[001] Настоящая заявка на патент испрашивает приоритет на основании предварительной заявки на патент США № 62/833529, поданной 12 апреля 2019 года; предварительной заявки на патент США № 62/911016, поданной 4 октября 2019 года; и предварительной заявки на патент США № 62/930240, поданной 4 ноября 2019 года. Полное содержание каждой из перечисленных заявок включено в настоящий документ посредством ссылки.
[002] Настоящее изобретение относится к новым соединениям пирролтриазина и их применению в качестве селективных ингибиторов активированных мутантных протеинкиназ KIT и PDGFRα. Описанные в настоящем документе соединения подходят для применения в фармацевтических композициях, таких как, например, композиции для лечения хронических заболеваний. Рецептор KIT принадлежит к семейству рецепторных тирозинкиназ III класса, которое также включает структурно родственный белок PDGFRα. Обычно фактор стволовых клеток связывается с KIT и активирует его путем индуцирования димеризации и аутофосфорилирования, что вызывает инициирование передачи сигнала ниже по каскаду. Однако в некоторых типах опухолей соматические активирующие мутации в KIT приводят к лиганд-независимой конститутивной онкогенной активности, в том числе в таких типах опухолей, как острый миелоидный лейкоз, меланома, внутричерепные герминогенные опухоли, медиастинальная B-клеточная лимфома, семинома и желудочно-кишечные стромальные опухоли. Также известно, что мутантный KIT влияет на активацию тучных клеток, что является обычным явлением и, возможно, необходимо для поддержания функционирования. Нарушение активации тучных клеток происходит, когда тучные клетки патологически чрезмерно продуцируются, или если их активация непропорциональна предполагаемой угрозе гомеостазу. Синдром активации тучных клеток относится к группе расстройств, вызванных различными причинами, которые проявляются эпизодическими мультисистемными симптомами в результате высвобождения тучными клетками медиаторов. Мастоцитоз представляет собой один из типов синдрома активации тучных клеток. Всемирная организация здравоохранения (ВОЗ) классифицирует мастоцитоз по 7 различным категориям: кожный мастоцитоз, вялотекущий системный мастоцитоз (ISM), тлеющий системный мастоцитоз (SSM), мастоцитоз с ассоциированным гематологическим новообразованием (SM-AHN), агрессивный системный мастоцитоз (ASM), лейкоз тучных клеток (MCL) и саркома тучных клеток.
[003] Системный мастоцитоз (SM) представляет собой клональное нарушение тучных клеток, характеризующееся повышенным содержанием тучных клеток с очаговыми и/или диффузными инфильтратами неопластических тучных клеток в коже, костном мозге, селезенке, печени, желудочно-кишечном тракте и других органах и повышенным выделением тучными клетками медиаторов. SM включает 5 подтипов мастоцитоза: вялотекущий SM (ISM), тлеющий SM (SSM), SM с ассоциированным гематологическим новообразованием нетучноклеточного происхождения (SM-AHN), агрессивный SM (ASM) и лейкоз тучных клеток (MCL). Последние три подкласса связаны с пониженной общей выживаемостью и объединены в группу продвинутого SM (AdvSM). ISM представляет собой хроническое заболевание, связанное с нормальной или почти нормальной продолжительностью жизни, а прогноз SSM является промежуточным. ISM и SSM классифицируют как непродвинутый SM (non-Adv SM).
[004] Во всех подтипах SM и у большинства пациентов с этим заболеванием неопластические тучные клетки имеют мутацию в положении D816 в экзоне 17 KIT, что приводит к лиганд-независимой активации киназной активности KIT. Тучным клеткам дикого типа требуется активность KIT для их дифференцировки и выживания, и, следовательно, как полагают, конститутивная активация KIT посредством мутации D816V является патогенным драйвером SM. В частности, мутации KIT D816V обнаруживают у от 90 до 98% пациентов с SM, при этом идентифицированы редкие варианты KIT D816Y, D816F и D816H. На основании таких данных KIT D816V считается основной терапевтической мишенью при SM.
[005] Хронические заболевания в виде вялотекущей SM и SSM характеризуются тяжелыми симптомами, в том числе зудом, приливами, спазмами желудочно-кишечного тракта, диареей, анафилаксией, болью в костях и остеопорозом. Такие симптомы могут серьезно подорвать здоровье, отрицательно сказываясь на качестве жизни. Не существует одобренных способов лечения ISM или SSM. Таким образом, было бы полезным обнаружение новых способов лечения, нацеленных на ISM или SSM.
[006] Соединения пирролтриазина, обладающие ингибирующей активностью в отношении мутантных KIT и PDGFRα, были описаны в WO2015/057873. В частности, примеры некоторых соединений, содержащих N-алкилпиразол, приведены в WO2015/057873 и обладают ингибирующей активностью в отношении мутантных KIT и PDGFRα, например, соединение 63 с N-этилпиразолом. Химические структуры таких соединений N-алкилпиразола, приведенные в качестве примеров в WO2015/057873, отличаются от структур соединений, предложенных в настоящем изобретении.
[007] Кроме того, хотя в WO2015/057873 описаны соединения пирролтриазина, обладающие ингибирующей активностью в отношении мутантных KIT и PDGFRα, свойства таких соединений совершенно отличаются от свойств соединений, предложенных в настоящем изобретении.
[008] Задачей настоящего изобретения является получение новых соединений с высокоселективной, сильной активностью против мутантных киназ KIT и PDGFRα для безопасного и эффективного лечения хронических заболеваний, таких как ISM и SSM, а также других заболеваний, опосредованных мутантным KIT или PDGFRA. При лечении таких расстройств, в частности, хронических расстройств, таких как ISM и SSM, любая новая терапия должна быть хорошо переносимой. В частности, существует потребность в новых соединениях, нацеленных на мутантные киназы KIT и PDGFRα , характеризующихся пониженными уровнями нежелательных побочных эффектов со стороны ЦНС, присущих другим известным соединениям пирролтриазина.
[009] Авторы настоящего изобретения обнаружили новые соединения, проявляющие высокую селективность и активность против мутантных киназ KIT и PDGFRα,обладающие при этом дополнительными желательными свойствами, такими как, например, незначительное проникновение или отсутствие проникновения в ЦНС, низкие концентрации несвязанных фракций в головном мозге и высокие уровни или активный транспорт из головного мозга, т.е. высокие коэффициенты эффлюкса из ЦНС. С учетом такого желательного соотношения свойств соединения согласно настоящему изобретению подходят, в частности, для лечения на периферии, особенно для длительного лечения на периферии, при этом побочные эффекты в ЦНС понижены или минимизированы.
[0010] Таким образом, соединения согласно настоящему изобретению направлены на обеспечение лекарственных средств, обладающих требуемой эффективностью, безопасностью и фармацевтическими свойствами для лечения KIT- и PDGFRA-опосредованных заболеваний. Более конкретно, соединения согласно настоящему изобретению демонстрируют совокупность полезных свойств, в том числе пониженный уровень проникновения в головной мозг, при одновременном сохранении эффективности и других требуемых фармацевтических свойств по сравнению с известными соединениями пирролтриазина, обладающими ингибирующей активностью в отношении мутантных KIT и PDGFRα.
Сокращения и определения
[0011] В настоящем документе следующие сокращения и термины имеют указанные значения:
[0012] Термин «KIT» относится к тирозинкиназе человека, которую можно назвать рецептором фактора роста тучных/стволовых клеток (SCFR), протоонкогеном c-KIT, тирозин-протеинкиназой Kit или CD117. В настоящем документе термин «нуклеотид KIT» включает ген KIT, мРНК KIT, кДНК KIT и их продукты амплификации, мутации, варианты и фрагменты. «Ген KIT» используют для обозначения гена, кодирующего полипептид с KIT-киназной активностью, например, последовательность которого расположена между нуклеотидами 55524085 и 55606881 хромосомы 4 эталонного генома человека hg19. «Транскрипт KIT» относится к продукту транскрипции гена KIT, один из примеров которого имеет последовательность эталонной последовательности NM_000222.2 в NCBI (Национальный центр биотехнологической информации). Термин «белок KIT» относится к полипептидной последовательности, продуцируемой путем трансляции нуклеотида KIT или его части.
[0013] Термин «PDGFRA» относится к тирозинкиназе человека, которую можно назвать тромбоцитарным фактором роста альфа. В настоящем документе термин «нуклеотид PDGFRA» включает ген PDGFRA, мРНК PDGFRA, кДНК KIT и их продукты амплификации, мутации, варианты и фрагменты. «Ген PDGFRA» используют для обозначения гена, кодирующего полипептид с PDGFRA-киназной активностью, например, последовательность которого расположена между нуклеотидами 54229089 и 54298247 хромосомы 4 эталонного генома 109, GRCh38.p12 в Homo sapiens Annotation Release. «Транскрипт PDGFRA» относится к продукту транскрипции гена PDGFRA, один из примеров которого имеет последовательность эталонной последовательности NM_006206.6 в NCBI. Термин «белок PDGFRA» или «PDGFRα» относится к полипептидной последовательности, продуцируемой путем трансляции нуклеотида PDGFRA или его части.
[0014] В настоящем документе «злокачественное заболевание» относится к заболеванию, при котором аномальные клетки делятся бесконтрольно и могут проникать в близлежащие ткани. Злокачественные клетки также могут распространяться на другие части тела через кровеносную или лимфатическую систему. Неограничивающими примерами злокачественных заболеваний являются карцинома, саркома, лейкоз и лимфома. Рак представляет собой неограничивающий пример злокачественного заболевания. Согласно некоторым вариантам реализации системный мастоцитоз является неограничивающим примером злокачественного заболевания.
[0015] Неограничивающие примеры рака включают стромальную опухоль желудочно-кишечного тракта (GIST), AML (острый миелоидный лейкоз), меланому, семиному, внутричерепные герминогенные опухоли и медиастинальную B-клеточную лимфому.
[0016] В настоящем документе термин «эозинофильное нарушение» относится к расстройству, при котором эозинофилы обнаруживают в различных частях тела в количестве, превышающем нормальное, и/или когда соотношение гиподенсных и нормоденсных эзозинофилов выше нормы (например, более 30%). Описанное в настоящем документе эозинофильное нарушение характеризуется переизбытком эозинофилов (эозинофилия). Повышенное количество эозинофилов вызывает воспаление тканей и повреждение органов. Чаще всего страдают сердце, легкие, кожа и нервная система, но может быть поврежден любой орган.
[0017] Эозинофильные нарушения диагностируют в зависимости от места, в котором повышен уровень эозинофилов:
Эозинофильная пневмония (легкие)
Эозинофильная кардиомиопатия (сердце)
Эозинофильный эзофагит (пищевод - EoE)
Эозинофильный гастрит (желудок - EG)
Эозинофильный гастроэнтерит (желудок и тонкий кишечник - EGE)
Эозинофильный энтерит (тонкий кишечник)
Эозинофильный колит (толстый кишечник - EC)
Гиперэозинофильный синдром (кровь и любой орган - HES)
[0018] В настоящем документе термин «субъект» или «пациент» относится к организмам, которые подлежат лечению с применением способов согласно настоящему изобретению. Такие организмы включают, но не ограничиваются ими, млекопитающих (например, мышей, обезьян, лошадей, крупный рогатый скот, свиней, собак, кошек и т.п.) и согласно некоторым вариантам реализации людей.
[0019] В настоящем документе выражение «терапевтически эффективное количество» относится к количеству активного агента, достаточному для достижения лечебных или требуемых результатов. Терапевтически эффективное количество может быть введено за одно или более введений, применений или в одной или более дозировок и не предназначено для ограничения конкретного состава или пути введения.
[0020] В настоящем документе выражение «эквивалентная масса его фармацевтически приемлемой соли» применительно к конкретному соединению включает массу, как соединения, так и связанной соли.
[0021] В настоящем документе выражение «его фармацевтически приемлемая соль», при применении в отношении активного агента, распределенного в виде солевой формы, относится к любой фармацевтически приемлемой солевой форме активного агента.
[0022] В настоящем документе термин «лечение» включает любой эффект, например, облегчение, уменьшение, модуляцию, улучшение или устранение, который приводит к улучшению состояния, заболевания, расстройства и т.п. или облегчению их симптомов.
[0023] Хотя активный агент можно вводить отдельно, согласно некоторым вариантам реализации активный агент можно вводить в виде фармацевтического состава, в котором указанный активный агент объединен с одним или более фармацевтически приемлемыми вспомогательными веществами или носителями. Например, активный агент можно получить для введения любым удобным способом для применения в медицине или ветеринарии. Согласно некоторым вариантам реализации соединение, включенное в фармацевтический препарат, может быть активным само по себе или может представлять собой пролекарство, например, способное превращаться в активное соединение в физиологических условиях.
[0024] Выражение «фармацевтически приемлемый» используют в настоящем документе для обозначения тех соединений, материалов, композиций и/или лекарственных форм, которые в рамках здравого медицинского суждения подходят для применения в контакте с тканями людей и животных, не вызывая чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соизмеримых с разумным соотношением польза/риск.
[0025] В настоящем документе «алкил» относится к одновалентному радикалу насыщенного линейного или разветвленного углеводорода, такому как линейная или разветвленная группа, состоящая из 1-12, 1-10 или 1-6 атомов углерода, обозначаемая в настоящем документе как C1-C12 алкил, C1-C10 алкил и C1-C6 алкил, соответственно. Например, C1 алкил представляет собой метил.
[0026] В настоящем документе термин «галоген» относится к радикалу любого галогена, например, -F, -Cl, -Br или -I.
[0027] В настоящем документе «галогеналкил» и «галогеналкокси» относятся к структурам алкила и алкокси, которые содержат в качестве заместителя одну или более групп галогенов или их комбинации. Например, термины «фторалкил» и «фторалкокси» включают галогеналкильные и галогеналкокси группы, соответственно, в которых галоген представляет собой фтор. «Галогеналкилен» относится к двухвалентному алкилу, например,
-CH2-, -CH2CH2- и -CH2CH2CH2-, в которых один или более атомов водорода заменены галогеном, и включает алкильные фрагменты, в которых все атомы водорода заменены галогеном.
[0028] В настоящем документе термин «циклоалкил» относится к циклическим, бициклическим, трициклическим или полициклическим неароматическим углеводородным группам, содержащим от 3 до 12 атомов углерода. Любой замещаемый кольцевой атом может содержать заместители (например, один или более заместителей). Циклоалкильные группы могут содержать конденсированные кольца или спиро-кольца. Конденсированные кольца представляет собой кольца, которые имеют общий атом углерода. Примеры циклоалкильных фрагментов включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил и циклогексил.
[0029] В настоящем документе термин «гетероциклил» относится к одновалентному радикалу гетероциклической кольцевой системы. Примеры гетероциклилов включают, но не ограничиваются ими, кольцевые системы, в которых каждое кольцо является неароматическим, при этом по меньшей мере одно кольцо содержит гетероатом, например, оксетанил, тетрагидрофуранил и тетрагидропиранил.
[0030] В настоящем документе подразумевают, что определение каждого термина, например, алкила, m, n и т.д., когда оно встречается более одного раза в какой-либо структуре, является независимым от его определения в другом месте в такой же структуре.
[0031] Некоторые соединения согласно настоящему изобретению могут существовать в определенных геометрических или стереоизомерных формах. В настоящем документе предполагают, что все такие соединения, в том числе цис- и транс-изомеры, R- и S-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и другие их смеси, находятся в пределах объема настоящего изобретения. Дополнительные асимметричные атомы углерода могут присутствовать в заместителе, таком как, например, алкильная группа. Предполагается, что все такие изомеры, а также их смеси, включены в настоящее изобретение.
[0032] Если, например, требуется конкретный энантиомер соединения согласно настоящему изобретению, его можно получить посредством асимметричного синтеза или путем получения производных с помощью хирального вспомогательного агента, при этом полученную диастереомерную смесь разделяют, а вспомогательную группу расщепляют с получением чистых требуемых энантиомеров. Альтернативно, если молекула содержит основную функциональную группу, такую как, например, амино, или кислотную функциональную группу, такую как, например, карбоксил, диастереомерные соли получают путем применения соответствующей оптически активной кислоты или основания с последующим разделением полученных таким образом диастереомеров с помощью фракционной кристаллизации или хроматографических методов, хорошо известных в данной области техники, и последующим выделением чистых энантиомеров.
[0033] Если не указано иное, когда описанное соединение названо или изображено в виде структуры без указания стереохимии и имеет один или более хиральных центров, подразумевается, что оно представляет все возможные стереоизомеры указанного соединения, а также его энантиомерные смеси.
[0034] «Энантиомерный избыток» или «энантиомерный избыток в %» композиции можно рассчитать с применением приведенного ниже уравнения. В показанном ниже примере композиция содержит 90% одного энантиомера, например, S- энантиомера, и 10% другого энантиомера, то есть R- энантиомера.
ее = (90-10)/100 = 80%.
[0035] Таким образом, считается, что композиция, содержащая 90% одного энантиомера и 10% другого энантиомера, имеет энантиомерный избыток 80%.
[0036] Описанные в настоящем документе соединения или композиции могут иметь энантиомерный избыток, соответствующий по меньшей мере 50%, 75%, 90%, 95% или 99% одной формы соединения, например, S-энантиомера. Другими словами, такие соединения или композиции имеют энантиомерный избыток S-энантиомера относительно R-энантиомера.
[0037] Описанные в настоящем документе соединения могут также иметь неприродные соотношения атомных изотопов у одного или более атомов, составляющих такие соединения. Например, указанные соединения могут быть помечены радиоактивными изотопами, такими как, например, дейтерий (2H), тритий (3H), углерод-13 (13C) или углерод-14 (14C). Предполагается, что все изотопные варианты описанных в настоящем документе соединений, радиоактивных или нет, включены в объем настоящего изобретения. Кроме того, подразумевается, что все таутомерные формы описанных в настоящем документе соединений лежат в пределах объема настоящего изобретения.
[0038] Описанные в настоящем документе соединения можно применять в форме свободного основания или в виде соли. Типичные соли включают гидробромидную, гидрохлоридную, сульфатную, бисульфатную, фосфатную, нитратную, ацетатную, валератную, олеатную, пальмитатную, стеаратную, лауратную, бензоатную, лактатную, фосфатную, тозилатную, цитратную, малеатную, фумаратную, сукцинатную, тартратную, нафтилатную, мезилатную, глюкогептонатную, лактобионатную и лаурилсульфонатную соль и т.п. (См., например, Berge et al. (1977) “Pharmaceutical Salts”, J. Pharm. Sci. 66:1-19.)
[0039] Некоторые описанные в настоящем документе соединения могут существовать в несольватированных формах, а также в сольватированных формах, в том числе гидратированных формах. В настоящем документе термин «гидрат» или «гидратированный» относится к соединению, образованному путем объединения воды с исходным соединением.
[0040] В общем случае, сольватированные формы эквивалентны несольватированным формам и включены в объем настоящего изобретения. Некоторые описанные в настоящем документе соединения могут существовать в нескольких кристаллических или аморфных формах. В общем случае, все физические формы эквивалентны для вариантов применения, рассматриваемых в настоящем документе, и находятся в пределах объема настоящего изобретения.
[0041] В настоящем изобретении предложены соединения формулы I и их фармацевтически приемлемые соли и/или сольваты любого из приведенных выше соединений. Неограничивающие варианты реализации настоящего изобретения включают:
[0042] Вариант реализации 1. Соединение формулы I:
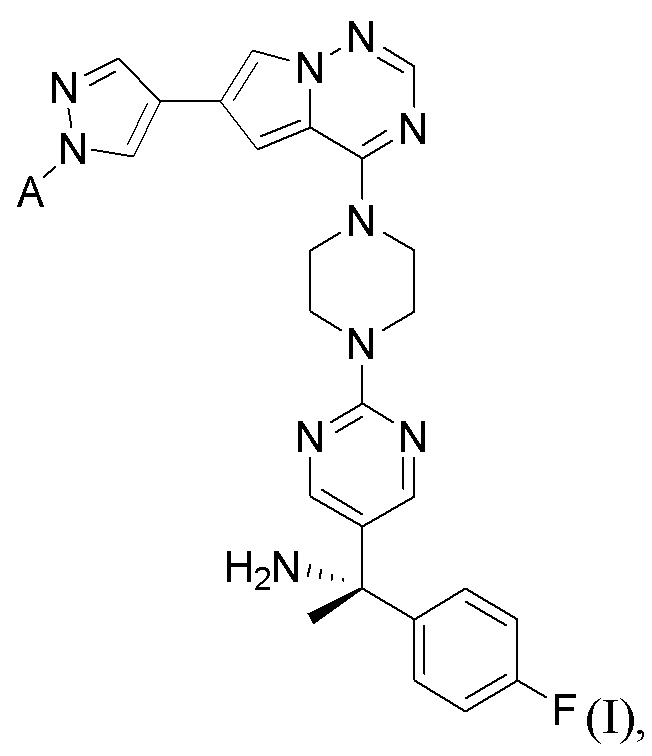
его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где:
A представляет собой
R1 выбран из атома водорода и метила;
R2 выбран из водорода и метила, или
R1 и R2 вместе образуют циклопропил;
R3 выбран из водорода и метила;
R4 выбран из водорода и метила, или
R3 и R4 вместе образуют циклопропил;
R5 выбран из водорода и метила;
R6 выбран из водорода и метила, или
R5 и R6 вместе образуют циклопропил, или
один из R2 или R4 вместе с R6 образуют циклобутил;
R7 представляет собой водород, или один из R2, R4 или R6 вместе с R7 образуют кольцо, выбранное из оксетана, тетрагидрофурана и тетрагидропирана, при этом указанный тетрагидрофуран или тетрагидропиран необязательно содержит гидроксил в качестве заместителя;
m равно 0 или 1; и
n равно 0 или 1.
[0043] Согласно некоторым вариантам реализации варианта 1, когда m равно 0, R1 и R2 отсутствуют. Согласно некоторым вариантам реализации варианта 1, когда n равно 0, R3 и R4 отсутствуют. Согласно некоторым вариантам реализации варианта 1 m + n = 1 или m и n не могут оба быть равными 0.
[0044] Следует отметить, что согласно настоящему изобретению, когда любые две группы R (например, R1 и R2) вместе образуют кольцевую структуру (например, циклопропил), предполагается, что они включают промежуточные атомы углерода и/или атом кислород в той же кольцевой структуре.
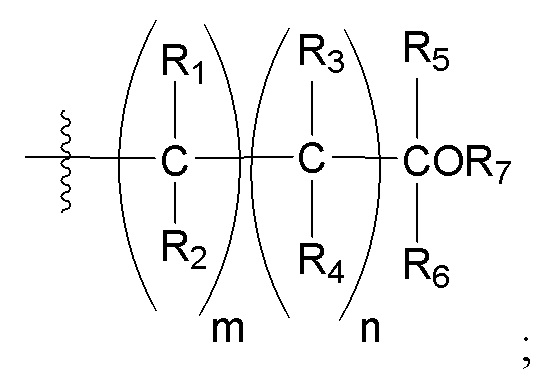
[0045] Вариант реализации 2. Соединение согласно варианту реализации 1, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где:
A представляет собой:
R3 выбран из водорода и метила;
R4 выбран из водорода и метила, или R3 и R4 вместе образуют циклопропил;
R5 выбран из водорода и метила; или
R4 и R6 совместно образуют циклобутил; или
R5 и R6 совместно образуют циклопропил; и
R7 представляет собой водород.
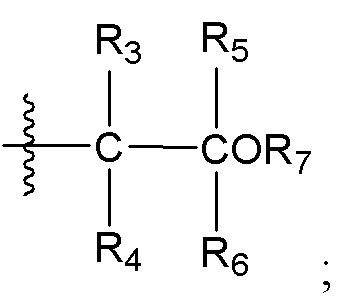
[0046] Вариант реализации 3. Соединение согласно варианту реализации 2, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где:
A представляет собой:
R3 выбран из водорода и метила;
R4 выбран из водорода и метила, или R3 и R4 вместе образуют циклопропил;
R5 выбран из водорода и метила; или
R5 и R6 вместе образуют циклопропил, и
R7 представляет собой водород.
[0047] Вариант реализации 4. Соединение согласно варианту реализации 1, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где:
A представляет собой
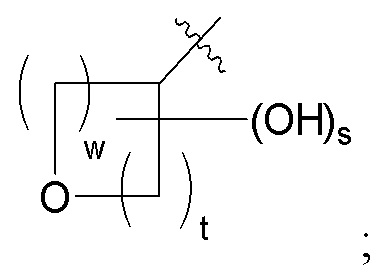
w равно 1 или 2;
t равно 1 или 2; и
s равно 0 или 1.
[0048] Вариант реализации 5. Соединение согласно любому из вариантов реализации 1-4, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что Kp указанного соединения составляет <0,39.
[0049] Согласно некоторым вариантам реализации варианта 5 Kp предложенного соединения составляет <0,39, как измерено в соответствии с процедурой, описанной в примере 4. Согласно некоторым вариантам реализации варианта 5 предложенное соединение, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений выбраны из соединений 1, 2, 3, 4, 5, 6, 7, 9, 10, 11, 12, 13, 14, 17, 18, 19, 20, 21 и 22.
[0050] Вариант реализации 6. Соединение согласно любому из вариантов реализации 1-4, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что Kp указанного соединения составляет <0,20.
[0051] Согласно некоторым вариантам реализации варианта 6 Kp предложенного соединения составляет ≤0,20, как измерено в соответствии с процедурой, описанной в примере 4. Согласно некоторым вариантам реализации варианта 6 предложенное соединение, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений выбраны из соединений 1, 3, 4, 5, 6, 9, 11, 13, 17, 18, 19, 20, 21 и 22.
[0052] Вариант реализации 7. Соединение согласно любому из вариантов реализации 1-6, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что Kp, uu указанного соединения составляет <0,2 в гомогенате головного мозга крысы.
[0053] Согласно некоторым вариантам реализации варианта 7 Kp uu предложенного соединения составляет <0,2 в гомогенате головного мозга крысы, как измерено в соответствии с процедурой, описанной в примере 4. Согласно некоторым вариантам реализации варианта 7 предложенное соединение, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений выбраны из соединений 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 16, 17, 18, 19, 20 и 22.
[0054] Вариант реализации 8. Соединение согласно любому из вариантов реализации 1-7, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что Kp uu указанного соединения составляет <0,1 в гомогенате головного мозга крысы.
[0055] Согласно некоторым вариантам реализации варианта 8 Kp uu предложенного соединения составляет <0,1 в гомогенате головного мозга крысы, как измерено в соответствии с процедурой, описанной в примере 4. Согласно некоторым вариантам реализации варианта 8 предложенное соединение, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений выбраны из соединений 1, 2, 3, 4, 5, 6, 7, 9, 11, 12, 17, 18, 19, 20 и 22.
[0056] Вариант реализации 9. Соединение согласно любому из вариантов реализации 1-8, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что Kp, uu указанного соединения составляет <0,05 в гомогенате головного мозга крысы.
[0057] Согласно некоторым вариантам реализации варианта 9 Kp uu предложенного соединения составляет <0,05 в гомогенате головного мозга крысы, как измерено в соответствии с процедурой, описанной в примере 4. Согласно некоторым вариантам реализации варианта 9 предложенное соединение, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений выбраны из соединений 1, 3, 4, 5, 6, 9, 17, 19, 20 и 22..
[0058] Вариант реализации 10. Соединение согласно любому из вариантов реализации 1-9, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что Kp uu указанного соединения составляет <0,1 в срезе головного мозга крысы.
[0059] Согласно некоторым вариантам реализации варианта 10 Kp uu предложенного соединения составляет <0,1 в срезе головного мозга крысы, как измерено в соответствии с процедурой, описанной в примере 4. Согласно некоторым вариантам реализации варианта 10 предложенное соединение, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений выбраны из соединений 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 20, 21 и 22.
[0060] Вариант реализации 11. Соединение согласно любому из вариантов реализации 1-10, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что Kp uu указанного соединения составляет <0,05 в срезе головного мозга крысы.
[0061] Согласно некоторым вариантам реализации варианта 11 Kp uu предложенного соединения составляет <0,05 в срезе головного мозга крысы, как измерено в соответствии с процедурой, описанной в примере 4. Согласно некоторым вариантам реализации варианта 11 предложенное соединение, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений выбраны из соединений 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 17, 18, 20 и 22.
[0062] Вариант реализации 12. Соединение согласно любому из вариантов реализации 1-11, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что у крысы клиренс несвязанной фракции (Clu) указанного соединения составляет <900 мл/мин/кг.
[0063] Согласно некоторым вариантам реализации варианта 12 у крысы Clu предложенного соединения составляет <900 мл/мин/кг, как измерено в соответствии с процедурой, описанной в примере 4. Согласно некоторым вариантам реализации варианта 12 предложенное соединение, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений выбраны из соединений 3, 4, 7 и 9.
[0064] Вариант реализации 13. Соединение согласно любому из вариантов реализации 1-12, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что у крысы клиренс несвязанной фракции (Clu) указанного соединения составляет <750 мл/мин/кг.
[0065] Согласно некоторым вариантам реализации варианта 13 у крысы Clu предложенного соединения составляет <750 мл/мин/кг, как измерено в соответствии с процедурой, описанной в примере 4. Согласно некоторым вариантам реализации варианта 13 предложенное соединение, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений выбраны из соединений 4, 7 и 9.
[0066] Вариант реализации 14. Соединение согласно любому из вариантов реализации 1-12, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что IC50 указанного соединения составляет для CYP3A4 <10 μМ.
[0067] Согласно некоторым вариантам реализации варианта 14 IC50 предложенного соединения составляет для CYP3A4 <10 μM, как измерено в соответствии с процедурой, описанной в примере 5. Согласно некоторым вариантам реализации варианта 14 предложенное соединение, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений представляет собой соединение 4.
[0068] Вариант реализации 15. Соединение согласно варианту реализации 1, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где A выбран из
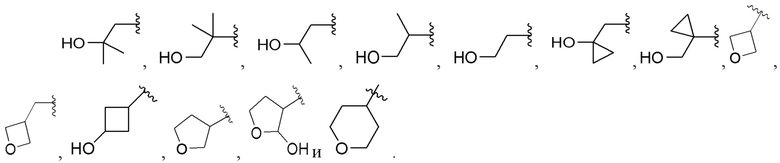
[0069] Вариант реализации 15-1. Соединение согласно варианту реализации 1, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где A выбран из
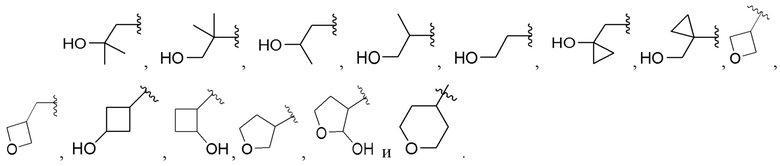
[0070] Вариант реализации 16. Соединение согласно варианту реализации 1, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где A выбран из
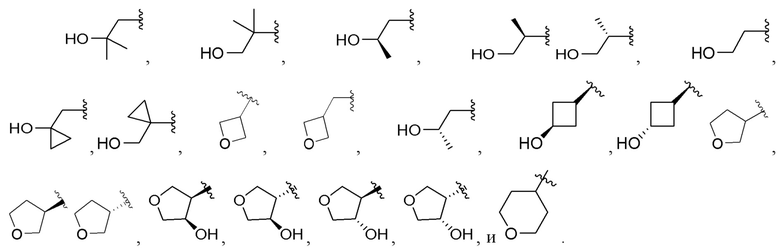
[0071] Вариант реализации 16-1. Соединение согласно варианту реализации 1, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где A выбран из
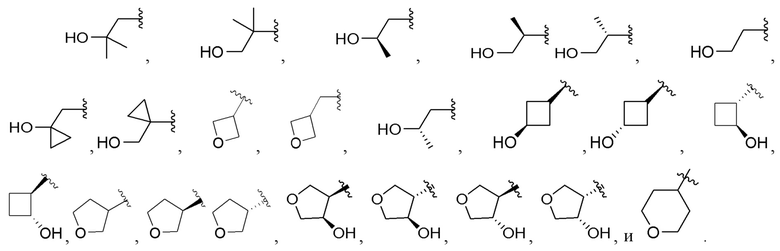
[0072] Вариант реализации 17. Соединение согласно любому из вариантов реализации 1-3, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где A выбран из
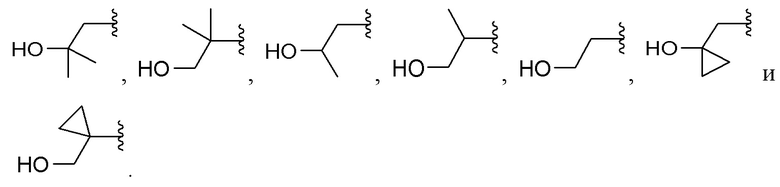
[0073] Вариант реализации 18. Соединение согласно любому из вариантов реализации 1-3 или 5, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где A выбран из
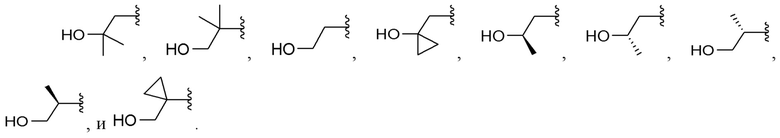
[0074] Вариант реализации 19. Соединение согласно любому из вариантов реализации 1-3 или 5-6, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где A выбран из
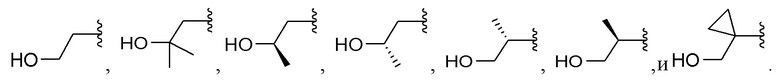
Вариант реализации 20. Соединение согласно любому из вариантов реализации 1-3 или 5-7, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, где A выбран из
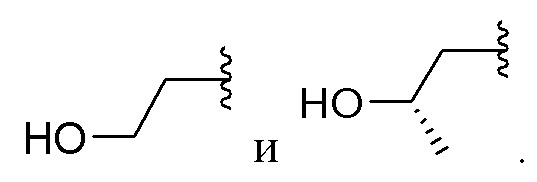
[0076] Вариант реализации 21. Соединение согласно любому из вариантов реализации 1-13, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что указанное соединение выбрано из 4, 7 и 9.
[0077] Вариант реализации 22. Соединение согласно любому из вариантов реализации 1-13, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что указанное соединение выбрано из 4 и 9.
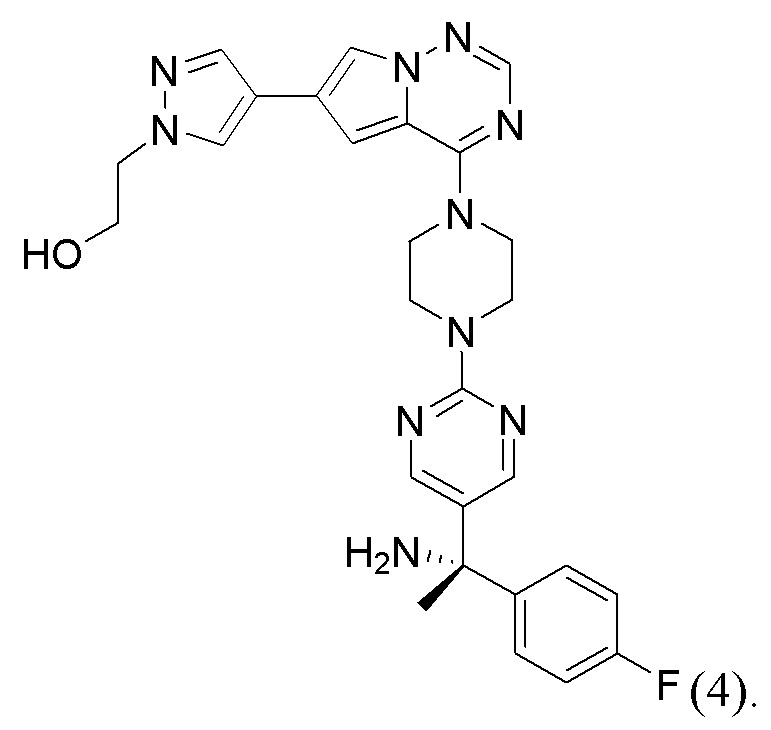
[0078] Вариант реализации 23. Соединение согласно любому из вариантов реализации 1-13, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что указанное соединение представляет собой
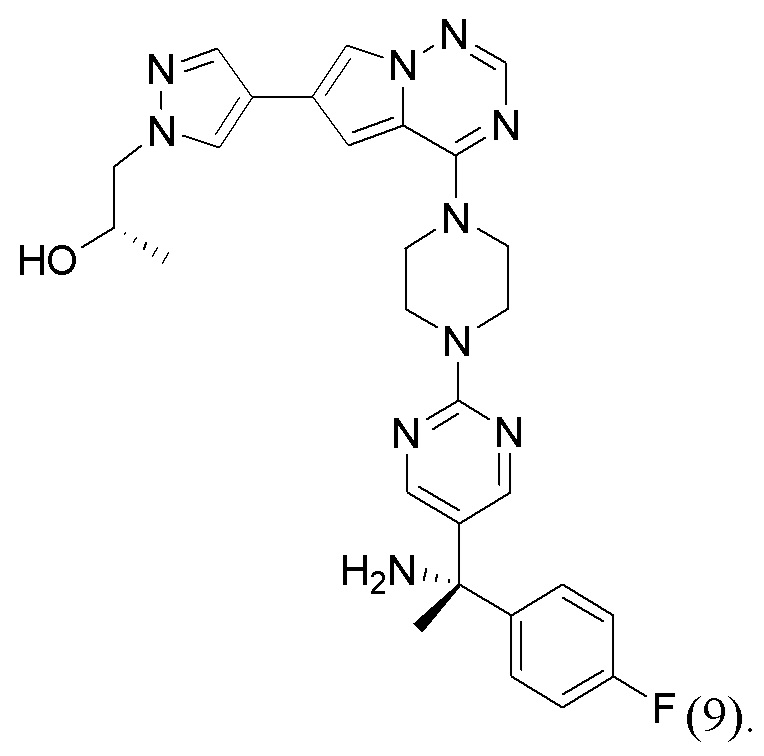
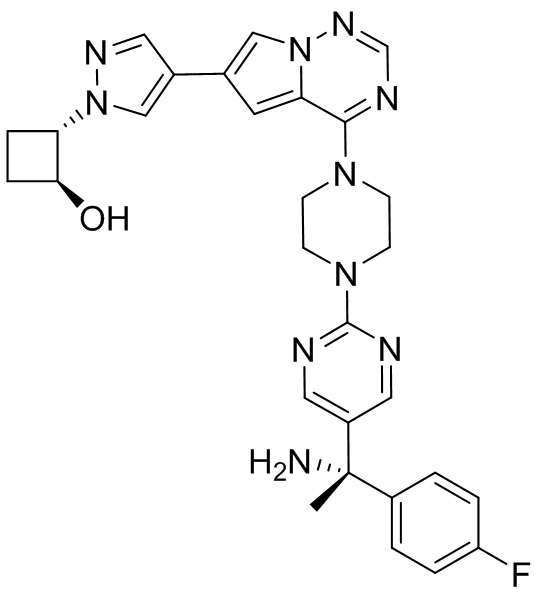
[0079] Вариант реализации 24. Соединение согласно любому из вариантов реализации 1-13, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что указанное соединение представляет собой
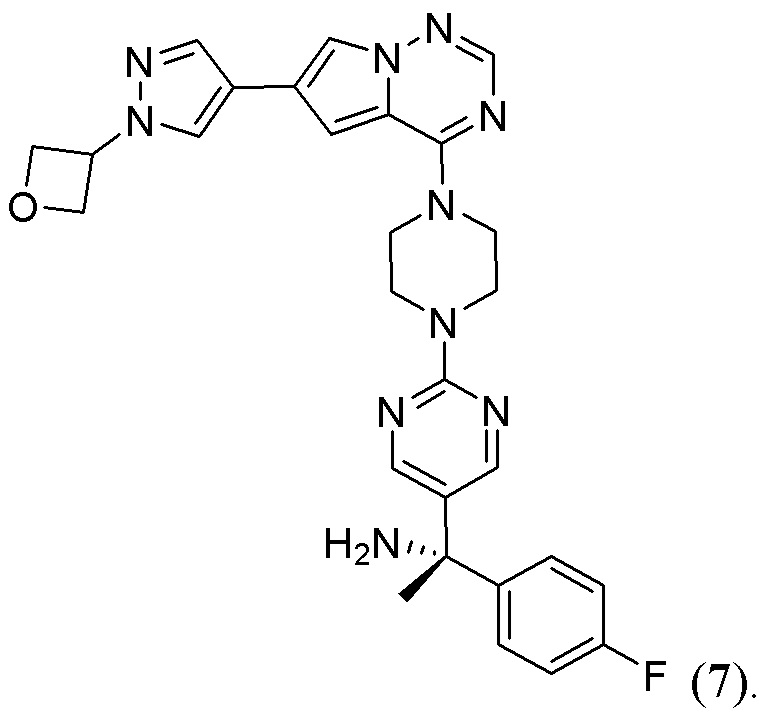
[0080] Вариант реализации 25. Соединение согласно любому из вариантов реализации 1-13, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений, отличающееся тем, что указанное соединение представляет собой
[0081] Вариант реализации 26. Фармацевтическая композиция, содержащая:
соединение согласно любому из вариантов реализации 1-25, его фармацевтически приемлемую соль и/или сольват любого из приведенных выше соединений; и
фармацевтически приемлемое вспомогательное вещество.
[0082] Вариант реализации 27. Способ лечения заболевания или состояния у пациента, нуждающегося в этом, отличающийся тем, что указанный способ включает введение пациенту соединения согласно любому из вариантов реализации 1-25, его фармацевтически приемлемой соли и/или сольвата любого из приведенных выше соединений, при этом указанное заболевание или состояние выбрано из системного мастоцитоза, желудочно-кишечных стромальных опухолей, острого миелоидного лейкоза, меланомы, семиномы, внутричерепных герминогенных опухолей, медиастинальной B-клеточной лимфомы, саркомы Юинга, диффузной B-крупноклеточной-лимфомы, дисгерминомы, миелодиспластического синдрома, назальной NK/Т-клеточной лимфомы, хронического миеломоноцитарного лейкоза и рака головного мозга.
[0083] Вариант реализации 28. Способ лечения заболевания или состояния, опосредованного мутантным KIT или PDGFRα, у пациента, нуждающегося в этом, отличающийся тем, что указанный способ включает введение пациенту соединения согласно любому из вариантов реализации 1-25, его фармацевтически приемлемой соли и/или сольвата любого из приведенных выше соединений.
[0084] Вариант реализации 29. Способ согласно варианту реализации 28, отличающийся тем, что указанное заболевание или состояние выбрано из системного мастоцитоза, желудочно-кишечных стромальных опухолей, острого миелоидного лейкоза, меланомы, семиномы, внутричерепных герминогенных опухолей, медиастинальной B-клеточной лимфомы, саркомы Юинга, диффузной B-крупноклеточной-лимфомы, дисгерминомы, миелодиспластического синдрома, назальной NK/Т-клеточной лимфомы, хронического миеломоноцитарного лейкоза и рака головного мозга.
[0085] Вариант реализации 30. Соединение согласно любому из вариантов реализации 1-25, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений для применения в качестве лекарственного средства для лечения заболевания или состояния у пациента, нуждающегося в этом, отличающееся тем, что указанное заболевание или состояние выбрано из системного мастоцитоза, желудочно-кишечных стромальных опухолей, острого миелоидного лейкоза, меланомы, семиномы, внутричерепных герминогенных опухолей, медиастинальной B-клеточной лимфомы, саркомы Юинга, диффузной B-крупноклеточной-лимфомы, дисгерминомы, миелодиспластического синдрома, назальной NK/Т-клеточной лимфомы, хронического миеломоноцитарного лейкоза и рака головного мозга.
[0086] Вариант реализации 31. Соединение согласно любому из вариантов реализации 1-25, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений для применения в качестве лекарственного средства для лечения заболевания или состояния, опосредованного мутантным KIT или PDGFRA, у пациента, нуждающегося в этом.
[0087] Вариант реализации 32. Соединение согласно варианту реализации 31, отличающееся тем, что указанное заболевание или состояние выбрано из системного мастоцитоза, желудочно-кишечных стромальных опухолей, острого миелоидного лейкоза, меланомы, семиномы, внутричерепных герминогенных опухолей, медиастинальной B-клеточной лимфомы, саркомы Юинга, диффузной B-крупноклеточной-лимфомы, дисгерминомы, миелодиспластического синдрома, назальной NK/Т-клеточной лимфомы, хронического миеломоноцитарного лейкоза и рака головного мозга.
[0088] Вариант реализации 33. Способ лечения эозинофильного нарушения, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения согласно любому из вариантов реализации 1-25, его фармацевтически приемлемой соли и/или сольвата любого из приведенных выше соединений.
[0089] Вариант реализации 34. Способ согласно варианту реализации 33, отличающийся тем, что эозинофильное нарушение выбрано из гиперэозинофильного синдрома, эозинофилии, эозинофильного энтерогастрита, эозинофильного лейкоза, эозинофильной гранулемы и болезни Кимуры.
[0090] Вариант реализации 35. Способ согласно варианту реализации 33, отличающийся тем, что эозинофильное нарушение представляет собой гиперэозинофильный синдром.
[0091] Вариант реализации 36. Способ согласно варианту реализации 33, отличающийся тем, что эозинофильное нарушение представляет собой эозинофильный лейкоз.
[0092] Вариант реализации 37. Способ согласно варианту реализации 36, отличающийся тем, что эозинофильный лейкоз представляет собой хронический эозинофильный лейкоз.
[0093] Вариант реализации 38. Способ согласно любому из вариантов реализации 33-37, отличающийся тем, что эозинофильное нарушение не поддается лечению с помощью иматиниба, сунитиниба и/или регорафениба.
[0094] Вариант реализации 39. Соединение согласно любому из вариантов реализации 1-25, его фармацевтически приемлемая соль и/или сольват любого из приведенных выше соединений для применения в качестве лекарственного средства для лечения эозинофильного нарушения.
[0095] Вариант реализации 40. Соединение согласно варианту реализации 39, отличающееся тем, что эозинофильное нарушение выбрано из гиперэозинофильного синдрома, эозинофилии, эозинофильного энтерогастрита, эозинофильного лейкоза, эозинофильной гранулемы и болезни Кимуры.
[0096] Вариант реализации 41. Соединение согласно варианту реализации 39, отличающееся тем, что эозинофильное нарушение представляет собой гиперэозинофильный синдром.
[0097] Вариант реализации 42. Соединение согласно варианту реализации 39, отличающееся тем, что эозинофильное нарушение представляет собой эозинофильный лейкоз.
[0098] Вариант реализации 43. Соединение согласно варианту реализации 42, отличающееся тем, что эозинофильный лейкоз представляет собой хронический эозинофильный лейкоз.
[0099] Вариант реализации 44. Способ согласно любому из вариантов реализации 39-43, отличающийся тем, что эозинофильное нарушение не поддается лечению с помощью иматиниба, сунитиниба и/или регорафениба.
[00100] Вариант реализации 45. Способ лечения нарушения тучных клеток, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения согласно любому из вариантов реализации 1-25, его фармацевтически приемлемой соли и/или сольвата любого из приведенных выше соединений.
[00101] Вариант реализации 46. Способ согласно варианту реализации 45, отличающийся тем, что нарушение тучных клеток опосредовано мутантным KIT или PDGFRα.
[00102] Вариант реализации 46-1. Способ согласно варианту реализации 45, отличающийся тем, что нарушение тучных клеток опосредовано KIT или PDGFRα дикого типа.
[00103] Вариант реализации 47. Способ согласно любому из вариантов реализации 46, отличающийся тем, что нарушение тучных клеток выбрано из синдрома активации тучных клеток (MCAS) и наследственной альфа-триптаземии (HAT).
[00104] Вариант реализации 48. Способ согласно варианту реализации 47, отличающийся тем, что MCAS выбран из синдрома активации моноклональных тучных клеток (MMAS), вторичного MCAS и идиопатического MCAS.
[00105] Вариант реализации 48-1. Способ согласно варианту реализации 27, отличающийся тем, что заболевание или состояние представляет собой системный мастоцитоз.
[00106] Вариант реализации 49. Способ согласно любому из вариантов реализации 48, отличающийся тем, что системный мастоцитоз выбран из вялотекущего системного мастоцитоза и тлеющего системного мастоцитоза.
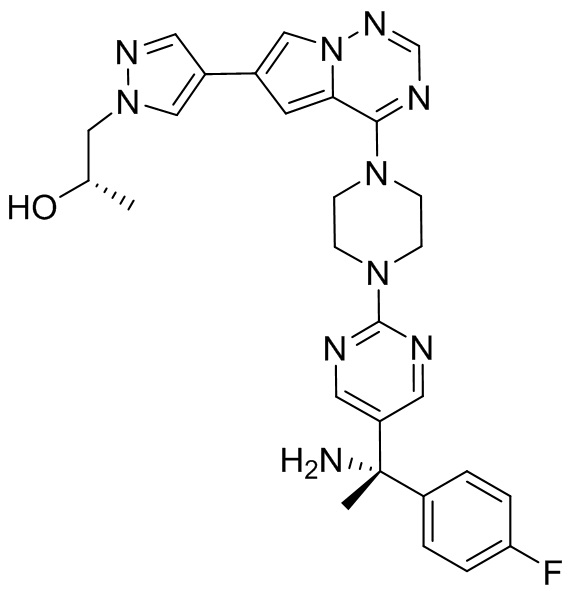
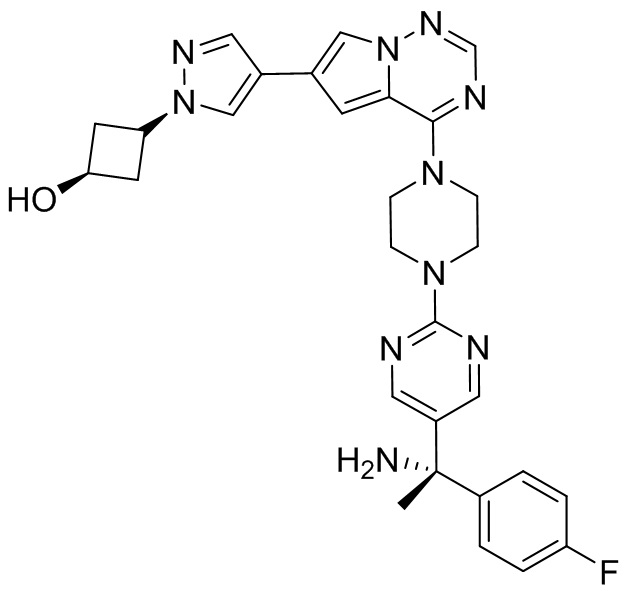
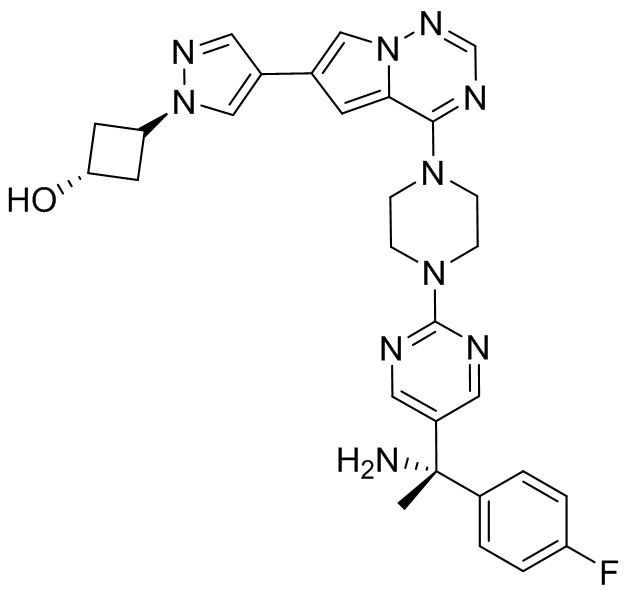
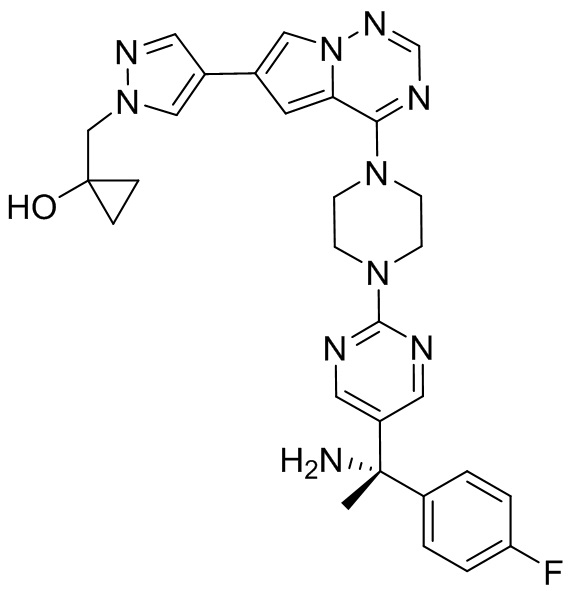
В таблице 1 перечислены соединения, полученные описанными в настоящем документе способами синтеза.
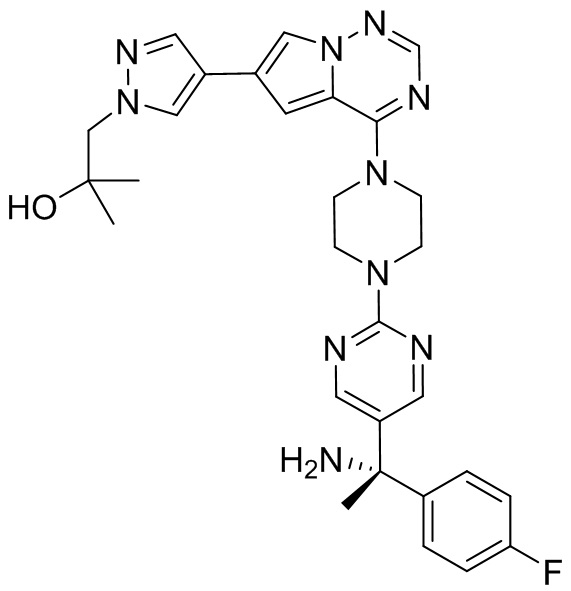
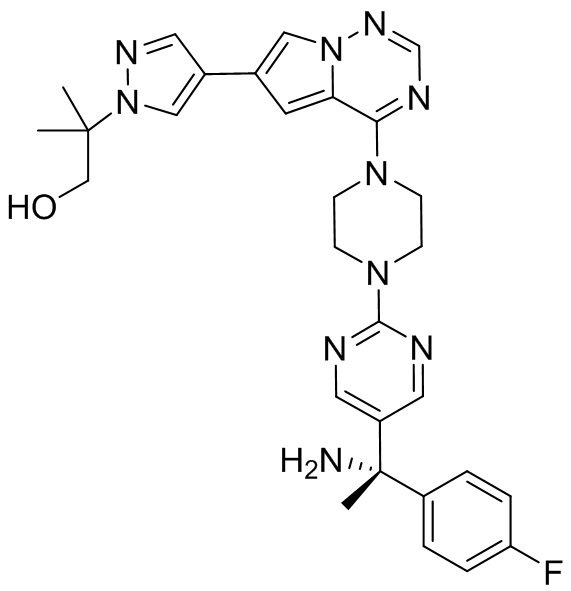
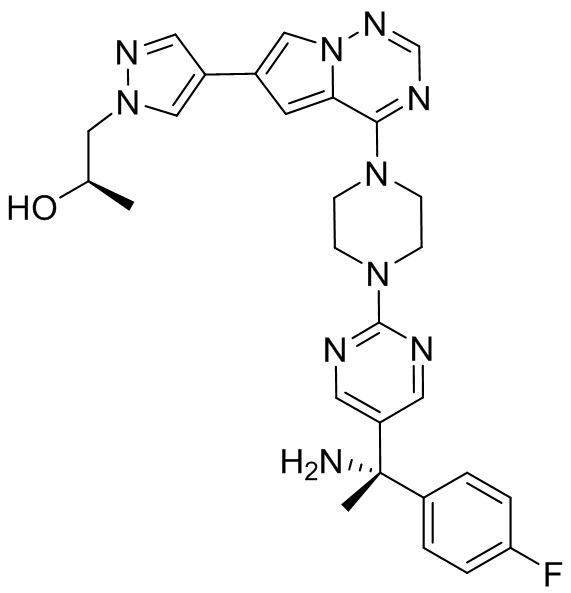
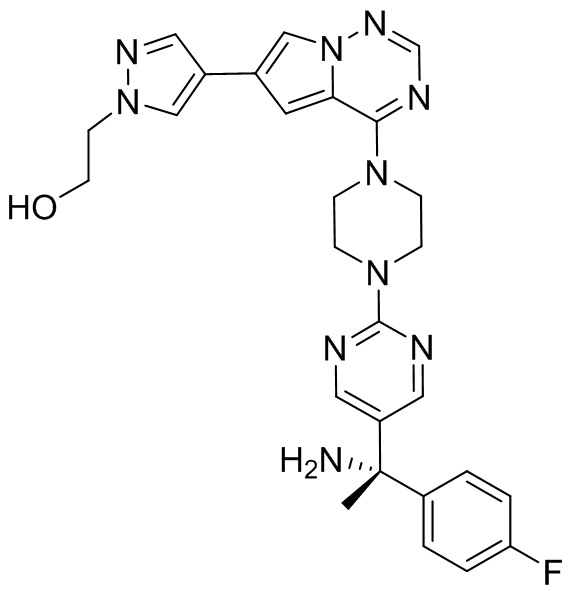
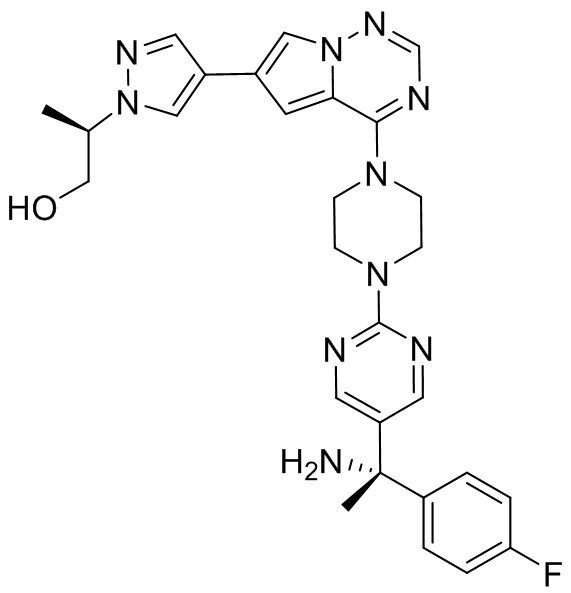
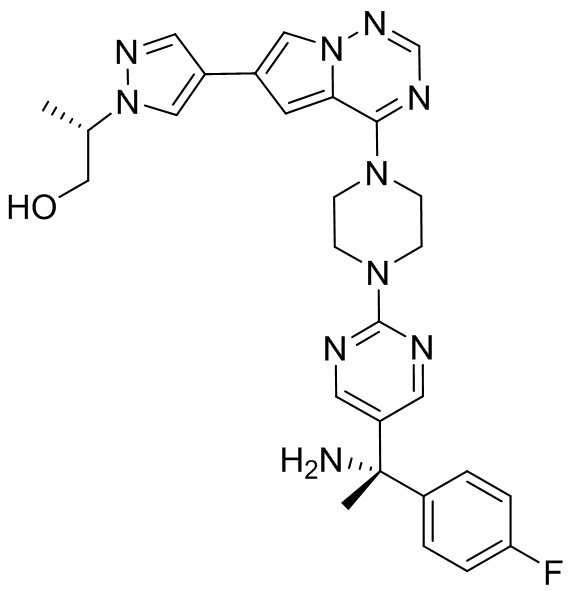
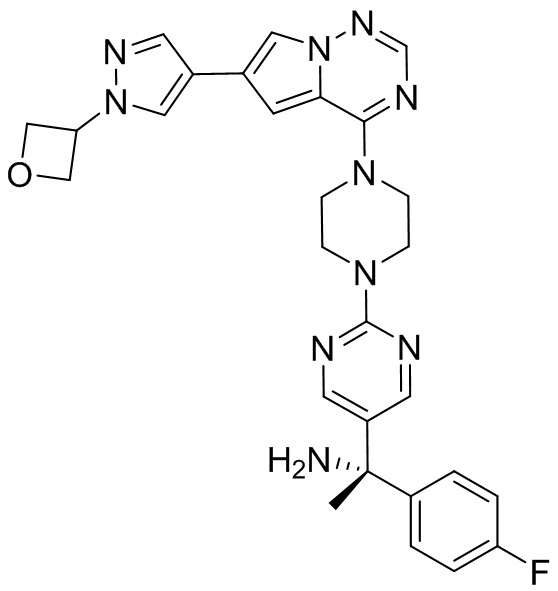
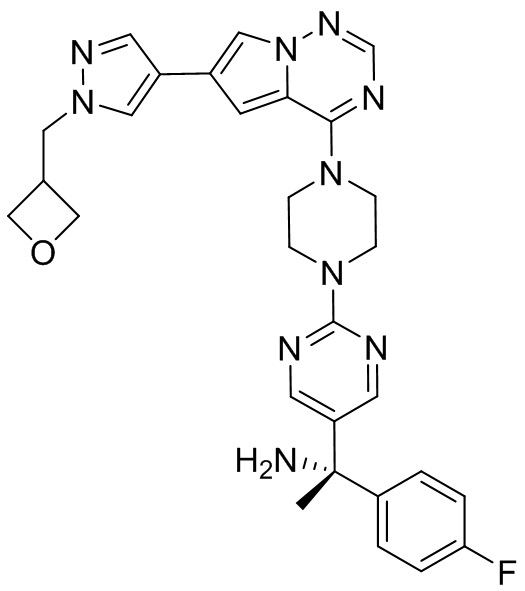
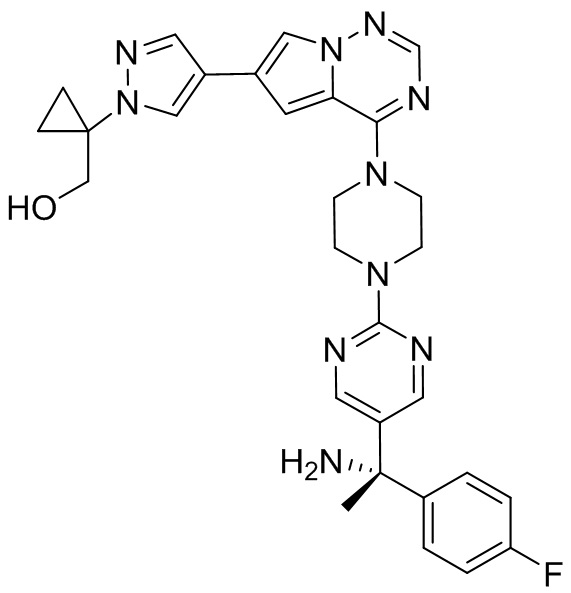
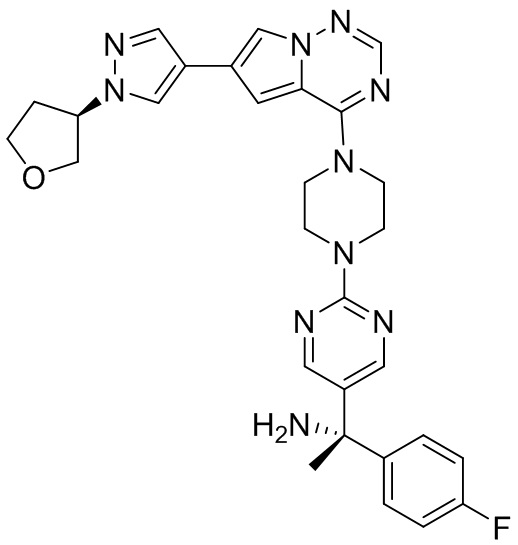
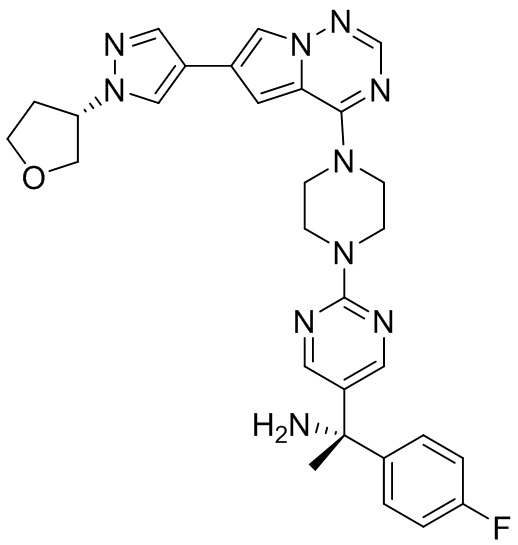
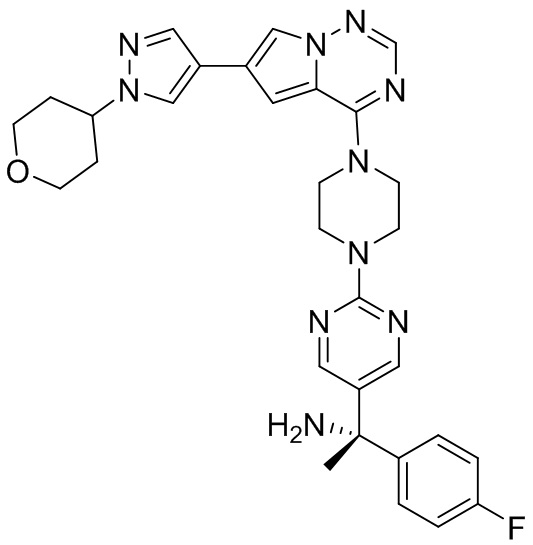
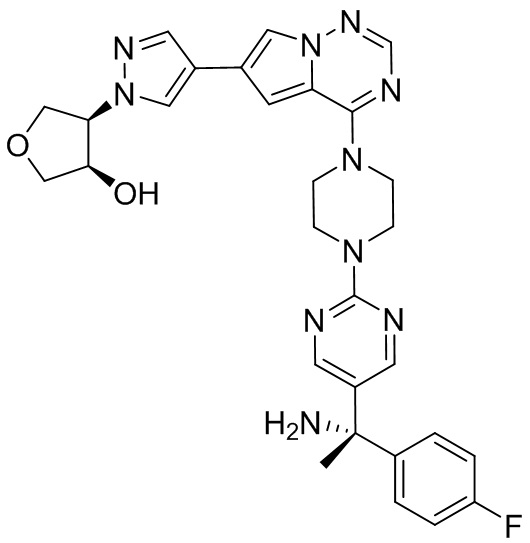
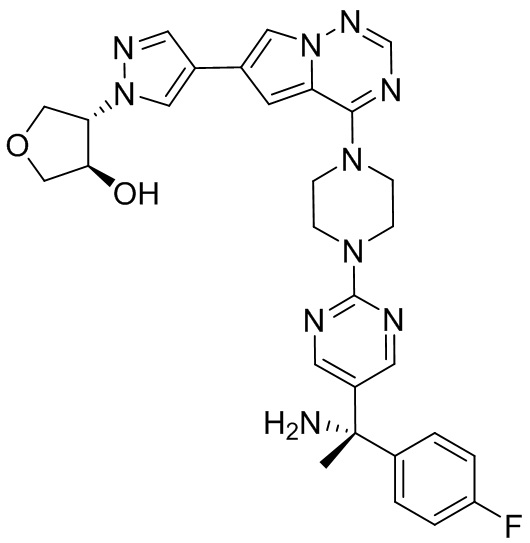
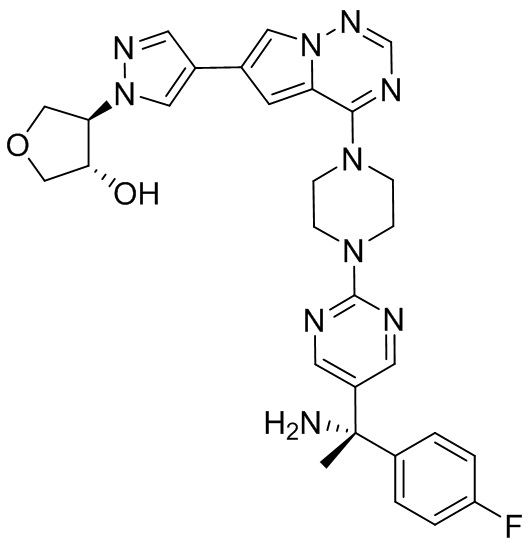
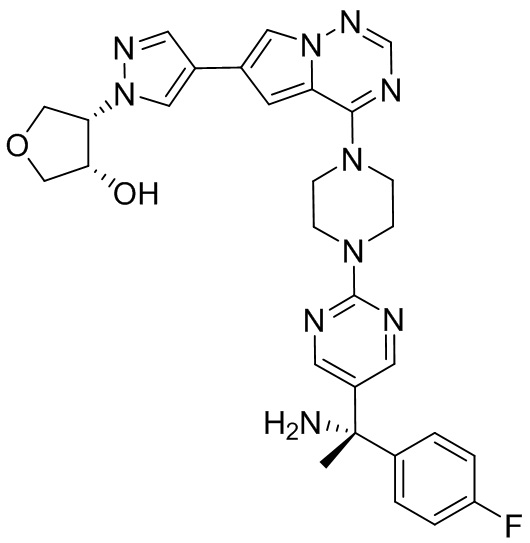
[00107] Соединения согласно настоящему изобретению представляют собой селективные ингибиторы KIT. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, представляют собой селективные ингибиторы KIT D816V. Соединения согласно настоящему изобретению представляют собой селективные ингибиторы PDGFRα. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, представляют собой селективные ингибиторы экзона 18 PDGFRα. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, представляют собой селективные ингибиторы PDGFRα D842V. В настоящем документе «селективный ингибитор KIT» или «селективный ингибитор PDGFRα» относится к соединению или его фармацевтически приемлемой соли или сольвату любого из приведенных выше соединений, которое селективно ингибирует протеинкиназу KIT или протеинкиназу PDGFRα по сравнению с другой протеинкиназой и проявляет по меньшей мере 2-кратную селективность в отношении протеинкиназы KIT или протеинкиназы PDGFRα по сравнению с другой киназой. Например, селективный ингибитор KIT или селективный ингибитор PDGFRA демонстрирует по меньшей мере 9-кратную селективность, 10-кратную селективность; по меньшей мере 15-кратную селективность; по меньшей мере 20-кратную селективность; по меньшей мере 30-кратную селективность; по меньшей мере 40-кратную селективность; по меньшей мере 50-кратную селективность; по меньшей мере 60-кратную селективность; по меньшей мере 70-кратную селективность; по меньшей мере 80-кратную селективность; по меньшей мере 90-кратную селективность; по меньшей мере 100-кратную, по меньшей мере 125-кратную, по меньшей мере 150-кратную, по меньшей мере 175-кратную или по меньшей мере 200-кратную селективность в отношении протеинкиназы KIT или киназы PDGFRα по сравнению с другой киназой. Согласно некоторым вариантам реализации селективный ингибитор KIT или селективный ингибитор PDGFRα проявляет по меньшей мере 150-кратную селективность по сравнению с другой киназой, например, VEGFR2 (рецептор 2 сосудистого эндотелиального фактора роста), SRC (нерецепторная протеин-тирозинкиназа) и FLT3 (Fms-подобная тирозинкиназа 3). Согласно некоторым вариантам реализации селективный ингибитор KIT или селективный ингибитор PDGFRα проявляет селективность по сравнению с PDGRFβ, CSF1R (рецептор 1 колониестимулирующего фактора) и FLT3. Согласно некоторым вариантам реализации селективный ингибитор KIT или селективный ингибитор PDGFRα демонстрирует селективность по сравнению с LCK (лимфоцит-специфическая протеинкиназа), ABL (ядерная протеин-тирозинкиназа), киназой 5, родственной (NIMA) (ген A никогда в митозе) (NEK5) (never-in-mitosis gene A (NIMA)-related kinase 5), и ROCK1 (rho-ассоциированная скрученная спиралью протеинкиназа-1) (rho-associated coil-coil-continuing protein kinase-1). Согласно некоторым вариантам реализации селективность в отношении протеинкиназы KIT или протеинкиназы PDGFRα по сравнению с другой киназой измеряют при клеточном анализе (например, при анализе клеток). Согласно некоторым вариантам реализации селективность в отношении протеинкиназы KIT или протеинкиназы PDGFRα по сравнению с другой киназой измеряют при биохимическом анализе (например, биохимическом анализе).
[00108] Соединения согласно настоящему изобретению являются селективными относительно ионных каналов. Согласно некоторым вариантам реализации селективный ингибитор KIT или селективный ингибитор PDGFRα обладает ограниченной способностью ингибировать потенциал-управляемые натриевые каналы человека (hNav 1.2).
[00109] Соединения согласно настоящему изобретению являются селективными в отношении мутантного KIT по сравнению с KIT дикого типа. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, являются селективными в отношении мутантного KIT в экзоне 17 по сравнению с KIT дикого типа.
[00110] Соединения согласно настоящему изобретению можно использовать для лечения заболеваний или состояний, связанных с активностью мутантного KIT или мутантного PDGFRA у людей или субъектов, не относящихся к людям. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, предназначены для применения в качестве лекарственного средства. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, предназначены для применения в терапии. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, предназначены для применения в производстве лекарственного средства. Согласно некоторым вариантам реализации в настоящем изобретении предложены способы лечения злокачественных новообразований, вызванных KIT, в том числе мастоцитоза (SM), GIST (желудочно-кишечных стромальных опухолнй), AML (острого миелоидного лейкоза), меланомы, семиномы, внутричерепных герминогенных опухолей и/или медиастинальной B-клеточной лимфомы. Кроме того, мутации в KIT были связаны с саркомой Юинга, DLBCL (диффузной B-крупноклеточной лимфомой), дисгерминомой, MDS (миелодиспластическим синдромом), NKTCL (назальной NK/T-клеточной лимфомой), CMML (хроническим миеломоноцитарным лейкозом) и раками головного мозга. Согласно некоторым вариантам реализации в настоящем изобретении предложены способы лечения саркомы Юинга, DLBCL, дисгерминомы, MDS, NKTCL, CMML и/или раков головного мозга. Мутации KIT также были обнаружены при раке щитовидной железы, колоректальном раке, раке эндометрия, раке мочевого пузыря, NSCLC и раке груди (проект AACR GENIE). Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, можно использовать для лечения синдрома активации тучных клеток (MCAS). Соединения согласно настоящему изобретению можно использовать для лечения системного мастоцитоза. Соединения согласно настоящему изобретению можно использовать для лечения продвинутого системного мастоцитоза. Соединения согласно настоящему изобретению можно использовать для лечения вялотекущего SM и тлеющего SM. Соединения согласно настоящему изобретению можно использовать для лечения GIST.
[00111] Соединения согласно настоящему изобретению можно использовать для лечения заболеваний или состояний, связанных с мутациями KIT в экзоне 9, экзоне 11, экзоне 14, экзоне 17 и/или экзоне 18 последовательности гена KIT. Соединения согласно настоящему изобретению можно использовать для лечения заболеваний или состояний, связанных с мутациями PDGFRA в экзоне 12, экзоне 14 и/или экзоне 18 последовательности гена PDGFRA. Согласно некоторым вариантам реализации в настоящем документе описаны способы лечения заболевания или состояния, связанного с по меньшей мере одной мутацией KIT в экзоне 9, экзоне 11, экзоне 14, экзоне 17 и/или экзоне 18 последовательности гена KIT. Согласно некоторым вариантам реализации описаны способы лечения заболевания или состояния, связанного с по меньшей мере одной мутацией PDGFRA в экзоне 12, экзоне 14 и/или экзоне 18 последовательности гена PDGFRA.
[00112] Соединения согласно настоящему изобретению могут быть активными против одной или более протеинкиназ KIT с мутациями в экзоне 17 последовательности гена KIT (например, мутациями белка KIT D816V, D816Y, D816F, D816K, D816H, D816A, D816G, D816E, D816I, D816F, D820A, D820E, D820G, D820Y, N822K, N822H, V560G, Y823D и A829P) и гораздо менее активными в отношении протеинкиназы KIT дикого типа. Согласно некоторым вариантам реализации в настоящем документе описаны способы лечения заболевания или состояния, связанного с по меньшей мере одной мутацией KIT, например, мутациями, выбранными из D816V, D816Y, D816F, D816K, D816H, D816A, D816G, D816E, D816I, D816F, D820A, D820E, D820G, D820Y, N822K, N822H, V560G, Y823D и A829P. Согласно некоторым вариантам реализации в настоящем документе описаны способы лечения заболевания или состояния, связанного с по меньшей мере одной мутацией KIT, такой как, например, мутации, выбранные из C809, C809G, D816H, D820A, D820G, N822H, N822K и Y823D.
[00113] Соединения согласно настоящему изобретению могут быть активными против одной или более протеинкиназ KIT с мутациями в экзоне 11 последовательности гена KIT (например, мутациями белка KIT del557-559insF, V559G/D). Согласно некоторым вариантам реализации в настоящем документе описаны способы лечения заболевания или состояния, связанного с по меньшей мере одной мутацией KIT, такой как, например, мутации, выбранные из L576P, V559D, V560D, V560G, W557G, Del 554-558EVQWK, del557-559insF, Del EVQWK554-558, Del EVQWKVVEEINGNNYVYI554-571, Del KPMYEVQWK550-558, Del KPMYEVQW550-557FL, Del KV558-559, Del KV558-559N, Del MYEVQW552-557, Del PMYE551-554, Del VV559-560, Del WKVVE557-561, Del WK557-558, Del WKVV557-560C, Del WKVV557-560F, DelYEVQWK553-558 и вставки K558NP.
[00114] Соединения согласно настоящему изобретению могут быть активными против одной или более протеинкиназ KIT с мутациями в экзоне 11/13 последовательности гена KIT (например, мутациями белка KIT V559D/V654A, V560G/D816V и V560G/822K). Согласно некоторым вариантам реализации в настоящем документе описаны способы лечения заболевания или состояния, связанного с одной или более мутациями KIT в экзоне 11/13).
[00115] Соединения согласно настоящему изобретению могут быть активными против одной или более протеинкиназ KIT с мутациями в экзоне 9 последовательности гена KIT. Согласно некоторым вариантам реализации в настоящем документе описаны способы лечения заболевания или состояния, связанного с по меньшей мере одной мутацией KIT в экзоне 9.
[00116] Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, не активны в отношении протеинкиназ KIT с мутациями V654A, N655T, T670I и/или N680.
[00117] Соединения согласно настоящему изобретению могут быть активными против одной или более протеинкиназ PDGFRα с мутациями. Согласно некоторым вариантам реализации в настоящем документе описаны способы лечения заболевания или состояния, связанного с по меньшей мере одной мутацией PDGFRA в экзоне 12 последовательности гена PDGFRA, такой как, например, мутации белка PDGFRα V561D, Del RV560-561, Del RVIES560-564, Ins ER561-562, SPDGHE566-571R, SPDGHE566-571K или Ins YDSRW582-586. Согласно некоторым вариантам реализации в настоящем документе описаны способы лечения заболевания или состояния, связанного с по меньшей мере одной мутацией PDGFRA в экзоне 14 последовательности гена PDGFRA, такой как, например, мутация белка PDGFRα N659K. Согласно некоторым вариантам реализации в настоящем документе описаны способы лечения заболевания или состояния, связанного с по меньшей мере одной мутацией PDGFRA в экзоне 18 последовательности гена PDGFRA, такой как, например, мутации белка PDGFRα D842V, D842Y, D842I, DI842-843IM, D846Y, Y849C, Del D842, Del I843, Del RD841-842, Del DIM842-845, Del DIMH842-845, Del IMHD843-846, Del MHDS844-847, RD841-842KI, DIMH842-845A, DIMH842-845V, DIMHD842-846E, DIMHD842-846S, DIMHD842-846N, DIMHD842-846G, IMHDS843-847T, IMHDS8843-847M или HDSN845-848P.
[00118] Соединения согласно настоящему изобретению могут быть активными против одного или более протеинкиназ PDGFRα с мутациями в экзоне 18 в последовательности гена PDGFRA (например, мутациями белка PDGFRα D842V, PDGFRα D842I или PDGFRα D842Y). Согласно некоторым вариантам реализации в настоящем документе описаны способы лечения заболевания или состояния, связанного с по меньшей мере одной мутацией PDGFRA в экзоне 18, такой как, например, мутация белка PDGFRα D842V.
[00119] Соединения согласно настоящему изобретению можно использовать для лечения эозинофильного нарушения. Согласно некоторым вариантам реализации эозинофильное нарушение опосредовано мутантным KIT или PDGFRα. Согласно некоторым вариантам реализации указанное эозинофильное нарушение опосредовано KIT или PDGFRα дикого типа. Согласно некоторым вариантам реализации в настоящем документе описаны способы лечения эозинофильного нарушения, включающие введение субъекту терапевтически эффективного количества соединений, предложенных в настоящем изобретении, или их фармацевтически приемлемой соли и/или сольвата любого из приведенных выше соединений. Согласно одному из вариантов реализации эозинофильное нарушение выбрано из гиперэозинофильного синдрома, эозинофилии, эозинофильного энтерогастрита, эозинофильного лейкоза, эозинофильной гранулемы и болезни Кимуры.
[00120] Согласно некоторым вариантам реализации эозинофильное нарушение выбрано из гиперэозинофильного синдрома, эозинофилии, эозинофильного энтерогастрита, эозинофильного лейкоза, эозинофильной гранулемы и болезни Кимуры. Другие эозинофильные нарушения включают эозинофильный эзофагит, эозинофильный гастроэнтерит, эозинофильный фасциит и синдром Чарджа-Стросса.
[00121] Согласно одному из вариантов реализации эозинофильное нарушение представляет собой гиперэозинофильный синдром. Согласно конкретному варианту реализации гиперэозинофильный синдром представляет собой идиопатический гиперэозинофильный синдром. Согласно одному из вариантов реализации эозинофильное нарушение представляет собой эозинофильный лейкоз. Согласно конкретному варианту реализации эозинофильный лейкоз представляет собой хронический эозинофильный лейкоз. Согласно еще одному варианту реализации эозинофильное нарушение не поддается лечению с помощью иматиниба, сунитиниба и/или регорафениба. Согласно конкретному варианту реализации эозинофильное нарушение не поддается лечению с помощью иматиниба.
[00122] Соединения согласно настоящему изобретению можно использовать для снижения количества эозинофилов у субъекта, нуждающегося в этом. Согласно некоторым вариантам реализации в настоящем документе описаны способы уменьшения количества эозинофилов у субъекта, нуждающегося в этом, включающие введение субъекту терапевтически эффективного количества соединения, предложенного в настоящем изобретении, или его фармацевтически приемлемой соли и/или сольвата любого из приведенных выше соединений.
[00123] Согласно одному из вариантов реализации предложенные способы снижают количество эозинофилов в крови, костном мозге, желудочно-кишечном тракте (например, пищеводе, желудке, тонком кишечнике и толстой кишке) или легких. Согласно еще одному варианту реализации описанный в настоящем документе способ позволяет уменьшить количество эозинофилов в крови. Согласно дополнительному варианту реализации описанный в настоящем документе способ позволяет уменьшить количество эозинофилов в легких. Согласно еще одному дополнительному варианту реализации описанный в настоящем документе способ позволяет уменьшить количество клеток-предшественников эозинофилов.
[00124] Согласно другому варианту реализации предложенные способы снижают (после введения) количество эозинофилов на по меньшей мере примерно 10%, по меньшей мере примерно 20%, по меньшей мере примерно 30%, по меньшей мере примерно 40%, по меньшей мере примерно 50%, по меньшей мере примерно 60%, по меньшей мере примерно 70%, по меньшей мере примерно 80%; по меньшей мере примерно 90%, по меньшей мере примерно 95% или по меньшей мере примерно 99%. Согласно конкретному варианту реализации описанный в настоящем документе способ позволяет уменьшить количество эозинофилов ниже предела обнаружения.
[00125] Согласно еще одному варианту реализации предложенные способы снижают (после введения) количество предшественников эозинофилов на по меньшей мере примерно 10%, по меньшей мере примерно 20%, по меньшей мере примерно 30%, по меньшей мере примерно 40%, по меньшей мере примерно 50%, по меньшей мере примерно 60%, по меньшей мере примерно 70%, по меньшей мере примерно 80%, по меньшей мере примерно 90%, по меньшей мере примерно 95% или по меньшей мере примерно 99%. Согласно конкретному варианту реализации описанный в настоящем документе способ позволяет уменьшить количество предшественников эозинофилов ниже предела обнаружения.
[00126] Соединения согласно настоящему изобретению можно использовать для лечения нарушений тучных клеток. Соединения согласно настоящему изобретению можно использовать для лечения мастоцитоза. Мастоцитоз подразделяют на две группы нарушений: (1) кожный мастоцитоз (CM) включает формы, которые ограничены кожей; и (2) системный мастоцитоз (SM) включает формы, при которых тучные клетки образуют инфильтраты во внекожных органах с поражением кожи или без него. SM дополнительно подразделят на пять форм: вялотекущий (ISM); тлеющий (SSM); агрессивный (ASM); SM с ассоциированным гематологическим заболеванием нетучных клеток (SM-AHNMD); и лейкоз тучных клеток (MCL).
[00127] Диагностика SM частично основана на гистологических и цитологических исследованиях костного мозга, показывающих инфильтрацию тучных клеток часто атипичной морфологии, которые часто аномально экспрессируют маркеры нетучных клеток (CD25 и/или CD2). Диагноз SM подтверждают, когда инфильтрация тучных клеток в костный мозг происходит в контексте одного из следующих факторов: (1) аномальная морфология тучных клеток (веретенообразные клетки); (2) повышенный уровень триптазы в сыворотке выше 20 нг/мл; или (3) наличие активирующих мутаций белка KIT, таких как, например, мутации в экзоне 17, такие как мутации D816, например, D816V.
[00128] Активирующие мутации в положении D816 обнаруживают в подавляющем большинстве случаев мастоцитоза (90-98%), при этом наиболее частыми мутациями являются D816V, D816H и D816Y. Мутацию D816V обнаруживают в петле активации домена протеинкиназы, при этом указанная мутация приводит к конститутивной активации киназы KIT.
[00129] Ни одно из лекарственных средств не одобрено для лечения непродвинутых форм системного мастоцитоза, ISM или SSM. Современные подходы к лечению таких хронических заболеваний включают неспецифические симптоматические способы лечения, которые имеет разную степень эффективности и не влияют на тяжесть MC. Для уменьшения устойчивых симптомов иногда используют циторедуктивные способы лечения, такие как кладрибин и интерферон альфа. С учетом текущего комплекса имеющихся средств и методов лечения сохраняется неудовлетворенная медицинская потребность в пациентах с ISM и SSM с умеренными и тяжелыми симптомами, которыми невозможно адекватно управлять с помощью доступных симптоматических способов лечения.
[00130] Соединения согласно настоящему изобретению можно использовать для лечения ISM или SSM. Согласно некоторым вариантам реализации пациент с ISM или SSM имеет симптомы, которые плохо поддаются контролю посредством по меньшей мере одного, по меньшей мере двух, по меньшей мере трех симптоматических методов лечения. Симптомы можно оценить с помощью инструмента в виде исхода, сообщаемого пациентом (PRO), например, Формы для оценки симптомов вялотекущего системного мастоцитоза (ISM-SAF) (ISPOR Europe 2019, Копенгаген, Дания, 2-6 ноября 2019 г.). Соединения согласно настоящему изобретению можно использовать для улучшения симптомов, связанных с ISM или SSM, например, уменьшения или устранения зуда, приливов, головных болей и/или событий со стороны желудочно-кишечного тракта, таких как рвота, диарея и боль в животе. Улучшение симптомов можно оценить с помощью ISM-SAF.
[00131] Соединения согласно настоящему изобретению можно использовать для лечения и других нарушений тучных клеток, таких как синдром активации тучных клеток (MCAS) и наследственная альфа-триптаземия (HAT) (Picard Clin. Ther. 2013, May 35(5) 548; Akin J.Allergy Clin. Immuno. 140(2)349 62). Соединения согласно настоящему изобретению можно использовать для лечения нарушений тучных клеток, связанных с мутациями KIT и PDGFRα. Соединения согласно настоящему изобретению можно использовать для лечения заболеваний тучных клеток, связанных с KIT и PDGFRα дикого типа.
[00132] Соединения согласно настоящему изобретению можно использовать для лечения синдрома активации тучных клеток (MCAS), который представляет собой иммунологическое состояние, при котором тучные клетки неадекватно и чрезмерно высвобождают химические медиаторы, что приводит к ряду хронических симптомов, иногда включающих анафилаксию или приступы, близкие к анафилаксии. В отличие от мастоцитоза, при котором у пациентов наблюдается аномально повышенное количество тучных клеток, пациенты с MCAS имеют нормальное количество тучных клеток, которые не функционируют должным образом и определяются как «гиперчувствительные». Типы MCAS включают первичный MCAS (синдром активации моноклональных тучных клеток (MMAS)), вторичный MCAS (MCAS, возникающий в результате другого заболевания) и идиопатический MCAS (MCAS, исключающий первичный или вторичный MCAS).
[00133] Соединения согласно настоящему изобретению можно использовать для лечения наследственной альфа-триптаземии (HAT) (сверхэкспрессии TPSAB1, вызывающей повышенный уровень триптазы).
[00134] Другие заболевания тучных клеток включают астму, опосредованную тучными клетками, анафилаксию (в том числе идиопатическую, Ig-E- и не-Ig-E-опосредованную), крапивницу (в том числе идиопатическую и хроническую), атопический дерматит, отек (ангионевротический отек), синдром раздраженного кишечника, мастоцитарный гастроэнтерит, мастоцитарный колит, зуд, хронический зуд, вторичный зуд на фоне хронической почечной недостаточности и сердечных, сосудистых, кишечных заболеваний, заболеваний головного мозга, почек, печени, поджелудочной железы, мышц, костей и кожи, связанных с тучными клетками. Согласно некоторым вариантам реализации заболевание тучных клеток не связано с мутантным KIT или мутантным PDGFRα.
[00135] Мутации KIT и PDGFRA широко изучались в GIST. Соединения согласно настоящему изобретению можно использовать для лечения GIST, связанной с мутациями KIT. Соединения согласно настоящему изобретению можно использовать для лечения неоперабельной или метастатической GIST. Примерно 80% метастатических GISTs имеют первичную активирующую мутацию либо во внеклеточном участке (экзон 9), либо в околомембранном (JM) домене (экзон 11) последовательности гена KIT. Многие опухоли, содержащие мутантный KIT, реагируют на лечение посредством таргетной терапии, такой как иматиниб, селективный ингибитор тирозинкиназы, который специфически ингибирует белки BCR-ABL, KIT и PDGFRA. Однако у большинства пациентов с GIST со временем возникает рецидив вследствие вторичной мутации в KIT, которая заметно снижает сродство к связыванию иматиниба. Такие мутации резистентности неизбежно возникают в кармане, связывающем аденозин-5-трифосфат (АТФ), (экзоны 13 и 14) или в активационной петле (экзоны 17 и 18) гена киназы. Из одобренных в настоящее время препаратов для лечения GIST ни один не является селективным таргетным препаратом. В настоящее время для лечения GIST одобрен иматиниб; после иматиниба используют ингибиторы мультикиназы. Во многих случаях указанные ингибиторы мультикиназы, такие как, например, сунитиниб, регорафениб и мидостаурин, только слабо ингибируют мутанты, устойчивые к иматинибу, и/или ингибиторы мультикиназы ограничены более сложным профилем безопасности и небольшим терапевтическим окном. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, можно использовать для лечения GIST у пациентов, получавших иматиниб. Соединения согласно настоящему изобретению можно использовать для лечения GIST в качестве терапии первой линии (1L), второй линии (2L), третьей линии (3L) или четвертой линии (4L).
[00136] Соединения согласно настоящему изобретению можно использовать для лечения GIST при наличии или отсутствии конкретных мутаций в KIT. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, способны лечить GIST при отсутствии в KIT определенных мутаций. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, не способны лечить GIST при наличии в KIT определенных мутаций. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, не обеспечивают клинического результата у пациентов, несущих мутации в KIT-ATP-связывающем кармане (мутации белка KIT V654A, N655T и/или T670I).
[00137] Соединения согласно настоящему изобретению можно использовать для лечения GIST, связанного с мутациями PDGFRA. У от 5 до 6% пациентов с неоперабельной метастатической GIST мутация активирующей петли в экзоне 18 генной последовательности PDGFRA в белковой аминокислоте 842 возникает как первичная мутация.
[00138] Соединения согласно настоящему изобретению также можно использовать при лечении AML. Пациенты с AML также несут мутации KIT, при этом большинство из указанных мутаций находятся в положении D816 белка KIT.
[00139] Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, вводят субъекту, нуждающемуся в этом. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, вводят в виде фармацевтического состава, в котором предложенное соединение объединяют с одним или более фармацевтически приемлемыми вспомогательными веществами или носителями. Таким образом, согласно некоторым вариантам реализации в настоящем документе описаны композиции, содержащие по меньшей мере одно соединение, выбранное из соединений формулы I, и их фармацевтически приемлемые соли и/или сольваты любого из приведенных выше соединений и необязательно дополнительно содержащие по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
[00140] Соединения согласно настоящему изобретению можно получить для введения любым удобным способом для применения в медицине или ветеринарии. Согласно некоторым вариантам реализации соединение, включенное в фармацевтические композиции, может быть активным само по себе или может представлять собой пролекарство, например, способное превращаться в активное соединение в физиологических условиях.
[00141] Выражение «фармацевтически приемлемый» используют в настоящем документе для обозначения тех соединений, веществ, композиций и/или лекарственных форм, которые в рамках здравого медицинского суждения подходят для применения в контакте с тканями людей и животных, не вызывая чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соизмеримых с разумным соотношением польза/риск.
[00142] Примеры фармацевтически приемлемых носителей включают: (1) сахара, такие как, например, лактоза, глюкоза и сахароза; (2) крахмалы, такие как, например, кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как, например, карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; (4) порошковый трагакант; (5) солод; (6) желатин; (7) тальк; (8) вспомогательные вещества, такие как, например, масло какао и воски для суппозиториев; (9) масла, такие как, например, арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как, например, пропиленгликоль; (11) полиолы, такие как, например, глицерин, сорбитол, маннитол и полиэтиленгликоль; (12) сложные эфиры, такие как, например, этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как, например, гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатные буферные растворы; (21) циклодекстрины, такие как, например, Captisol® (Каптизол); и (22) другие нетоксичные совместимые вещества, применяемые в фармацевтических составах.
[00143] Примеры фармацевтически приемлемых антиоксидантов включают: (1) водорастворимые антиоксиданты, такие как, например, аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфит натрия, сульфит натрия и т.п.; (2) маслорастворимые антиоксиданты, такие как, например, аскорбилпальмитат, бутилированный гидроксианизол (BHA), бутилированный гидрокситолуол (BHT), лецитин, пропилгаллат, альфа-токоферол и т.п.; и (3) агенты, образующие хелатные комплексы с металлами, такие как, например, лимонная кислота, этилендиаминтетрауксусная кислота (ЭДТК), сорбитол, винная кислота, фосфорная кислота и т.п.
[00144] Твердые лекарственные формы (например, капсулы, таблетки, пилюли, драже, порошки, гранулы и т.п.) могут включать один или более фармацевтически приемлемых носителей, таких как, например, цитрат натрия или дикальцийфосфат, и/или любые из следующих веществ: (1) наполнители или разбавители, такие как, например, крахмалы, лактоза, сахароза, глюкоза, маннитол и/или кремниевая кислота; (2) связующие вещества, такие как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или гуммиарабик; (3) увлажнители, такие как, например, глицерин; (4) вещества для улучшения распадаемости таблеток, такие как, например, агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия; (5) агенты, замедляющие растворение, такие как, например, парафин; (6) ускорители абсорбции, такие как, например, четвертичные аммониевые соединения; (7) смачивающие агенты, такие как, например, цетиловый спирт и моностеарат глицерина; (8) абсорбенты, такие как, например, каолин и бентонитовая глина; (9) смазывающие вещества, такие как, например, тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси; и (10) красители.
[00145] Жидкие лекарственные формы могут включать фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активному ингредиенту жидкие лекарственные формы могут содержать инертные разбавители, обычно применяемые в данной области техники, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (такие как, например, хлопковое, арахисовое, кукурузное масло, масло зародышей, оливковое, касторовое и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана и их смеси.
[00146] Наряду с активными соединениями суспензии могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар, трагакант и их смеси.
[00147] Наряду с активным соединением мази, пасты, кремы и гели могут содержать вспомогательные вещества, такие как, например, животные и растительные жиры, масла, воски, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремниевая кислота, тальк, оксид цинка или их смеси.
[00148] Наряду с активным соединением порошки и спреи могут содержать вспомогательные вещества, такие как, например, лактоза, тальк, кремниевая кислота, гидроксид алюминия, силикаты кальция и порошок полиамида или смеси перечисленных веществ. Спреи могут дополнительно содержать обычные пропелленты, такие как, например, хлорфторуглеводороды и летучие незамещенные углеводороды, такие как, например, бутан и пропан.
[00149] Неограничивающие примеры лекарственных форм для местного или трансдермального введения соединений согласно настоящему изобретению включают порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и ингаляторы. Активное соединение можно смешивать в стерильных условиях с фармацевтически приемлемым носителем и с любыми консервантами, буферами или пропеллентами, которые могут потребоваться.
[00150] При введении соединения согласно настоящему изобретению людям и животным в виде фармацевтического препарата, указанное соединение можно вводить само по себе или в виде фармацевтической композиции, содержащей, например, от 0,1 до 99,5% (например, от 0,5 до 90%) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
[00151] Составы можно вводить местно, перорально, трансдермально, ректально, вагинально, парентерально, интраназально, внутрилегочно, внутриглазным способом, внутривенно, внутримышечно, внутриартериальным способом, интратекально, внутрикапсульным способом, внутрикожно, внутрибрюшинно, подкожно, внутриподкожно или путем ингаляции.
[00152] Кроме того, соединения согласно настоящему изобретению можно вводить отдельно или в комбинации с другими соединениями, в том числе другими KIT- или PDGFRα-модулирующими соединениями или другими терапевтическими агентами. Согласно некоторым вариантам реализации соединение, предложенное в настоящем изобретении, можно вводить в комбинации с рипретинибом. Согласно некоторым вариантам реализации соединение, предложенное в настоящем изобретении, можно вводить в комбинации с одним или более соединениями, выбранными из иматиниба, сунитиниба, регорафениба, кабозантиниба, креноланиба, мидостаурина, брентуксимаб ведотина и маститиниба, для лечения заболевания или состояния, описанного в настоящем документе.
[00153] Соединения согласно настоящему изобретению можно вводить пациенту, которого ранее подвергали лечению с применением другого соединения или соединений. Соединения согласно настоящему изобретению можно использовать в качестве терапии первой линии (1L), второй линии (2L), третьей линии (3L) или четвертой линии (4L).
[00154] Согласно некоторым вариантам реализации соединение, предложенное в настоящем изобретении, вводят после предшествующего лечения с помощью иматиниба.
[00155] Соединения согласно настоящему изобретению можно вводить пациенту, которого ранее не подвергали лечению с помощью мидостаурина. Согласно некоторым вариантам реализации соединения, предложенные в настоящем изобретении, можно вводить пациенту, которому ранее проводили лечение с помощью мидостаурина.
ПРИМЕРЫ
Общие методы синтеза и промежуточные продукты
Определения
C Цельсия
Cs2CO3 карбонат цезия
ДХМ дихлорметан
ДИПА диизопропиламин
ДМФ диметилформамид
ДМСО диметилсульфоксид
ЭА этилацетат
EDCI 1-этил-3-(3-диметиламинопропил)карбодиимид
час часы
H2 газообразный водород
H2O вода
HCl соляная кислота
HOAc уксусная кислота
HOBT гидроксибензотриазол
ВЭЖХ высокоэффективная жидкостная хроматография
IC50 ингибирующая концентрация 50%
ИПС изопропиловый спирт
K2CO3 карбонат калия
KOAc ацетат калия
ЖХМС жидкостная хроматография/масс-спектрометрия
LiAlH4 литийалюминийгидрид
мин минуты
MsCl мезилхлорид
МТБЭ метил-трет-бутиловый эфир
MeOH метанол
N2 газообразный азот
NaOH гидроксид натрия
Na2SO4 сульфат натрия
NH4HCO3 формиат аммония
NMP N-метилпирролидинон
Pd/C палладий на углероде
Pd(dppf)Cl2 [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II)
ПЭ петролейный эфир
КТ комнатная температура
ТЭА триэтиламин
ТГФ тетрагидрофуран
TsCl тозилхлорид
[00156] Способы получения соединений согласно настоящему изобретению можно осуществлять в подходящих растворителях, которые легко могут быть выбраны специалистом в области органического синтеза. Подходящие растворители могут по существу не вступать в реакцию с исходными веществами (реагентами), промежуточными соединениями или продуктами при температурах, при которых проводят указанные реакции, например, при температурах, которые могут находиться в диапазоне от температуры замерзания растворителя до температуры кипения растворителя. Данная реакция может быть проведена в одном растворителе или в смеси более чем одного растворителя. В зависимости от конкретной стадии реакции специалист может выбрать подходящие растворители для конкретной стадии реакции.
[00157] Получение соединений согласно настоящему изобретению может включать защиту и снятие защиты с различных химических групп. Специалист в данной области техники может легко определить необходимость защиты и снятия защиты, а также выбор подходящих защитных групп. Химию защитных групп можно найти, например, в Wuts and Greene, Protective Groups in rganic Synthesis, 5th ed., John Wiley & Sons: New Jersey, (2014), которая полностью включена в настоящий документ посредством ссылки.
[00158] Реакции можно контролировать любым подходящим способом, известным в данной области техники. Например, образование продукта можно контролировать с применением спектроскопических методов, таких как спектроскопия ядерного магнитного резонанса (ЯМР) (например, 1H или 13C), инфракрасная (ИК) спектроскопия, спектрофотометрия (например, в УФ и видимой области), масс-спектрометрия (МС), или с помощью хроматографических методов, таких как высокоэффективная жидкостная хроматография (ВЭЖХ) или тонкослойная хроматография (ТСХ).
Аналитические инструменты и способы исследования соединений:
[00159] ЖХ-МС: Если не указано иное, все данные жидкостной хроматографии-масс-спектрометрии (ЖХ-МС) (образец, анализируемый в отношении чистоты и идентичности) были получены с помощью системы для жидкостной хроматографии Agilent model-1260 LC с применением масс-спектрометра Agilent model 6120, в котором используют ионизацию ES-API (ионизацию электрораспылением/при атмосферном давлении), оборудованного колонкой с обращенной фазой Agilent Poroshel 120 (EC-C18, размер частиц 2,7 мкм, размеры 3,0 × 50 мм) при 22,4 градусах Цельсия. Подвижная фаза состояла из смеси растворителя 0,1% муравьиной кислоты в H2O и 0,1% муравьиной кислоты в ацетонитриле. Использовали постоянный градиент в диапазоне от 95% водной/5% органической подвижной фазы до 5% водной/95% органической подвижной фазы в течение 4 минут. Скорость потока была постоянной и составляла 1 мл/мин.
[00160] Препаративная ЖХ-МС: Исследования с применением препаративной ВЭЖХ выполняли в системе для препаративного анализа Shimadzu Discovery VP®, оборудованной колонкой с обращенной фазой Luna 5u C18(2) 100A размером 250 × 21,2 мм с набивкой AXIA при 22,4 градуса Цельсия. Подвижная фаза состояла из смеси растворителя 0,1% муравьиной кислоты в H2O и 0,1% муравьиной кислоты в ацетонитриле. Использовали постоянный градиент в диапазоне от 95% водной/5% органической подвижной фазы до 5% водной/95% органической подвижной фазы в течение 25 минут. Скорость потока была постоянной и составляла 20 мл/мин. Реакции, проводимые в микроволновой печи, осуществляли в микроволновой установке Biotage Initiator.
[00161] Хроматография на силикагеле: Анализ методом хроматографии на силикагеле проводили либо на установке Teledyne Isco CombiFlash® Rf, либо на установке Biotage® Isolera Four.
[00162] Протонный ЯМР: Если не указано иное, все 1H ЯМР-спектры были получены с помощью прибора Varian 400MHz Unity Inova 400 MHz NMR (время выборки данных = 3,5 секунды с задержкой в 1 секунду; от 16 до 64 сканирований). При описании характеристик все протоны были представлены в растворителе ДМСО-d6 в миллионных долях (ppm) относительно остаточного ДМСО (2,50 ppm).
[00163] Специалист в данной области поймет, что возможны модификации градиента, длины колонки и скорости потока и что некоторые условия могут быть более подходящими для исследования соединения, чем другие, в зависимости от анализируемых химических веществ.
Пример 1: Способы синтеза
Получение промежуточных продуктов
[00164] Получение 1: (S)-1-(2-(4-(6-бромпирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)-1-(4-фторфенил)этан-1-амина (I-1)
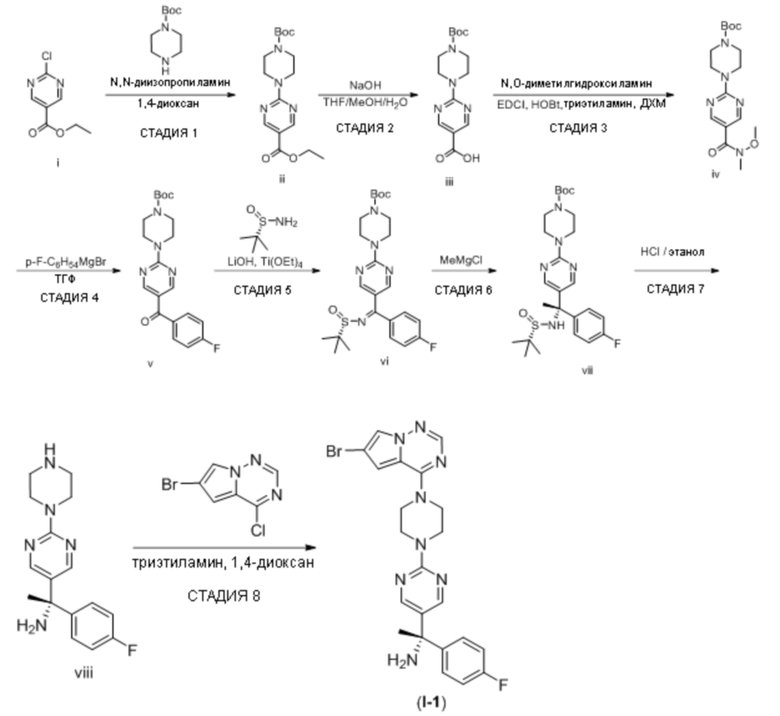
ТГФ
СТАДИЯ 7
[00165] Стадия 1: Синтез этил-2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)пиримидин-5-карбоксилата (ii): К раствору трет-бутилпиперазин-1-карбоксилата (i) (10,0 г, 53,7 ммоль) и диизопропилэтиламина (23,4 мл, 134,25 ммоль) в диоксане (80 мл) добавляли этил-2-хлорпиримидин-5-карбоксилат (10 г, 53,7 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 3 часов. Анализ с применением ЖХМС показал, что реакция завершилась. Реакционную смесь концентрировали с получением указанного в заголовке соединения (ii) (17 г, неочищенный продукт), которое непосредственно использовали на следующей стадии без дополнительной очистки. МС (ЭС+) рассчитано для C16H24N4O4: 336, найдено: 237, 281 [М-56+Н]+.
[00166] Стадия 2: Синтез 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)пиримидин-5-карбоновой кислоты (iii): К раствору этил-2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)пиримидин-5-карбоксилата (ii) (17 г, неочищенный продукт) в ТГФ/MeOH/H2O (300 мл) добавляли гидроксид натрия (4,3 г, 107,5 ммоль) и перемешивали реакционную смесь при 70°C в течение 2 часов. Анализ с применением ЖХМС показал, что реакция завершилась. Реакционную смесь охлаждали до комнатной температуры, подкисляли до pH ≈ 5-6 с помощью 1 M HCl и фильтровали. Твердое вещество собирали и высушивали с получением указанного в заголовке соединения (iii) (16 г, 96%) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без дополнительной очистки. МС (ЭС+) рассчитано для C14H20N4O4: 308, найдено: 253 [M -56+ H]+.
[00167] Стадия 3: Синтез трет-бутил-4-(5-(метокси(метил)карбамоил)пиримидин-2-ил)пиперазин-1-карбоксилата (iv): К суспензии 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)пиримидин-5-карбоновой кислоты (iii) (13,8 г, 44,8 ммоль), EDCI (12,8 г, 67,2 ммоль) и HOBT (7,2 г, 53,7 ммоль) в ДХМ (200 мл) добавляли ТЭА (25 мл, 179,2 ммоль) и перемешивали смесь при комнатной температуре в течение 1 часа с последующим добавлением N,O-диметилгидроксиламина (5 г, 53,7 ммоль). Реакционную смесь перемешивали в течение еще 3 часов. Анализ с применением ЖХМС показал, что реакция завершилась. Реакционную смесь промывали H2O (100 мл) и органическую фазу высушивали, фильтровали и концентрировали. Остаток очищали с применением хроматографии на силикагеле (петролейный эфир:этилацетат = 1:1) с получением указанного в заголовке соединения (iv) (11,2 г, 67%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C16H25N5O4: 351, найдено: 296 [M-56+H]+. 296 [M -56+ H]+.
[00168] Стадия 4: Синтез трет-бутил-4-(5-(4-фторбензоил)пиримидин-2-ил)пиперазин-1-карбоксилата (v): К раствору трет-бутил-4-(5-(метокси(метил)карбамоил)пиримидин-2-ил)пиперазин-1-карбоксилата (iv) (7,8 г, 22,22 ммоль) в сухом ТГФ (50 мл) добавляли C6H5MgFBr (1 M в ТГФ, 50 мл) при 0°C в атмосфере азота и перемешивали смесь при комнатной температуре в течение 3 часов. Анализ с применением ЖХМС показал, что реакция завершилась. Реакционную смесь гасили с помощью 1 М HCl и экстрагировали этилацетатом. Объединенные органические фазы промывали H2O и соляным раствором, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с применением хроматографии на силикагеле (петролейный эфир:ЭА = 5:1) с получением указанного в заголовке соединения (v) (7,2 г, 84%) в виде желтого твердого вещества. МС (ЭС+) рассчитано для C20H23FN4O3: 386, найдено: 331 [М-56+Н]+.
[00169] Стадия 5: Синтез трет-бутил(S,Z)-4-(5-(((трет-бутилсульфинил)имино)(4-фторфенил)метил)пиримидин-2-ил)пиперазин-1-карбоксилата (vi):Трет-бутил-4-(5-(4-фторбензоил)пиримидин-2-ил)пиперазин-1-карбоксилат (v) (20,0 г, 1,0 экв.), (S)-(-)-2-метил-2-пропансульфинамид (9,43 г, 1,5 экв.) и LiOH (0,64 г, 0,5 экв.) добавляли в реакционный сосуд с толуолом (160 мл). К указанной смеси добавляли изопропоксид титана (IV) (18,42 г, 1,25 экв.) и перемешивали реакционную смесь при температуре от 50 до 60°C в течение 1 часа. Затем реакционную смесь перегоняли для удаления 80 мл при одновременном добавлении дополнительного количества толуола (80 мл) при температуре от 40 до 60°C. Реакционную смесь охлаждали до температуры от 20 до 30°C и добавляли к раствору цитрата мононатрия (80 мл, 30% масс./масс. лимонная кислота при pH от 3 до 4). Смесь перемешивали 1,5 часа при температуре от 45 до 55°C, а затем разделяли фазы. Органическую фазу промывали бикарбонатом калия (40 мл, 25% масс./масс. водный раствор) и перегоняли органическую фазу для удаления 40 мл. Разбавляли раствор продукта (vi) тетрагидрофураном (30 мл) перед применением на следующей стадии непосредственно в виде раствора (приблизительно 15% масс./масс.).
[00170] Стадия 6: Синтез трет-бутил-4-(5-((S)-1-(((S)-трет-бутилсульфинил)амино)-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-карбоксилата. (vii): К раствору трет-бутил(S,Z)-4-(5-(((трет-бутилсульфинил)имино)(4-фторфенил)метил)пиримидин-2-ил)пиперазин-1-карбоксилата (vi) в толуоле/ТГФ (120 г, полученный на стадии 5) добавляли метилмагнийхлорид (27,8 г, 22% масс./масс. в ТГФ, 2,0 экв.) при 10°C в течение от 2 до 3 часов. Реакционную смесь оставляли перемешиваться 1,5 часа до завершения реакции. Реакционную смесь гасили путем добавления метанола (40 мл), а затем H2O (10 мл). Смесь перегоняли для удаления 100-110 мл, а затем промывали хлоридом аммония (80 мл, 20% масс/масс. в H2O). Органическую фазу промывали H2O (80 мл), разбавляли толуолом (60 мл) и перегоняли для удаления от 60 до 80 мл дистиллята. В раствор при температуре от 50 до 60°C загружали н-гептан (80 мл) и затем охлаждали до 42°C, после чего добавляли затравки (25-50 мг). Раствор выдерживали 30 минут, а затем охлаждали до температуры от 0 до 10°C в течение 30 минут. Твердую фазу выделяли посредством фильтрации, промывали смесью н-гептана и толуола (1:1, 30 мл), а затем н-гептаном (30 мл). Продукт высушивали с получением 9 г неочищенного продукта с диастереомерным избытком от 96,4 до 97,2% (vii).
[00171] Стадия 6a: Перекристаллизация неочищенного трет-бутил-4-(5-((S)-1-(((S)-трет-бутилсульфинил)амино)-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1- карбоксилата:Трет-бутил-4-(5-((S)-1-(((S)-трет-бутилсульфинил)амино)-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-карбоксилат (10,0 г) растворяли в изопропаноле (100 мл) и нагревали до температуры от 40 до 60°C, затем пропускали через осветительный фильтр, промывая/ополаскивая изопропанолом (20 мл). Полученный раствор перегоняли в вакууме при температуре от 40 до 60°C для удаления от 60 до 70 мл. Смесь разбавляли водой (45 мл) при температуре от 50 до 60°C и затем охлаждали до 40°C, после чего в нее вносили затравку в количестве от 25 до 50 мг. Смесь дополнительно охлаждали до температуры от 20 до 25°C и добавляли воду (20 мл). Твердую фазу выделяли посредством фильтрации, промывали смесью изопропанол/вода (1:1, 20 мл) и затем промывали суспензию смесью изопропанол/вода (1:2, 30 мл). После сушки получали 8,5 г продукта с диастереомерным избытком > 99,8% (vii).
[00172] Стадия 7: Синтез гидрохлорида (S)-1-(4-фторфенил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этан-1-амина (viii):Трет-бутил-4-(5-((S)-1-(((S)-трет-бутилсульфинил)амино)-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-карбоксилат (vii) (50 г, 1 экв) смешивали с этанолом (7,5 об.) и концентрированной соляной кислотой (11,2 М, 5,6 экв.). Реакционную смесь нагревали до температуры орошения. После того, как согласно ЖХМС реакция была завершена, смесь концентрировали до 5 объемов при атмосферном давлении. Концентрирование продолжали с добавлением этанола для поддержания 5 объемов до содержания H2O ≤ 3%. Концентрирование окончательно прекращали при 2 объемах с последующим охлаждением до температуры от 0 до 5°C в течение 30 минут. После фильтрации осуществляли сушку в вакууме с получением указанного в заголовке продукта (viii) (выход 92%).
[00173] Стадия 8: Синтез (S)-1-(2-(4-(6-бромпирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)-1-(4-фторфенил)этанамина (I-1): Смесь коммерчески доступного 6-бром-4-хлорпирроло[2,1-f][1,2,4]триазина (4,00 г, 17,2 ммоль) (например, Sigma Aldrich), гидрохлорида (S)-1-(4-фторфенил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанамина (viii) (5,81 г, 17,2 ммоль) и триэтиламина (7,20 мл, 51,6 ммоль) в диоксане (50 мл) перемешивали при комнатной температуре в течение ночи. Смесь концентрировали, затем очищали посредством колоночной флэш-хроматографии (ДХМ/MeOH = 20/1) с получением указанного в заголовке соединения (I-1) (8,0 г, выход 94%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C22H22BrFN8: 496, найдено: 497, 499 [М+Н]+.
[00174] Получение 2: (S)-1-(2-(4-(6-(1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил))пиримидин-5-ил)-1-(4-фторфенил)этанамина (I-2)
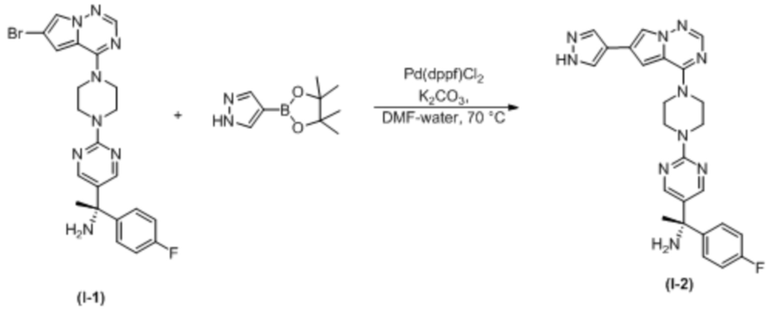
[00175] Смесь I-1 (3,0 г, 6,05 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (1,17 г, 6,05 ммоль), Pd(dppf)Cl2 (494 мг, 605 мкмоль) и K2CO3 (2,50 г, 18,2 ммоль) в ДМФ/H2O (40 мл/10 мл) продували с помощью N2 (газ) в течение 10 минут и перемешивали при 70°C в течение 16 часов в атмосфере N2. После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 10/1) с получением указанного в заголовке соединения (I-2) (2,0 г, выход 68%) в виде желтого твердого вещества. МС (ЭС+) рассчитано для C25H25FN10: 484, найдено: 485 [М+Н]+.
[00176] Получение 3: (S)-1-(4-фторфенил)-1-(2-(4-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина (I-3)
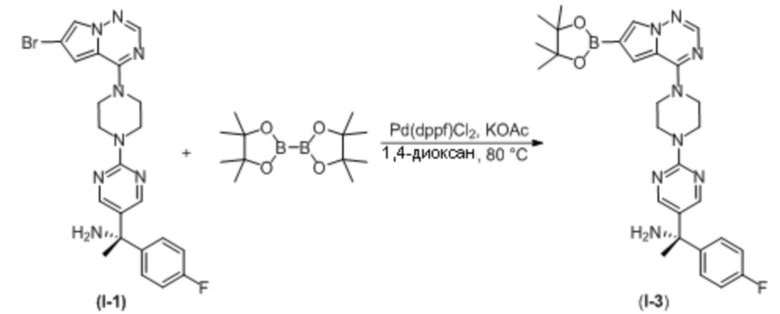
[00177] Смесь I-1 (1,0 г, 2,02 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (768 мг, 3,12 ммоль), Pd(dppf)Cl2 (165 мг, 202 мкмоль), dppf (167 мг, 303 мкмоль) и KOAc (395 мг, 4,04 ммоль) в 1,4-диоксане (30 мл) продували с помощью N2 (газ) в течение 10 минут и перемешивали при 80°C в течение 16 часов. После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 15/1) с получением указанного в заголовке соединения (I-3) (700 мг) в виде серого твердого вещества. МС (ЭС+) рассчитано для C28H34BFN8O2: 544, найдено: 545 [М+Н]+.
[00178] Получение соединений
[00179] Пример 1: (S)-1-(4-(4-(4-(5-(1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)-2-метилпропан-2-ол (1)
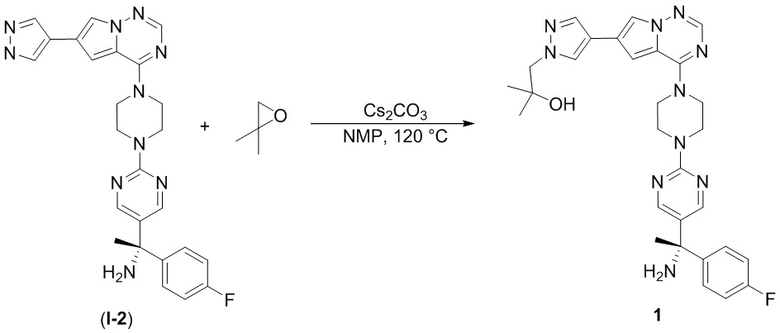
[00180] Смесь I-2 (полученная согласно способу получения 2) (200 мг, 0,412 ммоль), Cs2CO3 (269 мг, 0,83 ммоль) и 2,2-диметилоксиран (89,3 мг, 1,24 ммоль) в NMP (5 мл) перемешивали при 120°C в течение 10 часов. Реакционную смесь разбавляли EtOAc, промывали H2O и соляным раствором и высушивали над Na2SO4. Органическую фазу концентрировали в вакууме и очищали остаток с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (1) (74,5 мг, выход 32%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H33FN10O: 556, найдено: 557 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,03 (s, 1H), 8,02 (s, 1H), 7,87 (s, 1H), 7,84 (s, 1H), 7,49-7,45 (m, 2H), 7,25 (s, 1H), 7,13-7,08 (m, 2H), 4,76 (s, 1H), 4,12-4,07 (m, 4H), 4,02 (s, 2H), 3,93-3,90 (m, 4H), 2,44 (s, 2H), 1,73 (s, 3H), 1,10 (s, 6H).
[00181] Пример 2: (S)-2-(4-(4-(4-(5-(1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)-2-метилпропан-1-ол (2)
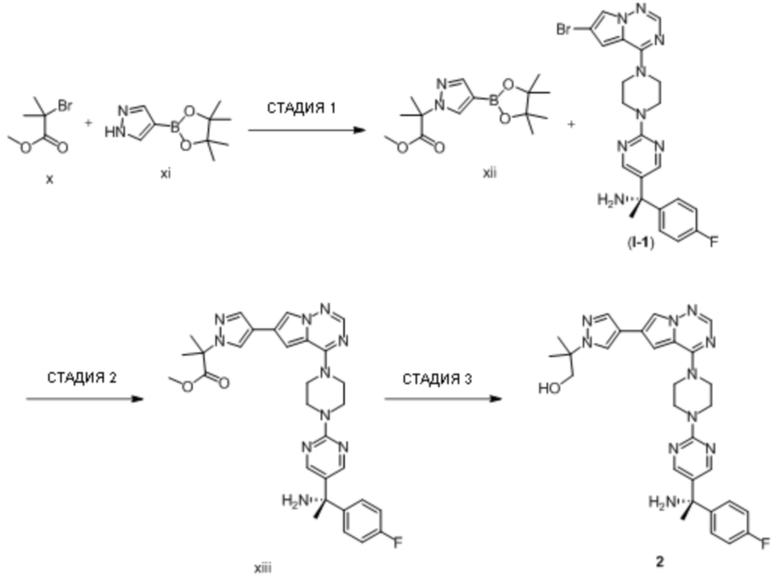
[00182] Стадия 1: Синтез метил-2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропаноата (xii): К раствору метил-2-бром-2-метилпропаноата (x) (3,0 г, 16,7 ммоль) и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (xi) (3,23 г, 16,7 ммоль) в NMP (20 мл) добавляли карбонат цезия (16,2 г, 50 ммоль) и иодид натрия (3,1 г, 16,7 ммоль) при комнатной температуре. Полученную смесь перемешивали при 120°C в течение 8 часов. Реакционную смесь разбавляли дихлорметаном и последовательно промывали H2O и соляным раствором. Органическую фазу концентрировали в вакууме и очищали остаток посредством колоночной флэш-хроматографии на силикагеле (петролейный эфир:этилацетат = 5/1) с получением указанного в заголовке соединения (xii) (1,5 г, выход 30%) в виде бесцветного масла.
[00183] Стадия 2: Синтез метил-(S)-2-(4-(4-(4-(5-(1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)-2-метилпропаноат (xiii): Смесь метил-2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропаноата (xii) (178 мг, 0,6 ммоль), I-1 (300 мг, 0,6 ммоль), Pd(dppf)Cl2 (99 мг, 0,12 ммоль) и K2CO3 (251 мг, 1,8 ммоль) в ДМФ/H2O (8 мл/2 мл) перемешивали при 70 оС в течение 4 часов в атмосфере N2 (г). После этого раствор разбавляли дихлорметаном, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 10/1) с получением указанного в заголовке соединения (xiii) (240 мг, выход 68%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C30H33FN10O2: 584, найдено: 585 [М+Н]+.
[00184] Стадия 3: Синтез (S)-2-(4-(4-(4-(5-(1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)-2-метилпропан-1-ола (2): К раствору (S)-метил-2-(4-(4-(4-(5-(1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)-2-метилпропаноата (xiii) (200 мг, 0,34 ммоль) в ТГФ (20 мл) добавляли LiAlH4 (100 мг, 3,4 ммоль) при 0°C и перемешивали полученную смесь при комнатной температуре в течение 6 часов. Реакционную смесь гасили H2O (100 мл) и 10% NaOH в H2O (300 мл), затем экстрагировали этилацетатом. Органическую фазу концентрировали в вакууме и очищали остаток с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (2) (90,5 мг, выход 47%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H33FN10O: 556, найдено: 557 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,18 (s, 1H), 8,01 (d, 1H, J = 1,6 Гц), 7,87 (s, 1H), 7,84 (s, 1H), 7,52-7,43 (m, 2H), 7,26 (d, 1H, J = 1,6 Гц), 7,16-7,07 (m, 2H), 4,99 (t, 1H, J = 5,6 Гц), 4,17-4,04 (m, 4H), 3,98-3,87 (m, 4H), 3,60 (d, 2H, J =5,6 Гц), 2,47 (s, 2H), 1,74 (s, 3H), 1,50 (s, 6H).
[00185] Пример 3: (R)1-{4-[4-(4-{5-[(S)-1-амино-1-(4-фторфенил)этил]пиримидин-2-ил}пиперазин-1-ил)-пирроло[2,1-f][1,2,4]триазин-6-ил]пиразол-1-ил}пропан-2-ол (3)
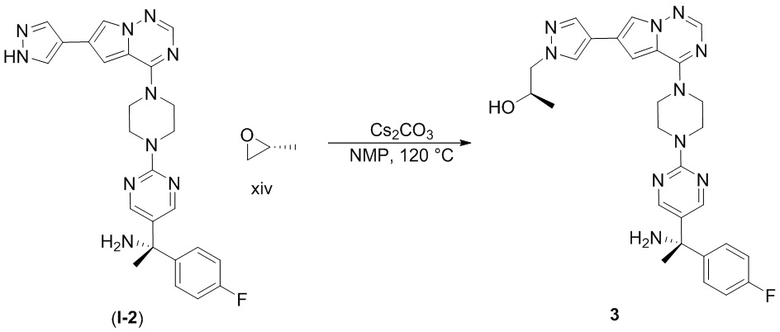
[00186] К раствору I-2 (полученному согласно способу получения 2) (200 мг, 412 мкмоль) и (2R)-2-метилоксирана (xiv) (71,4 мг, 1,23 ммоль) в NMP (3,0 мл) добавляли Cs2CO3 (400 мг, 1,23 ммоль) при комнатной температуре. Смесь перемешивали при 120 °C в течение 2 часов. После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (3) (90,0 мг, выход 40%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C28H31FN10O: 542, найдено: 543 [М+Н]+. 1H-ЯМР (400 МГц, ДМСО-d6) δ ppm 8,40 (s, 2H), 8,05 (s, 1H), 7,80 (d, 1H, J = 1,6 Гц), 7,87 (s, 1H), 7,83 (s, 1H), 7,46 (dd, 2H, J = 8,8, 5,6 Гц), 7,24 (s, 1H), 7,10 (t, 2H, J = 8,8 Гц), 4,96 (d, 1H, J = 4,8 Гц), 4,11-4,08 (m, 4H), 4,02-3,95 (m, 3H), 3,92-3,89 (m, 4H), 2,45 (s, 2H), 1,73 (s, 3H), 1,05 (d, 3H, J = 5,6 Гц).
[00187] Пример 4: (S)-2-(4-(4-(4-(5-(1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)этанол (4)
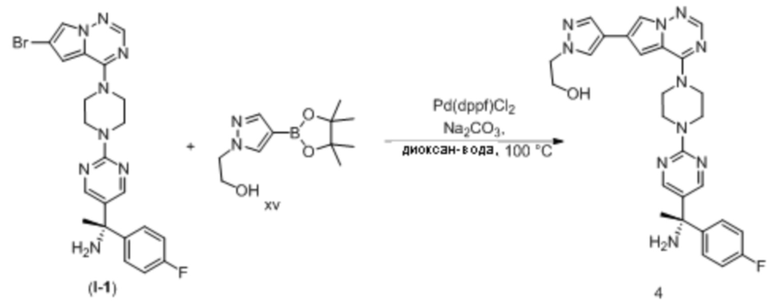
[00188] Реакционную смесь I-1 (полученную согласно способу получения 1) (500 мг, 1,00 ммоль), коммерчески доступный 2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)этанол (xv) (285 мг, 1,20 ммоль) (например, AstraTech), Pd(dppf)Cl2 (219 мг, 300 мкмоль) и Na2CO3 (317 мг, 3,00 ммоль) в диоксане/H2O (20 мл/2 мл) перемешивали при 100°C в течение ночи в атмосфере N2 (газ). Фазы разделяли и концентрировали органическую фазу в вакууме. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 15/1). Полученное вещество дополнительно очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10μм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (4) (154,0 мг, выход 29%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C27H29FN10O: 528, найдено: 529 [M+H]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,40 (s, 1H), 8,07 (s, 1H), 7,99 (s, 1H), 7,87 (s, 1H), 7,84 (s, 1H), 7,49-7,44 (m, 2H), 7,24 (s, 1H), 7,14-7,06 (m, 2H), 4,93 (t, 1H, J = 5,2 Гц), 4,17-4,13 (m, 2H), 4,12-4,07 (m, 4H), 3,94-3,88 (m, 4H), 3,89-3,71 (m, 2H), 2,45 (br, S,, 2H), 1,73 (s, 3H).
[00189] Пример 4A: гидрохлорид (S)-2-(4-(4-(4-(5-(1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)этанола
[00190] К раствору (S)-2-(4-(4-(4-(5-(1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)этанола (30 мг, 0,057 ммоль) в MeOH (5 мл) добавляли HCl/диоксан (0,05мл, 4,0 М) при комнатной температуре. Раствор перемешивали при комнатной температуре в течение 16 часов. Растворитель удаляли при пониженном давлении и лиофилизировали продукт с получением указанного в заголовке соединения (36,0 мг, выход 100%) в виде белого твердого вещества, чувствительного к влаге. МС (ЭС+) рассчитано для C29H31FN10O: 528, найдено: 529 [M+H]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 9,47 (s, 3H, br), 8,45 (s, 2H), 8,14 (s, 1H), 8,11 (s, 1H), 7,97 (s, 1H), 7,87 (s, 1H), 7,53-7,50 (m, 2H), 7,44 (s, 1H), 7,31-7,28 (m, 2H), 4,16-4,14 (m, 6H), 4,00-3,89 (m, 4H), 3,76-3,74 (m, 2H), 2,03 (s, 3H).
[00191] Пример 5: (R)-2-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)пропан-1-ол (5)
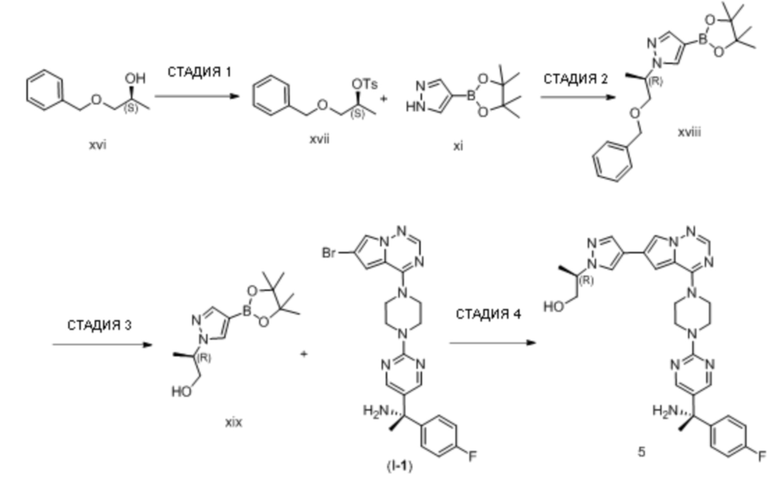
[00192] Стадия 1: Синтез (S)-1-(бензилокси)пропан-2-ил-4-метилбензолсульфоната (xvii): К раствору (S)-1-(бензилокси)пропан-2-ола (xvi) (5,0 г, 30,12 ммоль) и ТЭА (9,17 г, 90,36 ммоль) в ДХМ (80 мл) добавляли TsCl (6,30 г, 33,13 ммоль). Смесь перемешивали при комнатной температуре в течение 24 часов. Раствор разбавляли дихлорметаном, промывали H2O и промывали соляным раствором. Органическую фазу концентрировали и очищали остаток посредством колоночной флэш-хроматографии на силикагеле (петролейный эфир/этилацетат = 3/1) с получением указанного в заголовке соединения (xvii) (4,0 г, выход 42%) в виде бесцветного масла. МС (ЭС+) рассчитано для C17H20O4S: 320, найдено: 338 [M+18]+.
[00193] Стадия 2: Синтез (R)-1-(1-(бензилокси)пропан-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (xviii): Смесь (S)-1-(бензилокси)пропан-2-ил-4-метилбензолсульфоната (xvii) (2,0 г, 6,25 ммоль), 4-(4,4,5,5-тетраметил-1,3,2- диоксаборолан-2-ил)-1H-пиразола (xi) (1,22 г, 6,25 ммоль) и Cs2CO3 (4,08 мг, 12,5 ммоль) в NMP (12 мл) облучали посредством микроволнового излучения при 110 °C в течение 0,5 часа. После этого раствор разбавляли этилацетатом, промывали H2O и промывали соляным раствором. Органическую фазу концентрировали и очищали остаток посредством колоночной флэш-хроматографии на силикагеле (ПЭ/ЭА = 4/1) с получением указанного в заголовке соединения (xviii) (1,6 г, выход 75%) в виде бесцветного масла. МС (ЭС+) рассчитано для C19H27BN2O3: 342, найдено: 343 [М+Н]+.
[00194] Стадия 3: Синтез (R)-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропан-1-ола (xix): К раствору (R)-1-(1-(бензилокси)пропан-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (xviii) (800 мг, 2,34 ммоль) в MeOH (20 мл) добавляли Pd/C (800 мг) и HOAc (0,2 мл), раствор продували H2 (газ) в течение 5 минут, затем перемешивали при комнатной температуре в атмосфере H2 (газ) в течение 16 часов. После этого смесь фильтровали и концентрировали фильтрат с получением указанного в заголовке соединения в виде бесцветного масла (xix) (300 мг, выход 51%). МС (ЭС+) рассчитано для C12H21BN2O3: 252, найдено: 253 [М+Н]+.
[00195] Стадия 4: Синтез (R)-2-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)пропан-1-ола (5): Смесь ((R)-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропан-1-ола (xix) (150 мг, 595 мкмоль), I-1 (295 мг, 595 мкмоль), Pd(dppf)Cl2 (49 мг, 60 мкмоль) и K2CO3 (250 мг, 1,79 ммоль) в ДМФ/Н2О (4 мл/1 мл) продували с помощью N2 (газ) в течение 10 минут и перемешивали при 70 °C в течение 16 часов в атмосфере N2 (газ). Смесь экстрагировали EtOAc и концентрировали объединенные органические экстракты. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 10/1). Полученное вещество дополнительно очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (5) (148,1 мг, выход 46%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C28H31FN10O: 542, найдено: 543 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,11 (s, 1H), 8,00 (s, 1H), 7,87 (s, 1H), 7,83 (s, 1H), 7,48-7,44 (m, 2H), 7,25 (s, 1H), 7,14-7,08 (m, 2H), 4,98 (t, 1H, J = 5,2 Гц), 4,36-4,32 (m, 1H), 4,10-4,08 (m, 4H), 3,92-3,90 (m, 4H), 3,69-3,61 (m, 2H), 2,45 (s, 2H), 1,73 (s, 3H), 1,41 (d, 3H, J = 6,8 Гц).
[00196] Пример 6: (S)-2-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)пропан-1-ол (6)
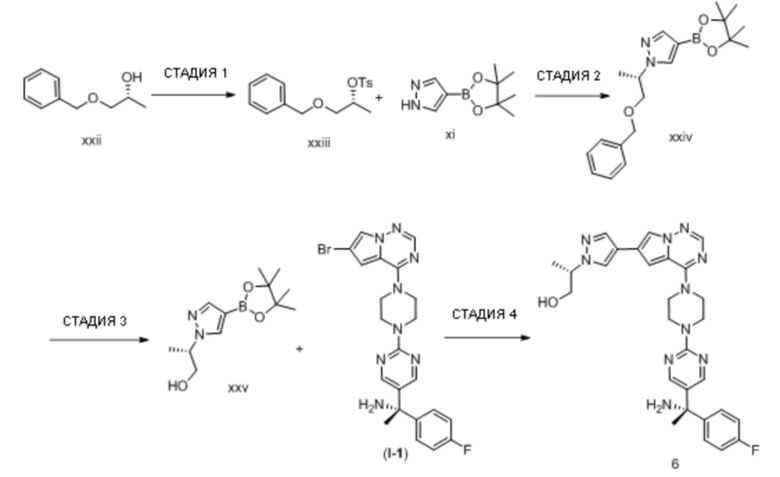
[00197] Стадия 1: Синтез (R)-1-(бензилокси)пропан-2-ил-4-метилбензолсульфоната (xxiii): К раствору (R)-1-(бензилокси)пропан-2-ола (xxii) (3,0 г, 18 ммоль) и ТЭА (5,48 г, 54,2 ммоль) в ДХМ (30 мл) добавляли TsCl (4,13 г, 21,7 ммоль). Полученную смесь перемешивали при 25°C в течение 16 часов. Затем смесь концентрировали в вакууме и очищали остаток с помощью колоночной флэш-хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1) с получением указанного в заголовке соединения (xxiii) (2,30 г, выход 39%) в виде желтого масла. МС (ЭС+) рассчитано для C17H20O4S: 320, найдено: 338 [M+18]+.
[00198] Стадия 2: Синтез (S)-1-(1-(бензилокси)пропан-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола. (xxiv): Смесь (R)-1-(бензилокси)пропан-2-ил-4-метилбензолсульфоната (xxiii) (2,20 г, 6,87 ммоль), 4-(4,4,5,5-тетраметил-1,3,2- диоксаборолан-2-ил)-1H-пиразола (xi) (2,00 г, 10,3 ммоль) и Cs2CO3 (2,24 г, 6,87 ммоль) в NMP (50 мл) перемешивали при 110°C в микроволновой печи в течение 16 часов. После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (петролейный эфир/этилацетат = от 5/1 до 4/1) с получением указанного в заголовке соединения (xxiv) (1,80 г, выход 39%) в виде желтого масла. МС (ЭС+) рассчитано для C19H27BN2O3: 342, найдено: 343 [М+Н]+.
[00199] Стадия 3: Синтез (S)-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропан-1-ола (xxv): В смесь (S)-1-(1-(бензилокси)пропан-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H- пиразола (xxiv) (0,90 г, 2,6 ммоль) в MeOH (20 мл) добавляли Pd/C (800 мг) и HOAc (0,2 мл). Полученную смесь продували H2 (газ) в течение 5 минут, затем перемешивали при комнатной температуре в атмосфере H2 (газ) в течение 16 часов. После этого смесь фильтровали и концентрировали с получением указанного в заголовке соединения (xxv) в виде желтого масла (500 мг, выход 75%). МС (ЭС+) рассчитано для C12H21BN2O3: 252, найдено: 253 [М+Н]+.
[00200] Стадия 4: Синтез (S)-2-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)пропан-1-ола (6): Смесь (S)-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропан-1-ола (xxv) (98 мг, 392 мкмоль), I-1 (130 мг, 261 мкмоль), K2CO3 (200 мг, 227 мкмоль) и Pd(dppf)Cl2 (20 мг, 27 мкмоль) в ДМФ/H2O (5 мл/1 мл) перемешивали при 70°C в атмосфере N2 (газ) в течение 4 часов. После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (6) (40,7 мг, выход 28%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C28H31FN10O: 542, найдено: 543 [М+Н]+. 1 H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,10 (s, 1H), 7,99 (s, 1H), 7,87 (s, 1H), 7,83 (s, 1H), 7,47 (dd, 2H, J = 8,8, 5,6 Гц), 7,24 (s, 1H), 7,11 (t, 2H, J = 8,8 Гц), 4,96 (t, 1H, J = 5,6 Гц), 4,38-4,35 (m, 1H), 4,11-4,08 (m, 4H), 3,92-3,90 (m, 4H), 3,70-3,60 (m, 2H), 2,43 (s, 1H), 1,73 (s, 3H), 1,41 (d, 3H, J = 6,8 Гц).
[00201] Пример 7: (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-(оксетан-3-ил)-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этан-1-амин (7)
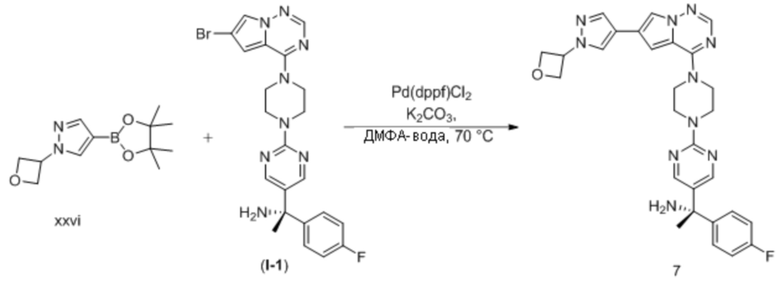
[00202] Смесь 1-(оксетан-3-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (xxvi) (600 мг, 2,4 ммоль), I-1 (1,19 г, 2,4 ммоль), Pd(dppf)Cl2 (391 мг, 0,48 ммоль) и K2CO3 (994 мг, 7,2 ммоль) в ДМФ/H2O (16 мл/4 мл) продували с помощью N2 в течение 10 минут и перемешивали при 70°C в течение 3 часов в атмосфере N2 (газ). После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Смесь очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 10/1). Полученное вещество дополнительно очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (7) (236,3 мг, выход 18%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C28H29FN10O: 540, найдено: 541 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,32 (s, 1H), 8,03 (d, 1H, J = 1,6 Гц), 7,99 (s, 1H), 7,88 (s, 1H), 7,52-7,44 (m, 2H), 7,29 (d, 1H, J = 1,6 Гц), 7,16-7,07 (m, 2H), 5,64-5,52 (m, 1H), 4,99-4,94 (m, 2H), 4,93-4,89 (m, 2H), 4,12-4,06 (m, 4H), 3,97-3,87 (m, 4H), 2,50 (br, s,, 2H), 1,74 (s, 3H).
[00203] Пример 8: (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-(оксетан-3-илметил)-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этан-1-амин (8)
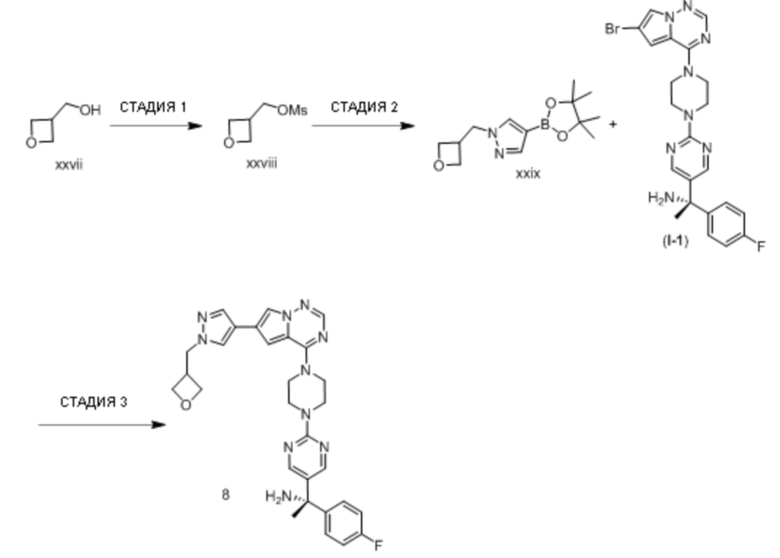
[00204] Стадия 1: Синтез оксетан-3-ил-метилметансульфоната (xxviii): К раствору оксетан-3-ил-метанола (xxvii) (300 мг, 3,40 ммоль) и ТЭА (1,03 г, 10,2 ммоль) в ДХМ (10 мл) добавляли MsCl (429 мг, 3,75 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем разбавляли дихлорметаном, промывали насыщенным раствором Na2CO3 и высушивали с помощью безводного Na2SO4. Растворитель выпаривали в вакууме с получением указанного в заголовке соединения (xxviii) (280 мг, выход 49%) в виде желтого масла.
[00205] Стадия 2: Синтез 1-(оксетан-3-илметил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (xxix): Смесь 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (xi) (280 мг, 1,44 ммоль), оксетан-3-ил-метилметансульфоната (xxviii) (275 мг, 1,66 ммоль) и Cs2CO3 (1,41 г, 4,33 ммоль) в ДМФ (20 мл) перемешивали при 70°C в течение 4 часов, затем разбавляли дихлорметаном и промывали соляным раствором. Органическую фазу упаривали в вакууме. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1) с получением указанного в заголовке соединения (xxix) (320 мг, выход 71%). МС (ЭС+) рассчитано для C13H21BN2O3: 264, найдено: 265 [М+Н]+.
[00206] Стадия 3: Синтез (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-(оксетан-3-илметил)-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этан-1-амина (8): Смесь I-1 (300 мг, 392 мкмоль), 1-(оксетан-3-илметил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (xxix) (239 мг, 904 мкмоль), K2CO3 (250 мг, 1,81 ммоль) и Pd(dppf)Cl2 (30 мг, 41 мкмоль) в ДМФ/H2O (10 мл/2 мл) перемешивали при 70°C в атмосфере N2 (газ) в течение 4 часов. После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (8) (40,2 мг, выход 12%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O: 554, найдено: 555 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,09 (s, 1H), 7,98 (s, 1H), 7,87 (s, 1H), 7,84 (s, 1H), 7,47 (dd, 2H, J = 8,8, 5,6 Гц), 7,22 (s, 1H), 7,11 (t, 2H, J = 8,8 Гц), 4,69-4,65 (m, 2H), 4,45-4,41 (m, 4H), 4,10-4,08 (m, 4H), 3,92-3,89 (m, 4H), 3,46-3,41 (m, 1H), 2,45 (s, 2H), 1,73 (s, 3H).
[00207] Пример 9: (S)-1-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этилпиримидин-2-ил}пиперазин-1-ил)-пирроло[2,1-f][1,2,4]триазин-6-ил]пиразол-1-ил}пропан-2-ол (9)
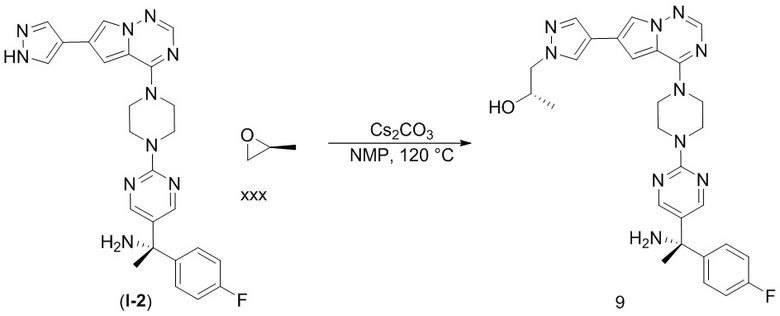
[00208] Смесь I-2 (220 мг, 455 мкмоль), (S)-2-метилоксирана (xxx) (79 мг, 1,37 ммоль) и Cs2CO3 (445 мг, 1,37 ммоль) в NMP (2 мл). Указанную смесь облучали при 120°C посредством микроволнового излучения в течение 1 часа. После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (9) (108 мг, выход 44%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C28H31FN10O: 542, найдено: 543 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,05 (s, 1H), 8,00 (s, 1H), 7,87 (s, 1H), 7,83 (s, 1H), 7,48-7,44 (m, 2H), 7,25 (s, 1H), 7,14-7,08 (m, 2H), 4,96 (d, 1H, J = 4,4 Гц), 4,10-4,08 (m, 4H), 4,02-3,98 (m, 3H), 3,92-3,90 (m, 4H), 2,44 (s, 2H), 1,73 (s, 3H), 1,06 (d, 3H, J = 5,6 Гц).
[00209] Пример 10: цис-3-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)циклобутанол (10)
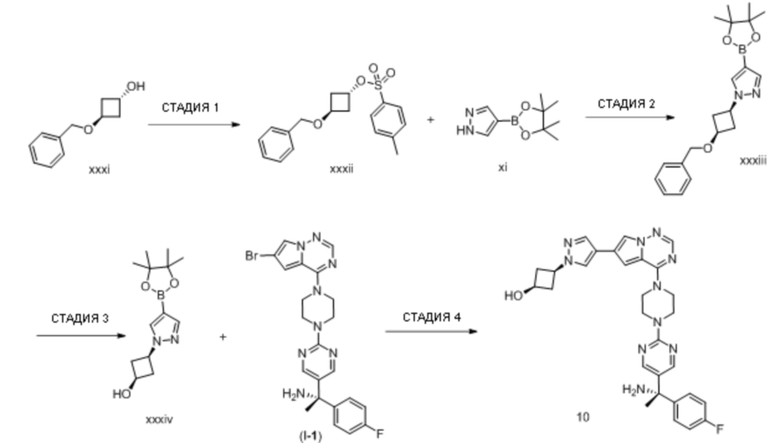
[00210] Стадия 1: Синтез транс-3-(бензилокси)циклобутил-4-метилбензолсульфоната (xxxii): К раствору транс-3-(бензилокси)циклобутанола (xxxi) (300 мг, 1,7 ммоль) в ДХМ (20 мл) добавляли TsCl (389 мг, 2,0 ммоль) и ТЭА (343 мг, 3,4 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Раствор разбавляли дихлорметаном, промывали H2O и соляным раствором, затем концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ПЭ/ЭА = 5/1) с получением указанного в заголовке соединения (xxxii) (315 мг, выход 56%) в виде бесцветного масла. МС (ЭС+) рассчитано для C18H20O4S: 332, найдено: 350 [M+18]+.
[00211] Стадия 2: Синтез цис-3-(бензилокси)циклобутил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (xxxiii): Смесь транс-3-(бензилокси)циклобутил-4-метилбензолсульфоната (xxxii) (315 мг, 0,95 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ила)-1H-пиразола (xi) (185 мг, 0,95 ммоль) и Cs2CO3 (619 мг, 1,9 ммоль) в NMP (5 мл) облучали при 110°C посредством микроволнового излучения в течение 0,5 часа. После этого раствор разбавляли этилацетатом и промывали H2O и соляным раствором. Органическую фазу концентрировали в вакууме и очищали остаток посредством колоночной флэш-хроматографии на силикагеле (ПЭ/ЭА = 4/1) с получением указанного в заголовке соединения (xxxiii) (190 мг, выход 56%) в виде бесцветного масла. МС (ЭС+) рассчитано для C20H27BN2O3: 354, найдено: 355 [М+Н]+.
[00212] Стадия 3: Синтез цис-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутанола (xxxiv): К раствору цис-3-(бензилокси)циклобутил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (xxxiii) (190 мг, 0,54 ммоль) в MeOH (5 мл) добавляли Pd/C (200 мг) и HOAc (5 капель), раствор продували H2 (газ) в течение 5 минут и перемешивали при комнатной температуре в атмосфере H2 (газ) в течение 16 часов. Смесь фильтровали и упаривали фильтрат досуха в вакууме с получением указанного в заголовке соединения (xxxiv) в виде бесцветного масла (85 мг, выход 60%). МС (ЭС+) рассчитано для C13H21BN2O3: 264, найдено: 265 [М+Н]+.
[00213] Стадия 4: Синтез (цис-3-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)циклобутанола (10): Смесь цис-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутанола (xxxiv) (55 мг, 0,21 ммоль), I-1 (104 мг, 0,21 ммоль), Pd(dppf)Cl2 (18 мг, 0,021 мкмоль) и K2CO3 (87 мг, 0,63) в ДМФ/H2O (4 мл/1 мл) продували с помощью N2 в течение 10 минут и перемешивали при 70°C в течение 16 часов в атмосфере N2 (газ). После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали непосредственно с помощью колоночной флэш-хроматографии (ДХМ/MeOH = 8/1). Полученное вещество дополнительно очищали посредством препаративной ВЭЖХ ((Подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм)) с последующей лиофилизацией с получением указанного в заголовке соединения (10) (14,6 мг, выход 13%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O: 554, найдено: 555 [М+Н]+. 1 H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,17 (s, 1H), 8,00 (s, 1H), 7,87-7,86 (m, 2H), 7,49-7,45 (m, 2H), 7,26 (s, 1H), 7,19-7,13 (m, 2H), 5,33 (d, 1H, J = 6,4 Гц), 4,38-4,31 (m, 1H), 4,13-4,06 (m, 4H), 3,99-3,96 (m, 1H), 3,94-3,88 (m, 4H), 2,79-2,71 (m, 2H), 2,34-2,31 (m, 2H), 1,73 (s, 3H).
[00214] Пример 11: транс-3- {4-[4-(4-{5-[1-амино-1-(4-фторфенил)этил]пиримидин-2-ил}пиперазин-1-ил)-пирроло[2,1-f][1,2,4]триазин-6-ил]пиразол-1-ил}циклобутанол (11)
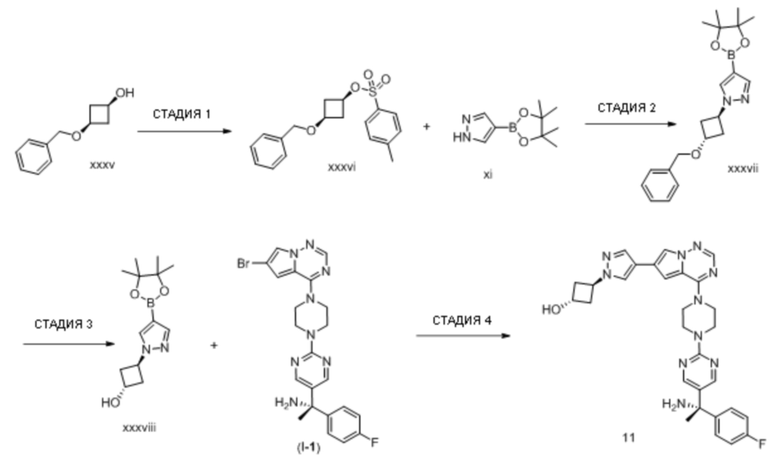
[00215] Стадия 1: Синтез 3-бензилокси-циклобутилового эфира цис-толуол-4-сульфоновой кислоты (xxxvi): К раствору цис-3-бензилокси-циклобутанола (xxxv) (500 мг, 2,81 ммоль) и ТЭА (851 мг, 8,43 ммоль) в ДХМ (10 мл) добавляли 4-метилбензолсульфонилхлорид (640 мг, 3,37 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 16 часов. Смесь разбавляли соляным раствором и экстрагировали ДХМ. Органический экстракт концентрировали. Остаток очищали непосредственно с помощью колоночной флэш-хроматографии на силикагеле (ПЭ/ЭА = 11/1) с получением указанного в заголовке соединения (xxxvi) (490 мг, выход 53%) в виде желтого твердого вещества. МС (ЭС+) рассчитано для C18H20O4S: 332, найдено: 350 [M+18]+.
[00216] Стадия 2: Синтез транс-1-(3-бензилоксициклобутил)-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-пиразола (xxxvii): Смесь 3-бензилокси-циклобутилового эфира цис-толуол-4-сульфоновой кислоты (xxxvi) (500 мг, 1,51 ммоль), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-пиразол (xi) (430 мг, 2,22 ммоль) и Cs2CO3 (1,47 г, 4,51 ммоль) в NMP (15 мл) облучали посредством микроволнового излучения при 120°C в течение 2 часов. После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ПЭ/ЭА = 1/1) с получением указанного в заголовке соединения (xxxvii) (227 мг, выход 42%) в виде желтого твердого вещества. МС (ЭС+) рассчитано для C20H27BN2O3: 354, найдено: 355 [М+Н]+.
[00217] Стадия 3: Синтез транс-3-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)пиразол-1-ил]циклобутанола (xxxviii): К раствору транс-1-(3-бензилоксициклобутил)-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-пиразола (xxxvii) (420 мг, 1,18 ммоль) в MeOH (10 мл) добавляли Pd/C (200 мг) и концентрированную HCl (0,5 мл). Реакционную смесь перемешивали в атмосфере H2 (газ) при комнатной температуре в течение 16 часов. Смесь фильтровали и концентрировали фильтрат с получением указанного в заголовке соединения (xxxviii) (250 мг, выход 80%) в виде твердого вещества. МС (ЭС+) рассчитано для C13H21BN2O3: 264, найдено: 265 [М+Н]+.
[00218] Стадия 4: Синтез транс-3-{4-[4-(4-{5-[1-амино-1-(4-фторфенил)этил]пиримидин-2-ил}пиперазин-1-ил)-пирроло[2,1-f][1,2,4]триазин-6-ил]пиразол-1-ил}циклобутанола (11): Смесь транс-3-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)пиразол-1-ил]циклобутанола (xxxviii) (200 мг, 0,76 ммоль), I-1 (376 мг, 0,76 ммоль), Pd(dppf)Cl2 (61,8 мг, 0,076 ммоль) и K2CO3 (313 мг, 2,27 ммоль) в диоксане/H2O (4 мл/1 мл) продували с помощью N2 (газ) в течение 10 минут и перемешивали при 70°C в течение 4 часов в атмосфере N2 (газ). После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали с применением флэш-хроматографии на силикагеле. Полученное вещество дополнительно очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (11) (27,2 мг, выход 6%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O: 554, найдено: 555 [М+Н]+. 1 H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,18 (s, 1H), 7,99 (d, 1H, J = 1,2 Гц), 7,87-7,85 (m, 2H), 7,49-7,45 (m, 2H), 7,24 (s, 1H), 7,15-7,08 (m, 2H), 5,24 (d, 1H, J = 5,2 Гц), 4,92-4,89 (m, 1H,), 4,50-4,43 (m, 1H), 4,17-4,10 (m, 4H), 3,96-3,90 (m, 4H), 2,67-2,61 (m, 2H), 2,44 (s, 2H), 2,42-2,37 (m, 2H), 1,73 (s, 3H).
[00219] Пример 12: (S)-1-((4-(4-(4-(5-(1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)метил)циклопропан-1-ол (12)
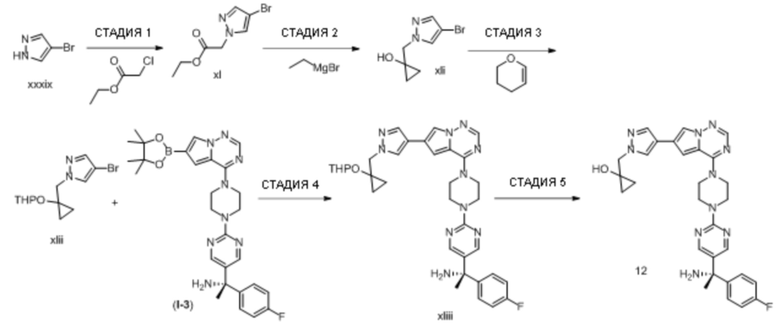
[00220] Стадия 1: Синтез этил-2-(4-бром-1H-пиразол-1-ил)ацетата (xl): Смесь 4-бром-1H-пиразола (xxxix) (8,0 г, 55 ммоль) и K2CO3 (15,2 г, 110 ммоль) в этил-2-хлорацетате (25 мл) перемешивали при 80°C в течение 15 часов. Реакционную смесь охлаждали, разбавляли этилацетатом и промывали H2O. Органическую фазу упаривали и очищали остаток с применением хроматографии на силикагеле (петролейный эфир/этилацетат = 5:1) с получением указанного в заголовке соединения (xl) (8,5 г, выход 66%) в виде бледно-желтого масла. МС (ЭС+) рассчитано для C7H9BrN2O2: 232, найдено: 233 [М+Н]+.
[00221] Стадия 2: Синтез этил-1-((4-бром-1H-пиразол-1-ил)метил)циклопропан-1-ола (xli): К раствору этил-2-(4-бром-1H-пиразол-1-ил)ацетата (xl) (7,0 г, 30 ммоль) и тетраизопропанолата титана (4,26 г, 15 ммоль) в безводном ТГФ (60 мл) добавляли по каплям раствор бромида этилмагния (3 M в гексане, 30 мл, 90 ммоль) при 60°C в течение 2 часов. После перемешивания при той же температуре в течение 2 часов реакционную смесь разбавляли этилацетатом и последовательно промывали 1N водным раствором HCl и H2O. Органическую фазу упаривали и очищали остаток с применением хроматографии на силикагеле (петролейный эфир/этилацетат = от 20:1 до 3:1) с получением указанного в заголовке соединения (xli) (1,3 г, выход 20%) в виде желтого твердого вещества. МС (ЭС+) рассчитано для C7H9BrN2O: 216, найдено: 217 [М+Н]+.
[00222] Стадия 3: Синтез 4-бром-1-[1-(тетрагидропиран-2-илокси)циклопропилметил]-1H-пиразола (xlii): К раствору 1-[(4-бром-1H-пиразол-1-ил)метил]циклопропан-1-ола (xli) (300 мг, 1,38 ммоль) и 3,4-дигидро-2H-пирана (348 мг 4,14 ммоль) в ДХМ (8 мл) добавляли пара-толуолсульфонат пиридиния (346 мг, 1,38 ммоль) при комнатной температуре. Смесь перемешивали в течение 4 часов, затем разбавляли соляным раствором и промывали ДХМ. Органическую фазу концентрировали и очищали остаток с применением хроматографии на силикагеле (ПЭ/ЭА = 10:1) с получением указанного в заголовке соединения (xlii) (200 мг, выход 48%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C12H17BrN2O2: 300, найдено: 217 [M-THP + H]+.
[00223] Стадия 4: Синтез 1-(4-фторфенил)-1-{2-[4-(6- {1-[1-(тетрагидропиран-2-илокси)циклопропилметил]-1H-пиразол-4-ил}-пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил]пиримидин-5-ил}этиламина (xliii): Смесь 4-бром-1-{[1-(оксан-2-илокси)циклопропил]метил}-1H-пиразола (xlii) (160 мг, 0,531 ммоль), I-3 (577 мг, 1,06 ммоль), Pd(dppf)Cl2 (77,5 мг, 106 мкмоль) и Na2CO3 (168 мг, 1,59 ммоль) в смеси 1,4-диоксана (3 мл), H2O (1 мл) и ДМФ (0,5 мл) перемешивали при 80°C в течение 3 часов в атмосфере N2 (газ). После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали с применением хроматографии на силикагеле (этилацетат/метанол = 4:1) с получением указанного в заголовке соединения (270 мг, выход 50%) в виде коричневого твердого вещества. МС (ЭС+) рассчитано для C34H39FN10O2: 621, найдено: 622 [М+Н]+.
[00224] Стадия 5: Синтез 1-{4-[4-(4-{5-[1-амино-1-(4-фторфенил)этил]пиримидин-2-ил}пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил]пиразол-1-илметил}циклопропанола (12): К раствору 1-(4-фторфенил)-1-{2-[4-(6-{1-[1-(тетрагидропиран-2-илокси)циклопропилметил]-1Н-пиразол-4-ил}-пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил]пиримидин-5-ил}этиламина (xliii) (200 мг, 0,32 ммоль) в MeOH (4 мл) добавляли п-толуолсульфоновую кислоту (180 мг, 1,04 ммоль) при комнатной температуре и перемешивали полученную смесь в течение 2 часов. Реакционную смесь концентрировали и очищали остаток с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (10 мМ NH4HCO3 и 0,025% NH3·H2O), B = ацетонитрил; градиент: от 51 до 56% B в течение 7 минут, прекращение через 15 минут; колонка: Agela Durashell C18 (L) 21,2 × 250 мм, 10 μм, 150 Å) с последующей лиофилизацией с получением указанного в заголовке соединения (12) (56 мг, выход 31%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O: 554, найдено: 555 [М+Н]+. 1H-ЯМР (400 МГц, ДМСО-d6) δ ppm 8,40 (s, 2H), 8,10 (s, 1H), 8,02 (s, 1H), 7,87 (s, 1H), 7,82 (s, 1H), 7,48-7,44 (m, 2H), 7,25 (s, 1H), 7,13-7,08 (m, 2H), 5,57 (s, 1H), 4,17 (s, 2H), 4,13-4,05 (m, 4H), 3,95-3,85 (m, 4H), 1,73 (s, 3H), 0,69-0,66 (m, 4H).
[00225] Пример 13: (S)-(1-(4-(4-(4-(5-(1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)циклопропил)метанол (13)
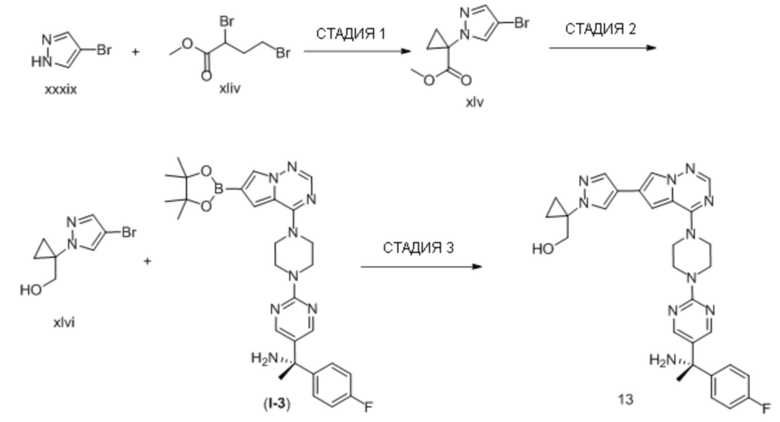
[00226] Стадия 1: Синтез метил-1-(4-бром-1H-пиразол-1-ил)циклопропанкарбоксилата (xiv): К раствору 4-бром-1H-пиразола (xxxix) (2,0 г, 13,70 ммоль) в ТГФ (50 мл) добавляли NaH (1,20 г, 30,14 ммоль) при 0°C. Раствор перемешивали при комнатной температуре в течение 1 часа, затем к раствору добавляли метил-2,4-дибромбутаноат (xliv) (3,53 г, 13,70 ммоль). Смесь перемешивали в течение 16 часов, затем разбавляли этилацетатом. Органическую фазу промывали H2O, промывали соляным раствором и концентрировали в вакууме. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (петролейный эфир/этилацетат = 2/1) с получением указанного в заголовке соединения (xiv) (570 мг, выход 17%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C8H9BrN2O2: 244, найдено: 245 [М+Н]+.
[00227] Стадия 2: Синтез (1-(4-бром-1H-пиразол-1-ил)циклопропил)метанола (xlvii): К раствору метил-1-(4-бром-1H-пиразол-1-ил)циклопропанкарбоксилата (xiv) (550 мг, 2,25 ммоль) в MeOH (15 мл) добавляли NaBH4 (257 мг, 6,75 ммоль) и перемешивали полученную смесь при 50°C в течение 36 часов. Реакционную смесь разбавляли дихлорметаном, последовательно промывали H2O и соляным раствором и концентрировали в вакууме. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ПЭ/ЭА = 1/1) с получением указанного в заголовке соединения (xlvii) (300 мг, выход 62%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C7H9BrN2O: 216, найдено: 217 [М+Н]+.
[00228] Стадия 3: Синтез (S)-(1-(4-(4-(4-(5-(1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)циклопропил)метанола (13): Смесь (1-(4-бром-1H-пиразол-1-ил)циклопропил)метанола (xlvii) (100 мг, 463 мкмоль), I-3 (полученного, как описано в способе получения 3) (380 мг, 695 мкмоль), Pd(t-Bu3P)2 (47 мг, 93 мкмоль) и Cs2CO3 (452 мг, 1,39 ммоль) в ТГФ /H2O (8 мл/2 мл) продували с помощью N2 (газ) в течение 10 минут и перемешивали при 80°C в течение 12 часов в атмосфере N2 (газ). После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии (ДХМ/MeOH = 10/1). Полученное вещество дополнительно очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (13) (57,3 мг, выход 22%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O: 554, найдено: 555 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,15 (s, 1H), 8,00 (d, 1H, J = 1,6 Гц), 7,87 (s, 1H), 7,83 (s, 1H), 7,48-7,44 (m, 2H), 7,27 (d, 1H, J = 1,6 Гц), 7,14-7,08 (m, 2H), 5,00 (t, 1H, J = 5,6 Гц), 4,10-4,08 (m, 4H), 3,92-3,90 (m, 4H), 3,66 (d, 2H, J = 5,6 Гц), 2,43 (s, 2H), 1,73 (s, 3H), 1,13-1,11 (m, 2H), 1,05-1,02 (m, 2H).
[00229] Пример 14: (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-((R)-тетрагидрофуран-3-ил)-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамин (14)
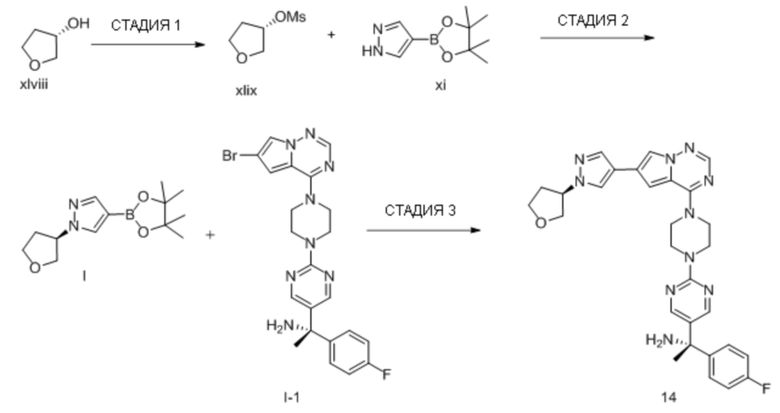
[00230] Стадия 1: Синтез (S)-тетрагидрофуран-3-ил-метансульфоната (xlix): К раствору тетрагидрофуран-3-ола (xlviii) (2,0 г, 22,7 ммоль) и ТЭА (4,6 г, 45,4 ммоль) в ДХМ (20 мл) добавляли MsCl (4,7 г, 25,0 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре 16 часов. Затем реакционную смесь разбавляли дихлорметаном, последовательно промывали H2O и соляным раствором, высушивали над безводным Na2SO4 и концентрировали досуха с получением указанного в заголовке соединения (xlix) (2,0 г, неочищенный продукт) в виде желтого масла.
[00231] Стадия 2: Синтез (R)-1-(тетрагидрофуран-3-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (l): К раствору (S)-тетрагидрофуран-3-ил-метансульфоната (xlviii) (1,9 г, 11,4 ммоль) в NMP (50 мл) добавляли 4-(4,4,5,5-тетраметил-[1,3, 2]диоксаборолан-2-ил)-1H-пиразол (xi) (3,3 г, 17,2 ммоль) и Cs2CO3 (11,2 г, 34,3 ммоль) при комнатной температуре. Смесь перемешивали при 120°C в течение 2 часов. Раствор разбавляли этилацетатом, последовательно промывали H2O и соляным раствором и концентрировали в вакууме. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ПЭ/ЭА = 4/1) с получением указанного в заголовке соединения (l) (2,3 г, выход 76%) в виде бесцветного масла. МС (ЭС+) рассчитано для C13H21BN2O3: 264, найдено: 265 [М+Н]+.
[00232] Стадия 3: Синтез (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-((R)-тетрагидрофуран-3-ил)-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина (14): Смесь (R)-1-(тетрагидрофуран-3-ил)-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-пиразола (l) (80 мг, 0,3 ммоль), I-1 (150 мг, 0,3 ммоль), Pd(dppf)Cl2 (50 мг, 0,06 ммоль) и K2CO3 (125 мг, 0,9 ммоль) в ДМФ (2 мл) и H2O (0,5 мл) перемешивали при 80°C в течение 16 часов в атмосфере N2 (газ). После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 16/1). Затем полученное вещество очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (14) (65 мг, выход 38%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O: 554, найдено: 555 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,16 (s, 1H), 8,01 (s, 1H), 7,87 (s, 2H), 7,49-7,45 (m, 2H), 7,26(s, 1H), 7,14-7,08 (m, 2H), 5,05-4,99 (m, 1H), 4,10-4,04 (m, 4H), 4,02-3,98 (m, 2H), 3,94-3,82 (m, 6H), 2,44-2,28 (m, 4H), 1,73 (s, 3H).
[00233] Пример 15: (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-((S)-тетрагидрофуран-3-ил)-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамин (15)
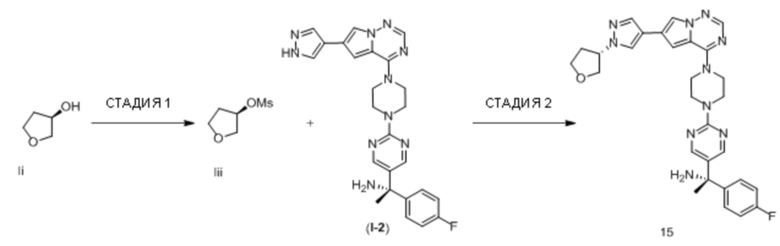
[00234] Стадия 1: Синтез (R)-тетрагидрофуран-3-ил-метансульфоната (lii): К раствору (R)-тетрагидрофуран-3-ола (li) (1,0 г, 11,4 ммоль) и ТЭА (2,3 г, 23 ммоль) в ДХМ (20 мл) добавляли MsCl (1,43 г, 12,5 ммоль) при комнатной температуре и перемешивали полученную смесь при комнатной температуре в течение 6 часов. Реакционную смесь разбавляли дихлорметаном, последовательно промывали H2O и соляным раствором и концентрировали с получением указанного в заголовке соединения (lii) (1,4 г, неочищенный продукт) в виде бесцветного масла.
[00235] Стадия 2: Синтез (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-((S)-тетрагидрофуран-3-ил)-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина (15): Смесь I-2 (300 мг, 0,62 ммоль), (R)-тетрагидрофуран-3-ил-метансульфоната (lii) (lii) (155 мг, 0,93 ммоль) и Cs2CO3 (600 мг, 1,89 ммоль) в NMP (10 мл) перемешивали при 120°C в течение 16 часов. После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали непосредственно с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (15) (133,5 мг, выход 39%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O: 554, найдено: 555 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,17 (s, 1H), 8,02 (d, 1H, J = 0,8 Гц), 7,88 (s, 2H), 7,52-7,44 (m, 2H), 7,26 (s, 1H), 7,11 (t, 2H, J = 17,6 Гц), 5,08-4,99 (m, 1H), 4,16-4,05 (m, 4H), 4,04-3,97 (m, 2H), 3,96-3,88 (m, 5H), 3,87-3,80 (m, 1H), 2,48-2,43 (m, 2H), 2,43-2,36 (m, 1H), 2,32-2,25 (m, 1H), 1,74 (s, 3H).
[00236] Пример 16: (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этан-1-амин (16)
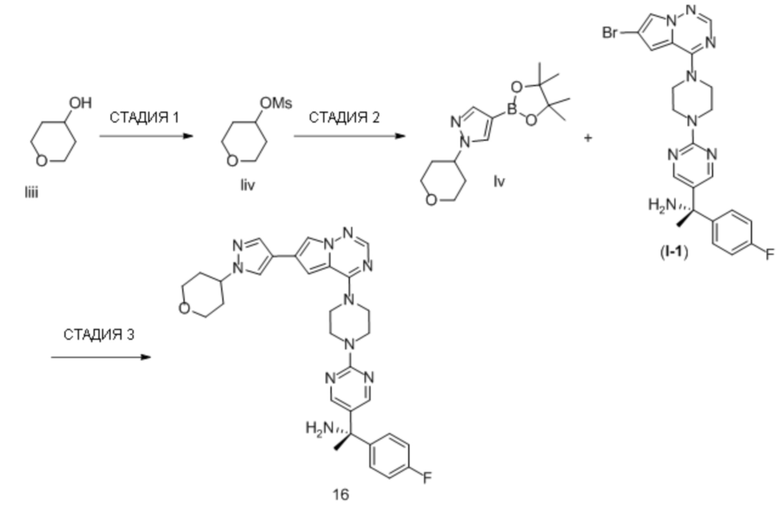
[00237] Стадия 1: Синтез тетрагидро-2H-пиран-4-ил-метансульфоната (liv): К раствору тетрагидро-2H-пиран-4-ола (liii) (3,20 г, 31,3 ммоль) и ТЭА (9,51 г, 94,0 ммоль) в ДХМ (100 мл) добавляли MsCl (5,38 г, 47,0 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем разбавляли дихлорметаном, промывали насыщенным водным раствором Na2CO3 и высушивали с помощью безводного Na2SO4. Растворитель удаляли с получением указанного в заголовке соединения (liv) (3,2 г, неочищенный продукт) в виде желтого масла.
[00238] Стадия 2: Синтез 1-(тетрагидро-2H-пиран-4-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (lv): Смесь тетрагидро-2H-пиран-4-илметансульфоната (liv) (3,20 г, 17,7 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (4,13 г, 21,3 ммоль) и Cs2CO3 (8,68 г, 26,6 ммоль) в NMP (50 мл) перемешивали при 80°C в течение 4 часов. Реакционную смесь разбавляли дихлорметаном и промывали соляным раствором. Органическую фазу упаривали в вакууме. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ПЭ/ЭА = 5/1) с получением указанного в заголовке соединения (lv) (1,2 г, выход 24%). МС (ЭС+) рассчитано для C14H23BN2O3: 278, найдено: 279 [М+Н]+.
[00239] Стадия 3: Синтез (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этан-1-амина (16): Смесь I-1 (300 мг, 603 мкмоль), 1-(тетрагидро-2H-пиран-4-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (lv) (210 мг, 754 мкмоль), K2CO3 (104 мг, 754 мкмоль) и Pd(dppf)Cl2 (30 мг, 41 мкмоль) в ДМФ/H2O (10 мл/2 мл) перемешивали при 70 °C в атмосфере N2 (газ) в течение 4 часов. После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 15% до 95% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (16) (40,2 мг, выход 6%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C30H33FN10O: 568, найдено: 569 [М+Н]+. 1H ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,19 (s, 1H), 7,99 (s, 1H), 7,88 (s, 1H), 7,86 (s, 1H), 7,48-7,45 (m, 2H,), 7,24 (s, 1H), 7,13-7,09 (m, 2H), 4,42-4,37 (m, 1H), 4,12-4,40 (m, 4H), 3,99-3,96 (m, 2H), 3,92-3,90 (m, 4H), 3,52-3,46 (m, 2H), 2,43 (s, 2H), 2,05-1,92 (m, 4H), 1,73 (s, 3H).
[00240] Пример 17: (3R,4R)-4-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)тетрагидрофуран-3-ол (17)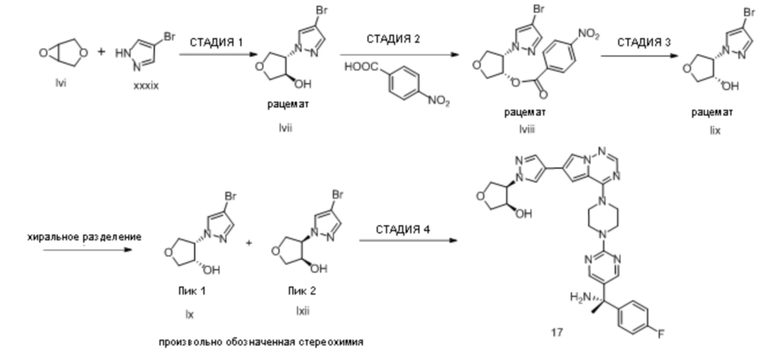
[00241] Стадия 1: Синтез rac-транс-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ола (lvii): Раствор 3,6-диоксабицикло[3.1.0]гексана (lvi) (5,2 г, 60,5 ммоль), 4-бром-1H-пиразола (xxxix) (8,8 г, 60,5 ммоль) и Cs2CO3 (39,3 г, 121 ммоль) в NMP (100 мл) перемешивали при 120°C в течение 16 часов. Раствор охлаждали и разбавляли дихлорметаном, затем промывали H2O и соляным раствором. Органическую фазу концентрировали и очищали посредством колоночной флэш-хроматографии на силикагеле (ПЭ/ЭА = 3/1) с получением указанного в заголовке соединения (lvii) (10 г, выход 71%) в виде бесцветного твердого вещества. МС (ЭС+) рассчитано для C7H9BrN2O2: 232, найдено: 233 [М + 18]+.
[00242] Стадия 2: Синтез rac-цис-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ил-4-нитробензоата (lviii): Смесь rac-транс-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ола (lvii) (2,7 г, 11,6 ммоль), 4-нитробензойной кислоты (1,95 г, 11,6 ммоль), диизопропилазодикарбоксилата (3,53 мг, 17,4 ммоль) и трифенилфосфина (4,57 г, 17,4 ммоль) в ТГФ (50 мл) перемешивали при комнатной температуре в течение 16 часов. Раствор разбавляли этилацетатом и промывали H2O и соляным раствором. Органическую фазу концентрировали и очищали посредством колоночной флэш-хроматографии на силикагеле (ПЭ/ЭА = 3/1) с получением указанного в заголовке соединения (lviii) (4 г, выход 90%) в виде бесцветного твердого вещества. МС (ЭС+) рассчитано для C14H12BrN3O5: 381, найдено: 382 [М+Н]+.
[00243] Стадия 3: Синтез (3S,4S)-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ола (lx) (пик 1) и (3R,4R)-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ола (lxi) (пик 2): Смесь rac-цис-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ил-4-нитробензоата (lvii) (4 г, 10,5 ммоль) и гидроксида лития (2,2 г, 52,5 ммоль) в MeOH/ТГФ/H2O (30 мл/30 мл/30 мл) перемешивали при комнатной температуре в течение 4 часов. Полученную смесь разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали в вакууме. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ПЭ/ЭА = 3/1 = 3/1) с получением rac-цис-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ола (1,3 г, выход 53%) в виде бесцветного твердого вещества. МС (ЭС+) рассчитано для C7H9BrN2O2: 232, найдено: 233 [М+Н]+. Полученное вещество подвергали хиральному разделению посредством сверхкритической флюидной хроматографии (SFC) (колонка: AD 20 × 250 мм, 10 мкм (Daicel); подвижная фаза: СО2/МеОН (0,2% аммиака в метаноле) = 60/40; Скорость потока: 80 г/мин) с получением пика 1 (lx) (500 мг) и пика 2 (lxi) (500 мг). Пик 1 произвольно обозначали как (3S,4S)-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ол, и пик 2 произвольно обозначали как (3R,4R)-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ол.
[00244] Стадия 4: Синтез (3R,4R)-4-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)тетрагидрофуран-3-ола (17): Смесь (3R,4R)-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ола (lxi) (70 мг, 0,3 ммоль) (пик 2, полученный на стадии 3), I-3 (328,3 мг, 0,6 ммоль), Pd[(t-Bu)3P]2 (31 мг, 0,06 ммоль) и Na2CO3 (96 мг, 0,9 ммоль) в диоксане/H2O (8 мл/2 мл.) перемешивали при 90 °C в течение 4 часов. После охлаждения раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 10/1) с получением указанного в заголовке соединения (17) (106,3 мг, выход 62%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O2: 570, найдено: 571, 554 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,15 (s, 1H), 8,03 (s, 1H), 7,88 (s, 1H), 7,87 (s, 1H), 7,52-7,42 (m, 2H), 7,28 (s, 1H), 7,17-7,10 (m, 2H), 5,33 (d, 1H, J = 4,8 Гц), 4,92-4,83 (m, 1H), 4,46-4,38 (m, 1H), 4,21-4,05 (m, 6H), 4,01 (m, 1H), 3,95-3,85 (m, 4H), 3,73 (m, 1H), 2,45 (s, 2H), 1,73 (s, 3H).
[00245] Пример 18: (3R,4S)-4-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)тетрагидрофуран-3-ол (18)
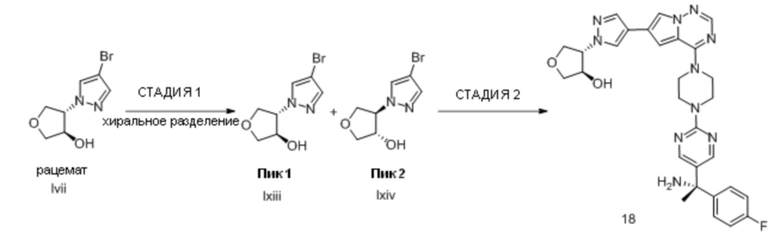
[00246] Стадия 1: Хиральное разделение rac-транс-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ола (lvii): rac-Транс-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ол (1,1 г) (полученный на стадии 1 примера 17) подвергали хиральному разделению с помощью SFC (колонка: AD 20 × 250 мм, 10μм (Daicel); подвижная фаза: СО2/МеОН (0,2% аммиака в МеОН) = 80/20; скорость потока: 80 г/мин) с получением пика 1 (lxiii) (400 мг) и пика 2 (lxiv) (500 мг). Пик 1 произвольно обозначали как (3R,4S)-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ол и пик 2 произвольно обозначали как (3S,4R)-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ол.
[00247] Стадия 2: Синтез (3R,4S)-4-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)тетрагидрофуран-3-ола (18): Смесь (3R,4S)-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ола (70 мг, 0,3 ммоль) (lxiii) (пик 1, полученный на стадии 1), I-3 (328,3 мг, 0,6 ммоль), Pd[(t-Bu)3P]2 (31 мг, 0,06 ммоль) и Na2CO3 (96 мг, 0,9 ммоль) в диоксане/H2O (8 мл/2 мл.) дегазировали с помощью N2 и перемешивали при 90°C в течение 4 часов. После этого раствор разбавляли дихлорметаном, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 10/1) с получением указанного в заголовке соединения (18) (55,6 мг, выход 33%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O2: 570, найдено: 571, 554 [M+H]+, [M+H-NH3]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,15 (s, 1H), 8,03 (s, 1H), 7,91 (s, 1H), 7,88 (s, 1H), 7,51-7,43 (m, 2H), 7,28 (s, 1H), 7,16-7,07 (m, 2H), 5,66 (d, 1H, J = 4 Гц), 4,75-4,67 (m, 1H), 4,51-4,42 (m, 1H), 4,20 (m, 1H), 4,15-3,99 (m, 6H), 3,96-3,85 (m, 4H), 3,63 (m, 1H), 2,47 (s, 2H), 1,73 (s, 3H).
[00248] Пример 19: (3S,4R)-4-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)тетрагидрофуран-3-ол (19)
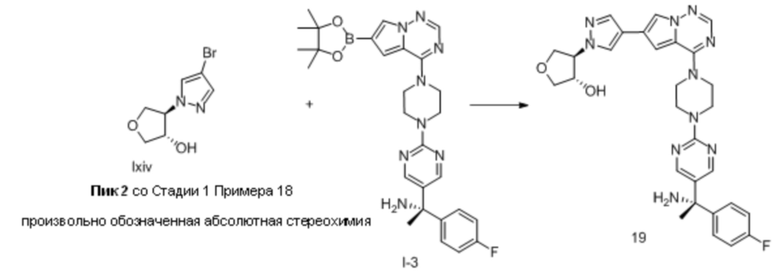
[00249] Смесь (3S,4R)-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ола (70 мг, 0,3 ммоль) (lxiv) (пик 2, полученный на стадии 1 примера 22), I-3 (328,3 мг, 0,6 ммоль), Pd[(t-Bu)3P]2 (31 мг, 0,06 ммоль) и Na2CO3 (96 мг, 0,9 ммоль) в диоксане/H2O (8 мл/2 мл) дегазировали с помощью N2 и перемешивали при 90°C в течение 4 часов. После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 10/1) с получением указанного в заголовке соединения (19) (51,9 мг, выход 31%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O2: 570, найдено: 571, 554 [M+H]+ и [M+H-NH3]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,15 (s, 1H), 8,03 (s, 1H), 7,91 (s, 1H), 7,88 (s, 1H), 7,52-7,43 (m, 2H), 7,28 (s, 1H), 7,19-7,06 (m, 2H), 5,66 (d, 1H, J = 4,4 Гц), 4,75-4,66 (m, 1H), 4,51-4,41 (m, 1H), 4,20 (m, 1H), 4,16-3,99 (m, 6H), 3,97-3,83 (m, 4H), 3,63 (dd, 1H, J = 9,6 Гц, J = 2,8 Гц), 2,54 (s, 2H), 1,73 (s, 3H).
[00250] Пример 20: (3S,4S)-4-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)тетрагидрофуран-3-ол (20)
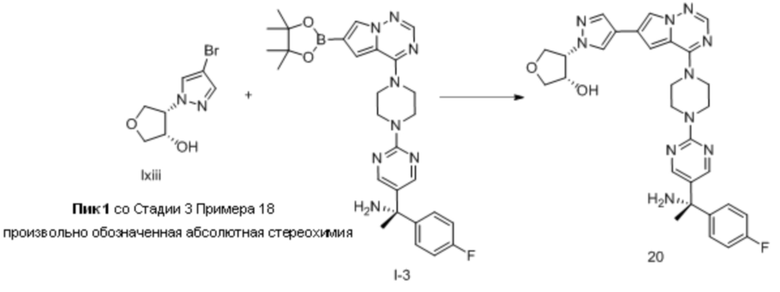
[00251] Смесь (3S,4S)-4-(4-бром-1H-пиразол-1-ил)тетрагидрофуран-3-ола (lxiii) (50 мг, 0,22 ммоль) (пик 1, полученный на стадии 3 примера 21), I-3 (234,5 мг, 0,44 ммоль), Pd[(t-Bu)3P]2 (22 мг, 0,044 ммоль) и Na2CO3 (68 мг, 0,66 ммоль) в диоксане/H2O (8 мл/2 мл) перемешивали при 90°C в течение 4 часов. После охлаждения раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 10/1) с получением указанного в заголовке соединения (20) (74,6 мг, выход 61%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O2: 570, найдено: 571, 554 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,15 (s, 1H), 8,03 (d, 1H, J = 1,6 Гц), 7,88 (s, 1H), 7,87 (s, 1H), 7,51-7,44 (m, 2H), 7,28 (d, 1H, J = 1,2 Гц), 7,17-7,10 (m, 2H), 5,33 (d, 1H, J = 5,6 Гц), 4,93-4,83 (m, 1H), 4,47-4,38 (m, 1H), 4,20-4,06 (m, 6H), 4,01 (m, 1H), 3,95-3,88 (m, 4H), 2,45 (s, 2H), 3,73 (m, 1H), 1,76 (s, 3H).
[00252] Пример 21: (1S,2S)-2-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)циклобутанол (21)
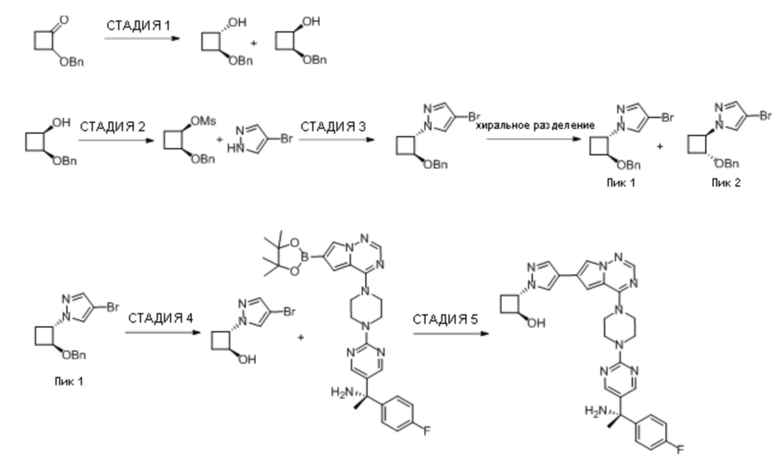
[00253] Стадия 1: Синтез транс-2-(бензилокси)циклобутанола и цис-2-(бензилокси)циклобутанола: К раствору 2-(бензилокси)циклобутанона (1,0 г, 5,7 ммоль) в MeOH (20 мл) добавляли NaBH4 (432 мг, 11,4 ммоль) при 0°C. Затем раствор перемешивали при комнатной температуре в течение 3 часов. Смесь разбавляли этилацетатом, промывали водой и соляным раствором, затем органическую фазу концентрировали и очищали посредством колоночной флэш-хроматографии на силикагеле (петролейный эфир/этилацетат = 5/1) с получением 400 мг пика 1 (произвольно обозначенного как цис-2-(бензилокси)циклобутанол) в виде бесцветного масла и 400 мг пика 2 (произвольно обозначенного как транс-2-(бензилокси)циклобутанол) в виде бесцветного масла. МС (ЭС+) рассчитано для C11H14O2: 178, найдено: 179 [М+Н]+.
[00254] Стадия 2: Синтез цис-2-(бензилокси)циклобутилметансульфоната: К раствору цис-2-(бензилокси)циклобутанола (270 мг, 1,52 ммоль) в ДХМ (10 мл) добавляли мезилхлорид (259 мг, 2,28 ммоль) и триэтиламин (459 мг, 4,56 ммоль) при 0°C. Смесь перемешивали при комнатной температуре в течение 3 часов. После этого раствор разбавляли дихлорметаном, промывали водой и соляным раствором, высушивали над безводным Na2SO4 и концентрировали с получением указанного в заголовке соединения (300 мг, выход 77%) в виде бесцветного масла. МС (ЭС+) рассчитано для C12H16O4S: 256, найдено: 274 [M+18]+.
[00255] Стадия 3: Синтез транс-2-(бензилокси)циклобутил)-4-бром-1H-пиразола: Смесь цис-2-(бензилокси)циклобутилметансульфоната (300 мг, 1,17 ммоль), 4-бром-1H-пиразола (171 мг, 1,17 ммоль) и Cs2CO3 (1,15 г, 3,51 ммоль) в ДМФ (8 мл) перемешивали при 100°C в течение 16 часов. После этого раствор разбавляли этилацетатом, промывали водой и соляным раствором, высушивали над безводным Na2SO4, концентрировали и очищали посредством колоночной флэш-хроматографии (ПЭ/ЭА = 5/1) с получением указанного в заголовке соединения (170 мг, 47 % выхода) в виде бесцветного масла. МС (ЭС+) рассчитано для C14H15BrN2O: 306, найдено: 307 [М+Н]+. Хиральное разделение транс-2-(бензилокси)циклобутил)-4-бром-1H-пиразола: транс-2-(бензилокси)циклобутил)-4-бром-1H-пиразол (600 мг) подвергали хиральному разделению с помощью SFC (колонка: IG 20 × 250 мм, 10 мкм (Daicel); подвижная фаза: СО2/МеОН (0,2% аммиака в метаноле) = 75/25; скорость потока: 4 г/мин) с получением пика 1 (250 мг) и пика 2 (250 мг). Пик 1 произвольно обозначали как 1-((1S,2S)-2-(бензилокси)циклобутил)-4-бром-1H-пиразол и пик 2 произвольно обозначали как 1-((1R,2R)-2-(бензилокси)циклобутил)-4-бром-1H-пиразол.
[00256] Стадия 4: Синтез (1S,2S)-2-(4-бром-1H-пиразол-1-ил)циклобутанола: Раствор 1-((1S,2S)-2-(бензилокси)циклобутил)-4-бром-1H-пиразола (250 мг, 820 мкмоль) в трифторуксусной кислоте (ТФУ) (2 мл) перемешивали при 80°C в течение 16 часов. После этого раствор концентрировали и очищали посредством колоночной флэш-хроматографии на силикагеле (петролейный эфир/этилацетат = 3/1) с получением указанного в заголовке соединения (120 мг, выход 68%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C7H9BrN2O: 216, найдено: 217 [М+Н]+.
[00257] Стадия 5: Синтез (1S,2S)-2-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)циклобутанола: Смесь (1S,2S)-2-(4-бром-1H-пиразол-1-ил)циклобутанола (120 мг, 556 мкмоль), I-3 (362 мг, 667 мкмоль), Pd(t-Bu3P)2 (50 мг, 99 мкмоль) и Cs2CO3 (362 мг, 1,12 ммоль) в диоксане/H2O (8 мл/2 мл) продували с помощью N2 в течение 10 минут и перемешивали при 90°C в течение 4 часа в атмосфере N2. После этого раствор разбавляли дихлорметаном, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 10/1). Полученное вещество дополнительно очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 32% до 62% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (52,6 мг, выход 17%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O: 554, найдено: 555 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,19 (s, 1H), 8,00 (d, 1H, J = 1,6 Гц), 7,88 (s, 2H), 7,48-7,44 (m, 2H), 7,26 (d, 1H, J = 1,6 Гц), 7,14-7,08 (m, 2H), 5,67 (d, 1H, J = 7,2 Гц), 4,46-4,39 (m, 1H), 4,34-4,26 (m, 1H), 4,10-4,06 (m, 4H), 3,92-3,90 (m, 4H), 2,44 (s, 2H), 2,16-2,10 (m, 2H), 1,89-1,79 (m, 1H), 1,73 (s, 3H), 1,62-1,52 (m, 1H).
[00258] Пример 22: (1R,2R)-2-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)циклобутанол (22)
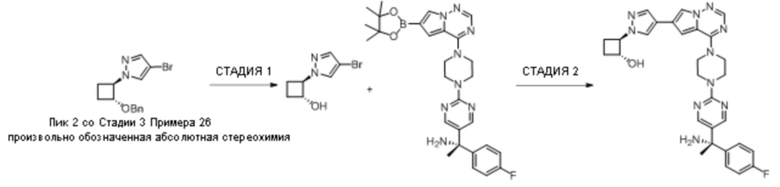
[00259] Стадия 1: Синтез (1R,2R)-2-(4-бром-1H-пиразол-1-ил)циклобутанола: Раствор 1-((1R,2R)-2-(бензилокси)циклобутил)-4-бром-1H-пиразола (250 мг, 820 мкмоль) (пик 2, полученный на стадии 3 примера 21) в ТФУ (2 мл) перемешивали при 80°C в течение 16 часов. После этого раствор концентрировали и очищали посредством колоночной флэш-хроматографии на силикагеле (петролейный эфир/этилацетат = 3/1) с получением указанного в заголовке соединения (120 мг, выход 68%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C7H9BrN2O: 216, найдено: 217 [М+Н]+.
[00260] Стадия 2: Синтез (1R,2R)-2-(4-(4-(4-(5-((S)-1-амино-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-ил)пирроло[2,1-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)циклобутанола: Смесь (1R,2R)-2-(4-бром-1H-пиразол-1-ил)циклобутанола (120 мг, 556 мкмоль), (S)-1-(4-фторфенил)-1-(2-(4-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина (362 мг, 667 мкмоль), Pd(t-Bu3P)2 (50 мг, 99 мкмоль) и Cs2CO3 (362 мг, 1,12 ммоль) в диоксан/H2O (8 мл/2 мл) продували с помощью N2 (газ) в течение 10 минут и перемешивали при 90°C в течение 4 часов в атмосфере N2 (газ). После этого раствор разбавляли этилацетатом, промывали H2O и соляным раствором и концентрировали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (ДХМ/MeOH = 10/1). Полученное вещество дополнительно очищали с помощью препаративной ВЭЖХ (подвижная фаза: A = H2O (0,1% NH4HCO3), B = ацетонитрил; градиент: B = от 30% до 60% в течение 18 минут; колонка: Xtimate™ 10 мкм 150A 21,2 × 250 мм) с последующей лиофилизацией с получением указанного в заголовке соединения (51,5 мг, выход 17%) в виде белого твердого вещества. МС (ЭС+) рассчитано для C29H31FN10O: 554, найдено: 555 [М+Н]+. 1H-ЯМР (400 МГц, 6d-ДМСО) δ ppm 8,41 (s, 2H), 8,19 (s, 1H), 8,00 (d, 1H, J = 1,6 Гц), 7,88 (s, 2H), 7,48-7,44 (m, 2H), 7,26 (d, 1H, J = 1,6 Гц), 7,14-7,08 (m, 2H), 5,67 (d, 1H, J = 7,2 Гц), 4,46-4,39 (m, 1H), 4,34-4,26 (m, 1H), 4,10-4,06 (m, 4H), 3,92-3,90 (m, 4H), 2,44 (s, 2H), 2,16-2,10 (m, 2H), 1,89-1,79 (m, 1H), 1,73 (s, 3H), 1,62-1,52 (m, 1H).
ПРИМЕР 2: Анализы биохимической ферментативной активности
[00261] Ферментативную активность PDGFRα и KIT контролировали с применением технологической платформы Perkin Elmer для оценки сдвига электрофоретической подвижности, EZReader 2. Пептидный субстрат с флуоресцентной меткой инкубировали в присутствии киназы и АТФ и в присутствии исследуемого соединения таким образом, что каждая доза исследуемого соединения приводила к фосфорилированию отражающей части пептида.
[00262] В рамках линейной стационарной фазы ферментативной реакции киназы смешанный пул фосфорилированных (продукт) и нефосфорилированных (субстрат) пептидов пропускали через микрожидкостную систему ридера PerkinElmer EZ Reader 2 при приложенной разности электрических потенциалов. Наличие фосфатной группы в пептиде-продукте обеспечивало разность массы и заряда относительно массы и заряда пептида-субстрата, что приводило к разделению пулов субстрата и продукта в образце (Perrin et al., Expert°pin Drug Discovery 2010, Jan 5(1):51-63).
[00263] При прохождении смеси пептида-продукта и пептида-субстрата через лазеры, установленные в приборе, указанные пулы обнаруживали (λex = 488 нм, λem = 568 нм) и разделяли на отдельные пики. Соотношение между такими пиками отражает активность соединения при данной концентрации в данной лунке в данных условиях.
Ингибирование биохимической ферментативной активности мутантных KIT (D816V) и PDGFRα (D842V)
[00264] Все исследуемые образцы растворяли в 100% ДМСО при исходной концентрации 10 мМ. Было проведено 100Х, 10-точечное, 4-кратное серийное разведение всех исследуемых соединений в 100% ДМСО, начиная с соответствующей концентрации, обычно 1 мМ. Объем 0,130 μл каждой концентрации переносили в соответствующую лунку 384-луночного аналитического планшета (Greiner 781 201) с применением нанолитрового дозатора TTPLabtech Mosquito. Затем с помощью Multidrop оставшиеся компоненты реакции добавляли к 0,130 μл соединения следующим образом:
[00265] Анализ D842V PDGFRα на кажущуюся константу Михаэлиса-Ментена (APPKM) для АТФ: В каждой лунке 384-луночного аналитического планшета инкубировали 7 нМ необработанного фермента в общем объеме 13 μл буфера (100 мМ HEPES pH 7,5, 0,015% Brij 35, 10 мМ MgCl2, 1 мМ ДТТ) с 1 μM CSKtide (5-FAM-AHA-KKKKDDIYFFFG-NH2) и 25 μM АТФ при 25°C в течение 90 минут в присутствии или отсутствии серии дозированных концентраций исследуемого соединения (конечная концентрация ДМСО 1%). Реакцию останавливали путем добавления 70 μл останавливающего буфера (100 мМ HEPES pH 7,5, 0,015% Brij 35, 35 мМ ЭДТК и 0,2% посадочного реагента 3, Caliper Lifesciences). Планшет считывали на Caliper EZReader 2.
[00266] Анализ KIT D816V на APPKM для АТФ: В каждой лунке 384-луночного аналитического планшета инкубировали 0,3 нМ необработанного фермента в общем объеме 13 μл буфера (100 мМ HEPES pH 7,5, 0,015% Brij 35, 10 мМ MgCl2, 1 мМ ДТТ) с 1 μM SRCtide (5-FAM-GEEPLYWSFPAKKK-NH2) и 20 μАТФ в 25°C в течение 60 минут в присутствии или отсутствии серии дозированных концентраций исследуемого соединения (конечная концентрация ДМСО 1 %). Реакцию останавливали путем добавления 70 μл останавливающего буфера (100 мМ HEPES pH 7,5, 0,015% Brij 35, 35 мМ ЭДТК и 0,2% посадочного реагента 3, Caliper Lifesciences). Планшет считывали на Caliper EZReader 2. Результаты, полученные в указанных экспериментах для соединений, полученных согласно приведенным примерам, суммированы ниже в таблице 2. Для биохимической активности D816V и D842V использовали следующие обозначения: ≤ 0,30 нМ = A; > 0,31 и <1 нМ = B; и ND = не определено. Для клеточной активности в клеточной линии HMC1.2 использовали следующие обозначения: А означает <4,5 нМ; B означает ≥ 4,6 и <10 нМ; и ND = не определено.
Таблица 2.
D816V
(нM)
D842V
(нM)
WO2015/057873 («63»)
[00267] Для справки: химическая структура соединения согласно примеру 63 в WO2015/057873:
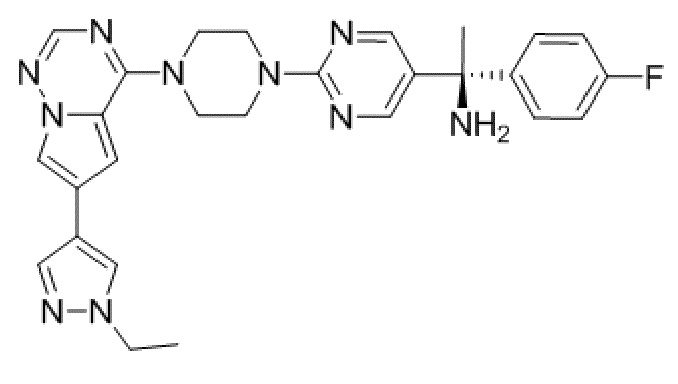 .
.
ПРИМЕР 3: Анализ аутофосфорилирования HMC1.2
[00268] 10000 клеток HMC1.2 инкубировали в 22 μл культуральной среды (среда Дульбекко, модифицированная по способу Исков (IMDM), не содержащая фенолового красного, без сыворотки) в каждой лунке 384-луночного планшета и без сыворотки в течение ночи в инкубаторе для тканевых культур (5% CO2, 37°C). Затем к клеткам добавляли 10-точечные серии дозируемых концентраций исследуемого соединения (2,5 μM-9,54 пM) в объеме 3,1 μл в каждую лунку (конечная концентрация ДМСО 0,25%). Через 90 минут в каждую лунку добавляли 6 μл 5Х лизирующего буфера AlphaLISA (Perkin Elmer) с добавкой коктейля ингибитора протеазы и фосфатазы (Cell Signaling Technologies) и встряхивали со скоростью 450 об/мин в течение 15 минут при 4°C. В каждую лунку добавляли 10 μл фосфо-Y719 c-KIT и общих антител c-KIT (конечная концентрация 15 нМ, Cell Signaling Technologies) и 50 μг/мл акцепторных гранул кроликов AlphaLISA (Perkin Elmer) и встряхивали со скоростью 300 об/мин при комнатной температуре в течение 2 часов. В каждую лунку добавляли 10 μл 100 μг/мл стрептавидиновых донорных гранул (Perkin Elmer), закрывали от света с помощью твердого черного адгезива и встряхивали со скоростью 300 об/мин при комнатной температуре в течение 2 часов. Сигнал флуоресценции получали на приборе Envision (Perkin Elmer) согласно протоколу AlphaScreen 384 well HTS. Данные нормализовали к контрольному ингибированию 0% и 100% и рассчитывали IC50 путем построения четырехпараметрической логистической кривой IC50.
В таблице показана активность соединений в клеточной линии тучноклеточного лейкоза, HMC 1.2. Такая клеточная линия содержит KIT, мутированный в положениях V560G и D816V, что приводит к конститутивной активации киназы. В данном анализе приведенные ниже соединения исследовали для измерения прямого ингибирования киназной активности KIT D816V путем измерения аутофосфорилирования KIT на тирозине 719 белка KIT. Результаты указанных экспериментов для соединений, полученных согласно приведенным примерам, приведены в таблице 2.
ПРИМЕР 4: Оценка проникновения в головной мозг крыс на основе соотношение концентраций в головном мозге и в плазме (Kp,мозг)
[00270] Для понимания процесса проникновения в головной мозг крыс Sprague-Dawley (SD) определяли соотношения концентраций предложенных соединений в головном мозге и в плазме. Равновесное распределение in vivo между кровью и головным мозгом у предклинических субъектов, таких как крысы, является широко применяемым параметром для оценки проникновения в головной мозг. Kp,мозг представляет собой соотношение концентраций в головном мозге и крови (Cмозг/Cплазма). Характеристики пассивной диффузии соединения, его сродство к мембранным переносчикам в области гемато-энцефалического барьера (ГЭБ) и относительные различия в сродстве к связыванию лекарственного препарата между белками плазмы и тканью головного мозга влияют на Kp,мозг. Соединения с Kp,мозг менее 0,1 имеют ограниченный доступ к ЦНС, тогда как считается, что соединения с Kp,мозг более 0,3-0,5 характеризуются хорошим проникновением в головной мозг, а соединения с Kp,мозг более 1 свободно пересекают ГЭБ (Expert°pin. Drug Delivery (2016) 13 (01): 85-92).
[00271] Проникновение в головной мозг соединений 4 и 63 измеряли у крыс Sprague-Dawley (3/соединение). Животным вводили указанное соединение посредством внутривенной инфузии со скоростью 1 мг/кг/час в течение 24 часов путем канюляции яремной вены. Через 24 часа кровь собирали путем кровоизвлечения из хвостовой вены или пункции сердца (под анестезией) и центрифугировали с получением образцов плазмы. Ткани головного мозга собирали и гомогенизировали с фосфатно-солевым буферным раствором (ФСБ). Концентрации исследуемых соединений измеряли в плазме и гомогенатах головного мозга посредством анализа методом ЖХ-МС/МС. В таблице 3A ниже приведены результаты измерения концентраций в плазме и головном мозге, а также Kp,мозг для соединения 4, полученного согласно приведенным примерам, описанным в настоящем документе, и соединения 63, описанного в WO2015/057873.
Таблица 3A.
[00272] Соединение 4 имеет очень низкий Kp,мозг (среднее значение = 0,17) по сравнению с соединением 63 (среднее значение = 1,8).
[00273] Связывание с белками плазмы крыс соединений 4 и 63 оценивали in vitro с применением метода равновесного диализа. Соединение 4 (10 мкМ) исследовали в 100% плазме в установке для диализа в течение 5 часов при 37°С. Образцы, отобранные со стороны донора и со стороны приемника, анализировали методом ЖХ-МС/МС. Фракции, связанные и несвязанные с белками плазмы, рассчитывали с применением следующих уравнений:
Связанная фракция (fb)*(%) = 100 × ([Донор]5час - [Приемник]5час)/[Донор]5час (Уравнение 1)
Несвязанная фракция (fu), p* (%) = 100 - % связанной фракции* (Уравнение 2)
где: [Донор]5час представляет собой измеренную концентрацию в доноре через 5 часов; [Приемник]5час представляет собой измеренную концентрацию в приемнике через 5 часов; fb* представляет собой связанную фракцию, определенную в плазме; fu,p* представляет собой рассчитанную несвязанную фракцию в плазме. В качестве положительного контроля использовали варфарин и хинидин.
[00274] fb для соединений 4 и 63 составляли 97,92% и 99,8%, соответственно. Таким образом, fu,p соединений 4 и 63 составили 2,08% и 0,2%, соответственно.
[00275] Аналогичным образом, связывание с белками в головном мозге крыс соединений 4 и 63 также оценивали in vitro с применением метода равновесного диализа. 1 мкМ соединения исследовали в гомогенате мозга в установке для диализа в течение 5 часов при 37°C. Образцы, отобранные со стороны донора и со стороны приемника, анализировали методом ЖХ-МС/МС. Фракции, связанные и несвязанные с белками головного мозга, рассчитывали с применением приведенных выше уравнений (уравнений 1 и 2). Вследствие значительного связывания с белками соединение 4 дополнительно разводили в 4 раза для анализа гомогената головного мозга. fu,мозг соединений 4 и 63 составляли 0,29% и 0,1%, соответственно.
Соотношения несвязанных фракций в головном мозге и плазме (Kpuu,мозг)
[00276] На основании полученных выше концентраций в головном мозге и плазме (таблица 3A) и полученных выше значений fu,мозг соотношения несвязанных фракций в головном мозге и плазме (Kpuu,мозг) рассчитывали для соединений 4 и 63 следующим образом:
[00277] Соединение 4 имеет самое низкое значение Kp,uu,мозг (среднее значение = 0,024) по сравнению с соединением 63 (среднее значение = 0,84). Концентрация несвязанного лекарственного препарата в ткани представляет собой свободный лекарственный препарат, доступный для оказания своего фармакологического действия в тканевом компартменте. Поскольку соединение 4 имеет очень низкий Kp,uu,мозг по сравнению с соединением 63, это означает, что количество соединения 4, доступного в головном мозгу и оказывающего свое фармакологическое действие, является очень низким по сравнению с соединением 63.
[00278] Альтернативно, связывание соединений 4 и 63 с белками головного мозга крыс оценивали in vitro, используя срезы головного мозга крысы (область полосатого тела) толщиной 300 мкм в инкубационном планшете. Значения fu,мозг соединений 4 и 63, определенные таким методом, составляли 0,329% и 0,057%, соответственно. В этом случае Kp,uu,мозг соединений 4 и 63 составляли 0,028 и 0,044, соответственно.
[00279] В таблице 3B приведены результаты определения Kp, Kp,uu (гомогенат головного мозга) и Kp,uu (срез головного мозга) для дополнительных соединений согласно настоящему изобретению, полученных в соответствии с приведенными примерами. Результаты в таблице 3B получали согласно описанным выше способам.
Таблица 3B.
Kp
Kp, uu
гомогенат
Kp,uu
срез головного мозга
*измерение невозможно из-за сильного связывания с белками
Оценка соединений как потенциального субстрата P-гликопротеина
[00280] Способность соединений, полученных согласно приведенным примерам, являться субстратами человеческого P-гликопротеина (P-gp), оценивали in vitro на монослоях клеток 1-Мадин-Дарби почек собак с мутацией множественной лекарственной устойчивости (MDR1-MDCK)) (клетки Мадин-Дарби почек собак), сверхэкспрессирующих P-gp, выращенных на проницаемых подложках. В качестве положительного контрольного ингибитора P-gp-опосредованного транспорта хинидина использовали элакридар. Более высокий коэффициент эффлюкса P-gp означает, что транспортер выталкивает указанное соединение из ткани головного мозга.
[00281] Оценка фармакокинетики после однократного внутривенного и перорального введения крысам: 3 крысы Sprague-Dawley использовали для каждого соединения для каждого пути введения (внутривенного или перорального). Для внутривенного введения 1 мг/кг (объем дозы = 5 мл/кг) каждого соединения вводили внутривенно путем инъекции в дорсальную вену стопы; тогда как в случае перорального введения 2,5 мг/кг (объем дозы = 5 мл/кг) вводили через желудочный зонд. Образцы крови отбирали из хвостовой вены перед введением дозы, через 0,083, 0,25, 0,5, 1, 2, 4 и 8 часов. Кроме того, образцы крови также отбирали через 24 часа путем пункции сердца (под анестезией с помощью изофлурана) для терминального кровоизвлечения. Все образцы крови анализировали для определения концентраций лекарственного препарата с помощью ЖХ/МС-МС. Фармакокинетические параметры, такие как Cmax, Tmax, AUClast (площадь под кривой «концентрация-время» от момента введения препарата до определения последней поддающийся количественному измерению концентрации), AUCinf (площадь под кривой «концентрация-время» от времени 0 до бесконечности), MRTlast (среднее время удержания препарата от момента введения препарата до определения последней поддающийся количественному измерению концентрации), MRTinf (среднее время удержания препарата от времени 0 до бесконечности), T1/2, Vss (стационарный объем распределения) и CL (клиренс), получали посредством некомпартментного анализа (NCA). Кроме того, клиренс несвязанной фракции (CLu) рассчитывали следующим образом:
Clu = Cl/f u,плазма.
% F рассчитывали следующим образом:
% F = [AUCinf(перорально)/доза]/[AUCinf(внутривенно)/доза] × 100
((Zhivkova & Doytchinova, Molecular Pharmaceuticals 10:3758-68 (2013)).
Таблица 3C.
Cl (Clu) (мл/мин/кг)
ПРИМЕР 5: Данные ингибирования CYP
[00282] Исследования in vitro микросом печени человека проводили согласно стандартному методу. Вкратце, семь различных концентраций исследуемого препарата или одну концентрацию положительного контрольного соединения совместно инкубировали с одной концентрацией маркерного субстрата для каждого фермента CYP450 в объединенных микросомах печени человека в течение от 5 до 10 минут, а затем завершали реакции путем добавления 0,1% муравьиной кислоты в ацетонитриле. Далее образцы анализировали с помощью ЖХ-МС/МС для количественной оценки маркерного субстрата, оставшегося после реакции, и определяли значения IC50 с применением нелинейной регрессии. Субстратами для CYP2C9, CYP2D6, CYP3A4 были диклофенак, декстрометорфан и мидазолам/тестостерон, соответственно. В таблице 4 приведены значения IC50 для CYP ингибирования соединений, полученных согласно приведенным примерам, для CYP2C9, CYP2D6 и CYP3A4.
Таблица 4.
Номер
(μM)
(μM)
мидазолам
(μM)
тестостерон
ПРИМЕР 6: Связывание с белками плазмы обезьяны при применении внутривенной инфузии, Kp обезьяны, Kp,uu (гомогенат/срез головного мозга) обезьяны
[00283] Обезьяне вводили однократную внутривенную болюсную дозу с последующей 2-часовой внутривенной инфузией исследуемого соединения (3 обезьяны/соединение). Кровь собирали из бедренной вены перед введением дозы, сразу после болюсного введения и в конце инфузии. Обезьяну умерщвляли после инфузии и собирали ткань головного мозга. Для определения соотношения концентраций исследуемого соединения в головном мозге и плазме (Kp) проводили токсикокинетическую оценку плазмы (полученной путем центрифугирования крови) и головного мозга (гомогенизированного в буфере). Kpuu рассчитывали с учетом fu,плазма и fu,мозг, как обсуждалось выше.
Таблица 5.
ПРИМЕР 7: Анализы биохимической активности KIT дикого типа
Пролиферация клеток UT-7 с анализом SCF-стимуляции как меры активности KIT дикого типа
[00284] Клетки UT-7 представляют собой клеточные линии мегакариобластного лейкоза человека, которые можно выращивать в культуре при зависимости от гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF) или фактора стволовых клеток (SCF). Клетки UT-7 реагируют на SCF-стимуляцию путем активации рецепторной тирозинкиназы KIT и последующей нисходящей передачи, что может поддерживать рост и пролиферацию клеток (Kuriu et al, 1999; Komatsu et al, 1991; Sasaki et al, 1995). Исследуемые соединения анализировали на их способность ингибировать SCF-стимулированную пролиферацию клеток UT-7.
[00285] Ингибирование SCF-стимулированной пролиферации клеток UT-7 оценивали с помощью теста CellTiter-Glo, посредством которого определяли количество присутствующего аденозинтрифосфата (АТФ), которое является показателем метаболически активных клеток и прямо пропорционально количеству жизнеспособных клеток в культуре. Способность исследуемых соединений ингибировать SCF-стимулированную пролиферацию клеток UT-7 определяли с применением 10-точечной кривой дозирования в диапазоне от 25 мкМ до 95,4 пM исследуемого соединения.
[00286] Клетки UT-7 поддерживали в IMDM с добавкой 10% FBS, 5 нг/мл GM-CSF и 100 единиц/мл пенициллин-стрептомицина и выращивали в инкубаторе для тканевых культур с увлажнением при 37°C. Клетки UT-7 один раз промывали IMDM, не содержащей сыворотки и GM-CSF. Затем клетки ресуспендировали в IMDM, содержащей 4% FBS и 50 нг/мл SCF, и высевали по 2500 клеток на лунку в объеме 22 мкл в 384-луночном микропланшете. Затем к клеткам в каждую лунку добавляли 10-точечные серии дозируемых концентраций исследуемых соединений (от 25,0 мкМ до 95,4 пМ) в объеме 3,1 мкл (конечная концентрация ДМСО 0,25%) и помещали в инкубатор для тканевых культур (5% CO2, 37°C) на 72 часов. После 3 дней с исследуемым соединением приготавливали свежий реагент CellTiter-Glo, и в каждую лунку добавляли 25 мкл реагента. Планшет перемешивали на планшетном шейкере путем встряхивания в течение 10 минут при комнатной температуре и скорости 300 об/мин. Планшет считывали на планшетном ридере EnVision, используя протокол для сверхчувствительного ридера люминесценции для 384-луночного планшета. Данные нормализовали к контрольному ингибированию 0% и 100% и рассчитывали IC50 путем построения четырехпараметрической логистической кривой IC50.
Анализ KIT дикого типа
[00287] Определение Kd. Для большинства анализов, включающих киназу KIT дикого типа, меченные киназой штаммы фага T7 получали в хозяине E. coli, полученном из штамма BL21. E. coli выращивали до фазы логарифмического роста, инфицировали фагом T7 и инкубировали при встряхивании при 32°C перед лизисом. Магнитные гранулы с стрептавидиновым покрытием обрабатывали с помощью биотинилированных низкомолекулярных лигандов в течение 30 минут при комнатной температуре для получения аффинных смол для анализов киназы. Лигандированные гранулы блокировали с помощью избытка биотина и промывали блокирующим буфером (SeaBlock (Pierce), 1% БСА, 0,05% Твин 20, 1 мМ ДТТ) для удаления несвязанного лиганда и уменьшения неспецифического связывания. Реакции связывания проводили путем объединения киназ, лигандированных аффинных гранул и исследуемых соединений в 1х связывающем буфере (20% SeaBlock, 0,17х ФСБ, 0,05% Твин 20, 6 мМ ДТТ). Исследуемые соединения получали в виде исходных материалов 111Х в 100% ДМСО. Kds определяли с применением 11-точечной серии 3-кратных разведений соединения с тремя контрольными точками ДМСО. Все соединения для измерений Kd распределяли путем акустического переноса (бесконтактное дозирование) в 100% ДМСО. Затем соединения разбавляли непосредственно в анализах с тем, чтобы конечная концентрация ДМСО составляла 0,9%. Все реакции проводили в полипропиленовом 384-луночном планшете. Конечный объем каждой пробы составлял 0,02 мл. Планшеты для анализа инкубировали при комнатной температуре при встряхивании в течение 1 часа, и промывали аффинные гранулы с помощью промывочного буфера (1х ФСБ, 0,05% Твин 20). Затем гранулы ресуспендировали в элюирующем буфере (1х ФСБ, 0,05% Твин 20, 0,5 мкМ небиотинилированного аффинного лиганда) и инкубировали при комнатной температуре при встряхивании в течение 30 минут. Концентрацию киназы в элюатах измеряли посредством количественной ПЦР.
[00288] Константы связывания (Kds). Константы связывания (Kds) рассчитывали с применением стандартной кривой доза-эффект, используя уравнение Хилла: Эффект = Фон + Сигнал - Фон 1 + (Угловой коэффициент Хилла Kd/Угловой коэффициент Хилла при введении дозы). Угловой коэффициент Хилла был установлен на уровне -1. Кривые были подобраны путем применения нелинейного метода наименьших квадратов с алгоритмом Левенберга-Марквардта.
[00289] Результаты, полученные в указанных экспериментах с применением KIT дикого типа для соединений, полученных согласно приведенным примерам, суммированы ниже в таблице 7. Для связывания KIT дикого типа использовали следующие обозначения: <10,0 нМ = A; > 10,1 нМ и <15 нМ = B; > 15,1 нМ и <20 нМ = C. Для ингибирования пролиферации использовали следующие обозначения: <90,0 нМ = A; > 90,1 нМ и <150 нМ = B; > 150,1 нМ и <200 нМ = C.
Таблица 7.
дикого типа
Kd (нM)
WO2015/
057873
(«63»)
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С KIT | 2014 |
|
RU2706235C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ТРИАЗОЛОПИРАЗИНА И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2643361C2 |
| ИНГИБИТОРЫ ФУРИНА | 2019 |
|
RU2799824C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2021 |
|
RU2826628C1 |
| БЕНЗОТИАЗОЛЬНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2018 |
|
RU2778370C2 |
| C26-СВЯЗАННЫЕ АНАЛОГИ РАПАМИЦИНА В КАЧЕСТВЕ ИНГИБИТОРОВ mTOR | 2019 |
|
RU2826559C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| С40-, С28- И С-32-СВЯЗАННЫЕ АНАЛОГИ РАПАМИЦИНА В КАЧЕСТВЕ ИНГИБИТОРОВ MTOR | 2019 |
|
RU2805211C2 |
| ИНГИБИТОРЫ GCN2 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2811403C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2805207C1 |
Изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где А представляет собой
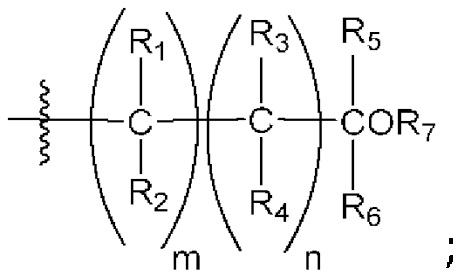
R1 выбран из водорода и метила; R2 выбран из водорода и метила или R1 и R2 вместе образуют циклопропил; R3 выбран из водорода и метила; R4 выбран из водорода и метила или R3 и R4 вместе образуют циклопропил; R5 выбран из водорода и метила; R6 выбран из водорода и метила, или R5 и R6 вместе образуют циклопропил, или один из R2 или R4 вместе с R6 образуют циклобутил; R7 представляет собой водород, или один из R2, R4, или R6 вместе с R7 образуют кольцо, выбранное из оксетана, тетрагидрофурана и тетрагидропирана, при этом указанный тетрагидрофуран или тетрагидропиран необязательно содержит гидроксил в качестве заместителя; m равно 0 или 1; и n равно 0 или 1. Изобретение также относится к фармацевтической композиции на основе указанного соединения. Технический результат – получено новое соединение и фармацевтическая композиция на его основе, которые могут найти применение в медицине для лечения KIT- и PDGFRα-опосредованных заболеваний, в частности желудочно-кишечных стромальных опухолей и системного мастоцитоза. 9 н. и 11 з.п. ф-лы, 7 табл., 22 пр.
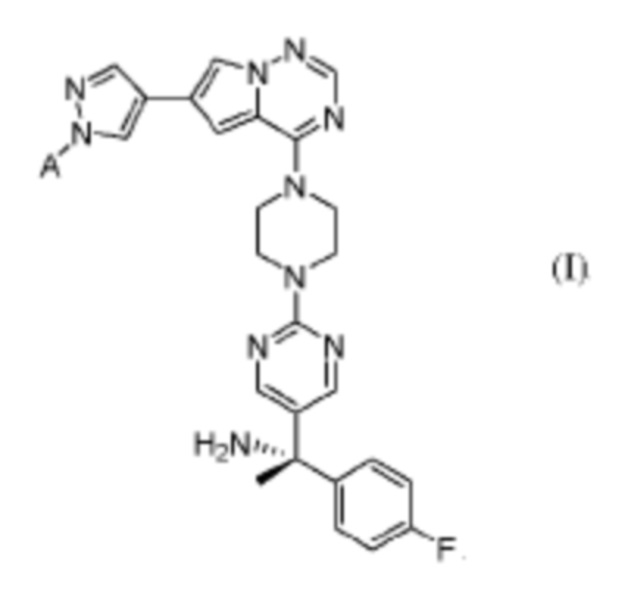
1. Соединение формулы I
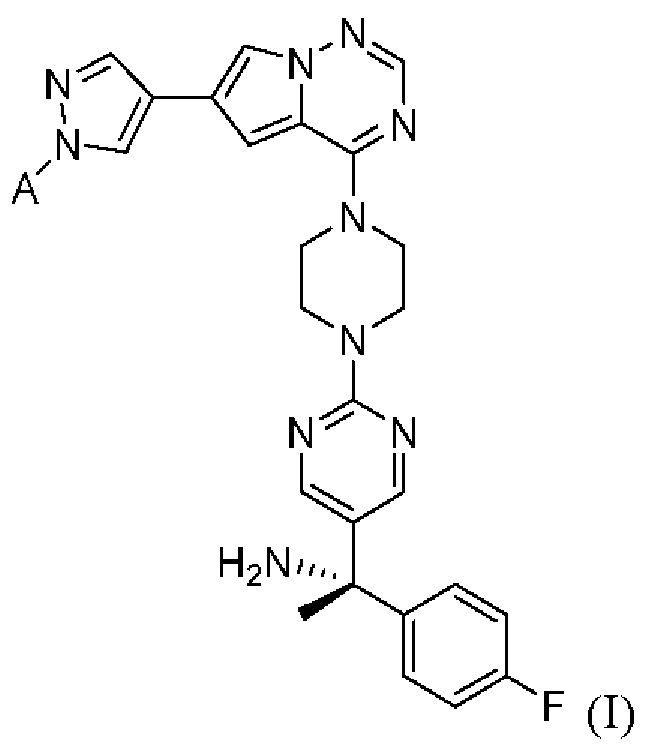
или его фармацевтически приемлемая соль,
где А представляет собой
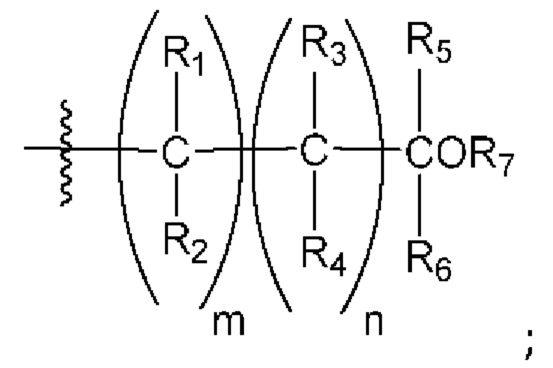
R1 выбран из водорода и метила;
R2 выбран из водорода и метила или
R1 и R2 вместе образуют циклопропил;
R3 выбран из водорода и метила;
R4 выбран из водорода и метила или
R3 и R4 вместе образуют циклопропил;
R5 выбран из водорода и метила;
R6 выбран из водорода и метила, или
R5 и R6 вместе образуют циклопропил, или
один из R2 или R4 вместе с R6 образуют циклобутил;
R7 представляет собой водород или один из R2, R4 или R6 вместе с R7 образуют кольцо, выбранное из оксетана, тетрагидрофурана и тетрагидропирана, при этом указанный тетрагидрофуран или тетрагидропиран необязательно содержит гидроксил в качестве заместителя;
m равно 0 или 1; и
n равно 0 или 1.
2. Соединение по п. 1 или его фармацевтически приемлемая соль,
где А представляет собой
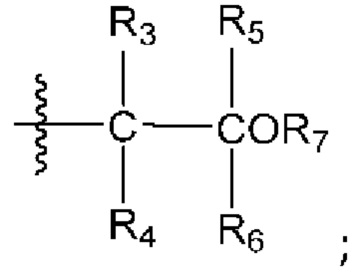
R3 выбран из водорода и метила;
R4 выбран из водорода и метила или
R3 и R4 вместе образуют циклопропил;
R5 выбран из водорода и метила;
R6 выбран из водорода и метила; или
R4 и R6 совместно образуют циклобутил; или
R5 и R6 совместно образуют циклопропил; и
R7 представляет собой водород.
3. Соединение по п. 2 или его фармацевтически приемлемая соль,
где А представляет собой
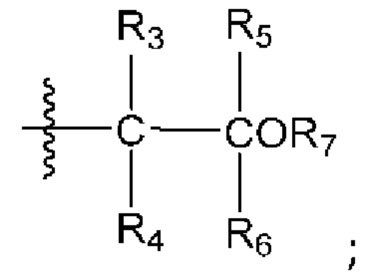
R3 выбран из водорода и метила;
R4 выбран из водорода и метила или
R3 и R4 вместе образуют циклопропил;
R5 выбран из водорода и метила;
R6 выбран из водорода и метила или
R5 и R6 вместе образуют циклопропил; и
R7 представляет собой водород.
4. Соединение по п. 1 или его фармацевтически приемлемая соль,
где А представляет собой
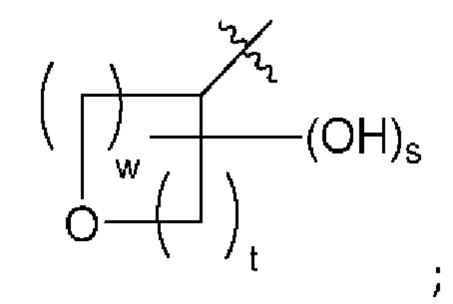
w равно 1 или 2;
t равно 1 или 2; и
s равно 0 или 1.
5. Соединение по п. 1 или его фармацевтически приемлемая соль, где А выбран из:
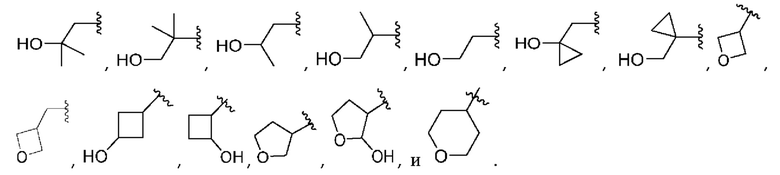
6. Соединение по п. 1 или его фармацевтически приемлемая соль, где А выбран из:
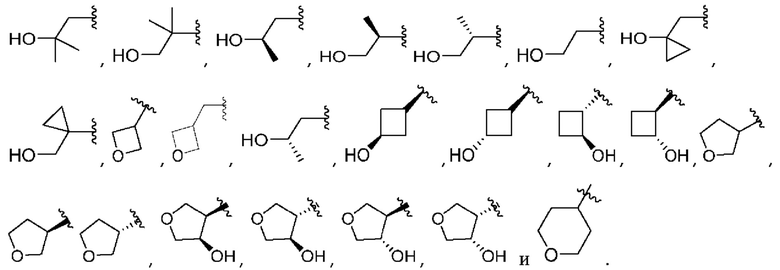
7. Соединение по п. 1 или его фармацевтически приемлемая соль, где А выбран из:
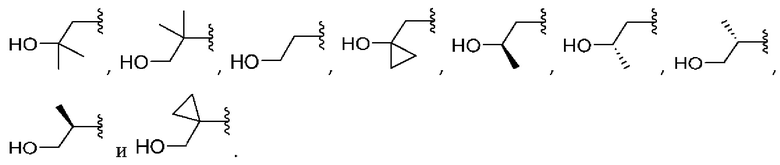
8. Соединение по п. 1 или его фармацевтически приемлемая соль, где А выбран из:

9. Соединение по п. 1 или его фармацевтически приемлемая соль, где А выбран из
10. Соединение или его фармацевтически приемлемая соль, где соединение представляет собой
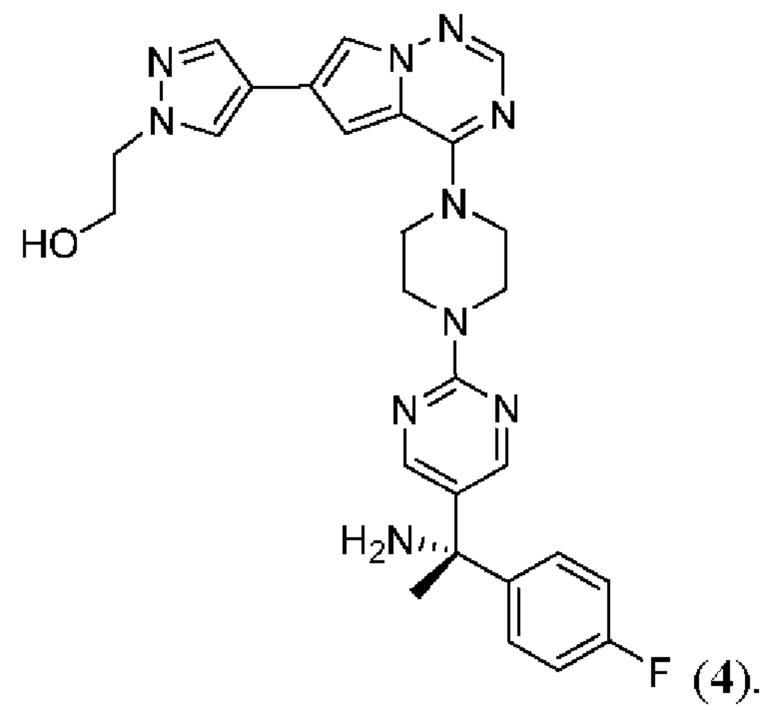
11. Соединение, представляющее собой
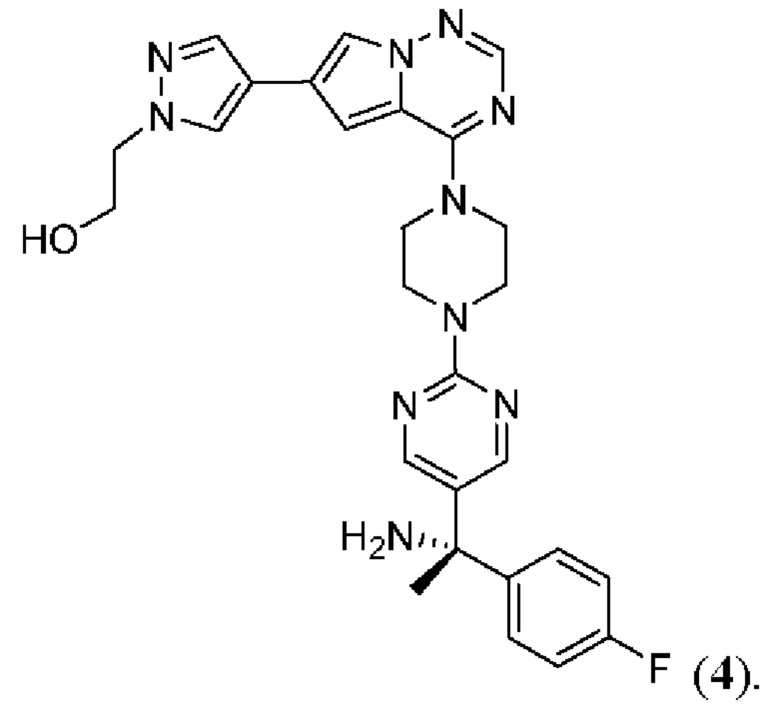
12. Соединение или его фармацевтически приемлемая соль, где соединение представляет собой
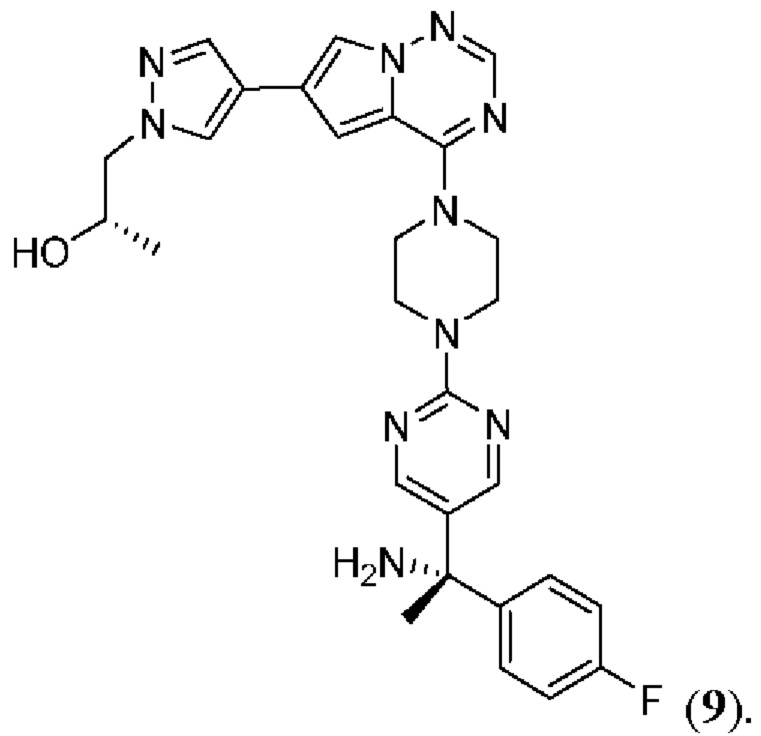
13. Соединение, представляющее собой
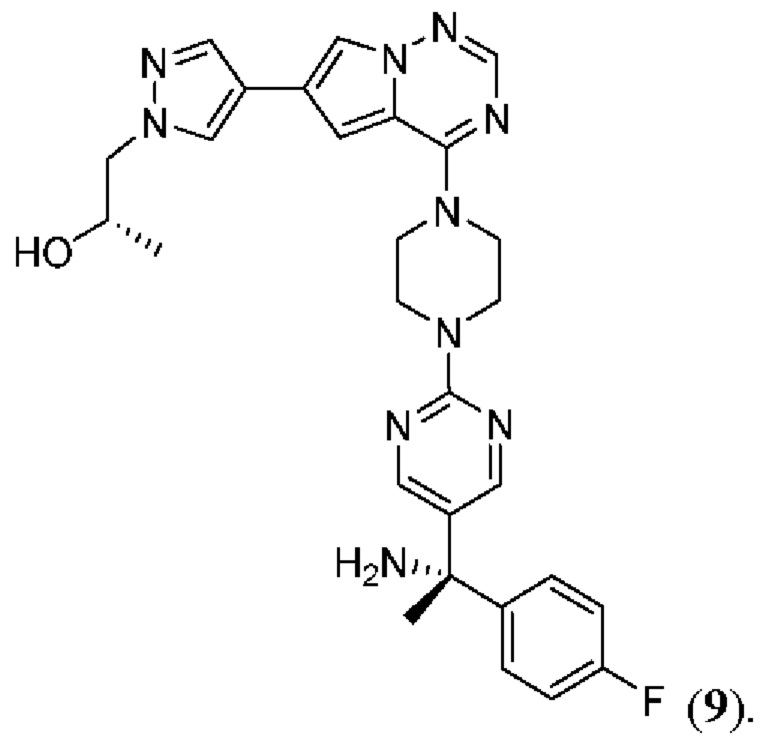
14. Соединение или его фармацевтически приемлемая соль, где соединение представляет собой
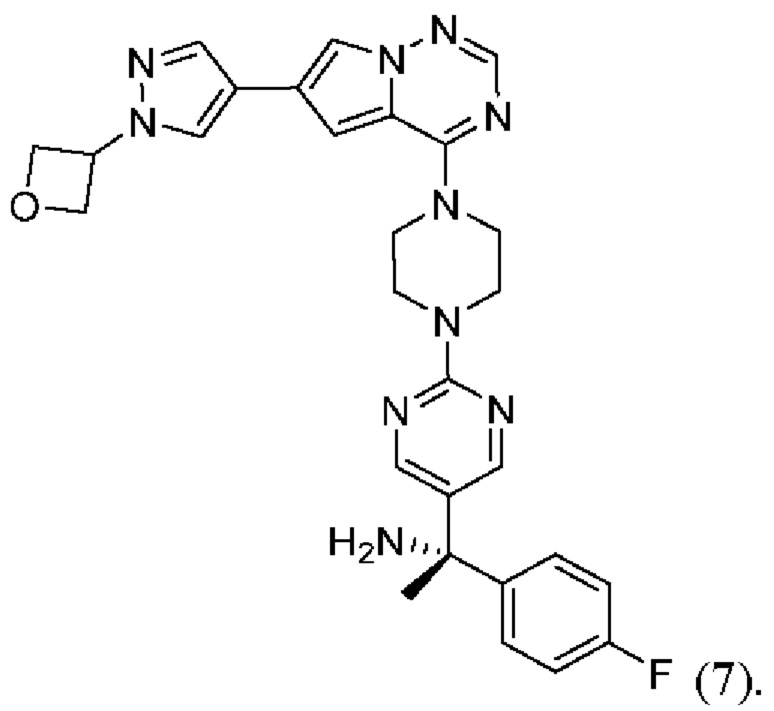
15. Соединение, представляющее собой
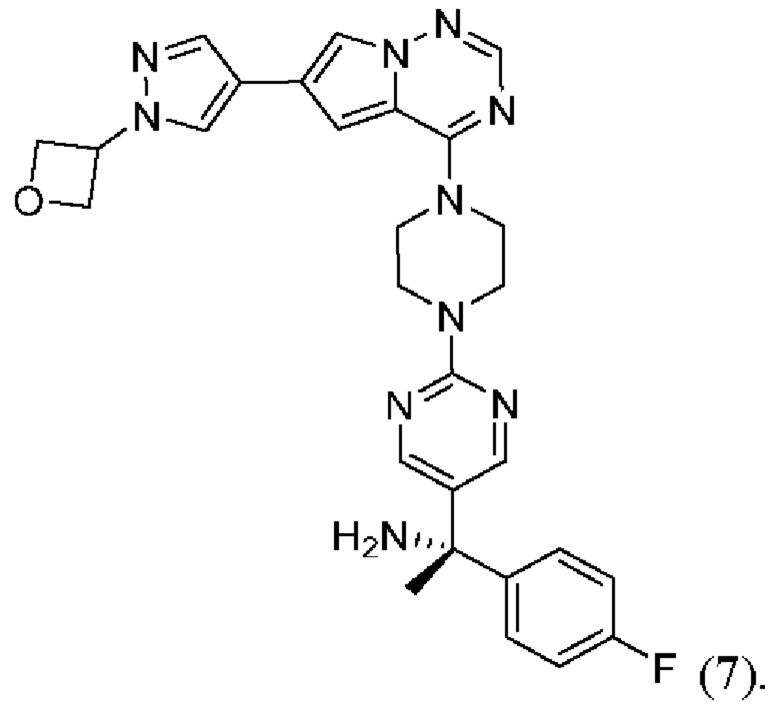
16. Фармацевтическая композиция для лечения KIT- и PDGFRα-опосредованных заболеваний, содержащая
терапевтически эффективное количество соединения по любому из пп. 1-15 или его фармацевтически приемлемой соли и
фармацевтически приемлемое вспомогательное вещество.
17. Применение соединения по любому из пп. 1-15 или его фармацевтически приемлемой соли для лечения KIT- и PDGFRα-опосредованного заболевания или состояния у пациента, нуждающегося в этом, где указанное заболевание или состояние выбрано из системного мастоцитоза, желудочно-кишечных стромальных опухолей, острого миелоидного лейкоза, меланомы, семиномы, внутричерепных герминогенных опухолей, медиастинальной В-клеточной лимфомы, саркомы Юинга, диффузной В-крупноклеточной-лимфомы, дисгерминомы, миелодиспластического синдрома, назальной NK/T-клеточной лимфомы, хронического миеломоноцитарного лейкоза и рака головного мозга.
18. Применение по п. 17, отличающееся тем, что указанное заболевание или состояние представляет собой системный мастоцитоз.
19. Применение по п. 18, отличающееся тем, что системный мастоцитоз выбран из вялотекущего системного мастоцитоза и тлеющего системного мастоцитоза.
20. Применение по п. 18, отличающееся тем, что системный мастоцитоз выбран из агрессивного системного мастоцитоза, системного мастоцитоза с заболеванием нетучных клеток и лейкоза тучных клеток.
| WO 2015057873 A1, 23.04.2015 | |||
| WO 2018183712 A1, 04.10.2018 | |||
| WO 2005117909 A2, 15.12.2005 | |||
| WO 2012027495 A1, 01.03.2012 | |||
| WO 2007065100 A1, 07.06.2007 | |||
| ПИРРОЛТРИАЗИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2000 |
|
RU2331640C2 |

