Это изобретение относится к имидазохинолиновым соединениям, которые имеют эфирную и арильную или алкенильную функциональные группы в положении 1, а также к фармацевтическим составам, содержащим такие соединения. Дополнительный аспект этого изобретения относится к использованию указанных выше соединений в качестве иммуномодуляторов, для стимулирования биосинтеза цитокина в организме животного, а также для лечения различных болезней, включая вирусные и опухолевые заболевания.
Первые надежные данные по 1Н-имидазол[4,5-с]хинолиновой циклической системе приведены Бакманом с сотр. в журнале J. Orq. Chem.. 15, 1278-1284 (1950), которые сообщили о синтезе 1-(6-метокси-8-хинолинил)-2-метил-1Н-имидазо[4,5-с]хинолина с целью его возможного применения в качестве противомалярийного средства. В последующем появились сообщения о синтезе различных замещенных 1Н-имидазо[4,5-с]хинолина. Например, Джайн с сотр., J. Med. Chem. 11, 87-92 (1968), синтезировали 1-[2-4-пиперидил)этил]-1Н-имидазо[4,5-с]хинолин для его использования в качестве возможного противосудорожного и сердечно-сосудистого средства. Кроме того, Баранов с сотр., Chem. Abs. 85, 94362 (1976), сообщили о синтезе нескольких 2-оксоимидазо[4,5-с]хинолинов, а Берени с сотр., J. Heterocyclic Chem 18, 1537-1540 (1981), также сообщили о синтезе нескольких 2-оксоимидазо[4,5-с]хинолинов.
Позже было установлено, что некоторые 1H-имидазо[4,5-с]хинолин-4-амины и их 1- и 2-замещенные производные могут быть использованы в качестве противовирусных агентов, бронхолитических средств и иммуномодуляторов. Эти соединения описаны среди прочих химических продуктов в патентах США №№4689338, 4698348, 4929624, 5037986, 5268376, 5346905 и 5389640; все эти патенты приведены в списке цитируемой литературы.
Большой интерес к имидазохинолиновым циклическим системам не прекращается и в настоящее время. Известны некоторые 1Н-имидазо[4,5-с]нафтиридин-4-амины, 1Н-имидазо[4,5-с]пиридин-4-амины и 1Н-имидазо[4,5-с]хинолин-4-амины, имеющие эфирную группу, содержащую заместитель в положении 1. Эти соединения описаны в патентах США №№5268376, 5389640, 5494916, а также в WO 99/29693.
В настоящее время существует потребность в соединениях, обладающих способностью модулировать иммунную реакцию за счет стимулирования биосинтеза цитокина или с использованием других механизмов.
Краткое изложение сущности изобретения
Найден новый класс соединений, способных стимулировать биосинтез цитокина в организме животных. В соответствии с этим данное изобретение предлагает соединения имидазо[4,5-с]хинолин-4-амина и тетрагидроимидазо[4,5-с]хинолин-4-амина, которые содержат заместитель с эфирной группой в 1-положении. Согласно данным ИК-спектроскопии эти соединения имеют формулу (I), (II), (III) и (IV). Ниже приведена общая структурная формула этих соединений
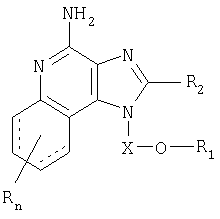
причем природа заместителей X, R1, R2 и R определена ниже для каждого класса соединений, имеющих формулу (I), (II), (III) и (IV).
Соединения, имеющие общую формулу (I), (II), (III) и (IV), являются полезными модификаторами иммунной реакции благодаря их способности стимулировать биосинтез цитокина иным образом модулировать иммунную реакцию при введении в организм животных. Это делает указанные выше соединения полезными средствами для лечения различных заболеваний, таких как вирусные инфекции и опухолевые заболевания, которые вызывают такое изменение в иммунной реакции.
В изобретении также приводятся фармацевтические составы, содержащие соединения, изменяющие иммунную реакцию, и сообщается о способах стимулирования биосинтеза цитокина в организме животного, лечении вирусной инфекции и/или опухолевых заболеваний у животного путем введения в его организм соединений формулы (I), (II), (III) и (IV).
Кроме того, приводятся способы синтеза соединений, являющихся предметом изобретения, а также промежуточных продуктов, используемых при синтезе этих соединений.
Подробное описание изобретения
Как указывалось выше, мы обнаружили некоторые соединения, которые стимулируют биосинтез цитокина и тем самым модифицируют иммунную реакцию в организме животных. Такие соединения имеют общие формулы (I), (II), (III) и (IV), показанные ниже.
Предлагаемые в изобретении имидазохинолиновые соединения, содержащие эфирную и арильную или алкенильную функциональные группы, находящиеся в положении 1, имеют формулу (1):
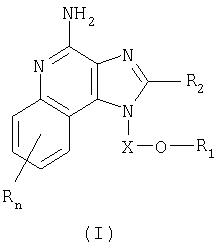
где Х представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
R1 выбран из группы, включающей:
- алкенил;
- арил;
- R4-арил;
R2 выбран из группы, включающей:
- атом водорода;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил; и
- алкил или алкенил, замещенные одним или большим количеством заместителей, выбранных из группы, включающей:
- ОН;
- атом галогена;
- N(R3)2;
- СО-N(R3)2;
- СО-С1-10алкил;
- СО-O-С1-10алкил;
- N3
- арил;
- гетероарил;
- гетероциклил;
- СО-арил; и
- СО-гетероарил;
каждый R4 представляет собой алкильную или алкенильную группы, причем обе эти группы могут быть разделены одной или несколькими -O-группами;
каждый R3 представляет собой независимо друг от друга атом водорода или С1-10алкильную группу;
каждая группа Y независимо друг от друга представляет собой -О- или S(O)0-2-;
значение n может изменяться от 0 до 4; и
каждый заместитель R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
В настоящем изобретении предлагаются также имидазохинолиновые соединения, содержащие эфирную группу в положении 1, причем заместитель с эфирной группой содержит также и алкинильную группу. Эти соединения имеют общую структурную формулу (II):
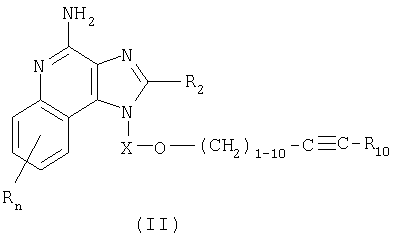
где
Х представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
заместитель R10 выбран из группы, включающей:
- атом водорода
- алкил;
- алкенил; и
- арил;
R2 выбран из группы, включающей:
- атом водорода;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил; и
- алкил или алкенил, замещенные одним или большим количеством заместителей, выбранных из группы, включающей:
- ОН;
- атом галогена;
- N(R3)2;
- СО-N(R3)2;
- CO-C1-10алкил;
- CO-O-C1-10алкил;
- N3
- арил;
- гетероарил;
- гетероциклил;
- СО-арил; и
- СО-гетероарил;
значение n находится в интервале от 0 до 4;
каждая группа Y независимо друг от друга представляет собой -О- или S(O)0-2-;
каждый R3 представляет собой независимо друг от друга атом водорода или С1-10алкильную группу; и
каждый R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
Настоящее изобретение включает в себя также тетрагидроимидазохинолиновые соединения, содержащие эфирную и арильную или алкенильную группы в положении 1. Такие тетрагидроимидазохинолиновые соединения имеют формулу (III):

где Х представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
заместитель R1 выбран из группы, включающей:
- арил;
- алкенил; и
- R4-арил;
R2 выбран из группы, включающей:
- атом водорода;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-арил;
- алкил-Y-алкенил; и
- алкил или алкенил, замещенные одним или большим количеством заместителей, выбранных из группы, включающей:
- ОН;
- атом галогена;
- N(R3)2;
- CO-N(R3)2;
- CO-C1-10алкил;
- СО-О-С1-10алкил;
- N3
- арил;
- гетероарил;
- гетероциклил;
- СО-арил; и
- СО-гетероарил;
R4 представляет собой алкильную или алкенильную группы, причем обе эти группы могут быть разделены одной или несколькими -О- группами;
каждый R3 представляет собой независимо друг от друга атом водорода или С1-10 алкильную группу;
каждая группа Y независимо друг от друга представляет собой -О- илиS(O)0-2-;
значение n может изменяться от 0 до 4; и
каждый R независимо выбран из группы, состоящей из C1-10 алкильной, С1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
Дополнительным классом представленных в настоящем изобретении соединений, модифицирующих иммунную реакцию, являются соединения, включающие эфирсодержащий заместитель, находящийся в положении 1, причем этот эфирсодержащий заместитель содержит также и алкинильную группу. Эти соединения имеют общую структурную формулу (IV):

где
Х представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
заместитель R10 выбран из группы, включающей:
- атом водорода
- алкил;
- алкенил; и
- арил;
R2 выбран из группы, включающей:
- атом водорода;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-арил;
- алкил-Y-алкенил; и
- алкил или алкенил, замещенные одним или большим количеством заместителей, выбранных из группы, включающей:
- ОН;
- атом галогена;
- N(R3)2;
- СО-N(R3)2;
- CO-C1-10алкил;
- CO-O-C1-10алкил;
- N3
- арил;
- гетероарил;
- гетероциклил;
- СО-арил; и
- СО-гетероарил;
каждый R3 представляет собой независимо друг от друга атом водорода или C1-10алкильную группу;
каждая группа Y независимо друг от друга представляет собой -О- или S(O)0-2-;
величина n находится в интервале от 0 до 4; и
каждый R независимо выбран из группы, состоящей из С1-10 алкильной, С1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
Получение соединений
Предлагаемые в изобретении соединения могут быть получены в соответствии с приведенной ниже схемой I реакции, в которой значения R, R2, Х и n определены выше, а заместитель R11 представляет собой алкильную группу, содержащую арильный заместитель, причем арильная группа может быть незамещенной или замещенной, или R11 является замещенной арильной группой, при условии, что если R11 является замещенной арильной группой, по крайней мере, один заместитель представляет собой сильную электроно-акцепторную группу, расположенную в орто- или пара-положении по отношению к эфирной связи.
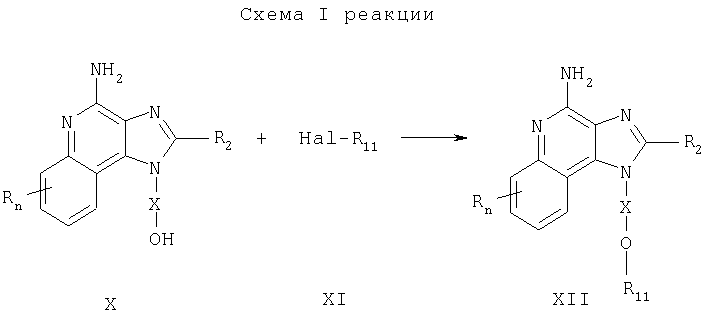
В соответствии со схемой I реакции 4-амино-1H-имидазо[4,5-с]хинолин-1-иловый спирт формулы Х подвергают алкилированию под действием галогенида формулы XI, в результате чего образуется 1Н-имидазо[4,5-с]хинолин-4-амин формулы XII, который является производным соединением продукта I. Спирт формулы Х обрабатывают гидридом натрия, диспергированным в подходящем растворителе, таком как N,N-диметилформамид, и в результате получают соответствующий алкоксид. После этого к реакционной смеси добавляют соответствующий галогенид и смесь выдерживают при температуре окружающей среды или при необходимости при слабом нагревании (около 50°С). Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
Известны многие соединения формулы X, см., например, Герстер, Патент США №4689338 и Герстер с сотр., Патент США №5605899, а также цитируемые в этих патентах работы. Другие соединения такого типа могут быть легко получены с помощью известных способов синтеза, см., например, Андре с сотр. Патент США №5578727; Герстер, Патент США №5175296, Николайдес с сотр., Патент США №5395937 и Герстер с сотр., Патент США №5741908 и цитируемую в этих патентах литературу. Многие галогениды формулы XI являются коммерческими продуктами; другие можно легко синтезировать по известным способам.
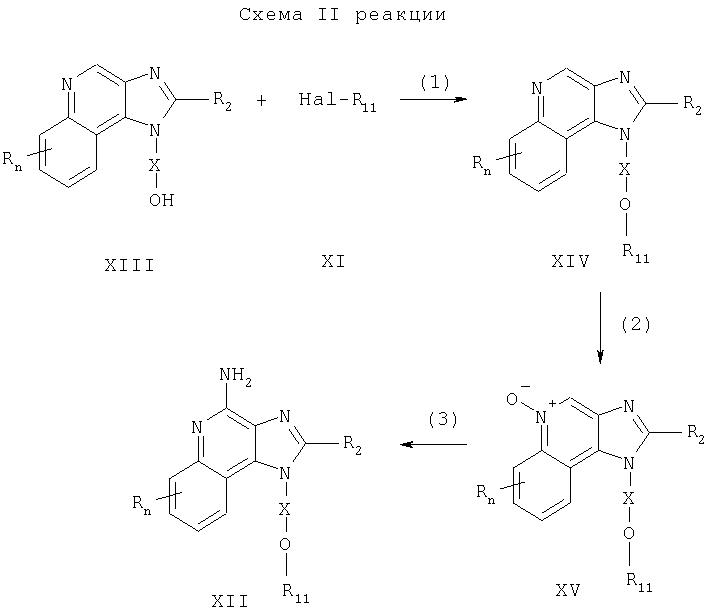
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой II реакции, в которой значения R, R2, R11, Х и n определены выше.
На стадии (1) процесса (схема II реакции) 1Н-имидазо[4,5-с]хинолин-1-иловый спирт формулы XIII подвергают алкилированию под действием галогенида формулы XI, в результате чего образуется 1H-имидазо[4,5-с]хинолин-1 -иловый эфир формулы XIV. Для получения алкоксида спирт XIII обрабатывают гидридом натрия, диспергированным в подходящем растворителе, таком как N,N-диметилформамид или тетрагидрофуран. К полученному алкоксиду добавляют галогенид. Альтернативный способ заключается в проведении реакции между спиртом и галогенидом в двухфазной смеси, состоящей из 50%-ного водного раствора гидроксида натрия и инертного растворителя, такого как дихлорметан, в присутствии катализатора межфазного переноса, такого как бензилтриметиламмоний хлорид. Реакцию можно проводить при температуре окружающей среды. Многие соединения формулы XIII известны, см., например, Герстер, Патент США №4689338, другие соединения такого типа могут быть легко получены с помощью известных синтетических способов, см., например, Герстер с сотр., Патент США №5605899 и Герстер с сотр., Патент США №5175296.
На стадии (2) процесса (схема II реакции) 1Н-имидазо[4,5-с]хинолин-1-иловый эфир формулы XIV под действием обычного окислителя, способного образовывать N-оксид, окисляют до 1H-имидазо[4,5-с]хинолин-5N-оксида формулы XV. Преимущественно раствор соединения формулы XIV в подходящем растворителе, таком как хлороформ или дихлорметан, окисляют при температуре окружающей среды под действием 3-хлорпероксибензойной кислоты.
На стадии (3) процесса (схема II реакции) 1H-имидазо[4,5-с]хинолин-5N-оксид формулы XV аминируют до 1H-имидазо[4,5-с]хинолин-4-амина формулы XII, который является производным соединением продукта I. Стадия (3) включает в себя (i) реакцию присоединения XV к ацилирующему агенту и (ii) взаимодействие образовавшегося продукта с аминирующим агентом. Часть (i) стадии (3) заключается во взаимодействии N-оксида формулы XV с ацилирующим агентом. Подходящими ацилирующими агентами являются алкил- или арилсульфонилхлориды (например, бензолсульфонилхлорид, метансульфонилхлорид, п-толуиленсульфонилхлорид). Предпочтительными ацилирующими агентами являются арилсульфонилхлориды, причем наиболее предпочтительным ацилирующим агентом является пара-толуиленсульфонилхлорид. В части (ii) стадии (3) продукт, полученный в части (i), обрабатывают избытком аминирующего агента. Подходящими аминирующими агентами являются аммиак (например, в виде гидроксида аммония) и соли аммония (например, карбонат аммония, бикарбонат аммония, фосфат аммония). Гидроксид аммония является предпочтительным аминирующим агентом. Реакцию преимущественно проводят, растворяя N-оксид формулы XV в инертном растворителе, таком как дихлорметан, добавляя к полученному раствору аминирующий агент и после этого медленно добавляя ацилирующий агент. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
Альтернативным способом можно проводить стадию (3), (i) обрабатывая N-оксид формулы XV изоцианатом и (ii) подвергая образовавшийся продукт гидролизу. Часть (i) заключается во взаимодействии N-оксида XV с изоцианатом, в котором изоцианатная группа связана с карбонильной группой. Предпочтительными изоцианатами являются трихлорацетилизоцианат и ароилизоцианаты, такие как бензоилизоцианат. Реакцию между изоцианатом и N-оксидом проводят в безводных условиях, добавляя изоцианат к раствору N-оксида в инертном растворителе, таком как хлороформ или дихлорметан. Часть (ii) заключается в проведении гидролиза продукта, образовавшегося в части (i). Гидролиз можно проводить с помощью обычных способов, таких, например, как нагревание в присутствии воды или низших спиртов, желательно в присутствии катализатора, такого как гидроксид щелочного металла или низший алкоксид. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
Соединения формулы I, в которой значения R, R2, Х и n определены выше, а заместитель R1 представляет собой замещенную фенильную группу, могут быть получены в соответствии со схемой III реакции. В этой схеме значение m находится в пределах от 0 до 3, а каждый заместитель R' независимым образом выбран из группы, содержащей следующие группы: алкил-, алкокси-, алкилтио-, галоидоалкил-, галоидоалкокси-, галоидоалкилтио-, атом галогена, нитро-, меркапто-, циано-, карбокси-, формил-, арил-, арилокси-, арилтио-, арилалкокси-, арилалкилтио-, гетероарил-, гетероарилокси-, гетероарилтио-, гетероарилалкокси-, гетероарилалкилтио-, амино-, алкиламино-, диалкиламино-, гетероциклил-, гетероциклоалкил-, алкилкарбонил-, алкенилкарбонил-, арилкарбонил-, алкоксикарбонил-, галоидоалкилкарбонил-, галоидоалкоксикарбонил-, алкилтиокарбонил-, арилоксикарбонил-, алканоилокси-, алканоилтио-, алканоиламино-, ароилокси- и ароиламино-.
В соответствии со схемой III реакции 4-амино-1Н-имидазо[4,5-с]хинолин-1-иловый спирт формулы Х подвергают конденсации с фенолом формулы XVI, в результате чего получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XVII, который является производным соединением продукта формулы I. Предпочтительно раствор соединения формулы Х и фенола в соответствующем растворителе, таком как N,N-диметилформамид, при температуре окружающей среды обрабатывают диэтилазодикарбоксилатом и трифенилфосфином. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
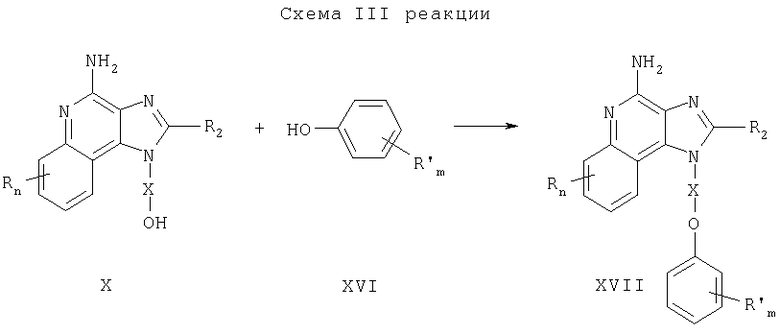
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой IV реакции, в которой значения R, R2, R11, X и n определены выше.
На стадии (1) процесса (схема IV реакции) гидроксильную группу 1Н-имидазо[4,5-с]хинолин-1-илового спирта формулы XIII защищают бензильной группой. Спирт формулы XIII обрабатывают гидридом натрия, диспергированным в подходящем растворителе, таком как N,N-диметилформамид, и получают соответствующий алкоксид. Этот алкоксид подвергают алкилированию под действием бензилбромида и в результате получают соединение формулы XVIII. Реакцию можно проводить при температуре окружающей среды.
На стадии (2) процесса (схема IV реакции) соединение формулы XVIII, используя метод, применяемый на стадии (2) процесса (схема II реакции), окисляют до 1H-имидазо[4,5-с]хинолин-5N-оксида формулы XIX.
На стадии (3) процесса (схема IV реакции) 1Н-имидазо[4,5-с]хинолин-5N-оксид формулы XIX хлорируют и в результате получают 4-хлор-1Н-имидазо[4,5-с]хинолин формулы XX. Предпочтительно раствор соединения формулы XIX в подходящем растворителе, таком как толуол, обрабатывают при температуре окружающей среды оксихлоридом фосфора.
На стадии (4) процесса (схема IV реакции) 4-хлор-1Н-имидазо[4,5-с]хинолин формулы XX обрабатывают фенолом и получают 4-фенокси-1H-имидазо[4,5-с]хинолин формулы XXI. Сначала фенол для получения феноксида обрабатывают гидридом натрия, диспергированным в соответствующем растворителе, таком как диглим. Затем феноксид при повышенной температуре обрабатывают соединением формулы XX.
На стадии (5) процесса (схема IV реакции) из соединения формулы XXI удаляют защитную бензильную группу и получают 4-фенокси-1H-имидазо[4,5-с]хинолин-1-иловый спирт формулы XXII. Реакцию предпочтительно проводят при температуре окружающей среды, медленно добавляя трифторметануксусную кислоту к раствору соединения формулы XXI в соответствующем растворителе, таком как дихлорметан.
На стадии (6) процесса (схема IV реакции) 4-фенокси-1H-имидазо[4,5-с]хинолин-1-иловый спирт формулы XXII подвергают алкилированию под действием галогенида Hal-R11 и в результате получают 4-фенокси-1H-имидазо[4,5-с]хинолин-1-иловый эфир формулы XXIII. Алкоксид соединения формулы XXII получают в присутствии катализатора переноса фаз, такого, например, как бензилтриметиламмонийхлорид, при добавлении спирта к двухфазной системе, состоящей из 50%-ного водного раствора гидроксида натрия и инертного растворителя, такого как дихлорметан. Затем проводят алкилирование алкоксида. Реакцию можно проводить при температуре окружающей среды.
На стадии (7) процесса (схема IV реакции) 4-фенокси-1H-имидазо[4,5-с]хинолин-1-иловый эфир формулы XXIII подвергают аминированию и получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XII, который является производным соединением продукта формулы I. Для проведения этой реакции соединение формулы XXIII смешивают с ацетатом аммония и эту смесь нагревают при температуре около 150°С. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
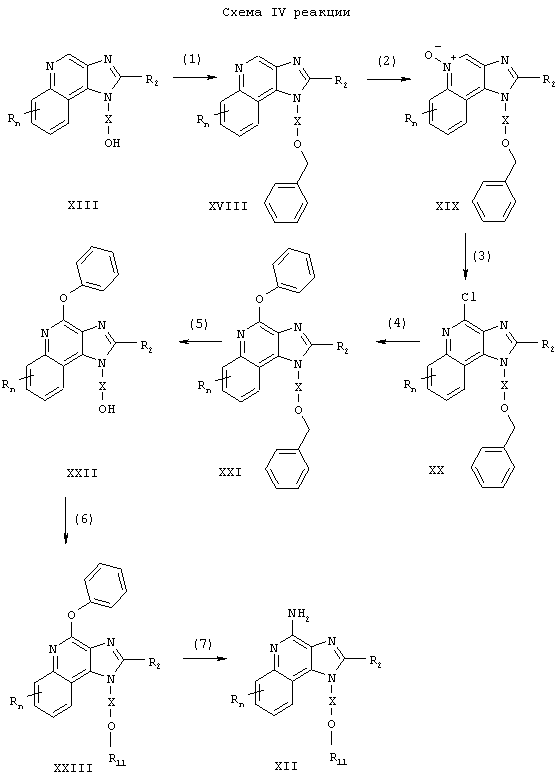
Предлагаемые в изобретении тетрагидроимидазохинолины могут быть получены в соответствии со схемой V реакции, в которой значения R, R2, R11, X и n определены выше.
В соответствии со схемой V реакции 4-амино-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-1 -иловый спирт формулы XXIV алкилируют с помощью галогенида XI до 6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амина формулы XXV, который является производным соединением продукта формулы III. Для получения алкоксида спирт формулы XXIV обрабатывают гидридом натрия, диспергированным в соответствующем растворителе, таком как N,N-диметилформамид. Затем алкоксид соединяют с галогенидом, и реакцию проводят при температуре окружающей среды. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
Известны многие тетрагидро-1H-имидазо[4,5-с]хинолины формулы XXIV, см., например, Николайдес с сотр., Патент США №5352784; другие подобные соединения можно получить с помощью известных синтетических способов, см., например Линдстром, Патент США №5693811 и цитируемую в нем литературу.
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой VI реакции, в которой значения R, R1, R2, Х и n определены выше.
На стадии (1) процесса (схема VI реакции) 4-хлор-3-нитрохинолин формулы XXVI обрабатывают амином формулы R1-O-X-NH2 для получения 3-нитрохинолин-4-амина формулы XXVII. Реакцию проводят, добавляя амин к раствору соединения формулы XXVI в соответствующем растворителе, таком как хлороформ или дихлорметан; в ряде случаев реакцию проводят при нагревании. Известны многие хинолины формулы XXVI (см., например, Патент США №4689338 и цитируемую в нем литературу).
На стадии (2) процесса (схема VI реакции) 3-нитрохинолин-4-амин формулы XXVII восстанавливают до хинолин-3,4-диамина формулы XXVIII. Предпочтительно восстановление проводят, используя обычный катализатор гетерогенного гидрирования, такой как нанесенный на активированный уголь палладий или нанесенная на активированный уголь платина. Реакцию обычно можно проводить в аппарате Парра в подходящем растворителе, таком как изопропиловый спирт или, предпочтительно, толуол.
На стадии (3) процесса (схема VI реакции) хинолин-3,4-диамин формулы XXVIII обрабатывают карбоновой кислотой или эквивалентным ей соединением и получают 1H-имидазо[4,5-с]хинолин формулы XXIX. Подходящими эквивалентами карбоновой кислоты являются сложные ортоэфиры и 1,1-диалкоксиалкилалканоаты. Карбоновую кислоту или ее эквивалент выбирают таким образом, чтобы они обеспечивали введение в соединение формулы XXIX желаемого заместителя R2. Например, при использовании для этой цели триэтилортоформиата в результате реакции будет получаться соединение, в котором R2 представляет собой атом водорода, а при использовании триметилортоацетата - R2 будет представлять собой группу СН3. Реакцию можно проводить в отсутствие растворителя или в инертном растворителе, таком как толуол. Реакцию проводят при достаточном нагревании, чтобы обеспечить удаление любого спирта или воды, образующихся в ходе этой реакции в качестве побочных продуктов. По желанию реакцию можно проводить в присутствии катализатора, такого как солянокислый пиридин.
Альтернативным образом стадию (3) можно проводить при (i) взаимодействии диамина формулы XXVIII с ацилгалогенидом формулы R2C(O)Cl и (ii) последующей циклизации полученного продукта. В части (i) ацилгалогенид добавляют к раствору диамина в подходящем растворителе, таком как ацетонитрил, пиридин или дихлорметан. Реакцию можно проводить при температуре окружающей среды. В части (ii) продукт, полученный в части (i), нагревают в пиридине в присутствии основания. Предпочтительно продукт, полученный в части (i), кипятят с обратным холодильником в этаноле в присутствии избытка триэтиламина или нагревают с метанольным раствором аммиака. Альтернативным образом, если часть (i) проводят в пиридине, часть (ii) после проведения анализа, который показывает, что реакция в части (i) полностью завершена, можно проводить просто нагреванием реакционной смеси.
На стадии (4) процесса (схема VI реакции) 1Н-имидазо[4,5-с]хинолин формулы XXIX окисляют, используя способ, применяемый на стадии (2) процесса (схема II реакции), и в результате получают 1H-имидазо[4,5-с]хинолин-5N-оксид формулы XXX.
На стадии (5) процесса (схема VI реакции) 1Н-имидазо[4,5-с]хинолин-5N-оксид формулы XXX, используя способ, применяемый на стадии (3) процесса (схема II реакции), подвергают аминированию и в результате получают 1H-имидазо[4,5-с]хинолин-4-амин формулы I.
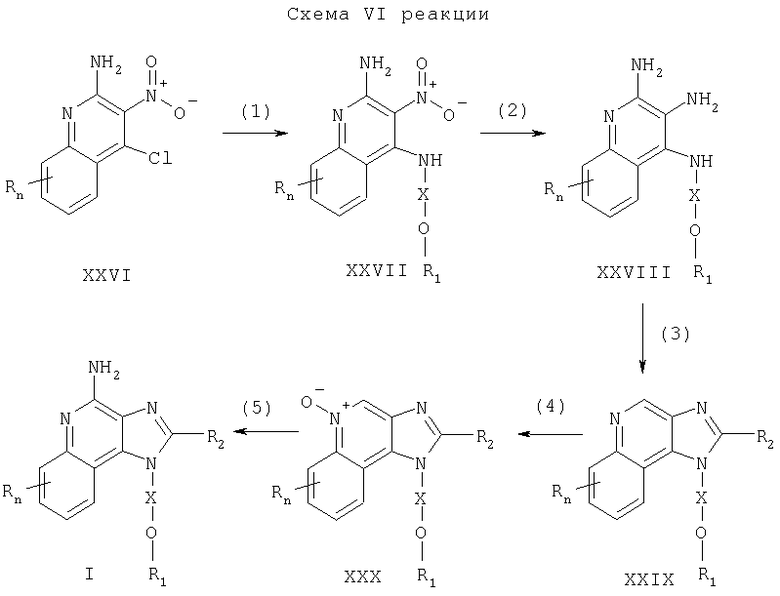
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой VII реакции, в которой значения R, R2, Х и n определены выше, а заместитель R12 представляет собой арильную группу, которая может быть либо незамещенной, либо содержать заместители, указанные выше.
На стадии (1) процесса (схема VII реакции) 1Н-имидазо[4,5-с]хинолин-1-иловый спирт формулы XIII подвергают алкилированию под действием галогенида формулы XXXI и получают 1H-имидазо[4,5-с]хинолин-1-иловый эфир формулы XXXII. Соединение XIII и галогенид формулы XXXI смешивают в двухфазной системе, состоящей из 50%-ного водного раствора гидроксида натрия и соответствующего растворителя, такого как дихлорметан, и проводят реакцию в присутствии катализатора переноса фаз, такого как бензилтриметиламмонийхлорид. Реакцию можно проводить при температуре окружающей среды.
На стадии (2) процесса (схема VII реакции) 1Н-имидазо[4,5-с]хинолин формулы XXXII окисляют, используя методику, применяемую на стадии (2) процесса (схема II реакции), и получают 1Н-имидазо[4,5-с]хинолин-5N-оксид формулы XXXIII.
На стадии (3) процесса (схема VII реакции) проводят реакцию между 1Н-имидазо[4,5-с]хинолин-5N-оксидом формулы XXXIII и трихлорацетилизоцианатом и получают 1Н-имидазо[4,5-с]хинолин-4-ил-трихлорацетамид формулы XXXIV. Предпочтительно изоцианат медленно добавляют при температуре окружающей среды к раствору 5N-оксида в соответствующем растворителе, таком как дихлорметан.
На стадии (4) процесса (схема VII реакции) 1Н-имидазо[4,5-с]хинолин-4-ил-трихлорацетамид формулы XXXIV подвергают гидролизу и в результате получают 1Н-имидазо[4,5-с]хинолин-4-амин формулы XXXV, который является производным соединением продукта формулы II. Гидролиз можно проводить, используя обычные способы, предпочтительно обрабатывая раствор соединения формулы XXXIV в метаноле метилатом натрия.
На стадии (5) процесса (схема VII реакции) проводят реакцию между 1H-имидазо[4,5-с]хинолин-4-амином формулы XXXV и галогенидом формулы Hal-R12 в присутствии соединения переходного металла, используемого в качестве катализатора. В результате получают 1Н-имидазо[4,5-с]хинолин-4-амин формулы XXXVI, который является производным соединением продукта формулы II. Предпочтительно взаимодействие соединения формулы XXXV с галогенидом проводят в присутствии иодида меди (I), дихлорбис(трифенилфосфина) палладия (II) и избытка триэтиламина в соответствующем растворителе, таком как N,N-диметилформамид или ацетонитрил. Реакцию предпочтительно проводят при повышенной температуре (60-80°С). Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
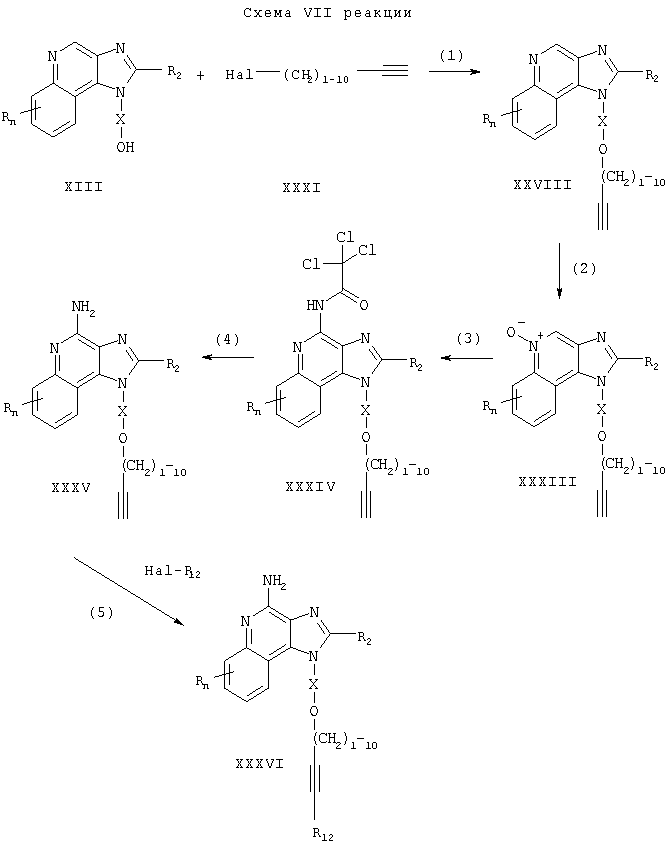
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой VIII реакции, в которой значения R, R2, R12, X и n определены выше. ВОС-трет-бутоксикарбонил.
На стадии (1) процесса (схема VIII реакции) амино-группу 1Н-имидазо[4,5-с]хинолин-4-амина формулы XXXV защищают двумя mpem-бутокси карбонильными группами. Реакцию между соединением XXXV и ди-трет-бутилдикарбонатом проводят в соответствующем растворителе, таком как N,N-диметилформамид, в присутствии 4-(диметиламино)пиридина и триэтиламина. Реакцию проводят при повышенной температуре (80-85°С).
На стадии (2) процесса (схема VIII реакции) защищенный 1Н-имидазо[4,5-с]хинолин-4-амин формулы XXXVII обрабатывают галогенидом формулы Hal-R12 в присутствии соединения переходного металла, используемого в качестве катализатора, и получают защищенный 1Н-имидазо[4,5-с]хинолин-4-амин формулы XXXVIII. Предпочтительно соединение формулы XXXVII обрабатывают галогенидом в присутствии иодида меди (I), дихлорбис(трифенилфосфина) палладия (II) и избытка триэтиламина в соответствующем растворителе, таком как N,N-диметилформамид или ацетонитрил. Реакцию предпочтительно проводят при температуре окружающей среды или повышенной температуре (40-80°С).
На стадии (3) процесса (схема VIII реакции) защитные группы удаляют в результате гидролиза в кислотных условиях и в результате получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XXXVI, который является производным соединением продукта II. Предпочтительно соединение XXXVIII обрабатывают трифторуксусной кислотой в подходящем растворителе, таком как дихлорметан. Реакцию можно проводить при температуре окружающей среды или пониженной температуре (0°С). Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
На стадии (4) процесса (схема VIII реакции) алкиновую (тройную) связь защищенного 1Н-имидазо[4,5-с]хинолин-4-амин формулы XXXVIII восстанавливают и в результате получают защищенный 1H-имидазо[4,5-с]хинолин-4-амин формулы XXXIX. Предпочтительно восстановление проводят, используя обычный катализатор гетерогенного гидрирования, такой как нанесенный на активированный уголь палладий или нанесенная на активированный уголь платина. Реакцию обычно можно проводить в аппарате Парра в подходящем растворителе, таком как метанол.
На стадии (5) процесса (схема VIII реакции) защитные группы соединения формулы XXXIX удаляют, используя способ, применяемый на стадии (3). В результате этой реакции получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XL, который является производным соединением продукта формулы I. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
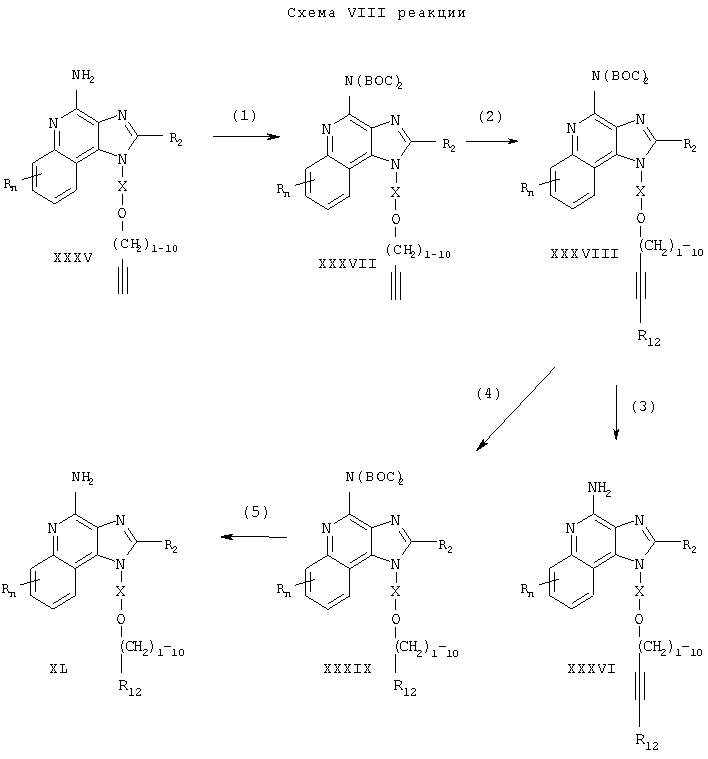
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой IX реакции, в которой значения R, R2, R12, Х и n определены выше. CBZ-бензилоксикарбонил.
На стадии (1) процесса (схема IX реакции) амино-группу 1Н-имидазо[4,5-с]хинолин-4-амина формулы XXXV защищают бензилоксикарбонильными группами. Реакцию между соединением формулы XXXV и дибензилдикарбонатом проводят в соответствующем растворителе, таком как N,N-диметилформамид. Реакцию проводят при комнатной или несколько повышенной температуре (40°С).
На стадии (2) процесса (схема IX реакции) защищенный 1H-имидазо[4,5-с]хинолин-4-амин формулы XLI обрабатывают галогенидом формулы Hal-R12 в присутствии соединения переходного металла, используемого в качестве катализатора, и получают защищенный 1Н-имидазо[4,5-с]хинолин-4-амин формулы XLII. Предпочтительно соединение формулы XLI обрабатывают галогенидом в присутствии иодида меди (I), дихлорбис(трифенилфосфина) палладия (II) и избытка триэтиламина в соответствующем растворителе, таком как N,N-диметилформамид или ацетонитрил. Реакцию предпочтительно проводят при температуре окружающей среды или повышенной температуре (40-80°С).
На стадии (3) процесса (схема IX реакции) защитные группы удаляют за счет гидролиза и в результате получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XXXVI, который является производным соединением продукта формулы II. Предпочтительно соединение формулы XLII обрабатывают метилатом натрия в подходящем растворителе, таком как метанол. Реакцию можно проводить при температуре окружающей среды. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
На стадии (4) процесса (схема IX реакции) защитные группы соединения формулы XLII удаляют в результате восстановления под действием водорода и проводят восстановление алкиновой (тройной) связи. В результате получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XL, который является производным соединением продукта формулы I. Предпочтительно реакцию восстановления проводят, используя в качестве катализатора гидроксид палладия, нанесенный на активированный уголь. Реакцию обычно можно проводить в аппарате Парра в подходящем растворителе, таком как метанол. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой Х реакции, в которой значения R, R1, R2, X и n определены выше.
На стадии (1) процесса (схема Х реакции) 2,4-дихлор-3-нитрохинолин формулы XLIII обрабатывают амином формулы R1-O-X-NH2 и получают 2-хлор-3-нитрохинолин-4-амин формулы XLIV. Реакцию можно проводить, добавляя амин к раствору соединения XLIII в соответствующем растворителе, таком как хлороформ или дихлорметан. В ряде случаев реакцию можно проводить при нагревании. Многие хинолины формулы XLIII либо являются известными продуктами, либо их можно получить, используя известные синтетические способы (см., например, Андре с сотр., Патент США №4988815 и цитируемую в нем литературу).
На стадии (2) процесса (схема Х реакции) 2-хлор-3-нитрохинолин-4-амин формулы XLIV восстанавливают, используя для этой цели способ, применяемый на стадии (2) процесса (Схема VI реакции). В результате получают 2-хлорхинолин-3,4-диамин формулы XLV.
На стадии (3) процесса (схема Х реакции) 2-хлорхинолин-3,4-диамин формулы XLV подвергают циклизации, используя способ, применяемый на стадии (3) процесса (Схема VI реакции), и в результате получают 4-хлор-1Н-имидазо[4,5-финолин формулы XLVI.
На стадии (4) процесса (схема Х реакции) 4-хлор-1H-имидазо[4,5-с]хинолин формулы XLVI аминируют до 1Н-имидазо[4,5-с]хинолин-4-амина формулы I. Реакцию проводят при нагревании (например, при 125-175°С) соединения формулы XLVI в герметичном реакторе в присутствии раствора аммиака в алканоле. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
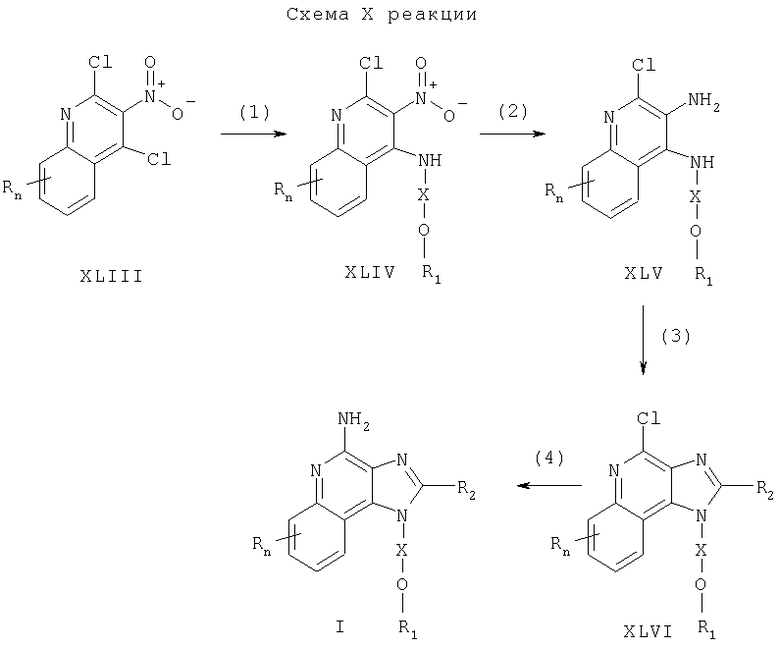
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой XI реакции, в которой значения R, R1, R2, X и n определены выше.
В соответствии со схемой XI реакции 1Н-имидазо[4,5-с]хинолин-4-амин формулы XLVII под действием галогенида формулы XLVIII алкилируют до 1H-имидазо[4,5-с]хинолин-4-амина формулы I. Соединение формулы XLVII обрабатывают гидридом натрия, диспергированным в соответствующем растворителе, таком как N,N-диметилформамид, и после этого к реакционной смеси добавляют галогенид. Реакцию проводят при повышенной температуре (˜100°С). Алкилирование протекает как по атому азота N1, так и по N3; однако целевой продукт, которым является 1-изомер, может быть легко отделен от 3-изомера с помощью обычных способов, например при использовании хроматографических колонок или с помощью перекристаллизации.
Многие 1Н-имидазо[4,5-с]хинолин-4-амины формулы XLVII либо являются известными продуктами, либо их можно получить, используя известные синтетические способы, см., например, Герстер, Патент США №5756747 и цитируемую в нем литературу.
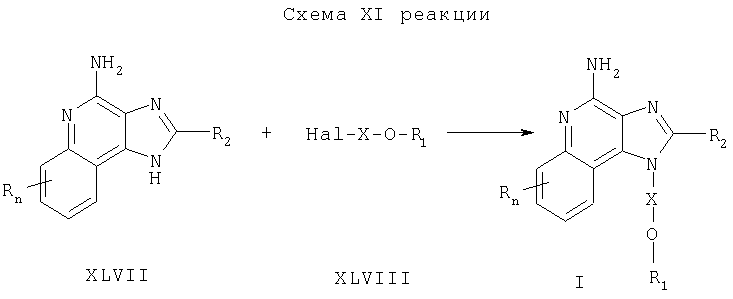
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой XII реакции, в которой значения R, R1, R2, Х и n определены выше.
На стадии (1) процесса (схема XII реакции) 4-нитротетразоло[1,5-а]хинолин-5-ол формулы XLIX хлорируют до 5-хлор-4-нитротетразоло[1,5-а]хинолин формулы L. Для проведения этой реакции могут быть использованы обычные хлорирующие агенты. Преимущественно реакцию проводят с использованием оксихлорида фосфора в подходящем растворителе, таком как N,N-диметилформамид. 4-Нитротетразоло[1,5-а]хинолин-5-олы формулы XLIX являются известными соединениями или могут быть получены с помощью известных синтетических способов (см., например, Герстер, Патент США №5741908 и цитируемую в нем литературу).
На стадии (2) процесса (схема XII реакции) проводят реакцию между 5-хлор-4-нитротетразоло[1,5-а]хинолином формулы L и амином формулы R1-O-X-NH2. В результате получают 4-нитротетразоло[1,5-а]хинолин-5-амин формулы LI. Реакцию можно проводить в присутствии триэтиламина, добавляя амин к раствору соединения LI в соответствующем растворителе, таком как дихлорметан.
На стадии (3) процесса (схема XII реакции) 4-нитротетразоло[1,5-а]хинолин-5-амин формулы LI восстанавливают по способу, применяемому на стадии (2) процесса (схема VI реакции), и в результате получают тетразоло[1,5-а]хинолин-4,5-диамин формулы LII.
На стадии (4) процесса (схема XII реакции), используя способ, применяемый на стадии (3) процесса (схема VI реакции), осуществляют циклизацию тетразоло[1,5-а]хинолин-4,5-диамина формулы LII до 6Н-имидазо[4,5-с]тетразоло[1,5-а]хинолина формулы LIII.
На стадии (5) процесса (схема XII реакции) 6Н-имидазо[4,5-с]тетразоло[1,5-а]хинолин формулы LIII восстанавливают до 1H-имидазо[4,5-с]хинолин-4-амина формулы I. Стадия (5) состоит из (i) взаимодействия соединения формулы LIII с трифенилфосфином и (ii) последующего гидролиза образующегося продукта. Для проведения части (i) нагревают смесь соединения формулы LIII с трифенилфосфином в соответствующем растворителе, таком как 1,2-дихлорбензол. Часть (ii) заключается в гидролизе продукта, образовавшегося в части (i). Гидролиз можно проводить с помощью обычных способов, таких как нагревание продукта в присутствии воды или низшего спирта и в ряде случаев в присутствии катализатора, такого как гидроксид щелочного металла или низший алкоксид щелочного металла. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
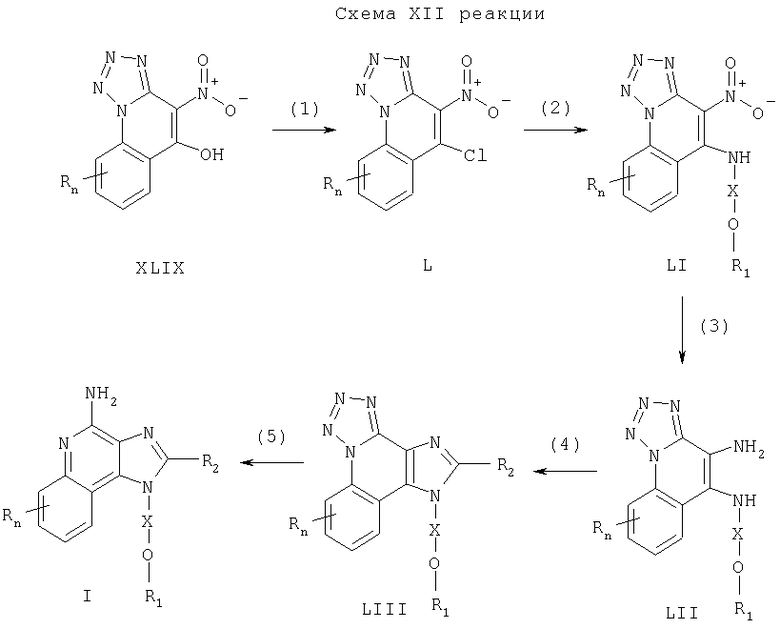
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой XIII реакции, в которой значения R, R2, R12, Х и n определены выше.
На стадии (1) процесса (схема XIII реакции) 1Н-имидазо[4,5-с]хинолин-1-иловый эфир формулы XXXII, используя способ, применяемый на стадии (5) процесса (схема VII реакции), обрабатывают галогенидом формулы Hal-R12 и получают 1H-имидазо[4,5-с-]хинолин-1-иловый эфир формулы LIV.
На стадии (2) процесса (схема XIII реакции) проводят окисление 1Н-имидазо[4,5-с]хинолин-1-илового эфира формулы LIV по способу, примененному на стадии (2) процесса (схема II реакции), и получают 1H-имидазо[4,5-с]хинолин-5N-оксид формулы LV.
Стадия (3) процесса (схема XIII реакции) заключается в аминировании продукта формулы LV по способу, изложенному в стадии (3) процесса (схема II реакции), и приводит к получению 1Н-имидазо[4,5-с]хинолин-4-амина формулы XXXVI, который является производным соединением продукта II. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
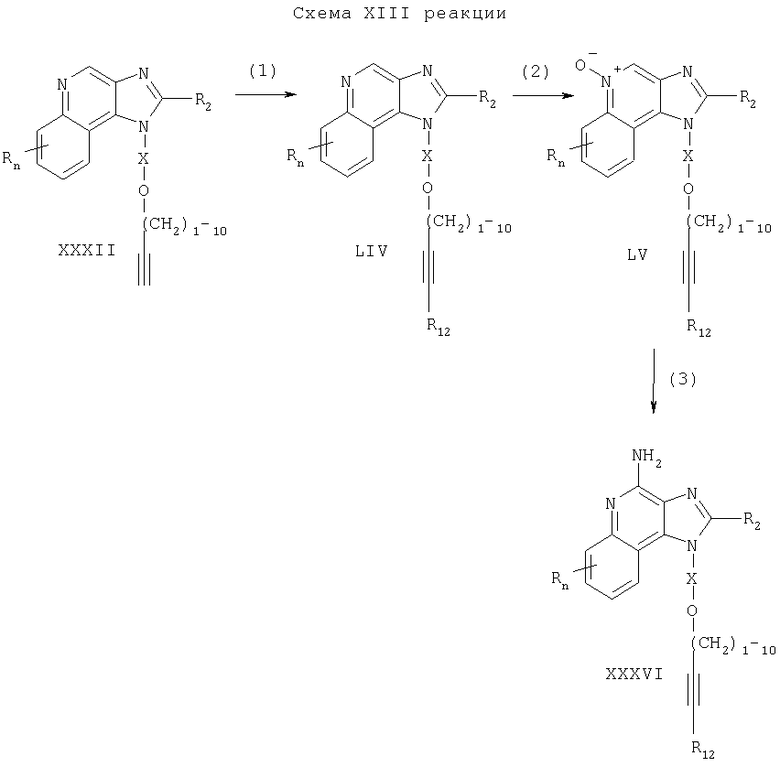
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой XIV реакции, в которой значения R, R2, R12, X и n определены выше.
На стадии (1) процесса (схема XIV реакции) восстанавливают тройную углерод-углеродную связь 1H-имидазо[4,5-с]хинолин-1 -илового эфира формулы LIV, используя способ, применяемый на стадии (4) процесса (схема VIII реакции), и в результате получают 1H-имидазо[4,5-с]хинолин-1-иловый эфир формулы LVI.
На стадии (2) процесса (схема XIV реакции) проводят окисление 1Н-имидазо[4,5-с]хинолин-1-илового эфира формулы LVI по способу, примененному на стадии (2) процесса (схема II реакции), и получают 1H-имидазо[4,5-с]хинолин-5N-оксид формулы LVII.
Стадия (3) процесса (схема XIV реакции) заключается в аминировании 1H-имидазо[4,5-с]хинолин-5N-оксида формулы LVII по способу, используемому на стадии (3) процесса (схема II реакции), и приводит к получению 1Н-имидазо[4,5-с]хинолин-4-амина формулы XL, который является производным соединением продукта формулы I. Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
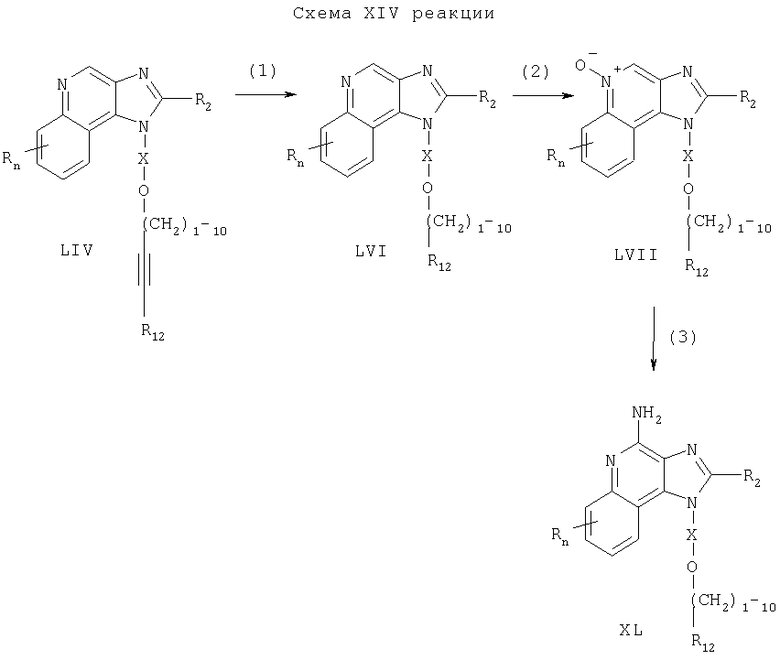
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой XV реакции, в которой значения R, R2, R12, Х и n определены выше.
На стадии (1) процесса (схема XV реакции) 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1 -иловый спирт формулы XXIV алкилируют в присутствии галогенида формулы Hal-(CH2)1-10-CH≡СН в соответствии с способом, используемым при проведении процесса по схеме V реакции. В результате получают 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-амин формулы LVIII, который является производным соединением продукта формулы IV.
На стадии (2) процесса (схема XV реакции) 6,7,8,9-тетрагидро-1H-имидазо[4,5-с]-хинолин-4-амин формулы LVIII алкилируют в присутствии галогенида формулы Hal-R12 в соответствии с способом, используемым на стадии (5) процесса (схема VII реакции). В результате получают 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-амин формулы LVIX, представляющий собой производное соединение продукта формулы IV.
Продукт или соль фармацевтического качества можно выделить из реакционной смеси с помощью обычных способов.
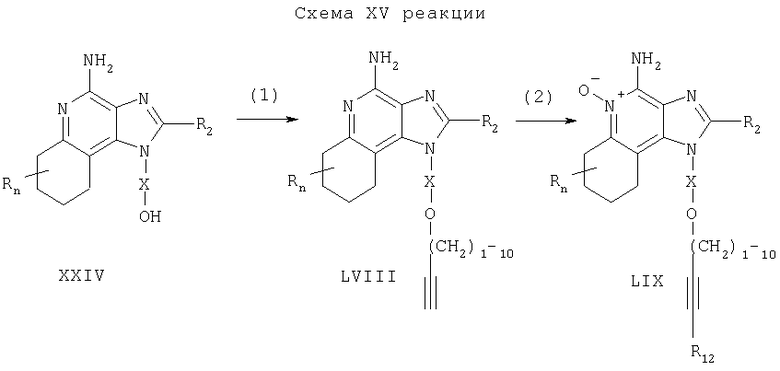
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой XVI реакции, в которой значения R, R1, R2, X и n определены выше.
На стадии (1) процесса (схема XVI реакции) проводят хлорирование 2,4-дигидрокси-3-нитро-6,7,8,9-тетрагидрохинолина формулы LX и получают 2,4-дихлор-3-нитро-6,7,8,9-тетрагидрохинолин формулы LXI. Для проведения реакции используют обычные хлорирующие агенты. Предпочтительно реакцию проводят, добавляя к соединению LX оксихлорид фосфора и нагревая полученную смесь до 55-65°C. Соединения формулы LX являются известными продуктами или могут быть получены с помощью известных синтетических способов (см., например, Николайдес с сотр., Патент США 5352784 и цитируемую в нем литературу).
На стадии (2) процесса (схема XVI реакции) проводят реакцию между 2,4-дихлор-3-нитро-6,7,8,9-тетрагидрохинолином формулы LXI и амином R1-O-X-NH2, в результате которой получают 2-хлор-3-нитро-6,7,8,9-тетрагидрохинолин-4-амин формулы LXH. Реакцию проводят, добавляя амин к раствору соединения LXI в соответствующем растворителе, таком как N,N-диметилформамид, и нагревая реакционную смесь до 55-65°С.
На стадии (3) процесса (схема XVI реакции) 2-хлор-3-нитро-6,7,8,9-тетрагидрохи-нолин-4-амин формулы LXII обрабатывают фенолом, используя методику, применяемую на стадии (4) процесса (схема IV реакции), и в результате получают 2-фенокси-3-нитро-6,7,8,9-тетрагидрохинолин-4-амин формулы LXIII.
На стадии (4) процесса (схема XVI реакции), используя методику, применяемую на стадии (2) процесса (схема VI реакции), проводят восстановление 2-фенокси-3-нитро-6,7,8,9-тетрагидрохинолин-4-амина формулы LXIII до 2-фенокси-6,7,8,9-тетрагидрохинолин-3,4-диамина формулы LXIV.
По методике, применяемой на стадии (3) процесса (схема VI реакции), на стадии (5) процесса (схема XVI реакции) осуществляют циклизацию 2-фенокси-6,7,8,9-тетрагидро-хинолин-3,4-диамина формулы LXIV до 4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо-[4,5-с]хинолина формулы LXV.
На стадии (6) процесса (схема XVI реакции), используя методику, применяемую на стадии (7) процесса (схема IV реакции), 4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо-[4,5-с]хинолин формулы LXV аминируют до 6,7,8,9-тетрагидро-1Н-имидазо-[4,5-с]хинолин-4-амина формулы III.
В изобретении сообщается также о синтезе новых соединений, представляющих интерес в качестве промежуточных продуктов, необходимых для получения соединений формулы (I), (II), (III) и (IV). Эти промежуточные соединения имеют структурные формулы (V)-(IX), детали которых приведены ниже.
Один класс промежуточных соединений имеет общую формулу (V):
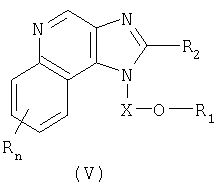
где Х представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
заместитель R1 выбран из группы, включающей:
- арил;
- алкенил;
- R4-арил; и
- (CH2)1-10-C≡C-R10;
R2 выбран из группы, включающей:
- атом водорода;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил; и
- алкил или алкенил, замещенные одним или большим количеством заместителей, выбранных из группы, включающей:
- ОН;
- атом галогена;
- N(R3)2;
- CO-N(R3)2;
- СО-С1-10алкил;
- CO-O-C1-10алкил;
- N3
- арил;
- гетероарил;
- гетероцикпил;
- СО-арил; и
- СО-гетероарил;
R4 представляет собой алкильную или алкенильную группы, причем обе эти группы могут быть разделены одной или несколькими -О- группами;
каждый заместитель R3 представляет собой независимо друг от друга атом водорода или C1-10алкильную группу;
каждый заместитель R10 выбран из группы, состоящей из атома водорода, алкильного, алкенильного и арильного радикала;
каждая группа Y независимо друг от друга представляет собой -О- или S(O)0-2-;
значение n может изменяться от 0 до 4; и
каждый заместитель R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
Другой класс промежуточных продуктов представляет собой имидазохинолин-4-феноксильные соединения формулы (VI):
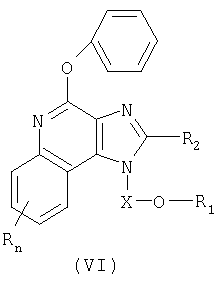
где Х представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
заместитель R1 выбран из группы, включающей:
- арил;
- алкенил;
- R4-арил; и
- (CH2)1-10-C≡C-R10;
R2 выбран из группы, включающей:
- атом водорода;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил; и
- алкил или алкенил, замещенные одним или большим количеством заместителей, выбранных из группы, включающей:
- ОН;
- атом галогена;
- N(R3)2;
- СО-N(R3)2;
- СО-C1-10алкил;
- CO-O-C1-10алкил;
- N3
- арил;
- гетероарил;
- гетероциклил;
- СО-арил; и
- СО-гетероарил;
R4 представляет собой алкильную или алкенильную группы, причем обе эти группы могут быть разделены одной или несколькими -О- группами;
каждый заместитель R3 представляет собой независимо друг от друга атом водорода или C1-10алкильную группу;
каждый заместитель R10 выбран из группы, состоящей из атома водорода, алкильного, алкенильного и арильного радикала;
каждая группа Y независимо друг от друга представляет собой -О- или S(O)0-2-;
значение n может изменяться от 0 до 4; и
каждый заместитель R независимо выбран из группы, состоящей из С1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы; или соль фармацевтического качества на основе этих групп.
Еще один класс промежуточных продуктов представляет собой имидазохинолин-N-оксидные соединения формулы (VII):
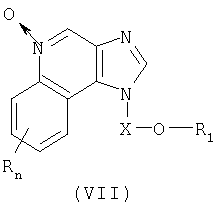
где
Х представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
заместитель R1 выбран из группы, включающей:
- арил;
- алкенил;
- R4-арил; и
- (CH2)1-10-C≡C-R10;
R4 представляет собой алкильную или алкенильную группы, причем обе эти группы могут быть разделены одной или несколькими -О- группами;
каждым заместитель R3 представляет собой независимо друг от друга атом водорода или С1-10алкильную группу;
каждый заместитель R10 выбран из группы, состоящей из атома водорода, алкильного, алкенильного и арильного радикала;
значение n может изменяться от 0 до 4; и
каждый заместитель R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
Дополнительный класс промежуточных соединений имеет общую формулу (VIII):
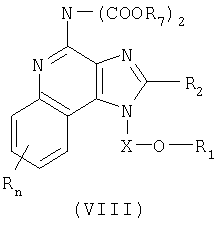
где
X представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
заместитель R1 выбран из группы, включающей:
- арил;
- алкенил;
- R4-арил; и
- (CH2)1-10-C≡C-R10;
R2 выбран из группы, включающей:
- атом водорода;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил; и
- алкил или алкенил, замещенные одним или большим количеством заместителей, выбранных из группы, включающей:
- ОН;
- атом галогена;
- N(R3)2;
- CO-N(R3)2;
- CO-C1-10алкил;
- CO-O-C1-10алкил;
- N3
- арил;
- гетероарил;
- гетероциклил;
- СО-арил; и
- СО-гетероарил;
R4 представляет собой алкильную или алкенильную группы, причем обе эти группы могут быть разделены одной или несколькими -О- группами;
каждый заместитель R3 представляет собой независимо друг от друга атом водорода или С1-10алкильную группу;
каждый заместитель R10 выбран из группы, состоящей из атома водорода, алкильного, алкенильного и арильного радикала;
каждая группа Y независимо друг от друга представляет собой -О- или S(O)0-2-;
значение n может изменяться от 0 до 4; и
каждый заместитель R независимо выбран из группы, состоящей из С1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы; и
заместитель R7 представляет собой трет-бутильную или бензильную группу; или соль фармацевтического качества на основе этих групп.
И последний класс промежуточных продуктов представляет собой имидазохинолин-4-хлор-соединения формулы (IX):
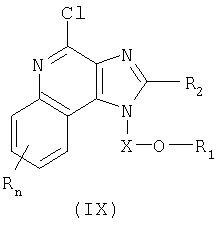
где
X представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
заместитель R1 выбран из группы, включающей:
- арил;
- алкенил;
- R4-арил; и
- (CH2)1-10-C≡C-R10;
R2 выбран из группы, включающей:
- атом водорода;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил; и
- алкил или алкенил, замещенные одним или большим количеством заместителей, выбранных из группы, включающей:
- ОН;
- атом галогена;
- N(R3)2;
- CO-N(R3)2;
- CO-C1-10алкил;
- CO-O-C1-10алкил;
- N3
- арил;
- гетероарил;
- гетероциклил;
- СО-арил; и
- СО-гетероарил;
R4 представляет собой алкильную или алкенильную группы, причем обе эти группы могут быть разделены одной или несколькими -О- группами;
каждый заместитель R3 представляет собой независимо друг от друга атом водорода или C1-10алкильную группу;
каждая группа Y независимо друг от друга представляет собой -О- или S(O)0-2-;
значение n может изменяться от 0 до 4; и
каждый заместитель R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
Используемые здесь термины «алкил», «алкиленил» и приставка «алк-» относятся как к линейным, так и разветвленным углеводородным цепям, а также к циклическим группам, например, циклоалкильным и циклоалкенильным группам. Если не указано иначе, эти группы содержат от 1 до 20 атомов углерода, а алкенильные группы от 2 до 20 атомов углерода. Предпочтительно эти группы содержат до 10 атомов углерода. Циклические группы могут быть либо моноциклическими, либо полициклическими и преимущественно содержать в цикле от 3 до 10 атомов углерода. Примерами циклических групп являются циклопропильная, циклопропилметильная, циклопентильная, циклогексильная и адамантильная группы.
Кроме того, алкильная и алкенильная часть групп -Х- могут быть либо незамещенными, либо содержать один или большее количество заместителей, причем эти заместители выбраны из группы, состоящей из алкильной, алкенильной, арильной, гетероарильной, гетероциклильной, арилалкильной, гетероарилалкильной и гетероциклоалкильной группировок.
Термин «галоидоалкил» включает группы, которые в качестве заместителя имеют один или большее количество галоидных атомов, включая перфторированные группы. Такое определение относится также к группам, которые включают приставку «гало». Примерами подходящих галоидоалкильных групп являются хлорметильная, трифторметильная и подобные группы.
Термин «арил», используемый в данном описании, включает в себя карбоциклические ароматические группы или циклические системы. Примеры арильных групп включают фенильную, нафтильную, бифенильную, флуоренильную и инденильную группы. Термин «гетероарил» относится к ароматическим циклам или циклическим системам, содержащим в кольце, по крайней мере, один гетероатом (например, О, S, N). Подходящие гетероарильные группы включают фурильную, тиенильную, пиридильную, хинолинильную, изохинолильную, индолильную, изоиндолильную, триазольную, пирролильную, тетразолильную имидазолильную, пиразолильную, оксазолильную, тиазолильную, бензофуранильную, бензотиофенильную, карбазолильную, бензоксазолильную, пиримидинильную, бензимидазолильную, хиноксалинильную, бензотиазолильную, нафтиридинильную, изоксазолильную, изотиазолильную, хиназолинильную, пуринильную и подобные группы.
В состав «гетероциклильных» соединений входят неароматические циклы или циклические системы, содержащие в кольце, по крайней мере, один гетероатом (например, атомы О, S, N). К этим соединениям относятся все полностью насыщенные и частично ненасыщенные производные указанных выше гетероарильных групп. Примерами гетероциклических групп являются пирролидинильная, тетрагидрофуранильная, морфолинильная, тиоморфолинильная, пиперидинильная, пиперазинильная, тиазолидинильная, имидазолидинильная, изотиазолидинильная и подобные группы.
Арильная, гетероарильная и гетероциклильная группы могут быть как незамещенными, так и содержащими один или более заместителей, независимо выбранных из группы, содержащей алкильную, алкоксильную, алкилтионильную, галоидоалкильную, галоидоалкоксильную, галоидоалкилтионильную группы, атом галогена, нитрильную, гидроксильную, меркапто- и цианогруппы, карбоксильную, формильную, арильную, арилоксильную, арилтионильную, арилалкоксильную, арилалкилтионильную, гетероарильную, гетероарилоксильную, гетероарилтионильную, гетероарилалкоксильную, гетероарилалкилтионильную, амино-, алкиламино-, диалкиламино-, гетероциклильную, гетероциклоалкильную, алкилкарбонильную, алкенилкарбонильную, алкоксикарбонильную, галоалкилкарбонильную, галоалкоксикарбонильную, алкилтиокарбонильную, арилкарбонильную, гетероарилкарбонильную, арилоксикарбонильную, гетероарилоксикарбонильную, арилтиокарбонильную, гетероарилтиокарбонильную, алканоилоксильную, алканоилтионильную, алканоиламино-, ароилоксильную, ароилтионильную, ароиламино-, алкиламиносульфонильную, алкилсульфонильную, арилсульфонильную, гетероарилсульфонильную, алкилкарбониламино-, алкенилкарбониламино-, арилкарбониламино, арилалкилкарбониламино-, гетероарилкарбониламино-, гетероарилалкилкарбониламино-, алкилсульфониламино-, алкенилсульфониламино-, арилсульфониламино-, арилалкилсульфониламино-, гетероарилсульфониламино-, гетероарилалкилсульфониламино-, алкиламинокарбониламино-, алкениламинокарбониламино-, ариламинокарбониламино-, арилалкиламинокарбониламино-, гетероариламинокарбониламино-, гетероарилалкилкарбониламино- группы; кроме того, в случае гетероциклильных групп - оксогруппы. В том случае, когда любые другие группы идентифицированы как «замещенные» или «иногда замещенные», эти группы также могут иметь в качестве заместителя одну или большее количество перечисленных выше группировок.
Обычно лишь некоторые заместители являются предпочтительными. Например, предпочтительным заместителем R1 является R4-арильная группа, а предпочтительными R10 группами являются алкильная и арильная группа, причем предпочтительной арильной группой является фенильная или замещенная фенильная группы. Предпочтительно, чтобы заместитель R отсутствовал (т.е. n=0). Предпочтительные R2 группы включают атом водорода, алкильные группы, содержащие от 1 до 4 атомов углерода (т.е., метильная, этильная, пропильная, изопропильная, н-бутильная, втор-бутильная, изобутильная и цикпопропилметильная группы), метоксиметильную и этоксиметильную группы. Для замещенных групп, таких как замещенные алкильные или замещенные арильные группы, предпочтительными заместителями являются атом галогена, нитрильная, нитро, карбоксильная, метоксильная, метилтиольная, трифторметильная и трифторметоксильная группы. Один или большее количество этих предпочтительных заместителей, если они вообще присутствуют, могут содержаться в предлагаемых в изобретении соединениях в любых комбинациях.
Данное изобретение включает описанные выше соединения в любой их фармацевтически доступной форме, включая изомеры, такие как диастереомеры и энантиомеры, соли, сольваты, полиморфные формы и т.п. В частности, если соединение является оптически активным, в изобретении описаны каждый энантиомер этого соединения, а также рацемическая смесь энантиомеров.
Фармацевтические составы и биологическая активность
Предлагаемые в изобретении фармацевтические составы содержат терапевтически эффективные количества описанных выше соединений в сочетании с фармацевтически приемлемым носителем.
Используемый здесь термин «терапевтически эффективное количество» означает количество соединения, достаточное для оказания терапевтического эффекта, такого как стимулирование образования цитокина, противоопухолевая активность и/или противовирусная активность. Хотя точное количество активного вещества, используемого в фармацевтическом составе, предлагаемом в изобретении, широко варьируется в зависимости от различных факторов, таких как физическая и химическая природа вещества, а также природа носителя и предполагаемая доза, полагают, что предлагаемые в изобретении составы будут содержать достаточное количество активного ингредиента, чтобы обеспечить дозу в пределах от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до приблизительно 5 мг соединения на 1 кг веса пациента. Составы могут использоваться в любых обычных лекарственных формах, таких как таблетки, лепешки, парентеральные составы, сиропы, кремы, мази, аэрозольные составы, чрезкожные пластыри, пластыри на слизистой оболочке и т.п.
Предлагаемые в изобретении соединения могут вводиться в организм в виде одного прописанного в рецепте терапевтического агента или в виде комбинации с одним или большим количеством других активных агентов, таких как дополнительные модификаторы иммунной реакции, противовирусные препараты, антибиотики и т.п.
Установлено, что предлагаемые в изобретении соединения стимулируют образование некоторых цитокинов в опытах, выполненных в соответствии с приведенными ниже способами испытания. Эти результаты показывают, что предлагаемые соединения представляют собой полезные модификаторы иммунной реакции, т.е. они способны модулировать иммунную реакцию целым рядом различных способов, оказывая тем самым помощь в излечении различных расстройств.
Цитокины, образующиеся при введении предлагаемых в данном изобретении соединений, обычно включают интерферон-о. (IFN-α) и/или фактор-α некроза опухоли (TNF-α), а также некоторые интерлейкины (IL). В частности, эти соединения вызывают образование IFN-α, TNF-α, IL-1, IL-6, IL-10 и IL-12, а также различных других цитокинов. Помимо других эффектов цитокины замедляют образование вирусов и рост опухолевых клеток, что делает предлагаемые в изобретении соединения полезными при лечении опухолей и вирусных заболеваний. Таким образом, настоящее изобретение предоставляет способ стимулирования биосинтеза цитокина при введении в организм животного эффективных количеств соединений или составов, предлагаемых в изобретении.
Установлено, что некоторые предлагаемые в изобретении соединения предпочтительно стимулируют участие IFN-α в изменении численности кроветворных клеток, таких как РВМС (одноядерные клетки периферийной крови), содержащих pDC2 клетки (предшественников дендритных клеток типа 2), не вызывая значительного сопутствующего воспалительного цитокинеза.
Помимо присущей им способности стимулировать образование цитокинов эти соединения оказывают влияние и на другие аспекты врожденной иммунной реакции. Например, в присутствии предлагаемых в изобретении соединений может быть стимулирована естественная активность клетки-убийцы, причем этот эффект может быть обусловлен образованием цитокина. Предлагаемые в изобретении соединения могут также активировать макрофаги, которые, в свою очередь, стимулируют секрецию оксида азота и образование дополнительных цитокинов. Кроме того, эти соединения могут вызывать разрастание и дифференциацию В-лимфоцитов.
Предлагаемые в изобретении соединения оказывают также влияние на приобретенную иммунную реакцию. Например, хотя, как полагают, предлагаемые соединения не оказывают непосредственного влияния на Т-лимфоциты или на образование Т-лимфоцитных цитокинов, эти соединения оказывают косвенное влияние на образование цитокина IFN-γ из фаг-помощника типа 1 Т-лимфоцита (Тh1), а образование Тh2 цикотинов IL-4, IL-5 и IL-13 замедляется при введении описываемых в изобретении соединений. Эти результаты показывают, что предлагаемые в данном изобретении соединения оказывают помощь при лечении заболеваний, при которых требуется увеличение количества Th1 и/или уменьшение количества Тh2. Учитывая способность предлагаемых в изобретении соединений замедлять иммунную реакцию Т-фаг-помощника типа 2, можно ожидать, что эти соединения окажутся полезными при лечении аллергических заболеваний, например, атопических дерматитов, астмы, аллергии, аллергических ринитов, а также в качестве вспомогательной вакцины для усиления иммунитета, а, возможно, и для лечения хронических грибковых заболеваний и хламидии.
Модифицирующее воздействие соединений на иммунную реакцию делает их полезными при лечении большого числа различных заболеваний. Вследствие своей способности стимулировать образование цитокинов, таких как IFN-α и/или TNF-α, предлагаемые в данном изобретении соединения особенно полезны при лечении вирусных заболеваний и опухолей. Эта иммуномодулирующая активность дает основания полагать, что предлагаемые в изобретении соединения могут быть полезными при лечении таких болезней (но не только их), как вирусные заболевания, например, остроконечная кондилома, обычные бородавки, подошвенные бородавки, гепатит В, гепатит С, простой герпес типа I и типа II, контагиозный моллюск, оспа, особенно натуральная оспа, ВИЧ, цитомегаловирус, вирус варицеллазостер, цервикальная интраэпителиальная неоплазия, человеческая вирусная папиллома и ассоциированные опухоли; грипп и парагрипп, грибковые заболевания, например, кандидоз, аспергиллез, криптококковые менингиты; заболевания, связанные с появлением новообразований, например, базалиома, лейкемия «волосистых» клеток, саркома Капоши, рак клеток почечного эпителия, рак клеток простого сквамозного эпителия, лейкемия миелопоэза, множественная миелома, меланома, неходжкинская лимфома, кожная лимфома и другие виды онкологических заболеваний; паразитарные заболевания, например, пневмоцистоз, криптоспоридиоз, гистоплазмоз, токсоплазмоз, трипаносомная инфекция, лейшманиоз; бактериальные инфекции, например, туберкулез и mycobacterium avium. С помощью предлагаемых в изобретении соединений можно, кроме того, лечить старческий кератоз, экзему, эозинофилию, эссенциальную тромбоцитаэмию, проказу, множественный склероз, синдром Оммена, дискоидную волчанку, болезнь Боуена и боуеноидный папулез, облысение, ингибирование образования келоидных швов после хирургических операций и других типов постхирургических шрамов. Кроме того, эти соединения могут стимулировать заживление ран, включая хронические раны. Они могут быть полезны также для лечения оппортунистических инфекций и опухолей, которые появляются после подавления иммунитета, например, у онкологических и ВИЧ больных, а также после операций по пересадке органов.
Эффективным количеством введенного соединения, которое предназначено для стимулирования биосинтеза цитокинов, является такое количество, которое оказывается достаточным, чтобы побудить один или большее число типов клеток, таких как моноциты, макрофаги, дендритные клетки и В-клетки, к образованию одного или большего числа цитокинов, таких как, например, IFN-α, TNF-α, IL-1, IL-6, IL-10 и IL-12, в количестве, превышающем фоновый уровень таких цитокинов. Точное эффективное количество соединения будет изменяться в широких пределах в зависимости от различных факторов, но ожидается, что эффективная доза этого соединения будет находиться в интервале от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно в интервале между приблизительно 10 мкг/кг до приблизительно 5 мг/кг.
Изобретение предлагает также способ лечения вирусной инфекции и онкологических заболеваний у животного путем введения в организм животного эффективного количества предлагаемого в изобретении соединения или составы на его основе. Эффективным количеством соединения для лечения или подавления вирусной инфекции является такое его количество, которое будет вызывать уменьшение одного или большего числа симптомов вирусной инфекции, таких как вирусные поражения, вирусную нагрузку, скорость образования вирусов и увеличивать их смертность по сравнению с ситуацией, наблюдаемой для контрольных животных, не принимающих эти соединения. Точное количество соединения будет изменяться в широких пределах в зависимости от различных факторов, но ожидается, что доза этого соединения должна находиться в интервале от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно в интервале между приблизительно 10 мкг/кг до приблизительно 5 мг/кг. Эффективным количеством соединения для лечения онкологического заболевания является такое его количество, которое будет вызывать уменьшение размера опухоли или числа опухолей. И в этом случае точное количество соединения будет изменяться в широких пределах в зависимости от различных факторов, но ожидается, что доза этого соединения должна находиться в интервале от приблизительно 100 нг/кг до 50 мг/кг, предпочтительно в интервале между приблизительно 10 мкг/кг до приблизительно 5 мг/кг.
Предлагаемое изобретение иллюстрируется ниже различными примерами. Эти примеры приведены только в качестве иллюстрации и ни в коей мере не ограничивают общие рамки изобретения.
Примеры
В приведенных ниже примерах некоторые соединения очищали с помощью полупрепаративной высокоэффективной жидкостной хроматографии. С этой целью использовали два способа, которые обсуждены ниже. В обоих случаях использовали прибор А-100 Gilson, снабженный интерфейсом серии 900 Series Intelligent Interface. Фракции, получаемые на полупрепаративном хроматографе, анализировали масс-спектрометрическим способом LC-APCI/MS. Соответствующие фракции соединяли вместе и подвергали лиофилизации для получения трифторуксуснокислой соли определенного соединения.
Способ А
Колонка: колонка Microsorb C18, 21,4×250 мм, размер частиц 8 мкм, размер пор 60 Ангстрем. Скорость потока 10 мл/мин. Градиент элюента: содержание компонента В в элюенте изменялось от 2 до 95% в течение 25 мин и элюент с 95%-ным содержанием компонента В использовался в течение 5 минут. Компонент А - 0,1%-ный раствор трифторуксусной кислоты в воде; компонент В - 0,1%-ный раствор трифторуксусной кислоты в ацетонитриле. Отбирают фракцию, имеющую максимум поглощения при 254 нм.
Способ В
Колонка: Phenomenex Capcell PakC18, 35×20 мм, размер частиц 5 мкм. Скорость потока 20 мл/мин. Градиент элюента: содержание компонента В в элюенте изменялось от 5 до 95% в течение 10 мин и элюент с 95%-ным содержанием компонента В использовался в течение 2 минут. Компонент А - 0,1%-ный раствор трифторуксусной кислоты в воде; компонент В - 0,1%-ный раствор трифторуксусной кислоты в ацетонитриле. Отбирают фракцию, имеющую максимум поглощения при 254 нм.
Пример 1
1[2-(2-Пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
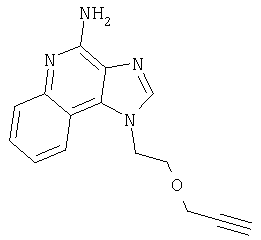
Часть А
28,5 г (0,133 моля) 2-(1Н-имидазо[4,5-с]хинолин-1-ил)-1-этанола добавляют несколькими порциями в течение 1 часа к смеси 240 мл 50%-ного водного раствора гидроксида натрия, 240 мл дихлорметана, 39,6 г (0,266 моля) 80%-ного пропаргилбромида и 2,46 г (0,013 моля) бензилтриметиламмонийхлорида. Полученную смесь перемешивают при температуре окружающей среды в течение 16 часов. Водный и органический слои разделяют, и водный слой экстрагируют дополнительным количеством дихлорметана. Органические слои объединяют, промывают водой, сушат над сульфатом магния и концентрируют при пониженном давлении. К полученному остатку добавляют диэтиловый эфир и смесь перемешивают. Твердый оранжевый продукт отфильтровывают и перекристаллизовывают из этилацетата. В результате получают 19,8 г желтого кристаллического 2-(1Н-имидазо[4,5-с]хинолин-1-ил)этил(2-пропинилового) эфира. Температура плавления этого вещества 124-126°C.
Анализ. Рассчитано для С15Н13N3О:%С 71,70; %Н, 5,21; %N, 16,72. Найдено: %С 71,85; %Н, 5,25; %N, 16,90.
1H ЯМР(300 МГц, ДМСО) δ 9,21 (синглет, 1Н); 8,44 (мультиплет, 1Н); 8,36 (синглет, 1Н); 8,18 (мультиплет, 1Н); 7,71 (мультиплет, 2Н); 4,93 (триплет, J=5,1 Гц, 2Н); 4,14 (дублет, J=2.4 Гц, 2Н); 3,98 (триплет, J=5,1 Гц, 2Н); 3,55 (триплет, J=2,2 Гц, 1Н);
HRMS (масс-спектр высокого разрешения) (ESI) Рассчитано для C15H13N3O (MH+) 252,1137, найдено 252,1141.
Часть В
19,7 г (78,4 ммоля) 2-(1H-имидазо[4,5-с]хинолин-1-ил)этил(2-пропинилового) эфира смешивают с хлороформом и охлаждают до 0°С. К смеси добавляют 15,7 г 3-хлорпероксибензойной кислоты (57-86%) и перемешивают в течение 0,5 часа. Смесь нагревают до температуры окружающей среды, при этом весь продукт переходит в раствор. Данные тонкослойной хроматографии показывают, что после этого в смеси еще присутствует некоторое количество исходного продукта. Поэтому к смеси добавляют дополнительное количество 3-хлорпероксибензойной кислоты (две порции по 4 г каждая). Через 0,5 часа после добавления второй порции кислоты, анализ способом тонкослойной хроматографии не обнаруживает в смеси исходного материала. Реакционную смесь экстрагируют 10%-ным раствором гидроксида натрия. Водные фракции несколько раз экстрагируют дихлорметаном. Органические слои объединяют, промывают водой, сушат над сульфатом магния и концентрируют при пониженном давлении до получения 18,5 г 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксида в виде желтого масла.
HRMS(ESI) Рассчитано для C15H14N3O2 (MH+) 268,1086, найдено 268,1098.
Часть С
15,5 г (82,2 ммоля) трихлорацетилизоцианата в атмосфере азота по каплям добавляют к смеси 18,3 г (68,5 ммоля) 1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-5N-оксида и 300 мл дихлорметана. Наблюдается интенсивное выделение двуокиси углерода. Через 0,5 часа весь материал переходит в раствор. Этот раствор перемешивают в течение приблизительно 1 часа и анализируют способом тонкослойной хроматографии (ТСХ). Анализ указывает на наличие в смеси малых количеств исходного материала. Добавляют еще 4,5 г трихлорацетилизоцианата. Через 1 час реакция согласно данным ТСХ завершается полностью. Летучие продукты удаляют при пониженном давлении и получают N-[1[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-ил}-2,2,2-трихлорацетамид в виде бледно-желтого твердого вещества.
Часть D
К смеси твердого продукта, образовавшегося в части С, и 200 мл метанола добавляют 150 мл дихлорметана. При этом весь продукт переходит в раствор. К раствору добавляют 50 г метилата натрия (в виде 25%-ного раствора в метаноле) и перемешивают его при температуре окружающей среды в течение ночи. Образовавшийся осадок отфильтровывают. Фильтрат концентрируют до общего объема около 100 мл и отфильтровывают вновь выпавший осадок. Твердый осадок объединяют вместе и сушат в вакуумной печи при 60°С в течение 16 часов. В результате получают 16,4 г твердого беловатого 1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина, температура плавления которого составляет 225-227°С.
Анализ. Рассчитано для C15H14N4O. (H2O)1/4:%C 66,53;%Н, 5,40;%N, 20,69. Найдено: %С 66,33; %Н, 5,18; %N, 21,12.
1H ЯМР(300 МГц, ДМСО) δ 8,13 (синглет, 1Н); 8,08 (широкий дублет, J=7,8 Гц, 1Н); 7,62 (широкий дублет, J=8,3 Гц, 1Н); 7,44 (широкий триплет, J=7,6 Гц, 2Н); 7,24 (широкий триплет, J=7,5 Гц, 2Н); 6,54 (синглет, 2Н); 4,81 (триплет, J=5,4 Гц, 2Н); 4,14 (дублет, J=2,4 Гц, 2Н); 3,93 (триплет, J=5,1 Гц, 2Н); 3,38 (триплет, J=2,4 Гц, 1Н)
HRMS (ESI) Рассчитано для C15H14N4O (MH+) 267,1246, найдено 267,1253.
Пример 2
2-{3-[2-(4-Амино-1H-имидазо[4,5-хинолин-1-ил)этокси]-1-пропинил}бензонитрил
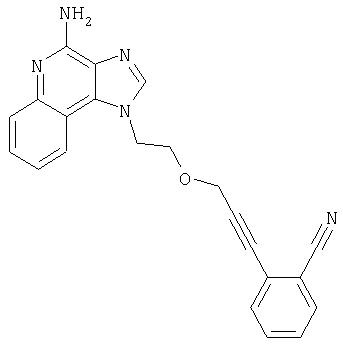
Часть А
К 16 г (60,1 ммоля) 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина добавляют 32,7 г (150 ммолей) ди-трет-бутилдикарбоната, 21 мл (150 молей) триэтиламина, 150 мл N,N-диметилформамида и 0,1 г 4-(диметиламино)пиридина и смесь нагревают до 80-85°С. Через 1 час смесь становится гомогенной и по данным ТСХ содержит очень небольшое количество исходного материала. Раствор нагревают еще в течение 1 часа, после чего разбавляют водой и этилацетатом. Водный и органический слои разделяют, и водный слой экстрагируют этилацетатом. Органические слои объединяют, промывают водой и рассолом, сушат над сульфатом магния и концентрируют при пониженном давлении до получения бледно-желто-оранжевого твердого продукта. После обработки этого вещества диэтиловым эфиром получают 22,6 г N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде беловатого твердого вещества, температура плавления которого составляет 139-142°С.
Анализ. Рассчитано для С15Н30N4O5: %С 64,36; %Н, 6,48; %N, 12,01. Найдено: %С 64,40; %Н, 6,43; %N,11,06.
1H ЯМР(300 МГц, ДМСО) δ 8,44 (мультиплет, 1Н); 8,35 (синглет, 1Н); 8,08 (мультиплет, 1Н); 7,73 (мультиплет, 2Н); 4,94 (триплет, J=4,9 Гц, 2Н); 4,12 (дублет, J=2,4 Гц, 2Н); 3,98 (триплет, J=5,1 Гц, 2Н); 3,31 (триплет, J=2,4 Гц, 1Н); 1,34 (синглет, 18Н)
HRMS(ESI) Рассчитано для С25Н31N4O5 (MH+) 467,2294, найдено 467,2307.
Часть В
В атмосфере азота 0,54 г (2,35 ммоля) 2-иодбензонитрила, 0,09 г (0,13 ммоля) дихлорбис(трифенилфосфин)палладия (II) и 0,05 г (0,26 ммоля) иодида меди (I) добавляют к смеси 1,0 г (2,14 ммоля) N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина и 25 мл безводного N,N-диметилформамида. Смесь выдерживают в течение 2 часов и затем медленно выливают в воду. Полученный осадок собирают и сушат при 35°С в течение 16 часов. В результате получают 1,18 г твердого 2-(3-{2-[-(4-(бис-третбутоксикарбонил)амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]-1-пропинил}бензонитрила.
1H ЯМР(300 МГц, ДМСО) δ 8,47 (дублет, J=6,8 Гц, 1Н); 8,39 (синглет, 1Н); 8,06 (дублет, J=7,8 Гц, 1Н); 7,87 (дублет, J=7,3 Гц, 1Н); 7,40-7,80 (мультиплет, 4Н); 7,34 (дублет, J=7,3 Гц, 1Н); 5,00 (широкий синглет, 2Н); 4,47 (широкий синглет, 2Н); 4,13 (синглет, 2Н); 1,31 (синглет, 18Н)
HRMS(ESI) Рассчитано для С32Н34N5O5 (MH+) 568,2560, найдено 568,2565.
Часть С
К раствору материала, полученного в части В, в 20 мл дихлорметана добавляют 20 мл трифторуксусной кислоты. Через 4 часа реакционную смесь разбавляют дихлорметаном, содержащим малое количество метанола, и 20%-ным раствором гидроксида натрия. Водный и органический слои разделяют, и водный слой экстрагируют дихлорметаном. Органические слои объединяют, сушат над сульфатом магния и концентрируют при пониженном давлении до получения желтого порошка. Этот материал очищают способом флеш-хроматографии, используя в качестве элюента 9/1 смесь дихлорметан/метанол, и получают 0,48 г белого порошкообразного 2-{3-[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]-1-пропинил}бензонитрила. Температура плавления этого вещества 180-183°С.
Анализ. Рассчитано для C22H17N5O.(H2O)2/5: %С 70,54; %Н, 4,79; %N, 18,70. Найдено: %С 70,61; %Н, 4,75; %N, 18,70.
1H ЯМР(300 МГц, ДМСО) δ 8,19 (синглет, 1Н); 8,12 (дублет, J=8,3 Гц, 1Н); 7,88 (дублет, J=7,8 Гц, 1Н); 7,55-7,75 (мультиплет, 3Н); 7,40-7,50 (мультиплет, 2Н); 7,24 (широкий триплет, J=7,5 Гц, 1Н); 6,68 (широкий синглет, 2Н); 4,81 (триплет, J=5,4 Гц, 2Н); 4,14 (дублет, J=2,4 Гц, 2Н); 4,87 (триплет, J=5,1 Гц, 2Н); 4,50(синглет, 2Н); 4,09 (триплет, J=5,1 Гц, 1Н).
Пример 3
1-{2-[(3-Фенил-2-пропинил)окси]этил}-1H-имидазо[4,5-с]хинолин-4-амин

Смесь 10 г (37,6 ммоля) 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина, 150 мл безводного N,N-диметилформамида и 6,23 г (45,1 ммоля) карбоната калия нагревают в атмосфере азота до 70°С. Добавляют 4,43 мл (39,5 ммоля) иодбензола, 0,53 г (0,75 ммоля) дихлорбис(трифенилфосфин)палладия (II) и 0,29 г (1,50 ммоля) иодида меди (I) и смесь перемешивают в течение 0,5 часа. Температуру повышают до 85°С. Через 1,5 часа проводят анализ способом высокоэффективной жидкофазной хроматографии (обратная фаза, ацетонитрил/вода с 0,1% трифторуксусной кислоты), который показывает, что реакция полностью завершена. Смесь охлаждают до температуры окружающей среды и отфильтровывают. Осадок дважды очищают способом флеш-хроматографии (95/5 дихлорметан/метанол) и получают 2,7 г белого твердого 1-{2-[(3-фенил-2-пропинил)окси]этил}-1Н-имидазо[4,5-с]-хинолин-4-амина. Температура плавления этого продукта 196-197°С.
Анализ. Рассчитано для C21H18N4O: %С 73,67; %Н, 5,30; %N, 16,36. Найдено: %С 73,29; %Н, 5,23; %N, 16,35.
1H ЯМР(300 МГц, ДМСО) δ 8.17 (синглет, 1Н); 8,12 (дублет, J=7,4 Гц, 2Н); 7,63 (двойной дублет, J=8,3, 0,9 Гц, 1Н); 7,44 (триплет, J=7,5 Гц, 1Н); 7,15-7,40 (мультиплет, 6Н); 6,60 (синглет, 2Н); 4,86 (триплет, J=5,1 Гц, 2Н); 4,39 (синглет, 2Н); 4,03 (триплет, J=5,1 Гц, 2Н)
HRMS(EI) Рассчитано для C21H18N40O (МН+) 342,1481, найдено 342,1490.
Пример 4
Солянокислый 1-{2-[(3-фенил-2-пропинил)окси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин
1,0 г (2,92 ммоля) 1-{2-[(3-фенил-2-пропинил)окси]этил}-1Н-имидазо[4,5-с]-хинолин-4-амина растворяют в смеси 15 мл метанола и 5 мл дихлорметана. К раствору добавляют 10 мл 1 М раствора хлористого водорода в диэтиловом эфире, и смесь перемешивают в течение 16 часов. В процессе перемешивания наблюдается выпадение осадка. Смесь концентрируют при пониженном давлении до получения твердого продукта. Этот материал перекристаллизовывают из ацетонитрила, содержащего малые количества метанола, и в результате получают 0,52 г беловатого кристаллического солянокислого 1-{2-[(3-фенил-2-пропинил)окси]этил}-1Н-имидазо{4,5-с]-хинолин-4-амина. Температура плавления этой соли 231-236°С.
Анализ. Рассчитано для C21H19CIN4O,(H2O)1/4: %C 65,79; %Н, 5,13; %N, 14,61. Найдено: %C 65,72; %Н, 5,0; %N, 14,73.
1H ЯМР(300 МГц, ДМСО) δ 8,49 (синглет, 1Н); 8,34 (дублет, J=8,3 Гц, 1Н); 7,81 (широкий дублет, J=8,3, 1Н); 7,72 (триплет, J=7,8 Гц, 1Н); 7,56 (триплет, J=7,8 Гц, 1Н); 7,30-7,40 (мультиплет, 3Н); 7,14 (двойной дублет, J=8,0, 1,5 Гц, 12); 4,94 (триплет, J=4,8 Гц, 2Н); 4,38 (синглет, 2Н); 4,05 (триплет, J=4,9 Гц, 2Н)
HRMS(EI) Рассчитано для C21H18N4O (MH+) 342,1481. найдено 342,1485.
Пример 5
1-{2-[3-(4-Метоксифенил-2-пропокси]этил}-1H-имидазо[4,5-с]хинолин-4-амин
Часть А
В атмосфере азота к 1,0 г (2,14 ммоля) N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина добавляют 0,8 мл (5,56 ммоля) триэтиламина, 0,51 г (2,18 ммоля) 4-иоданизола и 15 мл N,N-диметилформамида. К смеси добавляют 0,09 г (0,13 ммоля) дихлорбис(трифенилфосфин)палладия (II) и 0,05 г (0,26 ммоля) иодида меди (I) и перемешивают ее в течение 1 часа при температуре окружающей среды. Анализ смеси способом высокоэффективной жидкофазной хроматографии (обратная фаза, ацетонитрил/вода) показывает, что после этого времени реакция полностью завершена. Реакционную смесь распределяют между этилацетатом и водным раствором бикарбоната натрия. Органический слой промывают водой и затем рассолом, сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении до получения 0,95 г N,N-(бис-трет-бутоксикарбонил)-1-(2-{[3-(4-метоксифенил)-2-пропинил]окси}этил-1H-имидазо[4,5-с]хинолин-4-амина в виде твердого продукта оранжевого цвета.
HRMS(EI) Рассчитано для С32Н36N4O6 (MH+) 572,2635, найдено 572,2635.
Часть В
Смесь 0,75 г (1,31 ммоля) N,N-(бис-трет-бутоксикарбонил)-1-(2-{[3-(4-метокси-фенил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина, 25 мл этилацетата и 100 мг катализатора (5% Pd/C с 50% воды) гидрируют в аппарате Парра при давлении водорода 2,8 кг/см2 (40 фунт/дюйм2). При этом не протекает никакой реакции. Добавляют 150 мг оксида платины и 10 мл метанола и смесь гидрируют при 3,15 кг/см2 (45 фунт/дюйм2) в течение 1 часа. Сразу же отмечается расход водорода. Реакционную смесь отфильтровывают для удаления катализатора. Фильтрат концентрируют при пониженном давлении и получают желто-коричневую смолу, представляющую собой N,N-(бис-трет-бутоксикарбонил)-1-{2-[3-(4-метоксифенил)пропокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин.
HRMS(EI) Рассчитано для С32Н40N4O6 (МН+) 576,2948, найдено 576,2965.
Часть С
10 мл трифторуксусной кислоты добавляют к смеси материала, полученного в части В, и 10 мл дихлорметана. Полученный раствор перемешивают в течение 4 часов, после чего концентрируют при пониженном давлении. Остаток распределяют между 50%-ным водным раствором гидроксида натрия и дихлорметаном, содержащим небольшое количество метанола. Органическую фракцию сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении до получения желтовато-коричневого вспененного продукта. Этот продукт очищают способом флеш-хроматографии (9/1 дихлорметан/метанол) и получают светло-желтое стеклообразное вещество. Это вещество обрабатывают диэтиловым эфиром, после чего получают 0,41 г белого твердого 1-{2-[3-(4-метоксифенил)пропокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина. Температура плавления целевого продукта 116-118°С.
Анализ. Рассчитано для C22H24N4O2: %С 70,19; %Н, 6,43; %N, 14,88. Найдено: %С 69,79; %Н, 6,40; %N, 14,73.
1H ЯМР(300 МГц, ДМСО) δ 8,17 (синглет, 1Н); 8,12 (дублет, J=8,3 Гц, 2Н); 7,64 (дублет, J=8,3, 1Н); 7,45 (триплет, J=7,8 Гц, 1Н); 7,24 (триплет, J=7,6 Гц, 1Н); ); 6,80 (дублет, J=8,8, 2Н);); 6,66 (дублет, J=8,8, 2Н); 6,60 (синглет, 2Н); 4,80 (триплет, J=5,1 Гц, 2Н); 3,81 (триплет, J=4,9 Гц, 2Н); 3,66 (синглет, 3Н); 3,27 (триплет, J=6,1 Гц, 2Н); 2,32 (триплет, J=7,3 Гц, 2Н); 1,60 (мультиплет, 2Н).
Пример 6
N1,4-Диметил-3-{3-[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]пропил}-1-бензолсульфонамид
Часть А
В атмосфере азота к 1,7 г (6,35 ммоля) 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина добавляют 4,55 г (15,9 ммоля) дибензилдикарбоната, 1,8 мл триэтиламина (13,0 ммоля), 4-(диметиламино)пиридин и 20 мл N,N-диметилформамида. Реакционную смесь нагревают до 90°С, и при этой температуре смесь быстро становится гомогенной. Затем ее нагревают при 130°С в течение 4 часов. После этого реакционную смесь охлаждают и распределяют между дихлорметаном и водой. Водную фракцию экстрагируют дихлорметаном. Органические фракции соединяют вместе, сушат над сульфатом магния и затем концентрируют до общего объема около 10 мл. Концентрат оставляют стоять в течение выходных дней и затем разбавляют его толуолом. Образовавшийся осадок, который был отделен фильтрованием, был идентифицирован как исходный материал. Фильтрат разбавляют диэтиловым эфиром. После фильтрации образовавшегося осадка получают 1,1 г бензил-N-{1-[2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-ил}карбамата в виде белого твердого вещества.
1H ЯМР(300 МГц, ДМСО) δ 9,98 (синглет, 1Н); 8,34 (дублет, J=7,8 Гц, 1Н); 8,30 (синглет, 1Н); 7,97 (дублет, J=7,3 Гц, 1Н); 7,70 (триплет, J=7,8, 1Н); 7,58 (триплет, J =7,8 Гц, 1Н); 7,15-7,50 (мультиплет, 5Н); 5,21 (синглет, 2Н); 4,90 (триплет, J=5,1 Гц, 2Н); 4,14 (дублет, J=2,4 Гц, 2Н); 3,96 (триплет, J=4,9 Гц, 2Н); 3,38 (триплет, J=2,4 Гц, 2Н).
Часть В
В атмосфере азота к 0,37 г (0,91 ммоля) бензил-N-{1-[2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-ил}карбамата добавляют 0,3 г (0,96 ммоля) 3-иод-4-метил-1-бензолсульфонамида, 0,2 мл (1,36 ммоля) триэтиламина и 20 мл безводного ацетонитрила. К реакционному раствору добавляют 13 мг (0,018 ммоля) дихлорбис(трифенилфосфин)палладия (II) и 7 мг (0,036 ммоля) иодида меди (I) и раствор нагревают до приблизительно 45°С. Согласно данным высокоэффективной жидкостной хроматографии (с обратной фазой) реакция полностью завершается через 3 часа. Полученный раствор концентрируют при пониженном давлении, и остаток очищают способом флеш-хроматографии (от 98/2 до 95/5 дихлорметан/метанол), в результате чего получают 0,33 г бензил-N-(1-{2-[(3-{2-метил-5-[(метиламино)сульфонил]фенил}-2-пропинил)окси]этил}-1Н-имидазо[4,5-с]хинолин-4-ил)карбамата в виде бледно-желтого твердого вещества.
1H ЯМР(300 МГц, ДМСО) δ 9,96 (синглет, 1Н); 8,36 (мультиплет, 2Н); 7,96 (дублет, J=8,3 Гц, 1Н); 7,55-7,70 (мультиплет, 4Н); 7,48 (мультиплет, 2Н); 7,30 - 7,45 (мультиплет, 5Н); 5,21 (синглет, 2Н); 4,95 (триплет, J=4,6 Гц, 2Н); 4,40 (синглет, 2Н); 4,06 (триплет, J=5,1 Гц, 2Н); 2,54 (синглет, 3Н), 2,40 (дублет, J=4,9 Гц, 3Н) MS(CI) 584,476.
Часть С
0,08 г платины, нанесенной на активированный уголь (10%-ной), добавляют к смеси 0,3 г (0,51 ммоля) бензил-N-(1-{2-[(3-{2-метил-5-[(метиламино)сульфонил]фенил}-2-пропинил)окси]этил}-1Н-имидазо[4,5-с]хинолин-4-ил)карбамата и 10 мл метанола. Смесь подвергают гидрированию на аппарате Парра при давлении водорода 40 фунт/дюйм2 (2,8 кг/см2) в течение 16 часов. Данные анализа способом жидкостной хроматографии и масс-спектрометрии (хромасс-анализ) показывают, что в результате этой операции происходит восстановление тройной связи алкина, но не наблюдается никакого удаления феноксикарбонильной группы. К реакционной смеси добавляют 0,1 г палладия, нанесенного на активированный уголь (10%-ный), и гидрирование продолжают в тех же условиях в течение 8 часов, данные анализа способом жидкостной хроматографии и масс-спектроскопии показывают удаление лишь небольшого количества феноксикарбоксильной группы. К реакционной смеси добавляют 0,1 г палладия на активированном угле и реакционную смесь гидрогенирируют при давлении 40фунт-дюйм2 (2,8 кг/см2) в течение 16 часов. Согласно данным хромасс-анализа в результате этой реакции образуется один основной продукт, масса которого соответствует массе целевого продукта. Реакционную смесь фильтруют, и фильтрат промывают метанолом и дихлорметаном. Растворитель удаляют при пониженном давлении и в результате получают беловатый твердый продукт, перекристаллизация которого из ацетонитрила дает 0,11 г N1,4-диметил-3-{3-[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]пропил}-1-бензолсульфонамида в виде бледно-желтого кристаллического твердого вещества. Температура плавления этого продукта 207-209°С.
Анализ. Рассчитано для С23Н27N5O3S: %С 60,91; %Н, 6,00; %N, 15,44. Найдено: %С 60,87; %Н, 5,75; %N, 15,51.
1H ЯМР(300 МГц, ДМСО) δ 8,16 (синглет, 1Н); 8,12 (дублет, J=8,3 Гц, 2Н); 7,62 (дублет, J=8,3, 1Н); 7,53 (дублет, J=1,5 Гц, 1Н); 7,44 (широкий триплет, J=7,6 Гц, 1Н); 7,38 (мультиплет, 1Н); 7,24 (широкий триплет, J=7,6 Гц, 1Н); 7,16 (дублет, J=7,8Гц, 1Н); 7,02 (двойной дублет, J=7,8, 2,0 Гц, 1Н); 6,58 (синглет, 2Н); 4,80 (триплет, J=5,2 Гц, 2Н); 3,82 (триплет, J=5,2 Гц, 2Н); 3,31 (триплет, J=5,9 Гц, 2Н); 2,47 (синглет, 3Н); 2,37 (дублет, J=4,4 Гц, 2Н); 1,65 (мультиплет, 2Н).
HRMS(El) Рассчитано для С23Н27M5O3S (MH+)453,1835, найдено 453,1834.
Пример 7
Солянокислый 1-(2-{13-(2-изопропилфенил)-2-пропинил]окси}этил-1H-имидазо[4,5-финолин-4-амин
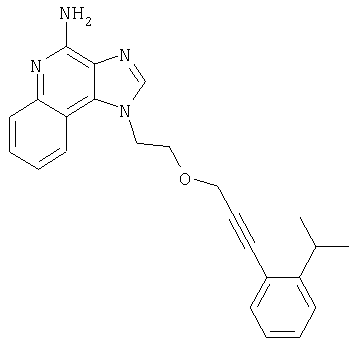
Смесь 0,50 г (1,88 ммоля) 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина, 0,65 г (2,63 ммоля) 2-иодизопропилбензола, 0,68 мл (4,88 ммоля) триэтиламина и 10 мл N,N,-диметилформамида нагревают в атмосфере азота до 60°С. К смеси добавляют 0,08 г дихлорбис(трифенилфосфин)палладия (II) и 0,04 г иодида меди (I). Согласно данным анализа способом тонкослойной хроматографии (9/1 дихлорметан/метанол) реакция завершается через 1,5 часа. Реакционную смесь концентрируют при пониженном давлении. Остаток очищают на хроматографической колонке, используя в качестве элюента 9/1 смесь дихлорметана с метанолом. Фракции продукта соединяют вместе и концентрируют при пониженном давлении. Остаток очищают на хроматографической колонке, используя в качестве элюента 9/1 смесь дихлорметана с метанолом, содержащим 0,5% концентрированного гидроксида аммония. Фракции продукта соединяют вместе и концентрируют при пониженном давлении до получения около 0,38 г твердого вещества. Это вещество в течение ночи перемешивают с 3,9 мл 1,0 М раствора соляной кислоты в диэтиловом эфире и затем концентрируют при пониженном давлении. Остаток перекристаллизовывают из смеси изопропанол/метанол, отфильтровывают и после сушки получают 0,24 г твердого солянокислого 1-(2-{[3-(2-изопропилфенил)-2-пропинил]окси}этил-1H-имидазо[4,5-с]хинолин-4-амина. Температура плавления этой соли 239-241°С.
Анализ. Рассчитано для C24H24N4O.HCl.(H2O)1/2: %C 67,06; %Н, 6,09; %N, 13,03. Найдено: %C 67,07; %Н, 6,00; %N, 13,09.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,54 (синглет, 1Н); 8,39 (дублет, J=8,1 Гц, 1Н); 7,85 (дублет, J=8,2, 1Н); 7,76 (триплет, J=7,2 Гц, 1Н); 7,59 (триплет, J=8,0 Гц, 1Н); 7,30-7,38 (мультиплет, 2Н); 7,11-7,19 (мультиплет, 2Н); 5,00 (триплет, J=4,7 Гц, 2Н); 4,47 (синглет, 2Н); 4,10 (триплет, J=4,7 Гц, 2Н); 3,16 (мультиплет, 1Н); 1,13 (дублет, J=6,9 Гц, 6Н)
ИК-спектр (в таблетке KBr): 3363, 3111, 2957, 1672, 753 см-1
HRMS(EI) Рассчитано для С24Н24N4O (M+) 384,1950, найдено 384,1943.
Пример 8
1-(2-{[3-(2,6-Диметилфенил)-2-пропинил]окси}этил-1H-имидазо[4,5-с]хинолин-4-амин
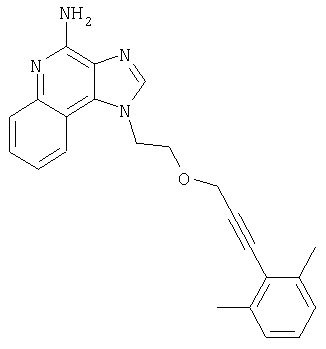
Используя основной способ, применяемый в примере 7, проводят реакцию между 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амином (0,50 г, 1,88 ммоля) и 2,6-диметилиодбензолом (0,61 г, 2,63 ммоля). Полученный продукт очищают на хроматографической колонке, используя в качестве элюента смесь 95/5 дихлорметан/метанол, и в результате получают 0,056 г твердого 1-(2-{[3-(2,6-диметилфенил)-2-пропинил]окси}этил-1Н-имидазо[4,5-с]хинолин-4-амина. Температура плавления этого продукта 200-201°С.
Анализ. Рассчитано для С23Н22N4O.(Н2O)2/5: %С 73,29; %Н, 6,07; %N, 14,86. Найдено: %С 73,36; %Н, 5,88; %N, 14,84.
1H ЯМР(300 МГц, ДМСО-d6) δ 8.19 (синглет, 1Н); 8,13 (дублет, J=8,1 Гц, 1Н); 7,62 (дублет, J=7,9,1Н); 7,44 (триплет, J=8,0 Гц, 1Н); 7,23 (триплет, J=7,9 Гц, 1Н); 7,09-7,14 (мультиплет, 1Н); 7,01 -7,03 (мультиплет, 2Н); 6,76 (синглет, 2Н); 4,87 (триплет, J=4,9 Гц, 2Н); 4,48 (синглет, 2Н); 4,05 (триплет, J=4,9 Гц, 2Н); 2,15 синглет, 6Н)
ИК-спектр (в таблетке KBr): 3379, 3065, 1659, 1530, 1483, 1107, 751 см-1
HRMS(EI) Рассчитано для C23H22N4O (M+) 370,1794, найдено 370,1789.
Пример 9
1-(2-{[3-(4-Феноксифенил)-2-пропинил]окси}этил-1H-имидазо[4,5-c]хинолин-4-амин
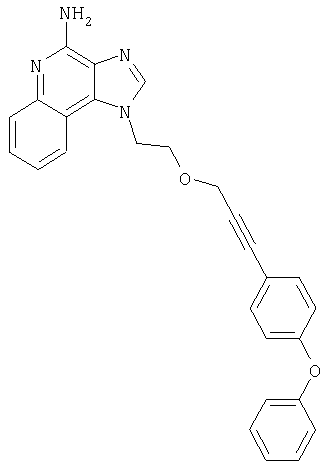
Используя основной способ, применяемый в примере 7, проводят реакцию между 1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-амином (0,50 г, 1,88 ммоля) и 4-иодфенилфениловым эфиром (0,78 г, 2,63 ммоля). Образующийся продукт очищают на хроматографической колонке, используя в качестве элюента смесь 95/5 дихлорметан/метанол, и в результате получают твердое вещество. Это вещество смешивают с водным раствором гидроксида натрия, чтобы удалить соли, и затем очищают на хроматографической колонке, используя в качестве элюента смесь 9/1 этилацетат/метанол. Полученный на этой стадии продукт подвергают дальнейшей хроматографической очистке, используя в качестве элюента смесь 99/1 этилацетат/метанол, и в результате получают 24 мг 1-(2-{[3-(4-феноксифенил)-2-пропинил]окси}этил-1Н-имидазо[4,5-с]хинолин-4-амина, температура плавления которого составляет 146-148°С.
Анализ. Рассчитано для С27H22N4O2.(Н2O)4/5: %С 72,24; %Н, 5,30; %N, 12,48. Найдено: %С 71,82; %Н, 4,85; %N, 12,35.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,18 (синглет, 1Н); 8,12 (дублет, J=7,4 Гц, 1Н); 7,62 (дублет, J=7,7, 1Н); 7,41-7,47 (мультиплет, 3Н); 7,18-7,27 (мультиплет, 4Н); 7,06 (двойной дублет, J=7,6, 1,0 Гц, 1Н); 6,90 (двойной дублет, J=6,7 Гц, 2Н); 6,71 (синглет, 2Н); 4,85 (триплет, J=5,1 Гц, 2Н); 4,37 (синглет, 2Н); 4,02 (триплет, J=5,0 Гц, 2Н)
ИК-спектр (в таблетке KBr): 3444, 3070, 2928, 1500, 1230 см-1.
Пример 10
1-(2-{3-[2-(Трифторметил)фенил]-2-пропинил}окси)этил]-1H-имидазо[4,5-с]хинолин-4-амин
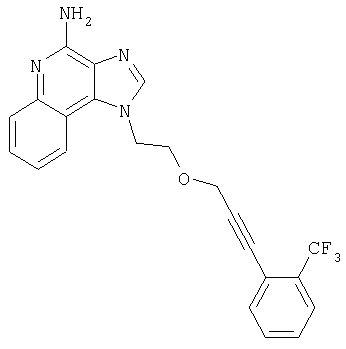
Используя основной способ, применяемый в примере 7, проводят реакцию между 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амином (0,50 г, 1,88 ммоля) и 2-иодбензотрифторидом (0,71 г, 2,63 ммоля). Реакционную смесь концентрируют при пониженном давлении. Полученный стеклообразный продукт обрабатывают 10 мл водного раствора бисульфита натрия и 20 мл метанола. Твердый остаток отфильтровывают. Фильтрат концентрируют при пониженном давлении до получения белого порошка. Этот материал промывают водой и сушат в печи течение 4 дней при 80°С. Получают около 0,33 г твердого вещества. Это вещество частично растворяют в смеси, состоящей из 17 мл дихлорметана и 17 мл метанола. Затем добавляют 3,24 мл 1,0 М раствора соляной кислоты в диэтиловом эфире, в результате чего смесь становится гомогенной. После концентрирования при пониженном давлении получают коричневый кристаллический остаток, который соединяют с 50/50 смесью ацетонитрила с этилацетатом, содержащим небольшое количество метанола. Добавляют 0,5 мл 20%-ного раствора гидроксида натрия. Смесь снова концентрируют до получения стеклообразного твердого продукта, который подвергают очистке на хроматографической колонке, используя в качестве элюента смесь 9/1 этилацетат/метанол. В результате получают 14 мг белого кристаллического 1-(2-{3-[2-(трифторметил)фенил]-2-пропинил}окси)этил]-1H-имидазо[4,5-с]хинолин-4-амина. Температура плавления этого продукта 154-155°С.
Анализ. Рассчитано для С22Н17F3N4O: %С 64,39; %Н, 4,18; %М, 13,65. Найдено: %С 64,39; %Н, 4,19; %N, 13,71.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,16 (синглет, 1Н); 8,11 (дублет, J=7,4 Гц, 1Н); 7,74 (дублет, J=7,3, 1Н); 7,56-7,64 (мультиплет, 3Н); 7,38-7,46 (мультиплет, 2Н); 7,22 (триплет, J=7,6 Гц, 1Н); 6,59 (синглет, 2Н); 4,87 (триплет, J=5,1 Гц, 2Н); 4,45 (синглет, 2Н); 4,04 (триплет, J=5,1 Гц, 2Н)
ИК-спектр (в таблетке KBr): 3375, 3102, 1657, 1583, 1530, 1484, 1320,1103, 765 см-1.
HRMS(EI) Рассчитано для C22H17F3N4O (M+) 410,1354, найдено 410,1350.
Пример 11
Трифторацетат 1-(2-{3-[4-(1H-1-пирролил)фенил]-2-пропокси}этил)-1H-имидазо[4,5-c]хинолин-4-амина
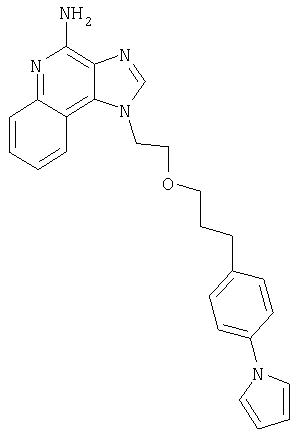
Часть А
50 г (174 ммолей) дибензилдикарбоната добавляют в атмосфере азота к смеси 16,4 г (61,6 ммоля) 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина и 200 мл безводного N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов, в процессе перемешивания смесь становится гомогенной. После этого реакционную смесь распределяют между этилацетатом и водой. Водный и органический слои разделяют, и водный слой экстрагируют этилацетатом. Органические слои объединяют, промывают водой и затем рассолом, сушат над сульфатом магния, отфильтровывают и концентрируют при пониженном давлении до получения полутвердого продукта. После обработки этого вещества диэтиловым эфиром получают 27,4 г белого твердого N,N-(бисбензилоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина.
Часть В
В атмосфере азота к 0,5 г (0,94 ммоля) N,N-(бисбензилоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина добавляют 5 мл безводного ацетонитрила, 0,34 мл (2,43 ммоля) триэтиламина и 0,28 г (1,03 ммоля) 1-(4-иодфенил)пиррола и полученную гомогенную смесь нагревают до 80°С. К реакционному раствору добавляют 0,007 г иодида меди (I) и 0,013 г дихлорбис(трифенилфосфин)палладия (II). Реакция завершается в течение 30 минут. Продукт очищают способом жидкостной хроматографии, используя смесь 4/6 гексан/этилацетат в качестве элюента, и получают стеклообразное твердое вещество. Этот материал очищают на второй колонке, используя смесь 9/1 гексан/этилацетат, и получают 0,229 г N,N-(бисбензилоксикарбонил)1-[2-({3-[4-(1Н-пиррол-1-ил)фенил]проп-2-инил}окси)этил]-1H-имидазо[4,5-с]хинолин-4-амина.
1H ЯМР(500 МГц, ДМСО-d6) δ 8,49 (дублет, J=7,7 Гц, 1Н); 8,44 (синглет, 1Н); 8,14 (дублет, J=7,9 Гц, 1Н); 7,75-7,77 (мультиплет, 2Н); 7,54 (дублет, J=5,1, 2Н); 7,40 (синглет, 2Н); 7,32 (дублет, J=6,8, 2Н); 7,24-7,27 (мультиплет, 6Н); 7,14-7,16 (мультиплет, 4Н); 6,29 (синглет, 2Н); 5,18 (синглет, 4Н); 5,00 (триплет, J=5,2 Гц, 2Н); 4,42 (синглет, 2Н); 4,10 (триплет, J=5,1 Гц, 2Н)
MS(CI) для C41H33N5O5 m/z 676 (MH+), 632, 524, 408.
Часть С
Материал, полученный в части В, 0,24 г гидроксида палладия (в виде 20%-ного продукта, нанесенного на активированный уголь) и 5 мл метанола загружают в аппарат Парра и проводят гидрирование при давлении водорода 45 фунт/дюйм2 (3,2 кг/см2) в течение 3-4 часов. Реакционную смесь фильтруют для удаления катализатора, остаток на фильтре промывают дополнительным количеством метанола и фильтрат концентрируют при пониженном давлении. После очистки остатка с помощью полупрепаративной высокоэффективной жидкостной хроматографии (способ В) получают 36,6 мг твердого трифторацетата 1-(2-{3-[4-(1H-1-пирролил)фенил]-2-пропокси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина. Температура плавления продукта 179-181°С.
Анализ. Рассчитано для С25Н25N5O.С2HF3O2: %С 61,71; %Н, 4,99; %N, 13,33. Найдено: %С 61,49; %Н, 4,89; %N, 13,23.
1H ЯМР(500 МГц, ДМСО-d6) δ 8,51 (синглет, 1Н); 8,38 (дублет, J=8,4 Гц, 1Н); 7,84 (дублет, J=8,4 Гц, 1Н);7,73 (триплет, J=7,3 Гц, 1Н); 7,56 (триплет, J=7,8 Гц, 1Н); 7,33 (дублет, J=8,4 Гц, 2Н); 7,26 (триплет, J=2,1 Гц, 2Н); 6,96 (дублет, J=8,4 Гц, 2Н); 6,24 (триплет, J=8,4 Гц, 1Н); 4,91 (триплет, J=5,0 Гц, 2Н); 3,85 (триплет, J=5,0 Гц, 2Н); 3,3-3,4 (мультиплет, 2Н) 2,35 (триплет, J=7,6 Гц, 2Н); 1,61 (мультиплет, 2Н);
ИК-спектр (в таблетке KBr): 2949, 1705, 1523, 1204, 1123, 721 см-1.
HRMS(EI) Рассчитано для С25Н25N5О (M+) 411,2059, найдено 411,2060.
Пример 12
Бис(трифторацетат) 3-{3-[2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)этокси]пропил}бензойной кислоты
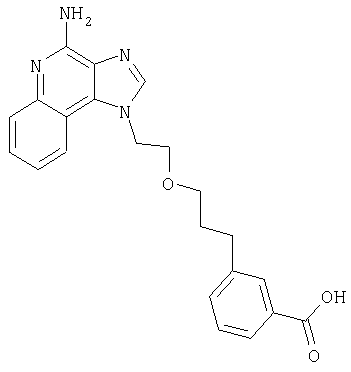
Часть А
2,82 г (6,04 ммоля) N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина добавляют к 2,245 г (6,64 ммоля) бензил-3-иодбензоата, 2,2 мл (15,7 ммоля) триэтиламина и 20 мл безводного ацетонитрила и полученную гомогенную смесь нагревают до 60°С. К реакционному раствору добавляют 0,05 г иодида меди (I) и 0,08 гдихлорбис(трифенилфосфин)палладия (II). Реакция завершается в течение 30 минут. Реакционную смесь концентрируют при пониженном давлении, и остаток очищают на хроматографической колонке, используя в качестве элюента сначала дихлорметан и затем смесь 98/2 дихлорметан/метанол. В результате получают 1,82 г бензил-3-{3-[2-(4-(бис-трет-бутоксикарбонил)амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]проп-1-инил}бензоата.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,46 (дублет, J=9,6 Гц, 1Н); 8,39 (синглет, 1Н); 8,05 (дублет, J=9,8 Гц, 1Н); 7,94-7,98 (мультиплет, 1Н); 7,84 (синглет, 1Н); 7,50-7,70 (мультиплет, 2Н); 7,36-7,49 (мультиплет, 7Н); 5,36 (синглет, 2Н); 4,98 (триплет, J=4,6 Гц, 2Н); 4,37 (синглет, 2Н); 4,06-4,13 (мультиплет, 2Н); 1,30 (синглет, 18Н);
MS(CI) для C39H40N4O7 m/z 677 (MH+), 577, 477.
Часть В
Раствор материала, полученного в части А, в метаноле соединяют с катализатором (1,0 г палладия (10%), нанесенного на активированный уголь) и проводят гидрирование при давлении 45 фунт/дюйм2 (3,2 кг/см2) при температуре окружающей среды в течение около 2,25 часа. Затем добавляют еще 0,3 г катализатора и гидрирование продолжают в течение 2 часов. Реакционную смесь фильтруют для удаления катализатора, остаток на фильтре тщательно промывают метанолом. Фильтрат концентрируют при пониженном давлении до получения около 1,2 г N,N-(бис-трет-бутоксикарбонила) 3-{3-[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]пропил}бензойной кислоты.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,50 (дублет, J=9,5 Гц, 1Н); 8,40 (синглет, 1Н); 8,07-8,10 (мультиплет, 1Н); 7,70-7,75 (мультиплет, 1Н); 7,65 (синглет, 1Н); 1,29 (синглет, 18Н); 7,29 (триплет, J=7,6 Гц, 1Н); 7,10 (дублет, J=7,8 Гц, 1Н); 4,94 (триплет, J=4,5 Гц, 2Н); 3,88 (триплет, J=4,5 Гц, 2Н); 3,32 (триплет, J=6,0 Гц, 2Н); 2,43 (триплет, J=7,0 Гц, 2Н); 1,62 (мультиплет, 2Н);
MS(CI) для С32Н38N4O7 m/z 591 (МН+), 491, 391.
Часть С
Материал, полученный в части В, в атмосфере азота соединяют с 10 мл безводного дихлорметана и 10 мл трифторуксусной кислоты. Реакционную смесь перемешивают в течение 1,5 часов, после чего концентрируют при пониженном давлении до получения масла, которое после сушки при температуре окружающей среды в высоком вакууме дает твердый продукт. Этот продукт обрабатывают эфиром. Полученный при этом белый порошок сушат в течение ночи в вакуумной печи при 65°С и в результате получают 1,19 г бис(трифторацетата) 3-{3-[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]пропил}-бензойной кислоты. Температура плавления продукта 138-140°С.
Анализ. Рассчитано для С22Н22N4O3.(С2HF3O2)2: %С 50,49; %Н, 3,91; %N, 9,06. Найдено: %С 50,37; %Н, 3,67; %N, 9,08.
1H ЯМР(300 МГц, ДМСО-d6) δ 9,07-9,14 (широкий синглет, 2Н); 8,51 (синглет, 1Н); 8,37 (дублет, J=7,8 Гц, 1Н); 7.82 (дублет, J=8,0 Гц, 1Н); 7,74 (мультиплет, 2Н); 7,64 (синглет, 1Н); 7,56 (триплет, J=7,1 Гц, 1Н); 7,30 (триплет, J=7,7 Гц, 1Н); 7,15 (дублет, J=7,6 Гц, 1Н); 4,91 (триплет, J=4,5 Гц, 2Н); 3,86 (триплет, J=4,4 Гц, 2Н); 3,42 (триплет, J=5,9 Гц, 2Н); 2,44 (триплет, J=7,4 Гц, 2Н); 1,64 (мультиплет, 2Н);
ИК-спектр (в таблетке КВг): 3367, 3104, 2372, 1685, 1204, 1146 см-1.
HRMS(EI) Рассчитано для C22H22N4O3 (М+) 390,1692, найдено 390,1690.
Пример 13
Трифторацетат 2-{3-[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]пропил}бензойной кислоты
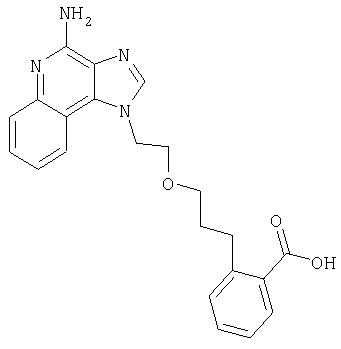
Часть А
Используя основной способ, применяемый в части А примера 12, в результате реакции между N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амином (2 г, 4,3 ммоля) и бензил-2-иодбензоатом (1,57 г, 4,71 ммоля) получают 1,79 г смеси моно- и ди-замещенных бензил-2-{3-[2-(4-амино-1H-имидазо[4,5-финолин-1-ил)этокси]проп-1-инил}бензоатов, содержащих в качестве заместителя защитные ВОС-группы.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,45 (дублет, J=7,9 Гц, 1Н); 8,39 (синглет, 1Н); 8,06-8,09 (мультиплет, 1Н); 7,85-7,88 (мультиплет, 1Н); 7,70-7,73 (мультиплет, 2Н); 7,47-7,51 (мультиплет, 2Н); 7,40-7,43 (мультиплет, 2Н); 7,28-7,37 (мультиплет, 3Н); 7,19 (мультиплет, 1Н); 5,23 (синглет, 2Н); 4,97 (триплет, J=5,0 Гц, 2Н); 4,27 (синглет, 2Н); 4,07 (триплет, J=4,9 Гц, 2Н); 1,30 (синглет, 18Н);
MS(CI) для С39Н40N4O7 m/z 677 (МН+), 577, 477.
Часть В
Используя основной способ, применяемый в части В примера 12, материал, полученный в части А этого примера, гидрируют и получают 0,041 г моно- и ди-замещенных 2-{3-[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]пропил}бензойной кислоты, содержащих в качестве заместителя защитные ВОС-группы.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,50 (дублет, J=7,3 Гц, 1Н); 8,39 (синглет, 1Н); 8,08 (дублет, J=7,9 Гц, 1Н); 7,71-7,75 (мультиплет, 3Н); 7,22-7,28 (мультиплет, 2Н); 6,90 (дублет, J=7,4 Гц, 1Н); 4,93 (триплет, J=4,6 Гц, 2Н); 3,87 (триплет, J=4,5 Гц, 2Н); 3,30 (триплет, J=5,6 Гц, 2Н); 2,73 (триплет, J=5,7 Гц, 2Н); 1,61 (мультиплет, 2Н); 1,28 (синглет, 18Н);
MS(CI) для C32H38N4O7 m/z 591 (МН+), 491, 391.
Часть С
Используя основной способ, применяемый в части С примера 12, материал, полученный в части В этого примера, подвергают гидролизу и получают 0,28 г 2-{3-[2-(4-амино-1H-имидазо[4,5-с]хинолин-1 -ил)этокси]пропил}бензойной кислоты. Температура плавления 186-188°С.
Анализ. Рассчитано для C22H22N4O3.C2HF3O2: %С 57,14; %Н, 4,59; %N, 11,11. Найдено: %С 56,81; %Н, 4,47; %N, 1108.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,90-9,20 (широкий синглет, 1Н); 8,50 (синглет, 1Н); 8,38 (дублет, J=10,1 Гц, 1Н); 7,84 (дублет, J=8,3 Гц, 1Н); 7,71-7,75 (мультиплет, 2Н); 7,56 (триплет, J=7,6 Гц, 1Н); 7,21-7,32 (мультиплет, 2Н); 6,88 (дублет, J=6,9 Гц, 2Н); 4,90 (триплет, J=4,8 Гц, 2Н); 3,84 (триплет, J=4,6 Гц, 2Н); 3,32 (мультиплет, 2Н); 2,72 (триплет, J=6,9 Гц, 2Н); 1,62 (мультиплет, 2Н);
ИК-спектр (в таблетке KBr): 3212, 2929, 1709, 1204, 1124, 747 см-1.
HRMS(EI) Рассчитано для С22Н22N4O3 (M+) 390,1692, найдено 390,1693.
Пример 14
Трифторацетат 4-{3-[2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)этокси]пропил}бензойной кислоты
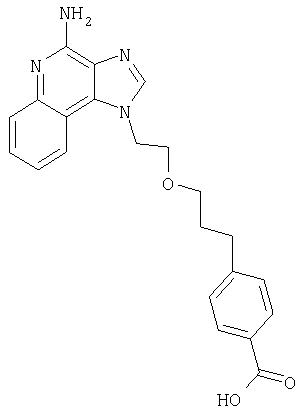
Часть А
Используя основной способ, применяемый в части А примера 12, в результате реакции между N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-финолин-4-амином (2,82 г, 6,04 ммоля) и бензил-4-иодбензоатом (2,25 г, 6,64 ммоля) получают 2,14 г смеси моно- и ди-защищенных бензил-4-{3-[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]проп-1-инил}бензоатов, содержащих в качестве заместителя защитные ВОС-группы.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,47 (дублет, J=7,2 Гц, 1Н); 8,40 (синглет, 1Н); 8,06 (дублет, J=6,5 Гц, 1Н); 7,87-7,89 (мультиплет, 1Н); 7,70-7,73 (мультиплет, 2Н); 7,36-7,49 (мультиплет, 5Н); 7,23-7,27 (мультиплет, 2Н); 5,35 (синглет, 2Н); 5,0 (триплет, J=4,5 Гц, 2Н); 4,40 (синглет, 2Н); 4,09 (триплет, J=4,5 Гц, 2Н); 1,30 (синглет, 18Н);
MS(CI) для С39Н40N4O7 m/z 677 (МН+), 577, 477.
Часть В
Используя основной способ, применяемый в части В примера 12, материал, полученный в части А этого примера, гидрируют и получают 1,86 г моно- и ди-замещенных 4-{3-[2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)этокси]пропил}бензойной кислоты, содержащих в качестве заместителя защитные ВОС-группы.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,51 (дублет, J=7,1 Гц, 1Н); 8,40 (синглет, 1Н); 8,07-8,10 (мультиплет, 1Н); 7,72-7,75 (мультиплет, 3Н); 7,01 (дублет, J=8,4 Гц, 1Н); 4,94 (триплет, J=4,7 Гц, 2Н); 3,88 (триплет, J=4,6 Гц, 2Н); 3,30 (мультиплет, 2Н); 2,38 (триплет, J=7,3 Гц, 2Н); 1,62 (мультиплет, 2Н); 1,29 (синглет, 18Н);
MS(CI) для C32H38N4O7 m/z 591 (MH+), 491, 391.
Часть С
Используя основной способ, применяемый в части С примера 12, материал, полученный в части В этого примера, подвергают гидролизу и получают 0,96 г трифторацетата 4-{3-[2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)этокси]пропил}бензойной кислоты. Температура плавления 235-237°С.
Анализ. Рассчитано для С22Н22N4O3.С2HF3O2: %С 57,14; %Н, 4,59; %N, 11,11. Найдено: %С 57,06; %Н, 4,47; %N, 11,03.
1H ЯМР(300 МГц, ДМСО-d6) δ 9,00-9,11 (широкий синглет, 1Н); 8,51 (синглет, 1Н); 8,37 (дублет, J=8,4 Гц, 1Н); 7,83 (дублет, J=6,0 Гц, 1Н); 7,71-7,76 (мультиплет, 3Н); 7,55 (триплет, J=9,7 Гц, 1Н); 7,01 (дублет, J=8,2 Гц, 2Н); 4,91 (триплет, J=5,0 Гц, 2Н); 3,84 (триплет, J=4,7 Гц, 2Н); 3,32 (триплет, J=5,8 Гц, 2Н); 2,38 (триплет, J=7,1 Гц, 2Н); 1,62 (мультиплет, 2Н);
ИК-спектр (в таблетке KBr): 3266, 3014, 2361, 1667, 1277, 1201, 1142 см-1.
HRMS(EI) Рассчитано для С22Н22N4O3 (M+) 390,1692, найдено 390,1697.
Пример 15
Дигидрохлорид 1-(2-{3-[3-(диметиламино)фенил]пропокси}этил)-1H-имидазо[4,5-с]хинолин-4-амина
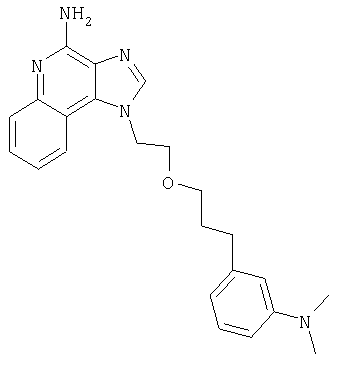
Часть А
Используя основной способ, применяемый в части А примера 12, только при температуре 80°С, в результате реакции между N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-амином (3 г, 6,43 ммоля) и 3-иод-N,N-диметиланилином (7,07 ммоль) получают 3,06 г смеси монозащищенного и незащищенного 1-[2-({3-[3-(диметиламино)фенил]проп-2-инил}окси)этил]-1H-имидазо[4,5-с]хинолин-4-амина.
Часть В
Используя основной способ, применяемый в части В примера 12, материал, полученный в части А этого примера, гидрируют и получают приблизительно 2,9 г смеси монозащищенного трет-бутоксикарбонильной группой и незащищенного 1-(2-{3-[3-(диметиламино)фенил]пропокси}этил]-1Н-имидазо[4,5-с]хинолин-4-амина.
Часть С
К материалу, полученному в части В, добавляют 30 мл 3 М раствора хлористого водорода в метаноле и смесь перемешивают при температуре окружающей среды в течение 19 часов. Образовавшийся осадок отфильтровывают. Фильтрат концентрируют при пониженном давлении, и остаток растворяют в небольшом количестве метанола, а затем нейтрализуют концентрированным раствором гидроксида аммония до достижения рН˜11. Полученный осадок очищают на хроматографической колонке, используя в качестве элюента смесь 95/5/1 дихлорметан/метанол/гидроксид аммония. Этот материал соединяют с системой хлористый водород/диэтиловый эфир. Полученный раствор концентрируют при пониженном давлении. Остаток обрабатывают диэтиловым эфиром. Образовавшееся твердое вещество отфильтровывают и после сушки получают 0,114 г дигидрохлорида 1-(2-{3-[3-(диметиламино)фенил]пропокси}этил)-1H-имидазо[4,5-с]хинолин-4-амина. Температура плавления этого продукта 180-183°С.
Анализ. Рассчитано для C23H27N5O.(HCl)2,1(H2O)2,1: %C 54,82; %Н, 6,66; %N, 13,89. Найдено: %C 54,60; %Н, 6,50; %N, 13,66.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,71-8,73 (широкий синглет, 2Н); 8,44 (синглет, 1Н); 8,35 (дублет, J=7,4 Гц, 1Н); 7,83 (дублет, J=8,0 Гц, 1Н); 7,72 (триплет, J=7,6 Гц, 1Н); 7,55 (триплет, J=6,8 Гц, 1Н); 7,15 (мультиплет, 1Н); 7,05 (мультиплет, 1Н); 6,96 (синглет, 1Н); 6,66 (дублет, J=8,1 Гц, 1Н); 4,88 (триплет, J=5,3 Гц, 2Н); 4,02 (триплет, J=3,7 Гц, 2Н); 3,37 (триплет, J=6,4 Гц, 2Н); 2,94 (синглет, 6Н); 2,40 (триплет, J=7,6 Гц, 2Н); 1,66 (мультиплет, 2Н);
ИК-спектр (в таблетке KBr): 3426, 3138, 2928, 1693, 1113 см-1.
HRMS(EI) Рассчитано для С23Н27N5O (М+) 389,2216, найдено 389,2217.
Пример 16
Солянокислый 2-(этоксиметил)-1-[2-(3-фенилпропокси)этил)-1H-имидазо[4,5-с]хинолин-4-амин
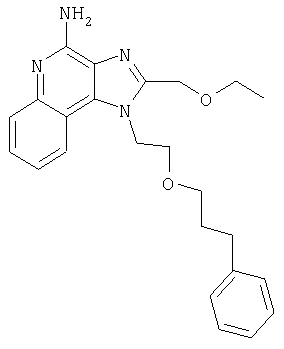
Часть А
3,50 г (12,9 ммоля) 2-[2-(Этоксиметил)-1H-имидазо[4,5-с]хинолин-1-ил]этанол медленно в течение 20 минут добавляют к суспензии 0,67 г (60% в минеральном масле, 16,77 ммоля) гидрида натрия в безводном N,N-диметилформамиде. Реакционную смесь перемешивают в течение 1 часа и затем добавляют 2,16 мл (14,19 ммоля) 1-бром-3-фенилпропана, после чего перемешивание продолжают в течение ночи. Реакционную смесь разбавляют этилацетатом, промывают водой и затем рассолом, сушат над сульфатом магния, отфильтровывают и концентрируют при пониженном давлении. Остаток очищают на хроматографической колонке, используя в качестве элюента этилацетат, и в результате получают 2,38 г 2-(этоксиметил)-1-[2-(3-фенилпропокси)этил]-1H-имидазо[4,5-с]хинолина в виде желтого масла.
MS(CI) для С24Н27N3O2 m/z 390 (МН+), 346.
Часть В
Материал, полученный в части А, соединяют с 50 мл хлороформа и охлаждают до 0°С. К охлажденной смеси добавляют 2,22 г 57-86%-ной 3-хлорпероксибензойной кислоты. Через 1 час реакционную смесь нагревают до температуры окружающей среды и распределяют между водным раствором бикарбоната натрия и дихлорметаном. Органическую фракцию сушат над сульфатом магния, фильтруют и затем концентрируют при пониженном давлении до получения коричневого твердого продукта, представляющего собой 2-(этоксиметил)-1-[2-(3-фенилпропокси)этил]-1H-имидазо[4,5-с]хинолин-5N-оксид.
Часть С
К смеси материала, полученного в части В, и 60 мл безводного дихлорметана в атмосфере азота добавляют 0,87 мл (7,33 ммоля) трихлорацетилизоцианата. Через 1 час реакционную смесь концентрируют при пониженном давлении и получают 2,2,2-трихлор-N-{2-(этоксиметил)-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]хинолин-1 -ил}ацетамид.
Часть D
К смеси продукта, полученного в части С, и 30 мл метанола добавляют 4,79 мл 25%-ного раствора метилата натрия в метаноле. Реакционную смесь перемешивают в течение ночи и затем концентрируют при пониженном давлении до получения коричневого масла. Это масло очищают на хроматографической колонке, используя в качестве элюента дихлорметан, содержащий 5% метанола, и получают светло-желтое масло. При обработке этого масла 1,0 М раствором соляной кислоты получают белое твердое вещество, которое отфильтровывают и сушат в течение ночи в вакуумной печи при 80°С. В результате получают 0,79 г белого твердого солянокислого 2-(этоксиметил)-1-[2-(3-фенилпропокси}этил)-1H-имидазо[4,5-с]хинолин-4-амина. Температура плавления этого продукта 128-134°С.
Анализ. Рассчитано для С24Н28N4O2.1,55 HCl: %С 62,53; %Н, 6,46; %N, 12,15. Найдено: %С 62,64; %Н, 6,47; %N, 11,91.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,14 (широкий дублет, J=8,3 Гц, 1Н); 7,63 (двойной дублет, J=8,3, 1,0 Гц, 1Н); 7,45 (мультиплет, 1Н); 7,24 (мультиплет, 1Н); 7,05-7,15 (мультиплет, 3Н); 6,90 (мультиплет, 2Н); 6,62 (синглет, 2Н); 4,80-4,90 (мультиплет, 4Н); 3,83 (триплет, J=5,4 Гц, 2Н); 3,56 (квартет, J=7,0 Гц, 2Н); 3,27 (триплет, J=6,1 Гц, 2Н); 2,37 (триплет, J=7,6 Гц, 2Н); 1,63 (мультиплет, 2Н); 1,16 (триплет, J=6,8 Гц, 3Н)
ИК-спектр (в таблетке KBr): 3267, 3023, 1681, 1108 см-1.
HRMS(EI) Рассчитано для C24H28N4O2 (M+) 404,2212, найдено 404,2215.
Пример 17
1-(1-{[(3-Хлорбензил)окси]метил}пропил)-1Н-имидазо[4,5-с]хинолин-4-амин
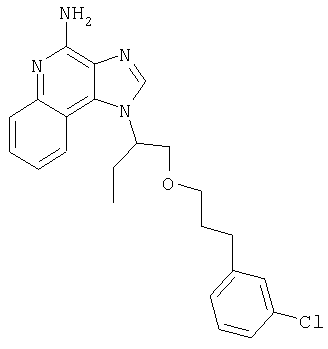
Часть А
3,0 г (12,43 ммоля) 2-этил-2-(1Н-имидазо[4,5-с]хинолин-1-ил)этанола добавляют к смеси 40 мл дихлорметана, 40 мл 50%-ного водного раствора гидроксида натрия, 0,01 г бензилтриметиламмонийхлорида и 2,81 г (13,67 ммоля) 3-хлорбензилбромида. Полученный раствор перемешивают при температуре окружающей среды в течение ночи. После этого согласно данным анализа способом тонкослойной хроматографии (5% метанола в дихлорметане) реакция полностью завершается. После этого реакционную смесь разбавляют 100 мл дихлорметана и 100 мл воды. Разделяют органический и водный слои. Водную фракцию экстрагируют дихлорметаном. Объединенные органические фракции промывают рассолом, сушат над сульфатом магния и концентрируют при пониженном давлении. Остаток подвергают очистке способом флеш-хроматографии (силикагель, элюент - этилацетат) и получают 4,22 г светло-оранжевого маслообразного 1-(1-{[(3-хлорбензил)окси]метил}пропил)-1Н-имидазо[4,5-с]хинолина.
1H ЯМР(300 МГц, ДМСО-d6) δ 9,22 (синглет, 1Н), 8,63 (синглет, 1Н), 8,55 (дублет, J=7,8 Гц, 1Н); 8,17 (двойной дублет, J=7,8, 1,5 Гц, 1Н); 7,69 (мультиплет, 2Н); 7,23 (двойной дублет, J=4,9, 1,5 Гц, 2Н); 7,08 (синглет, 1Н), 7,03 (мультиплет, 1Н), 5,40 (мультиплет, 1Н), 4,47 (синглет, 2Н); 3,34-4,07 (мультиплет, 2Н), 2,11 (мультиплет, 2Н); 0,88 (триплет, J=7,3 Гц, 3Н);
MS(CI) для С21Н20CIN3О m/z 366 (MH+), 322.
Часть В
К раствору материала, полученного в части А, в 60 мл хлороформа порциями добавляют 2,84 г 77%-ной хлорпероксибензойной кислоты. Согласно данным анализа способом тонкослойной хроматографии (10% метанола в дихлорметане) реакция полностью завершается через 2 часа. Реакционную смесь разбавляют хлороформом, промывают насыщенным водным раствором бикарбоната натрия и затем рассолом, сушат над сульфатом магния и затем концентрируют при пониженном давлении до получения 1-(1-{[(3-хлорбензил)окси]метил}пропил)-1Н-имидазо[4,5-с]хинолин-5N-оксида.
Часть С
К раствору материала, полученного в части В, в 80 мл дихлорметана добавляют 20 мл гидроксида аммония, а затем порциями 2,42 г хлористого тозила. Согласно данным анализа способом тонкослойной хроматографии (5% метанола в дихлорметане) реакция полностью завершается сразу же после добавления хлористого тозила. Реакционную смесь разбавляют дихлорметаном и насыщенным водным раствором бикарбоната натрия. Разделяют органический и водный слои. Органический слой промывают рассолом, сушат над сульфатом магния и концентрируют при пониженном давлении до получения светло-коричневого масла. Это масло подвергают очистке способом флеш-хроматографии (силикагель, элюент - дихлорметан, содержащий 5% метанола) и получают клейкий беловатый твердый продукт. Очистка этого продукта способом флеш-хроматографии (силикагель, элюент - дихлорметан, содержащий 5% метанола) дает розовато-белый твердый продукт. Этот продукт вновь подвергают очистке способом флеш-хроматографии (силикагель, элюент - этилацетат) и в результате получают около 1,0 г твердого беловатого 1-(1-[[(3-хлорбензил)окси]метил}пропил)-1H-имидазо[4,5-с]хинолин-4-амина, температура плавления которого составляет 60-62°С. Анализ.
Рассчитано для C21H21ClN4O.% Н2O: %С 65,41; %Н, 5,62; %N, 14,54. Найдено: %С 65,5; %Н, 5,62; %N, 14,61.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,37 (синглет, 1Н), 8,19 (дублет, J=8,3 Гц, 1Н); 7,62 (двойной дублет, J=8,3, 1,5 Гц, 1Н); 7,43 (двойной триплет, J=8,3, 1,5 Гц, 1Н); 7,18-7,28 (мультиплет, 3Н); 7,09 (мультиплет, 1Н); 6,52 (широкий синглет, 2Н); 5,24 (мультиплет, 1Н); 4,48 (синглет, 2Н), 4,01 (двойной дублет, J=10,5, 6,6 Гц, 2Н), 3,92 (двойной дублет, J=10,3, 4,4 Гц, 2Н), 2,10 (квартет, J=7,3 Гц, 2Н), 0,88 (триплет, J=7,3 Гц, 3Н)
MS(CI) для C21H21ClN4O m/z 381 (MH+), 185.
Пример 18
Трифторацетат 1-{2-[3-(2-аминофенил)пропокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина
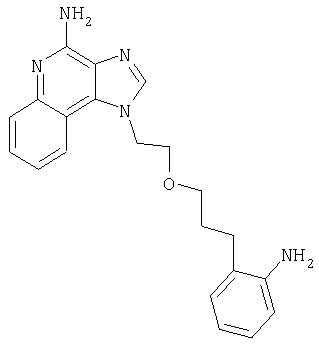
Часть А
0,50 г (1,07 ммоля) N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина, 0,39 мл (2,79 ммоля) триэтиламина и 10 мл безводного ацетонитрила смешивают в атмосфере азота. Полученный раствор нагревают до 80°С и добавляют к нему 0,26 мл (1,18 ммоля) 2-иоданилина, 0,012 г иодида меди (I) и 0,023 гдихлорбис(трифенилфосфин)палладия (II). Смесь выдерживают при 80°С в течение ночи. Ацетонитрил отгоняют при пониженном давлении, остаток очищают способом флеш-хроматографии (силикагель, элюент - дихлорметан, содержащий 3% метанола) и в результате получают 0,47 г твердого коричневого N,N-(бис-трет-бутоксикарбонил)-1-[2-{[3-(2-аминофенил)проп-2-инил]окси}этил)-1H-имидазо[4,5-с]хинолин-4-амина.
1H ЯМР(300 МГц, ДМСО-d6, D2O) δ 8,47 (дублет, J=3,6 Гц, 1Н); 8,37 (синглет, 1Н); 8,10 (дублет, J=9,6 Гц, 1Н); 7,75 (мультиплет, 2Н); 7,04 (триплет, J=7,2 Гц, 1Н); 6,80 (мультиплет, 1Н), 6,65 (дублет, J=8,3 Гц, 1Н); 6,45 (триплет, J=7,3 Гц, 1Н); 4,98 (триплет, J=4,4 Гц, 2Н); 4,36 (синглет, 2Н), 4,08 (триплет, J=4,9 Гц, 2Н); 1,31 (синглет, 18Н).
Часть В
К раствору N,N-(бис-трет-бутоксикарбонил)-1-[2-{[3-(2-аминофенил)проп-2-инил]-окси}этил)-1H-имидазо[4,5-с]хинолин-4-амина в метаноле добавляют катализатор (активированный уголь с нанесенной на него 5% платины). Смесь подвергают гидрированию в аппарате Парра при давлении водорода 50 фунт/дюйм2 (3,5 кг/см2) в течение ночи. После этого реакционную смесь фильтруют через слой Celite® и остаток на фильтре промывают дополнительным количеством метанола. Фильтрат концентрируют при пониженном давлении до получения беловатого твердого продукта. Этот материал очищают способом флеш-хроматографии на силикагеле, используя в качестве элюента сначала дихлорметан, затем смесь 99/1 дихлорметан/метанол, затем смесь этих же растворителей состава 98/2 и наконец - смесь 97/3 дихлорметан/метанол. В результате получают около 0,25 г светло-желтого маслообразного N,N-(бис-трет-бутоксикарбонил)-1-{2-13-(2-аминофенил)-пропокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,23 (двойной дублет, J=8,4, 0,9 Гц, 1Н), 8,16 (двойной дублет, J=8,4, 0,9 Гц, 1Н), 7,97 (синглет, 1Н), 6,96 (двойной триплет, J=7,5, 1,6 Гц, 2Н), 6,87 (двойной дублет, J=7,5, 1,4 Гц, 1Н), 6,62 (двойной триплет, J=7,3, 1,0 Гц, 1Н), 6,57 (двойной дублет, J=8,3, 1,1 Гц, 1Н), 5,29 (синглет, 1Н), 4,71 (триплет, J=5,3 Гц, 2Н); 3,91 (триплет, J=5,1 Гц, 2Н), 3,38 (триплет, J=6,0 Гц, 2Н), 2,39 (триплет, J=7,4 Гц, 2Н), 1,76 (мультиплет, 2Н), 1,41 (широкий синглет, 18Н).
MS(CI) для С31Н39N5O5 m/z 562 (MH+), 462, 362, 229.
Часть С
Раствор материала, полученного в части В, в 4 мл дихлорметана при перемешивании добавляют к охлажденному до 0°С раствору 2 мл трифторуксусной кислоты в 2 мл безводного дихлорметана. Реакционную смесь выдерживают в течение 2 часов на ледяной бане, а затем нагревают до температуры окружающей среды и выдерживают при этой температуре в течение ночи. После отгонки летучих продуктов из реакционной смеси остается розовое масло. Это масло растворяют в 3 мл этилацетата и к раствору по каплям добавляют около 1 мл триэтиламина, после чего смесь перемешивают в течение 1 часа. Полученный осадок отфильтровывают и получают 0,13 г белого твердого трифторацетата 1-{2-[3-(2-аминофенил)пропокси]этил}-1H-имидазо[4,5-с]хинолин-4-амина.
Анализ. Рассчитано для С21H23N5O.С2HF3O2: %С 58,10; %Н, 5,09; %N, 14,73. Найдено: %С 57,78; %Н, 4,97; %N, 14,59.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,87 (широкий синглет, 1Н), 8,49 (синглет, 1Н), 8,36 (дублет, J=7,8 Гц, 1Н); 7,83 (дублет, J=8,3 Гц, 1Н); 7,72 (триплет, J=7,3 Гц, 1Н), 7,56 (триплет, J=7,6 Гц, 1Н), 6,81 (триплет, J=7,6 Гц, 1Н), 6,51 (мультиплет, 2Н); 6,32 (триплет, J=6,8 Гц, 1Н), 4,90 (триплет, J=4,6 Гц, 2Н), 3,85 (триплет, J=4,9 Гц, 2Н), 3,33 (триплет, J=6,1 Гц, 2Н), 2,22 (триплет, J=736 Гц, 2Н), 1,55 (мультиплет, 2Н)
ИК-спектр (в таблетке KBr): 3414, 3335, 3253, 3019,1738, 1202, 1185, 1131 см-1.
HRMS(EI) Рассчитано для C21H23N5O (M+)361,1903, найдено 361,1903.
Пример 19
4-{[2-(4-Амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]метил}бензонитрил
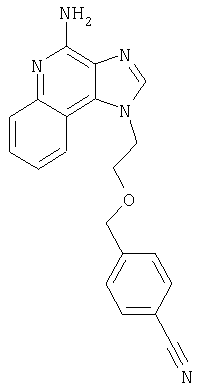
Часть А
1,5 г (7,0 ммоля) 2-(1Н-имидазо[4,5-с]хинолин-1-ил)этанола при перемешивании добавляют к смеси 1,79 г (9,1 ммоля) α-бром-п-толуилнитрила, 20 мл 50%-ного раствора гидроксида натрия, 20 мл дихлорметана и 0,06 г (0,3 ммоля) бензилтриметиламмонийхлорида. Реакцию продолжают в течение 18 часов и затем реакционную смесь разбавляют, добавляя к ней 20 мл дихлорметана и 20 мл воды. Разделяют органический и водный слои. Водную фракцию экстрагируют дополнительным количеством дихлорметана. Органические фракции соединяют вместе, промывают водой, сушат над сульфатом магния и концентрируют. Остаток подвергают очистке способом флеш-хроматографии (силикагель, элюент - смесь 9/1 дихлорметан/метанол) и получают 1,8 г 4-{[2-(1H-имидазо[4,5-с]хинолин-1-ил)этокси]метил}бензонитрила.
1H ЯМР(500 МГц, ДМСО-d6) δ 9,22 (синглет, 1Н), 8,41 (синглет, 1Н), 8,40 (дублет, J=1,1 Гц, 1Н); 8,17 (двойной дублет, J=7,6, 1,3 Гц, 1Н); 7,66 (двойной триплет, J=7,6, 1,3 Гц, 1Н), 7,56 (триплет, J=7,6 Гц, 1Н), 6,81 (триплет, J=7,6 Гц, 1Н), 6,51 (мультиплет, 2Н); 7,63 (дублет, J=8,3 Гц, 2Н), 7,25 (дублет, J=8,2 Гц, 2Н), 4,97 (триплет, J=5,1 Гц, 2Н), 4,53 (синглет, 2Н), 3,97 (триплет, J=5,5 Гц, 2Н);
MS(CI) m/z 329 (M+Н).
Часть В
1,6 г (5,5 ммоля) 60%-ной 3-хлорпероксибензойной кислоты медленно добавляют к раствору 1,8 г (5,5 ммоля) 4-{[2-(1H-имидазо[4,5-с]хинолин-1-ил)этокси-метил}-бензонитрила в 50 мл хлороформа. Реакционную смесь выдерживают в течение ночи (около 16 часов), после чего промывают последовательно 200 мл насыщенного раствора бикарбоната натрия и двумя порциями воды по 100 мл каждая, сушат над сульфатом магния, фильтруют и концентрируют до получения 1,4 г 1-{[2-[(4-цианобензил)окси]этил}-1Н-имидазо[4,5-с]хинолин-5N-оксида.
Часть С
К раствору 1,4 г (4,1 ммоля) 1-{[2-[(4-цианобензил)окси]этил}-1Н-имидазо[4,5-с]хинолин-5N-оксида в 25 мл дихлорметана по каплям добавляют 0,73 мл (6,1 ммоля) трихлорацетилизоцианата. Реакционную смесь выдерживают в течение ночи и затем концентрируют. Образующееся твердое вещество красного цвета растворяют в 100 мл метанола, к раствору по каплям добавляют 4 мл 25%-ного раствора метилата натрия в метаноле и снова выдерживают реакционную смесь в течение ночи. Образовавшийся твердый осадок отфильтровывают. В результате перекристаллизации этого продукта из изопропилового спирта и последующей флеш-хроматографической очистки (силикагель, 9/1 дихлорметан/метанол) получают 1,0 г белого твердого 4-{[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]метил}бензонитрила, температура плавления которого составляет 238,1-239,2°С.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,19 (синглет, 1Н), 8,07 (двойной дублет, J=8,2, 1,0 Гц, 1Н); 7,67 (дублет, J=8,4 Гц, 2Н), 7,62 (двойной дублет, J=8,4, 1,1 Гц, 1Н), 7,43 (двойной триплет, J=7,6, 1,3 Гц, 1Н), 7,30 (дублет, J=8,4 Гц, 2Н), 7,21 (двойной триплет, J=7,6, 1,3 Гц, 1Н), 6,56 (синглет, 2Н), 4,86 (триплет, J=5,1 Гц, 2Н), 4,55 (синглет, 2Н), 3,93 (триплет, J=5,1 Гц, 2Н);
ИК-спектр (в таблетке KBr): 3456, 3285, 3117, 3069, 2228, 1637, 1583, 1526, 1481, 1397, 1372, 1353, 1252, 1097, 884, 822, 760 см-1.
MS (El) m/e 343.1440 (343.1433. Рассчитано для C20H17N5O);
Анализ. Рассчитано для C20H17N5O: %С 69,96; %Н, 4,99; %N, 20,39. Найдено: %С 70,09; %Н, 4,90; %N, 20,16.
Пример 20
2-(Этоксиметил)-1-(2-{[6-(4-фенилбутокси)гексил]окси}этил)-1H-имидазо[4,5-c]хинолин-4-амин
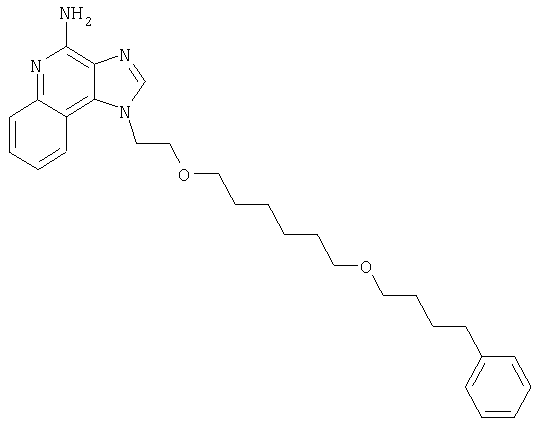
Часть А
Раствор 1,0 г (3,7 ммоля) 2-[2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил]этанола в 20 мл N,N-диметилформамида добавляют по каплям к суспензии гидрида натрия (0,19 г 60%-ной суспензии в минеральном масле, 4,8 ммоля) в 10 мл N,N-диметилформамида. Смесь выдерживают в течение 45 минут и затем по каплям добавляют к ней 1,6 г (5,1 ммоля) {4-[(6-бромгексил)окси]бутил}бензола. Реакционную смесь перемешивают при температуре окружающей среды в течение ночи, после чего распределяют ее между этилацетатом и водой. Разделяют органический и водный слои. Водную фракцию экстрагируют дополнительным количеством этилацетата. Органические фракции соединяют вместе, промывают водой, сушат над сульфатом магния и концентрируют. Остаток подвергают очистке способом флеш-хроматографии (силикагель, элюент - смесь 4/1 этилацетат/гексаны) и получают 0,81 г 2-(этоксиметил)-1-(2-{[6-(4-фенилбутокси)гексил]окси}этил)-1H-имидазо-[4,5-с]-хинолина в виде коричневого масла.
Часть В
0,47 г (1,6 ммоля) 60%-ной 3-хлорпероксибензойной кислоты медленно добавляют к раствору 0,81 г (1,6 ммоля) 2-(этоксиметил)-1-(2-{[6-(4-фенилбутокси)гексил]окси}этил)-1Н-имидазо[4,5-с]-хинолина в 15 мл хлороформа. Реакционную смесь выдерживают в течение ночи (около 16 часов), после чего промывают последовательно насыщенным раствором бикарбоната натрия и водой, сушат над сульфатом магния, фильтруют и концентрируют до получения 0,7 г 2-(этоксиметил)-1-(2-{[6-(4-фенилбутокси)-гексил]окси}этил)-1H-имидазо[4,5-с]-хинолина-5N-оксида в виде твердого вещества оранжевого цвета.
Часть С
К раствору 0,7 г (1,4 ммоля) 2-(этоксиметил)-1-(2-{[6-(4-фенилбутокси)-гексил]окси}этил)-1Н-имидазо[4,5-с]-хинолина-5N-оксида в 20 мл дихлорметана по каплям добавляют 0,25 мл (2,1 ммоля) трихлорацетилизоцианата. Реакционную смесь выдерживают в течение 2 часов и затем по каплям добавляют к ней 2,5 мл 25%-ного раствора метилата натрия в метаноле, после чего смесь выдерживают еще в течение ночи. Затем смесь подвергают фильтрации, и фильтрат концентрируют. Флеш-хроматографическая очистка фильтрата (силикагель, 97/3 этилацетат/метанол) дает 0,22 г бесцветного маслообразного 2-(этоксиметил)-1-(2-{[6-(4-фенилбутокси)гексил]окси}этил)-1Н-имидазо[4,5-с]-хинолин-4-амина.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,10 (дублет, J=7,9 Гц, 1Н); 7,62 (дублет, J=7,9 Гц, 1Н), 7,43 (триплет, J=7,3 Гц, 1Н), 7,28-7,12 (мультиплет, 6Н), 6,55 (синглет, 2Н), 4,79 (широкий синглет, 4Н), 3,82 (триплет, J=5,3 Гц, 2Н), 3,55 (квартет, J=7,0 Гц, 2Н), 3,33-3,22 (мультиплет, 6Н), 2,56 (триплет, J=7,2 Гц, 2Н), 1,62-1,33 (мультиплет, 8Н), 1,18-1,10 (мультиплет, 7Н);
MS (El) m/e 518.3263 (518.3256 Рассчитано для С31Н42N4O3);
Анализ. Рассчитано для С31Н42N4O3: %С 71,78; %Н, 8,16; %N, 10,80. Найдено: %С 71,20; %Н, 8,39; %N, 10,68.
Пример 21
1-{2-[3-(Бензилокси)пропокси]этил}-2-(этоксиметил)-1H-имидазо[4,5-с]хинолин-4-амин
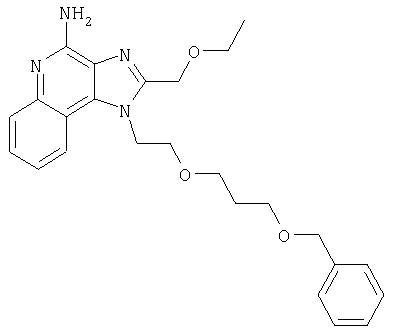
Часть А
Раствор 1,0 г (3,7 ммоля) 2-[2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил]этанола в N,N-диметилформамиде добавляют по каплям к суспензии гидрида натрия (0,19 г 60%-ной суспензии в минеральном масле, 4,8 ммоля) в 20 мл N,N-диметилформамида. Смесь выдерживают в течение 2 часов и затем по каплям добавляют к ней 0,72 мл (4,1 ммоля) бензил-3-бромпропилового эфира. Реакционную смесь перемешивают при 100°С в течение ночи, после чего выливают ее в ледяную воду и экстрагируют этилацетатом. Разделяют органический и водный слои. Органическую фракцию промывают водой, сушат над сульфатом магния и концентрируют. Остаток подвергают очистке способом флеш-хроматографии (силикагель, элюент - смесь 4/1 этилацетат/гексаны) и получают 0,45 г 1-{2-[3-(бензилокси)пропокси]этил}-2-(этоксиметил)-1H-имидазо[4,5-с]хинолина в виде коричневого масла.
Используя основные способы, применяемые в частях В и С примера 20, 1-{2-[3-(бензилокси)пропокси]этил}-2-(этоксиметил)-1H-имидазо[4,5-с]хинолин превращают в 1-{2-[3-(бензилокси)пропокси]этил}-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин. Очистка образовавшегося вещества способом флеш-хроматографии (силикагель, 95/5 этилацетат/метанол) позволяет получить целевой продукт в виде бесцветного масла.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,11 (двойной дублет, J=8,2, 0,8 Гц, 1Н); 7,62 (двойной дублет, J=8,3, 1,2 Гц, 1Н), 7,44 (двойной триплет, J=7,6, 1,3 Гц, 1Н), 7,30 (дублет, J=8,4 Гц, 2Н), 7,21 (двойной триплет, J=7,6, 1,2 Гц, 1Н), 7,32-7,19 (мультиплет, 6Н), 6,56 (синглет, 2Н), 4,85-4,77 (мультиплет, 4Н), 4,26 (синглет, 2Н), 3,84 (триплет, J=5,4 Гц, 2Н), 3,54 (квартет, J=7,0 Гц, 2Н), 3,40 (триплет, J=6,2 Гц, 2Н), 3,26 (триплет, J=6,2 Гц, 2Н), 1,63 (квинтет, J=6,3 Гц, 2Н), 1,15 (триплет, J=7,0 Гц, 3Н);
13С ЯМР (125 МГц, ДМСО-d6) δ 152,0, 149,5, 145,2, 138,5, 133,3, 128,1, 127,4, 127,3, 126,8, 126,3, 126,24, 121,0, 120,6, 114,8, 71,8, 69,0, 67,5, 66,3, 65,4, 64,4, 45,4, 29,4, 14,9;
ИК-спектр (в таблетке KBr): 3305, 3174, 2970, 2925, 2864, 1633, 1583, 1533, 1481, 1437, 1386, 1099, 754, 737, 698 см-1.
MS (El) m/e 434.2318 (434.2317 Рассчитано для С25H30N4O3).
Пример 22
1-[2-(3-(Фенилпропокси]этил-1Н-имидазо[4,5-с]хинолин-4-амин
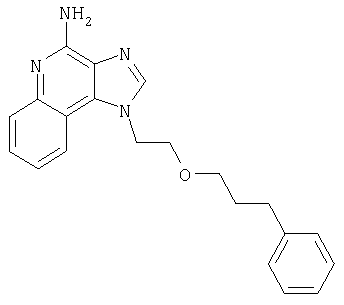
Часть А
В соответствии с основным способом, применяемым в примере 20 (части А-С), при взаимодействии 2-(1H-имидазо[4,5-с]хинолин-1-ил)этанола с (3-бромпропил)бензолом получают 1-[2-(3-(фенилпропокси]этил-1H-имидазо[4,5-с]хинолин-4-амин в виде белого твердого вещества.
1H ЯМР(300 МГц, ДМСО-d6) δ 8.17 (синглет, 1Н), 8,12 (дублет, J=7,2 Гц, 1Н); 7,64 (двойной дублет, J=8,3, 1,0 Гц, 1Н), 7,45 (мультиплет,1Н), 7,24 (мультиплет, 1Н), 7,16-7,08 (мультиплет, 3Н), 6,92-6,89 (мультиплет, 2Н), 6,60 (синглет, 2Н), 4,81 (триплет, J=5,1 Гц, 2Н), 3,82 (триплет, J=5,1 Гц, 2Н), 3,29 (триплет, J=6,1 Гц, 2Н), 2,38 (мультиплет, 2Н), 1,63 (мультиплет, 2Н), 1,56-1,25 (мультиплет, 8Н), 0,88 (триплет, J=7,2 Гц, 3Н);
13С ЯМР (75 МГц.CDCl3) δ 151,5, 144,9, 142,6, 141,4, 132,6, 128,3, 128,2, 127,4, 127,1, 125,8, 122,2, 119,8, 115,4, 70,4, 68,6, 47,6, 32,0, 30,9;
MS (El) m/e 347.1882 (347.18 27 Рассчитано для C21H22N4O).
Пример 23
1-(2-{[3-(3,4-Диметилфенил)-2-пропинил]окси}этил-1Н-имидазо[4,5-с]хинолин-4-амин
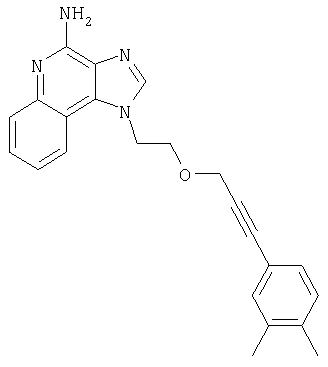
В атмосфере азота 0,5 г (1,9 ммоля) 1-[2-(2-пропинилокси]этил-1H-имидазо[4,5-с]-хинолин-4-амина добавляют к 0,036 г (0,2 ммоля) иодида меди (I), 0,5 г (2,1 ммоля) 4-иод-орто-ксилола и 10 мл пирролидина и смесь перемешивают при температуре окружающей среды. Добавляют 0,066 г (0,1 ммоля) дихлорбис(трифенилфосфин)палладия (II) и перемешивание продолжают при температуре окружающей среды в течение 1 часа. Проведенный после этого анализ способом тонкослойной хроматографии (для проявления используют 30%-ный раствор метанола в хлорформе) показывает наличие в реакционной смеси исходного материала. Поэтому реакционную смесь в течение ночи нагревают при 65°С. Пирролидин отгоняют при пониженном давлении, и остаток обрабатывают дихлорметаном, содержащим метанол. Нерастворимый продукт отфильтровывают и перекристаллизовывают из толуола (40 мл), в результате чего получают 0,1 г твердого 1-(2-{[3-(3,4-диметилфенил)-2-пропинил]окси}этил-1H-имидазо[4,5-с]хинолин-4-амина. Температура плавления этого продукта составляет 214-216°С.
Анализ. Рассчитано для С23Н22N4O: %С 74,57; %Н, 5,99; %N, 15,12. Найдено: %С 74,24; %Н, 5,98; %N, 15,08.
1H ЯМР(300 МГц, ДМСО-d6) δ (м.д.) 8,167 (синглет, 1Н), 8,49 (синглет, 1Н), 8,112 (дублет, J=7,3 Гц, 1Н); 7,628 (дублет, J=8,3 Гц, 1Н); 7,44 (триплет, J=7,3 Гц, 1Н), 7,232 (триплет, J=6,8 Гц, 1Н), 7,078 (дублет, J=7,8 Гц, 1Н), 7,024 (синглет, 1Н); 6,952 (дублет, J=7,9 Гц, 1Н), 6,586 (синглет, 2Н), 4,849 (триплет, J=5 Гц, 2Н), 4,365 (синглет, 2Н), 4,015 (триплет, J=5,6 Гц, 2Н), 2,197 (синглет, 3Н), 2,159 (синглет, 3Н).
Примеры 24-27
Соединения, показанные в таблице 1, получают в соответствии со схемой 1 процесса, используя следующий основной способ синтеза.
25 мг 2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)-2-этилэтанола помещают в ампулу объемом 7,4 мл и добавляют туда 1,75 эквивалента гидрида натрия (в виде 60%-ной суспензии в минеральном масле) и 1 мл N,N-дииметилформамида. Ампулу помещают на 10 мин в ультразвуковое перемешивающее устройство, и за это время при температуре окружающей среды образуется алкоксид. После этого к реакционной смеси добавляют 1,75 эквивалента галогенида и перемешивание осуществляют в течение 30-60 мин при температуре окружающей среды. Проводят масс-спектрометрический и хроматографический анализ реакционной смеси, чтобы установить образование целевого продукта. Реакционную смесь подвергают очистке способом полупрепаративной высокой эффективной жидкостной хроматографии. Полученные при этом фракции анализируют масс-спектрометрическим способом LC-APCI/MS. Соответствующие фракции соединяют вместе и подвергают лиофилизации для получения трифторуксуснокислой соли целевого продукта, структуру и молекулярную массу которого подтверждают с помощью масс-спектрометрии и ПМР-спектроскопии. В таблице 1 показана структура свободного основания, а также теоретическое (ТМ) и экспериментально определенное (ММ) значение молекулярной массы продукта.
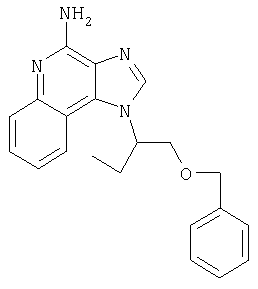
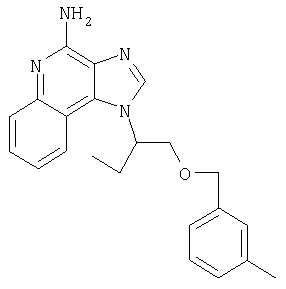
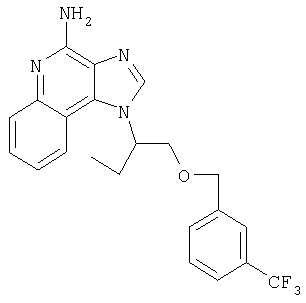
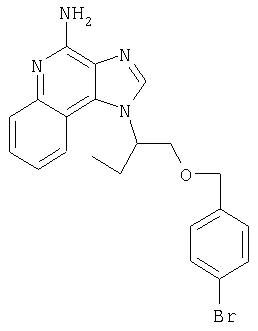
Примеры 28-41
Соединения, показанные в таблице 2, получают в соответствии со схемой I процесса, используя следующий основной способ синтеза.
25 мг 4-амино-1Н-имидазо[4,5-с]хинолин-1-илового спирта помещают в ампулу объемом 7,4 мл и добавляют туда 1,2 эквивалента гидрида натрия (в виде 60%-ной суспензии в минеральном масле) и 1 мл N,N-дииметилформамида. Ампулу помещают на 1 час в ультразвуковое перемешивающее устройство, и за это время при 50°С образуется алкоксид. После этого к реакционной смеси добавляют 1,2 эквивалента галогенида и перемешивание осуществляют в течение 1-2 часов при 50°С. Проводят масс-спектрометрический и хроматографический анализ реакционной смеси, чтобы установить образование целевого продукта. Реакционную смесь подвергают очистке способом полупрепаративной высокой эффективной жидкостной хроматографии. Полученные при этом фракции анализируют масс-спектрометрическим способом LC-APCI/MS. Соответствующие фракции соединяют вместе и подвергают лиофилизации для получения трифторуксуснокислой соли целевого продукта, структуру и молекулярную массу которого подтверждают с помощью масс-спектрометрии и ПМР-спектроскопии. В таблице 2 показана структура свободного основания, а также теоретическое (ТМ) и экспериментально определенное (ММ) значение молекулярной массы продукта.
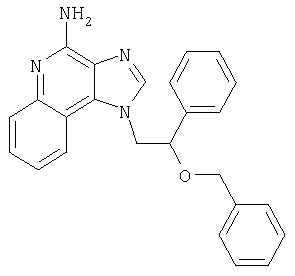
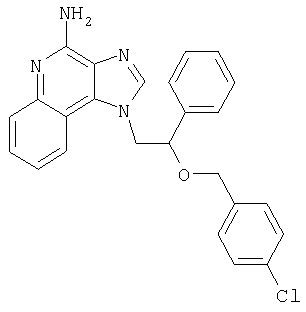
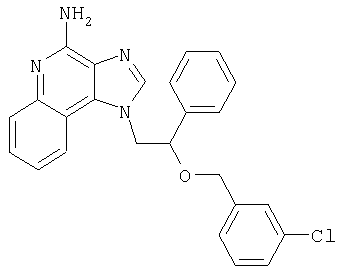
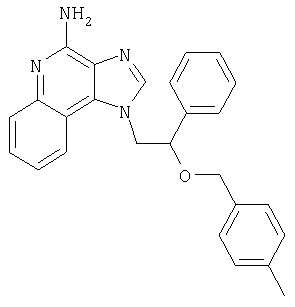
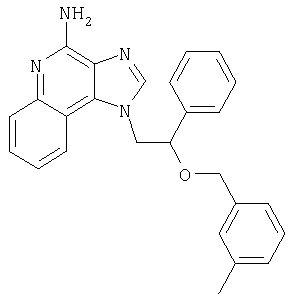
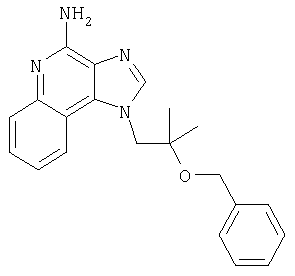
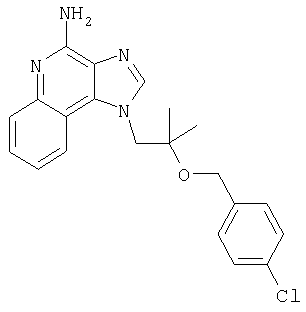

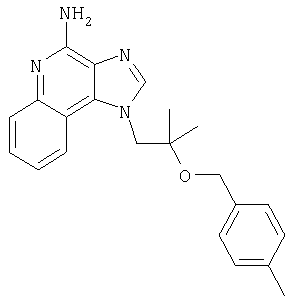

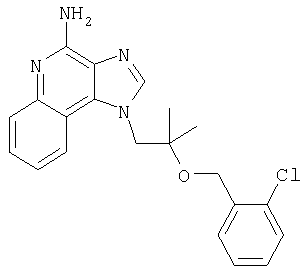
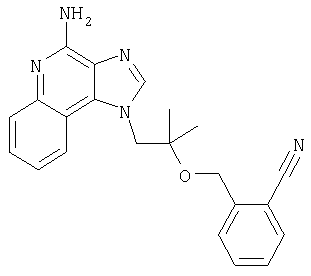
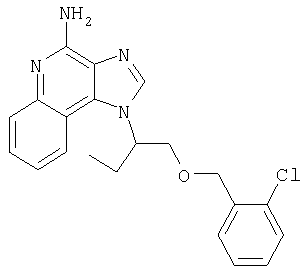
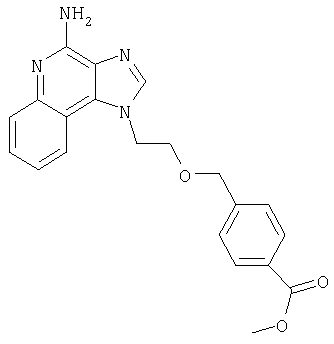
Примеры 42-88
Соединения, показанные в таблице 3, получают в соответствии со схемой 1 процесса, используя следующий основной способ синтеза.
25 мг 4-амино-1H-имидазо[4,5-с]хинолин-1-илового спирта помещают в ампулу объемом 7,4 мл и добавляют туда 1,2 эквивалента гидрида натрия (в виде 60%-ной суспензии в минеральном масле) и 1 мл N,N-дииметилформамида. Ампулу помещают на 15-30 минут в ультразвуковое перемешивающее устройство, и за это время при температуре окружающей среды образуется алкоксид. После этого к реакционной смеси добавляют 1,2 эквивалента галогенида и перемешивание осуществляют дополнительно в течение 15-120 минут при температуре окружающей среды. Проводят масс-спектрометрический и хроматографический анализ реакционной смеси, чтобы установить образование целевого продукта. Реакционную смесь подвергают очистке способом полупрепаративной высокой эффективной жидкостной хроматографии. Полученные при этом фракции анализируют масс-спектрометрическим способом LC-APCI/MS. Соответствующие фракции соединяют вместе и подвергают лиофилизации для получения трифторуксуснокислой соли целевого продукта, структуру и молекулярную массу которого подтверждают с помощью масс-спектрометрии и ПМР-спектроскопии. В таблице 3 показана структура свободного основания, а также теоретическое (ТМ) и экспериментально определенное (ММ) значение молекулярной массы или номинальной массы (NM) продукта.
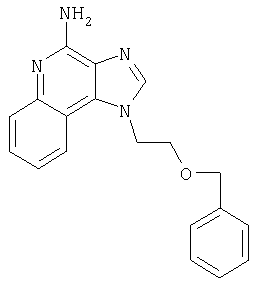
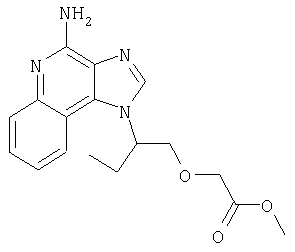
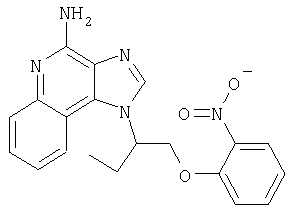
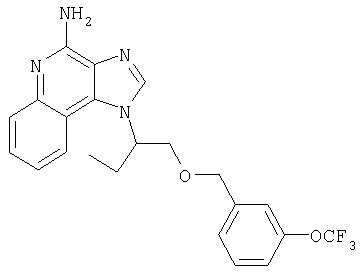
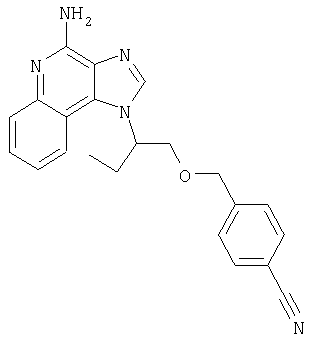
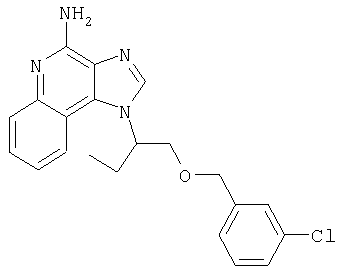

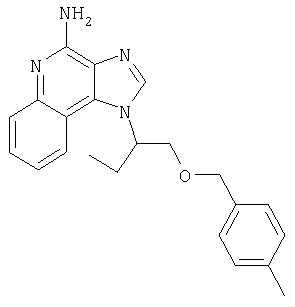
очистки
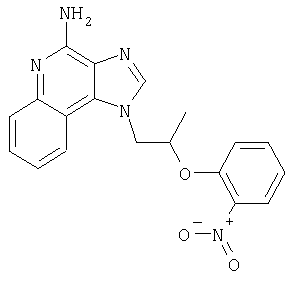
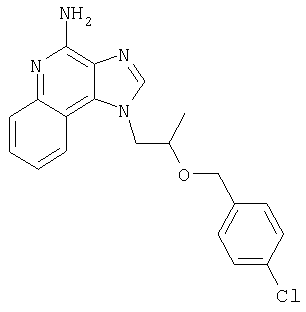
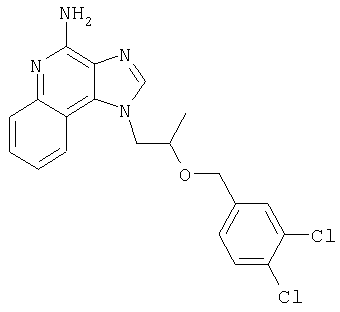
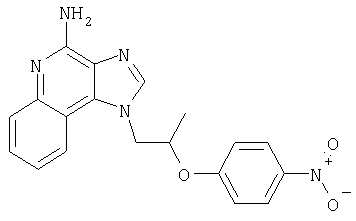
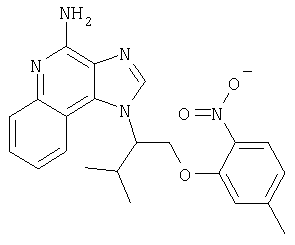
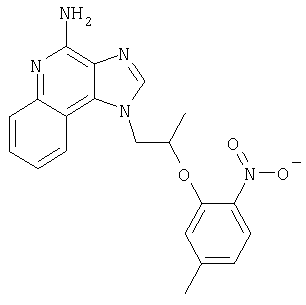
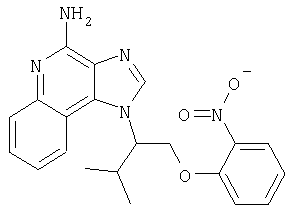
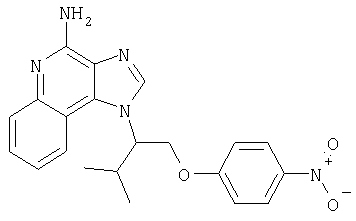
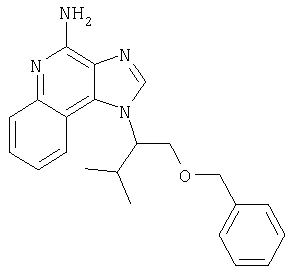
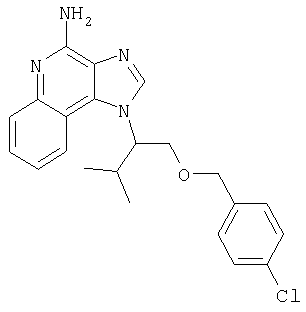
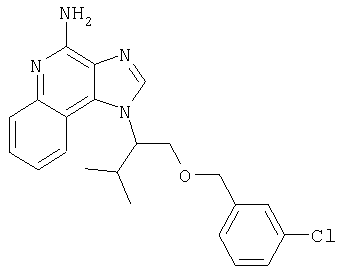
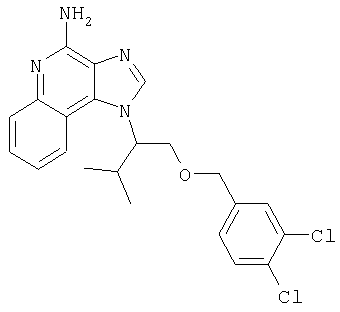
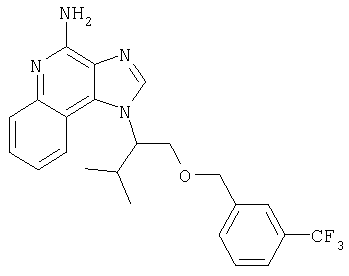

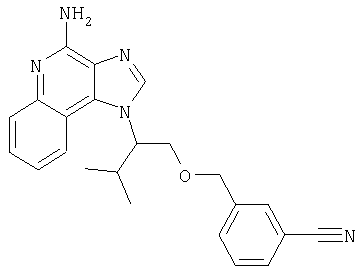
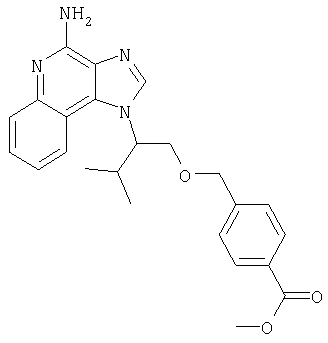

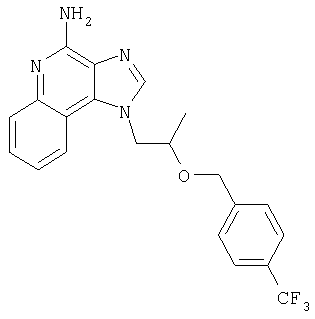
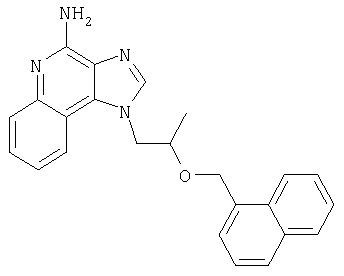
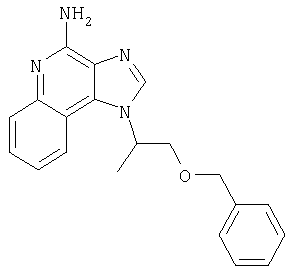
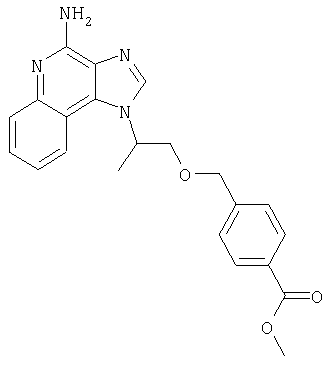
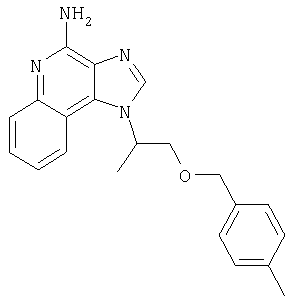
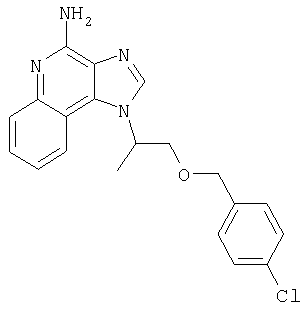
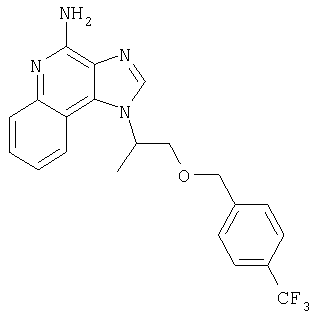
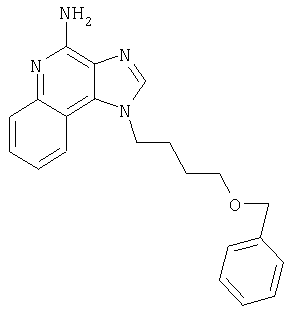
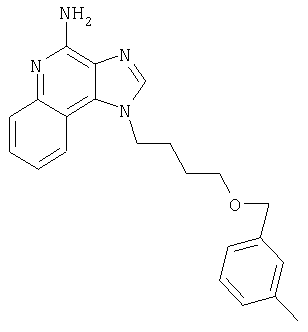
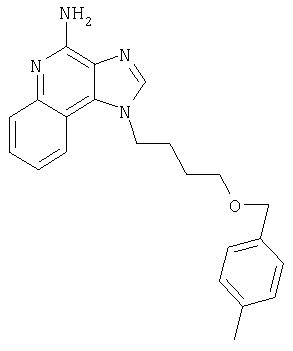
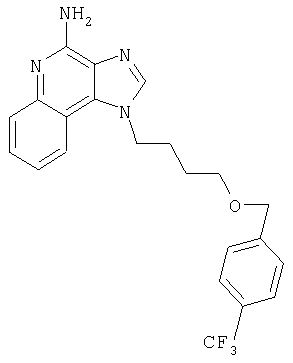
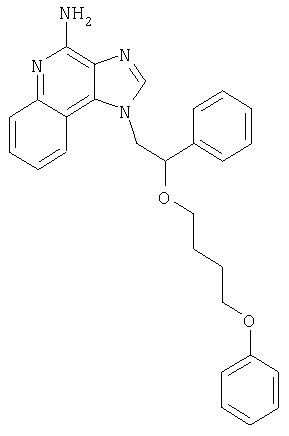
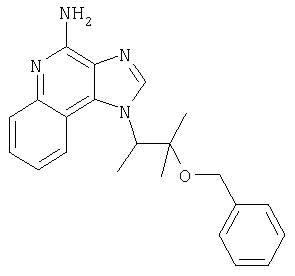

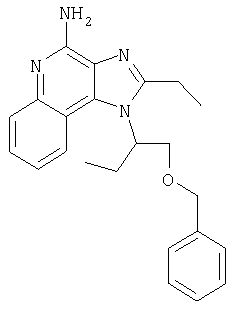
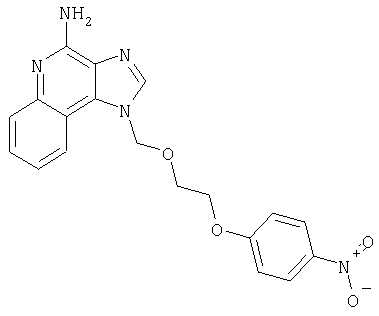
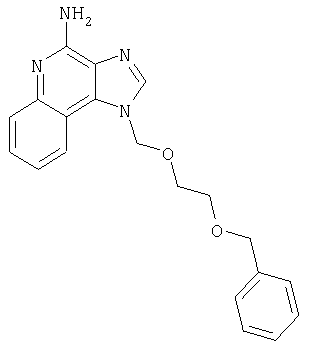
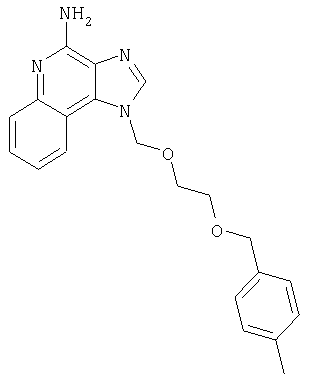
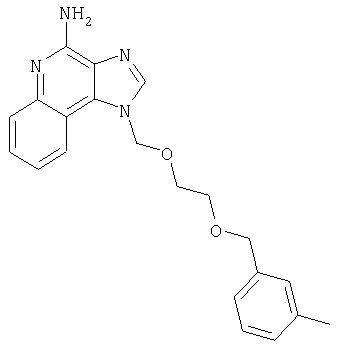
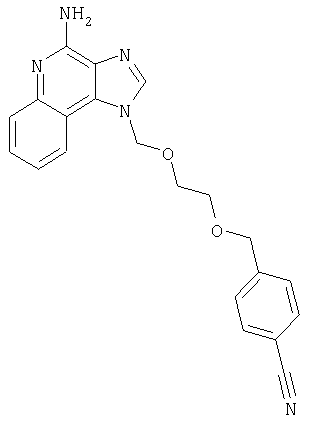
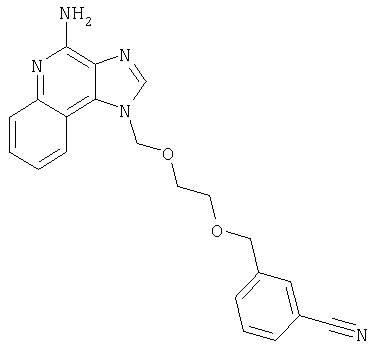
Примеры 89-96
Соединения, показанные в таблице 4, получают в соответствии со схемой V процесса, используя следующий основной способ синтеза.
25 мг 2-(4-амино-2-бутил-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)этанола помещают в ампулу объемом 7,4 мл и добавляют туда 1,2 эквивалента гидрида натрия (в виде 60%-ной суспензии в минеральном масле) и 1 мл N,N-дииметилформамида. Ампулу помещают на 15 минут в ультразвуковое перемешивающее устройство, и за это время при температуре окружающей среды образуется алкоксид. После этого к реакционной смеси добавляют 1,2 эквивалента галогенида и перемешивание осуществляют дополнительно в течение 15 минут при температуре окружающей среды. Проводят масс-спектрометрический и хроматографический анализ реакционной смеси, чтобы установить образование целевого продукта. Реакционную смесь подвергают очистке способом полупрепаративной высокой эффективной жидкостной хроматографии. Полученные при этом фракции анализируют масс-спектрометрическим способом LC-APCI/MS. Соответствующие фракции соединяют вместе и подвергают лиофилизации для получения трифторуксуснокислой соли целевого продукта, структуру и молекулярную массу которого подтверждают с помощью масс-спектрометрии и ПМР-спектроскопии. В таблице 4 показана структура свободного основания, а также теоретическое (ТМ) и экспериментально определенное (ММ) значение молекулярной массы продукта.


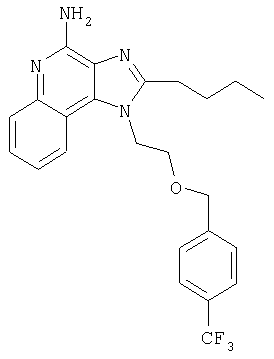
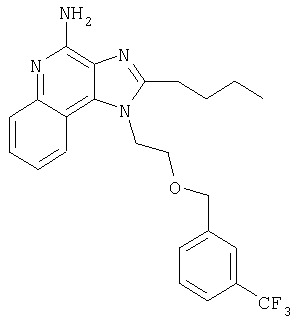
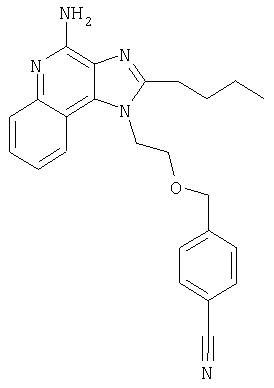
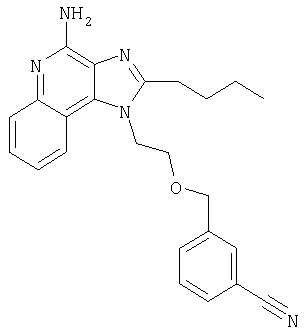
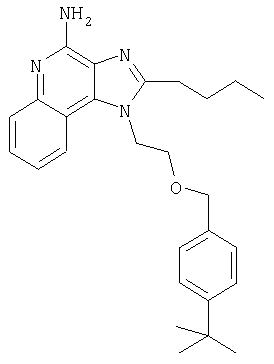
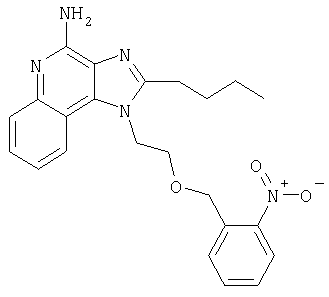
Примеры 97-100
Соединения, показанные в таблице 5, получают в соответствии со схемой III процесса, используя следующий основной способ синтеза.
Порцию раствора объемом 1 мл, полученного растворением 0,5 г 1-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)пропан-2-ола в 20 мл N,N-дииметилформамида, помещают в стеклянную ампулу объемом 7,4 мл, содержащую 2 эквивалента фенола. В эту ампулу добавляют также раствор 54 мг (2 эквивалента) трифенилфосфина в 1 мл N,N-дииметилформамида. Полученную суспензию подвергают воздействию ультразвука для растворения фенола, после чего к раствору добавляют 36 мг (2 эквивалента) чистого диэтилазодикарбоксилата. Реакционную смесь облучают ультразвуком в течение 30 минут и затем встряхивают при температуре окружающей среды в течение ночи. После этого реакционную смесь подвергают очистке способом полупрепаративной высокой эффективной жидкостной хроматографии (по способу А). Целевые продукты в примерах 99 и 100 получают в виде солей трифторуксусной кислоты. Структура и молекулярная масса продуктов были подтверждены с помощью масс-спектрометрии и ПМР-спектроскопии. В таблице 5 показана структура свободного основания, а также теоретическое значение молекулярной массы продукта (ТМ), а также номинальной молекулярной массы продукта (NM).
примера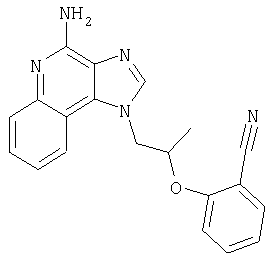

NM[M+H]+1=385
примера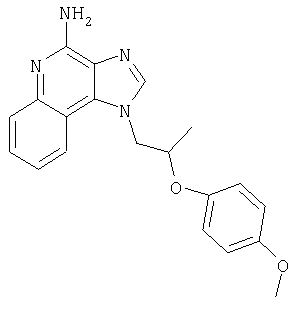
Примеры 101-104
Соединения, показанные в таблице 6, получают в соответствии со схемой III процесса, используя следующий основной способ синтеза.
Порцию раствора объемом 1 мл, полученного растворением 0,5 г 2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)-2-этилэтанола в 20 мл N,N-диметилформамида, помещают в ампулу объемом 15 мл, содержащую 2 эквивалента фенола. В эту ампулу добавляют также раствор 51 мг (2 эквивалента) трифенилфосфина в 1 мл N,N-дииметилформамида, а затем еще и 34 мг (2 эквивалента) чистого диэтилазодикарбоксилата. Реакционную смесь облучают ультразвуком в течение 2 минут и затем встряхивают при температуре окружающей среды в течение ночи. Проведенный после этого анализ с помощью высокоэффективной жидкостной хроматографии показывает, что реакция не завершена. Растворитель удаляют в вакууме. Полученный маслообразный продукт растворяют в 1 мл тетрагидрофурана, содержащего 2 эквивалента трифенилфосфина, и к раствору добавляют 2 эквивалента чистого диэтилазодикарбоксилата. Реакционную смесь встряхивают при комнатной температуре в течение ночи. Согласно данным анализа способом жидкостной хроматографии после этого реакция завершается полностью. Реакционную смесь подвергают очистке способом полупрепаративной высокой эффективной жидкостной хроматографии (по способу В). Полученные при этом фракции анализируют масс-спектрометрическим способом LC-APCI/MS. Соответствующие фракции соединяют вместе и подвергают лиофилизации для получения трифторуксуснокислой соли целевого продукта, структуру и молекулярную массу которого подтверждают с помощью масс-спектрометрии и ПМР-спектроскопии. В таблице 6 показана структура свободного основания, а также теоретическое значение молекулярной массы продукта (ТМ), а также номинальной молекулярной массы продукта (NM).
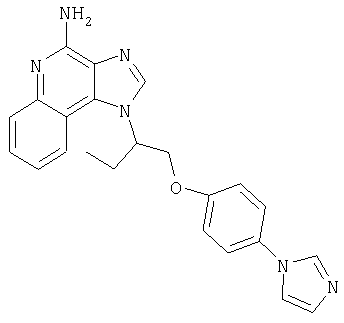
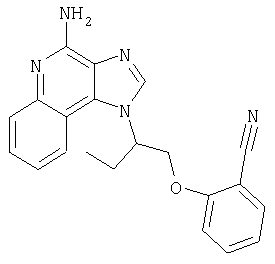
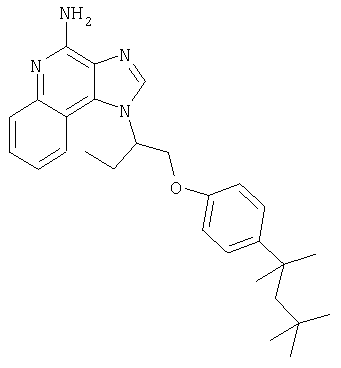
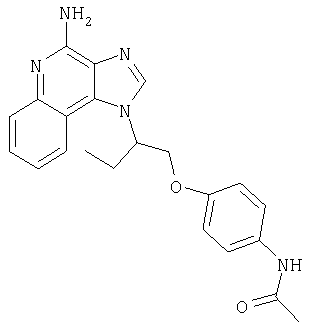
Пример 105
1-(2-Феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин
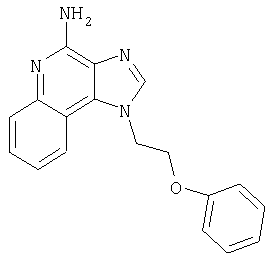
25 мг (0,108 моля) 2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этанола смешивают с 1 мл N,N-диметилформамидом, к смеси добавляют 12 мг (0,130 ммоля) фенола и 34 мг (0,130 ммоля) трифенилфосфина и полученную суспензию облучают ультразвуком в течение 1 минуты. К смеси дополнительно добавляют 23 мг (0,130 ммоля) диэтилазодикарбоксилата и встряхивают ее при температуре окружающей среды в течение 24 часов. Проведенный после этого анализ способом жидкостной хроматографии и масс-спектрометрии (хромасс-анализ) показывает, что основная часть исходного материала остается непрореагировавшей. К смеси добавляют дополнительно фенол, трифенилфосфин и диэтилазодикарбоксилат по одному эквиваленту каждого. Реакционную смесь подвергают воздействию ультразвука в течение 30 мин. Через 1 час согласно данным хромасс-анализа образуется целевой продукт. Растворитель удаляют, и остаток очищают способом полупрепаративной высокоэффективной жидкостной хроматографии (по способу А). Данные по молекулярной массе продукта: ТМ=304, NM[М+Н]+1=305.
Пример 106
1-[(1-Феноксиметил)пропил]-1H-имидазо[4,5-с]хинолин-4-амин
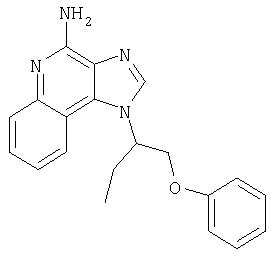
50 мг (0,195 ммоля) 2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)-2-этилэтанола смешивают с 2 мл N,N-диметилформамида, добавляют сначала 37 мг (0,390 ммоля) фенола и 102 мг трифенилфосфина, а затем 67 мг (0,390 ммоля) диэтилазодикарбоксилата. Полученный раствор облучают ультразвуком в течение 1 часа. Проведенный после этого хромасс-анализ показывает, что реакционная смесь содержит целевой продукт и небольшое количество исходного материала. Растворитель удаляют, и остаток очищают способом полупрепаративной высокоэффективной жидкостной хроматографии (по способу А). Измерение молекулярной массы продукта: ТМ=332, NM[М+Н]+1=333.
Пример 107
1-{(1R)-1-[(Проп-2-инилокси)метил]пропил}-1Н-имидазо[4,5-с]хинолин-4-амин
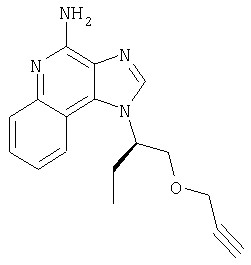
Часть А
413,8 г (1 эквивалент) неочищенного 4-хлор-3-нитрохинолина растворяют в 1,65 л дихлорметана. Раствор нагревают до кипения и фильтруют через слой абсорбента Celite®. Фильтрат при перемешивании охлаждают до 5°С и затем добавляют к нему одной порцией 305,4 мл (1,1 эквивалента) триэтиламина. Реакционную смесь перемешивают в течение 15 минут, после чего по каплям добавляют 250 мл (1,1 эквивалента) (R)-(-)-2-амино-1-бутанола, поддерживая при этом температуру реакционной смеси не выше 40°С. Смесь перемешивают при температуре окружающей среды в течение нескольких дней, затем ее охлаждают до -30°С. Образовавшийся желтый осадок отфильтровывают, промывают очень холодным дихлорметаном и сушат. Твердый продукт перемешивают в течение 1 часа с 1 л холодной смеси 80/20 вода/метанол, отфильтровывают, промывают холодной водой, дважды промывают очень холодным метанолом (порциями по 300 мл) и затем сушат на фильтре в течение ночи. В результате получают 475 г (2R)-2-{(3-нитрохинолин-4-ил)амино]бутан-1-ола.
Часть В
238 г (2R)-2-[(3-нитрохинолин-4-ил)амино]бутан-1-ола смешивают в стальном сосуде с 5 л изопропанола и 23,8 г катализатора (активированный уголь с нанесенными на него 5% платины) и подвергают гидрированию при давлении 50 фунт/дюйм2 (3,5 кг/см2) в течение 16 часов. Реакционную смесь фильтруют через слой абсорбента Celite® для удаления катализатора. Фильтрат концентрируют при пониженном давлении и получают 208,3 г (2R)-2-[(3-аминохинолин-4-ил)амино]бутан-1-ола в виде янтарного масла. Реакцию повторяют второй раз в тех же самых условиях.
Часть С
416,0 г (1 эквивалент) (2R)-2-[(3-аминохинолин-4-ил)амино]бутан-1-ола смешивают с 1,2 л (4 эквивалента) триэтилортоформиата и медленно нагревают до 145°С. Этанол, образующийся в ходе реакции, отгоняют. После того, как объем отогнанного этанола составляет около 500 мл, реакционную смесь охлаждают в атмосфере азота до 50°С. Избыток триэтилортоформиата отгоняют при пониженном давлении и в результате получают неочищенный (2R)-2-(1H-имидазо[4,5-с]хинолин-1-ил)бутан-1-ол.
Часть D
Смесь 434,3 г (2R)-2-(1Н-имидазо[4,5-хинолин-1-ил)бутан-1-ола и 1,2 л уксусного ангидрида медленно в течение около 2 часов нагревают до 100°С. После этого реакционную смесь оставляют остывать до температуры окружающей среды в течение ночи. К смеси добавляют 2,5 л метанола и нагревают ее до интенсивного кипения. Реакционную смесь кипятят с обратным холодильником в течение 2 часов, охлаждают до температуры окружающей среды и концентрируют при пониженном давлении. Остаток разбавляют водой и затем подщелачивают ее, добавляя бикарбонат натрия. Анализ способом тонкослойной хроматографии (20% метанола в этилацетате) показывает, что реакционная смесь состоит из двух продуктов и не содержит исходного материала. Образовавшееся масло экстрагируют этилацетатом. Органический слой промывают водой, сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении до получения 359,3 г остатка. Этот материал соединяют с 1,6 л уксусного ангидрида и кипятят с обратным холодильником в течение 1 часа. Затем смесь охлаждают в течение ночи до температуры окружающей среды и снова концентрируют при пониженном давлении. Анализ способом тонкослойной хроматографии показывает наличие только одного пятна от целевого продукта. Остаток разбавляют 1 л воды, подщелачивают ее до рН 8, добавляя насыщенный раствор бикарбоната натрия, и затем перемешивают в течение 1 часа. Образовавшийся осадок отфильтровывают, промывают водой и затем сушат в вакуумной печи в течение ночи при 60°С. В результате получают твердый (2R)-2-(1H-имидазо[4,5-с]хинолин-1-ил)бутилацетат коричневого цвета.
Часть Е
163,0 г (1,1 эквивалента) метилата натрия в виде 25%-ной суспензии в метаноле добавляют одной порцией к раствору 194,0 г (1 эквивалент) (2R)-2-(1H-имидазо[4,5-с]хинолин-1-ил)бутилацетата в 970 мл метанола. Реакционную смесь перемешивают при температуре окружающей среды в течение 3 часов и затем концентрируют при пониженном давлении. Остаток разбавляют 1 л воды, нейтрализуют раствор до рН 6-7, добавляя уксусную кислоту, и затем перемешивают при температуре окружающей среды в течение ночи. Полученный осадок отфильтровывают, промывают двумя порциями воды (по 200 мл каждая), сушат на фильтре воздухом и затем сушат в течение ночи в вакуумной печи при 50°С. В результате получают 145,5 г твердого (2R)-2-(1H-имидазо[4,5-с]хинолин-1-ил)бутан-1-ола.
Часть F
19 г (78,8 ммоля) (2R)-2-(1H-имидазо[4,5-с]хинолин-1-ил)бутан-1-ола добавляют к смеси 124 мл 50%-ного раствора гидроксида натрия, 150 мл дихлорметана, 0,73 г бензилтриметиламмонийхлорида и 11,4 мл (102 ммолей) бромистого пропаргила. Реакционную смесь перемешивают при температуре окружающей среды в течение ночи, после чего разбавляют ее дихлорметаном и водой. Водную фракцию многократно экстрагируют дихлорметаном. Органические фракции объединяют вместе, промывают водой, сушат над сульфатом магния, фильтруют и затем концентрируют при пониженном давлении. Остаток подвергают очистке на хроматографической колонке, используя в качестве элюента этилацетат. В результате получают 20,9 г жидкого коричневого 1-{(1R)-1-[(проп-2-инилокси)метил]-пропил}-1Н-имидазо[4,5-с]хинолина.
Часть G
К охлажденной до 0°С смеси материала, полученного в части F, и 250 мл хлороформа добавляют 15,0 г 57-86%-ной 3-хлорпероксибензойной кислоты. Через 0,5 часа реакционную смесь нагревают до температуры окружающей среды. Проводят анализ реакционной смеси способом тонкослойной хроматографии, после чего в две порции добавляют 3,75 г 3-хлорпероксибензойной кислоты. По окончании реакции (о чем судят на основании данных анализа способом тонкослойной хроматографии) реакционную смесь промывают водным раствором бикарбоната натрия. Водную фракцию экстрагируют этилацетатом. Органические фракции объединяют вместе, сушат над сульфатом магния, фильтруют и затем концентрируют при пониженном давлении до получения 1-{(1R)-1-[(пропинилокси)метил]пропил}-1H-имидазо[4,5-с|хинолин-5N-оксида в виде коричневого масла, которое затвердевает при стоянии в течение ночи.
Часть Н
К смеси материала, полученного в части G, и 300 мл безводного дихлорметана добавляют по каплям 10,7 мл трихлорацетилизоцианата. Согласно данным анализа способом тонкослойной хроматографии, который проводят через 1 час, реакция не завершается полностью. Поэтому добавляют еще 2 мл трихлорацетилизоцианата. Через 1 час реакционную смесь концентрируют при пониженном давлении и получают твердый 2,2,2-трихлор-N-(1-{(1R)-1-[2-пропинилокси)метил]пропил}-1Н-имидазо[4,5-с]хинолин-4-ил)-ацетамид в виде твердого вещества желтого цвета.
Часть I
57,5 мл 25%-ного раствора метилата натрия в метаноле добавляют к смеси материала, полученного в части Н, и 250 мл метанола. Через 0,5 часа реакционная смесь становится гомогенной, и ее перемешивают в течение ночи. Затем смесь концентрируют при пониженном давлении. Остаток очищают на хроматографической колонке, используя в качестве элюента смесь 80/20 дихлорметан/метанол. Полученный твердый продукт промывают диэтиловым эфиром, перекристаллизовывают из толуола и сушат в печи при 60°С в течение ночи. В результате -получают 9,77 г кристаллического твердого 1-{(1R)-1-[(проп-2-инилокси)метил]пропил}-1H-имидазо[4,5-с]хинолин-4-амина.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,37 (синглет, 1Н), 8,19 (дублет, J=8,3 Гц, 1Н), 7,65 (двойной дублет, J=8,3, 1,5 Гц, 1Н), 7,44 (широкий триплет, J=7,6 Гц, 1Н), 7,25 (широкий триплет, J=7,6 Гц, 1Н), 6,65 (синглет, 2Н), 5,23 (мультиплет, 1Н), 4,17 (дублет, J=2,0 Гц, 2Н), 3,90-4,10 (мультиплет, 2Н), 3,46 (триплет, J=2,4 Гц, 1Н), 2,07 (мультиплет, 2Н), 0,88 (триплет, J=7,3 Гц, 3Н).
Пример 108
1-((1R)-1-{[(3-Фенилпроп-2-инил)окси]метил}пропил)-1H-имидазо[4,5-с]хинолин-4-амин
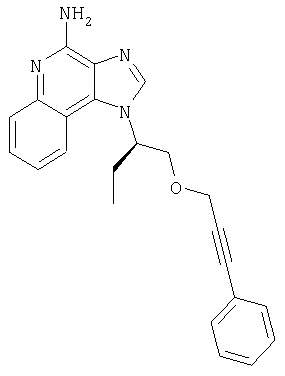
Часть А
0,80 г (1,25 ммоля) 1-{(1R)-1-[(проп-2-инилокси)метил]пропил}-1Н-имидазо[4,5-с]хинолин-4-амина в атмосфере азота добавляют к 60 мл безводного N,N-диметилформамида и смесь нагревают до 40°С, после чего добавляют к ней 3,98 г (13,9 ммоля) дибензилдикарбоната. За ходом реакции следят с помощью способа тонкослойной хроматографии и высокоэффективной жидкостной хроматографии.
Через 2 часа к смеси добавляют еще 1 г дибензилдикарбоната. Через 1 час после этого реакция завершается. Реакционную смесь разбавляют этилацетатом, промывают сначала водой и затем рассолом, сушат над сульфатом магния, фильтруют и фильтрат концентрируют при пониженном давлении до получения N,N-(бис-бензилоксикарбонил)-1-{(1R)-1-[(проп-2-инилокси)метил]пропил}-1H-имидазо[4,5-с]хинолин-4-амина в виде светло-коричневого масла. Это масло промывают гексаном для удаления избытка дибензилдикарбоната.
Часть В
Смесь 1,91 г (3,4 ммоля) N,N-(бис-бензилоксикарбонил)-1-{(1R)-1-[(проп-2-инил-окси)метил]пропил}-1H-имидазо[4,5-с]хинолин-4-амина, 30 мл безводного ацетонитрила и 0,71 мл (5,1 ммоля) триэтиламина нагревают до 70°С и к смеси добавляют 0,026 г иодида меди (I), 0,048 г дихлорбис(трифенилфосфин)палладия (II) и 0,40 мл (3,7 ммоля) иодбензола. Реакция завершается через 0,5 часа. Реакционную смесь разбавляют этилацетатом, промывают сначала водой и затем рассолом, сушат над сульфатом магния, фильтруют и концентрируют при понижженном давлении до получения коричневой жидкости. Этот материал очищают на хроматографической колонке, используя в качестве элюента смесь 39,5/59,5/1 этилацетат/гексан/триэтиламин, и в результате получают 2,1 г маслообразного продукта, который представляет собой смесь моно- и дизащищенного бензилоксикарбонильными группами 1-((1R)-1-{[(3-фенилпроп-2-инил)окси)метил]пропил}-1Н-имидазо[4,5-с]хинолин-4-амина.
Часть С
Часть материала (0,8 г), полученного на стадии В, смешивают с метанолом и 1,0 мл 25%-ного раствора метилата натрия в метаноле. Через 16 часов реакция по данным анализа способом тонкослойной хроматографии завершается. Реакционную смесь концентрируют при пониженном давлении. Полученное масло очищают на хроматографической колонке, элюируя продукт 5%-ным раствором метанола в дихлорметане. Полученное на этой стадии стеклообразное вещество сушат в течение ночи в высоком вакууме при температуре окружающей среды и получают 0,3 г 1-((1R)-1-{[(3-фенилпроп-2-инил)окси]метил}пропил)-1H-имидазо[4,5-с]хинолин-4-амина. Температура плавления этого продукта составляет 63-67°С.
Анализ. Рассчитано для С23Н22N4O: %С 74,57; %Н, 5,99; %N, 15,12. Найдено: %С 74,18;%Н,6,10;%М, 15,00.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,40 (синглет, 1Н), 8,21 (дублет, J=8,3 Гц, 1Н), 7,64 (двойной дублет, J=8,5, 1,2 Гц, 1Н), 7,43 (широкий триплет, J=7,6 Гц, 1Н), 7,25-7,40 (мультиплет, 5Н), 7,22 (широкий триплет, J=7,6 Гц, 1Н), 6,61 (синглет, 2Н), 5,26 (мультиплет, 1Н), 4,41 (синглет, 2Н), 3,95-4,20 (мультиплет, 2Н), 2,10 (мультиплет, 2Н), 0,90 (триплет, J=7,3 Гц, 3Н).
ИК-спектр (в таблетке KBr): 3306, 1634, 1526,1100, 755см-1.
HRMS(EI) Рассчитано для C23H22N4O (M+) 370,1794, найдено 370,1798.
Пример 109
1-{(1R)-1-[(3-Фенилпропокси)метил]пропил}-1Н-имидазо[4,5-с]хинолин-4-амин
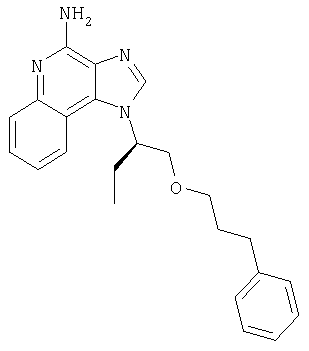
0,72 г гидроксида палладия (в виде 20%-ного продукта, нанесенного на активированный уголь) в атмосфере азота добавляют к раствору 1,3 г материала, полученного в части В примера 108, приблизительно в 20 мл метанола. Смесь гидрируют при давлении 50 фунт/дюйм2 (3,5 кг/см2) в течение 3,5 часов. После этого катализатор отфильтровывают. Фильтрат концентрируют при пониженном давлении. После очистки остатка на хроматографической колонке при использовании в качестве элюента 2,5%-ного раствора метанола в дихлорметане получают маслообразный продукт. Этот продукт обрабатывают диэтиловым эфиром, полученный твердый продукт отделяют и сушат. В результате получают 0,4 г белого кристаллического твердого 1-{(1R)-1-[(3-фенилпропокси)метил]пропил}-1Н-имидазо[4,5-с]хинолин-4-амина, температура плавления которого составляет 118-120°С.
Анализ. Рассчитано для С23Н26N4O: %С 73,77; %Н, 7,00; %N, 14,96. Найдено: %С 73,68; %Н, 7.17; %N, 14,72.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,39 (синглет, 1Н), 8,22 (дублет, J=7,8 Гц, 1Н), 7,65 (двойной дублет, J=8,3, 1,0 Гц, 1 Н), 7,43 (широкий триплет, J=7,6 Гц, 1 Н), 7,25-7,40 (мультиплет, 5Н), 7,44 (широкий триплет, J=7,7 Гц, 1Н), 7,05-7,30 (мультиплет, 4Н), 6,95 (широкий дублет, J=6,8 Гц, 2Н), 6,62 (синглет, 2Н), 5,20 (мультиплет, 1Н), 3,88 (мультиплет, 2Н), 3,36 (мультиплет, 2Н), 2,37 (широкий триплет, J=7,6 Гц, 2Н), 2,08 (мультиплет, 2Н), 1,63 (мультиплет, 2Н), 0,89 (триплет, J=7,3 Гц, 3Н).
ИК-спектр (в таблетке KBr): 3458, 3109, 1639, 1528, 1392, 1250, 760см-1.
HRMS(EI) Рассчитано для С23Н26N4O (M+) 374,2107, найдено 374,2104.
Примеры 110-112
Часть А
15 мл триэтиламина и около 0,1 моля неочищенного R-3-амино-2-метилпропан-1-ола добавляют к раствору 24,3 г (0,1 моля) 2,4-дихлор-3-нитрохинолина в 250 мл дихлорметана. Реакционную смесь кипятят с обратным холодильником до тех пор, пока согласно данным тонкослойной хроматографии состав реакционной смеси не перестает изменяться. После этого смесь упаривают досуха. Твердый желто-коричневый остаток измельчают и для удаления остатков исходного хинолина экстрагируют несколько раз гексаном, содержащим небольшое количество дихлорметана. Остаток перекристаллизовывают из изопропанола и получают 19,0 г желтого твердого R-3-[(2-хлор-3-нитрохинолин-4-ил)амино]-2-метилпропан-1-ола. 500 мг этого продукта вновь перекристаллизовывают из изопропанола и в результате получают желтое твердое кристаллическое вещество, температура плавления которого составляет 174-176°С.
Часть В
Смесь 10 г (33,8 ммоля) R-3-[(2-хлор-3-нитрохинолин-4-ил)амино]-2-метилпропан-1-ола, 350 мл изопропанола и около 1 г катализатора (активированный уголь с нанесенной на него 5% платины) подвергают гидрированию в аппарате Парра при начальном давлении водорода 50 фунт/дюйм2 (3,5 кг/см2). После прекращения поглощения водорода, реакционную смесь фильтруют для удаления катализатора. Фильтрат упаривают при пониженном давлении до получения неочищенного R-3-[(3-амино-2-хлорхинолин-4-ил)амино]-2-метилпропан-1-ола. К этому неочищенному промежуточному продукту добавляют 10,0 мл (61,5 ммоля) диэтоксиметилацетата. В результате наблюдается реакция, сопровождающаяся сильным разогревом. Образующийся раствор нагревают на паровой бане в течение 20 минут и затем реакционную смесь разбавляют водой и гидроксидом аммония. Полученный маслообразный продукт экстрагируют этилацетатом. Экстракты объединяют вместе, сушат над сульфатом магния и затем концентрируют при пониженном давлении. Образующееся твердое вещество смешивают со смесью этилацетат/гексан, отфильтровывают, промывают смесью этилацетат/гексан и сушат. В результате получают 6,0 г рыжевато/желтого твердого R-3-(4-хлор-1H-имидазо[4,5-с]хинолин-1-ил)амино]-2-метил-пропан-1-ола.
Часть С
1,0 г (3,6 ммоля) R-3-(4-хлор-1Н-имидазо[4,5-с]хинолин-1-ил)амино]-2-метил-пропан-1-ола и 30 мл 15%-ного метанольного раствора аммиака нагревают в стальном автоклаве при 150°С. Через определенное время реакционный сосуд охлаждают до температуры окружающей среды. К реакционной смеси добавляют избыток метанольного раствора гидроксида натрия, после чего смесь концентрируют при пониженном давлении до получения необходимого объема смеси. К остатку добавляют воду и концентрирование продолжают до получения твердого продукта. Этот продукт отфильтровывают, промывают водой и сушат, в результате чего получают белое твердое вещество. Этот материал перекристаллизовывают из смеси метанол/дихлорметан и получают бесцветный твердый R-3-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-1-ол, температура плавления которого составляет 258-261°С.
Анализ. Рассчитано для С14Н16N4О: %С 65,61; %Н, 6,29; %N, 21,86. Найдено: %С 65,50; %Н, 6,3; %N, 21,7.
Часть D
Соединения, показанные в таблице 7, получают в соответствии со схемой I процесса, используя следующий основной способ синтеза.
25 мг R-3-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)-2-метилпропан-1-ола помещают в ампулу объемом 7,4 мл и добавляют туда 1,2 эквивалента гидрида натрия (в виде 60%-ной суспензии в минеральном масле) и 1 мл N,N-дииметилформамида. Ампулу помещают в ультразвуковое перемешивающее устройство и облучают ультразвуком при 50°С в течение 15 минут. За это время происходит образование алкоксида. После этого к реакционной смеси добавляют 1,2 эквивалента галогенида, и перемешивание в ультразвуковом перемешивающем устройстве осуществляют в течение 2 часов при 50°С. Проводят масс-спектрометрический и хроматографический анализ реакционной смеси, чтобы установить образование целевого продукта. Реакционнную смесь подвергают очистке способом полупрепаративной высокой эффективной жидкостной хроматографии. Полученные при этом фракции анализируют масс-спектрометрическим способом LC-APCI/MS. Соответствующие фракции соединяют вместе и подвергают лиофилизации для получения трифторуксуснокислой соли целевого продукта, структуру и молекулярную массу которого подтверждают с помощью масс-спектрометрии и ПМР-спектроскопии. В таблице 7 показана структура свободного основания, а также теоретическое (ТМ) и экспериментально определенное (ММ) значение молекулярной массы продукта.
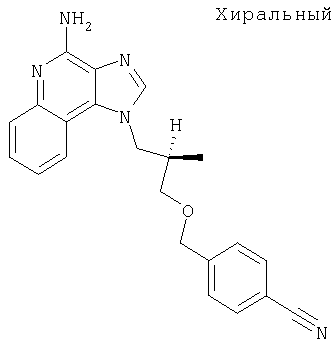

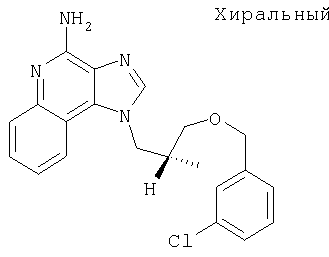
Пример 113
1-[(Бензилокси)метил]-1H-имидазо[4,5-c]хинолин-4-амин
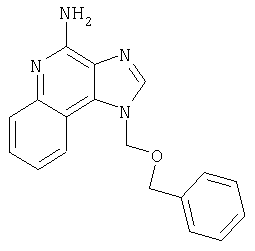
0,48 г (11,9 ммоля) 60%-ной суспензии гидрида натрия в минеральном масле добавляют к суспензии 2,0 г (10,9 ммоля) 1Н-имидазо[4,5-с]хинолин-4-амина в N,N-диметилформамиде. Реакционную смесь перемешивают при температуре окружающей среды в течение 3 часов, затем охлаждают на ледяной бане и добавляют 1,5 мл (10,9 ммоля) бензилхлорметилового эфира. Смесь перемешивают при температуре окружающей среды в течение 2 часов, после чего нагревают на паровой бане в течение 1 часа. Образовавшийся осадок отфильтровывают. Фильтрат разбавляют водой и отделяют масляную фазу. Масло смешивают с осадком и получают 2,1 г клейкого твердого вещества. Это вещество обрабатывают 5 мл кипящего этилацетата. Смесь охлаждают и осадок отфильтровывают. Фильтрат концентрируют при пониженном давлении. Полученный остаток дважды обрабатывают этилацетатом и затем присоединяют к ранее полученному осадку, в результате чего получают 0,8 г твердого продукта. После перекристаллизации этого продукта из приблизительно 5 мл этанола получают 0,6 г 1-[(бензилокси)метил]-1Н-имидазо[4,5-с]хинолин-4-амина, температура плавления которого составляет 168-172°С.
Анализ. Рассчитано для C18H16N4O: %C 71,0; %Н, 5,3; %N, 18,4. Найдено: %С 70,9; %Н, 5,3; %N, 18,4.
Пример 114
1-(2-{3-[4-(Диметиламино)фенил]пропокси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин
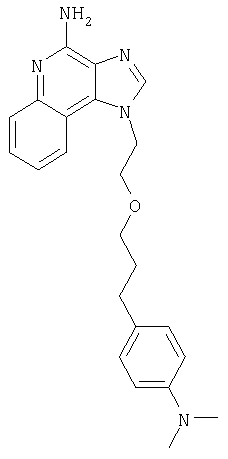
Часть А
Используя основной способ, применяемый в примере 12, часть А, 2,5 г (5,36 ммоля) N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина обрабатывают при 70°С 1,46 г (5,89 ммоля) 4-иод-N,N-диметиланилина. Реакция завершается в течение 30 минут. Раствор разбавляют этилацетатом, по три раза промывают водой, насыщенным водным раствором бикарбоната натрия и рассолом, сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Оставшийся твердый продукт очищают хроматографически на силикагеле, используя в качестве элюента смесь 98/2 дихлорметан/метанол, и в результате получают 0,883 г твердого коричневого трет-бутил-1-[2-({3-[4-(диметиламино)фенил]проп-2-инил}окси)этил]-1H-имидазо-[4,5-с]хинолин-4-илкарбамата.
MS(Cl) для C33H39N5O5 m/z 586 (MH+), 486, 386, 229.
Часть В
Используя основной способ, применяемый в примере 12, часть В, осуществляют гидрирование 0,883 г (1,507 ммоля) трет-бутил-1-[2-({3-[4-(диметиламино)фенил]проп-2-инил}окси)этил]-1H-имидазо[4,5-с]-хинолин-4-илкарбамата, в результате чего получают 0,783 г твердого коричневого трет-бутил-1-[2-{3-[4-(диметиламино)фенил]пропокси}-этил]-1H-имидазо[4,5-с]хинолин-4-илкарбамата.
MS(Cl) для С33Н43N5O5 m/z 590 (МН+), 490, 390, 229.
Часть С
Используя основной способ, применяемый в примере 12, часть С, проводят реакцию между 0,783 г (1,327 ммоля) трет-бутил-1-[2-{3-[4-(диметиламино)фенил]пропокси}этил]-1H-имидазо[4,5-с]хинолин-4-илкарбамата и 10 мл трифторуксусной кислоты. Полученный продукт дважды обрабатывают этиловым эфиром и в результате получают 0,634 г белого твердой соли трифторуксусной кислоты: (1-(2-{3-[4-(диметиламино)фенил]пропокси}-этил]-1Н-имидазо-[4,5-с]хинолин-4-амин).(трифторацетат)1,5. Температура плавления этого продукта составляет 137-140°С.
Анализ. Рассчитано для C23H27N5O.(C2HF3O2)1,5: %C 54,83; %Н, 5,22; %N, 12,30. Найдено: %C 54,67; %Н, 4,91; %N, 12,27.
1H ЯМР(300 МГц, ДМСО-d6) δ 9,04-9,11 (широкий синглет, 2Н), 8,49 (синглет, 1Н), 8,36 (дублет, J=7,3 Гц, 1Н), 7,83 (дублет, J=8,3 Гц, 1Н), 7,74 (триплет, J=8,3 Гц, 1Н), 7,56 (триплет, J=6,8 Гц, 1Н), 6,71 (дублет, J=7,8 Гц, 2Н), 6,60 (мультиплет, 2Н), 4,90 (триплет, J=4,9 Гц, 2Н), 3,83 (триплет, J=4,9 Гц, 2Н), 3,27 (триплет, J=5,9 Гц, 2Н), 2,28 (синглет, 6Н), 2,25 (триплет, J=7,8 Гц, 2Н), 1,54 (квинтет, J=6,4, 6,8, 2Н)
MS(Cl) для C23H27N5O m/z 390 (MH+), 229.
Пример 115
1-(2-{[(2Е)-3-Фенилпроп-2-енил]окси}этил)-1H-имидазо[4,5-с]хинолин-4-амин
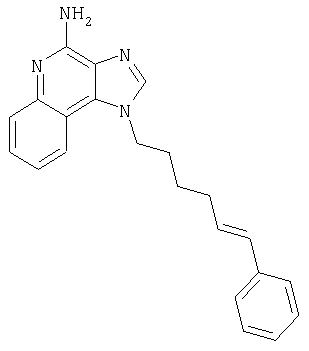
Часть А
В сухую круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 0,19 г (4,65 ммоля) 60%-ной суспензии гидрида натрия в минеральном масле и 2 мл гексана. После этого с помощью шприца в колбу добавляют 10 мл безводного диметилформамида и 0,902 г (4,23 ммоля) 2-(1Н-имидазо[4,5-с]хинолин-1-ил)этанола, и смесь нагревают при 60°С в течение 20 минут. По истечении этого времени к раствору с помощью шприца добавляют 0,65 мл (4,65 ммоля) хлорангидрида коричной кислоты. Через 50 минут выход целевого продукта достигает приблизительно 80%. Летучие продукты отгоняют при пониженном давлении и образующееся масло распределяют между дихлорметаном и водой. Водный слой экстрагируют дихлорметаном; органические фракции объединяют вместе, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Образовавшийся стеклообразный продукт подвергают хроматографической очистке на силикагеле (используя в качестве элюента смесь 95/5 дихлорметан/метанол) и сушат в вакуумной печи при 60°С в течение 15 часов. В результате получают 0,652 г 1-(2-{[(2Е)-3-фенилпроп-2-енил]окси}этил)-1H-имидазо[4,5-с]хинолина в виде стеклообразного твердого продукта.
MS(Cl) для C21H19N3O m/z 330 (MH+), 214.
Часть В
Используя основной способ, применяемый в примере 1, часть В, 0,652 г (1,98 ммоля) 1-(2-{[(2Б)-3-фенилпроп-2-енил]окси}этил)-1Н-имидазо[4,5-с]хинолина окисляют до 0,67 г 1-(2-{[(2Е)-3-фенилпроп-2-енил]окси}этил)-1H-имидазо[4,5-с]хинолин-5N-оксида. Это коричневое твердое вещество используют в дальнейшем без дополнительной очистки.
Часть С
В круглодонную колбу, снабженную магнитной мешалкой, при температуре окружающей среды загружают 0,67 г (1,98 ммоля) 1-(2-{[(2Е)-3-фенилпроп-2-енил]окси}этил)-1Н-имидазо[4,5-с]хинолин-5N-оксида, 15 мл дихлорметана и 7 мл 27%-ного водного раствора гидроксида аммония. К смеси несколькими порциями добавляют 0,415 г (2,18 ммоля) твердого п-толуолсульфонилхлорида и полученный раствор перемешивают. Реакция завершается в течение 20 минут; раствор распределяют между водной и органической фазами. Водный слой трижды экстрагируют дихлорметаном. Органические фракции объединяют вместе, трижды экстрагируют 5%-ным водным раствором бикарбоната натрия, промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Образовавшееся белое твердое вещество пять раз перекристаллизовывают из смеси метанол/вода и в результате получают 0,086 г 1-(2-{[(2Е)-3-фенилпроп-2-енил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде белого пушистого продукта. Температура плавления этого продукта составляет 183,7-184,3°С.
Анализ. Рассчитано для C21H20N4O: %С 73,23; %Н, 5,85; %N, 16,27. Найдено: %С 73,11;%Н, 5,81 ;%N, 16,10.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,19 (синглет, 1Н), 8,12 (дублет, J=7,3 Гц, 1Н), 7,62 (дублет, J=8,3 Гц, 1Н), 7,43 (триплет, J=8,3 Гц, 1Н), 7,19-7,31 (мультиплет, 6Н), 6,61 (синглет, 2Н), 6,33 (дублет, J=15,6 Гц, 1Н), 6,17 (двойной триплет, J=16,0, 5,2 Гц, 1 Н), 4,84 (триплет, J=4,9 Гц, 2Н). 4,07 (триплет, J=3,9 Гц, 2Н), 3,91 (триплет, J=5,4 Гц, 2Н)
MS(Cl) для C21H20N4O m/z 345 (МН+), 270, 229.
Пример 116
2-Октил-1-{2-[(3-фенилпроп-2-инил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин
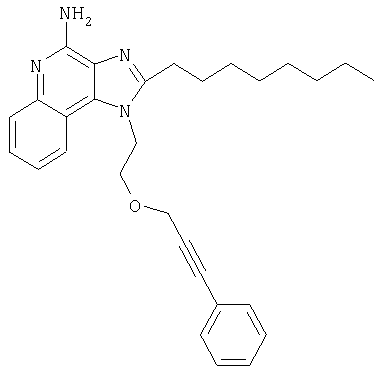
Часть А
Используя основной способ, применяемый в примере 1, часть А, проводят реакцию между 4,8 г (14,75 ммоля) 2-(2-октил-1H-имидазо[4,5-с]хинолин-1-ил)этанолом и бромистым пропаргилом (в виде 80%-ного раствора в толуоле) (4,93 мл, 44,25 ммоля), в результате которой получают 4,84 г твердого коричневого продукта - 2-октил-1-[2-(проп-2-инилокси)этил]-1H-имидазо[4,5-с]хинолина.
Часть В
Используя основной способ синтеза, применяемый в примере 12, часть А, 4,84 г (13,32 ммоля) 2-октил-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолина при 40°С обрабатывают 1,7 мл (14,65 ммоля) иодбензола. Реакция завершается в течение 45 минут. Летучие продукты удаляют при пониженном давлении, а оставшийся маслообразный продукт подвергают хроматографической очистке на силикагеле (в качестве элюента используют смесь 98/2 дихпорметан/метанол). В результате очистки получают 4,2 г бледно-желтого твердого 2-октил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолина.
MS(Cl) для С29Н33N3О m/z 440 (МН+), 291.
Часть С
Используя основной способ, применяемый в примере 1, часть В, 2,2 г (5,004 ммоля) 2-октил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолина окисляют до 2,28 г 2-октил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолин-5N-оксида, представляющего собой маслообразный продукт.
Часть D
Используя основной способ, применяемый в примере 115, часть С, проводят аминирование 2,22 г (4,83 ммоля) 2-октил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-5N-оксида. Образовавшийся в результате этой реакции твердый коричневый продукт обрабатывают диэтиловым эфиром и перекристаллизовывают из 2-пропанола, что приводит к получению 1,23 г 2-октил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолин-4-амина в виде белого кристаллического твердого вещества. Температура плавления этого продукта составляет 138-138,7°С.
Анализ. Рассчитано для С29Н34N4О: %С 76,62; %Н, 7,54; %N, 12,32. Найдено: %С 76,6; %Н, 7,49; %N, 12,19.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,07 (дублет, J=8,3 Гц, 1Н), 7,62 (дублет, J=8,3 Гц, 1Н), 7,41 (триплет, J=6,8 Гц, 1Н), 7,27-7,36 (мультиплет, 3Н), 7,18-7,24 (мультиплет, 3Н), 6,45 (синглет, 2Н), 4,78 (триплет, J=4,9 Гц, 2Н), 4,34 (синглет, 2Н), 4,00 (триплет, J=4,9 Гц, 2Н), 2,94 (триплет, J=7,8 Гц, 2Н), 1,83 (квинтет, J=7,3, 7,3 Гц, 2Н), 1,22-1,43 (мультиплет, 10Н), 0,85 (триплет, J=6,8 Гц, 3Н)
MS(Cl) для C29H34N4O m/z 455 (МН+), 283.
Пример 117
2-Октил-1-[2-(3-фенилпропокси)этил])-1Н-имидазо[4,5-с]хинолин-4-амин
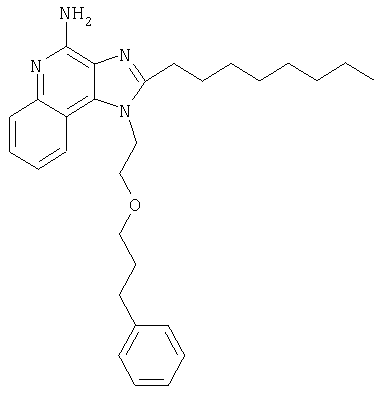
Часть А
Используя основной способ, применяемый в примере 12, часть В, осуществляют гидрирование 2-октил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолина (2,0 г, 4,55 ммоля) до 2-октил-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]хинолина (1,78 г), представляющего собой твердый белый продукт.
1H ЯМР(300 МГц, ДМСО-d6) δ 9,15 (синглет, 1Н), 8,41 (дублет, J=9,78 Гц, 1Н), 8,16 (дублет, J=9,8 Гц, 1Н), 7,63-7,71 (мультиплет, 2Н), 7,06-7,09 (мультиплет, 3Н), 6,81-6,84 (мультиплет, 2Н), 4,85 (триплет, J=4,9 Гц, 2Н), 3,84 (триплет, J=4,9 Гц, 2Н), 3,25 (триплет, J=4,9 Гц, 2Н), 3,04 (триплет, J=7,8 Гц, 2Н), 2,31 (триплет, J=8,3 Гц, 2Н), 1,91 (квинтет, J=7,3, 7,3 Гц, 2Н), 1,59 (квинтет, J=8,8, 5,8 Гц, 2Н), 1,25-1,49 (мультиплет, 10Н), 0,85 (триплет, J=7,3 Гц, 3Н).
Часть В
Используя основной способ, применяемый в примере 1, часть В, 1,78 г (4,03 ммоля) 2-октил-1-[2-(3-фенилпропокси)этил]-1H-имидазо[4,5-с]хинолина окисляют до 1,8 г маслообразного 2-октил-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксида.
Часть С
Используя основной способ, применяемый в примере 115, часть С, проводят аминирование 1,85 г (4,03 ммоля) 2-октил-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]-хинолин-5N-оксида. Образовавшийся в результате этой реакции твердый коричневый продукт обрабатывают диэтиловым эфиром и перекристаллизовывают из ацетонитрила, что приводит к получению 0,31 г 2-октил-1-[2-(3-фенилпропокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина в виде белого кристаллического твердого вещества. Температура плавления этого продукта составляет 103,8-104,5°С.
Анализ. Рассчитано для С29Н38N4O: %С 75,94; %Н, 8,35; %N, 12,22. Найдено: %С 75,71; %Н, 8,46; %N, 12,22.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,06 (дублет, J=7,8 Гц, 1Н), 7,62 (дублет, J=8,3 Гц, 1Н), 7.41 (триплет, J=7,8 Гц, 1Н), 7,21 (триплет, J=7,8 Гц, 1Н), 7,05-7,15 (мультиплет, 3Н), 6,90 (двойной дублет, J=5,4, 1,9 Гц, 2Н), 6,45 (синглет, 2Н), 4,73 (триплет, J=4,4 Гц, 2Н), 3,80 (триплет, J=4,9 Гц, 2Н), 3,24 (триплет, J=5,9 Гц, 2Н), 2,97 (триплет, J=7,8 Гц, 2Н), 2,39 (триплет, J=7,8 Гц, 2Н), 1,85 (квинтет, J=7,3, 7,8 Гц, 2Н), 1,62 (квинтет, J=6,8, 6,3 Гц, 2Н), 1,22-1,44 (мультиплет, 10Н), 0,84 (триплет, J=6,8 Гц, 3Н)
MS(Cl) для С29Н38N4O m/z 459 (МН+), 373, 285.
Пример 118
2-Метил-1-{2-[(3-фенилпроп-2-инил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин
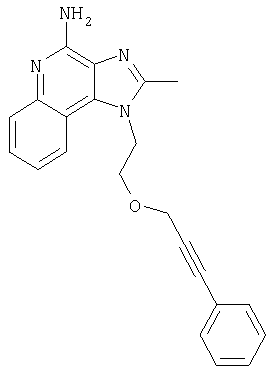
Часть А
Используя основной способ, применяемый в примере 1, часть А, проводят реакцию между 4,0 г (17,6 ммоля) 2-(2-метил-1H-имидазо[4,5-с]хинолин-1-ил)этанолом и бромистым пропаргилом (в виде 80%-ного раствора в толуоле) (5,9 мл, 52,8 ммоля), в результате которой получают 3,6 г темно-коричневого маслообразного продукта - 2-метил-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-}хинолина.
MS(Cl) для С16Н15N3О m/z 266 (MH+), 184.
Часть В
Используя основной способ, применяемый в примере 12, часть А, 3,6 г (13,57 ммоля) 2-метил-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолина обрабатывают при температуре окружающей среды 1,7 мл (14,92 ммоля) иодбензола. Реакция завершается через 20 часов. Раствор подщелачивают, добавляя к нему 5%-ный водный раствор бикарбоната натрия, и затем трижды экстрагируют дихлорметаном. Органические фракции соединяют вместе, трижды промывают водой, а затем рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Для окончательной очистки продукта его пропускают через силикагель (в качестве элюента используют смесь 95/5 дихлорметан/метанол) и перекристаллизовывают из ацетонитрила. В результате получают 1,94 г светло-желтого твердого 2-метил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолина.
MS(Cl) для С22Н19N3О m/z 342 (MH+), 228.
Часть С
Используя основной способ, применяемый в примере 1, часть В, 1,0 г (2,93 ммоля) 2-метил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолина окисляют до получения 1,3 г 2-метил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-5N-оксида, представляющего собой рыжеватое твердое вещество.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,94 (синглет, 1Н), 8,78 (дублет, J 8,3 Гц, 1Н), 8,48 (дублет. J=7,8 Гц, 1Н), 7,79 (мультиплет, 2Н), 7,26-7,35 (мультиплет, 3Н), 7,09-7,18 (мультиплет, 2Н), 4,86 (триплет, J=5,4 Гц, 2Н), 4,34 (синглет, 2Н), 4,04 (триплет, J=4,9 Гц, 2Н), 2,66 (синглет, 3Н).
Часть D
Используя основной способ, применяемый в примере 115, часть С, проводят аминирование 1,05 г (2,93 ммоля) 2-метил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-5N-оксида. Образовавшийся в процессе этой реакции твердый рыжеватый продукт обрабатывают диэтиловым эфиром и перекристаллизовывают из толуола, после чего подвергают хроматографической очистке на силикагеле (используя в качестве элюента смесь 98/2 дихлорметан/метанол). В результате получают 0,261 г 2-метил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолин-4-амина в виде белого кристаллического твердого вещества. Температура плавления этого продукта составляет 142,7-143,3°С.
Анализ. Рассчитано для С22Н20N4O: %С 74,14; %Н, 5,66; %N, 15,72. Найдено: %С 73,97; %Н, 5,77; %N, 15,77.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,08 (дублет, J=8,3 Гц, 1Н), 7,61 (дублет, J=8,3 Гц, 1Н), 7,41 (триплет, J=8,3 Гц, 1Н), 7,28-7,35 (мультиплет, 3Н), 7,12-7,24 (мультиплет, 3Н), 6,52 (синглет, 2Н), 4,77 (триплет, J=4,9 Гц, 2Н), 4,36 (синглет, 2Н), 4,02 (триплет, J=4,9 Гц, 2Н), 2,62 (синглет, 3Н)
MS(Cl) для C22H20N4O m/z 357 (МН+), 243, 199.
Пример 119
2-Метил-1-[2-(3-фенилпропокси)этил])-1Н-имидазо[4,5-с]хинолин-4-амин
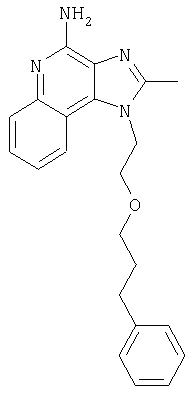
Часть А
Используя основной способ, применяемый в примере 12, часть В, осуществляют гидрирование 2-метил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]-хинолина (0,9 г, 2,636 ммоля) до 2-метил-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]хинолина (0,845 г), представляющего собой твердый белый продукт.
1H ЯМР(300 МГц, ДМСО-d6) δ 9,12 (синглет, 1Н), 8,44 (дублет, J=7,3 Гц, 1Н), 8,16 (дублет, J=7,8 Гц, 1Н), 7,65-7,70 (мультиплет, 2Н), 7,04-7,08 (мультиплет, 3Н), 6,79-6,83 (мультиплет, 2Н), 4,85 (триплет, J=4,9 Гц, 2Н), 3,85 (триплет, J=5,4 Гц, 2Н), 3,23 (триплет, J=6,4 Гц, 2Н), 2,70 (синглет, 3Н), 2,3 (триплет, J=7,8 Гц, 2Н), 1,58 (квинтет, J=6,36, 6,36 Гц, 2Н).
Часть В
Используя основной способ, применяемый в примере 1, часть В, 0,845 г (2,45 ммоля) 2-метил-1-[2-(3-фенилпропокси)этил]-1H-имидазо[4,5-с]хинолина окисляют до 0,88 г 2-метил-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксида, представляющего собой твердое стеклообразное вещество. Этот продукт в дальнейшем применяют без дополнительной очистки.
Часть С
Используя основной способ, применяемый в примере 115, часть С, проводят аминирование 0,88 г (2,45 ммоля) 2-метил-1-[2-(3-фенилпропокси)этил]-1H-имидазо[4,5-с]-хинолин-5N-оксида. Образовавшийся в результате этой реакции твердый коричневый продукт обрабатывают диэтиловым эфиром и перекристаллизовывают из толуола, что приводит к получению 0,596 г 2-метил-1-[2-(3-фенилпропокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина в виде белого порошка. Температура плавления этого продукта составляет 129,7-130,7°С.
Анализ. Рассчитано для C22H24N4O: %С 73,31; %Н, 6,71; %N, 15,54. Найдено: %С 73,21 ;%Н, 6,66; %N, 15,58.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,07 (дублет, J=8,3 Гц, 1Н), 7,62 (дублет, J=7,3 Гц, 1Н), 7,41 (триплет, J=7,3 Гц, 1Н), 7,22 (триплет, J=8,3 Гц, 1Н), 7,05-7,14 (мультиплет, 3Н), 6,88 (двойной дублет, J=6,8, 2,4 Гц, 2Н), 6,52 (синглет, 2Н), 4,73 (триплет, J=4,9 Гц, 2Н), 3,80 (триплет, J=4,9 Гц, 2Н), 3,24 (триплет, J=6,4 Гц, 2Н), 2,64 (синглет, 3Н), 2,38 (триплет, J=8,3 Гц, 2Н), 1,62 (квинтет, J=6,8, 6,4 Гц, 2Н)
MS(Cl) для C22H24N4O m/z 361 (МН+), 347, 199.
Пример 120
2-(Метоксиэтил)-1-{2-[(3-фенилпроп-2-инил]окси}этил)-1H-имидазо[4,5-c]хинолин-4-амин
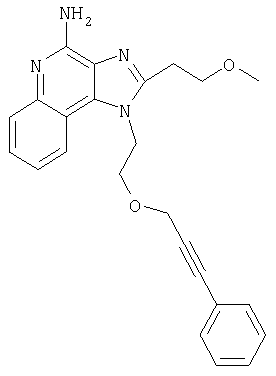
Часть А
Используя основной способ, применяемый в примере 1, проводят реакцию между 2,53 г (9,33 ммоля) 2-[2-(метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этанолом и бромистым пропаргилом (в виде 80%-ного раствора в толуоле) (3,11 мл, 27,9 ммоля), в результате которой получают 2,72 г маслообразного продукта - 2-(метоксиэтил)-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолина.
MS(Cl) для С18Н19N3O2 m/z 310 (MH+), 278, 196.
Часть В
Используя основной способ, применяемый в примере 12, часть А, 2,72 г (1,79 ммоля) 2-(метоксиэтил)-1-[2-(проп-2-инилокси)этил]-1H-имидазо[4,5-с]хинолина обрабатывают при температуре окружающей среды 1,1 мл (9,67 ммоля) иодбензола. Реакция завершается через 45 минут. Летучие продукты отгоняют при пониженном давлении и оставшийся маслообразный продукт распределяют между дихлорметаном и 5%-ным водным раствором бикарбоната натрия. Водный слой экстрагируют дихлорметаном. Органические фракции соединяют вместе, промывают рассолом, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении до получения твердого коричневого продукта. Этот продукт подвергают хроматографической очистке на силикагеле (элюент - смесь 95/5 дихлорметан/метанол) и обрабатывают гексаном. В результате получают 2,39 г желтого твердого 2-(метоксиэтил)-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолина.
MS(Cl) для С24Н23N3O2 m/z 386 (МН+), 354, 270.
Часть С
Используя основной способ, применяемый в примере 1, часть В, 1,19 г (3,097 ммоля) 2-(метоксиэтил)-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолина окисляют до получения 1,24 г 2-(метоксиэтил)-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолин-5N-оксида, представляющего собой стеклообразное твердое вещество.
Часть D
Используя основной способ, применяемый в примере 115, часть С, проводят аминирование 1,243 г (3,097 ммоля) 2-(метоксиэтил)-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолин-5N-оксида. Образовавшийся в результате этой реакции коричневый маслообразный продукт подвергают хроматографической очистке на силикагеле (используя в качестве элюента смесь 98/2 дихлорметан/метанол) и перекристаллизовывают из этилацетата и ацетонитрила, что приводит к получению 0,379 г 2-(метоксиэтил)-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина в виде белого твердого кристаллического вещества. Температура плавления этого продукта составляет 134,5-135,5°С.
Анализ. Рассчитано для C24H24N4O2: %С 71,98; %Н, 6,04; %N, 13,99. Найдено: %С 72,21; %Н, 5,98; %N, 14,29.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,09 (дублет, J=8,3 Гц, 1Н), 7,62 (дублет, J=8,3 Гц, 1Н), 7,41 (триплет, J=8,3 Гц, 1Н), 7,28-7,35 (мультиплет, 3Н), 7,18-7,24 (мультиплет, 3Н), 6,50 (синглет, 2Н), 4,82 (триплет, J=4,9 Гц, 2Н), 4,36 (синглет, 2Н), 4,01 (триплет, J=4,9 Гц, 2Н), 3,84 (триплет, J=6,8 Гц, 2Н), 3,29 (синглет, 3Н), 3,23 (триплет, J=6,8 Гц, 2Н),
MS(Cl) для C24H24N4O2 m/z 401 (MH+), 255, 183.
Пример 121
2-(2-Метоксиэтил)-1-[2-(3-фенилпропокси)этил])-1H-имидазо[4,5-с]хинолин-4-амин
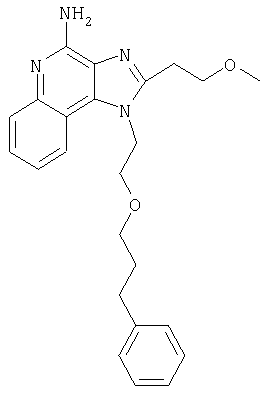
Часть А
Используя основной способ, применяемый в примере 12, часть В, осуществляют гидрирование 2-(2-метоксиэтил)-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]-хинолина (1,2 г, 3,11 ммоля) до 2-(2-метоксиэтил)-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]хинолина (1,01 г), представляющего собой маслообразный продуют.
MS(Cl) для С24Н27N3O2 m/z 390 (МН+), 235.
Часть В
Используя основной способ, применяемый в примере 1, часть В, 1,01 г (2,60 ммоля) 2-(2-метоксиэтил)-1-[2-(3-фенилпропокси)этил]-1H-имидазо[4,5-с]хинолина окисляют до 1,05 г 2-(2-метоксиэтил)-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксида, представляющего собой маслообразный продукт.
Часть С
Используя основной способ, применяемый в примере 115, часть С, проводят аминирование 1,05 г (2,601 ммоля) 2-(2-метоксиэтил)-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]-хинолин-5N-оксида. Образовавшийся в результате этой реакции твердый коричневый продукт подвергают хроматографической очистке на силикагеле (используя в качестве элюента смесь 98/2 дихлорметан/метанол) и перекристаллизовывают из смеси этилацетат/гексан и в результате получают 0,111 г твердого белого 2-(2-метоксиэтил)-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина. Температура плавления этого продукта составляет 103,8-104,5°С.
Анализ. Рассчитано для C24H28N4O2.(H2O)0,2: %C 70,63; %Н, 7,01; %N, 13,73. Найдено: %C 70,38; %Н, 6,80; %N, 13,57.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,09 (дублет, J=7,3 Гц, 1Н), 7,63 (дублет, J=8,3 Гц, 1Н), 7,42 (триплет, J=6,8 Гц, 1Н), 7,22 (триплет, J=7,8 Гц, 1Н), 7,08-7,15 (мультиплет, 3Н), 6,89 (дублет, J=5,4 Гц, 2Н), 6,49 (синглет, 2Н), 4,78 (триплет, J=4,9 Гц, 2Н), 3,86 (триплет, J=6,8 Гц, 2Н), 3,80 (триплет, J=5,4 Гц, 2Н), 3,30 (синглет, 3Н), 3,22-3,28 (мультиплет, 4Н), 2,39 (триплет, J=8,3 Гц, 2Н), 1,62 (квинтет, J=8,3, 6,4 Гц, 2Н)
MS(Cl) для C24H28N4O2 m/z 405 (МН+), 373, 235.
Пример 122
2-(Этоксиэтил)-1-{2-[(3-фенилпроп-2-инил]окси}этил)-1H-имидазо[4,5-c]хинолин-4-амин
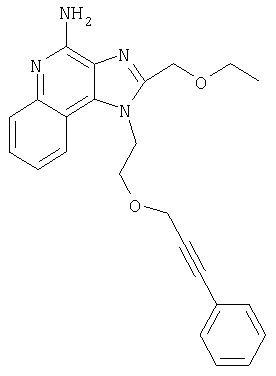
Часть А
Используя основной способ, применяемый в примере 1, часть А, проводят реакцию между 1,39 г (5,123 ммоля) 2-[2-(этоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]этанолом и бромистым пропаргилом (в виде 80%-ного раствора в толуоле) (1,7 мл, 15,37 ммоля), в результате которой получают 1,6 г маслообразного продукта - 2-(этоксиэтил)-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолина.
MS(Cl) для C18H19N3O2 m/z 310 (MH+), 371, 270.
Часть В
Используя основной способ, применяемый в примере 12, часть А, 1,5 г (4,13 ммоля) 2-(этоксиэтил)-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолина обрабатывают при 40°С иодбензолом (0,51 мл, 4,54 ммоля). Реакция завершается через 50 минут. Летучие продукты отгоняют при пониженном давлении и оставшийся маслообразный продукт распределяют между дихлорметаном и 5%-ным водным раствором бикарбоната натрия. Водный слой экстрагируют дихлорметаном. Органические фракции соединяют вместе, промывают рассолом, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении до получения коричневого маслообразного продукта. Этот продукт подвергают хроматографической очистке на силикагеле (элюент - смесь 95/5 дихлорметан/метанол) и в результате получают 1,25 г коричневого стеклообразного твердого 2-(этоксиэтил)-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолина.
MS(Cl) для С24Н23N3O2 m/z 386 (MH+), 342, 272.
Часть С
Используя основной способ, применяемый в примере 1, часть В, окисляют 0,655 г (1,70 ммоля) 2-(этоксиэтил)-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-хинолина и получают 0,68 г маслообразного 2-(этоксиэтил)-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-5N-оксида.
Часть D
Используя основной способ, применяемый в примере 115, часть С, проводят аминирование 0,682 г (1,700 ммоля) 2-(этоксиэтил)-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолин-5N-оксида. Образовавшийся в результате этой реакции коричневый твердый продукт подвергают хроматографической очистке на силикагеле (используя в качестве элюента смесь 98/2 дихлорметан/метанол), что приводит к получению 0,297 г 2-(этоксиэтил)-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолин-4-амина в виде белого гранулированного твердого вещества. Температура плавления этого продукта составляет 110,8-111,7°С.
Анализ. Рассчитано для C24H24N4O2.(H2O)0,1: %C 71,66; %Н, 6,06; %N, 13,93. Найдено: %C 71,56; %Н, 5,96; %N, 13,74.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,13 (дублет, J=7,8 Гц, 1Н), 7,63 (дублет, J=8,3 Гц, 1Н), 7,44 (триплет, J=6,8 Гц, 1Н), 7,28-7,36 (мультиплет, 3Н), 7,19-7,26 (мультиплет, 3Н), 6,67 (синглет, 2Н), 4,88 (триплет, J=5,4 Гц, 2Н), 4,38 (синглет, 2Н), 4,03 (триплет, J=5,9 Гц, 2Н), 3,55 (квинтет, J=6,8, 7,3 Гц, 2Н), 1,15 (триплет, J=6,8 Гц, 3Н)
Пример 123
2-Бутил-1-{2-[(3-фенилпроп-2-инил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин
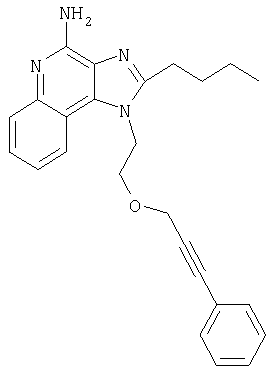
Часть А
Используя основной способ, применяемый в примере 1, часть А, проводят реакцию между 5,0 г (18,56 ммоля) 2-(2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)этанолом и бромистым пропаргилом (в виде 80%-ного раствора в толуоле) (6,3 мл, 55,62 ммоля), в результате которой получают 4,02 г твердого желтовато-коричневого продукта - 2-бутил-1-[2-(проп-2-инилокси)этил]-1H-имидазо[4,5-с]хинолина.
MS(Cl) для C19H21N3O m/z 308 (MH+), 268, 220.
Часть В
Используя основной способ, применяемый в примере 12, часть А, 4,0 г (13,08 ммоля) 2-бутил-1-[2-(проп-2-инилокси)этил]-1H-имидазо[4,5-хинолина при 90°С обрабатывают 1,6 мл (14,38 ммоля) иодбензола. Реакция завершается в течение 15 минут. Летучие продукты удаляют при пониженном давлении, а оставшийся маслообразный продукт подвергают хроматографической очистке на силикагеле (в качестве элюента используют смесь 98/2 дихлорметан/метанол), а затем перекристаллизовывают из смеси этилацетат/гексан. После очистки получают 3,1 г желтовато-коричневого твердого 2-бутил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолина.
Часть С
Используя основной способ, применяемый в примере 1, часть В, 1,0 г (2,61 ммоля) 2-бутил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолина окисляют до 1,0 г маслообразного 2-бутил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-5N-оксида.
Часть D
Используя основной способ, применяемый в примере 115, часть С, проводят аминирование 1,04 г (2,60 ммоля) 2-бутил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-5N-оксида. Образовавшийся в результате этой реакции твердый коричневый продукт обрабатывают диэтиловым эфиром, после чего дважды подвергают хроматографической очистке на силикагеле, используя в качестве элюента сначала смесь 8/2 дихлорметан/этилацетат, а затем смесь 98/2 дихлорметан/метанол. В результате очитки получают 0,450 г 2-бутил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина в виде белого порошка. Температура плавления этого продукта составляет 133-140°С.
Анализ. Рассчитано для С25Н26N4O.(Н2O)0,2: %С 74,67; %Н, 6,62; %N, 13,93. Найдено: %С 74,65; %Н, 6,60; %N, 14,00.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,08 (дублет, J=7,8 Гц, 1Н), 7,61 (дублет, J=7,3 Гц, 1Н), 7,41 (триплет, J=7,3 Гц, 1Н), 7,29-7,36 (мультиплет, 3Н), 7,17-7,24 (мультиплет, 3Н), 6,45 (синглет, 2Н), 4,78 (триплет, J=4,9 Гц, 2Н), 4,34 (синглет, 2Н), 4,01 (триплет, J=4,9 Гц, 2Н), 2,95 (триплет, J=8,3 Гц, 2Н), 1,81 (квинтет, J=7,3, 8,3 Гц, 2Н), 1,44 (секстет, J=7,3, 7,3, 7,8 Гц, 2Н), 0,93 (триплет, J=7,3 Гц, 3Н)
MS(Cl) для C25H26N4O m/z 399 (МН+), 283, 267.
Пример 124
2-Бутил-1-[2-(3-фенилпропокси)этил])-1Н-имидазо[4,5-с]хинолин-4-амин
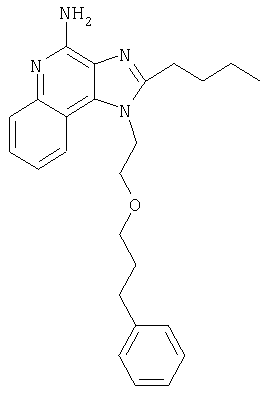
Часть А
Используя основной способ, применяемый в примере 12, часть В, осуществляют гидрирование 2-бутил-1-{2-[(3-фенилпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолина (2,4 г, 6,26 ммоля) до 2-бутил-1-[2-(3-фенилпропокси)этил]-1H-имидазо[4,5-с]хинолина (1,67 г), представляющего собой твердый белый продукт.
MS(Cl) для С25Н29N3О m/z 388 (MH+), 279.
Часть В
Используя основной способ, применяемый в примере 1, часть В, 1,68 г (4,34 ммоля) 2-бутил-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]хинолина окисляют до 1,75 г 2-бутил-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксида, представляющего собой стеклообразный твердый продукт.
MS(Cl) для С25Н29N3O2 m/z 404 (МН+), 388.
Часть С
Используя основной способ, применяемый в примере 115, часть С, проводят аминирование 1,75 г (4,34 ммоля) 2-бутил-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]-хинолин-5N-оксида. Образовавшийся в результате этой реакции твердый рыжеватый продукт перекристаллизовывают из ацетонитрила, что приводит к получению 0,572 г 2-бутил-1-[2-(3-фенилпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде желтовато-коричневого кристаллического твердого вещества. Температура плавления этого продукта составляет 80,8-81,3°С.
Анализ. Рассчитано для C25H30N4O.(H2O)0,3: %C 73,61; %Н, 7,56; %N, 13,73. Найдено: %С 73,3; %Н, 7,65; %N, 13,67.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,07 (дублет, J=8,3 Гц, 1Н), 7,62 (дублет, J=8,3 Гц, 1Н), 7,41 (триплет, J=7,3 Гц, 1Н), 7,21 (триплет, J=7,3 Гц, 1Н), 7,05-7,14 (мультиплет, 3Н), 6,89 (дублет, J=7,3 Гц, 2Н), 6,45 (синглет, 2Н), 4,74 (триплет, J=4,4 Гц, 2Н), 3,80 (триплет, J=4,9 Гц, 2Н), 3,24 (триплет, J=5,9 Гц, 2Н), 2,98 (триплет, J=7,8 Гц, 2Н), 2,39 (триплет, J=7,8 Гц, 2Н), 1,84 (квинтет, J=7,3, 8,3 Гц, 2Н), 1,62 (квинтет, J=7,8, 5,9 Гц, 2Н), 1,48 (секстет, J=7,3, 7,3, 7,8 Гц, 2Н), 0,95 (триплет, J=7,3 Гц, 3Н)
MS(Cl) для С25Н30N4O m/z 403 (МН+), 213.
Пример 125
1-[2-(Бензилокси)этил]-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин
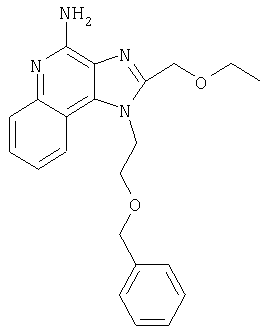
Часть А
Используя основной способ, применяемый в примере 1, часть В, проводят окисление 0,324 г (0,897 ммоля) 1-[2-(бензилокси)этил]-2-(этоксиметил)-1H-имидазо[4,5-с]хинолина, в результате чего получают 0,338 г 1-[2-(бензилокси)этил]-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-5N-оксида в виде коричневого масла.
Часть В
Используя основной способ, применяемый в примере 115, часть С, проводят аминирование 0,339 г (0,897 ммоля) 1-[2-(бензилокси)этил]-2-(этоксиметил)-1H-имидазо[4,5-с]хинолин-5N-оксида. Образовавшийся в результате этой реакции твердый желтовато-коричневый продукт перекристаллизовывают из ацетонитрила, что приводит к получению0,187 г 1-[2-(бензилокси)этил]-2-(этоксиметил)-1H-имидазо[4,5-с]хинолин-4-амина в виде белого порошка. Температура плавления этого продукта составляет 144,5-146,0°С.
Анализ. Рассчитано для C22H24N4O2: %С 70,19; %Н, 6,43; %N, 14,88. Найдено: %С 69,96; %Н, 6,29; %N, 15,09.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,08 (дублет, J=7,8 Гц, 1Н), 7,61 (дублет, J=8,3 Гц, 1Н), 7,43 (триплет, J=6,8 Гц, 1Н), 7,19-7,24 (мультиплет, 4Н), 7,11-7,14 (мультиплет, 2Н), 6,6 (синглет, 2Н), 4,87 (триплет, J=5,4 Гц, 2Н), 4,79 (синглет, 2Н), 4,44 (синглет, 2Н), 3,90 (триплет, J=5,4 Гц, 2Н), 3,90 (квинтет, J=6,8, 6,8 Гц, 2Н), 1,13 (триплет, J=6,8 Гц, 3Н)
MS(Cl) для C22H24N4O2 m/z 377 (MH+), 331, 241.
Пример 126
1-[2-(Бензилокси)этил]-2-бутил-1H-имидазо[4,5-с]хинолин-4-амин
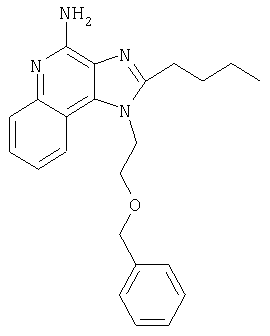
Часть А
Используя основной способ, применяемый в примере 1, часть В, проводят окисление 2,3 г (6,39 ммоля) 1-[2-(бензилокси)этил]-2-бутил-1H-имидазо[4,5-с]хинолина, в результате чего получают 2,4 г 1-[2-(бензилокси)этил]-2-бутил-1Н-имидазо[4,5-с]хинолин-5N-оксида в виде коричневого масла.
MS(Cl) для C23H25N3O2 m/z 376 (MH+), 360, 270.
Часть В
Используя основной способ, применяемый в примере 1, часть С, 2,4 г (6,39 ммоля) 1-[2-(бензилокси)этил]-2-бутил-1Н-имидазо[4,5-с]хинолин-5N-оксида обрабатывают трихлорацетилизоцианатом (1,45 г, 7,678 ммоля), в результате чего образуется 3,3 г коричневого маслообразного N-{1-[2-(бензилокси)этил]-2-бутил-1H-имидазо[4,5-с]хинолин-4-ил}-2,2,2-трихлорацетамида.
Часть С
3,3 г (6,39 ммоля) N-{1-[2-(бензилокси)этил]-2-бутил-1Н-имидазо[4,5-хинолин-4-ил}-2,2,2-трихлорацетамида подвергают гидролизу под действием метилата натрия (5 мл, 25%-ного раствора в метаноле) по способу, применяемому в примере 1, часть Д. Образовавшееся рыжеватое твердое вещество подвергают хроматографической очистке на силикагеле (элюент - смесь 98/2 дихлорметан/метанол), после чего перекристаллизовывают из метанола и сушат в вакууме при 60°С в течение 18 часов. В результате получают 0,174 г белого твердого 1-[2-(бензилокси)этил]-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амина, температура плавления которого составляет 133-135°С.
Анализ. Рассчитано для С23Н26N4О: %С 73,77; %Н, 7,00; %N, 14,96. Найдено: %С 73,51 ;%Н, 7,06; %N, 14,92.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,03 (дублет, J=7,3 Гц, 1Н), 7,60 (дублет, J=8,3 Гц, 1Н), 7,39 (триплет, J=6,8 Гц, 1Н), 7,17-7,24 (мультиплет, 4Н), 7,10-7,12 (мультиплет, 2Н), 6,45 (синглет, 2Н), 4,76 (триплет, J=5,4 Гц, 2Н), 4,41 (синглет, 2Н), 3,89 (триплет, J=4,9 Гц, 2Н), 2,94 (триплет, J=8,3 Гц, 2Н), 1,77 (квинтет, J=7,8, 7,8 Гц, 2Н), 1,40 (секстет, J=7,8, 7,3, 6,8 Гц, 2Н), 0,91 (триплет, J=7,3 Гц, 3Н)
MS(Cl) для C23H26N4O m/z 375 (MH+), 242,183.
Пример 127
1-[2-(Бензилокси)этил]-2-метил-1Н-имидазо[4,5-с]хинолин-4-амин
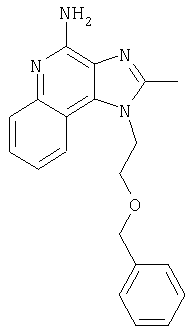
Часть А
Используя основной способ, применяемый в примере 1, часть В, проводят окисление 6 г (18,9 ммоля) 1-[2-(бензилокси)этил]-2-метил-1Н-имидазо[4,5-с]хинолина, в результате чего получают 6,3 г 1-[2-(бензилокси)этил}-2-метил-1Н-имидазо[4,5-с]хинолин-5N-оксида в виде коричневого твердого вещества.
Часть В
Используя основной способ, применяемый в примере 1, часть С, 6,3 г (18,9 ммоля) 1-[2-(бензилокси)этил]-2-метил-1Н-имидазо[4,5-с]хинолин-5N-оксида обрабатывают трихлорацетилизоцианатом (4,95 г, 26,27 ммоля), в результате чего образуется 10,4 г коричневого твердого N-{1-[2-(бензилокси)этил]-2-метил-1Н-имидазо[4,5-с]хинолин-4-ил}-2,2,2-трихлорацетамида.
Часть С
10,46 г (21,89 ммоля) N-{1-[2-(бензилокси)этил]-2-метил-1Н-имидазо[4,5-с]хинолин-4-ил}-2,2,2-трихлорацетамида подвергают гидролизу под действием метилата натрия (20 мл, 25%-ного раствора в метаноле) по способу, применяемому в примере 1, часть Д. Образовавшееся коричневое твердое вещество подвергают хроматографической очистке на силикагеле (элюент - смесь 98/2 дихлорметан/метанол) и сушат в вакууме при 60°С в течение 18 часов. В результате получают 1,036 г белого твердого 1-[2-(бензилокси)этил]-2-метил-1H-имидазо[4,5-с]хинолин-4-амина, температура плавления которого составляет 159-160°С.
Анализ. Рассчитано для C20H20N4O: %С 72,27; %Н, 6,06; %N, 16,85. Найдено: %С 72,17; %Н, 5,96; %N, 16,81.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,04 (дублет, J=7,3 Гц, 1Н), 7,59 (дублет, J=8,3 Гц, 1Н), 7,39 (триплет, J=8,3 Гц, 1Н), 7,15-7,27 (мультиплет, 4Н), 7,08-7,13 (мультиплет, 2Н), 6,49 (синглет, 2Н), 4,75 (триплет, J=5,4 Гц, 2Н), 4,43 (синглет, 2Н), 3,90 (триплет, J=5,4 Гц, 2Н), 2,61 (синглет, 3Н)
MS(Cl) для C20H20N4O m/z 333 (МН+), 243, 199.
Пример 128
1-[2-(Бензилокси)этил]-2-октил-1H-имидазо[4,5-c]хинолин-4-амин
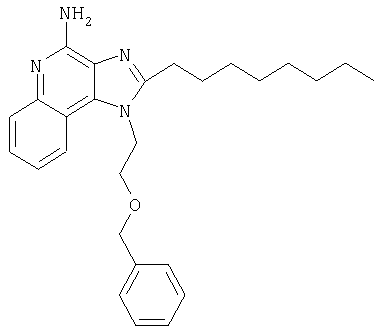
Часть А
Используя основной способ, применяемый в примере 1, часть В, проводят окисление 2,4 г (5,8 ммоля) 1-[2-(бензилокси)этил]-2-октил-1Н-имидазо[4,5-с]хинолина, в результате чего получают 2,5 г 1-[2-(бензилокси)этил]-2-октил-1H-имидазо[4,5-с]хинолин-5N-оксида в виде коричневого масла.
Часть В
Используя основной способ, применяемый в примере 115, часть С, проводят аминирование 2,50 г (5,80 ммоля) 1-[2-(бензилокси)этил]-2-октил-1Н-имидазо[4,5-с]хинолин-5N-оксида. Образовавшееся масло подвергают хроматограсрической очистке на силикагеле (элюент - смесь 98/2 дихлорметан/метанол) и перекристаллизовывают из ацетонитрила. В результате получают 0,75 г твердого белого 1-[2-(бензилокси)этил]-2-октил-1Н-имидазо[4,5-с]хинолин-4-амина, температура плавления которого составляет 110-111°С.
Анализ. Рассчитано для С27Н34N4O: %С 75,31; %Н, 7,96; %N, 13,01. Найдено: %С 75,20; %Н, 7,88; %N, 13,00.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,03 (дублет, J=7,8 Гц, 1Н), 7,60 (дублет, J=8,3 Гц, 1Н), 7,40 (триплет, J=7,3 Гц, 1Н), 7,17-7,26 (мультиплет, 4Н), 7,10-7,13 (мультиплет, 2Н), 6,45 (синглет, 2Н), 4,76 (триплет, J=4,9 Гц, 2Н), 4,41 (синглет, 2Н), 3,88 (триплет, J=4,9 Гц, 2Н), 2,93 (триплет, J=7,8 Гц, 2Н), 1,79 (квинтетет, J=7,3, 7,3 Гц, 2Н), 1,20-1,38 (мультиплет, 10Н), 0,85 (триплет, J=6,3 Гц, 3Н)
MS(Cl) для С27Н34N4O m/z 431 (МН+), 291, 214.
Пример 129
2-(2-Метоксиэтил)-1-(2-феноксиэтил)-1Н-имидазо[4,5-хинолин-4-амин
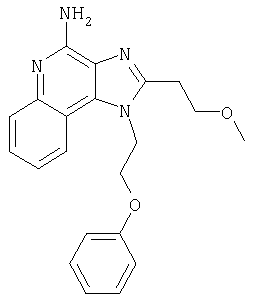
Часть А
17,6 мл (0,13 моля) 2-феноксиэтиламина по каплям в атмосфере азота добавляют к охлажденному на ледяной бане раствору 21,5 г (0,1 моля) 4-хлор-3-нитрохинолина и 21,5 мл (0,16 моля) триэтиламина в 500 мл дихлорметана. Реакционную смесь выдерживают в течение ночи при температуре окружающей среды, после чего добавляют к ней воду и отделяют водную фазу от органической фазы. Органическую фазу сушат над сульфатом магния, фильтруют и растворитель отгоняют в вакууме. К остатку добавляют гексан, и раствор охлаждают в холодильнике. Образовавшийся осадок удаляют с помощью вакуумной фильтрации и в результате получают 19,1 г 3-нитро-N-(2-фенилоксиэтил)хинолин-4-амина в виде твердого желтого вещества.
Часть В
В аппарат для проведения гидрирования загружают 6,0 г (19 ммолей) 3-нитро-N-(2-фенилоксиэтил)хинолин-4-амина, 1,5 г активированного угля с нанесенной на него платиной (5%) и 300 мл этилацетата. Смесь встряхивают в течение ночи при давлении водорода 40 фунт/дюйм2 (2,8 кг/см2), после чего смесь фильтруют и оставшийся на фильтре катализатор промывают этилацетатом. Фильтрат сушат над сульфатом магния, фильтруют и упаривают досуха в вакууме. К остатку добавляют гексан, и образовавшийся осадок отделяют с помощью вакуумной фильтрации. В результате получают 4,9 г бледно-желтого твердого N4-(2-фенилоксиэтил)хинолин-3,4-диамина.
Часть С
К охлажденному на ледяной бане раствору 2,0 г (7,2 ммоля) N4-(2-фенилоксиэтил)хинолин-3,4-диамина в 100 мл дихлорметана по каплям в течение 30 минут добавляют 0,86 мл (7,9 ммоля) хлорангидрида 3-метоксипропановой кислоты. Через несколько часов происходит образование осадка. Растворитель упаривают в вакууме почти досуха и добавляют 100 мл гексана. После вакуумной фильтрации получают 2,9 г солянокислой соли 3-метокси-N-{4-[(2-феноксиэтил)амино]хинолин-3-ил}пропанамида.
Часть D
Полученный в части С продукт (2,9 г) и 200 мл 7,5%-ного метанольного раствора аммиака загружают в автоклав. Автоклав закрывают и нагревают при 160°С в течение 6 часов. После этого реакционную смесь охлаждают до температуры окружающей среды и концентрируют в вакууме. Остаток распределяют между 150 мл дихлорметана и 150 мл воды. Отделяют водную и органическую фазы, и водную фазу экстрагируют 100 мл дихлорметана. Органические фракции соединяют вместе, сушат над сульфатом магния и фильтруют. Растворитель удаляют в вакууме и к остатку добавляют гексан до получения белого осадка. После вакуумной фильтрации получают 1,8 г белого твердого 2-(2-метоксиэтил)-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолина.
Часть Е
К раствору 1,8 г (5,2 ммоля) 2-(2-метоксиэтил)-1-(2-феноксиэтил)-1H-имидазо[4,5-с]хинолина в 100 мл хлороформа тремя порциями в течение 20 минут добавляют 1,5 г (8,7 ммоля) 60%-ной 3-хлорпероксибензойной кислоты. Реакционную смесь выдерживают в течение ночи при температуре окружающей среды, после чего промывают сначала насыщенным водным раствором бикарбоната натрия, а затем водой. Органический слой сушат над сульфатом магния и концентрируют в вакууме почти досуха. К остатку добавляют гексан и после вакуумной фильтрации образовавшегося осадка получают 1,6 г 2-(2-метоксиэтил)-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-5N-оксида в виде светло-желтого порошка.
Часть F
0,8 мл (6,6 ммоля) трихлорацетилизоцианата в атмосфере азота по каплям добавляют к раствору 1,6 г (4,4 ммоля) 2-(2-метоксиэтил)-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-5N-оксида в 100 мл дихлорметана, и реакционную смесь выдерживают при температуре окружающей среды в течение 2 часов. После этого к смеси добавляют 5 капель 7%-ного метанольного раствора гидроксида аммония и выдерживают смесь при температуре окружающей среды еще в течение 2,5 суток. По истечении этого времени добавляют 10%-ный раствор гидроксида натрия и разделяют водную и органическую фазы. Органическую фазу концентрируют и очищают способом флеш-хроматографии на силикагеле, используя в качестве элюента смесь 9:1 дихлорметан: метанол. Фракции, содержащие продукт, соединяют вместе, концентрируют в вакууме, растворяют в кипящем толуоле и обрабатывают активированным углем. Смесь фильтруют для удаления активированного угля и фильтрат охлаждают. Образовавшийся осадок отфильтровывают и после его сушки в вакуумной печи при 80°С получают 0,68 г 2-(2-метоксиэтил)-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде желтовато-коричневого порошка. Температура плавления этого продукта составляет 171,0-174,0°С.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,19 (дублет, J=8,1 Гц, 1Н), 7,64 (дублет, J=8,3 Гц, 1Н), 7,44 (триплет, J=7,5 Гц, 1Н), 7,29-7,20 (мультиплет, 3Н), 6,90 (триплет, J=7,4 Гц, 1Н), 6,82 (дублет, J=8,2 Гц, 2Н), 6,58 (синглет, 2Н), 5,01 (триплет, J=5,0 Гц, 2Н), 4,43 (триплет, J=5,0 Гц, 2Н), 3,87 (триплет, J=6,9 Гц, 2Н), 3,34 (синглет, 3Н), 3,30 (триплет, J=6,9 Гц, 2Н);
MS(Cl) m/e 363,1820 (363,1821, вычислено для С21Н23N4O2 М+Н);
Анализ. Рассчитано для C21H22N4O2: %С 69,59; %Н, 6,12; %N, 15,46. Найдено: %С 69,32; %Н, 6,17; %N, 15,48.
Пример 130
2-Изобутил-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин
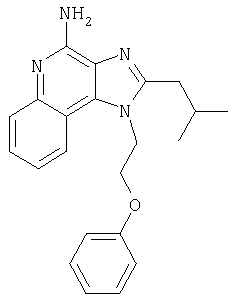
Смесь 1,5 г (5,4 ммоля) N4-(2-фенилоксиэтил)хинолин-3,4-диамина и 0,8 мл (6,4 ммоля) хлорангидрида изовалериановой кислоты обрабатывают таким же образом, как описано в примере 129, части С-Е. Образовавшийся в результате такой обработки продукт - 2-изобутил-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-5N-оксид (1,6 г, 4,5 ммоля) растворяют в 200 мл дихлорметана и к раствору добавляют 50 мл гидроксида аммония. Реакционную смесь охлаждают на ледяной бане и медленно в течение 20 мин добавляют к ней 0,85 г (4,5 ммоля) хлорангидрида п-толуолсульфоновой кислоты. Охлаждающую баню убирают, и смесь выдерживают при температуре окружающей среды в течение ночи. Разделяют водную и органическую фазы. Органическую фазу промывают последовательно 1%-ным водным раствором карбоната натрия (3 раза), водой и рассолом, после чего сушат над сульфатом натрия и упаривают в вакууме досуха. Добавляют гексан. Образовавшийся осадок собирают и перекристаллизовывают из ацетонитрила. В результате получают 0,96 г 2-изобутил-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде желтовато-коричневого порошка, температура плавления которого составляет 176,6-177,8°С.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,16 (дублет, J=8,2 Гц, 1Н), 7,63 (дублет, J=8,3 Гц, 1Н), 7,43 (триплет, J=7,6 Гц, 1Н), 7,28-7.20 (мультиплет, 3Н), 6,89 (триплет, J=7,3 Гц, 1Н), 6,81 (дублет, J=8,6 Гц, 2Н), 6,49 (синглет, 2Н), 4,98 (триплет, J=4,8 Гц, 2Н), 4,42 (триплет, J=4,8 Гц, 2Н), 2,89 (дублет, J=7,2 Гц, 2Н), 2,40-2,22 (мультиплет, 1Н), 1,02 (дублет, J=6,6 Гц, 6Н);
13С ЯМР(75 МГц, ДМСО-d6) 158,6, 153,9, 152,4, 145,5, 132,9, 130,1, 127,1, 126,9, 121,5, 120,8, 115,3, 114,7, 66,6, 44,4, 35,3, 27,1, 22,4;
MS(Cl) m/e 361,2017 (361,2028, вычислено для C22H25N4O М+Н);
Анализ. Рассчитано для C22H24N4O: %С 73,31; %Н, 6,71; %N, 15,54. Найдено: %С 73,33; %Н, 6,56; %N, 15,79.
Пример 131
2-Изопропил-1-(2-феноксиэтил)- 1Н-имидазо[4,5-с]хинолин-4-амин
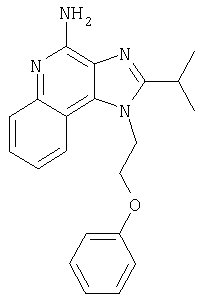
Смесь 2,0 г (7,2 ммоля) N4-(2-фенилоксиэтил)хинолин-3,4-диамина и 0,9 мл (8,6 ммоля) хлорангидрида изомасляной кислоты обрабатывают таким же образом, как описано в примере 130. Перекристаллизация образовавшегося продукта из ацетонитрила дает 0,82 г 2-изопропил-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде желтовато-коричневого порошка. Температура плавления этого продукта составляет 229-231°С.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,17 (дублет, J=7,5 Гц, 1Н), 7,65-7,62 (двойной дублет, J=8,3, 1,1 Гц, 1Н), 7,46-7,40 (двойной триплет, J=8,2, 1,1 Гц, 1Н), 7,29-7,20 (мультиплет, 3Н), 6,90 (триплет, J=7,3 Гц, 1Н), 6,81 (дублет, J=7,8 Гц, 2Н), 6,46 (синглет, 2Н), 5,01 (триплет, J=4,9 Гц, 2Н), 4,42 (триплет, J=4,9 Гц, 2Н), 3,54 (септет, J=6,8 Гц, 1Н), 1,41 (дублет, J=6,8 Гц, 6Н);
13С ЯМР(75 МГц, ДМСО-d6) 159,3, 158,5, 152,3, 145,4, 132,6, 130,1, 126,84, 126,78, 121,5, 120,7, 115,3, 114,6, 66,5, 44,1, 25,2, 21,8;
MS(Cl) m/e 347,1872 (347,1872, вычислено для C21H23N4O M+H);
Анализ. Рассчитано для C21H22N4O: %С 72,81; %Н, 6,40; %N, 16,17. Найдено: %С 72,48; %Н, 6,59; %N, 16,50.
Пример 132
2-Бутил-1-(2-феноксиэтил)-1H-имидазо[4,5-с]хинолин-4-амин
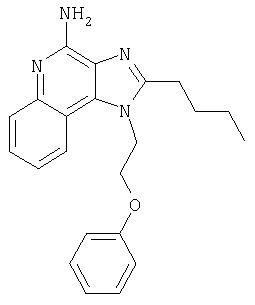
Смесь 2,0 г (7,2 ммоля) N4-(2-фенилоксиэтил)хинолин-3,4-диамина, 150 мл ксилола и 2,5 мл (14,3 ммоля) триметилортовалериата в атмосфере азота кипятят с обратным холодильником в течение 4 суток. После этого внешний нагрев увеличивают и отгоняют приблизительно 35 мл ксилола. Реакционную смесь медленно охлаждают до температуры окружающей среды, и в результате образуется осадок. После вакуумной фильтрации твердого продукта получают 2,4 г желтовато-коричневого твердого кристаллического 2-бутил-1-(2-феноксиэтил)-1H-имидазо[4,5-с]хинолина.
2-Бутил-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин обрабатывают таким же образом, как в примере 129, части Е и F. Перекристаллизация конечного продукта из ацетонитрила дает 0,93 г 2-бутил-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде белых игольчатых кристаллов. Температура плавления целевого продукта составляет 168,3-169,5°С.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,16 (дублет, J=8,1 Гц, 1Н), 7,63 (дублет, J=8,3 Гц, 1Н), 7,43 (триплет, J=7,6 Гц, 1Н), 7,28-7,20 (мультиплет, 3Н), 6,90 (триплет, J=7,4 Гц, 1Н), 6,82 (дублет, J=8,5 Гц, 2Н), 6,47 (синглет, 2Н), 4,97 (триплет, J=4,8 Гц, 2Н), 4,43 (триплет, J=4,8 Гц, 2Н), 3,00 (триплет, J=7,7 Гц, 2Н), 1,86 (мультиплет, 2Н), 1,47 (мультиплет, 2Н), 0,96 (триплет, J=7,3 Гц, 3Н);
13С ЯМР(75 МГц, ДМСО-d6) 158,5, 154,6, 152,3, 145,6, 132,9, 130,1, 126,8, 121,5, 120,7, 115,2, 114,6, 66,7, 44,4, 29,3, 26,2, 21,9, 13,6;
MS(Cl) m/e 361,2032 (361,2028, вычислено для C22H25N4O М+Н);
Анализ. Рассчитано для С22H24N4O: %С 73,31; %Н, 6,71; %N, 15,54. Найдено: %С 73,15;%H,6,69;%N,15,57.
Пример 133
1-(2-Феноксиэтил)-2-(феноксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин
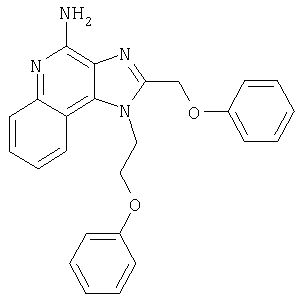
Используя методику, приведенную в примере 129, часть С, проводят реакцию между феноксиацетилхлоридом (1,2 мл, 8,6 ммоля) и N4-(2-феноксиэтил)хинолин-3,4-диамином (2,0 г, 7,2 ммоля). Образовавшийся на этой стадии продукт обрабатывают таким же образом, как и в примере 129, стадии D-F. Перекристаллизация конечного продукта из ацетонитрила дает 0,65 г желтовато-коричневого порошка, представляющего собой 1-(2-феноксиэтил)-2-(феноксиметил)-1Н-имидазо[4,5-с]хинолин-4-амин. Температура плавления этого продукта составляет 168,5-170,0°С.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,25 (дублет, J=7,9 Гц, 1Н), 7,64 (двойной дублет, J=8,3, 1,0 Гц, 1Н), 7,47 (мультиплет, 1Н), 7,38-7,14 (мультиплет, 7Н), 7,01 (триплет, J=7,3 Гц, 1Н), 6,89 (триплет, J=7,3 Гц, 1Н), 6,81 (дублет, J=7,8 Гц, 2Н), 6,69 (синглет, 2Н), 5,53 (синглет, 2Н), 5,29 (триплет, J=5,0 Гц, 2Н), 4,48 (триплет, J=5,0 Гц, 2Н);
13С ЯМР(75 МГц, ДМСО-d6) 158,5, 152,7, 149,2, 146,1, 134,1, 130,2, 130,1, 127,6, 127,0, 126,9, 122,0, 121,6, 121,5, 121,4, 115,3, 114,7, 66,6, 62,7, 45,0;
MS(Cl)m/e 411,1813 (411,1821, вычислено для С25Н23N4O2 М+Н);
Анализ. Рассчитано для C25H22N4O2: %С 73,15; %Н, 5,40; %N, 13,65. Найдено: %С 73,36; %Н, 5,30; %N, 13,66.
Пример 134
2-(4-Метоксибензил)-1-(2-феноксиэтил)-1H-имидазо[4,5-с]хинолин-4-амин
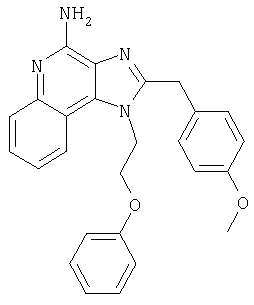
Используя методику, приведенную в примере 129, часть С, проводят реакцию между 4-метоксифенилацетилхлоридом (1,2 мл, 7,9 ммоля) и N4-(2-феноксиэтил)хинолин-3,4-диамином (2,0 г, 7,2 ммоля). Образовавшийся на этой стадии продукт обрабатывают таким же образом, как и в примере 129, стадии D-F. Перекристаллизация конечного продукта из ацетонитрила дает 1,1 г желтовато-коричневого твердого вещества, представляющего собой 2-(4-метоксибензил)-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин. Температура плавления этого продукта составляет 201,0-203,6°С.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,15 (дублет, J=8,1 Гц, 1Н), 7,63 (дублет, J=8,3 Гц, 1Н), 7,43 (триплет, J=7,6 Гц, 1Н), 7,26-7,18 (мультиплет, 5Н), 6,93-6,87 (мультиплет, 3Н), 6,74 (дублет, J=8,2 Гц, 2Н), 6,58 (синглет, 2Н), 4,89 (триплет, J=5,1 Гц, 2Н), 4,40 (синглет, 2Н), 4,24 (триплет, J=5,1 Гц, 2Н), 3,70 (синглет, 3Н);
MS(Cl) m/e 425,1948 (425,1978, вычислено для С26Н25N4O2 М+Н);
Анализ. Рассчитано для С26Н24N4O2: %С 73,57; %Н, 5,70; %N, 13,20. Найдено: %С 73,25; %Н, 5,93; %N, 13,06.
Пример 135
2-Циклопентил-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин
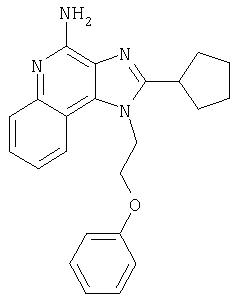
Используя методику, приведенную в примере 130, проводят реакцию между N4-(2-феноксиэтил)хинолин-3,4-диамином (2,0 г, 7,2 ммоля) и циклопентанкарбонилхлоридом (1,1 мл, 8,6 ммоля). Перекристаллизация конечного продукта из ацетонитрила дает 1,4 г желтовато-коричневого твердого вещества, представляющего собой 2-циклопентил-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин. Температура плавления этого продукта составляет 216,0-217,9°С.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,17 (дублет, J=8,1 Гц, 1Н), 7,63 (дублет, J=8,2 Гц, 1Н), 7,43 (триплет, J=7,6 Гц, 1Н), 7,28-7,20 (мультиплет, 3Н), 6,90 (триплет, J=7,3 Гц, 1Н), 6,81 (дублет, J=8,5 Гц, 2Н), 6,46 (синглет, 2Н), 5,02 (триплет, J=4,9 Гц, 2Н), 4,42 (триплет, J=4,9 Гц, 2Н), 3,60 (квинтет, J=8,2 Гц, 1Н), 2,18-1,67 (мультиплет, 8Н);
13С ЯМР(75 МГц, ДМСО-d6) 158,5, 158,3, 152,9, 144,6, 133,0, 130,1, 126,8, 121,5, 120,8, 115,3, 114,7, 66,5, 44,2, 36,1, 32,3, 25,3;
MS(Cl) m/e 373,2030 (373,2028, вычислено для С23Н25N4O М+Н);
Анализ. Рассчитано для С23Н24N4O: %С 74,17; %Н, 6,49; %N, 15,04. Найдено: %С 74,18; %Н, 6,59; %N, 15,08.
Пример 136
2-[(2-Метоксиэтокси)метил]-1-(2-феноксиэтил)-1H-имидазо[4,5-с]хинолин-4-амин
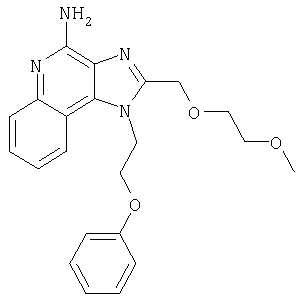
Используя методику, приведенную в примере 130, проводят реакцию между N4-(2-феноксиэтил)хинолин-3,4-диамином (2,0 г, 7,2 ммоля) и 2-(2-метоксиэтокси)-ацетилхлоридом (1,3 г, 8,6 ммоля). Перекристаллизация конечного продукта из метанола дает 1,6 г 2-[(2-метоксиэтокси)метил]-1-(2-феноксиэтил)-1Н-имидазо[4,5-финолин-4-амина в виде белых игольчатых кристаллов. Температура плавления этого продукта находится в интервале 170,0-171,5°С.
1H ЯМР(300 МГц, CDCl3) δ 8,06 (двойной дублет, J=8,3, 1,0 Гц, 1Н), 7,82 (двойной дублет, J=8,4, 1,0 Гц, 1Н), 7,55-7,50 (мультиплет, 1Н), 7,35-7,29 (мультиплет, 1Н), 7,26-7,18 (мультиплет, 2Н), 6,92 (триплет, J=7,4 Гц, 1Н), 6,79 (двойной дублет, J=8,7, 0,9 Гц, 2Н), 5,57 (синглет, 2Н), 5,07 (триплет, J=5,9 Гц, 2Н), 5,00 (синглет, 2Н), 4,47 (триплет, J=5,9 Гц, 2Н), 3,71 (мультиплет, 2Н), 3,55 (мультиплет, 2Н), 3,31 (синглет, 3Н);
13С ЯМР(75 МГц, ДМСО-d6) 158,9, 152,3, 150,3, 146,2, 135,2, 130,3, 128,3, 128,2, 127,6, 123,1, 122,2, 120,6, 116,1, 115,1, 72,1, 70,2, 66,6, 66,3, 59,3, 45,6;
MS(Cl) m/e 393,1912 (393,1927, вычислено для С22Н25N4O3 М+Н);
Анализ. Рассчитано для С22Н24N4O3: %С 67,33; %Н, 6,16; %N, 14,27. Найдено: %С 67,62; %Н, 6,24; %N, 14,37.
Пример 137
2-(Циклопропилметил)-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин
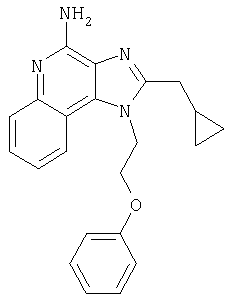
Используя методику, приведенную в примере 130, проводят реакцию между N4-(2-феноксиэтил)хинолин-3,4-диамином (1,7 г, 6,1 ммоля) и циклопропилацетилхлоридом (0,86 мл, 7,3 ммоля). Перекристаллизация конечного продукта из метанола дает 0,86 г белого твердого вещества, представляющего собой 2-(циклопропилметил)-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин. Температура плавления этого продукта составляет 191,7-192,6°С.
1H ЯМР(300 МГц, AMCO-d6) δ 8,17 (дублет, J=7,5 Гц, 1Н), 7,63 (двойной дублет, J=8,3, 1,1 Гц, 1Н), 7,46-7,41 (мультиплет, 1Н), 7,28-7,19 (мультиплет, 3Н), 6,89 (триплет, J=7,3 Гц, 1Н), 6,79 (дублет, J=7,8 Гц, 2Н), 6,49 (синглет, 2Н), 4,98 (триплет, J=5,0 Гц, 2Н), 4,42 (триплет, J=5,0 Гц, 2Н), 2,99 (дублет, J=6,7 Гц, 2Н), 1,40-1,26 (мультиплет, 1Н), 0,55 (мультиплет, 2Н), 0,32 (мультиплет, 2Н);
13С ЯМР(75 МГц, ДМСО-d6) 158,6, 154,1, 152,4, 145,5, 133,1, 130,1, 127,0, 126,9, 121,5, 120,8, 115,2, 114,7, 72,1, 66,6, 44,5, 31,1,10 9,0, 4,6;
Анализ. Рассчитано для С22Н22N4O. 0,1Н2O: %С 73,35; %Н, 6,21; %N, 15,55. Найдено: %С 73,23; %Н, 6,31; %N, 15,57.
Пример 138
2-(2-Циклопентилэтил)-1-(2-феноксиэтил)-1H-имидазо[4,5-с]хинолин-4-амин
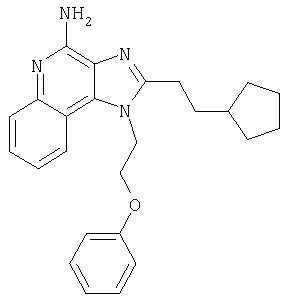
Используя методику, приведенную в примере 129, часть С, проводят реакцию между циклопентилпропионилхлоридом (1,3 мл, 8,6 ммоля) и N4-(2-феноксиэтил)хинолин-3,4-диамином (2,0 г, 7,2 ммоля). Полученный на последней стадии реакции продукт обрабатывают таким же образом, как указано в примере 129, части D-F. Перекристаллизация конечного продукта из ацетонитрила дает 0,44 г целевого вещества - 2-(циклопентилэтил)-1-(2-феноксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде белого порошка. Температура плавления этого продукта равна 165,0°С.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,17 (дублет, J=8,0 Гц, 1Н), 7,64 (двойной дублет, J=8,3, 0,8 Гц, 1Н), 7,44 (триплет, J=7,3 Гц, 2Н), 7,29-7,20 (мультиплет, 3Н), 6,90 (триплет, J=7,3 Гц, 1Н), 6,81 (дублет, J=7,9 Гц, 2Н), 6,60 (синглет, 2Н), 4,97 (триплет, J=4,6 Гц, 2Н), 4,44 (триплет, J=4,6 Гц, 2Н), 3,00 (триплет, J=7,6 Гц, 2Н), 1,91-1,77 (мультиплет, 5Н), 1,64-1,48 (мультиплет, 4Н), 1,20-1,14 (мультиплет, 2Н);
13С ЯМР(75 МГц, ДМСО-d6) 158,2, 155,0, 151,5, 144,7, 133,6, 129,9, 127,5, 127,4, 127,0, 122,6, 121,9, 119,5, 115,5, 114,5, 66,0, 45,7, 39,8, 32,3, 26,4, 24,9;
MS(Cl)m/e 401,2336 (401,2341, вычислено для C25H29N4O, M+H);
Анализ. Рассчитано для C25H18N4O. 0.1H2O: %C 74,97; %Н, 7,05; %N, 13,99. Найдено: %С 74,67; %Н, 7,11; %N, 13,97.
Пример 139
1-(2-Феноксиэтил)-2-тетрагидрофуран-3-ил-1H-имидазо[4,5-с]хинолин-4-амин
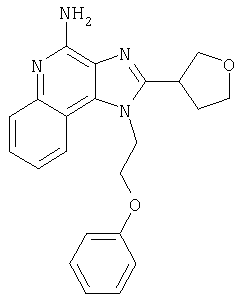
Используя методику, приведенную в примере 130, проводят реакцию между N4-(2-феноксиэтил)хинолин-3,4-диамином (1,6 г, 5,7 ммоля) и хлорангидридом тетрагидрофуран-3-карбоновой кислоты (0,98 мл, 7,3 ммоля). Перекристаллизация конечного продукта из ацетонитрила дает 0,3 г желтовато-коричневого твердого вещества, представляющего собой 1-(2-феноксиэтил)-2-тетрагидрофуран-3-ил-1H-имидазо[4,5-с]хинолин-4-амин. Температура плавления этого продукта составляет 235,9-36,3°С.
1H ЯМР(300 МГц, AMCO-d6) δ 8,18 (дублет, J=7,8 Гц, 1Н), 7,63 (двойной дублет, J=8,3, 1,0 Гц, 1Н), 7,44 (двойной триплет, J=7,6, 1,0 Гц, 1Н), 7,29-7,20 (мультиплет, 3Н), 6,90 (триплет, J=7,3 Гц, 1Н), 6,81 (дублет, J=798 Гц, 2Н), 6,49 (синглет, 2Н), 5,05 (триплет, J=4,9 Гц, 2Н), 4,42 (триплет, J=4,9 Гц, 2Н), 4,24 (мультиплет, 1Н), 4,04-3,98 (мультиплет, 3Н), 3,92-3,87 (мультиплет, 1Н), 2,50-2,30 (мультиплет, 2Н);
13С ЯМР(75 МГц, ДМСО-d6) 158,6, 155,2, 152,4, 145,5, 133,2, 130,1, 127,0, 126,9, 121,6, 120,3, 115,2, 114,7, 72,1, 68,0, 66,5, 44,4, 36,0, 32,4;
MS(Cl) m/e 375,1808 (375,1821, вычислено для С22Н23N4O2, М+Н);
Анализ. Рассчитано для C22H22N4O. 0,25H2O: %С 69,73; %Н, 5,98; %N, 14,78. Найдено: %С 769290 %Н, 5,91; %N, 14,90.
Пример 140
1-(2-Феноксиэтил)-2-фенил-1Н-имидазо[4,5-с]хинолин-4-амин
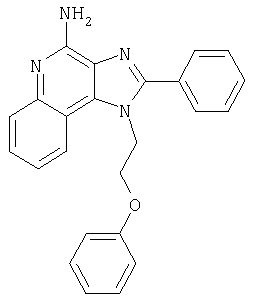
Используя методику, приведенную в примере 129, часть С, проводят реакцию между хлорангидридом бензойной кислоты (1,0 мл, 8,5 ммоля) и N4-(2-феноксиэтил)хинолин-3,4-диамином (2,0 г, 7,2 ммоля). Образовавшийся на этой стадии продукт обрабатывают таким же образом, как и в примере 129, стадии D-F. Перекристаллизация конечного продукта из метанола дает 0,74 г желтовато-коричневого твердого вещества, представляющего собой 1-(2-феноксиэтил)-2-фенил-1Н-имидазо[4,5-с]хинолин-4-амин. Температура плавления этого продукта находится в интервале 182,5-184,6°С.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,21 (дублет, J=7,9 Гц, 1Н), 7,83-7,79 (мультиплет, 2Н), 7,68-7,58 (мультиплет, 4Н), 7,48 (триплет, J=7,3 Гц. 1Н), 7,29 (триплет, J=7,3 Гц, 1Н), 7,16 (мультиплет, 2Н), 6,85 (триплет, J=7,3 Гц, 1Н), 6,68 (мультиплет, 4Н), 5,02 (триплет, J=5,1 Гц, 2Н), 4,33 (триплет, J=5,1 Гц. 2Н);
13С ЯМР(75 МГц, ДМСО-d6) 158,2, 153,6, 152,9, 146,0, 133,6, 131,1, 130,8, 130,3, 130,1, 129,3, 127,9, 127,5, 127,1, 121,9, 121,6, 121,2, 115,4, 114,7, 66,1, 45.6;
MS(Cl) m/e 381,1703 (381,1715, вычислено для C24H21N4O, M+H);
Анализ. Рассчитано для C24H20N4O. 0,25H2O: %C 74,88; %Н, 5,37; %N, 14,55. Найдено: %С 74,42; %Н, 5,10; %N, 14,48.
Пример 141
4-{[(2-(4-Амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутокси]метил}бензонитрил

Часть А
К перемешиваемой смеси 3,0 г (15,3 ммоля) α-бром-п-толуилнитрила, 40 мл 50%-ного раствора гидроксида натрия, 40 мл дихлорметана и 0,02 г (0,11 ммоля) бензилтриметиламмонийхлорида добавляют 3,0 г (12,4 ммоля) 2-(1Н-имидазо[4,5-с]хинолин-1-ил)-1-бутанола. Реакционную смесь выдерживают в течение 72 часов и затем разбавляют, добавляя к ней по 100 мл воды и дихлорметана. Разделяют водную и органическую фазы. Водную фазу экстрагируют дополнительным количеством (100 мл) дихлорметана. Органические фракции соединяют вместе, промывают водой, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток подвергают очистке с помощью флеш-хроматографии (силикагель, 9/1 дихлорметан/метанол, Rf 0,48). В результате получено 2,66 г 4-{[(2-(1Н-имидазо[4,5-с]хинолин-1-ил)бутокси]метил}бензонитрила.
Часть В
К раствору 2,6 г (7,3 ммоля) 4-{[(2-(1Н-имидазо[4,5-с]хинолин-1-ил)бутокси]-метил}бензонитрила в 70 мл хлороформа медленно добавляют 2,2 г (7,5 ммоля) 60%-ной 3-хлорпероксибензойной кислоты. Реакционную смесь выдерживают в течение 2 часов и затем промывают последовательно сначала насыщенным раствором бикарбоната натрия (200 мл) и дважды водой (по 100 мл). Промытый продукт сушат над сульфатом магния, фильтруют и концентрируют до получения 2,7 г соответствующего 5N-оксида.
Часть С
1,43 г (7,5 ммоля) хлорангидрида п-толуолсульфоновой кислоты медленно добавляют к охлажденной до 0°С смеси, содержащей продукт реакции, полученный в части В (2,7 г, 7,3 ммоля), 10 мл концентрированного раствора гидроксида аммония и 20 мл дихлорметана. Мониторинг реакции с помощью способа тонкослойной хроматографии (9:1 дихлорметан:метанол) показывает, что реакция завершается в течение нескольких минут. После этого реакционную смесь нагревают до температуры окружающей среды и разделяют водную и органическую фазы. Органическую фазу последовательно промывают водным раствором карбоната натрия (трижды), водой и рассолом, сушат над сульфатом натрия и концентрируют в вакууме. Очистка образовавшегося коричневого масла способом флеш-хроматографии (силикагель, 92/8 дихлорметан/метанол) с последующей многократной перекристаллизацией из смеси этилацетат/гексан дает 0,45 г желтовато-коричневого порошка 4-[[(2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)бутокси]метил}бензонитрила. Температура плавления очищенного продукта составляет 160,0-161,0°С.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,41 (синглет, 1Н), 8,20 (дублет, J=7,3 Гц, 1Н), 7,67 (мультиплет, 3Н), 7,44 (триплет, J=7,3 Гц, 1Н), 7,31-7,21 (мультиплет, 3Н), 6,72 (синглет, 2Н), 5,26 (широкий синглет, 1Н), 4,54 (синглет, 2Н), 4,02-3,91 (мультиплет, 2Н), 2,07 (мультиплет, 2Н), 0,87 (триплет, J=7,3 Гц, 3Н);
13С ЯМР(125 МГц, ДМСО-d6) 152,2, 145,2, 143,8, 140,1, 132,4, 132,0, 127,5, 126,6, 126,4, 121,0, 120,5, 118,7, 115,0, 110,0;
MS(El) m/e 371,1754 (371,1746, вычислено для C22H21N5O);
Анализ. Рассчитано для C22H21N5O: %С 71,14; %Н, 5,70; %N 18,85. Найдено: %С 70,78; %Н, 5,65; %N, 18,51.
Пример 142
4-({[(2R)-2-(4-Амино-1H-имидазо[4,5-хинолин-1-ил)бутил]окси}метил)бензонитрил
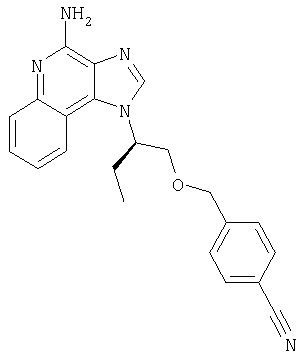
В соответствии с основной методикой, используемой в частях А и В примера 141, осуществляют превращение (2R)-2-(1H-имидазо[4,5-с]хинолин-1-ил)бутан-1-ола (1,36 г, 5,3 ммоля) в 1,60 г соответствующего 5N-оксидного продукта.
К раствору 1,60 г полученного 5N-оксида в 25 мл дихлорметана по каплям добавляют 0,77 мл (6,5 ммоля) трихлорацетилизоцианата. Реакционную смесь выдерживают в течение ночи и затем концентрируют в вакууме. Образовавшееся красное масло растворяют в 25 мл метанола и к раствору добавляют 4,0 мл 21%-ного раствора метилата натрия в метаноле. Смесь выдерживают в течение 2,5 суток. Растворитель удаляют в вакууме и оставшийся неочищенный продукт подвергают хроматографической очистке на силикагеле, используя смесь 92/8 дихлорметан/метанол в качестве элюента, а затем перекристаллизовывают из метилацетата. В результате получают белый твердый продукт, представляющий собой 4-{[((2R)-2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]окси}метил)-бензонитрил. Согласно данным жидкостной хроматографии (колонка: CHIRALCEL® OD-RH; элюент: 90/10/0,2 пентан/метанол/триэтиламин; скорость потока 2 мл/мин, время удерживания Rt 7,8 минуты) конечный продукт содержит более 99% энантиомера.
1H ЯМР(500 МГц, ДМСО-d6) δ 8,39 (синглет, 1Н), 8,20 (дублет, J=7,8 Гц, 1Н), 7,69 (дублет, J=8,1 Гц, 2Н), 7,63 (двойной дублет, J=8,3, 1,1 Гц, 1Н), 7,45-7,42 (мультиплет, 1Н), 7,31 (дублет, J=8,1 Гц, 2Н), 7,23 (мультиплет, 1Н), 6,58 (синглет, 2Н), 5,27 (широкий синглет, 1Н), 4,57 (синглет, 2Н), 4,03 (двойной дублет, J=10,3, 6,8 Гц, 1Н), 3,93 (двойной дублет, J=10,3, 3,9 Гц, 1Н), 2,09 (мультиплет, 2Н), 0,89 (триплет, J=7,3 Гц, 3Н);
Анализ. Рассчитано для C22H21N5O: %С 71,14; %Н, 5,70; %N 18,85. Найдено: %С 71,00; %Н, 5,66; %N, 18,64.
Пример 143
4-({[(2S)-2-(4-Амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]окси}метил)бензонитрил
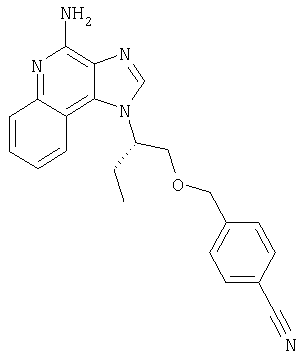
Превращение (2S)-2-(1Н-имидазо[4,5-с]хинолин-1-ил)бутан-1-ола (1,3 г) до целевого продукта проводят в соответствии с основной методикой, используемой в примере 142. Перекристаллизация конечного продукта из смеси этилацетат/гексан дает 0,2 г твердого белого 4-{[((2S)-2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)бутил]окси}метил)бензонитрила. Согласно данным жидкостной хроматографии (колонка: CHIRALCEL® OD-RH; элюент: 90/10/0,2 пентан/метанол/триэтиламин; скорость потока 2 мл/мин, время удерживания Rt 7,8 минуты) конечный продукт содержит более 99% энантиомера.
1H ЯМР(500 МГц, ДМСО-d6) δ 8,40 (синглет, 1Н), 8,20 (дублет, J=8,0 Гц, 1Н), 7,70 (дублет, J=8,2 Гц, 2Н), 7,63 (двойной дублет, J=8,3, 1,1 Гц, 1Н), 7,46-7,41 (мультиплет, 1Н), 7,31 (дублет, J=8,2 Гц, 2Н), 7,23 (мультиплет, 1H). 6,62 (синглет, 2Н), 5,27 (широкий синглет, 1Н), 4,57 (синглет, 2Н), 4,04 (двойной дублет, J=10,3, 6,7 Гц, 1Н), 3,93 (двойной дублет, J=10,3, 3,9 Гц, 1Н), 2,10 (мультиплет, 2Н), 0,88 (триплет, J=7,3 Гц, 3Н);
Анализ. Рассчитано для С22Н21N5О: %С 71,14; %Н, 5,70; %N 18,85. Найдено: %С 71,10; %Н, 5,98; %N, 18,96.
Пример 144
2-(2-Метоксиэтил)-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
10 мл (89,8 ммоля) пропаргилбромида (в виде 80%-ного раствора в толуоле) и 0,60 г (3,2 ммоля) бензилтриметиламмонийхлорида растворяют в 130 мл дихлорметана. Раствор обрабатывают 130 мл 50%-ного водного раствора гидроксида натрия и затем добавляют к нему 20,0 г (73,7 ммоля) 2-[2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]этанола. Смесь интенсивно перемешивают в течение 18 часов. Проведенный после этого анализ реакционной смеси способом тонкослойной хроматографии (9/1 хлороформ/метанол) указывают на полное завершение реакции. Реакционную смесь разбавляют 200 мл воды и разделяют водную и органическую фазы. Водную фракцию экстрагируют дополнительным количеством дихлорметана (3 порции по 150 мл). Органические фракции соединяют вместе, промывают 100 мл рассола, сушат над сульфатом натрия, отфильтровывают и концентрируют до получения 22,7 г 2-(2-метоксиэтил)-1-[2-(проп-2-инлокси)этил]-1Н-имидазо[4,5-с]хинолина в виде оранжевого твердого вещества.
1H ЯМР(300 МГц, ДМСО-d6) δ 9,15 (синглет, 1Н), 8,40 (мультиплет, 1Н), 8,15 (мультиплет, 1Н), 7,73-7,64 (мультиплет, 2Н), 4,89 (триплет, J=5,3 Гц, 2Н), 4,10 (дублет, J=2,4 Гц, 2Н), 3,95 (триплет, J=5,1 Гц, 2Н), 3,89 (триплет, J=6,9 Гц, 2Н), 3,36 (триплет, J=6,9 Гц, 2Н), 3,32 (синглет, 3Н), 3,27 (триплет, J=6,9 Гц, 2Н),
Часть В
22,7 г (73,4 ммоля) 2-(2-метоксиэтил)-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолина растворяют в 300 мл хлороформа и раствор охлаждают на ледяной бане. К охлажденному раствору малыми порциями в течение 30 минут добавляют 17,0 г (127,9 ммоля) 77%-ной 3-хлорпероксибензойной кислоты. Анализ способом тонкослойной хроматографии (9/1 хлороформ/метанол), проведенный через 30 минут после окончания добавления кислоты, указывает на наличие в реакционной смеси некоторого количества исходного материала. Поэтому к реакционной смеси добавляют еще 7,00 г (52,7 ммоля) 77%-ной 3-хлорпероксибензойной кислоты. Через 2 часа реакционную смесь нагревают до температуры окружающей среды и добавляют к ней 100 мл насыщенного раствора бикарбоната натрия. Разделяют водную и органическую фазы. Водный слой экстрагируют дополнительным количеством хлороформа (2 порции по 50 мл). Объединенные органические фракции промывают 100 мл воды, 100 мл рассола, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме до получения темно-оранжевого твердого вещества. Согласно данным ПМР-спектроскопии этот неочищенный продукт содержит менее 5% 3-хлорбензойной кислоты. В дальнейшем его используют без дополнительной очистки.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,56 (синглет, 1Н), 8,33 (дублет, J=7,7 Гц, 1Н), 7,99 (дублет, J=7,3 Гц, 1Н), 7,38-7,30 (мультиплет, 2Н), 4,40 (триплет, J=4,8 Гц, 2Н), 3,63 дублет, J=2,1 Гц, 2Н), 3,47 (триплет, J=4,9 Гц, 2Н), 3,40 (триплет, J=6,9 Гц, 2Н), 2,88 (триплет, J=2,0 Гц, 2Н), 2,84 (синглет, 3Н), 2,78 (триплет, J=6,3 Гц, 2Н),
Часть С
1,57 г (4,83 г ммоля) 2-(2-метоксиэтил)-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксида в атмосфере азота растворяют в 25 мл дихлорметана и к раствору с помощью шприца по каплям добавляют 0,80 мл (6,71 ммоля) трихлорацетилизоцианата. Реакционную смесь перемешивают в течение часа, после чего в вакууме удаляют летучие продукты. Остаток обрабатывают 15 мл метанола и в результате получают оранжевую суспензию. К этой суспензии с помощью шприца медленно добавляют 25%-ный раствор метилата натрия в метаноле. Суспензия превращается в темно-оранжевый раствор. Через 1,5 часа реакционную смесь медленно добавляют к 10 мл насыщенного раствора хлористого аммония. Метанол отгоняют в вакууме. Оставшийся водный раствор экстрагируют тремя порциями дихлорметана (по 10 мл каждая), и органические фракции соединяют вместе и промывают 10 мл воды и 10 мл рассола, после чего сушат над сульфатом натрия, фильтруют и концентрируют в вакууме до получения неочищенного оранжевого твердого вещества. Перекристаллизация из пропилацетата дает 0,78 г беловатых кристаллов 2-(2-метоксиэтил)-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина.
1H ЯМР(300 МГц, ДМСО-d6) δ 8.05 (дублет, J=7,3 Гц, 1Н), 7,61 (дублет, J=7,2 Гц, 1Н), 7,42 (триплет, J=7,8 Гц, 1Н), 7,23 (триплет, J=7,3 Гц, 1Н), 6,44 (широкий синглет, 2Н), 3,63 дублет, J=2,1 Гц, 2Н), 4,78 (триплет, J=5,2 Гц, 2Н), 4,11 (дублет, J=2,5 Гц, 2Н), 3,91 (триплет, J=5,5 Гц, 2Н), 3,83 (триплет, J=6,7 Гц, 2Н), 3,37 (триплет, J=2,6 Гц, 1Н), 3,30 (синглет, 3Н), 3,20 (триплет, J=6,8 Гц, 2Н);
MS(Cl) m/e 325 (M+H);
Анализ. Рассчитано для C18H20N4O2: %C 66,65; %Н, 6,21; %N 17,27. Найдено: %С 66,34; %H, 6,05; %N, 16,96.
Пример 145
2-Метил-1-(2-{[(2Е)-3-фенилпроп-2-енил]окси}этил)-1H-имидазо[4,5-с]хинолин-4-амин
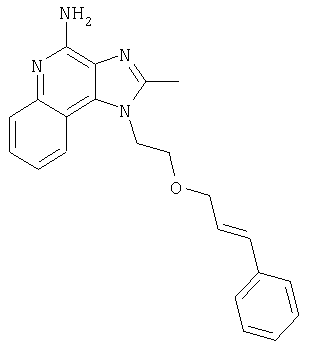
Часть А
Используя основной способ синтеза, применяемый в примере 1, часть В, проводят окисление 12,0 г (44,56 ммоля) 2-(2-метил-1Н-имидазо[4,5-с]хинолин-1-ил)этилацетата до 8,7 г 2-(2-метил-5-оксидо-1H-имидазо[4,5-с]хинолин-1-ил)этилацетата, представляющего собой коричневое твердое вещество. Этот материал используют в дальнейшем без дополнительной очистки.
Часть В
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 8,7 г (30,49 ммоля) 2-(2-метил-5-оксидо-1Н-имидазо[4,5-с]хинолин-1-ил)этилацетата, 80 мл безводного диметилформамида и 100 мл безводного толуола. К этой смеси коричневого цвета с помощью шприца при температуре окружающей среды добавляют 3,1 мл оксихлорида фосфора. Реакция протекает в течение нескольких минут, при этом наблюдается слабый разогрев реакционной смеси. Полностью реакция завершается через 30 минут. Летучие продукты удаляют при пониженном давлении. Оставшийся коричневый твердый продукт распределяют между дихлорметаном и 4%-ным водным раствором бикарбоната натрия, имеющим рН около 8. Водный слой 5 раз экстрагируют дихлорметаном. Органические фракции объединяют вместе, сушат над безводным сульфатом натрия, концентрируют при пониженном давлении, после чего продукт оставляют сушить при температуре окружающей среды в вакууме в течение ночи. В результате получают 9,2 г 2-(4-хлор-2-метил-1Н-имидазо[4,5-с]хинолин-1-ил)этилацетата в виде коричневого масла.
MS(Cl) для C15H14ClN3O2 m/z 304 (МН+), 262, 218.
Часть С
В круглодонную колбу, снабженную мешалкой, загружают 9,2 г (30,5 ммоля) 2-(4-хлор-2-метил-1Н-имидазо[4,5-с]хинолин-1-ил)этилацетата, 200 мл метанола и 0,4 г (3,0 ммоля) карбоната калия. Реакция полностью завершается при перемешивании при 26°С в течение 5 часов. Полученный раствор распределяют между рассолом и хлороформом. Органический слой удаляют, а водный слой шесть раз экстрагируют хлороформом. Органические фракции соединяют вместе, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении до получения общего объема смеси около 200 мл. При достижении такого объема наблюдается кристаллизация продукта. Колбу с реакционной смесью закрывают и выдерживают при температуре окружающей среды в течение 24 часов. Образующиеся мелкие белые кристаллы отфильтровывают и в результате получают 4,49 г 2-(4-хлор-2-метил-1H-имидазо[4,5-с]хинолин-1-ил)этанола.
MS(Cl) для С13Н12CIN3О m/z 262 (MH+), 218.
Часть D
В круглодонную колбу, снабженную мешалкой, загружают 3,9 г (14,9 ммоля) 2-(4-хлор-2-метил-1Н-имидазо[4,5-с]хинолин-1-ил)этанола, 125 мл дихлорметана, 125 мл 50%-ного водного раствора гидроксида натрия и 0,55 г (0,003 ммоля) бензилтриметиламмонийхлорида. Реакционную смесь интенсивно перемешивают при температуре окружающей среды, после чего добавляют к ней 8,8 г (44,71 ммоля) твердого бромангидрида коричной кислоты. Через 45 минут раствор становится прозрачным, и реакция завершается. Раствор выливают в 200 мл ледяной воды и органический слой отделяют. Водный слой четыре раза экстрагируют дихлорметаном. Органические фракции соединяют вместе, промывают рассолом, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Оставшееся оранжевое масло подвергают хроматографической очистке на силикагеле (в качестве элюента используют сначала дихлорметан, а затем смесь 98/2 дихлорметан/метанол). Полученное после такой очистки масло обрабатывают диэтиловым эфиром, и после фильтрования образовавшегося твердого вещества получают 4,22 г белого твердого 4-хлор-2-метил-1-(2-{[(2Е)-3-фенилпроп-2-енил]окси}этил)-1Н-имидазо[4,5-с]хинолина.
MS(Cl) для С22Н20CIN3О m/z 378 (МН+), 262, 228.
Часть Е
2,12 г (5,61 ммоля) 4-хлор-2-метил-1-(2-{[(2Е)-3-фенилпроп-2-енил]окси}этил)-1Н-имидазо[4,5-с]хинолина вместе с 70 мл 7%-ного метанольного раствора аммиака помещают в автоклав и в течение 16,5 часов нагревают при 150°С, после чего смесь охлаждают до температуры окружающей среды. Проведенный на этой стадии анализ указывает на то, что реакция не завершена полностью. Раствор концентрируют при пониженном температуре до общего объема около 10 мл, разбавляют 50 мл 7%-ного метанольного раствора аммиака и для завершения реакции нагревают в автоклаве при 150°С еще в течение 8,5 часов. По окончании нагревания раствор распределяют между дихлорметаном и насыщенным водным раствором бикарбоната натрия. Органическую фазу отделяют. Водный слой насыщают хлористым натрием и затем трижды экстрагируют дихлорметаном. Органические фракции объединяют вместе, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Оставшийся твердый коричневый продукт перекристаллизовывают из метанола и в результате получают 0,963 г твердого белого 2-метил-1-(2-{[(2Е)-3-фенилпроп-2-енил]окси}этил)-1H-имидазо[4,5-с]хинолин-4-амина. Температура плавления этого вещества составляет 111,8-112,5°С.
Анализ. Рассчитано для C22H22N4O: %С 73,72; %Н, 6,19; %N 15,63. Найдено: %С 73,48; %Н, 6,25; %N, 15,57.
1H ЯМР(300 МГц, ДМСО-d6) δ 8,08 (дублет, J=7,5 Гц, 1Н), 7,61 (дублет, J=8,1 Гц, 1Н), 7,40 (триплет, J=5,6 Гц, 1Н), 7,18-7,30 (мультиплет, 6Н), 6,51 (синглет, 2Н), 6,31 (дублет, J=16,2 Гц, 1Н), 6,17 (двойной триплет, J=15,6, 5,3 Гц, 1Н), 4,76 (триплет, J=5,0 Гц, 2Н), 4,05 (дублет, J=3,9 Гц, 2Н), 3,91 (триплет, J=5,6 Гц, 2Н), 2,64 (синглет, 3Н)
MS(Cl) для C22H22N4O m/z 259 (MH+), 243, 199.
Пример 146
2-Метил-1-{2-[(3-фенилпроп-2-инил]окси}этил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
Используя основной способ синтеза, применяемый в примере 115, часть С, в 2-(2-метил-5-оксидо-1Н-имидазо[4,5-с]хинолин-1-ил)этилацетат (8,47 г, 29,71 ммоля) вводят 4-аминогруппу. Образовавшееся коричневое масло обрабатывают ацетонитрилом и сушат, в результате чего получают 3,583 г желтовато-коричневого твердого 2-(4-амино-2-метил-1Н-имидазо[4,5-с]хинолин-1-ил)этилацетата.
MS(Cl) для C15H16ClN4O2 m/z 285 (МН+), 270, 199.
Часть В
В аппарат Парра загружают 3,61 г (12,64 ммоля) 2-(4-амино-2-метил-1Н-имидазо[4,5-с]хинолин-1-ил)этилацетата и 50 мл трифторуксусной кислоты и смесь продувают азотом. К полученному раствору добавляют 0,5 г оксида платины (IV). Процесс гидрирования завершается при температуре окружающей среды в течение 13 дней. Образовавшийся раствор фильтруют, и летучие продукты удаляют при пониженном давлении. Оставшееся коричневое масло распределяют между дихлорметаном и насыщенным водным раствором бикарбоната натрия, имеющим рН около 8. Разделяют органический и водный слои. Водный слой четыре раза экстрагируют дихлорметаном. Объединенные органические фракции сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Оставшийся белый твердый продукт перекристаллизовывают из смеси этилацетат/метанол (9/1) и после сушки получают 0,98 г твердого белого 2-(4-амино-2-метил-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)этанола.
MS(Cl) для C13H18N4O m/z 247 (MH+), 203.
Часть С
Используя основной способ синтеза, применяемый в примере 1, часть А, проводят реакцию между 0,763 г (3,098 ммоля) 2-(4-амино-2-метил-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)этанола и 1,1 мл (9,29 ммоля) 80%-ного раствора бромистого пропаргила в толуоле и в результате получают 0,42 г коричневого маслообразного 2-метил-1-[2-(проп-2-инилокси)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина.
MS(Cl) для C16H20N4O m/z 285 (MH+), 247, 183.
Часть D
Используя основной способ синтеза, применяемый в примере 12, часть А, при температуре окружающей среды проводят реакцию между 0,396 г (1,392 ммоля) 2-метил-1-[2-(проп-2-инилокси)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина и 0,17 мл (1,532 ммоля) иодбензола. После выдержки реакционной смеси в течение 18 часов реакция еще не была полностью завершена. Для завершения процесса раствор дополнительно нагревают в течение 3 часов при 50°С. Летучие продукты удаляют при пониженном давлении. Оставшееся после отгонки масло распределяют между дихлорметаном и 4%-ным водным раствором карбоната натрия. Органический слой отделяют. Водный слой трижды экстрагируют дихлорметаном. Объединенные органические фракции сушат над безводным сульфатом натрия, и летучие продукты удаляют при пониженном давлении. Оставшийся маслообразный продукт подвергают хроматографической очистке на силикагеле (элюент - смесь 95/5 дихлорметан/метанол). Полученный белый твердый продукт растворяют в 2 мл дихлорметана и раствор обрабатывают 2 мл 1М HCl в эфире. Летучие продукты отгоняют в вакууме и оставшийся твердый продукт перекристаллизовывают из метанола, в результате чего получают 0,1089 г солянокислой соли 2-метил-1-{2-[(3-фенилпроп-2-инил]окси}этил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина (HCl)1,9 в виде твердого желтовато-коричневого вещества.
Анализ. Рассчитано для C22H24N4O.(HCl)1,9.(H2O)0,7: %С, 59,74; %Н, 6,22; %N, 12,67; %Cl, 15,23. Найдено: %С, 59,72; %Н, 6,04; %N, 12,65; %Cl, 14/99.
1H ЯМР(300 МГц, ДМСО-d6) δ 7,93 (синглет, 2Н), 7,36-7,40 (мультиплет, 3Н), 7,28-7,30 (мультиплет, 2Н), 4,56 (триплет, J=5,0 Гц, 2Н), 4,35 (синглет, 2Н), 3,88 (триплет, J=5,3 Гц, 2Н), 2,92 (синглет, 3Н), 2,69 (синглет, 2Н), 2,60 (синглет, 3Н), 1,73 (синглет, 4Н)
MS(Cl) для C22H24N4O m/z 361 (MH+), 247, 199.
ОБРАЗОВАНИЕ ЦИТОКИНА В КЛЕТКАХ ЧЕЛОВЕКА
Определение активности полученных в изобретении соединений для стимулирования образования цитокина проводили с использованием клеток крови человека in vitro. Активность оценивали, исходя из количества интерферона и фактора-α некроза опухоли (IFN и TNF соответственно), секретируемых в культуральную среду, как описано в статье Тестермана с сотр. «Индукция кинина с помощью иммуномодуляторов Imiquimod и S-27609», опубликованной в журнале «Journal of Leukocyte Biology», 58, 365-372 (Сентябрь, 1995).
Подготовка культуры на основе клеток крови
Цельную кровь собирали венепункцией от здоровых доноров в специальные вакуумные контейнеры, соответствующие требованиям EDTA. Одноядерные клетки периферийной крови (РВМС) отделяли от цельной крови с помощью способа градиентного центрифугирования с использованием аппарата Histopaque®-1077. РВМС дважды промывали сбалансированным солевым раствором Hank (Ханка) и затем суспендировали в среде RPMI. Концентрации клеток крови в такой суспензии составляла (3-4)×106 клеток на 1 мл суспензии. Суспензию РВМС помещали в плоскодонные стерильные планшеты с 48 лунками (поставляемых компанией Costar, Кэмбридж, шт. Массачусетс или Becton Dickinson Labware, Линкольн Парк, шт. Нью-Джерси), в которых находились равные объемы среды RPMI, содержащей исследуемое соединение.
Подготовка соединения
Исследуемые соединения солюбилизировали в диметилсульфоксиде (ДМСО). Конечная концентрация ДМСО в приготовленных образцах, используемых для испытания, не должна превышать 1%..
Инкубация
Раствор применяемого для испытания соединения (60 мкмоль) вводили сначала в первую лунку, в которой находилась полная среда RPMI, а в остальные лунки вводили растворы после серийных трехкратных разбавлений. После этого добавляли суспензию РМВС в равных объемах с тем, чтобы концентрация исследуемого вещества находилась в заданных пределах (0,12-30 мкмоль). Конечная концентрация суспензии РМВС составляла (1,5-2)×106 клеток/мл. Лунки закрывали стерильными пластмассовыми крышками, аккуратно перемешивали образцы и выдерживали их в течение 18-24 часов при 37°С в атмосфере, содержащей 5% углекислого газа.
Отделение
После окончания инкубационного периода образцы подвергали центрифугированию в течение 5-10 минут при скорости вращения центрифуги 1000 об/мин (приблизительно 200 г) и температуре 4°С. Очищенный раствор, не содержащий клеток культуры, отбирали с помощью стерильной полипропиленовой пипетки и переносили в стерильную полипропиленовую ампулу. Образцы до проведения анализа хранили при температуре от -30 до -70°С. Образцы анализировали на интерферон-α и фактор-α некроза опухоли с помощью способа ELISA
Анализ интерферона (α) и фактора-α некроза опухоли по способу ELISA
Концентрацию интерферона (α) определяли по способу ELISA, используя набор человеческих мульти-частиц, полученный в PBL биомедицинской лаборатории, Нью-Брунсвик, шт. Нью-Джерси. Этот способ анализа позволяет определить содержание интерферона в единицах пикограмм/мл.
Концентрацию фактора-α некроза опухоли определяли с помощью набора ELISA, поставляемого фирмой Genzyme, Кембридж, шт. Массачусетс; R&D Systems, Миннеаполис, шт. Миннесота; или фирмой Pharmingen, Сан-Диего, шт. Калифорния. Этот способ анализа позволяет определить содержание интерферона в единицах пикограмм/мл.
В таблице 8 приведены минимальные концентрации соединений, при которых наблюдается образование интерферона и фактора некроза опухоли. Знак «*» означает, что при любой исследованной концентрации добавленного соединения образования интерферона и фактора некроза опухоли не наблюдалось. Обычно максимальная исследованная концентрация исследуемого вещества составляла 10 или 30 мкмоль.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2001 |
|
RU2315049C2 |
| ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2248975C2 |
| МОЧЕВИНОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2265020C2 |
| АМИДОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2000 |
|
RU2295343C2 |
| СОЕДИНЕНИЕ, ИМЕЮЩЕЕ ИНГИБИРУЮЩУЮ АКТИВНОСТЬ В ОТНОШЕНИИ КИНАЗ | 2021 |
|
RU2831403C1 |
| Способ получения производных 1,3-дигидро-2Н-имидазо/4,5-в/хинолин-2-онов или их фармацевтически приемлемых солей | 1986 |
|
SU1450746A3 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ НЕЙРОПРОТЕКТОРНЫМ ДЕЙСТВИЕМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2802457C1 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНЫ | 2001 |
|
RU2294934C2 |
| Способ получения производного хинолона | 1990 |
|
SU1836367A3 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ 2-АМИНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2795914C1 |
Изобретение относится к арилэфирзамещенным имидазохинолинам и тетрагидроимидазохинолинам, которые могут быть использованы для индуцирования биосинтеза цитокинов. Описываются имидазохинолины, имеющие формулу (I):
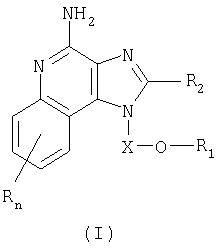
где Х представляет собой -CHR3-; R1 выбран из группы, включающей: - арил; - R4-арил; R2 выбран из группы, включающей: - атом водорода; - алкил; - арил; - тетрагидрофуран; - алкил-Y-алкил; - алкил-Y-арил; и - алкил, замещенный одним или большим количеством заместителей, выбранных из арила; R4 представляет собой алкильную или алкенильную группу, которая может быть разделена одной или несколькими -O-группами; каждый R3 представляет собой независимо друг от друга атом водорода или С1-10алкильную группу; каждая группа Y независимо друг от друга представляет собой -О; значение п равно 0; и каждый R независимо выбран из группы, состоящей из C1-10 алкильной, С1-10алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы; или соль фармацевтического качества на основе этих групп. Также описываются фармацевтические составы для индуцирования биосинтеза цитокинов, способы лечения вирусного заболевания, способы лечения опухолевого заболевания. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 20 н. и 19 з.п. ф-лы, 8 табл.
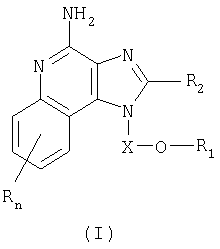
где Х представляет собой -CHR3-;
R1 выбран из группы, включающей арил; R4-арил;
R2 выбран из группы, включающей атом водорода; алкил; арил; тетрагидрофуран; алкил-Y-алкил; алкил-Y-арил; и алкил, замещенный одним или большим количеством заместителей, выбранных из арила;
R4 представляет собой алкильную или алкенильную группу, которая может быть разделена одной или несколькими -O-группами;
каждый R3 представляет собой независимо друг от друга атом водорода или С1-10алкильную группу;
каждая группа Y представляет собой -О;
значение n равно 0;
каждый R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
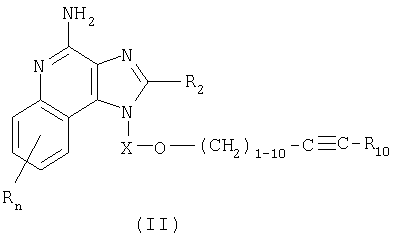
где X представляет собой -CHR3- или -CHR3-алкильную группу;
R10 выбран из группы, включающей атом водорода; арил;
R2 выбран из группы, включающей атом водорода; алкил; алкил-Y-алкил;
значение n равно 0;
каждая группа Y представляет собой -О-;
каждый R3 представляет собой независимо друг от друга атом водорода или C1-10алкильную группу; и
каждый R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
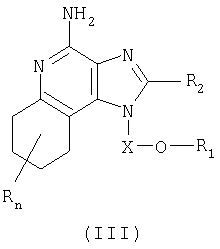
где Х представляет собой -CHR3- или -CHR3-алкильную группу;
R1 выбран из группы, включающей R4-арил;
R2 выбран из группы, включающей алкил;
R4 представляет собой алкильную группу;
каждый R3 представляет собой независимо друг от друга атом водорода или С1-10алкильную группу;
значение n равно 0;
каждый R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
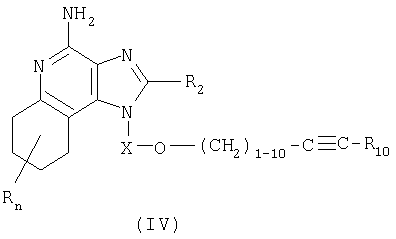
где Х представляет собой -CHR3- или -CHR3-алкильную группу;
R10 выбран из группы, включающей арил;
R2 выбран из группы, включающей алкил;
каждый R3 представляет собой независимо друг от друга атом водорода или С1-10алкильную группу;
значение n равно 0;
каждый R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
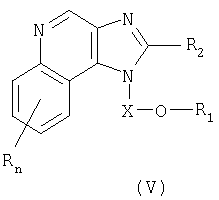
где Х представляет собой -CHR3- или -CHR3-алкильную группу;
R1 выбран из группы, включающей R4-арил;
- (CH2)1-10-C≡C-R10;
R2 выбран из группы, включающей атом водорода; алкил; алкил-Y-алкил;
R4 представляет собой алкильную или алкенильную группы, причем обе эти группы могут быть разделены одной или несколькими -О- группами;
каждый R3 представляет собой независимо друг от друга атом водорода или C1-10алкильную группу;
R10 выбран из группы, состоящей из атома водорода и арильного радикала;
каждая группа Y представляет собой -О-;
значение n равно 0;
каждый R независимо выбран из группы, состоящей из C1-10 алкильной, С1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
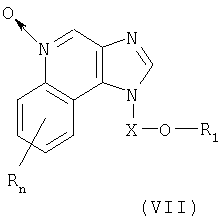
в которой Х представляет собой -CHR3- или -CHR3-алкильную группу;
R1 выбран из группы, включающей R4-арил;
- (CH2)1-10-C≡C-R10;
R4 представляет собой алкильную или алкенильную группы, причем обе эти группы могут быть разделены одной или несколькими -О- группами;
каждый R3 представляет собой независимо друг от друга атом водорода или C1-10алкильную группу;
каждый R10 выбран из группы, состоящей из атома водорода и арильного радикала;
значение n равно 0;
каждый R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
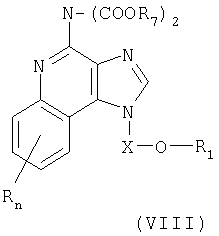
в которой Х представляет собой -CHR3- или -CHR3алкильную группу;
R1 выбран из группы, включающей R4-арил;
- (CH2)1-10-C≡C-R10;
R2 выбран из группы, включающей атом водорода;
R4 представляет собой алкильную группу;
каждый R3 представляет собой независимо друг от друга атом водорода или C1-10алкильную группу;
R10 выбран из группы, состоящей из атома водорода и арильного радикала;
значение n равно 0;
каждый R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
R7 представляет собой третбутильную или бензильную группу;
или соль фармацевтического качества на основе этих групп.
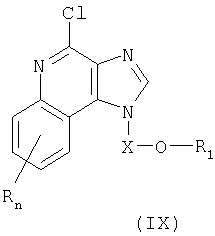
где Х представляет собой -CHR3- или -CHR3-алкильную группу;
R1 выбран из группы, включающей R4-арил;
R2 выбран из группы, включающей алкил;
R4 представляет собой алкильную или алкенильную группы;
каждый R3 представляет собой независимо друг от друга атом водорода или С1-10алкильную группу;
значение n равно 0;
каждый R независимо выбран из группы, состоящей из С1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы;
или соль фармацевтического качества на основе этих групп.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Термопластавтомат для переработки полиамидных полимерных материалов | 1961 |
|
SU145340A1 |

