Изобретение относится к ингибитору атеросклеротического утолщения внутренней оболочки сосудов, который представляет производное пиридина, обладающее сильным ингибирующим действием в отношении HMG-CoA редуктазы, а также сильным ингибирующим действием в отношении пролиферации клеток гладких мышц внутренней оболочки аорты (I-SMC), происходящей в процессе атеросклеротического утолщения внутренней оболочки сосудов, в отношении миграции клеток гладких мышц срединного слоя аорты (M-SMC) и в отношении адгезии кровяных клеток (таким, как лимфоциты, лейкоциты и макрофаги) к эндостелиальным клеткам.
Считается, что утолщение внутренней оболочки коронарной артерии является одной из основных причин инфаркта миокарда и стенокардии. Предполагается, что атеросклеротическое утолщение внутренней оболочки сосудов начинается с адгезии моноцитов или тромбоцитов к эндостелиальным клеткам, которое стимулируется цитокинезом и накоплением липидов и далее развивается под действием миграции M-SMC из срединного слоя во внутреннюю оболочку сосудов, пролиферации клеток гладких мышц во внутренней оболочке и увеличения внеклеточного материала вследствие патологической и пролиферативной активации или модуляции клеток гладких мышц. Подобная активация клеток стимулируется такими факторами риска, как гиперлипемия. Сообщалось, что ингибиторы HMG-CoA редуктазы подавляют атеросклеротическое утолщение внутренней оболочки сосудов в результате значительного уменьшения холестерина сыворотки в животной модели (Biochim Biophis Acta 960, 294-302 (1988)), но клинические испытания не подтвердили эффективности такого воздействия.
В качестве более эффективного ингибитора атеросклеротического утолщения внутренней оболочки сосудов необходимо лекарственное средство, способное оказывать непосредственное воздействие на такое атеросклеротическое поражение. В литературе сообщалось об ингибирующем действии, оказываемом на клетки гладких мышц лекарственным средством, противодействующим фактору роста тромбоцитов (PDGF), или лекарственным средством, противодействующим фактору роста фибробластов (FGF) (Lif. Science 28, 1641-1646, (1981); J. Pharm Exp. Ther 248, 1167-1174) (1989)), об ингибирующем действии кальциевых антагонистов на миграцию клеток гладких мышц под влиянием PDGF тромбоксана B4 и интерлейнина-1 (Atherosclerosis 72, 213 - 219 (1988)), об ингибирующем действии RGD пептидов на адгезию клеток (J. Ecll Biol., 112, 335-344 (1981)), об ингибирующем действии ингибитора синтеза тромбоксана на повторный атенов после РТСА (Atherosclerosis 18, 661-665, (1990)). Однако все эти воздействия лекарственных средств направлены против тромбоцитов или стимулирующих факторов, таких, как цитокинез, при этом не существует лекарственного средства, которое оказывало бы непосредственное воздействие на патологические клетки гладких мышц, пораженных атеросклерозом.
Фактор миграции клеток гладких мышц, выделяемый из этих клеток, оказывает более сильное стимулирующее действие на миграцию, чем PDGF, при этом считается, что он тесно связан с возникновением и прогрессированием атеросклероза (Atherosclerosis, 72, 213-226, (1991)), но не было обнаружено лекарственного средства, которое бы подавляло такую миграцию клеток гладких мышц. Кроме того, пролиферативное состояние I-SMC, которое не может подавлять простагландином I2 (Atherosclerosis 73, 67-69 (1988)), считается важным показателем атеросклероза. Однако не существует лекарственного средства, которое ингибировало бы такое пролиферативное состояние.
Сообщалось, что ингибиторы HMG-CoA редуктазы оказывают подавляющее действие на пролиферацию фибробластов, клеток гладких мышц и лимфоцитов (J. Biol. Chem, 259, 1546-1551 (1984); там же, 255, 5134-5140; Biochim. Biophys. Acta 1051, 138-143 (1990)), а также подавляют активацию лимфоцитов (Int. J. Immunopharmac, 11, 863-869 (1069)). Однако такое ингибирующее действие не является вполне удовлетворительным, поэтому существует потребность в лекарственном средстве, которое было бы более эффективным и более избирательным в отношении целевых клеток, в частности I-MG, макрофагов и моноцитов.
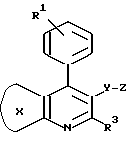
Изобретением предусматривается ингибитор атеросклеротического утолщения внутренней оболочки сосудов, который включает эффективное количество соединения формулы (I):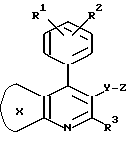 ,
,
в которой кольцо X представляет фенил, замещенный фенил или 5- или 6-членный гетероциклический арил;
R1 и R2 независимо друг от друга представляют водород, C1-C8-алкил, C3-C7-циклоалкил, C1-C3-алкокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, R20R21N- (где R20 и R21 независимо друг от друга представляют водород или C1-C3-алкил), трифторметил, трифторметокси, дифторметокси, фтор, хлор, бром, фенил, фенокси, бензилокси, гидрокси, триметилсилилокси, дифенил-трет-бутилсилилокси, гидроксиметил или -O/CH2/COR22 (где R22 - водород или C1-C3-алкил, а l - число, равное 1, 2 или 3); либо R1 и R2 вместе образуют -CH= CH-CH=CH- или метилендиокси, когда они находятся в орто-положении по отношению друг к другу;
R3 - водород, C1-C8-алкил, C2-C6-алкенил, C3-C7-циклоалкил, C5-C7-циклоалкенил или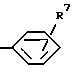 ,
,
где R7 - водород, C1-C8-алкил, C1-C8-алкокси, C1-C3-алкилтио, хлор, бром, фтор, хлорметил, трихлорметил, трифторметил, трифторметокси, трихлорметокси, дифторметокси, фенокси, бензилокси, гидрокси, триметилсилилокси, дифенил-трет-бутилсилилокси или гидроксиметил); или C1-C3-алкил, замещенный одной группой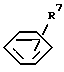 ,
,
(где R7 имеет указанные выше значения) и необязательно одним или двумя C1-C3-алкилами;
Y - -CH2-, -CH2CH2-, -CH=CH-, -CH2-CH=CH-, -CH=CH-CH2-, -C(CH3)=CH- или -CH=C(CH3)-;
Z - -O-CH-W-CH2-COP12,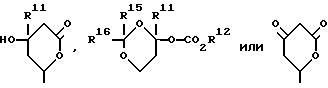 ,
,
(где O - -C(O)-, -C(OR13)2 или -CH(OH)-,
W - -C(O), =C(OR13)2 или -C(R11) (OH)=,
R11 - водород или C1-C3-алкил,
R12 - водород, R14 (где R14 представляет алкил химически или физиологически гидролизуемой части сложного алкилового эфира NHR23R24R25 (где R23, R24 и R25 - водород или C1-C4-алкил), натрий, калий или 1/2 кальций),
каждый R13 независимо представляет первичный или вторичный C1-C6-алкил, или два вместе образуют -(CH2)2- или -(CH2)3-,
R15 и R16 независимо друг от друга представляют атом водорода или C1-C3-алкил, либо R15 и R16 вместе образуют -(CH2)2- или (CH2)3-.
Такое соединение может иметь по крайней мере один или два асимметричных атома углерода и, следовательно, по меньшей мере два или более число оптических изомеров. Таким образом, соединение формулы (I) включает все оптические изомеры и их смеси.
Кроме того, среди соединений, в которых производный фрагмент карбоновой кислоты (-CO2R12) заместителя Z соединения формулы (I) изобретения имеет значение, отличное от -CO2R12 соединения, которые обладает способностью превращаться в карбоновую кислоту (соединение, в котором фрагмент -COR12 представляет -CO2H) под действием физиологического гидролиза после поглощения, считаются эквивалентными соединениями изобретения.
Соединения изобретения могут быть сильнодействующим ингибитором адгезии кровяных клеток (таких, как моноциты, макрофаги) к эндотелиальным клеткам и могут подавлять начальную стадию атеросклеротического утолщения внутренней оболочки сосудов. Кроме того, они способны подавлять миграцию клеток гладких мышц срединного слоя под действием PDGF и кондиционированной среды клеток гладких мышц (SMC-CM), которая может содержать фактор миграции клеток гладких мышц системы стимуляции производного движения. Кроме того, соединение по изобретению ингибирует поглощение 3H-тимидина клетками гладких мышц и, таким образом, подавляет репликацию ДНК, в результате чего достигается эффективное подавление пролиферации клеток. Такие воздействия являются более сильными в отношении I-SMC, чем в отношении M-SMC. Из вышесказанного следует, что соединения по изобретению подавляют пролиферацию клеток гладких мышц во внутренней оболочке аорты, которая представляет наиболее важную стадию атеросклеротического утолщения внутренней оболочки сосудов. В основе изобретения лежит открытие того, что соединения по изобретению оказывают ингибирующее действие в отношении атеросклеротического утолщения внутренней оболочки сосудов.
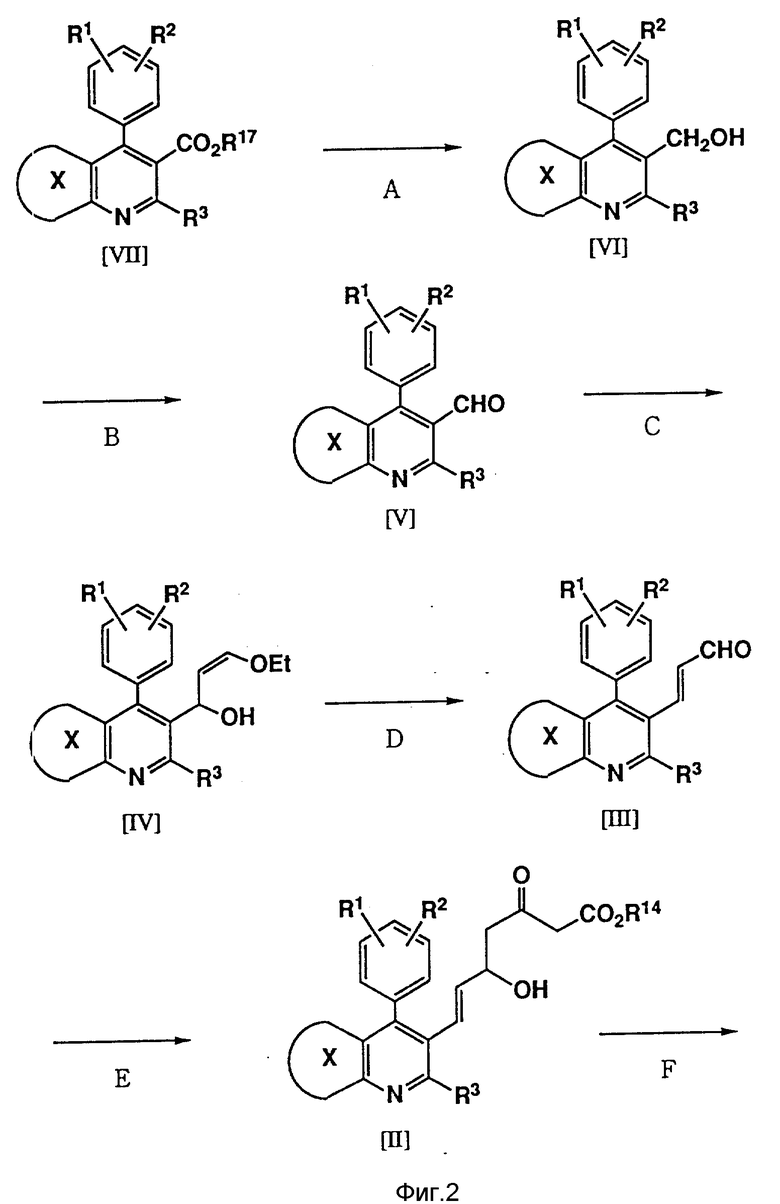
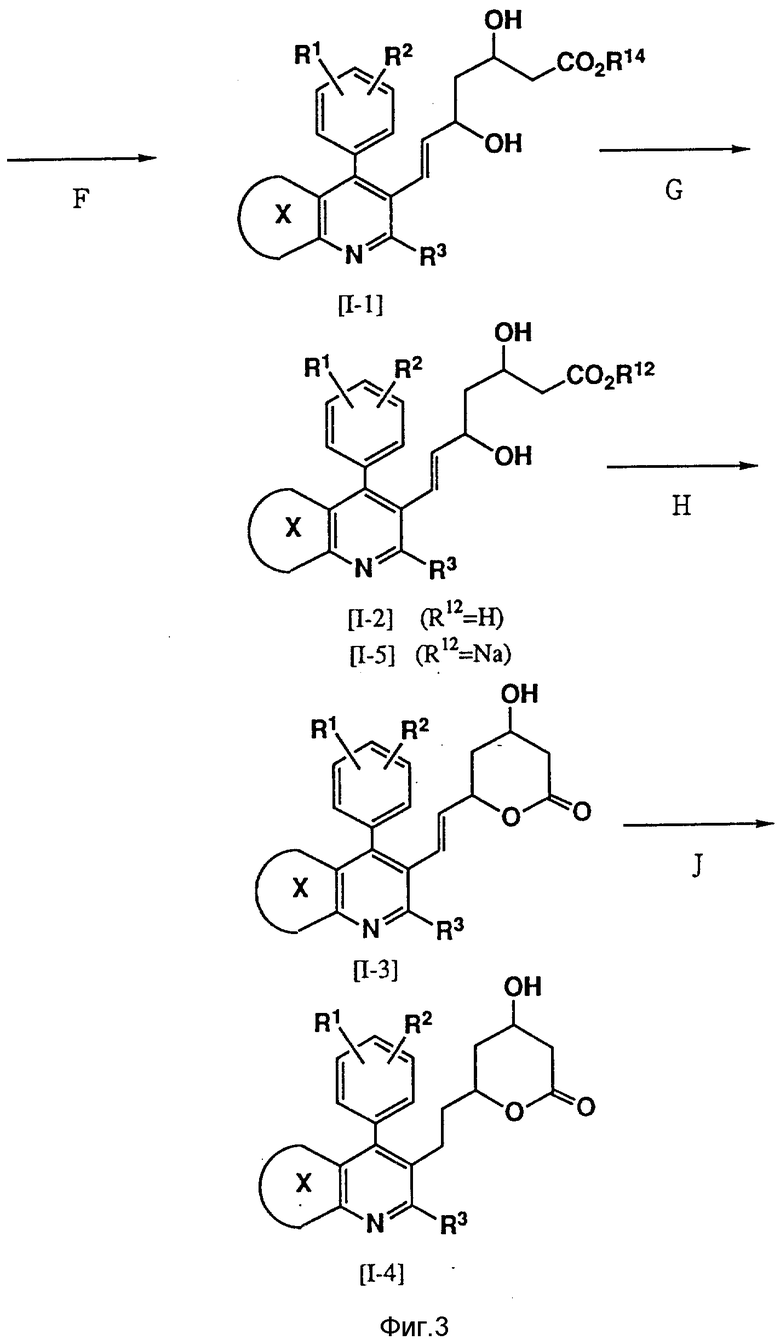
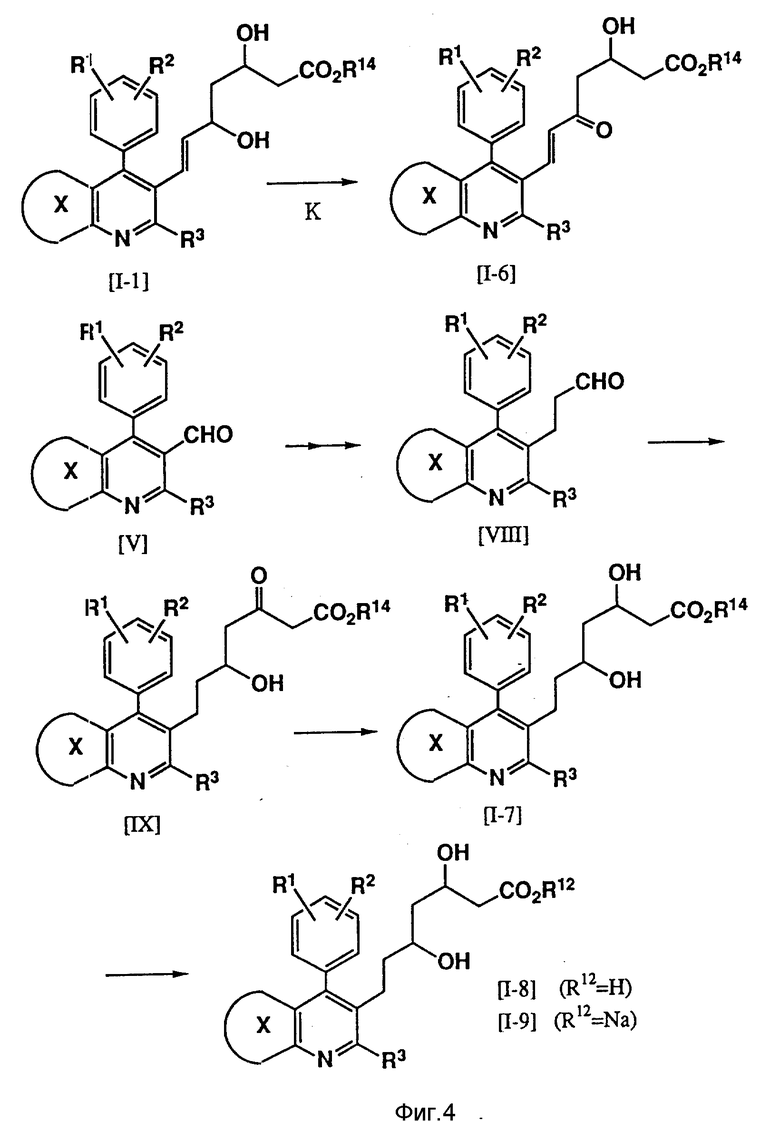
Соединения изобретения, то есть мевалоколактоны формулы (I), можно получить в результате осуществления следующих реакций (см. фиг. 1-4).
В формулах реакций R1, R2, R3, R12, R14 и кольцо X имеют значения, указанные выше для формулы (I), а R17 представляет С1-С4-низший алкил, такой, как метил, этил, н-пропил, изопропил или н-бутил.
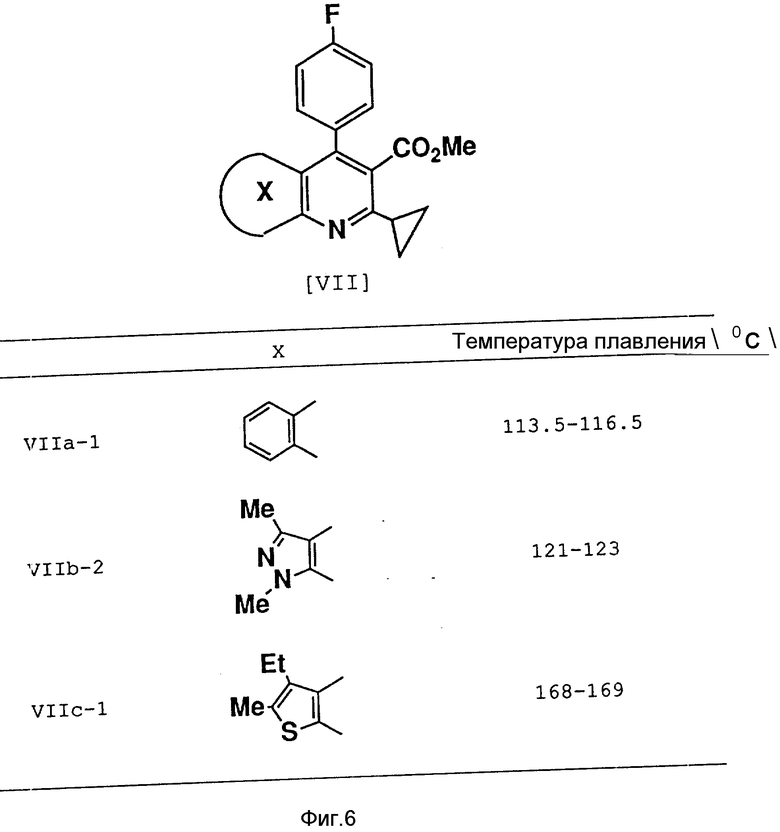
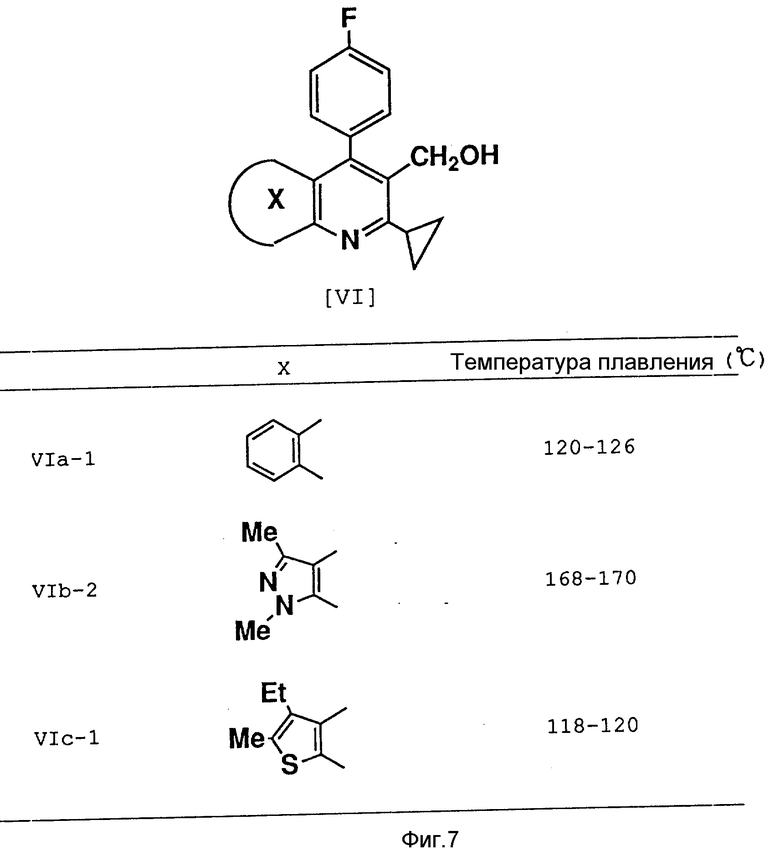
Соединения формул (VIIa), (VIIb) и (VIIc), относящиеся к соединениям формулы (I), можно получить в соответствии с методами, описанными в не прошедшей экспертизу в патентной публикации Японии N 279866 (1989), соответствующей европейскому патенту N 304063 и патенту США N 5011930, C 07 D 215/00), в нерассмотренной патентной публикации Японии N 275878 (1990), соответствующей европейскому патенту N 339358 и патенту США N 5024999) и в нерассмотренной патентной публикации Японии N 7290 (1991), соответствующей европейскому патенту N 367235 и патенту США N 5026698, A 61 K 31/435).
Стадия A представляет восстановление сложного эфира в первичный спирт, которое может осуществляться с помощью различных гидридов металлов, предпочтительно гидрида диизобутилалюминия, в таком растворителе, как тетрагидрофуран, толуол или метиленхлорид, при температуре от -20oC до 20oC, предпочтительно от -10oC до 10oC.
Стадия B представляет окисление первичного спирта в альдегид, которое может осуществляться с помощью различных оксидантов. Предпочтение отдается такому методу, при осуществлении которого реакция проводится с использованием хлорхромата пиридиния в метиленхлориде при 0 - 25oC, а также методу, в соответствии с которым реакция (реакция окисления Сверна) проводится с использованием оксалилхлорида, диметилсульфоксида и третичного амина (такого, как триэтиламин), и методу, в соответствии с которым реакция выполняется с использованием комплексного соединения триоксида серы и пиридина.
Стадия С представляет синтез производного 3-этокси-1-гидрокси-2-пропена, которое можно получить в результате взаимодействия соединения (V) с соединением лития, образуемым путем предварительной обработки цис-1-этокси-2-(три-н-бутилстаннил)этилена бутиллитием в тетрагидрофуране. В качестве температуры реакции предпочтение отдается низкой температуре в интервале от -60oC до -78oC.
Стадия D представляет синтез энала посредством кислотного гидролиза. При использовании кислотного катализатора, которым предпочтительно является паратолуолсульфокислота, хлористоводородная кислота или серная кислота, эта реакция может проводиться в смеси растворителей, состоящей из воды и тетрагидрофурана или этанола, при 10 - 25oC.
Производное 3-этокси-1-гидрокси-2-пропена, полученного на стадии C, можно без очистки использовать на стадии D, производя лишь удаление одновременно образовавшегося тетра-н-бутилолова.
Стадия E представляет реакцию присоединения эмали (III) и сложного эфира ацетоуксусной кислоты. Эта реакция предпочтительно проводится с использованием гидрида натрия и н-бутиллития, используемых в качестве основания, в тетрагидрофуране при температуре от -80oC до 0oC, предпочтительно от -30oC до -10oC.
Стадия F представляет реакцию, при осуществлении которой сложный эфир кетокарбоновой кислоты (II) восстанавливается с помощью различных восстановителей.
В соответствии с этой реакцией карбонильная группа восстанавливается с помощью борогидрида натрия, цианоборогидрида натрия, борогидрида цинка, дисиамилборана, диборана, третбутиламиноборана, пиридин-боранового комплекса, дициклогексилборана, тексилборана, 9-борабицикло (3.3.1) нонана, диизоцинокамфенилборана или три-втор-бутилборогидрида лития, в результате чего получают соответствующий сложный эфир дигидроксикарбоновой кислоты (I-1).
Эта реакция может осуществляться в растворителе, выбираемом из углеводорода, галогенированного углеводорода, C1-C4-спирта, простого эфира и смесей этих растворителей, при температуре от -100oC до 50oC, предпочтительно от -78oC до 30oC.
В противном случае можно использовать триалкилборан, такой, как три-н-бутилборан или триэтилборан и борогидрид натрия, при низкой температуре, как это описывается в журнале "J. Amer. Chem. Soc", 105, 593 (1983).
Кроме того, как указывается в "Tetrahedron Letters" 28, 155 (1987), можно предпочтительно получить эритропродукт, обладающий более сильным действием в биологическом отношении в результате использования алкоксидиалкилборана, такого, как метоксидиэтилборан или этоксидиэтилборан.
Эту реакцию можно осуществлять с использованием смеси растворителей, включающей С1-С4 спирт и тетрагидрофуран, при температуре от -80oC до -50oC, предпочтительно от -72oC до -68oC.
Стадия G представляет гидролиз сложного эфира, и эта реакция может осуществляться с использованием эквимолярного количества основания, предпочтительно гидроксида калия или гидроксида натрия, в смеси растворителей, включающей воду и метанол или этанол, при 10 - 25oC.
Полученная на этой стадии свободная кислота может взаимодействовать с приемлемым основанием с образованием соли.
Стадия H представляет дегидратацию свободной оксикислоты (I-2) с образованием мевалонолактона, и эта реакция может осуществляться в результате нагрева с обратным холодильником в бензоле или толуоле при одновременном удалении образовавшейся воды или в результате добавления приемлемого осушителя, такого, как молекулярное сито.
В противном случае эта реакция может осуществляться с использованием лактонообразующего вещества, такого, как карбодиимид, предпочтительно водорастворимый карбодиимид, например N-циклогексил-N'-[2'-(метилморфолин)этил] карбодиимидо- паратолуолсульфонат, в сухом метиленхлориде при 10 - 35oC, предпочтительно 20 - 25oC.
Стадия J представляет гидрогенизацию двойной связи, соединяющей мевалонолактоновую часть с гетероциклическим кольцом, которую можно осуществлять с использованием каталитического количества палладированного угля или родированного угля в таком растворителе, как метанол, этанол, тетрагидрофуран или ацетонитрил, при 0 - 50oC, предпочтительно 10 - 25oC.
Стадия K представляет реакцию получения α,β -ненасыщенного кетона в результате избирательного окисления сложного эфира дигидроксикарбоновой кислоты, которая может осуществляться с использованием активированного диоксида марганца в таком растворителе, как простой этиловый эфир, тетрагидрофуран, бензол или толуол, при 20 - 80oC, предпочтительно 40 - 80oC.
В противном случае соединение формулы (I-6) можно синтезировать из альдегида формулы (V) с помощью реакции сочетания Вадсворта - Эммонса (J. Amer. Chem. Soc, 107, 3731, (1985)).
Кроме того, это соединение можно синтезировать из энала формулы (III) (Tetrahedron Letters, 26, 2951, (1985)). Соединение формулы (I-7) также можно получить путем добавления двойного аниона сложного эфира цетоуксусной кислоты, производя эту операцию так же, как на стадии E, к альдегиду (VIII), синтезированному из альдегида формулы (V) с помощью непрерывной реакции Биттинга (0-8402131), в результате чего образуется сложный эфир кетокарбоновой кислоты (IX), и последующего восстановления карбоксильной группы так же, как на стадии F.
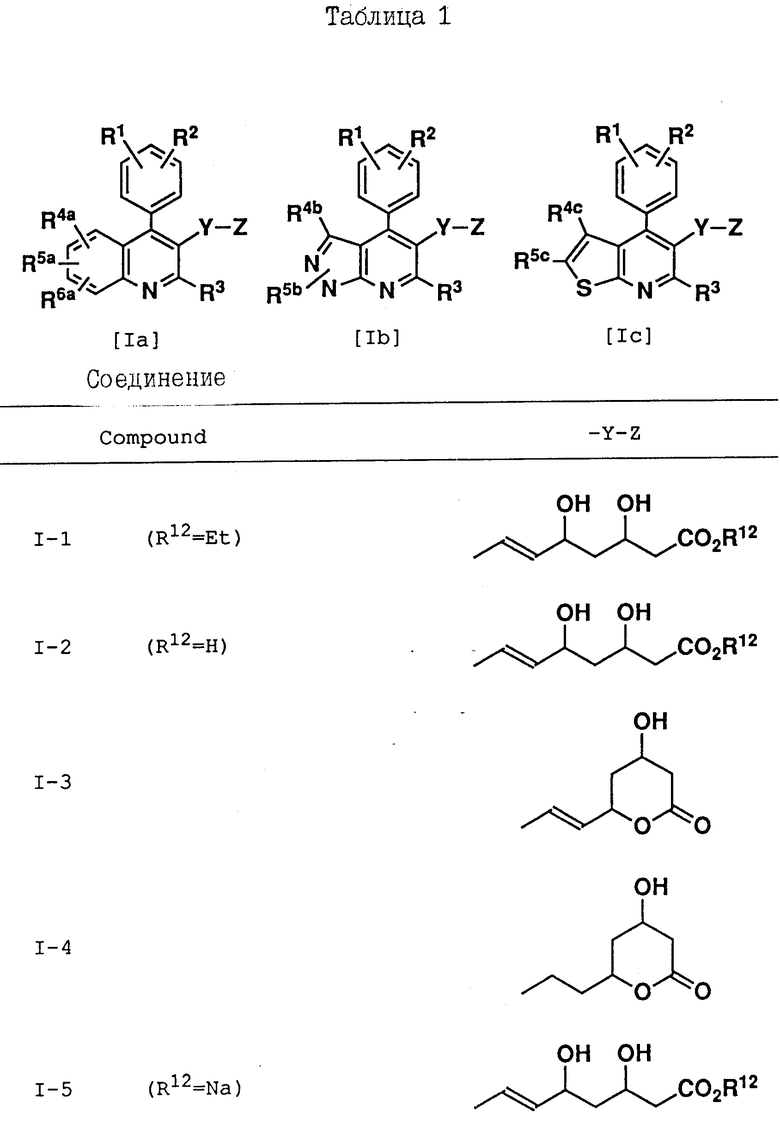
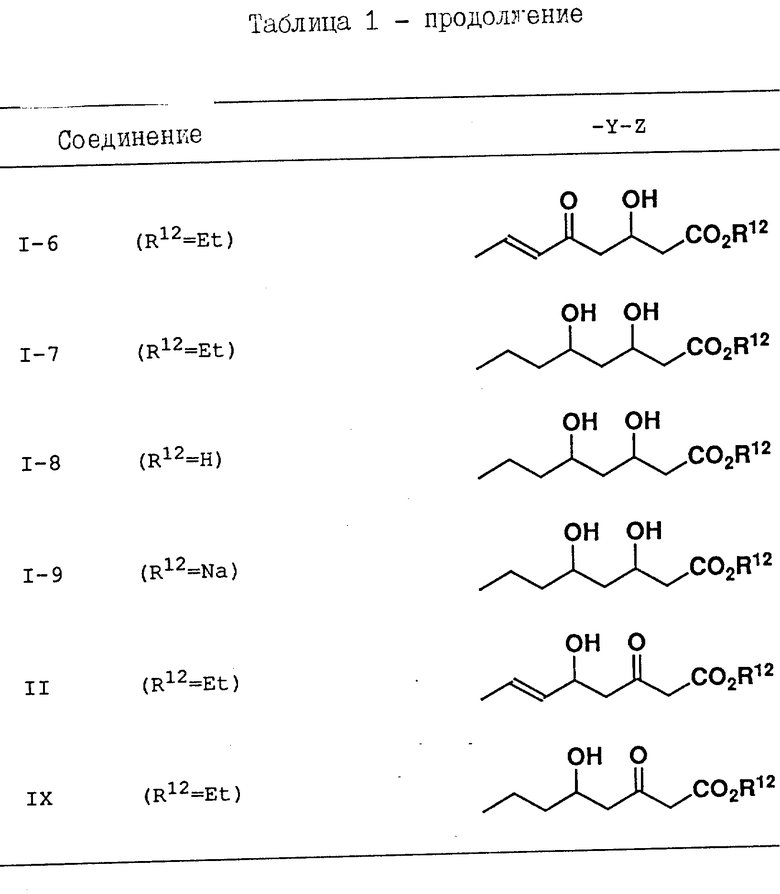
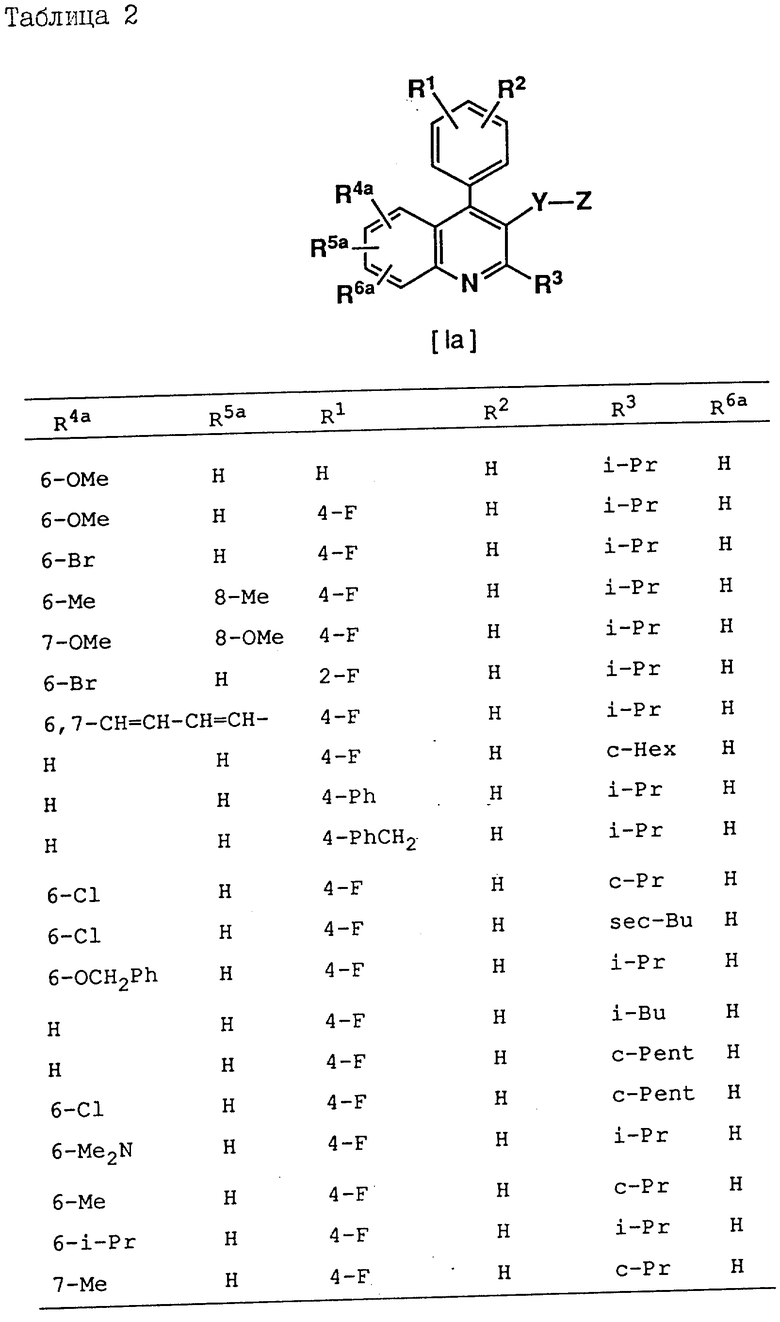
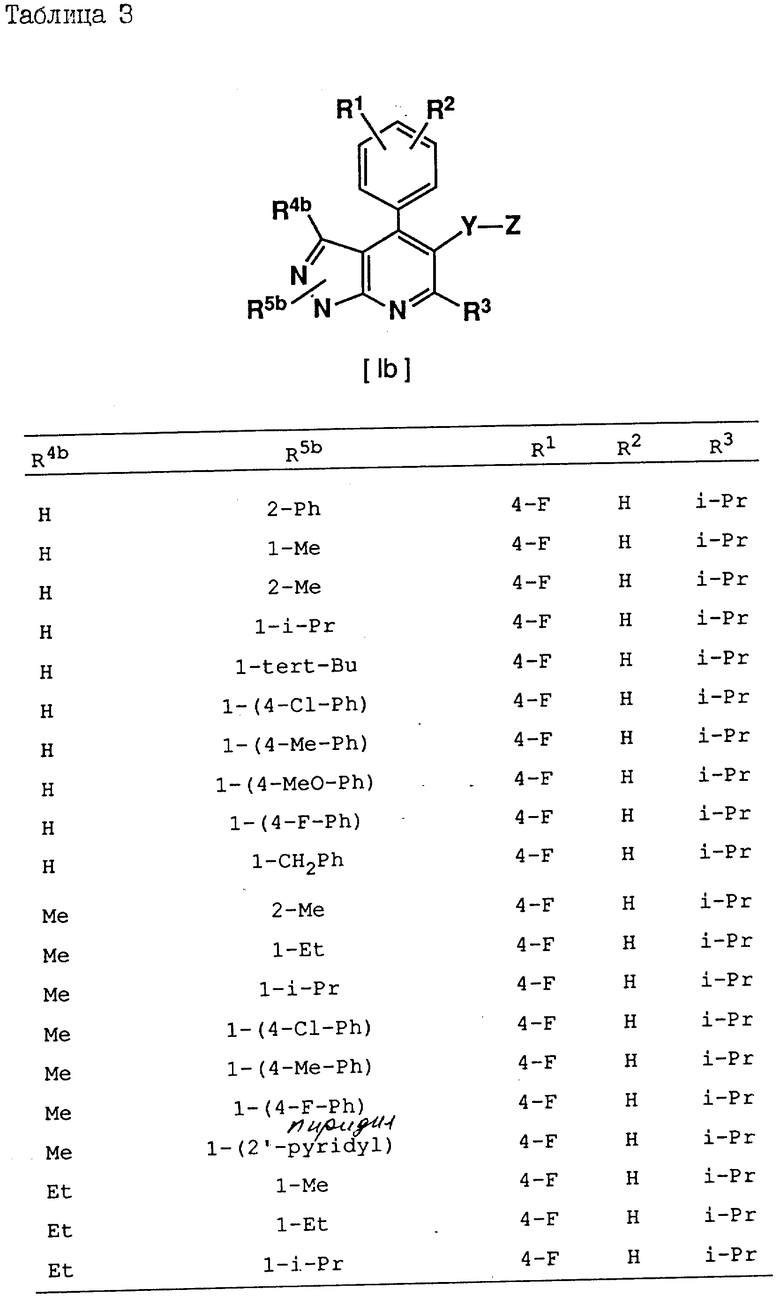
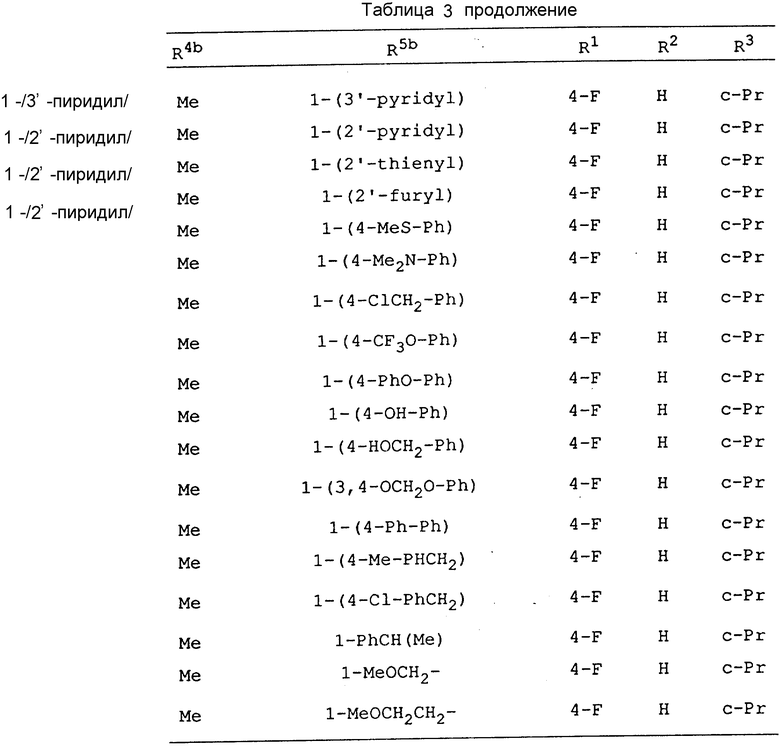
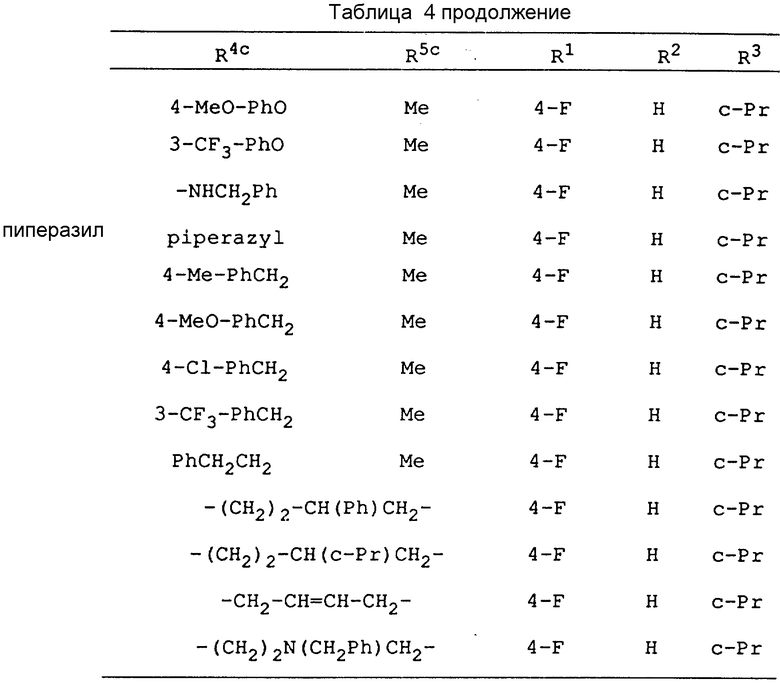
Соединения, приведенные в табл. 1 - 4, включая соединения, рассматриваемые в последующих примерах, являются всего лишь определенными примерами соединения по изобретению. Используемый в этих формулах заместитель -Y-Z приведен в табл. 1, а другие заместители представлены в табл. 2-4.
В следующих определениях заместителей различные символы обозначают следующие заместители, а именно n - представляет нормальный, i-изо, sec-вторичный, tert - третичный, c - цикло, Me - метил, Et - этил, Pr - пропил, Bu - бутил, Pent - пентил, Hex - гексил и Ph - фенил.
Кроме того, аналогичным образом можно получить фармакологически приемлемые соли, такие, как соли калия или соли полукальция, сложные эфиры, такие, как сложные метиловые эфиры, сложные н-пропиловые эфиры, сложные изопропиловые эфиры, сложные циклопропиловые эфиры, сложные н-бутиловые эфиры, сложные изобутиловые эфиры, сложные втор-бутиловые эфиры, сложные трет-бутиловые эфиры, сложные н-пептиловые эфиры, сложные изопентиловые эфиры или сложные н-гексиловые эфиры, и соли аммония, соли триметиламина, соли диэтиламина, соли пиперазина, соли морфолина, соли пиперидина, соли аурамина, соли диаурамина или соли триметамина, этих соединений.
Соединения по изобретению обладают не только высоким ингибирующим действием в отношении биосинтеза холестерина в том случае, когда HMG-CoA редуктаза выступает в качестве фермента, ограничивающего скорость роста, но и ингибирующим действием в отношении миграции M-MC, пролиферации 1-SMC и адгезии кровяных клеток к эндотериальным клеткам, как показывают результаты испытаний, приводимые ниже. Таким образом, соединения по изобретению являются полезными лечебными средствами против гиперлипемии, гиперлипопротеинами и атеросклероза.
Из этих соединений могут быть изготовлены различные приемлемые составы в зависимости от способа введения. Соединения по изобретению могут вводиться в форме свободных кислот или в форме физиологически гидролизуемых и приемлемых сложных эфиров или лактонов либо в форме фармацевтически приемлемых солей.
Фармацевтический состав по изобретению предпочтительно вводят перорально в виде самого соединения по изобретению или в виде порошков, гранул, таблеток и капсул, получаемых в результате смешения соединения по изобретению с фармацевтически приемлемым носителем, включая такое связующее, как гидроксипропилцеллюлоза, сироп, аравийская камедь, желатин, сорбит, трагант, поливинилпирролидон или карбоксиметилцеллюлозно-кальциевый комплекс, такой наполнитель, как лактоза, сахар, кукурузный крахмал, фосфат кальция, сорбит, глицин или порошкообразная кристаллическая целлюлоза, такую смазку, как стеарат магния, тальк, полиэтиленгликоль или диоксид кремния, и такой дезинтегратор, как картофельный крахмал.
Однако фармацевтический состав по изобретению не ограничивается пероральным введением и пригоден для парентерального введения. Например, он может вводиться в форме суппозитория, изготовленного с использованием масляной основы, такой, как масло какао, полиэтиленгликоль, ланолин или триглицерид жирной кислоты, трансдермальной лечебной основы, изготавливаемой из жидкого парафина, белого вазелина, высшего спирта, макрогеловой мази, гидрофильной мази или гидрогелевого основного материала, а также в форме состава для инъекций, изготавливаемого с использованием одного или нескольких материалов, выбираемых из группы, включающей полиэтиленгликоль, гидрогелевый основной материал, дистиллированную воду, дистиллированную воду для инъекций, и наполнитель, такой, как лактоза или кукурузный крахмал, либо в форме состава, предназначенного для введения через слизистую оболочку, такую, как слизистая оболочка глаза и слизистая оболочка рта.
Кроме того, соединения по изобретению можно соединять с основными ионообменными смолами, способными связывать желчные кислоты, которые при этом не адсорбируются желудочно-кишечным трактом.
Суточная доза соединения формулы I составляет 0,05 - 500 мг, предпочтительно 0,5 - 50 мг для взрослого человека. Ее введение производится один - три раза в день. Эта доза, несомненно, может изменяться в зависимости от возраста, веса и состоянии больного.
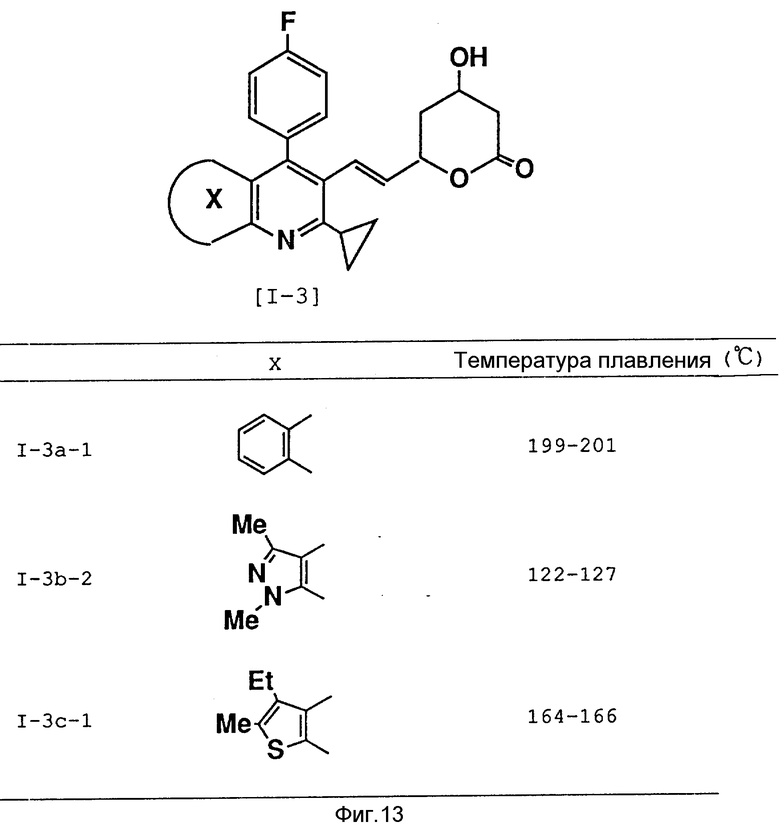
Ниже приводится более детальное описание изобретения со ссылкой на примеры, демонстрирующие ингибирующее действие соединений по изобретению в отношении атеросклеротического утолщения внутренней оболочки сосудов. Испытуемые соединения по соединению (испытуемые соединения I-3) и сравнительные соединения (правастатин, описываемый в публикации нерассмотренного патента Японии N 185275/1982 или в европейском патенте N 65835, и симвастатин, описываемый в публикации нерассмотренного патента Японии N 122373/1984 или в европейском патенте N 33536), имеют химические структуры представленные на фиг. 5.
Справочный пример
(E) - транс-6-/2'-[4''-/4'''/фторфенил/-1'',3''-диметил-6''- /1'''-метилетил/пиразоло/3,4-b/пиридин-5''-ил] этенил/4-гидрокси- 3,4,5,6-тетрагидро-2H-пиран-2-он (соединение I-3b-1)
Это соединение было получено с использованием в качестве исходного материала метил-2-циклопропил-5-этил-3-/4'-фторфенил/-6-метилтиено/2,3-b/ пиридин-3-ил-карбкосилата (соединение IIIb-1) в результате осуществления стадий A - H.
Аналогичным образом были получены испытуемые соединения 1, 2 и 3 из следующих промежуточных соединений (VIIa-1, VII-2 и VIIc-1) (см. фиг. 6):
Соединение VIIa-1
Метил-2-циклопропил-4-(4'-фторфенил)(хинолин-3-ил-карбкосилат.
Соединение VIIb-2
Метил-6-циклопропил-1,3-диметил-4-/4'-фторфенил/пиразоло-3,4- пиридин-5-ил-карбоксилат
Соединение VIIc-1
Метил-6-циклопропил-3-этил-4-/4'-фторфенил/-2-метилтиено-2,3- пиридин-5-ил-карбоксилат.
Испытуемое соединение 1 (I/3a-1)
(E)-транс-6-/2'[2''-циклопропил-4''/4'''-фторфенил/хинолин-3''- ил]этенил/-4-гидрокси-3,4,5,6-тетрагидро-2H-пиран-2-он
Испытуемое соединение 2 (I-3b-2)
(E)-транс-6-/2'[6''-циклопропил/1'', 3''-диметил-4''-/4'''- фторфенил/пиразоло[3,4-b] пиридин-5''-ил] этенил/-4-гидрокси-3,4,5,6- тетрагидро-2H-пиран-2-он
Испытуемое соединение 3(I-3c-1)
(E)-транс-6-(2'-/2''-циклопропил-5''-этил-3''-/4'''-фторфенил/- 6''-метилтиено[2,3-b] пиридин-3''-ил]этенил/-4-гидрокси-3,4,5,6- тетрагидро-2H-пиран-2-он
Стадия A
4-/4'-фторфенил/-5-гидроксиметил-1,3-диметил-6-/1'-метилэтил/- пиразоло[3,4-b]пиридин /соединение VI-b-1/
5,0 г (0,014 моля) соединения VIIb-1 растворяли в сухом толуоле, производя эту операцию в атмосфере азота, и охлаждали до 0oC в ледяной бане. К этому раствору по каплям добавляли 35 мл раствора толуола, содержащего 16 мас. % гидрида диизобутилалюминия, после чего эту смесь перемешивали в течение 2 ч при температуре 0oC. После подтверждения посредством тонкослойной хроматографии полного исчезновения соединения VIIb-1 для окончания реакции к указанной смеси добавляли при 0oC насыщенный раствор хлорида аммония. К реакционной смеси добавляли простой этиловый эфир, после чего отделяли органический слой. К желатинированному веществу добавляли водный раствор гидроксида натрия с целью его растворения, после чего полученный раствор экстрагировали простым этиловым эфиром. Экстракт простого этилового эфира собирали, сушили над безводным сульфатом магния и фильтровали, а растворитель отгоняли с образованием 3,9 г целевого продукта желтоватого цвета.
Выход: 88%, температура плавления: 174 - 175oC.
Аналогичным образом были получены соединения, представленные на фиг. 7.
Стадия B
[4-/4'-фторфенил/-1,3-диметил-6-/1'-метиленэтил/пиразоло[3,4-b] пиридин-5-ил]карбоксиальдегид (соединение Vb-1)
4,2 (19 ммолей) хлорхромата пиридиния, 0,69 г безводного ацетата натрия и 3,8 г (12 ммолей) соединения VIb-1 суспендировали в 50 мл сухого дихлорметана при комнатной температуре. Реакционный раствор перемешивали в течение одного часа, после чего добавляли к нему 100 мл простого этилового эфира и тщательно перемешивали. Реакционную смесь фильтровали под вакуумом через слой перита, после чего фильтрат выпаривали до сухого состояния при пониженном движении. Остаток очищали посредством хроматографии на колонках из силикагеля (элюент : хлороформ), в результате чего было получено 2,9 г целевого продукта желтоватого цвета.
Выход: 78%, температура плавления: 144-146oC.
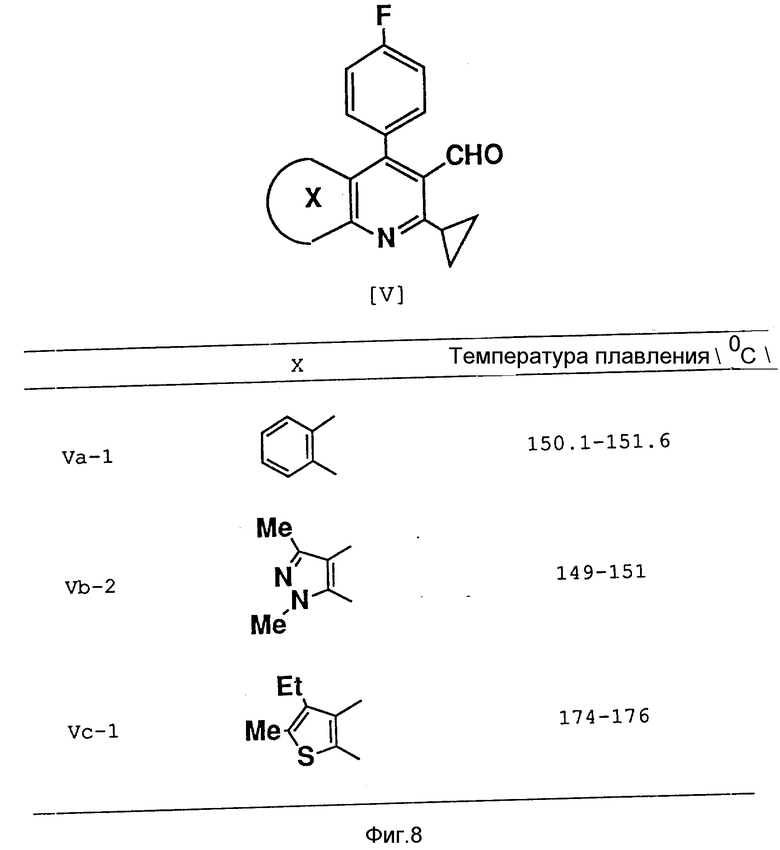
Аналогичным образом были получены соединения, представленные на фиг. 8.
Стадии C и D
(E)-3-[4'-/4''-фторфенил/-1', 3'-диметил-6'-/1''-метилэтил/- пиразоло[3,4-b]пиридин-5'-ил]пропенальдегид (соединение III-1)
Стадия C
1,45 г (40 ммолей) цис-1-этокси-2-(три-н-бутилстаннил) этилена растворяли в 50 мл сухого тетрагидрофурана, после чего полученный раствор охлаждали до -78oC в потоке азота. К этому раствору по каплям добавляли 26 мл (40 ммолей) раствора 15 мас.% н-бутиллития в н-гексане. Эту смесь перемешивали в течение 20 мин. Затем раствор, содержащий 2,5 г (8 ммолей) соединения Vb-1, растворенного в 20 мл сухого тетрагидрофурана, по каплям добавляли к вышеуказанной смеси. Реакционную смесь перемешивали в течение одного часа при температуре -78oC, после чего для окончания реакции добавляли 26 мл насыщенного раствора хлорида аммония. Органический слой экстрагировали простым диэтиловым эфиром, эфирный экстракт промывали насыщенным водным раствором хлорида, отгоняли при пониженном давлении, а остаток подвергали жидкостному разделению между н-гексаном и ацетонитрилом, после чего слой ацетонитрила перегоняли при пониженном давлении, при этом отгоняли растворитель, что позволило получить в основном чистое соединение IVb-1.
Стадия D
Соединение IVb-1, полученное на стадии C, растворяли в 70 мл тетрагидрофурана и к этому раствору добавляли 20 мл воды и 3 г паратолуолсульфокислоты. Эту смесь перемешивали в течение 2 ч при комнатной температуре. Реакционный раствор тщательно нейтрализовали водным раствором гидроксида натрия, а затем несколько раз экстрагировали простым диэтиловым эфиром. Экстракт промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. После этого растворитель отгоняли при пониженном давлении. Остаток очищали посредством хроматографии на колонках из силикагеля элюент: смесь этилацетат и н-гексана = 1/9 (в объемном отношении), в результате чего был получен целевой продукт желтого цвета.
Количество: 2,2 г (выход: 79%), температура плавления: 133-134oC.
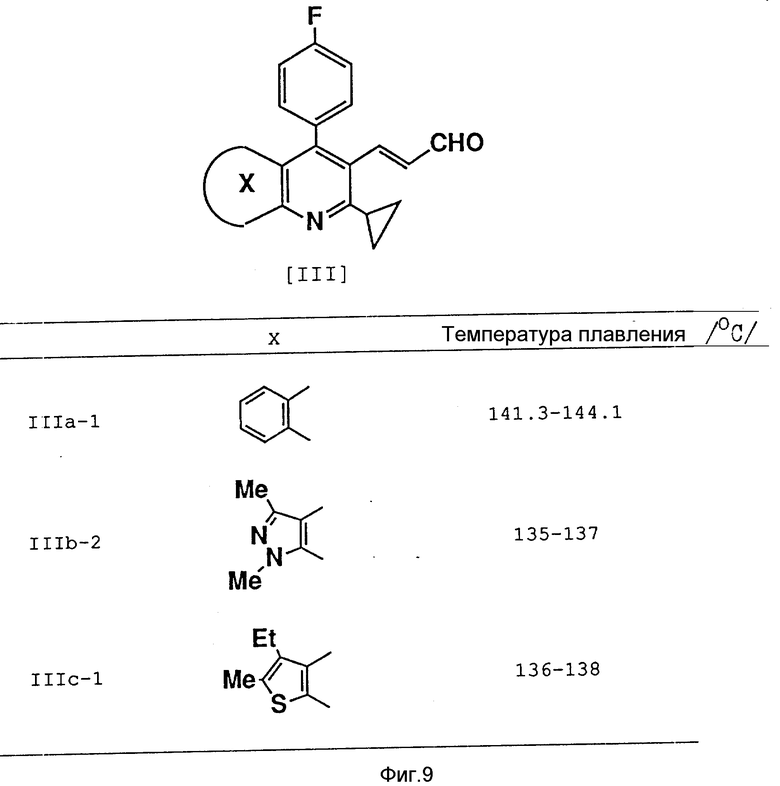
Аналогичным образом были получены соединения, представленные на фиг. 9.
Стадия E
Этил-/E/-7-[4'-/4''фторфенил/-1', 3'-диметил-6-'-/1''- метилэтил/-пиразоло[3,4-b]пиридин-5-ил]-5- гидрокси-3-оксогепта-6-эноат (соединение IIb-1)
1,25 г 60% гидрида натрия промывали высушенным петролейным эфиром, сушили в потоке азота, а затем суспендировали в 200 мл сухого тетрагидрофурана. Полученную суспензию охлаждали до -15oC в атмосфере азота, после чего к ней по каплям добавляли 3,9 мл (30 ммолей) этилацетоацетоната. Эту смесь перемешивали в течение 15 мин. Затем к ней по каплям добавляли 20 мл (30 ммоль) раствора 15 мас.% н-бутиллития в н-гексане и перемешивали смесь в течение 30 мин. Кроме того, в эту смесь по каплям добавляли раствор, содержащий 2,1 Г (6,1 ммоля) соединения IIIb-1, растворенного в сухом тетрагидрофуране, и перемешивали в течение одного часа. К реакционной смеси добавляли 10 мл насыщенного водного раствора хлорида аммония при температуре -15oC, после чего ее трижды экстрагировали простым диэтиловым эфиром. Эфирный раствор промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния, а затем выпаривали до сухого состояния при пониженном давлении. Остаток очищали посредством хроматографии на колонках из силикагеля (элюент: смесь этилацетата и хлороформа = 1/9 (в объемном отношении).
В результате было получено 2,5 г (выход: 89%) целевого продукта белого цвета. Температура плавления: 95 - 98oC.
Аналогичным образом были получены соединения, представленные на фиг. 10.
Стадия F
Этил-(E)-7-[4'-/4''-фторфенил/-1',3'-диметил-6'-/1''-метилэтил)- пиразоло[3,4-b]пиридил-5'-ил-3,5-дигидроксигепта-6-эноат (соединение I-1b-1)
2,32 г (4,96 ммоля) соединения IIb-1 растворяли в 20 мл этанола, выполняя эту операцию в атмосфере азота, после чего полученный раствор охлаждали до 0oC. Затем к нему добавляли 740 мг (20 ммолей) борогидрида натрия и перемешивали смесь в течение одного часа. К этой смеси добавляли 10% водный раствор хлористоводородной кислоты, что позволяло произвести тщательную нейтрализацию смеси. Эту смесь трижды экстрагировали простым этиловым эфиром. Эфирный раствор промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния, а затем выпаривали до сухого состояния при пониженном давлении. Остаточное масло очищали посредством хроматографии на колонках из силикагеля (элюент: смесь этанола и хлороформа = 3/97 (в объемном отношении), что позволило получить чистый целевой продукт в виде бесцветного вязкого масла.
Количество: 1,81 г (выход: 78%).
Спектр ЯМР (6 частей на миллион CDCl3): 1,28 (т., J = 8 Гц, 3H), 1,32 (д. , J = 8 Гц, 6H), 1,4 - 1,8 (м., 1H), 1,92 (с., 3H), 2,2 - 2,6 (м., 3H), 2,9 - 3,8 (м., 2H), 3,42 (7 гептет, J=8 Гц, 1H), 4,06 (с., 3H), 4,1 - 4,6 (м., 4H), 5,1 - 5,5 (м., 1H), 6,4 - 6,7 (м., 1H), 6,9 - 7,3 (м., 4H).
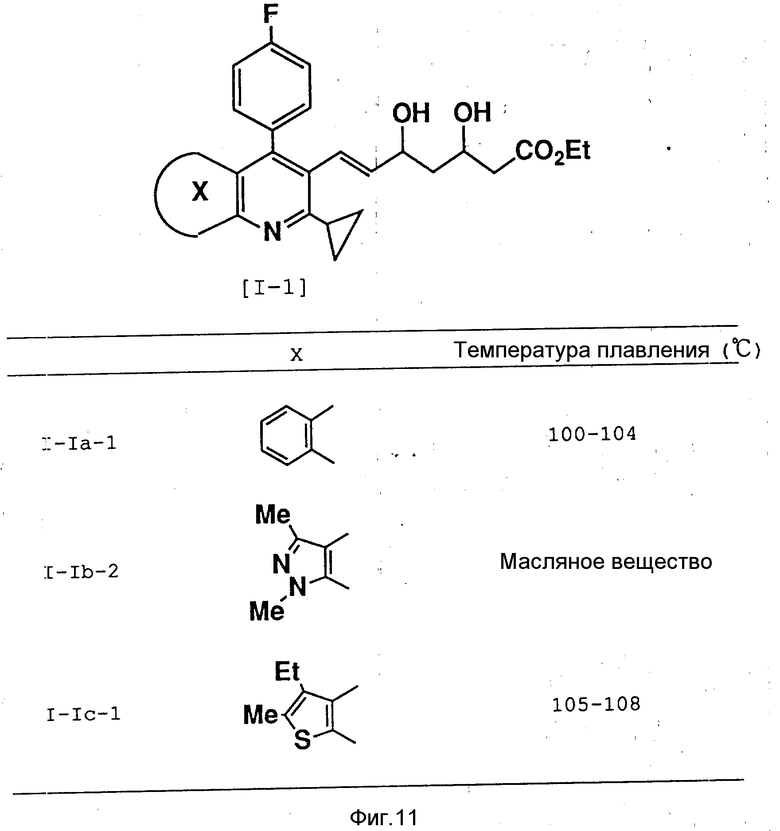
Аналогичным образом были получены следующие соединения, представленные на фиг. 11.
Стадия G
Натриевая соль (E)-7- [4''-фторфенил/-1', 3'-диметил-6'-1''- метилэтил/пиразоло [3,4-b] пиридин-5'-ил/-3,5-дигидроксигепте-6-эноевой кислоты (соединение I-5b-1)
200 мг (0,43 ммоля) соединения I-1b-1 растворяли в 2 мл этанола и к полученному раствору по каплям добавляли 0,85 мл 0,5 н. водного раствора гидроксида натрия. Эту смесь продолжали перемешивать при комнатной температуре в течение одного часа. Затем при пониженном давлении отгоняли этанол и добавляли к остатку 2 мл воды. Полученную смесь экстрагировали простым этиловым эфиром. Водный слой сушили вымораживанием, что позволило получить 180 мг (91%) гигроскопичного порошка желтоватого цвета.
Температура плавления: 258 - 264oC (разложение).
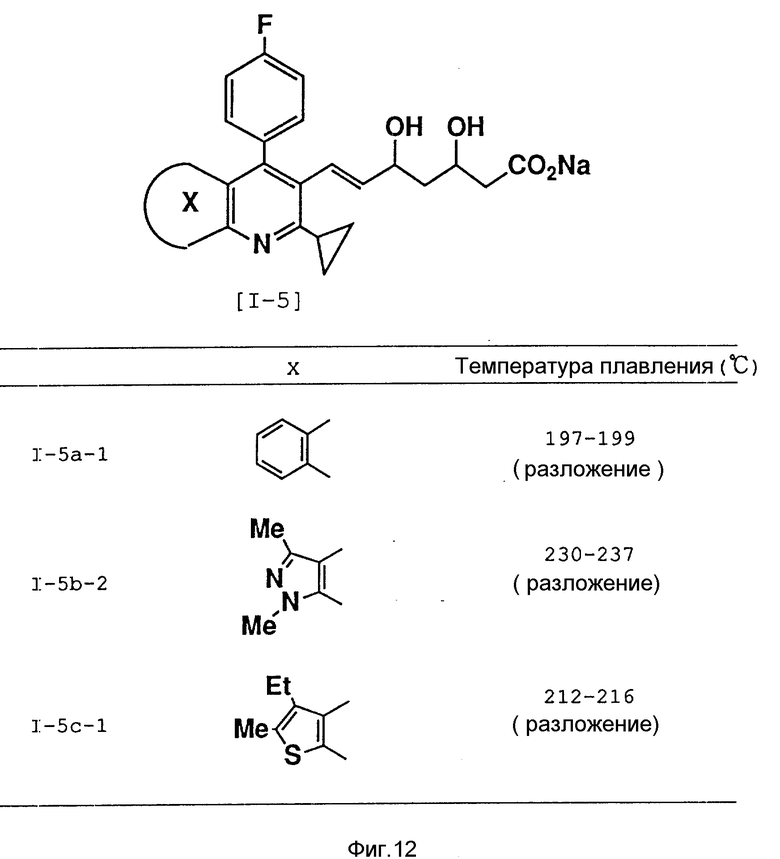
Аналогичным образом были получены соединения, представленные на фиг. 12.
(E)-7-[4'/4''-фторфенил/-1', 3'-диметил-6'-/1''-метилэтил/- пиразоло[3,4-b] пиридин-5'-ил/-3,5-дигидроксигепта-6-эноевая кислота (соединение I-2b-1)
0,25 г (0,53 ммоля) соединения I-1b-1 растворяли в 3 мл этанола и к полученному раствору по каплям добавляли 1,06 мл 0,5 н. водного раствора гидроксида натрия. Этанол отгоняли при пониженном давлении, а к остатку добавляли 3 мл дистиллированной воды. После этого смесь экстрагировали простым этиловым эфиром. Водный слой тщательно нейтрализовали 1% хлористоводородной кислотой, а затем экстрагировали простым этиловым эфиром. Эфирный слой сушили над безводным сульфатом магния и перегоняли при пониженном давлении с образованием целевого продукта.
Количество: 0,21 г (выход 90%).
Спектр импульсного ЯМР (DMCO - d6) δ частей на миллион: 1,29 (д., J = 7 Гц, 6H), 1,83 (с., 3H), 2,1 - 2,3 (м., 2H), 2,4 - 2,6 (м., 1H), 3,0 - 3,6 (м. , 4H), 3,96 (с., 3H), 4,3 - 4,8 (м., 2H), 5,2 - 5,6 (м., 1H), 6,3 - 6,6 (м., 1H), 7,2 - 7,4 (м., 4H), 11,5 - 12,0 (широкий синглет, 1H).
Стадия H
(E)-транс-6-/2'-[4''-/4'''-фторфенил/-1'', 3''-диметил-6''-1'''- метилэтил/пиразоло[3,4 - b)пиридин-5''-ил/этенил/-4-гидрокси-3,4,5,6- тетрагидро-2H-пиран-2-он-(соединение I-3b-1)
130 мг (0,29 ммоля) соединения I-2b-1 растворяли в 6 мл дихлорметана и к полученному раствору добавляли 125 мг (0,29 ммоля) N-циклогексил-N'-/2''-метилморфолиноэтил/ карбодиимид-паратолуолсульфоната. Полученную смесь перемешивали при комнатной температуре в течение 2 ч, после чего при пониженном давлении отгоняли растворитель до сухого состояния. Остаточное масло очищали посредством тонкослойной хроматографии на силикагеле (элюент: смесь гексана и этилацетата = 9/1 (в объемном отношении), что позволило получить чистый целевой продукт в виде бесцветного вязкого масла.
Количество: 47 мг (выход: 39%).
Спектр импульсного ЯМР (CDCl3) δ частей на миллион: 1,33 (д., J = 6,8 Гц, 6H), 1,4 - 1,5 (м., 1H), 1,6 - 1,7 (м., 2H), 1,93 (с., 3H), 2,5 - 2,6 (м. , 1H), 2,68 (двойной дублет, J = 18 Гц, J = 5 Гц, 1H), 3,39 (7 гептет, J = 6,8 Гц, 1H), 1,07 (с., 8H), 4,1 - 4,2 (м., 1H), 5,1 - 5,2 (м., 1H), 5,31 (двойной дублет, J = 16 Гц, J = 6 Гц, 1H), 6,61 (двойной дублет, J = 16 Гц, J = 1,5 Гц, 1H), 7,1 - 7,3 (м., 4H).
Аналогичным образом были получены следующие соединения, представленные на фиг. 13.
Пример 1. Ингибирующее действие в отношении миграции клеток гладких мышц срединного слоя аорты (M-SMC)
Ингибирующее действие соединений по изобретению в отношении миграции M-SMC определяли с помощью следующих методов.
Срезы срединного слоя грудной аорты самца крысы вида "Spragne Dawley" культивировали в среде Игла, модифицированной Дульбеко (DME), содержащей 10% сыворотки плода коровы (FBS), при температуре 37oC в атмосфере, состоящей из 95% воздуха и 5% углекислого газа, в течение 3 или 4 недель. Клетки гладких мышц срединного слоя аорты, выращенные из срезов срединного слоя артерии, инокулировали путем разделения плотности клеток в отношении 1:2 с целью получения стабильной субкультуры. После трех- или четырехкратной инокуляции соединенные клетки обрабатывали трипсином и суспендировали в вышеуказанной среде при плотности клеток, равной 500000 клеткам на мл. К взвеси клеток добавляли испытуемое соединение, растворенное в диметилсульфоксиде (DMCO), в результате чего конечная концентрация DMCO составила 0,2%, после чего взвесь клеток предварительно инкубировали при температуре 37oC в течение 30 мин. Для получения контрольной пробы добавляли один DMCO в такой же концентрации. В нижнее отделение камеры Бойдена, разделенной нитроцеллюлозной мембраной, вводили 10 мг/мл фактора роста тромбоцитов (PDGF) или среды DME, представляющей 10% среду DME, стандартизированную клетками гладких мышц (SMC-CM), полученную в результате 48-часовой стандартизации, которую использовали в качестве фактора миграции. В верхнее отделение указанной камеры вводили 1 мл взвеси клеток и производили инкубацию при температуре 37oC в течение 4 ч в условиях культивирования. Для получения холостой пробы в нижнее отделение вводили среду DME без фактора миграции. После 4-часовой инкубации удаляли клетки, прилипшие к верхней стороне нитроцеллюлозной мембраны, а клетки, переместившиеся к нижней стороне мембраны, фиксировали и окрашивали "Diff Quik". Число переместившихся клеток подсчитывали в 10 полях с помощью светового нитроскопа при 400 x поле зрения под большим увеличением.
Полученные результаты представляли в виде среднего значения числа клеток в трех камерах, полученного в каждом случае, а величину ингибирования (%) высчитывали в соответствии со следующей формулой:
Ингибирование (%) - {100 - (T-B) (C-B) • 100}
где
B - число клеток в холостой пробе, C - число клеток в контрольной пробе, T - число клеток в пробе, содержащей испытуемое соединение.
Результаты приведены в табл. 5. Соединения по изобретению продемонстрировали сильное ингибирующее действие в отношении миграции M-SMC по сравнению со сравнительным соединением.
Пример 2. Ингибирующее действие в отношении пролиферации клеток гладких мышц внутренней оболочки и срединного слоя аорты (1-SMC и SMC)
Ингибирующее действие соединений по изобретению в отношении пролиферации I-SMC и M-SMC определяли с помощью следующих методов.
При использовании среды DME, содержащей 10% сыворотки плода коровы и антибиотик, срезы срединного слоя аорты, полученные у здорового японского белого кролика, и срезы пораженного участка внутренней оболочки аорты, выделенные из срединного слоя, взятого у японского белого кролика, пораженного атеросклерозом, культивировали так же, как это описывалось в примере 1. После двух- и трехкратной инокуляции соединенные клетки обрабатывали трипсином и суспендировали в вышеуказанной среде, так что плотность клеток составляла 20000 клеток/мл. Затем производили посев 10000 клеток, которые культивировали в чашке с 24 ячейками. После инкубации в течение 6 ч вышеуказанную среду заменяли 0,5 мл контрольной среды или среды, содержащей испытуемое соединение. В это время подсчитывали число клеток, первоначально прикрепившихся к чашке, которое принимали за исходное число клеток (I). К среде добавляли испытуемое соединение, как это описано в примере 1. Для получения контрольной пробы вводили один DMCO, так чтобы конечная концентрация составила 0,2%. Замену среды производили через каждые 2 дня, при этом в первый, второй, третий, пятый и седьмой день клетки обрабатывали трипсином и суспендировали в изотоническом растворе, после чего определяли число клеток с помощью счетчика Каултера.
Результаты представлены в виде среднего числа клеток в трех ячейках, полученного в каждом случае, а величину ингибирования (%) процесса увеличения числа клеток со второго по пятый день высчитывали в соответствии со следующей формулой:
Ингибирование (%) = 100 = {(T5/T2/ /C5/C2/ • 100}
где
T2 и T5 - число клеток соответственно во второй и пятый день в среде, содержащей испытуемое соединение, C2 и C5 - число клеток соответственно во второй и пятый день в контрольной среде.
Полученные результаты приведены в табл.6. Соединения по изобретению продемонстрировали сильное ингибирующее действие в отношении пролиферации как I-SMC, так и M-SMC по сравнению со сравнительным соединением. Кроме того, ингибирующее действие с отношением пролиферации I-SMC было выше по своей эффективности, чем в отношении пролиферации M-SMC.
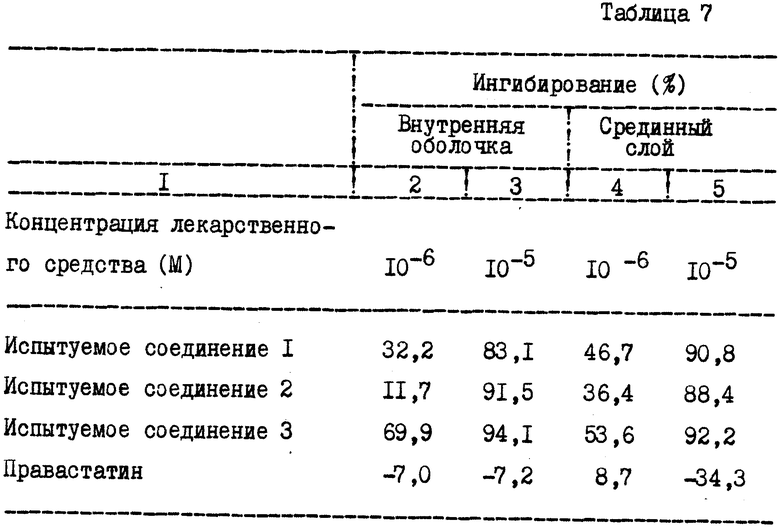
Пример 3. Ингибирующее действие в отношении поглощения 3H-тримидина клетками гладких мышц внутренней оболочки и срединного слоя аорты (I-SMC и M-SMC)
Ингибирующее действие соединения по изобретению в отношении поглощения 3H-тимидина клетками гладких мышц внутренней оболочки и срединного слоя аорты определяли с помощью следующих методов.
M-SMC и I-SMC получали из срединного слоя аорты здорового японского кролика и из внутренней оболочки аорты японского белого кролика, страдающего атеросклерозом, как это указывалось в примере 2. После трех- или четырехкратной инокуляции соединенные клетки обрабатывали трипсином и суспендировали в среде DME, содержащей 10% сыворотки плода коровы и антибиотик, так что плотность клеток составляла 40000 клеток/мл. 10000 клеток высевали в чашку с 48 ячейками. После культивирования в течение 4 дней вышеуказанную среду заменяли контрольной средой или средой, содержащей испытуемое соединение, аналогичное используемому в примере 2, после чего культивирование продолжали в течение 24 ч. Затем добавляли 1 μCi (37 MBg) 3H-тимидина и продолжали культивирование в течение 3 ч. Клетки трижды промывали фосфатно-солевым буферным раствором (PBS) и обрабатывали охлажденной 5% трихлоруксусной кислотой (TCA). Нерастворимую фракцию промывали охлажденной TCA и растворяли в 0,5 н. водном растворе гидроксида калия. Радиоактивность 3H измеряли с помощью жидкостного сцинтилляционного счетчика и определяли содержание белка.
Результаты высчитывали в виде величины радиоактивности на 1 мг белка и представляли в качестве среднего значения для 3 ячеек. Величину ингибирования (%) высчитывали в соответствии со следующей формулой:
Ингибирование (%) = 100 -T/C • 100
Полученные результаты приведены в табл. 7. Соединения по изобретению продемонстрировали сильное ингибирующее действие в отношении поглощения 3H-тимидина фракцией ДНК в клетках гладких мышц внутренней оболочки и срединного слоя аорты по сравнению со сравнительным соединением.
Пример 4. Ингибирующее действие в отношении адгезии лейкозных клеток (HL-60)
Ингибирующее действие соединений по изобретению в отношении адгезии клеток HL-60 определяли с помощью следующих методов. Клетки HL-60 культивировали в среде RPMI 1640, содержащей 10% сыворотки плода коровы и антибиотик, при температуре 37oC в атмосфере, состоящей из 95% воздуха и 5% углекислого газа, после чего эти клетки высевали в чашку с 6 ячейками в количестве 2 мл на каждую ячейку при плотности клеток, равной 1000000/мл. Испытуемое соединение, растворенное в DMCO, добавляли в таком количестве, что конечная концентрация DMCO достигала 0,2%, и продолжали инкубацию в течение 48 ч в качестве предварительной обработки. Затем 500000 клеток высевали в чашку с 24 ячейками, добавляли 0,02 мМ форбол-миристат-ацетата (TPA), растворенного в этаноле, в количестве 1/250 (в объемном отношении) культуральной среды и продолжали культивирование в течение 12 ч. Вслед за этим клетки промывали фосфатно-солевым буферным раствором, клетки, прикрепившиеся к ячейкам чашки, удаляли путем обработки трипсином и суспендировали в изотоническом растворе, после чего определяли число клеток с помощью счетчика Каултера. Контрольное испытание выполняли при добавлении одного DMCO.
Результаты представляли в виде среднего значения числа клеток в трех ячейках, полученного во время предварительной обработки, а величину ингибирования (%) высчитывали в соответствии со следующей формулой:
Ингибирование (%) адгезии = 100 - T/C•100
где
T - число клеток в среде, содержащей испытуемое соединение,
C - число клеток в контрольной среде.
Полученные результаты приведены в табл. 8. Соединения по изобретению продемонстрировали сильное ингибирующее действие в отношении адгезии клеток HL-60 под действием TPA по сравнению со сравнительным соединением.
Пример 5. Ингибирующее действие в отношении адгезии макрофагов
J 774 (774-МФ)
Ингибирующее действие соединений по изобретению в отношении адгезии клеток J774-МФ определяли с помощью следующих методов.
1000000 клеток высевали в чашку с 6 ячейками и культивировали в 1 мл среды DME, содержащей 10% сыворотки плода коровы и антибиотик, при температуре 37oC в атмосфере, состоящей из 95% воздуха и 5% углекислого газа, в течение 2 дней. Затем культивирование продолжали в контрольной среде, содержащей 0,2% DMCO, или в среде, содержащей испытуемое соединение, что представляло предварительную обработку. Затем клетки удаляли резиновым скребком и суспендировали в вышеуказанной среде. 200000 клеток высевали в чашку с 24 ячейками и культивировали в течение 12 ч. После этого клетки, прикрепившиеся к ячейкам чашки, удаляли резиновым скребком и с помощью счетчика Каултера определяли число клеток.
Результаты представляли в виде среднего значения числа клеток в трех ячейках, полученного в каждом случае во время предварительной обработки, а величину ингибирования (%) адгезии высчитывали в соответствии со следующей формулой:
Ингибирование (%) адгезии = 100 - T/C • 100
где
T - число клеток в среде, содержащей испытуемое соединение, C - число клеток в контрольной среде.
Полученные результаты приведены в табл. 9. Соединения по изобретению продемонстрировали сильное ингибирующее действие в отношении адгезии клеток J774-МФ.
Соединения по изобретению оказывают ингибирующее действие на HMG-CoA редуктазу и ингибируют атеросклеротическое утолщение внутренней оболочки сосудов, поэтому они являются полезными профилактическими средствами, предупреждающими возникновение коронарных болезней сердца, таких, как стенокардия, инфаркт миокарда, повторный атеноз после PTCA, сокращение цереброваскулярных сосудов после внутримозгового кровоизлияния и облитерирующий склеротический артериит.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРАЗОЛО(3,4-B)ПИРИДИНА | 1989 |
|
RU2022964C1 |
| ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ | 1994 |
|
RU2128043C1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕВАЛОНОЛАКТОНОВЫХ ПРОИЗВОДНЫХ | 1989 |
|
RU2045529C1 |
| ОПТИЧЕСКИ-АКТИВНЫЙ β-АМИНОАЛКОКСИБОРАНОВЫЙ КОМПЛЕКС, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ОПТИЧЕСКИ АКТИВНОЕ β-АМИНОСПИРТОВОЕ ПРОИЗВОДНОЕ ДЛЯ ЕГО ПОЛУЧЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ СПИРТОВ С УЧАСТИЕМ КОМПЛЕКСА | 1994 |
|
RU2126412C1 |
| ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ТЕРАПЕВТИЧЕСКИМ ЭФФЕКТОМ НА МНОЖЕСТВЕННУЮ МИЕЛОМУ | 2011 |
|
RU2583430C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ АМИДНОЕ СОЕДИНЕНИЕ | 2014 |
|
RU2681941C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СТАБИЛИЗИРОВАННАЯ ОСНОВНЫМ АГЕНТОМ | 1996 |
|
RU2142790C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПРОТИВОДЕЙСТВИЯ ТАХИКИНИНОВЫМ РЕЦЕПТОРАМ | 1994 |
|
RU2135471C1 |
| ПРОИЗВОДНОЕ УРАЦИЛА | 1992 |
|
RU2040523C1 |
| Способ получения производных 3(2Н)-пиридазинона | 1988 |
|
SU1584750A3 |

































Изобретение касается применения нижеуказанных соединений в качестве ингибитора пролиферации клеток гладкой мышцы аорты, миграции средних клеток гладкой мышцы аорты в ее интиму (внутреннюю оболочку) или адгезии кровяных клеток и эндотермию. Предложенные соединения, известные ранее, представляют собой:
9 табл., 13 ил.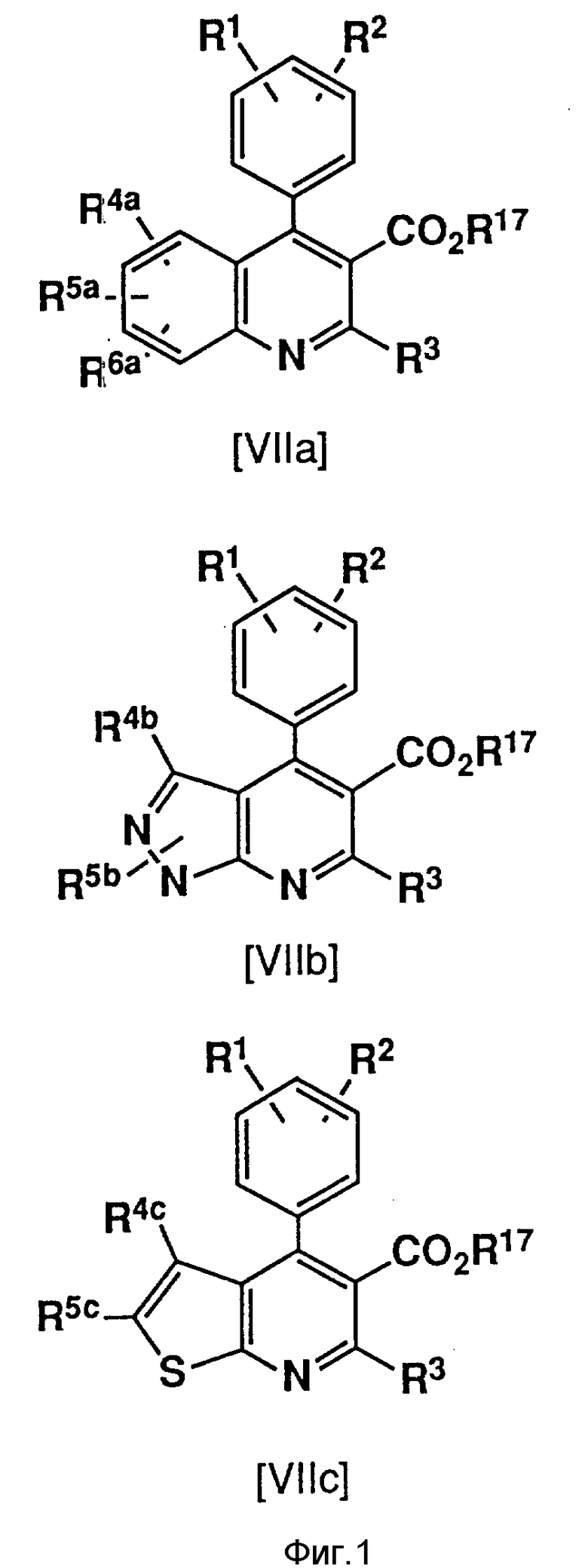
в которой кольцо X представляет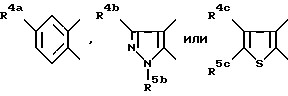
где R4a - водород, C1 - C6-алкил, фтор, хлор или бром;
R4b - С1 - С6-алкил или фенил;
R5b - С1 - C8-алкил;
R4c и R5c - водород или C1 - C8-алкил;
R1 - водород, фтор, хлор или бром;
R3 - C1 - C7-алкил или C3 - C7-циклоалкил;
Y - -CH2CH2- или -CH=CH-;
Z - -CH(OH)-CH2 - CH(OH)-CH2 -CO2R12,
где R12 представляет водород, алкил химически или физиологически гидролизуемого фрагмента сложного алкилового эфира, NH4, натрий, калий или 1/2 кальция,
или
в качестве ингибитора пролиферации клеток гладкой мышцы аорты, миграции срединных клеток гладкой мышцы аорты в ее интиму (внутреннюю оболочку) или адгезии кровяных клеток к эндотелию.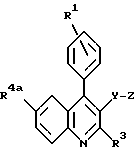
6. Применение по любому из пп.1 - 4, при котором указанным соединением является соединение формулы Ib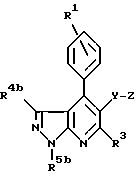
7. Применение по любому из пп.1 - 4, при котором указанным соединением является соединение формулы Ic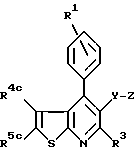
8. Применение по п.5, при котором в формуле Ia R1 - фтор, R3 - циклопропил, Y - -CH=CH-.
| Biochimical and Biophysical Reseorch Communications | |||
| Приспособление, увеличивающее число оборотов движущихся колес паровоза | 1919 |
|
SU146A1 |
| Изолирующее кольцо для патрона Эдисона, предохраняющее электрическую лампу накаливания от вывертывания | 1922 |
|
SU802A1 |
| The Gournol of Biological Chemisty | |||
| Способ нагрева эквипотенциального катода в электронных вакуумных реле | 1921 |
|
SU266A1 |
| Biochim Biophis Aeta | |||
| СКЛАДНАЯ НИВЕЛЛИРОВОЧНАЯ РЕЙКА | 1923 |
|
SU560A1 |

