Область изобретения
Настоящее изобретение относится к производному пиразола формулы (I), его фармацевтически приемлемой соли, его пролекарству, его гидрату, его стереоизомеру или его дейтерированной форме, где n, Y, Ra, R1, R2, R3, R4, R5 и R6 являются такими, как определено в настоящем описании далее, к фармацевтической композиции, содержащей соединение формулы (I) в качестве активного ингредиента, к способам их получения и к способам их применения. В частности, настоящее изобретение относится к соединению формулы (I) в качестве ингибитора фосфатидилинозитол-3-киназы (PI3K), которое можно использовать для лечения или предупреждения воспалительного, аутоиммунного, орфанного и гиперпролиферативного заболевания и нарушения.
Уровень техники, к которому относится изобретение
Семейство фосфоинозитол-3-киназы (PI3K), содержащие киназы липидов, подразделяют на три различных класса: класс I, класс II и класс III. Классификации основаны на первичной структуре, регуляции и специфичности к липидному субстрату in vitro. PI3K 1 класса наиболее детально исследованы, и они активируются рецепторами клеточной поверхности, такими как сопряженные с G-белком рецепторы (GPCR), факторы роста и инсулин. PI3K класса I далее подразделяются на два подкласса: IA и IB. Ферменты PI3K IA представляют собой гетеродимеры, состоящие из каталитической субъединицы p110 (α, β и δ) и регуляторной субъединицы (p85, p55, p50). Семейство ферментов PI3K IB состоит из одного члена - PI3-киназы γ. PI3K-α вовлечена в метаболизм глюкозы и передачу сигнала инсулина, в то время как PI3K-β вовлечена в активацию тромбоцитов при тромботических заболеваниях. Напротив, изоформы PI3K-δ и PI3K-γ в основном экспрессируются в гемопоэтических системах. Фармакологическое и генетическое вмешательство выявило, что PI3K-δ является необходимым для регуляции как врожденного, так и адаптивного иммунного ответа, включая экспрессию и активацию медиаторов воспаления, привлечение воспалительных клеток, ремоделирование дыхательных путей и нечувствительность к кортикостероидам при хроническом воспалительном заболевании дыхательных путей [Rommel C, et al. Nat. Rev. Immunol. 2007; 7(3):191-201; Medina-Tato DA, et. al. Immunology. 2007; 121(4):448-61 и Foster JG et.al. Pharmacol. Rev. 2012; 64(4):1027-54].
PI3K является подтвержденной мишенью, изученной различными фармацевтическими компаниями. Например, иделалисиб (Gilead), ингибитор PI3Kδ, был выпущен в продажу в 2014 году для лечения злокачественной опухоли; однако он имеет предостережение в черной рамке (гепатотоксичность, тяжелая диарея или колит) и риск DDI (рекомендован мониторинг для стероидов; не рекомендован с сальметеролом). GSK-2269557 (GlaxoSmithKline), специфический ингибитор PI3Kδ, находится на фазе II для астмы и хронического обструктивного заболевания легких (COPD) в качестве ингалируемого продукта. Дувелисиб (Infinity), двойной ингибитор PI3Kδ/γ, снят с продажи в январе 2015 года (фаза II, ингалируемый продукт против астмы и ревматоидного артрита). Мыши PI3K δ/γ K/O продемонстрировали тяжелое нарушение развития тимоцитов (оппортунистические инфекции). RV-1729 (RespiVert), другой двойной ингибитор PI3Kδ/γ, находится на фазе I для астмы и COPD в качестве ингалируемого терапевтического средства. AMG-319 (Amgen), TGR-1202/RP-5264 (Incozen/TG therapeutics) и INCB040093 (Incyte) находятся на различных стадиях разработки для лечения лимфоидной злокачественной опухоли.
Ингибиторы PI3K, предпочтительно ингибиторы PI3Kδ, описаны в WO2012/082997, WO2012/037226, WO2012/007493, WO2012/107465, WO2011/058027, WO2010/136491, WO2010/138589, WO2010/044401, WO2009/146406, WO2009/045174, WO2009/045175, WO2009/053716 и GB2431156.
Также в настоящее описание включены в качестве ссылок WO2012/104776, WO2010/005558, WO2010/114494, WO2009/100406, WO2009/034386 WO2008/116129, WO2005/000404 и WO2004035740. Однако ни в одной из цитированных ссылок не описаны производные пиразола, как описано далее.
Несмотря на значительный прогресс, остается неудовлетворенная необходимость и существенная возможность в отношении безопасных и перорально эффективных ингибиторов фосфатидилинозитол-3-киназы δ (PI3Kδ) для лечения и/или предупреждения воспалительного, аутоиммунного и гиперпролиферативного заболевания или нарушения, такого как аллергическая астма, тяжелая астма, резистентная к стероидам астма, COPD, псориаз, псориатический артрит, ревматоидный артрит, рассеянный склероз, системная красная волчанка или злокачественная опухоль. В результате тщательного исследования авторы настоящего изобретения идентифицировали безопасные и перорально эффективные ингибиторы PI3Kδ, которые можно использовать для указанной цели.
Сущность изобретения
Таким образом, настоящее изобретение относится производному пиразола формулы (I) или к его фармацевтически приемлемой соли, к фармацевтической композиции, содержащей соединение формулы (I) в качестве активного ингредиента, к способам их получения и к способам их применения. В частности, настоящее изобретение относится к соединению формулы (I), пригодному для лечения или предупреждения воспалительного и аутоиммунного заболевания или нарушения, ассоциированного с нарушением регуляции PI3Kδ.
Таким образом, один аспект настоящего изобретения относится к соединению формулы (I)
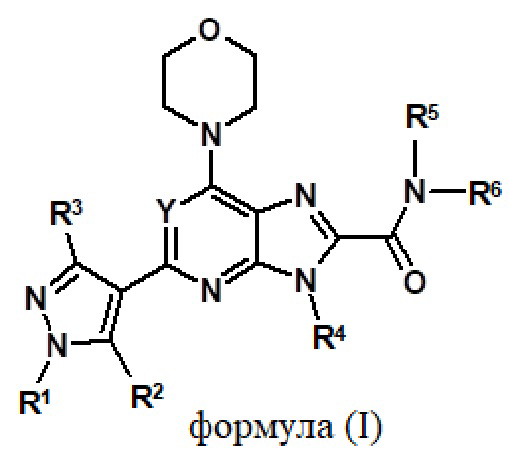
или его фармацевтически приемлемой соли, где:
Y обозначает N, CH, CF, CCl или CCH3;
R1, R2 и R3 независимо обозначают H, алкил, содержащий 1-3 атомов углерода, или галогенированный алкил, содержащий 1-3 атомов углерода;
R4 и R5 независимо обозначают H или необязательно замещенный алкил, содержащий 1-3 атомов углерода;
R6 обозначает алкил, циклоалкил или гетероциклил, где алкил, циклоалкил и гетероциклил являются необязательно замещенными; и
R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют необязательно замещенный гетероциклил, необязательно содержащий один или несколько гетероатом(ов), выбранный из N, O или S.
Другой аспект относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль в качестве ее активного ингредиента и один или несколько фармацевтически приемлемый эксципиент(ов).
Другой аспект относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или уменьшения тяжести заболевания или нарушения, отвечающих на ингибирование PI3Kδ.
Другой аспект относится к способу лечения или уменьшения тяжести заболевания или нарушения, отвечающих на ингибирование PI3Kδ у пациента, включающему введение указанному пациенту терапевтически эффективного количества соединения формулы (I), его фармацевтически приемлемой соли или их фармацевтической композиции.
Другой аспект относится к соединению формулы (1) или его фармацевтически приемлемой соли для применения для лечения или уменьшения тяжести заболевания или нарушения, отвечающего на ингибирование PI3Kδ.
В другом варианте осуществления предусматривается лекарственное средство для ингибирования PI3Kδ, содержащее соединение формулы (1) или его фармацевтически приемлемую соль в качестве активного ингредиента.
Настоящее изобретение включает следующие варианты осуществления [1]-[24] и [1a]-[23a].
[1] Соединение формулы (I) или его фармацевтически приемлемая соль, где Y обозначает N.
[2] Соединение формулы (I) или его фармацевтически приемлемая соль, где Y обозначает CH, CF, CCl или CCH3.
[3] Соединение формулы (I) или его фармацевтически приемлемая соль, где R1 и R4 независимо обозначают H, метил, этил, пропил или изопропил.
[4] Соединение формулы (I) или его фармацевтически приемлемая соль, где R2 и R3 обозначают H, метил или трифторметил.
[5] Соединение формулы (I) или его фармацевтически приемлемая соль, где R5 обозначает H, метил или этил, и R6 обозначает необязательно замещенный алкил, содержащий 1-6 атомов углерода.
[6] Соединение формулы (I) или его фармацевтически приемлемая соль, где R5 обозначает H, метил или этил, и R6 обозначает необязательно замещенный 5-6-членный циклоалкил.
[7] Соединение формулы (I) или его фармацевтически приемлемая соль, где R5 обозначает H, метил или этил, и R6 обозначает необязательно замещенный 5-6-членный гетероциклил.
[8] Соединение формулы (I) или его фармацевтически приемлемая соль, где R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют 4-6-членый гетероциклил, необязательно замещенный Ra, который выбран из алкила, арила, гетероарила, гетероциклила, -(CH2)pNRbRc, -NRbCORc, -NRcS(O)2Rc, (CH2)pC(O)ORd, -C(O)NRbRc, -C(O)Rd, -C(O)ORd, -ORd, где алкил, арил, гетероарил и гетероциклил являются необязательно замещенными; Rb, Rc, Rd независимо выбраны из H или необязательно замещенного алкила, содержащего 1-6 атомов углерода; и p представляет собой целое число 0, 1, 2 или 3.
[9] Соединение формулы (I) или его фармацевтически приемлемая соль, где R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют 6-членный гетероциклил, необязательно замещенный Ra, который выбран из алкила, арила, гетероарила, гетероциклила, -(CH2)pNRbRc, -NRbCORc, -NRbS(O)2Rc, (CH2)pC(O)ORd, -C(O)NRbRc, -C(O)Rd, -C(O)ORd, -ORd, где алкил, арил, гетероарил и гетероциклил являются необязательно замещенными; Rb, Rc, Rd независимо выбраны из H или алкила, содержащего 1-6 атомов углерода; и p представляет собой целое число 0, 1, 2 или 3.
[10] Соединение формулы (I) или его фармацевтически приемлемая соль, где R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют спирокольцо, содержащее 5-7 атомов углерода и по меньшей мере один N или O.
[11] Соединение формулы (I) или его фармацевтически приемлемая соль, где R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют конденсированное кольцо, содержащее 5-7 атомов углерода и по меньшей мере один N или O.
[12] Соединение формулы (I), которое выбрано из:
2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[2-(пиридин-3-ил)этил]-9H-пурин-8-карбоксамида (соединение № 1),
2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[2-(морфолин-4-ил)этил]-9H-пурин-8-карбоксамида (соединение № 2),
[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил](2-окса-7-азаспиро[3.5]нон-7-ил)метанона (соединение № 3),
(3-гидроксиазетидин-1-ил)[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 4),
{4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 5),
(9aR)-8-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}гексагидропиразинo[2,1-c][1,4]оксазин-4(3H)-она (соединение № 6),
4-(1-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперидин-4-ил)морфолин-3-она (соединение № 7),
2-(4-{[2-(1,3-диметил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперазин-1-ил)-2-метилпропанамида (соединение № 8),
[2-(1,3-диметил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(тетрагидро-2H-пиран-4-ил)пиперазин-1-ил]метанона (соединение № 9),
2-метил-2-(4-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперазин-1-ил)пропанамида (соединение № 10),
[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(тетрагидро-2H-пиран-4-ил)пиперазин-1-ил]метанона (соединение № 11),
[2-(1,3-диметил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(2-гидроксипропан-2-ил)пиперидин-1-ил]метанона (соединение № 12),
[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][(3S)-3-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 13),
[4-(2-гидроксипропан-2-ил)пиперидин-1-ил][2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 14),
(1,1-диоксидотиоморфолин-4-ил)[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 15),
[2-(3,5-диметил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 16),
{2-[1-метил-3-(трифторметил)-1H-пиразол-4-ил]-6-(морфолин-4-ил)-9H-пурин-8-ил}[4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 17),
[4-(морфолин-4-ил)пиперидин-1-ил]{6-(морфолин-4-ил)-2-[1-(пропан-2-ил)-1H-пиразол-4-ил]-9H-пурин-8-ил}метанона (соединение № 18),
[4-(морфолин-4-ил)пиперидин-1-ил]{6-(морфолин-4-ил)-2-[3-(трифторметил)-1H-пиразол-4-ил]-9H-пурин-8-ил}метанона (соединение № 19),
[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил](пиперазин-1-ил)метанона (соединение № 20),
[4-(диметиламино)пиперидин-1-ил][9-этил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 21),
[9-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 22),
[9-метил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 23),
[4-(диметиламино)пиперидин-1-ил][9-метил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 24),
[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(пиримидин-2-илкарбонил)пиперазин-1-ил]метанона (соединение № 25),
N-(1-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперидин-4-ил)ацетамида (соединение № 26),
[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(пиримидин-2-ил)пиперазин-1-ил]метанона (соединение № 27),
N-(1-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперидин-4-ил)метансульфонамида (соединение № 28),
1-(4-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперазин-1-ил)-2-(пиразин-2-ил)этанона (соединение № 29),
5,6-дигидроимидазо[1,2-a]пиразин-7(8H)-ил[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 30),
4-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперазин-2-она (соединение № 31),
[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил](4-фенилпиперазин-1-ил)метанона (соединение № 32),
N-циклогексил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (соединение № 33),
[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(метилсульфонил)пиперазин-1-ил]метанона (соединение № 34),
[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил](4-фенилпиперидин-1-ил)метанона (соединение № 35),
N-(циклогексилметил)-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (соединение № 36),
N-[(3S)-1-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пирролидин-3-ил]ацетамида (соединение № 37),
N-[(3R)-1-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пирролидин-3-ил]метансульфонамида (соединение № 38),
1-(4-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперазин-1-ил)этанона (соединение № 39),
[2-(1,5-диметил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 40),
[2-(1,3-диметил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 41),
[4-(морфолин-4-ил)пиперидин-1-ил][6-(морфолин-4-ил)-2-(1H-пиразол-4-ил)-9H-пурин-8-ил]метанона (соединение № 42),
трет-бутил 4-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперазин-1-карбоксилата (соединение № 43),
[(3S)-3-(диметиламино)пирролидин-1-ил][2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 44),
этил (4-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперазин-1-ил)ацетата (соединение № 45),
(4-метилпиперазин-1-ил)[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 46),
N-(1-метилпиперидин-4-ил)-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (соединение № 47),
N,N-диметил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (соединение № 48),
N-(1-бензилпиперидин-4-ил)-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (соединение № 49),
(1-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперидин-4-ил)уксусной кислоты (соединение № 50),
этил (1-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}пиперидин-4-ил)ацетата (соединение № 51),
N-(2-метоксиэтил)-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (соединение № 52),
[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил](пиперидин-1-ил)метанона (соединение № 53),
[4-(диметиламино)пиперидин-1-ил][6-(морфолин-4-ил)-2-(1H-пиразол-4-ил)-9H-пурин-8-ил]метанона (соединение № 54),
[4-(диметиламино)пиперидин-1-ил][6-(морфолин-4-ил)-2-(1-пропил-1H-пиразол-4-ил)-9H-пурин-8-ил]метанона (соединение № 55),
[4-(диметиламино)пиперидин-1-ил][2-(1-этил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 56),
[4-(диметиламино)пиперидин-1-ил][2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 57),
[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 58),
N-этил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[2-(морфолин-4-ил)этил]-9H-пурин-8-карбоксамида (соединение № 59),
N-метил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[2-(морфолин-4-ил)этил]-9H-пурин-8-карбоксамида (соединение № 60),
9-метил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[2-(морфолин-4-ил)этил]-9H-пурин-8-карбоксамида (соединение № 61),
[(3S)-3-гидроксипирролидин-1-ил][2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанона (соединение № 62),
2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[3-(морфолин-4-ил)пропил]-9H-пурин-8-карбоксамида (соединение № 63),
2-(1-метил-1H-пиразол-4-ил)-N-{2-[1-метилпирролидин-2-ил]этил}-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (соединение № 64),
{4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[6-(морфолин-4-ил)-2-(1H-пиразол-4-ил)-9H-пурин-8-ил]метанона (соединение № 65),
[2-(3-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 66),
9-метил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[3-(морфолин-4-ил)пропил]-9H-пурин-8-карбоксамида (соединение № 67),
2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[3-(2-оксопирролидин-1-ил)пропил]-9H-пурин-8-карбоксамида (соединение № 68),
N-этил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[3-(морфолин-4-ил)пропил]-9H-пурин-8-карбоксамида (соединение № 69),
N-[2-(4-метилпиперазин-1-ил)этил]-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (соединение № 70),
2-(1,3-диметил-1H-пиразол-4-ил)-N-{[3-(гидроксиметил)оксетан-3-ил]метил}-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (соединение № 71),
[6-(морфолин-4-ил)-2-(1H-пиразол-4-ил)-9H-пурин-8-ил](2-окса-7-азаспиро[3.5]нон-7-ил)метанона (соединение № 72),
9-метил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[3-(2-оксопирролидин-1-ил)пропил]-9H-пурин-8-карбоксамида (соединение № 73),
4-(1-{[6-(морфолин-4-ил)-2-(1H-пиразол-4-ил)-9H-пурин-8-ил]карбонил}пиперидин-4-ил)морфолин-3-она (соединение № 74),
N-{3-[цис-2,6-диметилморфолин-4-ил]пропил}-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (соединение № 75),
N-(3-гидрокси-2,2-диметилпропил)-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (соединение № 76),
4-(1-{[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]карбонил}пиперидин-4-ил)морфолин-3-она (соединение № 77),
[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил](2-окса-7-азаспиро[3.5]нон-7-ил)метанона (соединение № 78),
[6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил](2-окса-7-азаспиро[3.5]нон-7-ил)метанона (соединение № 79),
(9aR)-8-{[6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]карбонил}гексагидропиразинo[2,1-c][1,4]оксазин-4(3H)-она (соединение № 80),
[6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(тетрагидро-2H-пиран-4-ил)пиперазин-1-ил]метанона (соединение № 81),
2-(4-{[6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]карбонил}пиперазин-1-ил)-2-метилпропанамида (соединение № 82),
[3-(циклопропилметил)-5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 83),
[5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(2-гидроксипропан-2-ил)пиперидин-1-ил]метанона (соединение № 84),
1-{[5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]карбонил}пиперидин-4-карбоксамида (соединение № 85),
[5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил](4-гидроксипиперидин-1-ил)метанона (соединение № 86),
[5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 87),
[6-метил-7-(морфолин-4-ил)-5-(1,3,5-триметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 88),
[5-(1,3-диметил-1H-пиразол-4-ил)-6-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 89),
[6-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 90),
[6-хлор-5-(1,3-диметил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 91),
[6-хлор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 92),
[6-фтор-7-(морфолин-4-ил)-5-(1,3,5-триметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 93),
[5-(1,3-диметил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 94),
[5-(1,5-диметил-1H-пиразол-4-ил)-6-фтор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 95),
[5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 96),
[4-(диметиламино)пиперидин-1-ил][5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 97),
[6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 98),
[4-(диметиламино)пиперидин-1-ил][6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 99),
[4-(диметиламино)пиперидин-1-ил][3-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 100),
[4-(диметиламино)пиперидин-1-ил][5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 101),
[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 102),
5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-N-[2-(морфолин-4-ил)этил]-3H-имидазо[4,5-b]пиридин-2-карбоксамида (соединение № 103),
[5-(1,3-диметил-1H-пиразол-4-ил)-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 104),
6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-N-[2-(морфолин-4-ил)этил]-3H-имидазо[4,5-b]пиридин-2-карбоксамида (соединение № 105),
[3-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 106),
[5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(2-гидроксипропан-2-ил)пиперидин-1-ил]метанона (соединение № 107),
[6-фтор-3-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил](2-окса-7-азаспиро[3.5]нон-7-ил)метанона (соединение № 108),
{4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 109),
{4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[3-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 110),
[6-фтор-3-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 111),
{4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 112),
{4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[5-(1,3-диметил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 113),
{4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[5-(1,3-диметил-1H-пиразол-4-ил)-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 114),
5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-N-[3-(морфолин-4-ил)пропил]-3H-имидазо[4,5-b]пиридин-2-карбоксамида (соединение № 115),
5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-N-[3-(2-оксопирролидин-1-ил)пропил]-3H-имидазо[4,5-b]пиридин-2-карбоксамида (соединение № 116),
[4-(2-гидроксипропан-2-ил)пиперидин-1-ил][5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 117),
{4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[7-(морфолин-4-ил)-5-(1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 118),
{3-[(2R,6S)-2,6-диметилморфолин-4-ил]азетидин-1-ил}[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 119),
[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][3-(морфолин-4-ил)азетидин-1-ил]метанона (соединение № 120),
N-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-N-[1-(тетрагидро-2H-пиран-4-ил)пиперидин-4-ил]-3H-имидазо[4,5-b]пиридин-2-карбоксамида (соединение № 121),
8-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}-2,8-диазаспиро[4.5]декан-3-она (соединение № 122),
{6-фтор-7-(морфолин-4-ил)-5-[3-(трифторметил)-1H-пиразол-4-ил]-3H-имидазо[4,5-b]пиридин-2-ил}[4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 123),
6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-N-[3-(морфолин-4-ил)пропил]-3H-имидазо[4,5-b]пиридин-2-карбоксамида (соединение № 124),
[6-фтор-7-(морфолин-4-ил)-5-(1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (соединение № 125),
[6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][3-(морфолин-4-ил)азетидин-1-ил]метанона (соединение № 126),
{3-[(2R,6S)-2,6-диметилморфолин-4-ил]азетидин-1-ил}[6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 127),
4-(1-{[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]карбонил}азетидин-3-ил)пиперазин-2-она (соединение № 128),
1-метил-4-(1-{[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]карбонил}азетидин-3-ил)пиперазин-2-она (соединение № 129),
1-метил-4-(1-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}азетидин-3-ил)пиперазин-2-она (соединение № 130),
N-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-N-[1-(оксетан-3-ил)пиперидин-4-ил]-3H-имидазо[4,5-b]пиридин-2-карбоксамида (соединение № 131),
N-метил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[1-(оксетан-3-ил)пиперидин-4-ил]-9H-пурин-8-карбоксамида (соединение № 132),
[4-(морфолин-4-ил)пиперидин-1-ил][7-(морфолин-4-ил)-5-(1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанона (соединение № 133),
[4-(морфолин-4-ил)пиперидин-1-ил]{7-(морфолин-4-ил)-5-[3-(трифторметил)-1H-пиразол-4-ил]-3H-имидазо[4,5-b]пиридин-2-ил}метанона (соединение № 134),
[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(тетрагидро-2H-пиран-4-ил)пиперазин-1-ил]метанона (соединение № 135),
или их фармацевтически приемлемой соли.
[13] Соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии.
[14] Соединение формулы (I) или его фармацевтически приемлемая соль для применения для лечения аутоиммунного заболевания или нарушения.
[15] Соединение формулы (I) или его фармацевтически приемлемая соль для применения для лечения воспалительного заболевания или нарушения.
[16] Соединение формулы (I) или его фармацевтически приемлемая соль для применения для лечения гиперпролиферативного заболевания или нарушения.
[17] Соединение формулы (I) или его фармацевтически приемлемая соль для применения для лечения псориаза, псориатического артрита или ревматоидного артрита.
[18] Соединение формулы (I) или его фармацевтически приемлемая соль для применения для лечения аллергической астмы, тяжелой астмы, резистентной к стероидам астмы или COPD.
[19] Соединение формулы (I) или его фармацевтически приемлемая соль для применения для лечения системной красной волчанки, первичного синдрома иммунодефицита, опухоли или злокачественной опухоли.
[20] Фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, для применения для лечения аллергической астмs.
[21] Фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, для применения для лечения тяжелой астмы.
[22] Фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, для применения для лечения резистентной к стероидам астмы.
[23] Фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, для применения для лечения COPD.
[24] Фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, для применения для лечения ревматоидного артрита.
[1a] Производное пиразола формулы (I) или его фармацевтически приемлемая соль, где:
Y обозначает N, CH, CF, CCl или CCH3;
R1, R2 и R3 независимо обозначают H, алкил, содержащий 1-3 атомов углерода или галогенированный алкил, содержащий 1-3 атомов углерода;
R4 и R5 независимо обозначают H или необязательно замещенный алкил, содержащий 1-3 атомов углерода;
R6 обозначает алкил, циклоалкил или гетероциклил, где алкил, циклоалкил и гетероциклил являются необязательно замещенными; и
R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют необязательно замещенный гетероциклил, необязательно содержащий один или несколько гетероатом(ов), выбранных из N, O или S.
[2a] Соединение согласно [1a] или его фармацевтически приемлемая соль, где
Y обозначает N, CH, CF, CCl или CCH3;
R1, R2 и R3 независимо обозначают H, (C1-C3)алкильную группу или галогенированную (C1-C3)алкильную группу;
R4 обозначает H или (C1-C3)алкильную группу,
где (C1-C3)алкильная группа необязательно замещена (C3-C6)циклоалкильной группой;
R5 обозначает H или (C1-C3)алкильную группу;
R6 обозначает группу, имеющую формулу -X-R6a,
[где X обозначает связь или (C1-C3) алкиленильную группу,
R6a обозначает a (C3-C6)циклоалкильную группу, гетероарильную группу или 4-6-членную гетероциклическую группу, которая необязательно имеет 1-3 заместителя, независимо выбранных из группы заместителей A],
R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют 4-6-членное гетероциклическое кольцо, спиро- или конденсированное кольцо, содержащее 5-7 атомов углерода, и по меньшей мере один N или O,
где 4-6-членное гетероциклическое кольцо необязательно замещено группой, имеющей формулу -W-R6b
[где W обозначает группу, состоящую из связи, (C1-C3)алкиленильной группы, -NH-, -CO-, -(C1-C3) алкиленил-CO- или -CO-(C1-C3)алкиленила-,
R6b обозначает гидроксигруппу, (C1-C6)алкоксигруппу, аминогруппу, ди(C1-C3)алкиламиногруппу, (C1-C3)алкилкарбонильную группу, (C1-C3)алкилсульфонильную группу, гетероарильную группу, арильную группу или 4-6-членную гетероциклическую группу, которая необязательно имеет 1-3 заместителя, независимо выбранных из группы заместителей A],
группа заместителей A представляет собой группу, состоящую из (C1-C3)алкильной группы, арил-(C1-C3)алкильной группы, оксогруппы, гидрокси-(C1-C3)алкильной группы и оксетанильной группы.
[3a] Соединение согласно [1a] или [2a] или его фармацевтически приемлемая соль, где Y обозначает N.
[4a] Соединение согласно любому из [1a]-[3a] или его фармацевтически приемлемая соль, где Y обозначает CH, CF, CCl или CCH3.
[5a] Соединение согласно любому из [1a]-[4a] или его фармацевтически приемлемая соль, где R1 обозначает метильную группу, и R2 и R3 независимо обозначают H или метильную группу.
[6a] Соединение согласно любому из [1a]-[5a] или его фармацевтически приемлемая соль, где R4 обозначает H или метильную группу,
[7a] Соединение согласно любому из [1a]-[6a] или его фармацевтически приемлемая соль, где R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют азетидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо или пиперазиновое кольцо, где азетидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо и пиперазиновое кольцо являются необязательно замещенными тетрагидропиранильной группой, морфолинильной группой или 2,6-диметилморфолинильной группой.
[8a] Соединение согласно любому из [1a]-[7a], которое представляет собой {4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанон, или его фармацевтически приемлемая соль.
[9a] Соединение согласно любому из [1a]-[7a], которое представляет собой [5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанон, или его фармацевтически приемлемая соль.
[10a] Соединение согласно любому из [1a]-[7a], которое представляет собой [2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанон, или его фармацевтически приемлемая соль.
[11a] Соединение согласно любому из [1a]-[7a], которое представляет собой [5-(1,3-диметил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанон, или его фармацевтически приемлемая соль.
[12a] Соединение согласно любому из [1a]-[7a], которое представляет собой {4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[5-(1,3-диметил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанон, или его фармацевтически приемлемая соль.
[13a] Фармацевтическая композиция, содержащая соединение согласно любому из [1a]-[12a] или его фармацевтически приемлемую соль в качестве активного ингредиента и один или несколько фармацевтически приемлемый эксципиент(ов).
[14a] Фармацевтическая композиция согласно [13a] для лечения или уменьшения тяжести заболевания или нарушения, отвечающих на ингибирование фосфоинозитол-3-киназы δ(PI3Kδ).
[15a] Фармацевтическая композиция согласно [14a], где заболевание или нарушение представляет собой псориаз, псориатический артрит, ревматоидный артрит, аллергическую астму, тяжелую астму, резистентную к стероидам астму, COPD, системную красную волчанку, первичный синдром иммунодефицита или злокачественную опухоль.
[16a] Применение соединения согласно любому из [1a]-[12a] или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или уменьшения тяжести заболевания или нарушения, отвечающего на ингибирование PI3Kδ.
[17a] Применение согласно [16a], где заболевание или нарушение представляет собой псориаз, псориатический артрит, ревматоидный артрит, аллергическую астму, тяжелую астму, резистентную к стероидам астму, COPD, системную красную волчанку, первичный синдром иммунодефицита или злокачественную опухоль.
[18a] Способ лечения или уменьшения тяжести заболевания или нарушения, отвечающего на ингибирование PI3Kδ, у пациента, включающий введение указанному пациенту терапевтически эффективного количества соединения согласно любому из [1a]-[12a] или его фармацевтически приемлемой соли.
[19a] Способ согласно [18a], где заболевание или нарушение представляет собой псориаз, псориатический артрит, ревматоидный артрит, аллергическую астму, тяжелую астму, резистентную к стероидам астму, COPD, системную красную волчанку, первичный синдром иммунодефицита или злокачественную опухоль.
[20a] Соединение согласно любому из [1a]-[12a] или его фармацевтически приемлемая соль для применения для лечения или уменьшения тяжести заболевания или нарушения, отвечающего на ингибирование PI3Kδ.
[21a] Соединение согласно [20a] или его фармацевтически приемлемая соль, где заболевание или нарушение представляет собой псориаз, псориатический артрит, ревматоидный артрит, аллергическую астму, тяжелую астму, резистентную к стероидам астму, COPD, системную красную волчанку, первичный синдром иммунодефицита или злокачественную опухоль.
[22a] Лекарственное средство для ингибирования PI3Kδ, содержащее соединение согласно любому из [1a]-[12a] или его фармацевтически приемлемую соль в качестве активного ингредиента.
[23a] Лекарственное средство согласно [22a], где заболевание или нарушение представляет собой псориаз, псориатический артрит, ревматоидный артрит, аллергическую астму, тяжелую астму, резистентную к стероидам астму, COPD, системную красную волчанку, первичный синдром иммунодефицита или злокачественную опухоль.
Вышеупомянутые аспекты и варианты осуществления, и другие аспекты, задачи, признаки и преимущества настоящего изобретения станут понятны из приведенного далее подробного описания и прилагаемой формулы изобретения.
Подробное описание изобретения
Как используют в рамках изобретения применяются следующие определения, если прямо не указано иное.
Следует понимать, что, если не утверждается обратное, "соединение общей формулы (I) или его фармацевтически приемлемая соль" относится к и включает любые и все соединения, описываемые формулой (I), их варианты осуществления, а также подтипы, включая все соли, их пролекарства, гидраты, стереоизомеры и дейтериевые формы. Также следует отметить, что формула единственного числа включает множественное число указанного объекта, если контекст явно не указывает на иное.
Термин "галоген", как используют в рамках изобретения отдельно или в комбинации, относится к фтору, хлору, брому и йоду.
Термин "алкил", как используют в рамках изобретения отдельно или в комбинации, относится к прямой или разветвленной цепи, содержащей от 1 до 12 атомов углерода. Прямая или разветвленная алкильная группа присоединена к любой доступной точке с образованием стабильного соединения. В определенных вариантах осуществления прямая или разветвленная алкильная группа содержит 1-6, 1-4 или 1-3 атомов углерода, как например, метил, этил, пропил, изопропил, бутил, трет-бутил и т.п. "Замещенный алкил" обозначает алкил, который независимо является замещенным, если нет иных указаний, одним или несколькими, предпочтительно 1, 2 или 3, более предпочтительно 1 или 2 заместителями, связанными с любым доступным атомом, с образованием стабильного соединения, где заместители выбраны из, но не ограничиваются ими, галогена, ORd, -C(O)NRbRc, необязательно замещенного циклоалкила, арила, гетероарила или гетероциклила. "(C1-C3)алкильная группа" относится к линейной или разветвленной алкильной группе, имеющей 1-3 атома углерода. Ее примеры включают метильную группу, этильную группу, пропильную группу и изопропильную группу. "Галогенированная (C1-C3)алкильная группа" означает описанную выше (C1-C3)алкильную группу, замещенную одной группой галогена. Ее примеры включают хлорметильную группу, бромметильную группу, фторметильную группу, 2-хлорметильную группу, 2-бромметильную группу и 2-фторметильную группу. "(C1-C6)алкоксигруппа" относится к линейной или разветвленной алкоксигруппе, имеющей 1-6 атомов углерода. Ее примеры включают метоксигруппу, этоксигруппу, пропоксигруппу, бутоксигруппу и трет-бутоксигруппу. "Ди(C1-C3)алкиламиногруппа" означает аминогруппу, замещенную двумя описанными выше (C1-C3)алкильными группами. Ее примеры включают диметиламиногруппу, диэтиламиногруппу и диизопропиламиногруппу. "(C1-C3)алкилкарбонильная группа" означает карбонильную группу, замещенную одной из описанных выше (C1-C3)алкильных групп. Ее примеры включают метилкарбонильную группу (ацетильная группа), этилкарбонильную группу и пропилкарбонильную группу. "(C1-C3)алкилсульфонильная группа" означает сульфонильную группу, замещенную одной (C1-C3)алкильной группой. Ее примеры включают метилсульфонильную группу (метансульфонильная группа), этилсульфонильную группу и пропилсульфонильную группу. "Арил-(C1-C3)алкильная группа" означает описанную выше (C1-C3)алкильную группу, замещенную одной арильной группой. Ее примеры включают фенилметильную группу и 2-фенэтильную группу. "Гидрокси-(C1-C3)алкильная группа" означает описанную выше (C1-C3)алкильную группу, замещенную одной гидроксигруппой. Ее примеры включают гидроксиметильную группу и 2-гидроксиэтильную группу.
Термин "(C1-C3)алкиленильная группа", как используют в рамках изобретения, относится к прямой или разветвленной двухвалентной алкильной цепи. Ее примеры включают -CH2-, -(CH2)2-, -(CH2)3-, -CH(CH3)- и -C(CH3)2-.
Термин "циклоалкил", как используют в рамках изобретения, относится к насыщенным или ненасыщенным неароматическим моноциклическим, бициклическим или трициклическим углеродным кольцевым системам из 3-10, предпочтительно 3-8, более предпочтительно 3-6 членов на кольцо. Примеры циклоалкила включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, адамантил и т.п. "Замещенный циклоалкил" означает циклоалкил, который независимо является замещенным, если нет иных указаний, одним или несколькими, предпочтительно 1, 2 или 3, более предпочтительно 1 или 2, заместителями, присоединенными к любому доступному атому, с образованием стабильного соединения, где заместители выбраны из, но не ограничиваясь ими, галогена или необязательно замещенного алкила, содержащего 1-3 атомов углерода. "(C3-C6)циклоалкильная группа" обозначает циклопропильную группу, циклобутильную группу, циклопентильную группу или циклогексильную группу.
Термин "гетероциклил", как используют в рамках изобретения, относится к насыщенной или ненасыщенной неароматической моно- или полициклической циклоалкильной группе, в которой от 1 до 3 атомов углерода в кольце заменены гетероатомом, выбранным из кислорода, серы или азота. Также подразумевается, что гетероциклил включает окисленный атом S или N, как например, сульфинил, сульфонил и N-оксид третичного азота кольца. Также подразумевается, что гетероциклил включает соединения, в которых углерод кольца может быть оксо-замещенным, т.е. углерод кольца представляет собой карбонильную группу, такие как лактоны и лактамы. Также подразумевается, что гетероциклил включает конденсированную, мостиковую и спиро-кольцевую систему. Предпочтительно, гетероциклические кольца необязательно являются конденсированными с бензо- или 4-6-членным гетероарильным или гетероциклическим кольцом. Точкой присоединения гетероциклического кольца является атом углерода или азота, так чтобы кольцо оставалось стабильным. Примеры гетероциклила включают, но не ограничиваются ими, оксиранил, тиаранил, азиридинил, оксетанил, тиатанил, азетидинил, тетрагидрофуранил, тетрагидротиофенил, пирролидинил, тетрагидропиранил, пиранил, тетрагидротиопиранил, тиопиранил, пиперидинил, 1,4-диоксанил, 1,4-оксатианил, морфолинил, тиоморфолинил, 1,4-дитианил, пиперазинил, 1,4-азатианил, оксепанил, тиэпанил, азепанил, 1,4-диоксепанил, 1,4-оксатиэпанил, 1,4-оксаазепанил, 1,4-дитиэпанил, 1,4-тиаазепанил, 1,4-азафосфинанил, 1,4-диазепанил, 1,2-тетрагидротиазин-2-ил, 1,3-тетрагидротиазин-3-ил, тетрагидротиадиазинил, 1,2-тетрагидродиазин-2-ил, 1,3-тетрагидродиазин-1-ил, тетрагидроазепинил, хроманил, хроменил, изооксазолидинил, 1,3-оксазолидин-3-ил, изотиазолидинил, 1,3-тиазолидин-3-ил, 1,2-пиразолидин-2-ил, 1,3-пиразолидин-1-ил, 7-окса-1-аза-спиро[4,4]нонанил, 3-азабицикло[3.1.0]гексанил, индолинил, дигидроиндолинил, октагидро-1H-индолил, октагидро-2H-пиридо[1,2-a]пиразинил, 3-азабицикло[4.1.0]гептанил, 3,4-дигидро-2H-пиранил, 1,2,3,4-тетрагидропиридинил, 1,2,5,6-тетрагидропиридинил, или тетрагидро-1H-бензо[d]азепинил и т.д. "Замещенный гетероциклил" означает гетероциклил, который независимо является замещенным, если нет иных указаний, одним или несколькими, предпочтительно 1, 2 или 3, более предпочтительно 1 или 2, заместителями, присоединенными к любому доступному атому с образованием стабильного соединения, где заместители выбраны из, но не ограничиваются ими, галогена, алкила, арила, гетероарила, гетероциклила, -(CH2)pNRbRc, -NRbCORc, -NRbS(O)2Rc, (CH2)pC(O)ORd, -C(O)NRbRc, -C(O)Rd, -C(O)ORd, -ORd, где алкил, арил, гетероарил и гетероциклил являются необязательно замещенными; Rb, Rc, Rd независимо выбраны из H или необязательно замещенного алкила, содержащего 1-6 атомов углерода; и p представляет собой целое число 0, 1, 2 или 3. "4-6-членная гетероциклическая группа" означает насыщенную неароматическую моноциклоалкильную группу, в которой от 1 до 3 атомов углерода в кольца заменены по меньшей мере одним гетероатомом, выбранным из кислорода, серы или азота. Их примеры включают азетидинильную группу, пирролидинильную группу, пиперидинильную группу, пиперазинильную группу, оксетанильную группу, тетрагидрофуранильную группу, тетрагидропиранильную группу и морфолинильную группу.
Термин "арил", как используют в рамках изобретения отдельно или в комбинации, относится к моноциклическим и полициклическим ароматическим углеводородным кольцевым системам, содержащим требуемое количество атомов углерода, как описано выше. Репрезентативные примеры включают, но не ограничиваются ими, фенил, нафтил и т.д. "Замещенный арил" означает арил, который независимо является замещенным, если нет иных указаний, одним или несколькими, предпочтительно 1, 2 или 3, более предпочтительно 1 или 2, заместителями, присоединенными к любому доступному атому, с образованием стабильного соединения, где заместители выбраны из, но не ограничиваясь ими, галогена, -NO2, -CN, алкила, арила, гетероарила, гетероциклила, -(CH2)pNRbRc, -NRbCORc, -NRbS(O)2Rc, (CH2)pC(O)ORd, -C(O)NRbRc, -C(O)Rd, -C(O)ORd, -ORd, где алкил, арил, гетероарил и гетероциклил являются необязательно замещенными; Rb, Rc, Rd независимо выбраны из H или необязательно замещенного алкила, содержащего 1-6 атомов углерода; и p представляет собой целое число 0, 1, 2 или 3.
Термин "гетероарил", как используют в рамках изобретения отдельно или в комбинации, относится к моноциклическим или полициклическим ароматическим кольцевым системам, содержащим требуемое количество атомов углерода и по меньшей мере один гетероатом, выбранный из N, O или S. Полициклические кольцевые системы могут содержать ароматические части, в то время как другие части кольцевой системы могут быть полностью насыщенными или неароматическими. Репрезентативные примеры гетероарила включают, но не ограничиваются ими, пирролил, фуранил, тиофенил, тиенил, пиразолил, имидазолил, изоксазолил, оксазолил, изотиазолил, тиазолил, 1,2,3-триазолил, 1,2,4-триазолил, тетразолил, 1,3,5-оксадиазолил, 1,2,4-оксадиазолил, 1,2,3-оксадиазолил, 1,3,5-тиадиазолил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, 1,2,3-триазинил, пиразолo[3,4-b]пиридинил, циннолинил, птеридинил, пуринил, 6,7-дигидро-5H-[1]пиридинил, бензо[b]тиофенил, бензоксазолил, бензотиазолил, бензоизотиазолил, бензоизоксазолил, бензимидазолил, бензофуранил, изобензофуранил, изоиндолил, индолил, индолизинил, индазолил, изохинолинил, хинолинил, фталазинил, хиноксалинил, хиназолинил, бензоксазинил и т.п. "Замещенный гетероарил" означает гетероарил, который является независимо замещенным, если нет иных указаний, одним или несколькими, предпочтительно 1, 2 или 3, более предпочтительно 1 или 2 заместителями, присоединенными к любому доступному атому, с образованием стабильного соединения, где заместители выбраны из, но не ограничиваются ими, галогена, -NO2, -CN, алкила, арила, гетероарила, гетероциклила, -(CH2)pNRbRc, -NRbCORc, -NRbS(O)2Rc, (CH2)pC(O)ORd, -C(O)NRbRc, -C(O)Rd, -C(O)ORd, -ORd, где алкил, арил, гетероарил и гетероциклил являются необязательно замещенными; Rb, Rc, Rd независимо выбраны из H или необязательно замещенного алкила, содержащего 1-6 атомов углерода; и p представляет собой целое число 0, 1, 2 или 3.
В одном варианте осуществления настоящее изобретение относится к соединению формулы (I):
или его фармацевтически приемлемой соли, где:
Y обозначает N;
R1, R2 и R3 независимо обозначают H, алкил, содержащий 1-3 атомов углерода, или галогенированный алкил, содержащий 1-3 атомов углерода;
R4 и R5 независимо обозначают H или необязательно замещенный алкил, содержащий 1-3 атомов углерода;
R6 обозначает алкил, циклоалкил или гетероциклил, где алкил, циклоалкил и гетероциклил являются необязательно замещенными; и
R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют необязательно замещенный гетероциклил, необязательно содержащий один или несколько гетероатом(ов), выбранный из N, O или S.
Другой вариант осуществления относится к соединению формулы (I),
или его фармацевтически приемлемой соли, где:
Y обозначает CH, CCl, CF или CMe;
R1, R2 и R3 независимо обозначают H, алкил, содержащий 1-3 атомов углерода, или галогенированный алкил, содержащий 1-3 атомов углерода;
R4 и R5 независимо обозначают H или необязательно замещенный алкил, содержащий 1-3 атомов углерода;
R6 обозначает алкил, циклоалкил или гетероциклил, где алкил, циклоалкил и гетероциклил являются необязательно замещенными; и
R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют необязательно замещенный гетероциклил, необязательно содержащий один или несколько гетероатом(ов), выбранный из N, O или S.
Другой вариант осуществления относится к соединению формулы (1),
или его фармацевтически приемлемой соли, где:
Y обозначает N, CH, CF, CCl или CCH3;
R1, R2 и R3 независимо обозначают H, (C1-C3)алкильную группу или галогенированную (C1-C3)алкильную группу;
R4 обозначает H или (C1-C3)алкильную группу, где (C1-C3)алкильная группа необязательно замещена (C3-C6)циклоалкильной группой;
R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют азетидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо или пиперазиновое кольцо,
где азетидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо и пиперазиновое кольцо являются необязательно замещенными тетрагидропиранильной группой, морфолинильной группой или 2,6-диметилморфолинильной группой.
Другой вариант осуществления относится к соединению формулы (I) или его фармацевтически приемлемой соли, где R5 обозначает H, метил или этил, и R6 обозначает необязательно замещенный алкил, содержащий 1-6 атомов углерода, необязательно замещенный 5-6-членный циклоалкил и необязательно замещенный 4-6-членный гетероциклил, где необязательные заместители выбраны из, но не ограничиваются ими, галогена, -NO2, -CN, алкила, арила, гетероарила, гетероциклила, -(CH2)pNRbRc, -NRbCORc, -NRbS(O)2Rc, (CH2)pC(O)ORd, -C(O)NRbRc, -C(O)Rd, -C(O)ORd, -ORd, где алкил, арил, гетероарил и гетероциклил являются необязательно замещенными; Rb, Rc, Rd независимо выбраны из H или необязательно замещенного алкила, содержащего 1-6 атомов углерода; и p представляет собой целое число 0, 1, 2 или 3.
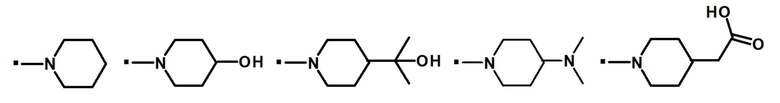
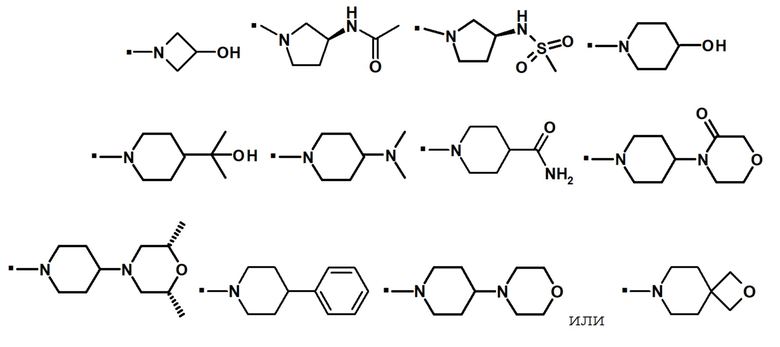
В конкретном варианте осуществления -NR5R6 выбран из, но не ограничивается ими
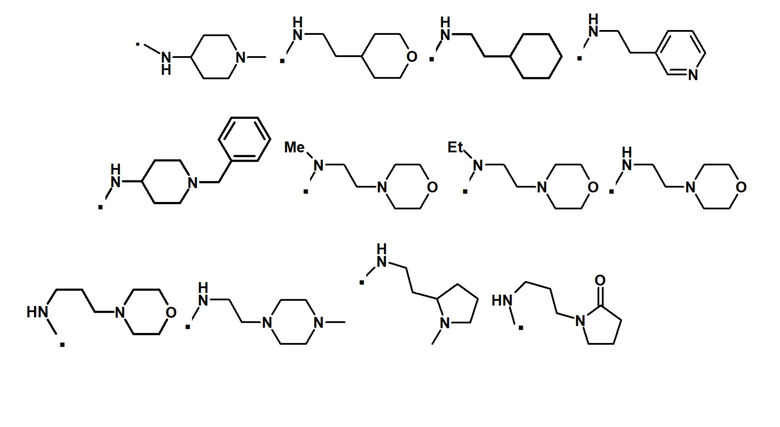
. обозначает точку присоединения.
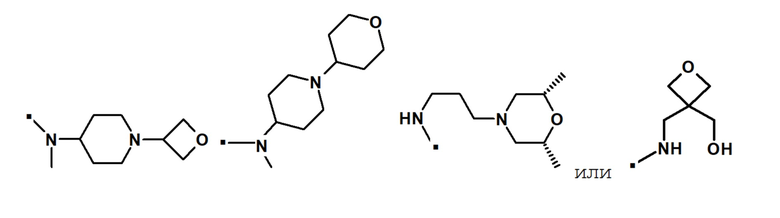
Другой вариант осуществления относится к соединению формулы (I) или его фармацевтически приемлемой соли, где R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют 4-6-членный гетероциклил, необязательно замещенный Rb, который выбран из алкила, арила, гетероарила, гетероциклила, -(CH2)pNRbRc, -NRbCORc, -NRbS(O)2Rc, (CH2)pC(O)ORd, -C(O)NRbRc, -C(O)Rd, -C(O)ORd, -ORd, где алкил, арил, гетероарил и гетероциклил являются необязательно замещенными; Rb, Rc, Rd независимо выбраны из H или алкила, содержащего 1-6 атомов углерода; и p представляет собой целое число 0, 1, 2 или 3.
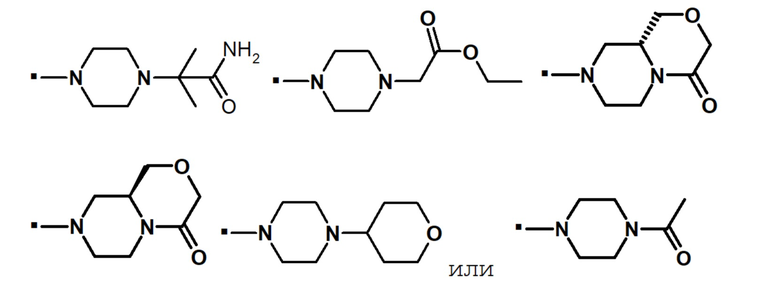
В конкретном варианте осуществления, -NR5R6 выбран из, но не ограничивается ими:
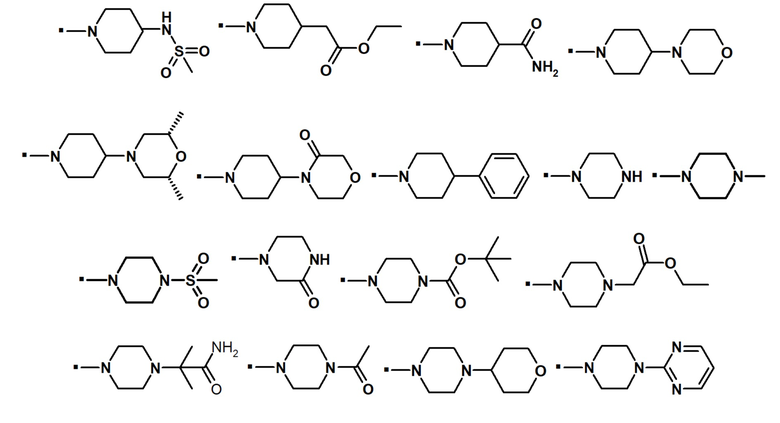
. обозначает точку присоединения.
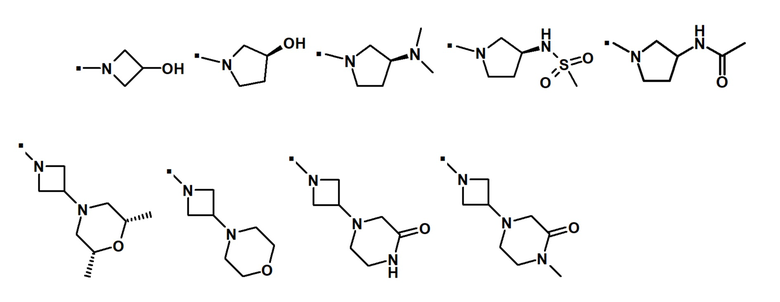
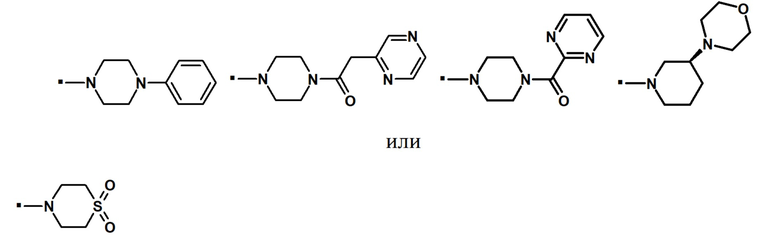
Другой вариант осуществления относится к соединению формулы (I) или его фармацевтически приемлемой соли, где R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют спиро- или конденсированное кольцо, содержащее 5-7 атомов углерода и по меньшей мере один гетероатом, выбранный из N, S или O, которое иллюстрируется, но не ограничивается ими:

. обозначает точку присоединения.
В предпочтительном варианте осуществления, -NR5R6 выбран из:
. обозначает точку присоединения.
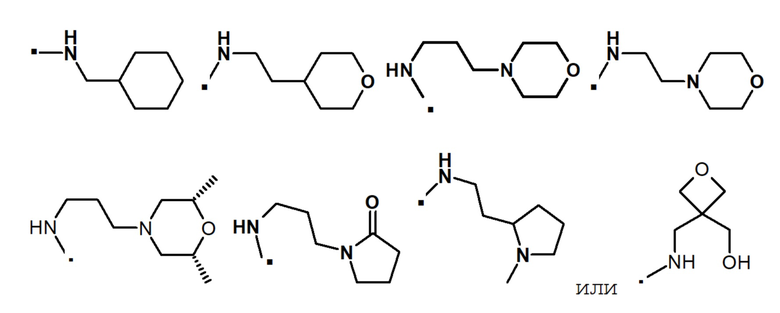
В другом предпочтительном варианте осуществления -NR5R6 выбран из:
. обозначает точку присоединения.
В другом предпочтительном варианте осуществления -NR5R6 выбран из:
. обозначает точку присоединения.
Соединения формулы (I) и их промежуточные соединения можно выделять в форме фармацевтически приемлемой соли. В описании "фармацевтически приемлемая соль" относится к солям, которые являются химически и/или физически совместимыми с другими ингредиентами, составляющими состав, и/или физиологически совместимыми с их реципиентом.
Иллюстративные соли включают, но не ограничиваются ими: хлорид, бромид, йодид, нитрат, сульфат, бисульфат, карбонат, бикарбонат, борат, фосфат, гидрофосфат, дигидрофосфат, ацетат, аскорбат, аспартат, бензоат, безилат, цитрат, формиат, фумарат, глюконат, глюкуронат, глутамат, лактат, малат, малеат, малонат, мезилат, метансульфонат, этансульфонат, бензолсульфонат, пара-толуолсульфонат, метилсульфат, никотинат, оксалат, пальмитат, памоат, стеарат, сахарат, сукцинат, салицилат, тартрат, тозилат, трифторацетат; катионы щелочных и щелочноземельных металлов, такие как натрий, литий, калий, кальций, магний и т.п.; катионы аммония, четвертичного аммония или аминов, такие как аммоний, тетраметиламмоний, тетраэтиламмоний, лизин, аргинин, бензатин, холин, трометамин, диоламин, глицин, меглумин, оламин и т.п. Более того, фармацевтически приемлемая соль может иметь один или несколько заряженных атомов и/или один или несколько противоионов. Соли могут существовать в различных кристаллических или полиморфных формах, все из которых входят в объем настоящего изобретения.
Фармацевтически приемлемую соль можно получать путем реакции соединения с подходящей органической или неорганической кислотой (если соединение представляет собой основание) или основанием (если соединение представляет собой кислоту) и выделения полученной таким образом соли. Соли можно осаждать с добавлением или без добавления сорастворителей и/или антирастворителей и собирать фильтрацией, или соли можно выделять путем выпаривания растворителя(ей). Фармацевтически приемлемую соль также можно получать in situ в ходе выделения и/или очистки соединения.
В описании пролекарство относится к соединению, которое преобразуется in vivo с образованием исходного соединения, где преобразование in vivo может происходить посредством различных механизмов, таких как гидролиз (желудочная кислота в физиологических условиях) или ферментативный гидролиз. Пролекарство представляет собой соединение, где амино или гидроксильная группа в соединении формулы (I) становится ацилированной, алкилированной, фосфорилированной, сульфурированной или гликолизированной, или где карбоксильная группа является этерифицированной или амидированной. Пролекарство соединения формулы (I) можно получать стандартным путем, например, если соединение формулы (I) содержит функциональную группу карбоновой кислоты, пролекарство может быть получено путем замены атома водорода кислотной группы на группу, такую как алкил или арил.
В описании гидрат относится к соединению, полученному путем связывания одной или нескольких молекул воды с соединением формулы (I). Молекула воды может быть связанной или свободно доступной на поверхности соединения формулы (I). Пример включает, но не ограничивается ими, моногидрат, дигидрат, тригидрат или тетрагидрат. В определенных вариантах осуществления соединение по настоящему изобретению может существовать в форме сольвата, где сольват относится к связыванию молекул растворителя с соединением формулы (I).
Соединение формулы (I) может содержать асимметричный или хиральный центр(ы), и, таким образом, существовать в различных стереоизомерных формах. Подразумевается, что все стереоизомерные формы соединений, описанных в настоящем описании, включая, но не ограничиваясь ими, диастереомеры, энантиомеры и атропизомеры, а также их смеси (например, рацемические смеси), составляют часть настоящего изобретения. В описании R и S используют для обозначения абсолютной конфигурации молекулы вокруг ее хирального центра(ов). Соединение формулы (I) с асимметричными центрами можно синтезировать (и/или выделять) в качестве смесей энантиомеров, индивидуальных энантиомеров или диастереомеров.
Чистый энантиомер можно получать с использованием способов, хорошо известных специалисту в данной области, например a) путем образования диастереомерных солей, которые можно разделять кристаллизацией; (b) путем образования диастереомерных производных или комплексов, которые можно разделять посредством кристаллизации, газожидкостной или жидкостной хроматографии; (c) посредством селективной реакции одного энантиомера со специфичным к энантиомеру реагентом, например, посредством ферментативной этерификации; (d) с использованием оптически активного исходного материала; (e) посредством асимметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей; или (f) путем преобразования одного стереоизомера в другой посредством асимметричного преобразования или инверсии.
Настоящее изобретение охватывает изотопно меченные соединения формулы (I). Предусматривается, что все изотопы какого-либо конкретного атома или элемента, как описано в настоящем описании, входят в объем настоящего изобретения. Примеры изотопов, которые могут быть включены в соединения по настоящему изобретению, включают, но не ограничиваясь ими, изотопы водорода (например, 2H или 3H), углерода (например, 13C или 14C), азота (например, 13N или 15N), кислорода (например, 15O, 17O или 18O), фосфора (например, 32P или 33P), серы (например, 35S), галогена (например, 18F, 36Cl, 123I или 125I). В предпочтительном варианте осуществления настоящее изобретение относится к дейтерированным (D или 2H) соединениям формулы (I). Изотопно меченные соединения формулы (I) можно получать согласно общей схеме и способам с использованием изотопно меченных реагентов. Изотопно меченные соединения по настоящему изобретению могут быть пригодными в анализах распределения в тканях соединений и/или субстратов. Такое применение изотопно меченных соединений хорошо известно специалисту в данной области и, таким образом, они входят в объем настоящего изобретения.
Метаболиты соединения формулы (I) также составляют часть настоящего изобретения, которая относится к соединениям, происходящим из соединения формулы (I), в клетке или организме, предпочтительно млекопитающем. Структура метаболитов соединений может быть понятна любому специалисту в данной области.
Соединения по настоящему изобретению можно использовать для лечения и/или предупреждения заболевания или нарушения, отвечающего на ингибирование PI3Kδ. Согласно литературе, в качестве орфанного заболевания может рассматриваться гиперпролиферативное, воспалительное и/или аутоиммунное заболевание или нарушение, когда речь идет о лечении и/или предупреждении с использованием такого соединения.
Таким образом, другой аспект относится к способу лечения или уменьшения тяжести заболевания или нарушения, отвечающего на ингибирование PI3Kδ, у пациента, включающему введение указанному пациенту терапевтически эффективного количества соединения формулы (I), его фармацевтически приемлемой соли или их фармацевтической композиции.
В описании терапевтически эффективное количество относится к количеству соединения формулы (I), достаточному для лечения или предупреждения конкретного заболевания или нарушения. Количество соединения, которое составляет эффективное количество, варьируется, в зависимости от различных факторов, включающих, например, используемое соединение, болезненное состояние и его тяжесть, возраст пациента, подвергаемого лечению, и т.п.
В описании пациент относится к человеку и другим животным. В этом контексте термины "индивидуум", "животное" и т.п. относятся к человеку, такому как мужчина и женщина, и не являющимся человеком позвоночным, например, млекопитающим, таким как не являющиеся человеком приматы, спортивные и коммерческие животные и домашние питомцы (например, животные семейства собачьих и кошачьих). Предпочтительно пациентом является человек.
В описании воспалительное заболевание относится к воспалению тканей и органов. Воспалительные заболевания или нарушения, ассоциированные с активацией PI3K, включают, но не ограничиваются ими, воспаление кожи, воспаление кожи вследствие воздействия радиации, аллергическую астму, тяжелую астму, резистентную к стероидам астму, COPD, аллергическое воспаление и хроническое воспаление.
В описании аутоиммунное заболевание относится к заболеванию, которое частично индуцируется иммунной реакцией организма против его собственных компонентов, например ДНК, липидов, белка и т.п. Аутоиммунное заболевание может быть органоспецифическим или не органоспецифическим. Примеры органоспецифических заболеваний включают, но не ограничиваются ими, инсулинзависимый диабет (тип I), глютеновую болезнь, псориаз, воспалительное заболевание кишечника, хронический активный гепатит, синдром поликистоза яичников, пернициозную анемию или анкилозирующий спондилит. Примеры не органоспецифических заболеваний включают, но не ограничиваются ими, ревматоидный артрит, рассеянный склероз, системную красную волчанку, псориатический артрит или миастению.
В описании гиперпролиферативное заболевание относится к опухоли или злокачественной опухоли. Примеры включают, но не ограничиваются ими, рак молочной железы, лимфому из клеток мантийной зоны, почечноклеточный рак, острый миелогенный лейкоз, хронический миелогенный лейкоз, рабдомиосаркому, рак яичника, рак эндометрия, рак шейки матки, немелкоклеточную карциному легкого, мелкоклеточную карциному легкого, аденокарциному, рак толстого кишечника, рак прямой кишки, карциному желудка, печеночно-клеточную карциному, меланому, рак поджелудочной железы, карциному предстательной железы, карциному щитовидной железы, анапластическую крупноклеточную лимфому, гемангиому, глиобластому, солидные опухоли, лимфоидную злокачественную опухоль или рак головы и шеи.
В описании орфанное заболевание относится к редким заболеваниям. Примеры включают, но не ограничиваются ими, эозинофильный гастроэнтерит (EGE) и эозинофильный гастрит (EG), эозинофильный колит, гиперэозинофильный синдром, эозинофильную пневмонию, синдром Черджа-Стросс или мастоцитоз.
В определенных вариантах осуществления заболевание или нарушение выбрано из, но не ограничивается ими, воспаления кожи вследствие воздействия радиации, тяжелой астмы, хронического обструктивного заболевания легких, аллергического воспаления, хронического воспаления, аллергического заболевания, ринита, синусита, пищевой аллергии, псориаза, воспалительного заболевания кишечника, хронического активного гепатита, синдрома поликистоза яичников, пернициозной анемии, ревматоидного артрита, рассеянного склероза, системной красной волчанки, первичного синдрома иммунодефицита (например, синдрома активированной PI3Kδ), миастении или злокачественной опухоли (например, рака молочной железы, яичника, шейки матки, желудка, легкого, меланомы, мелкоклеточного рака легкого и т.п.).
В определенных вариантах осуществления соединение по настоящему изобретению является более чем в 50 раз более селективным в отношении ингибирования активности PI3Kδ, чем в отношении ингибирования PI3Kα, PI3Kβ или PI3Kγ. В одном предпочтительном варианте осуществления соединение по настоящему изобретению является более чем в 100 раз более селективным в отношении ингибирования активности PI3Kδ, чем в отношении ингибирования PI3Kα, PI3Kβ или PI3Kγ, и даже в некоторых случаях более чем в 200 раз. Как очевидно из литературы, PI3Kδ необходима для регуляции как врожденного, так и адаптивного иммунного ответа, включая экспрессию и активацию медиаторов воспаления, привлечение воспалительных клеток, ремоделирование дыхательных путей и нечувствительность к кортикостероидам при хроническиом воспалительном заболевании дыхательных путей, таким образом, можно понимать, что соединения по настоящему изобретению предпочтительно можно использовать для лечения или предупреждения воспалительного и/или аутоиммунного заболевания или нарушения, выбранного из, но не ограничиваясь ими, псориаза, псориатического артрита, ревматоидного артрита, аллергической астмы, тяжелой астмы, резистентной к стероидам астмы или COPD.
В предпочтительном варианте осуществления предусматривается способ лечения или предупреждения псориаза.
В другом предпочтительном варианте осуществления предусматривается способ лечения или предупреждения псориатического артрита.
В другом предпочтительном варианте осуществления предусматривается способ лечения или предупреждения ревматоидного артрита.
В другом предпочтительном варианте осуществления предусматривается способ лечения или предупреждения хронического обструктивного заболевания легких.
В другом предпочтительном варианте осуществления предусматривается способ лечения или предупреждения аллергической астмы.
В другом предпочтительном варианте осуществления предусматривается способ лечения или предупреждения тяжелой астмы.
Другой аспект относится к соединению формулы (1) или его фармацевтически приемлемой соли для применения для лечения или уменьшения тяжести заболевания или нарушения, отвечающего на ингибирование PI3Kδ.
В другом варианте осуществления предусматривается лекарственное средство для ингибирования PI3Kδ, содержащее соединение формулы (1) или его фармацевтически приемлемую соль в качестве активного ингредиента.
Для терапии может потребоваться подходящая дозированная форма. Подходящие дозированные формы зависят от применения или пути введения. Следует понимать, что такие дозированные формы должны позволить соединению достигнуть клеток-мишеней. Также следует учитывать другие факторы, такие как токсичность и дозированные формы, которые замедляют проявление соединением или композицией их эффектов. Для способов и составов может быть рассмотрена справочная литература, например, The Science and Practice of Pharmacy, 21st edition, Lippincott, Willams and Wilkins, Philadelphia, Pa., 2005.
Таким образом, другой аспект относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль в качестве активного ингредиента и один или несколько фармацевтически приемлемый эксципиент(ов).
В описании эксципиент относится к любому ингредиенту в составе, отличному от соединения формулы (I) или его фармацевтически приемлемой соли. Примеры включают, но не ограничиваются ими, носитель, переносчик, растворитель, адъювант, смазывающее вещество, поверхностно-активное вещество, связующее вещество, буфер, разбавитель, вкусовую добавку, краситель, разрыхлитель, эмульгатор, суспендирующее вещество, пластификатор, солюбилизатор, наполнитель или объемообразующее вещество. Выбор эксципиента(ов) в большой степени зависит от таких факторов, как конкретный путь введения, эффект эксципиентов на растворимость, стабильность и профиль высвобождения, и природа дозированной формы. Соединение формулы (I) или его фармацевтически приемлемая соль могут в общем упоминаться как активный ингредиент(ы) в составе или фармацевтической композиции. Фармацевтическая композиция, пригодная для доставки соединения формулы (I), и способы ее получения будут понятны специалистам в данной области. Такие композиции и способы их получения могут быть найдены, например, в Remington's Pharmaceutical Sciences, 19th ed., (Mack Publishing Company, 1995).
В соответствии с настоящим изобретением, предпочтительным путем введения является пероральный, который включает таблетку, капсулу, пилюлю, порошок, составы с замедленным или немедленным высвобождением, раствор или суспензию. Другим предпочтительным путем введения может быть ингаляция. Также в объем настоящего изобретения входят такие пути введения, как внутривенный, подкожный, внутримышечный и т.п.
Количество активного ингредиента(ов) и эксципиента(ов), присутствующих в составе или фармацевтической композиции, можно определять стандартными способами, учитывая такие факторы, как IC50 соединения, биологическое время полужизни соединения, возраст, размер и масса тела пациента, и заболевание или нарушение, ассоциированные с пациентом.
Как правило, доза составляет приблизительно от 5 мг до 100 мг, bid, предпочтительно, от 5 мг до 50 мг, bid, более предпочтительно, от 5 мг до 25 мг, bid, у пациента, подвергаемого лечению. Можно использовать множество доз. Специалисту в данной области будет понятно, что дозу корректируют в соответствии со способами, хорошо известными в области терапии. Иными словами, максимальную переносимую дозу можно без труда определять, и также можно определять эффективное количество, обеспечивающее поддающуюся обнаружению терапевтическую пользу у пациента. Таким образом, хотя определенные дозы и режимы введения проиллюстрированы в настоящем описании, это никоим образом не ограничивает дозу и режим введения, которые могут быть предоставлены пациенту при применении настоящего изобретения на практике.
В предпочтительном варианте осуществления предусматривается фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль для лечения или уменьшения тяжести заболевания или нарушения, отвечающего на ингибирование PI3Kδ, где заболевание или нарушение выбрано из псориаза, псориатического артрита, ревматоидного артрита, аллергической астмы, тяжелой астмы, резистентной к стероидам астмы, COPD, системной красной волчанки, первичного синдрома иммунодефицита или злокачественной опухоли.
Когда желательно, соединение формулы (I) или его фармацевтически приемлемую соль можно использовать в комбинации с одним или несколькими β2-агонистами, кортикостероидами, антагонистами лейкотриенов, антихолинэргетиками, противоаллергическими средствами, антагонистами мускариновых рецепторов, модуляторами Treg, модуляторами точки контроля и лекарственными средствами против злокачественной опухоли.
В описании β2-агонист относится, но не ограничивается ими, к альбутеролу, сальбутамолу, тербуталину, фенотеролу, сальметеролу или формотеролу. Кортикостероид относится, но не ограничивается ими, к флунизолиду, беклометазону, триамцинолону, будезониду, флукатизону, мометазону, циклесониду или дексаметазону. Антагонист лейкотриена относится, но не ограничивается ими, к монтелукасту, зафирлукасту или пранлукасту. Антихолинергическое средство относится, но не ограничивается ими, к тиотропия бромиду, ипратропия бромиду или окситропия бромиду. Противоаллергическое средство относится, но не ограничивается ими, к цетиразину, азеластину, фексофенадину, левокабастину, лоратидину, фенирамину, доксиламину, деслоратидину или меклизину. Антагонисты мускариновых рецепторов относятся, но не ограничиваются ими, к толтеродину, оксибутинину или атропину. Средство против злокачественной опухоли относится, но не ограничивается ими, к цитотоксическому средству (например, бендамустин), противоопухолевому антибиотику (например, блеомицин), ингибитору микротрубочек (например, топотекан), антиметаболиту (например, метотрексат), ДНК-связывающему средству (например, цисплатин), биологическому средству (например, иматиниб), бисфосфонату (например, клодронат) или ингибитору PI3K (например, иделалисиб).
Далее предоставлены общие схемы и экспериментальные методики для получения соединений формулы (I) и их промежуточных соединений. Следует понимать, что методики, приведенные ниже, предоставлены для иллюстративных целей, и их не следует истолковывать как ограничивающие объем изобретения. Любые модификации методик, описанных в настоящем описании, другие методики синтеза и их модификации, можно использовать и адаптировать. Все такие модификации и альтернативные методики входят в сущность и объем настоящей заявки. Для цели определения структуры конечного продукта и промежуточных соединений авторы изобретения полагались на 1H-ЯМР, который относится к спектру протонного магнитного резонанса, и способ получения спектра масс, такой как ESI, и различные другие данные, такие как оптическое вращение. В рамках настоящей заявки химический сдвиг в 1H-ЯМР выражается в м.д. (в масштабе δ) относительно тетраметилсилана в качестве внутреннего стандарта, в то время как константа сопряжения (J) и множественность пика могут быть обозначены как синглет (с); дублет (д); дублет дублета (дд); триплет (т); мультиплет (м); уширенный (ушир.), уширенный синглет (ушир. с), триплет дублета (тд) и квинтет (квин). Для получения номенклатуры соединений и промежуточных соединений использовали ACD Labs 12.0 (версия 12.5), как описано в настоящем описании. Соединению формулы (I) соответствует соединение 1, как показано в подробном описании экспериментов ниже.
Подробное описание экспериментов
Соединение 1 можно получать различными способами, известными специалисту в данной области. Следует понимать, что настоящее изобретение не ограничивается этими примерами.
Например, соединение 1 (где R1 обозначает H или метил, R2, R3 и R4 обозначают H или алкил) можно получать путем амидирования сложноэфирного соединения 1a соединением амина 1b, как показано на схеме 1.
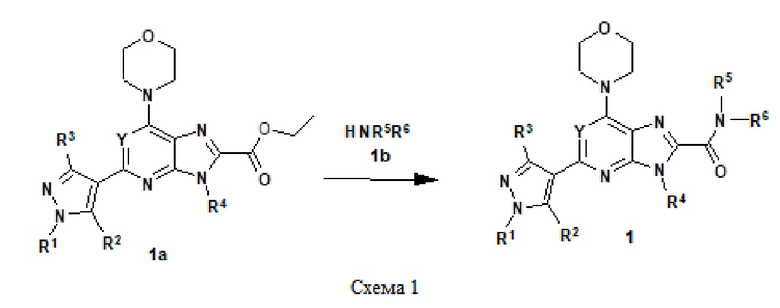
Эту реакцию можно проводить способом, описанным в Syn. Comm., 1982, 12; Org. Syn., 1979, 59, 49, или способами, эквивалентными им. В определенных вариантах осуществления реакцию можно проводить в присутствии соединений алкилалюминия, таких как триметилалюминий или триизобутилалюминий, в растворителе, таком как тетрагидрофуран или толуол. Реакцию можно проводить при температуре от 30°C до 150°C, и предпочтительно от 80°C до 120°C. Более конкретно, реакцию проводили следующим образом.
[Пример 1] К раствору сложноэфирного соединения 1a (1 экв.) и соединения амина 1b (2 экв.) в тетрагидрофуране (от 10 до 100 мл) капельно добавляли триметилалюминий в толуоле (2 M, 3 экв.) при комнатной температуре. После завершения добавления реакционную смесь энергично кипятили с обратным холодильником при приблизительно 100°C в течение приблизительно 18 часов. Реакционную смесь охлаждали до комнатной температуры, осторожно гасили капельным добавлением метанола, а затем добавлением дихлорметана. Добавляли воду и перемешивали в течение приблизительно 60 минут. Органический слой отделяли. Водный слой экстрагировали с использованием дихлорметана (от 100 до 300 мл три раза) и объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией (Combiflash) с использованием метанола и дихлорметана (от 5 до 15% метанол) в качестве элюента. Например, [2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил](2-окса-7-азаспиро[3.5]нон-7-ил)метанон (соединение № 3) получали с использованием этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксилата (120 мг) и 2-окса-7-азаспиро[3.5]нонана (122 мг).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,49 (с, 1H), 8,24 (с, 1H), 7,93 (с, 1H), 4,16-4,40 (м, 8H), 4,02 (ушир. с, 2H), 3,88 (с, 3H), 3,74-3,79 (м, 4H), 3,56-3,64 (м, 2H), 1,86 (д, J=4,27 Гц, 4H). Спектр масс (ESI): m/z 438,97 [M+H]
Аналогичным образом получали соединения, приведенные в таблице 1, с использованием 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксилата и соединения амина 1b, которое является коммерчески доступным (таблица 1) или которое синтезировали, как описано в настоящем описании далее.
Альтернативно сложноэфирное соединение 1a можно конвертировать в соответствующее кислотное соединение посредством гидролиза с использованием основания, такого как гидроксид лития, в растворителе, таком как тетрагидрофуран, вода или их комбинация. Кислотное соединение, полученное таким образом, можно конвертировать в соединение 1 с использованием методики, известной специалисту в данной области, например (3-гидроксиазетидин-1-ил)[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанон (соединение № 4) получали следующим образом.
[Пример 2] К раствору кислотного соединения (200 мг, 0,61 ммоль) в дихлорметане (15 мл) добавляли каталитическое количество диметилформамида и оксалилхлорида (0,2 мл) при 0°C. Затем реакционную смесь перемешивали при комнатной температуре в течение 2-3 часов. Реакционную смесь концентрировали в вакууме до сухого состояния и остаток отбирали в дихлорметан (10 мл), к нему добавляли триэтиламин (0,3 мл) и 3-гидроксиазетидин (200 мг, 1,82 ммоль) и перемешивали при комнатной температуре в течение 14 часов. К реакционной смеси добавляли воду и экстрагировали дихлорметаном (100 мл три раза). Объединенный органический слой сушили над безводным сульфатом натрия, концентрировали в вакууме. Продукт очищали колоночной хроматографией с использованием метанола и дихлорметана (от 10 до 15% метанол) в качестве элюента с получением 75 мг желаемого соединения в виде не совсем белого твердого вещества.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 12,40-14,27 (м, 1H), 8,24 (с, 1H), 7,92 (с, 1H), 5,79 (д, J=6,27 Гц, 1H), 4,76-4,88 (м, 1H), 4,55 (д, J=6,27 Гц, 1H), 4,09-4,48 (м, 6H), 3,88 (с, 3H), 3,79-3,84 (м, 1H), 3,73-3,79 (м, 4H). Спектр масс (ESI): m/z 384,89. [M+H]
Аналогично получали [(3S)-3-гидроксипирролидин-1-ил][2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанон (соединение № 62) (15 мг) в виде не совсем белого твердого вещества.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,41-13,66 (м, 1H), 8,24 (с, 1H), 7,93 (с, 1H), 4,88-5,08 (м, 1H), 4,09-4,53 (м, 6H), 3,96-4,01 (м, 1H), 3,89 (с, 3H), 3,77 (д, J=4,52 Гц, 4H), 3,47-3,66 (м, 2H), 1,77-2,06 (м, 2H). Спектр масс (ESI): m/z 399,18. [M+H]
Соединение 1 (где R4 обозначает метил)) можно получать путем реакции соединения 1 (где R4 обозначает H) с алкилгалогенидом, таким как метилйодид, в присутствии основания, такого как карбонат калия, карбонат натрия, карбонат цезия, в растворителе, таком как диметилформамид. Более конкретно, реакцию проводили следующим образом.
[Пример 3] К раствору [6-фтор-7-(морфолин-4-ил)-5-(1-метил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил](2-окса-7-азаспиро[3.5]нон-7-ил)метанона (50 мг, 0,1 ммоль) в диметилформамиде (3 мл) добавляли карбонат калия (41 мг, 0,27 ммоль), а затем метилйодид (19 мг, 0,13 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. К этой реакционной смеси добавляли насыщенный бикарбонат натрия и проводили экстракцию дихлорметаном (150 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием метанола и дихлорметана (5% метанол) в качестве элюента с получением 25 мг [6-фтор-3-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил](2-окса-7-азаспиро[3.5]нон-7-ил)метанона (соединение № 108).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,26 (ушир. с, 1H), 8,00 (ушир. с, 1H), 4,36 (ушир. с, 4H), 3,92 (ушир. с, 4H), 3,79 (ушир. с, 10H), 3,63 (ушир. с, 4H), 1,88 (ушир. с, 4H). Спектр масс (ESI): m/z 470,30. [M+H]
Аналогичным образом получали [3-(циклопропилметил)-5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанон (соединение № 83) с использованием [5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (200 мг, 0,39 ммоль), диметилформамида (10 мл), карбоната калия (134 мг, 0,97ммоль) и циклопропилметилбромида (68 мг, 0,50 ммоль).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,06 (с, 1H), 4,49 (д, J=13,64 Гц, 1H), 4,26 (д, J=13,14 Гц, 1H), 4,18 (д, J=7,33 Гц, 1H), 3,78-3,88 (м, 12H), 3,57 (ушир. с, 6H), 3,19 (т, J=12,00 Гц, 1H), 2,94 (т, J=12,25 Гц, 1H),2,5(ушир. м, 4H), 1,83-1,93 (м, 2H), 1,23 (с, 3H), 0,84-0,92 (м, 2H), 0,47 (д, J=6,82 Гц, 4H). Спектр масс (ESI): m/z 567,08. [M+H]
Аналогичным образом получали следующие соединения.
[5-(1,3-диметил-1H-пиразол-4-ил)-6-фтор-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(2-гидроксипропан-2-ил)пиперидин-1-ил]метанон (соединение № 107).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,05 (д, J=4,29 Гц, 1H), 4,57-4,66 (м, 1H), 4,29-4,40 (м, 1H), 4,18 (с, 1H), 3,71-3,90 (м, 14H), 2,99-3,09 (м, 1H), 2,72-2,86 (м, 1H), 2,48-2,54(с,3H), 1,82-1,89 (м, 1H), 1,72-1,79 (м, 1H), 1,46-1,58 (м, 1H), 1,15-1,39 (м, 2H), 1,06 (с, 6H). Спектр масс (ESI): m/z 500,34. [M+H]
[5-(1,3-диметил-1H-пиразол-4-ил)-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанон (соединение № 104).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,23 (с, 1H), 6,77 (с, 1H), 4,46-4,52 (м, 1H), 4,22-4,29 (м, 1H), 3,85-3,90 (м, 4H), 3,76-3,81 (м, 11H), 3,51-3,62 (м, 4H), 3,13-3,20 (м, 1H), 2,88-2,95 (м, 1H),2,45-2,5(м,7H), 1,87-1,95 (м, 1H), 1,78-1,85 (м, 1H), 1,38-1,51 (м, 2H). Спектр масс (ESI): m/z 509,26. [M+H]
{4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[3-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанон (соединение № 110).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,30 (с, 1H), 8,04 (д, J=0,76 Гц, 1H), 6,88 (с, 1H), 4,42-4,62 (м, 1H), 4,16-4,29 (м, 1H), 3,86-3,94 (м, 7H), 3,74-3,82 (м, 7H), 3,47-3,57 (м, 2H), 3,08-3,20 (м, 1H), 2,84-2,97 (м, 1H), 2,70-2,80 (м, 2H), 1,69-2,01 (м, 5H), 1,36-1,52 (м, 2H), 1,04 (дд, J=2,53, 6,32 Гц, 6H). Спектр масс (ESI): m/z 523,39. [M+H]
{4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[5-(1,3-диметил-1H-пиразол-4-ил)-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанон (соединение № 114).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,24 (с, 1H), 6,77 (с, 1H), 4,43-4,53 (м, 1H), 4,19-4,28 (м, 1H), 3,85-3,90 (м, 4H), 3,75-3,83 (м, 10H), 3,46-3,57 (м, 2H), 3,15 (ушир. с, 1H), 2,89 (ушир. с, 1H), 2,74 (д, J=10,54 Гц, 2H), 2,48-2,50 (м, 4H), 1,74-1,94 (м, 4H), 1,35-1,54 (м, 2H), 1,04 (дд, J=2,26, 6,27 Гц, 6H). Спектр масс (ESI): m/z 537,44. [M+H]
Соединение 1 (где R1 обозначает метил, R2, R3 и R4 обозначают H, и Y обозначает N, CH, CF или CCl) также можно получать согласно схеме 2.
Соединение 2b можно получать посредством реакции амидирования, описанной на схеме 1, выше. Соединение 1 можно синтезировать посредством реакции соединения 2b с моногидратом п-толуолсульфоновой кислоты (можно использовать камфорсульфоновую кислоту или каталитическое количество хлористоводородной кислоты) в подходящем растворителе, таком как метанол, этанол, толуол при температуре в диапазоне от 50 до 200°C, предпочтительно от 70 до 150°C. Более конкретно, реакцию проводили следующим образом.
[Пример 4] К раствору соединения 2b (1 экв.) в этаноле добавляли моногидрат п-толуолсульфоновой кислоты (1 экв.) и кипятили с обратным холодильником в течение 14 часов. Реакционную смесь охлаждали до комнатной температуры, переливали на насыщенный раствор бикарбоната натрия и экстрагировали дихлорметаном (от 250 до 500 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который затем растирали с гексаном.
Аналогичным образом получали 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[2-(пиридин-3-ил)этил]-9H-пурин-8-карбоксамид (соединение № 1) с использованием этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбоксилата и 2-(пиридин-3-ил)этанамина, с последующей реакцией с моногидратом п-толуолсульфоновой кислоты.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,61 (с, 1H), 8,77 (с, 1H), 8,37-8,53 (м, 2H), 8,23 (с, 1H), 7,93 (с, 1H), 7,68 (д, J=7,78 Гц, 1H), 7,25-7,39 (м, 1H), 4,29 (ушир. с, 4H), 3,88 (с, 3H), 3,77 (т, J=4,64 Гц, 4H), 3,55 (д, J=7,03 Гц, 2H), 2,91 (т, J=7,15 Гц, 2H). Спектр масс (ESI): m/z 431,10.[M+H]
Аналогичным образом получали соединения, приведенные в таблице 1, с использованием соединения амина 1b в соответствии с методикой, описанной в примере 4.
Соединение 2b (где R5 обозначает алкил, такой как метил или этил) также можно получать путем обработки соединения 2b (где R5 представляет собой H) алкилгалогенидом в подходящем растворителе, таком как диметилформамид. В частности, соединения получали следующим образом.
[Пример 5] К раствору соединения 2b (1 экв., где R5 представляет собой H) в диметилформамиде добавляли гидрид натрия (1,5 экв.) и реакционную смесь перемешивали при 0°C в течение 30 минут. К реакционной смеси добавляли алкилгалогенид (1,2 экв., R5I), а затем перемешивали при комнатной температуре в течение 2 часов. Добавляли воду, экстрагировали дихлорметаном, промывали водой, сушили над безводным сульфатом натрия и концентрировали в вакууме с получением соединения 2b (где R5 представляет собой алкил), которое очищали колоночной хроматографией.
Аналогичным образом получали N-этил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[2-(морфолин-4-ил)этил]-9H-пурин-8-карбоксамид (соединение № 59) с использованием этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбоксилата и 2-(морфолин-4-ил)этанамина с последующей обработкой этилйодидом и моногидратом п-толуолсульфоновой кислоты.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,46 (ушир. с, 1H), 8,23 (с, 1H), 7,93 (с, 1H), 4,24 (ушир. с, 4H), 4,02 (т, J=6,27 Гц, 1H), 3,84-3,93 (м, 4H), 3,75 (т, J=4,02 Гц, 4H), 3,53-3,62 (м, 6H), 2,51-2,59 (м, 1H), 2,45 (ушир. с, 2H), 2,30 (ушир. с, 2H), 1,13-1,28 (м, 4H). Спектр масс (ESI): m/z 470,20. [M+H].
Аналогично получали следующие соединения.
N-метил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[2-(морфолин-4-ил)этил]-9H-пурин-8-карбоксамид (соединение № 60),
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,46 (ушир. с, 1H), 8,23 (с, 1H), 7,93 (с, 1H), 4,24 (ушир. с, 4H), 4,02 (т, J=6,27 Гц, 1H), 3,84-3,93 (м, 4H), 3,75 (т, J=4,02 Гц, 4H), 3,53-3,62 (м, 6H), 2,51-2,59 (м, 1H), 2,45 (ушир. с, 2H), 2,30 (ушир. с, 2H), 1,13-1,28 (м, 4H). Спектр масс (ESI): m/z 456,10 [M+H].
N-этил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-N-[3-(морфолин-4-ил)пропил]-9H-пурин-8-карбоксамид (соединение № 69),
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,11 (ушир. с, 1H), 7,99 (ушир. с, 1H), 4,36 (ушир. с, 4H), 4,01-4,21 (м, 2H), 3,77-3,99 (м, 8H), 3,72 (ушир. с, 2H), 3,59 (ушир. с, 2H), 2,47 (ушир. с, 6H), 1,82-2,08 (м, 2H), 1,29 (д, J=1,26 Гц, 2H), 1,25 (с, 3H). Спектр масс (ESI): m/z 484,30 [M+H].
Соединение 1 (где Y представляет собой CF) можно получать согласно схеме 2. Более конкретно, реакцию проводили следующим образом.
[Пример 6] К раствору соединения 2a (1 экв.) и соединения амина 1b (4 экв.) в тетрагидрофуране (от 10 до 50 мл) добавляли триметилалюминий (4 экв.) при 0°C, перемешивали при комнатной температуре, а затем кипятили с обратным холодильником в течение 24 часов. Реакционную смесь охлаждали и распределяли между насыщенным бикарбонатом натрия и дихлорметаном (от 100 до 300 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием метанола и дихлорметана (от 5 до 15% метанола) в качестве элюента с получением соединения 2b. К раствору соединения 2b (1 экв.) в этаноле (от 10 до 100 мл) добавляли пара-толуолсульфоновую кислоту (1 экв.) и реакционную смесь перемешивали в течение 15 минут при 140°C в условиях микроволнового излучения. Реакционную смесь распределяли между насыщенным бикарбонатом натрия и дихлорметаном (от 200 до 500 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием метанола и дихлорметана (от 5 до 15% метанола; в некоторых случаях использовали 1% аммиак) в качестве элюента.
Аналогичным образом получали (9aR)-8-{[6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]карбонил}гексагидропиразинo[2,1-c][1,4]оксазин-4(3H)-он (соединение № 80) с использованием метил 6-фтор-5-(1-метил-1H-пиразол-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (120 мг) и (9aR)-гексагидропиразинo[2,1-c][1,4]оксазин-4(3H)-она (95,21 мг).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,53 (ушир. с, 1H), 8,20 (ушир. с, 1H), 7,93 (ушир. с, 1H), 5,22-5,42 (м, 1H), 4,50 (д, J=13,05 Гц, 2H), 4,09 (ушир. с, 2H), 3,92 (с, 3H), 3,92 (м,1H), 3,81 (д, J=8,53 Гц, 8H), 3,65 (д, J=8,78 Гц, 2H), 3,09-3,28 (м, 1H), 2,74-2,98 (м, 2H). Спектр масс (ESI): m/z 484,99.[M+H]
Аналогично получали соединения, приведенные в таблице 1, согласно методике, описанной в примере 6.
Соединение 1 (где Y обозначает CH, CF или N) также можно получать путем реакции соединения 3a с боронатным эфиром 3b, как показано на схеме 3.
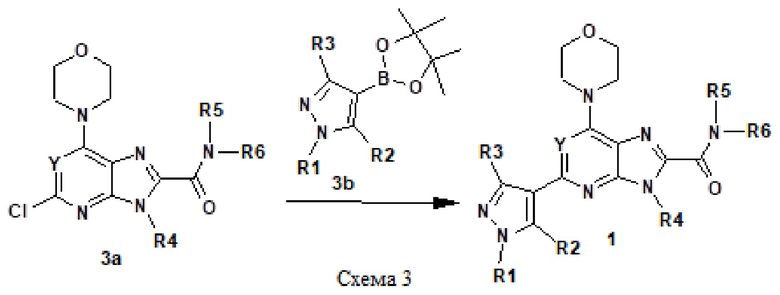
Реакцию сочетания Сузуки можно проводить согласно методике, известной специалисту в данной области. В частности, реакцию можно проводить в присутствии палладиевого катализатора, такого как тетракис(трифенилфосфин)палладий(0) или [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий(II), основания, такого как карбонат калия, карбонат цезия, карбонат натрия или бикарбонат натрия, в растворителе, таком как дихлорэтан, диметилформамид, диметилсульфоксид, толуол, н-пропанол, диоксан, ацетонитрил, вода или их комбинация. Реакцию можно проводить при температуре от 80 до 150°C, и предпочтительно от 100 до 150°C. Более конкретно, реакцию проводили следующим образом.
[Пример 7] К раствору соединения 3a (1 экв.) в 1,2-дихлорэтане (от 10 до 50 мл мл) добавляли соединение 3b (1,4 экв.; коммерчески доступное, таблица 2), карбонат цезия (2,5 экв.). Реакционную смесь продували с использованием аргона в течение 15 минут, а затем добавляли тетракис(трифенилфосфин)палладий(0) (0,1 экв.). Через 15 минут аргон удаляли и реакционной смеси позволяли кипеть с обратным холодильником при 110°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры; добавляли воду (от 40 до 250 мл) и экстрагировали с использованием дихлорметана (от 100 до 500 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией с использованием системы из от 50% этилацетата в гексане до чистого этилацетата. Наиболее чистые фракции собирали и концентрировали в вакууме с получением желаемых соединений. Альтернативно реакцию проводили с использованием соединения 3a (1 экв.), н-пропанола и воды (9:1, 20 мл), соединения 3b (1,3 экв.), бикарбоната натрия (3 экв.), тетракис(трифенилфосфин)палладия(0) (0,1 экв.) при 140°C в условиях микроволнового излучения в течение 1-4 часов. Также можно использовать карбонат натрия в ацетонитриле и воде.
Аналогичным образом получали N,N-диметил-2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксамид (соединение № 48) посредством реакции 2-хлор-N,N-диметил-6-(морфолин-4-ил)-9H-пурин-8-карбоксамида (80 мг) и пинаколового эфира 1-метил 4-пиразолбороновой кислоты (85 мг).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,39-13,59 (м, 1H), 8,21-8,27 (м, 1H), 7,89-7,94 (м, 1H), 4,13-4,38 (м, 4H), 3,88 (с, 3H), 3,76 (д, J=4,27 Гц, 4H), 3,45 (с, 3H), 3,05 (с, 3H). Спектр масс (ESI): m/z 357,14 [M+H]
Аналогичным образом получали соединения, приведенные в таблице 2, согласно методике, описанной в примере 7.
Соединение 1 (где Y обозначает CF или CCl) можно получать посредством реакции соединения 4a с боронатным эфиром 3b, как показано на схеме 4.
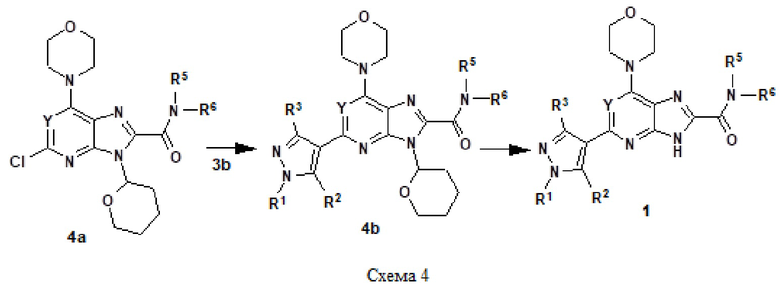
Реакцию сочетания Сузуки можно проводить по методике, описанной для схемы 3 выше. Соединение 4b можно конвертировать в соединение 1 согласно методике синтеза, известной специалисту в данной области, например, в процессе, описанном на схеме 2, выше. Более конкретно, реакции проводили следующим образом.
[Пример 8] К раствору соединения 4a (1 экв.) в н-пропаноле и воде (9:1, 20 мл), добавляли боронатный эфир 3b (1,4 экв.) и бикарбонат натрия (3 экв.) и реакционную смесь продували аргоном в течение 10 минут. Добавляли тетракис(трифенилфосфин)палладий(0) (0,1 экв.) и смесь нагревали в течение 1 часа в условиях микроволнового излучения при 140°C. Реакционную смесь фильтровали через целит; фильтрат упаривали в вакууме до сухого состояния. Остаток очищали флэш-хроматографией с использованием метанола и дихлорметана (от 5 до 15% метанол) в качестве элюента с получением соединения 4b. К раствору соединения 4b (1 экв.) в этаноле (от 10 до 100 мл) добавляли пара-толуолсульфоновую кислоту (1 экв.) и реакционную смесь перемешивали в течение 15 минут при 140°C в условиях микроволнового излучения. Реакционную смесь распределяли между бикарбонатом натрия и дихлорметаном (от 100 до 500 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием метанола и дихлорметана (от 5 до 15% метанол) в качестве элюента.
Аналогичным образом получали 6-фтор-7-(морфолин-4-ил)-5-(1,3,5-триметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанон (соединение № 93) с использованием [6-фтор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-5-(1,3,5-триметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (95 мг).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,42 (с, 1H), 4,94-5,05 (м, 1H), 4,43-4,53 (м, 1H), 3,66-3,86 (м, 11H), 3,57 (т, J=4,39 Гц, 4H), 3,17-3,27 (м, 1H), 2,84-2,94 (м, 1H), 2,47 (д, J=4,77 Гц, 5H), 2,19 (д, J=0,75 Гц, 3H), 2,09 (с, 3H), 1,89 (ушир. с, 2H), 1,30-1,54 (м, 2H). Спектр масс (ESI): m/z 527,15 (M+1). [M+H]
Сходные соединения, приведенные в таблице 2, получали согласно методике, описанной в примере 8.
Соединение 1 также можно получать посредством реакции сочетания кислотного соединения 5a с соединением амина 1b, как показано на схеме 5.
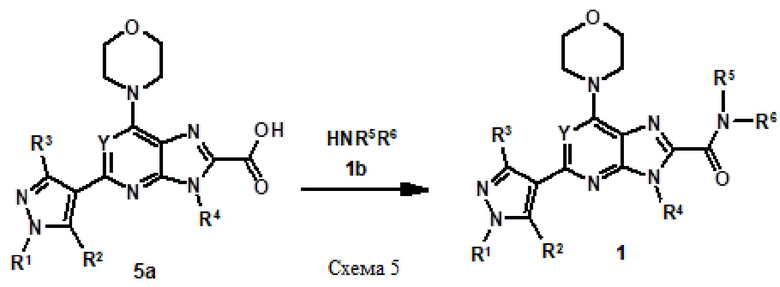
Эту реакцию сочетания можно проводить в растворителе, таком как диметилформамид, в присутствии агента реакции сочетания, такого как (гексафторфосфат бензотриазол-1-илокситрипирролидинофосфония), и основания, такого как N,N-диизопропилэтиламин. Более конкретно, реакцию проводили следующим образом.
[Пример 9] К раствору соединения 5a (1 экв.; справочный пример 4) в диметилформамиде (10 мл) добавляли соединение амина 1b (1,5 экв.) и N,N-диизопропилэтиламин (1,5 экв.) и реакционную смесь перемешивали в течение 30 минут при комнатной температуре. К реакционной смеси добавляли (гексафторфосфат бензотриазол-1-илокситрипирролидинoфосфония) (1,5 экв.), перемешивали при комнатной температуре в течение 12 часов. К ней добавляли воду и экстрагировали дихлорметаном. Органический слой промывали водой, сушили над сульфатом натрия, концентрировали в вакууме. Неочищенную смесь очищали флэш-хроматографией, элюируя продукт 5-15% метанолом в дихлорметане.
Аналогичным образом получали следующие соединения с использованием соответствующего кислотного соединения и соединения амина.
{3-[(2R,6S)-2,6-диметилморфолин-4-ил]азетидин-1-ил}[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанон (соединение № 119).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,33 (с, 1H), 8,23 (с, 1H), 7,97 (с, 1H), 6,83 (с, 1H), 4,60-4,70 (м, 1H), 4,36-4,44 (м, 1H), 4,04-4,13 (м, 1H), 3,90-3,99 (м, 5H), 3,88 (с, 3H), 3,80 (д, J=3,79 Гц, 4H), 3,52-3,61 (м, 2H), 3,17-3,24 (м, 1H), 2,68-2,83 (м, 2H), 1,57 (д, J=6,32 Гц, 2H), 1,07 (д, J=6,32 Гц, 6H). Спектр масс (ESI): m/z 418,03 [M+H]
N-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-N-[1-(тетрагидро-2H-пиран-4-ил)пиперидин-4-ил]-3H-имидазо[4,5-b]пиридин-2-карбоксамид (соединение № 121).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,40-13,58 (м, 1H), 8,24 (с, 1H), 7,93 (с, 1H), 4,84-4,95 (м, 1H), 4,07-4,48 (м, 4H), 3,89 (с, 5H), 3,76 (д, J=4,02 Гц, 4H), 3,30 (м, 4H), 2,92 (с, 5H), 2,00-2,13 (м, 2H), 1,57-1,90 (м, 6H), 1,35-1,52 (м, 2H). Спектр масс (ESI): m/z 510,05 [M+H].
4-(1-{[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]карбонил}азетидин-3-ил)пиперазин-2-он (соединение № 128).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,20-13,47 (м, 1H), 8,20-8,25 (м, 1H), 7,95-8,00 (м, 1H), 7,76-7,82 (м, 1H), 6,80-6,87 (м, 1H), 4,64-4,74 (м, 1H), 4,38-4,47 (м, 1H), 4,09-4,17 (м, 1H), 3,92-4,01 (м, 5H), 3,88 (с, 3H), 3,77-3,82 (м, 4H), 3,71-3,76 (м, 1H), 3,17-3,21 (м, 2H), 2,95-2,98 (м, 2H), 2,55-2,60 (м, 2H). Спектр масс (ESI): m/z 465,91 [M+H].
1-метил-4-(1-{[5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]карбонил}азетидин-3-ил)пиперазин-2-он (соединение № 129).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,33-13,34 (м, 1H), 8,22 (с, 1H), 7,97 (с, 1H), 6,83 (с, 1H), 4,61-4,75 (м, 1H), 4,38-4,47 (м, 1H), 4,05-4,17 (м, 1H), 3,94 (ушир. с, 4H), 3,88 (с, 3H), 3,80 (д, J=5,02 Гц, 4H), 3,15-3,18 (м, 1H), 3,03 (д, J=4,27 Гц, 2H), 2,83 (с, 3H), 2,63-2,70 (м, 2H). Спектр масс (ESI): m/z 479,98 [M+H].
1-метил-4-(1-{[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]карбонил}азетидин-3-ил)пиперазин-2-он (соединение № 130).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,58-13,67 (м, 1H), 8,24 (с, 1H), 7,92 (с, 1H), 4,63-4,73 (м, 1H), 4,38-4,47 (м, 1H), 4,16-4,38 (м, 2H), 4,08-4,16 (м, 1H), 3,93-4,00 (м, 1H), 3,88 (с, 3H), 3,76 (ушир. с, 4H), 3,25-3,30 (м, 2H), 3,03 (с, 2H), 2,83 (с, 3H), 2,61-2,71 (м, 2H). Спектр масс (ESI): m/z 480,98 [M+H].
N-метил-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-N-[1-(оксетан-3-ил)пиперидин-4-ил]-3H-имидазо[4,5-b]пиридин-2-карбоксамид (соединение № 131).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,13-13,31 (м, 1H), 8,23 (с, 1H), 7,98 (с, 1H), 6,84 (с, 1H), 4,92-5,14 (м, 1H), 4,49-4,60 (м, 2H), 4,35-4,46 (м, 2H), 3,91 (ушир. с, 4H), 3,88 (с, 3H), 3,79 (д, J=5,02 Гц, 4H), 2,94 (с, 2H), 2,77-2,83 (м, 2H), 1,67-1,91 (м, 6H). Спектр масс (ESI): m/z 480,97 [M+H].
Альтернативно можно использовать агент реакции сочетания, такой как 1-этил-3-(3-диметиламинопропил)карбодиимид и гидроксибензотриазол. Например, [2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанон (соединение № 58) получали следующим образом.
[Пример 10] К раствору соединения 5a (150 мг, 455,4 ммоль; справочный пример 4) в диметилформамиде (3 мл), добавляли триэтиламин(0,13 мл, 910,8 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид (173 мг, 910,8 ммоль), гидроксибензотриазол (123 мг, 910,8 ммоль) и морфолин пиперидин (116 мг,683,2 ммоль; AK Scientific)) и реакционную смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли воду и экстрагировали дихлорметаном (150 мл×2). Объединенные органические экстракты промывали рассолом (10 мл), сушили и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией, элюируя продукт 5-10% метанолом в дихлорметане, с получением [2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (152 мг).
Соединение 1 (где R1 представляет собой метил, R2, R3 и R4 представляют собой водород) также можно получать согласно схеме реакции 6, как показано ниже.
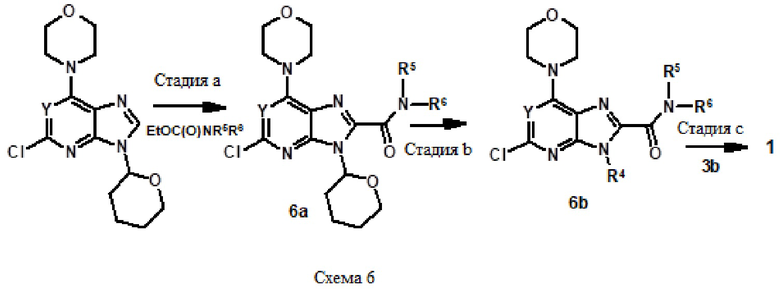
Согласно схеме 6, можно получать соединения по настоящему изобретению, в частности, когда Y обозначает N. Например, [2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанон (соединение №58) получали следующим образом:
Стадия a: синтез 6a (где -NR5R6 представляет собой морфолин пиперидинил).
К раствору 2-хлор-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурина (1 г, 3,09 ммоль) и карбамата (2,2 г, 9,28 ммоль; справочный пример 23) в тетрагидрофуране (70 мл) капельно добавляли диизопропиламид лития (2 M) в тетрагидрофуране (4,6 мл, 9,28 ммоль) при -78°C, а затем перемешивали при той же температуре в течение 30 минут и реакционной смеси позволяли достигнуть комнатной температуры в течение 90 минут. Реакционную смесь переливали в насыщенный раствор хлорида аммония (100 мл) и экстрагировали с использованием этилацетата (150 мл×2). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме до сухого состояния. Неочищенный продукт очищали флэш-хроматографией с использованием 2-5% метанола в дихлорметане в качестве градиентной системы с получением соединения 6a (1,07 г) в виде не совсем белого твердого вещества.
1H-ЯМР (400 МГц, CHCl3-d) δ м.д.: 5,67-5,84 (м, 1H), 4,57-4,85 (м, 1H), 3,95-4,55 (м, 5H), 3,80 (т, J=3,76 Гц, 4H), 3,52-3,77 (м, 7H), 2,80-3,18 (м, 2H), 2,48-2,64 (м, 5H), 2,45 (тд, J=3,45, 7,15 Гц, 1H), 1,98 (ушир. с, 4H), 1,96 (м, 4H).
Стадия b: синтез 6b
К раствору соединения 6a (400 мг, 0,7692 ммоль) в этаноле (15 мл) добавляли моногидрат п-толуолсульфоновой кислоты (146 мг, 0,7692 ммоль) и кипятили с обратным холодильником в течение 2 часов при 100°C. Проводили проверку посредством тонкослойной хроматографии, и она продемонстрировала еще не прореагировавший исходный материал, таким образом, вновь добавляли моногидрат п-толуолсульфоновой кислоты (43 мг,0,230 ммоль) и кипятили с обратным холодильником в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, переливали на насыщенный раствор бикарбоната натрия (100 мл) и экстрагировали дихлорметаном (150 мл×2). Объединенный органический экстракт сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток растирали в гексане и фильтровали через воронку Бюхнера, сушили в вакууме с получением соединения 6b (290 мг).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,57-14,14 (м, 1H), 4,88 (д, J=14,15 Гц, 1H), 4,46 (д, J=13,39 Гц, 1H), 4,01 (br s, 5H), 3,73 (т, J=4,55 Гц, 4H), 3,49-3,63 (м, 4H), 3,23 (т, J=11,62 Гц, 1H), 2,84-2,95 (м, 1H), 2,41-2,49 (м, 4H), 1,75-1,96 (м, 2H), 1,24-1,51 (м, 2H).
Стадия c: Синтез [2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона.
К раствору соединения 6b (200 мг, 0,4587 ммоль) в ацетонитриле (4 мл) добавляли соединение 3b (пинаколовый эфир 1-метил 4-пиразолбороновой кислоты; 143,11 г, 0,688 ммоль), раствор карбоната натрия (121,55 мг, 1,146 ммоль) в воде (2 мл) и дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) (18,714 мг, 0,022 ммоль) и реакционную смесь продували аргоном в течение 15 минут. Через 15 минут аргон удаляли и реакционной смеси позволяли кипеть с обратным холодильником при 140°C в течение 8-10 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли воду (80 мл) и экстрагировали с использованием смеси 10% метанол-дихлорметан (100 мл×2). Объединенный органический экстракт сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали с использованием флэш-хроматографии и элюирования продукта в 8-20% метаноле в дихлорметане с получением [2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (183 мг).
Синтез сложноэфирного соединения 1a и 5a
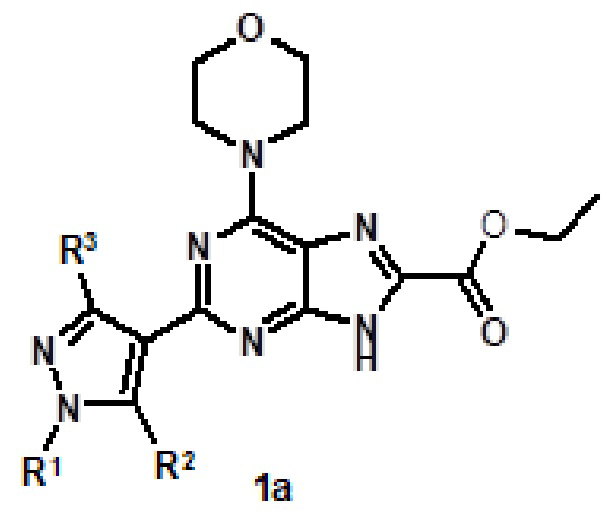
[Справочный пример 1] Синтез этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксилата

Стадия a: Синтез 2,6-дихлор-9-(тетрагидро-2H-пиран-2-ил)-9H-пурина.
К раствору 2,6-дихлор-9H-пурина (50 г, 264,55 ммоль) в этилацетате (500 мл) добавляли моногидрат п-толуолсульфоновой кислоты (1,36 г, 7,92 ммоль), а затем 3,4 дигидро-2H-пиран (55,36 г, 661,37 ммоль) через капельную воронку и нагревали при 70-80°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли аммиак (15 мл) и перемешивали в течение 15 минут. Добавляли воду (400 мл) и экстрагировали этилацетатом (300 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме до сухого состояния. Остаток растирали в гексане (500 мл), фильтровали и сушили в вакууме с получением 70 г 2,6-дихлор-9-(тетрагидро-2H-пиран-2-ил)-9H-пурина.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,96 (с, 1H), 5,75 (дд, J=2,01, 10,79 Гц, 1H), 3,99-4,12 (м, 1H), 3,69-3,83 (м, 1H), 2,20-2,34 (м, 1H), 1,93-2,07 (м, 2H), 1,69-1,84 (м, 1H), 1,39-1,67 (м, 2H).
Стадия b: синтез 2-хлор-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурина.
К раствору 2,6-дихлор-9-(тетрагидро-2H-пиран-2-ил)-9H-пурина (17 г, 62,24 ммоль) в метаноле (300 мл), добавляли морфолин (11,92 г, 136,92 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли в вакууме; добавляли воду (300 мл) и экстрагировали дихлорметаном (500 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме до сухого состояния с получением 20 г 2-хлор-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурина.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,42 (с, 1H), 5,60 (дд, J=2,13, 10,92 Гц, 1H), 4,01 (дд, J=1,76, 10,79 Гц, 2H), 3,64-3,77 (м, 7H), 2,51 (тд, J=1,76, 3,51 Гц, 1H), 2,11-2,25 (м, 1H), 1,90-2,01 (м, 2H), 1,69-1,81 (м, 1H), 1,53-1,62 (м, 2H).
Стадия c: синтез этил 2-хлор-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбоксилата.
К раствору 2-хлор-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурина (10 г, 30,88 ммоль) в сухом тетрагидрофуране (200 мл) добавляли N,N,N,N-тетраметилэтилендиамин (5,383 г, 46,32 ммоль) при комнатной температуре и охлаждали до -78°C. К реакционной смеси капельно добавляли н-бутиллитий (1,6 M, 29 мл, 46,32 ммоль), и ей позволяли нагреваться при -50°C в течение 1 часа. Реакционную смесь вновь охлаждали до -78°C и перемешивали в течение 5 минут. В круглодонной колбе этилхлорформиат (14,7 мл, 154,43 ммоль) отбирали в сухой тетрагидрофуран (100 мл) и охлаждали до -78°C. Реакционную смесь прямо переливали в раствор этилхлорформиата через жидкостную воронку и перемешивали в течение 2 минут. Реакционную смесь переливали в насыщенный раствор хлорида аммония (400 мл) и экстрагировали этилацетатом (500 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме до сухого состояния, после чего очищали колоночной хроматографией на силикагеле (размер пор 100-200) с использованием этилацетата и гексана (15-30%) в качестве градиентной системы. Наиболее чистые фракции собирали и концентрировали в вакууме с получением 7,5 г этил 2-хлор-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбоксилата.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 6,02-6,14 (м, 1H), 4,36-4,45 (м, 2H), 3,99-4,07 (м, 2H), 3,69-3,78 (м, 4H), 3,58-3,67 (м, 1H), 2,69-2,78 (м, 1H), 1,81-2,02 (м, 2H), 1,42-1,70 (м, 4H), 1,35 (т, 3H), 1,21-1,30 (м, 2H).
Стадия d: синтез этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбоксилата.
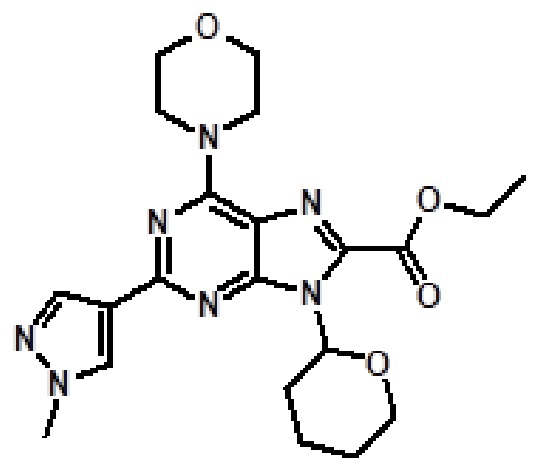
К раствору этил 2-хлор-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбоксилата (14 г, 35,36 ммоль) в 1,2-дихлорэтане (210 мл) добавляли пинаколовый эфир 1-метил 4-пиразолбороновой кислоты (9,595 г, 45,97 ммоль), карбонат цезия (28,751 г, 88,41 ммоль), тетракис(трифенилфосфин)палладий(0) (4,086 г, 3,53 ммоль) и реакционную смесь продували аргоном в течение 15 минут. Через 15 минут аргон удаляли и реакционной смеси позволяли кипеть с обратным холодильником при 110°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры; добавляли воду (400 мл) и экстрагировали дихлорметаном (500 мл, два раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который очищали колоночной хроматографией с использованием этилацетата и гексана (система от смеси 50% этилацетат-гексан до чистого этилацетата) в качестве системы градиента. Наиболее чистые фракции собирали и концентрировали в вакууме с получением 13,5 г этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбоксилата.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,30 (с, 1H), 7,99 (с, 1H), 6,11-6,23 (м, 1H), 4,40 (дд, J=1,25, 7,03 Гц, 3H), 4,16-4,35 (м, 3H), 4,00-4,10 (м, 1H), 3,90 (с, 3H), 3,72-3,81 (м, 4H), 3,58-3,69 (м, 1H), 3,02-3,15 (м, 1H), 1,94-2,06 (м, 1H), 1,82-1,92 (м, 1H), 1,52-1,71 (м, 3H), 1,35 (т, J=7,03 Гц, 3H).
Стадия e: синтез этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксилата.
К раствору этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбоксилата (10 г, 22,67 ммоль) в этаноле (200 мл) добавляли моногидрат п-толуолсульфоновой кислоты (4,31 г, 22,67 ммоль) и кипятили с обратным холодильником в течение 14 часов. Реакционную смесь охлаждали до комнатной температуры, выливали в насыщенный раствор бикарбоната натрия и экстрагировали дихлорметаном (500 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который затем очищали посредством растирания с гексаном (200 мл) с получением 5,1 г чистого этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксилата.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,67-14,03 (м, 1H), 8,26 (с, 1H), 7,95 (с, 1H), 4,38 (д, J=7,07 Гц, 3H), 4,18-4,34 (м, 2H), 3,89 (с, 3H), 3,77 (т, J=4,67 Гц, 4H), 1,34 (т, J=7,07 Гц, 3H).
Этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксилат также получали следующим образом.
Стадия a: синтез 2-хлор-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбальдегида
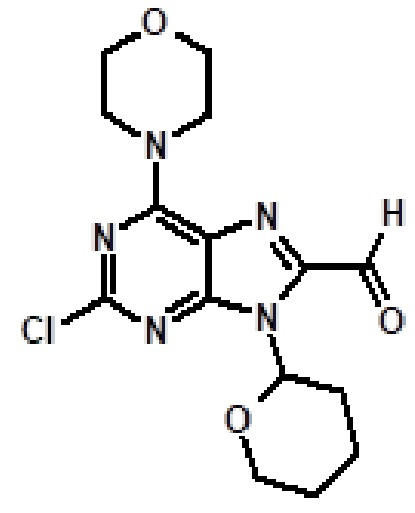
К смеси диизопропиламина (18,76 г, 185 ммоль) в тетрагидрофуране (100 мл) добавляли н-бутиллитий (1,6 M в гексане, 110 мл, 185 ммоль) при -78°C и перемешивали в течение 30 минут при 0°C. К этому свежеполученному диизопропиламиду лития капельно добавляли раствор 2-хлор-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурина (20 г, 61,9 ммоль) в тетрагидрофуране (100 мл) -78°C и перемешивали в течение 30 мнут. Добавляли диметилформамид (13,5 г, 185 ммоль) при -78°C и перемешивали при той же температуре в течение 2 часов. К реакционной смеси добавляли насыщенный раствор хлорида аммония (250 мл), экстрагировали этилацетатом (500 мл, три раза). Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали в вакууме с получением 19 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 9,93 (с, 1H), 5,57-5,63 (м, 1H), 4,03 (д, J=7,03 Гц, 2H), 3,67-3,75 (м, 8H), 1,57 (д, J=5,02 Гц, 4H), 0,81-0,96 (м, 2H).
Стадия b: синтез 2-хлор-6-(морфолин-4-ил)-9H-пурин-8-карбоновой кислоты.
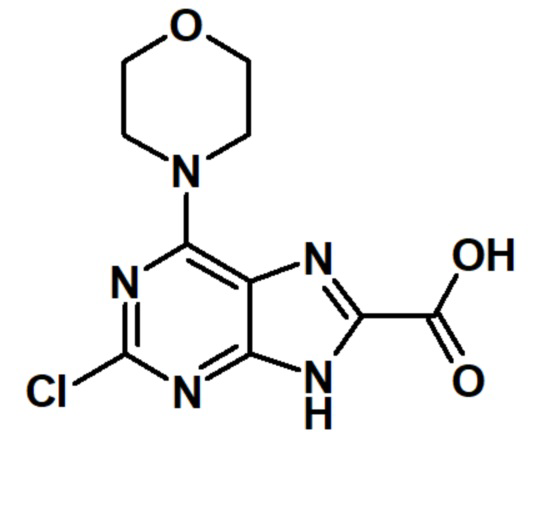
К раствору 2-хлор-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбальдегида (8 г, 22,7 ммоль) в этаноле (30 мл) добавляли нитрат серебра (4,86 г, 28,65 ммоль) и раствор гидроксида натрия (1,5 Н, 70 мл), а затем перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь фильтровали через слой целита, фильтрат концентрировали и остаток отбирали в воду и подкисляли добавлением гидроксида натрия (1 Н), а затем экстрагировали дихлорметаном (200 мл, два раза), водный слой подкисляли концентрированной хлористоводородной кислотой, объем уменьшали наполовину упариванием в вакууме с получением осадка, который отфильтровывали и сушили с получением 7 г 2-хлор-6-(морфолин-4-ил)-9H-пурин-8-карбоновой кислоты.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,71-14,31 (м, 1H), 4,40-4,74 (м, 1H), 3,67-4,00 (м, 4H), 3,34 (ушир. с, 4H).
Стадия c: синтез этил 2-хлор-6-(морфолин-4-ил)-9H-пурин-8-карбоксилата.
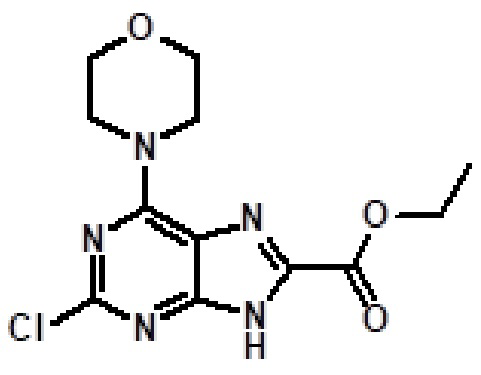
К раствору 2-хлор-6-(морфолин-4-ил)-9H-пурин-8-карбоновой кислоты (4 г, 14,1 ммоль) в этаноле (200 мл) добавляли тионилхлорид (20 мл) при 0°C и реакционную смесь кипятили с обратным холодильником в течение 12 часов. Реакционную смесь концентрировали в вакууме и остаток отбирали в воду, экстрагировали дихлорметаном (300 мл, два раза). Объединенный органический слой промывали рассолом, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией с использованием метанола и дихлорметана (5% метанол) в качестве элюента с получением 3 г этил 2-хлор-6-(морфолин-4-ил)-9H-пурин-8-карбоксилата.
Этил 2-хлор-6-(морфолин-4-ил)-9H-пурин-8-карбоксилат также получали следующим образом. К раствору этил 2-хлор-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбоксилата (10 г, 25,44 ммоль) в этаноле (200 мл), добавляли моногидрат пара-толуолсульфоновой кислоты (4,84 г, 25,44 ммоль) и кипятили с обратным холодильником в течение 14 часов. Реакционную смесь охлаждали до комнатной температуры, переливали в насыщенный водный раствор бикарбоната натрия и экстрагировали дихлорметаном (500 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия, концентрировали в вакууме с получением неочищенного продукта, и растирали с гексаном с получением 7,4 г указанного в заголовке соединения в качестве светло-желтого твердого вещества.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,37-15,08 (м, 1H), 4,32-4,43 (кв, J=7,07 Гц, 2H), 3,99-4,31 (м, 4H), 3,69-3,81 (м, 4H), 1,34 (т, J=7,07 Гц, 3H).
Стадия d: синтез этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоксилата.
К раствору этил 2-хлор-6-(морфолин-4-ил)-9H-пурин-8-карбоксилата (3 г, 9,6 ммоль) в диметилформамиде (30 мл) добавляли пинаколовый эфир 1-метил 4-пиразолбороновой кислоты (2,82 г, 13,5 ммоль), карбонат цезия (7,8 г, 24 ммоль) и тетракис(трифенилфосфин)палладий(0) (1,1 г, 0,96 ммоль) и реакционную смесь продували аргоном в течение 15 минут. Аргон удаляли и реакционной смеси позволяли кипеть с обратным холодильником при 110°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры; добавляли воду (100 мл), а затем экстрагировали дихлорметаном (300 мл, два раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного материала, который очищали колоночной хроматографией с использованием метанола и дихлорметана (5% метанол) в качестве градиентной системы с получением 1,8 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.:13,67-14,03 (м, 1H), 8,26 (с, 1H), 7,95 (с, 1H), 4,38 (д, J=7,07 Гц, 3H), 4,18-4,34 (м, 2H), 3,89 (с, 3H), 3,77 (т, J=4,67 Гц, 4H), 1,34 (т, J=7,07 Гц, 3H).
[Справочный пример 2] Синтез этил 6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата.
К раствору этил 6-фтор-5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (700 мг, 1,52 ммоль) в этаноле (10 мл) добавляли п-толуолсульфоновую кислоту (290 мг, 1,52 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 2 часов при 100°C. Реакционную смесь охлаждали до комнатной температуры; добавляли насыщенный бикарбонат натрия (50 мл) и экстрагировали дихлорметаном (250 мл, два раза). Объединенные органические экстракты сушили над безводным сульфатом натрия, концентрировали в вакууме, добавляли гексан (50 мл) и перемешивали в течение 5 минут, фильтровали, сушили в вакууме с получением 500 мг указанного в заголовке соединения.
[Справочный пример 3] Синтез этил 5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата
К раствору этил 5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (450 мг, 1,025 ммоль) в этаноле (5 мл) добавляли п-толуолсульфоновую кислоту (194 мг, 1,025 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 14 часов. Реакционную смесь охлаждали до комнатной температуры и добавляли насыщенный бикарбонат натрия (50 мл), экстрагировали дихлорметаном (250 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия, концентрировали в вакууме с получением неочищенного продукта, который растирали с использованием гексана (50 мл) и фильтровали, сушили в вакууме с получением 210 мг указанного в заголовке соединения в качестве желтого твердого вещества.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,68 (ушир. с, 1H), 8,27 (с, 1H), 8,01 (с, 1H), 6,85 (с, 1H), 4,37 (кв, J=7,03 Гц, 2H), 3,96 (ушир. с, 4H), 3,76-3,92 (м, 8H), 1,34 (т, J=7,03 Гц, 3H).
[Справочный пример 4] Синтез 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-карбоновой кислоты
К раствору этил 2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9-(тетрагидро-2H-пиран-2-ил)-9H-пурин-8-карбоксилата (5 г, 11,32 ммоль; справочный пример 1) в тетрагидрофуране и воде (8:2, 60 мл) добавляли гидроксид лития (1,42 г, 33,97 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь упаривали до сухого состояния, добавляли воду (15 мл), промывали этилацетатом (100 мл, два раза). Водный слой подкисляли с использованием концентрированной хлористоводородной кислоты (pH2). Выпавший осадок отфильтровывали и остаток промывали гексаном и сушили с получением указанного в заголовке соединения (2,5 г).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 12,42-13,44 (м, 1H), 8,16-8,23 (м, 1H), 7,86-7,94 (м, 1H), 4,17-4,40 (м, 4H), 3,88 (с, 3H), 3,75 (ушир. с, 4H). Спектр масс (ESI): m/z 330,7.
Синтез сложноэфирного соединения 2a
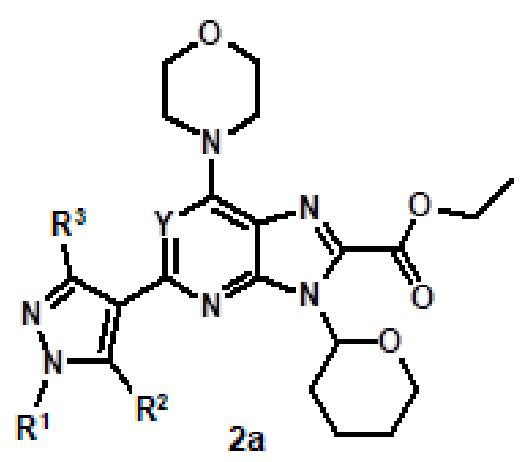
Общий способ синтез сложноэфирного соединения 2a.
К раствору этил 5-хлор-6-фтор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (1 экв.) в 1,2-дихлорэтане (10 мл), добавляли боронатный сложный эфир 3b (1,25 экв.) и карбонат цезия (2,5 экв.), и реакционную смесь продували аргоном в течение 10 минут, добавляли тетракис(трифенилфосфин)палладия (0) (0,075 экв.) и смесь нагревали в течение 1 часа в условиях микроволнового излучения при 140°C. Реакционную смесь фильтровали через слой целита; фильтрат упаривали в вакууме до сухого состояния. Остаток очищали флэш-хроматографией с использованием метанола и дихлорметана (5-15% метанол) в качестве элюирующей системы с получением сложноэфирного соединения 2a.
[Справочный пример 5] Синтез этил 5-(1-метил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата
К раствору этил 5-хлор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (1 г, 2,53 ммоль) в 1,2-дихлорэтане (10 мл) добавляли боронатный сложный эфир (791 мг, 3,8 ммоль, combiblocks) и фосфат калия (1,34 г, 6,34ммоль, acros) и реакционную смесь продували аргоном в течение 30 минут, добавляли тетракис(трифенилфосфин)палладий(0) (293 мг, 0,25 ммоль), и смесь перемешивали в течение 1 часа в условиях микроволнового излучения при 140°C. Реакционную смесь фильтровали через слой целита, упаривали в вакууме до сухого состояния. Остаток растворяли в дихлорметане (300 мл) и промывали водой (100 мл), рассолом (100 мл) и сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали колоночной хроматографией с использованием метанола и дихлорметана (5% метанол) в качестве элюента с получением 1,2 г указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,30 (с, 1H), 8,05 (с, 1H), 6,91 (с, 1H), 6,17-6,22 (м, 1H), 4,40 (дд, J=2,53, 7,07 Гц, 2H), 4,00-4,07 (м, 1H), 3,88-3,95 (м, 7H), 3,77-3,84 (м, 4H), 3,59-3,68 (м, 1H), 3,08-3,15 (м, 1H), 1,98-2,05 (м, 1H), 1,82-1,90 (м, 1H), 1,62-1,71 (м, 2H), 1,53-1,60 (м, 1H), 1,36 (т, J=7,07 Гц, 3H).
[Справочный пример 6] Синтез этил 5-(1,3-диметил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата
К раствору этил 5-хлор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (1 г, 2,53 ммоль) в 1,2-дихлорэтане (15 мл) добавляли пинаколовый эфир 1,3-диметил 4-пиразолбороновой кислоты (729 мг, 3,28 ммоль), карбонат цезия (2,053 г, 6,31 ммоль), тетракис(трифенилфосфин)палладий(0) (292 мг, 0,25 ммоль) и реакционную смесь продували аргоном в течение 15 минут. Через 15 минут аргон удаляли и реакционной смеси позволяли кипеть с обратным холодильником при 110°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли воду (400 мл) и экстрагировали дихлорметаном (500 мл, два раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который при очистке колоночной хроматографией с использованием системы от смеси 50% этилацетат-гексан до чистого этилацетата дал 1,4 г указанного в заголовке соединения.
Синтез амидного соединения 3a
Общий способ синтез амидного соединения 3a.
Стадия a: синтез амидного соединения формулы:
К раствору этил 5-хлор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (1 экв.) и соединения амина 1b (3 экв.) в тетрагидрофуране (20 мл) добавляли триметилалюминий (4 экв.) при комнатной температуре и реакционную смесь кипятили с обратным холодильником в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры и добавляли насыщенный раствор хлорида аммония (от 50 до 200 мл), распределяли с помощью дихлорметана (от 300 до 800 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием метанола и дихлорметана (5-10% метанол) в качестве элюента с получением желаемых соединений.
Стадия b: синтез амидного соединения формулы:
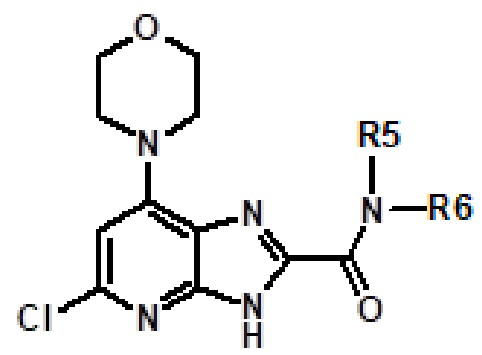
К раствору соединения (1 экв., стадия a) в этаноле (10 мл) добавляли п-толуолсульфоновую кислоту (1 экв.) и реакционную смесь кипятили с обратным холодильником 18 часов. Реакционную смесь охлаждали до комнатной температуры и распределяли между насыщенным бикарбонатом натрия (от 50 до 200 мл) и дихлорметаном (от 300 до 800 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием метанола и дихлорметана (5-15% метанол) в качестве элюента с получением желаемых соединений.
Стадия c: синтез амидного соединения формулы:
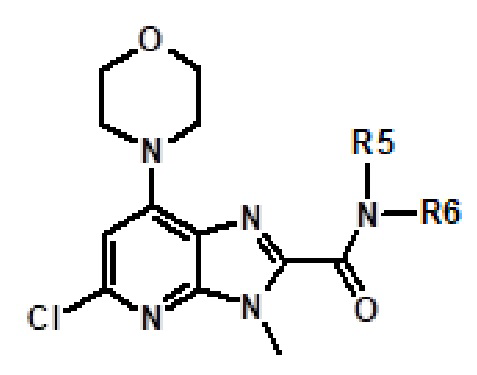
К раствору амидного соединения (1 экв., стадия b) в диметилформамиде (12 мл) добавляли карбонат калия (2,5 экв.), а затем метилйодид (1,5 экв.) при 0°C и ему позволяли нагреться до комнатной температуры в течение 2 часов. Добавляли воду (20-100 мл) и экстрагировали этилацетатом (100-400 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием метанола и дихлорметана (5-15% метанол) в качестве элюента с получением желаемого соединения.
Стадия d: синтез амидного соединения формулы:
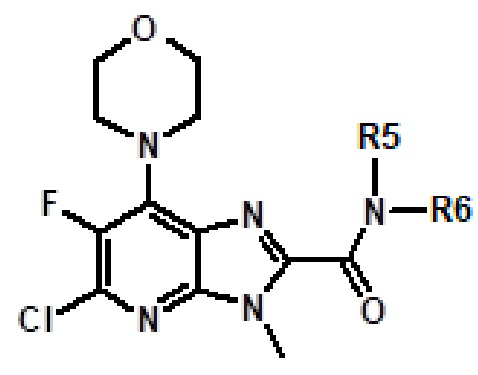
К раствору соединения (1 экв., стадия c) в ацетонитриле (110 мл) добавляли accuflor (2,3 экв.) при 0°C и ему позволяли нагреться до комнатной температуры в течение 2,5 часов. Добавляли насыщенный хлорид аммония (50-200 мл) и экстрагировали дихлорметаном (200-500 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием метанола и дихлорметана (5-15% метанол) в качестве элюента с получением желаемого соединения.
Соединение 3a также можно получать путем связывания 2-хлор-6-(морфолин-4-ил)-9H-пурин-8-карбонилхлорида (промежуточное соединение, не выделенное) с аминосоединением 1b, как показано ниже:
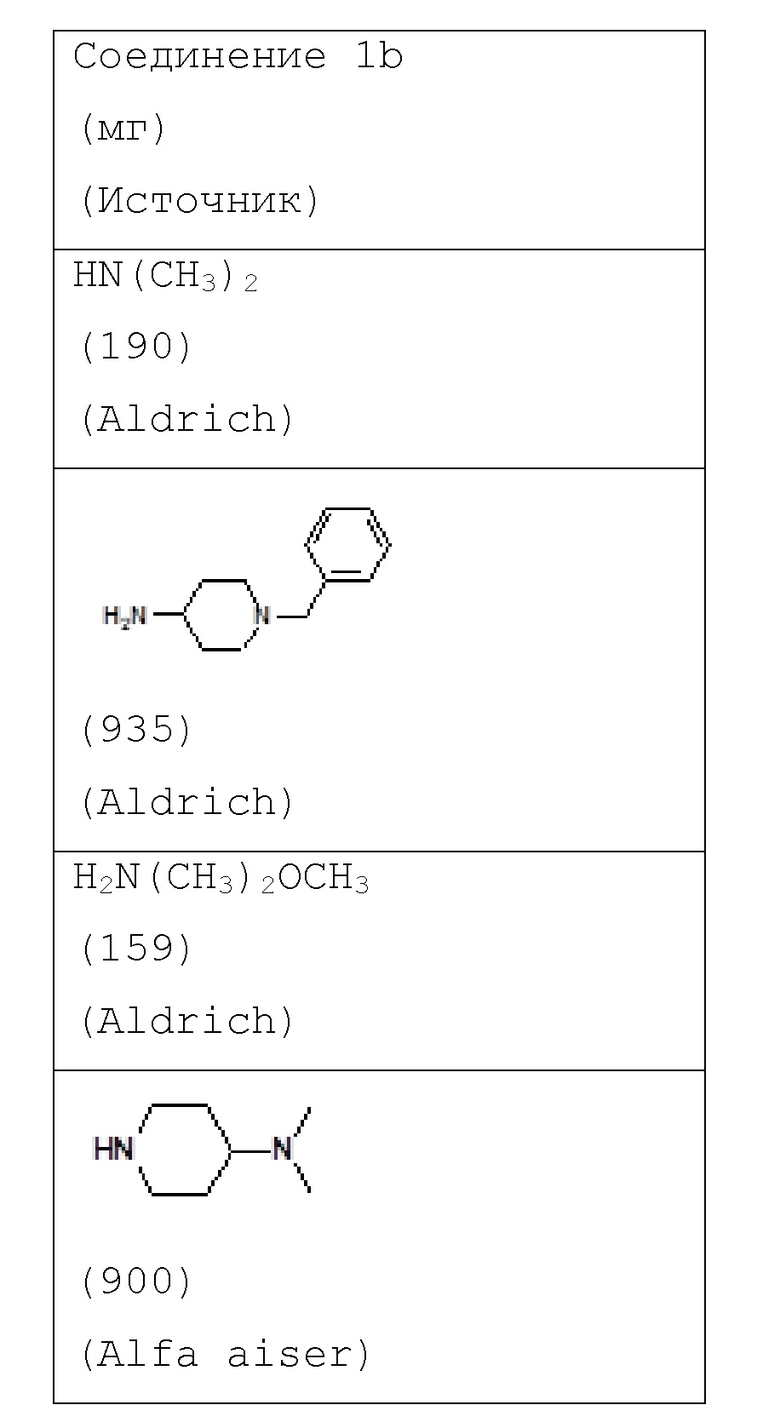
К раствору 2-хлор-6-(морфолин-4-ил)-9H-пурин-8-карбоновой кислоты (1 экв.) добавляли тионилхлорид (2,5 мл) и каталитическое количество диметилформамида, нагретого до 60-70°C в течение 2 часов. Реакционную смесь упаривали в вакууме с получением остатка. К остатку добавляли дихлорметан (20 мл), и к нему добавляли аминосоединение 1b, показанное выше (2 экв.), и триэтиламин (6 экв.) при 0°C, реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли дихлорметаном (300-900 мл) и промывали водой (100-300 мл). Органический слой сушили над безводным сульфатом натрия и концентрировали в вакууме, а затем очищали колоночной хроматографией с использованием метанола и дихлорметана (5-15% метанол) в качестве элюента.
[Справочный пример 7] Согласно описанной выше методике получали [5-хлор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанон (2 г) с использованием 5-хлор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-карбоновой кислоты (3,4 г, 12 ммоль), тионилхлорида (17 мл), дихлорметана (100 мл), 4-морфолинопиперидина (4,08 г, 24 ммоль) и триэтиламина (6,55 мл, 48,05 ммоль).
[Справочный пример 8] Синтез [2-хлор-6-(морфолин-4-ил)-9H-пурин-8-ил](2-окса-7-азаспиро[3.5]нон-7-ил)метанона
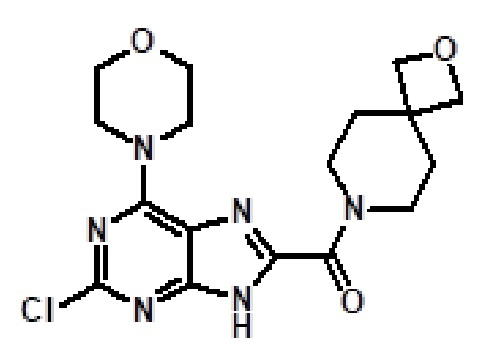
К раствору этил 2-хлор-6-(морфолин-4-ил)-9H-пурин-8-карбоксилата (240 мг, 0,77 ммоль) и 2-окса-7-азаспиро[3.5]нонана (195,96 мг, 1,54 ммоль) в тетрагидрофуране (10 мл), капельно добавляли триметилалюминий (2 M) в толуоле (0,96 мл, 1,93 ммоль) при комнатной температуре. Во время добавления внутренняя температура реакционной смеси возрастала. После завершения добавления реакционную смесь энергично кипятили с обратным холодильником при 110°C в течение 2-3 часов. Реакционную смесь охлаждали до комнатной температуры и осторожно гасили с использованием капельного добавления метанола, а затем добавления дихлорметана (100 мл). Добавляли воду (50 мл) и перемешивали в течение 10 минут, органический слой отделяли. Водный слой экстрагировали с использованием дихлорметана (200 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который при очистке колоночной хроматографией (Combiflash) с использованием метанола и дихлорметана (5% метанол) в качестве градиентной системы дал 170 мг указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,69-13,94 (м, 1H), 4,35 (с, 4H), 3,99 (ушир. с, 2H), 3,69-3,80 (м, 4H), 3,59-4,5 (ушир. с, 4H), 3,59 (ушир. с, 2H), 1,86 (д, J=5,52 Гц, 4H).
Аналогичным образом получали следующие соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,69-14,07 (м, 1H), 4,88 (д, J=13,05 Гц, 1H), 4,47 (д, J=13,55 Гц, 1H), 3,82-4,35 (м,4H), 3,73 (т, J=4,39 Гц, 5H), 3,45-3,57 (м, 2H), 3,21 (т, 1H), 2,82-2,92 (м, 1H), 2,74 (д, J=10,54 Гц, 2H), 1,75-1,91 (м, 4H), 1,31-1,50 (м, 2H), 1,04 (д, J=6,27 Гц, 6H).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,74-14,06 (м, 1H), 4,89-5,21 (м, 1H), 4,51-4,67 (м, 2H), 4,11-4,47 (м, 4H), 4,04 (с, 2H), 3,80 (т, J=5,02 Гц, 2H), 3,73 (т, J=4,52 Гц, 4H), 3,25 (д, J=4,52 Гц, 3H), 2,83-2,98 (м, 1H), 1,69 (ушир. с, 4H).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 4,41-4,49 (м, 1H), 4,20-4,41 (м, 2H), 4,07-4,16 (м, 1H), 3,79-4,05 (м, 2H), 3,71-3,76 (м, 5H), 3,70 (с, 3H), 3,51-3,60 (м, 4H), 3,10-3,21 (м, 1H), 2,86-2,98 (м, 1H), 2,42-2,49 (м, 4H), 1,75-1,98 (м, 2H), 1,34-1,49 (м, 2H).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 4,41-4,49 (м, 1H), 4,40-4,52 (т, 2H), 4,20-4,41 (м, 2H), 4,07-4,16 (м, 1H), 3,79-4,05 (м, 2H), 3,71-3,76 (м, 5H), 3,51-3,60 (м, 4H), 3,10-3,21 (м, 1H), 2,86-2,98 (м, 1H), 2,42-2,49 (м, 4H), 1,75-1,98 (м, 2H), 1,34-1,49 (м, 2H), 1,32 (кв, 2H).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 4,41-4,47 (м, 1H), 4,22-4,38 (м, 2H), 4,07-4,14 (м, 1H), 3,77-4,02 (м, 2H), 3,71-3,75 (м, 4H), 3,71 (с, 3H), 3,10-3,21 (м, 1H), 2,84-2,97 (м, 1H), 2,34-2,46 (м, 1H), 2,19 (с, 6H), 1,84-1,94 (м, 1H), 1,71-1,81 (м, 1H), 1,28-1,48 (м, 2H).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 4,41-4,49 (м, 1H), 4,27-4,40 (м, 2H), 4,19 (д, J=7,28 Гц, 2H), 4,00-4,07 (м, 2H), 3,78-4,00 (м, 1H), 3,73 (т, J=4,39 Гц, 4H), 3,09-3,21 (м, 1H), 2,89-3,00 (м, 1H), 2,33-2,45 (м, 1H), 2,18 (с, 6H), 1,83-1,94 (м, 1H), 1,71-1,81 (м, 1H), 1,34-1,42 (м, 2H), 1,32 (т, J=7,03 Гц, 3H).
[Справочный пример 9] Синтез [5-хлор-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона
К раствору [5-хлор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона (800 мг, 1,7 ммоль) в диметилформамиде (30 мл) добавляли метилйодид (289,68 мг, 2,04 ммоль, spectrochem) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов; добавляли воду (50 мл), экстрагировали дихлорметаном (250 мл, три раза). Объединенный органический слой промывали водой (200 мл, три раза), сушили над безводным сульфатом натрия и концентрировали в вакууме, а затем очищали колоночной хроматографией с использованием дихлорметана и метанола (5% метанол) в качестве элюента с получением 400 мг указанного в заголовке соединения в виде белого твердого вещества.
[Справочный пример 10] Синтез [5-хлор-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(диметиламино)пиперидин-1-ил]метанона
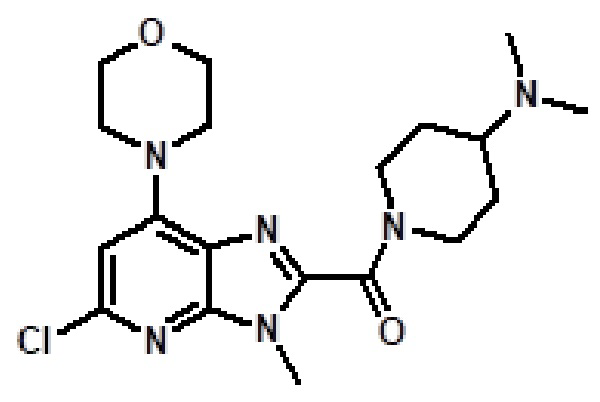
Стадия a: синтез этил 5-хлор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата
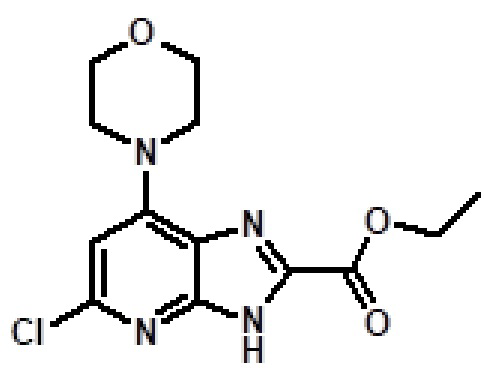
К раствору этил 5-хлор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (1,4 г, 3,54 ммоль) в этаноле (14 мл) добавляли п-толуолсульфоновую кислоту (670 мг, 3,54 ммоль) и реакционную смесь перемешивали в течение 15 минут при 140°C в условиях микроволнового излучения. Добавляли насыщенный бикарбонат натрия (50 мл), экстрагировали этилацетатом (250 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия, концентрировали в вакууме с получением 980 мг указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,82-14,11 (м, 1H), 6,58 (с, 1H), 4,39 (д, J=7,07 Гц, 2H), 3,95 (ушир. с, 4H), 3,73-3,79 (м, 4H), 1,34 (т, J=7,07 Гц, 3H).
Стадия b: синтез этил 5-хлор-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата
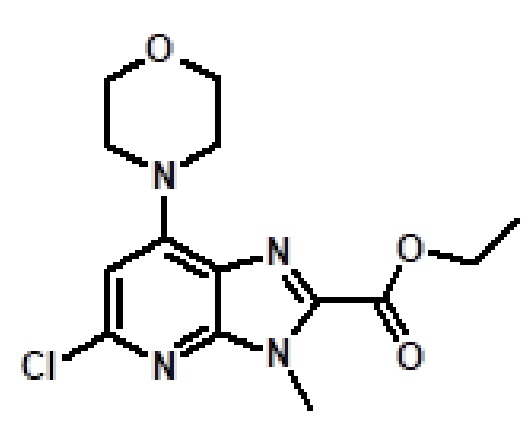
К раствору этил 5-хлор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (960 мг, 3,1 ммоль) в ацетоне (16 мл) добавляли метилйодид (886 мг, 6,2 ммоль) и карбонат калия (855 мг, 6,2 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь фильтровали и концентрировали до сухого состояния и очищали колоночной хроматографией (от 50% этилацетата в гексан до чистого этилацетата) с получением 820 мг указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 6,65 (с, 1H), 4,35-4,46 (м, 2H), 3,93-4,01 (м, 7H), 3,76 (д, J=4,55 Гц, 4H), 1,34 (с, 3H).
Стадия c: синтез [5-хлор-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(диметиламино)пиперидин-1-ил]метанона
К раствору этил 5-хлор-3-метил-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (380 мг, 1,17 ммоль) и 4-N,N-диметилпиперидина (300 мг, 2,34 ммоль) в тетрагидрофуране (10 мл) добавляли триметилалюминий (1,2 мл, 2,34 ммоль) при 0°C и реакционную смесь кипятили с обратным холодильником при 80°C в течение 18 часов. Реакционную смесь охлаждали, добавляли метанол (5 мл), концентрировали в вакууме до сухого состояния и очищали колоночной хроматографией с использованием метанола и дихлорметана (5% метанол) с получением 410 мг указанного в заголовке соединения.
[Справочный пример 11] Синтез 2-хлор-9-метил-6-(морфолин-4-ил)-9H-пурин-8-карбоновой кислоты
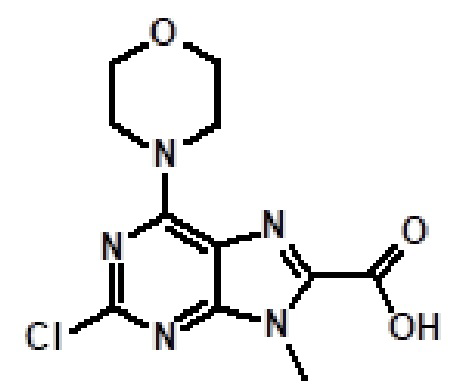
Стадия a: синтез 2-хлор-6-(морфолин-4-ил)-9H-пурина
К раствору 2,6-дихлор-9H-пурина (20 г, 105,82 ммоль) в этаноле (200 мл), добавляли морфолин (20,49 г, 232,80 ммоль) при комнатной температуре и кипятили с обратным холодильником в течение 1 часа при 80°C. Реакционную смесь охлаждали до комнатной температуры, растворитель удаляли в вакууме? и добавляли воду (300 мл), и экстрагировали дихлорметаном (300 мл×3). Объединенные органические экстракты (900 мл) сушили над безводным сульфатом натрия и концентрировали в вакууме до сухого состояния с получением 23 г 2-хлор-6-(морфолин-4-ил)-9H-пурина.
Стадия b: синтез 2-хлор-9-метил-6-(морфолин-4-ил)-9H-пурина
К раствору 2-хлор-6-(морфолин-4-ил)-9H-пурина (4 г, 16,73 ммоль) в ацетоне (60 мл) добавляли метилйодид (2,14 мл, 33,47 ммоль) при 0°C и кипятили с обратным холодильником в течение ночи. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Остаток промывали гексаном (100 мл) и сушили в вакууме с получением 3,9 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,17 (с, 1H), 3,85-4,94 (м, 4H), 3,60-3,82 (м, 4H), 3,60-3,82 (с, 3H).
Аналогично получали 2-хлор-9-этил-6-(морфолин-4-ил)-9H-пурин.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,24 (с, 1H), 4,15 (д, J=7,28 Гц, 2H), 3,75-4,15- (м, 4H) 3,64-3,78 (м, 4H), 1,37 (т, J=7,28 Гц, 3H).
Стадия c: синтез 2-хлор-9-метил-6-(морфолин-4-ил)-9H-пурин-8-карбальдегида

К смеси 2-хлор-9-метил-6-(морфолин-4-ил)-9H-пурина (3,3 г, 13,04 ммоль) в тетрагидрофуране (80 мл) добавляли тетраметилэтилендиамин (4,3 мл, 26,08 ммоль) при комнатной температуре и охлаждали до -78°C. После достижения этой температуры капельно добавляли н-бутиллитий (2,5 M, 13 мл, 32,60 ммоль), перемешивали в течение 60 минут, а затем добавляли диметилформамид (2 мл, 26,08 ммоль) при -78°C и перемешивание продолжали при той же температуре в течение 2 часов. К реакционной смеси добавляли насыщенный хлорид аммония (100 мл), экстрагировали этилацетатом (300 мл, три раза). Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали в вакууме с получением 3,1 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 9,87 (с, 1H), 4,40-4,74 (м, 2H), 3,91-3,71 (м, 2H), 3,91 (с, 3H), 3,77 (ушир. с, 4H).
Аналогично получали 2-хлор-9-этил-6-(морфолин-4-ил)-9H-пурин-8-карбальдегид.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 9,86 (с, 1H), 4,52-4,72 (м, 2H), 4,45 (д, J=7,28 Гц, 2H), 3,82-3,99 (м, 2H), 3,77 (ушир. с, 4H), 1,31 (т, J=7,15 Гц, 3H).
Стадия d: синтез 2-хлор-9-метил-6-(морфолин-4-ил)-9H-пурин-8-карбоновой кислоты
К раствору 2-хлор-9-метил-6-(морфолин-4-ил)-9H-пурин-8-карбальдегида (3,3 г, 11,74 ммоль) в этаноле (30 мл) добавляли нитрат серебра (2,5 г, 14,79 ммоль) и раствор гидроксида натрия (1,5 Н, 40 мл) и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь фильтровали через слой целита концентрировали и остаток отбирали в воду и подщелачивали добавлением гидроксида натрия (1 Н), экстрагировали дихлорметаном (60 мл, два раза), водный слой подкисляли концентрированной хлористоводородной кислотой, объем уменьшали наполовину упариванием в вакууме с получением осадка, который отфильтровывали и сушили с получением 2 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 12,70-15,36 (м, 1H), 3,93-4,85 (м, 4H), 3,89 (с, 3H), 3,74 (ушир. с, 4H).
Аналогично получали 2-хлор-9-этил-6-(морфолин-4-ил)-9H-пурин-8-карбоновую кислоту.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 12,70-15,36 (м, 1H), 4,53-4,71 (м, 2H), 4,40-4,52 (т, 2H), 3,82-4,07 (м, 2H), 3,74 (ушир. с, 4H), 1,32 (кв, 2H).
В соответствии с описанной выше методикой, 2-хлор-9-метил-6-(морфолин-4-ил)-9H-пурин-8-карбоновую кислоту конвертировали в амидное соединение 3a с использованием тионилхлорида (17 мл), диметилформамида (каталитическое количество), аминосоединения 1b (2 экв.) и триэтиламина (6 экв.)
Синтез амидного соединения 4a
Стадия a: синтез N-оксида 3H-имидазо[4,5-b]пиридина
К раствору 1-деазапурина (50 г, 420 ммоль) в этилацетате (450 мл) добавляли м-хлорпероксибензойную кислоту (55%, 171 г, 546 ммоль) порционно при 0°C и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь разбавляли гексаном (500 мл) и фильтровали. Образовавшееся твердое вещество сушили в вакууме с получением 75,86 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,43 (с, 1H), 8,21 (д, J=6,27 Гц, 1H), 7,63 (д, J=8,03 Гц, 1H), 7,23 (дд, J=6,27, 8,28 Гц, 1H).
Стадия b: синтез 7-хлор-3H-имидазо[4,5-b]пиридина
К перемешиваемому фосфорилхлориду (350 мл, 3,76 моль) в круглодонной колбе добавляли N-оксид 3H-имидазо[4,5-b]пиридина (47 г, 348 ммоль) порционно при 0°C и реакционную смесь нагревали при 115°C в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры и растворитель выпаривали при пониженном давлении до сухого состояния. Остаток выливали на раздробленный лед, и нейтрализовывали насыщенным раствором бикарбоната натрия, и экстрагировали этилацетатом (1 л, два раза), промывали водой (500 мл), рассолом (500 мл) и сушили над безводным сульфатом натрия, и концентрировали в вакууме с получением 31,5 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,52 (д, J=11,29 Гц, 2H), 8,30 (д, J=5,02 Гц, 1H), 8,08 (д, J=8,28 Гц, 1H), 7,40 (д, J=5,27 Гц, 1H), 7,31 (д, J=8,28 Гц, 1H).
Стадия c: синтез N-оксида 7-хлор-3H-имидазо[4,5-b]пиридина
К раствору 7-хлор-3H-имидазо[4,5-b]пиридина (31,3 г, 204 ммоль) в этилацетате (450 мл) добавляли м-хлорпероксибензойную кислоту (55%, 89,5 г, 286 ммоль) порционно при 0°C и реакционную смесь перемешивали при комнатной температуре в течение 7 часов. Реакционную смесь разбавляли гексаном (500 мл) и фильтровали. Образовавшееся твердое вещество сушили в вакууме с получением 48,3 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,52 (с, 1H), 8,23 (д, J=6,78 Гц, 1H), 7,87-7,93 (м, 2H), 7,68-7,75 (м, 1H), 7,52-7,59 (м, 1H), 7,40 (д, J=6,78 Гц, 1H).
Стадия c: синтез 5,7-дихлор-3H-имидазо[4,5-b]пиридина
К перемешиваемому фофорилхлориду (240 мл, 262 ммоль) в круглодонной колбе добавляли N-оксид 7-хлор-3H-имидазо[4,5-b]пиридина (48 г, 284 ммоль) порционно при 0°C и реакционную смесь нагревали при 115°C в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры. Растворитель выпаривали при пониженном давлении до сухого состояния. Остаток переливали на раздробленный лед, и нейтрализовывали насыщенным раствором бикарбоната натрия, и экстрагировали этилацетатом (1 л, два раза), промывали водой (500 мл), рассолом (500 мл) и сушили над безводным сульфатом натрия, фильтровали, концентрировали в вакууме, и очищали колоночной хроматографией на силикагеле (размер пор 100-200) с использованием метанола и дихлорметана (5% метанол) в качестве элюента с получением 22,8 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,59 (с, 1H), 7,90 (д, J=2,01 Гц, 1H), 7,59 (с, 1H).
Стадия d: синтез 5-хлор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридина
К раствору 5,7-дихлор-3H-имидазо[4,5-b]пиридина (17,75 г, 94,4 ммоль) в этаноле (180 мл) добавляли морфолин (82,1 г, 944 ммоль) и реакционную смесь нагревали при 140°C в течение 18 часов в стальном баллоне. Реакционную смесь охлаждали до комнатной температуры, переливали в воду и образовавшийся осадок отфильтровывали и сушили в вакууме с получением 16,2 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 12,72-13,12 (м, 1H), 8,11 (с, 1H), 6,50 (с, 1H), 3,88 (д, J=4,52 Гц, 4H), 3,70-3,76 (м, 4H).
Стадия e: синтез 5-хлор-6-фтор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридина

К раствору 5-хлор-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридина (4 г, 16,74 ммоль) в ацетонитриле и воде (9:1, 60 мл) добавляли selectfluor (14,8 г, 41,84 ммоль), нагревали при 150°C в условиях микроволнового излучения в течение 10 минут. Реакционную смесь переливали в насыщенный бикарбонат натрия (30 мл), экстрагировали этилацетатом (100 мл, два раза), промывали водой (50 мл), рассолом (50 мл) и сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали колоночной хроматографией с использованием метанола и дихлорметана (6% метанол) в качестве элюента с получением 1,15 г указанного в заголовке соединения (в качестве неочищенного соединения).
Стадия f: синтез 5-хлор-6-фтор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридина
К раствору соединения (1,13 г, 4,40 ммоль, стадия e) в этилацетате (15 мл), добавляли п-толуолсульфоновую кислоту (170 мг, 0,88 ммоль) и 3,4-дигидро-2H-пиран (923 мг, 10,99 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (300 мл), промывали насыщенным бикарбонатом натрия (100 мл), водой (100 мл), рассолом (100 мл) и сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией с использованием метанола и дихлорметана (1% метанол) в качестве элюента с получением 650 мг неочищенной смеси (F:Cl:H).
Стадия g: синтез этил 5-хлор-6-фтор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата
К раствору 5-хлор-6-фтор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридина (5,4 г, 15,81 ммоль) в тетрагидрофуране (150 мл) добавляли н-бутиллитий (1,6 M, 14,85 мл, 23,76 ммоль) при -78°C и реакционную смесь перемешивали в течение 1 часа при той же температуре и далее перемешивали в течение 1 часа при -40°C. Реакционную смесь переливали в раствор этилхлорформиата (13,68 г, 126,72 ммоль) в тетрагидрофуране при -78°C и реакционную смесь перемешивали в течение 10 минут. Затем ее переливали в насыщенный раствор хлорида аммония (75 мл) и экстрагировали этилацетатом (400 мл, три раза), промывали водой (200 мл), рассолом (200 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме. Соединение очищали колоночной хроматографией с использованием этилацетата и гексана (1:1) в качестве элюента с получением 1,9 г указанного в заголовке соединения и этил 5-хлор-6-хлор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата(0,91 г).
Стадия h: синтез соединения 4a (где Y представляет собой CF и CCl)
К раствору соединения 1b (2,5 экв.) в тетрагидрофуране (10-70 мл) добавляли триметилалюминий (2,5 экв.) при -10°C и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Затем этот раствор добавляли к раствору этил 5-хлор-6-фтор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата в тетрагидрофуране (1 экв. в 10-70 мл) и реакционную смесь кипятили с обратным холодильником при 100°C в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (50-200 мл) и метанолом (5-25 мл). Затем ее распределяли между водой (30-150 мл) и этилацетатом (200-600 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием метанола и дихлорметана (5-15% метанол) в качестве элюента с получением указанного в заголовке соединения.
[Справочный пример 12] Синтез [5,6-дихлор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона
К раствору 4-морфолинопиперидина (2,5 экв.) в тетрагидрофуране (8 мл), добавляли триметилалюминий (2,5 экв.) при -10°C и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Затем этот раствор добавляли к раствору этил 5,6-дихлор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата в тетрагидрофуране (1 экв., 8 мл) и реакционную смесь кипятили с обратным холодильником при 100°C в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (50 мл) и метанолом (5 мл). Затем ее распределяли между водой и этилацетатом. Органический слой отделяли, сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием метанола и дихлорметана в качестве элюента с получением указанного в заголовке соединения.
1H-ЯМР (400 МГц, DMSO-d6) δ 5,58-5,74 (м, 1H), 4,36-4,53 (м, 1H), 3,94-4,06 (м, 1H), 3,72-3,78 (м, 4H), 3,64-3,69 (м, 4H), 3,56 (ушир, с, 5H), 2,85-3,14 (м, 2H), 2,46 (д, J=3,76 Гц, 6H), 1,80-1,97 (м, 3H), 1,61-1,76 (м, 2H), 1,29-1,58 (м, 4H).
[Справочный пример 13] Синтез [5-хлор-6-метил-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона
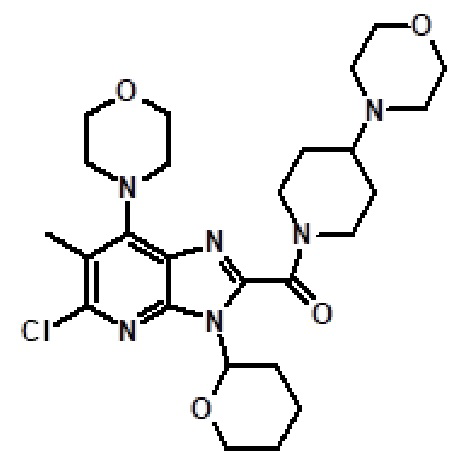
Стадия a: синтез этил 6-бром-5-хлор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата
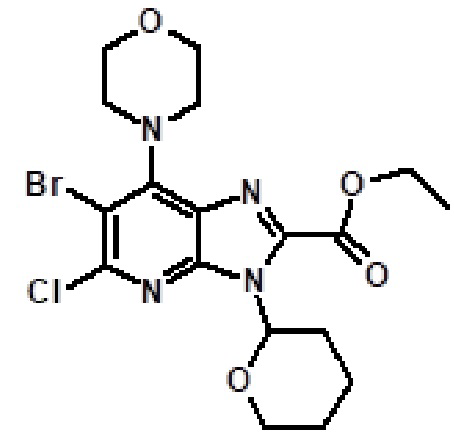
К раствору этил 5-хлор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (2,75 г, 6,96 ммоль) в диметилформамиде (22 мл), капельно добавляли N-бромсукцинимид (1,24 г, 6,96 ммоль) в диметилформамиде (8 мл) при 0°C и позволяли им перемешаться при комнатной температуре в течение 90 минут. Добавляли воду (50 мл) и экстрагировали этилацетатом (250 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием этилацетата и гексана (1:1) в качестве элюента с получением 2,65 г указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 6,22 (м, 1H), 4,42-4,56 (м, 2H), 4,15 (м, 1H), 3,85-3,95 (м, 4H), 3,66-3,82 (м, 5H), 2,90-3,02 (м, 1H), 2,02-2,11 (м, 1H), 1,92 (м, 1H), 1,67-1,85 (м, 2H), 1,57 (м, 2H), 1,47 (м, 3H). Спектр масс (ESI): m/z 472,95 (M+H) и 474,93 (M+2H).
Стадия b: синтез этил 5-хлор-6-метил-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата
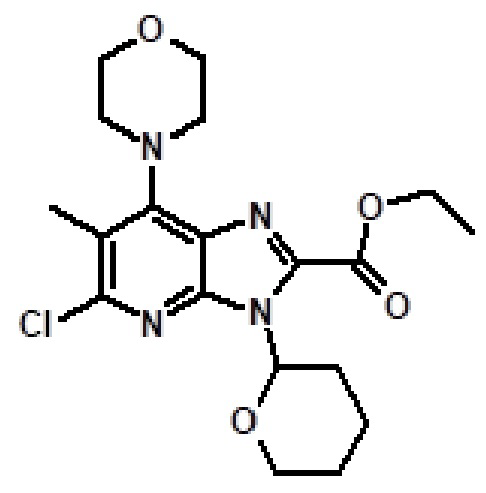
К раствору этил 6-бром-5-хлор-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (250 мг, 0,52 ммоль) в 1,2-дихлорэтане (5 мл) добавляли метилбороновую кислоту (126 мг, 2,1 ммоль) и карбонат цезия (514 мг, 1,58 ммоль) и реакционную смесь продували аргоном в течение 10 минут. Затем добавляли тетракис(трифенилфосфин)палладий(0) (61 мг, 0,05 ммоль) и смесь нагревали при 140°C в течение 90 минут. Реакционную смесь охлаждали, разбавляли дихлорметаном (50 мл) и концентрировали в вакууме до сухого состояния. Остаток очищали флэш-хроматографией с использованием метанола и дихлорметана (5% метанол) в качестве элюента с получением 110 мг указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 6,20-6,27 (м, 1H), 4,48 (м, 2H), 4,10-4,18 (м, 1H), 3,83-3,92 (м, 4H), 3,67-3,76 (м, 1H), 3,54-3,63 (м, 4H), 2,95-3,08 (м, 1H), 2,36 (с, 3H), 2,02-2,11 (м, 1H), 1,87-1,95 (м, 1H), 1,67-1,85 (м, 2H), 1,57-1,61 (м, 1H), 1,47 (м, 3H). Спектр масс (ESI): m/z 409,08 (M+H) и 411,06 (M+2H).
Стадия c: синтез [5-хлор-6-метил-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанона
К раствору 4-морфолинопиперидина (1,06 г, 6,23 ммоль) в тетрагидрофуране (10 мл), добавляли триметилалюминий (3,1 мл, 6,23 ммоль) при -10°C и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Затем этот раствор добавляли к раствору этил 5-хлор-6-метил-7-(морфолин-4-ил)-3-(тетрагидро-2H-пиран-2-ил)-3H-имидазо[4,5-b]пиридин-2-карбоксилата (850 мг, 2,08 ммоль) в тетрагидрофуране (5 мл) и реакционную смесь кипятили с обратным холодильником при 100°C в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (50 мл) и метанолом (5 мл). Затем ее распределяли между водой (50 мл) и этилацетатом (400 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали флэш-хроматографией с использованием метанола и дихлорметана (5% метанол) в качестве элюента с получением 750 мг указанного в заголовке соединения в виде не совсем белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 5,76-5,90 (м, 1H), 4,62-4,81 (м, 1H), 4,02-4,13 (м, 1H), 3,85 (м, 4H), 3,64-3,77 (м, 6H), 3,46-3,59 (м, 4H), 2,82-3,13 (м, 2H), 2,61-2,74 (м, 1H), 2,55 (ушир. с, 4H), 2,41-2,49 (м, 1H), 2,36 (с, 3H), 1,93-2,06 (м, 3H), 1,63-1,82 (м, 3H), 1,45-1,55 (м, 3H). Спектр масс (ESI): m/z 533,18 (M+H) и 535,11 (M+2H).
Синтез аминосоединения 1b
[Справочный пример 15] Синтез 2-окса-7-азаспиро[3.5]нонана
Стадия a: синтез соединения 15b
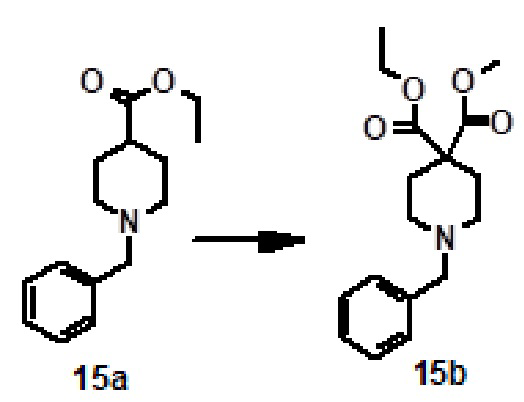
К раствору диизопропиламина (1,63 г, 16,18 ммоль) в тетрагидрофуране (20 мл) добавляли н-бутиллитий (1,6 M в гексане, 10 мл, 16,18 ммоль) при 0°C и перемешивали в течение 30 минут при комнатной температуре, а затем охлаждали при 0°C. Этот свежеполученный диизопропиламид лития капельно добавляли к раствору соединения 15a (2 г, 8,097 ммоль) в тетрагидрофуране (40 мл) при -78°C и перемешивали при -40°C в течение 1 часа. Реакционную смесь охлаждали до -78°C; добавляли метилхлорформиат (0,841 г, 8,90 ммоль). Перемешивание продолжали при той же температуре в течение 2 часов и при комнатной температуре в течение 3-4 часов. Добавляли насыщенный хлорид аммония (100 мл) и экстрагировали этилацетатом (100 мл, три раза). Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали в вакууме с получением 2,1 г соединения 15b.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 7,06-7,45 (м, 5H), 4,13 (кв, 2H), 3,63-3,69 (с, 3H), 3,41 (с, 2H), 2,33 (ушир. с, 4H), 1,99 (т, J=5,40 Гц, 4H), 1,02-1,35 (т, 3H).
Стадия b: синтез соединения 15c
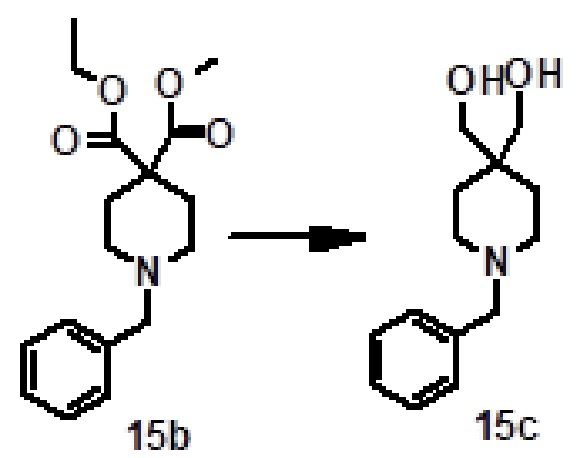
К раствору соединения 15b (2,2 г, 7,189 ммоль) в тетрагидрофуране (50 мл) добавляли алюмогидрид лития (0,683 г, 17,97 ммоль) порционно при 0°C и перемешивали в течение 2-3 часов при комнатной температуре. Затем к нему капельно добавляли насыщенный сульфат натрия (100 мл) при 0°C. Реакционную смесь фильтровали через целит и фильтрат концентрировали в вакууме с получением 1,3 г соединения 15c.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 6,89-7,43 (м, 5H), 4,30 (ушир. с, 2H), 3,42 (ушир. с, 2H), 3,33 (ушир. с, 2H), 2,28 (ушир. с, 4H), 1,35 (ушир. с, 4H).
Стадия c: синтез соединения 15d
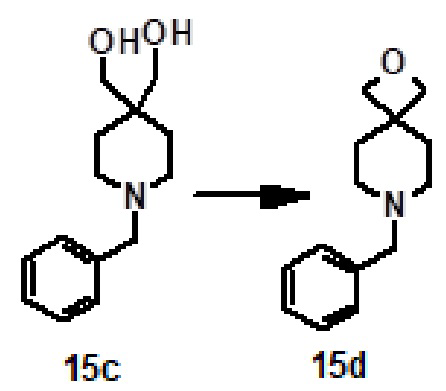
К раствору соединения 15c (2,8 г, 11,91 ммоль) в тетрагидрофуране (60 мл) добавляли н-бутиллитий (1,6 M в гексане, 7,4 мл, 11,91 ммоль) при 0°C и перемешивали при той же температуре в течение 30 минут. К нему капельно добавляли раствор п-толуолсульфонилхлорида (2,26 г, 11,91 ммоль) в тетрагидрофуране (10 мл) и перемешивание продолжали при 0°C в течение 1-2 часов. Затем к нему добавляли н-бутиллитий (1,6 M в гексане, 7,4 мл, 11,91 ммоль) и перемешивали в течение 30 минут при той же температуре, а затем перемешивали при 70°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и добавляли насыщенный хлорид аммония (100 мл) и экстрагировали этилацетатом (100 мл, два раза). Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали в вакууме с получением 1,8 г соединения 15d.
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 7,01-7,56 (м, 5H), 4,40 (с, 4H), 3,44 (с, 2H), 2,31 (ушир. с, 4H), 1,86 (т, J=5,14 Гц, 4H).
Стадия d: синтез 2-окса-7-азаспиро[3.5]нонана
К раствору соединения 15d (1,8 г, 8,29 ммоль) в метаноле (50 мл) добавляли палладий на угле (0,5 г) и перемешивали в атмосфере водорода (давление баллона) при комнатной температуре в течение 18-22 часов. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением 1 г 2-окса-7-азаспиро[3.5]нонана.
1H-ЯМР (400 МГц, CDCl3) δ м.д.: 4,26-4,59 (с, 4H), 2,63-2,88 (м, 4H), 1,73-1,96 (м, 4H).
[Справочный пример 16] Синтез N-(пиперидин-4-ил)ацетамида
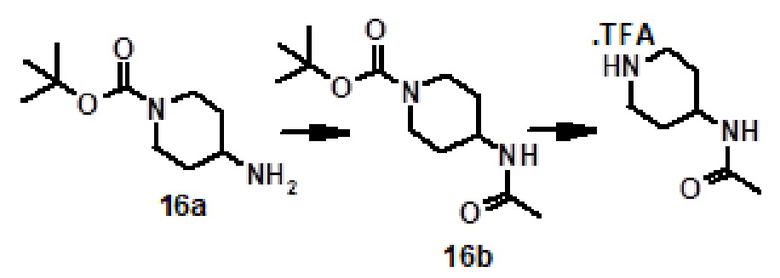
К раствору соединения 16a (1 г, 5 ммоль) в дихлорметане (15 мл), добавляли ацетилхлорид (0,3 мл, 5,5 ммоль) при 0°C и им позволяли перемешаться при комнатной температуре в течение ночи. Летучие вещества удаляли в вакууме и добавляли воду (30 мл), а затем насыщенный бикарбонат натрия (30 мл), а затем экстрагировали этилацетатом (250 мл, три раза). Объединенные органические экстракты промывали рассолом (100 мл), разделяли, сушили над сульфатом натрия и концентрировали в вакууме с получением 460 мг соединения 16b. К раствору соединения 16b (460 мг) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (1 мл) при комнатной температуре и перемешивали в течение ночи. Летучие вещества удаляли в вакууме и растирали с использованием диэтилового эфира (50 мл). Осадок отфильтровывали и сушили в вакууме с получением 380 мг N-(пиперидин-4-ил)ацетамида.
[Справочный пример 17] Синтез N-(пиперидин-4-ил)метансульфонамида
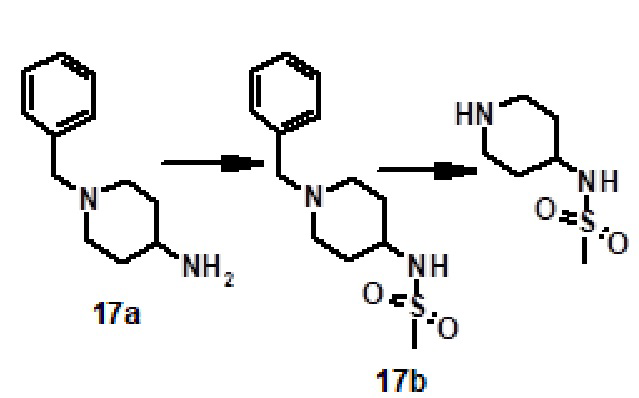
К раствору соединения 17a (1 г, 5,263 ммоль) в дихлорметане (10 мл) добавляли триэтиламин (1,07 мл, 7,81 ммоль) при 0°C, а затем метансульфонилхлорид (0,81 мл, 6,315 ммоль), и им позволяли перемешаться при комнатной температуре в течение ночи. Летучие вещества удаляли в вакууме и добавляли воду (30 мл), а затем экстрагировали дихлорметаном (150 мл, три раза). Объединенные органические экстракты промывали рассолом (75 мл), разделяли, сушили над сульфатом натрия и концентрировали в вакууме с получением 430 мг соединения 17b. К раствору этого соединения (430 мг) в метаноле (15 мл) добавляли палладий на угле (250 мг) и перемешивали в атмосфере водорода (давление баллона) при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением 200 мг N-(пиперидин-4-ил)метансульфонамида.
[Справочный пример 18] Синтез 1-(метилсульфонил)пиперазина
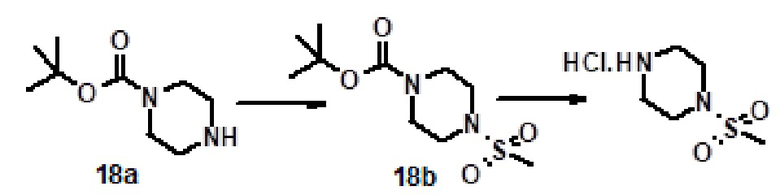
К раствору соединения 18a (2 г, 10,80 ммоль) в дихлорметане (40 мл) добавляли триэтиламин (3 мл, 21,60 ммоль) при 0°C, а затем метансульфонилхлорид (1 мл, 12,96 ммоль), и им позволяли перемешаться при комнатной температуре в течение ночи. Летучие вещества удаляли в вакууме и добавляли воду (50 мл), а затем проводили экстракцию дихлорметаном (300 мл, три раза). Объединенные органические экстракты промывали рассолом (200 мл), разделяли, сушили над сульфатом натрия и концентрировали в вакууме с получением 430 мг соединения 18b. К раствору этого соединения (2 г, 7,56 ммоль) в 1,4-диоксане (15 мл) добавляли 1,4-диоксанхлористоводородную кислоту при 0°C и им позволяли перемешаться при комнатной температуре в течение ночи. Летучие вещества удаляли в вакууме и растирали с диэтиловым эфиром (50 мл). Осадок отфильтровывали и сушили в вакууме с получением 1,7 г 1-(метилсульфонил)пиперазина.
[Справочный пример 19] Синтез 2-(пиперидин-4-ил)пропан-2-ола
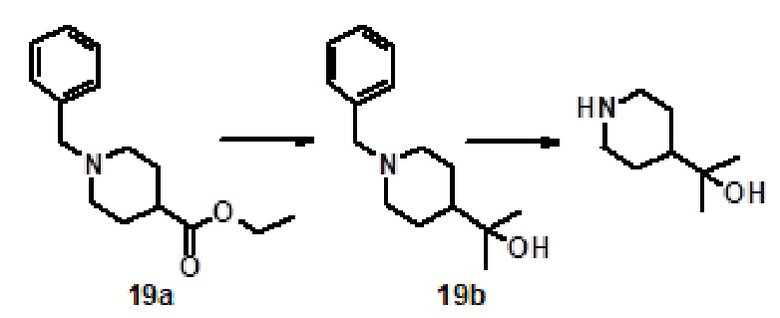
К раствору соединения 19a (10 г, 40,6 ммоль) в тетрагидрофуране (100 мл) капельно добавляли метилмагнийбромид (54 мл, 162,4 ммоль) при 0°C, и им позволяли перемешаться при комнатной температуре в течение ночи. К реакционной смеси добавляли насыщенный хлорид аммония (150 мл) и экстрагировали этилацетатом (300 мл, три раза). Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме с получением 7,5 г соединения 19b. К раствору этого соединения (7,5 г) в метаноле (150 мл) добавляли палладий на угле (2 г) и перемешивали в атмосфере водорода (давление баллона) при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением 5,3 г 2-(пиперидин-4-ил)пропан-2-ола.
[Справочный пример 20] Синтез 1-(тетрагидро-2H-пиран-4-ил)пиперазина
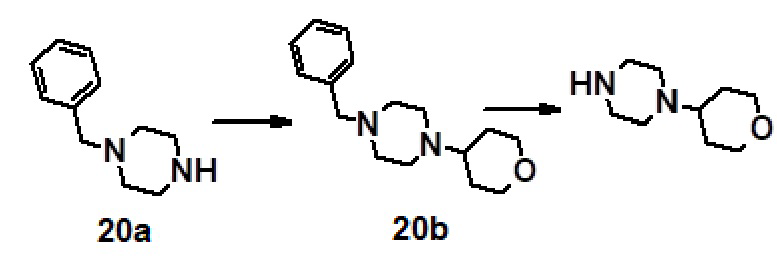
К раствору соединения 20a (12 г, 68,08 ммоль) в тетрагидрофуране (200 мл) добавляли 4-тетрагидропиранон (13,632 г, 136,16 ммоль) и п-толуолсульфоновую кислоту (389 мг, 2,04 ммоль), а затем добавляли уксусную кислоту (6 мл). Порционно добавляли триацетоксиборгидрид натрия (28,9 г, 136,16 ммоль) при 0°C и реакционной смеси позволяли перемешаться при комнатной температуре в течение 16 ч. Добавляли насыщенный бикарбонат натрия (150 мл) и экстрагировали дихлорметаном (300 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который при очистке флэш-хроматографией (5% метанол в дихлорметане в качестве элюента) дал 12,5 г соединения 20b. К раствору этого соединения (15 г) в метаноле (200 мл) добавляли палладий на угле (3 г), и им позволяли встряхиваться в аппарате Парра под давлением водорода (50 фунт./кв. дюйм (344,7 кПа)) в течение 6 часов при комнатной температуре. Реакционную смесь фильтровали и промывали метанолом. Фильтрат концентрировали в вакууме с получением 10 г 1-(тетрагидро-2H-пиран-4-ил)пиперазина. Аналогичным образом получали цис-2,6-диметил-4-(пиперидин-4-ил)морфолин и 4-(пиперидин-3-ил)морфолин.
[Справочный пример 21] Синтез 2-метил-2-(пиперазин-1-ил)пропанамида
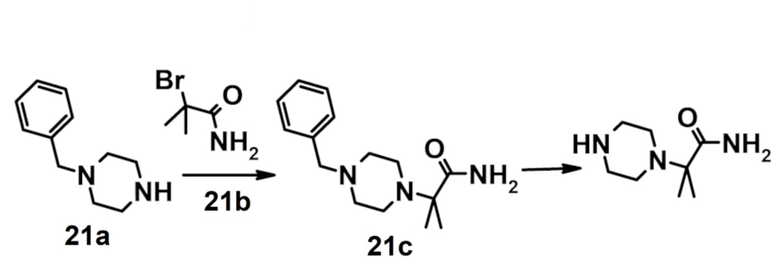
Раствор соединения 21a (2,5 г, 14,2 ммоль), соединения 21b (4,72 г, 28,41 ммоль) и карбоната цезия (9,3 г, 28,41 ммоль) в ацетонитриле (40 мл) нагревали при 100°C в течение 18 часов. Добавляли воду (25 мл) и экстрагировали этилацетатом (150 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который при очистке флэш-хроматографией (3% метанол в дихлорметане в качестве элюента) дал 3,05 г соединения 21c. К раствору этого соединения (3 г) в метаноле (60 мл) добавляли палладий на угле (1 г) и перемешивали в атмосфере водорода (давление баллона) при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением 2,25 г 2-метил-2-(пиперазин-1-ил)пропанамида.
[Справочный пример 22] Синтез (9aR)-гексагидропиразинo[2,1-c][1,4]оксазин-4(3H)-она
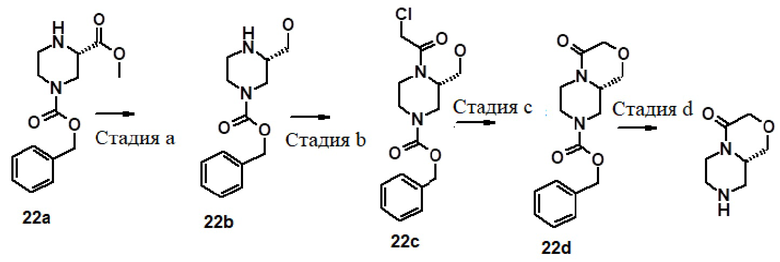
Стадия a: к раствору соединения 22a (10 г, 36,1 ммоль) в сухом тетрагидрофуране (100 мл) добавляли боргидрид лития (1,13 г, 54,51 ммоль) порционно при 0°C. Реакционной смеси позволяли перемешаться при комнатной температуре в течение 18 часов. Капельно добавляли этилацетат (200 мл), а затем добавляли воду (50 мл), а затем экстрагировали этилацетатом (250 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта. Колоночная хроматография на силикагеле (размер пор 100-200) с использованием 5% метанола в дихлорметане в качестве элюента дала 8,5 г соединения 22b.
Стадия b: к раствору соединения 22b (4,4 г, 17,6 ммоль) в дихлорметане (50 мл) капельно добавляли триэтиламин (5,9 мл, 52,8 ммоль) и хлорацетилхлорид (2 мл, 17,6 ммоль) при 0°C. Реакционной смеси позволяли перемешаться при комнатной температуре в течение 90 минут. Добавляли воду (30 мл) и экстрагировали этилацетатом (150 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением 4,8 г соединения 22c.
Стадия c: к раствору соединения 22c (4,8 г, 14,81 ммоль) в сухом тетрагидрофуране (45 мл) добавляли третичный бутоксид калия (2,48 г, 22,2 ммоль) порционно при 0°C. Реакционной смеси позволяли перемешаться при комнатной температуре в течение 18 часов. Добавляли воду (30 мл) и экстрагировали этилацетатом (150 мл, три раза). Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который при очистке колоночной хроматографией (размер пор 100-200) с использованием 5% метанола в дихлорметане в качестве элюента дал 2,5 г соединения 22d.
Стадия d: к раствору соединения 22d (2,5 г, 8,68 ммоль) в метаноле (20 мл) добавляли палладий на угле (500 мг) и перемешивали в атмосфере водорода (давление баллона) при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением 1,3 г (9aR)-гексагидропиразинo[2,1-c][1,4]оксазин-4(3H)-она.
[Справочный пример 23] Синтез этил 4-(морфолин-4-ил)пиперидин-1-карбоксилата
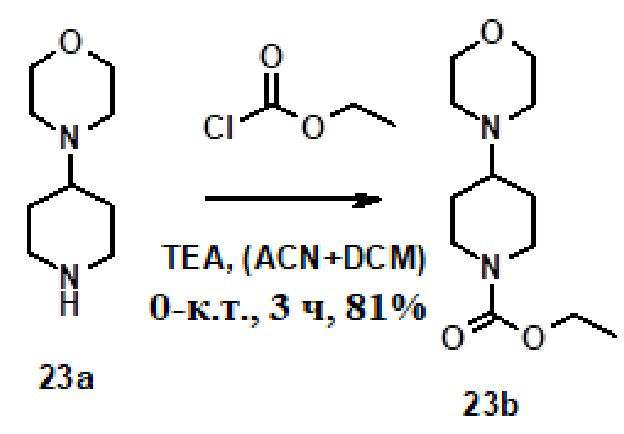
К раствору морфолинпиперидина 23a (5 г, 29,23 ммоль; AK Scientific) в ацетонитриле и дихлорметане (100:20 мл) добавляли триэтиламин (5,90 г, 58,47 ммоль) при комнатной температуре и охлаждали до 0°C. После достижения этой температуры добавляли этилхлорформиат (3,798 г, 35,076 ммоль) и перемешивали в течение 10 минут при той же температуре, а затем перемешивали в течение 2-3 часов при комнатной температуре. Реакционную смесь фильтровали, к фильтрату добавляли воду (400 мл) и экстрагировали дихлорметаном (500 мл×2). Объединенный органический экстракт сушили над безводным сульфатом натрия, концентрировали в вакууме с получением этил 4-(морфолин-4-ил)пиперидин-1-карбоксилата (5,8 г).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 3,86-4,09 (кв, 2H), 3,86-4,09 (ушир., 2H), 3,50-3,62 (м, 4H), 2,76 (ушир. с, 2H), 2,36-2,46 (м, 4H), 2,30 (тт, J=3,54, 11,01 Гц, 1H), 1,65-1,82 (м, 2H), 1,05-1,35 (м, 2H), 1,05-1,35 (т, 3H).
[Справочный пример 24] Синтез 4-(азетидин-3-ил)-1-метилпиперазин-2-она
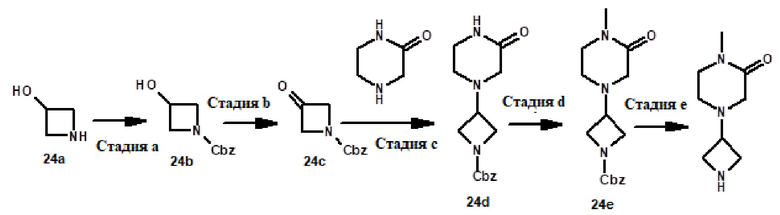
Стадия a: к раствору соединения 24a (12 г, 109,5 ммоль; Spectrochem) в тетрагидрофуране (500 мл) добавляли карбонат калия (45,417 г, 328,617 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. К описанной выше реакционной смеси капельно добавляли бензилхлорформиат (52,35 мл, 153,35 ммоль) при 0°C и реакционную смесь перемешивали при комнатной температуре в течение 12 часов, затем добавляли воду (250 мл), экстрагировали этилацетатом (500×3 мл), промывали рассолом (250 мл), сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали колоночной хроматографией на силикагеле с использованием 0-5% метанола в дихлорметане в качестве элюента с получением соединения 24b (52,9%; 18 г).
Стадия b: к раствору соединения 24b (9 г, 43,47 ммоль) в дихлорметане (500 мл) добавляли перйодинан Десс-Мартина (36,86 г, 86,95 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов, а затем фильтровали через воронку с фильтром из спеченого стекла. Фильтрат промывали водой (200 мл), рассолом (200 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме с получением соединения 24c (95,4%; 8,5 г).
Стадия c: к раствору соединения 24c (6,15 г, 30 ммоль) в тетрагидрофуране (100 мл) добавляли пиперазин-2-он (2 г, 20 ммоль; Spectrochem) и ледяную уксусную кислоту (1,8 г, 30 ммоль), п-толуолсульфоновую кислоту (344 мг, 2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли триацетоксиборгидрид натрия (7,42 г, 35 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь нейтрализовывали насыщенным раствором бикарбоната натрия, экстрагировали этилацетатом (200×3 мл), промывали рассолом (150 мл), сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали колоночной хроматографией с использованием 0-5% метанола в дихлорметане в качестве элюента с получением соединения 24d (3,5 г).
Стадия d: к раствору соединения 24d (2 г, 6,9 ммоль) в диметилформамиде (50 мл) добавляли гидрид натрия (414 мг, 10,35 ммоль) при 0°C и реакционную смесь перемешивали в течение 30 минут, добавляли метилйодид (1,175 г, 8,28 ммоль), перемешивали в течение 4 часов при комнатной температуре. К реакционной смеси добавляли воду (100 мл) и экстрагировали этилацетатом (250×3 мл), промывали водой (300 мл), рассолом (300 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме с получением соединения 24e (1,5 г).
Стадия e: к раствору соединения 24e (1,5 г, 4,95 ммоль) в метаноле (50 мл) добавляли палладий на угле (400 мг) и реакционную смесь перемешивали при 50 фунт./кв. дюйм (344,7 кПа) в аппарате Парра в течение 2 часов, затем фильтровали через целит и фильтрат концентрировали в вакууме с получением 4-(азетидин-3-ил)-1-метилпиперазин-2-она (800 мг).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 7,91-7,97 (м, 1H), 3,30-3,38 (м, 4H), 3,21-3,27 (м, 2H), 2,84 (ушир. с, 3H), 2,80-2,82 (м, 5H).
Аналогичным образом получали 4-(азетидин-3-ил)пиперазин-2-он с использованием соединения 24d.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,48 (с, 1H), 7,75 (ушир. с, 1H), 3,42 (квин, J=7,59 Гц, 4H), 3,12-3,15 (м, 3H), 2,80 (с, 2H), 2,38-2,46 (м, 2H).
Аналогично получали 4-(азетидин-3-ил)морфолин (3,0 г) и (2R,6S)-4-(азетидин-3-ил)-2,6-диметилморфолин (2,0 г).
[Справочный пример 25] Синтез N-метил-1-(тетрагидро-2H-пиран-4-ил)пиперидин-4-амина
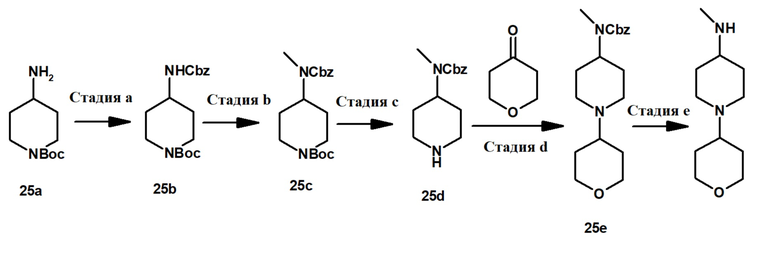
Стадия a: к раствору соединения 25a (10 г, 50 ммоль) в дихлорметане (200 мл) добавляли триэтиламин (10,1 г, 100 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. К описанной выше реакционной смеси капельно добавляли бензилхлорформиат (17 мл, 60 ммоль) при 0°C и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Затем ее разбавляли дихлорметаном, промывали водой (250 мл), рассолом (250 мл), сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали колоночной хроматографией на силикагеле с использованием 50-60% этилацетата в гексане в качестве элюента с получением соединения 25b (8 г).
Стадия b: к раствору соединения 25b (8 г, 23,95 ммоль) в тетрагидрофуране (150 мл) добавляли гидрид натрия (1,24 г, 31,13 ммоль) при 0°C и реакционную смесь перемешивали в течение 30 минут. К реакционной смеси добавляли метилйодид (5,099 г, 35,92 ммоль) и перемешивали в течение 12 часов при комнатной температуре. Добавляли воду (100 мл), экстрагировали этилацетатом (250×3 мл), промывали рассолом (300 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме с получением соединения 25c (8 г).
Стадия c: к раствору соединения 25c (4 г, 11,49 ммоль) в дихлорметане (50 мл) добавляли хлористоводородную кислоту (4 M) в диоксане (11 мл, 44 ммоль) и реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Ее концентрировали в вакууме, растирали с диэтиловым эфиром, нейтрализовывали насыщенным раствором бикарбоната натрия, экстрагировали дихлорметаном, сушили над безводным сульфатом натрия и концентрировали в вакууме с получением соединения 25d (2 г).
Стадия d: к раствору соединения 25d (2 г, 8,6 ммоль) в тетрагидрофуране (50 мл) добавляли соединение тетрагидро-4H-пиран-4-он (1,6 г, 17,2 ммоль) и ледяную уксусную кислоту (1 г, 17,2 ммоль), п-толуолсульфоновую кислоту (172 мг, 1 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. К ней добавляли триацетоксиборгидрид натрия (3,4 г, 17,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Затем ее нейтрализовывали насыщенным раствором бикарбоната натрия, экстрагировали этилацетатом (200×3 мл), промывали рассолом (150 мл), сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали колоночной хроматографией с использованием 0-5% метанола в дихлорметане в качестве элюента с получением соединения 25e (1,5 г).
Стадия e: к раствору соединения 25e (1,5 г, 4,5 ммоль) в метаноле (50 мл) добавляли палладий на угле (400 мг) и реакционную смесь перемешивали под давлением водорода 50 фунт./кв. дюйм (344,7 кПа) в аппарате Парра в течение 2 часов. Затем ее фильтровали через целит и фильтрат концентрировали в вакууме с получением указанного в заголовке соединения (800 мг).
1H-ЯМР (400 МГц, CHCl3-d) δ м.д.: 4,02 (дд, J=4,52, 11,29 Гц, 2H), 3,37 (дт, J=2,01, 11,80 Гц, 2H), 2,87-2,98 (м, 2H), 2,44-2,51 (м, 1H), 2,43 (с, 3H), 2,29-2,40 (м, 1H), 2,19 (дт, J=2,26, 11,54 Гц, 2H), 1,70-1,96 (м, 4H), 1,59 (дкв, J=4,52, 12,21 Гц, 2H), 1,28-1,43 (м, 2H).
Аналогичным образом получали N-метил-1-(оксетан-3-ил)пиперидин-4-амин с использованием оксетан-3-она (1,23 г, 17,2 ммоль) на стадии d выше.
1H-ЯМР (400 МГц, CHCl3-d) δ м.д.: 4,56-4,67 (м, 4H), 3,40-3,50 (м, 2H), 2,67-2,74 (м, 2H), 2,44 (с, 3H), 2,38-2,43 (м, 2H), 1,83-1,96 (м, 4H).
[Справочный пример 26] Синтез 2,8-диазаспиро[4.5]декан-3-она
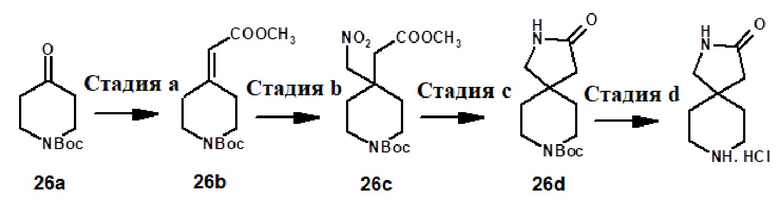
Стадия a: к раствору гидрида натрия (1,2 г, 30,15 ммоль) в диметилформамиде (30 мл) капельно добавляли триметилфосфеноацетат при 0°C и реакционную смесь перемешивали в течение 30 минут. Капельно добавляли раствор соединения 26a (5 г, 25,12 ммоль) в диметилформамиде (20 мл) при 0°C и реакционную смесь перемешивали в течение 6 часов при комнатной температуре. Затем добавляли воду и экстрагировали этилацетатом (300×3 мл), промывали водой (200 мл), рассолом (200 мл), сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали колоночной хроматографией с использованием 40-50% этилацетата в гексане в качестве элюента с получением соединения 26b (3 г).
Стадия b: к раствору соединения 26b (8 г, 31,37 ммоль) в ацетонитриле (100 мл) добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (6,2 мг, 40,78 ммоль) и нитрометан (2,48 г, 40,78 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 4 часов. Затем растворитель выпаривали в вакууме, остаток разбавляли этилацетатом (800 мл) и промывали водой (300×2 мл), рассолом (300 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме с получением соединения 26c (4,5 г).
Стадия c: к раствору соединения 26c (4,5 г, 14,15 ммоль) в метаноле (50 мл) добавляли палладий на угле (1 г) и реакционную смесь перемешивали под давлением водорода 50 фунт./кв. дюйм (344,7 кПа) в аппарате Парра в течение 2 часов. Затем ее фильтровали через целит и фильтрат концентрировали в вакууме с получением соединения 26d (3,5 г).
Стадия d: к раствору соединения 26d (3,5 г, 13,8 ммоль) в дихлорметане (50 мл) добавляли хлористоводородную кислоту (4 M) в диоксане (14 мл, 56 ммоль) и реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Ее концентрировали в вакууме и растирали с диэтиловым эфиром с получением 2,8-диазаспиро[4.5]декан-3-она (2 г).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 3,08-3,19 (м, 2H), 2,97-3,01 (м, 2H), 2,56-2,65 (м, 2H), 1,97-2,02 (м, 2H), 1,38-1,45 (м, 4H).
[Справочный пример 27] Синтез (2R,6S)-2,6-диметил-4-(пиперидин-4-ил)морфолина
Стадия a: к раствору соединения 27a (6 г, 33,89 ммоль) в тетрагидрофуране (90 мл) добавляли цис-2,6 диметилморфолин (4,596 г, 40,67 ммоль) и п-толуолсульфоновую кислоту (645 мг, 3,38 ммоль), а затем уксусную кислоту (5 мл, 67,79 ммоль). Через 5 минут порционно добавляли триацетосиборгидрид натрия (14,305 г, 67,79 ммоль) при 0°C и реакционной смеси позволяли перемешаться при комнатной температуре в течение ночи. Добавляли насыщенный раствор бикарбоната натрия и экстрагировали дихлорметаном. Органический сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который при очистке флэш-хроматографией (силикагель) с использованием 0-5% метанола в дихлорметане дал соединение 27b (7 г).
Стадия b: к раствору соединения 27b (7 г) в метаноле (100 мл) добавляли палладий на угле (2 г) и перемешивали в атмосфере водорода (давление баллона) при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением (2R,6S)-2,6-диметил-4-(пиперидин-4-ил)морфолина (4,1 г).
Аналогичным образом получали 4-(пиперидин-3-ил)морфолин (3,0 г) с использованием 1-бензилпиперидин-3-она (5 г, 26,45 ммоль) и морфолина (4,6 мл, 52,9 ммоль).
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 3,48-3,58 (м, 4H), 3,05 (д, J=11,54 Гц, 1H), 2,71-2,95 (м, 2H), 2,36-2,48 (м, 6H), 2,14-2,25 (м, 1H), 1,84 (д, J=2,01 Гц, 1H), 1,59-1,69 (м, 1H), 1,24-1,37 (м, 2H).
[Справочный пример 28] Синтез 4-(пиперидин-4-ил)морфолин-3-она
Стадия a: к раствору соединения 28a (3 г, 15,85 ммоль) в 1,2-дихлорэтане (60 мл), добавляли 2-аминоэтанол (0,96 г, 15,85 ммоль) и уксусную кислоту (1,14 г, 19,2 ммоль) при комнатной температуре. Через 5 минут порционно добавляли триацетоксиборгидрид натрия (10,05 г, 66,12 ммоль) и реакционной смеси позволяли перемешаться при комнатной температуре в течение 18 часов. Добавляли насыщенный раствор бикарбоната натрия и экстрагировали этилацетатом. Органический экстракт сушили над безводным сульфатом натрия и концентрировали в вакууме с получением соединения 28b (3,5 г).
Стадия b: к раствору соединения 28b (1 г, 4,27 ммоль) в 1,2-дихлорэтане (20 мл), капельно добавляли триэтиламин (1,19 мл, 8,54 ммоль) и хлорацетилхлорид (0,52 мл, 6,41 ммоль) при 0°C, и ему позволяли перемешаться при комнатной температуре в течение 2 часов. Добавляли насыщенный раствор бикарбоната натрия и экстрагировали дихлорметаном. Органический экстракт сушили над безводным сульфатом натрия и концентрировали в вакууме с получением соединения 28c (1,3 г).
Стадия c: к раствору соединения 28c (1,4 г, 4,5 ммоль) в диметилформамиде (10 мл), порционно добавляли гидрид натрия (110 мг, 4,95 ммоль) при -10°C и перемешивали в течение 60 минут, нагревали и перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь нагревали при 80°C в течение 15 часов, а затем в течение 48 часов при комнатной температуре. Реакционную смесь охлаждали до комнатной температуры и добавляли воду и экстрагировали дихлорметаном. Объединенный органический экстракт сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта, который при очистке колоночной хроматографией на силикагеле (размер пор 100-200) и с использованием системы 0-5% метанола и дихлорметана в качестве элюента дал соединение 28d (450 мг).
Стадия d: к раствору соединения 28d (2,5 г) в метаноле (30 мл) добавляли палладий на угле (500 мг) и перемешивали в атмосфере водорода (давление баллона) при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением 4-(пиперидин-4-ил)морфолин-3-она (1,5 г).
[Справочный пример 29] Синтез пиперазин-1-ил(пиримидин-2-ил)метанона
Стадия a: к раствору соединения 29a (500 мг, 2,688 ммоль; Spectrochem) в дихлорметане (15 мл), добавляли пиримидин-2 карбоновую кислоту (430 мг, 3,495 ммоль), гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиния (1,276 г, 3,36 ммоль), N,N-диизопропилэтиламин (1,55 мл, 9,406 моль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли дихлорметаном, промывали 10% раствором хлористоводородной кислоты, а затем бикарбонатом натрия и, наконец, водой. Органический слой сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенной смеси, которую растирали в гексане с получением чистого соединения 29b (450 мг).
Стадия b: к раствору соединения 29b (450 мг) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (2 мл) при комнатной температуре и перемешивали в течение ночи. Летучие вещества удаляли в вакууме и добавляли дихлорметан, промывали раствором бикарбоната натрия. Органический слой сушили над безводным сульфатом натрия и концентрировали в вакууме с получением пиперазин-1-ил(пиримидин-2-ил)метанона (250 мг).
Аналогичным образом получали 1-(пиперазин-1-ил)-2-(пиримидин-2-ил)этанон (400 мг) с использованием пиримидин-2 уксусной кислоты (573 мг, 3,49 ммоль), гексафторфосфата 3 оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиния (1,276 г, 3,36 ммоль) и N,N-диизопропилэтиламина (1,55 мл, 9,46 ммоль).
[Справочный пример 30] Синтез N-(пирролидин-3-ил)ацетамида
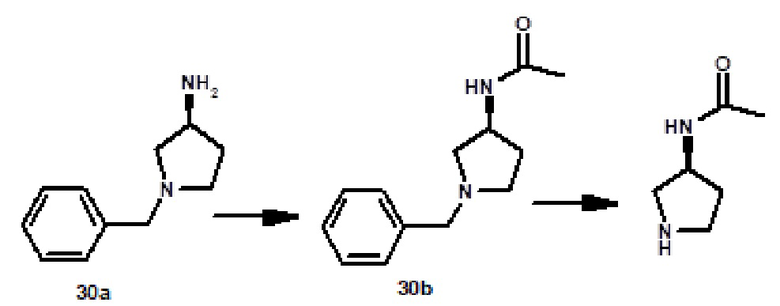
Стадия a: к раствору соединения 30a (500 мг, 2,83 ммоль; Aldrich) в дихлорметане (15 мл) добавляли уксусный ангидрид (0,3 мл, 1,12 ммоль) при 0°C и им позволяли перемешаться при комнатной температуре в течение 2 часов. Летучие вещества удаляли в вакууме и добавляли воду, а затем насыщенный бикарбонат натрия, а затем экстрагировали дихлорметаном. Объединенный органический экстракт промывали рассолом, сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали колоночной хроматографией с использованием 0-5% метанола в дихлорметане с получением соединения 30b (238 мг).
Стадия b: к раствору соединения 30b (238 мг) в метаноле (15 мл) добавляли палладий на угле (250 мг) и перемешивали в атмосфере водорода (давление баллона) при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением N-(пирролидин-3-ил)ацетамида (150 мг).
[Справочный пример 31] Синтез N-(пирролидин-3-ил)метансульфонамид
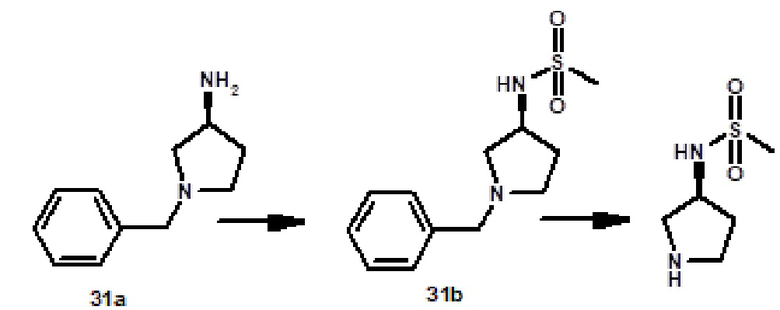
Стадия a: к раствору соединения 31a (500 мг, 2,83 ммоль) в дихлорметане (40 мл) добавляли триэтиламин (0,58 мл, 4,25 ммоль) при 0°C, а затем метансульфонилхлорид (0,43 мл, 3,4 ммоль), и им позволяли перемешаться при комнатной температуре в течение ночи. Летучие вещества удаляли в вакууме; добавляли воду и экстрагировали дихлорметаном. Органический экстракт промывали рассолом, сушили над безводным сульфатом натрия и концентрировали в вакууме с получением соединения 31b (750 мг).
Стадия b: к раствору соединения 31b (750 мг) в метаноле (15 мл) добавляли палладий на угле (250 мг) и перемешивали в атмосфере водорода (давление баллона) при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением N-(пирролидин-3-ил)метансульфонамида (450 мг).
Таблица 1
(400 МГц, DMSO-d6) δ м.д.
[M+H]
(мг)
(Источник)
(1a или 2a)
(мг)
328
(Aldrich)
208
(Синтезированное)
109
(Синтезированное)
155
(Синтезированное)
188
(Синтезированное)
187
(Синтезированное)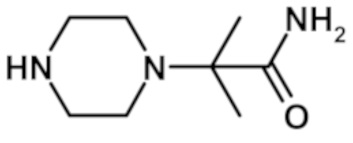
144
(Синтезированное)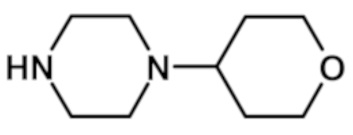
143
(Синтезированное)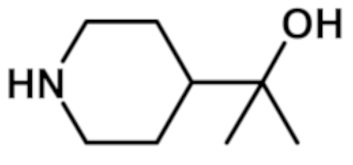
189
(Синтезированное)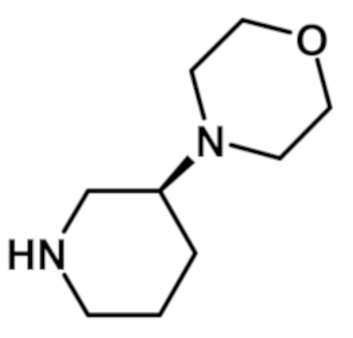
143
(Синтезированное)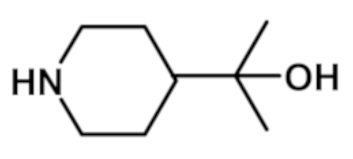
120
(Синтезированное)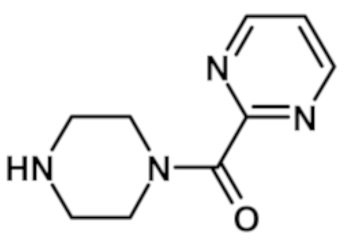
173
(Синтезированное)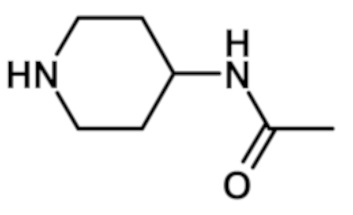
80
(Acros)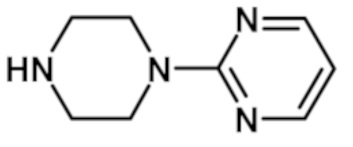
147
(Alfa aiser)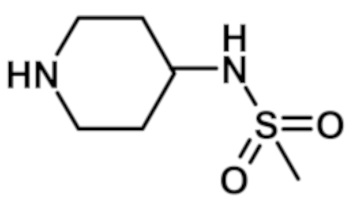
99
(Синтезированное)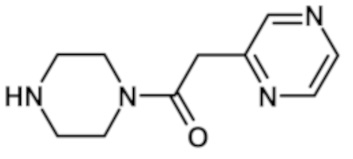
230
(Синтезированное)
56
(Spectrochem)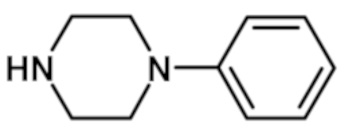
85
(Spectrochem)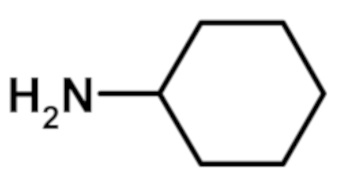
65
(Spectrochem)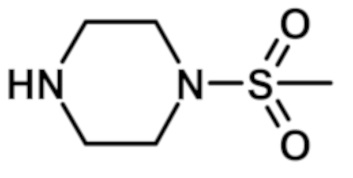
183
(Синтезированное)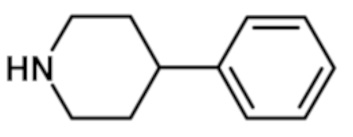
90
(Spectrochem)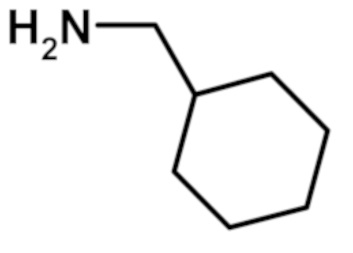
63
(Alfa aiser)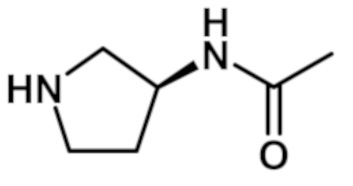
72
(Синтезированное)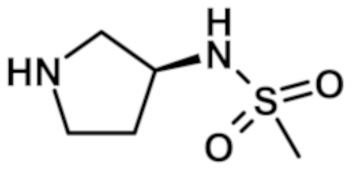
92
(Синтезированное)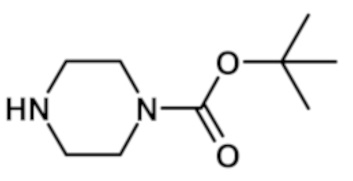
52
(Spectrochem)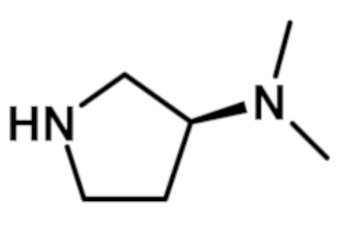
18
(Aldrich)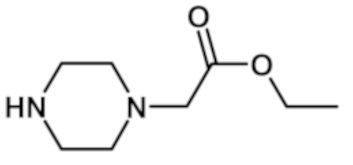
36
(Aldrich)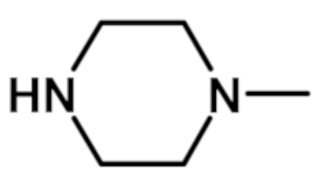
28
(Spectrochem)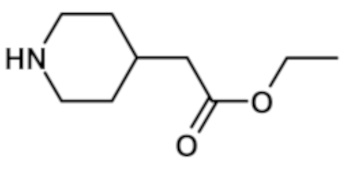
287
(Aldrich)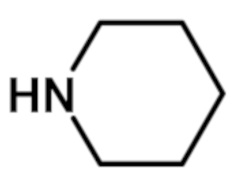
16
(Spectrochem)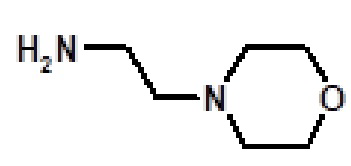
268
(Aldrich)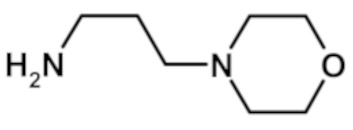
489
489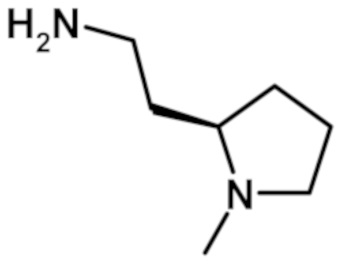
261
(Aldrich)
209
(TCI)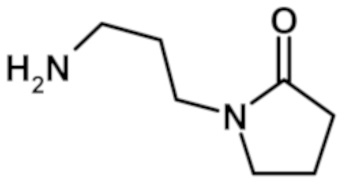
290
(Acros)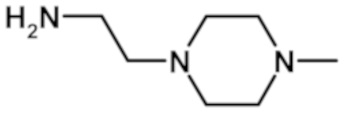
121
(Spectrochem)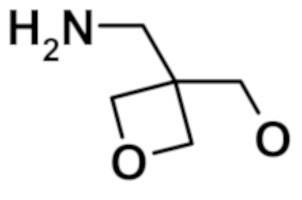
39,5
(Aldrich)
199
(Acros)
373
(Acros)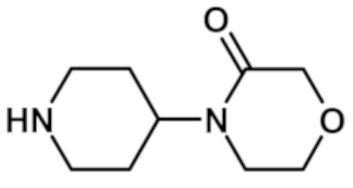
80
(Синтезированное)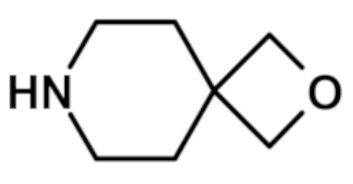
80
(Синтезированное)
95
(Синтезированное)
194
(Синтезированное)
310
(Синтезированное)
235
(Синтезированное)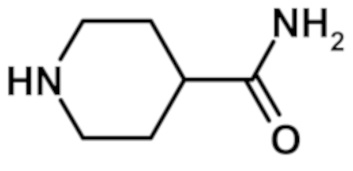
210
(Acros)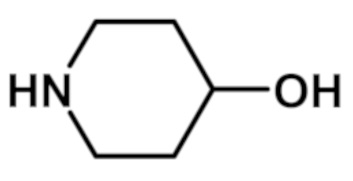
166
(Acros)
2,7
(Aldrich)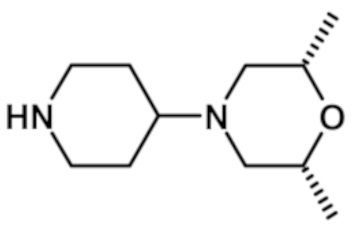
720
(Синтезированное)
587
(TCI)
514
(Acros)
194
(Синтезированное)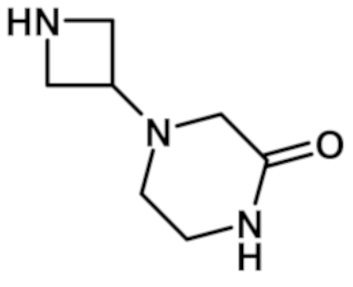
200
(Синтезированное)
96,25
(TCI)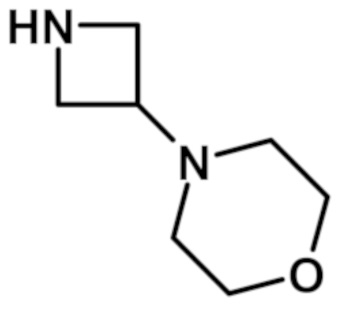
75
(Синтезированное)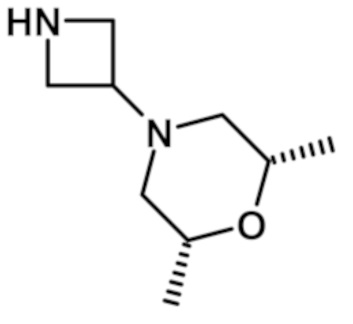
91
(Синтезированное)
*400 МГц, MeOH-d4 **400 МГц, CDCl3
# получали следующим образом: к раствору соединения 51 (60 мг, 0,125 ммоль) в тетрагидрофуране (8 мл) добавляли раствор гидроксида лития (21 мг, 0,5 ммоль) в воде (4 мл) и реакционную смесь перемешивали при комнатной температуре в течение 14 часов. Растворитель выпаривали до сухого состояния в вакууме. К остатку добавляли воду и промывали этилацетатом (50 мл, два раза). Водный слой подкисляли раствором хлористоводородной кислоты (1 Н, pH6), затем образовавшийся остаток фильтровали и сушили с получением 25 мг соединения 50.
1H-ЯМР (400 МГц, DMSO-d6) δ м.д.: 13,42-13,57 (м, 1H), 12,07-12,25 (м, 1H), 8,21-8,26 (м, 1H), 7,90-7,96 (м, 1H), 4,88-4,96 (м, 1H), 4,42-4,52 (м, 1H), 4,14-4,36 (м, 4H), 3,88 (с, 3H), 3,73-3,80 (м, 4H), 3,16-3,26 (м, 1H), 2,81-2,91 (м, 1H), 2,17-2,25 (м, 2H), 1,94-2,07 (м, 1H), 1,71-1,85 (м, 2H), 1,11-1,21 (м, 2H). Спектр масс (ESI): m/z 455,11[M+H].
Таблица 2
(400 МГц, DMSO-d6) δ м.д.
[M+H]
(мг)
(Источник)
(3a или 4a)
(мг)
71
(Combiblock)
89
(Combiblock)
81
(Aldrich)
84
(Combiblock) 69,15
69,15
(Aldrich)60,89
(Aldrich)
71,54
(Aldrich)64,71
(Aldrich)
33
(Aldrich)
71
(Combiblock)
71
(Aldrich)
62
(Combiblock)
226
(Combiblock)
32
(Aldrich)
61
(Aldrich)
69
(Combiblock) 85
85
(Aldrich) 80
80
(Aldrich)
75
(Aldrich)
67
(Aldrich)
81,70
(Combiblock) 86,29
86,29
(Aldrich)59,38
(Combiblock)
71,29
(Combiblock)
1,26
(Aldrich) 77
77
(Labex)
70
(Aldrich)
66
(Aldrich)
81
(Aldrich)112
(Aldrich)
66
(Aldrich)
78
(Combiblock)1,266
(Aldrich)
64
(Aldrich) 66
66
(Aldrich)
48
(Aldrich)75
(Aldrich)69
(Aldrich)68
(Aldrich)
86
(Aldrich)60
(Aldrich)
116
(Aldrich)
58
(Aldrich)
132
(Aldrich)
132
(Aldrich)
92
(Aldrich)
200
(Aldrich)
87
(Combiblock)
44
(Combiblock)
155
(Aldrich)
116
(Combiblock)
(Combiblock)
116
(Aldrich)
(* 400 МГц, CDCl3)
# Соединение 47 получали следующим образом: к раствору соединения 49 (260 мг, 0,57 ммоль) добавляли порошок палладия на угле (200 мг) и реакционную смесь перемешивали в течение 24 часов в атмосфере водорода с использованием баллона. Реакционную смесь фильтровали через целит и промывали метанолом (50 мл); фильтрат упаривали в вакууме и очищали колоночной хроматографией с использованием метанола и дихлорметана (5% метанол) в качестве элюента с получением 30 мг соединения 47.
Ингибиторы PI3K формулы (I), проиллюстрированные в настоящем описании, показаны в таблице 3, ниже.
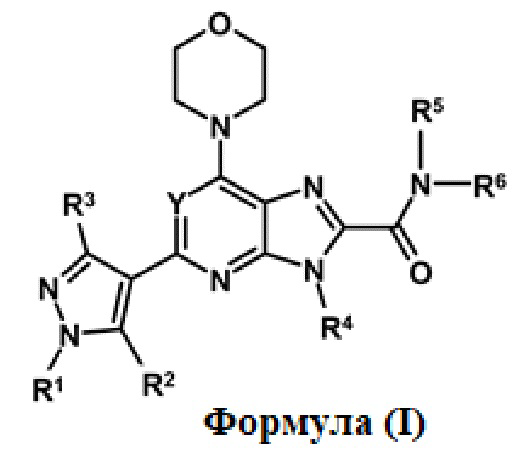
Таблица 3
IC50 (нМ)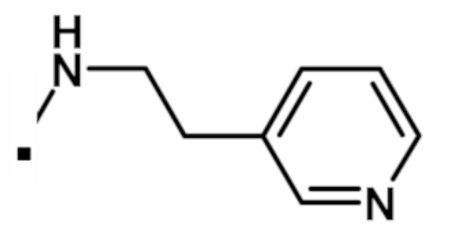
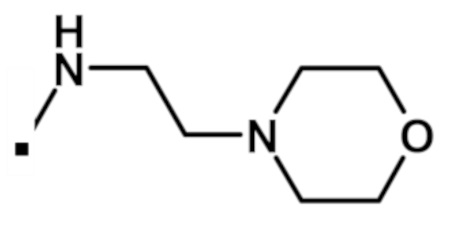
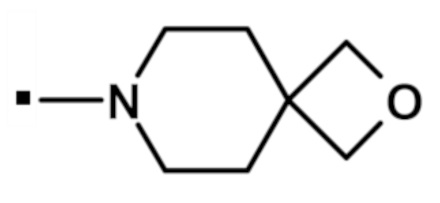
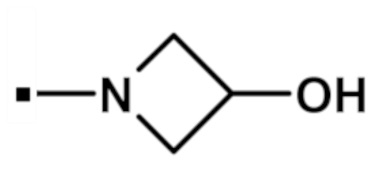
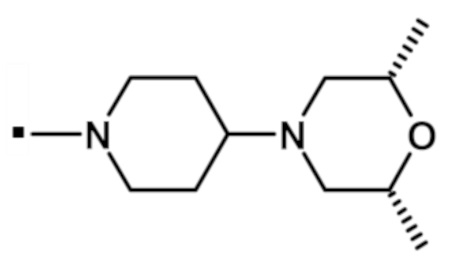
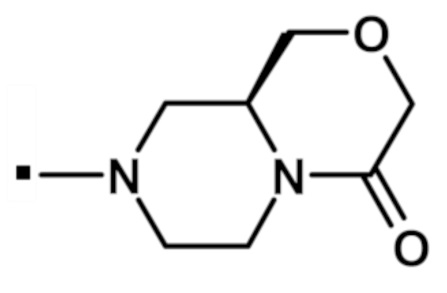
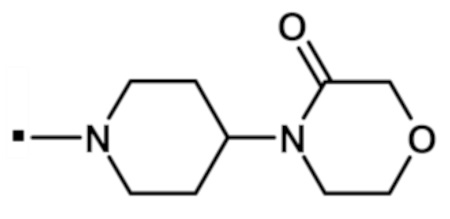
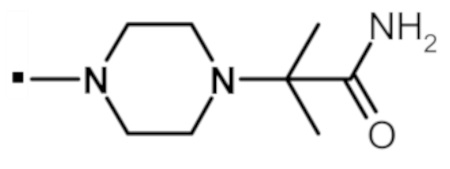
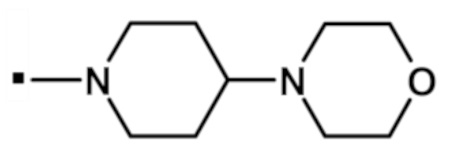
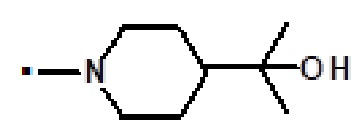
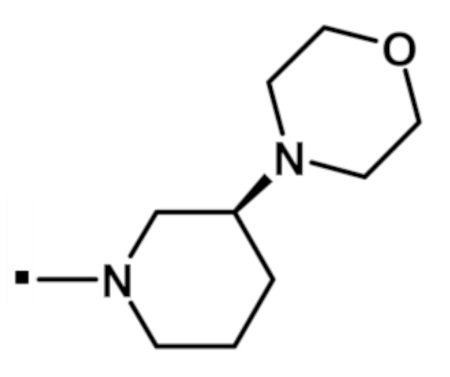
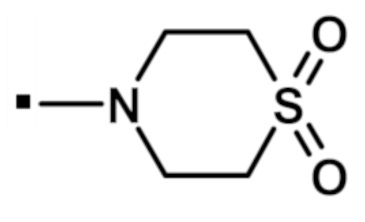

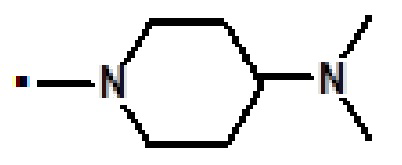
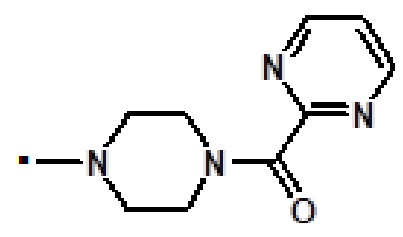
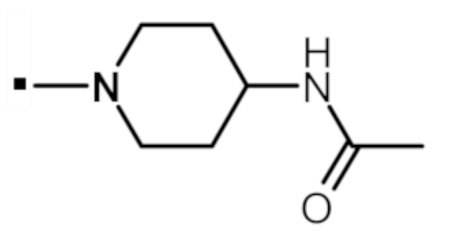
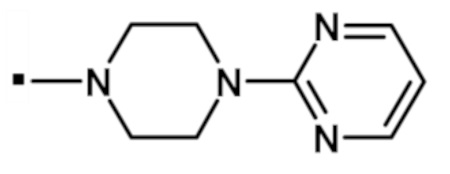
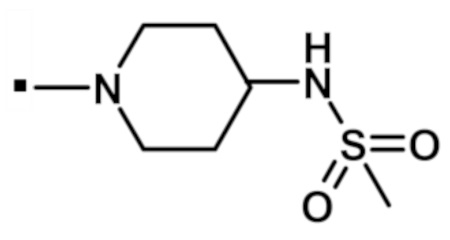
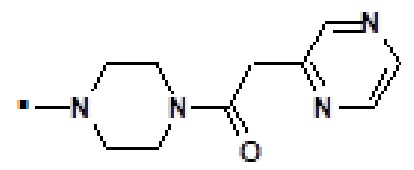
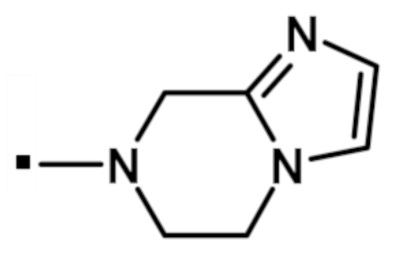
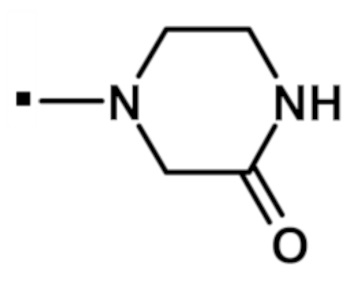
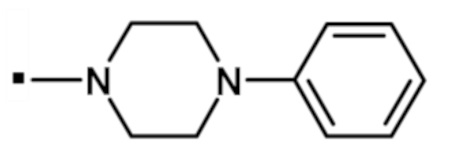
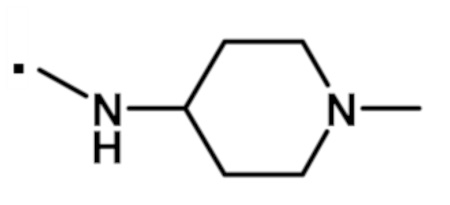
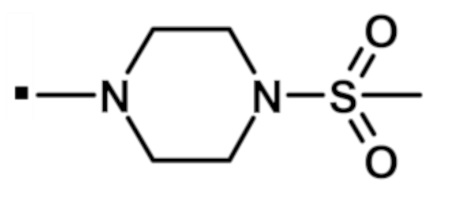
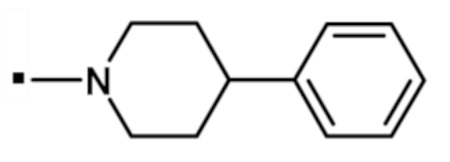
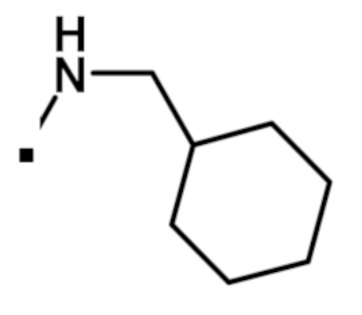
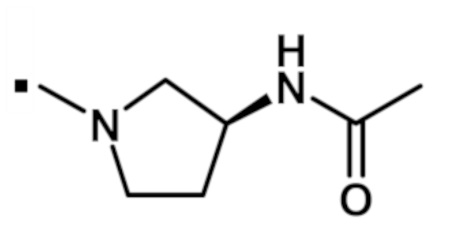
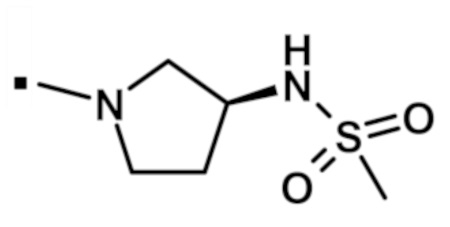
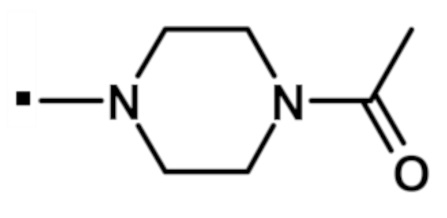
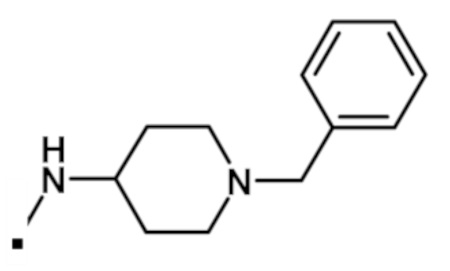
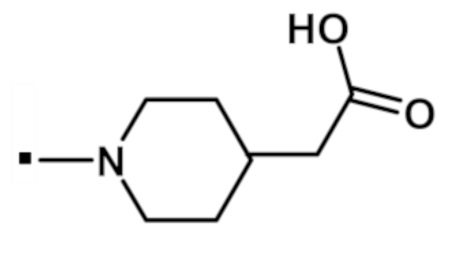
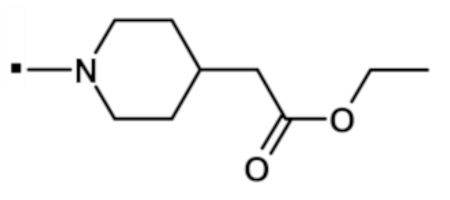
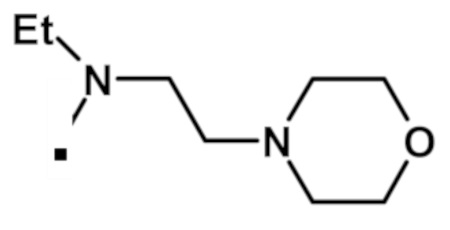
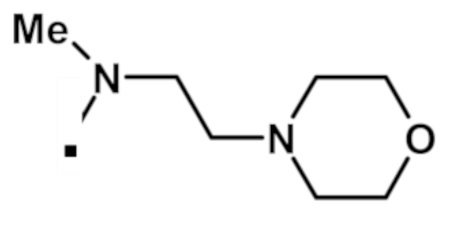
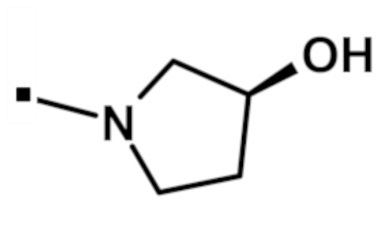
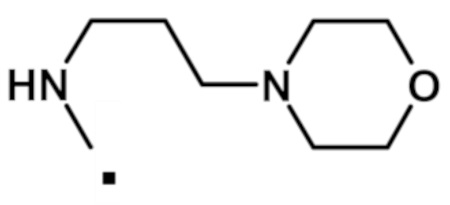
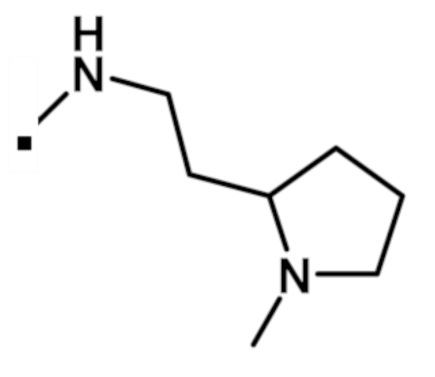
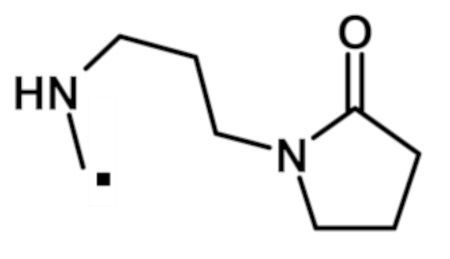
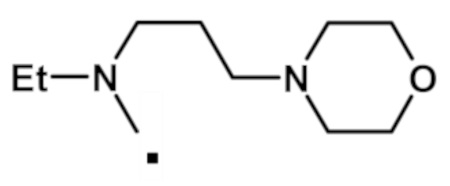

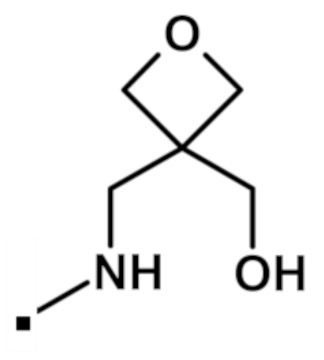
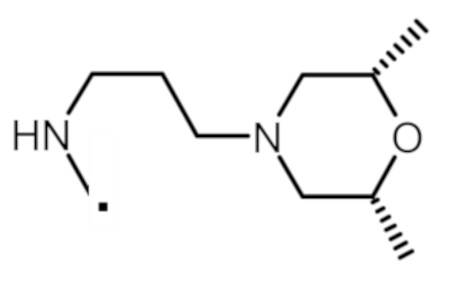
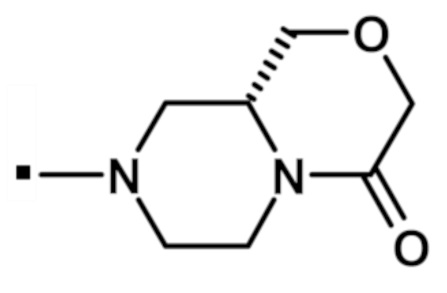
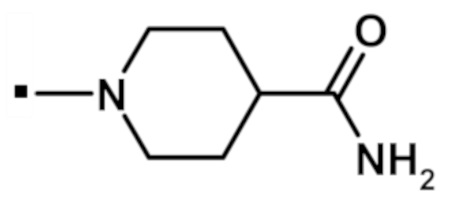
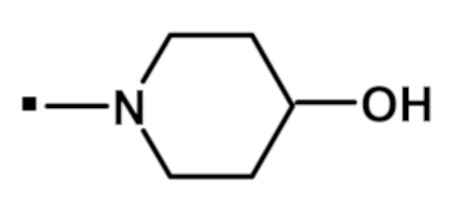
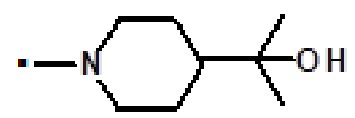
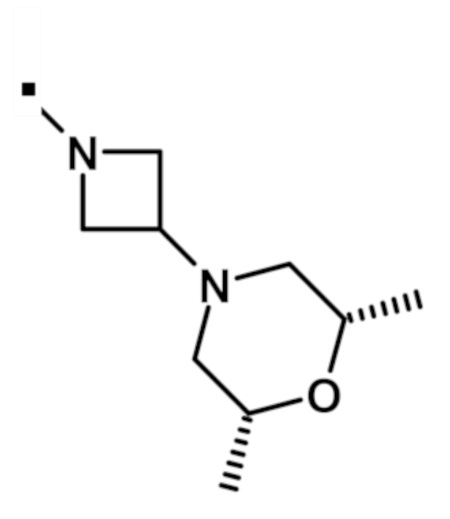
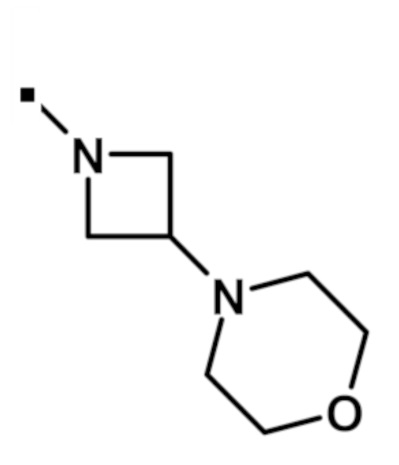
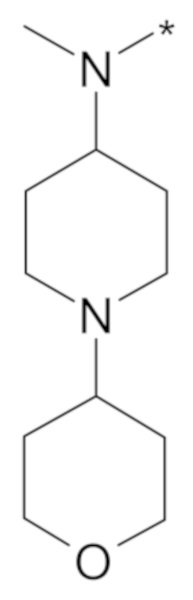
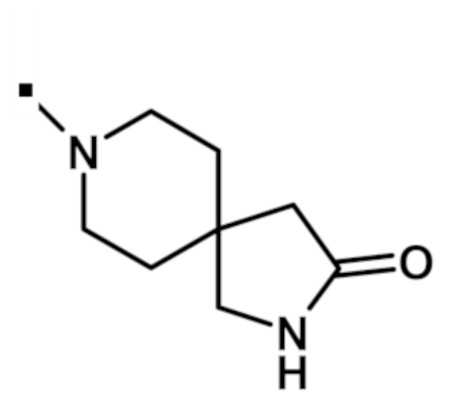
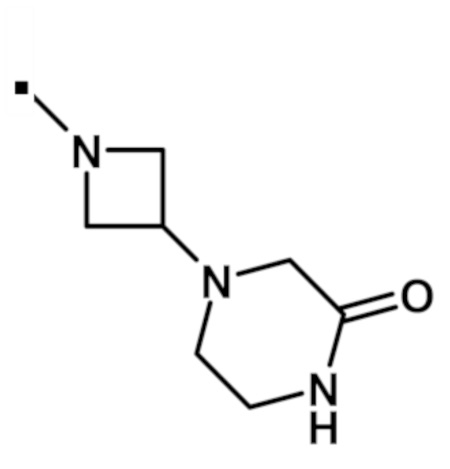
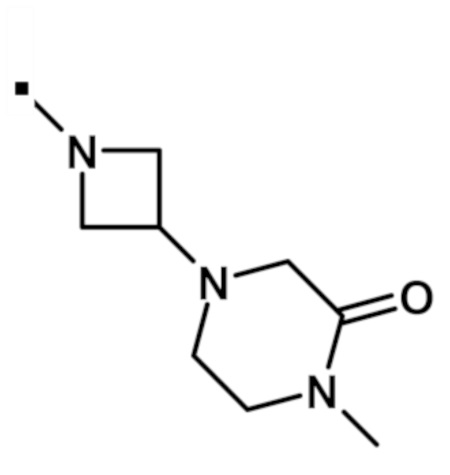
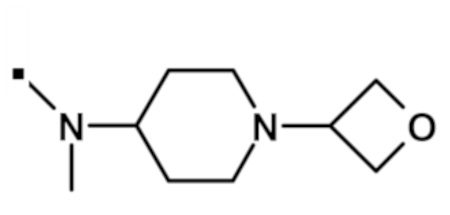
[Me(Метил); Et(Этил); iPr(изопропил); nPr(н-пропил); Cyp(циклопропил)]
Биологическая оценка
Можно использовать различные способы анализа активности PI3K. В дополнение к способам анализа, упомянутым ниже, специалисту в данной области будут известны другие способы анализа, которые можно использовать, и анализ можно модифицировать для конкретного применения. Такие способы анализа и их модификации являются частью сущности и объема настоящего изобретения.
(Пример испытания 1) Ферментный анализ in vitro
Для измерения киназной активности использовали набор для люминесцентного анализа киназы Kinase-Glo (от Promega). В этом анализе измеряют количество ATP, оставшегося в растворе после киназной реакции. Определение эффекта соединений на ингибирование PI3Kδ проводили добавлением 2,29 мкг/мл рекомбинантного фермента PI3Kδ (Proteros, Германия) к реакционной смеси, содержащей буфер для анализа (50 мМ HEPES, pH 7,4, 50 мМ NaCl, 0,05% CHAPS), дополненный 10 мМ MgCl2, 5 мМ DTT, 60 мкМ фосфатидилинозитолбисфосфатом (PIP2) и 10 мкМ ATP в отсутствие или в присутствии различных концентраций соединений в конечном объеме 15 мкл/лунка в 384-луночном планшете. Реакционную смесь инкубировали в течение 2 часов при комнатной температуре. В конце периода инкубации добавляли равный объем Kinase-Glo plus (Promega, V3772) на лунку и количественно определяли люминесценцию после инкубации в течение 10 минут при комнатной температуре в темноте. Результаты вычисляли путем количественного определения единиц люминесценции в исследуемых образцах относительно пустых растворов, не содержавших фермента.
В определенных вариантах осуществления соединения продемонстрировали IC50 для PI3Kδ менее 1000 нМ, в другом варианте осуществления величины IC50 находятся в диапазоне приблизительно от 100 нМ до 500 нМ, и в другом варианте осуществления они составляют даже менее 30 нМ, как показано в таблице 3 и 4.
Соединения по настоящему изобретению исследовали в отношении их селективности к PI3Kδ относительно PI3Kα, PI3Kβ и PI3Kγ согласно описано выше анализу с использованием специфических рекомбинантных ферментов (Proteros, Германия) для каждой киназы. Условия киназного анализа (PI3Kα, PI3Kβ, PI3Kγ и PI3Kδ) были следующими. Фермент: 2,29 мкг/мл; ATP: 10 мкМ; субстрат PIP2: 60 мкМ и время реакции: 2 часа.
Сокращенные обозначения:
HEPES: 2-[4-(2-гидроксиэтил)пиперазин-1-ил]этансульфоновая кислота
CHAPS: 3-[(3-хлорамидопропил)диметиламмонио]-1-пропансульфонат
MgCl2: хлорид магния
PIP2: фосфатидилинозитол 4,5-бисфосфат
DTT: дитиотреитол
ATP: аденозинтрифосфат
Таблица 4 (IC50 в нМ)
(Пример испытания 2) Индуцированное фитогемагглютинином (PHA) высвобождение интерферона (IFN)-гамма в спленоцитах мыши
Эффект соединений на индуцируемое митогеном высвобождение IFN-γ в спленоцитах мыши использовали для оценки их эффективности в системе анализа на клеточной основе [Blood (2010) 115: 2203-2213; Current Protocols in Immunology (2004) 3.12.1-3.12.20].
Спленоциты мыши получали из селезенки мышей C57BL/6 и высевали при плотности 0,25 миллионов клеток/лунка в 96-луночный планшет для культивирования тканей. Эффект соединений в отношении ингибирования высвобождения IFN-γ оценивали путем обработки спленоцитов различными концентрациями исследуемых соединений, а затем путем стимуляции посредством PHA (10 мкг/мл) в течение 48 часов. Высвобождение IFN-γ в супернатанте клеточной культуры количественно определяли посредством ELISA в соответствии с протоколом изготовителя (BD Biosciences, #555138).
Было обнаружено, что величины IC50 для соединений по настоящему изобретению составляют менее чем приблизительно 1,5 мкМ, предпочтительно менее чем приблизительно 1 мкМ, наиболее предпочтительно менее чем приблизительно 0,5 мкМ. Было обнаружено, что в предпочтительном варианте осуществления величины IC50 составляют даже менее 0,2 мкМ. Репрезентативные соединения приведены в таблице 4, выше.
(Пример испытания 3) Способы исследования терапевтического эффекта
(Пример испытания 3a) Модель индуцируемой овальбумином эозинофилии дыхательных путей на крысах Brown Norway
Следовали протоколам, аналогичным протоколам, описанным в Clin. Exp. Immunol., 2001; 126:9- 15 и J. Pharmacol. Exp. Ther., 2011; 337:145-54. Самцов крыс Brown Norway сенсибилизировали посредством внутрибрюшинной инъекции суспензии 1 мг овальбумина и 100 мг гидроксида алюминия (в стерильном 0,9% физиологическом растворе) на 0 и 7 сутки. На 14 сутки крысам вводили соединения через пероральный зонд. Через один час после перорального введения животных помещали в камеру из плексигласа и подвергали воздействию аэрозоля с 5% овальбумином в течение десяти минут. Соединения вводили на 14 сутки и 15 сутки либо один раз в сутки, либо два раза в сутки. Через сорок восемь часов после нагрузки овальбумином животных умерщвляли и собирали жидкость бронхоальвеолярного лаважа. Суспензии клеток обрабатывали и подсчитывали абсолютное количество эозинофилов.
В этой модели соединения по настоящему изобретению продемонстрировали эффективность, например, соединения № 3, 78 и 84 продемонстрировали ED50 46, 45 и 26% в дозе 3 мг/кг, соответственно. Было обнаружено, что величины ED50 составляют менее 3 мг/кг, bid, например, соединение № 96 имело ED50 2,5 мг/кг, bid. В предпочтительном варианте осуществления величины ED50 составляли даже менее 1,5 мг/кг, bid, например, соединения № 58 и 87 продемонстрировали ED50 0,3 мг/кг, bid, и 0,8 мг/кг, bid, соответственно.
(Пример испытания 3b) Модель хронической астмы, индуцированной клещом домашней пыли (HDM), на мышах Balb/c
Следовали протоколу, описанному в Am. J. Respir. Crit. Care Med., 2004; 169: 378-385. Самок мышей Balb/c подвергали воздействию очищенного экстракта HDM (Dermatophagoides pteronyssinus) интраназально (25 мкг белка в 20 мкл физиологического раствора) в течение 5 суток/неделя на протяжении вплоть до пяти последовательных недель. Соединения вводили перорально два раза в сутки с 3 недели по 5 неделю. Через сорок восемь часов после последнего воздействия HDM животных умерщвляли и жидкость бронхоальвеолярного лаважа собирали. Суспензии клеток обрабатывали и подсчитывали абсолютное количество эозинофилов.
Было обнаружено, что в этой модели соединения по настоящему изобретению являются эффективными. Было обнаружено, что величины ED50 составляют менее 2 мг/кг, bid, например, соединения № 5 и 58 продемонстрировали ED50 1,6 мг/кг, bid, и 0,1 мг/кг, bid, соответственно.
(Пример испытания 4) Способ исследования пероральной биодоступности (BA) у крыс и мышей
(Пример испытания 4a) Пероральная биодоступность (BA) у крыс
Самкам крыс Wistar (210±10 г) вводили исследуемое соединение в качестве раствора 2,0 мг/мл в носителе, содержащем полисорбат и декстрозу (pH 5,0), внутривенным путем или в качестве суспензии 1,0 мг/мл в метилцеллюлозе для перорального пути. Конечная доза составляла 3,0 мг/кг массы тела (внутривенная) или 10,0 мг/кг массы тела (пероральная). Образцы плазмы анализировали в отношении исследуемого соединения с использованием способа LC-MS/MS. Оценку фармакокинетических параметров проводили с использованием анализа моментов. Для оценки параметров PK использовали программное обеспечение WinNonlin 6.1 (Pharsight). Пероральную биодоступность вычисляли с использованием нормализованной к дозе пероральной и внутривенной экспозиций. Соединения, описанные в настоящем описании, продемонстрировали биодоступность, пригодную для применения в качестве пероральной терапии, например, биодоступность соединений № 3, 58 и 97 составляла 82, 88 и 123, соответственно.
(Пример испытания 4b) Пероральная биодоступность (BA) у мышей
Самцам мышей Swiss (23±3 г) вводили исследуемое соединение в качестве раствора 0,3 мг/мл в носителе, содержащем полисорбат и декстрозу (pH 5,0) внутривенным путем или в качестве суспензии 1,0 мг/мл в метилцеллюлозе для перорального пути. Конечная доза составляла 3,0 мг/кг массы тела (внутривенная) или 10,0 мг/кг массы тела (пероральная). Образцы плазмы анализировали в отношении исследуемого соединения с использованием способа LC-MS/MS. Оценку параметров PK проводили с использованием анализа моментов. WinNonlin 6.1 (Pharsight). Пероральную биодоступность вычисляли с использованием нормализованной к дозе пероральной и внутривенной экспозиций. Соединения, описанные в настоящем описании, продемонстрировали биодоступность, пригодную для применения в качестве пероральной терапии, например, биодоступность соединений № 3, 5, 58 и 103 составляла 73, 116, 100 и 112, соответственно.
(Пример испытания 5) Способ испытания растворимости
Получали раствор 10 ммоль/л исследуемого соединения в DMSO и 100 мкл исходных растворов 10 ммоль/л в DMSO распределяли в маркированные стеклянные пробирки в двух экземплярах, одну для первой жидкости Japanese Pharmacopeia (JP1) и вторую для второй жидкости Japanese Pharmacopeia (JP2). После выпаривания DMSO из каждой пробирки в каждую пробирку добавляли 500 мкл жидкости JP1 и JP2, соответственно. Эти пробирки обрабатывали ультразвуковым излучением в течение 1 минуты и помещали на устройство для встряхивания на 30 минут с интервалом 30 секунд каждые 5 минут. Пробирки помещали в темноту при комнатной температуре на 1 час и раствор фильтровали через мембранный фильтр. Фильтрат разбавляли в 2 раза и в 10 раз. Полученные исследуемые растворы анализировали и количественно определяли против стандартов с использованием UPLC (стандартный препарат - раствор 10 ммоль/л в DMSO, подвергнутый серийному разведению 50% водным раствором ацетонитрила с получением 2 растворов: стандартный раствор 100 мкмоль/л и стандартный раствор 5 мкмоль/л). Растворимость репрезентативных соединений является такой, как показано в таблице 5 ниже.
Таблица 5
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ SUV39H2 | 2016 |
|
RU2729187C1 |
| ПУРИНОВЫЕ ИНГИБИТОРЫ ЧЕЛОВЕЧЕСКОЙ ФОСФАТИДИЛИНОЗИТ 3-КИНАЗЫ ДЕЛЬТА | 2013 |
|
RU2658006C2 |
| 1Н-ПИРАЗОЛО[3,4-B]ПИРИДИНЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2013 |
|
RU2689141C2 |
| ПУРИНОВЫЕ ИНГИБИТОРЫ ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ ДЕЛЬТА ЧЕЛОВЕКА | 2013 |
|
RU2661896C2 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ТКБ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2742122C2 |
| ИНДАЗОЛ-3-КАРБОКСАМИДЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ СИГНАЛЬНОГО ПУТИ WNT/b-КАТЕНИНА | 2012 |
|
RU2627693C2 |
| МОДУЛЯТОРЫ ЯДЕРНЫХ РЕЦЕПТОРОВ | 2016 |
|
RU2715897C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2259202C2 |
| ЗАМЕЩЕННЫЕ [1,2,4]ТРИАЗОЛО[1,5-a]ПИРИМИДИН-7-ИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE2 | 2015 |
|
RU2659070C9 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PRMT5 | 2018 |
|
RU2797822C2 |
Изобретение относится к производному пиразола формулы (I) и фармацевтической композиции, содержащей соединение формулы (I) в качестве активного ингредиента, способам ее получения и способам ее применения. Технический результат: получены новые соединения, которые могут быть использованы в качестве ингибитора фосфатидилинозитол-3-киназы (PI3K), который можно использовать для лечения или предупреждения опосредованных заболеваний. 5 н. и 16 з.п. ф-лы, 5 табл.
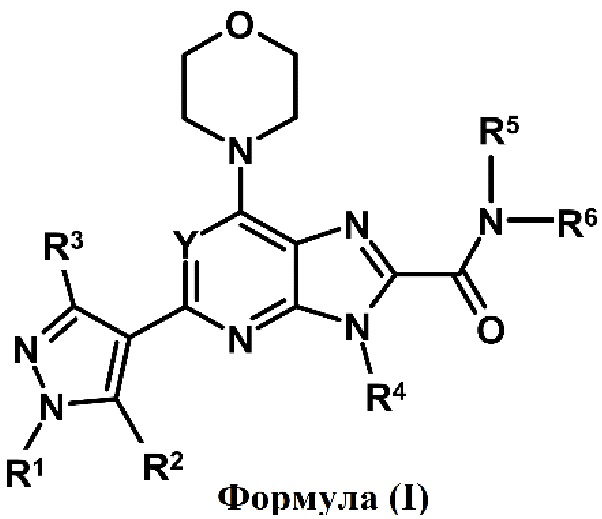
1. Соединение формулы (I)
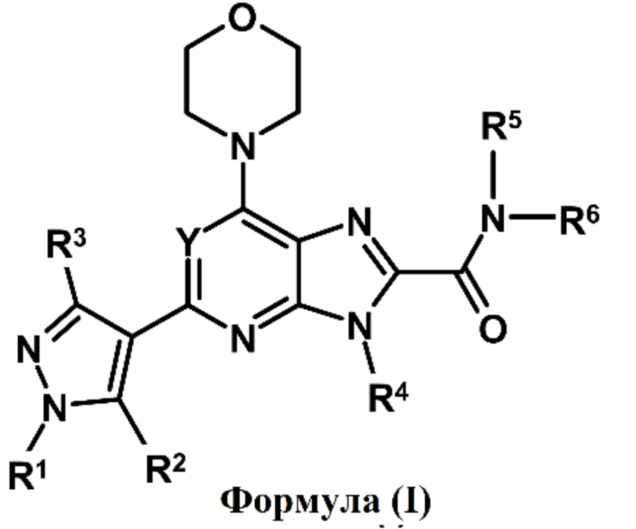
или его фармацевтически приемлемая соль, где:
Y обозначает N, CH или CF;
R1 представляет собой Н или метильную группу;
R2 и R3 независимо обозначают H, CF3 или метильную группу;
R4 обозначает H или метильную группу;
R5 и R6, взятые вместе с азотом, к которому они присоединены, образуют азетидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо или пиперазиновое кольцо,
где азетидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо и пиперазиновое кольцо необязательно замещены тетрагидропиранильной группой, морфолинильной группой или 2,6-диметилморфолинильной группой.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где Y обозначает N.
3. Соединение по п.1 или его фармацевтически приемлемая соль, где Y обозначает CH или CF.
4. Соединение по п.1 или его фармацевтически приемлемая соль, где R1 обозначает метильную группу и R2 и R3 независимо обозначают H или метильную группу.
5. Соединение по п.1 или его фармацевтически приемлемая соль, где R4 обозначает H или метильную группу,
6. Соединение по п.1 или его фармацевтически приемлемая соль, где R1 обозначает Н.
7. Соединение п.1, которое представляет собой {4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил]метанон, или его фармацевтически приемлемая соль.
8. Соединение по п.1, которое представляет собой [2-(1-метил-1H-пиразол-4-ил)-6-(морфолин-4-ил)-9H-пурин-8-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанон, или его фармацевтически приемлемая соль.
9. Соединение по п.1, которое представляет собой [5-(1,3-диметил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил][4-(морфолин-4-ил)пиперидин-1-ил]метанон, или его фармацевтически приемлемая соль.
10. Соединение по п.1, которое представляет собой {4-[цис-2,6-диметилморфолин-4-ил]пиперидин-1-ил}[5-(1,3-диметил-1H-пиразол-4-ил)-7-(морфолин-4-ил)-3H-имидазо[4,5-b]пиридин-2-ил]метанон, или его фармацевтически приемлемая соль.
11. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении фосфатидилинозитол-3-киназы (PI3K), содержащая эффективное количество соединения по любому из пп.1-10 или его фармацевтически приемлемую соль в качестве активного ингредиента и один или несколько фармацевтически приемлемый эксципиент(ов).
12. Фармацевтическая композиция по п.11 для лечения или уменьшения тяжести заболевания или нарушения, отвечающего на ингибирование фосфоинозитол-3-киназы δ (PI3Kδ).
13. Фармацевтическая композиция по п.12, где заболевание или нарушение представляет собой псориаз, псориатический артрит, ревматоидный артрит, аллергическую астму, тяжелую астму, резистентную к стероидам астму, COPD, системную красную волчанку, первичный синдром иммунодефицита или злокачественную опухоль.
14. Применение соединения по любому из пп.1-10 или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или уменьшения тяжести заболевания или нарушения, отвечающего на ингибирование PI3Kδ.
15. Применение по п.14, где заболевание или нарушение представляет собой псориаз, псориатический артрит, ревматоидный артрит, аллергическую астму, тяжелую астму, резистентную к стероидам астму, COPD, системную красную волчанку, первичный синдром иммунодефицита или злокачественную опухоль.
16. Способ лечения или уменьшения тяжести заболевания или нарушения, отвечающего на ингибирование PI3Kδ, у пациента посредством введения указанному пациенту терапевтически эффективного количества соединения по любому из пп.1-10 или его фармацевтически приемлемой соли.
17. Способ по п.16, где заболевание или нарушение представляет собой псориаз, псориатический артрит, ревматоидный артрит, аллергическую астму, тяжелую астму, резистентную к стероидам астму, COPD, системную красную волчанку, первичный синдром иммунодефицита или злокачественную опухоль.
18. Соединение по любому из пп.1-10 или его фармацевтически приемлемая соль для применения для лечения или уменьшение тяжести заболевания или нарушения, отвечающего на ингибирование PI3Kδ.
19. Соединение по п.18 или его фармацевтически приемлемая соль, где заболевание или нарушение представляет собой псориаз, псориатический артрит, ревматоидный артрит, аллергическую астму, тяжелую астму, резистентную к стероидам астму, COPD, системную красную волчанку, первичный синдром иммунодефицита или злокачественную опухоль.
20. Лекарственное средство для ингибирования PI3Kδ, содержащее соединение по любому из пп.1-10 или его фармацевтически приемлемую соль в качестве активного ингредиента.
21. Лекарственное средство по п.20, где заболевание или нарушение представляет собой псориаз, псориатический артрит, ревматоидный артрит, аллергическую астму, тяжелую астму, резистентную к стероидам астму, COPD, системную красную волчанку, первичный синдром иммунодефицита или злокачественную опухоль.
| WO 2010138589 A1, 02.12.2010 | |||
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2007 |
|
RU2443706C2 |
| Jeremy M Murray et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| ПРИСПОСОБЛЕНИЕ К ВИНТОРЕЗНОЙ МАШИНЕ ДЛЯ НАМОТКИ НА КАТУШКИ НАВИНТОВАННОЙ ПРОВОЛОКИ | 1927 |
|
SU7686A1 |
| Safina Brian S, Identification of GNE-293, a potent and selective PI3Kδ inhibitor: Navigating in vitro genotoxicity while improving | |||

