продукт 7* очищали посредством ЖХ/МС и ТЖХ, и использовали на следующей стадии без дополнительной очистки. МС м/з 352,42 (M+H)+ (352,20 рассчитанная).
Пример 8: Синтез 2-[2-[6-(бензилоксикарбониламино)гексаноиламино]этокси]этилметансульфоната (8*).
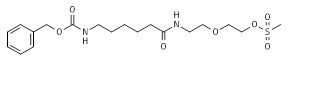
Раствор бензил N-[6-[2-(2-гидроксиэтокси)этиламино]-6-оксо-гексил]карбамата (7*, 17,25 г, 0,049 моль, пример 7) в метиленхлориде (115 мл) в аргоне охлаждали на ледяной-солевой бане до -5°C. Добавляли тиэтиламин (8,2 мл, 0,059 моль) и полученный раствор перемешивали при -5°C в течение 10 мин. Метансульфонилхлорид (4,5 мл, 0,059 моль) добавляли по каплям при поддержании температуры реакционной смеси между 0°C и -5°C. После завершения добавления реакционную смесь нагревали до комнатной температуры. Через 4 ч реакционную смесь промывали водой, насыщенным хлоридом натрия и снова водой. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Сырое вещество 8* очищали посредством ЖХ/МС и ТЖХ и использовали на следующей стадии без дополнительной очистки. МС м/з 430,52 (M+H)+ (430,18 рассчитанная).
Пример 9: Синтез соли метансульфоновой кислоты 2-[2-[6-(бензилоксикарбониламино)гексаноиламино]этокси]этил-(2-гидроксиэтил)диметиламмония (9*).
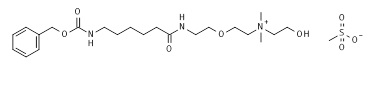
К 1 M раствору 2-[2-[6-(бензилоксикарбониламино)гексаноиламино]этокси]этилметансульфоната (8*, 18,6 г, 0,043 моль, пример 8) в ацетонитриле, 2-(диметиламино)этанол (4,24 г, 0,048 моль) добавляли в течение 20-30 минут. После завершения добавления полученный раствор дефлегмировали при непрерывном перемешивании до потребления всего алкилмезилата. Прогресс реакции отслеживали посредством ЖХ/МС и ТЖХ. Через 12 ч реакционную смесь охлаждали и ацетонитрил выпаривали в вакууме. Полученную смесь растворяли в 200 мл горячего метил трет-бутилового эфира. Раствору позволяли медленно остыть до комнатной температуры и затем позволяли стоять 12 ч при 0-5°C до завершения кристаллизации. Восстанавливали приблизительно 19,7 г (87,7% в виде соли метансульфоновой кислоты) белого твердого вещества, соответствующего целевому спирту 9*. Сырой материал очищали посредством ЖХ/МС и ТЖХ, и использовали на следующей стадии без дополнительной очистки. МС м/з 424,55 M+ (424,28 рассчитанная).
Пример 10: Синтез 6-(1,3-диоксоизоиндолил-2-ил)гексановой кислоты (10*).
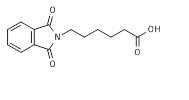
Перемешиваемую смесь 6-аминокапроевой кислоты (65,6 г, 0,5 моль), фталового ангидрида (74,1 г, 0,5 моль), и уксусной кислоты (115 мл) нагревали в колбе с обратным холодильником в течение 9 ч. Продукт, который кристаллизовался при охлаждении, выделяли фильтрацией, промывали несколько раз водой, и сушили в вакууме при 100°C для получения 124,2 г (95,1%) 6-(1,3-диоксоизоиндолин-2-ил)гексанoвой кислоты 10*, которая была гомогенной по ЖХ/МС и ТЖХ, и ее использовали на следующей стадии без дополнительной очистки. МС м/з 261,24 (M+H)+ (261,10 рассчитанная).
Пример 11: Синтез 2-[2-(2-гидроксиэтокси)этил]изоиндолин-1,3-диона (11*).
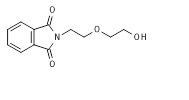
В 1 л трехгорлую круглодонную колбу, снабженную механической мешалкой, входом-выходом азота и насадкой Дина-Старка, снабженной обратным конденсатором, загружали раствор фталового ангидрида (88,87 г, 0,6 моль) и 2-(2-аминоэтокси)этанола (63,08 г, 0,6 моль) в толуоле (540 мл). Реакционную смесь дефлегмировали до того, как собирали 1 эквив воды в насадке Дана-Старка (приблизительно 24 ч). Через 24 ч толуол удаляли в вакууме и остаток растворяли в этилацетате (550 мл) и промывали последовательно 10% (об/масс) гидросульфата натрия, насыщенного хлорида натрия, насыщенного гидрокарбоната натрия и сушили над безводным сульфатом магния. Растворитель удаляли в вакууме и остаток кристаллизовали из метил трет-бутилового эфира. Восстанавливали приблизительно 130 г (92,1%) белого твердого вещества, соответствующего целевому спирту 11*. Сырой продукт очищали посредством ЖХ/МС и ТЖХ и использовали на следующей стадии без дополнительной очистки. МС м/з 235,27 (M+H)+ (235,08 рассчитанная).
Пример 12: Синтез 2-[2-(1,3-диоксоизоиндолин-2-ил)этокси]этилметансульфоната (12*).
Настоящий пример иллюстрирует способ получения метансульфонатного эфира N-защищенного 2-(2-аминоэтокси)этанола.
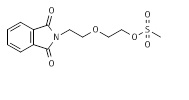
Раствор 2-[2-(2-гидроксиэтокси)этил]изоиндолин-1,3-диона (11*, 47,05 г, 0,2 моль, пример 11) в метиленхлориде (300 мл) в аргоне охлаждали на ледяной-солевой бане до -5°C. Добавляли триэтиламин (33,4 мл, 0,24 моль) и полученный раствор перемешивали при -5°C в течение 10 мин. Метансульфонилхлорид (18,6 мл, 0,24 моль) добавляли по каплям при поддержании температуры реакционной смеси между 0°C и -5°C. После окончания добавления реакционную смесь нагревали до комнатной температуры. Через 4 ч реакционную смесь промывали водой, насыщенным хлоридом натрия и снова водой. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Сырой материал оказался чистым по ЖХ/МС и ТЖХ и его использовали на следующей стадии без дополнительной очистки. МС м/з 313,17 (M+H)+ (313,06 рассчитанная).
Пример 13: Синтез соли метансульфоновой кислоты 2-[2-(1,3-диоксоизоиндолин-2-ил)этокси]этил-(2-гидроксиэтил)диметиламмония (13*).
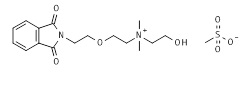
К приблизительно 1 M раствору 2-[2-(1,3-диоксоизоиндолин-2-ил)этокси]этилметансульфоната (12*, 32,53 г, 0,104 моль, пример 12) в ацетонитриле добавляли 2-(диметиламино)этанол (10,18 г, 0,114 моль) в течение 20-30 минут. После завершения добавления полученный раствор дефлегмировали при непрерывном перемешивании до потребления всего алкилмезилата. Прогресс реакции отслеживали посредством ЖХ/МС и ТЖХ. Через 12 ч реакционную смесь охлаждали и ацетонитрил выпаривали в вакууме. Полученную смесь растворяли в 150 мл горячего ацетонитрила и к полученному раствору последовательно добавляли 200 мл горячего метил трет-бутилового эфира. Раствору позволяли медленно остыть до комнатной температуры и затем позволяли стоять в течение ночи при 0-5°C до завершения кристаллизации. Восстанавливали приблизительно 50,7 г (92,6% в виде соли метансульфоновой кислоты) белого твердого вещества. Сырой материал, соответствующий целевому соединению 13*, очищали посредством ЖХ/МС и ТЖХ и использовали на следующей стадии без дополнительной очистки. МС м/з 307,07 M+ (307,17 рассчитанная).
Пример 14: Синтез соли метансульфоновой кислоты 2-[2-(1,3-диоксоизоиндолин-2-ил)этокси]этил-(3-гидроксипропил)диметиламмония (14*).
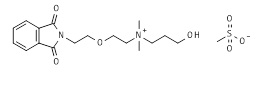
К приблизительно 1 M раствору 2-[2-(1,3-диоксоизоиндолин-2-ил)этокси]этилметансульфоната (12*, 30,41 г, 0,097 моль, пример 12) в ацетонитриле добавляли 3-(диметиламино)пропан-1-ол (11,01 г, 0,107 моль). Добавление 3-(диметиламино)пропан-1-ола занимало около 20-30 минут. Когда все было добавлено, полученный раствор дефлегмировали при непрерывном перемешивании до потребления всего алкилмезилата. Прогресс реакции отслеживали посредством ЖХ/МС и ТЖХ. Через 12 ч реакционную смесь охлаждали и ацетонитрил выпаривали в вакууме. Полученную смесь растворяли в 120 мл горячего ацетонитрила и к полученному раствору последовательно добавляли 200 мл горячего метил трет-бутилового эфира. Раствору позволяли медленно остыть до комнатной температуры и затем позволяли стоять в течение ночи при 0-5°C до завершения кристаллизации. Восстанавливали приблизительно 46,1 г (88,1%) белого твердого вещества. Сырое вещество, соответствующее целевому соединению 14*, было чистым по ЖХ/МС и ТЖХ, и его использовали на следующей стадии без дополнительной очистки. МС м/з 321,11 M+ (321,18 рассчитанная).
Пример 15: Эпокси-активация Сефарозы 6B.
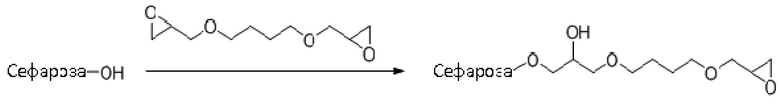
Сефарозу 6B (GE Healthcare Life Sciences) промывали на воронке со стеклянным фильтром с помощью 3×1000 мл порций воды и сушили отсасыванием. Затем сто грамм высушенной отсасыванием сефарозы 6B смешивали с 100 мл 1,4-бутандиол диглицидилового эфира (CAS N° 2425-79-8) и 100 мл 0,6 M раствора гидроксида натрия, содержащего 2 мг борогидрида натрия на миллилитр. Суспензию смешивали путем вращения в течении 8 ч при комнатной температуре и реакцию останавливали путем промывки геля на воронке со стеклянным фильтром с помощью 10×400 мл порций горячей (40-60°C) воды. Содержание оксирана в геле составило 67 мкмоль/г высушенного отсасыванием геля.
Пример 16: Анализ для определения количества эпоксигрупп.
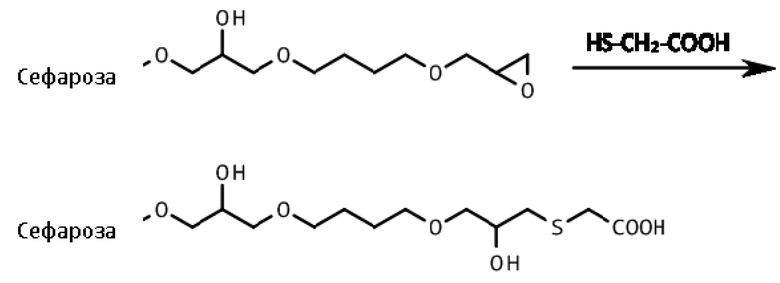
Эпокси-активированная Сефароза 6B реагировала с меркаптоуксусной кислотой и количество вносимых кислотных групп определяли путем титрации в соответствии со способом, описанным в Scoble, JA and Scopes, RK Journal of Chromatography A, 1996, 752, 67.
Пример 17: Синтез азид-функционализированной Сефарозы 6B
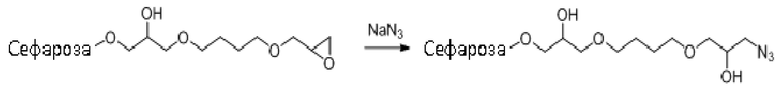
Десять граммов высушенной отсасыванием эпокси-активированной Сефарозы 6B (пример 15) добавляли в 50 мл 1 M раствора азида натрия в воде. Смесь инкубировали на лабораторном ротационном смесителе в течение 24 ч при комнатной температуре. Гель промывали 4×100 мл воды и держали в 20% (об./об.) этаноле до дальнейшего применения.
Пример 18: Анализ для определения количества азидо-групп.
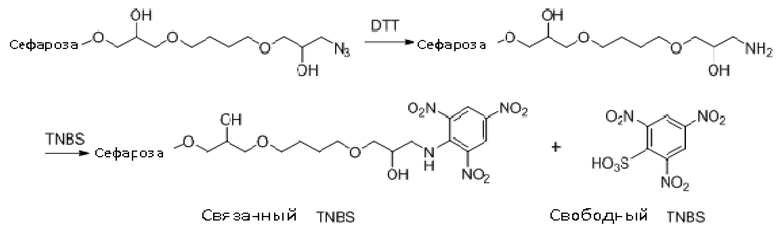
Азидогруппы азидо Сефарозы 6B количественно восстанавливали до аминогрупп посредством реакции с DTT (Handlon, AL and Oppenheimer, NJ, Pharmaceutical Research 1988, 5, 297.) и полученные аминогруппы количественно оценивали с TNBS в соответствии с опубликованной методикой (Antoni, G et al. Analytical Biochemistry 1983, 129, 60). Способ основан на реакции матрицы с избытком 2,4,6-тринитробензолсульфоновой (TNBS) и последующего количественного определения непрореагировавшего TNBS посредством реакции с глицином.
Пример 19: Определение свободных первичных аминогрупп посредством спектрофотометрического метода.
Количество первичных аминогрупп непосредственно количественно оценивали с использованием стандартной кривой, полученной после реакции L-глютаминовой кислоты с o-фтальальдегидом и N-ацетил-L-цистеином при комнатной температуре и pH 9,5 (Medina Hernandez, MJ et al. Microchemical Journal 1990, 42, 288). Производные N-ацетил-L-цистеина являются высокостабильными и не требуют четкого контроля времени реакции в отличие от 2-меркаптоэтанола. Были обнаружены линейные взаимоотношения между поглощением УФ при 335 нм и концентрацией аминогрупп.
Пример 20: Определение свободных вторичных аминогрупп посредством спектрофотометрического метода.
Количество вторичных аминогрупп непосредственно количественно оценивали с использованием реакции указанной группы соединений с нитропруссидом натрия и ацетальдегидом (Lin, CML and Wagner, C Analytical Biochemistry 1974, 60, 278).
Пример 21: Иммобилизация 2-[3-аминопропил(диметил)аммонио]этилгидрофосфата на эпокси-активированной Сефарозе 6B (Сепарационный материал 1).
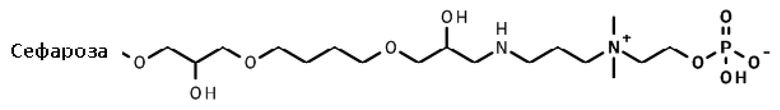
Фосфорилирование.
К 0,2 M раствору оксихлорида фосфора (3,7 г) в сухом ацетонитриле, выдерживаемом при 0°C - -10°C, добавляли триэтиламин (2,68 г). После добавления триэтиламина, приблизительно 0,2 M раствор бромида 3-(бензилоксикарбониламино)пропил-(2-гидроксиэтил)диметиламмония (2*, 5,81 г, пример 2) добавляли по каплям в течение временного промежутка 30 мин, при поддержании температуры реакционной смеси между 0°C и -5°C. После завершения добавления суспензию инкубировали в течение дополнительных 24 ч при комнатной температуре. Прогресс реакции отслеживали посредством ЖХ/МС и ТЖХ. Осадок гидрохлорида триэтиламина фильтровали и к полученному раствору добавляли воду (1,8 мл) при перемешивании, при поддержании температуры на 0-5°C. Удаление смеси вода-растворитель в вакууме давало фосфорилированный продукт, который был достаточно чистым для последующего депротектирования и иммобилизации.
Отщепление защитной группы.
К перемешиваемому раствору вышеуказанного фосфорилированного продукта в воде (50 мл) добавляли 10% палладий на активированном угле (10 моль%) при 25°C. Полученную смесь перемешивали в течение 24 ч при 25°C под H2 (1 атм) и затем фильтровали. Катализатор промывали водой (2×10 мл) и прозрачный раствор концентрировали при пониженном давлении для получения 2-[3-аминопропил(диметил)аммонио]этил гидрофосфата с высокой чистотой после фильтрации.
Иммобилизация.
Двадцать грамм высушенной отсасыванием эпокси-активированной Сефарозы 6B (пример 15) добавляли в раствор 2-[3-аминопропил(диметил)аммонио]этилгидрофосфата (40 мл). pH полученной суспензии доводили до 11,0 с помощью 1 M раствора гидроксида натрия. Смесь инкубировали на лабораторном ротационном миксере 24 ч при комнатной температуре. Гель промывали 4×100 мл автоклавированной воды и хранили в 20% (об./об.) этаноле до последующего применения.
Пример 22: Иммобилизация 2-[3-(6-аминогексаноиламино)пропилдиметиламмонио]этилгидрофосфата на эпокси-активированной Сефарозе 6B (Сепарационный материал 2).
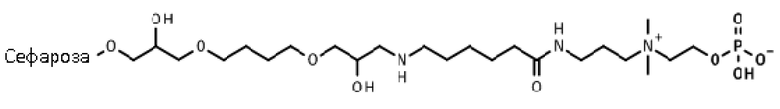
Фосфорилирование.
К приблизительно 0,2 M раствору оксихлорида фосфора (3,7 г) в сухом ацетонитриле, хранимом при между 0°C и -10°C, добавляли триэтиламин (2,68 г). После добавления триэтиламина, приблизительно 0,2 M раствор бромида 3-[6-(бензилоксикарбониламино)гексаноиламино]пропил-(2-гидроксиэтил)-диметиламмония (5*, 7,63 г, пример 5) добавляли по каплям в течение временного промежутка 30 мин при поддержании температуры реакционной смеси между 0°C и -5°C. После завершения добавления суспензию инкубировали в течение дополнительных 24 ч при комнатной температуре. Прогресс реакции отслеживали посредством ЖХ/МС и ТЖХ. Осадок гидрохлорида триэтиламина фильтровали и к полученному раствору добавляли воду (1,8 мл) при перемешивании, при поддержании температуры при 0-5°C. Удаление смеси воды-растворителя в вакууме давало фосфорилированный продукт, который был достаточно чистым для последующего депротектирования и иммобилизации.
Отщепление защитной группы.
К перемешиваемому раствору вышеуказанного фосфорилированного продукта в воде (50 мл) добавляли 10% палладий на активированном угле (10 моль%) при 25°C. Полученную смесь перемешивали в течение 24 ч при 25°C при H2 (1 атм) и затем фильтровали. Катализатор промывали водой (2×10 мл) и чистый раствор концентрировали при пониженном давлении для получения 2-[3-аминопропил(диметил)аммонио]этилгидрофосфата с высокой чистотой после фильтрации.
Иммобилизация.
Двадцать грамм высушенной отсасыванием эпокси-активированной Сефарозы 6B (пример 15) добавляли в раствор 2-[3-(6-аминогексаноиламино)пропилдиметиламмонио]этилгидрофосфата (40 мл). pH полученной суспензии доводили до 11,0 с 1 M раствором гидроксида натрия. Смесь инкубировали на лабораторном ротационном смесителе 24 ч при комнатной температуре. Гель промывали 4×100 мл автоклавированной воды и держали в 20% (об./об.) этаноле до последующего применения.
Пример 23: Иммобилизация 2-[2-[2-(6-аминогексаноиламино)этокси]этилдиметиламмонио]этилгидрофосфата на эпокси-активированной Сефарозе 6B (Сепарационный материал 3).
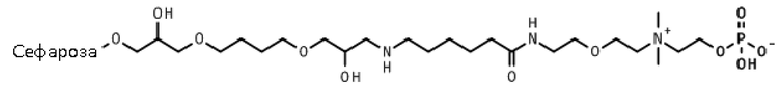
Фосфорилирование.
К приблизительно 0,2 M раствору оксихлорида фосфора (3,7 г) в сухом ацетонитриле, хранимом при от 0°C до -10°C, добавляли триэтиламин (2,68 г). После добавления триэтиламина, приблизительно 0,2 M раствор соли метансульфоновой кислоты 2-[2-[6-(бензилоксикарбониламино)гексаноиламино]этокси]этил-(2-гидроксиэтил)диметиламмония (9*, 8,36 г, пример 9) добавляли по каплям в течение временного диапазона 30 мин при поддержании температуры реакционной смеси между 0°C и -5°C. После завершения добавления суспензию инкубировали в течение дополнительных 24 ч при комнатной температуре. Прогресс реакции отслеживали посредством ЖХ/МС и ТЖХ. Осадок гидрохлорида триэтиламина фильтровали и к полученному раствору добавляли воду (1,8 мл) при перемешивании, при поддержании температуры на 0-5°C. Удаление смеси вода-растворитель в вакууме давало фосфорилированный продукт, который был достаточно чистым для последующего депротектирования и иммобилизации.
Отщепление защитной группы.
К перемешиваемому раствору вышеуказанного фосфорилированного продукта в воде (50 мл) добавляли 10% палладий на активированном угле (10 моль%) при 25°C. Полученную смесь перемешивали в течение 24 ч при 25°C под H2 (1 атм) и затем фильтровали. Катализатор промывали водой (2×10 мл) и прозрачный раствор концентрировали при пониженном давлении для получения 2-[3-аминопропил(диметил)аммонио]этилгидрофосфата с высокой чистотой после фильтрации.
Иммобилизация.
Двадцать грамм высушенной отсасыванием эпокси-активированной Сефарозы 6B (пример 15) добавляли в раствор 2-[2-[2-(6-аминогексаноиламино)этокси]этилдиметиламмонио]этилгидрофосфата (40 мл). pH полученной суспензии доводили до 11,0 с помощью 1 M раствора гидроксида натрия. Смесь инкубировали на лабораторном ротационном смесителе 24 ч при комнатной температуре. Гель промывали 4×100 мл автоклавированной воды и держали в 20% (об./об.) этаноле до последующего применения.
Пример 24: Иммобилизация 2-[2-(2-аминоэтокси)этилдиметиламмонио]этилгидрофосфата на эпокси-активированной Сефарозе 6B (Сепарационный материал 4).
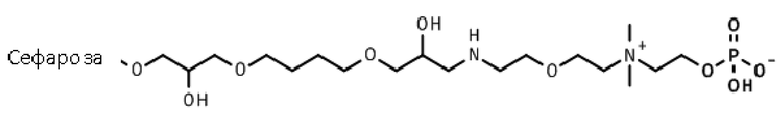
Фосфорилирование.
К приблизительно 0,2 M раствору оксихлорида фосфора (3,7 г) в сухом ацетонитриле, хранимом между 0°C и -10°C, добавляли триэтиламин (2,68 г). После добавления триэтиламина приблизительно 0,2 M раствор соли метансульфоновой кислоты 2-[2-(1,3-диоксоизоиндолин-2-ил)этокси]этил-(2-гидроксиэтил)диметиламмония (6,47 г, пример 13) добавляли по каплям в течение временного диапазона 30 мин при поддержании температуры реакционной смеси между 0°C и -5°C. После завершения добавления суспензию инкубировали в течение дополнительных 24 ч при комнатной температуре. Прогресс реакции отслеживали посредством ЖХ/МС и ТЖХ. Осадок гидрохлорида триэтиламина фильтровали и в полученный раствор добавляли воду (1,8 мл) при перемешивании, при поддержании температуры 0-5°C. Удаление смеси вода-растворитель в вакууме давало фосфорилированный продукт, который был достаточно чистым для последующего депротектирования и иммобилизации.
Отщепление защитной группы
К перемешиваемому раствору вышеуказанного фосфорилированного продукта в воде (50 мл) добавляли 1 M гидроксида натрия до того, как pH раствора достигнет 11,5. Полученную смесь перемешивали в течение 1 ч при 25°C и затем 6 M соляной кислоты добавляли до того как pH раствора достигала 2. Полученную смесь перемешивали в течение 12 ч при 25°C и затем pH реакционной смеси доводили до 7.
Иммобилизация.
Двадцать грамм высушенной отсасыванием эпокси-активированной Сефарозы 6B (пример 15) добавляли в раствор 2-[2-(2-аминоэтокси)этилдиметиламмонио]этилгидрофосфата (40 мл). pH полученной суспензии доводили до 11,0 с помощью 1 M раствора гидроксида натрия. Смесь инкубировали на лабораторном ротационном смесителе 24 ч при комнатной температуре. Гель промывали 4×100 мл автоклавированной воды и держали в 20% (об./об.) этаноле до дальнейшего применения.
Пример 25: Иммобилизация 3-[2-(2-аминоэтокси)этилдиметиламмонио]пропилгидрофосфата на эпокси-активированной Сефарозе 6B (Сепарационный материал 5).
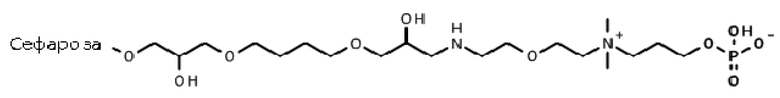
Фосфорилирование.
К приблизительно 0,2 M раствору оксихлорида фосфора (3,7 г) в сухом ацетонитриле, хранимому при от 0°C и -10°C, добавляли триэтиламин (2,68 г). После добавления триэтиламина, приблизительно 0,2 M раствор соли метансульфоновой кислоты 2-[2-(1,3-диоксоизоиндолин-2-ил)этокси]этил-(3-гидроксипропил)диметиламмония (14*, 6,7 г, пример 14) добавляли по каплям в течение временного промежутка 30 мин при поддержании температуры реакционной смеси от 0°C до -5°C. После завершения добавления суспензию инкубировали в течение дополнительных 24 ч при комнатной температуре. Прогресс реакции отслеживали посредством ЖХ/МС и ТЖХ. Осадок гидрохлорида триэтиламина фильтровали и к полученному раствору добавляли воду (1,8 мл) при перемешивании, поддерживая температуру на уровне 0-5°C. Удаление смеси воды-растворителя в вакууме давало фосфорилированный продукт, который был достаточно чистым для последующего депротектирования и иммобилизации.
Отщепление защитной группы.
К перемешиваемому раствору вышеуказанного фосфорилированного продукта в воде (50 мл) добавляли 1 M гидроксид натрия до того, как pH раствора достигала 11,5. Полученную смесь перемешивали в течение 1 ч при 25°C и затем 6 M соляную кислоту добавляли до того, как pH раствора достигала 2. Полученную смесь перемешивали в течение 12 ч при 25°C и затем pH реакционной смеси доводили до 7.
Иммобилизация.
Двадцать грамм высушенной отсасыванием эпокси-активированной Сефарозы 6B (пример 15) добавляли в раствор 3-[2-(2-аминоэтокси)этилдиметиламмонио]пропилгидрофосфата (40 мл). pH полученной суспензии доводили до 11,0 с помощью 1 M раствора гидроксида натрия. Смесь инкубировали на лабораторном ротационном смесителе 24 ч при комнатной температуре. Гель промывали 4×100 мл автоклавированной воды и хранили в 20% (об./об.) этаноле до дальнейшего применения.
Пример 26: Сравнительный пример
Иммобилизация (4-аминофенил) 2-(триметиламмонио)этил фосфата на эпокси-активированной Сефарозе 6B (Сепарационный материал 6).
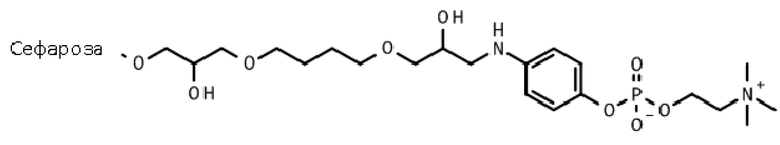
Иммобилизация.
Двадцать грамм высушенного отсасыванием эпокси-активированной Сефарозы 6B (пример 15) добавляли в раствор (4-аминофенил) 2-(триметиламмонио)этилфосфата (4,41 г в 40 мл воды). pH полученной суспензии доводили до 11,5 с помощью 1 M раствора гидроксида натрия. Смесь инкубировали на лабораторном ротационном миксере 24 ч при комнатной температуре. Гель промывали 4×100 мл автоклавированной воды и держали в 20% (об./об.) этаноле до дальнейшего применения.
Пример 27: Определение афинности связывания
Связывающий и промывочный буфер: 100 мM Tris, pH 8,0, 200 мM хлорида натрия, 2 мM хлорида кальция.
Элюирующий буфер: 100 мM Tris, pH 8,0, 200 мM хлорида натрия, 2 мM EDTA.
Все хроматографические операции проводили при комнатной температуре.
0,5 г каждого сепарационного материала упаковывали в небольшую колонку и уравновешивали с помощью связывающего буфера в количестве 12 объемов ионообменной смолы (6 мл). 40-50 мл человеческой сыворотки или плазмы (CРБ>0,1 мг/мл) наносили на каждую колонку при 1,2 мл/мин. Несвязавшийся материал отмывали с помощью связывающего буфера в количестве 60 объемов ионообменной смолы (30 мл), пока OD280 считывание не приближалось к исходному нулю. Аффинные связанные со смолой белки элюировали с помощью элюирующего буфера со скоростью 1,2 мл/мин и собирали фракции 3,6 мл. Собранные фракции хранили при 4°C до анализа посредством SDS-PAGE. Количественное определение человеческого С-реактивного белка (CРБ) проводили посредством ELISA.
Результаты суммированы в таблице 1.
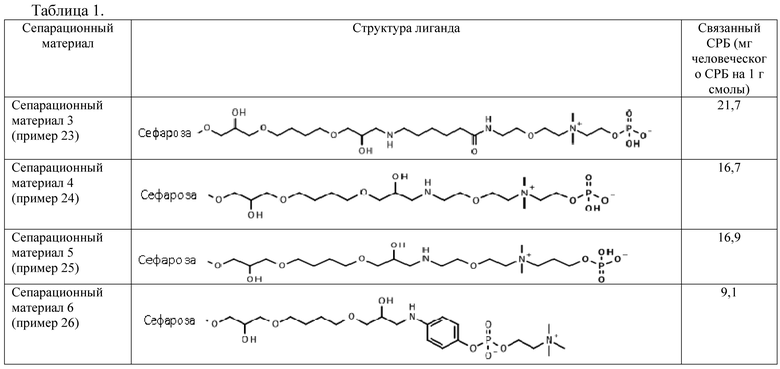
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ КОРИЧНОЙ КИСЛОТЫ, ПРОИЗВОДНОЕ КОРИЧНОЙ КИСЛОТЫ И ПОЛИСАХАРИДА, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО КОРИЧНОЙ КИСЛОТЫ И ГЛИКОЗАМИНОГЛИКАНА, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО КОРИЧНОЙ КИСЛОТЫ И ПОЛИАМИНОСАХАРИДА, СПОСОБ ПОЛУЧЕНИЯ СШИТОГО ПРОИЗВОДНОГО КОРИЧНОЙ КИСЛОТЫ И ПОЛИСАХАРИДА | 1995 |
|
RU2169136C2 |
| АНТИГИПЕРТЕНЗИВНЫЕ СОЕДИНЕНИЯ ДВОЙНОГО ДЕЙСТВИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2476427C2 |
| ВОДОРАСТВОРИМЫЕ АНАЛОГИ СС-1065 И ИХ КОНЪЮГАТЫ | 2007 |
|
RU2489423C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ, ВОЗДЕЙСТВУЮЩИЕ НА ГЛАЗА, И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ГЛАЗНЫХ ЗАБОЛЕВАНИЙ | 2020 |
|
RU2830634C1 |
| ЗАМЕЩЕННЫЕ ЖЕЛЧНЫМИ КИСЛОТАМИ ФЕНИЛАЛКЕНОИЛГУАНИДИНЫ, ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1999 |
|
RU2232769C2 |
| ПРОИЗВОДНЫЕ КАЛИХЕАМИЦИНА И ИХ КОНЪЮГАТЫ "АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО" | 2018 |
|
RU2732568C1 |
| СОЕДИНЕНИЯ И ПРОИЗВОДНЫЕ ЗАМЕЩЕННОГО ГИДРОКСИСТИЛЬБЕНА, ИХ СИНТЕЗ И ПРИМЕНЕНИЕ | 2020 |
|
RU2828710C1 |
| МАКРОЛИДЫ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ НА ИХ ОСНОВЕ | 2000 |
|
RU2243230C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА И ХИНОКСАЛИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ЦИСТЕИНИЛ-ЛЕЙКОТРИЕНА | 2012 |
|
RU2605929C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ТРАНСГЛУТАМИНАЗЫ ТКАНЕЙ | 2013 |
|
RU2652987C2 |
Изобретение относится к соединению формулы (I), сепарационному материалу формулы (II), колонке для экстракорпорального удаления С-реактивного белка (СРБ) и устройству ее содержащему, которые могут быть применены в медицине:
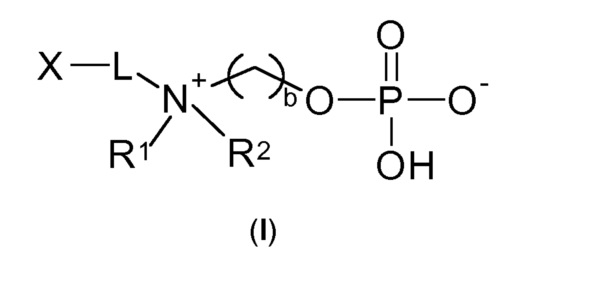
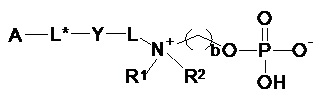 (II)
(II)
где b выбран из 2 и 3; X выбран из -SH, -NH2, -C≡CH, -CH=CH2, -N3 и -CHO; R1 и R2 выбраны из -CH3, -C2H5, -C3H7, -C4H9, -C5H11 и -C6H13, или R1 и R2 образуют вместе с атомом азота, с которым они соединены, гетероцикл, выбираемый из:
 ,
, 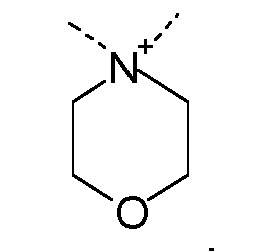 и
и 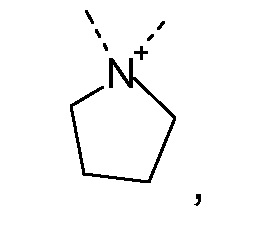
где атомы водорода могут быть заменены атомами фтора; -L- в формуле (I) выбран из
-(CH2)m-O-C(O)-NH-(CH2)p1-, -(CH2)m-O-(CH2)p1-, -(CH2)m-C(O)-NH-(CH2)p1-, -(CH2)m-NH-C(O)-(CH2)p1-, -(CH2)m-C(O)-NH-(CH2)n-O-(CH2)p1-, -(CH2)m-O-C(O)-NH-(CH2)n-O-(CH2)p1-, -(CH2)m-C(O)-NH-(CH2)n-C(O)-NH-(CH2)p1-O-(CH2)p2-, и -(CH2)m-O-C(O)-NH-(CH2)n-C(O)-NH-(CH2)p1-O-(CH2)p2-; m, n, p1, p2, o, r, q выбраны из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10; Y выбран из -CH(OH)-CH2-NH-, -CH(OH)-CH2-S-, -CH2-NH-, -NH-CH2-, -CH2-CH2-S-, -S-CH2-CH2-, 
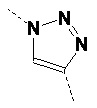 ,
,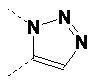 ,
, 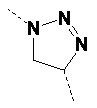 ,
,
,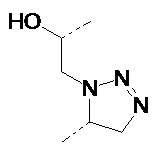 ,
, ,
,  и
и  ;
;
-L- в формуле (II) выбран из -La-, -La-Le-, -La-Lb-Le- и -La-Lb-Ld-Lc-Le-, где -La- выбран из -(CH2)m-, -(CH2-CH2-O)m-CH2-; -Lb- и -Lc- выбраны из -O-, -NH-C(O)-, -C(O)- NH-, -O-C(O)- NH- и -SO2-; -Ld- выбран из -(CH2)n-, -(CH2-CH2-O)n-CH2-; -Le- выбран из
-(CH2)p1-, -(CH2)p1-O-(CH2)p2-; -L*- выбран из -L*a-, -L*a-L*e- и -L*a-L*b-L*e-, где -L*a- выбран из -(CH2)o-, -(CH2-CH2-O)o-C2H4-, -(CH2-CH2-O)o-CH2- и -CH2-CH(OH)-CH2-; -L*e- выбран из -(CH2)q-, -C2H4-(O-CH2-CH2)q-, и CH2-(O-CH2-CH2)q-; -L*b- выбран из -O-(CH2)r-O-, -S-(CH2)r-S-, -SO2-, -S-, -O-, -NH-C(O)-, -C(O)-NH- и -S-S-; и A представляет собой твердую подложку из агарозы и сефарозы®. Предложены новое соединение, новый сепарационный материал, новая колонка и устройство, ее содержащее, для эффективного экстракорпорального удаления СРБ из биологической жидкости для профилактики или лечения иммунных дисфункций и сердечно-сосудистых заболеваний. 4 н. и 5 з.п. ф-лы, 27 пр., 1 табл.
1. Соединение общей формулы (I)
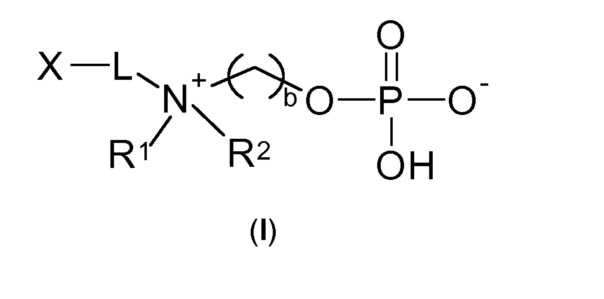
где b выбирают из 2 и 3;
R1 и R2 независимо друг от друга каждый выбирают из: -CH3, -C2H5, -C3H7, -C4H9, -C5H11 и -C6H13, или R1 и R2 могут образовывать вместе с атомом азота, с которым они соединены, гетероцикл, выбираемый из:
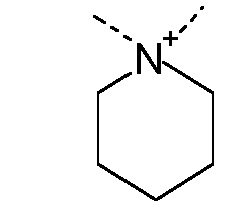 ,
, 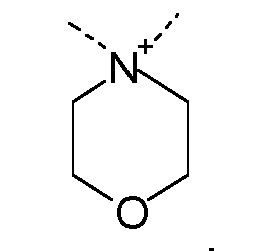 , и
, и 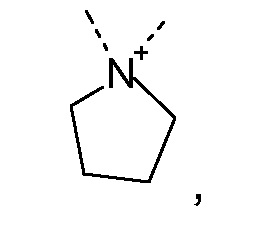
где один или более из атома(ов) водорода могут быть заменены атомом(ами) фтора;
X выбирают из: -SH, -NHR3, -C≡CH, -CH=CH2, -N3 и -CHO;
R3 представляет собой -H;
-L- выбирают из: -(CH2)m-O-C(O)-NH-(CH2)p1-, -(CH2)m-O-(CH2)p1-, -(CH2)m-C(O)-NH-(CH2)p1-, -(CH2)m-NH-C(O)-(CH2)p1-, -(CH2)m-C(O)-NH-(CH2)n-O-(CH2)p1-, -(CH2)m-O-C(O)-NH-(CH2)n-O-(CH2)p1-, -(CH2)m-C(O)-NH-(CH2)n-C(O)-NH-(CH2)p1-O-(CH2)p2-, и -(CH2)m-O-C(O)-NH-(CH2)n-C(O)-NH-(CH2)p1-O-(CH2)p2-;
и m, n, p1 и p2 независимо друг от друга выбирают из 1, 2, 3, 4, 5.
2. Соединение по п. 1, выбранное из:
2-[2-(2-аминоэтокси)этилдиэтиламмонио]этилгидрофосфата;
2-[4-[2-(2-аминоэтокси)этил]морфолин-4-иум-4-ил]этилгидрофосфата;
2-[1-[2-(2-аминоэтокси)этил]пиперидин-1-иум-1-ил]этилгидрофосфата;
2-[2-(2-аминоэтокси)этилдиметиламмонио]этилгидрофосфата;
2-[3-(6-аминогексаноиламино)пропилдиэтиламмонио]этилгидрофосфата;
2-[1-[2-[2-(6-аминогексаноиламино)этокси]этил]пиперидин-1-иум-1-ил]этилгидрофосфата;
2-[4-[2-[2-[3-(6-аминогексаноиламино)пропаноиламино]этокси]этил]морфолин-4-иум-4-ил]этилгидрофосфата;
2-[1-[2-[2-[6-(6-аминогексаноиламино)гексаноиламино]этокси]этил]пирролидин-1-иум-1-ил]этилгидрофосфата;
2-[2-аллилоксиэтил(диметил)аммонио]этилгидрофосфата;
2-[2-аллилоксиэтил(диэтил)аммонио]этилгидрофосфата;
2-[4-(2-аллилоксиэтил)морфолин-4-иум-4-ил]этилгидрофосфата;
2-[1-(2-аллилоксиэтил)пиперидин-1-иум-1-ил]этилгидрофосфата;
2-[2-[2-(6-аминогексаноиламино)этокси]этил-диметил-аммонио]этилгидрофосфата;
2-[2-[2-[3-(6-аминогексаноиламино)пропаноиламино]этокси]этил-диметил-аммонио]этилгидрофосфата;
2-[3-азидопропил(диметил)аммонио]этилгидрофосфата;
2-[диметил-[2-[2-(проп-2-иноксикарбониламино)этокси]этил]аммонио]этилгидрофосфата;
2-[2-[2-(аллилоксикарбониламино)этокси]этил-диметил-аммонио]этилгидрофосфата;
2-[2-[2-[6-(аллилоксикарбониламино)гексаноиламино]этокси]этил-диметил-аммонио]этилгидрофосфата;
2-[2-(6-аминогексаноиламино)этилдиметиламмонио]этилгидрофосфата;
2-[диметил-[3-[6-(проп-2-иноксикарбониламино)гексаноиламино]пропил]аммонио]этилгидрофосфата;
2-[3-(6-аминогексаноиламино)пропил-диметил-аммонио]этилгидрофосфата.
3. Сепарационный материал общей формулы (II):
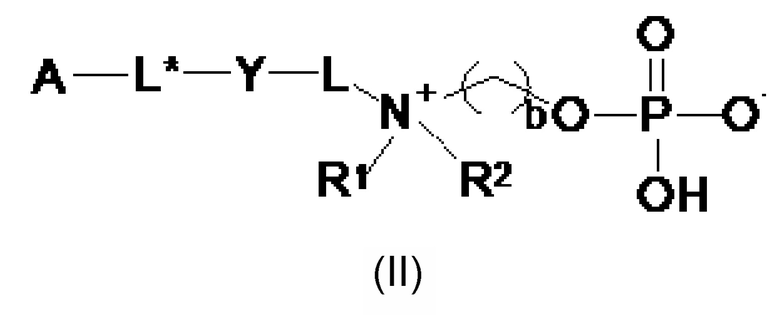
где
b выбирают из 2 и 3;
R1 и R2 независимо друг от друга выбирают из: -CH3, -C2H5, -C3H7, -C4H9, -C5H11 и -C6H13, или R1 и R2 могут образовывать вместе с атомом азота, с которым они соединены, гетероцикл, выбираемый из:
, , и
где один или более из атома(ов) водорода могут быть заменены атомом(ами) фтора;
Y выбирают из: -CH(OH)-CH2-N(R4)-, -CH(OH)-CH2-S-, -CH2-NH-, -NH-CH2-, -CH2-CH2-S-, -S-CH2-CH2-,
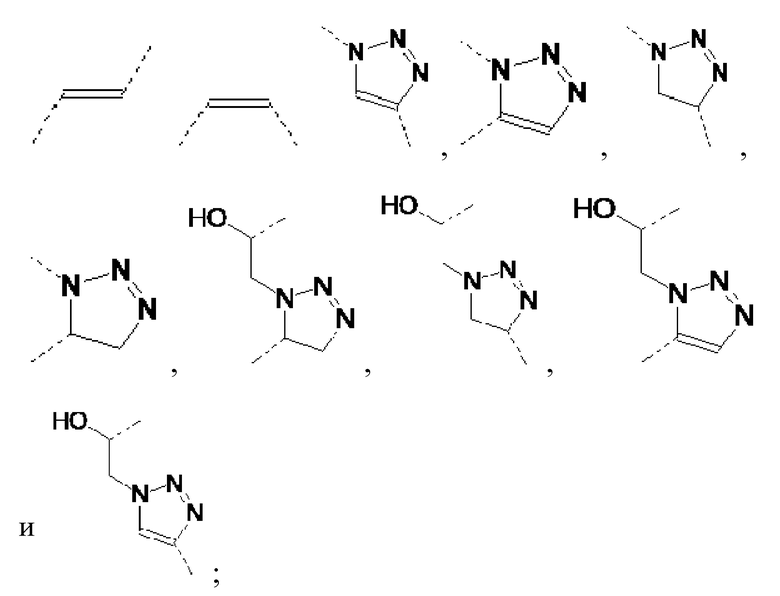
R4 представляет собой -H;
-L- выбирают из: -La-, -La-Le-, -La-Lb-Le- и -La-Lb-Ld-Lc-Le-, где
-La- выбирают из: -(CH2)m-, -(CH2-CH2-O)m-CH2-,
-Lb- и -Lc- независимо друг от друга выбирают из: -O-, -NH-C(O)-, -C(O)- NH-, -O-C(O)- NH- и -SO2-;
-Ld- выбирают из: -(CH2)n-, -(CH2-CH2-O)n-CH2-,
-Le- выбирают из: -(CH2)p1-, -(CH2)p1-O-(CH2)p2-,
-L*- выбирают из: -L*a-, -L*a-L*e- и -L*a-L*b-L*e-, где
-L*a- выбирают из: -(CH2)o-, -(CH2-CH2-O)o-C2H4-, -(CH2-CH2-O)o-CH2- и -CH2-CH(OH)-CH2-;
-L*e- выбирают из: -(CH2)q-, -C2H4-(O-CH2-CH2)q-, и -CH2-(O-CH2-CH2)q-;
-L*b- выбирают из: -O-(CH2)r-O-, -S-(CH2)r-S-, -SO2-, -S-, -O-, -NH-C(O)-, -C(O)-NH- и -S-S-;
m, n, p1, p2, o, r, q независимо друг от друга выбирают из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10; и
A представляет собой твердую подложку, выбираемую из группы, состоящей из: агарозы и сефарозы®.
4. Колонка для экстракорпорального удаления С-реактивного белка (СРБ), включающая сепарационный материал по п. 3.
5. Устройство для экстракорпорального удаления С-реактивного белка (СРБ), включающее колонку по п. 4.
6. Соединение по любому из пп. 1 и 2 или сепарационный материал по п. 3 для применения в экстракорпоральном удалении С-реактивного белка (СРБ) из биологической жидкости для профилактики и/или лечения иммунных дисфункций и сердечно-сосудистых заболеваний.
7. Соединение для применения или сепарационный материал для применения по п. 6, где биологическую жидкость выбирают из: крови, плазмы крови, перитонеальной жидкости и лимфатической жидкости.
8. Соединение для применения или сепарационный материал для применения по п. 6, где сердечно-сосудистые заболевания выбирают из группы, включающей инфаркт, инсульт, диабет, терминальную стадию болезни почек, почечную недостаточность, почечную недостаточность из-за гипертонии, повреждения эндотелия, деструкцию эндотелия, артериосклероз, тромбоз, атеросклероз, стеноз, рестеноз, атеросклеротические или тромботические заболевания, недостаточность кровотока, ишемические события, эмболию легких, стабильную и нестабильную стенокардию напряжения, ишемическую болезнь сердца, инфаркт миокарда, а также патологические результаты артериосклеротических или тромботических заболеваний.
9. Соединение для применения или сепарационный материал для применения по п. 6, где иммунные дисфункции выбирают из группы, включающей иммунные заболевания, аутоиммунные заболевания, реакции отторжения трансплантата, отторжение аллотрансплантата, отторжение ксенотрансплантата, отторжение трансплантат против хозяина, отторжение хозяин-против-трансплантата, сахарный диабет, ревматизм, ревматоидный артрит, псориатический артрит, анкилозирующий спондилит, рассеянный склероз, миастению гравис, вульгарный псориаз, болезнь Грейвса, синдром Гудпасчера, идиопатическую тромбоцитопеническую пурпуру (ITP), апластическую анемию, воспалительные заболевания кишечника (IBD), болезнь Крона (также известную как синдром Крона), язвенный колит, дилатационную кардиомиопатию (DCM), аутоиммунный тиреоидит, тиреоидит Хашимото, заместительную гормональную терапию (ЗГТ), остеоартрит и подагру.
| CN 102912640 A, 06.02.2013 | |||
| WO 2007076844 A1, 12.07.2007 | |||
| US 4181634 A1, 01.01.1980 | |||
| WO 1990012632 A1, 01.11.1990 | |||
| WO 2013176084 A1, 28.11.2013 | |||
| WO 2012160187 A1, 29.11.2012 | |||
| СПОСОБ ВЫДЕЛЕНИЯ И ОЧИСТКИ ТРОФОБЛАСТИЧЕСКОГО БЕТА-1-ГЛИКОПРОТЕИНА | 2007 |
|
RU2325171C1 |
| Lenore M | |||
| Martin et al, Tetrahedron Letters, 1996, vol | |||
| Пишущая машина | 1922 |
|
SU37A1 |

