Область изобретения
Настоящее изобретение относится к соединению и его применению, и в частности к соединению и его терапевтическому применению в лечении или профилактике состояний, связанных с субстратами, такими как нейромедиатор анандамид, которые расщепляются ферментом гидролазой амидов жирных кислот (FAAH, от англ. fatty acid amide hydrolase).
Предшествующий уровень техники
Фермент FAAH расщепляет амиды жирных кислот, такие как анандамид (N-арахидоноилэтаноламин), N-олеоилэтаноламин (ОЕА), N-пальмитоилэтаноламин (PEA) и олеамид. Анандамид, также известный как N-арахидоноилэтаноламин или АЕА, является нейромедиатором эндоканнабиноидной системы, который обнаруживают в органах животных и человека, особенно в головном мозге. Кроме того, было обнаружено, что анандамид связывается с ваниллоидным рецептором. Анандамид расщепляется ферментом гидролазой амидов жирных кислот (FAAH) на этаноламин и арахидоновую кислоту. Соответственно, ингибиторы FAAH приводят к повышенным уровням анандамида.
Анандамид является нейромедиатором эндоканнабиноидной системы и стимулирует каннабиноидные рецепторы. Каннабиноидные рецепторы, такие как СВ1 и СВ2, являются рецепторами, сопряженными с G-белком. СВ1 встречается в основном в центральной нервной системе, тогда как СВ2 встречается в основном в периферических тканях. Эндоканнабиноидная система вовлечена во все большее число физиологических функций как в центральной, так и в периферической нервной системе, а также в периферических органах. Было показано, что модуляция активности эндоканнабиноидной системы имеет потенциальный терапевтический эффект в широком диапазоне различных заболеваний и патологических состояний. Таким образом, эндоканнабиноидная система и в частности фермент FAAH стали терапевтической мишенью для разработки потенциальных методов лечения многих заболеваний. Эндоканнабиноидная система вовлечена в регуляцию аппетита, ожирение, нарушение обмена веществ, кахексию, анорексию, боль, воспаление, нейротоксичность, нейротравму, инсульт, рассеянный склероз, травму спинного мозга, болезнь Паркинсона, леводопа-индуцированную дискинезию, болезнь Хантингтона, синдром Жиля де ла Туретта, позднюю дискинезию, дистонию, боковой амиотрофический склероз, болезнь Альцгеймера, эпилепсию, шизофрению, тревогу, депрессию, бессонницу, тошноту, рвоту, алкогольные расстройства, привыкание к наркотическим средствам, таким как опиаты, никотин, кокаин, алкоголь и психостимуляторы, в гипертонию, циркуляторный шок, реперфузионное повреждение миокарда, атеросклероз, астму, глаукому, ретинопатию, рак, воспалительные заболевания кишечника, острые и хронические заболевания печени, такие как гепатит и цирроз печени, артрит и остеопороз. Эндоканнабиноидная система и связанные с ней состояния обсуждаются подробно в работе Pacher et al. (2006) Pharmacol. Rev. 58:389-462.
Для модуляции уровня субстратов эндогенной FAAH, таких как анандамид, которые, в свою очередь, модулируют эндоканнабиноидную систему, были разработаны ингибиторы фермента FAAH. Это позволяет лечить или предотвращать состояния и заболевания, связанные с эндоканнабиноидной системой, по меньшей мере частично.
Так как субстраты FAAH связываются с другими рецепторами, например ваниллоидными рецепторами, и/или участвуют в других сигнальных путях, ингибиторы FAAH могут также позволить лечить или предотвращать, по меньшей мере частично, состояния и заболевания, связанные с другими путями или системами, например с ваниллоидной системой.
В WO 2010/074588 описаны соединения, которые являются ингибиторами FAAH. В работе 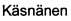 et al. (Heikki , Mikko J.
et al. (Heikki , Mikko J.  , Anna
, Anna  , Antti O. Kataja, Susanna M. Saario, Tapio Nevalainen, Ari M.P. Koskinen, and Antti Poso. Chem Med Chem 2010, 5(2), 213-231) раскрыты карбаматные соединения, которые являются ингибиторами FAAH. В частности, соединение 6b представляет собой ингибитор FAAH, который содержит структуру имидазола. Однако это соединение является слабым ингибитором FAAH, по сравнению со многими другими карбаматными соединениями, описанными в данном документе, и которые не содержат структуру имидазола.
, Antti O. Kataja, Susanna M. Saario, Tapio Nevalainen, Ari M.P. Koskinen, and Antti Poso. Chem Med Chem 2010, 5(2), 213-231) раскрыты карбаматные соединения, которые являются ингибиторами FAAH. В частности, соединение 6b представляет собой ингибитор FAAH, который содержит структуру имидазола. Однако это соединение является слабым ингибитором FAAH, по сравнению со многими другими карбаматными соединениями, описанными в данном документе, и которые не содержат структуру имидазола.
Сущность изобретения
В первом аспекте настоящее изобретение предлагает соединение, имеющее структуру, выбранную из следующих:
 ,
, 
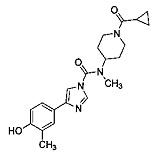 ,
,
или его фармацевтически приемлемую соль.
Было обнаружено, что соединения согласно изобретению модулируют активность фермента гидролазы амидов жирных кислот (FAAH). Кроме того, было показано, они являются относительно сильными и имеют относительно высокую периферическую селективность (т.е. они ингибируют FAAH в большей степени в периферических тканях по сравнению с тканью центральной нервной системы), и являются относительно метаболически стабильными. Также было показано, что соединения согласно настоящему изобретению обеспечивают лучшие результаты, относящиеся к одному или более свойствам, по сравнению с соединениями, описанными в публикации WO 2010/074588.
"Фармацевтически приемлемые соли" соединений согласно настоящему изобретению включают соли с неорганическими основаниями, соли с органическими основаниями, соли с неорганическими кислотами, соли с органическими кислотами и соли с основными или кислыми аминокислотами. Соли с кислотами, в частности, можно использовать в некоторых случаях. Типичные соли включают гидрохлоридную соль, ацетатную соль, трифторацетатную соль, метансульфонатную соль, 2-гидроксипропан-1,2,3-трикарбоксилатную соль, (2R,3R)-2,3-дигидроксисукцинатную соль, фосфатную соль, сульфатную соль, бензоатную соль, 2-гидроксибензоатную соль, S-(+)-манделатную соль, S-(-)-малатную соль, S-(-)пироглутаматную соль, пируватную соль, пара-толуолсульфонатную соль, 1-R-(-)-камфорсульфонатную соль, фумаратную соль, малеатную соль или оксалатную соль. Соединения согласно настоящему изобретению могут находиться в сольватированной (например гидратированной) форме или несольватированной (например негидратированной) форме. В сольватированной форме дополнительными растворителями могут быть спирты, такие как пропан-2-ол.
Общие способы получения солей хорошо известны специалистам в данной области техники. Фармацевтическая приемлемость солей будет зависеть от целого ряда факторов, включая технологические характеристики композиции и поведение in vivo, и специалист в данной области может легко оценить такие факторы, имеющие отношение к настоящему изобретению.
В соответствии со вторым аспектом изобретения предложена фармацевтическая композиция, содержащая соединение в соответствии с первым аспектом настоящего изобретения вместе с одним или более фармацевтически приемлемых эксципиентов.
Фармацевтические композиции согласно изобретению содержат соединения в соответствии с первым аспектом настоящего изобретения с любым фармацевтически приемлемым носителем, адъювантом или наполнителем. Фармацевтически приемлемыми носителями, адъювантами и наполнителями, которые могут быть использованы в фармацевтических композициях согласно настоящему изобретению, являются те, которые обычно используют в области фармацевтических препаратов, и они включают, но не ограничиваются ими, сахара, сахарные спирты, крахмалы, ионообменники, окись алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицерин, сорбиновую кислоту, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как сульфат протамина, гидрофосфат динатрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный оксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натриевую соль карбоксиметилцеллюлозы, полиакрилаты, воски, полиэтилен-полиоксипропиленовые блокполимеры, полиэтиленгликоль и ланолин.
Фармацевтические композиции согласно данному изобретению можно вводить перорально, парентерально, путем ингаляции, ректально, назально, трансбуккально, вагинально или с помощью имплантированного резервуара. Пероральное введение является предпочтительным. Фармацевтические композиции согласно настоящему изобретению могут содержать любые обычные нетоксичные фармацевтически приемлемые носители, адъюванты или наполнители. Термин парентеральный, используемый здесь, включает подкожную, внутрикожную, внутривенную, внутримышечную, внутрисуставную, внутрисиновиальную, интрастернальную, интратекальную, внутриочаговую и внутричерепную инъекцию или инфузию.
Фармацевтические композиции могут быть в форме стерильного инъекционного препарата, например, в виде стерильной инъекционной водной или масляной суспензии. Эта суспензия может быть приготовлена в соответствии с методами, известными в данной области, с использованием подходящих диспергирующих или смачивающих агентов (таких, как, например, Tween 80) и суспендирующих агентов. Стерильный инъекционный препарат может также быть стерильным инъекционным раствором или суспензией в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей, которые могут быть использованы, находятся маннит, вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла обычно используют в качестве растворителя или суспендирующей среды. С этой целью можно использовать любое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, являются полезными при получении инъекционных препаратов, как и природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных вариантах. Эти масляные растворы или суспензии могут также содержать спиртовой разбавитель с длинной цепью или диспергатор, такой как описан в Ph. Helv (в Фармакопее Швейцарии), или аналогичный спирт.
Фармацевтические композиции согласно данному изобретению можно вводить перорально в любой перорально приемлемой лекарственной форме, включая, но не ограничиваясь этим, капсулы, таблетки, порошки, гранулы и водные суспензии и растворы. Эти лекарственные формы получают в соответствии с методиками, хорошо известными в данной области фармацевтических препаратов. В случае таблеток для перорального применения, носители, которые обычно используют, включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие агенты, такие как стеарат магния. Для перорального введения в форме капсулы пригодные разбавители включают лактозу и высушенный кукурузный крахмал. Когда водные суспензии вводят перорально, активный ингредиент комбинируют с эмульгирующими и суспендирующими агентами. При желании, могут быть добавлены определенные подсластители и/или вкусовые добавки и/или красители.
Фармацевтические композиции согласно данному изобретению можно также вводить в форме суппозиториев для ректального введения. Эти композиции могут быть получены путем смешивания соединения согласно настоящему изобретению с подходящим нераздражающим эксципиентом, который является твердым при комнатной температуре, но жидким при ректальной температуре и, следовательно, будет плавиться в прямой кишке с высвобождением активных компонентов. Такие материалы включают в себя, но не ограничиваются этим, масло какао, пчелиный воск и полиэтиленгликоли.
Фармацевтические композиции согласно данному изобретению можно вводить с помощью назального аэрозоля или ингаляции. Такие композиции получают в соответствии с методиками, хорошо известными в данной области фармацевтических препаратов, и могут быть приготовлены в виде растворов в физиологическом растворе с использованием бензилового спирта или других подходящих консервантов, промоторов абсорбции для повышения биодоступности, фторуглеродов и/или других солюбилизирующих или диспергирующих агентов, известных в данной области техники.
Соединения согласно настоящему изобретению можно вводить в дозе от приблизительно 1 до приблизительно 20000 мкг/кг на дозу, например, от приблизительно 1 до приблизительно 10000 мкг/кг, от приблизительно 1 до приблизительно 5000 мкг/кг, от приблизительно 1 до приблизительно 3000 мкг/кг, от приблизительно 1 до приблизительно 2000 мкг/кг, от приблизительно 1 до приблизительно 1500 мкг/кг, от приблизительно 1 до приблизительно 1000 мкг/кг, от приблизительно 1 до приблизительно 500 мкг/кг, от приблизительно 1 до приблизительно 250 мкг/кг, от приблизительно 1 до приблизительно 100 мкг/кг, от приблизительно 1 до приблизительно 50 мкг/кг или от приблизительно 1 до приблизительно 25 мкг/кг на дозу в зависимости от состояния, подлежащего лечению или профилактике, а также от характеристик пациента, которому вводят это соединение. Во многих случаях эта доза может быть от приблизительно 1 до приблизительно 10 мкг/кг на дозу. В конкретных вариантах осуществления доза может составлять приблизительно 250 мкг/кг на дозу, приблизительно 100 мкг/кг, приблизительно 50 мкг/кг или приблизительно 10 мкг/кг на дозу. Режим дозирования для конкретного соединения может быть легко определен квалифицированным специалистом, имеющим доступ к данному раскрытию.
В одном конкретном варианте осуществления фармацевтическая композиция согласно настоящему изобретению дополнительно содержит один или более дополнительных активных фармацевтических ингредиентов. Соединение согласно настоящему изобретению можно вводить с одним или более дополнительных активных фармацевтических ингредиентов, таких как анандамид, олеоилэтаноламид или пальмитоилэтаноламид. Это может быть в виде одной композиции, содержащей соединение по изобретению и один или более дополнительных активных фармацевтических ингредиентов. В качестве альтернативы, это может быть в виде двух или более отдельных композиций, где соединение согласно настоящему изобретению содержится в одной композиции, и один или более дополнительных активных фармацевтических ингредиентов содержатся в одной или более отдельных композиций.
Поэтому введение соединений согласно настоящему изобретению можно осуществлять одновременно или со смещением во времени с одним или более дополнительных активных фармацевтических ингредиентов.
В третьем аспекте настоящее изобретение предлагает соединение в соответствии с первым аспектом изобретения или композицию в соответствии со вторым аспектом для применения в терапии.
В четвертом аспекте настоящее изобретение предлагает соединение в соответствии с первым аспектом настоящего изобретения или композицию в соответствии со вторым аспектом для применения в лечении или профилактике состояния, развитие или симптомы которого связаны с субстратом фермента FAAH.
Настоящее изобретение также предлагает применение соединения в соответствии с первым аспектом настоящего изобретения или композиции в соответствии со вторым аспектом для изготовления лекарственного средства для лечения или профилактики состояния, развитие или симптомы которого связаны с субстратом фермента FAAH.
Ряд состояний, развитие или симптомы которых связаны с субстратом фермента FAAH, известны специалисту в данной области. Некоторые из них описаны выше.
В пятом аспекте настоящее изобретение также относится к способу лечения или профилактики состояния, развитие или симптомы которого связаны с субстратом фермента FAAH, включающему введение пациенту, нуждающемуся в таком лечении или профилактике, терапевтически эффективного количества соединения в соответствии с первым аспектом изобретения или композиции в соответствии со вторым аспектом изобретения.
Для соединения в соответствии с четвертым аспектом или для способа в соответствии с пятым аспектом состояние представляет собой расстройство, связанное с эндоканнабиноидной системой.
В некоторых вариантах осуществления настоящего изобретения состояния, подлежащие лечению, могут быть выбраны из:
(i) боли, в частности острой или хронической нейрогенной боли, такой как мигрень и нейропатическая боль (например, диабетическая невропатическая боль, постгерпетическая невралгия, невралгия тройничного нерва); мигрени; острой или хронической воспалительной боли, такой как боль, связанная с воспалительными заболеваниями, такими как артрит, ревматоидный артрит, остеоартрит, остеопороз, спондилит, подагра, васкулит, болезнь Крона и синдром раздраженной толстой кишки; острой или хронической периферической боли; боли при раке;
(ii) головокружения, рвоты и тошноты, в частности, возникающих в результате химиотерапии;
(iii) расстройств пищевого поведения, в частности, расстройств аппетита, расстройств обмена веществ, анорексии и кахексии различной природы;
(iv) неврологических и психиатрических патологий, таких как тремор, дискинезии, дистонии, тошнота, рвота, аддиктивные расстройства (такие как привыкание к наркотическим средствам или алкоголю), спастичность, обсессивно-компульсивное поведение, синдром Туретта, все формы депрессии и тревоги любой природы и происхождения, бессонница, расстройства настроения и психозы, такие как шизофрения;
(v) острых и хронических нейродегенеративных заболеваний, таких как болезнь Паркинсона, болезнь Альцгеймера, старческое слабоумие, хорея Гентингтона, поражения, связанных с церебральной ишемией и черепно-мозговой и медуллярной травмой;
(vi) эпилепсии;
(vii) расстройств сна, включая апноэ во время сна;
(viii) сердечно-сосудистых заболеваний, таких как сердечная недостаточность, гипертония, циркуляторный шок, реперфузионное повреждение миокарда, сердечные аритмии, артериосклероз/атеросклероз, инфаркт миокарда, сердечная ишемия, васкулит и почечная ишемия;
(ix) рака, например, доброкачественных опухолей кожи, опухолей головного мозга и папиллом, опухолей предстательной железы и опухолей, относящихся к головному мозгу (глиобластом, медуллоэпителиом, медуллобластом, нейробластом, опухолей эмбрионального происхождения, астроцитом, астробластом, эпендимом, олигодендроглиом, опухоли сплетения, нейроэпителиом, эпифизарной опухоли, эпендимобластом, злокачественных менингиом, саркоматоза, злокачественных меланом и шванном);
(x) нарушений иммунной системы, в частности аутоиммунных заболеваний, таких как псориаз, красная волчанка, заболевания соединительных тканей или заболеваний коллагена, синдром Шегрена, анкилозирующий спондилит, недифференцированный спондилит, болезнь Бехчета, аутоиммунная гемолитическая анемия, рассеянный склероз, амилотрофический боковой склероз, амилоидоз, отторжение трансплантата, заболевания, влияющие на плазмацитарную линию, аллергические заболевания; гиперчувствительности немедленного или замедленного типа, аллергического ринита или конъюнктивита, контактного дерматита;
(xi) паразитарных, вирусных или бактериальных инфекционных заболеваний, таких как СПИД и менингит;
(xii) воспалительных заболеваний, в частности заболеваний суставов, таких как артрит, ревматоидный артрит, остеоартрит, спондилит, подагра, васкулит, болезни Крона, синдрома раздраженного/воспаленного кишечника, астмы;
(xiii) остеопороза;
(xiv) глазных заболеваний, таких как глазная гипертензия, ретинопатия и глаукома;
(xv) легочных заболеваний, включая заболевания дыхательных путей, бронхоспазм, кашель, астму, хронический бронхит, хроническую обструкцию дыхательных путей и эмфизему легких;
(xvi) желудочно-кишечных заболеваний, таких как синдром раздраженного/воспаленного кишечника, воспалительных кишечных расстройств, язв, диареи, недержания мочи и воспаления мочевого пузыря;
(xvii) острых и хронических заболеваний печени, таких как гепатит и цирроз печени;
(xviii) неврологических расстройств, таких как нейротравма, инсульт, рассеянный склероз, травма спинного мозга, болезнь Паркинсона, леводопа-индуцированная дискинезия, болезнь Хантингтона/хорея, синдром Жиля де ла Туретта, поздняя дискинезия, дистония, амиотрофический боковой склероз, болезнь Альцгеймера и эпилепсия.
Подробное описание изобретения
Далее изобретение будет описано более подробно только с помощью примера:
1. Методологии синтеза
Способы, используемые для синтеза соединений согласно настоящему изобретению проиллюстрированы общими схемами и конкретными примерами синтеза ниже. Все соединения и промежуточные продукты были охарактеризованы с помощью ядерного магнитного резонанса (ЯМР). Исходные материалы и реагенты, используемые при получении этих соединений, доступны от коммерческих поставщиков или могут быть получены способами, очевидными для специалистов в данной области. Эти общие схемы и конкретные примеры синтеза лишь иллюстрируют способы, с помощью которых могут быть синтезированы соединения согласно настоящему изобретению, и различные модификации этих схем и примеров синтеза могут быть сделаны и будут предложены специалистам в данной области техники со ссылкой на это раскрытие.
Соединения согласно настоящему изобретению были охарактеризованы температурой плавления и ЯМР. ЯМР-спектры регистрировали на спектрометре Bruker Avance III 600 МГц с растворителем, используемым в качестве внутреннего стандарта. Спектры 13С регистрировали на частоте 150 МГц и спектры 1Н регистрировали на частоте 600 МГц. Данные представлены в следующем порядке: приблизительный химический сдвиг (ppm - миллионных долей), число протонов, мультиплетность (br - широкий; d - дублет, m - мультиплет, s - синглет, t - триплет) и константа связывания (Гц).
Комнатная температура в следующих схемах означает температуру в пределах от 20°С до 25°С.
1.1. Общая схема синтеза N-Метил-4-(3-(сульфамоиламино)фенил)-N-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-1-карбоксамида (Соединение 1)

Фенил-4-(3-нитрофенил)-1Н-имидазол-1-карбоксилат

Фенилкарбонохлоридат (3,2 мл, 25,4 ммоль) добавляли к перемешиваемому раствору 4-(3-нитрофенил)-1Н-имидазола (4 г, 21,1 ммоль) и пиридина (Ру) (2,0 мл, 25,4 ммоль) в DCM (дихлорметан) (100 мл) при 0°С. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 ч. Добавляли воду и органический слой отделяли, сушили (MgSO4) и выпаривали в вакууме с получением твердого вещества бежевого цвета. Затем твердое вещество перекристаллизовывали из смеси пропан-2-ола и DCM, и продукт выделяли в виде бежевого твердого вещества. Фенил 4-(3-нитрофенил)-1Н-имидазол-1-карбоксилат (2,89 г, выход 44%).
N-Метил-4-(3-нитрофенил)-N-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-1-карбоксамид
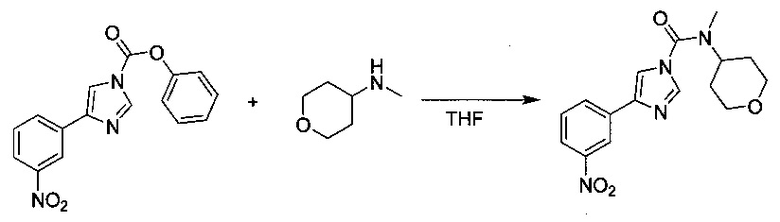
Раствор N-метилтетрагидро-2Н-пиран-4-амина (2,15 г, 18,7 ммоль) в тетрагидрофуране (THF) (6 мл) добавляли к перемешиваемому раствору фенил-4-(3-нитрофенил)-1Н-имидазол-1-карбоксилата (2,89 г, 9,3 ммоль) в THF (40 мл) при комнатной температуре. Желтый раствор оставляли перемешиваться при кипячении с обратным холодильником в течение ночи. Растворитель выпаривали в вакууме и продукт перекристаллизовывали из пропан-2-ола. N-метил-4-(3-нитрофенил)-N-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-1-карбоксамид (0,938 г, выход 30%).
4-(3-Аминофенил)-N-метил-N-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-1-карбоксамид
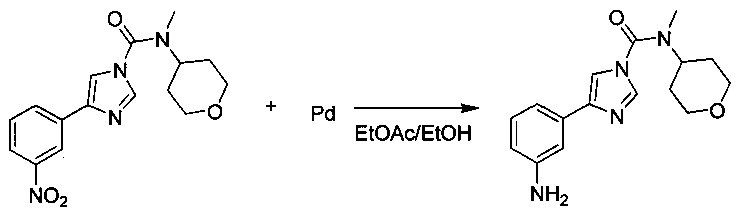
Смесь этанола (30,0 мл) и этилацетата (30 мл) добавляли к влажному палладию (0,151 г, 0,142 ммоль, 10% на активированном угле) в атмосфере аргона. К этой смеси добавляли N-метил-4-(3-нитрофенил)-N-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-1-карбоксамид (0,938 г, 2,84 ммоль) и суспензию оставляли перемешиваться при комнатной температуре в течение ночи в атмосфере водорода. Полученную серую суспензию фильтровали через целит и целит промывали DCM. Фильтрат выпаривали в вакууме, а бесцветный продукт перекристаллизовывали из пропан-2-ола. 4-(3-Аминофенил)-N-метил-N-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-1-карбоксамид (0,695 г, выход 81%).
N-Метил-4-(3-(сульфамоиламино)фенил)-N-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-1-карбоксамид (Соединение 1)

Сульфамоилхлорид (0,321 г, 2,78 ммоль) добавляли к перемешиваемой суспензии 4-(3-аминофенил)-N-метил-N-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-1-карбоксамида (0,695 г, 2,314 ммоль) и триэтиламина (0,481 мл, 3,47 ммоль) в DCM (12 мл) при комнатной температуре. Белую суспензию оставляли перемешиваться при комнатной температуре в течение ночи. Добавляли воду и органический слой разбавляли смесью DCM/пропан-2-ол 7:3. Органический слой отделяли и водный слой повторно экстрагировали. Объединенные органические слои сушили (MgSO4) и выпаривали в вакууме с получением прозрачного масла. Продукт отделяли с помощью колоночной хроматографии (силикагель, DCM/MeOH, 2%, 5%, 10%) и выделяли в виде бесцветного твердого вещества. Твердое вещество растирали со смесью пропан-2-ол/DCM. Твердое вещество перекристаллизовывали дважды из этанола и продукт сушили в высоком вакууме при 60°С в течение ночи. N-метил-4-(3-(сульфамоиламино)фенил)-N-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-1-карбоксамид (0,160 г, выход 18%), т.пл. 128°С.
ЯМР (ДМСО-d6):
1Н: 9.54 (1Н, s), 8.14 (1Н, s), 7.94 (1Н, s), 7.64 (1Н, s), 7.44 (1Н, d, J равно 7.7 Гц), 7.27 (1H, t, J равно 7.6 Гц), 7.15 (2Н, s), 7.05 (1Н, d, J равно 8.2 Гц), 4.10 (1Н, m), 3.93 (2Н, dd, J равно 4.0, 11.3 Гц), 3.36 (2Н, m), 2.95 (3Н, s), 1.86 (2Н, dq, J равно 4.1, 12.3 Гц), 1.70 (2Н, d, J равно 12.0 Гц).
13С: 151, 140.6, 139.9, 137.5, 134, 129, 118.6, 116.9, 114.6, 114.4, 66.3, 54.2, 31.6, 29.1.
1.2. Общая схема синтеза гидрохлорида 4-(4-метокси-3-метилфенил)-N-метил-N-(пиперидин-4-ил)-1Н-имидазол-1-карбоксамида (Промежуточное соединение 1)

1-(4-Метокси-3-метилфенил)этанон

Диметилсульфат (17,50 мл, 183 ммоль) добавляли к перемешиваемой суспензии 1-(4-гидрокси-3-метилфенил)этанона (25 г, 166 ммоль) и карбоната калия (28,8 г, 208 ммоль) в ацетоне (277 мл) при комнатной температуре. Суспензию оставляли перемешиваться при кипячении с обратным холодильником в течение ночи. Твердое вещество отделяли фильтрованием и промывали ацетоном, фильтрат выпаривали в вакууме. Органический остаток растворяли в EtOAc и промывали водой. Органический слой отделяли, сушили (MgSO4) и выпаривали в вакууме с получением желтого масла. Использовали без дополнительной очистки. 1-(4-Метокси-3-метилфенил)этанон (28,7 г).
2-Бром-1-(4-метокси-3-метилфенил)этанон

Раствор трибромида фенилтриметиламмония (30,2 г, 80 ммоль) в THF (122 мл) добавляли по каплям к раствору 1-(4-метокси-3-метилфенил)этанона (12 г, 73,1 ммоль) в THF (122 мл) при комнатной температуре. Желтую суспензию оставляли перемешиваться при комнатной температуре в течение часа. Твердое вещество отделяли фильтрованием и промывали THF. Фильтрат выпаривали в вакууме, а органический остаток растворяли в EtOAc и промывали водой. Органический слой сушили (MgSO4) и выпаривали в вакууме с получением фиолетового масла. Использовали без дополнительной очистки. 2-Бром-1-(4-метокси-3-метилфенил)этанон (27,9 г).
4-(4-метокси-3-метилфенил)-1Н-имидазол

Воду (4 мл) добавляли к перемешиваемой суспензии 2-бром-1-(4-метокси-3-метилфенил)этанона (27,9 г, 1 15 ммоль) и формамида (56,7 мл, 1423 ммоль) при комнатной температуре. Суспензию оставляли перемешиваться при температуре 140°С в течение 5 ч. Смесь выливали в 200 мл воды с получением темно-коричневого густого масла. Масло отделяли фильтрованием и промывали раствором HCl, фильтрат подщелачивали 50% NaOH с получением бежевого твердого вещества. Твердое вещество отделяли фильтрованием и промывали диэтиловым эфиром (5х) с получением не совсем белого твердого вещества. 4-(4-Метокси-3-метилфенил)-1Н-имидазол (5,2 г, выход 24%).
трет-Бутил-4-(метиламино)пиперидин-1-карбоксилат

Раствор метанамина (38,0 мл, 442 ммоль, 40% водный раствор) в метаноле (100 мл) добавляли к влажному палладию (1,602 г, 1,506 ммоль, 10% на активированном угле) при комнатной температуре в атмосфере аргона. К этой смеси порциями добавляли трет-бутил-4-оксопиперидин-1-карбоксилат (20 г, 100 ммоль) и смесь оставляли перемешиваться при 50°С, 20 бар, в течение 1 часа. Суспензию продували аргоном и фильтровали через целит, целит промывали DCM. Фильтрат выпаривали в вакууме с получением продукта в виде прозрачного масла. Масло растворяли в EtOAc и промывали водой. Органический слой сушили (MgSO4) и выпаривали в вакууме с получением прозрачного масла. Использовали без дополнительной очистки. трет-Бутил-4-(метиламино)пиперидин-1-карбоксилат (20 г, выход 93%).
трет-Бутил-4-(хлоркарбонил(метил)амино)пиперидин-1-карбоксилат

Раствор трет-бутил-4-(метиламино)пиперидин-1-карбоксилата (20 г, 93 ммоль) и основания Хюнига (35,9 мл, 205 ммоль) в THF (133 мл) добавляли по каплям к перемешиваемому фосгену (53,3 мл, 112 ммоль, 20% раствор в толуоле) при температуре 0°С с получением белой суспензии. Смесь оставляли перемешиваться при 0°С в течение 10 мин и при комнатной температуре в течение 2 ч. Суспензию выливали в смесь лед/вода и органический остаток экстрагировали EtOAc. Органический слой отделяли и промывали 1Н раствором HCl. Органический слой сушили (MgSO4) и выпаривали в вакууме с получением желтого масла. Масло растирали в порошок со смесью петролейного эфира (РЕ) и нескольких капель диэтилового эфира с получением бесцветного твердого вещества. Твердое вещество отделяли фильтрованием и промывали петролейным эфиром, трет-Бутил-4-(хлоркарбонил(метил)амино)пиперидин-1-карбоксилат (17,4 г, выход 67%).
трет-Бутил-4-(4-(4-метокси-3-метилфенил)-N-метил-N-имидазол-1-карбоксамидо)пиперидин-1-карбоксилат

Гидрид натрия (1,313 г, 32,8 ммоль, 60% дисперсия в масле) добавляли порциями к перемешиваемой суспензии 4-(4-метокси-3-метилфенил)-1H-имидазола (5,15 г, 27,4 ммоль) в THF (137 мл) при 0°С. Темно-голубой раствор оставляли перемешиваться при комнатной температуре в течение 30 мин, а затем добавляли трет-бутил-4-(хлоркарбонил(метил)амино)пиперидин-1-карбоксилат (1 1,36 г, 41,0 ммоль) при 0°С, получая темный раствор. Смесь оставляли перемешиваться при комнатной температуре в течение 2 ч. Добавляли воду при 0°С, а органический слой разбавляли смесью DCM/пропан-2-ол 7:3. Органический слой отделяли, сушили (MgSO4) и выпаривали в вакууме с получением твердого вещества бежевого цвета. Твердое вещество перекристаллизовывали из пропан-2-ола. трет-Бутил-4-(4-(4-метокси-3-метилфенил)-N-метил-1Н-имидазол-1-карбоксамидо)пиперидин-1-карбоксилат (9,39 г, выход 80%).
Гидрохлорид 4-(4-Метокси-3-метилфенил)-N-метил-N-(пиперидин-4-ил)-1Н-имидазол-1-карбоксамида

Трифторуксусную кислоту (TFA) (30 мл) осторожно добавляли к осадку трет-бутил-4-(4-(4-метокси-3-метилфенил)-N-метил-1Н-имидазол-1-карбоксамидо)-пиперидин-1-карбоксилата (9,39 г, 21,91 ммоль) при комнатной температуре. Желтый раствор оставляли перемешиваться при комнатной температуре в течение 1,5 часов. TFA выпаривали в вакууме, а затем подвергали азеотропной перегонке дважды с толуолом. Желтый остаток затем растворяли в этилацетате (30 мл) и добавляли по каплям 2М раствор хлористого водорода (32,9 мл, 65,7 ммоль) в диэтиловом эфире при 0°С с получением белой суспензии. Смесь оставляли перемешиваться при комнатной температуре в течение 30 мин, а затем твердое вещество отделяли фильтрованием и промывали EtOAc. 4-(4-Метокси-3-метилфенил)-N-метил-N-(пиперидин-4-ил)-1Н-имидазол-1-карбоксамида гидрохлорид (11,06 г).
1.3. Общая схема синтеза 4-(4-Гидрокси-3-метилфенил)-N-метил-N-(1-пропионилпиперидин-4-ил)-1Н-имидазол-1-карбоксамида (Соединение 2)

4-(4-Метокси-3-метил фенил)-N-метил-Н-(1-пропионилпиперидин-4-ил) 1Н-имидазол-1-карбоксамид

Пропионилхлорид (0,287 мл, 3,29 ммоль) добавляли к перемешиваемой суспензии гидрохлорида 4-(4-метокси-3-метилфенил)-N-метил-N-(пиперидин-4-ил)-1Н-имидазол-1-карбоксамида (промежуточное соединение 1) (1 г, 2,74 ммоль) и основания Хюнига (0,957 мл, 5,48 ммоль) в DCM (14 мл) при комнатной температуре. Розовый раствор оставляли перемешиваться при комнатной температуре в течение ночи. Добавляли воду и органический слой разбавляли DCM. Органический слой отделяли, сушили (MgSO4) и выпаривали в вакууме с получением не совсем белого твердого вещества. Твердое вещество перекристаллизовывали из пропан-2-ола. 4-(4-Метокси-3-метилфенил)-N-метил-N-(1-пропионилпиперидин-4-ил)-1Н-имидазол-1-карбоксамид (0,496 г, выход 45%).
4-(4-Гидрокси-3-метилфенил)-N-метил-N-(1-пропионилпиперидин-4-ил)-1Н-имидазол-1-карбоксамид (Соединение 2)

Трибромид бора (0,354 мл, 3,75 ммоль) добавляли к перемешиваемой суспензии 4-(4-метокси-3-метилфенил)-N-метил-N-(1-пропионилпиперидин-4-ил)-1Н-имидазол-1-карбоксамида (0,480 г, 1,248 ммоль) в безводном DCM (4 мл) при -78°С. Суспензию оставляли перемешиваться при -78°С в течение 15 мин и при комнатной температуре в течение 2 ч. Добавляли воду при температуре от -50°С, а затем органический слой разбавляли смесью DCM/пропан-2-ол 7:3. Органический слой отделяли; водный слой насыщали NaCl и повторно экстрагировали. Объединенный органический слой сушили (MgSO4) и выпаривали в вакууме с получением прозрачного масла. Продукт отделяли с помощью колоночной хроматографии (силикагель, DCM/MeOH, 2%, 5%, 10%) и выделяли в виде бесцветного твердого вещества. Твердое вещество перекристаллизовывали из пропан-2-ола. 4-(4-Гидрокси-3-метилфенил)-N-метил-N-(1-пропионилпиперидин-4-ил)-1Н-имидазол-1-карбоксамид (0,22 г, выход 45%), т.пл. 232°С.
ЯМР (ДМСО-d6):
1Н: 9.34 (1Н, s), 8.06 (1Н, d, J равно 1.2 Гц), 7.77 (1Н, d, J равно 1.2 Гц), 7.55 (1Н, d, J равно 1.6 Гц), 7.47 (1Н, dd, J равно 2, 8.3 Гц), 6.77 (1Н, d, J равно 8.3 Гц), 4.53 (1Н, d, J равно 12.5 Гц), 4.10 (1Н, m), 3.95 (1Н, d, J равно 13.5 Гц), 3.06 (1Н, mt, J равно 13.0 Гц), 2.91 (3Н, s), 2.56 (1Н, mt, J равно 12.8 Гц), 2.34 (2Н, q, J равно 7.5 Гц), 2.14 (3Н, s), 1.76 (3Н, m), 1.60 (1Н, dq, J равно 4.3, 12.3 Гц), 0.98 (3Н, t, J равно 7.5 Гц).
13С: 171.1, 154.8, 151.1, 141.1, 137.3, 127.3, 124.2, 123.8, 123.4, 114.6, 112.3, 55.1, 43.9, 40.3, 31.6, 28.6, 28, 25.5, 16.1, 9.5.
1.4 Общая схема синтеза N-(1-(циклопропанкарбонил)пиперидин-4-ил)-4-(4-гидрокси-3-метилфенил)-N-метил-1Н-имидазол-1-карбоксамида (Соединение 3)
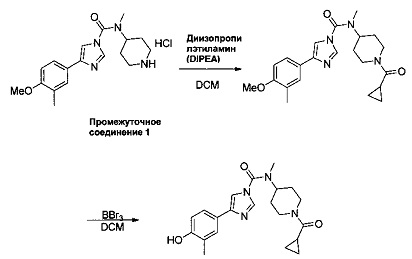
N-(1-(Циклопропанкарбонил)пиперидин-4-ил)-4-(4-метокси-3-метилфенил)-N-метил-1Н-имидазол-1-карбоксамид

Циклопропанкарбонилхлорид (1,5 мл, 16,44 ммоль) добавляли к перемешиваемой суспензии гидрохлорида 4-(4-метокси-3-метилфенил)-N-метил-N-(пиперидин-4-ил)-1Н-имидазол-1-карбоксамида (промежуточное соединение 1) (5 г, 13,70 ммоль) и основания Хюнига (4,8 мл, 27,4 ммоль) в DCM (70 мл) при комнатной температуре. Розовый раствор оставляли перемешиваться при комнатной температуре в течение ночи с получением розовой суспензии. Добавляли воду и органический слой разбавляли DCM. Органический слой отделяли, сушили (MgSO4) и выпаривали в вакууме с получением прозрачного масла, которое затвердевает в виде не совсем белого твердого вещества. Твердое вещество перекристаллизовывали из пропан-2-ола. N-(1-(Циклопропанкарбонил)пиперидин-4-ил)-4-(4-метокси-3-метилфенил)-N-метил-1Н-имидазол-1-карбоксамид (2,96 г, выход 55%).
N-(1-(Циклопропанкарбонил)пиперидин-4-ил)-4-(4-гидрокси-3-метилфенил)-N-метил-1Н-имидазол-1-карбоксамид (Соединение 3)
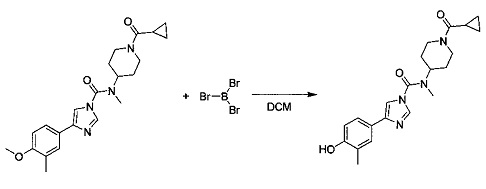
Трибромид бора (0,358 мл, 3,78 ммоль) добавляли к перемешиваемой суспензии N-(1-(циклопропанкарбонил)пиперидин-4-ил)-4-(4-метокси-3-метилфенил)-N-метил-1Н-имидазол-1-карбоксамида (0,500 г, 1,261 ммоль) в безводном DCM (4,20 мл) при -78°С. Суспензию оставляли перемешиваться при -78°С в течение 15 мин и при комнатной температуре в течение 2 ч. Добавляли воду при температуре -50°С, а затем органический слой разбавляли смесью DCM/пропан-2-ол 7:3. Органический слой отделяли; водный слой насыщали NaCl и повторно экстрагировали. Объединенный органический слой сушили (MgSO4) и выпаривали в вакууме с получением прозрачного масла. Продукт отделяли с помощью колоночной хроматографии (силикагель, DCM/MeOH, 2%, 5%, 10%) и выделяли в виде бесцветного твердого вещества. Твердое вещество перекристаллизовывали из смеси пропан-2-ола и DCM. N-(1-(Циклопропанкарбонил)пиперидин-4-ил)-4-(4-гидрокси-3-метилфенил)-N-метил-1Н-имидазол-1-карбоксамид (0,289 г, 57% выход), т.пл. 204°С.
ЯМР (ДМСО-d6):
1Н: 9.32 (1Н, s), 8.06 (1Н, d, J равно 1.2 Гц), 7.76 (1Н, d, J равно 1.2 Гц), 7.55 (1Н, d, J равно 1.6 Гц), 7.47 (1Н, dd, J равно 2, 8.2 Гц), 6.77 (1Н, d, J равно 8.2 Гц), 4.51 (1Н, d, J равно 12.0 Гц), 4.37 (1Н, d, J равно 13.0 Гц), 4.13 (1Н, m), 3.15 (1Н, t, J равно 13.0 Гц), 2.92 (3Н, s), 2.61 (1Н, mt, J равно 13.0 Гц), 2.14 (3Н, s), 2.0 (1Н, m), 1.85 (1Н, md), 1.77 (2Н, m), 1.63 (1Н, mq), 0.8-0.66 (4Н, m).
13С: 170.8, 154.8, 151.2, 141.1, 137.3, 127.3, 124.2, 123.8, 123.4, 114.6, 112.4, 55.2, 44, 40.8, 31.7, 28.9, 28, 16.1, 10.3, 7, 6.9.
2. Биологическая эффективность
Все процедуры на животных проводили в строгом соответствии с Европейской директивой по защите позвоночных животных, используемых для экспериментальных и других научных целей (86/609СЕЕ) и законодательством Португалии (Decreto-Lei 129/92, Portarias 1005/92 е 1131/97). Число используемых животных было минимально возможным в соответствии с действующими правилами и научной чистоты.
Испытания in vivo проводили в соответствии с протоколом, описанным ниже. Гомогенат мозга (BRh) указывает на ингибирование в ткани центральной нервной системы, в данном случае, мозга, и гомогенат печени (LVh) указывает на ингибирование в периферических тканях, в данном случае, печени. Контролем являлась реакционная смесь минус испытуемые соединения. Таким образом, низкое значение для тестируемого соединения указывает на сильный ингибитор. Значение 100 указывает на то, что никакого измеримого ингибирования не произошло.
Протоколы in vivo
Эксперименты на мышах
Обработка животных
Животными, используемыми в экспериментах, были самцы мышей линии NMRI (массой 27-44 г), полученные из Interfauna  (Испания). Мышей содержали по 5 животных на клетку при контролируемых условиях окружающей среды (12 ч свет/темнота и комнатная температура 22±1°С). Прием продуктов питания и водопроводной воды был разрешен вволю и все эксперименты проводили в дневное время. Животные всегда голодали в течение ночи перед введением соединений.
(Испания). Мышей содержали по 5 животных на клетку при контролируемых условиях окружающей среды (12 ч свет/темнота и комнатная температура 22±1°С). Прием продуктов питания и водопроводной воды был разрешен вволю и все эксперименты проводили в дневное время. Животные всегда голодали в течение ночи перед введением соединений.
Животным вводили соответствующую дозу соединения согласно изобретению перорально (р.о.) (8 мл/кг; соединение суспендировали в 0,5% карбоксиметилцеллюлозе (CMC) или растворяли в воде) или носитель (контроль) с использованием кривой иглы из нержавеющей стали для кормления (Perfectum, США). За пятнадцать минут до умерщвления животных анестезировали фенобарбиталом 60 мг/кг внутрибрюшинно. Фрагмент печени и головного мозга без мозжечка извлекали и помещали в пластиковые флаконы, содержащие мембранный буфер (3 мМ MgCl2, 1 мМ ЭДТА, 50 мМ Трис-HCl рН 7,4). Ткани хранили при -30°С до анализа.
Реагенты и растворы
Анандамид [этаноламин-1-3Н-] (40-60 Ки/ммоль) получали от American Radiochemicals. Все другие реагенты получали от Sigma-Aldrich. Optiphase Supermix получали от Perkin Elmer и активированный уголь получали от Sigma-Aldrich.
Подготовка тканей
Ткани размораживали на льду и гомогенизировали в 10 объемах мембранного буфера (3 мМ MgCl2, 1 мМ ЭДТА, 50 мМ Трис-HCl, рН 7,4) с использованием либо Potter-Elvejhem (мозг - 8 ударов при 500 оборотах в минуту), либо Heidolph Diax (печень - 2 удара в положении 5 в течение 20 сек с паузами по 30 сек).
Общее содержание белка в тканях определяли с помощью анализа белков BioRad (от BioRad) с использованием стандартной кривой БСА (50-250 мкг/мл).
Ферментный анализ
Реакционная смесь (общий объем 200 мкл) содержала: 2 мкМ АЕА (2 мкМ АЕА + 5 нМ 3Н-АЕА), 0,1% БСА без жирных кислот, 15 мкг белка (мозга) или 5 мкг белка (печени), в 1 мМ ЭДТА, 10 мМ Трис, рН 7,6. После 15-минутного периода предварительной инкубации при температуре 37°С реакцию инициировали добавлением раствора субстрата (немеченый АЕА + меченый АЕА+БСА). Реакцию проводили в течение 10 мин (мозг) или 7 мин (печень), и прекращали путем добавления 400 мкл суспензии активированного угля (8 г угля в 32 мл 0,5 М HCl при непрерывном перемешивании). После 30-минутного периода инкубации при комнатной температуре при перемешивании уголь осаждали центрифугированием в микроцентрифуге (10 мин при 13000 оборотах в минуту). 200 мкл надосадочной жидкости добавляли в 800 мкл сцинтилляционного коктейля Optiphase Supermix, предварительно распределенного в 24-луночные планшеты. Число импульсов в минуту (cpm) определяли с помощью сцинтилляционного счетчика MicrobetaTriLux.
В каждом анализе готовили холостые пробы (без белка).
Процент сохранившейся ферментативной активности вычисляли по отношению к контролю и после вычитания холостой пробы.
Эксперименты на крысах
Обработка животных
Самцов крыс линии Вистар (масса тела в диапазоне 190-230 г) получали от Harlan (Испания). Крыс содержали по 5 животных на клетку при контролируемых условиях окружающей среды (12 ч свет/темнота и комнатная температура 22±1°С). Прием продуктов питания и водопроводной воды был разрешен вволю, и все эксперименты проводили в дневное время.
Крысам вводили соответствующую дозу соединения согласно настоящему изобретению через зонд (объем введения равен 4 мл/кг массы тела) с использованием кривой иглы из нержавеющей стали для кормления (Perfectum, США). Носитель представлял собой 0,5% КМЦ в воде Midi Q. Крысы голодали в течение по меньшей мере 15 ч до экспериментов.
За пятнадцать минут до умерщвления животных анестезировали фенобарбиталом внутрибрюшинно 60 мг/кг массы тела. Собирали биопсии печени и образцы головного мозга (без мозжечка) и помещали в пластиковые флаконы, содержащие мембранный буфер (3 мМ MgCl2, 1 мМ ЭДТА, 50 мМ Трис-HCl, рН 7,4) и, в случае образцов печени, стеклянные бусины (2,5 мм BioSpec Products). Ткани хранили при -20°С до анализа.
Реагенты и растворы
Анандамид [этаноламин-1-3Н] получали от American Radiochemicals (удельная активность 60 Ки/ммоль). Все другие реагенты получали от Sigma-Aldrich. Optiphase Supermix получали от Perkin Elmer.
Подготовка тканей
Ткани оттаивали на льду; печень гомогенизировали в гомогенизаторе Precellys 24 Dual Tissue (Bertin Technologies) в течение 2 циклов по 5 сек с интервалом по 5 мин во льду, а мозг гомогенизировали с использованием измельчителя Heidolph Silent Crusher М (зонд 8 F/M) в течение примерно 45 секунд при максимальной скорости. Общее содержание белка в гомогенатах определяли с помощью анализа белков BioRad (от BioRad) с использованием стандартной кривой БСА (50-250 мкг/мл).
Ферментный анализ
Реакционная смесь (общий объем 200 мкл) содержала: 2 мкМ АЕА (2 мкМ АЕА + 5 нМ 3Н-АЕА), 0,1% БСА без жирных кислот, 15 мкг белка (мозга) или 1,5 мкг белка (печени), в 1 мМ ЭДТА, 10 мМ Трис, рН 7,6. После 15-минутного периода предварительной инкубации при температуре 37°С реакцию инициировали добавлением раствора субстрата (немеченый АЕА + меченый АЕА+БСА). Реакцию проводили в течение 7 мин для образцов печени и в течение 10 мин для образцов мозга, и прекращали путем добавления 400 мкл смеси хлороформ/метанол (1:1 об/об). Образцы реакции дважды вращали, оставляли на льду в течение 5 минут, а затем центрифугировали в микроцентрифуге (7 минут, 7000 оборотов в минуту). Двести мкл супернатантов добавляли к 800 мкл сцинтилляционного коктейля Optiphase Supermix, предварительно распределенного в 24-луночные планшеты. Число импульсов в минуту (cpm) определяли с помощью сцинтилляционного счетчика MicrobetaTriLux. В каждом анализе готовили холостые пробы (без белка).
Процент сохранившейся ферментативной активности вычисляли по отношению к контролю и после вычитания холостой пробы.
Анализ метаболической стабильности CYP
Анализ стабильности тестируемых соединений проводили на MLM (микросомах печени мыши) или HLM (микросомах печени человека) в присутствии и в отсутствие NADPH (восстановленного никотинамидадениндинуклеотидфосфата).
Стабильность измеряли с использованием инкубационной смеси (общий объем 100 мкл), содержащей 1 мг/мл белка, 5 мМ MgCl2 и 50 мМ K-фосфатного буфера. Образцы инкубировали в присутствии и в отсутствие 1 мМ NADPH. Реакционные смеси предварительно инкубировали 5 мин и реакцию инициировали с помощью тестируемого соединения (5 мкМ для HLM и 50 мкМ для MLM). Образцы инкубировали в течение 60 мин при встряхивании на водяной бане при 37°С. Реакцию останавливали добавлением 100 мкл ацетонитрила. Затем образцы центрифугировали, фильтровали и вводили супернатант в ВЭЖХ-МСД. Тестируемые соединения растворяли в ДМСО, конечная концентрация ДМСО в реакционной смеси была ниже 0,5% (об/об). В момент Т0 добавляли ацетонитрил перед добавлением соединения. Все эксперименты проводились с образцами в двух повторах.
Тестируемые соединения:
Соединение 1 представляющее собой (N-метил-4-(3-(сульфамоиламино)фенил)-N-(тетрагидро-2H-пиран-4-ил)-1Н-имидазол-1-карбоксамид).
Соединение 2 представляющее собой ((4-(4-Гидрокси-3-метилфенил)-N-метил-N-(1-пропионилпиперидин-4-ил)-1Н-имидазол-1-карбоксамид).
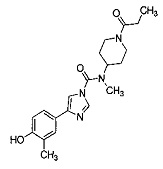
Соединение 3 представляющее собой ((N-(1-(Циклопропанкарбонил)пиперидин-4-ил)-4-(4-гидрокси-3-метилфенил)-N-метил-1Н-имидазол-1-карбоксамид).

Как видно из приведенной выше таблицы, соединения 1, 2 и 3 являются относительно эффективными соединениями с точки зрения ингибирования FAAH в печени.
Периферийную селективность можно рассчитать путем деления активности FAAH в печени на активность FAAH в головном мозге. При этом более низкое значение показывает, что соединение является периферически более селективным.
Результаты приведены в таблице ниже:

Эти результаты показывают, что соединения 2 и 3 являются наиболее периферически селективными соединениями, но все соединения проявляют относительно высокую периферическую селективность.
Дополнительные данные, относящиеся к активности FAAH в различных концентрациях, для соединений, приведены в таблице ниже:

Как видно из вышеприведенных данных, соединения 2 и 3 являются наиболее эффективными, поскольку они ингибируют активность FAAH даже при относительно низкой дозе. Однако все соединения являются относительно эффективными.
Кроме того, были проведены аналогичные эксперименты на крысах, которые показали следующие результаты:
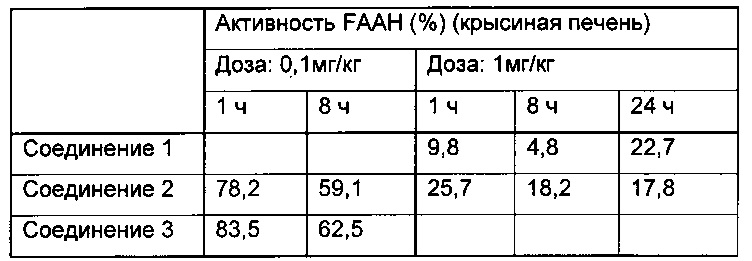
Как видно из вышеприведенных данных, все соединения показывают относительно хорошее ингибирование на крысиной печени и являются относительно эффективными.
Метаболическая стабильность
В приведенной ниже таблице показана метаболическая стабильность соединений. Данные по стабильности приведены в виде % оставшегося соединения через 1 ч воздействия в MLM или HLM. 100% означает полное отсутствие метаболической реакции и 0% соответствует полному ферментативному расщеплению. "CYP-" относится к отсутствию кофактора (NADPH), который является существенным для метаболических реакций CYP. Поэтому "CYP-" можно рассматривать в качестве контрольного значения. "CYP+" относится к наличию кофактора, и ферментативное расщепление может происходить в соответствии со стабильностью тестируемого соединения. Как можно видеть, все соединения являются метаболически стабильными.

| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ МОЧЕВИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФЕРМЕНТОВ | 2013 |
|
RU2685425C2 |
| НОВЫЕ СОЕДИНЕНИЯ - АНТАГОНИСТЫ РЕЦЕПТОРА НЕЙРОКИНИНА 1 | 2013 |
|
RU2631319C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2009 |
|
RU2553451C2 |
| Соединение формулы (I), фармацевтическая композиция, применение соединения формулы (I) для лечения гематологических и/или пролиферативных нарушений | 2016 |
|
RU2758259C2 |
| ТИАЗОЛОПИРИМИДИНЫ | 2012 |
|
RU2610840C2 |
| 2-(Азаиндол-2-ил)бензимидазолы в качестве ингибиторов PAD4 | 2012 |
|
RU2611010C2 |
| ФАРМАЦЕВТИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ Axl | 2011 |
|
RU2573834C2 |
| МОДИФИКАТОР СЛАДКОГО ВКУСА И АРОМАТА | 2013 |
|
RU2666534C2 |
| Бициклические конденсированные гетероарильные или арильные соединения в качестве модуляторов IRAK4 | 2016 |
|
RU2684324C1 |
Изобретение относится к области органической химии, а именно к гетероциклическим соединениям указанных ниже структур и к их фармацевтически приемлемым солям. Также изобретение относится к фармацевтической композиции на основе указанных соединений, их применению и способу лечения или профилактики состояния, развитие и симптомы которого связаны с субстратом фермента FAAH. Технический результат: получены новые гетероциклические соединения, обладающие полезными биологическими свойствами. 5 н. и 5 з.п. ф-лы.
 ,
, 

1. Соединение, имеющее структуру, выбранную из следующих:

или его фармацевтически приемлемая соль.
2. Соединение по п. 1, где фармацевтически приемлемая соль представляет собой гидрохлоридную соль, ацетатную соль, трифторацетатную соль, метансульфонатную соль, 2-гидроксипропан-1,2,2-трикарбоксилатную соль, (2R,3R)-2,3-дигидроксисукцинатную соль, фосфатную соль, сульфатную соль, бензоатную соль, 2-гидроксибензоатную соль, S-(+)-манделатную соль, S-(-)-малатную соль, S-(-)-пироглутаматную соль, пируватную соль, пара-толуолсульфонатную соль, 1-R-(-)-камфорсульфонатную соль, фумаратную соль, малеатную соль или оксалатную соль.
3. Фармацевтическая композиция для лечения или профилактики состояния, развитие или симптомы которого связаны с субстратом фермента FAAH (гидролаза амидов жирных кислот), содержащая эффективное количество соединения по п. 1 вместе с одним или более фармацевтически приемлемыми эксципиентами.
4. Фармацевтическая композиция по п. 3, где композиция предназначена для перорального введения.
5. Фармацевтическая композиция по п. 3, где композиция находится в форме стерильного инъекционного препарата.
6. Применение соединения по п. 1 или 2 для модуляции активности фермента FAAH.
7. Применение соединения по п. 1 или 2 в лечении или профилактике состояния, развитие или симптомы которого связаны с субстратом фермента FAAH.
8. Применение по п. 7, где состояние представляет собой расстройство, связанное с эндоканнабиноидной системой.
9. Применение по п. 8, где расстройство выбирают из регуляции аппетита, ожирения, нарушения обмена веществ, кахексии, анорексии, боли, воспаления, нейротоксичности, нейротравмы, инсульта, рассеянного склероза, травмы спинного мозга, болезни Паркинсона, леводопа-индуцированной дискинезии, болезни Хантингтона, синдрома Жиля де ла Туретта, поздней дискинезии, дистонии, амиотрофического бокового склероза, болезни Альцгеймера, эпилепсии, шизофрении, тревоги, депрессии, бессонницы, тошноты, рвоты, алкогольных расстройств, привыкания к наркотическим средствам, таким как опиаты, никотин, кокаин, алкоголь и психостимуляторы, гипертонии, циркуляторного шока, реперфузионного повреждения миокарда, атеросклероза, астмы, глазной гипертензии, глаукомы, ретинопатии, рака, воспалительного заболевания кишечника, острого и хронического заболевания печени, такого как гепатит и цирроз печени, артрита и остеопороза.
10. Способ лечения или профилактики состояния, развитие или симптомы которого связаны с субстратом фермента FAAH, включающий введение пациенту, нуждающемуся в таком лечении или профилактике, терапевтически эффективного количества соединения по п. 1 или композиции по п. 3.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Приспособление для беспыльного пересыпания мусора из ящиков в повозку | 1926 |
|
SU12589A1 |

