ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к аза-адамантановым производным, которые обладают способностью ингибировать 11-β-гидроксистероид-дегидрогеназу типа 1 (11β-HSD-1) и которые, таким образом, пригодны при лечении некоторых расстройств, которые можно предотвращать или лечить ингибированием этого фермента. Кроме того, настоящее изобретение относится к соединениям, способам их получения, фармацевтическим композициям, содержащим эти соединения, и к применению этих соединений при лечении некоторых расстройств. Предполагается, что соединения настоящего изобретения найдут применение при лечении состояний, таких как инсулиннезависимый сахарный диабет 2 типа (NIDDM), инсулинорезистентность, ожирение, нарушенная гликемия натощак, нарушение толерантности к глюкозы, липидные расстройства, такие как дислипидемия, гипертония, а также других заболеваний и состояний.
УРОВЕНЬ ТЕХНИКИ
Глюкокортикоиды представляют собой стрессовые гормоны с регуляторным эффектом на метаболизм углеводов, белков и жиров. Кортизол (или гидрокортизон у грызунов) представляет собой самый важный человеческий глюкокортикоид. 11-бета-гидроксил-стероид-дегидрогеназа или 11-бета-HSD1 (11β-HSD-1) является членом короткоцепного дегидрогеназного суперсемейства ферментов, который локально преобразует функционально инертный кортизон в активный кортизол дорецепторным образом. Учитывая, что этот фермент в большом количестве экспрессируется в метаболически важных тканях, таких как адипозная, мышечная и печеночная, которые становятся устойчивыми к действию инсулина при диабете 2 типа, ингибирование 11β-HSD-1 обеспечивает возможность восстановления понижающего концентрацию глюкозы действия инсулина в этих тканях без ухудшения центральной гипоталамо-гипофизарно-надпочечниковой оси (HPA). Другая важная 11-бета-гидроксил-стероид-дегидрогеназа, а именно 11-бета-HSD 2 типа (11β-HSD-2), которая преобразует кортизол в кортизон, представляет собой однонаправленную дегидрогеназу, которая расположена, в основном, в почках и защищает минералокортикоидные рецепторы от запрещенной активации под действием глюкокортикоидов.
Многочисленные наборы данных показывают, что 11β-HSD-1-опосредованная внутриклеточная выработка кортизола может иметь патогенную роль при ожирении, диабете 2 типа и его сопутствующих заболеваниях.
У людей лечение неспецифическим ингибитором карбеноксолоном улучшает восприимчивость инсулина у худых здоровых добровольцев и у людей с диабетом 2 типа (Walker В.R. Et al. (1995)). Таким же образом, активность 11β-HSD-1 снижается в печени и увеличивается в адипозной ткани индивидуумов с ожирением. Точно так же было обнаружено, что мРНК 11β-HSD-1 увеличивается как в висцеральной, так и в подкожной адипозной ткани пациентов с ожирением (Desbriere R. et al. (2006)) и положительно связана с индексом массы тела (BMI) и центральным ожирением у подростков индейцев Пима, белокожих людей и китайцев (Lindsat R.S. et al. (2003), Lee Z.S. et al. (1999)). Также было показано, что генная экспрессия 11β-HSD-1 и гексоза-6-фосфат-дегидрогеназы в адипозной ткани увеличивается у пациентов с сахарным диабетом 2 типа (Uckaya G. et al. (2008)). Было обнаружено, что экспрессия 11β-HSD-1 в скелетных мышцах человека положительно связана с инсулинорезистентностью (Whoewood С.В. et al. (2002). Увеличение экспрессии 11β-HSD-1 наблюдали также в диабетических мышечных трубочках (Abdallah В.М. et el. (2005)).
Были выполнены различные исследования в моделях на грызунах для подтверждения роли 11β-HSD-1 при диабете и ожирении. Например, сверхэкспрессия 11β-HSD-1, в частности, в адипозной ткани, вызывает развитие метаболического синдрома (непереносимость глюкозы, ожирение, дислипидемия и гипертония) у мышей (Masuzaki Н. et al. (2001)). И наоборот, при выключении гена 11β-HSD-1 полученные мыши демонстрировали устойчивость к инициированному питанием ожирению и улучшение сопутствующей дисрегуляции метаболизма глюкозы и липидов (Kotelevtsev Y. et al. (1997), Morton N.M. et al. (2001), Morton N.M. et al. (2004)). Кроме того, лечение мышей в модели диабета специфическими ингибиторами 11β-HSD-1 вызывало снижение выработки глюкозы печенью и общее увеличение чувствительности к инсулину (Alberts P. et al. (2003)).
Результаты доклинических и ранних клинических испытаний позволяют предположить, что лечение селективным и сильнодействующим ингибитором 11β-HSD-1 будет представлять собой эффективную терапию для диабета 2 типа, ожирения и метаболического синдрома.
Роль 11β-HSD-1 как важного регулятора содержания глюкокортикоида в печени и, следовательно, выработки глюкозы печенью, хорошо обоснована. Печеночная чувствительность к инсулину улучшается у здоровых людей-добровольцев, проходивших лечение неспецифическим ингибитором 11β-HSD-1 карбеноксолоном (Walker В.R. (1995)). Многие in vitro и in vivo (животные модели) исследования показали, что уровни мРНК и активность двух ключевых ферментов (PEPCK и G6PC) при глюкогенезе и гликогенолизе снижаются с уменьшением активности 11β-HSD-1. Данные из этих моделей подтверждают также, что ингибирование 11β-HSD-1 не вызывает гипогликемию, как было предсказано, поскольку базовые уровни PEPCK и G6P-азы регулируются независимо от глюкокортикоидов (Kotelevtsev Y. (1997)).
Было показано, что в поджелудочной железе кортизол ингибирует индуцированную глюкозой секрецию инсулина, а также увеличивает стресс, вызванный апоптозом бета-клеток. Ингибирование 11β-HSD-1 карбеноксолоном в изолированных мышиных панкреатических бета-клетках улучшает стимулированную глюкозой секрецию инсулина (Davani В. et al. (2000)). Недавно было показано, что 11β-HSD-1 в альфа-клетках регулирует секрецию глюкагона и, помимо этого, может действовать паракринным образом для ограничения секреции инсулина из бета-клеток (Swali A. et al. (2008)). Было показано, что уровни 11β-HSD-1 в островках из мышей линии ob/ob положительно регулируются глюкокортикоидами и снижаются под действием селективного ингибитора 11β-HSD-1 и антагониста глюкокортикоидного рецептора. Увеличенные уровни 11β-HSD-1 связаны с ухудшенной стимулированной глюкозой секрецией инсулина (GSIS) (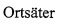 Н. et al. (2005)). У мышей с сахарным диабетом лечение троглитазоном улучшало метаболические патологии с 40% снижением экспрессии 11β-HSD-1 в островках (Duplomb L. et al. (2004)). Ингибирование кортизола может приводить к увеличению транскрипции гена инсулина и к нормализации первой фазы секреции инсулина (Shinozuka Y. et al (2001)).
Н. et al. (2005)). У мышей с сахарным диабетом лечение троглитазоном улучшало метаболические патологии с 40% снижением экспрессии 11β-HSD-1 в островках (Duplomb L. et al. (2004)). Ингибирование кортизола может приводить к увеличению транскрипции гена инсулина и к нормализации первой фазы секреции инсулина (Shinozuka Y. et al (2001)).
В скелетных мышцах человека экспрессия 11β-HSD-1 положительно связана с инсулинорезистентностью, и была также описана повышенная экспрессия 11β-HSD-1 в мышечных трубочках при диабете 2 типа (Abdallah В.М. et al. (2005)). Недавно было рассмотрено влияние кортизола при патологии мышц для модулирования их действия. Совсем недавно было показано, что направленное снижение или фармакологическое ингибирование 11β-HSD-1 в первостепенных скелетных мышцах человека предотвращает влияние кортизона на метаболизм глюкозы и окисление пальмитата (Salehzadeh F. et al. (2009)). Избыточная активность кортизола в мышцах приводит к мышечной атрофии, переключению типа волокон и слабому использованию глюкозы из-за инсулинорезистентности. Кортизол может иметь прямую роль в снижении поглощения глюкозы мышцами.
Ожирение представляет собой важный фактор при метаболическом синдроме, а также в большинстве (>80%) случаев диабета 2 типа, при этом сальниковый (висцеральный) жир имеет основное значение. Активность 11β-HSD-1 увеличивается как в висцеральной, так и в подкожной адипозной ткани индивидуумов с ожирением (Lindsay R.S. et al. (2003)). Активность кортизола в адипозной ткани, как известно, усиливает адипогенную программу. Было показано, что ингибирование активности 11β-HSD-1 в пре-адипоцитах снижает скорость дифференцировки на адипоциты (Bader Т. et al. (2002)). Предположительно, это приводит к сниженной экспансии (возможно, к снижению) сальникового жирового депо, то есть к уменьшению центрального ожирения (Bujalska I.J. et al. (1997) и (2006)). Внутрижировые концентрации кортизола связаны с адипозной гипертрофией, независимо от ожирения (Michailidou Z. et al. (2006)).
Известно также, что кортизол в координации с адренергической передачей сигналов увеличивает липолиз, что приводит к увеличению концентраций свободных жирных кислот в плазме, что, в свою очередь, представляет собой основную причину многих пагубных эффектов ожирения (Tomlinson J.W. et al. (2007)).
Адреналэктомия ослабляет эффект голодания с увеличением потребления пищи и гипоталамической экспрессией нейропептида Y. Это подтверждает роль глюкокортикоидов в промотировании потребления пищи и позволяет предположить, что ингибирование 11β-HSD-1 в мозге может усиливать чувство насыщения и, следовательно, снижать потребление пищи (Woods S.С. (1998)). Ингибирование 11β-HSD-1 низкомолекулярным ингибитором также снижает потребление пищи и увеличение веса при ожирении, индуцированном питанием, у мышей (Wang S. J. Y. et al. (2006)).
Таким образом, эффекты, рассмотренные выше, позволяют предположить, что эффективный ингибитор 11β-HSD-1 будет обладать активностью агента против ожирения.
Избыточное количество кортизола также может запускать образование триглицеридов и секрецию ЛПОНП в печени, что может способствовать гиперлипидемии и сопутствующей дислипидемии. Было показано, что трансгенные мыши 11β-HSD-1-/- имеют заметно более низкие концентрации триглицеридов в плазме и повышенные концентрации ЛПВП холестерина, что указывает на потенциальный атеропротективный фенотип (Morton N.М. et al. (2001)). В модели индуцированного питанием ожирения на мышах неселективный ингибитор 11β-HSD-1 снижает содержание свободных жирных кислот в плазме, а также триацилглицерина (Wang S.J. et al. (2006)). Сверхэкспрессия 11β-HSD-1 в печени увеличивает триглицериды в печени и свободные жирные кислоты в сыворотке с повышающей регуляцией печеночных липогенных генов (Paterson J.М. et al. (2004)). Было показано, что ингибирование 11β-HSD-1 улучшает триглицеридемию за счет снижения печеночной секреции ЛПОНП-триглицеридов, со сдвигом пути поглощения жирных кислот из триглицеридов в сторону окислительных тканей, в которых накопление липидов предотвращается за счет повышенного окисления липидов (Berthiaume М. et al. (2007)).
Атеросклеротическая модель мышей (АРОЕ -/-), которые подвержены атероме при употреблении пищи с высоким содержанием жира, были защищены от развития атеросклероза при лечении ингибиторами 11β-HSD-1 (Hermanowski-Vostaka A. et al. (2005)).
Ингибирование 11β-HSD-1 в зрелых адипоцитах предположительно ослабляет секрецию ингибитора плазминогенного активатора 1 (PAI-1) - независимого сердечнососудистого фактора риска (Halleux С.М. et al. (1999)). Более того, существует четкая корреляция между глюкокортикоидной активностью и сердечно-сосудистым фактором риска, что позволяет предположить, что снижение глюкокортикоидного эффекта будет иметь преимущество (Walker В.R. et al. (1998), Fraser R. et al. (1999)).
Связь между гипертонией и инсулинорезистентностью может быть объяснена повышенной активностью кортизола. Недавно полученные данные показали, что интенсивность сужения кровеносных сосудов кожи после локального нанесения глюкокортикоидов увеличивается у пациентов с существенно повышенным давлением (Walker В.R. et al. (1998)). Было показано, что глюкокортикоиды увеличивают экспрессию рецептора ангиотензина в сосудистых клетках и посредством этого стимулируют путь ренин-ангиотензина (Ullian М.Е. et al. (1996)), (Sato A. et al. (1994)). Недавно была подтверждена роль кортизола в NO-сигналинге и, следовательно, сужении сосудов (Liu Y. et al. (2009)). Эти открытия делают 11β-HSD-1 потенциальной мишенью для контролирования гипертонии и улучшения кровотока в заданных тканях.
В последнее десятилетие усилилось беспокойство из-за глюкокортикоид-индуцированного остеопороза в результате широкого применения экзогенных глюкокортикоидов (ГК). ГК-индуцированный остеопороз представляет собой наиболее распространенный и серьезный побочный эффект для пациентов, принимающих ГК. Снижение минеральной плотности костей (МПК) является максимальным в первые несколько месяцев применения ГК. Зрелые клетки, формирующие кость (остеобласты), считаются принципиальным местом действия ГК в скелете. Было подтверждено, что общая дифференцировка мезенхимальных стволовых клеток на линию остеобластов является чувствительной к ГК, а также к синтезу коллагена (Kim С.Н. et al. (1999)). Влияние ГК на этот процесс отличается в зависимости от стадии дифференцировки предшественников костных клеток. Наличие сигналинга интактного ГК является решающим для нормального развития и физиологии кости, в отличие от пагубного эффекта от воздействия высоких доз (Pierotti S. et al. (2008), Cooper M.S. et al. (2000)). Другие данные позволяют предположить роль 11β-HSD-1 в обеспечении достаточно высоких концентраций активного глюкокортикоида в остеокластах и, следовательно, в увеличении резорбции костей (Cooper М.S. et al. (2000)). Отрицательное влияние на образование костных узелков может быть блокировано неспецифическим ингибитором карбеноксолоном, что позволяет предположить важную роль 11β-HSD-1 в действии глюкокортикоидов (Bellows С.G. et al. (1998)).
Стресс и глюкокортикоиды влияют на когнитивную функцию (de Quervanian D.J. et al. (1998)). Фермент 11β-HSD-1 контролирует степень действия глюкокортикоида в мозге и, как известно, способствует нейротоксичности (Rajan V. et al. (1996)). Также было предположено, что ингибирование 11β-HSD-1 в мозге может приводить к снижению беспокойства (Tranche F. et al. (1999)). Следовательно, общая гипотеза заключается в том, что ингибирование 11β-HSD-1 в мозге человека предотвращает повторную активацию кортизона в кортизол и защищает от разрушительных эффектов, опосредованных глюкокортикоидами, оказываемых на выживание нейронов и другие аспекты нейронной функции, включая ухудшение когнитивной функции, депрессию и повышение аппетита.
Недавние данные позволяют предположить, что концентрации глюкокортикоида, направленного на рецепторы, и ферментов 11β-HSD-1 определяют предрасположенность к глаукоме (Stokes, J. et al. (2000)). Было показано, что прием карбексонолона, неспецифического ингибитора 11β-HSD-1, снижает внутриглазное давление на 20% у здоровых субъектов. Существуют данные, что изозим 11β-HSD-1 может модулировать стероид-регулируемый транспорт натрия через непигментированный эпителий (NPE), влияя посредством этого на внутриглазное давление (ВГД). Предположительно, 11β-HSD-1 играет роль в выработке внутриглазной жидкости, а не в ее оттоке, но в настоящее время неизвестно, обусловлено ли это взаимодействием с активацией глюкокортикоидного или минералокортикоидного рецептора, или их обоих (Rauz S. et al. (2001; 2003)).
Большое количество действий глюкокортикоидов представлено на примере пациентов с продолжительным увеличением глюкокортикоидов в плазме, так называемым «синдромом Кушинга». Эти пациенты имеют продолжительное увеличение глюкокортикоидов в плазме и демонстрируют ухудшенную переносимость глюкозы, диабет 2 типа, центральное ожирение и остеопороз. Эти пациенты также страдают от ухудшенного заживления ран и хрупкой кожи. Введение агониста глюкокортикоидного рецептора (RU 38486) пациентам с синдромом Кушинга реверсирует особенности метаболического синдрома (Neiman L.K. et al. (1985)).
Было показано, что глюкокортикоиды увеличивают риск инфекции и замедляют заживление открытых ран. Пациенты, проходившие лечение глюкокортикоидами, имеют в 2-5 раз более высокий риск осложнений при хирургическом вмешательстве. Глюкокортикоиды влияют на заживление ран за счет взаимодействия с выработкой или действием цитокинов и факторов роста таких как IGF, TGF-бета, EGF, KGF и PDGF (Beer Н.D. et al. (2000)). TGF-бета реверсирует вызванную глюкокортикоидами недостаточность заживления ран у крыс за счет регуляции PDGF в макрофагах (Pierce G.F. et al. (1989)). Было также показано, что глюкокортикоиды снижают синтез коллагена в коже крыс и мышей in vivo и в фибробластах крыс и человека (Oishi Y. et al., 2002).
Глюкокортикоиды также вовлечены в такие состояния как множественный синдром поликистозных яичников, бесплодие, нарушение памяти, расстройства сна, миопатия (Endocrinology, январь 2011 г.; 152(1):93-102. Эл. публикация 24 ноября 2010 г. PMID: 21106871) и мышечная дистрофия. Следовательно, способность воздействовать на ферменты, которые обладают действием на уровни глюкокортикоидов, предположительно обеспечивает возможность лечения этих состояний.
По данным патентной литературы и пресс-релизов различных компаний, существует много соединений, исследованных на ингибирование 11β-HSD-1 на различных стадиях процесса разработки лекарств.
Препарат INCB 13739 компании Incyte Corporation был продвинут дальше всего, до фазы ПЬ стадии клинических испытаний. Результаты фазы IIb испытаний для диабета 2 типа (28-дневные, плацебо-контролируемые, двухстадийные гиперинсулинемические клэмп-испытания) показали, что этот препарат безопасен и хорошо переносится без каких-либо серьезных побочных эффектов и гипогликемии.
Хотя эта молекула существенно улучшает печеночную чувствительность к инсулину, не было отмечено приемлемого улучшения содержания глюкозы в плазме. Эта молекула обладает положительным влиянием на факторы риска сердечнососудистых заболеваний, включая снижение ЛПНП, общего холестерина и триглицеридов, а также более умеренное увеличение ЛПВП. Препарат INCB 13739 в настоящее время исследуется в испытаниях IIb фазы с целью определения оптимальной дозы у T2D пациентов, концентрации глюкозы у которых не контролируются метформиновой монотерапией.
На доклинической стадии основной ингибитор Incyte INCB 13739 был испытан на макаках-резусах, и было показано, что он ингибирует адипозную 11β-HSD-1 (INCB 13739, селективный ингибитор 11β-гидроксистероид-дегидрогеназы типа 1 (11βHSD1) улучшает восприимчивость инсулина и снижает холестерин в плазме в течение 28 дней у пациентов с сахарным диабетом 2 типа).
Таким образом, эти данные дают веские основания предполагать, что соединения, которые ингибируют 11β-гидроксистероид-дегидрогеназу, будут пригодны при лечении ряда клинических состояний, связанных с экспрессией этого фермента. Кроме того, желательно, чтобы эти ингибиторы были селективными ингибиторами, которые за счет этого не мешали бы функционированию близкородственных ферментов, таких как 11β-HSD-2, которые, как известно, обеспечивают защитный эффект в организме.
ЗАДАЧИ ИЗОБРЕТЕНИЯ
Основная задача настоящего изобретения заключается в обеспечении соединений, которые представляют собой ингибиторы 11β-гидроксистероид-дегидрогеназы. Эти соединения предположительно пригодны при лечении состояний, связанных с 11β-гидроксистероид-дегидрогеназой, рассмотренных выше.
Дополнительная задача заключается в обеспечении фармацевтической композиции, содержащей соединение, которое представляет собой ингибитор 11β-гидроксистероид-дегидрогеназы, и фармацевтически приемлемое вспомогательное вещество, разбавитель или носитель.
Дополнительная задача заключается в обеспечении способа предупреждения или лечения состояния, связанного активностью 11β-гидроксистероид-дегидрогеназы, у млекопитающих.
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В настоящем изобретении представлены соединения формулы (I):
где: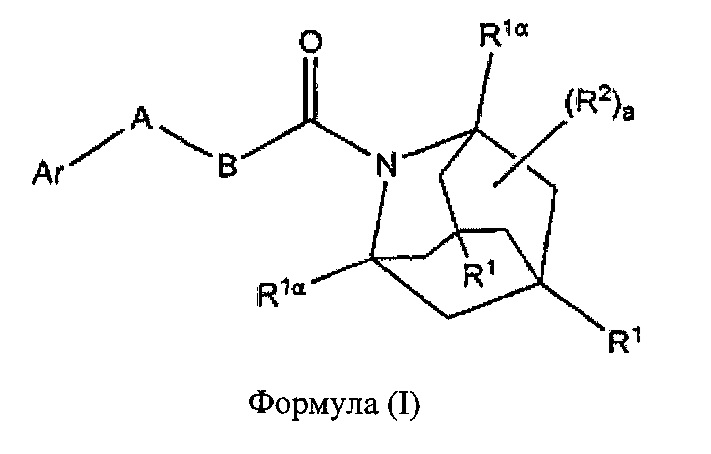
каждый R1, R1α и R2 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, OCH3, CH2OH, CH2CO2H, CH2CH2CO2H, CH2NH2, необязательно замещенного С1-С12алкила, необязательно замещенного C1-С12галоалкила, необязательно замещенного С2-С12алкенила, необязательно замещенного С2-С12алкинила, необязательно замещенного С2-С12гетероалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С3-С12циклоалкенила, необязательно замещенного С2-С12гетероциклоалкила, необязательно замещенного С2-С12гетероциклоалкенила, необязательно замещенного С6-С18арила, необязательно замещенного С1-С18гетероарила, необязательно замещенного С1-С12алкилокси, необязательно замещенного С2-С12алкенилокси, необязательно замещенного С2-С12алкинилокси, необязательно замещенного С2-С10гетероалкилокси, необязательно замещенного С3-С12циклоалкилокси, необязательно замещенного С3-С12циклоалкенилокси, необязательно замещенного С2-С12гетероциклоалкилокси, необязательно замещенного С2-С12гетероциклоалкенилокси, необязательно замещенного С6-С18арилокси, необязательно замещенного С1-С18гетероарилокси, необязательно замещенного С1-С12алкиламино, SR3, SO3H, SO2NR3R4, SO2R3, SONR3R4, SOR3, COR3, COOH, COOR3, CONR3R4, NR3COR4, NR3COOR4, NR3SO2R4, NR3CONR3R4 и NR3R4;
Ar представляет собой необязательно замещенную С1-С18гетерорарильную группу или необязательно замещенную С2-С12гетероциклоалкильную группу;
А выбран из группы, состоящей из S, SO, SO2, О и -CRaRb-;
В представляет собой группу формулы -(CRcRd)n-;
где каждый Ra, Rb, Rc и Rd независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, необязательно замещенного С1-С12алкила, необязательно замещенного С2-С10гетероалкила, необязательно замещенного C1-С12галоалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С6-С18арила, необязательно замещенного С1-С18гетероарила; SR3, SO3H, SO2NR3R4, SO2R3, SONR3R4, SOR3, COR3, COOH, COOR3, CONR3R4, NR3COR43, NR3COOR4, NR3SO2R4, NR3CONR3R4, NR3R4;
или любые два Ra, Rb, Rc и Rd у одного атома углерода, взятые вместе, могут образовывать заместитель формулы:
где каждый R3 и R4 независимо выбран из группы, состоящей из Н, необязательно замещенного С1-С12алкила, необязательно замещенного С2-С10гетероалкила, необязательно замещенного C1-С12галоалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С6-С18арила и необязательно замещенного С1-С18гетероарила;
R5 выбран из группы, состоящей из О, S и NR6;
R6 выбран из группы, состоящей из Н, OR7, необязательно замещенного C1-С12алкила, необязательно замещенного C1-С12галоалкила, необязательно замещенного С2-С12алкенила, необязательно замещенного С2-С12алкинила, необязательно замещенного C1-С12алкилокси, необязательно замещенного C1-С12галоалкилокси, необязательно замещенного С2-С10гетероалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С3-С12циклоалкенила, необязательно замещенного С2-С12гетероциклоалкила, необязательно замещенного С2-С12гетероциклоалкенила, необязательно замещенного С6-С18арила и необязательно замещенного С1-С18гетероарила;
R7 выбран из группы, состоящей из Н, необязательно замещенного необязательно замещенного C1-С12алкила, необязательно замещенного необязательно замещенного С2-С10гетероалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С6-С18арила и необязательно замещенного C1-С18гетероарила;
или любые два или более Ra, Rb, Rc и Rd могут объединяться с образованием кратной связи между соседними атомами углерода, такой как двойная или тройная связь, или циклического фрагмента, соединяющего атомы углерода, к которым они присоединены;
n представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3 и 4;
а представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 и 10;
или его фармацевтически приемлемая соль, N-оксид или пролекарство.
Как и в случае любой группы структурно родственных соединений, обладающих конкретной практической ценностью, некоторые варианты реализации переменных соединений Формулы (I) особенно пригодны при их конечном применении.
В некоторых вариантах реализации А представляет собой S. В некоторых вариантах реализации А представляет собой SO. В некоторых вариантах реализации А представляет собой SO2. В некоторых вариантах реализации А представляет собой О. В некоторых вариантах реализации А представляет собой CRaRb.
В некоторых вариантах реализации, где А представляет собой CRaRb, Ra и Rb, каждый независимо, выбраны из группы, состоящей из Н, СН3, СН2СН3, СН2СН2СН3, СН(СН3)2, (СН2)3СН3, Cl, Br, F, I, ОН, NO2, NH2, CN, SO3H, ОСН3, ОСН2СН2СН3, CF3 и OCF3. В некоторых вариантах реализации Ra представляет собой Н. В некоторых вариантах реализации Rb представляет собой Н. В некоторых вариантах реализации Ra и Rb являются различными, так что указанный атом углерода представляет собой хиральный углерод. В некоторых вариантах реализации один из Ra и Rb представляет собой Н, а другой представляет собой необязательно замещенный алкил.
В некоторых вариантах реализации Rb представляет собой Н, a Ra представляет собой необязательно замещенный алкил. В некоторых вариантах реализации Rb представляет собой Н, a Ra выбран из группы, состоящей из метила, этила, пропила, изопропила и бутила.
В представляет собой группу формулы -(CRcRd)n-. В некоторых вариантах реализации п равен 0. В некоторых вариантах реализации n равен 1. В некоторых вариантах реализации п равен 2.
В некоторых вариантах реализации Rc и Rd, каждый независимо, выбраны из группы, состоящей из Н, СН3, СН2СН3, СН2СН2СН3, СН(СН3)2, (СН2)3СН3, Cl, Br, F, I, OH, NO2, NH2, CN, SO3H, OCH3, OCH2CH2CH3, CF3 и OCF3. В некоторых вариантах реализации оба Rc и Rd представляют собой Н, так что В представляет собой СН2.
В некоторых вариантах реализации любые два или более Ra, Rb, Rc и Rd могут объединяться с образованием кратной связи между соседними углеродными атомами, такой как двойная или тройная связь, или циклического фрагмента, соединяющего углеродные атомы, к которым они присоединены.
В некоторых вариантах реализации два из Ra, Rb, Rc и Rd у соседних углеродных атомов объединены с образованием двойной связи. В некоторых вариантах реализации четыре из Ra, Rb, Rc и Rd у соседних углеродных атомов объединены с образованием тройной связи.
В некоторых вариантах реализации один из Ra и Rb и один из Rc и Rd, взятые вместе с углеродными атомами, к которым они присоединены, образуют циклический фрагмент. Примеры циклических фрагментов, которые могут быть образованы, включают циклопропил, циклобутил, циклопентил и циклогексил.
В некоторых вариантах реализации n=2, и один из Ra и Rb и один из Rc и Rd у углеродного атома, смещенного на два углеродных атома (у бета-углерода), взятые вместе с углеродными атомами, к которым они присоединены, и альфа-углеродным атомом образуют циклический фрагмент. Примеры циклических фрагментов, которые могут быть образованы, включают циклобутил, циклопентил и циклогексил.
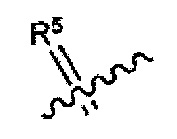
В некоторых вариантах реализации А представляет собой CRaRb, а В представляет собой СН2, это обеспечивает соединения формулы (II):
где R1, R1α, Ra, Rb, R2 и Ar являются такими, как описано выше.
Группа Ar может быть любым необязательно замещенным C1-C18гетероарильным фрагментом. Подходящие гетероарильные группы включают тиофен, бензотиофен, бензофуран, бензимидазол, бензоксазол, бензотиазол, бензизотиазол, нафто[2,3-b]тиофен, фуран, изоиндолизин, ксантолен, феноксатин, пиррол, имидазол, пиразол, пиридин, пиразин, пиримидин, пиридазин, тетразол, индол, изоиндол, 1Н-индазол, пурин, хинолин, изохинолин, фталазин, нафтиридин, хиноксалин, циннолин, карбазол, фенантридин, акридин, феназин, тиазол, изотиазол, фенотиазин, оксазол, изооксазол, фуразан, феноксазин, пиридил, хинолил, изохинолинил, индолил и тиенил. В каждом случае, в котором существует возможность нескольких центров замещения в гетероарильном кольце, подразумеваются все возможные точки присоединения. Лишь в качестве примера, если гетероарил представляет собой пиридильный фрагмент, он может представлять собой 2-пиридил, 3-пиридил или 4-пиридил.
В некоторых вариантах реализации Ar представляет собой группу формулы 3:
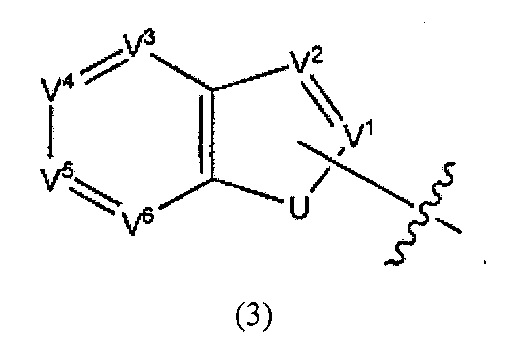
где каждый V1, V2, V3, V4, V5 и V6 независимо выбран из группы, состоящей из N и CR8;
U выбран из группы, состоящей из NR9, О, S и CR92,
где каждый R8 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, необязательно замещенного С1-С12алкила, необязательно замещенного С1-С12галоалкила, необязательно замещенного С2-С12алкенила, необязательно замещенного С2-С12алкинила, необязательно замещенного С2-С12гетероалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С3-С12циклоалкенила, необязательно замещенного С2-С12гетероциклоалкила, необязательно замещенного С2-С12гетероциклоалкенила, необязательно замещенного С6-C18арила, необязательно замещенного С1-C18гетероарила, необязательно замещенного С1-С12алкилокси, необязательно замещенного С2-С12алкенилокси, необязательно замещенного С2-С12алкинилокси, необязательно замещенного С2-С10гетероалкилокси, необязательно замещенного С3-С12циклоалкилокси, необязательно замещенного С3-С12циклоалкенилокси, необязательно замещенного С2-С12гетероциклоалкилокси, необязательно замещенного С2-С12гетероциклоалкенилокси, необязательно замещенного С6-С18арилокси, необязательно замещенного С1-С1гетероарилокси, необязательно замещенного C1-С12алкиламино, SR10, SO3H, SO2NR10R11, SO2R10, OSO2R10, SONR10R11, SOR10, COR10, COOH, COOR10, CONR10R11, NR10COR11, NR10COOR11, NR10SO2R11, NR10CONR10R11 и NR10R11;
где R9 выбран из группы, состоящей из Н, необязательно замещенного C1-С12алкила, необязательно замещенного С2-С12алкенила, необязательно замещенного С2-С12алкинила, необязательно замещенного С2-С12гетероалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С2-С12гетероциклоалкила, необязательно замещенного С6-C18арила, необязательно замещенного С1-С18гетероарила, SO3H, SO2NR10R11, SO2R10, SONR10R11, SOR10, COR10, COOH, COOR10 и CONR10R11;
где каждый R10 и R11 независимо выбран из группы, состоящей из Н, необязательно замещенного С1-С12алкила, необязательно замещенного С2-С10гетероалкила, необязательно замещенного С1-С12галолакила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С6-С18арила и необязательно замещенного С1-С18гетероарила.
В некоторых вариантах реализации Ar выбран из группы, состоящей из:
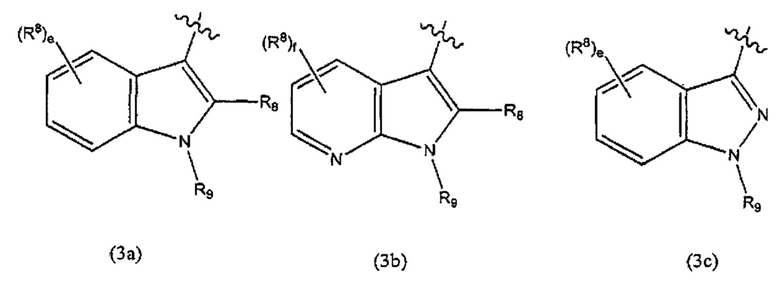
и
где R8 и R9 являются такими, как описано выше;
е представляет собой целое число, выбранное из группы, состоящей из 0, 1,2,3 и 4;
f представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2 и 3.
В некоторых вариантах реализации А представляет собой CRaRb, В представляет собой СН2, и Ar представляет собой группу формулы (3а), это обеспечивает соединения формулы (IVa):
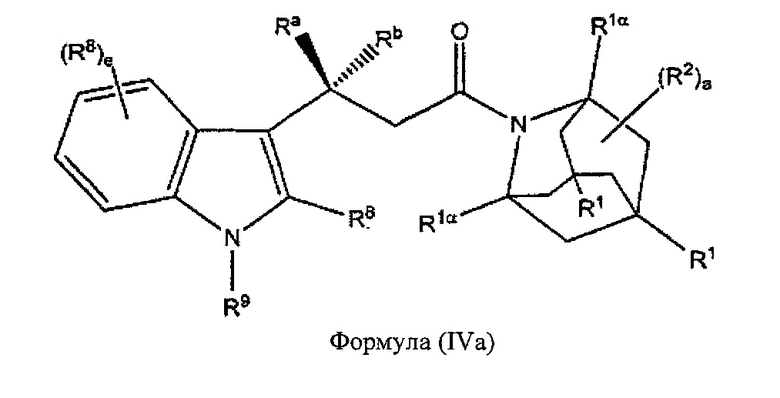
где R1, R1α, Ra, Rb, R2, R8, R9 и e являются такими, как описано выше.
В некоторых вариантах реализации А представляет собой CRaRb, В представляет собой СН2, и Ar представляет собой группу формулы (3b), это обеспечивает соединения формулы (IVb):
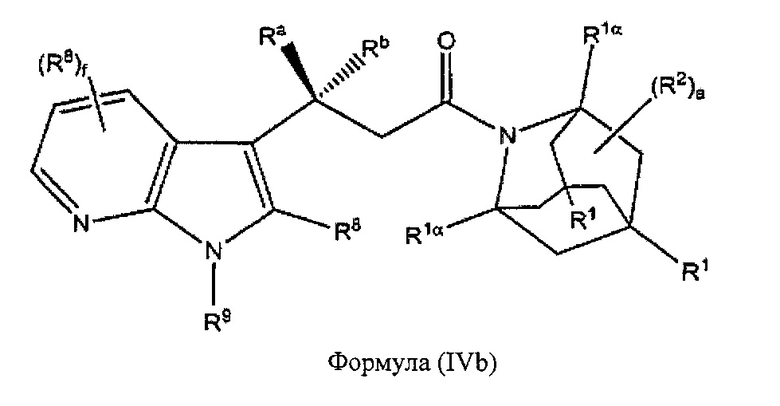
где R1, R1α, Ra, Rb, R2, R8, R9 и f являются такими, как описано выше.
В некоторых вариантах реализации А представляет собой CRaRb, В представляет собой СН2, и Ar представляет собой группу формулы (3с), это обеспечивает соединения формулы (IVc):
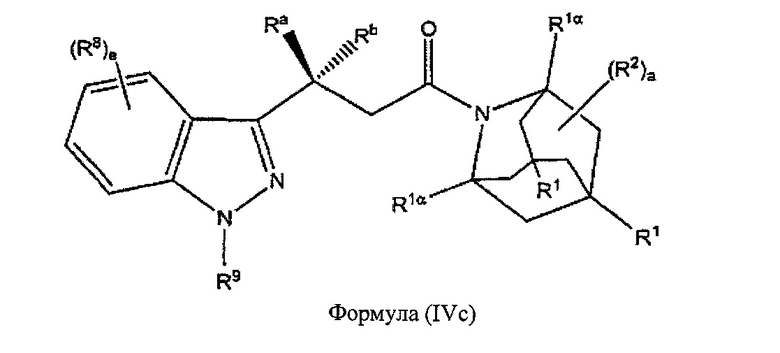
где R1, R1α, Ra, Rb, R2, R8, R9 и e являются такими, как описано выше.
В некоторых вариантах реализации А представляет собой CRaRb, В представляет собой СН2, и Ar представляет собой группу формулы (3d), это обеспечивает соединения формулы (IVd):
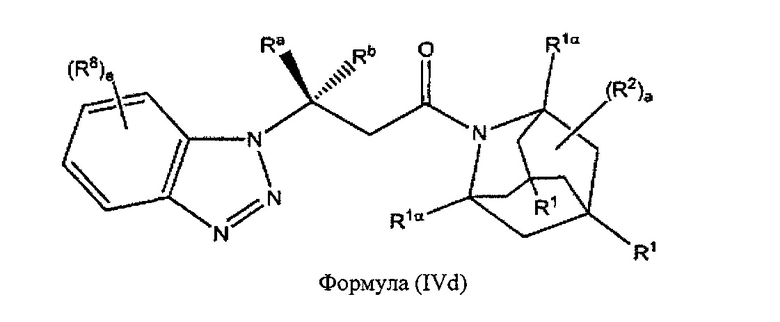
где R1, R1α, Ra, Rb, R2, R8 и e являются такими, как описано выше.
В некоторых вариантах реализации А представляет собой CRaRb, В представляет собой СН2, и Ar представляет собой группу формулы (3е), это обеспечивает соединения формулы (IVe):
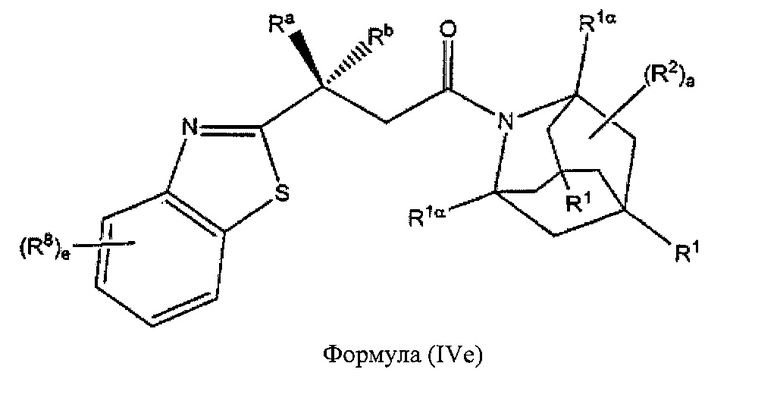
где R1, R1α, Ra, Rb, R2, R8 и e являются такими, как описано выше.
В некоторых вариантах реализации А представляет собой CRaRb, В представляет собой СН2, и Ar представляет собой группу формулы (3f), это обеспечивает соединения формулы (IVf):
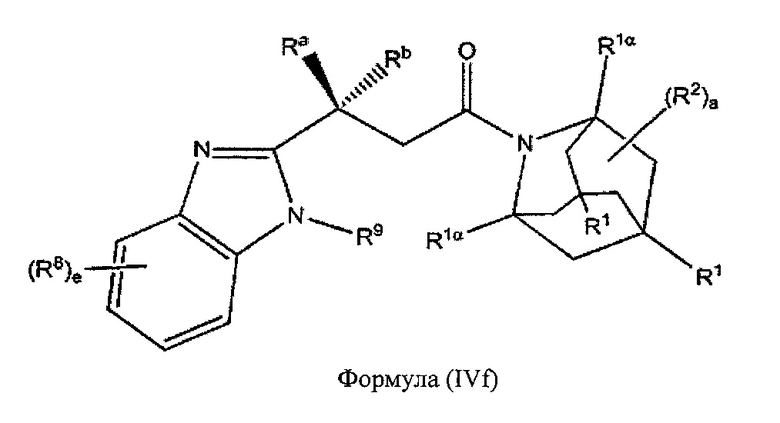
где R1, R1α, Ra, Rb, R2, R8, R9 и e являются такими, как описано выше.
В некоторых вариантах реализации А представляет собой CRaRb, В представляет собой СН2, и Ar представляет собой группу формулы (3g), это обеспечивает соединения формулы (IVg):
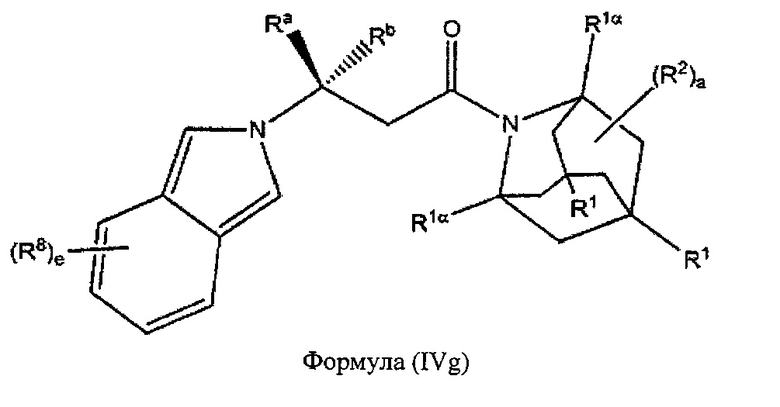
где R1, R1α, Ra, Rb, R2, R8 и e являются такими, как описано выше.
В некоторых вариантах реализации е равен 1. В некоторых вариантах реализации е равен 2. В некоторых вариантах реализации е равен 3. В некоторых вариантах реализации е равен 4. В тех случаях, где е равен 1, группа R8 может быть расположена в любом из 4, 5, 6 или 7 положении шестичленного кольца. В некоторых вариантах реализации, если е равен 1, то заместитель R8 расположен в 4 положении кольца. В некоторых вариантах реализации, если е равен 1, то заместитель R8 расположен в 5 положении кольца. В некоторых вариантах реализации, если е равен 1, то заместитель R8 расположен в 6 положении кольца. В некоторых вариантах реализации, если е равен 1, то заместитель R8 расположен в 7 положении кольца.
В некоторых вариантах реализации f равен 1. В некоторых вариантах реализации f равен 2. В некоторых вариантах реализации f равен 3. В некоторых вариантах реализации, в которых f равен 1, то заместитель R8 расположен в 4 положении кольца. В некоторых вариантах реализации, в которых f равен 1, то заместитель R8 расположен в 5 положении кольца. В некоторых вариантах реализации, в которых f равен 1, то заместитель R8 расположен в 6 положении кольца. В некоторых вариантах реализации, в которых f равен 1, то заместитель R8 расположен в 7 положении кольца.
В некоторых вариантах реализации соединений, описанных выше, каждый R1 независимо выбран из группы, состоящей из Н, ОН, F, Cl, Br, СН3, СН2СО2Н, CH2CH2CO2H, CO2H, CONH2, СН2ОН, CH2NH2, CN, ОСН3, О-циклопропила и OCHF2. В некоторых вариантах реализации один R1 представляет собой Н, а другой R1 представляет собой ОН. В некоторых вариантах реализации оба R1 представляют собой Н.
В некоторых вариантах реализации соединений, описанных выше, каждый R1α независимо выбран из группы, состоящей из Н, ОН, F, Cl, Br, СО2Н, CONH2, СН2ОН, CN, ОСН3 и OCHF2. В некоторых вариантах реализации один R1α представляет собой Н, а другой R1α представляет собой ОН. В некоторых вариантах реализации оба R1α представляют собой Н.
В некоторых вариантах реализации каждый R2 независимо выбран из группы, состоящей из Н, СН3, СН2СН3, СН2СН2СН3, СН(СН3)2, (СН2)3СН3, Cl, Br, F, I, ОН, NO2, NH2, CN, SO3H, ОСН3, ОСН2СН2СН3, CF3 и OCF3.
В некоторых вариантах реализации а равен 0. В некоторых вариантах реализации а равен 1. В некоторых вариантах реализации а равен 2. В некоторых вариантах реализации а равен 3. В некоторых вариантах реализации а равен 4. В некоторых вариантах реализации а равен 5. В некоторых вариантах реализации а равен 6. В некоторых вариантах реализации а равен 7. В некоторых вариантах реализации а равен 8. В некоторых вариантах реализации а равен 9. В некоторых вариантах реализации а равен 10.
В некоторых вариантах реализации соединений настоящего изобретения, содержащих группу R3, группа R3 выбрана из Н и C1-С12алкила. В некоторых вариантах реализации R3 представляет собой Н. В некоторых вариантах реализации R3 представляет собой метил.
В некоторых вариантах реализации соединений настоящего изобретения, содержащих группу R4, группа R4 выбрана из Н и C1-С12алкила. В некоторых вариантах реализации R4 представляет собой Н. В некоторых вариантах реализации R4 представляет собой метил.
В некоторых вариантах реализации соединений настоящего изобретения, содержащих группу R5, группа R5 выбрана из О и S. В некоторых вариантах реализации R5 представляет собой О. В некоторых вариантах реализации R5 представляет собой S.
В некоторых вариантах реализации соединений настоящего изобретения, содержащих группу R6, группа R6 выбрана из Н и C1-С12алкила. В некоторых вариантах реализации R6 представляет собой Н. В некоторых вариантах реализации R6 представляет собой метил.
В некоторых вариантах реализации соединений настоящего изобретения, содержащих группу R7, группа R7 выбрана из Н и C1-С12алкила. В некоторых вариантах реализации R7 представляет собой Н. В некоторых вариантах реализации R7 представляет собой метил.
R8 может быть выбран из широкого ряда возможных заместителей, рассмотренных выше. В некоторых вариантах реализации каждый R8 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, C1-С12алкила, C1-С12галоалкила, C1-С12алкоксила и C1-С12алоалкоксила. Иллюстративные заместители R8 включают Н, СН3, СН2СН3, СН2СН2СН3, СН(СН3)2, (СН2)3СН3, циклопропил, I, Br, F, I, ОН, NO2, NH2, CN, SO3H, OCH3, OCH(CH3)2, ОСН2СН2СН3, OSO2CF3, CF3 и OCF3.
R9 может быть выбран из широкого ряда возможных заместителей, рассмотренных выше. В некоторых вариантах реализации каждый R9 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, C1-С12алкила, C1-С12галоалкила, C1-С12алкоксила и C1-С12галоалкоксила. Иллюстративные заместители R9 включают CH3, СН2СН3, СН2СН2СН3, СН(СН3)2, (СН2)3СН3, I, Br, F, I, ОН, NO2, NH2, CN, SO3H, OCH3, OCH2CH2CH3, CF3 и OCF3.
Многие, если не все переменные, рассмотренные выше, могут быть необязательно замещены. Если переменная необязательно замещена, то в некоторых вариантах реализации каждый необязательный заместитель независимо выбран из группы, состоящей из галогена, =O, =S, -CN, -NO2, -CF3, -OCF3, алкила, алкенила, алкинила, галоалкила, галоалкенила, галоалкинила, гетероалкила, циклоалкила, циклоалкенила, гетероциклоалкила, гетероциклоалкенила, арила, гетероарила, циклоалкилалкила, гетероциклоалкилалкила, гетероарилалкила, арилалкила, циклоалкилалкенила, гетероциклоалкилалкенила, арилалкенила, гетероарилалкенила, циклоалкилгетероалкила, гетероциклоалкилгетероалкила, арилгетероалкила, гетероарилгетероалкила, гидрокси, гидроксиалкила, алкилокси, алкилоксиалкила, алкилоксициклоалкила, алкилоксигетероциклоалкила, алкилоксиарила,
алкилоксигетероарила, алкилоксикарбонила, алкиламинокарбонила, алкенилокси, алкинилокси, циклоалкилокси, циклоалкенилокси, гетероциклоалкилокси, гетероциклоалкенилокси, арилокси, фенокси, бензилокси, гетероарилокси, арилалкилокси, амино, алкиламино, ациламино, аминоалкила, ариламино, сульфониламино, сульфиниламино, сульфонила, алкилсульфонила, арилсульфонила, аминосульфонила, сульфинила, алкилсульфинила, арилсульфинила,
аминосульфиниламиноалкила, -С(=O)ОН, -C(=ORe, -C(=O)ORe, C(=O)NReRf, C(=NOH)Re, C(=NRe)NRfRg, NReRf, NReC(=O)Rf, NReC(=O)ORf, NReC(=O)NRfRg, NReC(=NRf)NRgRh, NReSO2Rf, -SRe, SO2NReRf, -ORe, OC(=O)NReRf, OC(=O)Re и ацила,
где Re, Rf, Rg и Rh, каждый независимо, выбраны из группы, состоящей из Н, C1-С12алкила, C1-С12галоалкила, С2-С12алкенила, С2-С12алкинила, C1-С10гетероалкила, С3-С12циклоалкила, С3-С12циклоалкенила, C1-С12гетероциклоалкила, C1-С12гетероциклоалкенила, С6-С18арила, С1-С18гетероарила и ацила, или любые два или более из Ra, Rb, Rc и Rd, взятые вместе с атомами, к которым они присоединены, образуют гетероциклическую кольцевую систему с 3-12 кольцевыми атомами.
В некоторых вариантах реализации каждый необязательный заместитель независимо выбран из группы, состоящей из: F, О, Br, =O, =S, -CN, -NO2, алкила, алкенила, гетероалкила, галоалкила, алкинила, арила, циклоалкила, гетероциклоалкила, гетероарила, гидрокси, гидроксиалкила, алкокси, алкиламино, аминоалкила, ациламино, фенокси, алкоксиалкила, бензилокси, алкилсульфонила, арилсульфонила, аминосульфонила, -C(O)ORa, СООН, SH и ацила.
В некоторых вариантах реализации каждый необязательный заместитель независимо выбран из группы, состоящей из: F, Br, О, =O, =S, -CN, метила, трифтор-метила, этила, 2,2,2-трифторэтила, изопропила, пропила, 2-этил-пропила, 3,3-диметил-пропила, бутила, изобутила, 3,3-диметил-бутила, 2-этил-бутила, пентила, 2-метил-пентила, пент-4-енила, гексила, гептила, октила, фенила, NH2, -NO2, фенокси, гидрокси, метокси, трифтор-метокси, этокси и метилендиокси.
Альтернативно, два необязательных заместителя у одного фрагмента, взятые вместе, могут быть объединены с образованием конденсированного циклического заместителя, присоединенного к фрагменту, который является необязательно замещенным. Соответственно, термин «необязательно замещенный» включает конденсированное кольцо, такое как циклоалкильное кольцо, гетероциклоалкильное кольцо, арильное кольцо или гетероарильное кольцо.
Кроме соединений формулы I, описанные варианты реализации относятся также к фармацевтически приемлемым солям, фармацевтически приемлемым N-оксидам, фармацевтически приемлемым пролекарствам и фармацевтически активным метаболитам таких соединений, а также к фармацевтически приемлемым солям таких метаболитов.
Настоящее изобретение относится также к фармацевтическим композициям, содержащим соединение настоящего изобретения и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество.
В дополнительном аспекте настоящего изобретения представлен способ предупреждения или лечения состояния у млекопитающего, включающий введение эффективного количества соединения настоящего изобретения. В одном варианте реализации состояние представляет собой состояние, которое можно лечить ингибированием 11β-HSD1.
В дополнительном аспекте настоящего изобретения представлено применение соединения настоящего изобретения для получения лекарственного средства для лечения состояния у млекопитающего. В одном варианте реализации состояние представляет собой состояние, которое можно лечить ингибированием 11β-HSD1.
В дополнительном аспекте настоящего изобретения представлено применение соединения настоящего изобретения при лечении состояния у млекопитающего. В одном варианте реализации состояние представляет собой состояние, которое можно лечить ингибированием 11β-HSD1.
В некоторых вариантах реализации состояние выбрано из группы, состоящей из диабета, гипергликемии, низкой переносимости глюкозы, гиперинсулинемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии, дислипидемии, ожирения, абдоминального ожирения, глаукомы, гипертонии, атеросклероза и его последствий, ретинопатии и других глазных расстройств, нефропатии, невропатии, миопатии, остеопороза, остеоартрита, деменции, депрессии, нейродегенеративного заболевания, психиатрических расстройств, синдрома поликистозных яичников, бесплодия, болезни Кушинга, синдрома Кушинга, вирусных заболеваний и воспалительных заболеваний.
В некоторых вариантах реализации состояние представляет собой диабет. В некоторых вариантах реализации состояние представляет собой диабет II типа.
В некоторых вариантах реализации соединение вводят в комбинации с адъювантом. В некоторых вариантах реализации адъювант выбран из группы, состоящей из ингибиторов дипептидил-пептидазы-IV (DP-IV); (b) инсулин-сенсибилизирующих агентов; (с) инсулина и инсулиномиметиков; (d) сульфонилмочевин и других стимуляторов секреции инсулина; (е) ингибиторов альфа-глюкозидазы; (f) GLP-1, аналогов GLP-1 и агонистов рецептора GLP-1; и их комбинаций.
В одном из других вариантов реализации соединение вводят в качестве замены для монотерапии или комплексной терапии, в случае неудачного лечения агентом, выбранным из группы, состоящей из ингибиторов дипептидил-пептидазы-IV (DP-IV); (b) инсулин-сенсибилизирующих агентов; (с) инсулина и инсулиномиметиков; (d) сульфонилмочевин и других стимуляторов секреции инсулина; (е) ингибиторов альфа-глюкозидазы; (f) GLP-1, аналогов GLP-1 и агонистов рецептора GLP-1; и их комбинаций.
В одном варианте реализации инсулин-сенсибилизирующий агент выбран из группы, состоящей из (i) PRAR-гамма-агонистов, (ii) PRAR-альфа-агонистов, (iii) PRAR-альфа/гамма двойных агонистов, (iv) бигуанидов и их комбинаций.
Эти и другие указания настоящего изобретения представлены в настоящем документе.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В этом описании используется ряд терминов, которые хорошо известны специалистам в данной области. Тем не мене, для ясности представлено определение ряда терминов.
Используемый в настоящем документе термин «незамещенный» означает, что не существует заместителя, или что единственные заместители представляют собой водород.
Используемый в настоящем документе термин «необязательно замещенный» обозначает, что группа может быть или не быть дополнительно замещена или конденсирована (с образованием конденсированной полициклической системы) с одной или более неводородными группами заместителей. В некоторых вариантах реализации группы заместителей представляют собой одну или более групп, независимо выбранных из группы, состоящей из галогена, =O, =S, -CN, -NO2, -CF3, - OCF3, алкила, алкенила, алкинила, галоалкила, галоалкенила, галоалкинила, гетероалкила, циклоалкила, циклоалкенила, гетероциклоалкила, гетероциклоалкенила, арила, гетероарила, циклоалкилалкила, гетероциклоалкилалкила, гетероарилалкила, арилалкила, циклоалкилалкенила, гетероциклоалкилалкенила, арилалкенила, гетероарилалкенила, циклоалкилгетероалкила, гетероциклоалкилгетероалкила, арилгетероалкила, гетероарилгетероалкила, гидрокси, гидроксиалкила, алкилокси, алкилоксиалкила, алкилоксициклоалкила, алкилоксигетероциклоалкила, алкилоксиарила, алкилоксигетероарила, алкилоксикарбонила, алкиламинокарбонила, алкенилокси, алкинилокси, циклоалкилокси, циклоалкенилокси, гетероциклоалкилокси, гетероциклоалкенилокси, арилокси, фенокси, бензилокси, гетероарилокси, арилалкилокси, амино, алкиламино, ациламино, аминоалкила, ариламино, сульфониламино, сульфиниламино, сульфонила, алкилсульфонила, арилсульфонила, аминосульфонила, сульфинила, алкилсульфинила, арилсульфинила, аминосульфиниламиноалкила, -С(=O)ОН, -C(=O)Re, -C(=O)ORe, C(=O)NReRf, C(=NOH)Re, C(=NRe)NRfRg, NReC(=O)Rf, NReC(=O)ORf, NReC(=O)NRfRg, NReC(=NRf)NRgRh, NReSO2Rf, -SRe, SO2NReRf, -ORe, OC(=O)NReRf, OC(=O)Re и ацила,
где Re, Rf, Rg и Rh, каждый независимо, выбраны из группы, состоящей из Н, C1-С12алкила, C1-C12алоалкила, С2-С12алкенила, С2-С12алкинила, C1-C10гетероалкила, С3-С12циклоалкила, С3-С12циклоалкенила, C1-C12гетероциклоалкила, C1-С18гетероциклоалкенила, С6-C18арила, C1-C18гетероарила и ацила, или любые два или более из Ra, Rb, Rc и Rd, взятые вместе с атомами, к которым они присоединены, образуют гетероциклическую кольцевую систему с 3-12 кольцевыми атомами.
В некоторых вариантах реализации каждый необязательный заместитель независимо выбран из группы, состоящей из: галогена, =O, =S, -CN, -NO2, -CF3, -OCF3, алкила, алкенила, алкинила, галоалкила, галоалкенила, галоалкинила, гетероалкила, циклоалкила, циклоалкенила, гетероциклоалкила, гетероциклоалкенила, арила, гетероарила, гидрокси, гидроксиалкила, алкилокси, алкилоксиалкила, алкилоксиарила, алкилоксигетероарила, алкенилокси, алкинилокси, циклоалкилокси, циклоалкенилокси, гетероциклоалкилокси, гетероциклоалкенилокси, арилокси, гетероарилокси, арилалкила, гетероарилалкила, арилалкилокси, амино, алкиламино, ациламино, аминоалкила, ариламино, сульфонила, алкилсульфонила, арилсульфонила, аминосульфонила, аминоалкила, -СООН, -SH и ацила.
Примеры особенно подходящих необязательных заместителей включают F, Cl, Br, I, СН3, СН2СН3, ОН, OCH3, CF3, OCF3, NO2, NH2 и CN.
В определениях ряда заместителей, представленных ниже, указано, что «группа может быть концевой группой или мостиковой группой». Это предназначено для обозначения, что использование термина предусмотрено для включения ситуации, в которой группа представляет собой линкер между двумя другими частями молекулы, а также случая, когда она представляет собой концевой фрагмент. Используя термин «алкил» в качестве примера, в некоторых публикациях применяется термин «алкилен» для мостиковой группы и, следовательно, в этих других публикациях существует разделение между терминами «алкил» (концевая группа) и «алкилен» (мостиковая группа). В настоящей заявке такое разделение не делается, и большинство групп могут быть как мостиковыми группами, так и концевыми группами.
«Ацил» означает группу R-C(=O)-, в которой группа R может быть алкилом, циклоалкилом, гетероциклоалкилом, арильной или гетероарильной группой, как описано в настоящем документе. Примеры ацила включают ацетил и бензоил. Эта группа может быть концевой группой или мостиковой группой. Если группа представляет собой концевую группу, то она связана с остальной частью молекулы через карбонильный атом углерода.
«Ациламино» означает группу R-C(=O)-NH-, в которой группа R может быть алкилом, циклоалкилом, гетероциклоалкилом, арильной или гетероарильной группой, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом азота.
«Алкенил» как группа или часть группы означает алифатическую углеводородную группу, содержащую по меньшей мере одну двойную углерод-углеродную связь и которая может быть прямой или разветвленной, предпочтительно содержащей 2-12 углеродных атомов, более предпочтительно 2-10 углеродных атомов, наиболее предпочтительно 2-6 углеродных атомов, в нормальной цепи. Эта группа может содержать множество двойных связей в нормальной цепи, а ориентация вокруг каждой из них независимо представляет собой Е или Z. Алкенильная группа предпочтительно представляет собой 1-алкенильную группу. Иллюстративные алкенильные группы включают, но не ограничиваясь этим, этенил, пропенил, бутенил, пентенил, гексенил, гептенил, октенил и ноненил. Эта группа может быть концевой группой или мостиковой группой.
«Алкенилокси» относится к группе алкенил-О-, в которой алкенил является таким, как описано в настоящем документе. Предпочтительные алкенилокси-группы представляют собой C1-С6алкенилокси-группы. Эта группа может быть концевой группой или мостиковой группой. Если группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом кислорода.
«Алкил» как группа или часть группы относится к прямой или разветвленной алифатической углеводородной группе, предпочтительно C1-C12алкилу, более предпочтительно C1-C10алкилу, наиболее предпочтительно C1-С6, если не указано иное. Примеры подходящих прямых или разветвленных C1-С6алкильных заместителей включают метил, этил, н-пропил, 2-пропил, н-бутил, втор-бутил, трет-бутил, гексил и тому подобные. Эта группа может быть концевой группой или мостиковой группой.
«Алкиламино» включает моно-алкиламино и диалкиламино, если не указано иное. «Моно-алкиламино» означает группу алкил-NH-, в которой алкил является таким, как описано в настоящем документе. «Диалкиламино» означает группу (алкил)2N-, в которой каждый алкил может быть таким же или другим, и каждый является таким, как описано в настоящем документе для алкила. Алкильная группа предпочтительно представляет собой C1-С6алкильную группу. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом азота.
«Алкиламинокарбонил» относится к группе формулы (алкил)х(Н)yNC(=O)-, в которой алкил является таким, как описано в настоящем документе, х равен 1 или 2, а сумма X+Y=2. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через карбонильный атом углерода.
«Алкилокси» относится к группе алкил-О-, в которой алкил является таким, как описано в настоящем документе. Предпочтительно, алкилокси представляет собой C1-С6алкилокси. Примеры включают, но не ограничиваясь этим, метокси и этокси. Эта группа может быть концевой группой или мостиковой группой.
«Алкилоксиалкил» относится к алкилокси-алкильной группе, в которой алкилокси и алкильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через алкильную группу.
«Алкилоксиарил» относится к алкилокси-арильной группе, в которой алкилокси и арильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через арильную группу.
«Алкилоксикарбонил» относится к группе алкил-O-С(=O)-, в которой алкил является таким, как описано в настоящем документе. Алкильная группа предпочтительно представляет собой С1-С6алкильную группу. Примеры включают, но не ограничиваясь этим, метоксикарбонил и этоксикарбонил. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через карбонильный атом углерода.
«Алкилоксициклоалкил» относится к алкилокси-циклоалкильной группе, в которой алкилокси и циклоалкильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через циклоалкильную группу.
«Алкилоксигетероарил» относится к алкилокси-гетероарильной группе, в которой алкилокси и гетероарильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через гетероарильную группу.
«Алкилоксигетероциклоалкил» относится к алкилокси-гетероциклоалкильной группе, в которой алкилокси и гетероциклоалкильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через гетероциклоалкильную группу.
«Алкилсульфинил» означает группу алкил-S-(=O)-, в которой алкил является таким, как описано в настоящем документе. Алкильная группа предпочтительно представляет собой C1-С6алкильную группу. Иллюстративные алкилсульфинильные группы включают, но не ограничиваясь этим, метилсульфинил и этилсульфинил. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом серы.
«Алкилсульфонил» относится к группе алкил-S(=O)2-, в которой алкил является таким, как описано выше. Алкильная группа предпочтительно представляет собой C1-C6алкильную группу. Примеры включают, но не ограничиваясь этим, метилсульфонил и этилсульфонил. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом серы.
«Алкинил» как группа или часть группы означает алифатическую углеводородную группу, содержащую тройную углерод-углеродную связь и которая может быть прямой или разветвленной, предпочтительно содержащей от 2 до 12 углеродных атомов, более предпочтительно 2-10 углеродных атомов, более предпочтительно 2-6 углеродных атомов в нормальной цепи. Иллюстративные структуры включают, но не ограничиваясь этим, этинил и пропинил. Эта группа может быть концевой группой или мостиковой группой.
«Алкинилокси» относится к группе алкинил-О-, в которой алкинил является таким, как описано в настоящем документе. Предпочтительные алкинилокси-группы представляют собой С1-С6алкинилокси-группы. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом кислорода.
«Аминоалкил» означает группу NH2-алкил-, в которой алкильная группа является такой, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через алкильную группу.
«Аминосульфонил» означает группу NH2-S(=O)2-. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом серы.
«Арил» как группа или часть группы означает (i) необязательно замещенный моноциклический или конденсированный полициклический карбоцикл (кольцевая структура, содержащая в качестве кольцевых атомов только атомы углерода), предпочтительно содержащий от 5 до 12 атомов в кольце. Примеры арильных групп включают фенил, нафтил и тому подобные; (ii) необязательно замещенный частично насыщенный бициклический ароматический карбоциклический фрагмент, в котором фенил и С5-7циклоалкильная или С5-7циклоалкенильная группа конденсированы вместе с образованием циклической структуры, такой как тетрагидронафтил, инденил или инданил. Эта группа может быть концевой группой или мостиковой группой. Как правило, арильная группа представляет собой С6-C18арильную группу.
«Арилалкенил» означает арил-алкенильную группу, в которой арил и алкенил являются такими, как описано в настоящем документе. Иллюстративные арилалкенильные группы включают фенилаллил. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через алкенильную группу.
«Арилалкил» означает арил-алкильную группу, в которой арильный и алкильный фрагменты являются такими, как описано в настоящем документе. Предпочтительные арилалкильные группы содержат C1-5алкильный фрагмент. Иллюстративные арилалкильные группы включают бензил, фенетил, 1-нафталинметил и 2-нафталинметил. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через алкильную группу.
«Арилалкилокси» относится к группе арил-алкил-О-, в которой алкил и арил являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом кислорода.
«Ариламино» включает как моно-ариламино, так и ди-ариламино, если не указано иное. Моно-ариламино означает группу формулы арилNH-, в которой арил является таким, как описано в настоящем документе. Ди-ариламино означает группу формулы (арил)2N-, в которой каждый арил может быть таким же или другим, и каждый является таким, как описано в настоящем документе для арила. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом азота.
«Арилгетероалкил» означает арил-гетероалкильную группу, в которой арильный и гетероалкильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через гетероалкильную группу.
«Арилокси» относится к группе арил-О-, в которой арил является таким, как описано в настоящем документе. Предпочтительно, арилокси представляет собой С6-С18арилокси, более предпочтительно С6-C10арилокси. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом кислорода.
«Арилсульфонил» означает группу арил-S(=O)2-, в которой арильная группа является такой, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом серы.
«Связь» представляет собой связь между атомами в соединении или молекуле. Связь может представлять собой одинарную связь, двойную связь или тройную связь.
«Циклоалкенил» означает неароматическую моноциклическую или полициклическую кольцевую систему, содержащую по меньшей мере одну двойную углерод-углеродную связь и предпочтительно имеющую от 5 до 10 углеродных атомов в кольце. Иллюстративные моноциклические циклоалкенильные кольца включают циклопентенил, циклогексенил или циклогептенил. Циклоалкенильная группа может быть замещена одной или более группами заместителей. Циклоалкенильная группа, как правило, представляет собой С3-C12алкенильную группу. Эта группа может быть концевой группой или мостиковой группой.
«Циклоалкил» относится к насыщенному моноциклическому или конденсированному, или спиро-полициклическому карбоциклу, предпочтительно содержащему от 3 до 9 углеродных атомов в кольце, такому как циклопропил, циклобутил, циклопентил, циклогексил и тому подобные, если не указано иное. Он включает моноциклические системы, такие как циклопропил и циклогексил, бициклические системы, такие как декалин, и полициклические системы, такие как адамантан. Циклоалкильная группа, как правило, представляет собой С3-C12алкильную группу. Эта группа может быть концевой группой или мостиковой группой.
«Циклоалкилалкил» означает циклоалкил-алкильную группу, в которой циклоалкильный и алкильный фрагменты являются такими, как описано в настоящем документе. Иллюстративные моноциклоалкилалкильные группы включают циклопропилметил, циклопентилметил, циклогексилметил и циклогептилметил. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через алкильную группу.
«Циклоалкилалкенил» означает циклоалкил-алкенильную группу, в которой циклоалкильный и алкенильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через алкенильную группу.
«Циклоалкилгетероалкил» означает циклоалкил-гетероалкильную группу, в которой циклоалкильный и гетероалкильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через гетероалкильную группу.
«Циклоалкилокси» означает группу циклоалкил-О-, в которой циклоалкил является таким, как описано в настоящем документе. Предпочтительно, циклоалкилокси представляет собой C1-С6циклоалкилокси. Примеры включают, но не ограничиваясь этим, циклопропанокси и циклобутанокси. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом кислорода.
«Циклоалкенилокси» относится к группе циклоалкенил-О-, в которой циклоалкенил является таким, как описано в настоящем документе. Предпочтительно, циклоалкенилокси представляет собой C1-С6циклоалкенилокси. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом кислорода.
Неудача лечения может быть определена как состояние, при котором содержание глюкозы в крове не натощак, составляющее менее 200 мг/дл, и содержание глюкозы в крови натощак (воздержание от еды в течение по меньшей мере 8 часов), составляющее менее 126 мг/дл, сохраняются после введения агента в его рекомендуемой дозе.
«Галоалкил» относится к алкильной группе, описанной в настоящем документе, в которой один или более водородных атомов заменены атомом галогена, выбранным из группы, состоящей из фтора, хлора, брома и йода. Галоалкильная группа, как правило, имеет формулу CnH(2n+1-m)Xm, где каждый X независимо выбран из группы, состоящей из F, О, Br и I. В группах этого типа n обычно равен от 1 до 10, более предпочтительно от 1 до 6, наиболее предпочтительно от 1 до 3. m обычно равен от 1 до 6, более предпочтительно от 1 до 3. Примеры галоалкила включают фторметил, дифторметил и трифторметил.
«Галоалкенил» относится к алкенильной группе, описанной в настоящем документе, в которой один или более водородных атомов заменены атомом галогена, независимо выбранным из группы, состоящей из F, Cl, Br и I.
«Галоалкинил» относится к алкинильной группе, описанной в настоящем документе, в которой один или более водородных атомов заменены атомом галогена, независимо выбранным из группы, состоящей из F, Cl, Br и I.
«Галоген» представляет собой хлор, фтор, бром или йод.
«Гетероалкил» относится к прямой или разветвленной алкильной группе, предпочтительно имеющей от 2 до 12 углеродных атомов, более предпочтительно от 2 до 6 углеродных атомов в цепи, в которой один или более углеродных атомов (и все сопряженные водородные атомы), каждый независимо, заменены гетероатомной группой, выбранной из S, О, Р и NR', где R' выбран из группы, состоящей из Н, необязательно замещенного C1-C12алкила, необязательно замещенного С3-C12циклоалкила, необязательно замещенного С6-C18арила и необязательно замещенного C1-C18гетероарила. Иллюстративные гетероалкилы включают алкильные эфиры, вторичные и третичные алкиламины, амиды, алкилсульфиды и тому подобные. Примеры гетероалкила включают также гидроксиC1-С6алкил, C1-С6алкилоксиС1-С6алкил, аминоC1-С6алкил, C1-С6алкиламиноС1-C6алкил и ди(С1-C6алкил)аминоС1-С6алкил. Эта группа может быть концевой группой или мостиковой группой.
«Гетероалкилокси» относится к группе гетероалкил-О-, в которой гетероалкил является таким, как описано в настоящем документе. Предпочтительно, гетероалкилокси представляет собой С2-C6гетероалкилокси. Эта группа может быть концевой группой или мостиковой группой.
«Гетероарил» отдельно или как часть группы относится к группам, содержащим ароматическое кольцо (предпочтительно, 5- или 6-членное ароматическое кольцо), имеющее один или более гетероатомов в качестве кольцевых атомов в ароматическом кольце, а остальные кольцевые атомы представляют собой углеродные атомы. Подходящие гетероатомы включают азот, кислород и серу. Примеры гетероарила включают тиофен, бензотиофен, бензофуран, бензимидазол, бензоксазол, бензотиазол, бензизотиазол, нафто[2,3-b]тиофен, фуран, изоиндолизин, ксантолен, феноксантин, пиррол, имидазол, пиразол, пиридин, пиразин, пиримидин, пиридазин, тетразол, индол, изоиндол, 1Н-индазол, пурин, хинолин, изохинолин, фталазин, нафтиридин, хиноксалин, циннолин, карбазол, фенантридин, акридин, феназин, тиазол, изотиазол, фенотиазин, оксазол, изооксазол, фуразан, феноксазин, 2-, 3- или 4-пиридил, 2-, 3-, 4-, 5- или 8-хинолил, 1-, 3-, 4- или 5-изохинолил, 1-, 2- или 3-индолил и 2- или 3-тиенил. Гетероарильная группа, как правило, представляет собой C1-C18гетероарильную группу. Эта группа может быть концевой группой или мостиковой группой.
«Гетероарилалкил» означает гетероарил-алкильную группу, в которой гетероарильный и алкильный фрагменты являются такими, как описано в настоящем документе. Предпочтительные гетероарилалкильные группы содержат низший алкильный фрагмент. Иллюстративные гетероарилалкильные группы включают пиридилметил. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через алкильную группу.
«Гетероарилалкенил» означает гетероарил-алкенильную группу, в которой гетероарильный и алкенильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через алкенильную группу.
«Гетероарилгетероалкил» означает гетероарил-гетероалкильную группу, в которой гетероарильный и гетероалкильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через гетероалкильную группу.
«Гетероарилокси» относится к группе гетероарил-О-, в которой гетероарил является таким, как описано в настоящем документе. Предпочтительно, гетероарилокси представляет собой C1-C18гетероарилокси. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом кислорода.
«Гетероциклический» относится к насыщенной, частично ненасыщенной или полностью ненасыщенной моноциклической, бициклической или полициклической кольцевой системе, содержащей по меньшей мере один гетероатом, выбранный из группы, состоящей из азота, серы и кислорода, в качестве кольцевого атома. Примеры гетероциклических фрагментов включают гетероциклоалкил, гетероциклоалкенил и гетероарил.
«Гетероциклоалкенил» относится к гетероциклоалкильной группе, описанной в настоящем документе, но содержащей по меньшей мере одну двойную связь. Гетероциклоалкенильная группа, как правило, представляет собой С2-С12гетероциклоалкенильную группу. Эта группа может быть концевой группой или мостиковой группой.
«Гетероциклоалкил» относится к насыщенному моноциклическому, бициклическому или полициклическому кольцу, содержащему по меньшей мере один гетероатом, выбранный из азота, серы, кислорода, предпочтительно от 1 до 3 гетероатомов по меньшей мере в одном кольце. Каждое кольцо предпочтительно является 3-10-членным, более предпочтительно 4-7-членным. Примеры подходящих гетероциклоалкильных заместителей включают пирролидил, тетрагидрофурил, тетрагидротиофуранил, пиперидил, пиперазил, тетрагидропиранил, морфолино, 1,3-диазапан, 1,4-диазапан, 1,4-оксазепан и 1,4-оксатиапан. Гетероциклоалкильная группа, как правило, представляет собой C1-C12гетероциклоалкильную группу. Эта группа может быть концевой группой или мостиковой группой.
«Гетероциклоалкилалкил» относится к гетероциклоалкил-алкильной группе, в которой гетероциклоалкильный и алкильный фрагменты являются такими, как описано в настоящем документе. Иллюстративные гетероциклоалкилалкильные группы включают (2-тетрагидрофурил)метил, (2-тетрагидротиофуранил)метил. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через алкильную группу.
«Гетероциклоалкилалкенил» относится к гетероциклоалкил-алкенильной группе, в которой гетероциклоалкильный и алкенильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через алкенильную группу.
«Гетероциклоалкилгетероалкил» означает гетероциклоалкил-гетероалкильную группу, в которой гетероциклоалкильный и гетероалкильный фрагменты являются такими, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через гетероалкильную группу.
«Гетероциклоалкилокси» относится к группе гетероциклоалкил-О-, в которой гетероциклоалкил является таким, как описано в настоящем документе. Предпочтительно, гетероциклоалкилокси представляет собой C1-C6гетероциклоалкилокси. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом кислорода.
«Гетероциклоалкенилокси» относится к группе гетероциклоалкенил-О-, в которой гетероциклоалкенил является таким, как описано в настоящем документе. Предпочтительно, гетероциклоалкенилокси представляет собой C1-C6гетероциклоалкенилокси. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом кислорода.
«Гидроксиалкил» относится к алкильной группе, описанной в настоящем документе, в которой один или более водородных атомов заменены группой ОН. Гидроксиалкильная группа, как правило, имеет формулу CnH(2n+1-x)(OH)x. В группах этого типа n, как правило, равен от 1 до 10, более предпочтительно от 1 до 6, наиболее предпочтительно от 1 до 3. х, как правило, равен от 1 до 6, более предпочтительно от 1 до 3.
«Сульфинил» означает группу R-S(=O)-, в которой группа R может быть ОН, алкилом, циклоалкилом, гетероциклоалкилом; арильной или гетероарильной группой, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом серы.
«Сульфиниламино» означает группу R-S(=O)-NH-, в которой группа R может быть ОН, алкилом, циклоалкилом, гетероциклоалкилом; арильной или гетероарильной группой, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом азота.
«Сульфонил» означает группу R-S(=O)2-, в которой группа R может быть ОН, алкилом, циклоалкилом, гетероциклоалкило; арильной или гетероарильной группой, как описано в настоящем документе. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом серы.
«Сульфониламино» означает группу R-S(=O)2-NH-. Эта группа может быть концевой группой или мостиковой группой. Если эта группа представляет собой концевую группу, то она связана с остальной частью молекулы через атом азота.
Следует понимать, что в семейство соединений Формулы (I) включены изомерные формы, в том числе диастереоизомеры, энантиомеры, таутомеры и геометрические изомеры в «Е» или «Z» конфигурации изомера, или смесь Е и Z изомеров. Также следует понимать, что некоторые изомерные формы, такие как диатереомеры, энантиомеры и геометрические изомеры, могут быть разделены физическими и/или химическими способами специалистами в данной области техники. Для тех соединений, в которых существует возможность геометрической изомерии, авторы настоящей заявки изобразили тот изомер, который предположительно представляет собой соединение, хотя подразумевается, что правильное структурное отнесение может представлять собой другой изомер.
Некоторые из соединений в описанных вариантах реализации могут существовать в виде единственных стереоизомеров, рацематов и/или смесей энантиомеров и/или диастереомеров. Все такие одиночные стереоизомеры, рацематы и их смеси подразумеваются входящими в рамки описанного и заявленного объекта изобретения.
Дополнительно, Формула (I) предназначена для охвата, где это применимо, сольватированных и несольватированных форм соединений. Следовательно, каждая формула включает соединения, имеющие указанную структуру, включая гидратированные, а также негидратированные формы.
Термин «фармацевтически приемлемые соли» относится к солям, которые сохраняют заданную биологическую активность описанных выше соединений, и включают фармацевтически приемлемые соли присоединения кислот и соли присоединения оснований. Подходящие фармацевтически приемлемые соли присоединения кислот соединений Формулы (I) могут быть получены из неорганической кислоты или из органической кислоты. Примеры таких неорганических кислот представляют собой хлороводородная, серная и фосфорная кислота. Соответствующие органические кислоты могут быть выбраны из алифатических, циклоалифатических, ароматических, гетероциклических карбоновых и сульфоновых классов органических кислот, примеры который представляют собой муравьиная, уксусная, пропановая, янтарная, гликолевая, глюконовая, молочная, яблочная, винная, лимонная, фумаровая, малеиновая, алкилсульфоновая, арилсульфоновая. Дополнительная информация о фармацевтически приемлемых солях представлена в публикации Remington Pharmaceutical Sciences, 19-ое издание, Mack Publishing Co., Истон, штат Пенсильвания, 1995. В случае твердых агентов специалистам в данной области понятно, что соединения настоящего изобретения, агенты и соли могут существовать в различных кристаллических или полиморфных формах, которые все подразумеваются входящими в рамки настоящего изобретения и определенных формул.
«Пролекарство» означает соединение, которое подвергается превращению в соединение формулы (I) в биологической системе, обычно метаболическими способами (например, гидролизом, восстановлением или окислением). Например, сложноэфирное пролекарство соединения формулы (I), содержащего гидроксильную группу, может быть преобразовано гидролизом in vivo в исходную молекулы. Подходящие сложные эфиры соединений формулы (I), содержащих гидроксильную группу, представляют собой, например, ацетаты, цитраты, лактаты, тартраты, малонаты, оксалаты, салицилаты, пропионаты, сукцинаты, фумараты, малеаты, метилен-бис-β-гидроксинафтоаты, гестисаты, изетионаты, ди-n-толуоилтартраты, метансульфонаты, этансульфонаты, бензолсульфонаты, n-толуолсульфонаты, циклогексилсульфаматы и хинаты. В качестве другого примера, сложноэфирное пролекарство соединения формулы (I), содержащего карбоксильную группу, может быть преобразовано гидролизом in vivo в исходную молекулу. (Примеры сложноэфирных пролекарств представляют собой те, которые описаны в публикации F.J. Leinweber, Drug Metab. Res., 18:379, 1987). Точно так же, ацильное пролекарство соединения формулы (I), содержащего аминогруппу, может быть преобразовано гидролизом in vivo в исходную молекулу (многие примеры пролекарств для этих и других функциональных групп, включая амины, описаны в публикации Prodrugs: Challenges and Rewards (части 1 и 2); ред. V. Stella, R. Borchardt, M. Hageman, R. Oliyai, H. Maag and J. Tilley; Springer, 2007).
Термин «терапевтически эффективное количество» или «эффективное количество» представляет собой количество, достаточное для благоприятного эффекта или заданных клинических результатов. Эффективное количество может быть введено за одно или более введений. Эффективного количества, как правило, достаточно для временного облегчения, улучшения, стабилизации, реверсирования, замедления или отсрочки прогрессирования болезненного состояния.
Конкретные соединения настоящего изобретения включают следующие:




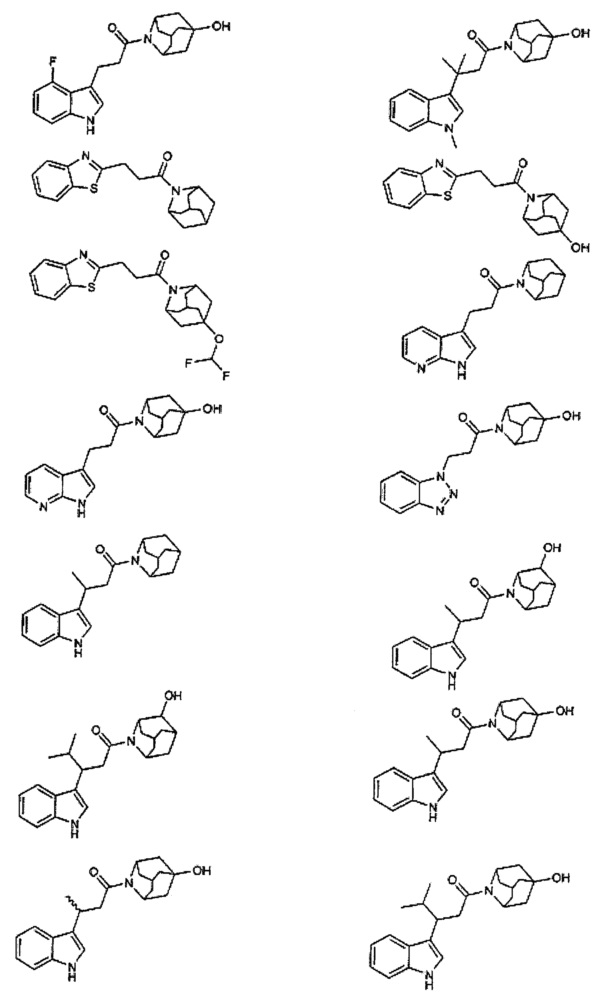



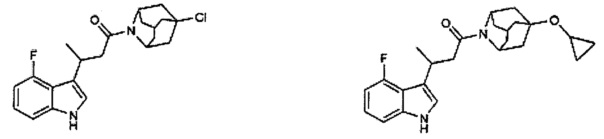

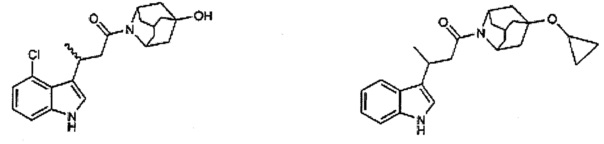
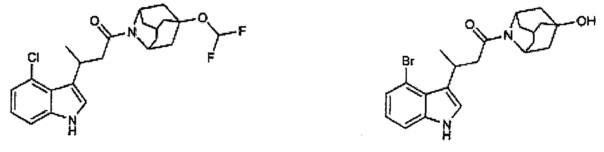

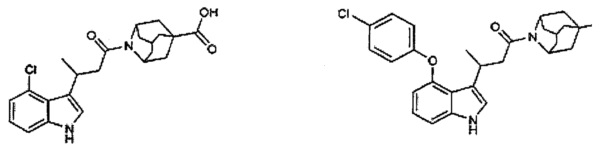
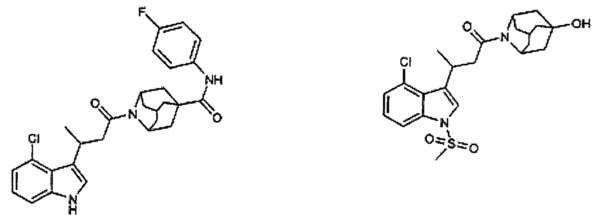


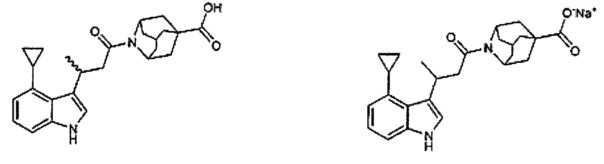
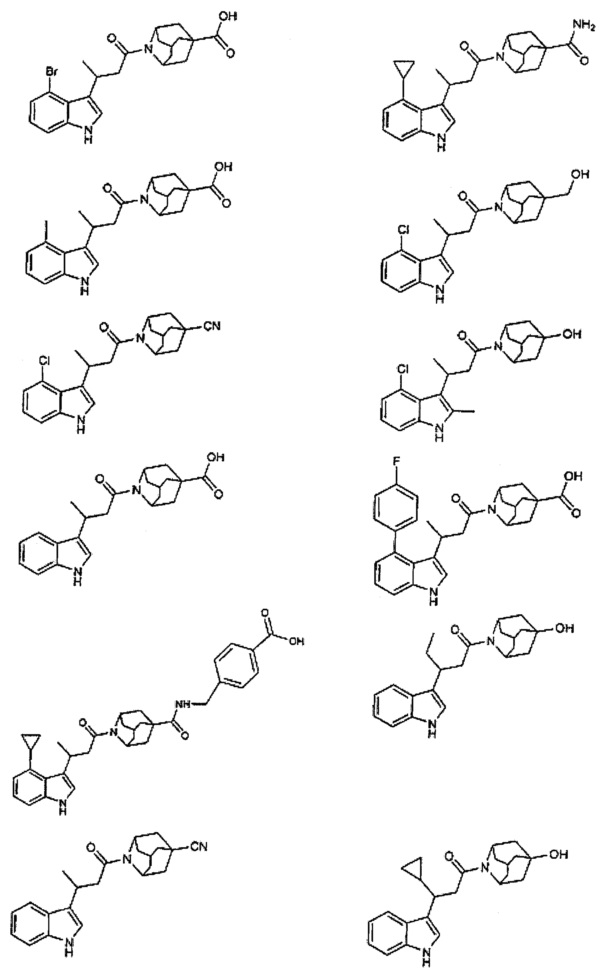
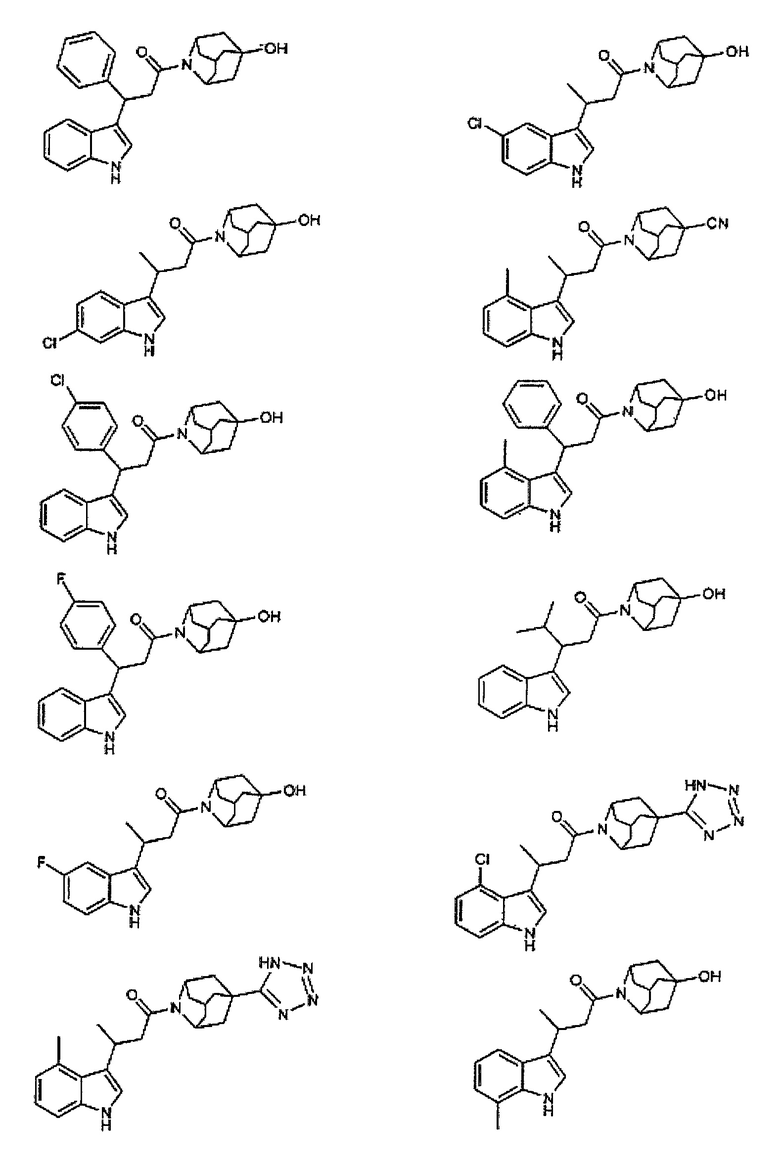
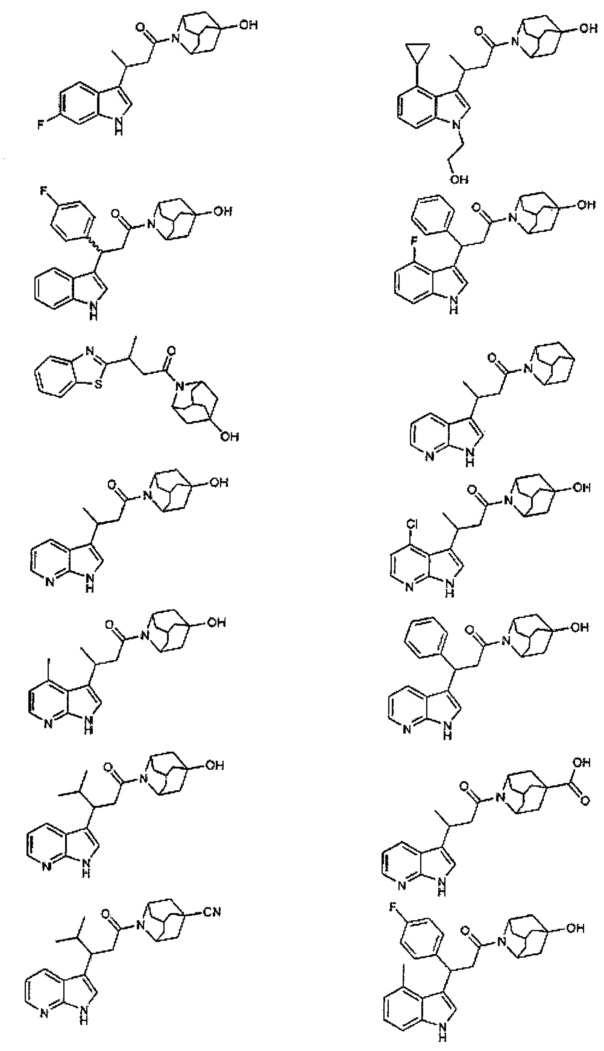
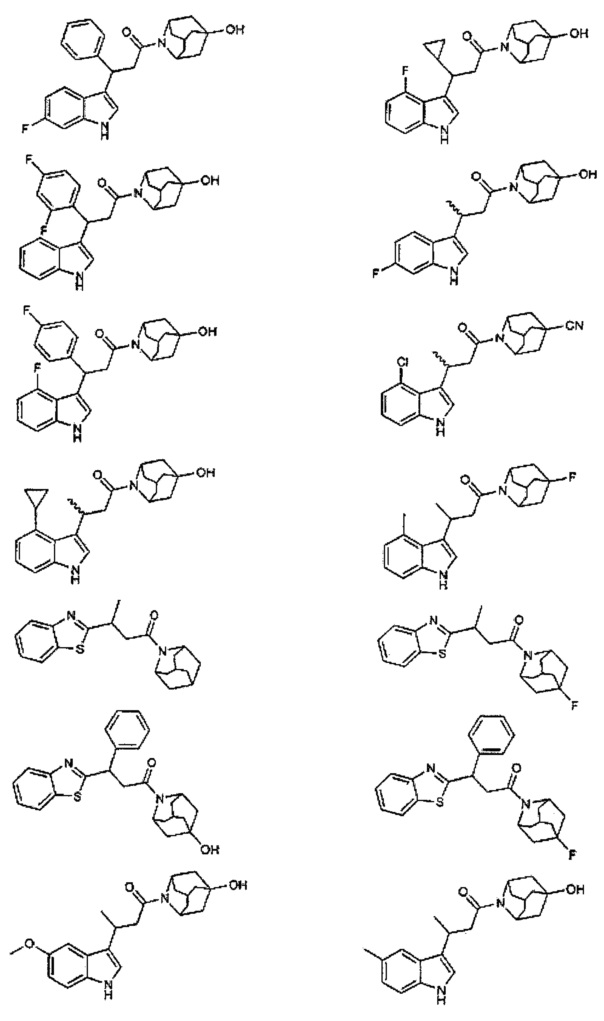
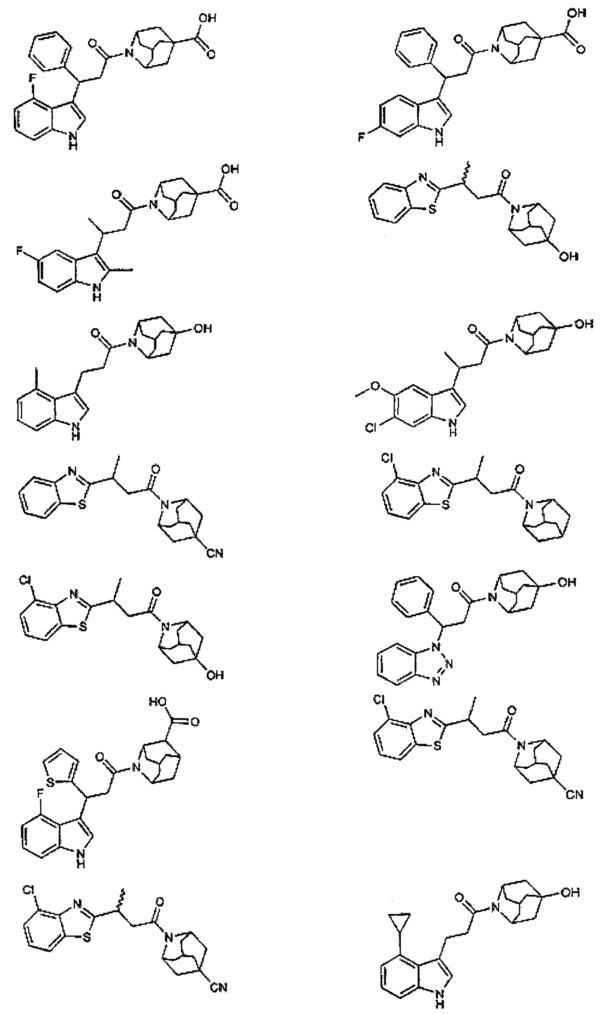
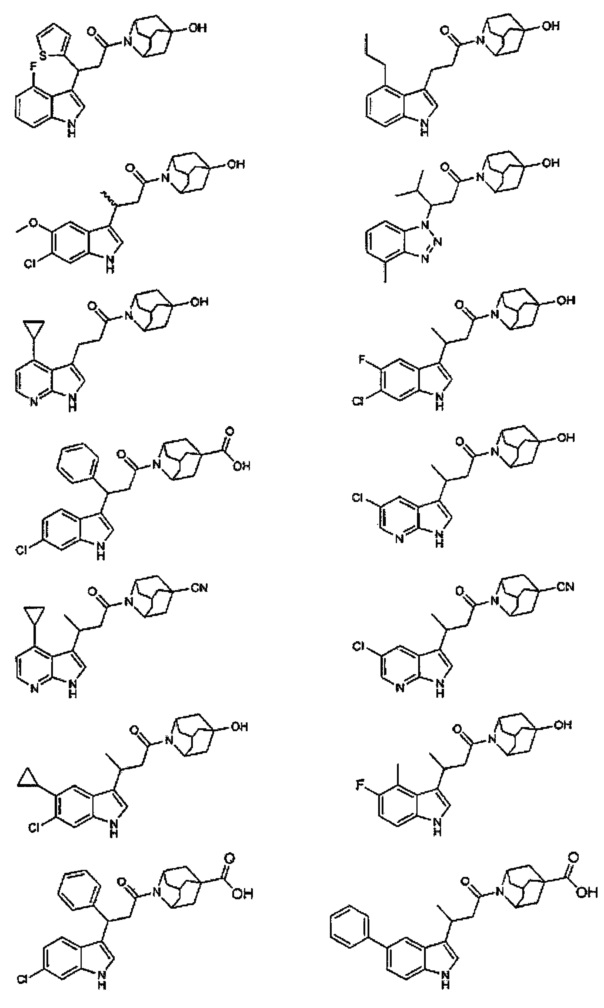
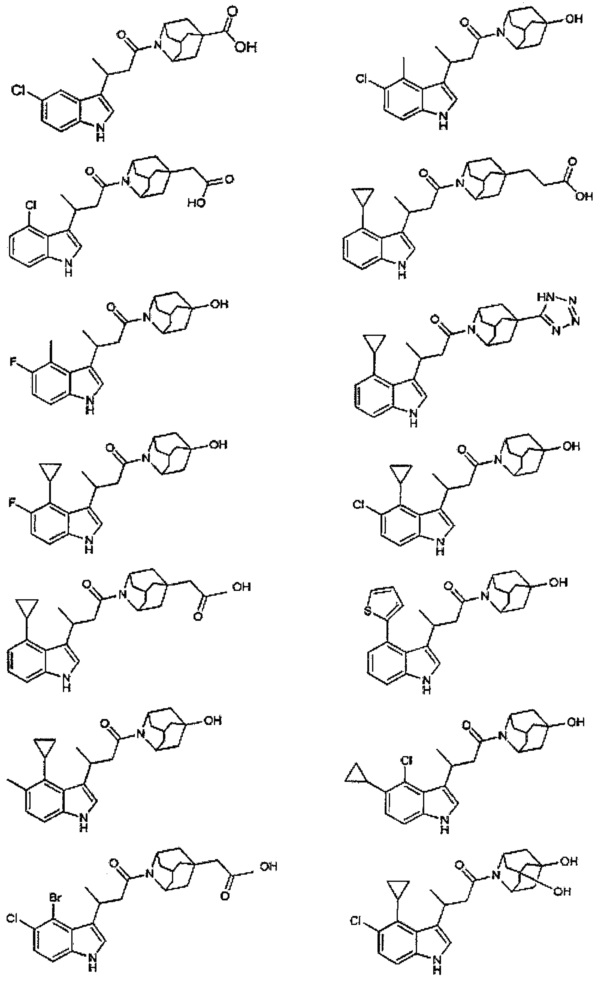
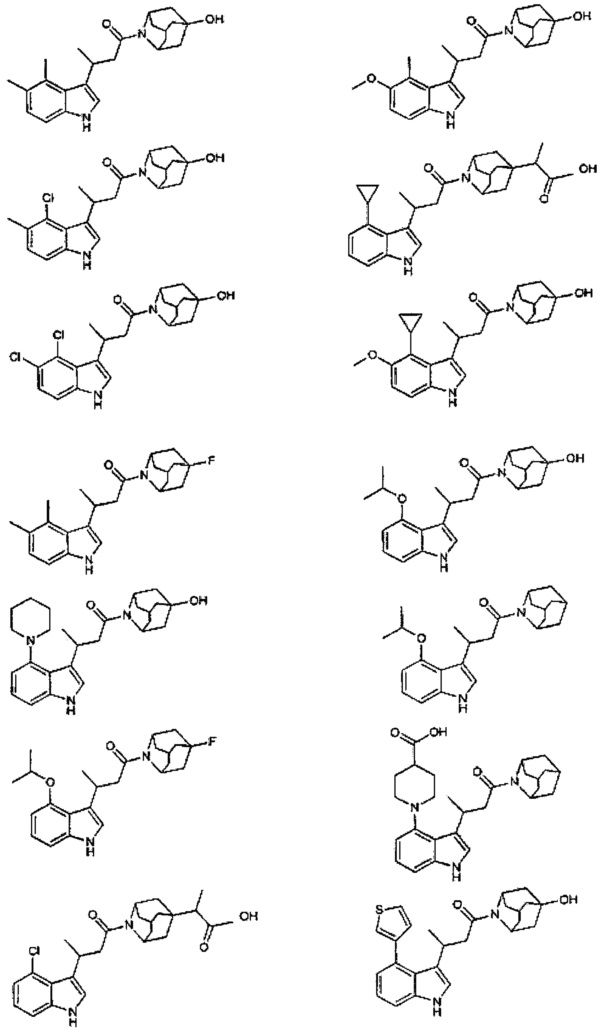
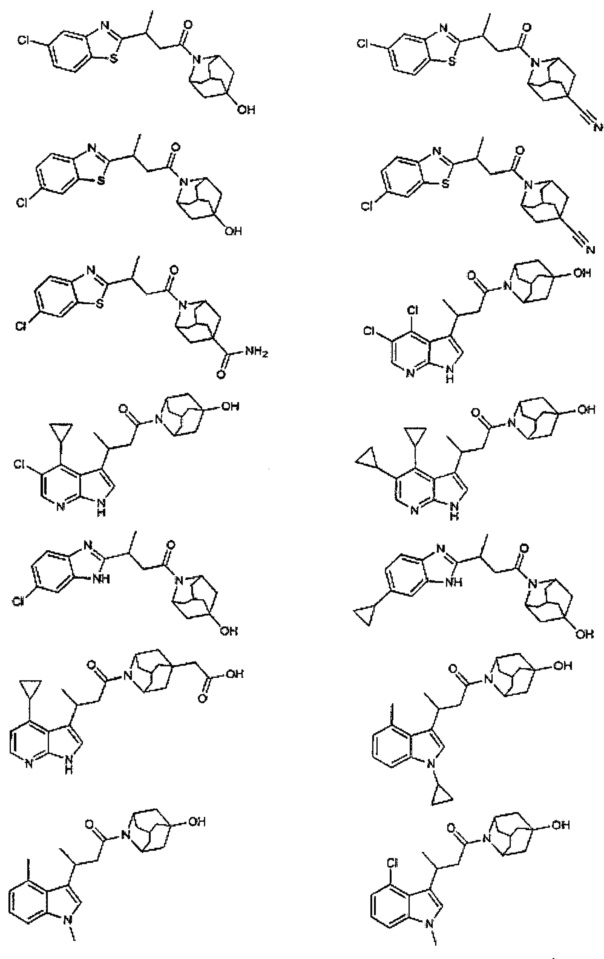
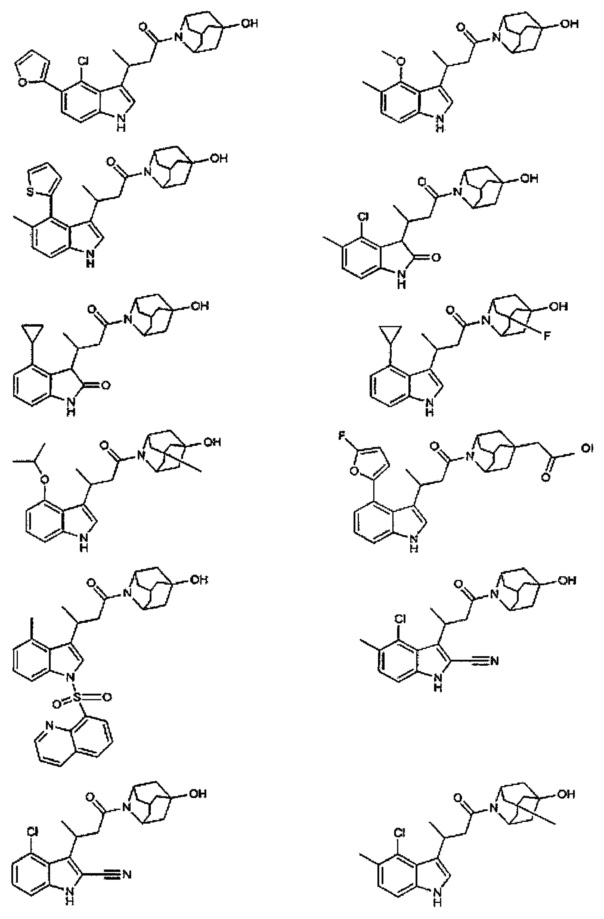
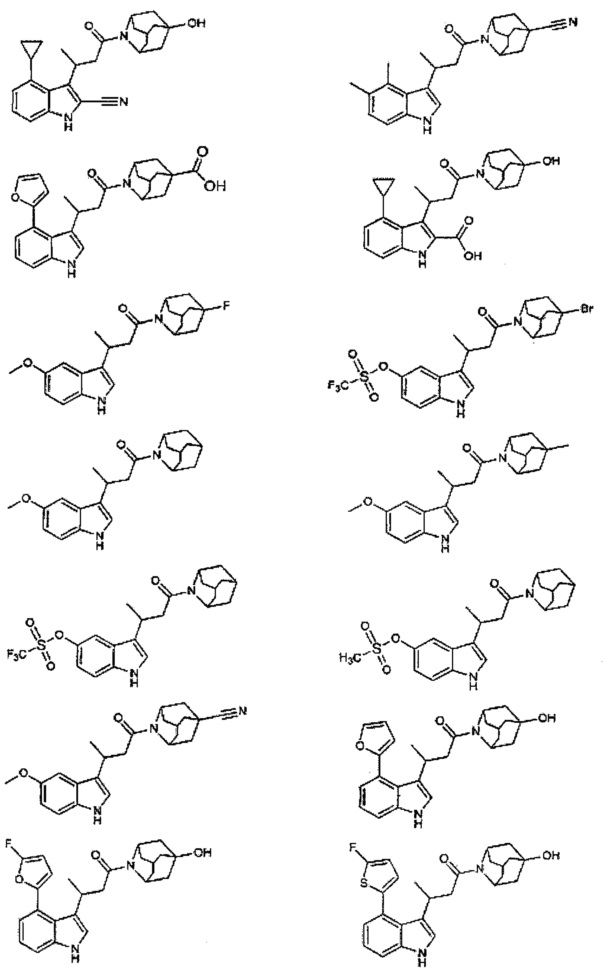
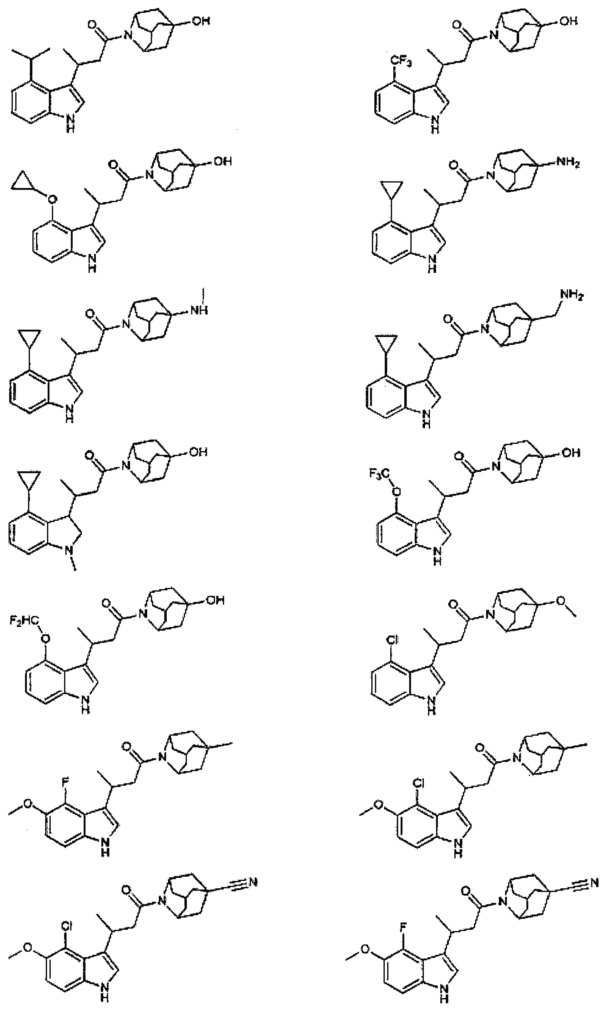
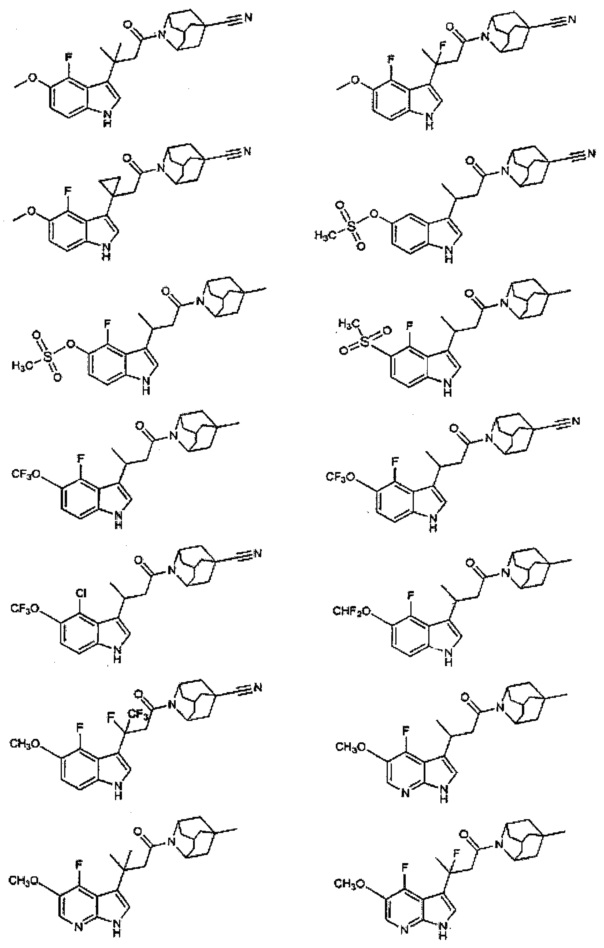
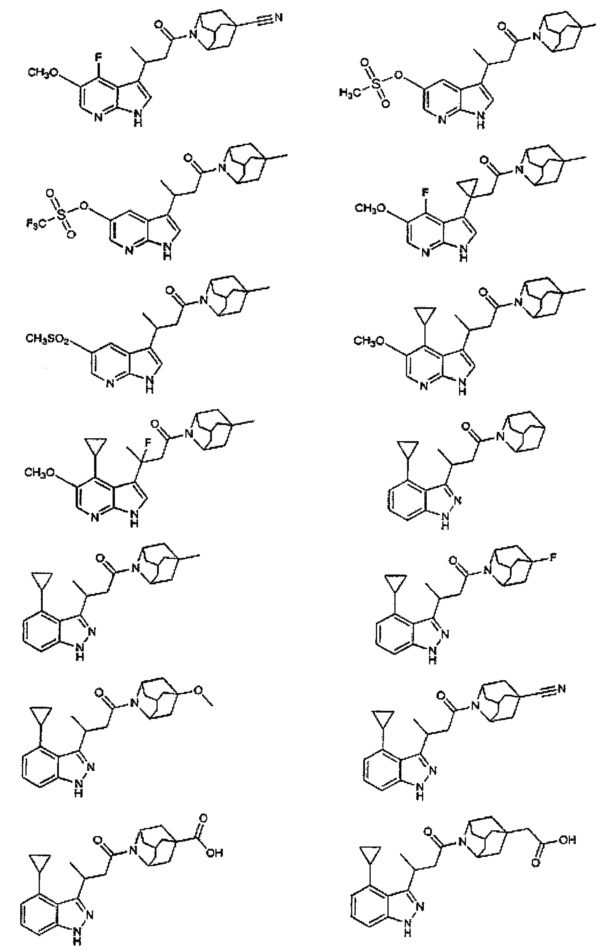
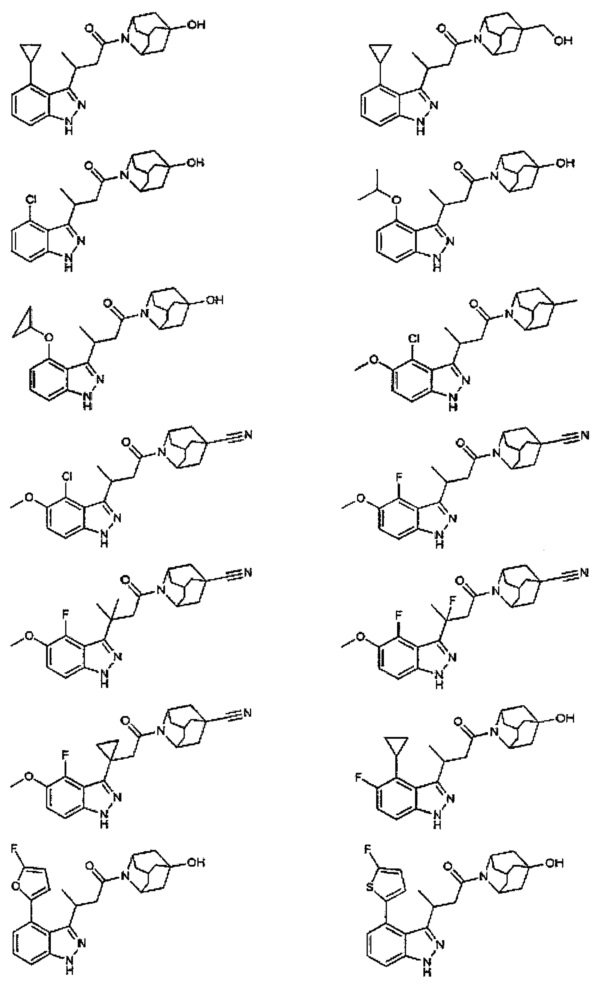
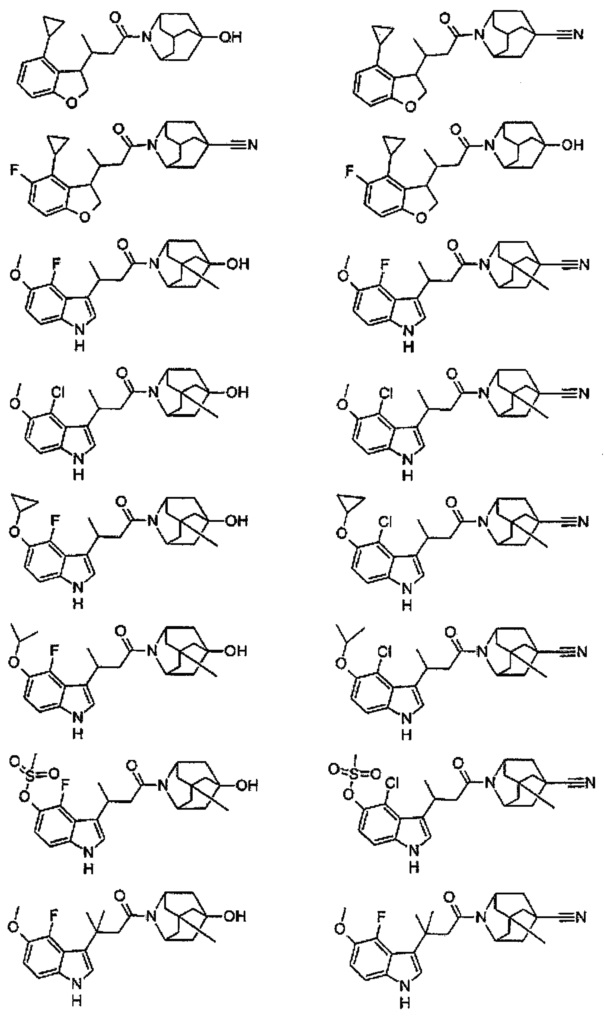
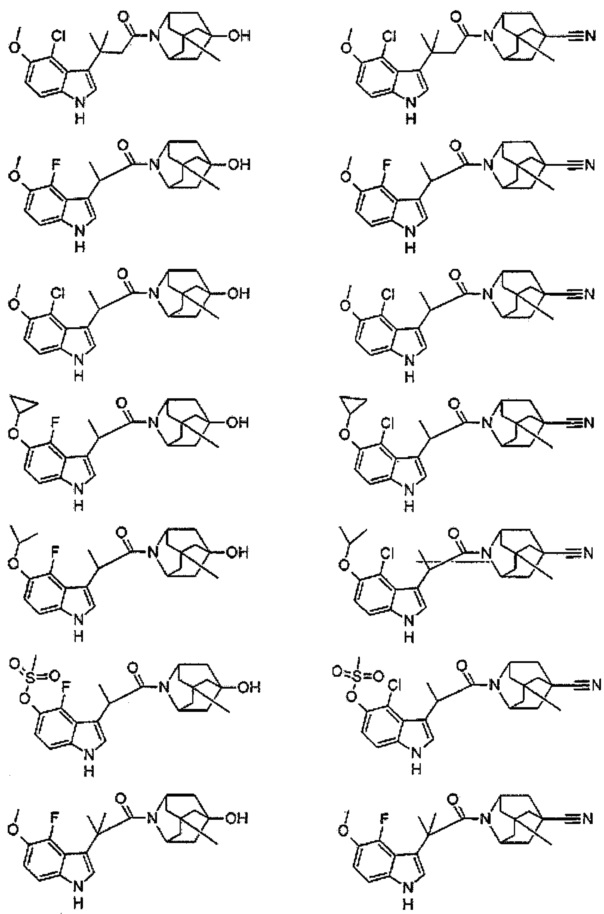
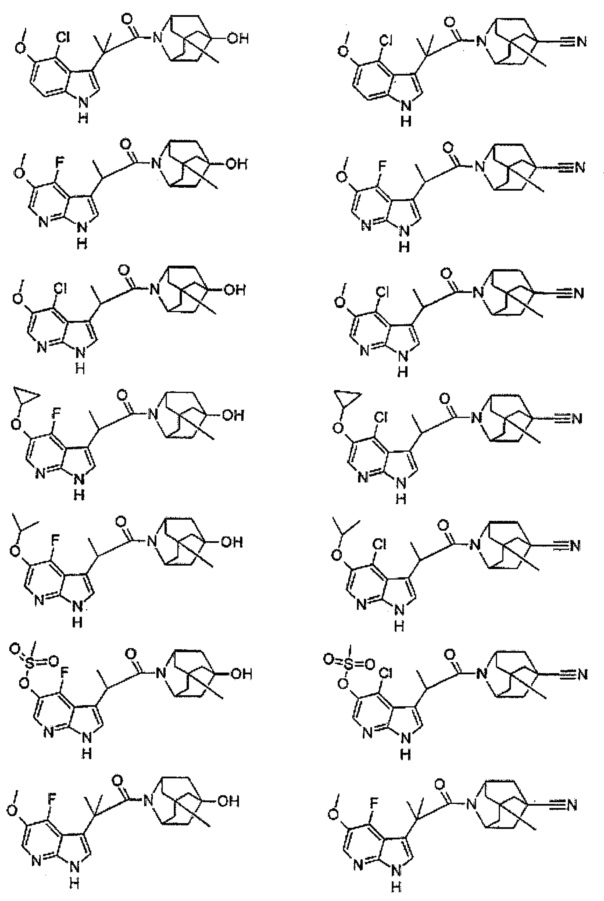
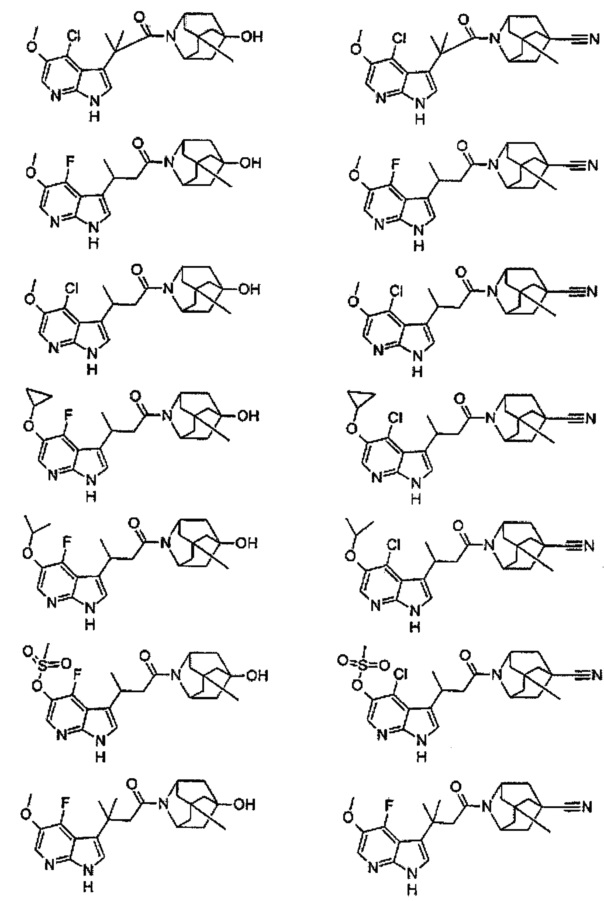
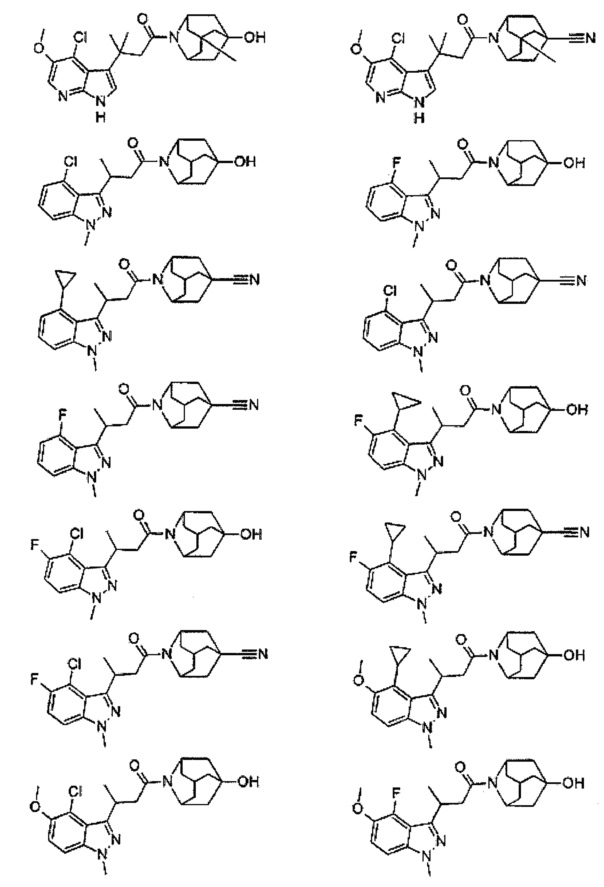
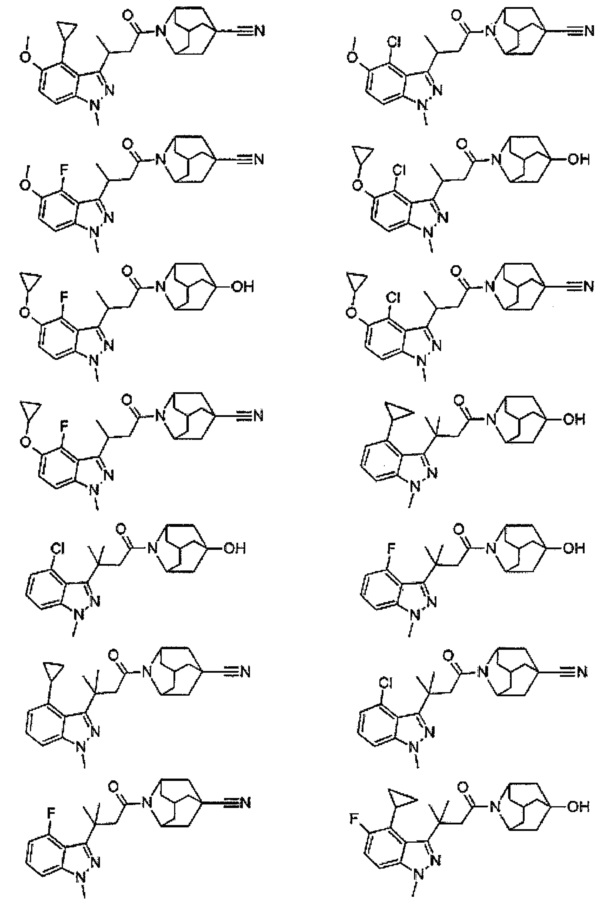
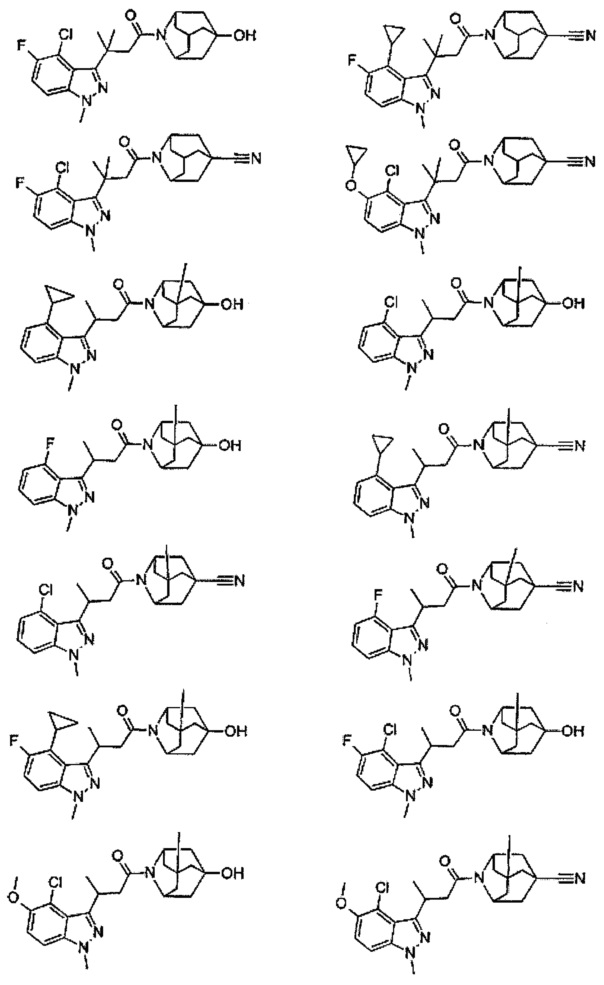
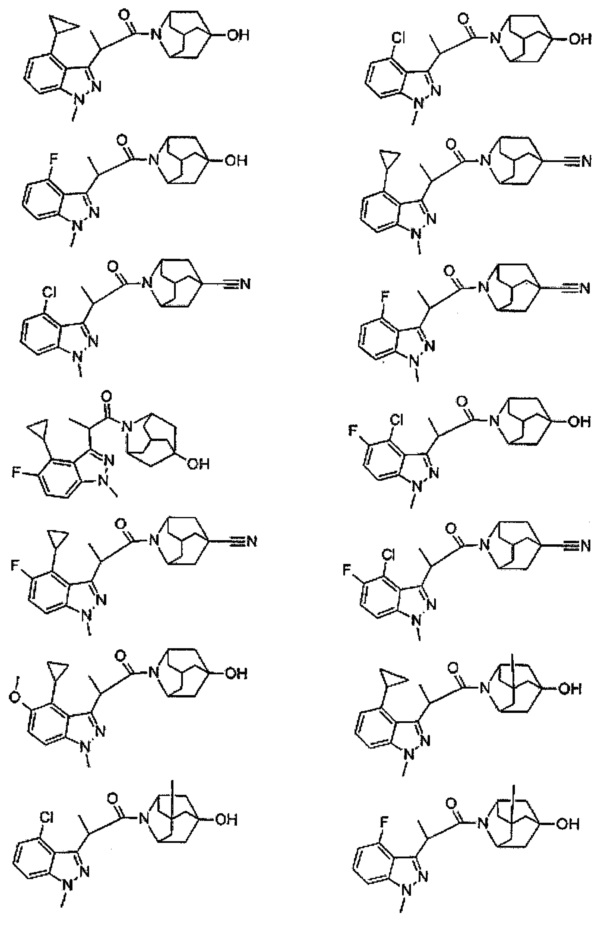
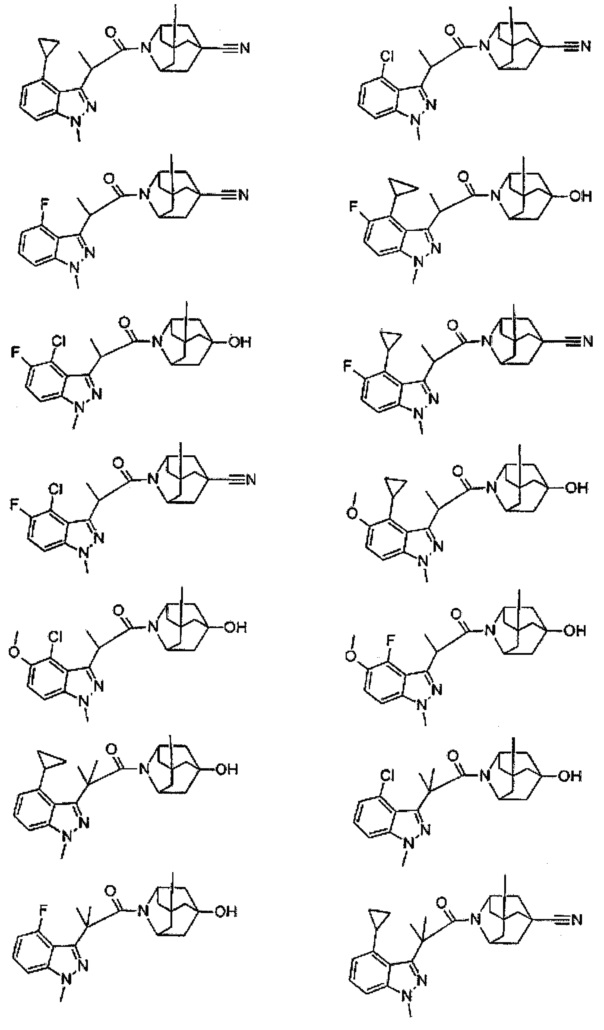
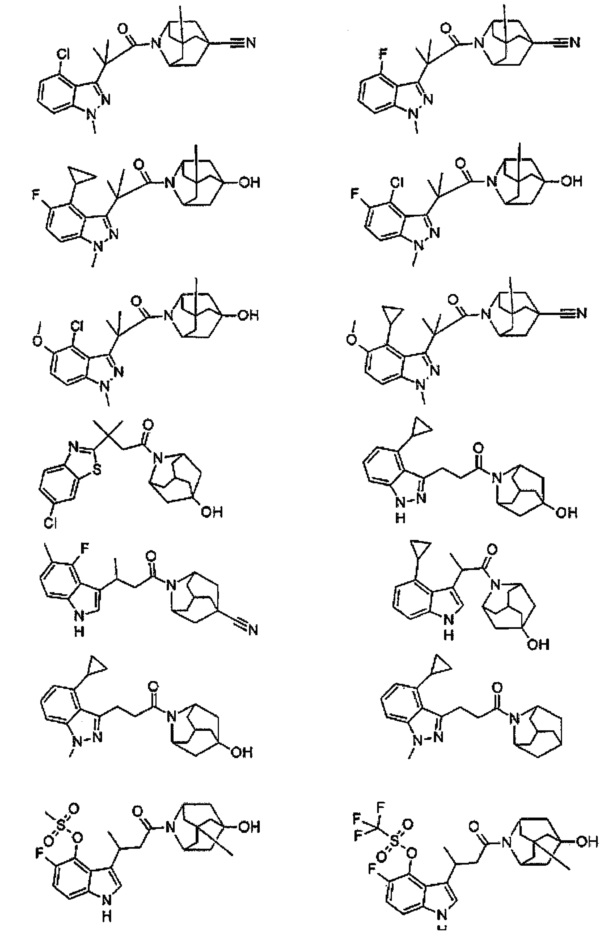
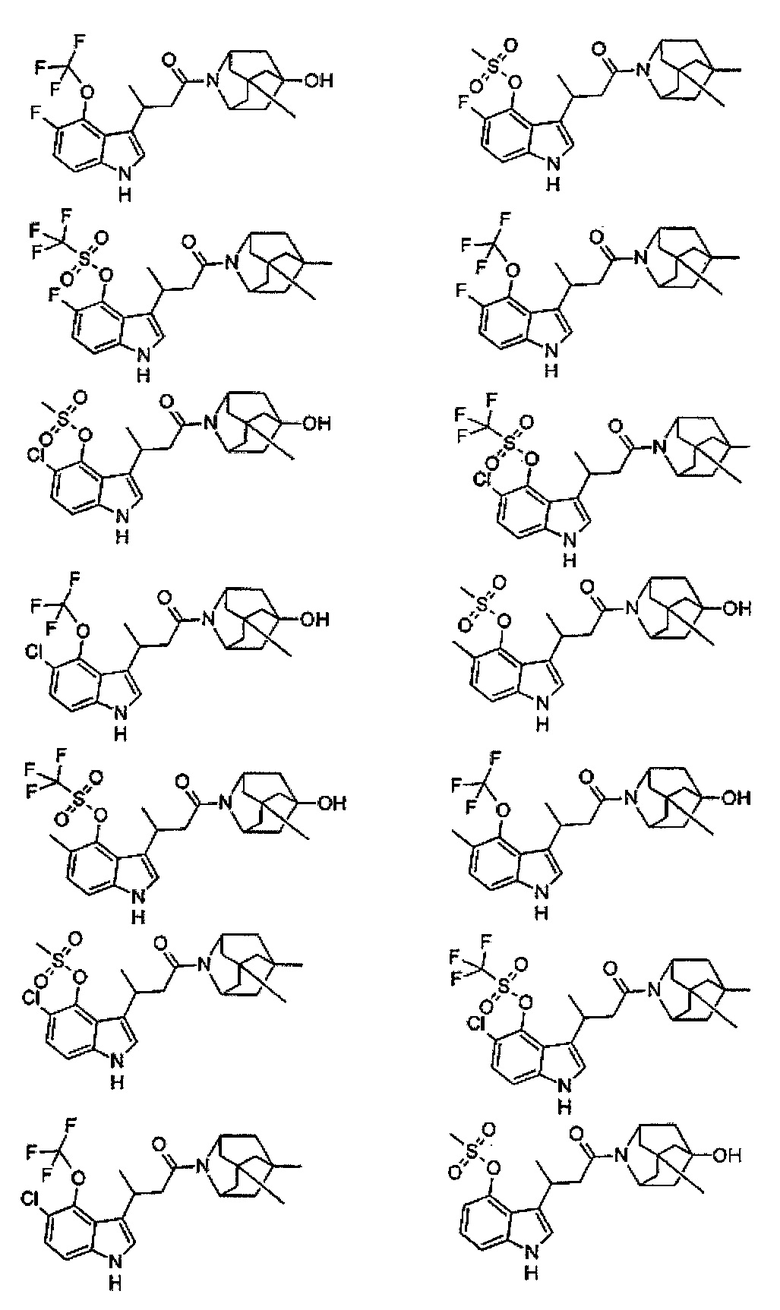
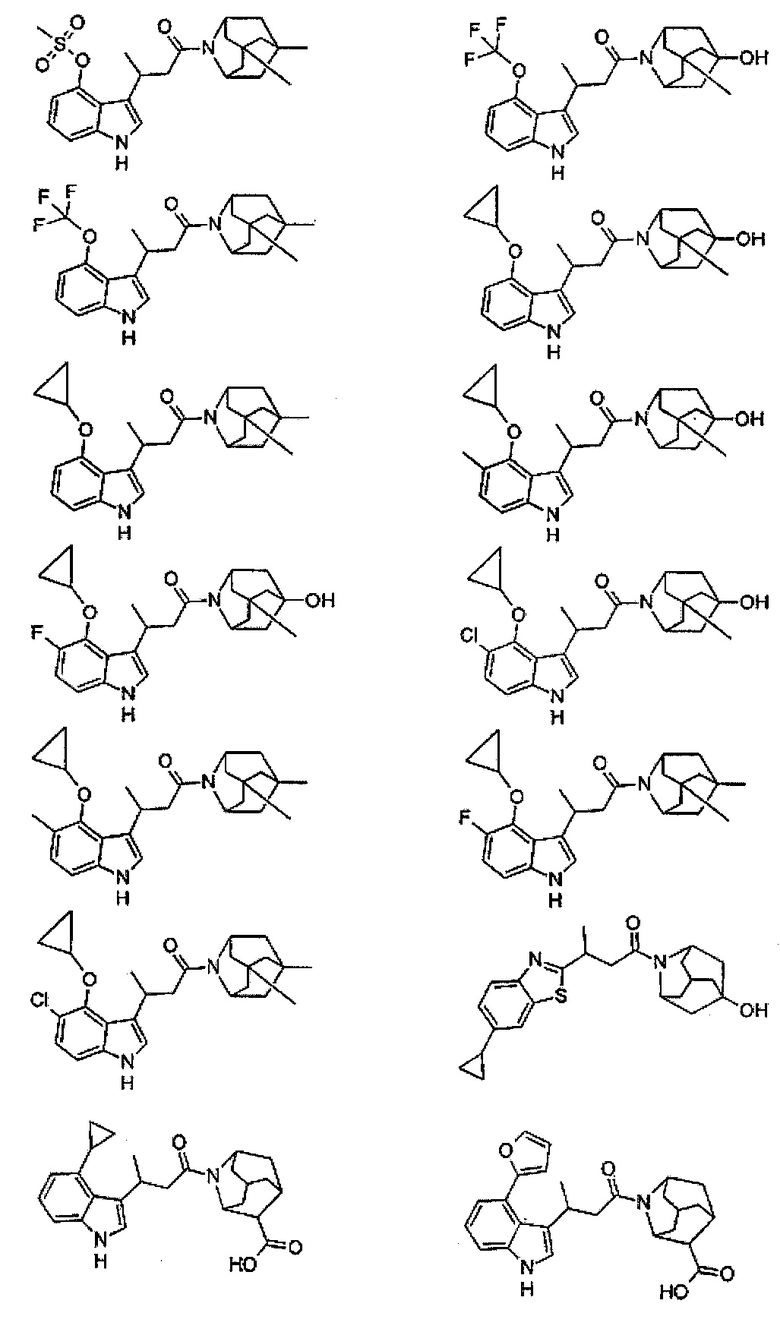
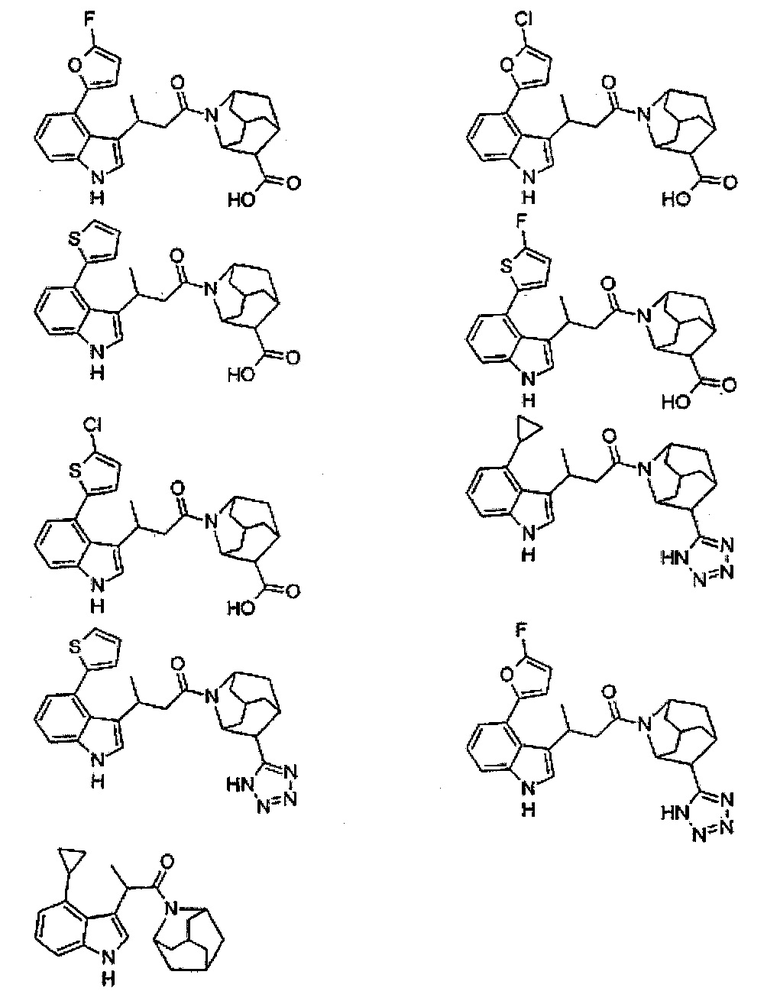
или их фармацевтически приемлемые соли или пролекарства.
Эти соединения обладают способностью ингибировать 11β-HSD1. Эта способность ингибировать 11β-HSD1 может быть обусловлена действием соединений напрямую и исключительно на 11β-HSD1 для модулирования/потенцирования биологической активности. Однако следует понимать, что эти соединения также могут по меньшей мере частично действовать на другие факторы, связанные с активностью 11β-HSD1.
Ингибирование 11β-HSD1 может быть выполнено многочисленными способами, известными в данной области техники. Например, если необходимо ингибирование 11β-HSD1 in vitro, то соответствующее количество соединения может быть добавлено в раствор, содержащий 11β-HSD1. В случае если необходимо ингибировать 11β-HSD1 в организме млекопитающего, то ингибирование 11β-HSD1 обычно подразумевает введение соединения млекопитающему, в организме которого находится 11β-HSD1.
Соответственно, соединения могут находить многочисленные применения, в которых может быть использована их способность ингибировать фермент 11β-HSD1 вышеупомянутого типа.
Соответственно, соединения настоящего изобретения предположительно обладают полезными терапевтическими свойствами, особенно в отношении диабета, гипергликемии, низкой переносимости глюкозы, гиперинсулинемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии, дислипидемии, ожирения, абдоминального ожирения, глаукомы, гипертонии, атеросклероза и его последствий, ретинопатии, нефропатии, невропатии, остеопороза, остеоартрита, деменции, депрессии, нейродегенеративного заболевания, психиатрических расстройств, болезни Кушинга, синдрома Кушинга, вирусных заболеваний и воспалительных заболеваний.
Введение соединений Формулы (I) людям может быть осуществлено любым из приемлемых способов энтерального введения, таких как пероральный или ректальный, или парентеральным введением, таким как подкожный, внутримышечный, внутривенный и интрадермальный способ. Инъекция может быть болюсом или в виде непрерывной или периодической инфузии. Активное соединение обычно включают в фармацевтически приемлемый носитель или разбавитель и в количестве, достаточном для доставки пациенту терапевтически эффективной дозы. В различных вариантах реализации соединение-активатор может быть селективно токсичным или более токсичным в отношении быстро пролиферирующих клеток, например, раковых опухолей, чем в отношении нормальных клеток.
При использовании соединений настоящего изобретения они могут быть введены в любой форме или любым способом, который обеспечивает биодоступность соединения. Специалисты в области составления препаратов могут легко выбрать подходящую форму и способ введения, в зависимости от конкретных характеристик выбранного соединения, состояния, подлежащего лечению, стадии состояния, подлежащего лечению, и других релевантных обстоятельств. Читателю следует обратиться к публикации Remington Pharmaceutical Sciences, 19-ое издание, Mack Publishing Co. (1995), где представлена дополнительная информация.
Соединения настоящего изобретения могут быть введены самостоятельно или в форме фармацевтической композиции в комбинации с фармацевтически приемлемым носителем, разбавителем или вспомогательным веществом. Соединения настоящего изобретения, сами будучи эффективными, обычно составляют в композицию и вводят в форме их фармацевтически приемлемых солей, поскольку эти формы обычно более устойчивы, легче кристаллизуются и обладают улучшенной растворимостью.
Однако соединения обычно используют в форме фармацевтических композиций, которые составлены в зависимости от заданного способа введения. Поэтому в некоторых вариантах реализации настоящего изобретения представлена фармацевтическая композиция, содержащая соединение Формулы (I) и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество. Эти композиции получают по хорошо известным в данной области техники способам.
В других вариантах реализации настоящего изобретения представлен фармацевтический пакет или набор, содержащий один или более контейнеров, наполненных одним или более ингредиентами фармацевтических композиций настоящего изобретения. В таком пакете или наборе может находиться контейнер, содержащий разовую дозу агента(-ов). Наборы могут включать композицию, содержащую эффективный агент в виде концентратов (включая лиофилизированные композиции), которые могут быть дополнительно разбавлены перед применением, или они могут быть представлены в концентрации, предназначенной для применения, при этом пробирки могут содержать одну или более доз. Для удобства в наборах могут быть представлены однократные дозы в стерильных пробирках, так чтобы врач мог использовать эти пробирки напрямую, при этом пробирки содержат заданное количество и концентрацию агента(-ов). Вместе с таким контейнером(-ами) могут быть представлены различные письменные материалы, такие как инструкции по применению, или памятка в форме, предписанной государственным органом, регулирующим производство, применение или продажу фармацевтических препаратов или биологических продуктов, и эта памятка отражает одобрение указанным агентством по производству, применению или продаже для введения людям.
Соединения настоящего изобретения могут быть использованы или введены в комбинации с одним или более дополнительным лекарством(-ами) для лечения упомянутого расстройства/заболевания. Компоненты могут быть введены в одном препарате или в различных препаратах. При необходимости введения в отдельных препаратах, соединения настоящего изобретения могут быть введены последовательно или одновременно с другим лекарством(-ами).
Помимо возможности введения в комбинации с одним или более дополнительными лекарствами, соединения настоящего изобретения могут быть использованы в комплексной терапии. В таком случае эти соединения обычно вводят в комбинации друг с другом. Таким образом, одно или более соединений настоящего изобретения могут быть введены одновременно (в виде комбинированного препарата) или последовательно для достижения заданного эффекта. Если терапевтические профили каждого соединения различны, то особенно желательно, чтобы комбинированный эффект от двух лекарств обеспечивал улучшенный терапевтический результат.
Фармацевтические композиции настоящего изобретения для парентеральной инъекции содержат фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для восстановления в стерильных растворах или дисперсиях для инъекций непосредственно перед применением. Примеры подходящих водных и неводных носителей, разбавителей, растворителей или сред включают воду, этанол, многоатомные спирты (такие как глицерин, пропиленгликоль, полиэтиленгликоль и тому подобные), а также их соответствующие смеси, растительные масла (такие как оливковое масло) и органические сложные эфиры для инъекций, такие как этилолеат.Надлежащая текучесть может поддерживаться, например, с помощью материалов покрытий, таких как лецитин, за счет поддержания уменьшенного размера частиц в случае дисперсий и за счет использования поверхностно-активных веществ.
Эти композиции также могут содержать адъюванты, такие как консерванты, смачивающие агенты, эмульгирующие агенты и диспергирующие агенты. Предотвращение действия микроорганизмов может быть обеспечено включением различных антибактериальных и противогрибковых агентов, например, парабена, хлорбутанола, фенола, сорбиновой кислоты и тому подобных. Также может быть необходимо включать изотонические агенты, такие как сахара, хлорид натрия и тому подобные. Пролонгированная абсорбция инъецируемой фармацевтической формы может быть обеспечена за счет включения агентов, которые вызывают отсрочку абсорбции, таких как моностеарат алюминия и желатин.
При необходимости и для более эффективного распределения соединения могут быть введены в системы с медленным высвобождением или направленной доставкой, такие как полимерные матрицы, липосомы и микросферы.
Композиции для инъекций могут быть стерилизованы, например, фильтрованием через фильтр, удерживающий бактерии, или путем введения стерилизующих агентов в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или в другой стерильной среде для инъекций непосредственно перед применением.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешано по меньшей мере с одним инертным, фармацевтически приемлемым вспомогательным веществом или носителем, таким как цитрат натрия или фосфат дикальция, и/или а) наполнителями или сухими разбавителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) связующими веществами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и гуммиарабик, с) увлажнителями, такими как глицерин, d) агентами для улучшения распадаемости таблеток, такими как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия, е) агентами для замедления растворения, такими как парафин, f) ускорителями абсорбции, такими как четвертичные аммониевые соединения, g) смачивающими агентами, такими как, например, цетиловый спирт и моностеарат глицерина, h) абсорбентами, такими как каолин и бентонитовая глина, и i) смазывающими веществами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, натрия лаурилсульфат и их смеси. В случае капсул, таблеток и пилюль лекарственная форма может содержать также буферные агенты.
Твердые композиции аналогичного типа также могут быть использованы в качестве наполнителей в мягких и твердых желатиновых капсулах, с помощью таких вспомогательных веществ как лактоза или молочный сахар, а также высокомолекулярный полиэтиленгликоли и тому подобные.
Твердые лекарственные формы таблеток, драже, капсул, пилюль и гранул могут быть приготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в области составления фармацевтических препаратов. Они могут необязательно содержать контрастные агенты и могут также иметь такой состав, что они высвобождают активный ингредиент(-ы) только или предпочтительно в определенной части кишечного тракта, необязательно замедленным образом. Примеры залитых композиций, которые могут быть использованы, включают полимерные вещества и воски.
Активные соединения также могут быть в микроинкапсулированной форме, если это уместно, с одним или более из вышеупомянутых вспомогательных веществ.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Помимо активных соединений, жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в данной области техники, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, масло зародышей, оливковое, касторовое и кунжутное масло), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбита, а также их смеси.
Помимо инертных разбавителей, пероральные композиции также могут содержать адюъванты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, ароматизаторы и отдушки.
Суспензии, помимо активных соединений, могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и эфиры сорбита, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также их смеси.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые могут быть приготовлены смешиванием соединений настоящего изобретения с подходящими не раздражающими вспомогательными веществами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при комнатной температуре, но жидкими при температуре тела и поэтому плавятся в прямой кишке или полости влагалища и высвобождают активное соединение.
Лекарственные формы для местного введения соединения настоящего изобретения включают порошки, пластыри, спреи, мази и средства для ингаляций. Активное соединение смешивают при стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами, буферами или газами-вытеснителями, которые могут потребоваться.
Количество введенного соединения предпочтительно лечит и снижает или облегчает указанное состояние. Терапевтически эффективное количество может быть легко определено лечащим диагностом с помощью стандартных приемов и по наблюдению результатов, полученных в аналогичных обстоятельствах. При определении терапевтически эффективного количества следует учитывать ряд факторов, включая, но не ограничиваясь этим, вид животного, его размер, возраст и общее состояние здоровья, конкретное рассматриваемое состояние, тяжесть этого состояния, реакция пациента на лечения, конкретное введенное соединение, способ введения, биодоступность введенного препарата, выбранный режим дозирования, применение других лекарственных средств и другие релевантные обстоятельства.
Предпочтительная доза находится в диапазоне от около 0,01 до 300 мг на килограмм массы тела в сутки. Более предпочтительная доза находится в диапазоне от 0,1 до 100 мг на килограмм массы тела в сутки, более предпочтительно от 0,2 до 80 мг на килограмм массы тела в сутки, еще более предпочтительно от 0,2 до 50 мг на килограмм массы тела в сутки. Подходящая доза может быть введена в нескольких субдозах в течение суток.
Соединение настоящего изобретения также может быть введено в комбинации (либо одновременно, либо последовательно) с адъювантом для улучшения характеристик соединения. Подходящие адъюванты могут включать (а) ингибиторы дипептидил-пептидазы-IV (DP-IV); (b) инсулин-сенсибилизирующие агенты; (iv) бигуаниды; (с) инсулин и инсулиномиметики; (d) сульфонилмочевины и другие стимуляторы секреции инсулина; (е) ингибиторы альфа-глюкозидазы; и (f) GLP-1, аналоги GLP-1 и агонисты рецептора GLP-1. Адъюванты могут быть частью той же композиции, или адъюванты могут быть введены отдельно (одновременно или последовательно). Порядок введения композиции и адъюванта обычно известен медицинскому персоналу и может варьироваться.
СИНТЕЗ СОЕДИНЕНИЙ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Агенты различных вариантов реализации могут быть получены с помощью реакционных последовательностей и схем синтеза, описанных ниже, с использованием методик, доступных в данной области техники, с применением легкодоступных исходных материалов. Получение конкретных соединений вариантов реализации подробно описано в следующих примерах, но специалистам понятно, что описанные химические реакции могут быть легко адаптированы для получения множества других агентов различных вариантов реализации. Например, синтез соединений, не представленных в примерах, может быть успешно выполнен с помощью модификаций, понятных специалистам в данной области, например, с помощью соответствующей защиты мешающих групп, заменой на другие подходящие реагенты, известные в данной области техники, или осуществлением стандартных модификаций условий реакции. Список подходящих защитных групп в органическом синтезе представлен в публикации T.W. Greene Protective Groups in Organic Synthesis, 3-е издание, John Wiley & Sons, 1991. Альтернативно, другие реакции, описанные в настоящем документе или известные в данной области техники, могут считаться применимыми для получения других соединений различных вариантов реализации.
Реагенты, пригодные для синтеза соединений, могут быть приобретены или получены по методикам, известным в данной области техники.
Символы, сокращения и условные обозначения в способах, схемах и примерах соответствуют тем, которые используются в современной научной литературе. В частности, но не для ограничения, в примерах и тексте описания могут быть использованы следующие сокращения.
- г (граммы)
- л (литры)
- Гц (герцы)
- моль (моли)
- RT (комнатная температура)
- мин. (минуты)
- МеОН (метанол)
- СНСl3 (хлороформ)
- ДХМ (дихлорметан)
- ДМСО (диметилсульфоксид)
- EtOAc (этилацетат)
- мг (миллиграммы)
- мл (миллилитры)
- psi (фунтов на квадратный дюйм)
- мМ (миллимолярный)
- МГц (мегагерцы)
- ч. (часы)
- ТСХ (тонкослойная хроматография)
- EtOH (этанол)
- CDCl3 (дейтерированный хлороформ)
- НСl (хлороводородная кислота)
- ДМФ (N,N-диметилформамид)
- ТГФ (тетрагидрофуран)
- K2СО3 (карбонат калия)
- Na2SO4 (сульфат натрия)
- RM (реакционная смесь)
Если не указано иное, все температуры выражены в °С (градусы по стоградусной шкале). Все реакции выполнены при комнатной температуре, если не упомянуто иное.
Все использованные растворители и реагенты имеются в продаже и были закуплены у компаний Sigma Aldrich, Fluka, Acros, Spectrochem, Alfa Aesar, Avra, Qualigens, Merck, Rankem и Leonid Chemicals.
Спектры 1Н ЯМР были записаны на приборе Bruker AV 300. Химические сдвиги выражены в миллионных долях (ppm, единицы δ). Константы взаимодействия представлены в единицах герц (Гц). Структуры расщепления описывают видимую мультиплетность и обозначены как s (синглет), d (дублет), t (триплет), q (квартет), m (мультиплет) или br (широкий).
Масс-спектры были получены на одноквадрупольном приборе ЖХМС модели 6120 производства Agilent technologies, с использованием атмосферной химической ионизации (APCI) или электрораспылительной ионизации (ESI), или комбинации этих двух источников.
Все образцы анализировали на системе SHIMADZU с насосом LC-20 AD, с детектором на диодной матрице SPD-M20A, с автоматическим пробоотборником SIL-20А.
СХЕМА СИНТЕЗА 1
Одна из схем получения некоторых соединений настоящего изобретения представлена ниже на схеме 1.
Схема синтеза 1
Синтез 4-оксотрицикло[3.3.1.13,7]дец-2-ил метансульфоната (Промежуточное соединение 1):
В 1000 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили метансульфоновую кислоту (416,0 г, 4328,8 ммоль) и исходный материал 1 (50,0 г, 333 ммоль). К этой смеси частями добавили азид натрия (23,0 г, 351 ммоль) за 2 часа. Затем реакционную смесь перемешивали при 20-25°С в течение 3 дней. После завершения реакции (реакцию контролировали по ТСХ), реакционную смесь погасили ледяной водой (3000 мл) и экстрагировали этилацетатом (1000X3 мл). Органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия и концентрировали с получением указанного в заголовке Промежуточного соединения 1 (54,0 г, выход = 66%).
Синтез бицикло[3.3.1]нон-6-ен-3-карбоновой кислоты (Промежуточное соединение 2):
В 2000 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили 1200 мл этанола и Промежуточное соединение 1 (54,0 г, 221,3 ммоль). Затем к этой реакционной смеси добавили гидроксид калия (84,0 г, 150 ммоль), затем добавили 950 мл воды. Реакционную смесь перемешивали при 110°С в течение 12 часов. После завершения реакции (реакцию контролировали по ТСХ), реакционную смесь концентрировали под вакуумом. Полученный неочищенный материал подкислили с помощью 1 н. НСl (рН = 2) и экстрагировали этилацетатом (250 X 3 мл). Органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия и концентрировали с получением Промежуточного соединения 2 (32,0 г, выход = 88%).
Синтез метил бицикло[3.3.1]нон-6-ен-3-илкарбамата (Промежуточное соединение 3)
В 500 мл круглодонную колбу, оснащенную магнитной мешалкой, под атмосферой азота загрузили толуол (100 мл), Промежуточное соединение 2 (16,0 г, 96 ммоль) и DPPA (28,8 г, 105 ммоль). Реакционную смесь охладили до 0°С, а затем добавили триэтиламин (15,4 г, 143,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь нагревали при 80°С в течение 8 часов и 12 часов - при комнатной температуре. К ней добавили 100 мл метанола и нагревали с дефлегматором в течение 12 часов. После завершения реакции смесь концентрировали под вакуумо. Полученное неочищенное вещество экстрагировали этилацетатом. Органический слой промыли 1 н. раствором НСl, насыщенным раствором NaHCO3, насыщенным солевым раствором, а затем высушили над безводным сульфатом натрия и концентрировали. Неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 6% EtOAc, с получением Промежуточного соединения 3 (8,0 г, выход = 42%).
Синтез метил 2-азатрицикло[3.3.1.13,7]декан-2-карбоксилата (Промежуточное соединение 4):
В 100 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили 50 мл дихлорметана и Промежуточное соединение 3 (5,0 г, 25,6 ммоль). К этой реакционной смеси добавили трифторметансульфоновую кислоту (19,2 г, 125,2 ммоль) при 0°С. Затем реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения реакции реакционную смесь погасили водой и экстрагировали дихлорметаном. Органический слой промыли насыщенным раствором бикарбоната натрия, насыщенным солевым раствором, а реакционную массу высушили над безводным сульфатом натрия и концентрировали с получением Промежуточного соединения 4 (4,3 г, выход = 86%).
Синтез 2-азатрицикло[3.3.1.13,7]декана (Промежуточное соединение 5):
В 50 мл герметично закрытую пробирку, оснащенную магнитной мешалкой, загрузили Промежуточное соединение 4 (3,0 г, 15 ммоль) в 1,4-диоксан, содержащий НСl (20 мл). Затем реакционную смесь перемешивали при 90°С в течение 8 часов. После завершения реакции (реакцию контролировали по ЖХМС) смесь концентрировали, затем растерли со смесью гексана : эфира (1:1) с получением Промежуточного соединения 5 (3,0 г, выход = 100%).
Синтез трет-бутил 5-гидрокси-2-азатрицикло[3.3.1.13,7] декан-2-карбоксилата (Промежуточное соединение 6):
В 250 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили Промежуточное соединение 5 (3,0 г, 21,6 ммоль), концентрированную азотную кислоту (30 мл) и H2SO4 (5 мл). Реакционную смесь перемешивали при 80°С в течение 12 часов. После завершения реакции (реакцию контролировали по ЖХ-МС) реакционную смесь погасили водой и подщелочили карбонатом натрия. Водный слой промыли ДХМ (100 мл), а полученный водный раствор разбавили ТГФ (200 мл) и охладили до 0°С. рН смеси довели до щелочного значения с помощью триэтиламина (5 мл). К этой реакционной смеси добавили Вос-ангидрид (6,0 г, 27,52 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения реакции (реакцию контролировали по ЖХ-МС) реакционную смесь экстрагировали этилацетатом (100 мл X 3). Объединенный органический слой промыли водой и насыщенным солевым раствором, а реакционную массу высушили над сульфатом натрия. Органический слой концентрировали с получением неочищенного промежуточного соединения, которое затем очистили силикагелевой колоночной хроматографией, элюируя 40% EtOAc, с получением Промежуточного соединения 6 (2,5 г, выход = 50%).
Синтез 2-азатрицикло[3.3.1.13,7]декан-5-ола (Промежуточное соединение 7):
В 100 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили Промежуточное соединение 6 (5,5 г, 21,5 ммоль) в ДХМ (30 мл). Реакционную смесь, полученную таким образом, охладили до 0°С и добавили к ней трифторуксусную кислоту (7,4 г, 65,2 ммоль), и перемешивали в течение 4 часов. После завершения реакции (реакцию контролировали по ЖХМС) реакционную смесь концентрировали, затем растерли со смесью гексана : эфира (1:1) с получением Промежуточного соединения 7 (3,4 г, выход = 100%).
Синтез метил 2-азатрицикло[3.3.1.13,7]декан-5-карбоксилата, Промежуточного соединения 8.
К 2-азатрицикло[3.3.1.13,7]декан-5-олу (Промежуточное соединение 7) (1,5 г, 5,9 ммоль, 1 экв.) по каплям добавляли 98% муравьиную кислоту (9 мл) при энергичном выделении газа в течение 30 минут, смесь добавили к быстро перемешиваемому 30% олеуму (36 мл), нагретому до 60°С. После завершения добавления медленно добавили 99% муравьиную кислоту (9 мл) за следующие 30 минут. Реакционную смесь перемешивали еще 1 час при 60°С (контролировали по ЖХМС). Реакционную смесь, полученную таким образом, затем медленно вылили в энергично перемешиваемый метанол (75 мл), охлажденный до 0°С. Смесь оставили медленно нагреваться до комнатной температуры при перемешивании реакционной смеси в течение 4-5 часов. Затем смесь концентрировали под вакуумом. Остаток вылили на лед (30 г) и подщелочили насыщенным раствором Nа2СО3. Водный слой экстрагировали 5% раствором метанола в ДХМ (3×100 мл). Объединенный органический слой промыли насыщенным солевым раствором и высушили над Na2SO4. Органический слой окончательно концентрировали с получением Промежуточного соединения 8 (550 мг, выход 50%) в виде маслянистой массы.
Синтез 2-трет-бутил 5-метил 2-азатрицикло[3.3.1.13,7]декан-2,5-дикарбоксилата, Промежуточного соединения 9.
Метил 2-азатрицикло[3.3.1.13,7]декан-5-карбоксилат, Промежуточное соединение 8 (0,32 г, 1,6 ммоль) добавили в ТГФ (5 мл) и охладили до 0°С. К реакционной смеси добавили триэтиламин (1,3 мл), затем добавили Вос-ангидрид (0,5 г, 1,96 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 6 часов. После завершения реакции (реакцию контролировали по ЖХ-МС) реакционную смесь экстрагировали этилацетатом (100 мл X 3). Объединенный органический слой промыли водой и насыщенным солевым раствором и высушили над сульфатом натрия. Органический слой концентрировали с получением неочищенного Промежуточного соединения 9, которое очистили силикагелевой колоночной хроматографией, элюируя 15% ЕtOАс, с получением Промежуточного соединения 9 (0,32 г, выход = 66%).
Синтез 2-(трет-бутоксикарбонил)-2-азатрицикло[3.3.1.13,7]декан-5-карбоновой кислоты, Промежуточного соединения 10.
К охлажденному до 0°С, перемешанному раствору 2-трет-бутил 5-метил 2-азатрицикло[3.3.1.13,7]декан-2,5-дикарбоксилата, Промежуточного соединения 9 (0,16 г, 0,5 ммоль), растворенного в метаноле (3 мл), ТГФ (1 мл) и воде (1 мл), добавили LiOH (50 мг, 2 ммоль), и полученную реакционную массу перемешивали при комнатной температуре в течение 6 часов. После завершения реакции (реакцию контролировали по ТСХ), растворитель, содержащийся в реакционной смеси, полностью удалили под вакуумом, а неочищенный остаток подкислили насыщенным раствором лимонной кислоты и экстрагировали этилацетатом (3×15 мл). Органический слой затем промыли насыщенным солевым раствором и высушили над сульфатом натрия, и окончательно концентрировали под вакуумом с получением Промежуточного соединения 10 в виде маслянистой массы, 0,16 г (95%).
Синтез трет-бутил 5-карбамоил-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилата, Промежуточного соединения 11.
В 50 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили 5 мл ацетонитрила и 2-(трет-бутоксикарбонил)-2-азатрицикло[3.3.1.13,7]декан-5-карбоновую кислоту, Промежуточное соединение 10 (0,16 г, 0,56 ммоль). Под атмосферой N2 к реакционной смеси добавили пиридин (60 мг, 0,6 ммоль) и Вос-ангидрид (0,148 г, 0,6 ммоль) и перемешивали в течение 1 часа. Через 1 час добавили твердый бикарбонат аммония (75 мг, 0,9 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 12 часов. После завершения реакции (реакцию контролировали по ТСХ) реакционную смесь концентрировали под вакуумом. Полученный неочищенный материал экстрагировали этилацетатом (25 мл X 3). Органический слой промыли раствором хлорида аммония и насыщенным раствором бикарбоната натрия. Затем его высушили над безводным сульфатом натрия и концентрировали с получением Промежуточного соединения 11 в виде маслянистой массы (0,14 г, выход = 87%).
Синтез 2-азатрицикло[3.3.1.13,7]декан-5-карбоксамида, Промежуточного соединения 12.
В 100 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили трет-бутил 5-карбамоил-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилат, Промежуточное соединение 11 (0,1 г, 0,0357 ммоль) в ДХМ (5 мл). Реакционную смесь охладили до 0°С и добавили трифторуксусный ангидрид (0,21 г, 0,17 ммоль), и перемешивали в течение 4 часов. После завершения реакции (реакцию контролировали по ЖХМС) реакционную смесь концентрировали, затем растерли со смесью гексана : эфира (1:1) с получением Промежуточного соединения 12 (0,09 г, выход = 90%).
Синтез трет-бутил 5-циано-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилата, Промежуточного соединения 13.
В 50 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили 5 мл ДХМ и mpem-бутил 5-карбамоил-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилат,
Промежуточное соединение 11 (0,8 г, 2,8 ммоль). Под атмосферой N2 добавили триэтиламин (1,15 г, 11,4 ммоль) и трифторуксусный ангидрид (2,4 г, 11,4 ммоль) и перемешивали в течение 6 часов. После завершения реакции (реакцию контролировали по ТСХ) реакционную смесь погасили раствором KHSO4 и экстрагировали ДХМ (50 мл X 3). Органический слой промыли насыщенным раствором бикарбоната натрия, затем насыщенным солевым раствором. Наконец, реакционную смесь высушили над безводным сульфатом натрия и концентрировали с получением неочищенного Промежуточного соединения 13, которое подвергли колоночной хроматограмме (10% EtOAc в петролейном эфире) в виде грязновато-белого твердого вещества (0,5 г, выход = 70%).
Синтез 2-азатрицикло[3.3.1.13,7]декан-5-карбонитрила, Промежуточного соединения 14.
В 50 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили трет-бутил 5-циано-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилат, Промежуточное соединение 13 (0,5 г, 1,98 ммоль) в ДХМ (5 мл). Затем реакционную смесь охладили до 0°С и добавили трифторуксусную кислоту (1,1 г, 9,54 ммоль), и перемешивали в течение 4 часов. После завершения реакции (реакцию контролировали по ЖХМС) реакционную смесь концентрировали, затем растерли со смесью гексана : эфира (1:1) с получением Промежуточного соединения 14 (0,5 г, выход = 97%).
Синтез трет-бутил 5-(гидроксиметил)-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилата, Промежуточного соединения 15.
В 50 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили 5 мл ТГФ, 2-трет-бутил 5-метил 2-азатрицикло[3.3.1.13,7]декан-2,5-дикарбоксилат,
Промежуточное соединение 9 (0,15 г, 0,5 ммоль) и охладили до 0°С. Под атмосферой N2 частями добавили LAH (30 мг, 0,8 ммоль) и перемешивали в течение 2 часов. После завершения реакции (реакцию контролировали по ТСХ) реакционную смесь погасили этилацетатом и промыли водой, затем 1 н. раствором НСl. Органический слой промыли насыщенным солевым раствором. Наконец, органический слой высушили над безводным сульфатом натрия и концентрировали с получением неочищенного Промежуточного соединения 15 в виде маслянистой массы (0,12 г, выход = 88%).
Синтез 2-азатрицикло[3.3.1.13,7]дец-5-илметанола, Промежуточного соединения 16.
В 50 мл круглодонную колбу, оснащенную магнитной мешалкой, добавили mpem-бутил 5-(гидроксиметил)-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилат, Промежуточное соединение 15 (0,12 г, 0,45 ммоль) в ДХМ (5 мл). Реакционную смесь охладили до 0°С, затем добавили трифторуксусную кислоту (0,26 г, 2,2 ммоль). Эту смесь перемешивали в течение 4 часов. После завершения реакции (реакцию контролировали по ЖХМС) реакционную смесь концентрировали, затем растерли со смесью гексана : эфира (1:1) с получением Промежуточного соединения 16 (0,12 г, выход = 97%) в виде маслянистой массы.
Синтез трет-бутил 5-метокси-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилата, Промежуточного соединения 17.
В 15 мл закрытую пробирку, оснащенную магнитной мешалкой, загрузили 5 мл ТГФ, mpem-бутил 5-гидрокси-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилат, Промежуточное соединение 6 (0,15 г, 0,5 ммоль). К этой смеси при 0°С добавили гидрид калия (47 мг, 1,1 ммоль), под атмосферой N2. Реакционную смесь затем перемешивали при комнатной температуре в течение 30 минут. Медленно добавили метилйодид (0,12 г, 0,8 ммоль) при 0°С, а полученную реакционную массу нагревали с дефлегматором при 60°С в герметичных условиях в течение 12 часов. После завершения реакции (реакцию контролировали по ТСХ) реакционную смесь погасили холодной водой и экстрагировали этилацетатом (25 мл X 3). Органический слой промыли раствором хлорида натрия, высушили над безводным сульфатом натрия и концентрировали с получением неочищенного Промежуточного соединения 17, которое подвергли колоночной хроматограмме (22% EtOAc в петролейном эфире) с получением маслянистой массы (0,1 г, выход = 70%).
Синтез 5-метокси-2-азатрицикло[3.3.1.13,7]декана, Промежуточного соединения 18:
В 50 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили трет-бутил 5-метокси-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилат, Промежуточное соединение 17 (0,1 г, 0,037 ммоль) в ДХМ (5 мл). Затем реакционную смесь охладили до 0°С и добавили трифторуксусную кислоту (0,22 г, 1,8 ммоль), и перемешивали в течение 4 часов. После завершения реакции (реакцию контролировали по ЖХМС) реакционную смесь концентрировали, затем растерли со смесью гексана : эфира (1:1) с получением Промежуточного соединения 18 (0,09 г, выход = 95%) в виде маслянистой массы.
Синтез трет-бутил 5-(циклопропилметокси)-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилата, Промежуточного соединения 19.
В 15 мл закрытую пробирку, оснащенную магнитной мешалкой, загрузили 5 мл ТГФ, трет-бутил 5-гидрокси-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилат, Промежуточное соединение 6 (50 мг, 0,2 ммоль). В пробирку добавили гидрид калия (20 мг, 0,5 ммоль) при 0°С, под атмосферой N2. Реакционную массу перемешивали при комнатной температуре в течение 30 минут. К этой смеси медленно добавили циклопропилметил-бромид (40 мг, 0,3 ммоль) при 0°С, а полученную реакционную массу нагревали с дефлегматором при 60°С в герметичных условиях в течение 12 часов. После завершения реакции (реакцию контролировали по ТСХ) реакционную смесь погасили холодной водой и экстрагировали этилацетатом (25 мл X 3). Органический слой промыли раствором хлорида натрия, высушили над безводным сульфатом натрия и концентрировали с получением неочищенного Промежуточного соединения 17, которое подвергли колоночной хроматограмме (18% ЕtOАс в петролейном эфире) с получением маслянистой массы (50 мг, выход = 80%).
Синтез 5-(циклопропилметокси)-2-азатрицикло[3.3.1.13,7]декана, Промежуточного соединения 20.
В 50 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили трет-бутил 5-(циклопропилметокси)-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилат,
Промежуточное соединение 19 (50 мг, 0,016 ммоль) в ДХМ (5 мл). Затем реакционную смесь охладили до 0°С и добавили трифторуксусную кислоту (0,099 г, 0,084 ммоль), и перемешивали в течение 4 часов. После завершения реакции (реакцию контролировали по ЖХМС) реакционную смесь концентрировали, затем растерли со смесью гексана : эфира (1:1) с получением Промежуточного соединения 20 (50 мг, выход = 95%) в виде маслянистой массы.
ПРИМЕР 1: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)пропан-1-он (1)
ПРИМЕР 1: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)пропан-1-он (1)
Исходный материал 2 (0,2 ммоль) добавили к Промежуточному соединению 5 (0,2 ммоль) в дихлорметане (ДХМ), затем добавили 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (EDCI) (0,26 ммоль) и 1-гидроксибензтриазол (HOBt) (0,23 ммоль). Реакционную смесь охладили до 0°С и поддерживали при той же температуре в течение 30 минут. Затем к реакционной смеси добавили триэтиламин (0,93 ммоль), а полученный раствор перемешивали при комнатной температуре в течение 15 часов. Затем реакционную массу разбавили смесью с равными соотношениями ДХМ и воды и промыли 1 н. раствором НСl, затем NaHCO3 и насыщенным солевым раствором. Органический слой отделили и высушили над безводным сульфатом натрия. Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили силикагелевой колоночной хроматографией, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (1) (9,5 мг, смолянистый материал). 1Н ЯМР (300 МГц, CDCl3): δ 7.91 (brs, 1H), 7.54 (d, 1Н), 7.28 (d, 1H), 7.12 (t, 1H), 7.02 (t, 1Н), 6.98 (s, 1H), 4.82 (s, 1H), 3.92 (s, 1H), 3.06 (t, 2Н), 2.61 (t, 2Н), 1.97-1.98 (m, 2Н), 1.60-1.75 (m, 10Н). ЖХ-МС (М+Н)+ = 309,2; чистота по ВЭЖХ = 92,94%.
ПРИМЕР 2: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(4-метил-1H-индол-3-ил)пропан-1-он (2)
Синтез 4-метил-1H-индола (Промежуточное соединение 21): В 100 мл круглодонную колбу, оснащенную магнитной мешалкой и обратным холодильником, загрузили 60 мл ДМФ. К перемешанному растворителю добавили исходный материал 3 (5 г, 33 ммоль), затем диметилформамид-диметилацеталь (13,1 мл, 99,2 ммоль). К этой смеси добавили пирролидин (3,2 мл, 39,6 ммоль) и нагревали реакционную смесь при 120°С под атмосферой азота в течение 21 часа. После завершения реакции смесь охладили до комнатной температуры, а растворитель удалили под пониженным давлением. Полученную неочищенную массу растворили в эфире (250 мл) и промыли водой (50 мл × 3), насыщенным солевым раствором (50 мл), а органический слой высушили над безводным сульфатом натрия и концентрировали. Полученный неочищенный материал растворили в этилацетате (50 мл). К нему добавили 10% Pd/C (1,0 г, 10% вес./вес.) и гидрогенировали во встряхивателе Парра в течение 2 часов. После завершения реакции (реакцию контролировали по ТСХ) смесь отфильтровали через слой целита. Фильтрат концентрировали с получением неочищенного продукта, который очистили колоночной хроматографией на силикагеле (120 меш), используя петролейный эфир (60-80) и этилацетат в качестве элюента, с получением Промежуточного соединения 21 (1,2 г).
Синтез 3-(4-метил-1H-индол-3-ил)пропановой кислоты (Промежуточное соединение 22): В 100 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили 2,5 мл уксусной кислоты. К перемешанному растворителю добавили уксусный ангидрид, 2,0 мл, затем добавили акриловую кислоту (1,8 мл, 27,4 ммоль). К этой перемешанной смеси добавили Промежуточное соединение 21 (1,2 г, 9,15 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 1 недели. После завершения реакции (реакцию контролировали по ТСХ) реакционную массу подщелочили с помощью 5 н. NaOH (5 мл) и промыли этилацетатом (100 мл X 2). Водный слой подкислили концентрированной НСl (3 мл) и экстрагировали этилацетатом (100 мл X 3). Объединенный этилацетатный слой промыли насыщенным Солевым раствором и концентрировали с получением Промежуточного соединения 22 (350 мг).
Синтез Соединения (2): Соединение (2) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат в качестве элюента, с получением Соединения (2). 1Н ЯМР (300 МГц, CDCl3): δ 7.94 (brs, 1H), 7.12 (d, 1Н), 6.99 (t, 1Н), 6.92 (s, 1H), 6.76 (d, 1H), 4.82 (s, 1H), 3.94 (s, 1Н), 3.21 (t, 2Н), 2.64 (s, 3Н), 2.58 (t, 2Н), 2.12-2.17 (m, 1Н), 1.64-1.76 (m, 11Н). ЖХ-МС (М+Н)+ = 323,2; чистота по ВЭЖХ = 71,84%.
ПРИМЕР 3: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1,4-диметил-1H-индол-3-ил)пропан-1-он (3)
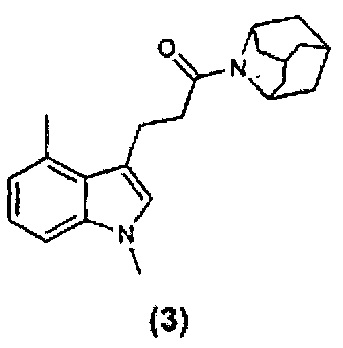
Синтез Соединения (3): Соединение (3) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат в качестве элюента, с получением Соединения (3). 1Н ЯМР (300 МГц, CDCl3): δ 6.98-7.05 (m, 2Н), 6.75-6.78 (m, 2Н), 4.82 (s, 1H), 3.93 (s, 1Н), 3.63 (s, 3Н), 3.16-3.22 (m, 2Н), 2.64 (s, 3Н), 2.55-2.61 (m, 2Н), 1.97-2.01 (m, 2Н), 1.75-1.79 (m, 2Н), 1.70-1.72 (m, 3Н), 1.59-1.65 (m, 5Н). ЖХ-МС (М+Н)+ = 337,2; чистота по ВЭЖХ = 79,30%.
ПРИМЕР 4: 3-(4-фтор-1Н-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)пропан-1-он (4)
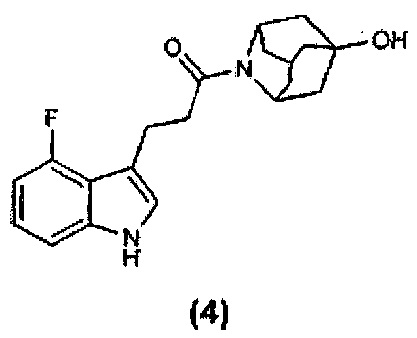
СХЕМА СИНТЕЗА 4
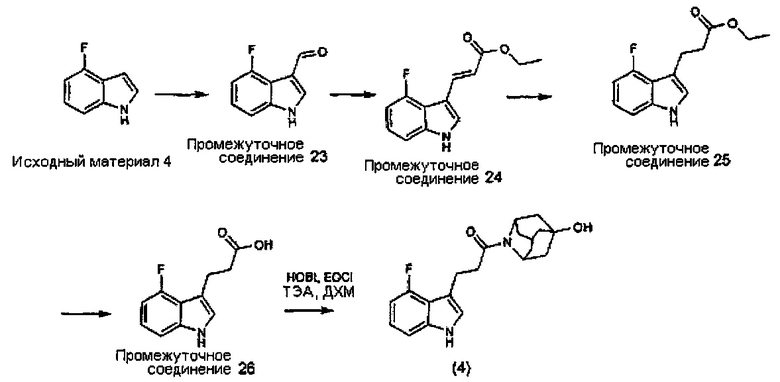
Синтез 4-фтор-1H-индол-3-карбальдегида (Промежуточное соединение 23):
В 25 мл круглодонную колбу, оснащенную магнитной мешалкой, добавили ДМФ (0,413 г) и POCl3 (0,623 г, 4 ммоль) при 0°С под атмосферой N2, и полученную смесь перемешивали в течение 30 минут при той же температуре. Затем к смеси добавили исходный материал 4 (500 мг, 3,7 ммоль) в ДМФ и перемешивали при 40°С в течение 1 часа. После завершения реакции реакционную смесь охладили до 0°С, погасили раствором NaOH и экстрагировали этилацетатом. Органические слои концентрировали с получением неочищенного материала, который затем очистили силикагелевой колоночной хроматографией, элюируя гексаном: EtOAc, с получением Промежуточного соединения 23 (230 мг) в виде коричневого материала. ЖХ-МС (М+Н)+ = 164,2.
Синтез этил (2E)-3-(4-фтор-1H-индол-3-ил)проп-2-еноата (Промежуточное соединение 24):
В 100 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили Промежуточное соединение 23 (0,23 г, 1,4 ммоль), этилмалонат (0,204 г, 1,5 ммоль) и пиперидин (0,011 г, 0,13 ммоль) в пиридине (10 мл). Полученную реакционную смесь нагревали при 110°С в течение 14 часов. После завершения реакции реакционную смесь концентрировали с получением неочищенного материала, который затем растворили в этилацетате и промыли водой и насыщенным солевым раствором. Затем органический слой концентрировали с получением неочищенного материала, который очистили силикагелевой колоночной хроматографией, элюируя гексаном : EtOAc, с получением Промежуточного соединения 24 (250 мг).
Синтез этил 3-(4-фтор-1H-индол-3-ил)пропаноата (Промежуточное соединение 25):
Промежуточное соединение 24 (0,24 г, 1,0 ммоль) растворили в EtOAc (10 мл) и добавили 10% Pd/C (50 мг). Полученную реакционную массу перемешивали под атмосферой Н2 (30 psi) в течение 4 часов. Реакционную массу отфильтровали через слой целита и концентрировали с получением Промежуточного соединения 25 (240 мг).
Синтез 3-(4-фтор-1H-индол-3-ил)пропановой кислоты (Промежуточное соединение 26):
Промежуточное соединение 25 (100 мг, 0,4 ммоль) растворили в EtOH : ТГФ : Н2О (5 мл : 5 мл : 1 мл). К этой смеси добавили NaOH (51 мг, 1,2 ммоль). Полученную реакционную смесь нагревали с дефлегматором в течение 4 часов. После завершения реакции (реакцию контролировали по ТСХ) реакционную смесь концентрировали, затем разбавили водой, подкислили (рН=1-2) с помощью 1 н. HCl, экстрагировали с помощью EtOAc и концентрировали с получением Промежуточного соединения 26 (90 мг).
Синтез Соединения (4): Соединение (4) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (4). 1Н ЯМР (300 МГц, CDCl3): δ 8.04 (s, 1Н), 6.97-7.07 (m, 2Н), 6.94 (s, 1H), 6.65-6.71 (m, 1Н), 5.01 (s, 1Н), 4.21 (s, 1Н), 3.08-3.13 (t, 2Н), 2.62-2.67 (t, 2Н), 2.24 (s, 1Н), 1.53-1.74 (m, 10Н). ЖХ-МС (М+Н)+ = 343,12; чистота по ВЭЖХ = 95,20%.
ПРИМЕР 5: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(6-фтор-1H-индол-3-ил)пропан-1-он (5)
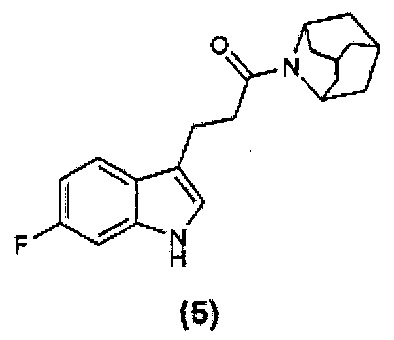
Синтез Соединения (5): Соединение (5) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этил ацетат в качестве элюента, с получением Соединения (5). 1Н ЯМР (300 МГц, CDCl3): δ 8.02 (brs, 1Н), 7.41-7.46 (dd, 1H), 6.95-6.99 (m, 2Н), 6.78-6.85 (m, 1H), 4.81 (s, 1Н), 3.90 (s, 1Н), 3.03-3.08 (t, 2Н), 2.63-2.68 (t, 2Н), 1.96-2.02 (m, 2Н), 1.56-1.76 (m, 10Н). ЖХ-МС (М+Н)+ = 327,3; чистота по ВЭЖХ = 95,25%.
ПРИМЕР 6: 3-(5-фтор-1H-индол-3-ил)-1-(4-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)пропан-1-он (6)
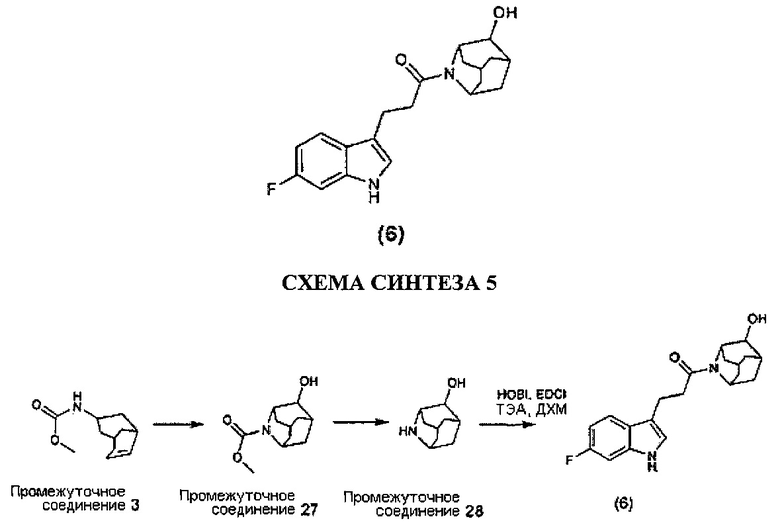
Синтез метил 4-гидрокси-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилата (Промежуточное соединение 27):
В 100 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили 25 мл дихлорметана. К нему добавили Промежуточное соединение 3 (0,5 г, 2,5 ммоль), затем добавили m-СРВА (0,69 г, 4,0 ммоль) при 0°С. Затем реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После завершения реакции реакционную смесь погасили с помощью водного раствора NaHCO3 и экстрагировали дихлорметаном. Органический слой концентрировали с получением Промежуточного соединения 27 (0,5 г).
Синтез 2-азатрицикло[3.3.1.13,7]декан-4-ола гидрохлоридной соли (Промежуточное соединение 28):
В 50 мл герметично закрытую пробирку, оснащенную магнитной мешалкой, загрузили Промежуточное соединение 27 (0,2 г, 0,9 ммоль) в 1,4-диоксане, содержащем HCl (20 мл). Затем реакционную смесь перемешивали при 90°С в течение 8 часов. После завершения реакции (реакцию контролировали по ЖХМС) смесь концентрировали, затем растерли со смесью гексана : эфира (1:1) с получением Промежуточного соединения 28 (0,2 г).
Синтез Соединения (6): Соединение (6) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (6). 1Н ЯМР (300 МГц, CDCl3): δ 7.97 (brs, 1Н), 7.17-7.23 (d, 1Н), 7.14-7.15 (d, 1Н), 7.02 (s, 1H), 6.84-6.90 (m, 1H), 4.67-4.73 (d, 1H), 3.83 (brs, 0.5Н), 3.68 (s, 1H), 3.35 (brs, 1H), 2.98-3.03 (t, 2Н), 2.56-2.64 (m, 2Н), 2.07-2.11 (m, 1H), 1.97 (m, 1H), 1.61-1.69 (m, 7Н). ЖХ-МС (М+Н)+ = 343,1; чистота по ВЭЖХ = 95,88%.
ПРИМЕР 7: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(5-фтор-1H-индол-3-ил)пропан-1-он (7)
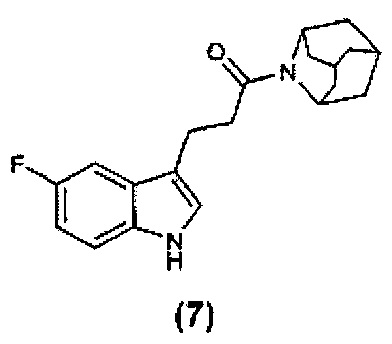
Синтез Соединения (7): Соединение (7) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат в качестве элюента, с получением Соединения (7). 1Н ЯМР (300 МГц, CDCl3): δ 7.94 (s, 1Н), 722 (d, 1H), 7.16 (s, 1H), 7.03 (s, 1H), 6.86-6.87 (t, 1H), 4.81 (s, 1Н), 3.91 (s, 1Н), 2.99-3.04 (t, 2Н), 2.57-2.62 (t, 2Н), 1.92-1.98 (m, 2Н), 1.58-1.75 (m, 10Н). ЖХ-МС (М+Н)+ = 327,2; чистота по ВЭЖХ = 96,53%.
ПРИМЕР 8: 3-(4-фтор-1-метил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)пропан-1-он (8)
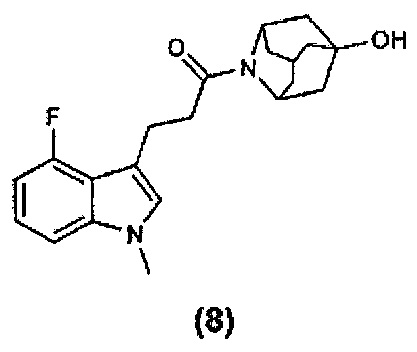
Синтез Соединения (8): Соединение (8) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (8). 1Н ЯМР (300 МГц, CDCl3): δ 6.96-7.07 (m, 2Н), 6.78 (s, 1H), 6.63-6.69 (m, 1H), 5.01 (s, 1Н), 4.21 (s, 1H), 3.64 (s, 3Н), 3.05-3.10 (t, 2Н), 2.59-2.64 (t, 2Н), 2.24 (s, 1H), 1.53-1.74 (m, 10Н). ЖХ-МС (М+Н)+ = 357,1; чистота по ВЭЖХ = 89,95%.
ПРИМЕР 9: 3-(5-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)пропан-1-он (9)
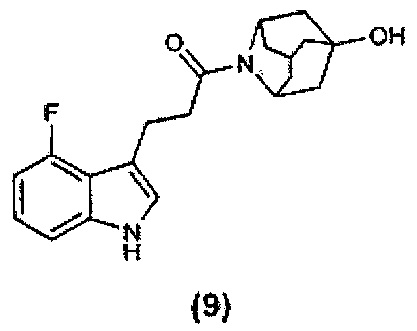
Синтез Соединения (9): Соединение (9) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (9). 1H ЯМР (300 МГц, CDCl3): δ 7.94 (brs, 1Н), 7.19-7.22 (d, 1Н), 7.15-7.16 (d, 1H), 7.02 (s, 1H), 6.83-6.90 (m, 1Н), 5.02 (s, 1Н), 4.01 (s, 1H), 2.99-3.04 (t, 2Н), 2.57-2.62 (t, 2Н), 2.26 (s, 1Н), 1.61-1.97 (m, 10Н). ЖХ-МС (М+Н)+ = 343,1; чистота по ВЭЖХ = 93,42%.
ПРИМЕР 10: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-метил-3-(1-метил-1H-индол-3-ил)бутан-1-он (10)
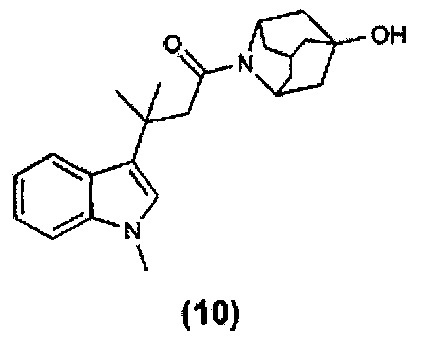
СХЕМА СИНТЕЗА 6
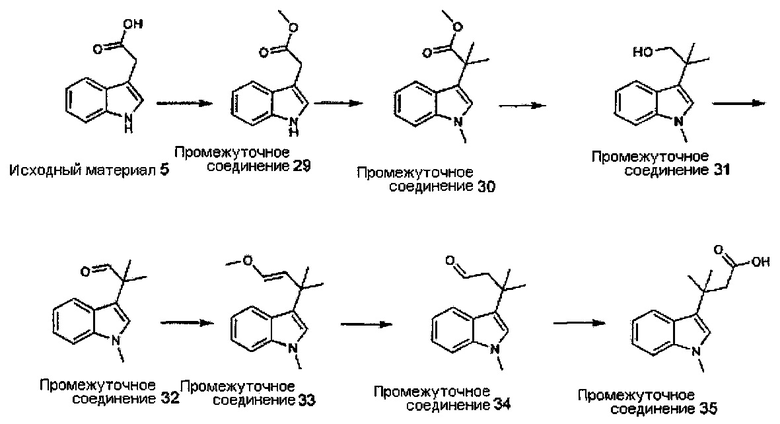
Синтез метил 1H-индол-3-ил ацетата (Промежуточное соединение 29):
В 100 мл кругло донную колбу, оснащенную магнитной мешалкой, загрузили 15 мл метанола. К перемешанному растворителю добавили исходный материал 5 (2,0 г, 11,41 ммоль). Полученную смесь охладили до 0°С и добавили концентрированную H2SO4 (0,5 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения реакции (реакцию контролировали по ТСХ) растворитель удалили из реакционной массы под пониженным давлением. Полученную неочищенную массу растворили в этилацетате (100 мл) и промыли водой (50 мл), раствором бикарбоната натрия (100 мл × 2) и насыщенным солевым раствором (50 мл). Затем органический слой высушили над безводным сульфатом натрия. Затем растворитель удалили под пониженным давлением. Продукт, Промежуточное соединение 29, получили в виде коричневого сиропообразного вещества (2,1 г). ЖХ-МС (М+Н)+ = 190,2.
Синтез метил 2-метил-2-(1-метил-1H-индол-3-ил)пропаноата (Промежуточное соединение 30):
В 100 мл 3-горлую круглодонную колбу, оснащенную магнитной мешалкой, загрузили 10 мл сухого ТГФ. К перемешанному растворителю добавили диизопропиламин (401,12 мг, 3,964 ммоль), а полученный раствор охладили до -78°С. Добавили н-BuLi (2,5 мл, 3,964 ммоль) и перемешивали в течение 1 часа при 0°С. Реакционную смесь снова охладили до -78°С и добавили Промежуточное соединение 29 (150 мг, 0,7928 ммоль). Затем реакционную смесь перемешивали в течение 1 часа. Затем добавили метилйодид. Затем полученную массу перемешивали при комнатной температуре в течение 15 часов. После завершения реакции (реакцию контролировали по ТСХ) реакционную массу погасили насыщенным раствором хлорида аммония и экстрагировали с помощью EtOAc (100 мл × 3). Объединенные органические слои промыли насыщенным солевым раствором и высушили, после чего растворитель удалили под пониженным давлением. Полученное неочищенное соединение очистили колоночной хроматографией на силикагеле (120 меш), используя петролейный эфир (60-80) и этилацетат в качестве элюента. Продукт, Промежуточное соединение 30, получили в виде коричневого сиропообразного вещества (150 мг). ЖХ-МС (М+Н)+ = 232,2.
Синтез 2-метил-2-(1-метил-1H-индол-3-ил)пропан-1-ола (Промежуточное соединение 31):
В 250 мл кругло донную колбу, оснащенную магнитной мешалкой, загрузили алюмогидрид лития (0,983 г, 25,951 ммоль) и добавили к этой смеси ТГФ (20 мл) при 0°С. К полученной суспензии добавили Промежуточное соединение 30 (2,0 г, 8,65 ммоль) в ТГФ (20 мл), а полученную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения реакции реакционную смесь разбавили EtOAc (50 мл), а затем погасили Na2SO4 (5 г). Полученную суспензию перемешивали при комнатной температуре в течение 1 часа, отфильтровали через целит и промыли этилацетатом. Полученный фильтрат концентрировали с получением Промежуточного соединения 31 (0,9 г). 1Н ЯМР (300 МГц, ДМСО-d6): δ 7.65-7.68 (d, 1Н), 7.34-7.36 (d, 1Н), 7.07-7.12 (t, 1Н), 7.03 (s, 1Н), 6.94-6.99 (t, 1Н), 4.53-4.57 (t, 1Н), 3.71 (s, 3Н), 3.54-3.56 (d, 2Н), 1.31 (s, 6Н).
Синтез 2-метил-2-(1-метил-1Н-индол-3-ил)пропаналя (Промежуточное соединение 32):
В 100 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили 30 мл ДХМ, к которому добавили пиридиния хлорхромат (2,466 г, 11,4419 ммоль), затем добавили Промежуточное соединение 31 (1,55 г, 7,627 ммоль) в 10 мл ДХМ. Полученную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения реакции растворитель удалили из реакционной массы под пониженным давление с получением неочищенного соединения. Неочищенную массу очистили колоночной хроматографией, используя 60-120 силикагель и 9:1 смесь петролейного эфира/этилацетата в качестве элюента, с получением Промежуточного соединения 32 (0,79 г). 1Н ЯМР (300 МГц, ДМСО-d6): δ 9.39 (s, 1Н), 7.40-7.44 (t, 1Н), 7.32 (s, 1Н), 7.13-7.18 (t, 1Н), 6.98-7.03 (t, 1Н), 3.77 (s, 3Н), 1.46 (s, Н).
Синтез 3-[(3E)-4-метокси-2-метилбут-3-ен-2-ил]-1-метил-1H-индола (Промежуточное соединение 33):
В 100 мл кругло донную колбу, оснащенную магнитной мешалкой, загрузили 20 мл сухого ТГФ и метоксиметил-трифенилфосфония хлорид (2,566 г, 7,487 ммоль), затем трет-бутоксид калия (2,295 г, 20,451 ммоль). Полученную массу перемешивали при комнатной температуре в течение 2 часов, а затем охладили до 0°С. К полученной выше реакционной массе добавили Промежуточное соединение 32 (1,37 г, 6,807 ммоль) в 10 мл ТГФ и перемешивали при комнатной температуре в течение 2 часов. После завершения реакции реакционную массу разбавили 10 мл воды и экстрагировали этилацетатом (100 мл × 3). Объединенные органические слои промыли насыщенным солевым раствором и высушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очистили колоночной хроматографией, используя 60-120 силикагель и 6% этилацетат в петролейном эфире в качестве элюента, с получением Промежуточного соединения 33. Выход: 1,12 г (71,8%). 1Н ЯМР (300 МГц, CDCl3): δ 7.73-7.80 (m, 1Н), 7.33 (s, 1Н), 7.16-7.21 (t, 1Н), 7.03-7.08 (t, 1Н), 6.81 (s, 1Н), 5.79-6.34 (m, 1Н), 64.58-5.15 (m, 1Н), 3.73-3.74 (d, 3Н), 3.49-3.53 (d, 3Н), 1.55 (s, 6Н).
Синтез 3-метил-3-(1-метил-1H-индол-3-ил)бутаналя (Промежуточное соединение 34):
В 100 мл кругло донную колбу, оснащенную магнитной мешалкой, загрузили 50,4 мл 1,4-диоксана и 12,76 мл воды. К этой смеси добавили Промежуточное соединение 33 (1,12 г, 4,884 ммоль), затем добавили п-толуолсульфоновую кислоту (0,0424 г, 0,2232 ммоль). Полученную смесь нагревали при 60°С в течение 16 часов. После завершения реакции реакционную смесь погасили 10 мл воды и экстрагировали этилацетатом (100 мл × 3), а объединенный органический слой промыли насыщенным раствором бикарбоната натрия, затем насыщенным солевым раствором и высушили над безводным сульфатом натрия, и концентрировали с получением неочищенного продукта. Неочищенный продукт очистили колоночной хроматографией, используя 60-120 силикагель и 8% этилацетата в петролейном эфире в качестве элюента, с получением Промежуточного соединения 34. 1Н ЯМР (300 МГц, ДМСО-d6): δ 9.47-9.49 (t, 1Н), 7.73-7.76 (d, 1Н), 7.37-7.40 (d, 1Н), 7.11-7.16 (t, 1Н), 7.10 (s, 1Н), 6.99-7.04 (t, 1Н), 3.72 (s, 3Н), 2.78 (s, 2Н), 1.49 (s, 6Н).
Синтез 3-метил-3-(1-метил-1Н-индол-3-ил)бутановой кислоты (Промежуточное соединение 35):
В 50 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили 10 мл ТГФ и охладили до -78°С, затем добавили 2-метил-2-бутен (3 мл) и перемешивали в течение 15 минут. В другую 100 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили Промежуточное соединение 34 (557 мг, 2,59 ммоль) и трет-бутанол (15 мл) и перемешивали при комнатной температуре, и добавили к нему полученный выше раствор ТГФ. Затем полученную массу охладили до 0°С, добавили к ней NaH2PO4 (1,42 г) в воде, затем добавили NaClO2 (0,35 г) в воде. Полученную смесь перемешивали при 0°С в течение 20 минут и погасили водой. рН реакционной смеси довели до значения 1-2, используя 1 н. HCl, а продукт экстрагировали этилацетатом и концентрировали с получением Промежуточного соединения 35 (480 мг). 1Н ЯМР (300 МГц, ДМСО-d6): δ 11.82 (s, 1H), 7.69-7.71 (d, 1H), 7.35-7.38 (d, 1H), 7.09-7.14 (t, 1H), 7.05 (s, 1H), 6.96-7.01 (t, 1H), 3.71 (s, 3H), 2.66 (s, 2H), 1.48 (s, 6H).
Синтез Соединения (10): Соединение (10) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (10). 1Н ЯМР (300 МГц, ДМСО-d6): δ 7.70-7.73 (d, 1Н), 7.34-7.37 (d, 1Н), 7.10 (t, 1Н), 7.04 (s, 1Н), 6.98-7.04 (t, 1Н), 4.75 (brs, 1Н), 4.54 (s, 1Н), 3.99 (brs, 1Н), 3.70 (s, 3Н), 2.60-2.65 (dd, 2Н), 2.02 (s, 1Н), 1.28-1.62 (m, 16Н). ЖХ-МС (М+Н)+ = 367,3; чистота по ВЭЖХ = 88,74%.
ПРИМЕР 11: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1,3-бензотиазол-2-ил)пропан-1-он (11)
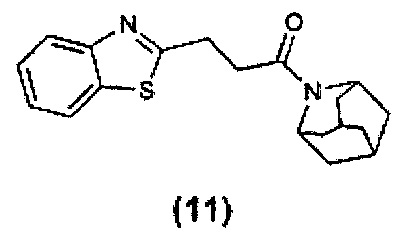
СХЕМА СИНТЕЗА 7
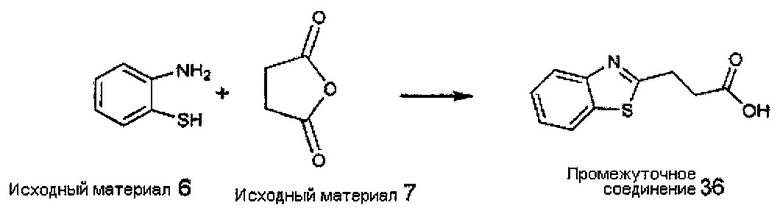
Синтез 3-(1,3-бензотиазол-2-ил)пропановой кислоты (Промежуточное соединение 36):
Исходный материал 7 (3,97 ммоль) в бензоле по каплям добавили к раствору исходного материала 6 (3,97 ммоль) в бензоле. Полученный раствор нагревали с дефлегматором в течение 2 часов. Через 2 часа реакционную массу охладили до комнатной температуры и экстрагировали 10% раствором гидроксида натрия. Водный слой подкислили с помощью концентрированной HCl (3 мл) при 0°С. Полученные твердые вещества отфильтровали и высушили при комнатной температуре с получением Промежуточного соединения 36 (660 мг).
Синтез Соединения (11): Соединение (11) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (11). 1Н ЯМР (300 МГц, CDCl3): δ 7.89-7.91 (d, 1Н), 7.76-7.78 (d, 1Н), 7.36-7.41 (t, 1Н), 7.26-7.31 (t, 1Н), 4.79 (s, 1Н), 4.01 (s, 1Н), 3.40-3.46 (t, 2Н), 2.82-2.88 (t, 2Н), 1.98-2.02 (m, 2Н), 1.66-1.81 (m, 10Н). ЖХ-МС (М+Н)+ = 327,3; чистота по ВЭЖХ = 94,43%.
ПРИМЕР 12: 3-(1,3-бензотиазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)пропан-1-он (12)
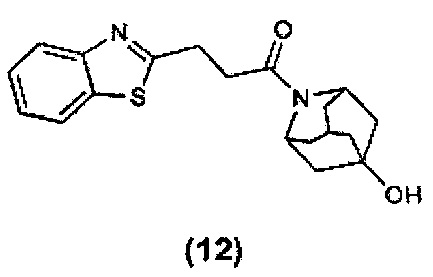
Синтез Соединения (12): Соединение (12) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (12). 1Н ЯМР (300 МГц, CDCl3): δ 7.91-7.93 (d, 1Н), 7.76-7.79 (d, 1Н), 7.37-7.43 (t, 1Н), 7.28-7.33 (t, 1Н), 4.99 (s, 1Н), 4.28 (s, 1Н), 3.42-3.47 (t, 2Н), 2.88-2.93 (t, 2Н), 2.28 (brs, 1Н), 1.51-1.79 (m, 10Н). ЖХ-МС (М+Н)+ = 343,1; чистота по ВЭЖХ = 99,27%.
ПРИМЕР 13: 3-(1,3-бензотиазол-2-ил)-1-[5-(дифторметокси)-2-азатрицикло[3.3.1.13,7]дец-2-ил]пропан-1-он (13)
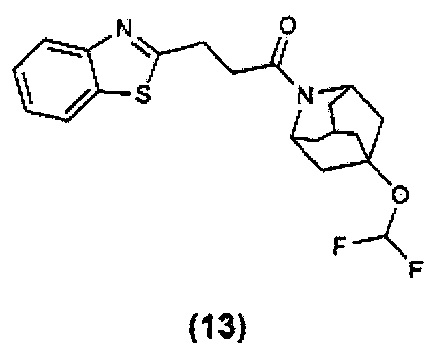
СХЕМА СИНТЕЗА 8
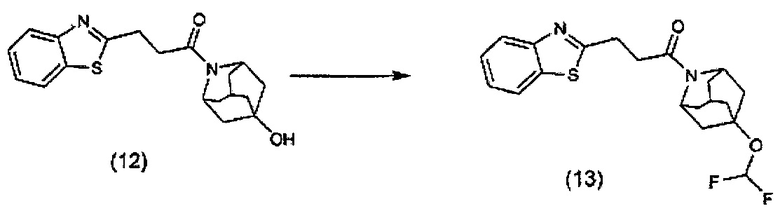
Синтез Соединения (13): К перемешанному раствору (80 мг, 0,28 ммоль) в MeCN (3 мл) добавили CuI (88 мг, 0,046 ммоль) и нагрели до 45°С. К этой смеси добавили дифтор(фторсульфонил)уксусную кислоту (23 мг, 0,46 ммоль). Полученную смесь оставили перемешиваться при той же температуре на 30 минут. После завершения реакции реакционную смесь погасили водой, экстрагировали EtOAc и концентрировали. Полученный неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя гексаном: EtOAc, с получением Соединения (13) (40 мг) в виде темно-желтого смолянистого материала. 1Н ЯМР (300 МГц, CDCl3): δ 7.87-7.89 (d, 1Н), 7.76-7.78 (d, 1Н), 7.35-7.40 (t, 1Н), 7.26-7.31 (t, 1Н), 5.97-6.48 (t, 1Н), 5.03 (brs, 1Н), 4.33 (brs, 1Н), 3.39-3.44 (t, 2Н), 2.85-2.89 (t, 2Н), 2.33 (brs, 1Н), 1.87-1.98 (m, 4Н), 1.57-1.70 (m, 6Н). ЖХ-МС (М+Н)+ = 393,2; чистота по ВЭЖХ = 89,63%.
ПРИМЕР 14: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1Н-пирроло[2,3-6]пиридин-3-ил)пропан-1-он (14)
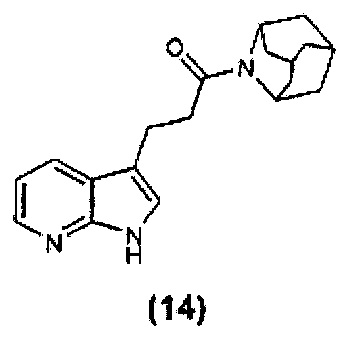
СХЕМА СИНТЕЗА 9
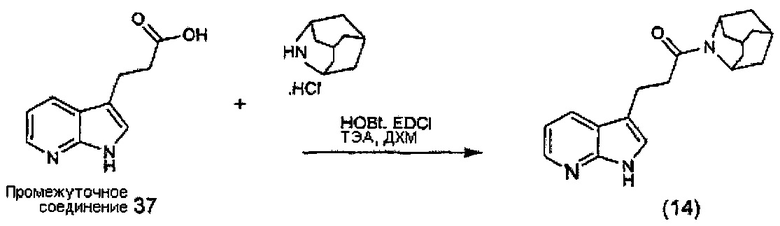
Синтез 3-(1H-пирроло[2,3-b]пиридин-3-ил)пропановой кислоты (Промежуточное соединение 37):
Промежуточное соединение 37 синтезировали по способу, использованному для получения Промежуточного соединения 26 (Схема 4).
Синтез Соединения (14): Соединение (14) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (14). 1H ЯМР (300 МГц, CDCl3): δ 8.99 (brs, 1Н), 8.22-8.21 (d, 1Н), 7.9-7.88 (d, 1Н), 7.07 (s, 1Н), 7.03-6.99 (t, 1Н), 4.81 (s, 1Н), 3.92 (s, 1H), 3.07-3.02 (t, 2H), 2.62-2.57 (t, 2H), 1.97-1.98 (m, 3H), 1.76-1.56 (m, 11Н). ЖХ-MC (M+H)+ = 310,2; чистота по ВЭЖХ = 98,28%.
ПРИМЕР 15: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-пирроло[2,3-b]пиридин-3-ил)пропан-1-он (15)
Синтез Соединения (15): Соединение (15) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (15). 1Н ЯМР (300 МГц, CDCl3): δ 9.02 (brs, 1Н), 8.20-8.22 (d, 1Н), 7.86-7.88 (d, 1Н), 7.07 (brs, 1Н), 6.99-7.03 (dd, 1Н), 5.00 (bs, 1Н), 4.10 (brs, 1Н), 3.02-3.07 (t, 2Н), 2.57-2.62 (t, 2Н), 2.22 (brs, 1Н), 1.45-1.73 (m, 10Н). ЖХ-МС (М+Н)+ = 326,1; чистота по ВЭЖХ = 98,04%.
ПРИМЕР 16: 3-(1H-бензотриазол-1-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)пропан-1-он (16)
СХЕМА СИНТЕЗА 10
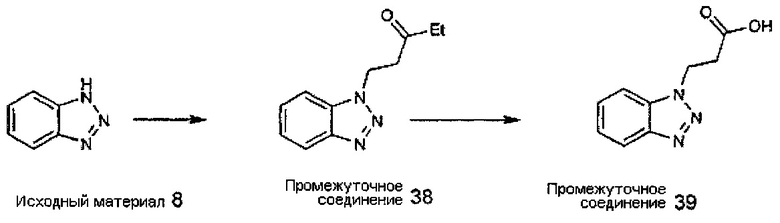
Синтез этил 3-(1Н-бензотриазол-1-ил)пропаноата (Промежуточное соединение 38):
Исходный материал 8 (4,1 ммоль) в сухом ТГФ (5 мл) охладили до 0°С, затем добавили NaH (6,0 ммоль). Реакционную смесь постепенно нагрели до комнатной температуры и оставили реагировать на 20 минут. Реакционную смесь снова охладили до 0°С, затем по каплям добавили этил 3-бромпропаноат (4,6 ммоль) в ТГФ (2,5 мл). Реакцию продолжали в течение 12 часов при комнатной температуре. Через 12 часов реакционную смесь погасили ледяной водой и экстрагировали этилацетатом. Органический слой высушили над безводным MgSO4 и концентрировали с получением Промежуточного соединения 38 (70 мг). 1Н ЯМР (300 МГц, CDCl3): δ 7.98-8.01 (1Н, d), 7.55-7.58 (d, 1Н), 7.41-7.46 (t, 1Н), 7.28-7.33 (t, 1H), 4.82-4.87 (t, 2H), 4.00-4.07 (t, 2H), 3.00-3.05 (t, 2H), 1.08-1.1 (t, 3H).
Синтез 3-(1Н-бензотриазол-1-ил)пропановой кислоты (Промежуточное соединение 39):
При 0°С LiOH (1,5 ммоль) в воде (1 мл) добавили к Промежуточному соединению 38 в растворителе ТГФ: МеОН (1:1, по 3 мл каждого). Реакцию продолжали в течение 12 часов при комнатной температуре. Через 12 часов реакционную смесь концентрировали, затем подкислили с помощью 1 н. HCl (рН=2). Реакционную смесь экстрагировали этилацетатом. Органический слой высушили над безводным MgSO4 и выпарили под пониженным давлением с получением Промежуточного соединения 39 (60 мг). 1Н ЯМР (300 МГц, CDCl3): δ 7.29-8.00 (4Н, m), 4.82-4.87 (t, 2Н), 3.09-3.14 (t, 2Н).
Синтез Соединения (16): Соединение (16) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (16). 1Н ЯМР (300 МГц, CDCl3): δ 7.23-7.97 (m, 4Н), 4.81-4.92 (m, 3Н), 4.10 (brs, 1Н), 3.01-3.05 (t, 2Н), 2.21 (brs, 1Н), 1.40-1.78 (m, 10Н). ЖХ-МС (М+Н)+ = 327,2; чистота по ВЭЖХ = 98,35%.
ПРИМЕР 17: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)бутан-1-он (17)
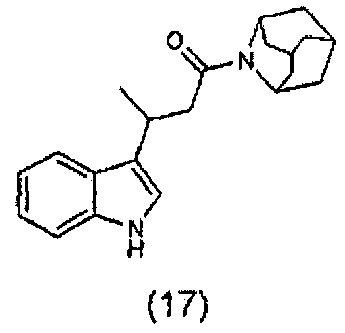
СХЕМА СИНТЕЗА 11
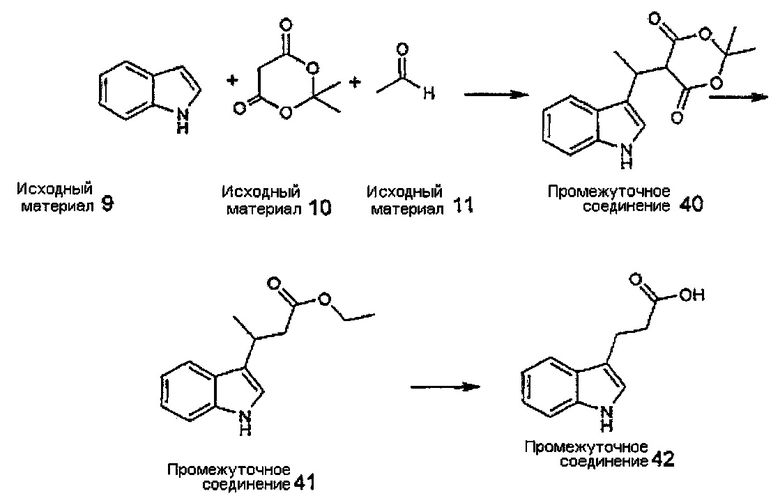
Синтез 5-[1-(1H-индол-3-ил)этил]-2,2-диметил-1,3-диоксан-4,6-диона (Промежуточное соединение 40):
В 100 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили исходный материал 9 (4,0 г, 34 ммоль), исходный материал 10 (4,92, 34 ммоль) и исходный материал 11 (3 г, 68 ммоль) в 75 мл ацетонитрила. Полученный раствор перемешивали при комнатной температуре в течение ночи. После завершения реакции (реакцию контролировали по ТСХ) растворитель удалили под пониженным давлением, а полученное неочищенное соединение очистили колоночной хроматографией на силикагеле (230-400 меш), используя петролейный эфир (60-80) и этилацетат в качестве элюента. Продукт (Промежуточное соединение 40) получили в виде коричневой жидкости (2,51 г). ЖХ-МС (М-Н)+ = 286.
Синтез этил 3-(1H-индол-3-ил)бутаноата (Промежуточное соединение 41):
В 100 мл кругло донную колбу, оснащенную магнитной мешалкой, загрузили Промежуточное соединение 40 (2,5 г, 8,7 ммоль) в 50 мл пиридина и 8 мл этанола. К этой смеси добавили порошок меди (0,4 г, 5 мол. %). Затем полученную реакционную массу нагревали с дефлегматором при 110°С в течение 3 часов. После завершения реакции (реакцию контролировали по ТСХ) растворитель удалили из реакционной массы, а реакционную массу разбавили 100 мл этилацетата. Затем реакционную массу промыли 50 мл 1,5 н. раствора HCl (2×25 мл) и насыщенным солевым раствором. Затем органический слой высушили над 10 г безводного MgSO4. Растворитель удалили под пониженным давлением, а полученное неочищенное соединение очистили колоночной хроматографией на силикагеле (230-400 меш), используя петролейный эфир (60-80) и этилацетат в качестве элюента. Продукт (Промежуточное соединение 41) получили в виде коричневой жидкости (0,380 г). ЖХ-МС (М+Н)+ = 232.
Синтез этил 3-(1H-индол-3-ил)бутановой кислоты (Промежуточное соединение 42):
В 50 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили 6 мл метанола и 2 мл воды. К перемешанному растворителю добавили Промежуточное соединение 41 (0,145 г, 0,62 ммоль) и KOH (0,098 г, 2,54 ммоль). Затем полученную реакционную массу нагревали с дефлегматором при 70°С в течение 3 часов. После завершения реакции (реакцию контролировали по ТСХ) растворитель удалили из реакционной массы, а реакционную массу разбавили 20 мл воды. Водный слой промыли 20 мл диэтилового эфира и подкислили с помощью 1 н. HCl до рН 5,5. Затем продукт экстрагировали этилацетатом, а растворитель удалили под пониженным давлением. Продукт (Промежуточное соединение 42) получили в виде коричневой жидкости (0,115 г). Продукт, полученный выше, использовали непосредственно на следующей стадии без какой-либо очистки.
Синтез Соединения (17): Соединение (17) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (17). 1Н ЯМР (300 МГц, CDCl3): δ 7.91 (brs, 1Н), 7.60-7.63 (d, 1Н), 7.27-7.30 (d, 1Н), 7.09-7.13 (t, 1Н), 7.01-7.03 (t, 1Н), 6.96 (s, 1Н), 4.80 (s, 1Н), 3.93 (s, 1Н), 3.54-3.61 (m, 1Н), 2.72-7.79 (m, 1Н), 2.45-2.50 (m, 1Н), 2.10 (brs, 1H), 1.88-1.97 (m, 1H), 1.66-1.74 (m, 5H), 1.47-1.59 (m, 5H), 1.38-1.41 (d, 3Н). ЖХ-МС (M+H)+ = 323,3; чистота по ВЭЖХ = 90,83%.
ПРИМЕР 18: 1-(4-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)бутан-1-он (18)
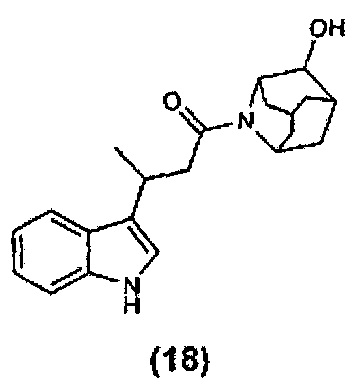
Синтез Соединения (18): Соединение (18) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (18). 1Н ЯМР (300 МГц, CDCl3): δ 7.92 (brs, 1Н), 7.75-7.54 (m, 1Н), 7.35-7.27 (m, 1Н), 7.14-7.01 (m, 2Н), 6.96 (brs, 1Н), 4.73-4.64 (d, 1Н), 3.89 (s, 1Н), 3.67 (s, 1Н), 3.37-3.60 (m, 1Н), 2.67-2.79 (m, 1Н), 2.59-2.46 (m, 1Н), 2.08-1.86 (m, 3Н), 1.75-1.60 (m, 5Н), 1.53-1.44 (m, 2Н), 1.38-1.42 (m, 3Н). ЖХ-МС (М+Н)+ = 339,2; чистота по ВЭЖХ = 98,80%.
ПРИМЕР 19: 1-(4-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1Н-индол-3-ил)-4-метилпентан-1-он (19)
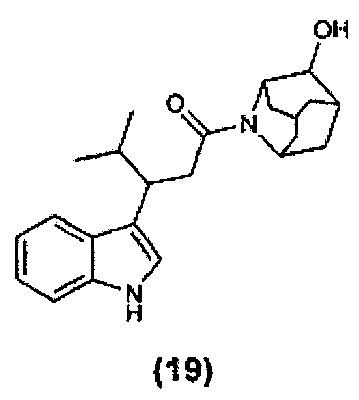
Синтез Соединения (19): Соединение (19) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (19). 1Н ЯМР (300 МГц, CDCl3): δ 8.06-7.98 (d, 1Н), 7.70-7.56 (d, 1Н), 7.36-7.24 (m, 1Н), 7.16-7.01 (m, 2Н), 6.98-6.91 (d, 1Н), 4.57-4.56 (d, 1Н), 3.83-3.39 (m, 2Н), 3.25-2.90 (m, 2Н), 2.80-2.66 (m, 1Н), 2.16-2.02 (m, 1Н), 1.98-1.66 (m, 4Н), 1.54-1.08 (m, 6Н), 0.961-0.89 (d, 6Н). ЖХ-МС (М+Н)+ = 367,3; чистота по ВЭЖХ = 98,74%.
ПРИМЕР 20: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)бутан-1-он (20)
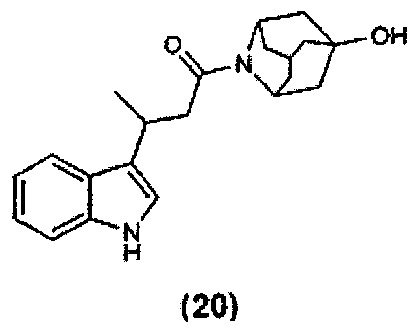
Синтез Соединения (20): Соединение (20) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (20). 1Н ЯМР (300 МГц, CDCl3): δ 7.99 (brs, 1Н), 7.59-7.62 (d, 1Н), 7.27-7.29 (d, 1Н), 7.08-7.13 (t, 1Н), 7.01-7.06 (t, 1Н), 6.96 (brs, 1Н), 4.98 (brs, 1Н), 4.09 (brs, 1Н), 3.56-3.61 (q, 1Н), 2.73-2.78 (t, 1Н), 2.43-2.50 (m, 1Н), 1.94-1.97 (m, 1Н), 1.42-1.68 (m, 10Н), 1.40 (d, 3Н). ЖХ-МС (М+Н)+ = 339,2; чистота по ВЭЖХ = 94,22%.
ПРИМЕР 21: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)бутан-1-он (21)
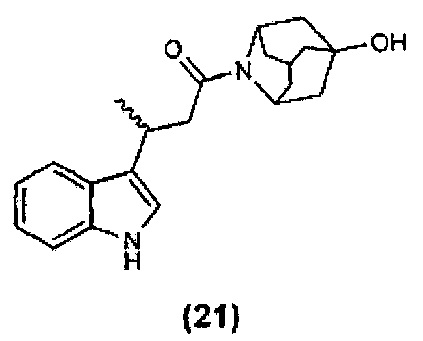
Синтез Соединения (21) (пик 1): Рацемат Соединения (20) разделили с помощью ВЭЖХ с получением энантиомера Соединения 21 (пик 1). 1Н ЯМР (300 МГц, CDCl3): δ 7.99 (brs, 1Н), 7.59-7.62 (d, 1Н), 7.27-7.29 (d, 1Н), 7.08-7.13 (t, 1Н), 7.01-7.06 (t, 1Н), 6.96 (brs, 1H), 4.98 (brs, 1H), 4.09 (brs, 1H), 3.56-3.61 (q, 1H), 2.73-2.78 (t, 1H), 2.43-2.50 (m, 1H), 1.94-1.97 (m, 1H), 1.42-1.68 (m, 10H), 1.40 (d, 3Н). ЖХ-МС (M+H)+ = 339,2; чистота по ВЭЖХ = 98,2%, хиральная чистота: (время удерживания = 19,9 мин.).
ПРИМЕР 22: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)бутан-1-он (22)
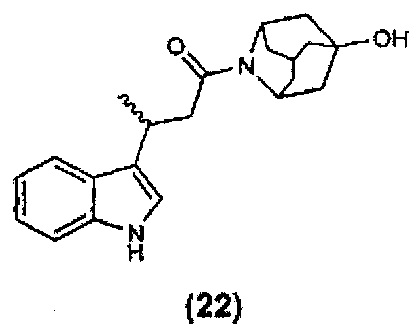
Синтез Соединения (22) (пик 1): Рацемат Соединения (20) разделили с помощью ВЭЖХ с получением энантиомера Соединения 22 (пик 2). 1H ЯМР (300 МГц, CDCl3): δ 7.99 (brs, 1Н), 7.59-7.62 (d, 1Н), 7.27-7.29 (d, 1Н), 7.08-7.13 (t, 1Н), 7.01-7.06 (t, 1Н), 6.96 (brs, 1Н), 4.98 (brs, 1Н), 4.09 (brs, 1Н), 3.56-3.61 (q, 1Н), 2.73-2.78 (t, 1Н), 2.43-2.50 (m, 1Н), 1.94-1.97 (m, 1Н), 1.42-1.68 (m, 10Н), 1.40 (d, 3Н). ЖХ-МС (М+Н)+ = 339,2; чистота по ВЭЖХ = 97,8%, хиральная чистота: (время удерживания = 22,28 мин.).
ПРИМЕР 23: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)-4-метилпентан-1-он (23)
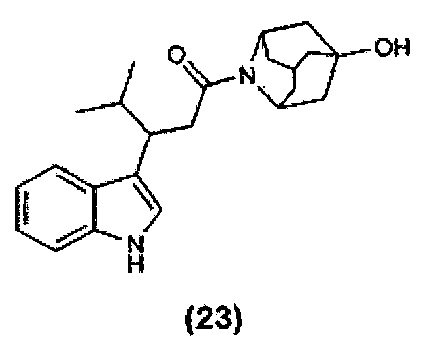
Синтез Соединения (23): Соединение (23) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (23). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.77 (s, 1Н), 7.50-7.53 (d, 1Н), 7.28-7.31 (d, 1Н), 7.07 (s, 1Н), 6.99-7.04 (t, 1Н), 6.90-6.95 (t, 1Н), 4.67 (s, 1Н), 4.55-4.62 (d, 1Н), 4.26-4.29 (d, 1Н), 3.22-3.26 (m, 1Н), 2.65-2.67 (m, 2Н), 1.98 (m, 2Н), 1.55-1.62 (d, 5Н), 1.36-1.48 (m, 3Н), 1.15-1.19 (m, 1Н), 1.01-1.06 (m, 1H), 0.77-0.87 (m, 6H). ЖХ-МС (M+H)+ = 367,2; чистота по ВЭЖХ = 85,41%.
ПРИМЕР 24: 1-(5-фтор-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)-4-метилпентан-1-он (24)
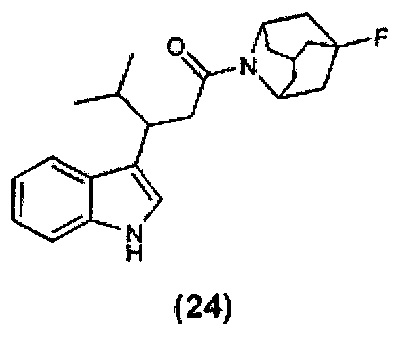
СХЕМА СИНТЕЗА 12
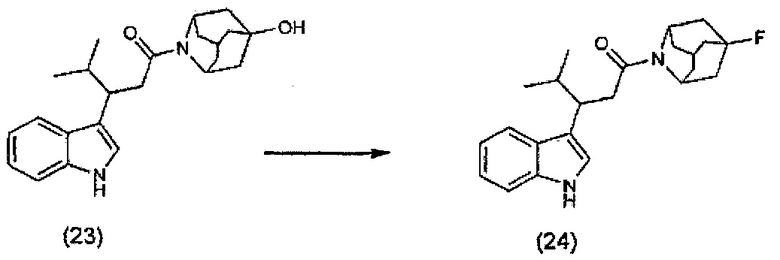
Синтез Соединения (24):
Под атмосферой N2 к перемешанному раствору соединения (23) (0,035 г, 0,09 ммоль) добавили DAST (0,015 г, 0,09 ммоль) при -78°С. Реакционную смесь перемешивали в течение 2 часов при той же температуре. После завершения реакции (реакцию контролировали по ТСХ) реакционную массу погасили раствором NaHSO3 и экстрагировали ДХМ (3×25 мл). Органический слой промыли насыщенным солевым раствором (15 мл) и концентрировали с получением неочищенного продукта. Неочищенный продукт загрузили на пластину для препаративной ТСХ (97:3, хлороформ: метанол) и собрали Соединение (24) (12 мг) в виде бледно-желтого твердого вещества. 1Н ЯМР (300 МГц, CDCl3): δ 7.94 (s, 1Н), 7.57-7.59 (d, 1Н), 7.25-7.30 (m, 1Н), 6.99-7.12 (m, 2Н), 6.93 (s, 1Н), 4.95 (s, 1Н), 4.09 (s, 1Н), 3.12-3.21 (m, 1Н), 2.72-2.82 (m, 1Н), 2.63-2.67 (m, 1Н), 2.08-2.25 (m, 3Н), 1.72-1.79 (m, 2Н), 1.61-1.69 (m, 7Н), 0.96-.98 (d, 3Н), 0.71-.81 (d, 3Н). ЖХ-МС (М+Н)+ = 369,1; чистота по ВЭЖХ = 96,16%.
ПРИМЕР 25: 3-(4-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-4-метилпентан-1-он (25)
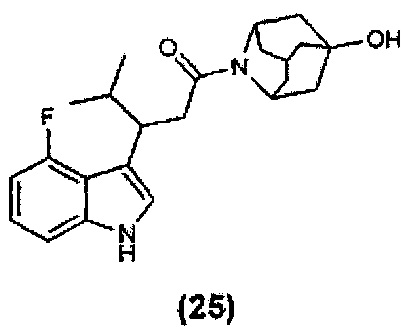
Синтез Соединения (25): Соединение (25) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (25). 1Н ЯМР (300 МГц, CDCl3): δ 8.29-8.26 (d, 1Н), 7.14-7.02 (m, 2Н), 6.98 (s, 1Н), 6.77-6.71 (t, 1Н), 4.93 (s, 1Н), 4.21 (s, 1Н), 3.25 (m, 1Н), 2.91-2.66 (m, 2Н), 2.25 (s, 1Н), 2.16-2.05 (m, 2Н), 1.72-1.34 (m, 9Н), 0.79-0.81 (d, 6Н). ЖХ-МС (М+Н)+ = 385,2; чистота по ВЭЖХ = 98,37%.
ПРИМЕР 26: 3-(4-фтор-1Н-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (26)
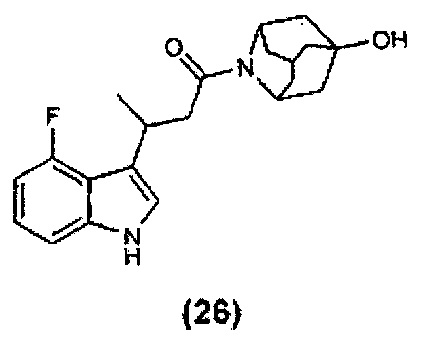
Синтез Соединения (26): Соединение (26) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (26). 1Н ЯМР (300 МГц, CDCl3): δ 8.15 (brs, 1Н), 7.00-7.09 (m, 3Н), 6.67-6.73 (t, 1Н), 4.99 (s, 1Н), 4.25 (s, 1Н), 3.59 (m, 1Н), 2.90 (s, 1Н), 2.63-2.70 (s, 1Н), 2.26-2.31 (m, 2Н), 1.85 (m, 3Н), 1.60-1.72 (m, 6Н), 1.35-1.40 (d, 3Н). ЖХ-МС (М+Н)+ = 357,2; чистота по ВЭЖХ = 97,11%.
ПРИМЕР 27: 3-(4-фтор-1Н-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (27)
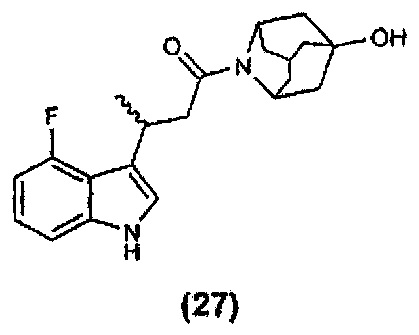
Синтез Соединения (27) (пик 1): Рацемат Соединения (26) разделили с помощью хиральной ВЭЖХ с получением энантиомера, Соединения (27) (пик 1). 1Н ЯМР (300 МГц, CDCl3): δ 8.15 (brs, 1Н), 7.00-7.09 (m, 3Н), 6.67-6.73 (t, 1Н), 4.99 (s, 1Н), 4.25 (s, 1Н), 3.59 (m, 1Н), 2.90 (s, 1Н), 2.63-2.70 (s, 1Н), 2.26-2.31 (m, 2Н), 1.85 (m, 3Н), 1.60-1.72 (m, 6Н), 1.35-1.40 (d, 3Н). ЖХ-МС (М+Н)+ = 357,2; чистота по ВЭЖХ = 99,60%; колонка: Chiralpak IA, 4,6 мм × 250 мм, подвижная фаза: гексаны : EtOH (8:2), хиральная чистота = 92,25% (время удерживания=12,52 мин.).
ПРИМЕР 28: 3-(4-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (28)
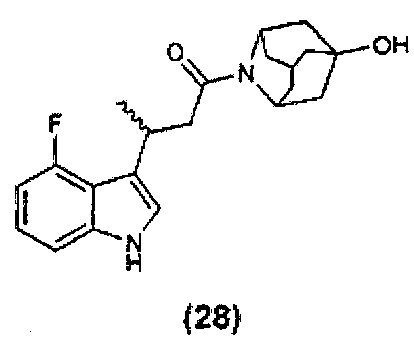
Синтез Соединения (28) (пик 2): Рацемат Соединения (26) разделили с помощью хиральной ВЭЖХ с получением энантиомера, Соединения (28) (пик 2). 1Н ЯМР (300 МГц, CDCl3): δ 8.15 (brs, 1Н), 7.00-7.09 (m, 3Н), 6.67-6.73 (t, 1Н), 4.99 (s, 1Н), 4.25 (s, 1Н), 3.59 (m, 1Н), 2.90 (s, 1Н), 2.63-2.70 (s, 1Н), 2.26-2.31 (m, 2Н), 1.85 (m, 3Н), 1.60-1.72 (m, 6Н), 1.35-1.40 (d, 3Н). ЖХ-МС (М+Н)+ = 357,2; чистота по ВЭЖХ = 94,43%; колонка: Chiralpak IA, 4,6 мм × 250 мм, подвижная фаза: гексаны : EtOH (8:2), хиральная чистота = 99,69% (время удерживания = 11,13 мин.).
ПРИМЕР 29: 3-(4-фтор-1H-индол-3-ил)-1-(5-метокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (29)
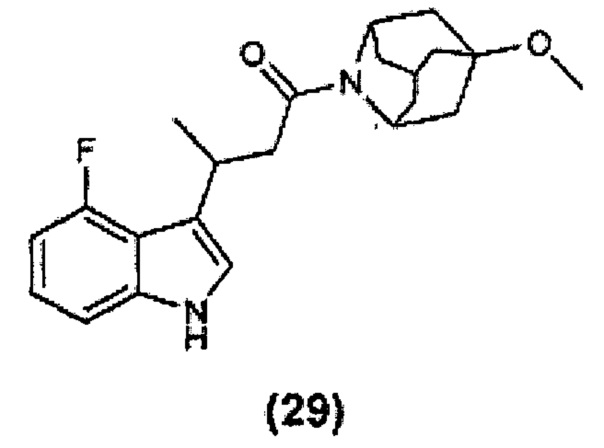
Синтез Соединения (29): Соединение (29) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (29). 1Н ЯМР (300 МГц, CDCl3): δ 8.06 (brs, 1H), 7.02-7.07 (m, 1Н), 6.97-7.01 (m, 1H), 6.94-6.95 (d, 1H), 6.66-6.72 (dd, 1H), 4.26 (brs, 1H), 4.26 (brs, 1H), 3.5-3.6 (m, 1Н), 3.05-3.12 (d, 3Н), 2.8-2.89 (m, 1H), 2.41-2.48 (m, 1H), 2.17-2.23 (m, 1H), 1.48-1.72 (m, 10Н), 1.36-1.38 (d, 3Н). ЖХ-МС (М+Н)+=371,2; чистота по ВЭЖХ = 97,80%.
ПРИМЕР 30: 3-(4-фтор-1H-индол-3-ил)-1-(5-метокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (30)
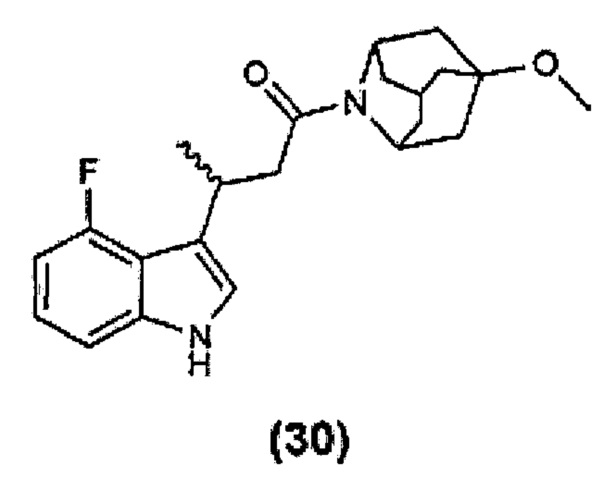
Синтез Соединения (30) (пик 1): Рацемат Соединения (29) разделили с помощью хиральной ВЭЖХ с получением энантиомера Соединения (30) (пик 1). 1H ЯМР (300 МГц, CDCl3): δ 8.06 (brs, 1), 7.02-7.07 (m, 1H), 6.97-7.01 (m, 1H), 6.94-6.95 (d, 1H), 6.66-6.72 (dd, 1H), 4.26 (brs, 1H), 4.26 (brs, 1H), 3.5-3.6 (m, 1H), 3.05-3.12 (d, 3H), 2.8-2.89 (m, 1H), 2.41-2.48 (m, 1H), 2.17-2.23 (m, 1H), 1.48-1.72 (m, 10H), 1.36-1.38 (d, 3Н). ЖХ-МС (M+H)+=371,2; чистота по ВЭЖХ = 99,56%; хиральная чистота = 99,75% (время удерживания = 13,52 мин., колонка: Chiral pack IA, 4,6 мм × 250 мм, подвижная фаза: МТБЭ: МеОН (98:02).
ПРИМЕР 31: 3-(4-фтор-1H-индол-3-ил)-1-(5-метокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (31)
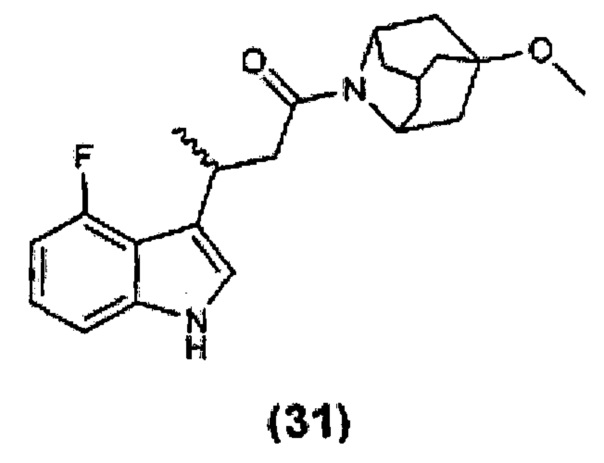
Синтез Соединения (31) (пик 2): Рацемат Соединения (29) разделили с помощью хиральной ВЭЖХ с получением энантиомера Соединения (31). 1Н ЯМР (300 МГц, CDCl3): δ 8.06 (brs, 1), 7.02-7.07 (m, 1Н), 6.97-7.01 (m, 1H), 6.94-6.95 (d, 1H), 6.66-6.72 (dd, 1H), 4.26 (brs, 1H), 4.26 (brs, 1H), 3.5-3.6 (m, 1H), 3.05-3.12 (d, 3H), 2.8-2.89 (m, 1H), 2.41-2.48 (m, 1H), 2.17-2.23 (m, 1H), 1.48-1.72 (m, 10H), 1.36-1.38 (d, 3Н). ЖХ-МС (M+H)+ = 371,2; чистота по ВЭЖХ = 95,79%; хиральная чистота = 99,88% (время удерживания = 16,37 мин., колонка: Chiral pack IA, 4,6 мм × 250 мм, подвижная фаза: МТБЭ: МеОН (98:02).
ПРИМЕР 32: 1-(5-хлор-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(4-фтор-1H-индол-3-ил)бутан-1-он (32)
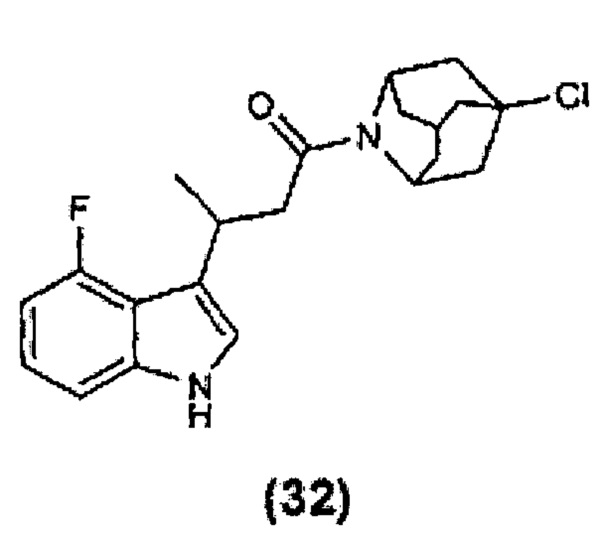
СХЕМА СИНТЕЗА 13
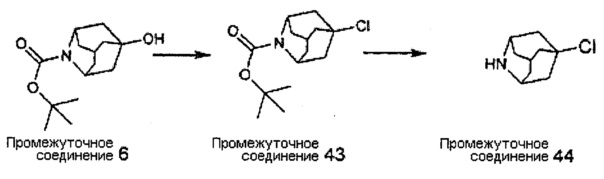
Синтез трет-бутил 5-хлор-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилата (Промежуточное соединение 43):
К перемешанному раствору Промежуточного соединения 6 (70 мг, 0,19 ммоль) в CCl4 (3 мл) добавили SOCl2 (1,5 мл) и нагревали реакционную смесь при 80°С в течение 15 часов. После завершения реакции (реакцию контролировали по ЖХ-МС) реакционную массу концентрировали с получением Промежуточного соединения 43 (60 мг).
Синтез 5-хлор-2-азатрицикло [3.3.1.13,7]декана, трифторуксуснокислой соли (Промежуточное соединение 44):
Промежуточное соединение 44 синтезировали по способу, использованному для получения Промежуточного соединения 20 (Схема 1).
Синтез Соединения (32): Соединение (32) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат в качестве элюента, с получением Соединения (32). 1Н ЯМР (300 МГц, CDCl3): δ 8.02-8.06 (d, 1Н), 7.01-7.09 (m, 2Н), 6.94 (s, 1Н), 6.67-6.73 (m, 1Н), 4.95 (s, 1H), 4.20 (s, 1H), 3.54-3.61 (m, 1H), 2.79-2.88 (m, 1Н), 2.40-2.49 (m, 1H), 2.12-2.26 (m, 4Н), 1.81-2.02 (m, 3Н), 1.58 (m, 4Н), 1.36-1.43 (d, 3Н). ЖХ-МС (М+Н)+ = 376,1; чистота по ВЭЖХ = 90,30%.
ПРИМЕР 33: 1-[5-(циклопропилметокси)-2-азатрицикло[3.3.1.13,7]дец-2-ил]-3-(4-фтор-1H-индол-3-ил)бутан-1-он (33)
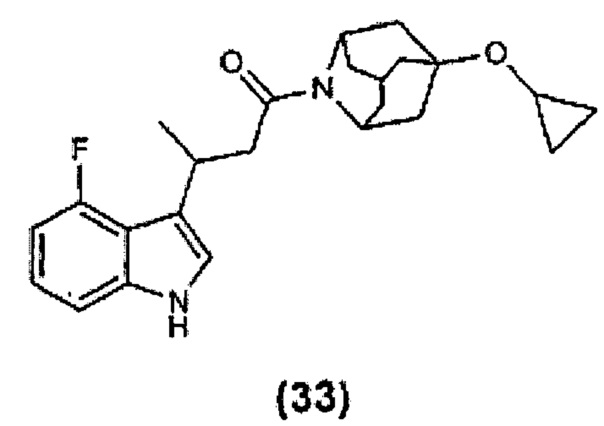
Синтез Соединения (33): Соединение (33) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (33). 1Н ЯМР (300 МГц, CDCl3): δ 8.07 (s, 1H), 6.97-7.07 (m, 2Н), 6.95-6.94 (d, 1Н), 6.66-6.72 (t, 1H), 4.97 (s, 1Н), 4.25 (s, 1H), 3.53-3.60 (m, 1Н), 3.00-312 (dd, 2Н), 2.79-2.89 (m, 1H), 2.40-2.50 (m, 1H), 2.16-2.25 (d, 1Н), 2.13-2.26 (d, 1H), 1.54-1.74 (m, 8Н), 1.36-1.38 (d, 3Н), 0.87-.89 (m, 1Н), 0.41-0.47 (m, 2Н), 0.07 (m, 2Н). ЖХ-МС (М+Н)+ = 411,2; чистота по ВЭЖХ = 96,10%.
ПРИМЕР 34: 3-(1H-индол-3-ил)-1-(5-метокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (34)
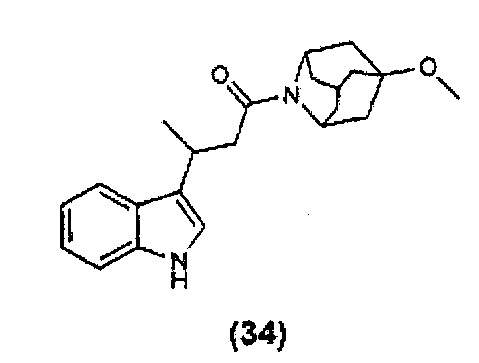
Синтез Соединения (34): Соединение (34) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (34). 1Н ЯМР (300 МГц, CDCl3): δ 7.95 (brs, Ш), 7.59-7.62 (d, 1H), 7.26-7.29 (d, 1Н), 7.07 (m, 2Н), 6.95 (s, 1Н), 4.99 (brs, 1H), 4.10 (brs, 1H), 3.50-3.67 (m, 1H), 3.00-3.12 (d, 3Н), 2.74-2.82 (m, 1Н), 2.42-2.52 (m, 1H), 2.15-2.12 (m, 1Н), 1.46-1.72 (m, 6Н), 1.36-1.38 (d, 3Н), 0.99-1.46 (m, 4Н). ЖХ-МС (М+Н)+ = 353,2; чистота по ВЭЖХ = 90,44%.
ПРИМЕР 35: 3-(4-хлор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (35)
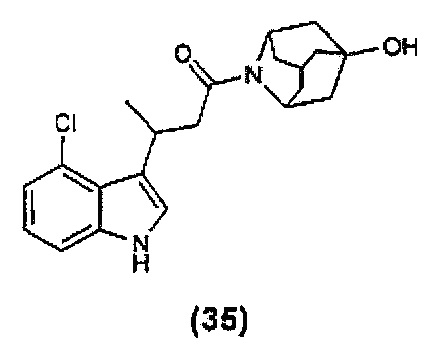
Синтез Соединения (35): Соединение (35) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (35). 1Н ЯМР (300 МГц, CDCl3): δ 8.44 (brs, 1H), 7.01-7.19 (m, 1H), 6.95-7.011 (m, 3Н), 4.99 (brs, 1Н), 4.23 (brs, 1Н), 3.99-4.06 (m, 1H), 2.82-2.87 (m, 1Н), 2.40-2.50 (m, 1Н), 1.34-2.22 (m, 14Н). ЖХ-МС (М+Н)+ = 373,2; чистота по ВЭЖХ = 93,64%.
ПРИМЕР 36: 3-(4-хлор-1Я-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (36)
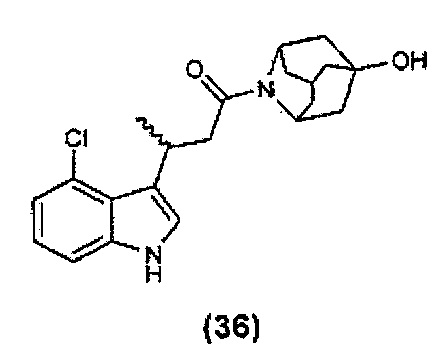
Синтез Соединения (36) (пик 1): Рацемат Соединения (35) разделили с помощью хиральной ВЭЖХ с получением энантиомера соединения (36) (пик 1). 1Н ЯМР (300 МГц, CDCl3): δ 8.44 (brs, 1Н), 7.01-7.19 (m, 1Н), 6.95-7.011 (m, 3Н), 4.99 (brs, 1H), 4.23 (brs, 1Н), 3.99-4.06 (m, 1Н), 2.82-2.87 (m, 1Н), 2.40-2.50 (m, 1Н), 1.34-2.22 (m, 14Н). ЖХ-МС (М+Н)+ = 373,2; чистота по ВЭЖХ = 91,31%; хиральная чистота = 98,36% (время удерживания = 17,89 мин.).
ПРИМЕР 37: 3-(4-хлор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (37)
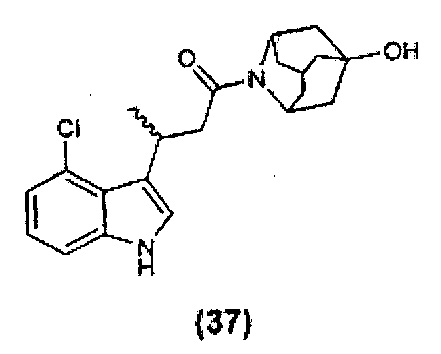
Синтез Соединения (37) (пик 2): Рацемат Соединения (35) разделили с помощью хиральной ВЭЖХ с получением энантиомера соединения (37) (пик 2). 1Н ЯМР (300 МГц, CDCl3): δ 8.44 (brs, 1H), 7.01-7.19 (m, 1Н), 6.95-7.011 (m, 3Н), 4.99 (brs, 1H), 4.23 (brs, 1Н), 3.99-4.06 (m, 1Н), 2.82-2.87 (m, 1H), 2.40-2.50 (m, 1Н), 1.34-2.22 (m, 14Н). ЖХ-МС (М+Н)+ = 373,2; чистота по ВЭЖХ = 97,96%; хиральная чистота = 99,11% (время удерживания = 15,94 мин.).
ПРИМЕР 38: 1-[5-(циклопропилметокси)-2-азатрицикло[3.3.1.13,7]дец-2-ил]-3-(1H-индол-3-ил)бутан-1-он (38)
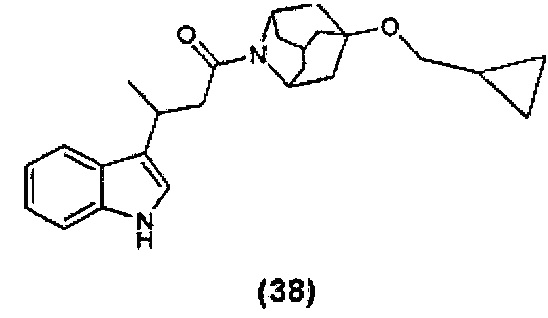
Синтез Соединения (38): Соединение (38) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (38). 1Н ЯМР (300 МГц, CDCl3): δ 7.92 (brs, 1H), 7.59-7.62 (d, 1H), 7.26-7.29 (d, 2Н), 7.01-7.02 (m, 2Н), 4.98 (brs, 1Н), 4.09 (brs, 1H), 3.45-3.61 (m, 1Н), 3.08-3.16 (dd, 2Н), 2.55-2.64 (m, 2Н), 2.09-2.36 (m, 2Н), 1.61-1.82 (m, 9Н), 1.41-1.43 (d, 3Н), 0.86-0.89 (m, 1Н), 0.41-0.47 (m, 2Н), 0.07 (m, 2Н). ЖХ-МС (М+Н)+ = 393,3; чистота по ВЭЖХ = 95,10%.
ПРИМЕР 39: 3-(4-хлор-1H-индол-3-ил)-1-[5-(дифторметокси)-2-азатрицикло[3.3.1.13,7]дец-2-ил]бутан-1-он (39)
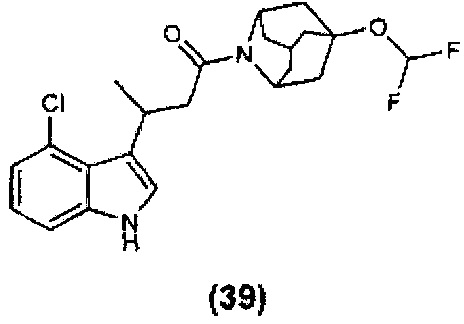
Синтез Соединения (39): Соединение (39) синтезировали по способу, использованному для получения Соединения (1) (Схема 2) и Соединения (13) (Схема 8). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат в качестве элюента, с получением Соединения (39). 1Н ЯМР (300 МГц, CDCl3): δ 8.22 (brs, 1H), 7.06 (brs, 2Н), 6.99-7.01 (m, 2Н), 5.9-6.4 (t, 1Н), 5.03 (brs, 1Н), 4.26 (brs, 1H), 4.03-4.04 (m, 1Н), 2.83-2.87 (m, 1H), 2.49-2.57 (m, 1Н), 2.28 (brs, 1Н), 1.93 (m, 1Н), 1.62-2.01 (m, 10Н), 1.38-1.41 (d, 3Н). ЖХ-МС (М+Н)+ = 423,2; чистота по ВЭЖХ = 92,19%.
ПРИМЕР 40: 3-(4-бром-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (40)
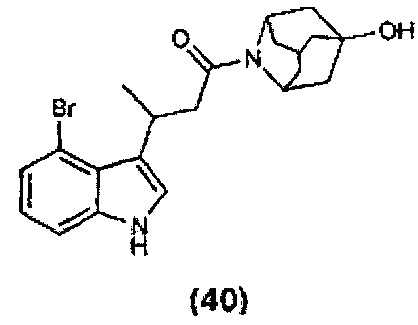
Синтез Соединения (40): Соединение (40) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (40). 1Н ЯМР (300 МГц, ДМСО-d6): δ 11.21 (brs, 1Н), 7.32-7.36 (m, 2Н), 7.14-7.17 (d, 1H), 6.92-6.97 (t, 1Н), 4.80 (brs, 1H), 4.66 (s, 1Н), 4.33 (bs, 1H), 3.99-4.06 (m, 1Н), 2.50-2.55 (m, 1H), 2.19 (brs, 1H), 1.69 (brs, 2Н), 1.62-2.01 (m, 8Н), 1.38-1.41 (d, 3Н). ЖХ-МС (М+Н)+ = 418,1; чистота по ВЭЖХ = 92,77%.
ПРИМЕР 41: 3-(4-циклопропил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (41)
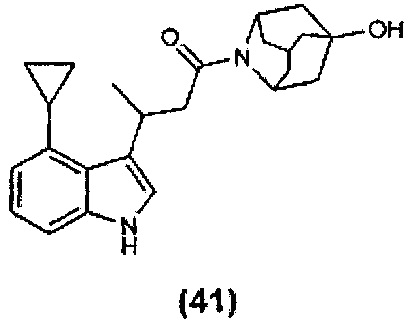
Синтез Соединения (41): Соединение (41) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (41). 1Н ЯМР (300 МГц, CDCl3): δ 7.97 (brs, 1H), 7.01-7.1 (d, 1H), 6.97-7.02 (m, 2Н), 6.66-6.68 (d, 1Н), 5.03 (brs, 1H), 4.19 (brs, 2Н), 2.77-2.83 (m, 1Н), 2.35-2.44 (m, 2Н), 2.24 (brs, 1H), 1.75 (brs, 2Н), 1.57-1.66 (m, 8Н), 1.36-1.38 (d, 3Н), 0.73-0.94 (m, 4Н). ЖХ-МС (М+Н)+ = 379,2; чистота по ВЭЖХ = 97,64%.
ПРИМЕР 42: 3-[4-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-4-оксобутан-2-ил]-1H-индол-4-карбонитрил (42)
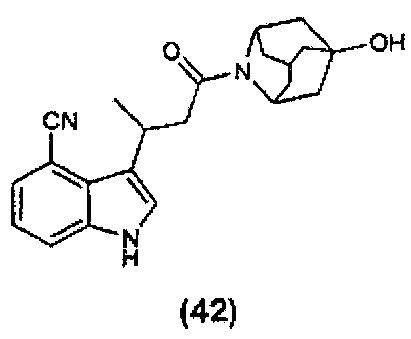
Синтез Соединения (42): Соединение (42) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (42). 1Н ЯМР (300 МГц, CDCl3): δ 8.97 (s, 1H), 7.47-7.50 (d, 1H), 7.38-7.40 (d, 1Н), 7.18 (m, 1H), 7.07-7.13 (m, 1Н), 4.95 (s, 1H), 4.30 (s, 1Н), 3.86-3.93 (m, 1Н), 2.81-2.91 (m, 1Н), 2.56-2.66 (m, 1Н), 2.23 (s, 1H), 1.57-1.74 (m, 10Н), 1.39-1.41 (d, 3Н). ЖХ-МС (М+Н)+ = 364,2; чистота по ВЭЖХ = 97,36%.
ПРИМЕР 41: 3-(4-циклопропил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (41)
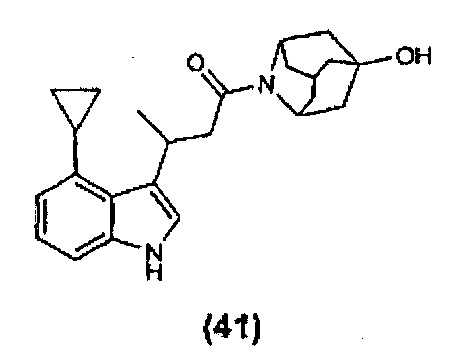
Синтез Соединения (41): Соединение (41) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (41). 1Н ЯМР (300 МГц, CDCl3): δ 7.97 (brs, 1H), 7.01-7.1 (d, 1Н), 6.97-7.02 (m, 2Н), 6.66-6.68 (d, 1Н), 5.03 (brs, 1Н), 4.19 (brs, 2Н), 2.77-2.83 (m, 1H), 2.35-2.44 (m, 2Н), 2.24 (brs, 1Н), 1.75 (brs, 2Н), 1.57-1.66 (m, 8Н), 1.36-1.38 (d, 3Н), 0.73-0.94 (m, 4Н). ЖХ-МС (М+Н)+ = 379,2; чистота по ВЭЖХ = 97,64%.
ПРИМЕР 42: 3-[4-(5-гидрокси-2-азатрицикло [3.3.1.13,7]дец-2-ил)-4-оксобутан-2-ил]-1H-индол-4-карбонитрил (42)
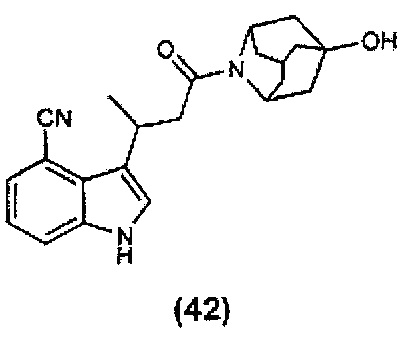
Синтез Соединения (42): Соединение (42) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (42). 1Н ЯМР (300 МГц, CDCl3): δ 8.97 (s, 1H), 7.47-7.50 (d, 1H), 7.38-7.40 (d, 1Н), 7.18 (m, 1H), 7.07-7.13 (m, 1Н), 4.95 (s, 1Н), 4.30 (s, 1Н), 3.86-3.93 (m, 1H), 2.81-2.91 (m, 1H), 2.56-2.66 (m, 1Н), 2.23 (s, 1Н), 1.57-1.74 (m, 10Н), 1.39-1.41 (d, 3Н). ЖХ-МС (М+Н)+ = 364,2; чистота по ВЭЖХ = 97,36%.
ПРИМЕР 43: 2-[3-(4-хлор-1H-индол-3-ил)бутаноил]-2-азатрицикло [3.3.1.13,7]декан-5-карбоновая кислота (43)
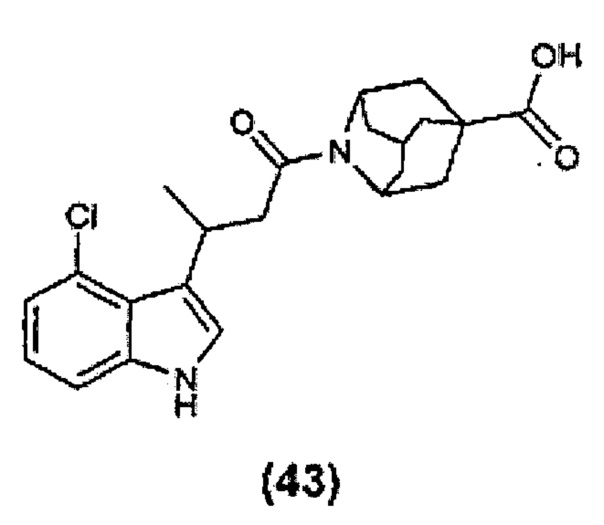
СХЕМА СИНТЕЗА 14
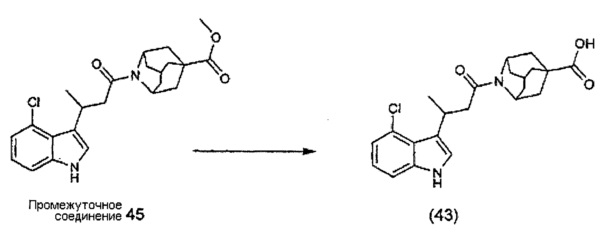
Синтез Соединения (43): Соединение (43) синтезировали по способу, использованному для получения Промежуточного соединения 26 (Схема 4). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (43). 1Н ЯМР (300 МГц, CDCl3): δ 12.25 (s, 1Н), 11.20 (s, 1Н), 7.29-7.31 (m, 2Н), 6.96-7.04 (m, 2Н), 4.78 (s, 1Н), 4.28 (s, 1H), 3.92-3.99 (m, 1H), 2.55-2.76 (m, 2Н), 2.11 (s, 1Н), 1.59-1.89 (m, 10Н), 1.26-1.28 (d, 3Н). ЖХ-МС (М+Н)+ = 401,1; чистота по ВЭЖХ = 89,33%.
ПРИМЕР 44: 3-[4-(4-хлорфенокси)-1H-индол-3-ил]-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (44)
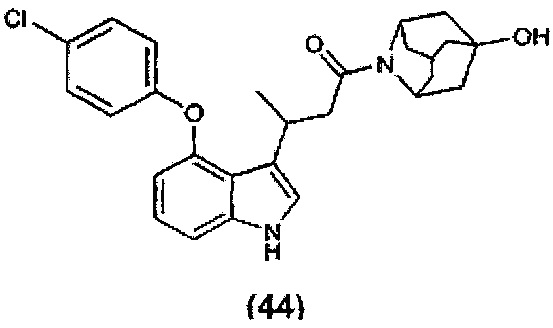
Синтез Соединения (44): Соединение (44) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (44). 1Н ЯМР (300 МГц, CDCl3): δ 8.12 (s, 1H), 7.21-7.24 (m, 2Н), 7.04-7.07 (m, 2Н), 6.94-7.01 (m, 2Н), 6.92-6.93 (m, 1Н), 6.40-6.43 (d, 1H), 4.94 (s, 1H), 4.11 (s, 1Н), 3.57-3.63 (m, 1Н), 2.78-2.85 (m, 1Н), 2.32-2.42 (m, 1Н), 2.16 (s, 1H), 1.66-1.69 (d, 3Н), 1.43-1.47 (m, 7Н), 1.38-1.39 (d, 3Н). ЖХ-МС (М+Н)+ = 465,2; чистота по ВЭЖХ = 99,73%.
ПРИМЕР 45: 2-[3-(4-хлор-1H-индол-3-ил)бутаноил]-N-(4-фторфенил)-2-азатрицикло[3.3.1.13,7]декан-5-карбоксамид (45)
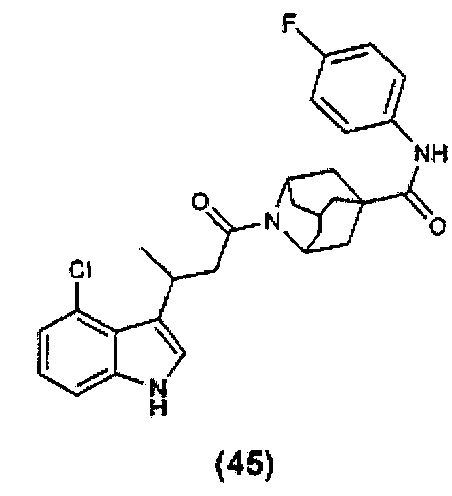
СХЕМА СИНТЕЗА 15
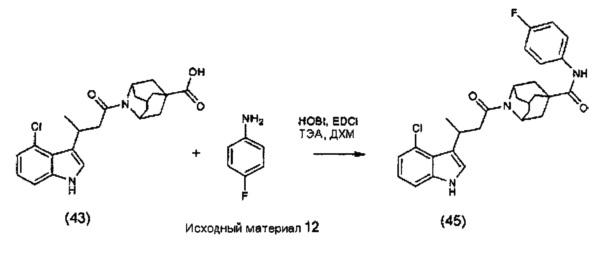
Синтез Соединения (45): Соединение (45) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (45). 1Н ЯМР (300 МГц, CDCl3): δ 8.21-8.24 (d, 1Н), 7.34-7.42 (m, 2Н), 7.19 (m, 2Н), 6.91-7.04 (m, 5Н), 4.93 (s, 1Н), 4.18 (s, 1H), 4.01-4.12 (m, 1H), 2.80-2.89 (m, 1H), 2.41-2.59 (m, 1Н), 2.19-2.26 (m, 1Н), 1.60-1.97 (m, 10Н), 1.37-1.40 (m, 3Н). ЖХ-МС (М+Н)+ = 494,2; чистота по ВЭЖХ = 98,06%.
ПРИМЕР 46: 3-[4-хлор-1-(метилсульфонил)-1H-индол-3-ил]-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (46)
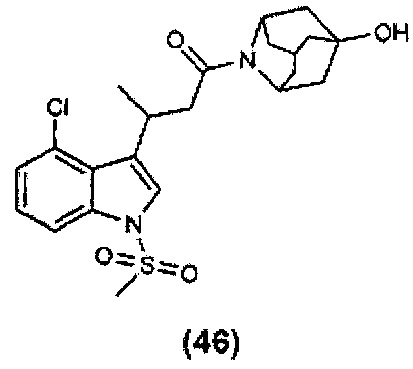
Синтез Соединения (46): Соединение (46) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (46). 1Н ЯМР (300 МГц, CDCl3): δ 7.74-7.77 (m, 1H), 7.20-7.22 (m, 3Н), 5.01 (s, 1H), 4.22 (s, 1Н), 4.08 (s, 1H), 3.02 (s, 3Н), 2.73-2.802 (m, 1H), 2.33-2.42 (m, 1Н), 2.28 (s, 1Н), 1.77 (s, 2Н), 1.60-1.66 (m, 9Н), 1.34-1.36 (d, 3Н). ЖХ-МС (М+Н)+ = 451,1; чистота по ВЭЖХ = 94,52%.
ПРИМЕР 47: 2-[3-(4-хлор-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоксамид (47)
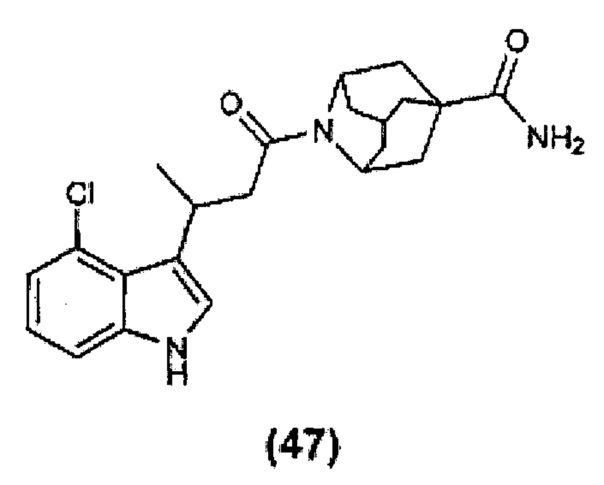
СХЕМА СИНТЕЗА 16
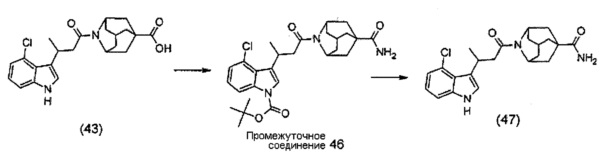
Синтез трет-бутил 3-(4-(5-карбамоил-2-азаадамантан-2-ил)-4-оксобутан-2-ил)-4-хлор-1Н-индол-1-карбоксилата (Промежуточное соединение 46): К перемешанному раствору соединения 43 (0,070 г, 0,17 ммоль) в MeCN (2 мл) добавили пиридин (0,016 г, 0,21 ммоль), затем добавили ди-трет-бутилдикарбонат (0,045 г, 0,21 ммоль) и перемешивали в течение 1 часа при комнатной температуре. К этому раствору добавили твердый бикарбонат аммония (0,021 г, 0,27 ммоль) и перемешивали при комнатной температуре в течение 12 часов. После завершения реакции реакционную смесь погасили Н2О и экстрагировали EtOAc, и концентрировали с получением Промежуточного соединения 46 (30 мг) в виде белого твердого вещества.
Синтез Соединения (47): К перемешанному раствору Промежуточного соединения 46 (0,030 г, 0,017 ммоль) в ДХМ (1 мл) добавили ТФК (0,013 г, 0,11 ммоль) при 0°С и перемешивали при комнатной температуре в течение 4 часов. После завершения реакции реакционную смесь концентрировали для удаления ДХМ и ТФК. Реакционную смесь затем разбавили Н2О и экстрагировали EtOAc, а затем концентрировали с получением неочищенного материала, который очистили с помощью силикагелевой колоночной хроматографии, элюируя смесью гексанов: EtOAc, с получением Соединения (47) (145 мг) в виде белого твердого вещества. 1Н ЯМР (300 МГц, CDCl3): δ 11.20 (s, 1Н), 7.29-7.31 (d, 2Н), 6.96-7.04 (m, 3Н), 6.81 (s, 1Н), 4.74 (s, 1H), 4.28 (s, 1H), 3.89-4.02 (m, 1H), 2.68-2.72 (m, 2Н), 2.10 (s, 1H), 1.62-1.85 (m, 10Н), 1.26-1.28 (d, 3Н). ЖХ-МС (М+Н)+ = 400,2; чистота по ВЭЖХ = 99,92%.
ПРИМЕР 48: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(4-метил-1H-индол-3-ил)бутан-1-он (48)
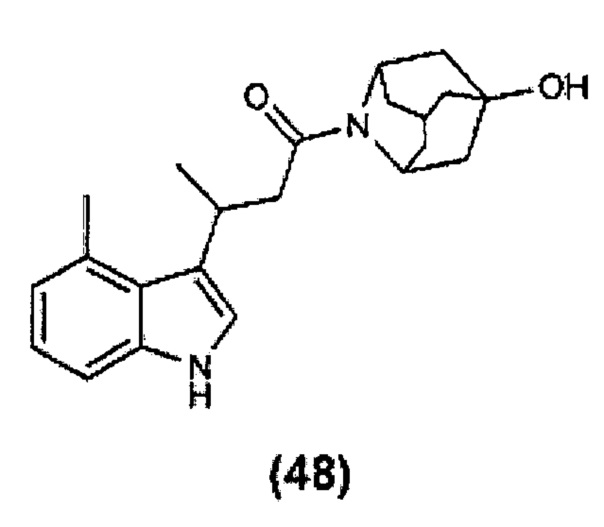
СХЕМА СИНТЕЗА 17
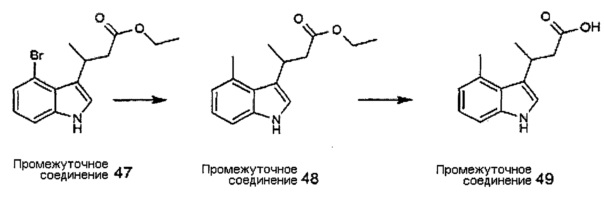
Синтез 3-(4-бром-1H-индол-3-ил)бутаноата (Промежуточное соединение 47):
Промежуточное соединение 47 синтезировали по способу, использованному для получения Промежуточного соединения 41 (Схема 11).
Синтез этил 3-(4-метил-1H-индол-3-ил)бутаноата (Промежуточное соединение 48): В 100 мл круглодонную колбу, оснащенную магнитной мешалкой и обратным холодильником, загрузили 25 мл толуола и 5 мл воды. К перемешанному растворителю добавили Промежуточное соединение 47 (4,4 г, 14,185 ммоль), затем добавили метилбороновую кислоту (1,696 г, 28,37 ммоль), трехосновной фосфат калия (10,535 г, 49,647 ммоль) и трициклогексилфосфин (0,397 г, 1,4185 ммоль). Полученную массу перемешивали при комнатной температуре, продувая аргоном, в течение 30 минут. Затем добавили ацетат палладия (0,159, 0,7092 ммоль) и перемешивали полученную смесь при 100°С в течение 16 часов. После завершения реакции реакционную массу разбавили 10 мл воды и экстрагировали этилацетатом (100 мл × 3), а объединенный органический слой промыли насыщенным солевым раствором и высушили над безводным сульфатом натрия и удалили растворитель из органического слоя под пониженным давлением с получением неочищенного соединения. Неочищенную массу очистили колоночной хроматографией, используя 60-120 силикагель и 8% этилацетата в петролейном эфире в качестве элюента, с получением Промежуточного соединения 48 (2,55 г).
Синтез 3-(4-метил-1H-индол-3-ил)бутановой кислоты (Промежуточное соединение 49):
Промежуточное соединение 49 синтезировали по способу, использованному для получения Промежуточного соединения 42 (Схема 11).
Синтез Соединения (48): Соединение (48) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (48). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.78 (s, 1Н), 7.12-7.15 (d, 2Н), 6.87-6.92 (t, 1H), 6.67-6.69 (d, 1Н), 4.80 (s, 1H), 4.64-4.65 (d, 1Н), 4.33 (s, 1H), 3.69-3.76 (m, 1H), 2.63-2.68 (m, 2Н), 2.60 (s, 3Н), 2.15-2.19 (d, 1H), 1.68 (s, 2Н), 1.42-1.63 (m, 8Н), 1.23-1.25 (d, 3Н). ЖХ-МС (М+Н)+ = 353,2; чистота по ВЭЖХ = 98,43%.
ПРИМЕР 49: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(4-метил-1H-индол-3-ил)бутан-1-он (49)
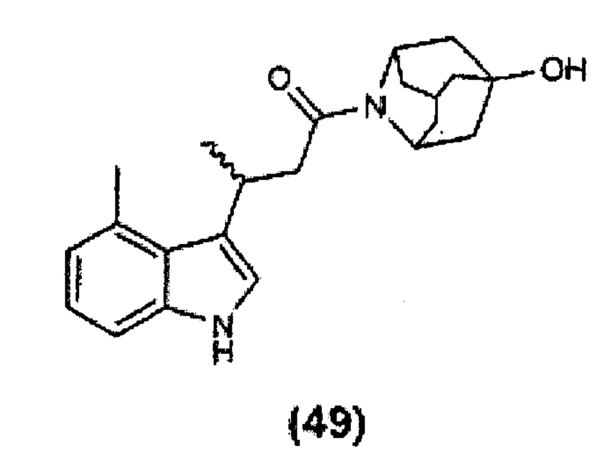
Синтез Соединения (49) (пик 1): Рацемат Соединения (48) разделили с помощью хиральной ВЭЖХ с получением энантиомера Соединения (49) (пик 1). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.78 (s, 1Н), 7.12-7.15 (d, 2Н), 6.87-6.92 (t, 1H), 6.67-6.69 (d, 1Н), 4.80 (s, 1Н), 4.64-4.65 (d, 1Н), 4.33 (s, Ш), 3.69-3.76 (m, 1Н), 2.63-2.68 (m, 2Н), 2.60 (s, 3Н), 2.15-2.19 (d, 1H), 1.68 (s, 2Н), 1.42-1.63 (m, 8Н), 1.23-1.25 (d, 3Н). ЖХ-МС (М+Н)+ = 353,2; чистота по ВЭЖХ = 95,87%; хиральная чистота = 100% (время удерживания = 17,45 мин.), колонка: Chiralpak IC 4,6 мм × 250 мм, подвижная фаза: гексан : IPA : ДХМ (75:15:10).
ПРИМЕР 50: 1-(5-гидрокси-2-азатрицикло [3.3.1.13,7]дец-2-ил)-3-(4-метил-1H-индол-3-ил)бутан-1-он (50)
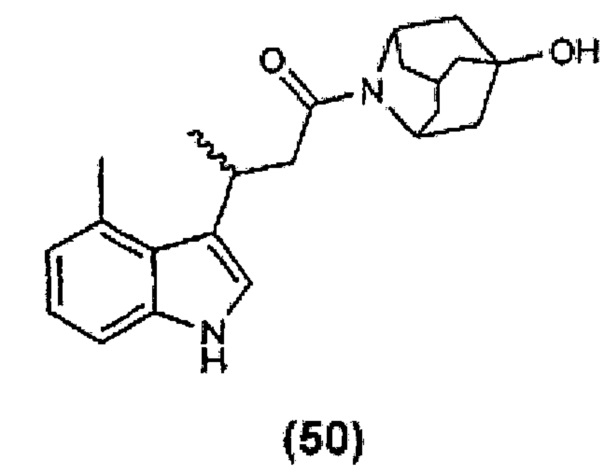
Синтез Соединения (50) (пик 2): Рацемат Соединения (48) разделили с помощью хиральной ВЭЖХ с получением энантиомера Соединения (50) (пик 2). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.78 (s, 1Н), 7.12-7.15 (d, 2H), 6.87-6.92 (t, 1H), 6.67-6.69 (d, 1H), 4.80 (s, 1H), 4.64-4.65 (d, 1H), 4.33 (s, 1H), 3.69-3.76 (m, 1H), 2.63-2.68 (m, 2H), 2.60 (s, 3H), 2.15-2.19 (d, 1H), 1.68 (s, 2H), 1.42-1.63 (m, 8H), 1.23-1.25 (d, 3Н). ЖХ-МС (M+H)+ = 353,2; чистота по ВЭЖХ = 98,64%; хиральная чистота = 100% (время удерживания = 21,17 мин.), колонка: Chiralpak IC 4,6 мм × 250 мм, подвижная фаза: гексан : IPA : ДХМ (75:15:10).
ПРИМЕР 51: 2-[3-(4-циклопропил-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (51)
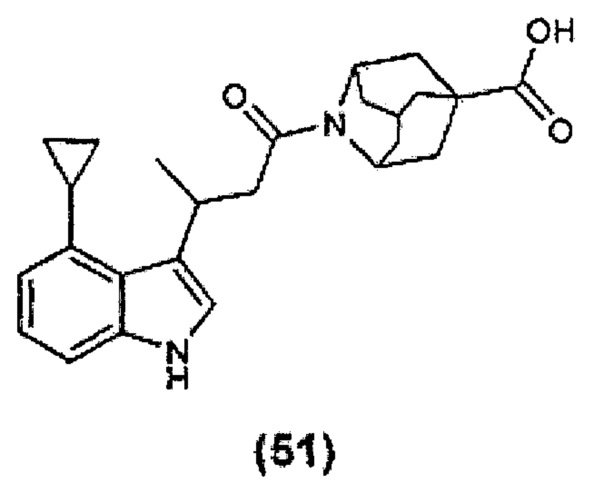
Синтез Соединения (51): Соединение (51) синтезировали по способу, использованному для получения Соединения 43 (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный ДХМ: МеОН в качестве элюента, с получением Соединения (51). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.23 (s, 1Н), 10.801 (s, 1Н), 7.11-7.17 (m, 2Н), 6.88-6.93 (t, 1Н), 6.55-6.58 (d, 1H), 4.75 (s, 1Н), 4.26 (s, 1H), 4.01-4.04 (m, 1Н), 3.37-3.41 (m, 2Н), 2.68-2.73 (m, 1H), 2.09 (s, 1Н), 1.62-1.88 (m, 13Н), 0.85-00.91 (m, 2Н), 0.70-0.77 (m, 2Н). ЖХ-МС (М+Н)+ = 407,2; чистота по ВЭЖХ = 91,19%.
ПРИМЕР 52: 2-[3-(4-циклопропил-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (52)
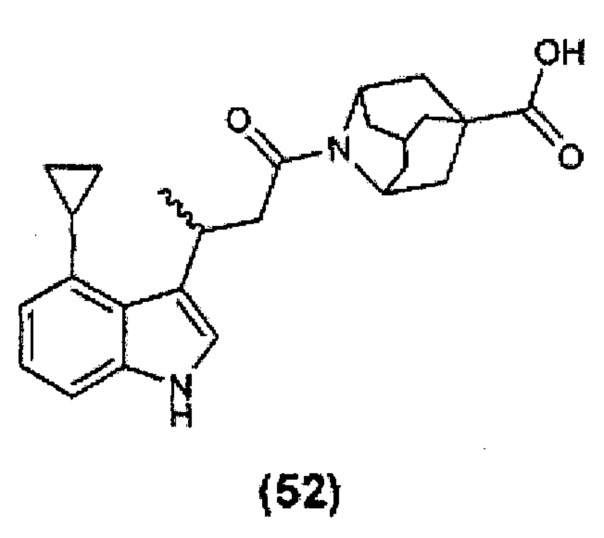
Синтез Соединения (52) (пик 1): Рацемат Соединения (51) разделили с помощью хиральной ВЭЖХ с получением энантиомера Соединения (52) (пик 1). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.23 (s, 1Н), 10.801 (s, 1Н), 7.11-7.17 (m, 2Н), 6.88-6.93 (t, 1H), 6.55-6.58 (d, 1Н), 4.75 (s, 1H), 4.26 (s, 1Н), 4.01-4.04 (m, 1Н), 3.37-3.41 (m, 2Н), 2.68-2.73 (m, 1H), 2.09 (s, 1Н), 1.62-1.88 (m, 13Н), 0.85-00.91 (m, 2Н), 0.70-0.77 (m, 2Н). ЖХ-МС (М+Н)+ = 407,3; чистота по ВЭЖХ = 89,77%; хиральная чистота = 100% (время удерживания = 8,29 мин.), колонка: Chiralpak IC 4,6 мм × 250 мм, подвижная фаза: гексан : IPA : ДХМ (75:15:10).
ПРИМЕР 53: 2-[3-(4-циклопропил-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (53)
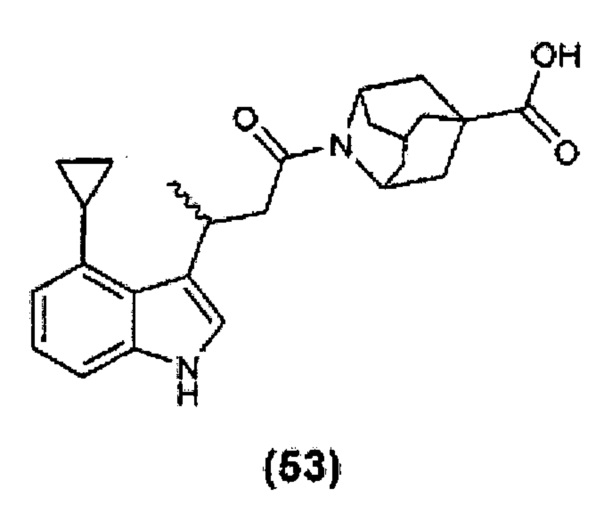
Синтез Соединения (53), пик 2: Рацемат Соединения (51) разделили с помощью хиральной ВЭЖХ с получением энантиомера Соединения (53) (пик 2). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.23 (s, 1H), 10.801 (s, 1Н), 7.11-7.17 (m, 2Н), 6.88-6.93 (t, 1Н), 6.55-6.58 (d, 1H), 4.75 (s, 1H), 4.26 (s, 1H), 4.01-4.04 (m, 1Н), 3.37-3.41 (m, 2Н), 2.68-2.73 (m, 1H), 2.09 (s, 1Н), 1.62-1.88 (m, 13Н), 0.85-00.91 (m, 2Н), 0.70-0.77 (m, 2Н). ЖХ-МС (М+Н)+ = 407,3; чистота по ВЭЖХ = 86,37%; хиральная чистота = 94,78% (время удерживания = 11,60 мин.), колонка: Chiralpak IC 4,6 мм × 250 мм, подвижная фаза: гексан : IPA : ДХМ (75:15:10).
ПРИМЕР 54: 2-[3-(4-циклопропил-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновой кислоты натриевая соль (натриевая соль) (54)
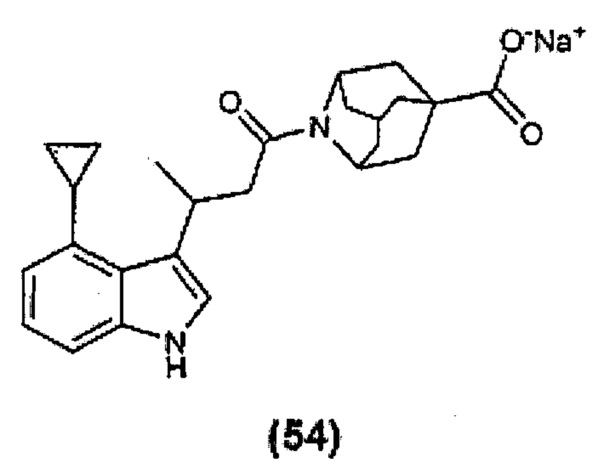
СХЕМА СИНТЕЗА 18
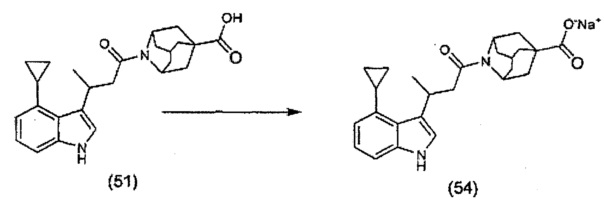
Синтез Соединения (54) (натриевая соль): К перемешанному раствору Соединения (51) (306 мг, 0,72 ммоль) в ТГФ: МеОН: Н2О (2 мл: 3 мл: 1 мл) добавили гидроксид натрия (26 мг, 0,65 ммоль) при 0°С. Полученную реакционную смесь оставили перемешиваться при комнатной температуре на 16 часов. Затем реакционную смесь концентрировали, затем растерли со смесью гексана: эфира с получением Соединения (54) (натриевой соли) (25 мг) в виде белого твердого вещества. 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.92 (S, 1Н), 7.12-7.15 (m, 2Н), 6.87-6.92 (t, 1Н), 6.54-6.57 (d, 1Н), 4.69 (s, 1H), 4.17 (s, 1Н), 4.02-4.03 (m, 1Н), 3.39-3.41 (m, 2Н), 2.61-2.7 (m, 2Н), 1.28-1.79 (m, 13Н), 0.89-0.92 (m, 2Н), 0.70-0.76 (m, 2Н). ЖХ-МС (М+Н)+ = 407,3; чистота по ВЭЖХ = 96,31%.
ПРИМЕР 55: 2-[3-(4-бром-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (55)
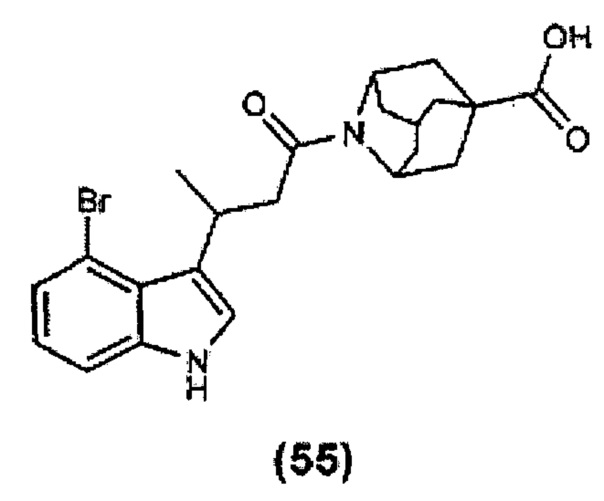
Синтез Соединения (55): Соединение (55) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный ДХМ: МеОН в качестве элюента, с получением Соединения (55). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.25 (s, 1H), 11.20 (s, 1H), 7.32-7.36 (m, 2Н), 7.14-7.17 (d, 1H), 6.92-6.97 (t, 1Н), 4.74 (s, 1H), 4.28 (s, 1H), 4.01-4.05 (m, 1H), 2.70-2.77 (m, 2Н), 2.11 (s, 2Н), 1.60-1.89 (m, 9Н), 1.26-1.28 (d, 3Н). ЖХ-МС (М+Н)+ = 445,1; чистота по ВЭЖХ = 90,50%.
ПРИМЕР 56: 2-[3-(4-циклопропил-1H-индол-3-ил)бутаноил]-2-азатрицикло [3.3.1.13,7]декан-5-карбоксамид (56)
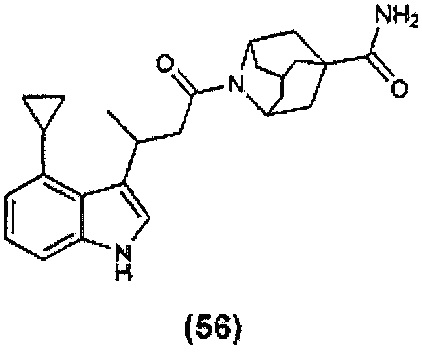
Синтез Соединения (56): Соединение (56)синтезировали по способу, использованному для получения Соединения (47) (Схема 16). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (56). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.79 (s, 1H), 9.50 (s, 2Н), 7.01-7.17 (m, 1Н), 6.88-6.93 (m, 1H), 6.79 (s, 1Н), 6.55-6.58 (d, 1H), 4.74 (s, 1H), 4.26 (s, 1H), 4.01-4.04 (d, 1Н), 2.6-2.8 (m, 3Н), 2.07-2.08 (m, 2Н), 1.15-1.98 (m, 12Н), 0.83-0.95 (m, 2Н), 0.70-0.95 (m, 2Н). ЖХ-МС (М+Н)+ = 406,2; чистота по ВЭЖХ = 88,73%.
ПРИМЕР 57: 2-[3-(4-метил-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (57)
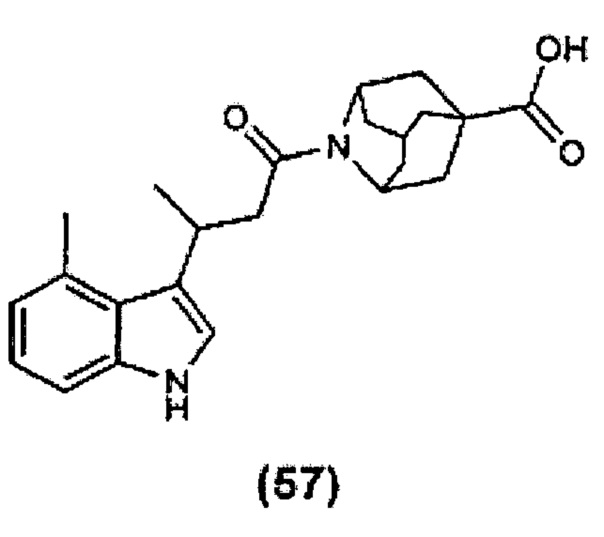
Синтез Соединения (57): Соединение (57) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (57). 1H ЯМР (300 МГц, ДМСО-d6): δ 12.25 (s, 1H), 10.78 (s, 1H), 7.12-7.15 (m, 2Н), 6.87-6.92 (t, 1Н), 6.67-6.69 (d, 1Н), 4.74 (s, 1H), 4.28 (s, 1Н), 3.76-3.80 (m, 1Н), 2.61-2.68 (m, 5Н), 2.08-2.11 (m, 1Н), 1.48-1.88 (m, 10Н), 1.24-1.26 (d, 3Н). ЖХ-МС (М+Н)+ = 381,2; чистота по ВЭЖХ = 99,05%.
ПРИМЕР 58: 3-(4-хлор-1H-индол-3-ил)-1-[5-(гидроксиметил)-2-азатрицикло[3.3.1.13,7]дец-2-ил]бутан-1-он (58)
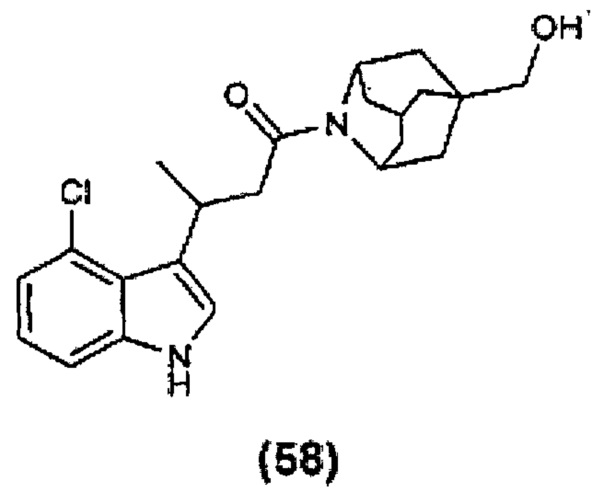
Синтез Соединения (58): Соединение (58) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (58). 1Н ЯМР (300 МГц, CDCl3): δ 11.19 (s, 1H), 7.28-7.31 (m, 2Н), 6.96-7.04 (m, 2Н), 4.72 (s, 1Н), 4.42-4.45 (m, 1H), 4.24 (s, 1H), 3.91-3,95 (m, 1Н), 2.99-3.01 (m, 2Н), 2.67-2.75 (m, 3Н), 2.07 (s, 1H), 1.42-1.63 (m, 9Н), 1.25-1.28 (d, 3Н). ЖХ-МС (М+Н)+ = 387,1; чистота по ВЭЖХ = 97,41%.
ПРИМЕР 59: 2-[3-(4-хлор-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбонитрил (59)
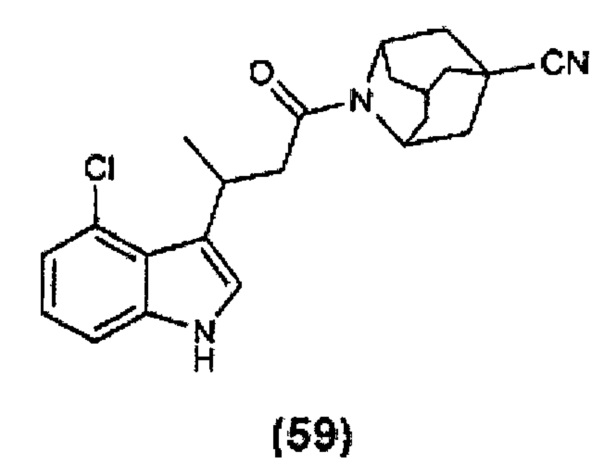
Синтез Соединения (59): Соединение (59) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (59). 1Н ЯМР (300 МГц, CDCl3): δ 8.10 (s, 1H), 7.21-7.25 (m, 1Н), 7.00-7.03 (m, 3Н), 4.90 (s, 1H), 4.02-4.12 (m, 2Н), 2.77-2.87 (m, 1H), 2.37-2.49 (m, 1H), 2.06-2.12 (m, 3Н), 1.60-1.94 (m, 8Н), 1.37-1.40 (m, 3Н). ЖХ-МС (М+Н)+ = 382,3; чистота по ВЭЖХ = 97,62%.
ПРИМЕР 60: 3-(4-хлор-2-метил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (60)
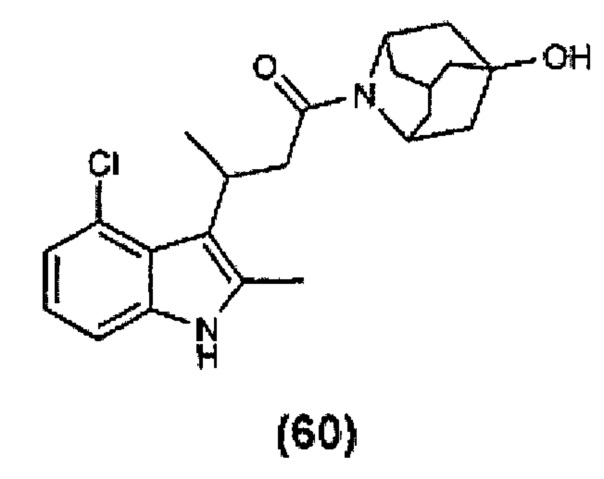
Синтез Соединения (60): Соединение (60) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (60). 1Н ЯМР (300 МГц, CDCl3): δ 7.86-7.89 (d, 1H), 7.08-7.10 (d, 1H), 6.99-7.03 (m, 1H), 6.88-6.94 (t, 1Н), 4.91-5.00 (m, 1Н), 4.00-4.33 (m, 2Н), 2.56-2.67 (m, 1Н), 2.40 (s, 3Н), 2.21-2.24 (m, 1H), 1.74-1.80 (m, 2Н), 1.54 (m, 9Н), 1.36-1.38 (d, 3Н). ЖХ-МС (М+Н)+ = 387,1; чистота по ВЭЖХ = 98,42%.
ПРИМЕР 61: 2-[3-(1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (61)
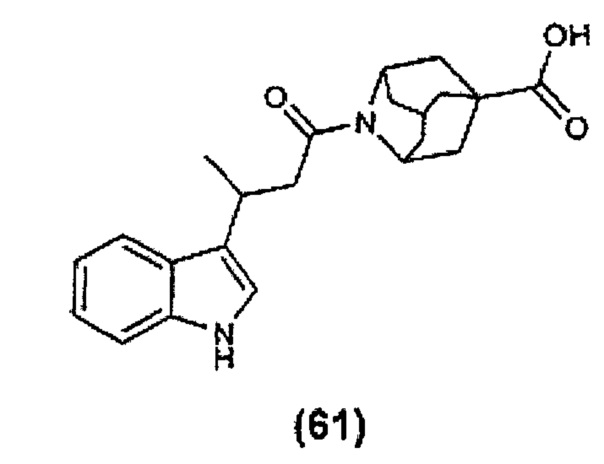
Синтез Соединения (61): Соединение (61) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (61). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.21 (s, 1H), 10.77 (s, 1Н), 7.51-7.54 (d, 1Н), 7.29-7.32 (m, 1Н), 7.12-7.13 (d, 1H), 7.01-7.06 (t, 1H), 6.92-6.97 (t, 1H), 4.72 (s, 1Н), 4.20 (s, 1Н), 3.39-3.45 (m, 1Н), 2.65-2.72 (m, 1H), 2.09 (s, 1H), 1.85 (s, 2Н), 1.73-1.77 (d, 3Н), 1.43-1.66 (m, 6Н), 1.29-1.31 (d, 3Н). ЖХ-МС (М+Н)+ = 367,2; чистота по ВЭЖХ = 93,96%.
ПРИМЕР 62: 2-{3-[4-(4-фторфенил)-1H-индол-3-ил]бутаноил}-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (62)
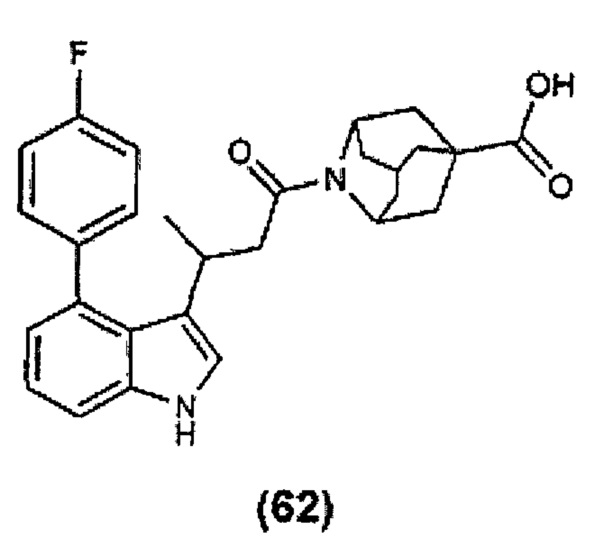
Синтез Соединения (62): Соединение (62) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (62). 1Н ЯМР (300 МГц, CDCl3): δ 8.30 (s, 1H), 7.31-7.39 (m, 2Н), 7.25-7.29 (d, 1H), 7.08-7.12 (t, 1H), 7.04-7.05 (m, 3Н), 6.82-6.85 (d, 1Н), 4.75 (s, 1H), 3.56-3.68 (t, 1H), 3.13-3.14 (d, 1H), 2.25-2.30 (m, 2Н), 1.88-1.92 (m, 3Н), 1.71-1.75 (m, 6Н), 1.58 (s, 3Н), 1.48-1.54 (d, 2Н). ЖХ-МС (М+Н)+ = 461,2; чистота по ВЭЖХ = 95,42%.
ПРИМЕР 63: 4-{[({2-[3-(4-циклопропил-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]дец-5-ил}карбонил)амино]метил}бензойная кислота (63)
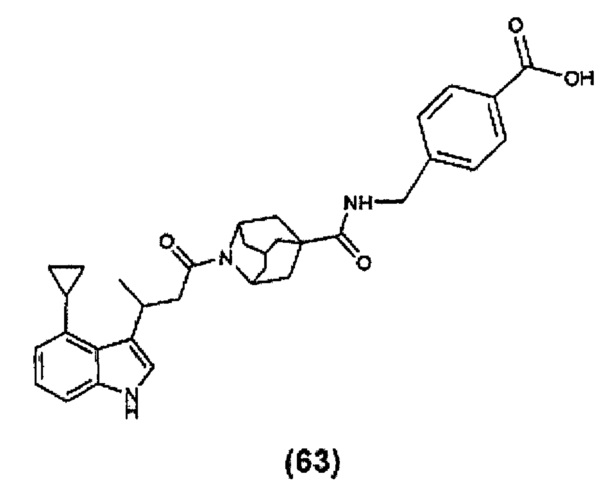
СХЕМА СИНТЕЗА 19
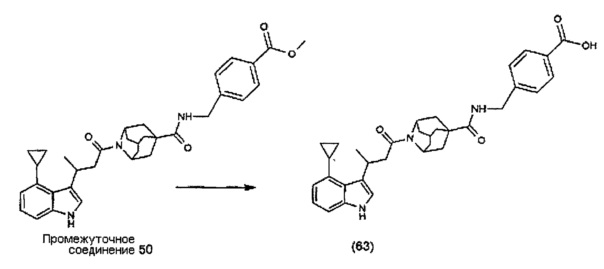
Синтез Соединения (63): Соединение (63) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (63). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.80 (s, 1Н), 10.81 (s, 1H), 8.12-8.20 (m, 1Н), 7.86-7.89 (d, 2Н), 7.28-7.31 (m, 2Н), 7.12-7.17 (t, 2Н), 6.88-6.93 (t, 1H), 6.55-6.58 (d, 1Н), 4.77 (s, 1Н), 4.30 (s, 2Н), 4.04 (s, 1H), 2.61-2.80 (m, 1Н), 2.50-2.54 (m, 2Н), 2.08-2.13 (t, 1Н), 1.86-1.91 (М, 4Н), 1.63-1.78 (m, 6Н), 1.50-1.55 (d, 1Н), 1.28-1.31 (d, 3Н), 0.89-0.91 (М, 2Н), 0.69-0.74 (m, 2Н). ЖХ-МС (М+Н)+ = 540,3; чистота по ВЭЖХ = 95,62%.
ПРИМЕР 64: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)пентан-1-он (64)
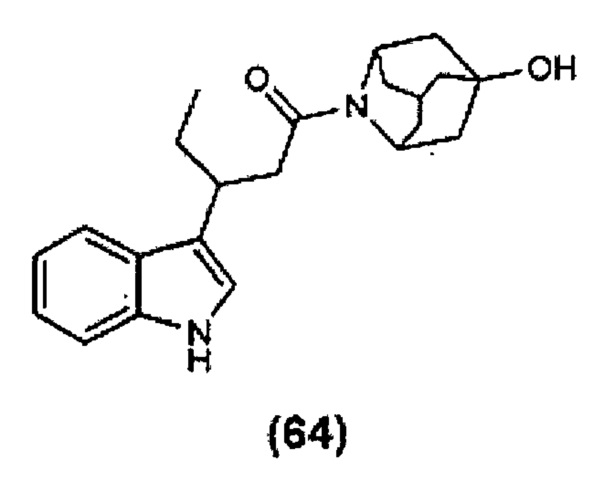
Синтез Соединения (64): Соединение (64) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (64). 1Н ЯМР (300 МГц, CDCl3): δ 8.03 (s, 1Н), 7.58-7.61 (d, 1Н), 7.26-7.29 (d, 1H), 7.07-7.12 (t, 1Н), 6.99-7.04 (t, 1H), 6.94 (s, 1Н), 4.93 (s, 1H), 4.01-4.04 (d, 1H), 3.25-3.35 (m, 1H), 2.70-2.79 (m, 1Н), 2.56-2.59 (m, 1Н), 2.03-2.17 (d, 1H), 1.75-1.85 (m, 2Н), 1.56-1.65 (m, 5Н), 1.31-1.45 (m, 3Н), 1.18-1.22 (m, 2Н), 0.77-0.82 (m, 3Н). ЖХ-МС (М+Н)+ = 353,2; чистота по ВЭЖХ = 94,01%.
ПРИМЕР 65: 2-[3-(1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбонитрил (65)
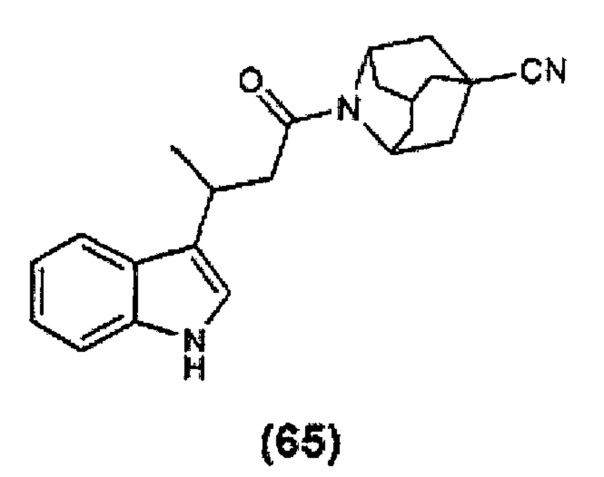
Синтез Соединения (65): Соединение (65) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (65). 1Н ЯМР (300 МГц, CDCl3): δ 7.92-7.95 (d, 1H), 7.58-7.61 (d, 1H), 7.27-7.32 (t, 1H), 7.09-7.13 (t, 1H), 7.01-7.06 (t, 1Н), 6.95 (d, 1H), 4.83-4.87 (d, 1H), 3.83-3.94 (d, 1Н), 3.55-3.62 (m, 1H), 2.69-2.77 (m, 1Н), 2.42-2.50 (m, 1H), 2.07-2.10 (m, 1H), 1.93-2.00 (m, 4H), 1.86 (s, 1H), 1.74-1.79 (d, 1H), 1.57-1.64 (m, 4H), 1.40-1.43 (m, 3Н). ЖХ-МС (M+H)+ = 348,2; чистота по ВЭЖХ = 91,69%.
ПРИМЕР 66: 3-циклопропил-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)пропа-1-он (66)
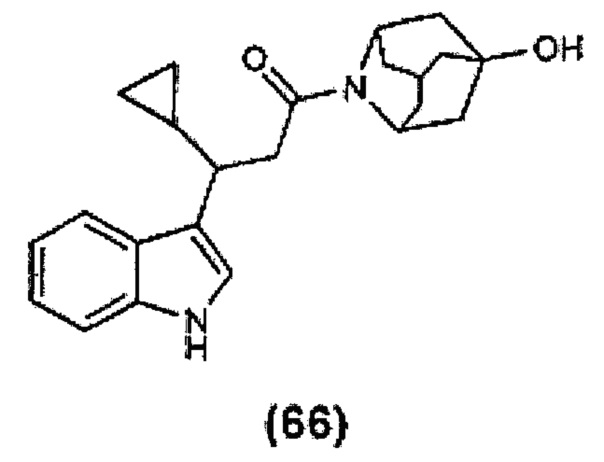
Синтез Соединения (66): Соединение (66) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (66). 1Н ЯМР (300 МГц, CDCl3): δ 8.01-8.05 (m, 1H), 7.59-7.62 (d, 1H), 7.26-7.29 (d, 1H), 7.07-7.12 (t, 1H), 6.98-7.04 (m, 2H), 4.93 (s, 1H), 4.60-4.61 (d, 1H), 4.08-4.11 (d, 1H), 2.84-2.91 (m, 1H), 2.65-2.78 (m, 2H), 2.04-2.18 (s, 1H), 1.65 (s, 2H), 1.57 (s, 2H), 1.44 (m, 2H), 1.32-1.34 (m, 3Н), 0.47-0.56 (M, 1H), 0.35-0.39 (m, 1H), 0.25-0.32 (m, 1H), 0.06-0.13 (m, 2H). ЖХ-МС (M+H)+ = 365,2; чистота по ВЭЖХ = 93,07%.
ПРИМЕР 67: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)-3-фенилпропан-1-он (67)
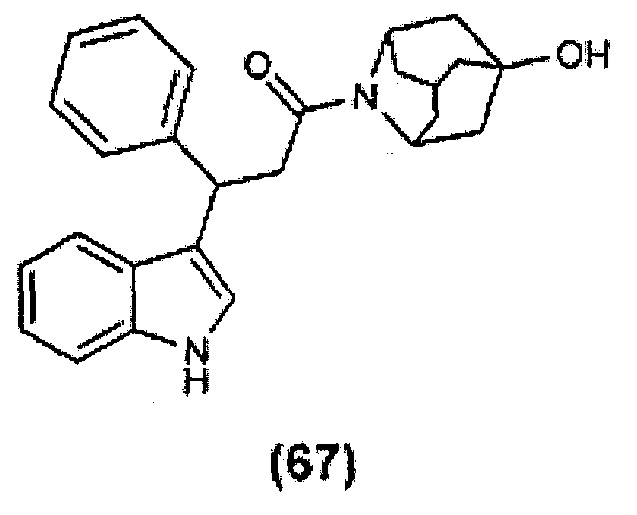
СХЕМА СИНТЕЗА 20
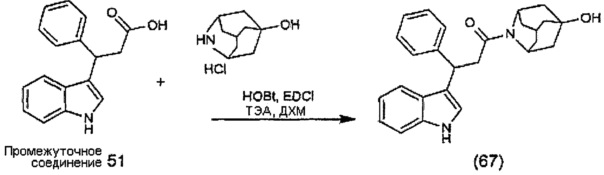
Синтез 3-(1Н-индол-3-ил)-3-фенилпропановой кислоты (Промежуточное соединение 51):
Промежуточное соединение 51 синтезировали по способу, использованному для получения Промежуточного соединения 42 (Схема 11).
Синтез Соединения (67): Соединение (67) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (67). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.85 (s, 1Н), 7.28-7.33 (m, 5H), 7.18-7.23 (t, 2H), 7.07-7.12 (t, 1H), 6.97-7.02 (t, 1H), 6.83-6.88 (t, 1H), 4.67-4.71 (m, 1H), 4.62-4.65 (m, 1H), 4.56 (s, 1H), 4.35 (s, 1H), 3.01-3.08 (m, 2H), 1.98-2.13 (d, 1H), 1.63 (s, 2H), 1.55 (s, 1H), 1.47-1.50 (d, 3H), 1.38-1.41 (m, 2H), 1.25-1.28 (m, 2H). ЖХ-МС (M+H)+ = 401,2; чистота по ВЭЖХ = 94,67%.
ПРИМЕР 68: 3-(5-хлор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрициклор[3.3.1.13,7]дец-2-ил)бутан-1-он (68)
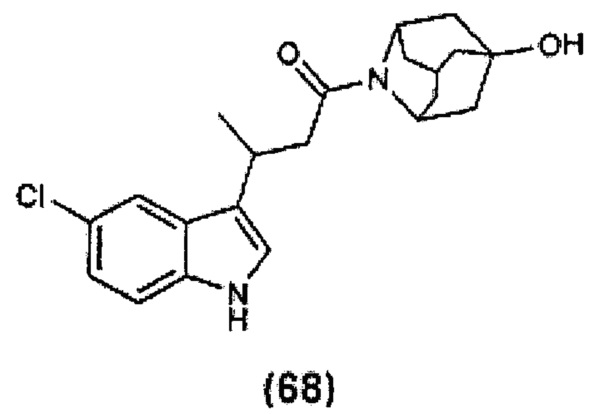
Синтез Соединения (68): Соединение (68) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (68). 1H ЯМР (300 МГц, CDCl3): δ 8.10 (s, 1H), 7.55-7.56 (d, 1H), 7.21-7.25 (d, 1H), 7.04-7.07 (d, 1H), 6.99 (s, 1H), 4.97 (s, 1H), 4.03-4.13 (m, 1H), 3.54-3.62 (m, 1H), 2.64-2.72 (d, 1H), 2.44-2.54 (d, 1H), 2.15-2.30 (m, 2H), 1.76 (s, 4H), 1.70 (s, 3H), 1.62 (s, 2H), 1.38-1.40 (d, 3Н). ЖХ-МС (M+H)+ = 373,1; чистота по ВЭЖХ = 92,50%.
ПРИМЕР 69: 3-(6-хлор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрициклор[3.3.1.13,7]дец-2-ил)бутан-1-он (69)
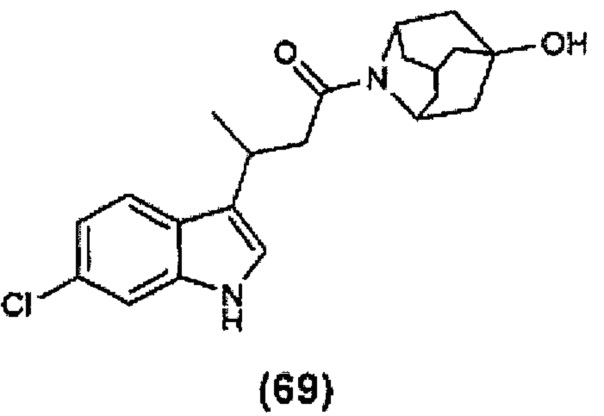
Синтез Соединения (69); Соединение (69) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (69). 1Н ЯМР (300 МГц, CDCl3): δ 8.03 (s, 1H), 7.49-7.51 (d, 1H), 7.27 (s, 1H), 6.95-7.01 (m, 2H), 4.91-5.01 (s, 1H), 4.08 (s, 1H), 3.56-3.58 (m, 1H), 2.69-2.73 (d, 1H), 2.54 (s, 1H), 2.11-2.27 (m, 1H), 1.8 (s, 1H), 1.68 (s, 9H), 1.37-1.40 (d, 3H). ЖХ-МС (M+H)+ = 373,1; чистота по ВЭЖХ = 91,91%.
ПРИМЕР 70: 2-[3-(4-метил-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбонитрил(70)
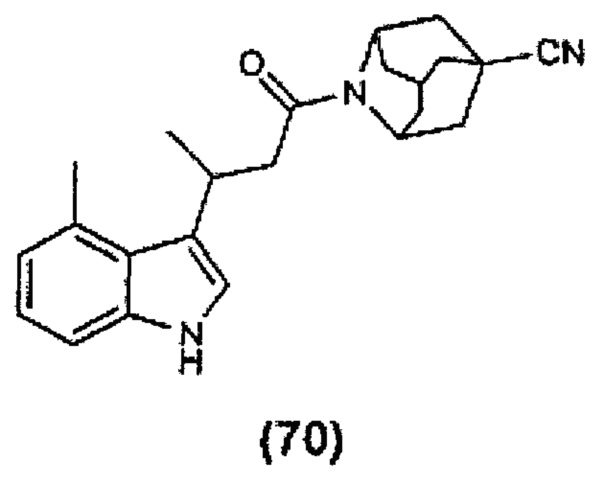
Синтез Соединения (70): Соединение (70) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (70). 1Н ЯМР (300 МГц, CDCl3): δ 7.94 (s, 1Н), 7.12-7.14 (d, 1H), 6.96-7.02 (m, 2H), 6.77-6.79 (d, 1H), 4.91 (s, 1H), 4.04 (s, 1H), 3.89 (s, 1H), 2.65 (s, 3H), 2.37-2.48 (m, 1H), 2.25-2.30 (m, 1H), 2.12 (s, 2H), 2.06 (s, 2H), 1.98-2.02 (d, 2H), 1.86-1.94 (s, 2H), 1.71-1.77 (m, 3H), 1.34-1.36 (d, 3H). ЖХ-МС (M+H)+ = 362,2; чистота по ВЭЖХ = 95,85%.
ПРИМЕР 71: 3-(4-хлорфенил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)пропан-1-он (71)
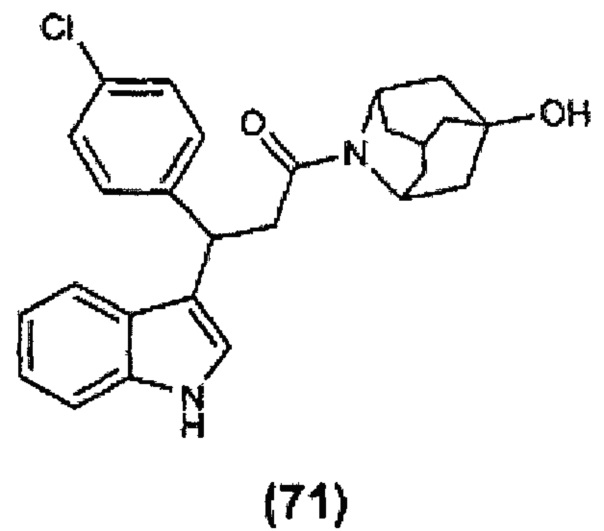
Синтез Соединения (71): Соединение (71) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (71). 1Н ЯМР (300 МГц, CDCl3): δ 8.01-8.13 (t, 1H), 7.25-7.28 (d, 2H), 7.18 (m, 4H), 7.05-7.10 (t, 1H), 6.93-6.96 (d, 2H), 4.92 (s, 1H), 4.79 (s, 1H), 4.11 (s, 1H), 2.97-3.05 (m, 2H), 2.15 (s, 1H), 1.69 (s, 7H), 1.45 (d, 3H). ЖХ-МС (M+H)+ = 435,1; чистота по ВЭЖХ = 95,96%.
ПРИМЕР 72: 1-(5-гидрокси-2-азатрицикло[3.3.11.13,7]дец-2-ил)-3-(4-метил-1H-индол-3-ил)-3-фенилпропан-1-он (72)
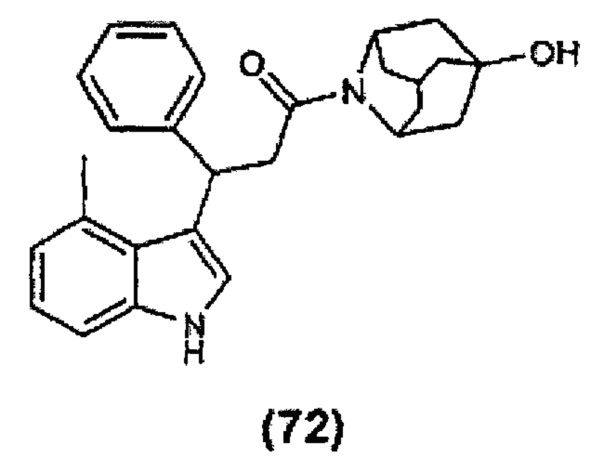
Синтез Соединения (72): Соединение (72) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (72). 1H ЯМР (300 МГц, CDCl3): δ 8.13-8.21 (d, 1Н), 7.14-7.15 (d, 4H), 7.04-7.11 (m, 2H), 6.99 (s, 1H), 6.90-6.96 (m, 1H), 6.64-6.66 (d, 1H), 5.08-5.13 (t, 1H), 4.92 (s, 1H), 4.13 (s, 1H), 2.89-3.02 (m, 2H), 2.4 (d, 3H), 2.08-2.21 (m, 1H), 1.81 (s, 1H), 1.68 (s, 2H), 1.39 (s, 2H), 0.78-0.90 (m, 3H). ЖХ-МС (M+H)+ = 415,2; чистота по ВЭЖХ = 99,51%.
ПРИМЕР 73: 3-(4-фторфенил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)пропан-1-он (73)
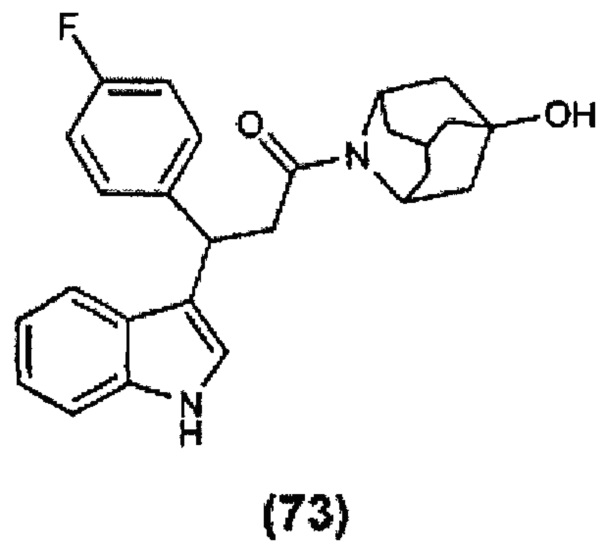
Синтез Соединения (73): Соединение (73) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (73). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.87 (s, 1H). 7.28-7.31 (m, 5Н), 6.98-7.05 (m, 3H), 6.84-6.89 (t, 1H), 4.58-4.71 (m, 3H), 4.36 (s, 1H), 2.97-3.12 (m, 2H), 2.08-2.13 (d, 1H), 1.64 (s, 2H), 1.41-1.55 (m, 6H), 1.20-1.35 (m, 2H). ЖХ-МС (М+H)+ = 419,2; чистота по ВЭЖХ = 95,83%.
ПРИМЕР 74: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-4-метил-3-(4-метил-1H-индол-3-ил)пентан-1-он (74)
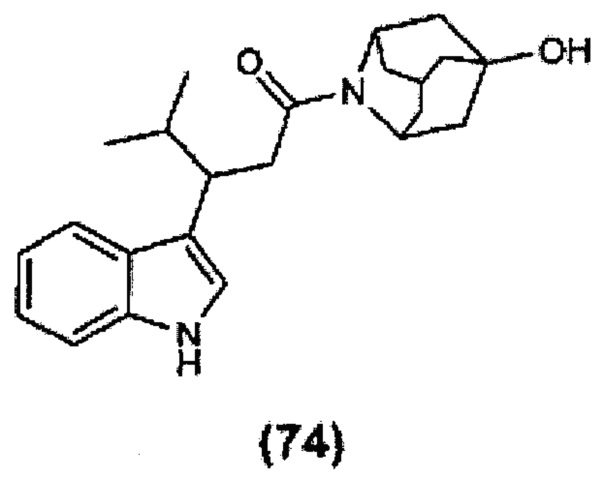
Синтез Соединения (74): Соединение (74) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (74). 1H ЯМР (300 МГц, ДМСО-d6): δ 10.77 (s, 1Н), 7.11-7.13 (d, 2H), 6.85-6.90 (t, 1H), 6.64-6.66 (d, 1H), 4.66 (s, 1H), 4.58-4.62 (d, 1H), 4.38-4.40 (d, 1H), 3.70 (s, 1H), 2.67-2.76 (m, 2H), 2.64 (s, 3H), 2.05-2.16 (d, 1H), 1.84-1.86 (m, 1H), 1.52-1.64 (m, 5H). 1.36-1.48 (m, 5H), 0.82-0.90 (m, 6H). ЖХ-МС (M+H)+ = 381,2; чистота по ВЭЖХ = 97,77%.
ПРИМЕР 75: 3-(5-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (75)
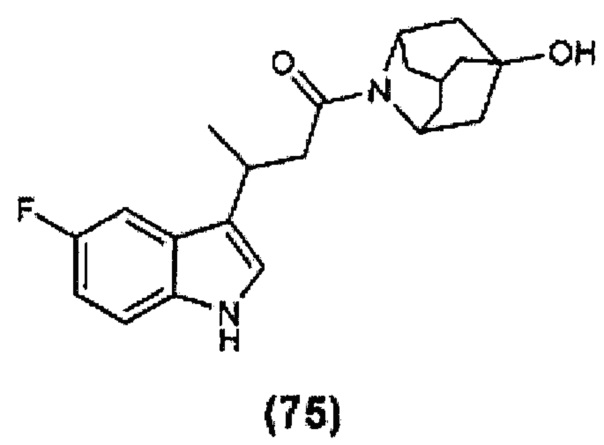
Синтез Соединения (75): Соединение (75) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (75). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.88 (s, 1H), 7.28-7.32 (m, 2H), 7.21-7.24 (m, 1H), 6.85-6.91 (m, 1H), 4.78 (s, 1H), 4.58-4.64 (d, 1H), 4.23-4.25 (d, 1H), 3.16-3.39 (m, 2H), 2.53-2.69 (m, 2H), 2.15-2.17 (m, 1H), 1.65 (s, 2H), 1.54-1.57 (m, 3H), 1.36-1.49 (m, 4H), 1.29-1.30 (d, 3H). ЖХ-МС (M+H)+ = 356,1; чистота по ВЭЖХ = 98,0%.
ПРИМЕР 76: 3-(4-хлор-1Н-индол-3-ил)-1-[5-(1Н-тетразол-5-ил)-2-азатрицикло[3.3.1.13,7]дец-2-ил]бутан-1-он (76)
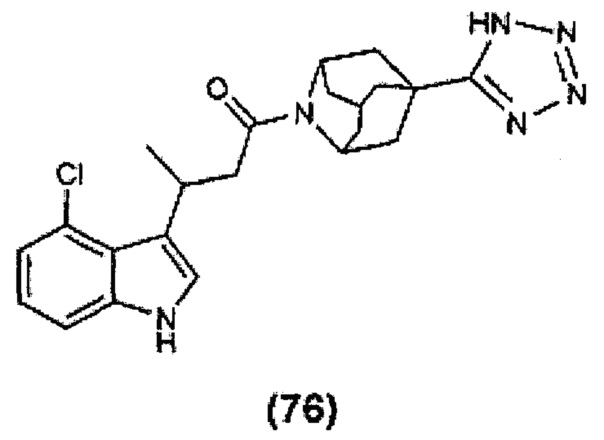
Синтез Соединения (76): Соединение (76) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (76). 1H ЯМР (300 МГц, CD3OD): δ 7.10-7.21 (m, 3H), 6.93 (s, 2H), 4.30-4.35 (d, 1H), 4.08 (s, 1H), 2.89 (s, 1H), 2.54-2.61 (m, 2H), 1.90-1.94 (m, 4H), 1.74-1.77 (m, 5H), 1.24-1.37 (m, 6H). ЖХ-МС (M+H)+ = 426,1; чистота по ВЭЖХ = 99,3 6%.
ПРИМЕР 77: 3-(4-метил-1Н-индол-3-ил)-1-[5-(1H-тетразол-5-ил)-2-азатрицикло[3.3.1.13,7]дец-2-ил]бутан-1-он (77)
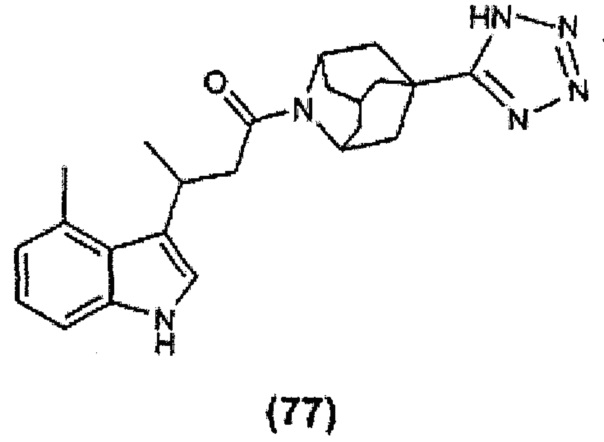
СХЕМА СИНТЕЗА 21
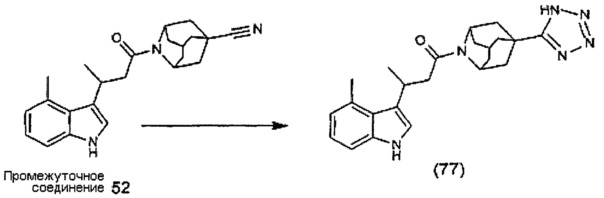
Синтез Соединения (77): К перемешанному раствору Промежуточного соединения 52 (20 мг, 0,05 ммоль) в толуоле (10 мл) добавили NaN3 (37 мг, 0,5 ммоль) вместе с триметилолова хлоридом (49 мг, 0,25 ммоль) под атмосферой N2. Полученную реакционную смесь нагревали при 110°С в течение 12 часов. После завершения реакции (контролировали по ТСХ) реакционную смесь погасили водой и экстрагировали этилацетатом (3×10 мл). Объединенный органический слой концентрировали с получением неочищенного продукта. Полученный неочищенный продукт очистили препаративной ТСХ, элюируя ДХМ: МеОН, с получением Соединения (77) (8 мг) в виде белого твердого вещества. 1Н ЯМР (300 МГц, CD3OD): δ 7.06-7.15 (m, 2Н), 6.84-6.94 (m, 1H), 6.67-6.72 (m. 1H), 4.69-4.81 (m, 1H), 4.30-4.35 (d, 1H), 3.92-3.98 (m, 1H), 2.82-2.90 (m, 1H), 2.68 (d, 3H), 2.57-2.64 (m, 1H), 2.21 (s, 1H), 1.96-2.11 (m, 5H), 1.8 (s, 3H). 1.70-1.74 (m, 2Н), 1.57-1.61 (d, 1H), 1.38-1.40 (d, 3H). ЖХ-MC (M+H)+ = 405,2; чистота по ВЭЖХ = 98,31%.
ПРИМЕР 78: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил-3-7-метил-1H-индол-3-ил)бутан-1-он (78)
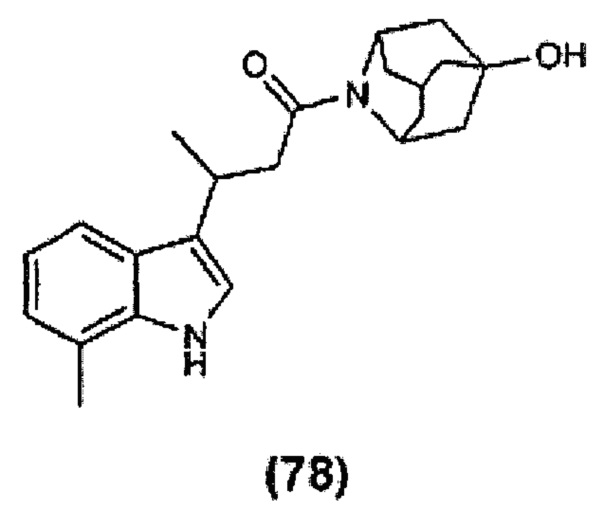
Синтез Соединения (78): Соединение (78) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (78). 1H ЯМР (300 МГц, CDCl3): δ 7.88 (s, 1Н), 7.44-7.46 (d, 1H), 6.90-7.03 (m, 3Н), 3.52-3.61 (m, 1H), 2.72-2.79 (m, 1H), 2.40-2.50 5 (m, 1H), 2.40 (m, 3Н), 2.22-2.23 (m, 1H), 1.70 (s, 3Н), 1.49-1.61 (m, 6H), 1.45 (s, 3Н), 1.40-1.43 (m, 3Н). ЖХ-МС (M+H)+ = 353,2; чистота по ВЭЖХ = 97,30%.
ПРИМЕР 79: 3-(6-фтор-1H-индол-3-ил)-1-(5-гидрокси-5-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (79)
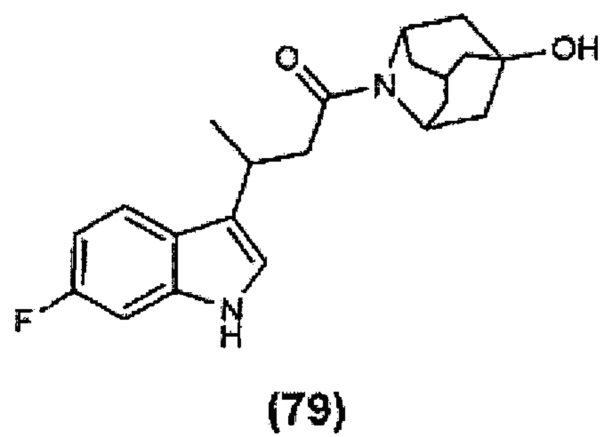
Синтез Соединения (79): Соединение (79) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (79). 1Н ЯМР (300 МГц, CDCl3): δ 7.99 (s, 1H), 7.55-7.60 (m, 1H), 6.98-7.04 (m, 2H), 6.84-6.91 (m, 1H), 5.04 (s, 1H), 4.15 (s, 1H), 3.56-3.68 (m, 1H), 2.74-2.82 (m, 1H), 2.48-2.55 (m, 1H), 2.29-2.18 (s, 1H), 1.76 (s, 2H), 1.67 (s, 2H), 1.57-1.62 (m, 4H), 1.48-1.50 (m, 2H), 1.43-1.46 (m. 3Н). ЖХ-МС (M+H)+ = 357,2; чистота по ВЭЖХ = 91,94%.
ПРИМЕР 80: 3-[4-циклопропил-1-(2-гидроксиэтил)-1H-индол-3-ил]-1-(5-гидрокси-2-азатрицикло [3.3.1.13,7]дец-2-ил)бутан-1-он (80)
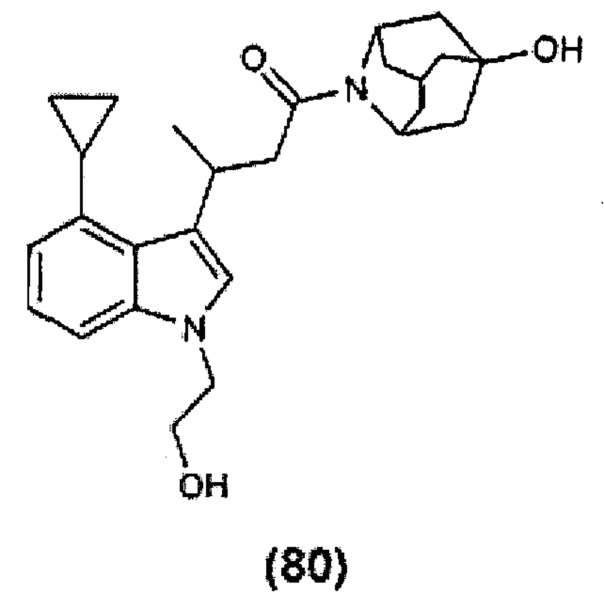
Синтез Соединения (80): Соединение (80) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (80). 1H ЯМР (300 МГц, CDCl3): δ 7.14-7.19 (m, 1H), 7.06-7.11 (m, 1H), 7.00 (s, 1H), 6.73-6.75 (d, 1H), 5.06 (s, 1H), 4.27 (s, 1H), 4.20-4.24 (m, 3H), 3.91-3.95 (m, 2H), 2.76-2.90 (m, 1H), 2.56-2.64 (m, 1H), 2.43-2.51 (m, 1H), 2.31 (s, 1H), 1.79 (s, 2H), 1.68 (s, 2H), 1.63-1.65 (d, 4H), 1.57 (s, 3H), 1.42-1.46 (m, 3H), 0.97-1.02 (m, 2H), 0.79-0.90 (m, 2H). ЖХ-МС (M+H)+ =423,2; чистота по ВЭЖХ = 97,26%.
ПРИМЕР 81: 3-(4-фторфенил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)пропан-1-он (81)
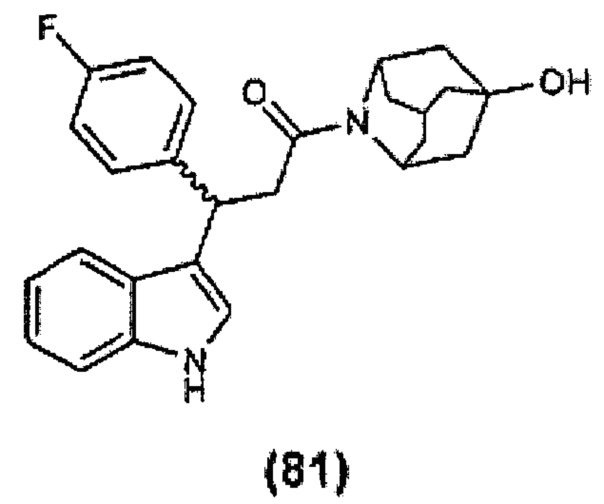
Синтез Соединения (81) (пик 1): Рацемат Соединения (73) разделили с помощью хиральной ВЭЖХ с получением энантиомера Соединения (81) (пик 1). 1H ЯМР (300 МГц, CDCl3): δ 10.87 (s, 1H), 7.20 (m, 5H), 7.00-7.05 (m, 3H), 6.83 (m, 1H), 4.66-4.68 (m, 1H), 4.58 (m, 2H), 4.40 (s, 1H), 3.10 (s, 2H), 2.08-2.13 (d, 1H), 1.61.23-1.55 (m, 8H), 1.11-1.15 (m, 2H). ЖХ-МС (M+H)+ = 419,2; чистота по ВЭЖХ = 94,26%; хиральная чистота = 100% (время удерживания = 8,57 мин.), хиральная колонка: Chiralpak IC 4,6 мм Х 250 мм, подвижная фаза: гексан :ЕЮН:ДХМ (75:15:10).
ПРИМЕР 82: 3-(4-фторфенил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-индол-3-ил)пропан-1-он (82)
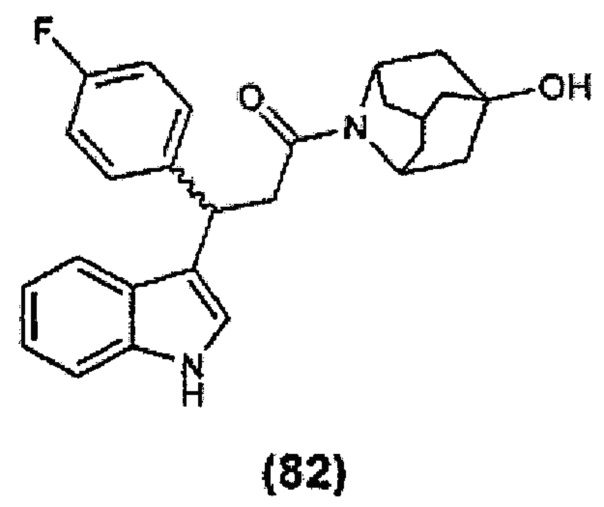
Синтез Соединения (82) (пик 2): Рацемат Соединения (73) разделили с помощью хиральной ВЭЖХ с получением энантиомера Соединения (82) (пик 2). 1Н ЯМР (300 МГц, CDCl3): δ 10.87 (s, 1Н), 7.20 (m, 5H), 7.00-7.05 (m, 3H), 6.83 (m, 1H), 4.66-4.68 (m, 1H), 4.58 (m, 2H), 4.40 (s, 1H), 3.10 (s, 2H), 2.08-2.13 (d, 1H), 1.61.23-1.55 (m, 8H), 1.11-1.15 (m, 2H). ЖХ-МС (M+H)+ = 419,2; чистота по ВЭЖХ = 97,71%; хиральная чистота = 100% (время удерживания = 10,56 мин.), хиральная колонка: Chiralpak IC 4,6 мм Х 250 мм, подвижная фаза: гексан:EtOH:ДХМ (75:15:10).
ПРИМЕР 83: 3-(4-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азабицикло[3.3.1.13,7]дец-2-ил)-3-фенилпропан-1-он (83)
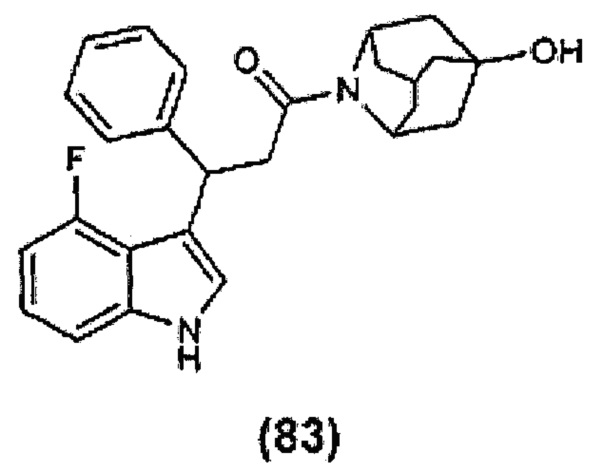
Синтез Соединения (83): Соединение (83) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (83). 1Н ЯМР (300 МГц, ДМСО-d6): δ 11.17 (s, 1H), 7.35-7.37 (m, 1H), 7.14-7.27 (m, 4H), 7.06-7.11 (m, 2H), 6.93-7.00 (m, 1H), 6.58-6.64 (m, 1H), 4.80-4.85 (t, 1H). 4.70 (s, 1H), 4.60-4.65 (d, 1H), 4.37 (s, 1H), 2.93-3.17 (m, 2H), 2.07-2.27 (m, 2H), 1.64 (s, 2H), 1.40-1.53 (m, 3Н), 1.36 (m, 2H), 1.23-1.28 (m, 2H). ЖХ-МС (M+H)+ = 419,2; чистота по ВЭЖХ = 96,72%.
ПРИМЕР 84: 3-(1,3-бензотиазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,73,7]дец-2-ил)бутан-1-он (84)
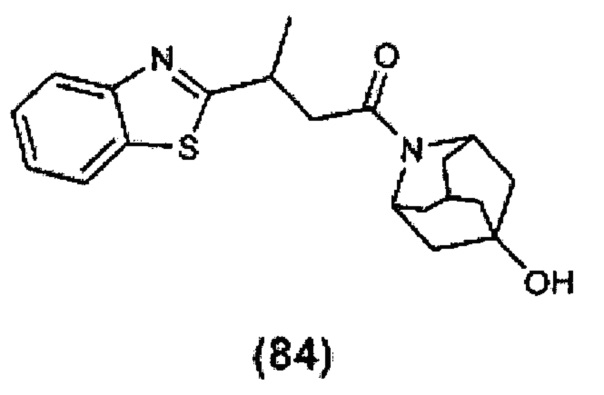
СХЕМА СИНТЕЗА 22
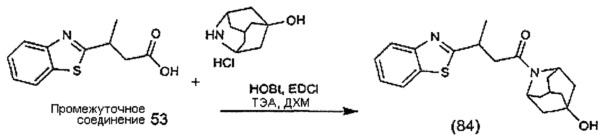
Синтез Соединения (84): Промежуточное соединение 53 синтезировали по способу, использованному для получения Промежуточного соединения 36 (Схема 7). Соединение (84) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением (84). 1Н ЯМР (300 МГц, CDCl3): δ 7.90-7.93 (d, 1Н), 7.76-7.79 (d, 1H), 7.36-7.41 (t, 1H), 7.26-7.31 (t, 1H), 4.96 (s, 1H), 4.33 (s, 1H), 3.84-3.86 (d, 1H), 3.04-3.11 (m, 1H), 2.58-2.73 (m, 1H), 2.23-2.27 (d, 1H), 1.72-1.76 (d, 4H), 1.55-1.65 (m, 6H), 1.45-1.47 (d, 3H). ЖХ-МС (М+Н)+ = 357,1; чистота по ВЭЖХ = 98,96%.
ПРИМЕР 85: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-пирроло[2,3-d]пиридин-3-ил)бутан-1-он (85)
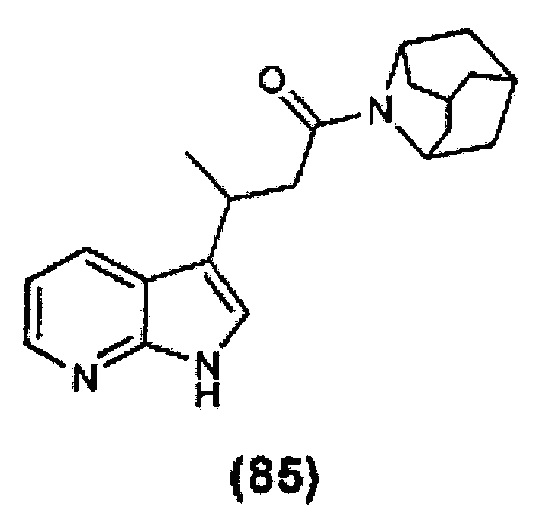
СХЕМА СИНТЕЗА 23
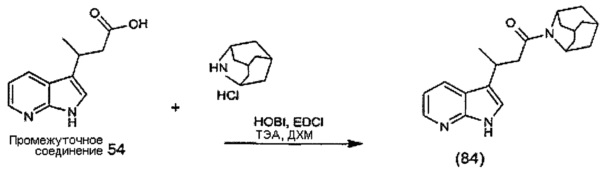
Синтез 3-(1H-пирроло[2,3-b]пиридин-3-ил)бутановой кислоты (Промежуточное соединение 54):
Промежуточное соединение 54 синтезировали по способу, использованному для получения Промежуточного соединения 42 (Схема 11)
Синтез Соединения (85): Соединение (85) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат в качестве элюента, с получением Соединения (85). 1Н ЯМР (300 МГц, CDCl3): δ 9.29 (s, 1H), 8.19-8.21 (m, 1H), 7.93-7.96 (m, 1H), 7.07 (s, 1H), 6.97-7.01 (m, 1H), 4.76 (s, 1H), 3.89 (s, 1H), 3.52-3.66 (m, 1H), 2.65-2.73 (m, 1H), 2.42-2.49 (m, 1H), 1.97 (s, 1H), 1.84 (s, 1H), 1.65-1.68 (m, 4H), 1.61-1.65 (m, 3H), 1.56-1.57 (d, 2H), 1.41-1.49 (m, 1H), 1.35 (d, 3Н). ЖХ-МС (M+H)+ = 324,2; чистота по ВЭЖХ = 94,58%.
ПРИМЕР 86: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(1H-пирроло[2,3-b]пиридин-3-ил)бутан-1-он (86)
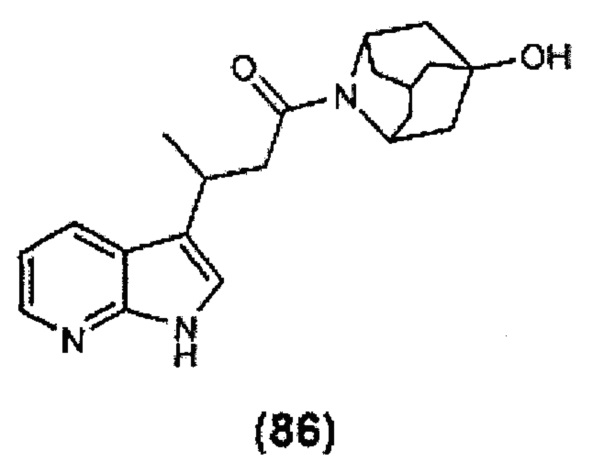
Синтез Соединения (86): Соединение (86) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (86). 1Н ЯМР (300 МГц, CDCl3): δ 8.82-9.01 (m, 1H), 8.19-8.21 (d, 1H), 7.94-7.96 (m, 1H), 7.05 (s, 1H), 6.98-7.02 (m, 1H), 4.96 (s, 1H), 4.08 (s, 1H), 3.55-3.62 (m, 1H), 2.65-2.72 (m, 1H), 2.42-2.55 (m, 1H), 2.09-2.29 (m, 1H), 1.69 (s, 3Н), 1.50 (s, 2H), 1.18-1.44 (m, 5H), 1.18 (s, 1H), 0.78-0.98 (m, 2H). ЖХ-МС (M+H)+ = 340,2; чистота по ВЭЖХ = 99,20%.
ПРИМЕР 87: 3-(4-хлор-1H-пирроло[2,3-b]пиридин-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (87)
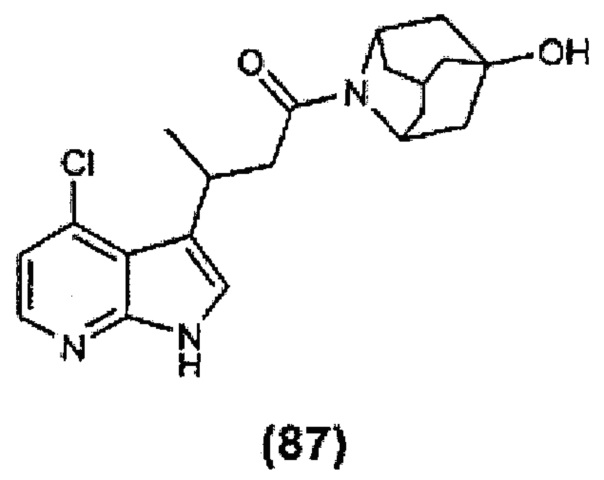
Синтез Соединения (87): Соединение (87) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (87). 1H ЯМР (300 МГц, ДМСО-d6): δ 11.77 (s, 1H), 8.09-8.11 (d, 1H), 7.38-7.43 (m, 1H), 7.10-7.11 (d, 1H), 4.80 (s, 1H), 4.66 (s, 1H), 4.35 (s, 1H), 3.84 (s, 1H), 2.69 (m, 3Н), 2.18 (s, 1H), 1.45-1.68 (m, 9H), 1.23-1.30 (m, 3Н). ЖХ-МС (М+Н)+ = 374,2; чистота по ВЭЖХ = 96,68%.
ПРИМЕР 88: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(4-метил-1H-пирроло[2,3-b]пиридин-3-ил)бутан-1-он (88)
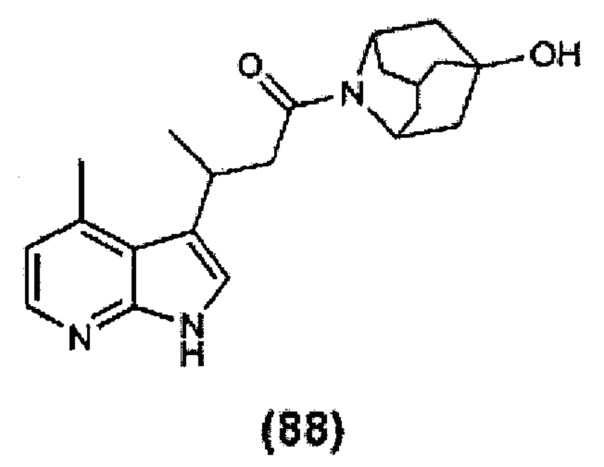
Синтез Соединения (88): Соединение (88) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (88). 1H ЯМР (300 МГц, CDCl3): δ 11.91-11.95 (d, 1H), 7.97-7.99 (d, 1H), 7.26-7.29 (d, 1H), 7.05-7.06 (d, 1H), 4.89-4.94 (d. 1H), 4.26 (s. 1H), 3.91 (s, 1H), 3.04-3.06 (m, 1H), 2.90 (s. 3H), 2.50-2.66 (m, 2H), 2.27 (s, 1H), 1.36-1.78 (m. 9H), 1.29-1.32 (d, 3H). ЖХ-МС (M+H)+ = 354,2; чистота по ВЭЖХ = 99,38%.
ПРИМЕР 89: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-метил-3-(1H-пирроло[2,3-b]пиридин-3-ил)пропан-1-он (89)
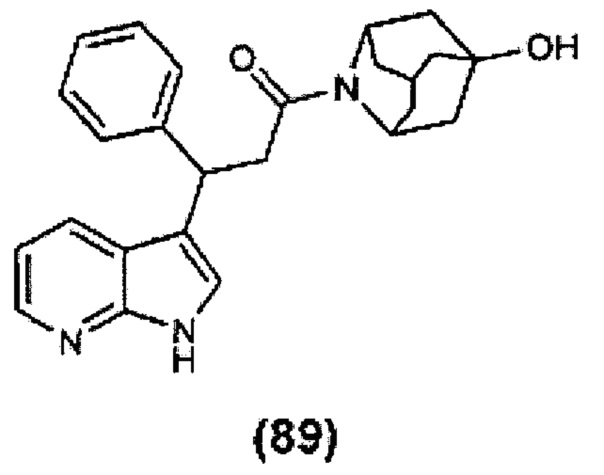
Синтез Соединения (89): Соединение (89) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир: этилацетат (1:4) в качестве элюента, с получением Соединения (89). ЖХ-МС (М+Н)+ = 402,2; чистота по ВЭЖХ = 95,83%.
ПРИМЕР 90: 1-(5-гидрокси-2-азатрицикло[3.3.1.13.7]дец-2-ил)-4-метил-3-(1H-пирроло[2,3-b]пиридин-3-ил)пентан-1-он(90)
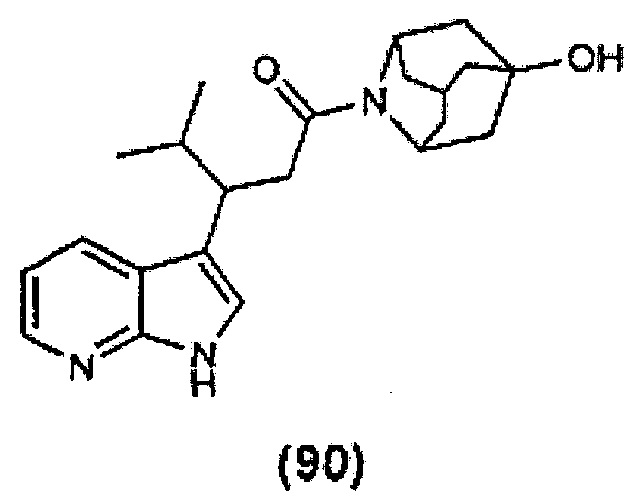
Синтез Соединения (90): Соединение (90) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя петролейный эфир : этилацетат (1:4) в качестве элюента, с получением Соединения (90). 1H ЯМР (300 МГц, CDCl3): δ 11.48-11.50 (d, 1H), 8.28-8.33 (t, 1H), 8.14-8.16 (d, 1H), 7.22 (m, 2H), 4.75-4.80 (d, 1H), 4.13 (s, 1H), 3.32-3.37 (m, 1H), 2.63-2.69 (m, 3H), 2.12-2.20 (d, 1H), 1.95-2.04 (m, 1H), 1.36-1.70 (m, 9H), 0.89-0.93 (m, 3H), 0.77-0.81 (m, 3H). ЖХ-МС (M+H)+ = 368,2; чистота по ВЭЖХ = 94,67%.
ПРИМЕР 91: 2-[3-(1H-пирроло[2,3-b]пиридин-3-ил)бутаноил]-2-азатрицикло[3.3.1.13.7]декан-5-карбоновая кислота (91)
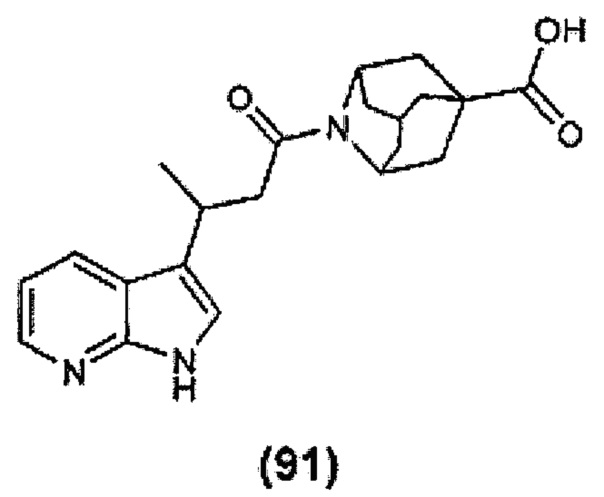
Синтез Соединения (91): Соединение (91) синтезировали по способу, использованному для получения Промежуточного соединения 26 (Схема 4). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (91). 1H ЯМР (300 МГц, CDCl3): δ 11.09 (s, 1H), 8.08-8.13 (m, 2H), 7.06-7.10 (m, 2H), 4.71 (s, 1H), 3.64-3.72 (m, 2H), 2.66-2.74 (t, 1H), 2.36-2.41 (m, 1H), 2.01-2.04 (d, 1H), 1.80-1.84 (d, 1H), 1.48-1.70 (m, 8H), 1.43-1.45 (d, 3H), 1.18-1.22 (m, 1H). ЖХ-МС (M+H)+ = 368,2; чистота по ВЭЖХ = 98,93%.
ПРИМЕР 92: 2-[4-метил-3-(1H-пирроло[2,3-b]пиридин-3-ил)пентаноил]-2-азатрицикло[3.3.1.13.7]декан-5-карбонитрил (92)
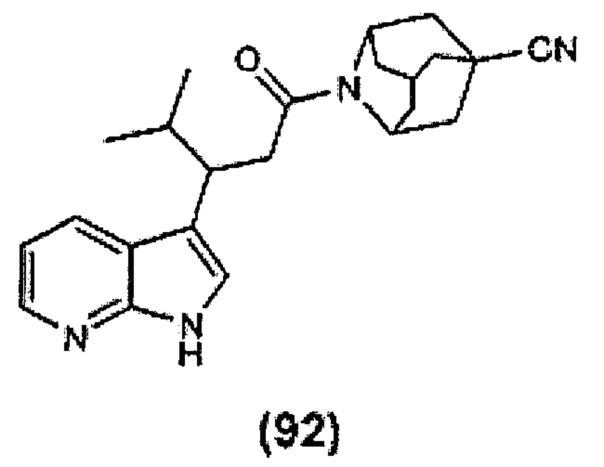
Синтез Соединения (92): Соединение (92) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (92). 1H ЯМР (300 МГц, CDCl3): δ 10.75-10.78 (d, 1H), 8.18-8.25 (m, 2H), 7.19-7.20 (m, 2H), 4.72 (s, 1H), 4.03 (s, 1H), 3.33-3.35 (m, 1H), 2.61-2.70 (m, 2H), 2.13 (s, 1H), 1.97-2.02 (m, 4H), 1.81-1.90 (m, 2H), 1.09-1.68 (m, 5H), 0.90-0.92 (m, 3H), 0.77-0.79 (d, 3H). ЖХ-МС (M+H)+ = 377,2; чистота по ВЭЖХ = 97,71%.
ПРИМЕР 93: 3-(4-фторфенил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13.7]дец-2-ил)-3-(4-метил-1H-индол-3-ил)пропан-1-он (93)
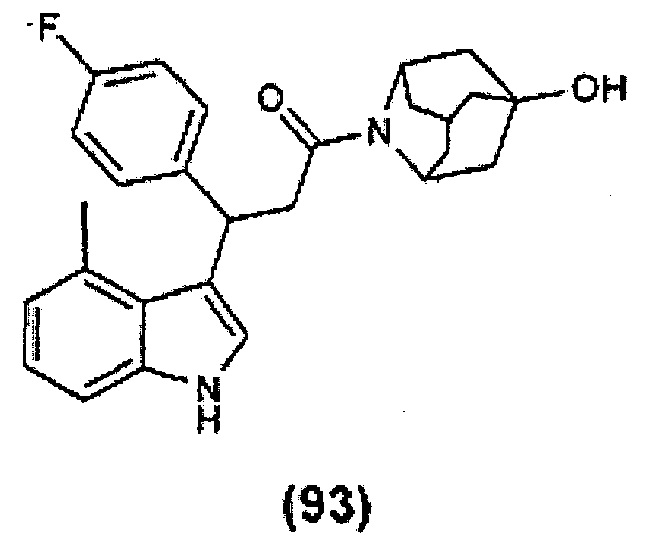
Синтез Соединения (93); Соединение (93) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (93). 1H ЯМР (300 МГц, ДМСО-d6): δ 10.91 (s, 1H), 7.36 (m, 1H), 7.18-7.23 (m, 2H), 7.12-7.15 (d, 1H), 7.01-7.06 (t, 2H), 6.85-6.902 (t, 1H), 6.58-6.60 (d, 1H), 4.99-5.02 (m, 1H), 4.74 (m, 1H), 4.58-4.66 (d, 1H), 4.36 (m, 1H), 2.95-2.97 (m, 2H), 2.42 (s, 3H), 2.07-2.17 (m, 1H), 1.28-1.65 (m, 10Н). ЖХ-МС (M+H)+ = 433,2; чистота по ВЭЖХ = 94,79%.
ПРИМЕР 94: 3-(6-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-фенилпропан-1-он (94)
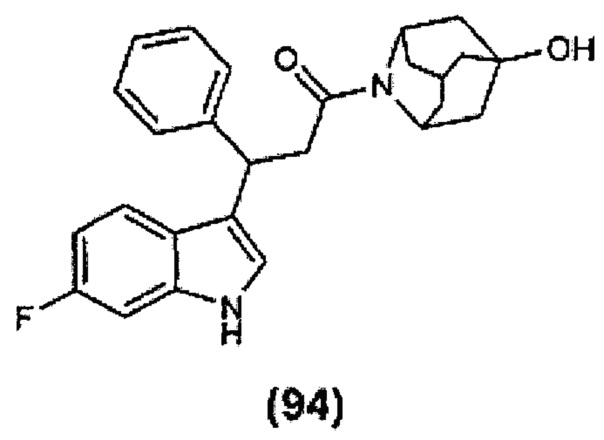
Синтез Соединения (94): Соединение (94) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (94). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.94 (s, 1H), 7.30-7.32 (m. 4H), 7.19-7.26 (m, 2H), 7.04-7.13 (m, 2H), 6.69-6.75 (t, 1H), 4.71 (s, 1H), 4.56-4.65 (m, 2H), 4.35 (s, 1H), 3.02-3.05 (m, 2H), 2.06-2.13 (d, 1H), 1.63 (s, 2H), 1.20-1.59 (m, 8H). ЖХ-МС (М+Н)+ = 419,1; чистота по ВЭЖХ = 93,86%.
ПРИМЕР 95: 3-циклопропил-3-(4-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)пропан-1-он (95)
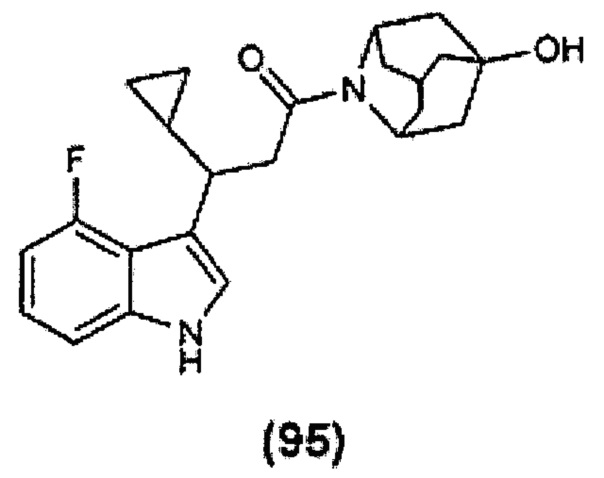
Синтез Соединения (95): Соединение (95) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (95). 1Н ЯМР (300 МГц, CDCl3): δ 8.13 (s, 1H), 6.93-7.08 (m, 3Н), 6.66-6.73 (m, 1H), 4.93 (s, 1H), 4.24 (s, 1H), 2.94-3.04 (m, 1H), 2.66-2.75 (m, 1H), 2.60 (s, 1H), 2.08-2.20 (d, 1H), 1.21-1.67 (m, 10Н), 0.76-0.97 (m, 1H), 0.49-0.53 (m, 1H), 0.26-0.32 (m, 2H), 0.05-0.07 (m, 1H). ЖХ-МС (M+H)+ = 383,1; чистота по ВЭЖХ = 96,02%.
ПРИМЕР 96: 3-(2,4-дифторфенил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13.7]дец-2-ил)-3-(1H-индол-3-ил)пропан-1-он (96)
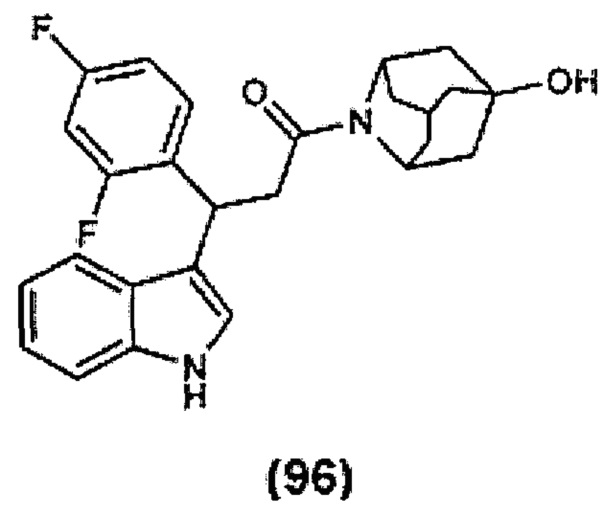
Синтез Соединения (96): Соединение (96) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (96). 1H ЯМР (300 МГц. ДМСО-d6): δ 10.90 (s, 1H), 7.36-7.41 (m, 1H), 7.32 (m, 2H), 7.29 (m, 1H), 7.11-7.15 (t, 1H), 6.97-7.05 (t, 1H), 6.88-6.95 (m, 2H), 4.93 (t, 1H), 4.69 (s, 1H), 4.62-4.64 (d, 1H), 4.40 (s, 1H), 2.72-3.11 (m, 2H), 2.14-2.27 (m, 1H), 1.24-1.66 (m, 10Н). ЖХ-МС (M+H)+ = 437,1; чистота по ВЭЖХ = 92,20%.
ПРИМЕР 97: 3-(6-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13.7]дец-2-ил)бутан-1-он, пик 1 (97)
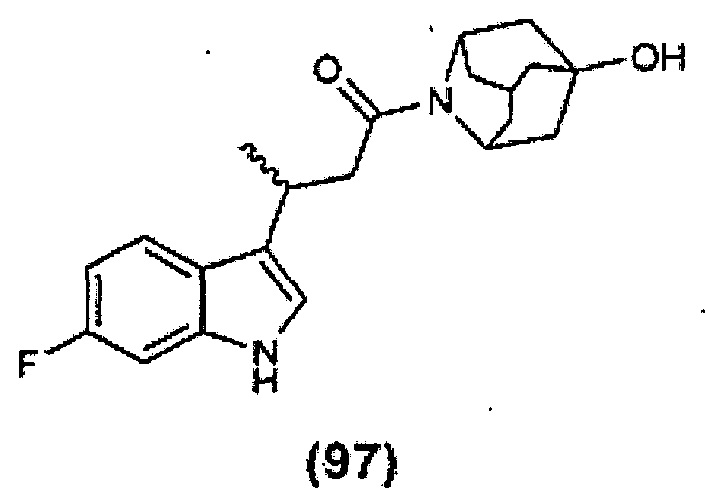
Синтез Соединения (97) (пик 1): Рацемет (79) разделили хиральной колоночной хроматографией с получением Соединения 97 (пик 1). 1Н ЯМР (300 МГц, CDCl3): δ 7.90 (s, 1Н), 7.48-7.53 (m, 1Н), 6.92-6.97 (m, 2H), 6.78-6.84 (t, 1Н), 4.97 (s, 1Н), 4.09 (s, 1Н), 3.52-3.96 (m, 1Н), 2.67-2.75 (m, 1Н), 2.41-2.48 (m, 1Н), 2.11-2.22 (d, 1Н), 1.60 (m, 6H), 1.36-1.39 (m, 6H). ЖХ-МС (М+H)+ = 357,1; чистота по ВЭЖХ = 93,22%; хиральная чистота: 100% (время удерживания = 15,45 мин.); колонка: Chiralpak IC, 4,6 мм Х 250 мм; подвижная фаза: гексаны: i-PrOH: ДХМ (7,5:1,5:1,0).
ПРИМЕР 98: 3-(6-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13.7]дец-2-ил)бутан-1-он, пик) (98)
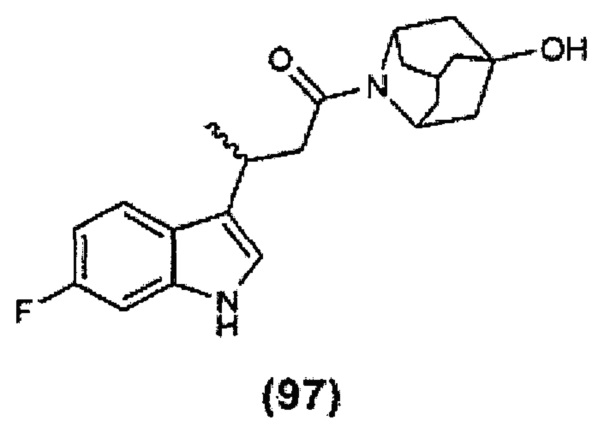
Синтез Соединения (98) (пик 2): Рацемет (79) разделили хиральной колоночной хроматографией с получением Соединения 98 (пик 2). 1Н ЯМР (300 МГц, CDCl3): δ 7.89 (s, 1Н), 7.48-7.53 (m, 1Н), 6.93-6.97 (m, 2H), 6.78-6.84 (t, 1Н), 4.97 (s, 1Н), 4.08 (s, 1Н), 3.52-3.59 (m, 1Н), 2.67-2.75 (m, 1Н), 2.41-2.48 (m, 1Н), 2.11-2.22 (d, 1Н), 1.54-1.69 (m, 9H), 1.36-1.38 (d, 3H). ЖХ-МС (М+Н)+ = 357,2; чистота по ВЭЖХ = 95,14%; хиральная чистота: 99,35% (время удерживания = 19,0 мин.); колонка: Chiralpak IC, 4,6 мм X 250 мм; подвижная фаза: гексаны: i-PrOH: ДХМ (7,5:1,5:1,0).
ПРИМЕР 99: 3-(4-фтор-1H-индол-3-ил)-3-(4-фторфенил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)пропан-1-он(99)
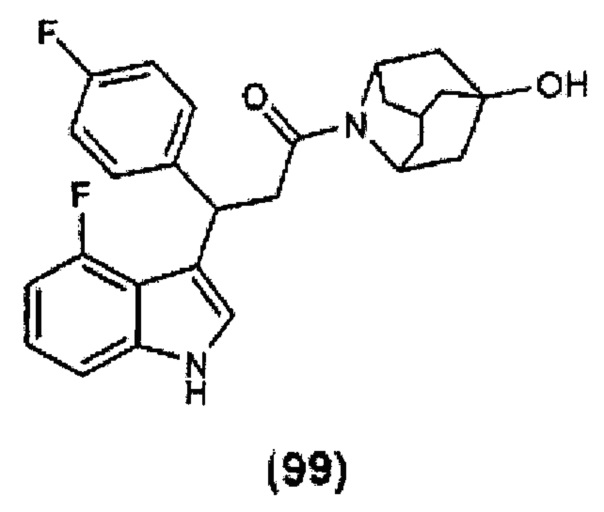
Синтез Соединения (99):
Соединение (99) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (99). 1Н ЯМР (300 МГц, ДМСО-d6): δ 11.19-11.23 (d, 1Н), 7.37 (s, 1H), 7.27-7.29 (t, 2H), 7.12-7.14 (d, 1Н), 6.93-7.05 (m, 3Н), 6.58-6.65 (m, 1Н), 4.80-4.84 (m, 1Н), 4.70 (s, 1Н), 4.59-4.65 (d, 1Н), 4.37 (s, 1Н), 2.96-3.11 (m, 2H), 2.08-2.16 (d, 1Н), 1.29-1.65 (m, 10Н). ЖХ-МС (M+H)+ = 437,1; чистота по ВЭЖХ = 99,01%.
ПРИМЕР 100: 2-[3-(4-хлор-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13.7]декан-5-карбонитрил, пик 1 (100):
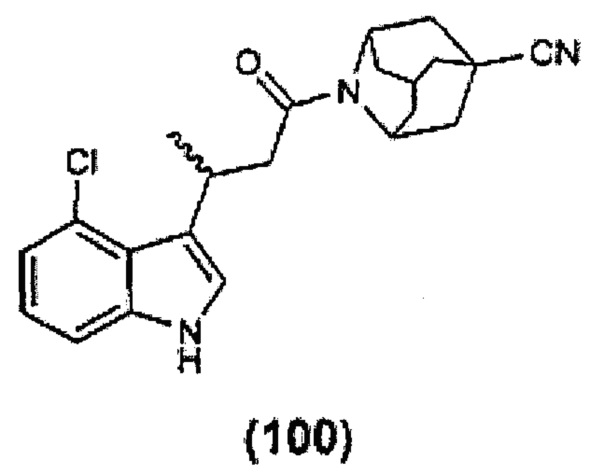
Синтез Соединения (100) (пик I): Рацемат Соединения (59) разделили с помощью препаративной хиральной колонки с получением Соединения (100) (пик 1). 1Н ЯМР (300 МГц, CDCl3): δ 8.06 (s, 1Н), 7.32 (m, 1Н), 7.00-7.02 (m, 3Н), 4.90 (s, 1Н), 3.96-4.11 (m, 2H), 2.77-2.87 (m, 1Н), 2.40-2.48 (m, 1Н), 1.60-2.12 (m, 11H), 1.37-1.40 (m, 3Н). ЖХ-МС (М+Н)+ = 382,1; чистота по ВЭЖХ = 98,74%; хиральная чистота: 100% (время удерживания = 16,17 мин.); колонка: Chiralpak IC, 4,6 мм Х 250 мм; подвижная фаза: гексаны: i-PrOH: ДХМ (7,5:1,5:1,0).
ПРИМЕР 101: 2-[3-(4-хлор-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13.7]декан-5-карбонитрил, пик 2 (101):
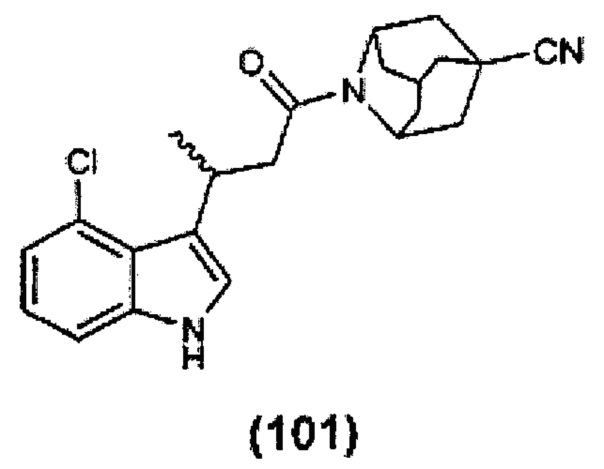
Синтез Соединения (101) (пик 2): Рацемат Соединения (59) разделили с помощью препаративной хиральной колонки с получением Соединения (101) (пик 2). 1Н ЯМР (300 МГц, CDCl3): δ 8.07 (s, 1Н). 7.00-7.04 (m, 4H), 4.90 (s, 1Н), 3.96-4.11 (m, 2H), 2.77-2.87 (m, 1Н), 2.40-2.48 (m, 1Н), 1.60-2.12 (m, 11H), 1.37-1.40 (m, 3Н). ЖХ-МС (M+H)+ =3 82,1; чистота по ВЭЖХ = 98,08%; хиральная чистота: 100% (время удерживания = 30,89 мин.); колонка: Chiralpak IC, 4,6 мм Х 250 мм; подвижная фаза: гексаны: i-PrOH: ДХМ (7,5:1,5:1,0).
ПРИМЕР 102: 3-циклопропил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он, пик 1 (102):
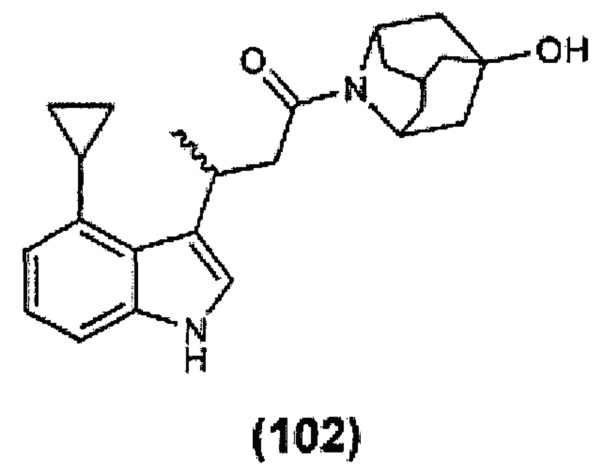
Синтез Соединения (102) (пик I): Рацемат Соединения (41) разделили с помощью препаративной хиральной колонки с получением Соединения (102) (пик 1). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.80 (s, 1Н), 7.12-7.16 (m, 2H), 6.88-6.963 (t, 1Н), 6.55-6.58 (d, 1Н), 4.81 (s, 1Н), 4.65 (d, 1Н), 4.32 (s, 1Н), 4.01-4.06 (m, 1Н), 2.67-2.72 (m, 2H), 2.17 (s, 1Н), 1.43-1.68 (m, 11H), 1.27-1.30 (d, 3Н), 0.89-0.95 (m, 2H), 0.73-0.86 (m, 2H). ЖХ-МС (M+H)+ = 379,2; чистота по ВЭЖХ = 98,53%; хиральная чистота: 100% (время удерживания = 15,36 мин.); колонка: Chiralpak IC, 4,6 мм Х 250 мм; подвижная фаза: гексаны: i-PrOH: ДХМ (7,5:1,5:1,0).
ПРИМЕР 103: 3-(4-циклопропил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он, пик 2 (103):
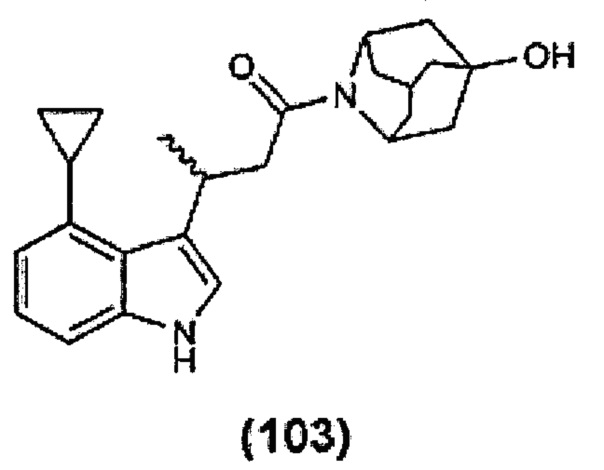
Синтез Соединения (103) (пик 2): Рацемат Соединения (41) разделили с помощью препаративной хиральной колонки с получением Соединения (103) (пик 2). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.80 (s, 1Н), 7.12-7.16 (m, 2H), 6.88-6.963 (t, 1Н), 6.55-6.58 (d, 1Н), 4.81 (s, 1Н), 4.65 (d, 1Н), 4.32 (s, 1Н), 3.97-4.05 (m. 1Н), 2.67-2.72 (m, 2H), 2.17 (s, 1Н), 1.43-1.68 (m, 11H), 1.27-1.30 (d, 3H), 0.83-0.95 (m, 2H), 0.73-0.74 (m. 2H). ЖХ-МС (M+H)+ = 379,2; чистота по ВЭЖХ = 99,43%; хиральная чистота: 100% (время удерживания = 22,17 мин.); колонка: Chiralpak IC, 4,6 мм Х 250 мм; подвижная фаза: гексаны: i-PrOH: ДХМ (7,5:1,5:1,0).
ПРИМЕР 104: 1-(5фтор-2-азатрицикло[3.3.1.13.7]дец-2-ил)-3(4-метил-1H-индол-3-ил)бутан-1-он (104):
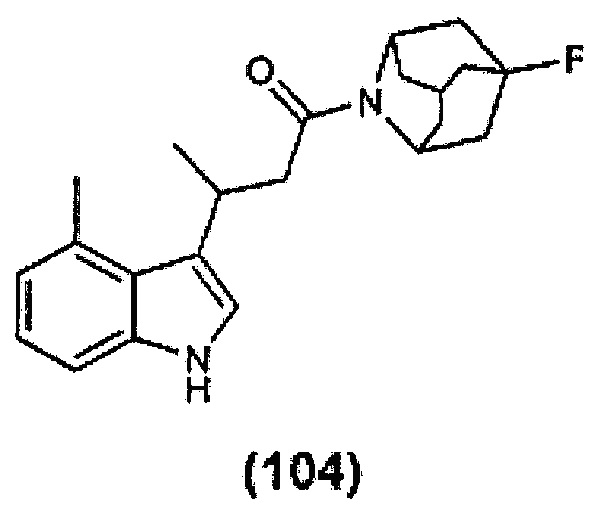
Синтез Соединения (104): Соединение (104) синтезировали по способу, использованному для получения Соединения (24) (Схема 12). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя гексан: EtOAc в качестве элюента, с получением Соединения (104). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.79 (s, 1H), 7.12-7.16 (m, 2H), 6.87-6.92 (mt, 1Н), 6.67-6.69 (d, 1Н), 4.92 (s, 1Н), 4.48 (s, 1Н), 3.73-3.80 (m, 1Н), 2.65-2.72 (m, 1Н), 2.61 (s, 3H), 2.27-2.36 (m, 2H), 1.42-1.93 (m, 10Н), 1.24-1.26 (d, 3H). ЖХ-МС (M+H)+ = 355,2; чистота по ВЭЖХ = 98,31%.
ПРИМЕР 105: 1-(2-азатрицикло[3.3.1.13.7]дец-2-ил)-3-(1,3-бензотиазол-2-ил)бутан-1-он (105):
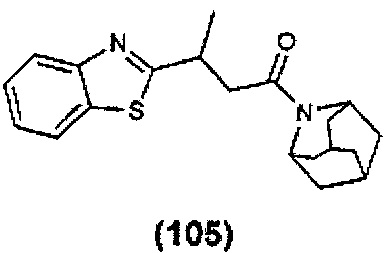
Синтез Соединения (105): Соединение (105) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя гексан: EtOAc в качестве элюента, с получением Соединения (105). 1Н ЯМР (300 МГц, CDCl3): δ 7.87-7.89 (d, 1H), 7.74-7.77 (d, 1H), 7.33-7.38 (tm, 1H), 7.23-7.28 (t, 1Н), 4.77 (s, 1Н), 4.05 (s, 1H), 3.65-3.89 (m, 1H), 2.96-3.03 (m, 1H), 2.55-2.57 (m, 1H), 1.66-2.01 (m, 12Н), 1.33-1.35 (d, 3Н). ЖХ-МС (М+Н)+ = 341,1; чистота по ВЭЖХ = 98,67%.
ПРИМЕР 106: 3-(1,3-бензотиазол-2-ил)-1-(5-фтор-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (196):
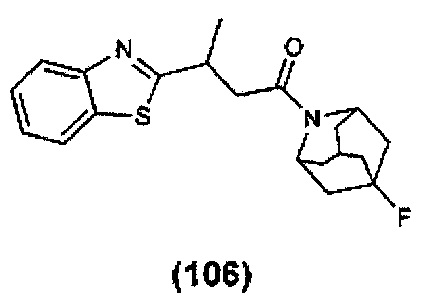
Синтез Соединения (106): Соединение (106) синтезировали по способу, использованному для получения Соединения (24) (Схема 12). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя гексан: EtOAc в качестве элюента, с получением Соединения (106). 1Н ЯМР (300 МГц, CDCl3): δ 7.85-7.88 (d, 1H), 7.75-7.78 (m, 1Н), 7.33-7.39 (m, 1Н), 7.23-7.29 (m, 1H), 5.04 (s, 1Н), 4.40 (s, 1Н), 3.79-3.88 (m, 1Н), 3.01-3.09 (m, 1Н), 2.53-2.61 (m, 1H), 2.36 (d, 1Н), 1.50-1.88 (m, 10Н), 1.42-1.47 (d, 3Н). ЖХ-МС (М+Н)+ = 359,1; чистота по ВЭЖХ = 95,97%.
ПРИМЕР 107: 3-(1,3-бензотиазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-фенилпропан-1-он (107):
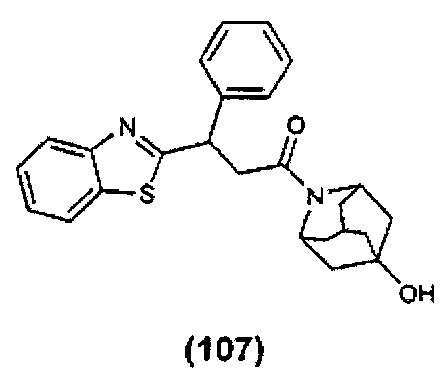
Синтез Соединения (107): Соединение (107) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (107). 1Н ЯМР (300 МГц, ДМСО-d6): δ 7.91-8.00 (m, 2Н), 7.22-7.50 (m, 7Н), 4.95-5.00 (m, 1H), 4.64-4.69 (m, 2Н), 4.43 (s, 1H), 3.52-3.59 (m, 1H), 2.95-3.04 (m, 1Н), 2.08-2.20 (m, 1Н), 1.23-1.79 (m, 10Н). ЖХ-МС (М+Н)+ = 419,1; чистота по ВЭЖХ = 99,70%.
ПРИМЕР 108: 3-(1,3-бензотиазол-2-ил)-1-(5-фтор-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-фенилпропан-1-он (108):
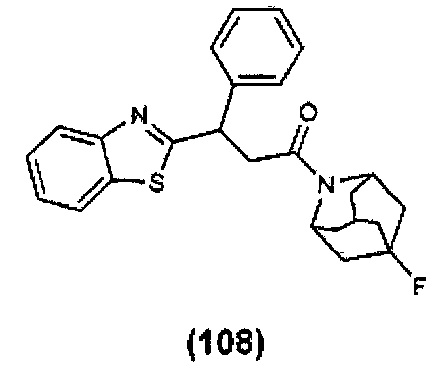
Синтез Соединения (108): Соединение (108) синтезировали по способу, использованному для получения Соединения (24) (Схема 12). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя гексан: EtOAc в качестве элюента, с получением Соединения (108). 1Н ЯМР (300 МГц, ДМСО-d6): δ 7.90-8.00 (m, 2Н), 7.22-7.50 (m, 7Н), 4.95-5.01 (m, 1Н), 4.82 (s, 1H), 4.60 (s, 1Н), 3.53-3.61 (m, 1Н), 2.96-3.09 (m, 1Н), 2.27-2.35 (m, 1H), 1.23-1.79 (m, 10Н). ЖХ-МС (М+Н)+ = 421,1; чистота по ВЭЖХ = 92,61%.
ПРИМЕР 109: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(5-метокси-1H-индол-3-ил)бутан-1-он (109):
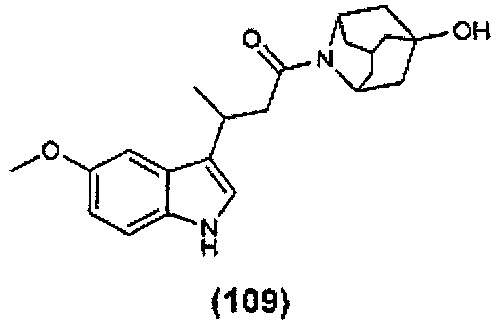
Синтез Соединения (109): Соединение (109) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (109). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.61 (s, 1Н), 7.18-7.22 (d, 1Н), 7.09 (s, 1H), 6.96 (s, 1H), 6.68-6.71 (m, 1Н), 4.79 (s, 1Н), 4.59-4.64 (d, 1H), 4.25-4.28 (m, 1Н), 3.74 (s, 3Н), 3.37-3.40 (m, 1H), 2.63-2.72 (m, 2Н), 2.07-2.37 (m, 1H), 1.27-1.65 (m, 13Н). ЖХ-МС (М+Н)+ = 369,2; чистота по ВЭЖХ = 98,14%.
ПРИМЕР 110: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(5-метил-1H-индол-3-ил)бутан-1-он (110):
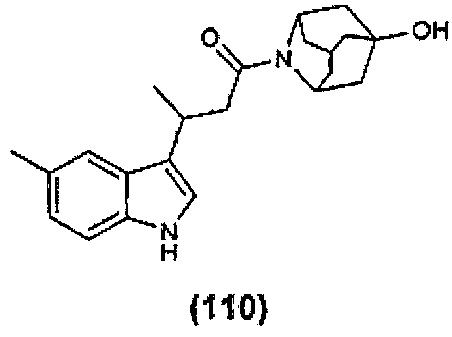
Синтез Соединения (110): Соединение (110) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (110). 1H ЯМР (300 МГц, ДМСО-d6): δ 10.62 (s, 1Н), 7.29 (s, 1H), 7.18-7.21 (d, 1Н), 7.06 (s, 1H), 6.85-6.88 (d, 1Н), 4.80 (s, 1H), 4.61-4.65 (d, 1H), 4.28 (s, 1H), 3.39 (m, 1H), 2.62-2.72 (m, 2H), 2.63 (s, 3H), 2.10-2.27 (m, 1H), 1.27-1.66 (m, 13H). ЖХ-МС (M+H)+ = 353,2; чистота по ВЭЖХ = 95,0%.
ПРИМЕР 111: 2-[3-(4-фтор-1H-индол-3-ил)-3-фенилпропаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (111):
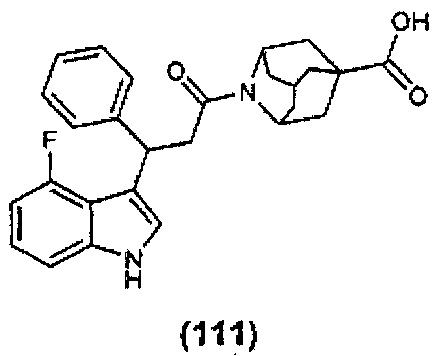
Синтез Соединения (111): Соединение (111) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (111). 1H ЯМР (300 МГц, ДМСО-d6): δ 12.17 (s, 1Н), 11.18 (s, 1Н), 7.37 (s, 1Н), 7.17-7.31 (m, 4Н), 7.05-7.14 (m, 2Н), 6.92-6.99 (m, 1Н), 6.57-6.64 (m, 1Н), 4.81-4.86 (t, 1H), 4.64 (s, 1Н), 4.31 (s, 1H), 3.00-3.07 (m, 2Н), 1.30-2.07 (m, 11Н). ЖХ-МС (М+Н)+ = 447,3; чистота по ВЭЖХ = 98,60%.
ПРИМЕР 112: 2-[3-(5-фтор-1H-индол-3-ил)-3-фенилпропаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (112):
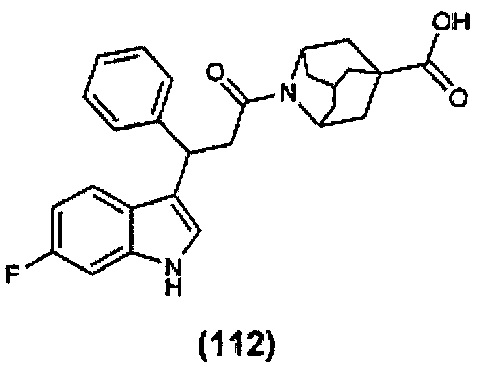
Синтез Соединения (112): Соединение (112) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (112). 1H ЯМР (300 МГц, ДМСО-d6): δ 12.19 (s, 1Н), 10.94 (s, 1Н), 7.30-7.33 (m, 4Н), 7.18-7.27 (m, 2Н), 7.03-7.13 (m, 2Н), 6.69-6.74 (m, 1Н), 4.61-4.66 (m, 2H), 4.29- (s, 1H), 3.02-3.05 (d, 2H), 2.72 (m, 2H), 1.32-2.08 (m, 9H). ЖХ-МС (M+H)+ = 447,2; чистота по ВЭЖХ = 96,93%.
ПРИМЕР 113: 3-(5-фтор-2-метил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (113):
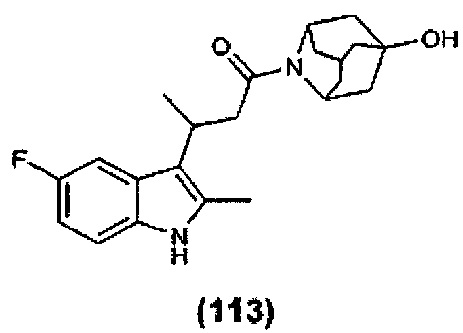
Синтез Соединения (113): Соединение (113) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (113). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.74 (s, 1H), 7.26-7.31 (m, 1H), 7.15-7.20 (m, 1H), 6.73-6.80 (m, 1Н), 4.72 (s, 1Н), 4.51-4.61 (m, 1H), 4.09 (s, 1H), 3.35-3.39 (m, 1H), 2.70-2.80 (m, 2Н), 2.29 (s, 3Н), 1.97-2.14 (d, 1Н), 0.74-1.60 (m, 13Н). ЖХ-МС (М+Н)+ = 371,2; чистота по ВЭЖХ = 97,40%.
ПРИМЕР 114: 3-(1,3-бензотиазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он, пик 1 (114):

Синтез Соединения (114) (пик 1): Рацемат Соединения (84) разделили с помощью препаративной хиральной ВЭЖХ колонки с получением Соединения (114) (пик 1). 1H ЯМР (300 МГц, CDCl3): δ 7.86-7.89 (d, 1H), 7.75-7.78 (d, 1H), 7.34-7.39 (t, 1H), 7.24-7.29 (t, 1Н), 4.96 (s, 1H), 4.31 (s, 1H), 3.79-3.86 (m, 1H), 2.99-3.07 (m, 1H), 2.54-2.62 (m, 1H), 2.22-2.27 (m, 1Н), 1.43-1.75 (m, 14Н). ЖХ-МС (М+Н)+ = 357,1; чистота по ВЭЖХ = 99,8%; хиральная чистота: 100% (время удерживания = 16,99 мин.); колонка: Chiralpak IC, 4,6 мм X 250 мм; подвижная фаза: гексаны: i-PrOH: ДХМ (7:2:1).
ПРИМЕР 115: 3-(1,3-бензотиазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он, пик 2 (115):
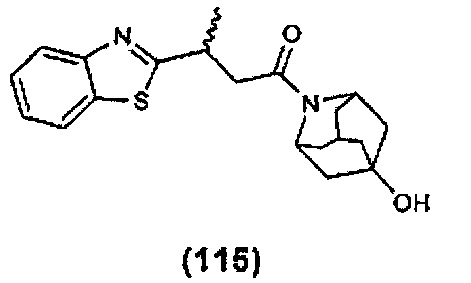
Синтез Соединения (115) (пик 2): Рацемат Соединения (84) разделили с помощью препаративной хиральной ВЭЖХ колонки с получением Соединения (115) (пик 2). 1Н ЯМР (300 МГц, CDCl3): δ 7.86-7.89 (d, 1H), 7.75-7.78 (d, 1Н), 7.34-7.39 (t, 1Н), 7.24-7.29 (t, 1H), 4.96 (s, 1Н), 4.31 (s, 1Н), 3.79-3.86 (m, 1Н), 2.99-3.07 (m, 1H), 2.54-2.62 (m, 1H), 2.22-2.27 (m, 1H), 1.43-1.75 (m, 14Н). ЖХ-МС (М+Н)+ = 357,1; чистота по ВЭЖХ = 99,85%; хиральная чистота: 100% (время удерживания = 23,47 мин.); колонка: Chiralpak IC, 4,6 мм X 250 мм; подвижная фаза: гексаны: i-PrOH: ДХМ (7:2:1).
ПРИМЕР 116: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(4-метил-1H-индол-3-ил)пропан-1-он (116):
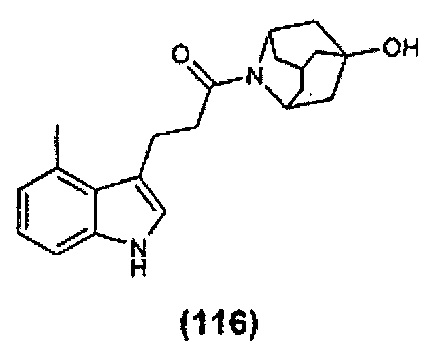
Синтез Соединения (116): Соединение (116) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (116). 1Н ЯМР (300 МГц, CDCl3): δ 7.90 (s, 1H), 7.10-7.13 (d, 1H), 6.96-7.01 (t, 1H), 6.92 (d, 1H), 6.76-6.78 (d, 1H), 5.02 (s, 1H), 4.13 (s, 1Н), 3.18-3.23 (m, 2Н), 2.64 (s, 3Н), 2.58-2.63 (m, 2Н), 2.24 (s, 1Н), 1.49-1.74 (m, 11Н). ЖХ-МС (М+Н)+ = 339,3; чистота по ВЭЖХ = 91,46%.
ПРИМЕР 117: 3-(6-хлор-5-метокси-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (117):
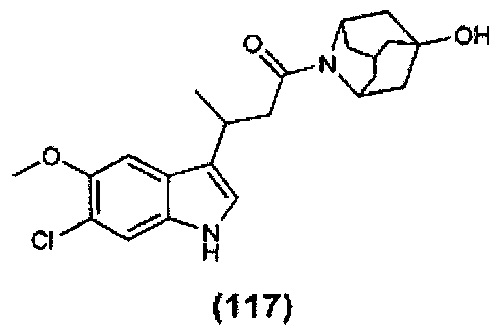
Синтез Соединения (117): Соединение (117) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (117). 1Н ЯМР (300 МГц, CDCl3): δ 7.85 (brs, 1H), 7.30-7.31 (d, 1Н), 7.09 (brs, 1H), 6.92-6.93 (m, 1H), 4.98 (brs, 1Н), 4.09 (brs, 1H), 3.87 (s, 3Н), 3.52-3.59 (m, 1H), 2.63-2.71 (m, 1H), 2.40-2.44 (m, 1Н), 2.10 (brs, 1Н), 1.41-1.70 (m, 10Н), 1.35-1.38 (d, 3Н). ЖХ-МС (М+Н)+ = 403,2; чистота по ВЭЖХ = 99,36%.
ПРИМЕР 118: 2-[3-(1,3-бензотиазол-2-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбонитрил (118):
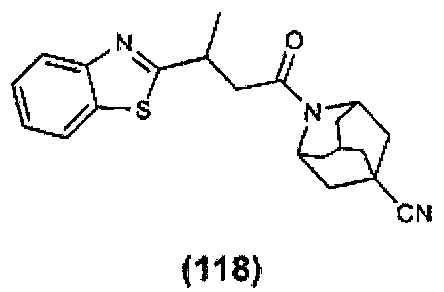
Синтез Соединения (118): Соединение (118) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (118). 1Н ЯМР (300 МГц, CDCl3): δ 7.84-7.89 (dd, 1H), 7.76-7.78 (d, 1Н), 7.34-7.40 (m, 1Н), 7.25-7.30 (m, 1H), 4.86 (brs, 1H), 4.23 (brs, 1H), 3.79-3.87 (m, 1Н), 3.00-3.09 (m, 1Н), 2.49-2.59 (m, 1Н), 1.90-2.18 (m, 8Н), 1.63-1.76 (m, 3Н), 1.45 (d, 3Н). ЖХ-МС (М+Н)+ = 366,2; чистота по ВЭЖХ = 93,68%.
ПРИМЕР 119: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(4-хлор-1,3-бензотиазол-2-ил)бутан-1-он (119):
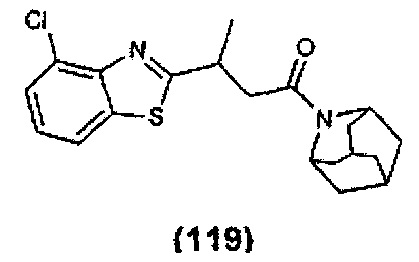
Синтез Соединения (119): Соединение (119) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя гексан: EtOAc в качестве элюента, с получением Соединения (119). 1Н ЯМР (300 МГц, CDCl3): δ 7.64-7.67 (d, 1Н), 7.36-7.39 (d, 1H), 7.15-7.21 (dd, 1Н), 4.74 (brs, 1H), 4.10 (brs, 1H), 3.85-3.92 (q, 1H), 3.04-3.11 (dd, 1H), 2.53-2.61 (dd, 1H), 1.98-2.02 (m, 2H), 1.65-1.79 (m, 10H), 1.45 (d, 3Н). ЖХ-МС (M+H)+ = 375,1; чистота по ВЭЖХ = 98,71%.
ПРИМЕР 120: 3-(4-хлор-1,3-бензотиазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (120):
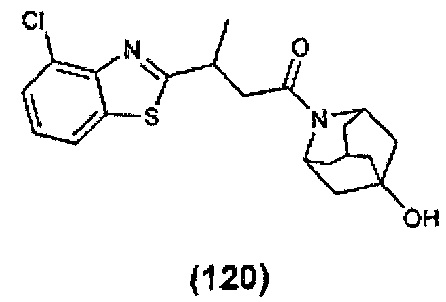
Синтез Соединения (120): Соединение (120) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (120). 1Н ЯМР (300 МГц, ДМСО-d6): δ 8.02-8.05 (d, 1Н), 7.56-7.58 (d, 1H), 7.36-7.41 (t, 1H), 4.65-4.72 (m, 2Н), 4.34 (s, 1Н), 3.80 (m, 1Н), 2.80-3.20 (m, 2Н), 1.45-2.22 (m, 11Н), 1.39-1.42 (d, 3Н). ЖХ-МС (М+Н)+ = 391,1; чистота по ВЭЖХ = 98,58%.
ПРИМЕР 121: 3-(1H-бензотриазол-1-ил)-1-(5-гидрокси-2-азатрицикло [3.3.1.13,7]дец-2-ил)-3-фенилпропан-1-он (121):
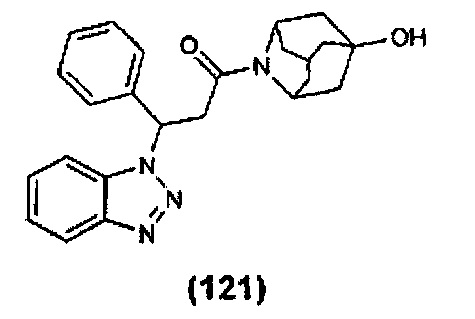
СХЕМА СИНТЕЗА 24
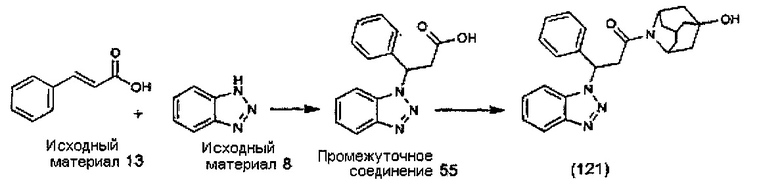
Синтез 3-(1H-бензотриазол-1-ил)-3-фенилпропановой кислоты (Промежуточное соединение 55):
В 30 мл пробирку АСЕ для работы под давлением, оснащенную магнитной мешалкой, загрузили исходный материал 13 (2 г, 13,4 ммоль) и исходный материал 8 (4,8 г, 40,4 ммоль). Реакционную смесь нагревали при 150°С в течение 12 часов. После завершения реакции смесь разбавили этил ацетатом и концентрировали. Полученный неочищенный продукт очистили с помощью колоночной комби-флэш-хроматографии, элюируя гексанами EtOAc, с получением Промежуточного соединения 55 (2,1 г).
Синтез Соединения (121): К перемешанному раствору Промежуточного соединения 55 (75 мг, 0,28 ммоль) в ТГФ (4 мл) добавили Промежуточное соединение 7 (43 мг, 0,28 ммоль) и HBTU (126 мг, 0,3 ммоль). Затем добавили DIPEA (108 мг, 0,84 ммоль) при 0°C. Полученную реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения реакции полученную массу сначала погасили водой, затем экстрагировали этилацетатом, а затем концентрировали. Полученный неочищенный продукт очистили препаративной ТСХ, элюируя гексанами: EtOAc, с получением соединения (121) (40 мг) в виде белого твердого вещества. 1Н ЯМР (300 МГц, ДМСО-d6): δ 7.99-8.02 (d, 1Н), 7.88-7.91 (d, 1H), 7.45-7.52 (m, 3Н), 7.24-7.39 (m, 3Н), 6.52-6.54 (d, 1Н), 4.66-4.67 (d, 1H), 4.62 (s, 1Н), 4.46 (s, 1H), 4.01-4.10 (m, 1H), 3.23-3.25 (m, 1Н), 2.16 (s, 1H), 1.23-1.67 (m, 11Н). ЖХ-МС (М+Н)+ = 403,2; чистота по ВЭЖХ = 93,95%.
ПРИМЕР 122: 2-[3-(4-фтор-1H-индол-3-ил)-3-(тиофен-2-ил)пропаноил]-2-азатрицикло[3.3.1.13,7]декан-4-карбоновая кислота (122):
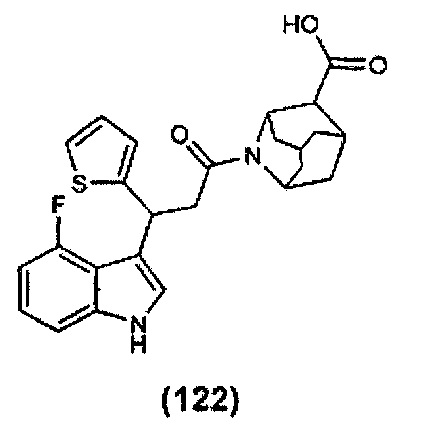
СХЕМА СИНТЕЗА 25
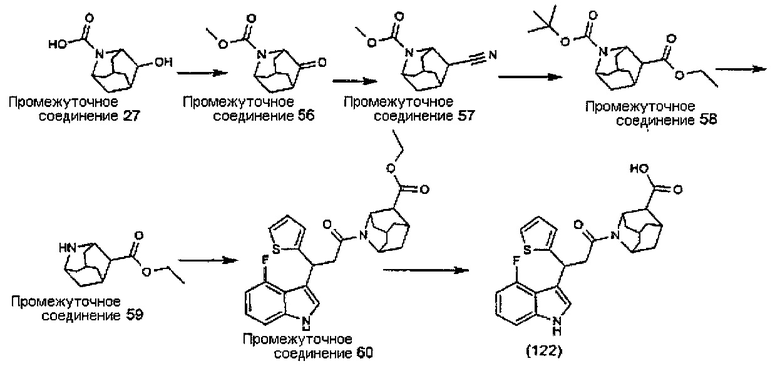
Синтез метил 4-оксо-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилата (Промежуточное соединение 56):
К перемешанному раствору Промежуточного соединения 27 (0,5 г, 2,3 ммоль) в ДХМ (10 мл) добавили РСС (1,01 г, 4,7 ммоль). Реакционную смесь затем перемешивали при комнатной температуре в течение 16 часов. После завершения реакции полученную массу погасили водой и экстрагировали ДХМ. Органический слой промыли 10% раствором NaHCO3 и концентрировали. Полученный неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя гексанами: EtOAc, с получением Промежуточного соединения 56 (230 мг).
Синтез метил 4-циано-2-азатрицикло[3.3.1.13,7]декан-2-карбоксилата (Промежуточное соединение 57):
К перемешанному раствору Промежуточного соединения 56 (0,23 г, 1,1 ммоль) и Tos MIC (0,3 г, 1,5 ммоль) в ДМЭ: EtOH (5 мл: 0,2 мл) добавили t-BuOK (0,37 г, 3,30 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь отфильтровали. Часть фильтрата концентрировали с получением Промежуточного соединения 57 (230 мг).
Синтез 2-трет-бутил 4-этил 2-азатрицикло[3.3.1.13,7]декан-2,4-дикарбоксилата (Промежуточное соединение 58):
В 100 мл круглодонную колбу, оснащенную магнитной мешалкой, загрузили Промежуточное соединение 57 (0,23 г, 1,04 ммоль) и 5 н. H2SO4 (25 мл). Реакционную смесь нагревали при 100°С в течение 36 часов. После завершения реакции (контролировали по ЖХ-МС) смесь охладили до 0°С. К охлажденной смеси добавили концентрированную HCl, затем добавили EtOH. Затем смесь нагревали при 90°С в течение 16 часов. Полученную массу погасили водой, подщелочили карбонатом натрия и промыли ДХМ. Водный слой разбавили ТГФ (40 мл), к которому добавили ТЭА (5 мл) и Boc-ангидрид (0,360 г, 1,4 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. Затем реакционную смесь экстрагировали этилацетатом и концентрировали. Полученный неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя гексанами: EtOAc, с получением Промежуточного соединения 58 (130 мг).
Синтез этил 2-азатрицикло[3.3.1.13,7]декан-4-карбоксилата трифторуксуснокислой соли (Промежуточное соединение 59):
К перемешанному раствору Промежуточного соединения 58 (0,13 г, 0,4 ммоль) в ДХМ (5 мл) добавили ТФК (0,1 г, 0,8 ммоль) при 0°C. Полученную реакционную смесь перемешивали в течение 4 часов при комнатной температуре. После завершения реакции (реакцию контролировали по ЖХ-МС) полученную массу концентрировали, затем растерли со смесью гексанов: эфира (1:1) с получением Промежуточного соединения 59 (130 мг).
Синтез этил 2-[3-(4-фтор-1H-индол-3-ил)-3-(тиофен-2-ил)пропаноил]-2-азатрицикло[3.3.1.13,7]декан-4-карбоксилата (Промежуточное соединение 60):
Промежуточное соединение 60 синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя гексаны: EtOAc в качестве элюента, с получением Промежуточного соединения 60.
Синтез Соединения (122):
Соединение (122) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). 1Н ЯМР (300 МГц, CDCl3): δ 8.30-9.30 (m, 1H), 7.00-7.02 (m, 2Н), 6.87-6.89 (m, 1H), 6.77-6.79 (m, 2Н), 6.62-6.65 (m, 1H), 6.46-6.56 (m, 1Н), 5.02-5.33 (m, 1Н), 4.34-4.73 (m, 1Н), 3.99-4.12 (m, 1H), 2.92-3.61 (m, 2Н), 1.45-2.64 (m, 11Н). ЖХ-МС (М+Н)+ = 453,2; чистота по ВЭЖХ = 98,96%.
ПРИМЕР 123: 2-[3-(4-хлор-1,3-бензотиазол-2-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбонитрил (123):
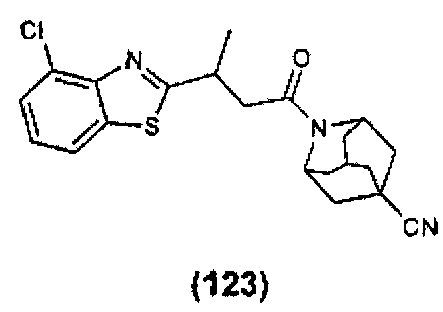
Синтез Соединения (123): Соединение (123) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя гексаны: EtOAc в качестве элюента, с получением Соединения (123). 1Н ЯМР (300 МГц, CDCl3): δ 7.65-7.67 (d, 1H), 7.36-7.40 (m, 1Н), 7.22 (m, 1H), 4.84 (s, 1H), 4.26 (s, 1Н), 3.84-3.91 (m, 1H), 3.11-3.20 (m, 1Н), 2.48-2.58 (m, 1Н), 1.55-2.15 (m, 11Н), 1.45-1.47 (m, 3Н). ЖХ-МС (М+Н)+ = 400,1; чистота по ВЭЖХ = 96,66%.
ПРИМЕР 124: 2-[3-(4-хлор-1,3-бензотиазол-2-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбонитрил (Соединение 124), пик 1:
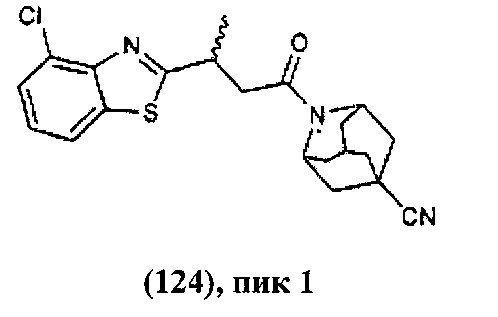
Синтез Соединения (124) (пик 1): Рацемическое соединение (123) разделили с помощью хиральной препаративной ВЭЖХ с получением Соединения (124) (пик 1). ЖХ-МС (М+Н)+ = 400,1; чистота по ВЭЖХ = 91,31%; хиральная колонка: Chiralpak IC, 4,6 мм X 250 мм; подвижная фаза: гексан: i-PrOH: ДХМ (7:2:1); время удерживания = 23,47 минуты.
ПРИМЕР 125: 2-[3-(4-хлор-1,3-бензотиазол-2-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбонитрил (Соединение 125), пик 2:
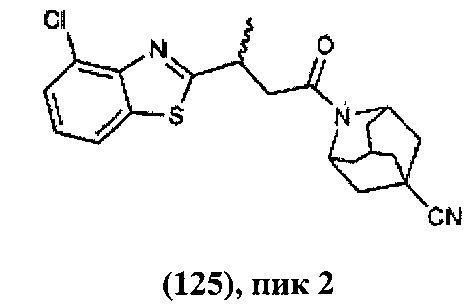
Синтез Соединения (125) (пик 2): Рацемическое соединение (123) разделили с помощью хиральной препаративной ВЭЖХ с получением Соединения (125), пик 2 (125). ЖХ-МС (М+Н)+ = 400,1; чистота по ВЭЖХ = 97,74%; хиральная колонка: Chiralpak IC, 4,6 мм X 250 мм; подвижная фаза: гексан: i-PrOH: ДХМ (7:2:1); время удерживания = 27,72 минуты.
ПРИМЕР 126: 3-(4-циклопропил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)пропан-1-он (126):
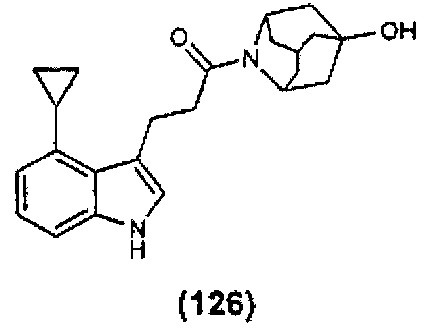
Синтез Соединения (126): Соединение (126) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (126). 1Н ЯМР (300 МГц, CDCl3): δ 8.00 (brs, 1H), 7.09-7.12 (d, 1Н), 6.96-7.01 (t, 1Н), 6.93-6.94 (d, 1Н), 6.64-6.66 (1H, d), 5.01 (brs, 1Н), 4.14 (brs, 1H), 3.31-3.36 (t, 2H), 2.65-2.70 (m, 2H), 2.32-2.41 (m, 1H), 2.23 (brs, 1H), 1.45-1.73 (m, 10H), 0.88-0.94 (m, 2H), 0.74-0.81 (m, 2H). ЖХ-МС (М+Н)+ = 365,2; чистота по ВЭЖХ = 98,22%.
ПРИМЕР 127: 3-(4-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(тиофен-2-ил)пропан-1-он (127):
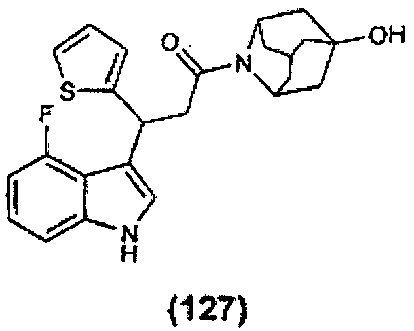
Синтез Соединения (127): Соединение (127) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (127). 1Н ЯМР (300 МГц, CDCl3): δ 8.34 (brs, 0.5Н), 8.30 (brs, 0.5Н), 6.99-7.06 (m, 2Н), 6.96-6.97 (m, 2Н), 6.81-6.83 (m, 2Н), 6.61-6.68 (m, 1Н), 5.14-5.20 (m, 1H), 4.94 (brs, 1H), 4.23 (brs, 1H), 3.02-3.20 (m, 2H), 2.14 (brs, 1H), 1.35-1.70 (m, 10H). ЖХ-МС (M+H)+ = 425,2; чистота по ВЭЖХ = 99,56%.
ПРИМЕР 128: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(4-пропил-1H-индол-3-ил)пропан-1-он (128):
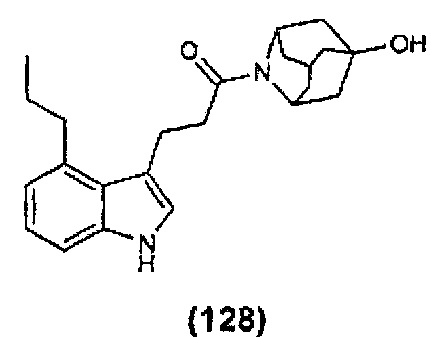
Синтез Соединения (128): Соединение (128) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (128). 1Н ЯМР (300 МГц, CDCl3): δ 7.99 (brs, 1Н), 7.10-7.13 (d, 1H), 6.99-7.04 (t, 1Н), 6.91-6.92 (d, 1Н), 6.80-6.82 (d, 1Н), 5.02 (brs, 1Н), 4.13 (brs, 1Н), 3.14-3.19 (t, 2H), 2.87-2.92 (t, 2H), 2.59-2.64 (m, 2H), 2.24 (brs, 1H), 1.53-1.74 (m, 12H), 0.93-0.98 (t, 3Н). ЖХ-МС (M+H)+ = 367,2; чистота по ВЭЖХ = 96,54%.
ПРИМЕР 129: 3-(6-хлор-5-метокси-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (129), пик 1:
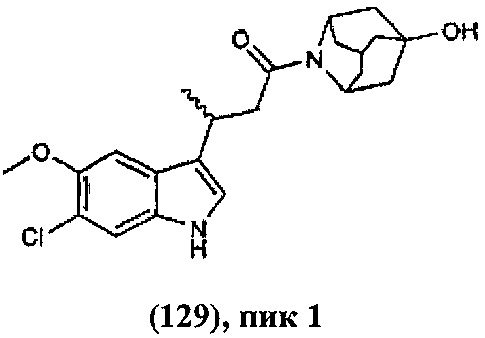
Синтез Соединения (129) (пик 1): Рацемат Соединения (117) разделили с помощью хиральной препаративной ВЭЖХ с получением Соединения (129) (пик 1). 1H ЯМР (300 МГц, CDCl3): δ 7.85 (brs, 1H), 7.30-7.31 (d, 1H), 7.09 (brs, 1H), 6.92-6.93 (m, 1H), 4.98 (brs, 1H), 4.09 (brs, 1H), 3.87 (s, 3Н), 3.52-3.59 (m, 1Н), 2.63-2.71 (m, 1H), 2.40-2.44 (m, 1H), 2.10 (brs, 1Н), 1.41-1.70 (m, 10Н), 1.35-1.38 (d, 3Н). ЖХ-МС (М+Н)+ = 403,2; чистота по ВЭЖХ = 99,32%; хиральная колонка: Chiralpak IC, 4,6 мм X 250 мм; подвижная фаза: гексан: i-PrOH: ДХМ (7:2:1); время удерживания = 11,65 минуты.
ПРИМЕР 130: 3-(6-хлор-5-метокси-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (130), пик 2:
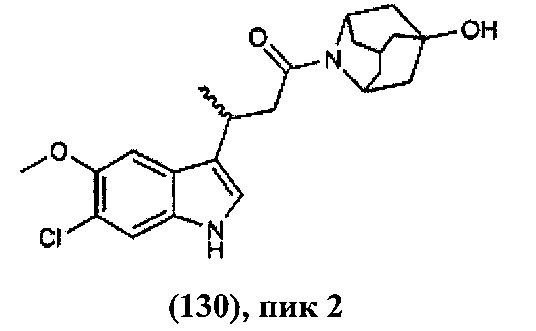
Синтез Соединения (130) (пик 2): Рацемат Соединения (117) разделили с помощью хиральной препаративной ВЭЖХ с получением Соединения (130) (пик 2). 1H ЯМР (300 МГц, CDCl3): δ 7.85 (brs, 1H), 7.30-7.31 (d, 1Н), 7.09 (brs, 1H), 6.92-6.93 (m, 1H), 4.98 (brs, 1H), 4.09 (brs, 1H), 3.87 (s, 3Н), 3.52-3.59 (m, 1Н), 2.63-2.71 (m, 1H), 2.40-2.44 (m, 1H), 2.10 (brs, 1Н), 1.41-1.70 (m, 10Н), 1.35-1.38 (d, 3Н). ЖХ-МС (М+Н)+ = 403,2; чистота по ВЭЖХ = 99,76%; хиральная колонка: Chiralpak IC, 4,6 мм X 250 мм; подвижная фаза: гексан: i-PrOH: ДХМ (7:2:1); время удерживания = 14,60 минуты.
ПРИМЕР 131: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-4-метил-3-(4-метил-1H-бензотриазол-1-ил)пентан-1-он (131):
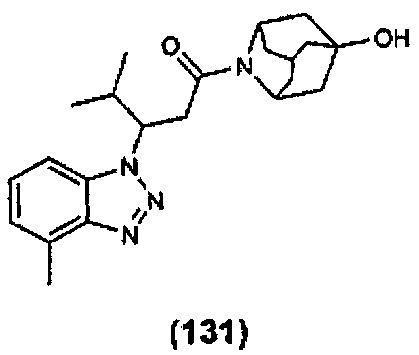
Синтез Соединения (131): Соединение (131) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (131). 1Н ЯМР (300 МГц, CDCl3): δ 7.37-7.40 (d, 1Н), 7.26-7.31 (t, 1H), 7.02-7.04 (d, 1H), 5.00-5.06 (m, 1H), 4.81 (brs, 1Н), 4.25 (brs, 1Н), 3.47-3.65 (m, 2Н), 2.72 (s, 3Н), 2.28-2.34 (m, 1Н), 2.10 (brs, 1H), 1.35-1.75 (М, 10Н), 0.96-0.98 (d, 3Н), 0.72-0.74 (d, 3Н). ЖХ-МС (М+Н)+ = 383,2; чистота по ВЭЖХ=94,65%.
ПРИМЕР 132: 3-(4-циклопропил-1H-пирроло[2,3-b]пиридин-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (132):
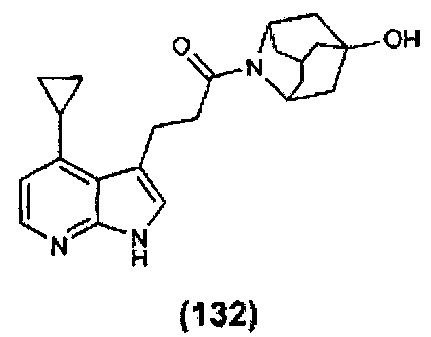
Синтез Соединения (132): Соединение (132) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (132). 1Н ЯМР (300 МГц, CDCl3): δ 9.30 (brs, 1H), 8.03-8.05 (d, 1H), 7.03 (s, 1Н), 6.47-6.49 (d, 1H), 5.02 (brs, 1H), 4.21 (brs, 1Н), 4.01 (brs, 1H), 2.73-2.78 (m, 1Н), 2.33-2.49 (m, 2Н), 2.26 (brs, 1Н), 1.42-1.75 (m, 10Н), 1.35-1.37 (d, 3Н). ЖХ-МС (М+Н)+ = 380,2; чистота по ВЭЖХ = 98,45%.
ПРИМЕР 133: 3-(6-хлор-5-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (133):
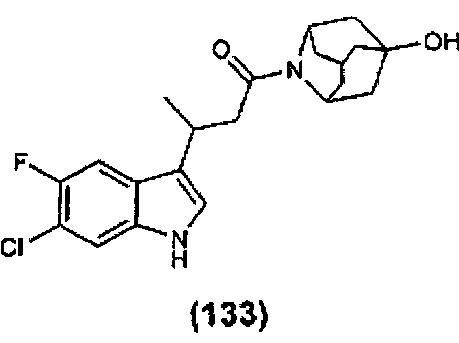
Синтез Соединения (133): Соединение (133) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (133). 1H ЯМР (300 МГц, CDCl3): δ 7.91 (brs, 1H), 7.31-7.34 (d, 1H), 7.28-7.30 (d, 1Н), 6.99-7.00 (d, 1Н), 4.98 (brs, 1H), 4.10 (brs, 1Н), 3.47-3.54 (q, 1H), 2.63-2.71 (m, 1Н), 2.40-2.44 (m, 1Н), 2.15 (brs, 1H), 1.40-1.71 (m, 10Н), 1.34-1.36 (d, 3Н). ЖХ-МС (М+Н)+ = 391,2; чистота по ВЭЖХ = 97,57%.
ПРИМЕР 134: 2-[3-(6-хлор-1H-индол-3-ил)-3-фенилпропаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (134):
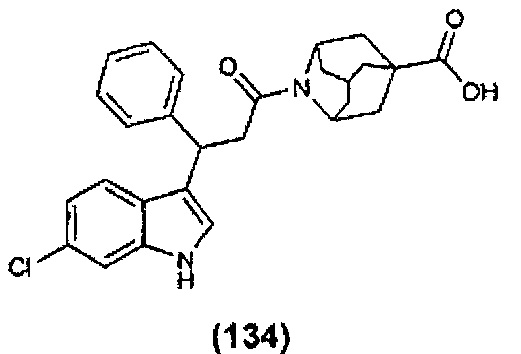
Синтез Соединения (134): Соединение (134) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (134). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.34 (brs, 1Н), 11.03 (s, 1H), 7.10-7.40 (m, 8Н), 6.84-6.91 (m, 1Н), 4.66 (brs, 2Н), 4.30 (brs, 1H), 3.01-3.10 (m, 2Н), 2.05 (brs, 1Н), 1.55-1.98 (m, 10Н). ЖХ-МС (М+Н)+ = 463,2; чистота по ВЭЖХ = 99,58%.
ПРИМЕР 135: 3-(5-хлор-1Я-пирроло[2,3-6]пиридин-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (135):
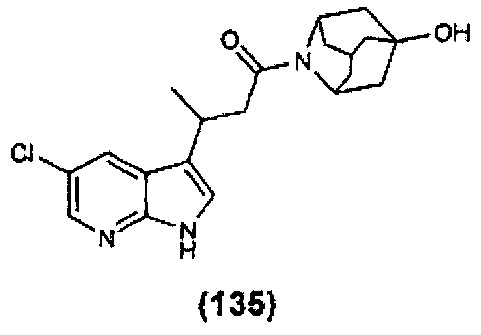
Синтез Соединения (135): Соединение (135) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (135). 1Н ЯМР (300 МГц, CDCl3): δ 9.15 (brs, 0.5Н), 9.01 (brs, 1H), 8.15-8.16 (d, 1Н), 7.91 (s, 1H), 7.09-7.10 (d, 1H), 4.97 (brs, 1H), 4.10 (brs, 1H), 3.51-3.58 (q, 1H), 2.61-2.68 (dd, 1H), 2.42-2.50 (dd, 1H), 2.24 (brs, 0.5H), 2.14 (brs, 0.5H), 1.44 (1.71 (m, 10H), .35-1.38 (d, 3Н). ЖХ-МС (M+H)+ = 374,2; чистота по ВЭЖХ = 98,36%. ПРИМЕР 136: 2-[3-(4-циклопропил-1H-пирроло[2,3-b]пиридин-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбонитрил (136):
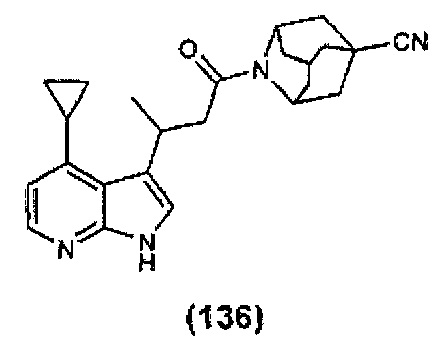
Синтез Соединения (136): Соединение (136) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (136). 1Н ЯМР (300 МГц, ДМСО-d6): δ 11.28 (brs, 1H), 7.99-8.00 (d, 1Н), 7.25 (s, 1H), 6.52-6.53 (d, 1Н), 4.72 (brs, 1H), 4.29 (brs, 1Н), 3.85-3.89 (m, 1H), 2.55-2.72 (m, 3Н), 2.09 (brs, 1H), 1.45-1.99 (m, 10Н), 1.28-1.30 (d, 3Н), 1.00-1.05 (m, 2Н), 0.83-0.88 (m, 2Н). ЖХ-МС (М+Н)+ = 389,3; чистота по ВЭЖХ = 98,69%.
ПРИМЕР 137: 2-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбонитрил (137):
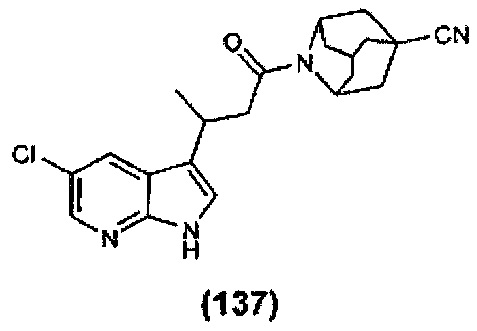
Синтез Соединения (137): Соединение (137) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (137). 1H ЯМР (300 МГц, CDCl3): δ 8.98 (brs, 0.5Н), 8.88 (brs, 0.5Н), 8.15-8.20 (m, 1H), 7.90 (s, 1H), 7.10 (s, 1H), 4.90 (brs, 1H), 4.00 (brs, 1H), 3.50-3.60 (m, 1H), 2.58-2.62 (dd, 1H), 2.40-2.45 (m, 1H), 2.13-2.16 (m, 1H), 1.60-1.93 (m, 10H), 1.38-1.40 (d, 3Н). ЖХ-МС (M+H)+ = 383,2; чистота по ВЭЖХ = 97,99%.
ПРИМЕР 138: 3-(6-хлор-5-циклопропил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (138):
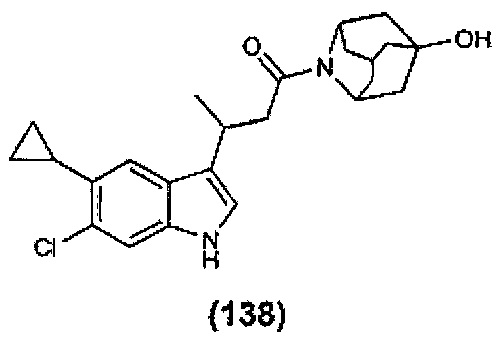
Синтез Соединения (138): Соединение (138) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (138). 1Н ЯМР (300 МГц, CDCl3): δ 7.98 (brs, 0.5Н), 7.95 (brs, 0.5Н), 7.29 (s, 1H), 7.23 (s, 1H), 6.88 (s, 1H), 4.96 (brs, 1H), 4.09 (brs, 1H), 3.46-3.52 (q, 1H), 2.64-2.72 (dd, 1H), 2.38-2.45 (dd, 1H), 2.21-2.24 (m, 1H), 2.08-2.14 (m, 2H), 1.40-1.70 (m, 9H), 1.34-1.36 (d, 3H), 0.83-0.95 (m, 2H), 0.58-0.63 (m, 2H). ЖХ-МС (M+H)+ = 413,3; чистота по ВЭЖХ = 95,14%.
ПРИМЕР 139: 3-(5-фтор-4-метил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он, Соединение (139):
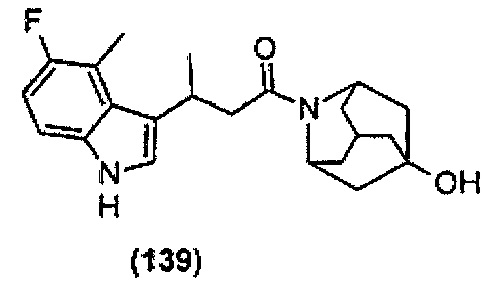
Синтез Соединения (139): Соединение (139) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (139). 1Н ЯМР (300 МГц, CDCl3): δ 7.89 (br s, 1H), 7.00-7.05 (dd, 1H), 6.98-6.99 (d, 1Н), 6.81-6.87 (t, 1Н), 5.04 (br s, 1H), 4.20 (br s, 1H), 3.84-3.93 (m, 1H), 2.65-2.71 (dd, 1H), 2.54-2.55 (d, 3H), 2.37-2.48 (dd, 1H), 2.26 (br s, 1H), 1.54-1.66 (m, 8H), 1.75 (br s, 2H), 1.39-1.43 (d, 3Н). ЖХ-МС (M+H)+ = 371,2; чистота по ВЭЖХ = 96,42%.
ПРИМЕР 140: 2-[3-(6-хлор-1H-индол-3-ил)-3-фенилпропаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (пик 1) (140):
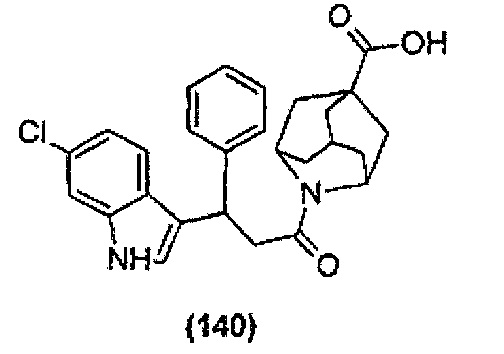
Синтез Соединения (140): Рацемат соединения (134) разделили с помощью хиральной препаративной ВЭЖХ с получением Соединения (140). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.34 (brs, 1Н), 11.03 (s, 1H), 7.10-7.40 (m, 8Н), 6.84-6.91 (m, 1Н), 4.66 (brs, 2Н), 4.30 (brs, 1Н), 3.01-3.10 (m, 2Н), 2.05 (brs, 1Н), 1.55-1.98 (m, 10Н). ЖХ-МС (М+Н)+ = 463,1; чистота по ВЭЖХ = 99,56%; хиральное время удерживания = 7,95 мин. [колонка: Chiralpak IC, подвижная фаза: гексан: IPA: ДХМ (7:2:1)].
ПРИМЕР 141: 2-[3-(6-хлор-1H-индол-3-ил)-3-фенилпропаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (пик 2) (141):
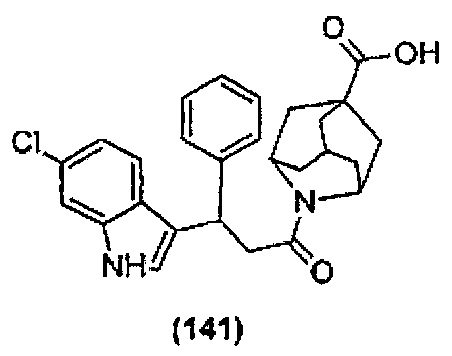
Синтез Соединения (141): Рацемат соединения (134) разделили с помощью хиральной препаративной ВЭЖХ с получением Соединения (141). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.34 (brs, 1Н), 11.03 (s, 1H), 7.10-7.40 (m, 8Н), 6.84-6.91 (m, 1Н), 4.66 (brs, 2Н), 4.30 (brs, 1Н), 3.01-3.10 (m, 2Н), 2.05 (brs, 1Н), 1.55-1.98 (m, 10Н). ЖХ-МС (М+Н)+ = 463,1; чистота по ВЭЖХ = 99,15%; хиральное время удерживания = 11,99 мин. [колонка: Chiralpak IC, подвижная фаза: гексан: IPA: ДХМ (7:2:1)].
ПРИМЕР 142: 2-[3-(5-фенил-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (142):
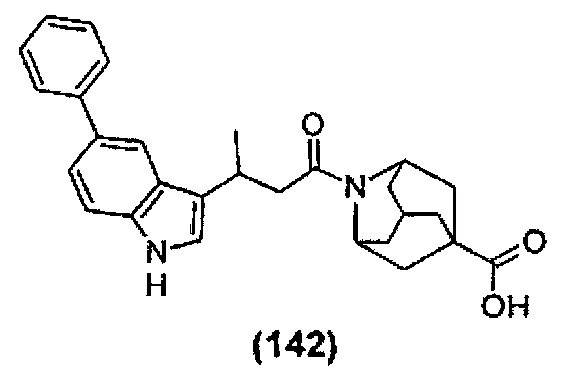
Синтез Соединения (142): Соединение (142) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (142). 1Н ЯМР (300 МГц, CDCl3): δ 7.96 (br s, 1H), 7.80 (br s, 1Н), 7.56-7.59 (d, 2Н), 7.32-7.39 (m, 4Н), 7.24-7.29 (dd, 1H), 6.97-6.99 (m, 1Н), 4.89 (br s, 1Н), 4.04 (br s, 1H), 3.61-3.63 (m, 1H), 2.75-2.81 (dd, 1H), 2.48-2.52 (dd, 1H), 1.54-2.09 (m, 11H), 1.41-1.44 (dd, 3Н). ЖХ-МС (M+H)+ = 443,2; чистота по ВЭЖХ = 98,51%.
ПРИМЕР 143: 2-[3-(5-хлор-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоновая кислота (143):
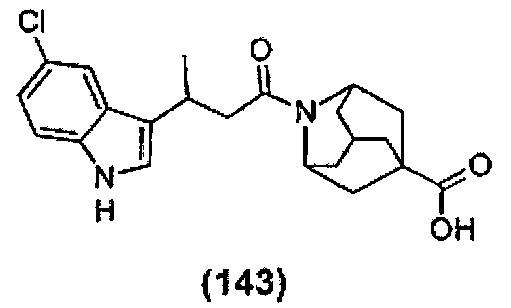
Синтез Соединения (143): Соединение (143) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (143). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.20 (br s, 1H), 11.00 (br s, 1H), 7.54 (br s, 1Н), 7.30-7.34 (dd, 1Н), 7.23 (br s, 1Н), 7.01-7.05 (m, 1H), 4.71 (br s, 1H), 4.18 (br s, 1H), 3.67-3.78 (m, 1H), 2.62-2.68 (m, 1H), 2.43-2.47 (m, 1H), 1.39-1.97 (m, 11H), 1.29-1.31 (d, 3Н). ЖХ-МС (M+H)+ = 401,2; чистота по ВЭЖХ = 97,21%.
ПРИМЕР 144: 3-(5-хлор-4-метил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (144):
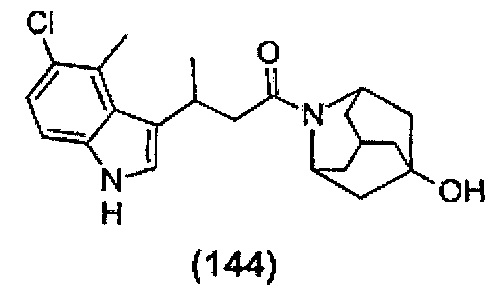
Синтез Соединения (144): Соединение (144) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (144). 1Н ЯМР (300 МГц, CDCl3): δ 7.95 (br s, 1Н), 7.09-7.12 (d, 1H), 7.03-7.06 (d, 1H), 6.93-6.98 (d, 1H), 5.03 (br s, 1H), 4.19 (br s, 1H), 3.88-3.90 (m, 1H), 2.68 (s, 3H), 2.62-2.65 (m, 1H), 2.37-2.45 (m, 1H), 2.27 (br s, 1H), 1.54-1.76 (m, 10H), 1.32-1.34 (d, 3Н). ЖХ-МС (M+H)+ = 387,2; чистота по ВЭЖХ = 97,47%.
ПРИМЕР 145: {2-[3-(4-хлор-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]дец-5-ил}уксусная кислота (145):
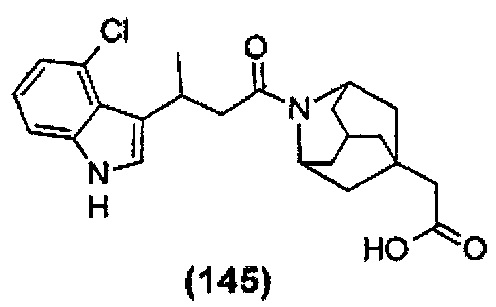
Синтез Соединения (145): Соединение (145) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (145). 1H ЯМР (300 МГц, CDCl3): δ 8.39 (br s, 1Н), 7.16-7.20 (m, 1H), 6.92-6.97 (m, 3Н), 4.88 (br s, 1H), 4.20 Br s, 1H), 3.98-4.13 (m, 1H), 2.78-2.94 (m, 1H), 2.52-2.59 (m, 1H), 2.08 (br s, 2H), 2.04 (br s, 1H), 1.52-1.68 (m, 10H), 1.37-1.39 (d, 3Н). ЖХ-МС (M+H)+ = 415,2; чистота по ВЭЖХ = 97,51%.
ПРИМЕР 146: 3-{2-[3-(4-циклопропил-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]дец-5-ил}пропановая кислота (146):
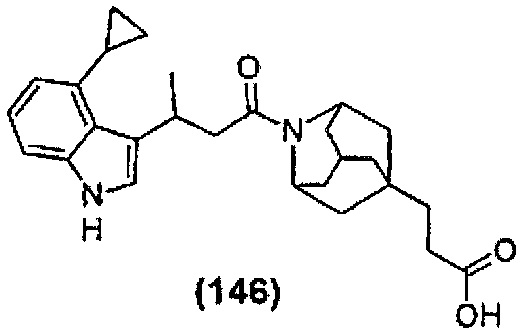
Синтез Соединения (146): Соединение (146) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (146). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.01 (br s, 1H), 10.81 (br s, 1Н), 7.17 (br s, 1H), 7.12-7.15 (d, 1H), 6.88-6.93 (t, 1H), 6.56-6.58 (d, 1H), 4.71 (br s, 1H), 4.20 (br s, 1H), 4.02-4.04 (m, 1H), 2.26-2.28 (m, 1H), 2.06-2.16 (m, 5H), 1.32-1.60 (m, 12H), 1.25-1.29 (d, 3Н). ЖХ-МС (M+H)+ = 435,2; чистота по ВЭЖХ = 98,38%.
ПРИМЕР 147: 3-(5-фтор-4-метил-1Н-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 1) (147):
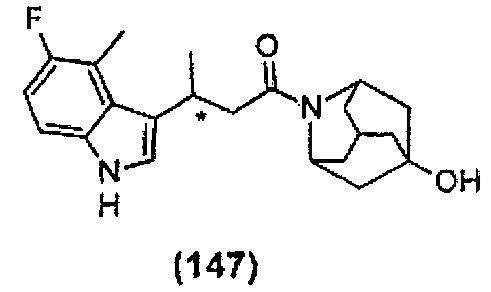
Синтез Соединения (147): Рацемат соединения (139) разделили с помощью хиральной препаративной колоночной хроматографии с получением Соединения (147). 1H ЯМР (300 МГц, CDCl3): δ 7.89 (br s, 1H), 7.00-7.05 (dd, 1Н), 6.98-6.99 (d, 1H), 6.81-6.87 (t, 1Н), 5.04 (br s, 1Н), 4.20 (br s, 1Н), 3.84-3.93 (m, 1H), 2.65-2.71 (dd, 1H), 2.54-2.55 (d, 3H), 2.37-2.48 (dd, 1H), 2.26 (br s, 1H), 1.54-1.66 (m, 8H), 1.75 (br s, 2H), 1.39-1.43 (d, 3H). ЖХ-МС (M+H)+ = 371,2; чистота по ВЭЖХ = 96,98%; хиральное время удерживания = 10,82 мин. [колонка: Chiralpak IC, подвижная фаза: гексан: ТГФ (7:3)].
ПРИМЕР 148: 3-(5-фтор-4-метил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 2) (148):
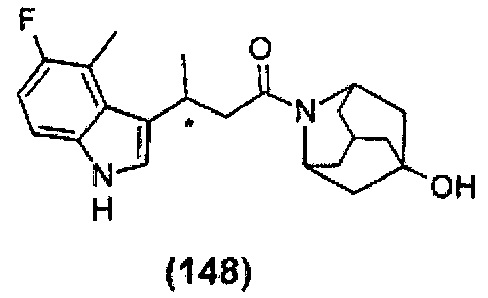
Синтез Соединения (148): Рацемат соединения (139) разделили с помощью хиральной препаративной колоночной хроматографии с получением Соединения (148). 1Н ЯМР (300 МГц, CDCl3): δ 7.89 (br s, 1H), 7.00-7.05 (dd, 1Н), 6.98-6.99 (d, 1H), 6.81-6.87 (t, 1H), 5.04 (br s, 1H), 4.20 (br s, 1H), 3.84-3.93 (m, 1H), 2.65-2.71 (dd, 1H), 2.54-2.55 (d, 3H), 2.37-2.48 (dd, 1H), 2.26 (br s, 1H), 1.54-1.66 (m, 8H), 1.75 (br s, 2H), 1.39-1.43 (d, 3H). ЖХ-МС (M+H)+ = 371,2; чистота по ВЭЖХ = 96,16%; хиральное время удерживания = 9,69 мин. [колонка: Chiralpak IC, подвижная фаза: гексан: ТГФ (7:3)].
ПРИМЕР 149: 3-(4-циклопропил-1H-индол-3-ил)-1-[5-(1H-тетразол-5-ил)-2-азатрицикло [3.3.1.13,7]дец-2-ил]бутан-1-он (149):
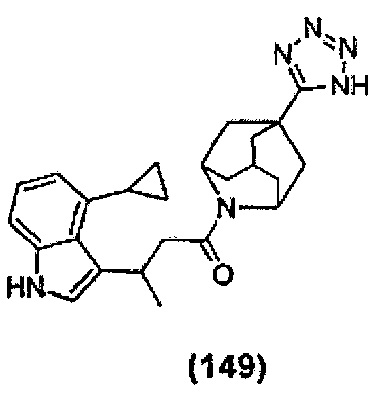
Синтез Соединения (149): Соединение (149) синтезировали по способу, использованному для получения Соединения (77) (Схема 21). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (149). 1H ЯМР (300 МГц, CDCl3): δ 8.38 (br s, 0.5Н), 8.25 (br s, 0.5Н), 7.02-7.07 (t, 1H), 6.98-7.00 (d, 1H), 6.89-6.94 (m, 1H), 6.56-6.66 (dd, 1H), 4.88-4.92 (d, 1H), 4.20 (br s, 1H), 3.47-3.50 (m, 1H), 2.78-2.85 (m, 1H), 2.46-2.59 (m, 1H), 2.29-2.34 (m, 1H), 2.17-2.19 (m, 1H), 1.61-2.01 (m, 10H), 1.32-1.38 (m, 3H), 0.72-0.79 (M, 2H), 0.63-0.65 (m, 2H). ЖХ-МС (M+H)+ = 431,2; чистота по ВЭЖХ = 92,97%.
ПРИМЕР 150: 3-(4-циклопропил-5-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (150):
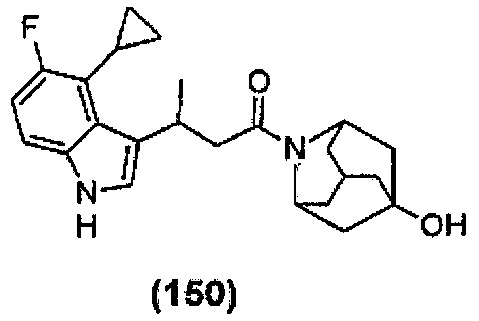
Синтез Соединения (150): Соединение (150) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (150). 1H ЯМР (300 МГц, CDCl3): δ 7.93 (br s, 1Н), 7.04-7.06 (d, 1H), 7.01-7.02 (d, 1H), 6.74-6.84 (dd, 1H), 5.02 (br s, 1H), 4.21-4.24 (m, 1H), 4.20 (br s, 1H), 2.68-2.75 (dd, 1H), 2.34-2.43 (dd, 1H), 2.22-2.25 (m, 1H), 2.00-2.09 (m, 1H), 1.52-1.75 (m, 10H), 1.31-1.34 (d, 3H), 0.99-1.03 (m, 2H), 0.83-0.87 (m, 2H). ЖХ-МС (M+H)+ = 397,2; чистота по ВЭЖХ = 97,0%.
ПРИМЕР 151: 3-(5-фтор-4-циклопропил-1Н-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 1) (151):
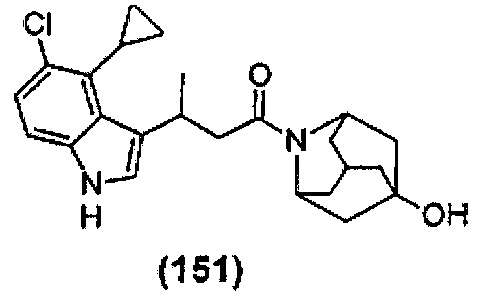
Синтез Соединения (151): Соединение (151) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (151). 1H ЯМР (300 МГц, ДМСО-d6): δ 11.04 (br s, 1Н), 7.29 (br s, 1H), 7.17-7.20 (d, 1Н), 6.98-7.00 (d, 1Н), 4.79 (br s, 1H), 4.64-4.65 (d, 1H, группа ОН), 4.31 (br s, 1H), 4.22-4.23 (m, 1H), 2.60-2.72 (m, 2H), 2.07-2.27 (m, 2H), 1.38-1.68 (m, 10H), 1.24-1.27 (d, 3H), 0.80-0.85 (m, 2H), 0.70-0.75 (m, 2H). ЖХ-МС (M+H)+ = 413,1; чистота по ВЭЖХ = 95,99%.
ПРИМЕР 152: {2-[3-(4-циклопропил-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]дец-5-ил}уксусная кислота (152):
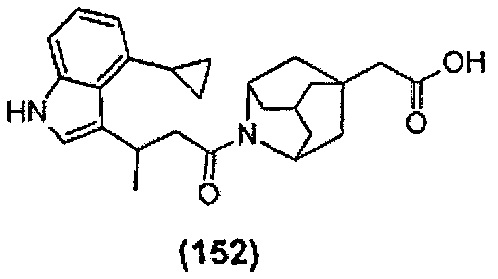
Синтез Соединения (152): Соединение (152) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (152). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.82 (br s, 1H), 7.17 (br s, 1H), 7.12-7.15 (d, 1H), 6.88-6.93 (t, 1H), 6.56-6.58 (d, 1H), 4.72 (br s, 1H), 4.22 (br s, 1H), 4.00-4.06 (m, 1H), 2.68-2.73 (m, 2H), 2.43-2.46 (m, 3H), 1.99-2.05 (m, 1H), 1.45-1.68 (m, 10H), 1.28-1.30 (d, 3H), 0.85-0.90 (m, 2H), 0.70-0.75 (m, 2H). ЖХ-МС (M+H)+ = 421,2; чистота по ВЭЖХ = 98,6%.
ПРИМЕР 153: 1-(5-гидрокси-2-азатрицикло [3.3.1.13,7]дец-2-ил)-3-[4-(тиофен-2-ил)-1H-индол-3-ил]бутан-1-он (153):
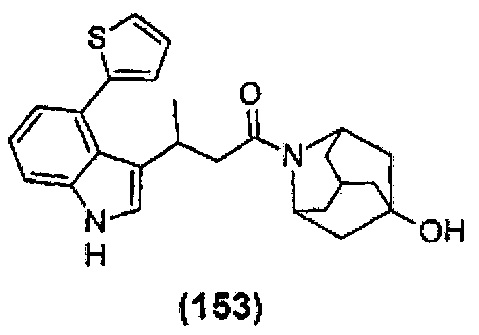
Синтез Соединения (153): Соединение (153) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (153). 1Н ЯМР (300 МГц, CDCl3): δ 8.16 (br s, 1H), 7.31-7.34 (d, 1Н), 7.23-7.26 (m, 1Н), 7.10-7.15 (t, 1H), 7.02-7.08 (m, 4Н), 4.89 (br s, 1Н), 3.80 (br s, 1H), 3.38-3.40 (m, 1H), 2.38-2.43 (m, 1H), 2.19-2.22 (m, 1H), 1.97-2.01 (m, 1H), 1.45-1.71 (m, 10H), 1.37-1.39 (d, 3Н). ЖХ-МС (M+H)+ = 421,1; чистота по ВЭЖХ = 96,43%.
ПРИМЕР 154: (2-{3-[4-(тиофен-2-ил)-1H-индол-3-ил]бутаноил}-2-азатрицикло[3.3.1.13,7]дец-5-ил)уксусная кислота (154):
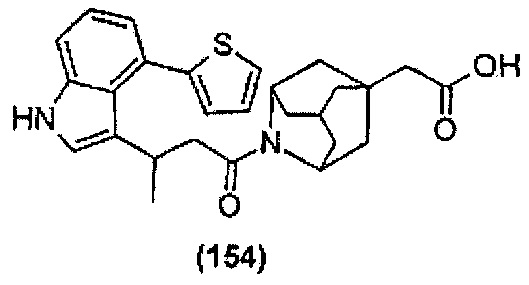
Синтез Соединения (154): Соединение (154) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (154). 1Н ЯМР (300 МГц, CDCl3): δ 8.68 (br s, 0.5Н), 8.57 (br s, 0.5H), 7.22-7.30 (m, 2H), 7.01-7.09 (m, 5H), 4.77 (br s, 1H), 3.46 (br s, 1H), 3.31-3.35 (m, 1H), 2.35-2.48 (m, 2H), 2.07 (br s, 1H), 1.40-1.78 (m, 13H). ЖХ-МС (M+H)+ = 463,1; чистота по ВЭЖХ = 92,86%.
ПРИМЕР 155: {2-[3-(4-циклопропил-1Н-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]дец-5-ил)уксусная кислота (пик 1) (147):
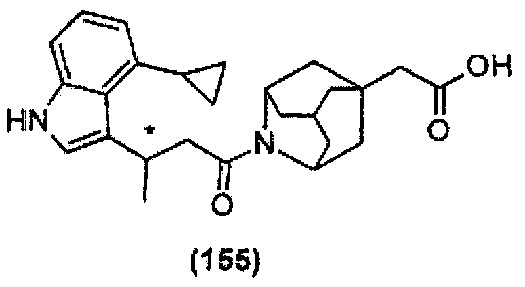
Синтез Соединения (155): Рацемическое соединение (152) разделили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (155). 1Н ЯМР (300 МГц, CDCl3): δ 7.98 (br s, 1H), 7.09-7.12 (d, 1H), 6.99-7.01 (d, 1H), 6.97 (br s, 1H), 6.66-6.78 (d, 1H), 4.89 (br s, 1H), 4.14-4.16 (m, 1H), 4.06 (br s, 1H), 2.75-2.82 (dd, 1H), 2.37-2.45 (m, 2H), 2.08 (s, 2H), 2.04 (br s, 1H), 1.52-1.71 (m, 10H), 1.36-1.38 (d, 3H), 0.90-0.95 (m, 2H), 0.76-0.81 (m, 2H). ЖХ-МС (M+H)+ = 421,2; чистота по ВЭЖХ = 98,67%; хиральное время удерживания = 9,07 мин. [колонка: Chiralpak IC, подвижная фаза: гексан: IPА: ДХМ (7,5:1,5:1)].
ПРИМЕР 156: {2-[3-(4-циклопропил-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]дец-5-ил}уксусная кислота (пик 2) (156):
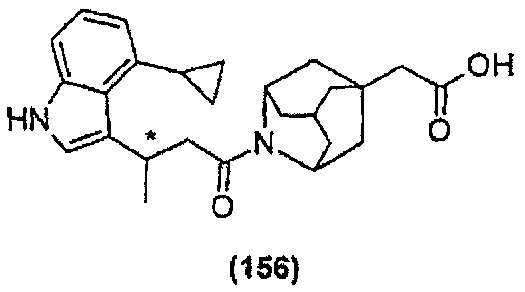
Синтез Соединения (156): Рацемическое соединение (152) разделили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (156). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10.82 (br s, 1Н), 7.17 (br s, 1H), 7.12-7.15 (d, 1H), 6.88-6.93 (t, 1H), 6.56-6.58 (d, 1H), 4.72 (br s, 1H), 4.22 (br s, 1H), 4.00-4.06 (m, 1H), 2.68-2.73 (m, 2H), 2.43-2.46 (m, 3H), 1.99-2.05 (m, 1H), 1.45-1.68 (m, 10H), 1.28-1.30 (d, 3H), 0.85-0.90 (m, 2H), 0.70-0.75 (m, 2H). ЖХ-МС (M+H)+ = 421,2; чистота по ВЭЖХ = 99,66%; хиральное время удерживания = 12,93 мин. [колонка: Chiralpak IC, подвижная фаза: гексан: IPA: ДХМ (7,5:1,5:1)].
ПРИМЕР 157: 3-(4-циклопропил-5-фтор-1Н-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 1) (157):
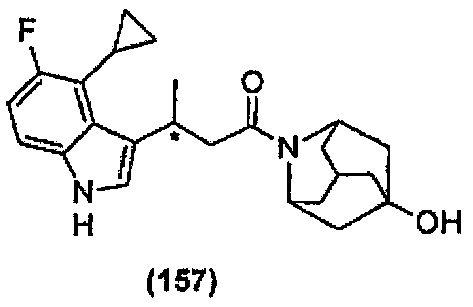
Синтез Соединения (157) (пик 1): Рацемическое соединение (150) разделили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (157). ЖХ-МС (М+Н)+ = 397,2; хиральное время удерживания = 10,98 мин. [колонка: Chiralpak IC, подвижная фаза: гексан: IPA: ДХМ (7,5:1,5:1)].
ПРИМЕР 158: 3-(4-циклопропил-5-фтор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 2) (158):
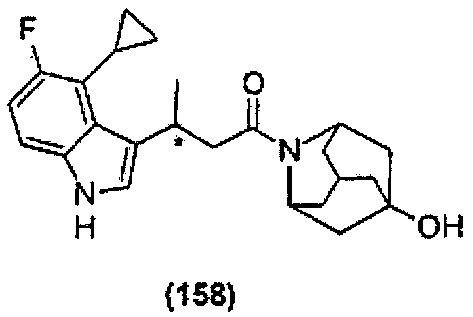
Синтез Соединения (158) (пик 2): Рацемическое соединение (150) разделили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (157). ЖХ-МС (М+Н)+ = 397,2; чистота по ВЭЖХ = 98,48%; хиральное время удерживания = 14,93 мин. [колонка: Chiralpak IC, подвижная фаза: гексан: IPA: ДХМ (7,5:1,5:1)].
ПРИМЕР 159: {2-[3-(4-фтор-1H-индол-3-ил)-3-(тиофен-2-ил)пропаноил]-2-азатрицикло[3.3.1.13,7]дец-5-ил}уксусная кислота (159):
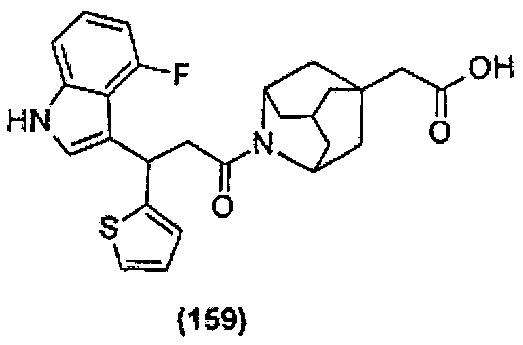
Синтез Соединения (159): Соединение (159) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (159). 1Н ЯМР (300 МГц, ДМСО-d6): δ 11.93 (br s, 1H), 11.21 (br s, 1H), 7.35 (s, 1H), 7.21-7.23 (m, 1H), 7.15-7.18 (d, 1H), 6.97-7.04 (m, 1H), 6.85-6.88 (m, 1H), 6.81-6.83 (m, 1H), 6.63-6.70 (dd, 1H), 5.09-5.11 (t, 1H), 4.65 (br s, 1H), 4.27 (br s, 1H), 3.10-3.17 (m, 1H), 2.95-3.05 (M, 1H), 2.01 (s, 2H), 1.97 (s, 2H), 1.40-1.66 (m, 10H). ЖХ-МС (M+H)+ = 467,1; чистота по ВЭЖХ = 99,63%.
ПРИМЕР 160: 3-(4-циклопропил-5-метил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (160):
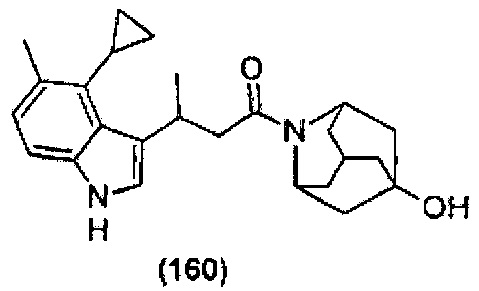
Синтез Соединения (160): Соединение (160) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (160). 1Н ЯМР (300 МГц, CDCl3): δ 7.84 (br s, 1H), 7.04-7.07 (d, 1H), 6.97 (br s, 1H), 6.88-6.91 (d, 1H), 5.00 (br s, 1H), 4.34-4.47 (m, 1H), 4.20 (br s, 1H), 2.67-2.73 (dd, 1H), 2.443 (s, 3H), 2.29-2.37 (dd, 1H), 2.15-2.22 (m, 1H), 2.00-2.05 (m, 1H), 1.45-1.78 (m, 10H), 1.33-1.36 (m, 3H), 1.04-1.10 (m, 2H), 0.64-0.69 (m, 2H). ЖХ-МС (M+H)+ = 393,3; чистота по ВЭЖХ = 98,30%.
ПРИМЕР 161: 3-(4-хлор-5-циклопропил-1Н-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (161):
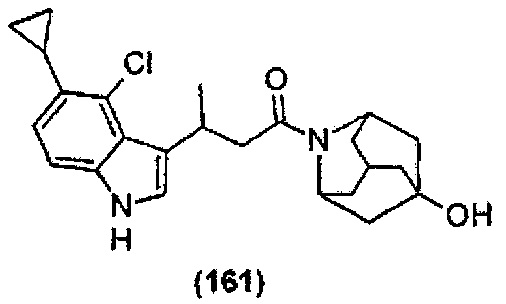
Синтез Соединения (161): Соединение (161) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (161). 1H ЯМР (300 МГц, CDCl3): δ 8.03 (br s, 1Н), 7.08-7.10 (d, 1H), 7.01-7.01 (d, 1H), 6.74-6.77 (d, 1H), 5.02 (br s, 1H), 4.26 (br s, 1H), 4.10-4.12 (m, 1H), 2.37-2.46 (m, 1H), 2.26-2.29 (m, 1H), 2.15-2.24 (m, 1H), 1.45-1.75 (m, 11H), 1.36-1.39 (d, 3H), 0.89-0.96 (m, 2H), 0.59-0.62 (m, 2H). ЖХ-МС (M+H)+ = 413,1; чистота по ВЭЖХ = 98,97%.
ПРИМЕР 162: {2-[3-(4-бром-5-хлор-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]дец-5-ил}уксусная кислота (162):
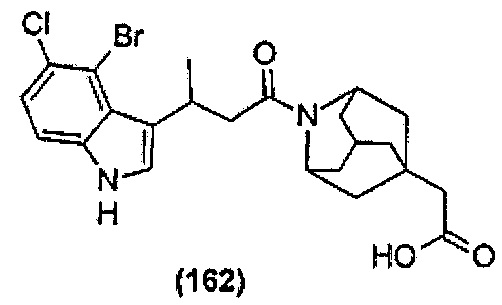
Синтез Соединения (162): Соединение (162) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (162). 1Н ЯМР (300 МГц, CDCl3): δ 8.70 (br s, 0.5Н), 8.62 (br s, 0.5H), 7.12-7.15 (d, 1H), 7.07-7.09 (d, 1H), 7.01 (s, 1H), 4.87 (br s, 1H), 4.15-4.21 (m, 1H), 4.01 (br s, 1H), 2.72-2.85 (m, 1H), 2.45-2.60 (m, 1H), 2.29- (s, 2H), 2.04 (s, 1H), 1.50-1.75 (m, 10Н). ЖХ-МС (M+H)+ = 493,2; чистота по ВЭЖХ = 91,99%.
ПРИМЕР 163: 3-(5-хлор-4циклопропил-1Н-индол-3-ил)-1-(5,7-дигидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (163):
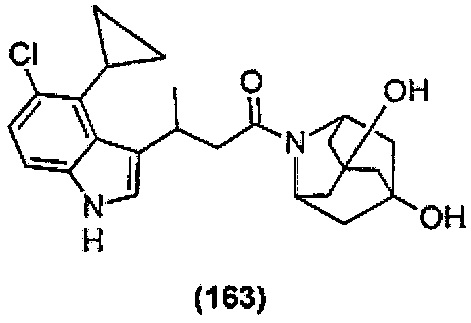
Синтез Соединения (163): Соединение (163) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (163). 1Н ЯМР (300 МГц, CDCl3): δ 8.01 (br s, 1H), 7.06 (s, 2Н), 7.01-7.02 (d, 1H), 4.27-4.42 (m, 1H), 3.50 (br s, 1H), 3.43 (br s, 1H), 2.62-2.77 (dd, 1H), 2.37-2.46 (dd, 1H), 2.11-2.13 (m, 1H), 1.83-1.98 (m, 4H), 1.62-1.68 (m, 4H), 1.55-1.58 (m, 2H), 1.33-1.35 (d, 3H), 1.12-1.16 (m, 2H), 0.76-0.84 (m, 2H). ЖХ-МС (M+H)+ = 429,1; чистота по ВЭЖХ = 99,90%.
ПРИМЕР 164: 3-(4,5-диметил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (164):
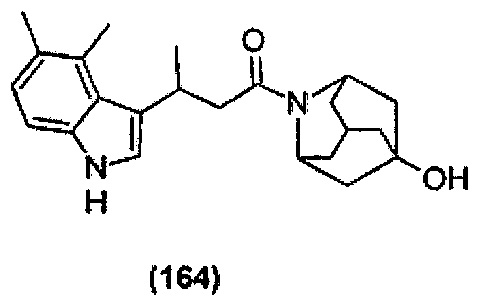
Синтез Соединения (164): Соединение (164) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (164). 1Н ЯМР (300 МГц, CDCl3): δ 7.92 (br s, 1H), 7.01-7.03 (d, 1H), 6.91-6.93 (d, 1H), 6.90 (s, 1H), 5.03 (br s, 1H), 4.18 (br s, 1H), 3.92-3.95 (m, 1H), 2.68-2.74 (dd, 1H), 2.54 (s, 3H), 2.35-2.43 (dd, 1H), 2.29 (s, 3H), 2.20-2.25 (m, 1H), 1.52-1.75 (m, 11H), 1.33-1.35 (d, 3Н). ЖХ-МС (M+H)+ = 367,2; чистота по ВЭЖХ = 97,04%.
ПРИМЕР 165: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(5-метокси-4-метил-1Н-индол-3-ил)бутан-1-он (165):
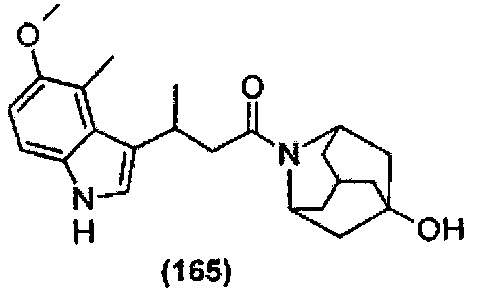
Синтез Соединения (165): Соединение (165) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (165). 1H ЯМР (300 МГц, CDCl3): δ 7.77 (br s, 1Н), 7.05-7.08 (d, 1H), 6.95-6.96 (d, 1H), 6.82-6.85 (d, 1H), 5.04 (br s, 1H), 4.19 (br s, 1H), 3.87-3.89 (m, 1H), 3.77 (s, 3H0, 2.67-2.73 (dd, 1H), 2.54 (s, 3H), 2.36-2.44 (dd, 1H), 2.24-2.26 (m, 1H), 1.55-1.75 (m, 10H), 1.33-1.35 (d, 3Н). ЖХ-МС (M+H)+ = 383,2; чистота по ВЭЖХ = 95,78%.
ПРИМЕР 166: 3-(4-хлор-5-метил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (166):
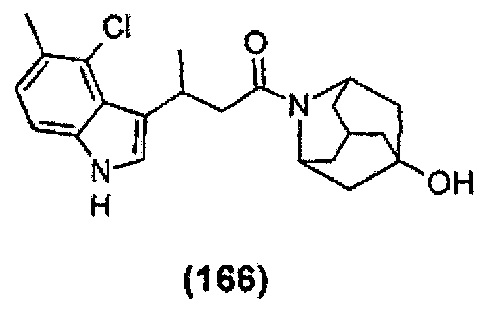
Синтез Соединения (166): Соединение (166) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (166). 1H ЯМР (300 МГц, CDCl3): δ 8.00 (br s, 1H), 7.08-7.10 (d, 1H), 6.98-7.00 (d, 1Н), 6.95 (s, 1H), 5.02 (br s, 1H), 4.25 (br s, 1H), 4.09-4.11 (m, 1H), 2.82-2.87 (m, 1H), 2.40-2.44 (m, 1H), 2.38 (s, 3H), 2.23-2.25 (m, 1H), 1.55-1.78 (m, 11H). ЖХ-МС (M+H)+ = 388,2; чистота по ВЭЖХ = 99,97%.
ПРИМЕР 167:2-{2-[3-(4-циклопропил-1Н-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]дец-5-ил}пропановая кислота (167):
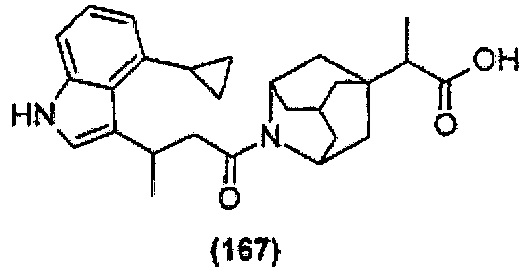
Синтез Соединения (167): Соединение (167) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (167). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12.01 (br s, 1H), 10.81 (s, 1Н), 7.16-7.17 (d, 1H), 7.12-7.15 (d, 1H), 6.88-6.93 (t, 1H), 6.55-6.58 (d, 1H), 4.72 (br s, 1H), 4.24 (br s, 1H), 4.02-4.04 (m, 1H), 2.67-2.723 (m, 2H), 2.38-2.43 (m, 1H), 2.05-2.07 (m, 1H), 1.99-2.02 (m, 1H), 1.46-1.74 (m, 10H), 1.28-1.30 (d, 3H), 0.95-0.98 (d, 3H), 0.83-0.88 (m, 2H), 0.70-0.74 (m, 2H). ЖХ-МС (M+H)+ = 435,2; чистота по ВЭЖХ = 96,54%.
ПРИМЕР 168: 3-(4,5-дихлор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (168):
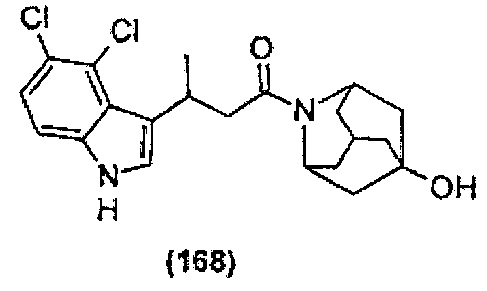
Синтез Соединения (168): Соединение (168) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (168). 1H ЯМР (300 МГц, CDCl3): δ 8.18 (br s, 1H), 7.10-7.13 (d, 2Н), 7.06-7.07 (d, 1H), 5.00 (br s, 1Н), 4.24 (br s, 1H), 4.04-4.07 (m, 1H), 2.78-2.84 (m, 1H), 2.37-2.47 (dd, 1H), 2.24-2.26 (m, 1H), 1.73-1.78 (m, 3H), 1.61-1.65 (m, 7H), 1.35-1.38 (d, 3Н). ЖХ-МС (M+H)+ = 407,1; чистота по ВЭЖХ = 99,28%.
ПРИМЕР 169: 3-(4,5-диметил-1Н-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 1) (169):
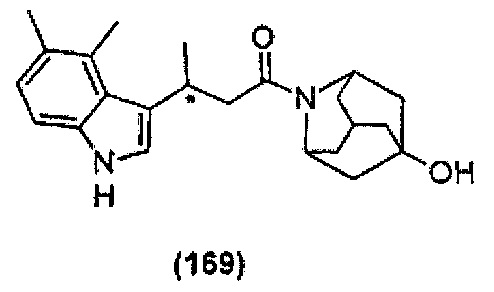
Синтез Соединения (169): Рацемическое соединение (164) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (169). 1Н ЯМР (300 МГц, CDCl3): δ 7.92 (br s, 1H), 7.01-7.03 (d, 1Н), 6.91-6.93 (d, 1H), 6.90 (s, 1H), 5.03 (br s, 1H), 4.18 (br s, 1H), 3.92-3.95 (m, 1H), 2.68-2.74 (dd, 1H), 2.54 (s, 3H), 2.35-2.43 (dd, 1H), 2.29 (s, 3H), 2.20-2.25 (m, 1H), 1.52-1.75 (m, 11H), 1.33-1.35 (d, 3Н). ЖХ-МС (M+H)+ = 367,2; чистота по ВЭЖХ = 93,27%; хиральное время удерживания = 20,93 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: IPA: ДХМ (7,5:1,5:1)].
ПРИМЕР 170: 3-(4,5-диметил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 2) (170):
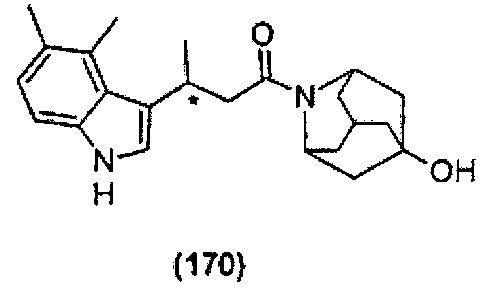
Синтез Соединения (170): Рацемическое соединение (164) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (170). 1Н ЯМР (300 МГц, CDCl3): δ 7.92 (br s, 1Н), 7.01-7.03 (d, 1H), 6.91-6.93 (d, 1H), 6.90 (s, 1H), 5.03 (br s, 1H), 4.18 (br s, 1H), 3.92-3.95 (m, 1H), 2.68-2.74 (dd, 1H), 2.54 (s, 3H), 2.35-2.43 (dd, 1H), 2.29 (s, 3H), 2.20-2.25 (m, 1H), 1.52-1.75 (m, 11H), 1.33-1.35 (d, 3Н). ЖХ-МС (M+H)+ = 367,2; чистота по ВЭЖХ = 99,81%; хиральное время удерживания = 25,61 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: IPA: ДХМ (7,5:1,5:1)].
ПРИМЕР 171: 3-(4-циклопропил-5-метокси-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (171):
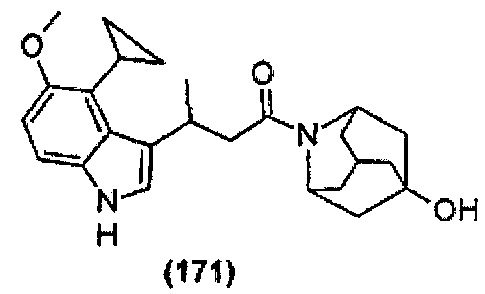
Синтез Соединения (171): Соединение (171) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (171). 1H ЯМР (300 МГц, CDCl3): δ 7.83 (br s, 1H), 7.06-7.09 (d, 1H), 6.98-6.99 (d, 1H), 6.78-6.81 (d, 1Н), 5.02 (br s, 1H), 4.26-4.29 (m, 1H), 4.19 (br s, 1H), 3.78 (s, 3H), 2.69-2.76 (dd, 1H), 2.31-2.39 (dd, 1H), 2.22-2.25 (m, 1H), 1.95-1.99 (m, 2H), 1.45-1.75 (m, 9H), 1.34-1.36 (m, 3H), 0.99-1.01 (m, 2H), 0.79-0.81 (m, 2H). ЖХ-МС (M+H)+ = 409,2; чистота по ВЭЖХ = 92,57%.
ПРИМЕР 172: 3-(4,5-дихлор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 1) (172):
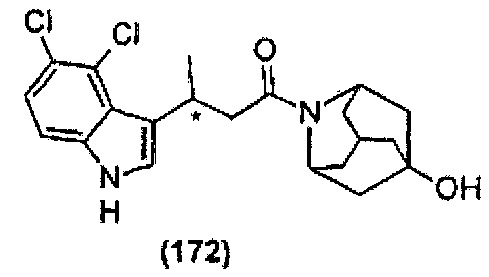
Синтез Соединения (172): Рацемическое соединение (168) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (172). 1Н ЯМР (300 МГц, CDCl3): δ 8.18 (br s, 1H), 7.10-7.13 (d, 2H), 7.06-7.07 (d, 1H), 5.00 (br s, 1H), 4.24 (br s, 1H), 4.04-4.07 (m, 1H), 2.78-2.84 (m, 1H), 2.37-2.47 (dd, 1H), 2.24-2.26 (m, 1H), 1.73-1.78 (m, 3H), 1.61-1.65 (m, 7H), 1.35-1.38 (d, 3Н). ЖХ-МС (M+H)+ = 407,0; чистота по ВЭЖХ = 94,74%; хиральное время удерживания = 8,15 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: IPA: ДХМ (7:2:1)].
ПРИМЕР 173: 3-(4,5-дихлор-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 2) (173):
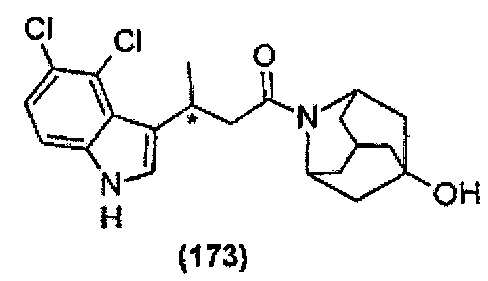
Синтез Соединения (173): Рацемическое соединение (168) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (173). 1Н ЯМР (300 МГц, CDCl3): δ 8.18 (br s, 1H), 7.10-7.13 (d, 2Н), 7.06-7.07 (d, 1Н), 5.00 (br s, 1H), 4.24 (br s, 1H), 4.04-4.07 (m, 1H), 2.78-2.84 (m, 1H), 2.37-2.47 (dd, 1H), 2.24-2.26 (m, 1H), 1.73-1.78 (m, 3H), 1.61-1.65 (m, 7H), 1.35-1.38 (d, 3Н). ЖХ-МС (M+H)+ = 407,0; чистота по ВЭЖХ = 99,34%; хиральное время удерживания = 11,31 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: IPA: ДХМ (7:2:1)].
ПРИМЕР 174: 3-(4-хлор-5-метил-1H-индол-3-ил)-1-(5-гидрокси-2-(азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 1) (174):
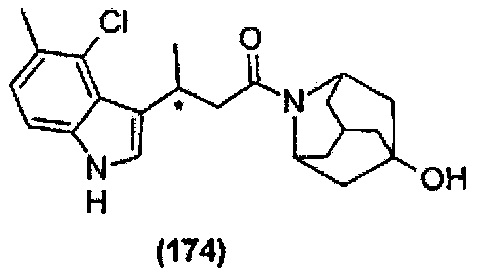
Синтез Соединения (174): Рацемическое соединение (166) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (174). 1Н ЯМР (300 МГц, CDCl3): δ 8.00 (br s, 1Н), 7.08-7.10 (d, 1H), 6.98-7.00 (d, 1H), 6.95 (s, 1H), 5.02 (br s, 1H), 4.25 (br s, 1H), 4.09-4.11 (m, 1H), 2.82-2.87 (m, 1H), 2.40-2.44 (m, 1H), 2.38 (s, 3H), 2.23-2.25 (m, 1H), 1.55-1.78 (m, 11Н). ЖХ-МС (M+H)+ = 387,1; чистота по ВЭЖХ = 95,57%; хиральное время удерживания = 15,30 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: ТГФ (7:3)].
ПРИМЕР 175: 3-(4-хлор-5-метил-1H-индол-3-ил)-1-(5-гидрокси-2-(азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 2) (175):
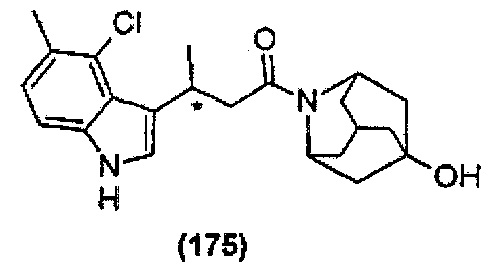
Синтез Соединения (175): Рацемическое соединение (166) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (175). 1Н ЯМР (300 МГц, CDCl3): δ 8.00 (br s, 1H), 7.08-7.10 (d, 1Н), 6.98-7.00 (d, 1H), 6.95 (s, 1H), 5.02 (br s, 1H), 4.25 (br s, 1H), 4.09-4.11 (m, 1H), 2.82-2.87 (m, 1H), 2.40-2.44 (m, 1H), 2.38 (s, 3H), 2.23-2.25 (m, 1H), 1.55-1.78 (m, 11Н). ЖХ-МС (M+H)+ = 387,1; чистота по ВЭЖХ = 98,33%; хиральное время удерживания = 17,15 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: ТГФ (7:3)].
ПРИМЕР 176: 3-(4,5-диметил-1H-индол-3-ил)-1-(5-фтор-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (176):
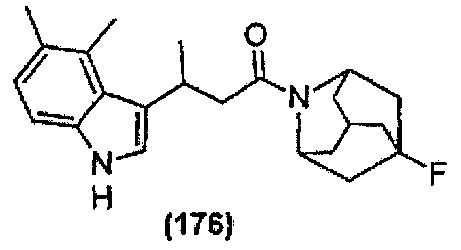
Синтез Соединения (176): Соединение (176) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (176). 1H ЯМР (300 МГц, CDCl3): δ 8.06 (br s, 1H), 8.02-8.04 (d, 1Н), 7.31-7.34 (d, 1H), 7.11-7.13 (d, 1H), 4.99-5.05 (m, 1H), 4.70-4.71 (m, 1H), 4.61-4.62 (m, 1H), 2.57-2.62 (m, 1H), 2.33-2.41 (m, 1H), 2.25 (s, 3H), 2.10 (s, 3H), 1.33-1.87 (m, 14H). ЖХ-МС (M-19)+ = 413,3; чистота по ВЭЖХ = 90,92%.
ПРИМЕР 177: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(пропан-2-илокси)-1H-индол-3-ил]бутан-1-он (177):
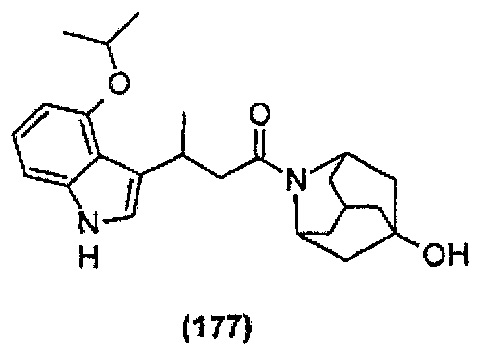
Синтез Соединения (177): Соединение (177) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (177). 1Н ЯМР (300 МГц, CDCl3): δ 7.91 (br s, 1Н), 7.03-7.08 (t, 1H), 6.91-6.93 (d, 1H), 6.90 (s, 1H), 6.46-6.49 (d, 1H), 5.06 (br s, 1H), 4.74-4.80 (m, 1H), 4.22 (br s, 1H), 3.76-3.88 (m, 1H), 2.97-3.06 (m, 1H), 2.52-2.56 (m, 1H), 2.22-2.27 (m, 1H), 1.55-1.77 (m, 10H), 1.42-1.45 (m, 9H). ЖХ-МС (M+H)+ = 397,2; чистота по ВЭЖХ = 90,56%.
ПРИМЕР 178: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(пропан-2-илокси)-1H-индол-3-ил]бутан-1-он (пик 1) (178):
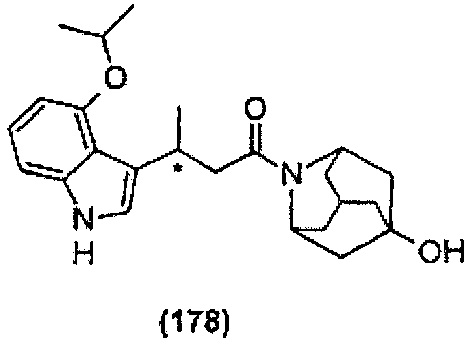
Синтез Соединения (178): Рацемическое соединение (177) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (178). ЖХ-МС (М+Н)+ = 397,2; чистота по ВЭЖХ = 99,12%. Хиральное время удерживания = 9,72 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: IPA: ДХМ (8:1:1)].
ПРИМЕР 179: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(пропан-2-илокси)-1H-индол-3-ил]бутан-1-он (пик 2) (179):
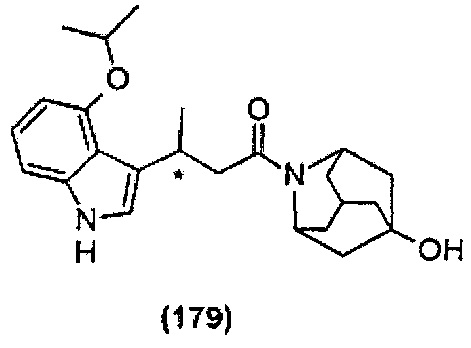
Синтез Соединения (179): Рацемическое соединение (177) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (179). ЖХ-МС (М+Н)+ = 397,2; чистота по ВЭЖХ = 99,65%. Хиральное время удерживания = 11,97 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: IPA: ДХМ (8:1:1)].
ПРИМЕР 180: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(пиперидин-1-ил)-1H-индол-3-ил]бутан-1-он (180):
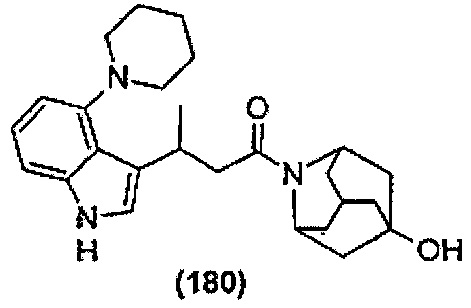
Синтез Соединения (180): Соединение (180) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (180). 1Н ЯМР (300 МГц, CDCl3): δ 7.90 (br s, 1Н), 7.00-7.04 (m, 2H), 6.90-6.94 (m, 1H), 6.73-6.76 (m, 1H), 5.03 (br s, 1H), 4.13 (br s, 1H), 3.76-3.79 (m, 1H), 3.64-3.66 (m, 1H), 3.15-3.25 (m, 2H0, 2.86-2.92 (m, 1H), 2.76-2.77 (m, 1H), 2.37-2.41 (m, 1H), 2.19-2.21 (m, 1H), 1.34-1.98 (m, 19H). ЖХ-МС (M+H)+ = 422,2; чистота по ВЭЖХ = 97,45%.
ПРИМЕР 181: 1-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(пропан-2-илокси)-1H-индол-3-ил]бутан-1-он (181):
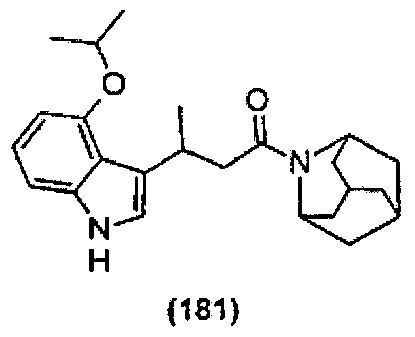
Синтез Соединения (181): Соединение (181) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (181). 1H ЯМР (300 МГц, CDCl3): δ 7.85 (br s, 1Н), 7.04-7.07 (m, 1H), 6.89-6.92 (m, 2H), 6.46-6.48 (m, 1H), 4.87 (br s, 1H), 4.75-4.79 (m, 1H), 4.04 (br s, 1H), 3.80-3.84 (m, 1H), 2.49-2.54 (m, 1H), 2.31-2.35 (m, 1H), 2.00-2.05 (m, 1H), 1.55-1.80 (m, 11H), 1.42-1.45 (m, 9H). ЖХ-МС (M+H)+ = 381,2; чистота по ВЭЖХ = 99,41%.
ПРИМЕР 182: 1-(5-фтор-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(пропан-2-илокси)-1H-индол-3-ил]бутан-1-он (182):
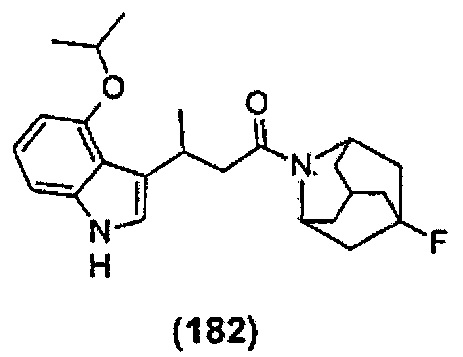
Синтез Соединения (182): Соединение (182) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (182). 1H ЯМР (300 МГц, CDCl3): δ 7.82 (br s, 1H), 6.97-7.02 (t, 1H), 6.84-6.86 (d, 1H), 6.83 (s, 1H), 6.40-6.42 (d, 1H), 5.07 (br s, 1H), 4.65-4.73 (m, 1H), 4.21 (br s, 1H), 3.71-3.78 (m, 1H), 2.90-3.00 (m, 1H), 2.39-2.50 (m, 1H), 2.21-2.24 (m, 1H), 1.53-1.90 (m, 10H), 1.35-1.39 (m, 9H). ЖХ-МС (M+H)+ = 399,2; чистота по ВЭЖХ = 93,52%.
ПРИМЕР 183: 1-{3-[4-(2-азатрицикло[3.3.1.13,7]дец-2-ил)-4-оксобутан-2-ил]-1H-индол-4-ил}пиперидин-4-карбоновая кислота (183):
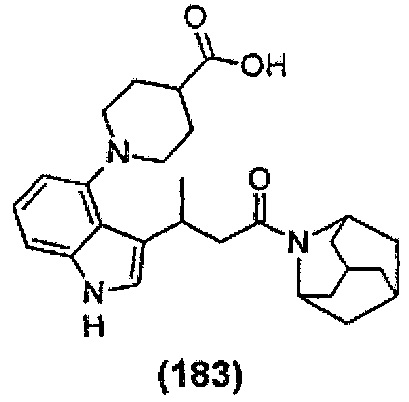
Синтез Соединения (183): Соединение (183) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (183). ЖХ-МС (М+Н)+ = 450,2; чистота по ВЭЖХ = 97,82%.
ПРИМЕР 184: 1-(5-фтор-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(пропан-2-илокси)-1H-индол-3-ил]бутан-1-он (пик 1) (184):
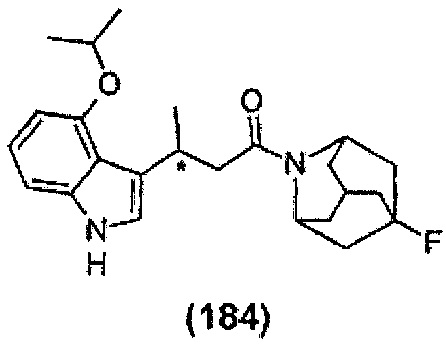
Синтез Соединения (184):
Рацемическое соединение (182) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (184). ЖХ-МС (М+Н)+ = 399,2; чистота по ВЭЖХ = 95,51%. Хиральное время удерживания = 13,60 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: IPA: ДХМ (8:1:1)].
ПРИМЕР 185: 1-(5-фтор-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(пропан-2-илокси)-1H-индол-3-ил]бутан-1-он (пик 2) (185):
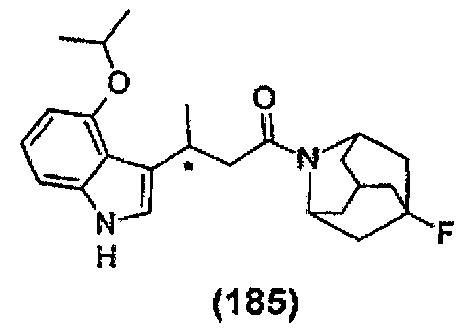
Синтез Соединения (185): Рацемическое соединение (182) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (185). ЖХ-МС (М+Н)+ = 399,2; чистота по ВЭЖХ = 97,38%. Хиральное время удерживания = 18,54 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: IPA: ДХМ (8:1:1)].
ПРИМЕР 186: 2-{2-[3-(4-хлор-1H-индол-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]дец-5-ил}пропановая кислота (186):
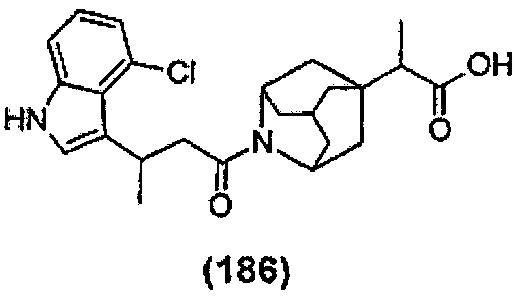
Синтез Соединения (186): Соединение (186) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (186). ЖХ-МС (М+Н)+ = 429,13; чистота по ВЭЖХ = 90,92%.
ПРИМЕР 187: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(тиофен-3-ил)-1H-индол-3-ил]бутан-1-он (187):
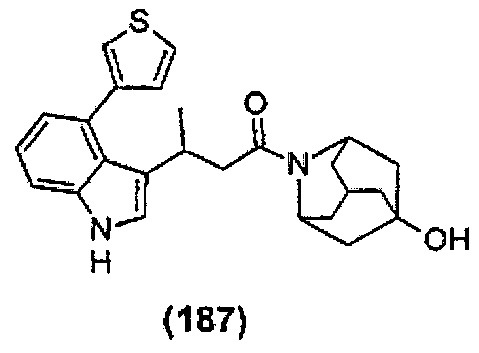
Синтез Соединения (187): Соединение (187) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (187). ЖХ-МС (М+Н)+ = 421,1; чистота по ВЭЖХ = 96,22%.
ПРИМЕР 188: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(тиофен-2-ил)-1H-индол-3-ил]бутан-1-он (пик 1) (188):
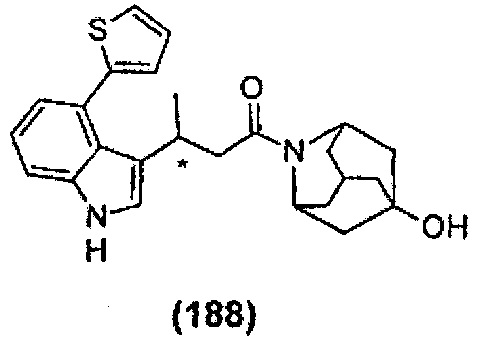
Синтез Соединения (188):
Рацемическое соединение (153) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (188). ЖХ-МС (М+Н)+ = 421,1; чистота по ВЭЖХ = 92,0%. Хиральное время удерживания = 6,57 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: ТГФ: EtOH (8:1:1)].
ПРИМЕР 189: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(тиофен-2-ил)-1H-индол-3-ил]бутан-1-он (пик 2) (189):
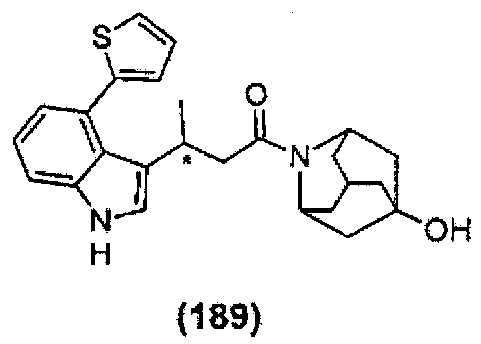
Синтез Соединения (189): Рацемическое соединение (153) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (189). ЖХ-МС (М+Н)+ = 421,1; чистота по ВЭЖХ = 93,52%. Хиральное время удерживания = 9,17 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: ТГФ: EtOH (8:1:1)].
ПРИМЕР 190: 3-(5-хлор-1,3-бензотиазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (190):
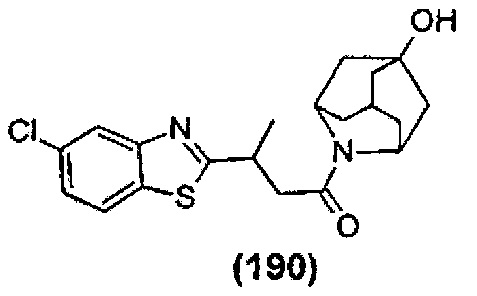
Синтез Соединения (190): Соединение (190) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (190). ЖХ-МС (М+Н)+ = 391,1; чистота по ВЭЖХ = 96,40%.
ПРИМЕР 191: 2-[3-(5-хлор-1,3-бензотиазол-2-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбонитрил (191):
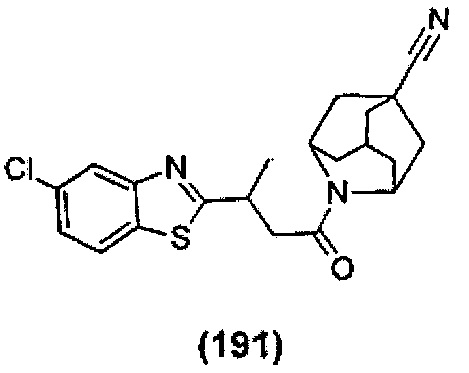
Синтез Соединения (191): Соединение (191) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (191). ЖХ-МС (М+Н)+ = 400,1; чистота по ВЭЖХ = 97,71%.
ПРИМЕР 192: 3-(6-хлор-1,3-бензотиазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (192):
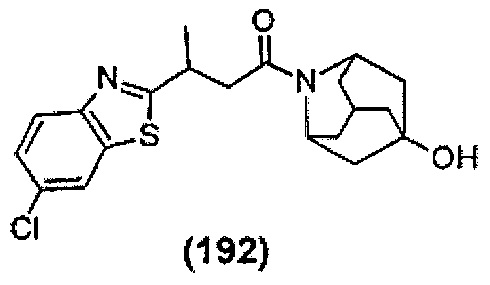
Синтез Соединения (192): Соединение (192) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (192). ЖХ-МС (М+Н)+ = 391,1; чистота по ВЭЖХ = 94,40%.
ПРИМЕР 193: 2-[3-(6-хлор-1,3-бензотиазол-2-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбонитрил (193):
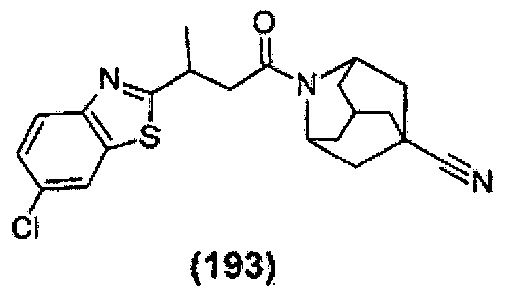
Синтез Соединения (193): Соединение (193) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (193). ЖХ-МС (М+Н)+ = 400,1; чистота по ВЭЖХ = 94,39%.
ПРИМЕР 194: 3-(6-хлор-1,3-бензотиазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 1) (194):
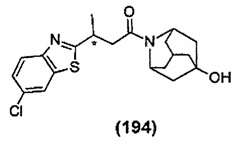
Синтез Соединения (194):
Рацемическое соединение (192) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (194). ЖХ-МС (М+Н)+ = 391,1; чистота по ВЭЖХ = 99,6%.
ПРИМЕР 195: 3-(6-хлор-1,3-бензотиазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 2) (195):
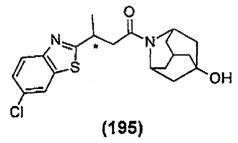
Синтез Соединения (195): Рацемическое соединение (192) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (195). ЖХ-МС (М+Н)+ = 391,1; чистота по ВЭЖХ = 99,56%.
ПРИМЕР 196: 2-[3-(6-хлор-1,3-бензотиазол-2-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]декан-5-карбоксамид (196):
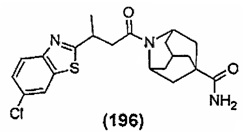
Синтез Соединения (196): Соединение (196) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (196). ЖХ-МС (М+Н)+ = 418,1; чистота по ВЭЖХ = 97,62%.
ПРИМЕР 197: 3-(4,5-дихлор-1H-пирроло[2,3-b]пиридин-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (197):
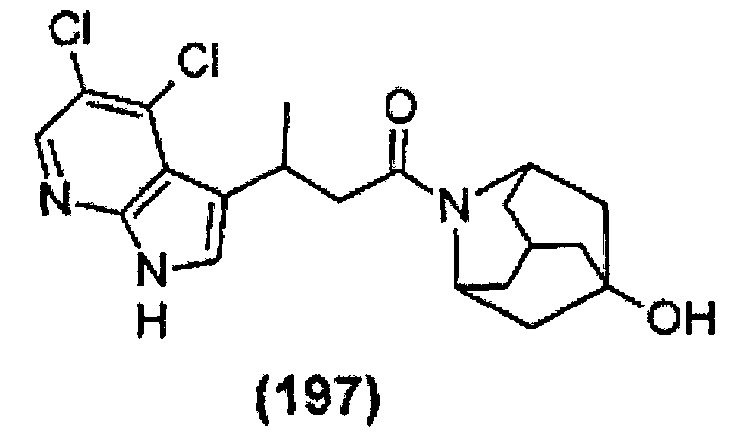
Синтез Соединения (197): Соединение (197) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (197). ЖХ-МС (М+Н)+ = 408,1; чистота по ВЭЖХ = 94,42%.
ПРИМЕР 198: 3-(5-хлор-4-циклопропил-1H-пирроло[2,3-b]пиридин-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (198):
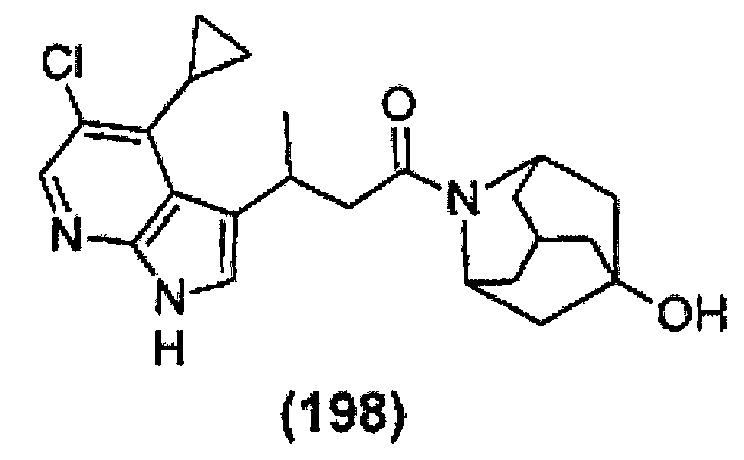
Синтез Соединения (198): Соединение (198) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (198). ЖХ-МС (М+Н)+ = 414,2; чистота по ВЭЖХ = 98,68%.
ПРИМЕР 199: 3-(5-хлор-4-циклопропил-1H-пирроло[2,3-b]пиридин-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 1) (199):
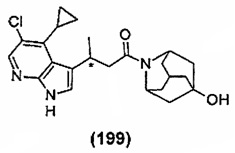
Синтез Соединения (199): Рацемическое соединение (198) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (199). ЖХ-МС (М+Н)+ = 414,2; чистота по ВЭЖХ = 94,06%. Хиральное время удерживания = 15,94 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: IPA: ДХМ (7,5:1,5:1)].
ПРИМЕР 200: 3-(5-хлор-4-циклопропил-1H-пирроло[2,3-b]пиридин-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (пик 2) (200):
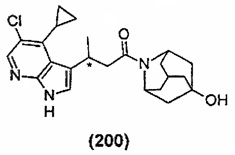
Синтез Соединения (200): Рацемическое соединение (198) очистили с помощью хиральной препаративной ВЭЖХ хроматографии с получением Соединения (200). ЖХ-МС (М+Н)+ = 414,2; чистота по ВЭЖХ = 98,40%. Хиральное время удерживания = 20,09 мин. [колонка: ChiralPak IC, подвижная фаза: гексан: IPA: ДХМ (7,5:1,5:1)].
ПРИМЕР 201: 3-(4,5-дициклопропил-1H-пирроло[2,3-b]пиридин-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (201):
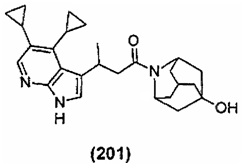
Синтез Соединения (201): Соединение (201) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (201). ЖХ-МС (М+Н)+ = 420,2; чистота по ВЭЖХ = 90,88%.
ПРИМЕР 202: 3-(6-хлор-1H-бензимидазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (202):
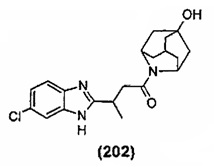
Синтез Соединения (202): Соединение (202) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (202). ЖХ-МС (М+Н)+ = 372,1; чистота по ВЭЖХ = 84,69%.
ПРИМЕР 203: 3-(6-циклопропил-1H-бензимидазол-2-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (203):
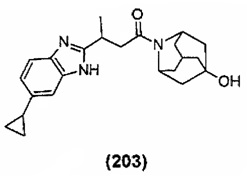
Синтез Соединения (203): Соединение (203) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (203). ЖХ-МС (М+Н)+ = 380,1; чистота по ВЭЖХ = 98,88%.
ПРИМЕР 204: {2-[3-(4-циклопропил-1H-пирроло[2,3-b]пиридин-3-ил)бутаноил]-2-азатрицикло[3.3.1.13,7]дец-5-ил}уксусная кислота (204):
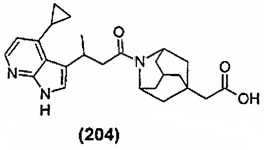
Синтез Соединения (204): Соединение (204) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (204). ЖХ-МС (М+Н)+ = 422,2; чистота по ВЭЖХ = 99,63%.
ПРИМЕР 205: 3-(1-циклопропил-4-метил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (205):
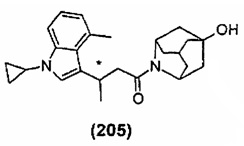
Синтез Соединения (205): Соединение (205) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (205). ЖХ-МС (М+Н)+ = 393,2; чистота по ВЭЖХ = 99,04%.
ПРИМЕР 206: 3-(1,4-диметил-1H-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (206):
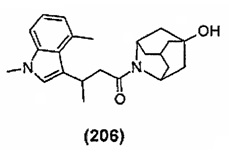
Синтез Соединения (206): Соединение (206) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (206). ЖХ-МС (М+Н)+ = 367,2; чистота по ВЭЖХ = 98,36%.
ПРИМЕР 207: 3-(4-хлор-1-метил-1Н-индол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (297):
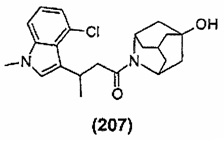
Синтез Соединения (207): Соединение (207) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (207). ЖХ-МС (М+Н)+ = 387,1; чистота по ВЭЖХ = 99,49%.
ПРИМЕР 208: 3-[4-хлор-5-(фуран-2-ил)-1H-индол-3-ил]-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (208):
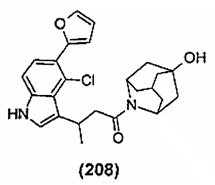
Синтез Соединения (208): Соединение (208) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (208). ЖХ-МС (М+Н)+ = 439,1; чистота по ВЭЖХ = 91,39%.
ПРИМЕР 209: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-(4-метокси-5-метил-1H-индол-3-ил)бутан-1-он (209):
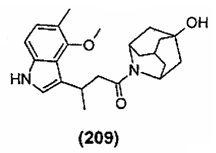
Синтез Соединения (209): Соединение (209) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (209). ЖХ-МС (М+Н)+ = 383,2; чистота по ВЭЖХ = 93,48%.
ПРИМЕР 210: 1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[5-метил-4-(тиофен-2-ил)-1H-индол-3-ил]бутан-1-он (210):
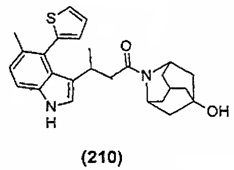
Синтез Соединения (210): Соединение (210) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (210). ЖХ-МС (М+Н)+ = 435,2; чистота по ВЭЖХ = 91,98%.
ПРИМЕР 211: 4-хлор-3-[4-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-4-оксобутан-2-ил]-5-метил-1,3-дигидро-2H-индол-2-он (211):
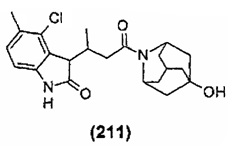
СХЕМА СИНТЕЗА 26
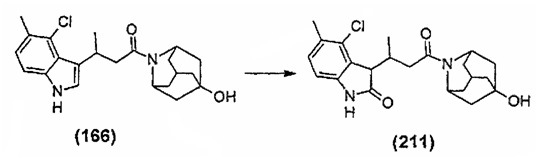
Синтез Соединения (211): К перемешанному раствору Соединения (166) (15 мг, 0,03 ммоль) в ДМФ (2 мл) добавили пиридиния трибромид (16 мг, 0,050 ммоль) при 0°С. Полученную реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакцию погасили Н2О (10 мл), затем экстрагировали эфиром (2×20 мл). Объединенные органические слои промыли насыщенным солевым раствором и концентрировали. Полученный неочищенный материал очистили препаративной ТСХ, элюируя ДХМ: МеОН (95:05), с получением Соединения (211) (5,5 мг, грязновато-белое твердое вещество).
Пример 212: 4-циклопропил-3-[4-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)-4-оксобутан-2-ил]-1,3-дигидро-2Н-индол-2-он (212):
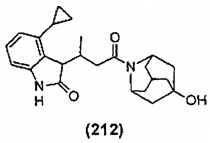
Синтез Соединения (212): Соединение (212) синтезировали по способу, использованному для получения Соединения (211) (Схема 26). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (212). 1Н ЯМР (300 МГц, CDCl3): δ 7.50 (br s, 1H), 7.07-7.12 (t, 1H), 6.59-6.62 (d, 1H), 6.48-6.51 (d, 1H), 5.11 (br s, 1H), 4.54 (br s, 1H), 3.81 (br s, 1H), 3.79-3.81 (m, 1H), 2.37-2.41 (m, 2H), 2.21-2.23 (m, 1H), 1.43-1.88 (m, 11H), 1.06-1.08 (m, 2H), 0.83-0.88 (M, 2H), 0.72-0.74 (d, 3H). ЖХ-МС (M+H)+ = 395,2; чистота по ВЭЖХ = 99,65%.
ПРИМЕР 213: 3-(4-циклопропил-1H-индол-3-ил)-1-(5-фтор-7-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)бутан-1-он (213):
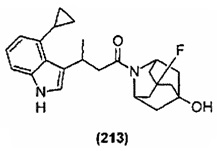
Синтез Соединения (213): Соединение (213) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (213). 1H ЯМР (300 МГц, CDCl3): δ 8.06 (br s, 1H), 7.18-7.20 (d, 1H), 7.05-7.10 (t, 1H), 7.04 (s, 1H), 6.74-6.76 (d, 1H), 5.26 (br s, 1H), 4.40 (br s, 1H), 4.23-4.25 (m, 1H), 2.85-2.90 (m, 1H), 2.45-2.53 (m, 2H), 1.99 (br s, 2H), 1.62-1.81 (m, 8H), 1.44-1.46 (d, 3H), 0.95-0.99 (m, 2H), 0.81-0.85 (m, 2H). ЖХ-МС (М+Н)+ = 397,3; чистота по ВЭЖХ = 93,84%.
ПРИМЕР 214: 1-(5-гидрокси-7-метил-2-азатрицикло[3.3.1.13,7]дец-2-ил)-3-[4-(пропан-2-илокси)-1H-индол-3-ил]бутан-1-он (214):
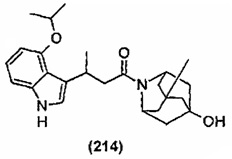
Синтез Соединения (214): Соединение (214) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (214). 1Н ЯМР (300 МГц, CDCl3): δ 7.86 (br s, 1H), 6.96-7.01 (t, 1H), 6.84-6.86 (d, 1H), 6.83 (s, 1H), 6.39-6.42 (d, 1H), 5.00 (br s, 1H), 4.67-4.72 (m, 1H), 4.20 (br s, 1H), 3.72-3.79 (m, 1H), 2.89-3.01 (m, 1H), 2.39-2.51 (m, 1H), 1.25-1.65 (m, 19H). ЖХ-МС (M+H)+ = 411,3; чистота по ВЭЖХ = 90,45%.
ПРИМЕР 215: 2-(2-(3-(4-(5-фторфуран-2-ил)-1Н-индол-3-ил)бутаноил)-2-азаадамантан-5-ил)уксусная кислота (215):
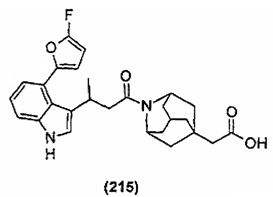
Синтез Соединения (215): Соединение (215) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). Неочищенный продукт получили выпариванием органического слоя под пониженным давлением и очистили с помощью силикагелевой колонки, используя ДХМ: МеОН в качестве элюента, с получением Соединения (215). ЖХ-МС (М+Н)+ = 465,4; чистота по ВЭЖХ = 99,37%.
ПРИМЕР 216: 1-(5-гидрокси-2-азаадамантан-2-ил)-3-(4-метил-1-(хинолин-8-илсульфонил)-1Н-индол-3-ил)бутан-1-он (216):
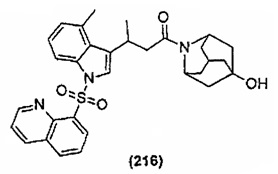
Синтез Соединения (216): Соединение (216) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). ЖХ-МС (М+Н)+ = 544,3; чистота по ВЭЖХ = 98,61%.
ПРИМЕР 217; 4-хлор-3-(4-(5-гидрокси-2-азаадамантан-2-ил)-4-оксобутан-2-ил)-5-метил-1H-индол-2-карбонитрил (217):
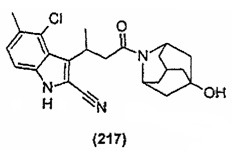
Синтез Соединения (217): Соединение (217) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). ЖХ-МС (М+Н)+ = 412,3.
ПРИМЕР 218: 4-хлор-3-(4-(5-гидрокси-2-азаадамантан-2-ил)-4-оксобутан-2-ил)-1Н-индол-2-карбонитрил (218):
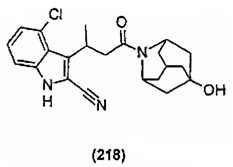
Синтез Соединения (218): Соединение (218) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). ЖХ-МС (М+Н)+ = 398,2; чистота по ВЭЖХ = 98,47%.
ПРИМЕР 219: 3-(4-хлор-5-метил-1Н-индол-3-ил)-1-(5-гидрокси-7-метил-2-азаадамантан-2-ил)бутан-1-он (219):
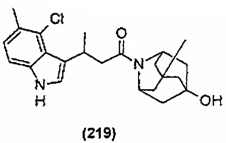
Синтез Соединения (219): Соединение (219) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). ЖХ-МС (М+Н)+ = 401,2; чистота по ВЭЖХ = 89,36%.
ПРИМЕР 220: 4-циклопропил-3-(4-(5-гидрокси-2-азаадамантан-2-ил)-4-оксобутан-2-ил)-1Н-индол-2-карбонитрил (220):
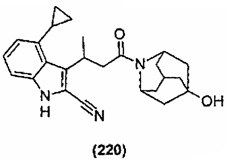
Синтез Соединения (220): Соединение (220) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). ЖХ-МС (М+Н)+ = 404,3; чистота по ВЭЖХ = 98,29%.
ПРИМЕР 221: 2-(3-(4,5-диметил-1Н-индол-3-ил)бутаноил)-2-азаадамантан-5-карбонитрил (221):
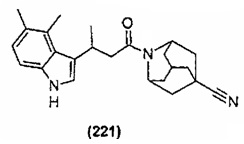
Синтез Соединения (221): Соединение (221) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). ЖХ-МС (М+Н)+ = 376,3; чистота по ВЭЖХ = 85,11%.
ПРИМЕР 222: 2-(3-(4-(фуран-2-ил)-1Н-индол-3-ил)бутаноил)-2-азаадамантан-5-карбоновая кислота (222):
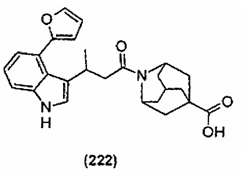
Синтез Соединения (222): Соединение (222) синтезировали по способу, использованному для получения Соединения (43) (Схема 14). ЖХ-МС (М+Н)+ = 433,3; чистота по ВЭЖХ = 98,85%.
ПРИМЕР 223: 4-циклопропил-3-(4-(5-гидрокси-2-азаадамантан-2-ил)-4-оксобутан-2-ил)-1Н-индол-2-карбоновая кислота (223):
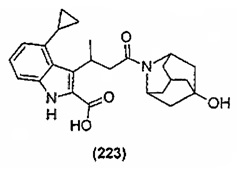
СХЕМА СИНТЕЗА 27
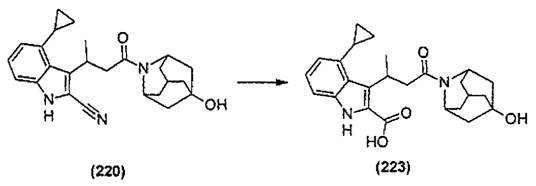
Синтез Соединения (223): Соединение 220 (0,070 г, 0,17 ммоль) поместили в закрытую пробирку и добавили к нему МеОН (3 мл), затем добавили 50% водный раствор KOH (2 мл). Затем реакционную смесь нагревали при 110°С в течение 24 часов и концентрировали с получением неочищенного материала, который разбавили H2O, подкислили 2 н. раствором HCl (рН=2), экстрагировали EtOAc и концентрировали с получением неочищенного продукта. Затем неочищенный продукт очистили с помощью силикагелевой колоночной хроматографии, элюируя смесью ДХМ: МеОН, с получением 30 мг Соединения (223) в виде коричневого твердого вещества. ЖХ-МС (М+Н)+ = 423,1; чистота по ВЭЖХ = 98,09%.
ПРИМЕР 224: 1-(5-фтор-2-азаадамантан-2-ил)-3-(5-метокси-1Н-индол-3-ил)бутан-1-он (224):
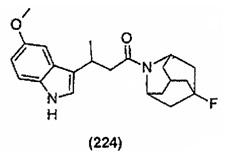
Синтез Соединения (224): Соединение (224) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). ЖХ-МС (М+Н)+ = 371,2; чистота по ВЭЖХ = 92,10%.
ПРИМЕР 225: 3-(4-(5-бром-2-азаадамантан-2-ил)-4-оксобутан-2-ил)-1Н-индол-5-ил трифторметансульфонат (225):
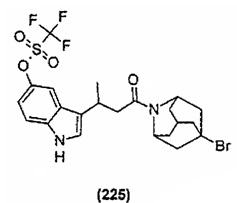
Синтез Соединения (225): Соединение (225) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). ЖХ-МС (М+Н)+ = 549,2; чистота по ВЭЖХ = 92,61%.
ПРИМЕР 226: 1-(2-азаадамантан-2-ил)-3-(5-метокси-1Н-индол-3-ил)бутан-1-он (226):
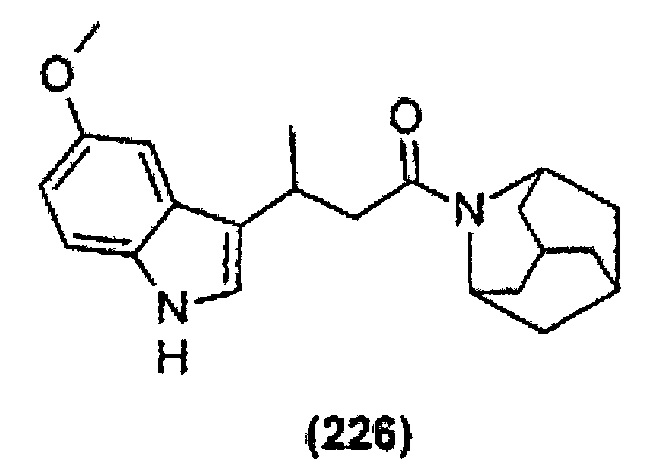
Синтез Соединения (226): Соединение (226) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). ЖХ-МС (М+Н)+ = 353,2; чистота по ВЭЖХ = 90,02%.
ПРИМЕР 227: 3-(5-метокси-1Н-индол-3-ил)-1-(5-метил-2-азаадамантан-2-ил)бутан-1-он (227):
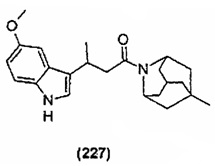
Синтез Соединения (227): Соединение (227) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). ЖХ-МС (М+Н)+ = 367,3; чистота по ВЭЖХ = 92,59%.
ПРИМЕР 228: 3-(4-(2-азаадамантан-2-ил)-4-оксобутан-2-ил)-1Н-индол-5-ил трифторметансульфонат (228):
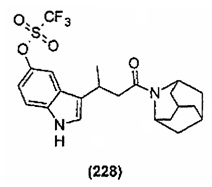
Синтез Соединения (228): Соединение (228) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). 1H ЯМР (300 МГц, ДМСО-d6): δ 11.24 (br s, 1H), 7.59 (s, 1H), 7.44-7.47 (d, 1H), 7.36 (s, 1H), 7.11-7.14 (d, 1H), 4.62 (br s, 1H), 4.01 (br s, 1H), 3.43-3.48 (m, 1H), 2.62-2.65 (m, 1H), 2.43.2.47 (m, 1H), 2.10 (s, 1Н), 2.00 (s, 1H), 1.44-1.86 (m, 10Н), 1.30-1.33 (d, 3Н). ЖХ-МС (М+Н)+ = 471,1; чистота по ВЭЖХ = 89,84%.
ПРИМЕР 229: 3-(4-(2-азаадамантан-2-ил)-4-оксобутан-2-ил)-1Н-индол-5-ил метансульфонат (229):
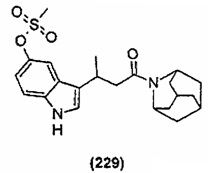
Синтез Соединения (229): Соединение (229) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). 1H ЯМР (300 МГц, CDCl3): δ 8.37 (br s, 1H), 7.50 (s, 1H), 7.23-7.26 (d, 1H), 6.98-7.05 (m, 2H), 4.74 (br s, 1H), 3.92 (br s, 1H), 3.51-3.58 (m, 1H), 3.08 (s, 1H), 2.68-2.75 (m, 1H), 2.41-2.48 (m, 1H), 1.96 (br s, 1H), 1.84 (br s, 1H), 1.45-1.72 (m, 10H), 1.39-1.42 (d, 3Н). ЖХ-МС (M+H)+ = 417,1; чистота по ВЭЖХ = 92,90%.
ПРИМЕР 230: 2-(3-(5-метокси-1Н-индол-3-ил)бутаноил)-2-азаадамантан-5-карбонитрил (230):
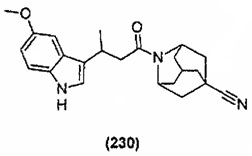
Синтез Соединения (230): Соединение (230) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). 1H ЯМР (300 МГц, CDCl3): δ 7.95 (br s, 1H), 7.22-7.28 (m, 1H), 7.09-7.12 (m, 1H), 6.99-7.00 (d, 1H), 6.83-6.89 (m, 1H), 4.91-4.95 (d, 1H), 3.97-4.03 (d, 1H), 3.86 (s, 3Н), 3.61-3.67 (m, 1H), 2.72-2.81 (m, 1H), 2.45-2.55 (m, 1H), 2.13-2.16 (m, 1H), 1.58-2.05 (m, 10Н), 1.48-1.51 (d, 3Н). ЖХ-МС (М+Н)+ = 378,3; чистота по ВЭЖХ = 99,49%.
ПРИМЕР 231: 1-(5-амино-2-азаадамантан-2-ил)-3-(4-циклопропил-1Н-индол-3-ил)бутан-1-он (231):
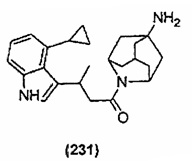
СХЕМА СИНТЕЗА 28
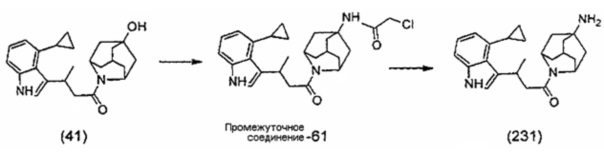
Синтез Промежуточного соединения 61: К перемешанному раствору соединения 41 (0,10 г, 0,26 ммоль) в ДХМ (5 мл) в АсОН (2 мл) добавили хлорацетонитрил (117 мг, 1,56 ммоль) при 0°С. Реакционную смесь затем обработали серной кислотой (0,75 мл). Реакционную смесь перемешивали при 0°С в течение 1 часа и продолжали перемешивать при комнатной температуре в течение 12 часов. После завершения реакции реакционную смесь погасили насыщенным водным раствором NaHCO3 и экстрагировали EtOAc с получением Промежуточного соединения 61 (90 мг). Его использовали на следующей стадии без какой-либо очистки.
Синтез Соединения 231: Перемешанный раствор Промежуточного соединения 61 (0,090 г, 0,19 ммоль) и тиомочевины (0,028 г, 0,38 ммоль) в EtOH (5 мл) при 0°С обработали уксусной кислотой (0,5 мл). Реакционную смесь нагревали с дефлегматором в течение 12 часов. После завершения реакции реакционную смесь погасили насыщенным раствором бикарбоната натрия, экстрагировали ДХМ и концентрировали с получением Соединения 231 (30 мг).
ПРИМЕР 232: 3-(4-циклопропил-1Н-индол-3-ил)-1-(5-(метиламино)-2-азаадамантан-2-ил)бутан-1-он (232)
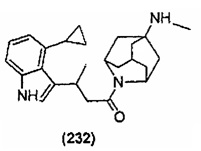
СХЕМА СИНТЕЗА 29
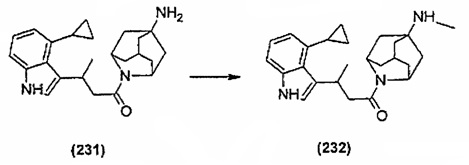
Синтез 232: К перемешанному раствору Соединения 231 (0,020 г, 0,053 ммоль) в ДМФ добавили K2CO3 (14,6 мг, 0,16 ммоль) при 0°С. К этому раствору добавили MeI и перемешивали при комнатной температуре в течение 2 часов. После завершения реакции реакционную смесь погасили H2O, экстрагировали EtOAc и концентрировали с получением неочищенного продукта, который очистили с помощью препаративной ТСХ с получением Соединения 232 (5 мг).
ПРИМЕР 233: 3-(4-циклопропил-1H-индазол-3-ил)-1-(5-гидрокси-2-азатрицикло[3.3.1.13,7]дец-2-ил)пропан-1-он (233):
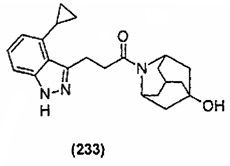
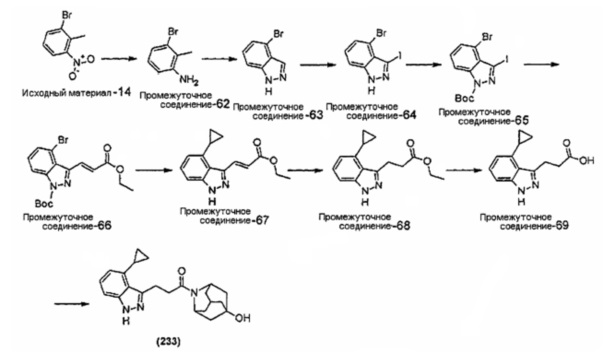
Синтез Промежуточного соединения 62: К перемешанному раствору исходного материала 14 (2,0 г, 8,6 ммоль) в ТГФ: МеОН (40 мл, 1:1) добавили активированный порошок Zn (3,71 г, 69,5 ммоль), затем насыщенный раствор NH4Cl и перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционную смесь концентрировали и разбавили Н2О, экстрагировали EtOAc и концентрировали с получением Промежуточного соединения 62 (1,7 г) в виде бледно-желтого твердого вещества.
Синтез Промежуточного соединения 63: К перемешанному раствору Промежуточного соединения 62 (1,8 г, 9,7 ммоль) в хлороформе (20 мл) добавили Ас2О (2,25 мл, 22,0 ммоль) при 0°С. Полученную реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Затем добавили KOAc (0,28 г, 2,9 ммоль), затем изоамилнитрил (2,44 г, 20 ммоль) и нагревали при 60°С в течение 18 часов. После завершения реакции реакционную смесь погасили H2O и концентрировали, к ней добавили концентрированную HCl (5 мл) и нагревали при 60°С в течение 2 часов. Затем реакционную смесь подщелочили 50% водным раствором NaOH и экстрагировали EtOAc, и концентрировали с получением Промежуточного соединения 63 (420 мг) в виде коричневого твердого вещества.
Синтез Промежуточного соединения 64: К перемешанному раствору Промежуточного соединения 63 (0,50 г, 2,5 ммоль) в ДМФ (5 мл) добавили KOH (0,28 г, 5,0 ммоль) и перемешивали при комнатной температуре в течение 20 минут. К реакционной смеси добавили йод (0,26 г, 5,0 ммоль) в ДМФ и продолжали реакцию при комнатной температуре в течение 12 часов. После завершения реакции реакционную смесь погасили Н2О, экстрагировали EtOAc и концентрировали с получением Промежуточного соединения 64 (750 мг) в виде коричневого твердого вещества, которое использовали на следующей стадии без какой-либо очистки.
Синтез Промежуточного соединения 65: К перемешанному раствору Промежуточного соединения 64 (0,75 г, 2,3 ммоль) в MeCN (5 мл) добавили ТЭА (0,696 г, 6,9 ммоль), DMAP (0,024 г, 0,2 ммоль) и (Вос)2O (1,00 г, 4,6 ммоль) при 0°С. Полученную реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После завершения реакции реакционную смесь погасили H2O, экстрагировали EtOAc и концентрировали с получением неочищенного материала. Неочищенный материал очистили с помощью силикагелевой колоночной хроматографии, элюируя смесью гексана: EtOAc, с получением Промежуточного соединения 65 (700 мг) в виде белого твердого вещества.
Синтез Промежуточного соединения 66: К перемешанному раствору Промежуточного соединения 65 (0,25 г, 0,7 ммоль) в ДМФ (5 мл) добавили ТЭА (0,31 мл, 2,3 ммоль), этилкротаноат (0,11 г, 1,1 ммоль) и TBAI (0,05 г, 0,14 ммоль) и продували газообразным аргоном в течение 15 минут. К реакционной смеси добавили PdCl2(dppf) (0,06 г, 0,07 ммоль). Полученную реакционную смесь выдерживали под микроволновым излучением при 100°С в течение 2 часов. После завершения реакции реакционную смесь отфильтровали через целит и концентрировали, полученный неочищенный материал очистили с помощью силикагелевой колоночной хроматографии, элюируя смесью гексанов: EtOAc, с получением Промежуточного соединения 66 (20 мг) в виде белого твердого вещества.
Синтез Промежуточного соединения 67: К перемешанному раствору Промежуточного соединения 66 (0,05 г, 0,12 ммоль) в диоксане: H2O (4 мл, 3:1) добавили циклопропилбороновую кислоту (0,02 г, 0,25 ммоль), Cs2CO3 (0,136 г, 0,42 ммоль) и продували газообразным аргоном в течение 10 минут. К реакционной смеси добавили PdCl2(dppf) (0,009 г, 0,012 ммоль). Полученную реакционную смесь выдерживали под микроволновым излучением при 120°С в течение 1 часа. После завершения реакции реакционную смесь погасили H2O, экстрагировали EtOAc и концентрировали. Полученную реакционную смесь очистили с помощью силикагелевой колоночной хроматографии, элюируя смесью гексана: EtOAc, с получением Промежуточного соединения 67 (30 мг) в виде коричневого смолянистого материала.
Синтез Промежуточного соединения 68: К перемешанному раствору Промежуточного соединения 67 (0,030 г, 0,11 ммоль) в ДМФ: МеОН (2 мл, 1:1) добавили NaBH4 (0,006 г, 0,17 ммоль), затем хлорид кобальта (0,002 г, 0,022 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 45 минут. После завершения реакции реакционную смесь погасили Н2О, экстрагировали EtOAc и концентрировали с получением Промежуточного соединения 68 (25 мг) в виде коричневого смолянистого материала.
Синтез Промежуточного соединения 69: Промежуточное соединение 69 синтезировали по способу, использованному для получения Промежуточного соединения 26 (схема 4).
Синтез Соединения (233): Соединение (233) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). 1H ЯМР (300 МГц, CDCl3): δ 7.22-7.33 (m, 2Н), 6.75-6.78 (m, 1H), 5.09 (br s, 1H), 4.40 (br s, 1H), 3.61-3.66 (t, 2H), 2.93-2.98 (t, 2H), 2.18-2.22 (t, 1H), 1.60-2.04 (m, 11H), 1.06-1.08 (m, 2H), 0.86-0.88 (m, 2H). ЖХ-МС (M+H)+ = 366,1.
ПРИМЕР 234: 2-(3-(4-фтор-5-метил-1H-индол-3-ил)бутаноил)-2-азаадамантан-5-карбонитрил (234):
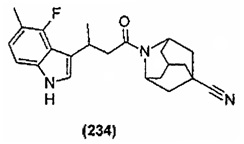
Синтез Соединения (234): Соединение (234) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). 1H ЯМР (300 МГц, CDCl3): δ 7.97 (br s, 1H), 7.02-7.06 (m, 1H), 6.93-6.99 (m, 2H), 4.96 (br s, 1H), 4.17-4.21 (m, 1H), 3.60-3.68 (m, 1H), 2.84-2.88 (m, 1H), 2.47-2.56 (m, 1H), 2.36 (s, 3Н), 2.10-2.13 (m, 1H), 1.61-2.05 (m, 10Н), 1.45-1.48 (d, 3H). ЖХ-МС (М+Н)+ = 380,2. Чистота по ВЭЖХ = 99,13%.
ПРИМЕР 235: 2-(4-циклопропил-1Н-индол-3-ил)-1-(5-гидрокси-2-азаадамантан-2-ил)пропан-1-он (235):
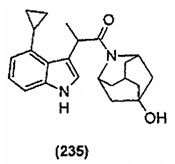
СХЕМА СИНТЕЗА 31
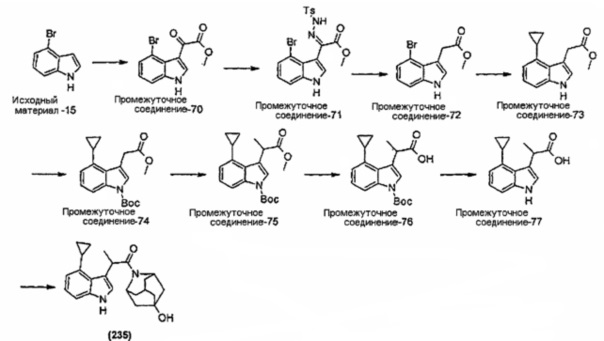
Синтез Промежуточного соединения 70: К перемешанному раствору исходного материала 15 (40 г, 206 ммоль) в эфире (400 мл) добавили оксалилхлорид (23,2 мл, 268 ммоль) при 0°С и перемешивали при комнатной температуре в течение 5 часов. Затем реакционную смесь отфильтровали и промыли эфиром с получением твердого материала (42 г), который обрабатывали МеОН (28 мл) в эфире (200 мл) при температуре от 0°С до комнатной температуры в течение 5 часов. После завершения реакции реакционную смесь разбавили гексанами, полученный осадок отфильтровали и высушили с получением Промежуточного соединения 70 (35 г) в виде желтого твердого вещества.
Синтез Промежуточного соединения 71: К перемешанному раствору Промежуточного соединения 70 (35 г, 129 ммоль) в МеОН (350 мл) добавили тозил-гидразин (23,1 г, 129 ммоль) и нагревали с дефлегматором в течение 4 часов. После завершения реакции реакционную смесь концентрировали с получением неочищенной смеси, которую разбавили H2O, экстрагировали ДХМ и концентрировали с получением Промежуточного соединения 69 (35 г) в виде бледно-желтого твердого вещества.
Синтез Промежуточного соединения 72: К перемешанному раствору Промежуточного соединения 71 (14 г, 31 ммоль) в ТГФ (140 мл) добавили NaBH4 (1,8 г, 46 ммоль) при 0°С и продолжали перемешивать при комнатной температуре в течение 6 часов. После завершения реакции реакционную смесь погасили Н2О, экстрагировали ДХМ и концентрировали. Полученный неочищенный продукт очистили с помощью силикагелевой колоночной хроматографии, элюируя смесью гексанов, EtOAc, с получением Промежуточного соединения 72 (3 г) в виде бледно-желтой жидкости.
Синтез Промежуточного соединения 73: Промежуточное соединение 73 синтезировали по способу, использованному для получения Промежуточного соединения 67 (Схема 30).
Синтез Промежуточного соединения 74: Промежуточное соединение 74 синтезировали по способу, использованному для получения Промежуточного соединения 65 (Схема 30).
Синтез Промежуточного соединения 75: Промежуточное соединение 75 синтезировали по способу, использованному для получения Промежуточного соединения 30 (Схема 6).
Синтез Промежуточного соединения 76: Промежуточное соединение 76 синтезировали по способу, использованному для получения Промежуточного соединения 26 (Схема 4).
Синтез Промежуточного соединения 77: Промежуточное соединение 77 синтезировали по способу, использованному для получения Промежуточного соединения 7 (Схема 1).
Синтез Соединения (233): Соединение (233) синтезировали по способу, использованному для получения Соединения (1) (Схема 2). 1Н ЯМР (300 МГц, CDCl3): δ 8.13 (br s, 1Н), 7.13-7.16 (d, 1Н), 6.96-7.04 (m, 2H), 6.75-6.78 (d, 1Н), 5.08 (br s, 1Н), 4.62-4.67 (m, 1Н), 4.32 (br s), 2.25-2.30 (m, 1Н), 2.05-2.08 (m, 1Н), 1.53-1.79 (m, 10Н), 1.46-1.49 (d, 3Н), 0.93-0.98 (m, 2H), 0.83-0.88 (m, 2H). ЖХ-МС (М+Н)+ = 365,2. Чистота по ВЭЖХ = 95,44%.
ПРИМЕР 236: 3-(4-циклопропил-1-метил-1Н-индазол-3-ил)-1-(5-гидрокси-2-азаадамантан-2-ил)пропан-1-он (236):
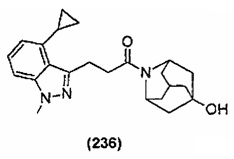
Синтез Соединения (236): Соединение (236) синтезировали по способу, использованному для получения Соединения (233) (Схема 30). ЖХ-МС (М+Н)+ = 380,3. Чистота по ВЭЖХ = 99,86%.
ПРИМЕР 237: 1-(2-азаадамантан-2-ил)-3-(4-циклопропил-1-метил-1Н-индазол-3-ил)пропан-1-он (237):
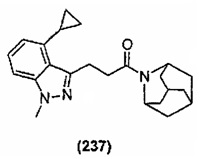
Синтез Соединения (237): Соединение (237) синтезировали по способу, использованному для получения Соединения (233) (Схема 30). ЖХ-МС (М+Н)+ = 364,3. Чистота по ВЭЖХ = 91,66%.
ПРИМЕР 238: 1-(2-азаадамантан-2-ил)-2-(4-циклопропил-1Н-индол-3-ил)пропан-1-он (238):
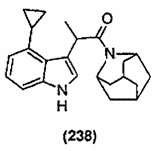
Синтез Соединения (238): Соединение (238) синтезировали по способу, использованному для получения Соединения (233) (Схема 27). ЖХ-МС (М+Н)+ = 349,2.
Биологическая активность In vitro анализ ингибирования HSD11β1:
Клетки СНО выдерживали в среде Игла, модифицированной по способу Дульбекко/питательной смеси F-12, содержащей 5% бычьей плодной сыворотки (об./об.) и 2 мМ глутамина. Клетки выращивали при 37°C с 5% CO2. Для временной экспрессии человеческого вектора экспрессии полной длины HSD11 β 1 (OriGene Technologies), клетки высеивали с плотностью 2×105 клеток/лунка в 6-луночном планшете. Трансфекцию выполнили с помощью реагента Turbofectin 8 (OriGene Technologies) no протоколу, предоставленному вместе с реагентом. Через 24 часа после трансфекции клетки трипсинизировали и слили вместе, затем выполнили повторное высевание в 96-луночный планшет с плотностью 40000 клеток/лунка. Через 24 часа после повторного высевания клетки инкубировали с 200 нМ кортизона + 500 мкМ НАДФН (или вместе с низкомолекулярными ингибиторами) в течение ночи. Ферментную активность или ингибирование ферментной активности измеряли по оценке превращения кортизона в кортизол по способу ЖХ/МС-МС. IC50 в нМ рассчитали по 8-точечной логарифмической шкале концентрации-ингибирования.
Результаты биологических испытаний представлены в таблице 1:
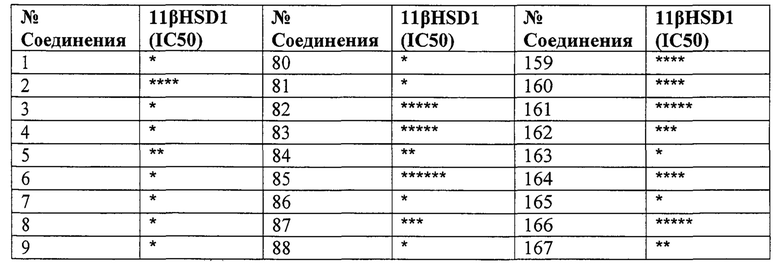
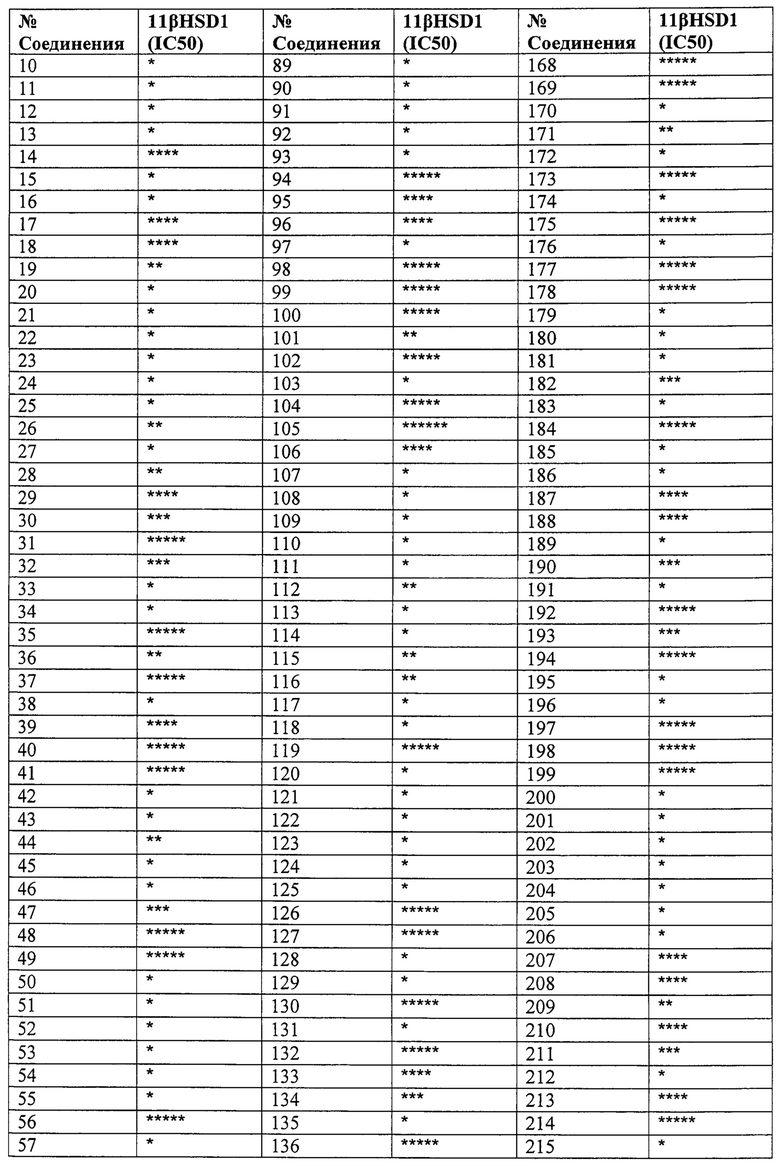
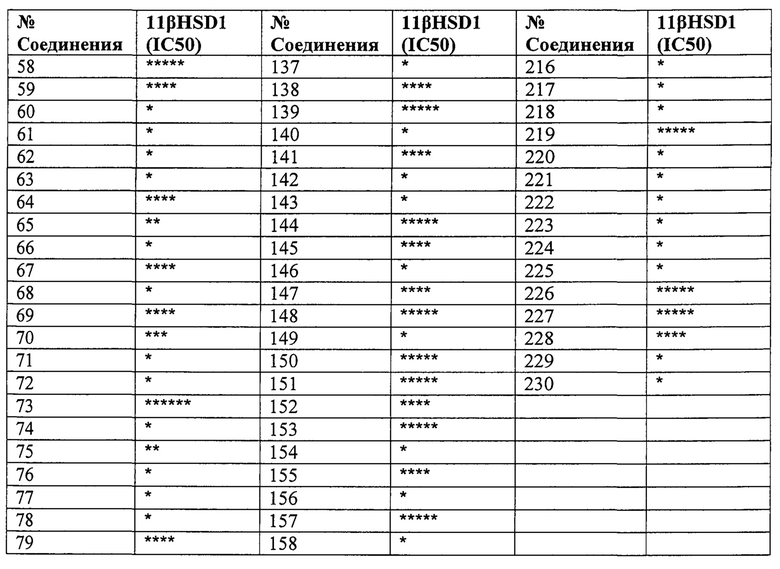
***** = <100 нМ
**** = 100 нМ < и <150 нМ
*** = 150нМ< и <200нМ
** = 200 нМ< и <250 нМ
* = 250 нМ<
| название | год | авторы | номер документа |
|---|---|---|---|
| ЦИКЛИЧЕСКИЕ АМИДНЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ 11-БЕТА-ГИДРОКСИСТЕРОИД-ДЕГИДРОГЕНАЗЫ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2669695C2 |
| ТРИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ГИРАЗЫ | 2012 |
|
RU2626979C2 |
| АГОНИСТЫ GPR40 | 2011 |
|
RU2627703C2 |
| МОДУЛЯТОРЫ РЕЦЕПТОРА НМДА И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2621049C2 |
| ФАРМАЦЕВТИЧЕСКИ АКТИВНЫЕ ПРОИЗВОДНЫЕ ПИРАЗОЛОТРИАЗИНА И/ИЛИ ПИРАЗОЛОПИРИМИДИНА | 2019 |
|
RU2818563C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОТРИАЗИНА И/ИЛИ ПИРАЗОЛОПИРИМИДИНА В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА ЦИКЛИНЗАВИСИМОЙ КИНАЗЫ | 2019 |
|
RU2809779C2 |
| АЗААДАМАНТАНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2450002C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНОЙ ТИРОЗИНКИНАЗЫ AXL/MER И CSF1R | 2019 |
|
RU2812631C2 |
| 6,7-ДИГИДРО-5H-ПИРИДО[2,3-C]ПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛКОВ BCLXL И ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2020 |
|
RU2832191C2 |
| Соединение диарилтиогидантоина в качестве антагониста андрогенового рецептора | 2018 |
|
RU2804108C9 |
Изобретение относится к соединениям формулы (I)
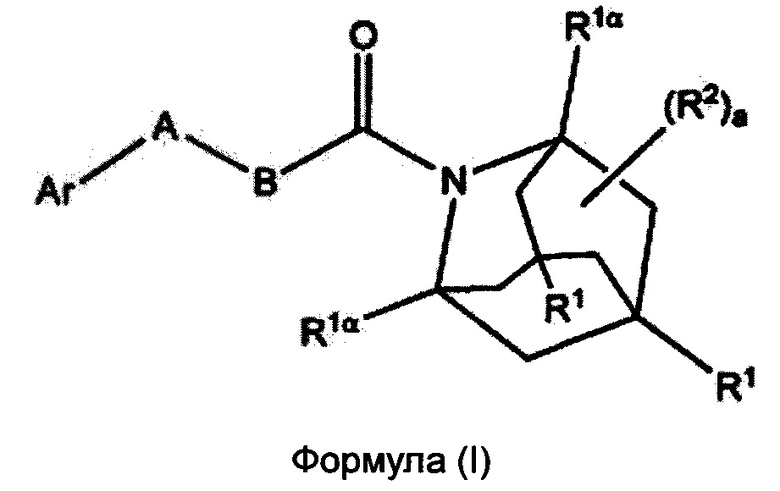
где: каждый R1, R1a и R2 независимо выбран из группы, состоящей из Н, галогена, ОН, CN, CF3, OCF3, ОСН3, СН2ОН, С3циклоалкилокси, СООН, CONR3R4 и тетразола; Аr представляет собой группу следующей формулы:
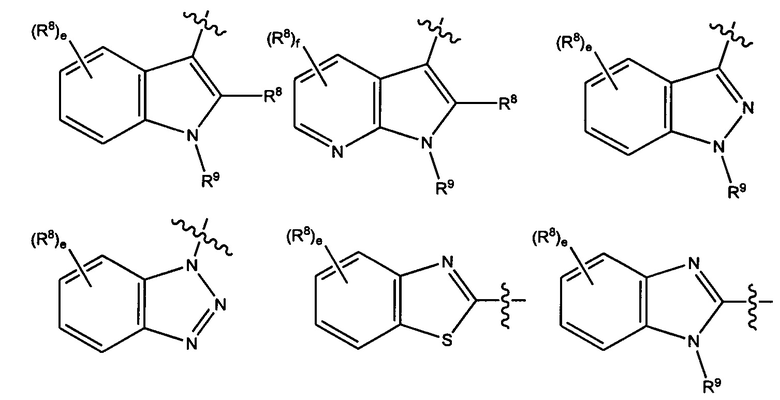
е представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3 и 4; f представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2 и 3; где каждый R8 независимо выбран из группы, состоящей из Н, галогена, метокси, OCF3, OCF2H, изопропокси, метила, пропила, 4-фторфенила, тиофена, фурана, 5-фторфурана, 5-фтортиофена, 5-хлортиофена, 5-хлорфурана, OSO2CF3, OSO2CH3, циклопропила и циклопропилокси; где R9 выбран из группы, состоящей из Н и метила; А представляет собой -CRaRb-; В представляет собой группу формулы -(CRcRd)n-; где каждый Ra и Rb независимо выбран из группы, состоящей из Н, метила, этила, изопропила, циклопропила, фенила, 4-хлорфенила, 4-фторфенила и тиофен-2-ила; где Rc и Rb - оба представляют собой Н; где каждый R3 и R4 представляет собой Н, n представляет собой целое число, выбранное из группы, состоящей из 0 и 1; а представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 и 10, которые способны ингибировать 11-β-гидроксистероид-дегидрогеназу типа 1 (11β-HSD-1) и которые, пригодны при лечении состояний, таких как инсулиннезависимый сахарный диабет 2 типа (NIDDM), инсулинорезистентность, ожирение, дислипидемия. 4 н. и 16 з.п. ф-лы, 1 табл., 238 пр.
1. Соединение формулы (I)
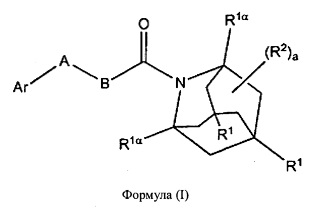
где:
каждый R1, R1α и R2 независимо выбран из группы, состоящей из Н, галогена, ОН, CN, CF3, OCF3, OCH3, СН2ОН, С3циклоалкилокси, СООН, CONR3R4 и тетразола;
Ar представляет собой группу следующей формулы:
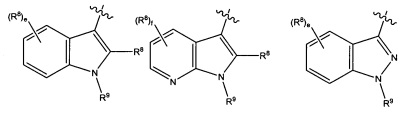
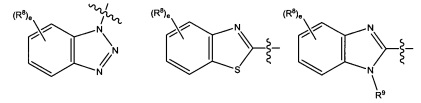
е представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3 и 4;
f представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2 и 3;
где каждый R8 независимо выбран из группы, состоящей из Н, галогена, метокси, OCF3, OCF2H, изопропокси, метила, пропила, 4-фторфенила, тиофена, фурана, 5-фторфурана,
5-фтортиофена, 5-хлортиофена, 5-хлорфурана, OSO2CF3, OSO2CH3, циклопропила и циклопропилокси;
где R9 выбран из группы, состоящей из Н и метила;
А представляет co6oH -CRaRb-;
В представляет собой группу формулы -(CRcRd)n-;
где каждый Ra и Rb независимо выбран из группы, состоящей из Н, метила, этила, изопропила, циклопропила, фенила, 4-хлорфенила, 4-фторфенила и тиофен-2-ила;
где Rc и Rb - оба представляют собой Н;
где каждый R3 и R4 представляет собой Н,
n представляет собой целое число, выбранное из группы, состоящей из 0 и 1;
а представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 и 10;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, отличающееся тем, что В представляет собой СН2.
3. Соединение по п. 1, отличающееся тем, что Ra и Rb различны.
4. Соединение по п. 3, отличающееся тем, что один из Ra и Rb представляет собой Н, а другой представляет собой метил.
5. Соединение по п. 4, отличающееся тем, что Rb представляет собой Н.
6. Соединение по п. 1, отличающееся тем, что Ra выбран из группы, состоящей из метила, этила, пропила и изопропила.
7. Соединение по п. 1, отличающееся тем, что заместитель R8 расположен в положении 4 или 5 шестичленного кольца.
8. Соединение по п. 1, отличающееся тем, что каждый R8 независимо выбран из группы, состоящей из СН3, Cl, Br, F и I.
9. Соединение по любому из пп. 1-8, отличающееся тем, что а равен 0.
10. Соединение по любому из пп. 1-9, отличающееся тем, что R1α представляет собой Н.
11. Соединение по любому из пп. 1-10, отличающееся тем, что каждый R1 независимо выбран из группы, состоящей из Н, ОН и OCH3.
12. Соединение по любому из пп. 1-11, отличающееся тем, что один R1 представляет собой Н, а другой R1 представляет собой ОН.
13. Соединение по п. 1, выбранное из группы, состоящей из:

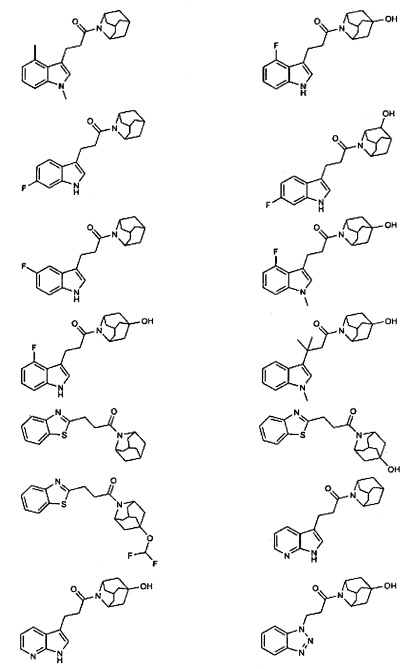
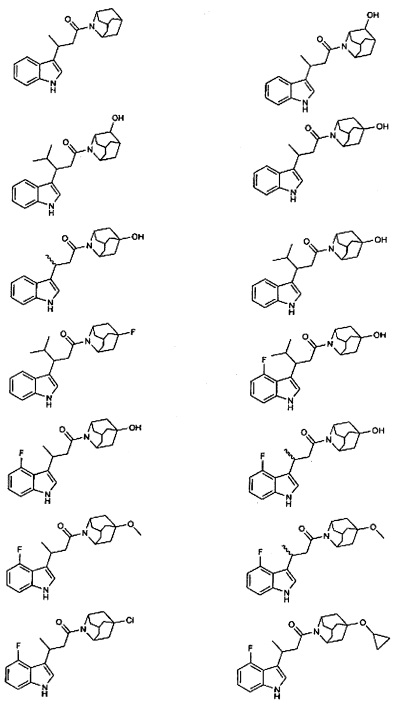
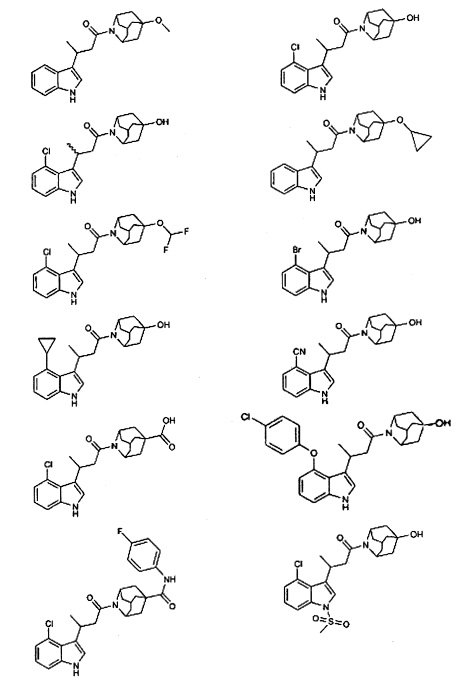
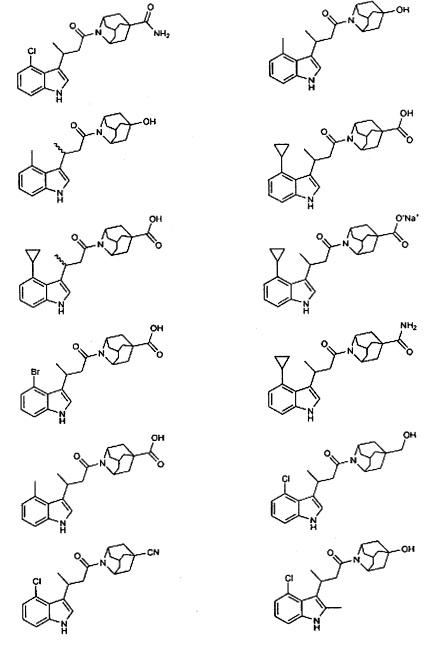
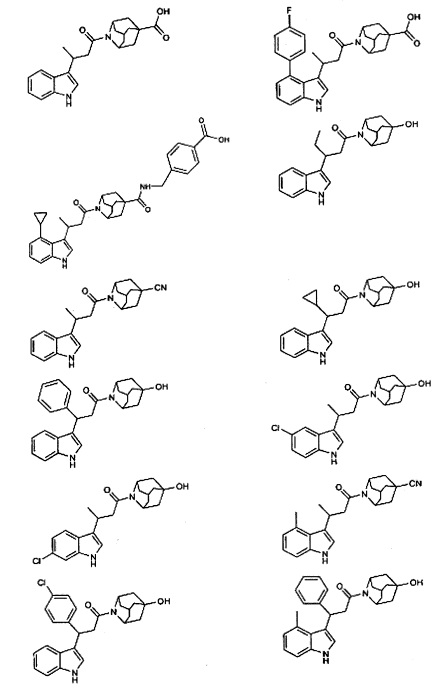
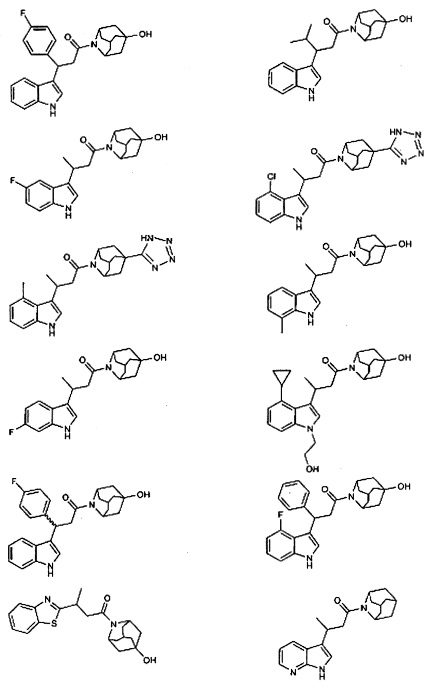
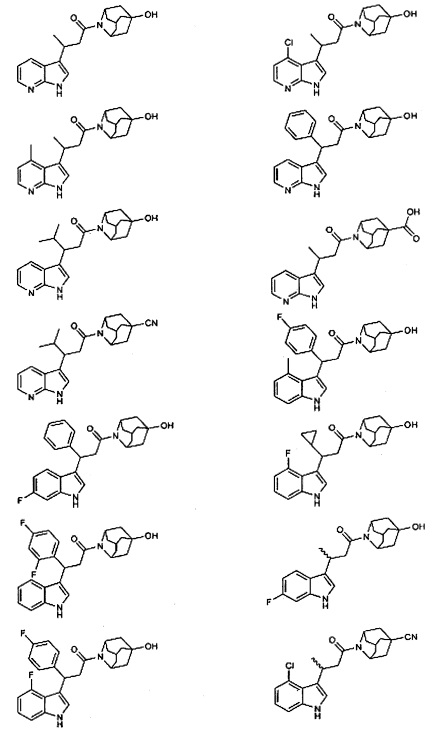
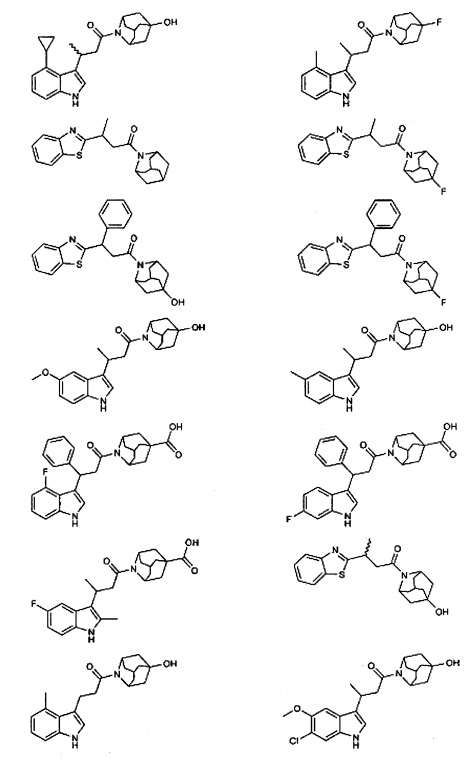
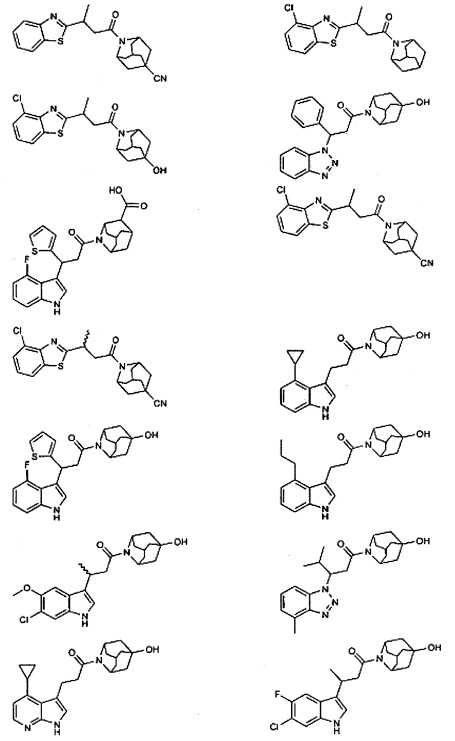
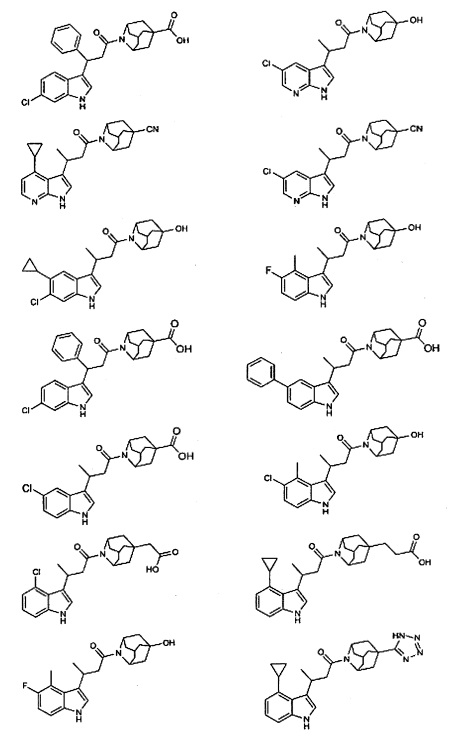
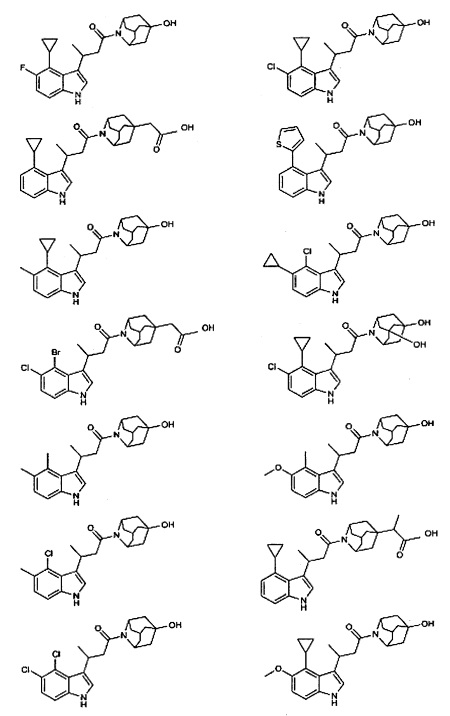
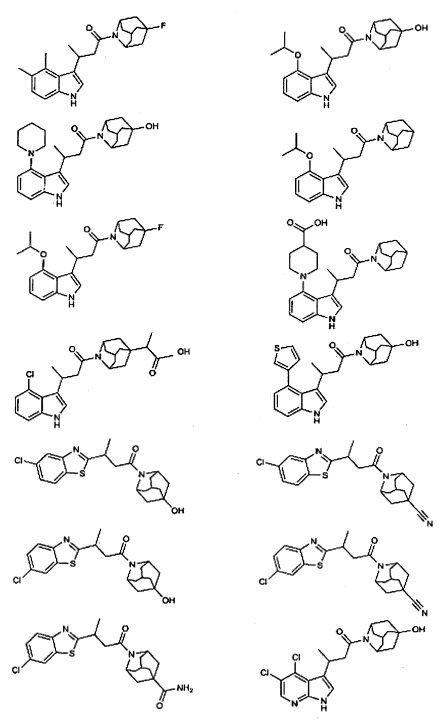
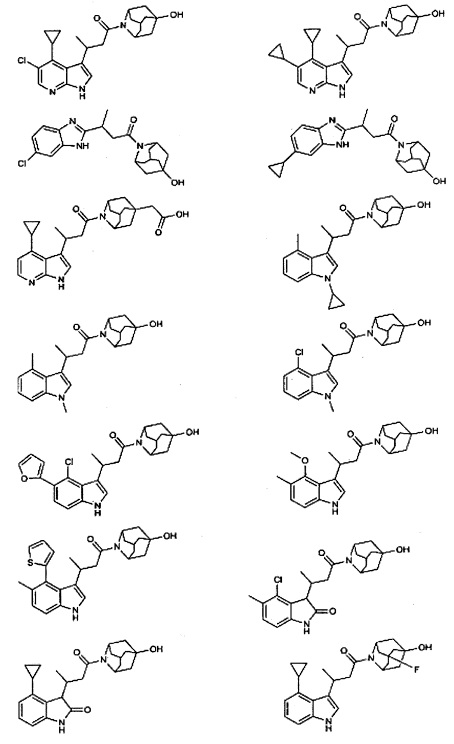
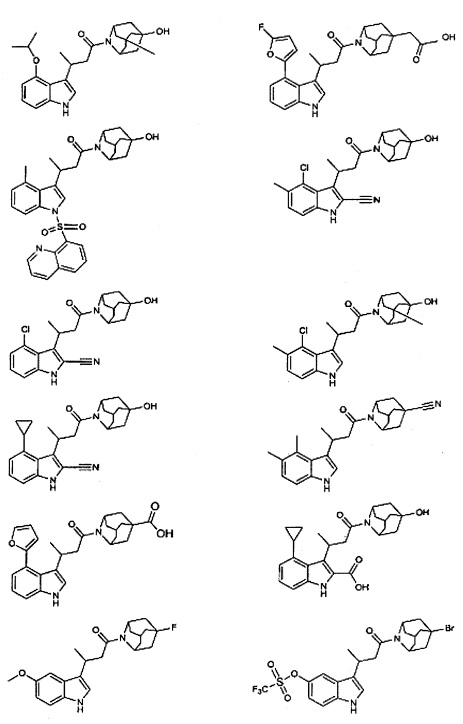
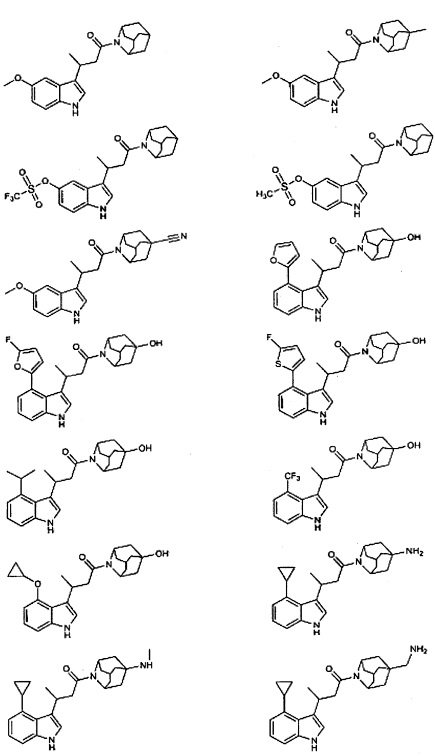
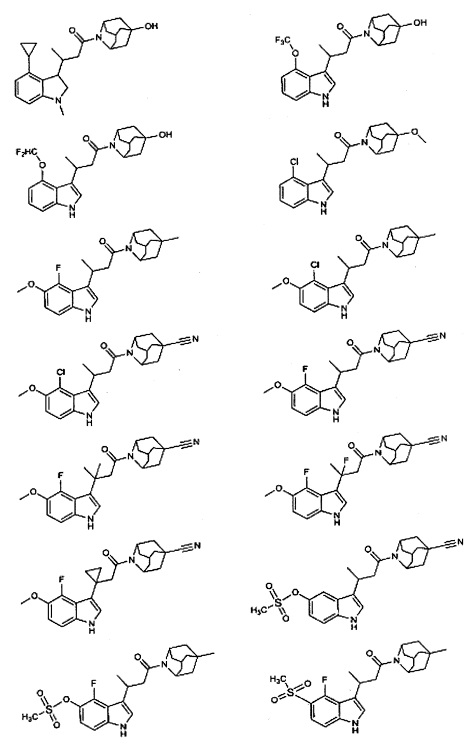
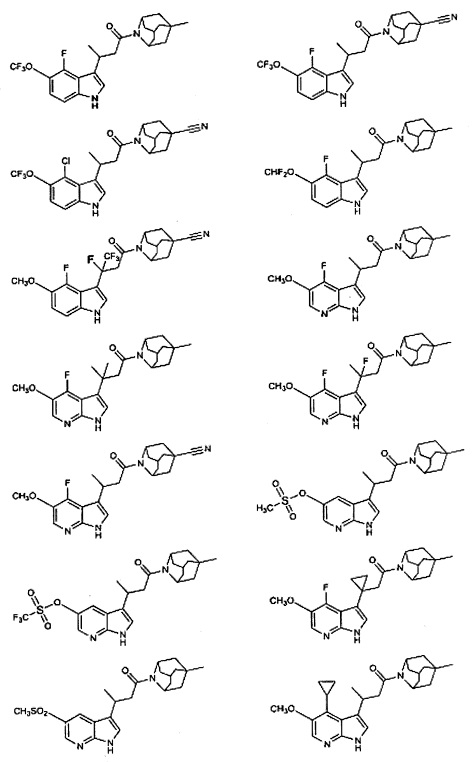
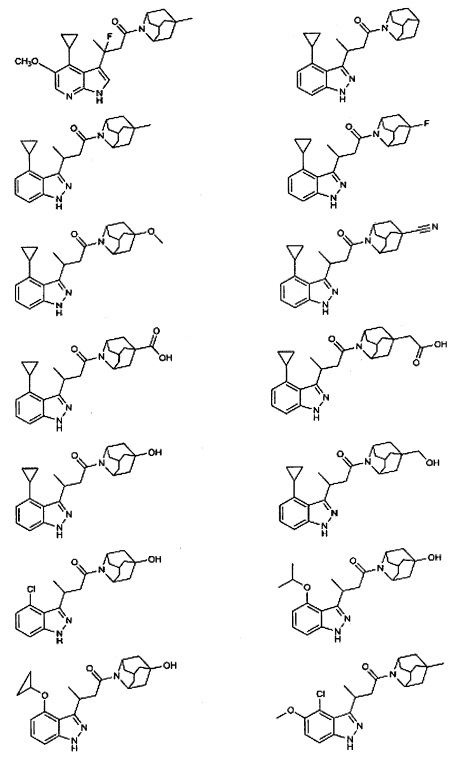
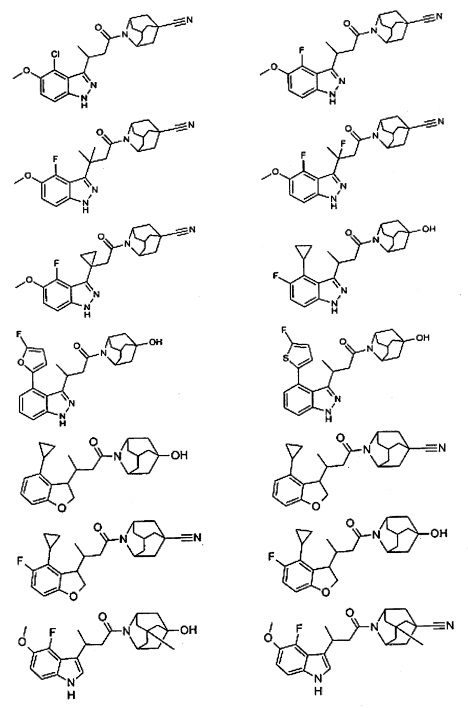
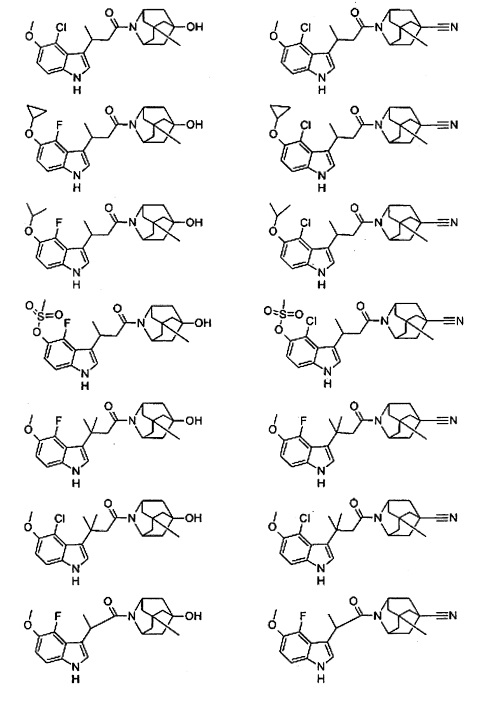
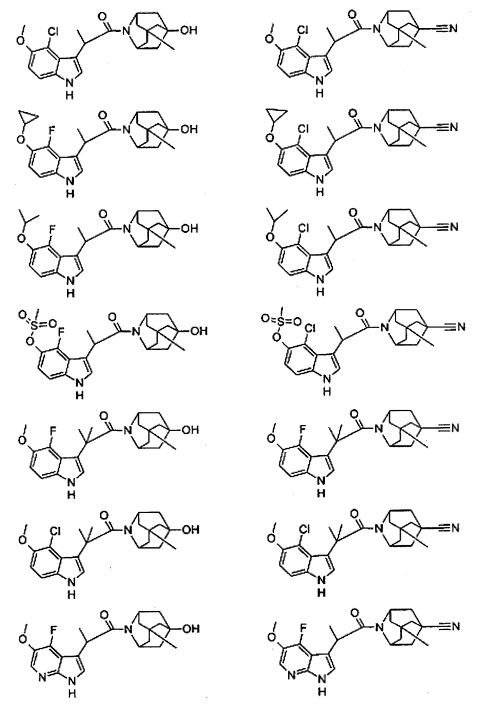
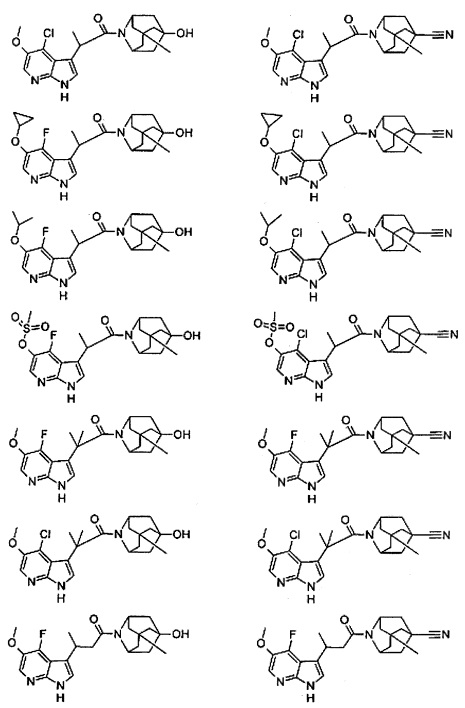
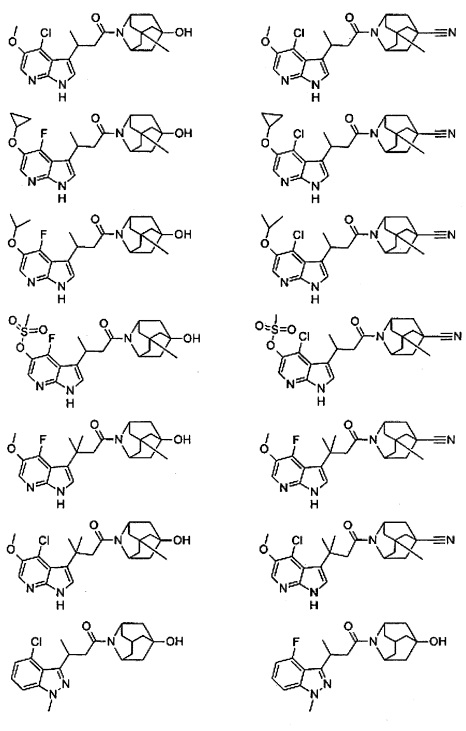
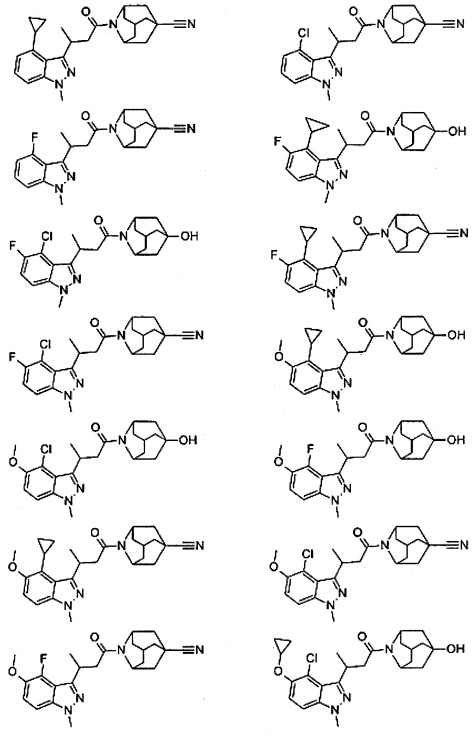
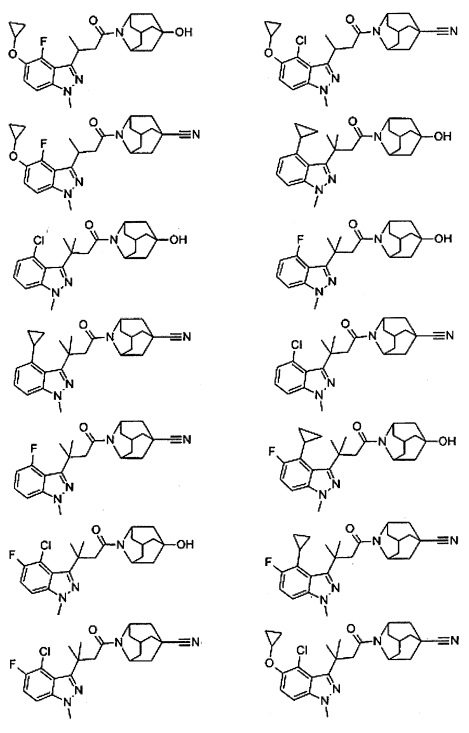
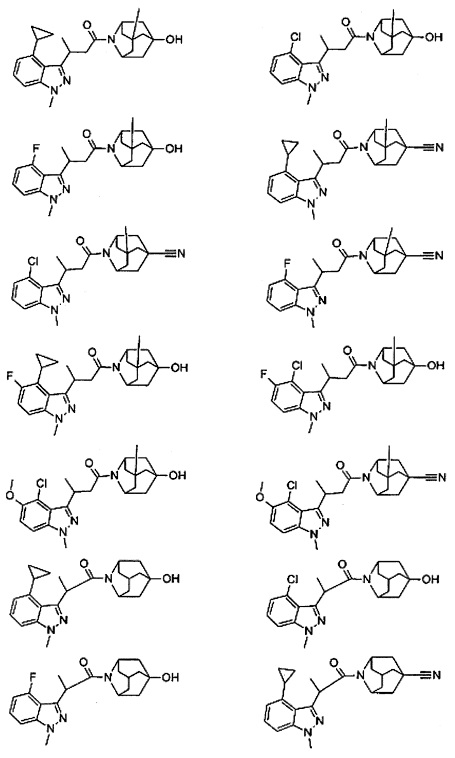
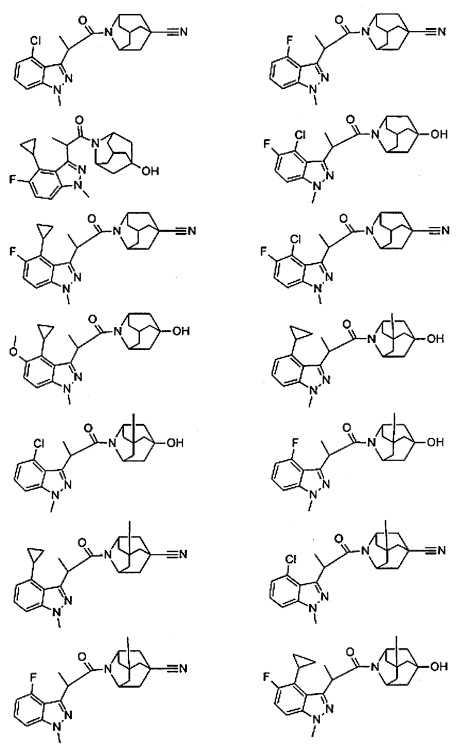
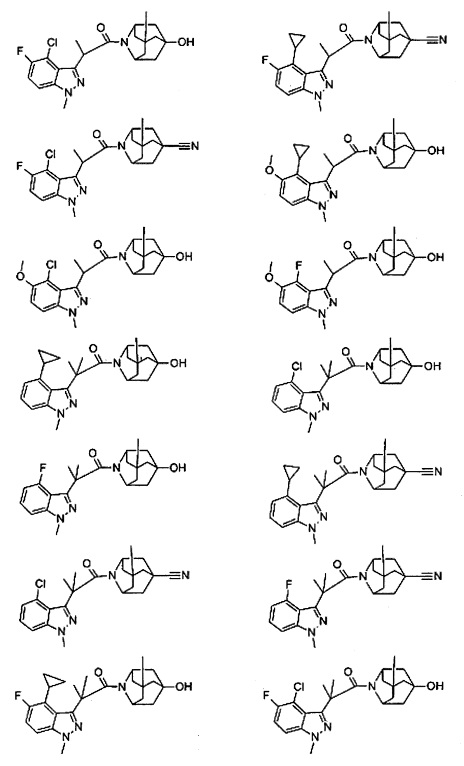
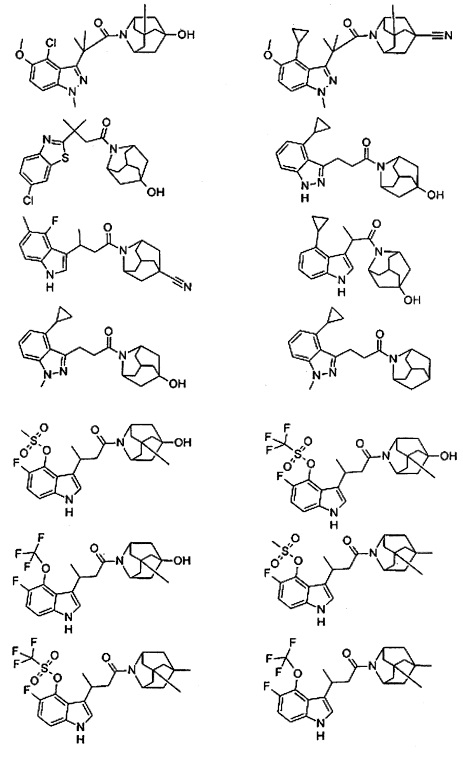
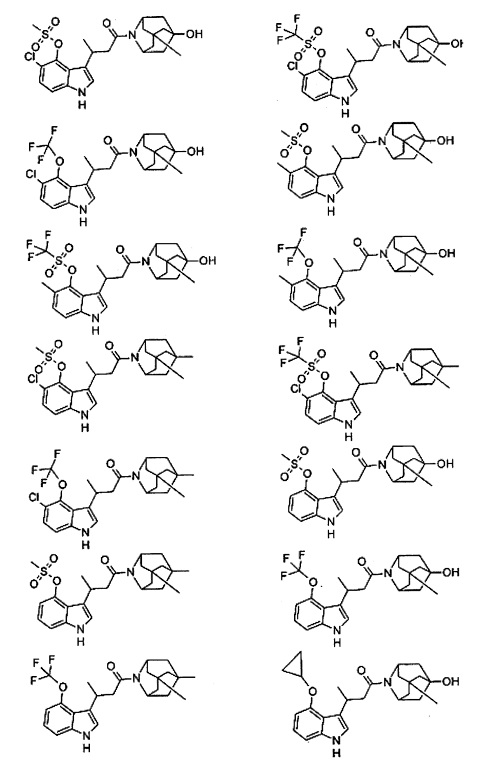
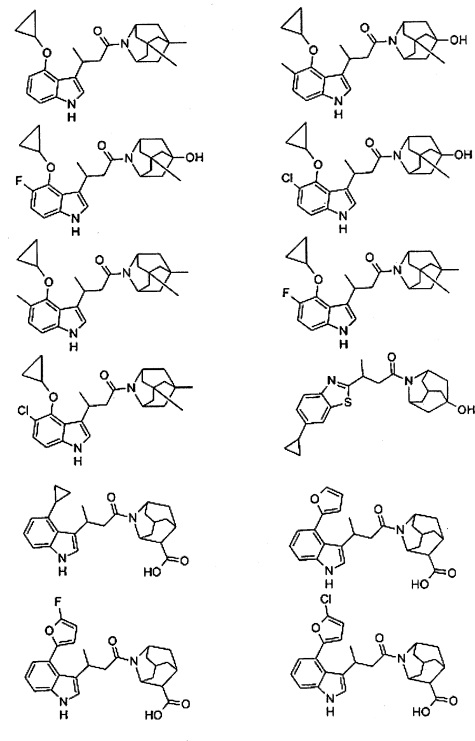
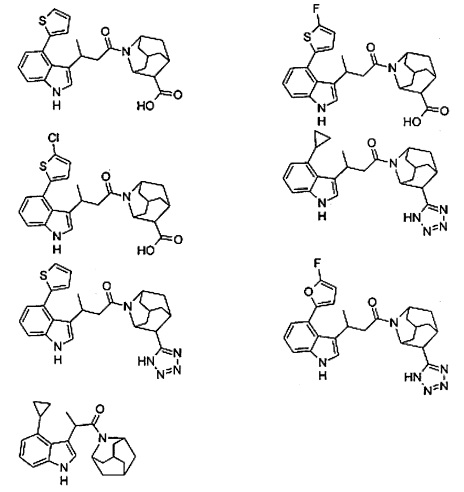
или его фармацевтически приемлемая соль.
14. Фармацевтическая композиция для лечения состояния, которое можно предупреждать или лечить ингибированием 11β-HSDl, содержащая эффективное количество соединения по любому из пп. 1-13 и фармацевтически приемлемый разбавитель, вспомогательное вещество или носитель.
15. Способ предупреждения или лечения состояния у млекопитающего, где указанное состояние можно предупреждать или лечить ингибированием 11β-HSD1, включающий введение эффективного количества соединения по любому из пп. 1-13.
16. Способ по п. 15, отличающийся тем, что указанное состояние представляет собой
диабет.
17. Способ по п. 16, отличающийся тем, что указанное состояние представляет собой диабет II типа.
18. Применение соединения по п. 1 для получения лекарственного средства для лечения состояния, которое можно предупреждать или лечить ингибированием 11β-HSD1.
19. Применение по п. 18, отличающееся тем, что указанное состояние представляет собой диабет.
20. Применение по п. 19, отличающееся тем, что указанное состояние представляет собой диабет II типа.
| WO 2008134221 A1, 06.11.2008 | |||
| US 2006100236 A1, 11.05.2006 | |||
| Камера для диатермии ультракороткими волнами | 1929 |
|
SU20227A1 |

