ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к гетероциклическому соединению, имеющему новую структуру, которое может быть использовано для предупреждения или лечения заболеваний, вызванных аномалиями в активности PRS (пролил-тРНК-синтетаза), к способу его получения и содержащей его фармацевтической композиции.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
PRS (пролил-тРНК-синтетаза) является одной из семейства аминоацил-тРНК-синтетаз (ARS) и служит для активации аминокислоты при синтезе белка. То есть ARS выполняет трансляционную функцию для образования аминоацил-аденилата (AA-AMP), а затем переносит активированную аминокислоту на 3-конец соответствующей тРНК. Поскольку ARS играет важную роль в синтезе белка, то, если ARS ингибируется, рост всех клеток подавляется. Таким образом, АРС признана в качестве перспективной цели в качестве терапевтического агента при лечении заболеваний, которые должны подавлять антибиотики или чрезмерную экспрессию клеток (Nature, 494: 121-125).
PRS присутствует или функционирует как комплекс мультисинтетазы (MSC) в виде EPRS (глутамил-пролил-тРНК-синтетаза). В частности, среди различных MSC EPRS функционирует как трансляционный глушитель, который подавляет продукцию VEGF (фактор роста эндотелия сосудов A), который является ключевым фактором ангиогенеза. Кроме того, сообщается, что EPRS тесно связана с различными солидными опухолями (Nat. Rev. Cancer, 2011, 11, 708-718).
Единственным веществом, известным в качестве ингибитора PRS, является галофугинон. Галофугинон является производным фебрифугина, получаемого из натуральных продуктов, и обладает противомалярийным действием и различными противовоспалительными эффектами. Его можно также использовать в качестве корма для животных. В настоящее время галофугинон клинически изучается как противораковый агент, противовоспалительный агент (J Immunol, 2014, 192 (5), 2167-76), терапевтический агент для лечения аутоиммунных заболеваний (Arthritis Rheumatol, 2014, 66 (5), 1195-207) и терапевтический агент для лечения фиброзных заболеваний (World J Gastroenterol, 2014, 20 (40), 14778-14786) (Bioorg Med. Chem., 2014, 22, 1993-2004).
Однако сообщалось, что галофугинон действует на различные мишени и обладает очень сильной токсичностью, и, кроме того, существует риск генотоксичности (EFSA Journal, 2003, 8: 1-45). Таким образом, обнаружение ингибиторов PRS, обладающих повышенной безопасностью для организма человека среди веществ, способных ингибировать PRS, подобных галофугинону, имеет значение с точки зрения разработки противоракового агента следующего поколения, который может быть использован в качестве противофиброзного агента, противовоспалительного агента, аутоиммунного терапевтического агента отдельно или в комбинации с существующим целевым противораковым средством.
В связи с этим авторы настоящего изобретения провели многочисленные исследования для разработки нового соединения с пониженной токсичностью при наличии ингибирующего фермент PRS действия и обнаружили, что соединение, имеющее новую структуру, которое описано далее, избирательно ингибирует PRS, и таким образом осуществили настоящее изобретение. Соединения, относящиеся к настоящему изобретению, сами по себе главным образом обладают ингибирующей фермент PRS активностью, но не исключают возможности проявления фармакологического действия в качестве эффективного агента посредством специальной среды организма или продуктов метаболического процесса после абсорбции в организм.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
Настоящее изобретение относится к гетероциклическому соединению, имеющему новую структуру, которое может быть использовано для предупреждения или лечения злокачественных новообразований, воспалительных заболеваний, аутоиммунных заболеваний или фиброза, к способу их получения и содержащей его фармацевтической композиции.
ТЕХНИЧЕСКОЕ РЕШЕНИЕ ЗАДАЧИ
Для достижения вышеуказанных целей в настоящем изобретении предложено соединение, представленное следующей химической формулой 1, или его фармацевтически приемлемая соль:
[Химическая формула 1]
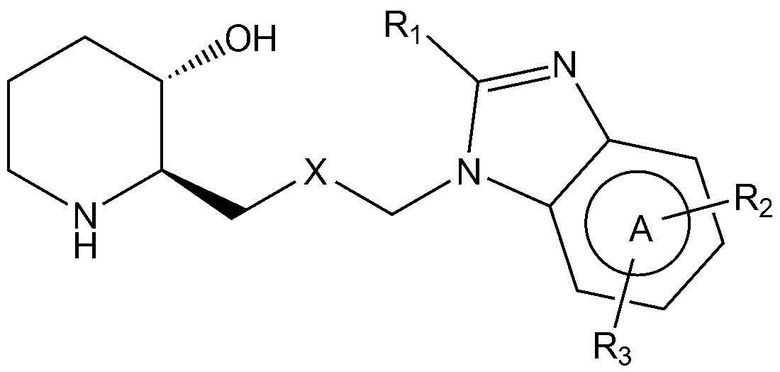
где:
A представляет собой бензольное кольцо или пиридиновое кольцо,
X представляет собой CO или CHOH,
R1 представляет собой водород или C1-4 гидроксиалкил,
R2 представляет собой фенил, пиразолил, пиридин-2-онил, пирролидинил или тиазолил,
где R2 является незамещенным или замещен одним или двумя заместителями, каждый из которых независимо выбран из группы, включающей C1-4 алкил, C1-4 алкокси, C1-4 галогеналкил, галоген и циано, и
R3 представляет собой водород или C1-4 алкил.
Предпочтительно, A, вместе с имидазольным кольцом, конденсированным с A, образует структуру 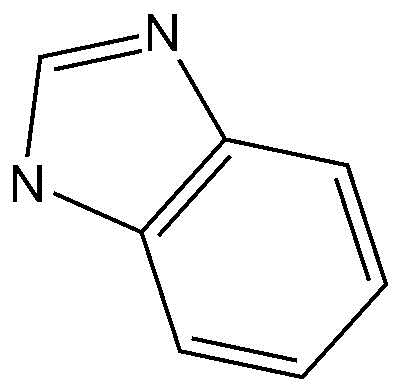 ,
, 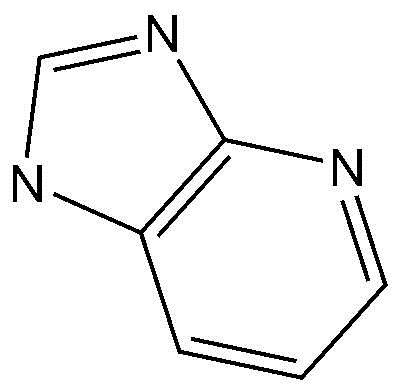 ,
, 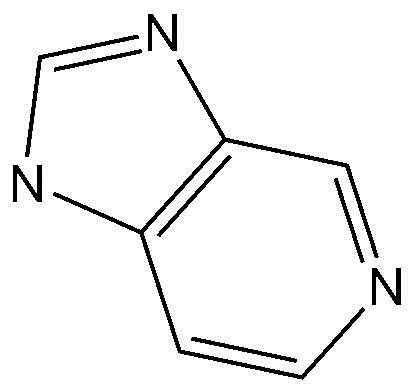 или
или  .
.
Также предпочтительно, R1 представляет собой водород или гидроксиметил.
Также предпочтительно, R2 представляет собой фенил, незамещенный или замещенный одним или двумя заместителями, каждый из которых независимо выбран из группы, включающей C1-4 алкил, C1-4 алкокси, C1-4 галогеналкил, галоген и циано; незамещенный пиразолил; незамещенный пиридин-2-онил; незамещенный пирролидинил; или незамещенный тиазолил.
Также предпочтительно, R2 является незамещенным или замещен одним или двумя заместителями, каждый из которых независимо выбран из метила, метокси, трифторметила, фтора, хлора и циано.
Также предпочтительно, R3 представляет собой водород или метил.
Также предпочтительно,
A представляет собой бензольное кольцо,
X представляет собой CO, или CHOH,
R1 представляет собой водород или C1-4 гидроксиалкил,
R2 представляет собой фенил, пиразолил, пиридин-2-онил или тиазолил,
где R2 является незамещенным или замещен одним или двумя заместителями, каждый из которых независимо выбран из группы, включающей C1-4 алкил, C1-4 алкокси, C1-4 галогеналкил, галоген и циано; и
R3 представляет собой водород или C1-4 алкил.
Также предпочтительно,
A, вместе с имидазольным кольцом, конденсированным с A, образует,
X представляет собой CO,
R1 представляет собой водород,
R2 представляет собой фенил,
где R2 замещен C1-4 галогеналкилом или галогеном, и
R3 представляет собой водород.
Также предпочтительно,
A, вместе с имидазольным кольцом, конденсированным с A, образует  ,
,
X представляет собой CO,
R1 представляет собой водород,
R2 представляет собой фенил или пирролидинил,
где R2 является незамещенным или замещен одним или двумя заместителями, каждый из которых независимо выбран из C1-4 галогеналкила и галогена, и
R3 представляет собой водород.
Также предпочтительно,
A, вместе с имидазольным кольцом, конденсированным с A, образует ,
X представляет собой CO или CHOH,
R1 представляет собой водород,
R2 представляет собой фенил,
где R2 замещен C1-4 галогеналкилом или галогеном, и
R3 представляет собой водород.
Характерные примеры соединений, представленных химической формулой 1, являются следующими:
1) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-он,
2) 1-(4-(3-фторфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
3) 1-(4-(3-хлорфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
4) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-(3-(трифторметил)фенил)-1H-имидазо[4,5-c]пиридин-1-ил)пропан-2-он,
5) 1-(4-(3-хлор-5-фторфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
6) 1-(4-(3,5-дихлорфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
7) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-(пирролидин-1-ил)-1H-имидазо[4,5-c]пиридин-1-ил)пропан-2-он,
8) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-он,
9) 1-(5-(2-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
10) 1-(5-(3-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
11) 1-(5-(4-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
12) 1-(5-(3-хлорфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
13) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-(3-(трифторметил)фенил)-1H-имидазо[4,5-b]пиридин-1-ил)пропан-2-он,
14) 1-(6-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
15) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(3-(трифторметил)фенил)-3H-имидазо[4,5-b]пиридин-3-ил)пропан-2-он,
16) 1-(5-(3-фторфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
17) 1-(5-(3-хлорфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
18) 1-(5-(2-хлорфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
19) 1-(5-(4-хлорфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
20) 3-(1-(3-((2R,3S)-3-гидроксипиперидин-2-ил)-2-оксопропил)-4-метил-1H-бензо[d]имидазол-5-ил)бензонитрил,
21) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-метил-5-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
22) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-(3-метоксифенил)-4-метил-1H-бензо[d]имидазол-1-ил)пропан-2-он,
23) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-метил-5-(тиазол-4-ил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
24) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-он,
25) 1-(6-(3-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
26) 1-(6-(2-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
27) 1-(6-(3-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
28) 1-(6-(4-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
29) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
30) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(м-толил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
31) 1-(6-(3,5-дихлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
32) 1-(6-(3-хлор-5-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
33) 1-(6-(3-хлор-4-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
34) 1-(6-(3-хлор-5-метилфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
35) 1-(6-(3-хлор-5-метоксифенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
36) 3-хлор-5-(1-(3-((2R,3S)-3-гидроксипиперидин-2-ил)-2-оксопропил)-1H-бензо[d]имидазол-6-ил)бензонитрил,
37) 1-(6-(1H-пиразол-4-ил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
38) 1-(6-(1H-пиразол-3-ил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
39) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(тиазол-4-ил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
40) 5-(1-(3-((2R,3S)-3-гидроксипиперидин-2-ил)-2-оксопропил)-1H-бензо[d]имидазол-6-ил)пиридин-2(1H)-он,
41) 1-(6-(3-хлорфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
42) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(3-(трифторметил)фенил)-1H-имидазо[4,5-b]пиридин-1-ил)пропан-2-он,
43) 1-(5-(2-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
44) 1-(5-(3-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
45) 1-(5-(4-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
46) 1-(5-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
47) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-(3-(трифторметил)фенил)-3H-имидазо[4,5-b]пиридин-3-ил)пропан-2-он,
48) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-он,
49) 1-(7-(3-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
50) 1-(7-(3-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
51) 1-(7-(4-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
52) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-(2-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
53) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
54) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-(4-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
55) 1-(2-(гидроксиметил)-5-фенил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
56) 1-(5-(3-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
57) 1-(2-(гидроксиметил)-6-фенил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
58) 1-(6-(2-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
59) 1-(6-(3-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
60) 1-(6-(4-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
61) 1-(2-(гидроксиметил)-6-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
62) (2R,3S)-2-(3-(6-(3-хлорфенил)-1H-бензо[d]имидазол-1-ил)-2-гидроксипропил)пиперидин-3-ол, и
63) (2R,3S)-2-(3-(5-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-гидроксипропил)пиперидин-3-ол.
Кроме того, соединения, представленные химической формулой 1, могут быть использованы в виде фармацевтически приемлемой соли, и в качестве соли может быть использована соль присоединения кислоты, образованная с фармацевтически приемлемой свободной кислотой. В качестве свободной кислоты можно использовать неорганическую кислоту и органическую кислоту. Примеры неорганической кислоты могут включать хлористоводородную кислоту, бромноватую кислоту, серную кислоту, фосфорную кислоту и тому подобное. Примеры органической кислоты могут включать лимонную кислоту, уксусную кислоту, молочную кислоту, малеиновую кислоту, глюконовую кислоту, метансульфоновую кислоту, янтарную кислоту, 4-толуолсульфоновую кислоту, глутаминовую кислоту, аспарагиновую кислоту или тому подобное.
Соли или сольваты соединений, представленных химической формулой 1, которые являются фармацевтически неприемлемыми, могут быть использованы в качестве промежуточных продуктов при получении соединения, представленного химической формулой 1, его фармацевтически приемлемой соли или сольвата.
Соединение, представленное химической формулой 1, в соответствии с настоящим изобретением, включает его фармацевтически приемлемые соли, а также все сольваты и гидраты, которые могут быть из него получены. Соли или сольваты соединения, представленного химической формулой 1, могут быть получены из соединений, представленных химической формулой 1, с использованием обычных методов области техники, к которой относится настоящее изобретение.
Кроме того, соединение, представленное химической формулой 1, в соответствии с настоящим изобретением, может быть получено в кристаллической форме или в некристаллической форме. Когда соединение, представленное химической формулой 1, получают в кристаллической форме, оно может быть необязательно гидратировано или сольватировано. Настоящее изобретение может включать не только стехиометрические гидраты соединения, представленного химической формулой 1, но также соединения, содержащие различное количество воды. Сольваты соединения, представленного химической формулой 1, в соответствии с настоящим изобретением включают как стехиометрические сольваты, так и нестехиометрические сольваты.
В настоящем изобретении предложен также способ получения соединения, представленного химической формулой 1, как показано на следующей реакционной схеме 1:
[Реакционная схема 1]
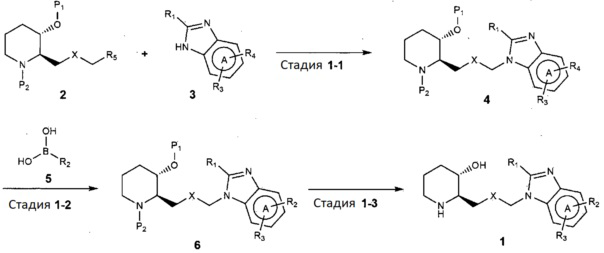
(на реакционной схеме 1, A, X, R1, R2 и R3 имеют значения, указанные выше, R4 и R5, каждый, независимо, представляют собой галоген, и P1 и P2, каждый, независимо, означают защитную группу. Защитная группа может представлять собой (трет-бутилдиметилсилил)окси или бензилоксикарбонил).
Стадия 1-1 представляет собой стадию получения соединения, представленного химической формулой 4, путем взаимодействия соединения, представленного химической формулой 2, с соединением, представленным химической формулой 3, в присутствии основания. В качестве основания могут быть использованы обычные неорганические основания и органические основания. Неограничивающие примеры органических оснований могут включать диизопропилэтиламин и триэтиламин. Неограничивающие примеры неорганических оснований могут включать карбонат калия, карбонат натрия, гидрокарбонат натрия, карбонат цезия или карбонат кальция. Также реакция может быть осуществлена в полярном растворителе, таком как метанол, этанол, бутанол, тетрагидрофуран, ацетон, толуол, диметилформамид, диметилформсульфоксид, хлороформ, диоксан, ацетонитрил, диэтиловый эфир или дихлорметан, при температуре от 20°C до 150°C в течение от 10 минут до 24 часов.
Стадия 1-2 представляет собой стадию получения соединения, представленного химической формулой 6, путем взаимодействия соединения, представленного химической формулой 4, и соединения, представленного химической формулой 5, с катализатором, выбранным из тетракис(трифенилфосфин)палладия, дихлорида (1,1՚-бис(дифенилфосфино)ферроцен)палладия или трис(дибензилиденацетон)дипалладия, в присутствии неорганических оснований, таких как карбонат калия, карбонат натрия, калия цезия или гидрокарбонат натрия. Реакция может быть осуществлена в полярном растворителе, таком как метанол, этанол, трет-бутанол, тетрагидрофуран, толуол, диоксан, диметилформамид, диметиловый эфир этиленгликоля или вода, при температуре от 70°C до 150°C в течение от 5 минут до 18 часов.
Стадия 1-3 представляет собой стадию получения соединения, представленного химической формулой 1, путем взаимодействия соединения, представленного химической формулой 6, в присутствии кислоты. Неограничивающие примеры кислоты могут включать хлористоводородную кислоту, бромноватую кислоту, фтористоводородную кислоту, трифторуксусную кислоту или тому подобное. Предпочтительно, в качестве реакционного растворителя может использоваться или не использоваться полярный органический растворитель. Предпочтительно, когда используется полярный органический растворитель, то можно использовать дихлорметан, хлороформ, толуол, диметилформамид, диоксан, тетрагидрофуран или тому подобное, и реакцию можно проводить при от комнатной температуры до 100°С в течение от 10 минут до 6 часов.
В качестве другого примера, соединение, представленное химической формулой 1, где R1 представляет собой водород, может быть получено, как показано на следующей реакционной схеме 2:
[Реакционная схема 2]
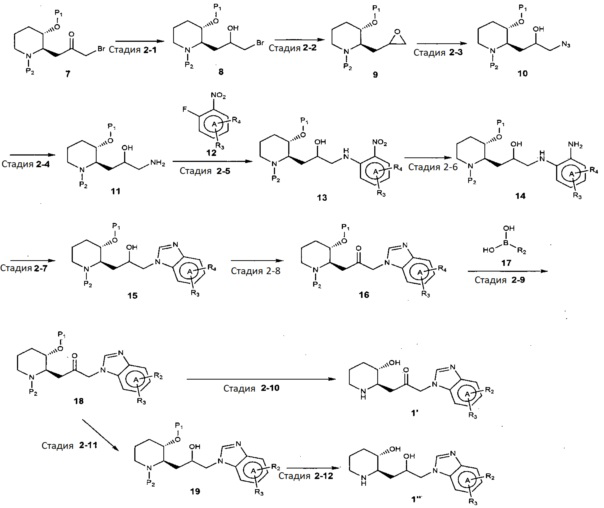
(на реакционной схеме 2, A, R2 и R3 имеют значения, указанные выше, R4 представляет собой галоген, и P1 и P2, каждый, независимо, означают защитную группу. Защитная группа может представлять собой трет-бутилдиметилсилил)окси или бензилоксикарбонил.)
Стадия 2-1 представляет собой стадию получения соединения, представленного химической формулой 8, путем взаимодействия соединения, представленного химической формулой 7, в присутствии основания. Соединение, представленное химической формулой 7, может быть получено в соответствии с известным способом (например, McLaughlin and Evans, J Org. Chem, 2010,75: 518-521), но им не ограничивается. В качестве основания могут быть использованы обычные неорганические основания, но их неограничивающие примеры могут включать боргидрид натрия, литийалюминийгидрид, карбонат натрия, формиат натрия, хлорид цезия, боран-тетрагидрофуран. Кроме того, реакция может быть осуществлена в полярном растворителе, таком как метанол, этанол, тетрагидрофуран, ацетон, толуол, диэтиловый эфир или дихлорметан, при температуре от -78°C до 20°C в течение от 10 минут до 12 часов.
Стадия 2-2 представляет собой стадию получения соединения, представленного химической формулой 9, путем взаимодействия соединения, представленного химической формулой 8, в присутствии основания. В качестве основания может быть использовано обычное неорганическое основание, и его неограничивающие примеры могут включать гидроксид калия, гидрид лития, фторид калия, гидрид натрия, этоксид натрия, карбонат калия или трет-бутоксид калия. Помимо этого, реакция может быть осуществлена в полярном растворителе, таком как метанол, тетрагидрофуран, ацетон, диоксан, диэтиловый эфир, дихлорметан, диметилформамид или ацетонитрил, при температуре от 0°C до 20°C в течение от 10 минут до 24 часов.
Стадия 2-3 представляет собой стадию получения соединения, представленного химической формулой 10, путем взаимодействия соединения, представленного химической формулой 9, в кислых условиях в присутствии азида натрия и/или триметилсилил азида. В качестве кислоты могут быть использованы обычные неорганические кислоты и органические кислоты, и их неограничивающие примеры могут включать хлорид аммония, тетрабутилхлорид аммония, п-толуолсульфоновую кислоту, уксусную кислоту, хлористоводородную кислоту или серную кислоту. Кроме того, реакцию можно осуществлять в полярном растворителе, таком как метанол, этанол, трет-бутанол, ацетон, диметилформамид, ацетонитрил или вода, при температуре от 20°C до 100°C в течение от 10 минут до 48 часов.
Стадия 2-4 представляет собой стадию получения соединения, представленного химической формулой 11, путем взаимодействия соединения, представленного химической формулой 10, в присутствии основания. В качестве основания могут быть использованы обычные неорганические основания, и их неограничивающие примеры могут включать боргидрид натрия, литийалюминийгидрид, палладий, никель или трифенилфосфин. Кроме того, реакция может быть осуществлена в полярном растворителе, таком как метанол, этанол, тетрагидрофуран, ацетон, толуол, диоксан, диметилформамид, ацетонитрил, диэтиловый эфир, дихлорметан или вода, при температуре от 20°C до 80°C в течение от 10 минут до 18 часов.
Стадия 2-5 представляет собой стадию получения соединения, представленного химической формулой 13, путем взаимодействия соединения, представленного химической формулой 11, и соединения, представленного химической формулой 12, в присутствии основания. В качестве основания могут быть использованы обычные неорганические основания и органические основания. Неограничивающие примеры органического основания могут включать диизопропилэтиламин или триэтиламин, и неограничивающие примеры неорганического основания могут включать карбонат калия, карбонат натрия, гидрокарбонат натрия, карбонат цезия или карбонат кальция. Кроме того, реакция может быть осуществлена в полярном растворителе, таком как метанол, этанол, бутанол, тетрагидрофуран, ацетон, толуол, диметилформамид, диметилформсульфоксид, при температуре от 20°C до 150°C в течение от 10 минут до 24 часов.
Стадия 2-6 представляет собой стадию получения соединения, представленного химической формулой 14, путем взаимодействия соединения, представленного химической формулой 13, в присутствии водорода и металла. Неограничивающие примеры металла могут включать палладий, никель или оксид платины. Кроме того, реакцию можно осуществлять в полярном растворителе, таком как метанол, этанол, изопропанол, тетрагидрофуран, диметилформамид, этилацетат, дихлорметан или вода, при температуре от 5°C до 50°C в течение от 10 минут до 12 часов.
Стадия 2-7 представляет собой стадию получения соединения, представленного химической формулой 15, путем взаимодействия соединения, представленного химической формулой 14, i) в присутствии триметил ортоформиата или триэтил ортоформиата, и пара-толуолсульфоновой кислоты или пиридиний пара-толуолсульфоната, или ii) в присутствии муравьиной кислоты. Реакция может быть осуществлена в полярном растворителе, таком как метанол, этанол, тетрагидрофуран, толуол, диоксан, диметилформамид, ацетон, хлороформ, этилацетат, дихлорметан или ацетонитрил, при температуре от 20°C до 120°C в течение от 10 минут до 12 часов.
Стадия 2-8 представляет собой стадию получения соединения, представленного химической формулой 16, путем взаимодействия соединения, представленного химической формулой 15, с окислителем. Неограничивающие примеры окислителя могут включать перйодинан Десса-Мартина, пероксид водорода или оксалил хлорид. Кроме того, реакцию можно осуществлять в полярном растворителе, таком как дихлорметан, диметилформамид, диметилформсульфоксид, толуол, хлороформ, тетрагидрофуран, ацетон, ацетонитрил диэтиловый эфир или этилацетат, при температуре от -78°C до 30°C в течение от 10 минут до 12 часов.
Стадия 2-9 представляет собой стадию получения соединения, представленного химической формулой 18, путем взаимодействия соединения, представленного химической формулой 16, с соединением, представленным химической формулой 17, в присутствии катализатора тетракис(трифенилфосфин)палладия, дихлорида (1,1՚-бис(дифенилфосфино)ферроцен)палладия или трис(дибензилиденацетон)дипалладия, и неорганического основания карбоната калия, карбоната натрия, цезия калия или гидрокарбоната натрия. Реакцию проводят в полярном растворителе, таком как метанол, этанол, трет-бутанол, тетрагидрофуран, толуол, диоксан, диметилформамид, диметиловый эфир этиленгликоля или вода, при температуре от 70°C до 150°C в течение от 5 минут до 18 часов.
Стадия 2-10 представляет собой стадию получения соединения, представленного химической формулой 1՚, путем взаимодействия соединения, представленного химической формулой 18, в присутствии кислоты. Неограничивающие примеры кислоты могут включать хлористоводородную кислоту, бромноватую кислоту, фтористоводородную кислоту, трифторуксусную кислоту или тому подобное. Предпочтительно, в качестве реакционного растворителя может использоваться или может не использоваться полярный органический растворитель. Предпочтительно, когда используется полярный органический растворитель, может быть использован дихлорметан, хлороформ, толуол, диметилформамид, диоксан, тетрагидрофуран или тому подобное, и реакцию можно осуществлять при от комнатной температуры до 100°C в течение от 10 минут до 6 часов.
Стадия 2-11 представляет собой стадию получения соединения, представленного химической формулой 19, путем взаимодействия соединения, представленного химической формулой 18, в присутствии основания. В качестве основания могут быть использованы обычные неорганические основания, и их неограничивающие примеры могут включать боргидрид натрия, литийалюминийгидрид, карбонат натрия, формиат натрия, хлорид цезия или боран-тетрагидрофуран. Кроме того, реакция может быть осуществлена в полярном растворителе, таком как метанол, этанол, тетрагидрофуран, ацетон, толуол, диэтиловый эфир или дихлорметан, при температуре от -78°C до 20°C в течение от 10 минут до 12 часов.
Стадия 2-12 представляет собой стадию получения соединения, представленного химической формулой 1ʺ, путем взаимодействия соединения, представленного химической формулой 19, в присутствии кислоты. Неограничивающие примеры кислоты могут включать хлористоводородную кислоту, бромноватую кислоту, фтористоводородную кислоту, трифторуксусную кислоту или тому подобное. Предпочтительно, в качестве реакционного растворителя может использоваться или может не использоваться полярный органический растворитель. Предпочтительно, когда используется полярный органический растворитель, может быть использован дихлорметан, хлороформ, толуол, диметилформамид, диоксан или тетрагидрофуран, и реакция может быть осуществлена при от комнатной температуре до 100°C в течение от 10 минут до 6 часов.
В качестве другого примера, соединение, представленное химической формулой 1, где R1 представляет собой C1-4 гидроксиалкил, может быть получено, например, как показано на следующей реакционной схеме 3:
[Реакционная схема 3]
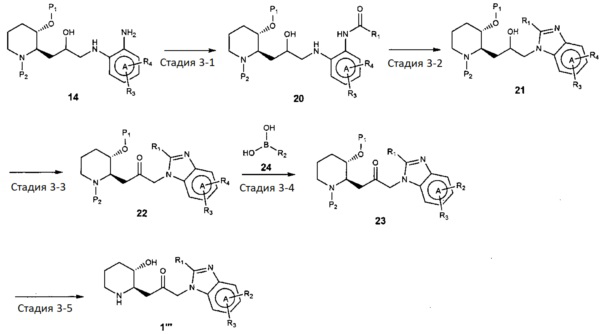
(на реакционной схеме 3, A, X, R2 и R3 имеют значения, указанные выше, R4 представляет собой галоген, и P1 и P2, каждый, независимо, означают защитную группу. Защитная группа может представлять собой (трет-бутилдиметилсилил)окси или бензилоксикарбонил).
Стадия 3-1 представляет собой стадию получения соединения, представленного химической формулой 20, путем взаимодействия соединения, представленного химической формулой 14, и R1-замещенной карбоновой кислоты (R1-COOH) в присутствии реагента амидного связывания гидрохлорида бис-(2-оксо-3-оксазолидинил)фосфорила, гидрохлорида 1-этил-(3-(3-диметиламино)пропил)-карбодиимида, гексафторфосфата бензотриазол-1-илокси-трис-(пирролидинo)фосфония, бензотриазол-ола, гексафторфосфата (бензотриазол-1-илокси)трис(диметиламино)фосфония или гексафторфосфата O-(бензотриазол-1-ил)-N,N,N,N՚-тетраметилурония, и основания триэтиламина, диизопропилэтиламина, пиридина, диметиланилина, диметиламинопиридина или гидроксида натрия. Реакция может быть осуществлена в полярном растворителе, таком как метанол, этанол, пропанол, тетрагидрофуран, толуол, диоксан, диметилформамид, дихлорметан, ацетонитрил или ацетон, при температуре от -20°C до 80°C в течение от 5 минут до 18 часов.
Стадия 3-2 представляет собой стадию получения соединения, представленного химической формулой 21, путем взаимодействия соединения, представленного химической формулой 20, i) в присутствии триметил ортоформиата или триэтил ортоформиата, и пара-толуолсульфоновой кислоты или пиридиний пара-толуолсульфоната, или ii) в присутствии муравьиной кислоты. Реакция может быть осуществлена в полярном растворителе, таком как метанол, этанол, тетрагидрофуран, толуол, диоксан, диметилформамид, ацетон, хлороформ, этилацетат, дихлорметан или ацетонитрил, при температуре от 20°C до 120°C в течение от 10 минут до 12 часов.
Стадия 3-3 представляет собой стадию получения соединения, представленного химической формулой 22, путем взаимодействия соединения, представленного химической формулой 21, с окислителем. Неограничивающие примеры окислителя могут включать перйодинан Десса-Мартина, пероксид водорода или оксалил хлорида. Кроме того, реакция может быть осуществлена в полярном растворителе, таком как дихлорметан, диметилформамид, диметилформсульфоксид, толуол, хлороформ, тетрагидрофуран, ацетон, ацетонитрил, диэтиловый эфир или этилацетат, при температуре от -78°C до 30°C в течение от 10 минут до 12 часов.
Стадия 3-4 представляет собой стадию получения соединения, представленного химической формулой 23, путем взаимодействия соединения, представленного химической формулой 22, с соединением, представленным химической формулой 24, в присутствии катализатора, такого как тетракис(трифенилфосфин)палладий, дихлорид (1,1՚-бис (дифенилфосфино)ферроцен)палладия или трис(дибензилиденацетон)дипалладий, и неорганического основания, такого как карбонат калия, карбонат натрия, цезия калия или гидрокарбонат натрия. Реакция может быть осуществлена в полярном растворителе, таком как метанол, этанол, трет-бутанол, тетрагидрофуран, толуол, диоксан, диметилформамид, диметиловый эфир этиленгликоля или вода, при температуре от 70°C до 150°C в течение от 5 минут до 18 часов.
Стадия 3-5 представляет собой стадию получения соединения, представленного химической формулой 1‴, путем взаимодействия соединения, представленного химической формулой 23, в присутствии кислоты. Неограничивающие примеры кислоты могут включать хлористоводородную кислоту, бромноватую кислоту, фтористоводородную кислоту, трифторуксусную кислоту или тому подобное. Предпочтительно, в качестве реакционного растворителя может использоваться или может не использоваться полярный органический растворитель. Предпочтительно, когда используется полярный органический растворитель, может быть использован дихлорметан, хлороформ, толуол, диметилформамид, диоксан, тетрагидрофуран или тому подобное, и реакция может быть осуществлена при от комнатной температуры до 100°C в течение от 10 минут до 6 часов.
Кроме того, настоящее изобретение относится к соединению, представленному химической формулой 16, или соединению, представленному химической формулой 21, в качестве промежуточного соединения, которое может быть использовано при получении соединения, представленного химической формулой 1.
[Химическая формула 16]
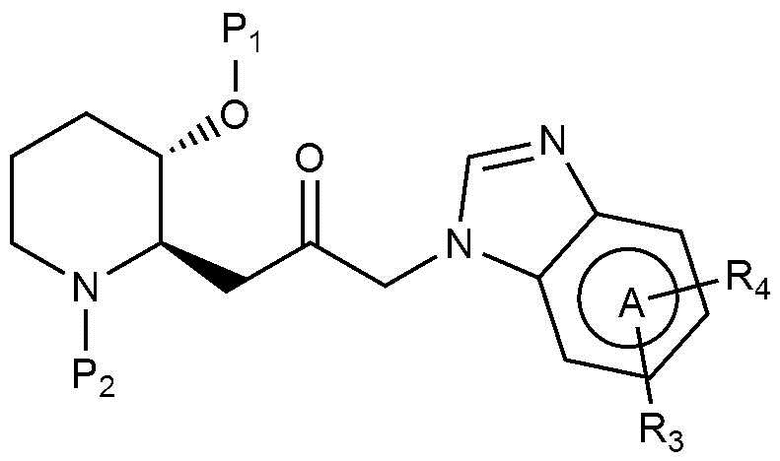
[Химическая формула 21]
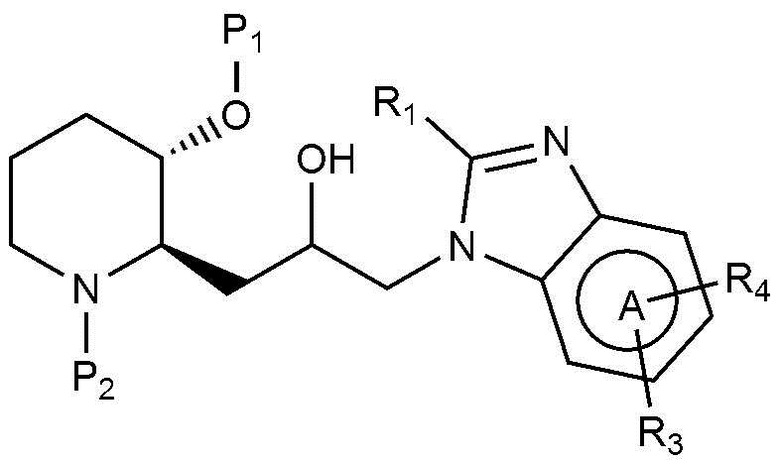
в химических формулах 16 и 21,
P1 и P2, каждый, независимо, представляют собой защитную группу,
A, R1 и R3 имеют значения, определенные в химической формуле 1, и
R4 представляет собой галоген.
Предпочтительно, P1 и P2, каждый, независимо, представляют собой (трет-бутилдиметилсилил)окси или бензилоксикарбонил.
Также предпочтительно, R4 представляет собой бром, хлор, или фтор.
Соединение, представленное химической формулой 16, может быть получено реакционным путем от стадии 2-5 до стадии 2-8 по вышеуказанной реакционной схеме 2. Также, соединение, представленное химической формулой 21, может быть получено на стадиях 3-1 и 3-2 в соответствии с вышеуказанной реакционной схемой 3.
Кроме того, настоящее изобретение относится к фармацевтической композиции для предупреждения или лечения заболеваний, вызванных аномальной активностью PRS (пролил-тРНК-синтетаза), содержащей соединение, представленное химической формулой 1, или его фармацевтически приемлемую соль.
Соединение, представленное химической формулой 1, в соответствии с настоящим изобретением может ингибировать ферментативную активность PRS и поэтому может быть использовано для предупреждения или лечения заболеваний, вызванных аномалиями в активности PRS (пролил-тРНК-синтетаза). Примеры заболеваний, вызванных аномалиями в активности PRS (пролил-тРНК-синтетаза), могут включать злокачественное новообразование, воспалительное заболевание, аутоиммунное заболевание и фиброз.
Как показано в примерах, которые будут описаны далее, соединение, представленное химической формулой 1, в соответствии с настоящим изобретением может значительно ингибировать ферментативную активность PRS и также ингибировать рост раковых клеток. Таким образом, это соединение может эффективно использоваться для предупреждения или лечения заболеваний.
Фармацевтическая композиция в соответствии с настоящим изобретением может быть изготовлена для перорального или парентерального введения в соответствии со стандартной фармацевтической практикой. Эти препараты могут содержать помимо активного ингредиента добавки, такие как фармацевтически приемлемый носитель, вспомогательное лекарственное вещество или разбавитель. Подходящие носители могут включать, например, физиологический раствор, полиэтиленгликоль, этанол, растительное масло и изопропилмиристат, а разбавитель может включать, например, лактозу, декстрозу, сахарозу, маннит, сорбит, целлюлозу и/или глицин, но ими не ограничивается. Кроме того, соединения по настоящему изобретению могут быть растворены в маслах, пропиленгликоле или других растворителях, которые обычно используются при получении инъекционных растворов. Кроме того, соединения по настоящему изобретению могут быть составлены в виде мазей или кремов для местного применения.
Предпочтительная доза соединения, представленного химической формулой 1, в соответствии с настоящим изобретением может варьироваться в зависимости от состояния и веса пациента, тяжести заболевания, типа лекарственного средства и пути и продолжительности введения, но она может быть соответствующим образом подобрана специалистами в данной области техники. Однако для достижения желаемых эффектов соединение по настоящему изобретению можно вводить ежедневно в дозе от 0,0001 до 100 мг/кг (массы тела) и, предпочтительно, от 0,001 до 100 мг/кг (массы тела). Введение может проводиться один раз в день или в разделенных дозах каждый день пероральным или парентеральным путем.
В зависимости от способа введения фармацевтическая композиция в соответствии с настоящим изобретением может содержать соединение, представленное химической формулой 1, или его фармацевтически приемлемую соль в количестве от 0,001 до 99 мас.%, предпочтительно, от 0,01 до 60 мас.%.
Фармацевтическую композицию по настоящему изобретению можно вводить млекопитающим, таким как крыса, мышь, домашнее животное, человек или тому подобное, различными путями. Введение может быть осуществлено всеми возможными способами, например, пероральным, ректальным, внутривенным, внутримышечным, подкожным, внутриматочным или интрацеребровентрикулярным введением.
ПОЛОЖИТЕЛЬНЫЙ ЭФФЕКТ
Как указано выше, соединение, представленное химической формулой 1, в соответствии с настоящим изобретением, может ингибировать ферментативную активность PRS и, таким образом, может эффективно использоваться для предупреждения или лечения заболеваний, вызванных аномалиями в активности PRS (пролил-тРНК-синтетаза), например, злокачественных новообразований, воспалительных заболеваний, аутоиммунных заболеваний и фиброза.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение описано более подробно с помощью примеров. Однако эти примеры приведены только для иллюстрации настоящего изобретения и не должны истолковываться как ограничивающие объем настоящего изобретения этими примерами.
Пример 1: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-она
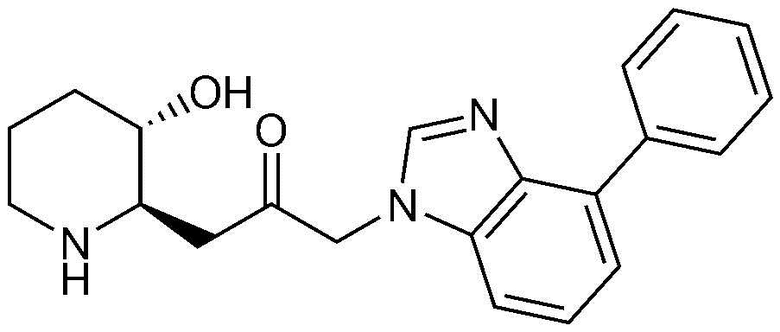
Стадия 1-1: Получение бензил (2R,3S)-2-(3-бром-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-бром-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (5 г, 10,32 ммоль) растворяли в смешанном растворителе из метанола и тетрагидрофурана (1:1) (20 мл, 0,5 M) и затем охлаждали до температуры 0°C. Затем добавляли боргидрид натрия (390 мг, 10,32 ммоль) и перемешивали при температуре 0°C в течение 30 минут. Температуру повышали до комнатной температуры, и смесь дополнительно перемешивали в течение 12 часов. Когда реакция завершалась, последующие реакции проводились без процедур обработки и очистки.
Стадия 1-2: Получение бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(оксирен-2-илметил)пиперидин-1-карбоксилата
К реакционному раствору бензил (2R,3S)-2-(3-бром-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата, полученному на стадии 1-1, добавляли гидроксид калия (203 мг, 3,61 ммоль), растворенный в небольшом количестве воды, и затем перемешивали при комнатной температуре в течение 1 часа. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и затем промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и затем концентрировали при пониженном давлении, и очищали с помощью хроматографии на колонке (гексан:этилацетат=4:1) с получением указанного в заголовке соединения (3,4 г, выход за две стадии: 81%).
Стадия 1-3: Получение бензил (2R,3S)-2-(3-азидо-2-гидроксипропил)-3-(трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(оксирен-2-илметил)пиперидин-1-карбоксилат (3,4 г, 8,38 ммоль), полученный на стадии 1-2, растворяли в смешанном растворителе из метанола и воды (8:1) (90 мл, 0,09 ммоль), к нему добавляли азид натрия (2,7 г, 41,92 ммоль) и хлорид аммония (1,3 г, 25,15 ммоль) и затем перемешивали при кипячении с обратным холодильником при температуре 70°C в течение 6 часов. Когда реакция завершалась, полученную смесь разбавляли этилацетатом и затем промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и затем концентрировали при пониженном давлении, и очищали с помощью хроматографии на колонке (гексан:этилацетат=3:1) с получением указанного в заголовке соединения (3,8 г, выход: 98%).
Стадия 1-4: Получение бензил (2R,3S)-2-(3-амино-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-азидо-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (1,5 г, 3,34 ммоль), полученный на стадии 1-3, растворяли в смешанном растворителе из тетрагидрофурана и воды (8:2) (50 мл, 0,08 M), к нему добавляли трифенилфосфин (1,8 г, 6,69 ммоль) и перемешивали при комнатной температуре в течение 6 часов. Когда реакция завершалась, полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и затем концентрировали при пониженном давлении, и очищали с помощью хроматографии на колонке (дихлорметан:метанол=9: 1) с получением указанного в заголовке соединения (1,2 г, выход: 81%).
Стадия 1-5: Получение бензил (2R,3S)-2-(3-((3-бром-2-нитрофенил)амино)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-амино-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (120 мг, 0,27 ммоль), полученный на стадии 1-4, растворяли в N,N-диметилформамиде (1 мл, 0,3 M), к нему добавляли 1-бром-3-фтор-2-нитробензол (59 мг, 0,27 ммоль) и диизопропилэтиламин (94 мг, 0,54 ммоль) и затем перемешивали при кипячении с обратным холодильником при температуре 80°C в течение 6 часов. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и затем концентрировали при пониженном давлении, и очищали с помощью хроматографии на колонке (гексан:этилацетат=2:1) с получением указанного в заголовке соединения (120 мг, выход: 71%).
Стадия 1-6: Получение бензил (2R,3S)-2-(3-((2-амино-2-бромфенил)амино)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-((3-бром-2-нитрофенил)амино)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (120 мг, 0,19 ммоль), полученный на стадии 1-5, растворяли в метаноле (6 мл, 0,03 M), к нему добавляли соответствующее количество никеля Ренея. После подсоединения баллона с водородом смесь перемешивали при комнатной температуре в течение 1 часа. Когда реакция завершалась, реакционный раствор фильтровали через целит и концентрировали при пониженном давлении. Последующие реакции проводились без процедуры очистки.
Стадия 1-7: Получение бензил (2R,3S)-2-(3-((4-бром-1H-бензо[d]имидазол-1-ил)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-((2-амино-2-бромфенил)амино)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (120 мг, 0,20 ммоль), полученный на стадии 1-6, растворяли в толуоле (1 мл, 0,3M), к нему добавляли пара-толуолсульфоновую кислоту (7 мг, 0,04 ммоль) и триэтилортоформиат (100 мкл, 0,42 ммоль) и затем перемешивали при температуре 40°C в течение 12 часов. Когда реакция завершалась, полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=1:2) с получением указанного в заголовке соединения (64 мг, выход: 55%).
Стадия 1-8: Получение бензил (2R,3S)-2-(3-(4-бром-1H-бензо[d]имидазол-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-((4-бром-1H-бензо[d]имидазол-1-ил)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (64 мг, 0,11 ммоль), полученный на стадии 1-7, растворяли в дихлорметане (1 мл, 0,1 M), к нему добавляли перйодинан Десса-Мартина (54 мг, 0,13 ммоль) и затем перемешивали при комнатной температуре в течение 3 часов. Когда реакция завершалась, полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (дихлорметан:метанол=15:1) с получением указанного в заголовке соединения (49 мг, выход: 77%).
Стадия 1-9: Получение бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(2-оксо-3-(4-фенил-1H-бензо[d]имидазол-1-ил)пропил)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-(4-бром-1H-бензо[d]имидазол-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (49 мг, 0,08 ммоль), полученный на стадии 1-8, растворяли в N,N-диметилформамиде (3 мл, 0,03 M), к нему добавляли фенилбороновую кислоту (15 мг, 0,12 ммоль), тетракис(трифенилфосфин)палладий(0) (19 мг, 0,02 ммоль) и 2M водный раствор карбонат натрия (300 мкл, 0,3 M) и перемешивали при температуре 120°C в течение 45 минут с использованием микроволнового устройства. Когда реакция завершалась, полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (100%-ный этилацетат) с получением указанного в заголовке соединения (42 мг, выход: 85%).
Стадия 1-10: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-она
Бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(2-оксо-3-(4-фенил-1H-бензо[d]имидазол-1-ил)пропил)пиперидин-1-карбоксилат (42 мг, 0,07 ммоль), полученный на стадии 1-9, растворяли в 6н водном растворе соляной кислоты (2 мл, 0,04 M) и затем перемешивали при кипячении с обратным холодильником в течение 1 часа. Когда реакция завершалась, реакционный раствор охлаждали до температуры 0°C, нейтрализовали (pH 7) карбонатом калия и экстрагировали смешанным раствором хлороформа и небольшого количества ацетона. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем перекристаллизовывали в диэтиловом эфире с получением указанного в заголовке соединения (17 мг, выход: 70%).
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,01 (с, 1H), 7,67 (д, 1H), 7,43 (м, 3H), 7,23 (м, 3H), 6,99 (д, 1H), 4,82 (дд, 2H), 4,64 (м, 1H), 2,77 (м, 1H), 2,68 (д, 1H), 2,25 (м, 1H), 2,22 (м, 2H), 1,78 (м, 1H), 1,68 (м, 1H), 1,48 (м, 1H), 1,25 (м, 1H), 1,12 (м, 1H)
Пример 2: Получение 1-(4-(3-фторфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
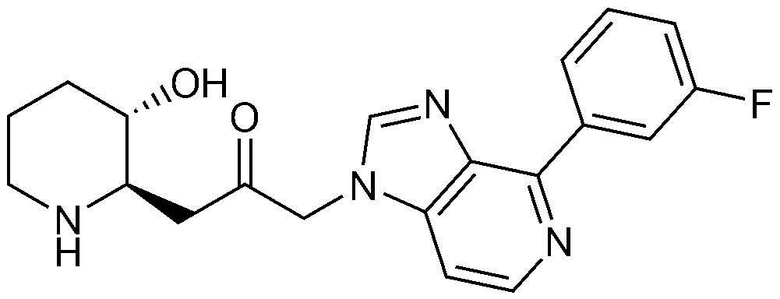
Стадия 2-1: Получение бензил (2R,3S)-3-(3-(4-бром-1H-имидазо[4,5-c]пиридин-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
4-Бром-1H-имидазо[4,5-c]пиридин (204 мг, 1,03 ммоль) растворяли в N,N-диметилформамиде (4 мл, 0,25 M), к нему добавляли карбонат калия (285 мг, 2,06 ммоль) и затем перемешивали при комнатной температуре в течение 10 минут. Затем добавляли бензил (2R,3S)-2-(3-бром-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (500 мг, 1,03 ммоль) и перемешивали при комнатной температуре в течение 3 часов. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (дихлорметан:метанол=10:1) с получением указанного в заголовке соединения (554 мг, выход: 89%).
Стадия 2-2: Получение бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(4-(3-фторфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-2-оксопропил)пиперидин-1-карбоксилата
Бензил (2R,3S)-3-(3-(4-бром-1H-имидазо[4,5-c]пиридин-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (91 мг, 0,15 ммоль), полученный на стадии 2-1, растворяли в N,N-диметилформамиде (3 мл, 0,05 M), к нему последовательно добавляли (3-фторфенил)бороновую кислоту (31 мг, 0,23 ммоль), тетракис(трифенилфосфин)палладий (35 мг, 0,03 ммоль) и 2M карбонат натрия (0,3 мл, 0,61 ммоль) и затем перемешивали при комнатной температуре в течение 5 минут. Затем смесь оставляли взаимодействовать при температуре 130°C в течение 45 минут с использованием микроволновой печи. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=1:2) с получением указанного в заголовке соединения (61 мг, выход: 65%).
Стадия 2-3: Получение 1-(4-(3-фторфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
Бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(4-(3-фторфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-2-оксопропил)пиперидин-1-карбоксилат (61 мг, 0,10 ммоль), полученный на стадии 2-2, растворяли в 6н водном растворе соляной кислоты (3 мл, 0,03 M) и затем перемешивали при кипячении с обратным холодильником в течение 1 часа. Когда реакция завершалась, реакционный раствор охлаждали до температуры 0°C, нейтрализовали (pH 7) карбонатом калия и затем экстрагировали смешанным раствором хлороформа и небольшого количества ацетона. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем перекристаллизовывали в диэтиловом эфире с получением указанного в заголовке соединения (19 мг, выход: 52%).
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,64 (д, 1H), 8,59 (д, 1H), 8,44 (д, 1H), 8,33 (с, 1H), 7,56-7,61 (м, 2H), 7,29-7,32 (м, 1H), 5,46 (м, 2H), 5,01 (с, 1H), 3,12 (шир. с, 1H), 3,00 (дд, 1H), 2,89 (д, 1H), 2,81 (с, 1H), 1,92 (м, 1H), 1,63 (д, 1H), 1,23-1,46 (м, 3H).
Пример 3: Получение 1-(4-(3-хлорфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
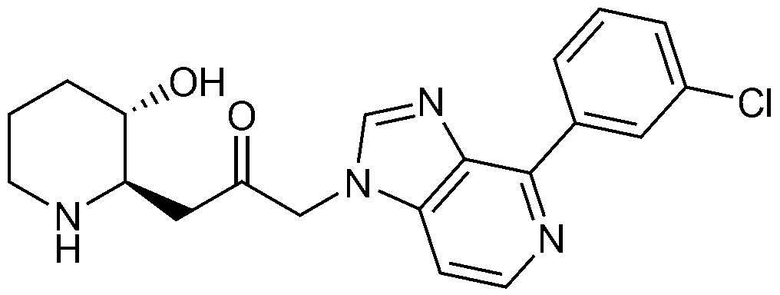
Указанное в заголовке соединение (18 мг, выход: 33%) получали таким же образом, как в примере 2, за исключением того, что вместо (3-фторфенил)бороновой кислоты использовали (3-хлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,86 (с, 1H), 8,74 (д, 1H), 8,44 (д, 1H), 8,34 (с, 1H), 7,52-7,60 (м, 3H), 5,42 (м, 2H), 4,92 (с, 1H), 3,07 (м, 1H), 2,98 (дд, 1H), 2,86 (д, 1H), 2,73 (м, 1H), 1,61 (д, 1H), 1,24-1,43 (м, 3H).
Пример 4: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-(3-(трифторметил)фенил)-1H-имидазо[4,5-c]пиридин-1-ил)пропан-2-она
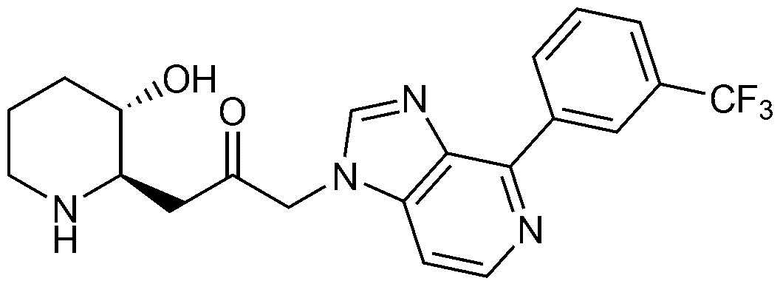
Указанное в заголовке соединение (22 мг, выход: 39%) получали таким же образом, как в примере 2, за исключением того, что вместо (3-фторфенил) бороновой кислоты использовали (3-трифторметил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 9,16 (с, 1H), 9,08 (д, 1H), 8,48 (д, 1H), 8,36 (с, 1H), 7,79-7,85 (м, 2H), 7,60 (д, 1H), 5,46 (м, 2H), 4,95 (с, 1H), 3,09 (шир. с, 1H), 2,99 (дд, 1H), 2,87 (д, 1H), 2,77 (с, 1H), 1,92 (м, 1H), 1,62 (д, 1H), 1,27-1,44 (м, 3H).
Пример 5: Получение 1-(4-(3-хлор-5-фторфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она

Указанное в заголовке соединение (19 мг, выход: 34%) получали таким же образом, как в примере 2, за исключением того, что вместо (3-фторфенил)бороновой кислоты использовали (3-хлор-5-фторметил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,78 (с, 1H), 8,59 (д, 1H), 8,45 (д, 1H), 8,38 (с, 1H), 7,62 (д, 1H), 7,55 (м, 1H), 5,46 (м, 1H), 4,92 (м, 1H), 3,09 (м, 1H), 2,98 (дд, 1H), 2,86 (д, 1H), 2,74 (м, 1H), 1,92 (м, 1H), 1,61 (д, 1H), 1,25-1,40 (м, 3H).
Пример 6: Получение 1-(4-(3,5-дихлорфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
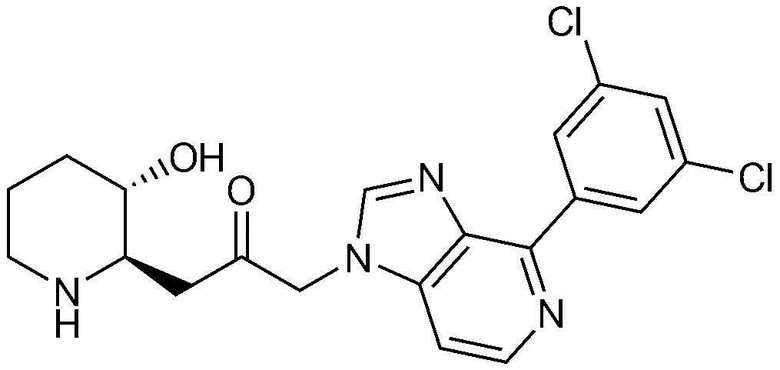
Указанное в заголовке соединение (20 мг, выход: 36%) получали таким же образом, как в примере 2, за исключением того, что вместо (3-фторфенил) бороновой кислоты использовали (3,5-дихлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,85 (д, 2H), 8,47 (д, 1H), 8,38 (с, 1H), 7,72 (м, 1H), 7,62 (м, 1H), 5,46 (м, 2H), 4,93 шир. с, 1H), 3,07 (м, 1H), 2,98 (дд, 1H), 2,86 (д, 1H), 2,75 (м, 1H), 1,92 (м, 1H), 1,61 (д, 1H), 1,25-1,41 (м, 3H).
Пример 7: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-(пирролидин-1-ил)-1H-имидазо[4,5-c]пиридин-1-ил)пропан-2-она
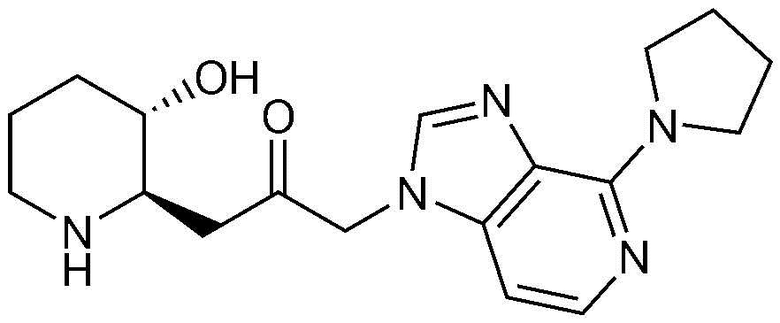
Стадия 7-1: Получение бензил (2R,3S)-2-(3-(4-бром-1H-имидазо[4,5-c]пиридин-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Указанное в заголовке соединение (554 мг, выход: 89%) получали исходя из 4-бром-1H-имидазо[4,5-c]пиридина (204 мг, 1,03 ммоль) таким же образом, как на стадии 2-1 примера 2.
Стадия 7-2: Получение бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(4-(пирролидин-1-ил)-1H-имидазо[4,5-c]пиридин-1-ил)-2-оксопропил)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-(4-бром-1H-имидазо[4,5-c]пиридин-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (100 мг, 0,17 ммоль), полученный на стадии 7-1, растворяли в этаноле (3 мл, 0,05 M), к нему последовательно добавляли пирролидин (27 мкл, 0,33 ммоль) и триэтиламин (93 мкл, 0,66 ммоль) и затем перемешивали при кипячении с обратным холодильником при температуре 80°C в течение 72 часов. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=1:2) с получением указанного в заголовке соединения (88 мг, выход: 89%).
Стадия 7-3: Получение (2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-(пирролидин-1-ил)-1H-имидазо[4,5-c]пиридин-1-ил)пропан-2-она
Бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(4-(пирролидин-1-ил)-1H-имидазо[4,5-c]пиридин-1-ил)-2-оксопропил)пиперидин-1-карбоксилат (88 мг, 0,15 ммоль), полученный на стадии 7-2, растворяли в 6н водном растворе соляной кислоты (3 мл, 0,05 M) и затем перемешивали при кипячении с обратным холодильником в течение 1 часа. Когда реакция завершалась, реакционный раствор охлаждали до температуры 0°C, нейтрализовали (pH 7) карбонатом калия и затем экстрагировали смешанным раствором хлороформа и небольшого количества ацетона. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем перекристаллизовывали в диэтиловом эфире с получением указанного в заголовке соединения (23 мг, выход: 45%).
1H-ЯМР (500 МГц, ДМСО-d6): δ 7,87 (с, 1H), 87,73 (д, 1H), 6,65 (д, 1H), 5,25 (м, 2H), 4,97 (с, 1H), 3,84 (с, 4H), 3,23 (с, 1H), 2,87-3,09 9m, 2H), 2,78 (с, 1H), 1,94 (м, 5H), 1,64 (д, 1H), 1,42 (м, 1H), 1,31 (с, 2H).
Пример 8: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-она
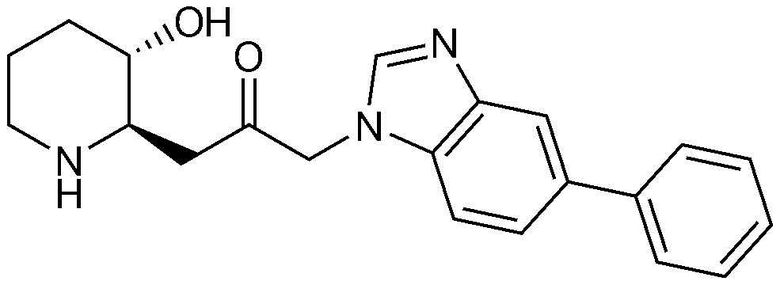
Указанное в заголовке соединение (25 мг, выход: 81%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 4-бром-1-фтор-2-нитробензол.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,08 (с, 1H), 7,89 (с, 1H), 7,69 (д, 2H), 7,52 (д, 2H), 7,45 (м, 2H), 7,33 (м, 1H), 5,34 (дд, 2H), 4,85 (д, 1H), 3,16 (м, 1H), 2,93 (м, 1H), 2,80 (м, 1H), 2,65 (м, 1H), 2,38 (м, 2H), 1,89 (м, 1H), 1,56 (м, 1H), 1,35 (м, 1H), 1,22 (м, 1H).
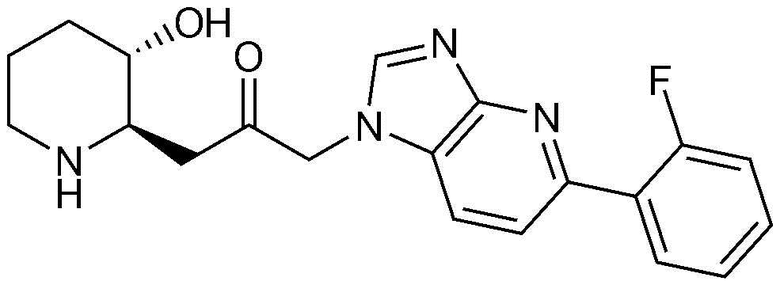
Пример 9: Получение 1-(5-(2-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
Стадия 9-1: Получение бензил (2R,3S)-2-(3-(5-бром-1H-имидазо[4,5-b]пиридин-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
5-Бром-1H-имидазо[4,5-b]пиридин (245 мг, 1,24 ммоль) растворяли в N,N-диметилформамиде (5 мл, 0,25 M), к нему добавляли карбонат калия (324 мг, 2,48 ммоль) и затем перемешивали при комнатной температуре в течение 10 минут. Затем добавляли бензил (2R,3S)-2-(3-бром-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (600 мг, 1,24 ммоль) и перемешивали при комнатной температуре в течение 3 часов. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (дихлорметан:метанол=10:1) с получением указанного в заголовке соединения (300 мг, выход: 40%).
Стадия 9-2: Получение бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(5-(2-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-2-оксопропил)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-(5-бром-1H-имидазо[4,5-b]пиридин-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (50 мг, 0,08 ммоль), полученный на стадии 9-1, растворяли в N,N-диметилформамиде (2 мл, 0,05 M), к нему последовательно добавляли (2-фторфенил)бороновую кислоту (18 мг, 0,13 ммоль), тетракис(трифенилфосфин)палладий (20 мг, 0,02 ммоль) и 2M раствор карбоната натрия (0,2 мл, 0,33 ммоль) и затем перемешивали при комнатной температуре в течение 5 минут. Затем смеси давали взаимодействовать при температуре 150°C в течение 30 минут с использованием микроволновой печи. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=1:2) с получением указанного в заголовке соединения (40 мг, выход: 78%).
Стадия 9-3: Получение 1-(5-(2-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
Бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(5-(2-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-2-оксопропил)пиперидин-1-карбоксилат (40 мг, 0,06 ммоль), полученный на стадии 9-2, растворяли в 6н водном растворе соляной кислоты (3 мл, 0,02 M) и затем перемешивали при кипячении с обратным холодильником в течение 1 часа. Когда реакция завершалась, реакционный раствор охлаждали до температуры 0°C, нейтрализовали (pH 7) карбонатом калия и затем экстрагировали смешанным раствором хлороформа и небольшого количества ацетона. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем перекристаллизовывали в диэтиловом эфире с получением указанного в заголовке соединения (17 мг, выход: 70%).
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,36 (с, 1H), 8,00 (д, 1H), 7,94 (т, 1H), 7,66 (д, 1H), 7,46 (м, 1H), 7,53 (м, 2H), 5,40 (дд, 2H), 4,86 (д, 1H), 3,03 (м, 1H), 2,95 (дд, 1H), 2,83 (д, 1H), 2,70 (м, 1H), 2,42 (м, 1H), 2,39 (м, 1H), 1,92 (д, 1H), 1,59 (д, 1H), 1,35 (m 1H), 1,23 (м, 1H).
Пример 10: Получение 1-(5-(3-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
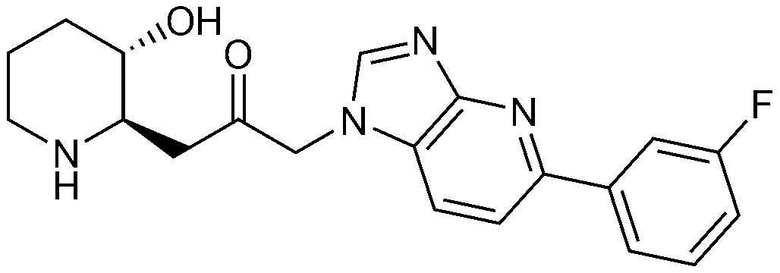
Указанное в заголовке соединение (18 мг, выход: 72%) получали таким же образом, как в примере 9, за исключением того, что вместо (2-фторфенил) бороновой кислоты использовали (3-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,34 (с, 1H), 7,99 (т, 2H), 7,93 (м, 2H), 7,54 (дд, 1H), 7,23 (дт, 1H), 5,40 (дд, 2H), 4,85 (д, 1H), 3,00 (м, 1H), 2,95 (дд, 1H), 2,83 (д, 1H), 2,69 (м, 1H), 2,45 (м, 1H), 2,38 (т, 1H), 1,92 (д, 1H), 1,59 (д, 1H), 1,35(м, 1H), 1,25 (м, 1H).
Пример 11: Получение 1-(5-(4-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
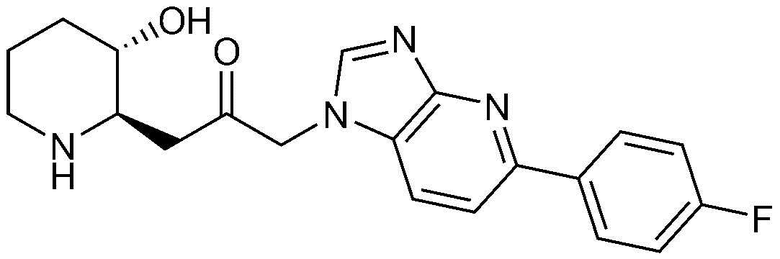
Указанное в заголовке соединение (17 мг, выход: 70%) получали таким же образом, как в примере 9, за исключением того, что вместо (2-фторфенил)бороновой кислоты использовали (4-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,32 (с, 1H), 8,17 (м, 2H), 7,89 (дд, 1H), 7,86 (дд, 1H), 7,33 (м, 2H), 5,39 (дд, 2H), 4,84 (д, 1H), 3,02 (м, 1H), 2,95 (дд, 1H), 2,83 (д, 1H), 2,69 (м, 1H), 2,43 (м, 1H), 2,38 (м, 1H), 1,92 (д, 1H), 1,59 (д, 1H), 1,35 (м, 1H), 1,25 (м, 1H).
Пример 12: Получение 1-(5-(3-хлорфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
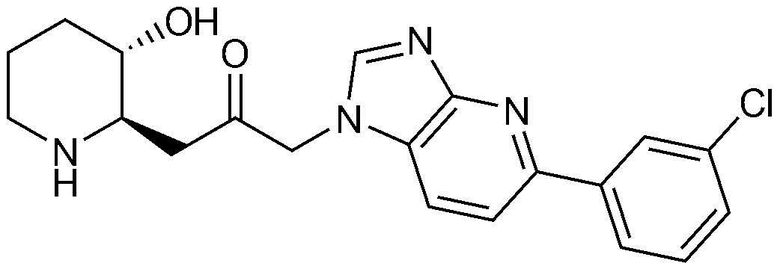
Указанное в заголовке соединение (19 мг, выход: 80%) получали таким же образом, как в примере 9, за исключением того, что вместо (2-хлорфенил)бороновой кислоты использовали (3-хлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,35 (с, 1H), 8,18 (с, 1H), 8,10 (д, 1H), 8,01 (д, 1H), 7,94 (д, 1H), 7,52 (т, 1H), 7,47 (д, 1H), 5,40 (дд, 2H), 4,85 (д, 1H), 3,05 (м, 1H), 2. 94 (дд, 1H), 2,83 (д, 1H), 2,69 (м, 1H), 2,43 (м, 1H), 2,38 (м, 1H), 1,92 (д, 1H), 1,59 (д, 1H), 1,35 (м, 1H), 1,25 (м, 1H).
Пример 13: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-(3-(трифторметил)фенил)-1H-имидазо[4,5-b]пиридин-1-ил)пропан-2-она
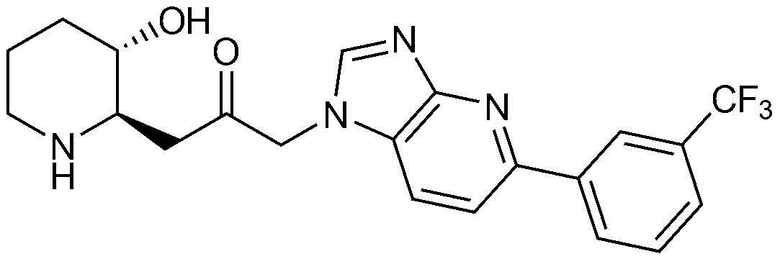
Указанное в заголовке соединение (20 мг, выход: 75%) получали таким же образом, как в примере 9, за исключением того, что вместо (2-фторфенил)бороновой кислоты использовали 3-(трифторметил)фенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,47 (с, 1H), 8,43 (м, 1H), 8,37 (д, 1H), 8,04 (м, 2H), 7,74 (м, 2H), 5,42 (дд, 2H), 4,84 (д, 1H), 3,03 (м, 1H), 2,96 (дд, 1H), 2,83 (д, 1H), 2,68 (м, 1H), 2,44 (м, 1H), 2,38 (м, 1H), 1,92 (д, 1H), 1,59 (д, 1H), 1,35 (м, 1H), 1,25 (м, 1H).
Пример 14: Получение 1-(6-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
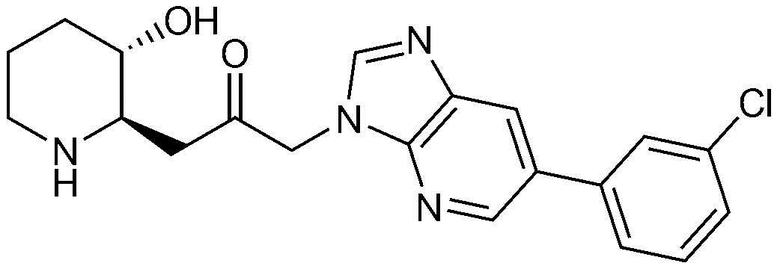
Стадия 14-1: Получение бензил (2R,3S)-2-(3-(6-бром-3H-имидазо[4,5-b]пиридин-3-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
6-Бром-3H-имидазо[4,5-b]пиридин (204 мг, 1,03 ммоль) растворяли в N,N-диметилформамиде (3 мл, 0,34 M), к нему добавляли карбонат калия (285 мг, 2,06 ммоль) и затем перемешивали при комнатной температуре в течение 10 минут. Затем при комнатной температуре в течение 3 часов добавляли бензил (2R,3S)-2-(3-бром-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (500 мг, 1,03 ммоль). Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (дихлорметан:метанол=10:1) с получением указанного в заголовке соединения (266 мг, выход: 43%).
Стадия 14-2: Получение бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(6-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-оксопропил)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-(6-бром-3H-имидазо[4,5-b]пиридин-3-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (75 мг, 0,13 ммоль), полученный на стадии 14-1, растворяли в N,N-диметилформамиде (2 мл, 0,07 M), к нему последовательно добавляли (3-хлорфенил)борную кислоту (29 мг, 0,19 ммоль), тетракис(трифенилфосфин)палладий (30 мг, 0,03 ммоль) и 2M карбонат натрия (0,25 мл, 0,5 ммоль) и затем перемешивали при комнатной температуре в течение 5 минут. Затем смеси давали взаимодействовать при температуре 150°C в течение 30 минут с использованием микроволновой печи. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=1:2) с получением указанного в заголовке соединения (62 мг, выход: 78%).
Стадия 14-3: Получение 1-(6-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
Бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(6-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-оксопропил)пиперидин-1-карбоксилат (62 мг, 0,1 ммоль), полученный на стадии 14-2, растворяли в 6н водном растворе соляной кислоты (3 мл, 0,03 M) и затем перемешивали при кипячении с обратным холодильником в течение 1 часа. Когда реакция завершалась, реакционный раствор охлаждали до температуры 0°C, нейтрализовали (pH 7) карбонатом калия и затем экстрагировали смешанным раствором хлороформа и небольшого количества ацетона. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем перекристаллизовывали в диэтиловом эфире с получением указанного в заголовке соединения (28 мг, выход: 75%).
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,65 (д, 1H), 8,41 (д, 1H), 8,35 (с, 1H), 7,84 (с, 1H), 7,74 (д, 1H), 7,48 (т, 1H), 7,44 (д, 1H), 5,36 (дд, 2H), 4,79 (д, 1H), 2,99 (м, 2H), 2,81 (д, 1H), 2,67 (м, 1H), 2,47 (м, 1H), 2,37 (м, 1H), 1,90 (м, 1H), 1,58 (д, 1H), 1,35 (м, 1H), 1,24 (м, 1H).
Пример 15: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(3-(трифторметил)фенил)-3H-имидазо[4,5-b]пиридин-3-ил)пропан-2-она
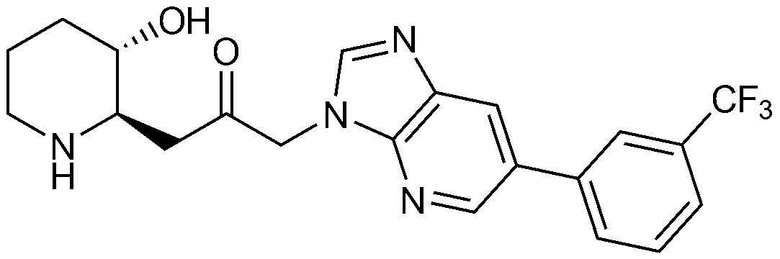
Указанное в заголовке соединение (33 мг, выход: 76%) получали таким же образом, как в примере 14, за исключением того, что вместо (3-хлорфенил)бороновой кислоты использовали (3-(трифторметил)фенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,70 (д, 1H), 8,48 (д, 1H), 8,38 (с, 1H), 8,09 (м, 2H), 7,75 (м, 1H), 5,37 (дд, 2H), 4,80 (д, 1H), 3,00 (м, 2H), 2,82 (д, 1H), 2,68 (м, 1H), 2,51 (м, 1H), 2,38 (м, 1H), 1,90 (м, 1H), 1,59 (д, 1H), 1,35 (м, 1H), 1,26 (м, 1H).
Пример 16: Получение 1-(5-(3-фторфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
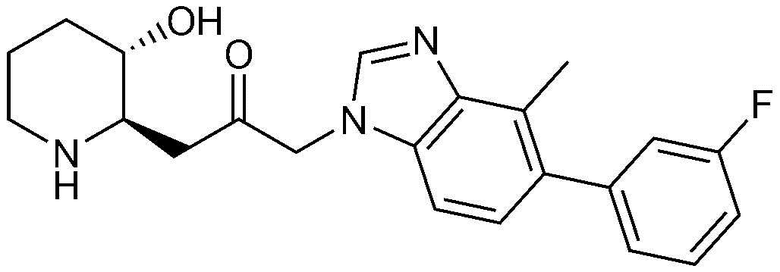
Стадия 16-1: Получение бензил (2R,3S)-2-(3-(5-бром-4-метил-1H-бензо[d]имидазол-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
5-Бром-4-метил-1H-бензо[d]имидазол (86,53 мг, 0,41 ммоль) растворяли в N,N-диметилформамиде (2 мл, 0,20 M), к нему добавляли карбонат калия (114,09 мг, 0,83 ммоль) и затем перемешивали при комнатной температуре в течение 10 минут. Затем добавляли бензил (2R,3S)-2-(3-бром-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (200 мг, 0,41 ммоль) и перемешивали при комнатной температуре в течение 3 часов. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=5:1) с получением указанного в заголовке соединения (217 мг, выход: 86%).
Стадия 16-2: Получение бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(5-(3-фторфенил)-4-метил-1H- бензо[d]имидазол-1-ил)-2-оксопропил)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-(5-бром-4-метил-1H-бензо[d]имидазол-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (50 мг, 0,08 ммоль), полученный на стадии 16-1, растворяли в N,N-диметилформамиде (1,5 мл, 0,05 M), к нему последовательно добавляли (3-фторфенил)бороновую кислоту (16,79 мг, 0,12 ммоль), тетракис(трифенилфосфин)палладий (23,11 мг, 0,02 ммоль) и 2M карбонат натрия (0,3 мл, 0,27 ммоль) и затем перемешивали при комнатной температуре в течение 5 минут. Затем смеси давали взаимодействовать при температуре 130°C в течение 45 минут с использованием микроволновой печи. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=1:3) с получением указанного в заголовке соединения (36,3 мг, выход: 72%).
Стадия 16-3: Получение 1-(5-(3-фторфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
Бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(5-(3-фторфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-2-оксопропил)пиперидин-1-карбоксилат (36,3 мг, 0,06 ммоль), полученный на стадии 16-2, растворяли в 6н водном растворе соляной кислоты (2 мл, 0,03 M) и затем перемешивали при кипячении с обратным холодильником в течение 1 часа. Когда реакция завершалась, реакционный раствор охлаждали до температуры 0°C, нейтрализовали (pH 7) карбонатом калия и затем экстрагировали смешанным раствором хлороформа и небольшого количества ацетона. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем перекристаллизовывали в диэтиловом эфире с получением указанного в заголовке соединения (11 мг, выход: 48%).
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,08 (д, 1H), 7,50 (д, 1H), 7,35 (д, 1H), 7,23-7,12 (м, 3H), 7,10 (с, 1H), 5,38-5,5,30 (м, 2H), 4,82 (д, 1H), 3,33 (с, 1H), 2,96 (д, 1H), 2,84 (д, 2H), 2,81 (с, 2H), 2,51 (с, 3H), 2,06 (с, 1H), 1,90 (д, 1H), 1,58 (д, 1H), 1,38-1,26 (м, 1H).
Пример 17: Получение 1-(5-(3-хлорфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
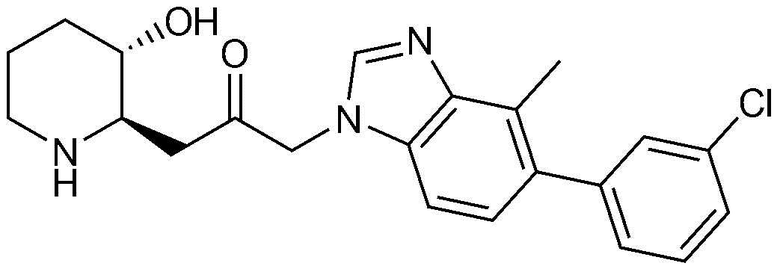
Указанное в заголовке соединение (11 мг, выход: 55%) получали таким же образом, как в примере 16, за исключением того, что вместо (3-фторфенил)бороновой кислоты использовали (3-хлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,08 (с, 1H), 7,50-7,43 (м, 3H), 7,37-7,34 (м, 2H) 7,10 (с, 1H) 5,39-5,30 (м, 2H), 4,81 (д, 1H), 3,01 (с, 1H), 2,96-2,92 (дд, 1H), 2,81 (д, 2H), 2,65 (д, 2H), 2,51 (с, 3H), 2,05 (с, 1H), 1,90 (д, 1H), 1,57 (д, 1 H), 1,38-1,26 (м, 1H).
Пример 18: Получение 1-(5-(2-хлорфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
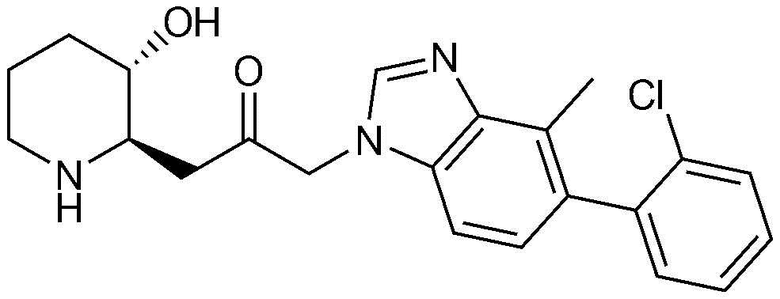
Указанное в заголовке соединение (4,0 мг, выход: 14,8%) получали таким же образом, как в примере 16, за исключением того, что вместо (3-фторфенил)бороновой кислоты использовали (2-хлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, MeOD): δ 8,11 (с, 1H), 7,65-7,62 (м, 1H), 7,57-7,50 (м, 1H) 7,37-7,28 (м, 3H) 7,04 (д, 1H), 3,49-3,48 (м, 1H), 3,11 (д, 1H), 3,00-2,98 (м, 1H), 2,62 (т, 1H), 2,57-2,55 (м, 2H), 2,35 (с, 3H), 2,04 (д, 1H), 1,77 (д, 1H), 1,58-1,56 (м, 1H), 1,42-1,40 (м, 1H).
Пример 19: Получение 1-(5-(4-хлорфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
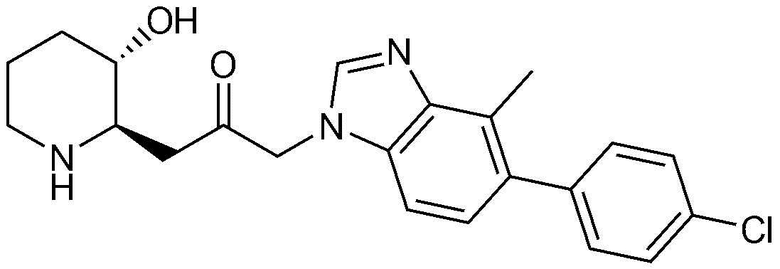
Указанное в заголовке соединение (4,0 мг, выход: 15,4%) получали таким же образом, как в примере 16, за исключением того, что вместо (3-фторфенил)бороновой кислоты использовали (4-хлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, MeOD): δ 8,10 (с, 1H), 7,42 (д, 2H), 7,34-7,32 (м, 3H), 7,15 (д, 1H), 3,48-3,44 (м, 1H), 3,16-3,07 (м, 1H), 2,93 (с, 1H), 2,86 (д, 1H), 2,53 (т, 2H), 2,51 (с, 3H), 2,03 (д, 1H), 1,73 (д, 1H), 1,55-1,53 (м, 1H), 1,39-37 (м, 1H).
Пример 20: Получение 3-(1-(3-((2R,3S)-3-гидроксипиперидин-2-ил)-2-оксопропил)-4-метил-1H-бензо[d]имидазол-5-ил)бензонитрила
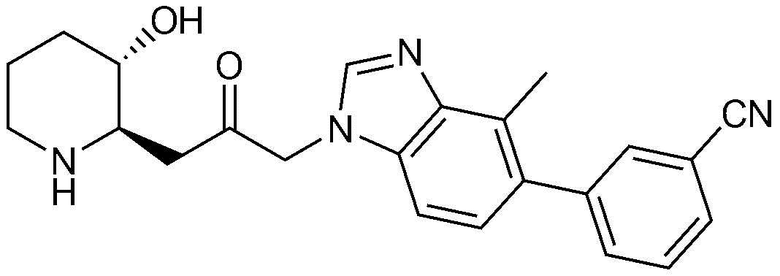
Указанное в заголовке соединение (3,5 мг, выход: 20,8%) получали таким же образом, как в примере 16, за исключением того, что вместо (3-фторфенил)бороновой кислоты использовали (3-цианофенил)бороновую кислоту.
1H-ЯМР (500 МГц, MeOD): δ 8,12 (с, 1H), 7,72-7,56 (м, 4H), 7,36 (д, 1H), 7,36 (д, 1H), 3,49-3,45 (м, 1H), 3,16-3,08 (м, 1H), 2,94 (с, 1H), 2,88 (д, 1H), 2,60-2,55 (м, 2H), 2,52 (с, 3H), 2,04 (д, 1H), 1,73 (д, 1H), 1,56-1,53 (м, 1H), 1,40-38 (м, 1H).
Пример 21: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-метил-5-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-она
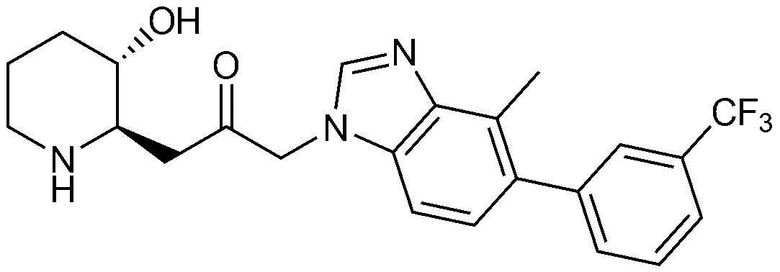
Указанное в заголовке соединение (8,6 мг, выход: 38,2%) получали таким же образом, как в примере 16, за исключением того, что вместо (3-фторфенил)бороновой кислоты использовали (3-(трифторметил)фенил)бороновую кислоту.
1H-ЯМР (500 МГц, MeOD): δ 8,12 (с, 1H), 7,65-7,61 (м, 4H), 7,36 (д, 1H), 7,18 (д, 1H), 3,22-3,07 (м, 2H), 2,92 (д, 2H), 2,87-2,84 (м, 2H), 2,57-2,55 (м, 2H), 2,52 (с, 3H), 2,04 (д, 1H), 1,72 (д, 1H), 1,54-1,52 (м, 1H), 1,41-1,36 (м, 1H).
Пример 22: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-(3-метоксифенил)-4-метил-1H-бензо[d]имидазол-1-ил)пропан-2-она
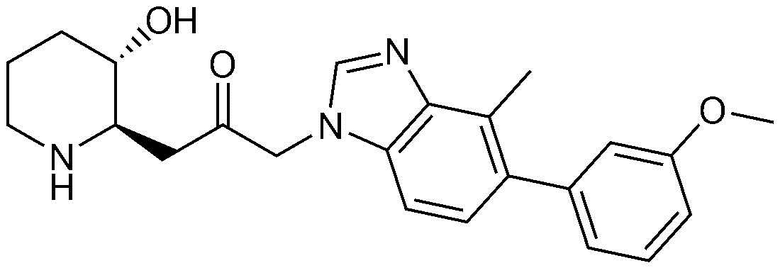
Указанное в заголовке соединение (5,0 мг, выход: 33,5%) получали таким же образом, как в примере 16, за исключением того, что вместо (3-фторфенил)бороновой кислоты использовали (3-метоксифенил)бороновую кислоту.
1H-ЯМР (500 МГц, MeOD): δ 8,09 (с, 1H), 7,35-7,29 (м, 2H), 7,16 (д, 1H), 6,91-6,87 (м, 3H), 3,82 (с, 3H), 3,51-3,44 (м, 1H), 3,23-3,07 (м, 1H), 2,93 (д, 1H), 2,88-2,87 (м, 1H), 2,59-2,54 (м, 2H), 2,51 (с, 3H), 2,03 (д, 1H), 1,73 (д, 1H), 1,55-1,52 (м, 1H), 1,41-1,37 (м, 1H).
Пример 23: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-метил-5-(тиазол-4-ил)-1H-бензо[d]имидазол-1-ил)пропан-2-она
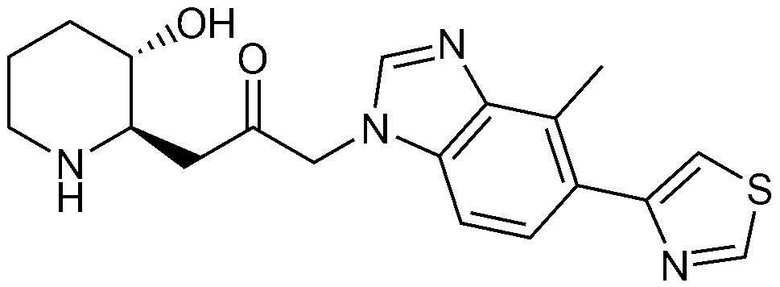
Указанное в заголовке соединение (10 мг, выход: 50%) получали таким же образом, как в примере 16, за исключением того, что вместо (3-фторфенил)бороновой кислоты использовали (тиазол-4-ил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,00 (с, 1H), 7,60 (м, 1H), 7,25 (д, 1H), 7,09 (т, 1H), 6,99 (д, 1H), 5,29 (м, 2H), 4,82 (д, 1H), 3,01 (м, 1H), 2,92 (дд, 1H), 2,82 (д, 1H), 2,67 (д, 2H), 2,54 (с, 3H), 2,41 (м, 2H), 1,90 (д, 1H), 1,58 (д, 1H), 1,21-1,37 (м, 2H).
Пример 24: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-она
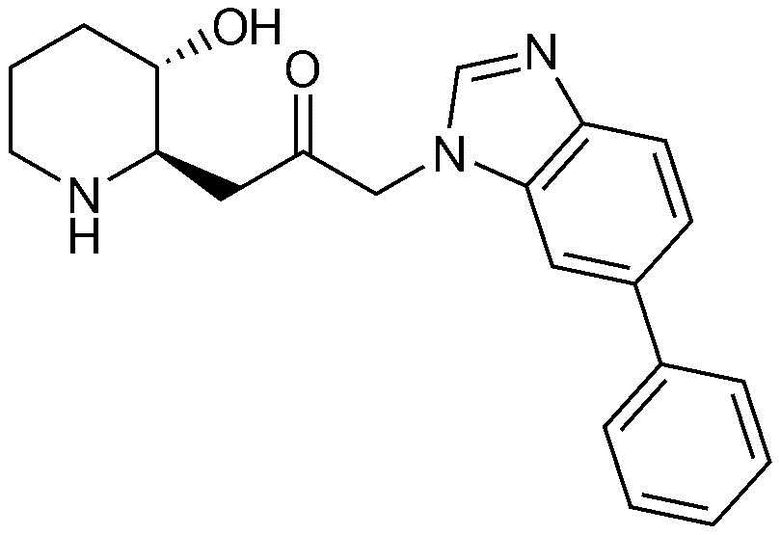
Указанное в заголовке соединение (22 мг, выход: 75%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера вместо 4-бром-3-фтор-2-нитробензола использовали 14-бром-1-фтор-2-нитробензол.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,06 (с, 1H), 7,77 (д, 1H), 7,66 (м, 3H), 7,52 (м, 1H), 7,43 (м, 2H), 7,33 (м, 1H), 5,37 (д, 2H), 4,92 (м, 1H), 3,05 (м, 1H), 2,97 (м, 1H), 2,81 (м, 1H), 2,68 (м, 1H), 2,36 (м, 2H), 1,85 (м, 1H), 1,55 (м, 1H), 1,36 (м, 1H), 1,22 (м, 1H)
Пример 25: Получение 1-(6-(3-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
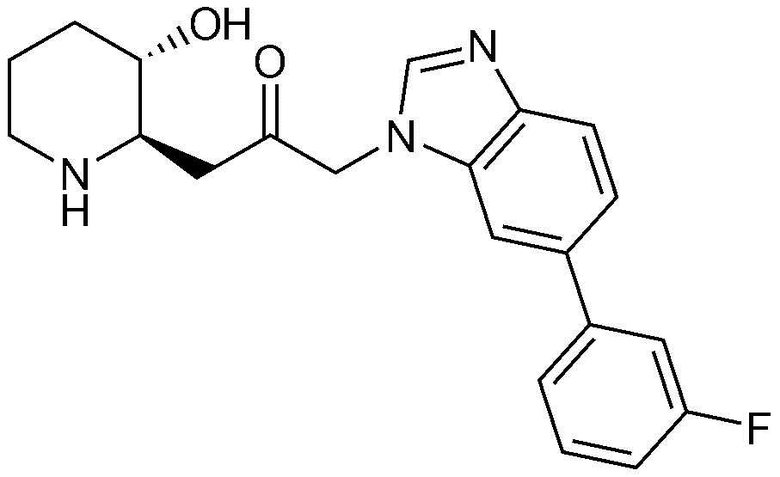
Указанное в заголовке соединение (19 мг, выход: 77%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 14-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (3-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6):δ 8,10 (с, 1H), 7,86 (д, 1H), 7,71 (д, 1H), 7,55 (м, 2H), 7,51 (м, 2H), 7,17 (м, 1H), 5,38 (дд, 2H), 4,82 (м, 1H), 3,01 (м, 1H), 2,98 (дд, 1H), 2,82 (д, 1H), 2,69 (м, 1H), 2,37 (м, 1H), 2,11 (м, 1H), 1,90 (д, 1H), 1,56 (м, 1H), 1,35 (м, 1H), 1,23 (м, 1H)
Пример 26: Получение 1-(6-(2-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
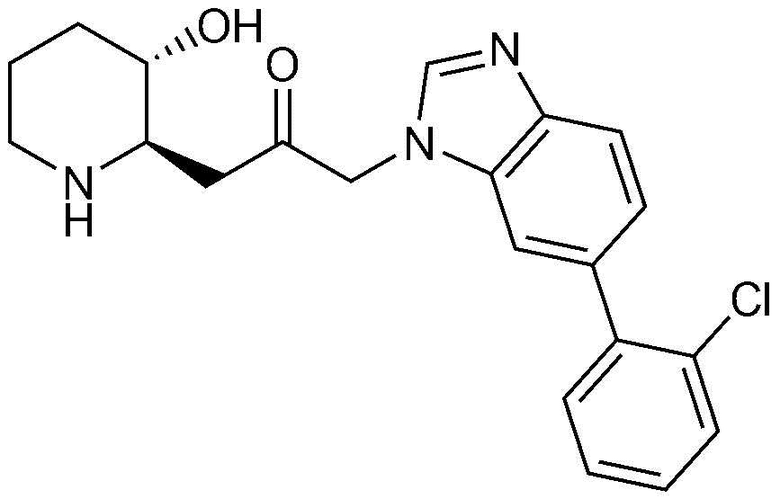
Указанное в заголовке соединение (17 мг, выход: 70%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера вместо 1-бром-3-фтор-2-нитробензола использовали 14-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (2-хлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,10 (с, 1H), 7,71 (д, 1H), 7,58 (с, 1H), 7,57 (д, 1H), 7,40 (м, 3H), 7,26 (д, 1H), 5,36 (дд, 2H), 4,77 (д, 1H), 2,99 (м, 1H), 2,94 (дд, 1H), 2,75 (д, 1H), 2,66 (м, 1H), 2,43 (дд, 1H), 2,33 (т, 1H), 1,88 (д, 1H), 1,55 (д, 1H), 1,33 (м, 1H), 1,22 (м, 1H).
Пример 27: Получение 1-(6-(3-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
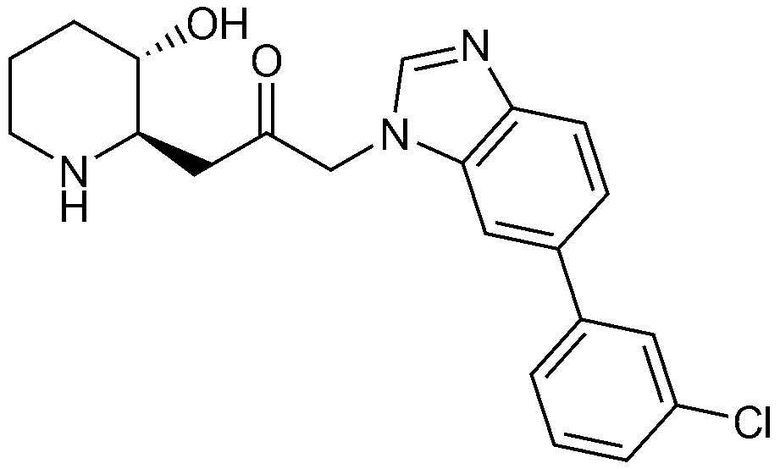
Указанное в заголовке соединение (18 мг, выход: 72%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера вместо 1-бром-3-фтор-2-нитробензола использовали 14-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (3-хлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,09 (с, 1H), 7,86 (с, 1H), 7,72 (д, 2H), 7,67 (д, 1H), 7,54 (д, 1H), 7,48 (т, 1H), 7,40 (д, 1H), 5,40 (дд, 2H), 4,83 (д, 1H), 3,01 (м, 1H), 2,89 (дд, 1H), 2,84 (д, 1H), 2,70 (м, 1H), 2,45 (м, 1H), 2,36 (м, 1H), 1,90 (м, 1H), 1,59 (д, 1H), 1,37 (м, 1H), 1,24 (м, 1H).
Пример 28: Получение 1-(6-(4-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
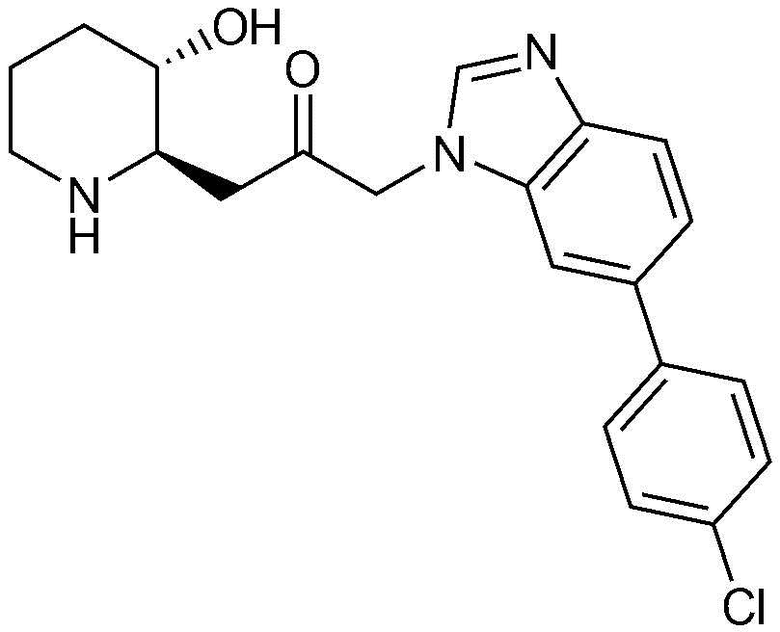
Указанное в заголовке соединение (17 мг, выход: 70%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера вместо 1-бром-3-фтор-2-нитробензола использовали 14-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (4-хлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,08 (с, 1H), 7,82 (с, 1H), 7,71 (m. 3H), 7,52 (м, 3H), 5,39 (дд, 2H), 4,82 (д, 1H), 3,01 (м, 1H), 2,95 (дд, 1H), 2,82 (д, 1H), 2,69 (м, 1H), 2,43 (м, 1H), 2,38 (м, 1H), 1,92 (д, 1H), 1,59 (д, 1H), 1,36 (м, 1H), 1,25 (м, 1H).
Пример 29: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-она
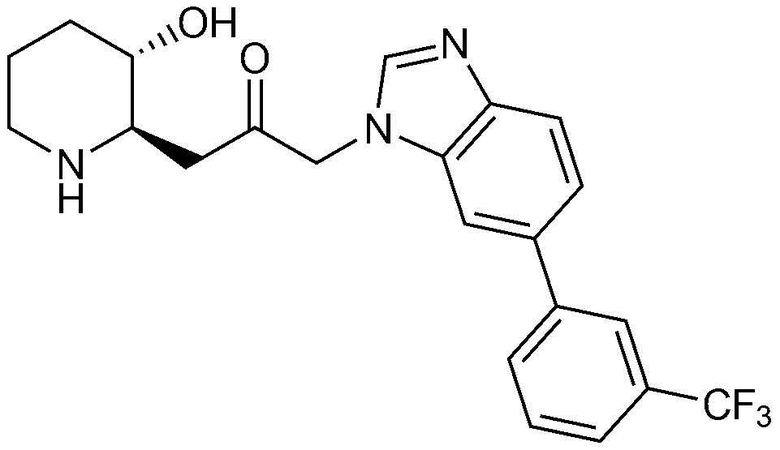
Указанное в заголовке соединение (18 мг, выход: 73%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера вместо 1-бром-3-фтор-2-нитробензола использовали 14-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (3-трифторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,11 (с, 1H), 7,99 (м, 1H), 7,96 (с, 1H), 7,75 (д, 1H), 7,70 (д, 2H), 7,58 (м, 1H), 5,39 (дд, 2H), 4,85 (д, 1H), 3,02 (м, 1H), 2,97 (дд, 1H), 2,82 (д, 1H), 2,71 (м, 1H), 2,44 (м, 1H), 2,38 (м, 1H), 1,91 (м, 1H), 1,58 (д, 1H), 1,34 (м, 1H), 1,25 (м, 1H).
Пример 30: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(м-толил)-1H-бензо[d]имидазол-1-ил)пропан-2-она
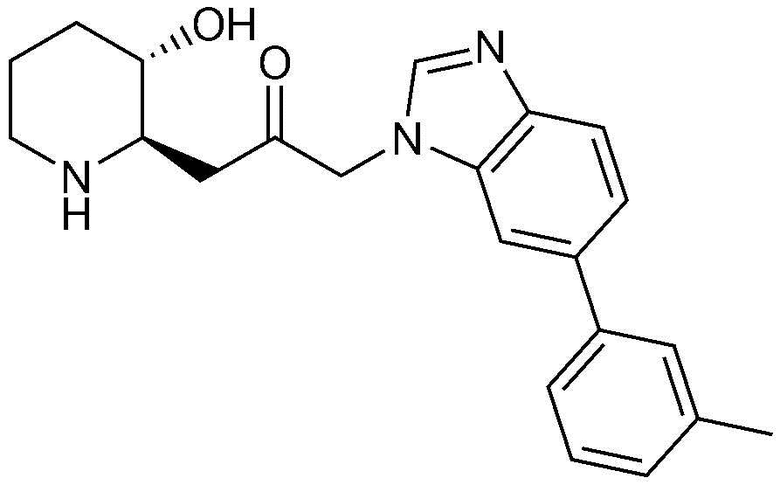
Указанное в заголовке соединение (20 мг, выход: 70%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера вместо 1-бром-3-фтор-2-нитробензола использовали 14-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (3-метилфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,06 (с, 1H), 7,78 (д, 1H), 7,69 (д, 1H), 7,49 (д, 1H), 7,45 (м, 2H), 7,34 (м, 1H), 7,15 (м, 1H), 5,41 (дд, 2H), 4,81 (м, 1H), 3,02 (м, 1H), 2,95 (дд, 1H), 2,82 (д, 1H), 2,68 (м, 1H), 2,45 (м, 2H), 2,36 (с, 3H), 1,90 (м, 1H), 1,57 (м, 1H), 1,35 (м, 1H), 1,24 (м, 1H).
Пример 31: Получение 1-(6-(3,5-дихлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
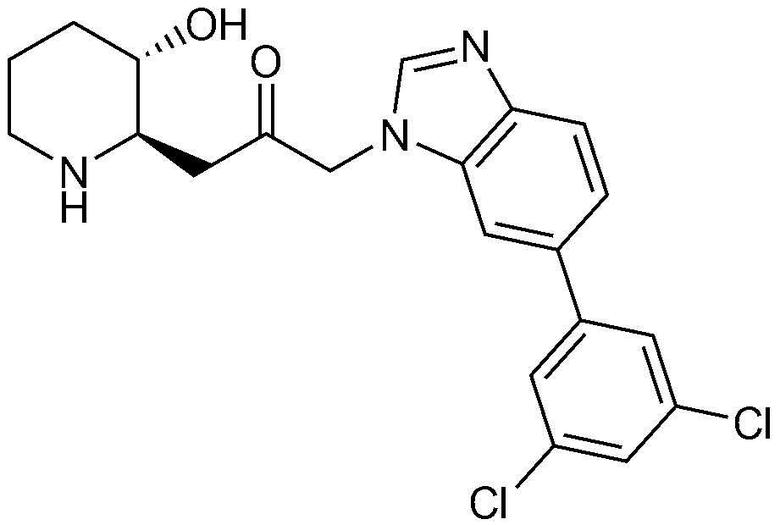
Указанное в заголовке соединение (19 мг, выход: 77%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера вместо 1-бром-3-фтор-2-нитробензола использовали14-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (3,5-дихлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,12 (с, 1H), 7,92 (с, 1H), 7,73 (м, 3H), 7,59 (м, 2H), 5,41 (дд, 2H), 4,89 (д, 1H), 3,05 (м, 1H), 2,99 (дд, 1H), 2,87 (д, 1H), 2,74 (м, 1H), 2,45 (м, 2H), 1,92 (д, 1H), 1,58 (д, 1H), 1,36 (м, 1H), 1,24 (м, 1H).
Пример 32: Получение 1-(6-(3-хлор-5-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
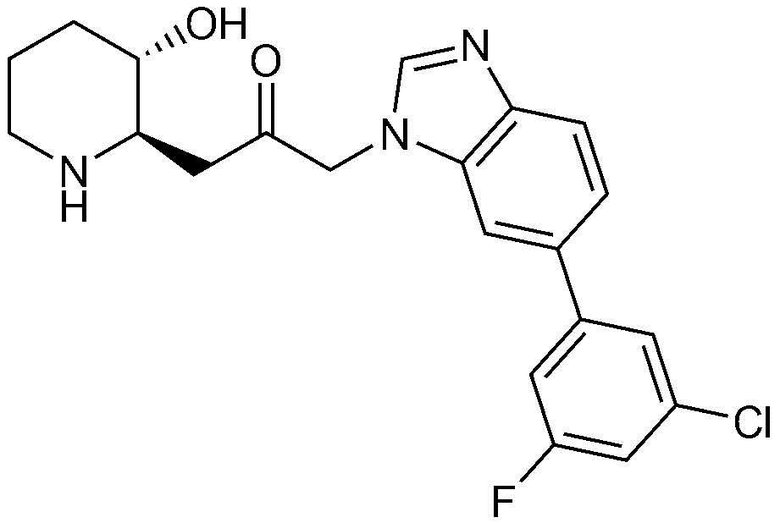
Указанное в заголовке соединение (17 мг, выход: 70%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали4-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (3-хлор-5-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,21 (с, 1H), 7,92 (с, 1H), 7,73 (д, 1H), 7,63 (с, 1H), 7,59 (дд, 1H), 7,39 (дд, 1H), 5,40 (дд, 2H), 4,87 (д, 1H), 3,03 (м, 1H), 2,99 (дд, 1H), 2,85 (д, 1H), 2,72 (м, 1H), 2,44 (м, 2H), 1,92 (с, 1H), 1,60 (д, 1H), 1,35 (м, 1H), 1,26 (м, 1H).
Пример 33: Получение 1-(6-(3-хлор-4-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
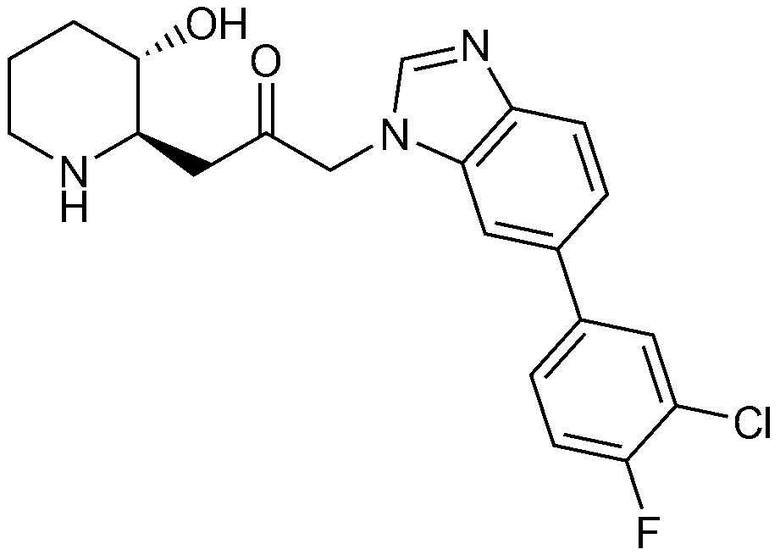
Указанное в заголовке соединение (18 мг, выход: 74%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 4-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали(3-хлор-4-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,09 (с, 1H), 7,86 (м, 2H), 7,69 (м, 2H), 7,51 (м, 2H), 5,39 (дд, 2H), 4,83 (д, 1H), 3,00 (м, 1H), 2,98 (дд, 1H), 2,83 (д, 1H), 2,70 (м, 1H), 2,41 (м, 2H), 1,91 (д, 1H), 1,59 (д, 1H), 1,34 (м, 1H), 1,27 (м, 1H).
Пример 34: Получение 1-(6-(3-хлор-5-метилфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
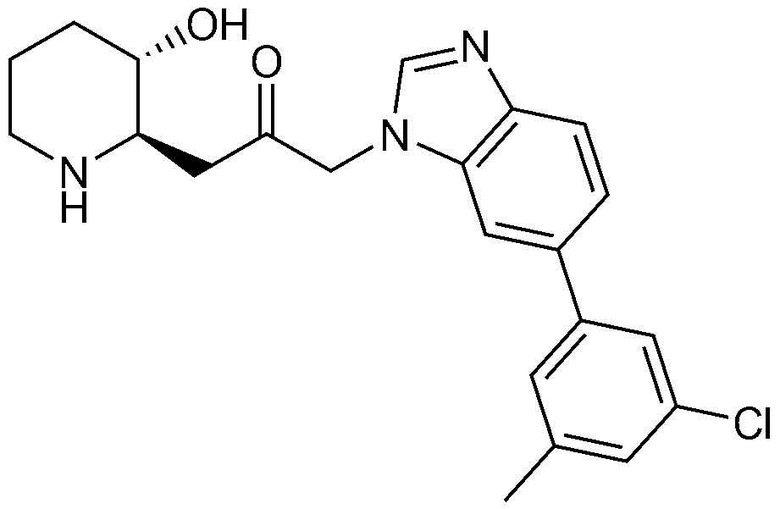
Указанное в заголовке соединение (17 мг, выход: 70%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 4-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали(3-хлор-5-метилфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,09 (с, 1H), 7,84 (с, 1H), 7,71 (д, 1H), 7,51 (м, 3H), 7,23 (с, 1H), 5,38 (дд, 2H), 4,88 (д, 1H), 3,05 (м, 1H), 2,89 (дд, 1H), 2,86 (д, 1H), 2,73 (м, 1H), 2,44 (м, 2H), 2,37 (с, 3H), 1,92 (д, 1H), 1,60 (д, 1H), 1,36 (м, 1H), 1,27 (м, 1H).
Пример 35: Получение 1-(6-(3-хлор-5-метоксифенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
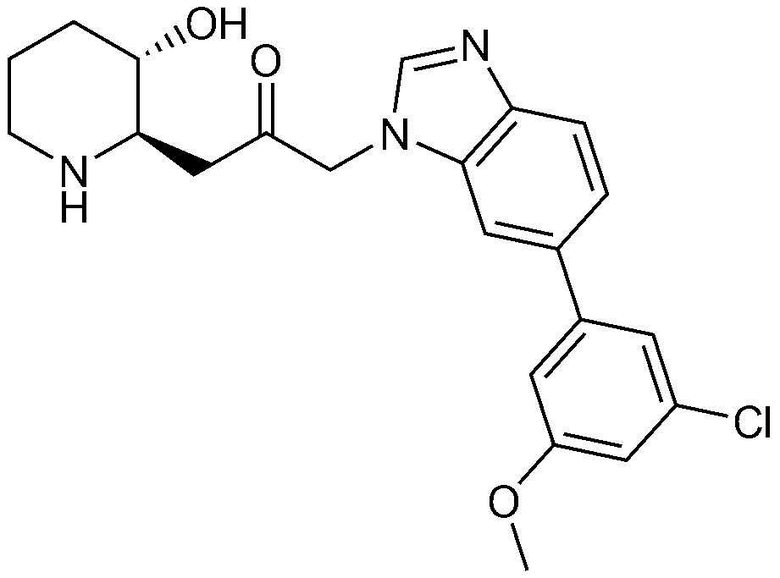
Указанное в заголовке соединение (19 мг, выход: 75%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 4-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (3-хлор-5-метоксифенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,09 (с, 1H), 7,85 (с, 1H), 7,70 (д, 1H), 7,55 (д, 1H), 7,35 (с, 1H), 7,19 (с, 1H), 7,00 (с, 1H), 5,40 (дд, 2H), 4,91 (д, 1H), 3,81 (с, 3H), 3,06 (м, 1H), 2,96 (дд, 1H), 2,87 (д, 1H), 2,75 (м, 1H), 2,40 (м, 1H), 1,89 (д, 1H), 1,60 (д, 1H), 1,36 (м, 1H), 1,27 (м, 1H).
Пример 36: Получение 3-хлор-5-(1-(3-((2R,3S)-3-гидроксипиперидин-2-ил)-2-оксопропил)-1H-бензо[d]имидазол-6-ил)бензонитрила
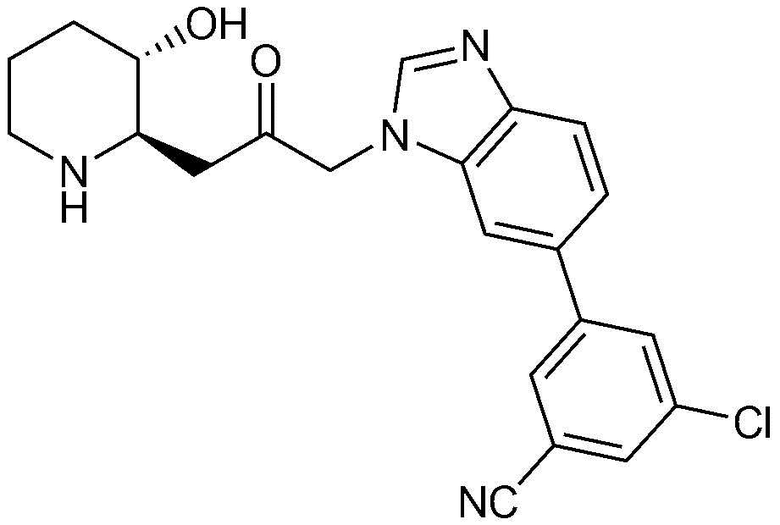
Указанное в заголовке соединение (17 мг, выход: 69%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 4-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (3-хлор-5-цианофенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,14 (м, 3H), 7,99 (м, 2H), 7,75 (д, 1H), 7,65 (д, 1H), 5,39 (дд, 2H), 4,90 (д, 1H), 3,05 (м, 1H), 3,00 (дд, 1H), 2,86 (д, 1H), 2,74 (м, 1H), 2,42 (м, 1H), 1,92 (д, 1H), 1,60 (д, 1H), 1,36 (м, 1H), 1,27 (м, 1H).
Пример 37: Получение 1-(6-(1H-пиразол-4-ил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он
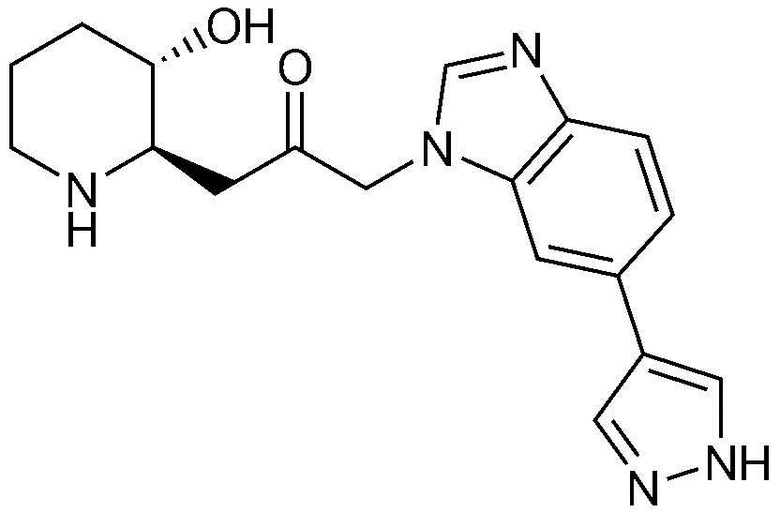
Указанное в заголовке соединение (10 мг, выход: 50%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 4-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)-1H-пиразол-1-карбоксилат.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,10 (с, 1H), 7,89 (с, 1H), 7,87 (с, 1H), 7,66 (с, 1H), 7,60 (д, 1H), 7,45 (д, 1H), 5,32 (дд, 2H), 4,88 (д, 1H), 3,04 (м, 1H), 2,97 (дд, 1H), 2,85 (д, 1H), 2,72 (м, 1H), 2,45 (м, 2H), 1,93 (м, 1H), 1,58 (м, 1H), 1,36 (м, 1H), 1,27 (м, 1H).
Пример 38: Получение 1-(6-(1H-пиразол-3-ил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она

Указанное в заголовке соединение (14 мг, выход: 60%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 4-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (1-(трет-бутокси-карбонил)-1H-пиразол-3-ил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,10 (с, 1H), 7,77 (м, 3H), 7,30 (м, 1H), 6,53 (д, 1H), 5,32 (дд, 2H), 4,88 (д, 1H), 3,04 (м, 1H), 2,97 (дд, 1H), 2,85 (д, 1H), 2,72 (м, 1H), 2,45 (м, 2H), 1,93 (м, 1H), 1,58 (м, 1H), 1,36 (м, 1H), 1,27 (м, 1H).
Пример 39: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(тиазол-4-ил)-1H-бензо[d]имидазол-1-ил)пропан-2-она
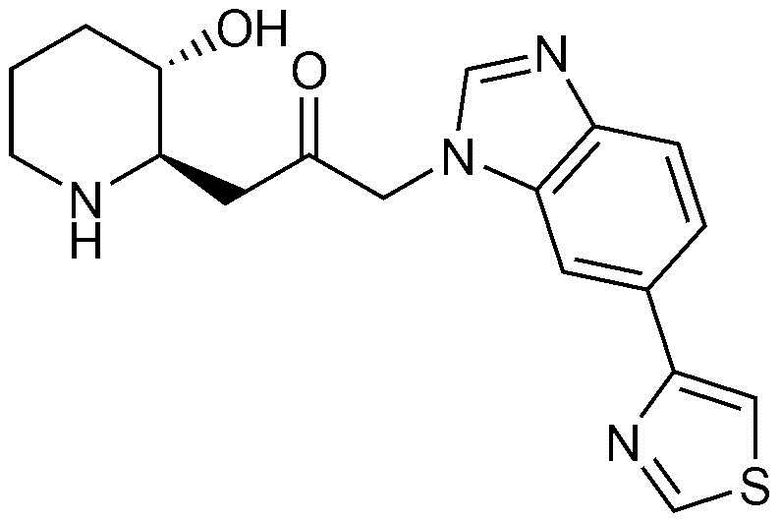
Указанное в заголовке соединение (15 мг, выход: 62%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 4-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали тиазол-4-ил бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 9,19 (с, 1H), 8,10 (м, 3H), 7,85 (д, 1H), 7,69 (д, 1H), 5,39 (дд, 2H), 4,82 (д, 1H), 3,01 (м, 1H), 2,99 (дд, 1H), 2,85 (д, 1H), 2,69 (м, 1H), 2,44 (м, 2H), 1,91 (м, 1H), 1,60 (д, 1H), 1,38 (м, 1H), 1,25 (м, 1H).
Пример 40: Получение 5-(1-(3-((2R,3S)-3-гидроксипиперидин-2-ил)-2-оксопропил)-1H-бензо[d]имидазол-6-ил)пиридин-2(1H)-она
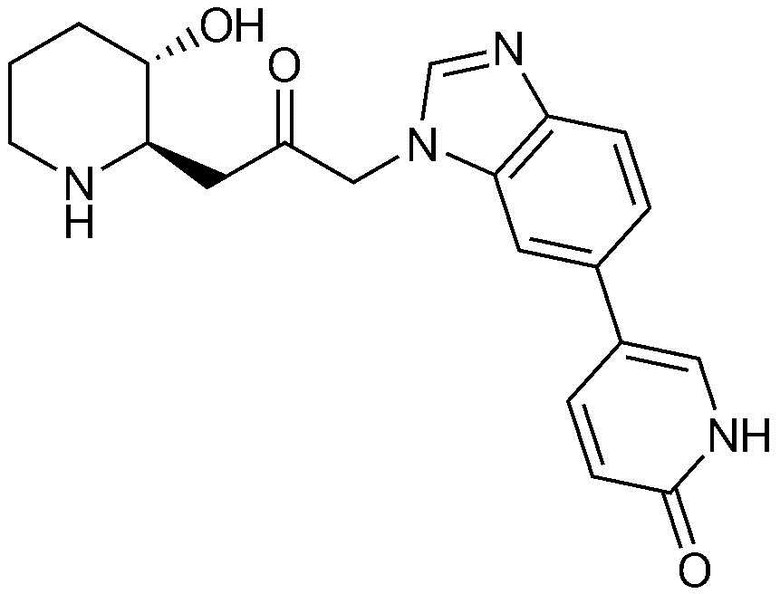
Указанное в заголовке соединение (13 мг, выход: 55%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 4-бром-2-фтор-1-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали(6-оксо-1,6-дигидропиридин-3-ил)борную кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,06 (д, 1H), 8,04 (с, 1H), 7,82 (дд, 1H), 7,67 (м, 2H), 7,41 (д, 1H), 6,50 (м, 1H), 5,33 (дд, 2H), 4,90 (д, 1H), 3,05 (м, 1H), 2,98 (дд, 1H), 2,85 (д, 1H), 2,73 (м, 1H), 2,42 (м, 2H), 1,90 (м, 1H), 1,61 (д, 1H), 1,38 (м, 1H), 1,29 (м, 1H).
Пример 41: Получение 1-(6-(3-хлорфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
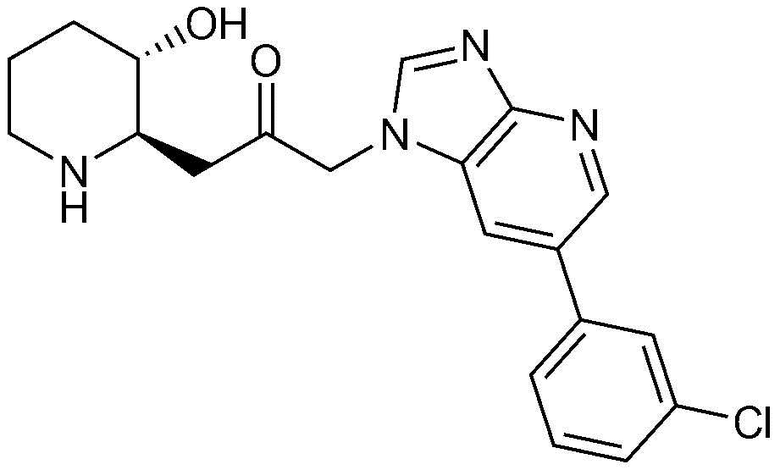
Стадия 41-1: Получение бензил (2R,3S)-2-(3-(6-бром-1H-имидазо[4,5-b]пиридин-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
6-Бром-3H-имидазо[4,5-b]пиридин (204 мг, 1,03 ммоль) растворяли в N,N-диметилформамиде (3 мл, 0,34 M), к нему добавляли карбонат калия (285 мг, 2,06 ммоль) и затем перемешивали при комнатной температуре в течение 10 минут. Затем добавляли бензил (2R,3S)-2-(3-бром-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (500 мг, 1,03 ммоль) и перемешивали при комнатной температуре в течение 3 часов. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (дихлорметан:метанол=10:1) с получением указанного в заголовке соединения (266 мг, выход: 43%).
Стадия 41-2: Получение бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(6-(3-хлорфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-2-оксопропил)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-(6-бром-1H-имидазо[4,5-b]пиридин-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (75 мг, 0,13 ммоль), полученный на стадии 41-1, растворяли в N,N-диметилформамиде (2 мл, 0,07 M), к нему последовательно добавляли (3-хлорфенил)бороновую кислоту (29 мг, 0,19 ммоль), тетракис(трифенилфосфин)палладий (30 мг, 0,03 ммоль) и 2M карбонат натрия (0,25 мл, 0,5 ммоль) и затем перемешивали при комнатной температуре в течение 5 минут. Затем смеси давали взаимодействовать при температуре 150°C в течение 30 минут с использованием микроволновой печи. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=1:2) с получением указанного в заголовке соединения (62 мг, выход: 78%).
Стадия 41-3: Получение 1-(6-(3-хлорфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
Бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(6-(3-хлорфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-2-оксопропил)пиперидин-1-карбоксилат (62 мг, 0,1 ммоль), полученный на стадии 41-2, растворяли в 6н водном растворе соляной кислоты (3 мл, 0,03 M) и затем перемешивали при кипячении с обратным холодильником в течение 1 часа. Когда реакция завершалась, реакционный раствор охлаждали до температуры 0°C, нейтрализовали (pH 7) карбонатом калия и затем экстрагировали смешанным раствором хлороформа и небольшого количества ацетона. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем перекристаллизовывали в диэтиловом эфире с получением указанного в заголовке соединения (28 мг, выход: 75%).
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,76 (д, 1H), 8,35 (с, 1H), 8,30 (д, 1H), 7,78 (с, 1H), 7,73 (д, 1H), 7,53 (т, 1H), 7,47 (д, 1H), 5,43 (дд, 2H), 4,86 (д, 1H), 3,04 (м, 1H), 2,99 (дд, 1H), 2,83 (д, 1H), 2,71 (м, 1H), 2,46 (м, 1H), 2,39 (м, 1H), 1,90 (м, 1H), 1,59 (д, 1H), 1,35 (м, 1H), 1,23 (м, 1H).
Пример 42: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(3-(трифторметил)фенил)-1H-имидазо[4,5-b]пиридин-1-ил)пропан-2-она
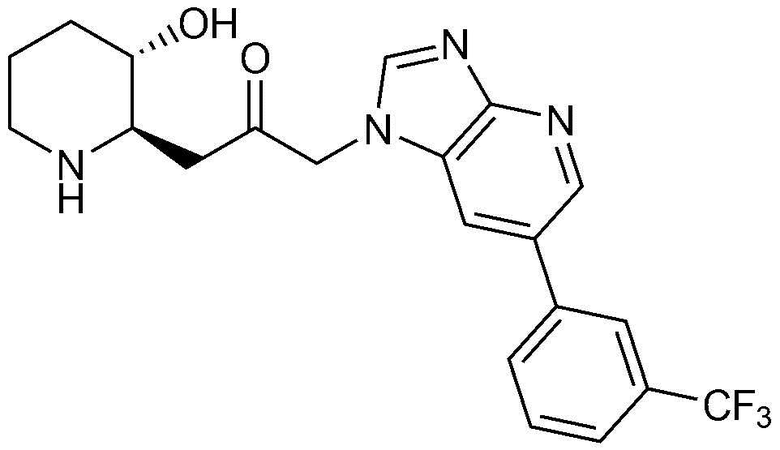
Указанное в заголовке соединение (33 мг, выход: 76%) получали таким же образом, как в примере 41, за исключением того, что вместо (3-хлорфенил)бороновой кислоты использовали (3-(трифторметил)фенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,80 (д, 1H), 8,37 (с, 1H), 8,34 (д, 1H), 8,07 (д, 1H), 8,03 (с, 1H), 7,76 (м, 2H), 5,45 (дд, 2H), 4,87 (д, 1H), 3,03 (м, 1H), 2,98 (дд, 1H), 2,81 (д, 1H), 2,71 (м, 1H), 2,46 (м, 1H), 2,38 (м, 1H), 1,91 (м, 1H), 1,58 (д, 1H), 1,36 (м, 1H), 1,24 (м, 1H).
Пример 43: Получение 1-(5-(2-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
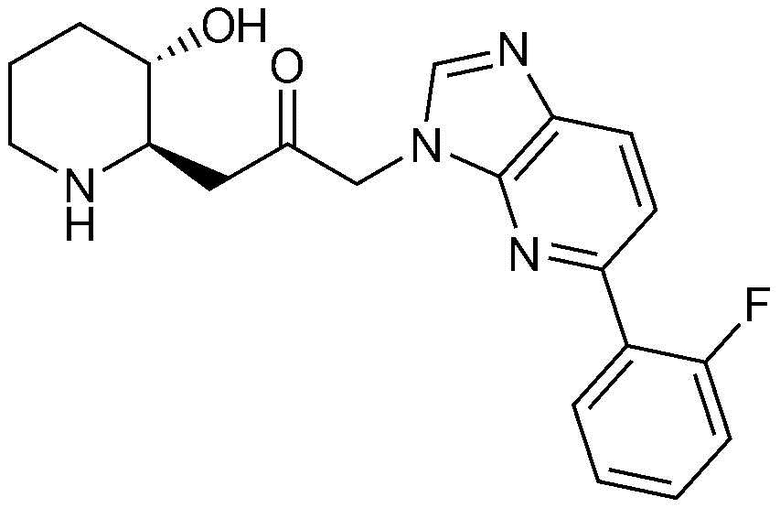
Стадия 43-1: Получение бензил (2R,3S)-2-(3-(5-бром-3H-имидазо[4,5-b]пиридин-3-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
5-Бром-1H-имидазо[4,5-b]пиридин (245 мг, 1,24 ммоль) растворяли в N,N-диметилформамиде (5 мл, 0,25 M), к нему добавляли карбонат калия (324 мг, 2,48 ммоль) и затем перемешивали при комнатной температуре в течение 10 минут. Затем добавляли бензил (2R,3S)-2-(3-бром-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (600 мг, 1,24 ммоль) и перемешивали при комнатной температуре в течение 3 часов. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (дихлорметан:метанол=10:1) с получением указанного в заголовке соединения (300 мг, выход: 40%).
Стадия 43-2: Получение бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(5-(2-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-оксопропил)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-(5-бром-3H-имидазо[4,5-b]пиридин-3-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (50 мг, 0,08 ммоль), полученный на стадии 43-1, растворяли в N,N-диметилформамиде (2 мл, 0,05 M), к нему последовательно добавляли (2-фторфенил)бороновую кислоту (18 мг, 0,13 ммоль), тетракис(трифенилфосфин)палладий (20 мг, 0,02 ммоль) и 2M карбонат натрия (0,2 мл, 0,33 ммоль) и затем перемешивали при комнатной температуре в течение 5 минут. Затем смеси давали взаимодействовать при температуре 150°C в течение 30 минут с использованием микроволновой печи. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=1:2) с получением указанного в заголовке соединения (40 мг, выход: 78%).
Стадия 43-3: Получение 1-(5-(2-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
Бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(5-(2-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-оксопропил)пиперидин-1-карбоксилат (40 мг, 0,06 ммоль), полученный на стадии 43-2, растворяли в 6н водном растворе соляной кислоты (3 мл, 0,02 M) и перемешивали при кипячении с обратным холодильником в течение 1 часа. Когда реакция завершалась, реакционный раствор охлаждали до температуры 0°C, нейтрализовали (pH 7) карбонатом калия и затем экстрагировали смешанным раствором хлороформа и небольшого количества ацетона. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем перекристаллизовывали в диэтиловом эфире с получением указанного в заголовке соединения (17 мг, выход: 70%).
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,37 (с, 1H), 8,19 (д, 1H), 7,95 (т, 1H), 7,71 (д, 1H), 7,48 (дд, 1H), 7,34 (м, 2H), 5,38 (дд, 2H), 4,77 (д, 1H), 3,01 (дд, 2H), 2,78 (д, 1H), 2,68 (м, 1H), 2,46 (м, 1H), 2,356 (т, 1H), 1,93 (м, 1H), 1,57 (д, 1H), 1,33 (м, 1H), 1,23 (м, 1H).
Пример 44: Получение 1-(5-(3-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
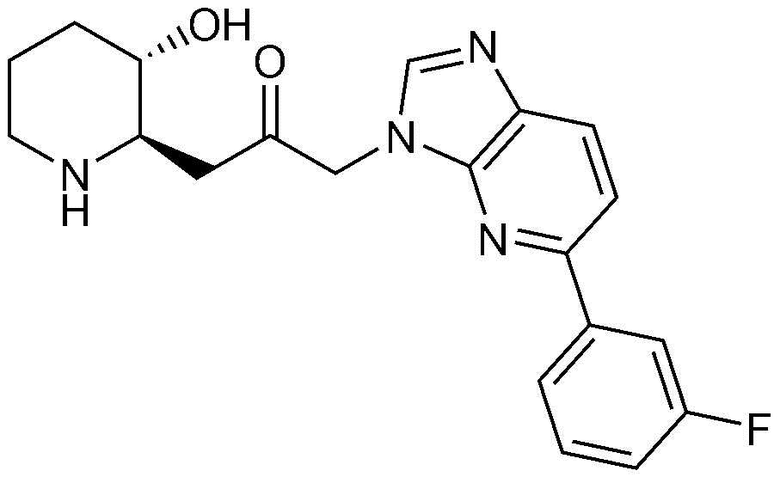
Указанное в заголовке соединение (18 мг, выход: 72%) получали таким же образом, как в примере 43, за исключением того, что вместо (2-фторфенил)бороновой кислоты использовали (3-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,34 (с, 1H), 8,18 (д, 1H), 7,96 (м, 3H), 7,53 (м, 2H), 7,24 (дт, 1H), 5,41 (дд, 2H), 4,80 (д, 1H), 3,02 (м, 2H), 2,82 (д, 1H), 2,70 (м, 1H), 2,54 (м, 1H), 2,38 (м, 1H), 1,92 (м, 1H), 1,59 (д, 1H), 1,37 (м, 1H), 1,27 (м, 1H).
Пример 45: Получение 1-(5-(4-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
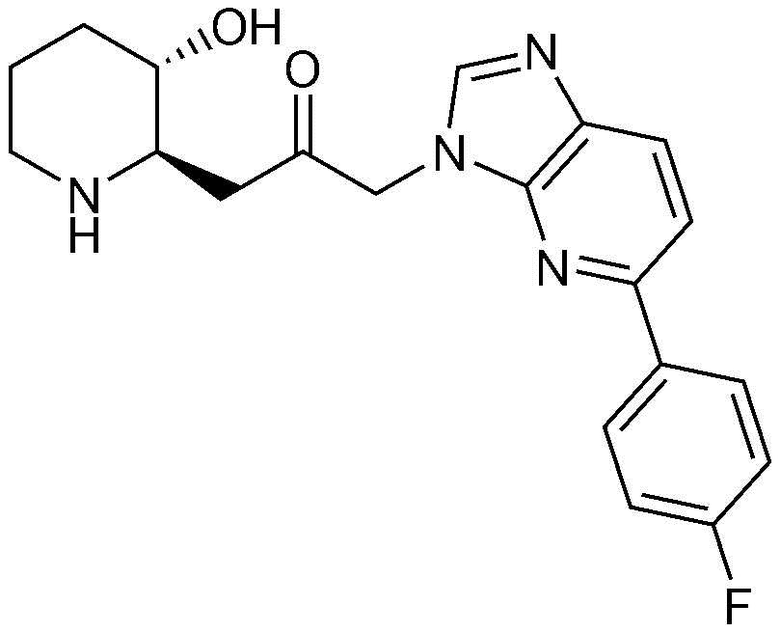
Указанное в заголовке соединение (17 мг, выход: 70%) получали таким же образом, как в примере 43, за исключением того, что вместо (2-фторфенил)бороновой кислоты использовали(4-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,32 (с, 1H), 8,17 (м, 3H), 7,87 (д, 1H), 7,31(т, 2H), 5,38 (дд, 2H), 4,78 (д, 1H), 3,02 (м, 2H), 2,81 (д, 1H), 2,68 (м, 1H), 2,48 (м, 1H), 2,36 (т, 1H), 1,92 (м, 1H), 1,58 (д, 1H), 1,34 (м, 1H), 1,24 (м, 1H).
Пример 46: Получение 1-(5-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
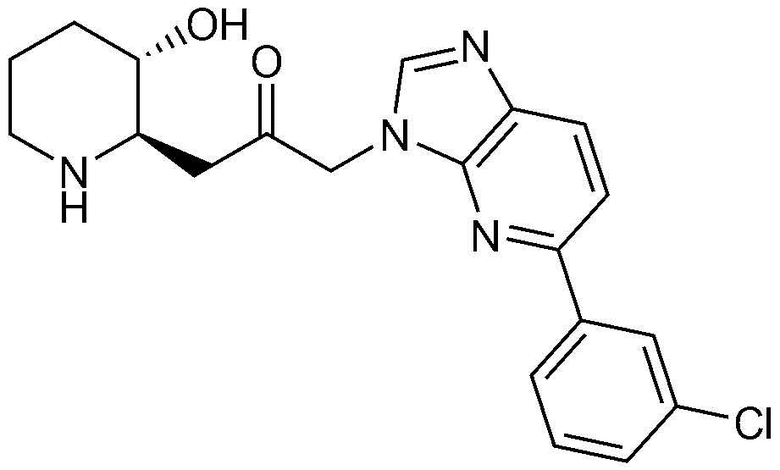
Указанное в заголовке соединение (19 мг, выход: 80%) получали таким же образом, как в примере 43, за исключением того, что вместо (2-фторфенил)бороновой кислоты использовали (3-хлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,34 (с, 1H), 8,18 (м, 2H), 8,10 (д, 1H), 7,95 (д, 1H), 7,51 (т, 1H), 7,48 (д, 1H), 5,41 (дд, 1H), 4,79 (д, 1H), 3,01 (м, 2H), 2,82 (д, 1H), 2,69 (м, 1H), 2,51 (м, 1H), 2,38 (м, 1H), 1,92 (м, 1H), 1,58 (д, 1H), 1,35 (м, 1H), 1,27 (м, 1H).
Пример 47: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-(3-(трифторметил)фенил)-3H-имидазо[4,5-b]пиридин-3-ил)пропан-2-она
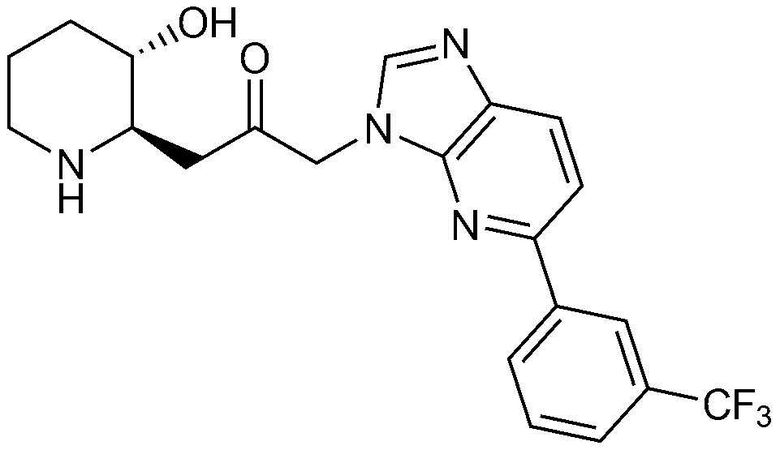
Указанное в заголовке соединение (20 мг, выход: 75%) получали таким же образом, как в примере 43, за исключением того, что вместо (2-фторфенил)бороновой кислоты использовали (3-(трифторметил)фенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,44 (д, 2H), 8,37 (д, 1H), 8,21 (дд, 1H), 8,02 (дд, 1H), 7,78 (д, 1H), 7,73 (т, 1H), 5,42 (дд, 2H), 4,78 (д, 1H), 3,03 (д, 2H), 2,80 (д, 1H), 2,68 (м, 1H), 2,52 (м, 1H), 2,36 (т, 1H), 1,92 (м, 1H), 1,58 (д, 1H), 1,34 (м, 1H), 1,24 (м, 1H).
Пример 48: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-она
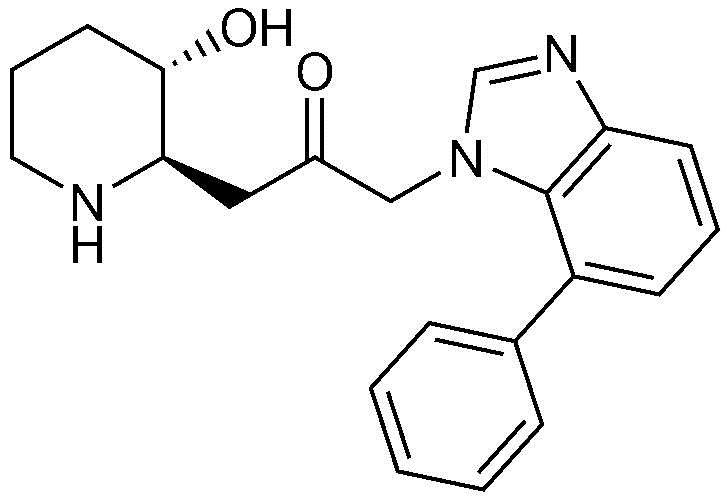
Указанное в заголовке соединение (12 мг, выход: 65%) получали таким же образом, как в примере 1, за исключением того, что вместо 1-бром-3-фтор-2-нитробензола использовали на стадии 1-5 примера 1 1-бром-2-фтор-3-нитробензол.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,04 (с, 1H), 7,70 (д, 1H), 7,45 (м, 3H), 7,27 (м, 3H), 7,01 (дд, 1H), 4,82 (д, 2H), 4,67 (м, 1H), 2,85 (м, 1H), 2,71 (м, 1H), 2,62 (м, 1H), 2,37 (м, 1H), 2,29 (м, 1H), 1,80 (м, 1H), 1,74 (м, 1H), 1,50 (м, 1H), 1,31 (м, 1H), 1,15 (м, 1H)
Пример 49: Получение 1-(7-(3-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
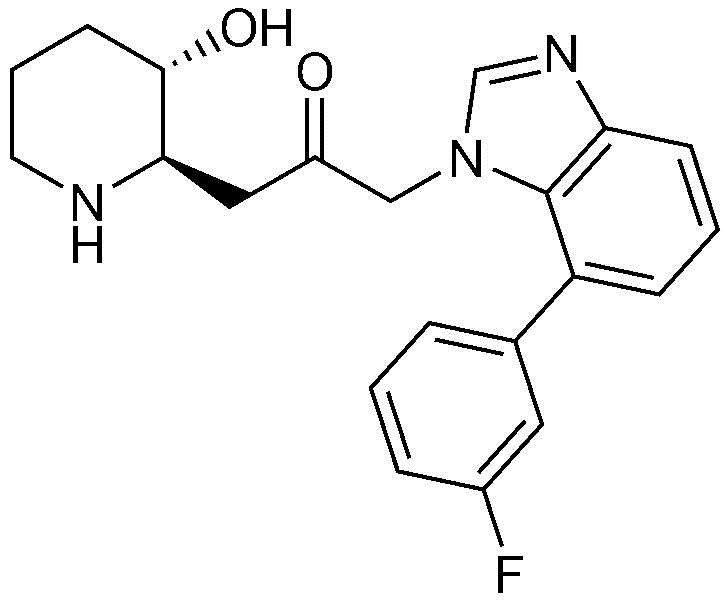
Указанное в заголовке соединение (27 мг, выход: 80%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 1-бром-2-фтор-3-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (3-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,06 (с, 1H), 7,72 (д, 1H), 7,48 (д, 1H), 7,28 (м, 2H), 7,15 (м, 2H), 7,04 (м, 1H), 4,87 (дд, 2H), 4,61 (д, 1H), 2,79 (м, 1H), 2,70 (д, 1H), 2,36 (д, 2H), 2,26 (м, 1H), 1,79 (м, 2H), 1,73 (м, 1H), 1,51 (д, 1H), 1,27 (м, 1H)
Пример 50: Получение 1-(7-(3-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
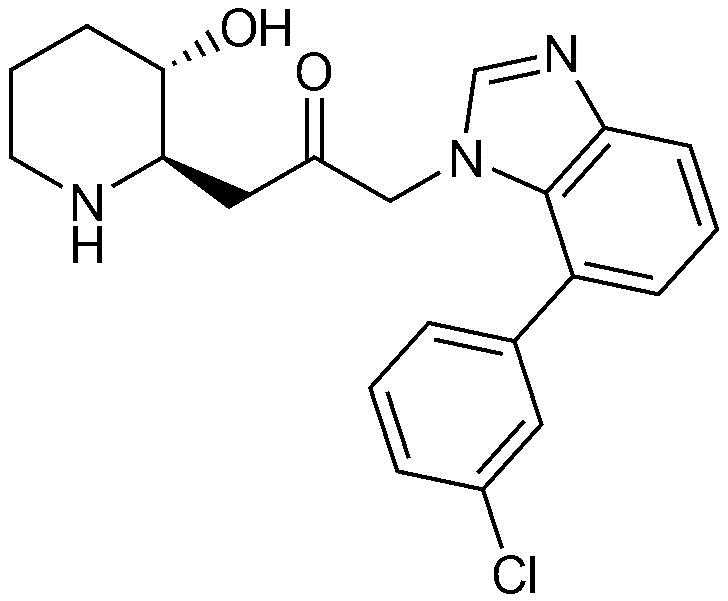
Указанное в заголовке соединение (22 мг, выход: 76%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 1-бром-2-фтор-3-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (3-хлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,04 (с, 1H), 7,70 (д, 1H), 7,49 (м, 1H), 7,45 (м, 1H), 7,32 (с, 1H), 7,23 (м, 2H), 7,02 (м, 1H), 4,85 (д, 2H), 4,58 (м, 1H), 2,77 (м, 1H), 2,69 (д, 1H), 2,34 (м, 2H), 2,26 (м, 1H), 1,75 (м, 2H), 1,50 (м, 1H), 1,27 (м, 1H), 1,13 (м, 1H)
Пример 51: Получение 1-(7-(4-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
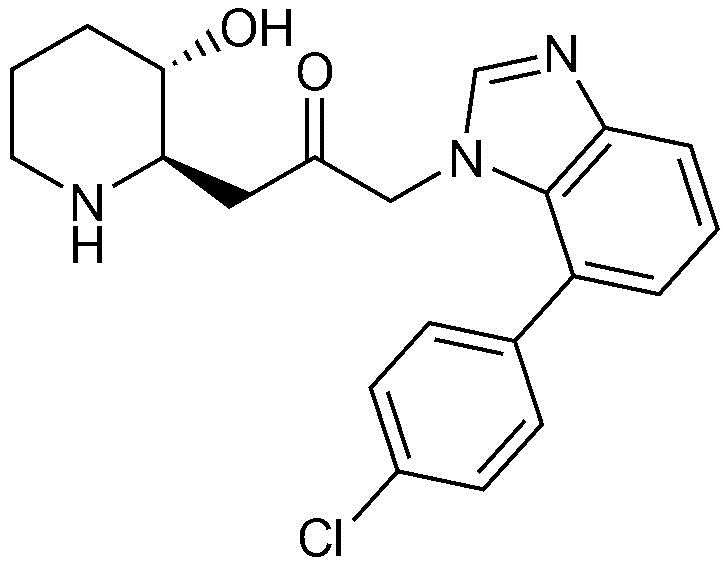
Указанное в заголовке соединение (18 мг, выход: 72%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 1-бром-2-фтор-3-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (4-хлорфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,04 (с, 1H), 7,69 (д, 1H), 7,48 (д, 2H), 7,28 (д, 2H), 7,24 (т, 1H), 6,98 (д, 1H), 4,86 (с, 2H), 4,60 (м, 1H), 2,79 (м, 1H), 2,70 (д, 1H), 2,36 (м, 2H), 2,25 (м, 1H), 1,76 (м, 2H), 1,50 (м, 1H), 1,28 (м, 1H), 1,25 (м, 1H)
Пример 52: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-(2-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-она
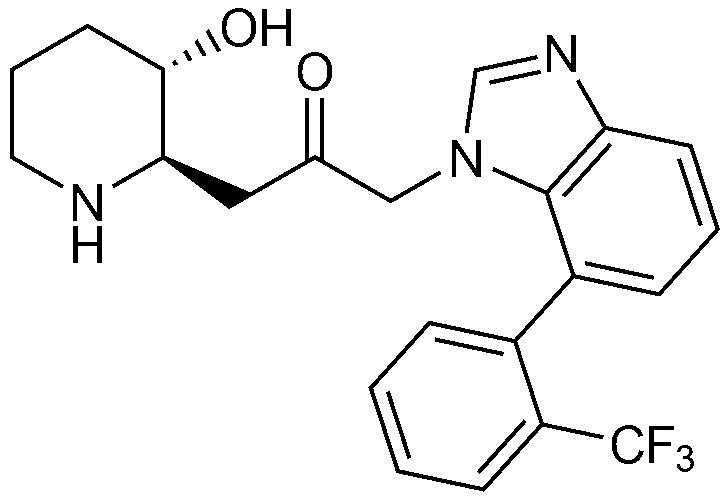
Указанное в заголовке соединение (11 мг, выход: 55%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 1-бром-2-фтор-3-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (2-трифторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 7,99 (д, 1H), 7,88 (м, 1H), 7,68 (м, 3H), 7,22 (м, 2H), 6,96 (д, 1H), 4,88 (т, 1H), 4,57 (м, 1H), 4,22 (т, 1H), 2,82 (м, 1H), 2,75 (м, 1H), 2,45 (м, 1H), 2,25 (м, 1H), 1,79 (м, 2H), 1,65 (м, 1H), 1,51 (м, 1H), 1,22 (м, 1H), 1,14 (м, 1H)
Пример 53: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-она
Указанное в заголовке соединение (19 мг, выход: 64%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 1-бром-2-фтор-3-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (3-трифторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,07 (с, 1H), 7,80 (д, 1H), 7,74 (д, 1H), 7,68 (т, 1H), 7,61 (м, 2H), 7,28 (т, 1H), 7,06 (д, 1H), 4,86 (д, 2H), 4,58 (м, 1H), 2,75 (м, 1H), 2,66 (д, 1H), 2,35 (д, 1H), 2,25 (м, 2H), 1,79 (д, 1H), 1,65 (м, 1H), 1,57 (м, 1H), 1,24 (м, 1H), 1,17 (м, 1H)
Пример 54: Получение 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-(4-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-она
Указанное в заголовке соединение (25 мг, выход: 75%) получали таким же образом, как в примере 1, за исключением того, что на стадии 1-5 примера 1 вместо 1-бром-3-фтор-2-нитробензола использовали 1-бром-2-фтор-3-нитробензол, и на стадии 1-9 примера 1 вместо фенилбороновой кислоты использовали (4-трифторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,06 (с, 1H), 7,78 (д, 2H), 7,73 (д, 1H), 7,50 (д, 2H), 7,27 (т, 1H), 7,04 (д, 1H), 4,86 (д, 2H), 4,53 (м, 1H), 2,74 (м, 1H), 2,68 (д, 1H), 2,31 (м, 2H), 2,20 (м, 1H), 1,79 (м, 1H), 1,68 (м, 1H), 1,49 (м, 1H), 1,25 (м, 1H), 1,12 (м, 1H)
Пример 55: Получение 1-(2-(гидроксиметил)-5-фенил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
Стадия 55-1: Получение бензил (2R,3S)-2-(3-((4-бром-2-нитрофенил)амино)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-амино-2-гидроксипропил)-3-(трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (980 мг, 2,21 ммоль), полученный на стадии 1-4 примера 1, растворяли в N,N-диметилформамиде (10 мл, 0,22 M), к нему добавляли N,N-диизопропилэтиламин (0,77 мл, 4,43 ммоль) и 4-бром-1-фтор-2-нитробензол (487 мг, 2,21 ммоль) и затем перемешивали при температуре 50°C в течение 6 часов. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=3:1) с получением указанного в заголовке соединения (580 мг, выход: 42%).
Стадия 55-2: Получение бензил (2R,3S)-2-(3-((4-бром-2-(2-((трет-бутилдифенилсилил)окси)ацетамидо)фенил)амино)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-((4-бром-2-нитрофенил)амино)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (550 мг, 0,88 ммоль), полученный на стадии 55-1, растворяли в метиловом спирте (6 мл, 0,15 M), к нему добавляли никель Ренея (1 мл), насыщали газообразным водородом и затем перемешивали при комнатной температуре. Когда реакция завершалась, органический слой сушили и затем растворяли в N,N-диметилформамиде (10 мл, 0,09 M), к нему добавляли 2-((трет-бутилдифенилсилил)окси)уксусную кислоту (280 мг, 0,88 ммоль), гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (502 мг, 1,32 ммоль), и диизопропилэтиламин (0,38 мл, 2,21 ммоль) и затем перемешивали при комнатной температуре в течение 3 часов. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=3:1) с получением указанного в заголовке соединения (365 мг, выход: 46%).
Стадия 55-3: Получение бензил (2R,3S)-2-(3-(5-бром-2-(((трет-бутилдифенилсилил)окси)метил)-1H-бензо[d]имидазол-1-ил)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-((4-бром-2-(2-((трет-бутилдифенилсилил)окси)ацетамидо)фенил)амино)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (163 мг, 0,18 ммоль), полученный на стадии 55-2, добавляли и растворяли в уксусной кислоте (6 мл, 0,03 M) и затем перемешивали при температуре 65°C в течение 3 часов. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=3:1) с получением указанного в заголовке соединения (144 мг, выход: 92%).
Стадия 55-4: Получение бензил (2R,3S)-2-(3-(5-бром-2-(((трет-бутилдифенилсилил)окси)метил)-1H-бензо[d]имидазол-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-(5-бром-2-(((трет-бутилдифенилсилил)окси)метил)-1H-бензо[d]имидазол-1-ил)-2-гидроксипропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (144 мг, 0,17 ммоль), полученный на стадии 55-3, растворяли в дихлорметане (5 мл, 0,04 M), к нему при температуре 0°C добавляли 1,1,1-триацетокси-1,1-дигидро-1,2-бензиодоксол-3(1H)-он (105 мг, 0,25 ммоль) и перемешивали при той же температуре в течение 1 часа, затем перемешивали при комнатной температуре в течение 3 часов. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=3:1) с получением указанного в заголовке соединения (126 мг, выход: 88%).
Стадия 55-5: Получение бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(2-(((трет-бутилдифенилсилил)окси)метил)-5-фенил-1H-бензо[d]имидазол-1-ил)-2-оксопропил)пиперидин-1-карбоксилата
Бензил (2R,3S)-2-(3-(5-бром-2-(((трет-бутилдифенилсилил)окси)метил)-1H-бензо[d]имидазол-1-ил)-2-оксопропил)-3-((трет-бутилдиметилсилил)окси)пиперидин-1-карбоксилат (63 мг, 0,07 ммоль), полученный на стадии 55-4, растворяли в N,N-диметилформамиде (2 мл, 0,04 M), к нему последовательно добавляли фенилбороновую кислоту (13 мг, 0,11 ммоль), тетракис(трифенилфосфин)палладий (17 мг, 0,02 ммоль) и 2M раствор карбоната натрия (0,15 мл, 0,29 ммоль) и затем перемешивали при комнатной температуре в течение 5 минут. Затем смеси давали взаимодействовать при температуре 150°C в течение 30 минут с использованием микроволновой печи. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (гексан:этилацетат=3:1) с получением указанного в заголовке соединения (26 мг, выход: 41%).
Стадия 55-6: Получение 1-(2-(гидроксиметил)-5-фенил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
Бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(2-(((трет-бутилдифенилсилил)окси)метил)-5-фенил-1H-бензо[d]имидазол-1-ил)-2-оксопропил)пиперидин-1-карбоксилат (26 мг, 0,03 ммоль), полученный на стадии 55-5, растворяли в 6н водном растворе соляной кислоты (3 мл, 0,02 M) и затем перемешивали при кипячении с обратным холодильником в течение 1 часа. Когда реакция завершалась, реакционный раствор охлаждали до температуры 0°C, нейтрализовали (pH 7) карбонатом калия и затем экстрагировали смешанным раствором хлороформа и небольшого количества ацетона. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем перекристаллизовывали в диэтиловом эфире с получением указанного в заголовке соединения (8 мг, выход: 70%).
1H-ЯМР (500 МГц, ДМСО-d6): δ 7,83 (с, 1H), 7,69 (д, 2H), 7,52 (м, 2H), 7,45 (т, 2H), 7,32 (т, 1H), 5,38 (дд, 2H), 4,82 (д, 1H), 3,00 (м, 1H), 2,98 (дд, 1H), 2,83 (д, 1H), 2,68 (м, 1H), 2,42 (м, 2H), 1,91 (м, 1H), 1,59 (д, 1H), 1,35 (м, 1H), 1,25 (м, 1H).
Пример 56: Получение 1-(5-(3-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
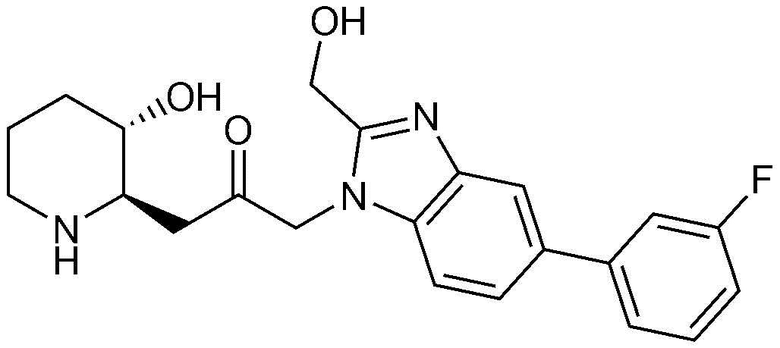
Указанное в заголовке соединение (10 мг, выход: 75%) получали таким же образом, как в примере 55, за исключением того, что на стадии 55-5 примера 55 вместо фенилбороновой кислоты использовали (3-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 7,90 (с, 1H), 7,54 (м, 5H), 7,15 (т, 1H), 5,39 (дд, 2H), 4,82 (д, 1H), 4,61 (с, 2H), 3,00 (м, 1H), 2,98 (дд, 1H), 2,83 (д, 1H), 2,68 (м, 1H), 2,42 (м, 2H), 1,91 (м, 1H), 1,35 (м, 1H), 1,23 (м, 1H).
Пример 57: Получение 1-(2-(гидроксиметил)-6-фенил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
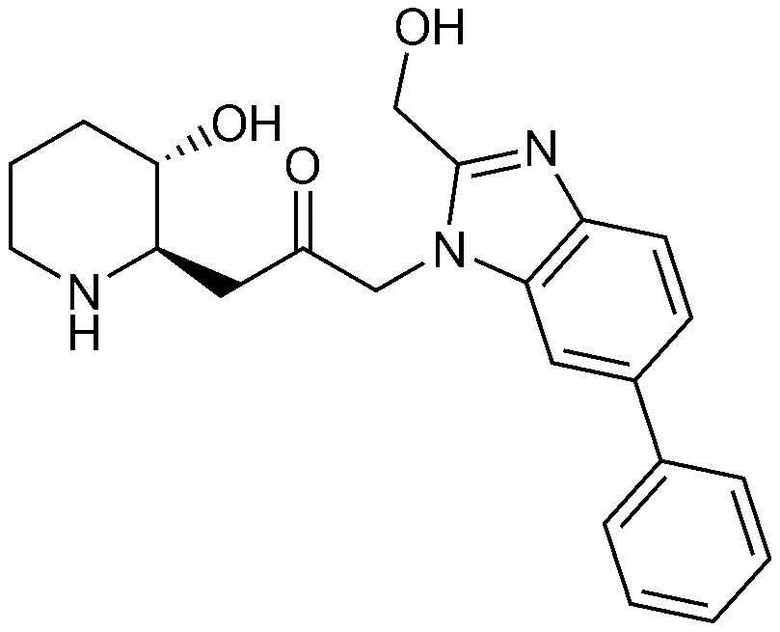
Указанное в заголовке соединение (10 мг, выход: 75%) получали таким же образом, как в примере 55, за исключением того, что на стадии 56-1 примера 55 вместо 4-бром-1-фтор-2-нитробензола использовали 4-бром-2-фтор-1-нитробензол.
1H-ЯМР (500 МГц, ДМСО-d6): δ 7,78 (с, 1H), 7,69 (д, 2H), 7,64 (м, 1H), 7,47 (м, 3H), 7,33 (м, 1H), 5,42 (дд, 2H), 4,81 (д, 1H), 4,60 (с, 2H), 2,99 (м, 1H), 2,82 (д, 1H), 2,69 (м, 1H), 2,41 (м, 2H), 1,91 (м, 1H), 1,59 (д, 1H), 1,34 (м,1H), 1,245 (м, 2H).
Пример 58: Получение 1-(6-(2-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
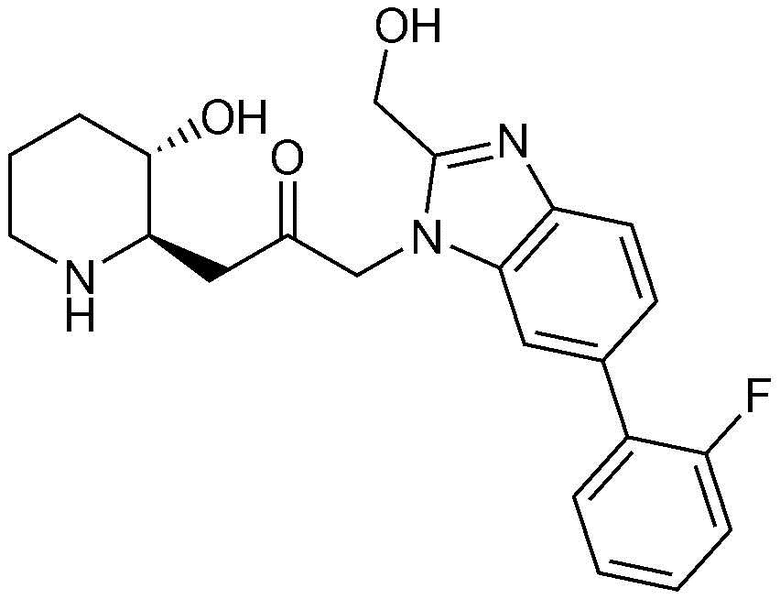
Указанное в заголовке соединение (10 мг, выход: 75%) получали таким же образом, как в примере 55, за исключением того, что на стадии 55-1 примера 55 вместо 4-бром-1-фтор-2-нитробензола использовали 4-бром-2-фтор-3-нитробензол, и на стадии 55-5 примера 55 вместо фенилбороновой кислоты использовали (2-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 7,66 (м, 2H), 7,53 (м, 1H), 7,39 (м, 1H), 7,34 (д, 1H), 7,29 (м, 2H), 5,39 (дд, 2H), 4,80 (д, 1H), 4,61 (с, 2H), 2,99 (м, 2H), 2,79 (м, 1H), 2,68 (м, 1H), 2,44 (дд, 1H), 2,36 (т, 1H), 1,90 (м, 1H), 1,58 (д, 1H), 1,33 (м, 1H), 1,26 (м, 1H).
Пример 59: Получение 1-(6-(3-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
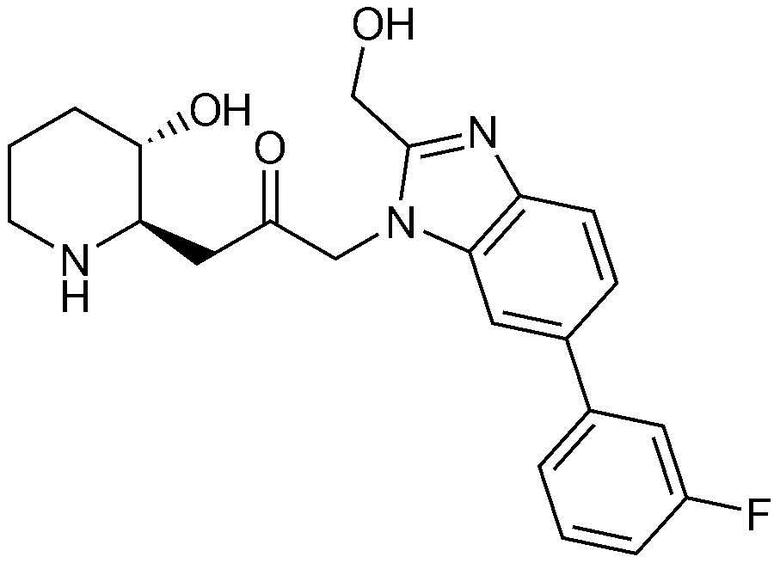
Указанное в заголовке соединение (16 мг, выход: 78%) получали таким же образом, как в примере 55, за исключением того, что на стадии 55-1 примера 55 вместо 4-бром-1-фтор-2-нитробензола использовали 4-бром-2-фтор-3-нитробензол, и на стадии 55-5 примера 55 вместо фенилбороновой кислоты использовали (3-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 7,84 (с, 1H), 7,64 (м, 1H), 7,54 (м, 3H), 7,17 (м, 1H), 5,43 (дд, 2H), 4,82 (д, 1H), 4,61 (с, 2H), 2,97 (м, 2H), 2,82 (д, 1H), 2,68 (м, 1h), 2,42 (м, 2H), 1,90 (м, 1H), 1,59 (д, 1H), 1,36 (м, 1h), 1,25 (м, 2H).
Пример 60: Получение 1-(6-(4-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
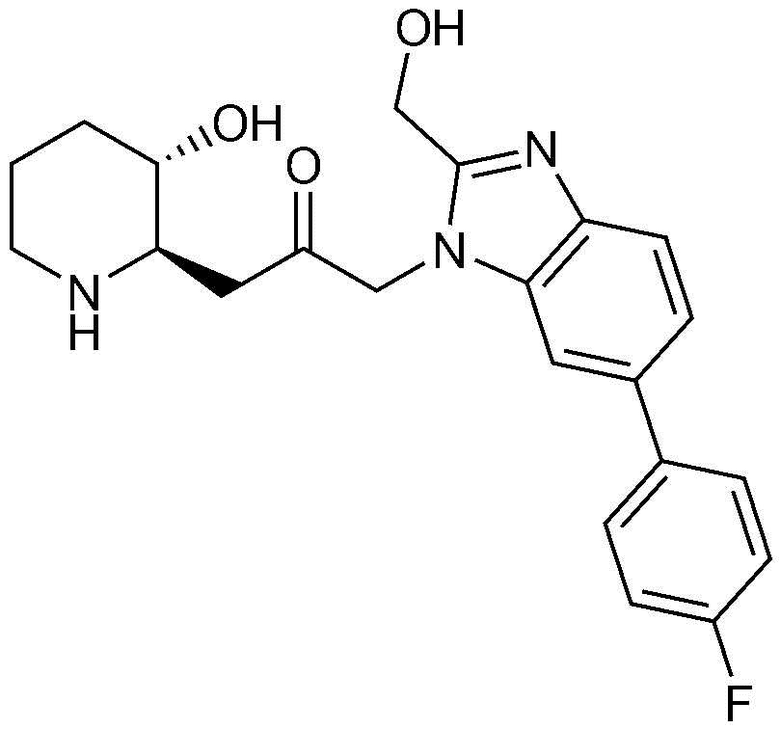
Указанное в заголовке соединение (13 мг, выход: 75%) получали таким же образом, как в примере 55, за исключением того, что на стадии 55-1 примера 55 вместо 4-бром-1-фтор-2-нитробензола использовали 4-бром-2-фтор-3-нитробензол, и на стадии 55-5 примера 55 вместо фенилбороновой кислоты использовали (4-фторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 7,82 (с, 1H), 7,72 (м, 2H), 7,65 (д, 1H), 7,47 (д, 1H), 7,29 (м, 2H), 5,44 (дд, 2H), 4,64 (с, 2H), 3,20 (м, 1H), 3,06 (дд, 1H), 2,93 (м, 2H), 2,63 (м, 2H), 1,92 (м, 1H), 1,68 (д, 1H), 1,47 (м, 1H), 1,33 (м, 1H).
Пример 61: Получение 1-(2-(гидроксиметил)-6-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-она
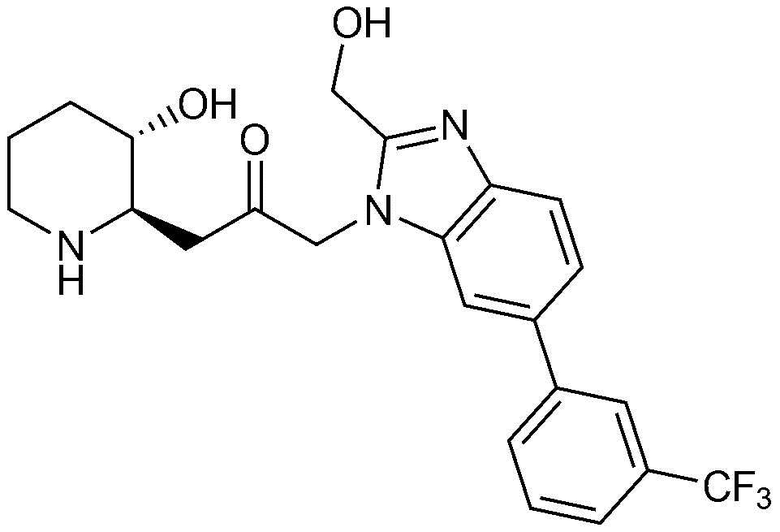
Указанное в заголовке соединение (14 мг, выход: 73%) получали таким же образом, как в примере 55, за исключением того, что на стадии 55-1 примера 55 вместо 4-бром-1-фтор-2-нитробензола использовали 4-бром-2-фтор-3-нитробензол, и на стадии 55-5 примера 55 вместо фенилбороновой кислоты использовали (3-трифторфенил)бороновую кислоту.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,00 (м, 3H), 7,69 (м, 3H), 7,57 (д, 1H), 5,46 (дд, 2H), 4,62 (с, 2H), 3,09 (м,1H), 3,03 (дд, 1H), 2,87 (д, 1H), 2,80 (м, 1H), 2,40 (м, 2H), 1,91 (м, 1H), 1,62 (д, 1H), 1,38 (м, 1H), 1,29 (м, 1H).
Пример 62: Получение (2R,3S)-2-(3-(5-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-гидроксипропил)пиперидин-3-ола
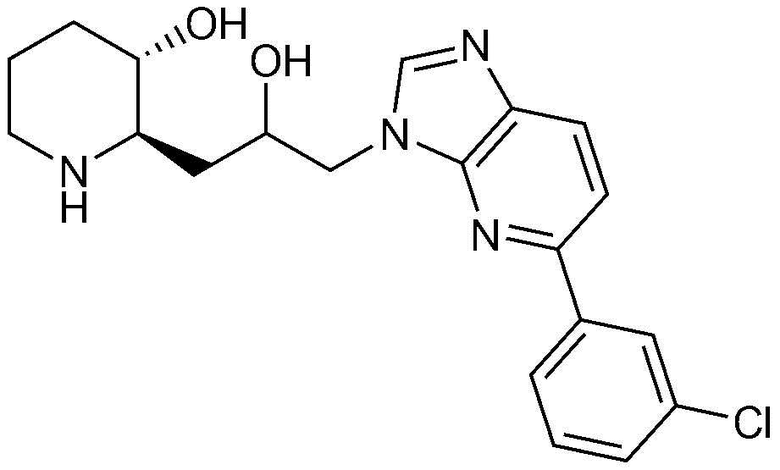
Стадия 62-1: Получение бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(5-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-гидроксипропил)пиперидин-1-карбоксилата
Бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(5-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-оксопропил)пиперидин-1-карбоксилат (110 мг, 0,17 ммоль) растворяли в метиловом спирте (5 мл, 0,03 M), к нему добавляли боргидрид натрия (20 мг, 0,52 ммоль) и затем перемешивали при температуре 0°C в течение 30 минут. Затем смесь перемешивали при комнатной температуре в течение 1 часа. Когда реакция завершалась, растворитель удаляли, и полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем очищали с помощью хроматографии на колонке (дихлорметан:метанол=10:1) с получением указанного в заголовке соединения (94 мг, выход: 85%).
Стадия 62-2: Получение (2R,3S)-2-(3-(5-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-гидроксипропил)пиперидин-3-ола
(2R,3S)-3-((трет-Бутилдиметилсилил)окси)-2-(3-(5-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-гидроксипропил)пиперидин-1-карбоксилат (37 мг, 0,06 ммоль), полученный на стадии 62-1, растворяли в 6н водном растворе соляной кислоты (3 мл, 0,02 M) и затем перемешивали при кипячении с обратным холодильником в течение 1 часа. Когда реакция завершалась, реакционный раствор охлаждали до температуры 0°C, нейтрализовали (pH 7) карбонатом калия и затем экстрагировали смешанным раствором хлороформа и небольшого количества ацетона. Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, и затем перекристаллизовывали в диэтиловом эфире с получением указанного в заголовке соединения (17 мг, выход: 76%).
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,42 (с, 1H), 8,21 (с, 1H), 8,16 (д, 1H), 8,12 (д, 1H), 7,93 (д, 1H), 7,53 (т, 1H), 7,49 (т, 1H), 4,64 (д, 1H), 4,37 (м, 1H), 4,22 (м, 2H), 2,97 (м, 1H), 2,80 (д, 1H), 2,44 (м, 1H), 2,38 (м, 1H), 1,85 (м, 2H), 1,56 (д, 1H), 1,37 (м, 2H), 1,21 (м, 1H).
Пример 63: Получение (2R,3S)-2-(3-(6-(3-хлорфенил)-1H-бензо[d]имидазол-1-ил)-2-гидроксипропил)пиперидин-3-ола
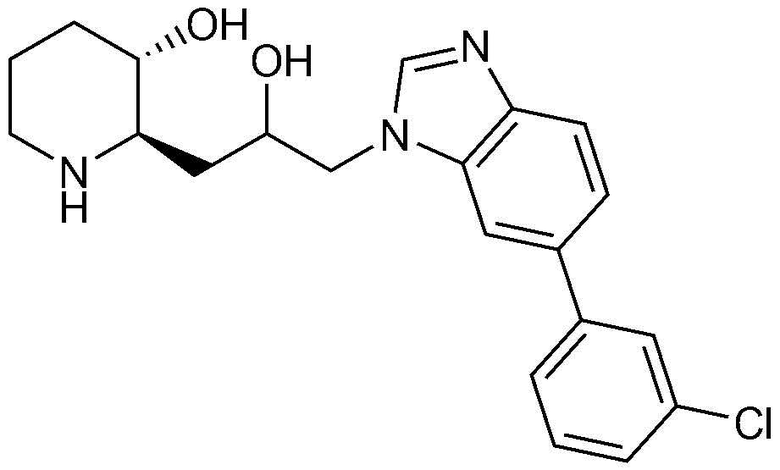
Указанное в заголовке соединение (14 мг, выход: 73%) получали таким же образом, как в примере 62, за исключением того, что вместо бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(5-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-оксопропил)пиперидин-1-карбоксилата использовали бензил (2R,3S)-3-((трет-бутилдиметилсилил)окси)-2-(3-(6-(3-хлорфенил)-1H-бензо[d]имидазол-1-ил)-2-оксопропил)пиперидин-1-карбоксилат.
1H-ЯМР (500 МГц, ДМСО-d6): δ 8,17 (с, 1H), 7,94 (с, 1H), 7,77 (с, 1H), 7,69 (д, 2H), 7,49 (м, 2H), 7,39 (м, 1H), 4,63 (д, 1H), 4,34 (дд, 1H), 4,16 (дд, 1H), 4,08 (м, 1H), 2,95 (м, 1H), 2,77 (д, 1H), 2,41 (м, 1H), 2,29 (м, 1H), 1,82 (м, 2H), 1,54(д, 1H), 1,36 (м, 1H), 1,24 (м, 2H).
Экспериментальный пример 1: Эксперимент по ингибированию ферментной активности PRS
Для подтверждения биологической активности соединений, полученных в примерах, рассчитывали % ингибирования или значения IC50 активности фермента PRS (фосфорибозилпирофосфат-синтетаза).
Конкретно, часть, соответствующую PRS в кДНК EPRS, субклонировали, и полученный высокочистый PRS-белок очищали и использовали в эксперименте. Соединения (1 мкМ), полученные в примерах, добавляли в реакционный буфер (20 мМ KPO4 (pH 7,4), 6 мМ MgAc, 5 мМ АТФ, 400 мг/мл тРНК, 0,5 мМ DTT, 20 мКи[3H]пролин (1 мКи/мл)) и давали реагировать при температуре 37°С в течение 5-10 минут. Реакцию прекращали с помощью бумаги 3М, которую предварительно сушили путем добавления 5% TCA. Радиоактивность измеряли с использованием жидкостного сцинтилляционного счетчика.
% Ингибирования и значения IC50 соответствующих соединений рассчитывали и анализировали с использованием Microsoft Excel или Sigma Plot 8.0. Результаты показаны в таблице 1 ниже. В таблице 1 результаты разделены на A, B и C в соответствии с диапазоном IC50. Случай, когда полученная IC50 равна 100 нм или менее, представлен как «A», случай, когда IC50 составляет от 100 до 500 нм, представлен как «B», а случай, когда IC50 составляет 500 нМ или выше, представлен как «C».
[Таблица 1]
Экспериментальный пример 2: Эксперимент по ингибированию роста раковых клеток
Клетки NCI-H460, клеточные линии рака легких, культивировали в 5% CO2, 37°C инкубаторе, используя колбу для культуры ткани объемом 75 см2. Для оценки использовали 96-луночные планшеты. Их готовили путем разного нанесения при концентрациях в пределах от 6000 до 12000 клеток/лунку в соответствии с темпом роста клеточных линий. Среду, содержащую 5% FBS, распределяли по 200 мкл/лунку и использовали. Среду культивировали в течение 24 часов. После подтверждения состояния ячейки и дозирующей формы 96-луночного планшета под микроскопом, они использовались для последующих экспериментов. Соединения оценивали в концентрациях 100, 30, 10, 3, 1, 0,3, 0,03, 0,01 мкМ. После удаления существующей среды, обрабатывали соединениями при различных концентрациях в количестве 200 мкл/лунку. Пластины, обработанные соединениями, дополнительно культивировали в течение 48 часов и измеряли способность клеток методом МТТ для расчета значений IC50.
% Ингибирование и значения IC50 соответствующих соединений были рассчитаны и проанализированы с использованием Sigma Plot 8.0. Результаты показаны в таблице 2 ниже. В таблице 2 результаты разделены на A, B и C в соответствии с диапазоном IC50. Случай, когда полученная IC50 составляет 3 мкМ или менее, представлен как «А», случай, когда IC50 составляет от 3 до 10 мкМ, представлен как «В», а случай, когда IC50 составляет 10 мкМ или выше, представлен как «C».
[Таблица 2]
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2733384C1 |
| 2-ПИРИДИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ALK5 И/ИЛИ ALK4 | 2012 |
|
RU2612958C2 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ТКБ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2742122C2 |
| БИАРИЛЬНЫЕ МОНОБАКТАМНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2746129C2 |
| СОЕДИНЕНИЯ, ЯВЛЯЮЩИЕСЯ АНТАГОНИСТАМИ А3-АДЕНОЗИНОВОГО РЕЦЕПТОРА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2737157C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2726622C2 |
| НОВЫЕ ЗАМЕЩЕННЫЕ БИАРИЛОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНДОЛАМИН-2,3-ДИОКСИГЕНАЗЫ (IDO) | 2018 |
|
RU2786586C2 |
| ИМИДАЗО[1, 2-B]ПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ КИНАЗ | 2013 |
|
RU2635917C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2012 |
|
RU2648997C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы 1 или к его фармацевтически приемлемой соли, где A представляет собой бензольное кольцо, или пиридиновое кольцо, X представляет собой CO, или CHOH, R1 представляет собой водород, или C1-4 гидроксиалкил, R2 представляет собой фенил, пиразолил, пиридин-2-онил, пирролидинил или тиазолил, где R2 является незамещенным или замещен одним или двумя заместителями, каждый из которых независимо выбран из группы, включающей C1-4 алкил, C1-4 алкокси, C1-4 галогеналкил, галоген и циано, и R3 представляет собой водород, или C1-4 алкил. Также изобретение относится к фармацевтической композиции на основе соединения формулы 1, промежуточным соединениям формул 16 и 21, а также к способам получения промежуточных соединений. Технический результат: получены новые гетероциклические соединения, полезные для предупреждения или лечения заболеваний, вызванных аномальной активностью PRS (пролил-тРНК-синтетаза). 5 н. и 10 з.п. ф-лы, 2 табл., 65 пр.
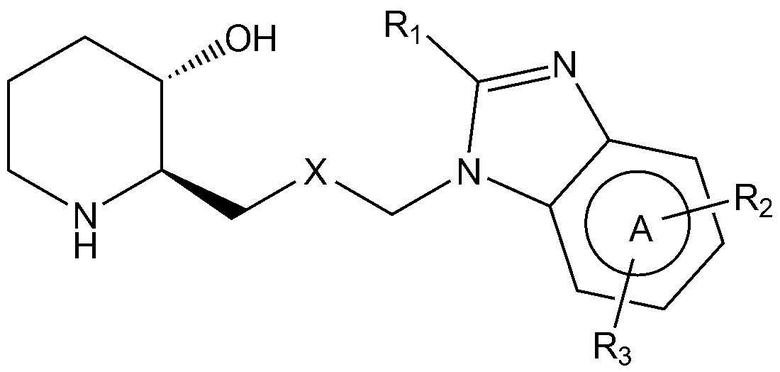
Формула 1
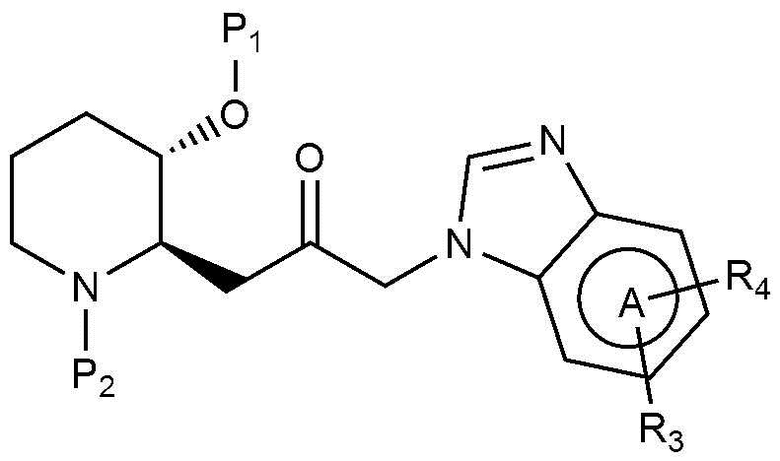
Формула 16
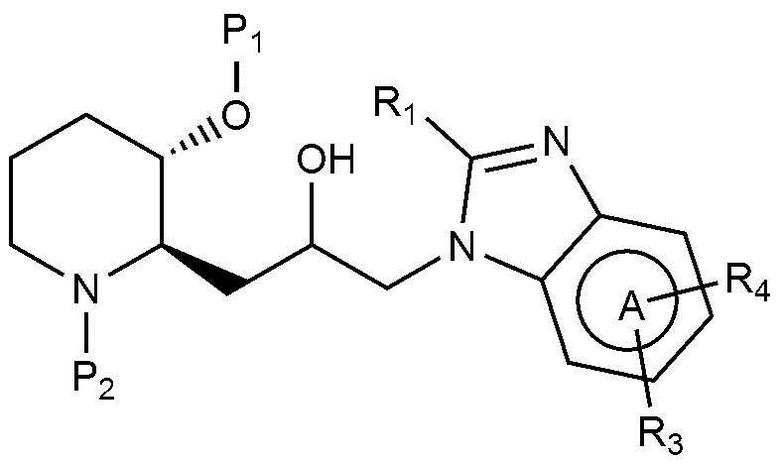
Формула 21
1. Соединение, представленное следующей химической формулой 1, или его фармацевтически приемлемая соль:
[Химическая формула 1]
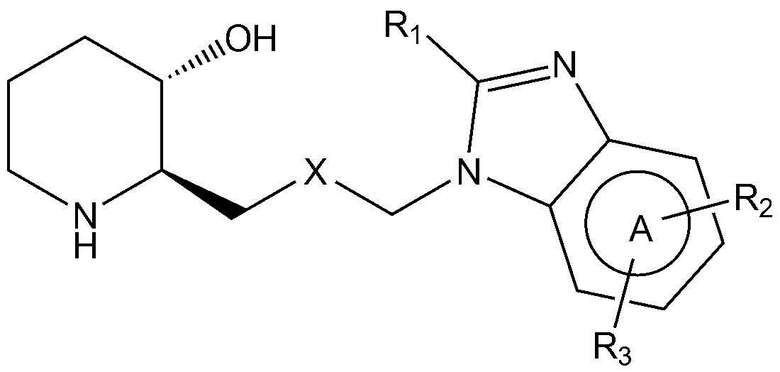
где:
A представляет собой бензольное кольцо, или пиридиновое кольцо,
X представляет собой CO, или CHOH,
R1 представляет собой водород, или C1-4 гидроксиалкил,
R2 представляет собой фенил, пиразолил, пиридин-2-онил, пирролидинил или тиазолил,
где R2 является незамещенным или замещен одним или двумя заместителями, каждый из которых независимо выбран из группы, включающей C1-4 алкил, C1-4 алкокси, C1-4 галогеналкил, галоген и циано, и
R3 представляет собой водород, или C1-4 алкил.
2. Соединение или его фармацевтически приемлемая соль по п.1, где
A, вместе с имидазольным кольцом, конденсированным с A, образует структуру 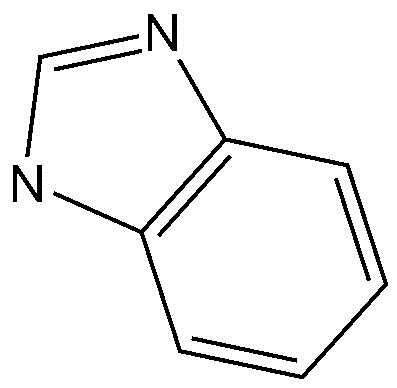 ,
, 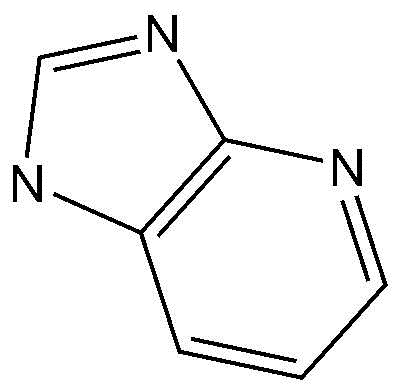 ,
, 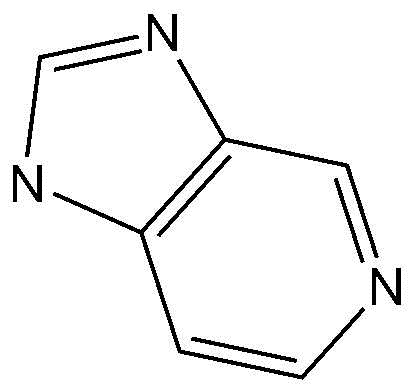 или
или 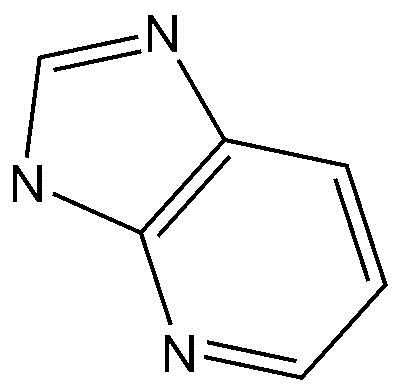 .
.
3. Соединение или его фармацевтически приемлемая соль по п.1, где
R1 представляет собой водород, или гидроксиметил.
4. Соединение или его фармацевтически приемлемая соль по п.1, где
R2 представляет собой фенил, незамещенный или замещенный одним или двумя заместителями, каждый из которых независимо выбран из C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, галогена и циано; незамещенного пиразолила; незамещенного пиридин-2-онила; незамещенного пирролидинила; или незамещенного тиазолила.
5. Соединение или его фармацевтически приемлемая соль по п.1, где
R2 является незамещенным или замещен одним или двумя заместителями, каждый из которых независимо выбран из метила, метокси, трифторметила, фтора, хлора и циано.
6. Соединение или его фармацевтически приемлемая соль по п.1, где
R3 представляет собой водород или метил.
7. Соединение или его фармацевтически приемлемая соль по п.1, где
A представляет собой бензольное кольцо,
X представляет собой CO или CHOH,
R1 представляет собой водород или C1-4 гидроксиалкил,
R2 представляет собой фенил, пиразолил, пиридин-2-онил или тиазолил,
где R2 является незамещенным или замещен одним или двумя заместителями, каждый из которых независимо выбран из группы, включающей C1-4 алкил, C1-4 алкокси, C1-4 галогеналкил, галоген и циано; и
R3 представляет собой водород или C1-4 алкил.
8. Соединение или его фармацевтически приемлемая соль по п.1, где
A, вместе с имидазольным кольцом, конденсированным с A, образует ,
X представляет собой CO,
R1 представляет собой водород,
R2 представляет собой фенил, или пирролидинил,
где R2 является незамещенным или замещен одним или двумя заместителями, каждый из которых независимо выбран из C1-4 галогеналкила и галогена, и
R3 представляет собой водород.
9. Соединение или его фармацевтически приемлемая соль по п.1, где
A, вместе с имидазольным кольцом, конденсированным с A, образует 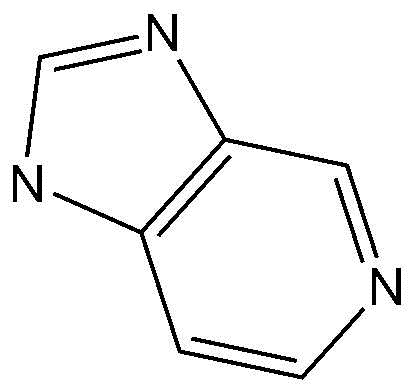 ,
,
X представляет собой CO,
R1 представляет собой водород,
R2 представляет собой фенил или пирролидинил,
где R2 является незамещенным или замещен одним или двумя заместителями, каждый из которых независимо выбран из группы, включающей C1-4 галогеналкил и галоген, и
R3 представляет собой водород.
10. Соединение или его фармацевтически приемлемая соль по п.1, где
A, вместе с имидазольным кольцом, конденсированным с A, образует ,
X представляет собой CO или CHOH,
R1 представляет собой водород,
R2 представляет собой фенил,
где R2 замещен C1-4 галогеналкилом или галогеном, и
R3 представляет собой водород.
11. Соединение или его фармацевтически приемлемая соль по п.1, где соединение, представленное химической формулой 1, выбрано из группы, включающей:
1) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-он,
2) 1-(4-(3-фторфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
3) 1-(4-(3-хлорфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
4) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-(3-(трифторметил)фенил)-1H-имидазо[4,5-c]пиридин-1-ил)пропан-2-он,
5) 1-(4-(3-хлор-5-фторфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
6) 1-(4-(3,5-дихлорфенил)-1H-имидазо[4,5-c]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
7) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-(пирролидин-1-ил)-1H-имидазо[4,5-c]пиридин-1-ил)пропан-2-он,
8) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-он,
9) 1-(5-(2-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
10) 1-(5-(3-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
11) 1-(5-(4-фторфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
12) 1-(5-(3-хлорфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
13) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-(3-(трифторметил)фенил)-1H-имидазо[4,5-b]пиридин-1-ил)пропан-2-он,
14) 1-(6-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
15) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(3-(трифторметил)фенил)-3H-имидазо[4,5-b]пиридин-3-ил)пропан-2-он,
16) 1-(5-(3-фторфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
17) 1-(5-(3-хлорфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
18) 1-(5-(2-хлорфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
19) 1-(5-(4-хлорфенил)-4-метил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
20) 3-(1-(3-((2R,3S)-3-гидроксипиперидин-2-ил)-2-оксопропил)-4-метил-1H-бензо[d]имидазол-5-ил)бензонитрил,
21) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-метил-5-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
22) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-(3-метоксифенил)-4-метил-1H-бензо[d]имидазол-1-ил)пропан-2-он,
23) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(4-метил-5-(тиазол-4-ил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
24) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-он,
25) 1-(6-(3-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
26) 1-(6-(2-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
27) 1-(6-(3-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
28) 1-(6-(4-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
29) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
30) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(м-толил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
31) 1-(6-(3,5-дихлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
32) 1-(6-(3-хлор-5-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
33) 1-(6-(3-хлор-4-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
34) 1-(6-(3-хлор-5-метилфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
35) 1-(6-(3-хлор-5-метоксифенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
36) 3-хлор-5-(1-(3-((2R,3S)-3-гидроксипиперидин-2-ил)-2-оксопропил)-1H-бензо[d]имидазол-6-ил)бензонитрил,
37) 1-(6-(1H-пиразол-4-ил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
38) 1-(6-(1H-пиразол-3-ил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
39) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(тиазол-4-ил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
40) 5-(1-(3-((2R,3S)-3-гидроксипиперидин-2-ил)-2-оксопропил)-1H-бензо[d]имидазол-6-ил)пиридин-2(1H)-он,
41) 1-(6-(3-хлорфенил)-1H-имидазо[4,5-b]пиридин-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
42) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(6-(3-(трифторметил)фенил)-1H-имидазо[4,5-b]пиридин-1-ил)пропан-2-он,
43) 1-(5-(2-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
44) 1-(5-(3-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
45) 1-(5-(4-фторфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
46) 1-(5-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
47) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(5-(3-(трифторметил)фенил)-3H-имидазо[4,5-b]пиридин-3-ил)пропан-2-он,
48) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-фенил-1H-бензо[d]имидазол-1-ил)пропан-2-он,
49) 1-(7-(3-фторфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
50) 1-(7-(3-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
51) 1-(7-(4-хлорфенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
52) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-(2-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
53) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
54) 1-((2R,3S)-3-гидроксипиперидин-2-ил)-3-(7-(4-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)пропан-2-он,
55) 1-(2-(гидроксиметил)-5-фенил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
56) 1-(5-(3-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
57) 1-(2-(гидроксиметил)-6-фенил-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
58) 1-(6-(2-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
59) 1-(6-(3-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
60) 1-(6-(4-фторфенил)-2-(гидроксиметил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
61) 1-(2-(гидроксиметил)-6-(3-(трифторметил)фенил)-1H-бензо[d]имидазол-1-ил)-3-((2R,3S)-3-гидроксипиперидин-2-ил)пропан-2-он,
62) (2R,3S)-2-(3-(6-(3-хлорфенил)-1H-бензо[d]имидазол-1-ил)-2-гидроксипропил)пиперидин-3-ол, и
63) (2R,3S)-2-(3-(5-(3-хлорфенил)-3H-имидазо[4,5-b]пиридин-3-ил)-2-гидроксипропил)пиперидин-3-ол.
12. Фармацевтическая композиция для предупреждения или лечения злокачественных новообразований, воспалительных заболеваний, аутоиммунных заболеваний или фиброза и для ингибирования ферментативной активности PRS, содержащая соединение по любому из пп.1-11 или его фармацевтически приемлемую соль в количестве от 0,001 до 99 мас.%, и фармацевтически приемлемый носитель, адъювант или разбавитель.
13. Соединение, представленное следующей химической формулой 16, или соединение, представленное следующей химической формулой 21:
[Химическая формула 16]
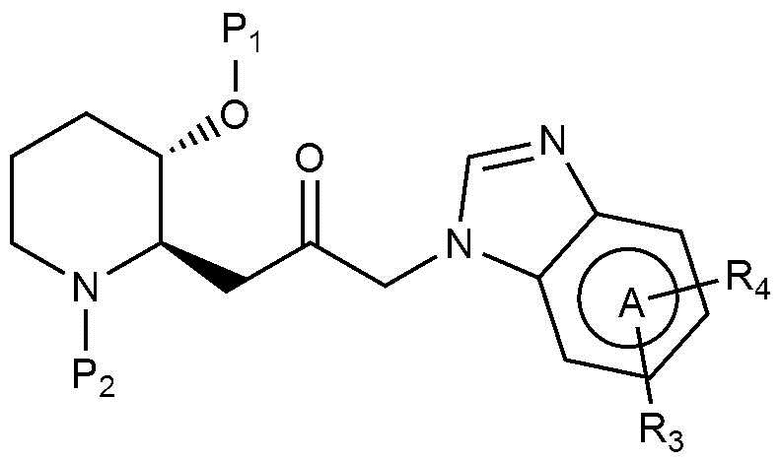
[Химическая формула 21]
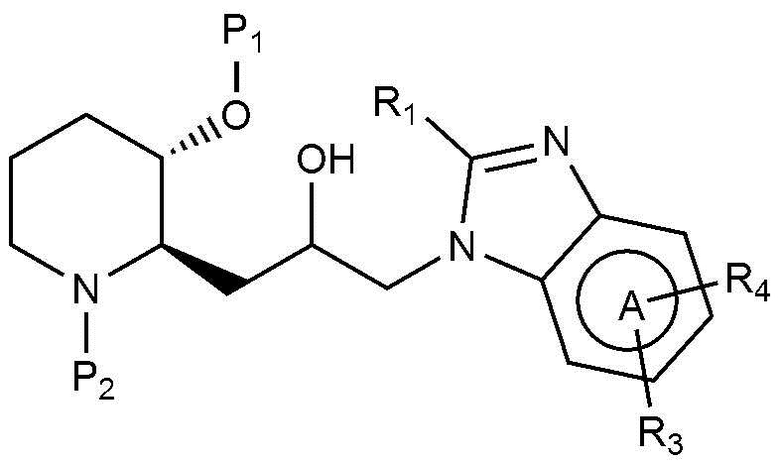
в химических формулах 16 и 21
P1 и P2, каждый, независимо, представляют собой (трет-бутилдиметилсилил)окси или бензилоксикарбонил,
A представляет собой бензольное кольцо или пиридиновое кольцо,
R1 представляет собой водород или C1-4 гидроксиалкил,
R3 представляет собой водород или C1-4 алкил, и
R4 представляет собой галоген.
14. Способ получения соединения, представленного следующей химической формулой 16 по п.13,
включающий следующие стадии:
1) взаимодействие соединения, представленного следующей химической формулой 11, с соединением, представленным следующей химической формулой 12, в присутствии основания с получением соединения, представленного следующей химической формулой 13;
2) взаимодействие соединения, представленного следующей химической формулой 13 в присутствии водорода и металла с получением соединения, представленного следующей химической формулой 14;
3) взаимодействие соединения, представленного следующей химической формулой 14, i) в присутствии триметил ортоформиата или триэтил ортоформиата и пара-толуолсульфоновой кислоты или пиридиний пара-толуолсульфоната, или ii) в присутствии муравьиной кислоты, с получением соединения, представленного следующей химической формулой 15; и
4) взаимодействие соединения, представленного следующей химической формулой 15, с окислителем с получением соединения, представленного следующей химической формулой 16:
[Химическая формула 11]
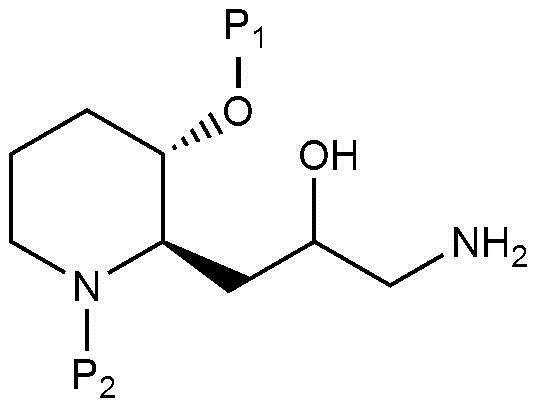
[Химическая формула 12]
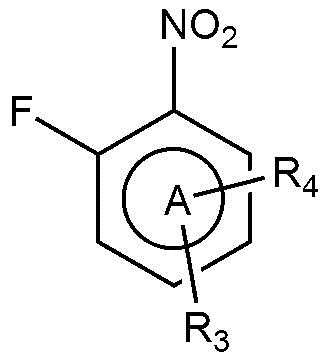
[Химическая формула 13]

[Химическая формула 14]
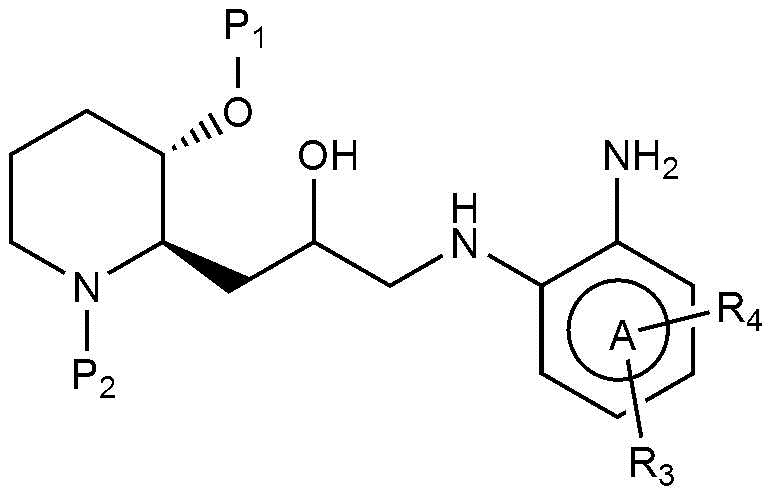
[Химическая формула 15]
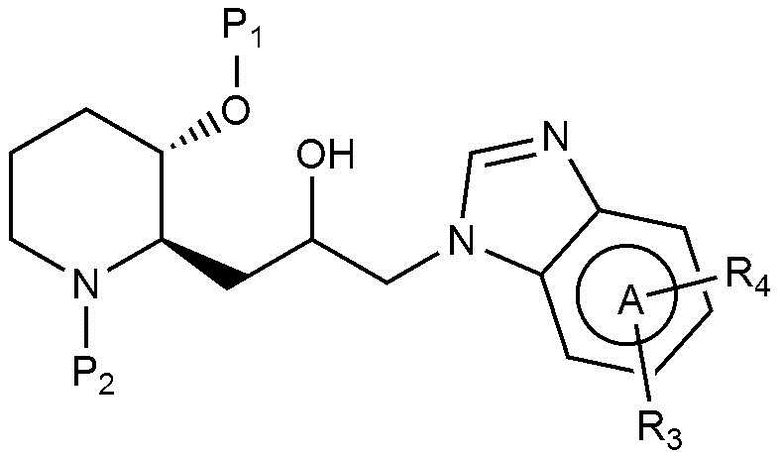
[Химическая формула 16]
в химических формулах 11-16
P1, P2, A, R3 и R4 имеют значения, описанные в п.13.
15. Способ получения соединения, представленного следующей химической формулой 21 по п.13,
включающий следующие стадии:
1) взаимодействие соединения, представленного химической формулой 14, и R1-замещенной карбоновой кислоты (R1-COOH) в присутствии реагента амидного связывания, выбранного из группы, включающей гидрохлорид бис-(2-оксо-3-оксазолидинил)фосфорила, гидрохлорид 1-этил-(3-(3-диметиламино)пропил)-карбодиимида, гексафторфосфат бензотриазол-1-илокси-трис-(пирролидинo)фосфония, бензотриазол-ол, гексафторфосфат (бензотриазол-1-илокси)трис(диметиламино)фосфония и гексафторфосфат O-(бензотриазол-1-ил)-N,N,N,N'-тетраметилурония, и основания, выбранного из группы, включающей триэтиламин, диизопропил этиламин, пиридин, диметиланилин, диметиламинопиридин и гидроксид натрия, с получением соединения, представленного химической формулой 20; и
2) взаимодействие соединения, представленного химической формулой 20, i) в присутствии триметил ортоформиата или триэтил ортоформиата и пара-толуолсульфоновой кислоты или пиридиний пара-толуолсульфоната, или ii) в присутствии муравьиной кислоты с получением соединения, представленного химической формулой 21.
[Химическая формула 14]
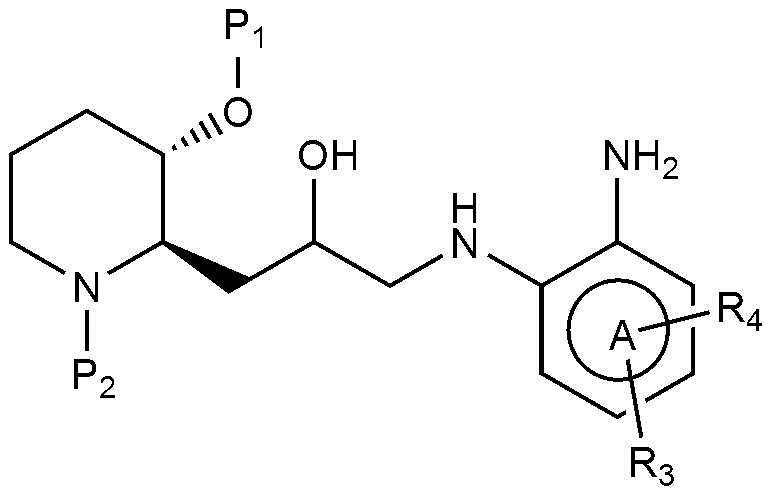
[Химическая формула 20]
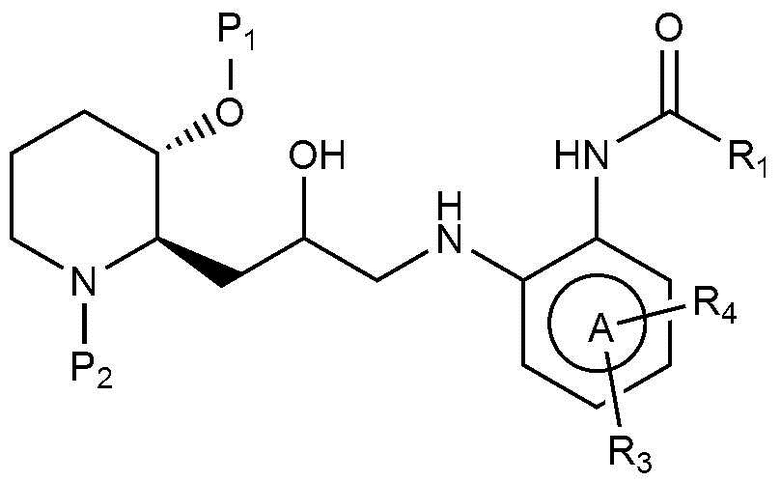
[Химическая формула 21]
в химических формулах 14, 20 и 21
P1, P2, A, R1, R3 и R4 имеют значения, описанные в п.13.
| WO 2010019210 A2, 18.02.2010 | |||
| ПРОИЗВОДНЫЕ БЕНЗИЛПИПЕРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2160259C2 |

